Attached files
| file | filename |
|---|---|
| 8-K - FORM 8-K - Armata Pharmaceuticals, Inc. | v448196_8k.htm |
Exhibit 99.1
Company Overview
We are a biotechnology company focused on the discovery, development and commercialization of novel phage therapeutics. Phage therapeutics use bacteriophages, a family of viruses, to kill pathogenic bacteria. Phages have powerful and highly selective mechanisms of action that permit them to target and kill specific bacteria. We believe that phages represent a promising means to treat bacterial infections, especially those that have developed resistance to current therapies, including the so-called multi-drug-resistant or “superbug” strains of bacteria.
Our goal is to be the leading developer of phage therapeutics. We are combining our expertise in the manufacture of drug-quality bacteriophages and our proprietary approach and expertise in identifying, characterizing and developing naturally occurring bacteriophages with that of our collaboration partners in bacteriophage biology, synthetic biology and manufacturing, to develop second-generation bacteriophage products.
The extensive use of antibiotics since their discovery in the 1940s has resulted in drug resistance among many disease-causing bacteria. According to the U.S. Centers for Disease Control and Prevention, or CDC, resistance to antibiotics threatens to reverse many of the key medical advances of the last half-century. Examples of clinically important microbes that are rapidly developing resistance to available antimicrobials include bacteria that cause skin, bone, lung and bloodstream infections (e.g., S. aureus and methicillin-resistant S. aureus, or MRSA), pneumonia and lung infections in both community and hospital settings and cystic fibrosis patients (e.g., A. baumanii, P. aeruginosa, and K. pneumoniae), meningitis (e.g., S. pneumonia), urinary tract and gastrointestinal infections (e.g., E. coli and C. difficile). As phages kill bacteria in ways entirely unlike the mechanisms used by traditional antibiotics, we believe that multi-drug resistant bacteria will be susceptible to phage therapy. Furthermore, should resistant bacteria emerge or evolve, we believe it will remain possible to identify phages that can effectively kill these resistant bacteria.
Our lead product candidate is AB-SA01, for the treatment of S. aureus infections, including MRSA. We also have another product candidate in earlier stage development, AB-PA01 for the treatment of P. aeruginosa infections, and an additional discovery program, AB-CD01 for the treatment of C. difficile infections.
We are developing our phage product candidates using a proprietary discovery and development platform, which is designed for rapid identification, characterization and manufacturing of multiple phage therapeutics. Each product candidate combines several carefully chosen phages, which target a specific disease-causing bacteria such as S. aureus, P. aeruginosa, and C. difficile. We believe that the combination of our platform, our manufacturing capability, our understanding of the regulatory and development requirements of bacteriophage therapeutics, and the clinical and scientific expertise of our collaboration partners may enable the rapid advancement of phage therapeutics through the clinic and the regulatory approval process.
In June 2013, we entered into a cooperative research and development agreement, or Research and Development Agreement, with the United States Army Medical Research and Materiel Command focusing on developing bacteriophage therapeutics to treat S. aureus, E. coli and P. aeruginosa infections. Under this Research and Development Agreement, we completed enrollment of a Phase 1 safety study of AB-SA01 for the treatment of wounds infected with S. aureus in July 2016. We expect to report top-line results by the end of the third quarter of 2016, with the complete study report expected by the end of 2016.
In September 2013, we entered into a license agreement, or the Leicester License Agreement, with the University of Leicester to develop a phage therapy to kill certain types of C. difficile. Pursuant to the Leicester License Agreement, we may be obligated to pay the University of Leicester a single digit royalty and an aggregate of up to £575,000 in milestone payments.
In November 2015, our Australian subsidiary, AmpliPhi Australia Pty Ltd, entered into a clinical trial research agreement with the University of Adelaide and the Queen Elizabeth Hospital, both of Adelaide, SA, Australia, to conduct a Phase 1 clinical trial titled “A Phase 1 Investigator Initiated Study to Evaluate the Safety, Tolerability and Preliminary Effectiveness of AB-SA01 in Patients with Chronic Rhinosinusitis Associated with S. aureus infection”. The University of Adelaide will sponsor the clinical trial while we will supply AB-SA01 and control the trial protocol. This clinical trial will primarily measure the safety and tolerability of AB-SA01 and will secondarily examine the presence of S. aureus and symptoms assessed by the patient as well as by the physician using standard questionnaires used by physicians to assess treatment efficacy. We plan to enroll nine patients, divided into three cohorts. The first cohort received a twice daily dose of AB-SA01 for seven days. The second cohort received the same dose twice daily for 14 days. The third cohort will receive a higher dose of AB-SA01 twice daily for 14 days. Patients will be monitored an additional 30 days following their last day of treatment. Patient screening for this clinical trial commenced in late 2015 and the first patient was dosed in January 2016. The first and second cohorts have been completed and the first subject in the third cohort has completed dosing. Two subjects remain to be dosed in the final cohort and we expect to report data from this first clinical trial in the second half of 2016. We are planning a Phase 2 trial in chronic rhinosinusitis patients, to commence in the second half of 2017.
In January 2016, we entered into an Asset Purchase Agreement with Novolytics Ltd., which we refer to as the Novolytics Purchase Agreement, to purchase certain tangible and intangible assets. Pursuant to the Novolytics Purchase Agreement, we acquired all rights, title and interest to two families of patents. The first patent family is titled “Anti-bacterial compositions” and has been granted in Australia and China with prosecution pending in the United States and other countries. The second patent family is titled “Novel bacteriophages” and the prosecution is pending in the United States and other countries. We also received clinical isolates for S. aureus which will bolster our libraries of clinically relevant strains. Additionally, we received know-how relating to certain formulation processes. We also have access to all previous dialogue between Novolytics and various regulatory organizations including the United Kingdom Medicines and Healthcare Products Regulatory Agency, or MHRA.
The Need for New Anti-Infective Therapies
The rapid and continuous emergence of antibiotic-resistant bacteria has become a global crisis. Despite this crisis, the number of novel anti-infective therapies currently in development is at historically-low levels. The CDC estimates that more than two million people in the United States acquire an antibiotic-resistant infection each year and more than 23,000 of these prove fatal. It is estimated that 50% of hospital-acquired infections are resistant to first-line anti-infective therapies. The cumulative annual cost for treating resistant bacterial infections in the United States alone is estimated to be $20 billion, while the global antibiotics market opportunity was estimated to be $40.3 billion in 2015.
The CDC’s latest report on the matter, Antibiotic Resistance Threats in the United States, 2013, notes that there are “potentially catastrophic consequences of inaction” and ranks C. difficile as belonging to the highest tier of threat, or “Urgent Threats.” Despite the potential market opportunity, only two New Drug Applications, or NDAs, for antibacterial drugs were approved by the FDA between 2010 and 2012 compared to 18 in the period between 1980 and 1984. One of the primary recommendations of the CDC is the development of new antimicrobials to diversify treatment options.
Product Candidates
AB-SA01: Infections Caused by S. aureus
By screening our proprietary library of phage samples against a panel of S. aureus bacteria, collected from around the world, we have selected a phage product candidate mix that has demonstrated, in in vitro studies, greater than 92% efficacy with high overlap against a global diversity panel that includes some of the most virulent isolates of S. aureus, including MRSA isolates. The three phage constituents of AB-SA01 were selected for their ability to target the greatest number of bacterial isolates in the collection and maximal complementation. Complementation, defined as the percentage of S. aureus isolates susceptible to more than one phage, is emphasized in product selection to reduce risk of the emergence of bacterial resistance.
In connection with our Research and Development Agreement with the U.S. Army Medical Research and Materiel Command, we are developing AB-SA01 to treat acute and chronic infections caused by S. aureus, including infections caused by MRSA strains of the same bacterium. MRSA infections are one of the most common causes of hospital-acquired (nosocomial) infections. The CDC estimates that more than 850,000 patients were treated for S. aureus infections of the skin or soft tissue in 2013 and, due to failure of first line treatment, more than 50% of these patients required a second-line treatment and approximately 35% of them required a third-line treatment. Global Data estimates the market for MRSA infection treatments alone was more than $2.7 billion in 2007. This market is forecasted to grow to more than $3.5 billion by 2019.
Also in connection with our Research and Development Agreement with the U.S. Army, we submitted a pre-IND briefing package to the FDA to obtain their feedback on our Chemistry, Manufacturing and Controls, or CMC, program and plans for our first human clinical trial of AB-SA01 for the treatment of S. aureus infections of wound and skin. The FDA concurred with our plan for progressing this bacteriophage product candidate into clinical trials, specifically agreeing with the proposed manufacturing process and product specifications and not requiring non-clinical toxicology data to initiate our first Phase 1 clinical trial. We initiated the Phase 1 clinical trial in May 2016 and completed enrollment in July 2016. We expect the complete study report to be available before the end of 2016.
In December 2015, we opened a clinical trial at the University of Adelaide Queen Elizabeth Hospital to evaluate the safety and preliminary efficacy of AB-SA01 in chronic rhinosinusitis patients infected with S. aureus. The first patient in this clinical trial was dosed in January 2016, and we have continued to dose additional patients and completed the first two cohorts in July 2016. We expect to complete enrollment of the last two patients during the third quarter of 2016 and report data from this first clinical trial by the end of 2016. We expect to initiate a Phase 2 trial of AB-SA01 in the second half of 2017 and to complete that trial within approximately 12 months thereafter.
AB-PA01: Lung Infections in Cystic Fibrosis (CF) Patients Caused by P. aeruginosa
We are initially developing AB-PA01 for the treatment of P. aeruginosa, the most prevalent bacterial infection in cystic fibrosis, or CF, patients and the one that leads to the highest mortality and is the primary cause of lung infection in approximately 80% of CF patients ages 25 to 34, causing an estimated 450 deaths per year in the United States. To develop our product candidates, we have created a global diversity panel of relevant clinical isolates (bacteria isolated from patients) from clinics around the globe. These diversity panels have been screened against our phage libraries, which are isolated and characterized according to our set of proprietary discovery protocols. We have demonstrated, in in vitro and in vivo studies, that our proprietary phage mix is able to effectively kill targeted bacteria. Furthermore, our phage mixes are selected to exhibit a high degree of overlap, defined as the number of bacteria targeted by more than one phage in the product. We believe that high overlap is an important factor in preventing bacteria from developing resistance to our phage product candidates.
Similar to work described above for S. aureus, we have tested over 400 clinical P. aeruginosa clinical isolates. As an example, initial host range testing was performed with a reference panel of 67 CF isolates. AB-PA01 showed an activity of 95.5% (64/67) with 87.5% (56/64) of the positives isolates hit by more than one phage in the mix.
In collaboration with Institut Pasteur (Paris, France) and also with the Brompton Hospital, Imperial College (London, United Kingdom), we have demonstrated in the preclinical studies that phages can effectively treat infections in animal models of acute P. aeruginosa lung infections. In one such study, we inoculated eight mice and treated them with either PBS (control group), our phage mix, or with an antibiotic.
Bacterial counts and the number of bacteriophage infection units detected by assay, or phage titers, were measured in these animals after 24 hours, and the results demonstrated that our phage mix effectively lowered the bacterial counts, or CFU, in the mouse lung to levels comparable to antibiotic treatment (PBS vs. antibiotic, p=0.0003; PBS vs. bacteriophage, p=0.0003). A p-value is a statistical measure of the probability that the difference in two values could have occurred by chance. The smaller the p-value, the lower the likelihood is that the difference occurred by chance, or the greater our confidence is that the results are statistically significant. Furthermore, it was evident that phage replicated to high levels in the infected lung.
An additional preclinical study conducted at the Institut Pasteur in mice (12 mice in each of the treatment and control groups) demonstrated the ability of our phage mix to reach the lung within two hours of being delivered by oral administration. The phage levels increased between two and six hours post-treatment, and the results were statistically significant (p-value <0.001). These results demonstrate that when orally administered in mice, phages not only reached the lungs, but were also able to infect and multiply in target bacteria.
In a separate in vivo study of acute P. aeruginosa infection of the mouse lung conducted at the Brompton Clinic, results demonstrated that our phage mix reduced CFU levels upon simultaneous intranasal administration (six mice in each of the treatment and control groups) and also when administered 24 hours post-bacterial infection (seven mice in the treatment group and eight mice in the control group) using a standard strain of P. aeruginosa, Pa01.
We were granted an advisory meeting with the MHRA in the first quarter of 2014 to discuss our plans and intend to move the AB-PA01 compound into additional preclinical testing in preparation for a Phase 1/2 clinical trial in CF patients. We also sought advice on the acceptability of CMC plans. The MHRA concurred with our approach and plans as presented, including a first-in-man dose ranging clinical trial in CF patients. We have completed product candidate selection and are currently conducting manufacturing process development and scale-up with the goal of initiating inhalation toxicology studies in the first quarter of 2017 and completing such studies within approximately six months thereafter. We plan to initiate a Phase 1 single-ascending dose study in CF patients during the second half of 2017 and currently expect to complete that study within approximately 12 months thereafter.
We are also currently evaluating our P. aeruginosa phages in preclinical animal models of chronic rhinosinusitis in collaboration with the University of Adelaide. Pending the outcome of this study, we also expect to move AB-PA01 into a chronic rhinosinusitis study in Australia in the second half of 2017. We expect the study to be similar in design to our current Phase 1 study of AB-SA01 in chronic rhinosinusitis, except the AB-PA01 study will target P. aeruginosa in chronic rhinosinusitis patients.
If we achieve successful proof of concept studies, we may consider developing this compound for the treatment of other acute and chronic lung infections, such as ventilator associated bacterial pneumonia, or VABP, and chronic obstructive pulmonary disease, or COPD. P. aeruginosa is the predominant pathogen in these indications.
AB-CD01: Gastrointestinal (GI) Infection Caused by C. difficile, or CDI
From 2000 through 2007, deaths in the United States from CDI increased over 400%. Over 90% of such deaths occur in hospitalized or confined patients over the age of 65. Global Data estimates that the major European Union and United States markets for CDI therapies grew to more than $314 million in 2011 and they are expected to grow to more than $500 million by 2019.
According to the CDC almost 250,000 people each year require hospitalization for CDI and at least 14,000 people die each year in the United States from CDI. The CDC also estimates that 20-40% of CDI recurs with standard antibiotic treatment. We are actively working with researchers at the University of Leicester to develop a phage therapeutic that targets and kills C. difficile. We believe that orally delivered phages are well suited to treat CDI. Within this collaboration, researchers at the University of Leicester have discovered phages that have been shown to be effective in vitro and in vivo against clinically-relevant strains of C. difficile isolated from around the world. These same researchers have also shown phage cocktails to be effective in preventing C. difficile biofilm formation in vitro. While current pathogenic strains of C. difficile are not yet antibiotic-resistant, the CDC has categorized C. difficile as an urgent threat and has stated that CDI requires urgent and aggressive action. We believe that there is a significant market opportunity for our product in treating this infection.
Preclinical studies are underway to select and optimize our phage cocktail and manufacturing strains as well as evaluate efficacy in animal models.
Prior Clinical Development
In 2010, our wholly owned subsidiary, Biocontrol Ltd, reported a double-blind placebo-controlled, randomized Phase 1/2 clinical trial targeting chronic ear infections (otitis) caused by P. aeruginosa. To our knowledge, this was the first randomized placebo-controlled efficacy trial of bacteriophage therapy. Results were published demonstrating decreasing levels of P. aeruginosa in the ear and improvement of clinical condition with a single input dose of 2.4 nanograms of bacteriophage preparation. While this was a small trial (n=24), changes from baseline at the end of the trial in the test group (n=12) were statistically significant for both clinical condition (p=0.001) and bacterial load (p=0.016). No significant changes were seen in the control group (n=12) compared to baseline at the end of the trial. Difference between test and control groups was statistically significant by analysis by covariance on day 21 for bacterial count (p=0.0365). These results will need to be validated in larger well-controlled trials.
Anti-Infective Therapeutics Market
The market opportunity for antibiotics is large, with the market estimated to reach $40.3 billion in annual sales globally in 2015. Almost one in every five deaths worldwide occurs as a result of infection and, according to the World Health Organization, or WHO, many bacterial infections will become difficult or impossible to cure as the efficacy of current antibiotic drugs wanes. Despite the advances in antimicrobial and vaccine development, infectious diseases still remain as the third-leading cause of death in the United States and the second-leading cause of death worldwide.
The number of new antibiotics approved by the FDA and other global regulatory authorities has declined consistently over the last two decades. According to the PEW Charitable Trusts report, as of March 2016 there are an estimated 37 new antibiotics in clinical development for the U.S. market. Historically, the success rate from Phase 1 to marketing approval is only 1 in 5 for infectious disease products. We therefore believe there is a need for new approaches to treat serious bacterial infections. Hospital-acquired (nosocomial) infections are a major healthcare problem throughout the world, affecting developed countries as well as resource-poor countries. The WHO reports that hospital-acquired infections are among the major causes of death and increased morbidity among hospitalized patients and estimates that more than 1.4 million people per year worldwide suffer from infectious complications from a hospital stay.
A recent CDC report also cites that in the United States, between 5 and 10% of all patients admitted to a hospital will be affected by a hospital-acquired infection during their stay, typically requiring extended stays and additional care. There is also a significant risk of death from such infections. In the United States, the CDC estimates that approximately 99,000 people die from hospital-acquired infections each year. The Cystic Fibrosis Foundation estimates that P. aeruginosa accounts for 10% of all hospital-acquired infections.
Compounding the above situations is the alarming and continuing rise in the prevalence of antibiotic-resistant bacterial infections. This, coupled with the lack of new antibiotics in current discovery and development pipelines, has generated a significant clinical management problem worldwide, leading to increases in morbidity and mortality due to these antibiotic-resistant bacteria as well as increases in healthcare costs.
The first of these antibiotic-resistant infections to reach epidemic proportions was caused by the Gram-positive bacterium S. aureus. S. aureus resistance to a broad range of antibiotics has necessitated the use of expensive and potentially toxic “drugs of last resort”, most notably vancomycin. Antibiotic-resistant forms of S. aureus, usually termed MRSA, VISA (vancomycin-intermediate S. aureus), or VRSA (vancomycin-resistant S. aureus), can be extremely challenging to treat. Although several antibiotics targeting S. aureus have been developed, rapidly developing bacterial resistance has been noted for all of these including linezolid, daptomycin and tigecycline. On the basis of historical evidence, resistance to these existing products is likely to increase over time, and this picture is further complicated by the reduced efficacy of conventional antibiotics against Staphylococcus biofilms.
Typically, S. aureus infection causes a variety of suppurative (pus-forming) infections and toxinoses (lesions) in humans. It causes superficial skin lesions such as boils, styes and furuncles; more serious infections such as pneumonia, mastitis, phlebitis, meningitis and urinary tract infections; and deep-seated infections, such as osteomyelitis and endocarditis. S. aureus is the leading cause of wound infections, in particular, hospital-acquired (nosocomial) infection of surgical wounds and infections associated with indwelling medical devices. S. aureus is the leading pathogen in healthcare-associated infections in the United States as a whole, accounting for 30.4% of surgical site infections, or SSI, and 15.6% of such infections overall.
Infections also occur in connection with CF, which is a genetic disease affecting primarily Caucasians of northern European descent. According to the Cystic Fibrosis Foundation, there are approximately 50,000 cases of CF in North America and Europe. P. aeruginosa opportunistically infects the mucous membranes, primarily the lungs, of CF patients and quickly grows out of control, resulting in pneumonia. P. aeruginosa infections are notoriously resistant to known antibiotics, and treatment may be further complicated by the formation of biofilms. Biofilms are organized structures of microorganisms growing on solid surfaces (such as lung tissue) and often limit access of antibiotics to the covered tissues. Since phages attack bacteria in a manner independent of chemical antibiotic resistance mechanisms and can infect bacteria growing in biofilms, we believe that P. aeruginosa infection among CF patients represents a compelling indication to pursue. The availability of Pseudomonas-specific phages along with validated animal models of P. aeruginosa lung infections has contributed to the development of our bacteriophage program in CF.
Anti-Infective Treatments with Bacteriophages
Background
The dramatic rise in antibiotic resistance, the appearance of an increasing number of new “superbugs” and the lack of new antibiotics in the pipeline has prompted calls to action from many of the world’s major health bodies such as the CDC and the WHO, who warn of an “antibiotic cliff” and a “post-antibiotic era.” In 2009, the European Antimicrobial Resistance Surveillance System, or EARSS, concluded that “the loss of effective antimicrobial therapy increasingly threatens the delivery of crucial health services in hospitals and in the community.” This conclusion was reinforced by The Antimicrobial Availability Task Force, or AATF, of the Infectious Diseases Society of America, or IDSA, and the European Centre for Disease Prevention and Control, or ECDC, in conjunction with the European Medicine Agency, or EMA. Clearly, there is a pressing need to find alternative antibacterial therapies.
Bacteriophage therapy has the potential to be an alternative method of treating bacterial infection. Phages are ubiquitous environmental viruses that grow only within bacteria. The name “bacteriophage” translates as “eaters of bacteria” and reflects the fact that as they grow, phages kill the bacterial host by multiplying inside and then bursting through the cell membrane in order to release the next generation of phages. Phages can differ substantially in morphology and each phage is active against a specific range of a given bacterial species. Phages were first discovered in 1915 at the Institut Pasteur and were shown to kill bacteria taken from patients suffering from dysentery. Furthermore, it was noted that phage numbers rose as patients recovered from infection, suggesting a direct association.
Life Cycle of a Bacteriophage
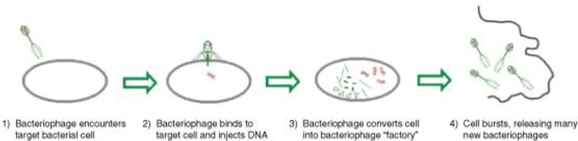
Until the discovery of effective antibiotics, phages were used as an effective means of combating bacterial infection. When broad-spectrum antibiotics came into common use in the early 1940s, phages were considered unnecessary, with antibiotics being seen for many years as the answer to bacterial disease. This attitude persisted until the development of the wide-ranging, and in some cases total, resistance to antibiotics seen within the last 10 years.
Phages have the potential to provide both an alternative to, and a synergistic approach with, antibiotic therapy. Since they use different mechanisms of action, phages are unaffected by resistance to conventional antibiotics. Phages containing certain enzymes also have the ability to disrupt bacterial biofilms, thus potentiating the effect of chemical antibiotics when used in combination with them.
Our Strategy
Our strategy is to use techniques of modern biotechnology and current state-of-the-art practices for drug development in concert with existing regulatory guidance to develop a pipeline of bacteriophage products that will destroy bacteria such as MRSA, which are resistant to antibiotics. Our business strategy will apply state-of-the-art techniques in molecular biology and in clinical trial design to build upon the long successful history of using phages therapeutically to treat and cure infections.
We supplement our internal resources with world-class scientific and medical collaborations throughout the world. For example, through a collaboration with The University of Adelaide in Australia, we conducted preclinical studies showing the ability of S. aureus phage preparations to kill clinical isolates from 61 patients demonstrating efficacy of greater than 90%. Furthermore, a S. aureus mixture was shown to be safe and efficacious in a preclinical sheep model of chronic rhinosinusitis. This program continues to progress and a clinical trial of patients at the University of Adelaide’s Queen Elizabeth Hospital with treatment refractory chronic rhinosinusitis patients infected with S. aureus commenced in late 2015 and the first patient was dosed in January 2016. More recently, we tested 90 S. aureus clinical isolates from chronic rhinosinusitis patients located in Belgium and showed similar efficacy to isolates obtained from Australian patients, highlighting the diverse geographic activity of our phage cocktail.
In collaboration with the U.S. Army, we completed enrollment of a Phase 1 safety study in July 2016 that we believe will support the further development of a treatment for S. aureus infections for wound and skin infections.
We collaborate with the Royal Brompton Hospital in London where we have demonstrated that a candidate phage product can survive nebulization, was effective in killing over 83% of recent clinical P. aeruginosa isolates, and in preclinical mouse models demonstrated that a phage mixture dose-dependently clears P. aeruginosa infection from the lung and reduced inflammation.
We have completed selection of the phages for drug product selection for AB-PA01, and in conjunction with the Brompton Hospital, we would expect to conduct a Phase 1/2 study using AB-PA01 to treat CF patients with P. aeruginosa lung infections.
Acquisitions
In January 2011, we completed the acquisition of Biocontrol Ltd, with the goal of developing their phage therapy programs using funding from the sale of our legacy gene therapy assets. Under the terms of our acquisition of Biocontrol Ltd, we issued 456,344 shares of our common stock to the stockholders of Biocontrol Ltd with a total fair value of approximately $8.6 million as of January 6, 2011, resulting in Biocontrol’s former stockholders owning approximately 50% of our outstanding equity securities at the time. As a condition to closing the acquisition, Biocontrol Ltd raised approximately £200,000 (US$310,000) in working capital for use by us.
In November 2012, we completed the acquisition of Special Phage Holdings Pty Ltd, a company based in Australia, which we refer to as SPH, with the goal of combining SPH’s research on addressing the rapidly escalating problem of antibiotic resistance through the development of a series of bacteriophage-based treatments into our own development programs. We acquired SPH in exchange for shares of our common stock pursuant to the terms of a Stockholder Sale Agreement and a Managers Warranty Deed.
In connection with our acquisition of SPH, we entered into certain other arrangements, including the repayment under a Loan Repayment Deed (as amended) of a $770,000 loan originally made by Cellabs Pty Ltd, or Cellabs, an Australian company, to SPH, a consulting agreement with Dr. Anthony Smithyman and the payment of $3,017 per month to Cellabs for our laboratory space in Australia through December 31, 2015. Under the terms of the Loan Repayment Deed, the loan from Cellabs to SPH was to be repaid and fully satisfied partly in cash and partly by issuing 40,000 shares of our common stock to Cellabs. As of December 31, 2015, $350,000 has been paid by us to Cellabs and all 40,000 shares have been issued. We paid the remaining balance of $200,000 under the terms of the Loan Repayment Deed in December 2013. The SPH acquisition also included several phage therapy projects which had reached the pre-clinical or animal study stage, including the Brompton Hospital CF study, the Adelaide University MRSA chronic rhinosinusitis study and the University of Leicester C. difficile project. We believe that acquisition of SPH brought substantial phage scientific expertise and know-how to us.
In January 2016, we entered the Novolytics Purchase Agreement, pursuant to which we acquired all rights, title and interest to two families of patents. The first patent family is titled “Anti-bacterial compositions” and has been granted in Australia and China, with prosecution pending in the United States and other countries. The second patent family is titled “Novel bacteriophages” and the prosecution is pending in the United States and other countries. We also received clinical isolates for S. aureus which will bolster our libraries of clinically relevant strains. Additionally, we received know-how relating to certain formulation processes. We also have access to all previous dialogue between Novolytics and various regulatory organizations including the MHRA.
In connection with the Novolytics Purchase Agreement, we paid cash to Novolytics to cover expenses incurred in connection with winding up its phage-related business, as well as warrants to the stockholders of Novolytics to purchase up to an aggregate of 170,000 shares of our common stock, each with an exercise price of $12.00 per share. Pursuant to the terms of the Novolytics Purchase Agreement, we granted certain registration rights covering the resale of the shares of common stock underlying such warrants.
Strategic Alliances and Research and License Agreements
As discussed below, we have established collaborations with the U.S. Army and the University of Leicester, which provide us with access to the considerable scientific, developmental, and regulatory capabilities of our collaborators. We believe that our collaborations contribute to our ability to rapidly advance our product candidates, build our product platform and concurrently progress a wide range of discovery and development programs.
Global R&D Agreement with U.S. Army
In June 2013, we entered into a Research and Development Agreement with the U.S. Army Medical Research and Materiel Command. The Research and Development Agreement focuses on developing bacteriophage therapeutics to treat at least three types of infections: S. aureus, E. coli and P. aeruginosa. The initial indication will be wounds and skin infections from S. aureus, which is the leading pathogen in healthcare-associated infections in the United States as a whole, accounting for 30.4% of surgical site infections.
We retain global regulatory ownership and commercial rights to all products developed by us under the Research and Development Agreement. The U.S. Army Medical Research and Materiel Command will have the right to retain a non-exclusive license to use any products developed by or on behalf of the U.S. Government for non-commercial uses. We also have the rights to exclusively license any intellectual property developed by the U.S. Army Medical Research and Materiel Command under the collaboration on terms to be agreed upon.
The Research and Development Agreement expires in June 2018 and can be terminated by either the U.S. Army Medical Research and Materiel Command or us upon 60 days’ written notice to the other party at any time.
University of Leicester Development Agreements
In April and September 2013, we entered into a collaboration agreement and a license agreement, respectively, with the University of Leicester to develop a phage therapy that targets and kills C. difficile.
Under these agreements, which we refer to collectively as the Leicester Development Agreements, we are funding the University of Leicester to carry out in vitro studies and animal model development work to identify bacteriophage to resolve C. difficile infections. We have licensed related patents, materials and know-how from the University of Leicester. Under the Leicester Development Agreements, the University of Leicester will provide the bacteriophage and act as overall project coordinator for preclinical studies. All rights, title and interest to any intellectual property developed under the Leicester Development Agreements belong to us. Under the Leicester License Agreement, we have exclusive rights to certain patents and materials owned by the University of Leicester, as well as non-exclusive licenses to related know-how.
The collaboration agreement expires in November 2018 and is terminable by either party upon (a) material breach by the other party, subject to a 90-day cure period, (b) the inability of the principal investigator to continue the collaboration or (c) our bankruptcy or winding up of our operations or, commencing on November 13, 2016, with 180 days’ notice.
Pursuant to the Leicester License Agreement, we paid an up-front fee and will pay the University of Leicester royalties based on product sales and make certain milestone payments based on product development. We are also required to pay minimum annual fees, which reduce future milestone payments. In the event that we sublicense a product created under the Leicester Development Agreements, we have agreed to pay the University of Leicester certain milestone payments or a certain percentage of any sublicense revenue received by us for the attainment of such milestone, as well as a certain percentage of all royalty payments we receive from any sublicensees.
The license agreement expires on the later of the expiration of the licensed patents or September 2028, and is terminable by us at any time upon 60 days’ notice, by the University of Leicester (a) if we legally challenge the validity or ownership of any of the licensed patents, (b) if we fail to pay the fees, milestones or royalties due under the license agreement or (c) if we fail to make substantial commercial process and agree with Leicester that we will be unable to do so. The license agreement is also terminable by either party upon the material breach by the other party (subject to a 30-day cure period) or upon the other party’s bankruptcy or insolvency.
License Agreement with United Kingdom Secretary of State for the Department of Health
In January 2011, upon completion of our acquisition of Biocontrol Ltd., we assumed a license agreement entered into in March 2007 between Biocontrol Ltd. and the Health Protection Agency, Centre for Emergency Preparedness and Response, to use certain intellectual property rights to develop treatments for bacterial biofilm infections. The agreement was subsequently assigned to the United Kingdom Secretary of State for the Department of Health, or DoH.
Under the license agreement, we have obtained exclusive rights to a patent portfolio related to the use of bacteriophages combined with biofilm-disrupting agents in treating biofilm infections. In consideration for the exclusive license, we may be required to pay to the DoH certain milestone payments in the aggregate of up to £10,000 per product, as well as single digit percentage royalty on net sales of products incorporating licensed intellectual property.
The license agreement shall remain in full force and effect until the expiration of the last patent exclusively licensed under the license agreement. If we default on any milestone or royalty payments, or upon breach by us of certain other terms of the license agreement, the DoH may either terminate the license agreement immediately upon written notice or modify the license to be non-exclusive upon 30 days’ written notice.
Intellectual Property
General
Our goal is to obtain, maintain and enforce patent protection for our product candidates, formulations, processes, methods and any other proprietary technologies, preserve our trade secrets and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, patent protection may not afford us with complete protection against competitors who seek to circumvent our patents.
We also depend upon the skills, knowledge, experience and know-how of our management and research and development personnel, as well as that of our advisors, consultants and other contractors. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently and will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
As of August 17, 2016, we owned or had exclusive license rights to a total of 65 patents and applications: five U.S. patents, seven U.S. patent applications, 37 foreign patents, and 16 foreign patent applications, expiring on various dates between 2024 and 2036. These patents and applications cover our lead phage-therapeutic programs and use thereof, the sequential use of bacteriophages in combination with conventional antibiotics, genetic sequence variations, biofilm disrupting agents, methods to reduce antibiotic resistance, methods to design therapeutic combination panels of bacteriophage, disinfection methods using bacteriophages, and bacteriophage mutants having increased bacterial host spectra.
US 7758856 and national patents within the EU deriving from PCT WO2004062677; Bacteriophage for the treatment of bacterial biofilms
Under an existing license from the United Kingdom Secretary of State for the Department of Health (DoH), we have exclusive rights to a patent portfolio related to the use of bacteriophages combined with biofilm-disrupting agents in treating biofilm infections. This portfolio includes one issued patent in the United States and a patent granted in Europe (EP1587520 is validated in France, Germany, Netherlands, Switzerland, Liechtenstein and the United Kingdom). Claims issued in these patents include those directed to compositions and methods related to agents that are able to facilitate the penetration of biofilms, and their combination with therapeutic bacteriophage preparations. The U.S. patent is expected to expire in December 2026 (absent any extensions). The foreign patents are expected to expire in January 2024 (absent any extensions).
US 7807149, US 8105579, US 8388946, continuation application and national filings deriving from PCT WO2005009451; Bacteriophage containing therapeutic agents
Through our wholly owned subsidiary, Biocontrol Ltd, we own three granted U.S. patents and one pending U.S. continuation patent application (US 13/757655) with claims directed generally to bacteriophage compositions, therapeutic methods of using bacteriophages, and methods of treating bacterial infections by sequentially administering bacteriophages in combination with conventional antibiotics. The pending U.S. continuation application relates generally to panels of bacteriophages with different strain specificities for bacterial infections. Corresponding patents have been granted in Australia (AU2004258731), Europe (EP1663265 and EP2570130 – both patents are validated in the United Kingdom, Switzerland, Liechtenstein, Germany, Spain, France, Italy and the Netherlands), Japan (JP5731727 and JP5856556) and Canada (CA2533352). Claims issued in these patents include those directed to therapeutic and non-therapeutic applications of bacteriophage and the sequential use of antibiotics to treat bacterial infections. U.S. patents are expected to expire from July 2024 to March 2027 (absent any extensions). The foreign patents are expected to expire in July 2024 to March 2027 (absent any extensions).
US 8475787, continuation application and national filings deriving from PCT WO2008110840; Beneficial effects of bacteriophage treatment
Through our wholly owned subsidiary, Biocontrol Ltd, we own one granted U.S. patent (8475787), and one pending continuation application (14/625049). This patent family broadly relates to bacteriophage-induced induction of antibiotic sensitivity in a bacterial target, such as P. aeruginosa. The granted U.S. patent is expected to expire in July 2029 (absent any extensions). Corresponding patents have been granted in Australia (AU2008224651) and Europe (EP2136826 – validated in the United Kingdom, Switzerland/Liechtenstein, Germany, Spain, France, Italy and the Netherlands), and Allowance Notices issued in respect of our Japanese applications JP2010/521428 and JP2014/157433. A related Canadian application (CA2680108) is currently pending. Foreign patents in this family are expected to expire in March 2028 (absent any extensions).
PCT WO2013/164640 (United Kingdom earliest priority filing 1207910.9); Therapeutic bacteriophage compositions
Through our wholly owned subsidiary, Biocontrol Ltd, we own a Patent Cooperation Treaty, or PCT, application relating to the design of effective bacteriophage combinations and elimination of antagonistic effects between said bacteriophage. The PCT application published on November 7, 2013, and following International Preliminary Examination a positive patentability opinion issued. National/regional phase applications are currently pending in the U.S. (US14/398384), Canada (CA2871986), Europe (EP2874635), Japan (JP2015/523850), and Australia (AU2013255583). Patents issuing from this PCT, if any, are expected to expire in May 2032 (absent any extensions).
PCT WO2009/044163 (United Kingdom earliest priority filing 0719438.4); Anti-bacterial compositions
Pursuant to the terms of the Asset Purchase Agreement with Novolytics Ltd., we acquired and currently own one U.S. continuation application (14/686315), relating to methods for killing/treating Staphylococcus aureus and MRSA, among other bacteria, using a combined bacteriophage K and bacteriophage P68 composition. A corresponding patent has been granted in Australia (AU2008306626) and China (CN101835384) and related applications are pending in Australia (AU2015264918), Japan (JP2015/007087), Canada (CA2700646) and Europe (EP2197284). The granted foreign patents are expected to expire October 2028 (absent any extensions).
PCT WO2013/068743 (United Kingdom priority filing 1119167.3); Novel bacteriophages
Pursuant to the terms of the Asset Purchase Agreement with Novolytics Ltd., we acquired and currently own a U.S. patent application (14/356869) relating to Staphylococcus aureus and MRSA therapeutics, and in particular Phage K mutants capable of targeting an increased number of Staphylococcus aureus strains when compared to wild-type Phage K, as well as uses of said mutant. Related applications are also pending in Australia (AU2012335397), Canada (CA2890450), Japan (JP 2014/533943) and Europe (EP2776559). Any granted patents will expire in November 2033.
US 15/237496 (converted from United States provisional filing 62/204915); Therapeutic bacteriophage compositions
We own U.S. patent application 15/237496, which is directed to our AB-SA01 bacteriophage panel, mutants thereof, and methods of treating Staphylococcus aureus infections (including MRSA) comprising the use of same. Corresponding foreign applications are being pursued by way of a parallel PCT application. Any granted patent is expected to expire in August 2036 (absent extensions).
Our success in preserving market exclusivity for our product candidates relies on patent protection, including extensions to this where appropriate, and on data exclusivity relating to an approved biologic. This may be extended by orphan drug and/or pediatric use protection where appropriate. Once any regulatory period of data exclusivity expires, depending on the status of our patent coverage, we may not be able to prevent others from marketing and selling biosimilar versions of our product candidates. We are also dependent upon the diligence of our appointed agents in national jurisdictions, acting for and on our behalf, which manage the prosecution of pending domestic and foreign patent applications and maintain granted domestic and foreign patents.
Competition
We operate in highly competitive segments of the biotechnology and biopharmaceutical markets. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions all seeking to develop novel treatment modalities for bacterial infections. Many of our competitors have significantly greater financial, product development, manufacturing and marketing resources than we do. Large pharmaceutical companies have extensive experience in clinical development and obtaining regulatory approval for drugs. In addition, many universities and private and public research institutes are active in antibacterial research, some in direct competition with us. We also may compete with these organizations to recruit scientists and clinical development personnel.
There are a handful of small biotechnology companies developing bacteriophage products to treat human diseases. Other than our ongoing clinical trials there is, to our knowledge, one corporate-sponsored clinical trial currently enrolling. A French biotechnology company, Pherecydes Pharma, is acting as clinical trial sponsor of a Phase 1/2 clinical trial in Europe of a phage therapy for the treatment of burn wounds infected with either E. coli and P. aeruginosa, referred to as PhagoBurn. This clinical trial is a randomized, multi-center open label study to assess tolerance and efficacy of local treatment with a bacteriophage cocktail. A multi-center clinical trial also sponsored by Pherecydes Pharma evaluating a bacteriophage cocktail versus placebo for diabetic foot ulcers, is listed on clinicaltrials.gov as active but not yet enrolling. To our knowledge, a small number of biotechnology companies, including Synthetic Genomics and LytPhage, Inc., as well as academic institutions, have earlier stage discovery programs utilizing synthetic biology approaches to genetically modify bacteriophages to remove or input genes to improve therapeutic properties such as increases to the bacterial host range to infect a larger number of bacterial strains and decrease the need for using multiple phages in a product.
A related approach to treating Staphylococcus infections is being pursued by Contrafect Corporation using a bacteriophage lysin (a hydrolytic enzyme produced by bacteriophages) to treat S. aureus bacteremia (infection in the blood). Contrafect has recently completed a Phase 1 intravenous single dose escalation study in healthy volunteers.
Our bacteriophage programs may compete with or be synergistic with currently approved antibiotics, and experimental approaches such as novel antibiotics, antimicrobial peptides, antimicrobial vaccines, metals, antisense, monoclonal antibodies and possibly microbiome manipulation. For example, Seres Therapeutics is developing a single-dose capsule (SER-109) consisting of bacterial spores to treat recurrent CDI (Clostridium difficile infection). In May 2015, Seres initiated a multi-center, randomized, placebo-controlled Phase 2 clinical trial, to assess the efficacy and safety of SER-109. SER-109, or similar products that may be in development by third parties, could prove to be competitive to or used in conjunction with a bacteriophage therapeutic approach.
Manufacturing and Supply
We have developed our own manufacturing capabilities at a facility in Ljubljana, Slovenia that is leased by our wholly owned subsidiary, AmpliPhi, Biotehnološke Raziskave in Razvoj, d.o.o. We believe that our facility complies with applicable cGMP regulations, which require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. Pharmaceutical product manufacturers and other entities involved in the manufacture and distribution of approved pharmaceutical products are required to register their establishments with the FDA, and certain state agencies, including the applicable government agency where the facility is located, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws.
After conducting a global search, we elected to proceed with establishing a wholly owned cGMP compliant manufacturing facility in Ljubljana, Slovenia. Upon final product selection, we plan to manufacture each of our product candidates in this facility. We have been able to access and hire highly skilled process development and phage manufacturing expertise and believe that we have control of our proprietary platform from phage identification through final product fill and finish. Our facility is comprised of approximately 4,000 sq. ft. of laboratory and office space, where we produce cGMP clinical trial supplies for our current and planned clinical trials. We believe this facility will be sufficient to meet our manufacturing needs through initial Phase 3 clinical trials. Our current formulation for AB-SA01 is intended for sinonasal or topical delivery via a nasal wash solution or dressed bandage. We plan to further optimize future formulations of our product candidates.
Our facility in Ljubljana, Slovenia is subject to inspection and regulation by JAZMP, the Slovenian agency that regulates and supervises pharmaceutical products in Slovenia. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved New Drug Application/Biologics License Application, including withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior regulatory approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further regulatory review and approval, including approval by the FDA.
Commercialization and Marketing
We have full worldwide commercial rights to all of our phage-based product candidates to treat drug-resistant bacterial infections, including our product candidates: AB-PA01 for the treatment of CF patients with P. aeruginosa lung infections; AB-SA01, for the treatment of S. aureus infections; and AB-CD01 for the prevention or treatment of C. difficile infections. We believe we can maximize the value of our company by retaining substantial global commercialization rights to these product candidates and, where appropriate, entering into partnerships to develop and commercialize our other product candidates. We plan to build a successful commercial enterprise using a sales team in the United States and possibly other major markets and with partners in other territories.
We have not yet established a sales, marketing or product distribution infrastructure because our lead candidates are still in early clinical development. We generally expect to retain commercialization and co-commercialization rights in the United States for all of our product candidates for which we receive marketing approvals. Subject to receiving marketing approvals, we intend to explore building the necessary marketing and sales infrastructure to market and sell our current product candidates. We also intend to explore the use of a variety of distribution agreements and commercial partnerships in those territories where we do not establish a sales force for any of our product candidates that obtain marketing approval.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing.
United States Product Development Process
In the United States, the FDA regulates biological products under the Federal Food, Drug and Cosmetic Act, or FDCA, and the Public Health Service Act, or the PHS Act, and related regulations. Biological products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process or approval process, or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a biological product may be marketed in the United States generally includes the following:
| · | completion of preclinical laboratory tests, animal studies and formulation studies according to good laboratory practice requirements, or GLP, or other applicable regulations; |
| · | submission to the FDA of an IND, which must become effective before human clinical trials may begin in the United States; |
| · | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as good clinical practices, or GCPs, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed biological product for its intended use or uses; |
| · | submission to the FDA of a Biologics License Application, or BLA, for a new biological product; |
| · | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with the FDA’s cGMP regulations, to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity; |
| · | potential FDA audit of the nonclinical study sites and clinical trial sites that generated the data in support of the BLA; and |
| · | FDA review and approval, or licensure, of the BLA which must occur before a biological product can be marketed or sold. |
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources even when approvals are inherently uncertain.
The strategies, nature, and technologies of bacteriophage products are different from the conventional antibiotic therapy products. From the regulatory requirements established to ensure the safety, efficacy and quality of bacteriophage preparations, there are several major points to consider during the development, manufacturing, characterization, preclinical study and clinical trial of bacteriophage. The major issues include:
| · | bacteriophage preparation design (single agent versus phage mixes and wild-type phage versus genetically engineered phage); |
| · | proof of concept in development of bacteriophage products; |
| · | selectivity of bacteriophage replication and targeting to specific species of bacteria; |
| · | relevant animal models in preclinical studies; and |
| · | clinical safety and efficacy. |
Before testing any compounds with potential therapeutic value in humans, the biological product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product biology, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the biological product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLP. The sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the IND on a clinical hold within that 30 day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, we cannot be certain that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such clinical trial.
Clinical trials involve the administration of the product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by the sponsor. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject inclusion and exclusion criteria and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA. Clinical trials must be conducted in accordance with GCP requirements. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, or ethics committee if conducted outside of the U.S., at or servicing each institution at which the clinical trial will be conducted. An IRB or ethics committee is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB or ethics committee also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. We intend to use third-party Clinical Research Organizations, or CROs, to administer and conduct our planned clinical trials and will rely upon such CROs, as well as medical institutions, clinical investigators and consultants, to conduct our trials in accordance with our clinical protocols. The failure by any of such third parties to meet expected timelines, adhere to our protocols or meet regulatory standards could adversely impact the subject product development program and we remain legally responsible for compliance with applicable laws and regulations governing the conduct of these clinical trials.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| · | Phase 1: The product candidate is initially introduced into healthy human subjects and tested primarily for safety and dosage tolerance. Absorption, metabolism, distribution and excretion may also be tested. |
| · | Phase 2: The product candidate is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product candidate for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| · | Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA and other regulatory authorities for approval of a marketing application. |
Post-approval studies, or Phase 4 clinical trials, may be requested by the FDA as a condition of approval and are conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggest that there may be a significant risk for human subjects. The FDA or the sponsor or, if used, its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB or ethics committee can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s or ethics committee’s requirements or if the pharmaceutical product has been associated with unexpected serious harm to patients. Suspension of a clinical trial due to safety risks attributed to the investigational product will result in termination of the trial and possibly others that are underway.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the physical characteristics of the product candidate as well as finalize a process for manufacturing the product candidate in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents or other impurities with the use of biological products, the PHS Act emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency, and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
United States Review and Approval Processes
In order to obtain approval to market a biological product in the United States, a BLA that provides data establishing to the FDA’s satisfaction the safety and effectiveness of the investigational product candidate for the proposed indication must be submitted to the FDA. The application includes all data available from nonclinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s manufacture and composition, and proposed labeling, among other things. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Each BLA must be accompanied by a significant user fee. The FDA adjusts the user fees on an annual basis.. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA has 60 days from its receipt of a BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that the application is sufficiently complete to permit substantive review. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. After the BLA is accepted for filing, the FDA reviews it to determine, among other things, whether the proposed product is safe and effective for its intended use, has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency, and purity. The FDA may refer applications for novel product candidates or those that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA may ultimately decide that the BLA does not satisfy the criteria for approval. If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including Fast Track designation, accelerated approval and priority review, that are intended to expedite the process for the development and FDA review of drugs that are intended for the treatment of serious or life threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs and biological products to patients earlier than under standard FDA review procedures.
To be eligible for a Fast Track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life threatening disease or condition and demonstrates the potential to address an unmet medical need, or if the drug or biological product qualifies as a qualified infectious disease product under the Generating Antibiotic Incentives Now Act, or GAIN Act. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. We intend to request Fast Track designation for our product candidates if applicable.
Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biological may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments.
As a condition of approval, the FDA may require a sponsor of a drug or biological product receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug or biological product may be subject to accelerated withdrawal procedures. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
A sponsor can also request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug or biological product that is intended, alone or in combination with one or more other drugs or biological products, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the biological product or drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs or biological products designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy. We intend to request “breakthrough therapy” designation for our product candidates if applicable.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of our drugs, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch Waxman Amendments. The Hatch Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one half the time between the effective date of an IND, and the submission date of an NDA or BLA, plus the time between the submission date of an NDA or BLA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension, and the extension must be applied for prior to expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
Pediatric exclusivity is a type of marketing exclusivity available in the U.S. under the Best Pharmaceuticals for Children Act, or BPCA, which provides for an additional six months of marketing exclusivity may be available if a sponsor conducts clinical trials in children in response to a written request from the FDA, or a Written Request. If the Written Request does not include clinical trials in neonates, the FDA is required to include its rationale for not requesting those clinical trials. The FDA may request studies on approved or unapproved indications in separate Written Requests. The issuance of a Written Request does not require the sponsor to undertake the described clinical trials.
Biologics Price Competition and Innovation Act of 2009
The Patient Protection and Affordable Care Act, which included the Biologics Price Competition and Innovation Act of 2009, or BPCIA, amended the PHSA to create an abbreviated approval pathway for two types of “generic” biologics—biosimilars and interchangeable biologic products, and provides for a twelve year data exclusivity period for the first approved biological product, or reference product, against which a biosimilar or interchangeable application is evaluated; however if pediatric clinical trials are performed and accepted by the FDA, the twelve year data exclusivity period will be extended for an additional six months. A biosimilar product is defined as one that is highly similar to a reference product notwithstanding minor differences in clinically inactive components and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product. An interchangeable product is a biosimilar product that may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The biosimilar applicant must demonstrate that the product is biosimilar based on data from (1) analytical studies showing that the biosimilar product is highly similar to the reference product; (2) animal studies (including toxicity); and (3) one or more clinical trials to demonstrate safety, purity and potency in one or more appropriate conditions of use for which the reference product is approved. In addition, the applicant must show that the biosimilar and reference products have the same mechanism of action for the conditions of use on the label, route of administration, dosage and strength, and the production facility must meet standards designed to assure product safety, purity and potency.
An application for a biosimilar product may not be submitted until four years after the date on which the reference product was first approved. The first approved interchangeable biologic product will be granted an exclusivity period of up to one year after it is first commercially marketed, but the exclusivity period may be shortened under certain circumstances. The FDA has issued a number of final and draft guidances in order to implement the law. The guidance documents provide FDA’s current thinking on approaches to demonstrating that a proposed biological product is biosimilar to a reference product. The FDA intends to issue additional guidance documents in the future, and has identified considerations in demonstrating interchangeability to a reference product, labeling and nonproprietary naming as several of the issues that it hopes to address in calendar year 2015. Nonetheless, the absence of final guidance documents covering all biosimilars issues does not prevent a sponsor from seeking licensure of a biosimilar under the BPCIA, and the FDA has already approved two biosimilar applications in the United States.
FDA Post-Approval Requirements
Maintaining substantial compliance with applicable federal, state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of new products continues after approval, particularly with respect to cGMP. We will rely on third parties for the production of commercial quantities of any products that we may commercialize. We and third party manufacturers of our products are required to comply with applicable requirements in the cGMPs, including quality control and quality assurance and maintenance of records and documentation. We cannot be certain that we or our present or future suppliers will be able to comply with the cGMP and other FDA requirements. Other post-approval requirements applicable to biological products include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products, such as viral vaccines, before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products.
Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements, by us or our suppliers, may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Biological product manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their facilities with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMPs and other laws. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Labeling, Marketing and Promotion
The FDA closely regulates the labeling, marketing and promotion of drugs and biological products, including direct-to-consumer advertising, promotional activities involving the internet, and industry-sponsored scientific and educational activities. While doctors are free to prescribe any product approved by the FDA for any use, a company can only make claims relating to safety and efficacy of a product that are consistent with FDA approval, and the company is allowed to actively market a product only for the particular use and treatment approved by the FDA. In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice and state and local governments.
International Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our future products. Our manufacturing facility in Ljubljana, Slovenia is subject to inspection and regulation by JAZMP, the Slovenian agency that regulates and supervises pharmaceutical products in Slovenia. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or a mutual recognition procedure. The centralized procedure, which is compulsory for medicinal products produced by biotechnology or those medicinal products containing new active substances for specific indications such as the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, viral diseases and designated orphan medicines and optional for other medicines which are highly innovative. Under the centralized procedure, a marketing application is submitted to the European Medicines Agency where it will be evaluated by the Committee for Medicinal Products for Human Use and a favorable opinion typically results in the grant by the European Commission of a single marketing authorization that is valid for all European Union member states within 67 days of receipt of the opinion. The initial marketing authorization is valid for five years, but once renewed is usually valid for an unlimited period.
Pricing and Reimbursement
Although none of our product candidates has been commercialized for any indication, if they are approved for marketing, commercial success of our product candidates will depend, in part, upon the availability of third-party reimbursement from payors at the federal, state and private levels. Third-party payors include government healthcare programs, such as Medicare and Medicaid, private health insurers and managed-care plans. We anticipate third party payors will provide reimbursement for our products. However, these third party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products. Our product candidates may not be considered cost effective. It is time consuming and expensive for us to seek reimbursement from third party payors. Reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
We expect that there will continue to be a number of federal and state proposals to implement governmental pricing controls and limit the growth of healthcare costs, including the cost of prescription drugs. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, ACA) enacted in March 2010, was expected to have a significant impact on the health care industry. ACA has resulted in expanded coverage for the uninsured and is expected to help contain overall healthcare costs. With regard to pharmaceutical products, among other things, ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the impact of ACA on pharmaceutical companies as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred. In addition, although the United States Supreme Court upheld the constitutionality of most of the ACA, some states have stated their intentions to not implement certain sections of ACA and some members of Congress are still working to repeal ACA. These challenges add to the uncertainty of the changes enacted as part of ACA.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the U.S. and generally tend to be significantly lower.
Employees
As of August 1, 2016, we had 32 full-time employees.
Facilities
Our principal offices occupy approximately 1,000 square feet of leased office space pursuant to a month-to-month sublease, located at 3579 Valley Centre Drive, Suite 100, San Diego, California 92130. We also lease approximately 700 square feet of lab space in Richmond, Virginia, approximately 5,000 square feet of lab space in Brookvale, Australia, and approximately 6,000 square feet of lab and office space in Ljubljana, Slovenia. We believe our facilities are adequate for our current and near-term needs.