Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - U.S. Stem Cell, Inc. | ex32-1.htm |
| EX-31.1 - EX-31.1 - U.S. Stem Cell, Inc. | ex31-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2015
|
|
OR
|
|
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from _____________ to ______________
Commission File Number 001-33718
U.S. STEM CELL, INC.
(Exact name of registrant as specified in its charter)
|
Florida
|
65-0945967
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
13794 NW 4th Street, Suite 212, Sunrise, Florida 33325
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code (954) 835-1500
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.001 Par Value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). x Yes o No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer o
|
Accelerated filer o
|
Non-accelerated filer o
|
Smaller reporting company x
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
As of June 30, 2015, the last day of registrant’s second fiscal quarter, the aggregate market value of the registrant’s common stock, $0.001 par value, held by non-affiliates, computed by reference to the closing sale price of the common stock reported on the OTCQB as of June 30, 2015, was approximately $4,092,862.57. For purposes of the above statement only, all directors, executive officers and 10% shareholders are assumed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares outstanding of the registrant’s Common Stock, $0.001 par value, as of March 8th, 2016 was 2,470,400.
Documents Incorporated By Reference None
U.S. STEM CELL, INC.
INDEX TO ANNUAL REPORT ON FORM 10-K
Fiscal Year Ended December 31, 2015
|
Page
|
||
|
PART I
|
||
|
Item 1.
|
2
|
|
|
Item 1A.
|
23
|
|
|
Item 1B.
|
33
|
|
|
Item 2.
|
33
|
|
|
Item 3.
|
34
|
|
|
Item 4.
|
34
|
|
|
PART II
|
||
|
Item 5.
|
35
|
|
|
Item 6.
|
37
|
|
|
Item 7.
|
37
|
|
|
Item 7A.
|
49
|
|
|
Item 8.
|
49
|
|
|
Item 9.
|
49
|
|
|
Item 9A.
|
50
|
|
|
Item 9B.
|
50
|
|
|
PART III
|
||
|
Item 10.
|
51
|
|
|
Item 11.
|
54
|
|
|
Item 12.
|
59
|
|
|
Item 13.
|
62
|
|
|
Item 14.
|
65
|
|
|
PART IV
|
||
|
Item 15.
|
67
|
|
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K includes “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995, as well as historical information. Such forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements, or industry results, to be materially different from anticipated results, performance or achievements expressed or implied by such forward-looking statements. When used in this Annual Report on Form 10-K, statements that are not statements of current or historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, the words “plan,” “intend,” “may,” “will,” “expect,” “believe,” “could,” “anticipate,” “estimate,” or “continue” or similar expressions or other variations or comparable terminology are intended to identify such forward-looking statements, although some forward-looking statements are expressed differently. We remind readers that forward-looking statements are merely predictions and therefore inherently subject to uncertainties and other factors and involve known and unknown risks that could cause the actual results, performance, levels of activity or our achievements or industry results, to be materially different from any future results, performance levels of activity or our achievements or industry results expressed or implied by such forward-looking statements. Such forward looking statements appear in Item 1- “Business” and Item 7-“Management’s Discussion and Analysis of Financial Condition and Results of Operations” as well as elsewhere in this Annual Report. Factors that could cause our actual results to differ materially from anticipated results expressed or implied by forward-looking statements include, among others:
• our ability to manage our business despite operating losses and cash outflows;
• our ability to obtain sufficient capital or strategic business arrangements to fund our operations and expansion plans, including meeting our financial obligations under various licensing and other strategic arrangements and the funding of our clinical trials for product candidates in our development programs;
• our ability to build and maintain the management and human resources infrastructure necessary to support the growth of our business;
• our ability to operate our subsidiary businesses successfully and grow such subsidiary businesses as anticipated;
• whether a large global market is established for our cellular-based products and services and our ability to capture a meaningful share of this market;
• scientific and medical developments beyond our control;
• our ability to obtain and maintain, as applicable, appropriate governmental licenses, accreditations or certifications or comply with healthcare laws and regulations or any other adverse effect or limitations caused by government regulation of our business;
• whether any of our current or future patent applications result in issued patents, the scope of those patents and our ability to obtain and maintain other rights to technology required or desirable for the conduct of our business; and
• our ability to complete our planned clinical trials (or initiate other trials) in accordance with our estimated timelines due to delays associated with obtaining sufficient capital to complete such trials, enrolling patients due to the novelty of the treatment, the size of the patient population and the need of patients to meet the inclusion criteria of the trial or otherwise.
The factors discussed herein, including those selected risks described in Item 1A. “Risk Factors” and elsewhere in this Annual Report on Form 10-K and in the Company’s other periodic filings with the Securities and Exchange Commission (the “SEC”) which are available for review at www.sec.gov under “Search for Company Filings” could cause actual results and developments to be materially different from those expressed or implied by such statements. All forward-looking statements attributable to us are expressly qualified in their entirety by these and other factors. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof.
Except as required by law, the Company undertakes no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise.
Unless otherwise indicated or the context otherwise requires, all references in this Form 10-K to “we,” “us,” “our,” “our company,” “U.S. Stem Cell” or the “Company” refer to U.S. Stem Cell, Inc. and its subsidiaries.
PART I
Item 1. BUSINESS
OVERVIEW
We are a biotechnology company focused on the discovery, development and, subject to regulatory approval, commercialization of autologous cell therapies for the treatment of chronic and acute heart damage. Our lead product candidate is MyoCell, an innovative clinical therapy designed to populate regions of scar tissue within a patient’s heart with autologous muscle cells, or cells from a patient’s body, for the purpose of improving cardiac function in chronic heart failure patients.
Biotechnology Product Candidates
Our business includes the development of proprietary cell therapy products as well as revenue generating physician and patient based regenerative medicine / cell therapy training services, cell collection and cell storage services, the sale of cell collection and treatment kits for humans and animals, and the operation of a cell therapy clinic. Management maintains that revenues and their associated cash in-flows generated from our businesses will, over time, provide funds to support our clinical development activities as they do today for our general business operations. We believe the combination of our own therapeutics pipeline combined with our revenue generating capabilities provides the Company with a unique opportunity for growth and a pathway to profitability.
US Stem Cell Training, (“SCT”), an operating division of U.S. Stem Cell, Inc., is a content developer of regenerative medicine / cell therapy informational and training materials for physicians and patients. SCT also provides in-person and online training courses which are delivered through in-person presentations at SCT’s state of the art facilities and globally at university, hospital and physician’s office locations as well as through online webinars. Additionally, SCT provides hands-on clinical application training for physicians and health care professionals interested in providing regenerative medicine / cell therapy procedures.
Vetbiologics, (“VBI”), an operating division of our company, is a veterinary regenerative medicine company committed to providing veterinarians with the ability to deliver the highest quality regenerative medicine therapies to dogs, cats and horses. VBI provides veterinarians with extensive regenerative medicine capabilities including the ability to isolate regenerative stem cells from a patient’s own adipose (fat) tissue directly on-site within their own clinic or stall-side.
US Stem Cell Clinic, LLC, (“SCC”), a partially owned 33.3% investment of our company, is a physician run regenerative medicine/cell therapy clinic providing cellular treatments for patients afflicted with neurological, autoimmune, orthopedic and degenerative diseases. SCC is operating in compliance with the FDA 1271s which allow for same day medical procedures to be considered the practice of medicine. We isolate stem cells from bone marrow and adipose tissue and also utilize platelet rich plasma.
U.S. Stem Cell’s comprehensive map of products and services:
U.S. Stem Cell, Inc. was incorporated in the State of Florida in August 1999 as Bioheart, Inc. In 2015, we changed our name to U.S. Stem Cell, Inc. Our principal executive offices are located at 13794 NW 4th Street, Suite 212, Sunrise, Florida 33325 and our telephone number is (954) 835-1500. Information about us is available on our corporate websites at www.U.S. Stem Cellus-stemcell.com, www.usstemcelltraining.com, www.vetbiologics.com and www.usstemcellclinic.com. We include our website addresses in the Annual Report on Form 10-K only as an interactive textual reference and do not intend it to be an active link to our website. The information on our websites is not incorporated by reference in the Annual Report on Form 10-K.
The Annual Report includes the following trademarks, service marks and trade names owned by the Company: U.S. Stem Cell, Inc. ™, US Stem Cell Training, Vetbiologics, US Stem Cell Clinic, LLC. ™, MyoCell ™ and AdipoCell ™. These trademarks, service marks and trade names are the property of U.S. Stem Cell, Inc. and its affiliates.
REGENERATIVE MEDICINE / CELL THERAPY INDUSTRY
Regenerative medicine is defined as the process of replacing or regenerating human cells, tissues or organs to restore normal function. Among the categories of therapeutic technology platforms within this field are cell therapy; tissue engineering; tools, devices and diagnostics; and aesthetic medicine. U.S. Stem Cell’s business model is focused on two of these areas. First, cell therapy, in which we introduce cells (adult, donor or patient, stem cell or differentiated) into the body to prevent and treat disease; and second, we are a provider of services and products to physicians and veterinaries who provide or seek to provide cellular therapies and direct patient care for individuals and animals who may benefit from cellular therapy.
All living complex organisms start as a single cell that replicates, differentiates (matures) and perpetuates in an adult organism through its lifetime. Cellular therapy is the process that uses cells to prevent, treat or cure disease, or regenerate damaged or aged tissue. To date, the most common type of cell therapy has been the replacement of mature, functioning cells such as through blood and platelet transfusions. Since the 1970s, first bone marrow and then blood and umbilical cord-derived stem cells have been used to restore bone marrow, as well as blood and immune system cells damaged by the chemotherapy and radiation that are used to treat many cancers. These types of cell therapies are standard practice world-wide and are typically reimbursed by insurance.
Within the field of cell therapy, research and development using stem cells to treat a host of diseases and conditions has greatly expanded. Stem cells (in either embryonic or adult forms) are primitive and undifferentiated cells that have the unique ability to transform into or otherwise affect many different cells, such as white blood cells, nerve cells or heart muscle cells. U.S. Stem Cell’s cell therapy development efforts are focused on the use of adult stem cells; those cells which are found in the muscle, fat tissue and peripheral blood.
There are two general classes of cell therapies: Patient Specific Cell Therapies (“PSCTs”) and Off-the-Shelf Cell Therapies (“OSCTs”). In PSCTs, cells collected from a person (“donor”) are transplanted, with or without modification, to a patient (“recipient”). In cases where the donor and the recipient are the same individual, these procedures are referred to as “autologous”. In cases in which the donor and the recipient are not the same individual, these procedures are referred to as “allogeneic.” Autologous cells offer a low likelihood of rejection by the patient and we believe the long-term benefits of these PSCTs can best be achieved with an autologous product. In the case of OSCT, donor cells are expanded many fold in tissue culture, and large banks of cells are frozen in individual aliquots that may result in treatments for as many as 10,000 people from a single donor tissue. By definition, OSCTs are always allogeneic in nature.
Various adult stem cell therapies are in clinical development for an array of human diseases, including autoimmune, oncologic, neurologic and orthopedic, among other indications. While no assurances can be given regarding future medical developments, we believe that the field of cell therapy holds the promise to better the human experience and minimize or ameliorate the pain and suffering from many common diseases and/or from the process of aging.
According to Robin R. Young’s Stem Cell Summit Executive Summary-Analysis and Market Forecasts 2014-2024, the United States stem cell therapy market is estimated to grow from an estimated $237 million in 2013 to more than $5.7 billion in 2020.
With approved cell therapy products currently being sold in the United States and abroad, and an increasing number of Phase 2 and Phase 3 trials with cell therapies underway, we believe the “promise” of cell therapy is becoming clearer. We contend that cell therapies, if approved, should cut health care costs as they aim to facilitate functional restoration of damaged tissues and not just abate or moderate symptoms. Safe and efficacious cell therapies for chronic diseases could potentially capture an increasing portion of future healthcare spending in the United States, driven both by favorable demographics and meaningful pharma-coeconomic benefit.
CELLULAR THERAPY PRODUCT DEVELOPMENT PIPELINE
Specific to cellular therapy, we are focused on the discovery, development and commercialization of autologous cellular therapies for the treatment of chronic and acute heart damage as well as vascular and autoimmune diseases.
In our pipeline, we have multiple product candidates for the treatment of heart damage, including MyoCell and Myocell SDF-1. MyoCell and MyoCell SDF-1 are autologous muscle-derived cellular therapies designed to populate regions of scar tissue within a patient’s heart with new living cells for the purpose of improving cardiac function in chronic heart failure patients.
MyoCell SDF-1 is intended to be an improvement to MyoCell. MyoCell SDF-1 is similar to MyoCell but the myoblast cells to be injected for use in MyoCell SDF-1 are modified prior to injection by an adenovirus vector or non-viral vector so that they will release extra quantities of the SDF-1 protein, which expresses angiogenic factors.
AdipoCell is a patient-derived cell therapy proposed for the treatment of lower limb ischemia and potentially, among other autoimmune diseases, rheumatoid arthritis. We hope to demonstrate that these product candidates are safe and effective complements to existing therapies for chronic and acute heart damage.
U.S. Stem Cell’s Clinical Development Pipeline Chart:
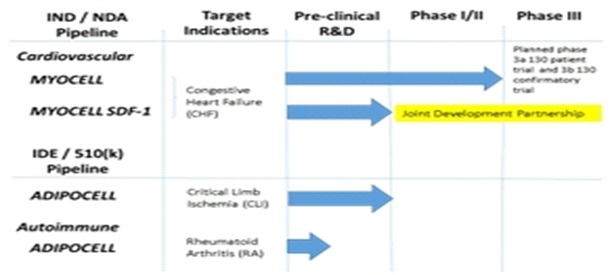
STATUS OF CELLULAR THERAPY PRODUCT DEVELOPMENT CLINICAL TRIALS.
MyoCell/MyoCell SDF-1
MyoCell is a regenerative, cellular therapy intended to improve cardiac function for those with congestive heart failure and is designed to be utilized months or even years after a patient has suffered severe heart damage due to a heart attack or other cause. We believe that MyoCell has the potential to become a leading treatment for severe, chronic damage to the heart due to its perceived ability to satisfy, at least in part, what we believe to be an unmet demand for more effective and/or more affordable therapies for chronic heart damage. MyoCell uses myoblasts, cells that are precursors to muscle cells, from the patient’s own body. The myoblasts are removed from a patient’s thigh muscle, isolated, grown through our proprietary cell culturing process, and injected directly in the scar tissue of a patient’s heart. A qualified physician performs this minimally invasive procedure using an endoventricular catheter. We entered into an agreement with Biosense Webster (a Johnson & Johnson company) to use its NOGA® Cardiac Navigation System along with its MyoStar™ injection catheter for the delivery of MyoCell in the MARVEL Trial.
When injected into scar tissue within the heart wall, myoblasts have been shown to be capable of engrafting in the damaged tissue and differentiating into mature skeletal muscle cells. In a number of clinical and animal studies, the engrafted skeletal muscle cells have been shown to express various proteins that are important components of contractile function. By using myoblasts obtained from a patient’s own body, we believe MyoCell is able to avoid certain challenges currently faced by other types of cell-based clinical therapies including tissue rejection and instances of the cells differentiating into cells other than muscle. Although a number of therapies have proven to improve the cardiac function of a damaged heart, no currently available treatment, to our knowledge, has demonstrated an ability to generate new muscle tissue within the scarred regions of a heart.
Our completed clinical trials of MyoCell to date have been primarily targeted to patients with severe, chronic damage to the heart, who are in Class II or Class III heart failure according to the New York Heart Association, or NYHA, heart failure classification system. The NYHA system classifies patients in one of four categories based on how limited they are during physical activity. NYHA Class II heart failure patients have a mild limitation of activity and are generally comfortable at rest or with mild exertion while NYHA Class III heart failure patients suffer from a marked limitation of activity and are generally comfortable only at rest.
We believe the market for treating patients in NYHA Class II or NYHA Class III heart failure is significant. According to the American Heart Association (“AHA”) Statistics and the European Society of Cardiology Task Force for the Treatment of Chronic Heart Failure, in the United States and Europe there are approximately 5.2 million and 9.6 million, respectively, patients with heart failure. The AHA Statistics further indicate that, after heart failure is diagnosed, the one-year mortality rate is high, with one in five dying and that 80% of men and 70% of women under age 65 who have heart failure will die within eight years.
We believe that approximately 60% of heart failure patients are in either NYHA Class II or NYHA Class III heart failure based upon a 1999 study entitled “Congestive Heart Failure Due to Diastolic or Systolic Dysfunction – Frequency and Patient Characteristics in an Ambulatory Setting” by Diller, PM, et. al.
MyoCell SDF-1 is intended to be an improvement to MyoCell. MyoCell SDF-1 is similar to MyoCell except that the myoblast cells to be injected for use in MyoCell SDF-1 will be modified prior to injection by an adenovirus vector or non-viral vector so that they will release extra quantities of the SDF-1 protein, which expresses angiogenic factors. AdipoCell is a patient-derived cell therapy proposed for the treatment of acute myocardial infarction, chronic heart ischemia, and lower limb ischemia. We hope to demonstrate that these product candidates are safe and effective complements to existing therapies for chronic and acute heart damage.
We have completed various clinical trials for MyoCell including the SEISMIC Trial, a 40-patient, randomized, multicenter, controlled, Phase II-a study conducted in Europe and the MYOHEART Trial, a 20-patient, multicenter, Phase I dose-escalation trial conducted in the United States. We were approved by the U.S. Food and Drug Administration, or the “FDA”, to proceed with a 330-patient, multicenter Phase II/III trial of MyoCell in North America and Europe, or the “MARVEL Trial”. We completed the MyoCell implantation procedure on the first patient in the MARVEL Trial on October 24, 2007. Thus far, 20 patients, including 6 control patients, have been treated. Initial results for the 20 patients were released at the Heart Failure Society of American meeting in September, 2009, showing a significant (35%) improvement in the 6 minute walk for those patients who were treated, and no improvement for those who received a placebo. On the basis of these results, we have applied for and received approval from the FDA to reduce the number of additional patients in the trial to 134, for a total of 154 patients. We are planning, on the basis of these results, to request the FDA to consider the MARVEL Trial a pivotal trial (pivotal from Phase II to Phase III) and to reduce the number of patients in the trial to 150. No assurances can be provided that this request will be approved. We have also initiated the MIRROR trial, which is a Phase III, double-blind placebo controlled study for centers outside the United States. The SEISMIC, MYOHEART, MARVEL and MIRROR Trials have been designed to test the safety and efficacy of MyoCell in treating patients with severe, chronic damage to the heart. We received approval from the FDA in July of 2009 to conduct a Phase I safety study on 15 patients of a combined therapy (MyoCell with SDF-1) called the REGEN trial, during the first quarter of 2010. Advancement of the MyoCell and MyoCell SDF-1 clinical development programs is contingent, among many factors, upon the Company obtaining access to sufficient funding to execute the necessary clinical trials to achieve proof of efficacy and regulatory authorization to market such products. The Company is also presently seeking a joint development partner for its MyoCell SDF-1 product candidate.
AdipoCell
U.S. Stem Cell has successfully completed various trials using adipose stem cells. We have completed the Phase 1 Angel Trial for AdipoCell (adipose derived stem cells) in congestive heart failure patients. Five patients were enrolled and treated in the second quarter of 2013. At the twelve (12) month time point, patients demonstrated a statistically significant average improvement in ejection fraction (“EF”) by echocardiogram.
At the three (3) month time point, 100% of the patients demonstrated either improvement or stayed the same. After three (3) months, patients showed an average absolute improvement of 3 percentage points in EF. The patients continued to improve from 3 months to 6 months with a statistically significant average absolute improvement of 10 percentage points (p=0.01) and at the 12 month follow up patients showed this same level of improvement (p=0.01).
The adipose cells have also been utilized in a phase I trial in Europe for critical limb ischemia (n=20). Patients enrolled in the trial were already on the list for amputation. The cells were directly injected into the affected limbs in an effort to prevent the amputation. Seventy-five percent of the patients were able to avoid amputation and progressed to wound healing. No adverse events or complications were reported or linked to the cell therapy.
We have also initiated several Institutional Review Board studies in 2013 using adipose derived stem cells for various indications including dry macular degeneration, degenerative disc disease, erectile dysfunction and chronic obstructive pulmonary disease.
In the second quarter of 2014, we announced the treatment of a patient in Honduras with congestive heart failure using AdipoCell and MyoCell. We believe that this was the first patient treated in the world using a combination of stem cells.
We have begun two clinical trials in India. The first cardiac patient has successfully been enrolled and treated in India using AdipoCell or adipose derived stem cells. The second trial will involve the combination of AdipoCell and MyoCell or muscle derived stem cells for congestive heart failure patients. These trials are active and ongoing.
We intend to focus our regulatory approvals with the FDA on two different indications: critical limb ischemia and rheumatoid arthritis. The Adipocell treatment will follow a device pathway with the US FDA and include the submission of an IDE.
Business Strategy
U.S Stem Cell’s mission is to advance to market novel regenerative medicine and cellular therapy products that substantially benefit humankind. Our business strategy is, to the extent possible, finance our clinical development pipeline through revenue (cash in-flows) generated through the marketing and sales of unique educational and training services, animal health products and personalized cellular therapeutic treatments.
A fundamental shift in venture capital investment strategies where, management believes, financial sponsorship is now directed toward commercial or near commercial enterprises has required U.S. Stem Cell to adapt its mission combining immediate revenue generating opportunities with longer-term development programs. Accordingly, U.S. Stem Cell has developed a multifaceted portfolio of revenue generating products and services in its US Stem Cell Training, Vetbiologics, and US Stem Cell Clinic, operating divisions that will, if successful, financially support its clinical development programs. Our goal is to maximize shareholder value through the generation of short-term profits that increase cash in-flows and decrease the need venture financings – a modern biotechnology company development strategy.
Today, we contend that U.S. Stem Cell is a combination of opportunistic business enterprises. We estimate that the products and services we offer through US Stem Cell Training, Vetbiologics, and US Stem Cell Clinics has the potential, although we cannot provide assurances as to if and when it will be accomplished, to drive up to $100 million dollars in cumulative peak annual revenues. What we are establishing is a foundation of value in the products and services we are and plan to sell from US Stem Cell Training, Vetbiologics, and US Stem Cell clinics. Our strategy is to expand the revenues generated from each of these operating divisions and to reinvest the profits we generate into our U. S. Stem Cell U.S. Stem Cell clinical development pipeline.
On January 29th, 2015 we announced an update and diversification of our clinical development pipeline. Our cardiovascular and vascular product candidates have been streamlined, putting our best opportunities at the forefront of our efforts. The MYOCELL and MYOCELL SDF-1 candidates will, in our opinion, advance forward in the treatment of chronic heart failure (CHF). We are in active prospective partnering discussion for the MYOCELL SDF-1 program. Partnering, we contend, will enhance our capabilities, reduce our development cost through cost sharing and potentially accelerate our time to approval and commercialization. We will apply our ADIPOCELL technology to the treatment of critical limb ischemia. Additionally, we have expanded and diversified our clinical development pipeline to include autoimmune disease, specifically applying ADIPOCELL to the treatment of Rheumatoid Arthritis (RA). We believe that updating and diversifying our clinical development programs increases the probability of our success, brings operational and fiscal clarity to our Company, and will ultimately enhance shareholder value.
We will continue to evaluate and act upon opportunities to increase our top line revenue position and that correspondingly increase cash in-flows. These opportunities include but are not limited to the development and marketing of new products and services, mergers and acquisitions, joint ventures, licensing deals and more.
Further, if the opportunity presents itself whereby the Company can raise additional capital at a reasonable fair market value, the Company will do so. Accordingly, we plan to continue in our efforts to restructure, equitize or eliminate legacy balance sheet issues that are obstacles to market capitalization appreciation and capital fund raising.
US STEM CELL TRAINING
US Stem Cell Training offers a variety of continuing medical education (CME) courses for physicians and other health care professionals. These courses include didactic lecture series and hands-on clinical techniques in the field of regenerative medicine. We are currently hosting these courses throughout the United States and in multiple countries. These courses are also available in an online format. Pricing currently ranges from $500-$7,500 depending on the location and modules.
U.S. STEM CELL, INC.
U.S. Stem Cell markets several products to physicians for in clinic regenerative medicine use. These products include equipment (centrifuges, heating block, laminar hood, autoclave) necessary to separate and obtain cellular medicine therapies. We are also providing a variety of materials necessary to obtain fat and/or bone marrow including cannulas, trocars, syringes and other supplies. U.S. Stem Cell also supplies laboratory kits for processing adipose and bone marrow tissue to obtain a mixture of cells for use in clinic. These kits include disposables and reagents and are prepared according to FDA cGMPs. U.S. Stem Cell also provides banking services to patients interested in storing their fat or bone marrow and the cells from this tissue. U.S. Stem Cell is a registered FDA tissue bank in good standing.
VETBIOLOGICS
Vetbiologics is focused on providing regenerative medicine therapies to veterinarians for use in both small and large animals. We provide a complete regenerative medicine package which includes training, equipment and supplies necessary for in clinic cell therapy. We sell kits for isolating stem cells from bone marrow and fat. We also provide kits for isolating platelet rich plasma. The kits include all of the disposables and reagents necessary and are produced according to FDA cGMPs. Vetbiologics is also working on several off the shelf type products including an allogeneic stem cell source.
US STEM CELL CLINIC, LLC.
US Stem Cell Clinic LLC, a partly owned investment, is offering in-clinic regenerative medicine treatments to patients suffering from degenerative diseases. Adipose stem cells can be obtained from the patient easily, abundantly, and with minimal patient discomfort. Clinical applications for patients can be performed in an office setting safely, legally, and ethically using autologous adipose-derived stem cells. Current applications include orthopedic conditions (tendon/ligament injuries, osteoarthritis, etc.), degenerative conditions (COPD, diabetes), neurological (MS, Parkinson’s, spinal cord injuries, autism, etc.) and auto-immune (RA, Crohn’s, colitis, lupus). Pricing depends on application and ranges from $5,000 to $12,000.
Patents and Proprietary Rights
We own or hold licenses or sublicenses to an intellectual property portfolio consisting of numerous patents and patent applications in the United States, and in foreign countries, for use in the field of heart muscle regeneration. References in this report to “our” patents and patent applications and other similar references include the patents and patent applications that are owned by us, and references to patents and patent applications that are “licensed” to us and other similar references refer to patents, patent applications and other intellectual property that are licensed or sublicensed to us.
Patent life determination depends on the date of filing of the application or the date of patent issuance and other factors as promulgated under the patent laws. Under the U.S. Drug Price Competition and Patent Term Restoration Act of 1984, as amended, a patent which claims a product, use or method of manufacture covering drugs and certain other products, including biologic products, may be extended for up to five years to compensate the patent holder for a portion of the time required for research and FDA review of the product. Only one patent applicable to an approved drug or biologic product is eligible for a patent term extension. This law also establishes a period of time following approval of a drug or biologic product during which the FDA may not accept or approve applications for certain similar or identical drugs or biologic products from other sponsors unless those sponsors provide their own safety and efficacy data.
MyoCell is no longer protected by patents, which means that competitors will be free to sell products that incorporate the same or similar technologies that are used in MyoCell without infringing our patent rights. As a result, MyoCell, if approved for use, may be vulnerable to competition. In addition, many of the patent and patent applications that have been licensed to us that pertain to our other product candidates do not cover certain countries within Europe.
Our commercial success will depend to a significant degree on our ability to:
|
·
|
defend and enforce our patents and/or compel the owners of the patents licensed to us to defend and enforce such patents, to the extent such patents may be applicable to our products and material to their commercialization;
|
|
·
|
obtain additional patent and other proprietary protection for MyoCell and our other product candidates;
|
|
·
|
obtain and/or maintain appropriate licenses to patents, patent applications or other proprietary rights held by others with respect to our technology, both in the United States and other countries;
|
|
·
|
preserve company trade secrets and other intellectual property rights relating to our product candidates; and operate without infringing the patents and proprietary rights of third parties.
|
In addition to patented intellectual property, we also rely on our own trade secrets and proprietary know-how to protect our technology and maintain our competitive position, since patent protection may not be available or applicable to our technology. Our policy is to require each of our employees, consultants and advisors to execute a confidentiality and inventions assignment agreement before beginning their employment, consulting or advisory relationship with us. The agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of the individual’s relationship with us except in limited circumstances. These agreements generally also provide that we shall own all inventions conceived by the individual in the course of rendering services to us. Moreover, some of our academic institution licensors, collaborators and scientific advisors have rights to publish data and information to which we have rights, which may impair our ability to protect our proprietary information or obtain patent protection in the future.
We work with others in our research and development activities and one of our strategies is to enter into collaborative agreements with third parties to develop our proposed products. Disputes may arise about inventorship and corresponding rights in know-how and inventions resulting from the joint creation or use of intellectual property by us and our licensors, collaborators, consultants and others. In addition, other parties may circumvent any proprietary protection we do have. As a result, we may not be able to maintain our proprietary position.
We are not currently a party to any litigation or other adverse proceeding related to our patents, patent licenses or intellectual property rights. However, if we become involved in litigation or any other adverse intellectual property proceeding, for example, as a result of an alleged infringement, or a third party alleging an earlier date of invention, we may have to spend significant amounts of money and time and, in the event of an adverse ruling, we could be subject to liability for damages, including treble damages, invalidation of our intellectual property and injunctive relief that could prevent us from using technologies or developing products, any of which could have a significant adverse effect on our business, financial condition and results of operation. In addition, any claims relating to the infringement of third party proprietary rights, or earlier date of invention, even if not meritorious, could result in costly litigation, lengthy governmental proceedings, divert management’s attention and resources and require us to enter royalty or license agreements which are not advantageous, if available at all.
See Item 1A. “Risk Factors — Risks Related to Our Intellectual Property” for a discussion of additional risks we face with respect to our intellectual property rights.
Primary MyoCath Patent
The Primary MyoCath Patent includes device claims that we believe covers, among other things, the structure of MyoCath. The Primary MyoCath Patent expires in the United States in September 2017.
In January 2000, we entered into a license agreement with Comedicus Incorporated pursuant to which Comedicus granted us a royalty-free, fully paid-up, non-exclusive and irrevocable license to the Primary MyoCath Patent in exchange for a payment of $50,000. This agreement was amended in August 2000 to provide us an exclusive license to the Primary MyoCath Patent in exchange for a payment of $100,000 and our loan of $250,000 to Comedicus. Pursuant to this amendment we also received the right, but not the obligation, with Comedicus’ consent, which consent is not to be unreasonably withheld, to defend the Primary MyoCath Patent against third party infringers.
In June 2003, we entered into agreements with Advanced Cardiovascular Systems, Inc., or ACS, originally a subsidiary of Guidant Corporation and now d/b/a Abbott Vascular, a division of Abbott Laboratories, pursuant to which we assigned our rights under the license agreement with Comedicus, as amended, and committed to deliver 160 units of MyoCath and sold certain of our other catheter related intellectual property, or, collectively, with the Primary MyoCath Patent (the Catheter IP), for aggregate consideration of $900,000. In connection with these agreements, ACS granted to us a co-exclusive, irrevocable, fully paid-up license to the Catheter IP for the life of the patents related to the Catheter IP.
ACS has the exclusive right, at its own expense, to file, prosecute, issue, maintain, license, and defend the Catheter IP, and the primary right to enforce the Catheter IP against third party infringers. If ACS fails to enforce the Catheter IP against a third party infringer within a specified period of time, we have the right to do so at our expense. The party enforcing the Catheter IP is entitled to retain any recoveries resulting from such enforcement. The asset purchase agreement only pertains to the Catheter IP developed or acquired by us prior to June 24, 2003.
Our subsequent catheter related developments and/or acquisitions, such as MyoCath II, were not sold or licensed to ACS.
MyoCell SDF-1 Patents
To develop our MyoCell SDF-1 product candidate, we rely primarily on patents. We had an agreement to license patents from Juventas. These patents relate to methods of repairing damaged heart tissue by transplanting myoblasts that express SDF-1 and other therapeutic proteins capable of recruiting other stem cells within a patient’s own body to the cell transplant area. We believe we will also need to, among other things, license some additional intellectual property to commercialize MyoCell SDF-1 in the form we believe may prove to be the most safe and/or effective.
MyoCath II Patents
In April 2006, we entered into an agreement with Tricardia, LLC pursuant to which Tricardia granted us a sublicenseable license to certain patents and patent applications in the United States, Australia, Canada, Europe and Japan covering the modified injection needle we intend to use as part of MyoCath II, or the MyoCath II Patents, in exchange for a one-time payment of $100,000. Our license covers and is exclusive with respect to products developed under the MyoCath II Patents for the delivery of therapeutic compositions to the heart. Unless earlier terminated by mutual consent of the parties, our agreement with Tricardia will terminate upon the expiration date of the last MyoCath II Patent. Tricardia has the obligation to take all actions necessary to file, prosecute and maintain the MyoCath II Patents. We are required to reimburse Tricardia, on a pro-rata basis with other licensees of Tricardia of the MyoCath II Patents, for all reasonable out-of-pocket costs and expenses incurred by Tricardia in prosecuting and maintaining the MyoCath II Patents. To the extent Tricardia determines not to initiate suit against any infringer, we have the right, but not the obligation, to commence litigation for such alleged infringement with respect to any jurisdiction or, in the alternative, the agreement will be automatically amended to exclude such jurisdiction.
GOVERNMENT REGULATION
The health care industry is one of the most highly regulated industries in the United States and abroad. Various governmental regulatory authorities, as well as private accreditation organizations, oversee and monitor the activities of individuals and businesses engaged in the development, manufacture and delivery of health care products and services. The following is a general description of certain current laws and regulations that are relevant to our business.
HCT/P Regulations
Manufacturing facilities that produce cellular therapies are subject to extensive regulation by the FDA. In particular, FDA regulations set forth requirements pertaining to establishments that manufacture human cells, tissues, and cellular and tissue-based products (“HCT/Ps”). Title 21, Code of Federal Regulations, Part 1271 provides for a unified registration and listing system, donor-eligibility, current Good Tissue Practices (“cGTP”), and other requirements that are intended to prevent the introduction, transmission, and spread of communicable diseases by HCT/Ps. More specifically, key elements of Part 1271 include:
• Registration and listing requirements for establishments that manufacture HCT/Ps;
• Requirements for determining donor eligibility, including donor screening and testing;
• cGTP requirements, which include requirements pertaining to the manufacturer’s quality program, personnel, procedures, manufacturing facilities, environmental controls, equipment, supplies and reagents, recovery, processing and process controls, labeling, storage, record-keeping, tracking, complaint files, receipt, pre-distribution shipment, distribution, and donor eligibility determinations, donor screening, and donor testing;
• Adverse reaction reporting;
• Labeling of HCT/Ps; and
• FDA inspection, retention, recall, destruction, and cessation of manufacturing operations.
U.S. Stem Cell and its affiliated entities currently collects, processes, stores and manufactures HCT/Ps, including the manufacture of cellular therapy products. Therefore, U.S. Stem Cell must comply with cGTP and with the current Good Manufacturing Practices (“cGMP”) requirements that apply to biological products. Cell and tissue based products may also be subject to the same approval standards, including demonstration of safety and efficacy, as other biologic and drug products if they meet certain criteria such as if the cells or tissues are more than minimally manipulated or if they are intended for a non-homologous use. Management believes that requirements pertaining to premarket approval, do not currently apply to U.S. Stem Cell because those entities are not currently investigating, marketing or selling cellular therapy products. If U.S. Stem Cell changes its business operations in the future, the FDA requirements that apply to U.S. Stem Cell may also change.
Federal Regulation of Clinical Laboratories
The Clinical Laboratory Improvement Amendments (“CLIA”) extends federal oversight to clinical laboratories that examine or conduct testing on materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of disease or for the assessment of the health of human beings. CLIA requirements apply to those laboratories that handle biological matter. CLIA requires that these laboratories be certified by the government, satisfy governmental quality and personnel standards, undergo proficiency testing, be subject to biennial inspections, and remit fees. The sanctions for failure to comply with CLIA include suspension, revocation, or limitation of a laboratory’s CLIA certificate necessary to conduct business, fines, or criminal penalties. Additionally, CLIA certification may sometimes be needed when an entity, such as U.S. Stem Cell, desire to obtain accreditation, certification, or license from non-government entities for cord blood collection, storage, and processing. However, to the extent that any of the activities of U.S. Stem Cell (for example, with regard to processing or testing blood and blood products) require CLIA certification, U.S. Stem Cell intends to obtain and maintain such certification and/or licensure.
Stem Cell Therapeutic and Research Act of 2005
The Stem Cell Therapeutic and Research Act of 2005 established a national donor bank of cord blood and created a national network for matching cord blood to patients. The National Marrow Donor Program (NMDP) carries out this legislation, which entails acting as the nation’s Cord Blood Coordinating Center and actively recruiting parents for cord blood donations. The NMDP also administers the National Cord Blood Inventory (NCBI), which has a goal of collecting 150,000 cord blood units that could be used to treat patients all over the United States. Importantly, the legislation also authorized federal funding to support the legislation’s goals for collecting cord blood units.
Pharmaceutical and Biologic Products
Government authorities in the United States, at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising promotion, distribution, marketing, import and export of biological products such as MyoCell. The process of obtaining required regulatory approvals and the subsequent compliance with appropriate statutes and regulations require the expenditure of substantial time and money, and there is no guarantee that we will successfully complete the steps needed to obtain regulatory approval of MyoCell or any future product candidates. In addition, these regulations may change and our product candidates may be subject to new legislation or regulations.
In the United States, pharmaceutical and biologic products, including cellular therapies, are subject to extensive pre- and post-market regulation by the U.S. FDA. The Federal Food, Drug, and Cosmetic Act (“FD&C Act”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products are approved for marketing under provisions of the Public Health Service Act, or PHS Act. However, because most biological products also meet the definition of “drugs” under the FD&C Act, they are also subject to regulation under FD&C Act provisions. The PHS Act requires the submission of a biologics license application (“BLA”), rather than a New Drug Application (“NDA”), for market authorization. However, the application process and requirements for approval of BLAs are similar to those for NDAs, and biologics are associated with similar approval risks and costs as drugs.
Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development in the U.S. typically involves preclinical laboratory and animal tests, the submission to the FDA of a notice of claimed investigational exemption or an investigational new drug application (“IND”), which must become effective before clinical testing can commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug or biologic for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
Submission of an IND may not result in FDA authorization to initiate a clinical trial if FDA raises concerns or questions about the design of the clinical trial or the preclinical or manufacturing information supporting it, including concerns that human research subjects will be exposed to unreasonable health risks. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations; good clinical practice, or GCP, as set forth in FDA guidance, which is meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND. Sponsors of clinical trials of FDA regulated products, including drugs and biologics, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial is not being conducted in accordance with FDA requirements, or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs or BLAs for marketing approval are typically conducted in four sequential phases, but the phases may overlap.
• Phase 1: Studies are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients, such as cancer patients when the drug or biologic is too toxic to be ethically given to healthy individuals.
• Phase 2: Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials.
• Phase 3: These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites. In most cases FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
• Phase 4: In some cases, FDA may condition approval of an NDA or BLA for a product candidate on the sponsor’s agreement to conduct additional clinical trials after NDA or BLA approval. In other cases, a sponsor may voluntarily carry out additional trials post approval to gain more information about the drug or biologic. Such post approval trials are typically referred to as Phase 4 studies.
After completion of the required clinical testing, an NDA or BLA is prepared and submitted to the FDA. FDA approval of the NDA or BLA is required before marketing of the product may begin in the U.S. The NDA or BLA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA or BLA is substantial. Under federal law, the submission of most NDAs or BLAs is additionally subject to a substantial application user fee, currently exceeding $2,169,000, and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees, currently exceeding $104,000 per product and $554,000 per establishment. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs and BLAs. Most such applications for standard review drug or biologic products are reviewed within ten to twelve months; most applications for priority review drugs or biologics are reviewed in six to eight months. FDA can extend these reviews by three months. Priority review can be applied to drugs or biologics that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. For biologics, priority review is further limited only for products intended to treat a serious or life-threatening disease relative to the currently approved products.
The FDA may refer applications for novel drug or biologic products, or drug or biologic products which present difficult questions of safety or efficacy, to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
Before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMP - a quality system regulating manufacturing - is satisfactory and the NDA or BLA contains data that provide substantial evidence that the drug or biologic is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in 2 or 6 months depending on the type of information included.
Additional Controls
The PHS Act also provides authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to prevent the introduction or spread of communicable diseases in the U.S. and between states.
Biosimilars
The Patient Protection and Affordable Care Act, or Affordable Care Act, signed into law on March 23, 2010, included a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, which created an abbreviated approval pathway for biological products shown to be highly similar to, or interchangeable with, an FDA-licensed reference biological product. This is conceptually similar to the established process for generic drug approval in that it attempts to minimize duplicative testing. Biosimilarity, which requires that there be no differences in conditions of use, route of administration, dosage form, and strength and there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, must be shown through analytical studies, animal studies, and at least one clinical study, absent a waiver by the Secretary. Interchangeability requires that a product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. No biosimilar or interchangeable products have been approved under the BPCIA to date. Complexities associated with the larger and often more complex structures of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against other biologics submitting under the abbreviated approval pathway for the same condition for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) eighteen months after the first interchangeable biosimilar is approved if there is no legal challenge, (iii) 18 months after the resolution in the first interchangeable applicant’s favor of a lawsuit challenging the reference biologics’ patents, or (iv) 42 months after the first interchangeable biosimilar’s application has been approved if a lawsuit is ongoing within the 42 month period.
Post-Approval Regulation
Once an NDA or BLA is approved, a product will be subject to certain post-approval requirements. For instance, FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. As a condition of NDA or BLA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the product drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. The requirement for a REMS can materially affect the potential market and profitability of the product.
Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA supplement or BLA supplement before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements and BLA supplements as it does in reviewing NDAs or BLAs. The FDA has broad enforcement authority under the FDC Act, and failure to abide by these regulations can result in enforcement action, including the issuance of a Warning Letter directing entities to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and federal civil and criminal investigations, prosecutions and penalties. State enforcement actions relating to promotional violations are also becoming more common.
Adverse experiences associated with the use of the drug must be reported to the FDA and could result in the imposition of market restrictions through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval. The FDA may also require a labeling change if it becomes aware of new safety information that it believes should be included in the labeling of a drug.
Current Good Manufacturing Practices (cGMP) Standards
The FDA Act and FDA regulations govern the quality control, manufacture, packaging, and labeling procedures of products regulated as a drug or biological product, including cellular therapies comprised of HCT/Ps. These laws and regulations include requirements for cGMP. These requirements are designed to ensure that a facility’s processes - and products resulting from those processes - meet defined safety requirements. The cGMP requirements, are federal regulations that govern the manufacture, processing, packaging and holding of drug and cell therapy products.
The objective of compliance with cGMP standards is to protect the public health and safety by ensuring that products (i) have the identity, strength, quality and purity that they purport or are represented to possess; (ii) meet their specifications; and (iii) are free of objectionable microorganisms and contamination.
A central focus of the cGMP requirements is to design and build quality into the manufacturing processes and the facilities in which products are produced and to ensure the consistency, product integrity, and reproducibility of results and product characteristics. This is done by implementing quality systems and processes including specifications and documentation.
In addition, drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs. Failure to comply with applicable FDA requirements can result in regulatory inspections and associated observations, warning letters, other requirements of remedial action, and, in the case of failures that are more serious, suspension of manufacturing operations, seizure, injunctions, product recalls, fines, and other penalties. We believe that our facilities are in material compliance with applicable existing FDA requirements.
Additionally, FDA, other regulatory agencies, or the United States Congress may be considering, and may enact laws or regulations regarding the use and marketing of stem cells, cell therapy products, or products derived from human cells or tissue. These laws and regulations can affect us directly or the business of some of U.S. Stem Cell’s clients and therefore the amount of business U.S. Stem Cell receives from these clients.
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or BLAs or supplements to NDAs or BLAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers or deferrals for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The Best Pharmaceuticals for Children Act, or BPCA, provides NDA holders a six-month extension of any exclusivity-patent or non-patent-for a drug if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition - generally a disease or condition that affects fewer than 200,000 individuals in the U.S. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Approval of Medical Devices
Medical devices are also subject to extensive regulation by the FDA. To be commercially distributed in the United States, medical devices must receive either 510(k) clearance or pre-market approval, or PMA, from the FDA prior to marketing. Devices deemed to pose relatively low risk are placed in either Class I or II, which requires the manufacturer to submit a pre-market notification requesting permission for commercial distribution, or 510(k) clearance. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, devices deemed not substantially equivalent to a previously 510(k) cleared device and certain other devices are placed in Class III which requires PMA. We anticipate that MyoCath will be classified as a Class III device.
To obtain 510(k) clearance, a manufacturer must submit a pre-market notification demonstrating that the proposed device is substantially equivalent in intended use and in safety and efficacy to a previously 510(k) cleared device, a device that has received PMA or a device that was in commercial distribution before May 28, 1976. The FDA’s 510(k) clearance pathway usually takes from four to twelve months, but it can last longer.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or efficacy, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require PMA. The FDA requires each manufacturer to make this determination, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance, the agency may retroactively require the manufacturer to seek 510(k) clearance or PMA. The FDA also can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA is obtained.
A product not eligible for 510(k) clearance must follow the PMA pathway, which requires proof of the safety and efficacy of the device to the FDA’s satisfaction. The PMA pathway is much more costly, lengthy and uncertain than the 510(k) approval pathway. A PMA application must provide extensive preclinical and clinical trial data and also information about the device and its components regarding, among other things, device design, manufacturing and labeling. As part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with quality system regulation requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. Upon acceptance by the FDA of what it considers a completed filing, the FDA commences an in-depth review of the PMA application, which typically takes from one to two years, but may last longer. The review time is often significantly extended as a result of the FDA asking for more information or clarification of information already provided.
If the FDA’s evaluation of the PMA application is favorable, and the applicant satisfies any specific conditions (e.g., changes in labeling) and provides any specific additional information (e.g., submission of final labeling), the FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the manufacturer. The PMA can include post-approval conditions that the FDA believes necessary to ensure the safety and efficacy of the device including, among other things, restrictions on labeling, promotion, sale and distribution. Failure to comply with the conditions of approval can result in an enforcement action, which could have material adverse consequences, including the loss or withdrawal of the approval.
Even after approval of a pre-market application, a new PMA or PMA supplement is required in the event of a modification to the device, its labeling or its manufacturing process.
Post-Approval Requirements
Even if regulatory clearances or approvals for our product candidates are obtained, our products and the facilities manufacturing our products will be subject to continued review and periodic inspections by the FDA. For example, as a condition of approval of a new drug application, the FDA may require us to engage in post-marketing
testing and surveillance and to monitor the safety and efficacy of our products. Holders of an approved new BLA, PMA or 510(k) clearance product are subject to several post-market requirements, including the reporting of certain adverse events involving their products to the FDA, provision of updated safety and efficacy information, and compliance with requirements concerning the advertising and promotion of their products.
In addition, manufacturing facilities are subject to periodic inspections by the FDA to confirm the facilities comply with cGMP requirements. In complying with cGMP, manufacturers must expend money, time and effort in the area of production and quality control to ensure full compliance. For example, manufacturers of biologic products must establish validated systems to ensure that products meet high standards of sterility, safety, purity, potency and identity. Manufacturers must report to the FDA any deviations from cGMP or any unexpected or unforeseeable event that may affect the safety, quality, or potency of a product. The regulations also require investigation and correction of any deviations from cGMP and impose documentation requirements.
In addition to regulations enforced by the FDA, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other federal, state and local regulations. Our research and development activities involve the controlled use of hazardous materials, chemicals, biological materials and radioactive compounds.
Other Health Care Regulations
Health Privacy Laws
The Administrative Simplification provisions of the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH Act”), require health care plans, health care providers and health care clearinghouses, collectively defined under HIPAA as “Covered Entities,” to comply with standards for the use and disclosure of health information within such organizations and with third parties. These include standards for:
• Common health care transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures;
• Unique identifiers for providers, employers, health plans and individuals; and
• Security and privacy of health information.
Although the obligations of HIPAA only apply directly to Covered Entities, any Covered Entity that uses third parties (referred to in HIPAA as “Business Associates”) to perform functions on its behalf involving the creation or use of certain patient health information is required to have a contract with the Business Associate that limits the use and disclosure of such information by the Business Associate.
HIPAA does not preempt, or override, state privacy laws that provide even more protection for individuals’ health information. These laws’ requirements could further complicate U.S. Stem Cell’s ability to obtain necessary research data from its collaborators. In addition, certain state privacy and genetic testing laws may directly regulate our research activities, affecting the manner in which we use and disclose individuals’ health information, potentially increasing the cost of doing business, and exposing us to liability claims. In addition, patients and research collaborators may have contractual rights that further limit our ability to use and disclose individually identifiable health information. Claims that we violated individuals’ privacy rights or breached its contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm the business.
While we believe that the current business operations of U.S. Stem Cell would not cause either of them to be considered a Covered Entity, there is a risk that due to conflicting interpretations of the regulations, U.S. Stem Cell may be deemed to be a Covered Entity. If U.S. Stem Cell is a Covered Entity, there is a risk of liability that U.S. Stem Cell may not be complying fully with all HIPAA requirements. U.S. Stem Cell has signed Business Associate Agreements where requested by U.S. Stem Cell’s customers who are Covered Entities, which would require compliance with certain privacy and security requirements relating to individually identifiable health information created or used in connection with such relationships. U.S. Stem Cell is in substantial compliance with such Business Associate Agreements. However, given the law’s complexity and the possibility that the regulations may change and may be subject to changing and even conflicting interpretation, U.S. Stem Cell’s ability to comply fully with all of the HIPAA requirements and requirements of its Business Associate Agreements is uncertain. Further, as a result of amendments the HITECH Act, U.S. Stem Cell’s compliance burden has increased and they will be subject to audit and enforcement by the federal government and, in some cases, by state authorities. Further, they are obligated to publicly disclose wrongful disclosures or losses of personal health information.
Fraud and Abuse Laws
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal prosecution, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of States also have statutes or regulations similar to the federal anti-kickback statute and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Affordable Care Act
In late March 2010, the Federal government enacted the comprehensive health care reform package, the Affordable Care Act. Among other provisions, the Affordable Care Act imposes individual and employer health insurance requirements, provides certain insurance subsidies (e.g., premiums and cost sharing), mandates extensive insurance market reforms, creates new health insurance access points (e.g., State and federal-based health insurance exchanges), expands the Medicaid program, promotes research on comparative clinical effectiveness of different technologies and procedures, and makes a number of changes to how products and services will be reimbursed by the Medicare program. Amendments to the Federal False Claims Act under the Affordable Care Act have made it easier for private parties to bring “qui tam” (whistleblower) lawsuits against companies, under which the whistleblower may be entitled to receive a percentage of any money paid to the government.
There are a number of provisions in the Affordable Care Act that may directly impact our customers and, therefore, indirectly affect us. For example, the Affordable Care Act expands the number of individuals that will be covered by either private or public health insurance, which may, in turn, increase the pool of potential purchasers for our customers’ products to the extent they are reimbursable by private or public health insurance. The Affordable Care Act also requires health insurance issuers in the individual and small group markets to cover certain “essential health benefits,” which include prescription drugs and which may increase coverage for our customers’ products. In addition, the Affordable Care Act reduces income and raises costs for our customers through, for instance, the imposition of drug price discounts for Medicare Part D enrollees in the “donut hole” and the imposition of an annual fee on prescription drug and biologic manufacturers. Such provisions may cause our customers to seek to restrain costs in other areas, including the services that we provide. The effective dates of the various provisions within the Affordable Care Act are staggered over the next several years, with some changes occurring immediately. Much of the interpretation of the Affordable Care Act will be subject to administrative rulemaking, the development of agency guidance, and court interpretation.
Other Applicable Laws
In addition to those described above, other federal and State laws and regulations that could directly or indirectly affect our ability to operate the business and/or financial performance include:
• state and local licensure, registration and regulation of laboratories, the processing and storage of human cells and tissue, and the development and manufacture of pharmaceuticals and biologics;
• other laws and regulations administered by the United States FDA, including the Federal Food, Drug, and Cosmetic Act and related laws and regulations and the Public Health Service Act and related laws and regulations;
• laws and regulations administered by the United States Department of Health and Human Services, including the Office for Human Research Protections;
• state laws and regulations governing human subject research;
• federal and state coverage and reimbursement laws and regulations, including laws and regulations administered by the Centers for Medicare & Medicaid Services and State Medicaid agencies;
• the federal Medicare and Medicaid Anti-Kickback Law and similar state laws and regulations;
• the federal physician self-referral prohibition commonly known as the Stark Law, and State equivalents of the Stark Law;
• Occupational Safety and Health Administration (“OSHA”) requirements;
• state and local laws and regulations dealing with the handling and disposal of medical waste; and
• the Intermediate Sanctions rules of the IRS providing for potential financial sanctions with respect to “Excess Benefit Transactions” with HUMC or other tax-exempt organizations.
Other Regulations
We are also subject to various local, State and federal laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including chemicals, micro-organisms and various radioactive compounds used in connection with our research and development activities. These laws include, but are not limited to, the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, there can be no assurances that accidental contamination or injury to employees and third parties from these materials will not occur. Our insurance program does not include environmental coverage.
Regulation in the European Union
In the European Union, or EU, medicinal products, including advanced therapy medicinal products, are subject to extensive pre- and post-market regulation by regulatory authorities at both the EU and national levels. Advanced therapy medicinal products comprise gene therapy products, somatic cell therapy products and tissue engineered products, which are cells or tissues that have undergone substantial manipulation and that are administered to human beings in order to regenerate, repair or replace a human tissue. We anticipate that our cell therapy products in development, including MyoCell , would be regulated as advanced therapy medicinal products in the EU.
Clinical Trials
Clinical trials of medicinal products in the EU must be conducted in accordance with EU and national regulations and the International Conference on Harmonization, or ICH, guidelines on Good Clinical Practices, or GCP. Additional GCP guidelines from the European Commission, focusing in particular on traceability, apply to clinical trials of advanced therapy medicinal products. If the sponsor of the clinical trial is not established within the EU, it must appoint an entity within the EU to act as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU countries the sponsor is liable to provide ‘no fault’ compensation to any study subject injured in the clinical trial.
Prior to commencing a clinical trial, the sponsor must obtain a clinical trial authorization from the competent authority, and a positive opinion from an independent ethics committee. The application for a clinical trial authorization must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. Currently, clinical trial authorization applications must be submitted to the competent authority in each EU member state in which the trial will be conducted. Under proposed new rules, there will be a centralized application procedure where one national authority takes the lead in reviewing the application and the other national authorities have only a limited involvement. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be notified to or approved by the relevant competent authorities and ethics committees.
The sponsor of a clinical trial must register the clinical trial in advance, and information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial will be made public as part of the registration. The results of the clinical trial must be submitted to the competent authorities and, with the exception of non-pediatric Phase 1 trials, will be made public at the latest within 12 months after the end of the trial.
During the development of a medicinal product, the European Medicines Agency, or EMA, and national medicines regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Scientific Advice Working Party of the Committee for Medicinal Products for Human Use, or CHMP. A fee is incurred with each scientific advice procedure. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future marketing authorization application of the product concerned. To date, we have not initiated any scientific advice procedures or other discussions with the EMA or any national regulatory authorities in the EU.
Marketing Authorizations
After completion of the required clinical testing, we must obtain a marketing authorization before we may place a medicinal product on the market in the EU. There are various application procedures available, depending on the type of product involved. All application procedures require an application in the common technical document, or CTD, format, which includes the submission of detailed information about the manufacturing and quality of the product, and non-clinical and clinical trial information. There is an increasing trend in the EU towards greater transparency and, while the manufacturing or quality information is currently generally protected as confidential information, the EMA and national regulatory authorities are now liable to disclose much of the non-clinical and clinical information in marketing authorization dossiers, including the full clinical study reports, in response to freedom of information requests after the marketing authorization has been granted. The EMA is currently considering a procedure under which clinical study reports would be posted on the agency’s website following the grant, denial or withdrawal of a marketing authorization application, subject to procedures for limited redactions and protection against unfair commercial use. A similar requirement is contained in a draft new Regulation on Clinical Trials that is expected to become applicable in mid-2016 or later.
The centralized procedure gives rise to marketing authorizations that are valid throughout the EU and, by extension (after national implementing decisions), in Norway, Iceland and Liechtenstein, which, together with the EU member states, comprise the European Economic Area, or EEA. Applicants file marketing authorization applications with the EMA, where they are reviewed by a relevant scientific committee, in most cases the CHMP. The EMA forwards CHMP opinions to the European Commission, which uses them as the basis for deciding whether to grant a marketing authorization. The centralized procedure is compulsory for medicinal products that (1) are derived from biotechnology processes, (2) contain a new active substance (not yet approved) indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders, viral diseases or autoimmune diseases and other immune dysfunctions, (3) are orphan medicinal products or (4) are advanced therapy medicinal products, such as cell therapy medicines. For medicines that do not fall within these categories, an applicant may voluntarily submit an application for a centralized marketing authorization to the EMA, as long as the CHMP agrees that (i) the medicine concerned contains a new active substance (not yet approved), (ii) the medicine is a significant therapeutic, scientific, or technical innovation, or (iii) if its authorization under the centralized procedure would be in the interest of public health.
For those medicinal products for which the centralized procedure is not available, the applicant must submit marketing authorization applications to the national medicines regulators through one of three procedures: (1) a national procedure, which results in a marketing authorization in a single EU member state; (2) the decentralized procedure, in which applications are submitted simultaneously in two or more EU member states; and (3) the mutual recognition procedure, which must be used if the product has already been authorized in at least one other EU member state, and in which the EU member states are required to grant an authorization recognizing the existing authorization in the other EU member state, unless they identify a serious risk to public health. A national procedure is only possible for one member state; as soon as an application is submitted in a second member state the mutual recognition or decentralized procedure will be triggered.
Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria: the seriousness of the disease (e.g. heavy disabling or life-threatening diseases) to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days.
Data Exclusivity
Marketing authorization applications for generic medicinal products do not need to include the results of pre-clinical and clinical trials, but instead can refer to the data included in the marketing authorization of a reference product for which regulatory data exclusivity has expired. If a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic marketing authorization applications referring to the data of that product may not be accepted by the regulatory authorities, and a further two years of market exclusivity, during which such generic products may not be placed on the market. The two-year period may be extended to three years if during the first eight years a new therapeutic indication with significant clinical benefit over existing therapies is approved.
There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials or manufacturing processes. For such products, the results of appropriate pre-clinical or clinical trials must be provided, and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and regulatory experience gained at the time.
Orphan Medicinal Products
The EMA’s Committee for Orphan Medicinal Products, or COMP, may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the EU. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria (for instance because in the meantime a new product was approved for the indication and no convincing data are available to demonstrate a significant benefit over that product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve any similar medicinal product, unless it offers a significant clinical benefit. This period may be reduced to 6 years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. The EMA is currently preparing guidance on assessing similarity of active ingredients for purposes of orphan exclusivity. It is possible that for biological products a narrow interpretation of similarity will be adopted.
Pediatric Development
In the EU, companies developing a new medicinal product must agree to a Pediatric Investigation Plan, or PIP, with the EMA and must conduct pediatric clinical trials in accordance with that PIP, unless a waiver applies ( e.g. because the relevant disease or condition occurs only in adults). The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Post-Approval Controls
The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, or QPPV, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new marketing authorization applications must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. Risk management plans and PSURs are routinely available to third parties requesting access, subject to limited redactions.
All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another.
Manufacturing
Medicinal products may only be manufactured in the EU, or imported into the EU from another country, by the holder of a manufacturing authorization from the competent national authority. The manufacturer or importer must have a qualified person, or QP, who is responsible for certifying that each batch of product has been manufactured in accordance with EU standards of good manufacturing practice, or GMP, before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with GMP.
Human Cells and Tissues
Human cells and tissues that are intended for human applications but that do not fall within the scope of rules governing medicinal products or medical devices are not subject to premarket review and approval, nor do they require extensive preclinical and clinical testing. However, there are EU rules governing the donation, procurement, testing and storage of human cells and tissues intended for human application, whether or not they are advanced therapy medicinal products. These rules also cover the processing, preservation and distribution of human cell and tissues that are not advanced therapy medicinal products. Establishments that conduct such activities must be licensed and are subject to inspection by regulatory authorities. Such establishments must implement appropriate quality systems and maintain appropriate records to ensure that cells and tissues can be traced from the donor to the recipient and vice versa. There are also requirements to report serious adverse events and reactions linked to the quality and safety of cells and tissues. More detailed rules may exist at the national level.
Named Patient Sales
The EU medicines rules allow individual member states to permit the supply of a medicinal product without a marketing authorization to fulfill special needs, where the product is supplied in response to a bona fide unsolicited order, formulated in accordance with the specifications of a healthcare professional and for use by an individual patient under his direct personal responsibility. This may in certain countries also apply to products manufactured in a country outside the EU and imported to treat specific patients or small groups of patients. In addition, advanced therapy medicinal products do not need a marketing authorization if they are prepared on a non-routine basis and are used within the same EU member state in a hospital in accordance with a medical prescription for an individual patient.
These exemptions may allow us to make limited sales of our products before we obtain a marketing authorization in the EU. However, the exemptions could also allow our competitors to make sales without having obtained a marketing authorization and without undergoing the expense of clinical trials, especially if those competitors have cell processing facilities in the relevant EU member state. Similarly, certain hospitals may be able to compete with us on the basis of these rules.
Pricing and Reimbursement
Governments influence the price of medicinal products in the EU through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Regulation in Other Countries
We intend to seek to market our products in jurisdictions outside the United States and the EU. Most of these jurisdictions have product approval and post-approval regulatory processes that are similar in principle to those in the United States or EU. Any such considerations are in the early stages.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the website at http://www.sec.gov. The public may also read and copy any document we file with the SEC at its Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549, on official business days during the hours of 10:00 am to 3:00 pm. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330.
Item 1A. Risk Factors
The risk factors required pursuant to Regulation S-K, Item 503(c) are not required for smaller reporting companies. Accordingly, the Company has determined to provide particular risk factors at this time. The risks and uncertainties described below are not the only ones facing us. Other events that we do not currently anticipate or that we currently deem immaterial also may affect our results of operations and financial condition. If any events described in the risk factors actually occur, our business, operating results, prospects and financial condition could be materially harmed. In connection with the forward looking statements that appear elsewhere in this annual report, you should also carefully review the cautionary statement referred to under “Cautionary Note Regarding Forward Looking Statements.”
SHOULD ONE OR MORE OF THE FOREGOING RISKS OR UNCERTAINTIES MATERIALIZE, OR SHOULD THE UNDERLYING ASSUMPTIONS OF OUR BUSINESS PROVE INCORRECT, ACTUAL RESULTS MAY DIFFER SIGNIFICANTLY FROM THOSE ANTICIPATED, BELIEVED, ESTIMATED, EXPECTED, INTENDED OR PLANNED.
Risks Related to Our Financial Position and Need for Additional Financing
We will need to secure additional financing in 2016 in order to continue to finance our operations. If we are unable to secure additional financing on acceptable terms, or at all, we may be forced to curtail or cease our operations.
As of December 31, 2015, we had cash and cash equivalents of $58,372 and an accumulated capital deficit of $122,076,374 As such, our existing cash resources are insufficient to finance even our immediate operations. Accordingly, we will need to secure additional sources of capital to develop our business and product candidates as planned. We are seeking substantial additional financing through public and/or private financing, which may include equity and/or debt financings, research grants and through other arrangements, including collaborative arrangements. As part of such efforts, we may seek loans from certain of our executive officers, directors and/or current shareholders. We may also seek to satisfy some of our obligations to the guarantors of our loan with Seaside National Bank & Trust, or the Guarantors, through the issuance of various forms of securities or debt on negotiated terms. On January 11, 2016, the Company renewed the loan with Seaside National Bank and Trust extend the maturity date to January 11, 2018, all other terms and conditions remain unchanged. However, financing and/or alternative arrangements with the Guarantors may not be available when we need it, or may not be available on acceptable terms.
If we are unable to secure additional financing in the near term, we may be forced to:
|
·
|
curtail or abandon our existing business plans;
|
|
·
|
reduce our headcount;
|
|
·
|
default on our debt obligations;
|
|
·
|
file for bankruptcy;
|
|
·
|
seek to sell some or all of our assets; and/or
|
|
·
|
cease our operations.
|
If we are forced to take any of these steps, any investment in our common stock may be worthless.
If we raise additional capital and/or secure alternative arrangements, with the Guarantors or otherwise, by issuing equity, equity-related or convertible securities, the economic, voting and other rights of our existing shareholders may be diluted, and those newly issued securities may be issued at prices that are a significant discount to current and/or then prevailing market prices. In addition, any such newly issued securities may have rights superior to those of our common stock. If we obtain additional capital through collaborative arrangements, we may be required to relinquish greater rights to our technologies or product candidates than we might otherwise have or become subject to restrictive covenants that may affect our business.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
Our independent registered public accounting firm issued its report dated March 8th, 2016 in connection with the audit of our financial statements as of December 31, 2015, which included an explanatory paragraph describing the existence of conditions that raise substantial doubt about our ability to continue as a going concern. In addition, our note to our financial statements for the year ended December 31, 2015 included an explanatory paragraph describing the existence of conditions that raise substantial doubt about our ability to continue as a going concern. If we are not able to continue as a going concern, it is likely that holders of our common stock will lose all of their investment. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We are a development stage life sciences company with a limited operating history and a history of net losses and negative cash flows from operations. We may never be profitable, and if we incur operating losses and generate negative cash flows from operations for longer than expected, we may be unable to continue operations.
We are a development stage life sciences company and have a limited operating history, limited capital, limited sources of revenue, and have incurred losses since inception. Our operations to date have been limited to organizing our company, developing and engaging in clinical trials of our MyoCell product candidate, expanding our pipeline of complementary product candidates through internal development and third party licenses, expanding and strengthening our intellectual property position through internal programs and third party licenses and recruiting management, research and clinical personnel. Consequently, it may be difficult to predict our future success or viability due to our lack of operating history. As of December 31, 2015, we have accumulated a deficit of approximately $122.1 million. Our MyoCell product candidate has not received regulatory approval or generated any material revenues and is not expected to generate any material revenues until commercialization of MyoCell, if ever.
Our ability to generate revenues from any of our product candidates will depend on a number of factors, including our ability to successfully complete clinical trials, obtain necessary regulatory approvals and implement our commercialization strategy. In addition, even if we are successful in obtaining necessary regulatory approvals and bringing one or more product candidates to market, we will be subject to the risk that the marketplace will not accept those products. We may, and anticipate that we will need to, transition from a company with a research and development focus to a company capable of supporting commercial activities and we may not succeed in such a transition.
Because of the numerous risks and uncertainties associated with our product development and commercialization efforts, we are unable to predict the extent of our future losses or when or if we will become profitable. Our failure to successfully commercialize our product candidates or to become and remain profitable could impair our ability to raise capital, expand our business, diversify our product offerings and continue our operations.
Risks Related to Product Development
All of our product candidates are in an early stage of development and we may never succeed in developing and/or commercializing them. We depend heavily on the success of our MyoCell product candidate. If we are unable to commercialize MyoCell or any of our other product candidates or experience significant delays in doing so, our business may fail.
|
·
|
We have invested a significant portion of our efforts and financial resources in our MyoCell product candidate and depend heavily on its success. MyoCell is currently in the clinical testing stage of development, although we have suspended work under our clinical trials as we seek to raise sufficient funds to complete the trials.
|
|
·
|
We need to devote significant additional research and development, financial resources and personnel to develop commercially viable products, obtain regulatory approvals and establish a sales and marketing infrastructure.
|
|
·
|
We are likely to encounter hurdles and unexpected issues as we proceed in the development of MyoCell and our other product candidates. There are many reasons that we may not succeed in our efforts to develop our product candidates, including the possibility that:
|
|
·
|
our product candidates will be deemed ineffective, unsafe or will not receive regulatory approvals;
|
|
·
|
our product candidates will be too expensive to manufacture or market or will not achieve broad market acceptance
|
|
·
|
others will hold proprietary rights that will prevent us from marketing our product candidates; or
|
|
·
|
our competitors will market products that are perceived as equivalent or superior.
|
Our approach of using cell-based therapy for the treatment of heart damage is risky and unproven and no products using this approach have received regulatory approval in the United States or Europe.
No company, to our knowledge, has yet been successful in its efforts to obtain regulatory approval in the United States or Europe of a cell-based therapy product for the treatment of heart damage. Cell-based therapy products, in general, may be susceptible to various risks, including undesirable and unintended side effects, unintended immune system responses, inadequate therapeutic efficacy or other characteristics that may prevent or limit their approval by regulators or commercial use. Many companies in the industry have suffered significant setbacks in advanced clinical trials, despite promising results in earlier trials. One of our competitors exploring the use of skeletal myoblasts ceased enrolling new patients in its European Phase II clinical trial based on the determination of its monitoring committee that there was a low likelihood that the trial would result in the hypothesized improvement in heart function. Although our clinical research to date suggests that MyoCell may improve the contractile function of the heart, we have not yet been able to demonstrate a mechanism of action and additional research is needed to precisely identify such mechanism.
If our clinical trials are unsuccessful or significantly delayed, or if we do not complete our clinical trials, we will not receive regulatory approval for or be able to commercialize our product candidates.
We cannot market any product candidate until regulatory agencies grant approval or licensure. In order to obtain regulatory approval for the sale of any product candidate, we must, among other requirements, provide the FDA and similar foreign regulatory authorities with preclinical and clinical data that demonstrate to the satisfaction of regulatory authorities that our product candidates are safe and effective for each indication under the applicable standards relating to such product candidate. The preclinical studies and clinical trials of any product candidates must comply with the regulations of the FDA and other governmental authorities in the United States and similar agencies in other countries.
Even if we achieve positive interim results in clinical trials, these results do not necessarily predict final results, and positive results in early trials may not be indicative of success in later trials. For example, MyoCell has been studied in a limited number of patients to date. Even though our early data has been promising, we have not yet completed any large-scale pivotal trials to establish the safety and efficacy of MyoCell. A number of participants in our clinical trials have experienced serious adverse events adjudicated or determined by trial investigators to be potentially attributable to MyoCell. There is a risk that safety concerns relating to our product candidates or cell-based therapies in general will result in the suspension or termination of our clinical trials.
We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent regulatory approval and/or commercialization of our product candidates, including the following:
|
·
|
the FDA or similar foreign regulatory authorities may find that our product candidates are not sufficiently safe or effective or may find our cell culturing processes or facilities unsatisfactory;
|
|
·
|
officials at the FDA or similar foreign regulatory authorities may interpret data from preclinical studies and clinical trials differently than we do;
|
|
·
|
our clinical trials may produce negative or inconclusive results or may not meet the level of statistical significance required by the FDA or other regulatory authorities, and we may decide, or regulators may require us, to conduct additional preclinical studies and/or clinical trials or to abandon one or more of our development programs;
|
|
·
|
the FDA or similar foreign regulatory authorities may change their approval policies or adopt new regulations;
|
|
·
|
there may be delays or failure in obtaining approval of our clinical trial protocols from the FDA or other regulatory authorities or obtaining institutional review board approvals or government approvals to conduct clinical trials at prospective sites;
|
|
·
|
we, or regulators, may suspend or terminate our clinical trials because the participating patients are being exposed to unacceptable health risks or undesirable side effects;
|
|
·
|
we may experience difficulties in managing multiple clinical sites;
|
|
·
|
enrollment in our clinical trials for our product candidates may occur more slowly than we anticipate, or we may experience high drop-out rates of subjects in our clinical trials, resulting in significant delays;
|
|
·
|
we may be unable to manufacture or obtain from third party manufacturers sufficient quantities of our product candidates for use in clinical trials; and
|
|
·
|
our product candidates may be deemed unsafe or ineffective, or may be perceived as being unsafe or ineffective, by healthcare providers for a particular indication.
|
In the SEISMIC Trial, we experienced delays attributable to slower than anticipated enrollment of patients. We may continue to experience difficulties in enrolling patients in our clinical trials, which could increase the costs or affect the timing or outcome of these trials and could prevent us from completing these trials.
Failures or perceived failures in our clinical trials would delay and may prevent our product development and regulatory approval process, make it difficult for us to establish collaborations, negatively affect our reputation and competitive position and otherwise have a material adverse effect on our business.
Healthcare reform could substantially reduce our revenues, earnings and cash flows.
We cannot predict how employers, private payors or persons buying insurance might react to the changes brought on by broad U.S. healthcare reform legislation or what form many of these regulations will take before implementation. The healthcare reform legislation, enacted in 2010, introduced healthcare insurance exchanges which provide a marketplace for eligible individuals and small employers to purchase healthcare insurance. While patients have begun receiving insurance coverage through these exchanges, the business and regulatory environment for these exchanges continues to evolve as the exchanges mature. Additionally, there is uncertainty about how the applicable state and federal agencies will enforce regulations relating to the exchanges. There is also a considerable amount of uncertainty as to the prospective implementation of the federal healthcare reform legislation and what similar measures might be enacted at the state level. There have been multiple attempts through legislative action and legal challenges to repeal or amend the Patient Protection and Affordable Care Act of 2010, as modified by the Health Reform Acts, including the case that was recently heard by the U.S. Supreme Court, King v. Burwell. Although the Supreme Court upheld the provision by the federal government of subsidies to individuals in federally facilitated healthcare exchanges in Burwell, which ultimately did not disrupt significantly the implementation of the healthcare reform legislation, we cannot predict whether other current or future efforts to repeal or amend these laws will be successful, nor can we predict the impact that such a repeal or amendment would have on our business and operations, or on our revenues and earnings. The enacted reforms as well as future legislative changes could have a material adverse effect on our results of operations.
We depend on third parties to assist us in the conduct of our preclinical studies and clinical trials, and any failure of those parties to fulfill their obligations could result in costs and delays and prevent us from obtaining regulatory approval or successfully commercializing our product candidates on a timely basis, if at all.
We engage consultants and CROs to help design, and to assist us in conducting, our preclinical studies and clinical trials and to collect and analyze data from those studies and trials. The consultants and contract research organizations we engage interact with clinical investigators to enroll patients in our clinical trials. As a result, we depend on these consultants and CROs to perform the studies and trials in accordance with the investigational plan and protocol for each product candidate and in compliance with regulations and standards, commonly referred to as “good clinical practice”, for conducting, recording and reporting results of clinical trials to assure that the data and results are credible and accurate and the trial participants are adequately protected, as required by the FDA and foreign regulatory agencies. We may face delays in our regulatory approval process if these parties do not perform their obligations in a timely or competent fashion or if we are forced to change service providers. The risk of delays is heightened for our clinical trials conducted outside of the United States, where it may be more difficult for us to ensure that studies are conducted in compliance with foreign regulatory requirements. Any third parties that we hire to conduct clinical trials may also provide services to our competitors, which could compromise the performance of their obligations to us. If these third parties do not successfully carry out their duties or meet expected deadlines, or if the quality, completeness or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical trial protocols or for other reasons, our clinical trials may be extended, delayed or terminated or may otherwise prove to be unsuccessful. If there are delays or failures in clinical trials or regulatory approvals as a result of the failure to perform by third parties, our development costs will increase, and we may not be able to obtain regulatory approval for our product candidates. In addition, we may not be able to establish or maintain relationships with these third parties on favorable terms, if at all. If we need to enter into replacement arrangements because a third party is not performing in accordance with our expectations, we may not be able to do so without undue delays or considerable expenditures or at all.
Our cell-based product candidates are based on novel technologies and the FDA and regulatory agencies in other countries have limited experience reviewing product candidates using these technologies.
We are subject to the risks of failure inherent in the development of product candidates based on new technologies. The novel nature of our product candidates creates significant challenges in regards to product development and optimization, government regulation, third party reimbursement and market acceptance. These include:
|
·
|
the scientific basis of our technology could be determined to be less sound than we believe;
|
|
·
|
the time and effort required to solve novel technical problems could delay the development of our product candidates;
|
|
·
|
the FDA and regulatory agencies in other countries have relatively limited experience with therapies based upon cellular medicine generally and, as a result, the pathway to regulatory approval for our cell-based product candidates may be more complex and lengthy; and
|
|
·
|
the healthcare community has relatively little experience with therapies based upon cellular medicine and, accordingly, following regulatory approval, if any, our product candidates may not become widely accepted by physicians, patients, third party payors or the healthcare community.
|
As a result, the development and commercialization pathway for our cell-based therapies may be subject to increased uncertainty, as compared to the pathway for new conventional drugs.
There is substantial uncertainty as to the coverage that may be available and the reimbursement rates that may be established for our product candidates. Any failure to obtain third party coverage or an adequate level of reimbursement for our product candidates will likely have a material adverse effect on our business.
If we successfully develop, and obtain necessary regulatory approvals for, our product candidates we intend to sell them initially in Europe and the United States. We have not yet submitted any of our product candidates to CMS or any private or governmental third party payor in the United States to determine whether or not our product candidates will be covered under private or public health insurance plans or, if they are covered, what coverage or reimbursement rates may be available. Although we believe hospitals may be entitled to some procedure reimbursement for MyoCell, we cannot assure you that such reimbursement will be adequate or available at all.
In Europe, the pricing of prescription pharmaceutical products and services and the level of government reimbursement generally are subject to governmental control. Reimbursement and healthcare payment systems in European markets vary significantly by country, and may include both government-sponsored healthcare and private insurance. In these countries, pricing negotiations with governmental authorities can take six to twelve months or longer after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct one or more clinical trials that compare the cost effectiveness of our product candidates to other available therapies. Conducting one or more clinical trials for this purpose would be expensive and result in delays in commercialization of our product candidates. We may not obtain coverage or reimbursement or pricing approvals from countries in Europe in a timely manner, or at all. Any failure to receive coverage or reimbursement or pricing approvals from one or more European countries could effectively prevent us from selling our product candidates in those countries, which could materially adversely affect our business.
In the United States, our revenues will depend upon the coverage and reimbursement rates and policies established for our product candidates by third party payors, including governmental authorities, managed-care providers, public health insurers, private health insurers and other organizations. These third party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new healthcare products approved for marketing by the FDA or regulatory agencies in other countries. As a result, significant uncertainty exists as to whether newly approved medical products will be eligible for coverage by third party payors or, if eligible for coverage, what the reimbursement rates will be for those products. Furthermore, cell-based therapies like MyoCell may be more expensive than pharmaceuticals due to, among other things, the higher cost and complexity associated with the research, development and production of these therapies. This, in turn, may make it more difficult for us to obtain adequate reimbursement from third party payors, particularly if we cannot demonstrate a favorable cost-benefit relationship. Third party payors may also deny coverage or offer inadequate levels of reimbursement for our potential products if they determine that the product has not received appropriate clearances from the FDA or other government regulators or is experimental, unnecessary or inappropriate. Accordingly, we cannot assure you that adequate third party coverage or reimbursement will be available for any of our product candidates to allow us to successfully commercialize these product candidates.
Coverage and reimbursement rates for our product candidates may be subject to increased restrictions both in the United States and in other countries in the future. Coverage policies and reimbursement rates are subject to change and we cannot guarantee that current coverage policies and reimbursement rates will be applicable to our product candidates in the future. U.S. federal, state and foreign agencies and legislatures from time to time may seek to impose restrictions on coverage, pricing, and reimbursement level of drugs, devices and healthcare services in order to contain healthcare costs.
Product liability and other claims against us may reduce demand for our products or result in substantial damages. We anticipate that we will need to obtain and maintain additional or increased insurance coverage, and we may not be able to obtain or maintain such coverage on commercially reasonable terms, if at all.
A product liability claim, a clinical trial liability claim or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have a material adverse effect on our business. Our business exposes us to potential liability risks that may arise from the clinical testing of our product candidates in human clinical trials and the manufacture and sale of any approved products. Any clinical trial liability or product liability claim or series of claims or class actions brought against us, with or without merit, could result in:
|
·
|
liabilities that substantially exceed our existing clinical trial liability insurance, or any clinical trial liability or product liability insurance that we may obtain in the future, which we would then be required to pay from other sources, if available;
|
|
·
|
an increase in the premiums we pay for our clinical trial liability insurance and any clinical trial liability or product liability insurance we may obtain in the future or the inability to renew or obtain clinical trial liability or product liability insurance coverage in the future on acceptable terms, or at all;
|
|
·
|
withdrawal of clinical trial volunteers or patients;
|
|
·
|
damage to our reputation and the reputation of our products, including loss of market share;
|
|
·
|
regulatory investigations that could require costly recalls or product modifications;
|
|
·
|
litigation costs; and
|
|
·
|
diversion of management’s attention from managing our business.
|
Claims may be made by consumers, healthcare providers, third party strategic collaborators or others selling our products if one of our products or product candidates causes, or appears to have caused, an injury. We may be subject to claims against us even if an alleged injury is due to the actions of others. For example, we rely on the expertise of physicians, nurses and other associated medical personnel to perform the medical procedures and processes related to our product candidates. If these medical personnel are not properly trained or are negligent in using our product candidates, the therapeutic effect of our product candidates may be diminished or the patient may suffer injury, which may subject us to liability. In addition, an injury resulting from the activities of our suppliers may serve as a basis for a claim against us.
We do not intend to promote, or to in any way support or encourage the promotion of, our product candidates for off-label or otherwise unapproved uses. However, if our product candidates are approved by the FDA or similar foreign regulatory authorities, we cannot prevent a physician from using them for any off-label applications. If injury to a patient results from such an inappropriate use, we may be subject to the expenses of litigation and may need to rely upon our product liability insurance.
These liabilities could prevent or interfere with our clinical efforts, product development efforts and any subsequent product commercialization efforts, all of which could have a material adverse effect on our business.
Risks Related to Our Intellectual Property
We hold limited patent and other intellectual property rights, and our success will be dependent in large part on safeguarding our existing intellectual property rights and obtaining patent and other proprietary protection for our product candidates.
We hold limited patent rights in our product candidates. Our MyoCath product candidate is protected by a patent, expiring in September 2017, in which we have an irrevocable co-exclusive license. Our MyoCell product candidate is no longer protected by patents, which means that competitors will be free to sell products that incorporate the same or similar technologies that are used in MyoCell without infringing our patent rights. As a result, MyoCell, if approved for use, may be vulnerable to competition in the form of products that use the same or similar technologies. We have previously licensed certain patents and patent applications relating to our MyoCell product candidate. These licenses have all lapsed as of the date of this report, although we have had discussions with the relevant licensor regarding a potential reinstatement of our rights in such licenses.
Our commercial success will depend to a significant degree on our ability to:
|
·
|
compel the owners of the patents licensed to us to defend and enforce such patents, to the extent such patents may be applicable to our products and material to their commercialization;
|
|
·
|
obtain new patent and other proprietary protection for MyoCell and our other product candidates;
|
|
·
|
obtain and/or maintain appropriate licenses to patents, patent applications or other proprietary rights held by others with respect to our technology, both in the United States and other countries;
|
|
·
|
preserve intellectual property rights relating to our product candidates; and
|
|
·
|
operate without infringing the patents and proprietary rights of third parties.
|
Failure to obtain adequate patent protection for our product candidates, or the failure to protect our existing patent rights, may impair our ability to be competitive. The availability of infringing products in markets where we have patent protection, or the availability of competing products in markets where we do not have adequate patent protection, could erode the market for our product candidates, negatively impact the prices we can charge for our product candidates, and harm our reputation if infringing or competing products are manufactured to inferior standards.
Our most important license agreement with respect to MyoCath is co-exclusive and the co-licensor of the intellectual property, a division of Abbott Laboratories, may also seek to commercialize MyoCath.
In June 2003, we assigned our exclusive license to the primary patent protecting MyoCath to ACS, originally a subsidiary of Guidant Corporation and now d/b/a Abbott Vascular, a division of Abbott Laboratories. In connection with this agreement, ACS granted to us a co-exclusive, irrevocable, fully paid-up license to this patent for the life of the patent. Because our license is co-exclusive with ACS, ACS may, parallel to our efforts, seek to commercialize MyoCath if MyoCath secures regulatory approval.
Accordingly, even if ACS does nothing to assist us to secure regulatory approval of MyoCath, ACS may become a direct competitor in the MyoCath manufacturing and supply business. In addition, pursuant to our agreement with ACS, we are prohibited from contracting with third parties for the distribution of MyoCath.
Our patents may not be valid or enforceable, and may be challenged by third parties.
We cannot assure you that any patents issued or licensed to us would be held valid by a court or administrative body or that we would be able to successfully enforce our patents against infringers, including our competitors. The issuance of a patent is not conclusive as to its validity or enforceability, and the validity and enforceability of a patent is susceptible to challenge on numerous legal grounds. Challenges raised in patent infringement litigation brought by or against us may result in determinations that patents that have been issued or licensed to us or any patents that may be issued to us or our licensors in the future are invalid, unenforceable or otherwise subject to limitations. In the event of any such determinations, third parties may be able to use the discoveries or technologies claimed in these patents without paying licensing fees or royalties to us, which could significantly diminish the value of our intellectual property and our competitive advantage. Even if our patents are held to be enforceable, others may be able to design around our patents or develop products similar to our products that are not within the scope of any of our patents.
In addition, enforcing the patents that have been licensed to us and any patents that may be issued to us in the future against third parties may require significant expenditures regardless of the outcome of such efforts. Our inability to enforce our patents against infringers and competitors may impair our ability to be competitive and could have a material adverse effect on our business.
If we are not able to protect and control unpatented trade secrets, know-how and other technological innovation, we may suffer competitive harm.
We rely to a large extent on unpatented technology, trade secrets, confidential information and proprietary know-how to protect our technology and maintain our competitive position, especially when we do not believe that patent protection is appropriate or can be obtained. Trade secrets are difficult to protect. In order to protect proprietary technology and processes, we rely in part on confidentiality and intellectual property assignment agreements with our employees, consultants and others. These agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of the individual’s relationship with us except in limited circumstances. These agreements generally also provide that we shall own all inventions conceived by the individual in the course of rendering services to us. These agreements may not effectively prevent disclosure of confidential information or result in the effective assignment to us of intellectual property, and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information or other breaches of the agreements. In addition, others may independently discover trade secrets and proprietary information that have been licensed to us or that we own, and in such case we could not assert any trade secret rights against such party.
Enforcing a claim that a party illegally obtained and is using trade secrets that have been licensed to us or that we own is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Costly and time-consuming litigation could be necessary to seek to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could have a material adverse effect on our business. Moreover, some of our academic institution licensors, collaborators and scientific advisors have rights to publish data and information to which we have rights. If we cannot maintain the confidentiality of our technologies and other confidential information in connection with our collaborations, our ability to protect our proprietary information or obtain patent protection in the future may be impaired, which could have a material adverse effect on our business.
The sale or issuance of our common stock to Magna Equities II, LLC upon the issuance of the common stock underlying the convertible promissory notes may cause substantial dilution and the resale of the shares of common stock by Magna Equities II, LLC into the public market, or the perception that such sales may occur, could cause the price of our common stock to fall.
On October 1, 2015, U.S. Stem Cell entered into a securities purchase agreement (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (“Magna”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, Magna shall purchase from the Company on the Closing Date a senior convertible note with an initial principal amount of $160,000 (the “Convertible Note”) for a purchase price of $100,000 (a 37.5% original issue discount). On December 3, 2015, the Convertible Note was amended and restated to reflect a final principal amount of $110,000.
In connection with the execution of the Purchase Agreement, the Company and Magna also entered into a registration rights agreement in which Magna Equities II, LLC may elect, upon effectiveness of the registration statement, to convert part or all of the outstanding principal and interest of the convertible promissory notes to the common stock registered hereunder, there will be substantial dilution to the current number of issued and outstanding shares and any sale of such stock may have an adverse effect upon our stock price. The number of shares ultimately offered for sale by Magna Equities II, LLC under this prospectus is dependent upon a number of factors, including the extent to which Magna Equities II, LLC converts the convertible promissory notes into shares of our common stock. Because the actual exercise price for the shares of common stock that Magna Equities II, LLC may receive upon conversion will fluctuate based on the market price of our common stock, we are not able to determine at this time the exact number of shares of our common stock that we will issue and, therefore, the exact number of shares we will ultimately register for resale under the Securities Act. At no time will Magna Equities II, LLC be entitled to convert any portion of the Convertible Note to the extent that after such conversion, Magna Equities II, LLC (together with its affiliates) would beneficially own more than 4.99% of our common stock (as calculated pursuant to Section 13(d) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and the rules and regulations thereunder). Moreover, there is an inverse relationship between the market price of our common stock and the number of shares of our common stock that may be sold following a conversion to common stock.. That is, the lower the market price, the more shares of our common stock that may be issued and sold. Accordingly, if the market price of our common stock decreases (whether such decrease is due to sales by Magna Equities II, LLC in the market or otherwise) and, in turn, the exercise price of our common stock provided in a conversion of the convertible promissory notes to common stock issued to Magna Equities II, LLC decreases, this could allow Magna Equities II, LLC to receive greater numbers of shares of our common stock. Although the number of shares of our common stock that our existing stockholders own will not decrease, the common stock owned by our existing stockholders will represent a smaller percentage of our total outstanding shares after any such issuances to Magna Equities II, LLC. Depending on market liquidity at the time, the issuance of a substantial number of shares of our common stock by Magna Equities II, LLC, and the resale of such shares by Magna Equities II, LLC into the public market, or the perception that such sales may occur, could cause the trading price of our common stock to decline, result in substantial dilution to existing stockholders and make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales.
In the prior registration statement (SEC File No. 200457), we registered 143,813 (post-split) shares of common stock underlying commitment shares, a draw down equity line, and convertible promissory notes. The number of shares and stock prices listed below reflect post-split calculations. Magna Equities II, LLC was issued 9,109 and 2,891 shares as of October 27, 2014 and December 23, 2014 respectively, representing 1.66% and 0.50% of the then current issued and outstanding shares of our common stock respectively. Our common stock price as of October 27, 2014 and December 23, 2014 was $20.50 and $12.80 respectively. Pursuant to the draw down equity line, we requested drawdowns from January 13, 2015 through August 27, 2015 (which includes issuing true up shares as per the terms and conditions of the Purchase Agreement of the equity line). During this period, we issued an aggregate of 968,514 shares (post-split) pursuant to ten draw down requests initiated solely by us. The percentage dilution for any one draw down or true up represented from 0.35% of the then issued and outstanding shares of common stock to 2.12% of the then issued and outstanding shares of common stock. The price of our common stock was $8.40 as of January 13, 2015 (the date of the initial draw down) to $4.28 as of August 27, 2015 (the date of the final true up issuance). Magna Equities II, LLC converted a portion of its convertible promissory notes on February 2, 2015, receiving 7,246 (post-split) shares of common stock (representing 1.19% of the then issued and outstanding shares of common stock). The remainder of the convertible promissory notes were converted to shares of common stock from August 27, 2015 through November 6, 2015. The percentage dilution for any one conversion during this period represented from 0.82% of the then issued and outstanding shares of common stock to 2.85% of the then issued and outstanding shares of common stock. The price of our common stock during this period was $4.00 as of August 27, 2015 to $1.40 as of October 27, 2015, the second to last conversion date) ($5.45 as of November 6, 2015 (the date of the final conversion). Consequently, during the period covered by the prior registration period, our stock price decreased from $20.50 to $1.40 ($5.45 as of the last conversion). During this time, our number of issued and outstanding shares increased from 548,186 to 1,615,772 shares, partly as a result of the issuances to Magna Equities II, LLC. Since Magna Equities II, LLC may convert all or part of its convertible promissory notes to common stock as registered hereunder, further dilution may occur similar to the prior registration. This further dilution may result in your additional dilution of the existing ownership interests of our common stockholders and may adversely affect the stock price of the shares of common stock.
Risks Related to Our Common Stock
Our common stock may be considered a “penny stock,” and thereby be subject to additional sale and trading regulations that may make it more difficult to sell.
Our common stock is considered to be a “penny stock.” It does not qualify for one of the exemptions from the definition of “penny stock” under Section 3a51-1 of the Exchange Act. Our common stock is a “penny stock” because it meets one or more of the following conditions (i) the stock trades at a price less than $5.00 per share; (ii) it is not traded on a “recognized” national exchange or (iii) it is not quoted on the NASDAQ Global Market, or has a price less than $5.00 per share. The principal result or effect of being designated a “penny stock” is that securities broker-dealers participating in sales of our common stock are subject to the “penny stock” regulations set forth in Rules 15-2 through 15g-9 promulgated under the Securities Exchange Act. For example, Rule 15g-2 requires broker-dealers dealing in penny stocks to provide potential investors with a document disclosing the risks of penny stocks and to obtain a manually signed and dated written receipt of the document at least two business days before effecting any transaction in a penny stock for the investor’s account. Moreover, Rule 15g-9 requires broker-dealers in penny stocks to approve the account of any investor for transactions in such stocks before selling any penny stock to that investor. This procedure requires the broker-dealer to (i) obtain from the investor information concerning his or her financial situation, investment experience and investment objectives; (ii) reasonably determine, based on that information, that transactions in penny stocks are suitable for the investor and that the investor has sufficient knowledge and experience as to be reasonably capable of evaluating the risks of penny stock transactions; (iii) provide the investor with a written statement setting forth the basis on which the broker-dealer made the determination in (ii) above; and (iv) receive a signed and dated copy of such statement from the investor, confirming that it accurately reflects the investor’s financial situation, investment experience and investment objectives. Compliance with these requirements may make it more difficult and time consuming for holders of our common stock to resell their shares to third parties or to otherwise dispose of them in the market or otherwise.
FINRA sales practice requirements may limit a shareholder’s ability to buy and sell our common shares.
In addition to the “penny stock” rules described above, FINRA has adopted rules that require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common shares, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our shares.
Rule 144 sales in the future may have a depressive effect on the company’s stock price as an increase in supply of shares for sale, with no corresponding increase in demand will cause prices to fall.
All of the outstanding shares of common stock held by the present officers, directors, and affiliate stockholders are “restricted securities” within the meaning of Rule 144 under the Securities Act of 1933, as amended. As restricted shares, these shares may be resold only pursuant to an effective registration statement or under the requirements of Rule 144 or other applicable exemptions from registration under the Securities Act of 1933 and as required under applicable state securities laws. Rule 144 provides in essence that a person who is an affiliate or officer or director who has held restricted securities for six months may, under certain conditions, sell every three months, in brokerage transactions, a number of shares that does not exceed the greater of 1.0% of a Company’s issued and outstanding common stock. There is no limit on the amount of restricted securities that may be sold by a non-affiliate after the owner has held the restricted securities for a period of six months if the Company is a current reporting company under the Securities Exchange Act of 1934. A sale under Rule 144 or under any other exemption from the Securities Act of 1933, if available, or pursuant to subsequent registration of shares of common stock of present stockholders, may have a depressive effect upon the price of the common stock in any market that may develop. In addition, if we are deemed a shell company pursuant to Section 12(b)-2 of the Act, our “restricted securities”, whether held by affiliates or non-affiliates, may not be re-sold for a period of 12 months following the filing of a Form 10 level disclosure or registration pursuant to the Securities Act of 1933.
Failure to achieve and maintain effective internal controls in accordance with section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our business and operating results.
It is time consuming, difficult and costly for us to develop and maintain the internal controls, processes and reporting procedures required by the Sarbanes-Oxley Act, and as our business develops, we may need to hire additional financial reporting, internal auditing and other finance staff in order to remain compliant. The cost of compliance will adversely affect our financial results, while, if we are unable to comply, we may not be able to obtain the independent accountant certifications that the Sarbanes-Oxley Act requires of publicly traded companies.
If we fail to comply in a timely manner with the requirements of Section 404 of the Sarbanes-Oxley Act regarding internal control over financial reporting or to remedy any material weaknesses in our internal controls that we may identify, such failure could result in material misstatements in our financial statements, cause investors to lose confidence in our reported financial information and have a negative effect on the trading price of our common stock.
Pursuant to Section 404 of the Sarbanes-Oxley Act and current SEC regulations, we are required to prepare assessments regarding internal controls over financial reporting and furnish a report by our management on our internal control over financial reporting. Failure to achieve and maintain an effective internal control environment or complete our Section 404 certifications could have a material adverse effect on our stock price.
In addition, in connection with our on-going assessment of the effectiveness of our internal control over financial reporting, we may discover “material weaknesses” in our internal controls as defined in standards established by the Public Company Accounting Oversight Board, or the PCAOB. A material weakness is a significant deficiency, or combination of significant deficiencies, that results in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected. The PCAOB defines “significant deficiency” as a deficiency that results in more than a remote likelihood that a misstatement of the financial statements that is more than inconsequential will not be prevented or detected.
In the event that a material weakness is identified, upon receiving sufficient financing or generating sufficient revenues, we will employ qualified personnel and adopt and implement policies and procedures to address any such material weaknesses. However, the process of designing and implementing effective internal controls is a continuous effort that requires us to anticipate and react to changes in our business and the economic and regulatory environments and to expend significant resources to maintain a system of internal controls that is adequate to satisfy our reporting obligations as a public company. We cannot assure you that the measures we will take will remediate any material weaknesses that we may identify or that we will implement and maintain adequate controls over our financial process and reporting in the future.
Any failure to complete our assessment of our internal control over financial reporting, to remediate any material weaknesses that we may identify or to implement new or improved controls, or difficulties encountered in their implementation, could harm our operating results, cause us to fail to meet our reporting obligations or result in material misstatements in our financial statements. Any such failure could also adversely affect the results of the periodic management evaluations of our internal controls and, in the case of a failure to remediate any material weaknesses that we may identify, would adversely affect the annual auditor attestation reports regarding the effectiveness of our internal control over financial reporting that are required under Section 404 of the Sarbanes-Oxley Act. Inadequate internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
The systems of internal controls and procedures that we have developed and implemented to date are adequate in a research and development business. The current transaction volume and limited transaction channels mean that operating management, financial management, board members and auditor can, and do, efficiently perform a very extensive and detailed transaction review to ensure compliance with the Company’s established procedures and controls. If our business grows rapidly, we may not be able to keep up with recruiting and training personnel, and enhancing our systems of internal control in line with the growth in transaction volumes and compliance risks which could result in loss of assets, profit, and ability to manage the daily operations of our Company.
Public disclosure requirements and compliance with changing regulation of corporate governance pose challenges for our management team and result in additional expenses and costs which may reduce the focus of management and the profitability of our company.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations promulgated thereunder, the Sarbanes-Oxley Act and SEC regulations, have created uncertainty for public companies and significantly increased the costs and risks associated with accessing the U.S. public markets. Our management team will need to devote significant time and financial resources to comply with both existing and evolving standards for public companies, which will lead to increased general and administrative expenses and a diversion of management time and attention from revenue generating activities to compliance activities.
SHOULD ONE OR MORE OF THE FOREGOING RISKS OR UNCERTAINTIES MATERIALIZE, OR SHOULD THE UNDERLYING ASSUMPTIONS PROVE INCORRECT, ACTUAL RESULTS MAY DIFFER SIGNIFICANTLY FROM THOSE ANTICIPATED, BELIEVED, ESTIMATED, EXPECTED, INTENDED OR PLANNED
Item 1B. Unresolved Staff Comments
This Item is not applicable to us as we are not an accelerated filer, a large accelerated filer, or a well-seasoned issuer.
Item 2. Properties
Our headquarters are located in Sunrise, Florida and consists of 4,860 square feet of space, which we lease at a current rent of approximately $82,620 per year. The lease expired in January 2013. In January 2013, the Company amended its facility lease to extend the term of the lease until April 30, 2013. In April 2013, the Company amended its facility lease to extend the term of the lease until August 15, 2013. In September 2013, the Company amended its facility lease to extend the term until July 31, 2016.
In addition to our corporate offices, at this location, we maintain:
|
·
|
our MyoCell cell culturing facility for supply within the United States; and
|
|
·
|
a fully equipped cell culturing laboratory where we perform experimental work in the areas of cell culturing, cell engraftment, and other advanced research projects related to our core business.
|
On February 4, 2016, the Company extended its facility lease to extend the term of the lease until August 31, 2019 at a monthly base rent of $7,306 plus a pro rata share of landlord’s operating expenses In Connection with the renewal, the Company was required to pay $11,072 security deposit.
Approximate annual future minimum lease obligations under this non-cancelable facilities operating lease agreement as of December 31, 2015 are as follows:
|
Year ending December 31,
|
||||
|
2016
|
$
|
87,674
|
||
|
2017
|
87,674
|
|||
|
2018
|
87,674
|
|||
|
2019
|
58,450
|
|||
|
Total
|
$
|
321,472
|
||
We believe the space available at our headquarters will be sufficient to meet the needs of our operations for the foreseeable future.
Item 3. Legal Proceedings
On November 10, 2014, the Company was served with a lawsuit by an alleged assignee and a guarantor to a Loan Guarantee, Payment and Security Agreement. These parties claim breach of that Agreement and damages of approximately $2.3 Million plus interest. The assignor and assignee also sued the Company’s directors and two past directors and an affiliate shareholder for breach of fiduciary duty, claiming damages as alleged creditors arising out of these parties’ alleged participation in Northstar Biotech Group, LLC, a secured creditor of the Company. On June 1, 2015, the Company settled the lawsuit. As part of the settlement, the Company recorded a gain of $1,169,058. The Company provided a note to the original guarantor in the amount of $1,697,762, to be paid over a five year period with a 0% interest rate.
On September 17, 2015, a product liability lawsuit was filed in Broward County, specifically Patsy Bade v. Bioheart, Inc. US Stem Cell Clinics LLC, Aleiandro Perez, ARNP, and Shareen Greenbaum, M.D., and on November 30, 2015, a product liability lawsuit was filed in Broward County, specifically Elizabeth Noble v. Bioheart, Inc. US Stem Cell Clinics LLC, Aleiandro Perez, ARNP, and Shareen Greenbaum, M.D. Both matters are ongoing and the Company intends to defend the matters rigorously. In the event, if ever, of a settlement or loss, management believes that any loss related to the claims will be covered by their insurance policy and that there is no indication that the insurance company will dispute any claim, if ever required.
The Company is subject to other legal proceedings that arise in the ordinary course of business. In the opinion of management, as of December 31, 2015, the amount of ultimate liability with respect to such matters, if any, in excess of applicable insurance coverage, is not likely to have a material impact on the Company’s business, financial position, results of operations or liquidity. However, as the outcome of litigation and other claims is difficult to predict significant changes in the estimated exposures could exist.
Item 4. Mine Safety Disclosures.
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed on the OTC Markets under the symbol “USRM”. For the periods indicated, the following table sets forth the high and low bid prices per share of common stock, as reported by the OTC Markets. These prices represent inter-dealer quotations without retail markup, markdown, or commission and may not necessarily represent actual transactions.
|
Fiscal Year 2014
|
||||||||
|
High
|
Low
|
|||||||
|
First Quarter
|
$ | 80.00 | $ | 9.00 | ||||
|
Second Quarter
|
$ | 55.00 | $ | 20.00 | ||||
|
Third Quarter
|
$ | 38.00 | $ | 23.00 | ||||
|
Fourth Quarter
|
$ | 25.00 | $ | 11.00 | ||||
|
Fiscal Year 2015
|
||||||||
|
High
|
Low
|
|||||||
|
First Quarter
|
$ | 22.90 | $ | 7.00 | ||||
|
Second Quarter
|
$ | 9.80 | $ | 4.00 | ||||
|
Third Quarter
|
$ | 8.80 | $ | 1.30 | ||||
|
Fourth Quarter
|
$ | 7.55 | $ | 0.25 | ||||
Holders
As of December 31, 2015, there were approximately 513 shareholders of record of our common stock.
Dividends
We have never declared or paid any cash dividends on our common stock or other securities and do not currently anticipate paying any cash dividends in the foreseeable future. The declaration and payment of dividends by us are subject to the discretion of our Board of Directors and the restrictions specified in our articles of incorporation and by applicable law. In addition, under the terms of the Northstar loan, we are restricted from paying cash dividends to our shareholders while the loan is outstanding. Any future determination to pay cash dividends will depend on our results of operations, financial condition, capital requirements, contractual restrictions and other factors deemed relevant by our Board of Directors.
Securities Authorized For Issuance Under Equity Compensation Plans
The following table provides certain information regarding our existing equity compensation plans as of December 31, 2015:
|
Number of
securities
to be issued upon
exercise of
outstanding
options
|
Weighted-
average
exercise
price of
outstanding
options
|
Number of
securities
remaining
available
for issuance under
equity
compensation
plans
|
||||||||||
|
Equity compensation plans approved by security holders (1)
|
555,820 | 6.43 | 7,383,070 | |||||||||
|
Equity compensation plans not approved by security holders (2)
|
139,367 | 182.26 | 0 | |||||||||
|
(1)
|
Consists of our 1999 Officers and Employees Stock Option Plan, 1999 Directors and Consultants Stock Option Plan and Omnibus Equity Compensation Plan, 2013 Omnibus Equity Compensation Plan.
|
|
|
(2)
|
Includes:
|
| · | 8,000 warrants in connection with a joint venture agreement dated March 10, 2014. The warrants are exercisable at $21.70 for four years vesting from June 8, 2014 through March 10, 2016. | ||
| · | 4,000 warrants in connection with use of certain intellectual property. The warrants are exercisable at $48.10 for four years vesting from July 6, 2014 through April 6, 2017. | ||
|
·
|
4,000 warrants in connection with the termination of a joint venture agreement. The warrants are exercisable at $15.70 for four years vesting May 27, 2015.
|
||
|
·
|
warrants issued in connection with our private placements in 2014 to purchase an aggregate of 41,592shares of our common stock at prices from $11.00 to $23.25 per share expiring ten years from the date of issuance.
|
||
|
·
|
Warrants issued in connection with our private placements in 2015 to purchase 1,444 shares of our common stock at $11.27 per share expiring ten years from date of issuance
|
||
|
·
|
Warrants issued in connection with settlement of debt to purchase 628 shares of our common stock at $40.00 per share expiring four years from date of issuance.
|
Recent Sales of Unregistered Securities
In 2015, we sold an aggregate of 1,444 shares of our common stock and warrants to purchase 1,444 shares of our common stock for aggregate gross cash proceeds of $16,270. The warrants are (i) exercisable solely for cash at an exercise price of $11.27 per share, (ii) non-transferable for six months following issuance and (iii) exercisable, in whole or in part, at any time during the period commencing on the date that is six months and one day following the date of issuance and ending on the tenth year anniversary of the date of issuance.
On January 7, 2016, we entered into Securities Purchase Agreements with Daniel James Management (“DJM”) for the sale of a 9.5% convertible notes in aggregate principal amount of $25,000 (the "DJM" Note"). The DJM Note is due on January 15, 2016. The DJM Note is convertible into common stock, at holder’s option, at a 47% discount to the lowest daily trading price of the common stock during the 10 trading day period prior to conversion.
In January and February 2016, the Company issued an aggregate of 192,500 shares of its common stock in settlement of $38,240 notes payable and $1,318 accrued interest; 16,753 shares for services of $13,110 and 14,606 shares in settlement of related party advances.
The issuance of such shares of our common stock was effected in reliance on the exemptions for sales of securities not involving a public offering, as set forth in Rule 506 promulgated under the Securities Act of 1933, as mended (the “Securities Act”) and in Section 4(2) of the Securities Act, based on the following: the investors confirmed to us that they were “accredited investors,” as defined in Rule 501 of Regulation D promulgated under the Securities Act and had such background, education and experience in financial and business matters as to be able to evaluate the merits and risks of an investment in the securities; (b) there was no public offering or general solicitation with respect to the offering; (c) the investors were provided with certain disclosure materials and all other information requested with respect to our company; (d) the investors acknowledged that all securities being purchased were “restricted securities” for purposes of the Securities Act, and agreed to transfer such securities only in a transaction registered under the Securities Act or exempt from registration under the Securities Act; and (e) a legend was placed on the certificates representing each such security stating that it was restricted and could only be transferred if subsequent registered under the Securities Act or transferred in a transaction exempt from registration under the Securities Act.
Issuer Purchases of Equity Securities
None.
Transfer Agent: Continental Stock Transfer & Trust Company, 17 Battery Place, 8th Floor, New York, NY 10004 acts as transfer agent for our common stock.
Item 6. Selected Financial Data
Not required under Regulation S-K for “smaller reporting companies.”
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following is management’s discussion and analysis (“MD&A”) of certain significant factors that have affected our financial position and operating results during the periods included in the accompanying financial statements, as well as information relating to the plans of our current management. This report includes forward-looking statements. Generally, the words “believes,” “anticipates,” “may,” “will,” “should,” “expect,” “intend,” “estimate,” “continue,” and similar expressions or the negative thereof or comparable terminology are intended to identify forward-looking statements. Such statements are subject to certain risks and uncertainties, including the matters set forth in this report or other reports or documents we file with the Securities and Exchange Commission from time to time, which could cause actual results or outcomes to differ materially from those projected. Undue reliance should not be placed on these forward-looking statements which speak only as of the date hereof. We undertake no obligation to update these forward-looking statements.
The following discussion and analysis should be read in conjunction with our financial statements and the related notes thereto and other financial information contained elsewhere in this Form 10-K
The Company’s MD&A is comprised of significant accounting estimates made in the normal course of its operations, overview of the Company’s business conditions, results of operations, liquidity and capital resources and contractual obligations. The Company did not have any off balance sheet arrangements as of December 31, 2014 or 2015.
The discussion and analysis of the Company’s financial condition and results of operations is based upon its financial statements, which have been prepared in accordance with generally accepted accounting principles generally accepted in the United States (or “GAAP”). The preparation of those financial statements requires us to make estimates and judgments that affect the reported amount of assets and liabilities at the date of its financial statements. Actual results may differ from these estimates under different assumptions or conditions.
Our Ability To Continue as a Going Concern
Our independent registered public accounting firm has issued its report dated March 8, 2016 in connection with the audit of our financial statements as of December 31, 2015 that included an explanatory paragraph describing the existence of conditions that raise substantial doubt about our ability to continue as a going concern. Our financial statements as of December 31, 2015 have been prepared under the assumption that we will continue as a going concern. If we are not able to continue as a going concern, it is likely that holders of our common stock will lose all of their investment. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Overview
We are a biotechnology company focused on the discovery, development and, subject to regulatory approval, commercialization of autologous cell therapies for the treatment of chronic and acute heart damage. Our lead product candidates are MyoCell and AdipoCell. MyoCell is an innovative clinical therapy designed to populate regions of scar tissue within a patient’s heart with autologous muscle cells, or cells from a patient’s body, for the purpose of improving cardiac function in chronic heart failure patients. AdipoCell is an innovative cell therapy with multiple possible treatment applications using autologous adipose cells. We are presently investigating the use of adipose cells in the treatment of critical limb ischemia and rheumatoid arthritis
Biotechnology Product Candidates
We are focused on the discovery, development and, subject to regulatory approval, commercialization of autologous cell therapies for the treatment of chronic and acute heart damage. In our pipeline, we have multiple product Candidates for the treatment of heart damage, including MyoCell, Myocell SDF-1 and AdipoCell. MyoCell and MyoCell SDF-1 are clinical muscle-derived cell therapies designed to populate regions of scar tissue within a patient’s heart with new living cells for the purpose of improving cardiac function in chronic heart failure patients. MyoCell SDF-1 is intended to be an improvement to MyoCell. MyoCell SDF-1 is similar to MyoCell except that the myoblast cells to be injected for use in MyoCell SDF-1 will be modified prior to injection by an adenovirus vector or non-viral vector so that they will release extra quantities of the SDF-1 protein, which expresses angiogenic factors. AdipoCell is a patient-derived cell therapy proposed for the treatment of acute myocardial infarction, chronic heart ischemia, and lower limb ischemia. We hope to demonstrate that these product candidates are safe and effective complements to existing therapies for chronic and acute heart damage.
Our most recent completed clinical trials of MyoCell are the SEISMIC Trial, a 40-patient, randomized, multicenter, controlled, Phase II-a study conducted in Europe and the MYOHEART Trial, a 20-patient, multicenter, Phase I dose-escalation trial conducted in the United States.
We were approved by the FDA, to proceed with a 330-patient, multicenter Phase II/III trial of MyoCell in North America and Europe, or the MARVEL Trial. We completed the MyoCell implantation procedure on the first patient in the MARVEL Trial on October 24, 2007. Thus far, 20 patients, including 6 control patients, have been treated. Initial results for the 20 patients were released at the Heart Failure Society of American meeting in September, 2009, showing a significant (35%) improvement in the 6 minute walk for those patients who were treated, and no improvement for those who received a placebo. On the basis of these results, we have applied for and received approval from the FDA to reduce the number of additional patients in the trial to 134, for a total of 154 patients. The SEISMIC, MYOHEART and MARVEL Trials have been designed to test the safety and efficacy of MyoCell in treating patients with severe, chronic damage to the heart. Upon regulatory approval of MyoCell, we intend to generate revenue in the United States from the sale of MyoCell cell-culturing services for treatment of patients by qualified physicians.
We received approval from the FDA in July of 2009 to conduct a Phase I safety study on 15 patients of a combined therapy (Myocell with SDF-1), which we believe was the first approval of a study combining gene and cell therapies. We initially commenced work on this study, called the REGEN Trial, during the first quarter of 2010. We suspended activity on the trial in 2010 while seeking additional funding necessary to conduct the trial. We are seeking to secure sufficient funds to reinitiate enrollment in the MARVEL and REGEN trials. If we successfully secure such funds, we intend to re-engage a contract research organization, or CRO, investigators and certain suppliers to advance such trials. We have initiated and enrolled our first patient in the MIRROR trial in 2013. The trial is very similar to the MARVEL trial but focuses on sites outside the US. We will continue enrollment in the MIRROR trial once we have secured sufficient funds.
We have completed the Phase 1 Angel Trial for AdipoCell (adipose derived stem cells). Five patients were enrolled and treated in the second quarter of 2013. At the twelve (12) month time point, patients demonstrated a statistically significant average improvement in ejection fraction (EF) by echocardiogram.
At the three (3) month time point, 100% of the patients demonstrated either improvement or stayed the same. After three (3) months, patients showed an average absolute improvement of 3 percentage points in EF. The patients continued to improve from 3 months to 6 months with a statistically significant average absolute improvement of 10 percentage points (p=0.01) and at the 12 month follow up patients showed this same level of improvement (p=0.01).
We have also initiated several Institutional Review Board studies in 2013 using adipose derived stem cells for various indications including dry macular degeneration, degenerative disc disease, erectile dysfunction and chronic obstructive pulmonary disease. In the second quarter of 2014, we announced the treatment of a patient in Honduras with congestive heart failure using AdipoCell and MyoCell. This was the first patient treated in the world using a combination of stem cells. We have begun two clinical trials in India. The first cardiac patient has successfully been enrolled and treated in India using AdipoCell or adipose derived stem cells. The second trial will involve the combination of AdipoCellTM and MyoCell® or muscle derived stem cells for congestive heart failure patients. These trials are active and ongoing.
MyoCath Product Candidate
The MyoCath is a deflecting tip needle injection catheter that has a larger (25 gauge) needle to allow for better flow rates and less leakage than systems that are 27 gauge. This larger needle allows for thicker compositions to be injected, which helps with cell retention in the heart. Also, the MyoCath needle has more fluoroscopic brightness than the normally used nitinol needle, enabling superior visualization during the procedure. Seeing the needle well during injections enables the physician who is operating the catheter to pinpoint targeted areas more precisely. The MyoCath is used to inject cells into cardiac tissue in therapeutic procedures to treat chronic heart ischemia and congestive heart failure. Investigators in our MARVEL Trial may use either our MyoCath catheters or Biosense Webster’s (a Johnson & Johnson company) NOGA® Cardiac Navigation System along with the MyoStar™ injection catheter for the delivery of MyoCell to patients enrolled in the trial. We are currently producing Myocath catheters with a contract manufacturer on an as needed basis.
We conduct operations in one business segment. We may organize our business into more discrete business units when and if we generate significant revenue from the sale of our product candidates. Our revenue since inception has been generated inside and outside the United States, and the majority of our long-lived assets are located in the United States.
Securities Purchase Agreements
On October 1, 2015, we entered into a securities purchase agreement dated as of the Closing Date (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (“Magna”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, Magna shall purchase from the Company on the Closing Date a senior convertible note with an initial principal amount of $160,000 (the “Convertible Note”) for a purchase price of $100,000 (a 37.5% original issue discount). Pursuant to the Purchase Agreement, on the Closing Date, the Company issued the Convertible Note to Magna.
The Convertible Note matures on August 1, 2016 and, in addition to the 37.5% original issue discount, accrues interest at the rate of 12% per annum. The Convertible Note is convertible at any time, in whole or in part, at Magna’s option into shares of the Company’s common stock, par value $0.01 per share (the “Common Stock”), at a fixed conversion price of $0.72 per share (subject to adjustment). This conversion price represents a discount of approximately 55% from the lowest trading price of the Common Stock during the five trading days prior to the Closing Date. If the Company has not properly filed a registration statement with the SEC on or prior to the date that is 70 calendar days after the Closing Date covering the resale by Magna of all of the shares of Common Stock issued or issuable upon conversion of the Convertible Note, then, from and after such date, the Convertible Note will be convertible at any time, in whole or in part, at Magna’s option into Common Stock at a conversion price equal to a 55% discount from the lowest trading price of the Common Stock during the five consecutive trading days ending and including the trading day immediately preceding the applicable date of conversion. At no time will Magna be entitled to convert any portion of the Convertible Note to the extent that after such conversion, Magna (together with its affiliates) would beneficially own more than 4.99% of the outstanding shares of Common Stock as of such date. The Convertible Note includes “full ratchet” and standard anti-dilution protection.
The Convertible Note includes customary event of default provisions, and provides for a default interest rate of 18%. Upon the occurrence of an event of default, Magna may require the Company to pay in cash the “Event of Default Redemption Price” which is defined in the Convertible Note to mean the greater of (i) the product of (A) the amount to be redeemed multiplied by (B) 140% (or 100% if an insolvency related event of default) and (ii) the product of (X) the conversion price in effect at that time multiplied by (Y) the product of (1) 140% (or 100% if an insolvency related event of default) multiplied by (2) the greatest closing sale price of the Common Stock on any trading day during the period commencing on the date immediately preceding such event of default and ending on the date the Company makes the entire payment required to be made under this provision.
The Company has the right at any time to redeem all, but not less than all, of the total outstanding amount then remaining under the Convertible Note in cash at a price equal to 140% of the total amount of the Convertible Note then outstanding.
The Purchase Agreement contains customary representations, warranties and covenants by, among and for the benefit of the parties. The Company also agreed to pay up to $10,000 of reasonable attorneys’ fees and expenses incurred by Magna in connection with the transaction. The Purchase Agreement also provides for indemnification of Magna and its affiliates in the event that Magna incurs losses, liabilities, obligations, claims, contingencies, damages, costs and expenses related to a breach by the Company of any of its representations, warranties or covenants under the Purchase Agreement.
On December 3, 2015, the Convertible Note was amended and restated to reflect a final principal amount of $110,000.
The issuance of the Convertible Note to Magna under the Note Purchase Agreement was exempt from the registration requirements of the Securities Act pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a) (2) of the Securities Act and Rule 506 of Regulation D promulgated under the Securities Act.
On December 3, 2015, we entered into a securities purchase agreement dated as of the Closing Date (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (“Magna”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, Magna shall purchase from the Company on the Closing Date a senior convertible note with an initial principal amount of $262,500 (the “Convertible Note”). Pursuant to the Purchase Agreement, on the Closing Date, the Company issued the Convertible Note to Magna.
The Convertible Note matures on December 3, 2016 and accrues interest at the rate of 12% per annum. The Convertible Note is convertible at any time, in whole or in part, at Magna’s option into shares of the Company’s common stock, par value $0.01 per share (the “Common Stock”), at a fixed conversion price of the lesser of (A) the product of (x) the lowest sale price of the Common Stock during the five (5) consecutive Trading Days ending and including the Trading Day immediately preceding the applicable Conversion Date and (y) sixty percent (60%), and (B) $0.70 (as adjusted for stock splits, stock dividends, stock combinations or other similar transactions). At no time will Magna be entitled to convert any portion of the Convertible Note to the extent that after such conversion, Magna (together with its affiliates) would beneficially own more than 4.99% of the outstanding shares of Common Stock as of such date. The Convertible Note includes “full ratchet” and standard anti-dilution protection.
The Convertible Note includes customary event of default provisions, and provides for a default interest rate of 18%. Upon the occurrence of an event of default, Magna may require the Company to pay in cash the “Event of Default Redemption Price” which is defined in the Convertible Note to mean the greater of (i) the product of (A) the amount to be redeemed multiplied by (B) 140% (or 100% if an insolvency related event of default) and (ii) the product of (X) the conversion price in effect at that time multiplied by (Y) the product of (1) 140% (or 100% if an insolvency related event of default) multiplied by (2) the greatest closing sale price of the Common Stock on any trading day during the period commencing on the date immediately preceding such event of default and ending on the date the Company makes the entire payment required to be made under this provision.
The Company has the right at any time to redeem all, but not less than all, of the total outstanding amount then remaining under the Convertible Note in cash at a price equal to 140% of the total amount of the Convertible Note then outstanding. In the prior registration statement (SEC File No. 200457), we registered 143,813 (post-split) shares of common stock underlying commitment shares, a draw down equity line, and convertible promissory notes. The number of shares and stock prices listed below reflect post-split calculations. Magna Equities II, LLC was issued 9,109 and 2,891 shares as of October 27, 2014 and December 23, 2014 respectively, representing 1.66% and 0.50% of the then current issued and outstanding shares of our common stock respectively. Our common stock price as of October 27, 2014 and December 23, 2014 was $20.50 and $12.80 respectively. Pursuant to the draw down equity line, we requested drawdowns from January 13, 2015 through August 27, 2015 (which includes issuing true up shares as per the terms and conditions of the Purchase Agreement of the equity line). During this period, we issued an aggregate of 968,514 shares (post-split) pursuant to ten draw down requests initiated solely by us. The percentage dilution for any one draw down or true up represented from 0.35% of the then issued and outstanding shares of common stock to 2.12% of the then issued and outstanding shares of common stock. The price of our common stock was $8.40 as of January 13, 2015 (the date of the initial draw down) to $4.28 as of August 27, 2015 (the date of the final true up issuance). Magna Equities II, LLC converted a portion of its convertible promissory notes on February 2, 2015, receiving 7,246 (post-split) shares of common stock (representing 1.19% of the then issued and outstanding shares of common stock). The remainder of the convertible promissory notes were converted to shares of common stock from August 27, 2015 through November 6, 2015. The percentage dilution for any one conversion during this period represented from 0.82% of the then issued and outstanding shares of common stock to 2.85% of the then issued and outstanding shares of common stock. The price of our common stock during this period was $4.00 as of August 27, 2015 to $1.40 as of October 27, 2015, the second to last conversion date) ($5.45 as of November 6, 2015 (the date of the final conversion). Consequently, during the period covered by the prior registration period, our stock price decreased from $20.50 to $1.40 ($5.45 as of the last conversion). During this time, our number of issued and outstanding shares increased from 548,186 to 1,615,772 shares, partly as a result of the issuances to Magna Equities II, LLC. Since Magna Equities II, LLC may convert all or part of its convertible promissory notes to common stock as registered hereunder, further dilution may occur similar to the prior registration. This further dilution may result in your additional dilution of the existing ownership interests of our common stockholders and may adversely affect the stock price of the shares of common stock.
Results of Operations Overview
Revenues
The Company’s primary source of revenue are from the sale of test kits and equipment, training services, patient treatments and laboratory services, and cell banking. Our revenue may vary substantially from quarter to quarter and from year to year. We believe that period-to-period comparisons of our results of operations are not meaningful and should not be relied upon as indicative of our future performance. We do not expect to generate substantial revenues until we obtain regulatory approval for and commercialize our product candidates, which we do not expect to occur before 2017
We recognized revenues of $2,191,177 in 2015 compared to revenues of $2,055,831 in 2014. Our revenue in 2015 was generated from the sale of test kits and equipment, training services, patient treatments and laboratory services, and cell banking. Our revenues for 2014 were generated from the sale of MyoCath Catheters, test kits and equipment, training services, patient treatments and laboratory services, and cell banking.
Cost of Sales
Cost of sales consists of the costs associated with the production of MyoCath and test kits, product costs, labor for production and training and lab and banking costs.
Cost of sales was $972,957 in the year ended December 31, 2015 compared to $844,941 in the year ended December 31, 2014. The increase is primarily due to costs of supplies.
Research and Development
Our research and development expenses consist of costs incurred in identifying, developing and testing our product candidates. These expenses consist primarily of costs related to our clinical trials, the acquisition of intellectual property licenses and preclinical studies. We expense research and development costs as incurred.
Clinical trial expenses include costs related to the culture and preparation of cells in connection with our clinical trials, costs of contract research, costs of clinical trial facilities, costs of delivery systems, salaries and related expenses for clinical personnel and insurance costs. Preclinical study expenses include costs of contract research, salaries and related expenses for personnel, costs of development biopsies, costs of delivery systems and costs of lab supplies.
Clinical trials and preclinical studies are time-consuming and expensive. Our expenditures on current and future preclinical and clinical development programs are subject to many uncertainties. We generally test our products in several preclinical studies and then conduct clinical trials for those product candidates that we determine to be the most promising. As we obtain results from clinical trials, we may elect to discontinue or delay trials for some product candidates in order to focus our resources on more promising product candidates. Completion of clinical trials may take several years or more, but the length of time generally varies substantially according to the type, size of trial and intended use of the product candidate.
Marketing, General and Administrative
Our marketing, general and administrative expenses primarily consist of the costs associated with our general management and clinical marketing and trade programs, including, but not limited to, salaries and related expenses for executive, administrative and marketing personnel, rent, insurance, legal and accounting fees, consulting fees, travel and entertainment expenses, conference costs and other clinical marketing and trade program expenses.
Stock-Based Compensation
Stock-based compensation which is included in the Marketing, General and Administrative above, reflects our recognition as an expense of the value of stock options and other equity instruments issued to our employees and non-employees over the vesting period of the options and other equity instruments. We have granted to our employees options to purchase shares of common stock at exercise prices as determined by our Board of Directors, with input from management.
In valuing our common stock, our Board of Directors considered a number of factors, including, but not limited to:
|
·
|
our financial position and historical financial performance;
|
|
|
·
|
the illiquidity of our capital stock;
|
|
|
·
|
arm’s length sales of our common stock;
|
|
|
·
|
the development status of our product candidates;
|
|
|
·
|
the business risks we face;
|
|
|
·
|
vesting restrictions imposed upon the equity awards;
|
|
|
·
|
an evaluation and benchmark of our competitors; and
|
|
|
·
|
the prospects of a liquidity event.
|
In April 1, 2013, the Board of Directors approved the establishment of the U.S. Stem Cell 2013 Omnibus Equity Compensation Plan, or the “2013 Omnibus Plan” (subsequently approved by the shareholders). The 2013 Omnibus Plan reserves up to hundred million shares of common stock for issuance, as amended. The 2013 Omnibus Plan was approved by the majority of shareholders at our annual meeting, dated February 2, 2015.
A summary of options at December 31, 2015 and activity during the year then ended is presented below:
|
Shares
|
Weighted-
Average
Exercise Price
|
Weighted-
Average
Remaining
Contractual
Term (in years)
|
||||||||||
|
Options outstanding at January 1, 2014
|
23,921
|
$
|
150.00
|
9.0
|
||||||||
|
Granted
|
43,148
|
$
|
23.00
|
10.0
|
||||||||
|
Exercised
|
—
|
$
|
||||||||||
|
Forfeited/Expired
|
(136
|
)
|
$
|
5,200.00
|
||||||||
|
Options outstanding at December 31, 2014
|
66,933
|
$
|
56.00
|
8.9
|
||||||||
|
Granted
|
489,116
|
$
|
1.98
|
10.0
|
||||||||
|
Exercised
|
—
|
|||||||||||
|
Forfeited/Expired
|
(229
|
)
|
$
|
5,103.28
|
||||||||
|
Options outstanding at December 31, 2015
|
555,820
|
$
|
6.43
|
9.6
|
||||||||
|
Options exercisable at December 31, 2015
|
511,660
|
$
|
5.60
|
9.7
|
||||||||
|
Available for grant at December 31, 2015
|
7,383,070
|
|||||||||||
The following information applies to options outstanding and exercisable at December 31, 2015:
|
Options Outstanding
|
Options Exercisable
|
||||||||||||||||||
|
Shares
|
Weighted-
Average
Remaining
Contractual
Term
|
Weighted-
Average
Exercise
Price
|
Shares
|
Weighted-
Average
Exercise
Price
|
|||||||||||||||
|
$
|
0.00 – $20.00
|
534,706
|
9.7
|
$
|
3.32
|
501,682
|
$
|
2.72
|
|||||||||||
|
$
|
20.01 – $30.00
|
19,849
|
8.5
|
$
|
26.96
|
8,713
|
$
|
26.94
|
|||||||||||
|
$
|
30.01 – $100.00
|
300
|
5.7
|
$
|
70.00
|
300
|
$
|
70.00
|
|||||||||||
|
$
|
>100.00
|
965
|
4.0
|
$
|
1,288.36
|
965
|
$
|
1,288.36
|
|||||||||||
|
555,820
|
9.6
|
$
|
6.43
|
511,660
|
$
|
5.60
|
|||||||||||||
On February 24, 2014, we issued an aggregate 15,000 options to purchase our common stock at $19.00 per share to officers, vesting at 25% immediately and the remainder over approximately 42 months, exercisable over 10 years. The aggregate fair value of $282,597, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 163.63% and Risk free rate: 2.75%.
On February 24, 2014, we issued an aggregate 4,800 options to purchase our common stock at $19.00 per share to officers, vesting immediately and exercisable over 10 years. The aggregate fair value of $90,431, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 163.63% and Risk free rate: 2.75%.
On May 12, 2014, we issued an aggregate 4,848 options to purchase our common stock at $27.20 per share to officers and employees, vesting over four years at anniversary and exercisable over 10 years. The aggregate fair value of $130,135, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 161.36% and Risk free rate: 2.66%.
On August 1, 2014, we issued an aggregate 15,000 options to purchase our common stock at $26.94 per share to officers and employees, vesting at 25% immediately and 75% over three years at anniversary and exercisable over 10 years. The aggregate fair value of $391,798, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 168.62% and Risk free rate: 2.52%.
On November 3, 2014, we issued an aggregate 700 options to purchase our common stock at $19.32 per share to officers and employees, vesting at 25% each year over four years at anniversary and exercisable over 10 years. The aggregate fair value of $12,739, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 2.36%.
On November 3, 2014, we issued an aggregate 2,800 options to purchase our common stock at $19.32 per share to board of directors, vesting immediately and exercisable over 10 years. The aggregate fair value of $54,957, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 2.36%.
On February 2, 2015, we issued an aggregate 7,000 options to purchase our common stock at $11.16 per share to members of the Board of Directors, vesting immediately and exercisable over 10 years. The aggregate fair value of $121,735, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 142.65% and Risk free rate: 1.68%.
On August 24, 2015, we issued 100 options to purchase our common stock at $5.31 per share to a consultant, vesting immediately and exercisable over 4 years. The aggregate fair value of $347, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 129.01% and Risk free rate: 1.39%.
On November 2, 2015, we issued an aggregate of 467,017 options to purchase our common stock at $1.713 per share to two officers, vesting immediately and exercisable over 10 years. The aggregate fair value of $738,405 determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 160.66% and Risk free rate: 1.57%.
On November 2, 2015, we issued an aggregate of 15,000 options to purchase our common stock at $6.24 per share to two officers, vesting over four years on each anniversary and exercisable over 10 years. The aggregate fair value of $23,512, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 160.66% and Risk free rate: 1.93%.
The fair value of all options vesting during the year ended December 31, 2015 and 2014 of $1,156,435 and $537,606, respectively, was charged to current period operations.
Warrants
A summary of common stock purchase warrants at December 31, 2015 and activity during the year then ended is presented below:
|
Shares
|
Weighted-
Average
Exercise
Price
|
Weighted-
Average
Remaining
Contractual
Term (in years)
|
||||||||||
|
Outstanding at January 1, 2014
|
118,134
|
$
|
220.00
|
6.3
|
||||||||
|
Issued
|
57,582
|
$
|
20.00
|
8.2
|
||||||||
|
Exercised
|
(11,918
|
)
|
$
|
10.00
|
||||||||
|
Expired
|
(13,178
|
)
|
$
|
80.00
|
||||||||
|
Outstanding at December 31, 2014
|
150,620
|
$
|
170.00
|
6.6
|
||||||||
|
Issued
|
2,072
|
$
|
19.98
|
8.18
|
||||||||
|
Exercised
|
$
|
|||||||||||
|
Expired
|
(13,325
|
)
|
$
|
24.00
|
||||||||
|
Outstanding at December 31, 2015
|
139,367
|
$
|
182.90
|
6.3
|
||||||||
|
Exercisable at December 31, 2015
|
133,822
|
$
|
99.98
|
6.3
|
||||||||
The following information applies to common stock purchase warrants outstanding and exercisable at December 31, 2015:
|
Warrants Outstanding
|
Warrants Exercisable
|
|||||||||||||||||||||
|
Shares
|
Weighted-
Average
Remaining
Contractual
Term
|
Weighted-
Average
Exercise
Price
|
Shares
|
Weighted-
Average
Exercise
Price
|
||||||||||||||||||
|
$
|
0.01 – $20.00
|
94,108
|
6.8
|
$
|
15.54
|
94,108
|
$
|
15.54
|
||||||||||||||
|
$
|
20.01 – $30.00
|
29,743
|
6.1
|
$
|
24.52
|
27,743
|
$
|
24.72
|
||||||||||||||
|
$
|
30.01 – $40.00
|
628
|
1.6
|
$
|
40.00
|
628
|
$
|
40.00
|
||||||||||||||
|
$
|
40.01 - $50.00
|
6,253
|
3.1
|
$ |
48.36
|
4,253
|
$ |
48.49
|
||||||||||||||
|
$
|
50.01 – $60.00
|
543
|
1.9
|
$
|
60.00
|
543
|
$
|
60.00
|
||||||||||||||
|
$
|
>60.00
|
8,092
|
4.2
|
$
|
2,823.67
|
6,547
|
$
|
1,675.28
|
||||||||||||||
|
139,367
|
6.3
|
$
|
182.26
|
133,822
|
$
|
99.98
|
||||||||||||||||
In conjunction with the authorized issuance of common stock, we issued 41,582 common stock purchase warrants during the year ended December 31, 2014.
During the year ended December 31, 2014, we issued an aggregate of 8,000 warrants in connection with a revenue share venture agreement dated March 10, 2014. The warrants are exercisable at $21.70 for four years vesting from June 8, 2014 through March 10, 2016 (term of agreement). During the year ended December 31, 2014, we charged $103,238 to current period operations.
During the year ended December 31, 2014, we issued an aggregate of 4,000 warrants in connection with use of certain intellectual property. The warrants are exercisable at $48.10 for four years vesting from July 6, 2014 through April 6, 2017. During the year ended December 31, 2014, we charged $32,878 to current period operations for the vesting portion.
During the year ended December 31, 2014, we issued an aggregate of 4,000 warrants in connection with the termination of a revenue share agreement. The warrants are exercisable at $15.70 for four years vesting May 27, 2015. During the year ended December 31, 2014, we charged $54,146 to current period operations.
On November 3, 2014, we extended an aggregate of 13,064 expiring warrants previously issued to our board of directors for additional three years. The aggregate change in fair value of $148,517, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 0.96% was charged to current period operations.
In conjunction with the authorized issuance of common stock, we issued 1,444 common stock purchase warrants during the year ended December 31, 2015. The warrants are exercisable at an exercise price of approximately $11.27 per share for ten years.
During the year ended December 31, 2015, we issued 628 warrants in connection with the settlement of debt. The warrants are exercisable at $40.00 for three years. The fair value of $14,886, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 164.06% and Risk free rate: 0.87%. In connection with the settlement, we recorded a gain of $10,221 in settlement of debt to current period operations.
Interest Expense
Interest expense during the year ended December 31, 2015 was $1,723,298 compared to $1,639,571 for the year ended December 31, 2014. Interest expense primarily consists of interest incurred on the principal amount of the Northstar loan, our former Bank of America loan, the Seaside National Bank loan, accrued fees and interest payable to the Guarantors, imputed interest on non-interest bearing debt, the amortization of debt discounts and non-cash interest incurred relating to our issued convertible notes payable. The debt discounts amortization and non-cash interest incurred during the year ended December 31, 2015 and 2014 was $1,396,989 and $1,252,680, respectively. On January 11, 2016, the Company renewed the loan with Seaside National Bank and Trust extend the maturity date to January 11, 2018, all other terms and conditions remain unchanged
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. While our critical accounting policies are described in Note 1 to our financial statements appearing elsewhere in this report, we believe the following policies are important to understanding and evaluating our reported financial results:
Revenue Recognition
At the time of each transaction, management assesses whether the fee associated with the transaction is fixed or determinable and whether or not collection is reasonably assured. The assessment of whether the fee is fixed or determinable is based upon the payment terms of the transaction. Collectability is assessed based on a number of factors, including past transaction history with the client and the creditworthiness of the client.
The Company’s primary source of revenue are from the sale of test kits and equipment, training services, patient treatments and laboratory services, and cell banking.
Revenues for test kits and equipment sold are not recorded until test kits and equipment are delivered. During 2014, the Company had revenue sharing arrangements for the sale of goods whereby the Company was the primary obligor, set pricing with the customers and bared all associated credit risks with the customers. Sales under revenue share arrangements are recorded as gross sales and any portion shared with third parties under such arrangements are classified as selling expense due to the nature of the marketing activities performed by the third party. All revenue sharing arrangements were terminated in November 2014. Revenues from trainings are recognized when the training occurs. Any cash received as a deposit for trainings are recorded by the company as a liability.
Patient treatments and laboratory services revenue are recognized when those services have been completed or satisfied.
Revenues for cell banking sales are accounted for as Multiple-Element Arrangements under ASC 605-10 which incorporates Accounting Standards Codification subtopic 605-25, Multiple-Element Arrangements (“ASC 605-25”). ASC 605-25 addresses accounting for arrangements that may involve the delivery or performance of multiple products, services and/or rights to use assets. Because the Company sells its services separately, on more than a limited basis and at a price within a narrow range, the Company was able to allocate revenue based on vendor-specific objective evidence of fair value (VSOE).
Stock-based compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. For employees and directors, the fair value of the award is measured on the grant date and for non-employees, the fair value of the award is generally re-measured on vesting dates and interim financial reporting dates until the service period is complete. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period. Stock-based compensation expense is recorded by the Company in the same expense classifications in the statements of operations, as if such amounts were paid in cash.
Derivative instrument liability
We account for derivative instruments in accordance with ASC 815, which establishes accounting and reporting standards for derivative instruments and hedging activities, including certain derivative instruments embedded in other financial instruments or contracts and requires recognition of all derivatives on the balance sheet at fair value, regardless of hedging relationship designation. Accounting for changes in fair value of the derivative instruments depends on whether the derivatives qualify as hedge relationships and the types of relationships designated are based on the exposures hedged. At December 31, 2015 and 2014, we did not have any derivative instruments that were designated as hedges.
Income taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss carry forwards that are available to be carried forward to future years for tax purposes. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. When it is not considered to be more likely than not that a deferred tax asset will be realized, a valuation allowance is provided for the excess. Although we have significant loss carry forwards available to reduce future income for tax purposes, no amount has been reflected on the balance sheet for deferred income taxes as any deferred tax asset has been fully offset by a valuation allowance.
Use of Estimates
The preparation of the financial statements in conformity with generally accepted accounting principles requires management to make certain estimates and assumptions, where applicable, that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements, as well as the reported amounts of revenues and expenses during the reporting period. While actual results could differ from those estimates, management does not expect such variances, if any, to have a material effect on the financial statements.
Research and Development Costs
The Company accounts for research and development costs in accordance with Accounting Standards Codification subtopic 730-10, Research and Development (“ASC 730-10”). Under ASC 730-10, all research and development costs must be charged to expense as incurred. Accordingly, internal research and development costs are expensed as incurred. Third-party research and development costs are expensed when the contracted work has been performed or as milestone results have been achieved as defined under the applicable agreement. Company-sponsored research and development costs related to both present and future products are expensed in the period incurred.
Depreciation
Depreciation is computed using the straight-line method over the assets’ expected useful lives.
Cash and Cash Equivalents
Cash and cash equivalents include cash on hand, deposits in banks with maturities of three months or less, and all highly liquid investments which are unrestricted as to withdrawal or use, and which have original maturities of three months or less.
Options and warrants issued
We allocate the proceeds received from equity financing and the attached options and warrants issued, based on their relative fair values, at the time of issuance. The amount allocated to the options and warrants is recorded as additional paid in capital.
Related Parties
For the purposes of these financial statements, parties are considered to be related if one party has the ability, directly or indirectly, to control the party or exercise significant influence over the party in making financial and operating decisions, or vice versa, or where our company and the party are subject to common control or common significant influence. Related parties may be individuals or other entities.
Results of Operations
We are a research and development stage company and our MyoCell product candidate has not received regulatory approval or generated any material revenues and is not expected to until 2016, if ever. We have generated substantial net losses and negative cash flow from operations since inception and anticipate incurring significant net losses and negative cash flows from operations for the foreseeable future as we continue clinical trials, undertake new clinical trials, apply for regulatory approvals, make capital expenditures, add information systems and personnel, make payments pursuant to our license agreements upon our achievement of certain milestones, continue development of additional product candidates using our technology, establish sales and marketing capabilities and incur the additional cost of operating as a public company.
Comparison of Years Ended December 31, 2015 and December 31, 2014
Revenues
We recognized revenues of $2,191,177 in 2015, revenues generated from the sale of, kits and equipment, services, MyoCath Catheters, AdipoCell, and laboratory services. In 2014, we recognized revenues of $2,055,831, revenues generated from the sales of kits, MyoCath Catheters and laboratory services.
Cost of Sales
Cost of sales was $972,957 in 2015 and $844,941 in 2014. The increase is primarily due to compensation paid in 2015 and increased lab supply costs
Research and Development
Research and development expenses were $10,000 in 2015, a decrease of $56,420 from research and development expenses of $66,420 in 2014. The decrease was primarily attributable to a decrease in the amount of available funds.
The timing and amount of our planned research and development expenditures is dependent on our ability to obtain additional financing.
Marketing, General and Administrative
Marketing, general and administrative expenses were $3,808,628 in 2015, a decrease of $860,804 from marketing, general and administrative expenses of $4,669,432 in 2014. The decrease in marketing, general and administrative expenses is attributable, in part, to reductions in legal fees, salaries and insurance expenses.
Interest Expense
Interest expense was $1,723,298 in 2015 compared to interest expense of $1,639,571 in 2014. Non cash interest comprised of amortization of debt discounts , warrants issued in connection with debt and non-cash interest totaled $1,396,989 in 2015 as compared to $1,252,680 in 2014.
Gain on settlement of debt
In 2015, we issued an amended and restated promissory note of $1,697,762 in settlement of settled $1,585,862 in guarantor fees, accrued interest of $373,469 and an outstanding note payable of $1,500,000 for a net gain of $1,960,082. In addition, we incurred a gain of $660,151 in open accounts payable.
During the year ended December 31, 2014, we settled an outstanding note payable and certain accounts payable by issuances of common stock. As such we realized a net $2,498,676 gain on extinguishment of debt during the year ended December 31, 2014.
Gain on change in fair value of derivative liabilities.
As of December 31, 2015 and 2014, we issued convertible notes and common stock purchase warrants with anti-dilutive provisions that had the possibility of exceeding our common shares authorized when considering the number of possible shares that may be issuable to satisfy settlement provisions of these agreements after consideration of all existing instruments that could be settled in shares. As such, we are required to determine the fair value of this derivative and mark to market each reporting period. For the year ended December 31, 2015, we incurred a $15,095 gain on change in fair value of our derivative liabilities compared to a gain of $425,953 the same period last year.
Inflation
Our opinion is that inflation has not had, and is not expected to have, a material effect on our operations.
Climate Change
Our opinion is that neither climate change, nor governmental regulations related to climate change, have had, or are expected to have, any material effect on our operations.
Liquidity and Capital Resources
In 2015, we continued to finance our considerable operational cash needs with cash generated from financing activities.
Operating Activities
Net cash used in operating activities was $844,690 in 2015 as compared to $1,108,647 of cash used in 2014. Our use of cash for operations in 2015 reflected a net loss generated during the period of $1,641,880, adjusted for non-cash items such as stock-based compensation of $1,152,195, amortization of debt discounts associated with our convertible notes of $818,404, non-cash interest payments of $528,915, common stock issued in settlement of ligation of $59,850, bad debt expense of $38,599 and depreciation expense of $5,326, net with gain on settlement of debt of $2,620,233 and gain on change in fair value of derivative liabilities of $15,095 . A net decrease in operating assets of $8,795 and a net increase in operating liabilities of $858,976 contributed to our use of operating cash in 2015.
Investing Activities
Net cash used in investing activities was $234,031 in 2015 compared to $57,835 in 2014. The primary increase is due to our re-purchase of our common stock of $221,996 as compared to $-0- in 2014.
Financing Activities
Net cash provided by financing activities was $1,100,419 in 2015 as compared to $1,156,929 in 2014. In 2015, we sold, in a private placement and put agreements, shares of common stock and warrants for aggregate net cash proceeds of approximately $582,808. In addition, we received an aggregate of $13,606 related party loans and advances and $770,052 from issuance of notes payable, net of repayments of $266,047.
Existing Capital Resources and Future Capital Requirements
Our MyoCell product candidate has not received regulatory approval or generated any material revenues. We do not expect to generate any material revenues or cash from sales of our MyoCell product candidate until commercialization of MyoCell, if ever. We have generated substantial net losses and negative cash flow from operations since inception and anticipate incurring significant net losses and negative cash flows from operations for the foreseeable future. Historically, we have relied on proceeds from the sale of our common stock and our incurrence of debt to provide the funds necessary to conduct our research and development activities and to meet our other cash needs.
At December 31, 2015, we had cash and cash equivalents totaling $58,372; our working capital deficit as of such date was $5,588,949. Our independent registered public accounting firm has issued its report dated March 8, 2016 in connection with the audit of our financial statements as of December 31, 2015 that included an explanatory paragraph describing the existence of conditions that raise substantial doubt about our ability to continue as a going concern.
As of December 31, 2015, we had $5,076,555 in outstanding debt.
On October 1, 2015, we entered into a securities purchase agreement dated as of the Closing Date (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (“Magna”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, Magna shall purchase from us t on the Closing Date a senior convertible note with an initial principal amount of $160,000 (the “Convertible Note”) for a purchase price of $100,000 (a 37.5% original issue discount). Pursuant to the Purchase Agreement, on the Closing Date, we issued the Convertible Note to Magna.
The Convertible Note matures on August 1, 2016 and, in addition to the 37.5% original issue discount, accrues interest at the rate of 12% per annum. The Convertible Note is convertible at any time, in whole or in part, at Magna’s option into shares of our common stock, par value $0.01 per share (the “Common Stock”), at a fixed conversion price of $0.72 per share (subject to adjustment). This conversion price represents a discount of approximately 55% from the lowest trading price of the Common Stock during the five trading days prior to the Closing Date. At no time will Magna be entitled to convert any portion of the Convertible Note to the extent that after such conversion, Magna (together with its affiliates) would beneficially own more than 4.99% of the outstanding shares of Common Stock as of such date. The Convertible Note includes “full ratchet” and standard anti-dilution protection.
The Convertible Note includes customary event of default provisions, and provides for a default interest rate of 18%. Upon the occurrence of an event of default, Magna may require us to pay in cash the “Event of Default Redemption Price” which is defined in the Convertible Note to mean the greater of (i) the product of (A) the amount to be redeemed multiplied by (B) 140% (or 100% if an insolvency related event of default) and (ii) the product of (X) the conversion price in effect at that time multiplied by (Y) the product of (1) 140% (or 100% if an insolvency related event of default) multiplied by (2) the greatest closing sale price of the Common Stock on any trading day during the period commencing on the date immediately preceding such event of default and ending on the date we make the entire payment required to be made under this provision.
We have the right at any time to redeem all, but not less than all, of the total outstanding amount then remaining under the Convertible Note in cash at a price equal to 140% of the total amount of the Convertible Note then outstanding.
The Purchase Agreement contains customary representations, warranties and covenants by, among and for the benefit of the parties. We also agreed to pay up to $10,000 of reasonable attorneys’ fees and expenses incurred by Magna in connection with the transaction. The Purchase Agreement also provides for indemnification of Magna and its affiliates in the event that Magna incurs losses, liabilities, obligations, claims, contingencies, damages, costs and expenses related to a breach by our company of any of its representations, warranties or covenants under the Purchase Agreement.
On December 3, 2015, the Convertible Note was amended and restated to reflect a final principal amount of $110,000.
The issuance of the Convertible Note to Magna under the Note Purchase Agreement was exempt from the registration requirements of the Securities Act pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a) (2) of the Securities Act and Rule 506 of Regulation D promulgated under the Securities Act.
On December 3, 2015, we entered into a securities purchase agreement dated as of the Closing Date (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (“Magna”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, Magna shall purchase from our company on the Closing Date a senior convertible note with an initial principal amount of $262,500 (the “Convertible Note”). Pursuant to the Purchase Agreement, on the Closing Date, we issued the Convertible Note to Magna.
Magna has completed the resale of securities registered for sale under the prior registration statement. On November 6, 2015, Magna converted the last portion of the promissory note described above and received 18,013 (post-split) shares of common stock (which was issued in electronic form). Magna has represented that these shares, representing all their holdings at the time, was sold into the public market on or before November 19, 2015.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
Recent Accounting Pronouncements
Refer to Note 1. Organization and Summary of Significant Accounting Policies in the notes to our financial statements for a discussion of recent accounting pronouncements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not required for smaller reporting companies.
Item 8. Financial Statements and Supplementary Data
Our Financial Statements begin on page F-1 of this Annual Report on Form 10-K and are incorporated herein by reference.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
(a) Evaluation of disclosure controls and procedures. Our management, with the participation of our Chief Executive Officer and Principal Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15 under the Securities Exchange Act of 1934 (the “Exchange Act”)) as of the end of the period covered by this Annual Report on Form 10-K. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected. Our Chief Executive Officer and Principal Financial Officer have concluded, based on their evaluation as of the end of the period covered by this report, that our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined In Exchange Act Rule 13a-15(f). The term “internal control over financial reporting” is defined as a process designed by, or under the supervision of, the registrant’s principal executive and principal financial officers, or persons performing similar functions, and effected by the registrant’s board of directors, management and other personnel,
|
|
· to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
|
|
· pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the registrant;
|
|
|
· provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the registrant are being made only in accordance with authorizations of management and directors of the registrant; and
|
|
|
· provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the registrant’s assets that could have a material effect on the financial statements.
|
Our internal control system is designed to provide reasonable assurance to our management and board of directors regarding the preparation and fair presentation of published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits of controls must be considered relative to their costs. In addition, because of changes in conditions, the effectiveness of internal control may vary over time.
As of December 31, 2015, management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the criteria in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992) (“COSO”) and have concluded our controls are effective.
The Company is a non-accelerated filer and is not subject to Section 404(b) of the Sarbanes Oxley Act. Accordingly, this Annual Report does not contain an attestation report of our independent registered public accounting firm regarding internal control over financial reporting, since the rules for smaller reporting companies provide for this exemption.
(b) Changes in internal control over financial reporting. There have been no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2015, that have materially affected or are reasonably likely to materially affect our internal control over financial reporting.
Item 9B. Other Information
None.
PART III
Item10. Directors, Executive Officers and Corporate Governance
Executive Officers and Directors
Set forth below is information regarding our executive officers and directors as of December 31, 2015
|
Mike Tomas
|
50
|
Director, President and Chief Executive Officer, Chief Financial Officer
|
|
William P. Murphy, Jr., M.D.
|
92
|
Director, Chairman of the Board
|
|
Mark P. Borman
|
61
|
Director
|
|
Charles A. Hart
|
55
|
Director
|
|
Sam Ahn
|
61
|
Director
|
|
Kristin Comella
|
39
|
Director, Chief Scientific Officer
|
|
Sheldon T. Anderson
|
65
|
Director
|
Our Bylaws provide that we shall have that number of directors determined by the majority vote of the board of directors. Currently we have seven directors. Each director will serve until our next annual shareholder meeting. Directors are elected for one-year terms. Our Board of Directors elects our officers at the regular annual meeting of the Board of Directors following the annual meeting of shareholders. Vacancies may be filled by a majority vote of the remaining directors then in office. Our directors and executive officers are as follows:
Executive Officers and Directors
Mike Tomas. Mike Tomas, President & CEO of U.S. Stem Cell Inc, is considered by many in the industry as one of the most experienced marketers and operating executives for IT/Communications and Biotech/Life Sciences private equity and venture groups portfolio companies. The son of a serial entrepreneur, he spent nearly 20 years driving the evolution of telecommunications technology in the U.S. and Mexico in leadership roles ranging from sales, marketing, customer service, telemarketing, engineering, and operations. Upon retiring as Chief Marketing Officer of Avantel, MCI/Worldcom's Global Ventures $1B investment with Banamex (at the time, the largest bank in Latin America), Mr. Tomás joined other former-MCI executives (including MCI CEO Jerry Taylor) and helped raise venture capital to form an integrated customer communications software solution that was named on Red Herring magazine’s “Top Ten to Watch” list.
Upon the successful sale of that company in 2001, Mr. Tomas helped launch The ASTRI Group, an early-stage private equity investment company providing capital, business development and strategic marketing support to emerging private companies. Mr. Tomas sits on the boards Easy Solutions Total Fraud Protection (authentication, fraud prevention and anti-phishing countermeasures), U.S. Stem Cell (adult stem cell development and applications), The IDEA Center (Miami Dade College’s entrepreneurial institute), Career Source Florida (appointed by Florida Governor Rick Scott to his statewide workforce investment board) and is the current chairman of Florida International University’s Global Entrepreneurship Center. Mr. Tomas is an inductee into the Miami-Dade College and WACE Halls of Fame for business, an FIU Torch Award winner--and winner of top communications, medical innovations, education and entrepreneurial awards. An avid athlete, Mr. Tomas is also a Miami-Dade County Sports Commissioner.
William P. Murphy, Jr., M.D. Dr. Murphy has served as a member of our Board of Directors since June 2003. Dr. Murphy founded Small Parts, Inc., a supplier of high quality mechanical components for design engineers, in 1964 and served as its Chairman until his retirement in April 2005. Small Parts, Inc. was acquired by Amazon.com, Inc. in March 2005. From October 1999 until October 2004, Dr. Murphy served as the Chairman and Chief Executive Officer of Hyperion, Inc., a medical diagnosis company which had an involuntary bankruptcy filed against it in December 2003. Dr. Murphy is the founder of Cordis Corporation (now Cordis Johnson & Johnson) which he led as President, Chairman and Chief Executive Officer at various times during his 28 years at Cordis until his retirement in October 1985. Cordis Johnson & Johnson is a leading firm in cardiovascular instrumentation.
Dr. Murphy received an M.D. in 1947 from the University of Illinois and a B.S. in pre-medicine from Harvard College in 1946. He also studied physiologic instrumentation at Massachusetts Institute of Technology, or MIT. After a two year rotating internship at St. Francis Hospital in Honolulu, he became a Research Fellow in Medicine at the Peter Bent Brigham Hospital in Boston where he was the dialysis engineer on the first clinical dialysis team in the United States. He continued as an Instructor in Medicine and then a research associate in Medicine at Harvard Medical School. Dr. Murphy is the author of numerous papers and owns 17 patents. He is the recipient of a number of honors, including the prestigious Lemelson-MIT Lifetime Achievement Award, the MIT Corporate Leadership Award, the Distinguished Service Award from North American Society of Pacing and Electrophysiology, and the Jay Malina Award from the Beacon Council of Miami, Florida. He is also a member of the Inventors Hall of Fame.
Mark P. Borman. Mr. Borman has served as a member of the Company’s Board of Directors since May 2009. He is a seasoned financial officer with more than 30 years of broad-based financial and investor relations experience. Mr. Borman brings small-company entrepreneurial passion and larger-company disciplines. In addition to the valuable experience he gained working with entrepreneurs and their startups from 2009 to present, Mr. Borman has experience with global, NASDAQ- and NYSE-listed companies in various executive and financial roles. He is currently a board member and Chief Financial Officer with private technology companies and has served on advisory and non-profit boards. During his career, Mr. Borman has held positions with, ADC Telecommunications, General Instrument Corporation, First Chicago Corporation, FMC Corporation, Price Waterhouse, and KPMG. Mr. Borman received his B.A. in Accounting from Michigan State University and his M.B.A. from the University of Chicago Graduate School of Business. He is an Audit Committee Financial Expert under SEC rules, a Certified Public Accountant and Chartered Financial Analyst. Mr. Borman is a Board Leadership Fellow and member of the National Association of Corporate Directors.
Charles A. Hart. Mr. Hart has served as a member of our Board of Directors since May 2009. Mr. Hart has more than 20 years of entrepreneurial experience. Mr. Hart founded Hart Masonry, Inc. in 1986 and has served as its President since then. He is also the Founder and President of Wildridge Enterprises. Mr. Hart is a member of the Board of Directors for Eagle Street Properties LLP.
Kristin Comella. Ms. Comella was appointed Chief Scientific Officer in September 2010. Ms. Comella has served as our Vice President of R&D and Corporate Development since December 2008 and has played a major role in managing our product development, manufacturing and quality systems since joining U.S. Stem Cell in 2004. Ms. Comella has 15 years of industry experience with expertise in regenerative medicine, training and education, research and product development, and currently serves on multiple advisory boards in the stem cell arena. Ms. Comella has many years of cell culturing experience including building and managing the stem cell laboratory at Tulane University's Center for Gene Therapy and developing stem cell therapies for osteoarthritis at Osiris Therapeutics. Ms. Comella holds an M.S. in Chemical Engineering from The Ohio State University and a B.S. in Chemical Engineering from the University of South Florida. On March 12, 2013, Kristin Comella was appointed to serve as a member of our Board of Directors.
Sheldon T. Anderson. Mr. Anderson is Chairman of the Florida Advisory Board of Northern Trust Corporation. From 1992 through December 31, 2012, Mr. Anderson served in a variety of executive capacities with Northern Trust Corporation, including his most recent position as Chairman and Chief executive Officer Southeast Region of Northern Trust Corporation. Mr. Anderson is the Chair-elect of the Beacon Council, Miami-Dade County's economic development agency. He is a Board member of the Miami-Dade College Foundation, Inc.; Museum of Contemporary Art (MOCA); the New World Symphony; Baptist Health Systems Governing Board and Carrollton School of the Sacred Heart. He is Past Chair and a member of the Advisory Council of the United Way of Miami-Dade County. Anderson is President of the Board of Cleveland Orchestra Miami / Miami Music Association and also serves on the Advisory Board of the University of Miami School of Law for Ethics& Public Service. He is a member of the Orange Bowl Committee and the President's Council of Florida International University. A Miami native, Sheldon holds a degree in International Studies from Ohio State University.
Family Relationships
There are no family relationships among our executive officers and directors.
Section16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than ten percent (10%) of our outstanding Common Stock, or the Reporting Persons, to file with the SEC initial reports of ownership on Form 3 and reports of changes in ownership of Common Stock on Forms 4 or 5. Such persons are required by SEC regulation to furnish us with copies of all such reports they file. Based solely on a review of Forms 3 and 4 furnished to us by the Reporting Persons or prepared on behalf of the Reporting Persons by the Company and on written representations from certain Reporting Persons that no Forms 5 were required, the Company believes that the Reporting Persons have complied with reporting requirements applicable to them.
Conflicts of Interest
Members of our management are associated with other firms involved in a range of business activities. Consequently, there are potential inherent conflicts of interest in their acting as officers and directors of our company. Although the officers and directors are engaged in other business activities, we anticipate they will devote an important amount of time to our affairs.
Our officers and directors are now and may in the future become shareholders, officers or directors of other companies, which may be formed for the purpose of engaging in business activities similar to ours. Accordingly, additional direct conflicts of interest may arise in the future with respect to such individuals acting on behalf of us or other entities. Moreover, additional conflicts of interest may arise with respect to opportunities which come to the attention of such individuals in the performance of their duties or otherwise. Currently, we do not have a right of first refusal pertaining to opportunities that come to their attention and may relate to our business operations.
Our officers and directors are, so long as they are our officers or directors, subject to the restriction that all opportunities contemplated by our plan of operation which come to their attention, either in the performance of their duties or in any other manner, will be considered opportunities of, and be made available to us and the companies that they are affiliated with on an equal basis. A breach of this requirement will be a breach of the fiduciary duties of the officer or director. If we or the companies with which the officers and directors are affiliated both desire to take advantage of an opportunity, then said officers and directors would abstain from negotiating and voting upon the opportunity. However, all directors may still individually take advantage of opportunities if we should decline to do so. Except as set forth above, we have not adopted any other conflict of interest policy with respect to such transactions.
Involvement in Certain Legal Proceedings
None of the following events have occurred during the past ten years and are material to an evaluation of the ability or integrity of any director or officer of the Company:
| 1. | A petition under the Federal bankruptcy laws or any state insolvency law was filed by or against, or a receiver, fiscal agent or similar officer was appointed by a court for the business or property of such person, or any partnership in which he was a general partner at or within two years before the time of such filing, or any corporation or business association of which he was an executive officer at or within two years before the time of such filing; | |||
|
2.
|
Such person was convicted in a criminal proceeding or is a named subject of a pending criminal proceeding (excluding traffic violations and other minor offenses);
|
|||
|
3.
|
Such person was the subject of any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from, or otherwise limiting, the following activities:
|
|||
|
|
a.
|
Acting as a futures commission merchant, introducing broker, commodity trading advisor, commodity pool operator, floor broker, leverage transaction merchant, any other person regulated by the Commodity Futures Trading Commission, or an associated person of any of the foregoing, or as an investment adviser, underwriter, broker or dealer in securities, or as an affiliated person, director or employee of any investment company, bank, savings and loan association or insurance company, or engaging in or continuing any conduct or practice in connection with such activity;
|
||
|
b.
|
Engaging in any type of business practice; or
|
|||
|
c.
|
Engaging in any activity in connection with the purchase or sale of any security or commodity or in connection with any violation of Federal or State securities laws or Federal commodities laws;
|
|||
|
|
4.
|
Such person was the subject of any order, judgment or decree, not subsequently reversed, suspended or vacated, of any Federal or State authority barring, suspending or otherwise limiting for more than 60 days the right of such person to engage in any activity described in paragraph (f)(3)(i) of this section, or to be associated with persons engaged in any such activity;
|
||
|
5.
|
Such person was found by a court of competent jurisdiction in a civil action or by the Commission to have violated any Federal or State securities law, and the judgment in such civil action or finding by the Commission has not been subsequently reversed, suspended, or vacated;
|
|||
|
6.
|
Such person was found by a court of competent jurisdiction in a civil action or by the Commodity Futures Trading Commission to have violated any Federal commodities law, and the judgment in such civil action or finding by the Commodity Futures Trading Commission has not been subsequently reversed, suspended or vacated;
|
|||
|
7.
|
Such person was the subject of, or a party to, any Federal or State judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of:
|
|||
|
|
a.
|
Any Federal or State securities or commodities law or regulation; or
|
||
|
b.
|
Any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order; or
|
|||
|
c.
|
Any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or
|
|||
|
8.
|
Such person was the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act (15 U.S.C. 78c(a)(26))), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act (7 U.S.C. 1(a)(29)), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member.
|
|||
Code of Ethics
As part of our system of corporate governance, our Board of Directors has adopted a code of ethics that is specifically applicable to our Chief Executive Officer and senior financial officers. This Code of Ethics for Senior Financial Officers, as well as our Code of Business Conduct and Ethics, applicable to all directors, officers and employees. If we make substantive amendments to the Code of Ethics for Senior Financial Officers or the Code of Business Conduct and Ethics or grant any waiver, including any implicit waiver, we will disclose the nature of such amendment or waiver on our website or in a report on Form 8-K within four days of such amendment or waiver.
Shareholder Recommendations for Board Nominees
There have been no material changes to the procedures by which security holders may recommend nominees to the Company’s Board of Directors.
Audit Committee
The Board of Directors has a separately-designated standing Audit Committee established in accordance with Section 3(a) (58) (A) of the Exchange Act. The members of our Audit Committee are Mr. Borman, who serves as Chairperson of the Audit Committee, Dr. Murphy, Mr. Anderson and Mr. Hart. Our Board of Directors has determined that Mr. Borman qualifies as a “financial expert” as that term is defined in the rules of the SEC implementing requirements of the Sarbanes-Oxley Act of 2002.
Item11. Executive Compensation.
Summary Compensation Table
The following table sets forth, for the fiscal years ended December 31, 2015 and 2014, the aggregate compensation awarded to, earned by or paid to our Chief Executive Officer and our two most highly compensated officers (other than the Chief Executive Officer), who were serving as executive officers as of December 31, 2015 , or the Named Executive Officers.
|
Name and
Principal
Position
|
Fiscal
Year
|
Salary
($)
|
Bonus
($)
|
Stock
Awards
($)
|
Option
Awards ($)
|
Non-
Equity
Incentive Plan
Compensation
($)
|
Change in
Pension
Value
and Non-
Qualified
Deferred
Compensation
Earnings
($)
|
All Other
Compensation
($)
|
Total
($)
|
|||||||||||||||||||||||||
|
Mike Tomas
CEO, President,
|
2015
|
$ | 585,463 | $ | 75,000 | $ | — | $ | 477,179 | (1)(2) | $ | 1,137,642 | ||||||||||||||||||||||
|
CFO and Director
|
2014
|
766,586 | 500,000 | (3) | — | $ | 587,895 | (4)(5)(6)(7)(8) | — | — | — | $ | 1,854,481 | |||||||||||||||||||||
|
Kristin Comella
Chief Science
|
2015
|
249,008 | 50,000 | — | $ | 284,739 | (1)(2) | 583,747 | ||||||||||||||||||||||||||
|
Officer and Director
|
2014
|
371,331 | 300,000 | (3) | — | 240,187 | (4)(5)(7)(8) | 911,518 | ||||||||||||||||||||||||||
|
(1)
|
On November 2, 2015, Mr. Tomas and Ms. Comella were granted 291,885 and 175,131, respectively, options to purchase the Company’s common stock at $1.71 per share for ten years, vesting immediately
|
|
(2)
|
On November 2, 2015, Mr. Tomas and Ms. Comella were granted 10,000 and 5,000 , respectively, options to purchase the Company’s common stock at $6.24 per share for ten years, vesting over four years on anniversary
|
|
(3)
|
On February 24, 2014, Mr. Tomas and Ms. Comella received $500,000 and $300,000, respectively, promissory notes for bonuses awarded. The promissory notes bear 5% interest per annum, unsecured and were due January 1, 2015
|
|
(4)
|
On February 24, 2014, Mr. Tomas and Ms. Comella were granted 10,000 and 5,000 respectively, options to purchase the Company’s common stock at $19.00 per share for ten years, vesting ¼ immediately, with remainder vesting 1/3 on August 1st and September 1st, respectively, for three years.
|
|
(5)
|
On February 24, 2014, Mr. Tomas and Ms. Comella, as members of the Company’s Board of Directors, were granted 800 and 400, respectively, options to purchase the Company’s common stock at $19.00 per share, vesting immediately.
|
|
(6)
|
On May 12, 2014, Mr. Tomas was granted 4,298 options to purchase the Company’s common stock at $27.16 per share for ten years vesting annually over four years.
|
|
(7)
|
On August 1, 2014, Mr. Tomas and Ms. Comella were granted 10,000 and 5,000, respectively, options to purchase the Company’s common stock at $26.90 per share for ten years with ¼ vesting immediately, with remainder vesting 1/3 annually on the anniversary.
|
|
(8)
|
On November 3, 2014, Mr. Tomas and Ms. Comella, as members of the Company’s Board of Directors, were granted 400 options to purchase the Company’s common stock at $19.32 per share, vesting immediately.
|
Our Stock Option Plans
In December 1999, the Board of Directors and shareholders adopted the 1999 Officers and Employees Stock Option Plan, or the Employee Plan, and the 1999 Directors and Consultants Stock Option Plan, or the Director Plan. The Employee Plan and the Director Plan are collectively referred to herein as the Plans. The Plans are administered by the Board of Directors and the Compensation Committee.
The objectives of the Plans include attracting and retaining key personnel by encouraging stock ownership in the Company by such persons. In February 2010, the Directors & Consultants Plan was amended to extend the termination date of the Plan to December 1, 2011.
On April 1, 2013, the Board of Directors approved, subject to shareholder approval, the establishment of the U.S. Stem Cell 2013 Omnibus Equity Compensation Plan, or the “2013 Omnibus Plan”. The 2013 Omnibus Plan reserves up to fifty thousand shares of common stock for issuance. On August 4, 2014, the Board of Directors approved to set the reserve to one hundred thousand shares of common stock for issuance and to close the 1999 Officers and Employees Stock Option Plan. On February 2, 2015, at the annual meeting of shareholders, the majority of shareholders approved the 2013 Omnibus Equity Compensation Plan. On November 2, 2015, the Board of Directors approved the increase of the reserve under the 2013 Omnibus Plan to five hundred million shares of common stock for issuance.
Employment Agreements
See page F-31
Outstanding Equity Awards at Fiscal Year End
The following table sets forth outstanding equity awards held by our Named Executive Officers as of December 31, 2015:
|
Number of Securities Underlying
|
Option
|
||||||||||||
|
Unexercised Options and Warrants
|
Exercise Price
|
Option Expiration
|
|||||||||||
|
Name
|
Exercisable (#)
|
Unexercisable (#)
|
($/per share)
|
Date
|
|||||||||
|
Mike Tomas
|
500
|
-
|
500.00
|
6/18/2020
|
|||||||||
|
500
|
-
|
16.94
|
8/12/2021
|
||||||||||
|
500
|
-
|
16.94
|
8/12/2022
|
||||||||||
|
2,000
|
2,000
|
16.94
|
8/6/2022
|
||||||||||
|
10,000
|
5,000
|
15.76
|
8/1/2023
|
||||||||||
|
400
|
—
|
16.54
|
9/1/2023
|
||||||||||
|
10,000
|
2,500
|
19.00
|
2/24/2024
|
||||||||||
|
800
|
-
|
19.00
|
2/24/2024
|
||||||||||
|
4,299
|
2,424
|
27.16
|
5/24/2024
|
||||||||||
|
10,000
|
5,000
|
26.90
|
8/1/2024
|
||||||||||
|
400
|
-
|
19.32
|
11/3/2024
|
||||||||||
|
291,885
|
-
|
1.71
|
11/2/2025
|
||||||||||
|
10,000
|
10,000
|
6.24
|
11/2/2025
|
||||||||||
|
Kristin Comella
|
10
|
-
|
710.00
|
4/18/2016
|
|||||||||
|
7
|
—
|
710.00
|
1/1/2017
|
||||||||||
|
7
|
—
|
710.00
|
10/16/2017
|
||||||||||
|
20
|
—
|
710.00
|
1/19/2019
|
||||||||||
|
3
|
—
|
740.00
|
3/13/2019
|
||||||||||
|
30
|
—
|
850.00
|
5/28/2019
|
||||||||||
|
250
|
—
|
16.94
|
8/12/2021
|
||||||||||
|
500
|
500
|
16.94
|
8/6//2022
|
||||||||||
|
5,000
|
2,500
|
15.76
|
8/1/2023
|
||||||||||
|
5,000
|
2,500
|
19.00
|
2/24/2024
|
||||||||||
|
5,000
|
2,500
|
26.90
|
8/1/2024
|
||||||||||
|
400
|
-
|
19.32
|
11/3/2024
|
||||||||||
|
1,000
|
-
|
11.16
|
2/2/2025
|
||||||||||
|
175,131
|
-
|
1.71
|
11/2/2025
|
||||||||||
|
5,000
|
-
|
6.24
|
11/2/2025
|
||||||||||
Options Exercises and Stocks Vested
Options exercised and stocks vested as at December 31, 2015 are as follows:
|
Option awards
|
Stock awards
|
|||||||||||||||
|
Name
|
Number of Shares Acquired
on Exercise (#)
|
Value Realized on
Exercise ($)
|
Number of Shares
Acquired on Vesting
(#)
|
Value
Realized on Investing ($)
|
||||||||||||
|
Mike Tomas, NEO
|
0 | 0 | 0 | 0 | ||||||||||||
|
Kristin Comella, NEO
|
0 | 0 | 0 | 0 | ||||||||||||
Grants of Plan-Based Awards
Grants of plan-based awards as at December 31, 2015 are as follows:
| Estimated future payouts | Estimated future payouts | All other | All other | Grant | ||||||||||||||||||
| under non-equity incentive | under equity incentive plan | stock | option | date | ||||||||||||||||||
|
plan awards
|
awards
|
awards: | awards: | fair | ||||||||||||||||||
| Number | Number | Exercise | value | |||||||||||||||||||
| of | of | or base | of | |||||||||||||||||||
| shares | securities | price of | stock | |||||||||||||||||||
| of stock | underlying | option | and | |||||||||||||||||||
| Grant | Threshold | Target | Maximum | Threshold | Target | Maximum | or units | options | awards | option | ||||||||||||
|
Name
|
date
|
($)
|
($)
|
($)
|
(#)
|
(#)
|
(#)
|
(#) | (#) |
($/Sh)
|
awards
|
|||||||||||
|
Mike Tomas, NEO
|
n/a
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
|||||||||||
|
Kristin Comella, NEO
|
n/a
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
0
|
|||||||||||
Reference – Grant Date - n/a = not applicable.
Non-Qualified Deferred Compensation
As at December 31, 2015 the Company had no formalized deferred compensation plan.
|
Name
|
Executive
contributions
in last FY ($)
|
Registrant
contributions in
last FY ($)
|
Aggregate
earnings in last
FY ($)
|
Aggregate
withdrawals/
distributions ($)
|
Aggregate
balance at last
FYE ($)
|
|||||||||||||||
|
Mike Tomas, NEO
|
0 | 0 | 0 | 0 | 0 | |||||||||||||||
|
Kristin Comella, NEO
|
0 | 0 | 0 | 0 | 0 | |||||||||||||||
Golden Parachute Compensation
As at December 31, 2015, the Company had no arrangements in place relating to the termination of employees.
|
Name
|
Cash
($)
|
Equity
($)
|
Pension/NQDC
($)
|
Perquisites/benefits
($)
|
Tax
reimbursement
($)
|
Other
($)
|
Total
($)
|
|||||||||||||||||||||
|
Mike Tomas, NEO
|
0 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||||
|
Kristin Comella, NEO
|
0 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||||
Compensation of Directors
Directors who provide services to the Company in other capacities has been previously reported under “Summary Compensation”. The following table summarizes compensation paid to or earned by our directors who are not Named Executive Officers for their service as directors of our company during the fiscal year ended December 31, 2015.
|
Name
|
Fees Earned or Paid in Cash ($)
|
Stock
Awards
($)
|
Option Awards (1)
($)
|
Non-Equity
Incentive Plan
Compensation ($)
|
Change in
Pension Value
and
Nonqualified
Deferred
Compensation
Earnings ($)
|
All other
Compensation
($)
|
Total (1)
($)
|
|||||||||||||||||||||
|
Mike Tomas, Director
|
0 | 0 | $ | 1,739 | 0 | 0 | 0 | $ | 1,739 | |||||||||||||||||||
|
Charles A. Hart, Director
|
0 | 0 | $ | 1,739 | 0 | 0 | 0 | $ | 1,739 | |||||||||||||||||||
|
William P. Murphy Jr., Director
|
0 | 0 | $ | 1,739 | 0 | 0 | 0 | $ | 1,739 | |||||||||||||||||||
|
Samuel S. Ahn, Director
|
0 | 0 | $ | 1,739 | 0 | 0 | 0 | $ | 1,739 | |||||||||||||||||||
|
Mark Bormon, Director
|
0 | 0 | $ | 1,739 | 0 | 0 | 0 | $ | 1,739 | |||||||||||||||||||
|
(1)
|
The values in the “Option Awards” and included within the “Total” columns above do not represent a cash payment of any kind. Rather these values represent the calculated Black-Scholes theoretical value of granted options. It is important to note that these granted options may or may not ever be exercised. Whether granted options are exercised or not will be based primarily, but not singularly, on the Company’s future stock price and whether the granted options become “in-the-money”. If these granted options are unexercised and expire, the cash value or benefit to the above noted individuals is $nil.
|
Pension Benefits
As of December 31, 2015, the Company had no pension or retirement plans.
|
Name
|
Plan name
|
Number of years
credited service
(#)
|
Present value of
accumulated benefit
($)
|
Payments during last
fiscal year ($)
|
||||||||||
|
Mike Tomas, NEO
|
not applicable
|
0 | 0 | 0 | ||||||||||
|
Kristin Comella, NEO
|
not applicable
|
0 | 0 | 0 | ||||||||||
Equity Compensation Plan Information
The following table sets forth information as of December 31, 2015 for all compensation plans under the Company’s Stock Option Plan:
|
Additional
|
||||||||||||||||||
|
No. of
|
Consideration
|
|||||||||||||||||
|
Shares of Common
|
to be Received
|
|||||||||||||||||
|
Stock Underlying
|
Upon Exercise
|
|||||||||||||||||
|
Unexercised
|
or Material
|
Value
|
||||||||||||||||
|
Common Stock
|
Conditions
|
Option
|
Realized
|
Option
|
||||||||||||||
|
Purchase Options
|
Date of
|
Required to
|
Exercise
|
if
|
Expiration
|
|||||||||||||
|
Name
|
Exercisable (#)
|
Grant
|
Exercise
|
Price ($)
|
Exercised ($)
|
Date
|
||||||||||||
|
Mike Tomas, NEO
|
375
|
9/18/2010
|
$
|
—
|
$
|
500.00
|
$
|
—
|
6/18/2020
|
|||||||||
|
250
|
8/12/2011
|
—
|
16.94
|
—
|
8/12/2021
|
|||||||||||||
|
250
|
1/16/2012
|
—
|
16.94
|
—
|
1/16/2022
|
|||||||||||||
|
1,250
|
8/6/2012
|
—
|
16.94
|
—
|
8/6/2022
|
|||||||||||||
|
10,000
|
8/01/13
|
—
|
16.94
|
—
|
8/1/2023
|
|||||||||||||
|
400
|
9/1/13
|
16.54
|
9/1/2023
|
|||||||||||||||
|
10,000
|
2/24/14
|
—
|
19.00
|
—
|
2/24/2024
|
|||||||||||||
|
4,298
|
5/12/14
|
27.16
|
5/12/2014
|
|||||||||||||||
|
10,000
|
8/01/14
|
—
|
26.90
|
—
|
8/1/2024
|
|||||||||||||
|
400
|
11/3/14
|
—
|
19.32
|
—
|
11/3/2024
|
|||||||||||||
|
400
|
9/1/2013
|
—
|
16.50
|
—
|
9/1/2023
|
|||||||||||||
|
1,000
|
2/2/15
|
-
|
11.16
|
-
|
2/2/2025
|
|||||||||||||
|
175,131
|
11/2/15
|
-
|
1.71
|
-
|
11/2/2025
|
|||||||||||||
|
10,000
|
11/2/15
|
-
|
6.24
|
-
|
11/2/2025
|
|||||||||||||
|
Kristin Comella, NEO
|
25
|
2/20/2005
|
—
|
710.00
|
—
|
2/19/2015
|
||||||||||||
|
9
|
4/19/2006
|
—
|
710.00
|
—
|
4/19/2016
|
|||||||||||||
|
6
|
1/2/2007
|
—
|
710.00
|
—
|
1/2/2017
|
|||||||||||||
|
7
|
10/17/2007
|
—
|
710.00
|
—
|
10/17/2017
|
|||||||||||||
|
20
|
1/9/2009
|
—
|
710.00
|
—
|
1/9/2019
|
|||||||||||||
|
3
|
3/13/2009
|
—
|
710.00
|
—
|
3/13/2019
|
|||||||||||||
|
30
|
5/29/2009
|
—
|
850.00
|
—
|
5/29/2019
|
|||||||||||||
|
125
|
8/12/2011
|
—
|
16.94
|
—
|
8/12/2021
|
|||||||||||||
|
188
|
8/06/2012
|
—
|
16.94
|
—
|
8/06/2022
|
|||||||||||||
|
5,000
|
8/01/13
|
—
|
15.76
|
—
|
8/1/2023
|
|||||||||||||
|
5,000
|
2/24/14
|
—
|
19.00
|
—
|
2/24/2024
|
|||||||||||||
|
5,000
|
8/01/14
|
—
|
26.90
|
—
|
8/1/2024
|
|||||||||||||
|
400
|
11/3/14
|
—
|
19.32
|
—
|
11/3/2024
|
|||||||||||||
|
1,000
|
2/2/15
|
-
|
11.16
|
-
|
2/2/2025
|
|||||||||||||
|
291,885
|
11/2/15
|
-
|
1.71
|
-
|
11/2/2025
|
|||||||||||||
|
5,000
|
11/2/15
|
-
|
6.24
|
-
|
11/2/2025
|
|||||||||||||
Director Compensation
As of December 31, 2015, we had seven directors that qualified for compensation. Our non-employee directors do not receive cash compensation for their services as directors. However, it is generally our policy to annually grant each non-employee director options to purchase shares of our common stock provided that he or she has served as a member of our Board of Directors for at least six months and one day of the twelve month period immediately preceding the date of grant. In addition, we reimburse non-employee directors for actual out-of-pocket expenses incurred.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth the beneficial ownership (1) of our common stock as of December 31, 2015, based on an aggregate of 1,813,691 common shares issued and outstanding and 678,805 shares issuance upon the conversion of securities, for each of our greater than 5% shareholders, directors, named executive officers that continue to serve as executive officers of U.S. Stem Cell and by all of our directors and named executive officers as a group. Unless otherwise indicated, the address of each of the individuals and entities named below is: c/o U.S. Stem Cell, Inc., 13794 NW 4th Street, Suite 212, Sunrise, Florida 33325. Except as noted below, to our knowledge, each person named in the table has sole voting and investment power with respect to all shares of our common stock beneficially owned by them.
|
|
(1)
|
The number and percentage of shares beneficially owned is determined in accordance with Rule 13d-3 under the Exchange Act, and the information is not necessarily indicative of beneficial ownership for any other purpose. The Company believes that each individual or entity named has sole investment and voting power with respect to the securities indicated as beneficially owned by them, subject to community property laws, where applicable, except where otherwise noted. The “Amount of Beneficial Ownership” in calculated based on total shares held plus warrants held (plus stock options entitled to exercise). The aggregate of these items, which totals 689,434,889, will be used as the denominator for the percentage calculation below.
|
|
Name and Address of Beneficial Owner
|
Amount of Beneficial Ownership
|
Percent of Class
|
||||||
|
Mike Tomas, President, CEO, CFO, and Director
|
340,563
|
(1)
|
14.9
|
|||||
|
Kristin Comella, Chief Scientific Officer and Director
|
187,762
|
(2)
|
8.2
|
|||||
|
William P. Murphy, Director**
|
51,969
|
(3)
|
2.3
|
|||||
|
Charles A. Hart, Director**
|
18,136
|
(4)
|
*
|
|||||
|
Sam Ahn, Director**
|
52,899
|
(5)
|
2.3
|
|||||
|
Mark P. Borman, Director
|
2,834
|
(6)
|
*
|
|||||
|
Sheldon T. Anderson, Director
|
5,282
|
(7)
|
*
|
|||||
|
All officers and directors as a group (7 persons)
|
167,228
|
(8)
|
28.8
|
|||||
|
Northstar Biotechnology Group, LLC
|
47,369
|
(9)
|
2.0
|
|||||
* less than 1%
** Excludes Northstar Biotechnology Group, LLC (“Northstar”), owned partly by certain directors and existing shareholders of the Company, including Dr. William P. Murphy Jr., Dr. Samuel Ahn and Charles Hart and controlled by Gregory Knutson.
|
(1)
|
Includes shares are held by The Astri Group over which Mr. Tomas has shared voting and investment power and includes (i) includes 8,279 shares of common stock and (ii) 332,284 shares of common stock issuable upon exercise of presently exercisable stock options .
|
|
(2)
|
Includes 187,762 shares of common stock issuable upon exercise of presently exercisable stock options.
|
|
(3)
|
Includes (i) 48,875 shares of common stock and (ii) 3,094 shares of common stock issuable upon exercise of presently exercisable stock options and warrants. Shares are directly owned by trusts controlled by Dr. Murphy and his spouse.
|
|
(4)
|
Includes (i) 9,194 shares of common stock and (ii) 8,942 shares of common stock issuable upon exercise of presently exercisable stock options and warrants.
|
|
(5)
|
Includes (i) 42,806 shares of common stock and (ii) 10,093 shares of common stock issuable upon exercise of presently exercisable stock options.
|
|
(6)
|
Includes (i) 24 shares of common stock and (ii) 2,810 shares of common stock issuable upon exercise of presently exercisable stock options
|
|
(7)
|
Includes (i) 1,941 shares of common stock and (ii) 3,341 shares of common stock issuable upon exercise of presently exercisable warrants.
|
|
(8)
|
Includes an aggregate of (i) 109,145 shares of common stock and (ii) 58,083 shares of common stock issuable upon exercise of presently exercisable stock options and warrants.
|
|
(9)
|
Excludes 20,000,000 shares of Series A Preferred Stock (non-convertible) with each share of Series A Preferred Stock having voting power of twenty-five common shares.
|
DESCRIPTION OF SECURITIES
The following statements relating to the capital stock set forth the material terms of our securities; however, reference is made to the more detailed provisions of, and such statements are qualified in their entirety by reference to, the Certificate of Incorporation, amendment to the Certificate of Incorporation and the By-laws, copies of which are filed as exhibits to this registration statement.
COMMON STOCK
The holders of our Common Stock are entitled to one vote per share on all matters to be voted on by our stockholders, including the election of directors. Our stockholders are not entitled to cumulative voting rights, and, accordingly, the holders of a majority of the shares voting for the election of directors can elect the entire board of directors if they choose to do so and, in that event, the holders of the remaining shares will not be able to elect any person to our board of directors.
On February 4, 2013, effective with the filing of the amendment to the Company’s Articles of Incorporation with the Florida Secretary of State (confirmed as filed on February 11, 2013), the Company amended its Articles of Incorporation to increase the authorized shares of capital stock of the Company to nine hundred and seventy million (970,000,000) shares of capital stock consisting of nine hundred and fifty million (950,000,000) shares of common stock and twenty million (20,000,000) shares of preferred stock, both $.001 par value respectively.
Effective May 19, 2014, the Company amended its articles of incorporation to increase the authorized shares of capital stock of the Company from nine hundred and fifty million (950,000,000) shares of common stock and twenty million (20,000,000) shares of preferred stock, both $.001 par value respectively, to two billion (2,000,000,000) shares of shares of common stock and twenty million (20,000,000) shares of preferred stock, both $.001 par value respectively.
On October 12, 2015, the Company filed an amendment to its Articles of Incorporation and effected a 1-for-1,000 reverse stock split of its issued and outstanding shares of common stock, $0.001 par value, effective November 19, 2015. The Financial Industry Regulatory Authority (“FINRA”) declared the ex-dividend date for the dividend date as November 4, 2015.
The holders of the Company’s Common Stock are entitled to receive ratably such dividends, if any, as may be declared from time to time by the board of directors, in its discretion, from funds legally available there for and subject to prior dividend rights of holders of any shares of our Preferred Stock which may be outstanding. Upon the Company’s liquidation, dissolution or winding up, subject to prior liquidation rights of the holders of our Preferred Stock, if any, the holders of our Common Stock are entitled to receive on a pro rata basis our remaining assets available for distribution. Holders of the Company’s Common Stock have no preemptive or other subscription rights, and there are no conversion rights or redemption or sinking fund provisions with respect to such shares. All outstanding shares of the Company’s Common Stock are, fully paid and not liable to further calls or assessment by the Company.
Preferred Stock
The Company is authorized to issue 20,000,000 shares of preferred stock, par value $0.001. The designations, rights, and preferences of such preferred stock are to be determined by the Board of Directors. Subsequently, 20,000,000 shares were designated as Series A Preferred Stock.
The Series A Preferred Stock collectively has voting rights equal to 25 votes on all matters presented to be voted by the holders of common stock per share of preferred stock and the right to convert to one share of common stock for each share of preferred stock. Northstar Biotechnology Group, LLC was issued an aggregate of 20,000,000 shares of Series A Preferred Stock.
DIVIDENDS
Dividends, if any, will be contingent upon our revenues and earnings, if any, capital requirements and financial conditions. The payment of dividends, if any, will be within the discretion of our Board of Directors. We presently intend to retain all earnings, if any, for use in its business operations and accordingly, the Board of Directors does not anticipate declaring any dividends prior to a business combination.
INDEMNIFICATION OF DIRECTORS AND OFFICERS.
We are incorporated under the laws of the State of Florida. Our articles of incorporation require us to indemnify and limit the liability of directors to the fullest extent permitted by the Florida Business Corporation Act, or the “FBCA”, as it currently exists or as it may be amended in the future.
Pursuant to the FBCA, a Florida corporation may indemnify any person who may be a party to any third party proceeding by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee, or agent of another entity, against liability incurred in connection with such proceeding (including any appeal thereof) if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
In addition, in accordance with the FBCA, a Florida corporation is permitted to indemnify any person who may be a party to a derivative action if such person acted in any of the capacities set forth in the preceding paragraph, against expenses and amounts paid in settlement not exceeding, in the judgment of the board of directors, the estimated expenses of litigating the proceeding to conclusion, actually and reasonably incurred in connection with the defense or settlement of such proceeding (including appeals), provided that the person acted under the standards set forth in the preceding paragraph. However, no indemnification shall be made for any claim, issue, or matter for which such person is found to be liable unless, and only to the extent that, the court determines that, despite the adjudication of liability, but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnification for such expenses which the court deems proper.
Any indemnification made under the above provisions, unless pursuant to a court’s determination, may be made only after a determination that the person to be indemnified has met the standard of conduct described above. This determination is to be made by a majority vote of a quorum consisting of the disinterested directors of the board of directors, by duly selected independent legal counsel, or by a majority vote of the disinterested shareholders. The board of directors also may designate a special committee of disinterested directors to make this determination. Notwithstanding the foregoing, a Florida corporation must indemnify any director, officer, employee or agent of a corporation who has been successful in the defense of any proceeding referred to above.
Generally, pursuant to the FBCA, a director of a Florida corporation is not personally liable for monetary damages to our company or any other person for any statement, vote, decision, or failure to act, regarding corporate management or policy, unless: (a) the director breached or failed to perform his duties as a director; and (b) the director’s breach of, or failure to perform, those duties constitutes (i) a violation of criminal law, unless the director had reasonable cause to believe his conduct was lawful or had no reasonable cause to believe his conduct was unlawful, (ii) a transaction from which the director derived an improper personal benefit, either directly or indirectly, (iii) an approval of an unlawful distribution, (iv) with respect to a proceeding by or in the right of the company to procure a judgment in its favor or by or in the right of a shareholder, conscious disregard for the best interest of the company, or willful misconduct, or (v) with respect to a proceeding by or in the right of someone other than the company or a shareholder, recklessness or an act or omission which was committed in bad faith or with malicious purpose or in a manner exhibiting wanton and willful disregard of human rights, safety, or property. The term “recklessness,” as used above, means the action, or omission to act, in conscious disregard of a risk: (a) known, or so obvious that it should have been known, to the directors; and (b) known to the director, or so obvious that it should have been known, to be so great as to make it highly probable that harm would follow from such action or omission.
Furthermore, under the FBCA, a Florida corporation is authorized to make any other further indemnification or advancement of expenses of any of its directors, officers, employees or agents under any bylaw, agreement, vote of shareholders or disinterested directors, or otherwise, both for actions taken in an official capacity and for actions taken in other capacities while holding such office. However, a corporation cannot indemnify or advance expenses if a judgment or other final adjudication establishes that the actions of the director, officer, employee, or agent were material to the adjudicated cause of action and the director, officer, employee, or agent (a) violated criminal law, unless the director, officer, employee, or agent had reasonable cause to believe his or her conduct was unlawful, (b) derived an improper personal benefit from a transaction, (c) was or is a director in a circumstance where the liability for unlawful distributions applies, or (d) engaged in willful misconduct or conscious disregard for the best interests of the corporation in a proceeding by or in right of the corporation to procure a judgment in its favor.
At present, there is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification is required or permitted and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
We maintain a liability insurance policy, pursuant to which our directors and officers may be insured against liability they incur for serving in their capacities as directors and officers of our company, including liabilities arising under the Securities Act or otherwise.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed hereby in the Securities Act and we will be governed by the final adjudication of such issue.
Amendment of our Bylaws
Our bylaws may be adopted, amended or repealed by the affirmative vote of a majority of our outstanding shares. Subject to applicable law, our bylaws also may be adopted, amended or repealed by our Board of Directors.
Item13. Certain Relationships and Related Transactions and Director Independence.
Certain Relationships and Related Party Transactions
Advances
As of December 31, 2015 and 2014, the Company officers and directors have provided advances in the aggregate of $106,505 and $98,759 respectively, for working capital purposes. The advances are unsecured, due on demand and non-interest bearing.
During the year ended December 31, 2014, the Company issued an aggregate of 10,232 shares of common stock in settlement of $298,759 of related party advances and outstanding accounts payable.
On November 4, 2015, the Company issued 3,411 shares of common stock in settlement of $5,860 of related party advances.
Notes payable-related party
Northstar Biotechnology Group, LLC
On February 29, 2012, a note issued to BlueCrest Master Fund Limited was assigned to Northstar Biotechnology Group, LLC (“Northstar”), owned partly by certain directors and existing shareholders of the Company, including Dr. William P. Murphy Jr., Dr. Samuel Ahn and Charles Hart. At the date of the assignment, the principal amount of the BlueCrest note was $544,267.
On March 30, 2012, the Company and Northstar agreed to extend until May 1, 2012 the initial payment date for any and all required monthly under the Note, such that the first of the four monthly payments required under the Note will be due and payable on May, 2012 and all subsequent payments will be due on a monthly basis thereafter commencing on June 1, 2012, and to waive any and all defaults and/or events of default under the Note with respect to such payments. The Company did not make the required payment, and as a result, was in default of the revised agreement The Company renegotiated the terms of the Note and Northstar agreed to suspend the requirement of principal payments by the Company and allow payment of interest-only in common stock.
On September 21, 2012, the Company issued 5,000 common stock purchase warrants to Northstar that was treated as additional interest expense upon issuance.
On October 1, 2012, the Company and Northstar entered into a limited waiver and forbearance agreement providing a recapitalized new note balance comprised of all sums due Northstar with a maturity date extended perpetually. The Company agreed to issue 5,000,000 shares of Series A Convertible Preferred Stock and 10,000 of common stock in exchange for $210,000 as payment towards outstanding debt, default interest, penalties, professional fees outstanding and due Northstar. In addition, the Company executed a security agreement granting Northstar a lien on all patents, patent applications, trademarks, service marks, copyrights and intellectual property rights of any nature, as well as the results of all clinical trials, know-how for preparing Myoblasts, old and new clinical data, existing approved trials, all right and title to Myoblasts, clinical trial protocols and other property rights.
In addition, the Company granted Northstar a perpetual license on products as described for resale, relicensing and commercialization outside the United States. In connection with the granted license, Northstar shall pay the Company a royalty of up to 8% on revenues generated.
Effective October 1, 2012, the effective interest rate was 12.85% per annum. The parties agreed, as of February 28, 2013, to reduce the interest rate to 7% per annum.
In connection with the consideration paid, Northstar waived, from the effective date through the earlier of termination or expiration of the agreement, satisfaction of the obligations as described in the forbearance agreement.
In 2012, 5,000,000 shares of Series A Convertible Preferred Stock were approved to be issued, which was subsequently increased to 20,000,000 shares of preferred stock as Series A Convertible Preferred Stock. In addition, the Company is obligated to issue additional preferred stock equal in lieu of payment of cash of accrued and unpaid interest on each six month anniversary of the effective date (October 1, 2012). In lieu of the initial two payments in preferred stock, the parties have determined to modify the voting rights of the Series A Convertible Preferred Stock from 20 votes per share on matters to be voted on by the common stock holders to 25 votes per share on matters to be voted on by the common stock holders and all prior and subsequent payments of interest will be in common stock. The Company is required to issue additional shares of its common stock (as amended), in lieu of cash, each six month anniversary of the effective date for any accrued and unpaid interest.
As described above, during the year ended December 31, 2013, the Company issued the 5,000,000 shares of Series A Convertible Preferred Stock and the 10,000 of common stock described above in exchange for the $210,000 as payment towards outstanding principle of the debt. In addition, the Company issued 15,000,000 shares of Series A Convertible Preferred Stock as a penalty in settlement of the terms of the forbearance agreement. The fair value of the Preferred Stock of $274,050 was included in interest expense for the year ended December 31, 2013.
On September 30, 2013, the Company issued 8,772 shares of its common stock as payment of $100,000 towards cash advances.
On December 24, 2013, the Company issued 3,916 shares of its common stock as payment of accrued interest through June 30, 2013 of $85,447.
On April 2, 2014, the Company issued 275 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,635 due April 1, 2014 per the forbearance agreement.
On September 17, 2014, limited waiver and forbearance agreement entered into on October 1, 2012 to provide that the perpetual license on products as described for resale, relicensing and commercialization outside the United States was amended as such to condition upon NorthStar providing certain financing, which financing the Company, in its sole discretion, could decline and retain the license.
On October 3, 2014, the Company issued 515 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,705 due October 1, 2014 per the forbearance agreement.
On April 3, 2015, the Company issued 1,363 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,635 due April 1, 2015 per the forbearance agreement.
On October 2, 2015, the Company issued 4,156 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,705 due October 1, 2015 per the forbearance agreement.
On October 7, 2015, the Company issued 34,522 shares of its common stock in settlement of $100,000 principal payment towards the outstanding debt.
As of December 31, 2015 and 2014, the principle of this note was $262,000 and $362,000, respectively.
U.S. Stem Cell Clinic, LLC
On November 6, 2015, the Company issued a 4% promissory note to U.S. Stem Cell Clinic, Inc for $30,000 due November 3, 2017 (See Note 3).
Officer and Director Notes
|
2015
|
2014
|
|||||||
|
Notes payable, Dr. Murphy
|
$
|
-
|
$
|
465,240
|
||||
|
Note payable, Beverly Murphy
|
50,000
|
50,000
|
||||||
|
Note payable, Mr. Tomas
|
252,250
|
331,354
|
||||||
|
Note payable, Mr. Tomas
|
375,000
|
375,000
|
||||||
|
Note payable, Mr. Tomas
|
500,000
|
500,000
|
||||||
|
Note payable, Ms. Comella
|
287,772
|
299,465
|
||||||
|
Total
|
$
|
1,465,022
|
$
|
2,021,059
|
||||
Notes payable, Dr. Murphy
At December 31, 2014, the Company has outstanding notes payable to Dr. Murphy with interest at 8% per annum due at maturity in aggregate $465,240. Of the outstanding balance, certain subordinated notes totaling $100,000 and $140,000 were previously due on November 30, 2012 and June 4, 2011 respectively, and are unsecured. The Company is not obligated to make payment until Northstar (or successor) Loan is paid off.
On June 20, 2014, the Company issued 10,333 shares of its common stock in settlement of $298,386 of related party note payable, $2,000 advances and accrued interest of $112,954.
On November 4, 2015, the Company issued 270,800 shares of common stock in settlement of $465,200 of outstanding notes payable to Dr. Murphy.
Note payable, Ms. Murphy
At December 31, 2015 and 2014, the Company has outstanding note payable of $50,000 due to Beverly Murphy with interest at 7% per annum due at maturity at October 15, 2015.
Note payable, Dr. Ahn
At December 31, 2013, the Company has outstanding note payable for $125,000 to Dr. Ahn with interest at 8% per annum due at maturity.
On June 20, 2014, the Company issued 4,046 shares of its common stock in settlement of $125,000 of related party note payable and accrued interest of $36,832. There were no amounts outstanding as of December 31, 2015 and 2014.
Notes payable, Mr. Tomas
In 2013, the Company issued a promissory note payable for previous advances and accrued compensation. The promissory note bears interest of 5% per annum and due on demand. During the year ended December 31, 2015 and 2014, the Company paid off $79,104 and $186,779 of the outstanding promissory note, respectively. The principle outstanding balance of this note as of December 31, 2015 is $252,250.
On August 1, 2013, the Company issued a $375,000 promissory note due on demand in settlement of accrued compensation. The promissory note bears interest of 5% per annum and is due on demand. The principle outstanding balance of this note as of December 31, 2015 is $375,000.
On July 1, 2014, the Company issued a $500,000 promissory note in settlement of accrued compensation. The promissory note bears interest of 5% per annum and was due on January 1, 2015. The principle outstanding balance of this note as of December 31, 2015 is $500,000.
Notes payable, Ms. Comella
On August 1, 2013, the Company issued a $125,000 promissory notes due on demand in settlement of accrued compensation. The promissory note bear interest of 5% per annum and due on demand. During the year ended December 31, 2014 and 2013, the Company paid off $97,718 and $27,282 of the outstanding promissory note. The principle outstanding balance of these notes as of December 31, 2015 is $-0-.
On July 1, 2014, the Company issued a $300,000 promissory note in settlement of accrued compensation. The promissory note bears interest of 5% per annum and due on January 1, 2015. During the years ended December 31, 2015 and 2014, the Company paid off $11,693 and $535 of the outstanding promissory note, respectively. The principle outstanding balance of this note as of December 31, 2015 is $287,772.
Transactions with Pavillion
During the years ended December 31, 2015 and 2014, the Company purchased $242,271 and $302,899 lab kits from Pavillion, Inc., respectively, a related party whose owner is related to an officer of the Company. As of December 31, 2015 and 2014, the Company had $74,793 and $23,862, respectively, in accounts payable owed to Pavillion.
Item14. Principal Accounting Fees and Services. Independent Registered Public Accounting Firm Fees
In December of 2012, U.S. Stem Cell engaged Fiondella, Milone, LaSaracina LLP (FML) to perform the 2012 audit. Aggregate fees billed to us for the fiscal years ended December 31, 2015 and 2014 by our independent registered public accounting firms are as follows:
|
Types of Fees
|
2015
|
2014
|
||||||
|
Audit Fees (1)
|
$
|
80,000
|
$
|
80,000
|
||||
|
Audit Related Fees
|
$ |
13,000
|
$ |
16,000
|
||||
|
Tax Fees
|
$ |
5,000
|
$ |
5,130
|
||||
|
All Other Fees
|
—
|
—
|
||||||
|
(1)
|
This category also includes advice on audit and accounting matters that arose during, or as a result of, the audit of the annual financial statements or the reviews of the interim financial statements.
|
Audit Committee Pre-Approval Policy
Consistent with policies of the SEC regarding auditor independence, the Audit Committee has responsibility for the appointment, compensation and oversight of the work of the independent auditor. As part of this responsibility, the Audit Committee has adopted, and our Board has ratified, an Audit and Non-Audit Services Pre-Approval Policy pursuant to which the Audit Committee is required to pre-approve the audit and non-audit services performed by our independent registered public accounting firm in order to assure that these services do not impair the auditor’s independence from us.
Prior to engagement of the independent auditor for the next year’s audit, the independent auditor and the Audit Committee will review a list of services and related fees expected to be rendered during that year within each of four categories of services to the Audit Committee for approval:
(i) Audit Services: Audit services include the annual financial statement audit (including required quarterly reviews), equity investment audits and other procedures required to be performed by the independent auditor to be able to form an opinion on our financial statements. Audit Services also include information systems and procedural reviews and testing performed in order to understand and place reliance on the systems of internal control, and consultations relating to the audit or quarterly review as well as the attestation engagement for the independent auditor’s report on management’s report on internal controls for financial reporting.
(ii) Audit-Related Services: Audit-related services are assurance and related services that are reasonably related to the performance of the audit or review of our financial statements, including due diligence related to potential business acquisitions/dispositions, accounting consultations related to accounting, financial reporting or disclosure matters not classified as “Audit Services,” assistance with understanding and implementing new accounting and financial reporting guidance from rulemaking authorities, financial audits of employee benefit plans, agreed-upon or expanded audit procedures related to accounting and/or billing records required to respond to or comply with financial, accounting or regulatory reporting matters and assistance with internal control reporting requirements.
(iii) Tax Services: Tax services include services such as tax compliance, tax planning and tax advice; however, the Audit Committee will not permit the retention of the independent registered public accounting firm in connection with a transaction initially recommended by the independent registered public accounting firm, the sole business purpose of which may be tax avoidance and treatment which may not be supported in the Internal Revenue Code and related regulations.
(iv) All Other Services: All other services are those permissible non-audit services that the Audit Committee believes are routine and recurring and would not impair the independence of the auditor and are consistent with the SEC’s rules on auditor independence.
Prior to engagement, the Audit Committee pre-approves the services and fees of the independent auditor within each of the above categories. During the year, it may become necessary to engage the independent auditor for additional services not previously contemplated as part of the engagement. In those instances, the Audit and Non-Audit Services Pre-Approval Policy requires that the Audit Committee specifically approve the services prior to the independent auditor’s commencement of those additional services. Under the Audit and Non-Audit Services Pre-Approval Policy, the Audit Committee may delegate the ability to pre-approve audit and non-audit services to one or more of its members provided the delegate reports any pre-approval decision to the Audit Committee at its next scheduled meeting. As of the date hereof, the Audit Committee has not delegated its ability to pre-approve audit services.
PART IV
Item 15. Exhibits, Financial Statement Schedules.
(a)(1) Financial Statements
See Item 8. “Financial Statements and Supplementary Data” for Financial Statements included with this Annual Report on Form 10-K.
(a)(2) Financial Statement Schedules
All schedules have been omitted because the required information is not applicable or the information is included in the financial statements or the notes thereto.
(a)(3) Exhibits
|
Exhibit No.
|
Exhibit Description
|
|
|
3.1(6)
|
Amended and Restated Articles of Incorporation of the registrant, as amended
|
|
|
3.2(9)
|
Articles of Amendment to the Articles of Incorporation of the registrant
|
|
|
3.3(37)
|
Articles of Amendment to the Articles of Incorporation of the registrant
|
|
|
3.4 (8)
|
Amended and Restated Bylaws
|
|
|
4.1(5)
|
Loan and Security Agreement, dated as of May 31, 2007 by and between BlueCrest Capital Finance, L.P. and the registrant
|
|
|
4.2(12)
|
Notice of Event of Default, from BlueCrest Venture Finance Master Fund Limited to the Company, dated January 28, 2009
|
|
|
4.3(12)
|
Notice of Acceleration, from BlueCrest Venture Finance Master Fund Limited to the Company, dated February 2, 2009
|
|
|
4.4(13)
|
Amendment to Loan and Security Agreement, between the Company and BlueCrest Venture Finance Master Fund Limited, dated as of April 2, 2009
|
|
|
4.5(13)
|
Grant of Security Interest (Patents), between the Company and BlueCrest Venture Finance Master Fund Limited, dated as of April 2, 2009
|
|
|
4.6(13)
|
Security Agreement (Intellectual Property), between the Company and BlueCrest Venture Finance Master Fund Limited, dated as of April 2, 2009
|
|
|
4.7(13)
|
Subordination Agreement, by Hunton & Williams, LLP in favor of BlueCrest Venture Finance Master Fund Limited, entered into and effective April 2, 2009
|
|
|
4.8(13)
|
Amended and Restated Promissory Note, dated April 2, 2009, by the Company to BlueCrest Venture Finance Master Fund Limited
|
|
|
4.9(13)
|
Warrant to purchase 1,315,542 shares of the registrant’s common stock, dated April 2, 2009, issued to BlueCrest Venture Finance Master Fund Limited
|
|
|
4.10(14)
|
Warrant to purchase 451,043 shares of the registrant’s common stock, dated April 2, 2009, issued to Rogers Telecommunications Limited
|
|
|
4.11(14)
|
Warrant to purchase 173,638 shares of the registrant’s common stock, dated April 2, 2009, issued to Hunton & Williams, LLP
|
|
|
4.12(4)
|
Warrant to purchase shares of the registrant’s common stock issued to Howard J. Leonhardt and Brenda Leonhardt
|
|
|
4.12(19)
|
10% Convertible Promissory Note Due July 23, 2010, in the amount of $20,000, payable to Dana Smith
|
|
|
4.13(19)
|
10% Convertible Promissory Note Due July 23, 2010, in the amount of $100,000, payable to Bruce Meyers
|
|
|
4.14(19)
|
Registration Rights Agreement, dated July 23, 2009
|
|
|
4.15(4)
|
Warrant to purchase shares of the registrant’s common stock issued to the R&A Spencer Family Limited Partnership
|
|
|
4.15(19)
|
Subordination Agreement, dated July 23, 2009
|
|
|
4.16(19)
|
Note Purchase Agreement, dated July 23, 2009
|
|
|
4.17(19)
|
Closing Confirmation of Conversion Election, dated July 23, 2009
|
|
|
4.20(6)
|
Warrant to purchase shares of the registrant’s common stock issued to Samuel S. Ahn, M.D.
|
|
|
4.23(7)
|
Warrant to purchase shares of the registrant’s common stock issued to Howard and Brenda Leonhardt
|
|
|
4.27(11)
|
Form of Warrant Agreement for October 2008 Private Placement
|
|
|
4.30(19)
|
10% Convertible Promissory Note Due July 23, 2010, in the amount of $100,000, payable to Bruce Meyers
|
|
4.31 (34)
|
Series A Convertible Preferred Stock
|
|
|
4.32 (35)
|
Amendment to the Series A Convertible Preferred Stock
|
|
|
10.1**(1)
|
1999 Officers and Employees Stock Option Plan
|
|
|
10.2**(1)
|
1999 Directors and Consultants Stock Option Plan
|
|
|
10.3(1)
|
Form of Option Agreement under 1999 Officers and Employees Stock Option Plan
|
|
|
10.4(3)
|
Form of Option Agreement under 1999 Directors and Consultants Stock Option Plan
|
|
|
10.5**(4)
|
Employment Letter Agreement between the registrant and Scott Bromley, dated August 24, 2006.
|
|
|
10.6(1)
|
Lease Agreement between the registrant and Sawgrass Business Plaza, LLC, as amended, dated November 14, 2006.
|
|
|
10.7(1)
|
Asset Purchase Agreement between the registrant and Advanced Cardiovascular Systems, Inc., dated June 24, 2003.
|
|
|
10.8(4)
|
Conditionally Exclusive License Agreement between the registrant, Dr. Peter Law and Cell Transplants International, LLC, dated February 7, 2000, as amended.
|
|
|
10.9(4)
|
Loan Guarantee, Payment and Security Agreement, dated as of June 1, 2007, by and between the registrant, Howard J. Leonhardt and Brenda Leonhardt
|
|
|
10.10(4)
|
Loan Guarantee, Payment and Security Agreement, dated as of June 1, 2007, by and between the registrant and William P. Murphy Jr., M.D.
|
|
|
10.11(4)
|
Loan Agreement, dated as of June 1, 2007, by and between the registrant and Bank of America, N.A.
|
|
|
10.13(4)
|
Warrant to purchase shares of the registrant’s common stock issued to Howard J. Leonhardt and Brenda Leonhardt
|
|
|
10.14(4)
|
Warrant to purchase shares of the registrant’s common stock issued to William P. Murphy, Jr., M.D.
|
|
|
10.16(4)
|
Material Supply Agreement, dated May 10, 2007, by and between the registrant and Biosense Webster
|
|
|
10.17(5)
|
Warrant to purchase shares of the registrant’s common stock issued to BlueCrest Capital Finance, L.P.
|
|
|
10.18(6)
|
Loan Guarantee, Payment and Security Agreement, dated as of September 12, 2007, by and between the registrant and Samuel S. Ahn, M.D.
|
|
|
10.19(6)
|
Loan Guarantee, Payment and Security Agreement, dated as of September 12, 2007, by and between the registrant and Dan Marino
|
|
|
10.21(6)
|
Loan Guarantee, Payment and Security Agreement, dated as of September 19, 2007, by and between the registrant and Jason Taylor
|
|
|
10.22(7)
|
Loan Guarantee, Payment and Security Agreement, dated as of October 10, 2007, by and between the registrant and Howard and Brenda Leonhardt
|
|
|
10.24(7)
|
Second Amendment to Loan Guarantee, Payment and Security Agreement, dated as of October 10, 2007, by and between the registrant and Howard and Brenda Leonhardt
|
|
|
10.25(7)
|
Second Amendment to Loan Guarantee, Payment and Security Agreement, dated as of October 10, 2007, by and between the registrant and William P. Murphy, Jr., M.D.
|
|
|
10.26**(10)
|
U.S. Stem Cell, Inc. Omnibus Equity Compensation Plan
|
|
|
10.28(11)
|
Form of Registration Rights Agreement for October 2008 Private Placement
|
|
|
10.29(19)
|
10% Convertible Promissory Note Due July 23, 2010, in the amount of $20,000, payable to Dana Smith
|
|
|
10.31(19)
|
Registration Rights Agreement, dated July 23, 2009
|
|
|
10.32(19)
|
Subordination Agreement, dated July 23, 2009
|
|
|
10.33(19)
|
Note Purchase Agreement, dated July 23, 2009
|
|
|
10.34(19)
|
Closing Confirmation of Conversion Election, dated July 23, 2009
|
|
|
10.35**(20)
|
Amended and Restated 1999 Directors and Consultants Stock Option Plan
|
|
|
10.36(21)
|
Preliminary Commitment Letter with Seaside National Bank and Trust, dated September 30, 2010.
|
|
10.37(22)
|
Loan Agreement with Seaside National Bank and Trust, dated October 25, 2010.
|
|
|
10.38(22)
|
Promissory Note with Seaside National Bank and Trust, dated October 25, 2010.
|
|
|
10.39(22)
|
Amended and Restated Loan and Security Agreement with BlueCrest Venture Finance Master Fund Limited, dated October 25, 2010.
|
|
|
10.40(23)
|
Form of Subscription Agreement, executed November 30, 2010.
|
|
|
10.41(23)
|
Form of Common Stock Purchase Warrant, issued November 30, 2010.
|
|
|
10.42(23)
|
Form of Registration Rights Agreement, dated November 30, 2010.
|
|
|
10.43(24)
|
Unsecured Convertible Promissory Note for $25,000, with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.44(24)
|
Promissory Note for $139,728.82 with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.45(24)
|
Securities Purchase Agreement with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.46(24)
|
Subordination Agreement, dated January 3, 2011.
|
|
|
10.47(24)
|
Notice of Conversion Election, dated January 3, 2011.
|
|
|
10.48(25)
|
Unsecured Convertible Promissory Note for $34,750, with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.49(25)
|
Promissory Note for $139,728.82 with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.50(25)
|
Securities Purchase Agreement with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.51(25)
|
Subordination Agreement, dated May 16, 2011.
|
|
|
10.52(26)
|
Promissory Note for $139,728.82 with Lotus Funding Group, LLC, dated June 15, 2011.
|
|
|
10.53(26)
|
Partial Assignment and Modification Agreement, dated June 15, 2011.
|
|
|
10.54(26)
|
Subordination Agreement, dated June 15, 2011.
|
|
|
10.55(27)
|
Promissory Note for $140,380.21 with Greystone Capital Partners, dated July 8, 2011.
|
|
|
10.56(27)
|
Partial Assignment and Modification Agreement, dated July 8, 2011.
|
|
|
10.57 (28)
|
Subordination Agreement, dated July 8, 2011.
|
|
|
10.58 (29)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated August 1, 2011.
|
|
|
10.59 (29)
|
Partial Assignment and Modification Agreement, dated August 1, 2011.
|
|
|
10.60 (29)
|
Subordination Agreement, dated August 1, 2011.
|
|
|
10.61 (30)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated September 1, 2011.
|
|
|
10.62 (30)
|
Partial Assignment and Modification Agreement, dated September 1, 2011.
|
|
|
10.63 (30)
|
Subordination Agreement, dated September 1, 2011.
|
|
|
10.64 (31)
|
Standby Equity Distribution Agreement dated as of November 2, 2011.
|
|
|
10.65 (31)
|
Registration Rights Agreement dated as of November 2, 2011.
|
|
|
10.66(32)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated January 3, 2012
|
|
|
10.67(32)
|
Term Note B Promissory Note for $139,728.82 with Greystone Capital Partners, dated January 3, 2012
|
|
|
10.68(32)
|
Unsecured Convertible Promissory Note for $63,000, with Asher Enterprises, Inc. dated April 2, 2012
|
|
|
10.69(32)
|
Unsecured Convertible Promissory Note for $125,000, with IBC Funds LLC., dated January 9, 2013
|
|
|
10.70(32)
|
Unsecured Convertible Promissory Note for $37,500, with Asher Enterprises, Inc. dated February 20, 2013
|
|
10.37(22)
|
Loan Agreement with Seaside National Bank and Trust, dated October 25, 2010.
|
|
|
10.38(22)
|
Promissory Note with Seaside National Bank and Trust, dated October 25, 2010.
|
|
|
10.39(22)
|
Amended and Restated Loan and Security Agreement with BlueCrest Venture Finance Master Fund Limited, dated October 25, 2010.
|
|
|
10.40(23)
|
Form of Subscription Agreement, executed November 30, 2010.
|
|
|
10.41(23)
|
Form of Common Stock Purchase Warrant, issued November 30, 2010.
|
|
|
10.42(23)
|
Form of Registration Rights Agreement, dated November 30, 2010.
|
|
|
10.43(24)
|
Unsecured Convertible Promissory Note for $25,000, with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.44(24)
|
Promissory Note for $139,728.82 with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.45(24)
|
Securities Purchase Agreement with Magna Group, LLC, dated January 3, 2011.
|
|
|
10.46(24)
|
Subordination Agreement, dated January 3, 2011.
|
|
|
10.47(24)
|
Notice of Conversion Election, dated January 3, 2011.
|
|
|
10.48(25)
|
Unsecured Convertible Promissory Note for $34,750, with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.49(25)
|
Promissory Note for $139,728.82 with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.50(25)
|
Securities Purchase Agreement with Magna Group, LLC, dated May 16, 2011.
|
|
|
10.51(25)
|
Subordination Agreement, dated May 16, 2011.
|
|
|
10.52(26)
|
Promissory Note for $139,728.82 with Lotus Funding Group, LLC, dated June 15, 2011.
|
|
|
10.53(26)
|
Partial Assignment and Modification Agreement, dated June 15, 2011.
|
|
|
10.54(26)
|
Subordination Agreement, dated June 15, 2011.
|
|
|
10.55(27)
|
Promissory Note for $140,380.21 with Greystone Capital Partners, dated July 8, 2011.
|
|
|
10.56(27)
|
Partial Assignment and Modification Agreement, dated July 8, 2011.
|
|
|
10.57 (28)
|
Subordination Agreement, dated July 8, 2011.
|
|
|
10.58 (29)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated August 1, 2011.
|
|
|
10.59 (29)
|
Partial Assignment and Modification Agreement, dated August 1, 2011.
|
|
|
10.60 (29)
|
Subordination Agreement, dated August 1, 2011.
|
|
|
10.61 (30)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated September 1, 2011.
|
|
|
10.62 (30)
|
Partial Assignment and Modification Agreement, dated September 1, 2011.
|
|
|
10.63 (30)
|
Subordination Agreement, dated September 1, 2011.
|
|
|
10.64 (31)
|
Standby Equity Distribution Agreement dated as of November 2, 2011.
|
|
|
10.65 (31)
|
Registration Rights Agreement dated as of November 2, 2011.
|
|
|
10.66(32)
|
Promissory Note for $139,728.82 with Greystone Capital Partners, dated January 3, 2012
|
|
|
10.67(32)
|
Term Note B Promissory Note for $139,728.82 with Greystone Capital Partners, dated January 3, 2012
|
|
|
10.68(32)
|
Unsecured Convertible Promissory Note for $63,000, with Asher Enterprises, Inc. dated April 2, 2012
|
|
|
10.69(32)
|
Unsecured Convertible Promissory Note for $125,000, with IBC Funds LLC., dated January 9, 2013
|
|
|
10.70(32)
|
Unsecured Convertible Promissory Note for $37,500, with Asher Enterprises, Inc. dated February 20, 2013
|
|
10.71(32)
|
Unsecured Convertible Promissory Note for $42,500, with Asher Enterprises, Inc. dated January 9, 2013
|
|
|
10.72**(33)
|
2013 U.S. Stem Cell, Inc. Omnibus Equity Compensation Plan
|
|
|
10.73 (34)
|
Securities Purchase Agreement, dated as of October 7, 2014, by and between Magna Holdings I, LLC and U.S. Stem Cell, Inc.
|
|
|
10.74(34)
|
Registration Rights Agreement, dated as of October 7, 2014, by and between Magna Holdings I, LLC and U.S. Stem Cell, Inc.
|
|
|
10.75(34)
|
Common Stock Purchase Agreement, dated as of October 23, 2014, by and between Magna Equities II, LLC and U.S. Stem Cell, Inc.
|
|
|
10.76(34)
|
Registration Rights Agreement, dated as of October 23, 2014, by and between Magna Equities II, LLC and U.S. Stem Cell, Inc.
|
|
|
10.77**(35)
|
2013 Omnibus Equity Compensation Plan Amendment One.
|
|
|
10.78 (36)
|
Senior Convertible Note, dated October 1, 2015
|
|
|
10.79 (36)
|
Securities Purchase Agreement, dated as of October 1, 2015, by and between Magna Equities II, LLC and U.S. Stem Cell, Inc.
|
|
|
10.80(36)
|
Registration Rights Agreement, dated as of October 1, 2015, by and between Magna Holdings I, LLC and U.S. Stem Cell, Inc.
|
|
|
10.81(38)
|
Senior Convertible Note, dated December 3, 2015
|
|
|
10.82 (38)
|
Amended and Restated Senior Convertible Note, dated December 3, 2015.
|
|
|
10.83 (38)
|
Securities Purchase Agreement, dated as of December 3, 2015, by and between Magna Equities II, LLC and U.S. Stem Cell, Inc.
|
|
|
10.84 (38)
|
Registration Rights Agreement, dated as of December 3, 2015, by and between Magna Holdings I, LLC and U.S. Stem Cell, Inc.
|
|
|
14.2(2)
|
Code of Business Conduct and Ethics
|
|
|
31.1*
|
||
|
32.1*
|
|
*
|
Filed herewith
|
|
|
**
|
Indicates management contract or compensatory plan.
|
|
|
(1)
|
Incorporated by reference to the Company’s Form S-1 filed with the Securities and Exchange Commission (the “SEC”) on February 13, 2007.
|
|
|
(2)
|
Incorporated by reference to Amendment No. 1 to the Company’s Form S-1 filed with the SEC on June 5, 2007.
|
|
|
(3)
|
Incorporated by reference to Amendment No. 2 to the Company’s Form S-1 filed with the SEC on July 12, 2007.
|
|
|
(4)
|
Incorporated by reference to Amendment No. 3 to the Company’s Form S-1 filed with the SEC on August 9, 2007.
|
|
|
(5)
|
Incorporated by reference to Amendment No. 4 to the Company’s Form S-1 filed with the SEC on September 6, 2007.
|
|
|
(6)
|
Incorporated by reference to Amendment No. 5 to the Company’s Form S-1 filed with the SEC on October 1, 2007.
|
|
|
(7)
|
Incorporated by reference to Post-effective Amendment No. 1 to the Company’s Form S-1 filed with the SEC on October 11, 2007.
|
|
|
(8)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on July 3, 2008.
|
|
|
(9)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on August 8, 2008.
|
|
|
(10)
|
Incorporated by reference to the Company’s Quarterly Report on Form 10-Q filed with the SEC on August 14, 2008.
|
|
(11)
|
Incorporated by reference to the Company’s Quarterly Report on Form 10-Q filed with the SEC on November 14, 2008.
|
|
|
(12)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on February 3, 2009.
|
|
|
(13)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on April 8, 2009.
|
|
|
(14)
|
Incorporated by reference to the Company’s Annual Report on Form 10-K filed with the SEC on April 15, 2009.
|
|
|
(15)
|
Incorporated by reference to the Company’s Annual Report on Form 10-K/A filed with the SEC on April 30, 2009.
|
|
|
(16)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on May 18, 2009.
|
|
|
(17)
|
Incorporated by reference to the Company’s Quarterly Report on Form 10-Q filed with the SEC on May 20, 2009.
|
|
|
(18)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on July 9, 2009.
|
|
|
(19)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on August 3, 2009.
|
|
|
(20)
|
Incorporated by reference to Exhibit 4.6 to the Company’s Post-Effective Amendment to Registration Statement on Form S-8/A, filed with the SEC on June 2, 2010.
|
|
|
(21)
|
Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on October 6, 2010.
|
|
|
(22)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on October 29, 2010.
|
|
|
(23)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on December 6, 2010.
|
|
|
(24)
|
Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on January 12, 2011.
|
|
|
(25)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on May 25, 2011
|
|
|
(26)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on June 21, 2011
|
|
|
(27)
|
Incorporated by reference to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 15. 2011
|
|
|
(28)
|
Incorporated by reference to the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 14, 2011
|
|
|
(29)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on January 13, 2012
|
|
|
(30)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on January 30, 2012
|
|
|
(31)
|
Incorporated by reference to the Company Registration Statement on Form S-1/A filed with the SEC on February 8, 2012
|
|
|
(32)
|
Incorporated by reference to the Company Annual Report on Form 10-K filed with the SEC on March 29, 2013
|
|
|
(33)
|
Incorporated by reference to the Company Quarterly Report on Form 10-Q filed with the SEC on May 9, 2013
|
|
|
(34)
|
Incorporated by reference to the Company’s Registration Statement on Form S-1/A filed with the SEC on December 12, 2014.
|
|
|
(35)
|
Incorporated by reference to the Company’s Definitive Proxy Statement on Schedule 14A filed with the SEC on December 19, 2014.
|
|
|
(36)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on October 2, 2015.
|
|
|
(37)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on November 4, 2015.
|
|
|
(38)
|
Incorporated by reference to the Company Current Report on Form 8-K filed with the SEC on December 4, 2015.
|
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
U.S. STEM CELL, INC.
|
|||
|
By:
|
/s/ Mike Tomas
|
||
|
Mike Tomas
Chief Executive Officer & President
|
|||
|
March 8, 2016
|
|||
|
By:
|
/s/ Mike Tomas
|
||
|
Mike Tomas
Chief Financial Officer (Principal Accounting Officer)
|
|||
|
March 8, 2016
|
|||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Each person whose signature appears below, hereby authorizes Mike Tomas, as attorney in fact to sign on his or her behalf, individually, in each capacity stated below, and to file all amendments or supplements to this annual report on Form 10-K.
|
SIGNATURE
|
TITLE
|
DATE
|
||
|
/s/ William P. Murphy, Jr., M.D.
|
Chairman of the Board
|
March 8, 2016
|
||
|
William P. Murphy, Jr., M.D.
|
||||
|
/s/ Mike Tomas
|
Chief Executive Officer, Chief Financial Officer, & Director
|
March 8, 2016
|
||
|
Mike Tomas
|
||||
|
/s/ Mark P. Borman
|
Director
|
March 8, 2016
|
||
|
Mark Borman
|
|
/s/ Kristin Comella
|
Director
|
March 8, 2016
|
||
|
Kristin Comella
|
||||
|
/s/ Charles A. Hart
|
Director
|
March 8, 2016
|
||
|
Charles A. Hart
|
||||
|
/s/ Samuel S. Ahn, MD, MBA
|
Director
|
March 8, 2016
|
||
|
Samuel S. Ahn, MD, MBA
|
||||
|
/s/ Sheldon Anderson
|
Director
|
March 8, 2016
|
||
|
Sheldon Anderson
|
As required under Item 15. Exhibits, Financial Statement Schedules, the exhibits filed as part of this report are provided in this separate section. The exhibits included in this section are as follows:
|
Exhibit No.
|
Description
|
|
|
31.1
|
||
|
32.1
|
FORM 10-K—ITEM 8
U.S. STEM CELL, INC.
INDEX TO FINANCIAL STATEMENTS
|
F-2
|
|
|
F-3
|
|
|
F-4
|
|
|
F-5
|
|
|
F-7
|
|
|
F-8
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of US Stem Cell, Inc.
13794 NW 4th Street, Suite 212,
Sunrise, Florida 33325
We have audited the accompanying balance sheets of US Stem Cell, Inc. (the “Company”) as of December 31, 2015 and 2014, and the related statements of operations, stockholders’ deficit, and cash flows for each of the years then ended. The Company’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits include consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of US Stem Cell, Inc. as of December 31, 2015 and 2014, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and used significant amounts of cash in its operations. In addition, at December 31, 2015 the Company’s current liabilities exceed its current assets by $5,588,949. Those conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding those matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/Fiondella, Milone & LaSaracina LLP
Glastonbury, Connecticut
March 8, 2016
U.S. STEM CELL, INC.
BALANCE SHEETS
DECEMBER 31, 2015 AND 2014
|
2015
|
2014
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 58,372 | $ | 36,674 | ||||
|
Accounts receivable, net
|
35,032 | 95,409 | ||||||
|
Inventory
|
17,406 | - | ||||||
|
Prepaid and other
|
4,832 | 9,255 | ||||||
|
Total current assets
|
115,642 | 141,338 | ||||||
|
Property and equipment, net
|
14,172 | 12,686 | ||||||
|
Other assets:
|
||||||||
|
Investments
|
89,139 | 40,597 | ||||||
|
Deposits
|
10,160 | 10,160 | ||||||
|
Total assets
|
$ | 229,113 | $ | 204,781 | ||||
|
LIABILITIES AND STOCKHOLDERS' DEFICIT
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable, including $104,089 and $56,953 to related parties, respectively
|
$ | 1,503,501 | $ | 1,991,979 | ||||
|
Accrued expenses
|
726,751 | 2,373,615 | ||||||
|
Advances, related party
|
106,505 | 98,759 | ||||||
|
Deferred revenue
|
71,961 | 13,000 | ||||||
|
Deposits
|
465,286 | 465,286 | ||||||
|
Promissory note, net of debt discount of $78,864 and $-0- respectively
|
71,136 | 1,500,000 | ||||||
|
Notes payable, related party
|
1,727,022 | 2,383,059 | ||||||
|
Notes payable, net of debt discount of $249,205 and $339,160, respectively
|
608,502 | 1,531,812 | ||||||
|
Derivative liabilities
|
423,927 | 741,271 | ||||||
|
Total current liabilities
|
5,704,591 | 11,098,781 | ||||||
|
Long term debt:
|
||||||||
|
Promissory note, long term portion, net of debt discount of $240,521
|
1,232,241 | - | ||||||
|
Notes payable, long term portion
|
983,727 | - | ||||||
|
Note payable, long term portion, related party
|
30,000 | - | ||||||
|
Total long term debt
|
2,245,968 | - | ||||||
|
Total liabilities
|
7,950,559 | 11,098,781 | ||||||
|
Commitments and contingencies
|
- | - | ||||||
|
Stockholders' deficit:
|
||||||||
|
Preferred stock, par value $0.001; 20,000,000 shares authorized, 20,000,000 issued and outstanding
|
20,000 | 20,000 | ||||||
|
Common stock, par value $0.001; 2,000,000,000 shares authorized, 1,813,689 and 581,433 shares issued and 1,728,478 and 581,433 outstanding as of December 31, 2015 and 2014, respectively
|
1,814 | 581 | ||||||
|
Additional paid in capital
|
114,555,110 | 109,519,913 | ||||||
|
Treasury stock, 85,211 shares
|
(221,996 | ) | - | |||||
|
Accumulated deficit
|
(122,076,374 | ) | (120,434,494 | ) | ||||
|
Total stockholders' deficit
|
(7,721,446 | ) | (10,894,000 | ) | ||||
|
Total liabilities and stockholders' deficit
|
$ | 229,113 | $ | 204,781 | ||||
See the accompanying notes to these financial statements.
U.S. STEM CELL, INC.
STATEMENTS OF OPERATIONS
|
Year Ended December 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Revenue:
|
||||||||
|
Products
|
$ | 1,356,864 | $ | 1,155,543 | ||||
|
Services
|
834,313 | 900,288 | ||||||
|
Total revenue
|
2,191,177 | 2,055,831 | ||||||
|
Cost of sales
|
972,957 | 844,941 | ||||||
|
Gross profit
|
1,218,220 | 1,210,890 | ||||||
|
Cost and operating expenses:
|
||||||||
|
Research and development
|
10,000 | 66,420 | ||||||
|
Marketing, general and administrative
|
3,808,628 | 4,669,432 | ||||||
|
Depreciation and amortization
|
5,326 | 4,490 | ||||||
|
Total operating expenses
|
3,823,954 | 4,740,342 | ||||||
|
Net loss from operations
|
(2,605,734 | ) | (3,529,452 | ) | ||||
|
Other income (expenses):
|
||||||||
|
Gain on settlement of debt
|
2,620,233 | 2,498,676 | ||||||
|
Gain on change of fair value of derivative liability
|
15,095 | 425,953 | ||||||
|
Income (loss) from equity investment
|
38,542 | (9,117 | ) | |||||
|
Other income
|
13,282 | - | ||||||
|
Interest expense
|
(1,723,298 | ) | (1,639,571 | ) | ||||
|
Total other income (expenses)
|
963,854 | 1,275,941 | ||||||
|
Net loss before income taxes
|
(1,641,880 | ) | (2,253,511 | ) | ||||
|
Income taxes (benefit)
|
- | - | ||||||
|
NET LOSS
|
$ | (1,641,880 | ) | $ | (2,253,511 | ) | ||
|
Net loss per common share, basic and diluted
|
$ | (1.80 | ) | $ | (4.61 | ) | ||
|
Weighted average number of common shares outstanding, basic and diluted
|
912,062 | 488,667 | ||||||
See the accompanying notes to these financial statements.
U.S. STEM CELL, INC.
STATEMENT OF STOCKHOLDERS' DEFICIT
TWO YEARS ENDED DECEMBER 31, 2015
|
Additional
|
||||||||||||||||||||||||||||||||||||
|
Preferred stock
|
Common stock
|
Paid in
|
Treasury
|
Subscription
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Stock
|
Receivable
|
Deficit
|
Total
|
||||||||||||||||||||||||||||
|
Balance, December 31, 2013
|
20,000,000 | $ | 20,000 | 379,787 | $ | 380 | $ | 104,198,527 | $ | - | $ | 215,000 | $ | (118,180,983 | ) | $ | (13,747,076 | ) | ||||||||||||||||||
|
Common stock issued in settlement of accounts payable
|
- | - | 48,585 | 48 | 1,071,107 | - | - | - | 1,071,155 | |||||||||||||||||||||||||||
|
Common stock issued in connection with settlement of other debt
|
- | - | 83,236 | 83 | 2,230,724 | - | - | - | 2,230,807 | |||||||||||||||||||||||||||
|
Common stock issued for services
|
- | - | 3,840 | 4 | 183,987 | - | - | - | 183,991 | |||||||||||||||||||||||||||
|
Common stock issued upon exercise of warrants
|
- | - | 11,918 | 12 | 135,988 | - | - | - | 136,000 | |||||||||||||||||||||||||||
|
Common stock issued as commitment shares in connection with equity purchase agreement
|
- | - | 12,000 | 12 | (12 | ) | - | - | - | - | ||||||||||||||||||||||||||
|
Proceeds from issuance of common stock
|
- | - | 42,067 | 42 | 694,583 | - | (215,000 | ) | - | 479,625 | ||||||||||||||||||||||||||
|
Reclassify fair value of derivative liability to equity upon debt extinguishment
|
- | - | - | - | 128,624 | - | - | - | 128,624 | |||||||||||||||||||||||||||
|
Fair value of vesting warrants issued in connection with revenue sharing and intellectual property
|
- | - | - | - | 190,262 | - | - | - | 190,262 | |||||||||||||||||||||||||||
|
Stock based compensation
|
- | - | - | - | 686,123 | - | - | - | 686,123 | |||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | (2,253,511 | ) | (2,253,511 | ) | |||||||||||||||||||||||||
|
Balance, December 31, 2014
|
20,000,000 | $ | 20,000 | 581,433 | $ | 581 | $ | 109,519,913 | $ | - | $ | - | $ | (120,434,494 | ) | $ | (10,894,000 | ) | ||||||||||||||||||
U.S. STEM CELL, INC.
STATEMENT OF STOCKHOLDERS' DEFICIT
TWO YEARS ENDED DECEMBER 31, 2015
|
Additional
|
||||||||||||||||||||||||||||||||||||
|
Preferred stock
|
Common stock
|
Paid in
|
Treasury
|
Subscription
|
Accumulated
|
|||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Stock
|
Receivable
|
Deficit
|
Total
|
||||||||||||||||||||||||||||
|
Balance, December 31, 2014
|
20,000,000 | $ | 20,000 | 581,433 | $ | 581 | $ | 109,519,913 | $ | - | $ | - | $ | (120,434,494 | ) | $ | (10,894,000 | ) | ||||||||||||||||||
|
Common stock issued in settlement of accounts payable and accrued interest
|
- | - | 67,944 | 68 | 236,818 | - | - | 236,886 | ||||||||||||||||||||||||||||
|
Common stock issued in settlement of guarantor fees
|
- | - | 24,353 | 24 | 170,077 | - | - | 170,101 | ||||||||||||||||||||||||||||
|
Common stock issued in connection with settlement of other debt
|
- | - | 1,037,645 | 1,038 | 2,818,666 | - | - | 2,819,704 | ||||||||||||||||||||||||||||
|
Proceeds from issuance of common stock
|
- | - | 95,664 | 96 | 582,712 | - | - | 582,808 | ||||||||||||||||||||||||||||
|
Common stock issued in settlement of litigation
|
- | - | 6,650 | 7 | 59,843 | - | - | 59,850 | ||||||||||||||||||||||||||||
|
Fair value of common stock warrants issued in settlement of debt
|
- | - | - | - | 14,886 | - | - | - | 14,886 | |||||||||||||||||||||||||||
|
Purchase of 85,211 shares of Company's common stock at average cost of $2.61 per share
|
- | - | - | - | - | (221,996 | ) | - | - | (221,996 | ) | |||||||||||||||||||||||||
|
Stock based compensation
|
- | - | - | - | 1,152,195 | - | - | - | 1,152,195 | |||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | (1,641,880 | ) | (1,641,880 | ) | |||||||||||||||||||||||||
|
Balance, December 31, 2015
|
20,000,000 | $ | 20,000 | 1,813,689 | $ | 1,814 | $ | 114,555,110 | $ | (221,996 | ) | $ | - | $ | (122,076,374 | ) | $ | (7,721,446 | ) | |||||||||||||||||
See the accompanying notes to these financial statements.
U.S. STEM CELL, INC.
STATEMENTS OF CASH FLOWS
|
Year ended December 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||||||
|
Net loss
|
$ | (1,641,880 | ) | $ | (2,253,511 | ) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||
|
Depreciation and amortization
|
5,326 | 4,490 | ||||||
|
Bad debt expense
|
38,599 | 28,732 | ||||||
|
Discount on convertible debt
|
818,404 | 546,970 | ||||||
|
Change in fair value of derivative liability
|
(15,095 | ) | (425,953 | ) | ||||
|
Gain on settlement of debt
|
(2,620,233 | ) | (2,498,676 | ) | ||||
|
Common stock issued in settlement of litigation
|
59,850 | |||||||
|
Non cash payment of interest
|
528,915 | 705,710 | ||||||
|
Change in fair value of warrant modifications
|
- | 148,517 | ||||||
|
Warrants issued in connection with revenue share agreement
|
- | 190,262 | ||||||
|
Related party notes payable issued for services rendered
|
- | 800,000 | ||||||
|
(Income) loss on equity investments
|
(38,542 | ) | 9,117 | |||||
|
Common stock issued in exchange for services
|
- | 38,750 | ||||||
|
Stock based compensation
|
1,152,195 | 537,606 | ||||||
|
(Increase) decrease in:
|
||||||||
|
Receivables
|
21,778 | (104,228 | ) | |||||
|
Inventory
|
(17,406 | ) | - | |||||
|
Prepaid and other current assets
|
4,423 | (8,471 | ) | |||||
|
Increase (decrease) in:
|
||||||||
|
Accounts payable, including related party accounts payable
|
434,490 | 858,437 | ||||||
|
Accrued expenses
|
365,525 | 313,601 | ||||||
|
Deferred revenue
|
58,961 | - | ||||||
|
Net cash used in operating activities
|
(844,690 | ) | (1,108,647 | ) | ||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||
|
Payments for investments
|
(10,000 | ) | (49,714 | ) | ||||
|
Purchase of treasury stock
|
(221,996 | ) | - | |||||
|
Acquisitions of property and equipment
|
(2,035 | ) | (8,121 | ) | ||||
|
Net cash used by investing activities
|
(234,031 | ) | (57,835 | ) | ||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||
|
Proceeds from issuance of common stock, net
|
582,808 | 479,625 | ||||||
|
Proceeds from related party advances
|
13,606 | 33,760 | ||||||
|
Proceeds from exercise of stock options and warrants
|
- | 136,000 | ||||||
|
Proceeds from notes payable
|
770,052 | 740,500 | ||||||
|
Repayments of notes payable
|
(266,047 | ) | (232,956 | ) | ||||
|
Net cash provided in financing activities
|
1,100,419 | 1,156,929 | ||||||
|
Net increase (decrease) in cash and cash equivalents
|
21,698 | (9,553 | ) | |||||
|
Cash and cash equivalents, beginning of period
|
36,674 | 46,227 | ||||||
|
Cash and cash equivalents, end of period
|
$ | 58,372 | $ | 36,674 | ||||
|
SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION
|
||||||||
|
Interest paid
|
$ | 321,846 | $ | 429,569 | ||||
|
Income taxes paid
|
$ | - | $ | - | ||||
|
Non cash financing activities:
|
||||||||
|
Common stock issued in settlement of notes payable
|
$ | 926,381 | $ | 1,367,927 | ||||
|
Common stock issued in settlement of accounts payable
|
$ | 236,886 | $ | 1,071,155 | ||||
|
Common stock issued in settlement of guarantor fees
|
$ | 170,101 | $ | - | ||||
|
Promissory note issued in exchange for subordinated debt and accrued expenses
|
$ | 1,697,762 | $ | - | ||||
|
Common stock issued in settlement of related party notes and advances payable
|
$ | 471,101 | $ | 862,880 | ||||
|
Note payable issued in settlement of legal fees
|
$ | 15,000 | $ | - | ||||
|
Common stock warrant issued in settlement of accounts payable
|
$ | 14,886 | $ | - | ||||
See the accompanying notes to these financial statements.
NOTE 1 — SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies applied in the presentation of the accompanying financial statements follows:
Basis and business presentation
U.S. Stem Cell, Inc. (the “Company”) was incorporated under the laws of the State of Florida in August, 1999. The Company is in the cardiovascular sector of the cell technology industry delivering cell therapies and biologics that help address congestive heart failure, lower limb ischemia, chronic heart ischemia, acute myocardial infarctions and other issues. Our business includes the development of proprietary cell therapy products as well as revenue generating physician and patient based regenerative medicine / cell therapy training services, cell collection and cell storage services, the sale of cell collection and treatment kits for humans and animals, and the operation of a cell therapy clinic. To date, the Company has not generated significant sales revenues to cover costs, incurred expenses, and as a result has sustained losses.
On October 12, 2015, the Company amended its Articles of Incorporation to change its name from Bioheart, Inc. to U.S. Stem Cell, Inc. and to implement a reverse stock split in the ratio of 1 share for every 1,000 shares of common stock. This amendment was approved and filed of record by the Florida Secretary of State on October 12, 2015, effective on October 19, 2015. FINRA has declared the Company’s 1-for-1,000 reverse stock split market effective as of November 4, 2015. In addition, the ticker symbol was BHRTD for 20 business days from November 4th 2015 at which point it will change to USRM. These financial statements have been retroactively restated to reflect the reverse stock split (see note 11).
Revenue Recognition
The Company recognizes revenue in accordance with Accounting Standards Codification subtopic 605-10, Revenue Recognition (“ASC 605-10”) which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred; (3) the selling price is fixed and determinable; and (4) collectability is reasonably assured. Determination of criteria (3) and (4) are based on management’s judgments regarding the fixed nature of the selling prices of the products delivered and the collectability of those amounts. Provisions for discounts and rebates to customers, estimated returns and allowances, and other adjustments are provided for in the same period the related sales are recorded.
At the time of each transaction, management assesses whether the fee associated with the transaction is fixed or determinable and whether or not collection is reasonably assured. The assessment of whether the fee is fixed or determinable is based upon the payment terms of the transaction. Collectability is assessed based on a number of factors, including past transaction history with the client and the creditworthiness of the client.
The Company’s primary source of revenue are from the sale of test kits and equipment, training services, patient treatments and laboratory services, and cell banking.
Revenues for test kits and equipment sold are not recorded until test kits and equipment are delivered. During 2014, the Company had revenue sharing arrangements for the sale of goods whereby the Company was the primary obligor, set pricing with the customers and bared all associated credit risks with the customers. Sales under revenue share arrangements are recorded as gross sales and any portion shared with third parties under such arrangements are classified as selling expense due to the nature of the marketing activities performed by the third party. All revenue sharing arrangements were terminated in November 2014. Revenues from trainings are recognized when the training occurs. Any cash received as a deposit for trainings are recorded by the company as a liability.
Patient treatments and laboratory services revenue are recognized when those services have been completed or satisfied.
Revenues for cell banking sales are accounted for as Multiple-Element Arrangements under ASC 605-10 which incorporates Accounting Standards Codification subtopic 605-25, Multiple-Element Arrangements (“ASC 605-25”). ASC 605-25 addresses accounting for arrangements that may involve the delivery or performance of multiple products, services and/or rights to use assets. Because the Company sells its services separately, on more than a limited basis and at a price within a narrow range, the Company was able to allocate revenue based on vendor-specific objective evidence of fair value (VSOE).
At December 31, 2015 and 2014, the Company had deferred revenues of $71,961 and $13,000, respectively.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Estimates
The preparation of the financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect certain reported amounts of assets and liabilities, revenues and expenses and certain disclosures. The most significant estimates are those used in determination of derivative liabilities and stock compensation. Accordingly, actual results could differ from those estimates.
Cash
The Company considers cash to consist of cash on hand and temporary investments having an original maturity of 90 days or less that are readily convertible into cash.
Accounts Receivable
Trade receivables are carried at their estimated collectible amounts. Trade credit is generally extended on a short-term basis; thus trade receivables do not bear interest. Trade accounts receivable are periodically evaluated for collectability based on past credit history with customers and their current financial condition.
Allowance for Doubtful Accounts
Any charges to the allowance for doubtful accounts on accounts receivable are charged to operations in amounts sufficient to maintain the allowance for uncollectible accounts at a level management believes is adequate to cover any probable losses. Management determines the adequacy of the allowance based on historical write-off percentages and the current status of accounts receivable. Accounts receivable are charged off against the allowance when collectability is determined to be permanently impaired. As of December 31, 2015 and 2014, allowance for doubtful accounts was $-0-.
Inventories
Inventories are stated at the lower of cost or market with cost being determined on a first-in, first-out (FIFO) basis. The Company writes down its inventory for estimated obsolescence or unmarketable inventory equal to the difference between the cost of inventory and the estimated market value based upon assumptions about future demand and market conditions. If actual market conditions are less favorable than those projected by management, additional inventory write-downs may be required. During 2015 not there were no inventory write-downs.
Investments
The Company has adopted Accounting Standards Codification subtopic 323-10, Investments-Equity Methods and Joint Ventures (“ASC 323-10) which requires the accounting for investments where the Company can exert significant influence, but not control of a joint venture or equity investment. The Company accounted for its 33 percent ownership of U.S. Stem Cell Clinic, LLC utilizing the equity method of accounting.
Long-Lived Assets
The Company follows FASB ASC 360-10-15-3, “Impairment or Disposal of Long-lived Assets,” which established a “primary asset” approach to determine the cash flow estimation period for a group of assets and liabilities that represents the unit of accounting for a long-lived asset to be held and used. Long-lived assets to be held and used are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The carrying amount of a long-lived asset is not recoverable if it exceeds the sum of the undiscounted cash flows expected to result from the use and eventual disposition of the asset. Long-lived assets to be disposed of are reported at the lower of carrying amount or fair value less cost to sell. The Company determined that there was no impairment on its long-lived assets during 2015 and 2014.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Property and Equipment
Property and equipment are stated at cost. When retired or otherwise disposed, the related carrying value and accumulated depreciation are removed from the respective accounts and the net difference less any amount realized from disposition, is reflected in earnings. For financial statement purposes, property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives of 3 to 15 years.
Income Taxes
The Company has adopted Accounting Standards Codification subtopic 740-10, Income Taxes (“ASC 740-10”) which requires the recognition of deferred tax liabilities and assets for the expected future tax consequences of events that have been included in the financial statement or tax returns. Under this method, deferred tax liabilities and assets are determined based on the difference between financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Temporary differences between taxable income reported for financial reporting purposes and income tax purposes consist primarily of derivative liability and stock compensation accounting versus tax differences.
Comprehensive Income
The Company does not have any items of comprehensive income in any of the periods presented.
Net Loss per Common Share, basic and diluted
The Company computes earnings (loss) per share under Accounting Standards Codification subtopic 260-10, Earnings Per Share (“ASC 260-10”). Net loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the year. Diluted earnings per share, if presented, would include the dilution that would occur upon the exercise or conversion of all potentially dilutive securities into common stock using the “treasury stock” and/or “if converted” methods as applicable.
The computation of basic and diluted income (loss) per share as of December 31, 2015 and 2014 excludes potentially dilutive securities when their inclusion would be anti-dilutive, or if their exercise prices were greater than the average market price of the common stock during the period.
Potentially dilutive securities excluded from the computation of basic and diluted net loss per share are as follows:
|
|
2015
|
2014
|
||||||
|
Convertible notes payable
|
677,540
|
901,796
|
||||||
|
Options to purchase common stock
|
555,820
|
66,933
|
||||||
|
Warrants to purchase common stock
|
139,367
|
150,620
|
||||||
|
Totals
|
1,372,727
|
1,119,349
|
||||||
Stock Based Compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. For employees and directors, the fair value of the award is measured on the grant date and for non-employees, the fair value of the award is generally re-measured on vesting dates and interim financial reporting dates until the service period is complete. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period. Stock-based compensation expense is recorded by the Company in the same expense classifications in the statements of operations, as if such amounts were paid in cash. As of December 31, 2015, there were outstanding stock options to purchase 555,820 shares of common stock, 511,660 shares of which were vested. (see Note 12)
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Treasury Stock
The Company use the cost method when it purchases its own common stock as treasury shares and displays treasury stock as a reduction of shareholders’ equity.
Concentrations of Credit Risk
The Company’s financial instruments that are exposed to a concentration of credit risk are cash and accounts receivable. Generally, the Company’s cash and cash equivalents in interest-bearing accounts does not exceed FDIC insurance limits. The financial stability of these institutions is periodically reviewed by senior management.
As of December 31, 2015, three customers represented 32%, 18% and 16%, an aggregate of 66% of the Company’s accounts receivable. As of December 31, 2014, two customers represented 38% and 17%, an aggregate of 55%, of the Company’s accounts receivable.
The Company’s revenues earned from sale of products and services for the year ended December 31, 2015 did not include any customers representing 10% or more of the Company’s total revenues. For the year ended December 31, 2014, Company’s revenues earned from sale of products and services included 10% of the Company’s total revenues from one customer.
Reliance on Key Personnel and Consultants
The Company has four full-time employees and one part-time employees. The Company is heavily dependent on the continued active participation of its two current executive officers, one employee and key consultants. The loss of any of the senior management or key consultants could significantly and negatively impact the business until adequate replacements can be identified and put in place.
Research and Development
The Company accounts for research and development costs in accordance with Accounting Standards Codification subtopic 730-10, Research and Development (“ASC 730-10”). Under ASC 730-10, all research and development costs must be charged to expense as incurred. Accordingly, internal research and development costs are expensed as incurred. Third-party research and development costs are expensed when the contracted work has been performed or as milestone results have been achieved as defined under the applicable agreement. Company-sponsored research and development costs related to both present and future products are expensed in the period incurred. The Company incurred research and development expenses of $10,000 and $66,420 for the years ended December 31, 2015 and 2014, respectively.
Fair Value
Accounting Standards Codification subtopic 825-10, Financial Instruments (“ASC 825-10”) requires disclosure of the fair value of certain financial instruments. The carrying value of cash and cash equivalents, accounts payable and accrued liabilities, and short-term borrowings, as reflected in the balance sheets, approximate fair value because of the short-term maturity of these instruments. All other significant financial assets, financial liabilities and equity instruments of the Company are either recognized or disclosed in the financial statements together with other information relevant for making a reasonable assessment of future cash flows, interest rate risk and credit risk. Where practicable the fair values of financial assets and financial liabilities have been determined and disclosed; otherwise only available information pertinent to fair value has been disclosed.
The Company follows Accounting Standards Codification subtopic 820-10, Fair Value Measurements and Disclosures (“ASC 820-10”) and Accounting Standards Codification subtopic 825-10, Financial Instruments (“ASC 825-10”), which permits entities to choose to measure many financial instruments and certain other items at fair value.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Derivative Instrument Liability
The Company accounts for derivative instruments in accordance with ASC 815, which establishes accounting and reporting standards for derivative instruments and hedging activities, including certain derivative instruments embedded in other financial instruments or contracts and requires recognition of all derivatives on the balance sheet at fair value, regardless of hedging relationship designation. Accounting for changes in fair value of the derivative instruments depends on whether the derivatives qualify as hedge relationships and the types of relationships designated are based on the exposures hedged. At December 31, 2015 and 2014, the Company did not have any derivative instruments that were designated as hedges.
At December 31, 2015 and 2014, the Company had outstanding convertible notes and warrants that contained embedded derivatives. These embedded derivatives include certain conversion features and reset provisions. (See Note 8 and Note 10).
Reclassification
Certain reclassifications have been made to prior periods’ data to conform with the current year’s presentation. These reclassifications had no effect on reported income or losses.
Recent Accounting Pronouncements
In November 2015, the FASB issued (ASU) 2015-17, Balance Sheet Classification of Deferred Taxes. Currently deferred taxes for each tax jurisdiction are presented as a net current asset or liability and net noncurrent asset or liability on the balance sheet. To simplify the presentation, the new guidance requires that deferred tax liabilities and assets for all jurisdictions along with any related valuation allowances be classified as noncurrent in a classified statement of financial position. This guidance is effective for interim and annual reporting periods beginning after December 15, 2016, and early adoption is permitted. The Company has adopted this guidance in the fourth quarter of the year ended December 31, 2015 on a retrospective basis. The adoption of this guidance did not have a material impact on the Company’s financial position, results of operations or cash flows, and did not have any effect on prior periods due to the full valuation allowance against the Company’s net deferred tax assets.
In January 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (ASU) 2016-01, which amends the guidance in U.S. GAAP on the classification and measurement of financial instruments. Changes to the current guidance primarily affect the accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. In addition, the ASU clarifies guidance related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. The new standard is effective for fiscal years and interim periods beginning after December 15, 2017, and upon adoption, an entity should apply the amendments by means of a cumulative-effect adjustment to the balance sheet at the beginning of the first reporting period in which the guidance is effective. Early adoption is not permitted except for the provision to record fair value changes for financial liabilities under the fair value option resulting from instrument-specific credit risk in other comprehensive income. The Company is currently evaluating the impact of adopting this guidance.
There were other various updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company’s financial position, results of operations or cash flows.
NOTE 2 – GOING CONCERN AND MANAGEMENT’S LIQUIDITY PLANS
The accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As shown in the accompanying financial statements during year ended December 31, 2015, the Company incurred net losses of $1,641,880 and used $844,690 in cash for operating activities. These factors among others may indicate that the Company will be unable to continue as a going concern for a reasonable period of time.
The Company’s existence is dependent upon management’s ability to develop profitable operations and to obtain additional funding sources. There can be no assurance that the Company’s financing efforts will result in profitable operations or the resolution of the Company’s liquidity problems. The accompanying statements do not include any adjustments that might result should the Company be unable to continue as a going concern.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
The Company’s ability to obtain additional debt financing and/or alternative arrangements, with the Guarantors or otherwise, may be limited by the amount of, terms and restrictions of our then current debt. Accordingly, until such time, we will generally be restricted from, among other things, incurring additional indebtedness or liens, with limited exceptions. Additional debt financing, if available, may involve restrictive covenants that limit or further limit our operating and financial flexibility and prohibit us from making distributions to shareholders.
NOTE 3 — INVESTMENTS
The investment recorded is comprised of a 33% ownership of U.S. Stem Cell Clinic, LLC, accounted for using the equity method of accounting. The investments in 2014 and 2015 of cash and expenses paid on U.S. Stem Cell Clinic, LLC’s behalf were an aggregate of $59,714. The Company’s 33% income (loss) earned by U.S. Stem Cell Clinic, LLC of $38,542 and $(9,117) for the year ended December 31, 2015 and 2014, respectively, (inception to date income of $29,425) was recorded as other income/expense in the Company’s Statement of Operations in the appropriate periods and increased the carrying value of the investment to $89,139.
At December 31, 2015 and 2014, accounts receivable for sales of test kits to U.S. Stem Cell Clinic, LLC was $5,946 and $-0-; revenues earned from sales to U.S. Stem Clinic, LLC for 2015 and 2014 were $182,922 and $-0-, respectively.
NOTE 4 — PROPERTY AND EQUIPMENT
Property and equipment as of December 31, 2015 and 2014 is summarized as follows:
|
2015
|
2014
|
|||||||
|
Laboratory and medical equipment
|
$
|
353,253
|
$
|
352,358
|
||||
|
Furniture, fixtures and equipment
|
130,410
|
125,634
|
||||||
|
Computer equipment
|
48,788
|
47,647
|
||||||
|
Leasehold improvements
|
362,046
|
362,046
|
||||||
|
894,497
|
887,685
|
|||||||
|
Less accumulated depreciation and amortization
|
(880,325
|
)
|
(874,999
|
)
|
||||
|
$
|
14,172
|
$
|
12,686
|
|||||
Property and equipment are recorded on the basis of cost. For financial statement purposes, property, plant and equipment are depreciated using the straight-line method over their estimated useful lives.
Expenditures for repair and maintenance which do not materially extend the useful lives of property and equipment are charged to operations. When property or equipment is sold or otherwise disposed of, the cost and related accumulated depreciation are removed from the respective accounts with the resulting gain or loss reflected in operations. Management periodically reviews the carrying value of its property and equipment for impairment in accordance with the guidance for impairment of long lived assets.
NOTE 5 — ACCRUED EXPENSES
Accrued expenses consisted of the following as of December 31, 2015 and 2014:
|
2015
|
2014
|
|||||||
|
Amounts payable to the Guarantors of the Company’s loan agreement with Bank of America and Seaside Bank, including fees and interest
|
$
|
64,199
|
$
|
1,533,217
|
||||
|
Interest payable on notes payable
|
467,664
|
685,575
|
||||||
|
Vendor accruals and other
|
147,244
|
120,836
|
||||||
|
Employee commissions, compensation, etc.
|
47,644
|
33,987
|
||||||
|
$
|
726,751
|
$
|
2,373,615
|
|||||
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
In June 2000, the Company had entered into an agreement with William Beaumont Hospital, or WBH, pursuant to which WBH granted the Company a worldwide, exclusive, non-sublicenseable license to two U.S. method patents covering the inducement of human adult myocardial cell proliferation in vitro, or the WBH IP. The term of the agreement was for the life of the patents, which expire in 2015. The Company did not use this license in any of their technologies or made any payments to WBH other than the initial payment to acquire the license. On April 2, 2014, the Company received confirmation from WBH that it has no obligation under the patent license agreement and WBH agreed to terminate the patent license agreement pursuant to a termination letter dated March 3, 2014. Accordingly, the Company recognized approximately $2,122,130 in settlement of debt which represents the accumulative accrual and related interest from past years under the 2000 patent license agreement during the year ended December 31, 2014.
During the year ended December 31, 2014, the Company issued an aggregate of 48,585 shares of its common stock in settlement of outstanding accounts payable and accrued expenses. In connection with the issuance, the Company incurred $120,656 net gain in settlement of debt.
During the year ended December 31, 2015, the Company issued an aggregate of 67,944 shares of its common stock in settlement of outstanding accounts payable and accrued interest. In connection with the issuance, the Company incurred $144,669 gain in settlement of debt.
During the year ended December 31, 2015, the Company incurred a gain of $513,940 in relief of accounts payable.
During the year ended December 31, 2015, the Company issued an aggregate of 24,353 shares of its common stock in settlement of accumulative outstanding amounts due to Guarantors of the Company of $961,124. In connection with the issuance, the Company incurred a $791,024 gain in settlement of debt.
During the year ended December 31, 2015, the Company settled an outstanding subordinated debt, related accrued interest and accounts payable due to the guarantor by issuing a five year, non-interest bearing note payable. (See Note 8). In connection with the note issuance, the Company settled $624,737 of outstanding guarantor fees.
NOTE 6 – STANDBY EQUITY DISTRIBUTION AGREEMENT
Greystone Capital Partners
On November 2, 2011, the Company and Greystone Capital Partners (“Greystone”) entered into a Standby Equity Distribution Agreement (the “Agreement”). Pursuant to the Agreement, Greystone has agreed to provide the Company with up to $1,000,000 of funding for the 24-month period following the date February 10, 2012, the registration statement of the Company’s common stock was declared effective by the SEC (the “Equity Line”).
During this 24-month period, commencing on the date on which the SEC first declared the registration statement effective, the Company may request a draw down under the Equity Line by which the Company would sell shares of its common stock to Greystone, which is obligated to purchase the shares under the Agreement.
For each share of the Company common stock purchased under the Agreement, Greystone will pay eighty percent (80%) of the average of the lowest daily volume weighted average price for five consecutive trading days immediately preceding Advance Notice (the “Valuation Period”) commencing the date an Advance Notice (the “Advance Notice”) is delivered to Greystone in a manner provided by the Agreement. Subject to certain limitations and floor price reductions, the Company may, at its sole discretion, issue a Put Notice to Greystone and Greystone will then be irrevocably bound to acquire such shares. The registration statement of the Company’s common stock pursuant to the Agreement was declared effective on February 10, 2012 and a Post-Effective Amendment was declared effective on May 7, 2013. On December 1, 2012, the parties to the Equity Line agreed that the Purchase Price be adjusted to seventy-five percent (75%) of the lowest daily volume weighted average price of the Common Stock as quoted by Bloomberg, LP, during the five (5) consecutive Trading Days (as such term is defined in the Equity Line) immediately subsequent to the date of the relevant Advance Notice.
No common stock were issued during the year ended December 31, 2014.
The Agreement automatically terminated on the first of April, 2014 (the first day of the month next following the second (2nd) anniversary of the Effective Date).
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
NOTE 7 – STOCK PURCHASE AGREEMENT
On October 23, 2014, the Company, entered into a common stock purchase agreement (the “Purchase Agreement”) with Magna Equities II, LLC, a New York limited liability company (the “Investor”). The Purchase Agreement provides that, upon the terms and subject to the conditions set forth therein, the Investor is committed to purchase up to $3,000,000 (the “Total Commitment”) worth of the Company’s common stock, $0.001 par value (the “Shares”), over the 24-month term of the Purchase Agreement.
From time to time over the term of the Purchase Agreement, commencing on the trading day immediately following the date on which the initial registration statement was declared effective by the Securities and Exchange Commission (the “Commission”), the Company may provide the Investor with a draw down notice to purchase a specified dollar amount of Shares, with each draw down subject to certain limitations.
The Company may not deliver any Draw Down Notice to the Investor if the Initial Purchase Price with respect to the Shares subject to such Draw Down Notice is less than $2.50 as of the date the applicable Draw Down Notice is received by the Investor (the “Draw Down Exercise Date”).
The applicable Initial Purchase Price, the “Initial Purchase Price”, is defined as a price equal to 93% of the lowest of (i) the arithmetic average of the three lowest daily volume weighted average prices for the Company’s common stock (the “VWAP”) during the 10 consecutive trading days ending on the trading day immediately preceding the applicable Draw Down Exercise Date, (ii) the arithmetic average of the three lowest closing sale prices for the Company’s common stock during the 10 consecutive trading days ending on the trading day immediately preceding the applicable Draw Down Exercise Date and (iii) the closing sale price for the Company’s common stock on the trading day immediately preceding the applicable Draw Down Exercise Date.
In 2014, the Company paid to the Investor as a commitment fee for entering into the Purchase Agreement equal an aggregate of to 12,000 shares of the Company’s common stock.
During the year ended December 31, 2015, the Company issued an aggregate of 87,812 shares of its common stock in exchange for $521,538 under the Purchase Agreement. (See Note 11)
NOTE 8 — NOTES PAYABLE
Notes payable were comprised of the following as of December 31, 2015 and 2014:
|
2015
|
2014
|
|||||||
|
Seaside Bank note payable.
|
$
|
980,000
|
$
|
980,000
|
||||
|
Hunton & Williams notes payable
|
384,972
|
384,972
|
||||||
|
Asher notes payable
|
-
|
151,000
|
||||||
|
Daniel James Management notes payable
|
75,000
|
75,000
|
||||||
|
Fourth Man, LLC notes payable
|
77,450
|
75,000
|
||||||
|
Magna Group notes payable
|
125,000
|
205,000
|
||||||
|
Power Up Lending Group notes payable
|
194,235
|
-
|
||||||
|
Equipment finance lease
|
4,777
|
-
|
||||||
|
Total notes payable
|
1,841,434
|
1,870,972
|
||||||
|
Less unamortized debt discount
|
(249,205
|
)
|
(339,160
|
)
|
||||
|
Total notes payable net of unamortized debt discount
|
1,592,229
|
1,531,812
|
||||||
|
Less current portion
|
(608,502
|
)
|
(1,531,812
|
)
|
||||
|
Long term portion
|
$
|
983,727
|
$
|
-
|
||||
Seaside Bank
On October 25, 2010, the Company entered into a Loan Agreement with Seaside National Bank and Trust for a $980,000 loan at 4.25% per annum interest that was used to refinance the Company’s loan with Bank of America. The obligation is guaranteed by certain shareholders of the Company. The Company renewed the loan with Seaside National Bank and Trust during the first quarter of 2016 to extend the maturity date to January 11, 2018.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Hunton & Williams Notes
At December 31, 2015 and 2014, the Company has two outstanding notes payable with interest at 8% per annum due at maturity. The two notes, $61,150 and $323,822, are payable in one balloon payment upon the date the Noteholder provides written demand, however the Company is not obligated to make payments until the Northstar (or successor) Loan is paid off.
Asher Notes
2014 Notes
During the year ended December 31, 2014, the Company entered into a Securities Purchase Agreements with Asher Enterprises, Inc. (“Asher”) or affiliates, for the sale of 8% convertible notes in aggregate principal amount of $334,000 (the “Asher Notes”).
The Asher Notes bear interest at the rate of 8% per annum. As of December 31, 2014, all interest and principal must be repaid nine months from the issuance date, with the last note being due August 12, 2015. The Notes are convertible into common stock, at Asher’s option, at a 45% discount to the average of the three lowest closing bid prices of the common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Asher Notes.
These embedded derivatives included certain conversion features and a reset provision. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Asher Notes and to fair value as of each subsequent reporting date, which at December 31, 2014 was $149,770. At the inception of the Asher Notes, the Company determined the aggregate fair value of $566,294 of the embedded derivatives.
2015 Notes
During the year ended December 31, 2015, the Company entered into Securities Purchase Agreements with Asher Enterprises, Inc. (“Asher”) or affiliates, for the sale of 8% convertible notes in aggregate principal amount of $180,000 (the “Asher Notes”). The Company incurred legal fees in the amount of $15,000 which were deducted from the proceeds of the notes.
The Asher Notes bear interest at the rate of 8% per annum. As of the year ended December 31, 2015, all interest and principal must be repaid nine months from the issuance date, with the last note being due February 6, 2016. The Asher Notes are convertible into shares of common stock, at Asher’s option, at a 45% discount to the average of the three lowest closing bid prices of the shares of common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Asher Notes. (see Note 10)
These embedded derivatives included certain conversion features and a reset provision. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Asher Notes and to fair value as of each subsequent reporting date, which at December 31, 2015 was $-0- (all Notes converted). At the inception of the Asher Notes, the Company determined the aggregate fair value of $211,575 of the embedded derivatives.
During the year ended December 31, 2015, $151,000 of notes plus accrued interest that were outstanding at December 31, 2014, and $180,000 of notes plus accrued interest that were issued during 2015, were converted into shares of the Company’s common stock (see Note 11).
The remaining aggregate Asher Notes unconverted principle balance as of December 31, 2015 was $-0-.
Daniel James Management
2014 Notes
During the year ended December 31, 2014, the Company entered into Securities Purchase Agreements with Daniel James Management (“Daniel”) for the sale of 8% to 9.5% convertible notes in aggregate principal amount of $135,000 (the “Daniel Notes”).
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
The Daniel Notes bear interest at the rate of 8% to 9.5% per annum. As of the year ended December 31, 2014, all interest and principal must be repaid one year from the issuance date, with the last note being due November 30, 2015. The Daniel Notes are convertible into common stock, at holder’s option, at a 47% discount to the average of the three lowest closing bid prices of the common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Daniel Note. These embedded derivatives included certain conversion features and reset provision.
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Daniel Notes and to fair value as of each subsequent reporting date which at December 31, 2014 was $95,866. At the inception of the Daniel Notes, the Company determined the aggregate fair value of $236,211 of the embedded derivatives.
2015 Notes
During the year ended December 31, 2015, the Company entered into Securities Purchase Agreements with Daniel James Management (“Daniel”) for the sale of 9.5% convertible note in aggregate principal amount of $125,000 (the “Daniel Notes”).
The Daniel Notes bear interest at the rate of 9.5% per annum. As of the year ended December 31, 2015, all interest and principal must be repaid one year from the issuance date, with the last note being due October 29, 2016. The Daniel Notes are convertible into common stock, at holder’s option, at a 47% discount to the average of the three lowest closing bid prices of the common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Daniel Notes. These embedded derivatives included certain conversion features and reset provision. (see Note 9).
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Daniel Notes and to fair value as of each subsequent reporting date which at December 31, 2015 was $120,263. At the inception of the Daniel Notes, the Company determined the aggregate fair value of $238,443 of the embedded derivatives.
During the year ended December 31, 2015, $75,000 of notes plus accrued interest that were outstanding at December 31, 2014, and $50,000 of notes plus accrued interest that were issued during 2015, were converted into shares of the Company’s common stock (see Note 11).
The remaining aggregate Daniel Notes unconverted principle balance as of December 31, 2015 was $75,000.
Fourth Man, LLC
2014 Notes
During the year ended December 31, 2014, the Company entered into Securities Purchase Agreements with Fourth Man, LLC. (“Fourth Man”), for the sale of an 8% to 9.5% convertible notes in the aggregate principal amount of $100,000 (the “Note”).
The Notes bears interest at the rate of 8% to 9.5% per annum. As of the year ended December 31, 2014, all interest and principal must be repaid one year from the issuance date, with the last note being due August 28, 2015. The Notes are convertible into common stock, at Fourth Man’s option, at a 47% discount to the average of the three lowest closing bid prices of the common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Fourth Man Notes. These embedded derivatives included certain conversion features and reset provision.
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Fourth Man Notes and to fair value as of each subsequent reporting date which at December 31, 2014 was $93,505. At the inception of the Fourth Man Notes, the Company determined the aggregate fair value of $168,501 of the embedded derivatives.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
2015 Notes
During the year ended December 31, 2015, the Company entered into Securities Purchase Agreements with Fourth Man, LLC. (“Fourth Man”), for the sale of a 9.5% convertible notes in the aggregate principal amount of $150,000 (the “Notes”).
The Notes bears interest at the rate of 8% to 9.5% per annum. As of the year ended December 31, 2015, all interest and principal must be repaid one year from the issuance date, with the last note being due December 2, 2016. The Notes are convertible into shares of common stock, at Fourth Man’s option, at a 47% discount to the lowest closing bid price of the common stock during the 10 trading day period prior to conversion. The Company has identified the embedded derivatives related to the Fourth Man Notes. These embedded derivatives included certain conversion features and reset provision. (see Note 10)
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Fourth Man Notes and to fair value as of each subsequent reporting date which at December 31, 2015 was $126,825. At the inception of the Fourth Man Notes, the Company determined the aggregate fair value of $356,225 of the embedded derivatives.
During the year ended December 31, 2015, $75,000 of notes plus accrued interest that were outstanding at December 31, 2014, and $72,550 of notes plus accrued interest that were issued during 2015, were converted into shares of the Company’s common stock (see Note 11).
The remaining aggregate Fourth Man, LLC Notes unconverted principle balance as of December 31, 2015 was $77,450.
Magna Group
2014 Note
During the year ended December 31, 2014, the Company entered into a Securities Purchase Agreement with Magna Capital Group (“Magna”) for the sale of a convertible note in aggregate principal amount of $307,500 (the “Magna Note”) and an original interest discount (“OID”) of $102,500. $40,000 of the outstanding principal amount of the Convertible Note (together with any accrued and unpaid interest with respect to such portion of the principal amount) will be automatically extinguished upon the filing of the registration statement, following the closing of the Securities Purchase Agreement. In addition, $62,500 of the outstanding principal amount of the Convertible Note (together with any accrued and unpaid interest with respect to such portion of the principal amount) will be automatically extinguished if (i) the registration statement is declared effective by the SEC on or prior to the earlier of (A) the 120th calendar day after October 7, 2014 and (B) the fifth business day after the date we are notified by the Securities and Exchange Commission, or the Commission, that the registration statement will not be reviewed or will not be subject to further review, and this prospectus is available for use by Magna for the resale by Magna of all of the shares of our common stock issued or issuable upon conversion of the Convertible Note and (ii) no event of default under the Convertible Note or an event that with the passage of time or giving of notice would constitute an event of default under the Convertible Note has occurred on or prior to such date. On November 21, 2014, the Company filed its registration statement and on December 22, 2014, was declared effective. As such, the principle amount of the note was reduced by an aggregate of $102,500.
The Convertible Note matured on August 7, 2015 and, in addition to the approximately 33.33% original issue discount, accrues interest at the rate of 12% per year. The Convertible Note is convertible at any time, in whole or in part, at Magna’s option into shares of Company common stock at a fixed conversion price of $10.35 per share, subject to adjustment pursuant to the “full ratchet” and standard anti-dilution provisions contained in the Convertible Note. This resulted in an embedded derivatives liability as a result of these anti-dilution provision. This conversion price represents a discount of approximately 45% from the lowest trading price the Company common stock during the five trading days prior to October 7, 2014, the date the Company issued the Convertible Note to Magna.
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivative as of the inception date of Magna Note and to fair value as of each subsequent reporting date which at December 31, 2014 was $252,211. At the inception of the Magna Note, the Company determined the aggregate fair value of $472,702 of the embedded derivative. At the dates of debt reductions, as described above, the Company reclassified the fair value of the related derivative liability of $128,624 to additional paid in capital.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
On August 6, 2015, the Company amended the securities purchase agreement with Magna amending the maturity date of its convertible note payable with Magna from August 7, 2015 to November 7, 2015. This amendment also changes the conversion price of the notes from $10.35 to the lesser of a) $6.00 or b) a 40% discount from the lowest trading price in the five trading days prior to conversion. Due to the reset provisions embedded in the initial convertible note, the Company determined the modification was immaterial.
The Company has identified the embedded derivatives related to the Magna Note. These embedded derivatives included certain conversion features and a reset provision. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of the 2014 Magna Notes and to fair value as of each subsequent reporting date which at December 31, 2015 was $-0- (Notes fully converted).
2015 Notes
On October 1, 2015, the Company entered into a securities purchase agreement with Magna Equities II, LLC. The purchase agreement provides that Magna shall purchase from the Company on the closing date a senior convertible note with an initial principal amount of $110,000 for a purchase price of $100,000.
The Note bears interest at the rate of 12% per annum. All interest and principal must be repaid on August 1, 2016 . The convertible note is convertible at any time, in whole or in part, at Magna’s option into shares of the Company’s common stock at a the lower of i) 60% discount to the lowest trading price of the common stock during the 5 trading day period prior to conversion or ii) $0.70 per share.
On December 3, 2015, the Company entered into a convertible promissory note with Magna Equities II, LLC. The note provides that Magna shall pay on the Company’s behalf certain accrued legal fees of $15,000. The December 3, 2015 convertible note bears interest at the rate of 12% per annum. All interest and principal must be repaid on August 1, 2016 . The convertible note is convertible at any time, in whole or in part, at Magna’s option into shares of the Company’s common stock at the lower of 60% of the lowest sale price of the Company’s common stock during five trading days preceding conversion date or $0.70 per share (subject to adjustment).
The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of Magna Notes and to fair value as of each subsequent reporting date which at December 31, 2015 was $164,600. At the inception of the Magna Note, the Company determined the aggregate fair value of $291,136 of the embedded derivatives.
During the year ended December 31, 2015, $205,000 of note that were outstanding at December 31, 2014, plus accrued interest, were converted into shares of the Company’s common stock (see Note 11).
The remaining Magna notes principle balance as of December 31, 2015 was $125,000.
On December 3, 2015, the Company entered into a securities purchase agreement with Magna Equities II, LLC. The purchase agreement provides that Magna shall purchase from the Company a senior convertible note with an initial principal amount of $262,500, subject to filing and becoming effective registration statement is declared effective by the SEC. As of December 31, 2015, the above described convertible note has not been funded.
PowerUp Lending Group, Ltd
In July 2015, the Company entered into two revenue based factoring agreements and received an aggregate of $180,000 (less origination fees of $3,590) in exchange for $243,000 of future receipts relating to monies collected from customers or other third party payors. Under the terms of the agreements, the Company is required to make daily payments equal to the greater of $1,464 or 20% of the Company’s daily cash or monetary sales receipts over the term of the agreements (ranging from 126 to 189 business days). The Company has recorded a debt discount which is being amortized to interest expense over the term of the agreements.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
On December 17, 2015, the Company entered into a revenue based factoring agreement and received an aggregate of $150,000 (less origination fees of $4,500) in exchange for $202,500 of future receipts relating to monies collected from customers or other third party payors. Under the terms of the agreements, the Company is required to make daily payments equal to $1,378 for 147 business days. The Company received net proceeds of $53,642 along with cancellation of the previous two revenue based factoring agreements described above. In connection with the cancellation of the July 2015 revenue based factoring agreements, the Company incurred a loss in settlement of debt of $8,679. The remaining principle balance of the PowerUp Lending Group notes payable at December 31, 2015 is $194,235
At December 31, 2015, the Company has recorded interest expense in the amount of $44,226 under the terms of the agreements. The remaining unamortized debt discount at December 31, 2015 is $51,164.
Equipment finance lease
On November 19, 2015, the Company entered into a financing lease for office equipment with an imputed interest of 7.75% payable at $116 per month over 48 months, secured by the leased equipment.
The remaining principle balance of the equipment finance lease as of December 31, 2015 was $4,777.
Promissory note
At December 31, 2014, a related party to one of the Company’s former officers provided notes in aggregate of $1,500,000. As of December 31, 2014, these notes were not considered as related party. The notes range from 4.75% to 8% per annum and are due upon payoff of the Northstar note payable described in Note 8.
On June 1, 2015, the Company issued an amended and restated promissory note of $1,697,762 in settlement of the $1,500,000 outstanding subordinated debt, related accrued interest of $373,469 and accumulated and unpaid guarantor fees of $624,737 (See Note 5).
The note is unsecured and non-interest bearing with four semi-annual payments of $75,000 beginning on December 31, 2015 with the remaining unpaid balance due June 1, 2020.
The Company imputed an interest rate of 5% and discounted the promissory note accordingly. The imputed debt discount of $368,615 is amortized to interest expense using the effective interest method. For the year ended December 31, 2015, the Company amortized $49,537 of debt discounts to current period operations as interest expense. The unamortized debt discount at December 31, 2015 is $319,385.
In connection with the settlement, the Company recorded a gain on settlement of debt of $1,169,058.
Summary:
The Company has identified the embedded derivatives related to the Asher, Daniel, Fourth Man LLC and Magna notes. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of these notes and to fair value as of each subsequent reporting date which at December 31, 2015 was $411,718. The fair value of the embedded derivatives at issuance of the Asher, Daniel, Fourth Man and Magna Notes, was determined using the Binomial Option Pricing Model based on the following assumptions: (1) dividend yield of 0%, (2) expected volatility of 111.98% to 175.1%, (3) weighted average risk-free interest rate of 0.17% to 0.57%, (4) expected lives of 0.75 to 1.00 years, and (5) estimated fair value of the Company’s common stock from $1.10 to $12.70 per share.
The initial fair value of the embedded debt derivative of $1,097,379 was allocated as a debt discount up to the proceeds of the notes ($568,464) with the remainder ($528,915) charged to current period operations as interest expense. For the year ended December 31, 2015, the Company amortized an aggregate of $768,241 of debt discounts to current period operations as interest expense.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
NOTE 9 — RELATED PARTY TRANSACTIONS
Advances
As of December 31, 2015 and 2014, the Company officers and directors have provided advances in the aggregate of $106,505 and $98,759 respectively, for working capital purposes. The advances are unsecured, due on demand and non-interest bearing.
During the year ended December 31, 2014, the Company issued an aggregate of 10,232 shares of common stock in settlement of $298,759 of related party advances and outstanding accounts payable.
On November 4, 2015, the Company issued 3,411 shares of common stock in settlement of $5,860 of related party advances.
Notes payable-related party
Northstar Biotechnology Group, LLC
On February 29, 2012, a note issued to BlueCrest Master Fund Limited was assigned to Northstar Biotechnology Group, LLC (“Northstar”), owned partly by certain directors and existing shareholders of the Company, including Dr. William P. Murphy Jr., Dr. Samuel Ahn and Charles Hart. At the date of the assignment, the principal amount of the BlueCrest note was $544,267.
On March 30, 2012, the Company and Northstar agreed to extend until May 1, 2012 the initial payment date for any and all required monthly under the Note, such that the first of the four monthly payments required under the Note will be due and payable on May, 2012 and all subsequent payments will be due on a monthly basis thereafter commencing on June 1, 2012, and to waive any and all defaults and/or events of default under the Note with respect to such payments. The Company did not make the required payment, and as a result, was in default of the revised agreement The Company renegotiated the terms of the Note and Northstar agreed to suspend the requirement of principal payments by the Company and allow payment of interest-only in common stock.
On September 21, 2012, the Company issued 5,000 common stock purchase warrants to Northstar that was treated as additional interest expense upon issuance.
On October 1, 2012, the Company and Northstar entered into a limited waiver and forbearance agreement providing a recapitalized new note balance comprised of all sums due Northstar with a maturity date extended perpetually. The Company agreed to issue 5,000,000 shares of Series A Convertible Preferred Stock and 10,000 of common stock in exchange for $210,000 as payment towards outstanding debt, default interest, penalties, professional fees outstanding and due Northstar. In addition, the Company executed a security agreement granting Northstar a lien on all patents, patent applications, trademarks, service marks, copyrights and intellectual property rights of any nature, as well as the results of all clinical trials, know-how for preparing Myoblasts, old and new clinical data, existing approved trials, all right and title to Myoblasts, clinical trial protocols and other property rights.
In addition, the Company granted Northstar a perpetual license on products as described for resale, relicensing and commercialization outside the United States. In connection with the granted license, Northstar shall pay the Company a royalty of up to 8% on revenues generated.
Effective October 1, 2012, the effective interest rate was 12.85% per annum. The parties agreed, as of February 28, 2013, to reduce the interest rate to 7% per annum.
In connection with the consideration paid, Northstar waived, from the effective date through the earlier of termination or expiration of the agreement, satisfaction of the obligations as described in the forbearance agreement.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
In 2012, 5,000,000 shares of Series A Convertible Preferred Stock were approved to be issued, which was subsequently increased to 20,000,000 shares of preferred stock as Series A Convertible Preferred Stock. In addition, the Company is obligated to issue additional preferred stock equal in lieu of payment of cash of accrued and unpaid interest on each six month anniversary of the effective date (October 1, 2012). In lieu of the initial two payments in preferred stock, the parties have determined to modify the voting rights of the Series A Convertible Preferred Stock from 20 votes per share on matters to be voted on by the common stock holders to 25 votes per share on matters to be voted on by the common stock holders and all prior and subsequent payments of interest will be in common stock. The Company is required to issue additional shares of its common stock (as amended), in lieu of cash, each six month anniversary of the effective date for any accrued and unpaid interest.
As described above, during the year ended December 31, 2013, the Company issued the 5,000,000 shares of Series A Convertible Preferred Stock and the 10,000 of common stock described above in exchange for the $210,000 as payment towards outstanding principle of the debt. In addition, the Company issued 15,000,000 shares of Series A Convertible Preferred Stock as a penalty in settlement of the terms of the forbearance agreement. The fair value of the Preferred Stock of $274,050 was included in interest expense for the year ended December 31, 2013.
On September 30, 2013, the Company issued 8,772 shares of its common stock as payment of $100,000 towards cash advances.
On December 24, 2013, the Company issued 3,916 shares of its common stock as payment of accrued interest through June 30, 2013 of $85,447.
On April 2, 2014, the Company issued 275 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,635 due April 1, 2014 per the forbearance agreement.
On September 17, 2014, limited waiver and forbearance agreement entered into on October 1, 2012 to provide that the perpetual license on products as described for resale, relicensing and commercialization outside the United States was amended as such to condition upon NorthStar providing certain financing, which financing the Company, in its sole discretion, could decline and retain the license.
On October 3, 2014, the Company issued 515 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,705 due October 1, 2014 per the forbearance agreement.
On April 3, 2015, the Company issued 1,363 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,635 due April 1, 2015 per the forbearance agreement.
On October 2, 2015, the Company issued 4,156 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,705 due October 1, 2015 per the forbearance agreement.
On October 7, 2015, the Company issued 34,522 shares of its common stock in settlement of $100,000 principal payment towards the outstanding debt.
As of December 31, 2015 and 2014, the principle of this note was $262,000 and $362,000, respectively.
U.S. Stem Cell Clinic, LLC
On November 6, 2015, the Company issued a 4% promissory note to U.S. Stem Cell Clinic, Inc for $30,000 due November 3, 2017 (See Note 3).
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Officer and Director Notes
|
2015
|
2014
|
|||||||
|
Notes payable, Dr. Murphy
|
$
|
-
|
$
|
465,240
|
||||
|
Note payable, Beverly Murphy
|
50,000
|
50,000
|
||||||
|
Note payable, Mr. Tomas
|
252,250
|
331,354
|
||||||
|
Note payable, Mr. Tomas
|
375,000
|
375,000
|
||||||
|
Note payable, Mr. Tomas
|
500,000
|
500,000
|
||||||
|
Note payable, Ms. Comella
|
287,772
|
299,465
|
||||||
|
Total
|
$
|
1,465,022
|
$
|
2,021,059
|
||||
Notes payable, Dr. Murphy
At December 31, 2014, the Company has outstanding notes payable to Dr. Murphy with interest at 8% per annum due at maturity in aggregate $465,240. Of the outstanding balance, certain subordinated notes totaling $100,000 and $140,000 were previously due on November 30, 2012 and June 4, 2011 respectively, and are unsecured. The Company is not obligated to make payment until Northstar (or successor) Loan is paid off.
On June 20, 2014, the Company issued 10,333 shares of its common stock in settlement of $298,386 of related party note payable, $2,000 advances and accrued interest of $112,954.
On November 4, 2015, the Company issued 270,800 shares of common stock in settlement of $465,200 of outstanding notes payable to Dr. Murphy.
Note payable, Ms. Murphy
At December 31, 2015 and 2014, the Company has outstanding note payable of $50,000 due to Beverly Murphy with interest at 7% per annum due at maturity at October 15, 2015.
Note payable, Dr. Ahn
At December 31, 2013, the Company has outstanding note payable for $125,000 to Dr. Ahn with interest at 8% per annum due at maturity.
On June 20, 2014, the Company issued 4,046 shares of its common stock in settlement of $125,000 of related party note payable and accrued interest of $36,832. There were no amounts outstanding as of December 31, 2015 and 2014.
Notes payable, Mr. Tomas
In 2013, the Company issued a promissory note payable for previous advances and accrued compensation. The promissory note bears interest of 5% per annum and due on demand. During the year ended December 31, 2015 and 2014, the Company paid off $79,104 and $186,779 of the outstanding promissory note, respectively. The principle outstanding balance of this note as of December 31, 2015 is $252,250.
On August 1, 2013, the Company issued a $375,000 promissory note due on demand in settlement of accrued compensation. The promissory note bears interest of 5% per annum and is due on demand. The principle outstanding balance of this note as of December 31, 2015 is $375,000.
On July 1, 2014, the Company issued a $500,000 promissory note in settlement of accrued compensation. The promissory note bears interest of 5% per annum and was due on January 1, 2015. The principle outstanding balance of this note as of December 31, 2015 is $500,000.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Notes payable, Ms. Comella
On August 1, 2013, the Company issued a $125,000 promissory notes due on demand in settlement of accrued compensation. The promissory note bear interest of 5% per annum and due on demand. During the year ended December 31, 2014 and 2013, the Company paid off $97,718 and $27,282 of the outstanding promissory note. The principle outstanding balance of these notes as of December 31, 2015 is $-0-.
On July 1, 2014, the Company issued a $300,000 promissory note in settlement of accrued compensation. The promissory note bears interest of 5% per annum and due on January 1, 2015. During the years ended December 31, 2015 and 2014, the Company paid off $11,693 and $535 of the outstanding promissory note, respectively. The principle outstanding balance of this note as of December 31, 2015 is $287,772.
Transactions with Pavillion
During the years ended December 31, 2015 and 2014, the Company purchased $242,271 and $302,899 lab kits from Pavillion, Inc., respectively, a related party whose owner is related to an officer of the Company. As of December 31, 2015 and 2014, the Company had $74,793 and $23,862, respectively, in accounts payable owed to Pavillion.
NOTE 10 — DERIVATIVE LIABILITIES
Reset warrants
On October 1, 2012, in connection with the forbearance agreement with Northstar as discussed in Note 9 above, the Company issued an aggregate of 15,000 common stock purchase warrants to purchase the Company’s common stock with an exercise price of $14.00 per share for ten years with anti-dilutive (reset) provisions.
The Company has identified embedded derivatives related to the issued warrants. These embedded derivatives included certain and anti-dilutive (reset) provisions. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date and to fair value as of each subsequent reporting date.
At December 31, 2015, the fair value of the reset provision related to the embedded derivative liability of $12,209 was determined using the Binomial Option Pricing model with the following assumptions: dividend yield: 0%; volatility: 178.53%; risk free rate: 2.09%; and expected life: 6.75 years. The Company recorded a gain on change in derivative liabilities of $137,710 during the year ended December 31, 2015.
Convertible notes
In 2014 and 2015, the Company issued convertible notes (see Note 8).
These notes are convertible into common stock, at holders’ option, at a discount to the market price of the Company’s common stock. The Company has identified the embedded derivatives related to these notes relating to certain anti-dilutive (reset) provisions. These embedded derivatives included certain conversion features. The accounting treatment of derivative financial instruments requires that the Company record fair value of the derivatives as of the inception date of these notes and to fair value as of each subsequent reporting date.
The fair value of the embedded derivatives at December 31, 2015, in the amount of $411,718, was determined using the Binomial Option Pricing Model based on the following assumptions: (1) dividend yield of 0%; (2) expected volatility of 178.53%, (3) weighted average risk-free interest rate of 0.49 to 0.65%, (4) expected lives of 0.42 to 0.92 years, and (5) estimated fair value of the Company’s common stock of $0.875 per share. The Company recorded a loss on change in derivative liabilities of $122,615 during the year ended December 31, 2015.
Based upon ASC 840-15-25 (EITF Issue 00-19, paragraph 11) the Company has adopted a sequencing approach regarding the application of ASC 815-40 to its outstanding convertible notes. Pursuant to the sequencing approach, the Company evaluates its contracts based upon earliest issuance date.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
NOTE 11 — STOCKHOLDERS’ EQUITY
Preferred stock
On August 17, 2012, the Board of Directors designated 5,000,000 shares of preferred stock as Series A Convertible Preferred Stock which was increased to 20,000,000 shares of preferred stock as Series A Convertible Preferred Stock (currently held by Northstar Biotechnology Group, LLC). Each share of preferred stock is convertible into equal number of common shares at the option of the holder; entitled to 20 votes on all matters presented to be voted by the holders of common stock; upon event of liquidation, entitled to amount equal to stated value plus any accrued and unpaid dividends or other fees before distribution to junior securities. In lieu of the initial two payments due to Northstar on April 1, 2013 and October 1, 2013, the parties have determined to modify the voting rights of the Series A Convertible Preferred Stock from 20 votes per share on matters to be voted on by the common stock holders to 25 votes per share on matters to be voted on by the common stock holders (see Note 9).
During the year ended December 31, 2013, the Company issued an aggregate of 20,000,000 shares of Series A Convertible Preferred Stock for principle payment and settlement of forbearance (see note 9 above).
Common stock
Effective May 19, 2014, the Company amended its articles of incorporation to increase the authorized shares of capital stock of the Company from nine hundred and fifty million (950,000,000) shares of common stock and twenty million (20,000,000) shares of preferred stock, both $.001 par value respectively, to two billion (2,000,000,000) shares of shares of common stock and twenty million (20,000,000) shares of preferred stock, both $.001 par value respectively.
On October 12, 2015, the Company filed an amendment to its Articles of Incorporation and effected a 1-for-1,000 reverse stock split of its issued and outstanding shares of common stock, $0.001 par value, effective November 19, 2015. The Financial Industry Regulatory Authority (“FINRA”) declared the ex-dividend date for the dividend date as November 4, 2015. All per share amounts and number of shares in the condensed financial statements and related notes have been retroactively restated to reflect the reverse stock split as if it had occurred on the first day of the first period presented resulting in the transfer of $580,852 from common stock to additional paid in capital at December 31, 2014.
During the year ended December 31, 2015, the Company issued an aggregate of 62,425 shares of its common stock in the amount of $211,546 for the settlement of outstanding accounts payable and accrued expenses. In connection with the issuance of the shares the Company recognized a gain on settlement of accounts payable and accrued expenses in the amount of $144,669 (see Note 5).
During the year ended December 31, 2015, the Company issued 6,650 shares of common stock in settlement of litigation. In connection with the issuances, the Company recognized a loss in the amount of $59,850, which is included in the marketing, general and administration expense in the Statement of Operations (see Note 13).
On April 3, 2015, the Company issued 1,363 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,635 due April 1, 2015 per the forbearance agreement on Northstar note (See Note 9).
On October 2, 2015, the Company issued 4,156 shares of its common stock in lieu of payment in cash of accrued and unpaid interest of $12,705 due October 1, 2015 per the forbearance agreement on Northstar note (See Note 9).
During the year ended December 31, 2015, the Company issued an aggregate of 763,434 shares of its common stock for the conversion of $949,017 of notes payable and related accrued interest. Upon conversion of the notes, the Company recorded an adjustment to the derivative liability in the amount of $1,399,628 (see Note 13).
On November 4, 2015, the Company issued 270,800 shares of common stock in settlement of $465,200 of outstanding notes payable to Dr. Murphy.
On November 4, 2015, the Company issued 3,411 shares of common stock in settlement of $5,860 of related party advances.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
During the year ended December 31, 2015, the Company purchased 85,211 shares of the Company’s common stock in the open market at an average cost of $2.61 per share.
During the year ended December 31, 2015, the Company issued an aggregate of 87,812 shares of common stock in exchange for $521,538 under the stock purchase agreement with Magna Equities II, LLC (see Note 7), and issued an aggregate of 7,852 shares of common stock in exchange for $61,270. In connection with the stock sale, the Company issued an aggregate of 1,444 warrants to purchase the Company’s common stock (see Note 12).
During the year ended December 31, 2015, the Company issued an aggregate of 24,353 shares of its common stock in settlement of accumulative outstanding amounts due to Guarantors of the Company of $961,125. In connection with the issuance, the Company incurred a $791,024 gain in settlement of debt.
During the year ended December 31, 2014, the Company issued 42,067 shares of common stock for proceeds of $479,625.
During the year ended December 31, 2014, the Company issued an aggregate of 48,585 shares of its common stock in settlement of outstanding accounts payable and accrued expenses. In connection with the issuance, the Company incurred $120,656 gain in settlement of debt.
During the year ended December 31, 2014, the Company issued an aggregate of 83,236 shares of its common stock for the conversion of $1,974,917 of notes payable and related accrued interest. In connection with the issuance, the Company incurred $255,890 gain in settlement of debt.
During the year ended December 31, 2014, the Company issued an aggregate of 3,840 shares of its common stock for services provided to the Company valued at $183,991.
During the year ended December 31, 2014, the Company issued an aggregate of 11,918 shares of its common stock in connection with the exercise of warrants.
During the year ended December 31, 2014 the Company issued Magna Group 12,000 shares of its common stock as a commitment fee for entering into the Purchase Agreement (See Note 7).
NOTE 12 — STOCK OPTIONS AND WARRANTS
Stock Options
In December 1999, the Board of Directors and shareholders adopted the 1999 Officers and Employees Stock Option Plan, or the Employee Plan, and the 1999 Directors and Consultants Stock Option Plan, or the Director Plan. The Employee Plan and the Director Plan are collectively referred to herein as the Plans. The Plans are administered by the Board of Directors and the Compensation Committee.
The objectives of the Plans include attracting and retaining key personnel by encouraging stock ownership in the Company by such persons. In February 2010, the Directors & Consultants Plan was amended to extend the termination date of the Plan to December 1, 2011.
On April 1, 2013, the Board of Directors approved, subject to shareholder approval, the establishment of the Bioheart 2013 Omnibus Equity Compensation Plan, or the “2013 Omnibus Plan”. The 2013 Omnibus Plan reserves up to fifty thousand shares of common stock for issuance. On August 4, 2014, the Board of Directors approved to set the reserve to one hundred thousand shares of common stock for issuance and to close the 1999 Officers and Employees Stock Option Plan. On February 2, 2015, at the annual meeting of shareholders, the majority of shareholders approved the 2013 Omnibus Equity Compensation Plan. On November 2, 2015, the Board of Directors approved the increase of the reserve under the 2013 Omnibus Plan to five hundred million shares of common stock for issuance.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
A summary of options at December 31, 2015 activity during the years ended December 31, 2015 and 2014 is presented below:
|
Shares
|
Weighted-
Average
Exercise Price
|
Weighted-
Average
Remaining
Contractual
Term (in years)
|
||||||||||
|
Options outstanding at January 1, 2014
|
23,921
|
$
|
150.00
|
9.0
|
||||||||
|
Granted
|
43,148
|
$
|
23.00
|
10.0
|
||||||||
|
Exercised
|
—
|
$
|
||||||||||
|
Forfeited/Expired
|
(136
|
)
|
$
|
5,200.00
|
||||||||
|
Options outstanding at December 31, 2014
|
66,933
|
$
|
56.00
|
8.9
|
||||||||
|
Granted
|
489,116
|
$
|
1.98
|
10.0
|
||||||||
|
Exercised
|
—
|
|||||||||||
|
Forfeited/Expired
|
(229
|
)
|
$
|
5,103.28
|
||||||||
|
Options outstanding at December 31, 2015
|
555,820
|
$
|
6.43
|
9.6
|
||||||||
|
Options exercisable at December 31, 2015
|
511,660
|
$
|
5.60
|
9.7
|
||||||||
|
Available for grant at December 31, 2015
|
7,383,070
|
|||||||||||
The following information applies to options outstanding and exercisable at December 31, 2015:
|
Options Outstanding
|
Options Exercisable
|
||||||||||||||||||
|
Shares
|
Weighted-
Average
Remaining
Contractual
Term
|
Weighted-
Average
Exercise
Price
|
Shares
|
Weighted-
Average
Exercise
Price
|
|||||||||||||||
| $ | 0.00 – $20.00 |
534,706
|
9.7
|
$
|
3.32
|
501,682
|
$
|
2.72
|
|||||||||||
| $ | 20.01 – $30.00 |
19,849
|
8.5
|
$
|
26.96
|
8,713
|
$
|
26.94
|
|||||||||||
| $ | 30.01 – $100.00 |
300
|
5.7
|
$
|
70.00
|
300
|
$
|
70.00
|
|||||||||||
| $ | >100.00 |
965
|
4.0
|
$
|
1,288.36
|
965
|
$
|
1,288.36
|
|||||||||||
|
555,820
|
9.6
|
$
|
6.43
|
511,660
|
$
|
5.60
|
|||||||||||||
On February 24, 2014, the Company issued an aggregate 15,000 options to purchase the Company’s common stock at $19.00 per share to officers, vesting at 25% immediately and the remainder over approximately 42 months, exercisable over 10 years. The aggregate fair value of $282,597, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 163.63% and Risk free rate: 2.75%.
On February 24, 2014, the Company issued an aggregate 4,800 options to purchase the Company’s common stock at $19.00 per share to officers, vesting immediately and exercisable over 10 years. The aggregate fair value of $90,431, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 163.63% and Risk free rate: 2.75%.
On May 12, 2014, the Company issued an aggregate 4,848 options to purchase the Company’s common stock at $27.20 per share to officers and employees, vesting over four years at anniversary and exercisable over 10 years. The aggregate fair value of $130,135, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 161.36% and Risk free rate: 2.66%.
On August 1, 2014, the Company issued an aggregate 15,000 options to purchase the Company’s common stock at $26.94 per share to officers and employees, vesting at 25% immediately and 75% over three years at anniversary and exercisable over 10 years. The aggregate fair value of $391,798, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 168.62% and Risk free rate: 2.52%.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
On November 3, 2014, the Company issued an aggregate 700 options to purchase the Company’s common stock at $19.32 per share to officers and employees, vesting at 25% each year over four years at anniversary and exercisable over 10 years. The aggregate fair value of $12,739, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 2.36%.
On November 3, 2014, the Company issued an aggregate 2,800 options to purchase the Company’s common stock at $19.32 per share to board of directors, vesting immediately and exercisable over 10 years. The aggregate fair value of $54,957, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 2.36%.
On February 2, 2015, the Company issued an aggregate 7,000 options to purchase the Company’s common stock at $11.16 per share to members of the Board of Directors, vesting immediately and exercisable over 10 years. The aggregate fair value of $121,735, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 142.65% and Risk free rate: 1.68%.
On August 24, 2015, the Company issued 100 options to purchase the Company’s common stock at $5.31 per share to a consultant, vesting immediately and exercisable over 4 years. The aggregate fair value of $347, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 129.01% and Risk free rate: 1.39%.
On November 2, 2015, the Company issued an aggregate of 467,016 options to purchase the Company’s common stock at $1.713 per share to two officers, vesting immediately and exercisable over 10 years. The aggregate fair value of $738,405 determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 160.66% and Risk free rate: 1.57%.
On November 2, 2015, the Company issued an aggregate of 15,000 options to purchase the Company’s common stock at $6.24 per share to two officers, vesting over four years on each anniversary and exercisable over 10 years. The aggregate fair value of $23,512, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 160.66% and Risk free rate: 1.93%.
The fair value of all options vesting during the year ended December 31, 2015 and 2014 of $1,156,435 and $537,606, respectively, was charged to current period operations.
As of December 31, 2015, the Company had approximately $473,898 of total unrecognized compensation cost related to non-vested awards granted under the Plan, which the Company expects to recognize over a weighted average period of 1.0 year.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Warrants
A summary of common stock purchase warrants at December 31, 2015 and activity during the years ended December 31, 2015 and 2014 is presented below:
|
Shares
|
Weighted-
Average
Exercise
Price
|
Weighted-
Average
Remaining
Contractual
Term (in years)
|
||||||||||
|
Outstanding at January 1, 2014
|
118,134
|
$
|
220.00
|
6.3
|
||||||||
|
Issued
|
57,582
|
$
|
20.00
|
8.2
|
||||||||
|
Exercised
|
(11,918
|
)
|
$
|
10.00
|
||||||||
|
Expired
|
(13,178
|
)
|
$
|
80.00
|
||||||||
|
Outstanding at December 31, 2014
|
150,620
|
$
|
170.00
|
6.6
|
||||||||
|
Issued
|
2,072
|
$
|
19.98
|
8.18
|
||||||||
|
Exercised
|
-
|
$
|
||||||||||
|
Expired
|
(13,325
|
)
|
$
|
24.00
|
||||||||
|
Outstanding at December 31, 2015
|
139,367
|
$
|
182.26
|
6.3
|
||||||||
|
Exercisable at December 31, 2015
|
133,822
|
$
|
99.98
|
6.3
|
||||||||
The following information applies to common stock purchase warrants outstanding and exercisable at December 31, 2015:
|
Warrants Outstanding
|
Warrants Exercisable
|
|||||||||||||||||||||
|
Shares
|
Weighted-
Average
Remaining
Contractual
Term
|
Weighted-
Average
Exercise
Price
|
Shares
|
Weighted-
Average
Exercise
Price
|
||||||||||||||||||
|
$
|
0.01 – $20.00
|
94,108
|
6.8
|
$
|
15.54
|
94,108
|
$
|
15.54
|
||||||||||||||
|
$
|
20.01 – $30.00
|
29,743
|
6.1
|
$
|
24.52
|
27,743
|
$
|
24.72
|
||||||||||||||
|
$
|
30.01 – $40.00
|
628
|
1.6
|
$
|
40.00
|
628
|
$
|
40.00
|
||||||||||||||
|
$
|
40.01 - $50.00
|
6,253
|
3.1
|
$ |
48.36
|
4,253
|
$ |
48.49
|
||||||||||||||
|
$
|
50.01 – $60.00
|
543
|
1.9
|
$
|
60.00
|
543
|
$
|
60.00
|
||||||||||||||
|
$
|
>60.00
|
8,092
|
4.2
|
$
|
2,823.67
|
6,547
|
$
|
1,675.28
|
||||||||||||||
|
139,367
|
6.3
|
$
|
182.26
|
133,822
|
$
|
99.98
|
||||||||||||||||
In conjunction with the authorized issuance of common stock, the Company issued 41,582 common stock purchase warrants during the year ended December 31, 2014.
During the year ended December 31, 2014, the Company issued an aggregate of 8,000 warrants in connection with a revenue share venture agreement dated March 10, 2014. The warrants are exercisable at $21.70 for four years vesting from June 8, 2014 through March 10, 2016 (term of agreement). During the year ended December 31, 2014, the Company charged $103,238 to current period operations.
During the year ended December 31, 2014, the Company issued an aggregate of 4,000 warrants in connection with use of certain intellectual property. The warrants are exercisable at $48.10 for four years vesting from July 6, 2014 through April 6, 2017. During the year ended December 31, 2014, the Company charged $32,878 to current period operations for the vesting portion.
During the year ended December 31, 2014, the Company issued an aggregate of 4,000 warrants in connection with the termination of a revenue share agreement. The warrants are exercisable at $15.70 for four years vesting May 27, 2015. During the year ended December 31, 2014, the Company charged $54,146 to current period operations.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
On November 3, 2014, the Company extended an aggregate of 13,064 expiring warrants previously issued to the Company’s board of directors for additional three years. The aggregate change in fair value of $148,517, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 152.87% and Risk free rate: 0.96% was charged to current period operations.
In conjunction with the authorized issuance of common stock, the Company issued 1,444 common stock purchase warrants during the year ended December 31, 2015. The warrants are exercisable at an exercise price of approximately $11.27 per share for ten years.
During the year ended December 31, 2015, the Company issued 628 warrants in connection with the settlement of debt. The warrants are exercisable at $40.00 for three years. The fair value of $14,886, determined using the Black Scholes option pricing model with the following assumptions: Dividend yield: 0%; Volatility: 164.06% and Risk free rate: 0.87%. In connection with the settlement, the Company recorded a gain of $10,221 in settlement of debt to current period operations.
NOTE 13 — COMMITMENTS AND CONTINGENCIES
Leases
The Company entered into several operating lease agreements for facilities and equipment. Terms of certain lease arrangements include renewal options, escalation clauses, payment of executory costs such as real estate taxes, insurance and common area maintenance. The landlord has filed a security interest against the assets of the Company.
In July 2013, the Company amended its facility lease to extend the term of the lease until July 31, 2016. Approximate annual future minimum lease obligations under non-cancelable operating lease agreements as of December 31, 2015 are as follows:
|
Year ending December 31,
|
||||
|
2016
|
$
|
43,830
|
||
Rent expense was $131,257 and $128,136 for the years ended December 31, 2015 and 2014, respectively.
Revenue Share Agreement
On March 10, 2014, the Company entered into a revenue sharing agreement (the Agreement) with Global Stem Cells, Group, LLC and its subsidiaries whereby both parties will participate in marketing for obtaining patients and provide physician training for stem cell treatments under the names of “Regenestem” and “Stem Cell Training,” respectively. In addition, each party will be responsible for selling equipment and kit to existing and previous customers. Profits are divided on a fifty/fifty basis with distribution within 10 days of the accounting for patients and physician training and 30 day with sales of equipment and kits. In November 2014, the Company terminated the Agreement.
In consideration of Global Stem Cell Group, LLC’s participation, the Company issued an aggregate of 8,000 warrants to purchase the Company’s common stock for four years at $21.70 per share with 2,000 warrants vesting 90 days from the effective date, 2,000 vesting on each anniversary date for three years. At termination of the revenue share agreement, the Company issued an aggregate of 4,000 warrants to purchase the Company’s common stock. During the year ended December 31, 2014, the Company charged $54,146 to current period operations. Because of the termination of the agreement during 2014, all services were considered final and rendered and the fair value of all 12,000 warrants issued under this agreement were recognized during 2014.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
William Beaumont Hospital
In June 2000, the Company entered into an agreement with William Beaumont Hospital, or WBH, pursuant to which WBH granted to the Company worldwide, exclusive, non-sublicenseable license to two U.S. method patents covering the inducement of human adult myocardial cell proliferation in vitro, or the WBH IP. The term of the agreement is for the life of the patents, which expire in 2015. The Company did not use this license in any of our technologies. The Company had not made any payments to WBH other than the initial payment to acquire the license. The Company has received confirmation from WBH that it has no obligation under the patent license agreement and WBH agreed to terminate the patent license agreement. (See Note 5)
Accordingly, the Company has recognized approximately $2,122,130 in settlement of debt which represents the accumulative accrual and related interest from past years under the 2000 patent license agreement during 2014.
Employment agreements
On July 28, 2014, the Company’s Board of Directors approved the 2014/2015 salary for Mike Tomas, Chief Executive Officer, at $525,000 per year, beginning July 1, 2014 with an incentive bonus ranging from $150,000 to $500,000. In addition, the Board of Directors will grant Mr. Tomas options to be determined on or before June 30, 2015. The Company’s Board of Directors approved a bonus of $500,000 and options to acquire 10,000 shares of the Company’s common stock for ten years with four year vesting and a cashless exercise provision at an exercise price equal to the five day average closing price of the Company’s common stock as of August 1, 2014. The cash bonus may be paid in the form a six month promissory note. On November 2, 2015, the Company granted Mr. Tomas - 291,885 options to acquire the Company’s stock with an exercise price of $1.713 per share, vesting immediately and expiring ten years from the date of issuance.
On July 28, 2014, the Company’s Board of Directors approved the 2014/2015 salary for Kristin Comella, Chief Scientific Officer, at $250,000 per year, beginning July 1, 2014 with an incentive bonus ranging from $100,000 to $300,000. In addition, the Board of Directors will grant Ms. Comella options to be determined on or before June 30, 2015. The Company’s Board of Directors approved a bonus of $300,000 and options to acquire 5,000 shares of the Company’s common stock for ten years with four year vesting and a cashless exercise provision at an exercise price equal to the five day average closing price of the Company’s common stock as of August 1, 2014. The cash bonus may be paid in the form a six month promissory note. On November 2, 2015, the Company granted Ms. Comella 175,131 options to acquire the Company’s stock with an exercise price of $1.713 per share, vesting immediately and expiring ten years from the date of issuance
Litigation
On March 19 2015, the Company settled a prospective dispute with a third party over the use of proprietary information through the issuance of 6,650 shares of common stock. (See Note 11)
On November 10, 2014, the Company was served with a lawsuit by an alleged assignee and a guarantor to a Loan Guarantee, Payment and Security Agreement. These parties claim breach of that Agreement and damages of approximately $2.3 Million plus interest. The assignor and assignee also sued the Company’s directors and two past directors and an affiliate shareholder for breach of fiduciary duty, claiming damages as alleged creditors arising out of these parties’ alleged participation in Northstar Biotech Group, LLC, a secured creditor of the Company. On June 1, 2015, the Company settled the lawsuit. As part of the settlement, the Company recorded a gain of $1,169,058. The Company provided a note to the original guarantor in the amount of $1,697,762, to be paid over a five year period with a 0% interest rate.
On September 17, 2015, a product liability lawsuit was filed in Broward County, specifically Patsy Bade v. Bioheart, Inc. US Stem Cell Clinics LLC, Aleiandro Perez, ARNP, and Shareen Greenbaum, M.D., and on November 30, 2015, a product liability lawsuit was filed in Broward County, specifically Elizabeth Noble v. Bioheart, Inc. US Stem Cell Clinics LLC, Aleiandro Perez, ARNP, and Shareen Greenbaum, M.D. Both matters are ongoing and the Company intends to defend the matters rigorously. In the event, if ever, of a settlement or loss, management believes that any loss related to the claims will be covered by their insurance policy and that there is no indication that the insurance company will dispute any claim, if ever required.
The Company is subject to other legal proceedings that arise in the ordinary course of business. In the opinion of management, as of December 31, 2015, the amount of ultimate liability with respect to such matters, if any, in excess of applicable insurance coverage, is not likely to have a material impact on the Company’s business, financial position, results of operations or liquidity. However, as the outcome of litigation and other claims is difficult to predict significant changes in the estimated exposures could exist.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
NOTE 14 — INCOME TAXES
The Company follows Accounting Standards Codification subtopic 740, Income Taxes (“ASC 740”) which requires the recognition of deferred tax liabilities and assets for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under such method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases using enacted tax rates in effect for the year in which the differences are expected to reverse.
The difference between income tax expense computed by applying the federal statutory corporate tax rate and actual income tax expense is as follows:
|
2015
|
2014
|
|||||||
|
Income taxes using U.S. federal statutory rate
|
$
|
(575,971
|
)
|
$
|
(766,194
|
)
|
||
|
State income taxes, net of federal benefit
|
(47,571
|
)
|
(90,051
|
)
|
||||
|
Return to Provision adjustments
|
116,025
|
-
|
||||||
|
Stock Option Expirations
|
-
|
66,043
|
||||||
|
Net Operating Loss adjustments
|
-
|
(144,824
|
)
|
|||||
|
Nontaxable Gain on Derivative Instrument
|
8,101
|
933,477
|
||||||
|
Change in Valuation Allowance
|
493,123
|
1,549
|
||||||
|
Other
|
6,293
|
-
|
||||||
|
$
|
-
|
$
|
—
|
|||||
At December 31, 2015, the significant components of the deferred tax assets (liabilities) are summarized below:
|
2015
|
2014
|
|||||||
|
Deferred tax assets:
|
||||||||
|
Stock Based Compensation
|
$ | 5,099,422 | $ | 4,794,777 | ||||
|
Net Operating Losses
|
36,323,004 | 36,117,831 | ||||||
|
Other
|
24,774 | 41,469 | ||||||
|
Total deferred tax assets
|
41,447,200 | 40,954,077 | ||||||
|
Deferred tax liabilities:
|
- |
—
|
||||||
|
Total deferred tax liabilities
|
- |
—
|
||||||
|
Valuation allowance
|
41,447,200 | 40,954,077 | ||||||
|
Net deferred tax assets
|
$ | - | $ |
—
|
||||
As of December 31, 2015 and December 31, 2014, the Company had U.S. federal net operating loss carryforwards of approximately $96.5 million and $95.8 million, respectively, which expire at various dates from 2020 through 2035. These net operating loss carryforwards may be used to offset future taxable income and thereby reduce the Company’s U.S. federal income taxes. Section 382 of the Internal Revenue Code of 1986 (the “Code”) imposes an annual limit on the ability of a corporation that undergoes a greater than 50% ownership change to use its net operating loss carry forwards to reduce its tax liability. If in the future the Company issues common stock or additional equity instruments convertible in common shares which result in an ownership change exceeding the 50% limitation threshold imposed by section 382 of the Code, the Company’s net operating loss carryforwards may be significantly limited as to the amount of use in a particular years. In addition, all or a portion of the Company’s net operating loss carryforwards may expire unutilized. As of December 31, 2015 and December 31, 2014, the Company had net operating loss carryforwards for state income tax purposes of approximately $97.5 million and $95.8 million, respectively, which expire at various dates from 2020 through 2035.
The Company has provided a full valuation allowance against its net deferred tax assets, since in the opinion of management based upon the earnings history of the Company; it is more likely than not that the benefits of these assets will not be realized.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
The Company complies with the provisions of FASB ASC 740-10 in accounting for its uncertain tax positions. ASC 740-10 addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740-10, the Company may recognize the tax benefit from an uncertain tax position only if it is more likely that not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. Management has determined that the Company has no significant uncertain tax positions requiring recognition under ASC 740-10.
The Company is subject to income tax in the U.S., and certain state jurisdictions. The Company has not been audited by the U.S. Internal Revenue Service, or any states in connection with income taxes. The Company’s tax years generally remain open to examination for all federal and state tax matters until its net operating loss carryforwards are utilized and the applicable statutes of limitation have expired. The federal and state tax authorities can generally reduce a net operating loss (but not create taxable income) for a period outside the statute of limitations in order to determine the correct amount of net operating loss which may be allowed as a deduction against income for a period within the statute of limitations.
The Company recognizes interest and penalties related to unrecognized tax benefits, if incurred, as a component of income tax expense.
NOTE 15 — FAIR VALUE MEASUREMENT
The Company adopted the provisions of Accounting Standards Codification subtopic 825-10, Financial Instruments (“ASC 825-10”) on January 1, 2008. ASC 825-10 defines fair value as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required or permitted to be recorded at fair value, the Company considers the principal or most advantageous market in which it would transact and considers assumptions that market participants would use when pricing the asset or liability, such as inherent risk, transfer restrictions, and risk of nonperformance. ASC 825-10 establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. ASC 825-10 establishes three levels of inputs that may be used to measure fair value:
Level 1 – Quoted prices in active markets for identical assets or liabilities.
Level 2 – Observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities; quoted prices in markets with insufficient volume or infrequent transactions (less active markets); or model-derived valuations in which all significant inputs are observable or can be derived principally from or corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3 – Unobservable inputs to the valuation methodology that are significant to the measurement of fair value of assets or liabilities.
All items required to be recorded or measured on a recurring basis are based upon level 3 inputs.
To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for disclosure purposes, the level in the fair value hierarchy within which the fair value measurement is disclosed and is determined based on the lowest level input that is significant to the fair value measurement.
Upon adoption of ASC 825-10, there was no cumulative effect adjustment to beginning retained earnings and no impact on the financial statements.
The carrying value of the Company’s cash and cash equivalents, accounts receivable, accounts payable, short-term borrowings (including convertible notes payable), and other current assets and liabilities approximate fair value because of their short-term maturity.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
As of December 31, 2015 or 2014, the Company did not have any items that would be classified as level 1 or 2 disclosures.
The Company recognizes its derivative liabilities as level 3 and values its derivatives using the methods discussed in notes 8 and 10. While the Company believes that its valuation methods are appropriate and consistent with other market participants, it recognizes that the use of different methodologies or assumptions to determine the fair value of certain financial instruments could result in a different estimate of fair value at the reporting date. The primary assumptions that would significantly affect the fair values using the methods discussed in Notes 8 and 10 are that of volatility and market price of the underlying common stock of the Company.
As of December 31, 2015 and 2014, the Company did not have any derivative instruments that were designated as hedges.
The derivative liability as of December 31, 2015, in the amount of $423,927 has a level 3 classification.
The following table provides a summary of changes in fair value of the Company’s Level 3 financial liabilities as of December 31, 2015:
|
Warrant
Liability
|
Debt
Derivative
|
|||||||
|
Balance, December 31, 2013
|
$ |
146,855
|
$
|
256,956
|
||||
|
Total (gains) losses
|
||||||||
|
Initial fair value of debt derivative at note issuance
|
—
|
1,443,708
|
||||||
|
Initial fair value of derivative relating to reset warrants
|
—
|
—
|
||||||
|
Mark-to-market at December 31, 2014:
|
3,065
|
(429,018
|
)
|
|||||
|
Transfers out of Level 3 upon increase in authorized shares
|
—
|
—
|
||||||
|
Transfers out of Level 3 upon conversion and settlement of notes
|
—
|
(680,295
|
)
|
|||||
|
Balance, December 31, 2014
|
$
|
149,920
|
$
|
591,351
|
||||
|
Total (gains) losses
|
||||||||
|
Initial fair value of debt derivative at note issuance
|
—
|
1,097,379
|
||||||
|
Mark-to-market at December 31, 2015:
|
(137,711
|
)
|
122,616
|
|||||
|
Transfers out of Level 3 upon conversion or payoff of notes payable
|
—
|
(1,399,628
|
)
|
|||||
|
Balance, December 31, 2015
|
$
|
12,209
|
$
|
411,718
|
||||
|
Net gain (loss) for the period included in earnings relating to the liabilities held at December 31, 2015
|
$
|
137,711
|
$
|
(122,616
|
)
|
|||
Fluctuations in the Company’s stock price are a primary driver for the changes in the derivative valuations during each reporting period. The Company’s stock price decreased approximately 22% from December 31, 2014 to December 31, 2015. As the stock price decreases for each of the related derivative instruments, the value to the holder of the instrument generally decreases. Additionally, stock price volatility is one of the significant unobservable inputs used in the fair value measurement of each of the Company’s derivative instruments.
Decreases in expected volatility would generally result in a lower fair value measurement. A 10 percent change in pricing inputs and changes in volatilities and correlation factors would result in less than a $70,000 change in our Level 3 fair value.
NOTE 16 — SUBSEQUENT EVENTS
Seaside Bank loan renewal
On January 11, 2016, the Company renewed the loan with Seaside National Bank and Trust extend the maturity date to January 11, 2018, all other terms and conditions remain unchanged. (see Note 8)
Office lease renewal
On February 4, 2016, the Company extended its facility lease to extend the term of the lease until August 31, 2019 at a monthly base rent of $7,306 plus a pro rata share of landlord’s operating expenses In Connection with the renewal, the Company was required to pay $11,072 security deposit.
U.S. STEM CELL, INC.
NOTES TO FINANCIAL STATEMENTS
DECEMBER 31, 2015 AND 2014
Approximate annual future minimum lease obligations under this non-cancelable facilities operating lease agreement as of December 31, 2015 are as follows:
|
Year ending December 31,
|
||||
|
2016
|
$
|
87,674
|
||
|
2017
|
87,674
|
|||
|
2018
|
87,674
|
|||
|
2019
|
58,450
|
|||
|
Total
|
$
|
321,472
|
||
Subsequent common stock issuances
In January, February and March 2016, the Company issued an aggregate of 625,352 shares of its common stock in settlement of $91,170 notes payable and $2,649 accrued interest; 16,753 shares for services of $13,110 and 14,606 shares in settlement of related party advances.
Subsequent financing
Magna Equities II, LLC
On December 3, 2015, the Company entered into a Securities Purchase Agreement with Magna Equities II, LLC for the sale of a 12% convertible note in the principal amount of $262,500 (the “Note”). The Note was subsequently funded in February 2016 upon effectiveness of the Company’s registration statement (see below).
The Note bears interest at the rate of 12% per annum. All interest and principal must be repaid on December 3, 2016. The Note is convertible into common stock, at Magna’s option, at the lower of i) 40% discount to the lowest sales price of the common stock during the 5 trading day period prior to conversion or ii) $0.70. In the event the Company prepays the Note in full, the Company is required to pay off all principal at 140%, interest and any other amounts.
On December 12, 2014, the Company filed a Registration Statement on Form S-1 to register 341,718 shares of common issuable upon the conversion of Magna Equity II, LLC convertible notes dated December 3, 2015 (as restated) for $110,000 and December 3, 2015 for $262,500. The latter note was funded in February 2016. The Registration Statement on Form S-1 was declared effective on February 12, 2016
Daniel James Management Inc.
On January 7, 2016, the Company entered into a Securities Purchase Agreement with Daniel James Management Inc., for the sale of a 9.5% convertible note in the principal amount of $25,000 (the “Note”).
The Note bears interest at the rate of 9.5% per annum. All interest and principal must be repaid on January 7, 2017. The Note is convertible into common stock, at Daniel James Management Inc.’s option, at a 47% discount to the lowest daily closing trading price of the common stock during the 10 trading day period prior to conversion. In the event the Company prepays the Note in full, the Company is required to pay off all principal at 150%, interest and any other amounts.
Effective registration statement
On February 12, 2016, the Company’s filed registration statement became effective registering common shares issuable upon the conversion of Magna Equity II, LLC convertible notes dated December 3, 2015 (as restated) for $110,000 and December 3, 2015 for $262,500.
Related party transactions
In February 2016, the Company reimbursed Dr. Murphy, a member of the Company’s board of directors, $100,000 for a previous advance of $50,000 and settlement of accrued interest on related party notes payable.
F-35