Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - TG THERAPEUTICS, INC. | v432524_ex23-1.htm |
| EX-32.1 - EXHIBIT 32.1 - TG THERAPEUTICS, INC. | v432524_ex32-1.htm |
| EX-32.2 - EXHIBIT 32.2 - TG THERAPEUTICS, INC. | v432524_ex32-2.htm |
| EX-31.2 - EXHIBIT 31.2 - TG THERAPEUTICS, INC. | v432524_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - TG THERAPEUTICS, INC. | v432524_ex31-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015.
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ________ to ________.
Commission File Number 1-32639
TG THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) |
36-3898269 (I.R.S. Employer Identification No.) |
3 Columbus Circle 15th Floor New York, New York (Address of principal executive offices) |
10019 (Zip Code) |
Registrant’s telephone number, including area code: (212) 554-4484
Securities registered pursuant to Section
12(b) of the Act:
Common Stock, Par Value $0.001 Per Share (Title of Class) |
The Nasdaq Capital Market (Name of Each Exchange on Which Registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act). (Check one):
| Large accelerated filer ¨ | Accelerated filer x |
| Non-accelerated filer ¨ | Smaller reporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ¨ No x
The aggregate market value of voting common stock held by non-affiliates of the registrant (assuming, for purposes of this calculation, without conceding, that all executive officers and directors are “affiliates”) was $570,132,000 as of June 30, 2015, based on the closing sale price of such stock as reported on the NASDAQ Capital Market.
There were 54,095,110 shares of the registrant’s common stock, $0.001 par value, outstanding as of March 1, 2016.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the 2016 Annual Meeting of Stockholders are incorporated by reference in Part III of this Annual Report on Form 10-K.
TG THERAPEUTICS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2015
TABLE OF CONTENTS
This Annual Report on Form 10-K contains trademarks and trade names of TG Therapeutics, Inc., including our name and logo. All other trademarks, service marks, or trade names referenced in this Annual Report on Form 10-K are the property of their respective owners.
SPECIAL CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
Certain matters discussed in this report, including matters discussed under the caption “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” may constitute forward-looking statements for purposes of the Securities Act of 1933, as amended, or the Securities Act, and the Securities Exchange Act of 1934, as amended, or the Exchange Act, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from the future results, performance or achievements expressed or implied by such forward-looking statements. The words "anticipate," "believe," "estimate," "may," "expect" and similar expressions are generally intended to identify forward-looking statements. Our actual results may differ materially from the results anticipated in these forward-looking statements due to a variety of factors, including, without limitation, those discussed under the captions “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report, as well as other factors which may be identified from time to time in our other filings with the Securities and Exchange Commission, or the SEC, or in the documents where such forward-looking statements appear. All written or oral forward-looking statements attributable to us are expressly qualified in their entirety by these cautionary statements. Such forward-looking statements include, but are not limited to, statements about our:
| · | expectations for increases or decreases in expenses; |
| · | expectations for the clinical and pre-clinical development, manufacturing, regulatory approval, and commercialization of our pharmaceutical product candidates or any other products we may acquire or in-license; |
| · | use of clinical research centers and other contractors; |
| · | expectations as to the timing of commencing or completing pre-clinical and clinical trials and the expected outcomes of those trials; |
| · | expectations for incurring capital expenditures to expand our research and development and manufacturing capabilities; |
| · | expectations for generating revenue or becoming profitable on a sustained basis; |
| · | expectations or ability to enter into marketing and other partnership agreements; |
| · | expectations or ability to enter into product acquisition and in-licensing transactions; |
| · | expectations or ability to build our own commercial infrastructure to manufacture, market and sell our drug candidates; |
| · | acceptance of our products by doctors, patients or payors; |
| · | ability to compete against other companies and research institutions; |
| · | ability to secure adequate protection for our intellectual property; |
| · | ability to attract and retain key personnel; |
| · | availability of reimbursement for our products; |
| · | estimates of the sufficiency of our existing cash and cash equivalents and investments to finance our operating requirements, including expectations regarding the value and liquidity of our investments; |
| · | stock price and its volatility; and |
| · | expectations for future capital requirements. |
The forward-looking statements contained in this report reflect our views and assumptions only as of the date this report is signed. Except as required by law, we assume no responsibility for updating any forward-looking statements.
We qualify all of our forward-looking statements by these cautionary statements. In addition, with respect to all of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
| 1 |
Unless the context requires otherwise, references in this report to “TG,” “Company,” “we,” “us” and “our” refer to TG Therapeutics, Inc. and our subsidiaries.
OVERVIEW
We are a biopharmaceutical company focused on the acquisition, development and commercialization of novel treatments for B-cell malignancies and autoimmune diseases. Currently, the company is developing two therapies targeting hematological malignancies. TG-1101 (ublituximab) is a novel, glycoengineered monoclonal antibody that targets a specific and unique epitope on the CD20 antigen found on mature B-lymphocytes. TG Therapeutics is also developing TGR-1202, an orally available PI3K delta inhibitor. The delta isoform of PI3K is strongly expressed in cells of hematopoietic origin and is believed to be important in the proliferation and survival of B-lymphocytes. Both TG-1101 and TGR-1202 are in clinical development for patients with hematologic malignancies. The Company also has pre-clinical programs seeking to develop IRAK4 (interleukin-1 receptor-associated kinase 4) inhibitors and anti-PD-L1 and anti-GITR antibodies.
We also actively evaluate complementary products, technologies and companies for in-licensing, partnership, acquisition and/or investment opportunities. To date, we have not received approval for the sale of any of our drug candidates in any market and, therefore, have not generated any product sales from our drug candidates.
CORPORATE INFORMATION
We were incorporated in Delaware in 1993. Our executive offices are located at 3 Columbus Circle, 15th Floor, New York, New York 10019. Our telephone number is 212-554-4484, and our e-mail address is info@tgtxinc.com.
We maintain a website with the address www.tgtherapeutics.com. We make available free of charge through our Internet website our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and any amendments to these reports, as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC. We are not including the information on our website as a part of, nor incorporating it by reference into, this report. You may read and copy any such reports and amendments thereto at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549 on official business days during the hours of 10:00 a.m. to 3:00 p.m. Please call the SEC at 1-800-SEC-0330 for information on the Public Reference Room. Additionally, the SEC maintains a website that contains annual, quarterly, and current reports, proxy statements, and other information that issuers (including us) file electronically with the SEC. The SEC’s website address is http://www.sec.gov.
PRODUCTS UNDER DEVELOPMENT
TG-1101 (ublituximab)
Overview
TG-1101 (ublituximab) is a chimeric, glycoengineered monoclonal antibody that targets a unique epitope on the CD20 antigen found on the surface of B-lymphocytes developed to aid in the depletion of circulating B-cells. We hold exclusive worldwide rights to develop and commercialize TG-1101 for all indications, except for the territories of France and Belgium which have been retained by LFB Biotechnologies (“LFB”), and South Korea and Southeast Asia which were licensed by us to Ildong Pharmaceutical Co. Ltd (“Ildong”) in November 2012.
Generally, anti-CD20 antibodies are believed to exert their B-cell depleting effects through three primary mechanisms: antibody dependent cell-mediated cytotoxicity (“ADCC”), complement dependent cytotoxicity (“CDC”), and direct or programmed cell death (“DCD” or “PCD”). TG-1101 has been specifically glycoengineered to enhance ADCC activity, which should enhance its ability to deplete B-cells and may improve its anti-cancer effects when compared to Rituxan®, the leading anti-CD20 monoclonal antibody, which had worldwide sales in 2014 of more than $8 billion.
Two single-agent, dose-escalation, Phase I studies were undertaken with TG-1101 to establish an optimal dose in patients with Non-Hodgkin’s Lymphoma (“NHL”) and Chronic Lymphocytic Leukemia (“CLL”). A two part first-in-human Phase I clinical trial was first completed in France in which TG-1101 was evaluated in relapsed or refractory CLL patients at doses as high as 450mg per infusion. Subsequently, a single-agent Phase I study was undertaken in the US enrolling patients with both NHL and CLL, dosing patients up to 1200mg per infusion. In both studies, single agent therapy with TG-1101 was deemed well tolerated by treating investigators and displayed promising clinical activity in relapsed and refractory patients.
| 2 |
In oncology settings, anti-CD20 therapy is generally used in combination with other anti-cancer agents where it demonstrates maximum activity as opposed to single agent usage. As a result, subsequent clinical development for TG-1101 has focused on combination therapy. Currently, our priority combination trials for TG-1101 are:
| · | The GENUINE Trial – a randomized controlled Phase 3 trial evaluating TG-1101 in combination with ibrutinib, for previously treated CLL patients with high risk cytogenetics; |
| · | The UNITY-CLL Trial – a randomized controlled Phase 3 trial evaluating TG-1101 in combination with TGR-1202, the Company’s development stage PI3Kδ inhibitor, for patients with front line and previously treated CLL; and |
| · | TG-1101 + TGR-1202 + Pembrolizumab for patients with CLL. |
Manufacturing of TG-1101 is currently performed by our partner, LFB Biotechnologies and we are in the process of bringing a secondary manufacturer on line.
Pre-Clinical Data Overview
The mechanism of action of anti-CD20 antibodies, including rituximab and TG-1101 has been elucidated and detailed in numerous academic and clinical studies. Upon conjugation of the antibody to the CD20 surface antigen, rituximab has been found to deplete B-lymphocytes through three primary mechanisms: ADCC, CDC, and DCD or PCD.
Antibody dependent cellular cytotoxicity, or ADCC, is a mechanism that is dependent on interactions between the Fc region of the antibody and the FcγR receptors on immune system effector cells, most notably the FcγRIIIA (CD16) receptor found on NK cells. These interactions trigger cells to release cytotoxic molecules and proteases resulting in B-cell death. TG-1101 is a third generation, type I chimeric IgG1 monoclonal antibody with a glycoengineered Fc region designed specifically to induce higher ADCC activity in comparison to rituximab, which has been demonstrated in pre-clinical models.
Clinical Data Overview and Recent Developments
Single Agent TG-1101 in Relapsed/Refractory CLL
A multicenter, open-label Phase I/Ib clinical trial of TG-1101 was completed which aimed to assess the safety, tolerability, and efficacy of TG-1101 in patients with relapsed or refractory CLL. This two part, first-in-man, dose escalating trial was conducted in nine centers in France with preliminary results presented at the 52nd and 53rd Annual American Society of Hematology (“ASH”) meetings, respectively.
The study regimen in Part 1 involved 21 patients in five dosing cohorts receiving infusions of TG-1101 at a dose ranging from 5mg to 450mg once weekly over the course of four weeks. Part 2 of this study included 12 patients and evaluated the safety and efficacy profile of TG-1101 when administered in an 8-dose regimen (150mg initial dose, followed by seven doses of 450mg).
TG-1101 was well tolerated with adverse events generally consistent with those exhibited by other anti-CD20 antibodies. In Part 2 of the study, rapid, near total blood lymphocyte depletion was observed in all patients. Overall response assessment at month four according to NCI-WG guidelines following treatment, TG-1101 was found to produce a durable partial response (“PR”) in 5/11 (45%) evaluable patients.
Single Agent TG-1101 in Relapsed/Refractory NHL & CLL
Our first US based trial entitled "An Open Label Phase I/II Trial of the Efficacy and Safety of TG-1101 in Patients with B-cell Non-Hodgkin’s Lymphoma who have Relapsed or are Refractory After CD20 Directed Antibody Therapy," was launched in the third quarter of 2012. In July 2014, this trial completed enrollment at 35 patients, of which 12 patients were included in the dose escalation component and 23 patients in various expansion cohorts. All enrolled patients were relapsed or refractory to Rituxan® or a Rituxan® containing regimen, and in most cases multiple other lines of therapy. Dr. Owen O'Connor, Professor of Medicine and Director, Center for Lymphoid Malignancies at New York Presbyterian Columbia Medical Center was the Principal Investigator for the multi-center study.
Data from this study was presented at the 50th American Society of Clinical Oncology (ASCO) 2014 Annual Meeting in Chicago, IL, and is summarized below:
| 3 |
TG-1101 was well tolerated at all dose levels tested in the 35 patients evaluable for safety, with Day 1 infusion related reactions (IRR) being the most frequently reported adverse event. The combined overall response rate (ORR) for the Phase 1 dose escalation component and expansion cohorts was 43% (30% PR, 13% CR) among the 30 rituximab relapsed/refractory patients evaluable for efficacy at the time of the presentation. TG-1101 displayed marked clinical activity as a single agent in a variety of lymphoma subtypes, reporting a 67% (4/6) response rate in patients with CLL and 44% (8/18) response rate in patients with indolent NHL (22% CR, 22% PR). Responses were durable, with a median duration of progression free survival (PFS) among patients who achieved SD or better not reached, and a median PFS for all patients on study of 34 weeks (n=30) at the time of the data presentation.
TG-1101 in Combination with TGR-1202 for Relapsed/Refractory NHL & CLL
In November 2013, we initiated a multi-center, Phase I study to evaluate the safety and efficacy of the combination of TG-1101 and TGR-1202, the Company's novel, once per day, PI3Kδ inhibitor, for patients with relapsed and/or refractory CLL and NHL. In this study, dosing of TGR-1202 commenced at 800mg (initial formulation) once per day (QD) with dose escalation proceeding in a 3+3 design. Dose-escalation up to 1200mg micronized formulation has been completed and expansion cohorts are now being evaluated at various doses. Additional cohorts were added to this study to explore the triple therapy combination of TG-1101, TGR-1202, and ibrutinib.
The MD Anderson Cancer Center is the lead center for the trial with Nathan Fowler, MD, Assistant Professor and Co-Director of Clinical Research in the Department of Lymphoma, as the Study Chair for the NHL patient group and Susan O’Brien, MD, formerly of MD Anderson and now Professor and Medical Director for Cancer Clinical Trials and Research at UC Irvine as the Study Chair for the CLL patient group.
Preliminary data from this study was presented at the 57th Annual American Society of Hematology (ASH) meeting held in December 2015 and is summarized below:
The combination of TG-1101 and TGR-1202 was well tolerated in the 71 patients evaluable for safety, with only 8% of patients discontinuing due to an adverse event. Notably, the only Grade 3/4 adverse event occurring in > 5% of patients was neutropenia. As of the data presentation, twenty-six patients had been on the combination of TG-1101 plus TGR-1202 for 6+ months, with no events of colitis reported. The combination displayed marked clinical activity in a variety of lymphoma subtypes, reporting an 80% (8/10) response rate in patients with CLL, a 71% (12/17) response rate in patients with indolent NHL, and a 35% (6/17) response rate in patients with DLBCL and Richter’s Transformation. The data from this study supports the current Phase 3 UNITY-CLL study of TG-1101 + TGR-1202 in CLL.
TG-1101 in Combination with Ibrutinib for Relapsed/Refractory MCL & CLL
In December 2013, we initiated a multi-center Phase 2 clinical trial to evaluate the safety and efficacy of the combination of TG-1101 and ibrutinib for patients with CLL and MCL. This is the first clinical trial evaluating the combination of TG-1101 and ibrutinib, an oral Bruton’s Tyrosine Kinase (BTK) inhibitor.
TG Therapeutics partnered with the US Oncology Network and other select centers throughout the United States on the study, with Jeff Sharman, MD, Medical Director for Hematology Research, US Oncology Network, as the Study Chair. This trial has completed enrollment.
Final data from this study was presented on the MCL cohort at the 57th Annual American Society of Hematology (ASH) meeting held in December 2015, and on the CLL cohort at the 13th International Congress on Malignant Lymphoma (ICML), held in June 2015 and is summarized below:
In the CLL cohort, TG-1101 in combination with ibrutinib was well tolerated in the 44 patients evaluable for safety, with day 1 infusion related reactions (IRR) being the most frequently reported adverse event (regardless of causality). In the MCL cohort, the combination was well tolerated in the 15 patients evaluable for safety, with fatigue being the most frequently reported adverse event (regardless of causality). Overall, in both CLL and MCL, aside from day 1 IRR, the addition of TG-1101 did not appear to alter the safety profile seen historically with single agent ibrutinib. Of the 59 patients treated, 40 CLL and 15 MCL patients were evaluable for response. The combination displayed marked clinical activity, reporting an 88% (35/40) response rate in patients with CLL, a 95% (19/20) response rate in those CLL patients with high-risk cytogenetics, and an 87% (13/15) response rate in patients with MCL. The data from this study supports the current Phase 3 GENUINE study of TG-1101 + ibrutinib in previously treated CLL patients with high-risk cytogenetics.
| 4 |
TG-1101 + Ibrutinib Phase 3 Study Program – The GENUINE Trial
We reached an agreement with the U.S. Food and Drug Administration (FDA) regarding a Special Protocol Assessment (SPA) on the design, endpoints and statistical analysis approach of a Phase 3 clinical trial for TG-1101 + ibrutinib for the treatment of previously treated CLL patients with high risk cytogenetics. The SPA provides agreement that the Phase 3 trial design adequately addresses objectives that would support the regulatory submission for drug approval.
The Phase 3 trial, named the GENUINE trial, is a randomized controlled clinical trial, with patients receiving either TG-1101 plus ibrutinib or ibrutinib alone. The trial will enroll approximately 330 patients, with the first 200 patients evaluated for overall response rate (ORR), and all patients followed for progression-free survival (PFS). As per the SPA, if the data is positive, we plan to use the ORR data from the trial as the basis for submission of a Biologics License Application (BLA) for accelerated approval for TG-1101, with the PFS assessment intended to support a filing for full approval.
TG-1101 in Combination with TGR-1202 Phase 3 Study Program – The UNITY-CLL Trial
In September 2015, we reached an agreement with the FDA regarding an SPA on the design, endpoints and statistical analysis approach of a Phase 3 clinical trial for the proprietary combination of TG-1101 plus TGR-1202, for the treatment of CLL. The SPA provides agreement that the Phase 3 trial design adequately addresses objectives that, if met, would support the regulatory submission for drug approval of both TG-1101 and TGR-1202 in combination.
The Phase 3 trial, called the UNITY-CLL trial, is a randomized controlled clinical trial that includes two key objectives: first, to demonstrate contribution of each agent in the TG-1101 + TGR-1202 regimen (the combination sometimes referred to as "1303"), and second, to demonstrate superiority in Progression Free Survival (PFS) over the standard of care to support the submission for full approval of the combination. The study will randomize patients into four treatment arms: TG-1101 + TGR-1202, TG-1101 alone, TGR-1202 alone, and an active control arm of obinutuzumab (GAZYVA®) + chlorambucil. An early interim analysis will assess contribution of each single agent in the TG-1101 + TGR-1202 combination regimen, which, if successful, will allow early termination of both single agent arms. A second interim analysis will be conducted following full enrollment into the study, which, if positive, we plan to utilize for accelerated approval. Assuming early termination of the TG-1101 and TGR-1202 single agent arms, the study will enroll approximately 450 patients.
TGR-1202
Overview
The phosphoinositide-3-kinases (“PI3Ks”) are a family of enzymes involved in various cellular functions, including cell proliferation and survival, cell differentiation, intracellular trafficking, and immunity. There are four isoforms of PI3K (alpha, beta, delta, and gamma), of which the delta (δ) isoform is strongly expressed in cells of hematopoietic origin, and often implicated in B-cell related lymphomas.
TGR-1202 is an orally available PI3K delta inhibitor with nanomolar potency to the delta isoform and high selectivity over the alpha, beta, and gamma isoforms. TGR-1202 has demonstrated activity in several pre-clinical models and primary cells from patients with various hematologic malignancies.
We hold exclusive rights to develop and commercialize TGR-1202 for all indications worldwide, except India which has been retained by Rhizen Pharmaceuticals, SA.
The Company’s Investigational New Drug (“IND”) application for TGR-1202 was accepted by the FDA in December 2012 and a first in-human Phase I clinical trial was initiated in January 2013.
Pre-Clinical Data Overview
In an enzyme based assay, TGR-1202 demonstrated potency and specificity towards PI3Kδ with >1000, 50 and 48-fold selectivity over the α, β, and γ PI3K isoforms.
| 5 |
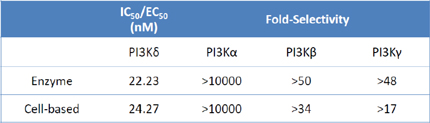 |
Figure 1: Potency
and |
Clinical Data Overview and Recent Developments
Initial clinical development of TGR-1202 was focused on establishing preliminary safety and efficacy in a wide variety of hematologic malignancies. Upon identification of safe and active doses of TGR-1202, a combination clinical trial program was opened, exploring TGR-1202 in combination with a variety of agents. In addition to the previously described study in combination with TG-1101 with or without the BTK inhibitor, ibrutinib, our current combination clinical trials for TGR-1202 are:
| · | TGR-1202 in combination with the anti-CD20 antibody, obinutuzumab (GAZYVA®) and chlorambucil in patients with CLL; |
| · | TGR-1202 in combination with the anti-CD30 antibody drug conjugate, brentuximab vedotin (ADCETRIS®), in patients with relapsed or refractory Hodgkin’s lymphoma; |
| · | TGR-1202 in combination with the BTK inhibitor, ibrutinib, in patients with previously treated CLL and MCL; and |
| · | TGR-1202 in combination with the JAK inhibitor, ruxolitinib (JAKAFI®), in patients with previously treated Myelofibrosis or Polycythemia Vera. |
Single Agent TGR-1202 in Patients with Relapsed/Refractory Hematologic Malignancies
In January 2013, the Company initiated a Phase I, open label, multi-center, first-in-human clinical trial of TGR-1202 in patients with hematologic malignancies. The study entitled TGR-1202-101, "A Phase I Dose Escalation Study Evaluating the Safety and Efficacy of TGR-1202 in Patients with Relapsed or Refractory Hematologic Malignancies," is being run in collaboration with the Sarah Cannon Research Institute in Nashville, TN with Howard “Skip” Burris, MD, Executive Director, Drug Development as the acting Study Chair. Enrollment is open to patients with relapsed or refractory NHL, CLL, and other select hematologic malignancies. As of February 2016, this study has closed to enrollment.
Data from this ongoing Phase I study was most recently presented at the 57th Annual American Society of Hematology (ASH) meeting held in December 2015, and is summarized below:
Overall, TGR-1202 was considered to be well-tolerated in the 81 patients evaluable for safety, with only 7% of patients discontinuing due to an adverse event. Limited Grade 3/4 adverse events were reported with anemia and neutropenia (each 9%) being the only grade 3/4 adverse events reported in greater than 5% of patients. Long-term safety has been well characterized with 47% (38 of 81) of patients on study more than 6 months, 27% (22 of 81) of patients on study more than 12 months, and the longest exposed to drug for more than 2.5 years. Of the 81 patients treated, 63 patients were evaluable for efficacy analysis, having been treated at therapeutic dose levels. TGR-1202 displayed marked clinical activity, reporting a 59% (10/17) response rate in patients with CLL (with a 94% nodal PR rate (16/17), and 75% (12/16) of follicular lymphoma patients demonstrated tumor reductions with a preliminary ORR of 38% (6/16).
TGR-1202 in Combination with obinutuzumab and chlorambucil in patients with CLL
In March 2014, the Company initiated a Phase I/Ib, open label, multi-center, clinical trial of TGR-1202 in combination with obinutuzumab and chlorambucil in patients with CLL, both treatment naïve and relapsed. The study entitled TGR-GA-106, " A Multi-center Phase I/Ib Study Evaluating the Efficacy and Safety of TGR-1202, a Novel PI3K Delta Inhibitor, in Combination with Obinutuzumab and Chlorambucil in Patients with Chronic Lymphocytic Leukemia (CLL)," is being led by Dr. Daruka Mahadevan of the West Clinic in Memphis, TN. As of February 2016, this study has completed enrollment.
Data from study was presented at the 57th Annual American Society of Hematology (ASH) meeting held in December 2015, and is summarized below:
| 6 |
Overall, the combination of TGR-1202, obinutuzumab and chlorambucil demonstrated acceptable tolerability, but notably differed from the toxicity profile observed with TGR-1202 and TG-1101 in patients with relapsed refractor CLL, specifically regarding neutropenia (78% vs. 30%), thrombocytopenia (78% vs. < 10%), and transaminase elevations (39% vs. 8%). TGR-1202 with obinutuzumab and chlorambucil reported a 95% (17/18) ORR in patients with CLL (with a 100% (15/15) ORR in those patients that were treatment naïve).
TGR-1202 Combination Trials
TGR-1202 is being evaluated in combination with the anti-CD30 antibody drug conjugate, brentuximab vedotin, in patients with relapsed or refractory Hodgkin’s lymphoma; in combination with the BTK inhibitor, ibrutinib, in patients with CLL and MCL; and in combination with the JAK inhibitor, ruxolitinib, in patients with Myelofibrosis or Polycythemia Vera. It is anticipated that preliminary results from these studies will be presented at future medical conferences.
TGR-1202 in Solid Tumors
In addition to the exploration of TGR-1202 in various hematologic malignancies, a study was opened in October 2015 to evaluate TGR-1202 as a single agent as well as in combination with various chemotherapies for the treatment of select solid tumors. The study, entitled TGR-1202-102, “A Phase I Study Evaluating the Safety and Efficacy of TGR-1202 Alone and in Combination with either nab-paclitaxel + Gemcitabine or with FOLFOX in Patients with Select Relapsed or Refractory Solid Tumors” is being run in collaboration with the Sarah Cannon Research Institute in Nashville, TN with Johanna Bendell, MD, Director of GI Oncology Research as the acting study chair.
Market Opportunity for TG-1101 & TGR-1202
Our lead products under development, TG-1101 and TGR-1202 are for the treatment of B-cell hematologic malignancies. Hematologic malignancies include cancers derived from the bone marrow and lymph tissue. The non-Hodgkin’s lymphomas (NHL) represent a heterogeneous subset of these malignancies. Underneath the single rubric of lymphoma exist some of the most aggressive growing cancers (Burkitt’s lymphoma, lymphoblastic lymphoma, diffuse large-B-cell lymphoma), as well as some of the most indolent (small lymphocytic lymphoma, follicular lymphoma, and marginal zone lymphoma). In the United States, NHL represents 4-5% of all new cancer cases, and is the eighth leading cause of cancer death. According to the American Cancer Society, it is estimated in 2016 that there will be 72,580 new cases in the United States, and 20,150 deaths from NHL, despite improvements in treatment. Chronic lymphocytic leukemia (CLL) affects mainly older adults and accounts for one third of all diagnosed cases of leukemia. In the US, an estimated 18,960 new cases of CLL will be reported in 2016 with deaths totaling 4,660 due to the disease according to American Cancer Society estimates. Despite improvements in therapy, up to one third of patients with aggressive NHL continue to die from their disease, and indolent lymphomas remain incurable in the absence of allogeneic stem cell transplant. The treatment paradigm for hematologic malignancies is well standardized in front line settings, with the anti-CD20 monoclonal antibody, rituximab, administered generally in combination with chemotherapeutic agents. While front line therapies are generally efficacious, there are numerous downsides, including a high rate of toxicity associated with exposure to chemotherapeutic agents. While initially responsive, most patients with hematologic malignancies will eventually relapse and require second, third, and sometimes more lines of therapy. As a result, there is a pressing need for new, innovative, targeted therapies for the treatment of this heterogeneous group of diseases.
Anti-CD20 antibodies have been approved and studied in a variety of diseases falling into several therapeutic areas including oncology, autoimmune disorders, and neurologic disease. NHL and CLL are the most common B-cell proliferative diseases for which rituximab, the first anti-CD20 antibody approved by the FDA, is the current gold standard treatment. While the addition of rituximab to chemotherapeutic treatment of NHL has dramatically improved patient outcomes, many patients will relapse or become refractory to rituximab containing regimens.
Rituximab resistance is becoming an increasing concern for clinicians as relapsing patients are exposed to multiple lines of rituximab containing regimens to treat recurrence of disease. It is estimated that over half of patients initially responsive to their first exposure to rituximab do not respond upon retreatment (Davis et al, 2000).
We believe these factors contribute to an immediate and sustained need for an anti-CD20 monoclonal antibody that is differentiated and potentially therapeutically superior to the gold standard rituximab in order to extend and enhance CD20 therapy as it stands today.
| 7 |
In addition to anti-CD20 therapy, novel targeted agents are now being introduced which target specific signaling pathways and enzymes known to exhibit aberrant activity and overexpression in B-cell malignancies such as Bruton’s Tyrosine Kinase (BTK), and Phosphoinositide-3-Kinase delta (PI3K delta). The PI3K/AKT/mTOR pathway has been the target of numerous pharmaceutical agents, both approved and in development, however only recently has the delta isoform of PI3K been identified as a potential target for the treatment of hematologic malignancies and other B-cell lymphoproliferative disorders. Idelalisib (ZYDELIG™), a PI3K delta specific inhibitor from Gilead Pharmaceuticals, was approved by the FDA in 2014 for patients with CLL and indolent NHL. Duvelisib (also known as IPI-145), a PI3K delta and gamma specific inhibitor under development by Infinity Pharmaceuticals has also shown preliminary activity in hematologic malignancies of both B- and T-cell origin. Other agents targeting kinases downstream of the B-cell receptor, such as the BTK inhibitor, ibrutinib, have displayed high rates of response in patients with relapsed and refractory B-cell malignancies and have been recently approved for these indications. While these agents have demonstrated high levels of single agent activity in B-cell disorders, their clinical activity has been shown to be greatly enhanced when utilized in combination with anti-CD20 agents.
As novel targeted agents gain FDA approval for the treatment of relapsed and refractory disease, it is anticipated that the size of this market will expand greatly as branded drugs enter use in multiple lines of therapy. Given the nature of the disease state for patients with hematologic malignancies, characterized by indolent disease progression and chronic relapses, the Company anticipates a great and growing need for novel agents that can be used alone or in combination with approved agents, and those currently under development to enhance the quality of life and extend the length of survival for patients suffering from hematologic malignancies.
IRAK4
Interleukin-1 Receptor Associated Kinase 4, referred to as IRAK4, is a key signaling kinase that becomes inappropriately activated in tumors that carry certain oncogenic mutations of MYD88, which can be found in most patients with Waldenström's Macroglobulinemia, a rare B-cell cancer, as well as in a sub-set of patients with Non-Hodgkin's Lymphoma and Chronic Lymphocytic Leukemia. Additionally, IRAK4 is a key component of signaling pathways which regulate immune and inflammatory processes suggesting that inhibition of IRAK4 may also be useful in the treatment of autoimmune related disorders. We hold global rights to develop and commercialize the IRAK4 program, which was licensed from Ligand Pharmaceuticals. Our IRAK4 program is currently in pre-clinical development. In April 2015, we presented pre-clinical data on the IRAK4 compounds at the 2015 American Association for Cancer Research (AACR) Annual Meeting held in Philadelphia, PA.
PD-L1 and GITR
In March 2015, we entered into a global collaboration agreement for the development and commercialization of anti-PD-L1 and anti-GITR antibody research programs in the field of hematological malignancies. Our anti-PD-L1 and anti-GITR programs are currently in pre-clinical development.
COSTS AND TIME TO COMPLETE PRODUCT DEVELOPMENT
The information below provides estimates regarding the costs associated with the completion of the current development phase and our current estimated range of the time that will be necessary to complete that development phase for our key pipeline products. We also direct your attention to the risk factors which could significantly affect our ability to meet these cost and time estimates found in this report in Item 1A under the heading “Risks Related to the Company’s Business and Industry.”
Product candidate |
Target indication |
Development status |
Completion of phase |
Estimated cost to complete phase |
|||||
| TG-1101 (ublituximab) |
In combination with ibrutinib in previously treated high-risk CLL patients | Phase III | 2017* | Approximately $10 million | |||||
| TG-1101 & TGR-1202 | In combination in CLL patients | Phase III | 2018** | Approximately $15 million | |||||
*Completion of phase for this study indicates completion of Part I of the study, the ORR endpoint, which, if successful, would support an accelerated approval
** Completion of phase for this study indicates completion of portion of study, which, if successful, would support an accelerated approval
Completion dates and costs in the above table are estimates due to the uncertainties associated with clinical trials and the related requirements of development. In the cases where the requirements for clinical trials and development programs have not been fully defined, or are dependent on the success of other trials, we cannot estimate trial completion or cost with any certainty. The actual spending on each trial during the year is also dependent on funding. We therefore direct your attention to Item 7 under the heading “Liquidity and Capital Resources.”
| 8 |
INTELLECTUAL PROPERTY AND PATENTS
General
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and elsewhere in the world.
We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors. This knowledge and experience we call “know-how.” To help protect our proprietary know-how which is not patentable, and for inventions for which patents may be difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all employees, consultants, advisors and other contractors to enter into confidentiality agreements which prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Patents and other proprietary rights are crucial to the development of our business. We will be able to protect our proprietary technologies from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents, supported by regulatory exclusivity or are effectively maintained as trade secrets. We have a number of patents and patent applications related to our compounds and other technology, but we cannot guarantee the scope of protection of the issued patents, or that such patents will survive a validity or enforceability challenge, or that any of the pending patent applications will issue as patents.
Generally, patent applications in the U.S. are maintained in secrecy for a period of 18 months or more. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we are not certain that we were the first to make the inventions covered by each of our pending patent applications or that we were the first to file those patent applications. The patent positions of biotechnology and pharmaceutical companies are highly uncertain and involve complex legal and factual questions. Therefore, we cannot predict the breadth of claims allowed in biotechnology and pharmaceutical patents, or their enforceability. To date, there has been no consistent policy regarding the breadth of claims allowed in biotechnology patents. Third parties or competitors may challenge or circumvent our patents or patent applications, if issued. If our competitors prepare and file patent applications in the U.S. that claim technology also claimed by us, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost, even if the eventual outcome is favorable to us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that before we commercialize any of our products, any related patent may expire or remain in existence for only a short period following commercialization, thus reducing any advantage of the patent. However, the life of a patent covering a product that has been subject to regulatory approval may have the ability to be extended through the patent restoration program, although any such extension could still be minimal.
If a patent is issued to a third party containing one or more preclusive or conflicting claims, and those claims are ultimately determined to be valid and enforceable, we may be required to obtain a license under such patent or to develop or obtain alternative technology. In the event of litigation involving a third party claim, an adverse outcome in the litigation could subject us to significant liabilities to such third party, require us to seek a license for the disputed rights from such third party, and/or require us to cease use of the technology. Further, our breach of an existing license or failure to obtain a license to technology required to commercialize our products may seriously harm our business. We also may need to commence litigation to enforce any patents issued to us or to determine the scope and validity of third-party proprietary rights. Litigation would involve substantial costs.
TG-1101
Pursuant to our license for TG-1101 (ublituximab) with LFB Biotechnologies, GTC Biotherapeutics, and LFB/GTC LLC, we have the exclusive commercial rights to a series of patents and patent applications in the U.S. and in multiple countries around the world, as well as a non-exclusive license to additional background patent rights. These patents and patent protections include composition of matter patents relating to the structure and mechanism of action for TG-1101 as well as method of use patents which cover use of TG-1101 in combination with various agents and for various therapeutic indications.
| 9 |
In the United States, we have, through our license agreement, access to 12 issued patent and 6 patent applications covering TG-1101 which expire between 2021 and 2033, excluding any patent term extensions, as well as granted and pending foreign counterpart patent filings related to these patent families. These patents include claims related to the manufacture and use of TG-1101. Additionally, we have over 30 granted patents outside the US, and over 25 patent applications pending worldwide also including claims directed to the composition of matter and methods of treatment with TG-1101 in various settings.
TGR-1202
Pursuant to our license for TGR-1202 with Rhizen, we have the exclusive commercial rights to a series of patent applications in the U.S. and abroad. The patent applications include composition of matter patents relating to the structure, mechanism of action, and formulation for TGR-1202 as well as method of use patents which cover use of TGR-1202 in combination with various agents and for various therapeutic indications. Our composition of matter patent for TGR-1202 has been issued in the United States, which affords patent protection until 2033, exclusive of patent term extensions. All other patent applications currently filed for TG-1202 are currently pending. Because the dates for any potential regulatory approval are currently unknown we cannot predict the expected expiration date, and it is possible that the life of these patents following regulatory approval could be minimal.
IRAK4
Pursuant to our license for the IRAK4 program with Ligand, we have the exclusive commercial rights to a patent family which covers the composition of matter and proposed methods of use for various therapeutic indications. All patent applications currently filed for the IRAK4 program are currently pending. Because the date for any potential regulatory approval is currently unknown we cannot predict the expected expiration date, and it is possible that the life of these patents following regulatory approval could be minimal.
The patent rights that we own or have licensed relating to our product candidates are limited in ways that may affect our ability to exclude third parties from competing against us if we obtain regulatory approval to market these product candidates. See “Item 1A – Risk Factors — Risks Related to the Company’s Intellectual Property.”
Proof of direct infringement by a competitor for method of use patents can prove difficult because the competitors making and marketing a product typically do not engage in the patented use. Additionally, proof that a competitor contributes to or induces infringement of a patented method of use by another can also prove difficult because an off-label use of a product could prohibit a finding of contributory infringement and inducement of infringement requires proof of intent by the competitor.
Moreover, physicians may prescribe such a competitive identical product for indications other than the one for which the product has been approved, or off-label indications, that are covered by the applicable patents. Although such off-label prescriptions may directly infringe or contribute to or induce infringement of method of use patents, such infringement is difficult to prevent or prosecute.
In addition, the limited patent protection described above may adversely affect the value of our product candidates and may inhibit our ability to obtain a corporate partner at terms acceptable to us, if at all.
Other Intellectual Property Rights
We depend upon trademarks, trade secrets, know-how and continuing technological advances to develop and maintain our competitive position. To maintain the confidentiality of trade secrets and proprietary information, we require our employees, scientific advisors, consultants and collaborators, upon commencement of a relationship with us, to execute confidentiality agreements and, in the case of parties other than our research and development collaborators, to agree to assign their inventions to us. These agreements are designed to protect our proprietary information and to grant us ownership of technologies that are developed in connection with their relationship with us. These agreements may not, however, provide protection for our trade secrets in the event of unauthorized disclosure of such information.
In addition to patent protection, we may utilize orphan drug regulations or other provisions of the Food, Drug and Cosmetic Act of 1938, as amended, or FDCA, to provide market exclusivity for certain of our drug candidates. Orphan drug regulations provide incentives to pharmaceutical and biotechnology companies to develop and manufacture drugs for the treatment of rare diseases, currently defined as diseases that exist in fewer than 200,000 individuals in the U.S., or, diseases that affect more than 200,000 individuals in the U.S. but that the sponsor does not realistically anticipate will generate a net profit. Under these provisions, a manufacturer of a designated orphan-drug can seek tax benefits, and the holder of the first FDA approval of a designated orphan product will be granted a seven-year period of marketing exclusivity for such FDA-approved orphan product.
| 10 |
Pursuant to these regulations, TG-1101 (ublituximab) has received Orphan-Drug designation from the FDA for the treatment of Marginal Zone Lymphoma (Nodal and Extranodal) in September 2013, for the treatment of CLL in August of 2010, and Orphan-Drug designation by the European Medicines Agency (“EMA”) for the treatment of CLL in November of 2009. We believe that TG-1101 may be eligible for additional orphan drug designations; however, we cannot assure you that TG-1101, or any other drug candidates we may acquire or in-license, will obtain such orphan drug designations. Additionally, upon FDA approval, we believe that TG-1101 would qualify as a New Chemical Entity, or NCE, which provides for five years of exclusivity following approval.
We cannot assure you that any other drug candidates we may acquire or in-license, will obtain such orphan drug designation or that we will be the first to receive FDA approval for such drugs so as to be eligible for market exclusivity protection.
LICENSING AGREEMENTS AND COLLABORATIONS
We have formed strategic alliances with a number of companies for the manufacture and commercialization of our products. Our current key strategic alliances are discussed below.
TG-1101
LFB Biotechnologies S.A.S, GTC Biotherapeutics, LFB/GTC LLC.
In January 2012, we entered into an exclusive license agreement with LFB Biotechnologies, GTC Biotherapeutics, and LFB/GTC LLC, all wholly-owned subsidiaries of LFB Group, relating to the development of TG-1101. Under the license agreement, we have acquired the exclusive worldwide rights (exclusive of France/Belgium) for the development and commercialization of TG-1101 (ublituximab). To date, we have made no payments to LFB Group and LFB Group is eligible to receive payments of up to an aggregate of approximately $31.0 million upon our successful achievement of certain clinical development, regulatory and sales milestones, in addition to royalty payments on net sales of TG-1101 at a royalty rate that escalates from mid-single digits to high-single digits. The license will terminate on a country by country basis upon the expiration of the last licensed patent right or 15 years after the first commercial sale of a product in such country, unless the agreement is earlier terminated (i) by LFB if the Company challenges any of the licensed patent rights, (ii) by either party due to a breach of the agreement, or (iii) by either party in the event of the insolvency of the other party.
Ildong Pharmaceutical Co. Ltd.
In November 2012, we entered into an exclusive (within the territory) sublicense agreement with Ildong relating to the development and commercialization of TG-1101 in South Korea and Southeast Asia. Under the terms of the sublicense agreement, Ildong has been granted a royalty bearing, exclusive right, including the right to grant sublicenses, to develop and commercialize TG-1101 in South Korea, Taiwan, Singapore, Indonesia, Malaysia, Thailand, Philippines, Vietnam, and Myanmar. To date, we have received $2 million in the form of an upfront payment from Ildong, and are eligible to receive sales based milestone payments up to an aggregate of $5 million and royalty payments on net sales of TG-1101 at a royalty rate that escalates from mid-teens to high-teens upon approval in South Korea and/or Southeast Asia. The license will terminate on a country by country basis upon the expiration of the last licensed patent right or 15 years after the first commercial sale of a product in such country, unless the agreement is earlier terminated (i) by Ildong if the Company challenges any of the licensed patent rights, (ii) by either party due to a breach of the agreement, or (iii) by either party in the event of the insolvency of the other party.
TGR-1202
In September 2014, we exercised our option to license the global rights to TGR-1202, thereby entering into an exclusive licensing agreement (the “TGR-1202 License”) with Rhizen Pharmaceuticals, S A (“Rhizen”) for the development and commercialization of TGR-1202. Prior to this, we had been jointly developing TGR-1202 in a 50:50 joint venture with Rhizen.
| 11 |
Under the terms of the TGR-1202 License, Rhizen received a $4.0 million cash payment and 371,530 shares of our common stock as an upfront license fee. With respect to TGR-1202, Rhizen will be eligible to receive regulatory filing, approval and sales based milestone payments in the aggregate of approximately $175 million, a small portion of which will be payable on the first New Drug Application (NDA) filing and the remainder on approval in multiple jurisdictions for up to two oncology indications and one non-oncology indication and attaining certain sales milestones. In addition, if TGR-1202 is co-formulated with another drug to create a new product (a "New Product"), Rhizen will be eligible to receive similar regulatory approval and sales based milestone payments for such New Product. Additionally, Rhizen will be entitled to tiered royalties that escalate from high single digits to low double digits on our future net sales of TGR-1202 and any New Product. In lieu of sales milestones and royalties on net sales, Rhizen shall also be eligible to participate in sublicensing revenue, if any, based on a percentage that decreases as a function of the number of patients treated in clinical trials following the exercise of the license option. Rhizen will retain global manufacturing rights to TGR-1202, provided that they are price competitive with alternative manufacturers. The license will terminate on a country by country basis upon the expiration of the last licensed patent right or any other exclusivity right in such country, unless the agreement is earlier terminated (i) by us for any reason, (ii) by either party due to a breach of the agreement.
IRAK4
In June 2014, we entered into an exclusive licensing agreement with Ligand Pharmaceuticals Incorporated ("Ligand") for the development and commercialization of Ligand's interleukin-1 receptor associated kinase-4 ("IRAK4") inhibitor technology, which currently is in preclinical development for potential use against certain cancers and autoimmune diseases. IRAK4 is a serine/threonine protein kinase that is a key downstream signaling component of the interleukin-1 receptor and multiple toll-like receptors.
Under the terms of the license agreement, Ligand received 125,000 shares of our common stock as an upfront license fee. Ligand will also be eligible to receive maximum potential milestone payments of approximately $207 million upon the achievement of specific clinical, regulatory and commercial milestone events. Additionally, Ligand will be entitled to royalties on our future net sales of licensed products containing IRAK4 inhibitors. The basic royalty rate for licensed products covered by Ligand's issued patents will be 6% for annual sales of up to $1 billion and 9.5% for annual sales in excess of that threshold. The license will terminate on a country by country basis upon the expiration of the last licensed patent right or 10 years after the first commercial sale of a product in such country, unless the agreement is earlier terminated by either party due to a breach of the agreement in the event of the insolvency of the other party.
PD-L1 and GITR
In March 2015, we entered into a Global Collaboration (the “Collaboration”) with Checkpoint Therapeutics, Inc. (“Checkpoint”), a subsidiary of Fortress Biotech, Inc. (“FBIO”) for the development and commercialization of Checkpoint’s anti-PD-L1 and anti-GITR antibody research programs in the field of hematological malignancies.
Under the terms of the Collaboration, we will make an up-front payment of $500,000 as well as make development and sales-based milestone payments up to an aggregate of $164 million, and will pay a tiered single digit royalty on net sales. The royalty term will terminate on a country by country basis upon the later of (i) ten years after the first commercial sale of any applicable licensed product in such country, or (ii) the expiration of the last-to-expire patent held by Dana Farber containing a valid claim to any licensed product in such country.
COMPETITION
Competition in the pharmaceutical and biotechnology industries is intense. Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies that are active in different but related fields represent substantial competition for us. Many of our competitors have significantly greater capital resources, larger research and development staffs and facilities and greater experience in drug development, regulation, manufacturing and marketing than we do. These organizations also compete with us to recruit qualified personnel, attract partners for joint ventures or other collaborations, and license technologies that are competitive with ours. To compete successfully in this industry we must identify novel and unique drugs or methods of treatment and then complete the development of those drugs as treatments in advance of our competitors.
The drugs that we are attempting to develop will have to compete with existing therapies. In addition, a large number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. Other companies have products or drug candidates in various stages of pre-clinical or clinical development to treat diseases for which we are also seeking to discover and develop drug candidates. Some of these potential competing drugs are further advanced in development than our drug candidates and may be commercialized earlier.
| 12 |
If approved, we expect TG-1101 to compete directly with Roche Group’s Rituxan® (rituximab) and Gazyva® (obinutuzumab), and Genmab and GlaxoSmithKline’s Arzerra® (ofatumumab) among others, each of which is currently approved for the treatment of various diseases including NHL and CLL. In addition, other pharmaceutical companies are developing anti-CD20 antibodies which, if approved, would potentially compete with TG-1101. New developments, including the development of other pharmaceutical technologies and methods of treating disease, occur in the pharmaceutical and life sciences industries at a rapid pace.
With respect to TGR-1202, if approved, we expect to compete directly with Gilead’s Zydelig™ (idelalisib), as well as with other PI3K delta inhibitors which are currently in development, which, if approved, would potentially compete with TGR-1202, such as Infinity Pharmaceuticals’ duvelisib (IPI-145). In addition, there are numerous other novel therapies targeting similar pathways to TGR-1202 in development, which would also compete with TGR-1202 in similar indications, such as the BTK inhibitor, ibrutinib (FDA approved for Mantle Cell Lymphoma, CLL, and Waldenstrom’s Macroglobulinemia marketed by AbbVie and Janssen), the BTK inhibitor acalabrutinib or ACP-196 (under clinical development by AstraZeneca), or the BCL-2 inhibitor venetoclax or ABT-199 (under clinical development by AbbVie and Roche), among others.
Additional information can be found under Item 1A - Risk Factors – Other Risks Related to Our Business within this report.
SUPPLY AND MANUFACTURING
We have limited experience in manufacturing products for clinical or commercial purposes. We currently do not have any manufacturing capabilities. We have established contract manufacturing relationships for the supply of TG-1101 as part of our license agreement with LFB Biotechnologies. We have also established contract manufacturing relationships for the supply of TGR-1202 as part of our licensing agreement with Rhizen, and contract manufacturing relationships to support our IRAK4 development program. As with any supply program, obtaining pre-clinical and clinical materials of sufficient quality and quantity to meet the requirements of our development programs cannot be guaranteed and we cannot ensure that we will be successful in this endeavor. In addition, we anticipate the need for the current scale of production for each of our products to be significantly expanded as we enter later stages of development. There can be no assurance given that such scale-up will be successful in providing pharmaceutical product that is of sufficient quantity, or of a quality that is consistent with our previously established specifications, or that meets the requirements set by regulatory agencies under which we may seek approval of our product candidates.
At the time of commercial sale, to the extent possible and commercially practicable, we would seek to engage a back-up supplier for each of our product candidates. Until such time, we expect that we will rely on a single contract manufacturer to produce each of our product candidates under current Good Manufacturing Practice, or cGMP, regulations. Our third-party manufacturers have a limited number of facilities in which our product candidates can be produced and will have limited experience in manufacturing our product candidates in quantities sufficient for commercialization. Our third-party manufacturers will have other clients and may have other priorities that could affect their ability to perform the work satisfactorily and/or on a timely basis. Both of these occurrences would be beyond our control.
We expect to similarly rely on contract manufacturing relationships for any products that we may in-license or acquire in the future. However, there can be no assurance that we will be able to successfully contract with such manufacturers on terms acceptable to us, or at all.
Contract manufacturers are subject to ongoing periodic and unannounced inspections by the FDA, the Drug Enforcement Administration and corresponding state agencies to ensure strict compliance with cGMP and other state and federal regulations. Our contractors outside of the United States face similar challenges from the numerous local and regional agencies and authorized bodies. We do not have control over third-party manufacturers’ compliance with these regulations and standards, other than through contractual obligations. If they are deemed out of compliance with cGMPs, product recalls could result, inventory could be destroyed, production could be stopped and supplies could be delayed or otherwise disrupted.
If we need to change manufacturers after commercialization, the FDA and corresponding foreign regulatory agencies must approve these new manufacturers in advance, which will involve testing and additional inspections to ensure compliance with FDA regulations and standards and may require significant lead times and delay. Furthermore, switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly or on terms acceptable to us, or at all.
| 13 |
GOVERNMENT AND INDUSTRY REGULATION
Numerous governmental authorities, principally the FDA and corresponding state and foreign regulatory agencies, impose substantial regulations upon the clinical development, manufacture and marketing of our drug candidates, as well as our ongoing research and development activities. None of our drug candidates have been approved for sale in any market in which we have marketing rights. Before marketing in the U.S., any drug that we develop must undergo rigorous pre-clinical testing and clinical trials and an extensive regulatory approval process implemented by the FDA under the FDCA. The FDA regulates, among other things, the pre-clinical and clinical testing, safety, efficacy, approval, manufacturing, record keeping, adverse event reporting, packaging, labeling, storage, advertising, promotion, export, sale and distribution of biopharmaceutical products.
The regulatory review and approval process is lengthy, expensive and uncertain. We are required to submit extensive pre-clinical and clinical data and supporting information to the FDA for each indication or use to establish a drug candidate’s safety and efficacy before we can secure FDA approval to market or sell a product in the U.S. The approval process takes many years, requires the expenditure of substantial resources and may involve ongoing requirements for post-marketing studies or surveillance. Before commencing clinical trials in humans, we must submit an IND to the FDA containing, among other things, pre-clinical data, chemistry, manufacturing and control information, and an investigative plan. Our submission of an IND may not result in FDA authorization to commence a clinical trial.
The FDA may permit expedited development, evaluation, and marketing of new therapies intended to treat persons with serious or life-threatening conditions for which there is an unmet medical need under its fast track drug development programs. A sponsor can apply for fast track designation at the time of submission of an IND, or at any time prior to receiving marketing approval of the new drug application, or NDA. To receive Fast Track designation, an applicant must demonstrate:
| · | that the drug is intended to treat a serious or life-threatening condition; |
| · | that the drug is intended to treat a serious aspect of the condition; and |
| · | that the drug has the potential to address unmet medical needs, and this potential is being evaluated in the planned drug development program. |
The FDA must respond to a request for fast track designation within 60 calendar days of receipt of the request. Over the course of drug development, a product in a fast track development program must continue to meet the criteria for fast track designation. Sponsors of products in fast track drug development programs must be in regular contact with the reviewing division of the FDA to ensure that the evidence necessary to support marketing approval will be developed and presented in a format conducive to an efficient review. Sponsors of products in fast track drug development programs ordinarily are eligible for priority review of a completed application in six months or less and also may be permitted to submit portions of a New Drug Application (“NDA”) to the FDA for review before the complete application is submitted.
Sponsors of drugs designated as fast track also may seek approval under the FDA’s accelerated approval regulations. Under this authority, the FDA may grant marketing approval for a new drug product on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence, to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. Approval will be subject to the requirement that the applicant study the drug further to verify and describe its clinical benefit where there is uncertainty as to the relation of the surrogate endpoint to clinical benefit or uncertainty as to the relation of the observed clinical benefit to ultimate outcome. Post-marketing studies are usually underway at the time an applicant files the NDA. When required to be conducted, such post-marketing studies must also be adequate and well-controlled. The applicant must carry out any such post-marketing studies with due diligence. Many companies who have been granted the right to utilize an accelerated approval approach have failed to obtain approval. Moreover, negative or inconclusive results from the clinical trials we hope to conduct or adverse medical events could cause us to have to repeat or terminate the clinical trials. Accordingly, we may not be able to complete the clinical trials within an acceptable time frame, if at all, and, therefore, could not submit the NDA to the FDA or foreign regulatory authorities for marketing approval.
In addition, sponsors may also apply to the FDA for Breakthrough Therapy Designation. The Breakthrough Therapy Designation is intended to expedite the development and review of a potential new drug for serious or life-threatening diseases where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy was enacted as part of the 2012 Food and Drug Administration Safety and Innovation Act.
| 14 |
Clinical testing must meet requirements for institutional review board oversight, informed consent and good clinical practices, and must be conducted pursuant to an IND, unless exempted.
For purposes of NDA approval, clinical trials are typically conducted in the following sequential phases:
| · | Phase 1: The drug is administered to a small group of humans, either healthy volunteers or patients, to test for safety, dosage tolerance, absorption, metabolism, excretion, and clinical pharmacology. |
| · | Phase 2: Studies are conducted on a larger number of patients to assess the efficacy of the product, to ascertain dose tolerance and the optimal dose range, and to gather additional data relating to safety and potential adverse events. |
| · | Phase 3: Studies establish safety and efficacy in an expanded patient population. |
| · | Phase 4: The FDA may require Phase 4 post-marketing studies to find out more about the drug’s long-term risks, benefits, and optimal use, or to test the drug in different populations. |
The length of time necessary to complete clinical trials varies significantly and may be difficult to predict. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Additional factors that can cause delay or termination of our clinical trials, or that may increase the costs of these trials, include:
| · | slow patient enrollment due to the nature of the clinical trial plan, the proximity of patients to clinical sites, the eligibility criteria for participation in the study or other factors; |
| · | inadequately trained or insufficient personnel at the study site to assist in overseeing and monitoring clinical trials or delays in approvals from a study site’s review board; |
| · | longer treatment time required to demonstrate efficacy or determine the appropriate product dose; |
| · | insufficient supply of the drug candidates; |
| · | adverse medical events or side effects in treated patients; and |
| · | ineffectiveness of the drug candidates. |
In addition, the FDA, equivalent foreign regulatory authority, or a data safety monitoring committee for a trial may place a clinical trial on hold or terminate it if it concludes that subjects are being exposed to an unacceptable health risk, or for futility. Any drug is likely to produce some toxicity or undesirable side effects in animals and in humans when administered at sufficiently high doses and/or for a sufficiently long period of time. Unacceptable toxicity or side effects may occur at any dose level at any time in the course of studies in animals designed to identify unacceptable effects of a drug candidate, known as toxicological studies, or clinical trials of drug candidates. The appearance of any unacceptable toxicity or side effect could cause us or regulatory authorities to interrupt, limit, delay or abort the development of any of our drug candidates and could ultimately prevent approval by the FDA or foreign regulatory authorities for any or all targeted indications.
Sponsors of drugs may apply for an SPA from the FDA. The SPA process is a procedure by which the FDA provides official evaluation and written guidance on the design and size of proposed protocols that are intended to form the basis for a new drug application. However, final marketing approval depends on the results of efficacy, the adverse event profile and an evaluation of the benefit/risk of treatment demonstrated in the Phase 3 trial. The SPA agreement may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of a substantial scientific issue essential to product safety or efficacy.
Before receiving FDA approval to market a product, we must demonstrate that the product is safe and effective for its intended use by submitting to the FDA an NDA or BLA containing the pre-clinical and clinical data that have been accumulated, together with chemistry and manufacturing and controls specifications and information, and proposed labeling, among other things. The FDA may refuse to accept an NDA/BLA for filing if certain content criteria are not met and, even after accepting an NDA/BLA, the FDA may often require additional information, including clinical data, before approval of marketing a product.
| 15 |
It is also becoming more common for the FDA to request a Risk Evaluation and Mitigation Strategy, or REMS, as part of a NDA/BLA. The REMS plan contains post-market obligations of the sponsor to train prescribing physicians, monitor off-label drug use, and conduct sufficient Phase 4 follow-up studies and registries to ensure the continued safe use of the drug.
As part of the approval process, the FDA must inspect and approve each manufacturing facility. Among the conditions of approval is the requirement that a manufacturer’s quality control and manufacturing procedures conform to cGMP. Manufacturers must expend significant time, money and effort to ensure continued compliance, and the FDA conducts periodic inspections to certify compliance. It may be difficult for our manufacturers or us to comply with the applicable cGMP, as interpreted by the FDA, and other FDA regulatory requirements. If we, or our contract manufacturers, fail to comply, then the FDA may not allow us to market products that have been affected by the failure.
If the FDA grants approval, the approval will be limited to those disease states, conditions and patient populations for which the product is safe and effective, as demonstrated through clinical studies. Further, a product may be marketed only in those dosage forms and for those indications approved in the NDA/BLA. Certain changes to an approved NDA/BLA, including, with certain exceptions, any significant changes to labeling, require approval of a supplemental application before the drug may be marketed as changed. Any products that we manufacture or distribute pursuant to FDA approvals are subject to continuing monitoring and regulation by the FDA, including compliance with cGMP and the reporting of adverse experiences with the drugs. The nature of marketing claims that the FDA will permit us to make in the labeling and advertising of our products will generally be limited to those specified in FDA approved labeling, and the advertising of our products will be subject to comprehensive monitoring and regulation by the FDA. Drugs whose review was accelerated may carry additional restrictions on marketing activities, including the requirement that all promotional materials are pre-submitted to the FDA. Claims exceeding those contained in approved labeling will constitute a violation of the FDCA. Violations of the FDCA or regulatory requirements at any time during the product development process, approval process, or marketing and sale following approval may result in agency enforcement actions, including withdrawal of approval, recall, seizure of products, warning letters, injunctions, fines and/or civil or criminal penalties. Any agency enforcement action could have a material adverse effect on our business.
Should we wish to market our products outside the U.S., we must receive marketing authorization from the appropriate foreign regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, companies are typically required to apply for foreign marketing authorizations at a national level. However, within the European Union, registration procedures are available to companies wishing to market a product in more than one European Union member state. Typically, if the regulatory authority is satisfied that a company has presented adequate evidence of safety, quality and efficacy, then the regulatory authority will grant a marketing authorization. This foreign regulatory approval process, however, involves risks similar or identical to the risks associated with FDA approval discussed above, and therefore we cannot guarantee that we will be able to obtain the appropriate marketing authorization for any product in any particular country.
Failure to comply with applicable federal, state and foreign laws and regulations would likely have a material adverse effect on our business. In addition, federal, state and foreign laws and regulations regarding the manufacture and sale of new drugs are subject to future changes. We cannot predict the likelihood, nature, effect or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
EMPLOYEES
As of March 1, 2016, we had forty full and part-time employees. None of our employees are represented by a collective bargaining agreement, and we have never experienced a work stoppage. We consider our relations with our employees to be good.
| 16 |
You should carefully consider the following risks and uncertainties. If any of the following occurs, our business, financial condition or operating results could be materially harmed. An investment in our securities is speculative in nature, involves a high degree of risk, and should not be made by an investor who cannot bear the economic risk of its investment for an indefinite period of time and who cannot afford the loss of its entire investment. You should carefully consider the following risk factors and the other information contained elsewhere in this Annual Report before making an investment in our securities.
Risks Related to Our Business and Industry
Because we have in-licensed our product candidates from third parties, any dispute with or non-performance by our licensors will adversely affect our ability to develop and commercialize the applicable product candidates.
Our product candidates have been in-licensed from third parties. Under the terms of our license agreements, the licensors generally will have the right to terminate such agreement in the event of a material breach by us. The licensors will also have the right to terminate the agreement in the event we fail to use diligent and reasonable efforts to develop and commercialize the product candidate worldwide.
If there is any conflict, dispute, disagreement or issue of non-performance between us and our licensing partners regarding our rights or obligations under the license agreements, including any such conflict, dispute or disagreement arising from our failure to satisfy payment obligations under such agreement, our ability to develop and commercialize the affected product candidate and our ability to enter into collaboration or marketing agreements for the affected product candidate may be adversely affected. Any loss of our rights under these license agreements would delay or completely terminate its product development efforts for the affected product candidate.
We do not have full internal development capabilities, and are thus reliant upon our partners and third parties to generate clinical, preclinical and quality data necessary to support the regulatory applications needed to conduct clinical trials and file for marketing approval.
In order to submit and maintain an IND, Biologics License Application (“BLA”), or New Drug Application (“NDA”) to the FDA, it is necessary to submit all information on the clinical, non-clinical, chemistry, manufacturing, controls and quality aspects of the product candidate. We rely on our third party contractors and our licensing partners to provide a significant portion of this data. If we are unable to obtain this data, or the data is not sufficient to meet the regulatory requirements, we may experience significant delays in our development programs. Additionally, an IND must be active in each division in which we intend to conduct clinical trials. Currently we do not have an active IND for any of the IRAK4 inhibitors, nor for our anti-PD-L1 and anti-GITR antibodies. Additionally, there can be no assurance given that any of the molecules under development in our IRAK4 inhibitor program or in our anti-PD-L1 and anti-GITR antibody research program will demonstrate sufficient pharmacologic properties during pre-clinical evaluation to advance to IND enabling studies, or that such IND enabling studies, if any are conducted, will provide data sufficient to support the filing of an IND, or that such IND, if filed, would be accepted by any FDA division under which we would seek to develop any product candidate. While we maintain an active IND for TG-1101 and TGR-1202 enabling the conduct of studies in the FDA’s Division of Hematology and Oncology, there can be no assurance that we will be successful in obtaining an active IND for TG-1101 or TGR-1202 in any other division under whose supervision we may seek to develop our product candidates, or that the FDA will allow us to continue the development of our product candidates in those divisions where we maintain an active IND.
We are highly dependent on the success of our product candidates and cannot give any assurance that these or any future product candidates will be successfully commercialized.
We are a development-stage biopharmaceutical company, and do not currently have any commercial products that generate revenues or any other sources of revenue. We may never be able to successfully develop marketable products. Our pharmaceutical development methods are unproven and may not lead to commercially viable products for any of several reasons.
If we are unable to develop, or receive regulatory approval for or successfully commercialize any of our product candidates, we will not be able to generate product revenues.
Because the results of preclinical studies and early clinical trials are not necessarily predictive of future results, any product candidate we advance into clinical trials may not have favorable results in later clinical trials, if any, or receive regulatory approval.
Pharmaceutical development has inherent risk. We will be required to demonstrate through adequate and well-controlled clinical trials that our product candidates are effective with a favorable benefit-risk profile for use in diverse populations for their target indications before we can seek regulatory approvals for their commercial sale. Success in early clinical trials does not mean that later clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety or efficacy despite having progressed through initial clinical testing. Companies frequently suffer significant setbacks in advanced clinical trials, even after earlier clinical trials have shown promising results. In addition, there is typically an extremely high rate of failure of pharmaceutical candidates proceeding through clinical trials.
| 17 |
We plan on conducting additional Phase I, II and III clinical trials for TG-1101 and TGR-1202. Early clinical results seen with TG-1101 and TGR-1202 in a small number of patients may not be reproduced in expanded or larger clinical trials. Additionally, individually reported outcomes of patients treated in clinical trials may not be representative of the entire population of treated patients in such studies. If the results from expansion cohorts or later trials are different from those found in the earlier studies of TG-1101 and TGR-1202, we may need to terminate or revise our clinical development plan, which could extend the time for conducting our development program and could have a material adverse effect on our business. Our IRAK4, anti-PD-L1 and anti-GITR programs are all in pre-clinical development and no assurance can be given that they will advance into clinical development. If the results from additional pre-clinical studies or early clinical trials differ from those found in earlier studies, our clinical development plans and timelines for this program could be adversely affected which could have a material adverse effect on our business. Many drugs fail in the early stages of clinical development for safety and tolerability issues, accordingly if our pre-clinical assets advance into clinical development, no assurance can be made that a safe and efficacious dose can be found.
If we are unable to successfully complete our clinical trial programs, or if such clinical trials take longer to complete than we project, our ability to execute our current business strategy will be adversely affected.
Whether or not and how quickly we complete clinical trials is dependent in part upon the rate at which we are able to engage clinical trial sites and, thereafter, the rate of enrollment of patients, and the rate we collect, clean, lock and analyze the clinical trial database. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the study, the existence of competitive clinical trials, and whether existing or new drugs are approved for the indication we are studying. We are aware that other companies are currently conducting or planning clinical trials that seek to enroll patients with the same diseases that we are studying. Certain clinical trials are designed to continue until a pre-determined number of events have occurred in the patients enrolled. Trials such as this are subject to delays stemming from patient withdrawal and from lower than expected event rates. They may also incur additional costs if enrollment is increased in order to achieve the desired number of events. If we experience delays in identifying and contracting with sites and/or in patient enrollment in our clinical trial programs, we may incur additional costs and delays in our development programs, and may not be able to complete our clinical trials in a cost-effective or timely manner. In addition, conducting multi-national studies adds another level of complexity and risk. We are subject to events affecting countries outside the U.S. Negative or inconclusive results from the clinical trials we conduct or unanticipated adverse medical events could cause us to have to repeat or terminate the clinical trials.
In September 2014, we announced a Phase 3 clinical trial for TG-1101 in previously treated patients with high-risk CLL, and in September 2015 we announced a Phase 3 clinical trial for the combination of TG-1101 + TGR-1202 for patients with CLL, each of which are being conducted pursuant to SPAs with the FDA. Many companies which have been granted SPAs and/or the right to utilize the FDA’s Fast Track or accelerated approval process have ultimately failed to obtain final approval to market their drugs. Since we are seeking approvals under SPAs for some of our product registration strategies, based on protocol designs negotiated with the FDA, we may be subject to enhanced scrutiny. Further, any changes or amendments to a protocol that is being conducted under SPA will have to be reviewed and approved by the FDA to verify that the SPA agreement is still valid. Additionally, even if the primary endpoint in a Phase 3 clinical trial is achieved, a SPA does not guarantee approval. The FDA may raise issues of safety, study conduct, bias, deviation from the protocol, statistical power, patient completion rates, changes in scientific or medical parameters or internal inconsistencies in the data prior to making its final decision. The FDA may also seek the guidance of an outside advisory committee prior to making its final decision. Even with “fast track” or “priority review” status which we intend to seek for our product candidates, such designations do not necessarily mean a faster development process or regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures.
Any product candidates we may advance into clinical development are subject to extensive regulation, which can be costly and time consuming, cause unanticipated delays or prevent the receipt of the required approvals to commercialize our product candidates.
The clinical development, manufacturing, labeling, storage, record-keeping, advertising, promotion, import, export, marketing and distribution of our product candidates or any future product candidates are subject to extensive regulation by the FDA in the United States and by comparable health authorities worldwide or in foreign markets. In the United States, we are not permitted to market our product candidates until we receive approval of a BLA or NDA from the FDA. The process of obtaining BLA and NDA approval is expensive, often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. Approval policies or regulations may change and the FDA has substantial discretion in the pharmaceutical approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons. In addition, the FDA may require post-approval clinical trials or studies which also may be costly. The FDA approval for a limited indication or approval with required warning language, such as a boxed warning, could significantly impact our ability to successfully market our product candidates. Finally, the FDA may require adoption of a Risk Evaluation and Mitigation Strategy (REMS) requiring prescriber training, post-market registries, or otherwise restricting the marketing and dissemination of these products. Despite the time and expense invested in clinical development of product candidates, regulatory approval is never guaranteed. Assuming successful clinical development, we intend to seek product approvals in countries outside the United States. As a result, we would be subject to regulation by the European Medicines Agency (“EMA”), as well as the other regulatory agencies in many of these countries, and other regulatory agencies around the world.
| 18 |
Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. As in the United States, the regulatory approval process in Europe and in other countries is a lengthy and challenging process. The FDA, and any other regulatory body around the world can delay, limit or deny approval of a product candidate for many reasons, including:
| · | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| · | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for any indication; |
| · | the FDA may not accept clinical data from trials which are conducted by individual investigators or in countries where the standard of care is potentially different from the United States; |
| · | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| · | we may be unable to demonstrate that a product candidate's clinical and other benefits outweigh its safety risks; |
| · | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| · | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a BLA, NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| · | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we or our collaborators contract for clinical and commercial supplies; or |
| · | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
In addition, recent events raising questions about the safety of certain marketed pharmaceuticals may result in increased cautiousness by the FDA and other regulatory authorities in reviewing new pharmaceuticals based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals. Regulatory approvals for our product candidates may not be obtained without lengthy delays, if at all. Any delay in obtaining, or inability to obtain, applicable regulatory approvals would prevent us from commercializing our product candidates.
Any product candidate we advance into clinical trials may cause unacceptable adverse events or have other properties that may delay or prevent their regulatory approval or commercialization or limit their commercial potential.
Unacceptable adverse events caused by any of our product candidates that we take into clinical trials could cause either us or regulatory authorities to interrupt, delay, modify or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications. This, in turn, could prevent us from commercializing the affected product candidate and generating revenues from its sale.
We have not completed testing of any of our product candidates for the treatment of the indications for which we intend to seek product approval in humans, and we currently do not know the extent that adverse events, if any, will be observed in patients who receive any of our product candidates. To date, clinical trials using TG-1101 and TGR-1202 have demonstrated a toxicity profile that was deemed acceptable by the investigators performing such studies. Such interpretation may not be shared by future investigators or by the FDA and in the case of TG-1101 and TGR-1202, even if deemed acceptable for oncology applications, it may not be acceptable for diseases outside the oncology setting, and likewise for any other product candidates we may develop. Additionally, the severity, duration and incidence of adverse events may increase in larger study populations. With respect to both TG-1101 and TGR-1202, the toxicity when manufactured under different conditions and in different formulations is not known, and it is possible that additional and/or different adverse events may appear upon the human use of those formulations and those adverse events may arise with greater frequency, intensity and duration than in the current formulation. Further, with respect to TGR-1202, although more than 80 patients have been dosed in the ongoing first-in-human dose-escalation Phase I single agent study, the full adverse effect profile of TGR-1202 is not known. It is unknown as the additional patients are exposed for longer durations to TGR-1202, whether greater frequency and/or severity of adverse events are likely to occur. Common toxicities of other drugs in the same class as TGR-1202 include high levels of liver toxicity, infections and colitis, the latter of which notably has presented with later onset, with incidence increasing with duration of exposure. To date, the incidence of these events has been limited for TGR-1202, however no assurance can be given that this safety and tolerability profile will continue to be demonstrated in the future as higher doses, longer durations of exposure, and multiple drug combinations are explored. If any of our product candidates cause unacceptable adverse events in clinical trials, we may not be able to obtain marketing approval and generate revenues from its sale, or even if approved for sale may lack differentiation from competitive products, which could have a material adverse impact on our business and operations.
| 19 |
Additionally, in combination clinical development, there is an inherent risk of drug-drug interactions between combination agents which may affect each component’s individual pharmacologic properties and the overall efficacy and safety of the combination regimen. Both TG-1101 and TGR-1202 are being evaluated in combination together, as well as with a variety of other active anti-cancer agents, which may cause unforeseen toxicity, or impact the severity, duration, and incidence of adverse events observed compared to those seen in the single agent studies of these agents. Further, with multi-drug combinations, it is often difficult to interpret or properly assign attribution of an adverse event to any one particular agent, introducing the risk that toxicity caused by a component of a combination regimen could have a material adverse impact on the development of our product candidates. There can be no assurances given that the combination regimens being studied will display tolerability or efficacy suitable to warrant further testing or produce data that is sufficient to obtain marketing approval.
If any of our product candidates receives marketing approval and we, or others, later identify unacceptable adverse events caused by the product, a number of significant negative consequences could result, including:
| · | regulatory authorities may withdraw their approval of the affected product; |
| · | regulatory authorities may require a more significant clinical benefit for approval to offset the risk; |
| · | regulatory authorities may require the addition of labeling statements that could diminish the usage of the product or otherwise limit the commercial success of the affected product; |
| · | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| · | we may choose to discontinue sale of the product; |
| · | we could be sued and held liable for harm caused to patients; |
| · | we may not be able to enter into collaboration agreements on acceptable terms and execute on our business model; and |
| · | our reputation may suffer. |
Any one or a combination of these events could prevent us from obtaining or maintaining regulatory approval and achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the affected product, which in turn could delay or prevent us from generating any revenues from the sale of the affected product.
We may experience delays in the commencement of our clinical trials or in the receipt of data from preclinical and clinical trials conducted by third parties, which could result in increased costs and delay our ability to pursue regulatory approval.
Delays in the commencement of clinical trials and delays in the receipt of data from preclinical or clinical trials conducted by third parties could significantly impact our product development costs. Before we can initiate clinical trials in the United States for our product candidates, we need to submit the results of preclinical testing, usually in animals, to the FDA as part of an IND, along with other information including information about product chemistry, manufacturing and controls and its proposed clinical trial protocol for our product candidates.
We plan to rely on preclinical and clinical trial data from third parties, if any, for the IND submissions for our product candidates. If receipt of that data is delayed for any reason, including reasons outside of our control, it will delay our plans for IND filings, and clinical trial plans. This, in turn, will delay our ability to make subsequent regulatory filings and ultimately, to commercialize our products if regulatory approval is obtained. If those third parties do not make this data available to us, we will likely, on our own, have to develop all the necessary preclinical and clinical data which will lead to additional delays and increase the costs of our development of our product candidates.
| 20 |
Before we can test any product candidate in human clinical trials the product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as in-vitro and animal studies to assess the potential safety and activity of the pharmaceutical product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices (“GLP”).
We must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the IND on a clinical hold within that 30-day time period. In such a case, we must work with the FDA to resolve any outstanding concerns before the clinical trials can begin. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such clinical trial.
The FDA may require that we conduct additional preclinical testing for any product candidate before it allows us to initiate the clinical testing under any IND, which may lead to additional delays and increase the costs of our preclinical development.
Even assuming an active IND for a product candidate, we do not know whether our planned clinical trials for any such product candidate will begin on time, or at all. The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
| · | obtaining regulatory clearance to commence a clinical trial; |
| · | identifying, recruiting and training suitable clinical investigators; |
| · | reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and trial sites, the terms of which can be subject to extensive negotiation, may be subject to modification from time to time and may vary significantly among different CROs and trial sites; |
| · | obtaining sufficient quantities of a product candidate for use in clinical trials; |
| · | obtaining institutional review board (“IRB”) or ethics committee approval to conduct a clinical trial at a prospective site; |
| · | identifying, recruiting and enrolling patients to participate in a clinical trial; |
| · | retaining patients who have initiated a clinical trial but may withdraw due to adverse events from the therapy, insufficient efficacy, fatigue with the clinical trial process or personal issues; and |
| · | unexpected safety findings. |
Any delays in the commencement of our clinical trials will delay our ability to pursue regulatory approval for our product candidates. In addition, many of the factors that cause, or lead to, a delay in the commencement of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate.
Delays in the completion of clinical testing could result in increased costs and delay our ability to generate product revenues.
Once a clinical trial has begun, patient recruitment and enrollment may be slower than we anticipate. Clinical trials may also be delayed as a result of ambiguous or negative interim results. Further, a clinical trial may be suspended or terminated by us, an IRB, an ethics committee or a Data Safety and Monitoring Committee overseeing the clinical trial, any of our clinical trial sites with respect to that site or the FDA or other regulatory authorities due to a number of factors, including:
| · | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| · | inspection of the clinical trial operations or clinical trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| · | unforeseen safety issues or any determination that the clinical trial presents unacceptable health risks; and |
| · | lack of adequate funding to continue the clinical trial. |
Changes in regulatory requirements and guidance also may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for re-examination, which may impact the costs, timing and successful completion of a clinical trial. If we experience delays in the completion of, or if we must terminate, any clinical trial of any product candidate that we advance into clinical trials, our ability to obtain regulatory approval for that product candidate will be delayed and the commercial prospects, if any, for the product candidate may be harmed. In addition, many of these factors may also ultimately lead to the denial of regulatory approval of a product candidate. Even if we ultimately commercialize any of our product candidates, other therapies for the same indications may have been introduced to the market during the period we have been delayed and such therapies may have established a competitive advantage over our product candidates.
| 21 |
We intend to rely on third parties to help conduct our planned clinical trials. If these third parties do not meet their deadlines or otherwise conduct the trials as required, we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all.
We intend to use CROs to assist in the conduct of our planned clinical trials and will rely upon medical institutions, clinical investigators and contract laboratories to conduct our trials in accordance with our clinical protocols. Our future CROs, investigators and other third parties may play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials.
There is no guarantee that any CROs, investigators and other third parties will devote adequate time and resources to our clinical trials or perform as contractually required. If any third parties upon whom we rely for administration and conduct of our clinical trials fail to meet expected deadlines, fail to adhere to its clinical protocols or otherwise perform in a substandard manner, our clinical trials may be extended, delayed or terminated, and we may not be able to commercialize our product candidates.
If any of our clinical trial sites terminate for any reason, we may experience the loss of follow-up information on patients enrolled in our ongoing clinical trials unless we are able to transfer the care of those patients to another qualified clinical trial site. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be jeopardized.
As all of our product candidates are still under development, manufacturing and process improvements implemented in the production of those product candidates may affect their ultimate activity or function.
Our product candidates are in the initial stages of development and are currently manufactured in small batches for use in pre-clinical and clinical studies. Process improvements implemented to date have changed, and process improvements in the future may change, the activity profile of the product candidates, which may affect the safety and efficacy of the products. No assurance can be given that the material manufactured from any of the optimized processes will perform comparably to the product candidates as manufactured to date and used in currently available pre-clinical data and or in early clinical trials reported in this or any previous filing. Additionally, future clinical trial results will be subject to the same level of uncertainty if, following such trials, additional process improvements are made. In addition, we are currently in the process of engaging a secondary manufacturer for TG-1101 to meet our current clinical and future commercial needs and anticipate engaging additional manufacturing sources for TGR-1202 to meet expanded clinical trial and commercial needs. No assurance can be given that the secondary manufacturing will be successful or that material manufactured by the secondary manufacturer will perform comparably to TG-1101 or TGR-1202 as manufactured to date and used in currently available pre-clinical data and or in early clinical trials reported in this or any previous filing. If the secondary manufacturer is not successful in replicating the product or experiences delays, or if regulatory authorities impose unforeseen requirements with respect to product comparability from multiple manufacturing sources, we may experience delays in clinical development.
If we fail to adequately understand and comply with the local laws and customs as we expand into new international markets, these operations may incur losses or otherwise adversely affect our business and results of operations.
We expect to operate a portion of our business in certain countries through subsidiaries or through supply and marketing arrangements. In those countries, where we have limited experience in operating subsidiaries and in reviewing equity investees, we will be subject to additional risks related to complying with a wide variety of national and local laws, including restrictions on the import and export of certain intermediates, drugs, technologies and multiple and possibly overlapping tax structures. In addition, we may face competition in certain countries from companies that may have more experience with operations in such countries or with international operations generally. We may also face difficulties integrating new facilities in different countries into our existing operations, as well as integrating employees hired in different countries into our existing corporate culture. If we do not effectively manage our operations in these subsidiaries and review equity investees effectively, or if we fail to manage our alliances, we may lose money in these countries and it may adversely affect our business and results of our operations.
If our competitors develop treatments for the target indications for which any of our product candidates may be approved, and they are approved more quickly, marketed more effectively or demonstrated to be more effective than our product candidates, our commercial opportunity will be reduced or eliminated.
We operate in a highly competitive segment of the biotechnology and biopharmaceutical market. We face competition from numerous sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies, and private and public research institutions. Many of our competitors have significantly greater financial, product development, manufacturing and marketing resources. Large pharmaceutical companies have extensive experience in clinical testing and obtaining regulatory approval for drugs. Additionally, many universities and private and public research institutes are active in cancer research, some in direct competition with us. We may also compete with these organizations to recruit scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
| 22 |
The cancer indications for which we are developing our products have a number of established therapies with which we will compete. Most major pharmaceutical companies and many biotechnology companies are aggressively pursuing new cancer development programs for the treatment of NHL, CLL, and other B-cell proliferative malignancies, including both therapies with traditional, as well as novel, mechanisms of action.
If approved, we expect TG-1101 to compete directly with Roche Group’s Rituxan® (rituximab) and Gazyva® (obinutuzumab or GA-101), and Genmab and GlaxoSmithKline’s Arzerra® (ofatumumab) among others, each of which is currently approved for the treatment of various diseases including NHL and CLL. In addition, a number of pharmaceutical companies are developing antibodies targeting CD20, CD19, and other B-cell associated targets, chimeric antigen receptor T-cell (CAR-T) immunotherapy, and other B-cell ablative therapy which, if approved, would potentially compete with TG-1101. New developments, including the development of other pharmaceutical technologies and methods of treating disease, occur in the pharmaceutical and life sciences industries at a rapid pace.
With respect to TGR-1202, there are several PI3K delta targeted compounds both approved, such as Gilead’s Zydelig™ (idelalisib), and in development, including, but not limited to, Infinity Pharmaceuticals’ duvelisib (IPI-145) which if approved we would expect to compete directly with TGR-1202. In addition, there are numerous other novel therapies targeting similar pathways to TGR-1202 both approved and in development, which could also compete with TGR-1202 in similar indications, such as the BTK inhibitor, ibrutinib (FDA approved for MCL, CLL, and WM and marketed by AbbVie and Janssen), the BTK inhibitor ACP-196 (under development by AstraZeneca), or the BCL-2 inhibitor ABT-199 (under clinical development by AbbVie and Roche).
These developments may render our product candidates obsolete or noncompetitive. Compared to us, many of our potential competitors have substantially greater:
| · | research and development resources, including personnel and technology; |
| · | regulatory experience; |
| · | pharmaceutical development, clinical trial and pharmaceutical commercialization experience; |
| · | experience and expertise in exploitation of intellectual property rights; and |
| · | capital resources. |
As a result of these factors, our competitors may obtain regulatory approval of their products more rapidly than us or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop products for the treatment of lymphoma or CLL that are more effective, better tolerated, more useful and less costly than ours and may also be more successful in manufacturing and marketing their products. Our competitors may succeed in obtaining approvals from the FDA and foreign regulatory authorities for their product candidates sooner than we do for our products.
We will also face competition from these third parties in recruiting and retaining qualified personnel, establishing clinical trial sites and enrolling patients for clinical trials and in identifying and in-licensing new product candidates.
We rely completely on third parties to manufacture our preclinical and clinical pharmaceutical supplies and we intend to rely on third parties to produce commercial supplies of any approved product candidate, and our commercialization of any of our product candidates could be stopped, delayed or made less profitable if those third parties fail to obtain approval of the FDA, fail to provide us with sufficient quantities of pharmaceutical product or fail to do so at acceptable quality levels or prices.
The facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA pursuant to inspections that will be conducted only after we submit a BLA or NDA to the FDA, if at all. We do not control the manufacturing process of our product candidates and are completely dependent on our contract manufacturing partners for compliance with the FDA’s requirements for manufacture of finished pharmaceutical products (good manufacturing practices, GMP). If our contract manufacturers cannot successfully manufacture material that conforms to our target product specifications, patent specifications, and/or the FDA’s strict regulatory requirements of safety, purity and potency, we will not be able to secure and/or maintain FDA approval for our product candidates. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If our contract manufacturers cannot meet FDA standards, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates. No assurance can be given that a long-term, scalable manufacturer can be identified or that they can make clinical and commercial supplies of our product candidates that meets the product specifications of previously manufactured batches, or is of a sufficient quality, or at an appropriate scale and cost to make it commercially feasible. If they are unable to do so, it could have a material adverse impact on our business.
| 23 |
In addition, we do not have the capability to package finished products for distribution to hospitals and other customers. Prior to commercial launch, we intend to enter into agreements with one or more alternate fill/finish pharmaceutical product suppliers so that we can ensure proper supply chain management once we are authorized to make commercial sales of our product candidates. If we receive marketing approval from the FDA, we intend to sell pharmaceutical product finished and packaged by such suppliers. We have not entered into long-term agreements with our current contract manufacturers or with any fill/finish suppliers, and though we intend to do so prior to commercial launch of our product candidates in order to ensure that we maintain adequate supplies of finished product, we may be unable to enter into such an agreement or do so on commercially reasonable terms, which could have a material adverse impact upon our business.
In most cases, our manufacturing partners are single source suppliers. It is expected that our manufacturing partners will be sole source suppliers from single site locations for the foreseeable future. Given this, any disruption of supply from these partners could have a material, long-term impact on our ability to supply products for clinical trials or commercial sale. If our suppliers do not deliver sufficient quantities of our product candidates on a timely basis, or at all, and in accordance with applicable specifications, there could be a significant interruption of our supply, which would adversely affect clinical development and commercialization of our products. In addition, if our current or future supply of any or our product candidates should fail to meet specifications during its stability program there could be a significant interruption of our supply of drug, which would adversely affect the clinical development and commercialization of the product.
We currently have no marketing and sales organization and no experience in marketing pharmaceutical products. If we are unable to establish sales and marketing capabilities or fail to enter into agreements with third parties to market and sell any products we may develop, we may not be able to effectively market and sell our products and generate product revenue.
We do not currently have the infrastructure for the sales, marketing and distribution of our biotechnology products, and we must build this infrastructure or make arrangements with third parties to perform these functions in order to commercialize our products. We plan to either develop internally or enter into collaborations or other commercial arrangements to develop further, promote and sell all or a portion of our product candidates.
The establishment and development of a sales force, either by us or jointly with a development partner, or the establishment of a contract sales force to market any products we may develop will be expensive and time-consuming and could delay any product launch, and we cannot be certain that we or our development partners would be able to successfully develop this capability. If we or our development partners are unable to establish sales and marketing capability or any other non-technical capabilities necessary to commercialize any products we may develop, we will need to contract with third parties to market and sell such products. We currently possess limited resources and may not be successful in establishing our own internal sales force or in establishing arrangements with third parties on acceptable terms, if at all.
If any product candidate that we successfully develop does not achieve broad market acceptance among physicians, patients, healthcare payors, and the medical community, the revenues that we generate from its sales will be limited.
Even if our product candidates receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payors, and the medical community. Coverage and reimbursement of our product candidates by third-party payors, including government payors, generally is also necessary for commercial success. The degree of market acceptance of any of our approved products will depend on a number of factors, including:
| · | the efficacy and safety as demonstrated in clinical trials; |
| · | the clinical indications for which the product is approved; |
| · | acceptance by physicians, major operators of cancer clinics and patients of the product as a safe and effective treatment; |
| · | the potential and perceived advantages of product candidates over alternative treatments; |
| · | the safety of product candidates seen in a broader patient group, including its use outside the approved indications; |
| · | the cost of treatment in relation to alternative treatments; |
| · | the availability of adequate reimbursement and pricing by third parties and government authorities; |
| 24 |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of adverse events; and |
| · | the effectiveness of our sales and marketing efforts. |
If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, hospitals, healthcare payors and patients, we may not generate sufficient revenue from these products and we may not become or remain profitable.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials, and will face an even greater risk if we sell our product candidates commercially. Although we are not aware of any historical or anticipated product liability claims against us, if we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to cease clinical trials of our drug candidates or limit commercialization of any approved products. An individual may bring a liability claim against us if one of our product candidates causes, or merely appears to have caused, an injury. If we cannot successfully defend our self against product liability claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| · | decreased demand for our product candidates; |
| · | impairment to our business reputation; |
| · | withdrawal of clinical trial participants; |
| · | costs of related litigation; |
| · | distraction of management’s attention from our primary business; |
| · | substantial monetary awards to patients or other claimants; |
| · | the inability to commercialize our product candidates; and |
| · | loss of revenues. |
We believe that we have obtained sufficient product liability insurance coverage for our clinical trials. We intend to expand our insurance coverage to include the sale of commercial products if marketing approval is obtained for any of our product candidates. However, we may be unable to obtain this product liability insurance on commercially reasonable terms and with insurance coverage that will be adequate to satisfy any liability that may arise. On occasion, large judgments have been awarded in class action or individual lawsuits relating to marketed pharmaceuticals. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
Reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell our products profitably.
We intend to seek approval to market our future products in both the United States and in countries and territories outside the United States. If we obtain approval in one or more foreign countries, we will be subject to rules and regulations in those countries relating to our product. In some foreign countries, particularly in the European Union, the pricing of prescription pharmaceuticals and biologics is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. In addition, market acceptance and sales of our product candidates will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for any of our product candidates and may be affected by existing and future healthcare reform measures.
Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which pharmaceuticals they will pay for and establish reimbursement levels. Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
| · | a covered benefit under its health plan; |
| · | safe, effective and medically necessary; |
| · | appropriate for the specific patient; |
| · | cost-effective; and |
| · | neither experimental nor investigational. |
| 25 |
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time consuming and costly process that could require that we provide supporting scientific, clinical and cost-effectiveness data for the use of our products to the payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If reimbursement of our future products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability. Additionally, while we may seek approval of our products in combination with each other, there can be no guarantee that we will obtain coverage and reimbursement for any of our products together, or that such reimbursement will incentivize the use of our products in combination with each other as opposed to in combination with other agents which may be priced more favorably to the medical community.
In both the United States and certain foreign countries, there have been a number of legislative and regulatory changes to the healthcare system that could impact our ability to sell our products profitably. In particular, the Medicare Modernization Act of 2003 revised the payment methodology for many products reimbursed by Medicare, resulting in lower rates of reimbursement for many types of drugs, and added a prescription drug benefit to the Medicare program that involves commercial plans negotiating drug prices for their members. Since 2003, there have been a number of other legislative and regulatory changes to the coverage and reimbursement landscape for pharmaceuticals. Most recently, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively, the “Affordable Care Act,” was enacted. The Affordable Care Act contains a number of provisions, including those governing enrollment in federal healthcare programs, the increased use of comparative effectiveness research on healthcare products, reimbursement and fraud and abuse changes, and a new regulatory pathway for the approval of biosimilar biological products, all of which will impact existing government healthcare programs and will result in the development of new programs. An expansion in the government’s role in the U.S. healthcare industry may further lower rates of reimbursement for pharmaceutical and biotechnology products.
There have been, and likely will continue to be, legislative and regulatory proposals at the federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare products and services. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may adversely affect:
| · | the demand for any products for which we may obtain regulatory approval; |
| · | our ability to set a price that we believe is fair for our products; |
| · | our ability to generate revenues and achieve or maintain profitability; |
| · | the level of taxes that we are required to pay; and |
| · | the availability of capital. |
In addition, governments may impose price controls, which may adversely affect our future profitability.
We will need to increase the size of our organization and the scope of our outside vendor relationships, and we may experience difficulties in managing this growth.
As of March 1, 2016, we had forty full and part time employees. Over time, we will need to expand our managerial, operational, financial and other resources in order to manage and fund our operations and clinical trials, continue research and development activities, and commercialize our product candidates. Our management and scientific personnel, systems and facilities currently in place may not be adequate to support our future growth. Our need to effectively manage our operations, growth, and various projects requires that we:
| · | manage our clinical trials effectively; |
| · | manage our internal development efforts effectively while carrying out our contractual obligations to licensors, contractors and other third parties; |
| · | continue to improve our operational, financial and management controls and reporting systems and procedures; and |
| · | attract and retain sufficient numbers of talented employees. |
We may utilize the services of outside vendors or consultants to perform tasks including clinical trial management, statistics and analysis, regulatory affairs, formulation development, chemistry, manufacturing, controls, and other pharmaceutical development functions. Our growth strategy may also entail expanding our group of contractors or consultants to implement these tasks going forward. Because we rely on a substantial number of consultants, effectively outsourcing many key functions of our business, we will need to be able to effectively manage these consultants to ensure that they successfully carry out their contractual obligations and meet expected deadlines. However, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for our product candidates or otherwise advance its business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, or at all. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may be unable to successfully implement the tasks necessary to further develop and commercialize our product candidates and, accordingly, may not achieve our research, development and commercialization goals.
| 26 |
If we fail to attract and keep key management and clinical development personnel, we may be unable to successfully develop or commercialize our product candidates.
We will need to expand and effectively manage our managerial, operational, financial and other resources in order to successfully pursue our clinical development and commercialization efforts for our product candidates and future product candidates. We are highly dependent on the development, regulatory, commercial and financial expertise of the members of our senior management. The loss of the services of any of our senior management could delay or prevent the further development and potential commercialization of our product candidates and, if we are not successful in finding suitable replacements, could harm our business. We do not maintain “key man” insurance policies on the lives of these individuals. We will need to hire additional personnel as we continue to expand our manufacturing, research and development activities.
Our success depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel and we may not be able to do so in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. Our industry has experienced a high rate of turnover of management personnel in recent years. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will impede significantly the achievement of our development objectives, our ability to raise additional capital, and our ability to implement our business strategy.
If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected.
In addition to FDA restrictions on the marketing of pharmaceutical and biotechnology products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical and medical device industries in recent years, as well as consulting or other service agreements with physicians or other potential referral sources. These laws include anti-kickback statutes and false claims statutes that prohibit, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or, in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally-financed healthcare programs, and knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and any practices we adopt may not, in all cases, meet all of the criteria for safe harbor protection from anti-kickback liability. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment. Any challenge to its business practices under these laws could have a material adverse effect on our business, financial condition, and results of operations.
We use biological and hazardous materials, and any claims relating to improper handling, storage or disposal of these materials could be time consuming or costly.
We use hazardous materials, including chemicals and biological agents and compounds, which could be dangerous to human health and safety or the environment. Our operations also produce hazardous waste products. Federal, state and local laws and regulations govern the use, generation, manufacture, storage, handling and disposal of these materials and wastes. Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our pharmaceutical development efforts.
In addition, we cannot entirely eliminate the risk of accidental injury or contamination from these materials or wastes. If one of our employees was accidentally injured from the use, storage, handling or disposal of these materials or wastes, the medical costs related to his or her treatment would be covered by our workers’ compensation insurance policy. However, we do not carry specific biological or hazardous waste insurance coverage and our property and casualty and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury, we could be held liable for damages or penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended, or operations otherwise affected.
| 27 |
All product candidate development timelines and projections in this report are based on the assumption of further financing.
The timelines and projections in this report are predicated upon the assumption that we will raise additional financing in the future to continue the development of our product candidates. In the event we do not successfully raise subsequent financing, our product development activities will necessarily be curtailed commensurate with the magnitude of the shortfall. If our product development activities are slowed or stopped, we would be unable to meet the timelines and projections outlined in this filing. Failure to progress our product candidates as anticipated will have a negative effect on our business, future prospects, and ability to obtain further financing on acceptable terms (if at all), and the value of the enterprise.
Risks Relating to Acquisitions
Acquisitions, investments and strategic alliances that we may make in the future may use significant resources, result in disruptions to our business or distractions of our management, may not proceed as planned, and could expose us to unforeseen liabilities.
We may seek to expand our business through the acquisition of, investments in and strategic alliances with companies, technologies, products, and services. Acquisitions, investments and strategic alliances involve a number of special problems and risks, including, but not limited to:
| · | difficulty integrating acquired technologies, products, services, operations and personnel with the existing businesses; |
| · | diversion of management’s attention in connection with both negotiating the acquisitions and integrating the businesses; |
| · | strain on managerial and operational resources as management tries to oversee larger operations; |
| · | difficulty implementing and maintaining effective internal control over financial reporting at businesses that we acquire, particularly if they are not located near our existing operations; |
| · | exposure to unforeseen liabilities of acquired companies; |
| · | potential costly and time-consuming litigation, including stockholder lawsuits; |
| · | potential issuance of securities to equity holders of the company being acquired with rights that are superior to the rights of holders of our common stock or which may have a dilutive effect on our stockholders; |
| · | risk of loss of invested capital; |
| · | the need to incur additional debt or use cash; and |
| · | the requirement to record potentially significant additional future operating costs for the amortization of intangible assets. |
As a result of these or other problems and risks, businesses we acquire may not produce the revenues, earnings, or business synergies that we anticipated, and acquired products, services, or technologies might not perform as we expected. As a result, we may incur higher costs and realize lower revenues than we had anticipated. We may not be able to successfully address these problems and we cannot assure you that the acquisitions will be successfully identified and completed or that, if acquisitions are completed, the acquired businesses, products, services, or technologies will generate sufficient revenue to offset the associated costs or other negative effects on our business.
Any of these risks can be greater if an acquisition is large relative to our size. Failure to effectively manage our growth through acquisitions could adversely affect our growth prospects, business, results of operations, financial condition and cash flows.
Risks Relating to Our Intellectual Property
Our success depends upon our ability to protect our intellectual property and proprietary technologies, and the intellectual property protection for our product candidates depends significantly on third parties.
Our commercial success depends on obtaining and maintaining patent protection and trade secret protection for our product candidates and their formulations and uses, as well as successfully defending these patents against third-party challenges. If any of our licensors or partners fails to appropriately prosecute and maintain patent protection for these product candidates, our ability to develop and commercialize these product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products. This failure to properly protect the intellectual property rights relating to these product candidates could have a material adverse effect on our financial condition and results of operations.
| 28 |
Currently, the composition of matter patent and several method of use patents for TG-1101 and TGR-1202 in various indications and settings have been applied for but have not yet been issued, or have been issued in certain territories but not under all jurisdictions in which such applications have been filed. While composition of matter patents have been granted in the US for TG-1101 and TGR-1202, no patents to date have been issued for our IRAK4 inhibitor and anti-PD-L1 and anti-GITR programs. There can be no guarantee that any of these patents for which an application has already been filed, nor any patents filed in the future for our product candidates will be granted in any or all jurisdictions in which there were filed, or that all claims initially included in such patent applications will be allowed in the final patent that is issued. The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or our partners will be successful in protecting our product candidates by obtaining and defending patents.
These risks and uncertainties include the following:
| · | the patent applications that we or our partners file may not result in any patents being issued; |
| · | patents that may be issued or in-licensed may be challenged, invalidated, modified, revoked or circumvented, or otherwise may not provide any competitive advantage; |
| · | as of March 16, 2013, the U.S. converted from a “first to invent” to a “first to file” system. If we do not win the filing race, we will not be entitled to inventive priority; |
| · | our competitors, many of which have substantially greater resources than we do, and many of which have made significant investments in competing technologies, may seek, or may already have obtained, patents that will limit, interfere with, or eliminate its ability to make, use, and sell our potential products either in the United States or in international markets; |
| · | there may be significant pressure on the U.S. government and other international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful as a matter of public policy regarding worldwide health concerns; and |
| · | countries other than the United States may have less restrictive patent laws than those upheld by United States courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products. |
If patents are not issued that protect our product candidates, it could have a material adverse effect on our financial condition and results of operations.
In addition to patents, we and our partners also rely on trade secrets and proprietary know-how. Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties, and confidential information and inventions agreements with employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect its rights. If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of this information may be greatly reduced.
Patent protection and other intellectual property protection are crucial to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate.
If we or our partners are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in that litigation would have a material adverse effect on our business.
Our commercial success also depends upon our ability and the ability of any of our future collaborators to develop, manufacture, market and sell our product candidates without infringing the proprietary rights of third parties. Numerous United States and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing products, some of which may be directed at claims that overlap with the subject matter of our intellectual property. For example, Roche has the Cabilly patents in the U.S. that block the commercialization of antibody products derived from a single cell line, like TG-1101. Also, Roche, Biogen Idec, and Genentech hold patents for the use of anti-CD20 antibodies utilized in the treatment of CLL in the U.S. While these patents have been challenged, to the best of our knowledge, those matters were settled in a way that permitted additional anti-CD20 antibodies to be marketed for CLL. If those patents are still enforced at the time we are intending to launch TG-1101, then we will need to either prevail in a litigation to challenge those patents or negotiate a settlement agreement with the patent holders. If we are unable to do so we may be forced to delay the launch of TG-1101 or launch at the risk of litigation for patent infringement, which may have a material adverse effect on our business and results of operations.
| 29 |
In addition, because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our product candidates or proprietary technologies may infringe. Similarly, there may be issued patents relevant to our product candidates of which we are not aware.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and biopharmaceutical industries generally. If a third party claims that we or any collaborators of ours infringe their intellectual property rights, we may have to:
| · | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| · | abandon an infringing product candidate or redesign its products or processes to avoid infringement; |
| · | pay substantial damages, including treble damages and attorneys’ fees, which we may have to pay if a court decides that the product or proprietary technology at issue infringes on or violates the third party’s rights; |
| · | pay substantial royalties, fees and/or grant cross licenses to our technology; and/or |
| · | defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
No assurance can be given that patents issued to third parties do not exist, have not been filed, or could not be filed or issued, which contain claims covering its products, technology or methods that may encompass all or a portion of our products and methods. Given the number of patents issued and patent applications filed in our technical areas or fields, we believe there is a risk that third parties may allege they have patent rights encompassing our products or methods.
Other product candidates that we may in-license or acquire could be subject to similar risks and uncertainties.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which typically are very expensive, time-consuming and disruptive of day-to-day business operations. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly. The adverse result could also put related patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by the U.S. Patent and Trademark Office (“PTO”) may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
We may be subject to claims that our consultants or independent contractors have wrongfully used or disclosed alleged trade secrets of their other clients or former employers to it.
As is common in the biotechnology and pharmaceutical industry, we engage the services of consultants to assist us in the development of our product candidates. Many of these consultants were previously employed at, may have previously been, or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management and day-to-day business operations.
| 30 |
Risks Relating to Our Finances and Capital Requirements
We will need to raise additional capital to continue to operate our business.
As of December 31, 2015, we had net cash, cash equivalents, investment securities and interest receivable of approximately $102.4 million. We believe that our cash and cash equivalents and investments will sustain our operations for more than 24 months from December 31, 2015. As a result, we will need additional capital to continue our operations beyond that time. Required additional sources of financing to continue our operations in the future might not be available on favorable terms, if at all. If we do not succeed in raising additional funds on acceptable terms, we might be unable to complete planned preclinical and clinical trials or obtain approval of any of our product candidates from the FDA or any foreign regulatory authorities. In addition, we could be forced to discontinue product development, reduce or forego sales and marketing efforts and forego attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity securities, which would have a dilutive effect to stockholders.
Currently, none of our product candidates have been approved by the FDA or any foreign regulatory authority for sale. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from cash on hand and amounts raised in future offerings or financings.
We have a history of operating losses, expect to continue to incur losses, and are unable to predict the extent of future losses or when we will become profitable, if ever.
We have not yet applied for or demonstrated an ability to obtain regulatory approval for or commercialize a product candidate. Our short operating history makes it difficult to evaluate our business prospects and consequently, any predictions about our future performance may not be as accurate as they could be if we had a history of successfully developing and commercializing pharmaceutical or biotechnology products. Our prospects must be considered in light of the uncertainties, risks, expenses and difficulties frequently encountered by companies in the early stages of operations and the competitive environment in which we operate.
We have never been profitable and, as of December 31, 2015, we had an accumulated deficit of $158.1 million. We have generated operating losses in all periods since we were incorporated. We expect to make substantial expenditures resulting in increased operating costs in the future and our accumulated deficit will increase significantly as we expand development and clinical trial efforts for our product candidates. Our losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and stockholders’ equity. Because of the risks and uncertainties associated with product development, we are unable to predict the extent of any future losses or when we will become profitable, if ever. Even if we achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis.
We have not generated any revenue from our product candidates and may never become profitable.
Our ability to become profitable depends upon our ability to generate significant continuing revenues. To obtain significant continuing revenues, we must succeed, either alone or with others, in developing, obtaining regulatory approval for and manufacturing and marketing our product candidates (or utilize early access programs to generate such revenue). To date, our product candidates have not generated any revenues, and we do not know when, or if, we will generate any revenue. Our ability to generate revenue depends on a number of factors, including, but not limited to:
| · | successful completion of preclinical studies of our product candidates; |
| · | successful commencement and completion of clinical trials of our product candidates and any future product candidates we advance into clinical trials; |
| · | achievement of regulatory approval for our product candidates and any future product candidates we advance into clinical trials (unless we successfully utilize early access programs which allow for revenue generation prior to approval); |
| · | manufacturing commercial quantities of our products at acceptable cost levels if regulatory approvals are obtained; |
| · | successful sales, distribution and marketing of our future products, if any; and |
| · | our entry into collaborative arrangements or co-promotion agreements to market and sell our products. |
If we are unable to generate significant continuing revenues, we will not become profitable and we may be unable to continue our operations without continued funding.
| 31 |
We will need substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our development programs or commercialization efforts.
We expect to spend substantial amounts on development, including significant amounts on conducting clinical trials for our product candidates, manufacturing clinical supplies and expanding our pharmaceutical development programs. We expect that our monthly cash used by operations will continue to increase for the next several years. We anticipate that we will continue to incur operating losses for the foreseeable future.
We will require substantial additional funds to support our continued research and development activities, as well as the anticipated costs of preclinical studies and clinical trials, regulatory approvals, and eventual commercialization. We anticipate that we will incur operating losses for the foreseeable future. We have based these estimates, however, on assumptions that may prove to be wrong, and we could expend our available financial resources much faster than we currently expect. Further, we will need to raise additional capital to fund our operations and continue to conduct clinical trials to support potential regulatory approval of marketing applications. Future capital requirements will also depend on the extent to which we acquire or in-license additional product candidates. We currently have no commitments or agreements relating to any of these types of transactions.
The amount and timing of our future funding requirements will depend on many factors, including, but not limited to, the following:
| · | the progress of our clinical trials, including expenses to support the trials and milestone payments that may become payable under our license agreements; |
| · | the costs and timing of regulatory approvals; |
| · | the costs and timing of clinical and commercial manufacturing supply arrangements for each product candidate; |
| · | the costs of establishing sales or distribution capabilities; |
| · | the success of the commercialization of our products; |
| · | our ability to establish and maintain strategic collaborations, including licensing and other arrangements; |
| · | the costs involved in enforcing or defending patent claims or other intellectual property rights; and |
| · | the extent to which we in-license or invest in other indications or product candidates. |
Until we can generate a sufficient amount of product revenue and achieve profitability, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements, as well as through interest income earned on cash balances. If we were to be unable to raise additional capital, we would have to significantly delay, scale back or discontinue one or more of our pharmaceutical development programs. We also may be required to relinquish, license or otherwise dispose of rights to product candidates or products that it would otherwise seek to develop or commercialize itself on terms that are less favorable than might otherwise be available.
Raising additional funds by issuing securities or through licensing or lending arrangements may cause dilution to our existing stockholders, restrict our operations or require us to relinquish proprietary rights.
We may raise additional funds through public or private equity offerings, debt financings or licensing arrangements. To the extent that we raise additional capital by issuing equity securities, the share ownership of existing stockholders will be diluted. Any future debt financing we enter into may involve covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, redeem our stock, make certain investments and engage in certain merger, consolidation or asset sale transactions, among other restrictions.
In addition, if we raise additional funds through licensing arrangements, it may be necessary to relinquish potentially valuable rights to our product candidates, or grant licenses on terms that are not favorable to us. If adequate funds are not available, our ability to achieve profitability or to respond to competitive pressures would be significantly limited and we may be required to delay, significantly curtail or eliminate the development of one or more of our product candidates.
Risks Related to Our Common Stock
We are controlled by current officers, directors and principal stockholders.
Our directors, executive officers, their affiliates, and our principal stockholders beneficially own approximately 55% percent of our outstanding voting stock, including shares underlying outstanding options and warrants. Our directors, officers and principal stockholders, taken as a whole, have the ability to exert substantial influence over the election of our Board of Directors and the outcome of issues submitted to our stockholders.
| 32 |
Our stock price is, and we expect it to remain, volatile, which could limit investors’ ability to sell stock at a profit.
The trading price of our common stock is likely to be highly volatile and subject to wide fluctuations in price in response to various factors, many of which are beyond our control. These factors include:
| · | publicity regarding actual or potential clinical results relating to products under development by our competitors or us; |
| · | delay or failure in initiating, completing or analyzing nonclinical or clinical trials or the unsatisfactory design or results of these trials; |
| · | achievement or rejection of regulatory approvals by our competitors or us; |
| · | announcements of technological innovations or new commercial products by our competitors or us; |
| · | developments concerning proprietary rights, including patents; |
| · | developments concerning our collaborations; |
| · | regulatory developments in the United States and foreign countries; |
| · | economic or other crises and other external factors; |
| · | period-to-period fluctuations in our revenues and other results of operations; |
| · | changes in financial estimates by securities analysts; and |
| · | sales of our common stock. |
We will not be able to control many of these factors, and we believe that period-to-period comparisons of our financial results will not necessarily be indicative of our future performance.
In addition, the stock market in general, and the market for biotechnology companies in particular, has experienced extreme price and volume fluctuations that may have been unrelated or disproportionate to the operating performance of individual companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of your stock.
We have never paid dividends on our common stock and do not anticipate paying any dividends for the foreseeable future. You should not rely on an investment in our stock if you require dividend income. Further, you will only realize income on an investment in our stock in the event you sell or otherwise dispose of your shares at a price higher than the price you paid for your shares. Such a gain would result only from an increase in the market price of our common stock, which is uncertain and unpredictable.
Certain anti-takeover provisions in our charter documents and Delaware law could make a third-party acquisition of us difficult. This could limit the price investors might be willing to pay in the future for our common stock.
Provisions in our amended and restated certificate of incorporation and restated bylaws could have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from attempting to acquire, or control us. These factors could limit the price that certain investors might be willing to pay in the future for shares of our common stock. Our amended and restated certificate of incorporation allows us to issue preferred stock without the approval of our stockholders. The issuance of preferred stock could decrease the amount of earnings and assets available for distribution to the holders of our common stock or could adversely affect the rights and powers, including voting rights, of such holders. In certain circumstances, such issuance could have the effect of decreasing the market price of our common stock. Our restated bylaws eliminate the right of stockholders to call a special meeting of stockholders, which could make it more difficult for stockholders to effect certain corporate actions. Any of these provisions could also have the effect of delaying or preventing a change in control.
On July 18, 2014, the Board of Directors declared a distribution of one right for each outstanding share of common stock. The rights may have certain anti-takeover effects. The rights will cause substantial dilution to a person or group that attempts to acquire us on terms not approved by the Board of Directors unless the offer is conditioned on a substantial number of rights being acquired. However, the rights should not interfere with any merger, statutory share exchange or other business combination approved by the Board of Directors since the rights may be terminated by us upon resolution of the Board of Directors. Thus, the rights are intended to encourage persons who may seek to acquire control of the Company to initiate such an acquisition through negotiations with the Board of Directors. However, the effect of the rights may be to discourage a third party from making a partial tender offer or otherwise attempting to obtain a substantial equity position in the equity securities of, or seeking to obtain control of, the Company. To the extent any potential acquirers are deterred by the rights, the rights may have the effect of preserving incumbent management in office.
| 33 |
Our corporate and executive office is located in New York, New York. Our New York facility consists of office space at 3 Columbus Circle, 15th Floor, New York, New York 10019. We are not currently under a lease agreement at 3 Columbus Circle. We are also currently leasing small office spaces in Cary, North Carolina and Kingsport, Tennessee to accommodate our clinical operations groups. We believe that our existing facilities are adequate to meet our current requirements. We do not own any real property.
We, and our subsidiaries, are not a party to, and our property is not the subject of, any material pending legal proceedings.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our common stock is listed on the Nasdaq Capital Market and trades under the symbol “TGTX”. Prior to May 30, 2013 our stock was listed on the Over the Counter Bulletin Board.
The following table sets forth the high and low closing sale prices of our common stock for the periods indicated.
| High | Low | ||||||
| Fiscal Year Ended December 31, 2015 | ||||||||
| Fourth Quarter | $ | 14.42 | $ | 10.22 | ||||
| Third Quarter | $ | 18.74 | $ | 9.76 | ||||
| Second Quarter | $ | 17.17 | $ | 13.24 | ||||
| First Quarter | $ | 18.82 | $ | 12.77 | ||||
| High | Low | |||||||
| Fiscal Year Ended December 31, 2014 | ||||||||
| Fourth Quarter | $ | 18.25 | $ | 9.38 | ||||
| Third Quarter | $ | 11.93 | $ | 7.37 | ||||
| Second Quarter | $ | 9.93 | $ | 4.51 | ||||
| First Quarter | $ | 7.59 | $ | 4.10 | ||||
Holders
The number of record holders of our common stock as of March 1, 2016 was 299.
Dividends
We have never declared or paid any cash dividends on our common stock and do not anticipate paying any cash dividends in the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors.
| 34 |
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information as of December 31, 2015, regarding the securities authorized for issuance under our equity compensation plan, the TG Therapeutics, Inc. Amended and Restated 2012 Incentive Plan.
| Equity Compensation Plan Information | ||||||||||||
| Plan Category | Number of securities to be issued upon exercise of outstanding options | Weighted-average exercise price of outstanding options | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders | — | $ | — | 4,164,631 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | — | $ | — | 4,164,631 | ||||||||
For information about all of our equity compensation plans see Note 5 to our Consolidated Financial Statements included in this report.
COMMON STOCK PERFORMANCE GRAPH
The following graph compares the cumulative total stockholder return on our common stock for the period from December 31, 2011(1) through December 31, 2015, with the cumulative total return over such period on (i) the U.S. Index of The Nasdaq Stock Market and (ii) the Biotechnology Index of The Nasdaq Stock Market. The graph assumes an investment of $100 on December 31, 2011, in our common stock (at the adjusted closing market price) and in each of the indices listed above, and assumes the reinvestment of all dividends. Measurement points are December 31 of each year.
(1) In connection with the Company having entered into and consummated an exchange transaction agreement (the “Exchange Transaction”) with Opus Point Partners, LLC (“Opus”) and TG Biologics, Inc. (formerly known as TG Therapeutics, Inc.) (“TG Bio”), we used the start date of December 31, 2011 to be in agreement with this transaction.
(2) $100 invested on 12/31/11 in stock or index, including reinvestment of dividends. Fiscal Years ending December 31.
| 35 |
ITEM 6. SELECTED FINANCIAL DATA
The following Statement of Operations Data for the years ended December 31, 2015, 2014, 2013, 2012 and 2011, and Balance Sheet Data as of December 31, 2015, 2014, 2013, 2012 and 2011, as set forth below are derived from our audited consolidated financial statements. This financial data should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data.”
| Years ended December 31, | ||||||||||||||||||||
| 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||||
| License revenue | $ | 152,381 | $ | 152,381 | $ | 152,381 | $ | 19,048 | $ | — | ||||||||||
| Research and development: | ||||||||||||||||||||
| Noncash stock expense associated with in-licensing agreements | — | 5,350,094 | — | 16,578,000 | 297,000 | |||||||||||||||
| Noncash compensation | 4,261,406 | 8,731,566 | 1,041,519 | 455,809 | — | |||||||||||||||
| Other research and development | 43,445,817 | 26,004,687 | 12,621,161 | 3,994,182 | 30,283 | |||||||||||||||
| Total research and development | 47,707,223 | 40,086,347 | 13,662,680 | 21,027,991 | 327,283 | |||||||||||||||
| General and administrative: | ||||||||||||||||||||
| Noncash compensation | 11,435,686 | 12,373,726 | 4,161,629 | 2,966,373 | 86,494 | |||||||||||||||
| Other general and administrative | 4,189,488 | 3,413,400 | 2,496,461 | 1,815,083 | 468,197 | |||||||||||||||
| Total general and administrative | 15,625,174 | 15,787,126 | 6,658,090 | 4,781,456 | 554,691 | |||||||||||||||
| Impairment of in-process research and development | — | — | 2,797,600 | 1,104,700 | — | |||||||||||||||
| Total costs and expenses | 63,332,397 | 55,873,473 | 23,118,370 | 26,914,147 | 881,974 | |||||||||||||||
| Operating loss | (63,180,016 | ) | (55,721,092 | ) | (22,965,989 | ) | (26,895,099 | ) | (881,974 | ) | ||||||||||
| Other (income) expense: | ||||||||||||||||||||
| Interest income | (174,653 | ) | (55,049 | ) | (30,822 | ) | (15,787 | ) | — | |||||||||||
| Other income | — | (95,427 | ) | (108,894 | ) | (272,232 | ) | — | ||||||||||||
| Interest expense | 972,736 | 930,701 | 952,888 | 905,744 | 7,097 | |||||||||||||||
| Change in fair value of notes payable | (1,029,453 | ) | (720,040 | ) | (3,300,951 | ) | (1,659,872 | ) | — | |||||||||||
| Total other (income) expense, net | (231,370 | ) | 60,185 | (2,487,779 | ) | (1,042,147 | ) | 7,097 | ||||||||||||
| Loss before income taxes | (62,948,646 | ) | (55,781,277 | ) | (20,478,210 | ) | (25,852,952 | ) | (889,071 | ) | ||||||||||
| Income taxes | — | — | — | 330,000 | — | |||||||||||||||
| Consolidated net loss | (62,948,646 | ) | (55,781,277 | ) | (20,478,210 | ) | (26,182,952 | ) | (889,071 | ) | ||||||||||
| Net loss attributable to non-controlling interest | — | — | — | (8,110,233 | ) | (35,997 | ) | |||||||||||||
| Net loss attributable to TG Therapeutics, Inc. and Subsidiaries | $ | (62,948,646 | ) | $ | (55,781,277 | ) | $ | (20,478,210 | ) | $ | (18,072,719 | ) | $ | (853,074 | ) | |||||
| Basic and diluted net loss per common share | $ | (1.38 | ) | $ | (1.64 | ) | $ | (0.81 | ) | $ | (1.38 | ) | $ | (0.44 | ) | |||||
Balance Sheet Information:
| December 31, | ||||||||||||||||||||
| 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||||
| Cash, cash equivalents, investment securities and interest receivable | $ | 102,416,894 | $ | 78,861,334 | $ | 45,431,532 | $ | 16,455,995 | $ | 9,748,491 | ||||||||||
| Total assets | 113,473,201 | 86,746,890 | 48,112,390 | 22,074,037 | 14,537,358 | |||||||||||||||
| Accumulated deficit | (158,133,926 | ) | (95,185,280 | ) | (39,404,003 | ) | (18,925,793 | ) | (853,074 | ) | ||||||||||
| Total equity | 101,573,302 | 80,101,884 | 40,054,492 | 15,550,301 | 9,636,202 | |||||||||||||||
| 36 |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion and analysis contains forward-looking statements about our plans and expectations of what may happen in the future. Forward-looking statements are based on a number of assumptions and estimates that are inherently subject to significant risks and uncertainties, and our results could differ materially from the results anticipated by our forward-looking statements as a result of many known or unknown factors, including, but not limited to, those factors discussed in “Item 1A. Risk Factors.” See also the “Special Cautionary Notice Regarding Forward-Looking Statements” set forth at the beginning of this report.
You should read the following discussion and analysis in conjunction with “Item 8. Financial Statements and Supplementary Data,” and our consolidated financial statements beginning on page F-1 of this report.
Overview
We are a biopharmaceutical company focused on the acquisition, development and commercialization of novel treatments for b-cell malignancies and autoimmune diseases. Currently, the company is developing two therapies targeting hematological malignancies. TG-1101 (ublituximab) is a novel, glycoengineered monoclonal antibody that targets a specific and unique epitope on the CD20 antigen found on mature B-lymphocytes. TG Therapeutics is also developing TGR-1202, an orally available PI3K delta inhibitor. The delta isoform of PI3K is strongly expressed in cells of hematopoietic origin and is believed to be important in the proliferation and survival of B-lymphocytes. Both TG-1101 and TGR-1202 are in clinical development for patients with hematologic malignancies. The Company also has a pre-clinical programs seeking to develop IRAK4 (interleukin-1 receptor-associated kinase 4) inhibitors and anti-PD-L1 and anti-GITR antibodies.
We also actively evaluate complementary products, technologies and companies for in-licensing, partnership, acquisition and/or investment opportunities. To date, we have not received approval for the sale of any of our drug candidates in any market and, therefore, have not generated any product sales from our drug candidates.
Our license revenues currently consist of license fees arising from our agreement with Ildong. We recognize upfront license fee revenues ratably over the estimated period in which we will have certain significant ongoing responsibilities under the sublicense agreement, with unamortized amounts recorded as deferred revenue.
We have not earned any revenues from the commercial sale of any of our drug candidates.
Our research and development expenses consist primarily of expenses related to in-licensing of new product candidates, fees paid to consultants and outside service providers for clinical and laboratory development, facilities-related and other expenses relating to the design, development, manufacture, testing and enhancement of our drug candidates and technologies. We expense our research and development costs as they are incurred. Research and development expenses for the years ended December 31, 2015, 2014 and 2013 were $43,445,817, $31,354,781 and $12,621,161, respectively, excluding non-cash compensation expenses related to research and development.
The following table sets forth the research and development expenses per project, exclusive of non-cash compensation expenses, for the periods presented.
| 2015 | 2014 | 2013 | ||||||||||
| TG-1101 | $ | 29,816,042 | $ | 15,410,925 | $ | 8,281,028 | ||||||
| TGR-1202 | 11,671,889 | 14,249,856 | 4,340,133 | |||||||||
| IRAK4 | 1,068,466 | 1,694,000 | — | |||||||||
| PD-L1/GITR | 500,300 | — | — | |||||||||
| Other | 389,120 | — | — | |||||||||
| Total | $ | 43,445,817 | $ | 31,354,781 | $ | 12,621,161 | ||||||
Our general and administrative expenses consist primarily of salaries and related expenses for executive, finance and other administrative personnel, recruitment expenses, professional fees and other corporate expenses, including investor relations, legal activities and facilities-related expenses.
| 37 |
Our results of operations include non-cash compensation expenses as a result of the grants of stock options and restricted stock. Compensation expense for awards of options and restricted stock granted to employees and directors represents the fair value of the award recorded over the respective vesting periods of the individual awards. The expense is included in the respective categories of expense in the consolidated statements of operations. We expect to continue to incur significant non-cash compensation expenses.
For awards of options and restricted stock to consultants and other third-parties, compensation expense is determined at the “measurement date.” The expense is recognized over the vesting period of the award. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record compensation expense based on the fair value of the award at the reporting date. The awards to consultants and other third-parties are then revalued, or the total compensation is recalculated based on the then current fair value, at each subsequent reporting date. This results in a change to the amount previously recorded in respect of the equity award grant, and additional expense or a reversal of expense may be recorded in subsequent periods based on changes in the assumptions used to calculate fair value, such as changes in market price, until the measurement date is reached and the compensation expense is finalized.
In addition, certain restricted stock issued to employees vest upon the achievement of certain milestones; therefore, the total expense is uncertain until the milestone is probable.
Our clinical trials will be lengthy and expensive. Even if these trials show that our drug candidates are effective in treating certain indications, there is no guarantee that we will be able to record commercial sales of any of our drug candidates in the near future. In addition, we expect losses to continue as we continue to fund in-licensing and development of new drug candidates. As we continue our development efforts, we may enter into additional third-party collaborative agreements and may incur additional expenses, such as licensing fees and milestone payments. In addition, we may need to establish the commercial infrastructure required to manufacture, market and sell our drug candidates following approval, if any, by the FDA, which would result in us incurring additional expenses. As a result, our quarterly results may fluctuate and a quarter-by-quarter comparison of our operating results may not be a meaningful indication of our future performance.
RESULTS OF OPERATIONS
Years Ended December 31, 2015, 2014 and 2013
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| License revenue | $ | 152,381 | $ | 152,381 | $ | 152,381 | ||||||
| Costs and expenses: | ||||||||||||
| Research and development: | ||||||||||||
| Noncash stock expense associated with in-licensing agreements | — | 5,350,094 | — | |||||||||
| Noncash compensation | 4,261,406 | 8,731,566 | 1,041,519 | |||||||||
| Other research and development | 43,445,817 | 26,004,687 | 12,621,161 | |||||||||
| Total research and development | 47,707,223 | 40,086,347 | 13,662,680 | |||||||||
| General and administrative: | ||||||||||||
| Noncash compensation | 11,435,686 | 12,373,726 | 4,161,629 | |||||||||
| Other general and administrative | 4,189,488 | 3,413,400 | 2,496,461 | |||||||||
| Total general and administrative | 15,625,174 | 15,787,126 | 6,658,090 | |||||||||
| Impairment of in-process research and development | — | — | 2,797,600 | |||||||||
| Total costs and expenses | 63,332,397 | 55,873,473 | 23,118,370 | |||||||||
| Operating loss | (63,180,016 | ) | (55,721,092 | ) | (22,965,989 | ) | ||||||
| Other expense (income), net | (231,370 | ) | 60,185 | (2,487,779 | ) | |||||||
| Net loss | $ | (62,948,646 | ) | $ | (55,781,277 | ) | $ | (20,478,210 | ) | |||
| 38 |
Years Ended December 31, 2015 and 2014
License Revenue. License revenue was $152,381 for each of the years ended December 31, 2015 and 2014. License revenue is related to the amortization of an upfront payment of $2.0 million associated with our license agreement with Ildong. The upfront payment from Ildong will be recognized as license revenue on a straight-line basis through December 2025, which represents the estimated period over which the Company will have certain ongoing responsibilities under the sublicense agreement.
Noncash Stock Expense Associated with In-licensing Agreements. Noncash stock expense associated with in-licensing agreements amounted to $5,350,094 for the year ended December 31, 2014. The expense during the year ended December 31, 2014 was recorded in conjunction with the stock issued to Rhizen of approximately $4,100,000 in connection with our license for TGR-1202, and approximately $1,200,000 for the common stock issued to Ligand Pharmaceuticals as an upfront payment for the license to the IRAK4 inhibitors program.
Noncash Compensation Expense (Research and Development). Noncash compensation expense (research and development) related to equity incentive grants totaled $4,261,406 for the year ended December 31, 2015, as compared to $8,731,566 during the comparable period in 2014. The decrease in noncash compensation expense was primarily related to milestone-based vesting of restricted stock grants to personnel during the year ended December 31, 2014 and a decrease in the measurement date fair value of certain consultant restricted stock during the period ended December 31, 2015.
Other Research and Development Expenses. Other research and development expenses increased by $17,441,130 from $26,004,687 for the year ended December 31, 2014 to $43,445,817 for the year ended December 31, 2015. The increase in other research and development expenses was due primarily to increases of approximately $11.7 million and $3.4 million for manufacturing and clinical trial expenses related to TG-1101 and TGR-1202, respectively, offset by a decrease of $4.0 million related to the upfront cash milestone payment to Rhizen to exercise the license option for TGR-1202 during the year ended December 31, 2014. We expect our other research and development costs to increase during 2016 as the enrollment of Phase 3 clinical trials increases and we prepare for launch.
Noncash Compensation Expense (General and Administrative). Noncash compensation expense (general and administrative) related to equity incentive grants decreased by $938,040 from $12,373,726 for the year ended December 31, 2014 to $11,435,686 during the year ended December 31, 2015. The decrease in noncash compensation expense was primarily related to milestone-based vesting of restricted stock grants to personnel during the year ended December 31, 2014.
Other General and Administrative Expenses. Other general and administrative expenses increased by $776,088 from $3,413,400 for the year ended December 31, 2014 to $4,189,488 for the year ended December 31, 2015. The increase was due primarily to increased personnel and other general and administrative costs. We expect our other general and administrative expenses to increase modestly during 2016.
Other Expense (Income), Net. Other income increased by $291,555 from $60,185 of expense for the year ended December 31, 2014 to $231,370 of income for the year ended December 31, 2015. The increase is mainly due to an increase in interest income for the year ended December 31, 2015.
Years Ended December 31, 2014 and 2013
License Revenue. License revenue was $152,381 for each of the years ended December 31, 2014 and 2013. License revenue is related to the amortization of an upfront payment of $2.0 million associated with our license agreement with Ildong. The upfront payment from Ildong will be recognized as license revenue on a straight-line basis through December 2025, which represents the estimated period over which the Company will have certain ongoing responsibilities under the sublicense agreement.
Noncash Stock Expense Associated with In-licensing Agreements. Noncash stock expense associated with in-licensing agreements was $5,350,094 for the year ended December 31, 2014, as compared to $0 for the year ended December 31, 2013. The expense during the year ended December 31, 2014 was recorded in conjunction with the stock issued to Rhizen of approximately $4,100,000 to exercise our option to license TGR-1202, and approximately $1,200,000 for the common stock issued to Ligand as an upfront payment for the license to the IRAK4 inhibitors program.
| 39 |
Noncash Compensation Expense (Research and Development). Noncash compensation expense (research and development) related to equity incentive grants increased by $7,690,047 from $1,041,519 for the year ended December 31, 2013 to $8,731,566 during the year ended December 31, 2014. The increase in noncash compensation expense was primarily related to milestone-based vesting of restricted stock grants to employees and consultants during the year ended December 31, 2014. We expect noncash compensation expense (research and development) to remain at a comparable level in 2015.
Other Research and Development Expenses. Other research and development expenses increased by $13,383,526 from $12,621,161 for the year ended December 31, 2013 to $26,004,687 for the year ended December 31, 2014. The increase in other research and development expenses was due primarily to increased research and development expenses associated with TG-1101 and TGR-1202 of approximately $7,100,000 and $3,800,000, respectively. In addition, during the year ended December 31, 2014 we expensed $4,000,000 related to the upfront cash milestone payment to Rhizen to exercise the license option for TGR-1202. We expect our other research and development costs to increase during 2015 as we commence multiple Phase 3 registration programs.
Noncash Compensation Expense (General and Administrative). Noncash compensation expense (general and administrative) related to equity incentive grants increased by $8,212,097 from $4,161,629 for the year ended December 31, 2013 to $12,373,726 during the year ended December 31, 2014. The increase in noncash compensation expense was primarily related to milestone-based vesting of restricted stock grants to personnel during the year ended December 31, 2014. We expect noncash compensation expense (general and administrative) to remain at a comparable level in 2015.
Other General and Administrative Expenses. Other general and administrative expenses increased by $916,939 from $2,496,461 for the year ended December 31, 2013 to $3,413,400 for the year ended December 31, 2014. The increase was due primarily to Delaware franchise taxes, legal fees and Nasdaq listing fees. We expect our other general and administrative expenses to increase modestly during 2015.
Impairment of in-process research and development. The impairments recognized in 2013 related to the in-process research and development intangible asset recorded in connection with the Exchange Transaction. As of December 31, 2013, as a result of expiring intellectual property, uncertain regulatory pathways, and market changes affecting the commercial potential for the Ariston in-process research and development asset (AST-726), the Company determined that the asset's carrying value was no longer recoverable. Impairment testing of our indefinite lived intangible assets is performed annually or when a triggering event occurs that could indicate a potential impairment.
Other Expense (Income), Net. Other expense (income) decreased by $2,547,964 from $2,487,779 of income for the year ended December 31, 2013 to $60,185 of expense for the year ended December 31, 2014. Other income for the year ended December 31, 2013 included a change in the fair value of notes payable of approximately $2,500,000 resulting from the valuation of the convertible notes payable to zero except for the conversion feature of the notes payable. See Note 4 for further details.
LIQUIDITY AND CAPITAL RESOURCES
Our primary sources of cash have been from the sale of equity securities, the upfront payment from our Sublicense Agreement with Ildong, and warrant and option exercises. We have not yet commercialized any of our drug candidates and cannot be sure if we will ever be able to do so. Even if we commercialize one or more of our drug candidates, we may not become profitable. Our ability to achieve profitability depends on a number of factors, including our ability to obtain regulatory approval for our drug candidates, successfully complete any post-approval regulatory obligations and successfully commercialize our drug candidates alone or in partnership. We may continue to incur substantial operating losses even if we begin to generate revenues from our drug candidates.
As of December 31, 2015, we had $102.4 million in cash and cash equivalents, investment securities, and interest receivable.
We anticipate that our cash and cash equivalents as of December 31, 2015 are sufficient to fund our anticipated operating cash requirements for more than 24 months from December 31, 2015. The actual amount of cash that we will need to operate is subject to many factors, including, but not limited to, the timing, design and conduct of clinical trials for our drug candidates. We are dependent upon significant financing to provide the cash necessary to execute our current operations, including the commercialization of any of our drug candidates.
Cash used in operating activities for the year ended December 31, 2015 was $44,690,272 as compared to $35,059,919 for the year ended December 31, 2014. The increase in cash used in operating activities was due primarily to increased expenditures associated with our clinical development programs for TG-1101 and TGR-1202.
| 40 |
For the year ended December 31, 2015, net cash used in investing activities was $24,685,869 as compared to $18,355,289 for the year ended December 31, 2014. The increase in net cash used in investing activities was primarily due to an increase in our investment in held-to-maturity treasury securities.
For the year ended December 31, 2015, net cash provided by financing activities of $68,723,686 related to our program under an At-the-Market Issuance Sales Agreement (the “ATM Program”), as well as proceeds from the exercise of warrants. For the year ended December 31, 2014, net cash provided by financing activities of $68,643,526 related to net proceeds from the issuance of Common Stock of our underwritten public offering in March 2014, as well as proceeds from the exercise of warrants, net of payment of notes payable of $677,778.
ATM Program
On June 21, 2013, we entered into an At-the-Market Issuance Sales Agreement (the “2013 ATM”) with MLV & Co. LLC (“MLV”) under which we could issue and sell shares of our common stock, having aggregate offering proceeds of up to $50.0 million, from time to time through MLV, acting as the sales agent. Under the agreement we would pay MLV a commission rate of up to 3.0% of the gross proceeds from the sale of any shares of common stock sold through MLV.
During the year ended December 31, 2014, we sold a total of 4,850,055 shares of common stock under this arrangement for aggregate total gross proceeds of approximately $50.0 million at an average selling price of $10.31 per share. Net proceeds were approximately $48.8 million after deducting commissions and other transaction costs. We have fully utilized the capacity under the 2013 ATM and, accordingly, no further sales can be made under the 2013 ATM.
In December 2014, we amended our 2013 ATM with MLV (the “2015 ATM”) such that we could issue and sell additional shares of our common stock, having an aggregate offering price of up to $75.0 million, from time to time through MLV, acting as the sales agent. Under the 2015 ATM, we would pay MLV a commission rate of up to 3.0% of the gross proceeds from the sale of any shares of common stock sold through MLV.
During the year ended December 31, 2015, we sold a total of 4,094,498 shares of common stock under the 2015 ATM for aggregate total gross proceeds of approximately $68.2 million at an average selling price of $16.66 per share, resulting in net proceeds of approximately $67.0 million after deducting commissions and other transaction costs.
Equity Financings
On March 11, 2014, we announced the pricing of an underwritten sale of 2,702,809 shares of our common stock at a price of $6.71 per share for gross proceeds of approximately $18.1 million. Net proceeds from this offering were approximately $16.8 million, net of underwriting discounts and offering expenses of approximately $1.3 million.
On July 18, 2013, we announced the pricing of an underwritten public offering of 5,700,000 shares of our common stock at a price of $6.15 per share for gross proceeds of approximately $35 million. We also granted to the underwriters a 30-day option to acquire an additional 855,000 shares to cover overallotments in connection with the offering, which they exercised. Total net proceeds from this offering, including the overallotment, were approximately $37.6 million, net of underwriting discounts and offering expenses of approximately $2.7 million.
OFF-BALANCE SHEET ARRANGEMENTS
We have not entered into any transactions with unconsolidated entities whereby we have financial guarantees, subordinated retained interests, derivative instruments or other contingent arrangements that expose us to material continuing risks, contingent liabilities, or any other obligations under a variable interest in an unconsolidated entity that provides us with financing, liquidity, market risk or credit risk support.
OBLIGATIONS AND COMMITMENTS
As of December 31, 2015, we have known contractual obligations, commitments and contingencies of $16.3 million related to our operating lease obligations.
| (in thousands) | Payment due by period | |||||||||||||||||||
| Total | Less
than 1 year | 1-3 years | 3-5 years | More than 5 years | ||||||||||||||||
| Contractual obligations | ||||||||||||||||||||
| Operating leases | $ | 16,335 | $ | 128 | $ | 1,947 | $ | 1,992 | $ | 12,268 | ||||||||||
| Total | $ | 16,335 | $ | 128 | $ | 1,947 | $ | 1,992 | $ | 12,268 | ||||||||||
| 41 |
Leases
On October 3, 2014, we entered into a Desk Space Agreement (the “Desk Agreement”) with Fortress, to occupy approximately 40% of the New York City office space recently leased by Fortress. This Desk Agreement requires us to pay our respective share of the average annual rent and other costs of the 15 year lease. We approximate an average annual rental obligation of $1.1 million under the Desk Agreement. Fortress does not expect to take possession of the space until early 2016.
Total rental expense was approximately $0.3 million, $0.1 million and $0.01 million for the years ended December 31, 2015, 2014 and 2013, respectively.
Future minimum lease commitments as of December 31, 2015, in the aggregate total approximately $16.3 million through December 31, 2031. The preceding table shows future minimum lease commitments, which include our office leases in New York, North Carolina and Tennessee, by period as of December 31, 2015.
CRITICAL ACCOUNTING POLICIES
The discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amount of assets and liabilities and related disclosure of contingent assets and liabilities at the date of our financial statements and the reported amounts of revenues and expenses during the applicable period. Actual results may differ from these estimates under different assumptions or conditions.
We define critical accounting policies as those that are reflective of significant judgments and uncertainties and which may potentially result in materially different results under different assumptions and conditions. In applying these critical accounting policies, our management uses its judgment to determine the appropriate assumptions to be used in making certain estimates. These estimates are subject to an inherent degree of uncertainty. Our critical accounting policies include the following:
Revenue Recognition. We recognize license revenue in accordance with the revenue recognition guidance of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”), or Codification. We analyze each element of our licensing agreement to determine the appropriate revenue recognition. The terms of the license agreement may include payments to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments over the period of significant involvement under the related agreements unless the fee is in exchange for products delivered or services rendered that represent the culmination of a separate earnings process and no further performance obligation exists under the contract. We recognize milestone payments as revenue upon the achievement of specified milestones only if (1) the milestone payment is non-refundable, (2) substantive effort is involved in achieving the milestone, (3) the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (4) the milestone is at risk for both parties. If any of these conditions are not met, we defer the milestone payment and recognize it as revenue over the estimated period of performance under the contract.
Stock Compensation. We have granted stock options and restricted stock to employees, directors and consultants, as well as warrants to other third parties. For employee and director grants, the value of each option award is estimated on the date of grant using the Black-Scholes option-pricing model. The Black-Scholes model takes into account volatility in the price of our stock, the risk-free interest rate, the estimated life of the option, the closing market price of our stock and the exercise price. We base our estimates of our stock price volatility on the historical volatility of our common stock and our assessment of future volatility; however, these estimates are neither predictive nor indicative of the future performance of our stock. For purposes of the calculation, we assumed that no dividends would be paid during the life of the options and warrants. The estimates utilized in the Black-Scholes calculation involve inherent uncertainties and the application of management judgment. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those equity awards expected to vest. As a result, if other assumptions had been used, our recorded stock-based compensation expense could have been materially different from that reported. In addition, because some of the options, restricted stock and warrants issued to employees, consultants and other third-parties vest upon the achievement of certain milestones, the total expense is uncertain. Compensation expense for such awards that vest upon the achievement of milestones is recognized when the achievement of such milestone becomes probable.
| 42 |
Total compensation expense for options and restricted stock issued to consultants is determined at the “measurement date.” The expense is recognized over the vesting period for the options and restricted stock. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record stock-based compensation expense based on the fair value of the equity awards at the reporting date. These equity awards are then revalued, or the total compensation is recalculated based on the then current fair value, at each subsequent reporting date. This results in a change to the amount previously recorded in respect of the equity award grant, and additional expense or a reversal of expense may be recorded in subsequent periods based on changes in the assumptions used to calculate fair value, such as changes in market price, until the measurement date is reached and the compensation expense is finalized.
In-process Research and Development. All acquired research and development projects are recorded at their fair value as of the date acquisition. The fair values are assessed as of the balance sheet date to ascertain if there has been any impairment of the recorded value. If there is an impairment the asset is written down to its current fair value by the recording of an expense.
Accruals for Clinical Research Organization and Clinical Site Costs. We make estimates of costs incurred in relation to external clinical research organizations, or CROs, and clinical site costs. We analyze the progress of clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of the amount expensed and the related prepaid asset and accrued liability. Significant judgments and estimates must be made and used in determining the accrued balance and expense in any accounting period. We review and accrue CRO expenses and clinical trial study expenses based on work performed and rely upon estimates of those costs applicable to the stage of completion of a study. Accrued CRO costs are subject to revisions as such trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known. With respect to clinical site costs, the financial terms of these agreements are subject to negotiation and vary from contract to contract. Payments under these contracts may be uneven, and depend on factors such as the achievement of certain events, the successful recruitment of patients, the completion of portions of the clinical trial or similar conditions. The objective of our policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical site costs are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Accounting Related to Goodwill. As of December 31, 2015 and 2014, there was $799,391 of goodwill on our consolidated balance sheets. Goodwill is reviewed for impairment annually, or when events arise that could indicate that an impairment exists. We test for goodwill impairment using a two-step process. The first step compares the fair value of the reporting unit with the unit's carrying value, including goodwill. When the carrying value of the reporting unit is greater than fair value, the unit’s goodwill may be impaired, and the second step must be completed to measure the amount of the goodwill impairment charge, if any. In the second step, the implied fair value of the reporting unit’s goodwill is compared with the carrying amount of the unit’s goodwill. If the carrying amount is greater than the implied fair value, the carrying value of the goodwill must be written down to its implied fair value.
We are required to perform impairment tests annually, at December 31, and whenever events or changes in circumstances suggest that the carrying value of an asset may not be recoverable. For all of our acquisitions, various analyses, assumptions and estimates were made at the time of each acquisition that were used to determine the valuation of goodwill and intangibles. In future years, the possibility exists that changes in forecasts and estimates from those used at the acquisition date could result in impairment indicators.
Accounting For Income Taxes. In preparing our consolidated financial statements, we are required to estimate our income taxes in each of the jurisdictions in which we operate. This process involves management estimation of our actual current tax exposure and assessment of temporary differences resulting from differing treatment of items for tax and accounting purposes. These differences result in deferred tax assets and liabilities. We must then assess the likelihood that our deferred tax assets will be recovered from future taxable income and, to the extent we believe that recovery is not likely, we must establish a valuation allowance. To the extent we establish a valuation allowance or increase this allowance in a period, we must include an expense within the tax provision in the consolidated statements of operations. Significant management judgment is required in determining our provision for income taxes, our deferred tax assets and liabilities and any valuation allowance recorded against our net deferred tax assets. We have fully offset our deferred tax assets with a valuation allowance. Our lack of earnings history and the uncertainty surrounding our ability to generate taxable income prior to the reversal or expiration of such deferred tax assets were the primary factors considered by management in maintaining the valuation allowance.
| 43 |
Fair Value of 5% Notes Payable. We measure certain financial assets and liabilities at fair value on a recurring basis in the financial statements. The hierarchy ranks the quality and reliability of inputs, or assumptions, used in the determination of fair value and requires financial assets and liabilities carried at fair value to be classified and disclosed in one of three categories.
We elected the fair value option for valuing the 5% Notes. We elected the fair value option in order to reflect in our financial statements the assumptions that market participants use in evaluating these financial instruments.
RECENTLY ISSUED ACCOUNTING STANDARDS
In May 2014, the FASB issued an update to ASC 606, Revenue from Contracts with Customers. This update to ASC 606 provides a five-step process to determine when and how revenue is recognized. The core principle of the guidance is that a Company should recognize revenue upon transfer of promised goods or services to customers in an amount that reflects the expected consideration to be received in exchange for those goods or services. This update to ASC 606 will also result in enhanced disclosures about revenue, providing guidance for transactions that were not previously addressed comprehensively, and improving guidance for multiple-element arrangements. This update to ASC 606 is effective for us beginning in fiscal 2018. We are currently evaluating the impact of this update on our consolidated financial statements.
In August 2014, the FASB issued Accounting Standards Update 2014-15, Presentation of Financial Statements—Going Concern, which requires that management of an entity should evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued or available to be issued. This update will become effective beginning January 1, 2017, with early adoption permitted. The provisions of this standard are not expected to significantly impact the Company.
In November 2015, the FASB issued Accounting Standards Update 2015-17, Balance Sheet Classification of Deferred Taxes. This update requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. The amendments in this update apply to all entities that present a classified statement of financial position. This update will become effective for public business entities for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The provisions of this standard are not expected to significantly impact the Company.
In February 2016, the FASB issued ASU No. 2016-02, “Leases (Topic 842),” which requires lessees to recognize assets and liabilities for the rights and obligations created by most leases on their balance sheet. The guidance is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early application is permitted. ASU 2016-02 requires modified retrospective adoption for all leases existing at, or entered into after, the date of initial application, with an option to use certain transition relief. The Company is currently evaluating the impact the standard may have on its consolidated financial statements and related disclosures.
Other pronouncements issued by the FASB or other authoritative accounting standards group with future effective dates are either not applicable or not significant to our consolidated financial statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK.
The primary objective of our investment activities is to preserve principal while maximizing our income from investments and minimizing our market risk. We currently invest in government and investment-grade corporate debt in accordance with our investment policy, which we may change from time to time. The securities in which we invest have market risk. This means that a change in prevailing interest rates, and/or credit risk, may cause the fair value of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the prevailing interest rate later rises, the fair value of our investment will probably decline. As of December 31, 2015, our portfolio of financial instruments consists of cash equivalents and short-term and long-term interest bearing securities, including government debt and money market funds. The average duration of all of our held-to-maturity investments held as of December 31, 2015, was less than 12 months. Due to the relative short-term nature of these financial instruments, we believe there is no material exposure to interest rate risk, and/or credit risk, arising from our portfolio of financial instruments.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
Our consolidated financial statements and the notes thereto, included in Part IV, Item 15(a), part 1, are incorporated by reference into this Item 8.
| 44 |
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES.
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures. As of December 31, 2015, management carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”)). Our disclosure controls and procedures are designed to provide reasonable assurance that information we are required to disclose in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in applicable rules and forms. Based upon that evaluation, our Chief Executive and Chief Financial Officers concluded that, as of December 31, 2015, our disclosure controls and procedures were effective.
Management's Report on Internal Control over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) or Rule 15d-15(f) under the Exchange Act). Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2015. In making this assessment, our management used the criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO Framework. Our management has concluded that, as of December 31, 2015, our internal control over financial reporting was effective based on these criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2015 was audited by CohnReznick LLP, our independent registered public accounting firm, as stated in their report appearing below, which expressed an unqualified opinion on the effectiveness of our internal control over financial reporting as of December 31, 2015.
Changes in Internal Control Over Financial Reporting. There were no changes in our internal control over financial reporting during the quarter ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the Effectiveness of Controls. Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our Company have been detected.
| 45 |
Report of Independent Registered Public Accounting Firm
To The Board of Directors and Stockholders
TG Therapeutics, Inc.
We have audited TG Therapeutics, Inc. and Subsidiaries’ internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). TG Therapeutics, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on TG Therapeutics, Inc.’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, TG Therapeutics, Inc. and Subsidiaries has maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015 based on the criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements of TG Therapeutics, Inc. and Subsidiaries as of December 31, 2015 and 2014, and for each of the three years in the period ended December 31, 2015 and our report dated March 15, 2016, expressed an unqualified opinion.
/s/ CohnReznick LLP
Roseland, New Jersey
March 15, 2016
| 46 |
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2016 Annual Meeting of Stockholders.
ITEM 11. EXECUTIVE COMPENSATION.
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2016 Annual Meeting of Stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2016 Annual Meeting of Stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2016 Annual Meeting of Stockholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2016 Annual Meeting of Stockholders.
| 47 |
ITEM 15. EXHIBITS and FINANCIAL STATEMENT SCHEDULES.
| 1. | Consolidated Financial Statements |
The following consolidated financial statements of TG Therapeutics, Inc. are filed as part of this report.
| Contents | Page |
| Report of Independent Registered Public Accounting Firm | F-1 |
| Consolidated Balance Sheets as of December 31, 2015 and 2014 | F-2 |
| Consolidated Statements of Operations for the years ended December 31, 2015, 2014 and 2013 | F-3 |
| Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2015, 2014 and 2013 | F-4 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2015, 2014 and 2013 | F-5 |
| Notes to Consolidated Financial Statements | F-6 |
| 2. | Consolidated Financial Statement Schedules |
All schedules are omitted as the information required is inapplicable or the information is presented in the consolidated financial statements or the related notes.
| 3. | Exhibits |
| Exhibit | |
| Number | Exhibit Description |
| 3.1 | Amended and Restated Certificate of Incorporation of TG Therapeutics, Inc. dated April 26, 2012 (incorporated by reference to Exhibit 3.2 to the Registrant’s Form 10-Q for the quarter ended June 30, 2012). |
| 3.2 | Certificate of Amendment to Amended and Restated Certificate of Incorporation of TG Therapeutics, Inc. dated June 9, 2014 (incorporated by reference to Exhibit 3.2 to the Registrant’s Form 10-Q for the quarter ended June 30, 2014). |
| 3.3 | Amended and Restated Bylaws of TG Therapeutics, Inc. dated July 18, 2014 (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on July 21, 2014). |
| 4.1 | Specimen common stock certificate (incorporated by reference to Exhibit 4.1 to the Registrant’s Form 10-K for the year ended December 31, 2011). |
| 4.2 | Form of warrant to purchase common stock of TG Therapeutics, Inc. (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K filed on November 13, 2012). |
| 4.3 | Form of Warrant issued to stockholders (incorporated by reference to Exhibit 10.34 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). |
| 4.4 | Stockholder Protection Rights Agreement, dated July 18, 2014 between TG Therapeutics, Inc. and American Stock Transfer & Trust Company, LLC, as Rights Agent (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K filed on July 21, 2014). |
| 10.1 | Amended and Restated Convertible Promissory Note, dated March 1, 2011 (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on March 7, 2011). |
| 10.2 | Employment Agreement, effective December 29, 2011, between the Registrant and Michael Weiss (incorporated by reference to Exhibit 10.30 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). † |
| 10.3 | Restricted Stock Subscription Agreement, effective December 29, 2011, between the Registrant and Michael Weiss (incorporated by reference to Exhibit 10.31 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). † |
| 48 |
| 10.4 | Amendment to Restricted Stock Agreement, dated July 12, 2013, by and between TG Therapeutics, Inc. and Michael S. Weiss (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on July 16, 2013). † |
| 10.5 | Amendment to Restricted Stock Agreements, dated December 31, 2014, by and between TG Therapeutics, Inc. and Michael S. Weiss (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on January 7, 2015). † |
| 10.6 | Employment Agreement, effective December 29, 2011, between the Registrant and Sean Power (incorporated by reference to Exhibit 10.32 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). † |
| 10.7 | Restricted Stock Subscription Agreement, effective December 29, 2011 between the Registrant and Sean Power (incorporated by reference to Exhibit 10.33 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). † |
| 10.8 | Amendment to Restricted Stock Agreement, dated July 12, 2013, by and between TG Therapeutics, Inc. and Sean A. Power (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on July 16, 2013). † |
| 10.9 | Amendment to Restricted Stock Agreements, dated December 31, 2014, by and between TG Therapeutics, Inc. and Sean A. Power (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on January 7, 2015). † |
| 10.10 | License Agreement, dated January 30, 2012, by and among the Registrant, GTC Biotherapeutics, Inc., LFB Biotechnologies S.A.S. and LFB/GTC LLC (incorporated by reference to Exhibit 10.35 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2011). * |
| 10.11 | TG Therapeutics, Inc. Amended and Restated 2012 Incentive Plan, dated May 14, 2012 (incorporated by reference to Exhibit 10.1 to the Registrant’s Form 10-Q/A for the quarter ended March 31, 2012). |
| 10.12 |
First Amendment to TG Therapeutics, Inc. Amended and Restated 2012 Incentive Plan, filed with the Registrant’s Definitive Proxy Statement for the Annual Meeting of Stockholders on June 4, 2015, filed on April 24, 2015, and incorporated herein by reference. |
| 10.13 | Sublicense Agreement between TG Therapeutics, Inc. and Ildong Pharmaceutical Co. Ltd., dated November 13, 2012 (incorporated by reference to Exhibit 10.37 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2012). * |
| 10.14 | License Agreement between TG Therapeutics, Inc. and Ligand Pharmaceuticals Incorporated, dated June 23, 2014 (incorporated by reference to Exhibit 10.1 to the Registrant’s Form 10-Q for the quarter ended June 30, 2014).* |
| 10.15 | Licensing Agreement between TG Therapeutics, Inc. and Rhizen Pharmaceuticals SA, dated September 22, 2014 (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on January 20, 2015). * |
| 10.16 | Collaboration Agreement between TG Therapeutics, Inc. and Checkpoint Therapeutics, Inc., dated March 3, 2015 (incorporated by reference to Exhibit 10.1 to the Registrant’s Form 10-Q for the quarter ended March 31, 2015). * |
| 23.1 | Consent of Independent Registered Public Accounting Firm |
| 31.1 | Certification of Principal Executive Officer |
| 31.2 | Certification of Principal Financial Officer |
| 32.1 | Certification of Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 32.2 | Certification of Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 101 | The following financial information from TG Therapeutics, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2015, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Stockholders’ Equity, (iv) Consolidated Statements of Cash Flows, (v) the Notes to Consolidated Financial Statements. |
| † | Indicates management contract or compensatory plan or arrangement. |
| * | Confidential treatment has been requested with respect to omitted portions of this exhibit. |
| 49 |
TG Therapeutics, Inc.
Consolidated Financial Statements
Report of Independent Registered Public Accounting Firm
To The Board of Directors and Stockholders
TG Therapeutics, Inc.
We have audited the accompanying consolidated balance sheets of TG Therapeutics, Inc. and Subsidiaries (the “Company”) as of December 31, 2015 and 2014, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2015. TG Therapeutics, Inc.’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of TG Therapeutics, Inc. and Subsidiaries as of December 31, 2015 and 2014, and their results of operations and cash flows for each of the three years in the period ended December 31, 2015, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), TG Therapeutics, Inc. and Subsidiaries’ internal control over financial reporting as of December 31, 2015 based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated March 15, 2016 expressed an unqualified opinion.
/s/ CohnReznick LLP
Roseland, New Jersey
March 15, 2016
| F-1 |
TG Therapeutics, Inc. and Subsidiaries
Consolidated Balance Sheets as of December 31, 2015 and 2014
| 2015 | 2014 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 55,061,329 | $ | 55,713,784 | ||||
| Short-term investment securities | 22,166,512 | 23,062,034 | ||||||
| Interest receivable | 186,021 | 85,516 | ||||||
| Prepaid research and development | 9,151,142 | 6,179,743 | ||||||
| Other current assets | 308,327 | 173,952 | ||||||
| Total current assets | 86,873,331 | 85,215,029 | ||||||
| Restricted cash | 579,143 | 575,012 | ||||||
| Long-term investment securities | 25,003,032 | — | ||||||
| Property, plant and equipment, net | 47,122 | 20,357 | ||||||
| Goodwill | 799,391 | 799,391 | ||||||
| Other assets | 171,182 | 137,101 | ||||||
| Total assets | $ | 113,473,201 | $ | 86,746,890 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | 9,346,068 | $ | 3,991,625 | ||||
| Accrued compensation | 818,472 | 702,000 | ||||||
| Current portion of deferred revenue | 152,381 | 152,381 | ||||||
| Notes payable | 211,549 | 275,190 | ||||||
| Total current liabilities | 10,528,470 | 5,121,196 | ||||||
| Deferred revenue, net of current portion | 1,371,429 | 1,523,810 | ||||||
| Total liabilities | 11,899,899 | 6,645,006 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.001 par value per share (10,000,000 shares authorized, none issued and outstanding as of December 31, 2015 and 2014) | — | — | ||||||
| Common stock, $0.001 par value per share (150,000,000 shares authorized, 54,095,110 and 44,974,248 shares issued, 54,053,801 and 44,932,939 shares outstanding at December 31, 2015 and 2014, respectively) | 54,095 | 44,974 | ||||||
| Contingently issuable shares | 6 | 6 | ||||||
| Additional paid-in capital | 259,887,464 | 175,476,521 | ||||||
| Treasury stock, at cost, 41,309 shares at December 31, 2015 and 2014 | (234,337 | ) | (234,337 | ) | ||||
| Accumulated deficit | (158,133,926 | ) | (95,185,280 | ) | ||||
| Total stockholders’ equity | 101,573,302 | 80,101,884 | ||||||
| Total liabilities and stockholders’ equity | $ | 113,473,201 | $ | 86,746,890 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-2 |
TG Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Operations for the Years Ended December 31, 2015, 2014 and 2013
| 2015 | 2014 | 2013 | ||||||||||
| License revenue | $ | 152,381 | $ | 152,381 | $ | 152,381 | ||||||
| Costs and expenses: | ||||||||||||
| Research and development: | ||||||||||||
| Noncash stock expense associated with in-licensing agreements | — | 5,350,094 | — | |||||||||
| Noncash compensation | 4,261,406 | 8,731,566 | 1,041,519 | |||||||||
| Other research and development | 43,445,817 | 26,004,687 | 12,621,161 | |||||||||
| Total research and development | 47,707,223 | 40,086,347 | 13,662,680 | |||||||||
| General and administrative: | ||||||||||||
| Noncash compensation | 11,435,686 | 12,373,726 | 4,161,629 | |||||||||
| Other general and administrative | 4,189,488 | 3,413,400 | 2,496,461 | |||||||||
| Total general and administrative | 15,625,174 | 15,787,126 | 6,658,090 | |||||||||
| Impairment of in-process research and development | — | — | 2,797,600 | |||||||||
| Total costs and expenses | 63,332,397 | 55,873,473 | 23,118,370 | |||||||||
| Operating loss | (63,180,016 | ) | (55,721,092 | ) | (22,965,989 | ) | ||||||
| Other (income) expense: | ||||||||||||
| Interest income | (174,653 | ) | (55,049 | ) | (30,822 | ) | ||||||
| Other income | — | (95,427 | ) | (108,894 | ) | |||||||
| Interest expense | 972,736 | 930,701 | 952,888 | |||||||||
| Change in fair value of notes payable | (1,029,453 | ) | (720,040 | ) | (3,300,951 | ) | ||||||
| Total other (income) expense, net | (231,370 | ) | 60,185 | (2,487,779 | ) | |||||||
| Net loss | (62,948,646 | ) | (55,781,277 | ) | (20,478,210 | ) | ||||||
| Basic and diluted net loss per common share | $ | (1.38 | ) | $ | (1.64 | ) | $ | (0.81 | ) | |||
| Weighted average shares used in computing basic and diluted net loss per common share | 45,646,414 | 34,068,926 | 25,413,964 | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-3 |
TG Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Stockholders’ Equity for the Years Ended December 31, 2015, 2014 and 2013
| Common stock | Contingently issuable | Additional
paid- in | Treasury Stock | Accumulated | ||||||||||||||||||||||||||||
| Shares | Amount | shares | capital | Shares | Amount | deficit | Total | |||||||||||||||||||||||||
| Balance at January 1, 2013 | 25,820,738 | $ | 25,821 | $ | 6 | $ | 34,534,805 | 13,526 | $ | (84,538 | ) | $ | (18,925,793 | ) | $ | 15,550,301 | ||||||||||||||||
| Issuance of common stock in connection with exercise of warrants | 1,013,009 | 1,013 | 2,279,760 | 2,280,773 | ||||||||||||||||||||||||||||
| Issuance of common stock in connection with cashless exercise of warrants | 3,024 | 3 | (4 | ) | (1 | ) | ||||||||||||||||||||||||||
| Issuance of restricted stock | 944,464 | 944 | (944 | ) | — | |||||||||||||||||||||||||||
| Issuance of common stock in public offering (net of offering costs of $2,664,970) | 6,555,000 | 6,555 | 37,641,725 | 37,648,280 | ||||||||||||||||||||||||||||
| Compensation in respect of restricted stock and stock options granted to employees, directors and consultants | 5,203,148 | 5,203,148 | ||||||||||||||||||||||||||||||
| Surrender of common stock for tax withholding | 27,783 | (149,799 | ) | (149,799 | ) | |||||||||||||||||||||||||||
| Net loss | (20,478,210 | ) | (20,478,210 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2013 | 34,336,235 | 34,336 | 6 | 79,658,490 | 41,309 | (234,337 | ) | (39,404,003 | ) | 40,054,492 | ||||||||||||||||||||||
| Issuance of common stock in connection with exercise of warrants | 1,560,826 | 1,561 | 3,555,063 | 3,556,624 | ||||||||||||||||||||||||||||
| Issuance of common stock in connection with exercise of options | 46,000 | 46 | 202,354 | 202,400 | ||||||||||||||||||||||||||||
| Issuance of restricted stock | 982,793 | 983 | (983 | ) | — | |||||||||||||||||||||||||||
| Forfeiture of restricted stock | (1,000 | ) | (1 | ) | 1 | — | ||||||||||||||||||||||||||
| Issuance of common stock in public offering (net of offering costs of $1,344,440) | 2,702,809 | 2,703 | 16,788,705 | 16,791,408 | ||||||||||||||||||||||||||||
| Issuance of common stock in At the Market offering (net of offering costs of $1,101,572) | 4,850,055 | 4,850 | 48,818,002 | 48,822,852 | ||||||||||||||||||||||||||||
| Compensation in respect of restricted stock and stock options granted to employees, directors and consultants | 21,105,292 | 21,105,292 | ||||||||||||||||||||||||||||||
| Common stock issued in connection with in-licensing agreements | 496,530 | 496 | 5,349,597 | 5,350,093 | ||||||||||||||||||||||||||||
| Net loss | (55,781,277 | ) | (55,781,277 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2014 | 44,974,248 | 44,974 | 6 | 175,476,521 | 41,309 | (234,337 | ) | (95,185,280 | ) | 80,101,884 | ||||||||||||||||||||||
| Issuance of common stock in connection with exercise of warrants | 2,946,703 | 2,946 | 1,064,393 | 1,067,339 | ||||||||||||||||||||||||||||
| Issuance of common stock in connection with cashless exercise of warrants | 2,915 | 3 | (3 | ) | — | |||||||||||||||||||||||||||
| Issuance of common stock in connection with conversion of notes payable | 522 | 1 | 6,924 | 6,925 | ||||||||||||||||||||||||||||
| Issuance of restricted stock | 1,992,535 | 1,993 | (1,993 | ) | — | |||||||||||||||||||||||||||
| Forfeiture of restricted stock | (31,166 | ) | (31 | ) | 31 | — | ||||||||||||||||||||||||||
| Issuance of common stock to related party for cash (See Note 9) | 114,855 | 115 | 749,890 | 750,005 | ||||||||||||||||||||||||||||
| Issuance of common stock in At-the-Market offering (net of offering costs of $1,310,591) | 4,094,498 | 4,094 | 66,894,609 | 66,898,703 | ||||||||||||||||||||||||||||
| Compensation in respect of restricted stock granted to employees, directors and consultants | 15,697,092 | 15,697,092 | ||||||||||||||||||||||||||||||
| Net loss | (62,948,646 | ) | (62,948,646 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2015 | 54,095,110 | $ | 54,095 | $ | 6 | $ | 259,887,464 | 41,309 | $ | (234,337 | ) | $ | (158,133,926 | ) | $ | 101,573,302 | ||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-4 |
TG Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows for the Years Ended December 31, 2015, 2014 and 2013
| 2015 | 2014 | 2013 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||||||
| Consolidated net loss | $ | (62,948,646 | ) | $ | (55,781,277 | ) | $ | (20,478,210 | ) | |||
| Adjustments to reconcile consolidated net loss to net cash used in operating activities: | ||||||||||||
| Gain on settlement of notes payable | — | (95,427 | ) | — | ||||||||
| Noncash stock compensation expense | 15,697,092 | 21,105,292 | 5,203,148 | |||||||||
| Noncash stock expense associated with in-licensing agreements | — | 5,350,094 | — | |||||||||
| Impairment of in-process research and development | — | — | 2,797,600 | |||||||||
| Depreciation | 15,452 | 3,931 | 1,127 | |||||||||
| Amortization of premium on investment securities | 536,142 | 193,581 | 975 | |||||||||
| Change in fair value of notes payable | (56,717 | ) | 210,661 | (2,414,569 | ) | |||||||
| Changes in assets and liabilities: | ||||||||||||
| Increase in restricted cash | (4,131 | ) | (575,012 | ) | — | |||||||
| (Increase) decrease in other current assets | (3,105,771 | ) | (4,563,067 | ) | 229,258 | |||||||
| Increase in accrued interest receivable | (100,505 | ) | (58,347 | ) | (27,169 | ) | ||||||
| Increase in other assets | (41,722 | ) | — | — | ||||||||
| Increase (decrease) in accounts payable and accrued expenses | 5,470,915 | (603,377 | ) | 4,034,605 | ||||||||
| Increase (decrease) in interest payable | — | (94,590 | ) | 66,506 | ||||||||
| Decrease in deferred revenue | (152,381 | ) | (152,381 | ) | (152,381 | ) | ||||||
| Net cash used in operating activities | (44,690,272 | ) | (35,059,919 | ) | (10,739,110 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||||||
| Purchases of property, plant and equipment | (42,217 | ) | (18,570 | ) | (5,681 | ) | ||||||
| Investment in held-to-maturity securities | (48,993,652 | ) | (18,336,719 | ) | (4,919,871 | ) | ||||||
| Proceeds from maturity of short-term securities | 24,350,000 | — | — | |||||||||
| Net cash used in investing activities | (24,685,869 | ) | (18,355,289 | ) | (4,925,552 | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||||||
| Proceeds from the exercise of warrants | 1,067,339 | 3,556,624 | 2,280,773 | |||||||||
| Proceeds from the exercise of options | — | 202,400 | — | |||||||||
| Payment of notes payable | — | (677,778 | ) | — | ||||||||
| Proceeds from sale of common stock, net | 67,760,517 | 65,614,260 | 37,648,280 | |||||||||
| Deferred financing costs paid | (104,170 | ) | (51,980 | ) | (85,121 | ) | ||||||
| Purchase of treasury stock | — | — | (149,799 | ) | ||||||||
| Net cash provided by financing activities | 68,723,686 | 68,643,526 | 39,694,133 | |||||||||
| NET (DECREASE) INCREASE IN CASH AND CASH EQUIVALENTS | (652,455 | ) | 15,228,318 | 24,029,471 | ||||||||
| Cash and cash equivalents at beginning of year | 55,713,784 | 40,485,466 | 16,455,995 | |||||||||
| CASH AND CASH EQUIVALENTS AT END OF YEAR | $ | 55,061,329 | $ | 55,713,784 | $ | 40,485,466 | ||||||
| NONCASH TRANSACTIONS | ||||||||||||
| Reclassification of deferred financing costs to additional paid-in capital | $ | (111,810 | ) | — | — | |||||||
| Conversion of convertible notes payable to common stock | $ | 6,924 | — | — | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-5 |
TG Therapeutics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
Unless the context requires otherwise, references in this report to “TG,” “Company,” “we,” “us” and “our” refer to TG Therapeutics, Inc. and our subsidiaries.
NOTE 1 – ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
DESCRIPTION OF BUSINESS
We are a biopharmaceutical company focused on the acquisition, development and commercialization of novel treatments for B-cell malignancies and autoimmune diseases. Currently, the Company is developing two therapies targeting hematologic malignancies. TG-1101 (ublituximab) is a novel, glycoengineered monoclonal antibody that targets a specific and unique epitope on the CD20 antigen found on mature B-lymphocytes. We are also developing TGR-1202, an orally available PI3K delta inhibitor. The delta isoform of PI3K is strongly expressed in cells of hematopoietic origin and is believed to be important in the proliferation and survival of B-lymphocytes. Both TG-1101 and TGR-1202 are in clinical development for patients with hematologic malignancies. The Company also has pre-clinical programs seeking to develop IRAK4 (interleukin-1 receptor-associated kinase 4) inhibitors and anti-PD-L1 and anti-GITR antibodies.
We also actively evaluate complementary products, technologies and companies for in-licensing, partnership, acquisition and/or investment opportunities. To date, we have not received approval for the sale of any of our drug candidates in any market and, therefore, have not generated any product sales from our drug candidates.
LIQUIDITY AND CAPITAL RESOURCES
We have incurred operating losses since our inception, and expect to continue to incur operating losses for the foreseeable future and may never become profitable. As of December 31, 2015, we have an accumulated deficit of $158.1 million.
Our major sources of cash have been proceeds from the private placement and public offering of equity securities. We have not yet commercialized any of our drug candidates and cannot be sure if we will ever be able to do so. Even if we commercialize one or more of our drug candidates, we may not become profitable. Our ability to achieve profitability depends on many factors, including our ability to obtain regulatory approval for our drug candidates; successfully complete any post-approval regulatory obligations; and successfully commercialize our drug candidates alone or in partnership. We may continue to incur substantial operating losses even if we begin to generate revenues from our drug candidates.
As of December 31, 2015, we had $102.4 million in cash and cash equivalents, investment securities, and interest receivable. We currently anticipate that our cash and cash equivalents to be sufficient to fund our anticipated operating cash requirements for more than 24 months from December 31, 2015. The actual amount of cash that we will need to operate is subject to many factors, including, but not limited to, the timing, design and conduct of clinical trials for our drug candidates. We are dependent upon significant future financing to provide the cash necessary to execute our current operations, including the commercialization of any of our drug candidates.
Our common stock is quoted on the Nasdaq Capital Market and trades under the symbol “TGTX.”
RECENTLY ISSUED ACCOUNTING STANDARDS
In May 2014, the Financial Accounting Standards Board (“FASB”) issued an update to Accounting Standards Codification (“ASC”) 606, Revenue from Contracts with Customers. This update to ASC 606 provides a five-step process to determine when and how revenue is recognized. The core principle of the guidance is that a Company should recognize revenue upon transfer of promised goods or services to customers in an amount that reflects the expected consideration to be received in exchange for those goods or services. This update to ASC 606 will also result in enhanced disclosures about revenue, providing guidance for transactions that were not previously addressed comprehensively, and improving guidance for multiple-element arrangements. This update to ASC 606 is effective for us beginning in fiscal 2018. We are currently evaluating the impact of this update on our consolidated financial statements.
| F-6 |
TG Therapeutics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
In August 2014, the FASB issued Accounting Standards Update 2014-15, Presentation of Financial Statements—Going Concern, which requires management of an entity to evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued or available to be issued. This update will become effective beginning January 1, 2017, with early adoption permitted. The provisions of this standard are not expected to significantly impact the Company.
In November 2015, the FASB issued Accounting Standards Update 2015-17, Balance Sheet Classification of Deferred Taxes. This update requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. The amendments in this update apply to all entities that present a classified statement of financial position. This update will become effective for public business entities for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The provisions of this standard are not expected to significantly impact the Company.
In February 2016, the FASB issued ASU No. 2016-02, “Leases (Topic 842),” which requires lessees to recognize assets and liabilities for the rights and obligations created by most leases on their balance sheet. The guidance is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early application is permitted. ASU 2016-02 requires modified retrospective adoption for all leases existing at, or entered into after, the date of initial application, with an option to use certain transition relief. The Company is currently evaluating the impact the standard may have on its consolidated financial statements and related disclosures.
Other pronouncements issued by the FASB or other authoritative accounting standards group with future effective dates are either not applicable or not significant to our consolidated financial statements.
USE OF ESTIMATES
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”) requires management to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the applicable reporting period. Actual results could differ from those estimates. Such differences could be material to the financial statements.
CASH AND CASH EQUIVALENTS
We treat liquid investments with original maturities of less than three months when purchased as cash and cash equivalents.
RESTRICTED CASH
We record cash pledged or held in trust as restricted cash. As of December 31, 2015, we have approximately $0.6 million of restricted cash pledged to secure a line of credit as a security deposit for a Desk Agreement (see Note 9).
INVESTMENT SECURITIES
Investment securities at December 31, 2015 and 2014 consist of short-term and long-term government securities. We classify these securities as held-to-maturity. Held-to-maturity securities are those securities in which we have the ability and intent to hold the security until maturity. Held-to-maturity securities are recorded at amortized cost, adjusted for the amortization or accretion of premiums or discounts. Premiums and discounts are amortized or accreted over the life of the related held-to-maturity security as an adjustment to yield using the effective interest method.
A decline in the market value of any investment security below cost, that is deemed to be other than temporary, results in a reduction in the carrying amount to fair value. The impairment is charged to operations and a new cost basis for the security is established. Other-than-temporary impairment charges are included in interest and other income (expense), net. Unrealized gains, if determined to be temporary, are included in accumulated other comprehensive income in equity. Dividend and interest income are recognized when earned.
| F-7 |
TG Therapeutics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
CREDIT RISK
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents, short-term investments and long-term investments. The Company maintains its cash and cash equivalents with high-credit quality financial institutions. At times, such amounts may exceed federally-insured limits.
REVENUE RECOGNITION
We recognize license revenue in accordance with the revenue recognition guidance of the FASB Accounting Standards Codification, or Codification. We analyze each element of our licensing agreement to determine the appropriate revenue recognition. The terms of the license agreement may include payments to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments over the period of significant involvement under the related agreements unless the fee is in exchange for products delivered or services rendered that represent the culmination of a separate earnings process and no further performance obligation exists under the contract. We recognize milestone payments as revenue upon the achievement of specified milestones only if (1) the milestone payment is non-refundable, (2) substantive effort is involved in achieving the milestone, (3) the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (4) the milestone is at risk for both parties. If any of these conditions are not met, we defer the milestone payment and recognize it as revenue over the estimated period of performance under the contract.
RESEARCH AND DEVELOPMENT COSTS
Generally, research and development costs are expensed as incurred. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and amortized over the period that the goods are delivered or the related services are performed, subject to an assessment of recoverability. We make estimates of costs incurred in relation to external clinical research organizations, or CROs, and clinical site costs. We analyze the progress of clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of the amount expensed and the related prepaid asset and accrued liability. Significant judgments and estimates must be made and used in determining the accrued balance and expense in any accounting period. We review and accrue CRO expenses and clinical trial study expenses based on work performed and rely upon estimates of those costs applicable to the stage of completion of a study. Accrued CRO costs are subject to revisions as such trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known. With respect to clinical site costs, the financial terms of these agreements are subject to negotiation and vary from contract to contract. Payments under these contracts may be uneven, and depend on factors such as the achievement of certain events, the successful recruitment of patients, the completion of portions of the clinical trial or similar conditions. The objective of our policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical site costs are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Prepaid research and development in our consolidated balance sheet includes, among other things, certain fees related to development and manufacturing services. These development and manufacturing agreements often require payments in advance of services performed or goods received. Accordingly, as of December 31, 2015 and 2014, we recorded approximately $7.7 million and $5.8 million, respectively, in prepaid research and development related to such advance agreements.
| F-8 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
INCOME TAXES
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, operating losses and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. If the likelihood of realizing the deferred tax assets or liability is less than “more likely than not,” a valuation allowance is then created.
We, and our subsidiaries, file income tax returns in the U.S. Federal jurisdiction and in various states. We have tax net operating loss carryforwards that are subject to examination for a number of years beyond the year in which they were generated for tax purposes. Since a portion of these net operating loss carryforwards may be utilized in the future, many of these net operating loss carryforwards will remain subject to examination. We recognize interest and penalties related to uncertain income tax positions in income tax expense.
STOCK - BASED COMPENSATION
We recognize all share-based payments to employees and non-employee directors (as compensation for service) as noncash compensation expense in the consolidated financial statements based on the fair values of such payments. Stock-based compensation expense recognized each period is based on the value of the portion of share-based payment awards that is ultimately expected to vest during the period. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
For share-based payments to consultants and other third-parties (including related parties), noncash compensation expense is determined at the “measurement date.” The expense is recognized over the vesting period of the award. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record compensation expense based on the fair value of the award at the reporting date. The awards to consultants and other third-parties (including related parties) are then revalued, or the total compensation is recalculated based on the then current fair value, at each subsequent reporting date.
In addition, because some of the options, restricted stock and warrants issued to employees, consultants and other third-parties vest upon achievement of certain milestones, the total expense is uncertain. Compensation expense for such awards that vest upon the achievement of milestones is recognized when the achievement of such milestone becomes probable.
BASIC AND DILUTED NET LOSS PER COMMON SHARE
Basic net loss per common share is calculated by dividing net loss applicable to common shares by the weighted-average number of common shares outstanding for the period. Diluted net loss per common share is the same as basic net loss per common share, since potentially dilutive securities from stock options, stock warrants and convertible preferred stock would have an antidilutive effect either because the Company incurred a net loss during the period presented or because such potentially dilutive securities were out of the money and the Company realized net income during the period presented. The amounts of potentially dilutive securities excluded from the calculation were 7,064,396, 8,890,796 and 10,618,584 at December 31, 2015, 2014 and 2013, respectively. During the years ended December 31, 2015, 2014 and 2013 the Company incurred a net loss, therefore, all of the dilutive securities are excluded from the computation of diluted loss per share.
LONG-LIVED ASSETS AND GOODWILL
Long-lived assets are reviewed for an impairment loss when circumstances indicate that the carrying value of long-lived tangible and intangible assets with finite lives may not be recoverable. Management’s policy in determining whether an impairment indicator exists, a triggering event, comprises measurable operating performance criteria as well as qualitative measures. If an analysis is necessitated by the occurrence of a triggering event, we make certain assumptions in determining the impairment amount. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized.
Goodwill is reviewed for impairment annually, or when events arise that could indicate that an impairment exists. We test for goodwill impairment using a two-step process. The first step compares the fair value of the reporting unit with the unit's carrying value, including goodwill. When the carrying value of the reporting unit is greater than fair value, the unit’s goodwill may be impaired, and the second step must be completed to measure the amount of the goodwill impairment charge, if any. In the second step, the implied fair value of the reporting unit’s goodwill is compared with the carrying amount of the unit’s goodwill. If the carrying amount is greater than the implied fair value, the carrying value of the goodwill must be written down to its implied fair value. We will continue to perform impairment tests annually, at December 31, and whenever events or changes in circumstances suggest that the carrying value of an asset may not be recoverable.
| F-9 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
NOTE 2 – CASH AND CASH EQUIVALENTS
The following tables summarize our cash and cash equivalents at December 31, 2015 and December 31, 2014:
| December 31, 2015 | December 31, 2014 | |||||||
| Money market funds | $ | 8,265,583 | $ | 12,364,537 | ||||
| Checking and bank deposits | 46,795,746 | 43,349,247 | ||||||
| Total | $ | 55,061,329 | $ | 55,713,784 | ||||
NOTE 3 – INVESTMENT SECURITIES
We record our investments as held-to-maturity. Held-to-maturity investments are recorded at amortized cost.
The following tables summarize our investment securities at December 31, 2015 and 2014:
| December 31, 2015 | ||||||||||||||||
| Amortized
cost, as adjusted | Gross
unrealized holding gains | Gross
unrealized holding losses | Estimated fair value | |||||||||||||
| Short-term investments: | ||||||||||||||||
| Obligations of domestic governmental agencies (maturing between January 2016 and December 2016) (held-to-maturity) | $ | 22,166,512 | $ | — | $ | 22,822 | $ | 22,143,690 | ||||||||
| Long-term investments: | ||||||||||||||||
| Obligations of domestic governmental agencies (maturing between January 2017 and December 2017) (held-to-maturity) | 25,003,032 | — | 85,846 | 24,917,186 | ||||||||||||
| Total short-term and long-term investment securities | $ | 47,169,544 | $ | — | $ | 108,668 | $ | 47,060,876 | ||||||||
| December 31, 2014 | ||||||||||||||||
| Amortized cost, as adjusted | Gross unrealized holding gains | Gross holding losses | Estimated fair value | |||||||||||||
| Short-term investments: | ||||||||||||||||
| Obligations of domestic governmental agencies (maturing between January 2015 and December 2015) (held-to-maturity) | $ | 23,062,034 | $ | 922 | $ | 5,806 | $ | 23,057,150 | ||||||||
| Total short-term investment securities | $ | 23,062,034 | $ | 922 | $ | 5,806 | $ | 23,057,150 | ||||||||
NOTE 4 – FAIR VALUE MEASUREMENTS
We measure certain financial assets and liabilities at fair value on a recurring basis in the financial statements. The fair value hierarchy ranks the quality and reliability of inputs, or assumptions, used in the determination of fair value and requires financial assets and liabilities carried at fair value to be classified and disclosed in one of the following three categories:
| · | Level 1 – quoted prices in active markets for identical assets and liabilities; |
| · | Level 2 – inputs other than Level 1 quoted prices that are directly or indirectly observable; and |
| · | Level 3 – unobservable inputs that are not corroborated by market data. |
| F-10 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
As of December 31, 2015 and 2014, the fair values of cash and cash equivalents, restricted cash, and notes and interest payable, current portion approximate their carrying value.
At the time of our merger (we were then known as Manhattan Pharmaceuticals, Inc. (“Manhattan”)) with Ariston Pharmaceuticals, Inc. (“Ariston”) in March 2010, Ariston issued $15.5 million of five-year 5% notes payable (the “5% Notes”) in satisfaction of several note payable issuances. The 5% Notes and accrued and unpaid interest thereon are convertible at the option of the holder into common stock at the conversion price of $1,125 per share. Ariston agreed to make quarterly payments on the 5% Notes equal to 50% of the net product cash flow received from the exploitation or commercialization of Ariston’s product candidates, AST-726 and AST-915. We have no obligations under the 5% Notes aside from a) 50% of the net product cash flows from Ariston’s product candidates, if any, payable to noteholders; and b) the conversion feature, discussed above.
The cumulative liability including accrued and unpaid interest of the 5% Notes was approximately $19.9 million at December 31, 2015 and $19.5 million at December 31, 2014. No payments have been made on the 5% Notes as of December 31, 2015.
In December 2011, we elected the fair value option for valuing the 5% Notes. The fair value option was elected in order to reflect in our financial statements the assumptions that market participants use in evaluating these financial instruments.
As of December 31, 2013, as a result of expiring intellectual property rights and other factors, it was determined that net product cash flows from AST-726 were unlikely. As we have no other obligations under the 5% Notes aside from the net product cash flows and the conversion feature, the conversion feature was used to estimate the 5% Notes’ fair value as of December 31, 2015 and 2014. The assumptions, assessments and projections of future revenues are subject to uncertainties, difficult to predict, and require significant judgment. The use of different assumptions, applying different judgment to inherently subjective matters and changes in future market conditions could result in significantly different estimates of fair value and the differences could be material to our consolidated financial statements.
The following tables provide the fair value measurements of applicable financial liabilities as of December 31, 2015 and 2014:
| Financial liabilities at fair value as of December 31, 2015 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| 5% Notes | $ | — | $ | — | $ | 211,549 | $ | 211,549 | ||||||||
| Totals | $ | — | $ | — | $ | 211,549 | $ | 211,549 | ||||||||
| Financial liabilities at fair value as of December 31, 2014 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| 5% Notes | $ | — | $ | — | $ | 275,190 | $ | 275,190 | ||||||||
| Totals | $ | — | $ | — | $ | 275,190 | $ | 275,190 | ||||||||
The Level 3 amounts above represent the fair value of the 5% Notes and related accrued interest.
| F-11 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
The following table summarizes the changes in Level 3 instruments for the years ended December 31, 2014 and 2015:
| Balance at January 1, 2014 | $ | 64,529 | ||
| Interest accrued on face value of 5% Notes | 930,701 | |||
| Change in fair value of Level 3 liabilities | (720,040 | ) | ||
| Balance at December 31, 2014 | 275,190 | |||
| Interest accrued on face value of 5% Notes | 972,736 | |||
| Conversion of 5% Notes | (6,924 | ) | ||
| Change in fair value of Level 3 liabilities | (1,029,453 | ) | ||
| Balance at December 31, 2015 | $ | 211,549 |
The change in the fair value of the Level 3 liabilities is reported in other (income) expense in the accompanying consolidated statements of operations.
Nonrecurring Fair Value Measurements
There were no nonrecurring fair value measurements during the year ended December 31, 2015. The following table summarizes the assets measured at fair value on a nonrecurring basis as of December 31, 2013:
| Fair value measurements as of December 31, 2013 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total
impairment charge for the year ended December 31, 2013 | |||||||||||||
| Assets: | ||||||||||||||||
| In-process research and development | $ | — | $ | — | $ | — | $ | (2,797,600 | ) | |||||||
| Total | $ | — | $ | — | $ | — | $ | (2,797,600 | ) | |||||||
As of December 31, 2013, as a result of expiring intellectual property rights, uncertain regulatory pathways, and market changes affecting the commercial potential for the Ariston in-process research and development asset (AST-726), we determined that the asset's carrying value was no longer recoverable. Accordingly, during the year ended December 31, 2013, we recorded a non-cash impairment charge of $2,797,600, which was recorded as impairment of in-process research and development in our consolidated statements of operations. The fair value of this asset was determined to be zero primarily as a result of the expiring intellectual property rights surrounding the asset. This fair value measurement technique is based on significant inputs not observable in the market and thus represents a Level 3 measurement within the fair value hierarchy.
NOTE 5 – STOCKHOLDERS’ EQUITY
Preferred Stock
Our amended and restated certificate of incorporation authorizes the issuance of up to 10,000,000 shares of preferred stock, $0.001 par value, with rights senior to those of our common stock, issuable in one or more series. Upon issuance, the Company can determine the rights, preferences, privileges and restrictions thereof. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of common stock.
Stockholder Rights Plan
On July 18, 2014, we adopted a stockholder rights plan. The stockholder rights plan is embodied in the Stockholder Protection Rights Agreement dated as of July 18, 2014 (the “Rights Agreement”), between us and American Stock Transfer & Trust Company, LLC, as rights agent (the “Rights Agent”).
Accordingly, the Board of Directors declared a distribution of one right (a “Right”) for each outstanding share of common stock, to stockholders of record at the close of business on July 28, 2014, for each share of common stock issued (including shares distributed from treasury) by us thereafter and prior to the Separation Time (as defined in the Rights Agreement), and for certain shares of common stock issued after the Separation Time. Following the Separation Time, each Right entitles the registered holder to purchase from us one one-thousandth (1/1,000) of a share of Series A Junior Participating Preferred Stock, par value $0.001 per share (the "Preferred Stock"), at a purchase price of $100.00 (the "Exercise Price"), subject to adjustment. The description and terms of the Rights are set forth in the Rights Agreement. Each one one-thousandth of a share of Preferred Stock has substantially the same rights as one share of common stock. Subject to the terms and conditions of the Rights Agreement, Rights become exercisable ten days after the public announcement that a “Person” has become an “Acquiring Person” (as each such term is defined in the Rights Agreement). Any Rights held by an Acquiring Person are void and may not be exercised.
| F-12 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
If a Person becomes an Acquiring Person, all holders of Rights, except the Acquiring Person, may purchase at the Right’s then-current exercise price, common stock having a market value equal to twice the exercise price. Moreover, at any time after a Person becomes an Acquiring Person (unless such Person acquires 50 percent or more of our common stock then outstanding, as more fully described in the Rights Agreement), the Board of Directors may exchange all (but not less than all) of the then outstanding Rights (other than rights owned by such Person, which would have become void) for shares of common stock at an exchange ratio of one share of common stock per Right, appropriately adjusted in order to protect the interests of holders of Rights.
The Rights Agreement was approved by our Board of Directors on July 18, 2014. The Rights will expire at the close of business on its ten year anniversary, unless earlier exchanged or terminated by us.
Common Stock
Our amended and restated certificate of incorporation authorizes the issuance of up to 150,000,000 shares of $0.001 par value common stock. At the annual shareholder meeting on June 6, 2014, an amendment to the Company's Certificate of Incorporation to decrease its authorized share capital by 350,000,000 shares from 500,000,000 to 150,000,000 was approved.
On July 18, 2013, we announced the pricing of an underwritten public offering of 5,700,000 shares of our common stock at a price of $6.15 per share for gross proceeds of approximately $35 million. We also granted to the underwriters a 30-day option to acquire an additional 855,000 shares to cover overallotments in connection with the offering. Total net proceeds from this offering, including the overallotment, were approximately $37.6 million, net of underwriting discounts and offering expenses of approximately $2.7 million. The shares were sold under a shelf registration statement on Form S-3 (File No. 333-189015) that was previously filed and declared effective by the SEC on June 17, 2013.
On March 11, 2014, we announced the pricing of an underwritten sale of 2,702,809 shares of our common stock at a price of $6.71 per share for gross proceeds of approximately $18.1 million. Net proceeds from this offering were approximately $16.8 million, net of underwriting discounts and offering expenses of approximately $1.3 million. The shares were sold under a shelf registration statement on Form S-3 (File No. 333-189015) that was previously filed and declared effective by the SEC on June 17, 2013.
On June 21, 2013, we entered into an At-the-Market Issuance Sales Agreement (the “2013 ATM”) with MLV & Co. LLC (“MLV”) under which we could issue and sell shares of our common stock, having an aggregate offering price of up to $50.0 million, from time to time through MLV, acting as the sales agent. Under the agreement we would pay MLV a commission rate of up to 3.0% of the gross proceeds from the sale of any shares of common stock sold through MLV.
During the year ended December 31, 2014, we sold a total of 4,850,055 shares of common stock under the 2013 ATM for aggregate total gross proceeds of approximately $50.0 million at an average selling price of $10.31 per share. Net proceeds were approximately $48.9 million after deducting commissions and other transactions costs.
In December 2014, we filed a shelf registration statement on Form S-3 (the “2015 S-3”), which was declared effective in January 2015. Under the 2015 S-3, the Company may sell up to a total of $250 million of its securities. In connection with the 2015 S-3, we amended our 2013 At-the-Market Issuance Sales Agreement with MLV (the “2015 ATM”) such that we may issue and sell additional shares of our common stock, having an aggregate offering price of up to $75.0 million, from time to time through MLV, acting as the sales agent. Under the 2015 ATM we pay MLV a commission rate of up to 3.0% of the gross proceeds from the sale of any shares of common stock sold through MLV.
| F-13 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
During the year ended December 31, 2015, we sold a total of 4,094,498 shares of common stock under the 2015 ATM for aggregate total gross proceeds of approximately $68.2 million at an average selling price of $16.66 per share, resulting in net proceeds of approximately $67.0 million after deducting commissions and other transaction costs.
We currently have two shelf registration statements on Form S-3 filed and declared effective by the SEC (File No. 333-189015 and File No. 333-201339). After deducting shares already sold, approximately $248 million of common stock remains available for sale under these shelf registration statements. We may offer the securities under our shelf registration statements from time to time in response to market conditions or other circumstances if we believe such a plan is in the best interests of our stockholders. We believe that these shelf registration statements provide us with the flexibility to raise additional capital to finance our operations as needed.
Treasury Stock
As of December 31, 2015 and 2014, 41,309 shares of common stock are being held in Treasury, at a cost of approximately $234,000, representing the fair market value on the date the shares were surrendered to the Company to satisfy employee tax obligations.
Equity Incentive Plans
In May 2012, we established the TG Therapeutics, Inc. Amended and Restated 2012 Incentive Plan (“2012 Incentive Plan”). Under the 2012 Incentive Plan, our compensation committee of board of directors is authorized to grant stock-based awards to directors, consultants, employees and officers. The 2012 Incentive Plan authorizes grants to purchase up to 6,000,000 shares of authorized but unissued common stock.
An amendment to the 2012 Incentive Plan was approved by stockholders in June 2015. Pursuant to this amendment, 6,000,000 shares were added to the 2012 Incentive Plan. As of December 31, 2015, no options were outstanding and up to an additional 4,164,631 shares may be issued under the 2012 Incentive Plan.
Stock Options
The following table summarizes stock option activity for the years ended December 31, 2015, 2014 and 2013:
Number of shares |
average exercise price | Weighted- average contractual |
Aggregate | |||||||||||||
| (in years) | ||||||||||||||||
| Outstanding at January 1, 2013 | 46,904 | $ | 61.08 | 9.44 | ||||||||||||
| Granted | — | — | ||||||||||||||
| Exercised | — | — | ||||||||||||||
| Forfeited | (313 | ) | 2,249.85 | |||||||||||||
| Expired | — | — | ||||||||||||||
| Outstanding at December 31, 2013 | 46,591 | 46.37 | 8.50 | $ | — | |||||||||||
| Granted | — | — | ||||||||||||||
| Exercised | (46,000 | ) | 4.40 | |||||||||||||
| Forfeited | — | — | ||||||||||||||
| Expired | (397 | ) | 4,457.57 | |||||||||||||
| Outstanding at December 31, 2014 | 194 | 971.70 | 3.50 | $ | — | |||||||||||
| Granted | — | — | ||||||||||||||
| Exercised | — | — | ||||||||||||||
| Forfeited | (152 | ) | 463.32 | |||||||||||||
| Expired | (42 | ) | 2,811.53 | |||||||||||||
| Outstanding at December 31, 2015 | — | $ | — | — | $ | — | ||||||||||
| Exercisable at December 31, 2015 | — | $ | — | — | $ | — | ||||||||||
| F-14 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
As of December 31, 2015, there are no unvested option awards and no unrecognized compensation cost related option awards.
Restricted Stock
Certain employees, directors and consultants have been awarded restricted stock. The restricted stock vesting consists of milestone and time-based vesting. The following table summarizes restricted share activity for the years ended December 31, 2015, 2014 and 2013:
Number of Shares | Weighted Average Grant Date Fair Value | |||||||
| Outstanding at January 1, 2013 | 6,614,243 | $ | 4.49 | |||||
| Granted | 944,464 | 4.13 | ||||||
| Vested | (523,750 | ) | 2.48 | |||||
| Forfeited | — | — | ||||||
| Outstanding at December 31, 2013 | 7,034,957 | 4.60 | ||||||
| Granted | 982,793 | 13.55 | ||||||
| Vested | (1,616,749 | ) | 6.53 | |||||
| Forfeited | (1,000 | ) | 6.60 | |||||
| Outstanding at December 31, 2014 | 6,400,001 | 5.86 | ||||||
| Granted | 1,992,535 | 12.89 | ||||||
| Vested | (1,001,455 | ) | 5.04 | |||||
| Forfeited | (31,166 | ) | 16.76 | |||||
| Outstanding at December 31, 2015 | 7,359,915 | $ | 7.83 | |||||
Total expense associated with restricted stock grants was $15,697,092, $20,726,512 and $5,146,743 during the years ended December 31, 2015, 2014 and 2013, respectively. As of December 31, 2015, there was approximately $22.9 million of total unrecognized compensation cost related to unvested time-based restricted stock, which is expected to be recognized over a weighted-average period of 2.2 years. This amount does not include, as of December 31, 2015, 411,172 shares of restricted stock outstanding which are milestone-based and vest upon certain corporate milestones; and 2,371,167 shares of restricted stock outstanding issued to non-employees. Milestone-based non-cash compensation expense will be measured and recorded if and when a milestone becomes probable. The expense for non-employee shares is determined at the “measurement date.” The expense is recognized over the vesting period of the award. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record compensation expense based on the fair value of the award at the reporting date.
Warrants
The following table summarizes warrant activity for the years ended December 31, 2015, 2014 and 2013:
Warrants | Weighted- Average exercise price | Aggregate intrinsic value | ||||||||||
| Outstanding at January 1, 2013 | 6,781,007 | $ | 1.58 | $ | 14,563,539 | |||||||
| Issued | — | — | ||||||||||
| Exercised | (1,018,068 | ) | 2.25 | |||||||||
| Expired | (43,992 | ) | 16.26 | |||||||||
| Outstanding at December 31, 2013 | 5,718,947 | 1.34 | $ | 14,809,030 | ||||||||
| Issued | — | — | ||||||||||
| Exercised | (1,560,826 | ) | 2.28 | |||||||||
| Expired | (9,893 | ) | 20.74 | |||||||||
| Outstanding at December 31, 2014 | 4,148,228 | 0.94 | $ | 61,792,184 | ||||||||
| Issued | — | — | ||||||||||
| Exercised | (2,950,115 | ) | 0.36 | |||||||||
| Expired | (11,364 | ) | 2.25 | |||||||||
| Outstanding at December 31, 2015 | 1,186,749 | $ | 2.37 | $ | 11,341,452 | |||||||
| F-15 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
Stock-Based Compensation
The fair value of stock options granted is estimated at the date of grant using the Black-Scholes pricing model. The expected term of options granted is derived from historical data and the expected vesting period. Expected volatility is based on the historical volatility of our common stock. The risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. We have assumed no expected dividend yield, as dividends have never been paid to stock or option holders and will not be paid for the foreseeable future. The Company did not grant any stock options during the years ended December 31, 2015, 2014 and 2013.
The following table summarizes stock-based compensation expense information about stock options and restricted stock for the years ended December 31, 2015, 2014 and 2013:
| 2015 | 2014 | 2013 | ||||||||||
| Stock-based compensation expense associated with restricted stock | $ | 15,697,092 | $ | 20,726,512 | $ | 5,146,743 | ||||||
| Stock-based compensation expense associated with stock options | — | 378,780 | 56,405 | |||||||||
| $ | 15,697,092 | $ | 21,105,292 | $ | 5,203,148 | |||||||
NOTE 6 – NOTES PAYABLE
The following is a summary of notes payable:
| December 31, 2015 | December 31, 2014 | |||||||||||||||||||||||
| Current portion, net | Non- current portion, net | Total | Current portion, net | Non- current portion, net | Total | |||||||||||||||||||
| Convertible 5% Notes Payable | $ | 211,549 | $ | - | $ | 211,549 | $ | 275,190 | $ | - | $ | 275,190 | ||||||||||||
| Totals | $ | 211,549 | $ | - | $ | 211,549 | $ | 275,190 | $ | - | $ | 275,190 | ||||||||||||
Convertible 5% Notes Payable
On March 8, 2010, Manhattan entered into an Agreement and Plan of Merger (the "Merger Agreement") by and among Manhattan, Ariston and Ariston Merger Corp., a Delaware corporation and wholly-owned subsidiary of Manhattan (the "Merger Sub"). Pursuant to the terms and conditions of the Merger Agreement, on March 8, 2010, the Merger Sub merged with and into Ariston (the "Merger"), with Ariston being the surviving corporation of the Merger. As a result of the Merger, Ariston became a wholly-owned subsidiary of Manhattan.
The 5% Notes and accrued and unpaid interest thereon are convertible at the option of the holder into common stock at the conversion price of $1,125 per share. Ariston agreed to make quarterly payments on the 5% Notes equal to 50% of the net product cash flow received from the exploitation or commercialization of Ariston’s product candidates, AST-726 and AST-915. We have no obligation under the 5% Notes aside from a) 50% of the net product cash flows from Ariston’s product candidates, if any, payable to noteholders; and b) the conversion feature, discussed above. Interest accrues monthly, is added to principal on an annual basis, every March 8, and is payable at maturity, which was March 8, 2015.
| F-16 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
The cumulative liability including accrued and unpaid interest of these notes was approximately $19.9 million at December 31, 2015 and $19.5 million at December 31, 2014. No payments have been made on the 5% Notes as of December 31, 2015.
In December 2011, we elected the fair value option for valuing the 5% Notes. The fair value option was elected in order to reflect in our financial statements the assumptions that market participants use in evaluating these financial instruments (See Note 4 for further details).
NOTE 7 – INCOME TAXES
We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are determined based on differences between the financial reporting and tax basis of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is established when necessary to reduce deferred tax assets to the amount expected to be realized. In determining the need for a valuation allowance, management reviews both positive and negative evidence, including current and historical results of operations, future income projections and the overall prospects of our business. Based upon management's assessment of all available evidence, we believe that it is more-likely-than-not that the deferred tax assets will not be realizable, and therefore, a valuation allowance has been established. The valuation allowance for deferred tax assets was approximately $86,359,000, $69,084,000 and $46,283,000 as of December 31, 2015, 2014 and 2013, respectively.
As of December 31, 2015, we have U.S. net operating loss carryforwards (“NOLs”) of approximately $195,895,000. For income tax purposes, these NOLs will expire in various amounts through 2035. The Tax Reform Act of 1986 contains provisions which limit the ability to utilize net operating loss carryforwards in the case of certain events including significant changes in ownership interests. The Exchange Transaction with TG Bio may have resulted in a “change in ownership” as defined by IRC Section 382 of the Internal Revenue Code of 1986, as amended. Additionally, stock issuance activities may have resulted in a “change in ownership” as defined by IRC Section 382 of the Internal Revenue Code of 1986, as amended. Accordingly, a substantial portion of the Company’s NOLs above may be subject to annual limitations in reducing any future year’s taxable income.
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets and deferred tax liabilities at December 31, 2015 and 2014 are presented below.
| 2015 | 2014 | |||||||
| Deferred tax assets (liabilities): | ||||||||
| Net operating loss carryforwards | $ | 68,700,379 | $ | 52,350,293 | ||||
| Research and development credit | 4,962,298 | 3,358,934 | ||||||
| Noncash compensation | 12,087,968 | 12,417,815 | ||||||
| Acquired in-process research and development | — | — | ||||||
| Foreign tax credit | — | — | ||||||
| Other | 608,205 | 956,727 | ||||||
| Deferred tax asset, excluding valuation allowance | 86,358,850 | 69,083,769 | ||||||
| Less valuation allowance | (86,358,850 | ) | (69,083,769 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
| F-17 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
There was no current or deferred income tax expense for the year ended December 31, 2015. Income tax expense differed from amounts computed by applying the US Federal income tax rate of 34% to pretax loss as follows:
| For the year ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Loss before income taxes, as reported in the consolidated statements of operations | $ | (62,948,646 | ) | $ | (55,781,277 | ) | $ | (20,478,210 | ) | |||
| Computed “expected” tax benefit | $ | (21,402,540 | ) | $ | (18,965,634 | ) | $ | (6,962,592 | ) | |||
| Increase (decrease) in income taxes resulting from: | ||||||||||||
| Expected benefit from state and local taxes | (672,306 | ) | (2,533,156 | ) | (917,810 | ) | ||||||
| Research and development credits | (1,603,364 | ) | (1,092,767 | ) | (250,000 | ) | ||||||
| Other | 566,310 | 35,459 | 43,026 | |||||||||
| Permanent difference related to contingent note payable | — | (244,814 | ) | (1,924,558 | ) | |||||||
| Impact of change in state tax rates on deferred taxes | 5,836,819 | — | — | |||||||||
| Change in the balance of the valuation allowance for deferred tax assets | 17,275,081 | 22,800,912 | 10,011,934 | |||||||||
| $ | — | $ | — | $ | — | |||||||
We file income tax returns in the U.S. Federal and various state and local jurisdictions. With certain exceptions, the Company is no longer subject to U.S. Federal and state income tax examinations by tax authorities for years prior to 2011. However, NOLs and tax credits generated from those prior years could still be adjusted upon audit.
The Company recognizes interest and penalties to uncertain tax position in income tax expense in the statement of operations. There was no accrual for interest and penalties related to uncertain tax positions for 2015 and 2014. We do not believe that there will be a material change in our unrecognized tax positions over the next twelve months. All of the unrecognized tax benefits, if recognized, would be offset by the valuation allowance.
NOTE 8 – LICENSE AGREEMENTS
Anti-PD-L1 and anti-GITR
On March 3, 2015, we entered into a Global Collaboration Agreement (the “Collaboration”) with Checkpoint Therapeutics, Inc. (“Checkpoint”), a subsidiary of Fortress Biotech, Inc. (“Fortress”), a related party, for the development and commercialization of Checkpoint’s anti-PD-L1 and anti-GITR antibody research programs in the field of hematological malignancies. Checkpoint retains the rights to develop and commercialize these antibodies in solid tumors.
Under the terms of the Collaboration, we made an up-front payment of $0.5 million, will make development and sales-based milestone payments up to an aggregate of $164 million, and will pay a tiered single digit royalty on net sales. The royalty term will terminate on a country by country basis upon the later of (i) ten years after the first commercial sale of any applicable licensed product in such country, or (ii) the expiration of the last-to-expire patent held by the Dana Farber Cancer Institute containing a valid claim to any licensed product in such country.
Michael Weiss, our Executive Chairman, Interim CEO and President is also the Executive Vice Chairman of Fortress and the Executive Chairman of Checkpoint (See Note 9).
TGR-1202
On September 22, 2014, we exercised our option to license the global rights to TGR-1202, thereby entering into an exclusive licensing agreement (the “TGR-1202 License”) with Rhizen Pharmaceuticals, SA (“Rhizen”) for the development and commercialization of TGR-1202. Prior to this, we had been jointly developing TGR-1202 in a 50:50 joint venture with Rhizen.
Under the terms of the TGR-1202 License, Rhizen received a $4.0 million cash payment and 371,530 shares of our common stock as an upfront license fee. With respect to TGR-1202, Rhizen will be eligible to receive regulatory filing, approval and sales-based milestone payments in the aggregate of approximately $175 million, a small portion of which will be payable on the first New Drug Application (NDA) filing and the remainder on approval in multiple jurisdictions for up to two oncology indications and one non-oncology indication and attaining certain sales milestones. In addition, if TGR-1202 is co-formulated with another drug to create a new product (a "New Product"), Rhizen will be eligible to receive similar regulatory approval and sales-based milestone payments for such New Product. Additionally, Rhizen will be entitled to tiered royalties on our future net sales of TGR-1202 and any New Product. In lieu of sales milestones and royalties on net sales, Rhizen shall also be eligible to participate in sublicensing revenue, if any, based on a percentage that decreases as a function of the number of patients treated in clinical trials following the exercise of the license option. Rhizen will retain global manufacturing rights to TGR-1202, provided that they are price competitive with alternative manufacturers.
| F-18 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
In connection with the TGR-1202 License, we recognized $4.1 million of noncash research and development expense during the year ended December 31, 2014 related to the issuance of the above mentioned common stock. In addition, we recognized $4.0 million of other research and development expense during the year ended December 31, 2014 related to the cash milestone payment.
IRAK4
On June 23, 2014, we entered into an exclusive licensing agreement with Ligand Pharmaceuticals Incorporated ("Ligand") for the development and commercialization of Ligand's interleukin-1 receptor associated kinase-4 ("IRAK4") inhibitor technology, which currently is in preclinical development for potential use against certain cancers and autoimmune diseases. IRAK4 is a serine/threonine protein kinase that is a key downstream signaling component of the interleukin-1 receptor and multiple toll-like receptors.
Under the terms of the license agreement, Ligand received 125,000 shares of our common stock as an upfront license fee. Ligand will also be eligible to receive maximum potential milestone payments of approximately $207 million upon the achievement of specific clinical, regulatory and commercial milestone events. Additionally, Ligand will be entitled to royalties on our future net sales of licensed products containing IRAK4 inhibitors. The basic royalty rate for licensed products covered by Ligand's issued patents will be 6% for annual sales of up to $1 billion and 9.5% for annual sales in excess of that threshold.
In connection with the license agreement, we recognized $1,211,250 of noncash research and development expense during the year ended December 31, 2014 in connection with the issuance of the above mentioned common stock.
Additionally, Opus Point Partners, LLC, who identified the opportunity and advised us on the transaction, will also be entitled to receive a 1% royalty for annual sales of up to $1 billion. Michael S. Weiss, our Executive Chairman and Interim Chief Executive Officer, is a Managing Member of Opus Point Partners, LLC.
TG-1101
In November 2012, we entered into an exclusive (within the territory) sublicense agreement with Ildong relating to the development and commercialization of TG-1101 in South Korea and Southeast Asia. Under the terms of the sublicense agreement, Ildong has been granted a royalty bearing, exclusive right, including the right to grant sublicenses, to develop and commercialize TG-1101 in South Korea, Taiwan, Singapore, Indonesia, Malaysia, Thailand, Philippines, Vietnam, and Myanmar.
An upfront payment of $2.0 million, which was received in December 2012, net of $0.3 million of income tax withholdings, is being recognized as license revenue on a straight-line basis over the life of the agreement, which is through the expiration of the last licensed patent right or 15 years after the first commercial sale of a product in such country, unless the agreement is earlier terminated, and represents the estimated period over which we will have certain ongoing responsibilities under the sublicense agreement. We recorded license revenue of approximately $152,000 for each of the years ended December 31, 2015, 2014 and 2013, and, at December 31, 2015, 2014 and 2013, have deferred revenue of approximately $1,524,000, $1,676,000 and $1,829,000, respectively, associated with this $2,000,000 payment (approximately $152,000 of which has been classified in current liabilities at December 31, 2015).
We may receive up to an additional $5.0 million in payments upon the achievement of pre-specified milestones. In addition, upon commercialization, Ildong will make royalty payments to us on net sales of TG-1101 in the sublicense territory.
| F-19 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
NOTE 9 – RELATED PARTY TRANSACTIONS
LFB Biotechnologies
On January 30, 2012, we entered into an exclusive license agreement with LFB Biotechnologies, GTC Biotherapeutics and LFB/GTC LLC, all wholly-owned subsidiaries of LFB Group, relating to the development of ublituximab (the “LFB License Agreement”). In connection with the LFB License Agreement, LFB Group was issued 5,000,000 shares of common stock, and a warrant to purchase 2,500,000 shares of common stock at a purchase price of $0.001 per share. In addition, on November 9, 2012, we nominated Dr. Yann Echelard to our Board of Directors as LFB Group’s nominee. LFB Group maintains the right to nominate a board member until such time as LFB Group owns less than 10% of the outstanding common stock.
In connection with the LFB License Agreement, LFB Group maintained the right to purchase at least $750,000 in additional shares of common stock at a purchase price per share as defined in a November 2012 securities exchange agreement. Accordingly, in February 2015, LFB Group purchased 114,855 shares of our common stock at a price of $6.53 per share for net proceeds of $750,000. In May 2015, LFB Group exercised its warrant to purchase 2,500,000 shares of common stock at a purchase price of $0.001 per share.
Under the terms of the LFB License Agreement, we utilize LFB Group for certain development and manufacturing services. We incurred approximately $9,300,000, $5,200,000 and $6,300,000 in expenses for such services during the years ended December 31, 2015, 2014 and 2013, respectively, which have been included in other research and development expenses in the accompanying consolidated statements of operations. As of December 31, 2015, and 2014, we had approximately $2.1 million and $0.1 million, respectively, recorded in accounts payable related to the LFB License Agreement. In conjunction with the development and manufacturing services discussed above, certain agreements between us and LFB Group require payments in advance of services performed or goods delivered. Accordingly, as of December 31, 2015 and 2014, we recorded $3.0 million and $1.9 million, respectively, in prepaid research and development for such advance payments.
Other Parties
In March 2014, we entered into a shared services agreement (the “Opus Shared Services Agreement”) with Opus Point Partners Management, LLC (“Opus”) in which the parties agreed to share a rented facility and costs for certain other services. Michael S. Weiss, our Executive Chairman and Interim Chief Executive Officer, is a Managing Member of Opus. During the years ended December 31, 2015 and 2014, we incurred expenses of approximately $0.3 million and $0.1 million, respectively, principally for rent, related to this Opus Shared Services Agreement. As of December 31, 2015 and 2014, we had approximately $0.1 million and $0.02 million, respectively, recorded in accounts payable related to this Opus Shared Services Agreement.
As discussed in Note 8 above, in connection with the licensing agreement with Ligand, Opus Point Partners, LLC, who identified the opportunity and advised us on the transaction, will be entitled to receive a 1% royalty for annual sales of up to $1 billion.
As discussed in Note 8 above, with regard to the Collaboration with Checkpoint, our Executive Chairman and Interim Chief Executive Officer is also the Executive Vice Chairman of Fortress and the Executive Chairman of Checkpoint. In addition, Mr. Weiss holds equity interests in TG, Fortress and Checkpoint.
On October 3, 2014, we entered into a Desk Space Agreement (the “Desk Agreement”) with Fortress, to occupy approximately 40% of the New York City office space recently leased by Fortress. This Desk Agreement requires us to pay our respective share of the average annual rent and other costs of the 15 year lease. We approximate an average annual rental obligation of $1.1 million under the Desk Agreement. Fortress does not expect to take possession of the space until early 2016.
In connection with the Desk Agreement, we paid approximately $0.1 million in advance rent, which is recorded in other current assets in the accompanying consolidated balance sheets as of December 31, 2015 and December 31, 2014. During the year ended December 31, 2015, we incurred expenses of approximately $0.2 million, principally for our share of project management services, related to this agreement, of which $0.1 million was recorded in accounts payable. Also in connection with this lease, in October 2014 we pledged $0.6 million to secure a line of credit as a security deposit for the Desk Agreement, which has been recorded as Restricted Cash in the accompanying consolidated balance sheets.
| F-20 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
In July 2015, we entered into a Shared Services Agreement (the “Shared Services Agreement”) with Fortress to share the cost of certain services such as facilities use, shared personnel costs and other shared overhead and administrative costs. This Shared Services Agreement requires us to pay our respective share of services utilized. In connection with the Shared Services Agreement, we incurred expenses of approximately $0.1 million for shared services as of December 31, 2015, which included only costs for shared personnel costs.
On September 1, 2015, we paid $25,000 to Checkpoint as an option fee for the exclusive right to enter into a collaboration for certain additional compounds licensed by Checkpoint. The option has not been exercised as of December 31, 2015. As of December 31, 2015, we had approximately $0.3 million recorded in accounts payable related to a sponsored research agreement.
For the year ended December 31, 2015, we incurred expenses of approximately $23,000 to AOI Communications, L.P. for manuscript services related to TG-1101. Our Executive Chairman and Interim Chief Executive Officer is the owner of AOI Communications, L.P.
NOTE 10 – COMMITMENTS AND CONTINGENCIES
As of December 31, 2015, we have known contractual obligations, commitments and contingencies of $16.3 million related to our operating lease obligations.
| (in thousands) | Payment due by period | |||||||||||||||||||
| Total | Less
than 1 year | 1-3 years | 3-5 years | More than 5 years | ||||||||||||||||
| Contractual obligations | ||||||||||||||||||||
| Operating leases | $ | 16,335 | $ | 128 | $ | 1,947 | $ | 1,992 | $ | 12,268 | ||||||||||
| Total | $ | 16,335 | $ | 128 | $ | 1,947 | $ | 1,992 | $ | 12,268 | ||||||||||
Leases
On October 3, 2014, we entered into a Desk Agreement with Fortress, to occupy approximately 40% of the New York City office space recently leased by Fortress. This Desk Agreement requires us to pay our respective share of the average annual rent and other costs of the 15 year lease. We approximate an average annual rental obligation of $1.1 million under the Desk Agreement. Fortress does not expect to take possession of the space until early 2016.
Total rental expense was approximately $0.3 million, $0.1 million and $0.01 million for the years ended December 31, 2015, 2014 and 2013, respectively.
Future minimum lease commitments as of December 31, 2015, in the aggregate total approximately $16.3 million through December 31, 2031. The preceding table shows future minimum lease commitments, which include our office leases in New York, North Carolina and Tennessee, by period as of December 31, 2015.
| F-21 |
| TG Therapeutics, Inc. and Subsidiaries |
| Notes to Consolidated Financial Statements |
NOTE 11 – QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
| 3 Months Ended | ||||||||||||||||
| March 31, 2015 | June 30, 2015 | September 30, 2015 | December 31, 2015 | |||||||||||||
| License revenue | $ | 38,095 | $ | 38,095 | $ | 38,096 | $ | 38,095 | ||||||||
| Total costs and expenses | 14,640,946 | 17,149,675 | 13,863,680 | 17,678,096 | ||||||||||||
| Net loss | $ | (14,577,735 | ) | $ | (17,103,183 | ) | $ | (13,655,916 | ) | $ | (17,611,812 | ) | ||||
| Basic and diluted net loss per common share | $ | (0.35 | ) | $ | (0.38 | ) | $ | (0.28 | ) | $ | (0.37 | ) | ||||
| 3 Months Ended | ||||||||||||||||
| March 31, 2014 | June 30, 2014 | September 30, 2014 | December 31, 2014 | |||||||||||||
| License revenue | $ | 38,095 | $ | 38,095 | $ | 38,096 | $ | 38,095 | ||||||||
| Total costs and expenses | 7,643,220 | 11,993,592 | 17,477,442 | 18,759,219 | ||||||||||||
| Net loss | $ | (7,547,249 | ) | $ | (11,986,430 | ) | $ | (17,451,169 | ) | $ | (18,796,429 | ) | ||||
| Basic and diluted net loss per common share | $ | (0.25 | ) | $ | (0.36 | ) | $ | (0.51 | ) | $ | (0.48 | ) | ||||
| F-22 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 15, 2016
| TG THERAPEUTICS, INC. | ||
| By: | /s/ Michael S. Weiss | |
| Michael
S. Weiss Executive Chairman, Interim Chief Executive Officer and President | ||
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Michael S. Weiss and Sean A. Power, his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and his name, place and stead, in any and all capacities, to sign any or all amendments to this annual report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the SEC, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent or any of his substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Form 10-K has been signed by the following persons on behalf of the Registrant on March 15, 2016, and in the capacities indicated:
| Signatures | Title | |
/s/ Michael S. Weiss Michael S. Weiss |
Executive Chairman, Interim Chief Executive Officer and President (principal executive officer) | |
/s/ Sean A. Power Sean A. Power |
Chief Financial Officer (principal financial and accounting officer) | |
/s/ Laurence N. Charney Laurence N. Charney |
Director | |
/s/ Yann Echelard Yann Echelard |
Director | |
/s/ Kenneth Hoberman Kenneth Hoberman |
Director | |
/s/ Daniel Hume Daniel Hume |
Director | |
/s/ William J. Kennedy William J. Kennedy |
Director | |
/s/ Mark Schoenebaum, M.D. Mark Schoenebaum, M.D. |
Director |
| F-23 |
EXHIBIT INDEX
| Exhibit | |
| Number | Exhibit Description |
| 23.1 | Consent of Independent Registered Public Accounting Firm |
| 31.1 | Certification of Principal Executive Officer |
| 31.2 | Certification of Principal Financial Officer |
| 32.1 | Certification of Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 32.2 | Certification of Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 101 | The following financial information from the Company’s Quarterly Report on Form 10-K for the period ended December 31, 2015, formatted in Extensible Business Reporting Language (XBRL): (i) the Condensed Consolidated Balance Sheets, (ii) the Condensed Consolidated Statements of Operations, (iii) the Condensed Consolidated Statement of Equity, (iv) the Condensed Consolidated Statements of Cash Flows, and (v) Notes to the Condensed Consolidated Financial Statements (filed herewith). |