Attached files
| file | filename |
|---|---|
| EX-32.2 - CERTIFICATION - CATALYST PHARMACEUTICALS, INC. | d298820dex322.htm |
| EX-32.1 - CERTIFICATION - CATALYST PHARMACEUTICALS, INC. | d298820dex321.htm |
| EX-31.2 - CERTIFICATION - CATALYST PHARMACEUTICALS, INC. | d298820dex312.htm |
| EX-31.1 - CERTIFICATION - CATALYST PHARMACEUTICALS, INC. | d298820dex311.htm |
| EX-23.1 - CONSENT - CATALYST PHARMACEUTICALS, INC. | d298820dex231.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
[Mark One]
| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2016
OR
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File No. 001-33057
CATALYST PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 76-0837053 | |
| (State of jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) | |
| 355 Alhambra Circle, Suite 1250 Coral Gables, Florida |
33134 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (305) 420-3200
Securities Registered Pursuant to Section 12(b) of the Act.
| Common Stock, par value $0.001 per share |
Nasdaq Capital Market | |
| (Title of each class) | (Name of exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act.: None
Indicate by check mark if registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if registrant is not required to file reports pursuant to Rule 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such report(s), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate web site, if any, every Interactive Data File required to be submitted and posted pursuant to rule 405 of Regulation S-T ((§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one):
| Large accelerated filer |
☐ |
Accelerated filer |
☒ | |||||
| Non-accelerated filer |
☐ |
(Do not check if a smaller reporting company) |
Smaller reporting company |
☐ | ||||
As of June 30, 2016, the last business day of the Registrant’s most recently completed second quarter, the aggregate market value of all voting, and non-voting common equity held by non-affiliates was $54,581,032.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date: 82,972,316 shares of common stock, $0.001 par value per share, were outstanding as of March 10, 2017.
Part III incorporates certain information by reference from the registrant’s definitive proxy statement for the 2016 annual meeting of stockholders. The proxy statement with respect to the 2017 annual meeting of stockholders will be filed no later than 120 days after the close of the registrant’s fiscal year ended December 31, 2016.
Table of Contents
| Page | ||||
| 1 | ||||
| 4 | ||||
| 40 | ||||
| 61 | ||||
| 61 | ||||
| 61 | ||||
| 61 | ||||
| 62 | ||||
| 62 | ||||
| 64 | ||||
| Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations |
65 | |||
| Item 7A. Quantitative and Qualitative Disclosures about Market Risk |
83 | |||
| 83 | ||||
| Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
83 | |||
| 83 | ||||
| 85 | ||||
| 86 | ||||
| 86 | ||||
| 86 | ||||
| Item 12. Security Ownership of Certain Beneficial Owners and Management |
86 | |||
| 86 | ||||
| 86 | ||||
| 87 | ||||
| 87 | ||||
| F-1 | ||||
| EXHIBITS FILED WITH FORM 10-K |
||||
| Ex. 23.1 Consent of Independent Registered Public Accounting Firm |
||||
| EX 31.1 Section 302 Certification of CEO |
||||
| EX 31.2 Section 302 Certification of CFO |
||||
| EX 32.1 Section 906 Certification of CEO |
||||
| EX 32.2 Section 906 Certification of CFO |
||||
Table of Contents
You are urged to read this Annual Report on Form 10-K (“Form 10-K”) in its entirety. This Form 10-K contains forward-looking statements that involve risks and uncertainties. Our actual results may differ significantly from the projected results discussed in these forward-looking statements. Factors that may cause such a difference include, but are not limited to, those discussed below and in Item 1A, “Risk Factors.”
“We,” “our,” “ours,” “us,” “Catalyst,” or the “Company,” when used herein, refers to Catalyst Pharmaceuticals, Inc., a Delaware corporation.
Forward-Looking Statements
This Annual Report on Form 10-K contains “forward-looking statements”, as that term is defined in the Private Securities Litigation Reform Act of 1995. These include statements regarding our expectations, beliefs, plans or objectives for future operations and anticipated results of operations. For this purpose, any statements contained herein that are not statements of historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, “believes”, “anticipates”, “proposes”, “plans”, “expects”, “intends”, “may”, and other similar expressions are intended to identify forward-looking statements. Such statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or other achievements to be materially different from any future results, performances or achievements expressed or implied by such forward-looking statements. Factors that might cause such differences include, but are not limited to, those discussed in the section entitled “Item 1A – Risk Factors” and those discussed in the section entitled “Item 7 – Management’s Discussion and Analysis of Financial Condition and Results of Operations – Caution Concerning Forward-Looking Statements.”
The successful development and commercialization of our current drug candidates is highly uncertain. We cannot reasonably estimate or know the nature, timing, or estimated expenses of the efforts necessary to complete the development of, or the period in which material net cash inflows are expected to commence due to the numerous risks and uncertainties associated with developing such products, including the uncertainty of:
| • |
our estimates regarding anticipated capital requirements and our need for additional financing; |
| • |
the risk that another pharmaceutical company will receive an approval for its formulation of 3,4-diaminopyridine (3,4-DAP) for the treatment of Lambert-Eaton Myasthenic Syndrome (LEMS), Congenital Myasthenic Syndromes (CMS), or any other indication, before we do; |
| • |
whether the clinical studies or trials that are required to be completed before the U.S. Food and Drug Administration (FDA) will accept a new drug application (NDA) submission for Firdapse® for the treatment of either LEMS or CMS will be successful; |
| • |
what additional supporting information, including any additional clinical studies or trials, will be required before the FDA will accept our NDA submission for Firdapse® for the treatment of either LEMS or CMS (or any other condition or disease); |
| • |
whether any NDA that we may submit to the FDA for Firdapse® will be accepted for filing, or whether even if any NDA that we submit for Firdapse® is accepted for filing by the FDA, whether any such NDA filing will be granted a priority review; |
1
Table of Contents
| • |
whether, even if the FDA accepts an NDA submission for Firdapse®, such product will be determined to be safe and effective and approved for commercialization; |
| • |
whether the receipt of breakthrough therapy designation for Firdapse® for LEMS will result in an expedited review of Firdapse® by the FDA or affect the likelihood that the product will be found to be safe and effective; |
| • |
whether as part of the FDA review of any NDA that we may submit for filing for Firdapse® , the tradename Firdapse®, which is the tradename used for the same product in Europe, will be approved for use for the product in the United States; |
| • |
whether, assuming Firdapse® is approved for commercialization, we will be able to develop or contract with a sales and marketing organization that can successfully market Firdapse® while maintaining full compliance with applicable federal and state laws, rules and regulations; |
| • |
whether any future trial we undertake evaluating Firdapse® for the treatment of myasthenia gravis patients with anti-MuSK antibodies will be successful and whether we can obtain the funding required for such trial; |
| • |
whether CPP-115 will be determined to be safe for humans; |
| • |
whether CPP-115 will be determined to be effective for the treatment of infantile spasms, post-traumatic stress disorder, Tourette’s Disorder or any other indication; |
| • |
whether we can successfully design and complete a bioequivalence study of our version of vigabatrin compared to Sabril® that is acceptable to the FDA; |
| • |
whether any such bioequivalence study, the design of which is acceptable to the FDA, will be successful; |
| • |
whether any abbreviated new drug application (ANDA) that we submit for a generic version of Sabril® will be accepted by the FDA for review and approved (and the timing of any such approval); |
| • |
the scope, rate of progress and expense of our clinical trials and studies, pre-clinical studies, proof-of-concept studies, and our other drug development activities, and whether our trials and studies will be successful; |
| • |
our ability to complete our trials and studies on a timely basis and within the budgets we establish for such trials and studies; |
| • |
the results of our clinical studies and trials, pre-clinical studies, proof-of-concept studies, and our other development activities, and the number of such studies and trials that will be required for us to seek and obtain approval of new drug applications, or NDAs, for our drug candidates; |
| • |
whether the third parties that assist us in our trials and studies perform as anticipated and within the budgets established for their activities; |
| • |
the ability of our third-party suppliers and contract manufacturers to maintain compliance with current Good Manufacturing Practices (cGMP); |
| • |
whether any of our drug candidates will ever be approved for commercialization; |
| • |
even if one or more of our drug candidates is approved for commercialization, whether we will be able to successfully commercialize those products and achieve sustained profitability; |
2
Table of Contents
| • |
whether our estimates of the pricing of our drug candidates, if approved, and our estimates of the size of the market for our drug candidates, will turn out to be accurate; |
| • |
the level of third-party payor reimbursement for any of our drug candidates that are commercialized; |
| • |
what pricing we will be able to achieve with respect to our drug products and the impact of public scrutiny and potential changes in the law that affect the pricing of drug products generally, and our products and orphan drugs in particular; |
| • |
changes in the healthcare industry occasioned by any future repeal and replacement of the Affordable Care Act, or changes in the healthcare industry generally; |
| • |
changes in the manner in which the FDA reviews NDAs; |
| • |
changes in the laws and regulations and regulatory guidance affecting our business, including any changes that may be made to the laws affecting orphan drugs (such as changes to the exclusivity that may be available for approved products under those laws); |
| • |
the market adoption of any of our drug candidates approved for commercialization by physicians and patients; |
| • |
our ability to obtain a sufficient commercial supply of our products; |
| • |
if one or more of our products are approved for commercialization, the costs, timing or estimated completion of any post-marketing studies that we may be obligated to complete; |
| • |
our expectations regarding licensing, acquisitions or strategic relationships; |
| • |
whether we can successfully protect any of our drug candidates under intellectual property laws; |
| • |
the expense of filing, and potentially prosecuting, defending and enforcing any patent claims and other intellectual property rights we may have for our drug candidates; |
| • |
our ability to attract and retain skilled employees; |
| • |
security breaches of our computer systems, or the computer systems of our contractors and/or vendors; |
| • |
the impact of potential employee, vendor or consultant misconduct; and |
| • |
changes in general economic conditions and interest rates. |
Our current plans and objectives are based on assumptions relating to the development of our current drug candidates. Although we believe that our assumptions are reasonable, any of our assumptions could prove inaccurate. The significant uncertainties inherent in the forward-looking statements we have made herein, which reflect our views only as of the date of this report, suggest that you should not place undue reliance upon such statements. We undertake no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise.
3
Table of Contents
| Business |
Overview
We are a biopharmaceutical company focused on developing and commercializing innovative therapies for people with rare debilitating diseases. We currently have three drug candidates in development:
Firdapse®
In October 2012, we licensed the North American rights to Firdapse®, a proprietary form of amifampridine phosphate, or chemically known as 3,4-diaminopyridine phosphate, from BioMarin Pharmaceutical Inc. (BioMarin). In August 2013, we were granted “breakthrough therapy designation” by the U.S. Food & Drug Administration (FDA) for Firdapse® for the treatment of patients with Lambert-Eaton Myasthenic Syndrome, or LEMS, a rare and sometimes fatal autoimmune disease characterized by muscle weakness. In March 2015, we were granted Orphan Drug Designation for Firdapse® for the treatment of patients with Congenital Myasthenic Syndromes, or CMS, and in August 2016, we were granted Orphan Drug Designation for Firdapse® for the treatment of patients with Myasthenia Gravis (MG).
The chemical entity, amifampridine (3,4-diaminopyridine or 3,4-DAP), has never been approved by the FDA for any indication. Because Firdapse® has been granted Orphan Drug designation for the treatment of LEMS, CMS and MG by the FDA, the product is also eligible to receive seven years of marketing exclusivity for any or all of these indications. Further, if we are the first pharmaceutical company to obtain approval for an amifampridine product, of which there can be no assurance, we will be eligible to receive five years of marketing exclusivity with respect to the use of this product for any indication, running concurrently with the seven years of orphan marketing exclusivity described above (if both exclusivities are granted).
We previously sponsored a multi-center, randomized, placebo-controlled Phase 3 trial evaluating Firdapse® for the treatment of LEMS. The Phase 3 trial, which involved 38 subjects, was designed as a randomized “withdrawal” trial in which all patients were treated with Firdapse® during a 7 to 91-day run-in-period followed by treatment with either Firdapse® or placebo over a two-week randomization period. The co-primary endpoints for this Phase 3 trial were the comparison of changes in patients randomized to continue Firdapse® versus those who transitioned to placebo that occurred in both the Quantitative Myasthenia Gravis Score (QMG), which measures muscle strength, and subject global impression score (SGI), on which the subjects rate their global impression of the effects of a study treatment during the two-week randomization period. In September 2014, we reported positive top-line results from this Phase 3 trial.
During 2014, we established an expanded access program (EAP) to make Firdapse® available to patients diagnosed with LEMS, CMS, or Downbeat Nystagmus in the United States, who meet the inclusion and exclusion criteria, with Firdapse® being provided to patients for free until sometime after new drug application (NDA) approval, should we receive such approval (of which there can be no assurance). We are informing neuromuscular physicians on the availability of the Firdapse® EAP and working with various rare disease advocacy organizations to inform patients and physicians about the program.
On December 17, 2015, we announced completion of the submission of an NDA for Firdapse® for the treatment of LEMS and CMS. However, on February 17, 2016, we announced that we had received a “refusal to file” (RTF) letter from the FDA regarding our NDA submission. In early April 2016, we met with the FDA to obtain greater clarity regarding what will be required by the FDA to accept the Firdapse® NDA for filing. Following the receipt of the formal minutes of that meeting, on April 26, 2016, we issued a press release reporting that the FDA has advised us that in addition to the results of our previously submitted multi-center, randomized, placebo-controlled Phase 3 trial, we will need to submit positive results from a second adequate and well-controlled study in patients with LEMS. Additionally, there is a requirement for several more short-term toxicology studies, which are currently in process.
4
Table of Contents
In October 2016, we announced that we had reached an agreement with the FDA under a Special Protocol Assessment (SPA) for the protocol design, clinical endpoints, and statistical analysis approach to be taken in our second Phase 3 study evaluating Firdapse® (amifampridine phosphate) for the symptomatic treatment of LEMS. A SPA is a process by which sponsors ask the FDA to evaluate the protocol of a proposed clinical trial to determine whether it adequately addresses scientific and regulatory requirements for the purpose identified by the sponsor. A SPA agreement indicates FDA concurrence with the adequacy and acceptability of specific critical elements of protocol design, endpoints and analysis. Additionally, it provides a binding agreement with FDA’s review division that a pivotal trial design, conduct, and planned analysis adequately addresses the scientific and regulatory objectives in support of a regulatory submission for drug approval. However, the FDA may rescind a SPA agreement when the division director determines that a substantial scientific issue essential to determining the safety or efficacy of the product has been identified after the trial has begun.
We are conducting our second Phase 3 trial evaluating Firdapse® for the treatment of LEMS (designated as LMS-003) at sites in Miami, Florida and Los Angeles, California. This double-blind, placebo-controlled withdrawal trial will include approximately 28 subjects, and will have the same co-primary endpoints as our first Phase 3 trial evaluating Firdapse® for the treatment of LEMS. Further, the FDA is allowing us to enroll patients from our expanded access program as study subjects in this second trial. Details of the Phase 3 clinical trial are available on www.clinicaltrials.gov (NCT02970162).
We initiated this trial in December 2016, and we expect to report top-line results from this trial during the second half of 2017. Assuming the results of this trial are successful, and our anticipated timeline for the completion of this trial is met, we expect to resubmit an NDA for Firdapse® for the treatment of LEMS in the second half of 2017. There can be no assurance as to the timing or requirements of this trial, whether this trial, along with the results of our first Phase 3 trial, will be sufficient for the FDA to accept for filing any NDA that we might resubmit in the future for Firdapse®, or whether Firdapse® will ever be approved for commercialization.
Our original NDA submission for Firdapse® included data and information (including data from a currently ongoing investigator treatment IND) providing evidence supporting the benefits of Firdapse® for treating certain types of CMS, and requested that CMS be included in our initial label for Firdapse® . To provide additional support for our submission of an NDA for Firdapse® for the treatment of CMS, in October 2015 we initiated a small blinded clinical trial at four academic centers of up to 10 subjects in the pediatric CMS population, ages 2 to 17. However, after considering comments from the FDA, we determined to enroll both adult and pediatric subjects with CMS in this trial and to expand the number of subjects to be evaluated in the trial to an aggregate of approximately 20 subjects. We also added a fifth trial site for this study. Details of this trial are available on www.clinicaltrials.gov (NCT02562066).
Based on currently available information, we expect to report top-line results from this study in the second half of 2017 and if the results of the study are successful, we hope to add the CMS indication to our labeling for Firdapse® (either as a part of our NDA resubmission for Firdapse® for LEMS or as a supplement to that resubmission). There can be no assurance that any trial we perform for Firdapse® for the treatment of CMS will be successful or whether any NDA that we may submit for Firdapse® for the treatment of CMS will be filed by the FDA for review and approved.
5
Table of Contents
In February 2016, we announced the initiation of an investigator-sponsored, randomized, double-blind, placebo-controlled, crossover Phase 2/3 clinical trial evaluating the safety, tolerability and potential efficacy of Firdapse® as a symptomatic treatment for patients with anti-MuSK antibody positive MG (MuSK-MG). MuSK-MG, an ultra-rare sub-population of myasthenia gravis (MG) patients, is a debilitating neuromuscular disease, and there are currently no FDA approved therapies for this specific form of MG. Seven patients participated in this proof-of-concept trial. We provided study drug, placebo and financial support for this study.
On March 15, 2017 we reported top-line results from this trial. Both of the co-primary efficacy endpoints of change from baseline (CFB) in total Quantitative Myasthenia Gravis (QMG) score (p=0.0003) and CFB in total Myasthenia Gravis Activities of Daily Living (MG-ADL) score (p=0.0006) were statistically and clinically significant in this trial. Several secondary efficacy measures also achieved statistical significance. Amifampridine phosphate was well tolerated in this population of patients.
We intend to discuss with the FDA conducting a registration trial in the United States evaluating Firdapse® for the treatment of patients with MuSK-MG. There can be no assurance that future clinical trials that we initiate to evaluate Firdapse® for this indication will be successful, or whether we can obtain the resources available to fund any such registration trial. Further, there can also be no assurance that the FDA will ever approve Firdapse® for this indication.
Finally, we may seek to evaluate Firdapse® for the treatment of other treatment-refractory types of MG or other rare, similar neuromuscular diseases, although we have not yet begun to develop clinical programs for these indications and all such programs are subject to the availability of funding. There can be no assurance that Firdapse® will be an effective treatment for other treatment-refractory types of MG or for any other rare, similar neuromuscular diseases.
Prior to the receipt of the RTF letter, we had been actively taking steps to prepare for the commercialization of Firdapse® in the United States. In light of the determination that we will have to complete a second adequate and well controlled study evaluating Firdapse® for the treatment of LEMS, we have placed most of these commercialization activities on hold in order to conserve cash. Notwithstanding, we are continuing to work with several rare disease advocacy organizations to help increase awareness of LEMS and CMS and to provide awareness and outreach support for the physicians who treat these rare diseases and the patients they treat.
CPP-115
We are developing CPP-115, a GABA aminotransferase inhibitor that, based on our preclinical studies to date, we believe is a more potent form of vigabatrin, and may have fewer side effects (e.g., visual field defects) than those associated with vigabatrin. We are hoping to develop CPP-115 for the treatment of refractory infantile spasms and possibly for the treatment of adult refractory patients with Tourette’s Disorder. CPP-115 has been granted Orphan Drug Designation by the FDA for the treatment of infantile spasms and Orphan Medicinal Product Designation in the European Union, or E.U., for West syndrome (a form of infantile spasms).
We are currently refining our development plan for this product. Once the refinement of our development plans is completed, and subject to the then availability of funding, we plan to take the steps to complete the work required to make our drug candidate Phase 2 ready. We are also working with one or more potential investigators who have expressed an interest in evaluating our product for particular indications (particularly infantile spasms). We are also continuing to seek a partner to work with us in furthering the development of CPP-115, although no agreements have been entered into to date.
There can be no assurance that we will ever successfully commercialize CPP-115.
6
Table of Contents
Generic Sabril®
During September 2015, we announced the initiation of a project to develop a generic version of Sabril® (vigabatrin). Sabril® is marketed by Lundbeck Inc. in the United States for the treatment of infantile spasms and complex partial seizures. There can be no assurance that we will be successful in these efforts or that any ANDA that we submit for vigabatrin will be accepted for review or approved. Further, while there can be no assurance, we are hopeful that any ANDA submission we make for vigabatrin will be one of the first ANDAs submitted for this product.
Capital Resources
Based on our current financial condition and forecasts of available cash, we believe that we have sufficient funds to support our operations through at least the next 12 months. However, we will require additional funding to support our operations beyond that time. There can be no assurance that we will obtain additional funding or that we will ever be in a position to commercialize any of our drug candidates. See Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Liquidity and Capital Resources” below for further information on our liquidity and cash flow.
Our Strategy
Our goal is to develop and commercialize novel prescription drugs targeting rare (orphan) diseases with an initial focus on neuromuscular and neurological diseases and disorders. Specifically, we intend to:
| • |
Pursue approval of Firdapse® for LEMS and CMS. We are continuing our efforts to seek approval to commercialize Firdapse® for LEMS. We are also taking steps that we hope will allow us to include CMS in our initial labeling for our product. |
| • |
Seek additional orphan drug indications for Firdapse®. Subject to the availability of funding, we plan to pursue the necessary clinical and non-clinical trials and studies to evaluate Firdapse® as a treatment for MuSK-MG and possibly for other neuromuscular indications. |
| • |
Continue required clinical and pre-clinical work on CPP-115. We hope to undertake the clinical and non-clinical studies required to make CPP-115 Phase 2 ready. We hope to develop CPP-115 for the treatment of infantile spasms and possibly for the treatment of adult refractory patients with Tourette’s Disorder. |
| • |
Generic Sabril®. We are taking the steps that we believe are necessary to develop and obtain approval of a generic version of Sabril® (vigabatrin tablets and sachets). Because of our prior experience developing our version of vigabatrin (CPP-109), we believe that we are well positioned to make this effort and, if we are successful, we hope to receive approval for one of the first generic versions of this product. |
| • |
Seek a partner for CPP-115 or generic Sabril®. We may seek a strategic partner to work with us in the development and future commercialization of CPP-115 and/or generic Sabril®. However, no arrangements have been entered into to date. |
| • |
Seek to acquire additional products. We continue to seek to acquire additional relatively late stage orphan drug opportunities to add to our product portfolio. However, no agreements have been entered into to date to acquire additional products and any such product acquisitions would be subject to the availability of funding. |
7
Table of Contents
Firdapse®
Product overview
Firdapse® is Catalyst’s and BioMarin’s (depending on market region) registered trade name for amifampridine phosphate tablets. Amifampridine is the WHO (World Health Organization) registered INN (International Nonproprietary Name) and United States Adopted Name (USAN) for the chemical entity, 3,4-diaminopyridine, often abbreviated as 3,4-DAP. Firdapse® contains the phosphate salt of amifampridine, hence the name “amifampridine phosphate.” We will refer to our drug by its proposed trade name in the United States (Firdapse®), by the INN/USAN (amifampridine), or by the specific salt in Catalyst’s product (amifampridine phosphate), throughout this Form 10-K.
In addition to the positive results we reported from our Phase 3 trial of amifampridine phosphate described below, clinical efficacy information for the symptomatic treatment of LEMS patients with amifampridine have been derived from five published randomized, double-blind, placebo-controlled studies and one published randomized, double-blind, active-control study in patients with LEMS. The data from the randomized controlled studies generally show statistically significant improvements across a number of measures of neurological function, including Quantitative Myasthenia Gravis (QMG) score and compound muscle action potential (CMAP), which have been demonstrated to be clinically relevant in patients with LEMS. Results of these studies suggest that amifampridine is more effective for the symptomatic treatment of LEMS compared with placebo or active investigational comparator (pyridostigmine). Additionally, data from multiple published uncontrolled investigations and case reports support the long-term benefits of treatment with amifampridine in patients with LEMS. In some cases, removal of patients from drug can lead to a recurrence of underlying symptoms, but with reintroduction of amifampridine improvement of muscle function is regained. Amifampridine has been recommended as first-line symptomatic treatment for LEMS by the European Federation of Neurological Societies (now known as the European Academy of Neurology). In December 2009, amifampridine phosphate received marketing approval from the European Commission (with the trade name Firdapse®) for the symptomatic treatment of patients with LEMS.
Safety data from clinical data published over the last 30 years in patients with LEMS or other neurological disorders treated with amifampridine show that amifampridine is well tolerated at doses £80 mg per day. Among the 1,279 patients or healthy subjects assessed in the literature, the most frequently reported adverse events (AEs) were perioral and peripheral paresthesias (unusual sensations like pins and needles), and gastrointestinal disorders (abdominal pain, nausea, diarrhea, and epigastralgia (pain around the upper part of the stomach)). These events were typically mild or moderate in severity, and transient, seldom requiring dose reduction or withdrawal from treatment.
Lambert-Eaton Myasthenic Syndrome
Lambert-Eaton Myasthenic Syndrome, or LEMS, is a rare autoimmune neuromuscular disorder characterized primarily by muscle weakness of the limbs. The disease is caused by an autoimmune reaction where antibodies are formed against voltage-gated calcium channels on nerve endings, which damages the channels. These calcium channels are responsible for the transport of charged calcium atoms that activate the biochemical machinery responsible for releasing acetylcholine. Acetylcholine is the neurotransmitter responsible for causing muscles to contract and the failure to release enough of this neurotransmitter results in muscle weakness in LEMS patients. Additionally, LEMS is often associated with an underlying malignancy, most commonly small-cell lung cancer (SCLC), and in some individuals, LEMS is the first symptom of such malignancy.
8
Table of Contents
LEMS generally affects the extremities, especially the legs. As LEMS most affects the parts of limbs closest to the trunk, difficulties with climbing stairs or rising from a sitting position are commonly noted. Physical exercise and high temperatures tend to worsen the symptoms. Other symptoms often seen include weakness of the muscles of the mouth, throat, and eyes. Individuals affected with LEMS also may have a disruption of the autonomic nervous system, including dry mouth, constipation, blurred vision, impaired sweating, and/or hypotension.
LEMS is managed by treating the symptoms or treating the underlying autoimmune attack on voltage-gated calcium channels. Unapproved treatments include steroids, azathioprine and intravenous immunoglobulin, which work by suppressing the immune system; and pyridostigmine and amifampridine, which enhance neuromuscular transmission. Plasma exchange has also been used to attempt to remove antibodies from the body. Firdapse® is a symptomatic treatment and does not alter the underlying autoimmune condition. As a voltage-gated potassium blocker, Firdapse® prevents charged potassium particles from leaving the nerve cells, which prolongs the period of depolarization. This allows more charged calcium atoms to enter the nerves, which enables the nerves to release acetylcholine and causes muscles to contract and to restore lost muscle strength in LEMS patients.
Based on currently available information, we estimate that there are approximately 3,000 LEMS patients in the United States. However, until an amifampridine product is finally approved by the FDA and awareness of the disease is increased, it is unlikely that the total number of LEMS patients in the United States can be determined with better certainty (as is typical of rare diseases), and the actual number of patients in the United States with LEMS may be higher or lower than our estimate. Some of the factors that affect the assessment of the size of the population with a rare disease such as LEMS include, without limitation, the number of patients actually diagnosed with the disease, the number of patients who were misdiagnosed with other diseases before it is determined that they have the disease, and the number of patients who have the disease whose doctors do not become aware of the availability of a treatment for the disease until after a product is approved or, even if they are aware of the product, are unwilling or unable to prescribe the product until it is approved and generally available in the commercial marketplace. In this case, we believe that some patients who have LEMS could have been misdiagnosed with another disease (such as MG) and that while there is an antibody test that positively identifies patients with LEMS, we believe that the test is not particularly well known or utilized at this time by neurologists. Further, we believe that many patients with small cell lung cancer, or SCLC, some of whom also have LEMS, are not being treated for LEMS because many of the oncology medical professionals who treat SCLC patients are generally not familiar with how to diagnose and treat LEMS. All of these factors are likely to affect the ultimate number of patients, either up or down, who are indicated and in need of treatment with an amifampridine product.
Congenital Myasthenic Syndromes
Congenital myasthenic syndromes, or CMS, is a rare neuromuscular disorder comprising a spectrum of genetic defects and is characterized by fatigable weakness of skeletal muscles with onset at or shortly after birth or early childhood; in rare cases symptoms may not manifest themselves until later in childhood. The severity and course of the genetic disease types are variable, ranging from minor symptoms to progressive disabling weakness; symptoms may be mild, but sudden severe exacerbations of weakness or even sudden episodes of respiratory insufficiency also occur.
Many patients with CMS may respond to unapproved pharmacologic intervention, including esterase inhibitors, amifampridine (i.e. 3,4-DAP), ephedrine, fluoxetine or quinidine, and albuterol, alone or in combinations. The particular therapy is dictated by the diagnosed CMS type, as drugs beneficial in treating one type of CMS can be detrimental in patients with another type of CMS.
9
Table of Contents
Congenital myasthenic syndrome(s) is rare, estimated at one-tenth that of MG, which in itself is rare. Based on currently available information, we estimate that there are between 1,000 and 1,500 CMS patients in the United States but that is subject to the same factors noted above with respect to LEMS.
Myasthenia Gravis
Myasthenia Gravis is a chronic autoimmune neuromuscular disorder that is characterized by fluctuating weakness of the voluntary muscle groups. The prevalence of MG in the United States is estimated to be about 20/100,000 population (equating to an estimate of approximately 64,000 patients in the United States). However, MG is probably under diagnosed and the prevalence may be higher. MG occurs in all races, both genders, and at any age. MG is not thought to be directly inherited nor is it contagious. It does occasionally occur in more than one member of the same family.
The voluntary muscles of the entire body are controlled by nerve impulses that arise in the brain. These nerve impulses travel down the nerves to the place where the nerves meet the muscle fibers. Nerve fibers do not actually connect with muscle fibers. There is a space between the nerve ending and muscle fiber; this space is called the neuromuscular junction. When the nerve impulse originating in the brain arrives at the nerve ending, it releases a chemical called acetylcholine. Acetylcholine travels across the space to the muscle fiber side of the neuromuscular junction where it attaches to many receptor sites. The muscle contracts when enough of the receptor sites have been activated by the acetylcholine. In MG, there can be as much as an 80% reduction in the number of these receptor sites. The reduction in the number of receptor sites is caused by an antibody that destroys or blocks the receptor site. Antibodies are proteins that play an important role in the immune system. They are normally directed at foreign proteins called antigens that attack the body. Such foreign proteins include bacteria and viruses. Antibodies help the body to protect itself from these foreign proteins. For reasons not well understood, the immune system of the person with MG makes antibodies against the receptor sites of the neuromuscular junction. Abnormal antibodies can be measured in the blood of many people with MG. The antibodies destroy the receptor sites more rapidly than the body can replace them. Muscle weakness occurs when acetylcholine cannot activate enough receptor sites at the neuromuscular junction.
Anti-MuSK antibody positive MG
About 15% of MG patients test negative for the acetylcholine receptor antibody. These patients have seronegative (SN) MG. Approximately 40-50% of these patients with SNMG (equating to an estimate of approximately 4,500 patients in the United States) test positive for the anti-MuSK antibody. MuSK is a protein that is required for the maintenance of the neuromuscular junction and patients with the anti-MuSK antibody are identified as having MuSK-MG. MuSK-MG is a clinically distinguishable, more severe form of MG. The disease is characterized by a predominance in females, a prevalent involvement of cranial and bulbar muscles, high incidence of respiratory crises and a resistance to treatment. Although many patients with MuSK-MG are presently treated with anticholinesterase inhibitors or immunosuppressants, such patients do not generally respond adequately to these treatments.
Strategic collaboration with BioMarin for Firdapse®
On October 26, 2012, we entered into a strategic collaboration with BioMarin for Firdapse®. The key components of the collaboration included our licensing of the exclusive North American rights to Firdapse® pursuant to a License Agreement, dated October 26, 2012, between us and BioMarin (the BioMarin License Agreement), and BioMarin making a $5.0 million investment in our common stock to advance the development of Firdapse®. We believe that we remain in compliance with the license agreement.
10
Table of Contents
Under the BioMarin License Agreement, we have agreed to make the following royalty payments:
(i) royalty payments to BioMarin for seven years from the first commercial sale equal to: (a) 7% of net sales (as defined in the license agreement) in North America in any calendar year for sales up to $100 million, and (b) 10% of net sales in North America in any calendar year in excess of $100 million; and
(ii) royalty payments to a third-party licensor of the rights sublicensed to us for seven years from the first commercial sale equal to 7% of net sales (as defined in the license agreement between BioMarin and the third party licensor) in North America in any calendar year.
Under the BioMarin License Agreement, we have also agreed to make certain milestone payments to such third-party licensor and to the former stockholders of Huxley Pharmaceuticals, Inc. (Huxley) that BioMarin is obligated to make. With respect to Firdapse®, the milestones aggregate approximately $2.6 million upon acceptance of an NDA for Firdapse® by the FDA for the treatment of LEMS, and approximately $7.2 million upon the unconditional approval by the FDA of an NDA for Firdapse® for the treatment of LEMS.
Firdapse® was approved by European Medicines Agency for the treatment of LEMS in December 2009, and BioMarin sells the product in the EU. BioMarin is also currently performing or has previously completed certain post-marketing studies of Firdapse® that they are required to conduct to support their continued approval of Firdapse® in the EU. We have agreed to pay one half of the costs of these studies. We have also shared the costs of a cardiac safety study and reproductive toxicity studies that have been completed and are studies that are required for approval of Firdapse® by the FDA.
On April 15, 2014, effective as of April 8, 2014, we and BioMarin entered into Amendment No. 1 to the License Agreement, amending in certain respects the BioMarin License Agreement, dated October 26, 2012. The amendment related to purchases of additional product by us from BioMarin, the sharing of data between the parties with respect to clinical trials and studies undertaken by each party, and the payment terms for certain joint studies.
Breakthrough therapy designation
Firdapse® for LEMS has been granted Breakthrough Therapy Designation by the FDA. A breakthrough therapy is defined as a drug that is “intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.”
A Breakthrough Therapy Designation conveys all of the fast track program features, as well as more intensive FDA guidance on an efficient drug development program. The FDA also has an organizational commitment to involve senior management in such guidance. The Breakthrough Therapy Designation is a distinct status from both accelerated approval and priority review, which can also be granted to the same drug if relevant criteria are met.
Orphan drug designation
The FDA has granted Orphan Drug Designation for amifampridine phosphate for the treatment of LEMS, CMS and MG, making the drug eligible to be granted seven-year marketing exclusivity for these indications if we are the first pharmaceutical company to obtain approval of an NDA for a product containing amifampridine as the active moiety for the treatment of LEMS, CMS or MG.
11
Table of Contents
In addition, the FDA has also granted Jacobus Pharmaceutical’s Orphan Drug Designation request for 3,4-diaminopyridine for the treatment of LEMS, which means that if Jacobus Pharmaceuticals were to be the first pharmaceutical company to obtain approval of an NDA for a product containing amifampridine as the active moiety for the treatment of LEMS, we would not be able to obtain FDA approval for that indication for seven years.
The first Phase 3 clinical trial
As part of our License Agreement with BioMarin, we took over a Phase 3 clinical trial that BioMarin had previously begun in the United States and Europe evaluating Firdapse® for the treatment of LEMS. The trial was designed as a randomized double-blind, placebo-controlled discontinuation trial in approximately 36 LEMS patients. After patients were treated with amifampridine phosphate for at least 91 days, they were randomly assigned to either continue on amifampridine phosphate or be discontinued to placebo over a 2-week period. They were then returned to open label amifampridine phosphate treatment for a two-year follow-up period.
On September 29, 2014, we reported top-line results from this trial. A summary of the results is as follows:
| • |
Primary endpoints: |
| • |
The primary endpoint of change in quantitative myasthenia gravis score, or QMG, at day 14 reached statistical significance (p=0.0452), with a worsening of 2.2 points observed in the placebo group and a worsening of 0.4 points observed in the treatment group. |
| • |
The primary endpoint of change in subject global impression, or SGI, at day 14 was highly statistically significant (p=0.0028), with a worsening of 2.6 points observed in the placebo group and a worsening of 0.8 points observed in the treatment group. |
| • |
Secondary endpoints: |
| • |
The secondary endpoint for the physician’s clinical global impression of improvement, or CGI-I, reached statistical significance (p=0.0267), with a worsening at day 14 of 1.1 points between the placebo group and the treatment group. |
| • |
The secondary endpoint of change in walking speed at day 14 was not statistically significant. |
| • |
Patient tolerance of Firdapse®: |
| • |
Firdapse® was generally safe and well tolerated. During the 91-day open label run-in period, treatment emergent adverse events occurred more frequently in treatment-naïve patients than in previously treated patients (approximately 10% of treatment naïve patients withdrew during this part of the study). During the placebo-controlled portion of the study, side effects occurring more frequently in the Firdapse® group were benign and consisted primarily of perioral and digital paresthesia and infections. No patients withdrew during this period. |
| • |
All subjects who were randomized into the trial elected to continue with Firdapse® in the two-year safety follow-up phase of the trial. |
The results of the Phase 3 trial were first presented in October 2014 at the 139th Annual Meeting of the American Neurological Association (ANA). They have subsequently been presented at the 2014 and 2015 annual meeting of the American Association of Neuromuscular and Electrodiagnostic Medicine (AANEM) and at the 2015 meeting of the American Academy of Neurology (AAN), and published in the May 2016 edition of Muscle & Nerve.
12
Table of Contents
Expanded access program
We currently operate an expanded access program (EAP) that makes Firdapse® available to all patients diagnosed with LEMS, CMS, or Downbeat Nystagmus in the United States who meet the inclusion and exclusion criteria, with Firdapse® being provided to patients at no cost until sometime after FDA approval, should we receive such approval (of which there can be no assurance). We are currently working with various rare disease advocacy organizations to inform physicians and patients as to the availability of our EAP.
NDA submission and Refusal-to-File Letter
On July 22, 2015, we announced that we had initiated a rolling submission of an NDA for Firdapse® for the treatment of LEMS and CMS, and on December 17, 2015, we announced the completion of that submission. On February 17, 2016, we announced that we had received an RTF letter from the FDA regarding our NDA submission. The RTF letter stated that after a preliminary review, the FDA has found that our application was not sufficiently complete and requests additional supporting information. The letter did not provide comment on the acceptability of the submitted clinical data, and no judgment was made in the letter on the efficacy or safety of Firdapse®.
On April 26, 2016, we announced that we had met with the FDA to discuss the FDA’s RTF letter. During that meeting, the FDA advised us that in addition to the results of our prior Phase 3 trial, we will need to submit positive results from an additional adequate and well-controlled study in patients with LEMS.
Ongoing clinical trials
Second Phase 3 trial evaluating Firdapse® for the treatment of LEMS
As required by the FDA, we are now conducting a second Phase 3 trial evaluating Firdapse® for the treatment of LEMS (designated as LMS-003) at sites in Miami, Florida and Los Angeles, California. This double-blind, placebo-controlled withdrawal trial will include approximately 28 subjects, and will have the same co-primary endpoints as our first Phase 3 trial evaluating Firdapse® for the treatment of LEMS. Further, the FDA is allowing us to enroll patients from our EAP as study subjects in this second trial. Details of this second Phase 3 clinical trial are available on www.clinicaltrials.gov (NCT02970162).
The protocol design, clinical endpoints, and statistical analysis approach being taken in our second Phase 3 study evaluating Firdapse® (amifampridine phosphate) for the symptomatic treatment of LEMS are based on the Special Protocol Assessment (SPA) that we received from the FDA and reported on in October 2016. A SPA is a process by which sponsors ask the FDA to evaluate the protocol of a proposed clinical trial to determine whether it adequately addresses scientific and regulatory requirements for the purpose identified by the sponsor. A SPA agreement indicates concurrence with the adequacy and acceptability of specific critical elements of protocol design, endpoints and analysis. Additionally, it provides a binding agreement with FDA’s review division that a pivotal trial design, conduct, and planned analysis adequately addresses the scientific and regulatory objectives in support of a regulatory submission for drug approval. However, FDA may rescind an SPA agreement when the division director determines that a substantial scientific issue essential to determining the safety or efficacy of the product has been identified after the trial has begun.
We initiated this second Phase 3 clinical trial in December 2016, and we expect to report top-line results from this trial during the second half of 2017. Assuming the results of this trial are successful, and our anticipated timeline for the completion of this trial is met, we expect to resubmit an NDA for Firdapse® for the treatment of LEMS in the second half of 2017. There can be no assurance as to the timing or requirements of this trial, whether this trial will be successful, whether this trial, along with the results of our first Phase 3 trial, will be sufficient for the FDA to accept for filing any NDA that we might resubmit in the future for Firdapse®, or whether Firdapse® will ever be approved for commercialization.
13
Table of Contents
Phase 3 clinical trial evaluating Firdapse® for the treatment of CMS
Our original NDA submission for Firdapse® included data and information (including data from a currently ongoing investigator treatment IND) providing evidence supporting the benefits of Firdapse® for treating certain types of CMS, and requested that CMS be included in our initial label for Firdapse®. To provide additional support for our submission of an NDA for Firdapse® for the treatment of CMS, in October 2015 we initiated a small blinded clinical trial at four academic centers of up to 10 subjects in the pediatric CMS population, ages 2 to 17. However, after considering comments from the FDA, we determined to enroll both adult and pediatric subjects with CMS in this trial and to expand the number of subjects to be evaluated in the trial to an aggregate of approximately 20 subjects. We also added a fifth trial site for this study. Details of this trial are available on www.clinicaltrials.gov (NCT02562066).
Based on currently available information, we expect to report top line results from this study in the second half of 2017 and if the results of the study are successful, we hope to add the CMS indication to our label for Firdapse® (either as a part of our NDA resubmission for Firdapse® for LEMS or as a supplement to that resubmission). There can be no assurance that any trial we perform for Firdapse® for the treatment of CMS will be successful or whether any NDA that we may submit for Firdapse® for the treatment of CMS will be filed by the FDA for review and approved.
Clinical trial evaluating Firdapse® for the treatment of MuSK-MG
In February 2016, we announced the initiation of an investigator-sponsored, randomized, double-blind, placebo-controlled, crossover Phase 2/3 clinical trial evaluating the safety, tolerability and potential efficacy of Firdapse® as a symptomatic treatment for patients with MuSK-MG. MuSK-MG, an ultra-rare sub-population of MG patients, is a debilitating neuromuscular disease, and there are currently no FDA approved therapies for this specific form of MG. Seven patients participated in this proof-of-concept trial. We provided study drug, placebo and financial support for this study.
On March 15, 2017 we reported top-line results from this trial. Both of the co-primary efficacy endpoints of change from baseline (CFB) in total Quantitative Myasthenia Gravis (QMG) score (p=0.0003) and CFB in total Myasthenia Gravis Activities of Daily Living (MG-ADL) score (p=0.0006) were statistically and clinically significant in this trial. Several secondary efficacy measures also achieved statistical significance. Amifampridine phosphate was well tolerated in this population of patients.
We intend to discuss with the FDA conducting a registration trial in the United States evaluating Firdapse® for the treatment of patients with MuSK-MG. There can be no assurance that future clinical trials that we initiate to evaluate Firdapse® for this indication will be successful, or whether we can obtain the resources available to fund any such registration trial. Further, there can also be no assurance that the FDA will ever approve Firdapse® for this indication.
Pre-commercialization efforts
Prior to the receipt of the RTF letter, we had been actively taking steps to prepare for the commercialization of Firdapse® in the United States, including the hiring of a Chief Commercial Officer. However, due to the receipt of an RTF letter, the need to complete a second Phase 3 trial evaluating Firdapse® for the treatment of LEMS, and the need to conserve cash, we underwent a reduction-in-force in May 2016 and terminated most of our commercial staff. As our second Phase 3 trial evaluating Firdapse® for the treatment of LEMS continues and approaches completion, we intend to renew our planning of our pre-commercialization efforts.
14
Table of Contents
Notwithstanding, we are continuing to work with several rare disease advocacy organizations to help increase awareness of LEMS, CMS, and our EAP and to provide awareness and outreach support for the physicians who treat these rare diseases and the patients they treat.
Future pricing of and access to Firdapse®
We have not yet established our pricing for Firdapse®. However, the independent market research that we have conducted to date indicates that we should be able to obtain typical orphan disease pricing for our product and that our product will likely be widely reimbursed by private and public payors for the indicated small populations of LEMS and CMS. There can be no assurance, however, as to the pricing of our product that we may be able to obtain or as to whether payors will agree to cover our product.
The pricing of pharmaceutical products, in general, and specialty drugs, in particular, has been a topic of concern in the U.S. Congress, where hearings have been held on the topic, and a topic of statements recently made by the President of the United States. There can be no assurance as to how this scrutiny on pricing of pharmaceutical products will impact future pricing of our products or orphan drugs or pharmaceutical products generally.
While our proposed pricing for Firdapse® has not been established, we recognize the importance of access to our medicines and, if Firdapse® is approved by the FDA, we expect to work with insurers to gain broad patient access in the U.S. market for the small patient populations of LEMS and CMS. We also expect to introduce and support comprehensive patient assistance programs and charitable access programs to assist eligible patients who on their own could not afford their medication.
There is a vocal group of neuromuscular physicians who have raised public concerns in a letter to the editor of a medical journal and some LEMS patients and neuromuscular physicians who have raised public concerns in interviews quoted in articles published in the press that LEMS patients may not be able to get amifampridine treatment. Their overarching concern appears to be that our product would be priced too high as an orphan drug if we were the first pharmaceutical company to receive an FDA approval for an amifampridine product, thereby giving us the seven-year orphan drug exclusivity and the five-year new chemical entity exclusivity for our product. Stories about their concerns have been published in several national publications and some in the press have sought to tie their expectations about the anticipated pricing of Firdapse® to stories about perceived abusive price increases of drug products by other pharmaceutical companies. This vocal group has also questioned the appropriateness of the provisions of the Orphan Drug Act that would grant us exclusivity if our product were to be the first amifampridine product approved by the FDA and whether this exclusivity should be eliminated from the law. We have directly responded to their concerns in a letter to the editor in this same medical publication. However, there can be no assurance as to the ultimate impact of these activities on us or our products.
Third-Party Reimbursement
Sales of pharmaceutical products depend in significant part on the availability of coverage and adequate reimbursement by third party payors, such as state and federal governments, including Medicare and Medicaid, managed care providers, private commercial insurance plans and pharmacy benefit management (PBM) plans. Decisions regarding the extent of coverage and the amount of reimbursement to be provided for Firdapse® are expected to be made on a plan-by-plan, and in some cases, on a patient-by-patient basis. Particularly given the rarity of LEMS and CMS, we anticipate that securing coverage and appropriate reimbursement from third-party payors will require targeted education. To that end, we expect to hire a dedicated team of field-based market access account managers and reimbursement experts focused on ensuring that clinically qualified patients have access to our product.
15
Table of Contents
Intellectual property protections for Firdapse®
Under the BioMarin License Agreement, we licensed two pending patents and certain trademarks for Firdapse®. One of the licensed patents is a pending composition of matter patent that, if issued, will protect Firdapse® until February 2027, which includes five years of patent term extension that is expected under the Patent Term Restoration Act. This application was initially rejected following an appeal to the Patent Trial and Appeal Board. The application was refiled with new claims. The new claims were the subject of an office action in which the claims were rejected. A response to the rejection was filed and a final rejection was issued. The application was refiled and is undergoing substantive examination. There can be no assurance that this patent will be issued. The second patent claims methods of administering Firdapse®. Substantive examination has begun on this patent application and a response is in progress. We may also pursue other patents in order to seek to protect the exclusivity of the drug, dosage forms and methods of administration.
No drug product containing amifampridine for any indication has been approved by the FDA. Therefore, our version of amifampridine, if we are the first to obtain approval of the product in the U.S., will be eligible for five-year new chemical entity exclusivity, which provides a five-year period of marketing exclusivity for all indications.
We have licensed the Firdapse® trademark from BioMarin. A trademark application for Firdapse® was allowed, but did not register due to the inability to show use of the mark in interstate shipment. The application was refiled and a Statement of Use was submitted and accepted by the Trademark Office, and the mark was registered in March 2015.
In January 2014, the FDA provisionally approved Firdapse® as a proprietary name for amifampridine phosphate tablets. This provisional approval by the FDA would not prevent the agency from rejecting the name Firdapse® at a later date as part of the NDA review and approval process.
CPP-115
Current status of our development efforts for CPP-115
We are developing CPP-115, a GABA aminotransferase inhibitor that, based on our preclinical studies to date, we believe is a more potent form of vigabatrin, and may have fewer side effects (e.g., visual field defects) than those associated with vigabatrin. We are hoping to develop CPP-115 for the treatment of refractory infantile spasms and possibly for the treatment of adult refractory patients with Tourette’s Disorder. CPP-115 has been granted Orphan Drug Designation by the FDA for the treatment of infantile spasms and Orphan Medicinal Product Designation in the European Union, or E.U., for West syndrome (a form of infantile spasms).
We are currently refining our development plan for this product. Once the refinement of our development plans are completed, and subject to the then availability of funding, we plan to take the steps to complete the work required to make our drug candidate Phase 2 ready. We are also working with one or more potential investigators who have expressed an interest in evaluating our product for particular indications (particularly infantile spasms). We are also continuing to seek a partner to work with us in furthering the development of CPP-115, although no agreements have been entered into to date.
There can be no assurance that we will ever successfully commercialize CPP-115.
16
Table of Contents
Product Overview
In August 2009, we licensed the exclusive worldwide rights to commercialize certain composition of matter patents relating to a new class of novel GABA aminotransferase inhibitors and derivatives of vigabatrin. We intend to develop these compounds for a broad range of neurological illnesses that could benefit from the inhibition of GABA aminotransferase. CPP-115 is our lead compound from this group of composition of matter patents.
The development efforts of CPP-115 were led by Dr. Richard B. Silverman, the Patrick G. Ryan/Aon Professor of Chemistry at Northwestern University (Northwestern). Dr. Silverman, who holds 75 patents, is the inventor of pregabalin, also known as Lyrica®, which is marketed by Pfizer. His goal in inventing the compound that became CPP-115 was to mimic the mechanism of action of vigabatrin, while making it both more potent and specific.
CPP-115 works by the same mechanism of action as vigabatrin; that is, the inhibition of GABA aminotransferase, which leads to increased brain GABA levels that reduce epileptogenesis. Due to these similarities, we believe that these two drugs will likely share certain biochemical features related to absorption, metabolism, and elimination, and our pre-clinical studies of CPP-115 to date support our expectations. However, based upon our pre-clinical studies of CPP-115 to date, we expect that there will be a significant reduction, and possibly elimination, of visual field defects (VFDs) from the use of CPP-115 compared to vigabatrin. However, there can be no assurance that this will ultimately prove to be the case.
Further, based on animal testing to date, CPP-115 has been shown to be at least 200 times more potent than vigabatrin in both in-vitro and animal model studies. The increased potency could enable the development of dosage forms potentially administrable by other routes of administration compared with the marketed oral, immediate release formulation of vigabatrin, Sabril®. Further, based on non-clinical testing completed to date, CPP-115 appears to have superior specificity to GABA aminotransferase and we believe, will have a better side effect profile (e.g. less VFDs) compared with Sabril®.
CPP-115 has been granted Orphan Drug Designation in the U.S. for the treatment of infantile spasms. CPP-115 has also been granted Orphan Medicinal Product Designation in the EU to treat West syndrome (a form of infantile spasms).
Mechanism of action for CPP-115
We believe that our drug candidate, CPP-115, will be an effective treatment for epilepsy because it increases endogenous GABA levels in the brain through the inhibition of GABA-aminotransferase (GABA-AT). GABA-AT is responsible for the eventual breakdown of GABA and helps to balance its inhibitory effects.
CPP-115 is a GABA analog that is readily absorbed and promptly available to the nervous system, producing effects that last for many hours after a single dose. Due to the fact that this drug is not “receptor active”, its administration does not appear to affect the baseline levels of dopamine, nor those variations in dopamine levels caused by normal stimuli.
Epilepsy and Infantile Spasms
Epilepsy is a brain disorder in which clusters of nerve cells, or neurons, in the brain sometimes signal abnormally. In epilepsy, the normal pattern of neuronal activity becomes disturbed, causing strange sensations, emotions, and behavior or sometimes convulsions, muscle spasms, and loss of consciousness.
17
Table of Contents
Epilepsy is a disorder with many possible causes. Anything that disturbs the normal pattern of neuron activity—from illness to brain damage to abnormal brain development—can lead to seizures. Epilepsy may develop because of an abnormality in brain wiring, an imbalance of nerve signaling chemicals called neurotransmitters, imbalance of sensitivity to neurotransmitters, or some combination of these factors. We intend to focus our development efforts for CPP-115 on its use as a treatment for refractory infantile spasms.
An infantile spasm is a specific type of seizure seen in an epilepsy syndrome of infancy and childhood. The onset of infantile spasms is usually in the first year of life, typically between 4-8 months. The seizures primarily consist of a sudden bending forward of the body with stiffening of the arms and legs; some children arch their backs as they extend their arms and legs. Spasms tend to occur upon awakening or after feeding, and often occur in clusters of up to 100 spasms at a time. Infants may have dozens of clusters and several hundred spasms per day. Infantile spasms usually stop by age five, but may be replaced by other seizure types.
In complex partial seizures, consciousness is altered. Patients may exhibit automatisms (automatic repetitive behavior) such as walking in a circle, sitting and standing, or smacking their lips together. Often accompanying these symptoms are the presence of unusual thoughts, such as the feeling of déjà vu, uncontrollable laughing, fear, visual hallucinations, and experiencing unusual unpleasant odors. These symptoms are thought to be caused by abnormal discharges in the temporal lobe.
According to the Epilepsy Foundation, there are about 3.0 million epilepsy patients in the United States, with approximately 150,000 new cases diagnosed in the U.S. each year. Worldwide, 65 million people are estimated to have epilepsy. The incidence of epilepsy appears to depend somewhat on the age of the individual. The risk of epilepsy from birth through age 20 is approximately 1%. Within this group, incidence is highest during the first year of life and increases somewhat at the onset of puberty. From age 20 to 55 it decreases again, but increases after age 55.
Anti-epileptic drugs work through a variety of mechanisms, including inhibition of sodium ion channels and the enhancement of GABA mechanisms. Although the different types of epilepsy vary greatly, in general, available medications can only control seizures in about two-thirds of patients. CPP-115, like vigabatrin, is a GABA-AT inhibitor, and we are developing it initially for refractory infantile spasms. Based on the historic use of vigabatrin in treating epilepsy, we believe that CPP-115 may ultimately work best as an adjunct therapy to existing drugs.
Vigabatrin has been marketed for decades in over 30 countries by Lundbeck and Sanofi-Aventis and their predecessors and licensees under the brand names Sabril®, Sabrilex® and Sabrilan® (hereinafter referred to as “Sabril®”) as an adjunct (add-on) treatment for adult epilepsy and as a primary treatment for the management of infantile spasms. The composition of matter patents for Sabril® in the U.S. expired many years ago. On August 21, 2009, the FDA approved two NDAs for Sabril® for the treatment of infantile spasms and as an adjunctive therapy for adult patients with refractory complex partial seizures who have failed treatments with several other anti-epileptic drugs. The NDAs are for different formulations of Sabril® and both NDAs are held by Lundbeck. Due to the risks of visual field damage associated with vigabatrin, Sabril® was approved under an FDA-mandated Risk Evaluation and Mitigation Strategy (REMS) program and is only available through a special restricted distribution program approved by the FDA. In 2016, the FDA authorized changes to the REMS program for Sabril® to make it less onerous and to make it easier for patients to obtain their medication.
In chronic use for the treatment of epilepsy, vigabatrin has been generally well tolerated with lower than average neurological side effects compared to other approved epilepsy therapies. The most common side effects reported have been drowsiness and fatigue. However, one clearly established adverse side effect is the development of peripheral visual field defects, or VFDs. These VFDs are manifest as a constriction of the peripheral field of vision (i.e., “tunnel vision”). VFDs occur in approximately 33% of users when cumulative dosage levels of vigabatrin approach approximately 1,500 grams.
18
Table of Contents
Our previous clinical and non-clinical studies of CPP-115
On November 1, 2010, we announced key results for our initial series of safety and efficacy evaluations in a number of animal and in-vitro laboratory studies. These results included superior visual safety of CPP-115, compared to vigabatrin, pharmacokinetic data supporting oral administration of CPP-115, pharmacologic target specificity, metabolic profile, and an absence of genotoxic, cardiovascular, respiratory, and liver enzyme side effects. It was also shown to be effective in multiple animal models for epilepsy and cocaine addiction.
On May 22, 2012, we reported positive results from a Phase 1a double-blind, placebo-controlled clinical trial evaluating the safety, tolerability and pharmacokinetic profile of CPP-115. The study evaluated single ascending doses ranging from 5 mg to 500 mg (a dose greater than ten times the predicted effective dose of 15-30 mg/day derived from animal data) of CPP-115 solution administered orally to 55 healthy volunteers. CPP-115 was found to be well tolerated with no side effects, rapidly absorbed and eliminated, and exhibited linear, dose dependent pharmacokinetics.
In December 2015 we announced top line results from a Phase 1b double-blind, placebo controlled safety and tolerance study of CPP-115 in six normal healthy adult male volunteers. The results showed significant increases in brain levels of the surrogate marker for potential efficacy, gamma-aminobutyric acid (GABA), a mechanism known to effectively treat epilepsy, infantile spams, and potentially Tourette’s Disorder. The main adverse effect of prolonged elevated brain GABA, somnolence, was also observed.
While the primary objective of this study was to obtain safety and tolerance data for CPP-115 administered over 14 days, brain GABA levels were measured as a surrogate marker of potential efficacy, since CPP-115 is a second-generation GABA aminotransferase inhibitor. Specifically, this study examined GABA levels in both the POC (Parietal-Occipital Cortex), a grey matter rich region thought to be associated with epilepsy, and which was previously studied for vigabatrin, and the SMA (Supplementary Motor Area), which is thought to be associated with Tourette’s Disorder. The maximum brain GABA increases, in both brain regions, ranged from about 150% to over 200% of baseline levels, as measured by magnetic resonance spectroscopy (MRS).
Previous clinical and pre-clinical studies of CPP-115 undertaken by others
An animal study reporting positive pre-clinical efficacy in a “rat multiple hit model” in which the use of CPP-115 was evaluated for the treatment of infantile spasms was published in the January 2014 issue of the journal, Epilepsia, The study was authored by Stephen W. Briggs, Tomonori Ono, MD, PhD, Solomon L. Moshe, MD and Aristea S. Galanopoulou, MD, PhD of the Saul R. Korey Department of Neurology, Dominick P. Purpura Department of Neuroscience, Laboratory of Developmental Epilepsy, The Comprehensive Epilepsy Center (CEC) at Montefiore Medical Center / Albert Einstein College of Medicine of Yeshiva University, Bronx, New York. The study concluded that (i) CPP-115 suppresses spasms in the multiple-hit model of infantile spasm, with onset of effect as early as the day after the first dose; (ii) the therapeutic doses of CPP-115 were well tolerated in developing rat pups; and (iii) CPP-115 showed efficacy for a longer duration at lower doses that were better tolerated than the previously tested therapeutic vigabatrin doses.
19
Table of Contents
In September 2016, the Journal of Epilepsy & Behavior Case Reports published a case report of a child treated with CPP-115 in an investigator-sponsored, investigational new drug protocol. Based on treatment with CPP-115, this particular child experienced a significant reduction of seizures, with no evidence of retinal dysfunction. According to the case report, prior to treatment with CPP-115, the patient had failed ten drugs and the ketogenic diet, and had approximately 100 seizures per day. One year after starting CPP-115 and coming off of clobazam and vigabatrin, the patient’s reported seizures have seen a marked reduction in frequency and his cognition and behavior have improved.
Northwestern University License Agreement
On August 27, 2009, we entered into a license agreement with Northwestern University (Northwestern), under which we acquired worldwide rights to commercialize new GABA aminotransferase inhibitors and derivatives of vigabatrin which had been discovered and patented by Northwestern. Under the terms of the license agreement, Northwestern granted us an exclusive worldwide license to United States composition of matter patents related to the new class of inhibitors and a patent application relating to derivatives of vigabatrin. This includes U.S. patent number 6,794,413 covering the composition of matter for CPP-115. We have designated the lead compound to be developed under this license as CPP-115.
Under our license agreement with Northwestern, we will be responsible for continued research and development of any resulting drug candidates. We have the right to terminate the agreement in whole or in part after August 27, 2012, upon written notice. As of December 31, 2016, we have paid Northwestern upfront payments, milestone fees and maintenance and patent fees aggregating $411,950 and we are obligated to pay certain additional fees and milestone payments in future years relating to our clinical development activities under this license or payable upon passage of time (the next milestone payment, in the amount of $300,000) is due on the earlier of completion of the first Phase 3 clinical trial of CPP-115 or August 27, 2018). We are also obligated to pay Northwestern royalties on any products resulting from the license agreement. We also have the right to enter into sub-license agreements, and if we do, a royalty on any sub-license fees will be payable to Northwestern.
Patent protection for CPP-115
In addition to the exclusively licensed U.S. Patent 6,794,413, in March 2015, the U.S. Patent & Trademark Office (US PTO) issued patent 8,969,413 for the method of use patent for CPP-115 for neurological and psychological uses. This patent will expire in 2032, subject to potential extensions allowed under the Patent Term Restoration Act. A continuation application was filed to capture additional methods of using CPP-115 for neurological and psychological conditions. This continuation application is undergoing substantive examination. Patents for the same coverage remain pending in the European Patent Office, Japan and Canada. There can be no assurance that the claims of this patent will be allowed, or if allowed, that such claims will provide adequate patent protection for CPP-115.
CPP-109
Generic Sabril®
In September 2015, we announced the launch of a program to develop CPP-109 as a generic version of Sabril®, which is marketed in the United States by Lundbeck. Lundbeck’s exclusivity for Sabril® expires on April 26, 2017. Because of our prior experience developing our version of vigabatrin (CPP-109), we believe we are well positioned to develop and seek approval for a generic version of Sabril®. We are also hopeful that if we are successful in obtaining approval to commercialize this product, that our product will be one of the first generic versions approved for this product.
20
Table of Contents
As part of our development of this product, we have obtained the reference listed drug and the active pharmaceutical ingredient, entered into an exclusive supply agreement for the vigabatrin active pharmaceutical ingredient with a manufacturer that has filed a DMF, validated the manufacturing process, and prepared a number of batches of vigabatrin for us on a commercial scale in the past, developed and validated quality control and stability test methods, and collected stability data showing that CPP-109 has an acceptable shelf life in two container closure systems. We are also taking the steps that will be required for us to submit one or more ANDAs for this generic product.
There can be no assurance that we will be successful in these efforts or that any ANDA that we may submit in the future for vigabatrin will be accepted for review or approved. There can also be no assurance that any bioequivalence studies that we design for this product will be acceptable to the FDA, and even if a study design is deemed acceptable, whether any such studies will be successful. Finally, any approved generic version of vigabatrin that we are approved to commercialize will, consistent with Sabril®, only be available subject to an FDA-mandated Risk Evaluation and Mitigation Strategy (REMS) program.
CPP-109 and CPP-115 for the treatment of Tourette’s Disorder and related license agreement
We, as a co-inventor with scientists at New York University (NYU) and the Feinstein Institute for Medical Research, have filed patent applications in the United States, European and Canadian Patent Offices for the use of GABA aminotransferase inhibitors, including CPP-109 and CPP-115, in the treatment of Tourette’s Disorder. We also have entered into a license agreement with NYU and the Feinstein Institute granting us worldwide rights with respect to such patent.
Tourette’s Disorder is a neurodevelopmental disorder characterized by multiple tics, which are repetitive, non-rhythmic involuntary movements and vocalizations that persist for more than one year. Onset occurs in most children by about age five to six years. Some of the more common simple motor tics include eye blinking, facial grimacing, shoulder shrugging, and head or neck jerking. Common simple vocal tics, include grunting, throat clearing, coughing and squeaking. Among the most dramatic and disabling complex tics are those that result in self-harm, such as punching or poking oneself, or complex vocal tics such as coprolalia (uttering swear words) or echolalia (repeating the words or phrases of others). Many patients with Tourette’s Disorder experience additional neurobehavioral problems including inattention, hyperactivity and impulsivity, and obsessive-compulsive symptoms such as intrusive thoughts/worries and repetitive rituals.
Tourette’s Disorder occurs in all ethnic groups; males are affected three to four times more often than females. The Centers for Disease Control and Prevention (CDC) reported in 2012 Tourette’s Disorder prevalence estimates from a national, telephone-based survey in which they asked parents if their children had received a diagnosis of the disorder. The trial found that approximately 1 out of every 360 children have Tourette’s Disorder, but it is thought that this underestimates the true occurrence of the disorder (the CDC notes that other studies using different methods have estimated the rate of Tourette’s Disorder at 1 out of every 162 children). The prevalence of Tourette’s Disorder in adults is expected to be significantly less than in children as tic symptoms generally abate in later adolescence or early adulthood.
Tourette’s Disorder is generally treated by a combination of therapy and psychiatric medication. Tics can be treated with medications such as clonidine (Catapres®), haloperidol (Haldol®), pimazide (Orap®), or fluphenazine (Prolixin®). Medications used to treat Obsessive Compulsive Disorder can also be used, such as clomipramine (Anafranil®), fluoxetine (Prozac®) and sertraline (Zoloft®), as well as stimulants used to treat ADHD, a disorder commonly comorbid with Tourette’s Disorder, such as methylphenidate (Ritalin®), pemoline (Cylert®) and dextroamphetamine (Dexadrine®).
21
Table of Contents
During June 2015, we announced the top-line results of an academic investigator proof-of-concept study evaluating the use of CPP-109 for the treatment of Tourette’s Disorder. The 8-week, four subject open label study was designed as an open label trial to evaluate the potential effect of GABA-AT inhibition as a mechanism for reducing tics in patients with treatment-refractory Tourette’s Disorder. The most common side-effects were daytime tiredness, and one subject experienced an increase in moodiness and obsessive-compulsive symptoms. Vigabatrin was used as a “research surrogate” in this study to demonstrate the utility of GABA-AT blockade, with the expectation that upon successfully demonstrating the utility of this mechanism, further development activities would focus on the potentially safer, more potent GABA-AT inhibitor, CPP-115.
We believe that the top-line results from this study suggest an encouraging signal of activity in adult treatment-refractory patients with Tourette’s Disorder. Further, we believe that CPP-109’s mechanism of action validates the potential for CPP-115 to be a candidate for the treatment of Tourette’s Disorder. However, there can be no assurance that CPP-115 will be effective for the treatment of adult refractory patients with Tourette’s Disorder or will ever be approved for this indication.
In February 2016, the US PTO issued U.S. Patent 9,254,274 for a method of treating Tourette’s Disorder using the entire class of GABA-aminotransferase inactivators, including CPP-115 and vigabatrin (marketed in the U.S. by Lundbeck as Sabril®). This patent is expected to expire no earlier than its twenty-year term in January 2033. The expiration of this patent could also be extended by up to 5 years under the patent term restoration act, depending on the review and approval of a new drug application for a new chemical entity claimed in this patent and upon the indication approved for that drug. A continuation in part application including new data was filed prior to the patent issuance. This application is undergoing substantive examination.
Intellectual Property Rights
Protection of our intellectual property and proprietary technology is a strategic priority for our business. We rely on a combination of patent, trademark, copyright and trade secret laws along with institutional know-how and continuing technological advancement, to develop and maintain our competitive position. Our ability to protect and use our intellectual property rights in the future development and commercialization of our products, operate without infringing the proprietary rights of others, and prevent others from infringing our proprietary rights, is crucial to our future success. See Item 1A. “Risk Factors — Risks Related to Our Intellectual Property.”
Manufacturing and Supply
We have no plans to build or acquire the manufacturing capability needed to manufacture any of our research materials or commercial products. We expect that our drug products and drug substances will be prepared by contractors with suitable capabilities for these tasks and that we will enter into appropriate supply agreements with these contractors at appropriate times in the development and commercialization of our products. Because we will use contractors to manufacture and supply our products, we will be reliant on such contractors. Further, the contractors selected would have to be inspected by the FDA and found to be in substantial compliance with federal regulations in order for a drug application for one of our drug candidates to be approved, and there can be no assurance that the contractors we select would pass such an inspection.
22
Table of Contents
Firdapse®
We have entered into agreements with a supplier of the active pharmaceutical ingredient (API) contained in Firdapse® for future requirements and we have contracted with third-party contract manufacturers who will manufacture Firdapse® tablets for us assuming Firdapse® is approved for commercialization.
Any NDA that we submit for Firdapse® must include a manufacturing plan. If the manufacturing plan and data are insufficient, any NDA we submit will not be approved. Before an NDA can be approved, our manufacturers must also demonstrate compliance with FDA’s current Good Manufacturing Practices (cGMPs) regulations and policies. Further, even if we receive approval of an NDA for Firdapse®, if our manufacturers do not follow cGMPs in the manufacture of our products, it may delay product launches or shipments and adversely affect our business.
Since we contract with third parties to manufacture our products, if the FDA approves an NDA for Firdapse®, our contract manufacturers will be required to comply with all applicable environmental laws and regulations that affect the manufacturing process. As a result, we do not believe that we will have any significant direct exposure to environmental issues.
CPP-115
We have entered into a contract to manufacture the API sufficient to meet the needs of our ongoing and planned pre-clinical and clinical studies of CPP-115. While we believe that we have ordered and obtained sufficient API for our planned upcoming studies, there can be no assurance of this.
CPP-109
In preparation for the potential future marketing of CPP-109 as a generic version of Sabril®, we have entered into supply agreements for the API of CPP-109. Additionally, our contract manufacturer of CPP-109 tablets previously developed a manufacturing process for vigabatrin tablets and prepared several commercial scale batches. Our current contract manufacturer also has, based on their experience with CPP-109 tablets, the necessary experience and capability to produce generic vigabatrin for oral solution product. Additionally, Catalyst has entered in to an agreement to package vigabatrin for oral solution. Finally, while we have not entered into a contract for commercial production of this product, we believe that our current contract manufacturer and packagers have the capability to produce the product for us for commercial distribution.
Sales and Marketing
We have not yet obtained regulatory approval for any of our drug candidates.
Until the receipt of an RTF letter regarding our NDA for Firdapse® for the treatment of LEMS, we had begun to hire a sales staff, including a Chief Commercial Officer. However, following the receipt of the RTF letter we put most of these activities on hold in order to conserve funds, and underwent a reduction-in-force in May 2016 in which we terminated most of our commercial staff.
As our second Phase 3 trial for Firdapse® continues and we get closer to acceptance of an NDA for this product, we intend to begin again to focus on our commercialization efforts. In that regard, we may restart our efforts to hire our own sales force or we may investigate alternate arrangements (such as retention of an outside sales force).
23
Table of Contents
In the future, we may also consider entering into arrangements with other pharmaceutical or biotechnology companies for the marketing and sale of Firdapse® in Canada or Mexico, where we have also licensed the product.
Competition
The pharmaceutical industry is intensely competitive, and any product candidate developed or licensed by us would likely compete with currently marketed and potentially new drugs and therapies even though they are not indicated for these conditions. There are many pharmaceutical companies, biotechnology companies, public and private universities, government agencies and research organizations that compete with us in developing various approaches to the treatment of orphan diseases. Many of these organizations have substantially greater financial, technical, marketing and manufacturing resources than we have.
Firdapse® for LEMS
LEMS is currently treated with unapproved drugs and therapies including steroids, azathioprine, other immunosuppressants and intravenous immunoglobulin, which work by suppressing the immune system, and pyridostigmine. Plasma exchange has also been used in an attempt to remove antibodies from the body. Further, one other product, guanidine HCl tablets, was approved many years ago (during a period when drugs were not required to be reviewed by the FDA for both safety and effectiveness) for use in the treatment of LEMS. However, this drug has significant side effects and is not currently viewed as an effective treatment for LEMS. Notwithstanding, drugs may be prescribed by physicians for the treatment of LEMS whether or not they are considered effective.
Another pharmaceutical company, Jacobus Pharmaceutical, has completed a clinical trial studying the safety and efficacy of its own formulation of amifampridine for the treatment of LEMS. Jacobus Pharmaceutical is a privately held company and there is little public information available about their development plans. While there can be no assurance, we believe that Firdapse® is further along in development than this other company’s version of amifampridine. Under the Orphan Drug Act of 1983, the first pharmaceutical product to get approval for an indication receives the orphan exclusivity under the statute. If this other pharmaceutical company is able to receive approval of an NDA for its formulation of amifampridine for the treatment of LEMS before we are able to receive approval of Firdapse® for the same indication, we would be barred from marketing Firdapse® in the United States during the seven-year orphan exclusivity period, which would have a severe adverse effect on our results of operations. In addition, if this other company were to receive five-year new chemical entity exclusivity for amifampridine for any indication prior to approval of Firdapse®, and FDA determined that our NDA was a 505(b)(2) NDA, we would be barred from marketing Firdapse® in the United States during this five-year exclusivity period for any indication. Further, we are aware that Jacobus Pharmaceutical has been making its 3,4-DAP product available to LEMS patients under compassionate use Investigational New Drug applications (INDs) for a number of years and, based on current information, we believe that approximately 200 LEMS patients may be receiving the drug under their program. If we are the first to obtain an approval for this product and its associated exclusivity and patent protection, we may not be able to stop Jacobus Pharmaceutical from continuing to supply its existing patients under compassionate use INDs.
Finally, we are aware that amifampridine has been available from compounding pharmacies for many years and may remain available, even if we are able to obtain FDA approval of Firdapse®. Compounded amifampridine, if it remains available, is likely to be substantially less expensive than Firdapse®. The Food and Drug Administration Modernization Act of 1997 included a new section, which clarified the status of pharmacy compounding under Federal law. Under section 503A, drug products that are compounded by a
24
Table of Contents
pharmacist or physician for an individual patient may be entitled to exemptions from three key provisions of the act: (1) the adulteration provision of section 501(a)(2)(B) (concerning the good manufacturing practice requirements); (2) the misbranding provision of section 502(f)(1) (concerning the labeling of drugs with adequate directions for use); and (3) the new drug provision of section 505 (concerning the approval of drugs under new drug or abbreviated new drug applications).
To qualify for these statutory exemptions, a compounded drug product must satisfy several requirements. One of these requirements restricted the universe of bulk drug substances that a compounder may use; i.e. that every bulk drug substance used in compounding: (1) must comply with an applicable and current USP or NF monograph, if one exists, as well as the current USP chapter on pharmacy compounding; (2) if such a monograph does not exist, the bulk drug substance must be a component of an FDA-approved drug; or (3) if a monograph does not exist and the bulk drug substance is not a component of an FDA-approved drug, it must appear on a list of bulk drug substances that may be used in compounding (i.e., the bulk drugs list). While Section 503 was ruled unconstitutional by the Supreme Court in 2002, the FDA has continued to aggressively oversee the practice of compounding under a compliance policy guide utilizing its discretion under the principles described above, and these principles were codified into a new section 503A passed by Congress as part of the Drug Quality and Security Act of 2013 (DQSA). While FDA has been aggressively enforcing section 503A since its enactment, compounders still may attempt to compound copies of approved drug products, under section 503A, so long as the prescriber makes a change to the compounded formulation that creates a difference between the commercially available drug and the compounded version for that particular patient.
The FDA’s Pharmacy Compounding Advisory Committee at its meeting on May 6-7, 1999 voted 7-4 against inclusion of 3,4-DAP on the bulk drugs list, largely based on the safety concerns and the commitment of Jacobus Pharmaceutical to make the drug available under compassionate use INDs, while pursuing FDA approval. Therefore, since 3,4-DAP does not meet the requirements codified in Section 503A described above, the individual or firm that compounds a drug product containing 3,4-DAP may be subject to a warning letter, seizure of product, injunction, and/or criminal prosecution for violations of the FD&C Act. After the re-enactment of section 503A, and the enactment of new section 503B of the DQSA (i.e., permitting sterile compounding for office use by outsourcing facilities), certain entities nominated 3,4 DAP as a bulk substance to be used in compounding under both reenacted section 503A and under the newly enacted section 503B. As of October 2015, FDA included 3,4-DAP in its Bulks Substance “List 3” under both section 503A and section 503B– which list includes bulk drug products that may not currently be used in compounding because there is insufficient clinical evidence to support their use.
We intend to take all available steps to try to enforce our marketing proprietary rights if we are the first company to obtain an approval for this product. We cannot determine with certainty what impact these factors will have on the market for our product. However, while there can be no assurance, we expect that despite these factors, we will be able to successfully market our product.
CPP-115 for Epilepsy
The market for epilepsy treatments is highly competitive. Large pharmaceutical companies, including Pfizer (Neurontin®, Lyrica®, Dilantin®, Zarontin®), J&J (Topamax®), UCB (Keppra®), Abbott (Depakote®), GSK (Lamictal®), Roche (Klonopin®), and Novartis (Trileptal®) sell, or are developing, epilepsy therapies. However, as stated earlier, approximately one-third of all epilepsy patients are refractory to treatment with any currently available epilepsy treatments. It is difficult to determine sales of products specifically for epilepsy as many of these products are used in other indications such as neuropathic pain, migraine, dementia, and bipolar disorders.
25
Table of Contents
Generic Sabril®
Sabril® is marketed by Lundbeck in the United States for infantile spasms and for refractory complex partial seizures. Lundbeck’s sales of Sabril® (tablets and sachets) were approximately $142 million in 2015 and $193 million in 2016. No generic version of Sabril® has been approved to date in the United States.
Factors affecting competition generally
In general, our ability to compete will depend in large part upon:
| • |
our ability to complete clinical development and obtain regulatory approvals for our drug candidates; |
| • |
the demonstrated efficacy, safety and reliability of our drug candidates; |
| • |
the timing and scope of regulatory approvals; |
| • |
product acceptance by physicians and other health care providers; |
| • |
protection of our proprietary rights and the level of generic competition; |
| • |
the speed at which we develop drug candidates; |
| • |
our ability to supply commercial quantities of a product to the market; |
| • |
our ability to obtain reimbursement from private and/or public insurance entities for product use in approved indications; |
| • |
our ability to recruit and retain skilled employees; and |
| • |
the availability of capital resources to fund development and commercialization activities, including the availability of funding from the federal government. |
Regulatory Matters
Government regulation and product approval
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, record-keeping, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our drugs must be approved by the FDA through the NDA process before they may be legally marketed in the United States.
In the United States, drugs are subject to rigorous regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations, as well as other federal and state statutes. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These
26
Table of Contents
sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • |
completion of pre-clinical laboratory tests, animal studies and formulation studies according to the FDA’s good laboratory practice, or GLP, regulations; |
| • |
submission of an investigational new drug application, or IND, which must become effective before human clinical trials may begin and which must include approval by an institutional review board, or IRB, at each clinical site before the trials are initiated; |
| • |
performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use conducted in compliance with federal regulations and good clinical practice, or GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; |
| • |
submission to, and acceptance by, the FDA of an NDA; |
| • |
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practice, or cGMP, regulations to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| • |
potential FDA audit of the non-clinical and clinical trial sites that generated the data in support of the NDA; and |
| • |
FDA review and approval of the NDA. |
United States drug development process
Once a pharmaceutical candidate is identified for development it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. Prior to beginning human clinical trials, an IND sponsor must submit an IND to the FDA. The IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some pre-clinical or non-clinical testing may continue even after the IND is submitted. In addition to including the results of the pre-clinical studies, the IND will also include a protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated, if the trial lends itself to an efficacy evaluation. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30–day time period, raises concerns or questions about the conduct of the trial. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may, at any time, impose a clinical hold on ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of one or more qualified investigators in accordance with federal regulations and GCP.
27
Table of Contents
Clinical trials must be conducted under protocols detailing the objectives of the trial and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND. Further, an Institutional Review Board (IRB) at each institution participating in the clinical trial must review and approve each protocol before any clinical trial commences at that institution. All research subjects must provide informed consent, and informed consent information must be submitted to the IRB for approval prior to initiation of the trial. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if adverse events or other certain types of other changes occur.
Human clinical trials are typically conducted in three phases. A fourth, or post-approval, phase may include additional clinical studies. These phases generally include the following, and may be sequential, or may overlap or be combined:
| • |
Phase 1 clinical trials involve the initial introduction of the drug into human subjects. These studies are designed to determine the safety of usually single doses of the compound and determine any dose limiting intolerance, as well as evidence of the metabolism and pharmacokinetics of the drug in humans. |
| • |
Phase 2 clinical trials usually involve studies in a limited patient population to evaluate the safety and efficacy of the drug for specific, targeted indications, to determine dosage tolerance and optimal dosage, and to identify possible adverse effects and safety risks. |
| • |
In Phase 3, if a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 (or occasionally Phase 1) studies, the Phase 3 studies will be conducted to further confirm clinical efficacy, optimal dosage and safety within an expanded population which may involve geographically diverse clinical trial sites. Generally, but not always, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA. |
| • |
Phase 4 clinical trials are studies required of or agreed to by a sponsor that are conducted after the FDA has approved a product for marketing. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of drugs approved under accelerated approval regulations. If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement. Failure to promptly conduct Phase 4 clinical trials where necessary could result in withdrawal of approval for products approved under accelerated approval regulations. |
While Phase 1, Phase 2, and Phase 3 tests are generally required for approval of an NDA, certain drugs may not require one or more steps in the process depending on other testing and the situation involved. Additionally, the FDA, an IRB, or the sponsor may stop testing at any time if results show patients being exposed to unnecessary health risks or overly dangerous side effects.
Pursuant to the 21st Century Cures Act, which was enacted on December 13, 2016, the manufacturer of an investigational drug for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access to such investigational drug. This requirement applies on the later of 60 calendar days after the date of enactment of the law or the initiation of a Phase 2 or Phase 3 trial of the investigational drug.
28
Table of Contents
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other requirements, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Special factors with respect to clinical trials and pre-clinical studies conducted by others
The primary focus of our product development efforts is on our own clinical trials and pre-clinical studies. However, we have in the past supported and will continue in the future to support pre-clinical studies and clinical trials and studies by academic investigators (including members of our scientific advisory committee and academic institutions with which they are affiliated) of the use of our drug candidates that we believe might further the understanding or increase the value of our drug candidates.
In some cases, in the past, we have provided unrestricted sponsorship funds for such studies and we may do so again in the future. In other cases, we have provided, and may in the future provide, alternative assistance to the investigator, most typically providing drug substance or dosage form as well as matching placebo. We expect to continue supporting investigator-sponsored studies in the future to the extent that they meet criteria acceptable to us. In all cases, we seek to assist investigators in designing their studies so that such studies are most appropriately conducted and, to the extent possible, to make sure that these investigator studies potentially complement, and do not adversely impact, our activities.
United States review and approval process
FDA approval of an NDA is required before marketing of the product may begin in the United States. The NDA must include the results of product development, pre-clinical studies and clinical studies, together with other detailed information, including information on the chemistry, manufacture and composition of the product. The FDA has 60 days from its receipt of the NDA to review the application to ensure that it is sufficiently complete for substantive review before accepting it for filing. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The submission of an NDA is also subject to the payment of a substantial application fee (for FDA fiscal year 2017 this fee was $2,038,100), although a waiver of such fee may be obtained under certain limited circumstances, including when the drug that is subject of the application has received Orphan Drug Designation for the indication sought. Further, the sponsor of an approved NDA is subject to annual product and establishment user fees, which for FDA fiscal year 2017 were $97,750 per product and $512,200 per establishment. Application, product, and establishment fees typically increase annually although they decreased slightly from fiscal year 2016 to fiscal year 2017. The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured to determine whether its manufacturing is cGMP–compliant to assure and preserve the product’s identity, strength, quality, purity and stability.
29
Table of Contents
If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA will issue a complete response letter. The complete response letter outlines the deficiencies in the submission and often requires additional testing or information in order for the FDA to reconsider the application. Even after submitting this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. With limited exceptions, the FDA may withhold approval of a NDA regardless of prior advice it may have provided or commitments it may have made to the sponsor.
Special Protocol Assessments
An SPA is a process in which sponsors may request to meet with the FDA to reach agreement on the design and size of certain clinical trials, clinical studies, or animal trials to determine if they adequately address scientific and regulatory requirements. As part of this process, sponsors submit specific questions about protocol design and scientific and regulatory requirements. After the FDA completes the review of an SPA request, the FDA may issue a SPA Letter, including an assessment of the protocol, agreement or non-agreement with the proposed protocol, and answers to the sponsor’s relevant questions.
An SPA agreement indicates concurrence by the FDA with the adequacy and acceptability of specific critical elements of overall protocol design (e.g., entry criteria, dose selection, endpoints, and planned analyses). These elements are critical to ensuring that the trial conducted under the protocol has the potential to support a future submitted application’s ability to meet regulatory requirements for approval. Feedback on these issues provides the greatest benefit to sponsors in planning late-phase development strategy. However, an SPA agreement does not indicate FDA concurrence on every protocol detail. Further, the FDA may rescind a SPA if the director of the FDA reviewing division determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the trial began. Thus, a SPA is not binding on the FDA if, for example, the Agency identifies a safety concern related to the product or its pharmacological class, if the FDA or the scientific community recognizes a paradigm shift in disease diagnosis or management, if the relevant data or assumptions provided by the sponsor in the SPA submission are found to be false or misstated, or if the sponsor fails to follow the protocol that was agreed upon with the FDA. The FDA retains significant latitude and discretion in interpreting the terms of a SPA agreement and the data and results from the applicable clinical trial.
Because an SPA provides for the evaluation of protocols for trials that have not been initiated, the conduct and results of the subsequent trial are not part of the evaluation. Therefore, the existence of an SPA agreement does not guarantee that the FDA will accept an NDA, or that the trial results will be adequate to support approval. Those issues are addressed during the review of a submitted application; however, it is hoped that trial quality will be improved by the SPA process.
Post-approval requirements and consideration
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. As a condition of NDA approval, the FDA may also require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for the healthcare professionals, and other Elements To Assure Safe Use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug.
30
Table of Contents
Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as drug manufacture, packaging, and labeling procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
The Hatch-Waxman Amendments
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims covering the applicant’s product or methods of using the product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown to be bioequivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a section viii statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
31
Table of Contents
A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Exclusivity
Upon NDA approval of a new chemical entity or NCE, which is a drug that contains no active moiety that has been approved by FDA in any other NDA, that drug receives five years of marketing exclusivity during which FDA cannot receive any ANDA seeking approval of a generic version of that drug. A drug may obtain a three-year period of exclusivity for a particular condition of approval, or change to a marketed product, such as a new formulation for the previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant. During this period of exclusivity, FDA cannot approve an ANDA for a generic drug that includes the change.
An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification, and, thus, no ANDA may be filed before the expiration of the exclusivity period.
Section 505(b)(2) New Drug Applications
Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2), or 505(b)(2), NDA, which enables the applicant to rely, in part, on FDA’s previous approval of a similar product, or published literature, in support of its application.
505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. If the Section 505(b)(2) applicant can establish that reliance on FDA’s prior findings of safety and effectiveness or published literature is scientifically appropriate, it may eliminate the need to conduct certain pre-clinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all, or some, of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. A Section 505(b)(2) NDA may be eligible for three years of marketing exclusivity to the same extent that a Section 505(b)(1) NDA is.
32
Table of Contents
Abbreviated new drug applications
Generic drugs may enter the market after the approval of an ANDA. The ANDA development process typically does not require new pre-clinical or clinical studies, but it does typically require one or more bioequivalence studies to show that the ANDA drug is bioequivalent to the previously approved brand name reference listed drug. Bioequivalence studies compare the bioavailability of the proposed drug product with that of the approved listed product containing the same active ingredient. Bioavailability is a measure of the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. A demonstration of bioequivalence means that the rate and extent of absorption of the ANDA drug is not significantly different from the rate and extent of absorption of the brand name reference listed drug when administered at the same molar dose under similar experimental conditions.
As noted above, generic drug products are generally introduced to the marketplace at the expiration of patent protection and non-patent market exclusivity for the reference listed drug. However, if an ANDA applicant is the first ANDA applicant to submit an ANDA containing a Paragraph IV certification, that ANDA may be eligible for a period of generic marketing exclusivity on approval. This exclusivity, which under certain circumstances must be shared with other ANDA applicants with Paragraph IV certifications, lasts for 180 days, during which the FDA cannot grant final approval to other ANDA sponsors of an application for a generic equivalent to the same reference drug. Under certain circumstances, eligibility for 180-day exclusivity may be forfeited.
Various types of changes to an approved ANDA must be requested in a prior approval supplement. In addition, some changes may only be approved only after new bioequivalence studies are conducted or other requirements are satisfied. In addition, the ANDA applicant must demonstrate that manufacturing procedures and operations conform to FDA cGMP requirements. Facilities, procedures, operations and/or testing of products are subject to periodic inspection by the FDA and other authorities. In addition, the FDA conducts pre-approval and post-approval reviews and inspections to determine whether the systems and processes are in compliance with cGMP and other FDA regulations.
There are also user fees for ANDA applicants, sponsors, and manufacturers. For fiscal year 2017, the fees are $70,480 per ANDA application, $35,240 per ANDA prior approval supplement, and facility fees of $258,646 per domestic final dosage form facility, $273,646 per foreign final dosage form facility, $44,234 per domestic active pharmaceutical ingredient facility, and $59,234 per foreign active pharmaceutical ingredient facility. These user fees typically increase each fiscal year.
Other regulatory requirements
In addition to regulation by the FDA and certain state regulatory agencies, we are also subject to a variety of foreign regulations governing clinical trials and the marketing of other products. Outside of the United States, our ability to market a product depends upon receiving a marketing authorization from the appropriate regulatory agencies. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. In any country, however, we will only be permitted to commercialize our products if the appropriate regulatory agency is satisfied that we have presented adequate evidence of safety, quality and efficacy. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing of the product in those countries. The regulatory approval and oversight process in other countries includes all of the risks associated with regulation by the FDA and certain state regulatory agencies as described above.
33
Table of Contents
Under the European Union regulatory system, applications for drug approval may be submitted either in a centralized or decentralized manner. Under the centralized procedure, a single application to the European Medicines Agency leads to an approval granted by the European Commission which permits marketing of the product throughout the European Union. The decentralized procedure provides for mutual recognition of nationally approved decisions and is used for products that do not comply with requirements for the centralized procedure. Under the decentralized procedure, the holders of national marketing authorization in one of the countries within the European Union may submit further applications to other countries within the European Union, who will be requested to recognize the original authorization based on an assessment report provided by the country in which marketing authorization is held.
Pharmaceutical pricing and reimbursement
In both US and foreign markets, our ability to commercialize our products successfully, and to attract commercialization partners for our products, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payors, including, in the United States, governmental payors such as Medicare and Medicaid, managed care organizations, private commercial health insurers and PBMs. Third party payors are increasingly challenging the prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic or other studies in order to further demonstrate the value of our products. Even with the availability of such studies, our products may be considered less safe, less effective or less cost-effective than alternative products, and third party payors may not provide coverage and reimbursement for our drug candidates, in whole or in part.
Political, economic and regulatory influences are subjecting the health care industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business, including the Patient Protection and Affordable Care Act of 2010 (the “Affordable Care Act”). In fact, there are currently efforts in Congress to repeal the Affordable Care Act and replace it with another law and President Trump has stated that he supports repeal of all or portions of the Affordable Care Act. As a result, there is great uncertainty as to what changes will be made to U.S. healthcare laws and there can be no assurance how changes to those laws may affect our business.
We anticipate that in the US, Congress, state legislatures, and private sector entities will continue to consider and may adopt healthcare policies intended to curb rising healthcare costs. These cost containment measures could include:
| • |
controls on government-funded reimbursement for drugs; |
| • |
controls on healthcare providers; |
| • |
controls on pricing of pharmaceutical products; |
| • |
challenges to the pricing of drugs or limits or prohibitions on reimbursement for specific products through other means; |
| • |
reform of drug importation laws; |
| • |
entering into contractual agreements with payors; and |
| • |
expansion of use of managed-care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person. |
34
Table of Contents
We are unable to predict what additional legislation, regulations or policies, if any, relating to the healthcare industry or third-party coverage and reimbursement may be enacted in the future or what effect such legislation, regulations or policies would have on our business. Any cost containment measures, including those listed above, or other healthcare system reforms that are adopted may have a material adverse effect on our business prospects.
Further, the pricing of pharmaceutical products generally, and particularly the pricing of orphan drugs, has recently received scrutiny from the press, from members of Congress in both parties, and from President Trump. Some members of the medical community have also weighed in the press on the potential pricing of orphan drugs generally and our product specifically. The impact of this scrutiny on us and on the pricing of orphan drugs and other pharmaceutical products generally cannot be determined with any certainty at this time.
Orphan Drug Exclusivity and Pediatric Exclusivity Designation
Some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act of 1983 (ODA), the FDA may grant Orphan Drug Designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. In the United States, Orphan Drug Designation must be requested before submitting an application for marketing approval. An Orphan Drug Designation does not shorten the duration of the regulatory review and approval process. The grant of an Orphan Drug Designation request does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and efficacy of a compound must be established through adequate and well-controlled studies. If a product which has been granted Orphan Drug Designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to an orphan drug exclusivity period, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as where an alternative product demonstrates clinical superiority to the product with orphan exclusivity. In addition, holders of exclusivity for orphan drugs are expected to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for the drug.
The orphan drug exclusivity contained in the ODA has been the subject of recent scrutiny from the press, from some members of Congress and from some in the medical community. There can be no assurance that the exclusivity granted in ODA to orphan drugs approved by the FDA will not be modified in the future, and as to how any such change might affect our products, if approved.
Pediatric exclusivity is another type of non-patent exclusivity in the U.S. and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the five-year and three-year non-patent and seven-year orphan exclusivities. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly responds to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied. If the FDA determines that information relating to the use of the new drug in the pediatric population may produce health benefits in the population, the clinical study is deemed to fairly respond to the FDA’s request and the reports of FDA-requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection covering the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve another application relying on the NDA sponsor’s data.
35
Table of Contents
The European Orphan Drug Regulation is considered for drugs intended to diagnose, prevent or treat a life-threatening or very serious condition afflicting five or fewer per 10,000 people in the EU, including compounds that for serious and chronic conditions would likely not be marketed without incentives due to low market return on the sponsor’s development investment. The medicinal product considered should be of significant benefit to those affected by the condition. Benefits of being granted Orphan Medicinal Product Designation are significant, including eight years of data exclusivity, two years of marketing exclusivity and a potential one-year extension of both. The EU Community and Member States may not accept or grant for ten years a new marketing authorization or application for another drug for the same therapeutic indication as the orphan drug, although the ten year period can be reduced to six years if, after the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of the marketing exclusivity. A supplementary protection certificate may extend the protection six months beyond patent expiration if that is later than the orphan drug exclusivity period. To apply for the supplementary protection, a pediatric investigation plan, or PIP, must be included in the market application. In Europe all drugs now seeking marketing authorization need to have a PIP agreed with the European Medicines Agency (EMA) before it can be approved, even if it is a drug being developed specifically for a pediatric indication. If a product is developed solely for use in the pediatric population, then a Pediatric Use Marketing Authorization, or PUMA, may provide eight years of data exclusivity and ten years of marketing exclusivity.
Breakthrough Therapy Designation
Breakthrough therapy designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. The criteria for breakthrough therapy designation require preliminary clinical evidence that demonstrates the drug may have substantial improvement on at least one clinically significant endpoint over available therapy. A breakthrough therapy designation conveys all of the fast track program features (see below for more details on fast track designation), as well as more intensive FDA guidance on an efficient drug development program. The FDA also has an organizational commitment to involve senior management in such guidance. Actions taken to expedite development may include the following actions, as appropriate:
| • |
holding meetings with the sponsor and review team throughout the development of the drug; |
| • |
providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the non-clinical and clinical data necessary for approval is as efficient as possible; |
| • |
taking steps to ensure that the design of the clinical trials is as efficient as practicable, when scientifically appropriate, such as by minimizing the number of patients exposed to a potentially less efficacious treatment; |
| • |
assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the cross-discipline members of the review team (i.e., clinical, pharmacology-toxicology, chemistry, manufacturing and control (CMC), compliance) for coordinated internal interactions and communications with the sponsor through the review division’s Regulatory Health Project Manager; and |
| • |
involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review. |
36
Table of Contents
Fast Track Designation and Accelerated Approval
FDA is required to facilitate the development, and expedite the review, of drugs that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug candidate may request that FDA designate the drug candidate for a specific indication as a fast track drug concurrent with, or after, the filing of the IND for the drug candidate. FDA must determine if the drug candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
Under the fast track program and FDA’s accelerated approval regulations, FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by FDA.
In addition to other benefits such as the ability to use surrogate endpoints and engage in more frequent interactions with FDA, FDA may initiate review of sections of a fast track drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Priority Review
Under FDA policies, a drug candidate is eligible for priority review, or review within a six to eight-month time frame from the time a complete NDA is submitted, if the drug candidate is intended for the treatment, diagnosis or prevention of a serious or life-threatening condition, demonstrates the potential to address an unmet medical need, or provides a significant improvement compared to marketed drugs.
Disclosure of clinical trial information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of results of these trials can be delayed in certain circumstances for up to two years after the date of completion of the clinical trial. Competitors may use this publicly-available information to gain knowledge regarding the progress of development programs.
37
Table of Contents
Anti-Kickback, False Claims Laws & the Prescription Drug Marketing Act
In addition to FDA restrictions on marketing of pharmaceutical products, other state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and patients, prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
The Centers for Medicare & Medicaid Services (CMS) has issued a final rule that requires manufacturers of approved prescription drugs to begin collecting and reporting information on payments or transfers of value to physicians and teaching hospitals, as well as investment interests held by physicians and their immediate family members. Manufacturers were required to begin collecting information in 2013, with the first reports filed in 2014. The information reported each year is made publicly available on a searchable website. Failure to submit required information may result in civil monetary penalties.
In addition, several states now require prescription drug companies to report expenses relating to the marketing and promotion of drug products and to report gifts and payments to individual physicians in these states. Other states prohibit various other marketing-related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, California, Connecticut, Nevada, and Massachusetts require pharmaceutical companies to implement compliance programs and/or marketing codes. Several additional states are considering similar proposals. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws face civil penalties.
As part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. The Prescription Drug Marketing Act, or the PDMA, imposes requirements and limitations upon the provision of drug samples to physicians, as well as prohibits states from licensing distributors of prescription drugs unless the state licensing program meets certain federal guidelines that include minimum standards for storage, handling, and record keeping. In addition, the PDMA sets forth civil and criminal penalties for violations.
38
Table of Contents
Our Employees
As of March 15, 2017 we had 18 employees. We also utilize the services of consultants, including several members of our Scientific Advisory Board. None of our employees are covered by a collective bargaining agreement. We believe our relationship with our employees and consultants is good.
Our Scientific Advisory Board
We rely on prominent scientists and physicians to advise us on the development of our drug candidates. All of our advisors are employed by organizations other than ours and may have commitments to or consulting or advisory agreements with other entities that may limit their availability to us. Our Scientific Advisory Board currently consists of the following members:
| • |
Jonathan Brodie, PhD, MD, is the chairman of our Scientific Advisory Board and Professor Emeritus of Psychiatry at New York University School of Medicine. Dr. Brodie completed his Bachelor of Science degree in chemistry as a Ford Foundation Scholar and his PhD in Physiological Chemistry (Organic Chemistry minor) at the University of Wisconsin-Madison. He was an NIH postdoctoral Fellow in Biochemistry at Scripps Clinic and Research Foundation and a tenured associate professor of Biochemistry at the School of Medicine at SUNY at Buffalo. He then received his MD degree at New York University School of Medicine and joined the faculty after completing his residency in psychiatry at NYU/Bellevue Medical Center. He has been a member of the Promotions and Tenure Committee of the School of Medicine and co-chairman of the Executive Advisory Committee of the General Clinical Research Center and the Protocol Review Committee of the Center for Advanced Brain Imaging (CABI) of Nathan Kline Institute. He also served as Interim Chairman of the Department of Psychiatry of the NYU School of Psychiatry at the NYU School of Medicine. For 15 years, he was the NYU Director of the Brookhaven National Laboratory/NYUSoM collaboration investigating the use of positron emitters and PET in neuroscience and psychiatry. In addition, Dr. Brodie serves as a psychopharmacology preceptor to psychiatry residents. As a clinician, he treats patients in general issues of adult psychiatry including anxiety and depression. |
| • |
Robert D. Fechtner, MD, is Professor and Chair of Ophthalmology at SUNY Upstate Medical University, Syracuse, New York. Dr. Fechtner received his Bachelor of Science degree in biomedical science and his medical degree from the University of Michigan. He completed his residency at Albert Einstein College of Medicine in New York. A fellowship in glaucoma followed at the University of California, San Diego, under a National Research Service Award from the National Institutes of Health. Dr. Fechtner is the Executive Vice President of the World Glaucoma Association and has published more than 100 scientific articles and book chapters. |
| • |
Eugene Laska, PhD, is a professor in the Department of Psychiatry at New York University and the former Director of the Statistical Sciences unit at the Nathan S. Kline Institute for Psychiatric Research. Dr. Laska was for 20 years the Director of the WHO Collaborating Center for Research and Training in Mental Health Program Management and has served as a statistical consultant to many pharmaceutical companies (including us) both large and small with regard to biostatistics and clinical trial design. He is a fellow of the American Statistical Association and the American Association for the Advancement of Science. |
| • |
Richard B. Silverman, Ph.D. is the Patrick G. Ryan/Aon Professor in the Department of Chemistry at Northwestern University. He is the inventor of Pfizer’s $4.5 billion/year Lyrica® (pregabalin), marketed worldwide for the treatment of epilepsy, neuropathic pain, fibromyalgia, pain from spinal cord injury, and (in Europe) for generalized anxiety disorder. He has received numerous awards, most recently American Chemical Society Creative Invention Award (2017), Fellow of the National Academy |
39
Table of Contents
| of Inventors (2014), Fellow of the American Academy of Arts & Sciences (2014), iCON Innovator Award of the iBIO Institute (2014), Northwestern University Trustee Medal for Faculty Innovation and Entrepreneurship (2014), Medicinal Chemistry Prize of the Israel Chemistry Society (2014), Fellow of the Royal Society of Chemistry (UK, 2013), Centenary Prize of the Royal Society of Chemistry (2013), Bristol-Myers Squibb-Edward E. Smissman Award of the American Chemical Society (2013), Sato Memorial International Award of the Pharmaceutical Society of Japan (2012), Fellow of the American Chemical Society (2011), E.B. Hershberg Award for Important Discoveries in Medicinally Active Substances from the American Chemical Society (2011), Perkin Medal from the Society of Chemical Industry (2009), Medicinal Chemistry Hall of Fame of the American Chemical Society (2009). Dr. Silverman holds 75 patents, has published over 350 peer-reviewed articles and has written five books over his almost 41-year career in academia. |
We may add additional members to or revise the makeup of our Scientific Advisory Board in the future to add personnel who will assist us in the future development of Firdapse®.
Available Information
We make available free of charge on or through our Internet website our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission (SEC). Our Internet address is www.catalystpharma.com. The content on our website is not, nor should it be deemed to be, incorporated by reference into this Form 10-K.
| Risk Factors |
Our business involves a high degree of risk. You should carefully consider the risks and uncertainties described below, and all of the other information contained in this Form 10-K in assessing the risks relating to ownership of our common stock. The risks described below could cause our business, results of operations, financial condition and prospects to materially suffer and the market price of our stock to decline.
Risks Related to our Business
We are a development stage company. Our limited operating history makes it difficult to evaluate our future performance.
We are a development stage company and, as such, we have a limited operating history upon which you can evaluate our current business and our prospects. The likelihood of our future success must be viewed in light of the problems, expenses, difficulties, delays and complications often encountered in the operation of a business without revenues, especially in the pharmaceutical industry, where failures of companies are common. We are subject to the risks inherent in the ownership and operation of a development stage company, including availability of capital, regulatory setbacks and delays, fluctuations in expenses, competition and government regulation. If we fail to address these risks and uncertainties our business, results of operations, financial condition and prospects would be adversely affected.
We have no products currently available and we have never had any products available for commercial sale.
We have had no revenues from product sales to date, currently have no products available for commercial sale, and have never had any products available for commercial sale. We expect to incur losses at least until we are in a position to commercialize Firdapse®, which may never occur. Our net loss was $18.1 million and $20.2 million for the years ended December 31, 2016 and December 31, 2015, respectively. We may never obtain approval of an NDA for any of our drug candidates and we may never achieve profitability.
40
Table of Contents
Our business will require additional capital.
Our business will require additional capital to meet our product development objectives. Based on currently available information, we estimate that we have sufficient working capital to support our operations through at least the next 12 months. The expectations described above are based on current information available to us. If the cost of our ongoing activities are greater than we expect, our assumptions may not prove to be accurate. There can be no assurance as to the exact amount of the funding we will require or as to whether any such required funding will be available to us when it is required.
We plan to raise additional funds in the future through public or private equity offerings, debt financings, corporate collaborations, or other means. We may also seek governmental grants to support our clinical and pre-clinical trials. However, there is no assurance that any such grants will be made available, and, if available, that we will qualify to receive any such grants. We may also seek to raise additional capital to fund additional product development efforts, even if we have sufficient funds for our planned operations.
Any sale by us of additional equity or convertible debt securities could result in dilution to our stockholders. There can be no assurance that any required additional funding will be available to us at all or available on terms acceptable to us. Further, to the extent that we raise funds through collaborative arrangements, it may be necessary to relinquish some rights to our technologies or grant sublicenses on terms that are not favorable to us. If we are not able to secure funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs, which could have an adverse effect on our business.
If we are not the first to obtain approval for Firdapse® for the treatment of LEMS, we may not be able to bring it to market in the United States.
Another pharmaceutical company, Jacobus Pharmaceutical, has completed its own clinical trial studying their own formulation of amifampridine (3,4-DAP) for the treatment of LEMS. Jacobus Pharmaceutical is a privately held company and there is little public information available about their development plans. While there can be no assurance, we believe that Firdapse® is further along in development and as a result we expect that we will be in a position to obtain the first approval of an NDA for 3,4-DAP. Under the Orphan Drug Act of 1983, the first pharmaceutical product to obtain approval for an indication receives the orphan exclusivity under the statute. If Jacobus Pharmaceutical receives approval of an NDA for its formulation of amifampridine for the treatment of LEMS before we are able to receive approval of Firdapse® for the same indication, we would be barred from marketing Firdapse® in the United States during the seven-year orphan exclusivity period, which would have a severe adverse effect on our results of operations. In addition, if Jacobus Pharmaceutical were to receive five-year new chemical entity exclusivity for amifampridine for any indication prior to approval of Firdapse®, we would be barred from marketing Firdapse® in the United States during this five-year exclusivity period.
The development of CPP-115 is at an early stage.
Our development of CPP-115 is at an early stage, and it is going to be several years before we are in a position to submit an NDA for CPP-115, assuming our future clinical trials of this product are successful. At the present time, there can be no assurance that we will ever submit an NDA for CPP-115 or successfully commercialize CPP-115.
41
Table of Contents
Our business is subject to substantial competition.
The biotechnology and pharmaceutical industries are highly competitive. Many of our competitors have substantially greater financial and other resources, larger research and development staffs and more experience developing products, obtaining FDA and other regulatory approvals of products and manufacturing and marketing products than we have. We compete against pharmaceutical companies that are developing or currently marketing therapies that will compete with our drug candidates. In addition, we compete against biotechnology companies, universities, government agencies, and other research institutions in the development of pharmaceutical products. While we believe that our drug candidates will offer advantages over many of the currently available competing therapies, our business could be negatively impacted if our competitors’ present or future offerings are more effective, safer or less expensive than ours, or more readily accepted by regulators, healthcare providers or third-party payors. Further, if we are permitted to commence commercial sales of our drug candidates, we may also compete with respect to manufacturing efficiency and marketing capabilities.
For example, amifampridine, the active ingredient in Firdapse®, despite not being FDA approved, has been available from compounding pharmacies and from Jacobus Pharmaceutical under compassionate use INDs for many years. Amifampridine from these sources can be expected to be substantially less expensive than Firdapse®. The FDA Pharmacy Compounding Advisory Committee, however, has previously issued a list of drugs which cannot be compounded, and amifampridine was included on that list. In addition, drugs that are not approved by FDA for the treatment of LEMS, such as a related aminopyridine drug, dalfampridine (Ampyra®), may nonetheless be prescribed by physicians for the treatment of LEMS.
For all of these reasons, we may not be able to compete successfully.
We face a risk of product liability claims and may not be able to obtain adequate insurance.
Our business exposes us to potential liability risks that may arise from the clinical testing, manufacture, and/or sale of our pharmaceutical products. Patients have received substantial damage awards in some jurisdictions against pharmaceutical companies based on claims for injuries allegedly caused by the use of pharmaceutical products used in clinical trials or after FDA approval. Liability claims may be expensive to defend and may result in large judgments against us. We currently carry liability insurance with an aggregate annual coverage limit of $15,000,000 per claim and $15,000,000 in the aggregate, with a deductible of $10,000 per occurrence. Our insurance may not reimburse us for certain claims or the coverage may not be sufficient to cover claims made against us. We cannot predict all of the possible harms or side effects that may result from the use of our current drug candidates, or any potential future products we may acquire and use in clinical trials or after FDA approval and, therefore, the amount of insurance coverage we currently hold may not be adequate to cover all liabilities we might incur. If we are sued for any injury allegedly caused by our products, our liability could exceed our ability to pay the liability. Whether or not we are ultimately successful in any adverse litigation, such litigation could consume substantial amounts of our financial and managerial resources, all of which could have a material adverse effect on our business, financial condition, results of operations, prospects and stock price.
42
Table of Contents
The obligations incident to being a public company place significant demands on our management.
As a public reporting company, we are required to comply with the Sarbanes-Oxley Act of 2002 and the related rules and regulations of the SEC, including periodic reports, disclosures and more complex accounting rules. As directed by Section 404 of Sarbanes-Oxley, the SEC adopted rules requiring public companies to include a report of management on a company’s internal control over financial reporting in their Annual Report on Form 10-K. Based on current rules, we are required to annually report under Section 404(a) of Sarbanes-Oxley regarding our management’s assessment as to the effectiveness of our internal control over financial reporting. Further, under Section 404(b) of Sarbanes-Oxley, our auditors are required to report on their assessment as to the effectiveness of our internal control over financial reporting. If we or our auditors are unable to conclude that we have effective internal control over our financial reporting, investors could lose confidence in the reliability of our financial statements, which could result in a decrease in the value of our common stock.
We are highly dependent on our small number of key personnel and advisors.
We are highly dependent on our officers and employees, on our Board of Directors and on our scientific advisors. The loss of the services of any of these individuals could significantly impede the achievement of our scientific and business objectives. Other than an employment agreement with Patrick J. McEnany, our Chairman, President and Chief Executive Officer with respect to his services, and the consulting agreements we have with several of our scientific advisors, we have no employment or retention agreements with our officers, directors or scientific advisors. If we lose the services of any of our existing officers, directors or scientific advisors, or if we were unable to recruit qualified replacements on a timely basis for persons who leave our employ, our efforts to develop our drug candidates might be significantly delayed. We do not carry key-man insurance on any of our personnel.
We have relationships with our scientific advisors and collaborators at academic and other institutions. Such individuals are employed by entities other than us and may have commitments to, or consulting advisory contracts with, such entities that may limit their availability to us. Although each scientific advisor and collaborator has agreed not to perform services for another person or entity that would create an appearance of a conflict of interest, conflicts may arise from the work in which other scientific advisors and/or collaborators are involved.
Risks Related to the Development of Our Drug Candidates
Our drug development efforts may fail.
Development of our pharmaceutical drug candidates is subject to risks of failure. For example:
| • |
our drug candidates may be found to be ineffective or unsafe, or fail to receive necessary regulatory approvals; |
| • |
our drug candidates may not be economical to market or take substantially longer to obtain necessary regulatory approvals than anticipated; or |
| • |
competitors may develop and market equivalent or superior products, including next generation products that act with the same mechanism of action as our drug candidates. |
As a result, our drug development activities may not result in any safe, effective and commercially viable products, and we may not be able to commercialize our products successfully. For example, for several years, we evaluated CPP-109 (our formulation of vigabatrin) for the treatment of cocaine addiction.
43
Table of Contents
However, CPP-109 failed to meet the primary and two key secondary endpoints in a Phase 2b trial for cocaine addiction, and we are no longer pursuing the evaluation of CPP-109 for addiction. Further, our lead compound, Firdapse®, is for very rare conditions for which there is no FDA-approved treatment. As such, the clinical development plan we pursued after consulting with FDA including the clinical endpoints, protocol design, and statistical analysis plan, may not allow the FDA to ultimately conclude that our Phase 3 trial of Firdapse® is adequate to establish the clinical benefit of the drug.
Our failure to develop safe, effective, and/or commercially viable products would have a material adverse effect on our business, prospects, results of operations and financial condition.
Failure can occur at any stage of our drug development efforts.
We will only obtain regulatory approval to commercialize our drug candidates if we can demonstrate to the satisfaction of the FDA (or the equivalent foreign regulatory authorities) in adequate and well-controlled clinical studies and trials that the drug is safe and effective for its intended use, that the clinical and other benefits outweigh the safety risks and that it otherwise meets approval requirements. As we have experienced in the past, a failure of one or more pre-clinical or clinical trials or studies can occur at any stage of drug development. We may experience numerous unforeseen events during, or as a result of, testing that could delay or prevent us from obtaining regulatory approval for, or commercializing our drug candidates, including but not limited to:
| • |
regulators or Institutional Review Boards (IRBs) may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| • |
conditions may be imposed upon us by the FDA regarding the scope or design of our clinical trials, or we may be required to resubmit our clinical trial protocols to IRBs for review due to changes in the regulatory environment; |
| • |
the number of subjects required for our clinical trials may be larger, patient enrollment may take longer, or patients may drop out of our clinical trials at a higher rate than we anticipate; |
| • |
we may have to suspend or terminate one or more of our clinical trials if we, regulators, or IRBs determine that the participants are being subjected to unreasonable health risks; |
| • |
our third-party contractors, clinical investigators or contractual collaborators may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner; |
| • |
the FDA may not accept clinical data from trials that are conducted at clinical sites in countries where the standard of care is potentially different from the United States; |
| • |
our tests may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional testing; and |
| • |
the costs of our pre-clinical and/or clinical trials may be greater than we anticipate. |
We rely on third parties to conduct our pre-clinical studies and clinical studies and trials, and if they do not perform their obligations to us we may not be able to obtain approval for our drug candidates.
We do not currently have the ability to independently conduct pre-clinical studies or clinical studies and trials for our drug candidates, and we typically rely on third parties such as third-party contract research and governmental organizations, medical institutions and clinical investigators (including academic clinical investigators), to conduct studies and trials of our drug candidates.
44
Table of Contents
Our reliance on third parties for development activities reduces our control over these activities. These third parties may not complete activities on schedule, or may not conduct our pre-clinical studies and our clinical studies and trials in accordance with regulatory requirements or our study design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be adversely affected, and our efforts to obtain regulatory approvals for and commercialize our drug candidates may be delayed.
If we conduct studies with other parties, we may not have control over all decisions associated with that trial. To the extent that we disagree with the other party on such issues as study design, study timing and the like, it could adversely affect our drug development plans.
Although we also rely on third parties to manage the data from our studies and trials, we are responsible for confirming that each of our studies and trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies will require us to comply with applicable regulations and standards, including Good Laboratory Practice (GLP) and Good Clinical Practice (GCP), for conducting, recording and reporting the results of such studies and trials to assure that the data and the results are credible and accurate and that the human study and trial participants are adequately protected. Our reliance on third-parties does not relieve us of these obligations and requirements, and we may fail to obtain regulatory approval for our drug candidates if these requirements are not met.
We will need to develop marketing, distribution and production capabilities or relationships to be successful.
In order to generate sales of any products we may develop, we must either acquire or develop an internal marketing force with technical expertise and with supporting documentation capabilities, or make arrangements with third parties to perform these services for us. The acquisition and development of a marketing and distribution infrastructure requires substantial resources and compete for available resources with our drug development efforts. To the extent that we enter into marketing and distribution arrangements with third parties, our revenues will depend on the efforts of others. If we fail to enter into such agreements, or if we fail to develop our own marketing and distribution channels, we would experience delays in product sales and incur increased costs.
We have no in-house manufacturing capacity and, to the extent we are successful in completing the development of our drug candidates, we will be obligated to rely on contract manufacturers. We cannot be sure that we will successfully manufacture any product we may develop, either independently or under manufacturing arrangements, if any, with third party manufacturers. Moreover, if any manufacturer should cease doing business with us or experience delays, shortages of supply or excessive demands on their capacity, we may not be able to obtain adequate quantities of product in a timely manner, or at all. Manufacturers, and in certain situations their suppliers, are required to comply with current NDA commitments and current good manufacturing practices (cGMP) requirements enforced by the FDA, and similar requirements of other countries. The failure by a manufacturer to comply with these requirements could affect its ability to provide us with product. Although we intend to rely on third-party contract manufacturers, we are ultimately responsible for ensuring that our products are manufactured in accordance with cGMP. In addition, if, during a preapproval inspection or other inspection of our third-party manufacturers’ facility or facilities, FDA determines that the facility is not in compliance with cGMP, any of our marketing applications that lists such facility as a manufacturer may not be approved or approval may be delayed until the facility comes into compliance with cGMP and completes a successful re-inspection by FDA.
45
Table of Contents
Any manufacturing problem, natural disaster affecting manufacturing facilities, or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Additionally, we will be reliant on third parties to supply the raw materials needed to manufacture our potential products. Any reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to future contract manufacture caused by problems at suppliers could delay shipment of products, increase our cost of goods sold and result in lost sales. If our suppliers were to be unable to supply us with adequate supply of our drug candidates, it could have a material adverse effect on our ability to commercialize our drug candidates.
We may not be able to sufficiently scale-up manufacturing of our drug candidates.
If our NDA for Firdapse® is approved, we will need to manufacture our product in larger quantities than we have in the past to launch the product and meet customer requirements. With respect to our other products, to date they have only been manufactured in small quantities for pre-clinical studies and clinical trials, and, in order to conduct large trials and commercialize these products, we will need to manufacture our products in larger quantities than we have in the past.
We may not be able to successfully increase in a sufficient manner the manufacturing capacity for our drug candidates, whether in collaboration with third-party manufacturers or on our own, in a timely or cost-effective manner or at all. If a contract manufacturer makes improvements in the manufacturing process for our drug candidates, we may not own, or may have to share, the intellectual property rights to those improvements. Significant scale-up of manufacturing may require additional validation studies, which are costly and which the FDA must review and approve. In addition, quality issues may arise during those scale-up activities because of the inherent properties of a drug candidate itself or of a drug candidate in combination with other components added during the manufacturing and packaging process, or during shipping and storage of the finished product or active pharmaceutical ingredients. If we are unable to successfully scale-up manufacture of any of our drug candidates in sufficient quality and quantity, the development of that drug candidate and regulatory approval or commercial launch for any resulting drug products may be delayed or there may be a shortage in supply, which could significantly harm our business.
We may encounter difficulties in managing our growth, which would adversely affect our results of operations.
If we are successful in obtaining approval to commercialize Firdapse® or any of our other drug candidates, we will need to significantly expand our operations, which could put significant strain on our management and our operational and financial resources. We currently have 18 employees and conduct many of our activities through outsourcing arrangements. To manage future growth, we will need to hire, train, and manage additional employees. Concurrent with expanding our operational and marketing capabilities, we will also need to increase our product development activities. We may not be able to support, financially or otherwise, future growth, or hire, train, motivate, and manage the required personnel. Our failure to manage growth effectively could limit our ability to achieve our goals.
Our success in managing our growth will depend in part on the ability of our executive officers to continue to implement and improve our operational, management, information and financial control systems and to expand, train and manage our employee base, and particularly to expand, train and manage a specially-trained sales force to market our products. We may not be able to attract and retain personnel on acceptable terms given the intense competition for such personnel among biotechnology, pharmaceutical and healthcare companies, universities and non-profit research institutions. Our inability to manage growth effectively could cause our operating costs to grow at a faster pace than we currently
46
Table of Contents
anticipate, and could have a material adverse effect on our business, financial condition, results of operations and prospects.
Pressure on drug product third-party payor coverage, reimbursement and pricing may impair our ability to be reimbursed for any of our drug candidates which we commercialize in the future at prices or on terms sufficient to provide a viable financial outcome.
The commercial success of Firdapse® will depend substantially on the extent to which the cost of Firdapse® will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities (such as Medicare and Medicaid), private health coverage insurers and other third party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize Firdapse®. Even if coverage is provided, the approved reimbursement amount may not be high enough to establish and maintain pricing sufficient to realize a meaningful return on our investment.
Our ability to commercialize Firdapse® or any other product candidate will depend in large part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third party payors, such as private health insurers and health maintenance organizations, decide which medications they will cover and establish reimbursement levels. The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications, which could affect our ability to sell our product candidate profitably. These payors may not view our products, if any, as cost-effective, and coverage and reimbursement may not be available to our customers, or may not be sufficient to allow our products, if any, to be marketed on a competitive basis. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated product revenues. If the prices for our products, if any, decrease or if governmental and other third party payors do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
There may also be delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Reimbursement rates may vary, by way of example, according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based on reimbursement levels already set for lower cost drugs or may be incorporated into existing payments for other services.
In addition, increasingly, third party payors are requiring higher levels of evidence of the benefits and clinical outcomes of new technologies and are challenging the prices charged. We cannot be sure that coverage will be available for any product candidate that we commercialize and, if available, that the reimbursement rates will be adequate. Further, the net reimbursement for drug products may be subject to additional reductions if there are changes to laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for any of our product candidates for which we obtain marketing approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
The pricing of pharmaceutical products, in general, and specialty drugs, in particular, has been a topic of concern in the U.S. Congress, where hearings on the topic have been held. It has also been a topic raised by President Trump, most recently in a meeting with pharmaceutical industry participants. There can be no assurance as to how this scrutiny on pricing of pharmaceutical products will impact future pricing of orphan drugs or pharmaceutical products generally or our products in particular.
47
Table of Contents
We cannot assess the impact on our business of the recent activities of a vocal group of neuromuscular physicians and some patients with LEMS.
There is a vocal group of neuromuscular physicians who have raised public concerns in a letter to the editor of a medical journal and some LEMS patients and neuromuscular physicians who have raised public concerns in interviews quoted in articles published in the press. Their overarching concern appears to be that LEMS patients may not be able to get amifampridine treatment because of the concern that it would be priced too high as an orphan drug if we are the first pharmaceutical company to receive an FDA approval for an amifampridine product, thereby giving us the seven-year orphan drug exclusivity and the five year new chemical entity exclusivity for our product. Articles about their concerns have been published in several national publications and some in the press have sought to tie their expectations about the anticipated pricing of Firdapse® to stories about perceived abusive price increases of drug products by other pharmaceutical companies. This vocal group has also questioned the appropriateness of the provisions of the Orphan Drug Act that would grant us exclusivity if our product were to be the first amifampridine product approved by the FDA, and whether this exclusivity should be eliminated from the law. We have responded to their concerns in a letter to the editor to the same medical journal. However, there can be no assurance as to the ultimate impact of these activities on us or our products.
Because the target patient populations for Firdapse® and our other drug candidates are small, we must achieve significant market share and obtain relatively high per-patient prices for our products to achieve meaningful gross margins.
Firdapse® and our other orphan drug candidates target diseases with small patient populations. A key component of the successful commercialization of a drug product for these indications includes identification of patients and a targeted prescriber base for the drug product. Due to small patient populations, we believe that we would need to have significant market penetration to achieve meaningful revenues and identifying patients and targeting the prescriber base are key to achieving significant market penetration. In addition, the per-patient prices at which we anticipate we may sell Firdapse® will need to be relatively high in order for us to generate an appropriate return for the investment in these product development programs and achieve meaningful gross margins. There can be no assurance that we will be successful in achieving a sufficient degree of market penetration and/or obtaining or maintaining high per-patient prices for Firdapse® for diseases with small patient populations. Further, even if we obtain significant market share for Firdapse®, if approved, because the potential target populations are very small, we may never achieve profitability despite obtaining such significant market share. Furthermore, because the potential target populations are very small, even if we do obtain significant market share for Firdapse®, we may never achieve profitability. Additionally, patients who discontinue therapy or do not fill prescriptions are not easily replaced by new patients, given the limited patient population.
Our internal computer systems, or those of our contract research organizations and other key vendors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Our internal computer systems and those of our contract research organizations and other key vendors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data from completed or ongoing clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.
48
Table of Contents
To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our drug candidates could be delayed.
Our employees and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee or consultant fraud or other misconduct. Misconduct by our employees or consultants could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee and consultant misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Risks Related to Government Regulation
We have not received regulatory approval in the United States or any foreign jurisdiction for the commercial sale of any of our drug candidates. The regulatory approval process is lengthy, and we may not be able to obtain all of the regulatory approvals required to manufacture and commercialize our drug candidates.
We do not currently have any products that have been approved for commercialization. We will not be able to commercialize our products until we have obtained the requisite regulatory approvals from applicable governmental authorities. To obtain regulatory approval of a drug candidate, we must demonstrate to the satisfaction of the applicable regulatory agency that such drug candidate is safe and effective for its intended uses. The type and magnitude of the testing required for regulatory approval varies depending on the drug candidate and the disease or condition for which it is being developed. In addition, in the U.S. we must show that the facilities used to manufacture our drug candidate are in compliance with cGMP requirements. We will also have to meet similar regulations in any foreign country where we may seek to commercialize our drug candidates. In general, these requirements mandate that manufacturers follow elaborate design, testing, control, documentation and other quality assurance procedures throughout the entire manufacturing process. The process of obtaining regulatory approvals typically takes several years and requires the expenditure of substantial capital and other resources. Despite the time, expense and resources invested by us in the approval process, we may not be able to demonstrate that our drug candidates are safe and effective, in which event we would not receive the regulatory approvals required to market them.
49
Table of Contents
The FDA and other regulatory authorities generally approve products for particular indications. Our drug candidates may not be approved for any or all of the indications that we request, which would limit the indications for which we can promote it and adversely impact our ability to generate revenues. We may also be required to conduct costly, post-marketing follow-up studies if FDA requests additional information.
The FDA and other regulatory bodies must approve trade names for products. The FDA typically conducts a thorough review of a proposed trade name, including an evaluation of potential confusion with other trade names. We have previously submitted a request for FDA approval of the trade name Firdapse®, which request has been conditionally approved.
If our pre-clinical studies or our clinical studies and trials are unsuccessful or significantly delayed, our ability to commercialize our products will be impaired.
Before we can obtain regulatory approval for the sale of our drug candidates, we may have to conduct, at our own expense, pre-clinical tests in animals in order to support the safety of our drug candidates. Pre-clinical testing is expensive, difficult to design and implement, can take several years to complete and is uncertain as to outcome. Our pre-clinical tests may produce negative or inconclusive results, and on the basis of such results, we may decide, or regulators may require us, to halt ongoing clinical trials or conduct additional pre-clinical testing.
In September 2014, we announced positive results from our Phase 3 clinical trial for Firdapse® . In October 2016, we announced that we had reached an agreement with the FDA under a SPA for the protocol design, clinical endpoints, and statistical analysis approach to be taken in our ongoing second Phase 3 study evaluating Firdapse® for the symptomatic treatment of LEMS. Even if our second Phase 3 trial of Firdapse® is successful, we may nevertheless fail to meet the safety and efficacy standards required by the FDA to obtain regulatory approval.
Additionally, future clinical trials for our drug candidates may not be successfully completed or may take longer than anticipated because of any number of factors, including potential delays in the start of the trial, an inability to recruit clinical trial participants at the expected rate, failure to demonstrate safety and efficacy, unforeseen safety issues, or unforeseen governmental or regulatory delays. Further, our drug candidates may not be found to be safe and effective, and may not be approved by regulatory authorities for the proposed indication. Further, regulatory authorities and IRBs that must approve and monitor the safety of each clinical study may suspend a clinical study at any time if the patients participating in such study are deemed to be exposed to any unacceptable health risk. We may also choose to suspend human clinical studies and trials if we become aware of any such risks. We might encounter problems in our clinical trials, such as problems associated with Visual Field Defects (VFDs) or other side effects that will cause us, regulatory authorities, or IRBs to delay or suspend such trial or study.
In other countries where Firdapse®, CPP-115 or any other product we develop or license may be marketed, we will also be subject to regulatory requirements governing human clinical studies, trials and marketing approval for drugs. The requirements governing the conduct of clinical studies, trials, product licensing, pricing and reimbursement varies widely from country to country.
We may face significant delays in our clinical studies and trials due to an inability to recruit patients for our clinical studies and trials or to retain patients in the clinical studies and trials we may perform.
We may encounter difficulties in our current and future clinical studies and trials recruiting patients, particularly since the conditions we are studying are rare, orphan conditions. We compete for study and trial subjects with others conducting clinical trials testing other treatments for the indications we are studying for our drug candidates. Further, unrelated third parties and investigators in the academic community have in the past and we expect will continue in the future to test our drug candidates. If these third-party tests are unsuccessful, or if they show significant health risk to the test subjects, our development efforts may also be adversely affected.
50
Table of Contents
Clinical trials in orphan diseases are often difficult to enroll given the small number of patients with these diseases. Completion of orphan clinical trials may take considerable more time than other trials, sometimes years, depending on factors such as type, complexity, novelty and intended use of a product candidate. As a result of the uncertainties described above, there can be no assurance that we will meet timelines that we establish for any of our clinical trials.
If our third-party suppliers or contract manufacturers do not maintain appropriate standards of manufacturing in accordance with cGMP and other manufacturing regulations, our development and commercialization activities could suffer significant interruptions or delays.
We rely, and intend to continue to rely, on third-party suppliers and contract manufacturers to provide us with materials for our clinical trials and commercial-scale production of our products. These suppliers and manufacturers must continuously adhere to cGMP as well as any applicable corresponding manufacturing regulations outside of the U.S. In complying with these regulations, we and our third-party suppliers and contract manufacturers must expend significant time, money and effort in the areas of design and development, testing, production, record-keeping and quality control to assure that our products meet applicable specifications and other regulatory requirements. Failure to comply with these requirements could result in an enforcement action against us, including warning letters, the seizure of products, suspension or withdrawal of approvals, shutting down of production and criminal prosecution. Any of these third-party suppliers or contract manufacturers will also be subject to inspections by the FDA and other regulatory agencies. If any of our third-party suppliers or contract manufacturers fail to comply with cGMP or other applicable manufacturing regulations, our ability to develop and commercialize our products could suffer significant interruptions and delays.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the product ourselves, including:
| • |
reliance on the third party for regulatory compliance and quality assurance; |
| • |
reliance on the continued financial viability of the third parties; |
| • |
limitations on supply availability resulting from capacity and scheduling constraints of the third parties; |
| • |
impact on our reputation in the marketplace if manufacturers of our products, once commercialized, fail to meet the demands of our customers; |
| • |
the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and |
| • |
the possible termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us. |
If any of our contract manufacturers fail to achieve and maintain appropriate manufacturing standards, patients using our drug candidates could be injured or die, resulting in product liability claims. Even absent patient injury, we may be subject to product recalls, product seizures or withdrawals, delays or failures in testing or delivery, cost overruns or other problems that could seriously harm our business or profitability.
51
Table of Contents
If we rely on a sole source of supply to manufacture our products we could be impacted by the viability of our supplier.
We intend to attempt to source our products from more than one supplier. We also intend to enter into contracts with any supplier of our products to contractually obligate them to meet our requirements. However, if we are reliant on a single supplier and that supplier cannot or will not meet our requirements (for whatever reason), our business could be adversely impacted.
Even if we obtain regulatory approvals, our drug candidates will be subject to ongoing regulatory review. If we fail to comply with continuing U.S. and applicable foreign regulations, we could lose those approvals, and our business would be severely harmed.
Even if we receive regulatory approval of any drugs we are developing or may develop, we will be subject to continuing regulatory review, including the review of clinical results which are reported after our drug candidates become commercially available approved drugs. As greater numbers of patients use a drug following its approval, side effects and other problems may be observed after approval that were not seen or anticipated during preapproval clinical studies and trials. In addition, the manufacturer, and the manufacturing facilities we use to make any approved drugs, will also be subject to periodic review and inspection by the FDA. The subsequent discovery of previously unknown problems with the drug, manufacturer or facility may result in restrictions on the drug, manufacturer or facility, including withdrawal of the drug from the market. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and criminal prosecutions.
As a condition of approval for some of our products, the FDA might require a Risk Evaluation and Mitigation Strategy (REMS) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and other Elements To Assure Safe Use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. For example, approved versions of vigabatrin, the active moiety in our CPP-109 product (which operates by the same mechanism of action as our CPP-115 product) were approved with an FDA-mandated REMS program due to the risks of visual field damage and are only available through a special restricted distribution program approved by the FDA. Accordingly, our ANDA for vigabatrin, if approved, will be subject to either the same REMS, or a comparable REMS that will need to be reviewed and approved by the FDA. If any of our products were to be approved with a REMS, the potential market and profitability of the drug could be materially affected.
Our product promotion and advertising is also subject to regulatory requirements and continuing regulatory review. In particular, the marketing claims we will be permitted to make in labeling or advertising regarding our marketed products will be limited by the terms and conditions of the FDA-approved labeling and available scientific data. We must submit copies of our advertisements and promotional labeling to the FDA at the time of initial publication or dissemination. If the FDA believes these materials or statements promote our products for unapproved indications, or with unsubstantiated claims, or if we fail to provide appropriate safety related information, the FDA could allege that our promotional activities misbrand our products. Specifically, the FDA could issue an untitled letter or warning letter, which may demand, among other things, that we cease such promotional activities and issue corrective advertisements and labeling to all recipients of the misbranded materials. The FDA also could take enforcement action including seizure of allegedly misbranded product, injunction or criminal prosecution against us and our officers or employees. If we repeatedly or deliberately fail to submit such advertisements and labeling to the agency, the FDA could withdraw our approvals. Moreover, the Department of Justice can bring civil or criminal actions against companies and executives that promote drugs or biologics for unapproved uses, based on the Federal Food, Drug, and Cosmetics Act, the False Claims Act and other federal laws governing the marketing and reimbursement for such products under federally supported healthcare programs such as Medicare and Medicaid. Monetary penalties in such cases have often been substantial, and civil penalties can include costly mandatory compliance programs and potential exclusion of a company’s products from federal healthcare programs.
52
Table of Contents
Enacted and future legislation or judicial action may increase the difficulty and cost for us to commercialize Firdapse® or any other drug candidate we develop and affect the prices we may obtain.
In the U.S., there have been a number of court cases, legislative and regulatory changes and other potential changes relating to the healthcare system that restrict or regulate post-approval activities, which may affect our ability to profitably sell Firdapse® or any other drug candidate for which we obtain marketing approval.
The Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for outpatient drug purchases by those covered by Medicare under a new Part D and introduced a reimbursement methodology based on average sales prices for Medicare Part B physician-administered drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies whereby they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, there is additional pressure to contain and reduce costs. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors. These cost reduction initiatives and other provisions of the MMA could decrease the coverage and reimbursement that we receive for any approved products, and could seriously harm our business. Manufacturers’ contributions to this area, including donut hole coverage (as described below) or potential excise taxes, are increasing and are subject to additional changes in the future.
In 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (together, the “Health Care Reform Law”), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Health Care Reform Law, among other things, revised the definition of Average Manufacturer Price used by the Medicaid Drug Rebate Program for reporting purposes, which could increase the amount of Medicaid drug rebates to states and extended the rebate program to beneficiaries enrolled in Medicaid managed care organizations. The Health Care Reform Law also imposed a significant annual fee on companies that manufacture or import branded prescription drug products and established an annual non-deductible fee on entities that sell branded prescription drugs or biologics to specified government programs in the U.S. The Health Care Reform Law also expanded the 340B drug discount program (excluding orphan drugs), including the creation of new penalties for non-compliance and included a 50% discount on brand name drugs for Medicare Part D participants in the coverage gap, or “donut hole.” The Health Care Reform Law increased the Medicaid rebates for line extensions or reformulated drugs, which could substantially increase our Medicaid rebate rate (in effect limiting reimbursement for these patients).
53
Table of Contents
Both President Trump and the Republican leadership in Congress have expressed their intention to eliminate the Health Care Reform Law and replace it with a still unknown new law. It is unknown what form any such modifications or any law proposed to replace the Health Care Reform Law would take, and how or any such new law may affect our business in the future.
Additionally, in response to controversies regarding pricing of pharmaceutical products, there has been a recent push to propose legislation, both on state and federal levels, that would require greater disclosure as to the reasoning behind drug prices and, in some cases, could give state or federal-level commissions the right to impose cost controls on certain drugs. These and other new provisions are likely to continue the pressure on pharmaceutical pricing, may require us to modify our business practices with healthcare practitioners, and may also increase our regulatory burdens and operating costs. In that regard, President Trump and members of Congress in both parties have recently expressed concerns about high drug prices. However, whether and to what extent any such positions will result in changes of the law, and how any such changes could impact of business, cannot be determined at this time.
Legislative and regulatory proposals also have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may subject us to more stringent product labeling and post-marketing testing and other requirements. Delays in feedback from the FDA may affect our ability to quickly update or adjust our label in the interest of patient adherence and tolerability. We cannot predict whether other legislative changes will be adopted or how such changes would affect the pharmaceutical industry generally and specifically the commercialization of Firdapse®.
If we fail to obtain or subsequently maintain orphan drug exclusivity or regulatory exclusivity for Firdapse® and our other orphan drug candidates, our competitors may sell products to treat the same conditions at greatly reduced prices, and our revenues would be significantly adversely affected.
In the U.S., orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. The company that first obtains FDA approval for a designated orphan drug for a given rare disease receives marketing exclusivity for use of that drug for the stated condition for a period of seven years, with an additional six months of exclusivity if the product also qualifies for pediatric exclusivity. Orphan drug exclusive marketing rights may be lost if the FDA later determines that the request for designation was materially defective, a subsequent product is deemed clinically superior, or if the manufacturer is unable to deliver sufficient quantity of the drug.
In the EU, the EMA’s Committee for Orphan Medicinal Products, or COMP, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the EU would be sufficient to justify the necessary investment in developing the medicinal product. An EU orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
54
Table of Contents
Because the extent and scope of patent protection for some of our drug products may be particularly limited, orphan drug designation is especially important for our products that are eligible for orphan drug designation. For eligible drugs, we plan to rely on the orphan exclusivity period to maintain a competitive position. However, if we do not obtain orphan drug exclusivity for our drug candidates or we cannot maintain orphan exclusivity for our drug candidates, our competitors may then sell the same drug to treat the same condition and our revenues will be reduced. Also, without strong patent protection, competitors may sell a generic version upon the expiration of orphan exclusivity, if our patent position is not upheld.
Even if we obtain orphan drug designation for our future drug candidates, we may not fulfill the criteria for exclusivity or we may not be the first to obtain marketing approval for any orphan indication. Further, even if we obtain orphan drug exclusivity for a particular product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve a drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. The FDA can discontinue Orphan Drug exclusivity after it has been granted if the orphan drug cannot be manufactured in sufficient quantities to meet demand.
Finally, there can be no assurance that the exclusivity provisions currently in the law may not be changed in the future and the impact of any such changes (if made) on us. The orphan drug exclusivity contained in the ODA has been the subject of recent scrutiny from the press, from some members of Congress and from some in the medical community. There can be no assurance that the exclusivity granted in ODA to orphan drugs approved by the FDA will not be modified in the future, and as to how any such change might affect our products, if approved.
Breakthrough Therapy Designation may not actually lead to a faster review process.
Under the Prescription Drug User Fee Act, the FDA has a goal of responding to NDAs for new molecular entities within 10 months of the date that the FDA files the NDA for standard review, but this timeframe is also often extended. We have in the past and we may in the future, seek approval of our drug candidates under programs designed to accelerate the FDA’s review and approval of NDAs. For example, there is a new category of drugs referred to as “breakthrough therapies,” which are defined as drugs intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. In our case, Firdapse® has been granted “breakthrough therapy designation” for the treatment of LEMS. In the future, we may request breakthrough designation or fast track designation from the FDA for our other drug candidates or for treatment of other diseases, but we cannot assure that we will obtain such designations. Moreover, even if we obtain breakthrough designation or fast track designation from the FDA, the designations do not guarantee FDA approval of our NDA, that the development program or review timeline will ultimately be shorter than if we had not obtained the designations, or that the FDA will not request additional information, including requesting additional clinical studies (although potentially a post-marketing requirement), during its review. Any request for additional information or clinical data could delay the FDA’s timely review of our NDA.
55
Table of Contents
Even though our second Phase 3 study is being conducted under a Special Protocol Assessment, or SPA, agreed to with the FDA, we cannot guarantee that the design of, or data collected from, that trial or any of our clinical trials will be sufficient to support filing or approval of an NDA.
In the context of a Phase 3 clinical trial, the purpose of a SPA is to reach agreement with the FDA on the protocol design and analysis that will form the primary basis of an efficacy claim: in other words, if the agreed-upon clinical trial protocol is followed, the clinical trial endpoints are achieved, and there is a favorable risk-benefit profile, the data may serve as the primary basis for an efficacy claim in support of an NDA. However, FDA may rescind a SPA if the director of the FDA reviewing division determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the trial began. Thus, a SPA is not binding on the FDA if, for example, the Agency identifies a safety concern related to the product or its pharmacological class, if FDA or the scientific community recognizes a paradigm shift in disease diagnosis or management, if the relevant data or assumptions provided by the sponsor in the SPA submission are found to be false or misstated, or if the sponsor fails to follow the protocol that was agreed upon with FDA. In addition, a SPA may be modified with the written agreement of the FDA and the trial sponsor. The FDA retains significant latitude and discretion in interpreting the terms of a SPA agreement and the data and results from the applicable clinical trial.
Risks Related to Our Intellectual Property
We are dependent on our relationships and license agreements, and we rely upon the patent rights granted to us pursuant to the license agreements.
All of our patent rights for Firdapse® are derived from our license agreement with BioMarin. Pursuant to this license agreement, we have licensed rights under BioMarin’s Firdapse® patent applications in the United States, which expire in 2022 and 2034. We may lose our rights to these patents and patent applications if we breach our obligations under the license agreement, including, without limitation, our financial obligations to BioMarin. If we violate or fail to perform any term or covenant of the license agreement, BioMarin may terminate the license agreement upon satisfaction of any applicable notice requirements and expiration of any applicable cure periods. Additionally, any termination of the license agreement, whether by us or by BioMarin, will not relieve us of our obligation to pay any license fees owing at the time of such termination. If we fail to retain our rights under the license agreement, we would not be able to commercialize Firdapse®, and our business, results of operations, financial condition and prospects would be materially adversely affected.
Most of our patent rights for CPP-115 are derived from our license agreement with Northwestern University. Pursuant to this license agreement, we have exclusive worldwide rights to two patents in the United States. These were filed and obtained by Northwestern relating to compositions of matter for a class of molecules, including CPP-115. Both patents expire in 2023. Additionally, we have licensed rights from Northwestern to a pending patent for derivatives of vigabatrin that are unrelated to CPP-115. These rights are subject to the right of Northwestern, under limited circumstances, to practice the covered inventions for or on its own behalf for research. We may lose our rights to these patents and patent applications if we breach our obligations under the license agreement, including, without limitation, our financial obligations, including milestone payments, to Northwestern. If we violate or fail to perform any term or covenant of the license agreement, Northwestern may terminate the license agreement upon satisfaction of any applicable notice requirements and expiration of any applicable cure periods. Additionally, any termination of the license agreement, whether by us or by Northwestern, will not relieve us of our obligation to pay any license fees owing at the time of such termination. If we fail to retain our rights under the license agreement, we would not be able to commercialize CPP-115, and our business, results of operations, financial condition and prospects would be materially adversely affected.
56
Table of Contents
If we obtain approval to market Firdapse® or CPP-115, our commercial success will depend in large part on our ability to use patents, especially those licensed to us by BioMarin and Northwestern, respectively, to exclude others from competing with our products. The patent position of emerging pharmaceutical companies like us can be highly uncertain and involve complex legal and technical issues. Until our licensed patents are interpreted by a court, either because we have sought to enforce them against a competitor or because a competitor has preemptively challenged them, we will not know the breadth of protection that they will afford us. Our patents may not contain claims sufficiently broad to prevent others from practicing our technologies or marketing competing products. Third parties may intentionally attempt to design around our patents or design around our patents so as to compete with us without infringing our patents. Moreover, the issuance of a patent is not conclusive as to its validity or enforceability, and so our patents may be invalidated or rendered unenforceable if challenged by others.
As a result of the foregoing factors, we cannot be certain how much protection from competition patent rights will provide us.
Our success will depend significantly on our ability to operate without infringing the patents and other proprietary rights of third parties.
While we are not currently aware of any third-party patents which we may infringe, there can be no assurance that we do not or will not infringe on patents held by third parties or that third parties will not claim that we have infringed on their patents. In the event that our technologies infringe or violate the patent or other proprietary rights of third parties, we may be prevented from pursuing product development, manufacturing or commercialization of our products that utilize such technologies. There may be patents held by others of which we are unaware that contain claims that our products or operations infringe. In addition, given the complexities and uncertainties of patent laws, there may be patents of which we are aware that we may ultimately be held to infringe, particularly if the claims of the patent are determined to be broader than we believe them to be. Adding to this uncertainty, in the U.S., patent applications filed in recent years are confidential for 18 months, while older applications are not publicly available until the patent issues. As a result, avoiding patent infringement may be difficult.
If a third-party claims that we infringe its patents, any of the following may occur:
| • |
we may be required to pay substantial financial damages if a court decides that our technologies infringe a competitor’s patent, which can be tripled if the infringement is deemed willful, or be required to discontinue or significantly delay development, marketing, selling and licensing of the affected products and intellectual property rights; |
| • |
a court may prohibit us from selling or licensing our product without a license from the patent holder, which may not be available on commercially acceptable terms or at all, or which may require us to pay substantial royalties or grant cross-licenses to our patents; and |
| • |
we may have to redesign our product so that it does not infringe others’ patent rights, which may not be possible or could require substantial funds or time and require additional studies. |
In addition, employees, consultants, contractors and others may use the proprietary information of others in their work for us or disclose our proprietary information to others. As an example, we do not currently have written agreements regarding confidentiality with several principal members of our Scientific Advisory Board. If our employees, consultants, contractors or others disclose our data to others or use data belonging to others in connection with our business, it could lead to disputes over the ownership of inventions derived from that information or expose us to potential damages or other penalties.
57
Table of Contents
The occurrence of any of these events could have a material adverse effect on our business, financial condition, results of operations or prospects.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
There is substantial history of litigation and other proceedings regarding patent and intellectual property rights in the pharmaceutical industry. We may be forced to defend claims of infringement brought by our competitors and others, and we may institute litigation against others who we believe are infringing our intellectual property rights. The outcome of intellectual property litigation is subject to substantial uncertainties and may, for example, turn on the interpretation of claim language by the court, which may not be to our advantage, or on the testimony of experts as to technical facts upon which experts may reasonably disagree.
Under our license agreements, we have the right to bring legal action against any alleged infringers of the patents we license. However, we are responsible for all costs relating to such potential litigation. We have the right to any proceeds received as a result of such litigation, but, even if we are successful in such litigation, there is no assurance we would be awarded any monetary damages.
Our involvement in intellectual property litigation could result in significant expense to us. Some of our competitors have considerable resources available to them and a strong economic incentive to undertake substantial efforts to stop or delay us from commercializing products. Moreover, regardless of the outcome, intellectual property litigation against or by us could significantly disrupt our development and commercialization efforts, divert our management’s attention and quickly consume our financial resources.
In addition, if third parties file patent applications or issue patents claiming technology that is also claimed by us in pending applications, we may be required to participate in interference proceedings with the U.S. Patent Office or in other proceedings outside the U.S., including oppositions, to determine priority of invention or patentability. Even if we are successful in these proceedings, we may incur substantial costs, and the time and attention of our management and scientific personnel will be diverted from product development or other more productive matters.
Risks Related to Our Common Stock
The trading price of the shares of our common stock has been and could in the future be highly volatile.
The market price of our common stock has fluctuated in the past and is likely to fluctuate in the future. Market prices for biopharmaceutical companies have historically been particularly volatile. Some of the factors that may cause the market price of our common stock to fluctuate include:
| • |
developments concerning our clinical studies and trials and our pre-clinical studies; |
| • |
status of regulatory requirements for approval of our drug candidates; |
| • |
announcements of product development successes and failures by us or our competitors; |
| • |
new products introduced or announced by us or our competitors; |
58
Table of Contents
| • |
adverse changes in the abilities of our third-party manufacturers to provide drug or product in a timely manner or to meet FDA requirements; |
| • |
changes in reimbursement levels; |
| • |
changes in financial estimates by securities analysts; |
| • |
actual or anticipated variations in operating results; |
| • |
expiration or termination of licenses (particularly our licenses from BioMarin and Northwestern), research contracts or other collaboration agreements; |
| • |
conditions or trends in the regulatory climate and the biotechnology and pharmaceutical industries; |
| • |
intellectual property, product liability or other litigation against us; |
| • |
changes in the market valuations of similar companies; |
| • |
changes in pharmaceutical company regulations or reimbursements as a result of healthcare reform or other legislation; |
| • |
changes in economic conditions; and |
| • |
sales of shares of our common stock, particularly sales by our officers, directors and significant stockholders, or the perception that such sales may occur. |
In addition, equity markets in general, and the market for emerging pharmaceutical and life sciences companies in particular, have experienced substantial price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of companies traded in those markets. Further, changes in economic conditions in the United States, Europe or globally could impact our ability to grow profitably. Adverse economic changes are outside our control and may result in material adverse impacts on our business or financial results. These broad market and industry factors may materially affect the market price of our shares, regardless of our own development and operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted against that company. Any such litigation that we become involved in could cause us to incur substantial costs and divert our management’s attention and resources, which could have a material adverse effect on our business, financial condition and results of operations.
Delaware law and our certificate of incorporation and by-laws contain provisions that could delay and discourage takeover attempts that stockholders may consider favorable.
Certain provisions of our certificate of incorporation and by-laws, and applicable provisions of Delaware corporate law, may make it more difficult for or prevent a third party from acquiring control of us or changing our Board of Directors and management. These provisions include:
| • |
the ability of our Board of Directors to issue preferred stock with voting or other rights or preferences; |
59
Table of Contents
| • |
limitations on the ability of stockholders to amend our charter documents, including stockholder supermajority voting requirements; |
| • |
the inability of stockholders to act by written consent or to call special meetings; |
| • |
requirements that special meetings of our stockholders may only be called by the Board of Directors; and |
| • |
advance notice procedures our stockholders must comply with in order to nominate candidates for election to our Board of Directors or to place stockholders’ proposals on the agenda for consideration at meetings of stockholders. |
On September 20, 2011, the Board of Directors approved the adoption of a stockholder rights plan, which was amended on September 19, 2016. The rights plan was implemented through our entry into a rights agreement with Continental Stock Transfer & Trust Company, as rights agent, and the declaration of a non-taxable dividend distribution of one preferred stock purchase right (each, a Right) for each outstanding share of our common stock. The dividend was paid on October 7, 2011 to holders of record as of that date. Each right is attached to and trades with the associated share of common stock. The rights will become exercisable only if a person acquires beneficial ownership of 17.5% or more of our common stock (or, in the case of a person who beneficially owned 17.5% or more of our common stock on the date the rights plan was adopted, such person acquires beneficial ownership of any additional shares of our common stock) or after the date of the Rights Agreement, commences a tender offer that, if consummated, would result in beneficial ownership by a person of 17.5% or more of our common stock. The rights will expire on September 20, 2019, unless the rights are earlier redeemed or exchanged.
The intent of the stockholder rights plan is to protect our stockholders’ interests by encouraging anyone seeking control of our company to negotiate with our Board of Directors. However, our stockholder rights plan could make it more difficult for a third party to acquire us without the consent of our Board of Directors, even if doing so may be beneficial to our stockholders. This plan may discourage, delay or prevent a tender offer or takeover attempt, including offers or attempts that could result in a premium over the market price of our common stock. This plan could reduce the price that stockholders might be willing to pay for shares of our common stock in the future. Furthermore, the anti-takeover provisions of our stockholder rights plan may entrench management and make it more difficult to replace management even if the stockholders consider it beneficial to do so.
In addition, Section 203 of the Delaware General Corporation Law generally prohibits us from engaging in a business combination with any person who owns 15% or more of our common stock for a period of three years from the date such person acquired such common stock, unless Board or stockholder approval is obtained. These provisions could make it difficult for a third party to acquire us, or for members of our Board of Directors to be replaced, even if doing so would be beneficial to our stockholders.
Any delay or prevention of a change of control transaction or changes in our Board of Directors or management could deter potential acquirers or prevent the completion of a transaction in which our stockholders could receive a substantial premium over the then current market price for their shares.
Future sales of our common stock may cause our stock price to decline.
As of March 10, 2017, we had 82,972,316 shares of our common stock outstanding, of which 6,042,623 shares were held by our officers and directors. We also had outstanding: (i) common stock purchase warrants to purchase an aggregate of 2,407,663 additional shares of our common stock at exercise prices ranging from $1.04 to $2.08 per share, (ii) stock options to purchase an aggregate of 6,088,333 shares at exercise prices ranging from $0.47 to $4.64 per share (2,636,661 of which are currently exercisable), and 26,667 restricted stock units that are subject to vesting. Sales of restricted shares or shares underlying stock options and common stock purchase warrants, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock.
60
Table of Contents
Our Board of Directors has the ability to issue “blank check” preferred stock.
Our Certificate of Incorporation authorizes the issuance of up to 5,000,000 shares of “blank check” preferred stock, with such designation rights and preferences as may be determined from time to time by our Board of Directors. Our Board of Directors is empowered, without stockholder approval, to issue shares of preferred stock with dividend, liquidation, conversion, voting or other rights which could adversely affect the voting power or other rights of the holders of our common stock. In the event of such issuances, the preferred stock could be utilized, under certain circumstances, as a method of discouraging, delaying or preventing a change in control of our company, pursuant to our stockholder rights plan. Although we have no present intention to issue any shares of our preferred stock, there can be no assurance that we will not do so in the future.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We have never declared or paid any cash dividends on our common stock or other securities, and we currently do not anticipate paying any cash dividends in the foreseeable future. Accordingly, investors should not invest in our common stock if they require dividend income. Our stockholders will not realize a return on their investment unless the trading price of our common stock appreciates, which is uncertain and unpredictable.
| Item 1B. |
None.
| Item 2. |
We currently operate our business in leased office space in Coral Gables, Florida. We currently lease approximately 5,200 square feet of space for which we pay annual rent of approximately $200,000.
| Item 3. |
From time to time we may become involved in legal proceedings arising in the ordinary course of business. We believe that there is no litigation pending at this time that could have, individually or in the aggregate, a material adverse effect on our results of operations, financial condition or cash flows.
| Item 4. |
Not applicable.
61
Table of Contents
| Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock trades on the Nasdaq Capital Market under the symbol “CPRX.” The following table sets forth the high and low closing sales prices per share of our common stock as reported on the Nasdaq Capital Market for the periods indicated.
| High | Low | |||||||
| Year Ended December 31, 2015 |
||||||||
| First Quarter |
$ | 4.93 | $ | 2.74 | ||||
| Second Quarter |
$ | 4.73 | $ | 3.16 | ||||
| Third Quarter |
$ | 5.74 | $ | 3.00 | ||||
| Fourth Quarter |
$ | 3.50 | $ | 2.33 | ||||
| Year Ended December 31, 2016 |
||||||||
| First Quarter |
$ | 2.36 | $ | 1.01 | ||||
| Second Quarter |
$ | 1.25 | $ | 0.56 | ||||
| Third Quarter |
$ | 1.25 | $ | 0.72 | ||||
| Fourth Quarter |
$ | 1.46 | $ | 0.96 | ||||
| Year ended December 31, 2017 |
||||||||
| First Quarter (through March 10, 2017) |
$ | 1.24 | $ | 1.09 | ||||
The closing sale price for the common stock on March 10, 2017 was $1.11. As of March 10, 2017, there were 44 holders of record of our common stock, which includes custodians who hold our securities for the benefit of others. We estimate that there are approximately 9,000 beneficial holders of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support operations and finance the growth and development of our business and do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our Board of Directors.
62
Table of Contents
Performance Graph
The graph below matches Catalyst Pharmaceuticals, Inc.’s cumulative 5-Year total shareholder return on common stock with the cumulative total returns of the NASDAQ Composite index, the Russell MicroCap index, and the NASDAQ Biotechnology index. The graph tracks the performance of a $100 investment in our common stock and in each index (with the reinvestment of all dividends) from 12/31/2011 to 12/31/2016.
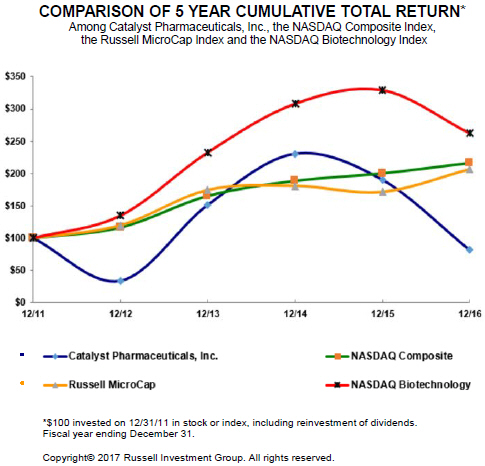
| 12/11 | 12/12 | 12/13 | 12/14 | 12/15 | 12/16 | |||||||||||||||||||
| Catalyst Pharmaceuticals, Inc. |
100.00 | 33.72 | 151.16 | 230.23 | 189.92 | 81.40 | ||||||||||||||||||
| NASDAQ Composite |
100.00 | 116.41 | 165.47 | 188.69 | 200.32 | 216.54 | ||||||||||||||||||
| Russell MicroCap |
100.00 | 119.75 | 174.37 | 180.73 | 171.41 | 206.32 | ||||||||||||||||||
| NASDAQ Biotechnology |
100.00 | 134.68 | 232.37 | 307.67 | 328.76 | 262.08 | ||||||||||||||||||
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
63
Table of Contents
| Selected Financial Data |
The selected statement of operations data for the years ended December 31, 2016, 2015, 2014, and the balance sheet data as of December 31, 2016 and 2015, have been derived from our audited financial statements included elsewhere in this Form 10-K. The selected statement of operations data for the years ended December 31, 2013 and 2012 and the selected balance sheet data at December 31, 2014, 2013 and 2012 have been derived from financial statements that are not included in this Form 10-K. Historical results are not necessarily indicative of future results. This selected financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| Statement of Operations Data: |
2016 | 2015 | 2014 | 2013 | 2012 | |||||||||||||||
| Revenues – government grant |
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Operating costs and expenses: |
||||||||||||||||||||
| Research and development |
11,369,941 | 11,801,342 | 10,117,774 | 8,096,774 | 2,659,597 | |||||||||||||||
| General and administrative |
7,910,260 | 8,597,010 | 4,473,654 | 2,214,884 | 2,561,543 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating cost and expenses |
19,280,201 | 20,398,352 | 14,591,428 | 10,311,658 | 5,221,140 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(19,280,201 | ) | (20,398,352 | ) | (14,591,428 | ) | (10,311,658 | ) | (5,221,140 | ) | ||||||||||
| Other income, net |
321,612 | 100,389 | 76,233 | 47,421 | 14,976 | |||||||||||||||
| Change in fair value of warrants liability |
886,137 | 65,005 | (993,866 | ) | (1,890,359 | ) | 1,129,778 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss before income taxes |
(18,072,452 | ) | (20,232,958 | ) | (15,509,061 | ) | (12,154,596 | ) | (4,076,386 | ) | ||||||||||
| Provision for income taxes |
— | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (18,072,452 | ) | $ | (20,232,958 | ) | $ | (15,509,061 | ) | $ | (12,154,596 | ) | $ | (4,076,386 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss per share—basic and diluted |
$ | (0.22 | ) | $ | (0.25 | ) | $ | (0.24 | ) | $ | (0.27 | ) | $ | (0.14 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average shares outstanding—basic and diluted |
82,875,281 | 80,858,393 | 64,142,534 | 45,452,447 | 30,033,108 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| As of December 31, | ||||||||||||||||||||
| Balance Sheet Data: |
2016 | 2015 | 2014 | 2013 | 2012 | |||||||||||||||
| Cash and cash equivalents, certificates of deposit and short-term investments |
$ | 40,405,817 | $ | 58,396,395 | $ | 39,275,123 | $ | 23,710,596 | $ | 15,417,208 | ||||||||||
| Working capital |
39,359,226 | 56,460,530 | 37,972,795 | 23,180,429 | 15,080,013 | |||||||||||||||
| Total assets |
41,706,853 | 60,101,570 | 43,908,086 | 25,369,554 | 16,789,245 | |||||||||||||||
| Warrants liability, at fair value |
122,226 | 1,008,363 | 2,794,891 | 1,819,562 | 498,587 | |||||||||||||||
| Total liabilities |
2,397,923 | 4,625,259 | 8,665,756 | 3,978,302 | 2,167,130 | |||||||||||||||
| Stockholders’ equity |
39,308,930 | 55,476,311 | 35,242,330 | 21,391,252 | 14,622,115 | |||||||||||||||
64
Table of Contents
| Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with “Selected Financial Data” and our financial statements and related notes appearing elsewhere in this Form 10-K. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties, and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including but not limited to those set forth under the caption “Risk Factors” in Item 1A of this Form 10-K.
Introduction
Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A) is intended to provide an understanding of our financial condition, changes in financial condition and results of operations. The discussion and analysis is organized as follows:
| • |
Overview. This section provides a general description of our business and information about our business that we believe is important in understanding our financial condition and results of operations. |
| • |
Basis of Presentation. This section provides information about key accounting estimates and policies that we followed in preparing our financial statements for the 2016 fiscal year. |
| • |
Critical Accounting Policies and Estimates. This section discusses those accounting policies that are both considered important to our financial condition and results of operations, and require significant judgment and estimates on the part of management in their application. All of our significant accounting policies, including the critical accounting policies, are also summarized in the notes to our accompanying financial statements. |
| • |
Results of Operations. This section provides an analysis of our results of operations for all three fiscal years presented in the accompanying statements of operations. |
| • |
Liquidity and Capital Resources. This section provides an analysis of our cash flows, capital resources, off-balance sheet arrangements and our outstanding commitments, if any. |
| • |
Caution Concerning Forward-Looking Statements. This section discusses how certain forward-looking statements made throughout this MD&A and in other sections of this report are based on management’s present expectations about future events and are inherently susceptible to uncertainty and changes in circumstance. |
Overview
We are a biopharmaceutical company focused on developing and commercializing innovative therapies for people with rare debilitating diseases. We currently have three drug candidates in development:
Firdapse®
In October 2012, we licensed the North American rights to Firdapse®, a proprietary form of amifampridine phosphate, or chemically known as 3,4-diaminopyridine phosphate, from BioMarin Pharmaceutical Inc. (BioMarin). In August 2013, we were granted “breakthrough therapy designation” by the U.S. Food & Drug Administration (FDA) for Firdapse® for the treatment of patients with Lambert-Eaton Myasthenic Syndrome, or LEMS, a rare and sometimes fatal autoimmune disease characterized by muscle weakness. In March 2015, we were granted Orphan Drug Designation for Firdapse® for the treatment of patients with Congenital Myasthenic Syndromes, or CMS, and in August 2016, we were granted Orphan Drug Designation for Firdapse® for the treatment of patients with Myasthenia Gravis (MG).
65
Table of Contents
The chemical entity, amifampridine (3,4-diaminopyridine or 3,4-DAP), has never been approved by the FDA for any indication. Because Firdapse® has been granted Orphan Drug designation for the treatment of LEMS, CMS and MG by the FDA, the product is also eligible to receive seven years of marketing exclusivity for any or all of these indications. Further, if we are the first pharmaceutical company to obtain approval for an amifampridine product, of which there can be no assurance, we will be eligible to receive five years of marketing exclusivity with respect to the use of this product for any indication, running concurrently with the seven years of orphan marketing exclusivity described above (if both exclusivities are granted).
We previously sponsored a multi-center, randomized, placebo-controlled Phase 3 trial evaluating Firdapse® for the treatment of LEMS. The Phase 3 trial, which involved 38 subjects, was designed as a randomized “withdrawal” trial in which all patients were treated with Firdapse® during a 7 to 91-day run-in-period followed by treatment with either Firdapse® or placebo over a two-week randomization period. The co-primary endpoints for this Phase 3 trial were the comparison of changes in patients randomized to continue Firdapse® versus those who transitioned to placebo that occurred in both the Quantitative Myasthenia Gravis Score (QMG), which measures muscle strength, and subject global impression score (SGI), on which the subjects rate their global impression of the effects of a study treatment during the two-week randomization period. In September 2014, we reported positive top-line results from this Phase 3 trial.
During 2014, we established an expanded access program (EAP) to make Firdapse® available to any patients diagnosed with LEMS, CMS, or Downbeat Nystagmus in the United States, who meet the inclusion and exclusion criteria, with Firdapse® being provided to patients for free until sometime after new drug application (NDA) approval, should we receive such approval (of which there can be no assurance). We are informing neuromuscular physicians on the availability of the Firdapse® EAP and working with various rare disease advocacy organizations to inform patients and physicians about the program.
On December 17, 2015, we announced completion of the submission of an NDA for Firdapse® for the treatment of LEMS and CMS. However, on February 17, 2016, we announced that we had received a “refusal to file” (RTF) letter from the FDA regarding our NDA submission. In early April 2016, we met with the FDA to obtain greater clarity regarding what will be required by the FDA to accept the Firdapse® NDA for filing. Following the receipt of the formal minutes of that meeting, on April 26, 2016, we issued a press release reporting that the FDA has advised us that in addition to the results of our previously submitted multi-center, randomized, placebo-controlled Phase 3 trial, we will need to submit positive results from a second adequate and well-controlled study in patients with LEMS. Additionally, there is a requirement for several more short-term toxicology studies, which are currently in process.
In October 2016, we announced that we had reached an agreement with the FDA under a Special Protocol Assessment (SPA) for the protocol design, clinical endpoints, and statistical analysis approach to be taken in our second Phase 3 study evaluating Firdapse® (amifampridine phosphate) for the symptomatic treatment of LEMS. A SPA is a process by which sponsors ask the FDA to evaluate the protocol of a proposed clinical trial to determine whether it adequately addresses scientific and regulatory requirements for the purpose identified by the sponsor. A SPA agreement indicates FDA concurrence with the adequacy and acceptability of specific critical elements of protocol design, endpoints and analysis.
66
Table of Contents
Additionally, it provides a binding agreement with FDA’s review division that a pivotal trial design, conduct, and planned analysis adequately addresses the scientific and regulatory objectives in support of a regulatory submission for drug approval. However, the FDA may rescind a SPA agreement when the division director determines that a substantial scientific issue essential to determining the safety or efficacy of the product has been identified after the trial has begun.
We are conducting our second Phase 3 trial evaluating Firdapse® for the treatment of LEMS (designated as LMS-003) at sites in Miami, Florida and Los Angeles, California. This double-blind, placebo-controlled withdrawal trial will include approximately 28 subjects, and will have the same co-primary endpoints as our first Phase 3 trial evaluating Firdapse® for the treatment of LEMS. Further, the FDA is allowing us to enroll patients from our expanded access program as study subjects in this second trial. Details of the Phase 3 clinical trial are available on www.clinicaltrials.gov (NCT02970162).
We initiated this trial in December 2016, and we expect to report top-line results from this trial during the second half of 2017. Assuming the results of this trial are successful, and our anticipated timeline for the completion of this trial is met, we expect to resubmit an NDA for Firdapse® for the treatment of LEMS in the second half of 2017. There can be no assurance as to the timing or requirements of this trial, whether this trial, along with the results of our first Phase 3 trial, will be sufficient for the FDA to accept for filing any NDA that we might resubmit in the future for Firdapse®, or whether Firdapse® will ever be approved for commercialization.
Our original NDA submission for Firdapse® included data and information (including data from a currently ongoing investigator treatment IND) providing evidence supporting the benefits of Firdapse® for treating certain types of CMS, and requested that CMS be included in our initial label for Firdapse® . To provide additional support for our submission of an NDA for Firdapse® for the treatment of CMS, in October 2015 we initiated a small blinded clinical trial at four academic centers of up to 10 subjects in the pediatric CMS population, ages 2 to 17. However, after considering comments from the FDA, we determined to enroll both adult and pediatric subjects with CMS in this trial and to expand the number of subjects to be evaluated in the trial to an aggregate of approximately 20 subjects. We also added a fifth trial site for this study. Details of this trial are available on www.clinicaltrials.gov (NCT02562066).
Based on currently available information, we expect to report top line results from this study in the second half of 2017 and if the results of the study are successful, we hope to add the CMS indication to our labeling for Firdapse® (either as a part of our NDA resubmission for Firdapse® for LEMS or as a supplement to that resubmission). There can be no assurance that any trial we perform for Firdapse® for the treatment of CMS will be successful or whether any NDA that we may submit for Firdapse® for the treatment of CMS will be filed by the FDA for review and approved.
In February 2016, we announced the initiation of an investigator-sponsored, randomized, double-blind, placebo-controlled, crossover Phase 2/3 clinical trial evaluating the safety, tolerability and potential efficacy of Firdapse® as a symptomatic treatment for patients with MuSK-MG. MuSK-MG, an ultra-rare sub-population of MG patients, is a debilitating neuromuscular disease, and there are currently no FDA approved therapies for this specific form of MG. Seven patients participated in this proof-of-concept trial. We provided study drug, placebo and financial support for this study.
On March 15, 2017 we reported top-line results from this trial. Both of the co-primary efficacy endpoints of change from baseline (CFB) in total Quantitative Myasthenia Gravis (QMG) score (p=0.0003) and CFB in total Myasthenia Gravis Activities of Daily Living (MG-ADL) score (p=0.0006) were statistically and clinically significant in this trial. Several secondary efficacy measures also achieved statistical significance. Amifampridine phosphate was well tolerated in this population of patients.
We intend to discuss with the FDA conducting a registration trial in the United States evaluating Firdapse® for the treatment of patients with MuSK-MG. There can be no assurance that future clinical trials that we initiate to evaluate Firdapse® for this indication will be successful, or whether we can obtain the resources available to fund any such registration trial. Further, there can also be no assurance that the FDA will ever approve Firdapse® for this indication.
67
Table of Contents
Finally, we may seek to evaluate Firdapse® for the treatment of other treatment-refractory types of MG or other rare, similar neuromuscular diseases, although we have not yet begun to develop clinical programs for these indications and all such programs are subject to the availability of funding. There can be no assurance that Firdapse® will be an effective treatment for other treatment-refractory types of MG or for any other rare, similar neuromuscular diseases.
Prior to the receipt of the RTF letter, we had been actively taking steps to prepare for the commercialization of Firdapse® in the United States. In light of the determination that we will have to complete a second adequate and well controlled study evaluating Firdapse® for the treatment of LEMS, we have placed most of these commercialization activities on hold in order to conserve cash. Notwithstanding, we are continuing to work with several rare disease advocacy organizations to help increase awareness of LEMS and CMS and to provide awareness and outreach support for the physicians who treat these rare diseases and the patients they treat.
CPP-115
We are developing CPP-115, a GABA aminotransferase inhibitor that, based on our preclinical studies to date, we believe is a more potent form of vigabatrin, and may have fewer side effects (e.g., visual field defects) than those associated with vigabatrin. We are hoping to develop CPP-115 for the treatment of refractory infantile spasms and possibly for the treatment of adult refractory patients with Tourette’s Disorder. CPP-115 has been granted Orphan Drug Designation by the FDA for the treatment of infantile spasms and Orphan Medicinal Product Designation in the European Union, or E.U., for West syndrome (a form of infantile spasms).
We are currently refining our development plan for this product. Once the refinement of our development plans is completed, and subject to the then availability of funding, we plan to take the steps to complete the work required to make our drug candidate Phase 2 ready. We are also working with one or more potential investigators who have expressed an interest in evaluating our product for particular indications (particularly infantile spasms). We are also continuing to seek a partner to work with us in furthering the development of CPP-115, although no agreements have been entered into to date.
There can be no assurance that we will ever successfully commercialize CPP-115.
Generic Sabril®
During September 2015, we announced the initiation of a project to develop a generic version of Sabril® (vigabatrin). Sabril® is marketed by Lundbeck Inc. in the United States for the treatment of infantile spasms and complex partial seizures. There can be no assurance that we will be successful in these efforts or that any ANDA that we submit for vigabatrin will be accepted for review or approved. Further, while there can be no assurance, we are hopeful that any ANDA submission we make for vigabatrin will be one of the first ANDAs submitted for this product.
Capital Resources
Based on our current financial condition and forecasts of available cash, we believe that we have sufficient funds to support our operations through at least the next 12 months. However, we will require additional funding to support our operations beyond that time. There can be no assurance that we will obtain additional funding or that we will ever be in a position to commercialize any of our drug candidates. See “Liquidity and Capital Resources” below for further information on this topic.
68
Table of Contents
Basis of presentation
Revenues – government grant
We are a development stage company and have no revenues from product sales to date. We will not have revenues from product sales until such time as we receive approval of our product candidates, successfully commercialize our products or enter into a licensing agreement which may include up-front licensing fees, of which there can be no assurance.
Research and development expenses
Our research and development expenses consist of costs incurred for company-sponsored research and development activities, as well as support for selected investigator-sponsored research. The major components of research and development costs include preclinical study costs, clinical manufacturing costs, clinical study and trial expenses, insurance coverage for clinical trials, consulting, scientific advisors and other third-party costs, salaries and employee benefits, stock-based compensation expense, supplies and materials and allocations of various overhead costs related to our product development efforts. To date, all of our research and development resources have been devoted to the development of CPP-109, CPP-115 and Firdapse®, and we expect this to continue for the foreseeable future.
Our cost accruals for clinical studies and trials are based on estimates of the services received and efforts expended pursuant to contracts with numerous clinical study and trial sites and clinical research organizations (CROs). In the normal course of our business we contract with third parties to perform various clinical study and trial activities in the on-going development of potential products. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events or milestones, the successful enrollment of patients, the allocation of responsibilities among the parties to the agreement, and the completion of portions of the clinical study or trial or similar conditions. The objective of our accrual policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to preclinical and clinical studies or trials are recognized based on our estimate of the degree of completion of the event or events specified in the specific study or trial contract. We monitor service provider activities to the extent possible; however, if we underestimate activity levels associated with various studies or trials at a given point in time, we could be required to record significant additional research and development expenses in future periods. Preclinical and clinical study and trial activities require significant up front expenditures. We anticipate paying significant portions of a study or trial’s cost before such begins, and incurring additional expenditures as the study or trial progresses and reaches certain milestones.
Selling and marketing expenses
We do not currently have any selling or marketing expenses. We had been incurring costs tied to our future sales and marketing efforts for Firdapse®. However, during the first quarter of 2016, following the receipt of the “refusal to file” letter, we put most of these activities on hold in order to conserve cash. Pre-commercialization costs are included in general and administrative expenses.
69
Table of Contents
General and administrative expenses
Our general and administrative expenses consist primarily of salaries and personnel expenses for accounting, corporate and administrative functions. Other costs include administrative facility costs, regulatory fees, insurance, pre-commercialization costs, and professional fees for legal, information technology, accounting and consulting services.
Stock-based compensation
We recognize expense for the fair value of all stock-based awards to employees, directors, scientific advisors and consultants in accordance with U.S. generally accepted accounting principles (GAAP). For stock options we use the Black-Scholes option valuation model in calculating the fair value of the awards.
Warrants Liability
We issued warrants to purchase shares of our common stock as part of the equity financing completed in October 2011. In accordance with GAAP, we have recorded the fair value of the warrants as a liability in the accompanying balance sheets at December 31, 2016 and 2015 using a Black-Scholes option-pricing model. We remeasure the fair value of the warrants liability at each reporting date until the warrants are exercised or have expired. Changes in the fair value of the warrants liability are reported in the statements of operations as income or expense. The fair value of the warrants liability is subject to significant fluctuation based on changes in the inputs to the Black-Scholes option-pricing model, including our common stock price, expected volatility, expected term, the risk-free interest rate and dividend yield. The market price for our common stock has been and may continue to be volatile. Consequently, future fluctuations in the price of our common stock may cause significant increases or decreases in the fair value of the warrants.
Income taxes
We have incurred operating losses since inception. As of December 31, 2016 and 2015, we had net operating loss carryforwards of approximately $56,255,000 and $49,922,000, respectively. Our net deferred tax asset has a 100% valuation allowance as of December 31, 2016 and 2015, as we believe it is more likely than not that the deferred tax asset will not be realized. The net operating loss carry-forwards will expire at various dates beginning 2023 through 2036. If an ownership change, as defined under Internal Revenue Code 382, occurs, the use of these carry-forwards may be subject to limitations.
As required by ASC 740, Income Taxes, we recognize the financial statement benefit of a tax position only after determining that the relevant tax authority would more likely than not sustain the position following an audit. For tax positions meeting the more-likely-than-not threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority.
Recent Accounting Pronouncements
For discussion of recently issued accounting standards, please see Note 2, ‘Basis of Presentation and Significant Accounting Policies”, in the financial statements included in this report.
Non-GAAP Financial Measures
We prepare our financial statements and notes thereto which accompany this report in accordance with U.S. GAAP. To supplement our financial results presented on a GAAP basis, we may use non-GAAP financial measures in our reports filed with the Commission
70
Table of Contents
and/or our communications with investors. Non-GAAP measures are provided as additional information and not as an alternative to our financial statements presented in accordance with GAAP. Our non-GAAP financial measures are intended to enhance an overall understanding of our current financial performance. We believe that the non-GAAP financial measures we present provide investors and prospective investors with an alternative method for assessing our operating results in a manner that we believe is focused on the performance of ongoing operations and provide a more consistent basis for comparison between periods.
The non-GAAP financial measures that we often present exclude from the calculation of net loss the expense (or the income) associated with the change in fair value of the liability-classified warrants. Further, we often report non-GAAP net loss per share, which is calculated by dividing non-GAAP net loss by the weighted average common shares outstanding.
Any non-GAAP financial measures that we report should not be considered in isolation or as a substitute for comparable GAAP accounting, and investors should read them in conjunction with our financial statements and notes thereto prepared in accordance with GAAP. Finally, the non-GAAP measures of net loss we may use may be different from, and not directly comparable to, similarly titled measures used by other companies.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires us to make judgments, estimates, and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue and expenses during the reporting periods. We continually evaluate our judgments, estimates and assumptions. We base our estimates on the terms of underlying agreements, our expected course of development, historical experience and other factors we believe are reasonable based on the circumstances, the results of which form our management’s basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
The accounting policies described below are not intended to be a comprehensive list of all of our accounting policies. In many cases, the accounting treatment of a particular transaction is specifically dictated by GAAP. There are also areas in which our management’s judgment in selecting any available alternative would not produce a materially different result. Our financial statements and the notes thereto included elsewhere in this report contain accounting policies and other disclosures as required by GAAP.
Preclinical study and clinical trial expenses
Research and development expenditures are charged to operations as incurred. Our expenses related to preclinical and clinical trials are based on actual and estimated costs of the services received and efforts expended pursuant to contracts with multiple research institutions and any CRO that conducts and manages our clinical trials. The financial terms of these agreements are subject to negotiation and will vary from contract to contract and may result in uneven payment flows. Generally, these agreements will set forth the scope of the work to be performed at a fixed fee or unit price. Payments under these contracts will depend on factors such as the successful enrollment of patients or the completion of clinical trial milestones. Expenses related to clinical trials generally are accrued based on contracted amounts applied to the level of patient enrollment and activity according to the protocol. If timelines or contracts are modified based upon changes in the clinical trial protocol or scope of work to be performed, we would be required to modify estimates accordingly on a prospective basis.
71
Table of Contents
Warrants Liability
We have issued warrants to purchase our common stock that may require us to purchase unexercised warrants for a cash amount equal to their fair value following the announcement of specified events defined as Fundamental Transactions (Fundamental Transactions) involving our company, which is deemed to occur if we are acquired in an all cash transaction or by a company that is not listed on a national securities exchange, or when the common stock is no longer listed on a national securities exchange. The cash settlement provisions require use of the Black-Scholes model in calculating the cash payment value in the event of a Fundamental Transaction. As a consequence of these provisions, the warrants are classified as a liability on our balance sheets. The cash settlement value at the time of any future Fundamental Transaction will depend upon the value of the following inputs at that time: the price per share of our common stock, the volatility of our common stock, the expected term of the warrants, the risk-free interest rate based on U.S. Treasury security yields, and our dividend yield. The fair value of the warrants is determined using a Black-Scholes model. The valuation of warrants is subjective and is affected by changes in inputs to the valuation model including the price per share of our common stock, the historical volatility of our stock price, risk-free rates based on U.S. Treasury security yields, the expected term of the warrants and our dividend yield. Changes in these assumptions can materially affect the fair value estimate. We could ultimately incur amounts to settle the warrant at a cash settlement value that is significantly different than the carrying value of the liability on our financial statements. We will continue to classify the fair value of the warrants as a liability until the warrants are exercised, expire, or are amended in a way that would no longer require these warrants to be classified as a liability. Changes in the fair value of the common stock warrants liability are recognized as income or loss in the changes in fair value of warrants liability line in the statement of operations.
Stock-based compensation
We recognize stock-based compensation for the fair value of all share-based payments, including grants of stock options and restricted stock units. For stock options, we use the Black-Scholes option valuation model to determine the fair value of stock options on the date of grant. This model derives the fair value of stock options based on certain assumptions related to expected stock price volatility, expected option life, risk-free interest rate and dividend yield. Expected volatility is based on reviews of historical volatility of our common stock. The estimated expected option life is based upon the simplified method. Under this method, the expected option life is presumed to be the mid-point between the vesting date and the end of the contractual term. We will continue to use the simplified method until we have sufficient historical exercise data to estimate the expected life of the options. The risk-free interest rate assumption is based upon the U.S. Treasury yield curve appropriate for the expected life of our stock options awards. For the years ended December 31, 2016, 2015 and 2014, the assumptions used were an estimated annual volatility of 100%, 102%, and 115%, average expected holding periods of two to six years, three to seven years, and three to seven years and risk-free interest rates of 0.76% to 2.15%, 1% to 2.13%, and 1.18% to 2.03%, respectively.
Results of Operations
Years Ended December 31, 2016 and 2015
Revenues
We had no revenues for the year ended December 31, 2016 or 2015.
72
Table of Contents
Research and Development Expenses
| Year |
Amount | Change from Prior Year |
Percentage of Total Operating Costs and Expenses |
|||||||||
| 2016 |
$ | 11,369,941 | (3.7 | )% | 59.0 | % | ||||||
| 2015 |
$ | 11,801,342 | 16.6 | % | 57.9 | % | ||||||
Our expenses, including stock-based compensation, for research and development for the year ended December 31, 2016 decreased 3.7% compared to amounts expended during the 2015 fiscal year. Research and development expenses in 2016 and 2015 consisted mainly of costs related to our Phase 3 trials of Firdapse® , our CMS trial, our Phase 1b trial of CPP-115, costs relating to other preclinical and clinical testing for Firdapse®, costs related to the operation of the Firdapse® Expanded Access Program, costs associated with the submission of our NDA filing for Firdapse®, costs relating to the manufacturing of Firdapse®, our share of the costs of the joint studies being conducted with BioMarin, and costs relating to our generic Sabril® program.
We expect that research and development expenses will increase in fiscal 2017 compared to fiscal 2016 as we continue our clinical development efforts for Firdapse®, including our second Phase 3 clinical trial for LEMS, our clinical trial for CMS in pediatric and adult populations, our clinical program for MuSK-MG, our Expanded Access Program and our generic Sabril® program.
Our research and development expenses for 2016 and 2015, include stock-based compensation relating to the value of stock options granted to certain employees. The amount of stock-based compensation recorded in 2016 and 2015 relating to our research and development activities was $590,857 and $378,548, respectively. The weighted-average grant-date fair value of the stock options granted in 2016 and 2015 was $0.62 and $2.28, respectively.
Selling and Marketing Expenses
We had no selling and marketing expenses during 2016 and 2015. In 2015 and 2016, we incurred pre-commercialization costs, tied to our preparation for future sales and marketing efforts, as we moved closer to the potential commercialization of Firdapse®. In the first quarter of 2016, following receipt of the “refusal to file” letter, these costs were substantially reduced, in order to conserve cash. These costs were for personnel, and their related activities, to develop both a sales force and a patient advocacy and assistance program so that we would be in a position to commence our selling efforts had we been successful in filing our NDA for Firdapse® and obtaining an approval. There can be no assurance that we will ever obtain an NDA approval for Firdapse®. Pre-commercialization costs have been included in general and administrative expenses.
General and Administrative Expenses
| Year |
Amount | Change from Prior Year |
Percentage of Total Operating Costs and Expenses |
|||||||||
| 2016 |
$ | 7,910,260 | (8.0 | )% | 41.0 | % | ||||||
| 2015 |
$ | 8,597,010 | 92.2 | % | 42.1 | % | ||||||
General and administrative expenses include, among other expenses, corporate and office expenses, legal, accounting and consulting fees, pre-commercialization costs and travel expenses for our administrative employees, consultants and members of our Board of Directors. Included in general and administrative expenses in the years 2016 and 2015, was stock-based compensation of $1,245,228 and $1,206,910, respectively. As discussed above, pre-commercialization costs are also included in general and administrative expenses, and amounted to $2,471,461 and $3,833,855 in 2016 and 2015, respectively.
73
Table of Contents
The 8.0% decrease in general and administrative expenses for the year ended December 31, 2016 when compared to the same period in 2015 was primarily due to our efforts to conserve cash after the receipt of the refusal to file letter, partly offset by increases in pre-commercialization expenses, payroll and benefits, during the first half of 2016, including approximately $600,000 in severance costs related to the reduction-in-force that occurred in May 2016. We expect that general and administrative costs will remain consistent for the next year as we continue our efforts to conserve cash.
Stock-Based Compensation
We issued stock options and other share-based payments to several of our employees, directors, and consultants in 2016 and 2015. Total stock-based compensation expense for the years ended December 31, 2016 and 2015 was $1,836,085 and $1,585,458, respectively. We regularly grant non-cash stock-based compensation to employees and directors as part of their compensation packages. The 2016 increase in expense from the prior year is primarily due to additional headcount and expense related to incentive grants to employees and directors.
Change in fair value of warrants liability
In connection with the October 2011 equity offering, we issued warrants to purchase an aggregate of 1,523,370 shares of common stock. The fair value of the warrants is recorded in the liability section of the balance sheet and was estimated at $122,000 and $1.0 million at December 31, 2016 and 2015, respectively. The fair value of the warrants liability is determined at the end of each reporting period with the resulting gains or losses recorded as the change in fair value of warrants liability in the statements of operations.
For the years ended December 31, 2016 and 2015, we recognized gains of $886,137 and $65,005, respectively, in connection with the change in the fair value of the warrants liability. The gains during 2016 and 2015 were principally a result of fluctuations in our common stock price. We believe that future changes in the fair value of the warrants liability will be due primarily to future fluctuations in the value of our common stock.
Other Income, net
We reported other income, net in all periods relating to our investment of funds received from offerings of our securities. Other income, net consists of interest income, dividend income and unrealized and realized gain (loss) on trading securities. The $221,000 increase in other income, net for the year ended December 31, 2016 as compared to the year ended December 31, 2015 was principally due to higher yields on investment balances from the proceeds of our offerings. These proceeds were used to fund our product-development activities and our operations. Substantially all such funds were invested in short-term interest bearing obligations and short-term bond funds.
Income taxes
We have incurred net operating losses since inception. Consequently, we have applied a 100% valuation allowance against our deferred tax asset as we believe that it is more likely than not that the deferred tax asset will not be realized.
74
Table of Contents
Net Loss
Our net loss was $18,072,452 in the year ended December 31, 2016 ($0.22 per basic and diluted share) as compared to $20,232,958 in the year ended December 31, 2015 ($0.25 per basic and diluted share).
Non-GAAP Net Loss
Our non-GAAP net loss, which excludes for 2016 a $886,137 gain associated with the change in the fair value of liability-classified warrants and excludes for 2015 a $65,005 gain associated with the change in the fair value of liability-classified warrants, was $18,958,589 ($0.23 per basic and diluted share), as compared to $20,297,963 ($0.25 per basic and diluted share) for 2015.
Years Ended December 31, 2015 and 2014
Revenues
We had no revenues for the year ended December 31, 2015 or 2014.
Research and Development Expenses
| Year |
Amount | Change from Prior Year |
Percentage of Total Operating Costs and Expenses |
|||||||||
| 2015 |
$ | 11,801,342 | 16.6 | % | 57.9 | % | ||||||
| 2014 |
$ | 10,117,774 | 25.0 | % | 69.3 | % | ||||||
Our expenses, including stock-based compensation, for research and development for the year ended December 31, 2015 increased 16.6% compared to amounts expended during the 2014 fiscal year. Research and development expenses in 2015 and 2014 consisted mainly of costs related to our Phase 3 trial of Firdapse® and our Phase 1b trial of CPP-115, costs relating to preclinical and clinical testing on both Firdapse® and CPP-115, costs related to the launch and operation of the Firdapse® Expanded Access Program, costs associated with the filing of our NDA for Firdapse®, costs relating to the manufacturing of Firdapse®, and our share of the costs of the joint studies being conducted with BioMarin.
Our research and development expenses for 2015 and 2014, include stock-based compensation relating to the value of stock options granted to certain employees. The amount of stock-based compensation recorded in 2015 and 2014 relating to our research and development activities was $378,548 and $133,862, respectively. The weighted-average grant-date fair value of the stock options granted in 2015 and 2014 was $2.28 and $2.45, respectively.
Selling and Marketing Expenses
We had no selling and marketing expenses during 2015 and 2014. We incurred pre-commercialization costs, tied to our preparation for future sales and marketing efforts, as we moved closer to the potential commercialization of Firdapse®. These costs are for personnel, and their related activities, to develop both a sales force and a patient advocacy and assistance program so that we are in a position to commence our selling efforts at such time as we are successful in obtaining an approval of any NDA that we may file for Firdapse®, of which there can be no assurance. Pre-commercialization costs have been included in general and administrative expenses.
75
Table of Contents
General and Administrative Expenses
| Year |
Amount | Change from Prior Year |
Percentage of Total Operating Costs and Expenses |
|||||||||
| 2015 |
$ | 8,597,010 | 92.2 | % | 42.1 | % | ||||||
| 2014 |
$ | 4,473,654 | 102.2 | % | 30.7 | % | ||||||
General and administrative expenses include, among other expenses, corporate and office expenses, legal, accounting and consulting fees, pre-commercialization costs and travel expenses for our administrative employees, consultants and members of our Board of Directors. Included in general and administrative expenses in the years 2015 and 2014, was stock-based compensation of $1,206,910 and $664,107, respectively. As discussed above, pre-commercialization costs are also included in general and administrative expenses, and amounted to $3,833,855 and $1,011,806 in the years 2015 and 2014, respectively.
The 92.2% increase in general and administrative expenses for the year ended December 31, 2015 when compared to the same period in 2014 was primarily due to increases in headcount and related expenses and pre-commercialization expenses, as we expanded our operations for the potential future commercialization of Firdapse®.
Stock-Based Compensation
We issued stock options and other share-based payments to several of our employees, directors, and consultants in 2015 and 2014. Total stock-based compensation expense for the years ended December 31, 2015 and 2014 was $1,585,458 and $777,969, respectively. We regularly grant non-cash stock-based compensation to employees and directors as part of their compensation packages. The 2015 increase in expense from the prior year is primarily due to additional expense related to the annual grant to employees and directors and initial grants made to new officers and employees.
Change in fair value of warrants liability
In connection with the October 2011 equity offering, we issued warrants to purchase an aggregate of 1,523,370 shares of common stock. The fair value of the warrants is recorded in the liability section of the balance sheet and was estimated at $1.0 million and $2.8 million at December 31, 2015 and 2014, respectively. The fair value of the warrants liability is determined at the end of each reporting period with the resulting gains or losses recorded as the change in fair value of warrants liability in the statements of operations.
For the years ended December 31, 2015 and 2014, we recognized a gain of $65,005 and a loss of $993,866, respectively, in connection with the change in the fair value of the warrants liability. The gains and losses during 2015 and 2014 were principally a result of fluctuations in our common stock price.
Other Income, net
We reported other income, net in all periods relating to our investment of funds received from offerings of our securities. Other income, net consists of interest income, dividend income and unrealized and realized gain (loss) on trading securities. The increase in other income, net for the year ended December 31, 2015 as compared to the year ended December 31, 2014 was principally due to higher average investment balances from the proceeds of our offerings, partially offset by lower interest rates. These proceeds were used to fund our product-development activities and our operations. Substantially all such funds were invested in short-term interest bearing obligations and short-term bond funds.
76
Table of Contents
Income taxes
We have incurred net operating losses since inception. Consequently, we have applied a 100% valuation allowance against our deferred tax asset as we believe that it is more likely than not that the deferred tax asset will not be realized.
Net Loss
Our net loss was $20,232,958 in the year ended December 31, 2015 ($0.25 per basic and diluted share), as compared to $15,509,061 in the year ended December 31, 2014 ($0.24 per basic and diluted share).
Non-GAAP Net Loss
Our non-GAAP net loss, which excludes for 2015 a $65,005 gain associated with the change in the fair value of liability-classified warrants and excludes for 2014 a $993,866 loss associated with the change in the fair value of liability-classified warrants, was $20,297,963 ($0.25 per basic and diluted share), as compared to $14,515,195 ($0.23 per basic and diluted share) for 2014.
Liquidity and Capital Resources
Our historical capital resource requirements have been the funding of working capital and preclinical and clinical testing of our product candidates. We have historically funded all of our requirements from equity issuances, government grants, and an investment by a strategic purchaser.
Since our inception, we have financed our operations primarily with the net proceeds of three private placements, an initial public offering (IPO), a secondary public offering and nine registered direct public offerings under our effective shelf registration statements. At December 31, 2016, we had cash and cash equivalents and short-term investments aggregating $40,405,817 and working capital of $39,359,226, as compared to cash and cash equivalents, certificates of deposit and short-term investments aggregating $58,396,395 and working capital of $56,460,530 at December 31, 2015. At December 31, 2016, substantially all of our cash and cash equivalents were deposited with one financial institution and our short-term investments were invested in a high-quality short-term bond fund. Throughout 2016, we had cash balances at certain financial institutions in excess of federally insured limits.
We have to date incurred operating losses, and we expect these losses to increase substantially in the future as we expand our drug development programs and prepare for the commercialization of our drug candidates. We anticipate using current cash on hand to finance these activities. It will likely be some time before we obtain the necessary regulatory approvals to commercialize one or more of our product candidates in the United States.
We currently believe that we have the cash resources to support our operations through at least the next 12 months. These expectations are based on current information available to us. If our costs are greater than we expect, our assumptions may not prove to be accurate.
At the present time, we will require additional funding for future studies or trials, other than those described as being on-going in this report. We will also require additional working capital to support our operations beyond that time. There can be no assurance as to the amount of any such funding that will be required for these purposes or whether any such funding will be available to us when it is required.
77
Table of Contents
In that regard, our future funding requirements will depend on many factors, including:
| • |
the scope, rate of progress and cost of our clinical trials and other product development activities; |
| • |
future clinical trial results; |
| • |
the terms and timing of any collaborative, licensing and other arrangements that we may establish; |
| • |
the cost and timing of regulatory approvals; |
| • |
the cost and delays in product development as a result of any changes in regulatory oversight applicable to our products; |
| • |
the cost and timing of establishing sales, marketing and distribution capabilities; |
| • |
the effect of competition and market developments; |
| • |
the cost of filing and potentially prosecuting, defending and enforcing any patent claims and other intellectual property rights; and |
| • |
the extent to which we acquire or invest in other products. |
We plan to raise additional funds to support our product development activities and working capital requirements through public or private equity offerings, debt financings, corporate collaborations or other means. We may also seek governmental grants to support our clinical trials and preclinical trials. We may also seek to raise capital to fund additional product development efforts even if we have sufficient funds for our planned operations. Any sale by us of additional equity or convertible debt securities could result in dilution to our stockholders. There can be no assurance that any such required additional funding will be available to us at all or available on terms acceptable to us. Further, to the extent that we raise additional funds through collaborative arrangements, it may be necessary to relinquish some rights to our technologies or grant sublicenses on terms that are not favorable to us. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs, which could have an adverse effect on our business.
On December 23, 2016, we filed a shelf registration statement with the SEC to sell up to $33,842,512 of common stock (the “2016 Shelf Registration Statement”). This shelf registration statement was declared effective by the SEC on January 9, 2017. We have made no sales under the 2016 Shelf Registration Statement.
As of the date of this Form 10-K, the full amount of our 2016 Shelf Registration Statement remains available for future sales. However, if our public float (the market value of our common stock held by non-affiliate stockholders) were to fall below $75 million, we would be subject to a further limitation under which we could sell no more than one-third (1/3) of our public float during any 12-month period. Further, the number of shares that we can sell at any one time may be limited under certain circumstances to 20% of the outstanding common stock under applicable NASDAQ marketplace rules.
On January 31, 2014, we filed a Shelf Registration Statement on Form S-3 (the 2014 Shelf Registration Statement) with the U.S. Securities and Exchange Commission (SEC) to sell up to $100 million of common stock. This registration statement (file No. 333-193699) was declared effective by the SEC on March 19, 2014. We completed two offerings under the 2014 Shelf Registration Statement:
78
Table of Contents
| • |
On April 3, 2014, we raised net proceeds of approximately $26.7 million from the sale of 13,023,750 shares of our common stock; and |
| • |
On February 4, 2015, we raised net proceeds of approximately $34.9 million from the sale of 11,500,000 shares of our common stock. |
Approximately $33.8 million remains available under the 2014 Shelf Registration Statement, which will expire on March 19, 2017. Now that the 2016 Shelf Registration Statement has become effective, we do not plan to issue any shares under the 2014 Shelf Registration Statement.
Contractual obligations and arrangements
As of December 31, 2016, we had the following contractual obligations. Further, we may owe in the future certain milestone or royalty payment obligations (as described below). Since we are not currently able to determine when or if these milestones will be achieved, or when or if the events triggering payment of the obligations will occur, they are not included in the following table.
| Payments Due by Period | ||||||||||||||||||||
| Total | Less than 1 year |
1-3 years | 4-5 years | After 5 years |
||||||||||||||||
| Operating lease obligations |
$ | 1,321,250 | $ | 207,421 | $ | 433,697 | $ | 460,109 | $ | 220,023 | ||||||||||
| License obligations |
300,000 | — | 300,000 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 1,621,250 | $ | 207,421 | $ | 733,697 | $ | 460,109 | $ | 220,023 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
We have entered into the following contractual arrangements:
| • |
Payments to BioMarin and others under our license agreement. We have agreed: (i) to pay BioMarin royalties for seven years from the first commercial sale of Firdapse® equal to 7% of net sales (as defined in our license agreement) in North America for any calendar year for sales up to $100 million, and 10% of net sales in North America in any calendar year in excess of $100 million; (ii) to pay to the third-party licensor of the rights sublicensed to us royalty payments for seven years from the first commercial sale of Firdapse® equal to 7% of net sales (as defined in the license agreement between BioMarin and the third-party licensor) in any calendar year; and (iii) to pay certain milestone payments that BioMarin is obligated to pay (approximately $2.6 million of which will be due upon acceptance by the FDA of an NDA for Firdapse® for the treatment of LEMS or CMS, and approximately $7.2 million of which will be due on the unconditional approval by the FDA of an NDA for Firdapse® for the treatment of LEMS or CMS). We have also agreed to share in the cost of certain post-marketing studies that are being conducted by BioMarin. |
| • |
Payments to Northwestern University under our license agreement. Under our license agreement with Northwestern, we have paid to date $411,590, had accrued liabilities of $152,500 at December 31, 2016 in the accompanying balance sheet, and owe certain milestone payments in future years if we do not cancel the license agreement. The next milestone payment of $300,000 is due in August 2018. |
| • |
Employment agreements. We have entered into an employment agreement with our Chief Executive Officer that requires us to make base salary payments of approximately $485,000 in 2017. The agreement expires in November 2018. |
79
Table of Contents
| • |
Lease for office space. We currently operate our business in leased office space in Coral Gables, Florida. We currently lease approximately 5,200 square feet of office space for which we pay annual rent of approximately $200,000. |
Off-Balance Sheet Arrangements
We currently have no debt or capital leases. We have operating leases for our office facilities. We do not have any off-balance sheet arrangements as such term is defined in rules promulgated by the SEC.
Cash Flows
Net cash used in operating activities was $17,963,503 and $18,015,221, respectively, for the years ended December 31, 2016 and 2015. During the year ended December 31, 2016, net cash used in operating activities was primarily attributable to our net loss of $18,072,452, decreases of $860,951 in accounts payable and $480,248 in accrued expenses and other liabilities, and a gain of $886,137 of non-cash change in fair value of warrants liability, which were partially offset by a decrease of $456,794 in prepaid expenses and other current assets and deposits. The loss included an additional $1,879,491 of non-cash expenses. During the year ended December 31, 2015, net cash used in operating activities was primarily attributable to our net loss of $20,232,958, an increase of $452,040 in prepaid expenses and other current assets and deposits, a decrease of $20,083 in accounts payable and a gain of $65,005 of non-cash change in fair value of warrants liability, which were partially offset with an increase of $1,134,939 in accrued expenses and other liabilities. The loss included an additional $1,619,926 of non-cash expenses. Such additional non-cash expenses consist of depreciation and stock-based compensation expense.
Net cash provided by (used in) investing activities was $3,552,565 and ($6,499), respectively, for 2016 and 2015. During 2016 such funds were primarily from redemption of certificates of deposit and short-term investments, partly offset by $96,061 of capital expenditures. During 2015 such funds were primarily used for capital expenditures, partly offset by proceeds from short-term investments.
Net cash provided by financing activities was $68,986 and $37,159,958, respectively, for 2016 and 2015. During 2016, net cash from financing activities consisted mostly of proceeds from exercise of stock options. During 2015, net cash from financing activities consisted mainly of the net proceeds from the sale of shares of common stock in underwritten and registered direct public offerings under our registration statements, as well as proceeds from exercise of stock options and warrants. Such funds are being used to fund our research and development costs and our general and administrative costs.
Caution Concerning Forward-Looking Statements
Some of the statements in this Form 10-K are “forward-looking statements”, as that term is defined in the Private Securities Litigation Reform Act of 1995. These include statements regarding our expectations, beliefs, plans or objectives for future operations and anticipated results of operations. For this purpose, any statements contained herein that are not statements of historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, “believes”, “anticipates”, “proposes”, “plans”, “expects”, “intends”, “may”, and other similar expressions are intended to identify forward-looking statements. Such statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. The forward-looking statements made in this Form 10-K are based on current expectations that involve numerous risks and uncertainties.
80
Table of Contents
The successful development of our product candidates is highly uncertain. We cannot reasonably estimate or know the nature, timing, or estimated expenses of the efforts necessary to complete the development of, or the period in which material net cash inflows are expected to commence due to the numerous risks and uncertainties associated with developing such products, including the uncertainty of:
| • |
our estimates regarding anticipated capital requirements and our need for additional financing; |
| • |
the risk that another pharmaceutical company will receive an approval for its formulation of 3,4-diaminopyridine (3,4-DAP) for the treatment of Lambert-Eaton Myasthenic Syndrome (LEMS), Congenital Myasthenic Syndromes (CMS), or any other indication, before we do; |
| • |
whether the clinical studies or trials that are required to be completed before the FDA will accept an NDA submission for Firdapse® for the treatment of either LEMS or CMS will be successful; |
| • |
what additional supporting information, including any additional clinical studies or trials, will be required before the U.S. Food and Drug Administration (FDA) will accept our new drug application (NDA) submission for Firdapse® for the treatment of either LEMS or CMS (or any other condition or disease); |
| • |
whether any NDA that we may submit to the FDA for Firdapse® will be accepted for filing, or whether even if any NDA that we submit for Firdapse® is accepted for filing by the FDA, whether any such NDA filing will be granted a priority review; |
| • |
whether, even if the FDA accepts an NDA submission for Firdapse®, such product will be determined to be safe and effective and approved for commercialization; |
| • |
whether the receipt of breakthrough therapy designation for Firdapse® for LEMS will result in an expedited review of Firdapse® by the FDA or affect the likelihood that the product will be found to be safe and effective; |
| • |
whether as part of the FDA review of any NDA that we may submit for filing for Firdapse® , the tradename Firdapse®, which is the tradename used for the same product in Europe, will be approved for use for the product in the United States; |
| • |
whether, assuming Firdapse® is approved for commercialization, we will be able to develop or contract with a sales and marketing organization that can successfully market Firdapse® while maintaining full compliance with applicable federal and state laws, rules and regulations; |
| • |
whether any future trial that we undertake evaluating Firdapse® for the treatment of MuSK-MG will be successful and whether we can obtain the funding required for such trial; |
| • |
whether CPP-115 will be determined to be safe for humans; |
| • |
whether CPP-115 will be determined to be effective for the treatment of infantile spasms, post-traumatic stress disorder, Tourette’s Disorder or any other indication; |
| • |
whether we can successfully design and complete a bioequivalence study of our version of vigabatrin compared to Sabril® that is acceptable to the FDA; |
| • |
whether any such bioequivalence study, the design of which is acceptable to the FDA, will be successful; |
| • |
whether any abbreviated new drug application (ANDA) that we submit for a generic version of Sabril® will be accepted by the FDA for review and approved (and the timing of any such approval); |
81
Table of Contents
| • |
the scope, rate of progress and expense of our clinical trials and studies, pre-clinical studies, proof-of-concept studies, and our other drug development activities, and whether our trials and studies will be successful; |
| • |
our ability to complete our trials and studies on a timely basis and within the budgets we establish for such trials and studies; |
| • |
the results of our clinical studies and trials, pre-clinical studies, proof-of-concept studies, and our other development activities, and the number of such studies and trials that will be required for us to seek and obtain approval of new drug applications, or NDAs, for our drug candidates; |
| • |
whether the third parties that assist us in our trials and studies perform as anticipated and within the budgets established for their activities; |
| • |
the ability of our third-party suppliers and contract manufacturers to maintain compliance with current Good Manufacturing Practices (cGMP); |
| • |
whether any of our drug candidates will ever be approved for commercialization; |
| • |
even if one or more of our drug candidates is approved for commercialization, whether we will be able to successfully commercialize those products and achieve sustained profitability; |
| • |
whether our estimates of the pricing of our drug candidates, if approved, and our estimates of the size of the market for our drug candidates, will turn out to be accurate; |
| • |
the level of third-party payor reimbursement for any of our drug candidates that are commercialized; |
| • |
what pricing we will be able to achieve with respect to our drug products and the impact of public scrutiny and potential changes in the law that affect the pricing of drug products generally, and our products and orphan drugs in particular; |
| • |
changes in the healthcare industry occasioned by any future repeal and replacement of the Affordable Care Act, or changes in the healthcare industry generally; |
| • |
changes in the manner in which the FDA reviews NDAs; |
| • |
changes in the laws and regulations and regulatory guidance affecting our business, including any changes that may be made to the laws affecting orphan drugs (such as changes to the exclusivity that may be available for approved products under those laws); |
| • |
the market adoption of any of our drug candidates approved for commercialization by physicians and patients; |
| • |
our ability to obtain a sufficient commercial supply of our products; |
| • |
if one or more of our products are approved for commercialization, the costs, timing or estimated completion of any post-marketing studies that we may be obligated to complete; |
82
Table of Contents
| • |
our expectations regarding licensing, acquisitions or strategic relationships; |
| • |
whether we can successfully protect any of our drug candidates under intellectual property laws; |
| • |
the expense of filing, and potentially prosecuting, defending and enforcing any patent claims and other intellectual property rights we may have for our drug candidates; |
| • |
our ability to attract and retain skilled employees; |
| • |
security breaches of our computer systems, or the computer systems of our contractors and/or vendors; |
| • |
the impact of potential employee, vendor or consultant misconduct; and |
| • |
changes in general economic conditions and interest rates. |
Our current plans and objectives are based on assumptions relating to the development of our current drug candidates. Although we believe that our assumptions are reasonable, any of our assumptions could prove inaccurate. The significant uncertainties inherent in the forward-looking statements we have made herein, which reflect our views only as of the date of this report, suggest that you should not place undue reliance upon such statements. We undertake no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise.
| Quantitative and Qualitative Disclosures About Market Risk |
Market risk represents the risk of changes in the value of market risk-sensitive instruments caused by fluctuations in interest rates, foreign exchange rates and commodity prices. Changes in these factors could cause fluctuations in our results of operations and cash flows.
Our exposure to interest rate risk is currently confined to our cash and short-term investments that are from time to time invested in highly liquid money market funds, short-term certificates of deposit and short-term, high-quality bond funds. The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. We do not use derivative financial instruments in our investment portfolio. Our cash and investments policy emphasizes liquidity and preservation of principal over other portfolio considerations.
| Financial Statements and Supplementary Data |
See the list of financial statements filed with this report under Item 15 below.
| Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Not applicable.
| Controls and Procedures |
Disclosure Controls and Procedures
We have carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and
83
Table of Contents
procedures. The term “disclosure controls and procedures”, as defined in Rules 13a-15(e) and 15(d)-15(e) under the Securities Exchange Act of 1934 (the “Exchange Act”), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure.
Based on such evaluation, our principal executive officer and principal financial officer have concluded that as of December 31, 2016, our disclosure controls and procedures were effective to ensure that the information required to be disclosed by us in the reports filed or submitted by us under the Securities Exchange Act of 1934, as amended, was recorded, processed, summarized or reported within the time periods specified in the rules and regulations of the SEC, and include controls and procedures designed to ensure that information required to be disclosed by us in such reports was accumulated and communicated to management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosures.
Management’s Annual Assessment of Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on our financial statements.
Internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements prepared for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our principal executive officer and our principal financial officer, management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2016 based on the 2013 framework in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and in accordance with the interpretive guidance issued by the SEC in Release No. 34-55929. Based on that evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2016.
During the fourth quarter of 2016, there were no changes in our internal control over financial reporting, as defined in Rule 13a-15(f) under the Securities and Exchange Act of 1934 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
84
Table of Contents
Our independent registered public accounting firm, Grant Thornton LLP, has issued a report on our internal control over financial reporting, which is included in Item 15 of this Annual Report on Form 10-K.
| Other Information |
Not applicable.
85
Table of Contents
| Directors and Executive Officers of the Registrant |
The information required by this item will be contained in our definitive proxy statement, or Proxy Statement, to be filed with the SEC in connection with our 2017 Annual Meeting of Stockholders. Our Proxy Statement for the 2017 Annual Meeting of Stockholders is expected to be filed not later than 120 days after the end of our fiscal year ended December 31, 2016 and is incorporated into this report by this reference.
We have adopted a code of ethics that applies to our chief executive officer, chief financial officer, and to all of our other officers, directors, employees and agents. The code of ethics is available on our website at www.catalystpharma.com. We intend to disclose future amendments to, or waivers from, certain provisions of our code of ethics on the above website within five business days following the date of such amendment or waiver.
| Executive Compensation |
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by this reference.
| Security Ownership of Certain Beneficial Owners and Management |
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by this reference.
| Certain Relationships and Related Transactions |
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by this reference.
| Principal Accounting Fees and Services |
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by this reference.
86
Table of Contents
| Exhibits and Financial Statement Schedules |
| (a) |
Documents filed as part of this report. |
1. The following financial statements of Catalyst Pharmaceuticals, Inc. and Report of Grant Thornton LLP, independent registered public accounting firm, are included in this report:
| • |
Reports of Grant Thornton LLP, Independent Registered Public Accounting Firm |
| • |
Balance Sheets as of December 31, 2016 and 2015 |
| • |
Statements of Operations for the years ended December 31, 2016, 2015 and 2014 |
| • |
Statement of Stockholders’ Equity for the period December 31, 2013 until December 31, 2016 |
| • |
Statements of Cash Flows for the years ended December 31, 2016, 2015 and 2014 |
| • |
Notes to Financial Statements |
2. List of financial statement schedules. All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
3. List of exhibits required by Item 601 of Regulation S-K. See part (b) below.
87
Table of Contents
| (b) |
Exhibits. |
| Exhibit No. | Description of Exhibit | |
| 2.1 |
Agreement and Plan of Merger, dated August 14, 2006, between the Company and Catalyst Pharmaceutical Partners, Inc., a Florida corporation (1) | |
| 3.1 |
Certificate of Incorporation (1) | |
| 3.2 |
Amendment to Certificate of Incorporation (1) | |
| 3.3 |
Amendment to Certificate of Incorporation (23) | |
| 3.4 |
Amendment to Certificate of Incorporation (26) | |
| 3.5 |
By-laws(1) | |
| 4.1 |
Specimen stock certificate for common stock (1) | |
| 4.2 |
Rights Agreement between the Company and Continental Stock Transfer and Trust Company(10) | |
| 4.3 |
Amendment to Rights Agreement between the Company and Continental Stock Transfer and Trust Company (26) | |
| 4.4 |
Form of Warrant to Purchase Common Stock issued in our October 2011 offering (11) | |
| 4.5 |
Form of Warrant to Purchase Common Stock issued in our May 2012 offering (14) | |
| 4.6 |
Form of Warrant to Purchase Common Stock issued in our August 2012 offering (15) | |
| 10.1 + |
Employment Agreement between the Company and Patrick J. McEnany (2) | |
| 10.2 + |
Amendment to Employment Agreement between the Company and Patrick J. McEnany (4) | |
| 10.3 + |
Amendment to Employment Agreement between the Company and Patrick J. McEnany (6) | |
| 10.4 + |
Amendment to Employment Agreement between the Company and Patrick J. McEnany (9) | |
| 10.5+ |
Amendment to Employment Agreement between the Company and Patrick J. McEnany (17) | |
| 10.6+ |
Amendment to Employment Agreement between the Company and Patrick J. McEnany (25) | |
| 10.7+ |
2006 Stock Incentive Plan (1) | |
| 10.8+ |
Amendment No. 1 to 2006 Stock Incentive Plan (7) | |
| 10.9+ |
Amendment No. 2 to 2006 Stock Incentive Plan (13) | |
| 10.10+ |
2014 Stock Incentive Plan (20) | |
| 10.11+ |
Amendment No. 1 to 2014 Stock Incentive Plan (24) | |
| 10.12 |
License Agreement between the Company and Northwestern University (5) | |
88
Table of Contents
Exhibit No. |
Description of Exhibit | |
| 10.13 |
Lease Agreement between the Company and 355 Alhambra Plaza, Ltd. (3) | |
| 10.14 |
First Amendment to Lease Agreement between the Company and 355 Alhambra Plaza, Ltd. (8) | |
| 10.15 |
License Agreement among the Company, New York University, and The Feinstein Institute for Medical Research (12) | |
| 10.16 |
Convertible Promissory Note and Note Purchase Agreement, dated as of October 26, 2012, between the Company and BioMarin Pharmaceutical, Inc. (16) | |
| 10.17 |
License Agreement, dated as of October 26, 2012, between the Company and BioMarin Pharmaceutical, Inc. (16) | |
| 10.18 |
Amendment No, 1 to License Agreement, dated April 8, 2014, between the Company and BioMarin Pharmaceutical, Inc. (21) | |
| 10.19 |
Termination Agreement, dated effective October 1, 2013, between the Company and Brookhaven Science Associates, LLC (18) | |
| 10.20 |
Second Amendment to Lease, dated as of February 4, 2014, between the Company and 355 Alhambra Circle LLC (19) | |
| 10.21 |
Third Amendment to Lease, dated effective as of March 16, 2015, between the Company and 355 Alhambra Circle LLC (22) | |
| 23.1 |
Consent of Independent Registered Public Accounting Firm* | |
| 31.1 |
Section 302 CEO Certification* | |
| 31.2 |
Section 302 CFO Certification* | |
| 32.1 |
Section 906 CEO Certification* | |
| 32.2 |
Section 906 CFO Certification* | |
| 101.INS |
XBRL Instance Document | |
| 101.SCH |
XBRL Taxonomy Extension Schema | |
| 101.CAL |
XBRL Taxonomy Extension Calculation Linkbase | |
| 101.DEF |
XBRL Taxonomy Extension Definition Linkbase | |
| 101.LAB |
XBRL Taxonomy Extension Label Linkbase | |
| 101.PRE |
XBRL Taxonomy Extension Presentation Linkbase | |
| (1) |
Filed by reference to the Company’s Registration Statement on Form S-1 (File No. 333-136039) |
| (2) |
Filed by reference to the Company’s Form 10-Q for the period ended September 30, 2006 |
| (3) |
Filed by reference to the Company’s Form 10-Q for the period ended March 31, 2007 |
| (4) |
Filed by reference to the Company’s Form 8-K dated December 23, 2008 |
89
Table of Contents
| Exhibit No. | Description of Exhibit | |
| (5) |
Filed by reference to the Company’s Form 8-K dated September 2, 2009 | |
| (6) |
Filed by reference to the Company’s Form 10-Q for the period ended September 30, 2009 | |
| (7) |
Filed by reference to the Company’s 2011 Annual Meeting Proxy Statement dated April 11, 2011 | |
| (8) |
Filed by reference to the Company’s Form 10-Q for the period ended June 30, 2011 | |
| (9) |
Filed by reference to the Company’s Form 8-K dated September 14, 2011 | |
| (10) |
Filed by reference to the Company’s Form 8-K dated September 20, 2011 | |
| (11) |
Filed by reference to the Company’s Form 8-K dated October 28, 2011 | |
| (12) |
Filed by reference to the Company’s Annual Report on Form 10-K for the period ended December 31, 2011 | |
| (13) |
Filed by reference to the Company’s 2012 Annual Meeting Proxy Statement dated April 17, 2012 | |
| (14) |
Filed by reference to the Company’s Registration Statement on Form S-1 (File No. 333-180617) | |
| (15) |
Filed by reference to the Company’s Form 8-K dated August 28, 2012 | |
| (16) |
Filed by reference to the Company’s Form 8-K dated October 26, 2012 | |
| (17) |
Filed by reference to the Company’s Form 8-K dated August 28, 2013 | |
| (18) |
Filed by reference to the Company’s Form 10-Q for the period ended September 30, 2013 | |
| (19) |
Filed by reference to the Company’s Form 8-K dated February 20, 2014 | |
| (20) |
Filed by reference to the Company’s Form 8-K dated March 4, 2014 | |
| (21) |
Filed by reference to the Company’s Form 8-K dated April 17, 2014 | |
| (22) |
Filed by reference to the Company’s 8-K dated March 23, 2015 | |
| (23) |
Filed by reference to the Company’s 2015 Annual Meeting Proxy Statement dated March 31, 2015 | |
| (24) |
Filed by reference to the Company’s 2016 Annual Meeting Proxy Statement dated April 29, 2016 | |
| (25) |
Filed by reference to the Company’s 8-K dated June 23, 2016 | |
| (26) |
Filed by reference to the Company’s 8-K dated September 19, 2016 |
| * |
Filed herewith |
| + |
Management contract or compensatory plan |
90
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has caused this Annual Report on Form 10-K to be signed by the undersigned, thereunto duly authorized, this 15th day of March, 2017.
| CATALYST PHARMACEUTICALS, INC. | ||
| By: |
/s/ Patrick J. McEnany | |
| Patrick J. McEnany, Chairman, | ||
| President and CEO | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons, in the capacities and on the dates indicated.
| Signature |
Title | Date | ||
| /s/ Patrick J. McEnany Patrick J. McEnany |
Chairman of the Board of Directors, President and Chief Executive Officer (Principal Executive Officer) | March 15, 2017 | ||
| /s/ Alicia Grande Alicia Grande |
Vice President, Treasurer, Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | March 15, 2017 | ||
| /s/ Charles B. O’Keeffe Charles B. O’Keeffe |
Director | March 15, 2017 | ||
| /s/ Philip H. Coelho Philip H. Coelho |
Director | March 15, 2017 | ||
| /s/ David S. Tierney, M.D. David S. Tierney, M.D. |
Director | March 15, 2017 | ||
| /s/ Donald A. Denkhaus Donald A. Denkhaus |
Director | March 15, 2017 | ||
| /s/ Richard Daly Richard Daly |
Director | March 15, 2017 | ||
91
Table of Contents
Years ended December 31, 2016, 2015, and 2014
| F-2 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
| F-8 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED
PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Catalyst Pharmaceuticals, Inc.
We have audited the internal control over financial reporting of Catalyst Pharmaceuticals, Inc. (a Delaware corporation) (the “Company”) as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Assessment of Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the financial statements of the Company as of and for the year ended December 31, 2016, and our report dated March 15, 2017 expressed an unqualified opinion on those financial statements.
/s/ GRANT THORNTON LLP
Miami, Florida
March 15, 2017
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED
PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Catalyst Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Catalyst Pharmaceuticals, Inc. (a Delaware corporation) (the “Company”) as of December 31, 2016 and 2015, and the related statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2016. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Catalyst Pharmaceuticals, Inc. as of December 31, 2016 and 2015, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 15, 2017 expressed an unqualified opinion thereon.
/s/ GRANT THORNTON LLP
Miami, Florida
March 15, 2017
F-3
Table of Contents
CATALYST PHARMACEUTICALS, INC.
| December 31, 2016 |
December 31, 2015 |
|||||||
| ASSETS |
||||||||
| Current Assets: |
||||||||
| Cash and cash equivalents |
$ | 13,893,064 | $ | 28,235,016 | ||||
| Certificates of deposit |
— | 3,717,229 | ||||||
| Short-term investments |
26,512,753 | 26,444,150 | ||||||
| Prepaid expenses and other current assets |
1,047,944 | 1,504,738 | ||||||
|
|
|
|
|
|||||
| Total current assets |
41,453,761 | 59,901,133 | ||||||
| Property and equipment, net |
244,204 | 191,549 | ||||||
| Deposits |
8,888 | 8,888 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 41,706,853 | $ | 60,101,570 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current Liabilities: |
||||||||
| Accounts payable |
$ | 933,176 | $ | 1,794,127 | ||||
| Accrued expenses and other liabilities |
1,161,359 | 1,646,476 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
2,094,535 | 3,440,603 | ||||||
| Accrued expenses and other liabilities, non-current |
181,162 | 176,293 | ||||||
| Warrants liability, at fair value |
122,226 | 1,008,363 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
2,397,923 | 4,625,259 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value, 5,000,000 shares authorized: none issued and outstanding at December 31, 2016 and 2015 |
— | — | ||||||
| Common stock, $0.001 par value, 150,000,000 shares authorized; 82,972,316 shares and 82,850,619 shares issued and outstanding at December 31, 2016 and 2015, respectively |
82,972 | 82,851 | ||||||
| Additional paid-in capital |
147,374,028 | 145,469,078 | ||||||
| Accumulated deficit |
(108,148,070 | ) | (90,075,618 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
39,308,930 | 55,476,311 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 41,706,853 | $ | 60,101,570 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
F-4
Table of Contents
CATALYST PHARMACEUTICALS, INC.
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Revenues |
$ | — | $ | — | $ | — | ||||||
| Operating costs and expenses: |
||||||||||||
| Research and development |
11,369,941 | 11,801,342 | 10,117,774 | |||||||||
| General and administrative |
7,910,260 | 8,597,010 | 4,473,654 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating costs and expenses |
19,280,201 | 20,398,352 | 14,591,428 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(19,280,201 | ) | (20,398,352 | ) | (14,591,428 | ) | ||||||
| Other income, net |
321,612 | 100,389 | 76,233 | |||||||||
| Change in fair value of warrants liability |
886,137 | 65,005 | (993,866 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(18,072,452 | ) | (20,232,958 | ) | (15,509,061 | ) | ||||||
| Provision for income taxes |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (18,072,452 | ) | $ | (20,232,958 | ) | $ | (15,509,061 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share - basic and diluted |
$ | (0.22 | ) | $ | (0.25 | ) | $ | (0.24 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average shares outstanding – basic and diluted |
82,875,281 | 80,858,393 | 64,142,534 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
F-5
Table of Contents
CATALYST PHARMACEUTICALS, INC.
STATEMENT OF STOCKHOLDERS’ EQUITY
for the years ended December 31, 2016, 2015 and 2014
| Preferred Stock |
Common Stock |
Additional Paid-In Capital |
Accumulated Deficit |
Total | ||||||||||||||||
| Balance at December 31, 2013 |
$ | — | $ | 54,133 | $ | 75,670,718 | $ | (54,333,599 | ) | $ | 21,391,252 | |||||||||
| Issuance of common stock, net |
— | 13,024 | 26,712,106 | — | 26,725,130 | |||||||||||||||
| Issuance of stock options for services |
— | — | 767,838 | — | 767,838 | |||||||||||||||
| Amortization of restricted stock for services |
— | — | 10,131 | — | 10,131 | |||||||||||||||
| Exercise of warrants for common stock |
— | 1,262 | 1,333,778 | — | 1,335,040 | |||||||||||||||
| Exercise of stock options for common stock |
— | 700 | 521,300 | — | 522,000 | |||||||||||||||
| Net loss |
— | — | — | (15,509,061 | ) | (15,509,061 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2014 |
— | 69,119 | 105,015,871 | (69,842,660 | ) | 35,242,330 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Issuance of common stock, net |
— | 11,527 | 34,862,342 | — | 34,873,869 | |||||||||||||||
| Issuance of stock options for services |
— | — | 1,510,018 | — | 1,510,018 | |||||||||||||||
| Amortization of restricted stock for services |
— | — | 75,440 | — | 75,440 | |||||||||||||||
| Exercise of warrants for common stock |
— | 1,178 | 3,616,083 | — | 3,617,261 | |||||||||||||||
| Exercise of stock options for common stock |
— | 1,027 | 389,324 | — | 390,351 | |||||||||||||||
| Net loss |
— | — | — | (20,232,958 | ) | (20,232,958 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2015 |
— | 82,851 | 145,469,078 | (90,075,618 | ) | 55,476,311 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Issuance of common stock, net |
— | 27 | (27 | ) | — | — | ||||||||||||||
| Issuance of stock options for services |
— | — | 1,760,591 | — | 1,760,591 | |||||||||||||||
| Amortization of restricted stock for services |
— | — | 75,494 | — | 75,494 | |||||||||||||||
| Exercise of stock options for common stock |
— | 94 | 68,892 | — | 68,986 | |||||||||||||||
| Net loss |
— | — | — | (18,072,452 | ) | (18,072,452 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2016 |
$ | — | $ | 82,972 | $ | 147,374,028 | $ | (108,148,070 | ) | $ | 39,308,930 | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
The accompanying notes are an integral part of these financial statements.
F-6
Table of Contents
CATALYST PHARMACEUTICALS, INC.
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Operating Activities: |
||||||||||||
| Net loss |
$ | (18,072,452 | ) | $ | (20,232,958 | ) | $ | (15,509,061 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation |
43,406 | 34,468 | 26,574 | |||||||||
| Stock-based compensation |
1,836,085 | 1,585,458 | 777,969 | |||||||||
| Change in fair value of warrants liability |
(886,137 | ) | (65,005 | ) | 993,866 | |||||||
| (Increase) decrease in: |
||||||||||||
| Prepaid expenses and other current assets and deposits |
456,794 | (452,040 | ) | (2,943,256 | ) | |||||||
| Increase (decrease) in: |
||||||||||||
| Accounts payable |
(860,951 | ) | (20,083 | ) | 963,421 | |||||||
| Accrued expenses and other liabilities |
(480,248 | ) | 1,134,939 | 2,748,704 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(17,963,503 | ) | (18,015,221 | ) | (12,941,783 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Investing Activities: |
||||||||||||
| Capital expenditures |
(96,061 | ) | (23,465 | ) | (57,323 | ) | ||||||
| Proceeds (purchase) of short-term investments |
(68,603 | ) | 18,812 | (8,979,900 | ) | |||||||
| Proceeds (purchase) of certificates of deposit |
3,717,229 | (1,846 | ) | 296,193 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
3,552,565 | (6,499 | ) | (8,741,030 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Financing Activities: |
||||||||||||
| Proceeds from issuance of common stock and warrants, net |
— | 34,873,869 | 26,725,130 | |||||||||
| Payment of employee withholding tax related to stock-based compensation |
(11,265 | ) | — | — | ||||||||
| Proceeds from exercise of warrants |
— | 1,895,738 | 1,316,503 | |||||||||
| Proceeds from exercise of options |
80,251 | 390,351 | 522,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
68,986 | 37,159,958 | 28,563,633 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
(14,341,952 | ) | 19,138,238 | 6,880,820 | ||||||||
| Cash and cash equivalents – beginning of period |
28,235,016 | 9,096,778 | 2,215,958 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents – end of period |
$ | 13,893,064 | $ | 28,235,016 | $ | 9,096,778 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing and financing activities: |
||||||||||||
| Exercise of liability classified warrants for common stock |
$ | — | $ | 1,721,523 | $ | 18,537 | ||||||
| Non-cash incentive received from lessor |
$ | — | $ | 131,175 | $ | — | ||||||
The accompanying notes are an integral part of these financial statements.
F-7
Table of Contents
CATALYST PHARMACEUTICALS, INC.
| 1. |
Organization and Description of Business |
Catalyst Pharmaceuticals, Inc. (the Company) is a development-stage biopharmaceutical company focused on developing and commercializing innovating therapies for people with rare debilitating diseases, including Lambert-Eaton Myasthenic Syndrome (LEMS), Congenital Myasthenic Syndromes (CMS) and infantile spasms. The Company (f/k/a Catalyst Pharmaceutical Partners, Inc.) was incorporated in Delaware in July 2006. It is the successor by merger to Catalyst Pharmaceutical Partners, Inc., a Florida corporation, which commenced operations in January 2002.
Since inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. The Company’s primary focus is on the development and commercialization of its drug candidates. The Company has incurred operating losses in each period from inception through December 31, 2016. The Company has been able to fund its cash needs to date through several public and private offerings of its common stock and warrants, through government grants, and through an investment by a strategic purchaser. See Note 11.
Capital Resources
On January 31, 2014, the Company filed a Shelf Registration Statement on Form S-3 (the 2014 Shelf Registration Statement) with the U.S. Securities and Exchange Commission (SEC) to sell up to $100 million of common stock. This registration statement (file No. 333-193699) was declared effective by the SEC on March 19, 2014. The Company has conducted two registered direct offerings under the 2014 Shelf Registration Statement. (See Note 11). Approximately $33.8 million remains available under the 2014 Shelf Registration Statement, which will expire on March 19, 2017.
On December 23, 2016, the Company filed a Shelf Registration Statement on Form S-3 (the 2016 Shelf Registration Statement) with the SEC to sell up to approximately $33.8 million of common stock. The 2016 Shelf Registration Statement was filed to continue the registration of the remaining unsold allotment under the 2014 Shelf Registration Statement beyond its expiration date. The 2016 Shelf Registration Statement (file No. 333-215315) was declared effective by the SEC on January 9, 2017. Now that the 2016 Shelf Registration Statement has become effective, the Company does not intend to issue any additional shares under the 2014 Shelf Registration Statement.
While there can be no assurance, based on currently available information, the Company estimates that it currently has sufficient resources to support its operations for at least the next 12 months.
The Company may raise required funds in the future through public or private equity offerings, debt financings, corporate collaborations, governmental research grants or other means. The Company may also seek to raise new capital to fund additional product development efforts, even if it has sufficient funds for its planned operations. Any sale by the Company of additional equity or convertible debt securities could result in dilution to the Company’s current stockholders. There can be no assurance that any such required additional funding will be available to the Company at all or available on terms acceptable to the Company. Further, to the extent that the Company raises additional funds through collaborative arrangements, it may be necessary to relinquish some rights to the Company’s drug candidates or grant sublicenses on terms that are not favorable to the Company. If the Company is not able to secure additional funding when needed, the Company may have to delay, reduce the scope of, or eliminate one or more research and development programs, which could have an adverse effect on the Company’s business.
F-8
Table of Contents
| 2. |
Basis of Presentation and Significant Accounting Policies |
| a. |
USE OF ESTIMATES. The preparation of financial statements in conformity with U.S. generally accepted accounting principles (U.S. GAAP) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates. |
| b. |
CASH AND CASH EQUIVALENTS. The Company considers all highly liquid instruments, purchased with an original maturity of three months or less to be cash equivalents. Cash equivalents consist mainly of money market funds. The Company has substantially all of its cash and cash equivalents deposited with one financial institution. These amounts at times may exceed federally insured limits. |
| c. |
CERTIFICATES OF DEPOSIT. The certificates of deposit are issued by a banking institution and are recorded at cost plus accrued interest. The original maturity was greater than three months but did not exceed one year. Interest income is recorded in the statement of operations as it is earned. All of the Company’s certificates of deposit were redeemed during the year ended December 31, 2016. Carrying value at December 31, 2015 approximates fair value. |
| d. |
SHORT-TERM INVESTMENTS. The Company invests in short-term investments in high credit-quality funds in order to obtain higher yields on its cash available for investments. As of December 31, 2016, and 2015, short-term investments consisted of a short-term bond fund. Such investments are not insured by the Federal Deposit Insurance Corporation. Short-term investments at December 31, 2016 and 2015 were accounted for as trading securities. Trading securities are recorded at fair value based on the closing market price of the security. For trading securities, the Company recognizes realized gains and losses and unrealized gains and losses to earnings. Unrealized and realized gain (loss) on trading securities for the years ended December 31, 2016, 2015 and 2014 were $58,861, ($29,430), and ($18,316), respectively and are included in other income, net in the accompanying statements of operations. |
| e. |
PREPAID EXPENSES AND OTHER CURRENT ASSETS. Prepaid expenses and other current assets consist primarily of prepaid research fees, prepaid insurance, prepaid pre-commercialization fees and prepaid subscription fees. Prepaid research fees consist of advances for the Company’s product development activities, including drug manufacturing, contracts for preclinical studies, clinical trials and studies, regulatory affairs and consulting. Such advances are recorded as expense as the related goods are received or the related services are performed. |
| f. |
PROPERTY AND EQUIPMENT. Property and equipment are recorded at cost. Depreciation is calculated to amortize the depreciable assets over their useful lives using the straight-line method and commences when the asset is placed in service. Leasehold improvements are amortized on a straight-line basis over the term of the lease or the estimated life of the improvement, whichever is shorter. Useful lives generally range from three years for computer equipment to three to six years for furniture and equipment, and from five to seven years for leasehold improvements. Expenditures for repairs and maintenance are charged to expenses as incurred. |
| g. |
OPERATING LEASES. The Company recognizes lease expense on a straight-line basis over the initial lease term. For leases that contain rent holidays, escalation clauses or tenant improvement allowances, the Company recognizes rent expense on a straight-line basis and records the difference between the rent expense and rental amount payable as deferred rent. As of December 31, 2016, and 2015, the Company had $199,256 and $194,386, respectively, of deferred rent and lease incentive in accrued expenses and other liabilities. |
| h. |
FAIR VALUE OF FINANCIAL INSTRUMENTS. The Company’s financial instruments consist of cash and cash equivalents, certificates of deposit, short-term investments, accounts payable and accrued expenses and other liabilities, and warrants liability. At December 31, 2016 and 2015, the fair value of these instruments approximated their carrying value. |
F-9
Table of Contents
| 2. |
Basis of Presentation and Significant Accounting Policies (continued) |
| i. |
FAIR VALUE MEASUREMENTS. Current Financial Accounting Standards Board (FASB) fair value guidance emphasizes that fair value is a market-based measurement, not an entity-specific measurement. Therefore, a fair value measurement should be determined based on the assumptions that market participants would use in pricing the asset or liability. As a basis for considering market participant assumptions in fair value measurements, current FASB guidance establishes a fair value hierarchy that distinguishes between market participant assumptions based on market data obtained from sources independent of the reporting entity (observable inputs that are classified within Levels 1 and 2 of the hierarchy) and the reporting entity’s own assumptions that it believes market participants would use in pricing assets or liabilities (unobservable inputs classified within Level 3 of the hierarchy). |
Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs may include quoted prices for similar assets and liabilities in active markets, as well as inputs that are observable for the asset or liability (other than quoted prices), such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals. Level 3 inputs are unobservable inputs for the asset or liability, which are typically based on an entity’s own assumptions, as there is little, if any, related market activity. In instances where the determination of the fair value measurement is based on inputs from different levels of the fair value hierarchy, the level in the fair value hierarchy within which the entire fair value measurement falls is based on the lowest level input that is significant to the fair value measurement in its entirety. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment, and considers factors specific to the asset or liability.
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||
| Balances as of December 31, 2016 |
Quoted Prices in Active Markets for Identical Assets/Liabilities (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Money market funds |
$ | 13,395,759 | $ | 13,395,759 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Short-term investments |
$ | 26,512,753 | $ | 26,512,753 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Warrants liability |
$ | 122,226 | $ | — | $ | — | $ | 122,226 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||
| Balances as of December 31, 2015 |
Quoted Prices in Active Markets for Identical Assets/Liabilities (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Money market funds |
$ | 25,157,601 | $ | 25,157,601 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Certificates of deposit |
$ | 3,717,229 | $ | — | $ | 3,717,229 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Short-term investments |
$ | 26,444,150 | $ | 26,444,150 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Warrants liability |
$ | 1,008,363 | $ | — | $ | — | $ | 1,008,363 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
F-10
Table of Contents
| 2. |
Basis of Presentation and Significant Accounting Policies (continued) |
| j. |
WARRANTS LIABILITY. In October 2011, the Company issued 1,523,370 warrants (the 2011 warrants) to purchase shares of the Company’s common stock in connection with a registered direct offering. The Company accounted for these warrants as a liability measured at fair value due to a provision included in the warrants agreement that provides the warrants holders with an option to require the Company (or its successor) to purchase their warrants for cash in an amount equal to their Black-Scholes Option Pricing Model (the Black-Scholes Model) value, in the event that certain fundamental transactions, as defined, occur. The fair value of the warrants liability is estimated using the Black-Scholes Model which requires inputs such as the expected term of the warrants, share price volatility and risk-free interest rate. These assumptions are reviewed on a quarterly basis and changes in the estimated fair value of the outstanding warrants are recognized each reporting period in the “Change in fair value of warrants liability” line in the statements of operations. As of both December 31, 2016, and 2015, 763,913 of the 2011 warrants remained outstanding. The 2011 warrants will expire on May 2, 2017. |
| k. |
RESEARCH AND DEVELOPMENT. Costs incurred in connection with research and development activities are expensed as incurred. These costs consist of direct and indirect costs associated with specific projects as well as fees paid to various entities that perform research related services for the Company. |
| l. |
STOCK-BASED COMPENSATION. The Company recognizes expense in the statements of operations for the fair value of all stock-based payments to employees, directors, scientific advisors and consultants, including grants of stock options and other share-based awards. For stock options, the Company uses the Black-Scholes option valuation model, the single-option award approach and the straight-line attribution method. Using this approach, compensation cost is amortized on a straight-line basis over the vesting period of each respective stock option, generally one to three years. The Company estimates forfeitures and adjusts this estimate periodically based on actual forfeitures. |
For the years ended December 31, 2016, 2015 and 2014, the Company recorded stock-based compensation expense as follows:
| 2016 | 2015 | 2014 | ||||||||||
| Research and development |
$ | 590,857 | $ | 378,548 | $ | 133,862 | ||||||
| General and administrative |
1,245,228 | 1,206,910 | 644,107 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation |
$ | 1,836,085 | $ | 1,585,458 | $ | 777,969 | ||||||
|
|
|
|
|
|
|
|||||||
| m. |
CONCENTRATION OF CREDIT RISK. The financial instruments that potentially subject the Company to concentration of credit risk are cash equivalents (i.e. money market funds) and short-term investments. The Company places its cash equivalents with high-credit quality financial institutions. These amounts at times may exceed federally insured limits. The Company has not experienced any credit losses in these accounts. |
| n. |
INCOME TAXES. The Company utilizes the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. |
The Company recognizes the financial statement benefit of a tax position only after determining that the relevant tax authority would more likely than not sustain the position following an audit. For tax positions meeting the more-likely-than-not threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority.
F-11
Table of Contents
| 2. |
Basis of Presentation and Significant Accounting Policies (continued) |
The Company is subject to income taxes in the U.S. federal jurisdiction and various state jurisdictions. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. The Company is not subject to U.S. federal, state and local tax examinations by tax authorities for years before 2013. If the Company were to subsequently record an unrecognized tax benefit, associated penalties and tax related interest expense would be reported as a component of income tax expense.
| o. |
COMPREHENSIVE INCOME (LOSS). U.S. generally accepted accounting principles require that all components of comprehensive income (loss) be reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is net income (loss), plus certain other items that are recorded directly into stockholders’ equity. For all periods presented, the Company’s net loss equals comprehensive loss, since the Company has no items which are considered other comprehensive income (loss). |
| p. |
NET INCOME (LOSS) PER SHARE. Basic loss per share is computed by dividing net loss for the period by the weighted average number of common shares outstanding during the period. The calculation of basic and diluted net loss per share is the same for all periods presented, as the effect of potential common stock equivalents is anti-dilutive due to the Company’s net loss position for all periods presented. The potential shares, which are excluded from the determination of basic and diluted net loss per share as their effect is anti-dilutive, are as follows, for the years ended December 31, 2016, 2015 and 2014: |
| 2016 | 2015 | 2014 | ||||||||||
| Stock options to purchase common stock |
4,660,000 | 4,250,000 | 3,884,610 | |||||||||
| Warrants to purchase common stock |
2,407,663 | 2,407,663 | 3,585,924 | |||||||||
| Unvested restricted stock |
26,667 | 53,334 | 80,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Potential equivalent common stock excluded |
7,094,330 | 6,710,997 | 7,550,534 | |||||||||
|
|
|
|
|
|
|
|||||||
Potentially dilutive stock options to purchase common stock as of both December 31, 2016 and December 31, 2015, have exercise prices ranging from $0.47 to $4.64. Potentially dilutive stock options to purchase common stock as of December 31, 2014, have exercise prices ranging from $0.47 to $3.12. Potentially dilutive warrants to purchase common stock as of December 31, 2016, 2015 and 2014 have exercise prices ranging from $1.04 to $2.08 and expire in periods between May 2017 and August 2017.
| q. |
SEGMENT INFORMATION. Management has determined that the Company operates in one reportable segment, which is the development and commercialization of pharmaceutical products. |
| r. |
RECLASSIFICATIONS. Certain prior year amounts in the financial statements have been reclassified to conform to the current year presentation. |
| s. |
RECENTLY ISSUED ACCOUNTING STANDARDS. In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The amendments in this ASU, require management to assess a company’s ability to continue as a going concern and to provide related disclosures in certain circumstances. The guidance was effective for the annual period ending after December 15, 2016 and subsequent interim and annual periods thereafter. The Company adopted this standard in the fourth quarter of 2016. The adoption of this standard did not have a material impact on the Company’s financial statements. |
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842), which requires an entity to recognize assets and liabilities arising from a lease for both financing and operating leases. The ASU will also require new qualitative and quantitative disclosures to help investors and other financial statement users better understand the amount, timing, and uncertainty of cash flows arising from leases. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, with early adoption permitted. The Company is currently evaluating the impact this accounting standard will have on its financial statements.
F-12
Table of Contents
| 2. |
Basis of Presentation and Significant Accounting Policies (continued) |
In March, 2016, the FASB issued ASU No. 2016-09, Compensation—Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which simplifies several aspects of the accounting for employee share-based payment transactions for both public and nonpublic entities including the accounting for income taxes, forfeitures, and statutory tax withholding requirements, as well as classification in the statement of cash flows. For public companies, the changes are effective for reporting periods (annual and interim) beginning after December 15, 2016. Early adoption is permitted. However, if early adoption is elected in an interim period, any adjustments should be reflected as of the beginning of the annual period that includes that interim period. The Company is currently evaluating the effect this standard will have on its financial statements.
| 3. |
Warrants Liability, at Fair Value |
The Company allocated approximately $1.3 million of proceeds from its October 2011 registered direct offering to the fair value of common stock purchase warrants issued in connection with the offering that are classified as a liability (the 2011 warrants). The 2011 warrants are classified as a liability because of provisions in such warrants that allow for the net cash settlement of such warrants in the event of certain fundamental transactions (as defined in the warrant agreement). The valuation of the 2011 warrants is determined using the Black-Scholes Model. This model uses inputs such as the underlying price of the shares issued when the warrant is exercised, volatility, risk free interest rate and expected life of the instrument. The Company has determined that the 2011 warrants liability should be classified within Level 3 of the fair value hierarchy by evaluating each input for the Black-Scholes Model against the fair value hierarchy criteria and using the lowest level of input as the basis for the fair value classification. There are six inputs: closing price of the Company’s common stock on the day of evaluation; the exercise price of the warrants; the remaining term of the warrants; the volatility of the Company’s common stock; annual rate of dividends; and the risk-free rate of return. Of those inputs, the exercise price of the warrants and the remaining term are readily observable in the warrants agreement. The annual rate of dividends is based on the Company’s historical practice of not granting dividends. The closing price of the Company’s common stock would fall under Level 1 of the fair value hierarchy as it is a quoted price in an active market. The risk-free rate of return is a Level 2 input, while the historical volatility is a Level 3 input in accordance with the fair value accounting guidance. Since the lowest level input is a Level 3, the Company determined the 2011 warrants liability is most appropriately classified within Level 3 of the fair value hierarchy. This liability is subject to a fair value mark-to-market adjustment each reporting period. The calculated value of the 2011 warrants liability was determined using the Black-Scholes option-pricing model with the following assumptions:
| December 31, 2016 | December 31, 2015 | |||||||
| Risk free interest rate |
0.85 | % | 0.79 | % | ||||
| Expected term |
0.33 years | 1.34 years | ||||||
| Expected volatility |
100 | % | 68 | % | ||||
| Expected dividend yield |
0 | % | 0 | % | ||||
| Expected forfeiture rate |
0 | % | 0 | % | ||||
The following table rolls forward the fair value of the Company’s warrants liability activity for the years ended December 31, 2016, 2015 and 2014:
| 2016 | 2015 | 2014 | ||||||||||
| Fair value, beginning of period |
$ | 1,008,363 | $ | 2,794,891 | $ | 1,819,562 | ||||||
| Issuance of warrants |
— | — | — | |||||||||
| Exercise of warrants |
— | (1,721,523 | ) | (18,537 | ) | |||||||
| Change in fair value |
(886,137 | ) | (65,005 | ) | 993,866 | |||||||
|
|
|
|
|
|
|
|||||||
| Fair value, end of period |
$ | 122,226 | $ | 1,008,363 | $ | 2,794,891 | ||||||
|
|
|
|
|
|
|
|||||||
F-13
Table of Contents
| 3. |
Warrants Liability, at Fair Value (continued) |
During 2016, none of the 2011 warrants were exercised. During 2015, 478,261 of the 2011 warrants were exercised, with proceeds to the Company of $621,739. During 2014, 12,696 of the 2011 warrants were exercised, with proceeds to the Company of $16,504. The Company recognizes the change in the fair value of the warrants liability as a non-operating income or loss in the accompanying statements of operations.
| 4. |
Prepaid Expenses and Other Current Assets |
Prepaid expenses and other current assets consist of the following as of December 31:
| 2016 | 2015 | |||||||
| Prepaid research fees |
$ | 334,565 | $ | 915,194 | ||||
| Prepaid insurance |
598,909 | 436,726 | ||||||
| Prepaid pre-commercialization fees |
35,500 | 90,248 | ||||||
| Prepaid subscriptions fees |
22,770 | 26,602 | ||||||
| Prepaid rent |
19,756 | 1,252 | ||||||
| Other |
36,444 | 34,716 | ||||||
|
|
|
|
|
|||||
| Total prepaid expenses and other current assets |
$ | 1,047,944 | $ | 1,504,738 | ||||
|
|
|
|
|
|||||
| 5. |
Property and Equipment |
Property and equipment, net consists of the following as of December 31:
| 2016 | 2015 | |||||||
| Computer equipment |
$ | 27,915 | $ | 27,915 | ||||
| Furniture and equipment |
177,061 | 102,533 | ||||||
| Leasehold improvements |
152,708 | 131,175 | ||||||
|
|
|
|
|
|||||
| 357,684 | 261,623 | |||||||
| Less: Accumulated depreciation |
(113,480 | ) | (70,074 | ) | ||||
|
|
|
|
|
|||||
| Total property and equipment, net |
$ | 244,204 | $ | 191,549 | ||||
|
|
|
|
|
|||||
Depreciation expense was $43,406, $34,468, and $26,574, respectively, for the years ended December 31, 2016, 2015 and 2014.
| 6. |
Accrued Expenses and Other Liabilities |
Accrued expenses and other liabilities consist of the following as of December 31:
| 2016 | 2015 | |||||||
| Accrued preclinical and clinical trial expenses |
$ | 623,855 | $ | 332,905 | ||||
| Accrued professional fees |
102,673 | 330,490 | ||||||
| Accrued compensation and benefits |
264,237 | 894,846 | ||||||
| Accrued license fees |
152,500 | 52,500 | ||||||
| Deferred rent and lease incentive |
18,094 | 18,093 | ||||||
| Other |
— | 17,642 | ||||||
|
|
|
|
|
|||||
| Current accrued expenses and other liabilities |
1,161,359 | 1,646,476 | ||||||
| Deferred rent and lease incentive—non-current |
181,162 | 176,293 | ||||||
|
|
|
|
|
|||||
| Non-current accrued expenses and other liabilities |
181,162 | 176,293 | ||||||
|
|
|
|
|
|||||
| Total accrued expenses and other liabilities |
$ | 1,342,521 | $ | 1,822,769 | ||||
|
|
|
|
|
|||||
F-14
Table of Contents
| 7. |
Commitments and Contingencies |
The Company has contracted with drug manufacturers and other vendors, including clinical research organizations (CRO) overseeing the clinical trials of the Company’s drug candidates, to assist in the execution of the Company’s preclinical and clinical trials, analysis, and the preparation of material necessary for the submission of new drug applications (NDAs) and abbreviated new drug applications (ANDAs) with the U.S. Food and Drug Administration (FDA). The contracts are cancelable at any time, but obligate the Company to reimburse the providers for any time or costs incurred through the date of termination.
The Company has executed a non-cancellable operating lease agreement for its corporate office. The lease has free and escalating rent payment provisions. The Company recognizes rent expense under such lease on a straight-line basis over the term of the lease. As of December 31, 2016, future minimum lease payments under the operating lease agreement are as follows:
| 2017 |
$ | 207,421 | ||
| 2018 |
213,644 | |||
| 2019 |
220,053 | |||
| 2020 |
226,655 | |||
| 2021 |
233,454 | |||
| Thereafter |
220,023 | |||
|
|
|
|||
| $ | 1,321,250 | |||
|
|
|
In March 2015, the Company amended the lease to its corporate offices to obtain additional space for its expanding operations. The Company now leases approximately 5,200 square feet and the lease term now expires on 2022, although the Company has the right to terminate the lease in November 2017 based on the payment of an early termination fee. In connection with the expansion, approximately $131,000 of tenant build-out costs were funded and paid by the landlord through lease incentives. The lease incentives are being amortized over the term of the lease as a reduction of rent expense. The lease provides for fixed increases in minimum annual rent payments, as well as rent free periods. The total amount of rental payments due over the lease term is being charged to rent expense on the straight-line method over the term of the lease. The differences between rent expense recorded and the amount paid is credited or charged to accrued expenses and other liabilities in the accompanying balance sheets. Rent expense was $201,920, $129,727, and $90,163 respectively, for the years ended December 31, 2016, 2015 and 2014.
There are no obligations under capital leases.
For commitments related to the Company’s license agreements with BioMarin (defined below) and Northwestern (defined below), see Note 8.
| 8. |
Agreements |
| a. |
LICENSE AGREEMENT WITH NORTHWESTERN UNIVERSITY. On August 27, 2009, the Company entered into a license agreement with Northwestern University (Northwestern), under which it acquired worldwide rights to commercialize new GABA aminotransferase inhibitors and derivatives of vigabatrin that have been discovered by Northwestern. Under the terms of the license agreement, Northwestern granted the Company an exclusive worldwide license to certain composition of matter patents related to the new class of inhibitors and a patent application relating to derivatives of vigabatrin. The Company has identified and designated the lead compound under this license as CPP-115. |
Under the license agreement with Northwestern, the Company is responsible for continued research and development of any resulting product candidates. As of December 31, 2016, the Company has paid $411,590 in connection with the license and has accrued license fees of $152,500 and $52,500 as of December 31, 2016, and 2015, respectively, in the accompanying balance sheets for expenses, maintenance fees and milestones. In addition, the Company is obligated to pay certain milestone payments in future years relating to clinical development activities with respect to CPP-115, and royalties on any products resulting from the license agreement. The next milestone payment of $300,000 is due on the earlier of successful completion of the first Phase 3 clinical trial for CPP-115 or August 27, 2018.
F-15
Table of Contents
| 8. |
Agreements (continued) |
| b. |
LICENSE AGREEMENT WITH NEW YORK UNIVERSITY AND THE FEINSTEIN INSTITUTE FOR MEDICAL RESEARCH. On December 13, 2011, the Company entered into a license agreement with New York University (NYU) and the Feinstein Institute for Medical Research (FIMR) under which it acquired worldwide rights to commercialize GABA aminotransferase inhibitors in the treatment for Tourette syndrome. The Company is obligated to pay certain milestone payments in future years relating to clinical development activities and royalties on any products resulting from the license agreement. |
| c. |
LICENSE AGREEMENT WITH BIOMARIN. On October 26, 2012, the Company entered into a strategic collaboration with BioMarin Pharmaceutical, Inc. (BioMarin) for Firdapse® under which: (i) the Company licensed the exclusive North American rights to Firdapse® pursuant to a License Agreement, dated as of October 26, 2012 (the License Agreement) between the Company and BioMarin, and (ii) BioMarin made a $5,000,000 investment in the Company to further the development of Firdapse®. |
As part of the License Agreement, the Company agreed: (i) to pay BioMarin royalties for seven years from the first commercial sale of Firdapse® equal to 7% of net sales (as defined in the license agreement) in North America for any calendar year for sales up to $100 million, and 10% of net sales in North America in any calendar year in excess of $100 million; (ii) to pay to the third-party licensor of the rights sublicensed to the Company royalty payments for seven years from the first commercial sale of Firdapse® equal to 7% of net sales (as defined in the license agreement between BioMarin and the third-party licensor) in any calendar year; and (iii) to pay certain milestone payments that BioMarin is obligated to pay (approximately $2.6 million of which will be due upon acceptance by the FDA of a filing of an NDA for Firdapse® for the treatment of LEMS, and approximately $7.2 million of which will be due on the unconditional approval by the FDA of an NDA for Firdapse® for the treatment of LEMS). The Company also agreed to share in the cost of certain post-marketing studies being conducted by BioMarin, and, as of both December 31, 2016, and 2015, the Company had paid BioMarin $3.8 million in the aggregate, related to expenses in connection with Firdapse® studies and trials.
On April 15, 2014, effective as of April 8, 2014, the Company and BioMarin entered into Amendment No. 1 to the License Agreement, amending in certain respects the License Agreement, dated October 26, 2012. The amendment related to purchases of additional product by the Company from BioMarin, the sharing of data between the parties with respect to clinical trials and studies undertaken by each party and the payment terms for certain joint studies.
| d. |
AGREEMENTS FOR DRUG DEVELOPMENT, PRECLINICAL AND CLINICAL STUDIES. The Company has entered into agreements with contract manufacturers for the manufacture of drug and study placebo for the Company’s trials and studies, and for commercial requirements if any product is approved for commercialization, with contract research organizations (CRO) to conduct and monitor the Company’s trials and studies and with various entities for laboratories and other testing related to the Company’s trials and studies. The contractual terms of the agreements vary, but most require certain advances as well as payments based on the achievement of milestones. Further, these agreements are cancellable at any time, but obligate the Company to reimburse the providers for any time or costs incurred through the date of termination. |
| 9. |
Related Party Transactions |
During the years ended December 31, 2016, 2015 and 2014, the Company paid approximately $10,000, $10,000, and $10,000, respectively, in consulting fees to members of the Company’s Scientific Advisory Board.
F-16
Table of Contents
| 9. |
Related Party Transactions (continued) |
During 2015, the Company entered into a consulting agreement with one of its directors to serve as interim chief commercial officer during the period that the Company sought to fill that position. The consulting arrangement ended in September 2015 and the director received a total of $45,000 in consulting fees for those services.
The Company has an employment agreement with its Chief Executive Officer. Under this agreement, the CEO will receive an annual base salary of approximately $485,000 in 2017. This agreement expires in November 2018.
| 10. |
Income Taxes |
Due to the ongoing operating losses and the inability to recognize any income tax benefit, there is no provision for income taxes in any period presented in these financial statements. Since inception, the Company has only generated pretax losses.
The reconciliation of income tax expense (benefit) computed at the statutory federal income tax rate of 34% to amounts included in the statements of operations is as follows:
| 2016 | 2015 | 2014 | ||||||||||
| Statutory rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State tax |
3.4 | % | 3.6 | % | 3.7 | % | ||||||
| Valuation allowance |
(44.7 | )% | (37.7 | )% | (35.5 | )% | ||||||
| Tax credit |
6.16 | % | 0.0 | % | 0.0 | % | ||||||
| Other |
1.14 | % | 0.1 | % | (2.2 | )% | ||||||
|
|
|
|
|
|
|
|||||||
| 0.0 | % | 0.0 | % | 0.0 | % | |||||||
|
|
|
|
|
|
|
|||||||
Deferred tax assets and liabilities reflect the net tax effects of net operating loss and tax credit carryovers and the temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2016 and 2015 are as follows:
| 2016 | 2015 | |||||||
| Net operating loss |
$ | 21,050,217 | $ | 18,684,232 | ||||
| Start-up cost |
12,823,113 | 9,997,709 | ||||||
| Tax credits |
6,960,838 | 3,167,766 | ||||||
| Deferred compensation |
1,555,425 | 2,368,352 | ||||||
| Other |
207,133 | 90,705 | ||||||
|
|
|
|
|
|||||
| Gross deferred tax asset |
42,596,726 | 34,308,764 | ||||||
| Valuation allowance |
(42,596,726 | ) | (34,308,764 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | 0 | $ | 0 | ||||
|
|
|
|
|
|||||
The Company’s deferred tax assets have been fully offset by a valuation allowance at December 31, 2016 and 2015 because the Company believes that it is more likely than not that the deferred tax asset will not be realized. The increase in the valuation allowance on the deferred tax assets was $8,287,962 and $9,413,647 for the years ended December 31, 2016 and 2015, respectively.
At December 31, 2016 and 2015, respectively, the Company had net operating loss carryforwards of approximately $56.3 million and $49.9 million available to reduce future taxable income, if any. The net operating loss carryforwards will expire at various dates beginning in 2023 and ending in 2036. If an ownership change, as defined under Internal Revenue Code Section 382, occurs, the use of these carry-forwards may be subject to limitation. The effective tax rate of 0% in all periods presented differs from the statutory rate of 34% due to the valuation allowance and because the Company had no taxable income.
F-17
Table of Contents
| 10. |
Income Taxes (continued) |
Beginning in 2010, the Company has received several orphan drug designations by the FDA for products currently under development. The orphan drug designations allow the Company to claim increased federal tax credits for certain research and development activities.
No interest or penalties were accrued through December 31, 2016. The Company’s policy is to recognize any related interest or penalties in income tax expense. The Company is not subject to U.S federal, state and local tax examinations by tax authorities for any years before 2013. The Company is not currently under income tax examinations by any tax authorities.
| 11. |
Stockholders’ Equity |
Preferred Stock
The Company has 5,000,000 shares of authorized preferred stock, $0.001 par value per share at December 31, 2016 and 2015. No shares of preferred stock were outstanding at December 31, 2016 and 2015.
Common Stock
During 2015, the Company’s stockholders approved an increase in the Company’s authorized common stock par value $0.001 per share, from 100,000,000 shares to 150,000,000 shares. At December 31, 2016 and 2015, 82,972,316 and 82,850,619 shares, respectively, of common stock were issued and outstanding. Each holder of common stock is entitled to one vote of each share of common stock held of record on all matters on which stockholders generally are entitled to vote.
2014 Shelf Registration Statement
On January 31, 2014, the Company filed a Shelf Registration Statement on Form S-3 (the 2014 Shelf Registration Statement) with the U.S. Securities and Exchange Commission (SEC) to sell up to $100 million of common stock. This registration statement (file No. 333-193699) was declared effective by the SEC on March 19, 2014. Approximately $33.8 million remains available under the 2014 Shelf Registration Statement, which will expire on March 19, 2017. The Company has to date conducted the following sales of its securities under the 2014 Shelf Registration Statement:
| (a) |
On April 3, 2014, the Company filed a prospectus supplement and offered for sale 13,023,750 shares of its common stock at a price of $2.21 per share in an underwritten public offering. The Company received gross proceeds in the public offering of approximately $28.8 million before underwriting commission and incurred expenses of approximately $2.1 million. |
| (b) |
On February 4, 2015, the Company filed a prospectus supplement and offered for sale 11,500,000 shares of its common stock at a price of $3.25 per share in an underwritten public offering. The Company received gross proceeds in the public offering of approximately $37.4 million before underwriting commission and incurred expenses of approximately $2.5 million. |
As of December 31, 2016, there is approximately $33.8 million available for future sale under the 2014 Shelf Registration Statement.
F-18
Table of Contents
| 11. |
Stockholders’ Equity (continued) |
2016 Shelf Registration Statement
On December 23, 2016, the Company filed a Shelf Registration Statement on Form S-3 (the 2016 Shelf Registration Statement) with the SEC to sell up to approximately $33.8 million of common stock. The 2016 Shelf Registration Statement was filed to continue the registration of the remaining unsold allotment under the 2014 Shelf Registration Statement beyond its expiration date. The 2016 Shelf Registration Statement (file No. 333-215315) was declared effective by the SEC on January 9, 2017, and, now that the 2016 Shelf Registration Statement has become effective, the Company does not intend to issue any additional shares under the 2014 Shelf Registration Statement.
If the Company’s public float (the market value of its common stock held by non-affiliate stockholders) were to fall below $75 million, the Company would be subject to a further limitation under which it could sell no more than one-third (1/3) of its public float during any 12-month period. Further, the number of shares that the Company can sell at any one time may be limited under certain circumstances to 20% of the outstanding common stock under applicable NASDAQ marketplace rules.
Warrant Exercises
No warrants were exercised during the year ended December 31, 2016. For the years ended December 31, 2015, and 2014, the Company issued an aggregate of 1,178,261 and 1,262,296 shares of its authorized but unissued common stock upon the exercise of previously issued common stock purchase warrants, raising gross proceeds of $1,895,738 and $1,316,503, respectively.
Stockholder Rights Plan
On September 20, 2011, the Board of Directors approved the Company’s adoption of a Stockholder Rights Plan. Under the Plan, a dividend of one preferred share purchase right (a Right) was declared for each share of common stock of the Company that was outstanding on October 7, 2011. Each Right entitles the holder to purchase from the Company one one-hundredth of a share of Series A Junior Preferred Stock at a purchase price of $7.80, subject to adjustment.
The Rights trade automatically with the common stock and will not be exercisable until a person or group has become an “acquiring person” by acquiring 17.5% or more of the Company’s outstanding common stock, or a person or group commences, or publicly announces a tender offer that will result in such a person or group owning 17.5% or more of the Company’s outstanding common stock. Upon announcement that any person or group has become an acquiring person, each Right will entitle all rightholders (other than the acquiring person) to purchase, for the exercise price of $7.80, a number of shares of the Company’s common stock having a market value equal to twice the exercise price. Rightholders would also be entitled to purchase common stock of the acquiring person having a value of twice the exercise price if, after a person had become an acquiring person, the Company were to enter into certain mergers or other transactions. If any person becomes an acquiring person, the Board of Directors may, at its option and subject to certain limitations, exchange one share of common stock for each Right.
The Rights have certain anti-takeover effects, in that they would cause substantial dilution to a person or group that attempts to acquire a significant interest in the Company on terms not approved by the Board of Directors. In the event that the Board of Directors determines a transaction to be in the best interests of the Company and its stockholders, the Board of Directors may redeem the Rights for $0.001 per share at any time prior to a person or group becoming an acquiring person.
On September 19, 2016, the Board of Directors unanimously approved, and on the same date the Company entered into Amendment No. 1 to the Stockholders Rights Plan (the “Amendment”). Under the terms of the Amendment, the outside expiration date of the rights plan has been extended from September 20, 2016 to September 20, 2019. Additionally, as part of the Amendment, the Board adopted a Certificate of Designation, Preferences and Rights of Series A Junior Participating Preferred Stock of the Company to increase the number of shares of Series A Junior Participating Preferred Stock of the Company available for issuance under the Rights Plan from 500,000 shares to 1.5 million shares.
F-19
Table of Contents
| 12. |
Stock Compensation Plans |
Stock Options
The Company may issue stock options, restricted stock, stock appreciation rights and restricted stock units (collectively, the “Awards”) to employees, directors, consultants and scientific advisors of the Company under the 2006 and 2014 Stock Incentive Plans (the 2006 Plan and the 2014 Plan or collectively, the Plans). At December 31, 2016, no shares remain available for future issuance under the 2006 Plan. Under the 2014 Plan, 6,000,000 shares were reserved for issuance and as of December 31, 2016, 2,076,667 shares remain available for future issuance.
The Company has granted stock options to employees, officers, directors, scientific advisors and consultants generally at exercise prices equal to the market price of the common stock at grant date. Option awards generally vest over a period of 1 to 3 years of continuous service and have contractual terms from 5 to 7 years. Certain awards provide for accelerated vesting if there is a change in control. The Company issues new shares as shares are required to be delivered upon exercise of outstanding stock options.
During the years ended December 31, 2016, 2015, and 2014, options to purchase 75,000, 265,000, and 580,000 shares of the Company’s common stock were exercised with gross proceeds to the Company of $80,251, $390,351, and $522,000, respectively. Further, during the years ended December 31, 2016, 2015, and 2014, options to purchase 50,000, 984,608, and 185,000 shares of the Company’s common stock were exercised on a “cashless” basis, resulting in the issuance of an aggregate of 20,030, 761,600, and 119,709 shares of the Company’s common stock, respectively.
During the years ended December 31, 2016, 2015 and 2014 the Company recorded non-cash stock-based compensation expense related to stock options totaling $1,760,591, $1,510,018, and $767,838, respectively.
During the years ended December 31, 2016 and 2015, the Company granted seven-year options to purchase an aggregate of 1,285,000 and 1,760,000 shares, respectively, of the Company’s common stock to certain of the Company’s officers, employees, directors, and consultants. During the year ended December 31, 2014, the Company granted five and seven-year options to purchase an aggregate of 1,305,000 shares of the Company’s common stock to certain of the Company’s officers, employees, directors and consultants.
Stock option activity under the Company’s Plans for the year ended December 31, 2016 is summarized as follows:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at beginning of year |
4,250,000 | $ | 2.23 | |||||||||||||
| Granted |
1,285,000 | 0.94 | ||||||||||||||
| Exercised |
(125,000 | ) | 0.83 | |||||||||||||
| Forfeited or cancelled |
(360,000 | ) | 3.06 | |||||||||||||
| Expired |
(390,000 | ) | 1.07 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding at end of year |
4,660,000 | $ | 1.94 | 4.66 | $ | 805,900 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable at end of year |
2,386,663 | $ | 2.02 | 3.44 | $ | 515,500 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
F-20
Table of Contents
| 12. |
Stock Compensation Plans (continued) |
Other information pertaining to stock option activity during the years ended December 31, 2016, 2015 and 2014 was as follows:
| 2016 | 2015 | 2014 | ||||||||||
| Weighted–average fair value of granted stock options |
$ | 0.62 | $ | 2.13 | $ | 2.41 | ||||||
| Total fair value of vested stock options |
$ | 1,634,562 | $ | 1,307,895 | $ | 409,476 | ||||||
| Total intrinsic value of exercised stock options |
$ | 42,000 | $ | 3,311,599 | $ | 1,339,100 | ||||||
The following table summarizes information about the Company’s options outstanding at December 31, 2016:
| Options Outstanding | Options Exercisable | |||||||||||||||||||||||
| Range of |
Number Outstanding |
Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
Number Exercisable |
Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
||||||||||||||||||
| $0.47 to $0.63 |
900,000 | 0.95 | $ | 0.47 | 875,000 | 0.95 | $ | 0.47 | ||||||||||||||||
| $0.64 to $0.82 |
1,060,000 | 6.45 | $ | 0.79 | — | — | — | |||||||||||||||||
| $0.83 to $2.67 |
1,260,000 | 5.65 | $ | 2.38 | 603,333 | 5.25 | $ | 2.41 | ||||||||||||||||
| $2.68 to $3.23 |
1,035,000 | 4.67 | $ | 3.11 | 751,665 | 4.67 | $ | 3.11 | ||||||||||||||||
| $3.24 to $4.64 |
405,000 | 5.03 | $ | 3.87 | 156,665 | 4.43 | $ | 3.94 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| 4,660,000 | 4.66 | $ | 1.94 | 2,386,663 | 3.44 | $ | 2.02 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
As of December 31, 2016, there was approximately $2,320,289 of unrecognized compensation expense related to non-vested stock option awards granted under the Plans. That cost is expected to be recognized over a weighted average period of approximately 4.66 years.
The Company utilizes the Black-Scholes option-pricing model to determine the fair value of stock options on the date of grant. This model derives the fair value of stock options based on certain assumptions related to the expected stock price volatility, expected option life, risk-free interest rate and dividend yield. Expected volatility is based on reviews of historical volatility of the Company’s common stock. The estimated expected option life is based upon estimated employee exercise patterns and considers whether and the extent to which the options are in-the-money. The Company estimates the expected option life for options granted to employees and directors based upon the simplified method. Under this method, the expected life is presumed to be the mid-point between the vesting date and the end of the contractual term. The Company will continue to use the simplified method until it has sufficient historical exercise data to estimate the expected life of the options. The risk-free interest rate assumption is based upon the U.S. Treasury yield curve appropriate for the estimated life of the stock options awards. The expected dividend rate is zero. Stock–based compensation expense also includes an estimate, which the Company makes at grant date, of the number of awards that are expected to be forfeited. The Company revises this estimate in subsequent periods if actual forfeitures differ from those estimates.
Assumptions used during the years were as follows:
| Year ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Risk free interest rate |
0.76% to 2.15 | % | 1.00% to 2.13 | % | 1.18% to 2.03 | % | ||||||
| Expected term |
2 to 6 years | 3 to 7 years | 3 to 7 years | |||||||||
| Expected volatility |
100 | % | 102 | % | 115 | % | ||||||
| Expected dividend yield |
— | % | — | % | — | % | ||||||
| Expected forfeiture rate |
— | % | — | % | — | % | ||||||
F-21
Table of Contents
| 12. |
Stock Compensation Plans (continued) |
Restricted Stock Units
Under the 2014 Plan, participants may be granted restricted stock units, each of which represents a conditional right to receive shares of common stock in the future. The restricted stock units granted under this plan generally vest ratably over a three to four-year period. Upon vesting, the restricted stock units will convert into an equivalent number of shares of common stock. The amount of expense relating to the restricted stock units is based on the closing market price of the Company’s common stock on the date of grant and is amortized on a straight-line basis over the requisite service period. Restricted stock unit activity during 2016, 2015, and 2014 was as follows:
| 2016 | 2015 | 2014 | ||||||||||||||||||||||
| Number of Restricted Stock Units |
Weighted Average Grant Date Fair Value |
Number of Restricted Stock Units |
Weighted Average Grant Date Fair Value |
Number of Restricted Stock Units |
Weighted Average Grant Date Fair Value |
|||||||||||||||||||
| Nonvested balance at beginning of year |
53,334 | $ | 2.83 | 80,000 | $ | 2.83 | — | $ | — | |||||||||||||||
| Granted |
— | — | — | — | 80,000 | — | ||||||||||||||||||
| Vested |
(26,667 | ) | 2.83 | (26,666 | ) | 2.83 | — | 2.83 | ||||||||||||||||
| Forfeited |
— | — | — | — | — | — | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Nonvested balance at end of year |
26,667 | $ | 2.83 | 53,334 | $ | 2.83 | 80,000 | $ | 2.83 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
During the years ended December 31, 2016, 2015 and 2014, the Company recorded non-cash stock-based compensation expense related to restricted stock units totaling $75,494, $75,440, and $10,131, respectively.
| 13. |
Benefit Plan |
The Company maintains an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code covering all eligible employees. Subject to certain dollar limits, eligible employees may contribute up to 15% of their pre-tax annual compensation to the plan. The Company has elected to make discretionary matching contributions of employee contributions up to 4% of an employee’s gross salary. For the years ended December 31, 2016, 2015 and 2014, the Company’s matching contributions were approximately $59,000, $69,000 and $44,000, respectively.
F-22
Table of Contents
| 14. |
Quarterly Financial Information (unaudited) |
The following table presents unaudited supplemental quarterly financial information for the years ended December 31, 2016 and 2015:
| Quarter Ended | ||||||||||||||||
| March 31, 2016 |
June 30, 2016 |
September 30, 2016 |
December 31, 2016 |
|||||||||||||
| Revenues |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Loss from operations |
(6,237,536 | ) | (4,814,452 | ) | (3,914,014 | ) | (4,314,199 | ) | ||||||||
| Change in fair value of warrants liability |
733,356 | 152,783 | (106,948 | ) | 106,946 | |||||||||||
| Net loss |
$ | (5,386,237 | ) | $ | (4,568,914 | ) | $ | (3,953,981 | ) | $ | (4,163,320 | ) | ||||
| Loss per share—basic and diluted |
$ | (0.07 | ) | $ | (0.06 | ) | $ | (0.05 | ) | $ | (0.05 | ) | ||||
| Quarter Ended | ||||||||||||||||
| March 31, 2015 |
June 30, 2015 |
September 30, 2015 |
December 31, 2015 |
|||||||||||||
| Revenues |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Loss from operations |
(4,291,915 | ) | (4,897,330 | ) | (5,017,428 | ) | (6,191,679 | ) | ||||||||
| Change in fair value of warrants liability |
(1,180,278 | ) | 333,956 | 521,731 | 389,596 | |||||||||||
| Net loss |
$ | (5,410,259 | ) | $ | (4,558,503 | ) | $ | (4,449,038 | ) | $ | (5,815,158 | ) | ||||
| Loss per share—basic and diluted |
$ | (0.07 | ) | $ | (0.06 | ) | $ | (0.05 | ) | $ | (0.07 | ) | ||||
Quarterly basic and diluted net loss per common share were computed independently for each quarter and do not necessarily total to the full year basic and diluted net loss per common share.
F-23