Attached files
Use these links to rapidly review the document
TABLE OF CONTENTS
GTx, Inc.
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
þ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF
THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2016
OR
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF
THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission file number 000-50549
GTx, Inc.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
62-1715807 (I.R.S. Employer Identification No.) |
|
| 175 Toyota Plaza 7th Floor Memphis, Tennessee (Address of principal executive offices) |
38103 (Zip Code) |
(901) 523-9700
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.001 per share | The NASDAQ Stock Market, LLC | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act:
| Large accelerated filer o | Accelerated filer o | |
| Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company ý | |
| Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý | ||
The aggregate market value of common stock held by non-affiliates of the registrant based on the closing sales price of the registrant's common stock on June 30, 2016 as reported on The NASDAQ Capital Market was $32,944,890.
There were 16,041,923 shares of registrant's common stock issued and outstanding as of March 17, 2017.
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant's definitive proxy statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, in connection with the Registrant's 2017 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements. The forward-looking statements are contained principally in the sections entitled "Risk Factors," "Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Business." These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements include statements about:
- •
- the implementation of our business strategies, including our ability to preserve or realize any significant value from our selective androgen
receptor modulator, or SARM, and selective androgen receptor degrader, or SARD, programs;
- •
- the therapeutic and commercial potential of, and our ability to advance the development of SARMs and our SARD program;
- •
- the timing, scope and anticipated initiation, enrollment and completion of our ongoing clinical trials and any other future clinical trials
that we may conduct;
- •
- our ability to establish and maintain potential new collaborative, partnering or other strategic arrangements for the development and
commercialization of our product candidates;
- •
- the anticipated progress of our preclinical and clinical programs, including whether our ongoing clinical trials will achieve clinically
relevant results;
- •
- the timing of regulatory discussions and submissions, and the anticipated timing, scope and outcome of related regulatory actions or guidance;
- •
- our ability to obtain and maintain regulatory approvals of our product candidates and any related restrictions, limitations, and/or warnings in
the label of an approved product candidate;
- •
- our ability to market, commercialize and achieve market acceptance for our product candidates;
- •
- our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others;
and
- •
- our estimates regarding the sufficiency of our cash resources, expenses, capital requirements and needs for additional financing, and our ability to obtain additional financing.
In some cases, you can identify forward-looking statements by terms such as "anticipates," "believes," "could," "estimates," "expects," "intends," "may," "plans," "potential," "predicts," "projects," "should," "will," "would," and similar expressions intended to identify forward-looking statements. Forward-looking statements reflect our current views with respect to future events, are based on assumptions, and are subject to risks, uncertainties and other important factors. We discuss many of these risks in this Annual Report on Form 10-K in greater detail in the section entitled "Risk Factors" under Part I, Item 1A below. Given these risks, uncertainties and other important factors, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our estimates and assumptions only as of the date of this Annual Report on Form 10-K. You should read this Annual Report on Form 10-K and the documents that we incorporate by reference in and have filed as exhibits to this Annual Report on Form 10-K, completely and with the understanding that our actual future results may be materially different from what we expect.
Except as required by law, we assume no obligation to update any forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in any forward-looking statements, even if new information becomes available in the future.
1
GTx, Inc., a Delaware corporation incorporated on September 24, 1997 and headquartered in Memphis, Tennessee, is a biopharmaceutical company dedicated to the discovery, development and commercialization of small molecules for the treatment of cancer, including treatments for breast and prostate cancer, and other serious medical conditions. Our current strategy is focused on the further development of selective androgen receptor modulators, or SARMs, a class of drugs that we believe have the potential to be used as a hormonal therapy for the treatment of advanced breast cancer, as well as the potential to treat other serious medical conditions where unmet medical needs in muscle-related diseases may benefit from increasing muscle mass, such as stress urinary incontinence, or SUI, and Duchenne muscular dystrophy, or DMD. In 2015, we entered into an exclusive worldwide license agreement with the University of Tennessee Research Foundation, or UTRF, to develop its proprietary selective androgen receptor degrader, or SARD, technology, which we believe has the potential to provide compounds that can degrade multiple forms of androgen receptor, or AR, by inhibiting tumor growth in patients with progressive castration-resistant prostate cancer, or CRPC, including those patients who do not respond or are resistant to current therapies.
Our lead SARM candidate, enobosarm (GTx-024), has to date been evaluated in 24 completed or ongoing clinical trials, including in six Phase 2 and two Phase 3 clinical trials, enrolling over 1,700 subjects, of which approximately 1,200 subjects were treated with enobosarm. Enobosarm is the generic name given to the compound by the USAN Council and the World Health Organization and is the first compound to receive the SARM stem in its name, recognizing enobosarm as the first in this new class of compounds. We announced in 2014 positive results from a Phase 2 proof-of-concept, open-label clinical trial evaluating a 9 mg oral daily dose of enobosarm for the treatment of patients with estrogen receptor, or ER, positive and AR positive metastatic breast cancer who have previously responded to hormonal therapy. During the second half of 2015, we commenced enrollment in both a Phase 2 clinical trial designed to evaluate the efficacy and safety of a 9 mg and 18 mg dose of enobosarm in patients whose advanced breast cancer is both ER positive and AR positive and a Phase 2 proof-of-concept clinical trial designed to evaluate the efficacy and safety of an 18 mg dose of enobosarm in patients with advanced AR positive triple-negative breast cancer, or TNBC. Both of these clinical trials are being conducted utilizing a Simon's two-stage trial design. The Phase 2 clinical trial evaluating enobosarm in patients with ER positive, AR positive advanced breast cancer has completed enrollment of both stages of the clinical trial for both dose cohorts. We announced in November 2016 that enobosarm achieved the pre-specified primary efficacy endpoint in the 9 mg dose cohort. We expect to report top-line clinical results from this clinical trial in the third quarter of 2017. In our trial evaluating enobosarm in patients with advanced AR positive TNBC, we anticipate having sufficient data from the first stage of this trial in the second quarter of 2017 to allow us to make a determination as to whether we will continue the clinical trial and enroll patients into the second stage of this trial. However, due to the slow rate of patient enrollment in this trial, our current capital resources may not be sufficient to enable us to complete the second stage of the TNBC trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone has been met.
We are also evaluating enobosarm and other compounds in our SARM portfolio for indications outside of oncology where unmet medical needs in muscle-related diseases may benefit from increasing muscle mass. In the first quarter of 2016, we initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI. This is the first clinical trial to evaluate a SARM for the treatment of SUI. We currently anticipate obtaining data from this clinical trial in the third
2
quarter of 2017 sufficient to enable us to determine if continued development of enobosarm in SUI is warranted. We have also evaluated several SARM compounds in preclinical models of DMD where a SARM's ability to increase muscle mass may prove beneficial to patients suffering from DMD, which is a rare disease characterized by progressive muscle degeneration and weakness.
With respect to SARDs, we believe this class of assets has the potential to treat prostate cancer, as well as other diseases such as benign prostatic hyperplasia and Kennedy's disease. We envision initially developing SARDs as a potentially novel treatment for men with CRPC, including those who do not respond or are resistant to currently approved therapies. Our evaluation of the SARD program is at an early stage. We are currently implementing an appropriate development program for SARDs and have selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic and toxicology studies, required to support potential initial human clinical trials. While we plan to initiate a first in human clinical trial during the second half of 2017, we will require additional funding to initiate and complete any such clinical trial.
We recently completed a Phase 2 clinical trial evaluating GTx-758 (Capesaris®), an oral nonsteroidal selective ER alpha agonist, as a secondary hormonal therapy in men with metastatic and high risk non-metastatic CRPC. We have determined to discontinue further development of GTx-758 and will not be making any further investments in this program.
We have discussions ongoing with several potential collaboration partners who have expressed interest in our SARM compounds for the treatment of breast cancer, SUI and/or DMD, as well as our SARD technology.
Scientific Background on Estrogen and Androgen Hormones,
Selective Hormone Receptor Modulators, and Selective Androgen Receptor Degraders
Estrogens and androgens are hormones that play critical roles in regulating the reproductive system and contributing to the homeostasis of the muscular, skeletal, cardiovascular, metabolic and central nervous systems.
Testosterone, the predominant androgen, is important for masculine physical characteristics, such as muscle size and strength and bone strength, as well as for mental well-being. Testosterone is converted into a more potent androgen, dihydrotestosterone, or DHT, which acts as the primary androgen in the prostate, sebaceous glands and hair follicles, and may cause unwanted effects like benign prostatic hyperplasia, or BPH, acne and hair loss. In aging men, there typically is a gradual decline in testosterone levels, which contributes to a loss of muscle mass and strength, erectile dysfunction, decreased sexual interest, depression and mood changes.
Estrogens and androgens perform their physiologic functions principally by binding to and activating their respective hormone receptors located in various tissues. Once a hormone binds with its receptor, this activates a series of cellular events that results in the hormone specific tissue effects.
Pharmaceuticals that target estrogen or androgen receptors have been used medically for over 50 years. The drugs that have been used to stimulate androgen receptors are either natural or synthetic hormones, known as anabolic/androgenic steroids. Steroids are generally believed to activate hormone receptors in all tissue types in a non-selective manner resulting in not only beneficial effects but also in unwanted clinical effects. Hair growth, acne and masculinization are also of concern in women who are exposed to exogenous testosterone. The lack of selectivity of testosterone and its conversion to DHT may result in unwanted side effects, such as the potential stimulation of latent into clinical prostate cancer, worsening of BPH, development or worsening of acne, or loss of hair. To date, no orally
3
available testosterone products have been approved for use in the United States. Those testosterone products that are available must be administered by intramuscular injections or by transdermal patches or gels that may not be convenient for patients and, in some cases, can result in inconsistent blood levels of testosterone.
There are also classes of small molecules that are not steroids that can bind to the same hormone receptors. These nonsteroidal small molecules may either stimulate or block hormone receptors depending on the type of tissue in which the receptor is found and the interaction of the small molecule with the receptor. A drug that has the ability to either block or stimulate the hormone receptor in this manner is called a selective hormone receptor modulator. A selective hormone receptor modulator may be able to mimic the beneficial, while minimizing the unwanted, effects of natural or synthetic steroid hormones.
A SARM is a small molecule that binds to and selectively modulates androgen receptors, the primary receptor to which testosterone binds. SARMs may be utilized in place of androgens for various medical conditions while avoiding the unwanted androgenic effects in the prostate in men or skin and hair in men and women. In previous studies, SARMs have been shown to decrease bone breakdown and increase muscle mass. In addition to the potential beneficial effects in muscle and bone, SARMs may provide a therapeutic option for some women with breast cancer. Although no SARMs have been commercialized to date, we believe that SARMs, without the harmful side effects of testosterone or other exogenous anabolic steroid therapies, can potentially be developed to treat a range of medical conditions, including:
- •
- androgen receptor positive breast cancer;
- •
- muscle loss conditions of chronic diseases, such as cancer, AIDS, chronic kidney disease, end-stage renal disease, and neurodegenerative
disorders;
- •
- muscle loss in acute conditions such as trauma, burns, and rehabilitation;
- •
- muscle loss conditions associated with aging, such as frailty and chronic sarcopenia;
- •
- the prevention and/or treatment of osteoporosis;
- •
- disorders of the central nervous system, such as low libido in both men and women;
- •
- low testosterone conditions, such as primary and secondary hypogonadism; and
- •
- disorders of male reproductive functions, such as infertility and erectile dysfunction.
SARDs are a novel class of drugs. The AR is a major driver of prostate tumor cell proliferation, and blocking its activity is a therapeutic target. Despite the use of therapies designed to inhibit the AR pathway in men with advanced prostate cancer, a significant number of men have tumors that do not respond to such therapeutic approaches and/or become resistant to them. This lack of response may be due to the presence of forms of the AR (splice variants and mutated) for which these therapies are not effective.
SARDs are designed to not only bind to androgen receptors, but also induce androgen receptor degradation and ultimately inhibit tumor cell growth. Selective AR degradation which targets the N-terminus may be an effective therapeutic strategy where a variant or mutated AR can be degraded
4
by the SARD. This ability to circumvent common drug resistance in prostate cancer patients may provide an important tool for effective new treatments.
The following table identifies the development phase and status for each of our clinical and preclinical product development programs:
| Product Candidate/ Proposed Indication |
Program |
Development Phase |
Status |
|||
|---|---|---|---|---|---|---|
| Enobosarm | ||||||
| Treatment of women with ER positive/AR positive advanced breast cancer (9 mg and 18 mg) | SARM | Phase 2 | Completed enrollment of a Phase 2 open-label clinical trial evaluating enobosarm in patients whose advanced breast cancer is both ER positive and AR positive. Previously announced clinical benefit was achieved in the 9 mg dose cohort in the ongoing clinical trial. Top-line clinical results for the trial expected in the third quarter of 2017. | |||
Enobosarm |
||||||
| Treatment of women with advanced AR positive TNBC (18 mg) | SARM | Phase 2 | Currently enrolling a Phase 2 open-label proof-of-concept clinical trial evaluating enobosarm in patients with advanced AR positive TNBC. | |||
Enobosarm |
||||||
| Treatment of postmenopausal women with SUI (3 mg) | SARM | Phase 2 | Currently enrolling a Phase 2 proof-of-concept clinical trial evaluating enobosarm in postmenopausal women with SUI with data expected in the third quarter of 2017. | |||
SARMs |
||||||
| Treatment of DMD | SARM | Preclinical | Preclinical research being evaluated by potential collaboration partners for the treatment of DMD. | |||
SARDs |
||||||
| Treatment of castration resistant prostate cancer | SARD | Preclinical | Selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic and toxicology studies, required to support potential initial human clinical trials. | |||
5
Enobosarm for the Potential Treatment of Breast Cancer
The treatment of breast cancer is one of the earliest examples of a targeted approach for cancer therapy. The development of therapeutic agents targeting the ER in breast cancer has served as a model for the development of other targeted therapies in oncology. The treatment for invasive breast cancer is guided, in part, by the characterization of receptor status in the tumor tissue which includes the presence or absence of ER, progesterone receptor, or PR, and human epidermal growth factor receptor 2, or HER2. Studies investigating the prevalence of receptor status in invasive breast cancer have demonstrated that 75-85% of tumors are ER positive and/or PR positive and 15-20% are HER2 positive. If there is a lack of expression of each of these three receptors, the breast cancer is known as TNBC, which is a more aggressive type of breast cancer with a worse prognosis than the receptor positive cancers.
Since the majority of breast cancers are receptor positive, historically, advances in the treatment for breast cancer were focused on targeting the ER through hormonal manipulation with selective ER modulators including ER antagonists, which block the proliferative action of estrogen, and aromatase inhibitors, which decrease the synthesis of estrogen in postmenopausal women. Unfortunately, as effective targeted approaches are not available for the treatment of TNBC, treatment is limited to cytotoxic chemotherapy.
Recent research has focused on identifying new potential therapeutic targets in both hormone receptor positive breast cancers and TNBC for several reasons. In ER positive patients, resistance to endocrine therapies is a clinical and scientific challenge leading researchers to investigate other targets that are linked to the ER function. In TNBC, therapeutic targets need to be identified to potentially improve outcomes for patients with this aggressive form of breast cancer either as first line therapy after chemotherapy or in conjunction with chemotherapy. One such target that has been identified in both ER positive and TNBCs is the AR. In fact, the AR is the most commonly expressed steroid receptor in breast cancer. Literature suggests that up to 90% of ER positive breast cancers and 10-15% of TNBCs express AR. Recent small studies have demonstrated that targeting the AR may be a viable treatment approach for advanced breast cancer.
To date, enobosarm has been evaluated in 24 completed or ongoing clinical trials, including in six Phase 2 and two Phase 3 clinical trials, enrolling over 1,700 subjects, of which approximately 1,200 subjects were treated with enobosarm. In our Phase 2 proof-of-concept clinical trial in patients with ER positive and AR positive metastatic breast cancer, we enrolled 22 postmenopausal women with ER positive metastatic breast cancer who have previously responded to hormonal therapy to assess clinical benefit at six months of enobosarm 9 mg once daily treatment. Clinical benefit was defined as those patients receiving treatment who have demonstrated (i) a complete response (disappearance of all targeted lesions), (ii) a partial response (at least a 30% decrease in the sum of the longest diameters of the targeted lesions), or (iii) stable disease (no disease progression from baseline). The primary endpoint was assessed in 17 AR positive patients, including one patient who had AR status determined outside the protocol specified window of time. Six of these 17 patients demonstrated clinical benefit at six months as stable disease, including the aforementioned patient, exceeding the pre-defined statistical threshold requiring that at least three of 14 patients with an AR positive metastatic lesion demonstrate clinical benefit. Seven patients in total (one patient with indeterminate AR status) achieved clinical benefit at six months as stable disease. The results also demonstrated that, after a median duration on study of 81 days, 41% of all patients (9/22) achieved clinical benefit as best response and also had increased prostate specific antigen, or PSA, which is an indicator of AR activity. No confirmed complete or partial responses were observed in the study, although one patient with liver metastases had a 27% reduction in a target tumor. Enobosarm was well tolerated throughout the clinical trial. The
6
most common adverse events, or AEs, reported were pain, fatigue, nausea, hot flash/night sweats, and arthralgia. The majority of AEs were Grade 1. There were two serious adverse events, or SAEs, reported during the study. One of the SAEs, bone pain of the chest cage, was assessed as possibly related to enobosarm.
Enobosarm for the Potential Treatment of Women Whose
Advanced Breast Cancer is Both ER Positive and AR Positive
Scientific Overview. Prior to the ability to characterize receptor status and the introduction of targeted therapies directed at the ER, it was known that hormonal manipulation through ovarian ablation, along with alterations of pituitary and adrenal function could lead to tumor responses in some patients with breast cancer. Hormonal manipulation with steroidal androgens was also used with success as a first line treatment prior to the introduction of treatment with tamoxifen and also after disease progression following treatment with tamoxifen. However, androgen treatment had limitations due to the virilizing side effects including body and facial hair growth, acne and deepening of voice. Presently, ER targeted therapies are the mainstay of treatment for hormone receptor positive breast cancer with androgens reserved for use after failure of anti-estrogen therapies. However, the virilizing side effects are still a major limitation for patient compliance and acceptance. Based on the historical success of androgens for the treatment of breast cancer along with our preclinical data demonstrating tumor growth inhibition in ER positive breast cancer, we initiated a Phase 2 proof-of-concept clinical trial to evaluate enobosarm in postmenopausal women with ER positive and AR positive metastatic breast cancer in the second quarter of 2013. Based on the positive results from this proof-of-concept clinical trial in 2015, as well as our preclinical data demonstrating tumor growth inhibition with enobosarm in animal models of disease, the extensive experience we have with enobosarm in over 1,200 clinical trial patients, and its favorable safety profile, we initiated an open-label clinical trial of enobosarm in 2015 to demonstrate the effectiveness and safety of enobosarm to treat women whose advanced breast cancer is both ER positive and AR positive.
Potential Market. Breast cancer is the most commonly diagnosed cancer in women with one in eight women developing invasive breast cancer during their lifetime. As of 2016, it was estimated there were more than 2.8 million women with a history of invasive breast cancer living in the United States. In 2017, an estimated 255,000 new cases of breast cancer will be diagnosed in women in the United States with approximately 6% to 8% of these women having metastatic disease at time of diagnosis. As studies investigating the prevalence of receptor status in invasive breast cancer have demonstrated that 75-85% of tumors are ER positive, anti-estrogen therapy has been noted to have the greatest global commercial impact than any other treatment intervention in oncology. However, despite the widespread use and success of ER targeted therapies, there is no cure for metastatic breast cancer and eventually approximately 20-30% of women diagnosed with invasive breast cancer will have a recurrence.
Clinical Trial. In 2015, we commenced enrollment in a Phase 2 clinical trial designed to evaluate the efficacy and safety of enobosarm in patients whose metastatic or locally advanced breast cancer is both ER positive and AR positive. This open-label, multinational clinical trial, which enrolled patients whose cancer has shown prior response to hormonal therapy but has subsequently progressed, is utilizing a Simon's two-stage clinical trial design. Patients receive orally-administered enobosarm (9 mg or 18 mg) daily for up to 24 months. The first stage of evaluation was assessed among the first 18 evaluable patients for each cohort to determine if clinical benefit was achieved at 24 weeks of treatment. Clinical benefit is defined as a complete response, partial response or stable disease as measured by standardized response evaluation criteria. At least 3 of 18 patients per cohort achieved clinical benefit at 24 weeks of treatment, and the trial has proceeded to the second stage of enrollment. In the second stage, if at least 9 of 44 evaluable patients achieve clinical benefit at week 24, the study will have successfully demonstrated its primary endpoint, and those patients achieving clinical benefit at
7
24 weeks of treatment will be able to continue treatment for a total of up to 24 months. The two dose cohorts in the trial are being treated independently for the purpose of assessing efficacy.
In September 2016, we announced that we had achieved clinical benefit for the first stage in the 9 mg cohort and were continuing enrollment into the second stage of the clinical trial for this cohort. In November 2016, we announced that patients treated with enobosarm 9 mg had achieved the pre-specified primary efficacy endpoint in the ongoing Phase 2 clinical trial in women with advanced ER positive AR positive breast cancer. The primary efficacy endpoint for the clinical trial requires that at least nine patients (out of a total of 44 evaluable patients) achieve clinical benefit at 24 weeks of treatment. To date, of the 40 patients in the 9 mg dose cohort whose AR status has been confirmed AR positive, 10 patients have demonstrated clinical benefit at week 24, 23 patients have discontinued either at or prior to week 24, and 7 patients remain on study and have not yet reached week 24. There are another 5 patients who have been enrolled to the 9 mg cohort whose AR status has not yet been confirmed. Of the 10 evaluable patients achieving clinical benefit, 2 had a partial response and 8 had stable disease. The majority of adverse events are grade 1 and 2, and the most common adverse events reported (occurring in >10% of patients) include nausea (31%), fatigue (18%), and arthralgias (13%). Elevations in transaminases (ALT and AST) during enobosarm treatment were mild with the majority being grade 1 or 2.
In November 2016, we also announced that a sufficient number of patients had also achieved clinical benefit in the first stage in the 18 mg cohort for us to continue enrollment into the second stage for that cohort. Enrollment for both of the second stages for the 9 mg and 18 mg dose cohorts was completed in the first quarter of 2017. The trial will continue as planned with a daily dose of either enobosarm 9 mg or 18 mg until 44 evaluable patients in each cohort have completed treatment to better characterize the clinical benefit response, evaluate secondary endpoints and describe the safety profile of the dose levels. We expect to report top-line clinical results from the clinical trial in the third quarter of 2017.
Enobosarm appears to be safe and generally well tolerated. The independent Safety Monitoring Committee established to monitor the safety of our two ongoing breast cancer clinical trials met on December 1, 2016, and recommended that the clinical trials continue as planned.
Enobosarm for the Potential Treatment of Women with Advanced AR Positive TNBC
Scientific Overview. Although the majority of breast cancers are determined to be hormone receptor positive (expressing ER, PR or HER2), up to 20% of women diagnosed with breast cancer will have TNBC which is characterized by a lack of expression of ER, PR or HER2. TNBC occurs more frequently in younger patients (less than 50 years of age) and generally exhibits a more aggressive pattern of progression along with lower survival rates. For those patients with advanced TNBC, standard treatment options are limited to cytotoxic chemotherapy. However, even after an initial response to chemotherapy, the duration of the response may be short and there may be a higher likelihood of visceral metastases, rapidly progressing disease, and inferior survival compared to hormone receptor positive breast cancer. Therefore, there is an emphasis on research focused towards identifying therapeutic targets in TNBC. One such target is the AR. Historically, the AR has been considered to be anti-proliferative and beneficial in hormone receptor positive breast cancers. In TNBC, data from peer-reviewed literature indicates that the presence of the AR and androgen synthesizing enzymes is associated with lower proliferation, lower tumor grade, better overall survival, and more favorable clinical outcomes, as compared to those patients with TNBC not expressing AR. The current literature also suggests that the AR biomarker, PSA, is a favorable prognostic marker in breast cancer. Based on these findings, research is focusing on the AR as a potential therapeutic target. We have studied SARMs in preclinical TNBC cell and animal models. This preclinical data suggests
8
that the growth of TNBC cells expressing AR was inhibited by AR agonists, but not by the AR antagonist bicalutamide, suggesting that using an AR agonist may be a potentially viable approach for the treatment of advanced AR positive TNBC. We believe that this data, coupled with the early clinical success of androgens in breast cancer, supports the clinical evaluation of enobosarm as a potential novel targeted therapy to treat advanced AR positive TNBC.
Potential Market. Breast cancer is the most commonly diagnosed cancer in women with one in eight women developing invasive breast cancer during their lifetime. As of 2016, it was estimated there were more than 2.8 million women with a history of invasive breast cancer living in the United States. In 2017, an estimated 255,000 new cases of breast cancer will be diagnosed in women in the United States with TNBC accounting for up to 20% of these newly diagnosed breast cancers each year with 10-15% of these TNBC patient tumors expressing the AR. To date, treatment of TNBC has been limited to chemotherapy due to the lack of expression of known therapeutic targets on these tumors. Although first line chemotherapy is effective initially for the treatment of TNBC, patients eventually relapse and second line therapies are needed. While this market is smaller than ER positive breast cancer, it is currently underserved and represents an unmet medical need.
Clinical Trial. We commenced enrollment in 2015 in a Phase 2 proof-of-concept clinical trial of enobosarm designed to evaluate the efficacy and safety of enobosarm in patients with advanced AR positive TNBC. This open-label, multinational clinical trial, which also utilizes a Simon's two-stage clinical trial design, is expected to enroll up to approximately 55 patients to obtain 41 evaluable patients, who will be administered an 18 mg oral daily dose of enobosarm, with clinical benefit being assessed at 16 weeks of treatment. There will be two stages of evaluation in the clinical trial, with the first stage assessment occurring following 16 weeks of treatment for the first 21 evaluable patients. If at least 2 of the 21 patients achieve clinical benefit, the trial is designed to enroll the second stage of the study. Clinical benefit is defined as a complete response, partial response or stable disease as measured by standardized response evaluation criteria. We anticipate having sufficient data from the first stage of this trial in the second quarter of 2017 to allow us to make a determination as to whether we will continue the clinical trial and enroll patients into the second stage of this study. However, due to the slow rate of patient enrollment in this trial, our current capital resources may not be sufficient to enable us to complete the second stage of the TNBC trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone has been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all.
Other SARM Clinical or Preclinical Development Programs
SARMs for the Potential Treatment of Postmenopausal Women with Stress Urinary Incontinence
Scientific Overview. SUI is the involuntary leakage of urine during activities such as coughing, laughing, sneezing, exercising or other movements that increase intra-abdominal pressure and thus increase pressure on the bladder. In women, physical changes resulting from pregnancy, childbirth, and menopause often contribute to stress incontinence predominantly through the weakening of the pelvic floor muscles. We view this as a unique opportunity given the enrichment of the pelvic floor muscles with androgen receptors and the demonstrated effects that our SARMs have on building muscle. We have completed a series of preclinical studies to determine the effect of some of our SARMs on pelvic floor muscle mass. These preclinical studies have shown that in ovariectomized mice (a well-accepted model that simulates a postmenopausal condition), there were statistically significant increases in pelvic floor muscle mass, compared to control groups, indicating that SARMs may potentially provide a treatment option for the numerous post-menopausal women suffering from SUI.
9
Potential Market. SUI affects up to 35% of adult women. Currently, there are no orally available, effective treatment options for SUI. Treatment is limited to physical therapy to strengthen the pelvic floor muscles, surgery to help augment or support the pelvic floor muscles, bulking agents injected into the urethra of the bladder and implantable devices which aim to minimize the leakage of urine under stress. Other than physical therapy, each of these other treatment modalities is invasive with risks and complications. There is clearly an unmet medical need for new safe and effective therapies in this space.
Clinical Trial. In the first quarter of 2016, we initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI. This is the first clinical trial to evaluate a SARM for the treatment of SUI. The rationale for evaluating enobosarm as a treatment for SUI in the proof-of-concept trial is supported by preclinical in vivo data demonstrating increases in pelvic floor muscle mass in animal models following treatment with our SARM compounds and safety data from clinical trials testing enobosarm 3 mg and other doses of enobosarm in more than 1,200 subjects. The trial is a single-arm, open-label proof-of-concept Phase 2 clinical trial evaluating the effects of orally administered enobosarm 3 mg in postmenopausal women with SUI. The primary endpoint of the trial is the change in frequency of daily stress urinary incontinence episodes from baseline to week 12. Secondary efficacy endpoints include accepted measurements of voiding, urethral pressure profile and change in pelvic floor muscles as measured by magnetic resonance imaging, or MRI. We currently anticipate obtaining data from this clinical trial in the third quarter of 2017 sufficient to enable us to determine if continued development of enobosarm in SUI is warranted. Continued development of enobosarm in SUI apart from our ongoing Phase 2 proof-of-concept clinical trial will require us to obtain additional funding.
SARMs for the Potential Treatment of Duchenne Muscular Dystrophy
Scientific Overview. We have evaluated several SARM compounds, including enobosarm, in preclinical models of DMD where a SARM's ability to increase muscle mass may prove beneficial to patients suffering from DMD, which is a rare genetic disorder characterized by progressive muscle degeneration and weakness. Symptom onset is in early childhood, usually between the ages of three and five, and the disease primarily affects boys. The DMD gene is the largest known gene in the human genome and, as a result, it is susceptible to mutations. These mutations can be inherited from a boy's mother, but approximately one-third of the mutations are spontaneous. The resulting disease is caused by the production of a dysfunctional, or completely non-functional, protein called dystrophin, which helps keep muscle cells intact. Until recently, boys with DMD did not survive much beyond their teen years, but with advances in cardiac and respiratory care, survival into the early thirties is becoming more common. DMD remains an unmet medical need and the U.S. Food and Drug Administration, or FDA, has recently issued guidance affirming FDA's interest in finding new treatment options for this disease. We believe that a SARM may be a viable therapeutic option for the treatment of DMD, including in combination with therapies that can potentially modify the underlying genetic defect.
Potential Market. The incidence of all the various manifestations of the disease is approximately 1 in 4,000 male births. Promising research is ongoing in the areas of modifying or correcting the genetic defect in DMD with some encouraging results. Other approaches include anti-inflammatory and anti-oxidant therapies, enhancement of utrophin expression and myostatin inhibitors; however, we believe there is still room for continued therapeutic advances.
Preclinical Development. Based on the extensive SARM data from our preclinical and clinical development efforts, we are undertaking preclinical studies and have initiated discussions with experts to better understand the potential of SARMs as a treatment for DMD. Our preclinical studies have continued to confirm beneficial effects from SARMs in mice genetically altered to simulate DMD, compared to control groups. DMD mice were treated with three different SARM compounds, including
10
enobosarm, and each cohort demonstrated increases in body weight, muscle mass, muscle performance (grip strength) and cardiac function compared to control groups. Based on our SARM data from these preclinical efforts, we have initiated discussions with potential collaboration partners to further develop a SARM for the treatment of DMD, and we will otherwise need to obtain additional funding in order to continue developing SARMs for the treatment of DMD.
SARDs for the Potential Treatment of Castration Resistant Prostate Cancer
Scientific Overview. In March 2015, we entered into an exclusive worldwide license agreement with the UTRF to develop SARD compounds that may be capable of degrading multiple forms of the AR. We believe SARDs have the potential to treat prostate cancer, as well as other diseases such as benign prostatic hyperplasia and Kennedy's disease. We envision initially developing SARDs as a potentially novel treatment for men with CRPC, including those who do not respond or are resistant to currently approved therapies. Although current therapies have improved overall survival in men with CRPC, approximately one-third of the CRPC patients do not respond to these therapies, due in part to the presence of splice variants, including AR-V7, as well as mutations in the androgen receptor. Splice variants of the androgen receptor have been identified in which the ligand binding domain, the binding site for androgens and necessary for the action of many of the current therapies, is lost. In addition, most patients who initially respond to available treatments eventually progress due to the emergence of resistance to these therapies. It is believed that CRPC growth remains highly dependent on androgen receptor activity, although the mechanisms which underlie this resistance are not fully understood. We believe a therapeutic agent that would safely degrade multiple forms of the androgen receptor, including those without the ligand binding domain, would be uniquely positioned to address this patient population.
Potential Market. In the United States alone, we believe there are approximately 80,000 men who have developed resistance to luteinizing hormone-releasing hormone, or LHRH, therapies and therefore have CRPC but who have not received chemotherapy. We believe there are approximately 36,000 men diagnosed each year with metastatic hormone sensitive prostate cancer. Zytiga® and XTANDI® are currently the only drugs approved for the treatment of metastatic CRPC in patients who have not yet received chemotherapy, although several other drugs are in clinical development for this indication. We believe new hormonal therapies in development, if approved, will be used prior to chemotherapy as physicians and patients look for treatment options capable of delaying cancer progression and possibly prolonging survival prior to chemotherapy.
Preclinical Development. Our evaluation of the SARD program is at an early stage. We are currently implementing an appropriate development program for SARDs and have selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic and toxicology studies, required to support potential initial human clinical trials. While we plan to initiate a first in human clinical trial during the second half of 2017, we will require additional funding to initiate and complete any such clinical trial.
Our objective is to discover, develop and commercialize small molecules for the treatment of cancer, including treatments for prostate and breast cancer, and other serious medical conditions. Key elements of our strategy to achieve these objectives are to:
Pursue Clinical Development of Enobosarm in Advanced Breast Cancer. Our current strategy is focused on further development of enobosarm, our lead product candidate, in two breast cancer indications targeting the androgen receptor. During the second half of 2015, we commenced enrollment
11
in two Phase 2 clinical trials of enobosarm. One trial is evaluating the efficacy and safety of enobosarm 9 mg and 18 mg doses in patients with ER positive, AR positive advanced breast cancer. Enrollment was complete for both dose cohorts in the first quarter of 2017. We have previously announced enobosarm achieved clinical benefit in the 9 mg dose cohort. We expect to report top-line clinical results from this clinical trial in the third quarter of 2017. The other Phase 2 clinical trial is evaluating the efficacy and safety of enobosarm 18 mg in patients with advanced AR positive TNBC. We anticipate having sufficient data from the first stage of this trial in the second quarter of 2017 to allow us to make a determination as to whether we will continue the clinical trial and enroll patients into the second stage of this study. However, due to the slow rate of patient enrollment in this trial, our current capital resources may not be sufficient to enable us to complete the second stage of the TNBC trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone has been met.
Pursue Development of SARMs for SUI. We are evaluating enobosarm for the treatment of SUI in a Phase 2 proof-of-concept clinical trial, which was initiated in the first quarter of 2016. We currently anticipate obtaining data from this clinical trial in the third quarter of 2017 to enable us to determine if continued development of enobosarm in SUI is warranted. Continued development of enobosarm in SUI apart from our ongoing Phase 2 proof-of-concept clinical trial will require us to obtain additional funding.
Pursue Development of SARMs for DMD. We are also evaluating enobosarm for the potential treatment of DMD. Based on our SARM data from these preclinical efforts, we have initiated discussions with potential collaboration partners to further develop a SARM for the treatment of DMD, and we will otherwise need to obtain additional funding in order to continue developing SARMs for the treatment of DMD.
Continue Evaluation of SARD Program. This class of assets is being evaluated as a potentially novel treatment for men with castration-resistant prostate cancer, including those who do not respond or are resistant to currently approved therapies. We are currently implementing an appropriate development program for SARDs and have selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic, and toxicology studies, required to support potential initial human clinical trials. While we plan to initiate a first in human clinical trial during the second half of 2017, we will require additional funding to initiate and complete any such clinical trial.
Pursue Licensing, Partnering or Sale of Certain Assets. Our ability to pursue the continued development of SARMs and our SARD program is contingent upon our ability to obtain additional funding. Accordingly, we are actively seeking additional funding through the licensing, partnering or sale of certain assets to provide us the necessary resources for the development of our preclinical and clinical product candidates.
Licenses and Collaborative Relationships
In addition to our internally-developed and discovered small molecules, we have established and intend to continue to pursue licenses from and collaborative relationships with pharmaceutical companies and academic institutions to further the development and potential commercialization of our product candidates. While we currently have no ongoing collaborations for the development and commercialization of our product candidates, our strategy includes selectively partnering or collaborating with leading pharmaceutical and biotechnology companies to assist us in furthering the development and potential commercialization of our product candidates.
12
In July 2007, we and the University of Tennessee Research Foundation, or UTRF, entered into a consolidated, amended and restated license agreement, or the SARM License Agreement, to consolidate and replace our two previously existing SARM license agreements with UTRF and to modify and expand certain rights and obligations of each of the parties under both license agreements. Pursuant to the SARM License Agreement, we were granted exclusive worldwide rights in all existing SARM technologies owned or controlled by UTRF, including enobosarm, and certain improvements thereto, and exclusive rights to certain future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University. Unless terminated earlier, the term of the SARM License Agreement will continue, on a country-by-country basis, for the longer of 20 years or until the expiration of the last valid claim of any licensed patent in the particular country in which a licensed product is being sold. UTRF may terminate the SARM License Agreement for our uncured breach or upon our bankruptcy.
Under the SARM License Agreement, we paid UTRF a one-time, upfront fee of $290,000 as consideration for entering into the SARM License Agreement. We are also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and mid-single-digit royalties on sublicense revenues. During the year ended December 31, 2007, we paid UTRF a sublicense royalty of approximately $1.9 million as a result of our previous collaboration with Merck & Co., Inc. We also agreed to pay all expenses to file, prosecute and maintain the patents relating to the licensed SARM technologies, and are obligated to use commercially reasonable efforts to develop and commercialize products based on the licensed SARM technologies. In December 2008, we and UTRF amended the SARM License Agreement, or the SARM License Amendment, to, among other things, clarify the treatment of certain payments that we may receive from our current and future sublicensees for purposes of determining sublicense fees payable to UTRF, including the treatment of payments made to us in exchange for the sale of our securities in connection with sublicensing arrangements. In consideration for the execution of the SARM License Amendment, we paid UTRF $494,000.
We and UTRF also entered into a license agreement, or the SARD License Agreement, in March 2015 pursuant to which we were granted exclusive worldwide rights in all existing SARD technologies owned or controlled by UTRF, including all improvements thereto. Under the SARD License Agreement, we are obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. We are also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed. Unless terminated earlier, the term of the SARD License Agreement will continue, on a country-by-country basis, until the expiration of the last valid claim of any licensed patent in the particular country in which a licensed patent is granted. UTRF may terminate the SARM License Agreement for our uncured breach or upon our bankruptcy.
We do not currently own or operate manufacturing facilities, and we rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates.
There are no complicated chemistries or unusual equipment required in the manufacturing process for either enobosarm or SARDs. The active ingredient in enobosarm is manufactured using a five-step synthetic process that uses commercially available starting materials for each step. Enobosarm drug
13
product is manufactured using conventional manufacturing technology without the use of novel excipients. We rely on third-party vendors for drug substance and drug product manufacturing, including drug substance for SARDs used in our preclinical studies.
The biotechnology and biopharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunities will be reduced or eliminated if our competitors develop and commercialize similar products that are safer, more effective, have fewer side effects or are less expensive than any products that we and/or our collaborators may develop.
There are other SARM product candidates in development that may compete with enobosarm and any future SARM product candidates, if approved for commercial sale. We are developing enobosarm for the treatment of patients with advanced AR positive breast cancer. To our knowledge, no other SARMs are currently in development for the treatment of advanced AR positive breast cancer; however, other companies with SARMs in development for muscle wasting and cachexia could enter into a breast cancer program in the future. For example, Radius Health, Inc. has stated that it may test its SARM compound, RAD140, in a breast cancer indication in the future. A number of other compounds targeting the androgen axis in breast cancer could compete with enobosarm if one or more are approved for commercial sale in the indications for which enobosarm is being developed. These compounds fall into two categories, androgen synthesis inhibitors, or ASIs, and androgen receptor antagonists, or ARAs. ASIs in development include orteronel being developed by Takeda Pharmaceuticals. ARAs in development include XTANDI® (enzalutamide) being developed by Medivation, Inc., which was recently acquired by Pfizer Inc., and Astellas Pharma, Inc., VT 464 being developed by Innocrin Pharmaceuticals Inc., and generic bicalutamide. Agents targeting pathways outside of the androgen axis also may compete with enobosarm in breast cancer as they are directed towards similar patient populations that may benefit from enobosarm.
Enobosarm for the Potential Treatment of Women Whose
Advanced Breast Cancer is Both ER Positive and AR Positive
In ER positive breast cancer, a number of targeted therapies are approved and/or are being developed to be used in combination with other hormonal agents. These therapies include CDK 4/6 inhibitors (palbociclib being developed by Pfizer has recently been approved by FDA, and ribociclib (Novartis) and abemaciclib (Eli Lilly and Company) are in Phase III trials), PI3K/AKT inhibitors (BKM120 and BYL719 being developed by Novartis, Taselisib being developed by Roche), and mTOR inhibitors (Everolimus being developed by Novartis has recently been approved by the FDA). In ER positive breast cancer, new selective estrogen receptor modulators and selective estrogen receptor degraders targeting the estrogen receptor are in development, including GDC-0910 (Roche), RAD 1901 (Radius Pharmaceuticals), and AZD9496 (Astra Zeneca).
14
Enobosarm for the Potential Treatment of Women with Advanced AR Positive TNBC
We are also developing enobosarm for the treatment of women with advanced AR positive TNBC. There are no currently approved therapies for this subset of patients, beyond chemotherapy. However, a number of approaches for the treatment of TNBC are currently under investigation. Agents also targeting the androgen axis include XTANDI® (enzalutamide) being developed by Pfizer and Astellas Pharma, orteronel (TAK-700) being developed by Takeda, VT 464 being developed by Innocrin and CR-1447 being developed by Curadis. Only a subset of the total TNBC population is AR positive; therefore, agents targeting TNBC as a whole may also compete with enobosarm if approved for commercial sale. These agents include: PI3K/AKT inhibitors (BKM120 and BYL719 being developed by Novartis, Taselisib being developed by Roche), IL6/JAK/Stat inhibitors (ruxolitinib being developed by Incyte), mTOR inhibitors (Everolimus being developed by Novartis), EGFR inhibitor (Neratinib being developed by Puma), and PARP inhibitors (Velaparib being developed by AbbVie), PD-1 inhibitors (pembrolizumab) being developed by Merck & Co. and MPDL3280A being developed by Roche.
SARMs for the Potential Treatment of Postmenopausal Women with Stress Urinary Incontinence
We initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI. There are a variety of treatments that may be used for SUI in women; however, currently, there are no available oral agents approved for the treatment of SUI. Behavioral modification and pelvic floor physical therapy are common initial treatment approaches. Bulking agents, including carbon coated beads (Durasphere® marketed by Coloplast Corp), calcium hydroxlapatite (Coaptite® marketed by BioForm Medical, Inc.) and silicon (Macroplastique® marketed by Cogentix Medical), can be injected into or around the urethra for treating intrinsic sphincter deficiency, a cause of SUI symptoms. Biologic bulking agents including patient-derived adipose stem cells and autologous muscle-derived stem cells (Cook Myosite) are being developed. Recently, an over-the-counter vaginal pessary (Impressa® marketed by Kimberly-Clark) has been approved for the temporary management of urine leakage in women with SUI. Finally, surgical procedures (e.g. sling; bladder neck suspension) have been demonstrated to be effective in some women.
SARMs for the Potential Treatment of Duchenne Muscular Dystrophy
We are also exploring the potential of SARMs to treat DMD. DMD is a rare genetic disorder which currently has no cure and leads to a progressive weakening of all the muscles in the body. A number of drugs are in various stages of development by pharmaceutical companies to meet the unmet medical need in DMD. These drugs may compete for patient enrollment during the clinical trial phase, should we be able to advance the development of SARMs as a potential treatment of DMD, or commercially if approved. The most advanced development is by those companies who are targeting the genetic mutation with exon skipping or codon blocking therapies including eteplirsen by Sarepta Therapeutics Inc. (which recently received FDA approval) and DS-1541b, by Daiichi Sankyo Co. Marathon Pharmaceuticals LLC recently received FDA approval for a glucocorticoid, deflazacort, which was recently acquired from Marathon by PTC Therapeutics. Santhera Pharmaceuticals has completed a Phase 3 trial with a synthetic analog of coenzyme Q10, idebenone. Eli Lilly and Company completed a Phase 3 trial with tadalafil, a PDE5 inhibitor, although the study did not meet its primary endpoint. Pfizer Inc. is developing its anti-myostatin monoclonal antibody, PF-06252616, and is currently in a Phase 2 trial. Bristol Myers Squibb Company is developing BMS 986089, an anti-myostatin adnectin, and currently has a Phase 2 trial ongoing. Italfarmco S.p.A. has a Phase 2 trial ongoing with givinostat, an HDAC inhibitor. Summit Therapeutics PLC has initiated a Phase 2 trial with ezutromid, an utrophin upregulator. Cardero Therapeutics Inc. is planning a Phase 2 trial with epicatechin, a flavanol. In addition, Akashi Therapeutics is developing two compounds for DMD, one of which is a SARM. Tarix Orphan is developing TXA127, an angiotensin 1-7 peptide. Fibrogen is developing FG-3019, a monoclonal antibody which inhibits connective tissue growth factor. Catabasis Pharmaceuticals Inc. is
15
developing CAT-1004, an NF-KB inhibitor. ReveraGen Biopharma Inc. plans to begin Phase 2 trials in DMD with VPB 15, a novel glucocorticoid. Capricor Therapeutics has initiated a Phase 1/2 trial with CAP 1002, cardiosphere derived cells.
SARDs for the Potential Treatment of CRPC
We have entered into an exclusive worldwide license agreement with UTRF to develop its proprietary SARD technology which we believe has the potential to provide compounds that can degrade multiple forms of AR by inhibiting tumor growth in patients with CRPC, including those patients who do not respond or are resistant to current therapies. Drugs in commercial development having potentially similar approaches to removing the AR by degradation include Arvinas Inc.'s ARV-330, which is a chimera with an AR binding moiety on one end and an E3 ligase recruiting element on the other that is in preclinical development for the treatment of advanced prostate cancer and Androscience Corporation's androgen receptor degrader enhancer, or ARD, which is currently in development for acne and alopecia with the potential for development as a treatment for prostate cancer. Additionally, Essa Pharma Inc. is beginning early studies with EPI-506, an AR antagonist that targets the N-terminal domain of the AR. C4 Therapeutics, Inc. is developing degronimids as means to degrade the AR through the ligand binding domain associated degradation. CellCentric is developing therapies that target the histone methyltransferase enzyme to lower AR levels and Oric Pharmaceuticals is targeting the glucocorticoid receptor as a means to impact men that have CRPC. In addition to this specific potential mechanistic competition, there are various products approved or under clinical development in the broader space of treating men with advanced prostate cancer who have metastatic CRPC which may compete with our proposed initial clinical objective for our SARD compounds. Pfizer and Astellas Pharma market XTANDI® (enzalutamide), an oral androgen receptor antagonist, for the treatment of metastatic CRPC in men previously treated with docetaxel as well as those that have not yet received chemotherapy. Zytiga®, sold by Johnson & Johnson, has been approved for the treatment of metastatic CRPC. Similarly, Johnson & Johnson acquired Aragon Pharmaceuticals, Inc., which developed a second generation anti-androgen apalutamide (ARN-509) that is currently being evaluated in Phase 3 studies in men with progressive, advanced prostate cancer. Bayer HealthCare and Orion Corporation are currently performing a Phase 3 study of darolutamide (ODM-201) in men with CRPC without metastases and with a rising PSA examining safety and efficacy by measuring metastatic free survival. In addition to targeting the androgen receptor, therapeutic approaches are being developed to target the progesterone receptor in these patients by Arno Therapeutics Inc.
We will be able to protect our technology from unauthorized use by third parties only to the extent it is covered by valid and enforceable patents or is effectively maintained as trade secrets. Patents and other proprietary rights are an essential element of our business.
For enobosarm and our other SARM compounds, we have an exclusive license from UTRF under its issued patents and pending patent applications in the United States, Canada, Australia, Japan, China and other countries in Asia, before the European Patent Office designating Germany, Great Britain, Spain, France, Italy, and other European Union countries, as well as in certain other countries outside those regions, covering the composition of matter of the active pharmaceutical ingredient for pharmaceutical products, pharmaceutical compositions and methods of synthesizing the active pharmaceutical ingredients. We have also exclusively licensed from UTRF issued and pending patent applications in the United States, Canada, Australia, Japan, China and other countries in Asia, before the European Patent Office designating Germany, Great Britain, Spain, France, Italy and other European Union countries, as well as in certain other countries outside those regions, related to
16
methods for treating muscle wasting disorders, including DMD and cancer cachexia, and for treating sarcopenia and increasing muscle performance, muscle size and muscle strength and increasing the strength of or mass of a bone and for treating bone related disorders, including bone frailty and osteoporosis. Issued patents for enobosarm composition of matter that we licensed from UTRF and issued in the United States expire in 2024. Issued patents for composition of matter for our other SARM compounds in the United States will expire from 2021-2029, depending on the specific SARM compound. The issued patents we licensed from UTRF and issued outside of the United States for enobosarm expire in 2025, and with respect to other SARM compounds, expire in 2023 and 2027, depending on the specific SARM compound. We have pending patent applications for enobosarm and our other SARM compounds that, if issued, would expire in the United States and in countries outside the United States in 2025 and 2027, depending on the specific SARM compound. We have issued patents in the United States, and issued patents and pending applications in countries outside the United States for enobosarm and certain other SARM compounds as a feed composition for animals. The patents in the United States will expire in 2025. Issued patents outside the United States, and patent applications, if issued, which are pending outside the United States, will expire in 2031. Patent applications which are pending in the United States and outside the United States using SARMs for SUI and pelvic floor disorders will expire in 2035, if the patents are issued. Patent applications which are pending in the United States using enobosarm for DMD will expire in 2024, if the patents are issued. Issued patents in the United States, and patent applications, if issued, which are pending in the United States, using other SARMs for DMD will expire in 2027.
We have our own issued patents and pending patent applications in the United States, Canada, Australia, Europe, Japan, China and other countries in Asia, as well as in certain other countries outside those regions, related to solid forms of enobosarm. Issued patents covering solid forms of enobosarm in the United States will expire in 2029. Issued patents and pending patent applications, if issued, in countries outside of the United States will expire in 2028. We have our own pending patent applications in the United States and as an International Application related to methods of treating breast cancer using our SARM compounds. Such patent applications, if issued, would expire in 2033 in the United States and outside of the United States. We have allowed claims in the United States directed to TNBC and AR positive breast cancer.
For our SARD compounds and methods of use thereof, we have filed certain patent applications and are the exclusive licensee of the SARD technology under a license agreement with UTRF executed in 2015. The patent applications will expire between 2036 and 2037, if the patents are issued.
We cannot be certain that any of our pending patent applications, or those of our licensors, will result in issued patents. In addition, because the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions, the patents we own and license, or any further patents we may own or license, may not prevent other companies from developing similar or therapeutically equivalent products. Patents also will not protect our product candidates if competitors devise ways of making or using these product candidates without legally infringing our patents. In recent years, several companies have been extremely aggressive in challenging patents covering pharmaceutical products, and the challenges have often been successful. We cannot be assured that our patents will not be challenged by third parties or that we will be successful in any defense we undertake. Failure to successfully defend a patent challenge could materially and adversely affect our business.
In addition, changes in patent laws, rules or regulations or in their interpretations in the United States and other countries by the courts may materially diminish the value of our intellectual property or narrow the scope of our patent protection, which could have a material adverse effect on our business and financial condition.
17
We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. We seek to protect our proprietary information by requiring our employees, consultants, contractors, outside scientific collaborators and other advisors to execute non-disclosure and confidentiality agreements and our employees to execute assignment of invention agreements to us on commencement of their employment. Agreements with our employees also prevent them from bringing any proprietary rights of third parties to us. We also require confidentiality or material transfer agreements from third parties that receive our confidential data or materials.
New Drug Development and Approval Process
Numerous governmental authorities in the United States and other countries extensively regulate the testing, clinical development, manufacturing and marketing of pharmaceutical products and ongoing research and development activities. In the United States, the FDA rigorously reviews pharmaceutical products under the Federal Food, Drug, and Cosmetic Act and applicable regulations. Non-compliance with FDA regulations can result in administrative and judicial sanctions, including warning or untitled letters, clinical holds, fines, recall or seizure of products, injunctions, total or partial suspension of production, refusal of the government to approve marketing applications or allow entry into supply contracts, refusal to permit import or export of products, civil penalties, criminal prosecution and other actions affecting a company and its products. The FDA also has the authority to revoke previously granted marketing authorizations.
To secure FDA approval, an applicant must submit extensive preclinical and clinical data, as well as information about product manufacturing processes and facilities and other supporting information to the FDA for each indication to establish a product candidate's safety and efficacy. The development and approval process takes many years, requires the expenditure of substantial resources and may be subject to delays or limitations of approval or rejection of an applicant's new drug application, or NDA. Even if the FDA approves a product, the approval is subject to post-marketing surveillance, adverse drug experience and other recordkeeping and reporting obligations, and may involve ongoing requirements for post-marketing studies. The FDA also has authority to place conditions on any approvals that could restrict the commercial applications, advertising, promotion or distribution of these products. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing.
Preclinical and Clinical Testing
Preclinical studies involve laboratory evaluation of product characteristics and animal studies to assess the biological activity and safety of the product. In some cases, long-term preclinical studies are conducted while clinical studies are ongoing. The FDA, under its Good Laboratory Practices regulations, regulates preclinical studies. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring these studies to be replicated. When the preclinical testing is considered adequate by the sponsor to demonstrate the safety and scientific rationale for initial human studies, the results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an Investigational New Drug application, or IND. The IND becomes effective, if not rejected by the FDA, within 30 days after the FDA receives the IND. The FDA may, either during the 30-day period after filing of an IND or at any future time, impose a clinical hold on proposed or ongoing clinical trials on various grounds, including that the study subjects are or would be exposed to an unreasonable and significant health risk. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA.
18
Clinical trials involve the administration of the investigational product candidates to humans under the supervision of a qualified principal investigator. Clinical trials must be conducted in accordance with Good Clinical Practices under protocols submitted to the FDA as part of the IND. In addition, each clinical trial must be approved and conducted under the auspices of an Investigational Review Board, or IRB, and with patient informed consent. The IRB typically considers, among other things, ethical factors and the safety of human subjects.
Clinical trials are conducted in three sequential phases, but the phases may overlap. Phase 1 clinical trials usually involve healthy human subjects. The goal of a Phase I clinical trial is to establish initial data about the safety, tolerability and pharmacokinetic properties of the product candidates in humans. In Phase 2 clinical trials, controlled studies are conducted on an expanded population of patients with the targeted disease. The primary purpose of these tests is to evaluate the initial effectiveness of the drug candidate on the intended target and to determine if there are any side effects or other risks associated with the drug and to determine the optimal dose of the drug from the safety and efficacy profile developed from the clinical study. Phase 3 trials involve even larger patient populations, often with several hundred or even several thousand patients, depending on the use for which the drug is being studied. Phase 3 trials are intended to establish the overall risk-benefit ratio of the drug and provide, if appropriate, an adequate basis for product labeling. During all clinical trials, physicians monitor the patients to determine effectiveness and to observe and report any reactions or other safety risks that may result from use of the drug candidate.
Product Formulation and Manufacture
Concurrent with clinical trials and preclinical studies, companies must develop information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product. In addition, manufacturers, including contract manufacturers, are required to comply with current applicable FDA Good Manufacturing Practice, or cGMP, regulations. The cGMP regulations include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. The manufacturing process must be capable of consistently producing quality batches of the product and the manufacturer must develop methods for testing the quality, purity and potency of the final drugs. Additionally, appropriate packaging must be selected and tested and chemistry stability studies must be conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life.
Compliance with cGMP regulations also is a condition of new drug application approval. The FDA must approve manufacturing facilities before they can be used in the commercial manufacture of drug products. In addition, manufacturing establishments are subject to pre-approval inspections and unannounced periodic inspections.
After the completion of the clinical trial phases of development, if the sponsor concludes that there is substantial evidence that the drug candidate is safe and effective for its intended use, the sponsor may submit a NDA to the FDA. The application must contain all of the information on the drug candidate gathered to that date, including data from the clinical trials, and be accompanied by a user fee.
Under the Prescription Drug User Fee Act, or PDUFA, submission of a NDA with clinical data requires payment of a fee, with some exceptions. In return, the FDA assigns a goal of six or ten months from filing of the application to return of a first "complete response," in which the FDA may approve the product or request additional information. There can be no assurance that an application will be approved within the performance goal timeframe established under PDUFA. The FDA initially
19
determines whether a NDA as submitted is acceptable for filing. The FDA may refuse to file an application, in which case the FDA retains one-half of the user fees. If the submission is accepted for filing, the FDA begins an in-depth review of the application. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation. The FDA is not bound by the recommendation of an advisory committee.
If the FDA evaluations of the NDA and the manufacturing facilities are favorable, the FDA may issue an approval letter authorizing commercial marketing of the drug candidate for specified indications. The FDA could also issue a "complete response" letter at the end of the review period. A "complete response" letter will be issued to let a company know that the review period for a drug is complete and that the application is not yet ready for approval. The letter will describe specific deficiencies and, when possible, will outline recommended actions the applicant might take to get the application ready for approval, including calling for additional clinical trial data.
Marketing Approval and Post-Marketing Obligations
If the FDA approves an application, the drug becomes available for physicians to prescribe. Periodic reports must be submitted to the FDA, including descriptions of any adverse reactions reported. The FDA may require post-marketing studies, also known as Phase IV studies, as a condition of approval. In addition to studies required by the FDA after approval, trials and studies are often conducted to explore new indications for the drug. The purpose of these trials and studies and related publications is to develop data to support additional indications for the drug, which must be approved by the FDA, and to increase its acceptance in the medical community. In addition, some post-marketing studies are done at the request of the FDA to develop additional information regarding the safety of a product.
The FDA may impose risk evaluation mitigation strategies, or REMS, on a product if the FDA believes there is a reason to monitor the safety of the drug in the marketplace. REMS could add training requirements for healthcare professionals, safety communications efforts, and limits on channels of distribution, among other things. The sponsor would be required to evaluate and monitor the various REMS activities and adjust them if need be. Whether a REMS would be imposed on a product and any resulting financial impact is uncertain at this time.
Any products manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including record keeping requirements, reporting of adverse experiences with the drug, drug sampling and distribution requirements, notifying the FDA and gaining its approval of certain manufacturing or labeling changes, complying with certain electronic records and signature requirements, and complying with FDA promotion and advertising requirements. Drug manufacturers and their subcontractors are required to register their establishments and are subject to periodic unannounced inspections for compliance with cGMP requirements. Also, newly discovered or developed safety or effectiveness data may require changes to a product's approved labeling, including the addition of new warnings and contraindications, or even in some instances revocation or withdrawal of the product's approval.
Approval Outside of the United States
In order to market any product outside of the United States, we must comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales and distribution of our products, which broadly reflect the issues addressed by the FDA above. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain
20
approval in other countries might differ from and be longer than that required to obtain FDA approval. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may negatively impact the regulatory process in other countries.
As in the United States, the marketing approval process in Europe and in other countries is a lengthy, challenging and inherently uncertain process. If we fail to comply with applicable foreign regulatory requirements, we may be subject to fines, suspension or withdrawal of marketing approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Generally the development and approval procedures are harmonized throughout the European Union: however, there is limited harmonization in relation to national pricing and reimbursement practices.
Under European Union regulatory systems, a company may not market a medicinal product without marketing authorization. There are three procedures for submitting a MAA in the EU: (1) the mutual recognition procedure (MRP); (2) the decentralized procedure (DCP) and (3) the centralized procedure (CP). The submission strategy for a given product will depend on the nature of the product, the target indication(s), the history of the product, and the marketing plan. The centralized procedure is compulsory for medicinal products which are produced by biotechnology processes, advanced therapy medicinal products and orphan drugs. Besides the products falling under the mandatory scope, the centralized procedure is also open for other innovative products that are new active substances or other medicinal products that constitute a significant therapeutic, scientific or technical innovation.
The centralized procedure leads to approval of the product in all 27 EU member states and in Norway, Iceland and Liechtenstein. Submission of one MAA thus leads to one assessment process and one authorization that allows access to all applicable markets within the entire EU. The process of the centralized procedure is triggered when the applicant sends the letter announcing the intent to submit a MAA (letter of intent). The letter of intent also initiates the assignment of the Rapporteur and Co-Rapporteur, who are the two appointed members of the Committee for Human Medicinal Products, or CHMP, representing two EU member states. However, in light of the United Kingdom's vote in 2016 to leave the European Union, the so-called Brexit vote, there may be changes forthcoming in the scope of the centralized approval procedure as the terms of that exit are negotiated between the UK and the European Union.
When using the MRP or DCP, the applicant must select which and how many EU member states in which to seek approval. In the case of an MRP, the applicant must initially receive national approval in one EU member state. This will be the so-called reference member state (RMS) for the MRP. Then, the applicant seeks approval for the product in other EU member states, the so-called concerned member states (CMS) in a second step: the mutual recognition process. For the DCP, the applicant will approach all chosen member states at the same time. To do so, the applicant will identify the RMS that will assess the submitted MAA and provide the other selected member states with the conclusions and results of the assessment.
When the application for marketing authorization is made, the competent authority responsible for granting a marketing authorization must verify whether the application complies with the relevant requirements, including compliance with the agreed pediatric investigational plan, or PIP. Assuming it does, the marketing authorization may be granted and the relevant results are included in the summary of product characteristics (SmPC) for the product, along with a statement indicating compliance with the agreed PIP. It is not necessary for the product actually to be indicated for use in the pediatric population (for example, if the results show that that would not be appropriate).
21
Drug Price Competition and Patent Term Restoration Act of 1984
Under the Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, a portion of a product's patent term that was lost during clinical development and application review by the FDA may be restored. The Hatch-Waxman Act also provides for a statutory protection, known as exclusivity, against the FDA's acceptance or approval of certain competitor applications. The Hatch-Waxman Act also provides the legal basis for the approval of abbreviated new drug applications, or ANDAs.
Patent term extension can compensate for time lost during product development and the regulatory review process by returning up to five years of patent life for a patent that covers a new product or its use. This period is generally one-half the time between the effective date of an IND and the submission date of a NDA, plus the time between the submission date of a NDA and the approval of that application. Patent term extensions, however, are subject to a maximum extension of five years, and the patent term extension cannot extend the remaining term of a patent beyond a total of 14 years. The application for patent term extension is subject to approval by the United States Patent and Trademark Office in conjunction with the FDA. It generally takes at least six months to obtain approval of the application for patent term extension.
The Hatch-Waxman Act also provides for a period of statutory protection for new drugs that receive NDA approval from the FDA. If a new drug receives NDA approval as a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active entity, then the Hatch-Waxman Act prohibits an ANDA or a NDA submitted pursuant to section 505(b)(2) of the Federal Food, Drug, and Cosmetics Act, where the applicant does not own or have a legal right of reference to all of the data required for approval to be submitted by another company for a generic version of such drug (505(b)(2) NDA), with some exceptions, for a period of five years from the date of approval of the NDA. The statutory protection provided pursuant to the Hatch-Waxman Act will not prevent the filing or approval of a full NDA, as opposed to an ANDA or 505(b)(2) NDA, for any drug, including, for example, a drug with the same active ingredient, dosage form, route of administration, strength and conditions of use. In order to obtain a NDA, however, a competitor would be required to conduct its own clinical trials, and any use of the drug for which marketing approval is sought could not violate another NDA holder's patent claims.
If NDA approval is received for a new drug containing an active ingredient that was previously approved by the FDA but the NDA is for a drug that includes an innovation over the previously approved drug, for example, a NDA approval for a new indication or formulation of the drug with the same active ingredient, and if such NDA approval was dependent upon the submission to the FDA of new clinical investigations, other than bioavailability studies, then the Hatch-Waxman Act prohibits the FDA from making effective the approval of an ANDA or 505(b)(2) NDA for a generic version of such drug for a period of three years from the date of the NDA approval. This three year exclusivity, however, only covers the innovation associated with the NDA to which it attaches. Thus, the three year exclusivity does not prohibit the FDA, with limited exceptions, from approving ANDAs or 505(b)(2) NDAs for drugs containing the same active ingredient but without the new innovation.
While the Hatch-Waxman Act provides certain patent restoration and exclusivity protections to innovator drug manufacturers, it also permits the FDA to approve ANDAs for generic versions of their drugs assuming the approval would not violate another NDA holder's patent claims. The ANDA process permits competitor companies to obtain marketing approval for a drug with the same active ingredient for the same uses but does not require the conduct and submission of clinical studies demonstrating safety and effectiveness for that product. Instead of safety and effectiveness data, an ANDA applicant needs only to submit data demonstrating that its product is bioequivalent to the
22
innovator product as well as relevant chemistry, manufacturing and product data. The Hatch-Waxman Act also instituted a third type of drug application that requires the same information as a NDA, including full reports of clinical and preclinical studies, except that some of the information from the reports required for marketing approval comes from studies which the applicant does not own or have a legal right of reference. This type of application, a 505(b)(2) NDA, permits a manufacturer to obtain marketing approval for a drug without needing to conduct or obtain a right of reference for all of the required studies.
If a competitor submits an ANDA or 505(b)(2) NDA for a compound or use of any compound covered by another NDA holder's patent claims, the Hatch-Waxman Act requires, in some circumstances, the applicant to notify the patent owner and the holder of the approved NDA of the factual and legal basis of the applicant's opinion that the patent is not valid or will not be infringed. Upon receipt of this notice, the patent owner and the NDA holder have 45 days to bring a patent infringement suit in federal district court and obtain a 30-month stay against the company seeking to reference the NDA. The NDA holder could still file a patent suit after the 45 days, but if they miss the 45-day deadline, they would not have the benefit of the 30-month stay. Alternatively, after this 45-day period, the applicant may file a declaratory judgment action, seeking a determination that the patent is invalid or will not be infringed. Depending on the circumstances, however, the applicant may not be able to demonstrate a controversy sufficient to confer jurisdiction on the court. The discovery, trial and appeals process in such suits can take several years. If such a suit is commenced, the Hatch-Waxman Act provides a 30-month stay on the approval of the competitor's ANDA or 505(b)(2) NDA. If the litigation is resolved in favor of the competitor or the challenged patent expires during the 30-month period, unless otherwise extended by court order, the stay is lifted and the FDA may approve the application. Under regulations issued by the FDA, and essentially codified under the Medicare prescription drug legislation, the patent owner and the NDA holder have the opportunity to trigger only a single 30-month stay per ANDA or 505(b)(2) NDA. Once the applicant of the ANDA or 505(b)(2) NDA has notified the patent owner and the NDA holder of the infringement, the applicant cannot be subjected to another 30-month stay, even if the applicant becomes aware of additional patents that may be infringed by its product.
Pharmaceutical Pricing and Reimbursement
We currently have no marketed products. In both domestic and foreign markets, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government authorities or programs, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Within the United States, if we obtain appropriate approval in the future to market any of our oral drug product candidates, those products could potentially be covered by various government health benefit programs as well as purchased by government agencies. The participation in such programs or the sale of products to such agencies is subject to regulation. The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement.
23
Medicaid is a joint federal and state program that is administered by the states for low income and disabled beneficiaries. Under the Medicaid Drug Rebate Program, participating manufacturers are required to pay a rebate for each unit of product reimbursed by the state Medicaid programs. The amount of the rebate for each product is set by law and may be subject to an additional discount if certain pricing increases more than inflation.
Medicare is a federal program that is administered by the federal government that covers individuals age 65 and over as well as those with certain disabilities. Oral drugs may be covered under Medicare Part D. Medicare Part D provides coverage to enrolled Medicare patients for self-administered drugs (i.e., drugs that do not need to be injected or otherwise administered by a physician). Medicare Part D is administered by private prescription drug plans approved by the U.S. government and each drug plan establishes its own Medicare Part D formulary for prescription drug coverage and pricing, which the drug plan may modify from time-to-time. The prescription drug plans negotiate pricing with manufacturers and may condition formulary placement on the availability of manufacturer discounts. Since 2011, manufacturers with marketed brand name drugs have been required to provide a 50% discount on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries reach the coverage gap in their drug benefits.
Drug products are subject to discounted pricing when purchased by federal agencies via the Federal Supply Schedule (FSS). FSS participation is required for a drug product to be covered and reimbursed by certain federal agencies and for coverage under Medicaid, Medicare Part B and the Public Health Service (PHS) pharmaceutical pricing program. FSS pricing is negotiated periodically with the Department of Veterans Affairs. FSS pricing is intended not to exceed the price that a manufacturer charges its most-favored non-federal customer for its product. In addition, prices for drugs purchased by the Veterans Administration, Department of Defense (including drugs purchased by military personnel and dependents through the TRICARE retail pharmacy program), Coast Guard, and PHS are subject to a cap on pricing (known as the "federal ceiling price") and may be subject to an additional discount if pricing increases more than the rate of inflation.
To maintain coverage of drugs under the Medicaid Drug Rebate Program, manufacturers are required to extend discounts to certain purchasers under the PHS pharmaceutical pricing program. Purchasers eligible for discounts include hospitals that serve a disproportionate share of financially needy patients, community health clinics and other entities that receive health services grants from the PHS.
The United States and state governments continue to propose and pass legislation designed to reduce the cost of healthcare. For example, in March 2010, the United States Congress enacted the Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act ("Healthcare Reform Act") which includes changes to the coverage and reimbursement of drug products under government health care programs. Modifications to or repeal of all or certain provisions of the Healthcare Reform Act are expected as a result of the outcome of the recent presidential election and Republicans maintaining control of Congress, consistent with statements made by Donald Trump and members of Congress during the presidential campaign and following the election. We cannot predict the ultimate content, timing or effect of any changes to the Healthcare Reform Act or other federal and state reform efforts. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results.
Although we currently have no products approved for commercial sale, we marketed FARESTON® through September 30, 2012 and the product was covered under various government health benefit programs as well as purchased by federal agencies. We could be subject to liability under federal laws regulating our participation in such programs or the sale of our product to such agencies if we failed to
24
comply with applicable requirements, including reporting prices for our products or offering products for sale at certain prices.
Regulations Pertaining to Sales and Marketing
Although we currently have no products approved for commercial sale, we may be subject to various federal and state laws pertaining to health care "fraud and abuse," including anti-kickback laws and false claims laws for activities related to our previous sales of FARESTON®, which we sold to a third party in 2012, or to future sales of any of our product candidates that may in the future receive regulatory and marketing approval. Anti-kickback laws generally prohibit a prescription drug manufacturer from soliciting, offering, receiving, or paying any remuneration to generate business, including the purchase or prescription of a particular drug. Although the specific provisions of these laws vary, their scope is generally broad and there may not be regulations, guidance or court decisions that apply the laws to particular industry practices. There is therefore a possibility that our practices might be challenged under such anti-kickback laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented, any claims for payment for reimbursed drugs or services to third party payors (including Medicare and Medicaid) that are false or fraudulent. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties, and/or exclusion from federal health care programs (including Medicare and Medicaid).
Laws and regulations have been enacted by the federal government and various states to regulate the sales and marketing practices of pharmaceutical manufacturers with marketed products. The laws and regulations generally limit financial interactions between manufacturers and health care providers and/or require disclosure to the government and public of such interactions. Many of these laws and regulations contain ambiguous requirements or require administrative guidance for implementation. Given the lack of clarity in laws and their implementation, our prior activities (when we marketed FARESTON®) or any future activities (if we obtain approval and/or reimbursement from federal healthcare programs for our product candidates) could be subject to the penalty provisions of the pertinent laws and regulations.
Since our inception in 1997, we have been focused on drug discovery and development programs. Research and development expenses include, but are not limited to, our expenses for personnel associated with our research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. Our research and development expenses were $17.2 million for the year ended December 31, 2016, $13.6 million for the year ended December 31, 2015, and $20.9 million for the year ended December 31, 2014.
As of December 31, 2016, we had 26 employees, 8 of whom were M.D.s, Pharm.D.s and/or Ph.D.s. None of our employees are subject to a collective bargaining agreement. We believe that we have good relations with our employees.
25
We file electronically with the U.S. Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We make available on our Web site at www.gtxinc.com, free of charge, copies of these reports as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Further, copies of these reports are located at the SEC's Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330. The SEC maintains a Web site that contains reports, proxy statements, and other information regarding our filings at www.sec.gov. The information provided on our Web site is not part of this report, and is therefore not incorporated by reference unless such information is otherwise specifically referenced elsewhere in this report.
On December 5, 2016, we effected a one-for-ten reverse stock split of our outstanding common stock, or the Reverse Stock Split. The primary purpose of the Reverse Stock Split was to enable us to regain compliance with the $1.00 minimum bid price requirement for continued listing on The NASDAQ Capital Market, which compliance was regained on December 20, 2016. At the effective time of the Reverse Stock Split, every ten shares of our issued and outstanding common stock was automatically combined and reclassified into one issued and outstanding share of common stock. No fractional shares of our common stock were issued in the Reverse Stock Split, but in lieu thereof, each holder of our common stock who would otherwise have been entitled to a fraction of a share of our common stock in the Reverse Stock Split received a cash payment. In addition, as a result of the Reverse Stock Split, proportionate adjustments were made to the per share exercise price and/or the number of shares issuable upon the exercise or vesting of all stock options, restricted stock units and warrants issued by GTx and outstanding immediately prior to the effective time of the Reverse Stock Split, which resulted in a proportionate decrease in the number of shares of our common stock reserved for issuance upon exercise or vesting of such stock options, restricted stock units and warrants, and, in the case of stock options and warrants, a proportionate increase in the exercise price of all such stock options and warrants. In addition, the number of shares reserved for issuance under our equity compensation plans immediately prior to the effective time of the Reverse Stock Split was reduced proportionately. Unless otherwise noted, all share and per share information included in this report has been retroactively adjusted to give effect to the Reverse Stock Split.
26
The following table sets forth information about our executive officers and other key clinical and regulatory officers as of March 17, 2017.
| Name | Age | Position(s) | ||
|---|---|---|---|---|
Executive Officers |
||||
Marc S. Hanover |
54 | Chief Executive Officer | ||
Robert J. Wills, Ph.D |
63 | Executive Chairman | ||
Henry P. Doggrell |
68 | Vice President, Chief Legal Officer and Secretary | ||
Diane C. Young, M.D |
60 | Vice President, Chief Medical Officer | ||
Jason T. Shackelford |
41 | Vice President, Finance and Accounting, and Principal Financial and Accounting Officer | ||
Other Key Clinical and Regulatory Officers |
||||
Jeffrey G. Hesselberg |
58 | Vice President, Regulatory Affairs | ||
Mary Ann Johnston, PharmD |
45 | Vice President, Clinical Development | ||
Executive Officers of the Registrant
Marc S. Hanover, a co-founder of GTx, served as our President and Chief Operating Officer from our inception in September 1997 until his appointment as our permanent Chief Executive Officer in February 2015, and served as our acting Principal Financial Officer from December 31, 2013 until his appointment as our interim Chief Executive Officer on April 3, 2014. He also previously served as a member of our Board of Directors from September 1997 to August 2011. Prior to joining GTx, Mr. Hanover was a founder of Equity Partners International, Inc., a private equity firm in Memphis, Tennessee, and participated as a founder and investor in three healthcare companies. From 1985 to 1997, Mr. Hanover was a Senior Vice President and a member of the Executive Management Committee of National Bank of Commerce in Memphis, Tennessee. Mr. Hanover holds a B.S. in Biology from the University of Memphis and an MBA in Finance from the University of Memphis.
Robert J. Wills, Ph.D., joined GTx as Executive Chairman of the Board of Directors and as the Chairman of the Board's Scientific and Development Committee on March 2, 2015. Prior to joining GTx, Dr. Wills served as Vice President, Alliance Manager for Johnson & Johnson (J&J) and was responsible for managing strategic alliances for J&J's Pharmaceutical Group worldwide since 2002. Prior to this, Dr. Wills spent 22 years in pharmaceutical drug development, 12 of which were at J&J and 10 of which were at Hoffmann-La Roche Inc. Before assuming his alliance management role at J&J, Dr. Wills served as Senior Vice President Global Development at J&J where he was responsible for its late stage development pipeline and was a member of several internal commercial and research and development operating boards. Since 2015, Dr. Wills has served as the chairman of the board of Cymabay Therapeutics Inc. (NASDAQ: CBAY). Dr. Wills holds a B.S. in Biochemistry and a M.S. in Pharmaceutics from the University of Wisconsin and a Ph.D. in Pharmaceutics from the University of Texas.
Henry P. Doggrell currently serves as our Vice President, Chief Legal Officer and Secretary, after joining GTx in October 2001 as General Counsel and Secretary. From April 1998 to August 2001, Mr. Doggrell was Senior Vice President, Corporate Affairs at Buckeye Technologies, Inc., a specialty cellulose company, where he was responsible for matters including corporate finance, investor relations, mergers and acquisitions, intellectual property and licensing and strategic development. From 1996 to 1998, Mr. Doggrell served as General Counsel and Secretary of Buckeye Technologies. Prior to joining
27
Buckeye Technologies, Mr. Doggrell was a partner of the Baker, Donelson, Bearman, Caldwell and Berkowitz law firm from 1988 to 1996, where he served as a member of the law firm management committee and Chair of the firm's Corporate Securities department. Mr. Doggrell holds a B.S. in Commerce from the University of Virginia and a JD from Vanderbilt University.
Diane C. Young, M.D. was appointed Vice President and Chief Medical Officer at GTx in July 2015. Dr. Young is a board-certified medical oncologist with 25 years of industry experience in clinical development and medical affairs, most recently with Novartis where she spent 12 years in global and regional leadership roles in oncology drug development. Prior to Novartis, Dr. Young spent 10 years with J&J, where she served as Vice President, Global Development at R. W. Johnson Pharmaceutical Research Institute (now Johnson & Johnson Research and Development). At Novartis, Dr. Young held senior leadership positions involved in the development, regulatory approval and medical affairs activities for several products, including Glivec®, Zometa®, Femara®, Sandostatin®, Tasigna®, Jakavi® and Afinitor®, all of which are treatments or supportive therapies for cancer patients.
Jason T. Shackelford currently serves as our Vice President, Finance and Accounting, after joining GTx in July 2007 as Director, Accounting and Corporate Controller, and has served as our principal accounting officer since December 31, 2013 and as our principal financial and accounting officer since April 3, 2014. Prior to joining GTx, Mr. Shackelford was a Senior Audit Manager at KPMG LLP. Mr. Shackelford is a Certified Public Accountant and holds a Bachelor of Business Administration and Master of Accountancy from the University of Mississippi.
Other Key Clinical and Regulatory Officers of the Registrant
Jeffrey G. Hesselberg has served as the Vice President, Regulatory Affairs since May 2007. He joined GTx from ICOS Corporation, where from 1996 to May 2007 he served as Manager, Associate Director, and then Director of Regulatory Affairs. Most recently, Mr. Hesselberg worked on the successful development, launch and commercialization of Cialis® (tadalafil) for the treatment of erectile dysfunction. From 1984 to 1996, Mr. Hesselberg worked for Immunex Corporation and the Puget Sound Blood Center. Mr. Hesselberg holds a B.S. in Molecular Biology from the University of Wisconsin — Madison and a MBA from the University of Washington.
Mary Ann Johnston, PharmD, was appointed Vice President, Medical Affairs in November 2012 and currently serves as Vice President, Clinical Development. Before that, she served as Director, Medical Affairs and Team Leader, Medical Science Liaisons, heading up the field-based medical organization since 2009. Prior to joining GTx, Dr. Johnston was Director, Medical Science Liaisons and Managed Markets at Actelion Pharmaceuticals specializing in pulmonary arterial hypertension. Before joining the pharmaceutical industry, Dr. Johnston practiced as a clinical specialist at the University of Texas Medical Branch in Galveston where she served as an adjunct professor for the University of Houston and University of Texas schools of pharmacy with a clinical practice focused in cardiology and critical care. Dr. Johnston holds a Doctor of Pharmacy degree from Samford University McWhorter School of Pharmacy and completed a postdoctoral residency at the Department of Veterans Affairs Medical Center in Tuscaloosa, Alabama.
We have identified the following additional risks and uncertainties that may have a material adverse effect on our business, financial condition or results of operations. Investors should carefully consider the risks described below before making an investment decision. Our business faces significant risks and the risks described below may not be the only risks we face. Additional risks not presently known to us or that we currently believe are immaterial may also significantly impair our business
28
operations. If any of these risks occur, our business, results of operations or financial condition could suffer, the market price of our common stock could decline and you could lose all or part of your investment in our common stock.
Risks Related to Our Financial Condition and Need for Additional Financing
We have incurred losses since inception, and we anticipate that we will incur continued losses for the foreseeable future.
As of December 31, 2016, we had an accumulated deficit of $531.2 million. Our net loss for the year ended December 31, 2016 was $17.7 million and we expect to incur significant operating losses for the foreseeable future as we continue our preclinical and clinical development activities and potentially seek regulatory approval of our product candidates. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity and working capital.
Our current product candidate, enobosarm (GTx-024), will require significant additional clinical development, financial resources and personnel in order to obtain necessary regulatory approvals for this product candidate and to develop it and our other SARMs into commercially viable products. A substantial portion of our efforts and expenditures were previously devoted to enobosarm 3 mg, which was the subject of our POWER 1 and POWER 2 Phase 3 clinical trials for the prevention and treatment of muscle wasting in patients with advanced non-small cell lung cancer, or NSCLC. The failure of the POWER trials to meet the primary statistical criterion for the co-primary endpoints agreed upon with the U.S. Food and Drug Administration, or FDA, significantly depressed our stock price and has harmed our future prospects. Our current strategy is focused on the further development of enobosarm for the treatment of patients with advanced androgen receptor, or AR, positive breast cancer. However, the development of enobosarm for the treatment of patients with advanced AR positive breast cancer is at a relatively early stage, is subject to the substantial risk of failure inherent in the development of early-stage product candidates, and will require significant additional financial resources and personnel in order for such development to continue. With regard to our remaining programs, our preclinical evaluation of our selective androgen receptor degrader, or SARD, technology, our preclinical evaluation of SARMs as a potential treatment of Duchenne muscular dystrophy, or DMD, and our clinical evaluation of enobosarm for the treatment of postmenopausal women with stress urinary incontinence, or SUI, will in each case require significant additional financial resources and personnel to continue our development of these programs. Because of the numerous risks and uncertainties associated with developing and commercializing small molecule drugs, we are unable to predict the extent of any future losses or when we will become profitable, if at all. In addition, we do not expect to obtain any regulatory approvals to market any of our product candidates, including enobosarm, for the foreseeable future, and it is possible that none of our product candidates will ever receive any regulatory approvals.
We have funded our operations primarily through public offerings and private placements of our securities, as well as payments from our former collaborators. We also previously recognized product revenue from the sale of FARESTON®, the rights to which we sold to a third party in the third quarter of 2012. Currently, we have no ongoing collaborations for the development and commercialization of our product candidates, and as a result of the sale of our rights and certain assets related to FARESTON®, we also currently have no sources of revenue.
If we are unable to raise substantial additional capital in the near term to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter, if we and/or any potential collaborators are unable to develop and commercialize SARMs or SARD technology, if development is further delayed or is eliminated, or if sales revenue from any SARM or
29
SARD products upon receiving marketing approval, if ever, is insufficient, we may never become profitable and we will not be successful.
We need to raise substantial additional capital in the near term and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our development programs and could cause us to discontinue our operations. We cannot be certain that additional capital will be available to us and, if substantial additional capital is not available to us when needed, we may not be able to continue as a going concern which may result in actions that could adversely impact our stockholders.
At December 31, 2016, we had cash, cash equivalents and short-term investments of $21.9 million. Based on our current business plan and assumptions, we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017. Accordingly, we will need to raise substantial additional capital in the near term order to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter. In addition, we have based our cash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding to sustain our operations even sooner than currently anticipated. We believe, based on our current estimates of clinical trial expenditures and enrollment status, that our existing capital resources will be adequate to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with estrogen receptor, or ER, positive and AR positive advanced breast cancer and our ongoing Phase 2 clinical trial of enobosarm in postmenopausal women with SUI. However, our existing capital resources will not be sufficient to allow us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC and we will otherwise need to raise substantial additional capital in order to continue developing enobosarm for any of these indications. If we determine that our existing capital resources are not sufficient to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all. Also, our clinical trials may continue to encounter technical, enrollment or other difficulties that could increase our development costs beyond our current estimates or delay our development timelines, and we could otherwise exhaust our available financial resources sooner than we expect. In any event, we need to raise substantial additional capital in order to:
- •
- potentially enroll the second stage of and complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced
AR positive TNBC;
- •
- undertake any further development of our SARMs beyond our ongoing Phase 2 clinical trials of enobosarm in breast cancer and SUI
and our ongoing preclinical development activities related to the development of SARMs as a potential treatment for DMD;
- •
- initiate and complete human clinical studies of our SARD program; and
- •
- fund our operations and to continue as a going concern.
Our future funding requirements will depend on many factors, including:
- •
- the scope, rate of progress and cost of our preclinical and clinical development programs, including our ongoing and any future clinical trials of enobosarm;
30
- •
- the terms and timing of any potential collaborative, licensing and other strategic arrangements that we may establish;
- •
- the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators, if any;
- •
- future clinical trial results;
- •
- the cost and timing of regulatory filings and/or approvals to commercialize our product candidates and any related restrictions, limitations,
and/or warnings in the label of an approved product candidate;
- •
- the effect of competing technological and market developments; and
- •
- the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
While we have been able to fund our operations to date, we currently have no ongoing collaborations for the development and commercialization of our product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We also do not have any commitments for future external funding. Accordingly, we expect to continue our efforts to seek additional funds through potential collaboration, partnering or other strategic arrangements, through public or private equity offerings or debt financings, or a combination of the foregoing.
In addition, the accompanying financial statements have been prepared assuming that we will continue as a going concern. Accordingly, the accompanying financial statements do not include any adjustments or charges that might be necessary should we be unable to continue as a going concern, such as charges related to impairment of our assets, the recoverability and classification of assets or the amounts and classification of liabilities or other similar adjustments. However, because we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017, there is doubt raised about our ability to continue as a going concern. While we believe that we have the ability to successfully implement plans to mitigate the conditions that may raise doubt about our ability to continue as a going concern within one year after the date of this report, such plans include reducing or delaying expenditures by postponing or discontinuing planned clinical or preclinical development and implementing cost saving measures related to other research and development and general and administrative expenditures, which plans, if implemented, would materially harm our business. In any event, if we are unable to raise additional funds in the near term to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter, we could be required to, among other things, make further reductions in our workforce, eliminate our ongoing AR positive TNBC clinical trial, discontinue further development of enobosarm and/or SARDs, liquidate all or a portion of our assets, and/or seek protection under the provisions of the U.S. Bankruptcy Code, all of which would have a material adverse effect on our business and stock price.
To the extent that we raise additional funds through potential collaboration, partnering or other strategic arrangements, it may be necessary to relinquish rights to some of our technologies or product candidates, or grant licenses on terms that are not favorable to us, any of which could result in the stockholders of GTx having little or no continuing interest in our SARMs and/or SARDs programs as stockholders or otherwise. To the extent we raise additional funds by issuing equity securities, our stockholders may experience significant dilution, particularly given our currently depressed stock price, and debt financing, if available, may involve restrictive covenants. For example, we completed a private
31
placement of common stock and warrants in March 2014, which was substantially dilutive, completed a subsequent private placement in November 2014 that represented additional dilution, and we again raised additional funds by issuing shares of common stock in a registered direct offering in October 2016. Our stockholders may experience additional, perhaps substantial, dilution should we again raise additional funds by issuing equity securities. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. Our ability to raise additional funds and the terms upon which we are able to raise such funds have been severely harmed by the failure of our two prior enobosarm POWER trials to meet the primary statistical criterion for the co-primary endpoints agreed upon with the FDA, and may in the future be adversely impacted by the uncertainty regarding the prospects of our development of enobosarm for the treatment of patients with advanced AR positive breast cancer and our ability to advance the development of enobosarm or SARDs, if at all. Our ability to raise additional funds and the terms upon which we are able to raise such funds may also be adversely affected by the uncertainties regarding our financial condition, the sufficiency of our capital resources, recent and potential future management turnover, and continued volatility and instability in the global financial markets. As a result of these and other factors, we cannot be certain that additional funding will be available on acceptable terms, or at all.
Risks Related to Development of Product Candidates
We are substantially dependent on the success of enobosarm and our failure to advance the development of enobosarm or to obtain regulatory approval of enobosarm would significantly harm our prospects.
Our current strategy is focused on the further development of SARMs. We have two ongoing Phase 2 clinical trials for the treatment of patients with advanced AR positive breast cancer and there continues to be a significant risk of failure inherent in the development of these product candidates. If the current clinical trials are successful, we will still need to conduct costly and time-consuming additional clinical trials of enobosarm for the treatment of patients with advanced AR positive breast cancer to determine whether enobosarm is an effective treatment for patients with advanced AR positive TNBC and ER positive/AR positive advanced breast cancer.
Preclinical studies, including studies of SARMs in animal models of disease, may not accurately predict the results of subsequent human clinical trials of enobosarm, including the results of our ongoing Phase 2 clinical trials of enobosarm in patients with advanced AR positive breast cancer. Furthermore, the positive results from our Phase 2 proof-of-concept clinical trial evaluating enobosarm 9 mg in women whose advanced breast cancer is both ER positive and AR positive does not ensure that our ongoing Phase 2 clinical trials of enobosarm in patients with advanced AR positive breast cancer will be successful or that any later trials will be successful. Likewise, the fact that we achieved clinical benefit in the 9 mg cohort for both the first and second stages of our ongoing Phase 2 clinical trial of enobosarm in patients whose advanced breast cancer is both ER positive and AR positive and achieved the first stage milestone in the 18 mg cohort in this trial does not ensure that either this trial or our ongoing Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC will be successful. A number of companies in the pharmaceutical industry, including us and those with greater resources and experience than we have, have suffered significant setbacks in Phase 3 and later-stage clinical trials, even after receiving encouraging results in earlier clinical trials. Due to the uncertain and time-consuming clinical development and regulatory approval process, we may not be successful in developing enobosarm for the treatment of patients with advanced AR positive breast cancer, or in developing or partnering any of our product candidates, and it is possible that none of our current product candidates will ever become commercial products.
A substantial portion of our efforts and expenditures have been devoted to enobosarm 3 mg, which was the subject of our POWER 1 and POWER 2 Phase 3 clinical trials evaluating enobosarm 3 mg for
32
the prevention and treatment of muscle wasting in patients with advanced NSCLC. We announced in August 2013 that these two Phase 3 clinical trials failed to meet the co-primary endpoints of lean body mass and physical function that were assessed statistically using responder analyses as required by the FDA. The failure of the POWER trials to meet the primary statistical criterion for the co-primary endpoints agreed upon with the FDA significantly depressed our stock price and has harmed our future prospects.
Our evaluation of our SARD program is at an early stage and to initiate and complete initial human clinical trials, we will require additional funding. In addition, our evaluation of SARMs as a potential treatment for SUI and DMD is at an early stage, and our ability to meaningfully advance development of SARMs as a potential treatment for SUI or DMD is subject to our ability to obtain additional funding, either through financing or by entering into new collaborative arrangements or other strategic transactions with third parties for any such further development.
Accordingly, our current strategy and near-term prospects are substantially dependent on the successful development of enobosarm for the treatment of patients with advanced AR positive breast cancer.
We and any potential collaborators will not be able to commercialize our product candidates if our preclinical studies do not produce successful results or if our clinical trials do not adequately demonstrate safety and efficacy in humans.
Significant additional clinical development and financial resources will be required to obtain necessary regulatory approvals for our product candidates and to develop them into commercially viable products. Preclinical and clinical testing is expensive, can take many years to complete and has an uncertain outcome. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. Typically, the failure rate for development candidates is high. If a product candidate fails at any stage of development, we will not have the anticipated revenues from that product candidate to fund our operations, and we will not receive any return on our investment in that product candidate. For example, we announced in August 2013 that our POWER 1 and POWER 2 Phase 3 clinical trials evaluating enobosarm for the prevention and treatment of muscle wasting in patients with advanced NSCLC failed to meet the co-primary endpoints of lean body mass and physical function that were assessed statistically using responder analyses as agreed upon with the FDA.
In addition, in the first quarter of 2015, we entered into an exclusive worldwide license agreement with the University of Tennessee Research Foundation, or UTRF, to develop its proprietary SARD technology. However, our evaluation of the SARD program is at an early stage and it is possible that we may determine not to move forward with any meaningful preclinical development of our SARD program. Even if we do determine to move forward with any meaningful preclinical development of our SARD program, to initiate and complete initial human clinical trials, we will require additional funding. Accordingly, as a result of our unsuccessful research and preclinical development and/or our inability to obtain sufficient funding to meaningfully advance preclinical development of our SARD program, we may fail to realize the anticipated benefits of our licensing of this program.
Significant delays in clinical testing could materially impact our product development costs. We do not know whether our ongoing clinical trials will need to be modified or will be completed on schedule, if at all. For example, our ongoing Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC is being conducted using a Simon's two-stage design, pursuant to which approximately half of the patients are enrolled in the first stage, and, upon achievement of a pre-specified minimal response rate, enrollment of the second stage would proceed. We have not commenced enrollment in
33
the second stage of the Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC. Due to the slow rate of patient enrollment in this trial, our current capital resources may not be sufficient to enable us to complete the second stage of the TNBC trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all. In any event, we or any potential collaborators may experience numerous unforeseen and/or adverse events during, or as a result of, preclinical testing and the clinical trial process that could delay or prevent our or our potential collaborators' ability to commercialize our product candidates, including:
- •
- regulators or institutional review boards may not authorize us or any potential collaborators to commence a clinical trial or conduct a
clinical trial at a prospective trial site, or we may experience substantial delays in obtaining these authorizations;
- •
- preclinical or clinical trials may produce negative or inconclusive results, which may require us or any potential collaborators to conduct
additional preclinical or clinical testing or to abandon projects that we expect to be promising;
- •
- even if preclinical or clinical trial results are positive, the FDA or foreign regulatory authorities could nonetheless require us to conduct
unanticipated additional clinical trials;
- •
- registration or enrollment in clinical trials may be slower than we anticipate, such as the slower than expected rate of enrollment we have
experienced in our ongoing Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, resulting in significant delays, additional costs and/or study terminations;
- •
- we or any potential collaborators may suspend or terminate clinical trials if the participating patients are being exposed to unacceptable
health risks;
- •
- regulators or institutional review boards may suspend or terminate clinical research for various reasons, including noncompliance with
regulatory requirements; and
- •
- our product candidates may not have the desired effects or may include undesirable side effects.
If any of these events were to occur and, as a result, we or any potential collaborators have significant delays in or termination of clinical trials, our costs could increase and our ability to generate revenue could be impaired, which would materially and adversely impact our business, financial condition and growth prospects.
If we or any potential collaborators observe serious or other adverse events during the time our product candidates are in development or after our products are approved and on the market, we or any potential collaborators may be required to perform lengthy additional clinical trials, may be required to cease further development of such product candidates, may be denied regulatory approval of such products, may be forced to change the labeling of such products or may be required to withdraw any such products from the market, any of which would hinder or preclude our ability to generate revenues.
In our Phase 2 clinical trials for enobosarm for the treatment of muscle wasting in patients with cancer and healthy older males and postmenopausal females, we observed mild elevations of hepatic enzymes, which in certain circumstances may lead to liver failure, in a few patients in both the placebo and enobosarm treated groups. Reductions in high-density lipoproteins, or HDL, have also been
34
observed in subjects treated with enobosarm. Lower levels of HDL could lead to increased risk of adverse cardiovascular events. In addition, in our Phase 2 proof-of-concept clinical trial evaluating enobosarm in a 9 mg daily dose for the treatment of patients with ER positive and AR positive metastatic breast cancer, bone pain of the chest cage, a serious adverse event, or SAE, was assessed as possibly related to enobosarm. Although doses up to 30 mg have been evaluated in short duration studies, doses of 9 mg and 18 mg currently being tested in our ongoing Phase 2 clinical trials may increase the risk or incidence of known potential side effects of SARMs, including elevations in hepatic enzymes and further reductions in HDL, in addition to the emergence of side effects that have not been seen to date.
In three Phase 2 clinical trials of GTx-758, we observed venous thromboembolic events (VTEs), or blood clots, in subjects treated with GTx-758 at the doses then being studied in these clinical trials (1000 mg and higher per day) and reported those events to the FDA. There were two deaths in subjects treated with GTx-758 and two deaths in subjects treated with Lupron Depot®. In February 2012, the FDA placed all of our then ongoing clinical studies of GTx-758 on full clinical hold, and we suspended further enrollment into these studies and notified clinical sites to discontinue treatment of subjects with GTx-758. In May 2012, the FDA notified us that it had removed the full clinical hold on GTx-758. In the third quarter of 2012, we initiated a Phase 2 clinical trial to evaluate GTx-758, at doses lower than those which were previously being tested in our discontinued Phase 2 clinical trials, as secondary hormonal therapy in men with metastatic castration-resistant prostate cancer, or CRPC, and in this trial, there was one reported incidence of a VTE and one reported incidence of a myocardial infarction, or MI, in patients enrolled in the 250 mg arm of the trial, resulting in the discontinuation of both patients from active treatment. We have determined to discontinue further development of GTx-758 and we do not expect to receive any return on our investment from this product candidate.
If the incidence of serious or other adverse events related to our product candidates increases in number or severity, if a regulatory authority believes that these or other events constitute an adverse effect caused by the drug, or if other effects are identified during clinical trials that we or any potential collaborators may conduct in the future or after any of our product candidates are approved and marketed:
- •
- we or any potential collaborators may be required to conduct additional preclinical or clinical trials, make changes in the labeling of any
such approved products, reformulate any such products, or implement changes to or obtain new approvals of our contractors' manufacturing facilities;
- •
- regulatory authorities may be unwilling to approve our product candidates or may withdraw approval of our products;
- •
- we may experience a significant drop in the sales of the affected products;
- •
- our reputation in the marketplace may suffer; and
- •
- we may become the target of lawsuits, including class action suits.
Any of these events could prevent approval or harm sales of the affected product candidates or products, or could substantially increase the costs and expenses of commercializing and marketing any such products.
35
Risks Related to Our Dependence on Third Parties
If we do not establish collaborations for our product candidates or otherwise raise substantial additional capital, we will likely need to alter, delay or abandon our development and any commercialization plans.
Our strategy includes selectively partnering or collaborating with leading pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of our product candidates and to provide funding for such activities. We face significant competition in seeking appropriate collaborators, and collaborations are complex and time consuming to negotiate and document. We may not be successful in entering into new collaborations with third parties on acceptable terms, or at all. In addition, we are unable to predict when, if ever, we will enter into any additional collaborative arrangements because of the numerous risks and uncertainties associated with establishing such arrangements. If we are unable to negotiate new collaborations, we may have to curtail the development of a particular product candidate, reduce, delay, or terminate its development or one or more of our other development programs, delay its potential commercialization or reduce the scope of our sales or marketing activities or increase our expenditures and undertake development or commercialization activities at our own expense. For example, we may have to cease further development of our enobosarm program if we are unable to raise sufficient funding for any additional clinical development of enobosarm through new collaborative arrangements or other strategic transactions with third parties or other financing alternatives. In this regard, if we decide to undertake any further development of our SARMs beyond our ongoing clinical trials and preclinical development, we would need to obtain additional funding for such development, either through financing or by entering into new collaborative arrangements or other strategic transactions with third parties for any such further development. Moreover, our ongoing Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC is being conducted using a Simon's two-stage design, pursuant to which approximately half of the patients are enrolled in the first stage, and, upon achievement of a pre-specified minimal response rate, enrollment of the second stage would proceed. We have not commenced enrollment of the second stage of the Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC. Due to the slow rate of patient enrollment in the TNBC trial, our current capital resources may not be sufficient to enable us to complete the second stage of this trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all. Likewise, to initiate and complete initial human clinical trials for our SARD program, we will require additional funding. In addition, our evaluation of SARMs as a potential treatment for SUI and DMD is at an early stage, and our ability to meaningfully advance development of SARMs as a potential treatment for SUI or DMD is subject to our ability to obtain additional funding. There can be no assurances that we will be successful in obtaining additional funding in any event. If we are not able to raise substantial additional capital, either through financing or by entering into new collaborative arrangements or other strategic transactions with third parties for the further development of our product candidates, we will not be able to advance the development of our product candidates or otherwise bring our product candidates to market and generate product revenues.
Any collaborative arrangements that we establish in the future may not be successful or we may otherwise not realize the anticipated benefits from these collaborations. In addition, any future collaborative arrangements may place the development and commercialization of our product candidates outside our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us.
We have in the past established and intend to continue to establish collaborations with third parties to develop and commercialize some of our current and future product candidates, and these collaborations may not be successful or we may otherwise not realize the anticipated benefits from
36
these collaborations. For example, in March 2011, we and Ipsen Biopharm Limited, or Ipsen, mutually agreed to terminate our collaboration for the development and commercialization of our toremifene-based product candidate. As of the date of this report, we have no ongoing collaborations for the development and commercialization of our product candidates. We may not be able to locate third-party collaborators to develop and market our product candidates, and we lack the capital and resources necessary to develop our product candidates alone.
Dependence on collaborative arrangements subjects us to a number of risks, including:
- •
- we may not be able to control the amount and timing of resources that our potential collaborators may devote to our product candidates;
- •
- potential collaborations may experience financial difficulties or changes in business focus;
- •
- we may be required to relinquish important rights such as marketing and distribution rights;
- •
- should a collaborator fail to develop or commercialize one of our compounds or product candidates, we may not receive any future milestone
payments and will not receive any royalties for the compound or product candidate;
- •
- business combinations or significant changes in a collaborator's business strategy may also adversely affect a collaborator's willingness or
ability to complete its obligations under any arrangement;
- •
- under certain circumstances, a collaborator could move forward with a competing product candidate developed either independently or in
collaboration with others, including our competitors; and
- •
- collaborative arrangements are often terminated or allowed to expire, which could delay the development and may increase the cost of developing our product candidates.
If third parties do not manufacture our product candidates in sufficient quantities, in the required timeframe, at an acceptable cost, and with appropriate quality control, clinical development and commercialization of our product candidates would be delayed.
We do not currently own or operate manufacturing facilities, and we rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins, if any, and our ability to develop product candidates and commercialize any product candidates on a timely and competitive basis.
We rely on third-party vendors for the manufacture of SARM and SARD drug substance. If the contract manufacturers that we are currently utilizing to meet our supply needs for enobosarm or any future SARM or SARD product candidates prove incapable or unwilling to continue to meet our supply needs, we could experience a delay in conducting any additional clinical trials of enobosarm or any future SARM or SARD product candidates. We may not be able to maintain or renew our existing or any other third-party manufacturing arrangements on acceptable terms, if at all. If our suppliers fail to meet our requirements for enobosarm or any future product candidates for any reason, we would be required to obtain alternate suppliers. Any inability to obtain alternate suppliers, including an inability to obtain approval from the FDA of an alternate supplier, would delay or prevent the clinical development and commercialization of our product candidates.
37
Use of third-party manufacturers may increase the risk that we will not have adequate supplies of our product candidates.
Reliance on third-party manufacturers entails risks, to which we would not be subject if we manufactured our product candidates ourselves, including:
- •
- reliance on the third party for regulatory compliance and quality assurance;
- •
- the possible breach of the manufacturing agreement by the third party because of factors beyond our control;
- •
- the possible termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or
inconvenient for us; and
- •
- drug product supplies not meeting the requisite requirements for clinical trial use.
If we are not able to obtain adequate supplies of our product candidates, it will be more difficult for us to develop our product candidates and compete effectively. Our product candidates and any products that we and/or our potential collaborators may develop may compete with other product candidates and products for access to manufacturing facilities.
Our present or future manufacturing partners may not be able to comply with FDA-mandated current Good Manufacturing Practice regulations, other FDA regulatory requirements or similar regulatory requirements outside the United States. Failure of our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates.
If third parties on whom we rely do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or successfully commercialize our product candidates.
We do not have the ability to independently conduct clinical trials for our product candidates, and we must rely on third parties, such as contract research organizations, or CROs, medical institutions, clinical investigators and contract laboratories to conduct our clinical trials. In addition, we rely on third parties to assist with our preclinical development of product candidates. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates.
Risks Related to Our Intellectual Property
If we lose our licenses from UTRF, we may be unable to continue our business.
We have licensed intellectual property rights and technology from UTRF used in substantially all of our business. Our license agreements with UTRF, under which we were granted rights to SARM compounds and technologies, including enobosarm, and more recently, to SARD compounds and technology, may be terminated by UTRF if we are in breach of our obligations under, or fail to
38
perform any terms of, the relevant agreement and fail to cure that breach. If one or both of these agreements are terminated, then we may lose our rights to utilize the SARM and/or SARD technology and intellectual property covered by those agreements to market, distribute and sell licensed products, which may prevent us from continuing our business and may cause us to cease operations altogether.
If some or all of our or our licensor's patents expire or are invalidated or are found to be unenforceable, or if some or all of our patent applications do not result in issued patents or result in patents with narrow, overbroad, or unenforceable claims, or claims that are not supported in regard to written description or enablement by the specification, or if we are prevented from asserting that the claims of an issued patent cover a product of a third party, we may be subject to competition from third parties with products in the same class of products as our product candidates or products with the same active pharmaceutical ingredients as our product candidates, including in those jurisdictions in which we have no patent protection.
Our commercial success will depend in part on obtaining and maintaining patent and trade secret protection for our product candidates, as well as the methods for treating patients in the product indications using these product candidates. We will be able to protect our product candidates and the methods for treating patients in the product indications using these product candidates from unauthorized use by third parties only to the extent that we or our exclusive licensor owns or controls such valid and enforceable patents or trade secrets.
Even if our product candidates and the methods for treating patients for prescribed indications using these product candidates are covered by valid and enforceable patents and have claims with sufficient scope, disclosure and support in the specification, the patents will provide protection only for a limited amount of time. Our and our licensor's ability to obtain patents can be highly uncertain and involve complex and in some cases unsettled legal issues and factual questions. Furthermore, different countries have different procedures for obtaining patents, and patents issued in different countries provide different degrees of protection against the use of a patented invention by others. Therefore, if the issuance to us or our licensor, in a given country, of a patent covering an invention is not followed by the issuance, in other countries, of patents covering the same invention, or if any judicial interpretation of the validity, enforceability, or scope of the claims in, or the written description or enablement in, a patent issued in one country is not similar to the interpretation given to the corresponding patent issued in another country, our ability to protect our intellectual property in those countries may be limited. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may materially diminish the value of our intellectual property or narrow the scope of our patent protection.
We may be subject to competition from third parties with products in the same class of products as our product candidates or products with the same active pharmaceutical ingredients as our product candidates in those jurisdictions in which we have no patent protection. Even if patents are issued to us or our licensor regarding our product candidates or methods of using them, those patents can be challenged by our competitors who can argue such patents are invalid or unenforceable, lack of utility, lack sufficient written description or enablement, or that the claims of the issued patents should be limited or narrowly construed. Patents also will not protect our product candidates if competitors devise ways of making or using these product candidates without legally infringing our patents. The Federal Food, Drug, and Cosmetic Act and FDA regulations and policies create a regulatory environment that encourages companies to challenge branded drug patents or to create non-infringing versions of a patented product in order to facilitate the approval of abbreviated new drug applications for generic substitutes. These same types of incentives encourage competitors to submit new drug applications that rely on literature and clinical data not prepared for or by the drug sponsor, providing another less burdensome pathway to approval.
39
We also rely on trade secrets to protect our technology, especially where we do not believe that patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our confidential information to competitors, and confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time-consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
If we infringe intellectual property rights of third parties, it may increase our costs or prevent us from being able to commercialize our product candidates.
There is a risk that we are infringing the proprietary rights of third parties because numerous United States and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields that are the focus of our development and manufacturing efforts. Others might have been the first to make the inventions covered by each of our or our licensor's pending patent applications and issued patents and/or might have been the first to file patent applications for these inventions. In addition, because patent applications take many months to publish and patent applications can take many years to issue, there may be currently pending applications, unknown to us or our licensor, which may later result in issued patents that cover the production, manufacture, synthesis, commercialization, formulation or use of our product candidates. In addition, the production, manufacture, synthesis, commercialization, formulation or use of our product candidates may infringe existing patents of which we are not aware. Defending ourselves against third-party claims, including litigation in particular, would be costly and time consuming and would divert management's attention from our business, which could lead to delays in our development or commercialization efforts. If third parties are successful in their claims, we might have to pay substantial damages or take other actions that are adverse to our business.
As a result of intellectual property infringement claims, or to avoid potential claims, we might:
- •
- be prohibited from selling or licensing any product that we and/or any potential collaborators may develop unless the patent holder licenses
the patent to us, which the patent holder is not required to do;
- •
- be required to pay substantial royalties or other amounts, or grant a cross license to our patents to another patent holder; or
- •
- be required to redesign the formulation of a product candidate so that it does not infringe, which may not be possible or could require substantial funds and time.
Risks Related to Regulatory Approval of Our Product Candidates
If we or any potential collaborators are not able to obtain required regulatory approvals, we or such collaborators will not be able to commercialize our product candidates, and our ability to generate revenue will be materially impaired.
Our product candidates and the activities associated with their development and commercialization are subject to comprehensive regulation by the FDA, other regulatory agencies in the United States and by comparable authorities in other countries, including the EMA. Failure to obtain regulatory approval for a product candidate will prevent us or any potential collaborator from commercializing the product candidate. We have not received regulatory approval to market any of our product candidates
40
in any jurisdiction, and we do not expect to obtain FDA, EMA or any other regulatory approvals to market any of our product candidates for the foreseeable future, if at all. The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the product candidates involved.
Changes in the regulatory approval policy during the development period, changes in or the enactment of additional regulations or statutes, or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an application. Even if the FDA or the EMA approves a product candidate, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product, and may impose ongoing requirements for post-approval studies, including additional research and development and clinical trials. Any FDA approval may also impose risk evaluation mitigation strategies, or REMS, on a product if the FDA believes there is a reason to monitor the safety of the drug in the market place. REMS may include requirements for additional training for health care professionals, safety communication efforts and limits on channels of distribution, among other things. The sponsor would be required to evaluate and monitor the various REMS activities and adjust them if need be. The FDA and EMA also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval.
Furthermore, the approval procedure and the time required to obtain approval varies among countries and can involve additional testing beyond that required by the FDA. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions.
The FDA, the EMA and other foreign regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. For example, in October 2009, we received a Complete Response Letter from the FDA regarding our NDA for toremifene 80 mg to reduce fractures in men with prostate cancer on ADT notifying us that the FDA would not approve our NDA as a result of certain clinical deficiencies identified in the Complete Response Letter. We have since discontinued our toremifene 80 mg development program, as well as other toremifene-based products. Although we evaluated the potential submission of a MAA to the EMA seeking marketing approval of enobosarm 3 mg in the EU for the prevention and treatment of muscle wasting in patients with advanced NSCLC, based on input from the MHRA, we determined that the data from the POWER trials was not sufficient to support the filing and approval of a MAA without confirmatory data from another Phase 3 clinical trial of enobosarm 3 mg. As a result of this input, we elected not to submit a MAA in the absence of such confirmatory data. In addition, since data from the two POWER trials failed to meet the primary statistical criterion pre-specified for the co-primary endpoints of lean body mass and physical function, the FDA would not accept a NDA for enobosarm 3 mg for the prevention and treatment of muscle wasting in patients with advanced NSCLC.
Additionally, there can be no assurance that the FDA will determine that the data from our ongoing or potential future clinical trials of enobosarm for the treatment of patients with advanced AR positive breast cancer will be sufficient for approval of these product candidates in any indications. For example, we may observe an unacceptable incidence of adverse events in our ongoing or potential future clinical trials of enobosarm, which could require us to abandon the development of enobosarm.
In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit, or prevent regulatory approval of a product candidate. Even if we submit an application to the FDA, the EMA and other foreign regulatory authorities for marketing approval of a product candidate, it may not result in any marketing approvals.
41
We do not expect to receive regulatory approval for the commercial sale of any of our product candidates that are in development for the foreseeable future, if at all. The inability to obtain approval from the FDA, the EMA and other foreign regulatory authorities for our product candidates would prevent us or any potential collaborators from commercializing these product candidates in the United States, the EU, or other countries. See the section entitled "Business — Government Regulation" under Part 1, Item 1 of this Annual Report on Form 10-K for additional information regarding risks associated with marketing approval, as well as risks related to potential post-approval requirements.
Risks Related to Commercialization
The commercial success of any products that we and/or any potential collaborators may develop will depend upon the market and the degree of market acceptance among physicians, patients, health care payors and the medical community.
Any products that we and/or any potential collaborators may develop, including enobosarm, may not gain market acceptance for its stated indication among physicians, patients, health care payors and the medical community. If these products do not achieve an adequate level of acceptance, we may not generate material product revenues or receive royalties to the extent we currently anticipate, and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
- •
- efficacy and safety results in clinical trials;
- •
- the prevalence and severity of any side effects;
- •
- potential advantages over alternative treatments;
- •
- whether the products we commercialize remain a preferred course of treatment;
- •
- the ability to offer our product candidates for sale at competitive prices;
- •
- relative convenience and ease of administration;
- •
- the strength of marketing and distribution support; and
- •
- sufficient third-party coverage or reimbursement.
If we are unable to establish sales and marketing capabilities or establish and maintain agreements with third parties to market and sell our product candidates, we may be unable to generate product revenue from such candidates.
We have limited experience as a company in the sales, marketing and distribution of pharmaceutical products. In the event one of our product candidates is approved, we will need to establish sales and marketing capabilities or establish and maintain agreements with third parties to market and sell our product candidates. We may be unable to build our own sales and marketing capabilities, and there are risks involved with entering into arrangements with third parties to perform these services, which could delay the commercialization of any of our product candidates if approved for commercial sale. In addition, to the extent that we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenues are likely to be lower than if we market and sell any products that we develop ourselves.
42
If we and/or any potential collaborators are unable to obtain reimbursement or experience a reduction in reimbursement from third-party payors for products we sell, our revenues and prospects for profitability will suffer.
Sales of products developed by us and/or any potential collaborators are dependent on the availability and extent of reimbursement from third-party payors, both governmental and private. Changes in the reimbursement policies of these third-party payors that reduce reimbursements for any products that we and/or any potential collaborators may develop and sell could negatively impact our future operating and financial results.
Medicare coverage and reimbursement of prescription drugs exists under Medicare Part D for oral drug products capable of self-administration by patients. Our oral drug product candidates would likely be covered by Medicare Part D (if covered by Medicare at all). In March 2010, the United States Congress enacted the Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act, or Healthcare Reform Act. This health care reform legislation, among other initiatives, implemented cost containment and other measures that could adversely affect revenues from sales of product candidates, including an increase in drug rebates manufacturers must pay under Medicaid for brand name prescription drugs and extension of these rebates to Medicaid managed care.
The future of the Healthcare Reform Act is currently uncertain. The Trump administration and Republican members of Congress recently introduced a plan to repeal and replace a number of provisions in the Healthcare Reform Act, including, for example, repeal of the individual mandate requiring most individuals to obtain health insurance or pay a tax penalty and significant changes to Medicaid coverage and funding. A repeal of significant portions of the Healthcare Reform Act would likely have a far-reaching effect on healthcare coverage and reimbursement. We cannot, however, predict the ultimate form, success or impact on the profitability of our product candidates of such a repeal and replace legislative initiative.
Economic pressure on state budgets may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for drugs. State Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization for use of drugs where supplemental rebates are not provided. Private health insurers and managed care plans are likely to continue challenging the prices charged for medical products and services, and many of these third-party payors may limit reimbursement for newly-approved health care products. In particular, third-party payors may limit the indications for which they will reimburse patients who use any products that we and/or any potential collaborators may develop or sell. These cost-control initiatives could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.
Similar cost containment initiatives exist in countries outside of the United States, particularly in the countries of the EU, where the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can extend well beyond the receipt of regulatory marketing approval for a product and may require us or any potential collaborators to conduct a clinical trial that compares the cost effectiveness of our product candidates or products to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in our or a potential collaborators' commercialization efforts. Third-party payors are challenging the prices charged for medical products and services, and many third-party payors limit reimbursement for newly-approved health care products. Recently budgetary pressures in many EU countries are also causing governments to consider or implement various cost-containment measures, such as price freezes, increased price cuts and rebates. If budget pressures continue, governments may
43
implement additional cost containment measures. Cost-control initiatives could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.
Another development that could affect the pricing of drugs would be if the Secretary of Health and Human Services allowed drug reimportation into the United States. The Medicare Prescription Drug, Improvement and Modernization Act of 2003 gives discretion to the Secretary of Health and Human Services to allow drug reimportation into the United States under some circumstances from foreign countries, including from countries where the drugs are sold at a lower price than in the United States. If the circumstances were met and the Secretary exercised the discretion to allow for the direct reimportation of drugs, it could decrease the price we or any potential collaborators receive for any products that we and/or any potential collaborators may develop, negatively affecting our revenues and prospects for profitability.
Health care reform measures could hinder or prevent our product candidates' commercial success.
Among policy makers and payors in the United States and elsewhere, there is significant interest in health care reform, as evidenced by the initial enactment of as well as the current proposed repeal and replacement of the Healthcare Reform Act in the United States. Aside from any repeal of the Healthcare Reform Act, federal and state legislatures within the United States and foreign governments will likely continue to consider changes to existing health care legislation. These changes adopted by governments may adversely impact our business by lowering the price of health care products in the United States and elsewhere. For example, there has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and legislative and administrative initiatives at the federal and state levels intended to, among other things, bring more transparency to drug pricing and modify government program reimbursement for drugs. We cannot predict what health care reform initiatives may be adopted in the future. Further federal, state and foreign legislative and regulatory developments are likely, and we expect ongoing initiatives to increase pressure on drug pricing, which could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.
We operate in a highly regulated industry and new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to health care availability, method of delivery or payment for health care products and services, or sales, marketing and pricing practices could negatively impact our business, operations and financial condition.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to our prior commercial sales of FARESTON® and the testing of our product candidates in human clinical trials, and we will face an even greater risk if we commercially sell any product that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
- •
- decreased demand for any product candidates or products;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial participants;
44
- •
- costs to defend the related litigation;
- •
- substantial monetary awards to trial participants or patients;
- •
- loss of revenue; and
- •
- the inability to commercialize any products for which we obtain or hold marketing approvals.
We have product liability insurance that covers our clinical trials and any commercial products up to a $25 million annual aggregate limit. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost, and we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise.
If our competitors are better able to develop and market products than any products that we and/or any potential collaborators may develop, our commercial opportunity will be reduced or eliminated.
We face competition from commercial pharmaceutical and biotechnology enterprises, as well as from academic institutions, government agencies and private and public research institutions. Our commercial opportunities will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than any products that we and/or any potential collaborators may develop. Competition could result in reduced sales and pricing pressure on our product candidates, if approved, which in turn would reduce our ability to generate meaningful revenue and have a negative impact on our results of operations. In addition, significant delays in the development of our product candidates could allow our competitors to bring products to market before us and impair any ability to commercialize our product candidates.
Various products are currently marketed or used off-label for some of the diseases and conditions that we are targeting in our pipeline, and a number of companies are or may be developing new treatments. These product uses, as well as promotional efforts by competitors and/or clinical trial results of competitive products, could significantly diminish any ability to market and sell any products that we and/or any potential collaborators may develop.
With respect to our SARM program, there are other SARM product candidates in development that may compete with enobosarm and any future SARM product candidates, if approved for commercial sale. We are developing enobosarm for the treatment of patients with advanced AR positive breast cancer. To our knowledge, no other SARMs are currently in development for the treatment of advanced AR positive breast cancer; however, other companies with SARMs in development for muscle wasting and cachexia could enter into a breast cancer program in the future. For example, Radius Health, Inc. has stated that it may test its SARM compound, RAD140, in a breast cancer indication in the future. A number of other compounds targeting the androgen axis in breast cancer could compete with enobosarm if one or more are approved for commercial sale in the indications for which enobosarm is being developed. These compounds fall into two categories, androgen synthesis inhibitors, or ASIs, and androgen receptor antagonists, or ARAs. ASIs in development include orteronel being developed by Takeda Pharmaceuticals. ARAs in development include XTANDI® (enzalutamide) being developed by Medivation Inc., which was recently acquired by Pfizer Inc., and Astellas Pharma, Inc., VT 464 being developed by Innocrin Pharmaceuticals Inc., and generic bicalutamide. Agents targeting pathways outside of the androgen axis also may compete with enobosarm in breast cancer as they are directed towards similar patient populations that may benefit from enobosarm. In ER positive breast cancer, a number of targeted therapies are being developed to be used in combination with other hormonal agents. These therapies include CDK 4/6 inhibitors (palbociclib being developed by Pfizer has recently been approved by FDA, and ribociclib (Novartis)
45
and abemaciclib (Lilly) are in Phase III trials), PI3K/AKT inhibitors (BKM120 and BYL719 being developed by Novartis, Taselisib being developed by Roche), and mTOR inhibitors (Everolimus being developed by Novartis (FDA approved)). In ER positive breast cancer, new selective estrogen receptor modulators and selective estrogen receptor degraders targeting the estrogen receptor are in development, including GDC-0910 (Roche), RAD 1901 (Radius Pharmaceuticals), and AZD9496 (Astra Zeneca). Additionally, we initiated a proof of concept study in advanced AR positive TNBC patients for which there are no currently approved therapies, beyond chemotherapy. However, a number of approaches for the treatment of TNBC are currently under investigation. Agents also targeting the androgen axis include XTANDI® (enzalutamide) being developed by Pfizer and Astellas Pharma, orteronel (TAK-700) being developed by Takeda, VT 464 being developed by Innocrin, and CR-1447 being developed by Curadis. Only a subset of the total TNBC population is AR positive; therefore, agents targeting TNBC as a whole may also compete with enobosarm if approved for commercial sale. These agents include: PI3K/AKT inhibitors (BKM120 and BYL719 being developed by Novartis, Taselisib being developed by Roche), IL6/JAK/Stat inhibitors (ruxolitinib being developed by Incyte), mTOR inhibitors (Everolimus being developed by Novartis), EGFR inhibitor (Neratinib being developed by Puma), and PARP inhibitors (Velaparib being developed by AbbVie), PD-1 inhibitors (pembrolizumab) being developed by Merck & Co. and MPDL3280A being developed by Roche.
We initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI. There are a variety of treatments that may be used for SUI in women; however, currently, there are no available oral agents approved for the treatment of SUI. Behavioral modification and pelvic floor physical therapy are common initial treatment approaches. Bulking agents, including carbon coated beads (Durasphere® marketed by Coloplast Corp), calcium hydroxlapatite (Coaptite® marketed by BioForm Medical, Inc.) and silicon (Macroplastique® marketed by Cogentix Medical), can be injected into or around the urethra for treating intrinsic sphincter deficiency, a cause of SUI symptoms. Biologic bulking agents including patient-derived adipose stem cells and autologous muscle-derived stem cells (Cook Myosite) are being developed. Recently, an over-the-counter vaginal pessary (Impressa® marketed by Kimberly-Clark) has been approved for the temporary management of urine leakage in women with SUI. Finally, surgical procedures (e.g. sling; bladder neck suspension) have been demonstrated to be effective in some women.
We are also exploring the potential of SARMs to treat DMD. DMD is a rare genetic disorder which currently has no cure and leads to a progressive weakening of all the muscles in the body. A number of drugs are in various stages of development by pharmaceutical companies to meet the unmet medical need in DMD. These drugs may compete for patient enrollment during the clinical trial phase, should we be able to advance the development of SARMs as a potential treatment of DMD, or commercially if approved. The most advanced development is by those companies who are targeting the genetic mutation with exon skipping or codon blocking therapies including eteplirsen by Sarepta Therapeutics Inc. (which recently received FDA approval) and DS-1541b, by Daiichi Sankyo Co. Marathon Pharmaceuticals LLC recently received FDA approval for a glucocorticoid, deflazacort, which was recently acquired from Marathon by PTC Therapeutics. Santhera Pharmaceuticals has completed a Phase 3 trial with a synthetic analog of coenzyme Q10, idebenone. Eli Lilly and Company completed a Phase 3 trial with tadalafil, a PDE5 inhibitor, although the study did not meet its primary endpoint. Pfizer Inc. is developing its anti-myostatin monoclonal antibody, PF-06252616, and is currently in a Phase 2 trial. Bristol Myers Squibb Company is developing BMS 986089, an anti-myostatin adnectin, and currently has a Phase 2 trial ongoing. Italfarmco S.p.A. has a Phase 2 trial ongoing with givinostat, an HDAC inhibitor. Summit Therapeutics PLC has initiated a Phase 2 trial with ezutromid, an utrophin upregulator. Cardero Therapeutics Inc. is planning a Phase 2 trial with epicatechin, a flavanol. In addition, Akashi Therapeutics is developing two compounds for DMD, one of which is a SARM. Tarix Orphan is developing TXA127, an angiotensin 1-7 peptide. Fibrogen is developing FG-3019, a monoclonal antibody which inhibits connective tissue growth factor. Catabasis Pharmaceuticals Inc. is
46
developing CAT-1004, an NF-KB inhibitor. ReveraGen Biopharma Inc. plans to begin Phase 2 trials in DMD with VPB 15, a novel glucocorticoid. Capricor Therapeutics has initiated a Phase 1/2 trial with CAP 1002, cardiosphere derived cells.
We have entered into an exclusive worldwide license agreement with UTRF to develop its proprietary SARD technology which we believe has the potential to provide compounds that can degrade multiple forms of the AR by inhibiting tumor growth in patients with CRPC, including those patients who do not respond or are resistant to current therapies. Drugs in commercial development having potentially similar approaches to removing the AR by degradation include Arvinas Inc.'s ARV-330, which is a chimera with an AR binding moiety on one end and an E3 ligase recruiting element on the other that is in preclinical development for the treatment of advanced prostate cancer and Androscience Corporation's androgen receptor degrader enhancer, or ARD, which is currently in development for acne and alopecia with the potential for development as a treatment for prostate cancer. Additionally, Essa Pharma Inc. is beginning early studies with EPI-506, an AR antagonist that targets the N-terminal domain of the AR. C4 Therapeutics, Inc. is developing degronimids as means to degrade the AR through the ligand binding domain associated degradation. CellCentric is developing therapies that target the histone methyltransferase enzyme to lower AR levels and Oric Pharmaceuticals is targeting the glucocorticoid receptor as a means to impact men that have CRPC. In addition to this specific potential mechanistic competition, there are various products approved or under clinical development in the broader space of treating men with advanced prostate cancer who have metastatic CRPC which may compete with our proposed initial clinical objective for our SARD compounds. Pfizer and Astellas Pharma market XTANDI® (enzalutamide), an oral androgen receptor antagonist, for the treatment of metastatic CRPC in men previously treated with docetaxel as well as those that have not yet received chemotherapy. Zytiga®, sold by Johnson & Johnson, has been approved for the treatment of metastatic CRPC. Similarly, Johnson & Johnson acquired Aragon Pharmaceuticals, Inc., which developed a second generation anti-androgen apalutamide (ARN-509) that is currently being evaluated in Phase 3 studies in men with progressive, advanced prostate cancer. Bayer HealthCare and Orion Corporation are currently performing a Phase 3 study of darolutamide (ODM-201) in men with CRPC without metastases and with a rising PSA examining safety and efficacy by measuring metastatic free survival. In addition to targeting the androgen receptor, therapeutic approaches are being developed to target the progesterone receptor in these patients by Arno Therapeutics Inc.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
Risks Related to Employees, Growth and Other Aspects of Operations
Management transition creates uncertainties and could harm our business.
Over the past few years, we have experienced significant changes in executive leadership, and more could occur. For example, on April 3, 2014, Marc S. Hanover was appointed as our interim Chief Executive Officer and on February 12, 2015, Mr. Hanover was appointed as our permanent Chief Executive Officer. Also, on March 2, 2015, Robert J. Wills was appointed as our Executive Chairman and effective July 13, 2015, Diane C. Young joined us as our Vice President, Chief Medical Officer.
47
Changes to company strategy, which can often times occur with the appointment of new executives, can create uncertainty, may negatively impact our ability to execute quickly and effectively, and may ultimately be unsuccessful. In addition, executive leadership transition periods are often difficult as the new executives gain detailed knowledge of our operations, and friction can result from changes in strategy and management style. Management transition inherently causes some loss of institutional knowledge, which can negatively affect strategy and execution. Until we integrate new personnel, and unless they are able to succeed in their positions, we may be unable to successfully manage and grow our business, and our results of operations and financial condition could suffer as a result. In any event, changes in our organization as a result of executive management transition may have a disruptive impact on our ability to implement our strategy and could have a material adverse effect on our business, financial condition and results of operations.
Our internal computer and information technology systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, or could otherwise face serious disruptions, which could result in a material disruption of our product development efforts.
Despite the implementation of security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, and telecommunication and electrical failures. Such events could cause interruptions of our operations. For instance, the loss of preclinical data or data from our ongoing and potential future clinical trials involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential, proprietary or protected health information, we could incur liability and the development of our product candidates could be delayed. In addition, our information technology and other internal infrastructure systems, including corporate firewalls, servers, leased lines and connection to the Internet, face the risk of systemic failure that could disrupt our operations. A significant disruption in the availability of our information technology and other internal infrastructure systems could cause delays in our research and development work and could otherwise adversely affect our business.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop or commercialize our product candidates.
Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. If we are not able to attract and keep senior management and key scientific personnel, we may not be able to successfully develop or commercialize our product candidates. All of our employees are at-will employees and can terminate their employment at any time.
In October 2013, we announced a reduction of approximately 60% of our workforce following our announcement that our POWER trials failed to achieve the results required by the FDA to file a NDA for enobosarm 3 mg for the prevention and treatment of muscle wasting in patients with advanced NSCLC. In addition, since our October 2013 workforce reduction, our former Chief Executive Officer, former Chief Financial Officer and former Chief Scientific Officer have resigned. Primarily as a result of our October 2013 workforce reduction, only 26 employees remained as employees of GTx as of December 31, 2016. Accordingly, we have been and are operating with a shortage of resources and may not be able to effectively conduct our operations with this limited number of employees. In addition, we announced past workforce reductions in each of December 2009 and June 2011, and our history of implementing workforce reductions, along with the potential for future workforce reductions, may negatively affect our ability to retain or attract talented employees. Further, to the extent we experience
48
additional management transition, competition for top management is high and it may take many months to find a candidate that meets our requirements. If we are unable to attract and retain qualified management personnel, our business could suffer.
If we are able to raise sufficient additional funds necessary to continue as a going concern and to pursue the development of our SARM and SARD programs, we may need to hire additional employees in order to grow our business. Any inability to manage future growth could harm our ability to develop and commercialize our product candidates, increase our costs and adversely impact our ability to compete effectively.
If we are able to raise sufficient additional funds necessary to continue as a going concern and to pursue the development of our SARM and SARD programs, we may need to hire experienced personnel to develop and commercialize our product candidates and to otherwise grow our business, and we may need to expand the number of our managerial, operational, financial and other employees to support that growth. Competition exists for qualified personnel in the biotechnology field. As of December 31, 2016, we had only 26 employees.
Future growth, if any, will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. Our future financial performance and our ability to develop and commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively.
Risks Related to Our Common Stock
The market price of our common stock has been volatile and may continue to be volatile in the future. This volatility may cause our stock price and the value of your investment to decline.
The market prices for securities of biotechnology companies, including ours, have been highly volatile and may continue to be so in the future. In this regard, the closing sale price for our common stock has varied between a high of $9.50 on November 18, 2016 and a low of $4.66 on January 15, 2016 in the twelve-month period ended December 31, 2016 (such prices as adjusted to give effect to the one-for-ten reverse stock split of our outstanding common stock effected on December 5, 2016, or the Reverse Stock Split). The market price of our common stock is likely to continue to be volatile and subject to significant price and volume fluctuations. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
- •
- new or continued delays in the initiation, enrollment and/or completion of our ongoing and any future clinical trials of enobosarm, or
negative, inconclusive or mixed results reported in any of our ongoing and any future clinical trials of enobosarm;
- •
- our ability to raise additional capital to carry through with our preclinical and clinical development plans, including to potentially complete
our ongoing Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, as well as our current and future operations, and the terms of any related financing arrangements;
- •
- reports of unacceptable incidences of adverse events observed in any of our ongoing clinical trials of enobosarm;
- •
- announcements regarding further cost-cutting initiatives or restructurings;
- •
- uncertainties created by our past and potential future management turnover;
49
- •
- our ability to enter into new collaborative, licensing or other strategic arrangements with respect to our product candidates;
- •
- the terms and timing of any future collaborative, licensing or other arrangements that we may establish;
- •
- the timing of achievement of, or failure to achieve, our and any potential collaborators' clinical, regulatory and other milestones, such as
the commencement of clinical development, the completion of a clinical trial or the receipt of regulatory approval;
- •
- announcement of FDA approval or non-approval of our product candidates or delays in or adverse events during the FDA review process;
- •
- actions taken by regulatory agencies with respect to our product candidates or our clinical trials, including regulatory actions requiring or
leading to a delay or stoppage of our ongoing clinical trials;
- •
- the commercial success of any product approved by the FDA or its foreign counterparts;
- •
- introductions or announcements of technological innovations or new products by us, our potential collaborators, or our competitors, and the
timing of these introductions or announcements;
- •
- market conditions for equity investments in general, or the biotechnology or pharmaceutical industries in particular;
- •
- regulatory developments in the United States and foreign countries;
- •
- changes in the structure or reimbursement policies of health care payment systems;
- •
- any intellectual property infringement lawsuit involving us;
- •
- actual or anticipated fluctuations in our results of operations;
- •
- changes in financial estimates or recommendations by securities analysts;
- •
- hedging or arbitrage trading activity that may develop regarding our common stock;
- •
- sales of large blocks of our common stock;
- •
- sales of our common stock by our executive officers, directors and significant stockholders;
- •
- The low trading volume of our common stock;
- •
- changes in accounting principles; and
- •
- additional losses of any of our key scientific or management personnel.
In addition, the stock markets in general, and the markets for biotechnology and pharmaceutical stocks in particular, have experienced significant volatility that has often been unrelated to the operating performance of particular companies. For example, negative publicity regarding drug pricing
50
and price increases by pharmaceutical companies, including as a result of statements on drug pricing by the Trump Administration, has negatively impacted, and may continue to negatively impact, the markets for biotechnology and pharmaceutical stocks. Likewise, as a result of significant changes in U.S. social, political, regulatory and economic conditions or in laws and policies governing foreign trade and health care spending and delivery, including the repeal and/or replacement of all or portions of the Healthcare Reform Act or greater restrictions on free trade stemming from Trump Administration policies, the financial markets could experience significant volatility that could also negatively impact the markets for biotechnology and pharmaceutical stocks. These broad market fluctuations may adversely affect the trading price of our common stock.
In the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Any such litigation brought against us could result in substantial costs, which would hurt our financial condition and results of operations and divert management's attention and resources, which could result in delays of our clinical trials or commercialization efforts.
Our executive officers, directors and largest stockholders have the ability to control all matters submitted to stockholders for approval.
As of December 31, 2016, our executive officers, directors and holders of 5% or more of our outstanding common stock, including their affiliated or associated entities, held approximately 76.8% of our outstanding common stock, and our executive officers and directors alone, including their affiliated or associated entities, held approximately 37.5% of our outstanding common stock as well as warrants to purchase up to an additional 2.5 million shares of common stock. As a result, these stockholders, acting together, have the ability to control all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of this group of stockholders may not always coincide with our interests or the interests of other stockholders.
If we fail to meet continued listing standards of The NASDAQ Stock Market LLC, our common stock may be delisted. Delisting could adversely affect the liquidity of our common stock and the market price of our common stock could decrease, and our ability to obtain sufficient additional capital to fund our operations and to continue as a going concern would be substantially impaired.
Our common stock is currently listed on The NASDAQ Capital Market. The NASDAQ Stock Market LLC, or NASDAQ, has minimum requirements that a company must meet in order to remain listed on The NASDAQ Capital Market. These requirements include maintaining a minimum closing bid price of $1.00 per share, or the Bid Price Requirement, and the closing bid price of our common stock has in the past been well below $1.00 per share. In this regard, on December 23, 2015, we received a letter from the staff, or Staff, of NASDAQ providing notification that, for the previous 30 consecutive business days, the closing bid price for our common stock was below the minimum $1.00 per share requirement for continued listing on The NASDAQ Capital Market, or the Bid Price Requirement. The notification had no immediate effect on the listing of our common stock. In accordance with NASDAQ listing rules, we were afforded 180 calendar days, or until June 20, 2016, to regain compliance with the Bid Price Requirement. On June 21, 2016, we received a letter from the Staff notifying us that we were eligible for an additional 180 calendar day period, or until December 19, 2016, to regain compliance with the minimum $1.00 Bid Price Requirement. In the letter, the Staff noted that our common stock had not regained compliance with the Bid Price Requirement during the initial 180-day compliance period that ended on June 20, 2016 and that we had submitted written notice of our intention to cure the Bid Price Requirement deficiency by effecting a reverse stock split prior to December 19, 2016, if necessary. On December 5, 2016, we effected the Reverse Stock Split, the primary purpose of which was to enable us to regain compliance with the Bid
51
Price Requirement, which compliance was regained on December 20, 2016. However, there can be no assurance that the market price of our common stock will remain in excess of the $1.00 minimum bid price for a sustained period of time. In any event, there can be no assurance that we will continue to meet the Bid Price Requirement, or any other NASDAQ continued listing requirement, in the future. If we fail to meet these requirements, including the Bid Price Requirement and requirements to maintain minimum levels of stockholders' equity or market values of our common stock, NASDAQ may notify us that we have failed to meet the minimum listing requirements and initiate the delisting process. If our common stock is delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease, and our ability to obtain sufficient additional capital to fund our operations and to continue as a going concern would be substantially impaired.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an "ownership change," generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation's ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes (such as research tax credits) to offset its post-change taxable income or taxes may be limited. We completed a study through December 31, 2014 to determine whether any Section 382 limitations exist and, as a result of this study and our analysis of subsequent ownership changes, we do not believe that any Section 382 limitations exist through December 31, 2016. Section 382 of the Internal Revenue Code is an extremely complex provision with respect to which there are many uncertainties and we have not established whether the IRS agrees with our determination. In any event, our recent registered direct offering of our common stock, future equity offerings and/or changes in our stock ownership, some of which are outside of our control, could in the future result in an ownership change and an accompanying Section 382 limitation. If a limitation were to apply, utilization of a portion of our domestic net operating loss and tax credit carryforwards could be limited in future periods and a portion of the carryforwards could expire before being available to reduce future income tax liabilities.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and our bylaws may delay or prevent an acquisition of us or a change in our management. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:
- •
- a classified Board of Directors;
- •
- a prohibition on actions by our stockholders by written consent;
- •
- the ability of our Board of Directors to issue preferred stock without stockholder approval, which could be used to institute a "poison pill"
that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our Board of Directors; and
- •
- limitations on the removal of directors.
52
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns 15% or more of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired 15% or more of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. Finally, these provisions establish advance notice requirements for nominations for election to our Board of Directors or for proposing matters that can be acted upon at stockholder meetings. These provisions would apply even if the offer may be considered beneficial by some stockholders.
If there are substantial sales of our common stock, the market price of our common stock could drop substantially, even if our business is doing well.
For the 12-month period ended December 31, 2016, the average daily trading volume of our common stock on The NASDAQ Capital Market was only 14,829 shares (as adjusted to give effect to the Reverse Stock Split). As a result, future sales of a substantial number of shares of our common stock in the public market, or the perception that such sales may occur, could adversely affect the then-prevailing market price of our common stock. As of December 31, 2016, we had 15,919,572 shares of common stock outstanding. In addition, as a result of the low trading volume of our common stock, which was exacerbated by the Reverse Stock Split, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the market price of our common stock in either direction. The price for our shares could, for example, decline significantly in the event that a large number of our common shares are sold on the market without commensurate demand, as compared to an issuer with a higher trading volume that could better absorb those sales without an adverse impact on its stock price.
In October 2016, we completed a registered direct offering in which we sold 1.7 million shares of our common stock (as adjusted to give effect to the Reverse Stock Split). In November 2014, we completed a private placement of 6.4 million shares of our common stock and warrants to purchase 6.4 million shares of our common stock (as adjusted to give effect to the Reverse Stock Split). Similarly, in March 2014 we completed a private placement of 1.2 million shares of our common stock and warrants to purchase 1.0 million shares of our common stock (as adjusted to give effect to the Reverse Stock Split). Pursuant to the terms of a registration rights agreement we entered into in connection with the March 2014 private placement, we filed a registration statement under the Securities Act registering the resale of the 1.2 million shares of common stock we issued to the investors in the March 2014 private placement, which include J.R. Hyde, III, our largest stockholder, as well as the 1.0 million shares of common stock underlying the warrants we issued to those investors (which warrants subsequently expired unexercised). Likewise, pursuant to the terms of the securities purchase agreement we entered into in connection with the November 2014 private placement, we filed registration statements under the Securities Act registering the resale of the 6.4 million shares of common stock we issued to the investors in the November 2014 private placement, which included J.R. Hyde, III, as well as the additional 6.4 million shares of common stock subject to the warrants we issued to the investors in the November 2014 private placement. Moreover, J.R. Hyde, III and certain of his affiliates, have rights under a separate registration rights agreement with us to require us to file resale registration statements covering an additional 790,000 shares of common stock held in the aggregate or to include these shares in registration statements that we may file for ourselves or other stockholders. If Mr. Hyde or his affiliates or any of our other significant stockholders, including the other investors in our 2014 private placements or in our 2016 registered direct offering of common stock, were to sell large blocks of shares in a short period of time, the market price of our common stock could drop substantially.
53
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We sublease approximately 26,000 square feet of office space located at 175 Toyota Plaza, Memphis, Tennessee, under an operating lease which expires on April 30, 2018. We believe that our facilities are currently adequate to meet our needs.
We are not currently involved in any material legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
54
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY
SECURITIES
Market for Registrant's Common Equity
Our common stock began trading on The NASDAQ Global Market under the symbol "GTXI" on February 3, 2004 and was transferred to The NASDAQ Capital Market on March 19, 2015. The following table presents, for the periods indicated, the high and low intraday sales prices per share of our common stock (as adjusted to give effect to the one-for-ten reverse stock split of our outstanding common stock effected on December 5, 2016) as reported on The NASDAQ Global Market prior to March 19, 2015 and The NASDAQ Capital Market subsequent to that date.
| |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | |||||||||||
| |
High | Low | High | Low | |||||||||
First Quarter |
$ | 8.00 | $ | 2.90 | $ | 8.40 | $ | 6.00 | |||||
Second Quarter |
8.00 | 5.00 | 15.90 | 6.50 | |||||||||
Third Quarter |
11.19 | 5.00 | 15.90 | 6.60 | |||||||||
Fourth Quarter |
9.90 | 5.14 | 11.70 | 6.20 | |||||||||
On March 17, 2017, the closing price of our common stock as reported on The NASDAQ Capital Market was $5.10 per share and there were approximately 81 holders of record of our common stock.
The rules of the SEC require that we include in our annual report to stockholders a line-graph presentation comparing cumulative stockholder returns on our common stock with a broad equity market index that includes companies whose equity securities are traded on the NASDAQ and either a published industry or line-of-business standard index or an index of peer companies selected by us. We have elected to use The NASDAQ Composite Index (which tracks the aggregate price performance of equity securities of companies traded on NASDAQ Stock Market) and The NASDAQ Biotechnology Index (consisting of a group of approximately 164 companies in the biotechnology sector) for purposes of the performance comparison that appears below.
The following graph shows the cumulative total stockholder return assuming the investment of $100.00 at the closing prices on December 31, 2011 on The NASDAQ Capital Market for: (1) our common stock; (2) The NASDAQ Composite Index and (3) The NASDAQ Biotechnology Index. All values assume reinvestment of the full amounts of all dividends. No dividends have been declared on our common stock. The closing sale price of our common stock on December 30, 2016 as reported on The NASDAQ Capital Market was $5.28.
The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
55
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among GTx Inc., the NASDAQ Composite Index, and the NASDAQ Biotechnology Index
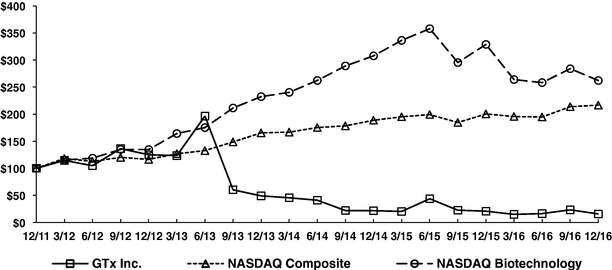
*$100 invested on 12/31/11 in stock or index, including reinvestment of dividends. Fiscal year ending December 31.
1 The material in this section is not "soliciting material," is not deemed filed with the SEC and is not to be incorporated by reference in any filing of GTx, Inc. under the Securities Act of 1933 or the Securities Exchange Act of 1934 whether made before or after the date hereof and irrespective of any general incorporation language in such filing.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any future earnings to fund the development and expansion of our business, and therefore we do not anticipate paying cash dividends on our common stock in the foreseeable future. Any future determination to pay dividends will be at the discretion of our Board of Directors.
56
ITEM 6. SELECTED FINANCIAL DATA
You should read the selected financial data below in conjunction with "Management's Discussion and Analysis of Financial Condition and Results of Operations" and the audited financial statements, notes thereto and other financial information included elsewhere in this Annual Report on Form 10-K. The following selected financial data have been derived from our audited historical financial statements, certain of which are included elsewhere in the Annual Report on Form 10-K. Historical results are not indicative of the results to be expected in the future.
| |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Years Ended December 31, | |||||||||||||||
| |
2016 | 2015 | 2014 | 2013 | 2012 | |||||||||||
| |
(in thousands, except per share data) |
|||||||||||||||
| Statement of Operations Data: | ||||||||||||||||
| Expenses: | ||||||||||||||||
Research and development expenses |
$ | 17,228 | $ | 13,607 | $ | 20,870 | $ | 32,318 | $ | 38,887 | ||||||
General and administrative expenses |
8,705 | 8,234 | 9,478 | 11,281 | 10,845 | |||||||||||
| | | | | | | | | | | | | | | | | |
| Total expenses | 25,933 | 21,841 | 30,348 | 43,599 | 49,732 | |||||||||||
| | | | | | | | | | | | | | | | | |
| Loss from operations | (25,933 | ) | (21,841 | ) | (30,348 | ) | (43,599 | ) | (49,732 | ) | ||||||
| Other income (expense), net | 46 | 57 | (259 | ) | 1,488 | (19 | ) | |||||||||
| Gain (loss) on change in fair value of warrant liability (a) | 8,163 | 3,081 | (8,804 | ) | - | - | ||||||||||
| | | | | | | | | | | | | | | | | |
| Loss from operations before income taxes | (17,724 | ) | (18,703 | ) | (39,411 | ) | (42,111 | ) | (49,751 | ) | ||||||
| Income tax benefit | - | - | - | - | 8,821 | |||||||||||
| | | | | | | | | | | | | | | | | |
| Net loss from continuing operations | (17,724 | ) | (18,703 | ) | (39,411 | ) | (42,111 | ) | (40,930 | ) | ||||||
| | | | | | | | | | | | | | | | | |
| Income from discontinued operations before income taxes | - | - | - | - | 22,676 | |||||||||||
| Income tax expense | - | - | - | - | (8,821 | ) | ||||||||||
| | | | | | | | | | | | | | | | | |
| Net income from discontinued operations | - | - | - | - | 13,855 | |||||||||||
| | | | | | | | | | | | | | | | | |
| Net loss | $ | (17,724 | ) | $ | (18,703 | ) | $ | (39,411 | ) | $ | (42,111 | ) | $ | (27,075 | ) | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Net loss per share — basic and diluted: (b) | ||||||||||||||||
Net loss from continuing operations |
$ | (1.22 | ) | $ | (1.33 | ) | $ | (4.82 | ) | $ | (6.68 | ) | $ | (6.52 | ) | |
Net income from discontinued operations |
- | - | - | - | 2.21 | |||||||||||
| | | | | | | | | | | | | | | | | |
| Net loss per share — basic | $ | (1.22 | ) | $ | (1.33 | ) | $ | (4.82 | ) | $ | (6.68 | ) | $ | (4.31 | ) | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Net loss per share — diluted | $ | (1.22 | ) | $ | (1.47 | ) | $ | (4.82 | ) | $ | (6.68 | ) | $ | (4.31 | ) | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
As of December 31, | |||||||||||||||
| |
2016 | 2015 | 2014 | 2013 | 2012 | |||||||||||
| |
(in thousands) |
|||||||||||||||
| Balance Sheet Data: | ||||||||||||||||
| Cash, cash equivalents and short-term investments (c) | $ | 21,869 | $ | 29,256 | $ | 49,295 | $ | 14,729 | $ | 56,089 | ||||||
| Working capital | 19,687 | 1,717 | 17,359 | 10,604 | 47,320 | |||||||||||
| Total assets | 24,502 | 32,031 | 50,651 | 15,605 | 57,774 | |||||||||||
| Accumulated deficit | (531,198 | ) | (513,474 | ) | (494,771 | ) | (455,360 | ) | (413,249 | ) | ||||||
| Total stockholders' equity | 19,891 | 1,859 | 17,829 | 10,684 | 47,701 | |||||||||||
- (a)
- The
gain (loss) on the change in fair value of warrant liability is related to the private placement of warrants completed in November 2014. See Note 6, Stockholders' Equity, for further information.
- (b)
- Net
loss per share — basic and diluted disclosures have been adjusted to give effect to the one-for-ten reverse stock split of our outstanding
common stock effected on December 5, 2016.
- (c)
- Cash, cash equivalents and short-term investments for the year ended December 31, 2016 includes the net proceeds of $13.7 million received from the registered direct offering of common stock completed in October 2016. Cash, cash equivalents and short-term investments for the year ended December 31, 2014 includes the net proceeds of $21.1 million and $42.8 million received from the private placements of common stock and warrants completed in March and November 2014, respectively. See Note 6, Stockholders' Equity, for further information.
57
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with our financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under Part I, Item 1A "Risk Factors" and elsewhere in this Annual Report on Form 10-K. See "Special Note Regarding Forward-Looking Statements" in this Annual Report on Form 10-K.
On December 5, 2016, we effected a one-for-ten reverse stock split of our outstanding common stock, or the Reverse Stock Split. At the effective time of the Reverse Stock Split, every ten shares of our issued and outstanding common stock was automatically combined and reclassified into one issued and outstanding share of common stock. No fractional shares of our common stock were issued in the Reverse Stock Split, but in lieu thereof, each holder of our common stock who would otherwise have been entitled to a fraction of a share of our common stock in the Reverse Stock Split received a cash payment. In addition, as a result of the Reverse Stock Split, proportionate adjustments were made to the per share exercise price and/or the number of shares issuable upon the exercise or vesting of all stock options, restricted stock units and warrants issued by GTx and outstanding immediately prior to the effective time of the Reverse Stock Split, which resulted in a proportionate decrease in the number of shares of our common stock reserved for issuance upon exercise or vesting of such stock options, restricted stock units and warrants, and, in the case of stock options and warrants, a proportionate increase in the exercise price of all such stock options and warrants. In addition, the number of shares reserved for issuance under our equity compensation plans immediately prior to the effective time of the Reverse Stock Split was reduced proportionately. Unless otherwise noted, all share and per share information included in this report has been retroactively adjusted to give effect to the Reverse Stock Split.
Business Overview
We are a biopharmaceutical company dedicated to the discovery, development and commercialization of small molecules for the treatment of cancer, including treatments for breast and prostate cancer, and other serious medical conditions. Our current strategy is focused on the further development of selective androgen receptor modulators, or SARMs, a class of drugs that we believe has the potential to be used as a hormonal therapy for the treatment of advanced breast cancer, as well as the potential to treat other serious medical conditions where unmet medical needs in muscle-related diseases may benefit from increasing muscle mass, such as stress urinary incontinence, or SUI, and Duchenne muscular dystrophy, or DMD. In 2015, we entered into an exclusive worldwide license agreement with the University of Tennessee Research Foundation, or UTRF, to develop its proprietary selective androgen receptor degrader, or SARD, technology, which we believe has the potential to provide compounds that can degrade multiple forms of androgen receptor, or AR, by inhibiting tumor growth in patients with progressive castration-resistant prostate cancer, or CRPC, including those patients who do not respond to or are resistant to current therapies.
Business Highlights
Our lead SARM candidate, enobosarm (GTx-024), has to date been evaluated in 24 completed or ongoing clinical trials, including in six Phase 2 and two Phase 3 clinical trials, enrolling over 1,700 subjects, of which approximately 1,200 subjects were treated with enobosarm. Enobosarm is the generic name given to the compound by the USAN Council and the World Health Organization and is
58
the first compound to receive the SARM stem in its name, recognizing enobosarm as the first in this new class of compounds. We announced in 2014 positive results from a Phase 2 proof-of-concept, open-label clinical trial evaluating a 9 mg oral daily dose of enobosarm for the treatment of patients with estrogen receptor, or ER, positive and AR positive metastatic breast cancer who have previously responded to hormonal therapy. During the second half of 2015, we commenced enrollment in both a Phase 2 clinical trial designed to evaluate the efficacy and safety of a 9 mg and 18 mg dose of enobosarm in patients whose advanced breast cancer is both ER positive and AR positive and a Phase 2 proof-of-concept clinical trial designed to evaluate the efficacy and safety of an 18 mg dose of enobosarm in patients with advanced AR positive triple-negative breast cancer, or TNBC. Both of these clinical trials are being conducted utilizing a Simon's two-stage trial design. The Phase 2 clinical trial evaluating enobosarm in patients with ER positive, AR positive advanced breast cancer has completed enrollment of both stages of the clinical trial for both dose cohorts. We announced in November 2016 that enobosarm achieved the pre-specified primary efficacy endpoint in the 9 mg dose cohort. We expect to report top-line clinical results from this clinical trial in the third quarter of 2017. In our trial evaluating enobosarm in patients with advanced AR positive TNBC, we anticipate having sufficient data from the first stage of this trial in the second quarter of 2017 to allow us to make a determination as to whether we will continue the clinical trial and enroll patients into the second stage of this study. However, due to the slow rate of patient enrollment in this trial, our current capital resources may not be sufficient to enable us to complete the second stage of the TNBC trial, in which case, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone has been met.
We are also evaluating enobosarm and other compounds in our SARM portfolio for indications outside of oncology where unmet medical needs in muscle-related diseases may benefit from increasing muscle mass. In the first quarter of 2016, we initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI. This is the first clinical trial to evaluate a SARM for the treatment of SUI. We currently anticipate obtaining data from this clinical trial in the third quarter of 2017 sufficient to enable us to determine if continued development of enobosarm in SUI is warranted. Continued development of enobosarm in SUI apart from our ongoing Phase 2 proof-of-concept clinical trial will require us to obtain additional funding. We have also evaluated several SARM compounds in preclinical models of DMD where a SARM's ability to increase muscle mass may prove beneficial to patients suffering from DMD, which is a rare disease characterized by progressive muscle degeneration and weakness.
With respect to SARDs, we believe this class of assets has the potential to treat prostate cancer, as well as other diseases such as benign prostatic hyperplasia and Kennedy's disease. We envision initially developing SARDs as a potentially novel treatment for men with CRPC, including those who do not respond or are resistant to currently approved therapies. Our evaluation of the SARD program is at an early stage. We are currently implementing an appropriate development program for SARDs and have selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic and toxicology studies, required to support potential initial human clinical trials. While we plan to initiate a first in human clinical trial during the second half of 2017, we will require additional funding to initiate and complete any such clinical trial.
Our ability to pursue the continued development of SARMs and our SARD program is contingent upon our ability to obtain additional funding. Accordingly, we are actively seeking additional funding through the licensing, partnering or sale of certain assets to provide us the necessary resources for the development of our preclinical and clinical product candidates. We have discussions ongoing with several potential collaboration partners who have expressed interest in our SARM compounds for the treatment of breast cancer, SUI, and/or DMD, as well as our SARD technology.
59
Financial Highlights
Our net loss for the year ended December 31, 2016 was $17.7 million. We expect to incur significant operating losses for the foreseeable future as we continue our preclinical and clinical development activities and potentially seek regulatory approval of our product candidates. We have funded our operations primarily through the sale of equity securities, collaboration and license agreements, and prior to September 2012, product revenue from sales of FARESTON®, the rights to which we sold to a third party in the third quarter of 2012. We currently have no ongoing collaborations for the development and commercialization of our product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We do not expect to obtain any regulatory approvals to market any of our product candidates, including enobosarm, for the foreseeable future, and it is possible that none of our product candidates will ever receive any regulatory approvals.
At December 31, 2016, we had cash, cash equivalents and short-term investments of $21.9 million compared to $29.3 million at December 31, 2015. On October 14, 2016, we completed a registered direct offering of our common stock, in which we sold 1.7 million shares of our common stock for net proceeds to us of approximately $13.7 million.
Based on our current business plan and assumptions, we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017. Accordingly, we will need to raise substantial additional capital in the near term in order to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter. In addition, we have based our cash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding to sustain our operations even sooner than currently anticipated. We believe, based on our current estimates of clinical trial expenditures and enrollment status, that our existing capital resources will be adequate to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with ER positive and AR positive advanced breast cancer and our ongoing Phase 2 clinical trial of enobosarm in postmenopausal women with SUI. However, our existing capital resources will not be sufficient to allow us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC and we will otherwise need to raise substantial additional capital in order to continue developing enobosarm for any of these indications. If we determine that our existing capital resources are not sufficient to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all. Also, our clinical trials may continue to encounter technical, enrollment or other difficulties that could increase our development costs beyond our current estimates or delay our development timelines, and we could otherwise exhaust our available financial resources sooner than we expect. In any event, we need to raise substantial additional capital in order to:
- •
- potentially enroll the second stage of and complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced
AR positive TNBC;
- •
- undertake any further development of our SARMs beyond our ongoing Phase 2 clinical trials of enobosarm in breast cancer and SUI and our ongoing preclinical development activities related to the development of SARMs as a potential treatment for DMD;
60
- •
- initiate and complete human clinical studies of our SARD program; and
- •
- fund our operations and to continue as a going concern.
In addition, the accompanying financial statements have been prepared assuming that we will continue as a going concern. Accordingly, the accompanying financial statements do not include any adjustments or charges that might be necessary should we be unable to continue as a going concern, such as charges related to impairment of our assets, the recoverability and classification of assets or the amounts and classification of liabilities or other similar adjustments. However, because we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017, there is doubt raised about our ability to continue as a going concern. While we believe that we have the ability to successfully implement plans to mitigate the conditions that may raise doubt about our ability to continue as a going concern within one year after the date of this report, such plans include reducing or delaying expenditures by postponing or discontinuing planned clinical or preclinical development and implementing cost saving measures related to other research and development and general and administrative expenditures, which plans, if implemented, would materially harm our business. In any event, if we are unable to raise additional funds in the near term to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter, we could be required to, among other things, make further reductions in our workforce, eliminate our ongoing AR positive TNBC clinical trial, discontinue further development of enobosarm and/or SARDs, liquidate all or a portion of our assets, and/or seek protection under the provisions of the U.S. Bankruptcy Code, all of which would have a material adverse effect on our business and stock price.
While we have been able to fund our operations to date, we currently have no ongoing collaborations for the development and commercialization of our product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We also do not have any commitments for future external funding. Accordingly, we expect to continue our efforts to seek additional funds through potential collaboration, partnering or other strategic arrangements, through public or private equity offerings or debt financings, or a combination of the foregoing. Our ability to raise additional funds and the terms upon which we are able to raise such funds have been severely harmed by the failure of our POWER 1 and POWER 2 Phase 3 clinical trials of enobosarm for the prevention and treatment of muscle wasting in patients with advanced non-small cell lung cancer, or NSCLC, to meet the primary statistical criterion for the co-primary endpoints agreed upon with the FDA, and may in the future be adversely impacted by the uncertainty regarding the prospects of our development of enobosarm for the treatment of patients with advanced AR positive breast cancer and our ability to advance the development of enobosarm or SARDs, if at all. Our ability to raise additional funds and the terms upon which we are able to raise such funds may also be adversely affected by the uncertainties regarding our financial condition, the sufficiency of our capital resources, recent and potential future management turnover, and continued volatility and instability in the global financial markets. As a result of these and other factors, we cannot be certain that additional funding will be available on acceptable terms, or at all.
Since our inception in 1997, we have been focused on drug discovery and development programs. Research and development expenses include, but are not limited to, our expenses for personnel and supplies associated with our research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. Assuming we raise additional capital in the near term to fund our operations through the fourth quarter of 2017, we
61
expect that our research and development expenses for fiscal year 2017 will remain relatively consistent as compared to fiscal year 2016 primarily due to our ongoing Phase 2 clinical trials of enobosarm in two different breast cancer indications targeting the androgen receptor and for the treatment of SUI and ongoing preclinical development of the SARD program.
There is a substantial risk that any development program may not produce revenue. Moreover, because of uncertainties inherent in drug development, including those factors described in Part I, Item 1A "Risk Factors" of this Annual Report on Form 10-K, we and/or potential future collaborators may not be able to successfully develop and commercialize any of our product candidates.
The successful development and commercialization of our product candidates is highly uncertain. We cannot reasonably estimate or know the nature, timing and estimated costs of the efforts necessary to complete the development and commercialization of, or the period in which material net cash inflows are expected to commence from, any of our product candidates due to the numerous risks and uncertainties associated with developing and commercializing drugs, including the uncertainty of:
- •
- the scope, rate of progress and cost of our preclinical and clinical development programs, including our ongoing and any future clinical trials
of enobosarm;
- •
- the terms and timing of any potential collaborative, licensing and other strategic arrangements that we may establish;
- •
- the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators, if any;
- •
- future clinical trial results;
- •
- the cost and timing of regulatory filings and/or approvals to commercialize our product candidates and any related restrictions, limitations,
and/or warnings in the label of an approved product candidate;
- •
- the effect of competing technological and market developments; and
- •
- the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
Any failure to complete the development of our product candidates in a timely manner could have a material adverse effect on our operations, financial position and liquidity. A discussion of the risks and uncertainties associated with completing our development efforts on schedule, or at all, and some consequences of failing to do so, are set forth under Part I, Item 1A "Risk Factors" of this Annual Report on Form 10-K.
General and Administrative Expenses
Our general and administrative expenses consist primarily of salaries and other related costs for personnel serving executive, finance, legal, human resources, information technology, and investor relations functions. General and administrative expenses also include facility costs, insurance costs, and professional fees for legal, accounting, and public relation services. We expect our general and administrative expenses for fiscal year 2017 to be relatively consistent with fiscal year 2016.
62
Critical Accounting Policies and Significant Judgments and Estimates
Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments related to revenue recognition, valuation of warrants, income taxes, intangible assets, long-term service contracts, share-based compensation, and other contingencies. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our financial statements appearing at the end of this Annual Report on Form 10-K, we believe that the following accounting policies are most critical to aid you in fully understanding and evaluating our reported financial results.
Warrant Liability
In November 2014, we issued warrants to purchase 6,430,948 shares of our common stock. At that time, we classified these warrants as a liability on our balance sheet since the warrants contained certain terms that could have required us (or our successor) to purchase the warrants for cash in an amount equal to the value (as calculated utilizing a contractually-agreed Black-Scholes-Merton option pricing valuation model) of the unexercised portion of the warrants in connection with certain change of control transactions occurring on or prior to December 31, 2016, with such cash payment capped at an amount equal to $1.25 per unexercised share underlying each warrant. As a result of the provision of the warrant requiring cash settlement upon certain change of control transactions, we were required to account for these warrants as a liability at fair value and the estimated warrant liability was required to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such provision on December 31, 2016. Effective March 25, 2016, each of the warrants was amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions. These warrants were no longer accounted for as a liability at March 31, 2016. We recorded a non-cash reclassification of the warrant fair value to stockholders' equity based on the warrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required.
Research and Development Expenses
Research and development expenses include, but are not limited to, our expenses for personnel, supplies, and facilities associated with research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. We expense these costs in the period in which they are incurred. We estimate our liabilities for research and development expenses in order to match the recognition of expenses to the period in which the actual services are received. As such, accrued liabilities related to third party research and development activities are recognized based upon our estimate of services received and degree of completion of the services in accordance with the specific third party contract.
63
Share-Based Compensation
We have stock option and equity incentive plans that provide for the purchase or acquisition of our common stock by certain of our employees and non-employees. We measure compensation expense for our share-based payments based on the fair value of the awards on the grant date and recognize the expense over the period during which an employee or non-employee director is required to provide service in exchange for the award.
The determination of the fair value of stock options on the date of grant include the expected life of the award, the expected stock price volatility over the expected life of the awards, and risk-free interest rate. We estimate the expected life of options by calculating the average of the vesting term and contractual term of the options. We estimate the expected stock price volatility based on the historical volatility of our common stock. The risk-free interest rate is determined using U.S. Treasury rates where the term is consistent with the expected life of the stock options. Expected dividend yield is not considered as we have not made any dividend payments and have no plans of doing so in the foreseeable future. The amount of share-based compensation expense recognized is reduced ratably over the vesting period by an estimate of the percentage of options granted that are expected to be forfeited or canceled before becoming fully vested. This estimate is adjusted periodically based on the extent to which actual forfeitures differ, or are expected to differ, from the previous estimate.
Share-based compensation also includes restricted stock units, or RSUs, granted to employees. We estimate the fair value of RSUs using the closing price of our stock on the grant date. The fair value of RSUs is amortized on a straight-line basis over the requisite service period of the awards. The amount of share-based compensation expense recognized is reduced ratably over the vesting period by an estimate of the percentage of RSUs granted that are expected to be forfeited or canceled before becoming fully vested.
The following table summarizes share-based compensation expense included within the statements of operations for the years ended December 31, 2016, 2015 and 2014:
| |
Years ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
| |
(in thousands) |
|||||||||
Research and development expenses |
$ | 1,260 | $ | 1,210 | $ | 2,512 | ||||
General and administrative expenses |
1,829 | 1,523 | 2,041 | |||||||
| | | | | | | | | | | |
Total share-based compensation |
$ | 3,089 | $ | 2,733 | $ | 4,553 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Share-based compensation expense recorded in the statement of operations as general and administrative expense for the years ended December 31, 2016, 2015 and 2014 included share-based compensation expense related to deferred compensation arrangements for our non-employee directors of $132,000, $113,000 and $125,000, respectively. At December 31, 2016, the total compensation cost related to non-vested stock options not yet recognized was approximately $3.7 million with a weighted average expense recognition period of 3.09 years. At December 31, 2016, the total compensation cost related to non-vested RSUs not yet recognized was approximately $2.0 million with a weighted average expense recognition period of 1.05 years.
Income Taxes
We account for deferred taxes by recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, at December 31, 2016 and 2015, net of the valuation allowance, the net deferred tax assets were reduced to zero.
64
Recent Accounting Pronouncements
In March 2016, the Financial Accounting Standards Board issued Accounting Standards Update 2016-09, Improvements to Employee Share Based Payment Accounting. This guidance addresses the income tax effects of stock-based payments and eliminates the windfall pool concept, as all of the tax effects related to stock-based payments will now be recorded at settlement (or expiration) through the income statement. The new guidance also permits entities to make an accounting policy election for the impact of forfeitures on the recognition of expense for stock-based payment awards. Forfeitures can be estimated or recognized when they occur. The standard is effective for annual periods beginning after December 15, 2016 and interim periods within that reporting period. We believe the adoption of this guidance will not have a material impact on our financial position or results of operations.
Research and Development Expenses
The following table identifies the research and development expenses for each of our clinical product candidates, as well as research and development expenses pertaining to our other research and development efforts, for each of the periods presented. Research and development spending for past periods is not indicative of spending in future periods.
| Proposed Candidate / Proposed Indication | Program | Years Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
2016 | 2015 | 2014 | |||||||||
| |
|
(in thousands) |
|||||||||||
| Enobosarm | |||||||||||||
| Treatment of women with ER positive / AR positive advanced breast cancer (9 mg and 18 mg) | SARM | $ | 7,316 | $ | 4,885 | $ | 3,506 | ||||||
Enobosarm |
|||||||||||||
| Treatment of women with advanced AR positive TNBC (18 mg) | SARM | 4,853 | 4,945 | 878 | |||||||||
Enobosarm |
|||||||||||||
| Treatment of postmenopausal women with SUI (3 mg) | SARM | 1,286 | - | - | |||||||||
Enobosarm |
|||||||||||||
| Prevention and treatment of muscle wasting in patients with advanced non-small cell lung cancer (3 mg) | SARM | - | - | 12,025 | |||||||||
GTx-758 |
|||||||||||||
| Secondary hormonal therapy in men with metastatic and non-metastatic CRPC | Selective ER alpha agonist | 699 | 1,667 | 4,201 | |||||||||
Other research and development |
3,074 |
2,110 |
260 |
||||||||||
| | | | | | | | | | | | | | |
Total research and development expenses |
$ |
17,228 |
$ |
13,607 |
$ |
20,870 |
|||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
65
Comparison of Years Ended December 31, 2016 and 2015
Research and development expenses increased 27% to $17.2 million for the year ended December 31, 2016 from $13.6 million for the year ended December 31, 2015.
Research and development expenses for enobosarm for the treatment of women with ER positive, AR positive advanced breast cancer increased for the year ended December 31, 2016 from the prior year due primarily to the timing and nature of activities related to conducting the ongoing Phase 2 clinical trial evaluating enobosarm 9 mg and enobosarm 18 mg in this indication, which commenced enrollment during the third quarter of 2015 and related to cash bonuses paid to employees upon the achievement of certain development milestones. The prior year period consisted primarily of expenses related to preparatory activities for the ongoing Phase 2 clinical trial for the treatment of women with ER positive and AR positive advanced breast cancer and expenses related to the previous Phase 2 proof-of-concept clinical trial evaluating enobosarm 9 mg in women who have previously responded to hormonal therapy for the treatment of their metastatic breast cancer.
Research and development expenses for enobosarm for the treatment of women with advanced AR positive TNBC decreased slightly for the year ended December 31, 2016 from the prior year due to the timing and nature of activities related to conducting the ongoing Phase 2 clinical trial, which commenced enrollment during the fourth quarter of 2015. The prior year period consisted primarily of expenses related to preparatory activities for this clinical trial.
Research and development expenses for enobosarm for the treatment of postmenopausal women with SUI during the year ended December 31, 2016 consisted of expenses related to the Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI that initiated enrollment in the first quarter of 2016.
Research and development expenses related to the completed Phase 2 clinical trial to evaluate GTx-758 as secondary hormonal therapy in men with metastatic CRPC decreased for the year December 31, 2016 compared to the prior year due to the timing of patient activities and related management expenses as this trial was initiated in the third quarter of 2012 and enrollment was completed during the first quarter of 2015. We have determined to discontinue further development of GTx-758 and will not be making any further investments in this program.
"Other research and development" expenses for the year ended December 31, 2016 increased from the prior year primarily due to the ongoing preclinical development of our SARD compounds, that was initiated in 2015, and activities relating to evaluating enobosarm and other compounds in our SARM portfolio for indications outside of oncology.
Comparison of Years Ended December 31, 2015 and 2014
Research and development expenses decreased 35% to $13.6 million for the year ended December 31, 2015 from $20.9 million for the year ended December 31, 2014.
Research and development expenses for enobosarm for the treatment of ER positive, AR positive advanced breast cancer during the year ended December 31, 2015 consisted of expenses for preparatory activities related to, and the initiation of, the Phase 2 clinical trial evaluating enobosarm 9 mg and 18 mg for the treatment of women whose advanced breast cancer is both ER positive and AR positive, which preparatory activities began in the fourth quarter of 2014, as well as expenses related to our Phase 2 proof-of-concept clinical trial evaluating enobosarm 9 mg for the treatment of AR positive and ER positive metastatic breast cancer in women who have previously responded to hormonal therapy for the treatment of their metastatic breast cancer. The prior year consisted primarily of expenses related
66
to our Phase 2 proof-of-concept clinical trial evaluating enobosarm 9 mg that began in the second quarter of 2013.
Research and development expenses for enobosarm for the treatment of women with advanced AR positive TNBC increased for the year ended December 31, 2015 from the prior year due to preparatory activities related to, and the initiation of, our Phase 2 proof-of-concept clinical trial of enobosarm 18 mg for the treatment of women with advanced AR positive TNBC, which preparatory activities began in the fourth quarter of 2014.
There were no research and development expenses for enobosarm for the prevention and treatment of muscle wasting in patients with advanced NSCLC for the year ended December 31, 2016 or 2015. As we previously announced in August 2013, data from our two POWER Phase 3 clinical trials evaluating enobosarm 3 mg daily for the prevention and treatment of muscle wasting in patients with advanced NSCLC failed to meet the primary statistical criterion pre-specified for the co-primary endpoints of lean body mass and physical function, and the FDA will not accept a new drug application for enobosarm for this indication. Additionally, we subsequently determined that data from the POWER trials is not sufficient to support the filing and approval of a marketing authorization application, or MAA, by the European Medicines Agency without confirmatory data from another Phase 3 clinical trial of enobosarm 3 mg and we do not intend to submit a MAA in the absence of such confirmatory data. Accordingly, we ceased spending on this indication. The year ended December 31, 2014 included expenses for activities related to satisfying the prerequisites necessary for our then-planned regulatory submission in Europe for enobosarm 3 mg, including conducting seven Phase 1 clinical trials.
Research and development expenses related to our Phase 2 clinical trial to evaluate GTx-758 as secondary hormonal therapy in men with metastatic CRPC decreased for the year ended December 31, 2015 from the prior year due to the timing of patient activities and related management expenses as this trial was initiated in the third quarter of 2012 and enrollment was completed during the first quarter of 2015.
Additionally, research and development expenses for each product candidate in the year ended December 31, 2014 included expenses related to cash bonuses and stock option and RSU grants made to employees as part of our efforts to retain essential employees continuing with us following our October 2013 workforce reduction.
"Other research and development" expenses for the year ended December 31, 2015 increased from the prior year primarily due primarily to initial activities to identify one or more potential lead SARD compounds that could potentially be advanced into preclinical and clinical development and activities related to evaluating enobosarm and other compounds in our SARM portfolio for indications outside of oncology.
General and Administrative Expenses
General and administrative expenses increased 6% to $8.7 million for the year ended December 31, 2016 from $8.2 million for the year ended December 31, 2015. The increase in the year ended December 31, 2016 from the prior year was due primarily to cash bonuses paid to employees upon the achievement of certain development milestones of the Phase 2 clinical trial of enobosarm for the treatment of women with ER positive, AR positive advanced breast cancer. This increase was offset by decreases in insurance and legal fees from the prior year period.
67
General and administrative expenses decreased 13% to $8.2 million for the year ended December 31, 2015 from $9.5 million for the year ended December 31, 2014. The decrease in the year ended December 31, 2015 from the prior year was due primarily to expenses in the prior year period related to cash bonuses and stock option and RSU grants made to the employees as part of our efforts to retain essential employees continuing with us following our October 2013 workforce reduction. Additionally, insurance and legal fees decreased from the prior year period.
Other Income (Expense), Net
Other income, net for the year ended December 31, 2016 was $46,000 and consisted of foreign currency transaction gains and losses, interest earned on our cash, cash equivalents and short-term investments, and other non-operating income or expense compared to other expense, net of $57,000 for the year ended December 31, 2015. Other expense for the year ended December 31, 2014 included an allocation of the total expenses related to the private placement of common stock and warrants completed in November 2014 as the warrants issued were accounted for as a liability. The remaining expenses were reflected as a reduction of equity.
Gain (Loss) on Change in Fair Value of Warrant Liability
Until March 2016, we recognized a warrant liability due to certain provisions of the warrants issued as part of the November 2014 private placement of common stock and warrants. The warrants were required to be accounted for as a liability at fair value and the fair value was to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such provision on December 31, 2016. The resulting non-cash gain or loss on the fair value revaluation at each balance sheet date was recorded as non-operating income in our statement of operations. When the warrants were revalued at fair value as of December 31, 2014, an increase in fair value of $8.8 million was recorded for the year then ended as a non-cash loss on the change in fair value of warrant liability. When the warrants were revalued at fair value as of December 31, 2015, the decrease in fair value for the year then ended of $3.1 million was recorded as a non-cash gain on the change in fair value of warrant liability in our statement of operations.
Effective March 25, 2016, each of the warrants was amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions. These warrants were no longer accounted for as a liability at March 31, 2016. The Company recorded a non-cash reclassification of the warrant fair value to stockholders' equity based on the warrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required. At that time a non-cash gain of $8.2 million was recorded on the change in fair value of the warrant liability in our statement of operations.
Liquidity and Capital Resources
We have financed our operations to date primarily through public offerings and private placements of our securities, as well as payments from our former collaborators. We have incurred significant losses since our inception in 1997 as we have devoted substantially all of our resources to research and development, including our clinical trials. As of December 31, 2016, we had an accumulated deficit of $531.2 million, which resulted primarily from:
- •
- our research and development activities associated with:
- •
- the preclinical development of our SARD program;
68
- •
- the preclinical and clinical development of our SARM compounds, including enobosarm;
- •
- the preclinical and clinical development of GTx-758 for the treatment of advanced prostate cancer;
- •
- the development of our discontinued toremifene 80 mg product candidate to reduce fractures and treat other estrogen deficiency
side effects of androgen deprivation therapy in men with prostate cancer, including two Phase 2 clinical trials, a Phase 3 clinical trial, and the preparation and submission of a NDA to
the FDA;
- •
- the development of our discontinued toremifene 20 mg product candidate for the prevention of prostate cancer in high risk men with
high grade prostatic intraepithelial neoplasia, including a Phase 2b clinical trial and a Phase 3 clinical trial; and
- •
- the preclinical development of other product candidates; and
- •
- general and administrative expenses.
We expect to incur significant operating losses for the foreseeable future as we continue our preclinical and clinical development activities and potentially seek regulatory approval of our product candidates. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity and working capital. We do not expect to obtain any regulatory approvals to market any of our product candidates, including enobosarm, for the foreseeable future, and it is possible that none of our product candidates will ever receive any regulatory approvals.
At December 31, 2016, we had cash, cash equivalents and short-term investments of $21.9 million, compared to $29.3 million at December 31, 2015 and $49.3 million at December 31, 2014.
On October 14, 2016, we completed a registered direct offering of our common stock consisting of 1.7 million shares of its common stock for net proceeds of approximately $13.7 million. The purchasers in the registered direct offering consisted of certain existing GTx stockholders and certain members of the GTx management team and board of directors.
On November 14, 2014, we completed a private placement of units consisting of an aggregate of 6.4 million shares of our common stock and warrants to purchase an aggregate of 6.4 million shares of our common stock for net proceeds of approximately $42.8 million. The purchasers in the private placement included certain existing GTx stockholders and certain members of the GTx management team and board of directors. The warrants became exercisable on May 6, 2015 and will continue to be exercisable for four years thereafter.
On March 6, 2014, we completed a private placement of units consisting of 1.2 million shares of common stock and warrants to purchase 1.0 million shares of our common stock for net proceeds of approximately $21.1 million. The purchasers in the private placement included an existing GTx stockholder and member of the GTx board of directors. The warrants, which had a one year term, expired unexercised on March 6, 2015.
69
The following table shows a summary of our cash flows for the periods indicated:
| |
Years Ending December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
| |
(in thousands) |
|||||||||
Net cash used in operating activities |
$ | (20,778 | ) | $ | (20,035 | ) | $ | (28,759 | ) | |
Net cash provided by (used in) investing activities |
2,151 | 16,211 | (31,220 | ) | ||||||
Net cash provided by financing activities |
13,481 | - | 63,330 | |||||||
| | | | | | | | | | | |
Net (decrease) increase in cash and cash equivalents |
$ | (5,146 | ) | $ | (3,824 | ) | $ | 3,351 | ||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net cash used in operating activities in all periods resulted primarily from funding our operations.
Net cash provided by investing activities for the year ended December 31, 2016 primarily resulted from the maturities of short-term investments of $37.6 million offset by the purchase of short-term investments of $35.4 million. Net cash provided by investing activities for the year ended December 31, 2015 primarily resulted from the maturities of short-term investments of $71.4 million offset by the purchase of short-term investments of $55.2 million. Net cash used in investing activities for the year ended December 31, 2014 primarily resulted from purchase of short-term investments of $41.9 million, partially offset by proceeds from the maturities of short-term investments of $10.7 million.
Net cash provided by financing activities for the year ended December 2016 reflected net proceeds of $13.7 million from the issuance of common stock related to the October 2016 registered direct offering, partially offset by $208,000 of employee withholding tax payments related to vested RSUs. There was no cash provided by or used in financing activities for the year ended December 31, 2015. Net cash provided by financing activities for the year ended December 2014 reflected aggregate net proceeds of $63.9 million from the issuance of common stock and warrants related to the March and November 2014 private placements, partially offset by $617,000 of employee withholding tax payments related to vested RSUs.
Based on our current business plan and assumptions, we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017. Accordingly, we will need to raise substantial additional capital in the near term in order to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter. In addition, we have based our cash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding to sustain our operations even sooner than currently anticipated. We believe, based on our current estimates of clinical trial expenditures and enrollment status, that our existing capital resources will be adequate to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with ER positive and AR positive advanced breast cancer and our ongoing Phase 2 clinical trial of enobosarm in postmenopausal women with SUI. However, our existing capital resources will not be sufficient to allow us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC and we will otherwise need to raise substantial additional capital in order to continue developing enobosarm for any of these indications. If we determine that our existing capital resources are not sufficient to enable us to complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, we may be unable or unwilling to enroll patients into the second stage of this trial even if we determine that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, we will need to obtain additional funding, which we may be unable to do in a timely manner or at all. Also, our clinical trials may continue to encounter technical,
70
enrollment or other difficulties that could increase our development costs beyond our current estimates or delay our development timelines, and we could otherwise exhaust our available financial resources sooner than we expect. In any event, we need to raise substantial additional capital in order to:
- •
- potentially enroll the second stage of and complete our ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced
AR positive TNBC;
- •
- undertake any further development of our SARMs beyond our ongoing Phase 2 clinical trials of enobosarm in breast cancer and SUI
and our ongoing preclinical development activities related to the development of SARMs as a potential treatment for DMD;
- •
- initiate and complete human clinical studies of our SARD program; and
- •
- fund our operations and to continue as a going concern.
Our estimate of the period of time or events through which our financial resources will be adequate to support our projected operating requirements is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed under Part I, Item 1A "Risk Factors" section of this Annual Report on Form 10-K. Because of the numerous risks and uncertainties associated with the development and potential commercialization of our product candidates and other research and development activities, including risks and uncertainties that could impact the rate of progress of our development activities, we are unable to estimate with certainty the amounts of increased capital outlays and operating expenditures associated with the future development of our product candidates, if any. Our future funding requirements will depend on many factors, including:
- •
- the scope, rate of progress and cost of our preclinical and clinical development programs, including our ongoing and any future clinical trials
of enobosarm;
- •
- the terms and timing of any potential collaborative, licensing and other strategic arrangements that we may establish;
- •
- the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators, if any;
- •
- future clinical trial results;
- •
- the cost and timing of regulatory filings and/or approvals to commercialize our product candidates and any related restrictions, limitations,
and/or warnings in the label of an approved product candidate;
- •
- the effect of competing technological and market developments; and
- •
- the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
While we have been able to fund our operations to date, we currently have no ongoing collaborations for the development and commercialization of our product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We also do not have any commitments for future external funding. Accordingly, we expect to continue our efforts to seek
71
additional funds through potential collaboration, partnering or other strategic arrangements, through public or private equity offerings or debt financings, or a combination of the foregoing.
In addition, the accompanying financial statements have been prepared assuming that we will continue as a going concern. Accordingly, the accompanying financial statements do not include any adjustments or charges that might be necessary should we be unable to continue as a going concern, such as charges related to impairment of our assets, the recoverability and classification of assets or the amounts and classification of liabilities or other similar adjustments. However, because we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements only into the fourth quarter of 2017, there is doubt raised about our ability to continue as a going concern. While we believe that we have the ability to successfully implement plans to mitigate the conditions that may raise doubt about our ability to continue as a going concern within one year after the date of this report, such plans include reducing or delaying expenditures by postponing or discontinuing planned clinical or preclinical development and implementing cost saving measures related to other research and development and general and administrative expenditures, which plans, if implemented, would materially harm our business. In any event, if we are unable to raise additional funds in the near term to fund our operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter, we could be required to, among other things, make further reductions in our workforce, eliminate our ongoing AR positive TNBC clinical trial, discontinue further development of enobosarm and/or SARDs, liquidate all or a portion of our assets, and/or seek protection under the provisions of the U.S. Bankruptcy Code, all of which would have a material adverse effect on our business and stock price.
To the extent that we raise additional funds through potential collaboration, partnering or other strategic arrangements, it may be necessary to relinquish rights to some of our technologies or product candidates, or grant licenses on terms that are not favorable to us, any of which could result in the stockholders of GTx having little or no continuing interest in our SARMs and/or SARDs programs as stockholders or otherwise. To the extent we raise additional funds by issuing equity securities, our stockholders may experience significant dilution, particularly given our currently depressed stock price, and debt financing, if available, may involve restrictive covenants. For example, we completed a private placement of common stock and warrants in March 2014, which was substantially dilutive, completed a subsequent private placement in November 2014 that represented additional dilution, and we again raised additional funds by issuing shares of common stock in a registered direct offering in October 2016. Our stockholders may experience additional, perhaps substantial, dilution should we again raise additional funds by issuing equity securities. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. Our ability to raise additional funds and the terms upon which we are able to raise such funds have been severely harmed by the failure of our two prior enobosarm POWER trials to meet the primary statistical criterion for the co-primary endpoints agreed upon with the FDA, and may in the future be adversely impacted by the uncertainty regarding the prospects of our development of enobosarm for the treatment of patients with advanced AR positive breast cancer and our ability to advance the development of enobosarm or SARDs, if at all. Our ability to raise additional funds and the terms upon which we are able to raise such funds may also be adversely affected by the uncertainties regarding our financial condition, the sufficiency of our capital resources, recent and potential future management turnover, and continued volatility and instability in the global financial markets. As a result of these and other factors, we cannot be certain that additional funding will be available on acceptable terms, or at all.
72
Contractual Obligations
At December 31, 2016, we had contractual obligations as follows:
| |
Payment Due by Period (in thousands) |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Contractual Obligations(1) | Total | Less than 1 year |
1-3 years | 4-5 years | More than 5 years |
|||||||||||
Operating lease obligations(2) |
$ | 634 | $ | 475 | $ | 159 | $ | - | $ | - | ||||||
- (1)
- This
table does not include any royalty obligations under our SARM and SARD license agreements with UTRF as the timing and likelihood of such payments are not known.
In addition to the minimum payments due under our SARM and SARD license agreements, we may be required to pay royalties on any net sales of product if we receive regulatory approval for a SARM,
including enobosarm, or SARD product candidate and successfully market the product. Additionally, if we sublicense rights under our SARM or SARD license agreements, we also are obligated to pay a
sublicense royalty on any licensing fee or milestone payments we may receive from a sublicensee.
- (2)
- Our long-term commitment under the operating lease consists of payments relating to a sublease for office space at 175 Toyota Plaza, Memphis, Tennessee. The sublease for the premises at 175 Toyota Plaza expires on April 30, 2018.
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet arrangements, including the use of standard finance, special purpose entities or variable interest entities.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk for changes in interest rates relates to our cash equivalents on deposit in highly liquid money market funds and investments in Federal Deposit Insurance Corporation insured certificates of deposit. The primary objective of our cash investment activities is to preserve principal while at the same time maximizing the income we receive from our invested cash without significantly increasing risk of loss. We do not use derivative financial instruments in our investment portfolio. The effect of a hypothetical decrease of ten percent in the average yield earned on our cash equivalents and short-term investments would have resulted in an immaterial decrease in our interest income for the year ended December 31, 2016.
In addition, we have exposure to fluctuations in certain foreign currencies in countries in which we conduct clinical trials. Most of our foreign expenses incurred are associated with initiating or conducting clinical trials for enobosarm and GTx-758 at clinical trial sites in Europe. Consequently, changes in exchange rates could result in material exchange losses and could unpredictably, materially and adversely affect our financial position, results of operations and cash flows. A hypothetical 10% increase or decrease in foreign exchange rates would result in an immaterial change in our financial assets and liabilities denominated in euros. This potential change is based on a sensitivity analysis performed on our financial position at December 31, 2016. Actual results may differ materially. We have elected not to hedge our exposure to foreign currency fluctuations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our financial statements and the reports of our independent registered public accounting firm are included in this Annual Report on Form 10-K beginning on page F-1. The index to these reports and our financial statements is included in Part IV, Item 15 below.
73
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended (the "Exchange Act")) that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC's rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow for timely decisions regarding required disclosures.
We have carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this report. Based on the evaluation of these disclosure controls and procedures, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures were effective.
Management's Report on Internal Control Over Financial Reporting
We, as management of GTx, Inc., are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with United States generally accepted accounting principles. Any system of internal control, no matter how well designed, has inherent limitations, including the possibility that a control can be circumvented or overridden and misstatements due to error or fraud may occur and not be detected. Also, because of changes in conditions, internal control effectiveness may vary over time. Accordingly, even an effective system of internal control will provide only reasonable assurance that the objectives of the internal control system are met.
Under the supervision and with the participation of management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2016 using the criteria for effective internal control over financial reporting as described in "Internal Control — Integrated Framework," issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on this evaluation, we concluded that, as of December 31, 2016, our internal control over financial reporting was effective. The effectiveness of our internal control over financial reporting has been audited by Ernst & Young LLP, independent registered public accounting firm.
Attestation Report of the Independent Registered Public Accounting Firm
Ernst & Young LLP, an independent registered public accounting firm, has issued an audit report on our internal control over financial reporting, which report is included elsewhere herein.
74
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the fourth quarter of 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
75
Certain information required by Part III is omitted from this Annual Report on Form 10-K because we will file our definitive proxy statement for our 2017 Annual Meeting of Stockholders with the U.S. Securities and Exchange Commission pursuant to Regulation 14A (the "2017 Proxy Statement") not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information included in the 2017 Proxy Statement is incorporated herein by reference.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
(1) The information required by this Item concerning our directors and nominees for director, including information with respect to our audit committee and audit committee financial experts, may be found under the section entitled "Proposal No. 1 — Election of Directors" and "Additional Information About the Board of Directors and Certain Corporate Governance Matters" appearing in the 2017 Proxy Statement. Such information is incorporated herein by reference.
(2) The information required by this Item concerning compliance with Section 16(a) of the Securities Exchange Act of 1934 may be found in the section entitled "Section 16(a) Beneficial Ownership Reporting Compliance" appearing in the 2017 Proxy Statement. Such information is incorporated herein by reference.
(3) The information required by this Item concerning our executive officers is set forth in the section entitled "Management — Executive Officers of the Registrant" in Part I, Item 1 of this Form 10-K.
(4) Our Board has adopted a Code of Business Conduct and Ethics applicable to all officers, directors and employees as well as Guidelines on Governance Issues. These documents are available on our Web site (www.gtxinc.com) under "Investors" at "Corporate Governance." We will provide a copy of these documents to any person, without charge, upon request, by writing to us at GTx, Inc., Chief Legal Officer, 175 Toyota Plaza, Suite 700, Memphis, Tennessee 38103. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding an amendment to, or waiver from, a provision of the Code of Business Conduct and Ethics by posting such information on our Web site at the address and the location specified above.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item concerning director and executive compensation is incorporated herein by reference to the information from the 2017 Proxy Statement under the sections entitled "Executive Compensation" and "Director Compensation."
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
(1) The information required by this Item with respect to security ownership of certain beneficial owners and management is incorporated herein by reference to the information from the 2017 Proxy Statement under the section entitled "Security Ownership of Certain Beneficial Owners and Management."
76
(2) The information required by this Item with respect to securities authorized for issuance under our equity compensation plans is incorporated herein by reference to the information from the 2017 Proxy Statement under the section entitled "Equity Compensation Plan Information."
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
(1) The information required by this Item concerning related party transactions is incorporated herein by reference to the information from the 2017 Proxy Statement under the section entitled "Related Party Transactions and Indemnification."
(2) The information required by this Item concerning director independence is incorporated herein by reference to the information from the 2017 Proxy Statement under the section entitled "Additional Information About the Board of Directors and Certain Corporate Governance Matters — Director Independence."
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item is incorporated herein by reference to the information from the 2017 Proxy Statement under the section entitled "Proposal No. 2 — Ratification of Appointment of Independent Registered Public Accounting Firm."
77
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)(1)
Index to Financial Statements
Page
|
Description
|
|
|---|---|---|
F-2 |
Management's Report on Internal Control Over Financial Reporting | |
F-3 |
Reports of Independent Registered Public Accounting Firm | |
F-5 |
Balance Sheets at December 31, 2016 and 2015 | |
F-6 |
Statements of Operations for the Years Ended December 31, 2016, 2015 and 2014 | |
F-7 |
Statements of Stockholders' Equity for the Years Ended December 31, 2016, 2015 and 2014 | |
F-8 |
Statements of Cash Flows for the Years Ended December 31, 2016, 2015 and 2014 | |
F-9 |
Notes to Financial Statements |
(a)(2) Financial statement schedules are omitted as they are not applicable.
(a)(3) See Item 15(b) below.
(b) Exhibits — The following exhibits are included herein or incorporated herein by reference:
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 2.1 | Asset Purchase Agreement dated as of September 28, 2012 between the Registrant and Strakan International S.à r.l. | 8-K | 000-50549 | 2.1 | 10/03/2012 | ||||||
3.1 |
Restated Certificate of Incorporation of GTx, Inc. |
S-3 |
333-127175 |
4.1 |
08/04/2005 |
||||||
3.2 |
Certificate of Amendment of Restated Certificate of Incorporation of GTx, Inc. |
8-K |
000-50549 |
3.2 |
05/06/2011 |
||||||
3.3 |
Certificate of Amendment of Restated Certificate of Incorporation of GTx, Inc. |
8-K |
000-50549 |
3.3 |
05/09/2014 |
||||||
3.4 |
Certificate of Amendment of Restated Certificate of Incorporation of GTx, Inc. |
10-Q |
000-50549 |
3.4 |
05/11/2015 |
||||||
3.5 |
Certificate of Amendment of Restated Certificate of Incorporation of GTx, Inc. |
8-K |
000-50549 |
3.1 |
12/05/2016 |
||||||
3.6 |
Amended and Restated Bylaws of GTx, Inc. |
8-K |
000-50549 |
3.2 |
07/26/2007 |
||||||
4.1 |
Reference is made to Exhibits 3.1, 3.2, 3.3, 3.4, 3.5 and 3.6 |
- |
- |
- |
- |
||||||
4.2 |
Specimen of Common Stock Certificate |
S-1 |
333-109700 |
4.2 |
12/22/2003 |
||||||
78
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 4.3 | Amended and Restated Registration Rights Agreement between Registrant and J. R. Hyde, III dated August 7, 2003 | S-1 | 333-109700 | 4.4 | 10/15/2003 | ||||||
4.4 |
Consent, Waiver and Amendment among Registrant, J. R. Hyde, III and Pittco Associates, L.P. dated December 3, 2007 |
S-3 |
333-148321 |
4.6 |
12/26/2007 |
||||||
4.5 |
Waiver and Amendment Agreement among Registrant, J.R. Hyde, III and Pittco Associates, L.P. dated March 6, 2014 |
10-K |
000-50549 |
4.5 |
03/12/2014 |
||||||
4.6 |
Amended and Restated Registration Rights Agreement among Registrant, J.R. Hyde, III and The Pyramid Peak Foundation, dated August 4, 2014 |
10-Q |
000-50549 |
4.6 |
08/05/2014 |
||||||
4.7 |
Consent, Waiver and Amendment Agreement between Registrant and J.R. Hyde, III and Pittco Associates, L.P., dated August 4, 2014 |
10-Q |
000-50549 |
4.8 |
08/05/2014 |
||||||
4.8 |
Form of Common Stock Warrant, issued by Registrant pursuant to the Purchase Agreement, dated November 9, 2014, between Registrant and the purchasers identified in Exhibit A therein |
10-K |
000-50549 |
4.9 |
03/16/2015 |
||||||
4.9 |
Form of Warrant Amendment Agreement entered into effective as of March 25, 2016 between Registrant and each holder of a Common Stock Warrant originally issued on November 14, 2014 |
10-Q |
000-50549 |
4.9 |
05/10/2016 |
||||||
10.1† |
Consolidated, Amended, and Restated License Agreement dated July 24, 2007, between Registrant and University of Tennessee Research Foundation |
10-Q |
000-50549 |
10.40 |
11/09/2007 |
||||||
10.2 |
First Amendment, dated December 29, 2008, to the Consolidated, Amended and Restated License Agreement dated July 24, 2007 between the Registrant and University of Tennessee Research Foundation |
10-K |
000-50549 |
10.47 |
03/03/2009 |
||||||
10.3* |
Form of Indemnification Agreement |
S-1 |
333-109700 |
10.12 |
12/22/2003 |
||||||
79
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 10.4*+ | Genotherapeutics, Inc. 1999 Stock Option Plan, as amended through December 10, 2009 (refiled to reflect reverse stock split effected on December 5, 2016), and Form of Stock Option Agreement | - | - | - | - | ||||||
10.5*+ |
GTx, Inc. 2000 Stock Option Plan, as amended through December 10, 2009 (refiled to reflect reverse stock split effected on December 5, 2016), and Form of Stock Option Agreement |
- |
- |
- |
- |
||||||
10.6*+ |
GTx, Inc. 2001 Stock Option Plan, as amended through November 3, 2009 (refiled to reflect reverse stock split effected on December 5, 2016), and Form of Stock Option Agreement |
- |
- |
- |
- |
||||||
10.7*+ |
GTx, Inc. 2002 Stock Option Plan, as amended through November 3, 2009 (refiled to reflect reverse stock split effected on December 5, 2016), and Form of Stock Option Agreement |
- |
- |
- |
- |
||||||
10.8* |
GTx, Inc. 2004 Equity Incentive Plan, as originally adopted, and Form of Stock Option Agreement |
S-1 |
333-109700 |
10.5 |
01/15/2004 |
||||||
10.9* |
GTx, Inc. 2004 Equity Incentive Plan, as amended effective April 30, 2008 |
8-K |
000-50549 |
10.6 |
05/06/2008 |
||||||
10.10*+ |
GTx, Inc. 2004 Equity Incentive Plan, as amended effective November 4, 2008 (refiled to reflect reverse stock split effected on December 5, 2016) and Form of Stock Option Agreement |
- |
- |
- |
- |
||||||
10.11* |
GTx, Inc. 2004 Non-Employee Directors' Stock Option Plan and Form of Stock Option Agreement, as originally adopted |
S-1 |
333-109700 |
10.6 |
01/15/2004 |
||||||
10.12* |
Amended and Restated GTx, Inc. 2004 Non-Employee Directors' Stock Option Plan, effective April 26, 2006 |
8-K |
000-50549 |
10.1 |
04/27/2006 |
||||||
10.13* |
Form of Stock Option Agreement under the Amended and Restated GTx, Inc. 2004 Non-Employee Directors' Stock Option Plan |
10-Q |
000-50549 |
10.35 |
08/09/2006 |
||||||
80
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 10.14*+ | Amended and Restated GTx, Inc. 2004 Non-Employee Directors' Stock Option Plan, as amended effective November 4, 2008 (refiled to reflect reverse stock split effected on December 5, 2016) | - | - | - | - | ||||||
10.15* |
GTx, Inc. 2013 Equity Incentive Plan, as originally adopted |
S-8 |
333-188377 |
99.1 |
05/06/2013 |
||||||
10.16*+ |
GTx, Inc. 2013 Equity Incentive Plan, as amended effective May 6, 2015 (refiled to reflect reverse stock split effected on December 5, 2016) |
- |
- |
- |
- |
||||||
10.17* |
Form of Stock Option Grant Notice and Option Agreement under the GTx, Inc. 2013 Equity Incentive Plan (Standard Form) |
10-Q |
000-50549 |
10.2 |
07/22/2013 |
||||||
10.18* |
Form of Retention Stock Option Grant Notice and Option Agreement under the GTx, Inc. 2013 Equity Incentive Plan |
10-Q |
000-50549 |
10.3 |
11/12/2013 |
||||||
10.19* |
Form of Retention Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement under the GTx, Inc. 2013 Equity Incentive Plan |
10-Q |
000-50549 |
10.4 |
11/12/2013 |
||||||
10.20* |
Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement under the GTx, Inc. 2013 Equity Incentive Plan |
10-Q |
000-50549 |
10.5 |
05/11/2015 |
||||||
10.21*+ |
GTx, Inc. 2013 Non-Employee Director Equity Incentive Plan, as originally adopted (refiled to reflect reverse stock split effected on December 5, 2016) |
- |
- |
- |
- |
||||||
10.22* |
Form of Stock Option Grant Notice and Option Agreement under the GTx, Inc. 2013 Non-Employee Director Equity Incentive Plan |
10-Q |
000-50549 |
10.4 |
07/22/2013 |
||||||
10.23* |
Employment Agreement dated February 12, 2015, between Registrant and Robert J. Wills |
10-Q |
000-50549 |
10.4 |
05/11/2015 |
||||||
10.24* |
Employment Agreement dated July 13, 2015, between Registrant and Diane C. Young |
10-Q |
000-50549 |
10.1 |
11/09/2015 |
||||||
81
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 10.25* | Amended and Restated Employment Agreement dated February 12, 2015, between Registrant and Marc S. Hanover | 10-K | 000-50549 | 10.25 | 03/16/2015 | ||||||
10.26* |
Amended and Restated Employment Agreement dated February 14, 2013, between Registrant and Henry P. Doggrell |
10-K |
000-50549 |
10.22 |
03/05/2013 |
||||||
10.27* |
Employment Agreement dated October 1, 2013 between Registrant and Jason T. Shackelford |
10-K |
000-50549 |
10.29 |
03/16/2015 |
||||||
10.28*+ |
Employment Agreement dated January 6, 2017 between Registrant and Jason T. Shackelford |
- |
- |
- |
- |
||||||
10.29* |
Form of Retention Benefits Letter Agreement for Mitchell S. Steiner and Marc S. Hanover |
10-Q |
000-50549 |
10.1 |
11/12/2013 |
||||||
10.30* |
Form of Retention Benefits Letter Agreement for Jason T. Shackelford and Henry P. Doggrell |
10-Q |
000-50549 |
10.2 |
11/12/2013 |
||||||
10.31* |
Amended and Restated GTx, Inc. Executive Bonus Compensation Plan, effective November 4, 2008 |
10-K |
000-50549 |
10.53 |
03/03/2009 |
||||||
10.32* |
2016 Compensation Information for Registrant's Executive Officers |
10-Q |
000-50549 |
10.1 |
05/10/2016 |
||||||
10.33* |
Directors' Deferred Compensation Plan, as amended and restated effective February 14, 2013 |
10-K |
000-50549 |
10.28 |
03/05/2013 |
||||||
10.34*+ |
Directors' Deferred Compensation Plan, as amended and restated effective February 18, 2016 (refiled to reflect reverse stock split effected on December 5, 2016) |
- |
- |
- |
- |
||||||
10.35* |
Non-Employee Director Compensation Policy of GTx, Inc., effective January 1, 2016 |
10-K |
000-50549 |
10.39 |
03/15/2016 |
||||||
10.36 |
Lease Agreement, dated March 7, 2001, between The University of Tennessee and TriStar Enterprises, Inc. |
S-1 |
333-109700 |
10.13 |
10/15/2003 |
||||||
10.37 |
Sublease Agreement dated October 1, 2000, as amended, between Registrant and TriStar Enterprises, Inc. |
S-1 |
333-109700 |
10.14 |
10/15/2003 |
||||||
82
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 10.38 | Sublease Agreement dated April 1, 2005, as amended, between Registrant and TriStar Enterprises, Inc. | 10-Q | 000-50549 | 10.27 | 07/27/2005 | ||||||
10.39 |
Sublease Agreement dated October 1, 2009 between Registrant and University of Tennessee Research Foundation |
10-K |
000-50549 |
10.55 |
03/15/2010 |
||||||
10.40 |
Memorandum of Understanding Concerning the Lease Agreement between The University of Tennessee Research Foundation and the Registrant as Amended July 20, 2009 |
10-Q |
000-50549 |
10.59 |
08/09/2011 |
||||||
10.41 |
Second Memorandum of Understanding Concerning the Lease Agreement between Registrant and The University of Tennessee Research Foundation as Amended July 20, 2009 |
10-Q |
000-50549 |
10.5 |
07/22/2013 |
||||||
10.42 |
Third Memorandum of Understanding, made effective as of October 1, 2013, Concerning the Lease Agreement between Registrant and The University of Tennessee Research Foundation as Amended July 20, 2009 |
10-Q |
000-50549 |
10.5 |
11/12/2013 |
||||||
10.43 |
Sublease Agreement, dated December 17, 2007, by and between the Registrant and ESS SUSA Holdings, LLC |
10-K |
000-50549 |
10.46 |
03/11/2008 |
||||||
10.44 |
First Amendment, dated July 21, 2008, to the Sublease and Parking Sublicense Agreements dated December 17, 2007 by and between the Registrant and ESS SUSA Holdings, LLC |
10-K |
000-50549 |
10.54 |
03/03/2009 |
||||||
10.45 |
Second Amendment to Sublease and Parking Sublicense Agreements dated January 1, 2011 by and between the Registrant and ESS SUSA Holdings, LLC |
10-K |
000-50549 |
10.57 |
03/08/2011 |
||||||
10.46 |
Lease agreement, dated April 13, 2015, between Registrant and Hertz Memphis Three LLC |
10-Q |
000-50549 |
10.1 |
08/10/2015 |
||||||
83
| |
|
Incorporation By Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
||||||||||
| Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||||
| 10.47 | Purchase Agreement, dated November 9, 2014, between Registrant and the purchasers identified in Exhibit A therein | 8-K | 000-50549 | 10.1 | 11/10/2014 | ||||||
10.48 |
Form of Subscription Agreement for October 2016 registered direct offering |
8-K |
000-50549 |
10.1 |
10/12/2016 |
||||||
23.1+ |
Consent of Independent Registered Public Accounting Firm |
- |
- |
- |
- |
||||||
24.1+ |
Power of Attorney (included on the signature pages hereto) |
- |
- |
- |
- |
||||||
31.1+ |
Certification of Chief Executive Officer, as required by Rule 13a-14(a) or Rule 15d-14(a) |
- |
- |
- |
- |
||||||
31.2+ |
Certification of Principal Financial Officer, as required by Rule 13a-14(a) or Rule 15d-14(a) |
- |
- |
- |
- |
||||||
32.1+ |
Certification of Chief Executive Officer, as required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350)(1) |
- |
- |
- |
- |
||||||
32.2+ |
Certification of Principal Financial Officer, as required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350)(1) |
- |
- |
- |
- |
||||||
101.INS+ |
XBRL Instance Document |
- |
- |
- |
- |
||||||
101.SCH+ |
XBRL Taxonomy Extension Schema Document |
- |
- |
- |
- |
||||||
101.CAL+ |
XBRL Taxonomy Extension Calculation Linkbase Document |
- |
- |
- |
- |
||||||
101.DEF+ |
XBRL Taxonomy Extension Definition Linkbase Document |
- |
- |
- |
- |
||||||
101.LAB+ |
XBRL Taxonomy Extension Labels Linkbase Document |
- |
- |
- |
- |
||||||
101.PRE+ |
XBRL Taxonomy Extension Presentation Linkbase Document |
- |
- |
- |
- |
||||||
- †
- Confidential
treatment has been granted with respect to certain portions of this exhibit. This exhibit omits the information subject to this
confidentiality request. Omitted portions have been filed separately with the SEC.
- *
- Indicates a management contract or compensation plan or arrangement.
84
- +
- Filed
herewith
- (1)
- This certification accompanies the Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing.
None provided.
85
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
GTx, Inc. |
|
||||
|
By |
/s/ Marc S. Hanover |
|
|||
|
Marc S. Hanover |
|
||||
|
Chief Executive Officer | Date: March 24, 2017 |
||||
|
(Principal Executive Officer) |
|
KNOW ALL PERSONS BY THESE PRESENT, that each person whose signature appears below constitutes and appoints Marc S. Hanover and Jason T. Shackelford, and each of them, acting individually, as his attorney-in-fact, each with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| /s/ Marc S. Hanover Marc S. Hanover |
Chief Executive Officer (Principal Executive Officer) |
March 24, 2017 | ||
/s/ Jason T. Shackelford Jason T. Shackelford |
Vice President, Finance and Accounting and Principal Financial and Accounting Officer (Principal Financial and Accounting Officer) |
March 24, 2017 |
||
/s/ Robert J. Wills Robert J. Wills, B.S., M.S., Ph.D. |
Executive Chairman of the Board of Directors |
March 24, 2017 |
||
/s/ Michael G. Carter Michael G. Carter, M. D. |
Director |
March 24, 2017 |
||
/s/ J. Kenneth Glass J. Kenneth Glass |
Director |
March 24, 2017 |
86
| /s/ J. R. Hyde, III J. R. Hyde, III |
Director | March 24, 2017 | ||
/s/ Garry A. Neil Garry A. Neil, M.D. |
Director |
March 24, 2017 |
||
/s/ Kenneth S. Robinson Kenneth S. Robinson, M.D. |
Director |
March 24, 2017 |
87
F-1
MANAGEMENT'S REPORT ON
INTERNAL CONTROL OVER FINANCIAL REPORTING
We, as management of GTx, Inc., are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with United States generally accepted accounting principles. Any system of internal control, no matter how well designed, has inherent limitations, including the possibility that a control can be circumvented or overridden and misstatements due to error or fraud may occur and not be detected. Also, because of changes in conditions, internal control effectiveness may vary over time. Accordingly, even an effective system of internal control will provide only reasonable assurance that the objectives of the internal control system are met.
Under the supervision and with the participation of management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2016 using the criteria for effective internal control over financial reporting as described in "Internal Control — Integrated Framework," issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on this evaluation, we concluded that, as of December 31, 2016, our internal control over financial reporting was effective. The effectiveness of our internal control over financial reporting has been audited by Ernst & Young LLP, independent registered public accounting firm who also audited the Company's financial statements included in this Annual Report on Form 10-K. Ernst & Young LLP's report on the Company's internal control over financial reporting is included in this Annual Report on the 10-K.
| /s/ Marc S. Hanover Marc S. Hanover Chief Executive Officer Principal Executive Officer |
/s/ Jason T. Shackelford Jason T. Shackelford Vice President, Finance and Accounting Principal Financial and Accounting Officer |
Memphis,
Tennessee
March 24, 2017
F-2
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of GTx, Inc.
We have audited GTx, Inc.'s internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), (the COSO criteria). GTx, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, GTx, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of GTx, Inc. as of December 31, 2016 and 2015, and the related statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2016 and our report dated March 24, 2017 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Memphis,
Tennessee
March 24, 2017
F-3
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of GTx, Inc.
We have audited the accompanying balance sheets of GTx, Inc. as of December 31, 2016 and 2015, and the related statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2016. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of GTx, Inc. at December 31, 2016 and 2015, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2016, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), GTx, Inc.'s internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 24, 2017 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Memphis,
Tennessee
March 24, 2017
F-4
GTx, Inc.
BALANCE SHEETS
(in thousands, except share and per share data)
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | |||||
ASSETS |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 8,910 | $ | 14,056 | |||
Short-term investments |
12,959 | 15,200 | |||||
Prepaid expenses and other current assets |
2,429 | 2,633 | |||||
| | | | | | | | |
Total current assets |
24,298 | 31,889 | |||||
Property and equipment, net |
81 | 5 | |||||
Intangible assets, net |
123 | 137 | |||||
| | | | | | | | |
Total assets |
$ | 24,502 | $ | 32,031 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||||
Current liabilities: |
|||||||
Accounts payable |
$ | 1,220 | $ | 382 | |||
Warrant liability |
- | 27,349 | |||||
Accrued expenses and other current liabilities |
3,391 | 2,441 | |||||
| | | | | | | | |
Total current liabilities |
4,611 | 30,172 | |||||
Commitments and contingencies |
|||||||
Stockholders' equity: |
|||||||
Common stock, $0.001 par value: 60,000,000 and 400,000,000 shares authorized at December 31, 2016 and December 31, 2015, respectively; 15,919,572 and 14,037,411 shares issued and outstanding at December 31, 2016 and December 31, 2015, respectively |
16 | 14 | |||||
Additional paid-in capital |
551,073 | 515,319 | |||||
Accumulated deficit |
(531,198 | ) | (513,474 | ) | |||
| | | | | | | | |
Total stockholders' equity |
19,891 | 1,859 | |||||
| | | | | | | | |
Total liabilities and stockholders' equity |
$ | 24,502 | $ | 32,031 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these financial statements.
F-5
GTx, Inc.
STATEMENTS OF OPERATIONS
(in thousands, except share and per share data)
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
Expenses: |
||||||||||
Research and development expenses |
$ | 17,228 | $ | 13,607 | $ | 20,870 | ||||
General and administrative expenses |
8,705 | 8,234 | 9,478 | |||||||
| | | | | | | | | | | |
Total expenses |
25,933 | 21,841 | 30,348 | |||||||
| | | | | | | | | | | |
Loss from operations |
(25,933 | ) | (21,841 | ) | (30,348 | ) | ||||
Other income (expense), net |
46 | 57 | (259 | ) | ||||||
Gain (loss) on change in fair value of warrant liability |
8,163 | 3,081 | (8,804 | ) | ||||||
| | | | | | | | | | | |
Net loss |
$ | (17,724 | ) | $ | (18,703 | ) | $ | (39,411 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share: |
||||||||||
Basic |
$ | (1.22 | ) | $ | (1.33 | ) | $ | (4.82 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
$ | (1.22 | ) | $ | (1.47 | ) | $ | (4.82 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average shares outstanding: |
||||||||||
Basic |
14,559,541 | 14,036,468 | 8,180,770 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
14,559,541 | 14,777,404 | 8,180,770 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
F-6
GTx, Inc.
STATEMENTS OF STOCKHOLDERS' EQUITY
For the Years Ended December 31, 2016, 2015 and 2014
(in thousands, except share data)
| |
Stockholders' Equity | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Common Stock | |
|
|
||||||||||||
| |
Additional Paid-in Capital |
Accumulated Deficit |
Total Stockholders' Equity |
|||||||||||||
| |
Shares | Amount | ||||||||||||||
Balances at January 1, 2014 |
6,318,539 | $ | 6 | $ | 466,038 | $ | (455,360 | ) | $ | 10,684 | ||||||
Issuance of common stock and warrants in March 2014 private placement, net of offering costs |
1,197,605 | 1 | 21,134 | - | 21,135 | |||||||||||
Issuance of common stock and warrants in November 2014 private placement, net of offering costs |
6,431,111 | 6 | 21,478 | - | 21,484 | |||||||||||
Vesting of restricted stock units, net of shares withheld for tax payments |
85,309 | 1 | (617 | ) | - | (616 | ) | |||||||||
Directors' deferred compensation |
- | - | 125 | - | 125 | |||||||||||
Share-based compensation |
- | - | 4,428 | - | 4,428 | |||||||||||
Net loss |
- | - | - | (39,411 | ) | (39,411 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balances at December 31, 2014 |
14,032,564 | 14 | 512,586 | (494,771 | ) | 17,829 | ||||||||||
Issuance of common stock under deferred compensation arrangements |
4,847 | - | - | - | - | |||||||||||
Directors' deferred compensation |
- | - | 113 | - | 113 | |||||||||||
Share-based compensation |
- | - | 2,620 | - | 2,620 | |||||||||||
Net loss |
- | - | - | (18,703 | ) | (18,703 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balances at December 31, 2015 |
14,037,411 | 14 | 515,319 | (513,474 | ) | 1,859 | ||||||||||
Issuance of common stock in October 2016 registered direct offering, net of offering costs |
1,728,395 | 2 | 13,690 | - | 13,692 | |||||||||||
Vesting of restricted stock units, net of shares withheld for tax payments |
154,170 | - | (208 | ) | - | (208 | ) | |||||||||
Directors' deferred compensation |
- | - | 132 | - | 132 | |||||||||||
Share-based compensation |
- | - | 2,957 | - | 2,957 | |||||||||||
Warrant liability reclassification |
- | - | 19,186 | - | 19,186 | |||||||||||
Settlement of fractional shares upon reverse stock split |
(404 | ) | - | (3 | ) | - | (3 | ) | ||||||||
Net loss |
- | - | - | (17,724 | ) | (17,724 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balances at December 31, 2016 |
15,919,572 | $ | 16 | $ | 551,073 | $ | (531,198 | ) | $ | 19,891 | ||||||
| | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
F-7
GTx, Inc.
STATEMENTS OF CASH FLOWS
(in thousands)
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
Cash flows from operating activities: |
||||||||||
Net loss |
$ | (17,724 | ) | $ | (18,703 | ) | $ | (39,411 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
(Gain) loss on change in fair value of warrant liability |
(8,163 | ) | (3,081 | ) | 8,804 | |||||
Private placement expenses recorded as other income (expense), net |
- | - | 297 | |||||||
Share-based compensation |
2,957 | 2,620 | 4,428 | |||||||
Directors' deferred compensation |
132 | 113 | 125 | |||||||
Depreciation and amortization |
28 | 43 | 102 | |||||||
Changes in assets and liabilities: |
||||||||||
Prepaid expenses and other assets |
204 | (1,458 | ) | (577 | ) | |||||
Accounts payable |
838 | (130 | ) | (296 | ) | |||||
Accrued expenses and other liabilities |
950 | 561 | (2,231 | ) | ||||||
| | | | | | | | | | | |
Net cash used in operating activities |
(20,778 | ) | (20,035 | ) | (28,759 | ) | ||||
| | | | | | | | | | | |
Cash flows from investing activities: |
||||||||||
Purchase of property and equipment |
(90 | ) | (4 | ) | (5 | ) | ||||
Purchase of short-term investments, held to maturity |
(35,404 | ) | (55,219 | ) | (41,905 | ) | ||||
Proceeds from maturities of short-term investments, held to maturity |
37,645 | 71,434 | 10,690 | |||||||
| | | | | | | | | | | |
Net cash provided by (used in) investing activities |
2,151 | 16,211 | (31,220 | ) | ||||||
| | | | | | | | | | | |
Cash flows from financing activities: |
||||||||||
Net proceeds from the issuance of common stock and warrants |
13,692 | - | 63,949 | |||||||
Tax payments related to shares withheld for vested restricted stock units |
(208 | ) | - | (617 | ) | |||||
Settlement of fractional shares upon reverse stock split |
(3 | ) | - | - | ||||||
Payments on capital lease and financed equipment obligations |
- | - | (2 | ) | ||||||
| | | | | | | | | | | |
Net cash provided by financing activities |
13,481 | - | 63,330 | |||||||
| | | | | | | | | | | |
Net (decrease) increase in cash and cash equivalents |
(5,146 | ) | (3,824 | ) | 3,351 | |||||
Cash and cash equivalents, beginning of period |
14,056 | 17,880 | 14,529 | |||||||
| | | | | | | | | | | |
Cash and cash equivalents, end of period |
$ | 8,910 | $ | 14,056 | $ | 17,880 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
F-8
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
1. Business and Going Concern
GTx, Inc. ("GTx" or the "Company"), a Delaware corporation incorporated on September 24, 1997 and headquartered in Memphis, Tennessee, is a biopharmaceutical company dedicated to the discovery, development and commercialization of small molecules for the treatment of cancer, including treatments for breast and prostate cancer, and other serious medical conditions.
The Company is developing selective androgen receptor modulators ("SARMs"), including its lead product candidate, enobosarm (GTx-024). SARMs are a class of drugs that the Company believes has the potential to be used as a novel hormonal therapy for the treatment of advanced breast cancer, as well as the potential to treat other serious medical conditions. The Company announced during the second quarter of 2014 positive results from a Phase 2 proof-of-concept, open-label clinical trial evaluating a 9 mg oral daily dose of enobosarm for the treatment of patients with estrogen receptor ("ER") positive and androgen receptor ("AR") positive metastatic breast cancer who have previously responded to hormonal therapy. The Company commenced enrollment in 2015 in a Phase 2 clinical trial designed to evaluate the efficacy and safety of enobosarm in patients whose advanced breast cancer is both ER positive and AR positive. During 2015, the Company also commenced enrollment in a Phase 2 proof-of-concept clinical trial designed to evaluate the efficacy and safety of enobosarm in patients with advanced AR positive triple-negative breast cancer ("TNBC").
The Company is also evaluating enobosarm and other compounds in its SARM portfolio for indications outside of oncology where unmet medical needs in muscle-related diseases may benefit from increasing muscle mass. In the first quarter of 2016, the Company initiated a Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with Stress Urinary Incontinence ("SUI"). The Company has also evaluated several SARM compounds, including enobosarm, in preclinical models of Duchenne Muscular Dystrophy ("DMD") where a SARM's ability to increase muscle mass may prove beneficial to patients suffering from DMD. Based on the Company's SARM data from these preclinical efforts, the Company has initiated discussions with potential collaboration partners to further develop a SARM for the treatment of DMD.
In March 2015, the Company entered into an exclusive license agreement with the University of Tennessee Research Foundation ("UTRF") to develop UTRF's proprietary selective androgen receptor degrader ("SARD") technology which may have the potential to provide compounds that can degrade multiple forms of AR to treat those patients who do not respond or are resistant to current therapies by inhibiting tumor growth in patients with progressive castration-resistant prostate cancer ("CRPC"). The Company is currently implementing an appropriate development program for SARDs and has selected lead SARD compounds that are undergoing further preclinical development, including formulation, pharmacokinetic and toxicology studies, required to support initial human clinical trials.
The Company's ability to pursue the continued development of SARMs and its SARD program is contingent upon its ability to obtain additional funding. Accordingly, the Company is actively seeking additional funding through the licensing, partnering or sale of certain assets to provide the Company with the necessary resources for the development of its preclinical and clinical product candidates.
Based on its current business plan and assumptions, the Company estimates that its current cash, cash equivalents and short-term investments together with interest thereon, will be sufficient to meet its projected operating requirements only into the fourth quarter of 2017. Accordingly, the Company will
F-9
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
need to raise substantial additional capital in the near term in order to fund its operations through and beyond the fourth quarter of 2017 and to continue as a going concern thereafter. Alternatively, the Company could modify its current business plan to preserve cash and continue as a going concern while evaluating future plans and activities. In addition, the Company has based its cash sufficiency estimates on its current business plan and its assumptions that may prove to be wrong. The Company could utilize its available capital resources sooner than it currently expects, and it could need additional funding to sustain its operations even sooner than currently anticipated. The Company believes, based on its current estimates of clinical trial expenditures and enrollment status, that the Company's existing capital resources will be adequate to enable it to complete its ongoing open-label Phase 2 clinical trial of enobosarm in patients with ER positive, AR positive advanced breast cancer and its ongoing Phase 2 clinical trial of enobosarm in postmenopausal women with SUI. However, the Company's existing capital resources will not be sufficient to allow it to complete its ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC and the Company will otherwise need to raise substantial additional capital in order to continue developing enobosarm for any of these indications. If the Company determines that its existing capital resources are not sufficient to enable it to complete its ongoing open-label Phase 2 clinical trial of enobosarm in patients with advanced AR positive TNBC, the Company may be unable or unwilling to enroll patients into the second stage of this trial even if the Company determines that the first stage milestone had been met. Accordingly, in order to enroll the second stage of and to complete this trial, the Company will need to obtain additional funding, which the Company may be unable to do in a timely manner or at all. Also, the Company's clinical trials may continue to encounter technical, enrollment or other difficulties that could increase its development costs beyond its current estimates or delay its development timelines, and the Company could otherwise exhaust its available financial resources sooner than the Company expects. In any event, the Company will need to raise substantial additional capital in order to:
- •
- potentially enroll the second stage of and complete the Company's ongoing open-label Phase 2 clinical trial of enobosarm in patients
with advanced AR positive TNBC;
- •
- undertake any further development of the Company's SARMs beyond its ongoing Phase 2 clinical trials of enobosarm in breast cancer
and SUI and its ongoing preclinical development activities related to the development of SARMs as a potential treatment for DMD;
- •
- initiate and complete human clinical studies of the Company's SARD program; and
- •
- fund the Company's operations and to continue as a going concern.
The Company has evaluated its capital resources and current business plans in accordance with the adoption of Accounting Standards Update (ASU) No. 2014-15, Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern, ("ASU 2014-15"), which is effective for the Company for the year ended December 31, 2016. ASU 2014-15 requires the assessment of an entity's ability to continue as a going concern for a period of one year after the date the entity's financial statements are issued and to provide related footnote disclosures, if necessary. As the Company currently estimates, based on its current business plan and assumptions, that its current cash, cash equivalents and short-term investments together with interest thereon, will be sufficient to meet its projected operating requirements only into the fourth quarter of 2017, the Company has evaluated its ability to continue as a going concern for one year after the date these financial statements are issued.
F-10
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
Pursuant to the guidance in ASU 2014-15, the Company believes that it has the ability to successfully implement plans to mitigate the conditions that may raise doubt about its ability to continue as a going concern as a significant portion of the Company's current business plan consists of uncommitted spending. The Company's plans for mitigation include reducing or delaying expenditures by postponing or discontinuing planned clinical or preclinical development and implementing cost saving measures related to other research and development and general and administrative expenditures. If the Company is unable to raise substantial additional capital through the licensing, partnering or sale of certain assets or through a third party financing, the Company believes it is probable that these plans could be effectively implemented to successfully mitigate the considerations regarding the Company's ability to continue as a going concern for one year after the date these financial statements are issued.
Therefore, these financial statements do not include any adjustments or charges that might be necessary should the Company be unable to continue as a going concern, such as charges related to impairment of its assets, the recoverability and classification of assets or the amounts and classification of liabilities or other similar adjustments.
2. Significant Accounting Policies
Basis of Presentation
The accompanying financial statements have been prepared in accordance with U.S. generally accepted accounting principles ("U.S. GAAP"). Additionally, GTx operates in one business segment.
On December 5, 2016, the Company effected a one-for-ten reverse stock split of its common stock through an amendment to its restated certification of incorporation. As of the effective time of the reverse stock split, every ten shares of the Company's issued and outstanding common stock were automatically combined and reclassified into one issued and outstanding share of common stock, without any change in par value per share. The amendment to the Company's restated certification of incorporation also reduced the number of authorized shares of common stock from 400,000,000 to 60,000,000 shares. The reverse stock split affected all shares of the Company's common stock outstanding immediately prior to the effective time of the reverse stock split. Additionally, as a result of the reverse stock split, proportionate adjustments were made to the per share exercise price and/or the number of shares issuable upon the exercise or vesting of all stock options, restricted stock units and warrants issued by the Company and outstanding immediately prior to the effective time, which resulted in a proportionate decrease in the number of shares of the Company's common stock reserved for issuance upon exercise or vesting of such stock options, restricted stock units and warrants, and, in the case of stock options and warrants, a proportionate increase in the exercise price of all such stock options and warrants. In addition, the number of shares reserved for issuance under the Company's equity compensation plans immediately prior to the effective time was reduced proportionately. No fractional shares were issued as a result of the reverse stock split. Stockholders who have otherwise been entitled to receive a fractional share received a cash payment in lieu thereof.
As the par value per share of the Company's common stock remained unchanged at $0.001 per share, a total of $144 was retroactively reclassified from common stock to additional paid-in capital in the Company's balance sheets and statements of stockholders' equity. All references to shares of common stock, all per share data, and all warrant, stock option and restricted stock unit ("RSU")
F-11
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
activity for all periods presented in these financial statements and notes to financial statements have been adjusted to reflect the reverse stock split on a retroactive basis.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual amounts and results could differ from those estimates.
Cash and Cash Equivalents
The Company considers highly liquid investments with initial maturities of three months or less to be cash equivalents.
Short-term Investments
At December 31, 2016 and 2015, short-term investments consisted of Federal Deposit Insurance Corporation ("FDIC") insured certificates of deposit with original maturities of greater than three months and less than one year.
Property and Equipment
Property and equipment is stated at cost. Amortization of leasehold improvements is recognized over the shorter of the estimated useful life of the leasehold improvement or the lease term. Depreciation is computed using the straight-line method over the estimated useful lives as follows:
| Office equipment | 3 to 5 years | ||||
Leasehold improvements |
3 to 7 years |
||||
Furniture and fixtures |
5 years |
||||
Computer equipment and software |
3 years |
Warrant Liability
In November 2014, the Company issued warrants to purchase 6,430,948 shares of its common stock. The Company classified these warrants as a liability on its balance sheet since the warrants contained certain terms that could have required the Company (or its successor) to purchase the warrants for cash in an amount equal to the value (as calculated utilizing a contractually-agreed Black-Scholes-Merton option pricing valuation model ("Black-Scholes Model")) of the unexercised portion of the warrants in connection with certain change of control transactions occurring on or prior to December 31, 2016, with such cash payment capped at an amount equal to $1.25 per unexercised share underlying each warrant. As a result of the provision of the warrants requiring cash settlement upon certain change of control transactions, the Company was required to account for these warrants as a liability at fair value and the estimated warrant liability was required to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such
F-12
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
provision on December 31, 2016. Effective March 25, 2016, each of the warrants was amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions. These warrants were no longer accounted for as a liability as of March 31, 2016. The Company recorded a non-cash reclassification of the warrant fair value to stockholders' equity based on the warrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required.
Fair Value of Financial Instruments and Warrant Liability
The carrying amounts of the Company's financial instruments (which include cash, cash equivalents, short-term investments, and accounts payable) and its prior warrant liability approximate their fair values. The fair value of the warrant liability was estimated using the Black-Scholes-Merton Model. See Note 6, Stockholders' Equity, for additional disclosure on the valuation methodology and significant assumptions. The Company's financial assets and liabilities are classified within a three-level fair value hierarchy that prioritizes the inputs used to measure fair value, which is defined as follows:
Level 1 — Quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date
Level 2 — Inputs other than quoted prices in active markets that are observable for the asset or liability, either directly or indirectly
Level 3 — Inputs that are unobservable for the asset or liability
There were no assets or liabilities measured at fair value on a recurring basis as of December 31, 2016. Liabilities measured at fair value on a recurring basis as of December 31, 2015 included only the Company's warrant liability of $27,349, which was classified within Level 3 of the hierarchy. A non-cash gain of $8,163 related to the change in the fair value of the warrant liability was recognized during the year ended December 31, 2016 in the Company's statement of operations.
As the Company has the positive intent and ability to hold its certificates of deposit classified as short-term investments until maturity, these investments have been classified as held to maturity investments and are stated at cost, which approximates fair value. The Company considers these to be Level 2 investments as the fair values of these investments are determined using third-party pricing sources, which generally utilize observable inputs, such as interest rates and maturities of similar assets.
Concentration of Risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist of cash and cash equivalents and short-term investments. The Company has established guidelines relating to diversification and maturities of its cash equivalents and short-term investments which are designed to manage risk. The Company's cash and cash equivalents consist of bank deposits, certificates of deposit, and money market mutual funds. Bank deposits may at times be in excess of FDIC insurance limits. The Company's short-term investments consist of FDIC insured certificates of deposit with original maturities of greater than three months and less than one year.
F-13
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
Research and Development Expenses
Research and development expenses include, but are not limited to, the Company's expenses for personnel, supplies, and facilities associated with research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. The Company expenses these costs in the period in which they are incurred. The Company estimates its liabilities for research and development expenses in order to match the recognition of expenses to the period in which the actual services are received. As such, accrued liabilities related to third party research and development activities are recognized based upon the Company's estimate of services received and degree of completion of the services in accordance with the specific third party contract.
Patent Costs
The Company expenses patent costs, including legal expenses, in the period in which they are incurred. Patent expenses are included in general and administrative expenses in the Company's statements of operations.
Income Taxes
The Company accounts for deferred taxes by recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, at December 31, 2016 and December 31, 2015, net of the valuation allowance, the net deferred tax assets were reduced to zero. See Note 8, Income Taxes, for further discussion.
Share-Based Compensation
The Company has stock option and equity incentive plans that provide for the purchase or acquisition of the Company's common stock by certain of the Company's employees and non-employees. The Company recognizes compensation expense for its share-based payments based on the fair value of the awards over the period during which an employee or non-employee is required to provide service in exchange for the award. See Note 3, Share-Based Compensation, for further discussion.
Other Income (Expense), Net
Other income (expense), net consists of foreign currency transaction gains and losses, interest earned on the Company's cash, cash equivalents and short-term investments, interest expense, and other non-operating income or expense. Other income (expense), net for the year ended December 31, 2014 also included expenses related to the private placement of common stock and warrants completed in November 2014 as the warrants issued were initially accounted for as a liability.
F-14
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
Basic and Diluted Net Loss Per Share
Basic and diluted net income (loss) per share attributable to common stockholders is calculated based on the weighted average number of common shares outstanding during the period. Diluted net income (loss) per share gives effect to the dilutive potential of common stock consisting of stock options, unvested RSUs and common stock warrants. For the year ended December 31, 2015, since the average market price of the shares underlying common stock warrants exceeded the exercise price of the warrants, and the presumed exercise of such warrants were dilutive to the net loss per share for the period, adjustments to net loss for the period were required to remove the change in fair value of the warrant liability.
The following table sets forth the computation of the Company's net loss per share is as follows:
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
Basic and diluted net loss per share |
||||||||||
Numerator: |
||||||||||
Net loss — basic |
$ | (17,724 | ) | $ | (18,703 | ) | $ | (39,411 | ) | |
Adjustments for the gain on change in fair value of the warrant liability |
- | (3,081 | ) | - | ||||||
| | | | | | | | | | | |
Net loss — diluted |
$ | (17,724 | ) | $ | (21,784 | ) | $ | (39,411 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Denominator: |
||||||||||
Weighted average shares outstanding — basic |
14,559,541 | 14,036,468 | 8,180,770 | |||||||
Dilutive warrants |
- | 556,372 | - | |||||||
Dilutive restricted stock units |
- | 184,379 | - | |||||||
Dilutive stock options |
- | 185 | - | |||||||
| | | | | | | | | | | |
Weighted average shares outstanding — diluted |
14,559,541 | 14,777,404 | 8,180,770 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share: |
||||||||||
Basic |
$ | (1.22 | ) | $ | (1.33 | ) | $ | (4.82 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
$ | (1.22 | ) | $ | (1.47 | ) | $ | (4.82 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average shares outstanding: |
||||||||||
Basic |
14,559,541 | 14,036,468 | 8,180,770 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
14,559,541 | 14,777,404 | 8,180,770 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average potential shares of common stock of 8,162,347, 838,745, and 2,462,877 were excluded from the calculation of diluted net loss per share for the years ended December 31, 2016, 2015 and 2014, respectively, as inclusion of the potential shares would have had an anti-dilutive effect on the net loss per share for the periods. At December 31, 2016, the Company had 15,919,572 shares of common stock outstanding.
F-15
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
Comprehensive Loss
For all periods presented, there were no differences between net loss and comprehensive loss.
Recent Accounting Pronouncements
In August 2014, the Financial Accounting Standards Board issued Accounting Standard Update 2014-15, Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. The new guidance is intended to define management's responsibility to evaluate whether there is substantial doubt about an organization's ability to continue as a going concern within one year of the date the financial statements are issued and to provide related footnote disclosure. This new guidance was effective for the year ended December 31, 2016 and interim periods thereafter.
Subsequent Events
The Company has evaluated all events or transactions that occurred after December 31, 2016 up through the date the financial statements were issued. There were no material recognizable or nonrecognizable subsequent events during the period evaluated.
3. Share-Based Compensation
Share-based payments include stock option and RSU grants under the Company's stock option and equity incentive plans and deferred compensation arrangements for the Company's non-employee directors.
The Company has granted and continues to grant to employees and non-employees options to purchase common stock under various plans at prices equal to the fair market value of its common stock on the dates the options are granted as determined in accordance with the terms of the applicable plan. The options have a term of ten years from the grant date and generally vest over three years from the grant date for director and non-employee options and over periods of up to five years from the grant date for employee options. Under the terms of the Company's stock option and equity incentive plans, employees generally have three months after the employment relationship ends to exercise all vested options except in the case of voluntary retirement, disability or death, where post-termination exercise periods are generally longer. The Company issues new shares of common stock upon the exercise of options. The Company estimates the fair value of stock option awards as of the date of the grant by applying the Black-Scholes Model. The application of this valuation model involves assumptions that are judgmental and highly sensitive in the determination of compensation expense.
The fair value of each stock option is amortized into compensation expense on a straight-line basis between the grant date for the award and each vesting date. The amount of share-based compensation expense recognized is reduced ratably over the vesting period by an estimate of the percentage of options granted that are expected to be forfeited or canceled before becoming fully vested.
Additionally, the Company periodically grants RSUs to its employees. The Company estimates the fair value of RSUs using the closing price of its common stock on the grant date. The fair value of the RSUs is amortized on a straight-line basis over the requisite service period of the awards. The amount of share-based compensation expense recognized is reduced ratably over the vesting period by an
F-16
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
estimate of the percentage of RSUs granted that are expected to be forfeited or canceled before becoming fully vested.
The following table summarizes share-based compensation expense included within the statements of operations for each of the three years in the period ended December 31, 2016:
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
Research and development expenses |
$ | 1,260 | $ | 1,210 | $ | 2,512 | ||||
General and administrative expenses |
1,829 | 1,523 | 2,041 | |||||||
| | | | | | | | | | | |
Total share-based compensation |
$ | 3,089 | $ | 2,733 | $ | 4,553 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Share-based compensation expense recorded in the statement of operations as general and administrative expense for the years ended December 31, 2016, 2015 and 2014 included share-based compensation expense related to deferred compensation arrangements for the Company's non-employee directors of $132, $113 and $125, respectively. See Note 9, Directors' Deferred Compensation Plan, for further discussion of deferred compensation arrangements for the Company's non-employee directors.
For the years ended December 31, 2016, 2015 and 2014, the weighted average grant date fair value per share of stock options granted was $5.45, $5.72 and $10.35, respectively. The key assumptions used in determining the grant date fair value of options granted in 2016, 2015 and 2014, and a summary of the methodology applied to develop each assumption is as follows:
| |
Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | 2014 | |||||||
Expected price volatility |
91.3% | 89.6% | 86.5% | |||||||
Risk-free interest rate |
2.0% | 1.6% | 2.3% | |||||||
Weighted average expected life in years |
6.9 years | 6.0 years | 6.9 years | |||||||
Dividend yield |
0% | 0% | 0% | |||||||
Expected Price Volatility — This is a measure of the amount by which a price has fluctuated or is expected to fluctuate. The Company based its determination of expected volatility on its historical stock price volatility. An increase in the expected price volatility will increase compensation expense.
Risk-Free Interest Rate — This is determined using U.S. Treasury rates where the term is consistent with the expected life of the stock options. An increase in the risk-free interest rate will increase compensation expense.
Expected Life — This is the period of time over which the options granted are expected to remain outstanding and is determined by calculating the average of the vesting term and the contractual term of the options. The Company has utilized this method due to the lack of historical option exercise information related to the Company's stock option and equity incentive plans. Options granted have a maximum term of ten years. An increase in the expected life will increase compensation expense.
F-17
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
Dividend Yield — The Company has not made any dividend payments nor does it have plans to pay dividends in the foreseeable future. An increase in the dividend yield will decrease compensation expense.
The following is a summary of stock option transactions for all of the Company's stock option and equity incentive plans for the three year period ended December 31, 2016:
| |
Number of Shares | Weighted Average Exercise Price Per Share |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at January 1, 2014 |
644,524 | $ | 65.79 | ||||
Options granted |
309,450 | 13.43 | |||||
Options forfeited or expired |
(143,537 | ) | 85.04 | ||||
Options exercised |
- | - | |||||
| | | | | | | | |
Options outstanding at December 31, 2014 |
810,437 | 42.39 | |||||
Options granted |
36,500 | 7.71 | |||||
Options forfeited or expired |
(48,628 | ) | 75.36 | ||||
Options exercised |
- | - | |||||
| | | | | | | | |
Options outstanding at December 31, 2015 |
798,309 | 38.80 | |||||
Options granted |
363,500 | 6.94 | |||||
Options forfeited or expired |
(71,829 | ) | 54.65 | ||||
Options exercised |
- | - | |||||
| | | | | | | | |
Options outstanding at December 31, 2016 |
1,089,980 | 27.13 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Options vested and expected to vest at December 31, 2016 |
1,049,750 | 27.82 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The following table summarizes information about stock options outstanding at December 31, 2016:
| Options Outstanding | Options Exercisable | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exercise Price
|
Number Outstanding |
Weighted Average Remaining Contractual Life (years) |
Weighted Average Exercise Price |
Number Exercisable |
Weighted Average Exercise Price |
|||||||||||
| $5.80 - $7.60 | 383,200 | 8.97 | $ | 6.98 | 14,586 | $ | 7.29 | |||||||||
| $7.80 - $18.80 | 383,600 | 7.11 | 14.89 | 131,619 | 17.69 | |||||||||||
| $26.50 - $204.00 | 323,180 | 3.46 | 65.54 | 290,883 | 68.39 | |||||||||||
| | | | | | | | | | | | | | | | | |
| 1,089,980 | 6.68 | 27.13 | 437,088 | 51.08 | ||||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
At December 31, 2016, the aggregate intrinsic value of all outstanding options was zero with a weighted average remaining contractual term of 6.68 years. Of the Company's outstanding options, 437,088 options were exercisable and had a weighted average remaining contractual term of 4.38 years and no aggregate intrinsic value. Additionally, the Company's vested and expected to vest options had a weighted average remaining contractual term of 6.61 years and no aggregate intrinsic value.
F-18
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
There were no options exercised during the years ended December 31, 2016 and 2015. At December 31, 2016, the total compensation cost related to non-vested options not yet recognized was $3,684, with a weighted average expense recognition period of 3.09 years. Shares available for future issuance under the Company's stock option and equity incentive plans were 702,043 at December 31, 2016. On January 1, 2017, shares available for future issuance under the 2013 equity incentive plan and 2013 non-employee director equity incentive plan increased by an aggregate of 686,783 shares in accordance with the automatic increase provisions of such plans.
During the year ended December 31, 2015, the Company granted 820,000 RSUs to employees, which had a weighted average grant date fair value per share of $7.20, of which a portion of each award vests annually over a three year period from the date of grant. During the year ended December 31, 2016, the Company granted 11,000 RSUs to employees, which had a weighted average grant date fair value per share of $6.40, and vest in full on January 1, 2018.
The following is a summary of the RSU transactions for all of the Company's equity incentive plans for the three year period ended December 31, 2016:
| |
Number of Shares | |||
|---|---|---|---|---|
Nonvested RSUs outstanding at January 1, 2014 |
122,500 | |||
RSUs granted |
- | |||
RSUs vested |
(122,500 | ) | ||
RSUs forfeited |
- | |||
| | | | | |
Nonvested RSUs outstanding at December 31, 2014 |
- | |||
RSUs granted |
820,000 | |||
RSUs vested |
- | |||
RSUs forfeited |
- | |||
| | | | | |
Nonvested RSUs outstanding at December 31, 2015 |
820,000 | |||
RSUs granted |
11,000 | |||
RSUs vested |
(184,001 | ) | ||
RSUs forfeited |
(62,000 | ) | ||
| | | | | |
Nonvested RSUs outstanding at December 31, 2016 |
584,999 | |||
| | | | | |
| | | | | |
| | | | | |
Nonvested RSUs expected to vest at December 31, 2016 |
571,639 | |||
| | | | | |
| | | | | |
| | | | | |
At December 31, 2016, the total compensation cost related to non-vested RSUs not yet recognized was $2,007, with a weighted average expense recognition period of 1.05 years. The number of RSUs vested during 2014 and 2016 included 37,191 and 29,829 shares, respectively, that were withheld on behalf of the Company's employees to satisfy the statutory tax withholding requirements.
F-19
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
4. Property and Equipment, Net
Property and equipment, net consisted of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | |||||
Computer equipment and software |
$ | 1,298 | $ | 1,435 | |||
Furniture and fixtures |
853 | 853 | |||||
Leasehold improvements |
355 | 355 | |||||
Office equipment |
211 | 211 | |||||
| | | | | | | | |
|
2,717 | 2,854 | |||||
Less: accumulated depreciation |
(2,636 | ) | (2,849 | ) | |||
| | | | | | | | |
|
$ | 81 | $ | 5 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Depreciation and amortization expense for the years ended December 31, 2016, 2015 and 2014 was $14, $27, and $88, respectively. Of these amounts, $2, $1 and $1, respectively, were included in research and development expenses in the statements of operations.
5. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consisted of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | |||||
| Clinical trials | $ | 2,628 | $ | 1,899 | |||
| General and administrative | 413 | 281 | |||||
| Research and development | 346 | 246 | |||||
| Employee compensation | 4 | 15 | |||||
| | | | | | | | |
| $ | 3,391 | $ | 2,441 | ||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
6. Stockholders' Equity
Authorized Capital
On December 5, 2016, the Company filed a Certificate of Amendment to the Company's Restated Certificate of Incorporation with the Secretary of State of the State of Delaware to effect a one-for-ten reverse stock split of its outstanding common stock and to effect a reduction in the number of authorized shares of common stock from 400,000,000 to 60,000,000 shares. The Company's certificate of incorporation currently authorizes the Company to issue 60,000,000 shares of common stock, $0.001 par value per share, and 5,000,000 shares of preferred stock, $0.001 par value per share. See Note 2, Significant Accounting Policies — Reverse Stock Split, for further discussion.
Common Stock and Associated Warrant Liability
On October 14, 2016, the Company completed a registered direct offering of its common stock. Under the terms of the offering, the Company sold 1,728,395 shares of its common stock for net proceeds of $13,692, after deducting offering expenses.
F-20
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
On November 14, 2014, the Company completed a private placement of units consisting of an aggregate of 6,431,111 shares of common stock and warrants to purchase an aggregate of 6,430,948 shares of its common stock for net proceeds of $42,814, after deducting offering expenses. The net proceeds from the private placement were allocated to the common stock and warrants based upon the fair value method. Similarly, the offering expenses were allocated between the common stock and warrants with the portion allocated to common stock offset against the proceeds allocated to stockholders' equity, whereas the portion allocated to the warrants was expensed immediately. The warrants have a per share exercise price of $8.50, became exercisable on May 6, 2015 and will continue to be exercisable for four years thereafter. Prior to May 6, 2015, each warrant was subject to net cash settlement if, at the time of any exercise, there was then an insufficient number of authorized and reserved shares of common stock to effect a share settlement of the warrant. Under the terms of the warrants, as of May 6, 2015, the net cash settlement feature of the warrants automatically became inoperative; accordingly, the warrants are exercisable only for shares of the Company's common stock. The warrants, however, also contained certain terms that could have required the Company (or its successor) to purchase the warrants for cash in an amount equal to the value (as calculated utilizing a contractually-agreed Black-Scholes Model) of the unexercised portion of the warrants in connection with certain change of control transactions occurring on or prior to December 31, 2016, with the cash payment capped at an amount equal to $1.25 per unexercised share underlying each warrant. Due to the provision of the warrants that could have required cash settlement upon certain change of control transactions, the Company was required to account for these warrants as a liability at fair value using the Black-Scholes Model and the estimated warrant liability was required to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such provision on December 31, 2016. Effective March 25, 2016, each of the warrants was amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions. These warrants were no longer accounted for as a liability at March 31, 2016. The Company recorded a non-cash reclassification of the warrant fair value to stockholders' equity based on the warrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required.
The fair value of the warrants on the March 25, 2016 modification date of $19,186 was estimated using the Black-Scholes Model with the following assumptions: expected volatility of 101%, risk-free interest rate of 1.1%, expected life of approximately 3.1 years and no dividends. The fair value of the warrants at December 31, 2015 of $27,349 was estimated using the Black-Scholes Model with the following assumptions: expected volatility of 98%, risk-free interest rate of 1.4%, expected life of approximately 3.4 years and no dividends. The decrease in fair value from December 31, 2015 to March 25, 2016 of $8,163 was recorded as a non-cash gain on the change in fair value of warrant liability in the Company's statement of operations for the year ended December 31, 2016.
On March 6, 2014, the Company completed a private placement of units consisting of an aggregate of 1,197,605 shares of common stock and warrants to purchase an aggregate of 1,017,964 shares of its common stock for net proceeds of $21,135, after deducting offering expenses. The net proceeds from the private placement were allocated to the common stock and warrants based upon their relative fair values. The warrants, which had a one year term, expired unexercised on March 6, 2015.
Each of these completed offerings included certain existing GTx stockholders and/or certain members of the GTx management team and/or board of directors.
F-21
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
7. License Agreements
University of Tennessee Research Foundation License Agreements
The Company and the University of Tennessee Research Foundation ("UTRF") are parties to a consolidated, amended and restated license agreement (the "SARM License Agreement") pursuant to which the Company has been granted exclusive worldwide rights in all existing SARM technologies owned or controlled by UTRF, including all improvements thereto, and exclusive rights to future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University. Under the SARM License Agreement, the Company is obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and mid single-digit royalties on sublicense revenues.
In accordance with the terms of the SARM License Agreement that the Company entered into with UTRF in July 2007, the Company paid a one-time up-front fee of $290, which was recorded as an intangible asset by the Company. This intangible asset, net at December 31, 2016 and 2015 was $123 and $137, respectively.
The Company and UTRF also entered into a license agreement in March 2015 pursuant to which the Company was granted exclusive worldwide rights in all existing SARD technologies owned or controlled by UTRF, including all improvements thereto (the "SARD License Agreement"). Under the SARD License Agreement, the Company is obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. The Company is also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed.
8. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax
F-22
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
purposes. The principal components of the Company's net deferred income tax assets and liabilities consisted of the following:
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2016 | 2015 | |||||
Deferred income tax assets: |
|||||||
Net federal and state operating loss carryforwards |
$ | 155,446 | $ | 146,433 | |||
Research and development credits |
13,928 | 13,245 | |||||
Share-based compensation |
6,876 | 7,088 | |||||
Depreciation and amortization |
43 | 58 | |||||
Other |
- | 37 | |||||
| | | | | | | | |
Total deferred tax assets |
176,293 | 166,861 | |||||
| | | | | | | | |
Deferred income tax liabilities: |
|||||||
Other |
336 | 251 | |||||
| | | | | | | | |
Total deferred tax liabilities |
336 | 251 | |||||
| | | | | | | | |
Net deferred tax assets |
175,957 | 166,610 | |||||
Valuation allowance |
(175,957 | ) | (166,610 | ) | |||
| | | | | | | | |
|
$ | - | $ | - | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Realization of deferred income tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, due to the Company's history of net operating losses, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $9,347, $8,101 and $9,648 in 2016, 2015 and 2014, respectively.
At December 31, 2016, the Company had net federal operating loss carryforwards of approximately $402,090, which expire from 2018 to 2036 if not utilized. The Company had state operating loss carryforwards of approximately $365,664, which expire from 2017 to 2036 if not utilized. The Company also had research and development credits at December 31, 2016 of approximately $13,928, which expire from 2020 to 2036 if not utilized.
Both of the net federal and state operating loss carryforwards include approximately $2,354 of deductions related to the exercise of stock options. This amount represents an excess tax benefit and has not been included in the gross deferred income tax asset reflected for net federal and state operating loss carryforwards. If utilized, the benefits from these deductions will be recorded as an adjustment to additional paid in capital.
The Company will recognize the impact of a tax position in the financial statements if that position is more likely than not of being sustained on audit based on the technical merits of the position. As of December 31, 2016, the Company had no unrecognized tax benefits. Utilization of the Company's net operating loss carryforwards may be subject to a substantial annual limitation due to ownership change limitations provided by Section 382 of the Internal Revenue Code of 1986, as amended, and similar state provisions. The annual limitations may result in the expiration of net operating loss carryforwards before utilization. The Company completed a study of its net operating losses through December 31,
F-23
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
2014 to determine whether such amounts are likely to be limited by Section 382. As a result of this study and its analysis of subsequent ownership changes, the Company does not currently believe any Section 382 limitation exists through December 31, 2016. However, any future ownership changes under Section 382 may limit the Company's ability to fully utilize these tax benefits. The Company has not yet conducted an in-depth study of its research and development credits, although the Company periodically reviews assumptions used in its calculations to reflect its best estimate of expected credit. An in-depth study may result in an increase or decrease to the Company's research and development credits and until such study is conducted of the Company's research and development credits, no amounts are being presented as an uncertain tax position. The Company's net deferred income tax assets have been fully offset by a valuation allowance. Therefore, future changes to the Company's unrecognized tax benefits would be offset by an adjustment to the valuation allowance and there would be no impact on the Company's balance sheet, statement of operations, or cash flows. The Company does not expect its unrecognized tax benefits to change significantly over the next 12 months.
The Company is currently open to audit under the statute of limitations by the Internal Revenue Service and the appropriate state income taxing authorities for all years due to the net loss carryforwards from those years. The Company is currently not under examination by the Internal Revenue Service or any other taxing authorities. The Company has not recorded any interest and penalties on any unrecognized tax benefits since its inception.
9. Directors' Deferred Compensation Plan
Non-employee directors may defer all or a portion of their fees under the Company's Directors' Deferred Compensation Plan until termination of their status as directors. Deferrals can be made into a cash account, a stock account, or a combination of both. Stock accounts will be paid out in the form of Company common stock, except that any fractional shares will be paid out in cash valued at the then current market price of the Company's common stock. Cash accounts and stock accounts under the Directors' Deferred Compensation Plan are credited with interest or the value of any cash and stock dividends, respectively. Non-employee directors are fully vested in any amounts that they elect to defer under the Directors' Deferred Compensation Plan.
For the years ended December 31, 2016, 2015 and 2014, the Company incurred non-employee director fee expense of $257, $229 and $247, respectively, of which $132, $113 and $125 was deferred into stock accounts and will be paid in common stock following separation from service as a director. At December 31, 2016, 54,605 shares of the Company's common stock had been credited to individual director stock accounts under the Directors' Deferred Compensation Plan, and no amounts had been credited to individual director cash accounts under the Directors' Deferred Compensation Plan.
10. 401(k) Plan
The Company sponsors a 401(k) retirement savings plan that is available to all eligible employees. The plan is intended to qualify under Section 401(k) of the Internal Revenue Code of 1986, as amended. The plan provides that each participant may contribute up to a statutory limit of their pre-tax compensation which was $18 for employees under age 50 and $24 for employees 50 and older in calendar year 2016. Employee contributions are held in the employees' name and invested by the plan's trustee. The plan also permits the Company to make matching contributions, subject to established limits. The Company elected to match a portion of employee's contributions to the plan in the amount of $200, $189 and $200 in 2016, 2015 and 2014, respectively.
F-24
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
11. Commitments and Contingencies
Operating Lease Commitments
Prior to April 30, 2015, the Company subleased office space under a sublease that was accounted for as an operating lease. Upon expiration of this lease, the Company entered into a new office lease with respect to the Company's current office space. The new office lease term commenced on May 1, 2015 with a three year term ending on April 30, 2018, with an option to extend the lease for an additional three years. Total rent expense under the operating leases was approximately $495, $501 and $513 for the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016, future annual minimum payments under operating lease arrangements were $475 and $159 for the year ended December 31, 2017 and 2018, respectively.
12. Quarterly Financial Data (Unaudited)
The following is a summary of the quarterly results of operations for the years ended December 31, 2016 and 2015:
| |
2016 Quarters Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | |||||||||
| Expenses: | |||||||||||||
Research and development expenses |
$ | 3,971 | $ | 4,058 | $ | 4,614 | $ | 4,585 | |||||
General and administrative expenses |
2,114 | 1,999 | 2,313 | 2,279 | |||||||||
| | | | | | | | | | | | | | |
| Total expenses | 6,085 | 6,057 | 6,927 | 6,864 | |||||||||
| | | | | | | | | | | | | | |
| Loss from operations | (6,085 | ) | (6,057 | ) | (6,927 | ) | (6,864 | ) | |||||
| Other income (expense), net | 28 | 5 | 13 | - | |||||||||
| Gain (loss) on change in fair value of warrant liability (a) | 8,163 | - | - | - | |||||||||
| | | | | | | | | | | | | | |
| Net loss | $ | 2,106 | $ | (6,052 | ) | $ | (6,914 | ) | $ | (6,864 | ) | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Net income (loss) per share: | |||||||||||||
| Basic | $ | 0.15 | $ | (0.43 | ) | $ | (0.49 | ) | $ | (0.44 | ) | ||
| | | | | | | | | | | | | | |
| Diluted | $ | 0.15 | $ | (0.43 | ) | $ | (0.49 | ) | $ | (0.44 | ) | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Weighted average shares outstanding: | |||||||||||||
Basic |
14,152,204 | 14,174,914 | 14,189,226 | 15,713,210 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Diluted |
14,344,816 | 14,174,914 | 14,189,226 | 15,713,210 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
F-25
GTx, Inc.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except share and per share data)
| |
2015 Quarters Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | |||||||||
| Expenses: | |||||||||||||
Research and development expenses |
$ | 2,948 | $ | 2,956 | $ | 3,824 | $ | 3,879 | |||||
General and administrative expenses |
2,111 | 2,005 | 2,039 | 2,079 | |||||||||
| | | | | | | | | | | | | | |
| Total expenses | 5,059 | 4,961 | 5,863 | 5,958 | |||||||||
| | | | | | | | | | | | | | |
| Loss from operations | (5,059 | ) | (4,961 | ) | (5,863 | ) | (5,958 | ) | |||||
| Other income (expense), net | 27 | 25 | 9 | (4 | ) | ||||||||
| Gain (loss) on change in fair value of warrant liability (a) | 2,648 | (43,016 | ) | 40,720 | 2,729 | ||||||||
| | | | | | | | | | | | | | |
| Net loss | $ | (2,384 | ) | $ | (47,952 | ) | $ | 34,866 | $ | (3,233 | ) | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Net income (loss) per share: | |||||||||||||
| Basic | $ | (0.17 | ) | $ | (3.42 | ) | $ | 2.48 | $ | (0.23 | ) | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Diluted | $ | (0.17 | ) | $ | (3.42 | ) | $ | (0.38 | ) | $ | (0.40 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Weighted average shares outstanding: | |||||||||||||
Basic |
14,033,587 | 14,037,411 | 14,037,411 | 14,037,411 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Diluted |
14,033,587 | 14,037,411 | 15,485,212 | 14,952,920 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (a)
- The gain (loss) on change in fair value of warrant liability is related to the private placement of warrants completed in November 2014. See Note 6, Stockholder's Equity, for further information.
F-26