UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of The Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): December 6, 2016
PUMA BIOTECHNOLOGY, INC.
(Exact Name of Registrant as Specified in its Charter)
| Delaware | 001-35703 | 77-0683487 | ||
| (State or other jurisdiction of incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) |
10880 Wilshire Boulevard, Suite 2150
Los Angeles, California 90024
(Address of principal executive offices) (Zip Code)
(424) 248-6500
(Registrant’s telephone number, including area code)
N/A
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ☐ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ☐ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ☐ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ☐ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
| Item 8.01 | Other Events. |
Managed Access Program
On December 6, 2016, Puma Biotechnology, Inc. (the “Company”) announced that it has initiated a Managed Access Program for PB272 (neratinib). Managed access programs provide physicians and patients access to medicines when there are limited or no other therapeutic options available.
The Company’s Managed Access Program for neratinib will enable participation from countries outside the United States, including European Union Member States, where permitted by applicable rules, procedures and regulatory authorities. The program will provide access to neratinib for the treatment of early stage HER2-positive breast cancer (extended adjuvant setting), HER2-positive metastatic breast cancer and HER2-mutated solid tumors. Patients must not be able to participate in any ongoing neratinib clinical trial to qualify for the Company’s managed access program. Patients in the managed access program will be given neratinib and will be instructed to take a prophylaxis during treatment to manage neratinib-related diarrhea, which the Company expects will consist of high dose loperamide and budesonide.
The Company has partnered with Caligor Opco LLC, which specializes in early access to medicines, to implement and oversee the Managed Access Program for neratinib.
Presentation of Interim Results of Phase II Trial of PB272 for ERBB2 (HER2) Mutant, HER2 Non-Amplified, Metastatic Breast Cancer at the 2016 San Antonio Breast Cancer Symposium
On December 7, 2016, the Company announced that updated interim results from an ongoing Phase II clinical trial of PB272 (neratinib), given as monotherapy and in combination with the anticancer drug fulvestrant, were presented at the 2016 CTRC-AACR San Antonio Breast Cancer Symposium (SABCS). The presentation entitled “Neratinib plus fulvestrant for ERBB2 mutant, HER2 non-amplified, estrogen receptor-positive, metastatic breast cancer: Preliminary analysis from the Phase II SUMMIT trial” was presented as a poster discussion by Dr. David Hyman, Director, Developmental Therapeutics at Memorial Sloan Kettering Cancer Center. The poster is available on the Company’s website (www.pumabiotechnology.com) under the Investors – Events & Webcasts tab.
Interim results from this trial were previously presented at the 2015 SABCS and included patients who were treated with neratinib monotherapy for metastatic breast cancer and whose tumors have a HER2 mutation. The presentation also discussed that a bidirectional cross-talk between hormone receptor and HER2 signaling pathways could lead to endocrine resistance due to activated HER2 signaling and ER-mediated tumor proliferation as a potential resistance mechanism to sustained HER2 inhibition. Preclinical xenograft data has demonstrated that the combination of an anti-estrogen with neratinib results in enhanced anti-tumor activity in preclinical models of estrogen receptor positive/HER2-positive breast tumors. Based on this, the SUMMIT study was amended to allow for the combination of neratinib plus fulvestrant in eligible postmenopausal hormone receptor-positive breast cancer patients. The presentation at SABCS included an update on both the neratinib monotherapy cohort and the neratinib plus fulvestrant cohort.
In the study, patients with HER2 mutant metastatic breast cancer were enrolled and received 240 mg of neratinib daily either as monotherapy or in combination with fulvestrant. All patients received loperamide (16 mg per day initially) prophylactically for the first cycle of treatment in order to reduce the neratinib-related diarrhea. For the 25 patients in the group who received neratinib monotherapy, 23 patients (92%) had HER2-negative disease, 19 patients (76%) were hormone receptor positive (estrogen receptor or progesterone receptor positive), and patients had received a median of 4 prior lines of therapy in the metastatic setting (range 0-8 prior regimens) before entering the trial. For the 17 patients in the trial who received neratinib plus fulvestrant, 15 patients (88%) had HER2-negative disease, 17 patients (100%) were hormone receptor positive (estrogen receptor or progesterone receptor positive), and patients had received a median of 4 prior lines of therapy in the metastatic setting (range 1-7 prior regimens) before entering the trial.
The interim efficacy results from the trial showed that for the 24 efficacy evaluable patients in the neratinib monotherapy cohort, 8 patients (33.3%) experienced an objective response, which included 3 patients with a complete response and 5 patients with partial responses. At week 8, 8 patients (33.3%) achieved an objective
response, with 2 patients achieving a complete response and 6 patients achieving a partial response. The secondary endpoints of the trial included confirmed objective response (complete response or partial response), clinical benefit rate and progression free survival (PFS). The results of the trial showed that 6 patients (25%) had a confirmed objective response, 10 patients (41.7%) demonstrated clinical benefit and the median progression free survival was 3.5 months.
For the 12 efficacy evaluable patients in the neratinib plus fulvestrant cohort, 7 patients (58.3%) experienced an objective response, which included 2 patients with a complete response and 5 patients with partial responses. At week 8, 5 patients (41.7%) achieved an objective response, with 2 patients achieving a complete response and 3 patients achieving a partial response. The secondary endpoints of the trial included confirmed objective response (complete response or partial response), clinical benefit rate and progression free survival (PFS). The results of the trial showed that 3 patients (25%) had a confirmed objective response, 7 patients (58.3%) demonstrated clinical benefit and the median progression free survival was 3.7 months. The progression free survival data may not be mature in the neratinib plus fulvestrant cohort as 4 of the 12 efficacy evaluable patients are continuing to receive study treatment without disease progression and an additional 5 patients have not yet had an assessment for efficacy.
The interim safety results of the study showed that the most frequently observed adverse event was diarrhea. For the 25 patients enrolled in the neratinib monotherapy arm, 6 patients (24%) reported grade 3 diarrhea. The median duration of grade 3 diarrhea for the patients in the neratinib monotherapy cohort was 1 day. No patient in the neratinib monotherapy cohort has permanently discontinued neratinib due to diarrhea and 5 patients (20%) have temporarily discontinued neratinib due to diarrhea and then restarted after the diarrhea subsided. For the 17 patients enrolled in the neratinib plus fulvestrant cohort, 2 of 17 patients (12%) experienced grade 3 diarrhea. The median duration of grade 3 diarrhea was 1 day and typically occurred during the first cycle of treatment. No patient (0%) in the neratinib plus fulvestrant cohort permanently discontinued neratinib due to diarrhea and 2 patients (12%) temporarily discontinued neratinib due to diarrhea and then restarted after the diarrhea subsided.
Presentation of Results of Biomarker Analysis of Phase II Trial of PB272 in Neoadjuvant Treatment of HER2-Positive Locally Advanced Breast Cancer at the 2016 San Antonio Breast Cancer Symposium
On December 7, 2016, the Company announced that a biomarker analysis of the NSABP FB-7 Phase II clinical trial of PB272 (neratinib) was presented at the 2016 SABCS. The presentation entitled “An exploratory correlative biomarker analysis of NSABP FB-7, a phase II randomized trial evaluating neoadjuvant therapy with weekly paclitaxel (P) plus neratinib (N) or trastuzumab (T) or neratinib and trastuzumab (N+T) followed by doxorubicin and cyclophosphamide (AC) with postoperative T in women with locally advanced HER2-positive breast cancer” was presented as a poster presentation. This trial was sponsored by the NSABP Foundation, Inc. The poster is available on the Company’s website under the Investors – Events & Webcasts tab.
The FB-7 trial is a randomized Phase II clinical trial for women with HER2-positive locally advanced stage IIB-IIIC invasive breast cancer. Patients were randomly assigned to receive trastuzumab (T) or neratinib (N) or the combination (T+N) with weekly paclitaxel (P) followed by standard doxorubicin and cyclophosphamide chemotherapy (AC) administered prior to surgery. 126 U.S., Canadian, and European patients were randomly assigned to Arm 1 (T+P followed by AC), Arm 2 (N+P followed by AC) or Arm 3 (T+N+P followed by AC). The primary endpoint of the trial was pathological complete response rate (pCR) in the breast and lymph nodes. The clinical safety and efficacy data from this trial was presented at the 2015 SABCS.
A key secondary endpoint of the FB-7 trial was to evaluate molecular and genetic markers for correlation with response. Pre-treatment core biopsy samples (n=59) and post treatment surgical samples (n=17) were obtained from a subset of patients treated in the FB-7 trial. pCR data were available for 51 patients from the biomarker cohort. After excluding low tumor content non-evaluable samples, correlative biomarker analysis was performed in 42 patients.
Expression levels and the activation status of EGFR/HER2 signaling proteins were investigated. The results of the phosphorylated HER2 (phosphoHER2) showed that median levels of phosphoHER2 were higher in the patients who achieved a pCR with neratinib (n=7) than in the patients who did not achieve a pCR who received either trastuzumab (n=8, p=0.07) or the combination of trastuzumab plus neratinib (n=4, p=0.035). There was not a significant difference in the median levels of phosphoHER2 in the patients who achieved a pCR with neratinib (n=7), trastuzumab (n=8, p=0.16) or the combination of trastuzumab plus neratinib (n=4, p=0.10).
The truncated form of HER2 known as p95HER2 was measured by the proprietary assay of Pierian Bioscience. p95HER2 represents a truncated form of the HER2 receptor that lacks the extracellular trastuzumab binding domain. It is believed to represent a mechanism of trastuzumab resistance. Median p95HER2 levels were higher in samples from patients who achieved a pCR with neratinib than in the patients who did not achieve a pCR who received either trastuzumab (p=0.027) or the combination of trastuzumab plus neratinib (p=0.009). There was not a significant difference in the median levels of p95HER2 in the patients who achieved a pCR with neratinib (n=7), trastuzumab (n=8, p=0.16) or the combination of trastuzumab plus neratinib (n=4, p=0.35).
The MammaPrint assay was performed on 59 samples to determine if there was any imbalance between arms. This assay is a genomic test that analyzes the activity of 70 genes and then calculates a recurrence score that is either low risk or high risk. The results of the MammaPrint showed that the patients in all three arms of the FB-7 trial were balanced with the median MammaPrint risk score being similar across arms. There were only three patients with a MammaPrint low score.
Presentation of Interim Results of Phase II CONTROL Trial of PB272 in Extended Adjuvant Treatment of HER2-Positive Early Stage Breast Cancer at the 2016 San Antonio Breast Cancer Symposium
On December 8, 2016, the Company announced that interim results from a Phase II clinical trial of PB272 (neratinib) were presented at the 2016 SABCS. The presentation entitled “Incidence and severity of diarrhea with neratinib plus intensive loperamide prophylaxis in patients with HER2-positive early-stage breast cancer (EBC): Interim analysis from the multicenter, open-label, phase II CONTROL trial” was presented as a poster presentation. The poster is available on the Company’s website under the Investors – Events & Webcasts tab.
The main adverse event that has been seen to date in clinical trials of neratinib is diarrhea and more specifically grade 3 diarrhea. In the Phase III ExteNET trial of neratinib as extended adjuvant treatment of HER2-positive early stage breast cancer that has previously been treated with adjuvant Herceptin, 95.4% of the patients experienced all grade diarrhea and 39.8% of the patients experienced grade 3 or higher diarrhea (there was one event of grade 4 diarrhea). The CONTROL trial is an international, open-label, phase II study investigating the use of loperamide prophylaxis with or without other agents in the prevention and reduction of neratinib-associated diarrhea and more specifically grade 3 diarrhea.
In the trial, patients with HER2-positive early-stage breast cancer who had completed trastuzumab-based adjuvant therapy received neratinib daily for a period of one year. High dose loperamide prophylaxis was given for the first 2 cycles (56 days) of treatment. Initially, the loperamide dosing used was 16 mg on day 1, then 12 mg on days 2 and 3 and then 6-8 mg on days 4-56 (original dosing). The protocol was later amended to simplify the regimen such that patients took 12 mg on days 1-14 and 8 mg on days 15-56 (modified dosing). The CONTROL trial has recently been expanded to include prophylaxis with the combination of loperamide and budesonide, a locally acting corticosteroid that the Company believes targets the inflammation identified in a preclinical model of neratinib-induced diarrhea.
The interim analysis of the trial presented in the poster included a total of 135 patients who received neratinib plus loperamide prophylaxis (28 patients taking the original dosing and 107 patients taking the modified dosing) and 40 patients who received neratinib plus loperamide prophylaxis for 2 cycles and budesonide for 1 cycle.
The results of the trial showed that the incidence of grade 3 diarrhea for the total 135 patients who received the loperamide prophylaxis was 28.1%. For the 28 patients who received loperamide using the original dosing regimen the grade 3 diarrhea rate was 25.0% and for the 107 patients who received the modified loperamide dosing regimen the grade 3 diarrhea rate was 29.0%. For the patients in the original dosing group, 71% of the patients who experienced grade 3 diarrhea were known to be non-compliant with the loperamide regimen and for the patients in the modified loperamide dosing regimen, 35% of the patients were known to be non-compliant with their loperamide dosing regimen. For the 135 patients who received the loperamide prophylaxis, the median number of grade 3 diarrhea episodes per patient was 1 and the median cumulative duration of grade 3 diarrhea was 3 days. For the 135 patients who received loperamide prophylaxis, 18.5% discontinued neratinib due to diarrhea.
For the 40 patients who received the combination of loperamide plus budesonide, the results of the trial showed that the incidence of grade 3 diarrhea was 15.0%. None of the patients who experienced grade 3 diarrhea were non-compliant with the loperamide plus budesonide regimen. The median number of grade 3 diarrhea episodes per patient was 1 and the median cumulative duration of grade 3 diarrhea was 2.5 days. For the 40 patients who received loperamide plus budesonide prophylaxis, 5.0% discontinued neratinib due to diarrhea. Further information is provided in Table 1 below:
Table 1: Characteristics of Treatment-Emergent Diarrhea
| Study | CONTROL | ExteNET | ||||||||||||||||||
| Loperamide cohort | Budesonide cohort |
Neratinib arm |
||||||||||||||||||
| Prophylaxis
|
Original | Modified | Loperamide | Loperamide + | Loperamide | |||||||||||||||
| schedule | schedule | total | budesonide | prn | ||||||||||||||||
| (n=28) | (n=107) | (N=135) | (N=40) | (N=1408) | ||||||||||||||||
| Diarrhea, % |
||||||||||||||||||||
| Any grade |
82.1 | 73.8 | 75.6 | 65.0 | 95.4 | |||||||||||||||
| Grade 1 |
35.7 | 21.5 | 24.4 | 32.5 | 22.9 | |||||||||||||||
| Grade 2 |
21.4 | 23.4 | 23.0 | 17.5 | 32.5 | |||||||||||||||
| Grade 3a |
25.0 | 29.0 | 28.1 | 15.0 | 39.8 | |||||||||||||||
| Grade 4 |
0 | 0 | 0 | 0 | 0.1 | |||||||||||||||
| Median cumulative duration, days |
||||||||||||||||||||
| Grade ³2 |
5.0 | 4.0 | 4.0 | 3.0 | 10.0 | |||||||||||||||
| Grade ³3b |
2.0 | 3.0 | 3.0 | 2.5 | 5.0 | |||||||||||||||
| Median diarrhea episodes/patient |
||||||||||||||||||||
| Any grade |
2 | 2 | 2 | 2 | 8 | |||||||||||||||
| Grade ³2 |
2 | 1 | 2 | 1 | 3 | |||||||||||||||
| Grade ³3b |
1 | 1 | 1 | 1 | 2 | |||||||||||||||
| Action taken, % |
||||||||||||||||||||
| Dose hold |
7.1 | 12.1 | 11.1 | 7.5 | 33.9 | |||||||||||||||
| Dose reduction |
10.7 | 7.5 | 8.1 | 5.0 | 26.4 | |||||||||||||||
| Discontinuation |
28.6 | 15.9 | 18.5 | 5.0 | 16.8 | |||||||||||||||
| Hospitalization |
0 | 1.9 | 1.5 | 0 | 1.4 | |||||||||||||||
| Duration of neratinib treatment, months |
||||||||||||||||||||
| Median |
9.7 | 7.4 | 7.5 | 1.8 | 11.6 | |||||||||||||||
| Range |
0.1–13.1 | 0.1–12.8 | 0.1–13.1 | 0.1–6.3 | 0.03–13.3 | |||||||||||||||
| a | Non-compliance with loperamide prophylaxis in patients with grade 3 diarrhea was 71% with the original loperamide schedule, 35% with the modified loperamide schedule, and 0% with loperamide prophylaxis plus budesonide. |
| b | No grade 4 events in the CONTROL study; one grade 4 event in the ExteNET study. |
In the ExteNET trial, higher grade (grade 2 and grade 3) diarrhea occurred early and persisted throughout the duration of the 12-month treatment period. In the CONTROL trial, in both the loperamide prophylaxis and loperamide plus budesonide prophylaxis arms, the results showed that higher grade diarrhea (grade 2 and 3) occurred early but did not typically recur. This is shown in more detail in Figure 1 below:
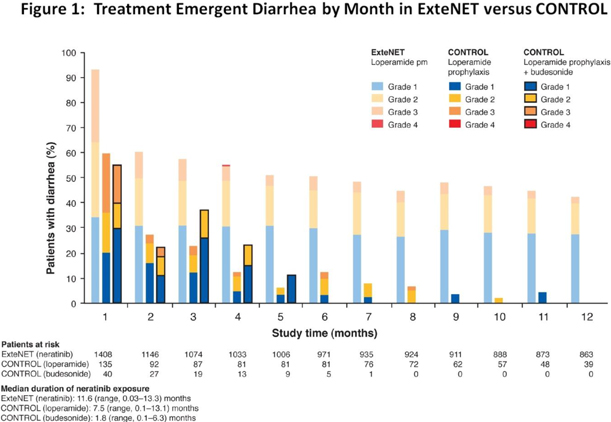
The grade 3 diarrhea rates seen in the loperamide cohort have increased over what was previously reported in December 2015 (n=50, grade 3 diarrhea rate 16%). During the course of the CONTROL trial there has been an increase in the proportion of patients previously treated with pertuzumab (mainly in the neoadjuvant setting). More specifically in the data reported in December 2015, 18% (9 of 50 patients) had previously received pertuzumab. In the current data set 40% (54 of 135 patients) of the patients in the combined loperamide prophylaxis arms received prior pertuzumab and 55% (22 of 40 patients) received prior pertuzumab in the budesonide arm.
For the 54 patients in the loperamide prophylaxis cohort who received prior pertuzumab, the grade 3 diarrhea rate was 35.2% (Table 2). For the 81 patients who did not receive prior pertuzumab, the grade 3 diarrhea rate was 23.5%. For the 22 patients in the budesonide cohort who received prior pertuzumab, the grade 3 diarrhea rate was 13.6%. For the 18 patients in the budesonide cohort who did not receive prior pertuzumab, the grade 3 diarrhea rate was 16.7%. This analysis suggests that prior pertuzumab exposure may have led to a higher rate of grade 3 diarrhea in CONTROL that was not effectively managed by loperamide prophylaxis alone but was more effectively managed by loperamide plus budesonide.
Table 2: Incidence of Grade 3 Diarrhea in CONTROL by Prior Pertuzumab Treatment
| Loperamide Cohort | Budesonide Cohort | |||||||||||||||
| Yes | No | Yes | No | |||||||||||||
| (n = 54) | (n = 81) | (n = 22) | (n = 18) | |||||||||||||
| Grade 3 Diarrhea |
35.2 | % | 23.5 | % | 13.6 | % | 16.7 | % | ||||||||
Forward-Looking Statements:
This Current Report on Form 8-K contains forward-looking statements, including statements regarding development of the Company’s drug candidates and the Managed Access Program for PB272 (neratinib) for the treatment of early
stage HER2-positive breast cancer (extended adjuvant setting), HER2-positive metastatic breast cancer and HER2-mutated solid tumors. All forward-looking statements included in this Current Report on Form 8-K involve risks and uncertainties that could cause the Company’s actual results to differ materially from the anticipated results and expectations expressed in these forward-looking statements. These statements are based on current expectations, forecasts and assumptions, and actual outcomes and results could differ materially from these statements due to a number of factors, which include, but are not limited to, the fact that the Company has no product revenue and no products approved for marketing, the Company’s dependence on PB272, which is still under development and may never receive regulatory approval, the challenges associated with conducting and enrolling clinical trials, the risk that the results of clinical trials may not support the Company’s drug candidate claims, even if approved, the risk that physicians and patients may not accept or use the Company’s products, the Company’s reliance on third parties to conduct its clinical trials and to formulate and manufacture its drug candidates, the Company’s dependence on licensed intellectual property, and the other risk factors disclosed in the periodic and current reports filed by the Company with the Securities and Exchange Commission from time to time, including the Company’s Annual Report on Form 10-K for the year ended December 31, 2015. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. The Company assumes no obligation to update these forward-looking statements, except as required by law.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| PUMA BIOTECHNOLOGY, INC. | ||||||
| Date: December 8, 2016 | By: | /s/ Alan H. Auerbach | ||||
| Alan H. Auerbach | ||||||
| President and Chief Executive Officer | ||||||