Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - REATA PHARMACEUTICALS INC | reta-ex312_8.htm |
| EX-32.2 - EX-32.2 - REATA PHARMACEUTICALS INC | reta-ex322_9.htm |
| EX-32.1 - EX-32.1 - REATA PHARMACEUTICALS INC | reta-ex321_7.htm |
| EX-31.1 - EX-31.1 - REATA PHARMACEUTICALS INC | reta-ex311_6.htm |
| EX-10.4 - EX-10.4 - REATA PHARMACEUTICALS INC | reta-ex104_349.htm |
| EX-10.3 - EX-10.3 - REATA PHARMACEUTICALS INC | reta-ex103_350.htm |
| EX-10.2 - EX-10.2 - REATA PHARMACEUTICALS INC | reta-ex102_352.htm |
| EX-10.1 - EX-10.1 - REATA PHARMACEUTICALS INC | reta-ex101_619.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-Q
(Mark One)
|
☒ |
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended September 30, 2016
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-37785
Reata Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in its Charter)
|
DELAWARE |
|
11-3651945 |
|
( State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer |
|
|
|
|
|
2801 Gateway Dr, Suite 150 |
|
75063 |
|
(Address of principal executive offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: (972) 865-2219
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☒ (Do not check if a small reporting company) |
|
Small reporting company |
|
☐ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of November 10, 2016, the registrant had 8,310,113 shares of Class A common stock, $0.001 par value per share, and 14,026,267 shares of Class B common stock, $0.001 par value per share, outstanding.
|
|
|
Page |
|
3 |
||
|
PART I. |
|
|
|
Item 1. |
4 |
|
|
|
4 |
|
|
|
5 |
|
|
|
6 |
|
|
|
7 |
|
|
Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
12 |
|
Item 3. |
32 |
|
|
Item 4. |
32 |
|
|
PART II. |
|
|
|
Item 1. |
33 |
|
|
Item 1A. |
33 |
|
|
Item 2. |
33 |
|
|
Item 3. |
33 |
|
|
Item 4. |
33 |
|
|
Item 5. |
33 |
|
|
Item 6. |
34 |
|
|
35 |
||
|
|
|
|
2
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains forward-looking statements that involve substantial risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. All statements, other than statements of historical facts, contained in this Quarterly Report on Form 10-Q, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans, and objectives of management, are forward-looking statements. The words “anticipate,” “believe,” “goals,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “potential,” “predict,” “project,” “seek,” “should,” “target,” “will,” “would,” “could,” “should,” and similar expressions are intended to identify forward-looking statements. These forward-looking statements include, but are not limited to, statements about:
|
• |
our expectations regarding the timing, costs, conduct, and outcome of our clinical trials, including statements regarding the timing of the initiation and availability of data from such trials; |
|
• |
the timing and likelihood of regulatory filings and approvals for our product candidates; |
|
• |
our expectations regarding the potential market size and the size of the patient populations for our product candidates, if approved for commercial use, and the potential market opportunities for commercializing our product candidates; |
|
• |
our expectations related to the use of our available cash; |
|
• |
estimates of our expenses, future revenue, capital requirements, and our needs for additional financing; |
|
• |
our ability to develop, acquire, and advance product candidates into, and successfully complete, clinical trials; |
|
• |
the initiation, timing, progress, and results of future preclinical studies and clinical trials, and our research and development programs; |
|
• |
the scope of protection we are able to establish and maintain for intellectual property rights covering our product candidates; |
|
• |
our ability to maintain and establish collaborations or obtain additional funding; |
|
• |
our ability to maintain and establish relationships with third parties, such as contract research organizations, suppliers, and distributors; |
|
• |
our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; |
|
• |
our ability to establish and maintain arrangements for manufacture of our product candidates; |
|
• |
the impact of governmental laws and regulations; |
|
• |
developments and projections relating to our competitors and our industry; and |
|
• |
other risks and uncertainties, including those described under the heading “Risk Factors” included in our final prospectus, or Final Prospectus, dated May 25, 2016, and filed with the U.S. Securities and Exchange Commission, or SEC, pursuant to Rule 424b under the Securities Act of 1933, or the Securities Act, on May 26, 2016. |
Any forward-looking statements in this Quarterly Report on Form 10-Q reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to be materially different from any future results, performance, or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those described under the heading “Risk Factors” in the Final Prospectus and discussed elsewhere in this Quarterly Report on Form 10-Q. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
You should read this Quarterly Report on Form 10-Q and the documents that we have filed as exhibits to this Quarterly Report on Form 10-Q completely and with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
3
PART I - FINANCIAL INFORMATION
Reata Pharmaceuticals, Inc.
(in thousands, except share data)
|
|
|
September 30, 2016 (unaudited) |
|
|
December 31, 2015 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
95,660 |
|
|
$ |
42,008 |
|
|
Federal income tax receivable |
|
|
— |
|
|
|
31,926 |
|
|
Prepaid expenses and other current assets |
|
|
4,260 |
|
|
|
3,325 |
|
|
Total current assets |
|
|
99,920 |
|
|
|
77,259 |
|
|
Property and equipment, net |
|
|
899 |
|
|
|
1,142 |
|
|
Other assets |
|
|
1,004 |
|
|
|
553 |
|
|
Total assets |
|
$ |
101,823 |
|
|
$ |
78,954 |
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities and stockholders’ deficit |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
1,296 |
|
|
$ |
3,531 |
|
|
Accrued direct research liabilities |
|
|
4,424 |
|
|
|
3,529 |
|
|
Other current liabilities |
|
|
4,756 |
|
|
|
4,030 |
|
|
Current portion of deferred revenue |
|
|
49,595 |
|
|
|
49,730 |
|
|
Total current liabilities |
|
|
60,071 |
|
|
|
60,820 |
|
|
Other long-term liabilities |
|
|
94 |
|
|
|
249 |
|
|
Deferred revenue, net of current portion |
|
|
253,947 |
|
|
|
291,041 |
|
|
Total noncurrent liabilities |
|
|
254,041 |
|
|
|
291,290 |
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ deficit: |
|
|
|
|
|
|
|
|
|
Common stock A, $0.001 par value: 500,000,000 shares authorized; issued and outstanding – 7,758,499 and 0 shares at September 30, 2016 and December 31, 2015 |
|
|
8 |
|
|
|
— |
|
|
Common stock B, $0.001 par value: 150,000,000 shares authorized; issued and outstanding – 14,577,568 and 15,998,106 shares at September 30, 2016 and December 31, 2015 |
|
|
15 |
|
|
|
16 |
|
|
Additional paid-in capital |
|
|
72,987 |
|
|
|
10,036 |
|
|
Shareholder notes receivable |
|
|
(81 |
) |
|
|
(81 |
) |
|
Accumulated deficit |
|
|
(285,218 |
) |
|
|
(283,127 |
) |
|
Total stockholders’ deficit |
|
|
(212,289 |
) |
|
|
(273,156 |
) |
|
Total liabilities and stockholders’ deficit |
|
$ |
101,823 |
|
|
$ |
78,954 |
|
See accompanying notes.
4
Unaudited Consolidated Statements of Operations
(in thousands, except share and per share data)
|
|
|
Three Months ended |
|
|
Nine Months ended |
|
||||||||||
|
|
|
September 30, |
|
|
September 30, |
|
||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
||||
|
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
License and milestone |
|
$ |
12,500 |
|
|
$ |
12,500 |
|
|
$ |
37,230 |
|
|
$ |
37,794 |
|
|
Other revenue |
|
|
51 |
|
|
|
— |
|
|
|
125 |
|
|
|
— |
|
|
Total collaboration revenue |
|
|
12,551 |
|
|
|
12,500 |
|
|
|
37,355 |
|
|
|
37,794 |
|
|
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
9,300 |
|
|
|
8,550 |
|
|
|
27,681 |
|
|
|
26,816 |
|
|
General and administrative |
|
|
4,039 |
|
|
|
2,980 |
|
|
|
11,783 |
|
|
|
9,203 |
|
|
Depreciation and amortization |
|
|
170 |
|
|
|
486 |
|
|
|
537 |
|
|
|
1,548 |
|
|
Total expenses |
|
|
13,509 |
|
|
|
12,016 |
|
|
|
40,001 |
|
|
|
37,567 |
|
|
Other income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Investment income |
|
|
62 |
|
|
|
9 |
|
|
|
113 |
|
|
|
25 |
|
|
Total other income |
|
|
62 |
|
|
|
9 |
|
|
|
113 |
|
|
|
25 |
|
|
(Loss) income before provision (benefit) for taxes on income |
|
|
(896 |
) |
|
|
493 |
|
|
|
(2,533 |
) |
|
|
252 |
|
|
Provision (benefit) for taxes on income |
|
|
1 |
|
|
|
(140 |
) |
|
|
(442 |
) |
|
|
(44 |
) |
|
Net (loss) income |
|
$ |
(897 |
) |
|
$ |
633 |
|
|
$ |
(2,091 |
) |
|
$ |
296 |
|
|
Net (loss) income per share—basic |
|
$ |
(0.04 |
) |
|
$ |
0.04 |
|
|
$ |
(0.11 |
) |
|
$ |
0.02 |
|
|
Net (loss) income per share—diluted |
|
$ |
(0.04 |
) |
|
$ |
0.04 |
|
|
$ |
(0.11 |
) |
|
$ |
0.02 |
|
|
Weighted-average number of common shares used in net (loss) income per share basic |
|
|
22,324,374 |
|
|
|
15,979,614 |
|
|
|
18,970,128 |
|
|
|
15,974,510 |
|
|
Weighted-average number of common shares used in net (loss) income per share diluted |
|
|
22,324,374 |
|
|
|
16,149,149 |
|
|
|
18,970,128 |
|
|
|
16,082,963 |
|
See accompanying notes.
5
Unaudited Consolidated Statements of Cash Flows
(in thousands)
|
|
|
Nine Months ended |
|
|||||
|
|
|
September 30, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
Operating activities |
|
|
|
|
|
|
|
|
|
Net (loss) income |
|
$ |
(2,091 |
) |
|
$ |
296 |
|
|
Adjustments to reconcile net (loss) income to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
537 |
|
|
|
1,548 |
|
|
Stock-based compensation expense |
|
|
1,451 |
|
|
|
1,019 |
|
|
Provision for deferred taxes on income |
|
|
— |
|
|
|
13,427 |
|
|
Loss on disposal of property and equipment |
|
|
— |
|
|
|
2 |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Prepaid expenses and other current assets |
|
|
(2,899 |
) |
|
|
(327 |
) |
|
Other assets |
|
|
(451 |
) |
|
|
282 |
|
|
Accounts payable |
|
|
(2,235 |
) |
|
|
665 |
|
|
Accrued direct research and other current liabilities |
|
|
2,869 |
|
|
|
1,576 |
|
|
Federal income tax receivable/payable |
|
|
31,926 |
|
|
|
(13,470 |
) |
|
Deferred revenue |
|
|
(37,230 |
) |
|
|
(37,095 |
) |
|
Net cash used in operating activities |
|
|
(8,123 |
) |
|
|
(32,077 |
) |
|
Investing activities |
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(281 |
) |
|
|
(257 |
) |
|
Net cash used in investing activities |
|
|
(281 |
) |
|
|
(257 |
) |
|
Financing activities |
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock from initial public offering, net of issuance costs |
|
|
64,705 |
|
|
|
— |
|
|
Payments on deferred offering costs |
|
|
(2,531 |
) |
|
|
(343 |
) |
|
Exercise of options and related tax withholdings |
|
|
(73 |
) |
|
|
15 |
|
|
Payment of capital lease obligation |
|
|
(45 |
) |
|
|
(45 |
) |
|
Net cash provided by (used in) financing activities |
|
|
62,056 |
|
|
|
(373 |
) |
|
Net increase (decrease) in cash and cash equivalents |
|
|
53,652 |
|
|
|
(32,707 |
) |
|
Cash and cash equivalents at beginning of year |
|
|
42,008 |
|
|
|
87,758 |
|
|
Cash and cash equivalents at end of period |
|
$ |
95,660 |
|
|
$ |
55,051 |
|
|
Supplemental disclosures |
|
|
|
|
|
|
|
|
|
Income taxes paid |
|
$ |
18 |
|
|
$ |
— |
|
|
Purchases of equipment in accounts payable and other current liabilities |
|
$ |
13 |
|
|
$ |
— |
|
|
Vested prepaid restricted stock |
|
$ |
348 |
|
|
$ |
348 |
|
See accompanying notes.
6
Notes to Unaudited Consolidated Financial Statements
1. Description of Business
Reata Pharmaceuticals, Inc., or the Company, is a clinical stage biopharmaceutical company located in Irving, Texas focused on identifying, developing, and commercializing product candidates to address rare and life-threatening diseases with few or no approved therapies by targeting molecular pathways involved in how cells produce energy and regulate inflammation. The Company operates as a single segment of business.
The Company’s lead product candidates, bardoxolone methyl and omaveloxolone, are members of a class of small molecules called antioxidant inflammation modulators and target an important transcription factor, called Nrf2, to restore mitochondrial function, reduce oxidative stress, and resolve inflammation. Bardoxolone methyl is in Phase 3 clinical development for the treatment of pulmonary arterial hypertension associated with connective tissue disease (CTD-PAH), and Phase 2 clinical development for the treatment of pulmonary hypertension due to interstitial lung disease and pulmonary arterial hypertension, each of which are subsets of pulmonary hypertension (PH). The Company began enrolling patients in its Phase 3 trial in CTD-PAH in October 2016. In addition, the Company recently met with the U.S. Food and Drug Administration and received guidance on endpoints and general design for a single, pivotal Phase 2/3 trial in chronic kidney disease caused by Alport syndrome. Omaveloxolone is in Phase 2 clinical development for the treatment of multiple diseases, including Friedreich’s ataxia, mitochondrial myopathies, and metastatic melanoma. Beyond its lead product candidates, the Company has several promising preclinical programs. The Company believes its product candidates and preclinical programs have the potential to improve clinical outcomes in numerous underserved patient populations.
The Company’s consolidated financial statements include the accounts of all majority-owned subsidiaries that are required to be consolidated. Accordingly, the Company’s share of net earnings and losses from these subsidiaries is included in the consolidated statements of operations. Intracompany profits, transactions, and balances have been eliminated in consolidation.
On January 6, 2016, the Company effected a 4.4-to-1 reverse split of its common stock, and an automatic conversion of its common stock into Class B common stock. Upon the effectiveness of the reverse stock split and conversion, (i) every 4.4 shares of outstanding common stock were combined into one share of Class B common stock, (ii) the number of shares of common stock for which each outstanding option to purchase common stock is exercisable was proportionally decreased on a 4.4-to-1 basis and converted into an option to purchase Class B common stock, and (iii) the exercise price of each outstanding option to purchase common stock was proportionately increased on a 4.4-to-1 basis. All of the outstanding common stock share numbers, common stock options, share prices, exercise prices and per share amounts have been adjusted in these consolidated financial statements, on a retroactive basis, to reflect this 4.4-to-1 reverse stock split for all periods presented. The par value per share was not adjusted as a result of the reverse stock split.
On May 11, 2016, the Company effected a 1.45-to-1 reverse split of its common stock. Upon the effectiveness of the reverse stock split, (i) every 1.45 shares of outstanding common stock were combined into one share of common stock of the same class, (ii) the number of shares of common stock for which each outstanding option to purchase common stock is exercisable was proportionally decreased on a 1.45-to-1 basis, and (iii) the exercise price of each outstanding option to purchase common stock was proportionately increased on a 1.45-to-1 basis. All of the outstanding common stock share numbers, common stock options, share prices, exercise prices and per share amounts have been adjusted in these consolidated financial statements, on a retroactive basis, to reflect this 1.45-to-1 reverse stock split for all periods presented. The par value per share was not adjusted as a result of the reverse stock split.
On May 25, 2016, the Company’s registration statement on Form S-1 (File No. 333-208843) relating to its initial public offering (IPO), of its common stock was declared effective by the U.S. Securities and Exchange Commission (SEC). The shares began trading on The NASDAQ Global Market on May 26, 2016. The public offering price of the shares sold in the offering was $11.00 per share. The IPO closed on June 1, 2016 for 6,325,000 shares of its Class A common stock, which included 825,000 shares of its Class A common stock issued pursuant to the over-allotment option granted to the underwriters. The Company received total proceeds from the offering of $60.9 million, net of underwriting discounts and commissions and offering expenses.
7
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements (continued)
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP) for interim financial information and with the instructions to Form 10-Q and Article 10 of Regulation S-X. Accordingly, they do not include all of the information and notes required by U.S. GAAP for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation have been included. Operating results for the nine months ended September 30, 2016 are not necessarily indicative of the results that may be expected for the year ending December 31, 2016. The consolidated balance sheet at December 31, 2015 has been derived from the audited consolidated financial statements at that date but does not include all of the information and footnotes required by U.S. GAAP for complete financial statements. For further information, refer to the annual consolidated financial statements and footnotes thereto of the Company.
Revenue Recognition
The Company’s revenue to date has been generated primarily through collaborative licensing agreements with AbbVie Ltd. (AbbVie) and Kyowa Hakko Kirin Co., Ltd. Revenues for periods shown consist of the recognition of deferred revenue from upfront payments and milestone payments received in 2012 and prior years. The Company has not generated any revenue based on the sale of products.
In June 2013, the Company entered into a research collaboration with a disease advocacy organization. Under the agreement, the Company may be provided milestone payments to fund research and development activities estimated over a two-year period. The Company recorded collaboration revenue totaling $700,000 related to milestone payments during the nine months ended September 30, 2015.
Research and Development Costs
AbbVie is not currently participating in the development of bardoxolone methyl for the treatment of PH and we are therefore incurring all costs for this program. With respect to its omaveloxolone programs and its collaboration agreement with AbbVie, the Company was responsible for a certain initial amount in early development costs before AbbVie began sharing development costs equally. As of April 2016, the Company had incurred all of these initial costs, after which payments from AbbVie with respect to research and development costs incurred by the Company were recorded as a reduction in research and development expenses. The Company’s expenses were reduced by $773,000 and $1,434,000 for AbbVie’s share of research and development costs for the three months and nine months ended September 30, 2016. Accordingly, as of September 30, 2016, the Company had receivables in the amount of $1,434,000 included in prepaid expenses and other current assets on the consolidated balance sheet. The Company received $661,000 from AbbVie in October 2016.
In September 2016, the Company and AbbVie mutually agreed that the Company would continue unilateral development of omaveloxolone. Therefore, AbbVie no longer co-funds the exploratory development costs of this program, but retains the right to opt back in at certain points in development. Depending upon what point, if any, AbbVie opts back into development, AbbVie may retain its right to commercialize a product outside the U.S. or the Company may be responsible for commercializing the product on a worldwide basis. Upon opting back in, AbbVie would be required to pay an agreed upon amount of all development costs accumulated up to the point of exercising their opt-in right, after which development costs incurred and product revenue worldwide would be split equally.
The Company bases its expense accruals related to clinical trials on its estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and contract research organizations that conduct and manage clinical trials on its behalf. The financial terms of these agreements vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing costs, the Company estimates the time period over which services will be performed and the level of effort to be expended in each period. If the Company does not identify costs that it has begun to incur or if the Company underestimates or overestimates the level of services performed or the costs of these services, its actual expenses could differ from its estimates.
8
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements (continued)
To date, the Company has not experienced significant changes in its estimates of accrued research and development expenses after a reporting period. However, due to the nature of estimates, the Company cannot assure that it will not make changes to its estimates in the future as the Company becomes aware of additional information about the status or conduct of its clinical trials and other research activities.
Stock-Based Compensation
The Company accounts for its equity-based compensation awards in accordance with Accounting Standard Codification ASC 718 Compensation—Stock Compensation (ASC 718). ASC 718 requires companies to recognize compensation expense using a fair value based method for costs related to stock-based payments, including stock options. The expense is measured based on the grant date fair value of the awards that are expected to vest, and the expense is recorded over the applicable requisite service period.
The Company uses the Black-Scholes option-pricing model to estimate the fair value of stock option awards, which takes into consideration various factors, including the exercise price of the award, the expected term of the award, the current price of the underlying shares, the expected volatility of the underlying share price based on peer companies, forfeitures rate, and the risk-free interest rate. Prior to the Company’s IPO of its common stock, the fair values of the shares of common stock underlying the Company’s share-based awards were estimated on each grant date using a probability-weighted expected return method. Following the close of its IPO in June 2016, the fair values of its common stock underlying its share-based awards were estimated using observable market prices.
Risks and Uncertainties
The Company has experienced losses and negative operating cash flows for many years since inception and has no marketed drug or other products. The Company’s ability to generate future revenue depends upon the results of its development programs, the success of which cannot be guaranteed. The Company will need to raise additional equity capital in the future in order to fund its operations.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Fair Value of Financial Instruments
The fair values of the Company’s stockholder notes receivable were approximately $166,000 and $138,000 at September 30, 2016 and December 31, 2015, respectively. The fair value was calculated using an income approach to estimate the present value of expected future cash flows to be received under the notes. The measurement is considered to be based primarily on Level 3 inputs used in the calculation, including the discount rate applied and the estimate of future cash flows.
Net Income (Loss) per Share
Basic and diluted net income (loss) per common share is calculated by dividing net income (loss) attributable to common stockholders by the weighted average number of common shares outstanding during the period, without consideration for common stock equivalents. The Company’s potentially dilutive shares, which include unvested restricted stock and options to purchase common stock, are considered to be common stock equivalents and are only included in the calculation of diluted net income (loss) per share when their effect is dilutive. For periods in which the Company reports a net loss attributable to common stockholders, diluted net loss per share attributable to common stockholders is the same as basic net loss per share attributable to common stockholders, since dilutive common shares are not assumed to have been issued if their effect is anti-dilutive.
The Company uses the two-class method to compute net income (loss) per common share attributable to common stockholders because the Company has issued securities, other than Class A and Class B common stock, that contractually entitle the holders to participate in dividends and earnings of the Company. The two-class method requires earnings for the period to be allocated between common stock and participating securities based upon their respective rights to receive distributed and undistributed earnings. Holders of restricted common stock are entitled to the dividend amount paid to common stockholders on an as-if-converted-to-common stock basis when declared by the Company’s Board of Directors. As a result, all restricted common stock are considered to be participating securities.
9
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements (continued)
Deferred offering costs, which primarily consist of direct incremental accounting, legal, and printing fees relating to the IPO, were initially capitalized. The deferred offering costs totaling $3,489,000 were subsequently offset against IPO proceeds upon the completion of the IPO on June 1, 2016.
Recent Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2016-02, Leases (Topic 842), which supersedes the leases in ASC 840, Leases. This ASU requires the recognition of lease assets and lease liabilities by lessees for those leases previously classified as operating leases. The ASU is effective for public companies for fiscal years, and for interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted. The Company will apply the guidance and disclosure provisions of the new standard upon adoption. The Company is currently evaluating this standard and has not yet determined what, if any, effect ASU No. 2016-02 will have on its consolidated results of operations or financial position.
In March 2016, the FASB issued ASU No. 2016-09, Compensation–Stock Compensation: Improvements to Employee Share-Based Payment Accounting, to simplify several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. The ASU is effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2016. Early adoption is permitted. The Company will apply the guidance and disclosure provisions of the new standard upon adoption. The Company is currently evaluating this standard and has not yet determined what, if any, effect ASU No. 2016-09 will have on its consolidated results of operations or financial position.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments, This update addresses eight specific cash flow issues with the objective of reducing the existing diversity in practice. The ASU is effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017. The Company is currently evaluating this standard and has not yet determined what, if any, effect ASU No. 2016-15 will have on its consolidated results of operations or financial position.
3. Income Taxes
The Company’s effective tax rate varies with the statutory rate due primarily to the impact of nondeductible stock-based compensation and the changes in valuation allowance related to certain deferred tax assets generated or utilized in the applicable period. The Company’s deferred tax assets have been fully offset by a valuation allowance at September 30, 2016, and the Company expects to maintain this valuation allowance until there is sufficient evidence that future earnings can be achieved, which is uncertain at this time.
4. Stock-Based Compensation
Stock Options
The following table summarizes stock-based compensation expense reflected in the consolidated statements of operations (in thousands):
|
|
|
Three Months ended |
|
|
Nine Months ended |
|
||||||||||
|
|
|
September 30, |
|
|
September 30, |
|
||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
||||
|
Research and development |
|
$ |
375 |
|
|
$ |
164 |
|
|
$ |
725 |
|
|
$ |
519 |
|
|
General and administrative |
|
|
341 |
|
|
|
162 |
|
|
|
726 |
|
|
|
500 |
|
|
|
|
|
716 |
|
|
|
326 |
|
|
|
1,451 |
|
|
|
1,019 |
|
10
Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements (continued)
The following table summarizes stock option activity as of September 30, 2016, and changes during the nine months ended September 30, 2016, under the 2007 Long Term Incentive Plan (the 2007 LTIP) and standalone option agreements:
|
|
|
Number of Options |
|
|
Weighted- Average Exercise Price |
|
||
|
Outstanding at January 1, 2016 |
|
|
550,675 |
|
|
|
16.11 |
|
|
Granted |
|
|
986,845 |
|
|
|
12.63 |
|
|
Exercised |
|
|
(26,382 |
) |
|
|
10.29 |
|
|
Forfeited |
|
|
(16,536 |
) |
|
|
11.62 |
|
|
Expired |
|
|
(19,709 |
) |
|
|
11.50 |
|
|
Outstanding at September 30, 2016 |
|
|
1,474,893 |
|
|
|
14.00 |
|
|
Exercisable at September 30, 2016 |
|
|
385,776 |
|
|
|
17.13 |
|
The total intrinsic value of all outstanding options and exercisable options at September 30, 2016 was $17,143,701 and $4,646,688, respectively.
5. Related-Party Transactions
The Company paid approximately $772,000 and $896,000 to certain stockholders for sponsored research, research and development consulting services, contract manufacturing services, regulatory and medical consulting services, license fees, and clinical study services during nine months ended September 30, 2016 and 2015, respectively. These amounts are recorded in research and development expense in the accompanying consolidated statements of operations.
6. Net (Loss) income per Share
The following table sets forth the computation of basic and diluted net (loss) income per share attributable to common stockholders:
|
|
|
Three Months ended |
|
|
Nine Months ended |
|
||||||||||
|
|
|
September 30, |
|
|
September 30, |
|
||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
||||
|
Numerator |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net (loss) income (in thousands) |
|
$ |
(897 |
) |
|
$ |
633 |
|
|
$ |
(2,091 |
) |
|
$ |
296 |
|
|
Denominator |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average number of common shares used in net (loss) income per share – basic |
|
|
22,324,374 |
|
|
|
15,979,614 |
|
|
|
18,970,128 |
|
|
|
15,974,510 |
|
|
Dilutive potential common shares |
|
|
— |
|
|
|
169,535 |
|
|
|
— |
|
|
|
108,453 |
|
|
Weighted-average number of common shares used in net (loss) income per share – diluted |
|
|
22,324,374 |
|
|
|
16,149,149 |
|
|
|
18,970,128 |
|
|
|
16,082,963 |
|
|
Net (loss) income per share – basic |
|
|
(0.04 |
) |
|
|
0.04 |
|
|
|
(0.11 |
) |
|
|
0.02 |
|
|
Net (loss) income per share – diluted |
|
|
(0.04 |
) |
|
|
0.04 |
|
|
|
(0.11 |
) |
|
|
0.02 |
|
The number of weighted average options that were not included in the diluted earnings per share calculation because the effect would have been anti-dilutive represented 1,474,893 shares for the three and nine months ended September 30, 2016 and 132,918 and 126,433 shares for the three months and nine months ended September 30, 2015, respectively.
11
Item 2. MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes and other financial information appearing in this Quarterly Report on Form 10-Q. Some of the information contained in this discussion and analysis or set forth elsewhere in this Quarterly Report on Form 10-Q, including information with respect to our plans and strategy for our business, operations, and product candidates, includes forward-looking statements that involve risks and uncertainties. Factors that may cause actual results to differ materially from current expectations include, among other things, those described under the heading “Risk Factors” and discussed elsewhere in this Quarterly Report on Form 10-Q.
Overview
We are a clinical stage biopharmaceutical company focused on identifying, developing, and commercializing product candidates to address rare and life-threatening diseases with few or no approved therapies by targeting molecular pathways that regulate cellular metabolism and inflammation. Our lead product candidates, bardoxolone methyl and omaveloxolone, are members of a class of small molecules called antioxidant inflammation modulators, or AIMs, and target an important transcription factor, called Nrf2, to restore mitochondrial function, reduce oxidative stress, and resolve inflammation. Bardoxolone methyl is currently being studied in a Phase 3 trial, known as CATALYST, for the treatment of pulmonary arterial hypertension, or PAH, associated with connective tissue disease, or CTD-PAH, as well as a Phase 2 trial, known as LARIAT, for the treatment of pulmonary hypertension due to interstitial lung disease, or PH-ILD, and PAH, each of which are subsets of pulmonary hypertension, or PH. We began enrolling patients in CATALYST in October 2016. In addition, we recently met with the U.S. Food and Drug Administration, or the FDA, and received guidance on endpoints and general design for a single, pivotal Phase 2/3 trial in chronic kidney disease, or CKD, caused by Alport syndrome. Omaveloxolone is being studied in Phase 2 trials for the treatment of multiple diseases, including Friedreich’s ataxia, or FA, mitochondrial myopathies, or MM, and metastatic melanoma, known as MOXIe, MOTOR, and REVEAL, respectively. Beyond our lead product candidates, we have several promising preclinical development programs. We believe that our product candidates and preclinical programs have the potential to improve clinical outcomes in numerous underserved patient populations.
To date, we have focused most of our efforts and resources on developing our product candidates and conducting preclinical studies and clinical trials. We have historically financed our operations primarily through revenue generated from our collaborations with AbbVie Ltd., or AbbVie, and Kyowa Hakko Kirin Co., Ltd., or KHK, and from sales of our securities. We have not received any payments or revenue from collaborations other than nonrefundable upfront, milestone, and cost sharing payments from our collaborations with AbbVie and KHK and reimbursements of expenses under the terms of our agreement with KHK. We have incurred losses in each year since our inception, other than in 2014. As of September 30, 2016, we had $95.7 million of cash and cash equivalents and an accumulated deficit of $285.2 million. We continue to incur significant research and development and other expenses related to our ongoing operations. Despite contractual product development commitments and the potential to receive future payments from our collaborators, we anticipate that, without taking into account deferred revenue, we will continue to incur losses for the foreseeable future, and we anticipate that our losses will increase as we continue our development of, and seek regulatory approval for, our product candidates. If we do not successfully develop and obtain regulatory approval of our existing product candidates or any future product candidates and effectively manufacture, market, and sell any products that are approved, we may never generate revenue from product sales. Furthermore, even if we do generate revenue from product sales, we may never again achieve or sustain profitability on a quarterly or annual basis. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Our failure to become and remain profitable could depress the market price of our Class A common stock and could impair our ability to raise capital, expand our business, diversify our product offerings, or continue our operations.
The probability of success for each of our product candidates and clinical programs and our ability to generate product revenue and become profitable depend upon a variety of factors, including the quality of the product candidate, clinical results, investment in the program, competition, manufacturing capability, commercial viability, and our collaborators’ ability to successfully execute our development and commercialization plans. We will also require additional capital through equity or debt financings in order to fund our operations and execute on our business plans, and there is no assurance that such financing will be available to us on commercially reasonable terms or at all. For a description of the numerous risks and uncertainties associated with product development and raising additional capital, see “Risk Factors” included in the Final Prospectus.
12
The chart below is a summary of our current clinical programs:

|
|
(1) |
We are currently designing a Phase 2/3 clinical trial, which we expect to initiate in the first half of 2017. |
|
|
(2) |
We continue to evaluate development of omaveloxolone for this indication. |
Bardoxolone Methyl
Bardoxolone methyl activates molecular pathways that promote the resolution of inflammation by restoring mitochondrial function, reducing oxidative stress, and inhibiting pro-inflammatory signaling. Bardoxolone methyl binds to Keap1, which activates Nrf2, a transcription factor that promotes normal mitochondrial function, increases production of antioxidant and detoxification enzymes, reduces oxidative stress, and reduces pro-inflammatory signaling. Bardoxolone methyl is currently being tested in a Phase 3 trial in CTD-PAH as well as Phase 2 trials in several forms of PH-ILD. In addition, we recently met with the FDA and received guidance on endpoints and general design characteristics of a single, pivotal Phase 2/3 trial in CKD caused by Alport syndrome and are in the process of designing that trial.
Although CTD-PAH and Alport syndrome have different causes and inflammatory stimuli, at a molecular level, mitochondrial dysfunction, inflammation, and proliferative signaling are common to the pathophysiology of both diseases. The anti-inflammatory and anti-fibrotic properties of bardoxolone methyl may therefore be relevant to preventing remodeling of the pulmonary vasculature in CTD-PAH as well as inhibiting structural alterations and glomerulosclerosis in Alport syndrome.
Bardoxolone Methyl in Pulmonary Hypertension
Pulmonary Hypertension Overview
PH is a multi-organ condition characterized by an abnormally high pressure in the network of arteries and veins that lead to and from the lungs due, in part, to narrowing of the pulmonary vasculature as a result of inflammation, remodeling, proliferation, and endothelial dysfunction. Mitochondrial dysfunction has also been implicated in PH, and impaired energetics of skeletal muscle is a common feature of PH. PH can be caused by a number of different underlying defects, which have been classified into five groups by the World Health Organization, or WHO. We are currently studying bardoxolone methyl in CATALYST in patients with WHO Group 1 CTD-PAH. We are also studying bardoxolone methyl in patients from certain subgroups of WHO Groups 3 and 5 PH-ILD.
While there are vasodilator therapies approved for the treatment of PAH, there are currently no therapies to treat PH-ILD. Further, current therapies for PAH are less effective in CTD-PAH, which makes up about 30% of the PAH population. CTD-PAH is a late and often fatal manifestation of many types of autoimmune disease, including systemic sclerosis, or scleroderma, systemic lupus erythematosus, mixed connective tissue disease, and others. PAH results in a progressive remodeling and fibrosis of the pulmonary vasculature, which increases pulmonary vascular resistance and ultimately leads to right ventricular heart failure and death. Patients with CTD-PAH are generally less responsive to existing therapies and have shorter survival than patients with other forms of PAH. In the United States, the five-year survival rate for CTD-PAH patients is approximately 44% compared to idiopathic PAH patients, who have a 68% five-year survival rate.
13
Currently approved therapies to treat PAH include endothelin receptor antagonists, nitric oxide pathway modulators, and prostacyclin pathway agonists, all of which are systemic vasodilators that directly modulate vasoconstrictive and vasodilatory pathways. All three classes of existing therapies do not specifically target the pulmonary vasculature and have systemic hemodynamic effects. These systemic hemodynamic effects can result in hypotension and syncope, or fainting, which generally limits their clinical effectiveness. These hemodynamic effects can be exacerbated when a patient is prescribed multiple vasodilators. In addition, clinically significant drug-drug interactions have been observed that can further limit the ability to deliver effective drug combinations.
Vasodilators approved for PAH also generally do not yield significant functional improvements in CTD-PAH patients because their disease involves more remodeling and fibrosis that is less affected by vasodilators. Recent research has indicated that PAH patients, and particularly CTD-PAH patients, experience mitochondrial dysfunction, which occurs in the pulmonary vasculature, heart, and other organ systems. Mitochondrial dysfunction promotes reduced energy production, inflammation, and tissue remodeling, which causes impaired cardiac and skeletal muscle function, fibrosis, and eventual death. As described in a recently published large meta-analysis performed at the University of Pennsylvania that analyzed data from eleven Phase 3 and Phase 4 clinical trials, CTD-PAH patients treated with vasodilator therapies have 6-minute walk distance, or 6MWD, improvements of only one third compared to the improvements seen in I-PAH patients.
Further, due to their vasodilatory mechanism, the efficacy of currently approved therapies is impacted by the number of other PAH therapies being administered to a patient, with each new therapy yielding lower marginal efficacy.
Bardoxolone methyl directly targets the bioenergetic and inflammatory components of PH. PH patients experience mitochondrial dysfunction, increased activation of NF-κB and related inflammatory pathways involved in reactive oxygen species, or ROS, signaling, cellular proliferation, and fibrosis. Bardoxolone methyl, through the combined effect of Nrf2 activation and NF-κB suppression, has the potential to inhibit inflammatory and proliferative signaling, suppress ROS production and signaling, reduce the production of enzymes related to fibrosis and tissue remodeling, and increase ATP production and cellular respiration. Bardoxolone methyl targets multiple cell types relevant to PH, including endothelial cells, smooth muscle cells, and macrophages. Additionally, unlike current therapies, bardoxolone methyl does not have systemic hemodynamic effects or drug-drug interactions in PH patients. Therefore, by addressing a novel pathway in PH, we believe that bardoxolone methyl may provide additional benefits beyond current PAH therapies, including increased functional capacity, potential effects beyond functional improvements, broader applicability to underserviced PH patients, and potential as a combination therapy with other current therapies.
Phase 3 Development
On October 6, 2016, the first patient was enrolled in CATALYST, an international, randomized, double-blind, placebo-controlled Phase 3 trial examining the safety, tolerability, and efficacy of bardoxolone methyl in patients with WHO Group 1 CTD-PAH when added to standard-of-care vasodilator therapy. Patients will be on up to two background therapies and will be randomized 1:1 to bardoxolone methyl or placebo. Patients will be enrolled at approximately 100 sites in the U.S., Canada, Australia, Japan, Mexico, Europe, Israel, and South America, and the study drug will be administered once daily for 24 weeks. Patients randomized to bardoxolone methyl will start at 5 mg and will dose-escalate to 10 mg at Week 4 unless contraindicated clinically. The primary endpoint is the change from baseline in 6MWD relative to placebo at Week 24. Secondary endpoints include time to first clinical improvement as measured by improvement in WHO functional class, increase from baseline in 6MWD by at least 10%, or decrease from baseline in creatine kinase (as a surrogate biomarker for muscle injury and inflammation) by at least 10%. The trial will enroll between 130 and 200 patients. To determine the final sample size, a pre-specified, blinded sample size re-calculation based on 6MWD variability and baseline characteristics will be conducted after 100 patients have been enrolled in the trial. Data from CATALYST are expected to be available during the first half of 2018.
During our interaction with the FDA in October 2015, the FDA noted that CATALYST, together with the Phase 2 data from our LARIAT trial in PAH patients and prior clinical trials with bardoxolone methyl, would provide adequate data for a New Drug Application, or NDA, review of the safety profile of bardoxolone methyl. Prior to this meeting, we had completed a series of clinical pharmacology studies, including a Thorough QT study, hepatic impairment study, food effect study, and three drug-drug interaction studies. The FDA recommended conducting a single additional clinical drug-drug interaction study and otherwise had no clinical trial, clinical pharmacology, or preclinical study requests.
In preparation for, and in advance of, the initiation of CATALYST, we analyzed data for all CTD-PAH patients treated with doses of up to 10 mg who had completed the 16-week treatment period (or terminated early) in the ongoing LARIAT trial. A total of 22 CTD-PAH patients, including patients from Cohorts 1, 2, and 3a, which are discussed below, met these criteria, with 15 randomized to bardoxolone methyl and seven randomized to placebo. Baseline characteristics of the 22 CTD-PAH patients can be found in the table below.
14
Baseline Characteristics for CTD-PAH Patients in LARIAT
|
|
Bard |
Placebo |
|
N |
15 |
7 |
|
Mean Age (years) |
56.4 |
52.4 |
|
Female/Male |
80%/20% |
100%/0% |
|
Mean Weight (kg) |
75.5 |
68.2 |
|
Mean BMI (kg/m^2) |
27.5 |
26.3 |
|
WHO Functional Class: |
|
|
|
II |
9 (60%) |
4 (57%) |
|
III |
6 (40%) |
3 (43%) |
|
Mean Time Since Diagnosis (years) |
2.9 |
3.0 |
|
Mean Baseline 6MWD (m) |
381 |
396 |
The LARIAT statistical analysis plan defined the treatment effect as the time-averaged change from baseline in 6MWD values using a longitudinal model to assess the average of all available 6MWD timepoints, improving the study’s sensitivity to detect a significant difference between the active drug and placebo groups. Change from baseline in 6MWD at Weeks 4, 8, 12, and 16 were analyzed using a mixed-model repeated measures, or MMRM, analysis to compare the difference between the active drug and placebo groups. The analysis showed that patients treated with bardoxolone methyl demonstrated a statistically significant mean time-averaged increase in 6MWD compared to baseline of 26.7 meters (p=0.001). Placebo-treated patients had a non-significant time-averaged mean change from baseline in 6MWD of 0.6 meters (p=0.96). The placebo-corrected time-averaged change in 6MWD was 26.1 meters (p=0.06).
Patients with moderate to severe anemia, which represents a small percentage of the patient population, will be excluded from CATALYST because treatment with iron supplementation or erythropoietin post-randomization can affect 6MWD values independent of study drug effect. Three CTD-PAH patients enrolled in LARIAT and included in the above analysis were anemic at screening (as defined by low hemoglobin values), and two of these patients, both randomized to placebo, received post-randomization anemia treatments. An analysis was conducted excluding patients with anemia at screening to estimate the treatment effect in patients who meet the final CATALYST eligibility criteria. MMRM analysis showed that CATALYST-eligible patients treated with bardoxolone methyl in LARIAT demonstrated a statistically significant mean time-averaged increase in 6MWD compared to baseline of 30.2 meters (p<0.001), and placebo-treated patients had a non-significant mean change from baseline in 6MWD of –10.1 meters (p=0.39) for a placebo-corrected change of 40.3 meters (p=0.009). The pooled standard deviation of change of 6MWD was 34.1 meters. The time-averaged change in 6MWD is shown in the table below.
Summary of Time-Averaged 6MWD Changes for CTD-PAH Patients in LARIAT
|
Treatment |
N |
All Patients |
N |
CATALYST-Eligible Patients |
||
|
Change from Baseline (m) |
Placebo-corrected (m) |
Change from Baseline (m) |
Placebo-corrected (m) |
|||
|
Placebo |
7 |
0.6 p=0.96 |
— |
5 |
–10.1 p=0.39 |
— |
|
Bardoxolone Methyl |
15 |
26.7 p=0.001 |
26.1 p=0.06 |
14 |
30.2 p < 0.001 |
40.3 p=0.009 |
The method of statistical analysis for the CATALYST primary endpoint is the placebo-corrected change from baseline in 6MWD at the end-of-treatment at 24 weeks. This method allows for greater separation in 6MWD values between active and placebo groups assuming improved efficacy over time, which was observed in the CTD-PAH patients in LARIAT as shown in the figure below.
15
Mean Change in 6MWD (+/- SEM) Over Time in CATALYST-Eligible Patients from LARIAT
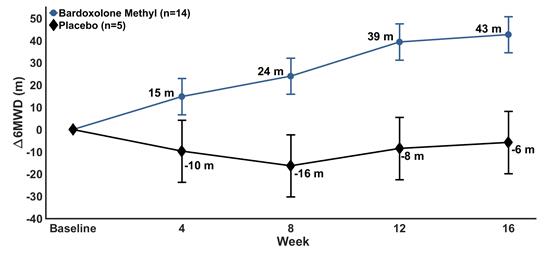
We performed an analysis applying the statistical methods for CATALYST to the available end-of-treatment (Week 16) change in 6MWD data from CTD-PAH patients in LARIAT as seen in the table below. Using MMRM to estimate change from baseline in 6MWD at Week 16, patients treated with bardoxolone methyl demonstrated a statistically significant mean increase of 38.2 meters (p<0.001). Placebo-treated patients had a non-significant mean change from baseline in 6MWD of 9.8 meters (p=0.44). The placebo-corrected change in 6MWD at Week 16 was 28.4 meters (p=0.07). Excluding patients with moderate to severe anemia at screening, the patients treated with bardoxolone methyl demonstrated a statistically significant mean increase in 6MWD compared to baseline of 42.7 meters (p<0.001). Placebo-treated patients had a non-significant mean change from baseline in 6MWD of –5.8 meters (p=0.68). The placebo-corrected change in 6MWD at Week 16 was 48.5 meters (p=0.005).
Summary of End-of-Treatment 6MWD Changes for CTD-PAH Patients in LARIAT
|
Treatment |
N |
All Patients |
N |
CATALYST Eligible Patients |
||
|
Change from Baseline |
Placebo-corrected |
Change from Baseline |
Placebo-corrected |
|||
|
Placebo |
7 |
9.8 p=0.44 |
— |
5 |
–5.8 p=0.68 |
— |
|
Bardoxolone Methyl |
15 |
38.2 p < 0.001 |
28.4 p=0.07 |
14 |
42.7 p < 0.001 |
48.5 p=0.005 |
With respect to safety, bardoxolone methyl continued to be well-tolerated. None of the 15 bardoxolone methyl treated patients discontinued early, whereas one of the seven placebo treated patients discontinued prematurely. The expanded data set shows no clinically meaningful differences in safety variables including vital signs and laboratory data. Bardoxolone methyl was combined with approved vasodilator therapies without increasing the risk of hypotensive events or exacerbating their adverse event profile.
CATALYST is designed to detect a minimum treatment effect of 12.5 meters assuming a standard deviation of 50 meters. The observed treatment effect in the LARIAT CTD-PAH subgroup analyses, both with and without the anemic patients included, was larger than the minimally detectable treatment effect in CATALYST. Further, the pooled standard deviation observed in LARIAT of 37 meters is lower than the estimated standard deviation of 50 meters in CATALYST.
Phase 2 Development
We initially tested bardoxolone methyl in PH patients in LARIAT, a randomized, placebo-controlled, double-blinded, dose-escalation Phase 2 trial evaluating the safety and efficacy of once daily, orally administered bardoxolone methyl in PH patients with PAH or PH-ILD. LARIAT is comprised of four separate cohort groups.
16
The primary endpoint of the LARIAT trial is change in 6MWD during a 16 week treatment period. All patients who complete the treatment period are eligible to continue into an extension trial to evaluate the intermediate and long-term safety and efficacy of bardoxolone methyl. Those patients who had been receiving placebo are converted to bardoxolone methyl in the extension trial. The initial treatment period for cohorts 1 and 2 has been completed and, as discussed above, data from cohorts 1 and 2 have been publicly presented, as has initial data from cohort 3. We also intend to use data from CTD-PAH patients to help support an application to the FDA for breakthrough status for the treatment of CTD-PAH, once we have enough data to do so.
Because bardoxolone methyl was active in patients with CTD-PAH, a fibrotic disease, we believe that bardoxolone methyl may be effective in PH-ILD patients. We have also begun enrolling patients with PH-ILD caused by CTD, idiopathic pulmonary fibrosis, non-specific interstitial pneumonia, and sarcoidosis in LARIAT cohorts 4a, 4b, 4c, and 4d, respectively. Data have not been presented from cohort 4. We previously reported that a serious adverse event, or SAE, involving a patient death had occurred in cohort 4a and that the investigator had initially reported it as possibly related to study drug. The investigator has since changed his evaluation to unlikely related. In addition, the Protocol Safety Review Committee that oversees safety for the LARIAT trial concluded that the SAE was unlikely treatment-related. We anticipate that data from PH-ILD patients in the LARIAT trial will be available in the second half of 2017.
The cohorts from the LARIAT trial are described below.
|
|
• |
Cohort 1. The first cohort began enrolling in May 2014 and consisted of PAH patients in the United States. Eligible patients must have had a baseline 6MWD of greater than or equal to 150 meters but less than or equal to 450 meters and must have been receiving at least one disease-specific PAH background therapy. Patients were randomized 3:1 in each dose group to either bardoxolone methyl at doses of 2.5 mg, 5 mg, 10 mg, or 20 mg, or placebo. |
|
|
• |
Cohort 2. The second cohort began enrolling in January 2015 and consisted of PAH patients in the United States. Eligible patients must have had a baseline 6MWD of greater than 450 meters and must have been receiving at least one disease-specific PAH background therapy. Patients were are randomized 3:1 in each dose group to bardoxolone methyl at doses of 5 mg or 20 mg, or placebo. |
|
|
• |
Cohort 3. The third cohort began enrolling in September 2015, and consists of PAH patients in the United States and potentially other countries, and is comprised of two sub-cohorts for CTD-PAH (cohort 3a) patients and non-CTD-PAH (cohort 3b) patients. Eligible patients must have a baseline 6MWD of greater than or equal to 150 meters and must be receiving zero to two disease-specific PAH background therapies. Patients are randomized 2:1 to bardoxolone methyl or placebo. Patients in the treatment group are titrated from 5 mg to 10 mg doses based on tolerability. |
|
|
• |
Cohort 4. The fourth cohort began enrolling in September 2015, consists of PH-ILD patients in the United States and potentially other countries, and is comprised of four sub-cohorts based on the patient’s underlying type of ILD: (a) PH-ILD caused by CTD, such as scleroderma and lupus, or CTD-PH-ILD; (b) PH-ILD caused by idiopathic pulmonary fibrosis, or IPF-PH-ILD; (c) PH-ILD caused by non-specific interstitial pneumonia, or NSIP-PH-ILD; and (d) PH-ILD caused by sarcoidosis, or SA-PH-ILD. Eligible patients must have a baseline 6MWD of greater than or equal to 150 meters. As no therapies are approved to treat these patients’ PH, no background therapies are required for enrollment. Patients are randomized 2:1 to bardoxolone methyl or placebo. Patients in the treatment group are titrated from 5 mg to 10 mg doses based on tolerability. |
Bardoxolone Methyl in Chronic Kidney Disease caused by Alport Syndrome
Alport Syndrome Overview
Alport syndrome is a rare and serious hereditary disease that affects approximately 12,000 children and adults in the United States and 40,000 globally. It is caused by mutations in the genes encoding type IV collagen, a major structural component of the glomerular basement membrane, or GBM, in the kidney. The abnormal expression of type IV collagen causes loss of GBM integrity, abnormal leakage of proteins through the GBM, and excessive reabsorption of protein in the proximal tubules of the kidney. Like other forms of CKD, excessive reabsorption of protein in the tubules induces oxidative stress and renal interstitial inflammation and fibrosis.
Patients with Alport syndrome are normally diagnosed with the disease in childhood to early adulthood and have average glomerular filtration rate, or GFR, declines of 4.0 mL/min/1.73 m2 per year. The progressive decline of GFR in Alport syndrome inexorably leads to renal failure and end-stage renal disease, or ESRD, with a median survival of approximately 55 years. Fifty percent of males with the most prevalent subtype of Alport syndrome require dialysis or kidney transplant by age 25. The incidence of renal failure in these patients increases to 90% by age 40 and nearly 100% by age 60. Similar to patients with other forms of CKD, Alport patients receiving dialysis are at increased risk for cardiovascular disease and infections, which are the most common causes of death in these patients. Currently, there are no approved therapies for the treatment of Alport syndrome.
17
The pathogenic role of inflammatory processes in Alport syndrome disease progression and declining renal function is similar to that of other chronic kidney diseases. The GBM defects and leaked proteins in Alport syndrome, the hyperglycemia in diabetes, and hypertension in cardiovascular disease all activate pro-inflammatory signaling pathways that normally detect cellular damage or pathogens. These signals induce mitochondrial dysfunction in which production of cellular energy, or ATP, is impaired in favor of production of pro-inflammatory ROS. ROS is a central feature of inflammation and activates pro-inflammatory signaling complexes including NF-κB and the NLRP3 complex referred to as the inflammasome. ROS-mediated activation of NF-κB and the inflammasome produce cytokines that promote inflammation in glomerular endothelial cells, mesangial cells, and podocytes while also recruiting activated macrophages and other inflammatory effector cells to the renal interstitium.
Chronic activation of pro-inflammatory pathways in kidney cells promotes GFR loss by at least three mechanisms. First, inflammation-associated ROS reduce the amount of nitric oxide available to the endothelial cells in the blood vessels of the glomerulus. This results in a decrease of the overall surface area of the glomerulus that is available for filtration, and thus decreases GFR. Second, inflammation-associated ROS cause contraction of mesangial cells in the kidney. The primary function of these cells is to remove debris and protein from the glomerular basement membrane, or GBM, allowing proper filtration to occur. Mesangial cell contraction reduces their function, and thus reduces GFR. Third, inflammation-associated ROS lead to fibrosis, which changes the structure of the mesangial cell layer and causes thickening of the GBM, contributing to decline of GFR.
Biological Rationale for Bardoxolone in Alport Syndrome
Bardoxolone methyl has the potential to address the underlying causes of GFR loss in Alport syndrome patients because it activates molecular pathways that promote the resolution of inflammation by restoring mitochondrial function, reducing oxidative stress, and inhibiting ROS-mediated pro-inflammatory signaling. Bardoxolone methyl binds to Keap1 and activates Nrf2, a transcription factor that increases cellular antioxidant and detoxification enzymes and promotes normal mitochondrial function by making reducing equivalents available for ATP production. This reduces mitochondrial ROS production and ROS-mediated activation of inflammatory signaling complexes. Through these effects, bardoxolone methyl restores mitochondrial production of ATP, increases production of antioxidant and detoxification enzymes, reduces oxidative stress, and reduces pro-inflammatory signaling. Bardoxolone methyl reverses endothelial dysfunction and pathogenic mesangial cell contraction, resulting in increased the surface area of the glomerulus and GFR. Additionally, bardoxolone methyl inhibits activation of inflammatory and pro-fibrotic pathways that lead to structural remodeling and glomerulosclerosis.
As a result, bardoxolone methyl and closely related structural analogs have been shown to improve renal function, reduce inflammation, and prevent injury, remodeling, and fibrosis in a number of animal models of renal injury and disease as seen in the table below. Specifically, bardoxolone methyl and analogs reverse endothelial dysfunction and mesangial cell contraction in response to angiotensin II, thereby increasing the surface area of the glomerulus and GFR. Further, data from animal models relevant to chronic renal disease demonstrate that the compounds are anti-fibrotic and have protective effects on the renal interstitium in response to high protein, pressure overload in the setting of hyperfiltration, and dyslipidemia.
18
Bardoxolone Methyl and Analogs are Active in Multiple Animal Models Relevant to Renal Disease
|
Model |
General Findings |
|
Protein overload1
|
-Reduced oxidative/nitrosative stress, interstitial inflammation, and fibrogenic mediators including TGF-β and α-SMA -Decreased proteinuria and tubular damage |
|
Angiotensin II-induced GFR decline2
|
-Increased inulin clearance by increasing Kf without affecting BP or renal plasma flow -Reduced whole glomeruli and mesangial cell contraction due to angiotensin II |
|
5/6 nephrectomy3
|
-Reversed endothelial dysfunction and impaired Nrf2 activity in arterial tissue -Ameliorated hyperfiltration-induced glomerulosclerosis and tubular injury -Suppressed renal inflammation and infiltration of lymphocytes and macrophages -Mitigated systolic blood pressure increase caused by chronic renal failure |
|
12-month monkey safety study4
|
-Improved kidney function for 12 months without adverse renal histological effects -Induced renal Nrf2 targets and suppressed megalin |
|
High-fat (HF) diet-induced CKD5
|
-Prevented HF diet-induced development of structural changes in the heart and kidneys -Prevented HF diet-induced renal corpuscle hypertrophy |
|
Lupus nephritis6
|
-Attenuated renal disease and reduced glomerulonephritis in 3 different models of lupus nephritis -Decreased proteinuria and BUN |
|
Ischemic-reperfusion7
|
-Prevented structural injury from ischemia acute kidney injury -Pre-treatment preserved renal function following ischemic surgery |
|
FeNTA-induced acute kidney injury8 |
-Prevented acute kidney injury and preserved renal function -Reduced severity of proximal tubule degeneration and necrosis |
|
Cisplatin-induced acute kidney injury9
|
-Protected against cisplatin-induced renal toxicity -Reduced proximal tubule degeneration, apoptosis, necrosis, and inflammation |
|
|
1 |
Zoja C, Corna D, Locatelli M, et al. Targeting Keap1-Nrf2 Pathway Ameliorates Renal Inflammation and Fibrosis in Mice with Protein-Overload Proteinuria. Poster American Society of Nephrology Meeting, 2010. |
|
|
2 |
Ding Z, Stidham RD, Bumeister R, Trevino I, Winters A, Sprouse M, Ding M, Ferguson DA, Meyer CJ, Wigley WC, Ma R. The synthetic triterpenoid, RTA 405, increases the glomerular filtration rate and reduces angiotensin II–induced contraction of glomerular mesangial cells. Kidney Intl 2013;83:845-854. |
|
|
3 |
Aminzadeh MA, Reisman SA, Vaziri ND, et al. The synthetic triterpenoid RTA dh404 (CDDO-dhTFEA) restores endothelial function impaired by reduced Nrf2 activity in chronic kidney disease. Redox Biol 2013;1:527-31. |
|
|
4 |
Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ. Bardoxolone Methyl Decreases Megalin and Activates Nrf2 in the Kidney. J Am Soc Nephrol 2012;23(10):1663-1673. |
|
|
5 |
Camer D, Yu Y, Szabo A, et al. Bardoxolone methyl prevents the development and progression of cardiac and renal pathophysiologies in mice fed a high-fed diet. Chem Biol Interact 2016;243:10-18. |
|
|
6 |
Wu T, Ye Y, Min SY, Zhu J, Khobahy E, Zhou J, Yan M, Hemachandran S, Pathak S, Zhou XJ, Andreeff M, Mohan C. Targeting multiple signaling axes and oxidative stress using a synthetic triterpenoid prevents murine lupus nephritis. Arthritis Rheumatol 2014;66(11):3129–3139. |
|
|
7 |
Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, Winterberg P, Chen B, Agarwal A, Lu CY. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARy, and HO-1 Targeting multiple signaling axes and oxidative stress using a synthetic triterpenoid prevents murine lupus nephritis. Am J Physiol Renal Physiol 2011;300:F1180–F1192. |
|
|
8 |
Tanaka Y, Aleksunes LM, Goedken MJ, Chen C, Reisman SA, Manautou JE, Klaassen CD. Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicology and Applied Pharmacology 2008;231:364–373. |
|
|
9 |
Aleksunes L, Reisman SA, Yeager RL, Goedken MJ, Klaassen CD. Nuclear Factor Erythroid 2-Related Factor 2 Deletion Impairs Glucose Tolerance and Exacerbates Hyperglycemia in Type 1 Diabetic Mice. Am Soc Pharmacology and Experimental Therapeutics 2010;333:140-151. |
Regulatory Interaction on Alport Syndrome
During a meeting with the FDA in October 2016, the FDA provided us with guidance on key elements of a single, pivotal clinical trial that would study the safety and efficacy of bardoxolone methyl in patients with CKD caused by Alport syndrome. We initially proposed a Phase 2 trial in Alport patients; however, the FDA suggested a potentially more efficient path to registration utilizing a single trial with estimated GFR, or eGFR, based endpoints.
19
We are in the process of designing the Phase 2/3 pivotal trial. Based on the guidance from the FDA, the trial will be an international, multi-center, double-blind, randomized, placebo-controlled trial. It will study the safety, tolerability, and efficacy of bardoxolone methyl in qualified patients with Alport syndrome from age 12 to 60 with eGFR values between 30 to 90 mL/min/1.73 m2 at up to 60 sites. The Phase 2 portion of the trial will be open-label and the primary endpoint will assess eGFR change in approximately 30 patients at 12 weeks. These patients will be followed for two years and will not be included in the Phase 3 portion of the trial. The Phase 3 portion will be designed to support registration. Up to 180 patients will be randomized 1:1 to either bardoxolone methyl or placebo. The eGFR change at one year will be measured after 48 weeks while the patient is on treatment, and after withdrawal of drug for four weeks (retained eGFR). After withdrawal, patients will be restarted on study drug with their original treatment assignments and will continue on study drug for a second year. The change from baseline in eGFR in bardoxolone methyl-treated patients relative to placebo will be measured again after two years. The eGFR change at two years will also be measured after 100 weeks while the patient is on treatment and after withdrawal of drug for four weeks (retained eGFR). If the trial is successful, the year one retained eGFR data could support accelerated approval under subpart H of the Federal Food, Drug, and Cosmetic Act, or the FD&C Act, and the year two retained eGFR data could support full approval under the FD&C Act.
Clinical Experience with Bardoxolone Methyl in CKD caused by Type 2 Diabetes
In addition to preclinical models of chronic renal disease, bardoxolone methyl has been studied in seven studies of patients with CKD from type 2 diabetes that enrolled approximately 2,600 patients. Reata conducted six Phase 2 studies that demonstrated significant improvements in renal function evidenced by increases in eGFR and creatinine clearance as well as reductions in uremic solutes BUN, uric acid, and phosphate in inverse correlation with eGFR. These studies included the BEAM study, a randomized, placebo-controlled 52-week Phase 2 study in diabetic CKD patients. BEAM enrolled primarily moderate or Stage 3 CKD patients and demonstrated that eGFR improvements were sustained for 52 weeks on treatment and that eGFR at week 56, four weeks after withdrawal of drug, was greater than both baseline and placebo eGFR values. The BEAM data demonstrated that a portion of the on treatment eGFR improvement was retained after withdrawal of drug and suggested that bardoxolone methyl had disease-modifying activity in CKD from type 2 diabetes.
On the basis of the Phase 2 results, Reata conducted BEACON, a large, multinational Phase 3 study in patients with Stage 4 CKD and type 2 diabetes. BEACON was designed to measure time to ESRD, and it enrolled only patients with severe or Stage 4 CKD. During October 2012, BEACON was terminated early in response to the independent data monitoring committee’s recommendation to stop the trial for safety concerns. At the time, nothing was known about the cause or timing of the safety events or whether these events might worsen. After the trial was terminated, analysis revealed that there was a small but significant imbalance in heart failure events of 5.0% on placebo and 8.8% on active drug, and the primary reason for the increase in heart failure events was fluid overload that occurred in the first four weeks after randomization. Patients with fluid overload events who were treated with intravenous diuretics resolved their symptoms, and there was no increase in risk for fluid overload, as compared to placebo, after the first four weeks of treatment.
Further investigation, including additional preclinical studies, indicated that bardoxolone methyl modulates the endothelin pathway. In Stage 4 and 5 CKD patients, the endothelin pathway is dysregulated and, under certain circumstances, activation of this pathway can put these patients at greater risk for fluid retention. Patients who have less compromised renal function do not have dysregulation of this pathway, and their kidneys are able to excrete excess fluid. Similar fluid overload events had been observed previously in late-stage CKD patients treated with other therapies, including endothelin receptor antagonists, or ERAs, which are vasodilating agents that are currently approved for the treatment of PAH. Post-hoc analysis from BEACON identified three major risk factors as predictors of fluid overload events: Stage 4 CKD, prior hospitalization for heart failure, and baseline elevation in B-type natriuretic peptide or BNP, a clinical chemistry measure of fluid overload. Patients without these risk factors showed no imbalance in heart failure events or mortality, which is consistent with the Phase 2 studies conducted by Reata that primarily enrolled Stage 3 CKD patients and did not show a risk of fluid overload.
Recently, Reata’s Asian development partner, KHK, announced interim results from their Phase 2 trial, TSUBAKI, showing that bardoxolone methyl treatment resulted in a significant improvement in measured GFR, as assessed by inulin clearance, after 16 weeks of treatment compared to placebo. Inulin clearance is the most rigorous technique to determine measured GFR. Moreover, the increase in inulin clearance is similar in magnitude to the changes in eGFR reported in other studies with bardoxolone methyl. In addition, in other CKD studies, bardoxolone methyl significantly increased creatinine clearance, significantly reduced uremic solutes (BUN, uric acid, and phosphate) in inverse correlation to eGFR increases, and numerically reduced renal SAEs and ESRD events. These data support that the increases in eGFR observed in seven CKD studies of bardoxolone methyl treatment reflect actual increases in GFR and support the use of eGFR as a reliable marker of renal function. Below is an overview of renal improvements that have been shown in clinical trials with bardoxolone.
20
Overview of Bardoxolone Methyl Studies Demonstrating Improvements in Renal Function
|
Study |
Phase/Country |
Patient Population |
Mean Placebo-corrected ∆eGFR (mL/min/1.73m2)a |
|
402-C-0903 (BEACON) |
3/Global |
CKD/Diabetes |
6.4 (p<0.001 vs PBO)c |
|
402-C-0804 (BEAM) |
2/US |
CKD/Diabetes |
8.6 (p<0.001 vs PBO) |
|
RTA402-005 (TSUBAKI) |
2/Japan |
CKD/Diabetes |
Data not yet publicly disclosed |
|
402-C-0902 |
2/US |
CKD/Diabetes |
6.5 (p<0.001)b |
|
402-C-0801 (Stratum 1) |
2a/US |
CKD/Diabetes |
6.7 (p<0.001)b |
|
402-C-0801 (Stratum 2) |
2b/US |
CKD/Diabetes |
7.2 (p<0.001)b,c |
|
402-C-1102 |
1/US |
CKD/Diabetes |
9.0 (p<0.05)b |
|
402-C-0501 |
1/US |
Cancer |
18.2 (p<0.0001)b |
|
402-C-0702 |
1/2/US |
Cancer |
32.2 (p=0.001)b |
|
402-C-1302 (LARIAT) |
2/US |
Pulmonary hypertension |
14.7 (p<0.001 vs PBO) |
|
|
a |
Unless noted, data are differences between mean eGFR changes from baseline for bardoxolone methyl versus placebo groups and p-values calculated comparing the difference in means between bardoxolone methyl and placebo groups. |
|
|
b |
Data are mean eGFR changes from baseline for bardoxolone methyl patients and p-values are calculated from two-sided paired t-tests comparing eGFR change to 0. |
|
|
c |
Study also demonstrated a significant increase in creatinine clearance. |
In addition to the eGFR increase data, two separate studies, BEAM and BEACON, which included approximately 600 patients treated for one year or longer, showed that increases in eGFR in CKD patients treated with bardoxolone methyl were sustained for at least one year. In BEACON, bardoxolone methyl patients had mean increases in eGFR through Week 48 of 5.5 mL/min/1.73 m2. In contrast, placebo-treated patients experienced a mean decline in eGFR of -0.9 mL/min/1.73 m2, corresponding to a relative difference between groups of 6.4 mL/min/1.73 m2 (p<0.001). The figure below shows the change in eGFR over time for both placebo and bardoxolone methyl patients in the BEACON trial demonstrating a durable response through at least one year.
One Year of Treatment with Bardoxolone Methyl in Stage 4 CKD Patients (BEACON)
Additionally, as shown in the table below, in BEACON, at 48 weeks, bardoxolone methyl significantly reduced the proportion of patients with an eGFR loss of 3, 5, or 7.5 mL/min/1.73 m2, which represents losses of approximately 13%, 22%, and 33%, respectively, of baseline eGFR. The proportion of patients with a loss of eGFR of 30% from baseline at any visit was reduced by 67% (p<0.001).
21
Percentage of Patients with Large Losses of Kidney Function in BEACON
|
Decliner Category |
BARD |
PBO |
p-value |
|
∆eGFR < -3.0 by WK48 |
20% |
67% |
p<0.001 |
|
∆eGFR < -5.0 by WK48 |
14% |
37% |
p<0.001 |
|
∆eGFR < -7.5 by WK48 |
8% |
16% |
p<0.01 |
|
∆eGFR < -30% (any visit) |
4% |
12% |
p<0.001 |
After one year of treatment, a residual eGFR increase from baseline was observed in bardoxolone methyl patients after cessation of drug for four weeks, while an eGFR decline from baseline was observed in placebo patients. These data suggest that the maladaptive structural deficits that contribute to declining kidney function may be improved over the course of longer-term treatment with bardoxolone methyl.
Retained eGFR Benefit After Withdrawal of Bardoxolone Methyl
|
|
Baseline eGFR |
Placebo-Corrected eGFR Change Post-Withdrawal |
p-value |
|
BEAM (n=172) |
|
|
|
|
Low Dose |
33 |
0.6 |
p>0.05 |
|
Mid Dose |
32 |
4.7 |
p<0.05 |
|
High Dose |
32 |
5.0 |
p<0.05 |
|
BEACON (n=498) |
|
|
|
|
20mg |
23 |
1.8 |
p<0.001 |
We believe that the improvements in renal function noted in many studies with bardoxolone methyl coupled with the BEACON data demonstrating reduced risk of loss of kidney function suggest that bardoxolone methyl may have the potential to prevent renal function decline, which may translate to a multi-year delay in disease progression to ESRD for patients with Alport syndrome.
Omaveloxolone
Omaveloxolone is a close structural analog of bardoxolone methyl that was developed to improve tissue distribution, including blood-brain barrier penetration. To date, omaveloxolone has been administered orally to patients with FA, MM, and solid tumors, and has been administered topically to patients receiving cataract surgery and suffering from radiation dermatitis. We believe that an omaveloxolone-induced increase in mitochondrial energy production could have beneficial effects on multiple organ systems, with the most profound effects being in skeletal muscle, the brain and other tissues with a high energy demand.
Omaveloxolone for the Treatment of Friedreich’s Ataxia
We are evaluating omaveloxolone in FA in MOXIe, a randomized, placebo-controlled, double-blind, dose-escalation Phase 2 trial to evaluate the safety and efficacy of omaveloxolone in up to 100 patients with FA at sites in the United States, Europe and Australia. Based on discussions with the FDA in 2014, MOXIe is designed in two parts, with the first primarily focused on the evaluation of safety of omaveloxolone in doses ranging from 2.5 mg to 300 mg and the second focused on efficacy. Data for multiple endpoints are being collected, with the primary efficacy endpoint being the change in peak work, as measured by exercise testing on a recumbent bicycle, and the secondary endpoint being assessment based on the neurological component of the Friedreich’s Ataxia Rating Scale, or FARS. Data from the first part of MOXIe are expected in the first half of 2017, and we expect to initiate the second part of MOXIe after receiving these data.
22
Omaveloxolone for the Treatment of Mitochondrial Myopathies
We are evaluating omaveloxolone in MOTOR, a randomized, placebo-controlled, double-blind, dose-escalation Phase 2 trial to evaluate the safety and efficacy of omaveloxolone in up to 100 patients with MM. MOTOR is being conducted at sites in the United States and Europe. Based on discussions with the FDA in 2014, MOTOR is designed in two parts, with the first primarily focused on the evaluation of safety of omaveloxolone in doses ranging from 2.5 mg to 160 mg and the second primarily focused on efficacy. Data for multiple endpoints are being collected, with the primary efficacy endpoint being the change in peak work, as measured by exercise testing on a recumbent bicycle, and the secondary endpoint being change in patients’ 6MWD. Data from the first part of MOTOR are expected in the first half of 2017, and we expect to initiate the second part of MOTOR after receiving these data.
Omaveloxolone for Immuno-Oncology
We are evaluating omaveloxolone in the REVEAL trial, an open-label, multi-center, dose-escalation Phase 1b/2 trial to evaluate the safety, pharmacodynamics, and efficacy of omaveloxolone, in combination with existing immunotherapies, in patients with metastatic melanoma at sites in the United States. We are using omaveloxolone in combination with checkpoint inhibitors to restore an immune response against the tumor in the presence of so-called myeloid-derived suppressor cells, which mask the tumor from the immune system by production of mitochondrial ROS. In REVEAL, patients receive omaveloxolone monotherapy for one week, followed by omaveloxolone in combination with the labeled treatment course of either Yervoy® or Opdivo ®. Data from REVEAL are expected in the second half of 2017.
Omaveloxolone for the Prevention of the Loss of Corneal Endothelial Cells during Cataract Surgery
We evaluated omaveloxolone in the GUARD trial, a multi-center, randomized, double-masked, vehicle-controlled, parallel group Phase 2 trial conducted at sites in the United States to evaluate a topical ophthalmic formulation of omaveloxolone in 307 patients undergoing cataract surgery. While the primary endpoint, which was loss of corneal endothelial cells, after cataract surgery, was not attained and no treatment effect was observed at the high dose, promising pharmacological activity was seen at the low dose of 0.5%. We continue to evaluate the best indications for which to develop and commercialize omaveloxolone.
Preclinical Programs
Additional AIM Indications
Our AIM pharmacology may provide therapeutic benefit for patients suffering from diseases other than those we are currently testing in clinical trials where inflammation, oxidative stress, mitochondrial dysfunction and tissue remodeling are implicated. We continue to evaluate disease models, preclinical information, and information from our clinical trials to determine where AIMs may be beneficial.
Neuroprotective Hsp90 Inhibitors
We are pursuing preclinical development of non-AIM neuroprotective Hsp90 inhibitors, including RTA 901, for the potential treatment of amyotrophic lateral sclerosis, diabetic neuropathy, spinocerebellar ataxia, and spinal bulbar muscular atrophy. Our Hsp90 inhibitors are highly potent and selective C-terminal inhibitors of Hsp90. Inhibition of Hsp90 may result in activation of Hsp70, a molecular chaperone that plays a critical role in the process through which a protein assumes its functional shape and that serves as a central gatekeeper for mitochondrial protein import. Mitochondria rely on Hsp70-dependent protein import mechanisms for almost all of their activity, including the production of ATP. There are also indications that Hsp70 activation may play a profound role in neuroprotection since nerve cells are high consumers of ATP and rely on Hsp70-dependent protein import for proper mitochondrial function.
In January 2016, the FDA informed us that they were imposing a clinical hold on our IND for RTA 901 pending resolution of some outstanding questions related to our animal toxicology studies, in which no adverse effects were identified. We are collaborating with the FDA to understand and resolve these outstanding questions.
RORγT Inhibitors
We are pursuing preclinical development of novel, small-molecule, orally bioavailable RORγT inhibitors. RORγT is the master regulator of human T Helper 17, or Th17, cellular differentiation, function, and cytokine production, and represents a compelling target for a variety of autoimmune and inflammatory conditions. Th17 cells produce cytokines, including IL-17, that play a critical role in driving immune-mediated inflammation and are implicated in the pathogenesis of certain autoimmune diseases. The efficacy of suppressing IL-17 as a means of treating these conditions has been demonstrated both in animal models and in humans.
23
Revenue
Our revenue to date has been generated primarily from licensing fees received under our collaborative license agreements and reimbursements for expenses. We currently have no approved products and have not generated any revenue from the sale of products to date. In the future, we may generate revenue from product sales, royalties on product sales, reimbursements for collaboration services under our current collaboration agreements, or license fees, milestones, or other upfront payments if we enter into any new collaborations or license agreements. We expect that our future revenue will fluctuate from quarter to quarter for many reasons, including the uncertain timing and amount of any such payments and sales.
Our license and milestone revenue has been generated primarily from our collaborative licensing agreements with AbbVie and KHK and consists of upfront payments and milestone payments. Under our revenue recognition policy, license revenue associated with upfront, non-refundable license payments received under the collaboration agreements with AbbVie and KHK are recognized ratably over the expected term of the performance obligations under the agreements, which extend through various periods beginning in 2017 and ending in 2026. License revenue recorded with respect to the collaboration agreements with AbbVie consists solely of the recognition of deferred revenue. License revenue recorded with respect to the collaboration agreements with KHK consists of the recognition of deferred revenue and reimbursement of supply costs.
We also have other license revenue, which consists of milestone payments from a disease advocacy organization in 2015, and other revenue, which consists of reimbursements from KHK for expenses incurred to obtain drug supplies.
Research and Development Expenses
The largest component of our total operating expenses has historically been our investment in research and development activities, including the clinical development of our product candidates. From our inception through September 30, 2016, we have incurred a total of $466.4 million in research and development expense, a majority of which relates to the development of bardoxolone methyl and omaveloxolone. We expect our research and development expense to continue to increase in the future as we advance our product candidates through clinical trials and expand our product candidate portfolio. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming and we consider the active management and development of our clinical pipeline to be crucial to our long-term success. The actual probability of success for each product candidate and preclinical program may be affected by a variety of factors, including the safety and efficacy data for product candidates, investment in the program, competition, manufacturing capability, and commercial viability.
Research and development expenses include:
|
|
• |
expenses incurred under agreements with clinical trial sites that conduct research and development activities on our behalf; |
|
|
• |
expenses incurred under contract research agreements and other agreements with third parties; |
|
|
• |
employee and consultant-related expenses, which include salaries, benefits, travel, and stock-based compensation; |
|
|
• |
laboratory and vendor expenses related to the execution of preclinical and non-clinical studies, and clinical trials; |
|
|
• |
the cost of acquiring, developing, manufacturing, and distributing clinical trial materials; and |
|
|
• |
facilities, depreciation, and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance, and other supply costs. |
Research and development costs are expensed as incurred. Costs for certain development activities such as clinical trials are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
We base our expense accruals related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and contract research organizations, or CROs, that conduct and manage clinical trials on our behalf. The financial terms of these agreements vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing costs, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
24
To date, we have not experienced significant changes in our estimates of accrued research and development expenses after a reporting period. However, due to the nature of estimates, we cannot assure you that we will not make changes to our estimates in the future as we become aware of additional information about the status or conduct of our clinical trials and other research activities.
Currently, AbbVie is not participating in the development of bardoxolone methyl for the treatment of PH and we are therefore incurring all costs for this program. With respect to our omaveloxolone programs and our collaboration agreement with AbbVie, we were responsible for a certain initial amount in early development costs before AbbVie began sharing development costs equally. In April 2016, we had incurred all of these initial costs, after which payments from AbbVie with respect to research and development costs incurred by us were recorded as a reduction in research and development expenses. Our expenses were reduced by $0.8 million for AbbVie’s share of research and development costs for the three months ended September 30, 2016 and we had related receivables from AbbVie in the amount of $1.4 million as of September 30, 2016. We received $0.6 million from AbbVie in October 2016.
In September 2016, we and AbbVie mutually agreed that we would continue unilateral development of omaveloxolone. Therefore, AbbVie no longer co-funds the exploratory development costs of this program, but retains the right to opt back in at certain points in development. Depending upon what point, if any, AbbVie opts back into development, AbbVie may retain its right to commercialize a product outside the U.S. or we may be responsible for commercializing the product on a worldwide basis. Upon opting back in, AbbVie would be required to pay an agreed upon amount of all development costs accumulated up to the point of exercising their opt-in right, after which development costs incurred and product revenue worldwide would be split equally.
The following table summarizes our research and development expenses incurred:
|
|
|
Three Months Ended September 30, |
|
|
Nine Months Ended September 30, |
|
||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
||||
|
|
|
(unaudited) |
|
|||||||||||||
|
|
|
(in thousands) |
|
|||||||||||||
|
Bardoxolone methyl |
|
$ |
4,508 |
|
|
$ |
1,565 |
|
|
$ |
11,725 |
|
|
$ |
4,107 |
|
|
Omaveloxolone |
|
|
725 |
|
|
|
2,329 |
|
|
|
3,539 |
|
|
|
9,651 |
|
|
RTA 901 |
|
|
303 |
|
|
|
1,978 |
|
|
|
1,880 |
|
|
|
4,473 |
|
|
Other research and development expenses |
|
|
3,764 |
|
|
|
2,678 |
|
|
|
10,537 |
|
|
|
8,585 |
|
|
Total research and development expenses |
|
$ |
9,300 |
|
|
$ |
8,550 |
|
|
$ |
27,681 |
|
|
$ |
26,816 |
|
The program-specific expenses summarized in the table above include costs that we directly allocate to our product candidates. Our other research and development expenses include research and development salaries, benefits, stock-based compensation and preclinical, research, and discovery costs, which we do not allocate on a program-specific basis.
General and Administrative Expenses
General and administrative expenses consist primarily of employee-related expenses for executive, operational, finance, legal, compliance, and human resource functions. Other general and administrative expenses include personnel expense, facility-related costs, professional fees, accounting and legal services, depreciation expense, other external services, and expenses associated with obtaining and maintaining our intellectual property rights.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development and potential commercialization of our product candidates. We have also incurred increased expenses associated with being a public company, including exchange listing and Securities and Exchange Commission requirements, director and officer insurance premium, legal, audit and tax fees, regulatory compliance programs, and investor relations costs. Additionally, if and when we believe the first regulatory approval of one of our product candidates appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially for the sales and marketing of our product candidates.
Other Income
Other income represents interest and gains earned on our cash and cash equivalents, which include money market funds.
25
Provision for taxes on income consists of net loss, taxed at federal tax rates and adjusted for certain permanent differences. We maintain a valuation allowance against the majority of our net deferred tax assets. Changes in this valuation allowance also affect the tax provision.
Results of Operations
Comparison of the three months ended September 30, 2016 and 2015 (unaudited)
The following table sets forth our results of operations for the three months ended September 30:
|
|
|
2016 |
|
|
2015 |
|
|
Change $ |
|
|
Change % |
|
||||
|
|
|
(unaudited) |
|
|||||||||||||
|
|
|
(in thousands, except percentage data) |
|
|||||||||||||
|
Consolidated Statements of Operations Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
License and milestone |
|
$ |
12,500 |
|
|
$ |
12,500 |
|
|
|
— |
|
|
|
— |
|
|
Other revenue |
|
|
51 |
|
|
|
— |
|
|
|
51 |
|
|
|
100 |
|
|
Total collaboration revenue |
|
|
12,551 |
|
|
|
12,500 |
|
|
|
51 |
|
|
|
— |
|
|
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
9,300 |
|
|
|
8,550 |
|
|
|
750 |
|
|
|
9 |
|
|
General and administrative |
|
|
4,039 |
|
|
|
2,980 |
|
|
|
1,059 |
|
|
|
36 |
|
|
Depreciation and amortization |
|
|
170 |
|
|
|
486 |
|
|
|
(316 |
) |
|
|
(65 |
) |
|
Total expenses |
|
|
13,509 |
|
|
|
12,016 |
|
|
|
1,493 |
|
|
|
12 |
|
|
Total other income |
|
|
62 |
|
|
|
9 |
|
|
|
53 |
|
|
|
589 |
|
|
(Loss) income before provision (benefit) for taxes on income |
|
|
(896 |
) |
|
|
493 |
|
|
|
(1,389 |
) |
|
|
(282 |
) |
|
Provision (benefit) for taxes on income |
|
|
1 |
|
|
|
(140 |
) |
|
|
141 |
|
|
|
101 |
|
|
Net loss |
|
$ |
(897 |
) |
|
$ |
633 |
|
|
|
(1,530 |
) |
|
|
(242 |
) |
Revenue
Revenue is consistent for the three months ended September 30, 2016, compared to the three months ended September 30, 2015, and primarily consists of the recognition of deferred revenue.
The following table summarizes the sources of our revenue for the three months ended September 30:
|
|
|
2016 |
|
|
2015 |
|
||
|
|
|
(unaudited) |
|
|||||
|
|
|
(in thousands) |
|
|||||
|
License and milestone |
|
|
|
|
|
|
|
|
|
AbbVie license agreement |
|
$ |
5,396 |
|
|
$ |
5,396 |
|
|
AbbVie collaboration agreement |
|
|
6,717 |
|
|
|
6,717 |
|
|
KHK agreement |
|
|
387 |
|
|
|
387 |
|
|
Total license and milestone |
|
|
12,500 |
|
|
|
12,500 |
|
|
Other revenue |
|
|
51 |
|
|
|
— |
|
|
Total collaboration revenue |
|
$ |
12,551 |
|
|
$ |
12,500 |
|
Research and Development Expenses
Research and development expenses increased by $0.8 million, or 9%, for the three months ended September 30, 2016, compared to the three months ended September 30, 2015. The increase was primarily due to increases of $2.9 million in clinical activities for bardoxolone methyl in PAH and PH-ILD, $0.4 million in non-clinical research development activities, and $0.7 million in personnel expense to support growth in our development activities, offset by decreases of $1.6 million related to clinical activities for omaveloxolone related to completion of clinical activities on our topical programs in 2015 and AbbVie’s share of development cost related to the omaveloxolone program in 2016 and $1.6 million related to IND directed preclinical activities for RTA 901 in 2015 compared to primarily toxicology work to resolve outstanding questions on the IND in 2016.
26
General and Administrative Expenses
General and administrative expenses increased by $1.1 million, or 36%, for the three months ended September 30, 2016, compared to the three months ended September 30, 2015. The increase was primarily due to increased insurance coverage and legal and accounting expenses in connection with being a public company and to expenses related to our overall growth in our development activities, including increases in commercial research consulting activities and personnel expenses.
Investment Income
Investment income was immaterial for the three months ended September 30, 2016 and 2015.
Provision (Benefit) for Taxes on Income
Provision (benefit) for taxes on income decreased by $0.1 million, or 101%, for the three months ended September 30, 2016, compared to the three months ended September 30, 2015, due to differences in income generated and changes in the valuation allowance.
Comparison of the nine months ended September 30, 2016 and 2015
The following table sets forth our results of operations for the nine months ended September 30:
|
|
|
2016 |
|
|
2015 |
|
|
Change $ |
|
|
Change % |
|
||||
|
|
|
(unaudited) |
|
|||||||||||||
|
|
|
(in thousands, except percentage data) |
|
|||||||||||||
|
Consolidated Statements of Operations Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
License and milestone |
|
$ |
37,230 |
|
|
$ |
37,794 |
|
|
|
(564 |
) |
|
|
(1 |
) |
|
Other revenue |
|
|
125 |
|
|
|
— |
|
|
|
125 |
|
|
|
100 |
|
|
Total collaboration revenue |
|
|
37,355 |
|
|
|
37,794 |
|
|
|
(439 |
) |
|
|
(1 |
) |
|
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
27,681 |
|
|
|
26,816 |
|
|
|
865 |
|
|
|
3 |
|
|
General and administrative |
|
|
11,783 |
|
|
|
9,203 |
|
|
|
2,580 |
|
|
|
28 |
|
|
Depreciation and amortization |
|
|
537 |
|
|
|
1,548 |
|
|
|
(1,011 |
) |
|
|
(65 |
) |
|
Total expenses |
|
|
40,001 |
|
|
|
37,567 |
|
|
|
2,434 |
|
|
|
6 |
|
|
Total other income |
|
|
113 |
|
|
|
25 |
|
|
|
88 |
|
|
|
352 |
|
|
(Loss) income before provision (benefit) for taxes on income |
|
|
(2,533 |
) |
|
|
252 |
|
|
|
(2,785 |
) |
|
|
(1,105 |
) |
|
(Benefit) provision for taxes on income |
|
|
(442 |
) |
|
|
(44 |
) |
|
|
(398 |
) |
|
|
(905 |
) |
|
Net loss |
|
$ |
(2,091 |
) |
|
$ |
296 |
|
|
|
(2,387 |
) |
|
|
(806 |
) |
Revenue
Revenue decreased by $0.4 million, or 1%, for the nine months ended September 30, 2016, compared to the nine months ended September 30, 2015. The decrease was primarily due to a decrease in milestone revenue from a disease advocacy organization received in 2015.
27
The following table summarizes the sources of our revenue for the nine months ended September 30:
|
|
|
2016 |
|
|
2015 |
|
||
|
|
|
(in thousands) |
|
|||||
|
License and milestone |
|
|
|
|
|
|
|
|
|
AbbVie license agreement |
|
$ |
16,073 |
|
|
$ |
16,015 |
|
|
AbbVie collaboration agreement |
|
|
20,004 |
|
|
|
19,931 |
|
|
KHK agreement |
|
|
1,153 |
|
|
|
1,148 |
|
|
Other revenue |
|
|
— |
|
|
|
700 |
|
|
Total license and milestone |
|
$ |
37,230 |
|
|
$ |
37,794 |
|
|
Other revenue |
|
|
125 |
|
|
|
— |
|
|
Total collaboration revenue |
|
$ |
37,355 |
|
|
$ |
37,794 |
|
Research and Development Expenses
Research and development expenses increased by $0.9 million, or 3%, for the nine months ended September 30, 2016, compared to the nine months ended September 30, 2015. The increase was primarily due to increases of $7.6 million in clinical activities for bardoxolone methyl in PAH and PH-ILD, $0.4 million in non-clinical research development activities, and $1.6 million in personnel expense to support growth in our development activities, offset by decreases of $6.1 million related to clinical activities for omaveloxolone related to completion of clinical activities on our topical programs in 2015 and AbbVie’s share of development cost related to the omaveloxolone program in 2016 and $2.6 million related to IND directed preclinical activities for RTA 901 in 2015 compared to primarily toxicology work to resolve outstanding questions on the IND in 2016.
General and Administrative Expenses
General and administrative expenses increased by $2.6 million, or 28%, for the nine months ended September 30, 2016, compared to the nine months ended September 30, 2015. The increase was primarily due to increased insurance coverage and legal and accounting expenses in connection with being a public company and to expenses related to our overall growth in our development activities, including increases in commercial research and investor relations consulting activities and personnel expenses.
Investment Income
Investment income was immaterial for the nine months ended September 30, 2016 and 2015.
(Benefit) Provision for Taxes on Income
(Benefit) provision for taxes on income increased by $0.4 million, or 905%, for the nine months ended September 30, 2016, compared to the nine months ended September 30, 2015, due to differences in income generated and changes in the valuation allowance.
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through the sale of preferred and common stock and collaboration and license agreements. To date, we have raised gross cash proceeds of $476.6 million through the sale of convertible preferred stock and received $750 million from payments under license and collaboration agreements. We also obtained $60.9 million in net proceeds from our initial public offering of our Class A common stock. We have not generated any revenue from the sale of any products. As of September 30, 2016, we had available cash and cash equivalents of approximately $95.7 million. Our cash and cash equivalents are invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation.
28
The following table sets forth the primary sources and uses of cash for each of the nine months ended September 30 set forth below:
|
|
|
2016 |
|
|
2015 |
|
||
|
|
|
(unaudited) |
|
|||||
|
|
|
(in thousands) |
|
|||||
|
Net cash (used in) provided by: |
|
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(8,123 |
) |
|
$ |
(32,077 |
) |
|
Investing activities |
|
|
(281 |
) |
|
|
(257 |
) |
|
Financing activities |
|
|
62,056 |
|
|
|
(373 |
) |
|
Net change in cash and cash equivalents |
|
$ |
53,652 |
|
|
$ |
(32,707 |
) |
Operating Activities
Net cash used in operating activities was $8.1 million for the nine months ended September 30, 2016, consisting primarily of net loss of $2.1 million adjusted for non-cash items including stock-based compensation expense of $1.5 million, depreciation expense of $0.5 million, and a net decrease in operating assets and liabilities of $8.0 million. The significant items in the change in operating assets and liabilities include an increase of prepaid expenses and other current assets of $2.9 million due to clinical trial prepayments and reimbursements due from KHK and AbbVie, a decrease in income tax receivable of $31.9 million due to tax refunds received, and a decrease in deferred revenue of $37.2 million. The decrease in deferred revenue relates to the timing of upfront payments and ratable recognition of revenue over the expected term of the performance obligations under our collaboration agreements with AbbVie and KHK, resulting in recognition of $37.2 million of license and milestone revenue.
Net cash used in operating activities was $32.1 million for the nine months ended September 30, 2015, consisting primarily of net income of $0.3 million adjusted for non-cash items including stock-based compensation expense of $1.0 million, depreciation expense of $1.5 million, decrease in provision for deferred taxes of $13.4 million, and net decrease in operating assets and liabilities of $48.3 million. The significant items in the change in operating assets and liabilities include an increase in accrued direct research and other current liabilities of $1.6 million due clinical trial activities, an increase in income tax receivable of $13.5 million due to carrying back 2015 losses to realize tax benefits, and a decrease in deferred revenue of $37.1 million. The decrease in deferred revenue relates to the timing of upfront payments and ratable recognition of revenue over the expected term of the performance obligations under our collaboration agreements with AbbVie and KHK, resulting in recognition of $37.1 million of license and milestone revenue.
Investing Activities
Net cash used in investing activities consisted of purchases and sales of property and equipment. Net cash used in investing activities for the nine months ended September 30, 2016 and 2015 was not significant.
Financing Activities
Net cash provided by financing activities was $62.1 million, primarily due to net proceeds of $62.2 million from the close of our initial public offering for the nine months ended September 30, 2016. Net cash used in financing activities for the nine months ended September 30, 2015 was not significant.
Operating Capital Requirements
To date, we have not generated any revenue from product sales. We do not know when or whether we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize one or more of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are subject to all the risks related to the development and commercialization of novel therapeutics, and we may encounter unforeseen expenses, difficulties, complications, delays, and other unknown factors that may adversely affect our business. We expect to incur additional costs associated with operating as a public company. We anticipate that we will need substantial additional funding in connection with our continuing operations.
29
We believe our existing cash and cash equivalents will be sufficient to enable us to fund our existing operating expenses from ongoing clinical trials and preclinical programs and capital expenditures through mid-2018. Our longer term liquidity requirements will require us to raise additional capital, such as through additional equity or debt financings. Our future capital requirements will depend on many factors, including the receipt of milestones under our current collaboration agreements and the timing of our expenditures related to existing and future clinical trials.
In addition, we may require additional capital in the shorter term for the further development of our existing product candidates and may also need to raise additional funds in the shorter term to pursue other development activities related to additional product candidates.
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance future cash needs through public or private equity or debt offerings. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back, or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional equity or debt securities, it could result in dilution to our existing stockholders or increased fixed payment obligations, and any such securities may have rights senior to those of our common stock. If we incur indebtedness, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely affect our ability to conduct our business. Any of these events could significantly harm our business, financial condition, and prospects.
Our forecast of the period through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
|
|
• |
the scope, rate of progress, results and cost of our clinical trials, preclinical testing, and other activities related to the development of our product candidates; |
|
|
• |
the number and characteristics of product candidates that we pursue; |
|
|
• |
the costs of development efforts for our product candidates that are not subject to reimbursement from our collaborators; |
|
|
• |
the costs necessary to obtain regulatory approvals, if any, for our product candidates in the United States and other jurisdictions, and the costs of post-marketing studies that could be required by regulatory authorities in jurisdictions where approval is obtained; |
|
|
• |
the continuation of our existing collaborations and entry into new collaborations and the receipt of any collaboration payments; |
|
|
• |
the time and unreimbursed costs necessary to commercialize products in territories in which our product candidates are approved for sale; |
|
|
• |
the revenue from any future sales of our products for which we are entitled to a profit share, royalties and milestones; |
|
|
• |
the level of reimbursement or third-party payor pricing available to our products; |
|
|
• |
the costs of obtaining third-party commercial supplies of our products, if any, manufactured in accordance with regulatory requirements; |
|
|
• |
the costs associated with being a public company; and |
|
|
• |
the costs we incur in the filing, prosecution, maintenance, and defense of our extensive patent portfolio and other intellectual property rights. |
If we cannot expand our operations or otherwise capitalize on our business opportunities because we lack sufficient capital, our business, financial condition, and results of operations could be materially adversely affected.
30
Contractual Obligations and Commitments
Contractual Obligations
As of September 30, 2016, our contractual obligations were as follows:
|
|
|
Payments due by period |
|
|||||||||
|
|
|
Less than 1 year |
|
|
1 to 3 years |
|
|
Total |
|
|||
|
|
|
(in thousands) |
|
|||||||||
|
Operating lease obligations |
|
$ |
592 |
|
|
$ |
660 |
|
|
$ |
1,252 |
|
|
Capital lease obligations |
|
|
45 |
|
|
|
— |
|
|
|
45 |
|
|
Total contractual obligations |
|
$ |
637 |
|
|
$ |
660 |
|
|
$ |
1,297 |
|
Clinical Trials
As of September 30, 2016, we have several on-going clinical trials in various stages. Under agreements with various CROs and clinical trial sites, we incur expenses related to clinical trials of our product candidates and potential other clinical candidates. The timing and amounts of these disbursements are contingent upon the achievement of certain milestones, patient enrollment, and services rendered or as expenses are incurred by the CROs or clinical trial sites. Therefore, we cannot estimate the potential timing and amount of these payments and they have been excluded from the table above.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, accrued research and development expenses, income taxes, and stock-based compensation. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. There have been no significant and material changes in our critical accounting policies during the nine months ended September 30, 2016, as compared to those disclosed in “Management’s Discussion and Analysis of Financial Condition and Results of Operations-Critical Accounting Policies and Significant Judgments and Estimates” in our Final Prospectus.
Off-Balance Sheet Arrangements
Since our inception, we have not had any relationships with unconsolidated organizations or financial partnerships, such as structured finance or special purpose entities that would have been established for the purpose of facilitating off-balance sheet arrangements, and we have not engaged in any other off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Recent Accounting Pronouncements
We are an “emerging growth company,” as defined in the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as public companies that are not emerging growth companies.
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842), which supersedes the leases in ASC 840, Leases. This ASU requires the recognition of lease assets and lease liabilities by lessees for those leases previously classified as operating leases. The ASU is effective for public companies for fiscal years, and for interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted. We will apply the guidance and disclosure provisions of the new standard upon adoption.
31
In March 2016, the FASB issued ASU No. 2016-09, Compensation–Stock Compensation: Improvements to Employee Share-Based Payment Accounting, to simplify several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. The ASU is effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2016. Early adoption is permitted. We will apply the guidance and disclosure provisions of the new standard upon adoption.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments, This update addresses eight specific cash flow issues with the objective of reducing the existing diversity in practice. The ASU is effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017. The Company is currently evaluating this standard and has not yet determined what, if any, effect ASU No. 2016-09 will have on its consolidated results of operations or financial position.
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations. We had cash and cash equivalents of $95.7 million at September 30, 2016, consisting primarily of funds in operating cash accounts. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate 1.0% increase in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect a sudden change in market interest rates to materially affect our operating results or cash flows.
We contract with research organizations and investigational sites globally. Generally, these contracts are denominated in U.S. dollars. However, we may be subject to fluctuations in foreign currency rates in connection with agreements not denominated in U.S. dollars. We do not hedge our foreign currency exchange rate risk. We do not expect a sudden change in foreign exchange rates to materially affect our operating results or cash flows.
Item 4. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of September 30, 2016. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of September 30, 2016, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the three months ended September 30, 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
32
We are not currently subject to any material legal proceedings.
In addition to the other information set forth in this Quarterly Report on Form 10-Q, you should carefully consider the risk factors and other cautionary statements described under the heading “Risk Factors” included in our Final Prospectus, which could materially affect our businesses, financial condition, or future results. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition, or future results. There has been no material changes in our risk factors from those described in the Final Prospectus.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
Unregistered Sales of Equity Securities
None.
Use of Proceeds from Initial Public Offering of Class A Common Stock
On May 25, 2016, our registration statement on Form S-1 (File No. 333-208843) relating to our initial public offering, or IPO, of our Class A common stock was declared effective by the SEC. The shares began trading on The NASDAQ Global Market on May 26, 2016. The public offering price of the shares sold in the offering was $11.00 per share. The IPO closed on June 1, 2016 and included 6,325,000 shares of Class A common stock, which included 825,000 shares of Class A common stock issued pursuant to the over-allotment option granted to the underwriters, for gross proceeds of approximately $69.6 million before deducting underwriters’ discounts and commissions and offering-related expenses. Net proceeds, after deducting underwriting discounts and commissions of $4.9 million and offering expenses of approximately $3.8 million, were $60.9 million. Citigroup Global Markets Inc., Cowen and Company, LLC, and Piper Jaffray & Co. acted as joint book-running managers of this offering.
There has been no material change in the planned use of proceeds from our IPO as described in our prospectus dated May 25, 2016, filed with the SEC pursuant to Rule 424(b)(4) of the Securities Act. We invested the funds received in highly liquid money market funds. The net proceeds from the IPO have been used and will be used, together with our cash and cash equivalents, to fund continued advancement of our bardoxolone methyl, omaveloxolone, and clinical trials and preclinical studies, and to provide funds for working capital and other general purposes. None of the offering proceeds were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning 10.0% or more of any class of our equity securities or to any other affiliates.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
None.
None.
33
|
Exhibit Number |
|
Description |
|
|
|
|
|
3.1 |
|
Thirteenth Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.7 to the Registrant’s Registration Statement on Form S-1, File No. 333-208803, filed with the Commission on May 16, 2016). |
|
|
|
|
|
3.2 |
|
Amended and Restated Bylaws (incorporated by reference to Exhibit 3.4 to the Registrant’s Registration Statement on Form S-1, File No. 333-208843, filed with the Commission on February 8, 2016). |
|
|
|
|
|
10.1+* |
|
Indemnification Agreement by and between the Registrant and Dawn C. Bir dated September 6, 2016. |
|
|
|
|
|
10.2+* |
|
Employment Agreement by and between the Registrant and Jason D. Wilson dated September 23, 2015. |
|
|
|
|
|
10.3+* |
|
Employment Agreement by and between the Registrant and Michael D. Wortley dated September 23, 2015. |
|
|
|
|
|
10.4+* |
|
Employment Agreement by and between the Registrant and Dawn C. Bir dated September 6, 2016. |
|
|
|
|
|
31.1* |
|
Certification of Principal Executive Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
31.2* |
|
Certification of Principal Financial Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
32.1* |
|
Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
32.2* |
|
Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
101.INS |
|
XBRL Instance Document |
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
101.PRE |
|
XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
|
+ |
Indicates management contract or compensatory plan. |
|
* |
Filed herewith. |
34
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Date: November 14, 2016 |
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ J. Warren Huff |
|
|
Name: |
|
J. Warren Huff |
|
|
Title: |
|
Chief Executive Officer and President |
35