Attached files
| file | filename |
|---|---|
| EX-23.2 - EX-23.2 - PLUS THERAPEUTICS, INC. | cytx-ex232_652.htm |
| EX-32.1 - EX-32.1 - PLUS THERAPEUTICS, INC. | cytx-ex321_7.htm |
| EX-31.2 - EX-31.2 - PLUS THERAPEUTICS, INC. | cytx-ex312_6.htm |
| EX-31.1 - EX-31.1 - PLUS THERAPEUTICS, INC. | cytx-ex311_8.htm |
| EX-23.1 - EX-23.1 - PLUS THERAPEUTICS, INC. | cytx-ex231_651.htm |
| EX-10.42 - EX-10.42 - PLUS THERAPEUTICS, INC. | cytx-ex1042_653.htm |
| EX-10.41 - EX-10.41 - PLUS THERAPEUTICS, INC. | cytx-ex1041_656.htm |
| EX-10.40 - EX-10.40 - PLUS THERAPEUTICS, INC. | cytx-ex1040_654.htm |
| EX-10.39 - EX-10.39 - PLUS THERAPEUTICS, INC. | cytx-ex1039_655.htm |
| EX-10.22 - EX-10.22 - PLUS THERAPEUTICS, INC. | cytx-ex1022_690.htm |
| EX-10.21 - EX-10.21 - PLUS THERAPEUTICS, INC. | cytx-ex1021_689.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 001-34375
CYTORI THERAPEUTICS, INC.
(Exact name of Registrant as Specified in Its Charter)
|
DELAWARE |
33-0827593 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) |
|
|
|
|
3020 CALLAN ROAD, SAN DIEGO, CALIFORNIA |
92121 |
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (858) 458-0900
Securities registered pursuant to Section 12(b) of the Act:
|
|
Title of each class |
|
Name of each exchange on which registered |
|
|
|
Common stock, par value $0.001 |
|
NASDAQ Stock Market LLC |
|
Securities registered pursuant to Section 12(g) of the Act:
Preferred Stock Purchase Rights
Indicate by check mark if the registrant is a well-known seasoned issuer as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one).
|
Large Accelerated Filer |
☐ |
|
Accelerated Filer |
☐ |
|
Non-Accelerated Filer |
☐ |
(Do not check if a smaller reporting company) |
Smaller reporting company |
☒ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the common stock of the registrant held by non-affiliates of the registrant on June 30, 2016, the last business day of the registrant’s most recently completed second fiscal quarter, was $42.3 million based on the closing sales price of the registrant’s common stock on June 30, 2016 as reported on the Nasdaq Capital Market, of $2.09 per share.
As of January 31, 2017, there were 21,966,424 shares of the registrant’s common stock outstanding.
|
|
|
Page |
|
|
PART I |
|
|
|
|
|
|
Item 1. |
3 |
|
|
Item 1A. |
17 |
|
|
Item 1B. |
52 |
|
|
Item 2. |
52 |
|
|
Item 3. |
52 |
|
|
Item 4. |
52 |
|
|
|
|
|
|
|
PART II |
|
|
|
|
|
|
Item 5. |
53 |
|
|
Item 6. |
55 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
56 |
|
Item 7A. |
67 |
|
|
Item 8. |
68 |
|
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
95 |
|
Item 9A. |
95 |
|
|
Item 9B. |
95 |
|
|
|
|
|
|
|
PART III |
|
|
|
|
|
|
Item 10. |
96 |
|
|
Item 11. |
101 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
108 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
109 |
|
Item 14. |
109 |
|
|
|
|
|
|
|
PART IV |
|
|
|
|
|
|
Item 15. |
111 |
|
|
Item 16. |
111 |
References to “Cytori,” “we,” “us” and “our” refer to Cytori Therapeutics, Inc. and its consolidated subsidiaries. References to “Notes” refer to the Notes to Consolidated Financial Statements included herein (refer to Item 8).
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This report contains certain statements that may be deemed “forward-looking statements” within the meaning of U.S. securities laws. All statements, other than statements of historical fact, that address activities, events or developments that we intend, expect, project, believe or anticipate and similar expressions or future conditional verbs such as will, should, would, could or may occur in the future are forward-looking statements. Such statements are based upon certain assumptions and assessments made by our management in light of their experience and their perception of historical trends, current conditions, expected future developments and other factors they believe to be appropriate.
These statements include, without limitation, statements about our anticipated expenditures, including research and development, sales and marketing, and general and administrative expenses; the potential size of the market for our products; future development and/or expansion of our products and therapies in our markets, our ability to generate product or development revenues and the sources of such revenues; our ability to effectively manage our gross profit margins; our ability to obtain regulatory approvals; expectations as to our future performance; portions of the “Liquidity and Capital Resources” section of this report, including our potential need for additional financing and the availability thereof; and the potential enhancement of our cash position through development, marketing, and licensing arrangements. Our actual results will likely differ, perhaps materially, from those anticipated in these forward-looking statements as a result of various factors, including: the early stage of our product candidates and therapies, the results of our research and development activities, including uncertainties relating to the clinical trials of our product candidates and therapies; our need and ability to raise additional cash; the outcome of our partnering/licensing efforts; our joint ventures, risks associated with laws or regulatory requirements applicable to us, market conditions, product performance, potential litigation, and competition within the regenerative medicine field, to name a few. The forward-looking statements included in this report are subject to a number of additional material risks and uncertainties, including but not limited to the risks described under the “Risk Factors” in Item 1A of Part I above, which we encourage you to read carefully
We encourage you to read the risks described under “Risk Factors” carefully. We caution you not to place undue reliance on the forward-looking statements contained in this report. These statements, like all statements in this report, speak only as of the date of this report (unless an earlier date is indicated) and we undertake no obligation to update or revise the statements except as required by law. Such forward-looking statements are not guarantees of future performance and actual results will likely differ, perhaps materially, from those suggested by such forward-looking statements.
This Annual report on Form 10-K refers to trademarks such as Cytori Cell Therapy, Habeo Cell Therapy, Celution, Celase, Intravase, Puregraft and StemSource. Solely for convenience, our trademarks and tradenames referred to in this Form 10-K may appear without the ® or ™ symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights to these trademarks and tradenames.
General
Our strategy is to build a profitable and growing specialty therapeutics company focused on rare and niche opportunities frequently overlooked by larger companies but requiring breadth of scope, expertise and focus often not possessed by or available to smaller companies. To meet this objective, we have, thus far, identified two therapeutic development platforms, discussed below, and candidate therapeutics in our pipeline that hold promise for millions of patients and significant market potential. Our current corporate activities fall substantially into one of two key areas related to our two therapeutic development platforms: Cytori Cell TherapyTM and Cytori NanomedicineTM.
Our Cytori Cell Therapy, or CCT, platform, is based on the scientific discovery that the human adipose or fat tissue compartment is a source of a unique mixed population of stem, progenitor and regenerative cells that may hold substantial promise in the treatment of numerous diseases. To bring this promise to patients, we are developing the processes and procedures via proprietary hardware- and software-based devices and single-use reagents and consumable sets, to enable doctors to have access to a variety of therapies at the bedside derived fundamentally from each patient’s own adipose tissue. Our lead product candidate is for the treatment of impaired hand function in scleroderma, and we have recently completed a U.S. pivotal clinical trial for this indication using our HabeoTM Cell Therapy product. We have additional CCT treatments in various stages of development. Further, our CCT platform is the subject of investigator-initiated trials conducted by our partners, licensees and other third parties, some of which are supported by us and/or
3
funded by government agencies and other funding sources. Currently, we internally manufacture or source our CCT-related products from third parties. We also have obtained regulatory approval to sell some of our CCT products, including our Celution devices and consumable kits, in certain markets outside the United States. In those markets, we have been able to further develop and improve our core technologies, gain expanded clinical experience and data and generate sales.
Our Cytori Nanomedicine platform features a versatile and novel protein-stabilized liposomal nanoparticle technology for drug encapsulation that has thus far provided the foundation to bring two promising drugs into early/late stage clinical trials. By encapsulating certain drugs, we can create both novel compounds and improve the performance via reformulated versions of existing drugs. Nanoparticle encapsulation is promising because it can help improve the trafficking and metabolism of many drugs, thus potentially enhancing the therapeutic profile and patient benefits. Our lead drug candidate, ATI-0918 is a generic version of liposomal encapsulated doxorubicin. Liposomal encapsulated doxorubicin is a heavily relied upon chemotherapeutic used in many cancer types on a global basis. We believe that data from a 60-patient European study of ATI-0918 has met the statistical criteria for bioequivalence to Caelyx®, the current reference listed drug in Europe. We intend that these bioequivalence data will serve as a basis for our planned regulatory submission to the European Medicines Agency, or EMA, for ATI-0918. Our second nanomedicine drug candidate is ATI-1123, a new chemical entity which is a nanoparticle-encapsulated form of docetaxel, also a standard chemotherapeutic drug used for many cancers. A phase I clinical trial of ATI-1123 has been completed, and we are investigating possible expansion of this trial to phase II, most likely in conjunction with a development partner. In addition, we are early in the long-term research and development of encapsulated regenerative medicine drugs, focused first on the treatment of scleroderma and related connective disorders. Finally, in connection with our acquisition of the ATI-0918 and ATI-1123 drug candidates, we have acquired know-how (including proprietary processes and techniques) and a scalable nanoparticle manufacturing plant in San Antonio, Texas from which we intend to test, validate and eventually manufacture commercial quantities of our nanoparticle drugs.
Development Pipeline
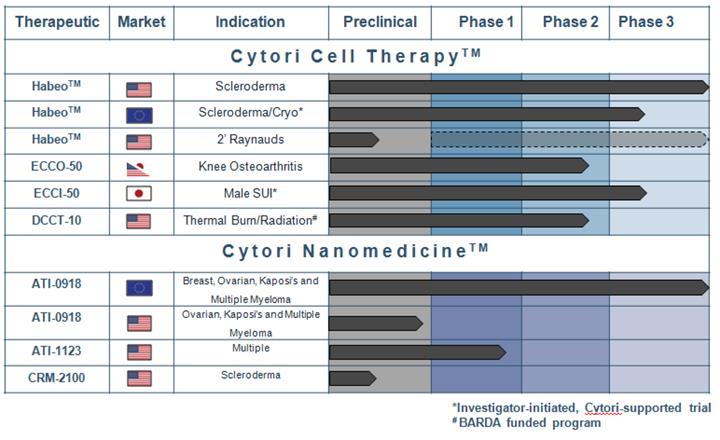
Cytori Cell Therapy
Our primary near-term goal is for Cytori Cell Therapy to be the first cell therapy to market for the treatment of impaired hand function in scleroderma, through Cytori-sponsored and supported clinical development efforts. The Cytori-sponsored Scleroderma Treatment with Celution Processed Adipose Derived Regenerative Cells, or STAR clinical trial, is a randomized, double-blind, placebo-controlled, Phase III pivotal clinical trial in the U.S. The purpose of the STAR trial is to evaluate the safety and efficacy of a single
4
administration of Habeo™ Cell Therapy (formerly named ECCS-50) in patients with scleroderma affecting the hands and fingers. We initiated the first sites for our STAR trial in July 2015 and we completed final enrollment of 88 patients in June 2016. We anticipate obtaining 48-week follow-up data in mid-2017. Once the study is unblinded and data are available, subjects randomized to the placebo arm will be given the option of being treated within a crossover arm of the study.
With respect to the remainder of our current cellular therapeutics clinical pipeline:
|
|
• |
We completed our Phase II Celution Prepared Adipose Derived Regenerative Cells in the Treatment of OsteoArthritis of the Knee, or ACT-OA clinical trial, in June 2015. The 48-week analysis was performed as planned and the top-line data are described in the “Osteoarthritis” section below. |
|
|
• |
In July 2015, a Japanese investigator-initiated study in men with stress urinary incontinence, or SUI, following prostatic surgery for prostate cancer or benign prostatic hypertrophy, called ADRESU, received approval to begin enrollment from the Japanese Ministry of Health, Labor and Welfare, or MHLW. In December 2016, we announced that the ADRESU trial had reached 50% enrollment. The Japan Agency for Medical Research and Development, or AMED, has provided partial funding for the ADRESU trial. |
|
|
• |
We are developing a treatment for thermal burns under a contract from the Biomedical Advanced Research Development Authority, or BARDA, a division of the U.S. Department of Health and Human Services. We submitted an Investigational Device Exemption, or IDE, application to the U.S. Food and Drug Administration, or FDA, in the fourth quarter of 2016 for a pilot clinical study in thermal burn, and we expect FDA’s final determination by mid-2017. If we receive FDA’s approval of the IDE, we will then seek approval of the pilot clinical study from BARDA as study sponsor. |
|
|
• |
We recently announced our intent to initiate clinical trials in secondary Raynaud’s Phenomenon, or SRP. This decision was based upon the encouraging Raynaud’s Condition Score data from the investigator-initiated, Phase I, open-label, 12-patient SCLERADEC I clinical trial assessing use of Cytori Cell Therapy in patients with impaired hand function due to systemic scleroderma. |
In addition to our targeted therapeutic development, we have continued to commercialize our Cytori Cell Therapy technology under select medical device approvals, clearances and registrations to customers in Europe, Japan and other regions. These customers are a mix of research customers evaluating new therapeutic applications of Cytori Cell Therapy and commercial customers, including our licensing partners, distributors, and end user hospitals, clinics and physicians, that use our Celution cell processing system (as further described in “Sales, Marketing and Service” below) mostly for treatment of patients in private pay procedures. In Japan, our largest commercial market, we gained increased utilization of our products in the private pay marketplace in 2016 due to several factors, including increased clarity around the November 2014 Regenerative Medicine Law (implemented in November 2015 as it relates to regenerative medicine products like Cytori Cell Therapy) and we project that our sales and market presence in Japan will continue to grow in 2017. The sale of Celution systems, consumables and ancillary products contribute a margin that partially offsets our operating expenses and will continue to play a role in fostering familiarity within the medical community with our technology.
Habeo Cell Therapy for Impaired Hand Function in Scleroderma and Secondary Raynaud’s Phenomenon
Scleroderma is a rare and chronic autoimmune disorder associated with fibrosis of the skin, and destructive changes in blood vessels and multiple organ systems as the result of a generalized overproduction of collagen. Scleroderma affects approximately 50,000 patients in the United States (women are affected four times more frequently than men) and is typically detected between the ages of 30 and 50. More than 90 percent of scleroderma patients have hand involvement that is typically progressive and can result in chronic pain, blood flow changes and severe dysfunction. A small number of treatments are occasionally used off-label for hand scleroderma but and they do little to modify disease progression or substantially improve symptoms. Treatment options are directed at protecting the hands from injury and detrimental environmental conditions as well as the use of vasodilators. When the disease is advanced, immunosuppressive and other medications may be used but are often accompanied by side effects.
The STAR trial is a 48-week, 19 site, randomized, double blind, placebo-controlled pivotal clinical trial of 88 patients in the U.S. for the treatment of impaired hand function in scleroderma. The trial evaluates the safety and efficacy of a single administration of Habeo Cell Therapy in patients with scleroderma affecting the hands and fingers. The STAR trial uses the Cochin Hand Function Scale, or CHFS, a validated measure of hand function, as the primary endpoint measured at 24 weeks and 48 weeks (approximately 6 and 12 months) after a single administration of Habeo Cell Therapy or placebo. Pending the 48 week results, patients in the placebo group will be eligible for crossover to the active arm of the trial after all patients have completed 48 weeks of follow-up. We anticipate study results in mid-2017. The STAR trial is predicated on a completed, investigator-initiated, 12-patient, open-label, Phase I pilot trial, termed SCLERADEC I, sponsored by Assistance Publique-Hôpitaux de Marseille, or AP-HM, in Marseille, France. The SCLERADEC I trial received partial support from Cytori. The six-month results were published in the Annals of the Rheumatic Diseases in May 2014 and demonstrated approximately a 50 percent improvement at six months across four important and validated
5
endpoints used to assess the clinical status in patients with scleroderma with impaired hand function. Two-year follow up data in the SCLERADEC I trial was presented at the Systemic Sclerosis World Congress in February 2016 and published in the journal Current Research in Translational Medicine in November 2016 and demonstrated sustained improvement in the following four key endpoints: CHFS, SHAQ, RCS, and hand pain, as assessed by a standard visual analogue scale.
Further, on December 5, 2016, we released topline results for three-year follow-up data showing sustained benefits materially consistent with those shown in two-year data.
In 2014, Drs. Guy Magalon and Brigitte Granel, under the sponsorship of AP-HM, submitted a study for review for a follow-up randomized, double-blind, placebo-controlled trial in France using Cytori Cell Therapy, to be supported by us. The trial, named SCLERADEC II, received approval from the French government in April 2015. Enrollment of this trial commenced in October 2015 and is ongoing. Enrollment is expected to be completed in 2017, approximately one year later than originally projected, due to delays in French regulatory approvals of participating sites. Patients will be followed at six-month post-treatment and compared with placebo treated patients. Pending the six-month results patients in the placebo group will be eligible for crossover using Habeo cells stored at the time of the initial procedure. This crossover arm will open after all patients have completed six-month follow up. We anticipate study results in 2018, however, the trial timeline is controlled in full by the sponsoring institution.
In November 2016, the US FDA Office of Orphan Products Development granted Cytori an orphan drug designation for cryopreserved or centrally processed ECCS-50 (Habeo) for scleroderma.
In January 2017, we announced our intention to broaden our investigation of Habeo Cell Therapy beyond systemic scleroderma to include secondary Raynaud’s Phenomenon, or SRP. This expansion of Cytori’s research and development efforts is based upon: (i) the 36-month follow-up data from the SCLERADEC I trial, which reported a 90 percent reduction in the Raynaud’s Condition Score, which assesses the frequency and severity of Raynaud’s attacks experienced by patients with Raynaud’s Phenomenon, or RP; (ii) earlier limited published data reporting an association between use of Habeo Cell Therapy and improvement in vascular architecture, hand color, and other direct and indirect indicators of vascular function, and (iii) our internal preclinical data regarding the potential role of Habeo Cell Therapy in the stabilization of the vascular endothelium, an important contributor to the vascular dysfunction found in patients with RP. SRP is a problem that affects millions of patients worldwide.
Osteoarthritis
Osteoarthritis is a disease of the entire joint involving the cartilage, joint lining, ligaments and underlying bone. The breakdown of tissue leads to pain, joint stiffness and reduced function. It is the most common form of arthritis and affects an estimated 13.9% of US adults over the age of 25, and 33.6% of U.S. adults over the age of 65. Current treatments include physical therapy, non-steroidal anti-inflammatory medications, viscosupplement injections, and total knee replacement. A substantial medical need exists as present medications have limited efficacy and joint replacement is a relatively definitive treatment for those with the most advanced disease.
ACT-OA, was a 94-patient, randomized, double-blind, placebo controlled study involving two doses of Cytori Cell Therapy, a low dose and a high dose, and was conducted over 48 weeks. The randomization was 1:1:1 between the control, low and high dose groups. The trial was completed in June 2015. The goal of this proof-of-concept trial was to help determine: (1) safety and feasibility of the ECCO-50 therapeutic for osteoarthritis, (2) provide dosing guidance and (3) explore key trial endpoints useful for a Phase III trial.
We completed top-line analysis of the final 48-week data in July 2016. A total of 94 patients were randomized (33 placebo, 30 low dose ECCO-50, 31 high dose ECCO-50). In general, a clear difference between low and high dose ECCO-50 was not observed and therefore the data for both groups have been combined. We evaluated numerous endpoints that can be summarized as follows:
|
|
• |
Intraarticular application of a single dose of ECCO-50 is feasible in an outpatient day-surgery setting; no serious adverse events were reported related to the fat harvest, cell injection or to the cell therapy. |
|
|
• |
Consistent trends were observed in most secondary endpoints at 12, 24 and 48 weeks in the target knee of the treated group relative to placebo control group; 12-week primary endpoint of single pain on walking question did not achieve statistical significance. |
|
|
• |
Consistent trends were observed in all six pre-specified MRI Osteoarthritis Knee Score (MOAKS) classification scores suggesting a lower degree of target knee joint pathological worsening at 48 weeks for the treated group relative to placebo control group. The differences against placebo favored ADRCs specifically in the number of bone marrow lesions, the percentage of the bone marrow lesion that is not a cyst, the size of the bone marrow lesions as a percentage of the total sub-region volume, percentage of full thickness cartilage loss, cartilage loss as a percentage of cartilage surface area and the size of the largest osteophyte. |
6
In summary, the ACT-OA Phase II trial demonstrated feasibility of same day fat harvesting, cell processing and intraarticular administration of autologous ADRCs (ECCO-50) with a potential for a beneficial effect of ECCO-50. The accumulated data and experienced gained will be critical in considering designs of further clinical trials in osteoarthritis and other potential indications. In addition, we are actively pursuing partnering and commercialization opportunities for ECCO-50 to further develop our knee osteoarthritis program and also to support our growing commercial sales into the knee osteoarthritis market in Japan.
Stress Urinary Incontinence
Another therapeutic target under evaluation by Cytori in combination with the University of Nagoya and the Japanese MHLW is stress urinary incontinence in men following surgical removal of the prostate gland, which is based on positive data reported in a peer reviewed journal resulting from the use of ADRCs prepared by our Celution System. The ADRESU trial is a 45 patient, investigator-initiated, open-label, multi-center, single arm trial that was approved by the Japanese MHLW in July 2015 and is being led by both Momokazu Gotoh, MD, Ph.D., Professor and Chairman of the Department of Urology and Tokunori Yamamoto, MD, Ph.D., Associate Professor Department of Urology at University of Nagoya Graduate School of Medicine. Trial enrollment began in September 2015, and in December 2016, the trial achieved 50% enrollment. This clinical trial is primarily sponsored and funded by the Japanese government, including a grant provided by AMED.
Cutaneous and Soft Tissue Thermal and Radiation Injuries
We are also developing Cytori Cell Therapy, or DCCT-10, for the treatment of thermal burns. In the third quarter of 2012, we were awarded a contract by BARDA valued at up to $106 million to develop a medical countermeasure for thermal burns. The total award under the BARDA contract has been intended to support all clinical, preclinical, regulatory and technology development activities needed to complete the FDA approval process for use of DCCT-10 in thermal burn injury under a device-based PMA regulatory pathway and to provide preclinical data in burn complicated by radiation exposure.
Pursuant to this contract, BARDA initially awarded us approximately $4.7 million over the initial two-year base period to fund preclinical research and continued development of our Celution System to improve cell processing. In August 2014, BARDA determined that Cytori had completed the objectives of the initial phase of the contract, and exercised its first contract option in the amount of approximately $12 million. In December 2014 and September 2016, BARDA exercised additional contract options pursuant to which it provided us with $2.0 million and $2.5 million in supplemental funds, respectively. These additional funds supported continuation of our research, regulatory, clinical and other activities required for submission of an IDE request to the FDA for RELIEF, a pilot clinical trial using DCCT-10 for the treatment of thermal burns. We submitted our IDE application to the FDA in the fourth quarter of 2016. Upon receipt of IDE approval, if granted, we anticipate that BARDA will provide funding to cover costs associated with execution of the clinical trial and related activities.
The latest BARDA contract modification, entered into in September 2016, is scheduled to terminate in April 2017, but is subject to a no-cost extension at our request and subject to BARDA’s approval. We are in active negotiations with BARDA regarding entry into a new contract or contract option, which, if executed, would provide funding for the proposed RELIEF pilot trial and related costs and expenses.
Other recent developments for Cytori Cell Therapy
|
|
• |
In April 2016, the European Commission, acting on the positive recommendation from the European Medicines Agency Committee for Orphan Medicinal Products, issued orphan drug designation to a broad range of Cytori Cell Therapy formulations when used for the treatment of systemic sclerosis under Community Register of Orphan Medicinal Products number EU/3/16/1643. |
|
|
• |
In February 2017, the U.S. FDA Division of Industry and Consumer Education, or DICE, granted us Small Business status for fiscal year 2017, thus entitling us to receive significant financial incentives, fee reductions, and fee waivers for selective FDA medical device regulatory filings. We anticipate that this grant of small business status will substantially reduce filing fees in 2017 for our planned pre-market authorization, or PMA, application for Habeo Cell Therapy, should the STAR Phase III data support filing of this application. |
Cytori Nanomedicine
In February 2017, we completed our acquisition of substantially all of the assets of Azaya Therapeutics, Inc., or Azaya, pursuant to the terms of an Asset Purchase Agreement, dated January 26, 2017 by and between us and Azaya. Pursuant to the terms of the agreement, we acquired equipment, inventory, certain intellectual property including, a portfolio of investigational therapies and related assets, and assumed certain liabilities, from Azaya in exchange for the issuance of $2.0 million of shares of our common stock, assumption of
7
approximately $1.9 million in Azaya’s trade payables and related charges, and the obligation to pay Azaya future milestones, earn-outs and licensing fees. The acquisition of Azaya brought two additional product candidates, ATI-0918 and ATI-1123, into the Cytori pipeline and we intend to develop and potentially commercialize both compounds.
ATI-0918 is a complex generic formulation of the market leading Doxil®/Caelyx®, which is a liposomal encapsulation of doxorubicin and approved for use in breast cancer, ovarian cancer, multiple myeloma, and Kaposi’s Sarcoma. The current approval pathway for ATI-0918 is to demonstrate bioequivalence to Caelyx® for approval in the EU and to Lipodox® in the U.S. A study to demonstrate ATI-0918’s bioequivalence to Caelyx®, for purposes of EMA approval, has been completed and we intend for these data to serve as the basis for our submission of a marketing authorization application for ATI-0918 to the EMA. We are also making plans to perform a bioequivalence study of ATI-0918 to the U.S. reference listed drug to serve as the basis for submission of an application for U.S. FDA approval. We currently anticipate that any U.S. bioequivalence trial for ATI-0918 would be funded by a development partner or licensee.
ATI-1123 is a liposomal formulation of docetaxel. Docetaxel is currently approved for non-small cell lung cancer, breast cancer, squamous cell carcinoma of the head and neck cancer, gastric adenocarcinoma, and hormone refractory prostate cancer. Its side effects include hair loss, bone marrow suppression, and allergic reactions. It is currently available as a generic drug. There is no form of docetaxel as a liposomal formulation. There is a protein (albumin) bound form of a similar chemotherapeutic drug, paclitaxel known as Abraxane®, which demonstrated some clinical advantages to paclitaxel. ATI-1123 has shown superiority to docetaxel in several animal models including some tumor types not amenable to treatment by docetaxel. A Phase I study of ATI-1123 has been completed in late stage refractory patients and has shown some activity in several tumor types (mostly stable disease). We are currently evaluating clinical scenarios to bring into Phase II studies in several indications.
Sales, Marketing and Service
Cytori Cell TherapyTM
We sell Celution cell processing systems, or Celution Systems, StemSource cell and tissue banking systems, or StemSource Systems, and surgical accessories and instrumentation to hospitals, clinics, physicians, researchers and other customers for commercial and research purposes, including performance of investigator-initiated studies. Our proprietary enzymatic reagents, which we market and sell under the brand names Celase® and Intravase®, are sold as part of our Celution Systems and StemSource Systems (with respect to Celase), or under certain circumstances, are sold separately.
We sell our Celution and StemSource Systems through a combination of a direct sales force, third-party distributors, independent sales representatives, and licensees. Our strategy is to grow and leverage our installed base of Celution and StemSource devices at cell processing facilities, clinics, hospitals and research labs to drive recurring sales of our proprietary disposables. To increase product familiarity and usage among current customers, we launch product enhancements, expand the approved indications for use, perform clinical and technical training, provide on-site case support, and facilitate facility-level licensing with regional and/or national regulatory bodies.
In Japan, we sell our products through our wholly owned subsidiary, Cytori Therapeutics, K.K., which has a direct sales capability. We currently intend to increase our direct sales personnel in Japan over time. In the Bahamas, Chile, Europe, South Korea, Russia and Vietnam, we sell our full product portfolio either directly to customers or through numerous third-party distributors. In the U.S., we are limited to selling only research reagents and surgical accessories and instrumentation directly to customers. Bimini Technologies, LLC, through its wholly owned subsidiary Kerastem Technologies, LLC, has a global exclusive license to sell our Celution cell processing systems for hair applications. Lorem Vascular has an exclusive license to sell our full product portfolio in all fields of use, excluding hair applications, in Australia, China, Hong Kong, Malaysia and Singapore.
In early 2016, we commenced the process of implementing a managed access program, or MAP, (also known as early access program or named patient program) for our Habeo Cell Therapy in conjunction with Idis Managed Access, part of Clinigen Group plc, or Idis, in select countries across Europe, the Middle East and Africa, or EMEA, for patients with impaired hand function due to scleroderma. Initially, we have focused on select countries within these regions and intend to expand our focus over time, depending on interest and participation in our MAP, our strategic focus, and other factors. Our MAP is intended to drive awareness of Habeo Cell Therapy in advance of anticipated commercial launch and also to provide useful pricing and clinical date. Though we have generated significant interest in the MAP, we have yet to treat a patient under it. We intend to continue to appropriately invest resources in our MAP.
8
As of December 31, 2016, we had three individuals in our global marketing team responsible for market assessments and business plans, competitive intelligence, distribution strategy, product management, social media and websites, forecasting, pricing and reimbursement, customer communication, relationship management and service. We create awareness of and demand for our products among physicians and researchers through digital advertising, e-marketing campaigns, and webinars, pre-clinical and clinical publications, patient advocacy group partnerships, sales collateral, and industry and medical society meetings.
As of December 31, 2016, we had three Cytori employees in our field service team responsible for providing Celution and StemSource installations, maintenance, training, troubleshooting, and hardware and software update/upgrade services to new and existing customers. This team also initiates and closes sites participating in Cytori-sponsored clinical trials.
For the year ended December 31, 2016, our sales were concentrated with respect to two distributors and three direct customers, which comprised 65% of our product revenue recognized. Two direct customers accounted for 57% of total outstanding accounts receivable (excluding receivables from BARDA) as of December 31, 2016.
Cytori Nanomedicine™
Our Cytori Nanomedicine pipeline includes both early and late stage nanomedicine product candidates, patented liposomal encapsulated docetaxel (ATI-1123) and generic liposomal encapsulated doxorubicin (ATI-0918), respectively. We are actively seeking regional and global partnerships with either pharmaceutical manufacturers or wholesale distributors for both of these product candidates, with priority on ATI-0918 in Europe where a generic form of liposomal doxorubicin is neither approved nor available.
Customers and Partners
In Japan, Europe, the Middle East, the Asia-Pacific region and Latin America, we offer our Cytori Systems and StemSource Systems through direct sales representatives, distributors and licensing partners, to hospitals, clinics and researchers, including for purposes of performing investigator-initiated and funded studies.
Pursuant to our Sale and Exclusive License/Supply Agreement, or Bimini Agreement, with Bimini, we granted Bimini a global exclusive license to our Cytori Cell Therapy devices and consumable products for hair applications excluding systemic or intravascular delivery of adipose-derived regenerative cells, or ADRCs. Bimini’s current focus is on the aesthetics cash-pay market. Through Kerastem, its wholly owned subsidiary, Bimini is conducting an FDA-approved Phase II clinical trial in the United States, called STYLE, to study the safety and feasibility of Kerastem’s solution for female and male pattern baldness. In September 2016, Bimini announced completion of its STYLE trial enrollment of 70 patients at four clinical trial sites within the United States. We anticipate that six- month follow-up data from this Phase II clinical trial will be available in mid-2017. Outside of the United States, Bimini is engaged in market development efforts in Europe and Japan for the hair market. The Kerastem Hair Therapy is CE mark approved in the EU for sales to patients with alopecia, or hair loss. Under the Bimini Agreement, Bimini is required, among other things, to pay an eight percent (8%) royalty on its net sales of our products for contemplated hair applications.
Pursuant to our Amended and Restated License/Supply Agreement, or Lorem Agreement, with Lorem Vascular Pte. Ltd., or Lorem Vascular, we granted Lorem Vascular an exclusive license in all fields of use (excluding hair applications subject to Bimini’s license) to our Cytori Cell Therapy products for sale into China, Hong Kong, Malaysia, Singapore and Australia. Under the Lorem Agreement, Lorem Vascular committed to pay up to $500 million in license fees in the form of revenue milestones. In addition, Lorem Vascular is required to pay us 30% of their gross profits in China, Hong Kong and Malaysia for the term of the Lorem Agreement. Lorem Vascular has certain minimum product purchase obligations, including purchase obligations triggered by achievement of applicable regulatory clearance for our products in China, which regulatory clearance was achieved as of April 2015. Lorem Vascular has partially satisfied these related product purchase obligations, and as a result, we are currently in discussions with Lorem Vascular regarding restructuring of its obligations and our rights under the Lorem agreement. We cannot guarantee that our restructuring discussions with Lorem Vascular will be successful. Should we be unable to conclude these negotiations to our satisfaction, a dispute may ensue. See, also, our discussions of the regulatory landscape in China for our products as well as discussions regarding our relationship with Lorem Vascular in the “Risk Factors” section and in the “Competition” and “Governmental Regulation” sections of this “Business” section below.
Refer to Note 2 of the Notes to Consolidated Financial Statements for a discussion of geographical concentration of sales.
9
Manufacturing
Cytori Cell Therapy
We currently manufacture or source our Cytori Cell Therapy products at our headquarters in San Diego, California and in Wales, in the United Kingdom. We believe that our manufacturing capabilities will be sufficient to enable us to meet anticipated demand for these products in 2017. We are, and the manufacturer of any future therapeutic products would be, subject to periodic inspection by regulatory authorities and distribution partners. The manufacturer of devices and products for human use is subject to regulation and inspection from time to time by the FDA for compliance with the FDA’s Quality System Regulation, or QSR, requirements, as well as equivalent requirements and inspections by state and non-U.S. regulatory authorities, such as our Notified Body in Europe and the California Food and Drug Branch.
We source the raw materials for the Celution device, Celution consumable kit and other products that we manufacture from a variety of sources. Most of these components are available from multiple vendors either as off-the-shelf items or as custom fabrication. We purchase our Celase and Intravase regents from Roche Diagnostics Corporation, or Roche. While we have significant inventory of these reagents in inventory, we do not have a second source to provide us with these reagents should our supply arrangement with Roche terminate or be suspended, or should Roche be unable to meet its supply obligations thereunder.
Cytori Nanomedicine
We are in the process of a facility re-start and validations at our recently acquired nanoparticle manufacturing facility located in San Antonio, Texas. Once validation is complete, the facility and processes are designed to comply with cGMP per FDA and EMA regulations to manufacture drug candidates for clinical, research, development and commercial use. Upon approval of our drug candidates, our manufacturing capabilities will encompass validated manufacturing processes for drug product as well as a quality assurance product release process with the ability to ultimately scale-up the process to meet increasing market demands. We believe our strategic investments in the analytical and manufacturing capabilities, including personnel from drug discovery through drug development, will allow us to advance our product candidates more quickly. Our San Antonio facility enables us to produce drug substance in a cost-effective manner while retaining control over the process and timing. As needed, the use of a qualified Clinical Manufacturing Organization may be utilized to perform various manufacturing processes as we deem appropriate to meet our operational objectives.
Our current principal suppliers for our Cytori Nanomedicines business are LGM Pharma, which supplies our active pharmaceutical ingredient, or API (doxorubicin HC1), as well as Lipoid, LLC and Dishman Netherlands, B.V., which supply us with other raw materials used in the manufacture of our ATI-0918 and ATI-1123 drug candidates. Each of these suppliers is currently a sole source supplier.
Competition
We compete primarily on the basis of the safety and efficacy of our therapies across a broad range of clinical indications to address significant unmet medical and market needs, supported by our brand name, pricing, products, published clinical data, regulatory approvals, and reimbursement. We believe that our continued success depends on our ability to:
|
|
• |
Develop and innovate our product and technology platforms; |
|
|
• |
Initiate new and advance existing clinical development programs; |
|
|
• |
Secure and maintain regulatory agency approvals; |
|
|
• |
Build and expand our commercial footprint; |
|
|
• |
Achieve improved economies of scale and scope; |
|
|
• |
Generate and protect intellectual property; |
|
|
• |
Hire and retain key talent; and |
|
|
• |
Successfully execute acquisition, licensing, and partnership activities. |
10
Cytori Cell Therapy
According to the Alliance for Regenerative Medicine, there over 700 companies worldwide and 801 clinical trials underway within the global regenerative medicine market. Per Allied Market Research, this market is projected to reach $30.2 billion by 2022 and to be dominated by the cell therapy segment.
Today, we compete directly against companies within the autologous adipose-derived cell therapy segment offering manual, semi-automated, or full automated cell processing and/or banking systems used with or without tissue dissociation reagents. Our primary competitors include, but are not limited to, Adisave, Biosafe Group, GID Group, Healeon Medical, Human Med AG, Medikan International, PNC International, SERVA Electrophoresis GmbH, and Tissue Genesis. None of these companies are conducting clinical trials for the treatment of hand dysfunction in scleroderma patients. However, they are engaged in a number of clinical trials around the world.
|
Company |
Clinical Trial |
||
|
Affiliation |
Location |
Indication |
|
|
Adisave |
Sponsor |
Canada |
Wounds and Soft Tissue Defects |
|
GID Group |
Sponsor |
U.S. |
Alopecia |
|
GID Group |
Sponsor |
U.S. |
Knee Osteoarthritis |
|
Healeon Medical |
Sponsor |
U.S. |
Alopecia |
|
Human Med AG |
Co-Collaborator |
France |
Knee Osteoarthritis |
|
Tissue Genesis |
Sponsor |
U.S. |
Critical Limb Ischemia |
A study published in 2016 reported that there were 570 medical clinics in the U.S. advertising and offering stem cell treatments, including those derived from adipose tissue, directly to patients. It is unclear whether the FDA will allow these clinics to continue to operate in this fashion and whether they will pose a threat to our business if and at such time that we obtain PMA approval to commercialize Habeo Cell Therapy in the U.S.
In the future, we also anticipate encountering competition from companies developing and offering drugs for the treatment of scleroderma including, but not limited to, Actelion Pharmaceuticals, Allergan, Apricus Biosciences, Bayer, Corbus Pharmaceuticals, Covis Pharma, CSL Behring, Genentech, and United Therapeutics. No companies today have approved drugs indicated for improving hand function in scleroderma patients while only Tracleer® (Bosentan) is approved in Europe for the prevention of new digital ulcers in scleroderma patients. Habeo Cell Therapy is expected to compete with or be used in conjunction with second and/or third line therapies including, but not limited to, phosphodiesterase inhibitors, botulinum toxin A, angiotensin II receptor blockers, ACE inhibitors, alpha blockers, selective serotonin reuptake inhibitors, topical nitrates, IV prostanoids, endothelin receptor antagonists, immunosuppressants, and surgical interventions.
Cytori Nanomedicine™
ATI-0918, our generic liposomal encapsulated doxorubicin product candidate is expected to face competition from both patented and generic nanomedicine products for the treatment of breast cancer (BC), ovarian cancer (OC), multiple myeloma (MM), and/or Kaposi’s Sarcoma (KS) in all geographies. New nanoparticle-doxorubicin monotherapies and drug combination therapies represent third generation approaches intended to be safer and more effective than today’s patented and generic pegylated liposomal doxorubicin.
|
U.S. |
||||
|
Company |
Product |
Formulation |
Stage |
Indications |
|
JNJ Janssen |
DOXIL |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, MM, KS |
|
Sun Pharma |
Lipodox |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, MM, KS |
|
Taiwan Liposome Co |
Doxisome |
Pegylated liposomal doxorubicin |
ANDA Submitted |
BC, OC, KS |
|
Teva Actavis |
Doxorubicin Liposome |
Pegylated liposomal doxorubicin |
ANDA Submitted |
BC, OC, MM, KS |
|
Celsion |
Thermodox |
Heat-sensitive liposomal doxorubicin |
Phase 1/2/3 |
Liver; Recurrent BC |
|
Supratek Pharma |
SP1049C |
Block copolymer doxorubicin |
Phase 1/2/3 |
Upper GI, MDR lung, BC |
|
Adocia |
DriveIn |
Hyaluronan nanoparticle doxorubicin |
Preclinical |
|
11
|
Europe |
||||
|
Company |
Product |
Formulation |
Stage |
Indications |
|
JNJ Janssen |
CAELYX |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS |
|
Teva |
Myocet |
Non-pegylated liposomal doxorubicin |
Commercial |
Breast (with cyclophosphamide) |
|
Taiwan Liposome Co |
Doxisome |
Pegylated liposomal doxorubicin |
MAA Submission H1 2017 |
BC, OC, KS |
|
InnoMedica |
Talidox |
Glycan targeted liposomal doxorubicin |
Phase 1/2 |
OC, KS |
|
Ceronco Biosciences |
CB001 |
Glucosylceramide-enriched liposomal doxorubicin |
Preclinical |
BC, OC, KS |
|
Rest of World |
|||||
|
Country |
Company |
Product |
Formulation |
Stage |
Indications |
|
China |
Shanghai F-Z |
Libaoduo |
Pegylated liposomal doxorubicin |
BE Study vs Lipodox Ongoing |
BC, OC, KS |
|
China |
CSPC |
Duomeisu |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS, MM, lymphoma |
|
Hong Kong |
NAL Pharma |
NAL1872 |
Pegylated liposomal doxorubicin |
Preclinical |
BC, OC, KS |
|
India |
Intas Pharma |
Pegadria |
Pegylated liposomal doxorubicin |
BE Study vs DOXIL Complete |
BC, OC, KS |
|
India |
Dr. Reddy's Labs |
Doxorubicin |
Pegylated liposomal doxorubicin |
BE Study vs Lipodox Ongoing |
BC, OC, KS |
|
India |
Alkem Labs |
Lipisol |
Pegylated liposomal doxorubicin |
Commercial |
|
|
India |
Celon Labs |
Lippod |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, MM, KS |
|
India |
Cipla |
Oncodox PEG |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, MM, KS |
|
India |
Natco Pharma |
Natdox-LP |
Pegylated liposomal doxorubicin |
Commercial |
OC |
|
India |
SRS Pharma |
Dox HCl Liposome |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS |
|
India |
Parenteral Drugs |
Doxopar |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS |
|
India |
Zuventus |
Rubilong |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS |
|
India |
Zydus Cadila |
Nudoxa |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, KS |
|
Philippines Sri Lanka Taiwan Thailand Vietnam |
TTY Biopharm |
Lipo-dox |
Pegylated liposomal doxorubicin |
Commercial |
BC, OC, MM, KS |
|
Philippines Sri Lanka Taiwan Thailand Vietnam |
TTY Biopharm |
CAELYX II |
Pegylated liposomal doxorubicin |
Development |
BC, OC, MM, KS |
|
Russia |
Oasmia |
Doxophos |
Nanoparticle doxorubicin |
MAA Submission in Dec 2015 |
BC |
Our ATI-1123 product candidate is expected to face competition from both Sanofi’s Taxotere, which is approved for 11 indications and available in 90 countries with a majority of sales from China, Japan, Korea, and Taiwan, and generic docetaxel which is available from major suppliers in the U.S., Europe and Japan including, but not limited to, Accord, Actavis, Dr. Reddy’s Labs, GLS Pharma, Hospira, Sun Pharma, Teva, and Winthrop. Further competition may result from advances made by companies currently developing nanoparticle-docetaxel products including, but not limited to, Adocia, Cristal Therapeutics, and Oasmia Pharmaceutical.
Research and Development
Research and development expenses were $16.2 million and $19.0 million for the years ended December 31, 2016 and 2015, respectively. These expenses have supported the basic research, product development and clinical activities necessary to bring our products to market.
Our research and development efforts in 2016 focused predominantly on the following areas:
|
• |
Completion of enrollment in the STAR (hand manifestation of scleroderma) trial and ongoing ACT-OA (knee osteoarthritis) trial expenses; |
|
• |
Support of ongoing preclinical and other research activities towards BARDA contract milestones; |
|
• |
Support of the investigator initiated trials ADRESU in Japan and SCLERADEC-II in France; |
|
• |
Planning and development of next generation Celution Cell Therapy products, including detailed product roadmaps for the device, consumables and accessories; |
12
|
• |
Development of new configurations and expanded functionality of our Celution® platform to address the current Japanese regulatory approval as a medical device (Japan Class I) and other markets; |
|
• |
Conduct ADRC viability and transport studies in support of clinical trial requirements; |
|
• |
Conduct presentation and publishing of research efforts related to ADRC characterization and potency to further establish scientific leadership in the field; and |
|
• |
Continued optimization and development of the Celution® System family of products and next-generation devices, single-use consumables and related instrumentation. |
Intellectual Property
Our success depends in large part on our ability to protect our proprietary technology, including the Celution® System product platform, and to operate without infringing on the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright and trademark laws, as well as confidentiality agreements, licensing agreements and other agreements, to establish and protect our proprietary rights. Our success also depends, in part, on our ability to avoid infringing patents issued to others. If we were judicially determined to be infringing on any third-party patent, we could be required to pay damages, alter our products or processes, obtain licenses or cease certain activities.
To protect our proprietary medical technologies, including the Celution® System platform and other scientific discoveries, we have a portfolio of over 100 issued patents worldwide. We currently have 34 issued U.S. patents and 68 issued international patents. Of the 34 issued U.S. patents, eight were issued in 2016. Of the 68 issued international patents, seven were issued in 2016. In addition, we have over 45 patent applications pending worldwide related to our Cytori Cell Therapy technology. We are seeking additional patents on methods and systems for processing adipose-derived stem and regenerative cells, on the use of adipose-derived stem and regenerative cells for a variety of therapeutic indications, including their mechanisms of actions, on compositions of matter that include adipose-derived stem and regenerative cells, and on other scientific discoveries. We are seeking additional patents on methods and systems for processing adipose-derived stem and regenerative cells, on the use of adipose-derived stem and regenerative cells for a variety of therapeutic indications, including their mechanisms of action, on compositions of matter that include adipose-derived stem and regenerative cells, and on other scientific discoveries. Regarding our Cytori Nanomedicine program, as part of our assert acquisition transaction with Azaya Therapeutics, we acquired Azaya Therapeutics’ patent portfolio consisting of two issued patents, and one pending patent application. Since the Azaya asset acquisition, we have filed one patent application relating to Cytori Nanomedicine, and intend to actively continue to enhance our nanomedicine portfolio.
We cannot assure that any of our pending patent applications will be issued, that we will develop additional proprietary products that are patentable, that any patents issued to us will provide us with competitive advantages or will not be challenged by any third parties or that the patents of others will not prevent the commercialization of products incorporating our technology. Furthermore, we cannot assure that others will not independently develop similar products, duplicate any of our products or design around our patents. U.S. patent applications are not immediately made public, so we might be surprised by the grant to someone else of a patent on a technology we are actively using.
There is a risk that any patent applications that we file and any patents that we hold or later obtain could be challenged by third parties and declared invalid or infringing of third party claims. For many of our pending applications, patent interference proceedings may be instituted with the U.S. Patent and Trademark Office, or the USPTO, when more than one person files a patent application covering the same technology, or if someone wishes to challenge the validity of an issued patent. At the completion of the interference proceeding, the USPTO will determine which competing applicant is entitled to the patent, or whether an issued patent is valid. Patent interference proceedings are complex, highly contested legal proceedings, and the USPTO’s decision is subject to appeal. This means that if an interference proceeding arises with respect to any of our patent applications, we may experience significant expenses and delay in obtaining a patent, and if the outcome of the proceeding is unfavorable to us, the patent could be issued to a competitor rather than to us. Third parties can file post-grant proceedings in the USPTO, seeking to have issued patent invalidated, within nine months of issuance. This means that patents undergoing post-grant proceedings may be lost, or some or all claims may require amendment or cancellation, if the outcome of the proceedings is unfavorable to us. Post-grant proceedings are complex and could result in a reduction or loss of patent rights. The institution of post-grant proceedings against our patents could also result in significant expenses.
Patent law outside the United States is uncertain and in many countries, is currently undergoing review and revisions. The laws of some countries may not protect our proprietary rights to the same extent as the laws of the United States. Third parties may attempt to oppose the issuance of patents to us in foreign countries by initiating opposition proceedings. Opposition proceedings against any of our patent filings in a foreign country could have an adverse effect on our corresponding patents that are issued or pending in the United States It may be necessary or useful for us to participate in proceedings to determine the validity of our patents or our competitors’ patents that have been issued in countries other than the United States. This could result in substantial costs, divert our
13
efforts and attention from other aspects of our business, and could have a material adverse effect on our results of operations and financial condition. We currently have pending patent applications or issued patents in Europe, Brazil, Mexico, India, Russia, Australia, Japan, Canada, China, Korea and Singapore, among others.
In addition to patent protection, we rely on unpatented trade secrets and proprietary technological expertise. We cannot assure you that others will not independently develop or otherwise acquire substantially equivalent techniques, somehow gain access to our trade secrets and proprietary technological expertise or disclose such trade secrets, or that we can ultimately protect our rights to such unpatented trade secrets and proprietary technological expertise. We rely, in part, on confidentiality agreements with our marketing partners, employees, advisors, vendors and consultants to protect our trade secrets and proprietary technological expertise. We cannot assure you that these agreements will not be breached, that we will have adequate remedies for any breach or that our unpatented trade secrets and proprietary technological expertise will not otherwise become known or be independently discovered by competitors.
Government Regulation – Medical Devices
As a medical company, we operate under stringent regulations and our companies and products are subject to a variety of distinct regulations around the world that are subject to modification or change.
Cytori Cell Therapy
Cytori Cell Therapy technology is regulated through a variety or agencies and approaches around the world. Our products must receive regulatory clearances or approvals from regulatory bodies in the European Union such as the EMA and the FDA and from other applicable governments prior to their sale or in some cases prior to clinical trials. This technology platform incorporates multiple elements including devices, reagents and software that in combination yield an autologous cellular product. As a result of the complex nature of our products and differing regulations through the world, there is no single unified of global set of regulatory requirements or common approach to regulation and is therefore region specific.
Cytori Cell Therapy technology is, and will be, subject to stringent government regulation in the United States by the FDA under the Federal Food, Drug and Cosmetic Act. The FDA regulates the design/development process, clinical testing, manufacture, safety, labeling, sale, distribution and promotion of medical devices and drugs. Included among these regulations are pre-market clearance and pre-market approval requirements, design control requirements, and the requirements to comply with Quality System Regulations/Good Manufacturing Practices. Other statutory and regulatory requirements govern, among other things, registration and inspection, medical device listing, prohibitions against misbranding and adulteration, labeling and post-market reporting. In the U.S., we must currently obtain FDA clearance or approval through the PMA application process, which requires clinical trials to generate clinical data supportive of safety and efficacy. Approval of a PMA could take four or more years from the time the process is initiated due to the requirement for clinical trials. Failure to comply with applicable requirements can result in application integrity proceedings, fines, recalls or seizures of products, injunctions, civil penalties, total or partial suspensions of production, withdrawals of existing product approvals or clearances, refusals to approve or clear new applications or notifications, and criminal prosecution.
Recently, the U.S. government enacted the 21st Century Cures Act, or the CURES Act, in the United States that has many provisions that could be favorable for us. However, the provisions of the CURES Act are broad and lack enough detail currently to determine its effect on our regulatory pathway. Further interpretation and implementation of the CURES Act must occur before any definitive assessments can be made.
Outside the U.S., the Cytori Cell Therapy family of products must also comply with the government regulations of each individual country in which the products are to be distributed and sold. These regulations vary in complexity and can be as stringent, and on occasion even more stringent, than FDA regulations in the United States. International government regulations vary from country to country and region to region. For example, regulations in some parts of the world only require product registration while other regions/countries require a complex product approval process. Due to the fact that there are new and emerging cell therapy and cell banking regulations that have recently been drafted and/or implemented in various countries around the world, the application and subsequent implementation of these new and emerging regulations have little to no precedent. Furthermore, the level of complexity and stringency is not always precisely understood today for each country, creating greater uncertainty for the international regulatory process. Furthermore, government regulations can change with little to no notice and may result in up-regulation of our product(s), thereby, creating a greater regulatory burden for our cell processing and cell banking technology products.
In Europe, Cytori Cell Therapy is approved as the Celution device and consumable product and is sold for commercial and research use. Expansion of use of Cytori Cell Therapy in Europe will likely require an expansion of our regulatory claims that would likely include disease-specific claims obtained through the completion of clinical trials. It is possible that Cytori Cell Therapy may be regulated as a device, similar to its regulatory pathway in the U.S., an advanced tissue medicinal product or ATMP, or some combination of the two in Europe. Cytori is current working with both European authorities and country-specific competent authorities to clarify the proper path for Cytori’s Habeo Cell Therapy in Europe.
14
Regulations in the Asia-Pacific and Japan regions are currently evolving for cell therapy products. For example, the Japan Diet enacted a regenerative medicine law in November of 2014 following sweeping changes in Japan’s medical device regulations in 2014. In China, the regulatory landscape for cell therapies such as ours is subject to increasing regulation, and success in this market will depend heavily on a firm understanding of applicable regulations and a commitment to pursuing appropriate regulatory approvals, including any required approvals from the National Health and Family Planning Commission of the People’s Republic of China, or NHFPC, and other governmental entities. These regulatory uncertainties further complicate the regulatory process in the Asia-Pacific region and may lengthen approval timelines and/or market entrance or penetration.
Regulatory Developments
China Regulatory Clearance
In April 2015, the State Food and Drug Administration of the People’s Republic of China, or CFDA, granted regulatory clearance for our Celution device, consumable kit and reagents necessary to allow the importation and sale of our products into the Chinese market, the world’s largest healthcare market. The Chinese market for our Celution products is subject to an exclusive license in favor of our partner, Lorem Vascular.
EU Orphan Designation
In April 2015, the European Commission, acting on the positive recommendation from the European Medicines Agency Committee for Orphan Medicinal Products, granted an orphan drug designation to Assistance Publique Hopitaux du Marseille (France), the sponsor institution for the SCLERADEC I and SCLERADEC II trials using Cytori Cell Therapy, for the treatment of systemic sclerosis.
In April 2016, the European Commission, acting on the positive recommendation from the European Medicines Agency Committee for Orphan Medicinal Products, issued orphan drug designation to a broad range of Cytori Cell Therapy formulations when used for the treatment of systemic sclerosis under Community Register of Orphan Medicinal Products number EU/3/16/1643.
In November 2016, the US FDA Office of Orphan Products Development (OOPD) granted Cytori an orphan drug designation for cryopreserved or centrally processed ECCS-50 Habeo for scleroderma.
Government Regulation – Nanoparticle Oncology Drugs
Our nanoparticle oncology drug products must receive regulatory approvals from the EMA and the FDA and, from other applicable governments prior to their sale.
Our current and future nanoparticle oncology drugs are, or will be, subject to stringent government regulation in the United States by the FDA under the Federal Food, Drug and Cosmetic Act. The FDA regulates the design/development process, clinical testing, manufacture, safety, labeling, sale, distribution, and promotion of oncology drugs. Included among these regulations are drug approval requirements and the current Good Manufacturing Practices, cGMP. Other statutory and regulatory requirements govern, among other things, cGMP inspection, prohibitions against misbranding and adulteration, labeling and post-market reporting. The recent CURES Act legislation regarding drugs in the United States has yet to be implemented and may yield additional regulatory requirements on therapeutic drugs while providing some relief in selected regulatory burdens. The FDA’s interpretation and implementation of the CURES Act has yet to be published.
Our nanoparticle oncology drugs must also comply with the government regulations of each individual country in which the products are to be distributed and sold. These regulations vary in complexity and can be as stringent, and on occasion even more stringent, than FDA regulations in the United States. International government regulations vary from country to country and region to region. For instance, our ATI-0918 drug candidate relies on an expedited approval process referred to as ‘bioequivalence’ or BE approved under an abbreviated new drug application, or ANDA. ANDA and BE products require a ‘reference drug’ and/or ‘reference listed drug’ ,or RLD, to show equivalence with. The reference drug may not be the same in all territories or countries, which could require different and unique BE clinical studies for some territories. Furthermore, the level of complexity and stringency is not always precisely understood today for each country, creating greater uncertainty for the international regulatory process. Additionally, government regulations can change with little to no notice and may result in the elimination of the BE regulatory pathway in some regions, creating increased regulatory burden.
Worldwide, the regulatory process can be lengthy, expensive, and uncertain with no guarantee of approval. Before any new drugs may be introduced to the U.S. market, the manufacturer generally must obtain FDA approval through either ANDA process for generic drugs off patent that allow for bioequivalence to and existing reference listed drug, or the lengthier new drug approval (NDA) process,
15
which typically requires multiple successful Phase III clinical trials to generate clinical data supportive of safety and efficacy along with extensive pharmacodynamic and pharmacokinetic preclinical testing to demonstrate safety. Approval of a ANDA could take four or more years from the time the process is initiated due to the requirement for clinical trials. NDA drugs could take significantly longer due to the additional preclinical requirements along with the typical requirement for two successful Phase III clinical trials.
Our lead ATI-0918 drug candidate is eligible for the ANDA regulatory pathway, while our ATI-0123 drug candidate is subject to the significantly lengthier NDA process. Changes to the reference listed drug (RLD) for drugs eligible for the ANDA process can result in significant delays in the regulatory process as BE clinical studies may need to be repeated for regions / countries that no longer recognize the RLD utilized in BE clinical studies. Failure to comply with applicable requirements can result in application integrity proceedings, fines, recalls or seizures of products, injunctions, civil penalties, total or partial suspensions of production, withdrawals of existing product approvals, refusals to approve new applications or notifications, and criminal prosecution.
Drugs are also subject to post-market reporting requirements for deaths or serious injuries when the drug may have caused or contributed to the death or serious injury, or serious adverse events. If safety or effectiveness problems occur after the drug reaches the market, the FDA may take steps to prevent or limit further marketing of the drug. Additionally, the FDA actively enforces regulations prohibiting marketing and promotion of drugs for indications or uses that have not been approved by the FDA.
We must comply with extensive regulations from foreign jurisdictions regarding safety, manufacturing processes and quality. These regulations, including the requirements for marketing and authorization, may differ from the FDA regulatory scheme in the United States.
Employees
As of December 31, 2016, we had 65 full-time employees. Of these full-time employees, seven were engaged in manufacturing, 31 were engaged in research and development, nine were engaged in sales and marketing and 18 were engaged in management, finance and administration. From time to time, we also employ independent contractors to support our operations. Our employees are not represented by any collective bargaining agreements and we have never experienced an organized work stoppage.
Corporate Information and Web Site Access to SEC Filings
We were initially formed as a California general partnership in July 1996, and incorporated in the State of Delaware in May 1997. We were formerly known as MacroPore Biosurgery, Inc., and before that as MacroPore, Inc. Our corporate offices are located at 3020 Callan Road, San Diego, CA 92121. Our telephone number is (858) 458-0900. We maintain an Internet website at www.cytori.com. Through this site, we make available free of charge our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) of the Securities Exchange Act of 1934, or the Exchange Act, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the U.S. Securities and Exchange Commission, or the SEC. In addition, we publish on our website all reports filed under Section 16(a) of the Exchange Act by our directors, officers and stockholders owning more than 10% of our outstanding common stock. These materials are accessible via the Investor Relations—Reports and Filings section of our website within the “SEC Filings” link. Some of the information is stored directly on our website, while other information can be accessed by selecting the provided link to the section on the SEC website, which contains filings for our company and its insiders.
The public can also obtain any documents that we file with the SEC at http://www.sec.gov. The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330.
16
In analyzing our company, you should consider carefully the following risk factors together with all of the other information included in this Annual Report on Form 10-K, including our audited Consolidated Financial Statements and the related notes and “Management’s Discussion and Analysis of Financial Conditions and Results of Operations”. If any of the risks described below occur, our business, operating results, and financial condition could be adversely affected and the value of our common stock could decline.
Risks Related to Our Business and Industry
Our success depends substantially upon the successful development and commercialization of our cellular therapeutics, and if we are unable to develop and commercialize our cellular therapeutic product candidates, especially Habeo Cell Therapy, our business could be seriously harmed.
Our success in large part is dependent upon our ability to develop our Cytori Cell Therapy products, and in particular, our Habeo Cell Therapy product. The success of Habeo Cell Therapy and any future cellular therapeutic products are highly dependent on meeting our primary endpoint in our U.S. Phase III STAR clinical trial. Further, if the primary endpoint in the currently enrolling French investigator-initiated SCLERADEC II trial is met, then the SCLERADEC II data would also be valuable to our regulatory and commercialization efforts within and outside the EU and could play a useful supporting role in any regulatory submissions to the U.S. FDA. If the STAR and/or SCLERADEC II clinical trial data are not deemed sufficient to support continued development and commercialization of Habeo Cell Therapy, our business will be significantly harmed. Further, even if the primary endpoints in these clinical trials are met, our ability to receive regulatory approval on a timely (or even possibly expedited) basis in the market in which we intend to market and sell Habeo Cell Therapy, and to receive the reimbursement coding, coverage and payment that we are currently anticipating, will likely be directly correlated to the reported efficacy of our Habeo Cell Therapy in the STAR trial, as well as SCLERADEC II clinical trial. There can be no assurance that such clinical data will meet these trials’ primary or secondary endpoints, or if met, that such data will support the regulatory approvals or reimbursement that we would seek for Habeo Cell Therapy, or any regulatory approvals or reimbursement at all.
Development and commercialization of our cellular therapeutics product candidates could be further materially harmed if we encounter difficulties such as:
|
|
• |
an inability to produce Habeo Cell Therapy or our other Cytori Cell Therapy product candidates at an appropriate cost or to scale for commercialization so as to meet customer demand for our cell therapy products; and |
|
|
• |
delayed, unexpected and/or adverse regulatory guidance, feedback or determinations, whether because of the novelty of our technology, changes in regulatory approval processes, or otherwise. |
We believe we must also continue to develop and manufacture enhanced and lower-cost versions of our Celution devices, reagents, and consumable kits. If we are not able to develop products capable of successfully competing in the marketplace, or if we experience disruptions and/or delays in our production of these products as required by the marketplace, our operations and commercialization efforts would be harmed. Further, there can be no assurance that we will be able to successfully develop and manufacture future generation Celution devices and other products in a manner that is cost-effective or commercially viable, or that development and manufacturing capabilities might not take much longer than currently anticipated to be ready for the market. Although we have been manufacturing the Celution 800 System and the StemSource 900-based Cell Bank since 2008, we cannot assure that we will be able to manufacture sufficient numbers of such products, or their successor products, to meet future demand, or that we will be able to overcome unforeseen manufacturing difficulties for this sophisticated equipment.
Our future success is in large part dependent upon our ability to successfully integrate and develop our Cytori Nanomedicine platform and commercialize our newly acquired ATI-0918 drug candidate, and any failure to do so could significantly harm our business and prospects.
In February 2017, we acquired substantially all of the assets of Azaya Therapeutics Inc, or Azaya, including Azaya’s two drug candidates, ATI-0918 and ATI-1123, and related manufacturing equipment and inventory. Our ability to successfully integrate, develop and commercialize these assets is subject to a number of risks, including the following:
|
|
• |
Azaya suspended its business, including its research and development efforts, at the end of 2015, so we must recommence the business, including (i) recalibration, revalidation and requalification of the acquired drug manufacturing equipment and manufacturing facility located in San Antonio, Texas; and (ii) hiring of substantial numbers of new employees to operate the Cytori Nanomedicines business. We may encounter unexpected issues and expenses in recommencing this business; |
17
|
|
• |
We do not have substantive drug development and commercialization experience, and thus we will be required to hire and rely on significant numbers of scientific, quality, regulatory and other technical personnel with the experience and expertise necessary to develop and commercialize our Cytori Nanomedicine drug candidates. We may be unable to identify, hire and retain personnel with the requisite experience to conduct the operations necessary to commercialize our ATI-0918 and ATI-1123 product candidates, in which case our business would be materially harmed; |
|
|
• |
ATI-0918, a complex generic liposomal formulation of doxorubicin, is very difficult to manufacture, and we can offer no assurances that we will (i) be able to manufacture this drug in accordance with all applicable laws and regulations; or (ii) demonstrate bioequivalence to Lipodox® (Sun Pharma) in the United States; or Caelyx® (Janssen, a Johnson & Johnson company) in Europe as required to obtain regulatory approvals within our currently anticipated timeframes, or at all; |
|
|
• |
We intend to find a commercialization partner to share or assume responsibility for commercialization, marketing and sales activities and related costs and expenses for our ATI-0918 drug candidate, as well as our ATI-1123 drug candidate. We do not currently have the financial resources to develop our ATI-1123 drug candidate internally, nor do we currently have the financial or human resources to market and sell ATI-0918 or ATI-1123 if and when commercialized, so if we are unable to find a suitable partner to take on these activities and costs, we may be forced to delay or suspend our development and commercialization activities, or procure additional capital to continue development of these drug candidates ourselves. There can be no assurance that we would obtain sufficient capital to fund the development and commercialization of our Cytori Nanomedicines program ourselves, or if we do obtain such capital, that our development and commercialization efforts would be successful; |
|
|
• |
Conduct of this newly acquired business will require significant capital, and to the extent that we incur unanticipated expenses or revenue downturns in our business, are unable to timely obtain sufficient additional capital on terms acceptable to us (or at all) to fund this business, our ability to commercialize our ATI-0918 drug candidate could be materially and adversely impacted; |
|
|
• |
New competitive products become commercially available before we launch ATI-0918; |
|
|
• |
It is possible that the EMA could change the reference drug for ATI-0918 in Europe from Caelyx. Though we deem this possibility to be unlikely, if the EMA were to change the reference drug, we could be required to conduct a bioequivalence trial to establish bioequivalence with the new reference drug, which would adversely affect our business and operations; and |
|
|
• |
We are not experienced in acquiring and integrating new businesses. |
If we are unable to successfully partner with other companies to commercialize our product candidates, our business could materially suffer.
A key part of our business strategy is to leverage strategic partnerships/collaborations to commercialize our product candidates. We do not have the financial, human or other resources necessary to develop, commercialize, launch or sell our therapeutic offerings in all of the geographies that we are targeting, and thus it is important that we identify and partner with third parties who possess the necessary resources to bring our products to market. We expect that any such partners will provide regulatory and reimbursement/pricing expertise, sales and marketing resources, and other expertise and resources vital to the success of our product offerings in their territories. We further expect, but cannot guarantee, that any such partnering arrangements will include upfront cash payments to us in return for the rights to develop, manufacture, and/or sell our products in specified territories, as well as downstream revenues in the form of milestone payments and royalties.
We are currently prioritizing our efforts to find a strategic partner for our Habeo Cell Therapy, formerly ECCS-50, which is specifically intended for treatment of hand dysfunction in scleroderma patients. For various reasons, including the novelty of our cellular therapeutic approach, the regulatory and reimbursement environments for Habeo Cell Therapy in certain markets, including Europe and the Asia-Pacific region, are complex and uncertain. There can be no assurance that regulatory agencies or authorities in the U.S., Europe, the Asia-Pacific region or elsewhere will grant conditional or full regulatory approval for Habeo Cell Therapy on the timeframes we anticipate, or at all, nor can we guarantee that government or commercial payers will grant us favorable reimbursement for use of Habeo Cell Therapy. Further, even if we receive regulatory approval and favorable reimbursement, there is no guarantee that a market will develop for Habeo Cell Therapy at our intended price points, or at all. These commercialization risks could affect prospective partners’ or collaborators’ willingness to enter into partnering arrangements on terms acceptable to us, or at all. Prospective partners may be unwilling to enter into an agreement with us unless and until we announce positive top-line data from our STAR clinical trial, which announcement is expected to occur in or around mid-2017. If the STAR and/or SCLERADEC II clinical trial data do not meet their primary endpoints, we anticipate that it will be difficult to thereafter find a commercialization partner for
18
our Habeo Cell Therapy on favorable terms, if at all. If we do conclude a partnering arrangement for our Habeo Cell Therapy prior to announcement of STAR clinical trial data, any such agreement may contain certain payment conditions, termination rights or other rights and obligations that would be triggered by positive or negative STAR data.
We are also prioritizing our efforts to find a strategic partner to help commercialize and sell our ATI-0918 drug candidate, initially in Europe, and secondarily, to fund development and commercialization of our ATI-1123 product candidate. We do not currently have the commercial expertise or resources to market and sell either ATI-0918 or ATI-1123. There can be no assurance that we will enter into partnering agreements for either ATI-0918 or ATI-1123 with suitable partners on terms acceptable to us, or at all. However, regardless of whether we enter into a partnering agreement for ATI-0918, we will still incur significant near-term costs and expenses in manufacturing, testing and validating it and in performing necessary regulatory and clinical work to ready our EMA marketing dossier for submission. If we cannot find a suitable partner for our ATI-0918 product candidate, our business could be significantly harmed.
We are also soliciting partnering interest in our ECCO-50 therapeutic for use in knee osteoarthritis, but we anticipate that our partnering efforts with respect to this indication will be subordinate to our Habeo Cell Therapy and ATI-0918 partnering efforts. Further, while consistent trends were observed in most secondary endpoints relative to the placebo group in our ACT-OA knee osteoarthritis trial, the 12-week endpoint of single pain on walking question did not achieve statistical significance, so there can be no assurance that our partnering efforts for our ECCS-50 therapeutics will be successful.
In addition, we may seek development and/or commercial partners for the other therapeutic indications set forth in our clinical pipeline, including:
|
|
• |
use of Cytori Cell Therapy in stress urinary incontinence, or SUI, in men following surgical removal of the prostate gland (this therapeutic indication is currently the subject of a Phase III, investigator-initiated trial in Japan, called ADRESU); and |
|
|
• |
development of Cytori Cell Therapy for Secondary Raynaud’s Phenomenon, or SRP (this therapeutic indication is currently in the pre-clinical stage). |
There can be no assurance that these male SUI and SRP pipeline indications will be attractive to prospective partners. The male SUI market is small (approximately $45.0 million), and the long-term viability of both indications, especially SRP, is in substantial part dependent upon receipt of positive STAR and/or SCLERADEC II clinical data.
Even if we succeed in securing partners for our lead or other product candidates, our partners may fail to develop or effectively commercialize our product candidates. Partnerships and collaborations involving our products and product candidates pose a number of risks, including the following:
|
|
• |
partners may not have sufficient resources or may decide not to devote the necessary resources due to internal constraints such as budget limitations, lack of human resources, or a change in strategic focus; |
|
|
• |
partners may believe our intellectual property is not valid or is unenforceable or unprotectable, or the product or product candidate infringes on the intellectual property rights of others; |
|
|
• |
partners may dispute their responsibility to conduct development and commercialization activities pursuant to the applicable collaboration, including the payment of related costs or the division of any revenues; |
|
|
• |
partners may decide to pursue a competitive product developed outside of the partnering arrangement; |
|
|
• |
partners may not be able to obtain, or believe they cannot obtain, the necessary regulatory approvals or reimbursement rates for the product candidates; and |
|
|
• |
partners may decide to terminate or not to renew their agreement with us for these reasons or other reasons. |
As a result, partnering agreements may not lead to development or commercialization of our lead product candidates or other product candidates in the most efficient manner or at all.
Our current business strategy is high-risk.
Our current business strategy is to aggressively develop and commercialize our Cytori Cell Therapy and Cytori Nanomedicine platforms, while simultaneously controlling expenses and preserving and growing our existing contract and
19
commercial sales revenues. We also believe that there are synergies between our existing cellular therapeutic technologies and our oncology drug assets that we can exploit and commercialize.
Our current business strategy is a high-risk strategy for a number of reasons including the following:
|
|
• |
current and anticipated clinical trials using Cytori Cell Therapy, including our current STAR clinical trial and the investigator-initiated SCLERADEC II trial, may not yield positive results; |
|
|
• |
research and development and commercialization of our cellular therapeutics and our oncology drug assets will require significant amounts of additional capital, and we cannot assure you that we will have access to sufficient capital, or find partners to provide capital, necessary to develop and bring our products to market; |
|
|
• |
our business model may be challenging for prospective business partners, as our therapeutic approach involves: |
|
|
o |
multiple procedures - liposuction followed by preparation and same-day administration of the autologous cellular therapeutic – for which there may not be existing reimbursement codes (or which reimbursement codes may not be deemed adequate by prospective partners); and |
|
|
o |
processing of cells via our Celution System (which to date has been regulated as a medical device), followed by administration of our Cytori Cell Therapy, which is considered to be a drug by FDA and other regulatory agencies. |
|
|
• |
our current installed base of Celution devices may pose potential risks to us if the operators of these devices (i) harm a patient during the course of treating the patient with Cytori Cell therapy; or (ii) treat patients “off label” in a manner that is competitive with us, creates channel conflict with us, or otherwise negatively impacts our business; |
|
|
• |
our Celution platform is a novel technology that may never receive regulatory or commercial approval for our intended therapeutic indications; |
|
|
• |
we may incur material costs and expenses in executing our business strategy that are not currently contemplated and that could cause our operating expenses to materially increase beyond current projections; |
|
|
• |
our Celution technology is potentially subject to different regulatory regimes in different territories, and we are not experienced in obtaining regulatory approvals for therapeutic indications, such as hand complications of scleroderma, of our Cytori Cell Therapy products; |
|
|
• |
we do not have an operating history as a drug development company, or prior experience with obtaining regulatory, reimbursement or other approvals for drug candidates such as ATI-0918 and ATI-1123; |
|
|
• |
our ATI-0918 and ATI-1123 drug candidates, if commercialized, will compete against established competitive drugs that are marketed and sold by large companies with significant human, technical and financial resources; |
|
|
• |
we are not experienced in acquiring and integrating new assets, such as those acquired from Azaya; |
|
|
• |
we are unfamiliar with the competitive landscape for our Cytori Nanomedicines product candidates, and as such key assumptions regarding customer acceptance and market share may not be realized; |
|
|
• |
our product candidates may never become commercially viable; |
|
|
• |
we may not be able to prevent other companies from depriving us of market share and profit margins by selling products based on our intellectual property and developments; and |
|
|
• |
the regenerative medicine industry is a very risky industry, and this has adversely affect our ability to attract investment capital and collaborators for our Cytori Cell Therapy. |
Our business is sensitive to general economic, business and industry conditions.
We are exposed to general economic, business and industry conditions, both in the United States and globally. Adverse global economic and financial conditions are difficult to predict and mitigate against, and therefore the potential impact is difficult to estimate. Negative trends in the economy, including trends resulting from an actual or perceived recession, tightening credit markets, such as significant reductions in available capital and liquidity from banks and other credit providers, substantial volatility in equity
20
and currency values worldwide, prolonged recessionary or slow growth periods, increased cost of commodities, including oil, actual or threatened military action by the United States, and threats of terrorist attacks in the United States and abroad, could cause a reduction of investment in and available funding for companies in certain industries, including ours and those of our customers. Thus, our business operations and ability to raise capital has been, and may in the future, be adversely affected by downturns in current credit conditions, financial markets and the global economy.
We face intense competition, and if our competitors market and/or develop products that are marketed more effectively, approved more quickly than our product candidates or demonstrated to be safer or more effective than our products, our commercial opportunities could be reduced or eliminated.
The life science industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary therapeutics. We face competition from a number of sources, some of which may target the same indications as our products or product candidates, including small and large, domestic and multinational, medical device, biotechnology and pharmaceutical companies, academic institutions, government agencies and private and public research institutions, many of which have greater financial resources, sales and marketing capabilities, including larger, well-established sales forces, manufacturing capabilities, experience in obtaining regulatory approvals for product candidates and other resources than we do.
We expect that product candidates in our pipeline, if approved, to compete on the basis of, among other things, product efficacy and safety, time to market, price, coverage and reimbursement by third-party payers, extent of adverse side effects and convenience of treatment procedures. One or more of our competitors may develop other products that compete with ours, obtain necessary approvals for such products from the FDA, EMA, MHLW or other agencies, if required, more rapidly than we do or develop alternative products or therapies that are safer, more effective and/or more cost effective than any products developed by us. The competition that we will encounter with respect to any of our product candidates that receive the requisite regulatory approval and classification and are marketed will have an effect on our product prices, market share and results of operations. We may not be able to differentiate any products that we are able to market from those of our competitors, successfully develop or introduce new products that are less costly or offer better results than those of our competitors, or offer purchasers of our products payment and other commercial terms as favorable as those offered by our competitors. In addition, competitors may seek to develop alternative formulations of or technological approaches to our product candidates and/or alternative cell therapy or drug delivery technologies that address our targeted indications.
Cytori Cell Therapy: Cytori Cell Therapy may face competition from cell therapies derived from autologous or allogeneic tissue sources such as adipose tissue, bone marrow and cord blood, and processed using alternative approaches, methods and technologies such as cryopreserved, cultured, expanded, manual, non-enzymatic, selectively isolated cell therapies, and other therapeutic approaches including those administered using oral, subcutaneous, topical and intravenous routes. If approved for the treatment of hand dysfunction and/or Raynaud’s Phenomenon in scleroderma patients, Habeo Cell Therapy will likely compete against other products and product candidates. Johns Hopkins University, in collaboration with Allergan, recently completed and reported results from a Phase III clinical trial evaluating injection of Botox into the hands of patients with scleroderma-associated Raynaud’s Syndrome. Further, Corbus Pharmaceuticals is conducting a Phase II clinical trial evaluating Resunab (JBT-101) in patients with diffuse cutaneous systemic sclerosis and has reported positive topline results showing a clear signal of clinical benefit. University of Pittsburgh, in collaboration with the NIAMS, is conducting a Phase II clinical trial evaluating Lipitor’s (atorvastatin) effect on blood vessel function and Raynaud symptoms in patients with early diffuse systemic sclerosis. Primus Pharmaceuticals is sponsoring a U.S. multi-center clinical trial to evaluate Diosmiplex in patients with Raynaud’s disease. Covis Pharmaceuticals has completed a Phase 2 clinical trial to evaluate Vascana in patients with Raynaud’s Phenomenon secondary to Connective Tissue Disease. Apricus Biosciences has completed a Phase 2 clinical trial for Vascana in patients with Raynaud’s Phenomenon secondary to systemic sclerosis. Stanford University, in collaboration with United Therapeutics, is sponsoring a Phase 2 clinical trial evaluating oral Orenitram (treprostinil) for the treatment of Calcinosis in patients with systemic sclerosis. Bayer is a collaborator in a Phase 2 clinical trial evaluating Adempas (riociguat) in patients with scleroderma-associated digital ulcers. Bristol-Myers Squibb and NIAID are collaborators in a Phase 2 clinical trial evaluating Abatacept in patients with diffuse cutaneous systemic sclerosis. Invtiva Pharma is sponsoring a Phase 2 clinical trial evaluating IVA337 in patients with diffuse cutaneous systemic sclerosis. The Catholic University of Korea is sponsoring a clinical trial evaluating autologous stromal vascular fraction injected into the fingers of patients with systemic sclerosis. Sanofi is sponsoring a Phase 2 clinical trial evaluating SAR156597 in patients with diffuse systemic sclerosis. Hoffman-La Roche is sponsoring Phase 3 clinical trials evaluating Actemra (tocilizumab) in patients with systemic sclerosis. Most of these studies use the primary and secondary outcome measures as used in our STAR clinical trial.
Our Cytori Cell Therapy may also face competition from lower price alternative cell therapies, including manually processed, or “home brewed” ADRCs that are harvested and used to treat patients for a wide range of indications. There are hundreds of stromal vascular fraction, or SVF, clinics within the United States alone, that purport to offer cell therapy treatments for ailments ranging from facial rejuvenation to stroke. Though FDA has indicated that it intends to regulate this “home brew” industry, if it fails to do so, then companies without FDA approvals may continue to offer cell therapy treatments on an “off-label,” unapproved basis at substantially
21
lower prices then we intend to command. Similar clinics existing in every other market in which we intend to compete. Further, it is possible that positive STAR or SCLERADEC II clinical data, if possible, could be used by our cheaper cost competitors to tout their own cell therapy offerings, which could significantly harm our business.
Cytori Nanomedicines: We may face competition for our ATI-0918 asset (which is intended for the treatment of breast and ovarian cancers, multiple myeloma, and Kaposi’s sarcoma) from multiple drug classes including antiretrovirals, chemotherapies, corticosteroids, histone deacetaylase inhibitors, hormone therapies, immunotherapies, and targeted therapies, as well as companies seeking approvals in Europe or the United States for their pegylated liposomal doxorubicin products. In particular, if a competitor is first to the European market with an EMA-approved generic liposomal doxorubicin that is bioequivalent to Caelyx, our projections and market assumptions for our ATI-0918 would have to be materially altered and our business could be harmed. Taiwan Liposome Company has reported their intent to file a Marketing Authorization Application, or MAA, with the EMA in the first half of 2017 for its generic Doxisome (TLC177) product which is ahead of our schedule for submitting our MAA for ATI-0918. In the United States, we may face competition for ATI-0918 from multiple generic formulations of pegylated liposomal doxorubicin. Sun Pharma’s Lipodox product is currently approved in the United States and both Taiwan Liposome Company (Doxisome) and Actavis have reported that they have filed ANDAs with the FDA. Shanghai F-Z (Libaoduo) and Dr. Reddy’s Labs are conducting ongoing bioequivalence studies versus Lipodox which they may decide to use to support FDA submissions for approval of their pegylated liposomal doxorubicin products.
Companies that currently have active development programs for nanoparticle-docetaxel products that may be future competitors for our ATI-1123asset include:
|
|
• |
Adocia’s DriveIn nanoparticle-docetaxel product candidate, which is in the preclinical stage; |
|
|
• |
Cristal Therapeutics’ CriPac nanoparticle-docetaxel, which is currently being evaluated in a Phase 1 clinical trial for the treatment of solid tumors; and |
|
|
• |
Oasmia’s Docecal, a formulation of docetaxel combined with a patented nanoparticle-based technology, XR17, which is currently being evaluated in a Phase 1 clinical trial. |
Competitors may have greater experience in developing therapies or devices, conducting clinical trials, obtaining regulatory clearances or approvals, manufacturing and commercialization. It is possible that competitors may obtain patent protection, approval, or clearance from the FDA or achieve commercialization earlier than we can, any of which could have a substantial negative effect on our business. Compared to us, many of our potential competitors have substantially greater:
|
|
• |
capital resources; |
|
|
• |
research and development resources and experience, including personnel and experience; |
|
|
• |
product development, clinical trial and regulatory resources and experience; |
|
|
• |
sales and marketing resources and experience; |
|
|
• |
manufacturing and distribution resources and experience; |
|
|
• |
name, brand and product recognition; and |
|
|
• |
resources, experience and expertise in prosecution and enforcement of intellectual property rights. |
As a result of these factors, our competitors may obtain regulatory approval of their products more quickly than we are able to or may obtain patent protection or other intellectual property rights that limit or block us from developing or commercializing our product candidates. Our competitors may also develop products that are more effective, more useful, better tolerated, subject to fewer or less severe side effects, more widely prescribed or accepted or less costly than ours and may also be more successful than we are in manufacturing and marketing their products. If we are unable to compete effectively with the marketed therapeutics of our competitors or if such competitors are successful in developing products that compete with any of our product candidates that are approved, our business, results of operations, financial condition and prospects may be materially adversely affected.
22
Our clinical trials may fail to demonstrate acceptable levels of safety and efficacy for Habeo Cell Therapy or any of our other product candidates, which could prevent or significantly delay their regulatory approval and commercialization.
Clinical testing of our products is a long, expensive and uncertain process, and the failure or delay of a clinical trial can occur at any stage. Many factors, currently known and unknown, can adversely affect clinical trials and the ability to evaluate a product candidate’s efficacy. During the course of treatment, patients can die or suffer other adverse events for reasons that may or may not be related to the proposed product being tested. Even if initial results of preclinical and nonclinical studies or clinical trial results are promising, we may obtain different results in subsequent trials or studies that fail to show the desired levels of safety and efficacy, or we may not obtain applicable regulatory approval for a variety of other reasons. For instance, the investigator-initiated 12-patient, open-label SCLERADEC I trial investigating use of Habeo Cell Therapy for hand complications of scleroderma, sponsored by the Assistance Publique Hôpitaux de Marseille, or AP-HM, located in Marseille, France, has reported strong clinical data suggesting safety and efficacy of a single treatment of Habeo Cell Therapyout to three years after treatment. However, there can be no assurances that our current STAR clinical trial or AP-HM’s currently enrolling SCLERADEC II clinical trial will be successful. These trials are testing broader human use of Habeo Cell Therapy in blinded, randomized, placebo-controlled trial settings, as opposed to SCLERADEC I’s open-label, single arm, uncontrolled, unblinded format. Many companies in our industry have suffered significant setbacks in advanced clinical trials, despite promising results in earlier trials. If our Phase III STAR clinical trial and the Phase III Cytori-supported SCLERADEC II clinical trial do not meet their primary endpoints, we will likely be unable to obtain regulatory approval for our Habeo Cell Therapy, and may be forced to abandon our scleroderma development program, which would severely affect our business.
Further, with respect to the conduct and results of clinical trials generally, in the United States, Europe, Japan and other jurisdictions, the conduct and results of clinical trials can be delayed, limited suspended, or otherwise adversely affected for many reasons, including, among others:
|
|
• |
clinical results may not meet prescribed endpoints for the studies or otherwise provide sufficient data to support the efficacy of our product candidates; |
|
|
• |
clinical and nonclinical test results may reveal side effects, adverse events or unexpected safety issues associated with the use of our product candidates; |
|
|
• |
lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and increased expenses associated with the services of our contract research organizations, or CROs, and other third parties; |
|
|
• |
inability to design appropriate clinical trial protocols; |
|
|
• |
slower than expected rates of subject recruitment and enrollment rates in clinical trials; |
|
|
• |
regulatory review may not find a product safe or effective enough to merit either continued testing or final approval; |
|
|
• |
regulatory review may not find that the data from preclinical testing and clinical trials justifies approval; |
|
|
• |
regulatory authorities may require that we change our studies or conduct additional studies which may significantly delay or make continued pursuit of approval commercially unattractive; |
|
|
• |
a regulatory agency may reject our trial data or disagree with our interpretations of either clinical trial data or applicable regulations; |
|
|
• |
the cost of clinical trials required for product approval may be greater than what we originally anticipate, and we may decide to not pursue regulatory approval for such a product; |
|
|
• |
a regulatory agency may identify problems or other deficiencies in our existing manufacturing processes or facilities or the existing processes or facilities of our collaborators, our contract manufacturers or our raw material suppliers; |
|
|
• |
a regulatory agency may change its formal or informal approval requirements and policies, act contrary to previous guidance, adopt new regulations or raise new issues or concerns late in the approval process; |
23
|
|
• |
a product candidate may be approved only for indications that are narrow or under conditions that place the product at a competitive disadvantage, which may limit the sales and marketing activities for such products or otherwise adversely impact the commercial potential of a product; and |
|
|
• |
a regulatory agency may ask us to put a clinical study on hold pending additional safety data; (and there can be no assurance that we will be able to satisfy the regulator agencies’ requests in a timely manner, which can lead to significant uncertainty in the completion of a clinical study). |
In addition, Cytori Cell Therapy is currently the subject of a number of investigator-initiated trials, including the SCLERADEC II clinical trial in France and the ADRESU clinical trial in Japan. While these investigator-initiated trials are useful to help enhance awareness and use of our cell therapy technologies and products, and to identify potential therapeutic targets, there are also associated risks. We do not control the design and conduct of these trials, thus any data integrity issues or patient safety arising out of any of these trials would be beyond our control, yet could adversely affect our reputation and damage the clinical and commercial prospects for our Cytori Cell Therapy product candidates.
We also face clinical trial-related risks with regard to our reliance on other third parties in the performance of many of the clinical trial functions, including CROs, that help execute our clinical trials, the hospitals and clinics at which our trials are conducted, the clinical investigators at the trial sites, and other third party service providers. Failure of any third-party service provider to adhere to applicable trial protocols, laws and regulations in the conduct of one of our clinical trials could adversely affect the conduct and results of such trial (including possible data integrity issues), which could seriously harm our business.
Our success depends in substantial part on our ability to obtain regulatory approvals for Habeo Cell Therapy and ATI-1123. However, we cannot be certain that we will receive regulatory approval for these product candidates or our other product candidates.
We have only a limited number of product candidates in development, and our business depends substantially on their successful development and commercialization. Our product candidates will require development, regulatory review and approval in multiple jurisdictions, substantial investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before we can generate any revenues from sales of our product candidates. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, whose regulations differ from country to country.
We are not permitted to market our product candidates in the United States until we receive approval from the FDA, or in any foreign countries until we receive the requisite approval from the regulatory authorities of such countries (including centralized marketing authorization from the European Medicines Agency), and we may never receive such regulatory approvals. Obtaining regulatory approval for a product candidate is a lengthy, expensive and uncertain process, and may not be obtained. Any failure to obtain regulatory approval of any of our product candidates would limit our ability to generate future revenues (and any failure to obtain such approval for all of the indications and labeling claims we deem desirable could reduce our potential revenue), would potentially harm the development prospects of our product candidates and would have a material and adverse impact on our business.
Even if we successfully obtain regulatory approvals to market our product candidates, our revenues will be dependent, in part, on our ability to commercialize such products as well as the size of the markets in the territories for which we gain regulatory approval. If the markets for our product candidates are not as significant as we estimate, our business and prospects will be harmed
Regarding to our two current lead commercialization candidates, Habeo Cell Therapy and ATI-0918:
|
|
• |
Though we believe that Habeo Cell Therapy will be regulated as an Advanced Therapeutic Medicinal Product, or ATMP, in Europe, it is possible that the EMA instead provides a medical device classification for Habeo Cell Therapy, in which case we will be unable to avail ourselves of the orphan drug designation granted to us covering use of Cytori Cell Therapy for systemic sclerosis, and will instead compete with other medical device manufacturers purporting to offer cellular therapeutics competitive with ours (and possibly at much lower price points than we currently contemplate for our therapy). Any classification of Cytori Cell Therapy as a medical device could make it difficult for us to identify pharmaceuticals companies willing to help us commercialize this product offering in Europe, and could also deter medical device companies from partnering with us given potential pricing and competitive concerns. |
|
|
• |
If Habeo Cell Therapy is classified as an ATMP in Europe, then we will be required to comply with applicable cGMP requirements, as interpreted and implemented at the national level in each country, which would take longer and cost more to get to market than if Habeo Cell Therapy were classified as a medical device, and would in turn increase the costs of commercializing Habeo Cell Therapy in these countries. Further, potential pharmaceutical |
24
|
|
partners may be wary of the medical device component of our cell therapy. These commercialization hurdles could increase the difficulties in finding suitable partners to help us commercialize this product offering in Europe. |
|
|
• |
The EMA has approved eight ATMPs in Europe to date, with application review periods ranging from approximately thirteen to thirty-five months. This wide range in review periods makes it difficult to predict whether and on what timeframe our Habeo Cell Therapy would receive EMA approval, if at all. |
|
|
• |
Given the novelty of our cellular therapeutics technology, we anticipate that we may face regulatory hurdles in other jurisdictions outside of the United States and Europe that could delay regulatory approval and commercial launch of Habeo Cell Therapy. |
|
|
• |
The reference drugs for ATI-0918, which are currently Lipodox® in the United States and Caelyx® in Europe, may change. |
|
|
• |
Though Azaya previously completed a European ATI-0918 60-patient bioequivalence trial, the EMA has not confirmed the adequacy of the trial for purposes of determining bioequivalence of ATI-0918 to Caelyx®. It is possible that the EMA could require us to conduct another bioequivalency trial for ATI-0918, which would cause us to incur significant delays and additional costs and expense and would materially and adversely affect our business. |
|
|
• |
Though it is our intent to expeditiously pursue regulatory review of ATI-0918 in Europe through submission of a marketing authorization application, or MAA, to the EMA, prior to submission of this application we must first conduct and complete certain activities, including chemistry, manufacturing and controls, or CMC, activities, for inclusion in the application, and we cannot guarantee that we will successfully complete these activities. |
|
|
• |
We intend to seek scientific advice from the EMA regarding required elements of the MAA before we submit the MAA, and if the EMA’s scientific advice requires us to conduct substantive additional work (including possible provision of substantial additional data or information), our submission of the MAA could be materially delayed, which in turn would materially push back our anticipated launch date for ATI-0918 in Europe. |
|
|
• |
If we are unable to satisfy the EMA’s requirements to issuance of the marketing authorization for ATI-0918, we will not be able to launch ATI-0918 in Europe, and our business would be materially harmed. |
If a product candidate is not approved in a timely fashion on commercially viable terms, or if development of any product candidate is terminated due to difficulties or delays encountered in the regulatory approval process, it could have a material adverse effect on our business, and we will become more dependent on the development of other proprietary products and/or our ability to successfully acquire other products and technologies. There can be no assurance that any product candidate will receive regulatory approval in a timely manner, or at all.
If our products candidate and technologies receive regulatory approval but do not achieve broad market acceptance, especially by physicians, the revenues that we generate will be limited.
The commercial success of any of our approved products or technologies will depend upon the acceptance of these products and technologies by physicians, patients and the medical community. The degree of market acceptance of these products and technologies will depend on a number of factors, including, among others:
|
|
• |
acceptance by physicians and patients of the product as a safe and effective treatment; |
|
|
• |
any negative publicity or political action related to our or our competitors’ products or technologies; |
|
|
• |
the relative convenience and ease of administration; |
|
|
• |
the prevalence and severity of adverse side effects; |
|
|
• |
demonstration to authorities of the pharmacoeconomic benefits; |
|
|
• |
demonstration to authorities of the improvement in burden of illness; |
|
|
• |
limitations or warnings contained in a product’s approved labeling; |
25
|
|
• |
payers’ level of restrictions and/or barriers to coverage; |
|
|
• |
the clinical indications for which a product is approved; |
|
|
• |
availability and perceived advantages of alternative treatments; |
|
|
• |
the effectiveness of our or any current or future collaborators’ sales, marketing and distribution strategies; and |
|
|
• |
pricing and cost effectiveness. |
Our Celution technology and products compete against cell-based therapies derived from alternate sources, such as bone marrow, umbilical cord blood and, potentially, embryos. Some of our competitors with products based on these other cell-based therapies have substantially greater financial, human and technical resources than we do. In addition, some of them have approved products with therapeutic claims, established revenues and broad market recognition. Physicians historically are slow to adopt new technologies like ours regardless of the perceived merits when older technologies, as the current standard of care, continue to be supported by established providers. Overcoming such inertia often requires significant marketing expenditures or definitive product performance and/or pricing superiority.
We face similar competitive pressures with our Cytori Nanomedicine product candidates. As a generic liposomal encapsulation of doxorubicin, ATI-0918, if approved and launched commercially, will potentially compete against Caelyx and Myocet® (manufactured by Teva) in Europe, and against Lipodox® in the United States. These existing competitive liposomal doxorubicin products have been on the market for many years, have gained widespread physician acceptance and are marketed by competitors with substantially greater resources than we have. Further, our ATI-1123 product candidate, if developed and commercialized, would compete against a number of established docetaxel drugs, including Taxotere® (Sanofi S.A.) and numerous existing generic docetaxel products.
We expect physicians’ inertia and skepticism to also be a significant barrier as we attempt to gain market penetration with our future products. We believe we will continue to need to finance lengthy time-consuming clinical studies to provide evidence of the medical benefit of our products and resulting therapies in order to overcome this inertia and skepticism.
Overall, our efforts to educate the medical community on the benefits of any of our products or technologies for which we obtain marketing approval from the FDA or other regulatory authorities and gain broad market acceptance may require significant resources and may never be successful. If our products and technologies do not achieve an adequate level of acceptance by physicians, pharmacists and patients, we may not generate sufficient revenue from these products to become or remain profitable.
Many potential applications of our product candidates are pre-commercial, which subjects us to development and marketing risks.
Our products candidates are at various stages of development. Successful development and market acceptance of our products is subject to developmental risks, including risk of negative clinical data from current and anticipated trials, failure of inventive imagination, ineffectiveness, lack of safety, unreliability, manufacturing hurdles, failure to receive necessary regulatory clearances or approvals, high commercial cost, preclusion or obsolescence resulting from third parties’ proprietary rights or superior or equivalent products, competition from copycat products and general economic conditions affecting purchasing patterns. There can be no assurance that we or our partners will successfully develop and commercialize our product candidates, or that our competitors will not develop competing technologies that are less expensive or superior. Failure to successfully develop and market our product candidates would have a substantial negative effect on our results of operations and financial condition.
Regarding our cell therapy products, we believe that our long-term viability and growth will depend in large part on our ability to establish the safety and efficacy of our cell therapies through clinical trials and studies. Though we generate revenues from commercial sales of our Celution products, there is no proven path for commercializing Cytori Cell Therapy in a way to earn a durable profit commensurate with the medical benefit. We have been engaged for a number of years in commercial sales of our Celution devices and consumable kits in Japan Europe and certain Asian markets, and our cell banking products in Japan, Europe, and certain Asian markets, but we have not achieved significant growth due in significant part to our inability thus far to obtain therapeutic, on-label use that is reimbursed by payers. Thus, we do not expect the market for our products to appreciably increase until we have positive clinical data from a validated, Phase III, controlled, randomized trial that reports safety and efficacy of our cellular therapeutic in a discrete disease state or condition. However, there can be no assurance that one or more clinical trials of our cell therapy product candidates will yield positive results.
26
Regarding our Cytori Nanomedicine program, our ATI-0918 generic drug candidate is pre-commercial. Our ATI-0918 bioequivalence trial results and accompanying manufacturing and other data are subject to review and feedback by the EMA prior to our submission of our marketing authorization application, or MAA, to the EMA. There can be no assurances that the EMA will view the results of the bioequivalence trial favorably. Further, we are required to complete certain manufacturing, drug stability and other activities before we submit our MAA to the EMA. There can be no assurance that the EMA will deem our MAA sufficient grant us marketing authorization within the timelines we currently project, or at all.
Our ATI-1123 drug candidate is in early clinical stages and is subject to all of the attendant risks of an early-stage drug. Should we wish to commercialize ATI-0918 in the United States, we believe we will need to conduct a clinical trial to demonstrate bioequivalence to the then-current reference drug in the United States (currently Lipodox®). Any such bioequivalency trial would be time and resource intensive, would take years to complete at considerable expense, and could ultimately fail to demonstrate ATI-0918’s bioequivalence to the reference drug. Also, we intend to find a partner to develop our ATI-1123 drug candidate, but and if we are unsuccessful in doing so, our ATI-1123 development program could be delayed or suspended.
If we or any party to a key collaboration, licensing, development, acquisition or similar arrangement fails to perform material obligations under such arrangement, or any arrangement is terminated for any reason, there could be an adverse effect on our business.
We are currently party to certain licensing, collaboration and acquisition agreements under which we may make or receive future payments in the form of milestone payments, maintenance fees, royalties and/or minimum product purchases. We are dependent on our collaborators to commercialize Cytori Cell Therapy in certain countries and in certain indications for us to realize any financial benefits from these collaborations. Our collaborators may not devote the attention and resources to such efforts to be successful. In addition, in the event that a party fails to perform under a key collaboration agreement, or if a key collaboration agreement is terminated, the reduction in anticipated revenues could delay or suspend our commercialization efforts in certain countries. Specifically, the termination of a key collaboration agreement by one of our collaborators could materially impact our ability to enter into additional collaboration agreements with new collaborators on favorable terms.
Risks relating to our current material collaborations (excluding our BARDA partnership, which is discussed below in these “Risk Factors”) include the following:
|
|
• |
Under our asset purchase agreement with Azaya, we are required to use commercial reasonable efforts to develop our ATI-0918 and ATI-1123 drug candidates, and we have future milestone, earn-out and other payments to Azaya tied to our commercialization and sale activities for these drug candidates. If we are unsuccessful in our efforts to develop our ATI-0918 and ATI-1223 drug assets, or if Azaya and we were to enter into a dispute over the terms of our agreement, then our business could be seriously harmed. |
|
|
• |
Lorem Vascular, is our exclusive licensee for our Cytori Cell Therapy products in all fields of use in China, Hong Kong, Singapore, Malaysia and Australia under the terms of the Lorem Agreement. Lorem Vascular is responsible for commercializing our Cytori Cell Therapy products in these territories. Lorem Vascular is relatively new company with no previous operating history, and has yet to generate meaningful revenues in its licensed territories. There can be no assurance that Lorem Vascular will be able to generate meaningful revenues in its licensed territories in the future. We are in ongoing discussions with Lorem Vascular regarding the terms of our collaboration, including the structure of the Lorem Agreement. If we are unable to agree with Lorem Vascular on revised terms to our collaboration, our relationship with them could suffer. A dispute may arise between us and Lorem Vascular that could lead to arbitration or other adversarial proceedings. Any such proceedings could cause significant diversion of management time and attention, cause us significant expense, and could potentially result in an outcome adverse to us. Further, any such dispute could negatively affect our ability to realize any sales or royalty revenues from Lorem Vascular’s commercial activities in the territories under its exclusive license. Even if we successfully restructure or otherwise revise our agreement with Lorem Vascular, there can be no assurance that Lorem Vascular will be able to successfully commercialize our Celution products in China or in the other territories subject to its license. Further, if Lorem Vascular fails to comply with any regulations applicable to its development, marketing and sale of our products, there can be no assurance that regulators would not try to hold us responsible for such activities. |
|
|
• |
Pursuant to the Bimini Agreement, we have, among other things, granted Bimini an exclusive, worldwide license to use and sell our Cytori Cell Therapy products in the alopecia (hair loss) field. Cytori and Bimini granted certain licenses to each other, and have certain license, royalty and other payment obligations under the Bimini agreement, as well as certain supply, development and non-competition obligations. If we and Bimini were to enter into a dispute regarding the terms of our agreement, our business could be harmed. |
27
If we or our distributors or collaborators fail to comply with regulatory requirements applicable to the development, manufacturing, and marketing of our products, regulatory agencies may take action against us or them, which could significantly harm our business.
Our products and product candidates, along with the clinical development process, the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for these products, are subject to continual requirements and review by the FDA and state and foreign regulatory bodies. Regulatory authorities subject a marketed product, its manufacturer and the manufacturing facilities to continual review and periodic inspections. We, our distributors and collaborators, and our and their respective contractors, suppliers and vendors, will be subject to ongoing regulatory requirements, including complying with regulations and laws regarding advertising, promotion and sales of products, required submissions of safety and other post-market information and reports, registration requirements, Clinical Good Manufacturing Practices (cGMP) regulations (including requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation), and the requirements regarding the distribution of samples to physicians and recordkeeping requirements. Regulatory agencies may change existing requirements or adopt new requirements or policies. We, our distributors and collaborators, and our and their respective contractors, suppliers and vendors, may be slow to adapt or may not be able to adapt to these changes or new requirements.
Failure to comply with regulatory requirements may result in any of the following:
|
|
• |
restrictions on our products or manufacturing processes; |
|
|
• |
warning letters; |
|
|
• |
withdrawal of the products from the market; |
|
|
• |
voluntary or mandatory recall; |
|
|
• |
fines; |
|
|
• |
suspension or withdrawal of regulatory approvals; |
|
|
• |
suspension or termination of any of our ongoing clinical trials; |
|
|
• |
refusal to permit the import or export of our products; |
|
|
• |
refusal to approve pending applications or supplements to approved applications that we submit; |
|
|
• |
product seizure; |
|
|
• |
injunctions; or |
|
|
• |
imposition of civil or criminal penalties. |
To the extent any of our customers fail to use our products in compliance with applicable regulations, regulators could try to hold us responsible if they believe we facilitated or were otherwise somehow responsible for our customer's non-compliance.
We currently sell our Celution Cell Therapy products in numerous markets outside of the United States for research and commercial use. These markets have different, and in some cases, less burdensome, regulatory schemes applicable to our products than in the United States. To the extent any of our customers, whether inside or outside the United States, use or further market our products in their home market or in other markets in a way that does not comply with applicable local regulations, regulators could try to hold us responsible if they believe we facilitated or were otherwise responsible for the customer’s actions. While we take measures in an effort to protect us against these types of risks, we cannot ensure you that such measures would prevent us from becoming subject to any such claims.
We and our products are subject to extensive regulation, and the requirements to obtain regulatory approvals in the United States and other jurisdictions can be costly, time-consuming and unpredictable. If we or our partners are unable to obtain timely regulatory approval for our product candidates, our business may be substantially harmed.
Cytori Cell Therapy: Our Celution system family of products and components of the Stemsource cell banks, must receive regulatory clearances or approvals from the FDA and from foreign regulatory bodies prior to commercial sale in those jurisdictions. Our Cytori Cell Therapy platform, including the Celution device, Celase and Intravase reagents, and consumable kits, is subject to
28
stringent government regulation in the United States by the FDA under the Federal Food, Drug and Cosmetic Act, and by the EMA and other regulatory agencies outside of the United States under their respective regulatory regimes.
The regulatory process for our cell therapy products can be lengthy, expensive, and uncertain. Before any new medical device may be introduced to the U.S. market, the manufacturer generally must obtain FDA clearance or approval through either the 510(k) pre-market notification process or the lengthier pre-market approval, or PMA, process. It generally takes from three to 12 months from submission to obtain 510(k) pre-market clearance, although it may take longer. Approval of a PMA could take four or more years from the time the process is initiated. The 510(k) and PMA processes can be expensive, uncertain, and lengthy, and there can be no assurance of ultimate clearance or approval. Our Celution® products under development today and in the foreseeable future will be subject to the lengthier PMA process. Securing FDA clearances and approvals may require the submission of extensive clinical data and supporting information to the FDA. Failure to comply with applicable requirements can result in application integrity proceedings, fines, recalls or seizures of products, injunctions, civil penalties, total or partial suspensions of production, withdrawals of existing product approvals or clearances, refusals to approve or clear new applications or notifications, and criminal prosecution.
For us to market our products in Europe, Canada, Japan and certain other non-U.S. jurisdictions, we need to obtain and maintain required regulatory approvals or clearances and must comply with extensive regulations regarding safety, manufacturing processes and quality. These regulations, including the requirements for approvals or clearances to market, may differ from the FDA regulatory scheme. International sales also may be limited or disrupted by political instability, price controls, trade restrictions and changes in tariffs. Additionally, fluctuations in currency exchange rates may adversely affect demand for our products by increasing the price of our products in the currency of the countries in which the products are sold.
Medical devices are also subject to post-market reporting requirements for deaths or serious injuries when the device may have caused or contributed to the death or serious injury, as well as for certain device malfunctions that would be likely to cause or contribute to a death or serious injury if the malfunction were to recur. If safety or effectiveness problems occur after the product reaches the market, the FDA may take steps to prevent or limit further marketing of the product. Additionally, the FDA actively enforces regulations prohibiting marketing and promotion of devices for indications or uses that have not been cleared or approved by the FDA. While we believe that our current activities are in compliance with FDA regulations relating to marketing and promotion, if regulators were to determine that our commercialization efforts, or those of our distributors, collaborators or customers, involve improper marketing and promotion of our products in violation of FDA regulations, our business could be substantially negatively affected.
There can be no assurance that we will be able to obtain the necessary 510(k) clearances or PMA approvals to market and manufacture our other products in the United States for their intended use on a timely basis, if at all. Delays in receipt of or failure to receive such clearances or approvals, the loss of previously received clearances or approvals or failure to comply with existing or future regulatory requirements could have a substantial negative effect on our results of operations and financial condition. In addition, there can be no assurance that we will obtain regulatory approvals or clearances in all of the other countries where we intend to market our products, or that we will not incur significant costs in obtaining or maintaining foreign regulatory approvals or clearances, or that we will be able to successfully commercialize current or future products in various foreign markets. Delays in receipt of approvals or clearances to market our products in foreign countries, failure to receive such approvals or clearances or the future loss of previously received approvals or clearances could have a substantial negative effect on our results of operations and financial condition.
Cytori Nanomedicines: The worldwide regulatory process for our Cytori Nanomedicines drug candidates can be lengthy and expensive, with no guarantee of approval.
Before any new drugs may be introduced to the U.S. market, the manufacturer generally must obtain FDA approval through either an abbreviated new drug application, or ANDA, process for generic drugs off patent that allow for bioequivalence to an existing reference listed drug, or RLD, or the lengthier new drug approval, or NDA, process, which typically requires multiple successful Phase III clinical trials to generate clinical data supportive of safety and efficacy along with extensive pharmacodynamic and pharmacokinetic preclinical testing to demonstrate safety. Our lead drug product under development (ATI-0918) is eligible ANDA process, while our ATI-1123 drug candidate is subject to the significantly lengthier NDA process. Approval of an ANDA could take four or more years from the time the process is initiated due to the requirement for clinical trials. NDA drugs could take significantly longer due to the additional preclinical requirements along with the typical requirement for two successful Phase III clinical studies.
In Europe, as in the United States, there are two regulatory steps to complete before a drug candidate is approved to be marketed in the European Union. These two steps are clinical trial application and marketing authorization application. Clinical trial applications are approved at the member state level, whereas marketing authorization applications are approved at both the member state and centralized levels. Both ATI-0918 and ATI-1123 will follow the centralized procedure for EMA regulatory approval. The centralized procedure allows the applicant to obtain a marketing authorization that is valid throughout the EU. Similar to the FDA process, the EMA centralized process requires bioequivalence data for generic drug candidates such as ATI-0918, and robust clinical data for non-generic drug candidates like ATI-01123 similar to clinical data required for NDA drug candidates.
There are numerous risks arising out of the regulation of our ATI-0918 and ATI-1123 drug candidates include the following:
29
|
|
• |
We can provide no assurances that our current and future oncology drugs will meet all of the stringent government regulation in the United States, by the FDA under the Federal Food, Drug and Cosmetic Act, and/or in international markets such as Europe, by the EMA under its Medicinal Products Directive, or Japan, by Japan’s Pharmaceuticals and Medical Devices Agency and Ministry of Health, Labor and Welfare under the Japanese Pharmaceutical Affairs Law. |
|
|
• |
We intend to seek regulatory of our ATI-0918 drug candidate via abbreviated approval processes referred to as bioequivalence or BE, approved under an abbreviated new drug application, or ANDA. There are no assurances that these abbreviated processes are or will be available in markets outside of the United States, or where available, that we will successfully obtain regulatory approvals via such abbreviated processes. |
|
|
• |
It is required for ANDA and BE drug candidates that there is a reference listed drug, or RLD, with which the drug candidate must demonstrate equivalence. There are no assurances that the reference drug for ATI-0918 will be the same in all territories or countries, which could require different and unique BE clinical studies for some territories where we currently intend to commercialize ATI-0918. Changes in the RLD may result in the nullification of BE clinical studies and can result in significant delays in the regulatory process as BE clinical studies may need to be repeated for jurisdictions that no longer recognize the reference drug utilized in BE clinical studies. |
|
|
• |
Our Cytori Nanomedicines drug candidates, if approved, will still be subject to post-market reporting requirements for deaths or serious injuries when the drug may have caused or contributed to the death or serious injury, or serious adverse events. There are no assurances that our drug products will not have safety or effectiveness problems occurring after the drugs reach the market. There are no assurances that regulatory authorities will not take steps to prevent or limit further marketing of the drug due to safety concerns. |
|
|
• |
It is possible that the new legislation in our priority markets, such as the newly enacted CURES Act in the United States, will yield additional regulatory requirements for therapeutic drugs for our Cytori Nanomedicine drug candidates (the FDA’s interpretation and implementation of the CURES Act has yet to be published). |
Changing, new and/or emerging government regulations may adversely affect us.
Cytori Cell Therapy: Government regulations can change without notice. Given the fact that we operate in various international markets, our access to such markets could change with little to no warning due to a change in government regulations that suddenly up-regulate our product(s) and create greater regulatory burden for our cell therapy and cell banking technology products.
Our ability to receive regulatory approvals for our Cytori Cell Therapy products and to sell into foreign markets is complex, due in part to by the nature of our Celution platform and manufacturing process. The platform consists of our Celution device that processes the patient’s own adipose (fat) tissue to create a heterogeneous mixture of regenerative cells. In the United States, this heterogeneous mixture of cells is subject to classification as a drug, but the FDA has made the determination that our Cytori Cell Therapy will be regulated as a Class III PMA medical device. However, foreign regulatory bodies must assess our particular platform and manufacturing process to make their own determination whether our Cytori Cell Therapy product candidates should receive medical device or drug classifications. For example, the European Commission has granted orphan drug designation for the use of Cytori Cell Therapy (currently branded as Habeo Cell Therapy) in treatment of system sclerosis. The EMA has not made a determination whether it would classify Habeo Cell Therapy as an ATMP or a medical device. Though we believe that Habeo Cell Therapy will be classified by the EMA as an ATMP, we cannot guarantee that the EMA will not arrive at a different determination at such time that we ask a determination to be made. Regardless of the EMA’s ultimate determination, we will also be required to comply with the particular regulatory requirements of each of the member states of the European Economic Area (comprised of 28 European Union, or EU, member states plus Iceland, Liechtenstein, and Norway) with respect to our cell therapy offerings, a process which we anticipate will require considerable time, effort and expense. We expect that regulatory bodies in other jurisdictions will engage in similar analyses of our Cytori Cell Therapy, and we cannot predict then outcomes of these analyses.
In Japan, the Japanese Diet recently passed the Act regarding Ensuring of Safety of Regenerative Medicine, or the Regenerative Medicine Law, and the revisions to the Pharmaceutical Affairs Law as applied to drugs, medical devices and regenerative medicine. The Regenerative Medicine Law initially caused some confusion for regenerative companies operating in Japan, but we believe that this law, as currently implemented, benefits Cytori and its customers by allowing an expedited path for our customers in Japan to obtain licenses under the Regenerative Medicine Law to treat patients with Cytori Cell Therapy. However, we cannot be certain that the Regenerative Medicine Law will not be repealed or that current interpretations and implementation of the Regenerative Medicine Law will not change in a manner adverse to our business. Further, we currently import and sell our products in Japan under Class I notifications that we obtained several years ago. However, at the request of Japanese regulators, we are in the process of obtaining Class III approvals for our Celution device and consumable kits. Though we are pursuing these Class III
30
approvals process without any anticipated interruption to our commercial activities, it is possible that other jurisdictions in which we currently sell may require similar heightened regulatory approvals but with potential restrictions on our ability to market and sell our Cytori Cell Therapy products in such territories during the application process and review period for the required regulatory approval(s).
Any regulatory review committees and advisory groups and any contemplated new guidelines may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval limitations or restrictions. As we advance our product candidates, we will be required to consult with these regulatory and advisory groups, and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of our product candidates. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a product candidate to market could decrease our ability to generate sufficient revenue to maintain our business. Divergence in regulatory criteria for different regulatory agencies around the globe could result in the repeat of clinical studies and/or preclinical studies to satisfy local territory requirements, resulting in the repeating of studies and/or delays in the regulatory process. Some territories may require clinical data in their indigenous population, resulting in the repeat of clinical studies in whole or in part. Some territories may object to the formulation ingredients in the final finished product and may require reformulation to modify or remove objectionable components; resulting in delays in regulatory approvals. Such objectionable reformulations include, but are not limited to, human or animal components, BSE/TSE risks, banned packaging components, prohibited chemicals, banned substances, etc. There can be no assurances that FDA or foreign regulatory authorities will accept our pre-clinical and/or clinical data.
Due to the fact that there are new and emerging cell therapy and cell banking regulations that have recently been drafted and/or implemented in various countries around the world, the application and subsequent implementation of these new and emerging regulations have little to no precedence. Therefore, the level of complexity and stringency is not known and may vary from country to country, creating greater uncertainty for the international regulatory process.
Anticipated or unanticipated changes in the way or manner in which the FDA or other regulators regulate products or classes/groups of products can delay, further burden, or alleviate regulatory pathways that were once available to other products. There are no guarantees that such changes in the FDA’s or other regulators’ approach to the regulatory process will not deleteriously affect some or all of our products or product applications.
Cytori Nanomedicines: Our nanotechnology technology is also subject to government regulations that are subject to change. Our lead product, ATI-0918 is regulated under bioequivalence rules that rely on a reference listed drug, or RLD, for equivalence in the United States and other jurisdictions. Government agencies can change the reference listed drug or reference drug without notice. These changes in the RLD could invalidate clinical studies and require the initiation of new clinical studies for determining equivalence to a newly assigned RLD. Furthermore, bioequivalence studies may need to be repeated in certain foreign entities as some governments may require additional confirmatory studies in their patient populations. These additional requirements could result in additional clinical studies or delays in the regulatory process. Other risks with the RLD criteria are in the criteria for demonstrating bioequivalence. Bioequivalence criteria may not be identical in all geographical regions, resulting in the requirement for new bioequivalence studies to demonstrate equivalence to a more stringent standard. Additionally, bioequivalence criteria rely on the products being “off patent” in the territory. Patent expiration dates may vary in different regions which may result in bioequivalence regulatory pathways being delayed in some territories. Current regulatory pathways such as the abbreviated new drug application, or ANDA, pathway, of we are currently relying on, are subject to change and may cease to be viable regulatory pathways in the future.
Our pipeline oncology products, such as ATI-1123, are being developed under existing government criteria, which are subject to change in the future. Clinical and/or pre-clinical criteria in addition to cGMP manufacturing requirements may change and impose additional regulatory burdens. Clinical requirements are subject to change which may result in delays in completing the regulatory process. Divergence in regulatory criteria for different regulatory agencies around the globe could result in the repeat of clinical studies and/or preclinical studies to satisfy local jurisdictional requirements, which would significantly lengthen the regulatory process and increase uncertainty of outcome. Some jurisdictions may require clinical data in their indigenous population, resulting in the repeat of clinical studies in whole or in part. Some jurisdictions may object to the formulation ingredients in the final finished product and may require reformulation to modify or remove objectionable components; resulting in delays in regulatory approvals. Such objectionable reformulations include, but are not limited to, human or animal components, bovine spongiform encephalopathy/ transmissible spongiform encephalopathy risks, banned packaging components, prohibited chemicals, banned substances, etc. There can be no assurance that the FDA or foreign regulatory authorities will accept our pre-clinical and/or clinical data.
We may have difficulty obtaining appropriate and sufficient pricing and reimbursement for our cell therapy products.
New and emerging cell therapy and cell banking technologies, such as those provided by the Cytori Cell Therapy family of products, may have difficulty or encounter significant delays in obtaining health care reimbursement in some or all countries around the world due to the novelty of our cell therapy and cell banking technology and subsequent lack of existing reimbursement
31
schemes/pathways. Therefore, the creation of new reimbursement pathways may be complex and lengthy with no assurances that such reimbursements will be successful. The lack of health insurance reimbursement or reduced or minimal reimbursement pricing may have a significant impact on our ability to successfully sell our cell therapy and cell banking technology product(s) into a county or region at pricing that is profitable and that adequately compensates Cytori for its development costs, which would negatively impact our operating results.
Habeo Cell Therapy, our lead indication, is intended to treat hand manifestations of systemic scleroderma, which is a rare, or orphan, disease. As such, we anticipate that Habeo Cell Therapy will be priced to reflect its orphan status in our prior target markets for this indication. In the United States and in Europe, we anticipate that this pricing will be supported by Habeo Cell Therapy meeting primary endpoints from the STAR and SCLERADEC II clinical trials. Further, in Europe, we expect that Habeo Cell Therapy will be classified as an ATMP with orphan drug status, and if we are the first ATMP approved for this indication in Europe, we will receive certain benefits, including market exclusivity (subject to certain caveats). Status as an approved ATMP with orphan drug designation could provide us with a strong platform from which to seek higher reimbursement. However, the level of reimbursement Habeo Cell Therapy will receive will be directly related to the quantity and quality of clinical evidence reported by these STAR and SCLERADEC II clinical trials. It is possible that our clinical trial data are sufficient to support regulatory approval of Habeo Cell Therapy, but not sufficient to support pricing at a level that makes Habeo Cell Therapy attractive to potential partners or to make it economically viable for us to directly commercialize Habeo Cell Therapy. Further, if the STAR and SCLERADEC II clinical trials are not successful, we may not be in a position to seek regulatory approval at all, and we may be required to suspend or abandon our Habeo Cell Therapy commercialization efforts.
Our European managed access program for Habeo Cell Therapy may not be successful, which in turn could adversely affect our Habeo Cell Therapy commercialization efforts.
Our managed access program, or MAP (also known as early access program or named patient program), is intended to provide access in select countries across Europe, the Middle East and Africa, or EMEA, to our Habeo Cell Therapy for patients with impaired hand function due to scleroderma in advance of anticipated commercialization of Habeo Cell Therapy. Our MAP will has faced and will continue to face numerous challenges, including the following:
|
|
• |
In most countries, patient access to Habeo Cell therapy will be provided on an ‘individual’ patient basis where physicians will make an application to their Competent Authority in each country on a patient-by-patient basis. This imposes a significant administrative burden on participating physicians, and requires them to navigate a process with which they are oftentimes unfamiliar. |
|
|
• |
In certain countries, hospitals and/or patients will be required to pay a portion of our procedure fees under our MAP. This payment obligation may limit the number of hospitals and patients who can afford to participate in our MAP. |
|
|
• |
Because Cytori is targeting an orphan indication in scleroderma where there is an established need for effective therapies, regulators in Europe have been willing to allow an approval trial based on limited data from the 12-patient, investigator initiated SCLERADEC I pilot trial. The lack of robust Phase II clinical data has also proven to be a hurdle to MAP acceptance. We believe that positive results from the STAR clinical trial and/or SCLERADEC II clinical trial will help drive interest in our MAP, but there is no guarantee that either trial will achieve positive results. |
Orphan drug designation may not ensure that we will enjoy market exclusivity in a particular market, and if we fail to obtain or maintain orphan drug designation or other regulatory exclusivity for some of our product candidates, our competitive position would be harmed.
A product candidate that receives orphan drug designation can benefit from potential commercial benefits following approval. Under the U.S. Orphan Drug Act, the FDA may designate a product candidate as an orphan drug if it is intended to treat a rare disease or condition, defined as affecting a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the European Union, or EU, the EMA’s Committee for Orphan Medicinal Products, or COMP, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than 10,000 persons in the EU. Currently, this designation provides market exclusivity in the U.S. and the European Union for seven years and ten years, respectively, if a product is the first such product approved for such orphan indication. This market exclusivity does not, however, pertain to indications other than those for which the drug was specifically designated in the approval, nor does it prevent other types of drugs from receiving orphan designations or approvals in these same indications. Further, even after an orphan drug is approved, the FDA can subsequently approve a drug with similar chemical structure for the same condition if the FDA concludes that the new drug is clinically superior to the orphan product or a market shortage occurs. In the EU, orphan exclusivity may be reduced to six years if the drug no longer satisfies the original designation criteria or can be lost altogether if the marketing authorization holder consents to a second orphan
32
drug application or cannot supply enough drug, or when a second applicant demonstrates its drug is “clinically superior” to the original orphan drug.
In April 2016, the European Commission, acting on the positive recommendation from the COMP, issued orphan drug designation to a broad range of Cytori Cell Therapy formulations when used for the treatment of systemic sclerosis. In November 2016, the U.S. FDA Office of Orphan Products Development granted us an orphan drug designation for cryopreserved or centrally processed ECCS-50 (Habeo) for scleroderma. Either or both of such orphan drug designations may fail to result in or maintain orphan drug exclusivity upon approval, which would harm our competitive position.
We generate 71% of our sales revenues from Japan, with 76% of those revenues generated by sales to four customers and 49% of these revenues generated by sales to one customer. This concentration of sales in one territory, and to one small group of customers in Japan, makes us vulnerable to the loss of our key customers and to adverse changes in the Japanese market.
In 2016, we generated approximately $3.3 million in sales revenues in Japan, representing 71% of our overall global sales revenues. 76% of the Japan sales revenues were from four key customers, and 49% of these sales revenues were from one key customer. We expect a relatively small number of customers to account for a majority of our revenues for the foreseeable future. This concentration of sales in one country, and in a small subset of customers within such country, represents a risk to our business. Our existing business in Japan, and our prospects for further growth of product sales in Japan, are subject to a number of risks, including the following:
|
|
• |
Existing laws and regulations pertaining to our business, including the Act regarding Ensuring of Safety of Regenerative Medicine, or the Regenerative Medicine Law, passed in 2013, may be repealed, or implemented, amended or superseded, in a manner that is adverse to our business; |
|
|
• |
Macroeconomic conditions in Japan may deteriorate, thus weakening demand for our cell therapy products, which are used in self-pay procedures in Japan; |
|
|
• |
Japanese regulatory authorities may take unexpected actions with respect to our cell therapy products, including with respect to required regulatory clearances and approvals in Japan, that could cause us to suspend or curtail our cell therapy sales operations in Japan; |
|
|
• |
Quality issues could arise, requiring product recalls or other actions that could cause us reputational damage and lost sales; |
|
|
• |
One or more of our key customers in Japan may decide to acquire competitive products, adopt other technological or therapeutics approaches to the conditions they treat, or otherwise reduce or cease their purchases of our products; |
|
|
• |
Our Cytori Cell Therapy product trials may not achieve statistical significance and thus could diminish the perceived value and efficacy of our technology; and |
|
|
• |
Our relatively small team in Japan may not be able to manage the needs of a growing business, and we may not able to hire and retain existing or new employees necessary to maintain and expand our business in Japan. |
Further, a loss of one or more of our key customers, a dispute or disagreement with one of these key customers, a significant deterioration in the financial condition of one of these key customers, or a significant reduction in the amount of our products ordered by any key customer could adversely affect our revenue, results of operations and cash flows.
If we experience an interruption in supply from a material sole source supplier, our business may be harmed
We acquire some our components and other raw materials from sole source suppliers. If there is an interruption in supply of our raw materials from a sole source supplier, there can be no assurance that we will be able to obtain adequate quantities of the raw materials within a reasonable time or at commercially reasonable prices. Interruptions in supplies due to pricing, timing, availability or other issues with our sole source suppliers could have a negative impact on our ability to manufacture products and product candidates, which in turn could adversely affect commercial sales of our commercially available Cytori Cell Therapy products, delay our development and commercialization efforts and cause us to potentially breach our supply or other obligations under our agreements with certain other counterparties.
33
We source our Celase and Intravase reagents, which are used to process patients’ autologous adipose (fat) tissue, under an exclusive manufacturing arrangement with Roche Diagnostics Corporation, or Roche. We do not have a second qualified supplier to manufacture these reagents, and we estimate that it would take approximately two years to qualify another manufacturing source for our reagents. Though we have significant inventory related to these reagents on hand which we believe are sufficient to satisfy currently anticipated internal and customer demand for at least the next three years, if our agreement with Roche were to terminate or if Roche were otherwise unable to manufacture sufficient volumes of the reagents to meet our customer demand, our business could be materially and adversely affected.
We are dependent on sole source suppliers to manufacture the API (active pharmaceutical ingredient) and certain other components of our Cytori Nanomedicines drug candidates. There are no assurances that these sole source suppliers will enter into supply agreements with us to provide contractual assurance to us around supply and pricing. Regardless whether a sole source supplier enters into a written supply arrangement with us, such supplier could still delay, suspend or terminate supply of raw materials to us for a number of reasons, including manufacturing or quality issues, payment disputes with us, bankruptcy or insolvency, or other occurrences.
If a sole source supplier ceases supply of raw materials necessary there is no guarantee that we will find an alternative supplier for the necessary raw materials on terms acceptable to us, or at all. Further the qualification process for a new vendor could take months or even years, and any such day in qualification could significantly harm our business.
We may engage in strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
From time to time we may consider strategic transactions, such as acquisitions of companies, asset purchases and out-licensing or in-licensing of products, product candidates or technologies. For example, in February 2017, we acquired intellectual property and a portfolio of investigational oncology therapies from Azaya Therapeutics. This acquisition materially impacted our liquidity and will materially increase our expenses (including a substantial increase in employee headcount). Further, growth of the Cytori Nanomedicine business will require significant management time and attention. Additional potential transactions that we may consider include a variety of different business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Any such transaction may require us to incur non-recurring or other charges, may increase our near and long-term expenditures and may pose significant integration challenges or disrupt our management or business, which could adversely affect our operations and financial results. For example, these transactions may entail numerous operational and financial risks, including:
|
|
• |
exposure to unknown liabilities; |
|
|
• |
disruption of our business and diversion of our management’s time and attention in order to develop acquired products, product candidates or technologies; |
|
|
• |
incurrence of substantial debt or dilutive issuances of equity securities to pay for acquisitions; |
|
|
• |
higher than expected acquisition and integration costs; |
|
|
• |
write-downs of assets or goodwill or impairment charges; |
|
|
• |
increased amortization expenses; |
|
|
• |
difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel; |
|
|
• |
impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership; and |
|
|
• |
inability to retain key employees of any acquired businesses. |
Accordingly, although there can be no assurance that we will undertake or successfully complete any additional transactions of the nature described above, any additional transactions that we do complete could have a material adverse effect on our business, results of operations, financial condition and prospects.
34
We are exposed to risks related to our international operations, and failure to manage these risks may adversely affect our operating results and financial condition.
We have operations in several regions around the world, including the United States, Japan, the Asia-Pacific region and Europe. Our global operations may be subject to risks that limit our ability to operate our business. We sell our products globally, which exposes us to a number of risks that can arise from international trade transactions, local business practices and cultural considerations, including, among others:
|
|
• |
political unrest, terrorism and economic or financial instability; |
|
|
• |
unexpected changes and uncertainty in regulatory requirements; |
|
|
• |
nationalization programs that may be implemented by foreign governments; |
|
|
• |
import-export regulations; |
|
|
• |
difficulties in enforcing agreements and collecting receivables; |
|
|
• |
difficulties in ensuring compliance with the laws and regulations of multiple jurisdictions; |
|
|
• |
changes in labor practices, including wage inflation, labor unrest and unionization policies; |
|
|
• |
longer payment cycles by international customers; |
|
|
• |
currency exchange fluctuations; |
|
|
• |
disruptions of service from utilities or telecommunications providers, including electricity shortages; |
|
|
• |
difficulties in staffing foreign branches and subsidiaries and in managing an expatriate workforce, and differing employment practices and labor issues; and |
|
|
• |
potentially adverse tax consequences. |
We also face risks associated with currency exchange and convertibility, inflation and repatriation of earnings as a result of our foreign operations. We are also vulnerable to appreciation or depreciation of foreign currencies against the U.S. dollar. Although we have significant operations in Asia, a substantial portion of transactions are denominated in U.S. dollars. As appreciation against the U.S. dollar increases, it will result in an increase in the cost of our business expenses abroad. Conversely, downward fluctuations in the value of foreign currencies relative to the U.S. dollar may make our products less price competitive than local solutions. From time to time, we may engage in currency hedging activities, but such activities may not be able to limit the risks of currency fluctuations.
We must maintain quality assurance certification and manufacturing approvals.
The manufacture of our products is, and the manufacture of any future drug and/or cell-related therapeutic products would be, subject to periodic inspection by regulatory authorities and distribution partners. The manufacture of drugs and devices products for human use is subject to regulation and inspection from time to time by the FDA for compliance with the FDA’s cGMP (current good manufacturing practices), Quality System Regulation, or QSR requirements, as well as equivalent requirements and inspections by state and non-U.S. regulatory authorities. There can be no assurance that the FDA or other authorities will not, during the course of an inspection of existing or new facilities, identify what they consider to be deficiencies in our compliance with QSRs or other requirements and request, or seek remedial action.
Failure to comply with such regulations or a potential delay in attaining compliance may adversely affect our manufacturing activities and could result in, among other things, injunctions, civil penalties, FDA refusal to grant pre-market approvals or clearances of future or pending product submissions, fines, recalls or seizures of products, total or partial suspensions of production and criminal prosecution. There can be no assurance that after such occurrences that we will be able to obtain additional necessary regulatory approvals or clearances on a timely basis, if at all. Delays in receipt of or failure to receive such approvals or clearances, or the loss of previously received approvals or clearances could have a substantial negative effect on our results of operations and financial condition.
35
BARDA may terminate or suspend its agreement with us, or suspend, delay or reduce its funding of our development hereunder, which could delay and/or adversely affect our business and our ability to further develop our Celution System.
In September 2012, we were awarded a contract, or the BARDA Agreement, with the Biomedical Advanced Research and Development Authority, or BARDA, a division of the U.S. Department of Health and Human Services. The objective of the BARDA Agreement is to develop our cell therapy technology for use as a new countermeasure for a combined injury involving thermal burn and radiation exposure that would be employed following a mass-casualty event. The original total value of the cost-plus-fixed-fee BARDA Agreement was up to an aggregate of $106 million, which aggregate potential value has decreased somewhat as we and BARDA have gained more insight into anticipated and actual budgets for different phases of our development work.
We have received over $20 million in cost-plus-fixed-fee funding from BARDA to fund our preclinical research and development of Cytori Cell Therapy for thermal burn, or DCCT-10, and to fund development of our Celution cell processing system. There are additional contract options under the BARDA Agreement to provide over $80 million in additional funds to:
|
|
• |
conduct a pilot clinical study, and related regulatory and other tasks; |
|
|
• |
conduct a pivotal clinical trial, and related clinical, regulatory, and other activities, with the objective of obtaining FDA approval for intravenous use of DCCT-10 in thermal burn injury; and |
|
|
• |
conduct of clinical, regulatory and other tasks required to develop and obtain FDA clearance for other characteristics suitable for use in thermal burn injury following a mass casualty event. |
The current contract modification to the BARDA Agreement executed by us and BARDA in September 2016 will expire in April 2017. We are in active discussions with BARDA regarding BARDA’s continued funding of our DCCT-10 development program, but there is no guarantee that we will reach agreement with BARDA regarding an extension of our existing contract modification, execution of a new contract modification, or execution of a new agreement. If we are unable to enter into a new contract or contract modification with BARDA, we may cease to receive funds from BARDA as soon April 2017. If this occurs, we would likely severely curtail, suspend or even terminate our DCCT-10 program, and our business would be harmed.
Further should we cease to receive BARDA funding, certain of our product development efforts, including development of our next generation Celution devices, could be harmed.
Further, we are currently in the process of seeking FDA approval of our IDE application for our proposed RELIEF Phase I clinical trial to assess the safety and feasibility of intravenous administration of DCCT-10 as a thermal burn countermeasure. If the FDA approves our IDE application, then BARDA’s approval and agreement to fund the trial will be required to proceed. There can be no assurance that BARDA will agree to fund the entire cost of the trial. If BARDA declines to fund the full costs of the trial, we may be required to terminate our DCCT-10 development program.
BARDA may suspend or terminate the BARDA Agreement, or decline to enter into a new agreement upon termination of the BARDA Agreement, for a number of reasons, including our failure to achieve key objectives or milestones or failure to comply with the operating procedures and processes approved by BARDA and its audit agency, the Defense Contract Audit Agency. There can be no assurance that we will be able to comply with BARDA’s operating procedures and processes, achieve the necessary clinical milestones or whether we will be able to successfully develop our DCCT-10 product candidate under the contract.
Our contract with BARDA will expire in September 2017. Though we intend to negotiate a new agreement with BARDA, there is no guarantee that we will be able to do so. Any new agreement with BARDA may be on terms less favorable to us than our current agreement.
Our current BARDA Agreement will expire in September 2017, and there is no guarantee that we will execute a new contract with BARDA. We anticipate that if the FDA approves our RELIEF pilot trial IDE application, and if BARDA agrees to fund this trial, that any BARDA funding for this trial would be awarded under our existing contract. However, we do not anticipate that any additional funds will be awarded to us under the current BARDA Agreement. Thus, it is likely that our current BARDA Agreement will expire with only approximately $30 million of the total original contract value of $106 million having been awarded to us. Any subsequent awards for a pivotal clinical trial of our DCCT-10 therapeutic, for regulatory activities in anticipation of FDA approval, and for other related development and commercialization activities, would be granted (if at all) under a new contract with BARDA. There can be no guarantee that BARDA and we will enter into a new agreement on terms acceptable to us, or at all. If we do enter into a new contract with BARDA, it might provide for lower funding caps and other material terms less favorable to us than the current BARDA Agreement. Further, we would expect that any contract with BARDA would be unlikely to fund the continued development of our latest generation Celution systems. If we do not enter into a new contract with BARDA when our current BARA Agreement
36
expires that provides for continued funding of our DCCT-10 development efforts, we will likely be required to suspend or terminate our thermal burn program.
The BARDA contract has certain contracting requirements that allow the U.S. Government to unilaterally control its contracts. If the U.S. Government suspends, cancels, or otherwise terminates our contract with them, we could experience significant revenue shortfalls, and our financial condition and business may be adversely affected.
Contracts with U.S. Government agencies typically contain termination provisions unfavorable to the other party, and are subject to audit and modification by the U.S. Government at its sole discretion, which will subject us to additional risks. These risks include the ability of the U.S. Government to unilaterally:
|
|
• |
audit or object to our contract-related costs and fees, and require us to reimburse all such costs and fees; |
|
|
• |
suspend or prevent us for a set period of time from receiving new contracts or extending our existing contracts based on violations or suspected violations of laws or regulations; |
|
|
• |
cancel, terminate or suspend our contracts based on violations or suspected violations of laws or regulations; |
|
|
• |
terminate our contracts if in the Government’s best interest, including if funds become unavailable to the applicable governmental agency; |
|
|
• |
reduce the scope and value of our contracts; and |
|
|
• |
change certain terms and conditions in our contracts. |
BARDA is able to terminate its contracts with us, either for its best interests or if we default by failing to perform in accordance with or to achieve the milestones set forth in the contract schedules and terms. Termination-for-convenience provisions generally enable us to recover only our costs incurred or committed and settlement expenses on the work completed prior to termination. Changes to, or an unexpected termination of, this contract could result in significant revenue shortfalls. If revenue shortfalls occur and are not offset by corresponding reductions in expenses, our business could be adversely affected. We cannot anticipate if, when or to what extent BARDA might revise, alter or terminate its contract with us in the future.
Under our contract with BARDA, our operations, and those of our contractors, are subject to audit by the U.S. Government, a negative outcome to which could adversely affect our financial conditions and business operations.
U.S. Government agencies, such as the Department of Health and Human Services, or DHHS, and the Defense Contract Audit Agency, or the DCAA, routinely audit and investigate government contractors and recipients of federal grants. These agencies evaluate a contractor’s performance under its contracts, cost structure and compliance with applicable laws, regulations and standards.
The DHHS and the DCAA also review the adequacy of, and a contractor’s compliance with, its internal control systems and policies, including the contractor’s purchasing, property, estimating, compensation and management information systems. Any costs found to be improperly allocated to a contract will not be reimbursed, while such costs already reimbursed must generally be repaid. If an audit identifies improper or illegal activities, we may be subject to civil and criminal penalties and administrative sanctions, including, but not limited to:
|
|
• |
termination of contracts; |
|
|
• |
forfeiture of profits; |
|
|
• |
suspension of payments; |
|
|
• |
fines; and |
|
|
• |
suspension or prohibition from conducting business with the U.S. Government. |
If we are unable to identify, hire and/or retain key personnel, we may not be able to sustain or grow our business.
Our ability to operate successfully and manage our potential future growth depends significantly upon our ability to attract, retain, and motivate highly skilled and qualified research, technical, clinical, regulatory, sales, marketing, managerial and financial
37
personnel. We compete for talent with numerous companies, as well as universities and non-profit research organizations. In the near term, we intend to hire a significant number of scientists, quality and regulatory personnel, and other technical staff with the requisite expertise to support and expand our Cytori Nanomedicines business. The manufacturing of these oncology drug assets is a highly complex process that requires significant experience and know-how. If we are unable to attract personnel with the necessary skills and experience to reestablish and expand our Cytori Nanomedicines business, which is currently conducted out of our San Antonio, Texas facility, our business could be harmed.
Our future success also depends on the personal efforts and abilities of the principal members of our senior management and scientific staff to provide strategic direction, manage our operations, and maintain a cohesive and stable environment. In particular, we are highly dependent on our executive officers, especially Marc Hedrick, M.D., our Chief Executive Officer, Tiago Girão, our Chief Financial Officer, and John Harris, our Vice President and General Manager of Cell Therapy. Given their leadership, extensive technical, scientific and financial expertise and management and operational experience, these individuals would be difficult to replace. Consequently, the loss of services of one or more of these named individuals could result in product development delays or the failure of our collaborations with current and future collaborators, which, in turn, may hurt our ability to develop and commercialize products and generate revenues. We have not entered into any employment agreements with our executive officers or key personnel, nor do we maintain key man life insurance on the lives of any of the members of our senior management. Although we have a stock option plan pursuant to which we provide our executive officers with various economic incentives to remain employed with us, these incentives may not be sufficient to retain them. The loss of key personnel for any reason or our inability to hire, retain, and motivate additional qualified personnel in the future could prevent us from sustaining or growing our business.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
The commercial use of our products and clinical use of our products and product candidates expose us to the risk of product liability claims. This risk exists even if a product or product candidate is approved for commercial sale by applicable regulatory authorities and manufactured in facilities regulated by such authorities. Our products and product candidates are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our products or our product candidates could result in injury to a patient or even death. For example, ATI-0918 is cytotoxic, or toxic to living cells, and, if incorrectly or defectively manufactured or labeled, or incorrectly dosed or otherwise used in a manner not contemplated by its label, could result in patient harm and even death. In addition, a liability claim may be brought against us even if our products or product candidates merely appear to have caused an injury.
Product liability claims may be brought against us by consumers, health care providers, pharmaceutical companies or others selling or otherwise coming into contact with our products or product candidates, if approved, among others. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in:
|
|
• |
the inability to commercialize our product candidates; |
|
|
• |
decreased demand for our product candidates, if approved; |
|
|
• |
impairment of our business reputation; |
|
|
• |
product recall or withdrawal from the market; |
|
|
• |
withdrawal of clinical trial participants; |
|
|
• |
costs of related litigation; |
|
|
• |
distraction of management’s attention from our primary business; |
|
|
• |
substantial monetary awards to patients or other claimants; or |
|
|
• |
loss of revenues. |
We have obtained product liability insurance coverage for commercial product sales and clinical trials with a $10 million per occurrence and annual aggregate coverage limit. Our insurance coverage may not be sufficient to cover all of our product liability related expenses or losses and may not cover us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost, in sufficient amounts or upon adequate terms to protect us against losses due to product liability. If we determine that it is prudent to increase our
38
product liability coverage, we may be unable to obtain this increased product liability insurance on commercially reasonable terms or at all. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and have a material adverse effect on our business, results of operations, financial condition and prospects.
Our employees, principal investigators, consultants and collaboration partners may engage in misconduct or other improper activities, including noncompliance with laws and regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state healthcare fraud and abuse laws and regulations, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of activity relating to pricing, discounting, marketing and promotion, sales commissions, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation, or, given we are a listed company the United States, breach of insider trading laws. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our operations are subject to anti-corruption laws, including the U.S. Foreign Corrupt Practices Act, the United Kingdom Bribery Act, and other anticorruption laws that apply in countries where we do business.
Anti-corruption laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under these anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
There is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws or other laws including trade related laws. If we are not in compliance with these laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of these laws by respective government bodies could also have an adverse impact on our reputation, our business, results of operations and financial condition.
A failure to adequately protect private health information could result in severe harm to our reputation and subject us to significant liabilities, each of which could have a material adverse effect on our business.
Throughout the clinical trial process, we may obtain the private health information of our trial subjects. There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. As part of the American Recovery and Reinvestment Act 2009, or ARRA, Congress amended the privacy and security provisions of the Healthcare Information Portability and Accountability Act, or HIPAA. HIPAA imposes limitations on the use and disclosure of an individual’s healthcare information by healthcare providers conducting certain electronic transactions, healthcare clearinghouses, and health insurance plans, collectively referred to as covered entities. The HIPAA amendments also impose compliance obligations and corresponding penalties for non-compliance on certain individuals and entities that provide services to or perform certain functions on behalf of healthcare providers and other covered entities involving the use or disclosure of individually identifiable health information, collectively referred to as business associates. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and extended enforcement authority to state attorneys general. The amendments also create notification requirements to federal regulators, and in some cases local and national media, for individuals whose health information has been inappropriately accessed or disclosed. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with certain encryption or other standards developed by the U.S. Department of Health and Human Services, or HHS. Most states have laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual
39
terms to ensure ongoing protection of personal information. Activities outside of the U.S. implicate local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for non-compliance. The EU’s Data Protection Directive, Canada’s Personal Information Protection and Electronic Documents Act and other data protection, privacy and similar national, state/provincial and local laws may also restrict the access, use and disclosure of patient health information abroad. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or to alleviate problems caused by such breaches.
We and our collaborators must comply with environmental laws and regulations, including those pertaining to use of hazardous and biological materials in our business, and failure to comply with these laws and regulations could expose us to significant liabilities.
We and our collaborators are subject to various federal, state and local environmental laws, rules and regulations, including those relating to discharge of materials into the air, water and ground, those relating to manufacturing, storage, use, transportation and disposal of hazardous and biological materials, and those relating to the health and safety of employees with respect to laboratory activities required for the development of our products and activities. In particular, our Cytori Nanomedicine products and processes involve the controlled storage, use and disposal of certain cytotoxic, or toxic to living cells, materials. Even if we and these suppliers and collaborators comply with the standards prescribed by law and regulation, the risk of accidental contamination or injury from hazardous materials, or other violations of applicable environmental laws, rules or regulations cannot be completely eliminated. In the event of any violation of such laws, rules or regulations, we could be held liable for any damages that result, and any liability could exceed the limits or fall outside the coverage of any insurance we may obtain and could exceed our financial resources. We may not be able to maintain insurance on acceptable terms, or at all. We may incur significant costs in complying with environmental laws, rules and regulations.
Increased information technology security threats and more sophisticated and targeted computer crime could pose a risk to our systems, networks, and products.
Increased global information technology security threats and more sophisticated and targeted computer crime pose a risk to the security of our systems and networks and the confidentiality, availability and integrity of our data and communications. While we attempt to mitigate these risks by employing a number of measures, including employee refreshers, monitoring of our networks and systems, and maintenance of backup and protective systems, our systems, networks and products remain potentially vulnerable to advanced persistent threats. Depending on their nature and scope, such threats could potentially lead to the compromising of confidential information and communications, improper use of our systems and networks, manipulation and destruction of data, defective products, production downtimes and operational disruptions, which in turn could adversely affect our reputation, competitiveness and results of operations.
The results of the United Kingdom’s referendum on withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business.
In June 2016, a majority of voters in the United Kingdom elected to withdraw from the European Union in a national referendum. The referendum was advisory, and the terms of any withdrawal are subject to a negotiation period that could last at least two years after the government of the United Kingdom formally initiates a withdrawal process. Nevertheless, the referendum has created significant uncertainty about the future relationship between the United Kingdom and the European Union, including with respect to the laws and regulations that will apply as the United Kingdom determines which European Union laws to replace or replicate in the event of a withdrawal. The referendum has also given rise to calls for the governments of other European Union member states to consider withdrawal. These developments, or the perception that any of them could occur, have had and may continue to have a material adverse effect on global economic conditions and the stability of global financial markets, and may significantly reduce global market liquidity and restrict the ability of key market participants to operate in certain financial markets. Any of these factors could depress economic activity and restrict our access to capital, which could have a material adverse effect on our business, financial condition and results of operations and reduce the price of our securities.
Risks Related to our Financial Position and Capital Requirements
The statements in this section, as well as statements described elsewhere in this annual report, or in other SEC filings, describe risks that could materially and adversely affect our business, financial condition and results of operations, which could also cause the trading price of our equity securities to decline. These risks are not the only risks that we face. Our business, financial condition and results of operations could also be affected by additional factors that are not presently known to us or that we currently consider to be immaterial to our operations
40
We have incurred losses since inception, we expect to incur significant net losses in the foreseeable future and we may never become profitable.
We have almost always had negative cash flows from operations and have incurred net operating losses each year since we started business. For the years ended December 31, 2016 and 2015, we incurred net loss of $22.0 million and $19.4 million, respectively, our net cash used in operating activities was $19.5 million and $20.5 million, respectively, and, at December 31, 2016, our accumulated deficit was $379.1 million. We expect to continue to incur net losses and negative cash flow from operating activities for at least the next year. As our focus on development of Cytori Cell Therapy, the Celution system platform and development of therapeutic applications for Cytori Cell Therapy has increased, losses have resulted primarily from expenses associated with research and development and clinical trial-related activities, as well as general and administrative expenses. While we have implemented and continue to implement cost reduction measures where possible, we nonetheless expect to continue operating in a loss position on a consolidated basis and expect that recurring operating expenses will be at even higher levels for at least the next year to perform clinical trial and other development activities for our Cytori Cell Therapy and Cytori Nanomedicines products and product candidates, including additional pre-clinical research, clinical trial-related activities, pre-commercialization activities (including regulatory and reimbursement analysis and market research), and marketing.
Our ability to generate sufficient revenues from any of our products, product candidates or technologies to achieve profitability will depend on a number of factors including, but not limited to:
|
|
• |
our ability to manufacture, test and validate our product candidates in compliance with applicable laws and as required for submission to applicable regulatory bodies, including manufacturing, testing and validation of our ATI-0918 drug candidate; |
|
|
• |
our or our partners’ ability to successfully complete clinical trials of our product candidates; |
|
|
• |
our ability to obtain necessary regulatory approvals for our product candidates; |
|
|
• |
our or our partners’ ability to negotiate and receive favorable reimbursement for our product candidates, including for our product candidates that have been granted or may be granted orphan drug status or otherwise command currently anticipated pricing levels; |
|
|
• |
our ability to negotiate favorable arrangements with third parties to help finance the development of, and market and distribute, our products and product candidates; and |
|
|
• |
the degree to which our approved products are accepted in the marketplace. |
Because of the numerous risks and uncertainties associated with our commercialization and product development efforts, we are unable to predict the extent of our future losses or when or if we will become profitable and it is possible we will never become profitable. If we do not generate significant sales from any of our product candidates that may receive regulatory approval, there would likely be a material adverse effect on our business, results of operations, financial condition and prospects which could result in our inability to continue operations.
We will need substantial additional funding to develop our products and for our future operations. If we are unable to obtain the funds necessary to do so, we may be required to delay, scale back or eliminate our product development activities or may be unable to continue our business.
We have had, and we will continue to have, an ongoing need to raise additional cash from outside sources to continue funding our operations to profitability, including our continuing substantial research and development expenses. We do not currently believe that our cash balance and the revenues from our operations will be sufficient to fund the development and marketing efforts required to reach profitability without raising additional capital from accessible sources of financing in the near future. Although it is difficult to predict future liquidity requirements, we believe that our $12.6 million in cash and cash equivalents on hand as of December 31, 2016 will be sufficient to fund our currently contemplated operations at least through June 2017. Our future capital requirements will depend on many factors, including:
|
|
• |
our ability to raise capital to fund our operations on terms acceptable to us, or at all; |
|
|
• |
our perceived capital needs with respect to development of our Cytori Cell Therapy and Cytori Nanomedicines development programs, and any delays in, adverse events of, and excessive costs of such programs beyond what we currently anticipate; |
|
|
• |
our ability to establish and maintain collaborative and other arrangements with third parties to assist in bringing our products to market and the cost of such arrangements at the time; |
41
|
|
• |
costs associated with the integration and operation of our newly acquired Cytori Nanomedicine business, including hiring of as many as 20 or more new employees to operate the Cytori Nanomedicine business, and costs of validation, requalification and recommencement of the Cytori Nanomedicine manufacturing operations at our San Antonio, Texas facility; |
|
|
• |
the cost of manufacturing our product candidates, including compliance with good manufacturing practices, or GMP, applicable to our product candidates; |
|
|
• |
expenses related to the establishment of sales and marketing capabilities for product candidates awaiting approval or products that have been approved; |
|
|
• |
the level of our sales and marketing expenses; |
|
|
• |
competing technological and market developments; and |
|
|
• |
our ability to introduce and sell new products. |
We have secured capital historically from grant revenues, collaboration proceeds, and debt and equity offerings. We will need to secure substantial additional capital to fund our future operations. We cannot be certain that additional capital will be available on terms acceptable to us, or at all. Our ability to raise capital was adversely affected when the FDA put a hold on our ATHENA cardiac trials in mid-2014, which had an adverse impact to stock price performance and our corresponding ability to restructure our debt. More recently, a continued downward trend in our stock price resulting from a number of factors, including (i) general economic and industry conditions, (ii) challenges faced by the regenerative medicine industry as a whole, (iii) the market’s unfavorable view of certain of our recent equity financings conducted in 2014 and 2015 (which financings were priced at a discount to market and included 100% warrant coverage), (iv) market concerns regarding our continued need for capital (and the effects of any future capital raising transactions we may consummate) (v) market perceptions of our ATHENA and ACT-OA clinical trial data; and (vi) our recent NASDAQ Stock Market LLC, or Nasdaq, listing deficiency issues and resultant 1-for-15 reverse stock split, have made it more difficult to procure additional capital on terms reasonably acceptable to us. Though our recent acquisition of the Cytori Nanomedicine business from Azaya Therapeutics, including our ATI-0918 and ATI-1123 drug candidates, appear to have been viewed favorably by our investors and the marketplace, we cannot assure you that this acquisition will not ultimately be viewed negatively and thus further hamper our efforts to attract additional capital. If we are unsuccessful in our efforts to raise any such additional capital, we may be required to take actions that could materially and adversely harm our business, including a possible significant reduction in our research, development and administrative operations (including reduction of our employee base), surrendering of our rights to some technologies or product opportunities, delaying of our clinical trials or regulatory and reimbursement efforts, or curtailing of or even ceasing operations.
Our financing plans include pursuing additional cash through use of our at-the-market, or ATM, offering program, or ATM, strategic corporate partnerships, licensing and sales of equity. In addition, in December 2016, we entered into a purchase agreement, or the Lincoln Park Purchase Agreement, with Lincoln Park Capital Fund, LLC, or Lincoln Park, pursuant to which we may direct Lincoln Park to purchase up to $20.0 million in shares of our common stock from time to time over a 30-month period, commencing upon the satisfaction of certain conditions, including that a registration statement be declared effective by the SEC. While we have an established history of raising capital through these platforms, and we are currently involved in negotiations with multiple parties, there is no guarantee that adequate funds will be available when needed from additional debt or equity financing, development and commercialization partnerships or from other sources or on terms acceptable to us. In addition, under current SEC regulations, at any time during which the aggregate market value of our common stock held by non-affiliates, or public float, is less than $75.0 million, the amount we can raise through primary public offerings of securities in any twelve-month period using shelf registration statements, including sales under our ATM offering program, is limited to an aggregate of one-third of our public float. As of December 31, 2016, our public float was 21.5 million shares, the value of which was $32.5 million based upon the closing price of our common stock of $1.51 on such date. The value of one-third of our public float calculated on the same basis was approximately $11.0 million.
Further, our Loan and Security Agreement with Oxford Finance, LLC, or Oxford, as further described below, requires us to maintain a minimum of $5.0 million in unrestricted cash and cash equivalents on hand to avoid an event of default under the Loan and Security Agreement. Based on our cash and cash equivalents on hand of approximately $12.6 million at December 31, 2016, and our obligation to make payments of principal of $590,000 plus accrued interest in monthly installments, we estimate that we must raise additional capital and/or obtain a waiver or restructure the Loan and Security Agreement on or before May 2017 to avoid defaulting under our $5 million minimum cash/cash equivalents covenant. If we are unable to avoid an event of default under the Loan and Security Agreement, our business could be severely harmed. See the Risk Factor below regarding the Loan and Security Agreement.
In addition to the funding sources previously mentioned, we continue to seek additional capital through product revenues and state and federal development programs, including additional funding opportunities though our current BARDA contract.
42
Our level of indebtedness, and covenant restrictions under such indebtedness, could adversely affect our operations and liquidity.
Under our Loan and Security Agreement with Oxford, as collateral agent and lender, Oxford made a term loan to us in an aggregate principal amount of $17,700,000, or the Term Loan, subject to the terms and conditions set forth in the Loan and Security Agreement.
The Term Loan accrues interest at a floating rate equal to the three-month LIBOR rate (with a floor of 1.00%) plus 7.95% per annum. Prior to January 2017, we made interest-only payments on the Term Loan. However, as of January 2017, we are required to make payments of principal (in the amount of $590,000 per month) and accrued interest in equal monthly installments of approximately $725,000 to amortize the Term Loan through June 1, 2019, the maturity date. All unpaid principal and accrued and unpaid interest with respect to the Term Loan is due and payable in full on June 1, 2019.
As security for our obligations under the Loan and Security Agreement, we granted a security interest in substantially all of our existing and after-acquired assets, subject to certain exceptions set forth in the Loan and Security Agreement and excluding our intellectual property assets, which are subject to a negative pledge by us. If we are unable to discharge these obligations, Oxford could foreclose on these assets, which would, at a minimum, have a severe material effect on our ability to operate our business.
Our indebtedness to Oxford could adversely affect our operations and liquidity, by, among other things:
|
|
• |
causing us to use a larger portion of our cash flow to fund interest and principal payments, reducing the availability of cash to fund working capital and capital expenditures and other business activities; |
|
|
• |
making it more difficult for us to take advantage of significant business opportunities, such as acquisition opportunities, and to react to changes in market or industry conditions; and |
|
|
• |
limiting our ability to borrow additional monies in the future to fund working capital and capital expenditures and for other general corporate purposes. |
The Loan and Security Agreement requires us to maintain at least $5 million in unrestricted cash and/or cash equivalents and includes certain reporting and other covenants, that, among other things, restrict our ability to (i) dispose of assets, (ii) change the business we conduct, (iii) make acquisitions, (iv) engage in mergers or consolidations, (v) incur additional indebtedness, (vi) create liens on assets, (vii) maintain any collateral account, (viii) pay dividends, (ix) make investments, loans or advances, (x) engage in certain transactions with affiliates, and (xi) prepay certain other indebtedness or amend other financing arrangements. If we fail to comply with any of these covenants or restrictions, such failure may result in an event of default, which if not cured or waived, could result in Oxford causing the outstanding loan amount ($17.7 million as of December 31, 2016) to become immediately due and payable. If the maturity of our indebtedness is accelerated, we may not have, or be able to timely procure, sufficient cash resources to satisfy our debt obligations, and such acceleration would adversely affect our business and financial condition.
In addition, our indebtedness under the Loan and Security Agreement is secured by a security interest in substantially all of our existing and after-acquired assets, excluding our intellectual property assets (which are subject to a negative pledge), and therefore, if we are unable to repay such indebtedness, Oxford could foreclose on these assets, which would, at a minimum, have a severe material effect on our ability to operate our business.
The report of our independent registered public accounting firm contains an emphasis paragraph regarding the substantial doubt about our ability to continue as a “going concern.”
The audit report of our independent registered public accounting firm covering the December 31, 2016 consolidated financial statements contains an explanatory paragraph that states that our recurring losses from operations, liquidity position, and debt service requirements raises substantial doubt about our ability to continue as a going concern. This going concern opinion could materially limit our ability to raise additional funds through the issuance of new debt or equity securities or otherwise. Future reports on our financial statements may also include an explanatory paragraph with respect to our ability to continue as a going concern. To date, our operating losses have been funded primarily from outside sources of invested capital and gross profits. We have had, and we will likely continue to have, an ongoing need to raise additional cash from outside sources to fund our future operations. However, no assurance can be given that additional capital will be available when required or on terms acceptable to us. If we are unsuccessful in our efforts to raise any such additional capital, we would be required to take actions that could materially and adversely affect our business, including significant reductions in our research, development and administrative operations (including reduction of our employee base), possible surrender or other disposition of our rights to some technologies or product opportunities, delaying of our clinical trials or curtailing or ceasing operations. We also cannot give assurance that we will achieve sufficient revenues in the future
43
to achieve profitability and cash flow positive operations to allow us to continue as a going concern. The perception that we may not be able to continue as a going concern may cause third parties to choose not to deal with us due to concerns about our ability to meet our contractual obligations, which could have a material adverse effect on our business.
We may not be able to access the full amounts available under the Lincoln Park Purchase Agreement, which could prevent us from accessing the capital we need to continue our operations, which could have an adverse effect on our business.
In December 2016, we entered into the Lincoln Park Purchase Agreement, pursuant to which we may direct Lincoln Park to purchase up to $20.0 million of shares of our common stock from time to time over a 30-month period, commencing upon the satisfaction of certain conditions, including that a registration statement be declared effective by the SEC. Thereafter, on any trading day selected by us, we may sell shares of common stock to Lincoln Park in amounts up to 100,000 shares per regular sale (such purchases, Regular Purchases) up to the aggregate commitment of $20.0 million. If the market price of our common stock is not below $2.00 per share on the purchase date, then the Regular Purchase amount may be increased to 150,000 shares. If the market price is not below $3.00 per share on the purchase date, then the Regular Purchase amount may be increased to 300,000 shares. Although there are no upper limits on the per share price Lincoln Park may pay to purchase our common stock, we may not sell more than $1.0 million in shares of common stock to Lincoln Park per any individual Regular Purchase. The purchase price of Regular Purchases will be based on the prevailing market prices of shares of our common stock, which shall be equal to the lesser of the lowest sale price of the common shares during the purchase date and the average of the three lowest closing sale prices of the common shares during the ten business days prior to the purchase date.
In addition to Regular Purchases, we may in our sole discretion direct Lincoln Park on each purchase date to make accelerated purchases on the following business day up to the lesser of (i) three times the number of shares purchased pursuant to such Regular Purchase or (ii) 30% of the trading volume on the accelerated purchase date at a purchase price equal to the lesser of (i) the closing sale price on the accelerated purchase date and (ii) 97% of the accelerated purchase date’s volume weighted average price (such purchases, Accelerated Purchases). We cannot submit an Accelerated Purchase notice if the market price of our common stock is below $1.00.
In addition to Regular Purchases and Accelerated Purchases described above, we may also direct Lincoln Park, on any business day that the closing price of our common stock is not below $1.00, to purchase additional amounts of our common stock, which we refer to as an Additional Purchase whereby, pursuant to each Additional Purchase we may sell up to $1.0 million of common stock in each Additional Purchase notice, provided, however, that (i) we may not deliver to Lincoln Park more than two separate Additional Purchase notices and (ii) at least 30 business days must pass between our delivery of the first Additional Purchase notice to Lincoln Park and our delivery of the second Additional Purchase notice. The purchase price for each such Additional Purchase shall be equal to the lower of (i) 97% of the purchase price under a Regular Purchase on the date we give notice for the related Additional Purchase, or (ii) $2.00 per share.
Depending on the prevailing market price of our common stock, we may not be able to sell shares to Lincoln Park for the maximum $20.0 million over the term of the Lincoln Park Purchase Agreement. For example, under the rules of the NASDAQ Capital Market, in no event may we issue more than 19.99% of our shares outstanding (which is approximately 4,315,814 shares based on 21,579,071 shares outstanding prior to the signing of the Lincoln Park Purchase Agreement) under the Lincoln Park Purchase Agreement unless we obtain stockholder approval or an exception pursuant to the rules of the NASDAQ Capital Market is obtained to issue more than 19.99%. This limitation will not apply if, at any time the exchange cap is reached and at all times thereafter, the average price paid for all shares issued and sold under the Lincoln Park Purchase Agreement is equal to or greater than $1.6674, which was the consolidated closing bid price of our common stock on December 22, 2016 including an increment for the commitment shares we issued and may issue to Lincoln Park. We are not required or permitted to issue any shares of common stock under the Purchase Agreement if such issuance would breach our obligations under the rules or regulations of the NASDAQ Capital Market. In addition, Lincoln Park will not be required to purchase any shares of our common stock if such sale would result in Lincoln Park’s beneficial ownership exceeding 9.99% of the then outstanding shares of our common stock. Our inability to access a portion or the full amount available under the Lincoln Park Purchase Agreement, in the absence of any other financing sources, could have a material adverse effect on our business.
The sale or issuance of our common stock to Lincoln Park may cause dilution and the sale of the shares of common stock acquired by Lincoln Park, or the perception that such sales may occur, could cause the price of our common stock to fall.
In December 2016, we entered into the Lincoln Park Purchase Agreement, pursuant to which Lincoln Park has committed to purchase up to $20.0 million of our common stock. Concurrently with the execution of the Lincoln Park Purchase Agreement, we issued 127,419 shares of our common stock to Lincoln Park as an initial fee for its commitment to purchase shares of our common stock under the Lincoln Park Purchase Agreement. Further, for each additional purchase by Lincoln Park, we will issue additional commitment shares in commensurate amounts up to a total of 382,258 shares based upon the relative proportion of the aggregate
44
amount of $20.0 million purchased by Lincoln Park. The purchase shares that may be sold pursuant to the Lincoln Park Purchase Agreement may be sold by us to Lincoln Park at our discretion from time to time over a 30-month period. The purchase price for the shares that we may sell to Lincoln Park under the Lincoln Park Purchase Agreement will fluctuate based on the price of our common stock. Depending on market liquidity at the time, sales of such shares may cause the trading price of our common stock to fall.
We generally have the right to control the timing and amount of any sales of our shares to Lincoln Park. Additional sales of our common stock, if any, to Lincoln Park will depend upon market conditions and other factors to be determined by us. Lincoln Park may ultimately purchase all, some or none of the shares of our common stock that may be sold pursuant to the Lincoln Park Purchase Agreement and, after it has acquired shares, Lincoln Park may sell all, some or none of those shares. Therefore, sales to Lincoln Park by us could result in substantial dilution to the interests of other holders of our common stock. Additionally, the sale of a substantial number of shares of our common stock to Lincoln Park, or the anticipation of such sales, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales.
Material weaknesses in our internal control over financial reporting have occurred in the past and could occur in the future.
We are required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting, as well as a statement that our independent registered public accounting firm has issued an attestation report on the effectiveness of our internal control over financial reporting.
We identified a material weakness in our internal control over financial reporting for the year ended December 31, 2013, which may have adversely affected investor confidence in us and, as a result, the value of our common stock. While no such material weakness was identified for the years ended December 31, 2016 or December 31, 2015, we cannot assure you that additional material weaknesses will not be identified in the future.
If we are unable to effectively remediate any material weaknesses in a timely manner, or if we identify one or more additional material weaknesses in the future, investors could lose confidence in the accuracy and completeness of our financial reports, which could have a material adverse effect on the price of our common stock.
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our budgeted expense levels are based in part on our expectations concerning future revenues from sales as well our assessment of the future investments needed to expand our commercial organization and support research and development activities. We may be unable to reduce our expenditures in a timely manner to compensate for any unexpected events or a shortfall in revenue. Accordingly, a shortfall in demand for our products or other unexpected events could have an immediate and material impact on our business and financial condition.
Our operating results have been and will likely continue to be volatile.
Our prospects must be evaluated in light of the risks and difficulties frequently encountered by emerging companies and particularly by such companies in rapidly evolving and technologically advanced biotech, pharmaceutical and medical device fields. From time to time, we have tried to update our investors’ expectations as to our operating results by periodically announcing financial guidance. However, we have in the past been forced to revise or withdraw such guidance due to lack of visibility and predictability of product demand.
Risks Relating to Our Intellectual Property
Our success depends in part on our ability to protect our intellectual property. It is difficult and costly to protect our proprietary rights and technology, and we may not be able to ensure their protection.
Our success depends in part on our ability to obtain and maintain patent, trademark and trade secret protection of our platform technology and current product candidates, including but not limited to our Cytori Cell Therapy and Cytori Nanomedicine products and product candidates, including Habeo Cell Therapy, ATI-0918 and ATI-1123, as well as successfully defending our intellectual property against third-party challenges. Our ability to stop unauthorized third parties from making using selling, offering to sell or importing our platform technology and/or our product candidates is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities.
45
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
|
|
• |
we, or Azaya Therapeutics, as the case may be, might not have been the first to file patent applications for the covered inventions; |
|
|
• |
it is possible that our pending patent applications will not result in issued patents; |
|
|
• |
it is possible that there are dominating patents to our products of which we are not aware; |
|
|
• |
it is possible that there are prior public disclosures that could invalidate our patents, of which we are not aware; |
|
|
• |
it is possible that others may circumvent our patents; |
|
|
• |
it is possible that there are unpublished applications or patent applications maintained in secrecy that may later issue with claims covering our products or technology similar to ours; |
|
|
• |
the claims of our patents or patent applications, if and when issued, may not cover our system or products, or our system or product candidates; |
|
|
• |
our owned or in-licensed issued patents may not provide us with any competitive advantages, or may be narrowed in scope, be held invalid or unenforceable as a result of legal administrative challenges by third parties; |
|
|
• |
others may be able to make or use compounds that are the same or similar to the ATI-1123 product but that are not covered by the claims of our patents; |
|
|
• |
we may not be able to detect infringement against our patents, which may be especially difficult for manufacturing processes or formulation patents, such as the patents/applications related to ATI-1123; |
|
|
• |
the API in ATI-0918 is commercially available in generic drug products; |
|
|
• |
we may not develop additional proprietary technologies for which we can obtain patent protection; or |
|
|
• |
the patents of others may have an adverse effect on our business. |
The patent positions of pharmaceutical, biopharmaceutical and medical device companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in patents in these fields has emerged to date in the United States. There have been recent changes regarding how patent laws are interpreted, and both the U.S. Patent and Trademark Office, or PTO, and Congress have recently made significant changes to the patent system. There have been three U.S. Supreme Court decisions that now show a trend of the Supreme Court which is distinctly negative on patents. The trend of these decisions along with resulting changes in patentability requirements being implemented by the U.S. Patent and Trademark Office could make it increasingly difficult for us to obtain and maintain patents on our products. We cannot accurately predict future changes in the interpretation of patent laws or changes to patent laws which might be enacted into law. Those changes may materially affect our patents, our ability to obtain patents and/or the patents and applications of our collaborators and licensors. The patent situation in these fields outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents we own or to which we have a license or third-party patents.
Intellectual property law outside the United States is uncertain and in many countries is currently undergoing review and revisions. The laws of some countries do not protect our patent and other intellectual property rights to the same extent as United States laws. Third parties may attempt to oppose the issuance of patents to us in foreign countries by initiating opposition proceedings. Opposition proceedings against any of our patent filings in a foreign country could have an adverse effect on our corresponding patents that are issued or pending in the United States. It may be necessary or useful for us to participate in proceedings to determine the validity of our patents or our competitors’ patents that have been issued in countries other than the United States. This could result in substantial costs, divert our efforts and attention from other aspects of our business, and could have a material adverse effect on our results of operations and financial condition. We currently have pending patent applications in Europe, Australia, Japan, Canada, China, South Korea, Brazil, South Africa, among other jurisdictions.
46
Our intellectual property related to Cytori Nanomedicine was acquired from Azaya. As ATI-0918 is a generic drug, we did not acquire any patents related to ATI-0918. We acquired two issued patents and one patent application related to ATI-1123 from Azaya, and intend to file additional patent applications around our ATI-1123 drug candidate. There is no guaranty that any patent applications we file on ATI-1123 will issue, or if issued, that we will be to use and enforce these patents as an effective component of our intellectual property strategy.
Failure to obtain or maintain patent protection or protect trade secrets, for any reason (or third-party claims against our patents, trade secrets, or proprietary rights, or our involvement in disputes over our patents, trade secrets, or proprietary rights, including involvement in litigation), could have a substantial negative effect on our results of operations and financial condition.
We may not be able to protect our trade secrets.
We may rely on trade secrets to protect our technology, especially with respect to the Cytori Nanomedicines products, as well as in areas where we do not believe patent protection is appropriate or obtainable. Trade secrets are difficult to protect, and we have limited control over the protection of trade secrets used by our collaborators and suppliers. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, state laws in the Unites States vary, and their courts as well as courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. If our confidential or proprietary information is divulged to or acquired by third parties, including our competitors, our competitive position in the marketplace will be harmed and our ability to successfully penetrate our target markets could be severely compromised.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the device, biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other device, biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management, which would adversely affect our financial condition.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, and we may be unable to protect our rights to our products and technology.
Litigation may be necessary to enforce or confirm the ownership of any patents issued or licensed to us, or to determine the scope and validity of third-party proprietary rights, which would result in substantial costs to us and diversion of effort on our part. If our competitors claim technology also claimed by us and prepare and file patent applications in the United States, we may have to participate in interference proceedings declared by the USPTO or a foreign patent office to determine priority of invention, which could result in substantial costs to and diversion of effort, even if the eventual outcome is favorable to us. Any such litigation or interference proceeding, regardless of outcome, could be expensive and time-consuming.
Successful challenges to our patents through oppositions, reexamination proceedings or interference proceedings could result in a loss of patent rights in the relevant jurisdiction. If we are unsuccessful in actions we bring against the patents of other parties, and it is determined that we infringe the patents of third-parties, we may be subject to litigation, prevented from commercializing potential products in the relevant jurisdiction and/or may be required to obtain licenses to those patents or develop or obtain alternative technologies, any of which could harm our business. Furthermore, if such challenges to our patent rights are not resolved in our favor, we could be delayed or prevented from entering into new collaborations or from commercializing certain products, which could adversely affect our business and results of operations.
Competitors or third parties may infringe on or upon our patents. We may be required to file patent infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable or that the third party’s technology does not in fact infringe upon our patents. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our related pending patent applications at risk of not issuing. Litigation may fail and, even if successful, may result in substantial costs and be a distraction to our management. We may not be able to prevent misappropriation of our proprietary rights, particularly in countries outside the United States where patent rights may be more difficult to enforce. Further, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential or sensitive information could be compromised by disclosure in the event of litigation. In addition, during the course of litigation there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
47
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations or otherwise have a material adverse effect on our business, results of operations, financial condition and prospects.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in that litigation would have a material adverse effect on our business.
Our commercial success will also depend, in part, on our ability to avoid infringing on patents issued by others. There may be issued patents of third parties of which we are currently unaware, that are infringed or are alleged to be infringed by our product candidate or proprietary technologies. Because some patent applications in the United States may be maintained in secrecy until the patents are issued, patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, and publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our owned and in-licensed issued patents or our pending applications, or that we or, if applicable, a licensor were the first to invent the technology. Our competitors may have filed, and may in the future file, patent applications covering our product candidates or technology similar to ours. Any such patent application may have priority over our patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our product candidates and/or proprietary technologies infringe their intellectual property rights. These lawsuits are costly and could adversely affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we or our commercialization partners are infringing the third party's patents and would order us or our partners to stop the activities covered by the patents. In addition, there is a risk that a court will order us or our partners to pay the other party damages for having violated the other party's patents.
If a third-party's patent was found to cover our products, proprietary technologies or their uses, we could be enjoined by a court and required to pay damages and could be unable to commercialize our product candidates or use our proprietary technologies unless we or they obtained a license to the patent. A license may not be available to us on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief which could prohibit us from making, using or selling our products, technologies or methods pending a trial on the merits, which could be years away.
Risks Relating to the Securities Markets and an Investment in Our Stock
The market price of our common stock may be volatile and fluctuate significantly, which could result in substantial losses for stockholders.
The market price of our common stock has been, and may continue to be, subject to significant fluctuations. Among the factors that may cause the market price of our common stock to fluctuate are the risks described in this “Risk Factors” section and other factors, including:
|
|
• |
fluctuations in our operating results or the operating results of our competitors; |
|
|
• |
the outcome of clinical trials involving the use of our products, including our sponsored trials; |
|
|
• |
changes in estimates of our financial results or recommendations by securities analysts; |
|
|
• |
variance in our financial performance from the expectations of securities analysts; |
|
|
• |
changes in the estimates of the future size and growth rate of our markets; |
|
|
• |
changes in accounting principles or changes in interpretations of existing principles, which could affect our financial results; |
|
|
• |
conditions and trends in the markets we currently serve or which we intend to target with our product candidates; |
|
|
• |
changes in general economic, industry and market conditions; |
|
|
• |
success of competitive products and services; |
48
|
|
• |
changes in market valuations or earnings of our competitors; |
|
|
• |
announcements of significant new products, contracts, acquisitions or strategic alliances by us or our competitors; |
|
|
• |
our continuing ability to list our securities on an established market or exchange; |
|
|
• |
the timing and outcome of regulatory reviews and approvals of our products; |
|
|
• |
the commencement or outcome of litigation involving our company, our general industry or both; |
|
|
• |
changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
|
|
• |
actual or expected sales of our common stock by the holders of our common stock; and |
|
|
• |
the trading volume of our common stock. |
In addition, the stock market in general, the Nasdaq markets and the market for cell therapy development companies in particular may experience a loss of investor confidence. A loss of investor confidence may result in extreme price and volume fluctuations in our common stock that are unrelated or disproportionate to the operating performance of our business, our financial condition or results of operations, which may materially harm the market price of our common stock and result in substantial losses for stockholders.
Future sales of our common stock may depress our share price.
As of December 31, 2016, we had 21,707,890 shares of our common stock outstanding. Sales of a number of shares of common stock in the public market, including pursuant to the Lincoln Park Purchase Agreement, or our ATM program, or the expectation of such sales, could cause the market price of our common stock to decline. We may also sell additional common stock or securities convertible into or exercisable or exchangeable for common stock in subsequent public or private offerings or other transactions, which may adversely affect the market price of our common stock.
We have granted demand registration rights for the resale of certain shares of our common stock to each of Astellas Pharma Inc. and Green Hospital Supply, Inc. pursuant to common stock purchase agreements previously entered into with each of these stockholders. An aggregate of approximately 300,000 shares of our common stock are subject to these demand registration rights. If we receive a written request from any of these stockholders to file a registration statement under the Securities Act of 1933, as amended, or the Securities Act, covering its shares of unregistered common stock, we are required to use reasonable efforts to prepare and file with the SEC within 30 business days of such request a registration statement covering the resale of the shares for an offering to be made on a continuous basis pursuant to Rule 415 under the Securities Act.
We have also granted registration rights to Azaya, with respect to the 1,173,241 shares of our common stock that we issued in the name of Azaya at the closing of our acquisition of the Cytori Nanomedicine assets. Under the terms of our asset purchase agreement with Azaya, we are required to use best efforts to have a registration statement covering these shares filed with the SEC, and are thereafter required to use commercially reasonable efforts to have the registration declared effective by the SEC. Though Azaya is subject to certain volume limitations regarding its sales of our common stock, once Azaya is able to sell these shares, any such sales could put pressure on our stock and depress our share price.
Our stockholders may experience substantial dilution in the value of their investment if we issue additional shares of our capital stock.
Our charter allows us to issue up to 75,000,000 shares of our common stock and to issue and designate the rights of, without stockholder approval, up to 5,000,000 shares of preferred stock. To raise additional capital, we may in the future sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock at prices that are lower than the prices paid by existing stockholders, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders, which could result in substantial dilution to the interests of existing stockholders.
We could be delisted from Nasdaq, which could seriously harm the liquidity of our stock and our ability to raise capital.
Following notice from Nasdaq staff in June 2015 and December 2015, we had a hearing in January 2016 relating to our noncompliance with the $1.00 minimum bid price per share requirement. The NASDAQ Hearing Panel granted us until May 31, 2016 to come into compliance with the minimum bid price requirement, including requirements relating to obtaining stockholders approval
49
of a reverse stock split that would bring our stock price above $1.00 per share for a minimum of 10 consecutive trading days. We transferred the listing of our common stock from the NASDAQ Global Market to the NASDAQ Capital Market in February 2016. In May 2016, we consummated a 1-for-15 reverse stock split pursuant to which the minimum bid price per share of our common stock rose above $1.00. Pursuant to a letter dated May 26, 2016, the Nasdaq staff delivered notice to us that we had regained compliance with Nasdaq’s minimum bid price rule. However, we may be unable to maintain compliance with our current minimum bid price obligation or the other listing requirements, which could cause us to lose eligibility for continued listing on the NASDAQ Capital Market or any comparable trading market. If we cease to be eligible to trade on the NASDAQ Capital Market:
|
|
• |
We may have to pursue trading on a less recognized or accepted market, such as the OTC Bulletin Board or the “pink sheets.” |
|
|
• |
The trading price of our common stock could suffer, including an increased spread between the “bid” and “asked” prices quoted by market makers. |
|
|
• |
Shares of our common stock could be less liquid and marketable, thereby reducing the ability of stockholders to purchase or sell our shares as quickly and as inexpensively as they have done historically. If our stock is traded as a “penny stock,” transactions in our stock would be more difficult and cumbersome. |
|
|
• |
We may be unable to access capital on favorable terms or at all, as companies trading on alternative markets may be viewed as less attractive investments with higher associated risks, such that existing or prospective institutional investors may be less interested in, or prohibited from, investing in our common stock. This may also cause the market price of our common stock to decline. |
We may be or become the target of securities litigation, which is costly and time-consuming to defend.
In the past, following periods of market volatility in the price of a company’s securities, the reporting of unfavorable news or continued decline in a company’s stock price, security holders have often instituted class action litigation. The market value of our securities has steadily declined over the past several years for a variety of reasons discussed elsewhere in this “Risk Factors” section, which heightens our litigation risk. If we become involved in this type of litigation, regardless of the outcome, we could incur substantial legal costs and our management’s attention could be diverted from the operation of our business, causing our business to suffer. Any adverse determination in any such litigation or any amounts paid to settle any such actual or threatened litigation could require that we make significant payments.
We may issue debt and equity securities or securities convertible into equity securities, any of which may be senior to our common stock as to distributions and in liquidation, which could negatively affect the value of our common stock.
In the future, we may attempt to increase our capital resources by entering into debt or debt-like financing that is unsecured or secured by up to all of our assets, or by issuing additional debt or equity securities, which could include issuances of secured or unsecured commercial paper, medium-term notes, senior notes, subordinated notes, guarantees, preferred stock, hybrid securities, or securities convertible into or exchangeable for equity securities. In the event of our liquidation, our lenders and holders of our debt and preferred securities would receive distributions of our available assets before distributions to the holders of our common stock. Because our decision to incur debt and issue securities in future offerings may be influenced by market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing or nature of our future offerings or debt financings. Further, market conditions could require us to accept less favorable terms for the issuance of our securities in the future.
If you hold warrants issued pursuant to our rights offering, you may be limited in your ability to engage in certain hedging transactions that could provide you with financial benefits.
In June 2016, we closed our rights offering to subscribe for units at a subscription price of $2.55 per unit, or the Rights Offering. Pursuant to the Rights Offering, we sold to our stockholders of record (as of May 20, 2016) an aggregate of 6,704,852 units consisting of 6,704,852 shares of common stock and 3,352,306 warrants, or Warrants, with each Warrant exercisable for one share of common stock at an exercise price of $3.06 per share.
Holders of Warrants were required to represent to us that they will not enter into any short sale or similar transaction with respect to our common stock for so long as they continue to hold Warrants. These requirements prevent our Warrant holders from pursuing certain investment strategies that could provide them greater financial benefits than they might have realized had they not been required to make this representation.
50
Absence of a public trading market for the Warrants may limit the ability to resell the Warrants.
The Warrants are listed for trading on Nasdaq under the symbol “CYTXW,” but there can be no assurance that a robust market will exist for the Warrants. Even if a market for the Warrants does develop, the price of the Warrants may fluctuate and liquidity may be limited. If the Warrants cease to be eligible for continued listing on Nasdaq, or if the market for the Warrants does not fully develop (or subsequently weakens), then purchasers of the Warrants may be unable to resell the Warrants or sell them only at an unfavorable price for an extended period of time, if at all. Future trading prices of the Warrants will depend on many factors, including:
|
|
• |
our operating performance and financial condition; |
|
|
• |
our ability to continue the effectiveness of the registration statement covering the Warrants and the common stock issuable upon exercise of the Warrants; |
|
|
• |
the interest of securities dealers in making and maintaining a market; and |
|
|
• |
the market for similar securities. |
The market price of our common stock may never exceed the exercise price of the Warrants issued in connection with the Rights Offering.
The Warrants issued pursuant to the Rights Offering became exercisable upon issuance and will expire thirty (30) months from the date of issuance. The market price of our common stock may never exceed the exercise price of the Warrants prior to their date of expiration. Any Warrants not exercised by their date of expiration will expire worthless and we will be under no further obligation to the Warrant holder.
The Warrants contain features that may reduce Warrant holders’ economic benefit from owning them.
The Warrants contain features that allow us to redeem the Warrants and that prohibit Warrant holders from engaging in certain investment strategies. We may redeem the Warrants for $0.01 per Warrant once the closing price of our common stock has equaled or exceeded $7.65 per share, subject to adjustment, for ten consecutive trading days, provided that we may not do so prior to the first anniversary of closing of the Rights Offering, and only upon not less than thirty (30) days’ prior written notice of redemption. If we give notice of redemption, Warrant holders will be forced to sell or exercise their Warrants or accept the redemption price. The notice of redemption could come at a time when it is not advisable or possible for Warrant holders to exercise the Warrants. As a result, Warrant holders may be unable to benefit from owning the Warrants being redeemed. In addition, for so long as Warrant holders continue to hold Warrants, they will not be permitted to enter into any short sale or similar transaction with respect to our common stock. This could prevent Warrant holders from pursuing investment strategies that could provide them greater financial benefits from owning the Warrants.
Since the Warrants are executory contracts, they may have no value in a bankruptcy or reorganization proceeding.
In the event a bankruptcy or reorganization proceeding is commenced by or against us, a bankruptcy court may hold that any unexercised Warrants are executory contracts that are subject to rejection by us with the approval of the bankruptcy court. As a result, holders of the Warrants may, even if we have sufficient funds, not be entitled to receive any consideration for their Warrants or may receive an amount less than they would be entitled to if they had exercised their Warrants prior to the commencement of any such bankruptcy or reorganization proceeding.
Our charter documents contain anti-takeover provisions.
Certain provisions of our amended and restated certificate of incorporation and amended and restated bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable. These provisions could also prevent or frustrate attempts by our stockholders to replace or remove members of our Board of Directors. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions:
|
|
• |
authorize our Board of Directors to issue without stockholder approval up to 5,000,000 shares of preferred stock, the rights of which will be determined at the discretion of the Board of Directors; |
|
|
• |
require that stockholder actions must be effected at a duly called stockholder meeting and cannot be taken by written consent; |
51
|
|
• |
establish advance notice requirements for stockholder nominations to our Board of Directors or for stockholder proposals that can be acted on at stockholder meetings; and |
|
|
• |
limit who may call stockholder meetings. |
We are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
We presently do not intend to pay cash dividends on our common stock.
We have never paid cash dividends in the past, and we currently anticipate that no cash dividends will be paid on the common stock in the foreseeable future. Furthermore, our Loan and Security Agreement with Oxford currently prohibits our issuance of cash dividends. This could make an investment in our common stock inappropriate for some investors, and may serve to narrow our potential sources of additional capital. While our dividend policy will be based on the operating results and capital needs of the business, it is anticipated that all earnings, if any, will be retained to finance the future expansion of our business.
If securities and/or industry analysts fail to continue publishing research about our business, if they change their recommendations adversely, or if our results of operations do not meet their expectations, our stock price and trading volume could decline.
The trading market for our common stock may be influenced by the research and reports that industry or securities analysts publish about us or our business. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. In addition, it is likely that in some future period our operating results will be below the expectations of securities analysts or investors. If one or more of the analysts who cover us downgrade our stock, or if our results of operations do not meet their expectations, our stock price could decline.
Item 1B. Unresolved Staff Comments
Not applicable.
We lease 77,585 square feet at 3020 and 3030 Callan Road, San Diego, California that we use for our corporate headquarters and manufacturing facilities. The related lease agreement, as amended, provides for a monthly rent that commenced at a rate of $1.80 per square foot, with an annual increase of $0.05 per square foot. The lease term is 88 months, commenced on July 1, 2010 and expiring on October 31, 2017.
Additionally, we entered into several lease agreements for international office locations. For these properties, we pay an aggregate of approximately $28,000 in rent per month. The lease for the property in Japan will expire in May 2017 and the lease for the property in the United Kingdom will expire in June 2019.
From time to time, we have been involved in routine litigation incidental to the conduct of our business. As of December 31, 2016, we were not a party to any material legal proceeding.
Item 4. Mine Safety Disclosures
Not applicable.
52
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Prices
From August 2000 (our initial public offering in Germany) until September 2007, our common stock was quoted on the Frankfurt Stock Exchange under the symbol “XMPA” (formerly XMP). In September 2007, our stock closed trading on the Frankfurt Stock Exchange. In December 2005, our common stock commenced trading on the NASDAQ Capital Market under the symbol “CYTX.” From December 2005 until February 2006, our common stock traded on the NASDAQ Capital Market, from February 2006 until February 2016, it traded on the NASDAQ Global Market, and since February 2016, it has traded on the NASDAQ Capital Market. Our common stock has, from time to time, traded on a limited, sporadic and volatile basis. The following tables show the high and low sales prices for our common stock for the periods indicated, as reported on the NASDAQ Global Market or the NASDAQ Capital Market, as applicable. These prices do not include retail markups, markdowns or commissions.
Common Stock
|
|
|
High |
|
|
Low |
|
||
|
2015 |
|
|
|
|
|
|
|
|
|
Quarter ended March 31, 2015 |
|
$ |
20.55 |
|
|
$ |
6.60 |
|
|
Quarter ended June 30, 2015 |
|
$ |
20.25 |
|
|
$ |
8.40 |
|
|
Quarter ended September 30, 2015 |
|
$ |
8.25 |
|
|
$ |
4.50 |
|
|
Quarter ended December 31, 2015 |
|
$ |
6.30 |
|
|
$ |
2.85 |
|
|
|
|
|
|
|
|
|
|
|
|
2016 |
|
|
|
|
|
|
|
|
|
Quarter ended March 31, 2016 |
|
$ |
3.30 |
|
|
$ |
1.95 |
|
|
Quarter ended June 30, 2016 |
|
$ |
5.25 |
|
|
$ |
2.00 |
|
|
Quarter ended September 30, 2016 |
|
$ |
2.25 |
|
|
$ |
1.83 |
|
|
Quarter ended December 31, 2016 |
|
$ |
2.00 |
|
|
$ |
1.36 |
|
All of our outstanding shares have been deposited with the Depository Trust & Clearing Corporation (DTCC) since December 9, 2005.
As of January 31, 2016, we had approximately 21 record holders of our common stock. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of individual stockholders represented by these record holders.
Dividends
We have never declared or paid any dividends on our common stock and do not anticipate paying any in the foreseeable future. Furthermore, our Loan and Security Agreement currently prohibits our issuance of cash dividends on common stock.
53
Equity Compensation Plan Information
The following table gives information as of December 31, 2016 about shares of our common stock that may be issued upon the exercise of outstanding options, warrants and rights and shares remaining available for issuance under all of our equity compensation plans:
|
Plan Category |
|
Number of securities to be issued upon exercise of outstanding options, warrants and rights |
|
|
Weighted-average exercise price of outstanding options, warrants and rights |
|
|
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column(a)) |
|
|||
|
|
|
(a) |
|
|
(b) |
|
|
(c) |
|
|||
|
Equity compensation plans approved by security holders (1) |
|
|
7,782 |
|
|
$ |
84.23 |
|
|
|
— |
|
|
Equity compensation plans not approved by security holders (2) |
|
|
230,748 |
|
|
$ |
56.75 |
|
|
|
— |
|
|
Equity compensation plans not approved by security holders (3) |
|
|
364,764 |
|
|
$ |
4.64 |
|
|
|
525,965 |
|
|
Equity compensation plans not approved by security holders (4) |
|
|
33,333 |
|
|
$ |
2.18 |
|
|
|
33,333 |
|
|
Total |
|
|
636,627 |
|
|
$ |
24.37 |
|
|
|
559,298 |
|
|
(1) |
The 1997 Stock Option and Stock Purchase Plan expired in October 2007. |
|
(2) |
The 2004 Stock Option and Stock Purchase Plan expired in August 2014. |
|
(3) |
See Notes to the Consolidated Financial Statements included elsewhere herein for a description of our 2014 Equity Incentive Plan. |
|
(4) |
See Notes to the Consolidated Financial Statements included elsewhere herein for a description of our 2015 New Employee Incentive Plan. |
Comparative Stock Performance Graph
The following graph shows how an initial investment of $100 in our common stock would have compared to an equal investment in the NASDAQ Composite Index and the NASDAQ Biotechnology Index during the period from December 31, 2010 through December 31, 2016. The performance shown is not necessarily indicative of future price performance.
54
Item 6. Selected Financial Data
The selected data presented below under the captions “Statements of Operations Data,” “Statements of Cash Flows Data” and “Balance Sheet Data” for, and as of the end of, each of the years in the two-year period ended December 31, 2016, are derived from, and should be read in conjunction with, our audited consolidated financial statements. The consolidated balance sheets as of December 31, 2016 and 2015, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2016, which have been audited by BDO USA, LLP as of December 31, 2016 and KPMG LLP as of December 31, 2015, which are independent registered public accounting firms, and their reports thereon, are included elsewhere in this Annual Report.
The information contained in this table should also be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and related notes thereto included elsewhere in this report:
Consolidated Statements of Operations and Comprehensive Loss (in thousands)
|
|
|
For the Years Ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Product revenues |
|
$ |
4,656 |
|
|
$ |
4,838 |
|
|
Cost of product revenues |
|
|
2,715 |
|
|
|
3,186 |
|
|
Gross profit |
|
|
1,941 |
|
|
|
1,652 |
|
|
Development revenues: |
|
|
|
|
|
|
|
|
|
Government contracts and other |
|
|
6,724 |
|
|
|
6,821 |
|
|
|
|
|
6,724 |
|
|
|
6,821 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
|
|
16,197 |
|
|
|
19,000 |
|
|
Sales and marketing |
|
|
3,611 |
|
|
|
2,662 |
|
|
General and administrative |
|
|
8,563 |
|
|
|
9,765 |
|
|
Change in fair value of warrant liabilities |
|
|
— |
|
|
|
(7,668 |
) |
|
Total operating expenses |
|
|
28,371 |
|
|
|
23,759 |
|
|
Operating loss |
|
|
(19,706 |
) |
|
|
(15,286 |
) |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
Loss on debt extinguishment |
|
|
— |
|
|
|
(260 |
) |
|
Interest income |
|
|
19 |
|
|
|
9 |
|
|
Interest expense |
|
|
(2,592 |
) |
|
|
(3,379 |
) |
|
Other income, net |
|
|
233 |
|
|
|
172 |
|
|
Total other expense |
|
|
(2,340 |
) |
|
|
(3,458 |
) |
|
Net loss |
|
$ |
(22,046 |
) |
|
$ |
(18,744 |
) |
|
Beneficial conversion feature for convertible preferred stock |
|
|
— |
|
|
|
(661 |
) |
|
Net loss allocable to common stockholders |
|
$ |
(22,046 |
) |
|
$ |
(19,405 |
) |
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share allocable to common stockholders |
|
$ |
(1.28 |
) |
|
$ |
(2.07 |
) |
|
Basic and diluted weighted average shares used in calculating net loss per share allocable to common stockholders |
|
|
17,290,933 |
|
|
|
9,386,488 |
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(22,046 |
) |
|
$ |
(18,744 |
) |
|
Other comprehensive income – foreign currency translation adjustments |
|
|
262 |
|
|
|
296 |
|
|
Comprehensive loss |
|
$ |
(21,784 |
) |
|
$ |
(18,448 |
) |
55
Consolidated Statements of Cash Flows (in thousands)
|
|
|
For the Years Ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Net cash used in operating activities |
|
$ |
(19,533 |
) |
|
$ |
(20,468 |
) |
|
Net cash provided by (used in) used in investing activities |
|
|
64 |
|
|
|
(613 |
) |
|
Net cash provided by financing activities |
|
|
17,609 |
|
|
|
20,797 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
82 |
|
|
|
— |
|
|
Net decrease in cash and cash equivalents |
|
|
(1,778 |
) |
|
|
(284 |
) |
|
Cash and cash equivalents at beginning of year |
|
|
14,338 |
|
|
|
14,622 |
|
|
Cash and cash equivalents at end of year |
|
$ |
12,560 |
|
|
$ |
14,338 |
|
Consolidated Balance Sheet Details (in thousands)
|
|
|
As of December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Cash and cash equivalents |
|
$ |
12,560 |
|
|
$ |
14,338 |
|
|
Working capital |
|
|
6,246 |
|
|
|
12,806 |
|
|
Total assets |
|
|
34,609 |
|
|
|
37,698 |
|
|
Deferred revenues |
|
|
97 |
|
|
|
105 |
|
|
Long-term deferred rent and other |
|
|
17 |
|
|
|
269 |
|
|
Long-term obligations, net of discount, less current portion |
|
|
11,008 |
|
|
|
16,681 |
|
|
Total stockholders’ equity |
|
|
10,986 |
|
|
|
12,206 |
|
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Overview
We develop cellular therapeutics uniquely formulated and optimized for specific diseases and medical conditions and related products. Lead therapeutics in our pipeline are currently targeted for impaired hand function in scleroderma, osteoarthritis of the knee, stress urinary incontinence, and deep thermal burns including those complicated by radiation exposure.
Our cellular therapeutics are collectively known by the trademarked name, Cytori Cell Therapy, and consist of a mixed population of specialized cells including stem cells that are involved in response to injury, repair and healing. These cellular therapeutics are extracted from an adult patient’s own adipose (fat) tissue using our fully automated Celution System, which includes a device, proprietary enzymes, and sterile consumable sets utilized at the point-of-therapeutic application or potentially at an off-site processing center. Cytori Cell Therapy can either be administered to the patient the same day or cryopreserved for future use.
Our primary near-term goal is for Cytori Cell Therapy to be the first cell therapy to market for the treatment of impaired hand function in scleroderma, through Cytori-sponsored and supported clinical development efforts. The STAR trial is a 48-week, randomized, double blind, placebo-controlled Phase III pivotal clinical trial of 80 patients in the U.S. The trial evaluates the safety and efficacy of a single administration of Cytori Cell Therapy (ECCS-50) in scleroderma patients affecting the hands and fingers. The first sites for the scleroderma study were initiated in July 2015 and completed enrollment of 88 patients in June 2016. We anticipate that we will receive 48-week follow-up data on this Phase III pivotal clinical trial in mid-2017.
With respect to the remainder of our clinical pipeline, we received Investigational Device Exemption, or IDE, approval from the U.S. Food and Drug Administration, or the FDA, in late 2014 for our Phase II ACT-OA osteoarthritis study and in early 2015 we initiated this study, and enrollment was completed in June 2015. The 48-week analysis was performed as planned and the top-line data are described in the “Osteoarthritis” section below. In July 2015, a Company-supported male stress urinary incontinence, or SUI, trial in Japan for male prostatectomy patients (after prostate surgery) received approval to begin enrollment from the Japanese Ministry of Health, Labor and Welfare, or MHLW. Patient enrollment is ongoing. Partial funding of this study is granted by AMED (Japan Agency for Medical Research and Development). The goal of this investigator-initiated trial is to gain regulatory approval in Japan of Cytori Cell Therapy for this indication. We are also developing a treatment for thermal burns combined with radiation injury under a contract from the Biomedical Advanced Research Development Authority, or BARDA, a division of the U.S. Department of Health and Human Services. We are also exploring other development opportunities in a variety of other conditions.
56
In addition to our targeted therapeutic development, we have continued to commercialize our Cytori Cell Therapy technology under select medical device approvals, clearances and registrations to research and commercial customers in Europe, Japan and other regions. Many of these customers are research customers evaluating new therapeutic applications of Cytori Cell Therapy. The sale of systems, consumables and ancillary products contributes a margin that partially offsets our operating expenses and will continue to play a role in fostering familiarity within the medical community with our technology. These sales have also facilitated the discovery of new applications for Cytori Cell Therapy by customers conducting investigator-initiated and funded research.
Lead Indication: Scleroderma
Scleroderma is a rare and chronic autoimmune disorder associated with fibrosis of the skin, and destructive changes in blood vessels and multiple organ systems as the result of a generalized overproduction of collagen. Scleroderma affects approximately 50,000 patients in the U.S. (women are affected four times more frequently than men) and is typically detected between the ages of 30 and 50. More than 90 percent of scleroderma patients have hand involvement that is typically progressive and can result in chronic pain, blood flow changes and severe dysfunction. The limited availability of treatments for scleroderma may provide some benefit but do little to modify disease progression or substantially improve symptoms. Treatment options are directed at protecting the hands from injury and detrimental environmental conditions as well as the use of vasodilators. When the disease is advanced, immunosuppressive and other medications may be used but are often accompanied by significant side effects.
In January 2015, the FDA granted IDE approval for a pivotal clinical trial, named the “STAR” trial, to evaluate Cytori Cell Therapy as a potential treatment for impaired hand function in scleroderma. The STAR trial is a 48-week, randomized, double blind, placebo-controlled pivotal clinical trial of 88 patients in the U.S. The trial evaluates the safety and efficacy of a single administration of Habeo Cell Therapy in patients with scleroderma affecting the hands and fingers. The STAR trial uses the Cochin Hand Function Scale, or CHFS, a validated measure of hand function, as the primary endpoint measured at six months after a single administration of Habeo Cell Therapy or placebo. Patients in the placebo group will be eligible for crossover to the active arm of the trial after all patients have completed 48 weeks of follow up. In February 2015, the FDA approved our request to increase the number of investigational sites from 12 to up to 20. The increased number of sites served to broaden the geographic coverage of the trial and facilitate trial enrollment. The enrollment of this trial began in August 2015 and was completed at 88 patients in June 2016. We anticipate that we will receive 48-week follow-up data on this Phase III pivotal clinical trial in mid-2017.
The STAR trial is predicated on a completed investigator-initiated pilot 12-patient, open-label Phase I trial performed in France termed SCLERADEC I. The SCLERADEC I trial received partial support from Cytori. The six-month results were published in the Annals of the Rheumatic Diseases in May 2014 and demonstrated approximately a 50 percent improvement at six months across four important and validated endpoints used to assess the clinical status in patients with scleroderma with impaired hand function. Patients perceived their health status to be improved as shown by a 45.2% and 42.4% decrease of the Scleroderma Health Assessment Questionnaire, or SHAQ, at month 2 (p=0.001) and at month 6 (p=0.001), respectively. A 47% and 56% decrease of the CHFS at month 2 and month 6 in comparison to baseline was observed (p<0.001 for both). Grip strength increased at month 6 with a mean improvement of +4.8±6.4 kg for the dominant hand (p=0.033) and +4.0±3.5 kg for the non-dominant hand (p=0.002). Similarly, an increase in pinch strength at month 6 was noted with a mean improvement of +1.0±1.1 kg for the dominant hand (p=0.009) and +0.8±1.2 kg for the non-dominant hand (p=0.050). Among subjects having at least one digital ulcer, or DU, at inclusion, total number of DU decreased, from 15 DUs at baseline, 10 at month 2 and 7 at month 6. The average reduction of the Raynaud’s Condition Score from baseline was 53.7% at month 2 (p<0.001) and 67.5% at month 6 (p<0.001). Hand pain showed a significant decrease of 63.6% at month 2 (p=0.001) and 70% at month 6 (p<0.001). One year results were published in September 2015 in the journal Rheumatology. Relative to baseline, the CHFS and the SHAQ improved by 51.3% and 46.8%, respectively (p<0.001 for both). The Raynaud’s score improved by 63.2% from baseline (p<0.001). Other findings at one-year included a 30.5% improvement in grip strength (p=0.002) and a 34.5% improvement in hand pain (p=0.052). In February 2016, two-year follow up data in the SCLERADEC I trial was presented at the Systemic Sclerosis World Congress, which demonstrated sustained improvement in the following four key endpoints: Cochin Hand Function Score (CHFS), Scleroderma Health Assessment Questionnaire, Raynaud’s Condition Score (which assesses severity of Raynaud’s Phenomenon), and hand pain, as assessed by a standard visual analogue scale. The major findings at 24 months following a single administration of ECCS-50 were as follows:
|
|
• |
Hand dysfunction assessed by the CHFS, showed a 62% reduction in hand dysfunction at two years (p<0.001). |
|
|
• |
Raynaud’s Condition Score decreased by an average of 89% over baseline at two years (p<0.001). |
|
|
• |
Hand pain, as measured by a 100 mm Visual Analogue Scale, and the Scleroderma Health Assessment Questionnaire (SHAQ) score at two years both showed improvement of 50% over baseline (p=0.01 and p<0.001, respectively). |
|
|
• |
Improvement of 20% in grip strength and 330% in pinch strength at two years (p=0.05 and p=0.004, respectively). |
|
|
• |
Continued reduction in the number of ulcers from 15 at baseline to 9 at one year and 6 at two years. |
57
In 2014, Drs. Guy Magalon and Brigitte Granel, under the sponsorship of the Assistance Publique - Hôpitaux de Marseille, submitted a study for review for a follow-up Phase III randomized, double-blind, placebo-controlled trial in France using Cytori Cell Therapy, to be supported by Cytori. The trial name is SCLERADEC II and was approved by the French government in April 2015. Enrollment of this trial commenced in October 2015 and is ongoing. Patients will be followed for a 6-month post-procedure.
In April 2016, the European Commission, acting on the positive recommendation from the European Medicines Agency Committee for Orphan Medicinal Products, issued orphan drug designation to a broad range of Cytori Cell Therapy formulations when used for the treatment of hand dysfunction and Raynaud’s Phenomenon in patients with scleroderma under Community Register of Orphan Medicinal Products number EU/3/16/1643. In November 2016, the US FDA Office of Orphan Products Development (OOPD) granted Cytori an orphan drug designation for cryopreserved or centrally processed ECCS-50 (HABEO) for scleroderma.
Osteoarthritis
Osteoarthritis is a disease of the entire joint involving the cartilage, joint lining, ligaments and underlying bone. The breakdown of tissue leads to pain, joint stiffness and reduced function. It is the most common form of arthritis and affects an estimated 13.9% of US adults over the age of 25, and 33.6% of U.S. adults over the age of 65. Current treatments include physical therapy, non-steroidal anti-inflammatory medications, viscosupplement injections, and total knee replacement. A substantial medical need exists as present medications have limited efficacy and joint replacement is a relatively definitive treatment for those with the most advanced disease.
In the later part of 2014, we received approval by the FDA to begin an exploratory U.S. IDE pilot (Phase II) trial of Cytori Cell Therapy (ECCO-50) in patients with osteoarthritis of the knee. The trial, called ACT-OA, is a 94-patient, randomized, double-blind, placebo controlled study involving two doses of Cytori Cell Therapy, a low dose and a high dose, and was conducted over 48 weeks. The randomization is 1:1:1 between the control, low and high dose groups. Enrollment on this trial began in February 2015 and was completed in June 2015. The goal of this proof-of-concept trial is to help determine: (1) safety and feasibility of the ECCO-50 therapeutic for osteoarthritis, (2) provide dosing guidance and (3) explore key trial endpoints useful for a Phase III trial.
Top-line analysis of the final 48-week data has recently been completed. The primary objective of this prospective, randomized, placebo controlled study was to evaluate the safety and feasibility of intraarticular injection of Celution prepared adipose-derived regenerative cells injected into knees of patients with chronic knee pain due to osteoarthritis. A total of 94 patients were randomized (33 placebo, 30 low dose ECCS-50, 31 high dose ECCS-50). In general, a clear difference between low and high dose ECCS-50 was not observed and therefore the data for both groups have been combined. Numerous endpoints were evaluated that can be summarized as follows:
|
|
• |
Intraarticular application of a single dose of ECCO-50 is feasible in an outpatient day-surgery setting; no serious adverse events were reported related to the fat harvest, cell injection or to the cell therapy. |
|
|
• |
Consistent trends observed in most secondary endpoints at 12, 24 and 48 weeks in the target knee of the treated group relative to placebo control group; 12-week primary endpoint of single pain on walking question did not achieve statistical significance. |
|
|
• |
Consistent trends observed in all 6 pre-specified MRI Osteoarthritis Knee Score (MOAKS) classification scores suggesting decrease in target knee joint pathologic features at 48 weeks for the treated group relative to placebo control group. The differences against placebo favored ADRCs specifically in the number of bone marrow lesions, the percentage of the bone marrow lesion that is not a cyst, the size of the bone marrow lesions as a percentage of the total sub-region volume, percentage of full thickness cartilage loss, cartilage loss as a percentage of cartilage surface area and the size of the largest osteophyte. |
In summary, the ACT-OA Phase II trial demonstrated feasibility of same day fat harvesting, cell processing and intraarticular administration of autologous ADRCs (ECCO-50) with a potential for a cell benefit effect. Additional analyses are ongoing. The accumulated data and experienced gained will be critical in considering designs of further clinical trials in osteoarthritis and other potential indications. As well, the multicenter nature of the trial in the United States provides relevant information as to optimizing commercialization.
Stress Urinary Incontinence
Another therapeutic target under evaluation by Cytori in combination with the University of Nagoya and the Japanese MHLW is stress urinary incontinence in men following surgical removal of the prostate gland, which is based on positive data reported in a peer reviewed journal resulting from the use of ADRCs prepared by our Celution System. The ADRESU trial is a 45 patient, investigator-initiated, open-label, multi-center, single arm trial that was approved by the Japanese MHLW in July 2015 and is being led by both Momokazu Gotoh, MD, Ph.D., Professor and Chairman of the Department of Urology and Tokunori Yamamoto, MD, Ph.D.,
58
Associate Professor Department of Urology at University of Nagoya Graduate School of Medicine. Trial enrollment began trial in September 2015, and in December 2016, the trial was 50% enrolled. This clinical trial is primarily sponsored and funded by the Japanese government, including a grant provided by AMED.
Cutaneous and Soft Tissue Thermal and Radiation Injuries
Cytori Cell Therapy is also being developed for the treatment of thermal burns combined with radiation injury. In the third quarter of 2012, we were awarded a contract valued at up to $106 million with BARDA to develop a medical countermeasure for thermal burns. The initial base period included $4.7 million over two years and covered preclinical research and continued development of Cytori’s Celution System to improve cell processing.
In 2014, an in-process review meeting was held with BARDA at which Cytori confirmed completion of the objectives of the initial phase of the contract. In August 2014, BARDA exercised contract option 1 in the amount of approximately $12 million. In December 2014 and September 2016, the option 1 was supplemented with an additional $2 million and $2.5 million in funds, respectively. This funded continuation of research, regulatory, clinical and other activities required for submission of an Investigational Device Exemption, or IDE, request to the FDA for a pilot clinical trial using Cytori Cell Therapy (DCCT-10) for the treatment of thermal burns. We anticipate that we will receive IDE approval in the first half of 2017 to execute this pilot clinical trial. Upon receipt of IDE approval, if granted, we anticipate that BARDA will provide funding to cover costs associated with execution of the clinical trial and related activities, currently estimated to be between $8.0 million and $12.0 million.
Our contract with BARDA contains two additional options to fund a pivotal clinical trial and additional preclinical work in thermal burn complicated by radiation exposure. These options are valued at up to $45 million and $23 million, respectively.
The total award under the BARDA contract is intended to support all clinical, preclinical, regulatory and technology development activities needed to complete the FDA approval process for use of DCCT-10 in thermal burn injury under a device-based PMA regulatory pathway and to provide preclinical data in burn complicated by radiation exposure.
Other Clinical Indications
Heart failure due to ischemic heart disease does not represent a current clinical target for us at this time. Our ATHENA and ATHENA II trials related to that indication were truncated and we have minimized expenses related to initiatives in this area. While the safety data from these trial programs will be used for regulatory support for our other indications and also for publication in peer reviewed forums, we are not actively pursuing indications related to these trials. The 12 month results of the ATHENA Trials were presented by the investigators at the Society of Cardiac Angiography and Interventions Annual Scientific Meeting on May 5, 2016 and data was published in the Catheterization and Cardiovascular Interventions journal in June 2016.
Results of Operations
Product revenues
Product revenues consisted of revenues primarily from the sale of our Cytori Cell Therapy-related products.
The following table summarizes the components for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Product revenues - third party |
|
$ |
4,656 |
|
|
$ |
4,838 |
|
A majority of our product revenue in 2016 and 2015 was derived from Japan. Two new regenerative medicine laws in Japan went into effect in November 2014, removing regulatory uncertainties and providing a clear path for us to offer Cytori Cell Therapy in Japan, where our technology is mainly being used in the aesthetics and orthopedic fields. Further, we expect continued demand from researchers at academic hospitals seeking to perform investigator-initiated and funded studies.
We experienced a decrease of $0.2 million in product revenue during the year ended December 31, 2016 as compared to the same period in 2015, due to decreased revenues in Asia Pacific of $0.7 million, primarily due to the opening order from Lorem Vascular in the second quarter of 2015 and lack of ongoing orders in subsequent periods and decreased revenue in EMEA of $0.3 million, but partially offset by increased revenues in Japan of $0.9 million due to continued adoption of Cytori Cell Therapy primarily in the aesthetic and osteoarthritis business.
59
The future: We expect to continue to generate a majority of product revenues from the sale of Cytori Cell Therapy-related products to researchers, clinicians, and distributors in EMEA, Japan, Asia Pacific, and the Americas. In Japan and EMEA, researchers will use our technology in ongoing and new investigator-initiated and funded studies focused on, but not limited to, hand scleroderma, Crohn’s disease, peripheral artery disease, erectile dysfunction, and diabetic foot ulcers. Habeo Cell Therapy for hand scleroderma will continue to be accessible to patients and physicians through a managed access program, or MAP, that we initiated in EMEA in 2016. In the Americas, Cytori’s partner, Kerastem, is utilizing the Cytori Cell Therapy technology as part of its FDA-approved STYLE trial for patients with alopecia, or hair loss. Overall, we expect 2017 product revenues to remain relatively consistent with 2016.
Cost of product revenues
Cost of product revenues relate primarily to Cytori Cell Therapy-related products and includes material, manufacturing labor, and overhead costs, as well as amortization of intangible assets. The following table summarizes the components of our cost of revenues for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Cost of product revenues |
|
$ |
2,128 |
|
|
$ |
2,745 |
|
|
Amortization of intangible assets |
|
|
546 |
|
|
|
362 |
|
|
Share-based compensation |
|
|
41 |
|
|
|
79 |
|
|
Total cost of product revenues |
|
$ |
2,715 |
|
|
$3,186 |
|
|
|
Total cost of product revenues as % of product revenues |
|
|
58 |
% |
|
|
66 |
% |
Cost of product revenues as a percentage of product revenues was 58% and 66% for the years ended December 31, 2016 and 2015, respectively. Fluctuation in this percentage is due to the product mix, distributor and direct sales mix, geographic mix, foreign exchange rates and allocation of overhead.
The future: We expect to continue to see variation in our gross profit margin as the product mix, distributor and direct sales mix and geographic mix comprising revenues fluctuate. We are investigating various pricing options for our cellular therapeutics, including orphan pricing for our Habeo Cell Therapy, which may help to increase our gross profit margins in 2017 and beyond.
Development revenues
Under our government contract with BARDA, we recognized a total of $6.7 million and $6.8 million in development revenues for the years ended December 31, 2016 and 2015, respectively which included allowable fees as well as cost reimbursements. During both of the years ended December 31, 2016 and 2015, we incurred $6.3 million in qualified expenditures. The decrease in revenues for the years ended December 31, 2016 as compared to the same periods in 2015 is primarily due to slight decreases in research and development activities related to our contact with BARDA.
The future: Our current contract with BARDA expires in April 2017. We are in the process of negotiating an extension of the current contract option (which will expire in mid-April) for initiation of a pilot clinical trial of DCCT-10 in thermal burn injury.
Research and development expenses
Research and development expenses relate to the development of a technology platform that involves using adipose tissue as a source of autologous regenerative cells for therapeutic applications as well as the continued development efforts related to our clinical trials.
Research and development expenses include costs associated with the design, development, testing and enhancement of our products, payment of regulatory fees, laboratory supplies, pre-clinical studies and clinical studies.
60
The following table summarizes the components of our research and development expenses for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
General research and development |
|
$ |
15,846 |
|
|
$ |
18,442 |
|
|
Share-based compensation |
|
|
351 |
|
|
|
558 |
|
|
Total research and development expenses |
|
$ |
16,197 |
|
|
$ |
19,000 |
|
The decrease in research and development expenses, excluding share-based compensation for the year ended December 31, 2016 as compared to the same period in 2015 is due to a decrease of approximately $2.6 million in clinical studies and related professional services as well as a decrease in salaries and benefits as a result of a decrease in the number of the U.S. clinical trials enrolling from two trials in 2015 to one trial in 2016.
The future: We expect aggregate research and development expenditures to increase in 2017 as we incur development costs in preparation of Habeo U.S. PMA filing submission, our development efforts of the recently acquired assets from Azaya Therapeutics, and ongoing activities of the U.S. STAR clinical trial.
Sales and marketing expenses
Sales and marketing expenses include costs of sales and marketing personnel, events and tradeshows, customer and sales representative education and training, primary and secondary market research, and product and service promotion. The following table summarizes the components of our sales and marketing expenses for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Sales and marketing |
|
$ |
3,444 |
|
|
$ |
2,552 |
|
|
Share-based compensation |
|
|
167 |
|
|
|
110 |
|
|
Total sales and marketing expenses |
|
$ |
3,611 |
|
|
$ |
2,662 |
|
Sales and marketing expenses excluding share-based compensation increased by approximately $0.9 million for the year ended December 31, 2016 as compared to the same period in 2015 due to increases in salary and related benefits expense and professional services mostly related to our operations in Japan, commercial planning activities for scleroderma in the U.S. and investments in the EMEA managed access program.
The future: We expect sales and marketing expenditures to slightly increase during the first half of 2017. These expenditures will have a greater increase in the second half of 2017 as we prepare for commercial readiness for hand scleroderma in the U.S. and knee osteoarthritis, aesthetics and stress urinary incontinence in Japan.
General and administrative expenses
General and administrative expenses include costs for administrative personnel, legal and other professional expenses, and general corporate expenses. The following table summarizes the general and administrative expenses for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
General and administrative |
|
$ |
8,042 |
|
|
$ |
8,471 |
|
|
Share-based compensation |
|
|
521 |
|
|
|
1,294 |
|
|
Total general and administrative expenses |
|
$ |
8,563 |
|
|
$9,765 |
|
|
General and administrative expenses excluding share-based compensation decreased by $0.4 million for the year ended December 31, 2016, as compared to the same period in 2015 primarily due to decreases in salary and related benefits expense and professional services consistent with our ongoing cost curtailment efforts.
61
The future: We expect general and administrative expenditures to increase significantly with the acquisition of Azaya assets and as we integrate its operations under the Cytori Therapeutics umbrella.
Share-based compensation expenses
Share-based compensation expenses include charges related to options and restricted stock awards issued to employees, directors and non-employees along with charges related to the employee stock purchases under the Employee Stock Purchase Plan, or ESPP. We measure stock-based compensation expense based on the grant-date fair value of any awards granted to our employees. Such expense is recognized over the requisite service period.
The following table summarizes the components of our share-based compensation for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Cost of product revenues |
|
$ |
41 |
|
|
$ |
79 |
|
|
Research and development-related |
|
|
351 |
|
|
|
558 |
|
|
Sales and marketing-related |
|
|
167 |
|
|
|
110 |
|
|
General and administrative-related |
|
|
521 |
|
|
|
1,294 |
|
|
Total share-based compensation |
|
$ |
1,080 |
|
|
$ |
2,041 |
|
The decrease in share-based compensation expenses for the year ended December 31, 2016 as compared to the same period in 2015 is primarily related to a lower annual grant activities caused by reductions in headcount and due to the decline in the stock price during 2016 as compared to the same period in 2015, and its corresponding impact on share-based compensation.
The future: We expect to continue to grant options and stock awards (which will result in an expense) to our employees, directors, and, as appropriate, to non-employee service providers. In addition, previously-granted options will continue to vest in accordance with their original terms. As of December 31, 2016, the total compensation cost related to non-vested stock options and stock awards not yet recognized for all our plans is approximately $1.0 million which is expected to be recognized as a result of vesting under service conditions over a weighted average period of 1.6 years.
Change in fair value of warrant liability
The following is a table summarizing the change in fair value of warrant liability for the years ended December 31, 2016 and 2015:
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Change in fair value of warrant liability |
|
$ |
— |
|
|
$ |
(7,668 |
) |
The decrease in fair value of our warrant liability for the year ended December 31, 2016 as compared to the same period in 2015 is due to the fact that all warrants with price reset features accounted for as liabilities were cashless exercised during the year ended December 31, 2015.
The future: We do not expect any further changes in fair value of warrant liability, as all of our outstanding warrants with exercise price reset features were settled during December 2015.
62
Financing items
The following table summarizes loss on debt extinguishment, interest income, interest expense, and other income and expense for the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Loss on debt extinguishment |
|
$ |
— |
|
|
$ |
(260 |
) |
|
Interest income |
|
|
19 |
|
|
|
9 |
|
|
Interest expense |
|
|
(2,592 |
) |
|
|
(3,379 |
) |
|
Other income, net |
|
|
233 |
|
|
|
172 |
|
|
Total |
|
$ |
(2,340 |
) |
|
$ |
(3,458 |
) |
|
|
• |
In connection with the Loan and Security Agreement, a loss on debt extinguishment was recorded that relates to the payoff of the prior loan obligations. |
|
|
• |
Interest expense decreased for the year ended December 31, 2016 as compared to the same period in 2015, due to partial pay down and refinance of principal loan balance in May 2015. |
|
|
• |
The changes in other income during the year ended December 31, 2016 as compared to the same periods in 2015 resulted primarily from changes in exchange rates related to transactions in foreign currency. |
The future: We expect interest expense in 2017 to decrease as we begin making payments on the principal balance of the Loan and Security Agreement.
Liquidity and Capital Resources
Short-term and long-term liquidity
The following is a summary of our key liquidity measures at December 31, 2016 and 2015 (in thousands):
|
|
|
As of December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Cash and cash equivalents |
|
$ |
12,560 |
|
|
$ |
14,338 |
|
|
|
|
|
|
|
|
|
|
|
|
Current assets |
|
$ |
18,747 |
|
|
$ |
21,243 |
|
|
Current liabilities |
|
|
12,501 |
|
|
|
8,437 |
|
|
Working capital |
|
$ |
6,246 |
|
|
$ |
12,806 |
|
We incurred net losses of $22.0 million and $18.7 million for the years ended December 31, 2016 and 2015, respectively. We have an accumulated deficit of $379.1 million as of December 31, 2016. Additionally, we have used net cash of $19.5 million and $20.5 million to fund our operating activities for the years ended December 31, 2016 and 2015, respectively.
To date, these operating losses have been funded primarily from outside sources of invested capital including our recently completed Lincoln Park Purchase Agreement, the Rights Offering (as defined below), our at-the-market or ATM offering program, the Loan and Security Agreement and gross profits. We have had, and we will likely continue to have, an ongoing need to raise additional cash from outside sources to fund our future clinical development programs and other operations.
On June 15, 2016, we closed the Rights Offering originally filed under a Form S-1 registration statement in April 2016. Pursuant to the Rights Offering, we sold an aggregate of 6,704,852 units consisting of a total of 6,704,852 shares of common stock and 3,352,306 warrants, with each warrant exercisable for one share of common stock at an exercise price of $3.06 per share, resulting in total net proceeds of $15.3 million.
63
During 2016, we sold 1,840,982 shares of our common stock under our ATM offering program, receiving total net proceeds of approximately $4.4 million. Although sales of our common stock have taken place pursuant to our ATM offering program, there can be no assurance that we will be successful in consummating future sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. In addition, under current SEC regulations, at any time during which the aggregate market value of our common stock held by non-affiliates, or public float, is less than $75.0 million, the amount we can raise through primary public offerings of securities in any twelve-month period using shelf registration statements, including sales under our ATM offering program, is limited to an aggregate of one-third of our public float. As of December 31, 2016, our public float was 21.5 million shares, the value of which was $32.5 million based upon the closing price of our common stock of $1.51 on such date. The value of one-third of our public float calculated on the same basis was approximately $11.0 million.
On December 22, 2016, we entered into the Lincoln Park Purchase Agreement and a registration rights agreement, with Lincoln Park pursuant to which we have the right to sell to Lincoln Park and Lincoln Park is obligated to purchase up to $20.0 million in amounts of shares, of our common stock, over the 30-month period commencing on the date that a registration statement, that we filed with the Securities and Exchange Commission (the “SEC”) in December 2016. We may direct Lincoln Park, at its sole discretion and subject to certain conditions, to purchase up to 100,000 shares of common stock on any business day but in no event will the amount of a single Regular Purchase exceed $1.0 million. The purchase price of shares of common stock related to the Regular Purchases will be based on the prevailing market prices of such shares at the time of sales. Our sales of shares of common stock to Lincoln Park under the Lincoln Park Purchase Agreement are limited to no more than the number of shares that would result in the beneficial ownership by Lincoln Park and its affiliates, at any single point in time, of more than 9.99% of the then outstanding shares of the common stock. There are no trading volume requirements or restrictions under the Lincoln Park Purchase Agreement. There is no upper limit on the price per share that Lincoln Park must pay for common stock under a Regular Purchase or an accelerated purchase and in no event will shares be sold to Lincoln Park on a day our closing price is less than the floor price of $0.50 per share as set forth in the Lincoln Park Purchase Agreement. On December 22, 2016, we issued to Lincoln Park 127,419 shares of common stock as commitment shares in consideration for entering into the Lincoln Park Purchase Agreement. We will issue up to an additional 382,258 shares of common stock on a pro rata basis to Lincoln Park only as and when shares are sold under the Lincoln Park Purchase Agreement to Lincoln Park. To date, we sold no shares under the Lincoln Park Purchase Agreement to Lincoln Park.
Pursuant to these securities transactions and related equity issuances, as well as anticipated gross profits and potential outside sources of capital, we believe we have sufficient cash to fund operations through at least through Q2 2017. We continue to seek additional capital through product revenues, strategic transactions, including extension opportunities under the awarded BARDA contract, and from other financing alternatives. However, there can be no assurance that we will be successful in securing additional resources when needed, on terms acceptable to us or at all. Therefore, there exists substantial doubt about our ability to continue as a going concern.
The accompanying consolidated financial statements have been prepared assuming we will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to our ability to continue as a going concern.
The following summarizes our contractual obligations and other commitments at December 31, 2016, and the effect such obligations could have on our liquidity and cash flow in future periods (in thousands):
|
|
|
Payments due by period |
|
|||||||||||||||||
|
Contractual Obligations |
|
Total |
|
|
Less than 1 year |
|
|
1 – 3 years |
|
|
3 – 5 years |
|
|
More than 5 years |
|
|||||
|
Long-term obligations |
|
$ |
18,789 |
|
|
$ |
7,080 |
|
|
$ |
11,709 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Interest commitment on long-term obligations |
|
|
3,162 |
|
|
|
1,311 |
|
|
|
1,851 |
|
|
|
— |
|
|
|
— |
|
|
Operating lease obligations |
|
|
1,847 |
|
|
|
1,782 |
|
|
|
65 |
|
|
|
— |
|
|
|
— |
|
|
Minimum purchase obligation |
|
|
6,567 |
|
|
|
1,074 |
|
|
|
2,547 |
|
|
|
2,946 |
|
|
|
— |
|
|
Clinical research study obligations |
|
|
3,329 |
|
|
|
3,220 |
|
|
|
109 |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
33,694 |
|
|
$ |
14,467 |
|
|
$ |
16,281 |
|
|
$ |
2,946 |
|
|
$ |
— |
|
64
Cash (used in) provided by operating, investing and financing activities for the years ended December 31, 2016 and 2015 is summarized as follows (in thousands):
|
|
|
Years Ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Net cash used in operating activities |
|
$ |
(19,533 |
) |
|
$ |
(20,468 |
) |
|
Net cash provided by (used in) investing activities |
|
|
64 |
|
|
|
(613 |
) |
|
Net cash provided by financing activities |
|
|
17,609 |
|
|
|
20,797 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
82 |
|
|
|
— |
|
|
Net decrease in cash and cash equivalents |
|
$ |
(1,778 |
) |
|
$ |
(284 |
) |
Operating activities
Net cash used in operating activities for the year ended December 31, 2016 was $19.5 million. Overall, our operational cash use decreased during the year ended December 31, 2016 as compared to the same period in 2015 due primarily to a decrease in losses from operations (when adjusted for non-cash items) of $3.3 million offset by working capital givebacks of approximately $2.1 million.
Investing activities
Net cash provided by investing activities for the year ended December 31, 2016 resulted from $0.1 million in proceeds from sale of assets offset by cash outflows for purchases of property and equipment of $0.1 million. This cash outflow for purchases of property and equipment was $0.5 million lower than the same period in 2015 due to cash outflow reduction efforts implemented throughout 2016.
Financing Activities
The net cash provided by financing activities for the year ended December 31, 2016 related primarily to a sale of common stock through our Rights Offering and ATM offering program. The cash inflow from financing activities was approximately $3.2 million lower than the same period in 2015, primarily due to the fact that there was $7.3 million less in capital raised during the year ended December 31, 2016 as compared to the same period in 2015, a decrease of $4.9 million in proceeds for exercised warrants, an increase of $0.2 million in Joint Venture purchase payments to Olympus Corporation, and $9.2 million decrease in principal payments on long-term obligations and loan fees.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements (as defined by applicable regulations of the SEC) that are reasonably likely to have a current or future material effect on our financial condition, results of operations, liquidity, capital expenditures or capital resources.
Critical Accounting Policies and Significant Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires us to make estimates and assumptions that affect the reported amounts of our assets, liabilities, revenues and expenses, and that affect our recognition and disclosure of contingent assets and liabilities.
While our estimates are based on assumptions we consider reasonable at the time they were made, our actual results may differ from our estimates, perhaps significantly. If results differ materially from our estimates, we will make adjustments to our financial statements prospectively as we become aware of the necessity for an adjustment.
We believe it is important for you to understand our most critical accounting policies. These are our policies that require us to make our most significant judgments and, as a result, could have the greatest impact on our future financial results.
65
Revenue Recognition
In accordance with the Securities and Exchange Commission’s guidance, we recognize revenue from product sales when the following fundamental criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery has occurred, (iii) the price to the customer is fixed or determinable and (iv) collection of the resulting accounts receivable is reasonably assured. For customers that have not developed a sufficient payment history with us or for whom a letter of credit is not in place at the time of the transaction, we defer revenues until collectability is reasonably assured.
For all sales, we use a binding purchase order or a signed agreement as evidence of an arrangement. If the other revenue recognition criteria are met, revenue for these product sales is recognized upon delivery to the customer, as all risks and rewards of ownership have been substantively transferred to the customer at that point. For sales to customers who arrange for and manage the shipping process, we recognize revenue upon shipment from our facilities. Shipping and handling costs that are billed to our customers are classified as revenue. The customer’s obligation to pay and the payment terms are set at the time of delivery and are not dependent on the subsequent use or resale of our products. For sales where all revenue recognition criteria are not met, revenue is deferred and related inventory remains on our books.
Accounts Receivable
Accounts receivable are recorded at the invoiced amount and do not bear interest. Amounts collected on accounts receivable are included in net cash provided by operating activities in the consolidated statements of cash flows. The Company maintains an allowance for doubtful accounts for estimated losses inherent in its accounts receivable portfolio. In establishing the required allowance, management considers historical losses adjusted to take into account current market conditions and our customers’ financial condition, the amount of receivables in dispute, and the current receivables aging and current payment patterns. Account balances are charged off against the allowance after all means of collection have been exhausted and the potential for recovery is considered remote.
Inventories
Inventories include the cost of material, labor, and overhead, and are stated at the lower of cost, determined on the first-in, first-out (FIFO) method, or market. We periodically evaluate our on-hand stock and make appropriate provisions for any stock deemed excess or obsolete. Manufacturing costs resulting from lower than “normal” production levels are expensed as incurred.
Impairment
We assess certain of our long-lived assets, such as property and equipment and intangible assets other than goodwill, for potential impairment when there is a change in circumstances that indicates carrying values of assets may not be recoverable. Such long-lived assets are deemed to be impaired when the undiscounted cash flows expected to be generated by the asset (or asset group) are less than the asset’s carrying amount. Any required impairment loss would be measured as the amount by which the asset’s carrying value exceeds its fair value, and would be recorded as a reduction in the carrying value of the related asset and a charge to operating expense.
66
Goodwill and Intangibles
Goodwill is reviewed for impairment annually or more frequently if indicators of impairment exist. We perform our impairment test annually during the fourth quarter. The impairment evaluation is performed assuming the we operate in a single operating segment and reporting unit. First we assess qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount as a basis for determining whether it is necessary to perform the two-step goodwill impairment test. If, after assessing qualitative factors, we determine it is not more likely than not that the fair value of a reporting unit is less than its carrying amount, then performing the two-step impairment test is unnecessary. If deemed necessary, a two-step test is used to identify the potential impairment and to measure the amount of goodwill impairment, if any. The first step is to compare the fair value of the reporting unit with its carrying amount, including goodwill. If the fair value of the reporting unit exceeds its carrying amount, goodwill is considered not impaired; otherwise, there is an indication that goodwill may be impaired and the amount of the loss, if any, is measured by performing step two. Under step two, the impairment loss, if any, is measured by comparing the implied fair value of the reporting unit goodwill with the carrying amount of goodwill. There was no indication of impairment of goodwill for all periods presented.
Separable intangible assets that have finite useful lives are amortized over their respective useful lives.
Share-based compensation
The estimated fair value of share-based awards exchanged for employee and non-employee director services are expensed over the requisite service period and over the period during which the employee and non-employee director is required to provide service in exchange for the award. For purposes of calculating stock-based compensation, we estimate the fair value of stock options and shares issued under the Employee Stock Purchase Plan using a Black-Scholes option-pricing model. The determination of the fair value of share-based payment awards utilizing the Black-Scholes model is affected by our stock price and a number of assumptions, including expected volatility, expected life, risk-free interest rate and expected dividends. The expected volatility is based on the historical volatility of our common stock over the most recent period commensurate with the estimated expected term of the stock options. The expected life of the stock options is based on historical and other economic data trended into the future. The risk-free interest rate assumption is based on observed interest rates appropriate for the expected terms of our stock options. The dividend yield assumption is based on our history and expectation of no dividend payouts. The fair value of restricted stock agreements granted is based on the market price of our common stock on the day of the grant.
Recent Accounting Pronouncements
See Note 2 to the Consolidated Financial Statements included elsewhere herein for disclosure and discussion of new accounting standards.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable.
67
Item 8. Financial Statements and Supplementary Data
68
PART I. FINANCIAL INFORMATION
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Cytori Therapeutics, Inc.
We have audited the accompanying consolidated balance sheet of Cytori Therapeutics, Inc. (the “Company”) as of December 31, 2016 and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for the year then ended. In connection with our audit of the consolidated financial statements, we have also audited the accompanying schedule of valuation and qualifying accounts listed in the accompanying index at Item 15. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements and schedule. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Cytori Therapeutics, Inc. at December 31, 2016, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
Also, in our opinion, the financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in material respects, the information set forth therein.
The accompanying consolidated financial statements and schedule have been prepared assuming that the Company will continue as a going concern. As described in Note 1 to the consolidated financial statements, the Company has suffered recurring losses and negative cash flows from operations that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ BDO USA, LLP
San Diego, California
March 24, 2017
69
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Cytori Therapeutics, Inc.:
We have audited the accompanying consolidated balance sheet of Cytori Therapeutics, Inc. and subsidiaries (the Company) as of December 31, 2015, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for the year ended December 31, 2015. In connection with our audit of the consolidated financial statements, we have also audited the accompanying schedule of valuation and qualifying accounts as of and for the year ended December 31, 2015. These consolidated financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Cytori Therapeutics, Inc. and subsidiaries as of December 31, 2015, and the results of their operations and their cash flows for the year ended December 31, 2015, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
The accompanying consolidated financial statements and financial statement schedule have been prepared assuming that the Company will continue as a going concern. As discussed in note 1 to the consolidated financial statements, the Company’s recurring losses from operations and liquidity position raises substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in note 1. The consolidated financial statements and financial statement schedule do not include any adjustments that might result from the outcome of this uncertainty.
/s/ KPMG LLP
San Diego, California
March 11, 2016
70
CYTORI THERAPEUTICS, INC.
(in thousands, except share and par value data)
|
|
|
As of December31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
12,560 |
|
|
$ |
14,338 |
|
|
Accounts receivable, net of reserves of $167 and $797 in 2016 and 2015, respectively |
|
|
1,242 |
|
|
|
1,052 |
|
|
Restricted cash |
|
|
350 |
|
|
|
— |
|
|
Inventories, net |
|
|
3,725 |
|
|
|
4,298 |
|
|
Other current assets |
|
|
870 |
|
|
|
1,555 |
|
|
Total current assets |
|
|
18,747 |
|
|
|
21,243 |
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment, net |
|
|
1,157 |
|
|
|
1,631 |
|
|
Restricted cash |
|
|
— |
|
|
|
350 |
|
|
Other assets |
|
|
2,336 |
|
|
|
1,521 |
|
|
Intangibles, net |
|
|
8,447 |
|
|
|
9,031 |
|
|
Goodwill |
|
|
3,922 |
|
|
|
3,922 |
|
|
Total assets |
|
$ |
34,609 |
|
|
$ |
37,698 |
|
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable and accrued expenses |
|
$ |
5,872 |
|
|
$ |
6,687 |
|
|
Current portion of long-term obligations, net of discount |
|
|
6,629 |
|
|
|
— |
|
|
Joint venture purchase obligation |
|
|
— |
|
|
|
1,750 |
|
|
Total current liabilities |
|
|
12,501 |
|
|
|
8,437 |
|
|
|
|
|
|
|
|
|
|
|
|
Deferred revenues |
|
|
97 |
|
|
|
105 |
|
|
Long-term deferred rent and other |
|
|
17 |
|
|
|
269 |
|
|
Long-term obligations, net of discount, less current portion |
|
|
11,008 |
|
|
|
16,681 |
|
|
Total liabilities |
|
|
23,623 |
|
|
|
25,492 |
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 7) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value; 5,000,000 shares authorized; 13,500 shares issued; no shares outstanding in 2016 and 2015 |
|
|
— |
|
|
|
— |
|
|
Common stock, $0.001 par value; 75,000,000 shares authorized; 21,707,890 and 13,003,893 shares issued and outstanding in 2016 and 2015, respectively |
|
|
22 |
|
|
|
13 |
|
|
Additional paid-in capital |
|
|
388,769 |
|
|
|
368,214 |
|
|
Accumulated other comprehensive income |
|
|
1,258 |
|
|
|
996 |
|
|
Accumulated deficit |
|
|
(379,063 |
) |
|
|
(357,017 |
) |
|
Total stockholders’ equity |
|
|
10,986 |
|
|
|
12,206 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
34,609 |
|
|
$ |
37,698 |
|
THE ACCOMPANYING NOTES ARE AN INTEGRAL PART OF THESE CONSOLIDATED FINANCIAL STATEMENTS
71
CYTORI THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except share and per share data)
|
|
|
For the Years Ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Product revenues |
|
$ |
4,656 |
|
|
$ |
4,838 |
|
|
Cost of product revenues |
|
|
2,715 |
|
|
|
3,186 |
|
|
Gross profit |
|
|
1,941 |
|
|
|
1,652 |
|
|
|
|
|
|
|
|
|
|
|
|
Development revenues: |
|
|
|
|
|
|
|
|
|
Government contracts and other |
|
|
6,724 |
|
|
|
6,821 |
|
|
|
|
|
6,724 |
|
|
|
6,821 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
|
|
16,197 |
|
|
|
19,000 |
|
|
Sales and marketing |
|
|
3,611 |
|
|
|
2,662 |
|
|
General and administrative |
|
|
8,563 |
|
|
|
9,765 |
|
|
Change in fair value of warrant liabilities |
|
|
— |
|
|
|
(7,668 |
) |
|
Total operating expenses |
|
|
28,371 |
|
|
|
23,759 |
|
|
Operating loss |
|
|
(19,706 |
) |
|
|
(15,286 |
) |
|
|
|
|
|
|
|
|
|
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
Loss on debt extinguishment |
|
|
— |
|
|
|
(260 |
) |
|
Interest income |
|
|
19 |
|
|
|
9 |
|
|
Interest expense |
|
|
(2,592 |
) |
|
|
(3,379 |
) |
|
Other income, net |
|
|
233 |
|
|
|
172 |
|
|
Total other expense |
|
|
(2,340 |
) |
|
|
(3,458 |
) |
|
Net loss |
|
$ |
(22,046 |
) |
|
$ |
(18,744 |
) |
|
Beneficial conversion feature for convertible preferred stock |
|
|
— |
|
|
|
(661 |
) |
|
Net loss allocable to common stockholders |
|
$ |
(22,046 |
) |
|
$ |
(19,405 |
) |
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share allocable to common stockholders |
|
$ |
(1.28 |
) |
|
$ |
(2.07 |
) |
|
Basic and diluted weighted average shares used in calculating net loss per share allocable to common stockholders |
|
|
17,290,933 |
|
|
|
9,386,488 |
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(22,046 |
) |
|
$ |
(18,744 |
) |
|
Other comprehensive income – foreign currency translation adjustments |
|
|
262 |
|
|
|
296 |
|
|
Comprehensive loss |
|
$ |
(21,784 |
) |
|
$ |
(18,448 |
) |
THE ACCOMPANYING NOTES ARE AN INTEGRAL PART OF THESE CONSOLIDATED FINANCIAL STATEMENTS
72
CYTORI THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR THE YEARS ENDED DECEMBER 31, 2016 AND 2015
(in thousands, except share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
Convertible |
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
other |
|
|
|
|
|
|
Total |
|
||||||||
|
|
|
preferred stock |
|
|
Common stock |
|
|
paid-in |
|
|
comprehensive |
|
|
Accumulated |
|
|
stockholders’ |
|
||||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
capital |
|
|
(loss) income |
|
|
deficit |
|
|
equity (deficit) |
|
||||||||
|
Balance at December 31, 2014 |
|
|
5,311 |
|
|
$ |
— |
|
|
|
6,623,225 |
|
|
$ |
7 |
|
|
$ |
331,864 |
|
|
$ |
700 |
|
|
$ |
(338,273 |
) |
|
$ |
(5,702 |
) |
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,041 |
|
|
|
— |
|
|
|
— |
|
|
|
2,041 |
|
|
Issuance of common stock under stock option plan and employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
15,437 |
|
|
|
— |
|
|
|
27 |
|
|
|
— |
|
|
|
— |
|
|
|
27 |
|
|
Conversion of Series A 3.6% Convertible Preferred Stock into common stock |
|
|
(5,311 |
) |
|
|
— |
|
|
|
680,943 |
|
|
|
1 |
|
|
|
(3 |
) |
|
|
— |
|
|
|
— |
|
|
|
(2 |
) |
|
Issuance of common stock under stock warrant agreement, net |
|
|
— |
|
|
|
— |
|
|
|
3,123,577 |
|
|
|
3 |
|
|
|
22,810 |
|
|
|
— |
|
|
|
— |
|
|
|
22,813 |
|
|
Sale of common stock, net |
|
|
— |
|
|
|
— |
|
|
|
2,560,711 |
|
|
|
2 |
|
|
|
10,699 |
|
|
|
— |
|
|
|
— |
|
|
|
10,701 |
|
|
Allocation of fair value for debt-related warrants |
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
776 |
|
|
|
— |
|
|
— |
|
|
|
776 |
|
|||
|
Foreign currency translation adjustment and accumulated other comprehensive income |
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
296 |
|
|
— |
|
|
|
296 |
|
||||
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
— |
|
|
|
(18,744 |
) |
|
|
(18,744 |
) |
||
|
Balance at December 31, 2015 |
|
|
— |
|
|
|
— |
|
|
|
13,003,893 |
|
|
|
13 |
|
|
|
368,214 |
|
|
|
996 |
|
|
|
(357,017 |
) |
|
|
12,206 |
|
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,080 |
|
|
|
— |
|
|
|
— |
|
|
|
1,080 |
|
|
Issuance of common stock under employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
30,744 |
|
|
|
— |
|
|
|
6 |
|
|
|
— |
|
|
|
— |
|
|
|
6 |
|
|
Sale of common stock, net |
|
|
— |
|
|
|
— |
|
|
|
8,673,253 |
|
|
|
9 |
|
|
|
19,469 |
|
|
|
— |
|
|
|
— |
|
|
|
19,478 |
|
|
Foreign currency translation adjustment and accumulated other comprehensive income |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
262 |
|
|
|
— |
|
|
|
262 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
(22,046 |
) |
|
|
(22,046 |
) |
|
Balance at December 31, 2016 |
|
|
— |
|
|
$ |
— |
|
|
|
21,707,890 |
|
|
$ |
22 |
|
|
$ |
388,769 |
|
|
$ |
1,258 |
|
|
$ |
(379,063 |
) |
|
$ |
10,986 |
|
THE ACCOMPANYING NOTES ARE AN INTEGRAL PART OF THESE CONSOLIDATED FINANCIAL STATEMENTS
73
CYTORI THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
|
For the Years Ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(22,046 |
) |
|
$ |
(18,744 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
1,182 |
|
|
|
1,093 |
|
|
Amortization of deferred financing costs and debt discount |
|
|
954 |
|
|
|
979 |
|
|
Joint Venture acquisition obligation accretion |
|
|
24 |
|
|
|
365 |
|
|
Provision for doubtful accounts |
|
|
— |
|
|
|
(105 |
) |
|
Provision for expired inventory |
|
|
172 |
|
|
|
— |
|
|
Change in fair value of warrants |
|
|
— |
|
|
|
(7,668 |
) |
|
Share-based compensation expense |
|
|
1,080 |
|
|
|
2,041 |
|
|
(Gain) loss on asset disposal |
|
|
(127 |
) |
|
|
8 |
|
|
Loss on debt extinguishment |
|
|
— |
|
|
|
260 |
|
|
Increases (decreases) in cash caused by changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
(179 |
) |
|
|
328 |
|
|
Inventories |
|
|
471 |
|
|
|
490 |
|
|
Other current assets |
|
|
633 |
|
|
|
(637 |
) |
|
Other assets |
|
|
(764 |
) |
|
|
363 |
|
|
Accounts payable and accrued expenses |
|
|
(673 |
) |
|
|
1,045 |
|
|
Deferred revenues |
|
|
(8 |
) |
|
|
3 |
|
|
Long-term deferred rent |
|
|
(252 |
) |
|
|
(289 |
) |
|
Net cash used in operating activities |
|
|
(19,533 |
) |
|
|
(20,468 |
) |
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(67 |
) |
|
|
(611 |
) |
|
Expenditures for intellectual property |
|
|
— |
|
|
|
(13 |
) |
|
Proceeds from sale of assets |
|
131 |
|
|
|
11 |
|
|
|
Net cash provided by (used in) investing activities |
|
|
64 |
|
|
|
(613 |
) |
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
Principal payments on long-term obligations |
|
|
— |
|
|
|
(25,032 |
) |
|
Proceeds from long-term obligations |
|
|
— |
|
|
|
17,700 |
|
|
Debt issuance costs and loan fees |
|
|
— |
|
|
|
(1,854 |
) |
|
Joint Venture purchase payments |
|
|
(1,774 |
) |
|
|
(1,623 |
) |
|
Proceeds from exercise of employee stock options and warrants |
|
|
— |
|
|
|
4,997 |
|
|
Proceeds from sale of common stock |
|
|
21,467 |
|
|
|
29,054 |
|
|
Costs from sale of common stock |
|
|
(2,084 |
) |
|
|
(2,370 |
) |
|
Dividends paid on preferred stock |
|
|
— |
|
|
|
(75 |
) |
|
Net cash provided by financing activities |
|
|
17,609 |
|
|
|
20,797 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
82 |
|
|
|
— |
|
|
Net decrease in cash and cash equivalents |
|
|
(1,778 |
) |
|
|
(284 |
) |
|
Cash and cash equivalents at beginning of period |
|
|
14,338 |
|
|
|
14,622 |
|
|
Cash and cash equivalents at end of period |
|
$ |
12,560 |
|
|
$ |
14,338 |
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flows information: |
|
|
|
|
|
|
|
|
|
Cash paid during period for: |
|
|
|
|
|
|
|
|
|
Interest |
|
$ |
1,618 |
|
|
$ |
1,994 |
|
|
Final payment fee on long-term debt |
|
$ |
— |
|
|
$ |
1,839 |
|
|
Supplemental schedule of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
Issuance costs paid in common stock |
|
$ |
189 |
|
|
$ |
— |
|
|
Conversion of preferred stock into common stock |
|
$ |
— |
|
|
$ |
10 |
|
|
Declared dividend related to preferred stock |
|
$ |
— |
|
|
$ |
3 |
|
|
Fair value of warrants allocated to additional paid-in capital |
|
$ |
— |
|
|
$ |
776 |
|
THE ACCOMPANYING NOTES ARE AN INTEGRAL PART OF THESE CONSOLIDATED FINANCIAL STATEMENTS
74
CYTORI THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2016
|
1. |
Organization and Operations |
The Company
Cytori Therapeutics, Inc. (NASDAQ: CYTX) develops cell therapies uniquely formulated and optimized for specific diseases and medical conditions with a primary focus on impaired hand function in scleroderma, in addition to our other pipeline areas, such as osteoarthritis of the knee, stress urinary incontinence, and full thickness thermal burns including those complicated by radiation exposure.
Principles of Consolidation
The accompanying consolidated financial statements include our accounts and those of our subsidiaries. All significant intercompany transactions and balances have been eliminated in consolidation.
We have five wholly-owned subsidiaries located in Japan, United Kingdom, Switzerland, India and Spain that have been established primarily to support our sales and marketing activities in these regions.
Reverse Stock Split
On May 10, 2016, following stockholder and Board approval, an amendment (the “Amendment”) to the Company’s amended and restated certificate of incorporation, as amended, was filed and declared effective, which Amendment effectuated a one-for-fifteen (1:15) reverse stock split of the Company’s (i) outstanding common stock, and (ii) common stock reserved for issuance upon exercise of outstanding warrants and options (the “1:15 Reverse Stock Split”). Upon effectiveness of the 1:15 Reverse Stock Split, the number of shares of the Company’s common stock (x) issued and outstanding decreased from approximately 200 million shares (as of May 10, 2016) to approximately 13.3 million shares; (y) reserved for issuance upon exercise of outstanding warrants and options decreased from approximately 16 million shares to approximately 1.1 million shares, and (z) reserved but unallocated under our current equity incentive plans (including the stockholder-approved share increase to the Company’s 2014 Equity Incentive Plan) decreased from approximately 6.5 million common shares to approximately 0.4 million common shares. In connection with the 1:15 Reverse Stock Split, the Company also decreased the total number of its authorized shares of common stock from 290 million to 75 million. The number of authorized shares of preferred stock remained unchanged. Following the 1:15 Reverse Stock Split, certain reclassifications have been made to the prior periods’ financial statements to conform to the current period's presentation. The Company adjusted stockholders’ equity to reflect the 1:15 Reverse Stock Split by reclassifying an amount equal to the par value of the shares eliminated by the split from common stock to the additional paid-in capital during the first quarter of fiscal 2016, resulting in no net impact to stockholders' equity on our consolidated balance sheets. The Company’s shares of common stock commenced trading on a split-adjusted basis on May 12, 2016. Proportional adjustments for the reverse stock split were made to the Company's outstanding stock options, warrants awards issued and available under and equity incentive plans for all periods presented.
Certain Risks and Uncertainties
Our prospects are subject to the risks and uncertainties frequently encountered by companies in the early stages of development and commercialization, especially those companies in rapidly evolving and technologically advanced industries such as the biotech/medical device field. Our future viability largely depends on our ability to complete development of new products and receive regulatory approvals for those products. No assurance can be given that our new products will be successfully developed, regulatory approvals will be granted, or acceptance of these products will be achieved. The development of medical devices for specific therapeutic applications is subject to a number of risks, including research, regulatory and marketing risks. There can be no assurance that our development stage products will overcome these hurdles and become commercially viable and/or gain commercial acceptance.
Liquidity and Going Concern
We incurred net losses of $22.0 million and $18.7 million for the years ended December 31, 2016 and 2015, respectively. We have an accumulated deficit of $379.1 million as of December 31, 2016. Additionally, we have used net cash of $19.5 million and $20.5 million to fund our operating activities for the years ended December 31, 2016 and 2015, respectively. These factors raise substantial doubt about the Company’s ability to continue as a going concern.
75
Further, our Loan and Security Agreement, or the Loan and Security Agreement, with Oxford Finance, LCC, or Oxford, as further described in Note 8, requires to maintain a minimum of $5.0 million in unrestricted cash and cash equivalents on hand to avoid an event of default under the Loan and Security Agreement. Based on our cash and cash equivalents on hand of approximately $12.6 million at December 31, 2016, and our obligation to make payments of principal of $0.6 million plus accrued interest in monthly installments, we estimate that we must raise additional capital and/or obtain a waiver or restructure the Loan and Security Agreement on or before May 2017 to avoid defaulting under our $5.0 million minimum cash/cash equivalents covenant.
To date, these operating losses have been funded primarily from outside sources of invested capital including our recently completed Lincoln Park Purchase Agreement with Lincoln Park Capital Fund, LLC (“Lincoln Park”) and the Rights Offering (each defined below), our at-the-market (“ATM”) equity facility, the Loan and Security Agreement and gross profits. We have had, and we will likely continue to have, an ongoing need to raise additional cash from outside sources to fund our future clinical development programs and other operations.
On June 15, 2016, we closed a rights offering originally filed under Form S-1 registration statement in April 2016 (the “Rights Offering”). Pursuant to the Rights Offering, we sold an aggregate of 6,704,852 units consisting of a total of 6,704,852 shares of common stock and 3,352,306 warrants, with each warrant exercisable for one share of common stock at an exercise price of $3.06 per share, resulting in total gross proceeds to Cytori of $17.1 million. See Note 11 for further discussion on the Rights Offering.
On December 22, 2016, we entered into a purchase agreement and a registration rights agreement, with Lincoln Park pursuant to which we have the right to sell to Lincoln Park and Lincoln Park is obligated to purchase up to $20.0 million in amounts of shares, of the Company’s common stock, over the 30-month period commencing on the date that a registration statement, which the Company filed with the Securities and Exchange Commission (the “SEC”) on December 30, 2016. See Note 11 for further discussion on the Lincoln Park agreement.
Pursuant to these securities transactions and related equity issuances, as well as anticipated gross profits and potential outside sources of capital, we believe we have sufficient cash to fund operations through Q2 2017. We continue to seek additional capital through product revenues, strategic transactions, including extension opportunities under our awarded U.S. Department of Health and Human Service’s Biomedical Advanced Research and Development Authority (“BARDA”) contract, and from other financing alternatives. Without additional capital, current working capital and cash generated from sales will not provide adequate funding for research, sales and marketing efforts and product development activities at their current levels. If sufficient capital is not raised, we will at a minimum need to significantly reduce or curtail our research and development and other operations, and this could negatively affect our ability to achieve corporate growth goals.
Should we be unable to raise additional cash from outside sources, this will have a material adverse impact on our operations.
The accompanying consolidated financial statements have been prepared assuming we will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to its ability to continue as a going concern.
Reclassifications
Certain immaterial reclassifications have been made to certain of the prior years’ consolidated financial statements to conform to the current year presentation.
|
2. |
Summary of Significant Accounting Policies |
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions affecting the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenue and expenses during the reporting period. Our most significant estimates and critical accounting policies involve recognizing revenue, reviewing goodwill and intangible assets for impairment, determining the assumptions used in measuring share-based compensation expense, measuring accretion expense related to our acquisition of the joint venture, and valuing allowances for doubtful accounts and inventory reserves.
Actual results could differ from these estimates. Management’s estimates and assumptions are reviewed regularly, and the effects of revisions are reflected in the consolidated financial statements in the periods they are determined to be necessary.
76
Cash and cash equivalents
We consider all highly liquid investments with maturities of three months or less at the time of purchase to be cash equivalents.
Cash and cash equivalents includes cash in readily available checking and savings accounts. We held no investments as of December 31, 2016 and 2015. We maintain our cash at insured financial institutions.
Restricted Cash
Restricted cash consists of cash invested in a certificate of deposit used as collateral for the issuance of a letter of credit pursuant to a lease agreement entered into on April 2, 2010 (amended November 4, 2011) for leasing of property at 3020 and 3030 Callan Road, San Diego, California. The lease agreement required us to execute a letter of credit for $0.4 million naming the landlord as a beneficiary. It is required by the landlord that we maintain $0.4 million as restricted cash for the duration of the lease, which expires October 31, 2017.
Accounts Receivable
Accounts receivable are recorded at the invoiced amount and do not bear interest. The Company periodically assesses the collectability of accounts receivable on a specific customer basis considering factors such as evaluation of collectability, historical collection experience, the age of accounts receivable and other currently available evidence of the collectability, and records an allowance for doubtful accounts for the estimated uncollectible amount. Account balances are charged off against the allowance after all means of collection have been exhausted and the potential for recovery is considered remote.
Inventories
Inventories include the cost of material, labor, and overhead related to Celution devices, consumable kits, and reagents, and are stated at the lower of cost, determined on the first-in, first-out (FIFO) method, or market. We periodically evaluate our on-hand stock and make appropriate provisions for any stock deemed excess or obsolete. Manufacturing costs resulting from lower than “normal” production levels are expensed as incurred.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation expense, which includes the amortization of capitalized leasehold improvements, is provided for on a straight-line basis over the estimated useful lives of the assets, or the life of the lease, whichever is shorter, and range from three to five years. When assets are sold or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts and the resulting gain or loss, if any, is included in operations. Maintenance and repairs are charged to operations as incurred.
Impairment
We assess certain of our long-lived assets, such as property and equipment and intangible assets other than goodwill, for potential impairment when there is a change in circumstances that indicates carrying values of assets may not be recoverable. Such long-lived assets are deemed to be impaired when the undiscounted cash flows expected to be generated by the asset (or asset group) are less than the asset’s carrying amount. Any required impairment loss would be measured as the amount by which the asset’s carrying value exceeds its fair value, and would be recorded as a reduction in the carrying value of the related asset and a charge to operating expense. We recognized no impairment losses during any of the periods presented in these financial statements.
Goodwill and Intangibles
Goodwill is reviewed for impairment annually or more frequently if indicators of impairment exist. We perform our impairment test annually during the fourth quarter. The impairment evaluation is performed assuming the Company operates in a single operating segment and reporting unit. First the Company assesses qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount as a basis for determining whether it is necessary to perform the two-step goodwill impairment test. If, after assessing qualitative factors, the Company determines it is not more likely than not that the fair value of a reporting unit is less than its carrying amount, then performing the two-step impairment test is unnecessary. If deemed necessary, a two-step test is used to identify the potential impairment and to measure the amount of goodwill impairment, if any. The first step is to compare the fair value of the reporting unit with its carrying amount, including goodwill. If the fair value of the reporting unit exceeds its carrying amount, goodwill is considered not impaired; otherwise, there is an indication that goodwill may be impaired and the amount of the loss, if any, is measured by performing
77
step two. Under step two, the impairment loss, if any, is measured by comparing the implied fair value of the reporting unit goodwill with the carrying amount of goodwill. There was no indication of impairment of goodwill for all periods presented.
Separable intangible assets that have finite useful lives are amortized over their respective useful lives.
As part of the May 2013 acquisition of the Joint Venture (see Note 4), we acquired intangible assets which consisted primarily of contractual license rights that had previously enabled the Joint Venture to conduct development and manufacturing activities pertaining to certain aspects of Cytori’s Celution technology. The useful life of the identifiable intangible assets was estimated based on the assumed future economic benefit expected to be received from the assets. The technology was valued at $9.4 million and is being amortized on a straight-line basis over a useful life of eleven years, commensurate with the expected cash flows. The amortization expense was $0.6 million and $0.4 million for the years ended December 31, 2016 and 2015, respectively, and was included in cost of product revenue on the consolidated statements of operations. The estimated aggregate amortization expense will be $1.2 million for 2017, $1.2 million for 2018 and $6.0 million thereafter. Accumulated amortization on the intangible assets was $1.2 million as of December 31, 2016 and $0.6 million as of December 31, 2015.
The changes in the carrying amounts of finite-life intangible assets and goodwill for the years ended December 31, 2016 and 2015 are as follows (in thousands):
|
|
|
December 31, 2016 |
|
|
|
Other intangibles, net: |
|
|
|
|
|
Beginning balance |
|
$ |
9,031 |
|
|
Increase |
|
|
— |
|
|
Amortization |
|
|
(584 |
) |
|
Ending balance |
|
|
8,447 |
|
|
Goodwill, net: |
|
|
|
|
|
Beginning balance |
|
|
3,922 |
|
|
Increase (decrease) |
|
|
— |
|
|
Ending balance |
|
|
3,922 |
|
|
Total goodwill and other intangibles, net |
|
$ |
12,369 |
|
|
|
|
December 31, 2015 |
|
|
|
Other intangibles, net: |
|
|
|
|
|
Beginning balance |
|
$ |
9,415 |
|
|
Increase |
|
|
13 |
|
|
Amortization |
|
|
(397 |
) |
|
Ending balance |
|
|
9,031 |
|
|
Goodwill, net: |
|
|
|
|
|
Beginning balance |
|
|
3,922 |
|
|
Increase (decrease) |
|
|
— |
|
|
Ending balance |
|
|
3,922 |
|
|
Total goodwill and other intangibles, net |
|
$ |
12,953 |
|
Warrant Liability
In connection with the October 2014 Securities Purchase Agreement, the Company issued common stock purchase warrants (the “October 2014 Warrants”) to certain institutional investors with certain exercise price reset features. Each warrant had an initial exercise price of $0.5771 per share, was exercisable six months and one day after the date of issuance and was to expire five years from the date on which it was initially exercisable. Pursuant to the second closing of the May 2015 Securities Purchase Agreement, the exercise price of these warrants was reset to $0.3263. The initial fair value of the liability associated with these warrants was $10.0 million and it decreased to $3.3 million as of December 17, 2015 when these warrants were cashless exercised by all holders.
In May 2015, the Company entered into a Securities Purchase Agreement with certain institutional investors pursuant to which the Company agreed to sell up to $25 million of units, with each unit consisting of one share of its common stock and one warrant to purchase one share of its common stock, in a registered direct offering. The May 2015 Securities Purchase Agreement contemplated two closings, the first of which occurred on May 8, 2015, the second of which occurred upon satisfaction of certain conditions precedent, including, but not limited to, receipt of required stockholder approval, on August 27, 2015. Each warrant issued at the initial closing (the “May 2015 Warrants”) had an initial exercise price of $1.02 per share, was exercisable six months and one day after the date of issuance and expires five years from the date on which it is initially
78
exercisable. Each warrant issued at the second closing (the “August 2015 Warrants”) had an initial exercise price of $0.401 per share, and was to expire five years from the date of issuance. The initial fair value of the liability associated with the May 2015 Warrants was $14.3 million and it decreased to $5.0 million as of December 17, 2015 when these warrants were cashless exercised by all holders. The initial fair value of the liability associated with the August 2015 Warrants was $1.6 million, and it decreased to $1.5 million as of December 17, 2015, when these warrants were cashless exercised by all holders.
On December 17, 2015, the Company and the holders of October 2014 Warrants agreed to amend the October 2014 Warrants pursuant to an Amendment to Common Stock Purchase Warrant (the “2014 Amendment”). Also on December 17, 2015, the Company and the holders of the May 2015 Warrants and the August 2015 Warrants (collectively the “2015 Warrants”) agreed to amend the 2015 Warrants pursuant to an Amendment to Series A-1 Warrant to Purchase Common Stock and Amendment to Series A-2 Warrant to Purchase Common Stock, respectively (the “2015 Amendment” and, together with the 2014 Amendment, the “Warrant Amendments”). The Warrant Amendments provide that the holders may exercise their warrants on a “cashless exercise” basis in whole on or prior to December 31, 2015, whereby each exercising holder of the amended 2015 Warrants would receive 0.75 shares for each warrant share exercised and each exercising holder of the amended 2014 Warrants would receive 0.69 shares for each warrant share exercised. In addition, the Warrant Amendments removed certain provisions which provided that the exercise price of the Warrants would be reset in the event of certain equity issuances by the Company for a price below the exercise price of the Warrants as of the time of such issuance. All warrants were cashless exercised on or before December 31, 2015.
The warrants were not traded in an active securities market and, as such, the estimated fair value as of their exercise date on December 17, 2015 was determined by using the Monte Carlo option pricing model. The 2014 and 2015 warrants were settled on or prior to December 31, 2015.
Revenue Recognition
Product Sales
We recognize revenue from product sales when the following fundamental criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery has occurred, (iii) the price to the customer is fixed or determinable and (iv) collection of the resulting accounts receivable is reasonably assured. We evaluate customers that have not developed a sufficient payment history with us or for whom a letter of credit is not in place at the time of the transaction and defer revenues until collectability is reasonably assured.
For all sales, we use a binding purchase order or a signed agreement as evidence of an arrangement. If the other revenue recognition criteria are met, revenue for these product sales is recognized upon delivery to the customer as all risks and rewards of ownership have been substantively transferred to the customer at that point. For sales to customers who arrange for and manage the shipping process, we recognize revenue upon shipment from our facilities. Shipping and handling costs that are billed to our customers are classified as revenue. The customer’s obligation to pay and the payment terms are set at the time of delivery and are not dependent on the subsequent use or resale of our products. For sales where all revenue recognition criteria are not met, revenue is deferred and related inventory remains on our books.
Concentration of Significant Customers & Geographical Sales
For the year ended December 31, 2016, our sales were concentrated with respect to two distributors and three direct customers, which comprised 65% of our product revenue recognized. Two direct customers accounted for 57% of total outstanding accounts receivable (excluding receivables from BARDA) as of December 31, 2016.
For the year ended December 31, 2015, our sales were concentrated with respect to one distributor and four direct customers, which comprised 63% of our product revenue recognized. Two direct customers accounted for 73% of total outstanding accounts receivable (excluding receivables from BARDA) as of December 31, 2015.
Product revenues, classified by geographic location, are as follows (in thousands):
|
|
|
Years ended December 31, |
|
|||||||||||||
|
|
|
2016 |
|
|
2015 |
|
||||||||||
|
|
|
Product Revenues |
|
|
% of Total |
|
|
Product Revenues |
|
|
% of Total |
|
||||
|
Americas |
|
$ |
936 |
|
|
|
20 |
% |
|
$ |
982 |
|
|
|
20 |
% |
|
Japan |
|
|
3,279 |
|
|
|
71 |
% |
|
|
2,394 |
|
|
|
50 |
% |
|
EMEA |
|
|
379 |
|
|
|
8 |
% |
|
|
675 |
|
|
|
14 |
% |
|
Asia Pacific |
|
|
62 |
|
|
|
1 |
% |
|
|
787 |
|
|
|
16 |
% |
|
Total product revenues |
|
$ |
4,656 |
|
|
|
100 |
% |
|
$ |
4,838 |
|
|
|
100 |
% |
79
Development Revenues
We earn revenue for performing tasks under research and development agreements with governmental agencies like BARDA. Revenues derived from reimbursement of direct out-of-pocket expenses for research costs associated with government contracts are recorded as government contract and other within development revenues. Government contract revenue is recorded at the gross amount of the reimbursement. The costs associated with these reimbursements are reflected as a component of research and development expense in our statements of operations. We recognized $6.7 million and $6.8 million in BARDA revenue for the years ended December 31, 2016 and 2015, respectively.
Research and Development
Research and development expenditures, which are charged to operations in the period incurred, include costs associated with the design, development, testing and enhancement of our products, regulatory fees, the purchase of laboratory supplies, and pre-clinical and clinical studies as well as salaries and benefits for our research and development employees.
Also included in research and development expenditures are costs incurred to support the government reimbursement contract, including $6.3 million and $6.3 million of qualified expenses that were incurred for the years ended December 31, 2016 and 2015, related to our government contract with BARDA.
Deferred Financing Costs and Other Debt-Related Costs
Deferred financing costs are capitalized, recorded as an offset to debt balances and amortized to interest expense over the term of the associated debt instrument using the effective interest method. If the maturity of the debt is accelerated because of default or early debt repayment, then the amortization would be accelerated.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income (loss) in the years in which those temporary differences are expected to be recovered or settled. Due to our history of losses, a full valuation allowance has been recognized against our deferred tax assets.
The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. For the years ended December 31, 2016 and 2015, the Company has not recorded any interest or penalties related to income tax matters. The Company does not foresee any material changes to unrecognized tax benefits within the next twelve months.
Share-Based Compensation
We recognize the fair value of all share-based payment awards in our statements of operations over the requisite vesting period of each award, which approximates the period during which the employee and non-employee director is required to provide service in exchange for the award. We estimate the fair value of these options using the Black-Scholes option pricing model using assumptions for expected volatility, expected term, and risk-free interest rate. Expected volatility is based primarily on historical volatility and is computed using daily pricing observations for recent periods that correspond to the expected term of the options. The expected term is calculated based on historical data for and applied to all employee awards as a single group as we do not expect (nor does historical data suggest) substantially different exercise or post-vesting termination behavior amongst our employee population. The risk-free interest rate is the interest rate for treasury instruments with maturities that approximate the expected term.
Segment Information
For the years ended December 31, 2016 and 2015, all of our financial results relate to cell therapy, therefore we report our results in one operating segment.
80
Loss Per Share
Basic per share data is computed by dividing net income or loss applicable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted per share data is computed by dividing net income or loss applicable to common stockholders by the weighted average number of common shares outstanding during the period increased to include, if dilutive, the number of additional common shares that would have been outstanding as calculated using the treasury stock method. Potential common shares were related entirely to outstanding but unexercised options and warrants for all periods presented.
We have excluded all potentially dilutive securities, including unvested performance-based restricted stock, from the calculation of diluted loss per share attributable to common stockholders for the years ended December 31, 2016 and 2015, as their inclusion would be antidilutive. Potentially dilutive securities excluded from the calculations of diluted loss per share were 4.2 million as of December 31, 2016, which includes 3.6 million outstanding warrants and 0.6 million options and restricted stock awards. Potentially dilutive securities excluded from the calculations of diluted loss per share were 12.3 million as of December 31, 2015.
Recent Accounting Pronouncements
In May 2016, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2016-12, Revenue from Contracts with Customers, the amendment of which addressed narrow-scope improvements to the guidance on collectability, noncash consideration, and completed contracts at transition as well as providing a practical expedient for contract modifications. In April 2016 and March 2016, the FASB issued ASU No. 2016-10 and ASU No. 2016-08, respectively, the amendments of which further clarified aspects of Topic 606: identifying performance obligations and the licensing and implementation guidance and intended to improve the operability and understandability of the implementation guidance on principal versus agent considerations. The FASB issued the initial release of Topic 606 in ASU No. 2014-09, which requires entities to recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. Entities may use a full retrospective approach or report the cumulative effect as of the date of adoption. On July 9, 2015, the FASB voted to defer the effective date by one year to December 15, 2017 for interim and annual reporting periods beginning after that date. Early adoption of ASU 2016-10 is permitted but not before the original effective date (annual periods beginning after December 15, 2017). We are currently in the process of evaluating our various contracts and revenue streams subject to this update but have not completed our assessment and, therefore, have not yet concluded on whether the adoption of this update will have a material effect on our consolidated financial statements and related disclosures.
In August 2014, the FASB issued ASU 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. ASU 2014-15 requires management to evaluate relevant conditions, events and certain management plans that are known or reasonably knowable that when, considered in the aggregate, raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued, for both annual and interim periods. ASU 2014-15 also requires certain disclosures around management’s plans and evaluation, as well as the plans, if any, that are intended to mitigate those conditions or events that will alleviate the substantial doubt. ASU 2014-15 is effective for annual reporting periods, and interim periods within those periods, ending after December 15, 2016. The Company adopted this guidance to assess going concern at December 31, 2016 and its liquidity disclosures reflect the requirements of the new standard.
In July 2015, FASB issued ASU 2015-11, Simplifying the Measurement of Inventory. This update applies to companies that measure inventory on a first in, first out, or FIFO, or average cost basis. Under this update, companies are to measure their inventory at the lower of cost and net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion. The amendments in this update are effective for annual reporting periods, and interim periods within those periods, beginning after December 15, 2016 with earlier application permitted as of the beginning of an interim or annual reporting period. The adoption of ASU 2015-11 will not have a material impact on our consolidated financial statements.
In February 2016, the FASB issued ASU 2016-02, Leases. Under this new guidance, at the commencement date, lessees will be required to recognize (i) a lease liability, which is a lessee’s obligation to make lease payments arising from a lease, measured on a discounted basis and (ii) a right-of-use asset, which is an asset that represents the lessee’s right to use, or control the use of, a specified asset for the lease term. This guidance is not applicable for leases with a term of 12 months or less. The new standard is effective for annual reporting periods, and interim periods within those periods, beginning after December 15, 2018, with early adoption permitted. We are currently evaluating the impact that this standard will have on our consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Improvements to Employee Share-Based Payment Accounting, which involves several aspects of the accounting for stock-based payment transactions, including the income tax consequences, classification of
81
awards as either equity or liabilities, and classification on the statement of cash flows. This new guidance will require all income tax effects of awards to be recognized as income tax expense or benefit in the income statement when the awards vest or are settled, as opposed to additional paid-in-capital where it is currently recorded. It also will allow an employer to repurchase more of an employee’s shares than it can today for tax withholding purposes without triggering liability accounting. All tax-related cash flows resulting from stock-based payments are to be reported as operating activities on the statement of cash flows. The guidance also allows a Company to make a policy election to either estimate the number of awards that are expected to vest or account for forfeitures as they occur. This new standard is effective for annual reporting periods, and interim periods within those periods, beginning after December 15, 2016, with early adoption permitted. We have elected to keep our policy consistent for the application of a forfeiture rate and, as such, the adoption of this standard will not have a material impact on our consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): Classification of certain cash receipts and cash payments, which addresses the following eight specific cash flow issues: Debt prepayment or debt extinguishment costs; settlement of zero-coupon debt instruments or other debt instruments with coupon interest rates that are insignificant in relation to the effective interest rate of the borrowing; contingent consideration payments made after a business combination; proceeds from the settlement of insurance claims; proceeds from the settlement of corporate-owned life insurance policies (including bank-owned life insurance policies); distributions received from equity method investees; beneficial interests in securitization transactions; and separately identifiable cash flows and application of the predominance principle. The new standard is effective for annual reporting periods, and interim periods within those periods, beginning after December 15, 2017, with early adoption permitted. We do not anticipate that the adoption of ASU 2016-15 will have a material impact on our consolidated financial statements.
In November 2016, the FASB issued Accounting Standards Update No. 2016-18, Restricted Cash, which requires entities to show the changes in the total of cash, cash equivalents, restricted cash and restricted cash equivalents in the statement of cash flows. As a result, entities will no longer present transfers between cash and cash equivalents and restricted cash and restricted cash equivalents in the statement of cash flows. The amendments in this update should be applied using a retrospective transition method to each period presented. This update is effective for annual periods beginning after December 15, 2017, and interim periods within those fiscal years with early adoption permitted, including adoption in an interim period. The adoption of this standard will change the presentation of our statement of cash flows to include our restricted cash balance. We are assessing whether to adopt the new guidance early in 2017.
In January 2017, the FASB issued Accounting Standards Update No. 2017-01, Clarifying the Definition of a Business, which clarifies and provides a more robust framework to use in determining when a set of assets and activities is a business. The amendments in this update should be applied prospectively on or after the effective date. This update is effective for annual periods beginning after December 15, 2017, and interim periods within those periods. Early adoption is permitted for acquisition or deconsolidation transactions occurring before the issuance date or effective date and only when the transactions have not been reported in issued or made available for issuance financial statements. We do not expect the adoption to have any significant impact on our consolidated financial statements, and we are in the process of determining whether to adopt the new guidance early.
In February 2017, the FASB recently issued ASU 2017-04, Simplifying the Test for Goodwill Impairment, to simplify how all entities assess goodwill for impairment by eliminating Step 2 from the goodwill impairment test. As amended, the goodwill impairment test will consist of one step comparing the fair value of a reporting unit with its carrying amount. An entity should recognize a goodwill impairment charge for the amount by which the reporting unit's carrying amount exceeds its fair value. This update is effective for annual periods beginning after December 15, 2019, and interim periods within those periods. Early adoption is permitted for interim or annual goodwill impairment tests performed on testing dates after January 1, 2017. We do not anticipate that the adoption of ASU 2017-04 will have a material impact on our consolidated financial statements.
|
3. |
Agreement with Lorem Vascular |
On October 29, 2013, we entered into an agreement with Lorem Vascular to commercialize Cytori Cell Therapy (OICH-D3) for the cardiovascular, renal and diabetes markets, in China, Hong Kong, Malaysia, Singapore and Australia (License/Supply Agreement), and a Common Stock Purchase Agreement. On January 30, 2014, we entered into the Amended and Restated License/Supply Agreement with Lorem Vascular (the “Restated Agreement”) which restated the License/Supply Agreement in its entirety and expanded the licensed field to all uses excepting alopecia (hair loss). Under the Restated Agreement, Lorem Vascular committed to pay up to $500 million in license fees in the form of revenue milestones. In addition, Lorem Vascular is required to pay us 30% of their gross profits in China, Hong Kong and Malaysia for the term of the agreement. In addition, Lorem Vascular has agreed to purchase the Cytori Celution® System and consumables under the Restated Agreement. Pursuant to the related Common Stock Purchase Agreement, Cytori sold Lorem Vascular 8.0 million shares of Cytori common stock at
82
$3.00 per share for a total of $24.0 million. The equity purchased was closed in two equal installments, in November 2013 and January 2014.
Lorem Vascular initially purchased approximately $1.8 million in Celution® devices and consumables in December 2013. In addition to this purchase, upon achieving regulatory clearance from the Chinese Food and Drug Administration (“CFDA”), Cytori’s license agreement with Lorem Vascular obligates Lorem Vascular to purchase an opening order of 23 Celution Systems and 1,100 Celution Consumable Sets. Class I regulatory clearance was granted in April 2015. There were no business transactions with Lorem Vascular during the year ended December 31, 2016. As of December 31, 2015, Lorem Vascular has partially satisfied this purchase order.
|
4. |
Transactions with Olympus Corporation |
Under our Joint Venture Termination Agreement (“Termination Agreement”), dated May 8, 2013, with Olympus Corporation (“Olympus”), we were required to pay Olympus a total purchase price of $6.0 million within two years of the date of the Termination Agreement. Pursuant to amendments to the Termination Agreement, dated April 30, 2015 and January 8, 2016, the Company’s repayment obligations were extended through May 8, 2016. We made payments under the Termination Agreement totaling approximately $4.2 million through December 31, 2015, as well as separate payments of $0.5 million each in January 2016 and April 2016, and paid the remaining balance of $0.8 million before the May 8, 2016 due date. There were no outstanding obligations to Olympus as of December 31, 2016.
|
5. |
Fair Value |
Measurements
Fair value measurements are market-based measurements, not entity-specific measurements. Therefore, fair value measurements are determined based on the assumptions that market participants would use in pricing the asset or liability. We follow a three-level hierarchy to prioritize the inputs used in the valuation techniques to derive fair values. The basis for fair value measurements for each level within the hierarchy is described below:
|
|
• |
Level 1: Quoted prices in active markets for identical assets or liabilities. |
|
|
• |
Level 2: Quoted prices for similar assets or liabilities in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations in which all significant inputs are observable in active markets. |
|
|
• |
Level 3: Valuations derived from valuation techniques in which one or more significant inputs are unobservable in active markets. |
As of December 31, 2016 and 2015, we did not have any assets or liabilities measured at fair value presented on our balance sheets.
The 2014 and 2015 warrants included exercise price reset features (down-round protection) and were accounted for as liabilities, with changes in the fair value included in net loss for the respective periods. Because some of the inputs to our valuation model were either not observable or were not derived principally from or corroborated by observable market data by correlation or other means, the warrant liability was classified as Level 3 in the fair value hierarchy. All of these warrants were cashless exercised on or before December 31, 2015.
The following table summarizes the final valuation pertaining to the warrants that were previously included in our Level 3 warrant liabilities (in thousands):
|
Warrant liability |
|
December 31, 2015 |
|
|
|
Balance as of December 31, 2014 |
|
$ |
9,793 |
|
|
Additions to warrant liability |
|
|
15,979 |
|
|
Exercised warrants |
|
|
(18,104 |
) |
|
Change in fair value |
|
|
(7,668 |
) |
|
Balance as of December 31, 2015 |
|
$ |
— |
|
Financial Instruments
We disclose fair value information about all financial instruments, whether or not recognized in the balance sheets, for which it is practicable to estimate fair value. The disclosures of estimated fair value of financial instruments at December 31, 2016 and 2015, were determined using available market information and appropriate valuation methods. Considerable judgment is
83
necessary to interpret market data and develop estimated fair value. The use of different market assumptions or estimation methods may have a material effect on the estimated fair value amounts.
The carrying amounts for cash and cash equivalents, accounts receivable, inventories, other current assets, accounts payable, accrued expenses and other liabilities approximate fair value due to the short-term nature of these instruments.
If quoted market prices are not available, we calculate the fair value of our fixed rate debt based on a currently available market rate assuming the loans are outstanding through maturity and considering the collateral. In determining the current market rate for fixed rate debt, a market spread is added to the quoted yields on federal government treasury securities with similar terms to the debt.
At December 31, 2016 and 2015, the aggregate fair value and the carrying value of the Company’s long-term debt were as follows (in thousands):
|
|
|
December 31, 2016 |
|
|
December 31, 2015 |
|
||||||||||
|
|
|
Fair Value |
|
|
Carrying Value |
|
|
Fair Value |
|
|
Carrying Value |
|
||||
|
Debt |
|
$ |
17,611 |
|
|
$ |
17,637 |
|
|
$ |
16,844 |
|
|
$ |
16,681 |
|
Carrying value is net of debt discount of $1.2 million and $2.1 million as of December 31, 2016 and 2015, respectively.
The fair value of debt is classified as Level 3 in the fair value hierarchy as some of the inputs, primarily the effective interest rate, to our valuation model are either not observable quoted prices or are not derived principally from or corroborated by observable market data by correlation or other means.
Nonfinancial Assets and Liabilities
We apply fair value techniques on a non-recurring basis associated with: (1) valuing potential impairment losses related to goodwill which are accounted for pursuant to the authoritative guidance for intangibles—goodwill and other; and (2) valuing potential impairment losses related to long-lived assets which are accounted for pursuant to the authoritative guidance for property, plant and equipment.
|
6. |
Composition of Certain Financial Statement Captions |
Inventories, net
As of December 31, 2016 and 2015, inventories, net, were comprised of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Raw materials |
|
$ |
885 |
|
|
$ |
1,009 |
|
|
Work in process |
|
|
1,021 |
|
|
|
816 |
|
|
Finished goods |
|
|
1,819 |
|
|
|
2,473 |
|
|
|
|
$ |
3,725 |
|
|
$ |
4,298 |
|
Other Current Assets
As of December 31, 2016 and 2015, other current assets were comprised of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Prepaid supplies, current |
|
$ |
734 |
|
|
$ |
995 |
|
|
Prepaid insurance |
|
|
83 |
|
|
|
300 |
|
|
Other receivables |
|
|
53 |
|
|
|
260 |
|
|
|
|
$ |
870 |
|
|
$ |
1,555 |
|
84
Property and Equipment, net
As of December 31, 2016 and 2015, property and equipment, net, were comprised of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Manufacturing and development equipment |
|
$ |
4,256 |
|
|
$ |
5,464 |
|
|
Office and computer equipment |
|
|
1,953 |
|
|
|
1,939 |
|
|
Leasehold improvements |
|
|
3,399 |
|
|
|
3,391 |
|
|
|
|
|
9,608 |
|
|
|
10,794 |
|
|
Less accumulated depreciation |
|
|
(8,451 |
) |
|
|
(9,163 |
) |
|
|
|
$ |
1,157 |
|
|
$ |
1,631 |
|
Depreciation expense totaled $0.7 million and $0.7 million for the years ended December 31, 2016 and 2015, respectively.
Other Assets
As of December 31, 2016 and 2015, other assets were comprised of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Prepaid supplies, long-term |
|
$ |
1,838 |
|
|
$ |
996 |
|
|
Deposits |
|
|
498 |
|
|
|
525 |
|
|
|
|
$ |
2,336 |
|
|
$ |
1,521 |
|
Accounts Payable and Accrued Expenses
As of December 31, 2016 and 2015, accounts payable and accrued expenses were comprised of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Accrued expenses |
|
$ |
1,752 |
|
|
$ |
2,022 |
|
|
Accounts payable |
|
|
1,332 |
|
|
|
1,009 |
|
|
Accrued payroll and bonus |
|
|
989 |
|
|
|
1,058 |
|
|
Accrued legal fees |
|
|
614 |
|
|
|
372 |
|
|
Accrued vacation |
|
|
502 |
|
|
|
573 |
|
|
Accrued R&D studies |
|
|
347 |
|
|
|
1,117 |
|
|
Deferred rent |
|
|
215 |
|
|
|
221 |
|
|
Accrued accounting fees |
|
|
121 |
|
|
|
315 |
|
|
|
|
$ |
5,872 |
|
|
$ |
6,687 |
|
|
7. |
Commitments and Contingencies |
We have entered into agreements with various research organizations for pre-clinical and clinical development studies, which have provisions for cancellation. Under the terms of these agreements, the vendors provide a variety of services including conducting research, recruiting and enrolling patients, monitoring studies and data analysis. Payments under these agreements typically include fees for services and reimbursement of expenses. The timing of payments due under these agreements is estimated based on current study progress. As of December 31, 2016, we have clinical research study obligations of $3.3 million, $3.2 million of which are expected to be complete within a year. Should the timing of the clinical trials change, the timing of the payment of these obligations would also change.
We lease facilities for our headquarters office location as well as international office locations. As of December 31, 2016, we have remaining lease obligations of $1.8 million, all of which is expected to be completed within a year. Rent expense, which includes common area maintenance, for the years ended December 31, 2016 and 2015 was $2.5 million and $2.5 million, respectively.
85
We are party to an agreement with Roche Diagnostics Corporation, our sole supplier of reagents, which requires us to make certain product purchase minimums. Pursuant to the agreement, as of December 31, 2016, we have a minimum purchase obligation as follows:
|
Years Ending December 31, |
|
|
Obligation |
|
|
2017 |
|
$ |
1,074 |
|
|
2018 |
|
|
1,074 |
|
|
2019 |
|
|
1,473 |
|
|
2020 |
|
|
1,473 |
|
|
2021 |
|
|
1,473 |
|
|
Total |
|
$ |
6,567 |
|
We are subject to various claims and contingencies related to legal proceedings. Due to their nature, such legal proceedings involve inherent uncertainties including, but not limited to, court rulings, negotiations between affected parties and governmental actions. Management assesses the probability of loss for such contingencies and accrues a liability and/or discloses the relevant circumstances, as appropriate. Management believes that any liability to us that may arise as a result of currently pending legal proceedings will not have a material adverse effect on our financial condition, liquidity, or results of operations as a whole.
|
8. |
Long-term Obligations |
On September 29, 2014 we entered into a 2nd Amendment to the 2013 Loan and Security Agreement (the “2013 Loan Agreement”) with Oxford and Silicon Valley Bank. Pursuant to the amended 2013 Loan Agreement, and we were provided a conditional waiver of principal payments subject to meeting certain capital raise requirements, which we achieved in October. The waiver of principal payments continued through April 1, 2015 and we were then required to make payments of principal and accrued interest in equal monthly installments sufficient to amortize the Term Loan through the maturity date.
On May 29, 2015, we entered into the Loan and Security Agreement, dated May 29, 2015 (the “Loan and Security Agreement”), with Oxford, pursuant to which Oxford funded an aggregate principal amount of $17.7 million (“Term Loan”), subject to the terms and conditions set forth in the Loan and Security Agreement. The Term Loan accrues interest at a floating rate of at least 8.95% per annum, comprised of three-month LIBOR rate with a floor of 1.00% plus 7.95%. Pursuant to the Loan and Security Agreement, we were previously required to make interest only payments through June 1, 2016 and thereafter we were required to make payments of principal and accrued interest in equal monthly installments sufficient to amortize the Term loan through June 1, 2019, the maturity date. On February 23, 2016, we received an acknowledgement and agreement from Oxford related to the positive data on our U.S. ACT-OA clinical trial. As a result, pursuant to the Loan and Security Agreement, the period for which we are required to make interest-only payments was extended from July 1, 2016 to January 1, 2017. All unpaid principal and interest with respect to the Term Loan is due and payable in full on June 1, 2019. At maturity of the Term Loan, or earlier repayment in full following voluntary prepayment or upon acceleration, we are required to make a final payment in an aggregate amount equal to approximately $1.1 million. In connection with the Term Loan, on May 29, 2015, we issued to Oxford warrants to purchase an aggregate of 94,441 shares of our common stock at an exercise price of $10.35 per share. These warrants became exercisable as of November 30, 2015 and will expire on May 29, 2025 and, following the authoritative accounting guidance, are equity classified.
In connection with the Loan and Security Agreement, we prepaid all outstanding amounts under our prior loan agreement with Oxford and Silicon Valley Bank, at which time the Company’s obligations under the prior loan agreement immediately terminated. We paid approximately $25.4 million to Oxford and Silicon Valley Bank, consisting of the then outstanding principal balance due of approximately $23.4 million, accrued but unpaid interest of approximately $0.2 million, final payment and other agency fees of approximately $1.8 million and other customary lender fees and expenses.
For Oxford, we accounted for this Term Loan as a debt modification. We retired $3.1 million of the principal of the previous loan and the corresponding unamortized fees were expensed. The remaining fees of $0.8 million were recorded as debt discount, and along with the new loan fees, are amortized as an adjustment of interest expense using the effective interest method. For Silicon Valley Bank, which did not participate in the Term Loan, the payoff of the loan was accounted for as debt extinguishment. Accordingly, a total loss on debt extinguishment of $0.3 million was recorded in 2015, which includes the unamortized fees and discounts along with final payment fees.
We allocated the aggregate proceeds of the Term Loan between the warrants and the debt obligations based on their relative fair values. The fair value of the warrants issued to Oxford was calculated utilizing the Black-Scholes option pricing model. The Black-Scholes option-pricing model incorporates various and highly sensitive assumptions including expected volatility, expected term and risk-free interest rates. The expected volatility is based on the historical volatility of the Company’s common
86
stock over the most recent period. The risk-free interest rate for period within the contractual life of the warrant is based on the U.S. Treasury yield in effect at the time of grant. We amortize the relative fair value of the warrants at the issuance date as a discount of $0.8 million over the term of the loan using the effective interest method, with an effective interest rate of 14.95%. The Term Loan is collateralized by a security interest in substantially all of the Company’s existing and subsequently acquired assets, subject to certain exceptions set forth in the Loan and Security Agreement and excluding its intellectual property assets, which are subject to a negative pledge. The minimum liquidity covenant is $5 million. As of December 31, 2016 we were in compliance with the debt covenants.
Additional details relating to the outstanding Term Loan as of December 31, 2016 and 2015 are presented in the following table (in thousands):
|
Year ended December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Origination Date |
|
Original Loan Amount |
|
|
Interest Rate** |
|
|
Current Monthly Payment* |
|
|
Original Term |
|
Remaining Principal (Face Value) |
|
||||
|
May 2015 |
|
$ |
17,700 |
|
|
|
8.95 |
% |
|
$ |
136 |
|
|
48 Months |
|
$ |
17,700 |
|
|
Year ended December 31, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Origination Date |
|
Original Loan Amount |
|
|
Interest Rate** |
|
|
Current Monthly Payment*** |
|
|
Original Term |
|
Remaining Principal (Face Value) |
|
||||
|
May 2015 |
|
$ |
17,700 |
|
|
|
8.95 |
% |
|
$ |
136 |
|
|
48 Months |
|
$ |
17,700 |
|
*Monthly payment as of December 2016, which reflects interest only
**3 month LIBOR rate with a floor of 1% plus 7.95%
As of December 31, 2016, the future contractual principal and final fee payments on all of our debt and capital lease obligations are as follows (as thousands):
|
Years Ending December 31, |
|
|
|
|
|
2017 |
|
$ |
7,080 |
|
|
2018 |
|
|
7,080 |
|
|
2019 |
|
|
4,629 |
|
|
Total |
|
$ |
18,789 |
|
|
Reconciliation of Face Value to Book Value as of December 31, 2016 |
|
|
|
|
|
Total debt and lease obligations, including final payment fee (Face Value) |
|
$ |
18,789 |
|
|
Less: Debt discount |
|
|
(1,152 |
) |
|
Total obligation |
|
$ |
17,637 |
|
Our interest expense for the years ended December 31, 2016 and 2015 was $2.6 million and $3.4 million, respectively. Interest expense is calculated using the effective interest method, therefore it is inclusive of non-cash amortization in the amount of $1.0 million and $1.0 million, respectively, related to the amortization of the debt discount related to the capitalized loan costs and accretion of final payment.
|
9. |
Income Taxes |
Due to our net losses for the years ended December 31, 2016 and 2015, and since we have recorded a full valuation allowance against deferred tax assets, there was no provision or benefit for income taxes recorded. We recorded an immaterial amount pertaining to current foreign income tax provision expense for the year ended December 31, 2016 and no components of current or deferred federal or state income tax provisions for the years ended December 31, 2015.
87
A reconciliation of the total income tax provision tax rate to the statutory federal income tax rate of 34% for the years ended December 31, 2016 and 2015 is as follows:
|
|
|
2016 |
|
|
2015 |
|
||
|
Income tax expense (benefit) at federal statutory rate |
|
|
(34.00 |
)% |
|
|
(34.00 |
)% |
|
Income tax expense (benefit) at state statutory rate |
|
|
(3.41 |
)% |
|
|
(4.40 |
)% |
|
Mark to market permanent adjustment |
|
|
0.00 |
% |
|
|
(13.91 |
)% |
|
Change in valuation allowance |
|
|
16.75 |
% |
|
|
(7.45 |
)% |
|
Change in state rate |
|
|
(0.06 |
)% |
|
|
(0.09 |
)% |
|
Permanent interest adjustments |
|
|
0.16 |
% |
|
|
6.25 |
% |
|
Stock compensation |
|
|
12.67 |
% |
|
|
20.43 |
% |
|
Transfer pricing |
|
|
0.00 |
% |
|
|
18.49 |
% |
|
Research credit |
|
|
(1.44 |
)% |
|
|
(2.37 |
)% |
|
Foreign rate differential |
|
|
0.79 |
% |
|
|
0.69 |
% |
|
NOLs expiring and adjustments to NOL |
|
|
6.00 |
% |
|
|
13.92 |
% |
|
Other, net |
|
|
2.54 |
% |
|
|
2.44 |
% |
|
|
|
|
0.00 |
% |
|
|
0.00 |
% |
The tax effects of temporary differences that give rise to significant portions of our deferred tax assets and deferred tax liabilities as of December 31, 2016 and 2015 are as follows (in thousands):
|
|
|
2016 |
|
|
2015 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Allowances and reserves |
|
$ |
573 |
|
|
$ |
673 |
|
|
Accrued expenses |
|
|
701 |
|
|
|
951 |
|
|
Deferred revenue and gain-on-sale |
|
|
33 |
|
|
|
39 |
|
|
Stock based compensation |
|
|
1,947 |
|
|
|
4,547 |
|
|
Net operating loss carryforwards |
|
|
125,182 |
|
|
|
119,000 |
|
|
Income tax credit carryforwards |
|
|
7,764 |
|
|
|
7,437 |
|
|
Property and equipment, principally due to differences in depreciation |
|
|
675 |
|
|
|
683 |
|
|
Other, net |
|
|
15 |
|
|
|
16 |
|
|
|
|
|
136,890 |
|
|
|
133,346 |
|
|
Valuation allowance |
|
|
(134,873 |
) |
|
|
(131,187 |
) |
|
Total deferred tax assets, net of allowance |
|
|
2,017 |
|
|
|
2,159 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Intangibles assets |
|
|
(2,017 |
) |
|
|
(2,159 |
) |
|
Total deferred tax liability |
|
|
(2,017 |
) |
|
|
(2,159 |
) |
|
Net deferred tax assets (liability) |
|
$ |
— |
|
|
$ |
— |
|
We have established a valuation allowance against our net deferred tax assets due to the uncertainty surrounding the realization of such assets. We periodically evaluate the recoverability of the deferred tax assets. At such time as it is determined that it is more likely than not that deferred assets are realizable, the valuation allowance will be reduced. We have recorded a full valuation allowance of $134.9 million as of December 31, 2016 as we do not believe it is more likely than not our net deferred tax assets will be realized. We increased our valuation allowance by approximately $3.7 million during the year ended December 31, 2016.
At December 31, 2016, we had federal, and state tax loss carry forwards of approximately $344.2 million, and $158.1 million. The federal and state net operating loss carry forwards begin to expire in 2019 and 2017, respectively, if unused. At December 31, 2016, we had federal and state tax credit carry forwards of approximately $4.9 million and $4.4 million, respectively, after reduction for uncertain tax positions. The Company has not performed a formal research and development credit study with respect to these credits. The federal credits will begin to expire in 2018, if unused, and the state credits carry forward indefinitely.
Pursuant to the Internal Revenue Code (“IRC”) of 1986, as amended, specifically IRC §382 and IRC §383, our ability to use net operating loss and R&D tax credit carry forwards (“tax attribute carry forwards”) to offset future taxable income is limited if we experience a cumulative change in ownership of more than 50% within a three-year testing period. We have not completed an ownership change analysis pursuant to IRC Section 382 for taxable years ended after December 31, 2007. If ownership changes
88
within the meaning of IRC Section 382 are identified as having occurred subsequent to 2007, the amount of remaining tax attribute carry forwards available to offset future taxable income and income tax expense in future years may be significantly restricted or eliminated. Further, our deferred tax assets associated with such tax attributes could be significantly reduced upon realization of an ownership change within the meaning of IRC §382.
We recognize tax benefits associated with the exercise of stock options directly to stockholders’ equity only when realized. Accordingly, deferred tax assets are not recognized for net operating loss carry forwards resulting from windfall tax benefits. At December 31, 2016, deferred tax assets do not include $1.3 million of excess tax benefits from stock-based compensation.
The Company follows the provisions of income tax guidance which provides recognition criteria and a related measurement model for uncertain tax positions taken or expected to be taken in income tax returns. The guidance requires that a position taken or expected to be taken in a tax return be recognized in the financial statements when it is more likely than not that the position would be sustained upon examination by tax authorities. Tax positions that meet the more likely than not threshold are then measured using a probability weighted approach recognizing the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate settlement. The Company has not recognized any liability for uncertain tax positions as of December 31, 2016 and 2015.
Following is a tabular reconciliation of the unrecognized tax benefits activity during the years ended December 31, 2016 and 2015 (in thousands):
|
|
|
2016 |
|
|
2015 |
|
||
|
Unrecognized Tax Benefits – Beginning |
|
$ |
1,987 |
|
|
$ |
1,852 |
|
|
Gross increases – tax positions in prior period |
|
|
1 |
|
|
|
— |
|
|
Gross decreases – tax positions in prior period |
|
|
(13 |
) |
|
|
— |
|
|
Gross increase – current-period tax positions |
|
|
87 |
|
|
|
135 |
|
|
Unrecognized Tax Benefits – Ending |
|
$ |
2,062 |
|
|
$ |
1,987 |
|
The unrecognized tax benefit amounts are reflected in the determination of the Company’s deferred tax assets. If recognized, none of these amounts would affect the Company’s effective tax rate, since it would be offset by an equal reduction in the deferred tax asset valuation allowance. The Company does not foresee material changes to its liability for uncertain tax benefits within the next twelve months.
The Company’s material tax jurisdictions are United States and California. The Company is currently not under examination by the Internal Revenue Service or any other taxing authority.
The Company’s tax years for 1998 (federal) and 1997 (CA) and forward can be subject to examination by the United States and California tax authorities due to the carry forward of net operating losses and research development credits.
|
10. |
Employee Benefit Plan |
We implemented a 401(k) retirement savings and profit sharing plan (the “Plan”) effective January 1, 1999. We may make discretionary annual contributions to the Plan, which is allocated to the profit sharing accounts based on the number of years of employee service and compensation. At the sole discretion of the Board of Directors, we may also match the participants’ contributions to the Plan. We made no discretionary or matching contributions to the Plan in 2016 or 2015.
|
11. |
Stockholders’ Equity |
Preferred Stock
We have authorized 5 million shares of $0.001 par value preferred stock. Our Board of Directors is authorized to designate the terms and conditions of any preferred stock we issue without further action by the common stockholders. There were 13,500 shares of Series A 3.6% Convertible Preferred Stock that had been issued at December 31, 2016 and 2015, none of which were outstanding as of either date.
All outstanding shares of the Series A 3.6% Convertible Preferred Stock were converted into common stock during the fourth quarter of 2014 and the first quarter of 2015 at the option of the holders. The fair value of the common stock into which the Series A 3.6% Convertible Preferred Stock was convertible on the date of issuance exceeded the proceeds allocated to the preferred stock, resulting in the beneficial conversion feature that we recognized as a dividend to the preferred stockholders and, accordingly, an adjustment to net loss to arrive at net loss allocable to common stockholders. Certain shares of Series A 3.6% Convertible Preferred Stock were not convertible until stockholder approval, which occurred in January 2015. As a result, a dividend for the beneficial conversion feature of $0.7 million was recorded during the quarter ended March 31, 2015.
89
In connection with the 3.6% Convertible Preferred Stock outstanding at December 31, 2014, we declared a cash dividend of $0.07 million. The cash dividend was paid in January and April 2015.
Common Stock
In May 2015, the Company entered into a Securities Purchase Agreement with certain institutional investors pursuant to which the Company agreed to sell up to $25.0 million of units, with each unit consisting of one share of its common stock and one warrant to purchase one share of its common stock, in a registered direct offering. The purchase and sale of the units took place in two separate closings. At the initial closing, which took place on May 8, 2015, the Company received approximately $17.4 million in net proceeds from the sale of units. The second closing occurred on August 27, 2015 upon satisfaction of certain conditions, including, without limitation, stockholder vote, and the Company received approximately $2.1 million in net proceeds from the sale of 500,000 units of the 1,000,000 units available for sale at the second closing.
On December 17, 2015, the Company and the holders of October 2014 warrants agreed to amend the October 2014 Warrants pursuant to an Amendment to Common Stock Purchase Warrant (the “2014 Amendment”). Also on December 17, 2015, the Company and the holders of the May 2015 Warrants and the August 2015 Warrants (collectively the “2015 Warrants”) agreed to amend the 2015 Warrants pursuant to an Amendment to Series A-1 Warrant to Purchase Common Stock and Amendment to Series A-2 Warrant to Purchase Common Stock, respectively (the “2015 Amendment” and, together with the 2014 Amendment, the “Warrant Amendments”). The Warrant Amendments provided that the holders may exercise their warrants on a “cashless exercise” basis in whole on or prior to December 31, 2015, whereby each exercising holder of the amended 2015 Warrants would receive 0.75 shares for each warrant share exercised and each exercising holder of the amended 2014 Warrants would receive 0.69 shares for each warrant share exercised. In addition, the Warrant Amendments removed certain provisions which provided that the exercise price of the Warrants would be reset in the event of certain equity issuances by the Company for a price below the exercise price of the Warrants at the time of such issuance. All 2014 Warrants and all 2015 Warrants were cashless exercised on or before December 31, 2015.
During 2016, we sold 1,840,982 shares of our common stock under an at-the-market offering program (“ATM”), receiving total net proceeds of approximately $4.4 million. During 2015, we sold 5,800,000 shares of our common stock under the ATM program, receiving total net proceeds of approximately $7.2 million.
Pursuant to a registration statement on Form S-1, originally filed on April 6, 2016, as amended, and declared effective by the SEC on May 26, 2016, and related prospectus (as supplemented), the Company registered, offered and sold to its participating stockholders of record as of the announced May 20, 2016 record date, one non-transferable subscription right for each share of common stock held by each stockholder as of the record date. Each right entitled the holder thereof to purchase one unit at the subscription price of $2.55 per unit, composed of one share of common stock and 0.5 of a warrant, with each whole warrant exercisable to purchase one share of common stock at an exercise price of $3.06 per share for 30 months from the date of issuance. Pursuant to the Rights Offering, which closed on June 15, 2016, the Company sold an aggregate of 6,704,852 units, resulting in total net proceeds to the Company of $15.3 million, respectively. The warrants issued pursuant to the Rights Offering are currently listed on NASDAQ under the symbol “CTYXW.” Based on the relevant authoritative accounting guidance, the warrants were equity classified at the issuance date. Upon notice to the warrant holders, the warrants may be redeemed by the Company at $0.01 per warrant prior to their expiration and exercise if the Company’s common stock closes above $7.65 per share for 10 consecutive trading days.
On December 22, 2016, we entered into the Lincoln Park Purchase Agreement pursuant to which we have the right to sell to Lincoln Park and Lincoln Park is obligated to purchase up to $20.0 million in amounts of shares, of our common stock, over the 30-month period commencing on the date that a registration statement, which we filed with the SEC in December 2016. We may direct Lincoln Park, at its sole discretion and subject to certain conditions, to purchase up to 100,000 shares of common stock on any business day but in no event will the amount of a single Regular Purchase (as defined in the Lincoln Park Purchase Agreement) exceed $1.0 million. The purchase price of shares of common stock related to the Regular Purchases will be based on the prevailing market prices of such shares at the time of sales. Our sales of shares of common stock to Lincoln Park under the Lincoln Park Purchase Agreement are limited to no more than the number of shares that would result in the beneficial ownership by Lincoln Park and its affiliates, at any single point in time, of more than 9.99% of the then outstanding shares of the common stock. There are no trading volume requirements or restrictions under the Lincoln Park Purchase Agreement. There is no upper limit on the price per share that Lincoln Park must pay for common stock under a Regular Purchase or an accelerated purchase and in no event will shares be sold to Lincoln Park on a day our closing price is less than the floor price of $0.50 per share as set forth in the Purchase Agreement. On December 22, 2016, we issued to Lincoln Park 127,419 shares of common stock with a market value on the date of issuance of approximately $0.2 million as commitment shares in consideration for entering into the Lincoln Park Purchase Agreement. We will issue up to an additional 382,258 shares of common stock on a pro rata basis to Lincoln Park only as and when shares are sold under the Lincoln Park Purchase Agreement to Lincoln Park. To date, we have sold no shares under the Lincoln Park Purchase Agreement with Lincoln Park.
90
|
12. |
Stock-based Compensation |
In August 2014, we adopted the 2014 Equity Incentive Plan (the “2014 Plan”), which provides our employees, directors and consultants the opportunity to purchase our common stock in the form of options (incentive or non-qualified), stock appreciation rights, restricted stock purchase rights, restricted stock bonuses, restricted stock units, performance shares, performance units, cash-based awards other stock-based awards, and deferred compensation awards. The 2014 Plan initially provides for issuance of 265,000 shares of our common stock. In May 2016, the Company amended the 2014 Plan to add 333,333 shares to its share pool. In August 2015, the Company amended the 2014 Plan to add 301,800 shares to its share pool. In addition, the amendment increased the number of “incentive stock options” which may be issued under the 2014 Plan by an identical amount.
On December 29, 2015, we adopted the 2015 New Employee Incentive Plan (the “2015 Plan”). Awards under the 2015 Plan may only be made to an employee who has not previously been an employee or member of the Board of any parent or subsidiary, or following a bona fide period of non-employment by the Company or a parent or subsidiary, if he or she is granted such award in connection with his or her commencement of employment with the Company or a subsidiary and such grant is an inducement material to his or her entering into employment with the Company or such subsidiary. The 2015 Plan provides for issuance of 66,666 shares.
As of December 31, 2016, there are 525,965 shares of common stock remaining and available for future issuances under the 2014 Plan, which is exclusive of securities to be issued upon an exercise of outstanding options, warrants, and rights.
Stock Options
Generally, options issued under the 2014 Plan, are subject to four-year vesting, and have a contractual term of 10 years. Most options contain one of the following two vesting provisions:
|
|
• |
12/48 of a granted award will vest after one year of service, while an additional 1/48 of the award will vest at the end of each month thereafter for 36 months, or |
|
|
• |
1/48 of the award will vest at the end of each month over a four-year period. |
A summary of activity for the year ended December 31, 2016 is as follows:
|
|
|
Options |
|
|
Weighted Average Exercise Price |
|
||
|
Balance as of January 1, 2016 |
|
|
573,727 |
|
|
$ |
44.85 |
|
|
Granted |
|
|
347,407 |
|
|
$ |
2.73 |
|
|
Expired |
|
|
(23,979 |
) |
|
$ |
104.11 |
|
|
Cancelled/forfeited |
|
|
(261,043 |
) |
|
$ |
32.07 |
|
|
Balance as of December 31, 2016 |
|
|
636,112 |
|
|
$ |
24.39 |
|
|
|
|
Options |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Term (years) |
|
|
Aggregate Intrinsic Value |
|
||||
|
Balance as of December 31, 2016 |
|
|
636,112 |
|
|
$ |
24.39 |
|
|
|
7.38 |
|
|
$ |
— |
|
|
Vested and expected to vest at December 31, 2016 |
|
|
598,135 |
|
|
$ |
25.72 |
|
|
|
7.27 |
|
|
$ |
— |
|
|
Exercisable at December 31, 2016 |
|
|
286,358 |
|
|
$ |
47.76 |
|
|
|
5.54 |
|
|
$ |
— |
|
There were no stock options exercised in 2016 or 2015.
The fair value of each option awarded during the year ended December 31, 2016 and 2015 was estimated on the date of grant using the Black-Scholes-Merton option valuation model based on the following weighted-average assumptions:
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Expected term |
|
6.0 years |
|
|
6.0 years |
|
||
|
Risk-free interest rate |
|
|
1.75 |
% |
|
|
1.58 |
% |
|
Volatility |
|
|
77.56 |
% |
|
|
75.07 |
% |
|
Dividends |
|
|
— |
|
|
|
— |
|
|
Resulting weighted average grant date fair value |
|
$ |
1.84 |
|
|
$ |
4.50 |
|
91
The weighted average risk-free interest rate represents the interest rate for treasury constant maturity instruments published by the Federal Reserve Board. If the term of available treasury constant maturity instruments is not equal to the expected term of an employee option, we use the weighted average of the two Federal Reserve securities closest to the expected term of the employee option.
The dividend yield has been assumed to be zero as we (a) have never declared or paid any dividends and (b) do not currently anticipate paying any cash dividends on our outstanding shares of common stock in the foreseeable future.
Restricted Stock Awards
Generally, restricted stock awards issued under the 2014 Plan are subject to a vesting period that coincides with the fulfillment of service requirements for each award and have a contractual term of 10 years. These awards are amortized to compensation expense over the estimated vesting period based upon the fair value of our common stock on the award date.
A summary of activity for the year ended December 31, 2016 is as follows:
|
|
|
Restricted Stock Awards |
|
|
Weighted Average Grant Date Fair Value |
|
||
|
Balance as of January 1, 2016 |
|
|
31,196 |
|
|
$ |
12.15 |
|
|
Vested/Released |
|
|
(11,568 |
) |
|
$ |
15.18 |
|
|
Cancelled/forfeited |
|
|
(19,113 |
) |
|
$ |
10.03 |
|
|
Balance as of December 31, 2016 |
|
|
515 |
|
|
$ |
64.52 |
|
The following summarizes the total compensation cost recognized for the stock options and restricted stock awards in the accompanying financial statements (in thousands):
|
|
|
Years ended December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Total compensation cost for share-based payment arrangements recognized in the statement of operations (net of tax of $0) |
|
$ |
1,080 |
|
|
$ |
2,041 |
|
As of December 31, 2016, the total compensation cost related to non-vested stock options and stock awards not yet recognized for all our plans is approximately $1.0 million, which is expected to be recognized as a result of vesting under service conditions over a weighted average period of 1.58 years.
To settle stock options and restricted stock awards, we will issue new shares of our common stock. At December 31, 2016, we have an aggregate of 49,708,768 shares authorized and available to satisfy option exercises under our plans.
|
13. |
Quarterly Information (unaudited) |
The following unaudited quarterly financial information includes, in management’s opinion, all the normal and recurring adjustments necessary to fairly state the results of operations and related information for the periods presented (in thousands):
|
|
|
For the three months ended |
|
|||||||||||||
|
|
|
March 31, 2016 |
|
|
June 30, 2016 |
|
|
September 30, 2016 |
|
|
December 31, 2016 |
|
||||
|
Product revenues |
|
$ |
1,333 |
|
|
$ |
1,126 |
|
|
$ |
731 |
|
|
$ |
1,466 |
|
|
Gross profit |
|
|
766 |
|
|
|
541 |
|
|
|
113 |
|
|
|
521 |
|
|
Development revenues |
|
|
1,585 |
|
|
|
1,699 |
|
|
|
1,879 |
|
|
|
1,561 |
|
|
Operating expenses |
|
|
(7,448 |
) |
|
|
(8,464 |
) |
|
|
(6,789 |
) |
|
|
(5,670 |
) |
|
Other expense, net |
|
|
(242 |
) |
|
|
(181 |
) |
|
|
(587 |
) |
|
|
(1,330 |
) |
|
Net income (loss) |
|
$ |
(5,339 |
) |
|
$ |
(6,405 |
) |
|
$ |
(5,384 |
) |
|
$ |
(4,918 |
) |
|
Beneficial conversion feature for convertible preferred stock |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net income (loss) allocable to common stock holders |
|
|
(5,339 |
) |
|
|
(6,405 |
) |
|
|
(5,384 |
) |
|
|
(4,918 |
) |
|
Basic and diluted net loss per share |
|
$ |
(0.41 |
) |
|
$ |
(0.43 |
) |
|
$ |
(0.26 |
) |
|
$ |
(0.24 |
) |
92
|
|
|
For the three months ended |
|
|||||||||||||
|
|
|
March 31, 2015 |
|
|
June 30, 2015 |
|
|
September 30, 2015 |
|
|
December 31, 2015 |
|
||||
|
Product revenues |
|
$ |
902 |
|
|
$ |
1,614 |
|
|
$ |
766 |
|
|
$ |
1,556 |
|
|
Gross profit |
|
|
305 |
|
|
|
318 |
|
|
|
264 |
|
|
|
765 |
|
|
Development revenues |
|
|
1,444 |
|
|
|
1,847 |
|
|
|
1,710 |
|
|
|
1,820 |
|
|
Operating expenses |
|
|
(22,745 |
) |
|
|
3,626 |
|
|
|
16 |
|
|
|
(4,656 |
) |
|
Other expense, net |
|
|
(961 |
) |
|
|
(1,342 |
) |
|
|
(470 |
) |
|
|
(685 |
) |
|
Net income (loss) |
|
$ |
(21,957 |
) |
|
$ |
4,449 |
|
|
$ |
1,520 |
|
|
$ |
(2,756 |
) |
|
Beneficial conversion feature for convertible preferred stock |
|
|
(661 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net income (loss) allocable to common stock holders |
|
|
(22,618 |
) |
|
|
4,449 |
|
|
|
1,520 |
|
|
|
(2,756 |
) |
|
Basic and diluted net loss per share |
|
$ |
(3.19 |
) |
|
$ |
0.45 |
|
|
$ |
0.15 |
|
|
$ |
(0.25 |
) |
|
14. |
Subsequent Events |
Azaya Therapeutics, Inc. Assets
On February 15, 2017 (the “Closing Date”), Cytori completed the acquisition from Azaya Therapeutics, Inc. (“Azaya”) of substantially all of the assets and the assumption of certain of liabilities, pursuant to an Asset Purchase Agreement. Pursuant to the Acquisition, Cytori has acquired the rights, title and interest in and to (i) Azaya’s ATI-0918 drug candidate, a generic bioequivalent formulation of DOXIL/CAELYX, a chemotherapy drug that is a liposomal encapsulation of doxorubicin (ATI-0918); (ii) Azaya’s ATI-1123 drug candidate, a liposomal formulation of docetaxel (ATI-1123); and (iii) certain equipment, inventory and other assets necessary to develop, manufacture, test and validate ATI-0918 and ATI-1123.
Under the terms of the Purchase Agreement, at the closing of the Acquisition, the Company (i) issued 1,173,241 of shares of its common stock, par value, $0.001 per share , in Azaya’s name, (A) 879,931 of which will be delivered to Azaya promptly after the Closing, and (B) 293,310 of which will be deposited into a 15-month escrow pursuant to a standard escrow agreement; and (ii) assumed the obligation to pay approximately $2.0 million of Azaya’s existing trade payables, which payments the Company intends to make at or within thirty days after the Closing. The price per share was $1.7047, which price was equal to the volume weighted average closing price of the shares on the Nasdaq Capital Market over the ten consecutive trading days ending on the trading date immediately prior to the date of the Closing Date.
In addition, as of the Closing Date, the Company assumed obligations to: (i) pay Azaya fixed commercialization milestone payments based upon achievement of certain net sales milestones for ATI-0918; (ii) make certain earn-out payments to Azaya equal to a mid-single-digit percentage of net sales of ATI-0918; and (iii) make certain earn-out payments to Azaya equal to a low single-digit percentage of net sales of any product (each a “Patented Product”), including ATI-1123, that practices a claim in the related patent assigned by Azaya to the Company (the “ATI-1123 Patent”). Cytori’s aggregate earn-out payment obligations to Azaya from global net sales of both ATI-0918 and any Patented Product will not exceed $100.0 million (the “Earn-Out Cap”).
Further, the Purchase Agreement provides that if Cytori enters into certain assignments, licenses or other transfers of rights to a Patented Product or the ATI-1123 Patent, the Company will pay Azaya a percentage in the low to mid-teens of the consideration received by the Company, provided, that Cytori’s aggregate payment obligation to Azaya for any such assignment, license or other transfer of rights will not exceed $50.0 million.
If the Company or its successors, sublicenses or transferees sells a competing product to ATI-0918 at any time prior to satisfaction of the Earn-Out Cap, other than because ATI-0918 fails to receive marketing authorization from the European Medicines Agency within a certain period of time or fails to generate a minimum threshold of net sales within a pre-determined amount of time, then 50% of the net sales of such competing product would be deemed to be net sales of ATI-0918 under the Purchase Agreement for purposes of calculating commercialization milestone payments and earn-out payments.
Both the Company and Azaya agreed to customary representations, warranties and covenants in the Purchase Agreement. Each party also agreed to customary indemnification obligations, provided, that Azaya’s maximum liability to the Company for breaches of Azaya’s representations and warranties in the Purchase Agreement and any ancillary agreements entered into in connection therewith, is limited to $3.9 million, subject to limited exceptions.
93
Lease Agreement
On February 27, 2017, Cytori entered into a Lease Agreement (the “Lease”) with 6262 Lusk Investors LLC, a California limited liability company (“Landlord”), for approximately 29,499 square feet of office space for the Company’s corporate headquarters in San Diego, California. The initial term of the Lease is 63 months, and may be extended upon mutual agreement of the Company and the Landlord. The Lease is scheduled to commence on November 1, 2017 date, unless the premises are earlier occupied by the Company or the commencement date is delayed to allow for substantial completion of tenant improvements.
Under the Lease, the Company will be obligated to pay base rent as follows (in thousands):
|
|
|
• |
Year 1: $761; |
|
|
|
• |
Year 2: $784; |
|
|
|
• |
Year 3: $807; |
|
|
|
• |
Year 4: $832; |
|
|
|
• |
Year 5: $857; |
|
|
|
• |
Months 61-63: $74 per month ($882 annualized base rent). |
In addition to the base rent, the Company will also be obligated under the Lease to make certain payments for operating expenses, property taxes, insurance, insurance deductibles and other amounts.
In connection with the Lease, the Company issued a letter of credit, or Letter of Credit, in favor of the Landlord in the initial principal amount of $0.1 million, which Letter of Credit will increase to $0.3 million on June 1, 2017, and to $0.5 million on the commencement date. The Letter of Credit will remain in effect for the term of the Lease.
The Company has agreed to customary indemnifications of the Landlord and its affiliates arising out of the Company’s use of the rented premises, breaches of the Company’s obligations under the Lease and similar matters (except to the extent arising out of the Landlord’s gross negligence or willful misconduct).
94
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
|
|
(a) |
Evaluation of Disclosure Controls and Procedures |
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports filed or furnished pursuant to the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by Rule 13a-15(b) under the Exchange Act, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended, as of the end of the period covered by this Annual Report on Form 10-K. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this Annual Report were effective.
|
|
(b) |
Management’s Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
|
• |
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
|
|
• |
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of management and our Board of Directors; and |
|
|
• |
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we have conducted an evaluation of the effectiveness of our internal control over financial reporting as of the end of the fiscal year covered by this annual report on Form 10-K based on the criteria set forth in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on our evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2016 based on the COSO criteria.
This report does not include an attestation report on internal control over financial reporting by the Company’s independent registered public accounting firm since the Company is a smaller reporting company under the rules of the SEC.
|
|
(c) |
Changes in Internal Control over Financial Reporting |
There have been no changes in our internal control over financial reporting during the quarter ended December 31, 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
95
Item 10. Directors, Executive Officers and Corporate Governance
The following table sets forth biographical information regarding our directors as of February 28, 2017
Directors and Business Experience
|
Name |
Age |
Position |
|
David M. Rickey |
61 |
Chairman of the Board |
|
Marc H. Hedrick, MD |
54 |
President and Chief Executive Officer and Director |
|
Richard J. Hawkins |
68 |
Director |
|
Paul W. Hawran |
64 |
Director |
|
Gary A. Lyons |
65 |
Director |
|
Ronald A. Martell |
55 |
Director |
|
Gail K. Naughton, Ph.D. |
61 |
Director |
David M. Rickey has served on our Board since November 1999 and has served as the Chairman of our Board since June 2013. Mr. Rickey was previously President and Chief Executive Officer of Applied Micro Circuits Corporation, or AMCC, a publicly-held company that provides high-performance, high-bandwidth silicon solutions for optical networks, from February 1996 to March 2005. Mr. Rickey served on the Board of AMCC from February 1996 to March 2005, and as its Chairman from August 2000 to March 2005. Mr. Rickey also served as a director of AMI Semiconductor, Inc. from 2000 to 2006 and was a director of Netlist, Inc. from 2005 to 2008, as well as several private technology companies. He holds a B.S. from Marietta College, a B.S. from Columbia University and an M.S. from Stanford University. Mr. Rickey’s qualifications to sit on our Board include his extensive executive experience and his service on other public company boards and committees.
Marc H. Hedrick, M.D. was appointed as Chief Executive Officer of the Company in April 2014. He was appointed as President of the Company in May 2004, and joined us as Chief Scientific Officer and Medical Director in October 2002. Dr. Hedrick has also served as a member of our Board since October 2002. In December 2000, Dr. Hedrick co-founded and served as President and Chief Executive Officer and Director of StemSource, Inc., a privately-held company specializing in stem cell research and development, which was acquired by us in 2002. He is a plastic surgeon and is a former Associate Professor of Surgery and Pediatrics at the University of California, Los Angeles, or UCLA. From 1998 until 2005, he directed the Laboratory of Regenerative Bioengineering and Repair for the Department of Surgery at UCLA. Dr. Hedrick earned his M.D. degree from University of Texas Southwestern Medical School, Dallas and an M.B.A. from UCLA Anderson School of Management. Dr. Hedrick’s qualifications to sit on our Board include his experience as a general, vascular and plastic surgeon; his academic appointments and achievements in the life sciences; his executive and managerial experience in stem cell research and scientific product development; and his foundational knowledge and experience of and contributions to our technology and operations. In addition, Dr. Hedrick has extensive global experience and familiarity with the cell therapy and regenerative medical industry.
Richard J. Hawkins has served on our Board since December 2007. In 1982, Mr. Hawkins founded Pharmaco, a clinical research organization, or CRO, that merged with the predecessor of PPD-Pharmaco in 1991 and is one of the largest CROs in the world today. In 1992, Mr. Hawkins co-founded Sensus Drug Development Corporation, or SDDC, a privately-held company focused on the treatment of drugs to treat endocrine disorders, which developed and received regulatory approval for SOMAVERT®, a growth hormone antagonist approved for the treatment of acromegaly, which is now marketed by Pfizer, Inc., and he served as Chairman of SDDC until 2000. In 1994, Mr. Hawkins co-founded Corning Biopro, a contract protein manufacturing firm, where he served on the Board until Corning BioPro’s sale to Akzo-Nobel, N.V., a publicly-held producer of paints, coatings and specialty chemicals, in 2000. In September 2003 Mr. Hawkins founded LabNow, Inc., a privately held company that develops lab-on-a-chip sensor technology, where he served as the Chairman and CEO until October 2009. Mr. Hawkins has served on the Board of SciClone Pharmaceuticals, Inc., a publicly-held specialty pharmaceutical company, since October 2004. In February 2011, Mr. Hawkins became CEO, and is currently CEO, of Lumos Pharma, Inc., a privately-held pharmaceutical company. He served on the Presidential Advisory Committee for the Center for Nano and Molecular Science and Technology at the University of Texas in Austin, and was inducted into the Hall of Honor for the College of Natural Sciences at the University of Texas. Mr. Hawkins graduated cum laude with a B.S. in Biology from Ohio University. Mr. Hawkins’s qualifications to sit on our Board include his executive experience working with life sciences companies, his extensive experience in pharmaceutical research and development, his knowledge, understanding and experience in the regulatory development and approval process and his service on other public company boards and committees.
96
Paul W. Hawran has served on our Board since February 2005. Mr. Hawran has held various executive, strategic, financial and operational positions in the health care industry for over 30 years. Mr. Hawran was a founder and President and CEO of Ascendant MDx, a molecular diagnostic testing company focused on women’s health care, through June 2013. Prior to Ascendant MDx, Mr. Hawran was the Chief Financial Officer of Sequenom, Inc., a publicly traded genetics company, from April 2007 to September 2009, served on their Board from August 2006 to February 2007 and was the Chairman of the Audit Committee of the Board. Mr. Hawran also served as a Founder, Executive Vice President and Chief Financial Officer of Neurocrine Biosciences, or Neurocrine, a publicly traded company engaged in pharmaceutical drug development, from May 1993 through September 2006, and as a Senior Advisor to Neurocrine from September 2006 through April 2007. Mr. Hawran was employed by SmithKline Beecham (now Glaxo SmithKline) from July 1984 to May 1993, most recently as Vice President and Treasurer. Prior to joining SmithKline Beecham in 1984, he held various financial positions at Warner Communications (now Time Warner) involving corporate finance and financial planning and forecasting. Mr. Hawran earned a B.S. in Finance from St. John's University and an M.S. in Taxation from Seton Hall University. He is a Certified Public Accountant (currently inactive) and is a member of the American Institute of Certified Public Accountants. Mr. Hawran’s qualifications to sit on our Board include his executive experience in life sciences industries, his extensive experience in strategic and corporate finance and planning, his status as an audit committee financial expert within the meaning of Item 407(d)(5) of SEC Regulation S-K and his service on other public company boards and committees
Gary A. Lyons has served on our Board since October 2013. Mr. Lyons has served on the Board of Neurocrine Biosciences, Inc., or Neurocrine, since 1993 and served as the President and Chief Executive Officer of Neurocrine from 1993 through January 2008. Prior to joining Neurocrine, Mr. Lyons held a number of senior management positions at Genentech, Inc., including Vice President of Business Development and Vice President of Sales. Mr. Lyons has served on the Boards of Rigel Pharmaceuticals, Inc., a publicly-held biotechnology company, since October 2005 (and as Chairman since November 2014); Vical Incorporated, a publicly-held biopharmaceutical company, since 1997; and Retrophin, Inc., a publicly-held biopharmaceutical company, since 2014 (and as Chairman since May 2016). Mr. Lyons was previously a director of PDL BioPharma, Inc., Poniard Pharmaceuticals, Inc., Neurogesx, KaloBios Pharmaceuticals, Inc. and Facet Biotech Corporation. Mr. Lyons holds a B.S. in Marine Biology from the University of New Hampshire and an M.B.A. from Northwestern University’s J.L. Kellogg Graduate School of Management. Mr. Lyons’ qualifications to sit on our Board include his executive experience working with life sciences companies, his extensive experience in pharmaceutical business development, his knowledge, understanding and experience in the regulatory development and approval process and his service on other public company boards and committees.
Ronald A. Martell has served on our Board since December 2016. Mr. Martell has more than 25 years’ experience building and managing unique businesses in the biotech industry. Mr. Martell is currently a founder of Achieve Life Sciences, ORCA BioSystems, Inc. and Cetya Therapeutics, Inc. Most recently he served as Chief Executive Officer of Sevion Therapeutics and Executive Chairman of KaloBios Pharmaceuticals, Inc. Prior to Sevion, Mr. Martell was President and CEO of NeurogesX and sold the company’s assets to Acorda Therapeutics. Prior to NeurogesX he was Chief Executive Officer of Poniard Pharmaceuticals. Before joining Poniard he served in the capacity of the Office of the CEO and as Senior Vice President of Commercial Operations at ImClone Systems. Mr. Martell built ImClone Systems' Commercial Operations and field sales force to market and commercialize Erbitux® with partners Bristol-Myers Squibb and Merck KGaA. Prior to joining ImClone Systems, Mr. Martell worked for 10 years at Genentech, Inc., or Genentech, in a variety of positions, the last of which was Group Manager, Oncology Products. At Genentech, he was responsible for the launch of Herceptin® for metastatic HER-2 positive breast cancer and Rituxan® for non-Hodgkin's lymphoma. Mr. Martell began his career at Roche Pharmaceuticals. Mr. Martell’s qualifications to sit on our Board include his executive experience working for life sciences companies, his extensive experience in pharmaceutical business development, his knowledge, understanding of and experience in developing and commercializing pharmaceutical products, and his service on other public company boards and committees.
Gail K. Naughton, Ph.D., has served on our Board since July 2014. Dr. Naughton is the founder of Histogen, Inc., or Histogen, a private regenerative medicine company developing innovative therapies based upon the products of cells grown under simulated embryonic conditions. She has served as Histogen’s Chief Executive Officer and Chairman of the Board since the company’s inception in 2007. Prior to that, Dr. Naughton held key management positions, including President, Chief Operating Officer and Director, at Advanced Tissue Sciences, a company which she co-founded and was co-inventor of the core technology. Dr. Naughton has also served on the Board of C.R. Bard, Inc. since July 2004. Dr. Naughton holds a B.S. in Biology from St. Francis College as well as a Master’s in Histology and a Ph.D. from New York University Medical Center. She also holds an EMBA from the Anderson School of Business at the University of California, Los Angeles. Dr. Naughton’s qualifications to sit on our Board include her extensive executive experience, her in-depth knowledge of the healthcare industry and regenerative medicine technology, and her service on other public company boards and committees.
97
Executive Officers and Business Experience
The following table sets forth biographical information regarding our executive officers as of February 28, 2017.
|
Name |
Age |
Position(s) |
|
Marc H. Hedrick, M.D.(1) |
54 |
President, Chief Executive Officer and Director |
|
Tiago Girão |
37 |
Vice President, Finance & Chief Financial Officer |
|
John Harris |
48 |
Vice President and General Manager of Cell Therapy |
|
Mark Marino, M.D. |
57 |
Senior Vice President and Chief Medical Officer |
|
Jeremy Hayden |
47 |
General Counsel, Chief Compliance Officer, Secretary and Vice President of Business Development |
|
|
(1) |
See “Directors and Business Experience” above for biographical information regarding Dr. Hedrick. |
Tiago Girão joined us as Vice President of Finance and Chief Financial Officer in September 2014. Mr. Girão joined us from NuVasive, Inc., or NuVasive, a publicly-held medical device company, where he last served as International Controller from February 2014 to August 2014. Prior to his position as International Controller, he served as NuVasive’s Director of Financial Reporting from March 2012 to February 2014. In his position as Director of Financial Reporting, Mr. Girao managed a team responsible for all corporate technical accounting and SEC-related matters for Nuvasive. Prior to joining NuVasive, Mr. Girão served as Senior Manager, Assurance at KPMG, LLP from October 2004 to March 2012. Prior to joining KPMG, Mr. Girão was a senior accountant for Ernst &Young in Brazil from October 2000 to August 2004. Mr. Girão is a certified public accountant with over 15 years’ experience in the accounting, finance and reporting for U.S. and public companies and substantial experience in global finance and operations.
John D. Harris has served as our Vice President and General Manager of Cell Therapy since he joined us in October 2015. Mr. Harris has over 20 years’ experience in medical device and biotechnology, most recently serving as the Vice President and General Manager of Becton Dickinson’s operations in Japan. Prior to Becton Dickinson, Mr. Harris held business development, product development, and marketing and sales leadership roles with Tyco Electronics (now TE Connectivity Corp.), Delphi Automotive, Sorenson Medical, Kimberly-Clark Healthcare and Ballard Medical Products. Mr. Harris is a member of the Board of Governors of the American Chamber of Commerce in Japan (ACCJ) and a member of the Executive Committee of the American Medical Device & Diagnostics Association, where he chairs the Regenerative Medicine Working Group. Mr. Harris holds Master of Business Administration and Bachelor of Arts degrees from the University of Utah.
Mark Marino, M.D. joined us as Senior Vice President of Medical Affairs in May 2016, and was also appointed as Chief Medical Officer of the Company in August 2016. Before joining us, Dr. Marino served as Senior Vice President of Early Clinical Development for Turing Pharmaceuticals from November 2015 to May 2016. Prior to Turing, Dr. Marino served as Executive Director of Clinical Development at Daiichi-Sankyo from September 2012 to February 2013, and then as Vice President of Clinical Development at Daiichi-Sankyo from February 2013 to November 2015. Prior to Daiichi-Sankyo, Dr. Marino held various senior clinical positions at Archimedes Pharma, Inc., MannKind Corporation and Hoffman-LaRoche from August 2006 to September 2012. Dr. Mario also previously served as Chief of the Department of Pharmacology at the Walter Reed Army Institute of Research as well as Associate Professor of Medicine at the Uniformed Services University of the Health Sciences and a staff physician at the Walter Reed Army Medical Center. Dr. Marino received his medical degree from the Albert Einstein School of Medicine and his specialty training in internal medicine at the Eisenhower Army Medical Center and sub-specialty training in Clinical Pharmacology at the Uniformed Services University of the Health Sciences.
Jeremy B. Hayden joined us as General Counsel and Vice President of Business Development in July 2015. Prior to joining us, Mr. Hayden served as Assistant General Counsel at Volcano Corporation, a publicly-held medical device company that was acquired by Koninklijke Philips N.V in early 2015. Prior to Volcano Corporation, Mr. Hayden practiced corporate and securities law at several national and international law firms, including Mintz Levin Cohn Ferris Glovsky & Popeo, P.C. and McKenna Long & Aldridge, LLP (now Dentons). Mr. Hayden received his A.B. in Politics from Princeton University and his J. D. from the University of Michigan Law School.
98
CORPORATE GOVERNANCE
During 2016:
|
|
• |
the Board held eleven meetings and took action via unanimous written consent six times; |
|
|
• |
the Audit Committee met eight times and took action via unanimous written consent one time; |
|
|
• |
the Compensation Committee met two times and took action via unanimous written consent one times; |
|
|
• |
the Governance and Nominating Committee met three times and did not take any actions via unanimous written consent; |
|
|
• |
the Executive Committee met one time did not take action via unanimous written consent; and |
|
|
• |
the sub-committee of the Executive Committee, comprised of our Chairman and our CEO, took action via unanimous written consent two times. |
Each member of the Board attended seventy-five percent (75%) or more of the aggregate of (i) the total number of Board meetings held during the period of such member’s service and (ii) the total number of meetings of committees of the Board on which such member served, during the period of such member’s service, other than Richard Hawkins, who attendance rate was slightly under 75% due to the fact that we were required to reschedule certain calendared Board and Committee meetings to dates and times that precluded Mr. Hawkins’ attendance.
All Board members are encouraged to attend our annual meetings of stockholders in person. However, in 2016, our stockholder meeting date did not coincide with our regularly scheduled quarterly Board meeting. Mr. Rickey, our Chairman, and Dr. Hedrick attended our 2016 Annual Meeting of Stockholders.
Board Independence
The Board has determined that Dr. Naughton and Messrs. Hawkins, Hawran, Lyons, Martell and Rickey are “independent” under the rules of the NASDAQ Stock Market. Under applicable SEC and the NASDAQ rules, the existence of certain “related person” transactions above certain thresholds between a director and the Company are required to be disclosed and preclude a finding by the Board that the director is independent. The Board is not able to consider Dr. Hedrick, our President and Chief Executive Officer, independent, as a result of his employment with us during his tenure as one of our directors.
Board of Directors Leadership Structure
Our bylaws and governance principles provide the Board with the flexibility to combine or separate the positions of Chairman and Chief Executive Officer. Historically, these positions have been separate. Our Board believes that the separation of these positions strengthens the independence of our Board and allows us to have a Chairman focused on the leadership of the Board while allowing our Chief Executive Officer to focus more of his time and energy on managing our operations. The Board currently believes this structure works well to meet the leadership needs of the Board and of the Company. Dr. Hedrick, our President and Chief Executive Officer, has comprehensive industry expertise and is able to devote substantial time to the Company, and Mr. Rickey, our Chairman, is able to devote focus on longer term and strategic matters, and to provide related leadership to the Board. As a result, we do not currently intend to combine these positions; however a change in this leadership structure could be made if the Board determined it was in the best long-term interests of stockholder based upon a departure of either our Chief Executive Officer or Chairman. For example, if the two roles were to be combined, we believe that the independence of the majority of our directors, and the three fully independent Board committees, would provide effective oversight of our management and the Company.
The Board’s Role in Risk Oversight
The Board’s role in risk oversight includes assessing and monitoring risks and risk management. The Board reviews and oversees strategic, financial and operating plans and holds management responsible for identifying and moderating risk in accordance with those plans. The Board fulfills its risk oversight function by reviewing and assessing reports from members of management on a regular basis regarding material risks faced by us Company and applicable mitigation strategy and activity. The reports cover the critical areas of operations, sales and marketing, development, regulatory and quality affairs, intellectual property, clinical development, legal and financial affairs. The Board and its Committees (described below) consider these reports; discuss matters with management and identify and evaluate any potential strategic or operational risks, and appropriate activity to address those risks.
99
Board Committees
The Board has standing Audit, Compensation, Executive, and Governance and Nominating Committees. All members of the Compensation Committee, Audit Committee, and Governance and Nominating Committee are independent directors.
Compensation Committee
The Compensation Committee currently consists of Mr. Lyons (Chairman), Dr. Naughton and Mr. Rickey. In May 2016, Tommy Thompson, a former director, stepped down as the Chairman (and a member) of our Compensation Committee. Mr. Lyons replaced Mr. Thompson as Chairman of the Compensation Committee, and Mr. Rickey joined the Compensation Committee to fill the vacancy created by Mr. Thompson’s departure. Each of the members of our Compensation Committee is independent as defined by NASDAQ, a “Non-Employee Director” as defined by rule 16b-3(b)(3)(i) of the Securities Exchange Act of 1934, as amended, and an “outside director” as defined by Section 162(m) of the Internal Revenue Code of 1986, as amended. The Committee Chairman is responsible for setting the Committee’s calendar and meeting agenda.
The Compensation Committee is responsible for developing and implementing compensation programs for our executive officers and other employees, subject only to the discretion of the full Board. More specifically, our Compensation Committee establishes base salary rates for each of the Company’s officers, and administers our 2004 Equity Incentive Plan, our 2014 Equity Incentive Plan, our Executive Management Incentive Compensation Plan, our 2011 Employee Stock Purchase Plan and our 2015 New Employee Incentive Plan. The Compensation Committee establishes the compensation and benefits for our Chief Executive Officer and other executive officers, and also reviews the relationship between our performance and our compensation policies as well as assessing any risks associated with our compensation policies. In addition, the Compensation Committee reviews, and advises the Board on director compensation matters and on, regional and industry-wide compensation practices and trends in order to assess the adequacy of our executive compensation programs. The charter of the Compensation Committee has been established and approved by the Board, and a copy of the charter has been posted on our website at www.cytori.com under Investor Relations – Corporate Governance.
Our CEO attends some of the meetings of the Compensation Committee upon invitation, but does not participate in the executive sessions of the Compensation Committee.
Audit Committee
Our Audit Committee currently consists of Mr. Hawran (Chairman), Mr. Hawkins and Mr. Lyons. At the outset of 2016, Mr. Hawran (Chairman), Mr. Thompson and Mr. Hawkins were the members of our Audit Committee. Upon Mr. Thompson’s departure in May 2016, Mr. Lyons joined the Audit Committee. The Audit Committee is comprised solely of independent directors, as defined by NASDAQ. The Board has determined that Mr. Hawran is an “audit committee financial expert” within the meaning of Item 407(d)(5) of SEC Regulation S-K. The charter of the Audit Committee has been established and approved by the Board, and a copy of the charter has been posted on our website at www.cytori.com under Investor Relations – Corporate Governance.
The Audit Committee selects our auditors, reviews the scope of the annual audit, approves the audit fees and non-audit fees to be paid to our auditors, and reviews our financial accounting controls with the staff and the auditors. The Audit Committee is also charged with review and oversight of management’s enterprise risk management assessment.
Governance and Nominating Committee
Our Governance and Nominating Committee currently consists of Mr. Hawkins (Chairman), Mr. Martell and Dr. Naughton. Mr. Martell replaced Mr. Lyons as a member of Governance and Nominating Committee in December 2016. The Governance and Nominating Committee is comprised solely of independent directors, as defined by NASDAQ. The Governance and Nominating Committee interviews, evaluates, nominates and recommends individuals for membership on the Board, evaluates the effectiveness of the Board and its serving members, and recommends the structure, responsibility and composition of the committees of the Board. The Committee is also responsible for recommending guidelines and policies for corporate governance for adoption by the Board. The charter of the Governance and Nominating Committee has been established and approved by the Board, and a copy of the charter has been posted on our website at www.cytori.com under Investor Relations – Corporate Governance.
Executive Committee
The Executive Committee is comprised of our Chief Executive Officer, Chairman of the Board, and Chairpersons of each committee of the Board. The Executive Committee currently consists of Dr. Hedrick, Mr. Rickey, Mr. Hawkins, Mr. Hawran, and Mr. Lyons.
100
The Executive Committee’s responsibilities, when such responsibilities are not discharged by our full Board, include to evaluate and approve the material terms of any financing transactions or business transactions as well as to authorize and approve accompanying the issuance of stock and/or other equity securities. The Executive Committee also would be able to act on behalf of the full Board in urgent or exigent circumstances wherein it would be very difficult or impossible to assemble the full Board between regularly scheduled meetings. In 2016, our Executive Committee acted as a special pricing committee of the Board with respect to our rights offering financing, consummated in June 2016. The Sub-Committee of the Executive Committee, consists of our Chairman of the Board and our Chief Executive Officer, has the authority to approve corporate expenditures presented by our management in excess of $250,000 up to a maximum of $1,000,000 for a single corporate transaction.
CODE OF BUSINESS CONDUCT AND ETHICS
We have adopted a Code of Business Conduct and Ethics that applies to all of our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer. This Code of Business Conduct and Ethics has been posted on our website at www.cytori.com. We intend to post amendments to this code, or any waivers of its requirements, on our website at www.cytori.com under Investor Relations – Corporate Governance, as permitted under SEC rules and regulations.
SECTION 16(a) BENEFICIAL OWNERSHIP REPORTING COMPLIANCE
Section 16(a) of the Securities Exchange Act of 1934 requires our directors, executive officers, and persons or entities who own more than ten percent of our common stock, to file with the SEC reports of beneficial ownership and changes in beneficial ownership of our common stock. Those directors, officers, and stockholders are required by regulations to furnish us with copies of all forms they file under Section 16(a). Based solely upon a review of the copies of such reports furnished to us and written representations from such directors, officers, and stockholders, we believe that all such reports required to be filed during 2016 were filed on a timely basis.
Item 11. Executive Compensation
Our named executive officers for fiscal year 2016 are:
|
|
• |
Marc H. Hedrick, M.D., our President and Chief Executive Officer; |
|
|
• |
Tiago Girao, our Chief Financial Officer; and |
|
|
• |
John Harris, our Vice President and General Manager of Cell Therapy. |
These individuals are collectively referred to in this discussion as the “named executive officers,” or “NEOs.” Investors are encouraged to read this discussion in conjunction with the compensation tables and related notes, which include more detailed information about the compensation of our NEOs for 2016 and 2015.
101
2016 Summary Compensation Table
The following table sets forth information concerning compensation earned during 2015 and 2016 for services rendered to us by our NEOs.
|
(a) |
|
(b) |
|
(c) |
|
|
(d) |
|
|
(e) |
|
|
(f) |
|
|
(g) |
|
|
(h) |
|
||||||
|
Name and Principal Position |
|
Year |
|
Salary |
|
|
Stock Awards(1) |
|
|
Option Awards(2) |
|
|
Non-Equity Incentive Plan Comp. (3) |
|
|
All Other Comp- ensation(4) |
|
|
Total |
|
||||||
|
Marc H. Hedrick, M.D., |
|
2016 |
|
$ |
450,000 |
|
|
— |
|
|
$ |
156,273 |
|
|
$ |
146,250 |
|
|
— |
|
|
$ |
752,523 |
|
||
|
President and Chief Executive Officer |
|
2015 |
|
$ |
450,000 |
|
|
$ |
80,172 |
|
|
$ |
115,200 |
|
|
$ |
200,475 |
|
|
— |
|
|
$ |
845,847 |
|
|
|
Tiago M. Girao, |
|
2016 |
|
$ |
265,000 |
|
|
— |
|
|
$ |
65,535 |
|
|
$ |
79,560 |
|
|
|
— |
|
|
$ |
410,095 |
|
|
|
VP of Finance, Chief Financial Officer and Chief Accounting Officer |
|
2015 |
|
$ |
265,000 |
|
|
$ |
40,086 |
|
|
$ |
57,600 |
|
|
$ |
69,563 |
|
|
|
— |
|
|
$ |
432,249 |
|
|
John Harris, |
|
2016 |
|
$ |
361,830 |
(5) |
|
— |
|
|
$ |
65,535 |
|
|
$ |
64,365 |
|
|
$125,249 |
(6) |
|
$ |
616,979 |
|
||
|
VP and General Manager of Cell Therapy |
|
2015 |
|
$ |
88,167 |
(5) |
|
— |
|
|
$ |
123,982 |
|
|
$ |
25,988 |
|
|
$ 19,647 |
(6) |
|
$ |
257,784 |
|
||
|
(1) |
This column represents the dollar amount of the aggregate grant date fair value of stock awards granted in 2015, computed in accordance with FASB ASC Topic 718. For information relating to the assumptions made by us in valuing the stock awards made to our NEOs in 2016, refer to Note 12 to our audited consolidated financial statements included in this Form 10-K. These amounts do not reflect the actual economic value that will be realized by our NEOs upon vesting of the stock awards or sale of the common stock underlying the stock. On May 26, 2015, the Compensation Committee granted performance-based RSUs and the grant date fair value in the table was calculated based on the probable achievement of the performance objectives applicable to such awards, which was estimated at “target” performance for this purpose. Had maximum achievement of the performance criteria been achieved, the full grant date fair value of the awards, assuming maximum achievement of the performance criteria, would have been 200% of the amount set forth in the table. |
|
(2) |
This column represents the dollar amount of the aggregate grant date fair value of option awards, computed in accordance with FASB ASC Topic 718. For information relating to the assumptions made by us in valuing the option awards made to our NEOs in 2016 and 2015, refer to Note 12 to our audited consolidated financial statements included in this Form 10-K. These amounts do not reflect the actual economic value that will be realized by our NEOs upon vesting of the stock options, exercise of the stock options, or sale of the common stock underlying the stock options. |
|
(3) |
The amounts in column (f) reflect the cash awards under our EMIC Plan, which is discussed in further detail below under the heading in the subsection entitled “Executive Management Incentive Compensation Plan” of the “Narrative Disclosure to Compensation Tables” below. |
|
(4) |
Dollar value of the perquisites and other personal benefits for Dr. Hedrick and Mr. Girao were less than $10,000 for the year reported. |
|
(5) |
We paid Mr. Harris in Japanese Yen. During 2015, and 2016 his salary was reported at the average exchange rate over the year, or 0.0083 and 0.0086 Japanese Yen to US dollar in 2015 and 2016, respectively. |
|
(6) |
Per the terms of his employment offer letter with us, Mr. Harris was eligible to receive a housing allowance while on assignment in Japan up to a maximum of 13,900,000 Japanese Yen per year, including direct payment by us of Mr. Harris’ local rent (not to exceed 1,100,000 Japanese Yen per month) and additional healthcare coverage. We paid these benefits in Japanese Yen, and we recorded them in 2015 and 2016 at the average exchange rate over the applicable year, or 0.0083 and 0.0086 Japanese Yen to U.S. dollar in 2015 and 2016, respectively. During 2015 and 2016, Mr. Harris’ rent expense was $18,260 and $111,994, respectively, and cost of his additional health care coverage was $1,387 and $13,255, respectively. |
Narrative Disclosures to Summary Compensation Table
Executive Compensation
In the process of determining compensation for our NEOs, the Compensation Committee considers the current financial position of the Company, the strategic goals of the Company and the performance of each of our NEOs. The Committee also benchmarks the various components (described below) of our compensation program for executives to compensation paid by other public companies in our defined peer group, compensation data from Radford Global Life Sciences Survey and BIOCOM Total Rewards Survey, historical review of all executive officer compensation, and recommendations from our CEO (other than for his own salary). From time to time the Committee engages the services of outside compensation consultants to provide compensation research, analysis and recommendations. The Committee has the sole authority to select, compensate and terminate its external advisors.
102
The Compensation Committee utilizes the following components of compensation (described further below) to strike an appropriate balance between promoting sustainable and excellent performance and discouraging any inappropriate short-sighted risk-taking behavior:
|
|
• |
Base salary; |
|
|
• |
Annual long-term equity compensation; |
|
|
• |
Personal benefits and perquisites; and |
|
|
• |
Acceleration and severance agreements tied to changes on control of the Company. |
Base Salaries
None of our NEOs received base salary increases for 2016. While the Compensation Committee had previously approved an increase in Mr. Girao’s annual base salary from $265,000 to $300,000 for fiscal year 2016, at Mr. Girao’s request, this salary increase was deferred. Commencing effective as of January 1, 2017, Mr. Girao’s annual base salary was increased from $265,000 to $300,000.
In connection with determination of executive compensation for fiscal year 2017, the Compensation Committee directed Barney & Barney, LLC, its independent compensation consultant, to prepare an updated senior management compensation assessment. The Compensation Committee reviewed this assessment at its normally scheduled meeting in January 2017. Based on this assessment and including other data points and information considered by the Compensation Committee in its discretion, the Compensation Committee approved the following NEO base salaries for fiscal year 2017, which base salaries went into effect in March 2017: Dr. Hedrick: $510,000; Mr. Harris: $360,500; Mr. Girao: $309,000. The increases to Dr. Hedrick’s and Mr. Girao’s base salaries were made to move such salaries closer to or within the 50th and 60th percentile range of base salary compensation for similarly situated executive at our peer companies, per our corporate compensation philosophy. Our compensation analysis indicates that Dr. Hedrick’s base salary is substantially closer to, but still below, this stated range, while Mr. Girao’s base salary is now within this stated range. Mr. Harris’ base salary remains above our stated range, but we believe that the Mr. Harris’ actual duties and responsibilities, combined with his experience and skills (including Japanese linguistic and business/cultural fluency) are appropriately reflected in his base salary and other compensation.
Barney & Barney did not provide any services to us in 2016 beyond its engagement as an advisor to the Compensation Committee on compensation matters. After review and consultation with Barney & Barney, the Compensation Committee has determined that Barney & Barney is independent and there is no conflict of interest resulting from retaining Barney & Barney currently or during the year ended December 31, 2016. In reaching these conclusions, the Compensation Committee considered the factors set forth in Exchange Act Rule 10C-1 and NASDAQ listing standards.
Annual Bonuses (Executive Management Incentive Compensation Plan)
Our Compensation Committee adopted the Cytori Therapeutics Executive Management Incentive Compensation, or EMIC, plan to increase the performance-based component of our executives’ compensation by linking their annual cash bonus payments to achievement of shorter-term performance goals. Target bonuses are reviewed annually and established as a percentage of the executives’ base salaries, generally based upon seniority of the officer and targeted at or near the median of the peer group (with reference to our corporate compensation philosophy) and relevant survey data (including the Radford Global Life Sciences Survey and BIOcom Total Rewards Survey). Each year the Compensation Committee establishes corporate and individual objectives and respective target percentages, taking into account recommendations from our Chief Executive Officer as it relates to executive positions other than the Chief Executive Officer’s compensation. Our Chief Executive Officer’s EMIC plan is set by the Compensation Committee to align entirely with our overall corporate objectives, while the other NEOs are also provided individual goals that constitute a portion of their overall EMIC plans. After each fiscal year-end, our Chief Executive Officer provides the Compensation Committee with a written evaluation showing actual performance as compared to corporate and/or individual objectives, and the Compensation Committee uses that information, along with the overall corporate performance, to determine what percentage of each executive’s bonus target will be paid out as a bonus for that year. Overall, we attempt to set the corporate and individual functional goals to be highly challenging yet attainable.
For 2016, the general corporate goals approved by the Board (upon recommendation of the Compensation Committee for purposes of executive compensation) were determined by the Compensation Committee to account for 100% of the target cash bonus amount payable under the EMIC plan for our Chief Executive Officer, Dr. Hedrick, and to account for 75% of the overall target bonus amount payable under the EMIC plans for our other NEOs. The Company’s general corporate objectives included clinical, financial and operational objectives, including the achievement of certain enrollment goals for our STAR clinical trial; the achievement of certain year-end cash objectives, revenue goals and business development objectives; and various operational objectives.
103
The following individual objectives for the NEOs other than Dr. Hedrick expanded upon their particular functions in the overall corporate objectives and were to weighted as 25% of their respective overall target bonus amounts.
Mr. Girão’s individual objectives included the achievement of certain investor-related, liquidity, and partner-related goals.
Mr. Harris’s individual objectives included achievement of certain revenue, product utilization and business development/partnering goals.
Our NEOs’ target bonuses for 2016 as a percentage of base salary were as follows: Dr. Hedrick, 50% (increased from 45% in 2015); Mr. Girao, 40% (increased from 30% in 2015); and Mr. Harris, 30% (unchanged from 2015, as Mr. Harris commenced employment with us in October 2015). The Compensation Committee, in its January 2017 meeting, evaluated our achievement in 2016 as compared to overall the corporate and individual objectives for the NEOs in the 2016 EMIC Plan described above. The Committee evaluated the overall results and then evaluated the NEOs’ achievement relative to their own functional objectives and the results are tabulated in the table below:
|
Officer and Position |
|
Target Bonus as a % of Salary |
|
|
% of Target Bonus Awarded |
|
|
Bonus Awarded as a % of Salary |
|
|
Amount of 2016 Bonus Payable in 2017(1) |
|
||||
|
Marc H. Hedrick, M.D. |
|
|
50 |
% |
|
|
65.0 |
% |
|
|
32.5 |
% |
|
$ |
109,688 |
|
|
President & CEO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tiago M. Girao, |
|
|
40 |
% |
|
|
66.3 |
% |
|
|
26.5 |
% |
(2) |
$ |
59,670 |
(2) |
|
Chief Financial Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
John Harris |
|
|
30 |
% |
|
|
61.3 |
% |
|
|
18.4 |
% |
|
$ |
48,274 |
|
|
VP & General Manager of Cell Therapy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
The 2016 bonus amounts are payable in 2017 in installments as follows: 50% of such amounts are payable on July 2, 2017, 25% of such amounts are payable on October 1, 2017 and the remaining 25% of such amounts are payable on January 1, 2018. |
|
|
(2) |
Mr. Girao’s 2016 bonus amount is based off of the increased base salary previously approved by the Compensation Committee for fiscal year 2016, but at Mr. Girao’s request, this salary increase was deferred. Commencing effective as of January 1, 2017, Mr. Girao’s annual base salary was increased from $265,000 to $300,000. |
As part of its determination of target executive compensation for fiscal year 2017, the Compensation Committee determined bonus targets for our NEOs in consultation with Barney & Barney and with reference to Barney & Barney’s senior management compensation assessment and other materials and information, as deemed necessary or appropriate by the Compensation Committee in its discretion. Upon completion of this review, the Compensation Committee approved target bonuses (as a percentage of base salary) for our NEOs for fiscal year 2017 as follows: Dr. Hedrick: 55%; Mr. Girao: 40%; Mr. Harris: 40%.
Long-Term Equity Compensation
We designed our long-term equity grant program to further align the interests of our executives with those of our stockholders and to reward the executives’ longer-term performance. Historically, the Compensation Committee has granted individual option grant awards, although from time-to-time, to further increase the emphasis on compensation tied to performance, the Compensation Committee may grant other equity awards as allowed by the 2014 Equity Incentive Plan. The Compensation Committee grants stock options, restricted stock, restricted stock units and similar equity awards permitted under our plans based on its judgment as to whether the complete compensation packages to our executives, including prior equity awards, are appropriate and sufficient to retain and incentivize the executives and whether the grants balance long-term versus short-term compensation. The Compensation Committee also considers our overall performance as well as the individual performance of each NEO, and the potential dilutive effect of restricted stock awards, and the dilutive and overhang effect of the equity grant awards, and recommendations from the Chief Executive Officer (other than with respect to his own equity awards).
Stock options are granted with an exercise price equal to the fair market value of our common stock on the date of grant.
In January 2016, our NEOs were granted stock options to acquire shares of our common stock at an exercise price equal to the fair market value of our common stock on the Nasdaq Stock Market as of the date of grant, vesting in accordance with our standard four-year vesting schedules. Specifically, Dr. Hedrick, Mr. Girao and Mr. Harris were granted (on a post-split basis reflecting the 1-for-15 reverse stock split that we consummated in May 2016) options to purchase 55,613, 23,322 and 23,322 shares of our common stock, respectively.
104
In March 2017, as part of its determination of target executive compensation for fiscal year 2017, the Compensation Committee assessed long-term incentive compensation for our NEOs in consultation with Barney & Barney and with reference to Barney & Barney’s senior management compensation assessment and other materials and information, as deemed necessary or appropriate by the Compensation Committee in its discretion. Upon completion of its review, the Compensation Committee granted stock options to our NEOs to acquire shares of our common stock at an exercise price equal to the fair market value of our common stock on the Nasdaq Stock Market as of the date of grant, such options to vest in accordance with our standard four-year vesting schedules (subject to the NEOs’ continued service as of the applicable vesting dates). Specifically, Dr. Hedrick, Mr. Girao and Mr. Harris were granted options to purchase 96,350, 31,100 and 31,100 shares of our common stock, respectively.
Personal Benefits and Perquisites
All of our executives are eligible to participate in our employee benefit plans, including medical, dental, vision, life insurance, short-term and long-term disability insurance, flexible spending accounts, 401(k), and an Employee Stock Purchase Program (ESPP). These plans are available to all full-time employees. In keeping with our philosophy to provide total compensation that is competitive within our industry, we offer limited personal benefits and perquisites to executive officers that include supplemental long-term disability insurance. You can find more information on the amounts paid for these perquisites to or on behalf of our NEOs in our 2016 Summary Compensation Table.
Company Acquisition / Post-Termination Compensation
We have entered into individual change of control and severance agreements, or CIC Agreements, with each of our NEOs. The CIC Agreements provide for certain severance benefits to be paid to each of our NEOs in the event of his involuntary termination without cause, or due to the executive’s resignation for good reason (including the Company’s material breach of its obligations, material reduction in duties, responsibilities, compensation or benefits, or relocation by more than 30 miles without prior consent), provided such termination or resignation occurs in connection with an acquisition of the Company. Upon such termination or resignation in the event of an acquisition, Dr. Hedrick would receive a lump sum payment of 18 times his monthly base salary, and 18 times his monthly COBRA payments, and Mr. Girão and Mr. Harris would each receive a lump sum payment of 12 times his monthly base salary, and 12 times his monthly COBRA payments. Notwithstanding the foregoing, these NEOs’ employment may be terminated for cause (including extended disability, repudiation of their CIC Agreements, conviction of a plea of no contest to certain crimes or misdemeanors, negligence that materially harms us, failure to perform material duties without cure, drug or alcohol use that materially interferes with performance, and chronic unpermitted absence) without triggering an obligation for us to pay severance benefits under the CIC Agreements.
In addition, under the CIC Agreements, any unvested stock options granted to each of the above named executive officers would vest in full upon (1) the date of the executive’s termination under the circumstances described above following entry into an acquisition agreement (subject to the actual consummation of the acquisition) or (2) consummation of an acquisition.
In all events, each NEO’s entitlement to the benefits described above is expressly conditioned upon his execution and delivery to us of a CIC Agreement and a general release of claims, in the form attached to each CIC Agreement.
105
Outstanding Equity Awards at December 31, 2016
The following table sets forth information regarding outstanding equity awards held by our NEOs as of December 31, 2016.
|
|
|
Option Awards |
|
Stock Awards |
||||||||||||||||
|
Name |
|
Option Grant Date (1) |
|
Number of Securities Underlying Unexercised Options (#) Exercisable(5) |
|
|
Number of Securities Underlying Unexercised Options (#) Un- Exercisable (2)(5) |
|
|
Option Exercise Price ($)(5) |
|
|
Option Expiration Date |
|
Number of Shares or Units of Stock That Have Not Vested (#) |
|
Market Value of Shares or Units of Stock That Have Not Vested ($) |
|||
|
Marc H. Hedrick, M.D., |
|
2/26/2007 |
|
|
3,332 |
|
|
— |
|
|
$ |
81.60 |
|
|
2/26/2017 |
|
— |
|
— |
|
|
President and Chief Executive Officer |
|
1/31/2008 |
|
|
4,000 |
|
|
— |
|
|
$ |
77.10 |
|
|
1/31/2018 |
|
— |
|
— |
|
|
|
|
1/29/2009 |
|
|
5,000 |
|
|
— |
|
|
$ |
72.00 |
|
|
1/29/2019 |
|
— |
|
— |
|
|
|
|
2/05/2010 |
|
|
7,333 |
|
|
— |
|
|
$ |
100.65 |
|
|
2/05/2020 |
|
— |
|
— |
|
|
|
|
1/27/2011 |
|
|
3,666 |
|
|
— |
|
|
$ |
83.55 |
|
|
1/27/2021 |
|
— |
|
— |
|
|
|
|
1/26/2012 |
|
|
7,666 |
|
|
— |
|
|
$ |
51.60 |
|
|
1/26/2022 |
|
— |
|
— |
|
|
|
|
1/31/2013 |
|
|
11,968 |
|
|
254 |
|
|
$ |
41.10 |
|
|
1/31/2023 |
|
— |
|
— |
|
|
|
|
1/31/2013 |
|
|
5,984 |
|
|
127 |
|
|
$ |
75.00 |
|
|
1/31/2023 |
|
— |
|
— |
|
|
|
|
4/11/2014 |
|
|
13,068 |
|
|
|
5,932 |
|
|
$ |
35.70 |
|
|
4/11/2024 |
|
— |
|
— |
|
|
|
8/21/2014 |
|
|
6,666 |
|
|
— |
(3) |
|
$ |
21.00 |
|
|
8/21/2024 |
|
— |
|
— |
|
|
|
|
1/30/2015 |
|
|
7,675 |
|
|
|
8,325 |
|
|
$ |
7.20 |
|
|
1/30/2025 |
|
— |
|
— |
|
|
|
1/04/2016 |
|
|
13,904 |
|
|
|
41,709 |
|
|
$ |
2.81 |
|
|
1/04/2026 |
|
|
|
|
|
Tiago M. Girao, |
|
9/2/2014 |
|
|
5,840(4) |
|
|
|
4,160 |
|
|
$ |
20.40 |
|
|
9/2/2024 |
|
— |
|
— |
|
VP of Finance Chief Financial Officer |
|
1/30/2015 |
|
|
3,841(4) |
|
|
|
4,159 |
|
|
$ |
7.20 |
|
|
1/30/2025 |
|
— |
|
— |
|
|
|
1/04/2016 |
|
|
5,831 |
|
|
|
17,491 |
|
|
$ |
2.81 |
|
|
1/04/2026 |
|
— |
|
— |
|
John Harris, VP and General Manager Cell Therapy |
|
11/11/2015 |
|
|
6,516(4) |
|
|
|
15,817 |
|
|
$ |
5.55 |
|
|
11/11/2025 |
|
— |
|
— |
|
|
|
1/04/2016 |
|
|
5,831(4) |
|
|
|
17,491 |
|
|
$ |
2.81 |
|
|
1/04/2026 |
|
— |
|
— |
|
|
(1) |
For a better understanding of this table, we have included an additional column showing the grant date of the stock options. |
|
|
(2) |
Unless otherwise provided, stock options are subject to four-year vesting, and have a contractual term of 10 years from the date of grant. Awards presented in this table contain one of the following two vesting provisions: |
|
|
• |
With respect to an initial stock option grant to an employee, 25% of the shares subject to the award vest on the one-year anniversary of the vesting start date, while an additional 1/48th of the remaining option shares vest at the end of each month thereafter for 36 consecutive months, or |
|
|
• |
With respect to stock option grants made to an employee after one full year of employment, 1/48th of the shares subject to the award vest at the end of each month over a four-year period, as measured from the vesting start date. |
|
|
(3) |
The August 2014 stock option awards vested to 50% of the shares subject to such awards after one year of service and the additional 50% vested on the second anniversary of the grant. |
|
|
(4) |
These options were granted during the first year of the NEO’s employment and thus were subject to the following vesting schedule: 25% of the shares subject to the award vest on the one-year anniversary of the vesting start date, while an additional 1/48th of the remaining option shares vest at the end of each month thereafter for 36 consecutive months. |
|
|
(5) |
We consummated a 1-for-15 reverse stock split in May 2016. The amounts set forth in this column reflect this 1-for-15 reverse stock split. |
Director Compensation
Generally, our Board believes that the level of director compensation should be based on time spent carrying out Board and committee responsibilities and be competitive with comparable companies. In addition, the Board believes that a significant portion of director compensation should align director interests with the long-term interests of stockholders. The Board makes changes in its director compensation practices only upon the recommendation of the Compensation Committee, and discussion and approval by the Board.
106
The following table summarizes director compensation awarded to, earned by or paid to our non-employee directors who served on our Board during fiscal year 2016.
|
(a) |
|
(b) |
|
|
(c) |
|
|
(d) |
|
|
(e) |
|
||||
|
|
|
Fees Earned or Paid in Cash(2) |
|
|
Stock Awards |
|
|
Option Awards(3)(4)(5) |
|
|
Total |
|
||||
|
Director Name(1) |
|
($) |
|
|
($) |
|
|
($) |
|
|
($) |
|
||||
|
David M. Rickey, Chairman |
|
$ |
66,667 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
76,749 |
|
|
Richard J. Hawkins |
|
$ |
55,000 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
65,082 |
|
|
Paul W. Hawran |
|
$ |
50,000 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
60,082 |
|
|
Gary A. Lyons |
|
$ |
60,000 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
70,082 |
|
|
Gail K. Naughton, Ph.D. |
|
$ |
50,000 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
60,082 |
|
|
Tommy G. Thompson(6) |
|
$ |
13,750 |
|
|
|
— |
|
|
$ |
10,082 |
|
|
$ |
23,832 |
|
|
Ronald A. Martell(7) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(1) |
Dr. Hedrick is not included in this table as he is an employee of ours and receives no extra compensation for his service as a director. The compensation received by Dr. Hedrick in his capacity as an employee is set forth in the 2016 Summary Compensation Table and further described in the “Narrative Disclosures to Summary Compensation Table” above |
|
|
|
(2) |
In fiscal year 2016, (i) each non-employee director received a $30,000 retainer for service on our Board; (ii) each Compensation Committee, Governance and Nominating Committee and Audit Committee member received a $10,000 retainer for Committee service; (iii) the Chairman of the Board received an additional annual stipend of $30,000; (iv) the Chairman of the Audit Committee received an additional annual stipend of $15,000; and (v) the Chairmen of the Compensation Committee and the Governance and Nominating Committee each received an additional annual stipend of $15,000, respectively. Executive Committee members were exempt from receiving committee fees. |
|
|
|
(3) |
Column (d) represents the grant date fair value of the option awards, computed in accordance with FASB ASC Topic 718, granted to our non-employee directors during 2016. For additional information on the valuation assumptions with respect to the 2016 grants, refer to Note 12 to our audited consolidated financial statements included in this Annual Report, regarding assumptions underlying valuation of equity awards. These amounts do not reflect the actual economic value that will be realized by our non-employee directors upon vesting of the stock options, exercise of the stock options or sale of the common stock underlying the stock. |
|
|
|
(4) |
On January 4, 2016, our non-employee directors were awarded options to purchase 3,588 shares of our common stock. These options vested on the first anniversary of the date of grant. These option amounts reflect a 1-for 15 reverse stock split consummated by us on May 10, 2016. |
|
|
|
(5) |
As of December 31, 2016, our non-employee directors held the following aggregate options: Mr. Rickey: 12,727 option shares; Richard Hawkins: 14,728 option shares; Paul Hawran: 12,728 option shares; Mr. Lyons: 6,654 option shares; Ronald Martell: None; Dr. Naughton: 6,654 option shares. |
|
|
|
(6) |
Mr. Thompson stepped down from our Board in May 2016. |
|
|
|
(7) |
Mr. Martell joined our Board in mid-December 2016, and did not receive any compensation for his brief service in 2016. |
|
Director Compensation Program
In October 2016, the Compensation Committee approved a Director Compensation Program for fiscal year 2017, as subsequently amended. The materials elements of the 2017 Director Compensation Program are as follows:
|
|
• |
$40,000 annual cash retainer for Board members (an increase from $30,000 in 2016); |
|
|
• |
$30,000 annual cash retainer for the Chairman of the Board (no change from 2016); |
|
|
• |
$20,000 annual cash retainer for the Chairman of the Audit Committee (no change from 2016); |
|
|
• |
$15,000 annual cash retainer for the Chairman of our Compensation Committee and Governance and Nominating Committee (no change from 2016); |
|
|
• |
$10,000 annual cash retainer for each non-Chairman committee member (no change from 2016); |
|
|
• |
Initial grants for new directors: Initial option grant, upon commencement of services, to purchase 50,000 shares of our common stock, vesting over two years in equal, annual installments as measured from the grant date; |
|
|
• |
Annual grants for existing directors: Recurring option grants to purchase 25,000 shares of our common stock, vesting in one installment on the first anniversary of the grant date. |
107
In January 2017, the Board granted options to our non-employee directors for fiscal year 2017 in accordance with the terms of the Director Compensation Program described immediately above, including approval of an initial option grant to Ron Martell in connection with his commencement of service as a Board member.
The Compensation Committee believes that these enhancements to the Director Compensation Program allow us to remain aligned with director compensation practices at our peer companies.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information under the heading “Equity Compensation Plan Information” in Part II, Item 5 is incorporated herein by reference.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information regarding ownership of our Common Stock as of February 28, 2017 (or earlier date for information based on filings with the SEC) by (a) each person known to us to own more than 5% of the outstanding shares of our Common Stock, (b) each director and nominee for director, (c) our President and Chief Executive Officer, VP of Finance and Chief Financial Officer and each other NEO named in the compensation tables in this Annual Report on Form 10-K and (d) all directors and executive officers as a group.
The information in this table is based solely on statements in filings with the SEC or other reliable information. We believe, based on information provided to us, that each of the stockholders listed below has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
A total of 23,568,403 shares of our common stock were issued and outstanding as of February 28, 2017.
|
Name and Address of Beneficial Owner (1) |
|
Number of Shares of Common Stock Owned (2) |
|
|
Number of Shares of Common Stock Subject to Awards/Warrants Exercisable Within 60 Days (3) |
|
|
Total Number of Shares of Common Stock Beneficially Owned (4) |
|
|
Percent Ownership |
|
||||
|
Sabby Management, LLC.(5) |
|
|
1,651,835 |
|
|
|
— |
|
|
|
1,651,835 |
|
|
|
7.0 |
% |
|
10 Mountainview Road, Suite 205 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Upper Saddle River, NJ 07458 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Marc H. Hedrick, M.D. |
|
|
78,133 |
|
|
|
111,739 |
|
|
|
189,872 |
|
|
* |
|
|
|
Tiago M. Girao |
|
|
14,084 |
|
|
|
20,067 |
|
|
|
34,151 |
|
|
* |
|
|
|
John D. Harris |
|
|
7,000 |
|
|
|
15,501 |
|
|
|
22,501 |
|
|
|
|
|
|
David M. Rickey |
|
|
95,231 |
|
|
|
22,935 |
|
|
|
118,166 |
|
|
* |
|
|
|
Richard J. Hawkins |
|
|
8,433 |
|
|
16, 405 |
|
|
|
24,838 |
|
|
* |
|
||
|
Paul W. Hawran |
|
|
8,236 |
|
|
|
12,727 |
|
|
|
20,963 |
|
|
* |
|
|
|
Gary A. Lyons |
|
|
4,357 |
|
|
|
7,604 |
|
|
|
11,961 |
|
|
* |
|
|
|
Ronald A. Martell |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
* |
|
|
|
Gail Naughton |
|
|
2,400 |
|
|
|
6,654 |
|
|
|
9,054 |
|
|
* |
|
|
|
All executive officers and directors as a group (11)(6) |
|
|
221,517 |
|
|
|
224,833 |
|
|
|
446,350 |
|
|
|
1.9 |
% |
*Represents beneficial ownership of less than one percent (1%) of the outstanding shares as of February 28, 2017.
|
(1) |
Unless otherwise indicated, the address of each of the named individuals is c/o Cytori Therapeutics, Inc., 3020 Callan Road, San Diego, CA 92121. |
|
(2) |
Unless otherwise indicated, represents shares of outstanding common stock owned by the named parties as of February 28, 2017. |
|
(3) |
Shares of common stock subject to stock options or warrants currently exercisable or exercisable within 60 days of February 28, 2017 are deemed to be outstanding for computing the percentage ownership of the person holding such options and the percentage ownership of any group of which the holder is a member, but are not deemed outstanding for computing the percentage of any other person. |
|
(4) |
The amounts and percentages of common stock beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial ownership of securities. Under the rules of the SEC, a person is deemed to be a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of such security, or “investment power,” which includes the power to dispose of or to direct the disposition of such security. A person is also deemed to be a beneficial owner of any securities for which that person has a right to acquire beneficial ownership within 60 days. |
108
|
(5) |
Based upon a Schedule 13G/A filed January 6, 2017, reporting beneficial ownership as of December 31, 2016. Sabby Healthcare Master Fund, Ltd. (“Sabby Healthcare”) has shared voting and dispositive power with respect to 1,132,643 shares. Sabby Volatility Warrant Master Fund, Ltd. (“Sabby Volatility”) has shared voting and dispositive power with respect to 519,192 shares. Sabby Management, LLC (“Sabby Management”) serves as the investment manager of Sabby Healthcare and Sabby Volatility and has shared voting and dispositive power with respect to 1,651,835 of these shares. Hal Mintz, in his capacity as manager of Sabby Management, has shared voting and dispositive power with respect to 1,651,835 of these shares. Each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over the securities owned except to the extent of their pecuniary interest therein. The address for Sabby Management is 10 Mountainview Road, Suite 205, Upper Saddle River, New Jersey 07458. The address for Mr. Mintz is c/o Sabby Management, LLC, 10 Mountainview Road, Suite 205, Upper Saddle River, New Jersey 07458. |
|
(6) |
This aggregate amount includes 14,844 shares owned (or subject to options that are exercisable within sixty days of February 28, 2017) by Jeremy Hayden, General Counsel and Vice President of Business Development. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
The following includes a summary of transactions since January 1, 2016 to which we have been a party in which the amount involved exceeded or will exceed $120,000, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest. We also describe below certain other transactions with our directors, executive officers and 5% stockholders. We believe the terms obtained or consideration that we paid or received, as applicable, in connection with the transactions described below were comparable to terms available or the amounts that would be paid or received, as applicable, from unaffiliated third parties.
Rights Offering
In June 2016, we consummated a rights offering, or Rights Offering, to our stockholders of record (as of May 20, 2016) to subscribe for units at a subscription price of $2.55 per unit. Pursuant to the Rights Offering, we sold an aggregate of 6,704,852 units consisting of a total of 6,704,852 shares of common stock and 3,352,306 warrants to our stockholders, or Warrants, with each Warrant exercisable for one share of common stock at an exercise price of $3.06 per share. Certain of our directors participated in the Rights Offering and along with other participants in the Rights Offering, purchased common stock and Warrants to purchase our common stock. The Warrants trade on the Nasdaq Stock Market under the symbol ”CYTXW.”
Director and Officer Indemnification
Our amended and restated certificate of incorporation, as amended, and our amended and restated bylaws, as amended, provide that we will indemnify each of our directors and officers to the fullest extent permitted by the Delaware General Corporation Law.
Stock Option Grants to Executive Officers and Directors
We have granted stock options to our executive officers and non-employee directors as more fully described elsewhere in this Annual Report.
The information under the heading “Board Independence” in Part III, Item 10 is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
On July 12, 2016, we notified KPMG, LLP, or KPMG, of its dismissal as our independent registered public accounting firm, effective as of that date. The decision to change independent registered public accounting firms was recommended by our Audit Committee and was approved by the Board.
On July 12, 2016, the Audit Committee appointed BDO USA, LLP, or BDO, as our independent registered public accounting firm for the fiscal year ending December 31, 2016, subject to completion of its standard client acceptance procedures (which were subsequently completed). The decision to engage BDO as our independent registered public accounting firm was recommended by the Audit Committee and approved by the Board.
109
The Audit Committee reviews and must pre-approve all audit and non-audit services performed by our independent registered public accounting firm, as well as the fees charged by it for such services. No fees charged by KPMG or BDO during 2016 were approved under the Regulation S-X Rule 2.01(c)(7)(i)(C) exception to the pre-approval requirement. In its review of non-audit service fees, the Audit Committee considers, among other things, the possible impact of the performance of such services on the accounting firm’s independence.
The following table shows the aggregate fees paid or accrued by us for the audit and other services provided by KPMG for fiscal years ended December 31, 2016 and 2015, and provided by BDO for the fiscal year ended December 31, 2016.
|
|
|
Fiscal Year Ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
BDO |
|
|
KPMG |
|
||||||
|
|
|
2016 |
|
|
2016 |
|
|
2015 |
|
|||
|
Audit Fees (1) |
|
$ |
281,204 |
|
|
$ |
261,400 |
|
|
$ |
470,000 |
|
|
Audit Related Fees (2) |
|
|
— |
|
|
|
— |
|
|
|
40,000 |
|
|
Tax Fees (3) |
|
|
35,000 |
|
|
|
4,823 |
|
|
|
58,000 |
|
|
Total |
|
$ |
316,204 |
|
|
$ |
266,223 |
|
|
$ |
568,000 |
|
|
(1) (1) |
|
Audit fees consist of fees for professional services performed by BDO USA, LLP and KPMG LLP for the audit of our annual financial statements included in this Form 10-K filing and review of financial statements included in our quarterly Form 10-Q filings, reviews of registration statements and issuances of consents, and services that are normally provided in connection with statutory and regulatory filings or engagements. |
|
(2) (2) |
|
Audit related fees consist of fees for assurance and related services, performed by BDO USA, LLP and KPMG LLP that are reasonably related to the performance of the audit or review of our financial statements. |
|
(3) (3) |
|
Tax fees consist of fees for professional services performed by BDO USA LLP and KPMG LLP with respect to tax compliance, tax advice, tax consulting and tax planning. |
110
PART IV
Item 15. Exhibits, Financial Statement Schedules
|
(a) (2) |
Financial Statement Schedules |
SCHEDULE II — VALUATION AND QUALIFYING ACCOUNTS
For the years ended December 31, 2016 and 2015
(in thousands)
|
|
|
Balance at beginning of year |
|
|
Additions (A) |
|
|
Deductions (B) |
|
|
Other (C) |
|
|
Balance at end of year |
|
|||||
|
Allowance for doubtful accounts |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2016 |
|
$ |
797 |
|
|
$ |
— |
|
|
$ |
(630 |
) |
|
$ |
— |
|
|
$ |
167 |
|
|
Year ended December 31, 2015 |
|
$ |
1,523 |
|
|
$ |
— |
|
|
$ |
(709 |
) |
|
$ |
(17 |
) |
|
$ |
797 |
|
|
|
(A) |
Includes charges to costs and expenses. |
|
|
(B) |
Deductions for uncollectible accounts receivable includes payments collected and devices recovered from customers. |
|
|
(C) |
Miscellaneous activity. |
|
(a) (3) |
Exhibits |
List of Exhibits required by Item 601 of Regulation S-K. See Item 15(b) below.
(b) Exhibits
The exhibits listed in the accompanying “Exhibit Index” are filed, furnished or incorporated by reference as part of this Annual Report, as indicated.
None.
111
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized.
|
CYTORI THERAPEUTICS, INC. |
|
|
|
|
|
By: |
/s/ Marc H. Hedrick, MD |
|
|
Marc. H. Hedrick, MD |
|
|
President & Chief Executive Officer |
|
|
March 24, 2017 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this annual report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
SIGNATURE |
|
TITLE |
|
DATE |
|
|
|
|
|
|
|
/s/ David M. Rickey |
|
Chairman of the Board of Directors |
|
March 24, 2017 |
|
David M. Rickey |
|
|
|
|
|
|
|
|
|
|
|
/s/ Marc H. Hedrick, MD |
|
President & Chief Executive Officer (Principal Executive Officer) |
|
March 24, 2017 |
|
Marc H. Hedrick, MD |
|
|
|
|
|
|
|
|
|
|
|
/s/ Tiago M. Girão |
|
VP of Finance and Chief Financial Officer (Principal Financial Officer) |
|
March 24, 2017 |
|
Tiago M. Girão |
|
|
|
|
|
|
|
|
|
|
|
/s/ Paul W. Hawran |
|
Director |
|
March 24, 2017 |
|
Paul W. Hawran |
|
|
|
|
|
|
|
|
|
|
|
/s/ Gail K. Naughton, PhD |
|
Director |
|
March 24, 2017 |
|
Gail K. Naughton, PhD |
|
|
|
|
|
|
|
|
|
|
|
/s/ Richard J. Hawkins |
|
Director |
|
March 24, 2017 |
|
Richard J. Hawkins |
|
|
|
|
|
|
|
|
|
|
|
/s/ Gary A. Lyons |
|
Director |
|
March 24, 2017 |
|
Gary A. Lyons |
|
|
|
|
|
|
|
|
|
|
|
/s/ Ronald A. Martell |
|
Director |
|
March 24, 2017 |
|
Ronald A. Martell |
|
|
|
|
|
|
|
|
|
|
112
EXHIBIT INDEX
|
CYTORI THERAPEUTICS, INC. |
|||||
|
|
|||||
|
Exhibit Number |
Exhibit Title |
Filed with this Form 10-K |
Incorporated by Reference |
||
|
Form |
File No. |
Date Filed |
|||
|
3.1 |
Composite Certificate of Incorporation. |
|
10-K |
000-32501 Exhibit 3.2 |
03/11/2016 |
|
|
|
|
|
|
|
|
3.2 |
Amended and Restated Bylaws of Cytori Therapeutics, Inc. |
|
10-Q |
000-32501 Exhibit 3.2 |
08/14/2003 |
|
|
|
|
|
|
|
|
3.3 |
Amendment to Amended and Restated Bylaws of Cytori Therapeutics, Inc. |
|
8-K |
001-34375 Exhibit 3.1 |
05/06/2014 |
|
|
|
|
|
|
|
|
3.4 |
Certificate of Designation of Preferences, Rights and Limitations of Series A 3.6% Convertible Preferred Stock |
|
8-K |
001-034375 Exhibit 3.1 |
10/08/2014 |
|
|
|
|
|
|
|
|
3.5 |
Certificate of Amendment to Amended and Restated Certificate of Incorporation, as amended |
|
8-K |
001-34375 Exhibit 3.1 |
05/10/2016 |
|
|
|
|
|
|
|
|
4.1 |
Warrant to Purchase Common Stock issued by the Company on October 14, 2008 in favor of Silicon Valley Bank, pursuant to the Loan and Security Agreement dated October 14, 2008. |
|
10-K |
000-32501 Exhibit 10.62 |
03/06/2009 |
|
|
|
|
|
|
|
|
4.2 |
Warrant to Purchase Common Stock issued by the Company on June 11, 2010 in favor of GE Capital Equity Investments, Inc., pursuant to the Amended and Restated Loan and Security Agreement dated June 11, 2010. |
|
8-K |
001-34375 Exhibit 10.73 |
06/17/2010 |
|
|
|
|
|
|
|
|
4.3 |
Warrant to Purchase Common Stock issued by the Company on June 11, 2010 in favor of Silicon Valley Bank, pursuant to the Amended and Restated Loan and Security Agreement dated June 11, 2010. |
|
8-K |
001-34375 Exhibit 10.74 |
06/17/2010 |
|
|
|
|
|
|
|
|
4.4 |
Warrant to Purchase Common Stock issued by the Company on June 11, 2010 in favor of Oxford Financial Corporation, pursuant to the Amended and Restated Loan and Security Agreement dated June 11, 2010. |
|
8-K |
001-34375 Exhibit 10.75 |
06/17/2010 |
|
|
|
|
|
|
|
|
4.5 |
Warrant to Purchase Common Stock issued by the Company on September 9, 2011 in favor of GE Capital Equity Investments, Inc., pursuant to the Amended and Restated Loan and Security Agreement dated September 9, 2011. |
|
8-K |
001-34375 Exhibit 10.84 |
09/15/2011 |
|
|
|
|
|
|
|
|
4.6 |
Warrant to Purchase Common Stock issued by the Company on September 9, 2011 in favor of Silicon Valley Bank, pursuant to the Amended and Restated Loan and Security Agreement dated September 9, 2011. |
|
8-K |
001-34375 Exhibit 10.85 |
09/15/2011 |
|
|
|
|
|
|
|
|
4.7 |
Warrant to Purchase Common Stock issued by the Company on September 9, 2011 in favor of Oxford Financial Corporation, pursuant to the Amended and Restated Loan and Security Agreement dated September 9, 2011. |
|
8-K |
001-34375 Exhibit 10.86 |
09/15/2011 |
|
|
|
|
|
|
|
113
|
4.8 |
Warrant to Purchase Common Stock issued by the Company on September 9, 2011 in favor of Oxford Financial Corporation, pursuant to the Amended and Restated Loan and Security Agreement dated September 9, 2011. |
|
8-K |
001-34375 Exhibit 10.87 |
09/15/2011 |
|
|
|
|
|
|
|
|
4.9 |
Warrant to Purchase Common Stock issued by the Company on June 28, 2013 in favor of Oxford Finance LLC pursuant to the Loan and Security Agreement dated June 28, 2013. |
|
10-Q |
001-34375 Exhibit 4.17 |
08/09/2013 |
|
|
|
|
|
|
|
|
4.10 |
Warrant to Purchase Common Stock issued by the Company on June 28, 2013 in favor of Oxford Finance LLC pursuant to the Loan and Security Agreement dated June 28, 2013. |
|
10-Q |
001-34375 Exhibit 4.18 |
08/09/2013 |
|
|
|
|
|
|
|
|
4.11 |
Warrant to Purchase Common Stock issued by the Company on June 28, 2013 in favor of Oxford Finance LLC pursuant to the Loan and Security Agreement dated June 28, 2013. |
|
10-Q |
001-34375 Exhibit 4.19 |
08/09/2013 |
|
|
|
|
|
|
|
|
4.12 |
Warrant to Purchase Common Stock issued by the Company on June 28, 2013 in favor of Oxford Finance LLC pursuant to the Loan and Security Agreement dated June 28, 2013. |
|
10-Q |
001-34375 Exhibit 4.20 |
08/09/2013 |
|
|
|
|
|
|
|
|
4.13 |
Warrant to Purchase Common Stock issued by the Company on June 28, 2013 in favor of Silicon Valley Bank pursuant to the Loan and Security Agreement dated June 28, 2013. |
|
10-Q |
001-34375 Exhibit 4.21 |
08/09/2013 |
|
|
|
|
|
|
|
|
4.14 |
Form of Warrant to Purchase Common Stock for Investors in the Units |
|
8-K |
001-34375 Exhibit 4.1 |
05/30/2014 |
|
|
|
|
|
|
|
|
4.15 |
Form of Warrant to Purchase Common Stock for Placement Agent of the Units |
|
8-K |
001-34375 Exhibit 4.2 |
05/30/2014 |
|
|
|
|
|
|
|
|
4.16 |
Form of Amendment to Warrant to Purchase Common Stock. |
|
8-K |
001-34375 Exhibit 4.1 |
09/08/2014 |
|
|
|
|
|
|
|
|
4.17 |
Form of Warrant to Purchase Common Stock. |
|
8-K |
001-34375 Exhibit 4.2 |
09/08/2014 |
|
|
|
|
|
|
|
|
4.18 |
Form of Warrant for Purchasers in the Units |
|
8-K |
001-034375 Exhibit 4.1 |
10/08/2014 |
|
|
|
|
|
|
|
|
4.19 |
Form of Initial Warrant to Purchase Common Stock |
|
8-K |
001-034375 Exhibit 4.1 |
05/05/2015 |
|
|
|
|
|
|
|
|
4.20 |
Form of Additional Warrant to Purchase Common Stock |
|
8-K |
001-034375 Exhibit 4.2 |
05/05/2015 |
|
|
|
|
|
|
|
|
4.21 |
Form of Pre-Funded Warrant to Purchase Common Stock |
|
8-K |
001-034375 Exhibit 4.3 |
05/05/2015 |
|
|
|
|
|
|
|
|
4.22 |
Amendment to Common Stock Purchase Warrant |
|
10-K |
001-34375 Exhibit 4.23
|
03/11/2015 |
|
|
|
|
|
|
|
|
4.23 |
Amendment to Series A-1 Warrant to Purchase Common Stock |
|
10-K |
001-34375 Exhibit 4.24
|
03/11/2015 |
|
|
|
|
|
|
|
|
4.24 |
Amendment to Series A-2 Warrant to Purchase Common Stock |
|
10-K |
001-34375 Exhibit 4.25
|
03/11/2015 |
|
|
|
|
|
|
|
|
4.25 |
Form of Non-Transferable Subscription Rights Certificate |
|
S-1/A |
333-210628 Exhibit 4.26 |
05/11/2016 |
|
|
|
|
|
|
|
|
4.26 |
Form of Series R Warrant underlying the Units |
|
S-1/A |
333-210628 Exhibit 4.27 |
05/11/2016 |
|
|
|
|
|
|
|
|
4.27 |
Form of Warrant Agreement between Cytori Therapeutics, Inc. and Broadridge Corporate Issuer Solutions, Inc. |
|
S-1/A |
333-210628 Exhibit 4.28 |
05/11/2016 |
|
|
|
|
|
|
|
|
10.1# |
Amended and Restated 1997 Stock Option and Stock Purchase Plan. |
|
10-K |
000-32501 Exhibit 10.1 |
03/30/2001 |
|
|
|
|
|
|
|
|
10.2# |
2004 Equity Incentive Plan of Cytori Therapeutics, Inc |
|
8-K |
000-32501 Exhibit 10.1 |
08/27/2004 |
|
|
|
|
|
|
|
|
10.3# |
Form of Options Exercise and Stock Purchase Agreement Relating to the 2004 Equity Incentive Plan. |
|
10-Q |
000-32501 Exhibit 10.23 |
11/15/2004 |
|
|
|
|
|
|
|
|
10.4# |
Form of Notice of Stock Options Grant Relating to the 2004 Equity Incentive Plan. |
|
10-Q |
000-32501 Exhibit 10.24 |
11/15/2004 |
|
|
|
|
|
|
|
|
10.5+ |
License & Royalty Agreement, effective August 23, 2007, by and between Olympus-Cytori, Inc. and Cytori Therapeutics, Inc. |
|
10-Q |
000-32501 Exhibit 10.49 |
11/13/2007 |
|
|
|
|
|
|
|
|
10.6 |
Common Stock Purchase Agreement, dated March 28, 2007, by and between Cytori Therapeutics, Inc. and Green Hospital Supply, Inc. |
|
10-Q |
000-32501 Exhibit 10.46
|
05/11/2007 |
|
|
|
|
|
|
|
|
10.7 |
Common Stock Purchase Agreement, dated February 8, 2008, by and between Green Hospital Supply, Inc. and Cytori Therapeutics, Inc. |
|
8-K |
000-32501 Exhibit 10.51 |
2/19/2008 |
|
|
|
|
|
|
|
|
10.8 |
Amendment No. 1, dated February 29, 2008, to Common Stock Purchase Agreement, dated February 8, 2008, by and between Green Hospital Supply, Inc. and Cytori Therapeutics, Inc. |
|
8-K |
000-32501 Exhibit 10.51 |
2/29/2008 |
|
|
|
|
|
|
|
|
10.9 |
Lease Agreement entered into on April 2, 2010, between HCP Callan Rd, LLC. and Cytori Therapeutics, Inc. |
|
10-Q |
001-34375 Exhibit 10.69 |
05/06/2010 |
|
|
|
|
|
|
|
|
10.10 |
Common Stock Purchase Agreement, dated December 6, 2010, by and among Cytori Therapeutics, Inc. and Astellas Pharma Inc. |
|
8-K |
001-34375 Exhibit 10.76 |
12/09/2010 |
|
|
|
|
|
|
|
|
10.11# |
Form of Notice and Restricted Stock Award Agreement for grants of performance-based restricted stock awards under the 2004 Equity Incentive Plan. |
|
8-K |
001-34375 Exhibit 10.1 |
03/04/2011 |
|
|
|
|
|
|
|
|
10.12 |
First Amendment to Lease Agreement entered into on November 4, 2011, between HCP Callan Rd, LLC. and the Company. |
|
10-Q |
001-34375 Exhibit 10.88 |
11/08/2011 |
|
|
|
|
|
|
|
|
10.13# |
2011 Employee Stock Purchase Plan |
|
DEF 14A |
001-34375 Appendix A |
05/02/2011 |
|
|
|
|
|
|
|
|
10.14+ |
Contract HHSO100201200008C dated September 27, 2012, by and between the Company and the U.S. Department of Health and Human Services Biomedical Advanced Research and Development Authority. |
|
8-K |
001-34375 Exhibit 10.90 |
10/03/2012 |
|
|
|
|
|
|
|
|
10.15 |
Joint Venture Termination Agreement dated May 8, 2013 by and between the Company and Olympus Corporation. |
|
10-Q |
001-34375 Exhibit 10.91 |
05/10/2013 |
|
|
|
|
|
|
|
|
10.16+ |
Puregraft Sale-License-Supply Agreement, dated July 30, 2013, by and between the Company and Bimini Technologies LLC. |
|
10-Q/A |
001-34375 Exhibit 10.93 |
11/12/2013 |
114
|
|
|
|
|
|
|
|
10.17+ |
Amended and Restated License and Supply Agreement dated January 30, 2014, by and between the Company and Lorem Vascular Pty. Ltd. |
|
8-K |
001-34375 Exhibit 10.94 |
02/04/2014 |
|
|
|
|
|
|
|
|
10.18 |
Sales Agreement, dated May 12, 2014, by and between Cytori Therapeutics, Inc. and Cowen and Company, LLC. |
|
8-K |
001-34375 Exhibit 10.1 |
05/12/2014 |
|
|
|
|
|
|
|
|
10.19 |
Contract HHSO100201200008C Amendment No. 1 dated August 18, 2014, by and between the Company and the U.S. Department of Health and Human Services Biomedical Advanced Research and Development Authority. |
|
8-K |
001-34375 Exhibit 10.99 |
08/19/2014 |
|
|
|
|
|
|
|
|
10.20 |
Form of Securities Purchase Agreement by and between Cytori Therapeutics, Inc. and the Purchasers (as defined therein), dated as of October 8, 2014. |
|
8-K |
001-034375 Exhibit 10.1 |
10/08/2014 |
|
|
|
|
|
|
|
|
10.21 |
Amendment of Solicitation/Amendment of Contract, effective December 17, 2014, by and between ASPR-BARDA and Cytori Therapeutics, Inc |
X |
|
|
|
|
|
|
|
|
|
|
|
10.22 |
Amendment of Solicitation/Modification of Contract, effective January 5, 2015, by and between ASPR-BARDA and Cytori Therapeutics, Inc |
X |
|
|
|
|
|
|
|
|
|
|
|
10.23 |
Amendment One to the Securities Purchase Agreement, dated March 16, 2015, between the Company and certain institutional investors |
|
10-Q |
001-034375 Exhibit 10.1 |
05/11/2015 |
|
|
|
|
|
|
|
|
10.24 |
Form of Securities Purchase Agreement, dated May 5, 2015, by and among Cytori Therapeutics, Inc. and the investors named therein |
|
8-K |
001-034375 Exhibit 10.1 |
05/05/2015 |
|
|
|
|
|
|
|
|
10.25 |
Placement Agency Agreement, dated May 5, 2015, by and between Cytori Therapeutics, Inc. and Mizuho Securities USA Inc. |
|
8-K |
001-034375 Exhibit 10.2 |
05/05/2015 |
|
|
|
|
|
|
|
|
10.26 |
Amendment One to Joint Venture Termination Agreement, dated April 30, 2015, by and between Cytori Therapeutics, Inc. and Olympus Corporation |
|
8-K |
001-034375 Exhibit 10.1 |
05/05/2015 |
|
|
|
|
|
|
|
|
10.27 |
Loan and Security Agreement, dated May 29, 2015, by and between Cytori Therapeutics, Inc. and Oxford Finance, LLC |
|
10-Q |
001-034375 Exhibit 10.4 |
08/10/2015 |
|
|
|
|
|
|
|
|
10.28 |
Amendment One to the Securities Purchase Agreement between the Company and certain institutional investors dated May 5, 2015 |
|
10-K |
001-034375 Exhibit 10.111 |
03/11/2016 |
|
|
|
|
|
|
|
|
10.29# |
2015 New Employee Incentive Plan |
|
8-K |
001-034375 Exhibit 10.1 |
01/05/2016 |
|
|
|
|
|
|
|
|
10.30# |
Form of Agreement for Acceleration and/or Severance |
|
10-K |
001-034375 Exhibit 10.113# |
03/11/2016 |
|
|
|
|
|
|
|
|
10.31# |
Form of Stock Option Agreement under the New Employee Incentive Plan. |
|
S-8 |
333-210211 Exhibit 99.4 |
03/15/2016 |
|
|
|
|
|
|
|
|
10.32# |
Form of Notice of Grant of Stock Option under the 2015 New Employee Incentive Plan. |
|
S-8 |
333-210211 Exhibit 99.5 |
03/15/2016 |
|
|
|
|
|
|
|
|
10.33# |
2014 Equity Incentive Plan of Cytori Therapeutics, Inc., as amended |
|
DEF-14A |
001-034375 Appendix A |
03/16/2016 |
|
|
|
|
|
|
|
|
10.34 |
Amendment Two to Joint Venture Termination Agreement, dated January 8, 2016. |
|
10-Q |
001-34375 Exhibit 10.4 |
05/10/2016 |
|
|
|
|
|
|
|
115
|
10.35 |
Amendment of Solicitation/Amendment of Contract, effective April 1, 2016, by and between ASPR-BARDA and Cytori Therapeutics, Inc. |
|
10-Q |
001-34375 Exhibit 10.1 |
08/05/2016 |
|
|
|
|
|
|
|
|
10.36 |
Amendment of Solicitation/Amendment of Contract, effective September 9, 2016, by and between ASPR-BARDA and Cytori Therapeutics, Inc. |
|
10-Q |
001-34375 Exhibit 10.1 |
11/09/2016 |
|
|
|
|
|
|
|
|
10.37 |
Purchase Agreement between Cytori Therapeutics, Inc. and Lincoln Park Capital Fund, LLC, dated December 22, 2016. |
|
8-K |
001-34375 Exhibit 10.1 |
12/29/2016 |
|
|
|
|
|
|
|
|
10.38 |
Registration Rights Agreement between Cytori Therapeutics, Inc. and Lincoln Park Capital Fund, LLC, dated December 22, 2016. |
|
8-K |
001-34375 Exhibit10.2 |
12/29/2016 |
|
|
|
|
|
|
|
|
10.39# |
Third Amendment to the Cytori Therapeutics, Inc. 2014 Equity Incentive Plan, dated January 26, 2017. |
X |
|
|
|
|
|
|
|
|
|
|
|
10.40† |
Asset Purchase Agreement by and between Cytori Therapeutics, Inc. and Azaya Therapeutics, Inc., effective Jan. 16, 2017. |
X |
|
|
|
|
|
|
|
|
|
|
|
10.41 |
Lease Agreement, dated February 27, 2017, by and between 6262 Lusk Investors LLC and Cytori Therapeutics, Inc. |
X |
|
|
|
|
|
|
|
|
|
|
|
10.42# |
First Amendment to the Cytori Therapeutics, Inc. 2015 New Employee Incentive Plan, dated Jan. 26, 2017. |
X |
|
|
|
|
|
|
|
|
|
|
|
23.1 |
Consent of BDO USA, LLP, Independent Registered Public Accounting Firm |
X |
|
|
|
|
|
|
|
|
|
|
|
23.2 |
Consent of KPMG, LLP, Independent Registered Public Accounting Firm |
X |
|
|
|
|
|
|
|
|
|
|
|
31.1 |
Certification of Principal Executive Officer Pursuant to Securities Exchange Act Rule 13a-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
X |
|
|
|
|
|
|
|
|
|
|
|
31.2 |
Certification of Principal Financial Officer Pursuant to Securities Exchange Act Rule 13a-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
X |
|
|
|
|
32.1 |
Certifications Pursuant to 18 U.S.C. Section 1350/ Securities Exchange Act Rule 13a-14(b), as adopted pursuant to Section 906 of the Sarbanes - Oxley Act of 2002 |
X |
|
|
|
|
|
|
|
|
|
|
|
101.INS |
XBRL Instance Document |
X |
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
XBRL Schema Document |
X |
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
XBRL Calculation Linkbase Document |
X |
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
XBRL Definition Linkbase Document |
X |
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
XBRL Label Linkbase Document |
X |
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
XBRL Presentation Linkbase Document |
X |
|
|
|
|
# |
Indicates management contract or compensatory plan or arrangement. |
|
+ |
Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
|
† |
Confidential treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
116