Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex312_12.htm |
| EX-32.2 - EX-32.2 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex322_7.htm |
| EX-32.1 - EX-32.1 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex321_11.htm |
| EX-31.1 - EX-31.1 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex311_9.htm |
| EX-23.1 - EX-23.1 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex231_10.htm |
| EX-10.40 - EX-10.40 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex1040_304.htm |
| EX-10.39 - EX-10.39 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex1039_305.htm |
| EX-10.38 - EX-10.38 - MELINTA THERAPEUTICS, INC. /NEW/ | cemp-ex1038_303.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from to
Commission File Number: 001-35405
CEMPRA, INC.
(Exact name of registrant specified in its charter)
|
Delaware |
2834 |
45-4440364 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
6320 Quadrangle Drive, Suite 360
Chapel Hill, NC 27517
(Address of Principal Executive Offices)
(919) 313-6601
(Telephone Number, Including Area Code)
Securities Registered Pursuant to Section 12(b) of the Exchange Act:
|
Title of Each Class
|
Name of Exchange on which Registered
|
|
Common Stock, $0.001 Par Value |
Nasdaq Global Market |
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
☒ |
Accelerated filer |
☐ |
|
|
|
|
|
|
Non-accelerated filer |
☐ (Do not check if a smaller reporting company) |
Smaller reporting company |
☐ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting stock held by non-affiliates of the registrant, as of June 30, 2016, was approximately $655.8 million. Such aggregate market value was computed by reference to the closing price of the common stock as reported on the Nasdaq Global Market on June 30, 2016. For purposes of making this calculation only, the registrant has defined affiliates as including only directors and executive officers and shareholders holding greater than 10% of the voting stock of the registrant as of June 30, 2016.
As of February 24, 2017 there were 52,392,905 shares of the registrant’s common stock, $0.001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant’s definitive Proxy Statement for its 2016 Annual Meeting of Stockholders are incorporated herein by reference, as indicated in Part III.
TABLE OF CONTENTS
|
|
|
|
Page |
||
|
|
|
1 |
|||
|
|
|
|
|
||
|
Item 1. |
|
1 |
|||
|
|
|
|
|
||
|
Item 1A. |
|
27 |
|||
|
|
|
|
|
||
|
Item 1B. |
|
58 |
|||
|
|
|
|
|
||
|
Item 2. |
|
58 |
|||
|
|
|
|
|
||
|
Item 3. |
|
58 |
|||
|
|
|
|
|
||
|
Item 4. |
|
58 |
|||
|
|
|
|
|
||
|
|
|
59 |
|||
|
|
|
|
|
||
|
Item 5. |
|
59 |
|||
|
|
|
|
|
||
|
Item 6. |
|
60 |
|||
|
|
|
|
|
||
|
Item 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operation |
61 |
||
|
|
|
|
|
||
|
Item 7A. |
|
75 |
|||
|
|
|
|
|
||
|
Item 8. |
|
76 |
|||
|
|
|
|
|
||
|
Item 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
76 |
||
|
|
|
|
|
||
|
Item 9A. |
|
76 |
|||
|
|
|
|
|
||
|
Item 9B. |
|
76 |
|||
|
|
|
|
|
||
|
|
|
77 |
|||
|
|
|
|
|
||
|
Item 10. |
|
77 |
|||
|
|
|
|
|
||
|
Item 11. |
|
78 |
|||
|
|
|
|
|
||
|
Item 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
79 |
||
|
|
|
|
|
||
|
Item 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
79 |
||
|
|
|
|
|
||
|
Item 14. |
|
79 |
|||
|
|
|
|
|
||
|
|
|
80 |
|||
|
|
|
|
|
||
|
Item 15. |
|
80 |
|||
|
|
|
||||
|
F-1 |
|||||
i
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. These forward-looking statements are subject to risks and uncertainties, including those set forth under “Item 1A. Risk Factors” and “Cautionary Statement” included in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report, that could cause actual results to differ materially from historical results or anticipated results.
Summary
We are a clinical-stage pharmaceutical company focused on developing differentiated anti-infectives for the acute care and community settings to meet critical medical needs in the treatment of infectious diseases. Our lead product, solithromycin, is being developed in oral capsules, intravenous, or IV, and suspension formulations, for the treatment of community-acquired bacterial pneumonia, or CABP, one of the most serious infections of the respiratory tract in adults and children, as well as for ophthalmic infections and other indications. We have applications under review for the IV and oral capsule formulations of solithromycin to treat CABP in adults, with the U.S. Food and Drug Administration, or FDA, and the European Medicines Agency, or EMA. In December 2016 we received a Complete Response Letter, or CRL, related to our New Drug Applications, or NDAs, for oral and IV solithromycin seeking additional clinical and manufacturing data prior to potential approval.
Despite many available antibiotics and the significant size of the market for antibiotics for CABP and other infections, we believe this market has critical unmet needs. The effectiveness of many antibiotics has declined worldwide due to increased bacterial resistance to currently available antibiotics. According to the Centers for Disease Control and Prevention, or CDC, macrolide resistance to Streptococcus pneumoniae, the pathogen that most frequently causes CABP, now averages 49% across the United States. These macrolide resistance rates exceed 90% in parts of Asia. According to the CDC, pneumonia is the most frequent cause of death from an infection in the United States.
We believe that solithromycin could be an effective antibiotic for CABP that addresses the current challenges posed by macrolide resistance.
Our second product, fusidic acid, is an antibiotic that has been used for decades outside the U.S., including in Western Europe, but has never been approved in the U.S. We have recently completed a successful Phase 3 study evaluating fusidic acid as an oral treatment of acute bacterial skin and skin structure infections, or ABSSSI, which are frequently caused by methicillin-resistant Staphylococcus aureus, or MRSA. We are exploring fusidic acid for the long-term oral treatment of refractory bone and joint infections, or BJI, including prosthetic joint infections, or PJI, caused by staphylococci, including S. aureus and MRSA. Currently, there is no optimal oral, chronic antibiotic for treating these infections.
Overview of Solithromycin
Solithromycin is a potent new fourth generation macrolide, the first fluoroketolide. We believe solithromycin is differentiated from other antibiotics because of its broad therapeutic potential and activity targeting pathogenic bacteria. Broad therapeutic potential means a drug that can be used in the treatment of several disease indications, such as bacterial pneumonia, cystic fibrosis, other respiratory tract infections, infections in children, gonorrhea, Helicobacter gastritis, eye infections, infections in pregnancy, and others. Targeting the bacteria responsible for CABP and other respiratory infections without causing major effects on the anaerobic gram-negative intestinal microflora is a benefit of the macrolide class.
We believe solithromycin’s potency comes from its unique chemical structure, which we believe provides greater ability to fight resistant bacteria. Increasingly, resistance is a major threat to the efficacy of currently available antibiotics, including currently approved macrolides. In our clinical and preclinical trials, incidence of resistance to CABP pathogens was rare, with no resistance seen to S. pneumoniae, the pathogen that most frequently causes CABP.
Our preclinical studies demonstrate that solithromycin has excellent organ and tissue distribution and intracellular activity, which allows it to reach bacteria at body sites that other antibiotics may not. Solithromycin is active against most CABP pathogens, including pneumococcal strains resistant to other macrolides. Our proposed treatment for CABP is a five to seven-day treatment regimen designed to maximize efficacy against CABP while minimizing toxicities, including well-described liver effects seen with other macrolides and solithromycin, and to support antibiotic stewardship measures by enabling the effective treatment of CABP with a single therapy. Solithromycin offers flexibility of dosing, whether IV, oral capsule, or oral suspension which we believe will be
1
attractive to both physicians and patients. These attributes of solithromycin make it a possible treatment for all age groups, including children.
Treatment guidelines from the Infectious Disease Society of America, or IDSA, recommend that outpatient CABP be treated with a macrolide, such as azithromycin, or with a fluoroquinolone, such as levofloxacin, or with the combination of a beta-lactam antibiotic, such as amoxicillin with a macrolide antibiotic. Macrolides are frequently used as first-line therapy as they target respiratory pathogens, reach high concentrations in the lung and in macrophages at the site of infection, and have anti-inflammatory properties that contribute to the patient’s recovery. However, in the last decade, CABP treatment failure due to pneumococcal macrolide resistance has led to more frequent use of fluoroquinolones, which have recognized serious side effects. Despite these side effects with fluoroquinolones, the need to treat CABP patients with an antibiotic that does not have resistance has driven increased use of levofloxacin (a fluoroquinolone).
There has not been a new IV and oral antibiotic approved in the U.S. to treat CABP in outpatients since 1999. Many of the antibiotics currently used to treat serious infections are difficult or inconvenient to administer, often requiring hospitalization for IV treatment. Many currently available IV antibiotics that are the standard of care have no oral formulations to allow discharge from the hospital on the same medication, forcing patients to switch to less favorable alternatives, and often another class of antibiotics, which can further promote the development of antibiotic resistance. Additionally, currently used cephalosporins and fluoroquinolones have broad spectrum activity, and eliminate essential intestinal microflora, which can result in serious consequences, such as C. difficile enterocolitis. In order to improve stewardship of antibiotics, and to provide more appropriate care for CABP patients that better aligns with IDSA treatment guidelines, we believe a new macrolide is needed.
Solithromycin Regulatory Status
In the second quarter of 2016, we submitted NDAs to the FDA seeking the approval of oral and IV solithromycin in adults for the treatment of CABP. The NDAs contained chemistry, manufacturing and controls, or CMC, information, and data from our preclinical and clinical development program, including data from two Phase 3 trials of solithromycin. The solithromycin development program was structured based on feedback and direct dialogue with the FDA and guidance published by the FDA. Because the FDA had designated each of oral and intravenous solithromycin as a Qualified Infectious Disease Product, or QIDP, for the indication of CABP, our solithromycin NDAs received a priority review in eight months.
In November 2016, a majority of the FDA’s Antimicrobial Drugs Advisory Committee, or AMDAC, voted (7-6) that efficacy results of solithromycin outweighed the risks for the treatment of CABP. Members of AMDAC voted unanimously (13-0) that there was substantial evidence of the efficacy of solithromycin for CABP. The AMDAC also voted (12-1) that the risk of hepatotoxicity with solithromycin had not been adequately characterized and discussed a variety of potential approaches to further characterize the existing liver safety information on solithromycin.
In December 2016, we received a CRL from the FDA on our NDAs. The CRL stated that the FDA could not approve the NDAs in their present form and noted that additional clinical safety information and the satisfactory resolution of manufacturing facility inspection deficiencies were required before the NDAs may be approved. The FDA noted the size of the safety database is limited to 920 patients who received solithromycin at the proposed dose and duration, and is too small to adequately characterize the nature and frequency of serious hepatic adverse effects. To address this deficiency, the FDA recommended a comparative study to evaluate the safety of solithromycin in patients with CABP. Specifically, the CRL recommended that we consider a study of approximately 9,000 patients exposed to solithromycin to enable exclusion of serious drug induced liver injury, or DILI, events occurring at a rate of approximately 1:3000 with a 95 percent probability. The CRL noted that while the FDA reserves comment on the proposed labeling until the NDAs are otherwise adequate, even in the absence of a case of Hy’s Law or of another form of serious DILI in future studies, labeling will need to include adequate information about the potential for hepatotoxicity, limiting use to patients who have limited therapeutic options and limitations regarding duration of therapy. A comprehensive plan for post-marketing safety assessment including an enhanced pharmacovigilance program would also be required. The FDA did not request any further information on solithromycin efficacy for CABP in the CRL.
In late February, we requested, and attended, a meeting with the FDA to discuss the issues identified in the CRL, including potential approaches to address the FDA’s request for additional clinical safety information, and the steps necessary to resolve the manufacturing facility inspection deficiencies noted in the CRL. At the meeting, the FDA reiterated their request for additional safety data prior to approval. Based on input from the FDA at the meeting, we are developing a protocol that will propose including fewer than 9,000 patients at the time we respond to the CRL, and will propose to deliver data from defined cohorts as the study progresses. We plan to discuss the protocol with the FDA to determine if it could support an initial approval in a limited group of patients with an urgent unmet need, while we continue to accumulate a larger post-approval safety database to support potential label expansions into broader CABP populations. If we and FDA agree on a protocol, we plan to seek non-dilutive funding to support the execution of the study.
2
In the second quarter of 2016, we completed our submission of a Marketing Authorization Application, or MAA, for IV and oral solithromycin to the EMA for the treatment of CABP. The EMA validated the MAA and began their formal review in the third quarter of 2016. In the fourth quarter of 2016, we received day 120 questions from the EMA and we are preparing responses to them. The EMA has suggested a post-approval clinical safety study, or PASS, in the EU and, we believe the EMA will request additional data prior to approval. We are currently assessing the path forward with the EMA that we believe would be most likely to achieve a positive benefit/risk assessment from them.
Solithromycin for CABP in Adults
We have completed two pivotal Phase 3 trials for solithromycin to treat CABP in adults, which were designed based on FDA guidance documents and comments from the FDA. We conducted the SOLITAIRE-ORAL trial to evaluate the treatment of CABP in adults with oral solithromycin and the SOLITAIRE-IV trial to evaluate the treatment of CABP in adults with IV solithromycin progressing to oral solithromycin. Both trials were randomized, double-blinded studies comparing solithromycin to a respiratory fluoroquinolone, moxifloxacin, and both studies demonstrated non-inferiority, or NI, for efficacy which means solithromycin was no worse than the comparator drug within a pre-defined statistical margin.
SOLITAIRE-ORAL was an active-controlled, global, multi-center trial that enrolled 860 adult patients with moderate to moderately severe CABP (pneumonia of PORT Class II, III and IV severity classification). Enrollment of PORT Class II pneumonia patients was limited to 50% of the study population. Patients were randomized to receive either oral solithromycin, as an 800 mg loading dose followed by 400 mg, once daily for a total of five days, while oral moxifloxacin was dosed at 400 mg once daily for seven days. The primary objective was demonstration of NI of early clinical response, or ECR, at 72 (-12/+36) hours, as specified by FDA guidance, defined as having improvement in at least two of the following four symptoms (without worsening of any); cough, shortness of breath, chest pain and sputum production in the ITT population. The study was designed to provide 90% power to demonstrate NI in ECR rate for solithromycin versus moxifloxacin utilizing a 10% NI margin. Secondary endpoints included the clinical success rate at the short term follow up visit 5 to 10 days following the last dose of study drug in the ITT and CE populations, the microbial ITT population, and a comparison of safety and tolerability of solithromycin compared to moxifloxacin.
As noted in the table below, in SOLITAIRE-ORAL, solithromycin met the primary objective of statistical NI (10% non-inferiority margin) of the ECR at 72 (-12/+36) hours after initiation of therapy compared to moxifloxacin in the ITT population. Solithromycin also met the secondary objectives of NI in clinical success at the short term follow up, or SFU, visit, 5-10 days after the end of therapy, both in the ITT and CE populations.
|
SOLITAIRE-ORAL Phase 3 Trial |
|
|
|
|
|
|
|
|
|
|
|
Early Clinical Response in ITT Populations (& Subgroups) |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
Moxifloxacin |
|
|
|
|
|
|
|
Population |
|
Success Rate % |
|
Success Rate % |
|
Difference |
|
|
95% CI |
|
|
ECR-ITT |
|
78.2 |
|
77.9 |
|
|
0.29 |
|
|
(-5.5, 6.1) |
|
ECR-PORT I/II |
|
80.5 |
|
80.7 |
|
|
-0.24 |
|
|
(-8.2, 7.7) |
|
ECR-PORT III/IV |
|
75.9 |
|
74.9 |
|
|
1.04 |
|
|
(-7.6, 9.7) |
3
In SOLITAIRE-ORAL, serious adverse events, or SAEs, occurred with equal frequency in both arms (<7% of patients) and no SAEs were considered study drug related. No patient in either arm of the study had treatment-emergent concomitant ALT and bilirubin elevation meeting Hy’s Law criteria. Overall, rates of AEs were comparable across treatment arms, while rates of Grade 3 ALT elevation (but not Grade 4 ALT) were higher in the solithromycin arm.
|
SOLITAIRE-ORAL Phase 3 Trial |
|
|
|
|
|
|
|
|
|
Adverse Events |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
|
Moxifloxacin |
|
||
|
|
|
800/400 mg QD |
|
|
400mg QD |
|
||
|
|
|
(n=424) |
|
|
(n=432) |
|
||
|
Headache |
|
|
4.5% |
|
|
|
2.5% |
|
|
Diarrhea* |
|
|
4.2% |
|
|
|
6.5% |
|
|
Nausea |
|
|
3.5% |
|
|
|
3.9% |
|
|
Emesis |
|
|
2.4% |
|
|
|
2.3% |
|
|
Dizziness |
|
|
2.1% |
|
|
|
1.6% |
|
|
ALTs**- Grade 3 |
|
|
4.6% |
|
|
|
2.1% |
|
|
Grade 4 |
|
|
0.5% |
|
|
|
1.2% |
|
|
|
|
|
|
|
|
|
|
|
|
* Not included in the diarrhea definition are 2 patients with C. difficile associated diarrhea, both of whom received moxifloxacin |
|
|||||||
|
** No patient in either arm of the study developed treatment emergent elevation of both ALT and bilirubin as defined by Hy's Law criteria. Observed ALT elevations reversible and asymptomatic. |
|
|||||||
SOLITAIRE-IV was an active-controlled, global, multi-center trial that enrolled 863 patients with moderate to moderately severe CABP (pneumonia of PORT Class II, III and IV severity classification). Enrollment of PORT Class II pneumonia patients was limited to 25% of the study population and 25% of the population were PORT IV patients. Patients were randomized to receive either intravenous solithromycin or moxifloxacin at a daily dose of 400 mg once each day for seven days with the ability for the physician to switch to oral solithromycin or oral moxifloxacin, at their discretion once pre-defined switch criteria were met, to complete the seven-day course of therapy. When switching to oral solithromycin, the first day of the switch was a loading dose of 800 mg followed by a 400 mg dose once a day for the remainder of the seven-day course. Patients randomized to moxifloxacin could be switched to 400 mg oral moxifloxacin once a day for the remainder of the seven-day course of treatment.
The primary objective was demonstration of NI of ECR at 72 (-12/+36) hours, as specified by FDA guidance, defined as having improvement in at least two of the following four symptoms (without worsening of any) in ITT population: cough, shortness of breath, chest pain and sputum production. The study was designed to provide 90% power to demonstrate NI in ECR rate for solithromycin versus moxifloxacin utilizing a 10% NI margin. Secondary endpoints included the clinical success rate at the SFU visit five to 10 days following the last dose of study drug in the ITT and CE populations, and a comparison of safety and tolerability of solithromycin compared to moxifloxacin. The pooled mITT population from both the oral and IV trials was also a co-primary endpoint for the oral Phase 3 trial and a secondary endpoint for the IV Phase 3 trial.
As noted in the table below, in SOLITAIRE-IV, solithromycin met all pre-defined endpoints for the FDA. In the ITT population (all randomized patients), solithromycin met the FDA primary objective of statistical NI (10% non-inferiority margin) compared to moxifloxacin at ECR at 72 [-12/+36] hours after initiation of therapy.
|
SOLITAIRE-IV Phase 3 Trial |
|
|
|
|
|
|
|
|
|
|
|
Early Clinical Response in ITT Populations |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
Moxifloxacin |
|
|
|
|
|
|
|
Population |
|
Success Rate % |
|
Success Rate % |
|
Difference |
|
|
95% CI |
|
|
ECR-ITT Population |
|
79.3 |
|
79.7 |
|
|
-0.46 |
|
|
(-6.1, 5.2) |
|
ECR-mITT Pooled P3 Studies (Co-1 objective) |
|
77.2 |
|
78.9 |
|
|
-1.70 |
|
|
(-7.4, 4.2) |
|
ECR-mITT* (2° objective) |
|
80.3 |
|
79.1 |
|
|
1.26 |
|
|
(-8.1, 10.6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
* mITT: microbiological ITT population, comprised of those patients in ITT population, in whom a pathogen was identified |
||||||||||
4
Additional secondary endpoints evaluated solithromycin at SFU five to 10 days after therapy in both the ITT and CE populations. The primary endpoint for the EMA was NI in the ITT-SFU and the CE-SFU populations limited to patients with PORT III/IV CABP at the SFU time point assessment of clinical success.
Clinical success rates as determined by investigators at the SFU visit were high for both the solithromycin and moxifloxacin groups in the ITT population. Clinical success rates were also high in the CE-SFU population for both the solithromycin and moxifloxacin groups, however, this CE outcome was skewed in favor of moxifloxacin by a blinded drug distribution delay which led to discontinuation of study drug, not related to safety or efficacy, in solithromycin patients only. Censoring these five patients, all of which fell on the solithromycin side, results in point estimates for CE population success at the SFU visit for solithromycin patients.
A total of 30 (6.9%) patients in the solithromycin group and 23 (5.4%) patients in the moxifloxacin group reported an SAE. Most of the SAEs were attributable to underlying respiratory or cardiovascular disease. Only three SAEs (two solithromycin, one moxifloxacin) were considered study drug related, all of which were allergic reactions and occurred during the first IV dose. One solithromycin patient met Hy’s Law criteria at baseline prior to exposure to solithromycin, but improved over the seven-day course of treatment. There were no concomitant ALT and bilirubin increases related to solithromycin administration and therefore no patient met Hy’s Law criteria post-baseline. Overall rates of non-infusion related adverse events were comparable between study arms.
|
SOLITAIRE-IV Phase 3 Trial |
|
|
|
|
|
|
|
|
|
Safety Outcomes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
|
Moxifloxacin |
|
||
|
|
|
(n=432) |
|
|
(n=426) |
|
||
|
Infusion-related AEs |
|
|
31.3% |
|
|
|
5.4% |
|
|
Study discontinuation due to infusion-related events |
|
10 |
|
|
1 |
|
||
|
Non-infusion related AEs leading to study drug discontinuation |
|
|
3.5% |
|
|
|
3.8% |
|
|
SOLITAIRE-IV Phase 3 Trial |
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-infusion-related Adverse Events (>2%) |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
|
Moxifloxacin |
|
|
|
|
|
||
|
|
|
400 mg QD (800 mg first oral dosing day) |
|
|
400mg QD |
|
|
|
|
|
||
|
|
|
(n=432) |
|
|
(n=426) |
|
|
|
|
|
||
|
Diarrhea* |
|
|
4.4% |
|
|
|
5.9% |
|
|
|
|
|
|
Headache |
|
|
3.5% |
|
|
|
4.2% |
|
|
|
|
|
|
Nausea |
|
|
3.2% |
|
|
|
1.6% |
|
|
|
|
|
|
Hypokalemia |
|
|
2.5% |
|
|
|
2.1% |
|
|
|
|
|
|
Dizziness |
|
|
2.5% |
|
|
|
1.2% |
|
|
|
|
|
|
Insomnia |
|
|
2.1% |
|
|
|
1.2% |
|
|
|
|
|
|
Hypertension |
|
|
1.4% |
|
|
|
2.3% |
|
|
|
|
|
|
ALTs**- Grade 3 |
|
|
8.2% |
|
|
|
3.4% |
|
|
|
|
|
|
Grade 4 |
|
|
0.7% |
|
|
|
0.5% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Not included in the diarrhea definition is 1 patient with C. difficile associated diarrhea, who received moxifloxacin (2 patients on moxifloxacin in the Oral study also had C. difficile colitis) |
||||||||||||
|
** No patient in either arm of the study developed treatment emergent elevation of both ALT and bilirubin as defined by Hy's Law criteria. Observed ALT elevations reversible and asymptomatic. |
||||||||||||
|
|
Data from preclinical, Phase 1 and Phase 2 studies of solithromycin to treat CABP are available in annual reports from prior years. |
Solithromycin for Gonorrhea
The second indication we are pursuing for solithromycin is uncomplicated urethritis, or gonorrhea. Given the high prevalence of gonorrhea in the United States and increasing resistance of the pathogen to recommended antibiotics, the FDA has designated oral solithromycin as a QIDP for the treatment of uncomplicated gonococcal infections. The current standard of treatment for bacterial urethritis/gonorrhea is combination therapy including both an intramuscular injection of ceftriaxone and oral azithromycin (1000 mg). There is no oral formulation of ceftriaxone and gonococcal resistance to azithromycin is now too high to allow it to be used in
5
monotherapy for the oral treatment of gonorrhea. Oral cefixime (Suprax) had been recommended as an alternative for treatment of patients as well as for treatment of their potentially infected partners. However, as of August 2012, the CDC, no longer recommends cefixime for the treatment of gonorrhea, which leaves no oral treatment option.
In our Phase 2 open-label study with solithromycin completed in 2013 in men and women with suspected gonococcal infection, microbiological eradication of gonococci was achieved in 100% of all evaluable patients at all body sites.
We have analyzed the data from the initial patient cohort of 262 patients in SOLITAIRE-U, a phase 3 study evaluating a single 1000 mg dose of oral solithromycin for treatment of uncomplicated genitourinary gonorrhea, or GC, with or without concomitant chlamydia infection, compared with intramuscular ceftriaxone (500 mg) plus oral azithromycin, or CTX/AZI, (1000 mg). While solithromycin demonstrated high success rates of 80.5% in the microbiological intent to treat, or mITT, population (defined as achievement of a negative urethral or cervical swab culture at Day seven to eight among those patients who had culture confirmation of GC infection at baseline) and 91.3% in the microbiologically evaluable, or ME, population (comprised of those patients with a positive baseline culture who returned for their follow-up evaluation), and showed a 100% success rate for females in the ME population, solithromycin did not demonstrate non-inferiority, NI, to standard of care treatment given the pre-specified 10% NI margin in the mITT population. The success rates for CTX/AZI in the mITT and ME populations were 84.5% and 100%, respectively.
Given the limited number of females and adolescents in SOLITAIRE-U, the National Institute of Allergy and Infectious Disease, or NIAID, agreed to fund an expansion of the trial to enroll up to 76 women and adolescents (age 15-17) under a cooperative research and development agreement. Enrollment of this trial expansion has been much slower than anticipated.
We believe that the small number of solithromycin treatment failures observed in SOLITAIRE-U could be reduced in an approval-enabling study with an adjustment to the dosing regimen, as we believe the treatment failures were most likely related to the duration of study drug exposure at the site of infection. We plan to discuss our next steps with the GC program with NIAID and the FDA, as resistance to existing therapies for GC has created an urgent unmet medical need. In SOLITAIRE-U, no GC isolates demonstrated solithromycin resistance at baseline, and there was no emergence of solithromycin resistance in the isolates obtained at follow-up cultures.
We plan to meet with the FDA in the second half of 2017 to discuss the protocol for a clinical trial with this adjusted dosing regimen, which could enable approval, if successful.
Solithromycin for CABP in Children and for Bioterror Pathogens—Collaboration with BARDA
In May 2013, we entered into an agreement with the Biomedical Advanced Research Development Authority of the U.S. Department of Health and Human Services, or BARDA, for the evaluation and development of solithromycin for the treatment of bacterial infections in pediatric populations and infections caused by bioterror threat pathogens, specifically anthrax and tularemia. BARDA is funding the development of an oral suspension formulation of solithromycin for pediatric use in CABP. Safety and pharmacokinetic evaluations have been completed, testing oral capsules, IV, and oral suspension in pediatric patients ranging from newborns to 17 years old in Phase 1a and Phase 1b trials.
We initiated a Phase 2/3 trial in the fourth quarter of 2016. The pediatric study plan to obtain regulatory approval was submitted and accepted by both the FDA and EMA. There is an urgent need for a new oral and intravenous antibiotic for use in pediatric infections. Concurrent with this program, and included in the BARDA funding, support is provided for solithromycin manufacturing, including the optimization of the commercial pediatric suspension product. In September 2016, the BARDA contract was modified to support increased manufacturing work related to the development of a second supply source for solithromycin.
BARDA has also funded pilot studies in non-human primates to test the efficacy of solithromycin in treating bioterror pathogens such as tularemia and anthrax. Solithromycin was effective in treating both of these infections in these studies.
Solithromycin for NASH
We have evaluated the effects of solithromycin in a proof-of-principle study in patients with nonalcoholic steatohepatitis, or NASH. NASH is a progressive form of non-alcoholic fatty liver disease, or NAFLD, where hepatic steatosis progresses to liver cell injury, inflammation and fibrosis, which can eventually lead to cirrhosis and liver cancer. To date, no single therapy has been approved for treating NAFLD/NASH. According to the National Institutes of Health, or NIH, up to 16 million Americans, or two to five percent of the U.S. population, has NASH.
6
Solithromycin has been demonstrated to have potent anti-inflammatory properties in addition to its antibacterial properties. As a result, we evaluated solithromycin in a diabetic mouse model of NASH to investigate its potential effects. In this preclinical study, solithromycin demonstrated potential anti-NASH and anti-hyperglycemic effects.
In September 2016, we announced interim results showing anti-NASH effects in the first six NASH patients dosed with solithromycin in an exploratory development program. Based on the safety profile and activity seen in the first six patients, we continued the study to obtain data from up to 15 NASH patients. In the first quarter of 2017, four additional patients had completed treatment and undergone end-of-treatment liver biopsies, and we have evaluated the data from the cohort of 10 patients.
While data from the initial six patients were promising, the overall efficacy from patients receiving a reduced dose of 200 mg three times a week (after a 200 mg loading dose), including the second cohort of four patients, is unclear. Therefore, we have elected to suspend the NASH development program for solithromycin at this time.
Solithromycin for COPD
Based on safety data from several of the initial patients dosed in an exploratory study evaluating the effect of long term systemic solithromycin administration on airway inflammation in chronic obstructive pulmonary disease, or COPD, we have closed the study.
Solithromycin for Ophthalmic Conditions
Many of the pathogens that cause CABP are the same as, or similar to, the pathogens that cause eye infections and we are developing an ophthalmic formulation of solithromycin as a potential treatment for bacterial conjunctivitis and other ophthalmic conditions. According to IMS, there are approximately 19 million antibiotic prescriptions written annually to treat ophthalmic conditions, and approximately 80% of these prescriptions are macrolides or fluoroquinolones. Because of increasing antimicrobial resistance to existing antibiotic therapies, we believe solithromycin could meet an unmet medical need in a significant market. Additionally, we believe that an ophthalmic formulation of solithromycin would have little to no systemic exposure. We plan to request a pre-IND meeting with the FDA in 2017 to discuss moving our ophthalmic program forward into clinical trials.
Overview of Fusidic Acid
Fusidic acid is an antibiotic that we are developing exclusively in the U.S. for ABSSSI, and we are exploring its use for the long-term oral treatment for refractory bone and joint infections, including PJI, which are frequently caused by staphylococci, including S. aureus, MRSA, coagulase negative staphylococci and other gram-positive bacteria. We have developed a novel and proprietary dosing regimen of fusidic acid, which is an approved antibiotic that has been sold by Leo Laboratories, Ltd. primarily for staphylococcal infections, including skin, soft tissue and bone infections, for several decades in Europe and other locations outside the U.S. and has a long-established safety and efficacy profile. However, fusidic acid has never been approved for use in the U.S.
7
Fusidic Acid for ABSSSI
Fusidic acid successfully completed a Phase 2 clinical trial in patients with ABSSSI demonstrating a tolerability profile and efficacy comparable to linezolid (sold under the brand name Zyvox®), which is one of the few oral antibiotics currently approved for the treatment of MRSA approved by the FDA. In the first quarter of 2017, we reported results from a Phase 3 trial evaluating the safety and efficacy of fusidic acid in 716 patients with ABSSSI.
The double-blind Phase 3 study was conducted at 62 sites in the United States. Patients randomized to treatment with oral fusidic acid received a loading dose of 1500 mg every 12 hours for two doses, followed by 600 mg every 12 hours thereafter, until the end of a 10-day course of therapy. Patients randomized to treatment with the active comparator, oral linezolid, received 600 mg every 12 hours for 10 days. Randomization was 1:1 and was stratified by type of infection (cellulitis, wound infection, major cutaneous abscess), by age and by prior use of an antibiotic within 36 hours prior to randomization. Overall, 67.5 % of study subjects had an infection associated with intravenous drug abuse. Less than 5% of study subjects received an antibiotic prior to randomization.
|
ITT Population |
Fusidic Acid N=359 |
Linezolid N=357 |
|
Sex, n (%) |
|
|
|
Male |
244 (68.0) |
218 (61.1) |
|
Female |
115 (32.0) |
139 (38.9) |
|
Infection Type, n (%) |
|
|
|
Major Cutaneous Abscess |
46 (12.8) |
47 (13.2) |
|
Cellulitis |
92 (25.6) |
92 (25.8) |
|
Wound Infection |
221 (61.6) |
218 (61.1) |
|
Prior Antibiotic Usage, n (%) |
|
|
|
Yes |
17 (4.7) |
18 ( 5.0) |
|
No |
342 (95.3) |
339 ( 95.0) |
|
Recent or Ongoing IV Drug Abuse, n (%) |
|
|
|
Yes |
245 (68.2) |
238 (66.7) |
|
No |
114 (31.8) |
119 (33.3) |
The primary objective of the study was to demonstrate non-inferiority of fusidic acid compared to linezolid for early clinical response, or ECR, defined as the proportion of patients alive and achieving a ≥ 20% reduction from baseline in lesion size at 48-72 hours after the start of study drug, without receiving rescue antibiotics, in the intent to treat, or ITT, patient population. In the study, 87.2 % of ITT patients receiving fusidic acid demonstrated ECR, compared to 86.6 percent of ITT patients receiving linezolid (treatment difference 0.6%, 95% confidence interval, or CI, -4.6, +5.9), demonstrating non-inferiority to linezolid. Fusidic acid also showed comparable efficacy to linezolid in investigator-assessed clinical response in the ITT and clinically evaluable, or CE, populations at end of treatment, or EOT, and post-therapy evaluation, or PTE, (7-14 days post-EOT) visits.
|
|
Fusidic Acid |
Linezolid |
Treatment Difference (95% CI) |
|
ITT Population |
|
|
|
|
ECR (Primary Endpoint) |
87.2 % (313/359) |
86.6 % (309/357) |
+0.6 (-4.6, +5.9) |
|
Clinical Success at EOT |
91.9 % (330/359) |
89.6 % (320/357) |
+2.3 (-2.2, +6.8) |
|
Clinical Success at PTE |
88.6 % (320/359) |
88.5 % (316/357) |
+0.1 (-4.9, +5.0) |
|
CE Populations |
|
|
|
|
Clinical Success at EOT (CE-EOT) |
97.1 % (303/312) |
97.3 % (288/296) |
-0.2 (-3.1, +2.8) |
|
Clinical Success at PTE (CE-PTE) |
95.7 % (292/305) |
96.9 % (283/292) |
-1.2 (-4.5, +2.2) |
Microbiological response rates by pathogen were high in both treatment groups in both the microbiological ITT, or mITT, and microbiologically-evaluable, or ME, patient populations (patients with isolation of a baseline pathogen, who were also clinically evaluable). The most common pathogens identified were Staphylococcus aureus, Streptococcus anginosus group species, Streptococcus pyogenes and Clostridium species. Notably, the microbiological success rate among fusidic acid recipients in each ME population with methicillin-resistant S. aureus, or MRSA, infection was 100 % (99/99) at both the EOT and PTE visits.
8
Fusidic acid was well tolerated in the study. The rates of treatment-emergent adverse events, or TEAEs, were comparable between treatment groups (37.9% fusidic acid, 36.1% linezolid). The most common TEAEs in both treatment groups were gastrointestinal events (22.8% fusidic acid, 18.2% linezolid). SAEs occurred in six fusidic acid recipients and eight linezolid recipients, and were considered study-drug related in one fusidic acid recipient (vomiting) and in two linezolid recipients (one drug induced liver injury, one vomiting). Adverse events led to study drug discontinuation in 2.2% of fusidic acid recipients, and 2.0% of linezolid recipients. There was one death in the study, an event due to illicit drug overdose and aspiration which occurred in a patient receiving linezolid. Rates of treatment-emergent ALT elevation to >3x ULN occurred in 1.0% of fusidic acid recipients and 0.7% of linezolid patients.
We plan to submit the full data from this study for presentation at an upcoming scientific forum.
Our strategy with fusidic acid is to meet with the FDA and review the best next steps to gain approval. For ABSSSI, we expect that two Phase 3 studies would be required for approval. Once we have input from the FDA on the path forward we will decide how to proceed.
Fusidic Acid for BJI
Like ABSSSI, bone and joint infections, including prosthetic joint infections, are often caused by staphylococci, including MRSA. In December 2012, we initiated a Phase 2 trial of fusidic acid for treatment of primarily staphylococcal infections of infected prosthetic joint infections, hip and knee joints. Based on the results of the 14 patients enrolled in this study, we concluded that the fusidic acid in combination with rifampin was generally comparable to intravenous standard of care antibiotics.
We believe fusidic acid has the potential to be used in hospital and community settings on both a short-term and chronic basis. Since bone and joint infections are primarily treated with a combination of IV and oral drugs, we believe that fusidic acid would enable out-patient treatment of many patients who would otherwise require hospitalization and/or intravenous therapy, which we also believe would provide pharmacoeconomic advantages, be well received by doctors and be more convenient for patients.
In early 2016, we also began a study of fusidic acid in patients with refractory bone and joint infection, which continues to enroll. Should we choose to pursue an indication for fusidic acid in patients with PJI, the FDA has granted orphan drug designation for fusidic acid for the treatment of PJI.
Our Earlier Stage Pipeline Programs
Our earlier stage programs include developing other uses for solithromycin and fusidic acid, as well as the development of analogs from our macrolide platform for non-infectious disease programs.
In the future we may pursue secondary indications for solithromycin to treat other infections in cystic fibrosis patients, Helicobacter gastritis, malaria and tuberculosis. Solithromycin has also been demonstrated to have in vitro activity against enterococci, including vancomycin-resistant enterococci.
We have developed our own proprietary macrolide compounds which we intend to use to develop drugs with no antibiotic effect and to replace use of older macrolides in inflammatory conditions and other indications such as diabetic gastroparesis. Several compounds have been identified through our screening programs that could potentially address therapeutic needs in the areas of inflammation, diabetic gastroparesis and cancer.
We are conducting early drug discovery studies for the use of macrolides in treating diabetic gastroparesis, which is related to a lack of neural response in the gastrointestinal tract of diabetic patients, and gastroesophageal reflux disease, or GERD, both likely to be helped by addressing motilin function. Motilin is a naturally occurring peptide that causes the stomach to contract to initiate the migratory motor complex that empties the stomach. Erythromycin and related antibiotics have known activity as motilin agonists. Through our discovery program, we have identified compounds that are active in the motilin receptor binding assay, as well as in rabbit duodenal strip contraction assays. These compounds are being optimized chemically for pharmacokinetic properties and oral bioavailability.
The Limitations Associated with Antibiotics
The widespread use of antibiotics has led to development of resistant strains of bacteria, which limits the effectiveness of existing drugs. This led the World Health Organization to state in 2010 that antibiotic resistance is one of the three greatest threats to human health. The CDC estimates that more than 70% of U.S. hospital infections are resistant to at least one of the antibiotics most commonly used to treat them. The CDC also estimates that each year more than 2,000,000 people are sickened with antibiotic-
9
resistant infections, with at least 23,000 dying as a result. Antibiotic-resistant infections also increase the costs to the U.S. healthcare system.
Antibiotic resistance is primarily caused by genetic mutations in bacteria selected by exposure to antibiotics where the drug does not kill all of the bacteria. In addition to mutated bacteria being resistant to the drug used for treatment, many bacterial strains can also be cross-resistant, meaning that the use of a particular treatment to address one kind of bacteria can result in resistance to other kinds of bacteria as well as to other antibiotics. As a result, the effectiveness of many antibiotics has declined, limiting physicians’ options to treat serious infections and creating a global health issue. For example, it is estimated that in the U.S. approximately 49% of pneumococci, the primary pathogen involved in respiratory tract infections, are resistant to azithromycin and other macrolides commonly used to treat them.
In addition to resistance issues, current antibiotic therapies also have other limitations, including serious side effects. These side effects may include: hepatotoxicity, severe allergic reaction, decreased blood pressure, nausea and vomiting, suppression of platelets, pain and inflammation at the site of injection, muscle, renal and ototoxicity, optic and peripheral neuropathies and headaches. Some of these side effects may be significant enough to require that therapy be discontinued or not used. As a result, some treatments require clinicians to closely monitor patients’ blood levels and other parameters, increasing the expense and inconvenience of treatment.
Further, many of the existing antibiotics used to treat serious infections are difficult or inconvenient to administer. Many drugs are given twice daily for seven to 14 days or more and patients can be hospitalized for much or all of this period or require in-home IV therapy. While IV treatment can deliver the drug more rapidly and in a larger dose than is possible orally, once a patient is stabilized, a switch to oral treatment allows for more convenient and cost-effective out-patient treatment. We believe that there is a need for new antibiotics that have improved potency and pharmacokinetics, effectiveness against resistant bacterial strains, improved side effect profiles and more flexible administration formulations.
Solithromycin Market Opportunity
Community Acquired Bacterial Pneumonia
Respiratory tract infections can range from severe diseases such as CABP, to similar infections of the respiratory tract such as pharyngitis (which is usually referred to as strep throat), bronchitis, chronic sinusitis and middle ear infections (which are especially common in children). CABP is one of the most common serious infectious diseases of the respiratory tract and is the most frequent cause of death due to bacterial infections in the U.S. There are 1.6 million fatal cases of pneumococcal disease annually worldwide which is more than the deaths caused annually by breast or prostate cancer. There are approximately five to six million cases of CABP in the U.S. every year, approximately one million of which require hospitalization. Typical bacteria that cause CABP include Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis and Staphylococcus aureus. These four bacteria account for approximately 85% of CABP cases. Other organisms, called atypical bacteria, may be involved in CABP and include Legionella pneumophila, Chlamydophila pneumoniae and Mycoplasma pneuomoniae.
Given the serious nature of these infections, and the length of time it takes to test for the pathogen causing the infection, physicians typically cannot wait for the results of diagnostic tests. Accordingly, under routine care to diagnose CABP, physicians must treat empirically with an antibiotic or combination of antibiotics that has the broadest activity against the most likely pathogen, including both typical and atypical bacteria are utilized.
The most recent CABP treatment guidelines from the American Thoracic Society, or ATS, and IDSA offer tiered approaches to treatment, with overarching guidance that empiric therapy should target both typical and atypical pneumonia pathogens. For generally healthy outpatients in the setting of low pneumococcal resistance to macrolides, use of macrolide monotherapy or doxycycline was recommended. Since pneumococcal macrolide resistance is now found in approximately 50% of isolates across the U.S., macrolide monotherapy would no longer be considered advisable. Alternatively, monotherapy with a respiratory fluoroquinolone, or with the combination of a beta-lactam antibiotic plus a macrolide, is recommended. These latter recommendations also apply for hospitalized patients who do not require admission to intensive care units. For patients with severe pneumonia requiring ventilatory support or ICU care, it is recommended that empiric therapy be expanded to include coverage for Pseudomonas and MRSA infection, along with the more common CABP pathogens.
In recent years, the effectiveness of earlier generation macrolides, including azithromycin, for treatment of CABP has declined due to increasing pneumococcal resistance. The most recently approved macrolide, telithromycin (Ketek), was effective against macrolide-resistant pneumococci, but the antibiotic was associated with serious side effects, resulting in limitation of its use. For these reasons, respiratory fluoroquinolones, such as levofloxacin and moxifloxacin, are now commonly used for serious CABP infections. Although these drugs are efficacious, they have been associated with serious side effects including C. difficile enterocolitis, tendonitis, hepatotoxicity, chronic disability syndromes and central nervous system effects. Beta-lactam antibiotics such as cephalosporins, which are commonly used in CABP, also have limitations, including absence of activity against the atypical pathogens, Legionella and
10
Mycoplasma. In addition, the cephalosporins most commonly utilized in treatment of CABP, ceftriaxone and ceftaroline, are only available in intravenous formulations.
As a result of the limitations of current therapies for CABP, we believe there is an opportunity to introduce solithromycin, a fourth generation macrolide that is more potent and effective against bacteria that are resistant to older generations of macrolides, while retaining the traditional safety and anti-inflammatory properties that macrolides are known to exhibit. To date, we believe our clinical trials of solithromycin to treat CABP have demonstrated that solithromycin is potent and effective against resistant bacteria and is well tolerated. We also believe that developing IV and oral formulations will provide flexibility to physicians to treat patients according to the severity of their disease and transition some patients from IV to oral, enabling them to leave the hospital sooner. If approved by the FDA, solithromycin would be the first macrolide approved with both IV and oral capsule and suspension formulations since azithromycin was approved more than 20 years ago.
Gonorrhea
In addition to CABP, there is a large public health need for a new and effective oral treatment for treating bacterial urethritis. Resistance has emerged to cefixime, which was the only remaining oral therapy for gonococci infections, such as bacterial urethritis. The development of solithromycin for bacterial urethritis would present an additional market opportunity for solithromycin and all of the patient data gathered in any trials would also contribute to the safety data base for CABP development.
Ophthalmic
There were approximately 19 million antibiotic prescriptions in 2016 for ophthalmic conditions. Macrolides and fluoroquinolones comprise approximately 60% of the total prescriptions. For conditions such as bacterial conjunctivitis specifically, macrolides and fluoroquinolones make up approximately 80% of the market. The common bacterial pathogens that cause bacterial conjunctivitis are similar to those that cause community acquired bacterial pneumonia.
Fusidic Acid Market Opportunity
We are developing fusidic acid exclusively in the U.S. for ABSSSI (which includes a subset of skin infections such as cellulitis, wound infections, major cutaneous abscess and burn infections). There are approximately 3.3 million patients treated annually in the hospital for ABSSSIs and, according to the IDSA, MRSA infections account for approximately 60% of skin infections seen in U.S. emergency departments, or EDs. Cellulitis accounts for 41% of ABSSSIs with traumatic and surgical wounds accounting for another 24%. MRSA is prevalent in ABSSSI and suspected in 25% of patients. Inadequate treatment of MRSA ABSSSI due to antibiotic resistance is likely a factor in relapse. Eighty percent of the patients are treated empirically, with MRSA expected in slightly over half.
Between 7-10% of all hospital admissions are for ABSSSI. ABSSSIs are frequently treated in an ED. An oral treatment option in the ED could enable some patients to avoid hospitalization and could also reduce hospital length of stay for some patients who could transition to oral therapy. The mean cost to treat ABSSSI in the hospital is over eight thousand dollars with an average length of stay of nearly five days. Currently, while some newer branded products are available, generic vancomycin is most commonly used. Many patients require a multi-drug regimen as oral monotherapy is an unmet need. About 4 million patients require second line treatment.
According to the IDSA, MRSA infections account for approximately 60% of skin infections seen in U.S. emergency rooms. Linezolid is available in both IV and oral formulations and is one of two oral antibiotics approved by the FDA for MRSA. However, linezolid has significant side effects, which include irreversible peripheral neuritis, or the inflammation of nerves and thrombocytopenia, or a relative decrease of platelets in blood. In July 2011, the FDA published a drug safety communication letter regarding the use of linezolid in patients on serotonergic drugs such as selective serotonin reuptake inhibitors, or SSRIs, (including Prozac, Paxil and Zoloft), which are taken for depression, bipolar disease, schizophrenia and other psychiatric disorders. Given the widespread use of SSRIs and some of the other side effects associated with linezolid, we do not believe that linezolid is an option for many patients. In 2015 a second oral drug, tedizolid (Sivextro®), was approved for ABSSSI caused by susceptible Gram-positive organisms including MRSA. Tedizolid does not have the same contraindications or drug interactions as linezolid but is only approved as a six-day IV and/or oral treatment course. Phase 1 studies conducted with tedizolid in healthy adults that were treated for 21 days showed a possible dose and duration effect on hematologic parameters beyond six days of treatment. The most common adverse drug reactions in clinical trials from patients treated with tedizolid were nausea, headache, diarrhea, vomiting, and dizziness. We believe there is an opportunity to develop an oral drug that is effective against MRSA and has a safety profile that supports out-patient use, use for chronic indications and use in children.
11
Competition
Our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions.
In many cases, however, we believe that competition often will be determined by antibiotic class and any limitations of that antibiotic class in general, and the antibiotic in particular, in treating a particular disease or population. We believe that the key competitive factors that will affect the development and commercial success of solithromycin, fusidic acid and any other product candidates that we develop are efficacy, safety and tolerability profile, convenience in dosing, price and reimbursement.
Solithromycin
We anticipate that, if approved for CABP, solithromycin will compete with other antibiotics that demonstrate CABP activity. These include well established, widely prescribed drugs, including generic versions, such as azithromycin, clarithromycin, moxifloxacin, levofloxacin, linezolid and ceftriaxone. Among macrolides, azithromycin (Zithromax, Z-Pak) is the class leader, and a generic drug. Azithromycin is available as oral tablets, lyophilized vials for IV and a powder for suspension, which has allowed dosing of all age groups and has been used broadly including for simple respiratory tract infections and in combination with ceftriaxone to treat CABP.
Azithromycin and other macrolides are used broadly in COPD for their anti-inflammatory properties to lower the dose of steroids, in bacterial urethritis to treat gonorrhea and chlamydia infections, and many other infections. Azithromycin is among the most widely used antibiotics. Ceftaroline was approved in CABP, but only the IV formulation is available; no oral or powder for suspension for pediatric use is available. For IV use in CABP, ceftaroline must be co-administered with azithromycin or another macrolide because it does not provide adequate coverage for atypical bacteria, such as Legionella or Mycoplasma. Patients can stay in the hospital for IV therapy or be discharged on a different, lower potency second generation cephalosporin.
Fluoroquinolones, such as levofloxacin and moxifloxacin, could be used in CABP and are available in oral and IV formulations. They are not approved for pediatric use because of safety. Levofloxacin is now generic and because of its lower potency, its use in CABP has been taken over by the branded moxifloxacin (Avelox). Prescribing information for fluoroquinolones notes several undesirable effects such as tendonitis, achilles tendon rupture, C. difficile colitis, hepatotoxicity and central nervous system side effects. These adverse events limit the use of these drugs in CABP. We believe that a few companies have Phase 3 CABP programs including Melinta Therapeutics, Inc. (a new fluoroquinolone), Paratek Pharmaceuticals, Inc (a new tetracycline) and Nabriva Therapeutics AG (a new pleuromutilin analog).
In the area of bacterial urethritis, gonococcus has become resistant to older macrolides and other drugs, including fluoroquinolones. Only intramuscular ceftriaxone is currently available for treating these patients.
Fusidic Acid
We anticipate that, if approved, fusidic acid will compete with other antibiotics that demonstrate MRSA activity. These include well established, widely prescribed drugs, including generic versions, such as vancomycin, linezolid, tedizolid, daptomycin, tigecycline and ceftaroline.
Intellectual Property
Due to the length of time and expense associated with bringing new products to market, biopharmaceutical companies have traditionally placed considerable importance on obtaining and maintaining patent protection for significant new technologies, products and processes. The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the U.S., a patent’s term may be lengthened by Patent Term Adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office, or USPTO, in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent. In the U.S. and certain other countries, the patent’s term may also be lengthened by patent term extension or restoration, which compensates a patentee for administrative delays in granting a regulatory approval by the FDA, or similar agency in other countries.
While we pursue patent protection and enforcement of solithromycin, fusidic acid and our other product candidates, and aspects of our technologies when appropriate, we also rely on trade secrets, know-how and continuing technological advancement to develop and maintain our competitive position. To protect this competitive position, we regularly enter into confidentiality and proprietary information agreements with third parties, including employees, independent contractors, suppliers and collaborators. Our employment policy requires each new employee to enter into an agreement containing provisions generally prohibiting the disclosure of confidential information to anyone outside of our company and providing that any invention conceived by an employee within the
12
scope of his or her employment duties is our exclusive property. We have a similar policy with respect to independent contractors, generally requiring independent contractors to enter into agreements containing provisions generally prohibiting the disclosure of confidential information to anyone outside of our company and providing that any invention conceived by an independent contractor within the scope of his or her services is our exclusive property with the exception of contracts with universities and colleges that may be unable to make such assignments. Furthermore, our know-how that is accessed by third parties through collaborations and research and development contracts and through our relationships with scientific consultants is generally protected through confidentiality agreements with the appropriate parties.
Further, we seek trademark protection in the U.S. and internationally where available and when appropriate. We have a registered trademark in the U.S. for the CEMPRA mark, which we use in connection with our pharmaceutical research and development services, and which we plan to use with our proposed products. We have filed applications at the U.S. Patent and Trademark Office for the SOLITHERA mark, which we plan to use with our proposed solithromycin product. We also have filed applications at the U.S. Patent and Trademark Office for the FUSIDIC ACID, STRAFEX, and STAFREL marks. We plan to use the FUSIDIC ACID mark with our proposed sodium fusidate product. The remaining marks may be used with the sodium fusidate product or other proposed products.
We have applied, and are applying, for patents directed to our three main areas of focus: (1) macrolide and ketolide antibiotics, (2) fusidic acid antibiotics, and (3) macrolides and ketolides for non-antibiotic uses, both in the U.S. and, when appropriate, in other countries. As of December 31, 2016, our owned and in-licensed patent portfolio consisted of 22 issued patents in the U.S., more than 150 patents issued in other countries, and approximately 140 additional patent applications pending in the U.S. and worldwide. The exclusively in-licensed portfolio also includes a pending continuing U.S. patent application, and divisional applications pending in Canada, Europe and Hong Kong. Prosecution is ongoing in each of those patent applications. Each of the foregoing patents and applications ultimately arise from a PCT international application filed on March 5, 2004, which claims the priority benefit of U.S. provisional applications filed on March 10, 2003, and May 6, 2003.
Solithromycin
Solithromycin is a new chemical entity developed from the macrolide library of compounds licensed from Optimer (now owned by Merck) and is covered by a series of patents and patent applications, which claim, among other things, the composition of matter of solithromycin.
Most of our portfolio consists of intellectual property that we own ourselves or that we exclusively license from Optimer. The intellectual property licensed from Optimer primarily relates to solithromycin and related compounds, and to other macrolide and ketolide compounds. Internally, we typically develop those compounds further and refine them to determine commercial applicability.
As noted in the table below, with respect to solithromycin and a broad group of other macrolide antibiotic compounds, our U.S. patent portfolio consists of four issued U.S. patents, which we exclusively license from Optimer. We have filed company-owned patent applications in the U.S., Australia, Brazil, Canada, China, Europe, Hong Kong, Israel, India, Japan, Malaysia, Mexico, New Zealand, the Russian Federation, South Africa, and South Korea claiming two new crystalline forms of solithromycin.
We have also filed additional company-owned patent applications in the U.S., Australia, Brazil, Canada, China, Europe, Hong Kong, Israel, India, Japan, Malaysia, Mexico, New Zealand, the Russian Federation, Singapore, South Africa, South Korea, and Taiwan covering solithromycin and related compounds, with the objective of increasing the breadth of solithromycin coverage, particularly in the U.S., Europe, South America, and Asia. Those applications claim pharmaceutical compositions, pharmaceutical formulations, methods for treating particular infections and other diseases, manufacturing processes, and compounds related to solithromycin.
We have also filed, or are preparing to file, U.S. and international patent applications covering solithromycin and other macrolides and ketolides for treating eye diseases, including topical formulations for ocular delivery, and for treating diseases other than infection, including inflammatory diseases, cystic fibrosis, and motilin receptor-mediated diseases to increase the breadth of coverage for solithromycin and other macrolides and ketolides in the U.S., Europe, and Asia.
We have engaged, and continue to engage, in research efforts to exploit the potential of the in-licensed Optimer inventions, including solithromycin and related compounds, in new therapy areas, and to discover new forms and formulations of solithromycin and related compounds. Our research efforts have indicated that solithromycin may also be useful in treating particular bacterial infections that may be considered to be generally untreatable with macrolide antibiotics, including bacterial infections arising from one or more resistant strains. In addition, alternative physical forms and alternative formulations of solithromycin and related compounds are being developed. If we are able to obtain issued patents for those forms and formulations, and the treatment methods,
13
then we will have several years of additional coverage above and beyond the expiration of the patents covering the chemical composition of solithromycin.
With respect to solithromycin and a broad group of other macrolide antibiotic compounds, a list of U.S. patents which we exclusively license from Optimer:
|
Title |
|
Country |
|
Patent No. |
|
Issue Date |
|
Expiration Date |
|
Antibacterial Agents |
|
USA |
|
US 7,601,695 |
|
13-Oct-09 |
|
29-Jan-25 |
|
Antibacterial Agents |
|
USA |
|
US 8,012,943 |
|
6-Sep-11 |
|
5-Mar-24 |
|
Novel Antibacterial Agents |
|
USA |
|
US 8,343,936 |
|
1-Jan-13 |
|
5-Mar-24 |
|
Novel Antibacterial Agents |
|
USA |
|
US 9,200,026 |
|
1-Dec-15 |
|
5-Mar-24 |
|
Novel Antibacterial Agents |
|
European Patent Convention |
|
EP 2 664 331 |
|
16-Sep-15 |
|
5-Mar-24 |
|
Novel Antibacterial Agents |
|
Canada |
|
CA 2529817 |
|
12-Feb-13 |
|
5-Mar-24 |
With respect to processes for manufacturing solithromycin and related 1,2,3-triazole-containing macrolides and ketolides, a list of U.S. patents which we license non-exclusively from The Scripps Research Institute (“TSRI”):
|
Title |
|
Country |
|
Patent No. |
|
|
Issue Date |
|
Expiration Date |
|
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
7375234 |
|
|
20-May-08 |
|
30-Sep-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
7763736 |
|
|
27-Jul-10 |
|
30-May-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
8129542 |
|
|
6-Mar-12 |
|
30-May-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
8580970 |
|
|
12-Nov-13 |
|
30-May-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
8877939 |
|
|
4-Nov-14 |
|
30-May-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
9040716 |
|
|
26-May-15 |
|
30-May-23 |
|
Copper-Catalyzed Ligation of Azides and Acetylenes |
|
USA |
|
|
9302997 |
|
|
5-Apr-16 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
Canada |
|
|
2487424 |
|
|
4-Jan-11 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
European Patent Convention |
|
|
1507769 |
|
|
26-May-10 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
European Patent Convention |
|
|
2226316 |
|
|
13-Jan-16 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
Japan |
|
|
4638225 |
|
|
3-Dec-10 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
Singapore |
|
|
108420 |
|
|
29-Dec-06 |
|
30-May-23 |
The in-licensed portfolio also includes the following issued patents, which are licensed exclusively:
|
Title |
|
Country |
|
Patent No. |
|
|
Issue Date |
|
Expiration Date |
|
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
Australia |
|
|
2003240482 |
|
|
25-Jun-09 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
Australia |
|
|
2009202299 |
|
|
16-Jun-11 |
|
30-May-23 |
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
China |
|
ZL03817917.2 |
|
|
26-May-10 |
|
30-May-23 |
|
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
South Korea |
|
10-1048279 |
|
|
5-Jul-11 |
|
30-May-23 |
|
|
Copper-Catalysed Ligation of Azides and Acetylenes |
|
South Korea |
|
10-1138643 |
|
|
16-Apr-12 |
|
30-May-23 |
|
14
The following patents have been issued claiming two new crystalline forms of solithromycin:
|
Title |
|
Country |
|
Patent No. |
|
|
Issue Date |
|
Expiration Date |
|
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
USA |
|
|
8975386 |
|
|
10-Mar-15 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
USA |
|
|
8759500 |
|
|
24-Jun-14 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
Australia |
|
|
2011232627 |
|
|
30-Jun-16 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
Australia |
|
|
2016203986 |
|
|
19-Jan-17 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
EPC |
|
|
2550286 |
|
|
9-Dec-15 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
Japan |
|
|
5711352 |
|
|
13-Mar-15 |
|
22-Mar-31 |
|
Crystalline Forms of a Macrolide, and Uses Therefor |
|
New Zealand |
|
|
602544 |
|
|
3-Mar-15 |
|
22-Mar-31 |
Fusidic Acid
The original patents covering the composition of matter for fusidic acid have expired. The table below highlights our U.S. patent portfolio and we are continuing to increase the breadth of our fusidic acid coverage in the U.S. and Asia, and have filed patent applications covering fusidic acid in the U.S., Canada and Japan. Our pending patent applications claim new dosing protocols and uses of fusidic acid, and new formulations and packaging. The novel loading dose regimen that has been developed to overcome pre-existing limitations on a broader, more effective use of fusidic acid in the treatment of bacterial infections, including infections not previously considered to be susceptible to fusidic acid, like urethritis. We have also filed patent applications covering new formulations of fusidic acid for direct bronchial and pulmonary delivery, and formulations and packaging of fusidic acid dosage units to overcome the storage limitations of fusidic acid.
In addition to filed patent applications claiming new dosing protocols and formulations of fusidic acid for treating infections, we plan to obtain regulatory exclusivity for the first use of fusidic acid through approval with the FDA. We are not aware of any competing applications before the FDA seeking approval for fusidic acid. Therefore, we believe that, if the FDA approves an NDA of ours for fusidic acid before the FDA approves an NDA or other application for fusidic acid use filed by any competitor, pursuant to amendments to Section 505 of the Food, Drug and Cosmetic Act enacted in 2008, we will have at least five years of regulatory exclusivity in the U.S. for the first approved indication for fusidic acid. We believe that the 2008 amendments will also provide us with three years of exclusivity for any additional uses.
With respect to fusidic acid, our U.S. patent portfolio consists of the following issued patents:
|
Title |
|
Country |
|
Patent No. |
|
|
Issue Date |
|
Expiration Date |
|
|
Fusidic Acid Dosing Regimens for Treatment of Bacterial Infections |
|
USA |
|
|
8450300 |
|
|
28-May-13 |
|
9-Dec-29 |
|
Methods of Treating Urethritis and Related Infections Using Fusidic Acid |
|
USA |
|
|
8247394 |
|
|
21-Aug-12 |
|
24-Feb-31 |
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval, if any, of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. Subject to certain limitations, the patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application, up to a total of five years. Only one patent applicable to an approved drug is eligible for the extension. The application for such extension must be submitted prior to the expiration of the patent and within 60 days of the drug’s approval. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, we may apply for restoration of patent term for
15
one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the Federal Food, Drug, and Cosmetic Act, or FDCA can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA. The FDCA provides a five-year period of non-patent data exclusivity within the U.S. to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of exclusivity in the U.S. Pediatric exclusivity, if granted, provides an additional six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study. The current pediatric exclusivity provision was reauthorized in September 2007 as part of the FDAAA.
We believe that both solithromycin and fusidic acid will benefit from the marketing incentives of the GAIN Act, enacted in 2012. This legislation rewards a Qualified Infectious Disease Product with five years of additional exclusivity (added to the five years of Hatch-Waxman exclusivity) when its NDA is approved. Pursuant to the GAIN Act, in 2013, the FDA designated each of the oral and IV formulations of solithromycin as a QIDP for the indication of CABP, which was designated a qualified infections disease pathogen by the FDA in 2013. The NDA also receives priority review, which reduces the standard 12-month review time by four months. The FDA also has designated the oral form as a QIDP for the treatment of uncomplicated gonococcal infections.
Fusidic acid has been approved for oral use in many countries, including Western countries, outside the U.S. for more than three decades to treat ABSSSI, as well as other types of infections caused by staphylococci and ß-hemolytic streptococci, but it has never been approved in the U.S. This is because of the general lack of intellectual property protection that was available for the molecule until recently. Significant patent protection expired in the 1980’s, and antibiotics were not eligible for Hatch-Waxman Act data exclusivity, which affords a five-year period of data exclusivity upon approval of a new chemical entity, or NCE, in the U.S. In November 1997, the FDA Modernization Act, or FDAMA, repealed section 507 of the Federal, Food, Drug, and Cosmetic Act, or FDCA, under which marketing applications for antibiotics were previously approved. This law made antibiotics, like other drugs, eligible for Hatch-Waxman Act exclusivity. However, fusidate/fusidic acid was the subject of a marketing application received by FDA under Section 507 of the FDCA before November 21, 1997, the effective date of FDAMA. Antibiotics for which marketing applications were submitted before that date, even if the application was not approved, as was the case with fusidic acid, are known as “old” antibiotics. Old antibiotics were not eligible for the exclusivity provisions afforded by FDAMA. Consequently, although fusidic acid had never been approved in the U.S., as an old antibiotic, it was not eligible for the five-year NCE exclusivity. The passage of Public Law (PL) 110-379 on October 8, 2008, allowed old antibiotics such as fusidic acid to obtain five-year NCE exclusivity upon NDA approval, thereby making development of fusidic acid for the U.S. feasible. In response to our question based on unclear language in PL 110-379 regarding other exclusivities, we received notification from the FDA in January 2011 that old antibiotics such as fusidic acid would also be eligible for orphan and pediatric exclusivity. In October 2013, the FDA granted orphan drug designation for fusidic acid for the treatment of PJI. In December 2013, PJI was classified as a very rare disease by the National Institutes of Health, or NIH. In addition, the GAIN Act extends the NCE data exclusivity period for QIDPs such as fusidic acid from five years to 10 years. Finally, our loading dose regimen, which received a U.S. patent in May 2013, provides patent protection to 2029.
Collaborations and Commercial Agreements
Optimer Pharmaceuticals, Inc. (now owned by Merck). In March 2006, we entered into a Collaborative Research and Development and License Agreement with Optimer, a biotechnology company focused on discovering, developing and commercializing innovative anti-infective products. Under this agreement, we obtained access to a library of over 500 macrolide compounds, including solithromycin. Optimer was acquired by Cubist in October 2013, which in turn was acquired by Merck in 2015. Optimer granted us an exclusive license to these compounds in all countries of the world except ASEAN countries, with the
16
right to sublicense, under Optimer’s patents and know-how related to certain macrolide and ketolide antibiotics and related proprietary technology. The exclusivity of our license is potentially subject to the U.S. government’s right to obtain a non-exclusive, irrevocable, royalty-free, paid-up right to practice and have practiced certain patents worldwide. As partial consideration for granting us such license, we issued shares of our common stock to Optimer. We also have an obligation to make additional payments upon achievement of specified development, regulatory and commercialization milestones. The aggregate amount of such milestone payments we may need to pay is based in part on the number of products developed under the agreement. The aggregate amount would be $27.5 million if four products are developed and gain FDA approval. Additional limited milestone payments would be due if we develop more than four products. In July 2010 and July 2012, we made $0.5 million and $1.0 million milestone payments, respectively to Optimer after our successful completion of the Phase 1 and Phase 2 trials for oral solithromycin, respectively. We will owe a milestone payment of $9.5 million upon FDA approval of solithromycin. We are also obligated to make tiered, mid-single-digit royalty payments to Optimer based on annual net sales of licensed products outside the ASEAN countries, which royalties are subject to reduction in the event additional licenses are obtained from third parties in order to practice our rights under the agreement and/or we are required to grant a compulsory license to a third party.
The agreement also includes the grant of an exclusive license to Optimer and its affiliates, with rights of sublicense, under our patents and other intellectual property in any products covered by the agreement to permit Optimer to develop and/or commercialize such products in ASEAN countries. In consideration of such license, Optimer will pay us $1.0 million in milestone payments for the first two products that receive regulatory approval or have a first commercial sale in any ASEAN country, as well as tiered, mid-single-digit royalty payments based on net sales of such products, which royalties are subject to reduction in the event additional licenses are obtained from third parties in order to practice Optimer’s rights under the agreement and/or Optimer is required to grant a compulsory license to a third party. The agreement also included a collaborative research program, to be performed by the parties, which was completed on March 31, 2008.
The Optimer patents and know-how existing as of the effective date of the agreement and improvements thereof remain the property of Optimer. Except for such improvements, any know-how or inventions developed by Optimer pursuant to the agreement or that relate to the licensed products (except those generated by using grant monies provided by the U.S. government) vest in us subject to the license we granted to Optimer. The Optimer license provides Optimer with the initial responsibility to prosecute the Optimer patents relating to macrolide antibiotics. We will be responsible for prosecuting any patents controlled by us that relate to macrolide antibiotics other than the Optimer patents described above. We will have the first right to prosecute patents claiming joint inventions. We have the first right to control any proceeding involving alleged infringement of Optimer patents with respect to rights granted to us under the agreement and Optimer has such right regarding alleged infringement of our patents with respect to rights granted to Optimer under the agreement. Should we exercise our right to control any proceeding involving alleged infringement of Optimer patents, we will be responsible for the costs of these proceedings.
Subject to certain exceptions, on a country-by-country and product-by-product basis, a party’s rights and obligations under the agreement continue until the later of: (i) the expiration of the last to expire patent rights of a covered product in the applicable country or (ii) ten years from the first commercial sale of a covered product in the applicable country. As a result, the final expiration date of the Optimer license is indeterminable until the last such patents issue and results of potential patent extensions are known, or each of the first commercial sales are made, as applicable. Upon expiration of the agreement with respect to a particular product and country, the licenses granted in the agreement with respect to such product and country will remain in effect and convert to a perpetual, unrestricted, fully-paid, royalty-free, worldwide license. Either party may terminate the agreement (i) in the event of a material breach by the other party, subject to prior notice and the opportunity to cure, (ii) in the event the other party fails to use diligent efforts to develop and commercialize products in its respective territory, or if the other party makes a determination not to develop and commercialize at least one product under the agreement, or (iii) in the event of the other party’s bankruptcy. In the case of these terminations, the terminating party can elect that all licenses granted by the other party survive, subject to continuing royalty, payment and other obligations. Additionally, either party may terminate the agreement for any reason upon 30 days’ prior written notice, in which case the non-terminating party can elect that all licenses granted by the other party survive, subject to continuing royalty, payment, and other obligations.
The Scripps Research Institute. Effective June 12, 2012, we entered into a license agreement with The Scripps Research Institute, or TSRI, whereby TSRI licensed rights to the company, with rights of sublicense, to make, use, sell, and import products for human or animal therapeutic use that use or incorporate one or more macrolides as an active pharmaceutical ingredient and is covered by certain patent rights owned by TSRI claiming technology related to copper-catalyzed ligation of azides and acetylenes. The rights licensed to us are exclusive as to the People’s Republic of China (excluding Hong Kong), South Korea and Australia, and are non-exclusive in all other countries worldwide, except the member-nations of the Association of Southeast Asian Nations, which are not included in the territory of the license. Under the terms of the agreement with TSRI, we paid a one-time only, non-refundable license issue fee in the amount of $350,000 which was charged to research and development expense in the second quarter of 2012. Our rights under the agreement are subject to certain customary rights of the U.S. government that arise or result from TSRI’s receipt of research support from the U.S. government.
17
We are also obligated to pay annual maintenance fees to TSRI in the amount of (i) $50,000 each year for the first three years (beginning on the first anniversary of the agreement), and (ii) $85,000 each year thereafter (beginning on the fourth anniversary of the agreement). Each calendar year’s annual maintenance fees will be credited against sales royalties due under the agreement for such calendar year. Under the terms of the agreement, we must pay TSRI low single-digit percentage royalties on the net sales of the products covered by the TSRI patents for the life of the TSRI patents, a low single-digit percentage of non-royalty sublicensing revenue received with respect to countries in the nonexclusive territory and a mid-single-digit percentage of sublicensing revenue received with respect to countries in the exclusive territory, with the sublicensing revenue royalty in the exclusive territory and the sales royalties subject to certain reductions under certain circumstances. TSRI is eligible to receive milestone payments of up to $1.1 million with respect to regulatory approval in the exclusive territory and first commercial sale, in each of the exclusive territory and nonexclusive territory, of the first licensed product to achieve those milestones that is based upon each macrolide covered by the licensed patents. Each milestone is payable once per each macrolide. Each milestone payment made to TSRI with respect to a particular milestone will be creditable against any payment due to TSRI with respect to any sublicense revenues received in connection with the achievement of such milestone. Pursuant to the terms of the Optimer Agreement, any payments made to TSRI under this license for territories subject to the Optimer Agreement can be deducted from any sales-based royalty payments due under the Optimer Agreement up to a certain percentage reduction of the royalties due to Optimer.
Under the terms of the agreement, we are also required to pay additional fees on royalties, sublicensing and milestone payments if we, an affiliate with TSRI, or a sublicensee challenges the validity or enforceability of any of the patents licensed under the agreement. Such increased payments would be required until all patent claims subject to challenge are invalidated in the particular country where such challenge was mounted.
The term of the license agreement (and the period during which we must pay royalties to TSRI in a particular country for a particular product) will end, on a country-by-country and product-by-product basis, at such time as no patent rights licensed from TSRI cover a particular product in the particular country.
TSRI may terminate the agreement in the event (i) we fail to cure any non-payment or default on our indemnity or insurance obligations, (ii) we declare insolvency or bankruptcy, (iii) if we are convicted of a felony relating to the development, manufacture, use, marketing, distribution or sale of any products licensed under the agreement, (iv) we fail to cure any underreporting or underpayment by a certain amount in any 12-month period, or (v) we fail to cure any default on any other obligation under the agreement. We may terminate the agreement with or without cause upon written notice. In the event of such termination, (i) all licenses granted to us will terminate except in the case of any sublicensee that was not the cause of the termination, is not in default on its obligations under its sublicense, and that pays any unpaid amounts owed by us under the agreement with respect to the sublicense, and (ii) we may complete any work in progress and sell any completed inventory on hand for a period of time after termination.
Biomedical Advanced Research and Development Authority. In May 2013, we entered into an agreement with the Biomedical Advanced Research and Development Authority of the U.S. Department of Health and Human Services, or BARDA, for the evaluation and development of solithromycin for the treatment of bacterial infections in pediatric populations and infections caused by bioterror threat pathogens, specifically anthrax and tularemia.
The agreement is a cost plus fixed fee development contract, with a base performance segment valued at approximately $18.7 million, and four option work segments that BARDA may request in its sole discretion. If all four option segments are requested, the cumulative value of the agreement would be approximately $68.2 million and the estimated period of performance would be until approximately May 2018. Three of the options are cost plus fixed fee arrangements, and one option is a cost sharing arrangement for which we are responsible for a designated portion of the costs associated with that work segment. The period of performance for the base performance segment was May 2013 through February 2016.
BARDA exercised the second option in November 2014. The value of the second option work segment is approximately $16.0 million and the estimated period of performance is November 2014 through April 2017. In February 2016, BARDA exercised the third option work segment of the agreement, which is intended to fund a Phase 2/3 study of intravenous, oral capsule and oral suspension formulations of solithromycin in pediatric patients from two months old to 17 years with CABP. This option is a cost sharing arrangement under which BARDA will contribute $25.5 million and we will be responsible for an additional designated portion of the costs associated with the work segment. In September 2016, the contract was modified to increase the third option work segment by $8.0 million for increased manufacturing work related to the development of a second supply source for solithromycin. The amendment raises the value of the third option work segment to approximately $33.5 million. The estimated period of performance of this option work segment runs through May 2018.
18
Toyama Chemical Co., Ltd. We have global rights (other than the Association of South East Asian Nations, or ASEAN, countries, which are Brunei Darussalam, Cambodia, Indonesia, Laos, Malaysia, Myanmar (Burma), the Philippines, Singapore, Thailand and Vietnam) to solithromycin. We have licensed solithromycin to Toyama Chemical Co., Ltd., or Toyama, for development and commercialization in Japan while retaining the rights to the rest of the world. Toyama has successfully completed a Phase 1 trial in healthy Japanese volunteers, a Phase 1 trial to measure solithromycin levels in the upper respiratory tract, and a Phase 2 trial in CABP. In December 2016, Toyama initiated Phase 3 trials in patients infected with CABP and other respiratory infections. Toyama and we are sharing the results of our respective development activities.
In May 2013, we entered into a license agreement with Toyama whereby we licensed to Toyama the exclusive right, with the right to sublicense, to make, use and sell any product in Japan that incorporates solithromycin as its sole API for human therapeutic uses, other than for ophthalmic indications or any condition, disease or affliction of the ophthalmic tissues. Toyama also has a nonexclusive license in Japan and certain other countries, with the right to sublicense, to manufacture or have manufactured API for solithromycin for use in manufacturing such products, subject to limitations and obligations of the concurrently executed supply agreement discussed below. Toyama has granted us certain rights to intellectual property generated by Toyama under the license agreement with respect to solithromycin or licensed products for use with such products outside Japan or with other solithromycin-based products inside or outside Japan.
As consideration for the execution of the license agreement, Toyama paid us an upfront payment of $10.0 million. Toyama is also obligated to pay us up to an aggregate of $60.0 million in milestone payments, depending on the achievement of various regulatory, patent, development and commercial milestones. The first of these milestones was achieved in the third quarter of 2014 for which we received a payment of $10.0 million from Toyama. In March 2015, we recognized a $10.0 million milestone from Toyama based on the Japan Patent Office issuing a Decision of Allowance for our patent covering certain crystal forms of solithromycin in Japan, which payment was received in April 2015. In October 2016, we received a $10.0 million milestone payment when Toyama decided to progress to Phase 3 studies. Under the terms of the license agreement, Toyama must also pay us a royalty equal to a low-to-high first double decimal digit percentage of net sales, subject to downward adjustment in certain circumstances.
The term of the license agreement (and the period during which Toyama must pay royalties under the license agreement) will end, on a product-by-product basis, at the later of: (i) such time as no patent rights under the agreement cover a particular licensed product in Japan; (ii) 15 years after such product is first launched in Japan, or (iii) the first commercial sale in Japan by a third party of a generic equivalent of such licensed product.
Toyama may terminate the license agreement (i) at any time, with or without cause, upon advance notice to us, (ii) upon the occurrence of any serious adverse effect in any human clinical trial of any licensed product that would significantly impact the long term commercial viability of a licensed product in Japan, or (iii) upon our failure to obtain the issuance of certain patents or file for U.S. regulatory approvals by certain dates, or to continue certain key clinical trials. We may terminate the license agreement if Toyama or any of its sublicensees is convicted of a felony relating to the development, manufacture, use, marketing, distribution or sale of a licensed product, or upon Toyama’s failure to (i) initiate certain clinical trials in Japan by certain dates, (ii) obtain regulatory approval in Japan within a certain period of completing certain clinical trials in Japan, (iii) launch and commercialize approved licensed products in Japan within a certain period of approval, (iv) use commercially reasonable efforts to market and sell licensed products, or (v) achieve expected benchmarks for net sales of licensed products. Either party may terminate the license agreement due to the other party’s insolvency or for uncured material breach.
As part of the license agreement, Toyama and we also entered into a supply agreement, whereby we will be the exclusive supplier (with certain limitations) to Toyama and its sublicensees of API for solithromycin for use in licensed products in Japan, as well as the exclusive supplier to Toyama and its sublicensees of finished forms of solithromycin to be used in clinical trials in Japan. Pursuant to the supply agreement, Toyama will pay us for such clinical supply of finished product and all supplies of API for solithromycin for any purpose, other than the manufacture of products for commercial sale in Japan, at prices equal to our costs. All API for solithromycin supplied by us to Toyama for use in the manufacture of finished product for commercial sale in Japan will be ordered from us at prices determined by our manufacturing costs, and which may, depending on such costs, equal, exceed, or be less than such costs. The supply agreement will continue until the expiration or termination of the license agreement. Either party may terminate the supply agreement for an uncured material breach or in the event of insolvency of the other party, with Toyama’s right to terminate for our breach subject to certain further conditions in the case of our failure to supply API for solithromycin or clinical supply.
FUJIFILM Finechemicals Co., Ltd. In January 2016, we entered into a supply agreement with FUJIFILM Finechemicals Co., Ltd., or FFFC, which is intended to provide us with solithromycin in sufficient quantities and at reasonable prices to ensure we meet our obligation to Toyama under the supply agreement. We are subject to a minimum purchase obligation for a designated number of years in the event of the successful completion of a manufacturing facility to be built and validation studies to be conducted by FFFC that could run to $80 million in the aggregate, which expense would be reduced by any supply sold to Toyama. The agreement’s initial
19
term runs until December 16, 2025. After the end of the initial term, and at the end of each year thereafter, the term will automatically extend for an additional year unless either party gives written notice to the other of its intent to terminate within a designated period of time prior to the expiration of the term, in which case the agreement will terminate at the end of such term. The parties may at any time terminate the agreement by mutual written consent. Each party has the right to terminate the agreement immediately if there is a product failure, the other party becomes involved in bankruptcy, insolvency or similar proceedings or materially breaches the agreement and such breach remains uncured for a period of time following notice of the breach. A violation by us of the minimum purchase obligation is considered a material breach. A product failure is not considered a material breach by us. We have the right to terminate the agreement upon written notice if there is a supply failure. We also may terminate in the event that FFFC cannot provide us with solithromycin for more than a designated period of time. Upon termination, any unfulfilled binding portion of the forecast must be delivered by FFFC and paid for by us. We also may elect to purchase the remaining inventory of FFFC’s solithromycin and any remaining raw materials. If FFFC terminates the agreement for a material breach by us and, prior to such termination, (i) FFFC has constructed a facility in Japan for the primary purpose of manufacturing API for us under the agreement and (ii) such facility is completed and fully operational and qualified for the manufacture of API for delivery thereunder, then, except to the extent otherwise agreed to by the parties, we may be subject to declining penalties that could aggregate as much as $17.5 million.
Macrolide Pharmaceuticals, Inc.
In January 2016, we entered into an Option and License Agreement with Macrolide Pharmaceuticals, Inc. (“MP”), pursuant to which MP granted us an exclusive option to license certain of MP’s patents and know-how involving macrolides, including specifically novel methods of synthesizing solithromycin (the “Compound”). Under the agreement, we will support research at MP focused on developing a novel, cost-competitive manufacturing approach to solithromycin. The option will run until the later to occur of (i) the earlier of (a) the date that we first obtains FDA approval for any product incorporating the Compound as an API, or (b) January 27, 2019, or (ii) the date that is six months after the earlier of (a) MP’s satisfaction of certain milestones, or (b) we terminate of MP’s obligations under the evaluation program. Under the evaluation program called for in the agreement, MP will conduct research activities for the manufacture of the Compound, which activities we will evaluate to determine whether to exercise the option to license.
Upon execution of the agreement, we paid MP a non-refundable, non-creditable initial license fee of $0.4 million. For conducting the evaluation program, we paid MP a non-refundable, non-creditable fee in the amount of $0.4 million. In addition, we will pay MP the expected reasonable, documented, direct compensation-related costs of employees and advisors necessary to conduct MP’s portion of the evaluation program in the aggregate amount of $1.5 million, which we will pay in 18 equal consecutive non-refundable, non-creditable monthly installments of $83,333, beginning with the first monthly anniversary of entry into the agreement. Further, we will pay MP up to an aggregate of $1.0 million upon the satisfaction of certain performance milestones.
If we exercise the option, the license will be exclusive and worldwide (other than Association of Southeast Asian Nations) and for any and all uses in human and non-human animals, and with the right to sublicense. We may, in our discretion, exercise the option for a reduced portion of the territory and, if we make this election, may increase as it wishes within the territory, and as many times as it wishes, provided such increase is made within 60 months of our exercise of the option.
If we exercise the option, we will pay MP a non-refundable, non-creditable license fee of $1.0 million, of which $0.5 million will be paid within 15 business days of exercise, and $0.5 million will be paid in the form of “deemed royalty” payments (up to such amount) equal to a fraction of a percent of net sales of licensed products. We will pay tiered royalties of a fraction of a percent on designated levels of annual net sales of license products. Further, we will pay a non-refundable, non-creditable additional royalty equal to a fraction of a percent on the net sales of licensed products of a designated amount sold by us, our sublicensees, and product partners, but the royalty will not exceed $1.0 million in the aggregate. Royalties will be paid on a country-by-country basis and product-by-product basis until the date on which there are no valid claims of any licensed MP patent covering a product in the applicable country.
If we exercise the option, the agreement’s term will run on a country by country and product by product basis until the date on which there are no valid claims in the licensed MP patents covering a particular product in a particular country.
Manufacturing
We do not own or operate manufacturing facilities for the production of solithromycin, fusidic acid or other product candidates that we might develop, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently depend on third-party contract manufacturers for all of our required raw materials, API and finished products for our pre-clinical research and clinical trials. We employ internal resources and third-party consultants to manage our manufacturing contractors.
To date, we have ordered pre-clinical and clinical supplies under short-term contract orders. We do not have long-term contracts for the commercial supply of solithromycin. If solithromycin is approved for treatment of CABP by the FDA, we intend to enter into
20
agreements with third-party contract manufacturers for the commercial production of solithromycin. We believe there are a number of qualified manufacturers who could supply clinical and commercial quantities of solithromycin.
Solithromycin API and Oral
We have employed the services of Wockhardt Limited, or Wockhardt, to produce solithromycin API and finished oral product. We have modified our arrangement with Wockhardt, with whom we had entered into an API Manufacturing and Supply Agreement as of January 30, 2013, referred to as the 2013 Agreement. Under the 2013 Agreement, we were obligated to purchase from Wockhardt at least 70% of our total annual purchases of solithromycin in any year for clinical or commercial use in humans. Pursuant to the agreement, we were allowed to develop alternative sources of solithromycin for the other 30% of our annual needs. The 2013 Agreement’s initial term ran until December 31, 2019.
As part of the modification of our arrangement with Wockhardt, in November 2015, we entered into a new three-year API Manufacturing and Supply Agreement, pursuant to which we will submit to Wockhardt a projection of the anticipated volume of solithromycin we will order in each of the next 12 months. The first three months of the forecast are binding and we must order between a certain range of the three-month volume projected. The remainder of our projection is nonbinding and there is no minimum order. Reasonably in advance of placing a purchase order, we will work in good faith with Wockhardt to agree in writing on the purchase price of the solithromycin to be supplied under such order. We have the right to terminate the agreement at any time after we or one of our licensees decides to cease clinical development or sales of solithromycin upon 90 days’ notice to Wockhardt. Due to the lack of any minimum purchase requirement, other than a three-month supply, and due to our ability to terminate the agreement on notice, we believe that our non-cancelable obligations under this agreement are not material. The no minimum purchase requirement, negotiable purchase price and terminable upon notice terms of this agreement are similar to the terms for the other supply sources for API for solithromycin that we have developed to date.
In connection with the entry into the supply agreement with Wockhardt discussed above, we and Wockhardt mutually terminated the 2013 Agreement effective as of November 4, 2015.
In our NDAs, a Wockhardt facility located in Ankleshwar, India, was filed as the manufacturer of the solithromycin API used in the capsule and IV drug product formulations. The facility received an FDA inspection in December of 2015 that resulted in multiple observations regarding the facility’s compliance with current good manufacturing practices, or cGMP. The FDA issued an Import Alert (#66-40) for products from the facility in August 2016 and issued a Warning Letter regarding the facility’s GMP compliance deficiencies in December 2016. In the CRL we received related to our NDAs, the FDA stated that during a recent inspection of the Wockhardt facility, FDA field investigators conveyed deficiencies to representatives of the facility. Satisfactory resolution of these deficiencies is required prior to approval of the NDAs. Wockhardt’s senior management has communicated to Cempra their progress to date in resolving the deficiencies. As Wockhardt works to regain an acceptable cGMP compliance status for the Ankleshwar facility, the technical transfer to an alternate API manufacturing site that meets all technical requirements and compliance expectations is underway. The alternate API manufacturer is the Uquifa facility located in Jiutepec, Mexico.
We had been developing various additional supply sources for the API of solithromycin, including manufacturing activities at Uquifa, which we began in the second quarter of 2014. We have accelerated our API manufacturing activities with Uquifa and expect to provide data from Uquifa in our response to the CRL.
Solithromycin IV
We have employed the services of Hospira to produce our finished solithromycin IV product. In July 2013, we entered into a development and supply agreement with Hospira, whereby Hospira will assist us in the development of a reconstitutable form of solithromycin (in glass vials) for IV administration and will provide our supply of that product for development purposes. If we receive regulatory approval for such form of solithromycin, we will purchase from Hospira at least 80% of our requirements of such product for commercial sale as a human pharmaceutical product in the U.S., the European Union, Canada, Norway and Switzerland (the “Territory”). We will pay one price for clinical supplies of solithromycin and another price for commercial supplies. Beginning in 2014, Hospira may increase the price of the product for commercial use once annually by the increase in Hospira’s manufacture of the product or the annual increase of a specified inflation index. The per unit price of our commercial supply will decrease if we purchase specified volumes in a given year. Additionally, we will pay Hospira certain development fees, with specified amounts becoming payable at defined stages of the development of the product. Each year during which we sell the product, we are required to purchase a specified minimum percentage of our forecasted amount of product required for that year. We must supply Hospira at no cost the active pharmaceutical ingredient for the product. If Hospira fails to supply a specified percentage of product, we may purchase all or a portion of our requirements of the product from an alternative supplier until Hospira remedies the supply failure. Unless earlier terminated, the agreement will remain in effect until the end of the third year after the first commercial sale of the product in the Territory. Thereafter, the agreement will automatically renew for an indefinite period. Beginning one year after the first commercial sale of the product in the Territory, either party may terminate the agreement at will upon 24 months’ notice. Prior to the completion
21
of the development of the product or the submission of an application for regulatory approval in the Territory, either party may terminate the development project or the agreement if such party determines the development of intravenous solithromycin under the agreement is not clinically, commercially or technically feasible.
In our NDAs, a Hospira facility in McPherson, Kansas was filed as the manufacturer of the sterile IV drug product vials for Cempra. The facility received an FDA inspection in June of 2016 that resulted in multiple observations regarding the facility’s compliance with cGMP. Hospira recently received a warning letter related to this facility. We would evaluate the results of any potential future re-inspection on this facility to determine our next steps. In the meantime, we are moving forward to qualify another supplier for IV solithromycin. In the CRL we received related to our NDAs, the FDA stated that during a recent inspection of the Hospira facility, FDA field investigators conveyed deficiencies to representatives of the facility. Satisfactory resolution of these deficiencies is required prior to approval of the NDAs. Hospira’s senior management has communicated to Cempra their progress to date in resolving these deficiencies. As Hospira works to regain an acceptable GMP compliance status, we are working to identify an alternative sterile manufacturing site that will meet all technical requirements and compliance expectations.
Fusidic Acid
We have a long term supply arrangement with Ercros, S.A., or Ercros, in Madrid, Spain, in which Ercros agrees to exclusively supply us with fusidic acid in the U.S., and we agree to exclusively obtain our supply of fusidic acid for commercial sale from Ercros, subject to a right to develop a second source for limited supply quantities. The supply agreement with Ercros will continue until at least March 2029, subject to earlier termination for our uncured material breach or our bankruptcy or insolvency. In addition, the exclusivity restrictions on Ercros are subject to termination if we fail to file with the FDA an NDA for the sale of fusidic acid prior to December 31, 2017. We believe Ercros is one of only two currently known manufacturers that can produce fusidic acid compliant with the purity required for human use. Fusidic acid is difficult to produce at the required purity levels because of its complex fermentation process. We believe the only other manufacturer of fusidic acid with sufficient purity is Leo Laboratories, which is using its manufacturing capacity for its own needs. We have yet to identify a viable alternate source of fusidic acid but continue to research alternatives. We intend to utilize a third-party manufacturer to produce the finished dosing formulation of fusidic acid.
Government Regulation and Product Approval
Government authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record keeping, promotion, advertising, distribution, marketing and export and import of products such as those we are developing. Solithromycin, fusidic acid and any other antibiotic product candidate that we develop must be approved by the FDA through the NDA process before they may be legally marketed in the U.S.
U.S. Drug Development Process
In the U.S., the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act, or FDCA, and implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a drug may be marketed in the U.S. generally involves the following:
|
|
• |
Completion of pre-clinical laboratory tests, animal studies and formulation studies according to good laboratory practices, or GLP, or other applicable regulations; |
|
|
• |
Submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
|
|
• |
Performance of adequate and well-controlled human clinical trials according to the FDA’s current good clinical practices, or cGCP, to establish the safety and efficacy of the proposed drug for its intended use; |
|
|
• |
Submission to the FDA of an NDA for a new drug; |
22
|
|
• |
Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with the FDA’s cGMPs to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
|
|
• |
FDA review and approval of the NDA. |
Each new clinical protocol must be submitted to the FDA review, and to an Institutional Review Board, or IRB, for approval. Protocols detail, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
U.S. Review and Approval Processes
The results of product development, pre-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances.
In addition, under the FDAAA, all NCEs prior to approval are referred to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions, unless the Secretary of Health and Human Services provides in the action letter on the drug application a summary of the reasons why it was not referred. An advisory committee is a panel of experts who provide advice and recommendations when requested by the FDA on matters of importance that come before the agency. The FDA is not bound by the recommendation of an advisory committee.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves post-approval clinical trials designed to further assess a drug safety and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Post-Approval Requirements
Any drug product for which we receive FDA approval will be subject to continuing regulation by the FDA, including, among other things, record keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, cGMP requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
23
The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems (quality or safety) occur after the product reaches the market. Later discovery of previously unknown quality, safety, or other problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, warning letters, holds on clinical trials, product recalls or seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.
In addition, from time to time, legislation is drafted, introduced and passed in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. For example, in September 2007, the FDAAA was enacted giving the FDA enhanced post-market authority, including the authority to require post-market studies and clinical trials, labeling changes based on new safety information and compliance with a risk evaluation and mitigation strategy. Failure to comply with any requirements under the new law may result in significant penalties. The law also authorized significant civil money penalties for the dissemination of false or misleading direct-to-consumer advertisements and allows the FDA to require companies to submit direct-to-consumer television drug advertisements for FDA review prior to public dissemination. Additionally, the law expanded the clinical trial registry so that sponsors of all clinical trials, except for Phase 1 clinical trials, are required to submit certain clinical trial information for inclusion in the clinical trial registry data bank. In addition to this legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Other U.S. Health Care Laws and Compliance Requirements
In the U.S., our activities are subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Under the Veterans Health Care Act, or VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including U.S. Department of Veterans Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes purport to require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives, as well as prohibiting pharmacies and other health care entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing, and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Foreign Regulation
In addition to regulations in the U.S., we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products to the extent we choose to sell any products outside of the U.S. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
24
E.U. member states require both regulatory clearances by the national competent authority and a favorable ethics committee opinion prior to the commencement of a clinical trial. Under the E.U. regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, conducted by the EMA, provides for the grant of a single marketing authorization that is valid for all E.U. member states. The centralized procedure is compulsory for medicines produced by certain biotechnological processes, products with a new active substance indicated for the treatment of certain diseases such as neurodegenerative disorder or diabetes and products designated as orphan medicinal products and optional for those products which are highly innovative or for which a centralized process is in the interest of patients. The decentralized procedure of approval provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one-member state, known as the reference member state. Under the decentralized approval procedure, an applicant submits an application, or dossier, and related materials (draft summary of product characteristics, draft labeling and package leaflet) to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states.
We presented to and received feedback from several E.U. member countries before submitting our MAA to the EMA. As part of this, our PIP was accepted by the EMA for the suspension formulation of solithromycin to treat CABP in pediatric patients.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. In the U.S. and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend considerably on the availability of reimbursement from third-party payors. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain FDA approvals. Our products may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the U.S. government enacted legislation providing a prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through prescription drug plans operating pursuant to this legislation. These plans will likely negotiate discounted prices for our products. In March 2010, the Patient Protection and Affordable Care Act became law, which substantially changed the way healthcare is financed by both governmental and private insurers. We anticipate that this legislation will result in additional downward pressure on coverage and the price that we receive for any approved product. Federal, state and local governments in the U.S. continue to consider legislation to limit the growth of health care costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of our particular drug products to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the U.S. has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more
25
products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Corporate History and Information
We were formed as Cempra Holdings, LLC, a limited liability company under the laws of the State of Delaware, on May 16, 2008. Cempra Holdings, LLC was formed in connection with a reorganization whereby the stockholders of Cempra Pharmaceuticals, Inc., a corporation formed under the laws of the State of Delaware on November 18, 2005, exchanged their shares of Cempra Pharmaceuticals, Inc. stock for shares of Cempra Holdings, LLC, pursuant to a merger of a subsidiary of Cempra Holdings, LLC with and into Cempra Pharmaceuticals, Inc., as a result of which Cempra Pharmaceuticals, Inc. became a wholly owned subsidiary of Cempra Holdings, LLC.
On February 2, 2012, Cempra Holdings, LLC converted from a Delaware limited liability company to a Delaware corporation and was renamed Cempra, Inc. As a result of the corporate conversion, the holders of common shares of Cempra Holdings, LLC became holders of shares of common stock of Cempra, Inc. and the holders of preferred shares of Cempra Holdings, LLC became holders of shares of common stock of Cempra, Inc. Holders of options to purchase common shares of Cempra Holdings, LLC became holders of options to purchase shares of common stock of Cempra, Inc. Holders of notes convertible into preferred shares of Cempra Holdings, LLC and associated warrants exercisable for preferred shares of Cempra Holdings, LLC became holders of shares of common stock and warrants to purchase shares of common stock of Cempra, Inc.
We have two subsidiaries, Cempra Pharmaceuticals, Inc. and CEM-102 Pharmaceuticals, Inc. Our primary executive offices are located at 6320 Quadrangle Drive, Suite 360, Chapel Hill, NC 27517-8149, and our telephone number is (919) 313-6601. Our website address is http://www.cempra.com. The information contained in, or that can be accessed through, our website is not part of this report.
We make available, free of charge through our website, our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports as soon as is reasonably practicable after such material is electronically filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC, but other information on our website is not incorporated into this report. The SEC maintains an Internet site that contains these reports at www.sec.gov. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330.
Employees
As of February 27, 2017, we had 45 employees. None of our employees is subject to a collective bargaining agreement. We consider our relationship with our employees to be good.
26
This report contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed in this report. Factors that could cause or contribute to these differences include, but are not limited to, those discussed below and elsewhere in this report and in any documents incorporated in this report by reference.
If any of the following risks, or other risks not presently known to us or that we currently believe to not be significant, develop into actual events, then our business, financial condition, results of operations or prospects could be materially adversely affected. If that happens, the market price of our common stock could decline, and stockholders may lose all or part of their investment.
Risks Related to our Business
We may change our plans for the development of solithromycin for the treatment of CABP or any of our other drug candidates or any of the indications we are pursuing for our drug candidates.
In late December, the FDA issued a complete response letter, or CRL, to our NDAs for solithromycin for the treatment of CABP. The FDA issues complete response letters to indicate that the review cycle for an application is complete and that the application is not ready for approval in its present form. The FDA determined that the risk of hepatotoxicity of solithromycin had not been adequately characterized. The FDA noted the size of the safety database is limited to 920 patients who received solithromycin at the proposed dose and duration, and is too small to adequately characterize the nature and frequency of serious hepatic adverse effects. To address this deficiency, the FDA is recommending a comparative study to evaluate the safety of solithromycin in patients with CABP. Specifically, the CRL recommends that we consider a study of approximately 9,000 patients exposed to solithromycin to enable exclusion of serious drug induced liver injury, or DILI, events occurring at a rate of approximately 1:3000 with a 95% probability. In addition, the CRL noted that while the FDA reserves comment on the proposed labeling until the NDAs are otherwise adequate, even in the absence of a case of Hy’s Law or of another form of serious DILI in future studies, labeling will need to include adequate information about the potential for hepatotoxicity, limiting use to patients who have limited therapeutic options and limitations regarding duration of therapy. A comprehensive plan for post-marketing safety assessment including an enhanced pharmacovigilance program will also be required. Further, the CRL stated that during recent inspections of the Wockhardt and Hospira manufacturing facilities, the FDA field investigator conveyed deficiencies to representatives of the facilities. Satisfactory resolution of these deficiencies is required prior to approval of the NDAs. Details on these deficiencies were not provided in the CRL.
While we have met with the FDA to obtain more clarity on the findings in the CRL, additional meetings will be required. In addition, the work to address the manufacturing issues could be lengthy and costly and is not within our control. Further, we are assessing the implications of the label warning on the marketability of solithromycin. In light of this, we are still determining the future of our development program for solithromycin. Depending on our resources, as well as any further insight into responding to the CRL and the development plans for solithromycin, including the cost and duration of responding to the CRL, as well as the anticipated market for solithromycin, we may choose to alter, reduce or terminate our development plans for solithromycin.
Given our need to conserve resources, we could change our plans for the other indications we are pursuing for solithromycin or our plans and research and development activities for any of our other product candidates.
Given the uncertainty at this time regarding our development plans for our drug product candidates, the other risks cited in this Section 1A generally do not reflect any possible changes to our current development plans for those product candidates. Consequently, the other risk factors should not be understood to indicate any decision by us to necessarily continue with our current development plans.
We may not realize the expected benefits of our recently initiated cost-saving initiatives.
As a result of the receipt of the CRL and the resulting uncertainty regarding our development plans for solithromycin, reducing costs to conserve our financial resources is a key focus of our management. In late February 2017, we initiated a reduction to our workforce. Personnel reductions were initiated across our entire organization that have resulted in a remaining workforce of 45 full-time employees. The principal objective of the reduction in workforce is to enable us to conserve our financial resources as we determine whether and how to respond to the CRL, as well as determine the future of the other indications we are pursuing for solithromycin and the development programs for our other drug candidates.
We expect to record an aggregate charge related to one-time termination benefits of approximately $3.5 million to be recorded in the first quarter of 2017. If we experience unanticipated inefficiencies or incremental costs in connection with restructuring activities, such as unanticipated inefficiencies or other negative impacts caused by reducing headcount, we may be unable to
27
meaningfully realize our expected cost savings and we may incur expenses in excess of what we anticipate. Either of these outcomes could prevent us from meeting our strategic objectives and could adversely impact our results of operations and financial condition.
We may undertake additional cost-saving initiatives, but there is a risk that we might not achieve the desired savings and efficiencies with any future initiatives. Further, the termination of any program we currently are pursuing could result in termination fees and other fixed costs that could have a material adverse impact on our results of operations and financial condition.
Our efforts to explore external late-stage assets and other potential strategic business opportunities to determine the best use of our resources and clinical programs to deliver value to patients and shareholders may not result in any definitive transaction or deliver the expected benefits, and may create a distraction for our management and uncertainty that may adversely affect our operating results and business.
In February 2017, as a consequence of the solithromycin CRL we received, and subsequent discussions with the FDA, resulting in the delay of the potential approval of solithromycin, we initiated companywide cost and personnel reductions. The principal objective of the reductions is to enable us to conserve our financial resources as we evaluate our path forward on our existing pipeline and potential business development opportunities. No timetable has been set for completion of this evaluation process, and there can be no assurance that any transaction will result. Strategic alternatives we may pursue could include, but are not limited to, joint ventures or partnering or other collaboration agreements, licensing arrangements, or another transaction intended to maximize shareholder value, such as a merger, a sale of our company or some or all of its assets, or another strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms.
There are various uncertainties and risks relating to our evaluation and negotiation of possible strategic alternatives and our ability to consummate a definitive transaction, including:
|
|
• |
expected benefits may not be successfully achieved; |
|
|
• |
evaluation and negotiation of a proposed transaction may distract management from focusing our time and resources on our current operations, which could have a material adverse effect on our operating results and business; |
|
|
• |
the process of evaluating proposed transactions may be time consuming and expensive and may result in the loss of business opportunities; |
|
|
• |
perceived uncertainties as to our future direction may result in increased difficulties in retaining key employees, particularly senior management; |
|
|
• |
even if we negotiate a definitive agreement, successful integration or execution of the strategic alternative will be subject to additional risks; |
|
|
• |
the current market price of our common stock may reflect a market assumption that a transaction will occur, and during the period in which we are considering a transaction, the market price of our common stock could be highly volatile; and |
|
|
• |
a failure to complete a transaction could result in a negative perception by investors of our company generally and could cause a decline in the market price of our common stock, as well as lead to greater volatility in the market price of our common stock, all of which could adversely affect our ability to access the equity and financial markets, as well as our ability to explore and enter into different strategic alternatives. |
We may not reduce our operating expenses as much as planned, which could negatively impact our cost-saving initiatives.
As part of our recently enacted corporate restructuring, we intend to reduce our research and corporate expenses while we determine whether and how to respond to the CRL, as well as determine the future of the other indications we are pursuing for solithromycin and the development programs for our other drug candidates, and consider potential business development opportunities. We might not be successful in reducing these expenses as much as we plan or to any significant degree, which could prevent us from meeting our strategic objectives and have a material adverse impact on our results of operations and financial condition.
We have incurred significant operating losses since inception and anticipate that we will incur continued losses for the foreseeable future.
As of December 31, 2016, we had an accumulated deficit of approximately $437.0 million. We have no product revenues, but do have revenue from contract research and upfront and milestone fees paid in connection with a license agreement. We have funded our operations to date from the private sale of equity and debt, our IPO and public offerings of our common stock. We expect to incur substantial additional losses over at least the next few years as we pursue our research, development, pre-clinical testing, clinical trial and commercialization activities, especially those related to solithromycin, especially to address the CRL, and fusidic acid, and we expect these losses to continue for a period of time until the approval of and any resulting launch of solithromycin, if any. In addition,
28
we also expect to incur additional costs operating as a public company. The amount of future losses and when, if ever, we will achieve profitability are uncertain.
If we are unable to commercialize solithromycin for the treatment of CABP or experience significant delays in doing so, our business, financial condition and results of operations will be materially adversely affected.
Our ability to generate product revenues from our main product candidate solithromycin for the treatment of CABP will depend heavily on the successful development and eventual commercialization of solithromycin for such indication, if approved.
In May 2016, we announced the completion of the NDAs for solithromycin for the treatment of CABP. In late December 2016, the FDA issued a Complete Response Letter, or CRL, to our NDAs for solithromycin for the treatment of CABP. The FDA issues complete response letters to indicate that the review cycle for an application is complete and that the application is not ready for approval in its present form. The FDA determined that the risk of hepatotoxicity had not been adequately characterized. The FDA noted the size of the safety database is limited to 920 patients who received solithromycin at the proposed dose and duration, and is too small to adequately characterize the nature and frequency of serious hepatic adverse effects. To address this deficiency, the FDA is recommending a comparative study to evaluate the safety of solithromycin in patients with CABP. Specifically, the CRL recommends that we consider a study of approximately 9,000 patients exposed to solithromycin to enable exclusion of serious drug induced liver injury, or DILI, events occurring at a rate of approximately 1:3000 with a 95% probability.
In addition, the CRL noted that while the FDA reserves comment on the proposed labeling until the NDAs are otherwise adequate, even in the absence of a case of Hy’s Law or of another form of serious DILI in future studies, labeling will need to include adequate information about the potential for hepatotoxicity, limiting use to patients who have limited therapeutic options and limitations regarding duration of therapy. A comprehensive plan for post-marketing safety assessment including an enhanced pharmacovigilance program will also be required.
Further, the CRL stated that during recent inspections of the Wockhardt and Hospira manufacturing facilities, the FDA field investigator conveyed deficiencies to representatives of the facilities. Satisfactory resolution of these deficiencies is required prior to approval of the NDAs.
While we have met and expect to continue dialogue with the FDA to discuss the CRL and are working to address the issues raised in the CRL, approval of our NDAs may not be received for a number of reasons, including the following:
|
|
• |
we may not prove the safety of solithromycin if there is a higher rate of DILI events than that set out in the CRL; |
|
|
• |
our third party manufacturers may not provide proper manufacturing capabilities that meet regulatory approval; and |
|
|
• |
the FDA could raise other issues not set out in the CRL. |
Our MAA for solithromycin for the treatment of CABP remains under review in the EU. If the MAA is not approved by the EMA, we will not receive marketing authorization for solithromycin in the EU and will be unable to sell solithromycin in the EU.
If our current applications for marketing approval in the U.S. and the EU are denied, we may need to conduct additional clinical trials at significant delay and cost or abandon development of solithromycin altogether. The failure to achieve regulatory approval of solithromycin for the treatment of CABP could materially adversely affect our business, financial condition and results of operations.
Even if we receive regulatory approval, any required post-marketing safety studies may fail to demonstrate safety, the labeling approved by the FDA may be so onerous as to prevent doctor and patient acceptance of solithromycin or other issues may arise with the administration, marketing or manufacture of solithromycin that could have a material adverse effect on our business, financial condition and results of operations.
Our planned commercialization of solithromycin for the treatment of CABP could be delayed or denied if our third party manufacturers do not meet regulatory requirements.
Each component of our planned solithromycin products, both the oral and intravenous formulations that are the subjects of our NDAs for the treatment of CABP on file with both the FDA and the EMA, as well as the manufacturer of each component, must meet all applicable regulatory requirements in order to receive regulatory approval as well as maintain regulatory compliance after approval. This occurred in 2016 when the FDA imposed an import restriction on a Wockhardt facility in India that manufactures solithromycin for us and the FDA cited in the CRL that there were deficiencies at the Wockhardt and Hospira facilities regarding their compliance with cGMP.
29
In the future, any of the following could have a material adverse impact on our business, financial condition, results of operations or prospects:
|
|
• |
If any manufacturer we employ for the production of solithromycin cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, in which event we would not receive approval from the FDA for solithromycin for the treatment of CABP; |
|
|
• |
Even if our manufacturers are successful in ultimately meeting FDA requirements for our NDAs, compliance issues that arise prior to receipt of approval of the NDAs could cause significant and expensive delays prior to receiving approval; |
|
|
• |
A delay in receiving approval of the NDAs could negatively impact any pre-launch activities for solithromycin; |
|
|
• |
If approval is received, the failure of our third party manufacturers to continue to meet applicable regulatory requirements could result in the loss of those manufacturing sources, which could cause shortages of commercial product. |
These same risks apply to our current application to the EMA for solithromycin for the treatment of CABP.
We are heavily dependent on the success of solithromycin and, to a lesser extent, fusidic acid, which are still under clinical development. The FDA and foreign regulatory approval process is lengthy, time consuming and inherently unpredictable and if we are ultimately unable to obtain regulatory approval for solithromycin or fusidic acid our business will be substantially harmed.
We have no products that have been approved for sale. We cannot commercialize, market, or sell solithromycin or fusidic acid in the U.S. without FDA approval. FDA approval for either product, if received, is at least one year away. To commercialize solithromycin outside of the U.S., we would need applicable foreign regulatory approval, for which we have submitted an MAA to the EMA. The clinical development of solithromycin and fusidic acid for any indication is susceptible to the risk of failure inherent in any stage of drug development, including failure to achieve efficacy across a broad population of patients, the occurrence of severe adverse events and the FDA or any applicable foreign regulatory authority determining that a drug product is not approvable.
The process required to obtain approval for commercialization from the FDA and similar foreign authorities is unpredictable, and typically takes many years following the commencement of clinical trials depending on numerous factors. In addition, approval policies, regulations, or the type and amount of clinical data necessary to obtain regulatory approval may change during the course of a product’s clinical development. We may fail to obtain regulatory approval for solithromycin, fusidic acid or any other product candidates for many reasons, including the following:
|
|
• |
we may not be able to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for any indication; |
|
|
• |
the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval, and/or the FDA may require additional, expensive trials; |
|
|
• |
we may not be able to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
|
|
• |
we may not be able to demonstrate that a product candidate is non-inferior or superior to the current standard of care, future competitive therapies in development, or over placebo in any indications for which the FDA requires a placebo-controlled trial; |
|
|
• |
the data collected from clinical trials of any product candidates that we develop may not be sufficient to support the submission of a new drug application, or NDA, or other submission or to obtain regulatory approval in the U.S. or elsewhere; |
|
|
• |
the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval; |
|
|
• |
the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
|
|
• |
the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from pre-clinical studies or clinical trials; |
|
|
• |
the FDA or comparable foreign regulatory authorities may not accept data generated at our clinical trial sites; |
30
|
|
• |
the FDA or comparable foreign regulatory authorities may fail to approve the clinical practices of the third-party clinical research organizations, or CROs, we use for clinical trials; and |
|
|
• |
the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we or our collaborators enter into agreements for clinical and commercial supplies. |
This lengthy approval process as well as the unpredictability of future clinical trial results may prevent us from obtaining regulatory approval to market solithromycin, fusidic acid or any future product candidates for any indication, which would significantly harm our business, financial condition, results of operations and prospects.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing is expensive, can take many years to complete and its outcome is highly uncertain. Failure can occur at any time during the clinical trial process due to inadequate performance of a drug or inadequate adherence by patients or investigators to clinical trial protocols. Pursuant to FDA guidelines, new drugs must show non-inferiority or superiority to existing approved treatments. We conducted our solithromycin for CABP clinical trials pursuant to proposed guidelines published by the FDA. While we believed we had completed all the clinical trials necessary to support the NDA for solithromycin for CABP and have a sufficient database of both efficacy and safety, the FDA disagreed with our assessment and is requiring additional clinical data to support approval, specifically lack of liver toxicity. This has added to the duration and cost of the development of solithromycin for CABP, and the liver toxicity trial could take longer and be more expensive than we currently estimate. Any additional trials, for whatever reason, would add to the time and cost of solithromycin’s development. We face these same risks with any other indication we are pursuing for solithromycin as well as for our other product candidates.
In addition, the results of pre-clinical studies and early clinical trials of product candidates may not be predictive of the results of later-stage clinical trials. For example, the positive results of our Phase 3 trial for fusidic acid for the treatment of ABSSSI that we announced in late February 2017 may not be repeated in subsequent trials. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience than us, have suffered significant setbacks in Phase 2 and Phase 3 clinical trials despite achieving successful results in earlier stage trials. The failure to obtain positive results in any of our Phase 2 or Phase 3 clinical trials could seriously impair the development prospects, and even prevent regulatory approval, of solithromycin or fusidic acid or any candidate in our existing proprietary macrolide library. Even with positive clinical trial results, there is risk that regulators will not accept the clinical trial findings or will require additional trials or other data, as we recently experienced with the CRL for solithromycin as a treatment for CABP.
Further, regulatory approvals in foreign countries are subject to risks associated with different regulatory requirements, including clinical trial guidance, and regulatory schemes, including, for example, multiple country regulation within the European Union. As a result, clinical trial results and other regulatory processes undertaken by us within the U.S. may not be accepted in foreign countries, which would add to the cost and time to develop our product candidates in foreign countries.
We have no experience as a company in bringing a drug to regulatory approval or to the market.
As a company, we have never obtained regulatory approval for, or commercialized, a drug. It is possible that the FDA may refuse to accept any or all of our planned NDAs for substantive review or may conclude after review of our data that our application is insufficient to obtain regulatory approval of solithromycin, fusidic acid or any future product candidates, as it did in issuing the CRL for our NDAs for solithromycin for the treatment of CABP in December 2016. As with the CRL, if the FDA does not accept or approve any or all of our future NDAs, it may require that we conduct additional clinical, pre-clinical or manufacturing validation studies, which may be time-consuming and costly, and submit that data before it will reconsider our applications. Depending on the extent of these or any other FDA required studies, approval of any NDA or application that we submit may be significantly delayed, possibly for several years, or may require us to expend more resources than we have planned or have available. We estimate that we will not be able to complete a response to the CRL until the second half of 2017 at the earliest. Any delay in obtaining, or an inability to obtain, regulatory approvals would prevent us from commercializing solithromycin for CABP or any other indication or fusidic acid, generating revenues and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve any NDA we submit, including our current NDAs for solithromycin for CABP. If any of these outcomes occur, we may be forced to abandon our current and any future NDAs for either solithromycin or fusidic acid or both, which would materially adversely affect our business and could potentially cause us to cease operations. We face similar risks for any approval in a foreign jurisdiction.
31
We might not successfully differentiate solithromycin from telithromycin (Ketek®), a macrolide found to cause severe side effects, including liver failure.
Ketek is a macrolide antibiotic that the FDA approved in 2004 for the treatment of multi-drug resistant pneumococci and other CABP bacteria. Soon after release, however, Ketek was found to cause reversible visual disturbances, exacerbate myasthenia gravis (a neurological disorder characterized by improper muscle regulation) and cause liver failure. These effects led the FDA to require the drug label for Ketek to include a strengthened warning section regarding specific drug-related adverse events and contributed to Ketek being withdrawn in 2007 for the treatment of all infections other than CABP. Through ongoing research, we have developed multiple ways to differentiate solithromycin from Ketek. Our research suggests these side effects may be caused by the pyridine moiety, which forms a part of the structure of Ketek. We have demonstrated that pyridine inhibits the action of nicotinic acid acetylcholine receptors that could result in the side effects caused by Ketek. Solithromycin and older generation macrolides, including azithromycin and clarithromycin, that have been widely marketed do not have a pyridine component.
Because of the Ketek experience, we believe solithromycin, which is a macrolide, was carefully scrutinized by the FDA as part of its review of our NDAs for solithromycin as a treatment for CABP. Another ketolide, cethromycin (Restanza) that is being developed by Advanced Life Sciences, received a complete response letter from the FDA in 2009, and it appears that development of the drug has not continued. The FDA attributed its rejection to the company’s trial design, which enrolled patients with less severe CABP and was conducted prior to the release of the FDA’s updated trial design guidance. Also, the FDA placed a partial clinical hold on one of our Phase 1 clinical trials for oral solithromycin over concern about possible toxicity related to solithromycin, and subsequently converted the partial clinical hold into a full clinical hold in April 2010. At the time, the FDA had concerns that solithromycin, as a fluoroketolide, may have similar toxicity issues as Ketek. While we addressed the FDA’s concerns and were allowed to proceed with the trial, which we successfully completed, we believe that the clinical hold indicates the scrutiny that the FDA applied to our NDAs for solithromycin due to the Ketek experience.
If our prior research is proven to be incorrect or if in a study of liver toxicity in response to the CRL or in other research solithromycin demonstrates similar side effects to Ketek or poses high risk of liver injury or any other SAE, the FDA might not approve solithromycin. Even if we receive approval and future studies show no adverse events, the CRL stated the FDA will require a warning on the label (as stated in the CRL) and might withdraw approval, require us to conduct additional clinical trials, or require additional warnings on product labeling, which could limit the available market for solithromycin, any of which would significantly harm our ability to generate revenues from solithromycin.
Even if the FDA approves solithromycin, physicians may not be convinced that solithromycin is a safe and effective treatment for CABP and other infections, especially with a label warning that the CRL stated will be required. If physicians believe solithromycin demonstrates characteristics similar to Ketek or poses other unacceptably high risks, they might not prescribe solithromycin, which would negatively affect our revenues.
The results of any ongoing or future study or trial involving solithromycin, if negative, could have an adverse effect on FDA and other regulatory approval of solithromycin as a treatment for CABP as well our commercialization efforts for solithromycin and market acceptance of the same.
If the results of any of our ongoing or future studies of solithromycin should be negative, it could have an adverse effect on the planned NDA and potential approval of solithromycin as a treatment for CABP, including a delay in or lack of approval, which was the case with our NDAs for solithromycin for CABP. If after approval were received, negative results in any study or trial of solithromycin could have a negative effect on our commercialization and market acceptance of solithromycin, including downward pressure on pricing.
Our dependence upon third parties for the manufacture and supply of solithromycin, fusidic acid and any future product candidates may cause delays in, or prevent us from, successfully developing and commercializing our products.
We do not currently have nor do we plan to build the infrastructure or capability internally to manufacture solithromycin or fusidic acid for use in the conduct of our clinical trials or for commercial supply. We have contracted with companies in India, Japan and North America to provide commercial supplies of solithromycin. Similarly, in July 2013, we contracted with Hospira Worldwide, Inc., or Hospira (now owned by Pfizer), to provide us with clinical and commercial supplies of the intravenous form of solithromycin. Under the terms of the agreement, if Hospira fails to supply a specified percentage of product to us, we may seek an alternate supplier. We aim to have several sources of oral and IV solithromycin in several locations worldwide and to that end we are working on developing at least one additional manufacturing source in North America for IV solithromycin. However, if any of these entities were unable to provide our needed supply of oral or intravenous solithromycin, we may not be able to negotiate an agreement with another source on acceptable terms or in a timely fashion, if at all.
32
In addition, regulatory requirements could pose barriers to the manufacture of our API and finished product for solithromycin and Fusidic acid. Our third-party manufacturers are required to comply with the FDA’s current good manufacturing practices, or cGMP, regulations. As a result, the facilities used by any of our current and future manufacturers to manufacture solithromycin and fusidic acid must be approved by the FDA after we submit an NDA to the FDA and before approval of solithromycin and fusidic acid. Similar regulations apply to manufacturers of our products for use or sale in foreign countries. We do not control the manufacturing process of solithromycin or fusidic acid and are completely dependent on these third-party manufacturing partners for compliance with the applicable regulatory requirements for the manufacture of solithromycin and fusidic acid API and their finished product. If our manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA and any applicable foreign regulatory authority, they will not be able to secure the applicable approval for their manufacturing facilities. If these facilities are unable to comply with the FDA’s cGMP requirements, or otherwise are not approved for the commercial manufacture of solithromycin or fusidic acid, we may need to find alternative manufacturing facilities, which would result in significant delays of up to several years in obtaining approval for solithromycin or fusidic acid. In addition, our manufacturers will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. Failure by any of our manufacturers to comply with applicable cGMP regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply, and criminal prosecutions, any of which could have a material adverse impact on our business, financial condition, results of operations or prospects.
As part of its review of our NDAs for solithromycin for CABP, the FDA evaluated the CMC section of those NDAs. The FDA placed an import alert on a Wockhardt manufacturing facility in August 2016, several months after our NDAs had been submitted and accepted for review by the FDA. The FDA’s concerns with Wockhardt’s operations and facilities are related to the GMP quality systems at Wockhardt. In the CRL issued by the FDA on our NDAs, the FDA stated that during recent inspections of the Wockhardt and Hospira manufacturing facilities, the FDA field investigator conveyed deficiencies to representatives of the facilities. Satisfactory resolution of these deficiencies is required prior to approval of the NDAs. Details on these deficiencies were not provided in the CRL. In December 2016, the FDA issued a warning letter to Wockhardt regarding cGMP compliance deficiencies at their Ankleshwar facility. There can be no assurance if or when Wockhardt will be able to resolve these issues.
Similarly, one of two facilities in India at which Wockhardt produced capsules of solithromycin for clinical trials, was audited by the Medicines and Healthcare Products Regulatory Agency, or MHRA, the regulatory agency of Great Britain, which took issue with Wockhardt’s practices at the plant and as a result the capsules of solithromycin produced at that plant could not be imported into Europe for use in our Phase 3 clinical trial. We had some capsules on hand and also produced new capsules, but the incident caused a several month delay in having drug available for our Phase 3 oral solithromycin trials in Europe, which, however, was planned to be in advance of the onset of flu season in Europe so the delay had minimal impact on the trial timeline. However, similar experiences could occur with more significant impact on our development program or commercialization of solithromycin.
As part of our earlier commercialization plans for the anticipated demand for solithromycin for the treatment of CABP, we have been developing various additional supply sources for the API of solithromycin. This includes manufacturing activities at Uquifa in Jiutepec, Mexico, which we began in the second quarter of 2014. We have accelerated our API manufacturing activities with Uquifa and expect to provide data from Uquifa to the FDA to address the issues noted in the CRL related to manufacturing. However, as we experienced, the issues cited by the FDA at the Wockhardt facility adversely affected our NDAs and similar issues, if experienced, could have similar consequences on any future applications.
We employ the services of Ercros S.A., or Ercros, to produce fusidic acid’s API and intend to utilize a third-party manufacturer to produce the finished dosing formulation of fusidic acid. We have a long-term exclusive supply arrangement with Ercros to produce the fusidic acid we need in which Ercros agrees to exclusively supply us with fusidic acid in the U.S., and we agree to obtain our supply of fusidic acid for commercial sale exclusively from Ercros, subject to a right to develop a second source for limited supply quantities. We believe Ercros is one of only two currently known manufacturers that can produce fusidic acid compliant with the purity required for human use. The second manufacturer is not available as a supplier to us. Fusidic acid is difficult to produce at the required purity levels because of its complex fermentation process. As such, there are underlying risks associated with its manufacture, which could include cost overruns, new impurities, difficulties in scaling up or reproducing manufacturing processes and lack of timely availability of raw materials. We have yet to identify a viable second source of fusidic acid but continue to research alternatives. If Ercros cannot supply sufficient quantities of fusidic acid to make clinical supplies, it would harm our ability to develop fusidic acid. We may not be able to locate a second manufacturer or, if we do, we may not be able to negotiate an agreement on favorable terms, if at all. On June 20, 2012, the FDA issued a Warning Letter to Ercros, citing cGMP violations at the Ercros facility that manufactures the API for fusidic acid. Although some of the alleged violations may be related to products other than fusidic acid, the FDA’s issuance of a Warning Letter signifies FDA concerns with cGMP compliance at the Ercros facility. We believe Ercros is actively working with FDA to resolve these issues. However, if Ercros is unable to satisfactorily address the FDA’s concerns in a timely manner, the FDA may take further enforcement actions that could significantly jeopardize our supply of fusidic acid API for use in clinical trials or later commercialization. For example, the FDA might issue an import alert, which could preclude us from importing
33
fusidic acid API manufactured at the Ercros facility. Particularly in light of the unavailability of alternative suppliers for fusidic acid API, this could significantly impact our ability to develop and commercialize fusidic acid.
Finally, we also could experience manufacturing delays if our third-party manufacturers give greater priority to the supply of other products over our product candidates or otherwise do not satisfactorily perform according to the terms of the agreement between us. If Hospira, Ercros, or any alternate supplier of API or finished drug product for solithromycin or fusidic acid experiences any significant difficulties in its respective manufacturing processes, does not comply with the terms of the agreement between us or does not devote sufficient time, energy and care to providing our manufacturing needs, we could experience significant interruptions in the supply of solithromycin or fusidic acid, which could impair our ability to supply solithromycin or fusidic acid at the levels required for our clinical trials and commercialization, if approved, and prevent or delay their successful development and commercialization.
In addition, our reliance on foreign suppliers poses risks due to possible shipping delays, import restrictions and foreign regulatory regimes. Finally, any manufacturing facility is at risk of natural or man-made disaster, which could significantly reduce our clinical and commercial supplies of drug product.
These same risks apply to procuring comparator API or other comparator supplies needed for clinical trials in which we may compare our product candidates to currently approved drugs.
If we fail to obtain additional financing, we may not be able to complete the development and commercialization of solithromycin or fusidic acid.
We need substantial amounts of cash to complete the clinical development and commercialization of solithromycin and fusidic acid, especially to address the issues raised in the CRL and, if approved by the FDA, the commercial launch of solithromycin for CABP. Prior to the receipt of the CRL, and in conjunction with completing the second of two pivotal clinical trials and the submission of our NDAs for solithromycin for the treatment of CABP during the fourth quarter of 2015, and throughout the majority of 2016 we engaged in certain additional clinical and commercial activities, and accelerated others, including building inventory of solithromycin in preparation for commercial launch in the U.S. (which prior to approval is expensed as research and development expense in accordance with GAAP) and supporting certain investigator-led studies of solithromycin in additional indications. In addition, in 2016 we began to engage in additional sales and marketing preparation activities focused on solithromycin for the treatment of CABP, including hiring additional commercial management personnel, engaging in pricing research and other market research, and began building our specialty antibiotic sales force.
As a consequence of the solithromycin complete response letter received, and subsequent discussions with the FDA, resulting in a delay of the potential approval of solithromycin, we recently initiated companywide cost and personnel reductions. These actions have resulted in an approximately 67% reduction in our workforce from 136 to 45 employees, and significant reductions in external spending related to commercial preparedness and non-essential activities. The principal objective of the reductions is to enable us to conserve our financial resources as we evaluate the best path forward with our existing pipeline and potential business development opportunities.
Based on current assumptions, we believe that our existing cash and equivalents will enable us to fund our current operating expenses and capital requirements for at least the next 12 months from the filing date of this report. Such operating and capital requirements do not contemplate incremental expenses associated with a full scale commercial launch of solithromycin or any additional clinical trials with any of our product candidates. However, we have based our estimates on assumptions that may prove to be wrong, and we may use our available capital resources sooner than we currently expect. Moreover, changing circumstances may cause us to consume capital significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. For example, our clinical trials may encounter technical, enrollment or other issues that could cause our development costs to increase more than we expect and we may be required to conduct additional trials requested by the FDA that could increase our costs significantly. We would also need to raise additional funds sooner if we choose to initiate clinical trials more rapidly than we presently anticipate or if we elect to conduct additional trials for alternate indications. In any event, the costs to develop and launch solithromycin and to develop fusidic acid will be significant and we will need to raise additional capital to execute a safety study for solithroymycin and to support any development for other indications for solithromycin, as well as to continue development activities to obtain regulatory approval of and to commercialize fusidic acid.
34
We may raise additional capital from the issuance of equity and/or debt securities, collaborations with third parties, out-licensing of rights to our product candidates and other means, or a combination of any of the above. Securing additional financing, however, will require a substantial amount of time and attention from our management and may divert a disproportionate amount of their attention away from our day-to-day activities, which may adversely affect our management’s ability to conduct our day-to-day operations. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital when required or on acceptable terms, we may be required to:
|
|
• |
significantly delay, scale back or discontinue the development or commercialization of solithromycin and/or fusidic acid; |
|
|
• |
seek collaborators for solithromycin and/or fusidic acid at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; and |
|
|
• |
relinquish or license, potentially on unfavorable terms, our rights to solithromycin and/or fusidic acid that we otherwise would seek to develop or commercialize ourselves. |
If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing discovery, development and commercialization efforts, and our ability to generate revenues and achieve or sustain profitability will be substantially harmed.
The commercial success of solithromycin, fusidic acid and any other product candidates that we develop, if approved in the future, will depend upon attaining significant market acceptance of these products among physicians and payors.
As a company, we have never commercialized a product candidate for any indication. Even if solithromycin, fusidic acid or any other product candidate that we develop is approved by the appropriate regulatory authorities for marketing and sale, physicians may not prescribe our approved products, which would prevent us from generating revenues or becoming profitable. Efforts to educate the medical community and third party payors on the benefits of our product candidates may require significant resources and may not be successful. If a product candidate is approved but does not achieve an adequate level of market acceptance, we may not generate significant revenues and we may not become profitable. Market acceptance of solithromycin, fusidic acid and any other product candidates that we develop by physicians, patients and payors will depend on a number of factors, many of which are beyond our control, including:
|
|
• |
the clinical indications for which the product is approved; |
|
|
• |
acceptance by physicians and payors of each product as a safe and effective treatment; |
|
|
• |
limitations or warnings contained in a product’s FDA-approved labeling, as will be the case if solithromycin is approved for CABP; |
|
|
• |
prevalence and severity of adverse side effects; |
|
|
• |
the cost of treatment in relation to alternative treatments, including numerous generic drug products, such as azithromycin, levofloxacin and vancomycin; |
|
|
• |
the relative convenience and ease of administration of solithromycin in the treatment of CABP and fusidic acid in the treatment of ABSSSI and/or refractory bone and joint infections; |
|
|
• |
the availability and efficacy of competitive drugs; |
|
|
• |
our ability to recruit and retain a sales force, if necessary; |
|
|
• |
the effectiveness of our or any third-party partner’s sales force and marketing efforts; |
|
|
• |
our ability to forecast demand and maintain sufficient supplies of our drug products; |
|
|
• |
our ability to manufacture or obtain commercial quantities of our drug products; |
|
|
• |
the strength of our sales and marketing and distribution support; |
|
|
• |
the effectiveness of our marketing and advertising campaigns; |
|
|
• |
our ability to deliver our products on a timely basis; |
|
|
• |
the extent to which bacteria develop resistance to any antibiotic product candidate that we develop, thereby limiting its efficacy in treating or managing infections; |
|
|
• |
our ability to establish and maintain pricing sufficient to realize a meaningful return on our investment; |
|
|
• |
the extent to which the product is approved for inclusion on formularies of hospitals and managed care organizations; |
35
|
|
• |
whether the product is designated under physician treatment guidelines as a first-line therapy or as a second- or third-line therapy for particular infections; |
|
|
• |
the availability of adequate reimbursement by third parties, such as insurance companies and other health care payors, and/or by government health care programs, including Medicare and Medicaid; |
|
|
• |
adverse publicity about a product or favorable publicity about competitive products; and |
|
|
• |
potential product liability claims. |
Even if the medical community accepts that solithromycin and fusidic acid are safe and efficacious for their approved indications, physicians may not immediately be receptive to the use or may be slow to adopt solithromycin as an accepted treatment for CABP and fusidic acid as an accepted treatment for ABSSSI and/or bone and joint infections. While we believe each of solithromycin and fusidic acid has significant advantages, we cannot assure you that any labeling approved by the FDA will permit us to promote solithromycin or fusidic acid as being safe or superior to competing products. If either or both of solithromycin or fusidic acid are approved but do not achieve an adequate level of acceptance by physicians and payors, we may not generate sufficient or any revenues from these products and we may not become profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of solithromycin and fusidic acid may require significant resources and may never be successful.
The successful commercialization of our product candidates will depend on the pricing we are able to achieve for our product candidates, both inside and outside the U.S.
Our ability to successfully commercialize our product candidates will be dependent on whether we can obtain adequate pricing for any particular product candidate. Pricing may be substantially dependent on our ability to obtain reimbursement from third party payors, both in the U.S. and in foreign countries. Outside the U.S., certain countries, including a number of European Union members, set prices and reimbursement for pharmaceutical products, or medicinal products as they are commonly referred to in the E.U., with limited participation from those marketing the products. We cannot be sure that any prices and reimbursement will be acceptable to us or our strategic commercial partners. If the regulatory authorities in these foreign jurisdictions set prices or reimbursement that are not commercially attractive for us or our strategic commercial partners, our revenues from sales by us or our collaborators, and the potential profitability of our product candidates, in those countries would be negatively affected. Further, through contractual or other arrangements, the price we may be able to obtain in foreign countries may be dependent on the price we can achieve in the U.S.
Our estimates of the market for and commercialization of solithromycin as a treatment for CABP or for any other product candidate may be inaccurate or vary significantly over the potential market size.
The potential market opportunities for solithromycin and any other product candidate are difficult to estimate precisely. Our estimates of the potential market opportunities are predicated on many assumptions, including industry knowledge and publications, third party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve the exercise of significant judgment on the part of our management, are inherently uncertain and the reasonableness of these assumptions has not been assessed by an independent source. If any of the assumptions proves to be inaccurate, the actual markets for our product candidates could be smaller than our estimates of the potential market opportunities.
In addition, our estimates regarding the timing and amount of acceptance of any product candidate, and the pricing achievable for any product candidate may prove incorrect. Further, our plans for commercialization of any product candidate may not materialize in the time or manner we anticipate and may be adversely impacted by any required label warnings, as will be the case if solithromycin is approved for CABP, or any perceived safety or efficacy concerns. Finally, we may underestimate the demand for a product candidate, which could lead to lack of commercial quantities when needed and result in market backlash against the product candidate. Any of these occurrences could have a material adverse effect on our plans for commercialization of and the generation of any revenue from any product candidate.
If we believe regulatory approval and a sufficient market are likely for solithromycin for CABP, we would expect to build our own marketing and sales organization for solithromycin for CABP, but have no experience as a company in marketing drug products. If we are unable to successfully establish our own marketing and sales capabilities, or enter into agreements with third parties to market and sell our products after they are approved, we may not be able to generate product revenues.
If we believe regulatory approval of and a sufficient market for solithromycin for CABP are likely, we would expect to build our U.S. sales organization for the marketing, sales and distribution of solithromycin as a treatment for CABP, the size and nature of which would be determined by approved labeling and the potential market for solithromycin in light of that label. The establishment and development of our own sales force will be expensive and time consuming and could delay the planned launch of solithromycin, and we cannot be certain that we will be able to successfully develop this capability. The timing of building any sales force will be dependent on many factors, including the anticipated approval date and our financial resources. We may seek one or more licensing partners to handle some or all of the sales and marketing of solithromycin for CABP in the U.S. In order to successfully
36
commercialize any other products, we must develop these capabilities on our own or make arrangements with third parties for the marketing, sales and distribution of our products. There also may be certain markets within the U.S. for solithromycin for which we may seek a co-promotion arrangement. If we are not successful in building our own sales force, we may not be able to enter into arrangements with third parties to sell solithromycin or fusidic acid on favorable terms or at all. We would not have control over a third-party sales organization and would be dependent on that organization for successfully selling any of our products. Such a third-party organization may not devote the necessary manpower, time, resources or priority to our products, which would negatively impact our results of operations. In the event we are unable to develop our own marketing and sales force or collaborate with a third-party marketing and sales organization, we would not be able to commercialize solithromycin, fusidic acid or any other product candidates that we develop, which would negatively impact our ability to generate product revenues. Further, whether we commercialize products on our own or rely on a third party to do so, our ability to generate revenue will be dependent on the effectiveness of the sales force. In addition, to the extent we rely on third parties to commercialize our approved products, we will likely receive less revenues than if we commercialized these products ourselves.
A failure to maintain optimal inventory levels to meet commercial demand for any product that may be approved, including solithromycin, could harm our reputation and subject us to financial losses.
Because accurate product planning is necessary to ensure that we maintain optimal inventory levels for any product candidate that might be approved, including solithromycin, significant differences between our estimates and judgments and future actual demand for any approved products and the shelf life of inventory may result in significant charges for excess inventory or purchase commitments in the future. If we are required to recognize charges for excess inventories, such charges could have a material adverse effect on our financial condition and results of operations. Our ability to maintain optimal inventory levels also depends on the performance of third-party contract manufacturers. If our manufacturers are unsuccessful in either obtaining raw materials, if we are unable to release inventory on a timely and consistent basis, if we fail to maintain an adequate level of product inventory, if inventory is destroyed or damaged, or if our inventory reaches its expiration date, patients might not have access to our products, sales could be lost, our reputation and brands could be harmed, and physicians may be less likely to prescribe our products in the future, each of which could have a material adverse effect on our business, financial condition, results of operations and cash flows.
We are obligated to provide Toyama with clinical and commercial supply of solithromycin at prices determined by our manufacturing costs, which could negatively impact our results of operations in the event we cannot provide those supplies on our own and have to purchase them outside of our contracted suppliers. To provide this supply, we entered into a supply agreement with Fujifilm, which contains a minimum purchase requirement, which if triggered, could have a material adverse impact on our results of operations.
Pursuant to the terms of our supply agreement with Toyama, we are the exclusive supplier (with certain limitations) to Toyama and its sublicensees of API for solithromycin for use in licensed products in Japan, as well as the exclusive supplier to Toyama and its sublicensees of finished forms of solithromycin to be used in clinical trials in Japan. Pursuant to the supply agreement, Toyama will pay us for such clinical supply of finished product and all supplies of API for solithromycin for any purpose, other than the manufacture of products for commercial sale in Japan, at prices equal to our costs. All API for solithromycin supplied by us to Toyama for use in the manufacture of finished product for commercial sale in Japan will be ordered from us at prices determined by our manufacturing costs, and which may, depending on such costs, equal, exceed, or be less than such costs. The supply agreement will continue until the expiration or termination of the license agreement. In the event we cannot provide Toyama’s supplies under our own contracts with manufactures, we would have to either reduce our own supply of solithromycin or purchase it outside of our contracted manufacturers, which would negatively impact our results of operations. In January 2016, we entered into an API supply agreement with Fujifilm to provide a supply source in Japan to meet our obligations under the supply agreement with Toyama, but circumstances could occur that might render this source insufficient for our purposes. In the event that supply validation studies are completed and Fujifilm has constructed a facility to provide us the supply, we are subject to a minimum purchase requirement for a period of time that could run to an aggregated approximately $80 million, although such expense would be offset by sales to Toyama, if any.
Future legislation, and/or regulations and policies adopted by the FDA or other regulatory health authorities may increase the time and cost required for us to conduct and complete clinical trials for solithromycin, fusidic acid or other product candidates that we develop.
The FDA has established regulations, guidelines and policies to govern the drug development and approval process, as have foreign regulatory authorities. Any change in regulatory requirements due to the adoption by the FDA and/or foreign regulatory authorities of new legislation, regulations, or policies may require us to amend existing clinical trial protocols or add new clinical trials to comply with these changes. Such amendments to existing protocols and/or clinical trial applications or the need for new ones, may significantly impact the cost, timing and completion of the clinical trials.
37
In particular, drugs being tested and/or developed for the treatment of CABP, including solithromycin, are subject to proposed guidelines published by the FDA in 2009 (with new guidelines proposed in November 2011 and again in 2014). We have conducted our clinical trials to date according to the standards established by the 2009 and 2011 proposed guidelines. While we expected the FDA to revise the proposed guidelines for CABP, we could not delay development of solithromycin and began the Phase 3 oral trial in December 2012 and the Phase 3 IV-to-oral trial in December 2013, which was before the FDA issued revised proposed guidelines in 2014. While the 2014 proposed guidelines did not impose any new requirements on our Phase 3 trials, the FDA could further revise the guidelines. Any new proposed guidelines may require us to conduct additional clinical trials, re-run previously completed trials to gather data at different endpoints or according to different protocols, or otherwise materially alter our planned clinical development of solithromycin. Any such regulatory change may materially increase our costs, delay the completion of our clinical trials, and otherwise impact our ability to obtain regulatory approval for our product candidate. Furthermore, the FDA’s guidance documents are not binding on the FDA. As a result, the FDA may not accept the results of clinical trials we conduct even if they were to follow the FDA’s most recent guidance.
In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process, particularly in our areas of focus, may significantly delay or prevent regulatory approval, as well as impose more stringent product labeling and post-marketing testing and other requirements.
Bacteria might develop resistance to solithromycin or fusidic acid, which would decrease the efficacy and commercial viability of that product.
Drug resistance is primarily caused by the genetic mutation of bacteria resulting from sub-optimal exposure to antibiotics where the drug does not kill all of the bacteria. While antibiotics have been developed to treat many of the most common infections, the extent and duration of their use worldwide has resulted in new mutated strains of bacteria resistant to current treatments. We are developing solithromycin and fusidic acid to treat patients infected with drug-resistant bacteria. With respect to solithromycin, which is a next generation macrolide, resistance issues associated with earlier generations of macrolides have led to a decrease in their use for treating serious respiratory tract infections such as CABP. If physicians, rightly or wrongly, associate the resistance issues of earlier generation macrolides with solithromycin, physicians might not prescribe solithromycin for treating a broad range of infections. Similarly, resistance to fusidic acid has developed outside the U.S. Our in vitro studies have shown that the reason for resistance to the oral formulation is that it was not dosed optimally. We believe that overuse of topical formulations of fusidic acid also contributed to development of resistance outside the U.S. If fusidic acid is improperly dosed, or if our studies incorrectly attributed an increase in resistance to inappropriate dosing, bacteria might develop resistance to fusidic acid in the U.S. If these bacteria develop resistance to solithromycin or fusidic acid, the efficacy of these products would decline, which would negatively affect our potential to generate revenues from these products.
Delays in clinical trials are common and have many causes, and any such delays could result in increased costs to us and jeopardize or delay our ability to obtain regulatory approval and commence product sales as currently contemplated.
We may experience delays in clinical trials of our product candidates. Our planned clinical trials might not begin on time, may be interrupted or delayed once commenced, might need to be redesigned, might not enroll a sufficient number of patients or might not be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including the following:
|
|
• |
delays in obtaining regulatory approval to commence a trial; |
|
|
• |
imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
|
|
• |
delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
|
|
• |
delays in obtaining required institutional review board, or IRB, approval at each site; |
|
|
• |
delays in identifying, recruiting and training suitable clinical investigators; |
|
|
• |
delays in recruiting suitable patients to participate in a trial; |
|
|
• |
delays in having patients complete participation in a trial or return for post-treatment follow-up; |
|
|
• |
clinical sites dropping out of a trial to the detriment of enrollment; |
|
|
• |
time required to add new sites; |
|
|
• |
delays in obtaining sufficient supplies of clinical trial materials, including suitable active pharmaceutical ingredient, or API, whether of our product candidates or comparator drugs; or |
|
|
• |
delays resulting from negative or equivocal findings of the data safety monitoring board, or DSMB, for a trial. |
38
We were subject to such a delay in 2008 when the FDA placed a partial clinical hold on our Phase 2 clinical trial for oral solithromycin over concern about possible toxicity related to solithromycin. The FDA converted the partial clinical hold into a full clinical hold in April 2010. At the time, the FDA had concerns that solithromycin, as a fluoroketolide, may have similar toxicity issues as Ketek. While we addressed the FDA’s concerns and were allowed to proceed with the trial, which we successfully completed, the trial was delayed by approximately 12 months. We could experience one or more such delays with other trials.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. In addition, the timing of our clinical trials may be dependent on a specific disease seasonality, as, for example, were our trials for solithromycin, which were dependent on the onset, degree and timing of the CABP season, which tends to occur in the winter months in each hemisphere. We could encounter delays in our ongoing and future clinical trials of solithromycin (for indications other than CABP), fusidic acid or any other product if participating physician investigators encounter unresolved ethical issues associated with enrolling patients in clinical trials of solithromycin (for indications others than CABP), fusidic acid or any other product in lieu of prescribing approved antibiotics that have established safety and efficacy profiles. Any of these delays in completing our clinical trials could increase our costs, slow down our product development and approval process and jeopardize our ability to commence product sales and generate revenues.
We may be required to suspend or discontinue clinical trials due to adverse side effects or other safety risks that could preclude approval of solithromycin or fusidic acid or any of our future product candidates.
Our clinical trials may be suspended or terminated at any time for a number of reasons. A clinical trial may be suspended or terminated by us, our collaborators, the FDA or other regulatory authorities due to a failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, presentation of unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using the investigational drug, changes in governmental regulations or administrative actions, lack of adequate funding to continue the clinical trial, or negative or equivocal findings of the DSMB or the IRB for a clinical trial. An IRB may also suspend or terminate our clinical trials for failure to protect patient safety or patient rights. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe the clinical trials are not being conducted in accordance with applicable regulatory requirements or present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate any clinical trial of any product candidates that we are developing, the commercial prospects of such product candidates will be harmed and our ability to generate product revenues, if at all, from any of these product candidates will be delayed or eliminated. Any of these occurrences may harm our business, financial condition, results of operations and prospects significantly.
We recently completed a Phase 3 clinical trial of fusidic acid for the treatment of ABSSSI, and we have an ongoing exploratory study for bone and joint infections, but there is no guarantee that the results of our Phase 3 trial for ABSSSI or any other completed trial will demonstrate safety and efficacy to the satisfaction of the FDA or that the results from the ongoing exploratory study for bone and joint infections or any other ongoing or future study will be consistent with the results of prior studies, including the Phase 3 trial for ABSSSI.
While we have completed Phase 2 and Phase 3 clinical trials comparing fusidic acid to linezolid for the treatment of ABSSSI the results of our completed Phase 2 and Phase 3 trial for the treatment of ABSSSI were not powered to show statistical superiority. Comparisons to results from other reported clinical trials, including our completed Phase 2 and Phase 3 clinical trials for the treatment of ABSSSI, can assist in evaluating the potential efficacy of fusidic acid for the treatment of ABSSSI and refractory bone and joint infections; however, there are many factors that affect the outcome for patients in clinical trials, some of which are not apparent in published reports, and results from different trials often cannot be reliably compared. Therefore, there is no assurance that the results of any other trials we conduct for fusidic acid in the treatment of ABSSSI or refractory bone and joint infections will demonstrate safety or efficacy comparable to the results of trials conducted to date or will be sufficient to attain FDA approval.
In December 2012, we initiated a Phase 2 clinical trial of fusidic acid for the treatment of prosthetic joint infections. In October 2013, the FDA granted orphan drug designation to fusidic acid for the treatment of PJI and we will work to have orphan drug designation granted for fusidic acid for refractory bone and joint infections. There is no published FDA guidance for clinical trials for PJI or for bone and joint infections. Further, we need to determine the impact of the orphan drug designation for PJI and possible designation for bone and joint infections on our clinical development plan and the Phase 2 clinical trial data to support an NDA. In the Phase 2 PJI trial we noted that although oral fusidic acid plus rifampin had similar efficacy to intravenous vancomycin, rifampin significantly diminished the blood levels of fusidic acid. We concluded the Phase 2 trial prior to completion because we demonstrated that fusidic acid in combination with rifampin was generally comparable to intravenous standard of care antibiotics. We believe that the proper dosing of fusidic acid is without rifampin and that the loading dose and maintenance dose that we had tested in the ABSSSI
39
Phase 2 trial was optimal for a Phase 3 refractory bone and joint infection trial. We have met with the FDA to discuss the development plan for fusidic acid for ABSSSI and bone and joint infections. Based on that meeting, our plan involves testing fusidic acid for long-term suppressive therapy of refractory bone and joint infections, including PJI. Also based on our discussions with the FDA, in November 2015, we began a Phase 3 trial for the treatment of ABSSSI, for which we reported topline results in February 2017, and began a refractory bone and joint infection study in January2016 to determine fusidic acid’s safety and efficacy. With the ABSSSI study now completed, we plan to meet with the FDA to discuss the next steps required to bring fusidic acid to patients in the United States. However, the FDA is not bound by discussions and if the FDA subsequently believes that the plan as discussed and developed is inadequate, it could delay or prevent our ability to receive regulatory approval or commercialize fusidic acid for the treatment of ABSSSI and/or refractory bone and joint infections.
Fusidic acid is not well absorbed in animals, which could impair our ability to obtain FDA approval.
As required by FDA regulations, we conducted pre-clinical studies of fusidic acid to determine its level of absorption in animals. The studies indicated that fusidic acid is not very well absorbed and has a short half-life in animals, resulting in minimum exposure levels which limited the ability to test fusidic acid in animal models. Fusidic acid, the API in fusidic acid, has been used for several decades in humans outside the U.S. and we believe there is sufficient human clinical trial data for fusidic acid to overcome the lack of absorption in animal studies. Despite this human data, and while all of our pre-clinical tests were benign and indicated no safety or tolerability issues, our limited ability to test fusidic acid in animal models may adversely affect our ability to obtain FDA approval.
Even if the FDA approves solithromycin for the treatment of CABP and fusidic acid for the treatment of ABSSSI and bone and joint infections, adverse effects discovered after approval could adversely affect the market for those products.
If we obtain regulatory approval for solithromycin, fusidic acid or any other product candidate that we develop, and we or others later discover that our products cause adverse effects, a number of potentially significant negative consequences could result, including:
|
|
• |
regulatory authorities may withdraw their approval of the product; |
|
|
• |
regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
|
|
• |
we may be required to change the way the product is administered, conduct additional clinical studies, implement a risk evaluation and mitigation strategy, or REMS, or restrict the distribution of the product; |
|
|
• |
we could be sued and held liable for harm caused to patients and our liability insurance may not adequately cover those claims; and |
|
|
• |
our reputation may suffer. |
Any of these events could prevent us from maintaining market acceptance of the affected product candidate and could substantially increase the costs of, or prevent altogether, the commercialization of our product candidates.
We continue to have negative cash flows from operations since inception and might not be able to generate sufficient cash to service our existing indebtedness to Comerica Bank, the level of which indebtedness could have a material adverse effect on our business, financial condition, results of operations and prospects.
On July 10, 2015, we entered into a loan and security agreement with Comerica Bank, or Comerica, pursuant to which we could and did borrow $20.0 million in a term loan and, if the FDA approves our planned NDA, for solithromycin, we may also borrow, under a revolver, an aggregate amount equal to the lesser of (i) up to 75% of our eligible inventory and 80% of eligible accounts receivable or (ii) $10.0 million. If the FDA approves our planned NDA for solithromycin, we may convert the term loan to the revolver, in which event the revolver would have a maximum amount available to us of $25.0 million. Amounts borrowed under the term loan may be repaid and reborrowed at any time without penalty or premium. The term loan is interest-only through April 30, 2016, followed by an amortization period of 36 months of equal monthly payments of principal plus interest, beginning on May 1, 2016 and continuing on the same day of each month thereafter until paid in full. Amounts available to be borrowed under the revolver may also be repaid and reborrowed at any time without penalty or premium prior to December 31, 2017, at which time all advances under the revolver shall be immediately due and payable in full. Our ability to make payments on this indebtedness depends on our ability to generate cash in the future. We expect to experience negative cash flow for the foreseeable future as we fund our operations and capital expenditures. There can be no assurance that we will be in a position to repay this indebtedness when due or obtain extensions of the maturity date. We anticipate that we will need to secure additional funding in order for us to be able to satisfy our obligations when due. We cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If that additional funding involves the sale of equity securities or convertible securities, it would result in the issuance of additional shares of our capital stock, which would result in dilution to our stockholders.
40
Moreover, this level of debt could have important consequences to you as an investor in our securities. For example, it could:
|
|
• |
make it more difficult for us to satisfy our obligations with respect to payments owed to our licensors; |
|
|
• |
limit our flexibility in planning for the development, clinical testing, approval and marketing of our products; |
|
|
• |
place us at a competitive disadvantage compared to any of our competitors that are less leveraged than we are; |
|
|
• |
increase our vulnerability to both general and industry-specific adverse economic conditions; and |
|
|
• |
limit our ability to obtain additional funds. |
In addition, the loan is secured by substantially all of our personal property assets except our intellectual property and our stock in our subsidiaries. In the event we fail to make timely payments or breach any of our representations or other obligations in the agreement, or upon any circumstance or occurrence that has a material adverse effect on the loan collateral, our business operations, properties, assets, prospects or condition, or our ability to perform our obligations under the loan agreement, Comerica Bank can declare the loan in default. Upon an event of default, the loan principal and accrued interest would become immediately due and payable and Comerica Bank would be entitled to enforce its security interest in our assets.
The addition of further debt to our current debt levels could make it more difficult for us to repay our indebtedness and meet our other obligations and would intensify the leverage-related risks that we now face.
A substantial portion of our future revenues may be dependent upon our strategic partnerships.
Our success will depend in significant part on our ability to attract and maintain strategic partners and strategic relationships to support the development and commercialization of our product candidates. We currently expect that a substantial portion of our future revenues may be dependent upon our strategic partnership with Toyama and our ability to enter into strategic relationships in other territories. Under the license agreement we entered into in May 2013 with Toyama, Toyama has significant development and commercialization responsibilities with respect to solithromycin in Japan. If Toyama or any of our other strategic partners were to terminate their agreements with us, fail to meet their obligations or otherwise decrease their level of efforts, allocation of resources or other commitments under these agreements with us, our future revenues could be negatively impacted and the development and commercialization of product candidates could be negatively impacted and/or interrupted. In addition, if some or any of the development, regulatory and commercial milestones are not achieved or if certain net sales thresholds are not achieved, as set forth in the Toyama agreement or any agreements with other strategic partners, we will not fully realize the expected economic benefits of those agreements. Further, the achievement of certain of the milestones under our partnership agreements will depend on factors that are outside of our control and most are not expected to be achieved for several years, if at all. Any failure to successfully maintain our strategic partnership agreements could materially and adversely affect our ability to generate revenues.
Strategic partners may cease to pursue their own development of our product candidates or cease funding and other activities required by our agreements with those strategic partners for reasons beyond our control.
In May 2013, we entered into a license agreement with Toyama under which Toyama is to initiate certain clinical trials, obtain regulatory approval and launch and commercialize approved licensed products in Japan. If the results of Toyama’s studies are disappointing or inconclusive, if Toyama were to breach its obligations under the license agreement, or if Toyama decides to cease developing solithromycin for any reason, the development of solithromycin in Japan could be materially harmed, and any negative clinical results could materially harm our own development efforts for solithromycin. In addition, the loss of milestone payments from Toyama called for under the license agreement could have a material adverse impact on our capital resources and ability to conduct our operations. These same risks will apply to any other strategic partnership into which we may enter in the future.
We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be delayed in obtaining, or may ultimately not be able to obtain, regulatory approval for or commercialize solithromycin, fusidic acid or any other product candidates.
We have relied, and plan to continue to rely, on various CROs to recruit patients, monitor and manage data for our on-going clinical programs for solithromycin and fusidic acid, as well as for the execution of our pre-clinical and non-clinical studies. We control only certain aspects of our CROs’ activities; nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with the FDA’s current good clinical practices, or cGCPs, which are regulations and guidelines enforced by the FDA for all of our products in clinical development. The FDA enforces these cGCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable, and the FDA may require us to perform additional clinical trials before deciding whether to approve our product candidates. We cannot assure you that, upon
41
inspection, the FDA will determine that any of our clinical trials comply with cGCPs. In addition, to evaluate the safety and effectiveness of solithromycin and fusidic acid to a statistically significant degree our clinical trials will require an adequately large number of test subjects. Any clinical trial that a CRO conducts abroad on our behalf is subject to similar regulation. Accordingly, if our CROs fail to comply with these regulations or recruit a sufficient number of patients, we may have to repeat clinical trials, which would delay the regulatory approval process.
In addition, our CROs are not our employees and we cannot control whether or not they devote sufficient time and resources to our on-going clinical, non-clinical and pre-clinical programs. Our CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other drug development activities, which could impede their ability to devote appropriate time to our clinical programs. If our CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize solithromycin, fusidic acid or any other product candidates that we seek to develop. As a result, our financial results and the commercial prospects for solithromycin, fusidic acid or any other product candidates that we seek to develop would be harmed, our costs could increase and our ability to generate revenues could be delayed or ended.
We typically engage one or more CROs on a project-by-project basis for each study or trial. While we have developed and plan to maintain our relationships with CROs that we have previously engaged, we also expect to enter into agreements with other CROs to obtain additional resources and expertise in an attempt to accelerate our progress with regard to on-going clinical, non-clinical and pre-clinical programs and, specifically, the compilation of clinical trial data for submission with an NDA for each of solithromycin and fusidic acid. Switching or entering into new relationships with CROs involves substantial cost and requires extensive management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Although we try to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition, results of operations or prospects.
The timing of the milestone, support and royalty payments we are required to make to Optimer Pharmaceuticals, Inc., The Scripps Research Institute and Macrolide Pharmaceuticals, Inc. is uncertain and could adversely affect our cash flows and results of operations.
In March 2006, we entered into a Collaborative Research and Development and License Agreement with Optimer Pharmaceuticals, Inc., or Optimer (now owned by Merck), pursuant to which we acquired an exclusive license to certain patent applications and other intellectual property related to a series of compounds, including solithromycin, to develop and commercialize licensed products outside of the Association of South East Asian Nations, or ASEAN, countries (Brunei Darussalam, Cambodia, Indonesia, Laos, Malaysia, Myanmar (Burma), the Philippines, Singapore, Thailand and Vietnam). We have an obligation to make additional payments upon achievement of specified development, regulatory and commercialization milestones. The aggregate amount of such milestone payments we may need to pay is based in part on the number of products developed under the agreement. The aggregate amount (including our two milestone payments to date) would be $27.5 million if four products are developed and gain FDA approval. Additional limited milestone payments would be due if we develop more than four products. We will also pay tiered mid-single-digit royalties based on the amount of annual net sales of solithromycin (or related licensed compounds), if and when approved by regulatory authorities. We have already paid a $0.5 million milestone in 2010 and a $1.0 million milestone in 2012. Optimer can elect to receive certain milestone payments in cash or in shares of our common stock having an equivalent fair market value. The timing of our achievement of these events and corresponding milestone payments to Optimer is subject to factors relating to the clinical and regulatory development and commercialization of solithromycin (or related licensed compounds), many of which are beyond our control. We may become obligated to make a milestone payment when we do not have the cash on hand to make such payment, which could require us to delay our clinical trials, curtail our operations, scale back our commercialization and marketing efforts or seek funds to meet these obligations on terms unfavorable to us. If we were unable to make a milestone payment, we would be in material breach of the agreement, in which event Optimer could terminate the agreement, which would result in the loss of our rights to develop and commercialize solithromycin, which would seriously harm our ability to generate revenues or achieve profitability.
We also must pay The Scripps Research Institute various milestone and annual payments, which, while significantly lower than amounts potentially due to Optimer, could become due when we do not have the cash on hand to make such payment, which could require us to delay our clinical trials, curtail our operations, scale back our commercialization and marketing efforts or seek funds to meet these obligations on terms unfavorable to us.
In January 2016, we entered into an Option and License Agreement with Macrolide Pharmaceuticals, Inc., or MP, pursuant to which MP granted us an exclusive option to license certain of MP’s patents and know-how involving macrolides, including
42
specifically novel methods of synthesizing solithromycin (the “Compound”). Under the agreement, we will support research at MP focused on developing a novel, cost-competitive manufacturing approach to solithromycin. Under the evaluation program called for in the agreement, MP will conduct research activities for the manufacture of the Compound, which activities we will evaluate to determine whether to exercise the option to license. For conducting the evaluation program, we will pay MP the expected reasonable, documented, direct compensation-related costs of employees and advisors necessary to conduct MP’s portion of the evaluation program in the aggregate amount of $1.5 million, which we will pay in 18 equal consecutive non-refundable, non-creditable monthly installments of $83,333, beginning with the first monthly anniversary of entry into the agreement. Further, we will pay MP up to an aggregate of $1.0 million upon the satisfaction of certain performance milestones. The timing of the milestone payments to MP are subject to factors relating to the Compound, which are beyond our control. Further, the monthly support payments or a milestone payment, if triggered, could be due when we do not have the cash on hand to make such payment, which could require us to seek funds to meet these obligations on terms unfavorable to us. If we were unable to make a support or milestone payment, we would be in material breach of the agreement, in which event MP could terminate the agreement, which would result in the loss of our rights to the synthetic version of solithromycin, which could harm our ability to reduce production costs and develop an alternate supply of solithromycin, which could adversely affect our business and results of operations
Our loan agreement with Comerica Bank contains covenants that impose restrictions on our operations that may adversely impact the operation of our business.
Our loan agreement with Comerica Bank contains customary restrictive covenants, including restrictions on our ability to incur additional debt, transfer or place a lien or security interest on our assets, merge with or acquire other companies, redeem any shares of our capital stock or pay cash dividends to our stockholders. These restrictions may inhibit our ability to conduct our business and to provide distributions to our stockholders. Future debt securities or other financing arrangements could contain similar or more restrictive negative covenants than the Comerica Bank loan.
If approved, solithromycin and fusidic acid will face significant competition from branded and generic antibiotics and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. If solithromycin or fusidic acid is approved, we will have competitors both in the U.S. and internationally, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies. Many of these companies have greater financial and other resources, such as larger research and development staffs and more experienced marketing and manufacturing organizations. As a result, these companies may obtain regulatory approval more rapidly and may be more effective in selling and marketing their products. They also may invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make solithromycin, fusidic acid or any other product candidates that we develop obsolete. As a result, our competitors may succeed in commercializing antibiotics before we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
If approved, both solithromycin and fusidic acid will face competition from currently commercially available antibiotics, as well as any competing products that may be developed in the future. In July 2012, the United States Congress passed, and President Obama signed, the Food and Drug Administration Safety and Innovation Act, which included the Generating Antibiotic Incentives Now Act, or the GAIN Act. The GAIN Act is intended to provide incentives for the development of new, qualified infectious disease products. These incentives might result in more competition in the market for new antibiotics and might cause pharmaceutical and biotechnology companies with more resources than we have to shift their efforts towards the development of products that could be competitive with our product candidates. Existing approved products that will compete with solithromycin include azithromycin (sold under the brand name Zithromax ® by Pfizer Inc. and available as a generic), clarithromycin (sold under the brand name Biaxin ® by Abbott Laboratories and available as a generic), moxifloxacin (sold under the brand name Avelox ® by Bayer AG), levofloxacin (sold under the brand name Levaquin by Johnson & Johnson and available as a generic), linezolid (sold under the brand name Zyvox by Pfizer Inc.), ceftriaxone (sold under the brand name Rocephin ® by F. Hoffman-La Roche Ltd and available as a generic) and ceftaroline (sold under the brand name Teflaro ® by Forest Laboratories, Inc.). There are two drugs in development to treat CABP: omadacycline, being developed by Paratek Pharmaceuticals, and lefamulin, being developed by Nabriva Therapeutics. Existing approved products that will compete with fusidic acid include vancomycin (available as a generic), linezolid (sold under the brand name Zyvox by Pfizer Inc.), daptomycin (sold under the brand name Cubicin by Cubist Pharmaceuticals, Inc., which was acquired by Merck), quinupristin/dalfopristin (sold under the brand name Synercid ® by Sanofi-Aventis and Monarch Pharmaceuticals, Inc.), tigecycline (sold under the brand name Tygacil® by Pfizer Inc.), telavancin (sold under the brand name Vibativ ® by Theravance, Inc. and Astellas Pharma, Inc.) and ceftaroline (sold under the brand name Teflaro by Forest Laboratories, Inc., now Allergan). Several antibiotics have been approved by the FDA in the past several years. Dalbavancin (Dalvance) and ceftazidime-avibactam (Avycaz) for Allergan, ceftolozane-tazobactam (Zerbaxa) and tedizolid (Sivextro) for Merck, and oritavancin (Orbactiv) for The Medicines Company. None of these approvals are for CABP. Omadacycline (Paratek Pharmaceuticals) and lefamulin (Nabriva Therapeutics), and dalafloxcacin (Melinta) all are expected to be pursued as a possible treatment for ABSSSI and other indications. Ceftobiprole has
43
been approved in Canada and is being developed in the U.S. for MRSA. Generic antibiotics are typically sold at lower prices than branded antibiotics and are generally preferred by managed care providers of health services.
If we are unable to demonstrate the advantages of solithromycin or fusidic acid over competing drugs and drug candidates, we will not be able to successfully commercialize solithromycin or fusidic acid and our results of operations will suffer.
Reimbursement may not be available for solithromycin, fusidic acid or any other product candidates that we develop, which could make it difficult for us to sell our products profitably.
Market acceptance and sales of solithromycin, fusidic acid or any other product candidates that we develop will depend on reimbursement policies and may be affected by health care reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will pay for and establish reimbursement levels. We cannot be sure that reimbursement will be available for solithromycin, fusidic acid or any other product candidates that we develop. Also, we cannot be sure that the amount of reimbursement available, if any, will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize solithromycin, fusidic acid or any other product candidates that we develop.
Specifically, in both the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. In the U.S., the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. In addition, this legislation provided authority for limiting the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policies and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors. In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, PPACA, became law in the U.S. The goal of PPACA is to reduce the cost of health care and substantially change the way health care is financed by both governmental and private insurers. While we cannot predict what ultimate impact on federal reimbursement policies this legislation will have in general or on our business specifically, the PPACA may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of solithromycin or fusidic acid or any future products. Members of the U.S. Congress and some state legislatures had sought to overturn at least portions of the legislation including those on the mandatory purchase of insurance. However, on June 28, 2012, the United States Supreme Court upheld the constitutionality of these provisions. Members of the U.S. Congress have since proposed a number of legislative initiatives, including repeal of all or portions of the PPACA. We cannot predict the outcome or impact of current proposals or whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
The availability of numerous generic antibiotics at lower prices than branded antibiotics, such as solithromycin or fusidic acid if either were approved for commercial introduction, may also substantially reduce the likelihood of reimbursement for such products. We expect to experience pricing pressures in connection with the sale of solithromycin, fusidic acid and any other products that we develop, due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative proposals. If we fail to successfully secure and maintain reimbursement coverage for our products or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our products and our business will be harmed.
If we are not successful in retaining or attracting as necessary highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceuticals industries depends in large part on our ability to retain or attract as necessary highly qualified managerial, scientific, medical and sales and marketing personnel. This will be especially true in light of the significant reduction in our workforce that we undertook in late February 2017; the retention of the remaining employees may be difficult. While we have provided all of these remaining employees with retention bonuses, there can be no assurance that any of these employees will remain in our employment. Historically, in order to induce valuable employees to remain with us, we have provided stock options that vest over time. The retention bonuses we recently granted consist of stock options and restricted stock units. The value to employees of stock options and restricted stock units will be significantly affected by movements in our stock price that we cannot control and may at any time be insufficient to counteract more lucrative offers from other companies.
44
Our executive and scientific team has expertise in many different aspects of drug discovery and development and in selling and marketing antibiotics in both the inpatient and outpatient markets. We conduct our operations at our facility in Chapel Hill, North Carolina, which is part of the Research Triangle consisting of Raleigh, Durham and Chapel Hill. This region is headquarters to other biopharmaceutical companies and many academic and research institutions and, as a result, at any given time there may be a shortage of experienced scientists and medical and sales and marketing personnel. Competition for skilled personnel in our area and elsewhere in the U.S. is very intense and competition for experienced scientists may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all.
Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice. We had a change in senior management in December 2016 and do not have a permanent Chief Executive Officer. We have only a short-term at-will employment agreement with our Acting Chief Executive Officer, David Zaccardelli, and we do not have employment agreements with David S. Moore, our President and Chief Commercial Officer, Mark W. Hahn, our Chief Financial Officer, David W. Oldach, our Chief Medical Officer, John D. Bluth, our Executive Vice President of Investor Relations and Corporate Communications, or any other employee. As a result, all employees are employed on an at-will basis, which means that any of these employees could leave our employment at any time, with or without notice, and may go to work for a competitor. Even though Dr. Zaccardelli has entered into an employment agreement with us, he could leave at any time. We do have change in control severance agreements with Mr. Moore, Mr. Hahn, Dr. Oldach, and Mr. Bluth, but any of these individuals could leave our employment at any time. While our agreements with Dr. Oldach and Messrs. Moore, Hahn and Bluth contain non-compete provisions, those provisions do not prevent any of these executives from leaving our employ. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results or financial condition. Our success also depends on our ability to continue to attract, retain and motivate highly skilled scientific and medical personnel.
Other biotechnology and pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles and longer histories than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we offer. If we are unable to continue to attract and retain high quality personnel, our ability to discover, develop and commercialize drug candidates will be limited.
Whether our current product candidates are successful or not, our future growth will depend on our ability to identify, develop, acquire or in-license products and if we do not successfully identify develop, acquire or in-license additional product candidates or integrate them into our operations, we may have limited growth opportunities.
An important part of our business strategy is to continue to develop a pipeline of product candidates by developing, acquiring or in-licensing products, businesses or technologies that we believe are a strategic fit with our focus developing anti-infectives to treat infectious diseases. However, these business activities may entail numerous operational and financial risks, including:
|
|
• |
inability to successfully identity new products candidates; |
|
|
• |
difficulty or inability to secure financing to fund development activities for such development, acquisition or in-licensed products or technologies; |
|
|
• |
incurrence of substantial debt or dilutive issuances of securities to pay for development, acquisition or in-licensing of new products; |
|
|
• |
disruption of our business and diversion of our management’s time and attention; |
|
|
• |
higher than expected development, acquisition or in-license and integration costs; |
|
|
• |
exposure to unknown liabilities; |
|
|
• |
difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel; |
|
|
• |
inability to retain key employees of any acquired businesses; |
|
|
• |
difficulty in managing multiple product development programs; and |
|
|
• |
inability to successfully develop new product candidates or clinical failure of new product candidates. |
We have limited resources to identify and execute the development, acquisition or in-licensing of products, businesses and technologies and integrate them into our current infrastructure. We may compete with larger pharmaceutical companies and other competitors in our efforts to establish new collaborations and in-licensing opportunities. These competitors likely will have access to greater financial resources than us and may have greater expertise in identifying and evaluating new opportunities. Moreover, we may
45
devote resources to potential development, acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts.
We may need to grow our organization if we make progress on the development of any of our product candidates, and we may experience difficulties in managing this growth, which could disrupt our operations.
As of February 28, 2017, we had 45 employees. As our development and commercialization plans and strategies progress and develop, we may need to expand our employee base for managerial, operational, financial and other resources, including sales and marketing resources in preparation for any commercial launch of a product. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. Also, our management may need to divert a disproportionate amount of its attention away from their day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations which may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Future growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage any future growth, our expenses may increase more than expected, our ability to generate and/or grow revenues could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize solithromycin, fusidic acid and our other product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth in our organization.
Even if we obtain FDA approval of solithromycin, or any other product candidate, we may never obtain approval or commercialize our products outside of the U.S., which would limit our ability to realize their full market potential. If foreign approval is obtained, there are risks in conducting business in international markets.
In order to market solithromycin or any other products outside of the U.S., we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and require additional pre-clinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays.
In addition, our failure to obtain regulatory approval in the U.S. or any foreign country may delay or have negative effects on the process for regulatory approval in other countries. We do not know what impact if any the issuance of the CRL may have on our MAA filed with the EMA. We do not have any product candidates approved for sale in the U.S. or any foreign country and we do not have experience as a company in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in a foreign country or to obtain and maintain required approvals, our potential market for solithromycin or other products will be reduced and our ability to realize the full market potential of our products will be harmed. We do not intend to commercialize fusidic acid outside the U.S. because of the widespread use of fusidic acid in Europe and Australia.
If approved for commercialization in a foreign country, we intend to enter into agreements with third parties to market solithromycin whenever it may be approved and wherever we have the right to market it. Consequently, we expect that we will be subject to additional risks related to entering into international business relationships, including:
|
|
• |
potentially reduced protection for intellectual property rights; |
|
|
• |
the potential for so-called parallel importing, which is what happens when a local seller, faced with high or higher local prices, opts to import goods from a foreign market (with low or lower prices) rather than buying them locally; |
|
|
• |
unexpected changes in tariffs, trade barriers and regulatory requirements; |
|
|
• |
economic weakness, including inflation, or political instability in particular foreign economies and markets; |
|
|
• |
compliance with laws for employees traveling abroad; |
|
|
• |
foreign taxes, including withholding of payroll taxes; |
|
|
• |
foreign currency fluctuations, which could result in increased operating expenses and reduced revenues; |
|
|
• |
workforce uncertainty in countries where labor unrest is more common than in the U.S.; |
46
|
|
• |
production shortages resulting from any events affecting API and/or finished drug product supply or manufacturing capabilities abroad; |
|
|
• |
business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires; and |
|
|
• |
failure to comply with Office of Foreign Asset Control rules and regulations and the Foreign Corrupt Practices Act. |
These and other risks may materially adversely affect our ability to attain or sustain revenue from international markets.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with federal and state health care fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a Code of Conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
To raise additional funds to support our business operations, we may issue equity or debt securities. Debt securities could contain restrictive covenants that may adversely impact the operation of our business. The issuance of equity securities or convertible debt securities would result in dilution to our stockholders.
The incurrence of indebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. For example, in July 2015, we entered into a loan agreement for $20.0 million with Comerica Bank that contains restrictive covenants, including restrictions on our ability to incur additional debt, transfer or place a lien or security interest on our assets, including our intellectual property, merge with or acquire other companies, redeem any shares of our capital stock or pay cash dividends to our stockholders. Future debt securities or other financing arrangements could contain similar or more restrictive negative covenants than the Comerica Bank loan. In addition, the sale of equity securities or convertible debt securities would result in the issuance of additional shares of our capital stock, which would result in dilution to our stockholders.
Our limited operating history makes it difficult to evaluate our business and prospects.
We began operations in 2006. Our operations to date have been limited to financing and staffing our company, conducting product development activities for solithromycin and fusidic acid, engaging in commercial launch preparation activities for solithromycin, performing research and development with respect to our proprietary macrolide library. We have not yet demonstrated an ability as a company to obtain regulatory approval for or commercialize a product candidate. Consequently, the ability to predict our future performance may not be as accurate as it could be if we had a history of successfully developing and commercializing pharmaceutical products.
Government funding for any current or future development programs may be withheld, delayed or terminated for reasons beyond our control, or if we fail to carry out our contractual obligations or cease development of a product candidate.
We have an agreement with BARDA pursuant to which we are pursuing the evaluation and development of solithromycin for the treatment of bacterial infections in pediatric populations and infections caused by bioterror threat pathogens, specifically anthrax and tularemia. Funding for any government-sponsored or government-funded program is subject to withholding, delay or termination for reasons beyond our control. Further, funding could be reprioritized due to national or international developments. Epidemics, such as the recent crisis with Ebola, could cause government sponsors, including BARDA, to shift funding away from our program to address what the sponsor views as more pressing needs. BARDA also has the right to terminate its agreement with us at any time if the contracting officer determines that it is in the government’s interest to do so.
47
In addition, if we fail to abide by our contractual obligations to any governmental entity, including BARDA, the entity generally would be able to terminate the agreement. Further, if we were to cease development of any product candidate for which we had a contract with a governmental entity, it would generally lead to the termination of such a contract, which could have adverse monetary implications for us, depending on the terms of the agreement. In the case of our agreement with BARDA, if we were to cease developing solithromycin, we would be in breach of the agreement and would lose the funding from BARDA and could, depending on the status of the work options at the time, could require us to pay our share of the one cost-sharing work option.
If we market any of our product candidates that receive approval in a manner that violates applicable health care laws, including laws prohibiting off-label promotion, disclosure laws or other similar laws, we may be subject to civil or criminal penalties.
Any regulatory approval of drug products is limited to those specific diseases and indications for which a product is deemed to be safe and effective by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. While physicians may choose to prescribe drugs for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical studies and approved by the regulatory authorities, our ability to promote the products is limited to those indications that are specifically approved by the FDA. Regulatory authorities in the United States generally do not regulate the behavior of physicians in their choice of treatments, and such off-label uses by healthcare professionals are common. Regulatory authorities do, however, restrict communications by pharmaceutical companies on the subject of off-label use. If we are not able to obtain FDA approval for any desired future indications for solithromycin, fusidic acid or any other product candidates that may be approved, our ability to market and sell such products will be limited and our business may be adversely affected.
In addition, in recent years, several states and localities have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state or make periodic public disclosures on sales, marketing, pricing, clinical trials, health care provider payments and other activities. Additionally, the federal government has enacted the Physician Payment Sunshine Act which requires pharmaceutical manufacturers to report annually to the Secretary of Health and Human Services payments or other transfers of value made by that entity to physicians and teaching hospitals. If any of our product candidates are approved, we will be required to report certain information with respect to such payments. We also expect to have to comply with similar reporting obligations in foreign countries. We will need to expend significant efforts to establish, maintain and enhance such reporting systems and processes in order to comply with these regulations. Failure to comply with the reporting requirements would result in significant civil monetary penalties. The Affordable Care Act also includes various provisions designed to strengthen significantly fraud and abuse enforcement, such as increased funding for enforcement efforts and the lowering of the intent requirement of the federal anti-kickback statute and criminal health care fraud statute such that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it.
Risks Related to Our Industry
We are subject to extensive and costly government regulation.
Antibiotics, including those we are developing and plan to develop in the future, are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the U.S. Department of Health and Human Services, the U.S. Department of Justice, state and local governments and their respective foreign equivalents. The FDA regulates the research, development, pre-clinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import and export of pharmaceutical products. If any products we develop are tested or marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not we have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding U.S. regulation. Government regulation substantially increases the cost and risk of researching, developing, manufacturing and selling products. Our failure to comply with these regulations could result in significant fines or the inability of our product candidates to obtain and maintain regulatory approval, which would have a materially adverse effect on our business, financial condition, results of operations and prospects.
Even if we obtain regulatory approval for solithromycin, fusidic acid or any of our future product candidates, we will still face extensive regulatory requirements and our products may face future development and regulatory difficulties.
Even if regulatory approval in the U.S. is obtained, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, the labeling ultimately approved for solithromycin will include, and for fusidic acid may include, restrictions on use. Solithromycin, fusidic acid or any of our other product candidates will also be subject to ongoing FDA requirements governing the labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, record keeping and reporting of safety and other post-market information. The holder of an approved NDA is subject to obligations to monitor and report adverse events and instances
48
of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical trials. Legal requirements have also been enacted to require disclosure of clinical trial results on publicly available databases.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing, requiring new warnings or other labeling changes to limit use of the drug, requiring that we conduct additional clinical trials, imposing new monitoring requirements, or requiring that we establish risk evaluation and mitigation strategies, or REMS. Advertising and promotional materials must comply with FDA rules in addition to other potentially applicable federal and state laws. The distribution of product samples to physicians must comply with the requirements of the Prescription Drug Marketing Act. Sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veteran’s Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. If we or our third party collaborators fail to comply with applicable regulatory requirements, a regulatory agency may:
|
|
• |
conduct an investigation into our practices and any alleged violation of law; |
|
|
• |
issue warning letters or untitled letters asserting that we are in violation of the law; |
|
|
• |
seek an injunction or impose civil or criminal penalties or monetary fines; |
|
|
• |
suspend or withdraw regulatory approval; |
|
|
• |
suspend any ongoing clinical trials; |
|
|
• |
refuse to approve pending applications or supplements to applications filed by us; |
|
|
• |
suspend or impose restrictions on operations, including costly new manufacturing requirements; |
|
|
• |
seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall; or |
|
|
• |
refuse to allow us to enter into supply contracts, including government contracts. |
The occurrence of any event or penalty described above may force us to expend significant amounts of time and money and may significantly inhibit our ability to bring to market or continue to market our products and generate revenues. Similar regulations apply in foreign jurisdictions.
Product liability lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our products.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates despite obtaining appropriate informed consents from our clinical trial participants, and will face an even greater risk if we commercialize solithromycin or fusidic acid in the U.S. or other additional jurisdictions or if we engage in the clinical testing of new product candidates or commercialize any additional products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. The liver toxicity issues raised by the FDA in its CRL for our NDAs for solithromycin for the treatment of CABP and the label warning, if solithromycin is approved, may heighten the risk of lawsuits if injuries appear to be caused by solithromycin. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
|
|
• |
decreased demand for our products or product candidates that we may develop; |
|
|
• |
loss of revenue; |
|
|
• |
injury to our reputation; |
49
|
|
• |
withdrawal of clinical trial participants; |
|
|
• |
initiation of investigations by regulators; |
|
|
• |
costs to defend the related litigation; |
|
|
• |
a diversion of management’s time and our resources; |
|
|
• |
substantial monetary awards to trial participants or patients; |
|
|
• |
product recalls, withdrawals or labeling, marketing or promotional restrictions; |
|
|
• |
exhaustion of any available insurance and our capital resources; |
|
|
• |
the inability to commercialize our products or product candidates; and |
|
|
• |
a decline in our stock price. |
Although we maintain general liability insurance of up to $2.0 million in the aggregate and clinical trial liability insurance of $10.0 million in the aggregate for each of solithromycin and fusidic acid, this insurance may not fully cover potential liabilities. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. In addition, inability to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the development and commercial production and sale of our products, which could adversely affect our business, financial condition, results of operations, and prospects.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical, biological and radioactive materials and viruses. In addition, our operations produce hazardous waste products. Federal, state and local laws and regulations in the U.S. govern the use, manufacture, storage, handling and disposal of hazardous materials. We may incur significant additional costs to comply with applicable laws in the future. We also cannot predict the impact on our business of new or amended environmental laws or regulations, or any changes in the way existing and future laws and regulations are interpreted or enforced. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials, and we may incur liability as a result of any such contamination or injury. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources, and we do not carry liability insurance covering the use of hazardous materials. If we fail to comply with applicable requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs, or capital expenditures for control equipment or operational changes necessary to achieve or maintain compliance. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which adversely affect our business, financial condition, results of operations, and prospects.
Risks Related to our Intellectual Property
Our ability to pursue the development and commercialization of solithromycin depends upon the continuation of our licenses from Optimer and The Scripps Research Institute.
Our agreement with Optimer (now owned by Merck) provides us with a worldwide exclusive license to develop and sell solithromycin outside of ASEAN countries. We are obligated to use our diligent efforts to develop and commercialize products licensed from Optimer. We have other obligations to Optimer under the license related to progress reporting, payment terms and confidentiality. If we do not continue to use diligent efforts to develop and commercialize solithromycin, if we are unable to make the required milestone and royalty payments under the agreement or if we otherwise materially breach the agreement, our rights to develop and commercialize solithromycin would terminate and revert to Optimer. In addition, either we or Optimer may terminate the agreement upon the uncured material breach of the agreement or upon the other party’s bankruptcy. If our agreement with Optimer is terminated by Optimer, we would lose our rights to develop and commercialize solithromycin, which would adversely affect our business, financial condition, results of operations, and prospects.
Our agreement with The Scripps Research Institute, or TSRI, provides us with a license to make, use, sell, and import products for human or animal therapeutic use that use or incorporate one or more macrolides as an active pharmaceutical ingredient and is covered by certain patent rights owned by TSRI claiming technology related to copper-catalyzed ligation of azides and acetylenes, with exclusive rights as to the People’s Republic of China (excluding Hong Kong), South Korea and Australia, and non-exclusive rights in all other countries worldwide, except the member-nations of the Association of Southeast Asian Nations (which are not
50
included in the license with TSRI). We are obligated to use commercially reasonable efforts to develop and obtain regulatory approvals to market and sell one or more licensed products. TSRI may terminate the agreement due to our insolvency, our conviction for a felony relating to the development, manufacture, use, marketing, distribution or sale of a licensed product, or upon an uncured breach of the agreement by us, including failure to make any required payment. If our agreement with TSRI is terminated by TSRI, we could lose our rights to synthesize and/or manufacture solithromycin under the licensed TSRI technology, which could adversely affect our business, financial condition, results of operations, and prospects.
Another party could develop a fusidic acid product and achieve FDA regulatory exclusivity in the U.S. before we do, potentially preventing our ability to commercialize fusidic acid.
We will rely partly on FDA regulatory exclusivity to protect our proprietary rights for fusidic acid, our fusidic acid product, in the U.S. fusidic acid has been approved and sold for several decades in Europe and countries outside the U.S., but it has never been approved in the U.S. We believe this was due to the lack of regulatory exclusivity that was available for the molecule until the passage of Public Law 110-379 on October 8, 2008, which allowed old antibiotics such as fusidic acid to obtain five-year new chemical entity, or NCE, exclusivity upon NDA approval. This exclusivity will be granted to the first fusidic acid product that receives NDA approval. During the exclusivity period, for a minimum of four years the FDA will not accept an application filed by a third party that relies on any data contained in the approved NDA. Although we are not aware of another party currently developing fusidic acid for use in the U.S. for any indication, if another party were to do so and obtain NDA approval before we do, we would not be able to obtain approval for Fusidic acid for any disease until after any period of regulatory exclusivity if our NDA relies on data contained in the previously approved NDA. In that event, we may not be able to commercialize fusidic acid, which would harm our ability to generate revenue and achieve profitability.
Our competitive position may be harmed if a competitor obtains orphan drug exclusivity for the treatment of prosthetic joint infections or refractory bone and joint infections before we do. Even if we were to obtain orphan drug exclusivity, a competitor could obtain approval of a different drug for the treatment of prosthetic joint infections or refractory bone and joint infections or for the same drug upon a showing that its drug is clinically superior to ours, which would harm our business.
Orphan drug designation is an important element of our competitive strategy for fusidic acid. The company that obtains the first FDA approval for a drug that is designated as an orphan drug for a rare disease receives a type of marketing exclusivity known as “orphan drug exclusivity.” Orphan drug exclusivity prevents FDA approval of applications by others for the same drug and the designated orphan disease or condition for seven years from the date of NDA approval. If the orphan indication is the first NDA approved for the drug, the drug is also eligible for the five-year Hatch-Waxman exclusivity for NCEs. Orphan and Hatch-Waxman exclusivities run concurrently. The FDA has designated fusidic acid as an orphan drug for the treatment of PJI. We will work to have orphan drug designation granted for fusidic acid for refractory bone and joint infections.
The FDA may approve a subsequent application from another entity for the orphan indication of prosthetic joint infections or refractory bone and joint infections if it determines that the application is for a different drug. The FDA may also approve a subsequent application for fusidic acid for an indication other than prosthetic joint infections or refractory bone and joint infections. Orphan exclusivity does not block the same drug from being approved for another indication; however, Hatch-Waxman exclusivity could block submission for a period of at least four years after approval if the subsequent application references data in the earlier NDA.
The FDA may approve a subsequent application from another entity for the same drug for the same designated and approved orphan indication during the orphan exclusivity period if it determines that the subsequent product is clinically superior, or that the holder of the initial orphan drug approval cannot assure the availability of sufficient quantities of the drug to meet the public’s need.
If we do not receive orphan exclusivity for fusidic acid for the treatment of prosthetic joint infections or refractory bone and joint infections, our business would be negatively affected. In addition, even if we do obtain orphan exclusivity for fusidic acid, the FDA may permit other companies to market other drugs for the same condition or use. In addition, the FDA may approve another fusidic acid product for prosthetic joint infections or refractory bone and joint infections during our period of orphan drug exclusivity if it can be demonstrated that the drug is clinically superior to our drug, or if we are unable to supply sufficient product to meet the public’s need. This could create a more competitive market for us.
If our efforts to protect the proprietary nature of the intellectual property related to solithromycin, fusidic acid, and our other product candidates are not adequate, we may not be able to compete effectively in our market.
Our commercial success will depend in part on our ability to obtain additional patents and protect our existing patent position as well as our ability to maintain adequate protection of other intellectual property for solithromycin, fusidic acid and any future products in the U.S. and other countries. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability. The patent positions of pharmaceutical companies are highly uncertain. The legal principles applicable to patents are in
51
transition due to changing court precedent and legislative action and we cannot assure you that the historical legal standards surrounding questions of validity will continue to be applied or that current defenses relating to issued patents in these fields will be sufficient in the future. Changes in patent laws in the U.S. such as the America Invents Act of 2011 may affect the scope, strength and enforceability of our patent rights or the nature of proceedings which may be brought by us related to our patent rights. In addition, the laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the U.S., and we may encounter significant problems in protecting our proprietary rights in these countries. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies, solithromycin, fusidic acid and any future products are covered by valid and enforceable patents or are effectively maintained as trade secrets.
These risks include the possibility that:
|
|
• |
the patent applications that we licensed or have filed on our own may fail to result in issued patents in the U.S. or in foreign countries; |
|
|
• |
patents issued or licensed to us or our partners may be challenged, discovered to have been issued on the basis of insufficient or incorrect information and/or held to be invalid or unenforceable; |
|
|
• |
the scope of any patent protection may be too narrow to exclude other competitors from developing or designing around these patents; |
|
|
• |
we or our licensors were not the first to make the inventions covered by each of our issued patents and pending patent applications; |
|
|
• |
we or our licensors were not the first to file patent applications for these inventions; |
|
|
• |
we may fail to comply with procedural, documentary, fee payment and other similar provisions during the patent application process, which can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights; |
|
|
• |
future product candidates may not be patentable; |
|
|
• |
others will claim rights or ownership with regard to patents and other proprietary rights which we hold or license; |
|
|
• |
delays in development, testing, clinical trials and regulatory review may reduce the period of time during which we could market our product candidates under patent protection; and |
|
|
• |
we may fail to timely apply for patents on our technologies or products. |
While we apply for patents covering both our technologies and potential products, including solithromycin and fusidic acid, as we deem appropriate, many biopharmaceutical companies and university and research institutions already have filed patent applications or have received patents in our areas of product development. These entities’ applications, patents and other intellectual property rights may conflict with patent applications to which we have rights and could prevent us from obtaining patents or could call into question the validity of any of our patents, if issued, or could otherwise adversely affect our ability to develop, manufacture or commercialize antibiotic candidates. In addition, if third parties file patent applications in the technologies that also claim technology to which we have rights, we may have to participate in interference, derivation or other proceedings with the U.S. Patent and Trademark Office, or USPTO, or applicable foreign patent regulatory authorities, as applicable, to determine our rights in the invention, which may be time-consuming and expensive. Moreover, issued patents may be challenged during post-grant proceedings brought by a third party or the USPTO, or in foreign countries, or in the courts. These proceedings may result in loss of patent claims or adverse changes to the scope of the claims. Patent applications may also be challenged during pre-grant proceedings. If we are unsuccessful in defending any such opposition, only part of such patent would issue or the patent might not issue at all.
If we or our licensors or partners fail to obtain and maintain patent protection for our product candidates, or our proprietary technologies and their uses, companies may be dissuaded from collaborating with us. In such event, our ability to commercialize solithromycin, fusidic acid and our other product candidates may be threatened, we could lose our competitive advantage and the competition we face could increase, all of which could adversely affect our business, financial condition, results of operations, and prospects.
If we are sued for infringing intellectual property rights of third parties, litigation will be costly and time consuming and could prevent us or delay us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our not infringing the patents and proprietary rights of other parties and not breaching any collaboration or other agreements we have entered into with regard to our technologies and products. Numerous third-party U.S. and non-U.S. issued patents and pending applications exist in the areas of antibacterial treatment, including compounds, formulations, treatment methods and synthetic processes that may be applied towards the synthesis of antibiotics. Although no legal
52
action has been commenced or threatened against us by a third party for infringing intellectual property rights, we cannot provide assurances that we or our partners will be free to manufacture or market our product candidates as planned, or that we or our licensors’ and partners’ patents will not be opposed or litigated by third parties.
There is a substantial amount of litigation involving intellectual property in the biopharmaceutical industry generally. If a third party asserts that we infringe its patents or other proprietary rights, we could face a number of risks that could adversely affect our business, financial condition, results of operations, and prospects, including:
|
|
• |
infringement and other intellectual property claims, which would be costly and time consuming to defend, whether or not we are ultimately successful, which in turn could delay the regulatory approval process, consume our capital and divert management’s attention from our business; |
|
|
• |
substantial damages for past infringement, which we may have to pay if a court determines that our products or technologies infringe a competitor’s patent or other proprietary rights; |
|
|
• |
a court prohibiting us from selling or licensing our technologies or future products unless the third party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; |
|
|
• |
if a license is available from a third party, we may have to pay substantial royalties or lump sum payments or grant cross licenses to our patents or other proprietary rights to obtain that license; and |
|
|
• |
redesigning our products so they do not infringe, which may not be possible or may require substantial monetary expenditures and time. |
Although we are not currently party to any legal proceedings relating to our intellectual property, in the future, third parties may file claims asserting that our technologies, processes or products infringe on their intellectual property. We cannot predict whether third parties will assert these claims against us or our partners or against the licensors of technology licensed to us, or whether those claims will harm our business. In addition, the outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. If we or our partners were to face infringement claims or challenges by third parties relating to our product candidates, an adverse outcome could subject us to significant liabilities to such third parties, and force us or our partners to curtail or cease the development of some or all of our product candidates, which could adversely affect our business, financial condition, results of operations, and prospects.
We may be required to file lawsuits or take other actions to protect or enforce our patents or the patents of our licensors, which could be expensive and time consuming.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally.
In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents, or those of our licensors, do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents, or those of our licensors, at risk of being invalidated, held unenforceable or interpreted narrowly and could put our patent applications, or those of our licensors, at risk of not issuing. Moreover, we may not be able to prevent, alone or with our licensors, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the U.S. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, if securities analysts or investors perceive public announcements of the results of hearings, motions or other interim proceedings or developments to be negative, the price of our common stock could be adversely affected. The occurrence of any of the above could adversely affect our business, financial condition, results of operations, and prospects.
The intellectual property protection for our product candidates is dependent on third parties.
With respect to patents and patent applications relating to solithromycin or other compounds licensed from Optimer (now owned by Merck), Optimer retains rights in ASEAN countries. Generally, we do not have the right to prosecute and maintain any applications in those countries, unless Optimer elects not to file, prosecute or maintain any or all of such patent applications. Our potential future licensors also may retain the right to prosecute and maintain the patent rights that they license to us. If Optimer or other licensors fail to appropriately prosecute and maintain patent protection for any of our product candidates in those countries
53
controlled by them, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products in those countries.
With respect to inventions that are jointly made by us and one of our licensors, partners or potential partners, we would need to determine, with our licensors, partners or potential partners, who would be responsible for the prosecution of patents relating to any joint inventions should they arise. In addition, we may be required to cede control of prosecution of our patents to partners or potential partners in order to consummate a partnering transaction. If any of our licensors or partners fails to appropriately prosecute and maintain patent protection for any of our product candidates in those countries controlled by them, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products in those countries.
If we are unable to protect the confidentiality of certain information, the value of our product candidates and technology could be materially adversely affected.
We also rely on trade secrets, know-how and continuing technological advancement to develop and maintain our competitive position. To protect this competitive position, we regularly enter into confidentiality and proprietary information agreements with third parties, including employees, independent contractors, suppliers and collaborators. We cannot, however, ensure that these protective arrangements will be honored by third parties, and we may not have adequate remedies if these arrangements are breached. In addition, enforcement of claims that a third party has illegally obtained and is using trade secrets, know-how and technological advancements is expensive, time consuming and uncertain. Non-U.S. courts are sometimes less willing than U.S. courts to protect this information. Moreover, our trade secrets, know-how and technological advancements may otherwise become known or be independently developed by competitors in a manner providing us with no practical recourse against the competing parties. If any such events were to occur, they could adversely affect our business, financial condition, results of operations, and prospects.
We have not yet registered our trademarks in all of our potential markets, and failure to secure those registrations could adversely affect our business.
We have filed applications with the USPTO for marks for our two current product candidates; however, we cannot guarantee that either application will be allowed, or whether the USPTO will ultimately issue a trademark registration in respect to those applications. In addition, although we are not currently aware of any oppositions to or cancellations of our registered trademarks or pending applications, it is possible that one or more of the applications could be subject to opposition or cancellation after the marks are registered. The registrations will be subject to use and maintenance requirements. We have not yet registered all of our trademarks in all of our potential markets and there are names or symbols other than “Cempra” that may be protectable marks for which we have not sought registration. Failure to secure those registrations could adversely affect our business. We cannot assure you that opposition or cancellation proceedings will not be filed against our trademarks or that our trademarks would survive such proceedings.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Such claims may lead to material costs for us, or an inability to protect or use valuable intellectual property rights, which could adversely affect our business, financial condition, results of operations, and prospects.
Risks Related to Ownership of Our Common Stock
We are subject to a putative securities class action and a shareholder derivative lawsuit, which may require significant management time and attention and significant legal expenses and may result in unfavorable outcomes, which could have a material adverse effect on our business, financial condition, results of operations and cash flows.
On November 4, 2016, a securities class action lawsuit was commenced in the United States District Court, Middle District of North Carolina, Durham Division, naming our company and certain of our officers as defendants, and alleging violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by the defendants between May 1, 2016 and November 1, 2016, or the Class Period. The plaintiff seeks to represent a class comprised of purchasers of our common stock during the Class Period and seeks damages, costs and expenses and such other relief as determined by the Court. Two substantially similar lawsuits were filed in the United States District Court, Middle District of North Carolina on November 22, 2016 and December 30, 2016. We believe we have meritorious defenses and intend to defend the lawsuit vigorously. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants.
54
On December 21, 2016, a shareholder derivative lawsuit was commenced in the North Carolina Durham County Superior Court, naming certain of our former and current officers and directors as defendants and our company as a nominal defendant, and asserting claims for breach of fiduciary duty, unjust enrichment, abuse of control, gross mismanagement, and corporate waste. A substantially similar lawsuit was filed in the North Carolina Durham County Superior Court on February 16, 2017. The complaint is based on similar allegations as asserted in the securities lawsuits described above, and seeks unspecified damages and attorneys' fees. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants. We believe we have meritorious defenses and intend to defend these lawsuits vigorously.
While we believe that we have meritorious defenses to the claims in these lawsuits and intend to vigorously defend the cases, these lawsuits could divert management’s attention from our ordinary business operations. Further, the outcome of this litigation is difficult to predict and quantify, and the defense against this litigation could be costly. The ultimate resolution of these cases could result in payments of monetary damages or other costs, materially and adversely affect our business, financial condition, results of operations and cash flows, or adversely affect our reputation, and consequently, could negatively impact the trading price of our common stock.
We have various insurance policies related to the risks associated with our business, including directors’ and officers’ liability insurance policies. However, there is no assurance that our insurance coverage will be sufficient or that our insurance carriers will cover all claims in that litigation. If we are not successful in our defense of the claims asserted in these cases and those claims are not covered by insurance or exceed our insurance coverage, we may have to pay damage awards, indemnify our officers from damage awards that may be entered against them and pay the costs and expenses incurred in defense of, or in any settlement of, such claims.
In addition, it is possible that similar lawsuits may be filed in the future in the same or other courts that name the same or additional defendants, in which case we could be similarly materially and adversely affected by such additional litigation.
The trading market for our common stock may not provide our stockholders with adequate liquidity.
Prior to February 3, 2012, there had not been a public market for our common stock. Our common stock has at time been thinly traded and may be so again. We cannot assure you that an active trading market for our common stock will be maintained. You may not be able to sell your shares quickly or at the market price if trading in our common stock is not active.
The market price of our common stock may be highly volatile, and you could lose all or part of your investment.
Our stock began trading on the Nasdaq Global Market on February 3, 2012. Between that date and February 24, 2017, it has traded between $2.55 and $46.99. Our stock price could be subject to wide fluctuations in response to a variety of factors, which include:
|
|
• |
the inability to address the issues raised in the CRL issued by the FDA for our NDAs for solithromycin for the treatment of CABP or unanticipated delays in addressing the issues raised in the CRL; |
|
|
• |
adverse regulatory decisions, including any for our applications for solithromycin as a treatment for CABP; |
|
|
• |
our ability to realize the benefits from our cost-savings initiatives; |
|
|
• |
our ability to retain key employees and other staff to adequately manage our operations; |
|
|
• |
our ability to identify and negotiate any strategic business opportunities; |
|
|
• |
market perception of risks associated with solithromycin; |
|
|
• |
market perception of, and our experience in dealing with, the impact of our recent reduction in force and our ability to retain staff sufficient to operate our company effectively; |
|
|
• |
unanticipated serious safety concerns related to the use of solithromycin, fusidic acid or any of our other product candidates; |
|
|
• |
developments concerning our manufacturing sources for solithromycin; |
|
|
• |
our cash position and investor concerns about our need for additional financing as well as our ability to raise capital and the timing and dilutive impact of any future financing transactions; |
|
|
• |
developments concerning our other sources of manufacturing supply and any commercialization partners; |
|
|
• |
any delay in enrollment of our ongoing Phase 3 clinical trial for solithromycin for gonorrhea or any other clinical trial or study we undertake; |
|
|
• |
adverse results or delays in clinical trials; |
55
|
|
• |
any delay in filing our NDA for fusidic acid and any adverse development or perceived adverse development with respect to the FDA’s review of the NDA, including without limitation the FDA’s issuance of a “refusal to file” letter or a request for additional information; |
|
|
• |
changes in laws or regulations applicable to our products or product candidates, including but not limited to clinical trial requirements for approvals; |
|
|
• |
a decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
|
|
• |
the inability to obtain adequate product supply for solithromycin, fusidic acid or any other approved drug product, or the inability to do so at acceptable prices; |
|
|
• |
the introduction of new products or technologies offered by us or our competitors; |
|
|
• |
the effectiveness of our or our potential partners’ commercialization efforts; |
|
|
• |
the inability to effectively manage our growth; |
|
|
• |
actual or anticipated variations in quarterly operating results; |
|
|
• |
the failure to meet or exceed the estimates and projections of the investment community; |
|
|
• |
the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
|
|
• |
the overall performance of the U.S. equity markets and general political and economic conditions; |
|
|
• |
announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
|
|
• |
disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
|
|
• |
additions or departures of key scientific or management personnel; |
|
|
• |
adverse market reaction to any indebtedness we may incur or securities we may issue in the future; |
|
|
• |
significant lawsuits, including patent or stockholder litigation; |
|
|
• |
changes in the market valuations of similar companies; |
|
|
• |
the trading volume of our common stock; |
|
|
• |
effects of natural or man-made catastrophic events or other business interruptions; and |
|
|
• |
other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the NASDAQ Global Market and the stock of biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
At January 31, 2017, our executive officers, directors and entities affiliated with certain of our directors beneficially owned approximately 12.6% of our outstanding voting common stock. Therefore, these stockholders have the ability to influence us through their ownership position. These stockholders may be able to determine the outcome of all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders.
If we fail to establish and maintain proper internal controls, our ability to produce accurate financial statements or comply with applicable regulations could be impaired.
SEC rules that implement Section 404 of the Sarbanes-Oxley Act require us to make a formal assessment of the effectiveness of our internal controls over financial reporting for that purpose. We first became subject to this requirement for our Annual Report on Form 10-K for the year ended December 31, 2013. While we have concluded that our internal control over financial reporting was effective as of December 31, 2015, there can be no assurance that we will be able to so conclude in the future or that we will not
56
identify one or more material weaknesses in our internal controls in connection with future evaluations. Additionally, in the past we had opted to rely on the exemptions provided in the JOBS Act regarding independent auditor assessments of internal controls over financial reporting. Beginning with the year ended December 31, 2015, when we ceased to be an “emerging growth company,” however, we now must provide our independent registered accounting firm’s assessment of our internal controls over financial reporting in each Annual Report on Form 10-K. Investors may lose confidence in our operating results, our stock price could decline and we may be subject to litigation or regulatory enforcement actions if (i) in the future we are unable to conclude that our internal control over financial reporting is effective, (ii) we identify material weaknesses in our internal control over financial reporting, which could result in financial statement errors which, in turn, could require us to restate our operating results or (iii) our independent auditors are unwilling or unable to provide us with an attestation report on the effectiveness of internal control over financial reporting as required by Section 404 of the Sarbanes-Oxley Act. Any of these events could cause investors to lose confidence in our operating results, our stock price could decline and we may be subject to litigation or regulatory enforcement actions. In addition, if we are unable to meet the requirements of Section 404 of the Sarbanes-Oxley Act, we may not be able to remain listed on the NASDAQ Global Market.
We might not be able to maintain the listing of our common stock on the Nasdaq Global Market.
Our common stock began listing on the Nasdaq Global Market on February 3, 2012, under the symbol “CEMP.” We might not be able to maintain the listing standards of that exchange. If we fail to maintain the listing requirements, our common stock might trade on the Nasdaq Capital Market, or move to the OTC Bulletin Board or in the “pink sheets” maintained by Pink OTC Markets, Inc. The OTC Bulletin Board and the “pink sheets” are generally considered to be markets that are less efficient and less broad than the Nasdaq Capital Market.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. A limited number of securities and industry analysts currently publish research on our company. If one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which might cause our stock price and trading volume to decline.
Our ability to use our net operating loss carry-forwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, referred to as the Internal Revenue Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carry-forwards and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We believe that, with the financing transactions that have occurred throughout our history, we may have triggered one or more “ownership change” limitations. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carry-forwards to offset U.S. federal taxable income may be subject to limitations, which potentially could result in increased future tax liability to us.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our capital shares. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. In addition, under our loan and security agreement with Comerica Bank, we are prohibited from declaring or paying any cash dividends. Any future determination related to dividend policy will be made at the discretion of our board of directors and will depend on then-existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if an acquisition would benefit our stockholders, and could also make it more difficult to remove our current management. These provisions include:
|
|
• |
authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
|
|
• |
limiting the removal of directors by the stockholders; |
57
|
|
• |
creating a staggered board of directors; |
|
|
• |
prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of stockholders; |
|
|
• |
eliminating the ability of stockholders to call a special meeting of stockholders; and |
|
|
• |
establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management. In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by the board of directors. This provision could have the effect of discouraging, delaying or preventing someone from acquiring us or merging with us, whether or not it is desired by or beneficial to our stockholders. Any provision of our certificate of incorporation or bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock.
Item 1B. Unresolved Staff Comments
None.
We lease approximately 32,182 square feet of office space for our headquarters in Chapel Hill, North Carolina under an agreement that expires in March 2021.
On November 4, 2016, a securities class action lawsuit was commenced in the United States District Court, Middle District of North Carolina, Durham Division, naming our company and certain of our officers as defendants, and alleging violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by the defendants between May 1, 2016 and November 1, 2016 (the “Class Period”). The plaintiff seeks to represent a class comprised of purchasers of our common stock during the Class Period and seeks damages, costs and expenses and such other relief as determined by the Court. Two substantially similar lawsuits were filed in the United States District Court, Middle District of North Carolina on November 22, 2016 and December 30, 2016, respectively. We believe we have meritorious defenses and intend to defend the lawsuits vigorously. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants.
On December 21, 2016, a shareholder derivative lawsuit was commenced in the North Carolina Durham County Superior Court, naming certain of our former and current officers and directors as defendants and our company as a nominal defendant, and asserting claims for breach of fiduciary duty, unjust enrichment, abuse of control, gross mismanagement, and corporate waste. A substantially similar lawsuit was filed in the North Carolina Durham County Superior Court on February 16, 2017. The complaints are based on similar allegations as asserted in the securities lawsuits described above, and seeks unspecified damages and attorneys' fees. We believe we have meritorious defenses and intend to defend the lawsuits vigorously. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants.
Other than as described above, we are not a party to any legal proceedings and we are not aware of any claims or actions pending or threatened against us. In the future, we might from time to time become involved in litigation relating to claims arising from our ordinary course of business.
Item 4. Mine Safety Disclosures
Not applicable
58
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is traded under the symbol “CEMP” and is quoted on the NASDAQ Global Market. Our common stock began trading on the NASDAQ Global Market on February 3, 2012.
On February 24, 2017, the closing price for the common stock as reported on the NASDAQ Global Market was $4.05.
As of February 24, 2017, there were 23 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers. We believe that, when our record holders and stockholders whose shares are held in nominee or street name by brokers are combined, we have in excess of 2,000 stockholders.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently do not plan to declare dividends on shares of our common stock in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. The payment of cash dividends in the future, if any, will be at the discretion of our board of directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors our board deems relevant.
Pursuant to the terms of the Comerica Bank loan, for as long as the Comerica Bank loan is outstanding, we may not pay any cash dividends on our common stock.
Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” in this report.
Stock Performance Graph
The following performance graph shall not be deemed to be “soliciting material” or “filed” or incorporated by reference in future filings with the SEC, or subject to the liabilities of Section 18 of the Exchange Act except as shall be expressly set forth by specific reference in such filing. The performance graph compares the performance of our common stock to the Nasdaq Biotechnology Index and the Nasdaq Composite Index. The graph covers the most recent five-year period ended December 31, 2016. The graph assumes that the value of the investment in our common stock and each index was $100.00 at February 6, 2012, the date our common stock first traded after our IPO, and that all dividends are reinvested.
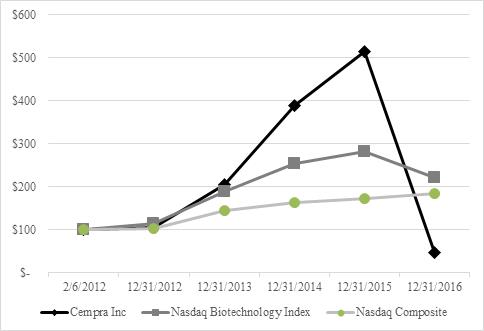
59
|
$100 investment in stock or index |
|
Ticker |
|
February 6, 2012 |
|
|
December 31, 2012 |
|
|
December 31, 2013 |
|
|
December 31, 2014 |
|
|
December 31, 2015 |
|
|
December 31, 2016 |
|
||||||
|
Cempra, Inc. |
|
CEMP |
|
$ |
100.00 |
|
|
$ |
106.00 |
|
|
$ |
204.00 |
|
|
$ |
388.00 |
|
|
$ |
514.00 |
|
|
$ |
46.00 |
|
|
NASDAQ Biotechnology Index |
|
NBI |
|
$ |
100.00 |
|
|
$ |
114.00 |
|
|
$ |
189.00 |
|
|
$ |
254.00 |
|
|
$ |
283.00 |
|
|
$ |
221.00 |
|
|
NASDAQ Composite Index |
|
IXIC |
|
$ |
100.00 |
|
|
$ |
104.00 |
|
|
$ |
144.00 |
|
|
$ |
163.00 |
|
|
$ |
172.00 |
|
|
$ |
185.00 |
|
Item 6. Selected Financial Data
The consolidated statement of income data set forth below with respect to the fiscal years ended December 31, 2016, 2015, 2014 and the consolidated balance sheet data at December 31, 2016 and 2015 are derived from the audited consolidated financial statements included in Item 8 of this Annual Report and should be read in conjunction with those financial statements and notes thereto. The consolidated statement of income data for the fiscal years ended December 31, 2013 and 2012 and the consolidated balance sheet data at December 31, 2014, 2013 and 2012 are derived from audited consolidated financial statements not included herein.
|
Consolidated Statement of Operations Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, |
|
|||||||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||||
|
|
|
(in thousands, except per share data) |
|
|||||||||||||||||
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue |
|
$ |
18,016 |
|
|
$ |
27,308 |
|
|
$ |
15,216 |
|
|
$ |
7,813 |
|
|
$ |
- |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
81,686 |
|
|
|
93,353 |
|
|
|
62,539 |
|
|
|
41,300 |
|
|
|
16,869 |
|
|
General and administrative |
|
|
53,538 |
|
|
|
22,871 |
|
|
|
12,077 |
|
|
|
9,433 |
|
|
|
6,069 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses |
|
|
135,224 |
|
|
|
116,224 |
|
|
|
74,616 |
|
|
|
50,733 |
|
|
|
22,938 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(117,208 |
) |
|
|
(88,916 |
) |
|
|
(59,400 |
) |
|
|
(42,920 |
) |
|
|
(22,938 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other income (expense), net |
|
|
(753 |
) |
|
|
(2,197 |
) |
|
|
(2,249 |
) |
|
|
(2,114 |
) |
|
|
(1,289 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
(117,961 |
) |
|
|
(91,113 |
) |
|
|
(61,649 |
) |
|
|
(45,034 |
) |
|
|
(24,227 |
) |
|
Accretion of redeemable convertible preferred shares |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(313 |
) |
|
Net loss attributable to common shareholders |
|
|
(117,961 |
) |
|
|
(91,113 |
) |
|
|
(61,649 |
) |
|
|
(45,034 |
) |
|
|
(24,540 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted loss per share |
|
$ |
(2.34 |
) |
|
$ |
(2.09 |
) |
|
$ |
(1.81 |
) |
|
$ |
(1.53 |
) |
|
$ |
(1.23 |
) |
|
Shares used in computation of basic and diluted loss per share |
|
|
50,313,614 |
|
|
|
43,565,518 |
|
|
|
34,130,901 |
|
|
|
29,449,716 |
|
|
|
19,882,585 |
|
|
Consolidated Balance Sheet Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, |
|
|||||||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||||||
|
Balance sheet data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and equivalents |
|
$ |
231,553 |
|
|
$ |
153,765 |
|
|
$ |
99,113 |
|
|
$ |
96,503 |
|
|
$ |
70,109 |
|
|
Working capital |
|
|
208,774 |
|
|
|
144,086 |
|
|
|
86,766 |
|
|
|
87,675 |
|
|
|
65,029 |
|
|
Total assets |
|
|
238,515 |
|
|
|
162,140 |
|
|
|
105,311 |
|
|
|
99,008 |
|
|
|
70,738 |
|
|
Total debt |
|
|
15,327 |
|
|
|
19,702 |
|
|
|
18,472 |
|
|
|
14,739 |
|
|
|
9,850 |
|
|
Total shareholders' equity |
|
|
183,348 |
|
|
|
117,665 |
|
|
|
61,021 |
|
|
|
69,975 |
|
|
|
57,770 |
|
60
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this report. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors. We discuss factors that we believe could cause or contribute to these differences below and elsewhere in this report, including those set forth under “Item 1A. Risk Factors.”
Overview
We are a clinical-stage pharmaceutical company focused on developing differentiated anti-infectives for the acute care and community settings to meet critical medical needs in the treatment of bacterial infectious diseases. Our lead product, solithromycin, has completed two Phase 3 clinical trials, for which we submitted NDAs for both oral and IV formulations for the treatment of CABP in April 2016. We have also filed a marketing authorization application, or MAA, with the European Medicines Agency, or EMA. In anticipation of potential approval on our PDUFA dates, throughout 2015 and 2016, we began preparations for potential commercial launch including building the commercial leadership team and implementing systems and processes to support the potential launch of solithromycin. In December 2016, we received a complete response letter, or CRL, from the FDA on our NDAs. The CRL stated that the FDA could not approve the NDAs in their present form and noted that additional clinical safety information and the satisfactory resolution of manufacturing facility inspection deficiencies were required before the NDAs may be approved. We recently met with the FDA to discuss the CRL and the FDA reiterated their request for additional clinical safety data prior to approval. Based on input from the FDA at the meeting, we are developing a protocol that will propose including fewer than 9,000 patients at the time we respond to the CRL, and will propose to deliver data from defined cohorts as the study progresses. We plan to discuss the protocol with the FDA to determine if it could support an initial approval in a limited group of patients with an urgent unmet need, while we continue to accumulate a larger post-approval safety database to support potential label expansions into broader CABP populations. If we and the FDA agree on a protocol, we plan to seek non-dilutive funding to support the execution of the study.
Our second product, fusidic acid, is an antibiotic that has been used for decades outside the U.S., including in Western Europe, but has never been approved in the U.S. We have recently completed a successful Phase 3 study evaluating fusidic acid as an oral treatment of acute bacterial skin and skin structure infections, or ABSSSI, which are frequently caused by methicillin-resistant Staphylococcus aureus, or MRSA, and we are exploring its use for the long-term oral treatment of refractory bone and joint infections, or BJI, including prosthetic joint infections, or PJI, caused by staphylococci, including S. aureus and MRSA. Currently, there is no optimal oral, chronic antibiotic for treating these infections.
In February 2017, as a consequence of the solithromycin CRL we received, and subsequent discussions with the FDA, resulting in the delay of the potential approval of solithromycin, we initiated companywide cost and personnel reductions. These actions have resulted in an approximately 67% reduction in our workforce from 136 to 45 employees, and significant reductions in external spending related to commercial preparedness and non-essential activities. The principal objective of the reductions is to enable us to conserve our financial resources as we evaluate our path forward on our existing pipeline and potential business development opportunities. As we progress our internal programs, we are also actively engaged in a process to evaluate and assess external late-stage assets and other potential strategic business opportunities to determine the best use of our significant cash resources and clinical programs to deliver value to patients and shareholders through internal and/or potential external opportunities. In connection with the reduction, we expect to record an aggregate charge related to one-time termination benefits of approximately $3.5 million in 2017.
Financial Overview
Revenue
To date, we have not generated revenue from the sale of any products. All of our revenue to date has been derived from (1) a government contract and (2) the receipt of proceeds under our license and supply agreements with Toyama Chemical Co., Ltd., or Toyama, a portion of which has been recognized as revenue in accordance with generally accepted accounting principles in the U.S., or U.S. GAAP.
In May 2013, we entered into an agreement with the Biomedical Advanced Research and Development Authority of the U.S. Department of Health and Human Services, or BARDA, for the evaluation and development of solithromycin for the treatment of bacterial infections in pediatric populations and infections caused by bioterror threat pathogens, specifically anthrax and tularemia.
The agreement is a cost plus fixed fee development contract, with a base performance segment valued at approximately $18.7 million with contract modifications, and four option work segments that BARDA may request at its sole discretion pursuant to the agreement. If all four option segments are requested, the cumulative value of the agreement, as amended to date, would be
61
approximately $68.2 million and the estimated period of performance would be until approximately May 2018. Three of the options are cost plus fixed fee arrangements and one option is a cost sharing arrangement without fixed fee, for which we are responsible for a designated portion of the costs associated with that work segment. The period of performance for the base performance segment was May 2013 through February 2016.
BARDA exercised the second option in November 2014. The value of the second option work segment is approximately $16.0 million and the estimated period of performance is November 2014 through April 2017.
In February 2016, BARDA exercised the third option work segment of the agreement, which is intended to fund a Phase 2/3 study of intravenous, oral capsule and oral suspension formulations of solithromycin in pediatric patients from two months old to 17 years with community acquired bacterial pneumonia. This option work segment is a cost-sharing arrangement under which BARDA will contribute $25.5 million and we will be responsible for an additional designated portion of the costs associated with the work segment. In September 2016, the contract was modified to increase the third option work segment by $8.0 million for increased manufacturing work related to the development of a second supply source for solithromycin. The amendment raises the value of the third option work segment to approximately $33.5 million. The estimated period of performance of this option work segment runs through May 2018.
Under the agreement, we are reimbursed and recognize revenue as allowable costs are incurred plus a portion of the fixed-fee earned. We consider fixed-fees under cost reimbursable contracts to be earned in proportion to the allowable costs incurred in performance of the work as compared to total estimated contract costs, with such costs incurred representing a reasonable measurement of the proportional performance of the work completed. Since inception of the agreement through December 31, 2016, we recognized $39.2 million in revenue under this agreement.
In May 2013, Cempra Pharmaceuticals, Inc., our wholly owned subsidiary, entered into a license agreement with Toyama, whereby we licensed to Toyama the exclusive right, with the right to sublicense, to make, use and sell any product in Japan that incorporates solithromycin as its sole active pharmaceutical ingredient, or API, for human therapeutic uses, other than for ophthalmic indications or any condition, disease or affliction of the ophthalmic tissues. Toyama also has a nonexclusive license in Japan and certain other countries, with the right to sublicense, to manufacture or have manufactured API for solithromycin for use in manufacturing such products, subject to limitations and obligations of the concurrently executed supply agreement discussed below. Toyama has granted us certain rights to intellectual property generated by Toyama under the license agreement with respect to solithromycin or licensed products for use with such products outside Japan or with other solithromycin-based products inside or outside Japan.
Following execution of the agreement, we received a $10.0 million upfront payment from Toyama. Toyama is also obligated to pay us up to an aggregate of $60.0 million in milestone payments, depending on the achievement of various regulatory, patent, development and commercial milestones. The first of these milestones was achieved in the third quarter of 2014 for which we received a payment of $10.0 million from Toyama. The second $10.0 million milestone was recognized in the first quarter of 2015 which is based on the Japan Patent Office issuing a Decision of Allowance for our patent covering certain crystal forms of solithromycin in Japan. We received payment for the second milestone in April 2015. In October 2016, we received the third $10.0 million milestone which was triggered by Toyama’s decision to progress to a Phase 3 trial of solithromycin in Japan following successful completion of a Phase 2 trial. Toyama must also pay us a royalty equal to a low-to-high first double decimal digit percentage of net sales, subject to downward adjustment in certain circumstances. Cumulatively, through December 31, 2016, we have recognized $23.0 million in revenue under this agreement with the remaining $17.0 million received being recorded as deferred revenue. Substantially all of this deferred revenue would be recognized upon FDA approval of solithromycin in the United States commercial launch in the United States and one additional country. As part of the license agreement, we also entered into a supply agreement with Toyama, whereby we will be the exclusive supplier (with certain limitations) to Toyama and its sublicensees of API for solithromycin for use in licensed products in Japan, as well as the exclusive supplier to Toyama and its sublicensees of finished forms of solithromycin to be used in its clinical trials in Japan. Pursuant to the supply agreement, Toyama will pay us for such clinical supply of finished product and all supplies of API for solithromycin for any purpose, other than the manufacture of products for commercial sale in Japan, at prices equal to our costs. All API for solithromycin supplied by us to Toyama for use in the manufacture of finished product for commercial sale in Japan will be ordered from us at prices determined by our manufacturing costs, and which may, depending on such costs, equal, exceed, or be less than such costs. Either party may terminate the supply agreement for uncured material breach or insolvency of the other party, with Toyama’s right to terminate for our breach subject to certain further conditions in the case of our failure to supply API for solithromycin or clinical supply, but otherwise the supply agreement will continue until the expiration or termination of the license agreement.
In the future, we anticipate generating revenue from a combination of sales of our products, if approved, through our own sales force in the U.S. for solithromycin, and third parties elsewhere, and license fees, milestone payments and royalties in connection with strategic collaborations regarding any of our product candidates. We expect that any revenue we generate will fluctuate from quarter
62
to quarter. If we or our strategic partners fail to complete the development of solithromycin or fusidic acid in a timely manner or obtain regulatory approval for them, or if we fail to develop our own sales force or find one or more strategic partners for the commercialization of approved products, our ability to generate future revenue, and our financial condition and results of operations would be materially adversely affected.
Research and Development Expenses
Since our inception, we have focused our resources on our research and development activities, including conducting pre-clinical studies and clinical trials, manufacturing development efforts and activities related to regulatory filings for our product candidates. We recognize our research and development expenses as they are incurred. Our research and development expenses consist primarily of:
|
|
• |
employee-related expenses, which include salaries, benefits and share-based compensation expense; |
|
|
• |
fees paid to consultants and clinical research organizations, or CROs, in connection with our clinical trials, and other related clinical trial costs, such as for investigator grants, patient screening, laboratory work and statistical compilation and analysis; |
|
|
• |
costs related to acquiring and manufacturing clinical trial materials and costs for developing additional manufacturing sources for and the manufacture of pre-approval inventory of solithromycin; |
|
|
• |
costs related to compliance with regulatory requirements; |
|
|
• |
consulting fees paid to third parties related to non-clinical research and development; |
|
|
• |
research supplies; and |
|
|
• |
license, research and milestone payments related to in-licensed technologies. |
Our direct research and development expenses consist principally of external costs, such as fees paid to investigators, consultants, central laboratories and CROs in connection with our clinical trials, and related clinical trial fees. Our internal resources, employees and infrastructure are not directly tied to any individual research project and are typically deployed across multiple projects. Through our clinical development programs, we are advancing solithromycin and fusidic acid in parallel primarily for the treatment of CABP and uncomplicated gonorrhea (for solithromycin) and ABSSSI and refractory bone and joint infections (for fusidic acid) as well as for other indications. Through our pre-clinical development programs, we are seeking to develop macrolide product candidates for non-antibacterial indications. The following table sets forth costs incurred on a program-specific basis for solithromycin and fusidic acid, excluding personnel-related costs. Macrolide research includes costs for discovery programs. All employee-related expenses for those employees working in research and development functions are included in “Research and development personnel cost” in the table, including salary, bonus, employee benefits and share-based compensation. We do not allocate insurance or other indirect costs related to our research and development function to specific product candidates. Those expenses are included in “Indirect research and development expense” in the table.
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
|
|
(in thousands) |
|
|||||||||
|
Direct research and development expense by program: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Solithromycin |
|
$ |
49,755 |
|
|
$ |
73,435 |
|
|
$ |
51,319 |
|
|
Fusidic acid |
|
|
13,522 |
|
|
|
7,093 |
|
|
|
2,903 |
|
|
Macrolide research |
|
|
2,275 |
|
|
|
317 |
|
|
|
258 |
|
|
Research and development personnel cost |
|
|
15,109 |
|
|
|
11,485 |
|
|
|
6,907 |
|
|
Total direct research and development expense |
|
|
80,661 |
|
|
|
92,330 |
|
|
|
61,387 |
|
|
Indirect research and development expense |
|
|
1,025 |
|
|
|
1,023 |
|
|
|
1,152 |
|
|
Total research and development expense |
|
$ |
81,686 |
|
|
$ |
93,353 |
|
|
$ |
62,539 |
|
63
The successful development of our clinical and pre-clinical product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing or costs of the efforts that will be necessary to complete the remainder of the development of any of our clinical or pre-clinical product candidates or the period, if any, in which material net cash inflows from these product candidates may commence. This is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
|
|
• |
the scope, rate of progress and expense of our ongoing, as well as any additional, clinical trials required and other research and development activities; |
|
|
• |
future clinical trial results; |
|
|
• |
the costs and the timing of our regulatory submissions and any regulatory approvals; and |
|
|
• |
changes in regulations governing drug approval, manufacturing, marketing and reimbursement. |
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those which we currently anticipate or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
We have completed two pivotal trials for solithromycin in CABP, including one with oral solithromycin and one with IV solithromycin progressing to oral solithromycin. We also are conducting a Phase 2/3 trial for solithromycin in pediatric patients with CABP which is funded by BARDA and a Phase 3 trial for solithromycin in patients with uncomplicated gonorrhea.
While we are conducting an exploratory study of fusidic acid for long-term suppressive therapy of refractory bone and joint infections, including PJI, we have recently concluded our Phase 3 trial for fusidic acid in ABSSSI, suspended our exploratory development program of solithromycin in NASH, and closed our study of solithromycin in COPD. As a result, we expect our research and development expenses to temporarily trend lower. However, following our discussions with the FDA with respect to the approval path for fusidic acid and solithromycin, we could decide to increase our research and development expenses.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs, including share-based compensation, for employees in executive, operational, commercial, finance and human resources functions. Other significant general and administrative expenses include professional fees for accounting, legal, and information technology services, facilities costs, expenses associated with obtaining and maintaining patents, and costs of commercial preparation activities.
We expect our general and administrative expenses to trend downward during 2017, driven primarily by reductions in personnel and expenses related to commercial preparations.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and the disclosure of contingent assets and liabilities in our financial statements. We evaluate our estimates and judgments, including those related to accrued expenses and share-based compensation, on an ongoing basis. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
Our significant accounting policies are described in more detail in the notes to our audited consolidated financial statements included in this report. We believe the following accounting policies to be most critical to the judgments and estimates used in preparation of our financial statements and such policies have been reviewed and discussed with our audit committee.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate accrued expenses. This process involves reviewing open contracts and purchase orders, communicating with applicable vendor personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for
64
services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued expenses include:
|
|
• |
fees paid to CROs in connection with pre-clinical or clinical trials; |
|
|
• |
marketing, market research and other commercial support vendors; |
|
|
• |
fees paid to investigative sites in connection with clinical trials; |
|
|
• |
milestone payments; and |
|
|
• |
unpaid salaries, wages and benefits. |
We accrue our expenses related to clinical trials based on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we will adjust the accrual accordingly. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. We do not currently anticipate the future settlement of existing accruals to differ materially from our estimates.
Revenue Recognition
Our revenue generally consists of research related revenue under federal contracts, supply revenue and licensing revenue related to non-refundable upfront fees, milestone payments and royalties earned under license agreements. Revenue is recognized when the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products and/or services has occurred; (3) the selling price is fixed or determinable; and (4) collectability is reasonably assured.
For arrangements that involve the delivery of more than one element, each product, service and/or right to use assets is evaluated to determine whether it qualifies as a separate unit of accounting. This determination is based on whether the deliverable has “stand-alone value” to the customer. The consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the fair value of each deliverable. The consideration allocated to each unit of accounting is recognized as the related goods and services are delivered, limited to the consideration that is not contingent upon future deliverables. When an arrangement is accounted for as a single unit of accounting, we determine the period over which the performance obligations will be performed and revenue recognized. Management exercises significant judgment in the determination of whether a deliverable has stand-alone value, is considered to be a separate unit of accounting, and in estimating the relative fair value of each deliverable in the arrangement.
Milestone payments are recognized when earned, provided that (i) the milestone event is substantive; (ii) there is no ongoing performance obligation related to the achievement of the milestone earned; and (iii) it would result in additional payments. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement, and the related risk associated with the achievement of the milestone. Contingent-based payments the Company may receive under a license agreement will be recognized when received.
Valuation of Financial Instruments
Share-Based Compensation
In accordance with FASB ASC Topic 718, Stock Compensation, as modified or supplemented, we measure compensation cost for share-based payment awards granted to employees and non-employee directors at fair value using the Black-Scholes option-pricing model. We recognize compensation expense on a straight-line basis over the service period for awards expected to vest. Share-based compensation cost related to share-based payment awards granted to non-employees is adjusted each reporting period for changes in the fair value of our shares until the measurement date. The measurement date is generally considered to be the date when all services have been rendered or the date that options are fully vested.
65
We calculate the fair value of share-based compensation awards using the Black-Scholes option-pricing model. The Black-Scholes option-pricing model requires the use of subjective assumptions, including share price volatility, the expected life of options, risk-free interest rate and the fair value of the underlying common shares on the date of grant. In developing our assumptions, we take into account the following:
|
|
• |
we do not have sufficient history to estimate the volatility of our common share price. We calculate expected volatility based on reported data for selected reasonably similar publicly traded companies for which the historical information is available. For the purpose of identifying peer companies, we consider characteristics such as industry, market capitalization, length of trading history, similar vesting terms and in-the-money option status. We plan to continue to use the guideline peer group volatility information until the historical volatility of our common shares is relevant to measure expected volatility for future option grants; |
|
|
• |
we determine the risk-free interest rate by reference to implied yields available from U.S. Treasury securities with a remaining term equal to the expected life assumed at the date of grant; |
|
|
• |
the assumed dividend yield is based on our expectation of not paying dividends in the foreseeable future; |
|
|
• |
we determine the average expected life of options based on the mid-point between the vesting date and the contractual term; and |
|
|
• |
we estimate forfeitures based on our historical analysis of actual option forfeitures. |
Results of Operations
Comparison of Years Ended December 31, 2016 and December 31, 2015
The following table summarizes the results of our operations for each of the years ended December 31, 2016 and 2015, together with the changes in those items in dollars:
|
|
|
Year Ended December 31, |
|
|
Dollar |
|
||||||
|
|
|
2016 |
|
|
2015 |
|
|
Change |
|
|||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract research |
|
$ |
13,677 |
|
|
$ |
12,448 |
|
|
$ |
1,229 |
|
|
License |
|
|
4,339 |
|
|
|
10,000 |
|
|
|
(5,661 |
) |
|
Supply |
|
|
- |
|
|
|
4,860 |
|
|
|
(4,860 |
) |
|
Total revenue |
|
|
18,016 |
|
|
|
27,308 |
|
|
|
(9,292 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense (1) |
|
|
81,686 |
|
|
|
93,353 |
|
|
|
(11,667 |
) |
|
General and administrative expense (1) |
|
|
53,538 |
|
|
|
22,871 |
|
|
|
30,667 |
|
|
Other income (expense), net |
|
|
(753 |
) |
|
|
(2,197 |
) |
|
|
1,444 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Includes the following stock-based compensation expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense |
|
$ |
2,942 |
|
|
$ |
1,948 |
|
|
$ |
994 |
|
|
General and administrative expense |
|
|
11,381 |
|
|
|
3,940 |
|
|
|
7,441 |
|
Contract revenue
For the twelve months ended December 31, 2016, contract research revenue increased $1.2 million as we had a third work segment under the BARDA contract commence during 2016.
License revenue
License revenue of $4.3 million was recognized in 2016 upon receipt of the $10.0 million development milestone payment which was triggered by Toyama’s decision to progress to a Phase 3 trial of solithromycin in Japan. License revenue of $10.0 million was recognized in 2015 upon receipt of the development milestone payment from the Decision of Allowance issued by the Japan Patent Office for our patent covering certain crystal forms of solithromycin in Japan.
Supply revenue
For the twelve months ended December 31, 2016, we did not recognize any revenue through our supply agreement with Toyama as compared to $4.9 million for the twelve months ended December 31, 2015.
66
Research and Development Expense
For the twelve months ended December 31, 2016, our research and development expenses decreased to $81.7 million compared to $93.3 million for the twelve months ended December 31, 2015. The decrease of $11.6 million is primarily related to the following:
|
|
• |
a decrease in solithromycin clinical trial expenses of $29.0 million related to completion of the IV-to-Oral Phase 3 clinical trial; |
|
|
• |
a decrease of $6.7 million in the purchase of API ordered for commercial quantities in preparation for the planned commercial launch of solithromycin; |
|
|
• |
a decrease of $3.6 million in purchases of API for Toyama’s clinical trials in Japan; |
|
|
• |
a decrease of $2.4 million in purchases of clinical trial supplies for fusidic acid; |
|
|
• |
an increase in fusidic acid clinical trial expenses of $8.6 million primarily related to the first Phase 3 ABSSSI trial and the ongoing refractory bone and joint study; |
|
|
• |
an increase in regulatory expenses of $8.4 million for submission fees related to the NDA and MAA filings as well as the preparation for and execution of the FDA Antimicrobial Drugs Advisory Committee meeting; |
|
|
• |
an increase in BARDA related expenses of $4.1 million as developmental activity increases; |
|
|
• |
an increase of $3.4 million related to process development and compound synthesis expenses incurred developing additional supply sources for solithromycin API; |
|
|
• |
an increase in employee cost of $2.6 million primarily from increased headcount; |
|
|
• |
an increase of $2.0 million in expenses related to research on pre-clinical programs; and |
|
|
• |
an increase of $1.0 million related to stock compensation expense. |
General and Administrative Expense
For the twelve months ended December 31, 2016, our general and administrative costs increased to $53.5 million compared to $22.9 million for the twelve months ended December 31, 2015. The increase of $30.6 million is related to the following:
|
|
• |
an increase in employee costs of $11.8 million primarily related to increased employee headcount in commercial, medical affairs and support staff in preparation for potential commercial launch; |
|
|
• |
an increase in professional services of $9.4 million primarily related to consulting fees for commercial readiness activities; |
|
|
• |
an increase of $7.4 million related to stock compensation expense, $4.2 million of which is related to the acceleration of expense due to the change in employment status from CEO to a consultant with continued service; |
|
|
• |
an increase in trade fees and related traveling expenses of $1.1 million; |
|
|
• |
an increase in office expenses and facility fees of $0.5 million; and |
|
|
• |
an increase in tax expenses of $0.4 million. |
Other Income (Expense), Net
Net other expense decreased $1.4 million for the twelve months ended December 31, 2016 compared to the twelve months ended December 31, 2015 due to an increased balance and a higher rate of return on cash equivalents, as well as a lower interest rate on the July 2015 Note compared to the December 2011 Note.
67
Comparison of Years Ended December 31, 2015 and December 31, 2014
The following table summarizes the results of our operations for each of the years ended December 31, 2015 and 2014, together with the changes in those items in dollars and as a percentage:
|
|
|
Year Ended December 31, |
|
|
Dollar |
|
||||||
|
|
|
2015 |
|
|
2014 |
|
|
Change |
|
|||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract research |
|
$ |
12,448 |
|
|
$ |
9,609 |
|
|
$ |
2,839 |
|
|
License |
|
|
10,000 |
|
|
|
4,339 |
|
|
|
5,661 |
|
|
Supply |
|
|
4,860 |
|
|
|
1,268 |
|
|
|
3,592 |
|
|
Total revenue |
|
|
27,308 |
|
|
|
15,216 |
|
|
|
12,092 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense (1) |
|
|
93,353 |
|
|
|
62,539 |
|
|
|
30,814 |
|
|
General and administrative expense (1) |
|
|
22,871 |
|
|
|
12,077 |
|
|
|
10,794 |
|
|
Other income (expense), net |
|
|
(2,197 |
) |
|
|
(2,249 |
) |
|
|
52 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Includes the following stock-based compensation expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense |
|
$ |
1,948 |
|
|
$ |
811 |
|
|
$ |
1,137 |
|
|
General and administrative expense |
|
|
3,940 |
|
|
|
2,294 |
|
|
|
1,646 |
|
Contract revenue
For the twelve months ended December 31, 2015, contract research revenue increased $2.8 million as we had two concurrent work segments under the BARDA contract running throughout 2015, while only one segment ran through the majority of 2014.
License revenue
License revenue of $10.0 million was recognized in 2015 upon receipt of the development milestone payment from the Decision of Allowance issued by the Japan Patent Office for our patent covering certain crystal forms of solithromycin in Japan. License revenue of $4.3 million was recognized in 2014 upon receipt of the $10.0 million development milestone payment upon the approval from the Pharmaceuticals and Medical Devices Agency (PMDA) for Phase 2 studies in Japan.
Supply revenue
For the twelve months ended December 31, 2015, we have recognized $4.9 million related to payments for clinical supply of finished product and supply of API under our supply agreement with Toyama as compared to $1.3 million for the twelve months ended December 31, 2014.
Research and Development Expense
For the twelve months ended December 31, 2015, our research and development expenses increased to $93.3 million compared to $62.5 million for the twelve months ended December 31, 2014. The increase of $30.8 million is primarily related to the following:
|
|
• |
an increase in purchases of initial quantity of solithromycin API in preparation for commercial launch of $20.8 million; |
|
|
• |
an increase in employee cost of $4.6 million primarily from increased headcount and stock compensation expense; |
|
|
• |
an increase in Toyama expenses of $3.3 million related to the purchase of API for clinical studies; and |
|
|
• |
an increase in fusidic acid expenses of $2.4 million primarily related to clinical trial supplies for the Phase 3 ABSSSI trial. |
General and Administrative Expense
For the twelve months ended December 31, 2015, our general and administrative costs increased to $22.9 million compared to $12.1 million for the twelve months ended December 31, 2014. The increase of $10.8 million is related to the following:
|
|
• |
an increase in professional services of $5.8 million primarily related to consulting fees for commercial readiness activities; |
68
|
|
• |
an increase in employee cost of $3.5 million from increased headcount and stock compensation expense; |
|
|
• |
an increase in trade fees of $1.0 million; and |
|
|
• |
an increase in office expenses and facility fees of $0.5 million. |
Other Income (Expense), Net
Other income (expense) remained consistent for the years ended December 31, 2015 and December 31, 2014.
Liquidity and Capital Resources
Sources of Liquidity
Since our inception through December 31, 2016, we have funded our operations principally with $618.6 million from the sale of debt and equity instruments (common and preferred), $39.2 million of research funding from our BARDA contract, and $40.0 million of licensing and milestone payments. As of December 31, 2016, we had cash and equivalents to fund operations of approximately $231.6 million.
Cash Flows
The following table sets forth the major sources and uses of cash for the periods set forth below:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
|
|
|
|
|
|
(in thousands) |
|
|
|
|
|
|
|
Net cash provided by (used in): |
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(87,080 |
) |
|
$ |
(87,849 |
) |
|
$ |
(49,092 |
) |
|
Investing activities |
|
|
(9 |
) |
|
|
(46 |
) |
|
|
(52 |
) |
|
Financing activities |
|
|
164,877 |
|
|
|
142,547 |
|
|
|
51,754 |
|
|
Net increase in cash and equivalents |
|
$ |
77,788 |
|
|
$ |
54,652 |
|
|
$ |
2,610 |
|
Operating Activities. Cash used in operating activities of $87.1 million during the year ended December 31, 2016 was primarily a result of our $118.0 million net loss, offset by changes in operating assets and liabilities of $16.4 million and non-cash items of $14.5 million. Cash used in operating activities of $87.8 million during the year ended December 31, 2015 was primarily a result of our $91.1 million net loss and changes in operating assets and liabilities of $3.2 million offset by non-cash items of $6.5 million. Cash used in operating activities of $49.1 million during the year ended December 31, 2014 was primarily a result of our $61.6 million net loss, offset by changes in operating assets and liabilities of $8.7 million and non-cash items of $3.8 million.
Investing Activities. Net cash used in investing activities was $9,000, $46,000 and $52,000 for the years ended December 31, 2016, 2015 and 2014, respectively, primarily related to our purchases of equipment.
Financing Activities. Net cash provided by financing activities was $164.9 million for the year ended December 31, 2016, $142.5 million for the year ended December 31, 2015, and $51.8 million for the year ended December 31, 2014. The cash provided by financing activities of $164.9 million in the year ended December 31, 2016 consisted primarily of net proceeds of $93.8 million and $75.1 million from the public offering in January 2016 and at-the-market (ATM) offering from May through July 2016, respectively, net of $4.4 million of principal payments on the debt refinanced with Comerica Bank. The cash provided by financing activities of $142.5 million in the year ended December 31, 2015 consisted primarily of net proceeds of $138.8 million from the public offering in January 2015, $3.0 million from proceeds from the exercise of stock options and warrants, $1.0 million in the debt refinance with Comerica Bank, offset by debt issuance costs of $0.3 million. The cash provided by financing activities of $51.8 million in the year ended December 31, 2014 consisted primarily of net proceeds of $48.5 million from the ATM offering and $3.0 million of proceeds from the amendment of the December 2011 Note.
Funding Requirements
To date, we have not generated any product revenue from our clinical stage product candidates or from any other source. We do not know when, or if, we will generate any product revenue. We do not expect to generate product revenue unless and until we obtain marketing approval of and commercialize solithromycin and/or fusidic acid or any of our other product candidates. At the same time, we expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval and engage in commercial readiness activities for, solithromycin and fusidic acid and our other product candidates. In addition, subject to obtaining regulatory approval of any of our product candidates, we expect to
69
incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We will need substantial additional funding in connection with our continuing operations.
Based on current assumptions, we believe that our existing cash and equivalents will enable us to fund our current operating expenses and capital requirements for at least the next 12 months from the filing date of this report. Such operating and capital requirements do not contemplate incremental expenses associated with a full scale commercial launch of solithromycin or any additional clinical trials with any of our product candidates or any funds from future financings or partnerships beyond the Toyama relationship and the BARDA contract. We will need to obtain additional financing for the continued development of solithromycin and fusidic acid and our other product candidates and to support the commercialization of solithromycin and/or any of our other product candidates should any receive regulatory approval. We have based our estimates on assumptions that may prove to be wrong, and we may use our available capital resources sooner than we currently expect. Due to the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures necessary to complete the development of our product candidates.
Our future capital requirements will depend on many factors, including:
|
|
• |
the scope, progress costs, and results of pre-clinical development, laboratory testing and clinical trials for any of our product candidates including any pre or post approval safety studies for solithromycin and any additional clinical trials for fusidic acid; |
|
|
• |
the costs, timing and outcome of regulatory review of our product candidates; |
|
|
• |
the costs and timing of commercialization readiness activities for any of our product candidates, including developing manufacturing sources and building our inventory of commercial product, in anticipation of regulatory approval; |
|
|
• |
the costs and timing of commercialization activities, including product sales, marketing, manufacturing and distribution, for any of our product candidates for which we receive regulatory approval; |
|
|
• |
the costs of commercial and clinical supplies of any of our drug candidates; |
|
|
• |
obtaining milestone payments from Toyama; |
|
|
• |
receipt of payments under the BARDA contract; |
|
|
• |
our ability to establish collaborations on favorable terms; |
|
|
• |
the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; |
|
|
• |
the acceptance in the medical community of any of our product candidates for which we receive approval; |
|
|
• |
revenue if any, and the timing of the related payment, from the sale of our product candidates, should any receive regulatory approval; |
|
|
• |
obtaining a commercially viable price for any of our product candidates, should any receive regulatory approval; |
|
|
• |
the availability of adequate coverage and reimbursement from federal, state and private healthcare payors for any of our product candidates, should any receive regulatory approval; |
|
|
• |
reimbursement and medical policy changes that may adversely affect the pricing, profitability or commercial appeal of any of our product candidates, should any receive regulatory approval; |
|
|
• |
our ability to enter into any license agreements for the distribution of our product candidates outside the U.S.; |
|
|
• |
the extent to which we acquire or invest in businesses, products and technologies; and |
|
|
• |
our ability to obtain government or other third-party funding. |
Until we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity offerings, debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. We do not anticipate any substantial product revenue for the foreseeable future. To the extent that we raise additional capital through the sale of equity or convertible debt securities, our stockholders’ ownership interests will be diluted, and the terms of any securities may include liquidation or other preferences that adversely affect our stockholders’ rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or declaring dividends, such as those imposed under the Comerica loan. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other
70
collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
We will need additional financing to continue development activities to obtain regulatory approval of and to commercialize solithromycin, fusidic acid and our other product candidates. We plan, as noted, to seek partners as well as equity or debt financings or other sources of third-party funding, including government grants to support the continued development and commercialization of solithromycin, fusidic acid and our other product candidates. If we are unable to raise additional funds when needed, whether on favorable terms or not, we may be required to delay, limit, reduce or terminate our development of our product candidates, or our commercialization efforts, or to grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations and Commitments
The following table summarizes our significant contractual obligations and commercial commitments at December 31, 2016 and the effects such obligations are expected to have on our liquidity and cash flows in future periods (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
More |
|
|
|
|
|
|
|
|
|
Less than |
|
|
1 - 3 |
|
|
4 - 5 |
|
|
Than |
|
||||
|
|
|
Total |
|
|
1 Year |
|
|
years |
|
|
years |
|
|
5 years |
|
|||||
|
Loan and Security Agreement |
|
|
15,556 |
|
|
|
6,667 |
|
|
|
8,889 |
|
|
|
- |
|
|
|
- |
|
|
Operating lease |
|
|
2,359 |
|
|
|
744 |
|
|
|
1,573 |
|
|
|
42 |
|
|
|
- |
|
|
Accrued Severance |
|
|
1,999 |
|
|
|
1,606 |
|
|
|
393 |
|
|
|
- |
|
|
|
- |
|
|
Interest on Loan and Security Agreement |
|
|
1,209 |
|
|
|
803 |
|
|
|
406 |
|
|
|
- |
|
|
|
- |
|
|
Macrolide Pharmaceuticals Inc. |
|
|
583 |
|
|
|
583 |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
The Scripps Research Institute |
|
|
545 |
|
|
|
85 |
|
|
|
255 |
|
|
|
170 |
|
|
|
35 |
|
|
Total (1) |
|
$ |
22,251 |
|
|
$ |
10,488 |
|
|
$ |
11,516 |
|
|
$ |
212 |
|
|
$ |
35 |
|
|
(1) |
Minimum purchase obligations under the supply agreement with FFFC was excluded from the above table as the conditions of the agreement make the committed amount undeterminable at this time |
In July 2015, we entered into a Loan and Security Agreement (the “Loan and Security Agreement”) with Comerica Bank (“Comerica”). The Loan and Security Agreement provides that we may borrow up to $20.0 million in a term loan (the “Term Loan”) and, upon FDA approval of our planned NDA for solithromycin, we may also borrow an aggregate amount equal to the lesser of (i) up to 75% of our eligible inventory and 80% of eligible accounts receivable or (ii) $10.0 million (the “Revolver”). After FDA approval of our planned NDA for solithromycin, we may convert the Term Loan to the Revolver, in which event the Revolver would have a maximum amount available to us of $25.0 million. The Loan and Security Agreement specifies the criteria for determining eligible inventory and eligible accounts receivable and sets forth ongoing limitations and conditions precedent to our ability to borrow under the Revolver.
At closing, we received the full $20.0 million under the Term Loan and paid a facility fee of $0.1 million for the Term Loan and a facility fee of $0.2 million for the Revolver. We immediately used proceeds from the Term Loan to pay all of our $17.7 million outstanding principal and interest and $1.2 million in end of term and prepayment fees under the loan and security agreement (“December 2011 Note”) with Hercules Technology Growth Capital, Inc. (“Hercules”) and terminated the December 2011 Note. We recorded a charge of $0.3 million on the early extinguishment of the December 2011 Note.
Amounts borrowed under the Term Loan may be repaid and reborrowed at any time without penalty or premium. The Term Loan was interest-only through April 30, 2016, followed by an amortization period of 36 months of equal monthly payments of principal plus interest, beginning on May 1, 2016 and continuing on the same day of each month thereafter until paid in full. Any amounts borrowed under the Term Loan bear interest at a floating interest rate equal to the 30 Day LIBOR rate plus 5.2%. Amounts available to be borrowed under the Revolver may also be repaid and reborrowed at any time without penalty or premium prior to December 31, 2017, at which time all advances under the Revolver shall be immediately due and payable in full. Any amounts borrowed under the Revolver will bear interest at the 30 Day LIBOR rate plus 4.2%. Once available, the Revolver is subject to an annual unused facility fee equal of 0.25%. Under the Loan and Security Agreement, we are subject to certain covenants including maintaining a minimum unrestricted cash balance of $15.0 million and continuing the development or commercially launching solithromycin.
In December 2016, we entered into a Retirement and Consulting Agreement with then current CEO, Dr. Prabhavathi Fernandes, whereby, for one year, subject to monthly extensions by mutual agreement, she will provide consulting services to us for up to 20
71
hours per week. For her consulting work, we will pay Dr. Fernandes $35,000 per month. In addition, all of Dr. Fernandes’s stock options will continue to vest during the consulting period. We recognized $4.2 million of accelerated stock compensation expense in the fourth quarter of 2016 due to the change in employment status from CEO to a consultant with continued service.
Additionally, Dr. Fernandes is entitled to the severance payments and benefits described in her employment agreement. In consideration of her waiver of the notice period provided under her employment agreement, we paid Dr. Fernandes $45,000. In lieu of her pro-rated annual bonus due under her employment agreement upon a termination of employment, we paid Dr. Fernandes an annual bonus for 2016 in the amount of $280,260. From the effective date of the release, we will continue to pay Dr. Fernandes her base salary for 18 months, at the current annual rate of $540,000. In addition, we will pay Dr. Fernandes an amount equal to one and one half times her Target Bonus (as defined in her employment agreement), based upon the average percentage of achievement of target objectives for the prior three years, which amount is $420,390, payable in 18 equal monthly payments. From the effective date of the release, upon the conclusion of the consulting period, or upon an earlier change in control of the company, all of Dr. Fernandes’s then outstanding and unvested stock options will become fully vested.
Effective June 12, 2012, Cempra Pharmaceuticals, Inc., our wholly owned subsidiary, entered into a license agreement with The Scripps Research Institute, or TSRI, whereby TSRI licensed to us rights, with rights of sublicense, to make, use, sell, and import products for human or animal therapeutic use that use or incorporate one or more macrolides as an active pharmaceutical ingredient and is covered by certain patent rights owned by TSRI claiming technology related to copper-catalysed ligation of azides and acetylenes. The rights licensed to us are exclusive as to the People’s Republic of China (excluding Hong Kong), South Korea and Australia, and are non-exclusive in all other countries worldwide, except the member-nations of the Association of Southeast Asian Nations, which are not included in the territory of the license. Under the terms of the agreement with TSRI, we paid a one-time only, non-refundable license issue fee in the amount of $0.4 million. Our rights under the agreement are subject to certain customary rights of the U.S. government that arise or result from TSRI’s receipt of research support from the U.S. government.
We are also obligated to pay annual maintenance fees to TSRI in the amount of (i) $50,000 each year for the first three years (beginning on the first anniversary of the agreement), and (ii) $85,000 each year thereafter (beginning on the fourth anniversary of the agreement). Each calendar year’s annual maintenance fees will be credited against sales royalties due under the agreement for such calendar year. Under the terms of the agreement, we must pay TSRI low single-digit percentage royalties on the net sales of the products covered by the TSRI patents for the life of the TSRI patents, a low single-digit percentage of non-royalty sublicensing revenue received with respect to countries in the nonexclusive territory and a mid-single-digit percentage of sublicensing revenue received with respect to countries in the exclusive territory, with the sublicensing revenue royalty in the exclusive territory and the sales royalties subject to certain reductions under certain circumstances. TSRI is eligible to receive milestone payments of up to $1.1 million with respect to regulatory approval in the exclusive territory and first commercial sale, in each of the exclusive territory and nonexclusive territory, of the first licensed product to achieve those milestones that is based upon each macrolide covered by the licensed patents. Each milestone is payable once per each macrolide. Each milestone payment made to TSRI with respect to a particular milestone will be creditable against any payment due to TSRI with respect to any sublicense revenues received in connection with the achievement of such milestone. Any payments made to TSRI under the TSRI license for territories subject to the Optimer agreement, discussed below, can be deducted pursuant to the terms of the license agreement we have with Optimer from any sales-based royalty payments due under the Optimer agreement up to a certain percentage reduction of the royalties due to Optimer.
Under the terms of the TSRI agreement, we are also required to pay additional fees on royalties, sublicensing and milestone payments if we, an affiliate, or a sub licensee challenges the validity or enforceability of any of the patents licensed under the agreement. Such increased payments would be required until all patent claims subject to challenge are invalidated in the particular country where such challenge was mounted.
The term of the TSRI license agreement (and the period during which we must pay royalties to TSRI in a particular country for a particular product) will end, on a country-by-country and product-by-product basis, at such time as no patent rights licensed from TSRI cover a particular product in the particular country. We have included in the table above the annual payments due TSRI, but have not included any other payments because we cannot estimate if, when or in what amounts such payments will become due under the agreement.
In March 2006, we entered into a Collaborative Research and Development and License Agreement with Optimer. Under the terms of the Optimer agreement, we acquired exclusive rights to further develop and commercialize certain Optimer technology worldwide, excluding ASEAN countries. As partial consideration for this license, during 2007 and 2006, we issued to Optimer an aggregate of 125,646 common shares with a total value of $0.2 million. We also have an obligation to make additional payments upon achievement of specified development, regulatory and commercialization milestones. The aggregate amount of such milestone payments we may need to pay is based in part on the number of products developed under the agreement. The aggregate amount of such payments would be $27.5 million if four products are developed and gain FDA approval. A milestone payment is due only once on a product regardless of the number of indications it may be approved for. Additional limited milestone payments would be due if
72
we develop more than four products. In July 2010, we made a $0.5 million milestone payment to Optimer after our successful completion of the Phase 1 trial for oral solithromycin and in July 2012 we made a $1.0 million milestone payment upon completion of our discussions with the FDA for the protocol for our proposed pivotal Phase 3 trial for oral solithromycin. Optimer can elect to receive certain milestone payments in cash or in shares of our common stock having an equivalent fair market value. We are also obligated to make tiered, mid-single-digit royalty payments to Optimer based on annual net sales of licensed products outside the ASEAN countries, which royalties are subject to reduction in the event additional licenses are obtained from third parties in order to practice our rights under the agreement and/or we are required to grant a compulsory license to a third party. We have not included these payments in the table above because we cannot estimate if, when or in what amounts such payments will become due under this agreement.
We enter into contracts in the normal course of business with clinical research organizations for clinical trials and clinical supply manufacturing and with vendors for pre-clinical research studies, research supplies and other services and products for operating purposes. These contracts generally provide for termination on notice and therefore we believe that our non-cancelable obligations under these agreements are not material.
In January 2016, we entered into a supply agreement with FUJIFILM Finechemicals Co., Ltd., or FFFC, which is intended to provide us with solithromycin in sufficient quantities and at reasonable prices to ensure we meet our obligation to Toyama under the supply agreement. We are subject to a minimum purchase obligation for a designated number of years after the successful completion of a manufacturing facility to be built and validation studies to be conducted by FFFC that could run to $80 million in the aggregate, which expense would be reduced by any supply sold to Toyama. The agreement’s initial term runs until December 16, 2025. After the end of the initial term, and at the end of each year thereafter, the term will automatically extend for an additional year unless either party gives written notice to the other of its intent to terminate within a designated period of time prior to the expiration of the term, in which case the agreement will terminate at the end of such term. The parties may at any time terminate the agreement by mutual written consent. Each party has the right to terminate the agreement immediately if there is a product failure, the other party becomes involved in bankruptcy, insolvency or similar proceedings or materially breaches the agreement and such breach remains uncured for a period of time following notice of the breach. A violation by us of the minimum purchase obligation is considered a material breach. A product failure is not considered a material breach by us. We have the right to terminate the agreement upon written notice if there is a supply failure. We also may terminate in the event that FFFC cannot provide us with solithromycin for more than a designated period of time. Upon termination, any unfulfilled binding portion of the forecast must be delivered by FFFC and paid for by us. We also may elect to purchase the remaining inventory of FFFC’s solithromycin and any remaining raw materials. If FFFC terminates the agreement for a material breach by us and, prior to such termination, (i) FFFC has constructed a facility in Japan for the primary purpose of manufacturing API for us under the agreement and (ii) such facility is completed and fully operational and qualified for the manufacture of API for delivery thereunder, then, except to the extent otherwise agreed to by the parties, we may be subject to declining penalties that could aggregate as much as $17.5 million.
On January 29, 2016, Cempra Pharmaceuticals, Inc. entered into an Option and License Agreement with Macrolide Pharmaceuticals, Inc. (“MP”), pursuant to which MP granted us an exclusive option to license certain of MP’s patents and know-how involving macrolides, including specifically novel methods of synthesizing solithromycin (the “Compound”). Under the agreement, we will support research at MP focused on developing a novel, cost-competitive manufacturing approach to solithromycin. The option will run until the later to occur of (i) the earlier of (a) the date that we first obtain FDA approval for any product incorporating the Compound as an active pharmaceutical ingredient, or API, or (b) January 27, 2019, or (ii) the date that is six months after the earlier of (a) MP’s satisfaction of certain milestones, or (b) we terminate of MP’s obligations under the evaluation program. Under the evaluation program called for in the agreement, MP will conduct research activities for the manufacture of the Compound, which activities we will evaluate to determine whether to exercise the option to license.
Upon execution of the agreement, we paid MP a non-refundable, non-creditable initial license fee of $0.4 million. For conducting the evaluation program, we will pay MP a non-refundable, non-creditable fee in the aggregate amount of $0.4 million within five business days of entry into the agreement. In addition, we will pay MP the expected reasonable, documented, direct compensation-related costs of employees and advisors necessary to conduct MP’s portion of the evaluation program in the aggregate amount of $1.5 million, which we will pay in 18 equal consecutive non-refundable, non-creditable monthly installments of $83,333, beginning with the first monthly anniversary of entry into the agreement. Further, we will pay MP up to an aggregate of $1.0 million upon the satisfaction of certain performance milestones.
If we exercise the option, the license will be exclusive and worldwide (other than Association of Southeast Asian Nations) and for any and all uses in human and non-human animals, and with the right to sublicense. We may, in our discretion, exercise the option for a reduced portion of the territory and, if we have elected a reduction the territory, may increase as we wish within the territory, and as many times as we wish, provided such increase is made within 60 months of our exercise of the option.
73
If we exercise the option, we will pay MP a non-refundable, non-creditable license fee of $1.0 million, of which $0.5 million will be paid within 15 business days of exercise, and $0.5 million will be paid in the form of “deemed royalty” payments (up to such amount) equal to a fraction of a percent of net sales of licensed products. We will pay tiered royalties of a fraction of a percent on designated levels of annual net sales of license products. Further, we will pay a non-refundable, non-creditable additional royalty equal to a fraction of a percent on the net sales of licensed products of a designated amount sold by us, our sublicensees, and product partners, but the royalty will not exceed $1.0 million in the aggregate. Royalties will be paid on a country-by-country basis and product-by-product basis until the date on which there are no valid claims of any licensed MP patent covering a product in the applicable country.
If we exercise the option, the agreement’s term will run on a country by country and product by product basis until the date on which there are no valid claims in the licensed MP patents covering a particular product in a particular country.
Net Operating Losses
As of December 31, 2016, we had federal net operating loss carryforwards of approximately $353.5 million and state net operating loss carryforwards of approximately $244.8 million. The net operating loss carryforwards begin to expire in 2026 and 2021 for federal and state tax purposes, respectively. We also had federal research and development credit carryforwards of approximately $11.6 million which begin to expire in 2026, federal orphan drug credits carryforwards of approximately $3.1 million which begin to expire in 2033, federal charitable contribution carryforwards of approximately $0.1 million which began to expire in 2017, and state credit carryforwards of approximately $0.8 million, which begin to expire in 2018.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events including significant changes in ownership interests. If our net operating loss carryforwards are limited, and we have taxable income which exceeds the permissible yearly net operating loss carryforward, we would incur a federal income tax liability even though net operating loss carryforwards would be available in future years.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements as defined under SEC rules.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, 2014-09, Revenue from Contracts with Customers. This new guidance clarifies the principles for recognizing revenue and develops a common revenue standard for U.S. GAAP. The guidance outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes the most current revenue recognition guidance. This guidance, as amended by ASU 2015-14, is effective for fiscal years and interim periods within those years beginning after December 15, 2017, which is effective for us for the year ending December 31, 2018. In March 2016, the FASB issued ASU 2016-08, Revenue from Contracts with Customers: Principal versus Agent Considerations, which clarifies the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued ASU 2016-10, Revenue from Contracts with Customers: Identifying Performance Obligations and Licensing, which clarifies an entity’s identification of its performance obligations in a contract. The update also clarifies the guidance regarding an entity’s evaluation of the nature of its promise to grant a license of intellectual property and whether or not that revenue is recognized over time or at a point in time. In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts with Customers: Narrow-Scope Improvements and Practical Expedients, which amends the guidance in the new revenue standard on collectability, non-cash consideration, presentation of sales tax, and transition. In December 2016, the FASB issued ASU 2016-20, Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers which increases shareholders’ awareness of the proposals and expedites improvements to Update 2014-09. The amendments are intended to address implementation issues that were raised by stakeholders and provide additional practical expedients to reduce the cost and complexity of applying the new revenue standard. These pronouncements have the same effective date as the new revenue standard.
In December 2016, we received a Complete Response Letter (“CRL”) from the FDA which outlined a number of steps we would be required to complete prior to commercial approval of solithromycin. Based on the nature of the items outlined in the CRL, the launch of solithromycin for the treatment of community acquired bacterial pneumonia, has been delayed. As a result, we have determined that we will not early adopt the new revenue recognition guidance in 2017. We have evaluated our contract research agreement with BARDA, and do not anticipate a material impact on our financial statements. We are currently evaluating our license agreement with Toyama to determine the impact that the implementation of this standard will have on our financial statements, if any. We plan to use the full retrospective method of adoption effective January 1, 2018.
74
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements-Going Concern. The guidance addresses management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. We have adopted this guidance as of December 31, 2016.
In February 2016, the FASB issued ASU 2016-02, Leases. The new guidance will increase transparency and comparability among organizations by recognizing lease assets and lease liabilities on the balance sheet and disclosing key information about leasing arrangements. This guidance is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. We are currently evaluating the impact that the implementation of this standard will have on our consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation. The new guidance simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. This guidance is effective for fiscal years beginning after December 15, 2016, and interim periods within those fiscal years. We are currently evaluating the impact that the implementation of this standard will have on our consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows: Classification of Certain Cash Receipts and Cash Payments. ASU 2016-15 will make eight targeted changes to how cash receipts and cash payments are presented and classified in the statement of cash flows. This new guidance is effective for fiscal years beginning after December 15, 2017. The adoption of this standard is not expected to have a material impact on our consolidated financial statements.
In January 2017, the FASB issued ASU 2017-01, Business Combinations: Clarifying the Definition of a Business which revises the definition of a business with the objective of adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. This new guidance is effective for fiscal years beginning after December 15, 2017. We are currently evaluating the impact that the implementation of this standard will have on our consolidated financial statements.
Cautionary Statement
We operate in a highly competitive environment that involves a number of risks, some of which are beyond our control. The following statement highlights some of these risks. For more detail, see “Item 1A. Risk Factors”.
Statements contained in this Form 10-K that are not historical facts are or might constitute forward-looking statements under the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Although we believe the expectations reflected in such forward-looking statements are based on reasonable assumptions, our expectations might not be attained. Forward-looking statements involve known and unknown risks that could cause actual results to differ materially from expected results. Factors that could cause actual results to differ materially from our expectations expressed in the report include, among others: risks related to the costs, timing, regulatory review and results of our studies and clinical trials and those of our strategic partners; the results of studies of our product candidates conducted by others; our and our strategic partners’ ability to obtain necessary FDA and foreign regulatory approval of our product candidates; differences between historical studies on which we have based our planned clinical trials and actual results from our trials; our anticipated capital expenditures, our estimates regarding our capital requirements, and our need for future capital; our liquidity and working capital requirements; our ability to commercialize and launch, whether on our own or with a strategic partner, any product candidate that receives regulatory approval; our expectations regarding our revenues, expenses and other results of operations; the unpredictability of the size of the markets for, and market acceptance of, any of our products, including solithromycin and fusidic acid; our ability to produce and sell any approved products and the price we are able to realize for those products; our need to obtain additional funding and our ability to obtain future funding on acceptable terms; our dependence on the success of solithromycin and fusidic acid; the possible impairment of, or inability to obtain, intellectual property rights and the cost of obtaining such rights from third parties; our ability to retain and hire necessary employees and to staff our operations appropriately; our ability to compete in our industry and innovation by our competitors; our ability to stay abreast of and comply with new or modified laws and regulations that currently apply or become applicable to our business; estimates and estimate methodologies used in preparing our financial statements; the future trading prices of our common stock and the impact of securities analysts’ reports on these prices; and the risks set out in our filings with the SEC.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, an immediate 10.0% change in interest rates would not have a material effect on the fair market value of our portfolio. Accordingly, we would not
75
expect our operating results or cash flows to be affected to any significant degree by the effect of a sudden change in market interest rates on our investment portfolio.
We do not believe that our cash and equivalents have significant risk of default or illiquidity. While we believe our cash and equivalents do not contain excessive risk, we cannot provide absolute assurance that in the future our investments will not be subject to adverse changes in market value. In addition, we maintain significant amounts of cash and equivalents at one or more financial institutions that are in excess of federally insured limits.
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation has had a material effect on our results of operations during 2016, 2015 or 2014.
Item 8. Financial Statements and Supplementary Data
The financial statements required to be filed pursuant to this Item 8 are appended to this report. An index of those financial statements is found in Item 15.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e)) are designed only to provide reasonable assurance that information to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosure. As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer (our principal executive officer) and Chief Financial Officer (our principal financial officer), of the effectiveness of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b). Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report to provide the reasonable assurance discussed above.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2016.
The effectiveness of our internal control over financial reporting as of December 31, 2016, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included in this Annual Report on Form 10-K.
Changes in Internal Control over Financial Reporting
No change to our internal control over financial reporting occurred during the last fiscal quarter ended December 31, 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
76
Item 10. Directors, Executive Officers and Corporate Governance
We have adopted a written code of ethics and business conduct that applies to our directors, executive officers and all employees. We intend to disclose any amendments to, or waivers from, our code of ethics and business conduct that are required to be publicly disclosed pursuant to rules of the SEC by filing such amendment or waiver with the SEC. This code of ethics and business conduct can be found in the corporate governance section of our website, www.cempra.com.
Executive Officers
As of February 24, 2017, our executive officers are Dr. David S. Zaccardelli, our Acting Chief Executive Officer, David S. Moore, our President and Chief Commercial Officer, Mark W. Hahn, our Chief Financial Officer, Dr. David W. Oldach, our Chief Medical Officer, and John D. Bluth, our Executive Vice President of Investor Relations and Corporate Communications. Information for each is provided below.
|
Name |
|
Age |
|
Business Experience For Last Five Years |
|
David S. Zaccardelli, Pharm.D. |
|
52 |
|
Dr. Zaccardellli joined our Board in August 2016. Prior to joining our Board, Dr. Zaccardelli served, from 2004 until 2016, in several senior management roles at United Therapeutics, including chief operating officer, chief manufacturing officer and executive vice president, pharmaceutical development and operations. Prior to joining United Therapeutics, Dr. Zaccardelli founded and led a startup company focused on contract pharmaceutical development services, from 1997 through 2003. From 1988 to 1996, Dr. Zaccardelli worked at Burroughs Wellcome & Co. and Glaxo Wellcome, Inc. in a variety of clinical research positions. He also served as director of clinical and scientific affairs for Bausch & Lomb Pharmaceuticals from 1996 to 1997. Dr. Zaccardelli received his doctor of pharmacy from the University of Michigan. |
|
David S. Moore |
|
43 |
|
Mr. Moore joined us in January 2014 as our Chief Commercial Officer and assumed the additional role of President in December 2016. From July 2013 to December 2013, Mr. Moore was Chief Business Officer of Ocera Therapeutics where he was responsible for developing the commercial plans for an orphan-designated advanced liver disease product for both the community and acute care markets. Mr. Moore was Chief Business Officer of Tranzyme Pharma from December 2012 to July 2013, and Vice President, Commercial Operations from August 2011 to January 2013, during which time he was responsible for building the commercial organization as well as in- and out-licensing clinical-stage assets. Between January 1998 and July 2011, Mr. Moore held increasing levels of responsibility in the Ortho-McNeil and Janssen divisions of Johnson & Johnson. Mr. Moore received his B.Sc. in Biology from Towson University and an M.B.A. from Lehigh University, and a second graduate degree in Health Policy Excellence from Thomas Jefferson University. |
|
Mark W. Hahn |
|
54 |
|
Mr. Hahn has been our Executive Vice President and Chief Financial Officer since February 2010. From 2008 to 2009, Mr. Hahn was the Chief Financial Officer of Athenix Corp., an agricultural biotechnology company, leading its merger with Bayer CropScience, where he served as Finance Director into 2010. Mr. Hahn has been the chief financial officer of various companies including GigaBeam Corporation, a telecommunications equipment company, from 2007 to 2008; BuildLinks, Inc., a software company, from 2002 to 2007; PerformaWorks, Inc., a software company, from 2001 to 2002; and Charles & Colvard, Ltd., a consumer products company, from 1996 to 2001. Mr. Hahn also served in various capacities, culminating in Senior Manager, at Ernst & Young and its predecessors from 1984 until 1996. Mr. Hahn holds a B.B.A. in accounting and finance from the University of Wisconsin-Milwaukee and is a certified public accountant in the State of Maryland and North Carolina. |
77
|
Name |
|
Age |
|
Business Experience For Last Five Years |
|
David W. Oldach, M.D. |
|
59 |
|
Dr. Oldach joined us in February 2011. From 2006 to 2011, Dr. Oldach directed clinical research at Gilead Sciences, Inc., where his drug development experience ranged from IND/first-in-human trial development and execution through NDA-supportive Phase 3 protocol development and execution. Dr. Oldach received his Medical Degree, Magna Cum Laude, from the University of Maryland School of Medicine and completed a residency in Internal Medicine at the Massachusetts General Hospital. He completed an Infectious Disease Fellowship at Johns Hopkins University School of Medicine, serving under John Bartlett. His academic clinical research included studies in community-acquired pneumonia and surgical infections, as well as HCV pathogenesis. At the time of his transition from academic medicine to industry, Dr. Oldach was a tenured Associate Professor of Medicine at the University of Maryland School of Medicine and served as the Infectious Diseases Section Chief in the Baltimore Veterans Administration Hospital. |
|
John D. Bluth |
|
44 |
|
Mr. Bluth joined us in August 2016. Prior to joining Cempra, Mr. Bluth was senior vice president of investor relations and corporate communications and served on the executive committee at PowerSecure International, Inc., a leading provider of energy technologies and services to electric utilities and their large industrial, commercial, institutional and municipal customers, a positon he held since 2012. From 2009 to 2012, he was senior vice president of investor relations and group communications at German-based Elster Group, one of the world's largest electricity, gas and water measurement and control providers. Mr. Bluth headed investor relations and corporate communications for biotechnology companies, CV Therapeutics, from 2002 to 2009, and Aviron Inc., from 1999 to 2002. Before joining Aviron, Mr. Bluth led the west coast healthcare practice for Fleishman-Hillard, an international public relations firm, where he worked from 1996 to 1999. |
The other information required by this Item concerning our directors is incorporated by reference from the section captioned “Proposal No. 1—Election of Directors” and “Corporate Governance” contained in our proxy statement related to the 2017 Annual Meeting of Stockholders scheduled to be held on May 17, 2017 which we intend to file with the SEC within 120 days of the end of our fiscal year pursuant to General Instruction G(3) of Form 10-K. The information required by this Item concerning compliance with Section 16(a) of the Exchange Act by our directors, executive officers and persons who own more than 10% of our outstanding common stock is incorporated by reference from the section captioned “Section 16(a) Beneficial Ownership Reporting Compliance” contained in the proxy statement.
Item 11. Executive Compensation
The information required by this Item concerning directors and executive compensation is incorporated by reference from the section captioned “Director Compensation,” “Executive Compensation – Summary Compensation Table” “Executive Compensation – Compensation Discussion and Analysis,” “Executive Compensation – Grants of Plan Based Awards,” “Executive Compensation – Option Exercises and Stock Vested,” “Executive Compensation – Outstanding Equity Awards at Fiscal Year End 2016” “Executive Compensation – Compensation Committee Interlocks and Insider Participation,” and “Executive Compensation – Compensation Committee Report” contained in the proxy statement.
78
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth the indicated information as of December 31, 2016 with respect to our equity compensation plans:
|
|
|
Number of securities to be issued upon exercise of outstanding options, warrants and rights, or vesting of restricted shares |
|
|
Weighted- average exercise price of outstanding options, warrants, rights and restricted shares |
|
|
Number of securities remaining available for future issuance under equity compensation plans |
|
|||
|
Plan Category |
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation plans approved by our shareholders |
|
|
3,929,258 |
|
|
$ |
12.77 |
|
|
|
2,900,230 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation plans not approved by our shareholders |
|
|
- |
|
|
$ |
- |
|
|
|
- |
|
|
Total |
|
|
3,929,258 |
|
|
|
|
|
|
|
2,900,230 |
|
Our equity compensation plans consist of the Sixth Amended and Restated 2006 Stock Plan and the 2011 Equity Incentive Plan, both of which were approved by our stockholders.
The other information required by this Item is incorporated by reference to the information under the section captioned “Security Ownership of Certain Beneficial Owners and Management” contained in the proxy statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference to the information under the section captioned “Certain Relationships and Related Transactions” and “Corporate Governance and Board Matters—Independence of Directors” contained in the proxy statement.
Item 14. Principal Accounting Fees and Services
The information required by this Item is incorporated by reference to the information under the section captioned “Audit and Audit Committee Matters” contained in the proxy statement.
79
Item 15. Exhibits, Financial Statement Schedules
|
|
Page |
|
(a) The following documents are filed as part of this report: |
|
|
(1) Financial Statements: |
|
|
|
F-2 |
|
|
F-3 |
|
|
F-4 |
|
|
F-5 |
|
|
F-6 |
|
|
F-7 |
(2) Financial Statement Schedules:
All financial statement schedules have been omitted because they are not applicable, not required or the information required is shown in the financial statements or the notes thereto.
(3) Exhibits. The following exhibits are included herein or incorporated herein by reference:
|
No. |
Description |
Registrant's Form |
Dated |
Exhibit Number |
Filed Herewith |
|
1.1 |
Sales Agreement, dated May 6, 2016, between Cempra, Inc. and Cowen and Company, LLC |
8-K |
05/06/2016 |
1.1 |
|
|
2.1 |
Form of Plan of Conversion of Cempra Holdings, LLC. |
S-1 |
10/12/2011 |
2.1 |
|
|
3.1 |
Certificate of Incorporation of Cempra, Inc. |
S-1/A |
1/13/2012 |
3.1 |
|
|
3.2 |
Form of Bylaws of Cempra, Inc. |
S-1 |
10/12/2011 |
3.2 |
|
|
4.1 |
Specimen of Common Stock Certificate of Cempra, Inc. |
S-1/A |
11/22/2011 |
4.1 |
|
|
4.2 |
Form of Registration Rights Agreement by and among Cempra, Inc. and certain of its stockholders, to be effective upon the corporate conversion. |
S-1 |
10/12/2011 |
4.2 |
|
|
10.1 |
Forms of Indemnification Agreements by and between Cempra, Inc. and its directors. |
S-1 |
10/12/2011 |
10.1 |
|
|
10.2 |
Cempra, Inc. Sixth Amended and Restated 2006 Stock Plan. |
S-1/A |
1/13/2012 |
10.2 |
|
|
10.3 |
Cempra, Inc. 2011 Equity Incentive Plan, as amended May 21, 2015, and Form of Stock Option Agreement thereunder. |
S-1/A |
1/13/2012 |
10.3 |
|
|
10.4* |
Collaborative Research and Development and License Agreement dated March 31, 2006, by and between Cempra Pharmaceuticals, Inc. and Optimer Pharmaceuticals, Inc. |
S-1 |
10/12/2011 |
10.4 |
|
|
10.5* |
Supply Agreement effective March 15, 2011, by and among CEM-102 Pharmaceuticals, Inc., Ercros S.A. and Gyma Laboratories of America, Inc. |
S-1 |
10/12/2011 |
10.5 |
|
|
10.6 |
Office Lease Agreement dated November 9, 2011 between Cempra Pharmaceuticals, Inc. and Property Reserve, Inc. |
S-1/A |
11/22/2011 |
10.6 |
|
|
10.9* |
License Agreement, effective June 12, 2012, between The Scripps Research Institute and Cempra Pharmaceuticals, Inc. |
10-Q |
8/08/2012 |
10.9 |
|
80
|
No. |
Description |
Registrant's Form |
Dated |
Exhibit Number |
Filed Herewith |
|
10.10 |
2011 Equity Incentive Plan, as amended May 23, 2013. |
10-Q |
7/31/2013 |
10.3 |
|
|
10.11* |
Exclusive License and Development Agreement by and between Cempra Pharmaceuticals, Inc. and Toyama Chemical Co., Ltd., dated May 8, 2013. |
10-Q |
7/31/2013 |
10.13 |
|
|
10.12* |
Supply Agreement by and between Cempra Pharmaceuticals, Inc. and Toyama Chemical Co., Ltd., dated May 8, 2013. |
10-Q |
7/31/2013 |
10.14 |
|
|
10.13* |
Contract by and between Cempra, Inc. and the Biomedical Advanced Research and Development Authority, dated May 24, 2013. |
10-Q |
7/31/2013 |
10.15 |
|
|
10.14* |
Development and Supply Agreement by and between Cempra Pharmaceuticals, Inc. and Hospira Worldwide, Inc. effective as of July 1, 2013. |
10-Q/A |
11/08/2013 |
10.18 |
|
|
10.15 |
Amendment No. 1, effective as of September 26, 2013 to Exclusive License And Development Agreement by and between Cempra Pharmaceuticals, Inc. and Toyama Chemical Co., Ltd, dated May 8, 2013. |
10-Q |
10/29/2013 |
10.19 |
|
|
10.17 |
Form of Employment Agreement by and between Cempra, Inc. and Prabhavathi B. Fernandes, Ph.D. |
8-K |
8/13/2013 |
10.16 |
|
|
10.18 |
Form of Change in Control Severance Agreement by and between Cempra, Inc. and Prabhavathi B. Fernandes, Ph.D. |
8-K |
8/13/2013 |
10.17 |
|
|
10.19 |
First Amendment, dated May 17, 2013, to Office Lease Agreement dated November 9, 2011 between Cempra Pharmaceuticals, Inc., and Property Reserve, Inc. |
10-K |
2/28/2014 |
10.21 |
|
|
10.20 |
Second Amendment, dated August 13, 2013, to Office Lease Agreement dated November 9, 2011 between Cempra Pharmaceuticals, Inc., and Property Reserve, Inc. |
10-K |
2/28/2014 |
10.22 |
|
|
10.22 |
Third Amendment, dated March 31, 2014, to Office Lease Agreement dated November 9, 2011 between Cempra, Inc. and Property Reserve, Inc. |
10-Q |
4/29/2014 |
10.24 |
|
|
10.23 |
Change in Control Severance Agreement, dated May 23, 2014, by and between Cempra, Inc. and Mark W. Hahn. |
8-K |
5/29/2014 |
10.25 |
|
|
10.25 |
Amendment, dated November 13, 2014, to Contract by and between Cempra Pharmaceuticals, Inc. and the Biomedical Advanced Research and Development Agency, dated May 24, 2013. |
10-K |
2/29/2015 |
10.27 |
|
|
10.26 |
Change in Control Severance Agreement, dated May 23, 2014, by and between Cempra, Inc. and David Moore |
10-Q |
4/30/2015 |
10.1 |
|
|
10.27 |
Loan and Security Agreement, dated as of July 10, 2015, by and among Comerica Bank and Cempra, Inc., Cempra Pharmaceuticals, Inc. and CEM-102 Pharmaceuticals, Inc. |
8-K |
7/16/2015 |
10.1 |
|
|
10.28 |
Form of Change in Control Severance Agreement by and between Cempra, Inc. and David W. Oldach, M.D. |
8-K |
10/19/2015 |
10.1 |
|
|
10.29 |
Amendment to Form of Employment Agreement by and between Cempra, Inc. and Prabhavathi B. Fernandes, Ph.D. |
8-K |
10/19/2015 |
10.2 |
|
|
10.30 |
Amendment to Form of Change in Control Severance Agreement by and between Cempra, Inc. and Prabhavathi Fernandes, Ph.D. |
8-K |
10/19/2015 |
10.3 |
|
|
10.31 |
Amendment to Form of Change in Control Severance Agreement by and between Cempra, Inc. and Mark W. Hahn |
8-K |
10/19/2015 |
10.4 |
|
81
|
No. |
Description |
Registrant's Form |
Dated |
Exhibit Number |
Filed Herewith |
|
10.32 |
Amendment to Form of Change in Control Severance Agreement by and between Cempra, Inc. and David Moore |
8-K |
10/19/2015 |
10.5 |
|
|
10.34* |
Option and License Agreement, dated January 29, 2016, between Cempra Pharmaceuticals, Inc. and Macrolide Pharmaceuticals, Inc. |
10-K |
02/25/2016 |
10.34 |
|
|
10.35* |
API Manufacturing and Supply Agreement, entered into January 18, 2016, by and between Cempra, Inc. and FUJIFILM Finechemicals Co., Ltd. |
10-K |
02/25/2016 |
10.35 |
|
|
10.36* |
Amendment, dated February 29, 2016, to Contract by and between Cempra Pharmaceuticals, Inc. and the Biomedical Advanced Research and Development Agency, dated May 24, 2013, as amended. |
10-Q |
05/02/2016 |
10.36 |
|
|
10.37 |
Amendment, dated September 26, 2016, to contract by and between Cempra Pharmaceuticals, Inc. and the Biomedical Advanced Research and Development Agency, dated May 24, 2013, as amended. |
10-Q |
10/27/2016 |
10.1 |
|
|
10.38 |
Form of Change in Control Severance Agreement, dated August 1, 2016, by and between Cempra, Inc. and John D. Bluth |
|
|
|
X |
|
10.39 |
Executive Employment Agreement, dated December 9, 2016, between Cempra, Inc. and David S. Zaccardelli. |
|
|
|
X |
|
10.40 |
Retirement and Consulting Agreement, dated December 9, 2016, between Cempra, Inc. and Prabhavathi Fernandes. |
|
|
|
X |
|
21.1 |
List of subsidiaries of Cempra Holdings, LLC. |
S-1 |
10/12/2011 |
21.1 |
|
|
23.1 |
Consent of Independent Registered Public Accounting Firm |
|
|
|
X |
|
31.1 |
Certification of the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
X |
|
31.2 |
Certification of the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
X |
|
32.1 |
Certification of the Chief Executive Officer Pursuant to 18 U.S. C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
X |
|
32.2 |
Certification of the Chief Financial Officer Pursuant to 18 U.S. C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
X |
|
101 |
Financials in XBRL format. |
|
|
|
X |
|
* |
The Registrant has received confidential treatment with respect to portions of this exhibit. Those portions have been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request. |
82
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
|
CEMPRA, INC. |
||
|
|
|
|
|
By: |
|
/s/ David S. Zaccardelli, Pharm.D. |
|
|
|
David S. Zaccardelli, Pharm.D. Acting Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form 10-K has been signed by the following persons on behalf of the Registrant and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
/s/ David S. Zaccardelli, Pharm.D |
|
Acting Chief Executive Officer and Director (Principal Executive Officer) |
|
February 28, 2017 |
|
David S. Zaccardelli, Pharm.D. |
|
|
||
|
/s/ Mark W. Hahn |
|
Executive Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) |
|
February 28, 2017 |
|
Mark W. Hahn |
|
|
||
|
/s/ Garheng Kong, M.D., Ph.D. |
|
Chairman of the Board |
|
February 28, 2017 |
|
Garheng Kong, M.D., Ph.D. |
|
|
||
|
/s/ Dov Goldstein, M.D. |
|
Director |
|
February 28, 2017 |
|
Dov Goldstein, M.D. |
|
|
||
|
/s/ John H. Johnson |
|
Director |
|
February 28, 2017 |
|
John H. Johnson |
|
|
||
|
/s/ Richard Kent, M.D. |
|
Director |
|
February 28, 2017 |
|
Richard Kent, M.D. |
|
|
||
|
/s/ P. Sherrill Neff P. |
|
Director |
|
February 28, 2017 |
|
P. Sherrill Neff |
|
|
||
|
/s/ Michael R. Dougherty |
|
Director |
|
February 28, 2017 |
|
Michael R. Dougherty |
|
|
||
|
/s/ David N. Gill |
|
Director |
|
February 28, 2017 |
|
David N. Gill |
|
|
||
|
|
|
|
|
|
|
|
|
|
CONSOLIDATED FINANCIAL STATEMENTS
CEMPRA, INC.
|
|
Page |
|
F-2 |
|
|
F-3 |
|
|
F-4 |
|
|
F-5 |
|
|
F-6 |
|
|
F-7 |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Cempra, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of shareholders’ equity, and of cash flows present fairly, in all material respects, the financial position of Cempra Inc. and its subsidiaries at December 31, 2016 and December 31, 2015 and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control Over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our audits (which were integrated audits in 2016 and 2015). We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Raleigh, North Carolina
February 28, 2017
F-2
CEMPRA, INC.
(In thousands, except share and per share data)
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2016 |
|
|
2015 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current assets |
|
|
|
|
|
|
|
|
|
Cash and equivalents |
|
$ |
231,553 |
|
|
$ |
153,765 |
|
|
Receivables |
|
|
6,162 |
|
|
|
7,639 |
|
|
Prepaid expenses |
|
|
579 |
|
|
|
573 |
|
|
Total current assets |
|
|
238,294 |
|
|
|
161,977 |
|
|
Furniture, fixtures and equipment, net |
|
|
48 |
|
|
|
90 |
|
|
Deposits |
|
|
173 |
|
|
|
73 |
|
|
Total assets |
|
$ |
238,515 |
|
|
$ |
162,140 |
|
|
Liabilities |
|
|
|
|
|
|
|
|
|
Current liabilities |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
15,657 |
|
|
$ |
9,635 |
|
|
Accrued expenses |
|
|
2,929 |
|
|
|
1,475 |
|
|
Accrued payroll and benefits |
|
|
4,267 |
|
|
|
2,337 |
|
|
Current portion of long-term debt |
|
|
6,667 |
|
|
|
4,444 |
|
|
Total current liabilities |
|
|
29,520 |
|
|
|
17,891 |
|
|
Deferred revenue |
|
|
16,987 |
|
|
|
11,326 |
|
|
Long-term debt |
|
|
8,660 |
|
|
|
15,258 |
|
|
Total liabilities |
|
$ |
55,167 |
|
|
$ |
44,475 |
|
|
Commitments and Contingencies (Notes 4 and 8) |
|
|
|
|
|
|
|
|
|
Shareholders' Equity |
|
|
|
|
|
|
|
|
|
Preferred stock; $.001 par value; 5,000,000 shares authorized; no shares issued or outstanding at December 31, 2016 and December 31, 2015 |
|
|
- |
|
|
|
- |
|
|
Common stock; $.001 par value; 80,000,000 shares authorized; 52,392,905 and 43,990,751 issued and outstanding at December 31, 2016 and December 31, 2015, respectively |
|
|
52 |
|
|
|
44 |
|
|
Additional paid-in capital |
|
|
620,279 |
|
|
|
436,643 |
|
|
Accumulated deficit |
|
|
(436,983 |
) |
|
|
(319,022 |
) |
|
Total shareholders’ equity |
|
$ |
183,348 |
|
|
$ |
117,665 |
|
|
Total liabilities and shareholders’ equity |
|
$ |
238,515 |
|
|
$ |
162,140 |
|
The accompanying notes are an integral part of these consolidated financial statements
F-3
CEMPRA, INC.
Consolidated Statements of Operations
(In thousands, except share and per share data)
|
|
|
Years Ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract research |
|
$ |
13,677 |
|
|
$ |
12,448 |
|
|
$ |
9,609 |
|
|
License |
|
|
4,339 |
|
|
|
10,000 |
|
|
|
4,339 |
|
|
Supply |
|
|
- |
|
|
|
4,860 |
|
|
|
1,268 |
|
|
Total revenue |
|
|
18,016 |
|
|
|
27,308 |
|
|
|
15,216 |
|
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
81,686 |
|
|
|
93,353 |
|
|
|
62,539 |
|
|
General and administrative |
|
|
53,538 |
|
|
|
22,871 |
|
|
|
12,077 |
|
|
Total costs and expenses |
|
|
135,224 |
|
|
|
116,224 |
|
|
|
74,616 |
|
|
Loss from operations |
|
|
(117,208 |
) |
|
|
(88,916 |
) |
|
|
(59,400 |
) |
|
Other income (expenses) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
475 |
|
|
|
9 |
|
|
|
134 |
|
|
Interest expense |
|
|
(1,228 |
) |
|
|
(2,206 |
) |
|
|
(2,383 |
) |
|
Other income (expense), net |
|
|
(753 |
) |
|
|
(2,197 |
) |
|
|
(2,249 |
) |
|
Net loss |
|
$ |
(117,961 |
) |
|
$ |
(91,113 |
) |
|
$ |
(61,649 |
) |
|
Basic and diluted net loss per share |
|
$ |
(2.34 |
) |
|
$ |
(2.09 |
) |
|
$ |
(1.81 |
) |
|
Basic and diluted weighted average shares outstanding |
|
|
50,313,614 |
|
|
|
43,565,518 |
|
|
|
34,130,901 |
|
The accompanying notes are an integral part of these consolidated financial statements
F-4
CEMPRA, INC.
Consolidated Statements of Shareholders’ Equity
(In thousands, except share data)
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
Total |
|
|||||||
|
|
|
Common Stock |
|
|
Paid-In |
|
|
Accumulated |
|
|
Shareholders' |
|
||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||
|
Balance as of December 31, 2013 |
|
|
33,200,341 |
|
|
$ |
33 |
|
|
$ |
236,202 |
|
|
$ |
(166,260 |
) |
|
$ |
69,975 |
|
|
Share-based compensation |
|
|
- |
|
|
|
- |
|
|
|
3,105 |
|
|
|
- |
|
|
|
3,105 |
|
|
Issuance of common shares upon exercise of options and warrants |
|
|
182,078 |
|
|
|
- |
|
|
|
351 |
|
|
|
- |
|
|
|
351 |
|
|
Issuance of common stock, net of offering costs of $0.1 million |
|
|
4,092,525 |
|
|
|
4 |
|
|
|
48,434 |
|
|
|
- |
|
|
|
48,438 |
|
|
Reclassification of additional paid-in capital to warrant liability |
|
|
- |
|
|
|
- |
|
|
|
801 |
|
|
|
- |
|
|
|
801 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(61,649 |
) |
|
|
(61,649 |
) |
|
Balance as of December 31, 2014 |
|
|
37,474,944 |
|
|
$ |
37 |
|
|
$ |
288,893 |
|
|
$ |
(227,909 |
) |
|
$ |
61,021 |
|
|
Share-based compensation |
|
|
- |
|
|
|
- |
|
|
|
5,888 |
|
|
|
- |
|
|
|
5,888 |
|
|
Issuance of common shares upon exercise of options and warrants |
|
|
478,307 |
|
|
|
1 |
|
|
|
3,038 |
|
|
|
- |
|
|
|
3,039 |
|
|
Issuance of common stock, net of offering costs of $0.2 million |
|
|
6,037,500 |
|
|
|
6 |
|
|
|
138,824 |
|
|
|
- |
|
|
|
138,830 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(91,113 |
) |
|
|
(91,113 |
) |
|
Balance as of December 31, 2015 |
|
|
43,990,751 |
|
|
$ |
44 |
|
|
$ |
436,643 |
|
|
$ |
(319,022 |
) |
|
$ |
117,665 |
|
|
Share-based compensation |
|
|
- |
|
|
|
- |
|
|
|
14,323 |
|
|
|
- |
|
|
|
14,323 |
|
|
Issuance of common shares upon exercise of options and warrants |
|
|
95,180 |
|
|
|
- |
|
|
|
493 |
|
|
|
- |
|
|
|
493 |
|
|
Issuance of common stock, net of offering costs of $0.3 million |
|
|
8,306,974 |
|
|
|
8 |
|
|
|
168,820 |
|
|
|
- |
|
|
|
168,828 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(117,961 |
) |
|
|
(117,961 |
) |
|
Balance as of December 31, 2016 |
|
|
52,392,905 |
|
|
$ |
52 |
|
|
$ |
620,279 |
|
|
$ |
(436,983 |
) |
|
$ |
183,348 |
|
The accompanying notes are an integral part of these consolidated financial statements
F-5
CEMPRA, INC.
Consolidated Statements of Cash Flows
(In thousands)
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Operating activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(117,961 |
) |
|
$ |
(91,113 |
) |
|
$ |
(61,649 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation |
|
|
51 |
|
|
|
69 |
|
|
|
77 |
|
|
Share-based compensation |
|
|
14,323 |
|
|
|
5,888 |
|
|
|
3,105 |
|
|
Change in fair value of warrant liabilities |
|
|
- |
|
|
|
- |
|
|
|
(120 |
) |
|
Amortization of debt issuance costs |
|
|
69 |
|
|
|
400 |
|
|
|
767 |
|
|
Loss on extinguishment of debt |
|
|
- |
|
|
|
153 |
|
|
|
- |
|
|
Changes in operating assets and liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Receivables |
|
|
1,477 |
|
|
|
(5,289 |
) |
|
|
(724 |
) |
|
Prepaid expenses |
|
|
(6 |
) |
|
|
2,815 |
|
|
|
(2,980 |
) |
|
Deposits |
|
|
(100 |
) |
|
|
273 |
|
|
|
(13 |
) |
|
Accounts payable |
|
|
6,022 |
|
|
|
(2,259 |
) |
|
|
5,620 |
|
|
Accrued expenses |
|
|
1,454 |
|
|
|
473 |
|
|
|
611 |
|
|
Accrued payroll and benefits |
|
|
1,930 |
|
|
|
741 |
|
|
|
553 |
|
|
Deferred revenue |
|
|
5,661 |
|
|
|
- |
|
|
|
5,661 |
|
|
Net cash used in operating activities |
|
|
(87,080 |
) |
|
|
(87,849 |
) |
|
|
(49,092 |
) |
|
Investing activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of furniture, fixtures and equipment |
|
|
(9 |
) |
|
|
(46 |
) |
|
|
(52 |
) |
|
Net cash used in investing activities |
|
|
(9 |
) |
|
|
(46 |
) |
|
|
(52 |
) |
|
Financing activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from borrowing on long-term debt |
|
|
- |
|
|
|
20,000 |
|
|
|
3,000 |
|
|
Payments on long-term debt |
|
|
(4,444 |
) |
|
|
(18,995 |
) |
|
|
- |
|
|
Payment of debt issuance costs |
|
|
- |
|
|
|
(327 |
) |
|
|
(35 |
) |
|
Proceeds from exercise of stock options and warrants |
|
|
493 |
|
|
|
3,038 |
|
|
|
351 |
|
|
Proceeds from issuance of common stock, net of underwriting discounts |
|
|
169,112 |
|
|
|
139,044 |
|
|
|
48,535 |
|
|
Payment of offering costs |
|
|
(284 |
) |
|
|
(213 |
) |
|
|
(97 |
) |
|
Net cash provided by financing activities |
|
|
164,877 |
|
|
|
142,547 |
|
|
|
51,754 |
|
|
Net change in cash and equivalents |
|
|
77,788 |
|
|
|
54,652 |
|
|
|
2,610 |
|
|
Cash and equivalents at beginning of the period |
|
|
153,765 |
|
|
|
99,113 |
|
|
|
96,503 |
|
|
Cash and equivalents at end of the period |
|
$ |
231,553 |
|
|
$ |
153,765 |
|
|
$ |
99,113 |
|
|
Supplemental cash flow information |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
1,177 |
|
|
$ |
1,527 |
|
|
$ |
1,579 |
|
|
Non-cash investing and financing activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclassification of warrant liability to additional paid-in capital |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
801 |
|
The accompanying notes are an integral part of these consolidated financial statements
F-6
CEMPRA, INC.
December 31, 2016
Notes to Consolidated Financial Statements
1. Description of Business
Cempra, Inc. (the “Company” or “Cempra”, previously known as Cempra Holdings, LLC) is the successor entity of Cempra Pharmaceuticals, Inc. which was incorporated on November 18, 2005 and commenced operations in January 2006. Cempra is located in Chapel Hill, North Carolina, and is a pharmaceutical company developing medicines to treat drug-resistant bacterial infections in the hospital and community.
The Company expects to continue to incur losses and require additional financial resources to advance its products to either commercial stage or liquidity events. There can be no assurance that the Company will be able to obtain additional financial resources such as debt or equity financing or generate revenues from collaborative partners, on terms acceptable to the Company, on a timely basis or at all. The failure of the Company to obtain sufficient funds on acceptable terms when needed could have a material adverse effect on the Company’s business, results of operations and financial condition. Based on current assumptions, the Company believes that its existing cash and equivalents will enable it to fund its current operating expenses and capital requirements for at least the next 12 months from the filing date of this report.
2. Basis of Presentation
Principles of Consolidation and Basis of Presentation
The accompanying consolidated financial statements include the accounts and results of operations of Cempra and its wholly owned subsidiaries. The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of these consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash Equivalents and Concentrations of Risks
The Company considers all highly liquid investments purchased with an original maturity of three months or less at the date of purchase to be cash equivalents. Cash deposits are all in financial institutions in the U.S. The Company maintains cash in accounts which are in excess of federally insured limits. The Company places cash and equivalents in financial institutions with high credit ratings.
Receivables
Receivables consist of amounts billed and amounts earned but unbilled under the Company’s licensing agreements or its contract with the Biomedical Advanced Research and Development Authority of the U.S. Department of Health and Human Services (“BARDA”). Receivables under the BARDA contract are recorded as qualifying research activities are conducted and invoices from the Company’s vendors are received. Unbilled receivables are also recorded based upon work estimated to be complete for which the Company has not received vendor invoices. The Company carries its accounts receivable at cost less an allowance for doubtful accounts. On a periodic basis, the Company evaluates its accounts receivable and establishes an allowance based on its history of collections and write-offs and the current status of all receivables. The Company does not accrue interest on trade receivables. If accounts become uncollectible, they will be written off through a charge to the allowance for doubtful accounts. The Company has not recorded an allowance for doubtful accounts as management believes all receivables are fully collectible.
Furniture, Fixtures and Equipment, net
Furniture, fixtures and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line method. Furniture, fixtures, equipment and leasehold improvements are amortized over the shorter of the lease term or the estimated useful life of the related asset. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to income. Repairs and maintenance costs are expensed as incurred.
F-7
Furniture, fixtures and equipment are reviewed for impairment when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. If there is an impairment, a loss is recognized.
Income Taxes
Income taxes are computed using the asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the Company’s financial statements. In estimating future tax consequences, the Company considers all expected future events other than enactment of changes in tax laws or rates. A valuation allowance is recorded, if necessary, to reduce net deferred tax assets to their realizable values if management does not believe it is more likely than not that the net deferred tax assets will be realized.
Segment and Geographic Information
Operating segments are defined as components of an enterprise engaging in business activities for which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company operates and manages its business as one operating segment and all of the Company’s operations are in North America.
Intellectual Property
The Company’s policy is to file patent applications to protect technology, inventions and improvements that are considered important to the development of its business. The patent positions of technology companies, including the Company, are uncertain and involve complex legal and factual questions for which important legal principles are largely unresolved. The Company accounts for its intellectual property under the guidance of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 350, Intangibles—Goodwill and Other. Patent costs since inception have been expensed as incurred.
Research and Development Expenses
Research and development (“R&D”) expenses include direct and indirect R&D costs. Direct R&D consists principally of external costs, such as fees paid to investigators, consultants, central laboratories and clinical research organizations, including costs incurred in connection with clinical trials, and related clinical trial fees and all employee-related expenses for those employees working in research and development functions, including stock-based compensation for R&D personnel. Indirect R&D costs include insurance or other indirect costs related to the Company’s research and development function to specific product candidates. R&D costs are expensed as incurred. Expenses paid but not yet incurred are recorded in prepaid expenses. The Company expenses pre-approval inventory as R&D until regulatory approval is received.
Clinical Trial Accruals
As part of the process of preparing financial statements, the Company is required to estimate its expenses resulting from its obligation under contracts with vendors and consultants and clinical site agreements in connection with conducting clinical trials. The Company’s objective is to reflect the appropriate clinical trial expenses in its financial statements by matching those expenses with the period in which services and efforts are expended. The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. The Company determines accrual estimates through discussions with applicable personnel and outside service providers as to the progress of trials, or the services completed. During the course of a clinical trial, the Company adjusts its rate of clinical trial expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date in its financial statements based on facts and circumstances known at that time. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of status and timing of services performed relative to the actual status and timing of services performed may vary and may result in the Company reporting amounts that are too high or too low for any particular period. The Company’s clinical trial accrual is dependent upon the timely and accurate reporting of fee billings and pass-through expenses from contract research organizations and other third-party vendors as well as the timely processing of any change orders from the contract research organizations.
Revenue Recognition
The Company’s revenue generally consists of research related revenue under federal contracts, supply revenue and licensing revenue related to non-refundable upfront fees, milestone payments and royalties earned under license agreements. Revenue is recognized when the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products and/or services has occurred; (3) the selling price is fixed or determinable; and (4) collectability is reasonably assured.
F-8
For arrangements that involve the delivery of more than one element, each product, service and/or right to use assets is evaluated to determine whether it qualifies as a separate unit of accounting. This determination is based on whether the deliverable has “stand-alone value” to the customer. The consideration that is fixed or determinable is then allocated to each separate unit of accounting based on the relative selling prices of each deliverable. The consideration allocated to each unit of accounting is recognized as the related goods and services are delivered, limited to the consideration that is not contingent upon future deliverables. When an arrangement is accounted for as a single unit of accounting, the Company determines the period over which the performance obligations will be performed and revenue recognized.
Milestone payments are recognized when earned, provided that (i) the milestone event is substantive; (ii) there is no ongoing performance obligation related to the achievement of the milestone earned; and (iii) it would result in additional payments. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement, and the related risk associated with the achievement of the milestone. Contingent-based payments the Company may receive under a license agreement will be recognized when received.
Net Loss Per Share
Basic net loss per share is computed by dividing the net loss by the weighted average number of common stock shares outstanding during the period. Diluted net loss per share is computed by dividing the net loss by the weighted average number of common stock shares adjusted for the dilutive effect of common stock equivalent shares outstanding during the period. Common stock equivalents consist of convertible senior notes (using the “as if converted” method), stock options, restricted stock shares and stock warrants. Common equivalent shares are excluded from the computation in periods in which they have an anti-dilutive effect on earnings per share.
Share-Based Compensation
The Company accounts for share-based compensation following the provisions of FASB ASC Topic 718, Stock Compensation. The Company measures compensation cost for share-based payment awards granted to employees and non-employee directors at fair value using the Black-Scholes option-pricing model. The Company recognizes compensation expense on a straight-line basis over the service period for awards expected to vest. Compensation cost related to share-based payment awards granted to non-employees is adjusted each reporting period for changes in the fair value of the Company’s shares until the measurement date. The measurement date is generally considered to be the date when all services have been rendered or the date on which options are fully vested.
The Company recorded the following share-based compensation expense in accordance with ASC Topic 718 (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Research and development |
|
$ |
2,942 |
|
|
$ |
1,948 |
|
|
$ |
811 |
|
|
General and administrative |
|
|
11,381 |
|
|
|
3,940 |
|
|
|
2,294 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
$ |
14,323 |
|
|
$ |
5,888 |
|
|
$ |
3,105 |
|
Allocations to research and development and general and administrative expense are based upon the department to which the associated employee reported. No related tax benefits of the share-based compensation expense have been recognized.
Valuation Assumptions for Stock Option Plans
The employee share-based compensation expense recognized was determined using the Black-Scholes option-pricing model. Option-pricing models require the input of subjective assumptions and these assumptions can vary over time.
The weighted-average assumptions used to determine the fair value of each option grant are as follows:
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Estimated dividend yield |
|
|
0.0 |
% |
|
|
0.0 |
% |
|
|
0.0 |
% |
|
Expected share price volatility |
|
|
76.7 |
% |
|
|
74.3 |
% |
|
|
82.3 |
% |
|
Risk-free interest rate |
|
|
1.6 |
% |
|
|
1.7 |
% |
|
|
2.0 |
% |
|
Expected life of option (in years) |
|
|
5.5 |
|
|
|
5.6 |
|
|
|
5.8 |
|
|
Weighted-average fair value per share |
|
$ |
15.55 |
|
|
$ |
17.00 |
|
|
$ |
8.58 |
|
F-9
Expected stock price volatility is based on an average of the Company’s volatility and several peer public companies due to the Company’s limited history. For purposes of identifying peer companies, the Company considered characteristics such as industry, market capitalization, length of trading history, similar vesting terms and in-the-money option status. The risk-free rate is based on the U.S. Treasury yield curve during the expected life of the option. The dividend yield percentage is zero because the Company neither currently pays dividends nor intends to do so during the expected option term. The expected term represents the average time that options are expected to be outstanding. Due to a lack of term length data, the Company elected to use the mid-point between the vesting date and the contractual term as the expected term for employee options and contractual life for non-employees options. This is in accordance with the simplified method prescribed in SEC Staff Accounting Bulletin No. 107, Share-Based Payment, as the options meet the criteria of “plain-vanilla” options.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2014-09, Revenue from Contracts with Customers. This new guidance clarifies the principles for recognizing revenue and develops a common revenue standard for U.S. GAAP. The guidance outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes the most current revenue recognition guidance. This guidance, as amended by ASU 2015-14, is effective for fiscal years and interim periods within those years beginning after December 15, 2017, which is effective for the Company for the year ending December 31, 2018. In March 2016, the FASB issued ASU 2016-08, Revenue from Contracts with Customers: Principal versus Agent Considerations, which clarifies the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued ASU 2016-10, Revenue from Contracts with Customers: Identifying Performance Obligations and Licensing, which clarifies an entity’s identification of its performance obligations in a contract. The update also clarifies the guidance regarding an entity’s evaluation of the nature of its promise to grant a license of intellectual property and whether or not that revenue is recognized over time or at a point in time. In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts with Customers: Narrow-Scope Improvements and Practical Expedients, which amends the guidance in the new revenue standard on collectability, non-cash consideration, presentation of sales tax, and transition. In December 2016, the FASB issued ASU 2016-20, Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers which increases shareholders’ awareness of the proposals and expedites improvements to Update 2014-09. The amendments are intended to address implementation issues that were raised by stakeholders and provide additional practical expedients to reduce the cost and complexity of applying the new revenue standard. These pronouncements have the same effective date as the new revenue standard.
In December 2016, the Company received a Complete Response Letter (“CRL”) from the FDA which outlined a number of steps the Company would be required to complete prior to commercial approval of solithromycin. Based on the nature of the items outlined in the CRL, the launch of solithromycin for the treatment of community acquired bacterial pneumonia, has been delayed. As a result, the Company has determined that it will not early adopt the new revenue recognition guidance in 2017. The Company has evaluated the contract research agreement with BARDA, and does not anticipate a material impact on the financial statements. The Company is currently evaluating the license agreement with Toyama to determine the impact that the implementation of this standard will have on the financial statements, if any. The Company plans to use the full retrospective method of adoption effective January 1, 2018.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements-Going Concern. The guidance addresses management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. The Company has adopted this guidance as of December 31, 2016.
In February 2016, the FASB issued ASU 2016-02, Leases. The new guidance will increase transparency and comparability among organizations by recognizing lease assets and lease liabilities on the balance sheet and disclosing key information about leasing arrangements. This guidance is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation. The new guidance simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. This guidance is effective for fiscal years beginning after December 15, 2016, and interim periods within those fiscal years. The adoption of this standard is not expected to have a material impact on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows: Classification of Certain Cash Receipts and Cash Payments. ASU 2016-15 will make eight targeted changes to how cash receipts and cash payments are presented and classified in the statement of cash flows. This new guidance is effective for fiscal years beginning after December 15, 2017. The adoption of this standard is not expected to have a material impact on the Company’s consolidated financial statements.
F-10
In January 2017, the FASB issued ASU 2017-01, Business Combinations: Clarifying the Definition of a Business which revises the definition of a business with the objective of adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. This new guidance is effective for fiscal years beginning after December 15, 2017. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s consolidated financial statements.
3. Fair Value of Financial Instruments
The carrying values of cash equivalents, receivables, prepaid expenses, and accounts payable at December 31, 2016 approximated their fair values due to the short-term nature of these items.
The Company’s valuation of financial instruments is based on a three-tiered approach, which requires that fair value measurements be classified and disclosed in one of three tiers. These tiers are: Level 1, defined as quoted prices in active markets for identical assets or liabilities; Level 2, defined as valuations based on observable inputs other than those included in Level 1, such as quoted prices for similar assets and liabilities in active markets, or other inputs that are observable or can be corroborated by observable input data; and Level 3, defined as valuations based on unobservable inputs reflecting the Company’s own assumptions, consistent with reasonably available assumptions made by other market participants.
At December 31, 2016 and December 31, 2015, the Company held money market funds classified as Level 1 financial instruments of $228.5 million and $129.0 million, respectively. The Term Loan (defined and discussed in Note 7), which is classified as a Level 2 liability, has a variable interest rate and, accordingly, its carrying value approximates its fair value. At December 31, 2016, the carrying value was $15.3 million. There were no transfers between levels of the fair value hierarchy for any assets or liabilities measured at fair value in the twelve months ended December 31, 2016.
4. Significant Agreements and Contracts
License Agreements
Optimer Pharmaceuticals, Inc.
In March 2006, the Company, through its wholly owned subsidiary, Cempra Pharmaceuticals, Inc., entered into a Collaborative Research and Development and License Agreement (“Optimer Agreement”) with Optimer Pharmaceuticals, Inc. (“Optimer”) which was acquired by Cubist Pharmaceuticals, Inc. in October 2013, which was in turn acquired by Merck in January 2015. Under the terms of the Optimer Agreement, the Company acquired exclusive rights to further develop and commercialize certain Optimer technology worldwide, excluding member nations of the Association of Southeast Asian Nations.
In exchange for this license, during 2006 and 2007, the Company issued an aggregate of 125,646 common shares with a total fair value of $0.2 million to Optimer. These issuances to Optimer were expensed as incurred in research and development expense.
In July 2010, the Company paid a $0.5 million milestone payment to Optimer after the successful completion of its first solithromycin Phase 1 program. In July 2012, the Company paid a $1.0 million milestone payment after the successful completion of its first solithromycin Phase 2 program. Both milestones were expensed as incurred in research and development expense. Under the terms of the Optimer Agreement, the Company will owe Optimer additional payments, contingent upon the achievement of various development, regulatory and commercialization milestone events. One such milestone event would be owed upon FDA approval of solithromycin which would result in a payment to Optimer of $9.5 million. The aggregate amount of such milestone payments the Company may need to pay is based in part on the number of products developed under the agreement and would total $27.5 million (including the two milestone payments made to date and the milestone payment for FDA approval) if four products are developed and gain FDA approval. The Company will also pay tiered mid-single-digit royalties based on the amount of annual net sales of its approved products.
The Scripps Research Institute
In June 2012, the Company entered into a license agreement with The Scripps Research Institute (“TSRI”), whereby TSRI licensed to the Company rights, with rights of sublicense, to make, use, sell, and import products for human or animal therapeutic use that use or incorporate one or more macrolides as an active pharmaceutical ingredient and is covered by certain patent rights owned by TSRI claiming technology related to copper-catalysed ligation of azides and acetylenes. The rights licensed to the Company are exclusive as to the People’s Republic of China (excluding Hong Kong), South Korea and Australia, and are non-exclusive in all other countries worldwide, except the member-nations of the Association of Southeast Asian Nations, which are not included in the territory
F-11
of the license. Under the terms of the agreement with TSRI, the Company paid a one-time only, non-refundable license issue fee in the amount of $0.4 million which was charged to research and development expense in the second quarter of 2012.
The Company is also obligated to pay annual maintenance fees to TSRI in the amount of (i) $50,000 each year for the first three years (beginning on the first anniversary of the agreement), and (ii) $85,000 each year thereafter (beginning on the fourth anniversary of the agreement). Each calendar year’s annual maintenance fees will be credited against sales royalties due under the agreement for such calendar year. Under the terms of the agreement, the Company must pay TSRI low single-digit percentage royalties on the net sales of the products covered by the TSRI patents for the life of the TSRI patents, a low single-digit percentage of non-royalty sublicensing revenue received with respect to countries in the nonexclusive territory and a mid-single-digit percentage of sublicensing revenue received with respect to countries in the exclusive territory, with the sublicensing revenue royalty in the exclusive territory and the sales royalties subject to certain reductions under certain circumstances. TSRI is eligible to receive milestone payments of up to $1.1 million with respect to regulatory approval in the exclusive territory and first commercial sale, in each of the exclusive territory and nonexclusive territory, of the first licensed product to achieve those milestones that is based upon each macrolide covered by the licensed patents. Each milestone is payable once per each macrolide. Each milestone payment made to TSRI with respect to a particular milestone will be creditable against any payment due to TSRI with respect to any sublicense revenues received in connection with the achievement of such milestone. Pursuant to the terms of the Optimer Agreement, any payments made to TSRI under this license for territories subject to the Optimer Agreement can be deducted from any sales-based royalty payments due under the Optimer Agreement up to a certain percentage reduction of the royalties due to Optimer.
Under the terms of the agreement, the Company is also required to pay additional fees on royalties, sublicensing and milestone payments if the Company, an affiliate with TSRI, or a sublicensee challenges the validity or enforceability of any of the patents licensed under the agreement. Such increased payments would be required until all patent claims subject to challenge are invalidated in the particular country where such challenge was mounted. In December 2014, the Company paid a $0.2 million milestone payment to TSRI in relation to license and milestone payments received under the license agreement with Toyama (discussed below).
The term of the license agreement (and the period during which the Company must pay royalties to TSRI in a particular country for a particular product) will end, on a country-by-country and product-by-product basis, at such time as no patent rights licensed from TSRI cover a particular product in the particular country.
TSRI may terminate the agreement in the event (i) the Company fails to cure any non-payment or default on its indemnity or insurance obligations, (ii) the Company declares insolvency or bankruptcy, (iii) the Company is convicted of a felony relating to the development, manufacture, use, marketing, distribution or sale of any products licensed under the agreement, (iv) the Company fails to cure any underreporting or underpayment by a certain amount in any 12-month period, or (v) the Company fails to cure any default on any other obligation under the agreement. The Company may terminate the agreement with or without cause upon written notice. In the event of such termination, (i) all licenses granted to the Company will terminate except in the case of any sublicensee that was not the cause of the termination, is not in default on its obligations under its sublicense, and that pays any unpaid amounts owed by the Company under the agreement with respect to the sublicense, and (ii) the Company may complete any work in progress and sell any completed inventory on hand for a period of time after termination.
Biomedical Advanced Research and Development Authority
In May 2013, the Company entered into an agreement with BARDA, for the evaluation and development of the Company’s lead product candidate solithromycin for the treatment of bacterial infections in pediatric populations and infections caused by bioterror threat pathogens.
The agreement is a cost plus fixed fee development contract, with a base performance segment valued at approximately $18.7 million with contract modifications, and four option work segments that BARDA may request at its sole discretion pursuant to the agreement. If all four option segments are requested, the cumulative value of the agreement, as amended to date, would be approximately $68.2 million and the estimated period of performance would be until approximately May 2018. Three of the options are cost plus fixed fee arrangements and one option is a cost sharing arrangement without fixed fee, for which the Company is responsible for a designated portion of the costs associated with that work segment. The period of performance for the base performance segment was May 2013 through February 2016.
BARDA exercised the second option in November 2014. The value of the second option work segment is approximately $16.0 million and the estimated period of performance is November 2014 through April 2017.
In February 2016, BARDA exercised the third option work segment of the agreement which is intended to fund a Phase 2/3 study of intravenous, oral capsule and oral suspension formulations of solithromycin in pediatric patients from newborn to 17 years with community acquired bacterial pneumonia. This option work segment is a cost-sharing arrangement under which BARDA will
F-12
contribute $25.5 million and the Company will be responsible for an additional designated portion of the costs associated with the work segment. In September 2016, the contract was modified to increase the third option work segment by $8.0 million for increased manufacturing work related to the development of a second supply source for solithromycin. The amendment raises the value of the third option work segment to approximately $33.5 million. The estimated period of performance of this option work segment runs through May 2018.
Under the agreement, the Company is reimbursed and recognizes revenue as allowable costs are incurred plus a portion of the fixed-fee earned. The Company considers fixed-fees under cost reimbursable agreements to be earned in proportion to the allowable costs incurred in performance of the work as compared to total estimated agreement costs, with such costs incurred representing a reasonable measurement of the proportional performance of the work completed. For the years ended December 31, 2016, 2015 and 2014, the Company recognized $13.7, $12.4 and $9.6 million in revenue under this agreement, respectively.
The agreement provides the U.S. government the ability to terminate the agreement for convenience or to terminate for default if the Company fails to meet its obligations as set forth in the statement of work. The Company believes that if the government were to terminate the agreement for convenience, the costs incurred through the effective date of such termination and any settlement costs resulting from such termination would be allowable costs.
Toyama Chemical Co., Ltd.
In May 2013, Cempra Pharmaceuticals, Inc., the Company’s wholly owned subsidiary, entered into a license agreement with Toyama Chemical Co., Ltd. (“Toyama”), whereby the Company licensed to Toyama the exclusive right, with the right to sublicense, to make, use and sell any product in Japan that incorporates solithromycin, the Company’s lead compound, as its sole active pharmaceutical ingredient, or API, for human therapeutic uses, other than for ophthalmic indications or any condition, disease or affliction of the ophthalmic tissues. Toyama also has a nonexclusive license in Japan and certain other countries, with the right to sublicense, to manufacture or have manufactured API for solithromycin for use in manufacturing such products, subject to limitations and obligations of the concurrently executed supply agreement discussed below. Toyama granted the Company certain rights to intellectual property generated by Toyama under the license agreement with respect to solithromycin or licensed products for use with such products outside Japan or with other solithromycin-based products inside or outside Japan.
Following execution of the agreement, the Company received a $10.0 million upfront payment from Toyama. Toyama is also obligated to pay the Company up to an aggregate of $60.0 million in milestone payments, depending on the achievement of various regulatory, patent, development and commercial milestones. Under the terms of the license agreement, Toyama must also pay the Company a royalty equal to a low-to-high first double decimal digit percentage of net sales, subject to downward adjustment in certain circumstances. In August 2014, the Company received a $10.0 million milestone payment from Toyama (“August 2014 Milestone”), which was triggered by Toyama’s progress of its solithromycin clinical development program in Japan. The payment was made following Toyama’s receipt of regulatory clearance to begin a Phase 2 trial of solithromycin in Japan following successful completion of a Phase 1 trial. In March 2015, the Company recognized a $10.0 million milestone from Toyama based on the Japan Patent Office issuing a Decision of Allowance for the Company’s patent covering certain crystal forms of solithromycin in Japan, which payment was received in April 2015. In October 2016, the Company received the third $10.0 million milestone from Toyama (“October 2016 Milestone”), which was triggered by Toyama’s progress of solithromycin clinical development program in Japan.
As part of the license agreement, Toyama and the Company also entered into a supply agreement, whereby the Company will be the exclusive supplier (with certain limitations) to Toyama and its sublicensees of API for solithromycin for use in licensed products in Japan, as well as the exclusive supplier to Toyama and its sublicensees of finished forms of solithromycin to be used in Phase 1 and Phase 2 clinical trials in Japan. Pursuant to the supply agreement, which is an exhibit to the license agreement, Toyama will pay the Company for such clinical supply of finished product and all supplies of API for solithromycin for any purpose, other than the manufacture of products for commercial sale in Japan, at prices equal to the Company’s cost. All API for solithromycin supplied by the Company to Toyama for use in the manufacture of finished product for commercial sale in Japan will be ordered from the Company at prices determined by the Company’s manufacturing costs, and which may, depending on such costs, equal, exceed, or be less than such costs. Either party may terminate the supply agreement for uncured material breach or insolvency of the other party, with Toyama’s right to terminate for the Company’s breach subject to certain further conditions in the case of the Company’s failure to supply API for solithromycin or clinical supply, but otherwise the supply agreement will continue until the expiration or termination of the license agreement. Through December 31, 2016, the Company has recognized $6.1 million in revenue under this supply agreement.
The Company has determined that there are six deliverables under this agreement including (1) the license to develop and commercialize solithromycin in Japan, (2) the obligation of the Company to conduct Phase 3 studies and obtain regulatory approval in the United States and one other territory, (3) participation in a Joint Development Committee, or JDC, (4) participation in a Joint Commercialization Committee, or JCC, (5) the right to use the Company’s trademark, and (6) a supply agreement. The amounts
F-13
received under the license agreement have been allocated to the deliverables based on their relative fair values and will be recognized into income when the revenue recognition criteria have been achieved.
Milestone payments are recognized when earned, provided that (i) the milestone event is substantive; (ii) there is no ongoing performance obligation related to the achievement of the milestone earned; and (iii) it would result in additional payments. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement, and the related risk associated with the achievement of the milestone. Contingent-based payments the Company may receive under a license agreement will be recognized when received.
Royalties are recorded as earned in accordance with the contract terms when third party sales can be reliably measured and collectability is reasonably assured.
The Company recognized $4.3 million in revenue associated with the delivery of the license in May 2013. Additionally, because the milestone event triggering the August 2014 Milestone and October 2016 Milestone payments were considered non-substantive for accounting purposes, these milestone payments are being recognized into revenue proportionately to the six deliverables in the agreement using the same allocation as the upfront payment. Therefore, $4.3 million of the August 2014 Milestone payment was recognized into revenue in August 2014 and $4.3 million of the October 2016 Milestone payment was recognized into revenue in October 2016. The remainder of the upfront and milestone payments which aggregate to $17.0 million are recorded as deferred revenue at December 31, 2016 and will be recognized as revenue when the revenue recognition criteria of each deliverable has been met. The Company also recognized a $10.0 million milestone based on the Japan Patent Office issuing a Decision of Allowance for the Company’s patent covering certain crystal forms of solithromycin in Japan. The March 2015 milestone payment is considered substantive for accounting purposes, and therefore the $10.0 million milestone was recognized in its entirety as revenue in March 2015.
FUJIFILM Finechemicals Co., Ltd.
On January 18, 2016, Cempra Pharmaceuticals, Inc. entered into an API manufacturing and supply agreement with FUJIFILM Finechemicals Co., Ltd. (“FFFC”), which will provide the Company with solithromycin in sufficient quantities and at reasonable prices to help ensure it meets its obligation under the May 8, 2013 supply agreement with Toyama. The Company will use reasonable efforts to ensure that the solithromycin supplied by FFFC is for use as the active pharmaceutical ingredient in a human drug product to be used or sold in Japan.
The Company is subject to a minimum purchase obligation for a designated number of years after the successful completion of the manufacturing facility and validation studies by FFFC. Each calendar month, the Company will submit to FFFC a projection of the anticipated volume of solithromycin that it will order for the next designated period (as set forth in the agreement) (or, if earlier, the final calendar month of the current term). Several months of each forecast are binding and the remaining months are non-binding, provided that the quantity of solithromycin ordered for any month is between designated percentages of the quantity specified in the initial forecast and between designated percentages of the most recent previous forecast.
The price of each shipment of solithromycin will be equal to the total number of kilograms in such shipment multiplied by the per-kilogram transfer price as set forth in the agreement.
For the term of the agreement plus an additional five years or until the expiration of the patents identified in the agreement, FFFC is prohibited from supplying, selling or distributing solithromycin to, or enabling the manufacture of solithromycin by, any third party for any purpose. The Company is not precluded from developing one or more alternative or additional sources of solithromycin.
The agreement’s initial term runs until December 16, 2025. After the end of the initial term, and at the end of each year thereafter, the term will automatically extend for an additional year unless either party gives written notice to the other of its intent to terminate within a designated period of time prior to the expiration of the term, in which case the agreement will terminate at the end of such term. The parties may at any time terminate the agreement by mutual written consent. Each party has the right to terminate the agreement immediately if there is a product failure, the other party becomes involved in bankruptcy, insolvency or similar proceedings or materially breaches the agreement and such breach remains uncured for a period of time following notice of the breach. A violation by the Company of the minimum purchase obligation is considered a material breach. A product failure is not considered a material breach by the Company. The Company has the right to terminate the agreement upon written notice if there is a supply failure. The Company also may terminate in the event that FFFC cannot provide the Company with solithromycin for more than a designated period of time. Upon termination, any unfulfilled binding portion of the forecast must be delivered by FFFC and paid for by the Company. The Company also may elect to purchase the remaining inventory of FFFC’s solithromycin and any remaining raw
F-14
materials. If FFFC terminates the agreement for a material breach by the Company and, prior to such termination, (i) FFFC has constructed a facility in Japan for the primary purpose of manufacturing API for the Company under the agreement and (ii) such facility is completed and fully operational and qualified for the manufacture of API for delivery thereunder, then, except to the extent otherwise agreed to by the parties, the Company will pay FFFC an amount equal to (a) the remaining book value of the facility less (b) the product of the number of kilograms of API ordered by the Company under the agreement prior to such termination times a designated dollar amount, provided that if the total direct costs incurred by FFFC in the construction of the facility, net of any tax credits, tax refunds, government subsidies, or similar financial, monetary, or in-kind benefits provided by any governmental agency or authority, do not equal or exceed a designated dollar amount, then the remaining book value will be reduced by a pro rata amount, based on ratios set forth in the agreement, and (z) no amount will be payable if the agreement terminates after December 31, 2025; provided, however, that if FFFC manufactures any product or performs any activities (other than the manufacture of API for the Company under the agreement) in, by, or using the facility prior to such termination and makes any profit thereby, the total amount of such profits will be subtracted from the total payment amount due from the Company to FFFC.
Macrolide Pharmaceuticals, Inc.
On January 29, 2016, Cempra Pharmaceuticals, Inc. entered into an Option and License Agreement with Macrolide Pharmaceuticals, Inc. (“MP”), pursuant to which MP granted the Company an exclusive option to license certain of MP’s patents and know-how involving macrolides, including specifically novel methods of synthesizing solithromycin (the “Compound”). Under the agreement, the Company will support research at MP focused on developing a novel, cost-competitive manufacturing approach to solithromycin. The option will run until the later to occur of (i) the earlier of (a) the date that the Company first obtains FDA approval for any product incorporating the Compound as an API, or (b) January 27, 2019, or (ii) the date that is six months after the earlier of (a) MP’s satisfaction of certain milestones, or (b) the Company’s termination of MP’s obligations under the evaluation program. Under the evaluation program called for in the agreement, MP will conduct research activities for the manufacture of the Compound, which activities the Company will evaluate to determine whether to exercise the option to license.
Upon execution of the agreement, the Company paid MP a non-refundable, non-creditable initial license fee of $0.4 million. For conducting the evaluation program, the Company paid MP a non-refundable, non-creditable fee in the amount of $0.4 million. In addition, the Company will pay MP the expected reasonable, documented, direct compensation-related costs of employees and advisors necessary to conduct MP’s portion of the evaluation program in the aggregate amount of $1.5 million, which the Company will pay in 18 equal consecutive non-refundable, non-creditable monthly installments of $83,333, beginning with the first monthly anniversary of entry into the agreement. Further, the Company will pay MP up to an aggregate of $1.0 million upon the satisfaction of certain performance milestones.
If the Company exercises the option, the license will be exclusive and worldwide (other than Association of Southeast Asian Nations) and for any and all uses in human and non-human animals, and with the right to sublicense. The Company may, in its discretion, exercise the option for a reduced portion of the territory and, if the Company makes this election, may increase as it wishes within the territory, and as many times as it wishes, provided such increase is made within 60 months of the Company’s exercise of the option.
If the Company exercises the option, it will pay MP a non-refundable, non-creditable license fee of $1.0 million, of which $0.5 million will be paid within 15 business days of exercise, and $0.5 million will be paid in the form of “deemed royalty” payments (up to such amount) equal to a fraction of a percent of net sales of licensed products. The Company will pay tiered royalties of a fraction of a percent on designated levels of annual net sales of license products. Further, the Company will pay a non-refundable, non-creditable additional royalty equal to a fraction of a percent on the net sales of licensed products of a designated amount sold by the Company, its sublicensees, and product partners, but the royalty will not exceed $1.0 million in the aggregate. Royalties will be paid on a country-by-country basis and product-by-product basis until the date on which there are no valid claims of any licensed MP patent covering a product in the applicable country.
If the Company exercises the option, the agreement’s term will run on a country by country and product by product basis until the date on which there are no valid claims in the licensed MP patents covering a particular product in a particular country.
5. Receivables
Receivables consist of amounts billed and amounts earned but unbilled under the Company’s licensing agreements or its contract with BARDA. At December 31, 2016, the Company’s receivables consisted primarily of earned but unbilled receivables under the BARDA agreement. At December 31, 2015, the Company’s receivable consisted primarily of earned but unbilled receivables under the BARDA agreement and API purchases made for Toyama’s clinical development of solithromycin in Japan.
F-15
6. Accrued Expenses
Accrued expenses are comprised of the following as of (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Accrued severance |
|
$ |
1,999 |
|
|
$ |
- |
|
|
Franchise tax |
|
|
570 |
|
|
|
436 |
|
|
Accrued professional fees |
|
|
145 |
|
|
|
642 |
|
|
Deferred rent |
|
|
85 |
|
|
|
76 |
|
|
Accrued interest |
|
|
80 |
|
|
|
99 |
|
|
Other accrued expenses |
|
|
50 |
|
|
|
222 |
|
|
Total accrued expenses |
|
$ |
2,929 |
|
|
$ |
1,475 |
|
7. Long-term Debt
In July 2015, the Company entered into a Loan and Security Agreement (the “Loan and Security Agreement”) with Comerica Bank (“Comerica). The Loan and Security Agreement provides that the Company may borrow up to $20.0 million in a term loan (the “Term Loan”) and, upon FDA approval of its planned New Drug Application for solithromycin, the Company may also borrow an aggregate amount equal to the lesser of (i) up to 75% of its eligible inventory and 80% of eligible accounts receivable or (ii) $10.0 million (the “Revolver”). After FDA approval of the Company’s planned New Drug Application for solithromycin, the Company may convert the Term Loan to the Revolver, in which event the Revolver would have a maximum amount available to the Company of $25.0 million. The Loan and Security Agreement specifies the criteria for determining eligible inventory and eligible accounts receivable and sets forth ongoing limitations and conditions precedent to the Company’s ability to borrow under the Revolver. The Company granted Comerica a security interest in substantially all of its personal property assets, excluding its intellectual property and its stock in its subsidiaries, to secure its outstanding obligations under the Loan and Security Agreement. The Company is also obligated to comply with various other customary covenants, including, among other things, restrictions on its ability to: dispose of assets, make acquisitions, be acquired, incur indebtedness, grant liens, make distributions to its stockholders, make investments, enter into certain transactions with affiliates, or pay down subordinated debt, subject to specified exceptions.
At closing, the Company received the full $20.0 million under the Term Loan and paid a facility fee of $0.1 million for the Term Loan and a facility fee of $0.2 million for the Revolver. The Company immediately used proceeds from the Term Loan to pay all of its $17.7 million outstanding principal and interest and $1.2 million in end of term and prepayment fees under the loan and security agreement (“December 2011 Note”) with Hercules Technology Growth Capital, Inc. (“Hercules”) and terminated the Hercules loan. The Company recorded a charge of $0.3 million on the early extinguishment of the December 2011 Note.
Amounts borrowed under the Term Loan may be repaid and reborrowed at any time without penalty or premium. The Term Loan is interest-only through April 30, 2016, followed by an amortization period of 36 months of equal monthly payments of principal plus interest, beginning on May 1, 2016 and continuing on the same day of each month thereafter until paid in full. Any amounts borrowed under the Term Loan bear interest at a floating interest rate equal to the 30 Day LIBOR rate plus 5.2%. Amounts available to be borrowed under the Revolver may also be repaid and reborrowed at any time without penalty or premium prior to December 31, 2017, at which time all advances under the Revolver shall be immediately due and payable in full. Any amounts borrowed under the Revolver will bear interest at the 30 Day LIBOR rate plus 4.2. Once available, the Revolver is subject to an annual unused facility fee equal of 0.25%. Under the Loan and Security Agreement, the Company is subject to certain covenants including maintaining a minimum unrestricted cash balance of $15.0 million and continuing the development or commercially launching solithromycin.
In December 2011, the Company entered into the $20.0 million December 2011 Note with Hercules and borrowed $10.0 million upon closing. Borrowings under the December 2011 Note bore interest at the greater of (i) 9.55%, or (ii) the sum of 9.55% plus the prime lending rate, as published by the Wall Street Journal, minus 3.25% per annum. In connection with the initial closing of the December 2011 Note, the Company entered into a warrant agreement with Hercules. In May 2013, the Company amended its December 2011 Note, increasing the initial loan amount to $15.0 million, and receiving an additional $5.2 million upon closing. In March 2014, the Company amended the December 2011 Note providing the Company the ability to request, at any time prior to December 26, 2014, another borrowing in the aggregate amount of $3.0 million. This amendment also provided for the Company to make interest only payments through May 31, 2015. In June 2014, the Company borrowed the additional $3.0 million and amended the December 2011 Note to provide the Company the ability to borrow up to an additional $10.0 million. Warrants associated with the December 2011 Note were reclassified to additional paid-in capital in the second quarter of 2014. The Hercules loan was terminated and paid using proceeds from the Comerica Term Loan.
F-16
In connection with the initial closing of the December 2011 Note, the Company entered into a warrant agreement with Hercules (the “First Hercules Warrant”), under which Hercules has the right to purchase 39,038 shares of the Company’s common stock. The exercise price of the First Hercules Warrant was initially $10.25 per share, subject to adjustment in the event of a merger, reclassification, subdivision or combination of shares or stock dividend and subject also to antidilution protection. In connection with the May 2013 amendment to the loan agreement, the exercise price of the First Hercules Warrant was reduced to the lower of (a) $6.11, and (b) the effective price per share of the Company’s common stock issued or issuable in any offering of the Company’s equity or equity-linked securities that occurred prior to June 1, 2014, provided that such offering was effected principally for equity or debt-financing purposes. Since the May 2013 amendment to the warrant resulted in a variable exercise price, the fair value of the warrant as of the date of the amendment was reclassified from additional paid-in capital to a warrant liability. The Company did not offer any common stock between the amendment date and June 1, 2014 at a price below $6.11, therefore, the exercise price of the First Hercules Warrant became fixed at $6.11, which resulted in the warrant liability being reclassified to additional paid-in capital in the second quarter of 2014.
Additionally, in connection with the May 2013 amendment of the December 2011 Note, the Company entered into a warrant agreement with Hercules (the “Second Hercules Warrant”), under which Hercules has the right to purchase an aggregate number of shares of the Company’s common stock equal to the quotient derived by dividing $0.6 million by the exercise price then in effect, which is defined as the lower of (a) $6.11, and (b) the effective price per share of the Company’s common stock issued or issuable in any offering of the Company’s equity or equity-linked securities prior to June 1, 2014, provided that such offering was effected principally for equity or debt-financing purposes. The Second Hercules Warrant expires on May 31, 2023. Proceeds equal to the fair value of the Second Hercules Warrant were recorded as a liability at the date of issuance. The Company did not offer any common stock between the amendment date and June 1, 2014 at a price below $6.11, therefore, the exercise price of the Second Hercules Warrant became fixed at $6.11, which resulted in the warrant being fixed at 99,759 shares of common stock and the warrant liability being reclassified to additional paid-in capital in the second quarter of 2014.
In December 2014, Hercules exercised the First Hercules Warrant of 39,038 shares and the Second Hercules Warrant of 99,759 shares in a cashless exercise which resulted in 97,931 shares issued. The exercise price was deemed to be $20.75, the average of the closing prices over a five-day period ending three days before the day the current fair market value of the common stock was determined.
Scheduled Maturities:
Scheduled maturities of long-term debt are as follows (in thousands):
|
Year Ending December 31: |
|
|
|
|
|
2017 |
|
|
6,667 |
|
|
2018 |
|
|
6,667 |
|
|
2019 |
|
|
2,222 |
|
|
Total |
|
|
15,556 |
|
|
Less: Unamortized discount |
|
|
(229 |
) |
|
Less: Current portion of long-term debt |
|
|
(6,667 |
) |
|
Long-term debt |
|
$ |
8,660 |
|
8. Commitments and Contingencies
Future minimum lease payments required under non-cancellable operating leases as of December 31, 2016 are as follows (in thousands):
|
|
|
Operating |
|
|
|
|
|
Leases |
|
|
|
2017 |
|
$ |
744 |
|
|
2018 |
|
|
762 |
|
|
2019 |
|
|
503 |
|
|
2020 |
|
|
308 |
|
|
2021 and thereafter |
|
|
42 |
|
|
Total minimum lease payments |
|
$ |
2,359 |
|
F-17
Rent expense for the years ended December 31, 2016, 2015 and 2014 was $0.7 million, $0.4 million, and $0.2 million, respectively.
See Note 4—Significant Agreements and Contracts for contingencies related to the Optimer Agreement and the TSRI agreement.
Legal Proceedings
On November 4, 2016, a securities class action lawsuit was commenced in the United States District Court, Middle District of North Carolina, Durham Division, naming the Company and certain of the Company’s officers as defendants, and alleging violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by the defendants between May 1, 2016 and November 1, 2016 (the “Class Period”). The plaintiff seeks to represent a class comprised of purchasers of the Company’s common stock during the Class Period and seeks damages, costs and expenses and such other relief as determined by the Court. Two substantially similar lawsuits were filed in the United States District Court, Middle District of North Carolina on November 22, 2016 and December 30, 2016, respectively. The Company believes it has meritorious defenses and intends to defend the lawsuits vigorously. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants.
On December 21, 2016, a shareholder derivative lawsuit was commenced in the North Carolina Durham County Superior Court, naming certain of the Company’s former and current officers and directors as defendants and the Company as a nominal defendant, and asserting claims for breach of fiduciary duty, unjust enrichment, abuse of control, gross mismanagement, and corporate waste. A substantially similar lawsuit was filed in the North Carolina Durham County Superior Court on February 16, 2017. The complaints are based on similar allegations as asserted in the securities lawsuits described above, and seeks unspecified damages and attorneys' fees. The Company believes it has meritorious defenses and intends to defend the lawsuits vigorously. It is possible that similar lawsuits may yet be filed in the same or other courts that name the same or additional defendants.
Other than as described above, the Company is not a party to any legal proceedings and is not aware of any claims or actions pending or threatened against the Company. In the future, the Company might from time to time become involved in litigation relating to claims arising from its ordinary course of business.
Accrued Severance
In December 2016, the Company entered into a Retirement and Consulting Agreement with then current CEO, Dr. Prabhavathi Fernandes, whereby, for one year, subject to monthly extensions by mutual agreement, she will provide consulting services to the Company for up to 20 hours per week. For her consulting work, the Company will pay Dr. Fernandes $35,000 per month. In addition, all of Dr. Fernandes’s stock options will continue to vest during the consulting period. The Company recognized $4.2 million of accelerated stock compensation expense in the fourth quarter of 2016 due to the change in employment status from CEO to a consultant with continued service.
Additionally, Dr. Fernandes is entitled to the severance payments and benefits described in her employment agreement. In consideration of her waiver of the notice period provided under her employment agreement, the Company will pay Dr. Fernandes $45,000. In lieu of her pro-rated annual bonus due under her employment agreement upon a termination of employment, the Company will pay Dr. Fernandes an annual bonus for 2016 in the amount of $280,260. From the effective date of the release, the Company will continue to pay Dr. Fernandes her base salary for 18 months, at the current annual rate of $540,000. In addition, the Company will pay Dr. Fernandes an amount equal to one and one half times her Target Bonus (as defined in her employment agreement), based upon the average percentage of achievement of target objectives for the prior three years, which amount is $420,390, payable in 18 equal monthly payments. From the effective date of the release, upon the conclusion of the consulting period, or upon an earlier change in control of the Company, all of Dr. Fernandes’s then outstanding and unvested stock options will become fully vested.
9. Shareholders’ Equity
Common Stock
In May 2016, the Company entered into an at-the-market (“ATM”) sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) under which the Company may, at its discretion, from time to time sell shares of its common stock, with a sales value of up to $150.0 million. The Company has provided Cowen with customary indemnification rights, and Cowen is entitled to a commission at a fixed commission rate of 3.0% of the gross proceeds per share sold. Sales of the shares under the Sales Agreement are to be made in transactions deemed to be “at the market offerings” as defined in Rule 415 under the Securities Act of 1933, as amended.
F-18
The Company began the sale of ATM shares in May 2016. Through December 31, 2016, the Company sold 4,140,307 shares of common stock under the Sales Agreement resulting in net proceeds of $75.1 million after deducting commissions and expenses of $2.3 million.
During January 2016, the Company completed a public offering of 4,166,667 shares of common stock, at a price of $24.00 per share, resulting in net proceeds to the Company of approximately $93.8 million after deducting underwriting discounts and expenses of approximately $6.2 million.
During 2016, the Company issued 95,180 shares of common stock at a weighted average exercise price of $5.17 per share upon the exercise of option grants.
During January 2015, the Company completed a public offering issuing 6,037,500 shares of common stock, at a price of $24.50 per share, resulting in net proceeds to the Company of approximately $138.8 million after deducting underwriting discounts and commissions, and expenses of approximately $9.1 million
During 2015, the Company issued 417,999 shares of common stock at a weighted average exercise price of $6.40 per share for the exercise of option grants and 60,309 shares of common stock at a weighted average exercise price of $6.00 per share for the exercise of warrants issued.
In March 2013, the Company entered into an at-the-market (“ATM”) sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) under which the Company could, at its discretion, from time to time sell shares of its common stock, with a sales value of up to $25.0 million. The Company provided Cowen with customary indemnification rights, and Cowen was entitled to a commission at a fixed commission rate of 3.0% of the gross proceeds per share sold. Sales of the shares under the Sales Agreement were to be made in transactions deemed to be “at the market offerings” as defined in Rule 415 under the Securities Act of 1933, as amended. In October 2014, the Company and Cowen amended the Sales Agreement (the “Amended Agreement”) to increase the aggregate gross sales proceeds that could be raised to $50 million.
The Company began the sale of ATM shares in July 2014 and sold an aggregate of 4,092,525 shares of common stock under the Sales Agreement in 2014 resulting in net proceeds of $48.5 million after deducting commissions of $1.5 million. The Sales Agreement was terminated on January 5, 2015.
The following table presents common stock reserved for future issuance for the following equity instruments as of December 31, 2016:
|
Warrants to purchase common stock (1) |
|
|
94,912 |
|
|
Options and restricted stock units: |
|
|
|
|
|
Outstanding under the 2006 Stock Plan |
|
|
451,525 |
|
|
Outstanding under the 2011 Equity Incentive Plan |
|
|
3,382,821 |
|
|
Available for future grants under the 2011 Equity Incentive Plan |
|
|
2,900,230 |
|
|
Total common stock reserved for future issuance |
|
|
6,829,488 |
|
|
(1) |
The Warrants to purchase common stock are exercisable at a price of $6.00 per share and expire in August 2018. |
10. Share-Based Compensation
The Company adopted the 2006 Stock Plan in January 2006 (“the 2006 Plan”). The 2006 Plan provided for the granting of incentive share options, nonqualified share options and restricted shares to Company employees, representatives and consultants. As of December 31, 2016, there were options for an aggregate of 451,525 shares issued and outstanding under the 2006 Plan.
The Company’s board of directors adopted the 2011 Equity Incentive Plan in October 2011 (the “2011 Plan”), which, as amended in May 2015, authorizes the issuance of up to 6,601,735 shares under the 2011 Plan, with an automatic annual increase discussed below. As of December 31, 2016, there were 2,900,230 options shares available under the 2011 Plan. The number of shares of common stock reserved for issuance under the 2011 Plan automatically increases on January 1 of each year, continuing through January 1, 2021, by 4% of the total number of shares of the Company’s common stock outstanding on December 31st of the preceding calendar year.
F-19
Upon adoption of the 2011 Plan, the Company eliminated the authorization for any unissued shares previously reserved under the Company’s 2006 Plan. The stock awards previously issued under the 2006 Plan remain in effect in accordance with the terms of the 2006 Plan.
The following table summarizes the Company’s 2006 and 2011 Plan stock option activity:
|
|
|
|
|
|
|
Weighted |
|
|
Weighted |
|
|
|
|
|
||
|
|
|
|
|
|
|
Average |
|
|
Average |
|
|
Aggregate |
|
|||
|
|
|
Number of |
|
|
Exercise |
|
|
Contractual |
|
|
Intrinsic |
|
||||
|
|
|
Options |
|
|
Price |
|
|
Term (in years) |
|
|
Value(1) |
|
||||
|
Outstanding - December 31, 2013 |
|
|
1,832,851 |
|
|
$ |
5.26 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
753,250 |
|
|
|
12.35 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(39,980 |
) |
|
|
2.16 |
|
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
(60,021 |
) |
|
|
10.28 |
|
|
|
|
|
|
|
|
|
|
Expired |
|
|
(1,541 |
) |
|
|
7.22 |
|
|
|
|
|
|
|
|
|
|
Outstanding - December 31, 2014 |
|
|
2,484,559 |
|
|
|
7.34 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
790,850 |
|
|
|
26.73 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(417,999 |
) |
|
|
6.40 |
|
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
(43,645 |
) |
|
|
15.71 |
|
|
|
|
|
|
|
|
|
|
Expired |
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
|
|
|
Outstanding - December 31, 2015 |
|
|
2,813,765 |
|
|
$ |
12.80 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
1,151,158 |
|
|
|
24.35 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(95,180 |
) |
|
|
5.17 |
|
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
(78,225 |
) |
|
|
23.78 |
|
|
|
|
|
|
|
|
|
|
Expired |
|
|
(7,172 |
) |
|
|
19.48 |
|
|
|
|
|
|
|
|
|
|
Outstanding - December 31, 2016 |
|
|
3,784,346 |
|
|
|
16.26 |
|
|
|
7.14 |
|
|
$ |
286,608 |
|
|
Exercisable - December 31, 2016 |
|
|
2,368,790 |
|
|
|
12.77 |
|
|
|
6.21 |
|
|
|
286,608 |
|
|
Vested and expected to vest at December 31, 2016 (2) |
|
|
3,722,615 |
|
|
$ |
16.14 |
|
|
|
7.12 |
|
|
$ |
286,608 |
|
|
(1) |
Intrinsic value is the excess of the fair value of the underlying common shares as of December 31, 2016 over the weighted-average exercise price. |
|
(2) |
The number of stock options expected to vest takes into account an estimate of expected forfeitures. |
The following table summarizes certain information about all options outstanding as of December 31, 2016:
|
|
|
Options Outstanding |
|
|
Options Exercisable |
|
||||||||||
|
Exercise Price |
|
Number of Options |
|
|
Weighted Average Remaining Contractual Term (in years) |
|
|
Number of Options |
|
|
Weighted Average Remaining Contractual Term (in years) |
|
||||
|
$2.09 - $6.63 |
|
|
723,291 |
|
|
|
4.16 |
|
|
|
723,291 |
|
|
|
4.16 |
|
|
$6.64 - $11.35 |
|
|
827,697 |
|
|
|
6.68 |
|
|
|
647,130 |
|
|
|
5.94 |
|
|
$12.38 - $18.61 |
|
|
739,442 |
|
|
|
7.68 |
|
|
|
348,239 |
|
|
|
7.03 |
|
|
$19.25 - $31.13 |
|
|
1,280,166 |
|
|
|
8.68 |
|
|
|
547,646 |
|
|
|
8.46 |
|
|
$32.05 - $43.43 |
|
|
213,750 |
|
|
|
8.04 |
|
|
|
102,484 |
|
|
|
7.72 |
|
|
|
|
|
3,784,346 |
|
|
|
|
|
|
|
2,368,790 |
|
|
|
|
|
In 2016, the Company issued time-vested Restricted Stock Units (RSUs) from the 2011 Equity Incentive Plan to certain employees, subject to continuous service with the Company at the vesting time. When vested, the RSU represented the right to be issued the number of shares of the Company’s common stock that is equal to the number of RSUs granted.
F-20
A summary of the activity related to the Company’s RSUs is as follows:
|
|
|
Number of |
|
|
Weighted |
|
||
|
|
|
Restricted |
|
|
Average |
|
||
|
|
|
Stock Units |
|
|
Grant-Date |
|
||
|
|
|
Outstanding |
|
|
Fair Value |
|
||
|
Balance - December 31, 2015 |
|
|
- |
|
|
$ |
- |
|
|
Granted |
|
|
50,000 |
|
|
|
7.70 |
|
|
Exercised |
|
|
- |
|
|
|
- |
|
|
Forfeited |
|
|
- |
|
|
|
- |
|
|
Expired |
|
|
- |
|
|
|
- |
|
|
Balance - December 31, 2016 |
|
|
50,000 |
|
|
|
7.70 |
|
|
Vested at December 31, 2016 |
|
|
- |
|
|
$ |
- |
|
During the years ended December 31, 2016, 2015 and 2014, the Company recorded $14.3 million, $5.9 million, and $3.1 million in share-based compensation expense, respectively. As of December 31, 2016, approximately $13.7 million of total unrecognized compensation cost related to unvested share options is expected to be recognized over a weighted-average period of 2.57 years, and approximately $0.3 million of total unrecognized compensation cost related to unvested restricted stock units is expected to be recognized over a weighted-average period of .94 years.
During the year ended December 31, 2016, the Company received $0.5 million in cash proceeds from the exercise of stock options through the course of the year.
Other information pertaining to the Company’s stock option awards is as follows (in thousands, except per share data):
|
|
|
Fiscal Years Ended |
|
|||||||||
|
|
|
December 31, |
|
|
December 31, |
|
|
December 31, |
|
|||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Weighted average grant date fair value per share of options |
|
$ |
15.55 |
|
|
$ |
17.00 |
|
|
$ |
8.58 |
|
|
Total intrinsic value of options exercised |
|
$ |
1,283 |
|
|
$ |
11,970 |
|
|
$ |
313 |
|
11. Income Taxes
No provision for U.S. Federal or state income taxes has been recorded as the Company has incurred net operating losses since inception.
Significant components of the Company’s deferred income tax assets as of December 31, 2016 and 2015 consist of the following (in thousands):
|
|
|
2016 |
|
|
2015 |
|
||
|
Non-current |
|
|
|
|
|
|
|
|
|
Deferred income tax assets |
|
|
|
|
|
|
|
|
|
Tax loss carryforwards |
|
$ |
121,953 |
|
|
$ |
86,600 |
|
|
Contribution carryforwards |
|
|
24 |
|
|
|
27 |
|
|
Tax credits |
|
|
15,212 |
|
|
|
12,719 |
|
|
Start-up costs & other intangibles |
|
|
5,984 |
|
|
|
5,661 |
|
|
Share-based compensation |
|
|
7,161 |
|
|
|
3,315 |
|
|
Deferred revenue |
|
|
4,033 |
|
|
|
4,049 |
|
|
Other assets |
|
|
566 |
|
|
|
350 |
|
|
Total net deferred income taxes, non-current |
|
|
154,933 |
|
|
|
112,721 |
|
|
Less valuation allowance |
|
|
(154,933 |
) |
|
|
(112,721 |
) |
|
Total net deferred income tax |
|
$ |
- |
|
|
$ |
- |
|
At December 31, 2016 and 2015, the Company provided a full valuation allowance against its net deferred tax assets since at that time, the Company could not assert that it was more likely than not that these deferred tax assets would be realized. There was an increase in the valuation allowance in the current year in the amount of $42.2 million.
F-21
The table below summarizes changes in the deferred tax valuation allowance (in thousands):
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Balance at beginning of year |
|
$ |
112,721 |
|
|
$ |
76,286 |
|
|
$ |
52,858 |
|
|
Charges to costs and expenses |
|
|
42,212 |
|
|
|
36,435 |
|
|
|
23,428 |
|
|
Write-offs |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at end of year |
|
$ |
154,933 |
|
|
$ |
112,721 |
|
|
$ |
76,286 |
|
As of December 31, 2016, the Company had federal net operating loss carryforwards of approximately $353.5 million and state net operating loss carryforwards of approximately $244.8 million. The net operating loss carryforwards begin to expire in 2026 and 2021 for federal and state tax purposes, respectively. The Company's federal and state net operating loss carryforwards include approximately $9.6 million of excess tax benefits related to deductions from the exercise of stock options. The tax benefit of these deductions has not been recognized in deferred tax assets. If utilized, the benefits from these deductions will be recorded as adjustments to additional paid-in capital. The Company also had federal research and development credit carryforwards of approximately $11.6 million which begin to expire in 2026, federal orphan drug credits carryforwards of approximately $3.1 million which begin to expire in 2033, federal charitable contribution carryforwards of approximately $0.1 million which begin to expire in 2017, and state credit carryforwards of approximately $0.8 million, which begin to expire in 2018.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events including significant changes in ownership interests. If the Company’s net operating loss carryforwards are limited, and the Company has taxable income which exceeds the permissible yearly net operating loss carryforward, the Company would incur a federal income tax liability even though net operating loss carryforwards would be available in future years.
The reasons for the difference between actual income tax expense (benefit) for the years ended December 31, 2016, 2015 and 2014 and the amount computed by applying the statutory federal income tax rate to losses before income tax (benefit) are as follows:
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||||||||||||||
|
|
|
Amount |
|
|
% of Pretax Earnings |
|
|
Amount |
|
|
% of Pretax Earnings |
|
|
Amount |
|
|
% of Pretax Earnings |
|
||||||
|
United States federal tax at statutory rate |
|
$ |
(40,107 |
) |
|
|
34 |
% |
|
$ |
(30,978 |
) |
|
|
34 |
% |
|
$ |
(20,961 |
) |
|
|
34 |
% |
|
State taxes (net of federal benefit) |
|
|
(1,895 |
) |
|
|
2 |
% |
|
|
(1,593 |
) |
|
|
2 |
% |
|
|
(1,239 |
) |
|
|
2 |
% |
|
Nondeductible expenses |
|
|
1,196 |
|
|
|
(1 |
%) |
|
|
1,380 |
|
|
|
(1 |
%) |
|
|
890 |
|
|
|
(1 |
%) |
|
Credits |
|
|
(2,493 |
) |
|
|
2 |
% |
|
|
(4,959 |
) |
|
|
5 |
% |
|
|
(3,329 |
) |
|
|
5 |
% |
|
Adjustment for state rate change |
|
|
1,142 |
|
|
|
(1 |
%) |
|
|
722 |
|
|
|
(1 |
%) |
|
|
1,249 |
|
|
|
(2 |
%) |
|
Other, net |
|
|
(55 |
) |
|
|
0 |
% |
|
|
(1,007 |
) |
|
|
1 |
% |
|
|
(38 |
) |
|
|
0 |
% |
|
Change in valuation allowance |
|
|
42,212 |
|
|
|
(36 |
%) |
|
|
36,435 |
|
|
|
(40 |
%) |
|
|
23,428 |
|
|
|
(38 |
%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Provision for income taxes |
|
$ |
- |
|
|
|
0 |
% |
|
$ |
- |
|
|
|
0 |
% |
|
$ |
- |
|
|
|
0 |
% |
During 2016, North Carolina enacted legislation to reduce the state corporate income tax rate from 4% to 3% for tax years 2017 and beyond. As a result of the new enacted tax rate, the Company adjusted its deferred tax assets as of December 31, 2016 by applying the lower rate, which resulted in a decrease in the deferred tax assets and a corresponding decrease to the valuation allowance of approximately $ 1.1 million.
As of December 31, 2016 and 2015, the Company had no unrecognized tax benefits. The Company’s policy is to recognize interest and penalties related to uncertain tax positions in the provision for income taxes. As of December 31, 2016 and 2015, the Company had no accrued interest or penalties related to uncertain tax positions.
The Company has analyzed its filing positions in all significant federal and state jurisdictions where it is required to file income tax returns, as well as open tax years in these jurisdictions. With few exceptions, the Company is no longer subject to United States Federal, state, and local tax examinations by tax authorities for years before 2013 although carryforward attributes that were generated prior to 2013 may still be adjusted upon examination by the taxing authorities if they either have been or will be used in a future period. No income tax returns are currently under examination by taxing authorities.
F-22
12. Net Loss Per Share
Basic and diluted net loss per common share was determined by dividing net loss by the weighted average common shares outstanding during the period. The Company’s potentially dilutive shares, which include redeemable warrants, common share options and restricted stock units, have not been included in the computation of diluted net loss per share for all periods as the result would be antidilutive.
The following potentially dilutive securities have been excluded from the computation of diluted weighted average shares outstanding, as they would be antidilutive:
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Warrants outstanding |
|
|
94,912 |
|
|
|
132,464 |
|
|
|
330,585 |
|
|
Stock options outstanding |
|
|
3,560,102 |
|
|
|
2,910,694 |
|
|
|
2,411,891 |
|
|
Restricted stock units |
|
|
3,005 |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
3,658,019 |
|
|
|
3,043,158 |
|
|
|
2,742,476 |
|
13. Selected Quarterly Data (unaudited, in thousands, except for loss per share data)
|
|
|
Quarter Ended |
|
|||||||||||||
|
|
|
March 31, |
|
|
June 30, |
|
|
September 30, |
|
|
December 31, |
|
||||
|
|
|
2016 |
|
|
2016 |
|
|
2016 |
|
|
2016 |
|
||||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract research |
|
$ |
2,679 |
|
|
$ |
3,420 |
|
|
$ |
3,972 |
|
|
$ |
3,606 |
|
|
License |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
4,339 |
|
|
Supply |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Total revenue |
|
$ |
2,679 |
|
|
$ |
3,420 |
|
|
$ |
3,972 |
|
|
$ |
7,945 |
|
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
23,529 |
|
|
|
16,018 |
|
|
|
21,096 |
|
|
|
21,043 |
|
|
General and administrative |
|
|
8,324 |
|
|
|
11,988 |
|
|
|
15,021 |
|
|
|
18,205 |
|
|
Loss from operations |
|
|
(29,174 |
) |
|
|
(24,586 |
) |
|
|
(32,145 |
) |
|
|
(31,303 |
) |
|
Interest income |
|
|
98 |
|
|
|
104 |
|
|
|
128 |
|
|
|
145 |
|
|
Interest expense |
|
|
(330 |
) |
|
|
(323 |
) |
|
|
(295 |
) |
|
|
(280 |
) |
|
Net loss |
|
$ |
(29,406 |
) |
|
$ |
(24,805 |
) |
|
$ |
(32,312 |
) |
|
$ |
(31,438 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(0.61 |
) |
|
$ |
(0.51 |
) |
|
$ |
(0.62 |
) |
|
$ |
(0.60 |
) |
|
Shares used in calculating basic and diluted net loss per share |
|
|
47,853 |
|
|
|
48,898 |
|
|
|
52,073 |
|
|
|
52,389 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended |
|
|||||||||||||
|
|
|
March 31, |
|
|
June 30, |
|
|
September 30, |
|
|
December 31, |
|
||||
|
|
|
2015 |
|
|
2015 |
|
|
2015 |
|
|
2015 |
|
||||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contract research |
|
$ |
2,076 |
|
|
$ |
3,171 |
|
|
$ |
2,497 |
|
|
$ |
4,704 |
|
|
License |
|
|
10,000 |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Supply |
|
|
1,887 |
|
|
|
1,883 |
|
|
|
- |
|
|
|
1,090 |
|
|
Total revenue |
|
$ |
13,963 |
|
|
$ |
5,054 |
|
|
$ |
2,497 |
|
|
$ |
5,794 |
|
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
26,118 |
|
|
|
23,676 |
|
|
|
23,541 |
|
|
|
20,018 |
|
|
General and administrative |
|
|
4,650 |
|
|
|
5,732 |
|
|
|
5,848 |
|
|
|
6,641 |
|
|
Loss from operations |
|
|
(16,805 |
) |
|
|
(24,354 |
) |
|
|
(26,892 |
) |
|
|
(20,865 |
) |
|
Interest income |
|
|
2 |
|
|
|
2 |
|
|
|
- |
|
|
|
5 |
|
|
Interest expense |
|
|
(614 |
) |
|
|
(617 |
) |
|
|
(679 |
) |
|
|
(296 |
) |
|
Net loss |
|
$ |
(17,417 |
) |
|
$ |
(24,969 |
) |
|
$ |
(27,571 |
) |
|
$ |
(21,156 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(0.41 |
) |
|
$ |
(0.57 |
) |
|
$ |
(0.63 |
) |
|
$ |
(0.48 |
) |
|
Shares used in calculating basic and diluted net loss per share |
|
|
42,671 |
|
|
|
43,686 |
|
|
|
43,911 |
|
|
|
43,976 |
|
F-23
14. Subsequent Event
In February 2017, as a consequence of the solithromycin complete response letter the Company received from the FDA, and subsequent discussions with the FDA, resulting in the delay of the potential approval of solithromycin, the Company initiated companywide cost and personnel reductions. These actions have resulted in an approximately 67% reduction in the Company’s workforce from 136 to 45 employees, and significant reductions in external spending related to commercial preparedness and non-essential activities. The principal objective of the reductions is to enable the Company to conserve its financial resources as it evaluates its path forward on its existing pipeline and potential business development opportunities. As the Company progresses its internal programs, it is also actively engaged in a process to evaluate and assess external late-stage assets and other potential strategic business opportunities to determine the best use of its significant cash resources and clinical programs to deliver value to patients and shareholders through internal and/or potential external opportunities. In connection with the reduction, the Company expects to record an aggregate charge related to one-time termination benefits of approximately $3.5 million in 2017.
F-24