Attached files
| file | filename |
|---|---|
| EX-21 - EX-21 - UNITED THERAPEUTICS Corp | a2227374zex-21.htm |
| EX-31.3 - EX-31.3 - UNITED THERAPEUTICS Corp | a2227374zex-31_3.htm |
| EX-32.1 - EX-32.1 - UNITED THERAPEUTICS Corp | a2227374zex-32_1.htm |
| EX-23.1 - EX-23.1 - UNITED THERAPEUTICS Corp | a2227374zex-23_1.htm |
| EX-32.3 - EX-32.3 - UNITED THERAPEUTICS Corp | a2227374zex-32_3.htm |
| EX-32.2 - EX-32.2 - UNITED THERAPEUTICS Corp | a2227374zex-32_2.htm |
| EX-31.1 - EX-31.1 - UNITED THERAPEUTICS Corp | a2227374zex-31_1.htm |
| EX-31.2 - EX-31.2 - UNITED THERAPEUTICS Corp | a2227374zex-31_2.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| (Mark One) | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
|
For the fiscal year ended December 31, 2015 |
||
OR |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
|
For the transition period from to |
||
Commission file number 0-26301
United Therapeutics Corporation
(Exact Name of Registrant as Specified in Its Charter)
| Delaware (State or Other Jurisdiction of Incorporation or Organization) |
52-1984749 (I.R.S. Employer Identification No.) |
|
1040 Spring Street, Silver Spring, MD (Address of Principal Executive Offices) |
20910 (Zip Code) |
(301) 608-9292
Registrant's Telephone Number, Including Area Code
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
|---|---|---|
| Common Stock, par value $.01 per share and associated preferred stock purchase rights |
NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of "large accelerated filer," "accelerated filer," and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ý | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the Common Stock held by non-affiliates of the registrant, based on the closing price on June 30, 2015, as reported by the NASDAQ Global Select Market was approximately $6,657,883,891.
The number of shares outstanding of the issuer's common stock, par value $0.01 per share, as of February 12, 2016, was 45,352,746.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's definitive proxy statement for the registrant's 2016 annual meeting of shareholders scheduled to be held on June 21, 2016, are incorporated by reference in Part III of this Form 10-K.
2
United Therapeutics Corporation is a biotechnology company focused on the development and commercialization of innovative products to address the unmet medical needs of patients with chronic and life-threatening diseases.
Our key therapeutic products and product candidates include:
- •
- Prostacyclin Analogues. Prostacyclin analogues are
stable synthetic forms of prostacyclin, an important molecule produced by the body that has powerful effects on blood vessel health and function. Our lead product is Remodulin®
(treprostinil) Injection (Remodulin), which is administered subcutaneously (under the skin) or intravenously (in the vein) for the treatment of pulmonary arterial hypertension (PAH) to diminish
symptoms associated with exercise. The United States Food and Drug Administration (FDA) approved Remodulin for subcutaneous and intravenous administration in 2002 and 2004, respectively. Outside the
United States, Remodulin is approved in 39 countries, most of which have approved both routes of administration. We are developing new technologies to make Remodulin delivery more convenient, such as
implantable pump systems for intravenous Remodulin and pre-filled, semi-disposable pumps for subcutaneous Remodulin. In 2009, the FDA approved Tyvaso® (treprostinil) Inhalation Solution
(Tyvaso), an inhaled prostacyclin therapy for the treatment of PAH. In December 2013, the FDA approved Orenitram® (treprostinil) Extended-Release Tablets (Orenitram), an oral prostacyclin
analogue for the treatment of PAH, which commenced sales during the second quarter of 2014. Our wholly-owned subsidiary, Lung Biotechnology PBC, is developing another oral prostacyclin analogue for
the treatment of PAH called esuberaprost.
- •
- Phosphodiesterase Type 5 (PDE-5) Inhibitor. PDE-5
inhibitors act to inhibit the degradation of cyclic guanosine monophosphate (cyclic GMP) in cells. Cyclic GMP is activated by nitric oxide (NO), a naturally occurring substance in the body that
mediates the relaxation of vascular smooth muscle. Our PDE-5 inhibitor is Adcirca® (tadalafil) tablets (Adcirca), a once-daily oral therapy for the treatment of PAH. We acquired exclusive
U.S. commercialization rights to Adcirca from Eli Lilly and Company (Lilly) in 2008. In 2009, the FDA approved Adcirca for the treatment of PAH.
- •
- Monoclonal Antibody (MAb). MAbs are antibodies that bind to cancerous tumors and destroy the cancer cells through a mechanism called antibody-dependent cell mediated cytotoxicity. In March 2015, the FDA approved Unituxin® (dinutuximab) Injection (Unituxin), in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma who achieve at least a partial response to prior first-line multiagent, multimodality therapy. We commenced U.S. sales of Unituxin in the third quarter of 2015. We received European Commission approval during the third quarter of 2015, and plan to commence commercial sales in individual European countries following pricing and reimbursement approvals on a country-by-country basis.
During the fourth quarter of 2015, we sold the rights to our glycobiology antiviral platform under terms that entitle us to milestone and royalty payments from the buyer in the event the program is successful. Additionally, during the fourth quarter of 2015, we terminated our license agreement with Pluristem Ltd. (Pluristem) relating to the development of a cell-based product for the treatment of PAH using Pluristem's PLacental eXpanded (PLX) cells.
We generate revenues from sales of Remodulin, Tyvaso, Adcirca, Orenitram and Unituxin (which we refer to as our commercial products). We commenced sales of Orenitram and Unituxin during the second quarter of 2014 and third quarter of 2015, respectively. We expect that sales of our existing
3
commercial products will continue to be our primary sources of revenues for the next several years. Our sales and marketing staff supports the availability of our commercial products in the countries in which they are approved. These efforts are supplemented by contracted distributors.
Finally, we are engaged in early-stage research and development into a number of organ transplantation-related technologies.
United Therapeutics was incorporated in Delaware in June 1996. Our principal executive offices are located at 1040 Spring Street, Silver Spring, Maryland 20910 and at 55 T.W. Alexander Drive, Research Triangle Park, North Carolina 27709.
Unless the context requires otherwise or unless otherwise noted, all references in this Annual Report on Form 10-K to "United Therapeutics" and to the "company", "we", "us" or "our" are to United Therapeutics Corporation and its subsidiaries.
Our Products
Our product portfolio includes the following:
Product
|
Mode of Delivery | Indication | Current Status | Our Territory | ||||
|---|---|---|---|---|---|---|---|---|
| Remodulin | Continuous subcutaneous | PAH | Commercial in the U.S., most of Europe*, Argentina, Brazil, Canada, Chile, China, Israel, Japan, Mexico, Peru, Saudi Arabia, South Korea, Taiwan and Venezuela | Worldwide | ||||
Remodulin |
Continuous intravenous |
PAH |
Commercial in the U.S., most of Europe*, Argentina, Canada, China, Israel, Japan, Mexico, Peru, Saudi Arabia, South Korea and Switzerland |
Worldwide |
||||
Tyvaso |
Inhaled |
PAH |
Commercial in the U.S. and Israel |
Worldwide |
||||
Adcirca |
Oral |
PAH |
Commercial in the U.S. |
United States |
||||
Orenitram |
Oral |
PAH |
Commercial in the U.S. |
Worldwide |
||||
Unituxin |
Intravenous |
High-risk neuroblastoma |
Commercial in the U.S.; also approved in Europe |
Worldwide |
||||
Remodulin Implantable System |
Continuous intravenous via implantable pump |
PAH |
Submitted to FDA for approval |
United States, United Kingdom, Canada, France, Germany, Italy and Japan |
4
Product
|
Mode of Delivery | Indication | Current Status | Our Territory | ||||
|---|---|---|---|---|---|---|---|---|
| Orenitram Combination Therapy | Oral | PAH | Phase III | Worldwide | ||||
Esuberaprost in Combination with Tyvaso |
Oral |
PAH |
Phase III |
North America, Europe, Mexico, South America, Egypt, India, South Africa and Australia |
||||
Ex-Vivo Lung Perfusion |
Pre-transplant service providing extended preservation and assessment of donor lungs |
End-stage lung disease |
Phase II/III |
United States |
||||
Tyvaso |
Inhaled |
Pulmonary hypertension associated with idiopathic pulmonary fibrosis |
Phase II |
Worldwide |
||||
Gene Therapy |
Intravenous |
PAH |
Phase I/II |
United States |
||||
Remodulin |
Subcutaneous via pre-filled, semi-disposable pump |
PAH |
Preclinical |
Worldwide |
- *
- We have obtained approval for subcutaneous and intravenous Remodulin in 24 member countries of the European Economic Area (EEA), as well as other non-EEA countries in Europe, and have received pricing approval in most of these countries.
Products to Treat Cardiopulmonary Diseases
Pulmonary Arterial Hypertension
PAH is a life-threatening disease that affects the blood vessels in the lungs and is characterized by increased pressure in the pulmonary arteries, which are the blood vessels leading from the heart to the lungs. The elevated pressure in the pulmonary arteries strains the right side of the heart as it pumps blood to the lungs. This eventually leads to right heart failure and, ultimately, death. PAH is characterized by structural changes in blood vessel walls, aggregation of platelets and alteration of smooth muscle cell function. We believe that PAH affects about 500,000 individuals worldwide. We have seen increases in the number of people diagnosed with the disease, but due to the rarity of the disease and the complexity of diagnosing it, only a small fraction of patients with PAH are being treated.
5
Currently, FDA-approved therapies for PAH focus on three distinct molecular pathways that have been implicated in the disease process: the prostacyclin pathway, the NO pathway, and the endothelin (ET) pathway. The classes of drugs that target these three pathways are:
- •
- Prostacyclin Analogues and IP Prostacyclin Receptor
Agonists. Patients with PAH have been shown to have reduced levels of prostacyclin, a naturally occurring substance that relaxes the
pulmonary blood vessels, prevents platelet aggregation and inhibits the proliferation of smooth muscle cells in the pulmonary vessels. Therefore, drugs that mimic the action of prostacyclin, known as
prostacyclin analogues, are established PAH treatments. Another class of therapy, called IP prostacyclin receptor agonists, has recently been developed recently to address PAH through the prostacyclin
pathway. As compared with prostacyclin analogues, which broadly mimic the effect of prostacyclin, IP prostacyclin receptor agonists bind selectively to the IP receptor, one of several prostacyclin
receptors.
- •
- PDE-5 Inhibitors and Guanylate Cyclase (sGC)
Stimulators. Patients with PAH have also been shown to have reduced levels of the enzyme responsible for producing NO, a naturally
occurring substance in the body that causes relaxation of the pulmonary blood vessels. NO produces this effect by increasing intracellular levels of cyclic GMP. Therefore, another established
therapeutic approach has been to inhibit the degradation of cyclic GMP using drugs known as PDE-5 inhibitors. In addition, sGC is an enzyme found in the endothelial cells and the receptor for
NO. When NO binds to sGC, the enzyme enhances production of cyclic GMP. As a result, sGC stimulators are also approved to treat PAH.
- •
- Endothelin Receptor Antagonists. PAH patients have also been shown to have elevated levels of endothelin-1, a naturally occurring substance in the body that causes constriction of, and structural changes to, the pulmonary blood vessels. Therefore, another established therapeutic approach has been to block the action of endothelin with drugs that are known as endothelin receptor antagonists (ETRAs).
Because any or all of the three pathways may be therapeutic targets in a patient, these classes of drugs are used alone or in combination to treat patients with PAH. We currently market drugs in two of these classes. Remodulin, Tyvaso and Orenitram are prostacyclin analogues, and Adcirca is a PDE-5 inhibitor.
Remodulin
One of our lead products for treating PAH is Remodulin, the active pharmaceutical ingredient of which is a prostacyclin analogue known as treprostinil. We sell Remodulin to specialty pharmaceutical distributors in the United States and to pharmaceutical distributors internationally. We recognized approximately $572.8 million, $553.7 million and $491.2 million in Remodulin net product sales, representing 39 percent, 43 percent and 44 percent of our total revenues for the years ended December 31, 2015, 2014 and 2013, respectively. The FDA approved Remodulin as a continuous subcutaneous infusion therapy in 2002, and as a continuous intravenous infusion therapy in 2004. Remodulin is indicated to treat patients with PAH (World Health Organization (WHO) Group 1), which includes multiple etiologies such as idiopathic and heritable PAH, as well as PAH associated with connective tissue diseases, to diminish symptoms associated with exercise. Studies establishing effectiveness included patients with New York Heart Association (NYHA) Functional Class II-IV (moderate to severe) symptoms. In 2006, the FDA expanded its approval to include transition of patients to Remodulin from Flolan®, the first FDA-approved prostacyclin therapy for PAH. In 2007, the results of a prospective, open-label study demonstrated that stable patients with PAH can be safely transitioned from Flolan to intravenous Remodulin using a rapid switch protocol.
Outside of the United States, Remodulin is approved for the treatment of PAH in 39 countries by continuous subcutaneous administration and in 35 countries by continuous intravenous administration.
6
Applications for approval of both subcutaneous and intravenous Remodulin are under review in other countries. We continue to work toward commercializing Remodulin in new territories.
We believe Remodulin has many qualities that make it an appealing alternative to competitive therapies. Remodulin is stable at room temperature, so it does not need to be cooled during infusion and patients do not need to use cooling packs or refrigeration to keep it stable. Treprostinil is highly soluble, which enables us to produce Remodulin in concentrated solutions. This allows therapeutic concentrations of Remodulin to be delivered at very low flow rates via miniaturized infusion pumps for both subcutaneous and intravenous infusion. Remodulin can be continuously infused for up to 48 hours intravenously or 72 hours subcutaneously before refilling the external infusion pump, and is packaged as an aqueous solution so patients do not have to reconstitute the drug before refilling their pumps.
In 2008, the FDA approved Teva Pharmaceuticals USA, Inc.'s (Teva) version of generic epoprostenol (the active ingredient in Flolan) for the treatment of PAH via intravenous delivery. Also in 2008, the FDA approved another intravenous but non-generic (temperature stable) version of epoprostenol, which is currently marketed by Actelion Pharmaceuticals Ltd (Actelion) under the name Veletri®. Actelion also markets Tracleer® and Opsumit®, both ETRAs, as well as an inhaled prostacyclin called Ventavis® and the IP prostacyclin receptor agonist known as Uptravi®. Flolan and generic epoprostenol are not stable at room temperature, but Veletri may be stable at room temperature depending on its concentration. Flolan, generic epoprostenol, and Veletri have shorter half-lives than Remodulin, require mixing and daily pump refills, and are not administered with miniaturized infusion pumps. None of these competitive products may be administered via subcutaneous infusion.
We settled patent litigation with two generic drug companies that filed abbreviated new drug applications (ANDAs) with the FDA to market a generic version of Remodulin in the United States. The first such settlement permits Sandoz Inc. (Sandoz) to launch its generic version of Remodulin in the United States in June 2018 (or earlier in certain circumstances). The second permits Teva to launch its generic version of Remodulin in the United States in December 2018 (or earlier in certain circumstances). For further detail, see the section below entitled Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity—Remodulin, Tyvaso and Orenitram Proprietary Rights—Generic Competition.
There are serious adverse events associated with Remodulin. When infused subcutaneously, Remodulin causes varying degrees of infusion site pain and reaction (redness and swelling) in most patients. Patients who cannot tolerate the infusion site pain related to the use of subcutaneous Remodulin may instead use intravenous Remodulin. Intravenous Remodulin is delivered continuously through a surgically implanted central venous catheter, similar to Flolan, Veletri and generic epoprostenol. Patients who receive therapy through implanted venous catheters have a risk of developing blood stream infections and a serious systemic infection known as sepsis. Other common side effects associated with both subcutaneous and intravenous Remodulin include headache, diarrhea, nausea, jaw pain, vasodilation and edema.
International Regulatory Review of Subcutaneous and Intravenous Remodulin
Remodulin is approved in 39 countries outside the United States. In 35 of these countries, it is approved for both subcutaneous and intravenous use. In the other four countries, Remodulin is approved for subcutaneous use only.
We used the mutual recognition process, described more fully below in Governmental Regulation—Marketing Pharmaceutical Products Outside the United States, to obtain approval of subcutaneous Remodulin in most countries in the European Union (EU) in 2005. Our reference member state for the mutual recognition process was the French regulatory agency, L'Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM). In 2011, we received regulatory approval for intravenous
7
Remodulin by ANSM, which allows us to market intravenous Remodulin in the EEA countries where subcutaneous Remodulin has already been approved and where we have obtained pricing approval and approval of our risk management plan (RMP).
In Europe, an RMP is routinely required as part of the regulatory approval process for new medicines and also for significant variations involving a change to the route of administration, formulation or indication. For intravenous Remodulin, we have implemented an RMP focused on minimizing the known risks of central venous catheter-related blood stream infections associated with intravenous administration. To date, our RMP for intravenous Remodulin has been approved in 22 EEA countries, with pricing approval in 16 of these.
In March 2013, the China Food and Drug Administration approved intravenous and subcutaneous Remodulin for PAH in the People's Republic of China. In March 2014, Japan's Ministry of Health, Labor and Welfare approved Remodulin for the treatment of PAH by subcutaneous and intravenous administration. Remodulin is sold in Japan under the brand name Treprost®. In the second and third quarters of 2014, we commenced sales of Remodulin to our distributors in China and Japan, respectively.
Remodulin Implantable System
In 2009, we entered into an agreement with Medtronic, Inc. (Medtronic) providing us exclusive rights in the United States, the United Kingdom, Canada, France, Germany, Italy and Japan to develop Medtronic's proprietary intravascular infusion catheter to be used with its SynchroMed® II implantable infusion pump and related infusion system components (together referred to as the Remodulin Implantable System) in order to deliver Remodulin to treat PAH. If the Remodulin Implantable System is successful, it could reduce many of the patient burdens and other complications associated with the use of external pumps to administer prostacyclin analogues. With our funding, Medtronic completed the DelIVery clinical trial, which studied the safety of the Remodulin Implantable System while administering Remodulin. The primary objective was to demonstrate a rate of catheter-related complications below 2.5 per 1,000 patient-days while using the Remodulin Implantable System to deliver Remodulin. In September 2013, Medtronic informed us that this primary objective was met (p<0.0001).
In order to launch the Remodulin Implantable System in the United States, Medtronic and we are pursuing parallel regulatory filings relating to the device and the drug, respectively. In December 2014, Medtronic submitted a premarket approval application (PMA) seeking FDA approval for the catheter and labeling changes for the SynchroMed II pump. Medtronic is entirely responsible for responding to any FDA requests for additional information concerning the use of the Remodulin Implantable System with Remodulin. In March 2015, the FDA requested that Medtronic amend its PMA to reflect an amendment to the SynchroMed II PMA separately submitted by Medtronic's neuromodulation business unit. Medtronic submitted an amendment to its PMA, which was accepted for review by FDA in January 2016, with FDA action expected in 2016.
In January 2015, we submitted a supplemental NDA with new labeling requesting FDA approval to allow the use of Remodulin with the Remodulin Implantable System. The FDA issued a refuse-to-file letter in March 2015, which meant that we would need to address FDA comments and resubmit our filing. The FDA also indicated that our submission would be treated as a new NDA. We resubmitted our filing as a new NDA in December 2015, and we expect a ten-month review period (October 2016).
In April 2015, the FDA filed a consent decree requiring Medtronic to stop manufacturing, designing and distributing SynchroMed II implantable infusion pump systems, except in limited circumstances, citing violations of the quality system regulation for medical devices. The consent decree will remain in effect until the FDA has determined that Medtronic has met all the provisions listed in the consent decree. It is unclear how this consent decree will impact our program to develop and
8
commercialize the Remodulin Implantable System, and we anticipate further insight into the potential impact on our program in 2016 when the FDA responds to Medtronic's PMA filing.
Subcutaneous Remodulin Administered via Pre-Filled, Semi-Disposable Pump
In December 2014, we entered into an exclusive agreement with DEKA Research & Development Corp. (DEKA) to develop a pre-filled, semi-disposable pump system for subcutaneous delivery of Remodulin. Under the terms of the agreement, we are funding all of the development costs related to the semi-disposable pump system and will pay product fees and a single-digit royalty to DEKA based on commercial sales of the system and the Remodulin sold for use with the system. Currently, we are undertaking engineering, design and development work to optimize the DEKA pump to deliver Remodulin in pre-filled reservoirs, and intend to conduct human factor studies in healthy volunteers before submitting an application to the FDA to approve the pre-filled DEKA pump. We do not anticipate that the FDA will require us to conduct clinical trials in patients. Our goal is to be in a position to receive FDA approval for this delivery system by the end of 2018.
Tyvaso
We commenced commercial sales of Tyvaso in the United States in 2009. We sell Tyvaso to the same specialty pharmaceutical distributors in the United States that distribute Remodulin. For the years ended December 31, 2015, 2014 and 2013, we recognized approximately $470.1 million, $463.1 million and $438.8 million in Tyvaso net product sales, representing 32 percent, 36 percent and 39 percent, respectively, of our total revenues.
Tyvaso, which contains the active ingredient treprostinil, is administered four times a day by inhaling up to nine breaths during each treatment session, which takes approximately three minutes. Tyvaso is required to be administered using our proprietary Tyvaso Inhalation System, which consists of an ultra-sonic nebulizer that provides a dose of Tyvaso on a breath-by-breath basis. A single ampule containing Tyvaso is emptied into the Tyvaso Inhalation System once per day, so the Tyvaso Inhalation System only needs to be cleaned once daily.
Tyvaso was generally well tolerated in our trials. The most common adverse events were transient cough, headache, nausea, dizziness and flushing. We completed an open-label study in the United States to investigate the clinical effects of switching patients from Ventavis to Tyvaso. Patients in this study saved an average of approximately 1.4 hours per day when administering Tyvaso compared to Ventavis.
Ventavis is the only other FDA-approved inhaled prostacyclin analogue and is marketed by Actelion in the United States and by Bayer Schering Pharma AG (Bayer) in Europe. The active ingredient in Ventavis is iloprost. Patients need to inhale Ventavis six to nine times per day via a nebulizer. According to its package insert, each Ventavis inhalation consists of four to ten minutes of continuous inhalation via the nebulizer. Ventavis can cause a decrease in systemic (body-wide) blood pressure if the drug is administered at too high a dose.
We are developing further enhancements intended to make the Tyvaso Inhalation System easier to use. In addition, we are studying Tyvaso in combination with esuberaprost, as discussed below. Finally, we are currently planning a phase II study of Tyvaso in patients with pulmonary hypertension associated with idiopathic pulmonary fibrosis.
Regulatory Approval of Tyvaso
In 2009, the FDA approved Tyvaso for the treatment of PAH in WHO Group 1 patients to improve exercise capacity using the Tyvaso Inhalation System. Studies establishing effectiveness included predominately patients with NYHA Functional Class III symptoms. Tyvaso is approved in Israel, where we commenced commercial sales during the second quarter of 2015.
9
In June 2010, the FDA granted orphan drug designation for Tyvaso. Such a designation, coupled with FDA approval of the product for the orphan indication, confers an exclusivity period through July 2016, during which the FDA may not approve any application to market the same drug for the same indication, except in limited circumstances.
In April 2004, the European Medicines Agency (EMA) designated Tyvaso an orphan medicinal product for the treatment of both PAH and chronic thromboembolic pulmonary hypertension. EMA orphan drug designation confers a ten-year exclusivity period commencing with marketing approval. We filed a Marketing Authorization Application (MAA) in December 2008 for Tyvaso with the EMA using the centralized filing process, but withdrew our MAA from consideration by the EMA due to the EMA's major objection related to findings of non-compliance with good clinical practices at two clinical sites. The EMA stated that these findings would preclude a recommendation for approval of Tyvaso in the EU, although the EMA had no major objections at the time of withdrawal related to the safety or efficacy of Tyvaso. At present, we are evaluating the resubmission of Tyvaso for EMA approval as a standalone treatment of PAH, based on experience with the drug since approval by the FDA.
Orenitram
Orenitram is an extended-release, oral tablet form of treprostinil, which we launched commercially in the United States during the second quarter of 2014. Orenitram is the only FDA-approved, orally administered prostacyclin analogue. We sell Orenitram to the same specialty pharmaceutical distributors in the United States that distribute Remodulin and Tyvaso. For the years ended December 31, 2015 and 2014, we recognized approximately $118.4 million and $41.3 million in Orenitram net product sales, representing eight percent and three percent, respectively, of our total revenues.
Regulatory Approval of Orenitram
In December 2013, the FDA approved Orenitram for the treatment of PAH in WHO Group 1 patients to improve exercise capacity. The primary study that established efficacy (FREEDOM-M) included predominately patients with WHO functional class II-III symptoms and etiologies of idiopathic or heritable PAH (75%) or PAH associated with connective tissue disease (19%). The most common side effects observed were headache, nausea and diarrhea. FREEDOM-M was a 12-week monotherapy study of Orenitram (meaning patients were not on any background PAH therapy), which met its primary endpoint of improvement in six-minute walk distance at week 12. Analysis of the FREEDOM-M results demonstrated that patients receiving Orenitram improved their six-minute walk distance by a median of approximately 23 meters (p=0.0125, Hodges-Lehmann estimate and non-parametric analysis of covariance in accordance with the trial's pre-specified statistical analysis plan) as compared to patients receiving the placebo. In January 2016, the FDA approved amended labeling describing the transition of patients from intravenous or subcutaneous Remodulin to Orenitram.
Orenitram Combination Therapy
In addition to the successful monotherapy study noted above, we also conducted two unsuccessful phase III studies of Orenitram in combination with other approved therapies. We believe that in order for Orenitram to reach its full commercial potential, we need to complete further studies to support an amendment to Orenitram's label to indicate that Orenitram delays morbidity and mortality (also known as "time to clinical worsening") in patients who are on an approved oral background therapy. As such, we are enrolling up to 610 patients in a phase IV clinical trial called FREEDOM-EV, which began in 2012. FREEDOM-EV is a placebo-controlled study of patients who enter the study on an approved background therapy, and one of the two primary endpoints of the study is the time to clinical worsening. The other primary endpoint is change in six-minute walk distance from baseline to week 24.
10
We expect to seek approval of Orenitram in Europe upon completion of the FREEDOM-EV study. In 2005, the EMA announced that Orenitram had been designated an orphan medicinal product for the treatment of PAH. A request for orphan drug designation for Orenitram for PAH is pending before the FDA.
Adcirca
We began selling Adcirca in 2009. Adcirca is a PDE-5 inhibitor, the active pharmaceutical ingredient of which is tadalafil. Tadalafil is also the active pharmaceutical ingredient in Cialis®, which is marketed by Lilly for the treatment of erectile dysfunction. We acquired the commercial rights to Adcirca for the treatment of PAH in the United States from Lilly in 2008. We sell Adcirca at prices established by Lilly, which are at parity with Cialis pricing and are typically set at a discount from an average wholesale price to pharmaceutical wholesalers. For the years ended December 31, 2015, 2014 and 2013, we recognized approximately $278.8 million, $221.5 million and $177.0 million in Adcirca net product sales, representing 19 percent, 17 percent and 16 percent, respectively, of our total revenues.
Patients with PAH have been shown to have reduced levels of the enzyme responsible for producing NO, a naturally occurring substance in the body that has the effect of relaxing vascular smooth muscle cells. NO relaxes pulmonary blood vessels by increasing intracellular levels of cyclic GMP. Because cyclic GMP is degraded by PDE-5, an established therapeutic approach in the treatment of PAH is to use PDE-5 inhibitors to increase levels of cyclic GMP in blood vessels and improve cardiopulmonary function in PAH patients.
In September 2014, Gilead announced the results of a study of ambrisentan (an ETRA) and tadalafil in PAH patients as a first-line combination treatment, compared to treating PAH patients with only ambrisentan or tadalafil. In the study, first-line treatment with both therapies reduced the risk of clinical failure compared to a monotherapy treatment by 50 percent (p=0.0002). Based on these results, in October 2015, the FDA approved an update to the NDA for Letairis® (ambrisentan), permitting the use of Letairis in combination with tadalafil for PAH to reduce the risks of disease progression and hospitalization for worsening PAH, and to improve exercise ability.
Prior to the approval of Adcirca, Revatio®, which is marketed by Pfizer Inc. (Pfizer), was the only PDE-5 inhibitor approved for the treatment of PAH. Sildenafil citrate, the active ingredient in Revatio, is also the active ingredient in Viagra®, which is marketed by Pfizer for the treatment of erectile dysfunction. In 2012, several companies launched generic formulations of sildenafil citrate. Revatio and generic sildenafil citrate are dosed three times daily. Adcirca is dosed once daily.
FDA Approval of Adcirca
In 2009, the FDA approved Adcirca with a recommended dose of 40 mg, making it the only once-daily PDE-5 inhibitor for the treatment of PAH. Adcirca is indicated to improve exercise ability in patients with PAH (WHO Group 1), which encompasses patients with various etiologies, such as idiopathic and heritable PAH as well as PAH associated with connective tissue diseases. Studies establishing effectiveness included predominately patients with NYHA Functional Class II-III symptoms. Headaches were the most commonly reported side effect.
Commercial Rights to Adcirca
In 2008, we entered into several agreements with Lilly, including a license agreement and a manufacturing and supply agreement. Pursuant to the license agreement, Lilly granted us an exclusive license to develop, market, promote and commercialize Adcirca for the treatment of pulmonary hypertension in the United States. Pursuant to the manufacturing and supply agreement, Lilly manufactures Adcirca and distributes it on our behalf via its wholesaler network, in the same manner that it distributes its own pharmaceutical products. See Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity below for more details on these agreements.
11
Esuberaprost
We have the exclusive right to develop and market a modified-release formulation of beraprost in North America, Europe, and certain other territories for the treatment of cardiovascular indications, pursuant to our license agreement with Toray Industries, Inc. (Toray), which is described below under Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity—Toray Amended License Agreement. Beraprost is a chemically stable, orally bioavailable prostacyclin analogue. Like natural prostacyclin and treprostinil, beraprost is believed to dilate blood vessels and prevent both platelet aggregation and proliferation of smooth muscle cells surrounding blood vessels, via a unique profile of pulmonary vascular receptor selectivity.
In July 2012, we completed a phase I safety trial of esuberaprost, a reformulated, single-isomer version of beraprost, and the data suggested that dosing esuberaprost four times a day was safe. We believe that esuberaprost and treprostinil have differing prostacyclin receptor-binding profiles and thus could provide benefits to certain groups of patients with differing sets of safety and efficacy profiles. We also believe that inhaled treprostinil and esuberaprost have complimentary pharmacokinetic and pharmacodynamic profiles, which indicate that they should provide greater efficacy in combination. As a result, in 2013 we began enrolling a phase III study called BEAT (BEraprost 314d Add-on to Tyvaso) to evaluate the clinical benefit and safety of esuberaprost in combination with Tyvaso for patients with PAH who show signs of deterioration on inhaled treprostinil or have a less than optimal response to inhaled treprostinil treatment. We intend to enroll 240 patients in the study, which will have a primary endpoint of time to clinical worsening.
Products to Treat Cancer
Unituxin
In March 2015, the FDA approved our Biologics License Application (BLA) for Unituxin, in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma who achieve at least a partial response to prior first-line multiagent, multimodality therapy. We commenced U.S. sales of Unituxin in the third quarter of 2015. We received European Commission approval during the third quarter of 2015, and plan to commence commercial sales in individual European countries following pricing and reimbursement approvals on a country-by-country basis. For the year ended December 31, 2015, we recognized approximately $20.4 million in Unituxin net product sales, representing one percent of our net revenues.
We previously received orphan drug designation for Unituxin from both the FDA and the EMA. Orphan designation, coupled with FDA approval of our BLA for Unituxin, confers an exclusivity period through March 2022, during which the FDA may not approve any application to market the same drug for the same indication, except in limited circumstances. See Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity—Unituxin Proprietary Rights below for more details.
Under our BLA approval for Unituxin, the FDA has imposed certain post-marketing requirements and post-marketing commitments on us. We are conducting additional clinical and non-clinical studies to satisfy these requirements and commitments. While we believe we will be able to complete these studies, any failure to satisfy these requirements or commitments could result in penalties, including fines or withdrawal of Unituxin from the market, unless we are able to demonstrate good cause for the failure.
Organ Transplantation Research and Development
We are engaged in a number of research and development activities in xenotransplantation, regenerative medicine and ex-vivo lung perfusion, all of which are intended to increase the supply of
12
transplantable organs and tissues. These activities are principally focused on lungs, but are also being applied to other organs such as hearts and kidneys. For additional detail, see Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations—Major Research and Development Projects—Organ Transplantation.
Sales and Marketing
Our marketing strategy for our commercial products is to use our sales and marketing teams to reach out to the prescriber community to: (1) increase PAH awareness; (2) increase understanding of the progressive nature of PAH; and (3) increase awareness of our commercial products and how they fit into the various stages of disease progression and treatment. Our sales and marketing teams consisted of approximately 160 employees as of December 31, 2015. We have divided our domestic PAH sales force into two teams. One team sells Remodulin and Orenitram, while the other team sells Tyvaso and Adcirca. We have a separate team that is responsible for marketing Unituxin.
Distribution of Commercial Products
United States Distribution of Remodulin, Tyvaso, Orenitram, and Unituxin
We distribute Remodulin, Tyvaso and Orenitram throughout the United States through two contracted specialty pharmaceutical distributors: Accredo Health Group, Inc. (Accredo) and CVS Caremark (Caremark). These distributors are required to maintain certain minimum inventory levels in order to ensure an uninterrupted supply to patients who are prescribed our therapies. We compensate Accredo and Caremark on a fee-for-service basis for certain ancillary services in connection with the distribution of these products. If any of our distribution agreements expire or terminate, we may, under certain circumstances, be required to repurchase any unsold Remodulin, Tyvaso or Orenitram inventory held by our distributors.
These specialty pharmaceutical distributors are responsible for assisting patients with obtaining reimbursement for the cost of our treprostinil-based products and providing other support services. Under our distribution agreements, we sell each of our treprostinil-based products to these distributors at a transfer price that we establish. We have generally increased the price of Tyvaso by 4.9 percent annually, with the last such price increase becoming effective on January 1, 2015. We have not increased the price of Remodulin since 2010. We have also established patient assistance programs in the United States, which provide our treprostinil-based products to eligible uninsured or under-insured patients at no charge. Accredo and Caremark assist us with the administration of these programs.
In the second quarter of 2015, we entered into an exclusive distribution agreement with ASD Specialty Healthcare, Inc. (ASD), an affiliate of AmerisourceBergen Corporation, to distribute Unituxin in the United States. Under this agreement, we sell Unituxin to ASD at a transfer price that we establish, and we pay ASD fees for services provided in connection with the distribution and support of Unituxin.
United States Distribution of Adcirca
We sell Adcirca to pharmaceutical wholesalers at a discount from an average wholesale price. Under our manufacturing and supply agreement with Lilly (see Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity below for more details), Lilly manufactures Adcirca and distributes it via its wholesaler network, which includes Accredo and Caremark, in the same manner that it distributes its own pharmaceutical products. Under the terms of this agreement, we take title to Adcirca upon completion of its manufacture by Lilly. Adcirca is shipped to customers in accordance with purchase orders received by Lilly. When customers take delivery of Adcirca, Lilly sends an invoice and collects the amount due from the customer subject to customary discounts and rebates, if any. Although Lilly provides these services on our behalf, we maintain the risk of loss as it pertains to
13
inventory, product returns and non-payment of invoices. The manufacturing and supply agreement will continue in effect until expiration or termination of the license agreement. Lilly retains authority under the license agreement for all regulatory activities with respect to Adcirca, as well as its retail pricing, which has been and is expected to remain at price parity with Cialis. Since receiving FDA approval of Adcirca, Lilly has generally increased the net wholesale price of Adcirca two or three times each year. During 2014, Lilly increased the net wholesale price of Adcirca by 9.1 percent in July and by 9.9 percent in December. During 2015, Lilly increased the net wholesale price of Adcirca by 9.9 percent in May and by 9.9 percent in December. We have also established a patient assistance program in the United States, which provides Adcirca to eligible uninsured or under-insured patients at no charge.
International Distribution of Remodulin
We currently sell subcutaneous and intravenous Remodulin outside the United States to various distributors, each of which has exclusive distribution rights in one or more countries within Europe, Israel and the Middle East, Asia and South and Central America. We also distribute Remodulin in Canada through a specialty pharmaceutical wholesaler. In some of the European markets where we are not licensed to market Remodulin, such as in Spain and the United Kingdom, we sell (but do not market) Remodulin on a named-patient basis in which therapies are approved for individual patients by a national medical review board, hospital or health plan on a case-by-case basis. We continue to work on expanding our sales of Remodulin into new territories through our existing network of distributors.
Patents and Other Proprietary Rights, Strategic Licenses and Market Exclusivity
Our success depends in part on our ability to obtain and maintain patent protection for our products, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others in the United States and worldwide. Many of these proprietary rights stem from licenses and other strategic relationships with third parties. In addition to intellectual property rights, U.S. and international regulatory authorities often provide periods of market exclusivity for manufacturers of biopharmaceutical products.
Patents provide the owner with a right to exclude others from practicing an invention. Patents may cover the active ingredients, uses, formulations, doses, administrations, delivery mechanisms, manufacturing processes and other aspects of a product. The period of patent protection for any given product generally depends on the expiration date of various patents and may differ from country to country according to the type of patents, the scope of coverage and the remedies for infringement available in a country. Most of our commercial products and investigational products are protected by patents that expire on varying dates.
Significant legal questions exist concerning the extent and scope of patent protection for biopharmaceutical products and processes in the United States and elsewhere. Accordingly, there is no certainty that patent applications owned or licensed by us will be issued as patents, or that our issued patents will afford meaningful protection against competitors. Once issued, patents are subject to challenge through both administrative and judicial proceedings in the United States and other countries. Such proceedings include re-examinations, inter partes reviews, post-grant reviews and interference proceedings before the U.S. Patent and Trademark Office, as well as opposition proceedings before the European Patent Office. Litigation may be required to enforce, defend or obtain our patent and other intellectual property rights. Any administrative proceeding or litigation could require a significant commitment of our resources and, depending on outcome, could adversely affect the scope, validity or enforceability of certain of our patent or other proprietary rights.
14
Remodulin, Tyvaso and Orenitram Proprietary Rights
We have a number of issued patents and pending patent applications covering the stable prostacyclin analogue known as treprostinil, which is the active pharmaceutical ingredient in Remodulin, Tyvaso and Orenitram.
In January 1997, we acquired patents covering the use of treprostinil for PAH from GlaxoSmithKline PLC (formerly Glaxo Wellcome, Inc.) (Glaxo) in exchange for certain payments including a royalty on sales of any product containing treprostinil. All of these patents expired in October 2014, as did our royalty payment obligation to Glaxo.
In October 1997, we filed patent applications for a new synthesis method for treprostinil in the United States, Europe and various other countries. These applications resulted in the grant of three patents in the United States, all of which expire in October 2017, as well as patents granted in a number of other countries which expire in October 2018.
We continue to conduct research into new methods to synthesize treprostinil and have filed a number of additional patent applications relating to production of treprostinil, several of which have already been granted in the United States. One such patent was granted, expiring in 2028, and is listed in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book (see Orange Book below), for Remodulin, Tyvaso and Orenitram.
In addition to the treprostinil patents noted above, we have other patents specific to our individual treprostinil-based products, including the following:
- •
- Remodulin. We have been granted three U.S. patents
covering an improved diluent for Remodulin, which expire in 2028 and 2029. We have another patent covering intravenous administration of Remodulin with certain diluents, which expires in 2024. All
four of these patents are listed in the Orange Book.
- •
- Tyvaso. We have been granted two U.S. patents, as well as
patents in other countries, for Tyvaso that cover methods of treating PAH by inhaled delivery. These patents will expire in the United States in 2018 and in various countries throughout the world in
2020.
- •
- Orenitram. Our patents for Orenitram cover methods of use for treating PAH, orally administered formulations, controlled moisture storage and production methods, as well as those covering controlled release formulations licensed to us by Supernus Pharmaceuticals Inc. (Supernus). These patents will expire in the United States between 2024 and 2031 and in various countries throughout the world between 2024 and 2027.
We have additional pending U.S. and international patent applications relating to Remodulin, Tyvaso and Orenitram.
Orange Book
In seeking approval of a drug through an NDA or BLA or upon issuance of new patents following approval of an NDA or BLA, applicants are required to submit to the FDA each patent that has claims covering the applicant's product or a method of using the product. Each of the patents submitted is then published in the Orange Book. See Governmental Regulation—Hatch-Waxman Act below for further details. Remodulin currently has six unexpired Orange Book-listed patents with expiration dates ranging from 2017 to 2029. Tyvaso currently has four unexpired Orange Book listed patents with expiration dates ranging from 2017 to 2028. Orenitram currently has nine unexpired Orange Book listed patents with expiration dates ranging from 2017 to 2031. Additional patent applications are pending, and if granted, may be eligible for listing in the Orange Book.
15
Regulatory Exclusivity
In June 2010, the FDA granted orphan drug designation for Tyvaso. This designation confers an exclusivity period through July 2016, during which the FDA may not approve any application to market the same drug for the same indication, except under limited circumstances. As a result of FDA approval of our NDA for Orenitram as a new dosage form, Orenitram has three years of market exclusivity for PAH expiring in December 2016. A request for orphan drug designation for Orenitram is pending with the FDA.
Remodulin was formerly protected in the EU by data protection regulations, which prevent the grant of an abbreviated marketing approval for a product containing treprostinil for the treatment of PAH for a period of either six or ten years from the date of the grant of the first marketing authorization in the EU. In those countries where protection runs for six years, that period expired prior to 2015, while in those countries where protection runs for ten years, that period expired in February 2015.
Generic Competition
In September 2015, we settled litigation with Sandoz relating to Sandoz's ANDA seeking FDA approval to market a generic version of Remodulin before the expiration of certain of our U.S. patents in October 2017, September 2028, December 2028, and March 2029. Under the terms of this settlement, Sandoz will be permitted to market its generic version of Remodulin in the United States beginning in June 2018, although Sandoz may be permitted to enter the market earlier under certain circumstances. We also recently settled litigation with Teva relating to a similar ANDA to market a generic version of Remodulin. That settlement permits Teva to market its generic version of Remodulin in the United States beginning in December 2018, although Teva may be permitted to enter the market earlier under certain circumstances. We are engaged in litigation against Watson Laboratories, Inc. (Watson), contesting its ANDA to market a generic version of Tyvaso before the expiration of certain of our U.S. patents in November 2018 and December 2028. Finally, in October 2015 SteadyMed Ltd. (SteadyMed) filed a petition for inter partes review seeking to invalidate one of our patents that expires in December 2028 and covers a method of making treprostinil, which is the active ingredient in Remodulin, Tyvaso and Orenitram. SteadyMed has announced that it is developing a product called Trevyent™, which is a single-use, pre-filled pump intended to deliver a two-day supply of treprostinil subcutaneously using SteadyMed's PatchPump® technology. In January 2016, SteadyMed announced that Trevyent has been granted orphan drug designation by the FDA for the treatment of PAH. These matters are discussed further in Item 3—Legal Proceedings of this Annual Report on Form 10-K, and in Note 19—Litigation, to our consolidated financial statements included with this Annual Report on Form 10-K. In February 2016, we received notice that Actavis Laboratories FL, Inc. (Actavis) filed an ANDA seeking FDA approval to market a generic version of the 2.5 mg strength of Orenitram. For further details, please see Part II, Item 9B—Other Information.
As a result of our settlements with Sandoz and Teva, we expect to see generic competition from these companies for Remodulin beginning in June 2018 and December 2018, respectively. This increased competition could reduce our sales and profits. In addition, while we intend to vigorously enforce our intellectual property rights relating to Tyvaso and Orenitram, we may not prevail in defending our patent rights, and additional challenges from other ANDA filers or other challengers may surface with respect to these products. Our patents could be invalidated, found unenforceable or found not to cover one or more generic forms of Remodulin, Tyvaso or Orenitram. If any ANDA filer receives approval to sell a generic version of Remodulin, Tyvaso or Orenitram and/or prevail in any patent litigation, the affected product(s) would become subject to increased competition, which could reduce our sales and profits.
16
Certain patents for Revatio, a PDE-5 inhibitor marketed by Pfizer for treatment of PAH, expired in 2012, leading several manufacturers to launch generic formulations of sildenafil citrate, the active ingredient in Revatio. Generic sildenafil's lower price relative to Adcirca could lead to pressure from payers to use generic products within the same class of therapy initially, which could erode Adcirca's market share and limit its potential sales. Although we believe Adcirca's once-daily dosing regimen provides a significant competitive advantage over generic sildenafil's multiple dosing regimen, government payers and private insurance companies may favor the use of less expensive generic sildenafil over Adcirca. Thus far, we have not observed any measurable impact of generic sildenafil on sales of Adcirca; however, circumstances could change over time and our revenues could be adversely impacted. The U.S. patent for Adcirca for the treatment of pulmonary hypertension will expire in November 2017, following which we expect to see generic competition for Adcirca.
Patent expiration and generic competition for any of our commercial PAH products could have a significant, adverse impact on our revenues and profits, and is inherently difficult to predict. For additional discussion, refer to the risk factor entitled, Our intellectual property rights may not effectively deter competitors from developing competing products that, if successful, could have a material adverse effect on our revenues and profits, contained in Item 1A—Risk Factors included in this Annual Report on Form 10-K.
Supernus License
In 2006, we entered into an exclusive license agreement with Supernus to use certain of its technologies in producing Orenitram. Under the agreement, we paid Supernus certain amounts upon the achievement of specified milestones based on the development and commercial launch of Orenitram for PAH, and we would be obligated to make additional milestone payments if we develop Orenitram for a second indication. In addition, the agreement provides that we will pay a single-digit royalty based on net worldwide sales. This royalty will be paid for approximately twelve years commencing with the first product sale and is subject to adjustments as specified in the agreement. The royalties commenced in the second quarter of 2014 with the first sale of Orenitram.
Lilly Agreements Related to Adcirca
In 2008, we entered into several agreements with Lilly regarding Adcirca, including a license agreement and a manufacturing and supply agreement.
License Agreement
Under the terms of the license agreement, Lilly granted us an exclusive license for the right to develop, market, promote and commercialize Adcirca for the treatment of pulmonary hypertension in the United States. We agreed to pay Lilly royalties equal to five percent of our net product sales of Adcirca, as a pass through of Lilly's third-party royalty obligations, for so long as Lilly is required to make such payments.
Lilly retained the exclusive rights to develop, manufacture and commercialize pharmaceutical products containing tadalafil, the active pharmaceutical ingredient in Adcirca, for the treatment of pulmonary hypertension outside of the United States and for the treatment of other diseases worldwide. Lilly retained authority for all regulatory activities with respect to Adcirca and for setting the wholesale price of Adcirca, which has been and is expected to continue to be at price parity with Cialis.
The license agreement will continue in effect until the later of: (1) expiration, lapse, cancellation, abandonment or invalidation of the last claim to expire within a Lilly patent covering the commercialization of Adcirca for the treatment of pulmonary hypertension in the United States; or
17
(2) expiration of any government-conferred exclusivity rights to use Adcirca for the treatment of pulmonary hypertension in the United States.
We have the right to terminate the license agreement upon six months written notice to Lilly. Lilly has the right to terminate in the event of a change of control of our company. Either party may terminate upon a material breach by the other party of the license agreement or the manufacturing and supply agreement, described above.
The U.S. patent for Adcirca for the treatment of pulmonary hypertension will expire in November 2017.
Manufacturing and Supply Agreement
Under the terms of the manufacturing and supply agreement, Lilly agreed to manufacture Adcirca and distribute it on our behalf via its pharmaceutical wholesaler network, in the same manner that it distributes its own pharmaceutical products. Under the terms of this agreement, we take title to Adcirca upon its manufacture by Lilly. Adcirca is shipped to customers, generally pharmaceutical wholesalers, in accordance with customers' purchase orders received by Lilly. Lilly invoices and collects amounts due from the customer subject to customary discounts and rebates, if any, and remits the collections to us. Although Lilly is providing these services on our behalf, we maintain the risk of loss as it pertains to inventory, product returns and nonpayment of sales invoices. The manufacturing and supply agreement will continue in effect until expiration or termination of the license agreement.
We also agreed to purchase Adcirca at a fixed manufacturing cost. The agreement provides a mechanism, generally related to the increase in the national cost of pharmaceutical manufacturing, pursuant to which Lilly may raise the manufacturing cost of Adcirca.
Unituxin Proprietary Rights
In 2010, we entered into a Cooperative Research and Development Agreement (CRADA) with the National Cancer Institute (NCI) of the United States National Institutes for Health (NIH) to collaborate on the late-stage development and regulatory approval process for Unituxin for children with high-risk neuroblastoma and patients with other forms of cancer. In lieu of a royalty payment to the NCI, we have an ongoing obligation to provide the NCI with Unituxin for its studies free of charge. We previously received orphan drug designation for Unituxin from both the FDA and the EMA. Orphan designation, coupled with FDA approval of our BLA in March 2015, confers an exclusivity period through March 2022, during which the FDA may not approve any application to market the same drug for the same indication, except in limited circumstances. For further details, refer to the section above entitled Products to Treat Cancer—Unituxin.
Medtronic Agreement
In 2009, we entered into an exclusive agreement with Medtronic, which was amended in 2011, to collaborate on the development and commercialization of Medtronic's proprietary intravascular infusion catheter to be used with Medtronic's Synchromed II implantable infusion pump and related infusion system components (together referred to as the Remodulin Implantable System) in order to deliver Remodulin for the treatment of PAH in the United States, United Kingdom, Canada, France, Germany, Italy and Japan. Under the amended agreement, we have been working together at our expense to develop the Remodulin Implantable System, conduct a clinical trial (which was completed in 2013) and obtain regulatory approval for the use of Remodulin with the Remodulin Implantable System. If this development program is successful, our agreement provides that, upon commercialization, we will purchase infusion pumps and supplies from Medtronic and will also pay a royalty to Medtronic based on net product sales of Remodulin for use in the Remodulin Implantable System within the exclusive territories, subject to certain adjustments specified in the agreement. The
18
Remodulin Implantable System will be exclusive to Remodulin so long as we purchase a minimum percentage of our annual requirement for implantable pump systems from Medtronic. We will be solely responsible for all marketing and promotion of the Remodulin Implantable System for the delivery of Remodulin for the treatment of PAH in the exclusive territories. For further details, refer to the section above entitled Products to Treat Cardiopulmonary Diseases—Pulmonary Arterial Hypertension—Remodulin—Remodulin Implantable System.
Toray Amended License Agreement
In 2000, we licensed from Toray the exclusive right to develop and market beraprost for cardiovascular indications. Beraprost is a chemically stable oral prostacyclin analogue in a sustained release formulation, which is approved to treat PAH in Japan and certain other countries. This license gives us exclusive rights to develop beraprost and its variants (including esuberaprost) throughout North America, Europe, and certain other territories. We are currently developing esuberaprost under this license agreement.
Pursuant to a March 2007 amendment to our license agreement with Toray, we issued 200,000 shares of our common stock to Toray. Toray has the right to request that we repurchase these shares (which have since split into 400,000 shares) upon 30 days prior written notice at the price of $27.21 per share. The 2007 amendment also provided for certain milestone payments during the development period and upon receipt of regulatory approval for beraprost in the United States or the EU.
In 2011, we amended our license agreement with Toray to reduce the royalty rates in exchange for a total of $50.0 million in equal, non-refundable payments to Toray over the five-year period ending in 2015. As of December 31, 2015, this obligation was fully satisfied. Toray has the right to terminate the license agreement in the event of a change of control of our company under certain circumstances. For further details, refer to the section above entitled Products to Treat Cardiopulmonary Diseases—Pulmonary Arterial Hypertension—Esuberaprost.
DEKA Agreement
In December 2014, we entered into an exclusive agreement with DEKA to develop a pre-filled, semi-disposable pump system for subcutaneous delivery of Remodulin. Under the terms of the agreement, we are funding the development costs related to the semi-disposable pump system and will pay product fees and a single-digit royalty to DEKA based on commercial sales of the system and the Remodulin sold for use with the system. Our goal is to be in a position to receive FDA approval for this delivery system by the end of 2018.
Other
We are party to various other license agreements relating to therapies and technologies under development. These license agreements require us to make payments based on a percentage of sales if we are successful in commercially developing these therapies, and may require other payments upon the achievement of certain milestones.
Research & Development Expenditures
We are engaged in research and development and have incurred substantial expenses for these activities. These expenses generally include the cost of acquiring or inventing new technologies and products, as well as new product development (both preclinical and clinical studies and manufacturing cost for unapproved products). Research and development expenses during the years ended December 31, 2015, 2014 and 2013 totaled approximately $245.1 million, $242.5 million and $299.3 million, respectively. See Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations—Major Research and Development Projects for additional information
19
regarding expenditures related to major research and development projects. Research and development expense is significantly impacted by fluctuations in our stock price, due to the cash payment obligations created by our share-based compensation programs. For further details, see Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations—Operating Expenses—Share-Based Compensation.
Production and Supply
We produce our primary supply of Remodulin, Tyvaso, Orenitram and Unituxin at our own facilities. In particular, we synthesize treprostinil, the active ingredient in Remodulin and Tyvaso, and treprostinil diolamine, the active ingredient in Orenitram, at our facility in Silver Spring, Maryland. We also produce finished Tyvaso, Remodulin, and Unituxin at our Silver Spring facility. We produce Orenitram and we package, warehouse and distribute Remodulin, Tyvaso, Orenitram and Unituxin, at our facility in Research Triangle Park, North Carolina.
We maintain a two-year inventory of Remodulin, Tyvaso and Orenitram based on expected demand, and we also contract with third-party contract manufacturers to supplement our capacity, in order to mitigate the risk that we might not be able to produce sufficient quantities to meet patient demand. For example, Baxter Pharmaceutical Solutions, LLC (Baxter) is approved by the FDA, the EMA and various other international regulatory agencies to produce Remodulin for us. In the case of Tyvaso, we rely on Catalent Pharma Solutions, Inc. (Catalent) to serve as an additional producer of Tyvaso, and we rely entirely on Minnetronix Inc. to manufacture the nebulizer used in our Tyvaso Inhalation System. We are working to obtain FDA approval of third party contract manufacturers to serve as additional producers of Orenitram and Unituxin.
Although we believe that additional third parties could provide similar products, services and materials, there are few companies that could replace our existing third-party producers and suppliers. A change in supplier or producer could cause a delay in the production, distribution and research efforts associated with our respective products or result in increased costs. See also Item 1A—Risk Factors included in this Annual Report on Form 10-K.
Competition
Many drug companies engage in research and development to commercialize products to treat cardiovascular diseases and cancer. For the treatment of PAH, we compete with many approved products in the United States and the rest of the world, including the following:
- •
- Flolan, Veletri and generic epoprostenol. Flolan
(epoprostenol) is a prostacyclin that is delivered by intravenous infusion. Glaxo began marketing Flolan in the United States in 1996, and the generic exclusivity period for Flolan expired in 2007. In
2008, the FDA approved Teva's version of generic epoprostenol for the treatment of PAH. In 2010, Actelion commenced sales of Veletri, which is another version of intravenous epoprostenol;
- •
- Ventavis and Ilomedin®. Approved in 2004 in
the United States and in 2003 in Europe, Ventavis (iloprost) is an inhaled prostacyclin analogue. Ventavis is currently marketed by Actelion in the United States and by Bayer in Europe. Iloprost is
also marketed by Bayer in certain countries outside the United States in an intravenous form known as Ilomedin;
- •
- Tracleer. Tracleer (bosentan), an oral ETRA therapy for
the treatment of PAH, was approved in 2001 in the United States and in 2002 in Europe. Tracleer is marketed worldwide by Actelion;
- •
- Letairis. Approved in 2007 in the United States, Letairis (ambrisentan) is an oral ETRA therapy marketed by Gilead for the treatment of PAH. In 2008, Glaxo received marketing authorization from the EMA for Letairis in Europe, where it is known as Volibris®;
20
- •
- Revatio and generic sildenafil citrate. Approved in 2005
in the United States, Revatio (sildenafil citrate) is an oral PDE-5 inhibitor therapy marketed by Pfizer. Revatio contains sildenafil citrate, the same active ingredient as Viagra. In the fourth
quarter of 2012, several companies began marketing generic formulations of sildenafil citrate;
- •
- Opsumit. Approved in October 2013 in the United States
and December 2013 in the EU, Opsumit (macitentan) is an oral ETRA marketed by Actelion for the treatment of PAH;
- •
- Adempas®. Approved in August 2013 in the
United States and March 2014 in the EU, Adempas (riociguat) is a soluble guanylate cyclase stimulator, which targets a similar vasodilatory pathway as PDE-5 inhibitors and is approved for chronic
thromboembolic pulmonary hypertension and PAH. Adempas is an oral therapy marketed by Bayer; and
- •
- Uptravi. Approved in the United States in December 2015, Uptravi (selexipag) is an oral IP prostacyclin receptor agonist marketed by Actelion. Actelion submitted an MAA for EMA approval of Uptravi in December 2014, and in January 2016 received a positive opinion from the Committee for Medicinal Products for Human Use recommending European Commission approval for Uptravi. Actelion also has applications pending in various other jurisdictions. Nippon Shinyaku Co., Ltd. holds the right to market Uptravi in Japan, where it submitted an NDA in January 2016.
There are also a variety of investigational PAH therapies in the later stages of development, including the following:
- •
- Ralinepag, an oral IP prostacyclin receptor agonist being developed by Arena
Pharmaceuticals, Inc. (Arena). Arena commenced a phase II clinical trial of ralinepag in 2014; and
- •
- Trevyent, a formulation of treprostinil being developed by SteadyMed Ltd. (SteadyMed) for delivery via its pre-filled, disposable PatchPump®. SteadyMed announced that it plans to submit an NDA and MAA for Trevyent in the second half of 2016. In January 2016, SteadyMed announced that Trevyent has been granted orphan drug designation by the FDA for the treatment of PAH.
Oral non-prostacyclin therapies (such as PDE-5 inhibitors and ETRAs) are commonly prescribed as first-line treatments for the least severely ill PAH patients (NYHA Class II patients). As patients progress in their disease severity (NYHA Class III and IV), less convenient approved therapies, such as inhaled prostacyclin analogues (such as Tyvaso) or infused prostacyclin analogues (such as Remodulin) are commonly added. Orenitram is the first approved oral prostacyclin therapy for PAH in the United States. We anticipate that it will face competition with existing oral PAH therapies, and will be regarded as a less invasive and more convenient alternative therapy to Tyvaso and Remodulin. The use of available oral therapies could delay many patients' need for inhaled or infused prostacyclin therapy. As a result, the availability of oral therapies affects demand for our inhaled and infused products.
Orenitram faces competition from Uptravi, which is indicated to delay disease progression and reduce the risk of hospitalization for PAH. This indication may provide a competitive advantage against Orenitram, which is indicated to improve exercise capacity. As noted above, however, Uptravi is an oral IP prostacyclin receptor agonist, a new class of therapy that addresses PAH through the prostacyclin pathway. While prostacyclin analogues such as Orenitram broadly mimic the effect of prostacyclin, IP prostacyclin receptor agonists bind selectively to the IP receptor, one of several prostacyclin receptors. In addition, Orenitram's label allows physicians flexibility to titrate each patient's dosing up to a level according to tolerability, without any stated maximum. By contrast, Uptravi's label limits uptitration to a specific maximum dose.
21
We will also likely face competition from generic pharmaceutical companies in the future. For example, two generic companies filed ANDAs requesting FDA approval to market a generic version of Remodulin. We settled patent litigation with both companies, and our settlement agreements will allow Sandoz and Teva to launch generic versions of Remodulin in June 2018 and December 2018, respectively. Generic companies have filed ANDAs requesting FDA approval to market generic versions of Tyvaso and Orenitram. For details, see the sections below entitled Governmental Regulation—Hatch-Waxman Act, Item 3—Legal Proceedings, and Part II, Item 9B—Other Information. In addition, certain Revatio patents expired in 2012, leading several manufacturers to launch generic formulations of sildenafil citrate, which physicians could prescribe for the treatment of PAH. Generic sildenafil citrate's lower price, relative to Adcirca, could erode Adcirca's market share and limit its growth potential. Although we believe Adcirca's once-daily dosing regimen is an appealing alternative to generic sildenafil citrate's dosing regimen of three times per day, we expect government payers and private insurance companies to favor over time the use of the less expensive generic sildenafil citrate instead of Adcirca. The U.S. patent for Adcirca for the treatment of pulmonary hypertension will expire in November 2017, following which we expect to see generic competition for Adcirca.
We compete with the developers, manufacturers and distributors of all of the PAH products noted above for customers, funding, access to licenses, personnel, third-party collaborators, product development and commercialization. Almost all of these companies have substantially greater financial, marketing, sales, distribution and technical resources, and more experience in research and development, product development, manufacturing and marketing, clinical trials and regulatory matters, than we have.
Governmental Regulation
Pharmaceutical Product Approval Process
The research, development, testing, manufacture, promotion, marketing, distribution, sampling, storage, approval, labeling, record keeping, post-approval monitoring and reporting, and import and export of pharmaceutical products (drugs or biological products, hereinafter collectively drugs) are extensively regulated by governmental agencies in the United States and in other countries. In the United States, failure to comply with requirements under the Federal Food, Drug, and Cosmetic Act (FDC Act), the Public Health Service Act (PHSA), and other federal statutes and regulations, may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Satisfaction of FDA pre-market approval requirements typically takes many years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Drugs are subject to rigorous regulation by the FDA in the United States, the EMA in the EU and similar regulatory authorities in other countries. The steps ordinarily required before a new drug may be marketed in the United States, which are similar to steps required in most other countries, include:
- •
- Preclinical laboratory tests, preclinical studies in animals, formulation studies and the submission to the FDA of an investigational
new drug application (IND) for a new drug, which must become effective before clinical testing may commence;
- •
- Clinical studies in healthy volunteers;
- •
- Clinical studies in patients to explore safety, efficacy and dose-response characteristics;
- •
- Adequate and well-controlled clinical trials to establish the safety and efficacy of the drug for each indication;
22
- •
- The submission of an NDA or BLA to the FDA; and
- •
- FDA review and approval of the NDA or BLA prior to any commercial sale or shipment of the drug.
Preclinical tests include laboratory evaluation of product chemistry and formulation, as well as animal studies to explore toxicity and for proof-of-concept. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. In the United States, the results of preclinical testing are submitted to the FDA as part of an IND, along with other information including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted. Absent FDA objection within 30 days after submission of an IND, the IND becomes effective and the clinical trial proposed in the IND may begin. At any time during this 30-day period or at any time thereafter, the FDA may halt, temporarily or permanently, a clinical trial. The IND process may be extremely costly and may substantially delay development of our products. Moreover, positive results of preclinical tests will not necessarily indicate positive results in clinical trials.
Clinical trials involve the administration of the investigational new drug or biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (1) in compliance with federal regulations; (2) in compliance with good clinical practices (GCP), an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; and (3) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be approved by an institutional review board (IRB). An IRB may also require the clinical trial at a site to be halted temporarily or permanently for failure to comply with the IRB's requirements, or may impose other conditions.
Clinical trials in support of an NDA or a BLA are typically conducted in sequential phases, but the phases may overlap. During phase I, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase II usually involves studies in a limited patient population to assess the efficacy of the drug in specific, targeted indications, assess tolerance and optimal dosage and identify possible adverse effects and safety risks. If a compound is found to be potentially effective and to have an acceptable safety profile in phase II evaluations, then a meeting may be requested at the end of phase II to determine the safety of proceeding to phase III. Phase III trials, also called pivotal studies, major studies or advanced clinical trials, are undertaken to demonstrate clinical efficacy and safety in a larger number of patients, typically at geographically diverse clinical study sites, and to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. Phase IV studies are often conducted following marketing approval, in order to meet regulatory requirements or to provide additional data relating to the use of the drug.
After successful completion of the required clinical testing, an NDA or a BLA is typically submitted to the FDA in the United States, and an MAA is typically submitted to the EMA in the EU. FDA approval of the NDA or BLA is required before marketing of the product may begin in the United States. The NDA or BLA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product's pharmacology, chemistry, manufacture, and controls.
23
The cost of preparing and submitting an NDA or BLA is substantial. Under federal law, the submission of most NDAs and BLAs is additionally subject to a substantial application fee, currently exceeding $2.3 million, and the manufacturer and/or sponsor of an approved NDA or BLA is also subject to annual product and establishment fees, currently exceeding $114,000 per product and $585,000 per establishment. These fees are typically increased annually. However, the application fees may be waived for orphan drugs if certain requirements are met.
The FDA has 60 days from its receipt of an NDA or a BLA to determine whether the application will be accepted for filing based on the agency's threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA may instead ask for additional information, in which case, the application must be amended and resubmitted with the requested information. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for non-priority drugs are reviewed within ten to twelve months, while most applications for priority review drugs are reviewed in six to eight months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. For biologics, priority review is further limited to drugs intended to treat a serious or life-threatening disease. The review process may be extended by the FDA for three additional months to consider certain information submitted during FDA review, including information intended to clarify information already provided or to address any deficiencies identified in the submission. The FDA may also refer applications for novel pharmaceutical products or pharmaceutical products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. During the review process, the FDA also reviews the drug's product labeling to ensure that appropriate information is communicated to health care professionals and consumers. In addition, before approving an NDA or a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with the FDA's current Good Manufacturing Practices (cGMP) and GCP is satisfactory and the NDA or BLA contains data that provide substantial evidence that the pharmaceutical product is safe and effective for purposes of the indication studied.
In the United States, after the FDA evaluates the NDA or BLA and the manufacturing facilities, the FDA may issue either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those conditions have been addressed to the FDA's satisfaction in a resubmission of the NDA or BLA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. A Class 1 resubmission may contain only limited information such as labeling, safety updates, stability updates, or minor analysis updates or clarifying information and is subject to a two-month review period. All other resubmissions are categorized as Class 2 and are subject to a six-month review period. Even after such a resubmission, the FDA may decide that the application does not satisfy the regulatory criteria for approval.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA or BLA approval, the FDA may require a risk evaluation and mitigation strategy (REMS) to help ensure that the benefits of the drug outweigh the potential risks. A REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use (ETASU). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. To continue marketing our products
24
after approval, applicable regulations require us to maintain a positive risk-benefit profile, maintain regulatory applications through periodic reports to regulatory authorities, fulfill pharmacovigilance requirements, maintain manufacturing facilities according to cGMP requirements, and successfully complete regulatory agency inspections, among other requirements. Our manufacturing facilities are subject to continual review and periodic inspections. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated drugs and other products are required to register and disclose certain clinical trial information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial. This clinical trial information is then made public as part of the sponsor's registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Competitors may use this publicly-available information to gain knowledge regarding the progress of development programs.
Orphan Drugs
Under the Orphan Drug Act, an applicant can request the FDA to designate a product as an "orphan drug" in the United States if the drug is intended to treat a rare disease or condition affecting fewer than 200,000 people in the United States. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA or BLA applicant to receive orphan drug designation and FDA approval for a particular active ingredient to treat a particular disease via a particular delivery method is entitled to a seven-year exclusive marketing period in the United States. During the seven-year period, the FDA may not approve any other application to market the same drug for the same disease, except in limited circumstances such as (1) a showing of clinical superiority to the product with orphan drug exclusivity, meaning that it has greater effectiveness or safety, or provides a major contribution to patient care (such as a change in delivery system); or (2) or the inability of the NDA or BLA holder for the product with orphan drug exclusivity to assure availability of sufficient quantities of the drug to meet the needs of patients with the rare disease or condition. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA or BLA application user fee.
The FDA granted orphan drug designation for the active ingredient treprostinil for the treatment of PAH as a continuous infusion. However, this designation does not preclude us from seeking orphan drug designation for other treprostinil formulations or routes of administration to treat PAH, or for treprostinil used to treat other orphan diseases. In order for the FDA to grant orphan drug designation for other formulations or routes of administration of treprostinil to treat PAH, we must demonstrate that such new formulation or route of administration is clinically superior to the formulation or route of administration previously granted orphan drug designation. The FDA has granted orphan drug designation for Tyvaso and Unituxin. A request for orphan drug designation for Orenitram is pending.
Pediatric Information
Under the Pediatric Research Equity Act of 2007 (PREA), NDAs, BLAs and supplements to NDAs and BLAs must contain data to assess the safety and effectiveness of the drug for the claimed indication(s) in all relevant pediatric subpopulations and to support dosing and administration for each such pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals for
25
submission of data or full or partial waivers. Unless otherwise required by regulation, the PREA does not apply to any drug for an indication for which orphan drug designation has been granted.
The Best Pharmaceuticals For Children Act (BPCA) provides NDA holders a six-month extension of any exclusivity, patent or non-patent, for a drug if certain conditions are met. Conditions for exclusivity include the FDA's determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the requested time frame. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Hatch-Waxman Act
The Hatch-Waxman Act (also known as the Drug Price Competition and Patent Term Restoration Act) was passed in 1984 to encourage research and development of new drugs and competition between brand and generic pharmaceutical companies. It created a faster approval process for generic drugs, called the ANDA, while providing protection to brand pharmaceuticals by extending their patent protection, in some cases, to compensate for patent life lost during the product development and approval process and providing periods of market exclusivity to encourage continuing research on, for example, new uses, strengths or dosage forms for existing drugs.
In seeking approval of a drug through an NDA, applicants are required to submit to the FDA each patent whose claims cover the applicant's product or FDA-approved method of using this product. Upon approval of a drug, each of the patents listed in the application is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA. Generally, an ANDA provides for marketing of a drug product that has the same active ingredients in the same strength(s), route of administration, and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. ANDA applicants are not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as "generic equivalents" to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA's Orange Book. Specifically, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product's listed patents or that such patents are invalid is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired. Alternatively, for a patent covering an approved method of use, an ANDA applicant may submit a statement to the FDA that the company is not seeking approval for the covered use.
If the ANDA applicant has submitted a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
26
The ANDA application also will not receive approval until any non-patent exclusivity, such as exclusivity for obtaining approval of an NDA for a new chemical entity, has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active moiety, during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV certification, in which case the submission may be made four years following the original product approval. Following approval of an application to market a drug that contains previously approved active ingredients in a new dosage form, route of administration or combination, or for a new condition of use that was required to be supported by new clinical trials conducted by or for the sponsor, the FDC Act provides for an exclusivity period of three years, during which the FDA cannot grant effective approval of an ANDA for such new condition of use, dosage form or strength that meets certain statutory requirements. Both of the five-year and three-year exclusivity periods, as well as any unexpired patents listed in the Orange Book for the listed drug, can be extended by six months if the FDA grants the NDA sponsor a period of pediatric exclusivity based on studies submitted by the sponsor in response to a written request.
The Hatch-Waxman Act provides that patent terms may be extended to compensate for some of the patent life that is lost during the FDA regulatory review period for a product. This extension period would generally be one-half the time between the effective date of an IND and the submission date of an NDA, plus all of the time between the submission date of an NDA and its approval, subject to a maximum extension of five years. Similar patent term extensions are available under European laws. Following FDA approval, we filed a patent term extension application with the United States Patent and Trademark Office for our patent covering the method of treating PAH using treprostinil. The application was approved in February 2005 with the maximum patent term extension of five years, and the patent expired in October 2014.
We have received Paragraph IV certification letters from Watson and Actavis advising that they have submitted ANDAs to the FDA requesting approval to market generic versions of Tyvaso and Orenitram, respectively. We have also settled litigation relating to Paragraph IV certification letters from Sandoz and Teva advising that each has submitted an ANDA to the FDA requesting approval to market generic versions of Remodulin. Under our settlement agreements with Sandoz and Teva, they will be permitted to market their generic versions of Remodulin in the Unites States beginning in June 2018 and December 2018 respectively, or earlier in certain circumstances. For further details, please see Note 19—Litigation, to our consolidated financial statements, Item 3—Legal Proceedings and Part II, Item 9B—Other Information.
Section 505(b)(2) New Drug Applications
Most drug products (other than biological products) obtain FDA marketing approval pursuant to an NDA submitted under Section 505(b)(1) of the Food, Drug and Cosmetic Act (FDCA), or an ANDA. A third alternative is a special type of NDA submitted under Section 505(b)(2) of the FDCA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA's finding of safety and efficacy data for an existing product, or published literature, in support of its application.
Section 505(b)(2) NDAs may provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA in which the applicant relies, at least in part, on information from studies made to show whether a drug is safe or effective that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use. A Section 505(b)(2) applicant may eliminate the need to conduct certain preclinical or clinical studies, if it can establish that reliance on studies conducted for a previously-approved product is scientifically appropriate. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the labeled indications for which
27
the referenced product has been approved, as well as for any new indication for which the Section 505(b)(2) NDA applicant has submitted data.
To the extent that the Section 505(b)(2) applicant is relying on prior FDA findings of safety and efficacy, the applicant is required to certify to the FDA concerning any patents listed for the previously approved product in the Orange Book to the same extent that an ANDA applicant would. Thus, approval of a Section 505(b)(2) NDA can be delayed until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Other Regulatory Requirements
Once an NDA or a BLA is approved, the product will be subject to continuing regulations. For instance, the FDA closely regulates the post-approval marketing, labeling and advertising of prescription drugs, including the standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Pharmaceutical products may be marketed only for their approved indications and in accordance with the provisions of their approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting promotion of off-label uses, and a company that is found to have engaged in off-label promotion may be subject to significant liability.
Certain changes to the conditions established in an approved application, including changes in indications, labeling, equipment, or manufacturing processes or facilities, will require submission and FDA approval of an NDA or BLA or supplement thereto before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing supplements as it does in reviewing NDAs or BLAs.
Adverse event reporting and submission of periodic reports continue to be required following FDA approval of an NDA or a BLA. The FDA also may require post-marketing testing, including phase IV clinical studies, risk minimization action plans, and surveillance to monitor the effects of an approved product or may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as drug manufacture, packaging, and labeling procedures must continue to conform to cGMP requirements. Manufacturers and certain of their contractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies, to assess compliance with cGMP requirements. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMP requirements. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards or if previously unrecognized problems are subsequently discovered. Later discovery of previously unknown problems with a product, including adverse events or problems with manufacturing processes of unanticipated severity or frequency, or failure to comply with regulatory requirements, may also result in (1) revisions to the approved labeling to add new safety information; (2) imposition of post-market studies or clinical trials to assess new safety risks; or (3) imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things, (1) restrictions on the marketing or manufacturing of the product; (2) fines, warning letters or holds on post-approval clinical trials; (3) refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; (4) product seizure or detention, or refusal to permit the import or export of products; or (5) injunctions or the imposition of civil or criminal penalties.
28
Marketing Pharmaceutical Products Outside the United States
Outside of the United States, our ability to market our products is also contingent upon receiving marketing authorizations from regulatory authorities. The foreign regulatory approval process may include some or all of the risks associated with the FDA review and approval process set forth above, and the requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country.
In the EU, marketing authorizations may be submitted through a centralized body or through a decentralized/mutual recognition or a national level process. The centralized procedure is mandatory for the approval of certain products, such as officially designated orphan medicines and medicines derived from biotechnology and high technology processes, and may be available at the applicant's option for other products that are a significant therapeutic, scientific or technical innovation or for which approval would be in the interest of public health. The centralized procedure provides for the grant of a single marketing authorization that is valid in the EEA, which consists of the EU member countries and Norway, Iceland, and Lichtenstein. The decentralized/mutual recognition procedures are available for all medicinal products that are not subject to the centralized procedure. Each EU member country has its own procedure for approval. A company may use the decentralized procedure to submit applications for marketing authorization in more than one EU country simultaneously for a product that has not previously been authorized in an EU country. In addition, the mutual recognition procedure provides for mutual recognition of national approval decisions, changes existing procedures for national approvals and establishes procedures for coordinated EU actions on products, suspensions and withdrawals. Under this procedure, the holder of a national marketing authorization for which mutual recognition is sought may submit an application to one or more EU member countries, certify that the dossier is identical to that on which the first approval was based, or explain any differences and certify that identical dossiers are being submitted to all EU member countries for which recognition is sought. Within 90 days of receiving the application and assessment report, each EU member country is required to decide whether to recognize approval. The procedure encourages member states to work with applicants and other regulatory authorities to resolve disputes concerning mutual recognition. Arbitration may be initiated when member countries fail to reach agreement. Following receipt of marketing authorization in an EU member country, the applicant is then usually (depending on the country) required to engage in pricing discussions and negotiations with a separate prescription pricing authority in that country. Commercial sales typically only commence in a country once pricing approval has been obtained.
To secure European regulatory approvals for subcutaneous Remodulin for PAH, we used the mutual recognition process. Under the rules then applicable, centralized filing was not required and we perceived the decentralized/mutual recognition procedure to be the most effective means for approval. We filed our first MAA in France in February 2001. Review of our application was completed in 2005. As a result, Remodulin was approved in 23 member countries of the EEA under the mutual recognition process described above. We withdrew applications in Spain, the United Kingdom and Ireland and are currently evaluating resubmitting applications in Spain and Ireland. In December 2011, we received approval for intravenous Remodulin in all of the 23 EEA member nations where subcutaneous Remodulin is approved.
To secure European regulatory approval for Tyvaso, we submitted an MAA to the EMA via the centralized process in 2008. Regulations in Europe have changed since we made our initial filing for Remodulin and all therapies for orphan diseases must now use the centralized process. In February 2010, we withdrew our MAA from consideration by the EMA, and do not currently intend to resubmit it as a standalone treatment for PAH due to the EMA's major objection related to findings of non-compliance with good clinical practice at two clinical sites. The EMA stated that these findings would preclude a recommendation for approval of Tyvaso in the EU. The EMA had no major objections at the time of withdrawal related to the safety or efficacy of Tyvaso.
29
Biologics
Biological products used for the prevention, treatment, or cure of a disease, or condition, of a human being are subject to regulation under the FDC Act and the PHSA. Biological products are approved for marketing via a BLA that follows an application process and approval requirements that are very similar to those for NDAs. To help reduce the increased risk of the introduction of adventitious agents, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The PHSA also provides authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to prevent the introduction, or spread, of communicable diseases in the United States.
After a BLA is approved, the product may also be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official lot release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer's tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products, such as viral vaccines, before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products. As with drugs, after approval of biologics, manufacturers must address any safety issues that arise, are subject to recalls or a halt in manufacturing, and are subject to periodic inspection after approval.
The Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 (PPACA), included a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, which created an abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product. This is conceptually similar to the Hatch-Waxman Act in that it attempts to minimize duplicative testing. Biosimilarity requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency must be shown through analytical studies, animal studies, and at least one clinical study absent a waiver. In addition, only minor differences in clinically inactive components are allowable in biosimilar products. An interchangeable biological product is a biosimilar to an FDA-approved reference product that meets additional standards for interchangeability. Interchangeability requires that a product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, intricacies associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation that are still being addressed by the FDA. In 2015, the FDA approved the first biosimilar in the United States and published a proposed rule and guidance documents relating to various provisions of the BPCI Act. For example, the FDA addressed the naming conventions and labeling of biosimilar products, including whether and how such labeling should include information from reference product labeling and identify a product's approval through the biosimilar pathway. The FDA suggested that shared non-proprietary names may not be appropriate for all biological products, noting that the naming conventions could improve pharmacovigilance, prevent inadvertent substitution among products which have not been determined to be interchangeable, avoid inaccurate perceptions of the safety and effectiveness of biological products based on their licensure pathway, and provide a consistent mechanism for healthcare professionals to
30
identify biological products. The FDA also released guidance recommendations relating to the clinical, scientific, and quality data required to demonstrate biosimilarity.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against other biologics submitted under the abbreviated approval pathway for the lesser of (1) one year after first commercial marketing; (2) eighteen months after approval of the initial application if there is no legal challenge; (3) eighteen months after the resolution in the applicant's favor of a lawsuit challenging the biologics' patents if an application has been submitted; or (4) 42 months after the application has been approved if a lawsuit is ongoing within the 42 month period.
Since the passage of the BPCI Act, there have been proposals to modify the exclusivity provisions. In the budget for fiscal year 2016, the Obama administration reasserted a proposal from prior years to cut the 12-year period of exclusivity down to seven years. The administration also reasserted a proposal to prohibit additional periods of exclusivity due to minor changes in product formulations. In October 2015, the United States agreed to the Trans-Pacific Partnership (TPP), an agreement with 11 other countries that addresses a variety of trade and economic issues. The TPP includes a provision that would require the signatory countries to provide a minimum of five years of exclusivity, and in some instances, eight years of exclusivity, to biological products. To come into effect, the TPP will require a requisite number of signatory countries to ratify the agreement; in the United States, such ratification, if it occurs, will be performed by Congress. It is possible that Congress could seek to harmonize the exclusivity periods in the TPP and the BPCI Act, or take other measures to modify or eliminate periods of exclusivity for biosimilar and interchangeable products.
Because biologically sourced raw materials are subject to unique contamination risks, their use may be restricted in some countries.
Cell and Tissue Based Biologics
Manufacturers of cell and tissue based products must comply with the FDA's current good tissue practices (cGTP), which are FDA regulations that govern the methods used in, and the facilities and controls used for, the manufacture of such products. The primary intent of the cGTP requirements is to ensure that cell and tissue based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable diseases. Cell and tissue based products may also be subject to the same approval standards, including demonstration of safety and efficacy, as other biologic and drug products, if they meet certain criteria such as if the cells or tissues are more than minimally manipulated or if they are intended for a non-homologous use (a use different from the cell's origin). In 2015, the FDA published guidance documents relating to topics such as donor screening, adverse reaction reporting, and the applicability of premarket approval and clearance requirements to cell and tissue based products. Following the numerous public comments on these draft guidance documents, the FDA scheduled a public hearing for April 2016.
U.S. Regulation of Medical Devices
Medical devices also may be subject to FDA approval and extensive regulation under the FDC Act. Under the FDC Act, medical devices are classified into one of three classes: Class I, Class II, or Class III. The classification of a device into one of these three classes generally depends on the degree of risk associated with the medical device and the extent of control needed to ensure safety and effectiveness.
Class I devices are those for which safety and effectiveness can be assured by adherence to a set of general controls. These general controls include compliance with the applicable portions of the FDA's Quality System Regulation (QSR), which sets forth good manufacturing practice requirements; facility
31
registration and product listing; reporting of adverse medical events; truthful and non-misleading labeling; and promotion of the device only for its cleared or approved intended uses. Class II devices are also subject to these general controls and to any other special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. Most Class I devices require review and clearance by the FDA through the so-called 510(k) pre-market notification procedure. A Class III device requires approval of a premarket approval application (PMA), an expensive, lengthy and uncertain process that can require many years to complete. Most Class II and Class III medical devices may only be marketed in the United States if the FDA has approved a PMA application for the device or cleared the device in response to a 510(k) submission. There is also an alternative pathway to approval for low or moderate risk devices that are not classified and for which no predicate device exists, known as de novo classification.
When 510(k) clearance is sought, a sponsor must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to a previously marketed device, also referred to as a "predicate" device. If the FDA agrees that the proposed device is substantially equivalent to the predicate device, then 510(k) clearance to market will be granted. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require pre-market approval. In recent years, the FDA has issued guidance recommendations relating to aspects of the FDA's 510(k) review process, including the use of multiple predicates and the content of 510(k) summaries, and the criteria the FDA uses to determine whether a 510(k) satisfies the minimum threshold of acceptability to be accepted for substantive review.
Clinical trials are almost always required to support a PMA and are sometimes required for a 510(k) pre-market notification. These trials generally require FDA approval by submitting an application for an investigational device exemption, or IDE application. An IDE application must be supported by preclinical data, such as animal and laboratory testing results, which show that the device is safe to test in humans and that the study protocols are scientifically sound. Studies of devices that pose a significant risk require approval from both the FDA and an Institutional Review Board (IRB) prior to initiation of the study. A "nonsignificant" risk device study does not require submission of an IDE application to the FDA but does require IRB approval prior to initiation of the study. Nonsignificant risk device studies must comply with abbreviated IDE requirements.
Both before and after a medical device is commercially distributed, manufacturers and marketers of the device have ongoing responsibilities under FDA regulations. The FDA reviews design and manufacturing practices, labeling and record keeping, and manufacturers' required reports of adverse experiences and other information to identify potential problems with marketed medical devices. Device manufacturers are subject to periodic and unannounced inspection by the FDA for compliance with the QSR, current good manufacturing practice requirements that govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, servicing, labeling, storage, installation, and distribution of all finished medical devices intended for human use.
If the FDA finds that a manufacturer has failed to comply or that a medical device is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, ranging from a public warning letter to more severe actions such as:
- •
- fines, injunctions, and civil penalties;
- •
- recall or seizure of products;
- •
- operating restrictions, partial suspension or total shutdown of production;
- •
- refusing requests for 510(k) clearance or PMA approval of new products;
- •
- withdrawing 510(k) clearances or PMA approvals already granted; and
32
- •
- criminal prosecution.
The FDA also has the authority to require repair, replacement or refund of the cost of a medical device under certain circumstances.
The FDA also administers certain controls over the import and export of medical devices to and from the United States. Additionally, each foreign country subjects such medical devices to its own regulatory requirements. In the EU, a single regulatory approval process has been created, and approval is represented by the CE Mark.
A combination product is a product composed of a combination of a drug, device, or biological product. Each combination product is assigned to a specific FDA center based on which constituent part of the combination product provides the primary mode of action. In 2015, the FDA released draft guidance recommendations relating to the application of cGMP requirements to combination products. The nebulizer used with our Tyvaso Inhalation System was included in our NDA for Tyvaso as a drug-device combination product, and was cleared by the FDA subject to compliance with the QSR as it applies to combination products. In 2012, we received FDA approval for a modified Tyvaso Inhalation System using an updated nebulizer (TD-100) based on the results of the completion of the QSR compliance commitments.
Government Reimbursement of Pharmaceutical Products
In the United States, many independent third-party health plans, and government health care programs, pay for patient use of our commercial products. Medicare is the federal program that provides health care benefits to senior citizens and certain disabled and chronically ill persons. Medicaid is the federal program jointly funded and administered by the states to provide health care benefits to certain indigent persons. Unituxin is administered entirely as an in-patient therapy and would typically be reimbursed under Medicare Part A. However, because Unituxin is indicated for treatment of a pediatric cancer, Medicare beneficiaries are unlikely to receive this treatment. The purchase prices for Remodulin and Tyvaso are reimbursed within the Medicare Part B program. The Medicare Part B contractors who administer the program provide reimbursement for Remodulin and Tyvaso according to statutory guidelines. In return for the inclusion of our commercial products Adcirca and Orenitram in the Medicare Part D program, we have agreed to pay rebates to Medicare Part D plan sponsors that reimburse these products. The state Medicaid programs also reimburse the cost of our commercial products at reimbursement rates established by statutory guidelines. Because Remodulin, Tyvaso, Adcirca, Orenitram and Unituxin are covered and reimbursed by state Medicaid programs, we are mandated to pay a rebate to those state Medicaid programs. We are required by government contract to sell our commercial products under contracts with the Department of Veterans Affairs, Department of Defense, Public Health Service and numerous other federal agencies as well as certain hospitals that are designated as 340B covered entities (entities designated by federal programs to receive drugs at discounted prices) at prices that are significantly below the price we charge to our specialty distributors. These programs and contracts are highly regulated and impose restrictions on our business. Failure to comply with these regulations and restrictions could result in a loss of our ability to continue receiving reimbursement for our drugs, exclusion of our products from reimbursement under the federal healthcare programs, or debarment, and expose us to liability under federal and state false claims laws. We estimate that between 35-50% of Remodulin, Tyvaso, Adcirca and Orenitram sales are reimbursed under the Medicare and Medicaid programs.
Anti-Kickback, False Claims Laws and The Prescription Drug Marketing Act
In addition to FDA restrictions on marketing pharmaceutical, biological and medical device products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical and medical device industries in recent years. These laws include
33
anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of, or referring an individual for the furnishing of, any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
The federal False Claims Act prohibits any person from, among other things, knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement material to a false claim. Many pharmaceutical and other healthcare companies have been prosecuted under the False Claims Act for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, companies have been prosecuted under the False Claims Act on the basis of allegations relating to marketing practices, including off-label promotion. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Sanctions under these federal and state laws may include civil penalties, exclusion of a manufacturer's products from reimbursement under government programs, criminal fines, and imprisonment.
As part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. The Prescription Drug Marketing Act (PDMA) imposes requirements and limitations upon the distribution of drugs and drug samples, and prohibits states from licensing distributors of prescription drugs unless the state licensing program meets certain federal guidelines that include minimum standards for storage and handling, as well as record keeping requirements for information regarding sample requests and distribution. The PDMA sets forth civil and criminal penalties for violations. In addition, PDMA requires manufacturers and distributors to submit similar drug sample information to FDA.
Patient Protection and Affordable Care Act of 2010
PPACA is intended to expand healthcare coverage within the United States. Several provisions of the law, which have varying effective dates, have impacted us and have increased certain of our costs. PPACA imposes an annual fee on pharmaceutical manufacturers, based on the manufacturer's sale of branded pharmaceuticals and biologics (excluding orphan drugs) to certain U.S. government programs during the preceding year; expands the 340B drug discount program (excluding orphan drugs) including the creation of new penalties for non-compliance; and includes a 50% discount on brand name drugs for Medicare Part D participants in the coverage gap, or "donut hole." Effective beginning in 2010, the law also revised the definition of "average manufacturer price" for reporting purposes, which could increase the amount of the Medicaid drug rebates paid to states.
As noted above under Governmental Regulation—Biologics, the PPACA also created a regulatory pathway for the abbreviated approval of biological products that are demonstrated to be "biosimilar" or "interchangeable" with an FDA-approved biological product. In addition, PPACA imposes new annual reporting requirements for pharmaceutical, biological and device manufacturers with regard to
34
payments or other transfers of value made to physicians and teaching hospitals. In addition, pharmaceutical, biological and device manufacturers are required to report annually investment interests held by physicians and their immediate family members during the preceding calendar year. Such information was required to be made publicly available by the Secretary of Health and Human Services in a searchable format beginning on September 30, 2014. Failure to submit required information may result in civil monetary penalties of up to $150,000 per year (and up to $1 million per year for "knowing failures") for all payments, transfers of value or ownership or investment interests not reported in an annual submission. Further, the PPACA amends the intent requirement of the federal anti-kickback and criminal health care fraud statute. A person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
State Pharmaceutical and Medical Device Marketing Laws
If not preempted by the PPACA, several jurisdictions, including the District of Columbia, Maine, Massachusetts, Minnesota, Vermont and West Virginia, require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to healthcare practitioners in those jurisdictions. Some of these jurisdictions also prohibit various marketing related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, certain states, such as California, Connecticut, Nevada, and Massachusetts, require pharmaceutical companies to implement compliance programs or marketing codes and several other states are considering similar proposals. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws face civil penalties or other civil enforcement action.
Employees
We had approximately 750 employees as of February 7, 2016. The success of our business is highly dependent on attracting and retaining highly talented and qualified personnel.
Industry Segments and Geographic Areas
Since March 2011, our core business has been pharmaceuticals, in which we closely monitor the revenues and gross margins generated by our commercial products. We sell our products in the United States and throughout the rest of the world. The information required by Item 101(b) and 101(d) of Regulation S-K relating to financial information about industry segments and geographical areas, respectively, is contained in Note 17—Segment Information to our consolidated financial statements included in this Annual Report on Form 10-K.
Corporate Website
Our Internet website address is http://www.unither.com. Our filings on Form 10-K, Form 10-Q, Form 3, Form 4, Form 5, Form 8-K and any and all amendments thereto are available free of charge through this internet website as soon as reasonably practicable after they are filed with or furnished to the Securities and Exchange Commission (SEC). They are also available through the SEC at http://www.sec.gov/edgar/searchedgar/companysearch.html.
35
EXECUTIVE OFFICERS OF THE REGISTRANT
The following is a list, as of February 17, 2016, setting forth certain information regarding our executive officers. Each executive officer holds office until the first meeting of the Board of Directors after the annual meeting of shareholders, and until his or her successor is elected and qualified or until his or her earlier resignation or removal. Each executive officer's employment will end pursuant to the terms of his or her employment contract.
Name
|
Age | Position | |||
|---|---|---|---|---|---|
Martine A. Rothblatt, Ph.D., J.D., M.B.A. |
61 | Chairman, Co-Chief Executive Officer and Director | |||
Roger Jeffs, Ph.D. |
54 | President, Co-Chief Executive Officer and Director | |||
David Zaccardelli, Pharm.D. |
51 | Executive Vice President and Chief Operating Officer | |||
James C. Edgemond |
48 | Chief Financial Officer and Treasurer | |||
Paul A. Mahon, J.D. |
52 | Executive Vice President, General Counsel and Corporate Secretary | |||
Martine A. Rothblatt, Ph.D., J.D., M.B.A., founded United Therapeutics in 1996 and has served as Chairman and Chief Executive Officer since its inception. In January 2015, she became United Therapeutics' Co-Chief Executive Officer upon the promotion of Roger Jeffs to Co-Chief Executive Officer. Prior to United Therapeutics, she founded and served as Chairman and Chief Executive Officer of SiriusXM Satellite Radio. She is a co-inventor on four of our patents pertaining to treprostinil.
Roger Jeffs, Ph.D., received his undergraduate degree in chemistry from Duke University and his Ph.D. in pharmacology from the University of North Carolina. Dr. Jeffs joined United Therapeutics in September 1998 as Director of Research, Development and Medical. He was promoted to Vice President of Research, Development and Medical in 2000 and to President and Chief Operating Officer in 2001. In January 2015, Dr. Jeffs was promoted to Co-Chief Executive Officer. From 1993 to 1995, Dr. Jeffs worked at Burroughs Wellcome & Company where he was a member of the clinical research team that developed Flolan, the first FDA-approved therapy for patients with PAH. From 1995 to 1998, Dr. Jeffs worked at Amgen, Inc. where he served as the worldwide clinical leader of the Infectious Disease Program. Dr. Jeffs currently leads our global clinical, commercial, manufacturing, regulatory, medical affairs, pharmacovigilance and business development efforts.
David Zaccardelli, Pharm.D., received his doctor of pharmacy from the University of Michigan. Dr. Zaccardelli joined United Therapeutics in 2004 as Vice President, Pharmaceutical Development. He was promoted to Senior Vice President, Pharmaceutical Development in 2006, to Executive Vice President, Pharmaceutical Development in 2007, Executive Vice President, Pharmaceutical Development & Operations in April 2008 and to Chief Manufacturing Officer and Executive Vice President, Pharmaceutical Development in November 2008. In January 2015, Dr. Zaccardelli was promoted to Executive Vice President and Chief Operating Officer. From 1988 to 1996, Dr. Zaccardelli worked at Burroughs Wellcome & Company and Glaxo Wellcome, Inc. in a variety of clinical research positions. He also served as Director of Clinical and Scientific Affairs for Bausch & Lomb Pharmaceuticals, Inc. from 1996 to 1997. Dr. Zaccardelli founded and led a startup company focused on contract pharmaceutical development services from 1997 through 2003.
James C. Edgemond joined United Therapeutics in January 2013 as Treasurer and Vice President, Strategic Financial Planning. Mr. Edgemond was promoted to Chief Financial Officer and Treasurer in March 2015. Prior to joining United Therapeutics, he was Vice President, Corporate Controller and Treasurer of Clark Construction Group from November 2008 through January 2013. He also served in a variety of roles at The Corporate Executive Board Company from 1998 to 2008, including most recently as Executive Director, Finance from 2005 to 2008. He began his career as a public accountant
36
at KPMG Peat Marwick LLP, where he served in a variety of roles from 1990 through 1998, including most recently as a Senior Manager.
Paul A. Mahon, J.D., has served as General Counsel and Corporate Secretary of United Therapeutics since its inception in 1996. In June 2001, Mr. Mahon joined United Therapeutics full-time as Senior Vice President, General Counsel and Corporate Secretary. In November 2003, Mr. Mahon was promoted to Executive Vice President, General Counsel and Corporate Secretary. Prior to June 2001, he served United Therapeutics, beginning with its formation in 1996, in his capacity as principal and managing partner of a law firm specializing in technology and media law.
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of Section 21E of the Securities Exchange Act of 1934 (the Exchange Act) and the Private Securities Litigation Reform Act of 1995. These statements, which are based on our beliefs and expectations as to future outcomes, include, among others, statements relating to the following:
- •
- Expectations of revenues, expenses, profitability, and cash flows, including our expectation that Orenitram®
(treprostinil) Extended Release Tablets (Orenitram) cost of product sales as a percentage of its net product sales will become comparable to our other treprostinil-based commercial products;
- •
- The sufficiency of current and future working capital to support operations;
- •
- Our ability to obtain financing on terms favorable to us or at all;
- •
- The value of our common stock and our ability and plans repurchase common stock under our $500 million share repurchase
program, which commenced in January 2016;
- •
- The maintenance of domestic and international regulatory approvals;
- •
- Our ability to maintain attractive pricing for our products, in light of increasing competition and pressure from government and other
payers to decrease the costs associated with healthcare;
- •
- The expected volume and timing of sales of our existing commercial products—Remodulin® (treprostinil)
Injection (Remodulin), Tyvaso® (treprostinil) Inhalation Solution (Tyvaso), Orenitram, Adcirca® (tadalafil) Tablets (Adcirca) and Unituxin® (dinutuximab) Injection
(Unituxin)—and potential future commercial products such as esuberaprost;
- •
- The timing and outcome of clinical studies, other research and development efforts, and related regulatory filings and approvals,
including: (1) our plans to complete our FREEDOM-EV study of Orenitram and our BEAT study of esuberaprost and Tyvaso, and achieve a time to clinical worsening endpoint in each of these studies;
(2) our aim to obtain approval from the U.S. Food and Drug Administration (FDA) for Orenitram as a combination therapy following our FREEDOM-EV study; (3) our plan to file for approval
of Orenitram in Europe upon the successful completion of the FREEDOM-EV study; (4) our phase III clinical trial of esuberaprost in combination with Tyvaso; (5) our collaboration
with DEKA Research & Development Corp. (DEKA) to develop a pre-filled, semi-disposable pump system for subcutaneous Remodulin; and (6) pending regulatory filings by
Medtronic, Inc. (Medtronic) and us with respect to the Remodulin Implantable System, as well as the consent decree relating to Medtronic's implantable pump;
- •
- The outcome of potential future regulatory actions, including audits and inspections, by the FDA and international regulatory agencies;
37
- •
- The impact of competing therapies on sales of our commercial products, including (1) the impact of generic products such as
(a) generic sildenafil, which launched in 2012; (b) generic tadalafil, which may become available following patent expiry in November 2017; and (c) generic forms of subcutaneous
and intravenous treprostinil, which we expect two generic companies will launch in June 2018 and December 2018; and (2) newly-developed therapies, such as Uptravi® (selexipag);
- •
- The expectation that we will be able to produce sufficient quantities and maintain adequate inventories of our commercial products,
through both our in-house production capabilities and third-party production sites, and our ability to obtain and maintain related approvals by the FDA and other regulatory agencies;
- •
- The adequacy of our intellectual property protections and the validity and expiration dates of the patents we own or license;
- •
- Our expectations regarding our ability to defend our intellectual property relating to Remodulin, Tyvaso and Orenitram against generic
and other challenges, including but not limited to our ongoing litigation with Watson Laboratories, Inc. (Watson) related to Tyvaso, an Abbreviated New Drug Application (ANDA) filed by Actavis
Laboratories FL, Inc. (Actavis) seeking approval to market a generic version of Orenitram, and the petition by SteadyMed Ltd. (SteadyMed) seeking to invalidate one of our patents
covering treprostinil, which is the active ingredient in Remodulin, Tyvaso and Orenitram;
- •
- Any statements that include the words "believe," "seek," "expect," "anticipate," "forecast," "project," "intend," "estimate,"
"should," "could," "may," "will," "plan," or similar expressions;
- •
- Other statements contained or incorporated by reference in this Annual Report on Form 10-K that are not historical facts; and
- •
- The statements identified as forward-looking statements may appear in Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations or elsewhere in this Annual Report on Form 10-K.
These statements are subject to risks and uncertainties and our actual results may differ materially from anticipated results. Factors that may cause such differences include, but are not limited to, those discussed below. We undertake no obligation to publicly update forward-looking statements, whether as a result of new information, future events or otherwise.
We rely heavily on sales of Remodulin, Tyvaso, Orenitram and Adcirca to generate revenues and support our operations.
Sales of our current PAH therapies (Remodulin, Tyvaso, Orenitram and Adcirca) comprise substantially all of our revenues. A wide variety of events, many of which are described in other risk factors below, could cause sales of these products to decline, or to grow more slowly than expected. For instance, we would be unable to sell any of these products if their regulatory approvals were withdrawn. Any substantial change in the prescribing practices or dosing patterns of patients using Remodulin, Tyvaso, Orenitram or Adcirca due to combination or competing therapies, side effects, adverse events, deaths or any other reasons could decrease related revenues. We also face potential generic competition. For example, during the fourth quarter of 2012, generic sildenafil became commercially available, which could negatively affect future demand for Adcirca. We could also face generic competition for Adcirca following patent expiry in November 2017. We also settled our patent litigation with Sandoz and Teva relating to Remodulin, and have agreed that Sandoz and Teva will be permitted to launch their generic versions of Remodulin in the United States in June 2018 and December 2018,
38
respectively, although they may be permitted to launch earlier under certain circumstances. We are also defending our intellectual property related to Tyvaso and Orenitram against generic challenges by two additional generic companies, and another company has filed a petition challenging the validity of one of our patents relating to Remodulin, Tyvaso and Orenitram. In addition, we rely on third parties to produce, market, distribute and sell all of our commercial products. The inability of any one of these third parties to perform these functions satisfactorily could result in a reduction in sales. In addition, any failure to effectively manage our internal production processes could result in an inability to meet patient demand. Because we are highly dependent on sales of Remodulin, Tyvaso, Adcirca and Orenitram, a reduction in sales of any one of these products could have a material adverse impact on our operations.
If our products fail in clinical trials, we will be unable to obtain or maintain FDA and international regulatory approvals and will be unable to sell those products.
To obtain regulatory approvals from the FDA and international regulatory agencies such as the European Medicines Agency (EMA), we must conduct clinical trials demonstrating that our products are safe and effective. In the past, several of our product candidates failed or were discontinued at various stages in the development process. Moreover, we may need to amend ongoing trials or the FDA and/or international regulatory agencies may require us to perform additional trials beyond those we planned. Such occurrences could result in significant delays and additional costs, and related clinical trials may be unsuccessful. Approval of a New Drug Application (NDA) or Biologics License Application (BLA) could be subject to delays if the FDA determines that it cannot review or approve the application as submitted. In such a case, the FDA would issue a refuse-to-file letter or a complete response letter outlining deficiencies in the submission, and the FDA may require substantial additional studies, testing or information in order to complete its review of the application. We may fail to address any of these deficiencies adequately and consequently would be unable to obtain FDA approval to market the product candidate.
In addition, we are enrolling a phase IV clinical trial called FREEDOM-EV, which is a study of Orenitram in combination with other approved therapies for pulmonary arterial hypertension (PAH). One primary endpoint of the study is time to clinical worsening. The primary endpoint of our phase III study of esuberaprost in combination with Tyvaso is also time to clinical worsening. We have not previously conducted a study with time to clinical worsening as its primary endpoint. Our inexperience with this type of trial design may impact our ability to conduct these trials appropriately and achieve positive results, or complete the trials within our anticipated timetable. In particular, failure to prove the efficacy of Orenitram in combination with other PAH therapies could materially limit the commercial potential of Orenitram and impede our growth.
The length of time that it takes for us to complete clinical trials and obtain regulatory approval for marketing varies by product, product use and country. Furthermore, we cannot predict with certainty the length of time it will take to complete necessary clinical trials or obtain regulatory approval of our future products.
Our clinical trials may be discontinued, delayed or disqualified for various reasons. These reasons include:
- •
- The drug is ineffective, or physicians and/or patients believe that the drug is ineffective;
- •
- We fail to reach agreement with the FDA or non-U.S. regulatory agencies regarding the scope or design of our clinical trials;
- •
- Patients do not enroll in our studies at the rate we expect;
- •
- We are unable to obtain approval from institutional review boards to conduct clinical trials at their respective sites;
39
- •
- Ongoing or new clinical trials conducted by drug companies in addition to our own clinical trials reduce the availability of patients
for our trials;
- •
- Other investigational or approved therapies are viewed as more effective or convenient by physicians or patients;
- •
- Our clinical trial sites, contracted clinical trial administrators or clinical studies conducted entirely by third parties do not
adhere to trial protocols and required quality controls under FDA good clinical practice (GCP) regulations and similar regulations outside the United States;
- •
- Patients experience severe side effects during treatment or die during our trials because of adverse events related to the trial drug,
advanced disease, or other medical complications; and
- •
- The results of our clinical trials conducted in countries outside of the United States are not acceptable to the United States or other countries, and the results of our clinical trials conducted in the United States are not acceptable to regulators in other countries.
In addition, the FDA and its international counterparts have substantial discretion over the approval process for pharmaceutical products. As such, these regulatory agencies may not agree that we have demonstrated the requisite level of product safety and efficacy to grant approval.
We may not compete successfully with established and newly developed drugs or products, or the companies that develop and market them.
We compete with well-established drug companies for, among other things, funding, licenses, expertise, personnel, clinical trial patients and investigators, consultants and third-party collaborators. We also compete with these companies for market share. Most of these competitors have substantially greater financial, marketing, manufacturing, sales, distribution and technical resources, and a larger number of approved products, than we do. These competitors also possess greater experience in areas critical to success such as research and development, clinical trials, sales and marketing and regulatory matters. There are numerous treatments that compete with our commercial therapies, as well as several other therapies under development. For the treatment of PAH, we compete with a number of approved products in the United States and worldwide, including the following: Adempas®, Flolan®, Ilomedin®, Letairis®, Opsumit®, Revatio®, Tracleer®, Uptravi®, Veletri®, Volibris®, Ventavis®, generic epoprostenol and generic sildenafil citrate. Patients and doctors may perceive these competing products, or products developed in the future, as safer, more effective, more convenient and/or less expensive than our therapies. Alternatively, doctors may reduce the prescribed doses of our products if they prescribe them in combination with our competitors' products. In addition, many competing PAH therapies are less invasive than Remodulin and the use of these products may delay or prevent initiation of Remodulin therapy. Any of these circumstances could negatively impact our operating results.
Development of new products or technologies by others may make our products obsolete or seemingly inferior.
Other companies may introduce new products that may render all or some of our technologies and products obsolete or noncompetitive. For example, Uptravi was recently approved by the FDA for the treatment of PAH, and will compete directly with Orenitram. Our commercial therapies may also have to compete with investigational products currently in development, such as Trevyent™, which is a single-use, pre-filled pump being developed by SteadyMed to deliver a two-day supply of treprostinil subcutaneously using its PatchPump® technology. In January 2016, SteadyMed announced that Trevyent has been granted orphan drug designation by the FDA for the treatment of PAH. In addition, alternative approaches to treating chronic diseases, such as gene therapy, cell therapy or transplantation technologies, may make our products obsolete or noncompetitive. If introduced into the market, investigational therapies for PAH could be used in combination with, or as a substitute for, our therapies. If this occurs, doctors may reduce or discontinue the use of our products for their patients.
40
Sales of our products are subject to reimbursement from government agencies and other third parties. Pharmaceutical pricing and reimbursement pressures may negatively impact our sales.
The commercial success of our products depends, in part, on the availability of reimbursements by governmental payers such as Medicare and Medicaid, and private insurance companies. An estimated 35-50% of Remodulin, Tyvaso, Adcirca and Orenitram sales in the United States are reimbursed under the Medicare and Medicaid programs. In the United States, the European Union and other potentially significant markets for our products such as China and Japan, government payers and/or third-party payers are increasingly attempting to limit or regulate the price of medicinal products and frequently challenge the pricing of new and expensive drugs. Our prostacyclin analogue products (Remodulin, Tyvaso and Orenitram) and our oncology product (Unituxin) are expensive therapies. Consequently, it may be difficult for our distributors to obtain adequate reimbursement for our products from third-party payers to motivate such distributors to support our products. Alternatively, third-party payers may reduce the amount of reimbursement for our products based on changes in pricing of other therapies for the same disease. If third-party payers do not approve our products for reimbursement, or limit reimbursements, patients and physicians could choose competing products that are approved for reimbursement or provide lower out-of-pocket costs.
In the United States, the federal government and others are increasingly focused on analyzing the impact of various regulatory programs on the federal deficit, which could result in increased pressure on federal programs to reduce costs. In addition, financial pressures may cause the federal government or other third-party payers to seek cost containment more aggressively through mandatory discounts or rebates on our products, policies requiring the automatic substitution of generic products, more rigorous requirements for initial reimbursement approvals for new products or other similar measures. For example, there have been proposals to reduce reimbursement rates and/or adopt mandatory rebates under Medicare Part B, which covers Remodulin and Tyvaso. A reduction in the availability or extent of reimbursement from government health care programs could have a material adverse effect on our business and results of our operations.
In Europe, the success of our commercial products and future products depends largely on obtaining and maintaining government reimbursement at acceptable levels. In many European countries, patients are unlikely to use prescription drugs that are not reimbursed by their governments. Countries in Europe are under increasing pressure to reduce the cost of health care. Changes to current reimbursement policies may adversely affect our ability to sell our products or sell our products on a profitable basis. In many markets outside the United States, governments control the prices of prescription pharmaceuticals through the implementation of reference pricing, price cuts, rebates, revenue-related taxes and profit control. Furthermore, international governments expect prices of prescription pharmaceuticals to decline over the life of the product or as prescription volumes increase. In addition, in December 2011, we received marketing approval for the intravenous use of Remodulin in most of the countries that are members of the European Economic Area (EEA); however, we are in the process of obtaining approval of our risk management plan on a country-by-country basis, and must obtain pricing approval in each of these member countries before we can market Remodulin. Similarly, we received European Commission approval for Unituxin during the third quarter of 2015, and must obtain pricing and reimbursement approvals on a country-by-country basis before launching in individual countries in Europe. Delays in obtaining these approvals, or failure to obtain satisfactory pricing approvals, could impact our future sales growth. Additionally, in granting pricing approval for the intravenous use of Remodulin, a member country may approve a lower reimbursement price for intravenous Remodulin than for subcutaneous Remodulin, or reduce the reimbursement price for both methods of administering Remodulin. Any regulatory action reducing the reimbursement rates for intravenous and subcutaneous Remodulin could have a material adverse effect on our revenues, results of operations and our business.
41
Our production strategy exposes us to significant risks.
We must be able to produce sufficient quantities of our commercial products to satisfy the growing demand for our products. We produce Remodulin, Orenitram, Tyvaso and Unituxin, including the active ingredient in each of these products, at our own facilities and rely on third parties for additional production capacity. We rely on Minnetronix, Inc. as the sole manufacturer of the Tyvaso Inhalation System, and on Eli Lilly and Company (Lilly) as the sole manufacturer of Adcirca. In addition, if the Remodulin Implantable System is approved, we will be reliant on Medtronic as the sole manufacturer of the Synchromed II infusion system.
We substantially rely on third parties to adhere to and maintain production processes in accordance with all applicable regulatory requirements. If any of these critical third-party production and supply arrangements are interrupted for compliance issues or other reasons, we may not have sufficient inventory to meet future demand. In addition, any change in suppliers and/or service providers could interrupt the production of our commercial products and impede the progress of our commercial launch plans and clinical trials.
In addition, our internal production process also subjects us to risks as we engage in increasingly complex production processes. For example, Remodulin, Tyvaso and Unituxin must be formulated in a sterile environment, which is challenging to maintain on a commercial scale. In addition, Unituxin is a monoclonal antibody. As with all biologic products, monoclonal antibodies are inherently more difficult to produce than our treprostinil-based products and involve increased risk of viral and other contaminants. Finally, we have limited experience producing Orenitram and Unituxin on a commercial scale, and currently all Orenitram and Unituxin production is performed internally. It could take substantial time to establish an FDA-approved contract manufacturer as a back-up supplier of our newest products, Orenitram and Unituxin, or this process may not be successful at all.
Additional risks we face with our production strategy include the following:
- •
- We and our third-party producers are subject to the FDA's current Good Manufacturing Practices, current Good Tissue Practices and
similar international regulatory standards. We are limited in our ability to exercise control over regulatory compliance by our third-party producers;
- •
- As we expand our production operations to include new elements of the production process or new products, we may experience difficulty
designing and implementing processes and procedures to ensure compliance with applicable regulations;
- •
- Even if we and our third-party producers are in compliance with applicable domestic and international drug production regulations, the
sterility and quality of the products being produced could be substandard and, therefore, such products would be unavailable for sale or use or subject to recalls;
- •
- If we had to replace our own production operations or a third-party producer, the FDA and its international counterparts would require
new testing and compliance inspections. Furthermore, a new producer would have to be familiarized with the processes necessary to produce and commercially validate our products, as producing our
treprostinil-based and biologic products is complex;
- •
- We may be unable to contract with needed producers on satisfactory terms or at all; and
- •
- The supply of materials and components necessary to produce and package our products may become scarce or unavailable. Disruptions to the supply of these materials could delay the production and subsequent sale of such products. Any products produced with substituted materials or components would be subject to approval from the FDA and international regulatory agencies before they could be sold. The timing of any such regulatory approval is difficult to predict.
42
Any of these factors could disrupt sales of our commercial products, delay clinical trials or commercialization of new products, result in product liability claims and product recalls, and entail higher costs. Interruptions in our production process could be significant given the length of time and complexity involved in obtaining necessary regulatory approvals for alternative arrangements, through either third parties or internal manufacturing processes.
We rely in part on third parties to perform activities that are critical to our business. Our ability to generate commercial sales or conduct clinical trials could suffer if our third-party suppliers and service providers fail to perform.
Third parties assist us in: (1) producing our commercial products; (2) conducting clinical trials, preclinical studies and other research and development activities; (3) obtaining regulatory approvals; (4) conducting pharmacovigilance-related and product complaint activities, including drug safety, reporting adverse events and product complaints; and (5) marketing and distributing our products. In addition, we rely on independent third party manufacturers for the availability of pumps and ancillary supplies necessary for the delivery of subcutaneous and intravenous Remodulin, and in most cases we have no contracts with these manufacturers. The involvement of third parties is necessary because we do not possess the internal capacity, and in certain cases the expertise, to perform all of these functions. Accordingly, the success of these third parties in performing their contractual obligations is critical to our operations.
For risks relating to the involvement of third parties in our production process, see the risk factor above, entitled Our production strategy exposes us to significant risks.
We rely on Accredo Health Group, Inc. and CVS Caremark to distribute and sell Remodulin, Tyvaso and Orenitram in the United States. These distributors are also partially responsible for negotiating reimbursements from third-party payers for the cost of our therapies. We also rely on ASD Specialty Healthcare, Inc. to distribute and sell Unituxin in the United States. We also rely on various distributors to market, distribute and sell Remodulin and Unituxin outside the United States. From time-to-time, we increase the price of products sold to our U.S.-based and international distributors. Our price increases may not be fully reimbursed by third-party payers. If our distributors do not achieve acceptable profit margins on our products, they may reduce or discontinue the sale of our products. Furthermore, if our distributors devote fewer resources to sell our products or are unsuccessful in their sales efforts, our revenues may decline materially. Outside the United States, we are substantially reliant on our international distributors to maintain regulatory approvals for our products and to market and sell our products in compliance with applicable laws and regulations.
We rely on Lilly to manufacture and supply Adcirca for us, and we use Lilly's pharmaceutical wholesaler network to distribute Adcirca. If Lilly is unable to manufacture or supply Adcirca or its distribution network is disrupted, it could delay, disrupt or prevent us from selling Adcirca. In addition, Lilly has the right to determine the price of Adcirca, which generally moves in parity with the price Lilly sets for Cialis® (both of these products contain the same active ingredient). Changes in Lilly's prices could adversely impact demand or reimbursement for Adcirca, particularly in light of the commercial availability of generic sildenafil, the active ingredient in Revatio, which could be prescribed in lieu of Adcirca.
In addition, any change in service providers could interrupt the distribution of our commercial products and our other products and services, and impede the progress of our clinical trials, commercial launch plans and related revenues.
We rely heavily on third-party contract research organizations, contract laboratories, clinical investigative sites and other third-parties to conduct our clinical trials, preclinical studies and other research and development activities. In addition, the success of certain products we are developing will depend on clinical trials sponsored by third parties. Failure by any third party to conduct or assist us in
43
conducting clinical trials in accordance with study protocols, quality controls and GCP, or other applicable U.S. or international requirements or to submit associated regulatory filings, could limit or prevent our ability to rely on results of those trials in seeking regulatory approvals.
We rely heavily on Medtronic for the success of our program to develop an implantable pump to deliver intravenous Remodulin (the Remodulin Implantable System). Medtronic has completed a clinical study in this regard, and submitted a Premarket Approval Application (PMA) seeking FDA approval for the Remodulin Implantable System. We rely on Medtronic to respond to FDA requests for additional information with respect to its PMA, and following approval we will rely on Medtronic to manufacture the Remodulin Implantable System and to maintain appropriate quality controls relating to the system. We also note that Medtronic has received a consent decree requiring the company to stop manufacturing, designing and distributing SynchroMed II implantable infusion pump systems, except in limited circumstances, citing violations of the quality system regulation for medical devices. The consent decree will remain in effect until the FDA has determined that Medtronic has met all the provisions listed in the consent decree. It is unclear how this consent decree will impact our program to develop and commercialize the Remodulin Implantable System, and we anticipate further clarity in 2016 when we anticipate FDA will respond to Medtronic's PMA filing. As such, we can provide no assurances as to the timing or likelihood of the Remodulin implantable pump program's success. Similarly, we rely heavily on DEKA for the development of a pre-filled, semi-disposable pump system for subcutaneous Remodulin.
We are reliant on third parties to supply pumps and other supplies necessary to deliver Remodulin. There are a limited number of pumps available in the market, and the discontinuation of any particular pump could have a material, adverse impact on our Remodulin revenues if a viable supply of an alternate pump is not available.
Our operations must comply with extensive laws and regulations in the United States and other countries, including FDA regulations. Failure to obtain approvals on a timely basis or to achieve continued compliance could delay, disrupt or prevent the commercialization of our products.
The products we develop must be approved for marketing and sale by regulatory agencies and, once approved, are subject to extensive regulation. Our research and development efforts must comply with extensive regulations, including those promulgated by the FDA and the United States Department of Agriculture. The process of obtaining and maintaining regulatory approvals for new drugs is lengthy, expensive and uncertain. The regulatory approval process is particularly uncertain for our lung transplantation programs, which include the development of xenotransplantation, regenerative medicine and cell-based products. The manufacture, distribution, advertising and marketing of our products are also subject to extensive regulation, including strict pharmacovigilance and adverse event and medical device reporting requirements. Any future product approvals we receive could be accompanied by significant restrictions on the use or marketing of a given product. Furthermore, our product candidates may fail to receive marketing approval on a timely basis, or at all. If granted, product approvals can be withdrawn for failure to comply with regulatory requirements, such as post-marketing requirements and post-marketing commitments, or upon the occurrence of adverse events subsequent to commercial introduction.
Discovery of previously unknown problems with our marketed products or problems with our manufacturing, regulatory, compliance, research and development, pharmacovigilance and adverse event reporting, marketing or sales activities could result in regulatory restrictions on our products up to and including withdrawal of our products from the market. If we fail to comply with applicable regulatory requirements, we could be subject to penalties that may consist of fines, suspension of regulatory approvals, product recalls, seizure of our products and/or criminal prosecution. In addition, our reputation could be harmed as a result of any such regulatory restrictions or actions, and patients and
44
physicians may avoid the use of our products even after we have resolved the issues that led to such regulatory action.
We are subject to ongoing regulatory review of our currently marketed products.
After our products receive regulatory approval, they remain subject to ongoing regulatory requirements, which can impact, among other things, product labeling, manufacturing practices, pharmacovigilance and adverse event and medical device reporting, complaint processing, storage, distribution, advertising and promotion, and record keeping. If we do not comply with applicable regulations, the range of possible sanctions may include: (1) adverse publicity; (2) product recalls or seizures; (3) fines; (4) total or partial suspensions of production and/or distribution; (5) suspension of marketing applications; and (6) enforcement actions, including injunctions and civil suits or criminal prosecution. Further, the FDA often requires post-marketing testing and surveillance to monitor the effects of approved products. The FDA and comparable international regulatory agencies may condition approval of our product candidates on the completion of such post-marketing clinical studies. These post-marketing studies may suggest that a product causes undesirable side effects or may present a risk to the patient. If data we collect from post-marketing studies suggest that one of our approved products may present an unacceptable safety risk, regulatory authorities could withdraw the product's approval, suspend production or place other marketing restrictions on that product. If regulatory sanctions are applied or if regulatory approval is delayed or withdrawn, our operating results and the value of our company may be adversely affected.
Regulatory approval for our currently marketed products is limited by the FDA and other regulators to those specific indications and conditions for which clinical safety and efficacy have been demonstrated.
Any regulatory approval of our products is limited to specific diseases and indications for which our products have been deemed safe and effective by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. If we are not able to obtain FDA approval for any desired future indications for our products, our ability to effectively market and sell our products may be reduced.
While physicians may choose to prescribe drugs for uses that are not described in the product's labeling and for uses that differ from those approved by regulatory authorities (called "off-label" uses), our ability to promote the products is limited to those indications that are specifically approved by the FDA. Although U.S. regulatory authorities generally do not regulate the behavior of physicians, they do restrict communications by companies on the subject of off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, failure to follow FDA rules and guidelines relating to promotion and advertising can result in the FDA's refusal to approve a product, suspension or withdrawal of an approved product from the market, product recalls, fines, disgorgement of money, operating restrictions, civil lawsuits, injunctions or criminal prosecution.
We must comply with various laws in jurisdictions around the world that restrict certain marketing practices in the pharmaceutical and medical device industries. Failure to comply with such laws could result in penalties and have a material adverse effect on our business, financial condition and results of operations.
There are various laws in jurisdictions around the world that restrict particular marketing practices in the pharmaceutical and medical device industries. These laws include, but are not limited to, anti-kickback and false claims statutes, the Foreign Corrupt Practices Act and the UK Bribery Act. Our business activities may be subject to challenge under these laws, and any penalties imposed upon us could have a material adverse effect on our business and financial condition. Furthermore, we have significantly expanded our sales and marketing staff. Any expansion of sales and marketing efforts can increase the risks of noncompliance with these laws. Finally, the growth in our operations outside the United States, both directly and through third-party distributors, also has increased these risks.
45
In the United States, the federal health care program anti-kickback statute prohibits, among other activities, knowingly and willfully offering, paying, soliciting, or receiving compensation to induce, or in return for, the purchase, lease, order or arranging the purchase, lease or order of any health care product or service reimbursable under any federally financed health-care program. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers on the other. The exemptions and safe harbors for this statute are narrow, and practices that involve compensation intended to induce prescriptions, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not always meet all of the criteria for safe harbor protection.
The federal False Claims Act prohibits any person from knowingly presenting or causing to be presented a false claim or knowingly making or causing a false statement material to a false claim. Several pharmaceutical and health care companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the free product. Other companies have been prosecuted for causing false claims to be submitted because of these companies' marketing of a product for unapproved and non-reimbursable uses. Potential liability under the federal False Claims Act includes mandatory treble damages and significant per-claim penalties, currently set at $5,500 to $11,000 per false claim. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs; furthermore, in several states, these statutes and regulations apply regardless of the payer. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer's product from reimbursement under government programs, debarment, criminal fines, and imprisonment.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (PPACA), also imposed new reporting requirements for pharmaceutical, biologic and device manufacturers with regard to payments or other transfers of value made to physicians and teaching hospitals. In addition, pharmaceutical, biologic and device manufacturers, with certain exceptions, are required to report and disclose investment interests held by physicians and their immediate family members during the preceding calendar year. Failure to submit required information may result in civil monetary penalties of up to $150,000 per year (and up to $1.0 million per year for "knowing failures") for all payments, transfers of value or ownership or investment interests not reported in an annual submission.
Further, the PPACA amends the intent requirement of the federal anti-kickback and criminal health care fraud statutes. This amendment provides that a person or entity no longer needs to have knowledge of these statutes or specific intent to violate them. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
If not preempted by this federal law, several states currently require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual physicians in those states. Depending on the state, legislation may prohibit various other marketing related activities, or require the posting of information relating to clinical studies and their outcomes. In addition, certain states, such as California, Nevada, Connecticut and Massachusetts, require pharmaceutical companies to implement compliance programs or marketing codes and several other states are considering similar proposals. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws will face civil penalties.
46
Government health care reform could increase our costs, which would adversely affect our revenue and results of operations.
Our industry is highly regulated and changes in law may adversely impact our business, operations or financial results. The PPACA is a broad measure intended to expand health care coverage within the United States, primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. The reforms imposed by the law will significantly impact the pharmaceutical industry; however, the full effects of the PPACA will be unknown until all of these provisions are implemented and the Centers for Medicare and Medicaid Services and other federal and state agencies issue applicable regulations or guidance. Moreover, in the coming years, additional changes could be made to governmental health care programs that could significantly impact the success of our products or product candidates.
Reports of actual or perceived side effects and adverse events associated with our products, such as sepsis, could cause physicians and patients to avoid or discontinue use of our products in favor of alternative treatments.
Reports of side effects and adverse events associated with our products could have a significant adverse impact on the sale of our products. An example of a known risk associated with intravenous Remodulin is sepsis, which is a serious and potentially life-threatening infection of the bloodstream caused by a wide variety of bacteria. Intravenous prostacyclin analogues, such as intravenous Remodulin, are infused continuously through a catheter placed in a large vein in the patient's chest, and sepsis is a known risk associated with this type of delivery. As a result, sepsis is included as a risk in the Remodulin package insert, and the occurrence of sepsis is familiar to physicians who prescribe intravenously administered therapies. Concerns about bloodstream infections may affect a physician's decision to prescribe or a patient's willingness to use intravenous Remodulin.
Negative attention from special interest groups may impair our business.
As is common with pharmaceutical and biotechnology companies, our early-stage research and development involves animal testing, which we conduct both directly and through contracts with third parties. Notwithstanding the vital role of animal research in the drug discovery and development process, certain special interest groups categorically object to the use of animals for research purposes. Historically, our research and development activities have not been the subject of significant animal rights media attention. However, research activities with animals have been the subject of adverse attention, generally including demonstrations near facilities operated by other companies in our industry. Any negative attention, threats or acts of vandalism directed against our animal research activities in the future could impede the operation of our business.
If any of the license or other agreements under which intellectual property rights are licensed to, or were acquired by us, are breached or terminated, our right to continue to develop, produce and sell the products covered by such agreements could be impaired or lost.
Our business depends upon our continuing ability to exploit our intellectual property rights in the drugs and other products that have been discovered and initially developed by others and those which we have commercialized and are developing further. These intellectual property rights have either been licensed to us or have been acquired by us. Under each of our product license agreements, we are granted a license to intellectual property owned by others that covers a drug or other product. Under each of our purchase agreements, we have rights to certain intellectual property. We may be required to license other intellectual property owned by third parties to continue to develop and commercialize our products.
47
This dependence on intellectual property developed by others involves the following risks:
- •
- We may be unable to obtain rights to intellectual property that we determine we need for our business at a reasonable cost or at all;
- •
- If any of our product licenses or purchase agreements are terminated, we may lose our rights to develop, make and sell the products to
which such licenses or agreements relate;
- •
- Our license and purchase agreements generally provide the licensor or seller with the right to terminate the agreement in the event of
a breach; for example, if we fail to pay royalties and other fees timely and do not cure the failure within a stated time period; and
- •
- If a licensor of intellectual property that we have rights to breaches its obligation or otherwise fails to maintain the intellectual property licensed, we may lose any ability to prevent others from developing or marketing similar products that are covered by such intellectual property. In addition, we may be forced to incur substantial costs to maintain the intellectual property ourselves or take legal action seeking to force the licensor to do so.
Certain agreements under which we acquired or licensed intellectual property rights may restrict our ability to develop related products in certain countries or for particular diseases and may impose other restrictions that affect our ability to develop and market related products in the most effective manner.
When we acquire or license intellectual property rights to drugs and other products that have been discovered and initially developed by others, these rights are frequently limited. For instance, our rights to market Adcirca are geographically limited to the United States. Furthermore, we cannot undertake any additional investigational work with respect to Adcirca in other indications of pulmonary hypertension without Lilly's prior approval. Provisions in our license and purchase agreements may impose other restrictions that affect our ability to develop and market products to which the intellectual property relates. For example, Lilly also has authority over all regulatory activities relating to Adcirca and has the right to determine the price at which we sell the drug.
Our intellectual property rights may not effectively deter competitors from developing competing products that, if successful, could have a material adverse effect on our revenues and profits.
The period under which our commercial and developmental therapies are protected by our patent rights is limited. Three of our U.S. patents covering our current methods of synthesizing and producing treprostinil, the active ingredient in Remodulin, Tyvaso and Orenitram, expire in October 2017, and a fourth will expire in 2028. We recently settled patent litigation with Sandoz and Teva, which will permit them to launch generic versions of Remodulin in the United States in June 2018 and December 2018, respectively, although they may be permitted to enter the market earlier under certain circumstances. We also have been granted one patent in the European Union and one patent in Japan, each of which covers our treprostinil synthesis and production methods and will expire in October 2018. Our three U.S. patents covering an improved diluent for Remodulin will expire in 2028 and 2029. Our U.S. patent covering intravenous administration of Remodulin with certain diluents expires in 2024. Our patents for Tyvaso covering methods of treating PAH by inhaled delivery will expire in the United States and in various countries throughout the world in 2018 and 2020, respectively. Our patents for Orenitram covering methods of use for treating PAH, orally administered formulations, controlled moisture storage and production methods and controlled release formulations will expire in the United States between 2024 and 2031 and in various countries throughout the world in 2024. The U.S. patent for Adcirca for the treatment of pulmonary hypertension will expire in November 2017.
We continue to conduct research into new methods to synthesize treprostinil and have pending U.S. and international patent applications and patents relating to such methods. However, we cannot be sure that these additional patents will effectively deter or delay competitors' efforts to bring new
48
products to market, or that additional patent applications will result in new patents. Upon the expiration of any of our patents, competitors may develop generic versions of our products and may market those generic versions at a lower price to compete with our products. Competitors may also seek to design around our patents prior to their expiration in an effort to develop competing products that do not infringe our patents. Prior to the expiration of our patents, third parties may challenge the validity of our patents, through patent litigation, proceedings before the U.S. Patent and Trademark Office or other applicable patent filing office, or other means.
The scope of any patent we hold may not deter competitors from developing a product that competes with the product we sell that is covered by the patent. Patent laws of foreign jurisdictions may not protect our patent rights to the same extent as the patent laws of the United States. In addition, we may be forced to incur substantial costs to defend the intellectual property rights conferred by our patents. Furthermore, our suppliers who have granted us exclusive rights may have inadequate intellectual property protections. Competitors also may attempt to invalidate our existing patents before they expire.
In addition to patent protection, we also rely on trade secrets to protect our proprietary know-how and other technological advances that we do not disclose to the public. We enter into confidentiality agreements with our employees and others to whom we disclose trade secrets and other confidential information. These agreements may not necessarily prevent our trade secrets from being used or disclosed without our authorization and confidentiality agreements may be difficult, time-consuming and expensive to enforce or may not provide an adequate remedy in the event of unauthorized disclosure. In addition, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such third party, or those to whom they communicate such technology or information, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our business and competitive position could be harmed.
The validity, enforceability and scope of certain of our patents covering Remodulin, Tyvaso and Orenitram are currently being challenged as a result of ANDA filings by generic drug companies and a petition for inter partes review. The outcome of current or future challenges with respect to the validity, enforceability, or scope of our patents could significantly reduce revenues from Remodulin, Tyvaso and Orenitram.
Both Sandoz and Teva filed ANDAs seeking FDA approval to market generic versions of Remodulin, and Watson has filed an ANDA seeking FDA approval to market a generic version of Tyvaso. We recently settled our litigation with Sandoz and Teva, which will permit them to launch their generic Remodulin products in the United States in June 2018 and December 2018, respectively, although they may be permitted to enter the market earlier under certain circumstances, and we have filed a lawsuit against Watson in the U.S. District Court for the District of New Jersey alleging patent infringement. In addition, in October 2015, SteadyMed filed a petition for inter partes review with the Patent Trial and Appeal Board of the United States Patent and Trademark Office seeking to invalidate one of our patents covering a method of making treprostinil that expires in 2028 and is listed in the Orange Book for Remodulin, Tyvaso, and Orenitram. In February 2016, we were notified that Actavis has filed an ANDA seeking FDA approval to market a generic version of Orenitram. For details on the status of these matters, please see Note 19—Litigation, to our consolidated financial statements, Item 3—Legal Proceedings and Part II, Item 9B—Other Information.
We may not prevail in our defense of our patent rights, and additional challenges from other ANDA filers or other competitors may surface with respect to Remodulin, Tyvaso and Orenitram. Our existing patents could be invalidated, found unenforceable or found not to cover one or more generic forms of Remodulin, Tyvaso or Orenitram. If any ANDA filer were to receive approval to sell a generic version of Remodulin, Tyvaso or Orenitram and/or prevail in any patent litigation, the affected product would become subject to increased competition and our revenue would decrease.
49
Third parties may allege that our patents are invalid, or that our products or services infringe their patents and other intellectual property rights, which could result in the payment of royalties. Payment of royalties would negatively affect our profits; furthermore, if we chose to contest these allegations, we could be subject to costly and time-consuming litigation or could lose the ability to continue to sell the related products.
Third parties may seek to invalidate or otherwise challenge our patents, through patent litigation and/or initiating proceedings, including re-examinations, inter partes reviews, post-grant reviews and interference proceedings, before the U.S. Patent and Trademark Office. We may initiate litigation to enforce or defend our patents or intellectual property rights; however, litigation can be time consuming, distracting to our operations, costly and may conclude unfavorably for us. In addition, the outcome of patent infringement litigation often is difficult to predict. If we are unsuccessful with respect to any future legal action in the defense of our patents and our patents are invalidated or determined to be unenforceable, our business could be negatively impacted. Even if our patents are determined to be valid or enforceable, it is possible that a competitor could circumvent our patents by effectively designing around the claims of our patents. Accordingly, our patents may not provide us with any competitive advantage.
To the extent third-party patents to which we currently do not hold licenses are necessary for us to manufacture, use or sell our products, we would need to obtain necessary licenses to prevent infringement. In the case of products or services that utilize intellectual property of strategic collaborators or other suppliers, such suppliers may have an obligation to secure the needed license to these patents at their cost. Otherwise, we would be responsible for the cost of these licenses. Royalty payments and other fees under these licenses would erode our profits from the sale of related products and services. Moreover, we may be unable to obtain these licenses on acceptable terms or at all. If we fail to obtain a required license or are unable to alter the design of the product to avoid infringing a third-party patent, we would be unable to continue to manufacture or sell related products.
If a third party commences legal action against us for infringement, or institutes proceedings challenging the validity of our patents, we could be compelled to incur significant costs to defend the action and our management's attention could be diverted, whether or not the action were to have any merit. We cannot be certain that we could prevail in the action, and an adverse judgment or settlement resulting from the action could require us to pay substantial amounts in damages for infringement or substantial amounts to obtain a license to continue to use the intellectual property that is the subject of the infringement claim.
We may not maintain adequate insurance coverage to protect us against significant product liability claims.
The testing, manufacturing, marketing, and sale of drugs and diagnostics involve product liability risks. We may not be able to maintain our current product liability insurance at an acceptable cost, if at all. In addition, our insurance coverage may not be adequate for all potential claims. If claims or losses significantly exceed our liability insurance coverage, we may experience financial hardship or potentially be forced out of business.
If we fail to attract and retain key management and qualified scientific and technical personnel, we may not be able to achieve our business objectives.
Members of our management team, including our founder, Chairman and Co-Chief Executive Officer, Dr. Martine Rothblatt, and our President and Co-Chief Executive Officer, Dr. Roger Jeffs, play a critical role in defining our business strategy and maintaining our corporate culture. The loss of the services and leadership of Dr. Rothblatt, Dr. Jeffs or any other members of our senior management team could have an adverse effect on our business. We do not maintain key person life insurance on our senior management team members. In addition, effective succession planning is important to our long-term success. Failure to identify, hire and retain suitable successors for members of our senior
50
management team and to transfer knowledge effectively could impede the achievement of our business objectives. Our future success also depends on our ability to attract and retain qualified scientific and technical personnel. Competition for skilled scientific and technical personnel in the biotechnology and pharmaceutical industries is intense. Furthermore, our compensation arrangements may not be sufficient to attract new qualified scientific and technical employees or retain such core employees. If we fail to attract and retain such employees, we may not be successful in developing and commercializing new therapies for PAH and other diseases.
Improper handling of hazardous materials used in our activities could expose us to significant remediation liabilities.
Our research and development and manufacturing activities involve the controlled use of chemicals and hazardous substances and we are expanding these activities in both scale and location. In addition, patients may dispose of our products using means we do not control. Such activities subject us to numerous federal, state, and local environmental and safety laws and regulations that govern the management, storage and disposal of hazardous materials. Compliance with current and future environmental laws and regulations can require significant costs; furthermore, we can be subject to substantial fines and penalties in the event of noncompliance. The risk of accidental contamination or injury from these materials cannot be completely eliminated. Furthermore, once chemical and hazardous materials leave our facilities, we cannot control the manner in which such hazardous waste is disposed of by our contractors. In the event of an accident, we could be liable for substantial civil damages or costs associated with the cleanup of the release of hazardous materials. Any related liability could have a material adverse effect on our business.
We may encounter substantial difficulties managing our growth relative to product demand.
We have spent considerable resources building and expanding our offices, laboratories and production facilities. However, our facilities could be insufficient to meet future demand for our products. Conversely, we may have excess capacity at our facilities if future demand falls short of our projections, or if we do not receive regulatory approvals for the products we intend to produce at our facilities. Constructing our facilities is expensive and our ability to satisfactorily recover our investment will depend on sales of the products manufactured at these facilities in sufficient volume. If we do experience substantial sales growth, we may have difficulty managing inventory levels as marketing new therapies is complicated and gauging future demand can be difficult and uncertain until we possess sufficient post-launch sales experience.
If we need additional financing and cannot obtain it, our product development and sales efforts may be limited.
In January 2016, we entered into a Credit Agreement (the 2016 Credit Agreement) with Wells Fargo Bank, National Association (Wells Fargo), as administrative agent and a swingline lender, and various other lender parties, providing for an unsecured revolving credit facility of up to $1.0 billion (the Revolving Facility). The Revolving Facility will mature five years after the closing date of the 2016 Credit Agreement, subject to the lenders' ability to extend the maturity date by one year if we request such an extension in accordance with the terms of the 2016 Credit Agreement.
Notwithstanding the 2016 Credit Agreement, we may be required to seek additional sources of financing to meet unplanned or planned expenditures. Unplanned expenditures could be significant and may result from necessary modifications to product development plans or product offerings in response to difficulties encountered with clinical trials. We may also face unexpected costs in preparing products for commercial sale, or in maintaining sales levels of our currently marketed therapeutic products. In addition, the 2016 Credit Agreement contains affirmative and negative covenants that, among other things, limit our ability to incur additional indebtedness. If we are unable to obtain additional funding
51
on commercially reasonable terms or at all, we may be compelled to delay clinical studies, curtail operations or obtain funds through collaborative arrangements that may require us to relinquish rights to certain products or potential markets.
We may require additional financing to meet significant future obligations. For example, awards granted under our Share Tracking Award Plans (which we collectively refer to as the STAP) entitle participants to receive in cash an amount equal to the appreciation in the price of our common stock, which is calculated as the positive difference between the closing price of our common stock on the date of exercise and the date of grant. Consequently, our STAP may require significant future cash payments to participants to the extent the price of our common stock appreciates and the number of vested STAP awards increases over time. If we do not have sufficient funds to meet such obligations or the ability to secure alternative sources of financing, we could be in default, face litigation and/or lose key employees, which could have a material adverse effect on our business.
We may not be able to generate sufficient cash to service our indebtedness, which may have a material adverse effect on our financial position, results of operations and cash flows. In addition, we may be forced to take other actions to satisfy our obligations in connection with our indebtedness, which actions may not be successful.
We may borrow up to $1.0 billion under the 2016 Credit Agreement. Our ability to make payments on or refinance our debt obligations, including any future debt that we may incur, will depend on our financial condition and operating performance, which are subject to prevailing economic and competitive conditions and to certain financial, business, legislative, regulatory and other factors beyond our control. We may be unable to maintain a level of cash flows from operating activities sufficient to permit us to pay the principal, premium, if any, and interest on our indebtedness. Our inability to generate sufficient cash flows to satisfy our debt obligations would materially and adversely affect our financial position and results of operations.
If we cannot repay or refinance our debt as it becomes due, we could be forced to take disadvantageous actions, including reducing or delaying investments and capital expenditures, disposing of material assets or operations, seeking additional debt or equity capital or restructuring or refinancing our indebtedness. We may not be able to effect any such alternative measures, if necessary, on commercially reasonable terms or at all and, even if successful, such actions may not be sufficient for us to meet any such debt service obligations. In addition, our ability to withstand competitive pressures and to react to changes in our industry could be impaired.
In addition, the 2016 Credit Agreement contains restrictive covenants that limit our ability to take certain actions including, among other things, our ability to incur additional indebtedness, grant liens, merge or consolidate; liquidate, wind up or dissolve; or sell all or substantially all of our assets. Our failure to comply with the covenants in the 2016 Credit Agreement could result in an event of default which, if not cured or waived, could result in the acceleration of all amounts due under the 2016 Credit Agreement.
Information technology security breaches and other disruptions could compromise our information and expose us to legal responsibility which would cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our suppliers, customers and business partners, and personally identifiable information. The secure maintenance of this information is critical to our operations and business strategy. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Such breaches could compromise sensitive and confidential information stored on our networks and expose such information to public disclosure, loss or theft. Any access, disclosure or
52
other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, disruption of our operations, and damage to our reputation which could adversely affect our business.
Risks Related to Our Common Stock
The price of our common stock can be highly volatile and may decline.
The price of common stock can be highly volatile within the pharmaceutical and biotechnology sector. Consequently, there can be significant price and volume fluctuations in the market that may not relate to operating performance. The following table sets forth the high and low closing prices of our common stock for the periods indicated:
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
January 1, 2015 - December 31, 2015 |
$ | 188.56 | $ | 119.57 | |||
January 1, 2014 - December 31, 2014 |
$ | 136.16 | $ | 86.14 | |||
January 1, 2013 - December 31, 2013 |
$ | 114.51 | $ | 51.64 | |||
The price of our common stock could decline sharply due to the following factors, among others:
- •
- Failure to meet our estimates or expectations, or those of securities analysts;
- •
- Quarterly and annual financial results;
- •
- Timing of enrollment and results of our clinical trials;
- •
- Announcements by us or others regarding generic or other challenges to the intellectual property relating to our products, including
developments with respect to the ANDA filed by a generic drug company relating to certain of our Tyvaso patents and to our pending lawsuit defending our patent rights, and the inter partes review
petition filed by SteadyMed challenging the validity of one of the patents listed in the Orange Book for Remodulin, Tyvaso and
Orenitram;
- •
- Physician, patient, investor or public concerns regarding the efficacy and/or safety of products marketed or being developed by us or
by others;
- •
- Changes in, or new legislation and regulations affecting reimbursement of, our therapeutic products by Medicare, Medicaid or other
government payers, and changes in reimbursement policies of private health insurance companies, and negative publicity surrounding the cost of high-priced therapies;
- •
- Announcements by us or others of technological innovations or new products or announcements regarding our existing products, including
in particular the development of new, competing PAH therapies;
- •
- Substantial sales of our common stock by us or our existing shareholders;
- •
- Future issuances of common stock by us or any other activity which could be viewed as being dilutive to our shareholders;
- •
- Rumors among, or incorrect statements by, investors and/or analysts concerning our company, our products, or our operations;
- •
- Failure to obtain or maintain regulatory approvals from the FDA or international regulatory agencies;
53
- •
- Discovery of previously unknown problems with our marketed products, or problems with our production, regulatory, compliance,
promotional, marketing or sales activities that result in regulatory penalties or restrictions on our products, up to the withdrawal of our products from the market;
- •
- Accumulation of significant short positions in our common stock by hedge funds or other investors or the significant accumulation of
our common stock by hedge funds or other institutional investors with investment strategies that may lead to short-term holdings; and
- •
- General market conditions.
We may fail to meet third-party projections for our revenues or profits.
Many securities analysts publish quarterly and annual projections of our revenues and profits. Such projections are inherently subject to uncertainty. As a result, actual revenues and profits may fail to meet these projections. Even minor variations in reported revenues and profits compared to securities analysts' expectations could have a significant adverse impact on the price of our common stock.
Sales or issuances of our common stock may depress our stock price.
The price of our common stock could decline if: (1) we issue common stock to raise capital or to acquire a license or business; (2) our shareholders transfer ownership of our common stock, or sell substantial amounts of our common stock in the public market; or (3) our investors become concerned that substantial sales of our common stock may occur. A decrease in the price of our common stock could make it difficult for us to raise capital or fund acquisitions through the issuance of our stock.
Our share repurchases may affect the value of our common stock.
In recent years, our Board of Directors has authorized several programs to repurchase our common stock, including a $500.0 million share repurchase program effective during the one-year period commencing January 1, 2016. The price of our common stock may, in part, reflect expectations that we will use all of the funds authorized under our repurchase program to repurchase shares or that additional repurchase programs will be authorized once the current program terminates. Our current share repurchase program does not obligate us to acquire any specific number of shares and any further repurchase programs are subject to the approval of our Board of Directors. If we fail to meet analyst or investor expectations regarding repurchase programs, our stock price may decline.
Provisions of Delaware law and our amended and restated certificate of incorporation, fourth amended and restated by-laws, shareholder rights plan and employment and license agreements, among other things, could prevent or delay a change of control or change in management that may be beneficial to our public shareholders.
Certain provisions of Delaware law and our amended and restated certificate of incorporation, fourth amended and restated by-laws and shareholder rights plan may prevent, delay or discourage:
- •
- A merger, tender offer or proxy contest;
- •
- The assumption of control by a holder of a large block of our securities; and/or
- •
- The replacement or removal of current management by our shareholders.
For example, our amended and restated certificate of incorporation divides our Board of Directors into three classes. Members of each class are elected for staggered three-year terms. This provision may make it more difficult for shareholders to replace the majority of directors. It may also deter the accumulation of large blocks of our common stock by limiting the voting power of such blocks.
54
Non-competition and all other restrictive covenants in most of our employment agreements will terminate upon a change of control that is not approved by our Board.
Similarly, a change of control, under certain circumstances, could also result in an acceleration of the vesting of outstanding STAP awards and stock options. This, together with any increase in our stock price resulting from the announcement of a change of control, could make an acquisition of our company significantly more expensive to the purchaser. We also have a broad-based change of control severance program, under which employees may be entitled to severance benefits in the event they are terminated without cause (or they terminate their employment for good reason) following a change of control. This program could also increase the cost of acquiring our company.
We enter into certain license agreements that generally prohibit our counterparties or their affiliates from taking necessary steps to acquire or merge with us, directly or indirectly throughout the term of these agreements, plus a specified period thereafter. We are also party to certain license agreements that restrict our ability to assign or transfer the rights licensed to us to third parties, including parties with whom we wish to merge, or those attempting to acquire us. These agreements often require that we obtain prior consent of the counterparties to these agreements if we are contemplating a change of control. If these counterparties withhold consent, related agreements could be terminated and we would lose related license rights. For example, both Lilly and Toray have the right to terminate our license agreements relating to Adcirca and esuberaprost, respectively, in the event of certain change of control transactions. These restrictive change of control provisions could impede or prevent mergers or other transactions that could benefit our shareholders.
Because we do not intend to pay cash dividends, our shareholders must rely on stock appreciation for any return on their investment in us.
We have never declared or paid cash dividends on our common stock. Furthermore, we do not intend to pay cash dividends in the future and our 2016 Credit Facility contains covenants that may restrict us from doing so. As a result, the return on an investment in our common stock will depend entirely upon the future appreciation in the price of our common stock. There can be no assurances that our common stock will provide a return to investors.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Maryland—We own a 232,000 square foot combination laboratory and office building complex in Silver Spring, Maryland that serves as our co-headquarters and is used for the synthesis of treprostinil, the active ingredient in Remodulin and Tyvaso, and treprostinil diolamine, the active ingredient in Orenitram, as well as the production of Remodulin and Tyvaso and Unituxin. We also own several other buildings in Silver Spring used principally for office and laboratory space.
North Carolina—We own a 380,000 square foot combination manufacturing facility and office building in Research Triangle Park, North Carolina (RTP facility), which serves as our co-headquarters and is occupied by our clinical research and development, commercialization and our logistics and manufacturing personnel. We warehouse and distribute Remodulin, Tyvaso and Orenitram and produce Orenitram at this location. In 2012, we acquired a 132-acre property containing approximately 312,000 square feet of building space adjacent to our RTP facility, which we use for our research, development and production facilities relating to our lung regeneration program, office space and for future expansion.
55
Europe—We own an office building near London, England which serves as our European headquarters. In Germany, we lease a warehouse where we maintain inventory of components for our Tyvaso Inhalation System. The German facility includes office and laboratory space.
District of Columbia—We own two adjacent buildings in Washington, D.C., which serve as office space.
Florida—We own office buildings in Satellite Beach and Melbourne, Florida.
We believe that these facilities, along with various other owned and leased facilities, are adequate for our current operations and that additional land and facilities for future expansion are reasonably available.
Please refer to Note 19—Litigation, to our consolidated financial statements contained elsewhere in this Annual Report on Form 10-K, which is incorporated herein by reference.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
56
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock (and associated preferred stock purchase rights) trades on the NASDAQ Global Select Market under the symbol "UTHR". The table below sets forth the high and low closing prices for our common stock for the periods indicated:
| |
2015 | 2014 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
High | Low | High | Low | |||||||||
January 1 - March 31 |
$ | 179.51 | $ | 124.93 | $ | 113.39 | $ | 90.67 | |||||
April 1 - June 30 |
$ | 188.56 | $ | 159.69 | $ | 107.81 | $ | 86.14 | |||||
July 1 - September 30 |
$ | 179.15 | $ | 131.24 | $ | 136.16 | $ | 86.44 | |||||
October 1 - December 31 |
$ | 160.91 | $ | 119.57 | $ | 134.80 | $ | 122.11 | |||||
Number of Holders
As of February 12, 2016, there were 38 holders of record of our common stock.
Dividend Policy
We have never paid and have no present intention to pay cash dividends on our common stock in the foreseeable future and our 2016 Credit Facility contains covenants that may restrict us from doing so. We intend to retain any earnings for use in our business operations.
Issuer Purchases of Equity Securities
We did not repurchase any of our outstanding equity securities during the three months ended December 31, 2015, as the authorization under our previous share repurchase program was fully exhausted in August 2015. On October 15, 2015, we announced that our Board of Directors authorized a new share repurchase program for up to $500.0 million in aggregate repurchases, which is effective from January 1, 2016 through December 31, 2016.
57
Comparison of Five-Year Total Cumulative Shareholder Return
The following chart shows the performance from December 31, 2010 through December 31, 2015 of our common stock, compared with an investment in the stocks represented in each of the NASDAQ U.S. Benchmark TR Index and the NASDAQ ICB: 4577 Pharmaceutical Stock Index, assuming the investment of $100 at the beginning of the period and the reinvestment of dividends, if any.
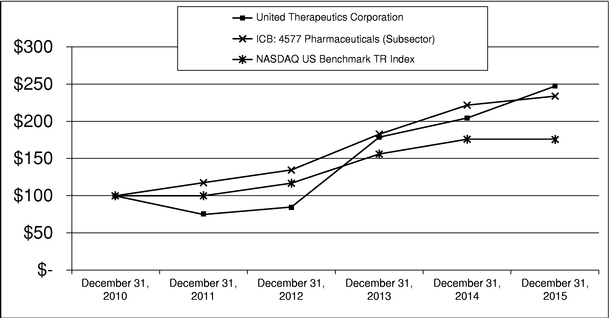
ITEM 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data should be read in conjunction with our consolidated financial statements and the notes accompanying the consolidated financial statements and Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations included in this Annual Report on Form 10-K. The historical results are not necessarily indicative of results to be expected for future periods. The following information is presented in thousands, except per share data.
| |
Year Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||
Consolidated Statements of Operations Data: |
||||||||||||||||
Revenues |
$ | 1,465,761 | $ | 1,288,519 | $ | 1,116,984 | $ | 916,076 | $ | 743,183 | ||||||
Operating income |
$ | 699,015 | $ | 538,800 | $ | 292,499 | $ | 421,646 | $ | 317,782 | ||||||
Income from continuing operations |
$ | 651,639 | $ | 340,074 | $ | 174,560 | $ | 304,442 | $ | 217,243 | ||||||
Income from discontinued operations, net of tax |
$ | — | $ | — | $ | — | $ | — | $ | 625 | ||||||
Net income |
$ | 651,639 | $ | 340,074 | $ | 174,560 | $ | 304,442 | $ | 217,868 | ||||||
Net income per common share: |
||||||||||||||||
Basic(1) |
$ | 14.17 | $ | 7.06 | $ | 3.49 | $ | 5.84 | $ | 3.81 | ||||||
Diluted(1) |
$ | 12.72 | $ | 6.28 | $ | 3.28 | $ | 5.71 | $ | 3.67 | ||||||
58
| |
As of December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||
Consolidated Balance Sheet Data: |
||||||||||||||||
Cash, cash equivalents and marketable investments |
$ | 991,774 | $ | 818,197 | $ | 1,142,037 | $ | 784,931 | $ | 747,378 | ||||||
Total assets |
$ | 2,184,445 | $ | 1,884,410 | $ | 2,087,567 | $ | 1,626,595 | $ | 1,518,079 | ||||||
Other liabilities |
$ | 143,974 | $ | 114,526 | $ | 95,582 | $ | 354,977 | $ | 346,132 | ||||||
Total stockholders' equity |
$ | 1,588,552 | $ | 1,242,356 | $ | 1,259,274 | $ | 1,083,981 | $ | 948,488 | ||||||
- (1)
- Refer to Note 11—Stockholders' Equity—Earnings Per Common Share to our consolidated financial statements contained in this Annual Report on Form 10-K for the computation of basic and diluted net income per share.
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with our consolidated financial statements and related notes to the consolidated financial statements included in this Annual Report on Form 10-K. The following discussion contains forward-looking statements made pursuant to the safe harbor provisions of Section 21E of the Securities Exchange Act of 1934 and the Private Securities Litigation Reform Act of 1995. These statements are based on our expectations about future outcomes and are subject to risks and uncertainties that could cause actual results to differ materially from anticipated results. Factors that could cause or contribute to such differences include those described under Part I, Item 1A—Risk Factors included in this Annual Report on Form 10-K and factors described in other cautionary statements, cautionary language and risk factors set forth in other documents we filed with the Securities and Exchange Commission. We undertake no obligation to publicly update forward-looking statements, whether as a result of new information, future events or otherwise.
Overview
Our key therapeutic products and product candidates include:
- •
- Prostacyclin analogues (Remodulin®, Tyvaso®, Orenitram® and
esuberaprost): stable synthetic forms of prostacyclin, an important molecule produced by the body that has powerful effects on blood
vessel health and function;
- •
- Phosphodiesterase type 5 (PDE-5) inhibitor
(Adcirca®): a molecule that acts to inhibit the degradation of cyclic guanosine monophosphate (cyclic GMP) in cells. Cyclic
GMP is activated by nitric oxide (NO), a naturally occurring substance in the body that mediates the relaxation of vascular smooth muscle;
- •
- Monoclonal antibody for oncologic applications
(Unituxin®): an antibody that binds to cancerous tumors and destroys the cancer cells through a mechanism called
antibody-dependent cell mediated cytotoxicity; and
- •
- Organ transplantation: engineered lungs and lung tissue, which we are developing using xenotransplantation and regenerative medicine technologies, for transplantation in patients suffering from PAH and other lung diseases. Although our primary focus is on engineered lungs, we are also developing technology for other engineered organs, such as kidneys and hearts. Through our wholly-owned subsidiary, Lung Biotechnology PBC, we are also developing technologies to improve outcomes for lung transplant recipients and to increase the supply of donor lungs through ex-vivo lung perfusion.
59
We concentrate substantially all of our research and development efforts on the preceding key therapeutic products and product candidates.
We currently market and sell the following commercial products:
- •
- Remodulin (treprostinil) Injection
(Remodulin). Remodulin, a continuously-infused formulation of the prostacyclin analogue treprostinil, is approved by the U.S. Food and
Drug Administration (FDA) for subcutaneous (under the skin) and intravenous (in the vein) administration. Remodulin is indicated to diminish symptoms associated with exercise in World Health
Organization (WHO) Group 1 PAH patients. Remodulin has also been approved in various countries outside of the United States.
- •
- Tyvaso (treprostinil) Inhalation Solution
(Tyvaso). Tyvaso, an inhaled formulation of treprostinil, is approved by the FDA to improve exercise ability in WHO Group 1 PAH
patients.
- •
- Orenitram (treprostinil) Extended-Release Tablets
(Orenitram). In December 2013, the FDA approved Orenitram, a tablet dosage form of treprostinil, for the treatment of PAH in WHO Group 1
PAH patients to improve exercise capacity. We commenced sales of Orenitram during the second quarter of 2014.
- •
- Adcirca (tadalafil) Tablets (Adcirca). We acquired
exclusive commercialization rights to Adcirca, an oral PAH therapy, in the United States from Eli Lilly and Company (Lilly). Adcirca is approved by the FDA to improve exercise ability in WHO Group 1
PAH patients.
- •
- Unituxin (dinutuximab) Injection (Unituxin). In March 2015, the FDA approved Unituxin in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma who achieve at least a partial response to prior first-line multiagent, multimodality therapy. We commenced U.S. sales of Unituxin in the third quarter of 2015. We received European Medicines Agency (EMA) approval during the third quarter of 2015.
Revenues
Our net product sales consist entirely of sales of our five commercial products: Remodulin, Tyvaso, Adcirca, Orenitram and Unituxin.
We have entered into separate, non-exclusive distribution agreements with Accredo Health Group, Inc. (Accredo) and CVS Caremark (Caremark) in the United States, to distribute Remodulin, Tyvaso and Orenitram. We also sell Remodulin to distributors internationally. In the second quarter of 2015, we entered into an exclusive distribution agreement with ASD Specialty Healthcare, Inc. (ASD), an affiliate of AmerisourceBergen Corporation, to distribute Unituxin in the United States. We sell Adcirca through Lilly's pharmaceutical wholesaler network at a wholesale price determined by Lilly, which Lilly generally increases two or three times per year. Most recently, Lilly increased the price of Adcirca by 9.9 percent effective May 14, 2015 and by another 9.9 percent effective December 1, 2015.
Under our distribution agreements, we sell Unituxin and each of our treprostinil-based products to these distributors at a transfer price that we establish. We pay Accredo, Caremark and ASD fees for services provided in connection with the distribution and support of these products. Historically, we have generally increased the price of Tyvaso annually by 4.9 percent, and the last price increase became effective on January 1, 2015. The price of Remodulin has not been increased since 2010. We have not increased the price of Orenitram or Unituxin since their launch in the second quarter of 2014 and the third quarter of 2015, respectively.
We require our distributors to maintain reasonable levels of inventory reserves of our treprostinil-based products as the interruption of Remodulin, Tyvaso or Orenitram therapy can be life threatening. Our distributors typically place monthly orders based on estimates of future demand and contractual
60
minimum inventory requirements. As a result, net product sales of Remodulin, Tyvaso and Orenitram can vary depending on the timing and magnitude of these orders and may not precisely reflect patient demand.
We recognize revenues net of: (1) estimated rebates; (2) prompt pay discounts; (3) allowances for sales returns; and (4) distributor fees. We estimate our liability for rebates based on an analysis of historical levels of rebates to both Medicaid and commercial third-party payers after considering the impact of sales trends, changes in government and commercial rebate programs and any anticipated changes in our products' pricing. In addition, we determine our obligation for prescription drug discounts required for Medicare Part D patients within the coverage gap based on estimates of the number of Medicare Part D patients and the period such patients will remain within the coverage gap. We provide prompt pay discounts to customers that pay amounts due within a specific time period and base related estimates on observed historical customer payment behavior. We derive estimates relating to our allowance for returns of Adcirca from actual return data accumulated since the drug's launch in 2009. We also compare patient prescription data for Adcirca to product sales on a quarterly basis to ensure a reasonable relationship between prescription and sales trends. To date, we have not identified any unusual patterns in the volume of prescriptions relative to sales that would warrant reconsideration of our methodology for estimating Adcirca returns. Remodulin, Tyvaso and Orenitram are distributed in the United States under separate contracts with substantially similar terms, which include exchange rights in the event that product is damaged during shipment or expires. The allowance for exchanges for Remodulin and Tyvaso is based on the historical rate of product exchanges, which has been negligible and immaterial. Furthermore, we anticipate minimal exchange activity in the future for Remodulin, Tyvaso and Orenitram since we typically sell these products with a remaining shelf life in excess of one year and our distributors generally carry a thirty- to sixty-day supply of our products at any given time. As a result, we do not record reserves for exchanges for Remodulin, Tyvaso and Orenitram at the time of sale. Lastly, we pay our distributors for contractual services rendered and accrue for related fees based on contractual rates applied to the estimated units of service provided by distributors for a given financial reporting period.
Generic Competition
We have settled litigation with Sandoz and Teva relating to abbreviated new drug applications (ANDAs) seeking FDA approval to market generic versions of Remodulin before the expiration of certain of our U.S. patents. Under the terms of our settlement agreements, Sandoz and Teva will be permitted to market their generic versions of Remodulin in the United States beginning in June 2018 and December 2018, respectively, although they may be permitted to enter the market earlier under certain circumstances. We are engaged in litigation with Watson Laboratories, Inc. (Watson), contesting its ANDA to market a generic version of Tyvaso before the expiration of certain of our U.S. patents in November 2018 and December 2028. Finally, SteadyMed Ltd. (SteadyMed) has recently filed a petition for inter partes review seeking to invalidate one of our patents that expires in December 2028 and covers treprostinil, which is the active ingredient in Remodulin, Tyvaso and Orenitram. SteadyMed has announced that it is developing a product called Trevyent™, which is a single-use, pre-filled pump being developed to deliver a two-day supply of treprostinil subcutaneously using its PatchPump® technology. In January 2016, SteadyMed announced that Trevyent has been granted orphan drug designation by the FDA for the treatment of PAH. In February 2016, we received notice that Actavis Laboratories FL, Inc. (Actavis) filed an ANDA seeking FDA approval to market a generic version of the 2.5 mg strength of Orenitram. For further details, please see Note 19—Litigation, to our consolidated financial statements, Item 3—Legal Proceedings and Part II, Item 9B—Other Information.
As a result of our settlements with Sandoz and Teva, we expect to see generic competition for Remodulin from these companies beginning in June 2018 and December 2018, respectively. This increased competition could reduce our net product sales and profits. In addition, while we intend to
61
vigorously enforce our intellectual property rights relating to our products, there can be no assurance that we will prevail in defending our patent rights, or that additional challenges from other ANDA filers or other challengers will not surface with respect to our products. Our patents could be invalidated, found unenforceable or found not to cover one or more generic forms of Remodulin, Tyvaso or Orenitram. If any ANDA filer were to receive approval to sell a generic version of Remodulin, Tyvaso or Orenitram and/or prevail in any patent litigation, the affected product(s) would become subject to increased competition which could reduce our sales.
Certain patents for Revatio®, a PDE-5 inhibitor marketed by Pfizer, Inc. for treatment of PAH, expired in 2012, leading several manufacturers to launch generic formulations of sildenafil citrate, the active ingredient in Revatio. Generic sildenafil's lower price relative to Adcirca could lead to pressure from payers to use generic products within the same class of therapy initially, which could erode Adcirca's market share and limit its potential sales. Although we believe Adcirca's once-daily dosing regimen provides a significant advantage over generic sildenafil's multiple dosing regimen, government payers and private insurance companies may favor the use of less expensive generic sildenafil over Adcirca. Thus far, we have not observed any measurable impact of generic sildenafil on sales of Adcirca; however, circumstances could change over time and our revenues could be adversely impacted. The U.S. patent for Adcirca for the treatment of pulmonary hypertension will expire in November 2017, following which we expect to see generic competition for Adcirca.
Patent expiration and generic competition for any of our commercial PAH products could have a significant, adverse impact on our revenues and profits, and is inherently difficult to predict. For additional discussion, please refer to the risk factor entitled, Our intellectual property rights may not effectively deter competitors from developing competing products that, if successful, could have a material adverse effect on our revenues and profits, contained in Part I, Item 1A—Risk Factors included in this Annual Report on Form 10-K.
Operating Expenses
Since our inception, we have devoted substantial resources to our various clinical trials and other research and development efforts, which are conducted both internally and through third parties. From time to time, we also license or acquire additional technologies and compounds to be incorporated into our development pipeline.
Our operating expenses include the following costs:
Research and Development
Our research and development expenses primarily include costs associated with the research and development of products and post-marketing research commitments. These costs generally include share-based compensation and salary-related expenses for research and development functions, professional fees for preclinical and clinical studies, costs associated with clinical manufacturing, facilities-related expenses and regulatory costs. Expenses also include costs for third-party arrangements, including upfront fees and milestone payments required under license arrangements for therapies under development.
Selling, General and Administrative
Our selling, general and administrative expenses primarily include costs associated with the commercialization of approved products and general and administrative costs to support our operations. Selling expenses generally include share-based compensation, salary-related expenses, product marketing and sales operations costs, and other costs incurred to support our sales efforts. General and administrative expenses include our core corporate support functions such as human resources, finance and legal, external costs such as insurance premiums, legal fees, grants to non-affiliated, not-profit organizations, and other professional service fees.
62
Cost of Product Sales
Cost of product sales comprise: (1) costs to produce and acquire products sold to customers; (2) royalty payments under license agreements granting us rights to sell related products; and (3) direct and indirect distribution costs incurred in the sale of products. We acquired the rights to sell our commercial products through license and assignment agreements with the original developers of these products. These agreements obligate us to pay royalties based on specified percentages of our net product sales from related products. We paid GlaxoSmithKline PLC (Glaxo) a royalty of ten percent of net product sales of our treprostinil-based products (Remodulin, Tyvaso and Orenitram) until October 2014, when the patents we acquired from Glaxo expired. We no longer have any royalty obligations for Remodulin or Tyvaso, and our only remaining royalty obligation on Orenitram sales is a single-digit royalty relating to technology used in its formulation. We pay a five percent royalty to Lilly on net product sales of Adcirca. We have no royalty obligation for sales of Unituxin.
We produce our primary supply of Remodulin, Tyvaso, Orenitram and Unituxin at our own facilities. In particular, we synthesize treprostinil, the active ingredient in Remodulin and Tyvaso, and treprostinil diolamine, the active ingredient in Orenitram, at our facility in Silver Spring, Maryland. We also produce finished Tyvaso, Remodulin, and Unituxin at our Silver Spring facility. We produce Orenitram and we package, warehouse and distribute Remodulin, Tyvaso, Orenitram and Unituxin, at our facility in Research Triangle Park, North Carolina. We intend to use our own facilities to produce our primary supply of Remodulin, Tyvaso, Unituxin and Orenitram. We utilize third-party contract manufacturers to supplement our Remodulin and Tyvaso production capacity and mitigate the risk of shortages and we are working to obtain FDA approval of a third party to serve as an additional producer of Orenitram and Unituxin. We engage a third-party contract manufacturer to produce the Tyvaso Inhalation System. We began selling Orenitram during 2014. Typical of the initial commercial activities of a newly-launched product, Orenitram's cost of product sales as a percentage of its net product sales is significantly higher than that of our other commercial products. We expect that as Orenitram's sales increase, its cost of product sales as a percentage of total revenue will decrease to levels similar to our other treprostinil-based commercial products.
Lilly manufactures Adcirca. We take title to Adcirca upon its manufacture and bear any losses related to the storage, distribution and sale of Adcirca.
Share-Based Compensation
We have granted awards under our share tracking award plans (STAP) and stock options under our equity incentive plans. Our operating expenses and net income are often materially impacted by the recognition of share-based compensation expense (benefit) associated with STAP awards and potential stock option grants containing a market or performance condition, as the fair value of these awards varies with the changes in our stock price. The fair values of STAP awards and potential stock option grants are measured using inputs and assumptions under the Black-Scholes-Merton model that can materially impact the amount of share-based compensation expense (benefit) for a given period.
We account for STAP awards as liabilities because they are settled in cash. As such, we must re-measure the fair value of STAP awards at the end of each financial reporting period until the awards are no longer outstanding. Changes in our STAP-related liability resulting from such re-measurements are recorded as adjustments to share-based compensation expense (benefit) and can create substantial volatility within our operating expenses from financial reporting period to period. The following factors, among others, have a significant impact on the amount of share-based compensation expense (benefit) recognized in connection with the STAP from period to period: (1) volatility in the price of our common stock (specifically, increases in the price of our common stock will generally result in an increase in our STAP liability and related compensation expense, while decreases in our stock price will
63
generally result in a reduction in our STAP liability and related compensation expense); (2) changes in the number of outstanding awards; and (3) changes in the number of vested and unvested awards.
In June 2015, our shareholders approved the United Therapeutics Corporation 2015 Stock Incentive Plan (the 2015 Plan). Following the approval of the 2015 Plan, which authorizes the grant of up to 6,150,000 shares of our common stock, we have ceased granting STAP awards and modified our equity compensation programs to grant stock options to employees and non-employee directors who previously received STAP awards. No further awards will be granted under our STAP plan or our previous equity plan, the Amended and Restated Equity Incentive Plan (the 1999 Plan).
Through December 31, 2014, we were contractually obligated to award stock options each year to our Chairman and Co-Chief Executive Officer, Dr. Rothblatt, based on a formula tied to the growth (if any) in our market capitalization. These awards were granted at year-end under the 1999 Plan, and vested immediately upon grant. We accrued compensation expense for Dr. Rothblatt's estimated stock option grant when we determined that it was probable that the performance criteria would be met. In 2015, Dr. Rothblatt's long term incentive compensation program is similar to our other employees in that she will be eligible for an annual grant of performance-based stock options based on the achievement of our annual corporate milestones, which vest over a four-year period from the grant date. Accordingly, we did not record any share-based compensation expense in 2015 for awards to Dr. Rothblatt as her annual grant of performance-based stock options will be granted in March 2016 based upon 2015 performance.
Major Research and Development Projects
Our major research and development projects focus on: (1) the use of prostacyclin analogues and other therapies to treat cardiopulmonary diseases; (2) monoclonal antibodies to treat cancer; and (3) organ transplantation technologies.
Cardiopulmonary Disease Projects
Remodulin Implantable System
In 2009, we entered into an agreement with Medtronic, Inc. (Medtronic) providing us exclusive rights in the United States, the United Kingdom, Canada, France, Germany, Italy and Japan to develop Medtronic's proprietary intravascular infusion catheter to be used with its SynchroMed® II implantable infusion pump and related infusion system components (together referred to as the Remodulin Implantable System) in order to deliver Remodulin for the treatment of PAH. If the Remodulin Implantable System is successful, it could reduce many of the patient burdens and other complications associated with the use of external pumps to administer prostacyclin analogues. With our funding, Medtronic completed the DelIVery clinical trial, in order to study the safety of the Remodulin Implantable System while administering Remodulin. The primary objective was to demonstrate a rate of catheter-related complications below 2.5 per 1,000 patient-days while using the Remodulin Implantable System to deliver Remodulin. In September 2013, Medtronic informed us that this primary objective was met (p<0.0001).
In order to launch the Remodulin Implantable System in the United States, we are pursuing parallel regulatory filings with Medtronic relating to the drug and the device, respectively. In December 2014, Medtronic submitted a premarket approval application (PMA) seeking FDA approval for the catheter and labeling changes for the SynchroMed II pump. Medtronic is entirely responsible for responding to any FDA requests for additional information concerning the use of the Remodulin Implantable System with Remodulin. In March 2015, the FDA requested that Medtronic amend its PMA to reflect an amendment to the SynchroMed II PMA separately submitted by Medtronic's neuromodulation business unit. Medtronic submitted an amendment to its PMA, which was accepted for review by FDA in January 2016, with FDA action expected in 2016.
64
In January 2015, we submitted a supplemental NDA with new labeling requesting FDA approval to allow the use of Remodulin with the Remodulin Implantable System. The FDA issued a refuse-to-file letter in March 2015, which meant we would need to address FDA comments and resubmit our filing. The FDA also indicated that our submission will be treated as a new NDA. We resubmitted our filing as a new NDA in December 2015, and we expect a ten-month review period (October 2016).
In April 2015, the FDA filed a consent decree requiring Medtronic to stop manufacturing, designing and distributing SynchroMed II implantable infusion pump systems, except in limited circumstances, citing violations of the quality system regulation for medical devices. The consent decree will remain in effect until the FDA has determined that Medtronic has met all the provisions listed in the consent decree. It is unclear how this consent decree will impact our program to develop and commercialize the Remodulin Implantable System, and we anticipate further insight regarding the potential impact on our program in 2016 when the FDA responds to Medtronic's PMA filing.
Subcutaneous Remodulin Administered via Pre-Filled, Semi-Disposable Pump
In December 2014, we entered into an exclusive agreement with DEKA Research & Development Corp. (DEKA) to develop a pre-filled, semi-disposable pump system for subcutaneous delivery of Remodulin. Under the terms of the agreement, we will fund the development costs related to the semi-disposable pump system and will pay product fees and a single-digit royalty to DEKA based on commercial sales of the system and the Remodulin sold for use with the system. Currently, we are undertaking engineering, design and development work to optimize the DEKA pump to deliver Remodulin in pre-filled reservoirs, and intend to conduct human factor studies in healthy volunteers before submitting an application to the FDA to approve the pre-filled DEKA pump. We do not anticipate that the FDA will require us to conduct clinical trials in patients. Our goal is to be in a position to receive FDA approval for this delivery system by the end of 2018.
Tyvaso
We are developing further enhancements intended to make the Tyvaso Inhalation System easier to use. In addition, we are studying Tyvaso in combination with esuberaprost, as discussed below, and we are planning a phase II study of Tyvaso in patients with pulmonary hypertension associated with idiopathic pulmonary fibrosis.
Orenitram
In December 2013, the FDA approved Orenitram for the treatment of PAH in WHO Group 1 patients to improve exercise capacity. The primary study that supported efficacy of Orenitram was a 12-week monotherapy study (FREEDOM-M) in which PAH patients were not on any approved background therapy.
We believe that in order for Orenitram to reach its full commercial potential, we need to complete further studies to support an amendment to Orenitram's label to indicate that Orenitram delays morbidity and mortality (also known as "time to clinical worsening") in patients who are on an approved oral background therapy. As such, we are enrolling up to 610 patients in a phase IV clinical trial called FREEDOM-EV, which began in 2012. FREEDOM-EV is a placebo-controlled study of patients who enter the study on an approved background therapy, and one of the two primary endpoints of the study is the time to clinical worsening.
We expect to seek approval of Orenitram in Europe upon completion of the FREEDOM-EV study. In 2005, the EMA announced that Orenitram had been designated an orphan medicinal product for the treatment of PAH. Our request for orphan drug designation for Orenitram for PAH is pending before the FDA.
65
Esuberaprost
We have been studying various formulations of beraprost since 2000, under a license agreement with Toray Industries, Inc. (Toray). In July 2012, we completed a phase I safety trial of esuberaprost, a reformulated, single-isomer version of beraprost, and the data suggested that dosing esuberaprost four times a day was safe. We believe that esuberaprost and treprostinil have differing prostacyclin receptor-binding profiles and thus could provide benefits to certain groups of patients with differing sets of safety and efficacy profiles. We also believe that inhaled treprostinil and esuberaprost have complimentary pharmacokinetic and pharmacodynamic profiles, which indicate that they should provide greater efficacy in combination. As a result, in 2013 we began enrolling a phase III study called BEAT (BEraprost 314d Add-on to Tyvaso) to evaluate the clinical benefit and safety of esuberaprost in combination with Tyvaso for patients with PAH who show signs of deterioration on inhaled treprostinil or have a less than optimal response to inhaled treprostinil treatment. We intend to enroll 240 patients in the study, which will have a primary endpoint of time to clinical worsening.
Cancer-Related Projects
Unituxin
In March 2015, the FDA approved our Biologics License Application (BLA) for Unituxin, in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma who achieve at least a partial response to prior first-line multiagent, multimodality therapy. We commenced U.S. sales of Unituxin in the third quarter of 2015. We received European Commission approval during the third quarter of 2015, and plan to commence commercial sales in individual European countries following pricing and reimbursement approvals on a country-by-country basis.
We previously received orphan drug designation for Unituxin from both the FDA and the EMA. Orphan designation, coupled with FDA approval of our BLA, confers an exclusivity period through March 2022, during which the FDA may not approve any application to market the same drug for the same indication, except in limited circumstances. In lieu of a royalty payment to the NCI, we have an ongoing obligation to provide the NCI with Unituxin for its studies free of charge.
Under our BLA approval for Unituxin, the FDA has imposed certain post-marketing requirements and post-marketing commitments on us. We are conducting additional clinical and non-clinical studies to satisfy these requirements and commitments. While we believe we will be able to complete these studies, any failure to satisfy these requirements or commitments could result in penalties, including fines or withdrawal of Unituxin from the market, unless we are able to demonstrate good cause for the failure.
Organ Transplantation
We are engaged in research and development into a variety of technologies designed to increase the supply of transplantable organs and tissues and improve outcomes for transplant recipients. These programs include preclinical research and development of alternative tissue sources through tissue and organ xenotransplantation, as well as regenerative medicine to create engineered organs and organ tissues. Our xenotransplantation efforts are supported in part by a multi-year research and development collaboration with Synthetic Genomics Inc. (SGI), where SGI will develop engineered primary pig cells with modified genomes for use in our xenotransplantation program.
In May 2014 and September 2015, we also completed two separate $50.0 million investments in the preferred stock of SGI for a total investment, as of December 31, 2015, of $100.0 million.
We are also conducting a phase II clinical trial in the United States to study the use of ex-vivo lung perfusion technology originally developed in Canada (where it is already used commercially) to
66
provide extended preservation and assessment of donated lungs that are initially rejected for transplantation.
In 2014, we completed the construction of a laboratory facility in Silver Spring, Maryland devoted to performing ex-vivo lung perfusion on a fee-for-service basis. In June 2015, we entered into a collaboration agreement with the Mayo Clinic in Jacksonville, Florida (Mayo) to build and operate a second such facility. We are responsible for nearly all costs associated with the construction of the facility on Mayo's campus, as well as the ongoing operating expenses for the facility. We expect to commence construction of the facility during the first quarter of 2016 and complete construction in late 2017.
Future Prospects
The extent of our future success is dependent on, among other things, how well we achieve the following objectives: (1) in the near term, continued sales growth of our current commercial products (including, in particular, Orenitram) by increasing our market share and launching enhancements designed to improve patient care, such as new delivery systems for Remodulin; (2) in the medium term, augmenting our near-term product growth through: (a) the successful launch of Orenitram for use in combination with other oral therapies following positive FREEDOM-EV results, and (b) the launch of esuberaprost in combination with Tyvaso following positive results of the BEAT study; and (3) in the long term, supplementing our oral, inhaled and infused PAH therapy revenues by introducing transplantable cells, tissues and organs that may prove effective in treating PAH and other end-stage diseases.
Our ability to achieve these objectives and sustain our growth and profitability will depend on many factors, including among others: (1) the timing and outcome of preclinical research, clinical trials and regulatory approvals for products we develop; (2) the timing of and the degree of success related to the commercial launch of new products; (3) the demand for our products; (4) the price of our products and the reimbursement of our products by public and private health insurance organizations; (5) the competition we face within our industry; (6) our ability to effectively manage our business in an increasingly complex legal and regulatory environment; (7) our ability to defend against generic competition and challenges to our patents, including the ongoing challenges to our Remodulin, Tyvaso and Orenitram patents and the expected launch of generic versions of Remodulin in the United States in June 2018 and December 2018 by Sandoz and Teva, respectively; and (8) the risks identified in Item 1A—Risk Factors, included in this Annual Report on Form 10-K.
We will need to construct additional facilities to support the development and commercialization of our products and services. The design and construction of the additional facilities will need to comply with stringent regulatory requirements, some of which have not yet been developed or adopted by the relevant government agencies. The extent to which we fully develop any of these facilities will depend on the progress of our preclinical and clinical development in various earlier stage programs.
We operate in a highly competitive market in which a small number of pharmaceutical companies control a majority of available PAH therapies. These pharmaceutical companies are well established in the market and possess greater financial, technical and marketing resources than we do. In addition, there are a number of investigational products in late-stage development that, if approved, may erode the market share of our existing commercial therapies and make market acceptance more difficult to achieve for any therapies we attempt to market in the future.
Financial Position
Cash and cash equivalents and marketable investments (both current and long-term) at December 31, 2015 and December 31, 2014 were $991.8 million and $818.2 million, respectively. The increase of $173.6 million in unrestricted cash and cash equivalents and marketable securities resulted
67
primarily from $382.8 million of cash generated from operations and a $350.0 million cash inflow from investing activities related to the sale of the Rare Pediatric Priority Review Voucher (PPRV) that we received from the FDA in connection with the approval of Unituxin. These increases were partially offset by the use of $394.5 million to repurchase shares of our common stock and the use of $133.2 million to settle early conversions of our Convertible Notes. Cash equivalents and marketable investments include long-term marketable investments of $38.0 million and $122.7 million at December 31, 2015 and December 31, 2014, respectively. The $84.7 million decrease in long-term marketable investments was due to the funding requirements for our share repurchase program and settlements of early conversions of our Convertible Notes.
Accounts receivable at December 31, 2015 and December 31, 2014 were $192.8 million and $162.3 million, respectively. The increase of $30.5 million reflected an approximately 14 percent increase in net product sales during the year ended December 31, 2015, compared to the year ended December 31, 2014, and the timing of invoicing and cash collections.
Other assets at December 31, 2015 and December 31, 2014 were $154.3 million and $97.9 million, respectively. The increase of $56.3 million was primarily due to our $50.0 million investment in SGI.
Convertible notes at December 31, 2015 and December 31, 2014 were $5.4 million and $126.4 million, respectively. The decrease of $121.0 million was due to early conversions of $133.2 million of principal of our Convertible Notes during the year ended December 31, 2015, net of amortization of $12.1 million for the unamortized discount of which $9.1 million was related to the early conversions of our Convertible Notes. Refer to Note 8—Debt—Convertible Notes Due 2016 to the consolidated financial statements contained in this Annual Report on Form 10-K for details.
Other liabilities at December 31, 2015 and December 31, 2014 were $144.0 million and $114.5 million, respectively. The increase of $29.4 million was due primarily to an increase of $39.0 million in the liability attributed to our outstanding STAP awards during 2015 as a result of a higher stock price at December 31, 2015 as compared to December 31, 2014, partially offset by the aggregation of individually immaterial items.
Additional paid-in capital at December 31, 2015 and December 31, 2014 was $1,790.6 million and $1,376.1 million, respectively. The increase of $414.5 million primarily consisted of $76.7 million in proceeds from stock option exercises and related tax benefits and $324.7 million related to the common stock issued in connection with the early conversion of $133.2 million of principal of our Convertible Notes based on the value of the closing price of our common stock on the date the shares were issued. Refer to Note 11—Stockholders' Equity—Equity Incentive Plan and Note 8—Debt—Convertible Notes Due 2016 to the consolidated financial statements contained in this Annual Report on Form 10-K for further details.
Treasury stock at December 31, 2015 and December 31, 2014 was $1,902.1 million and $1,185.8 million, respectively. The increase of $716.3 million primarily consisted of: (1) $394.5 million in expenditures to repurchase approximately 2.4 million shares of our common stock; and (2) $321.8 million reflecting the value of approximately 2.0 million shares we received under our note hedge in connection with the early conversion of $133.2 million of principal of our Convertible Notes based on the closing price of our common stock on the date the shares were received. Refer to Note 11—Stockholders' Equity—Share Repurchases and Note 8—Debt—Convertible Notes Due 2016 to the consolidated financial statements contained in this Annual Report on Form 10-K for further details.
68
Results of Operations
Years ended December 31, 2015 and 2014
Revenues
The following table presents the components of total revenues (dollars in thousands):
| |
Year Ended December 31, | |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2015 | 2014 | ||||||||
Net product sales: |
||||||||||
Remodulin |
$ | 572,795 | $ | 553,728 | 3.4 | % | ||||
Tyvaso |
470,069 | 463,067 | 1.5 | % | ||||||
Adcirca |
278,829 | 221,471 | 25.9 | % | ||||||
Orenitram |
118,434 | 41,267 | 187.0 | % | ||||||
Unituxin |
20,443 | — | NM | (1) | ||||||
Other |
5,191 | 8,986 | (42.2 | )% | ||||||
| | | | | | | | | | | |
Total revenues |
$ | 1,465,761 | $ | 1,288,519 | 13.8 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- Calculation is not meaningful.
Revenues for the year ended December 31, 2015 increased by $177.2 million compared to the same period in 2014. The growth in revenues primarily resulted from: (1) a $77.2 million increase in Orenitram net product sales due to an increase in the number of patients being treated with Orenitram, which we launched in the second quarter of 2014; (2) a $57.4 million increase in Adcirca net product sales due to price increases, which were determined by Lilly, and to a lesser extent by an increase in the number of Adcirca bottles sold; and (3) $20.4 million in net product sales of Unituxin, which we launched in the third quarter of 2015.
For the years ended December 31, 2015 and 2014, approximately 72 percent and 74 percent, respectively, of total revenues were derived from net product sales of Remodulin, Tyvaso and Orenitram to our U.S.-based specialty pharmaceutical distributors. Remaining revenues were derived primarily from net product sales of Adcirca and Unituxin and net product sales of Remodulin to our international distributors.
We recognize revenues net of: (1) estimated rebates; (2) prompt pay discounts; (3) allowances for sales returns; and (4) distributor fees. These are referred to as gross-to-net deductions and are based on historical experiences and contractual and statutory requirements. The tables below include a reconciliation of the accounts associated with these deductions (in thousands):
| |
Year Ended December 31, 2015 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Rebates | Prompt Pay Discounts |
Allowance for Sales Returns |
Distributor Fees |
Total | |||||||||||
Balance, January 1, 2015 |
$ | 31,616 | $ | 3,285 | $ | 4,028 | $ | 557 | $ | 39,486 | ||||||
Provisions attributed to sales in: |
||||||||||||||||
Current period |
171,653 | 33,508 | 2,640 | 9,830 | 217,631 | |||||||||||
Prior periods |
44 | — | 339 | (255 | ) | 128 | ||||||||||
Payments or credits attributed to sales in: |
||||||||||||||||
Current period |
(123,855 | ) | (29,645 | ) | — | (7,253 | ) | (160,753 | ) | |||||||
Prior periods |
(34,840 | ) | (3,245 | ) | (1,724 | ) | (286 | ) | (40,095 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Balance, December 31, 2015 |
$ | 44,618 | $ | 3,903 | $ | 5,283 | $ | 2,593 | $ | 56,397 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
69
| |
Year Ended December 31, 2014 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Rebates | Prompt Pay Discounts |
Allowance for Sales Returns |
Distributor Fees |
Total | |||||||||||
Balance, January 1, 2014 |
$ | 22,475 | $ | 2,500 | $ | 2,862 | $ | 1,092 | $ | 28,929 | ||||||
Provisions attributed to sales in: |
||||||||||||||||
Current period |
116,813 | 27,096 | 1,671 | 7,854 | 153,434 | |||||||||||
Prior periods |
6,622 | — | 429 | 278 | 7,329 | |||||||||||
Payments or credits attributed to sales in: |
||||||||||||||||
Current period |
(85,833 | ) | (23,998 | ) | — | (7,139 | ) | (116,970 | ) | |||||||
Prior periods |
(28,461 | ) | (2,313 | ) | (934 | ) | (1,528 | ) | (33,236 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Balance, December 31, 2014 |
$ | 31,616 | $ | 3,285 | $ | 4,028 | $ | 557 | $ | 39,486 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Research and Development Expense
The table below summarizes research and development expense by major project and non-project components (dollars in thousands):
| |
Year Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2015 | 2014 | ||||||||
Project and non-project: |
||||||||||
Cardiopulmonary |
$ | 130,097 | $ | 131,843 | (1.3 | )% | ||||
Share-based compensation expense |
87,713 | 72,714 | 20.6 | % | ||||||
Other |
27,288 | 37,992 | (28.2 | )% | ||||||
| | | | | | | | | | | |
Total research and development expense |
$ | 245,098 | $ | 242,549 | 1.1 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Share-based compensation. The increase in share-based compensation of $15.0 million for the year ended December 31, 2015, as compared to the same period in 2014, corresponded to a 21 percent appreciation in the price of our common stock during the year ended December 31, 2015, compared to a 15 percent appreciation in the price of our common stock price during the year ended December 31, 2014.
Other. The decrease in other research and development expenses of $10.7 million for the year ended December 31, 2015, as compared to the same period in 2014, was primarily attributable to a $6.4 million decrease in expenditures for our development of Unituxin, which was approved by the FDA in March of 2015, and a $3.9 million decrease in research and development expenditures not allocated to specific projects.
Selling, General and Administrative Expense
The table below summarizes selling, general and administrative expense by major categories (dollars in thousands):
| |
Year Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2015 | 2014 | ||||||||
Category: |
||||||||||
General and administrative |
$ | 174,570 | $ | 186,312 | (6.3 | )% | ||||
Sales and marketing |
94,297 | 82,000 | 15.0 | % | ||||||
Share-based compensation expense |
183,745 | 112,975 | 62.6 | % | ||||||
| | | | | | | | | | | |
Total selling, general and administrative expense |
$ | 452,612 | $ | 381,287 | 18.7 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
70
General and administrative. The decrease in general and administrative expenses of $11.7 million for the year ended December 31, 2015, as compared to the same period in 2014, was attributable to: (1) a $12.7 million decrease due to timing of grants to non-affiliated, non-profit organizations that provide financial assistance to patients with PAH; and (2) a $9.4 million decrease in legal expenses resulting from the April 2015 closure of an investigation by the Office of Inspector General of the Department of Health and Human Services related to our marketing practices; partially offset by (3) a $10.0 million increase in salaries and other compensation-related expenses driven by the general expansion of our business.
Sales and marketing. The increase in sales and marketing expenses of $12.3 million for the year ended December 31, 2015. as compared to the same period in 2014, was driven by: (1) an $8.6 million increase in marketing activities for all of our commercial products, primarily for our most recently approved PAH product, Orenitram, and our first oncology product, Unituxin; and (2) a $3.7 million increase in salaries and other compensation-related expenses driven by the expansion of our personnel in connection with the growth of our commercial product portfolio.
Share-based compensation. The increase in share-based compensation of $70.8 million for the year ended December 31, 2015, as compared to the same period in 2014, corresponded to a 21 percent appreciation in the price of our common stock during the year ended December 31, 2015, compared to the approximately 15 percent appreciation in our stock price during the same period in 2014.
Cost of Product Sales
The table below summarizes cost of product sales by major category (dollars in thousands):
| |
Year Ended December 31, | |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2015 | 2014 | ||||||||
Category: |
||||||||||
Cost of product sales |
$ | 60,240 | $ | 121,518 | (50.4 | )% | ||||
Share-based compensation expense |
8,796 | 4,365 | 101.5 | % | ||||||
| | | | | | | | | | | |
Total cost of product sales |
$ | 69,036 | $ | 125,883 | (45.2 | )% | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cost of Product Sales. The decrease in cost of product sales of $61.3 million for the year ended December 31, 2015 as compared to the same period in 2014, resulted primarily from the expiration of our royalty obligation to Glaxo in October 2014. During the twelve months ended December 31, 2014, we incurred $72.5 million in royalty expense related to this obligation. This decrease was partially offset by: (1) an increase in the cost of product sales of $5.6 million and $3.0 million relating to Orenitram and Adcirca, respectively due to increased sales of these products in 2015; and (2) $3.2 million in cost of product sales relating to Unituxin which was commercially launched during the third quarter of 2015.
Share-based compensation. The increase in share-based compensation of $4.4 million for the year ended December 31, 2015 as compared to the same period in 2014, corresponded to a 21 percent appreciation in the price of our common stock during the year ended December 31, 2015, compared to the approximately 15 percent appreciation in our stock price during the same period in 2014.
Gain on Sale of Intangible Asset
In September 2015, we sold the PPRV we received from the FDA in connection with the approval of Unituxin for $350.0 million in cash. The proceeds from the sale of the PPRV were recognized as a gain on the sale of an intangible asset, as the PPRV did not have a carrying value on our consolidated balance sheet at the time of sale.
71
Income Tax Expense
The provision for income taxes was $392.8 million for the year ended December 31, 2015 compared to $185.1 million for the same period in 2014. The increase in the provision for income taxes corresponded to the increase in income before income taxes. For the years ended December 31, 2015 and December 31, 2014, the effective tax rates were approximately 38 percent and 35 percent, respectively. For additional details refer to Note 13—Income Taxes to the consolidated financial statements contained in this Annual Report on 10-K.
Years ended December 31, 2014 and 2013
Revenues
The following table presents the components of total revenues (dollars in thousands):
| |
Year Ended December 31, | |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2014 | 2013 | ||||||||
Net product sales: |
||||||||||
Remodulin |
$ | 553,728 | $ | 491,179 | 12.7 | % | ||||
Tyvaso |
463,067 | 438,793 | 5.5 | % | ||||||
Adcirca |
221,471 | 176,972 | 25.1 | % | ||||||
Orenitram |
41,267 | — | NM | (1) | ||||||
Other |
8,986 | 10,040 | (10.5 | )% | ||||||
| | | | | | | | | | | |
Total revenues |
$ | 1,288,519 | $ | 1,116,984 | 15.4 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- Calculation is not meaningful.
Revenues for the year ended December 31, 2014 increased by $171.5 million compared to the same period in 2013. The growth in revenues primarily resulted from: (1) a $62.5 million increase in Remodulin net product sales due to an increase in the number of patients being treated with Remodulin; (2) a $44.5 million increase in Adcirca net product sales due to price increases, which were determined by Lilly, and to a lesser extent by an increase in the number of Adcirca bottles sold; (3) a $24.3 million increase in Tyvaso net product sales mostly due to a price increase on January 1, 2014; and (4) $41.3 million in net product sales of Orenitram, which we launched during the second quarter of 2014.
For the years ended December 31, 2014 and 2013, approximately 74 percent and 76 percent, respectively, of total revenues were derived from net product sales of Remodulin, Tyvaso and Orenitram to our U.S.-based specialty pharmaceutical distributors. Remaining revenues were derived primarily from net product sales of Adcirca and net product sales of Remodulin and Tyvaso to our international distributors.
72
The table below includes a reconciliation of the accounts associated with estimated rebates, prompt-pay discounts, allowances for sales returns and distributor fees (in thousands):
| |
Year Ended December 31, 2014 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Rebates | Prompt Pay Discounts |
Allowance for Sales Returns |
Distributor Fees |
Total | |||||||||||
Balance, January 1, 2014 |
$ | 22,475 | $ | 2,500 | $ | 2,862 | $ | 1,092 | $ | 28,929 | ||||||
Provisions attributed to sales in: |
||||||||||||||||
Current period |
116,813 | 27,096 | 1,671 | 7,854 | 153,434 | |||||||||||
Prior periods |
6,622 | — | 429 | 278 | 7,329 | |||||||||||
Payments or credits attributed to sales in: |
||||||||||||||||
Current period |
(85,833 | ) | (23,998 | ) | — | (7,139 | ) | (116,970 | ) | |||||||
Prior periods |
(28,461 | ) | (2,313 | ) | (934 | ) | (1,528 | ) | (33,236 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Balance, December 31, 2014 |
$ | 31,616 | $ | 3,285 | $ | 4,028 | $ | 557 | $ | 39,486 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| |
Year Ended December 31, 2013 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Rebates | Prompt Pay Discounts |
Allowance for Sales Returns |
Distributor Fees |
Total | |||||||||||
Balance, January 1, 2013 |
$ | 15,207 | $ | 2,115 | $ | 3,350 | $ | 1,281 | $ | 21,953 | ||||||
Provisions attributed to sales in: |
||||||||||||||||
Current period |
81,938 | 24,154 | 1,254 | 7,008 | 114,354 | |||||||||||
Prior periods |
997 | — | (1,530 | ) | 3 | (530 | ) | |||||||||
Payments or credits attributed to sales in: |
||||||||||||||||
Current period |
(59,225 | ) | (21,654 | ) | — | (5,916 | ) | (86,795 | ) | |||||||
Prior periods |
(16,442 | ) | (2,115 | ) | (212 | ) | (1,284 | ) | (20,053 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Balance, December 31, 2013 |
$ | 22,475 | $ | 2,500 | $ | 2,862 | $ | 1,092 | $ | 28,929 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Research and Development Expense
The table below summarizes research and development expense by major project and non-project components (dollars in thousands):
| |
Year Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2014 | 2013 | ||||||||
Project and non-project: |
||||||||||
Cardiopulmonary |
$ | 131,843 | $ | 116,137 | 13.5 | % | ||||
Share-based compensation expense |
72,714 | 134,706 | (46.0 | )% | ||||||
Other |
37,992 | 48,505 | (21.7 | )% | ||||||
| | | | | | | | | | | |
Total research and development expense |
$ | 242,549 | $ | 299,348 | (19.0 | )% | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cardiopulmonary. The increase in cardiopulmonary program expenses of $15.7 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, resulted from a $20.1 million increase in expenses related to our esuberaprost program, partially offset by a $7.9 million decrease in expenses related to our self-injectable treprostinil program, which we terminated during 2014.
Share-based compensation. The decrease in share-based compensation of $62.0 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, resulted from the approximately 15 percent appreciation in the price of our common stock during the year ended
73
December 31, 2014, compared to the approximately 112 percent appreciation in the price of our common stock price during the year ended December 31, 2013.
Other. The decrease in other research and development expenses of $10.5 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, was attributable to a $7.5 million decrease in research and development expenditures not allocated to specific projects and a $1.6 million decrease in expenditures for our development of Unituxin.
Selling, General and Administrative Expense
The table below summarizes selling, general and administrative expense by major category (dollars in thousands):
| |
Year Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2014 | 2013 | ||||||||
Category: |
||||||||||
General and administrative |
$ | 186,312 | $ | 140,235 | 32.9 | % | ||||
Sales and marketing |
82,000 | 73,871 | 11.0 | % | ||||||
Share-based compensation expense |
112,975 | 179,904 | (37.2 | )% | ||||||
| | | | | | | | | | | |
Total selling, general and administrative expense |
$ | 381,287 | $ | 394,010 | (3.2 | )% | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
General and administrative. The increase in general and administrative expenses of $46.1 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, was driven by the following: (1) an $18.2 million increase in consulting and professional fees primarily driven by our patent litigation and our response to a subpoena issued by the OIG relating to our marketing practices; (2) an $8.7 million increase in grants to non-affiliated, non-profit organizations that provide financial assistance to patients with PAH; and (3) $5.4 million and $7.5 million increases in operating expenses and salaries and other compensation-related expenses, respectively, associated with the general expansion of our business and the reclassification of certain staff from research and development to a general and administrative classification.
Sales and marketing. The increase in sales and marketing expenses of $8.1 million reflects the following increases: (1) $5.3 million increase in salaries and other compensation-related expenses as we expanded our sales personnel during 2014; and (2) a $2.8 million increase in marketing activities.
Share-based compensation. The decrease in share-based compensation of $66.9 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, corresponded to the approximately 15 percent appreciation in the price of our common stock during the year ended December 31, 2014, compared to the approximately 112 percent appreciation in our stock price during the year ended December 31, 2013.
74
Cost of Product Sales
The table below summarizes cost of product sales by major category (dollars in thousands):
| |
Year Ended December 31, | |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Change |
|||||||||
| |
2014 | 2013 | ||||||||
Category: |
||||||||||
Cost of product sales |
$ | 121,518 | $ | 124,952 | (2.7 | )% | ||||
Share-based compensation (benefit) expense |
4,365 | 6,175 | (29.3 | )% | ||||||
| | | | | | | | | | | |
Total cost of product sales |
$ | 125,883 | $ | 131,127 | (4.0 | )% | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cost of Product Sales. Cost of product sales decreased by $3.4 million for the year ended December 31, 2014, compared to the year ended December 31, 2013. The decrease resulted primarily from the expiration of our royalty obligation to Glaxo in October 2014.
Share-based compensation. The decrease in share-based compensation of $1.8 million for the year ended December 31, 2014, compared to the year ended December 31, 2013, corresponded to the approximately 15 percent appreciation in the price of our common stock during the year ended December 31, 2014, compared to the approximately 112 percent appreciation in our stock price during the year ended December 31, 2013.
Income Tax Expense
The provision for income taxes was $185.1 million for the year ended December 31, 2014 compared to $104.3 million for the year ended December 31, 2013. The increase in the provision for income taxes corresponded to the increase in income before income taxes. For the years ended December 31, 2014 and December 31, 2013, the effective tax rates were approximately 35 percent and 37 percent, respectively. For additional details refer to Note 13—Income Taxes to the consolidated financial statements contained in this Annual Report on 10-K.
Liquidity and Capital Resources
We have funded our operations principally through sales of our commercial products and, from time-to-time, third-party financing arrangements. We believe that our current liquidity is sufficient to fund ongoing operations and future business plans as we expect demand for our commercial products to continue to grow. Furthermore, our customer base remains stable and we believe it presents minimal credit risk. However, any projections of future cash flows are inherently subject to uncertainty and we may seek other forms of financing. In January 2016, we entered into a credit agreement providing a five-year, unsecured, revolving line of credit of up to $1.0 billion. See Unsecured Revolving Credit Facility below for further details.
Cash Flows
2015 Compared to 2014
Operating Activities
Our operating assets and liabilities consist primarily of accounts receivable, inventories, accounts payable and accrued expenses, which include share-based compensation arrangements. During the periods presented in the accompanying financial statements, the combination of revenue growth and profitable operations has resulted in positive cash flows provided by operations.
75
Net cash provided by operating activities was $382.8 million for the year ended December 31, 2015, compared to $355.3 million for the year ended December 31, 2014. The $27.5 million increase in cash flows from operations was primarily due to the following year-over-year changes:
- (1)
- Increases
in cash provided by operating activities:
- •
- $177.2 million increase in revenues during the year ended December 31, 2015 compared to the same period in
2014, which resulted in higher cash collections;
- •
- $23.7 million increase in cash flows due to an increase in accounts payable and accrued expenses at
December 31, 2015 compared to the prior year;
- •
- $14.2 million increase in cash flows due to a decrease in payments for inventory during the year ended
December 31, 2015 due to increased sales; and
- •
- $14.9 million increase in cash flows due to the aggregation of individually immaterial changes to operating assets
and liabilities.
- (2)
- Decreases
in cash provided by operating activities:
- •
- $104.7 million increase in cash paid to settle STAP awards. Cash paid to settle STAP awards exercised during the
years ended December 31, 2015 and December 31, 2014 was $248.8 million and $144.1 million, respectively; and
- •
- $97.8 million increase in cash paid for income taxes. Cash paid for income taxes during the years ended December 31, 2015 and December 31, 2014 was $293.3 million and $195.6 million, respectively.
Investing
Net cash provided by investing activities was $503.6 million for the year ended December 31, 2015, compared to $338.5 million for the year ended December 31, 2014. The $165.1 million increase in net cash provided by investing activities reflects $350.0 million of cash from the sale of our PPRV in September 2015, partially offset by a decrease of cash provided from the net maturities of held-to-maturity investments of $173.3 million. Cash provided from net maturities of held-to-maturity investments during the year ended December 31, 2015 was $257.6 million, compared to $430.9 million of cash provided from the net maturities of held-to-maturity investments during the same period in 2014. Due to the funding requirements for our share repurchase program and settlements of early conversions of our Convertible Notes, we decreased the amount of cash we were reinvesting in held-to-maturity investments.
Financing
Net cash used in financing activities was $446.9 million for the year ended December 31, 2015, compared to $576.5 million for the year ended December 31, 2014. The $129.6 million decrease reflects a decrease of $88.6 million in repurchases of our common stock, due to the completion of our repurchase program in August 2015, and a decrease of $44.7 million in principal payments of debt during the year ended December 31, 2015 compared to the year ended December 31, 2014. as a result of less early conversion requests on our Convertible Notes.
76
2014 Compared to 2013
Operating
Net cash provided by operating activities was $355.3 million for the year ended December 31, 2014, compared to $425.3 million for the year ended December 31, 2013. The $70.0 million decrease in cash flows from operations was primarily due to the following year-over-year changes:
- (1)
- Increase
in cash provided by operating activities:
- •
- $171.5 million increase in revenues during the year ended December 31, 2014 compared to the same period in
2013, which resulted in higher cash collections.
- (2)
- Decreases
in cash provided by operating activities:
- •
- $88.2 million increase in cash paid to settle STAP awards. Cash paid to settle STAP awards exercised during the
years ended December 31, 2014 and December 31, 2013 was $144.1 million and $55.9 million, respectively;
- •
- $53.4 million increase in cash paid for income taxes. Cash paid for income taxes during the years ended
December 31, 2014 and December 31, 2013 was $195.6 million and $142.1 million, respectively;
- •
- $25.7 million decrease in cash flow due to the timing of cash collections in accounts receivable;
- •
- $14.3 million decrease in cash flow due to an increase in cash payments of our accounts payable resulting in a
lower accounts payable and accrued expenses balance at December 31, 2014 compared with the prior year; and
- •
- $59.9 million decrease in cash flows due to the aggregation of individually immaterial changes to operating assets and liabilities.
Investing
Net cash provided by investing activities was $338.5 million for the year ended December 31, 2014, compared to net cash used in investing activities of $295.0 million for the year ended December 31, 2013. The increase of $633.4 million in cash provided by investing activities reflects $430.9 million of cash provided from the net maturities of held-to-maturity investments during the year ended December 31, 2014, compared to $232.3 million in net purchases of held-to-maturity investments during the same period in 2013. Due to the funding requirements in 2014 for our ongoing share repurchase programs and settlements of early conversions of our Convertible Notes, we did not reinvest the proceeds from our maturing investments. This increase in cash from maturing investments was partially offset by a $15.5 million increase in capital expenditures relating primarily to the completion of facilities used in our lung transplantation programs.
Financing
Net cash used in financing activities was $576.5 million for the year ended December 31, 2014, compared to $5.1 million for the year ended December 31, 2013. The $571.4 million increase in cash used in financing activities reflects an increase of $440.6 million in repurchases of our common stock and an increase of $176.5 million in principal payments of debt, partially offset by a $45.1 million increase in proceeds and tax benefits from the exercise of stock options during the year ended December 31, 2014, compared to the year ended December 31, 2013.
77
Unsecured Revolving Credit Facility
In January 2016, we entered into a Credit Agreement (the 2016 Credit Agreement) with Wells Fargo Bank, National Association (Wells Fargo), as administrative agent and a swingline lender, and various other lender parties, providing for an unsecured revolving credit facility of up to $1.0 billion (the Revolving Facility), which is available to refinance certain of our existing indebtedness and/or for working capital and other general corporate purposes. The Revolving Facility will mature five years after the closing date of the 2016 Credit Agreement, subject to the lenders' ability to extend the maturity date by one year if we request such an extension in accordance with the terms of the 2016 Credit Agreement.
At our option, amounts borrowed under the Revolving Facility will bear interest at either the LIBOR rate or a fluctuating base rate, in each case, plus an applicable margin determined on a quarterly basis based on our consolidated ratio of total indebtedness to EBITDA (as calculated in accordance with the 2016 Credit Agreement).
The 2016 Credit Agreement contains customary events of default and customary affirmative and negative covenants. As of January 29, 2016, we were in compliance with such covenants and we had not drawn any amounts on the Revolving Facility. In addition, Lung Biotechnology PBC is our only subsidiary that guarantees our obligations under the 2016 Credit Agreement though, from time to time, one or more of our other subsidiaries may be required to guarantee such obligations.
Secured Line of Credit
In September 2013, we entered into a one-year Credit Agreement (the 2013 Credit Agreement) with Wells Fargo for a $75.0 million revolving loan facility. In July 2015, we amended the Credit Agreement solely to extend its maturity to September 30, 2017. We used this facility for general corporate purposes. At our option, amounts borrowed under the Credit Agreement bore interest at either the one-month LIBOR rate plus a 0.50 percent margin, or a fluctuating base rate excluding any margin. In addition, we were subject to a monthly commitment fee at a rate of 0.06 percent per annum based on the average daily unused balance of the facility. Amounts borrowed under the Credit Agreement were secured by certain of our marketable investments. As of December 31, 2015, we had no outstanding balance on the line of credit. In January 2016, we terminated and repaid in full all obligations under the 2013 Credit Agreement when we entered into the 2016 Credit Agreement.
Convertible Senior Notes
In October 2011, we issued the Convertible Notes with an aggregate principal value of $250.0 million. Please see Note 8—Debt, to our consolidated financial statements contained elsewhere in this Annual Report on Form 10-K, for a description of the Convertible Notes. As of December 31, 2015, the outstanding principal balance of our Convertible Notes was $5.6 million, which is due on September 15, 2016.
Mortgage Financing
In December 2010, we entered into a Credit Agreement with Wells Fargo and Bank of America, N.A., pursuant to which we obtained a $70.0 million mortgage loan (the 2010 Credit Agreement). The 2010 Credit Agreement matured in December 2014 and we repaid in full the outstanding $66.5 million principal balance.
Share Tracking Award Plans
Awards granted under our STAP entitle participants to receive in cash an amount equal to the appreciation in our common stock, which is calculated as the increase in the closing price of our
78
common stock between the grant date and the exercise date. Cash requirements associated with the exercise of awards will likely be significant, with the actual requirements dependent on future stock price fluctuation and STAP award exercise activity. At December 31, 2015, the aggregate liability associated with vested STAP awards was $194.4 million, and the aggregate liability associated with all STAP awards was $354.8 million. We review the potential future cash requirements of the STAP program at least annually. Based on our review, we believe we currently have sufficient cash and cash equivalents and borrowing capacity to fund any STAP awards that could be exercised during 2016 and beyond. Following the adoption of the 2015 Plan, which is discussed above under Operating Expenses—Share-Based Compensation, we discontinued the issuance of STAP awards and modified our compensation programs to provide for future awards in the form of stock options instead of STAP awards.
Share Repurchases
From time to time, our Board of Directors authorizes plans to repurchase shares of our common stock. In June 2014, our Board of Directors authorized the repurchase of up to $500.0 million of our common stock. This program became effective on August 1, 2014, and remained open for one year. In the aggregate, we repurchased approximately 3.3 million shares of common stock under this program for $500.0 million.
In October 2015, our Board of Directors authorized a new program for the repurchase of up to $500.0 million of our common stock in open or privately negotiated transactions, at our discretion. This program is effective from January 1, 2016 to December 31, 2016. During the month ended January 31, 2016, we repurchased approximately 280,000 shares of our common stock at an aggregate cost of $37.8 million. We currently have sufficient cash and cash equivalents, borrowing capacity and, if needed, marketable investments, to fund additional repurchases of our common stock under this program.
Toray License Obligations
Pursuant to a March 2007 amendment to our license agreement with Toray, we issued 200,000 shares of our common stock to Toray. Toray has the right to request that we repurchase these shares (which have since split into 400,000 shares) upon 30 days prior written notice at the price of $27.21 per share. To date, Toray has not notified us that it intends to require us to repurchase these shares. In 2011, we amended our license agreement with Toray to reduce the royalty rates in exchange for a total of $50.0 million in equal, non-refundable payments to Toray over the five-year period ending in 2015. As of December 31, 2015, we have fulfilled our $50.0 million royalty obligation to Toray.
Obligations Under License and Assignment Agreements
We pay Lilly a five percent royalty on net product sales of Adcirca and we pay a single-digit percentage royalty based on net product sales of Orenitram, under our license agreement with Supernus Pharmaceuticals Inc. We have entered into other license rights arrangements under which we are required to make milestone payments upon the achievement of certain developmental and commercialization objectives and royalty payments upon the commercialization of related licensed technology.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements within the meaning of Item 303(a)(4) of Regulation S-K.
79
Contractual Obligations
At December 31, 2015, we had the following contractual obligations (in thousands):
| |
Payments Due by Period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Less than 1 year |
2 - 3 Years | 4 - 5 Years | More than 5 Years |
|||||||||||
Convertible Notes |
$ | 5,600 | $ | 5,600 | $ | — | $ | — | $ | — | ||||||
Mortgage and other loans |
3,703 | 3,495 | 138 | 70 | — | |||||||||||
Operating lease obligations |
10,534 | 3,716 | 6,083 | 735 | — | |||||||||||
Obligations under the STAP(1) |
460,450 | 267,603 | 191,301 | 1,546 | — | |||||||||||
Obligations under the SERP(2) |
71,067 | 14,651 | — | 4,580 | 51,836 | |||||||||||
Purchase commitments |
15,821 | 14,717 | 1,076 | 28 | — | |||||||||||
Milestone payments under license and acquisition agreements(3) |
33,021 | 6,136 | 8,345 | 11,470 | 7,070 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total |
$ | 600,196 | $ | 315,918 | $ | 206,943 | $ | 18,429 | $ | 58,906 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
- (1)
- Estimated
based on the intrinsic value of outstanding STAP awards vested and expected to vest, assuming that unvested awards will be exercised immediately
upon vesting. Refer to Note 7—Share Tracking Award Plans to our consolidated financial statements included in this Annual Report on
Form 10-K for further details.
- (2)
- Consists
of actuarially derived, estimated future payouts of benefits. Refer to Note 14—Employee Benefit
Plans—Supplemental Executive Retirement Plan to our consolidated financial statements included in this Annual Report on Form 10-K for further details.
- (3)
- Based on our estimates of the timing and probability of achieving milestones specified under our various license and acquisition agreements. Amount includes $21.4 million of contingent consideration and future payments that are recorded within other liabilities (current and non-current) on the consolidated balance sheet as of December 31, 2015. All other amounts have not been recorded as of December 31, 2015, because required milestones have not been met. The amounts and timing of future milestone payments may vary depending on when related milestones will be attained, if at all.
Summary of Critical Accounting Policies and Estimates
We prepare our consolidated financial statements in conformity with generally accepted accounting principles in the United States (GAAP). GAAP requires that we make estimates and assumptions that affect the amounts and timing reported in our consolidated financial statements. As we become aware of updated information or new developments, these estimates and assumptions may change and materially impact reported amounts. We consider the following accounting policies to be critical to our consolidated financial statements because they require the use of our judgment and estimates (including those that are forward-looking) in their application.
Revenue Recognition
Remodulin, Tyvaso, Orenitram and Unituxin
We market Remodulin, Tyvaso, Orenitram and Unituxin to specialty distributors in the United States and other distributors internationally under materially similar contractual arrangements. Net product sales of Remodulin, Tyvaso, Orenitram and Unituxin are recognized when title and risk of ownership pass to our distributors upon satisfactory delivery to our distributors' facilities—i.e., when all of our performance obligations under these distributor arrangements have been satisfied. We record
80
sales of Remodulin, Tyvaso, Orenitram and Unituxin net of: (1) estimated rebates; (2) prompt payment discounts; (3) service fees we pay to distributors; and (4) an allowance for return rights. Determining sales allowances involves the use of significant estimates and judgment and may involve the use of information from external sources.
We derive our provisions for rebates from an analysis of historical levels of rebates to both state Medicaid agencies and commercial third-party payers by product, relative to sales of each product. In addition, we determine our obligation for prescription drug discounts required for Medicare Part D Orenitram patients within the coverage gap based on estimations of the number of Medicare Part D Orenitram patients and the period that such patients will remain within the coverage gap. In formulating our estimates, we also consider the impact of anticipated changes in product prices, sales trends and changes to government rebate programs, particularly as they relate to eligibility requirements and/or rebate pricing. We analyze rebate data separately for Remodulin, Tyvaso and Orenitram, as these therapies have different routes of administration to treat PAH patients at different stages in the disease continuum and therefore, rebate eligibility and pricing requirements can differ for each therapy.
We estimate prompt pay discounts based on observed payment behavior. Our distributors have routinely taken advantage of these discounts and we expect them to continue to do so.
We pay our distributors for contractual services rendered and accrue for related fees based on contractual rates applied to the estimated units of service provided by distributors for a given financial reporting period.
Our distributors do not possess return rights for Remodulin, Tyvaso and Orenitram; however, the sales terms for Unituxin include return rights that extend throughout the distribution channel. We provide exchange rights for all products in the event that product is damaged during shipment or expires. Exchanges for damaged product are highly infrequent. In the event that Remodulin, Tyvaso, Orenitram or Unituxin has been damaged during shipment and we have been promptly notified as required under our distribution agreements, we do not recognize revenue on that shipment until damaged product has been replaced. Replacement of damaged product generally occurs within several days after notification of the damage. Furthermore, the number of product exchanges due to expiration has been minimal because we sell Remodulin, Tyvaso and Orenitram with a remaining shelf life in excess of one year and our distributors typically carry a thirty- to sixty-day supply of our treprostinil-based products at any given time. In addition, we closely track inventory levels held by our distributors. Except for contractual minimum inventory levels to prevent shortages of treprostinil-based drug supply, we do not require, nor do we provide incentives for our distributors to assume, inventory levels of Remodulin, Tyvaso, Orenitram or Unituxin beyond what would be considered reasonable and customary in the ordinary course of business.
The financial effects of exchange rights for Remodulin, Tyvaso, Orenitram and Unituxin have been immaterial and we expect the volume of exchanges to be consistent with historical levels. Specifically, exchanges of Remodulin, Tyvaso, Orenitram and Unituxin have comprised substantially less than one percent of the volume of the units that we sell. Because historical and anticipated future exchanges of Remodulin, Tyvaso, Orenitram and Unituxin have been and are expected to be immaterial, we do not record a reserve for estimated exchange rights in the period of sale. Lastly, we closely monitor product exchange data for all of these therapies to ensure that our assumptions continue to be reasonable, appropriate and current.
Adcirca
Adcirca is manufactured for us by Eli Lilly and Company (Lilly) and distributed through Lilly's pharmaceutical wholesaler network. Specifically, Lilly handles all of the administrative functions associated with the sale of Adcirca on our behalf, including the receipt and processing of customer
81
purchase orders, shipment to customers, and invoicing and collection of customer payments. In addition, the sales terms for Adcirca include return rights that extend throughout the distribution channel. We recognize sales of Adcirca on a gross basis (net of allowances) upon delivery to customers due to the following factors: (1) we are responsible for the acceptability of the product purchased by wholesalers; (2) we bear all inventory risk, as title and risk of loss pass to us at the shipping point from Lilly's manufacturing facility; (3) we assume credit risk if Lilly is unable to collect amounts due from customers; and (4) we assume the risk and cost of a product recall, if required.
We recognize sales of Adcirca net of: (1) estimated government-based and commercial payer rebates; (2) prompt pay discounts; (3) allowances for product returns; and (4) wholesaler fees. We estimate our liability for rebates based on an analysis of historical levels of rebates to both Medicaid and commercial third-party payers and we consider the impact of sales trends, changes in government and commercial rebate programs and anticipated changes in Adcirca's pricing. In addition, for Adcirca patients, we determine our obligation for prescription drug discounts required by Medicare Part D for patients within the coverage gap based on estimations of the number of patients and the period that such patients will remain within the coverage gap. We base our estimates for prompt pay discounts on observed customer payment behavior and expectations regarding the future utilization of such discounts. To date, we have not identified any unusual patterns in the volume of prescriptions relative to sales that would warrant reconsideration of, or adjustment to, the methodology we currently employ to estimate our allowance for returns. Lastly, wholesaler fees are based on contractual percentages of sales to wholesalers.
Share-Based Compensation
Our share-based awards are classified as either equity (stock options and our employee stock purchase plan) or as liabilities (STAP awards). We recognize related share-based compensation expense based on the fair value of the options granted to purchase stock and on outstanding STAP awards. We estimate the fair value of all share-based awards using the Black-Scholes-Merton valuation model. Valuation models, like the Black-Scholes-Merton model, require the use of subjective assumptions that could materially impact the estimation of fair value and related compensation expense to be recognized. These assumptions include, among others, the expected volatility of our stock price, the expected term of awards and the expected forfeiture rate. Developing these assumptions requires the use of judgment.
Pension Benefit Obligation
Accounting for our Supplemental Executive Retirement Plan (SERP) requires that we recognize in our consolidated balance sheet a liability equal to the unfunded status of the SERP (the total estimated projected benefit obligation, as we do not fund the SERP) and measure our projected benefit obligation as of the end of our fiscal year. Estimating the SERP obligation involves the use of judgment and estimates. The SERP obligation and related pension expense are derived from actuarial valuations that are developed using a number of assumptions. A key assumption underlying the valuation is the discount rate. The discount rate should be representative of the rate associated with high-quality, fixed-income debt securities. We must consider prevailing economic conditions and outlook, the state of the credit markets and other economic factors when determining an appropriate discount rate to employ. Changes in the discount rate can significantly increase or decrease our SERP obligation. For instance, a reduction in the discount rate would increase our projected benefit obligation and result in an actuarial loss. Consequently, we could be required to recognize additional pension expense in our consolidated statements of operations related to the actuarial loss in future periods if certain thresholds are met. Other actuarial assumptions include participant demographics such as the expected date of retirement, rate of salary increases and withdrawal rates, among other factors. Not only can actual experience differ
82
from actuarial assumptions, but changes in any of these assumptions can also materially affect the measurement of the SERP obligation.
Income Taxes
Income taxes are accounted for in accordance with the asset and liability method. Accordingly, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their tax bases. Deferred tax assets and liabilities are measured using the enacted tax rates that are expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Deferred tax assets are reduced by a valuation allowance when, in our opinion, it is more likely than not that some or all of the deferred tax assets will not be realized. Evaluating whether deferred assets will be realized requires us to review forecasts of earnings and taxable income, among other considerations. Accordingly, the evaluation of deferred tax assets requires us to make significant judgments and forward-looking assessments regarding the amounts and availability of future taxable income.
Financial statement recognition of a tax position taken or expected to be taken in a tax return is determined based on a more likely than not threshold of that position being sustained. If the tax position meets this threshold, the benefit to be recognized is measured as the largest amount that is more than 50 percent likely to be realized upon ultimate settlement. Accounting for uncertain tax positions involves considerable judgment in assessing the future tax consequences of amounts that have been recognized in our financial statements or tax returns. The ultimate resolution of uncertain tax positions could result in amounts different from those recognized in our consolidated financial statements.
Recently Issued Accounting Standards
See Note 3—Recently Issued Accounting Standards, to our consolidated financial statements for information on our anticipated adoption of recently issued accounting standards.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As of December 31, 2015, we have invested $160.0 million in corporate-debt securities and federally-sponsored agencies. The market value of these investments varies inversely with changes in prevailing market interest rates. In general, as interest rates increase, the market value of a debt investment would be expected to decrease. Conversely, as interest rates decrease, the market value of a debt investment would be expected to increase. To date, we have not experienced significant volatility in the value of these investments. However, to address market risk, we invest in debt securities with terms no longer than three years and hold these investments to maturity so that they can be redeemed at their stated or face value. At December 31, 2015, our investments in debt securities issued by corporations and federally-sponsored agencies had a weighted average stated interest rate of approximately 0.98 percent and a weighted average maturity of 1.0 years. Many of our investments may be called by their respective issuers prior to maturity.
During sustained periods of instability and uncertainty in the financial markets, we may be subjected to additional investment-related risks that could materially affect the value and liquidity of our investments. In light of these risks, we actively monitor market conditions and developments specific to the securities and security classes in which we invest. In addition, we believe that we maintain a conservative investment approach in that we invest exclusively in unstructured, highly-rated securities with relatively short maturities that we believe reduce our exposure to undue risks. While we believe we take prudent measures to mitigate investment related risks, such risks cannot be fully eliminated, as circumstances can occur that are beyond our control.
83
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
UNITED THERAPEUTICS CORPORATION
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
The
Board of Directors and Shareholders
United Therapeutics Corporation
We have audited the accompanying consolidated balance sheets of United Therapeutics Corporation as of December 31, 2015 and 2014, and the related consolidated statements of operations, comprehensive income, stockholders' equity, and cash flows for each of the three years in the period ended December 31, 2015. Our audits also included the financial statement schedule listed in the Index at Item 15(a)(2). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of United Therapeutics Corporation at December 31, 2015 and 2014, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the Standards of the Public Company Accounting Oversight Board (United States), United Therapeutics Corporation's internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 25, 2016 expressed an unqualified opinion thereon.
| /s/ Ernst & Young LLP |
McLean,
Virginia
February 25, 2016
F-2
Report of Independent Registered Public Accounting Firm on
Internal Control over Financial Reporting
The
Board of Directors and Shareholders
United Therapeutics Corporation
We have audited United Therapeutics Corporation's internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). United Therapeutics Corporation's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that: (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion United Therapeutics Corporation maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of United Therapeutics Corporation as of December 31, 2015 and 2014 and the related consolidated statements of operations, comprehensive income, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2015 and our report dated February 25, 2016, expressed an unqualified opinion thereon.
| /s/ Ernst & Young LLP |
McLean,
Virginia
February 25, 2016
F-3
UNITED THERAPEUTICS CORPORATION
Consolidated Balance Sheets
(In thousands, except share and per share data)
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Assets |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 831,798 | $ | 397,697 | |||
Marketable investments |
121,974 | 297,842 | |||||
Accounts receivable, net of allowance of none for 2015 and 2014 |
192,827 | 162,287 | |||||
Inventories, net |
81,334 | 66,927 | |||||
Other current assets |
47,402 | 32,836 | |||||
| | | | | | | | |
Total current assets |
1,275,335 | 957,589 | |||||
Marketable investments |
38,002 | 122,658 | |||||
Goodwill and other intangible assets, net |
28,378 | 29,465 | |||||
Property, plant, and equipment, net |
495,774 | 478,421 | |||||
Deferred tax assets, net |
192,676 | 198,329 | |||||
Other assets |
154,280 | 97,948 | |||||
| | | | | | | | |
Total assets |
$ | 2,184,445 | $ | 1,884,410 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Liabilities and Stockholders' Equity |
|||||||
Current liabilities: |
|||||||
Accounts payable and accrued expenses |
$ | 103,433 | $ | 85,382 | |||
Convertible notes |
5,387 | 126,414 | |||||
Share tracking awards plan |
274,542 | 282,101 | |||||
Other current liabilities |
57,462 | 10,413 | |||||
| | | | | | | | |
Total current liabilities |
440,824 | 504,310 | |||||
Other liabilities |
143,974 | 114,526 | |||||
| | | | | | | | |
Total liabilities |
584,798 | 618,836 | |||||
Commitments and contingencies—Note 9 |
|||||||
Temporary equity |
11,095 | 23,218 | |||||
Stockholders' equity: |
|||||||
Preferred stock, par value $.01, 10,000,000 shares authorized, no shares issued |
— | — | |||||
Series A junior participating preferred stock, par value $.01, 100,000 shares authorized, no shares issued |
— | — | |||||
Common stock, par value $.01, 245,000,000 shares authorized, 68,987,919 and 65,988,561 shares issued, and 45,760,845 and 47,107,709 shares outstanding at December 31, 2015 and 2014, respectively |
690 | 660 | |||||
Additional paid-in capital |
1,790,620 | 1,376,141 | |||||
Accumulated other comprehensive loss |
(20,401 | ) | (16,734 | ) | |||
Treasury stock, 23,227,074 and 18,880,852 shares at December 31, 2015 and 2014, respectively |
(1,902,110 | ) | (1,185,825 | ) | |||
Retained earnings |
1,719,753 | 1,068,114 | |||||
| | | | | | | | |
Total stockholders' equity |
1,588,552 | 1,242,356 | |||||
| | | | | | | | |
Total liabilities and stockholders' equity |
$ | 2,184,445 | $ | 1,884,410 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
See accompanying notes to consolidated financial statements.
F-4
UNITED THERAPEUTICS CORPORATION
Consolidated Statements of Operations
(In thousands, except per share data)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Revenues: |
||||||||||
Net product sales |
$ | 1,460,570 | $ | 1,279,533 | $ | 1,106,944 | ||||
Other |
5,191 | 8,986 | 10,040 | |||||||
| | | | | | | | | | | |
Total revenues |
1,465,761 | 1,288,519 | 1,116,984 | |||||||
Operating expenses: |
||||||||||
Research and development |
245,098 | 242,549 | 299,348 | |||||||
Selling, general and administrative |
452,612 | 381,287 | 394,010 | |||||||
Cost of product sales |
69,036 | 125,883 | 131,127 | |||||||
| | | | | | | | | | | |
Total operating expenses |
766,746 | 749,719 | 824,485 | |||||||
Operating income |
699,015 | 538,800 | 292,499 | |||||||
Other (expense) income: |
||||||||||
Interest expense |
(4,735 | ) | (17,592 | ) | (18,058 | ) | ||||
Gain on sale of intangible asset |
350,000 | — | — | |||||||
Other, net |
157 | 3,972 | 4,462 | |||||||
| | | | | | | | | | | |
Total other income (expense), net |
345,422 | (13,620 | ) | (13,596 | ) | |||||
Income before income taxes |
1,044,437 | 525,180 | 278,903 | |||||||
Income tax expense |
(392,798 | ) | (185,106 | ) | (104,343 | ) | ||||
| | | | | | | | | | | |
Net income |
$ | 651,639 | $ | 340,074 | $ | 174,560 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net income per common share: |
||||||||||
Basic |
$ | 14.17 | $ | 7.06 | $ | 3.49 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
$ | 12.72 | $ | 6.28 | $ | 3.28 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted average number of common shares outstanding: |
||||||||||
Basic |
46,000 | 48,176 | 50,076 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
51,221 | 54,155 | 53,231 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-5
UNITED THERAPEUTICS CORPORATION
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(In thousands)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Net income |
$ | 651,639 | $ | 340,074 | $ | 174,560 | ||||
Other comprehensive (loss) income: |
||||||||||
Foreign currency translation loss |
(5,296 | ) | (4,789 | ) | (1,193 | ) | ||||
Defined benefit pension plan: |
||||||||||
Prior service cost arising during period, net of tax |
— | (2,415 | ) | — | ||||||
Actuarial gain arising during period, net of tax |
826 | 2,999 | 2,075 | |||||||
Less: amortization of actuarial gain and prior service cost included in net periodic pension cost, net of tax |
877 | 904 | 1,020 | |||||||
| | | | | | | | | | | |
Total defined benefit pension plan, net |
1,703 | 1,488 | 3,095 | |||||||
Unrealized loss on available-for-sale securities, net of tax |
(74 | ) | (250 | ) | (128 | ) | ||||
| | | | | | | | | | | |
Other comprehensive (loss) gain, net of tax |
(3,667 | ) | (3,551 | ) | 1,774 | |||||
| | | | | | | | | | | |
Comprehensive income |
$ | 647,972 | $ | 336,523 | $ | 176,334 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-6
UNITED THERAPEUTICS CORPORATION
Consolidated Statements of Stockholders' Equity
(In thousands, except share data)
| |
Common Stock | |
Accumulated Other Comprehensive Income/(Loss) |
|
|
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-in Capital |
Treasury Stock |
Retained Earnings |
Stockholders' Equity |
||||||||||||||||||
| |
Shares | Amount | ||||||||||||||||||||
Balance, December 31, 2012 |
62,082,007 | $ | 621 | $ | 1,015,835 | $ | (14,957 | ) | $ | (470,998 | ) | $ | 553,480 | $ | 1,083,981 | |||||||
Net income |
— | — | — | — | — | 174,560 | 174,560 | |||||||||||||||
Foreign currency translation adjustments |
— | — | — | (1,193 | ) | — | — | (1,193 | ) | |||||||||||||
Unrealized gain on available-for-sale securities |
— | — | — | (128 | ) | — | — | (128 | ) | |||||||||||||
Defined benefit pension plan |
— | — | — | 3,095 | — | — | 3,095 | |||||||||||||||
Shares issued under employee stock purchase plan |
55,070 | 1 | 2,734 | — | — | — | 2,735 | |||||||||||||||
Equity component—2016 convertible notes (Note 10) |
— | — | (34,155 | ) | — | — | — | (34,155 | ) | |||||||||||||
Repurchase of shares |
— | — | — | — | (42,439 | ) | — | (42,439 | ) | |||||||||||||
Exercise of stock options |
876,115 | 8 | 26,611 | — | — | — | 26,619 | |||||||||||||||
Tax benefit from exercises of non-qualified stock options |
— | — | 9,299 | — | — | — | 9,299 | |||||||||||||||
Share-based compensation |
— | — | 36,900 | — | — | — | 36,900 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2013 |
63,013,192 | 630 | 1,057,224 | (13,183 | ) | (513,437 | ) | 728,040 | 1,259,274 | |||||||||||||
Net income |
— | — | — | — | — | 340,074 | 340,074 | |||||||||||||||
Foreign currency translation adjustments |
— | — | — | (4,789 | ) | — | — | (4,789 | ) | |||||||||||||
Unrealized (loss) on available-for-sale securities |
— | — | — | (250 | ) | — | — | (250 | ) | |||||||||||||
Defined benefit pension plan |
— | — | — | 1,488 | — | — | 1,488 | |||||||||||||||
Shares issued under employee stock purchase plan |
45,657 | 1 | 3,329 | — | — | — | 3,330 | |||||||||||||||
Conversion of 2016 convertible notes (Note 10) |
1,467,343 | 15 | 192,966 | — | (189,311 | ) | — | 3,670 | ||||||||||||||
Equity component—2016 convertible notes (Note 10) |
— | — | 11,056 | — | — | — | 11,056 | |||||||||||||||
Repurchase of shares |
— | — | — | — | (483,077 | ) | — | (483,077 | ) | |||||||||||||
Exercise of stock options |
1,462,369 | 14 | 50,154 | — | — | — | 50,168 | |||||||||||||||
Tax benefit from exercises of non-qualified stock options |
— | — | 30,845 | — | — | — | 30,845 | |||||||||||||||
Share-based compensation |
— | — | 30,567 | — | — | — | 30,567 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2014 |
65,988,561 | 660 | 1,376,141 | (16,734 | ) | (1,185,825 | ) | 1,068,114 | 1,242,356 | |||||||||||||
Net income |
— | — | — | — | — | 651,639 | 651,639 | |||||||||||||||
Foreign currency translation adjustments |
— | — | — | (5,296 | ) | — | — | (5,296 | ) | |||||||||||||
Unrealized (loss) on available-for-sale securities |
— | — | — | (74 | ) | — | — | (74 | ) | |||||||||||||
Defined benefit pension plan |
— | — | — | 1,703 | — | — | 1,703 | |||||||||||||||
Shares issued under employee stock purchase plan |
36,198 | — | 3,954 | — | — | — | 3,954 | |||||||||||||||
Conversion of 2016 convertible notes (Note 10) |
1,977,577 | 20 | 324,701 | — | (321,801 | ) | — | 2,920 | ||||||||||||||
Equity component—2016 convertible notes (Note 10) |
— | — | 3,020 | — | — | — | 3,020 | |||||||||||||||
Repurchase of shares |
— | — | — | — | (394,484 | ) | — | (394,484 | ) | |||||||||||||
Exercise of stock options |
985,583 | 10 | 39,301 | — | — | — | 39,311 | |||||||||||||||
Tax benefit from exercises of non-qualified stock options |
— | — | 37,426 | — | — | — | 37,426 | |||||||||||||||
Share-based compensation |
— | — | 6,077 | — | — | — | 6,077 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2015 |
68,987,919 | $ | 690 | $ | 1,790,620 | $ | (20,401 | ) | $ | (1,902,110 | ) | $ | 1,719,753 | $ | 1,588,552 | |||||||
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-7
UNITED THERAPEUTICS CORPORATION
Consolidated Statements of Cash Flows
(In thousands)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Cash flows from operating activities: |
||||||||||
Net income |
$ | 651,639 | $ | 340,074 | $ | 174,560 | ||||
Adjustments to reconcile net income to net cash provided by operating activities: |
||||||||||
Depreciation and amortization |
32,921 | 32,245 | 31,259 | |||||||
Share-based compensation expense |
280,254 | 190,054 | 320,786 | |||||||
Gain on sale of intangible asset |
(350,000 | ) | — | — | ||||||
Amortization of debt discount and debt issue costs |
5,941 | 12,456 | 12,601 | |||||||
Amortization of discount or premium on investments |
2,132 | 5,231 | 4,501 | |||||||
Other |
(515 | ) | 6,493 | 3,182 | ||||||
Excess tax benefits from share-based compensation |
(37,426 | ) | (30,845 | ) | (9,299 | ) | ||||
Changes in operating assets and liabilities: |
||||||||||
Accounts receivable |
(30,540 | ) | (35,689 | ) | (10,027 | ) | ||||
Inventories |
(6,794 | ) | (21,032 | ) | (12,394 | ) | ||||
Accounts payable and accrued expenses |
16,972 | (6,753 | ) | 7,507 | ||||||
Other assets and liabilities |
(181,823 | ) | (136,975 | ) | (97,409 | ) | ||||
| | | | | | | | | | | |
Net cash provided by operating activities |
382,761 | 355,259 | 425,267 | |||||||
| | | | | | | | | | | |
Cash flows from investing activities: |
||||||||||
Purchases of property, plant and equipment, net |
(49,792 | ) | (47,439 | ) | (31,910 | ) | ||||
Purchases of held-to-maturity investments |
(62,781 | ) | (118,672 | ) | (762,198 | ) | ||||
Maturities of held-to-maturity investments |
320,369 | 549,576 | 529,900 | |||||||
Gain on sale of intangible asset |
350,000 | — | — | |||||||
Purchase of investments under the cost method, net |
(54,217 | ) | (45,000 | ) | (30,766 | ) | ||||
| | | | | | | | | | | |
Net cash provided by (used in) investing activities |
503,579 | 338,465 | (294,974 | ) | ||||||
| | | | | | | | | | | |
Cash flows from financing activities: |
||||||||||
Principal payments of debt |
(133,150 | ) | (177,800 | ) | (1,320 | ) | ||||
Payments to repurchase common stock |
(394,484 | ) | (483,077 | ) | (42,439 | ) | ||||
Proceeds from line of credit |
— | 140,000 | — | |||||||
Payments on the line of credit |
— | (140,000 | ) | — | ||||||
Proceeds from exercise of stock options |
39,311 | 50,168 | 26,611 | |||||||
Issuance of stock under employee stock purchase plan |
3,954 | 3,329 | 2,734 | |||||||
Excess tax benefits from share-based compensation |
37,426 | 30,845 | 9,299 | |||||||
| | | | | | | | | | | |
Net cash used in financing activities |
(446,943 | ) | (576,535 | ) | (5,115 | ) | ||||
| | | | | | | | | | | |
Effect of exchange rate changes on cash and cash equivalents |
(5,296 | ) | (3,750 | ) | (319 | ) | ||||
Net increase in cash and cash equivalents |
434,101 | 113,439 | 124,859 | |||||||
Cash and cash equivalents, beginning of year |
397,697 | 284,258 | 159,399 | |||||||
| | | | | | | | | | | |
Cash and cash equivalents, end of year |
$ | 831,798 | $ | 397,697 | $ | 284,258 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplemental cash flow information: |
||||||||||
Cash paid for interest |
$ | 1,015 | $ | 5,453 | $ | 5,518 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cash paid for income taxes |
$ | 293,331 | $ | 195,564 | $ | 142,140 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Non-cash investing and financing activities: |
||||||||||
Acquisitions—non-cash consideration |
— | $ | 5,200 | $ | — | |||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Non-cash additions to property, plant and equipment |
$ | 1,078 | $ | 3,150 | $ | 9,018 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Issuance of common stock upon conversion of convertible notes |
$ | 321,801 | $ | 189,311 | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-8
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements
1. Organization and Business Description
United Therapeutics Corporation is a biotechnology company focused on the development and commercialization of innovative products to address the unmet medical needs of patients with chronic and life-threatening diseases.
We have approval from the United States Food and Drug Administration (FDA) to market the following therapies: Remodulin® (treprostinil) Injection (Remodulin), Tyvaso® (treprostinil) Inhalation Solution (Tyvaso), Adcirca® (tadalafil) Tablets (Adcirca), Orenitram® (treprostinil) Extended-Release Tablets (Orenitram) and Unituxin® (dinutuximab) Injection (Unituxin). Remodulin has also been approved in various countries outside the United States, and Unituxin was granted marketing authorization by the European Medicines Agency in August 2015. Tyvaso is also approved in Israel. We commenced commercial sales of Orenitram and Unituxin in the U.S. during the second quarter of 2014 and the third quarter of 2015, respectively.
As used in these notes to the consolidated financial statements, unless the context otherwise requires, the terms "we", "us", "our", and similar terms refer to United Therapeutics Corporation and its consolidated subsidiaries.
2. Summary of Significant Accounting Policies
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements of United Therapeutics Corporation and its wholly owned subsidiaries have been prepared in accordance with accounting principles generally accepted in the United States (GAAP). All intercompany balances and transactions have been eliminated in consolidation.
In the operating section of our statement of cash flows, we reclassified the prior period amounts within "current and deferred income tax expense" to "other assets and liabilities" to conform with the current period presentation.
Use of Estimates
The preparation of the consolidated financial statements in accordance with GAAP requires our management to make estimates and assumptions that affect reported amounts of assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. We base our estimates on assumptions regarding historical experience, currently available information and anticipated developments that we believe are reasonable and appropriate. However, because the use of estimates involves an inherent degree of uncertainty, actual results could differ from those estimates. Our significant accounting policies that require use of subjective and/or complex judgment and estimates impact the following financial statement areas: revenue recognition, share-based compensation, marketable investments, fair value measurements (including those relating to our acquisitions), income taxes, goodwill and other intangible assets, and obligations related to our Supplemental Executive Retirement Plan.
Fair Value of Financial Instruments
The carrying amounts of cash and cash equivalents, accounts receivables, accounts payable, and accrued expenses approximate fair value because of their short maturities. The fair values of our
F-9
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
marketable investments and 1.0 percent Convertible Senior Notes due September 15, 2016 (Convertible Notes) are reported in Note 4—Investments and Note 5—Fair Value Measurements, respectively.
Fair Value Measurements
Fair value is a market-based measurement, not an entity-specific measurement. The objective of a fair value measurement is to estimate the price to sell an asset or transfer a liability in an orderly transaction between market participants at the measurement date under current market conditions. Such transactions to sell an asset or transfer a liability are assumed to occur in the principal market for that asset or liability, or in the absence of the principal market, the most advantageous market for the asset or liability.
Assets and liabilities subject to fair value measurement disclosures are required to be classified according to a three-level fair value hierarchy with respect to the inputs (or assumptions) used to determine fair value. Observable inputs such as unadjusted quoted market prices for identical assets or liabilities are given the highest priority within the hierarchy (Level 1). When observable inputs are unavailable, fair value is measured using unobservable inputs—i.e., inputs that a reporting entity believes market participants would use in pricing that are developed based on the best information available. Unobservable inputs are given the lowest priority within the hierarchy (Level 3). The level in which an asset or liability is disclosed within the fair value hierarchy is based on the lowest level input that is significant to the related fair value measurement in its entirety. The guidance under the fair value measurement framework applies to other existing accounting guidance in the Financial Accounting Standard Board (FASB) codification that requires or permits fair value measurements. Refer to related disclosures at Note 5—Fair Value Measurements to these consolidated financial statements.
Cash Equivalents
Cash equivalents consist of highly liquid investments with maturities of three months or less from the date of acquisition and include money market funds, commercial paper, and certificates of deposit.
Marketable Investments
Substantially all of our marketable investments are debt securities that we classify as held-to-maturity because of our positive intent and ability to hold the securities until maturity. Held-to-maturity securities are classified as either current or non-current assets on our consolidated balance sheets based on their contractual maturity dates and are recorded at amortized cost, adjusted for the amortization of discounts or premiums. Related discounts and premiums are amortized over the term of these securities as an adjustment to yield using the effective interest method.
We monitor our investment portfolio for impairment quarterly or more frequently if circumstances warrant. In the event that the carrying value of an investment exceeds its fair value and the decline in value is determined to be other-than-temporary, we record an impairment charge within earnings attributable to the estimated credit loss. In determining whether a decline in the value of an investment is other-than-temporary, we evaluate currently available factors that may include, among others: (1) general market conditions; (2) the duration and extent to which fair value has been less than the carrying value; (3) the investment issuer's financial condition and business outlook; and (4) our
F-10
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
assessment as to whether it is more likely than not that we will be required to sell a security prior to recovery of its amortized cost basis.
Trade Receivables
Trade receivables consist of short-term amounts due from customers and are stated at the amount we expect to collect. We establish an allowance for doubtful accounts, if any, based on our assessment of the collectability of specific customer accounts.
Inventories
Inventories are stated at the lower of cost (first-in, first-out method) or market (current replacement cost) and consist of the following, net of reserves (in thousands):
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Raw materials |
$ | 23,093 | $ | 21,317 | |||
Work-in-progress |
22,494 | 15,994 | |||||
Finished goods |
35,747 | 29,616 | |||||
| | | | | | | | |
Total inventories |
$ | 81,334 | $ | 66,927 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Goodwill and Other Intangible Assets
The carrying amount of goodwill is not amortized but is subject to annual impairment testing. We conduct our impairment testing of goodwill annually during the fourth quarter, or more frequently, if impairment indicators exist. Initially, we evaluate various pertinent qualitative factors to assess whether it is more likely than not that the fair value of a reporting unit to which goodwill has been assigned is less than its carrying value. Such qualitative factors can include, among others: (1) industry and market conditions; (2) present and anticipated sales and cost factors; and (3) overall financial performance. If we conclude based on our qualitative assessment that it is more likely than not that the fair value of a reporting unit is less than its carrying value, we then measure the fair value of the reporting unit and compare its fair value to its carrying value (Step 1 of the goodwill impairment test). If the carrying amount of the reporting unit exceeds its fair value, then the amount of an impairment loss, if any, is measured as the excess of the recorded amount of goodwill over its implied fair value (Step 2 of the goodwill impairment test). The Company used a qualitative assessment for its goodwill impairment testing for 2015 and 2014. The Company's evaluation of goodwill completed during the years ended December 31, 2015 and 2014 resulted in no impairment losses.
Indefinite-lived intangible assets are not amortized but are evaluated annually or more frequently for impairment if impairment indicators exist. The Company's indefinite-lived intangible assets include purchased in-process research and development projects, which were measured at their estimated fair values as of their acquisition dates. The Company used a qualitative assessment for its indefinite-lived intangible asset impairment testing. The Company's evaluation of indefinite-lived intangible assets completed during the years ended December 31, 2015 and 2014 resulted in no impairment losses.
Intangible assets subject to amortization are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an intangible asset may not be recoverable.
F-11
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Impairment losses are measured and recognized to the extent the carrying value of such assets exceeds their fair value. The Company recorded no impairment losses during the years ended December 31, 2015 and 2014.
Goodwill and other intangible assets comprise the following (in thousands):
| |
As of December 31, 2015 | As of December 31, 2014 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Gross | Accumulated Amortization |
Net | Gross | Accumulated Amortization |
Net | |||||||||||||
Goodwill |
$ | 10,264 | $ | — | $ | 10,264 | $ | 10,264 | $ | — | $ | 10,264 | |||||||
Other intangible assets: |
|||||||||||||||||||
Technology, patents and trade names |
6,494 | (4,691 | ) | 1,803 | 6,494 | (4,100 | ) | 2,394 | |||||||||||
In-process, research and development |
15,500 | — | 15,500 | 15,500 | — | 15,500 | |||||||||||||
Customer relationships and non-compete agreements |
4,369 | (3,558 | ) | 811 | 4,369 | (3,062 | ) | 1,307 | |||||||||||
Contract-based |
1,270 | (1,270 | ) | — | 1,270 | (1,270 | ) | — | |||||||||||
| | | | | | | | | | | | | | | | | | | | |
Total |
$ | 37,897 | $ | (9,519 | ) | $ | 28,378 | $ | 37,897 | $ | (8,432 | ) | $ | 29,465 | |||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Related amortization expense for the years ended December 31, 2015, 2014 and 2013, was $1.1 million, $1.4 million and $2.6 million, respectively. As of December 31, 2015, aggregate amortization expense relating to definite lived intangible assets for each of the five succeeding years and thereafter is estimated as follows (in thousands):
Year Ended December 31,
|
|
|||
|---|---|---|---|---|
2016 |
$ | 621 | ||
2017 |
456 | |||
2018 |
125 | |||
2019 |
125 | |||
2020 |
125 | |||
Thereafter |
1,162 | |||
| | | | | |
|
$ | 2,614 | ||
| | | | | |
| | | | | |
| | | | | |
In September 2015, we sold the Rare Pediatric Priority Review Voucher (PPRV) we received from the FDA in connection with the approval of Unituxin for $350.0 million in cash. The proceeds from the sale of the PPRV were recognized as a gain on the sale of an intangible asset, as the PPRV did not have a carrying value on our consolidated balance sheet at the time of sale.
F-12
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Property, Plant and Equipment
Property, plant and equipment is recorded at cost and depreciated over its estimated useful life using the straight-line method. The estimated useful lives of property, plant and equipment by major category are as follows:
| Land improvements | 15 Years | |
| Buildings | 25 - 39 Years | |
| Building improvements | 10 - 39 Years | |
| Furniture, equipment and vehicles | 3 - 20 Years | |
| Leasehold improvements | Remaining lease term, or the estimated useful life of the improvement, whichever is shorter |
Property, plant and equipment consists of the following (in thousands):
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Land and land improvements |
$ | 61,091 | $ | 46,141 | |||
Buildings, building improvements and leasehold improvements |
418,245 | 413,066 | |||||
Buildings under construction |
26,458 | 17,379 | |||||
Furniture, equipment and vehicles |
144,862 | 136,805 | |||||
| | | | | | | | |
|
650,656 | 613,391 | |||||
Less—accumulated depreciation |
(154,882 | ) | (134,970 | ) | |||
| | | | | | | | |
Property, plant and equipment, net |
$ | 495,774 | $ | 478,421 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Depreciation expense for the years ended December 31, 2015, 2014 and 2013 was $31.8 million, $30.8 million and $28.6 million, respectively.
Buildings under construction consists of direct costs relating to our construction projects and includes capitalized interest.
Treasury Stock
Repurchased treasury stock is recorded at cost, including commissions and fees. Treasury stock acquired from the convertible note hedge on our Convertible Notes is recorded at the fair value on the acquisition date closing price of our common stock. The cost of treasury shares sold is determined using the first-in, first-out method. Related gains and losses on sales of treasury stock are recognized as adjustments to stockholders' equity.
Revenue Recognition
Remodulin, Tyvaso, Orenitram and Unituxin
We sell Remodulin, Tyvaso, Orenitram and Unituxin to distributors under similar contractual arrangements. We recognize sales of these products when title and risk of ownership pass to our
F-13
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
distributors upon satisfactory delivery—i.e., when all of our performance obligations under our distribution agreements have been satisfied. We record sales of these products net of various product sales allowances in the period that associated revenues are recognized. These sales allowances include estimated rebates, prompt payment discounts, sales returns and service fees paid to our distributors. Calculating these sales allowances involves the use of significant estimates and judgments and information obtained from external sources.
We derive our provisions for rebates from an analysis of historical levels of rebates to both state Medicaid agencies and commercial third-party payers by product, relative to sales of each product. In addition, for Orenitram patients, we determine our obligation for prescription drug discounts required by Medicare Part D for patients within the coverage gap based on estimations of the number of patients and the period that such patients will remain within the coverage gap. In formulating our estimates, we also consider the impact of anticipated changes in our product pricing, if any, sales trends and government rebate programs, particularly as they relate to eligibility requirements and/or rebate pricing.
We estimate prompt pay discounts based on observed payment behavior. Our distributors have routinely taken advantage of these discounts and we expect them to continue to do so.
Our distributors do not possess return rights for Remodulin, Tyvaso and Orenitram; however, the sales terms for Unituxin include return rights that extend throughout the distribution channel. We provide exchange rights for all products in the event that product is damaged during shipment or expires. Exchanges for damaged product are highly infrequent. In the event that Remodulin, Tyvaso, Orenitram, or Unituxin has been damaged during shipment and we have been promptly notified as required under our distribution agreements, we do not recognize revenue on that shipment until damaged product has been replaced. Replacement of damaged product generally occurs within several days after notification of the damage. Furthermore, the number of product exchanges due to expiration has been minimal because we sell Remodulin, Tyvaso and Orenitram with a remaining shelf life in excess of one year and our distributors typically carry a thirty- to sixty-day supply of our treprostinil-based products at any given time. In addition, we closely track inventory levels held by our distributors. Except for contractual minimum inventory levels to prevent shortages of treprostinil-based drug supply, we do not require, nor do we provide incentives for our distributors to assume, inventory levels of Remodulin, Tyvaso, Orenitram or Unituxin beyond what would be considered reasonable and customary in the ordinary course of business.
We pay our distributors for contractual services rendered and accrue for related fees based on contractual rates applied to the estimated units of service provided by distributors for a given financial reporting period.
The financial effects of exchange rights for Remodulin, Tyvaso, Orenitram and Unituxin have been immaterial and we expect the volume of exchanges to be consistent with historical levels. Specifically, exchanges of Remodulin, Tyvaso, Orenitram and Unituxin have comprised substantially less than one percent of the volume of the units that we sell. Because historical and anticipated future exchanges of Remodulin, Tyvaso, Orenitram and Unituxin have been and are expected to be immaterial, we do not record a reserve for estimated exchange rights in the period of sale. Lastly, we closely monitor product exchange data for all of these therapies to ensure that our assumptions continue to be reasonable, appropriate and current.
F-14
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Adcirca
Adcirca is manufactured for us by Eli Lilly and Company (Lilly) and distributed through Lilly's pharmaceutical wholesaler network. Specifically, Lilly handles all of the administrative functions associated with the sale of Adcirca on our behalf, including the receipt and processing of customer purchase orders, shipment to customers, and invoicing and collection of customer payments. In addition, the sales terms for Adcirca include return rights that extend throughout the distribution channel. We recognize sales of Adcirca on a gross basis (net of allowances) upon delivery to customers due to the following factors: (1) we are responsible for the acceptability of the product purchased by wholesalers; (2) we bear all inventory risk, as title and risk of loss pass to us at the shipping point from Lilly's manufacturing facility; (3) we assume credit risk if Lilly is unable to collect amounts due from customers; and (4) we assume the risk and cost of a product recall, if required.
We recognize sales of Adcirca net of: (1) estimated government-based and commercial payer rebates; (2) prompt pay discounts; (3) allowances for product returns; and (4) wholesaler fees. We estimate our liability for rebates based on an analysis of historical levels of rebates to both Medicaid and commercial third-party payers and we consider the impact of sales trends, changes in government and commercial rebate programs and anticipated changes in Adcirca's pricing. In addition, for Adcirca patients, we determine our obligation for prescription drug discounts required by Medicare Part D for patients within the coverage gap based on estimations of the number of patients and the period that such patients will remain within the coverage gap. We base our estimates for prompt pay discounts on observed customer payment behavior and expectations regarding the future utilization of such discounts. We derive our allowance for returns of Adcirca based on historical return rates accumulated since the commercial launch of Adcirca in 2009. To date, we have not identified any unusual patterns in the volume of prescriptions relative to sales that would warrant reconsideration of, or adjustment to, the methodology we currently employ to estimate our allowance for returns. Lastly, wholesaler fees are based on contractual percentages of sales to wholesalers.
Research and Development
Research and development costs are expensed as incurred except for refundable payments made in advance of services to be provided to us. Related expenses consist of internal labor and overhead, costs to acquire pharmaceutical products and product rights for development, materials used in clinical trials and amounts paid to third parties for services and materials relating to drug development and clinical trials.
We recognize the following as research and development expense in the period related costs are incurred:
- •
- Costs associated with in-house or contracted production activities prior to receiving FDA approval for such facilities, or for major
unproven changes to our production processes;
- •
- Costs incurred in licensing the rights to technologies in the research and development stage that have no alternative future uses; and
- •
- Up-front payments made in connection with arrangements to obtain license and distribution rights to pharmaceutical product candidates prior to regulatory approval, absent any alternative future uses.
F-15
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Share-Based Compensation
Our share tracking award plans require cash settlement upon exercise and are classified as a liability. Accordingly, the fair value of related cash-settled awards is re-measured at each reporting date until awards are exercised or are otherwise no longer outstanding. Related changes in the fair value of outstanding cash-settled awards at each financial reporting date are recognized as adjustments to share-based compensation expense.
Generally, the fair value of a stock option grant is measured on its grant date and related compensation expense is recognized ratably over the requisite service period. For stock option awards that vest immediately upon issuance, compensation expense is recognized in its entirety based on the grant-date fair value. We issue new shares of our common stock upon the exercise of stock options.
We measure the fair value of stock to be purchased through our employee stock purchase plan at the beginning of an offering period, or grant date, and recognize related compensation expense ratably over the requisite service period (the offering period). We issue new shares of our common stock upon the end of each offering period, or exercise date.
Income Taxes
Income taxes are accounted for in accordance with the asset and liability method. Accordingly, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their tax bases. Deferred tax assets and liabilities are measured using the enacted tax rates that are expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in the period that includes the enactment date. Deferred tax assets are reduced by a valuation allowance when, in our judgment, it is more likely than not that some or all of the deferred tax assets will not be realized.
Financial statement recognition of a tax position taken or expected to be taken in a tax return is determined based on a more likely than not threshold of that position being sustained. If the tax position meets this threshold, the benefit to be recognized is measured as the largest amount that is more than 50 percent likely to be realized upon ultimate settlement. It is our policy to record interest and penalties related to uncertain tax positions as a component of income tax expense.
Earnings (Loss) per Share
Basic earnings per share is computed by dividing net income by the weighted average number of shares of common stock outstanding during the period. Diluted earnings per common share is computed by dividing net income by the weighted average number of shares of common stock outstanding during the period, plus the potential dilutive effect of other securities if such securities were converted or exercised. During periods in which we incur net losses, both basic and diluted loss per share is calculated by dividing the net loss by the weighted average shares outstanding—potentially dilutive securities are excluded from the calculation because their effect would be anti-dilutive.
F-16
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Concentrations of Credit Risk, Products, Revenues and Customers
Concentration of credit risk
Financial instruments that are exposed to credit risk consist of cash, money market funds, commercial paper, marketable investments, and trade receivables. We maintain our cash and money market funds with financial institutions that are federally insured. While balances deposited in these institutions often exceed Federal Deposit Insurance Corporation limits, we have not experienced any losses on related accounts to date. Furthermore, we limit our risk exposure by maintaining funds in financial institutions that we believe are creditworthy and financially sound. Our investments in marketable debt securities have been issued by corporate entities and federally-sponsored enterprises with high credit ratings. We mitigate investment risks by investing in highly-rated securities with relatively short maturities that we believe do not subject us to undue investment or credit risk. In addition, our investment policy does not provide for investments in complex or structured financial instruments. At any given time, our trade receivables are concentrated among a small number of principal customers. If any of these financial institutions, issuers or customers fail to perform their obligations under the terms of these financial instruments, our maximum exposure to potential losses would be equal to amounts reported on our consolidated balance sheets.
Concentration of products, revenues, and customers
In the United States, through 2013 we sold Remodulin, Tyvaso, and Orenitram to three specialty pharmaceutical distributors: Accredo Health Group Inc. (Accredo), CuraScript Inc. (CuraScript) and CVS Caremark. In December 2013, the operations of CuraScript were integrated into Accredo's operations as a result of the 2012 acquisition of Medco Health Solutions, Inc., the parent company of Accredo, by Express Scripts, Inc., the parent company of CuraScript, and we consolidated our distribution agreements with CuraScript and Accredo into one contract for each product. During the years ended December 31, 2015, 2014 and 2013, net product sales of Remodulin, Tyvaso and Orenitram to these distributors accounted for 72 percent, 74 percent and 76 percent, respectively, of our total revenues. During the years ended December 31, 2015, 2014 and 2013, net product sales of Remodulin accounted for 39 percent, 43 percent and 44 percent, respectively, of our total revenues, while net product sales of Tyvaso during this period comprised 32 percent, 36 percent and 39 percent, respectively of our total revenues. During the years ended December 31, 2015 and 2014, net product sales of Orenitram accounted for 8 percent and 3 percent, respectively, of our total revenues.
At December 31, 2015 and 2014, 50 percent and 52 percent, respectively, of our accounts receivable was due from U.S.-based specialty distributors.
During the years ended December 31, 2015, 2014 and 2013, we derived 55 percent, 58 percent and 57 percent of our total revenues from one customer. Estimated total revenues from that customer were as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Accredo Health Group, Inc. |
$ | 807,012 | $ | 744,765 | $ | 632,599 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-17
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
3. Recently Issued Accounting Standards
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (ASU 2014-09). ASU 2014-09 will eliminate transaction-specific and industry-specific revenue recognition guidance under current GAAP and replace it with a principle-based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. ASU 2014-09 also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09 is effective for annual reporting periods beginning after December 15, 2016. Early application is not permitted. ASU 2014-09 allows for either full retrospective or modified retrospective adoption. On July 9, 2015, the FASB issued ASU No. 2015-14, Revenue from Contracts with Customers (Topic 606); Deferral of the Effective Date, which (1) delays the effective date of ASU 2014-09 by one year to annual periods beginning after December 15, 2017; and (2) allows early adoption of the ASU by all entities as of the original effective date for public entities. We are evaluating the transition method we will elect and the effects of the adoption of this ASU on our financial statements.
In April 2015, the FASB issued ASU No. 2015-03, Simplifying the Presentation of Debt Issuance Costs (ASU 2015-03), which requires that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. ASU 2015-03 requires retrospective adoption and will be effective for us beginning in our first quarter of 2016. Early adoption is permitted. We do not expect the adoption of ASU 2015-03 to have a material impact on our financial statements.
In July 2015, the FASB issued ASU No. 2015-11, Simplifying the Measurement of Inventory (ASU 2015-11), which requires that inventory be measured at the lower of cost or net realizable value for entities using first-in, first-out or average cost methods. ASU 2015-11 should be applied prospectively and will be effective for fiscal years beginning after December 15, 2016, and for interim periods within those fiscal years, with early adoption permitted. We are evaluating the effect of adoption on our financial statements.
In November 2015, the FASB issued ASU No. 2015-17, Balance Sheet Classification of Deferred Taxes (ASU 2015-17), which requires deferred tax assets and liabilities to be classified as noncurrent in a classified statement of financial position. The guidance is effective for financial statements issued for annual periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2018. Earlier application is permitted for all entities as of the beginning of an interim or annual reporting period. This amendment may be applied either prospectively or retrospectively to all periods presented.
We early adopted the provisions of ASU 2015-17 in the fourth quarter of 2015 on a retrospective basis. The adoption of this ASU will simplify the presentation of deferred income taxes and reduce complexity without decreasing the usefulness of information provided to users of financial statements. The adoption resulted in an $18.1 million and $16.6 million decrease in other current assets and a corresponding increase to deferred tax assets, net in our consolidated balance sheets at December 31, 2015 and 2014, respectively. The adoption had no impact on our results of operations or cash flows.
F-18
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
4. Investments
Marketable Investments
Held-to-Maturity Investments
Marketable investments classified as held-to-maturity consist of the following (in thousands):
As of December 31, 2015
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Government-sponsored enterprises |
$ | 53,254 | $ | 3 | $ | (119 | ) | $ | 53,138 | ||||
Corporate notes and bonds |
106,722 | 37 | (67 | ) | 106,692 | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 159,976 | $ | 40 | $ | (186 | ) | $ | 159,830 | ||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Reported under the following captions on the consolidated balance sheet: |
|||||||||||||
Current marketable investments |
$ | 121,974 | |||||||||||
Noncurrent marketable investments |
38,002 | ||||||||||||
| | | | | | | | | | | | | | |
|
$ | 159,976 | |||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
As of December 31, 2014
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Government-sponsored enterprises |
$ | 127,212 | $ | 118 | $ | (39 | ) | $ | 127,291 | ||||
Corporate notes and bonds |
293,288 | 260 | (108 | ) | 293,440 | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 420,500 | $ | 378 | $ | (147 | ) | $ | 420,731 | ||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Reported under the following captions on the consolidated balance sheet: |
|||||||||||||
Current marketable investments |
$ | 297,842 | |||||||||||
Noncurrent marketable investments |
122,658 | ||||||||||||
| | | | | | | | | | | | | | |
|
$ | 420,500 | |||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
F-19
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
4. Investments (Continued)
The following table summarizes gross unrealized losses and the length of time marketable investments have been in a continuous unrealized loss position (in thousands):
| |
As of December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||||||||
| |
Fair Value |
Gross Unrealized Loss |
Fair Value |
Gross Unrealized Loss |
|||||||||
Government-sponsored enterprises: |
|||||||||||||
Continuous unrealized loss position less than one year |
$ | 48,138 | $ | (119 | ) | $ | 15,293 | $ | (39 | ) | |||
Continuous unrealized loss position greater than one year |
— | — | — | — | |||||||||
| | | | | | | | | | | | | | |
|
48,138 | (119 | ) | 15,293 | (39 | ) | |||||||
Corporate notes and bonds: |
|||||||||||||
Continuous unrealized loss position less than one year |
63,840 | (67 | ) | 86,824 | (97 | ) | |||||||
Continuous unrealized loss position greater than one year |
— | — | 3,443 | (11 | ) | ||||||||
| | | | | | | | | | | | | | |
|
63,840 | (67 | ) | 90,267 | (108 | ) | |||||||
| | | | | | | | | | | | | | |
Total |
$ | 111,978 | $ | (186 | ) | $ | 105,560 | $ | (147 | ) | |||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
We attribute the unrealized losses on held-to-maturity securities as of December 31, 2015 and 2014, to the variability in related market interest rates. We do not intend to sell these securities, nor is it more likely than not that we will be required to sell them prior to the end of their contractual terms. Furthermore, we do not believe that these securities expose us to undue market risk or counterparty credit risk. As such, we do not consider these securities to be other than temporarily impaired.
The following table summarizes the contractual maturities of held-to-maturity marketable investments (in thousands):
| |
As of December 31, 2015 | ||||||
|---|---|---|---|---|---|---|---|
| |
Amortized Cost |
Fair Value |
|||||
Due in less than one year |
$ | 121,974 | $ | 121,932 | |||
Due in one to two years |
38,002 | 37,898 | |||||
Due in three to five years |
— | — | |||||
Due after five years |
— | — | |||||
| | | | | | | | |
Total |
$ | 159,976 | $ | 159,830 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Investments Held at Cost
As of December 31, 2015, we maintain in the aggregate, non-controlling equity investments of approximately $137.2 million in privately-held corporations, including a $100.0 million investment in the preferred stock of Synthetic Genomics Inc. (SGI), which we purchased in two separate $50.0 million transactions in May 2014 and September 2015. We account for these investments under the cost method since we do not have the ability to exercise significant influence over these companies and their
F-20
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
4. Investments (Continued)
fair values are not readily determinable. The fair value of these investments has not been estimated at December 31, 2015, as we have not identified any events or developments indicating that their carrying amounts may be impaired. We include these investments within other assets on our accompanying consolidated balance sheets.
In addition to the SGI investments noted above, we entered into a separate multi-year research and development collaboration agreement with SGI in May 2014, whereby SGI will develop engineered primary pig cells with modified genomes for use in our xenotransplantation program. This collaboration was initially focused primarily on lungs and was expanded in September 2015 to include an additional focus on kidneys. Under this agreement, each party assumes its own research and development costs and SGI may receive royalties and milestone payments from development and commercialization of organs.
5. Fair Value Measurements
Assets and liabilities subject to fair value measurements are required to be disclosed within a fair value hierarchy. The fair value hierarchy ranks the quality and reliability of inputs used to determine fair value. Accordingly, assets and liabilities carried at, or permitted to be carried at, fair value are classified within the fair value hierarchy in one of the following categories based on the lowest level input that is significant in measuring fair value:
Level 1—Fair value is determined by using unadjusted quoted prices that are available in active markets for identical assets and liabilities.
Level 2—Fair value is determined by using inputs other than Level 1 quoted prices that are directly or indirectly observable. Inputs can include quoted prices for similar assets and liabilities in active markets or quoted prices for identical assets and liabilities in inactive markets. Related inputs can also include those used in valuation or other pricing models such as interest rates and yield curves that can be corroborated by observable market data.
Level 3—Fair value is determined by using inputs that are unobservable and not corroborated by market data. Use of these inputs involves significant and subjective judgment.
We account for certain assets and liabilities at fair value and rank these assets within a fair value hierarchy (Level 1, Level 2 or Level 3). Our other current assets and our other current liabilities have
F-21
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
5. Fair Value Measurements (Continued)
fair values that approximate their carrying values. Assets and liabilities subject to fair value measurements are as follows (in thousands):
| |
As of December 31, 2015 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Level 1 | Level 2 | Level 3 | Balance | |||||||||
Assets |
|||||||||||||
Money market funds(1) |
$ | 496,434 | $ | — | $ | — | $ | 496,434 | |||||
Federally-sponsored and corporate debt securities(2) |
— | 159,830 | — | 159,830 | |||||||||
| | | | | | | | | | | | | | |
Total assets |
$ | 496,434 | $ | 159,830 | $ | — | $ | 656,264 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Liabilities |
|||||||||||||
Convertible notes due 2016(3) |
$ | 15,998 | $ | — | $ | — | $ | 15,998 | |||||
Contingent consideration(4) |
— | — | 9,400 | 9,400 | |||||||||
| | | | | | | | | | | | | | |
Total liabilities |
$ | 15,998 | $ | — | $ | 9,400 | $ | 25,398 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| |
As of December 31, 2014 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Level 1 | Level 2 | Level 3 | Balance | |||||||||
Assets |
|||||||||||||
Money market funds(1) |
$ | 298,416 | $ | — | $ | — | $ | 298,416 | |||||
Federally-sponsored and corporate debt securities(2) |
— | 420,731 | — | 420,731 | |||||||||
| | | | | | | | | | | | | | |
Total assets |
$ | 298,416 | $ | 420,731 | $ | — | $ | 719,147 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Liabilities |
|||||||||||||
Convertible notes due 2016(3) |
$ | 388,153 | $ | — | $ | — | $ | 388,153 | |||||
Contingent consideration(4) |
— | — | 11,502 | 11,502 | |||||||||
| | | | | | | | | | | | | | |
Total liabilities |
$ | 388,153 | $ | — | $ | 11,502 | $ | 399,655 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Included
in cash and cash equivalents on the accompanying consolidated balance sheets.
- (2)
- Included
in current and non-current marketable investments on the accompanying consolidated balance sheets. The fair value of these securities is
principally measured or corroborated by trade data for identical securities in which related trading activity is not sufficiently frequent to be considered a Level 1 input or comparable
securities that are more actively traded. See also Note 4—Investments—Marketable Investments—Held-to-Maturity
Investments to these consolidated financial statements.
- (3)
- Included in convertible notes on the accompanying consolidated balance sheets. The fair value of our Convertible Notes is estimated using Level 1 observable inputs since our Convertible Notes are trading with sufficient frequency such that we believe related pricing can be used as the primary basis for measuring their fair value. As of December 31, 2015 and December 31, 2014, the fair value of the Convertible Notes was substantially higher than their book value. This was primarily due to the excess conversion value of the notes compared to the notes' par value, and the fact that any such excess would be paid in shares of our common stock.
F-22
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
5. Fair Value Measurements (Continued)
- (4)
- Included in other liabilities on the accompanying consolidated balance sheets. The fair value of contingent consideration has been estimated using probability weighted discounted cash flow models (DCF). The DCF incorporates Level 3 inputs including estimated discount rates that we believe market participants would consider relevant in pricing and the projected timing and amount of cash flows, which are estimated and developed, in part, based on the requirements specific to each acquisition agreement. We analyze and evaluate these fair value measurements quarterly to determine whether valuation inputs continue to be relevant and appropriate or whether current period developments warrant adjustments to valuation inputs and related measurements.
6. Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses consist of the following by major categories (in thousands):
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Accounts payable |
$ | 7,502 | $ | 6,995 | |||
Accrued expenses: |
|||||||
Sales related (royalties, rebates and fees) |
52,832 | 38,095 | |||||
Payroll related |
31,437 | 28,019 | |||||
Research related |
7,635 | 7,500 | |||||
Other |
4,027 | 4,773 | |||||
| | | | | | | | |
Total accrued expenses |
95,931 | 78,387 | |||||
| | | | | | | | |
Total accounts payable and accrued expenses |
$ | 103,433 | $ | 85,382 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
7. Share Tracking Award Plans
We previously issued awards under the United Therapeutics Corporation Share Tracking Awards Plan, adopted in June 2008 (2008 STAP) and the United Therapeutics Corporation 2011 Share Tracking Awards Plan, adopted in March 2011 (2011 STAP). We refer to the 2008 STAP and the 2011 STAP collectively as the "STAP" and awards granted and/or outstanding under either of these plans as "STAP awards." STAP awards convey the right to receive in cash an amount equal to the appreciation of our common stock, which is measured as the increase in the closing price of our common stock between the dates of grant and exercise. STAP awards expire on the tenth anniversary of the date of grant, and in most cases they vest in equal increments on each anniversary of the grant date over a four-year period. The STAP liability includes vested awards and awards that are expected to vest. We recognize expense for awards that are expected to vest during the vesting period. We discontinued the issuance of STAP awards on June 26, 2015, when our shareholders approved the United Therapeutics Corporation 2015 Stock Incentive Plan (the 2015 Plan), a broad-based stock incentive plan enabling us to grant stock options and other forms of equity compensation to our employees. See Note 11—Stockholders' Equity to these consolidated financial statements for information on the 2015 Plan.
The aggregate balance of the STAP liability was $354.8 million and $322.7 million at December 31, 2015 and 2014, respectively, of which $80.2 million and $40.6 million, respectively, has been classified as
F-23
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
7. Share Tracking Award Plans (Continued)
non-current liabilities under the caption "Other liabilities" on our consolidated balance sheets based on their vesting terms.
Estimating the fair value of STAP awards requires the use of certain inputs that can materially impact the determination of fair value and the amount of compensation expense (benefit) we recognize. Inputs used in estimating fair value include the price of our common stock, the expected volatility of the price of our common stock, the risk-free interest rate, the expected term of STAP awards, the expected forfeiture rate and the expected dividend yield. The fair value of the STAP awards is measured each financial reporting period because the awards are settled in cash.
A description of the key inputs, requiring estimates, used in determining the fair value of the awards is provided below:
Expected volatility—Volatility is a measure of the amount the price of our common stock has fluctuated (historical volatility) or is expected to fluctuate (expected volatility) during a period. We use historical volatility based on weekly price observations of our common stock during the period immediately preceding an award that is equal to its expected term up to a maximum period of five years. We believe the volatility in the price of our common stock over the preceding five years generally provides a reliable projection of future long-term volatility.
Risk-free interest rate—The risk-free interest rate is the average interest rate consistent with the yield available on a U.S. Treasury note with a term equal to the expected term of an award.
Expected term—The expected term reflects the estimated time period we expect an award to remain outstanding. For the years ended December 31, 2015 and 2014, we used historical data to develop this input. Prior to 2014, we applied the simplified method to develop an estimate of the expected term. The change in methodologies for calculating the expected term of an award did not have a significant impact to our consolidated financial statements.
Expected forfeiture rate—The expected forfeiture rate is an estimated percentage of awards granted that are expected to be forfeited or canceled on an annual basis prior to becoming fully vested. We derive our estimate based on historical forfeiture experience for similar classes of employees.
Expected dividend yield—We do not pay cash dividends on our common stock and do not expect to do so in the future. Therefore, the dividend yield is zero.
The table below presents the assumptions used to measure the fair value of STAP Awards:
| |
As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Expected volatility |
35.3 | % | 34.0 | % | 32.7 | % | ||||
Risk-free interest rate |
1.4 | % | 1.3 | % | 1.1 | % | ||||
Expected term of awards (in years) |
3.4 | 4.0 | 3.9 | |||||||
Expected forfeiture rate |
8.8 | % | 9.3 | % | 10.1 | % | ||||
Expected dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||
F-24
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
7. Share Tracking Award Plans (Continued)
A summary of the status and activity of the STAP is presented below:
| |
Number of Awards |
Weighted- Average Exercise Price |
Weighted Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in 000s) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at January 1, 2015 |
7,716,424 | $ | 62.59 | ||||||||||
Granted |
1,655,388 | 159.68 | |||||||||||
Exercised |
(2,281,711 | ) | 56.71 | ||||||||||
Forfeited |
(244,938 | ) | 95.37 | ||||||||||
| | | | | | | | | | | | | | |
Outstanding at December 31, 2015 |
6,845,163 | $ | 86.86 | 7.3 | $ | 487,010 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Exercisable at December 31, 2015 |
2,115,571 | $ | 67.79 | 6.1 | $ | 188,683 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Expected to vest at December 31, 2015 |
4,306,092 | $ | 95.34 | 7.9 | $ | 271,767 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The weighted average grant-date fair value of STAP awards granted during the years ended December 31, 2015, 2014 and 2013 was $58.52, $33.82 and $24.78, respectively.
Share-based compensation expense recognized in connection with the STAP is as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Research and development |
$ | 87,372 | $ | 72,269 | $ | 134,355 | ||||
Selling, general and administrative |
178,100 | 82,937 | 143,407 | |||||||
Cost of product sales |
8,706 | 4,283 | 6,124 | |||||||
| | | | | | | | | | | |
Share-based compensation expense before taxes |
274,178 | 159,489 | 283,886 | |||||||
Related income tax benefit |
(103,431 | ) | (56,560 | ) | (106,693 | ) | ||||
| | | | | | | | | | | |
Share-based compensation expense, net of taxes |
$ | 170,747 | $ | 102,929 | $ | 177,193 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Share-based compensation capitalized as part of inventory |
$ | 7,063 | $ | 2,027 | $ | 1,593 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Cash paid to settle STAP exercises during the years ended December 31, 2015, 2014 and 2013 was $248.8 million, $144.1 million, and $55.9 million, respectively.
8. Debt
Unsecured Revolving Credit Facility
In January 2016, we entered into a Credit Agreement (the 2016 Credit Agreement) with Wells Fargo Bank, National Association (Wells Fargo), as administrative agent and a swingline lender, and various other lender parties, providing for an unsecured revolving credit facility of up to $1.0 billion (the Revolving Facility), which is available to refinance certain of our existing indebtedness and/or for working capital and other general corporate purposes. The Revolving Facility will mature five years after the closing date of the 2016 Credit Agreement, subject to the lenders' ability to extend the
F-25
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
8. Debt (Continued)
maturity date by one year if we request such an extension in accordance with the terms of the 2016 Credit Agreement.
At our option, amounts borrowed under the Revolving Facility will bear interest at either the LIBOR rate or a fluctuating base rate, in each case, plus an applicable margin determined on a quarterly basis based on our consolidated ratio of total indebtedness to EBITDA (as calculated in accordance with the 2016 Credit Agreement).
The 2016 Credit Agreement contains customary events of default and customary affirmative and negative covenants. As of January 29, 2016, we were in compliance with such covenants and we had not drawn any amounts on the Revolving Facility. In addition, Lung Biotechnology PBC is our only subsidiary that guarantees our obligations under the 2016 Credit Agreement though, from time to time, one or more of our other subsidiaries may be required to guarantee such obligations.
Secured Line of Credit
In September 2013, we entered into a credit agreement (the 2013 Credit Agreement) with Wells Fargo Bank, National Association (Wells Fargo) providing us a $75.0 million revolving loan facility. In July 2015, we amended the credit agreement solely to extend its maturity to September 30, 2017. At our option, amounts borrowed under the 2013 Credit Agreement bore interest at either the one-month LIBOR rate plus a 0.50 percent margin, or a fluctuating base rate excluding any margin. In addition, we were subject to a monthly commitment fee of 0.06 percent per annum on the average daily unused balance of the facility. Amounts borrowed under the 2013 Credit Agreement were secured by certain of our marketable investments. As of December 31, 2015, we had no outstanding balance on the facility. In January 2016, we terminated and repaid in full all obligations under the 2013 Credit Agreement when we entered into the 2016 Credit Agreement.
Convertible Notes Due 2016
In October 2011, we issued $250.0 million in aggregate principal value 1.0 percent Convertible Senior Notes due September 15, 2016 (Convertible Notes). The Convertible Notes are unsecured, unsubordinated debt obligations that rank equally with all of our other unsecured and unsubordinated indebtedness. We pay interest semi-annually on March 15 and September 15 of each year. The initial conversion price is $47.69 per share and the number of underlying shares used to determine the aggregate consideration upon conversion is approximately 5.2 million shares.
Conversion can occur: (1) any time after June 15, 2016; (2) during any calendar quarter that follows a calendar quarter in which the price of our common stock exceeds 130 percent of the conversion price for at least 20 days during the 30 consecutive trading-day period ending on the last trading day of the quarter; (3) during the ten consecutive trading-day period following any five consecutive trading-day period in which the trading price of the Convertible Notes is less than 95 percent of the closing price of our common stock multiplied by the then-current number of shares underlying the Convertible Notes; (4) upon specified distributions to our shareholders; (5) in connection with certain corporate transactions; or (6) in the event that our common stock ceases to be listed on the NASDAQ Global Select Market, the NASDAQ Global Market or the New York Stock Exchange, or any of their respective successors. The closing price of our common stock exceeded 130 percent of the conversion price of the Convertible Notes for more than 20 trading days during the 30 consecutive trading day period ended December 31, 2015. Consequently, the Convertible Notes are
F-26
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
8. Debt (Continued)
convertible at the election of their holders. As the Convertible Notes have a maturity date of September 15, 2016, they are classified as a current liability on our consolidated balance sheet at December 31, 2015.
At December 31, 2015, the aggregate conversion value of the Convertible Notes exceeded their par value by $12.8 million using a conversion price of $156.61, the closing price of our common stock on December 31, 2015.
Upon conversion, holders of our Convertible Notes are entitled to receive: (1) cash equal to the lesser of the par value of the notes or the conversion value (the number of shares underlying the Convertible Notes multiplied by the then current conversion price per share); and (2) to the extent the conversion value exceeds the par value of the notes, shares of our common stock. In the event of a change in control, as defined in the indenture under which the Convertible Notes have been issued, holders can require us to purchase all or a portion of their Convertible Notes for 100 percent of the notes' par value plus any accrued and unpaid interest.
During the year ended December 31, 2015, we settled conversion requests representing $133.2 million in principal value of our Convertible Notes. We paid $133.2 million in principal and issued 2.0 million shares of our common stock during the settlement process. We received 2.0 million shares of our common stock under our convertible note hedge (discussed below under Convertible Note Hedge and Warrant Transactions) from Deutsche Bank AG London (DB London) which we placed into our treasury stock account. We recognized a $3.7 million extinguishment loss with the settlement of these conversions. As of December 31, 2015, 117,000 shares of our common stock could be issued upon future conversions of our outstanding Convertible Notes.
The terms of the Convertible Notes provide for settlement wholly or partially in cash. Consequently, we are required to account for their liability and equity components separately so that the subsequent recognition of interest expense reflects our non-convertible borrowing rate. Accordingly, as of the date of issuance, we estimated the fair value of the Convertible Notes without consideration of the conversion option (Liability Component). The excess of the proceeds received over the estimated fair value of the Liability Component totaling $57.9 million has been recorded as the conversion option (Equity Component) and a corresponding offset has been recognized as a discount to the Convertible Notes to reduce their net carrying value. A portion of the Equity Component equal to the unamortized discount as of December 31, 2015 has been reclassified to temporary equity because one of the contingent conversion criteria had been met at December 31, 2015, as disclosed above. Refer to Note 10—Temporary Equity. We are amortizing the discount over the five-year period ending September 15, 2016 (the expected life of the Liability Component) using the effective interest method and an effective rate of interest of 6.7 percent, which corresponded to our estimated non-convertible borrowing rate at the date of issuance.
Interest expense incurred in connection with our convertible notes consisted of the following (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Contractual coupon rate of interest |
$ | 404 | $ | 2,151 | $ | 2,500 | ||||
Discount amortization |
3,020 | 11,057 | 11,178 | |||||||
| | | | | | | | | | | |
Interest expense—convertible notes |
$ | 3,424 | $ | 13,208 | $ | 13,678 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-27
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
8. Debt (Continued)
The carrying value of our convertible notes consisted of the following (in thousands):
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Principal balance |
$ | 5,600 | $ | 138,750 | |||
Discount, net of accumulated amortization of $1,085 and $19,819 |
(213 | ) | (12,336 | ) | |||
| | | | | | | | |
Carrying amount |
$ | 5,387 | $ | 126,414 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Convertible Note Hedge and Warrant Transactions
In connection with the issuance of our Convertible Notes, we entered into separate convertible note hedge and warrant transactions with DB London to reduce the potentially dilutive impact of the conversion of our convertible notes. Pursuant to the convertible note hedge, we purchased call options to acquire up to approximately 5.2 million shares of our common stock with a strike price of $47.69. The call options become exercisable upon any conversions and the maturity of the Convertible Notes, and will terminate upon the maturity of the Convertible Notes or the first day the Convertible Notes are no longer outstanding, whichever occurs first. The call options will offset on a share for share basis, any shares of our common stock that we issue upon any conversion or at the maturity of our Convertible Notes. As of December 31, 2015, approximately 117,000 shares of our common stock remained under the call options after the settlement of $133.2 million of conversion requests during the year ended December 31, 2015. We also sold DB London warrants to acquire up to approximately 5.2 million shares of our common stock with a strike price of $67.56. The warrants will expire incrementally on a series of expiration dates subsequent to the maturity date of our Convertible Notes. Both the convertible note hedge and warrant transactions will be settled on a net-share basis. To the extent that the price of our common stock exceeds the strike price of the warrants on any or all of the series of related incremental expiration dates, we will be required to issue shares of our common stock to DB London.
Mortgage Financing—Wells Fargo Bank
In December 2010, we entered into a Credit Agreement with Wells Fargo and Bank of America, N.A., pursuant to which we obtained a $70.0 million mortgage loan (the 2010 Credit Agreement). The 2010 Credit Agreement matured in December 2014 and we repaid the outstanding $66.5 million principal balance in full.
Interest Expense
Details of interest expense presented on our consolidated statements of operations are as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Interest expense |
$ | 4,735 | $ | 17,592 | $ | 18,117 | ||||
Less: interest capitalized |
— | — | (59 | ) | ||||||
| | | | | | | | | | | |
Total interest expense |
$ | 4,735 | $ | 17,592 | $ | 18,058 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-28
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
9. Commitments and Contingencies
Operating Leases
We lease facilities and equipment under operating lease arrangements that have terms expiring at various dates through 2020. Certain lease arrangements include renewal options and escalation clauses. In addition, various lease agreements to which we are party require that we comply with certain customary covenants throughout the term of these leases. If we are unable to comply with these covenants and cannot reach a satisfactory resolution in the event of noncompliance, these agreements could terminate.
Future minimum lease payments under non-cancelable operating leases as of December 31, 2015 are as follows (in thousands):
Year Ending December 31,
|
|
|||
|---|---|---|---|---|
2016 |
$ | 3,716 | ||
2017 |
3,447 | |||
2018 |
2,636 | |||
2019 |
498 | |||
2020 |
237 | |||
Thereafter |
— | |||
| | | | | |
Total |
$ | 10,534 | ||
| | | | | |
| | | | | |
| | | | | |
Total rent expense was $3.8 million, $3.6 million and $3.5 million for the years ended December 31, 2015, 2014 and 2013, respectively.
Milestone Payments
We are party to certain license agreements as described in Note 15—Assignment and License Agreements and acquisition agreements. Generally, these agreements require that we make milestone payments in cash upon the achievement of certain product development and commercialization goals and payments of royalties upon commercial sales.
Future milestone payments based on our estimates of the timing and probability of achieving milestones specified under these arrangements are as follows (in thousands):
Year Ending December 31,
|
(1) | |||
|---|---|---|---|---|
2016 |
$ | 6,136 | ||
2017 |
4,640 | |||
2018 |
3,705 | |||
2019 |
9,711 | |||
2020 |
1,759 | |||
Thereafter |
7,070 | |||
| | | | | |
Total |
$ | 33,021 | ||
| | | | | |
| | | | | |
| | | | | |
- (1)
- The amounts and timing of future milestone payments may vary depending on when related milestones will be attained, if at all.
F-29
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
10. Temporary Equity
Temporary equity includes securities that: (1) have redemption features that are outside our control; (2) are not classified as an asset or liability; (3) are excluded from permanent stockholders' equity; and (4) are not mandatorily redeemable. Amounts included in temporary equity relate to securities that are redeemable at a fixed or determinable price.
Components comprising the carrying value of temporary equity include the following (in thousands):
| |
As of December 31, 2015 |
As of December 31, 2014 |
|||||
|---|---|---|---|---|---|---|---|
Reclassification of Equity Component(1) |
$ | 213 | $ | 12,336 | |||
Common stock subject to repurchase(2) |
10,882 | 10,882 | |||||
| | | | | | | | |
Total |
$ | 11,095 | $ | 23,218 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
- (1)
- Represents
the reclassification of the Equity Component equal to the unamortized debt discount of our Convertible Notes as of December 31, 2015 and
2014, respectively, from additional paid-in capital to temporary equity. Our Convertible Notes were convertible at the election of their holders as noted above in
Note 8—Debt—Convertible Notes Due 2016.
- (2)
- In connection with our license agreement with Toray Industries Inc. (Toray), we issued 200,000 shares of our common stock (which have since split into 400,000 shares) to Toray in 2007, and provided Toray the right to require us to repurchase the shares at a price of $27.21 per share.
11. Stockholders' Equity
Equity Incentive Plans
As of December 31, 2015, we have two shareholder-approved equity incentive plans: the United Therapeutics Corporation Amended and Restated Equity Incentive Plan (the 1999 Plan) and the United Therapeutics Corporation 2015 Stock Incentive Plan (the 2015 Plan). Although the terms of the 1999 Plan and the 2015 Plan contemplate a variety of awards, to date all awards granted under these plans have been in the form of stock options. The 2015 Plan was approved by our shareholders in June 2015 and provides for the issuance of up to 6,150,000 shares of our common stock pursuant to awards granted under the 2015 Plan. During the year ended December 31, 2015, we granted zero stock options under the 1999 plan and 172,250 stock options under the 2015 Plan. During the year ended December 31, 2014, we granted 723,869 stock options under the 1999 Plan. No further awards will be granted under the 1999 Plan.
Employee Stock Options
We estimate the fair value of stock options using the Black-Scholes-Merton valuation model. Option-pricing models, including the Black-Scholes-Merton model, require the use of judgment and subjective assumptions that can materially impact the estimation of fair value and share-based compensation.
F-30
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
11. Stockholders' Equity (Continued)
Inputs included in estimating the fair value of a stock option include the price of our common stock, the expected volatility of our common stock, risk-free interest rate, the expected term of stock option awards, expected forfeiture rate and the expected dividend yield.
A description of the key inputs, requiring estimates, used in determining the fair value of stock options is provided below:
Expected volatility—Volatility is a measure of the amount the price of our common stock has fluctuated (historical volatility) or is expected to fluctuate (expected volatility) during a period. We use historical volatility based on weekly price observations of our common stock during the period immediately preceding a stock option grant that is equal to the expected term of the grant (up to a maximum of five years). We believe the volatility of the price of our common stock measured over the preceding five years provides a reliable projection of future long-term volatility.
Risk-free interest rate—The risk-free interest rate is the average interest rate consistent with the yield available on a U.S. Treasury note with a term equal to the expected term of a given stock option grant.
Expected term—The expected term reflects the estimated time period we expect an option grant to remain outstanding. We use historical data to develop this input.
Expected forfeiture rate—The expected forfeiture rate is the estimated percentage of options granted that are expected to be forfeited or canceled on an annual basis prior to becoming fully vested. We derive our estimate based on historical forfeiture experience for similar classes of employees.
Expected dividend yield—We do not pay dividends on our common stock and do not expect to do so in the future. Therefore, the dividend yield is assumed to be zero.
The following weighted-average assumptions were used in estimating the fair value of stock options granted to employees:
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Expected volatility |
33.1% | 32.6% | |||||
Risk-free interest rate |
2.0% | 1.7% | |||||
Expected term of options (in years) |
5.8 | 5.0 | |||||
Expected forfeiture rate |
1.5% | 0.0% | |||||
Expected dividend yield |
0.0% | 0.0% | |||||
F-31
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
11. Stockholders' Equity (Continued)
A summary of the status and activity of employee stock options is presented below:
| |
Options | Weighted- Average Exercise Price |
Weighted Average Remaining Contractual Term (in Years) |
Aggregate Intrinsic Value (in 000s) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at January 1, 2015 |
4,054,771 | $ | 76.83 | ||||||||||
Granted |
172,250 | 173.25 | |||||||||||
Exercised |
(984,583 | ) | 39.90 | ||||||||||
Forfeited |
— | — | |||||||||||
| | | | | | | | | | | | | | |
Outstanding at December 31, 2015 |
3,242,438 | $ | 93.17 | 6.6 | $ | 208,643 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Exercisable at December 31, 2015 |
3,070,188 | $ | 88.68 | 6.4 | $ | 208,559 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Expected to vest at December 31, 2015 |
169,737 | $ | 173.53 | 9.5 | $ | 73 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The weighted average fair value of an employee stock option granted during each of the years in the three-year period ended December 31, 2015, was $60.70, $40.70 and $36.10, respectively. The total fair value of employee stock options that vested for each of the years in the three-year period ended December 31, 2015 was $0.0 million, $29.5 million and $36.1 million, respectively.
Total share-based compensation expense relating to employee stock options is as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Research and development |
$ | 3 | $ | — | $ | — | ||||
Selling, general and administrative |
4,894 | 29,460 | 36,097 | |||||||
Related income tax benefit |
(1,847 | ) | (10,429 | ) | (13,566 | ) | ||||
| | | | | | | | | | | |
Share-based compensation expense, net of taxes |
$ | 3,050 | $ | 19,031 | $ | 22,531 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
As of December 31, 2015, the unrecognized compensation cost was $5.4 million. As of December 31, 2014, all employee stock options were fully vested; consequently, there were no amounts of unrecognized compensation cost remaining.
Employee and non-employee stock option exercise data is summarized below (dollars in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Number of options exercised |
985,583 | 1,462,369 | 876,115 | |||||||
Cash received from options exercised |
$ | 39,311 | $ | 50,168 | $ | 26,619 | ||||
Total intrinsic value of options exercised |
$ | 120,319 | $ | 108,425 | $ | 37,530 | ||||
Tax benefits realized from options exercised |
$ | 37,426 | $ | 30,845 | $ | 9,299 | ||||
F-32
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
11. Stockholders' Equity (Continued)
Employee Stock Purchase Plan
In June 2012, our shareholders approved the United Therapeutics Corporation Employee Stock Purchase Plan (ESPP), which has been structured to comply with Section 423 of the Internal Revenue Code. The ESPP provides eligible employees the right to purchase shares of our common stock at a discount through elective accumulated payroll deductions at the end of each offering period. Offering periods, which began in September 2012, occur in consecutive six-month periods commencing on September 5th and March 5th of each year. Eligible employees may contribute up to 15 percent of their base salary, subject to certain annual limitations as defined in the ESPP. The purchase price of the shares is equal to the lower of 85 percent of the closing price of our common stock on either the first or last trading day of a given offering period. In addition, the ESPP provides that no eligible employee may purchase more than 4,000 shares during any offering period. The ESPP has a 20-year term and limits the aggregate number of shares that can be issued to 3.0 million.
We estimate the fair value of the option to purchase shares of our common stock under the ESPP using the same methodology that we employ in valuing our stock options and STAP awards.
Earnings Per Common Share
Basic earnings per share is computed by dividing net income by the weighted average number of shares of common stock outstanding during the period. Diluted earnings per share is computed by dividing net income by the weighted average number of shares of common stock outstanding during the period, adjusted for the potential dilutive effect of other securities if such securities were converted or exercised.
The components of basic and diluted earnings per share comprised the following (in thousands, except per share amounts):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Numerator: |
||||||||||
Net income |
$ | 651,639 | $ | 340,074 | $ | 174,560 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Denominator: |
||||||||||
Weighted average outstanding shares—basic |
46,000 | 48,176 | 50,076 | |||||||
Effect of dilutive securities(1): |
||||||||||
Convertible notes |
915 | 2,630 | 1,736 | |||||||
Warrants |
3,009 | 1,910 | 276 | |||||||
Stock options and employee stock purchase plan |
1,297 | 1,439 | 1,143 | |||||||
| | | | | | | | | | | |
Weighted average shares—diluted |
51,221 | 54,155 | 53,231 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Earnings per common share: |
||||||||||
Basic |
$ | 14.17 | $ | 7.06 | $ | 3.49 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted |
$ | 12.72 | $ | 6.28 | $ | 3.28 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Stock options and warrants excluded from calculation(2) |
3,800 | 9,273 | 11,210 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- Calculated using the treasury stock method.
F-33
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
11. Stockholders' Equity (Continued)
- (2)
- Certain stock options and warrants have been excluded from the computation of diluted earnings per share because their impact would be anti-dilutive. Under our convertible note hedge agreement, we are entitled to receive shares required to be issued to investors upon conversion of our Convertible Notes. Since related shares used to compute dilutive earnings per share would be anti-dilutive, they have been excluded from the calculation above.
Share Repurchases
In June 2014, our Board of Directors authorized the repurchase of up to $500.0 million of our common stock. This program became effective on August 1, 2014, and remained open for one year. During the years ended December 31, 2015, and 2014, we repurchased 2,368,645 and 887,114 shares of our common stock, respectively, at an aggregate cost of $394.5 million and $105.5 million, respectively, under this repurchase program. We completed this repurchase program during the quarter ended September 30, 2015.
In October 2015, our Board of Directors authorized a new program for the repurchase of up to $500.0 million of our common stock in open or privately negotiated transactions, at our discretion. This program is effective from January 1, 2016 through December 31, 2016. During the month ended January 31, 2016, we repurchased approximately 280,000 shares of our common stock at an aggregate cost of $37.8 million.
Shareholder Rights Plan
In June 2008, we entered into an Amended and Restated Rights Agreement with The Bank of New York as Rights Agent (the Plan), which amended and restated our original Rights Agreement dated December 17, 2000. The Plan, as amended and restated, extended the expiration date of the Preferred Share Purchase Rights (Rights) from December 29, 2010 to June 26, 2018, and increased the purchase price of each Right from $64.75 to $400.00, respectively. Each Right entitles holders to purchase one one-thousandth of a share of our Series A Junior Participating Preferred Stock. Rights are exercisable only upon our acquisition by another company, or commencement of a tender offer that would result in ownership of 15 percent or more of the outstanding shares of our voting stock by a person or group (as defined under the Plan) without our prior express written consent. As of December 31, 2015, we have not issued any shares of our Series A Preferred Stock.
F-34
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
12. Accumulated Other Comprehensive Loss
The following table includes changes in accumulated other comprehensive (loss) income by component, net of tax (in thousands):
| |
Defined Benefit Pension Plan(1) |
Foreign Currency Translation Losses |
Unrealized Gains and (Losses) on Available-for- Sale Securities |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance, January 1, 2015 |
$ | (6,957 | ) | $ | (9,858 | ) | $ | 81 | $ | (16,734 | ) | ||
Other comprehensive income (loss) before reclassifications |
826 | (5,296 | ) | (74 | ) | (4,544 | ) | ||||||
Amounts reclassified from accumulated other comprehensive gain |
877 | — | — | 877 | |||||||||
| | | | | | | | | | | | | | |
Net current-period other comprehensive income (loss) |
1,703 | (5,296 | ) | (74 | ) | (3,667 | ) | ||||||
| | | | | | | | | | | | | | |
Balance, December 31, 2015 |
$ | (5,254 | ) | $ | (15,154 | ) | $ | 7 | $ | (20,401 | ) | ||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Refer to Note 14—Employee Benefit Plans—Supplemental Executive Retirement Plan, which identifies the captions within our consolidated statement of operations where reclassification adjustments were recognized and their associated tax impact.
13. Income Taxes
Components of income tax expense (benefit) consist of the following (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Current: |
||||||||||
Federal |
$ | 320,726 | $ | 137,993 | $ | 120,030 | ||||
State |
33,978 | 19,051 | 20,099 | |||||||
Foreign |
806 | 1,252 | 2,164 | |||||||
| | | | | | | | | | | |
Total current |
355,510 | 158,296 | 142,293 | |||||||
Deferred |
||||||||||
Federal |
(2,846 | ) | (2,945 | ) | (37,713 | ) | ||||
State |
7,279 | 463 | (9,059 | ) | ||||||
Foreign |
194 | (225 | ) | (1,055 | ) | |||||
| | | | | | | | | | | |
Total deferred |
4,627 | (2,707 | ) | (47,827 | ) | |||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Other non-current |
||||||||||
Federal |
29,678 | 27,115 | 7,797 | |||||||
State |
2,990 | 2,383 | 1,907 | |||||||
Foreign |
(7 | ) | 19 | 173 | ||||||
| | | | | | | | | | | |
Total other |
32,661 | 29,517 | 9,877 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Total income tax expense |
$ | 392,798 | $ | 185,106 | $ | 104,343 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
F-35
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
13. Income Taxes (Continued)
Presented below is a reconciliation of income taxes computed at the statutory federal tax rate to income tax expense as reported (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Federal tax provision computed at 35% |
$ | 365,476 | $ | 183,813 | $ | 97,616 | ||||
State tax provision, net of federal tax provision |
28,760 | 12,865 | 8,320 | |||||||
General business credits |
(6,904 | ) | (12,195 | ) | (13,346 | ) | ||||
Incentive stock option expense |
— | (181 | ) | (304 | ) | |||||
Section 199 deduction |
(21,774 | ) | (11,735 | ) | (10,861 | ) | ||||
Nondeductible compensation expense |
29,280 | 13,000 | 22,813 | |||||||
Nondeductible expenses |
(2,040 | ) | (461 | ) | 105 | |||||
| | | | | | | | | | | |
Total income tax expense |
$ | 392,798 | $ | 185,106 | $ | 104,343 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Components of the net deferred tax asset are as follows (in thousands):
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Deferred tax assets: |
|||||||
General business credits |
$ | — | $ | 2,186 | |||
Impairment losses on investments |
294 | 291 | |||||
License fees capitalized for tax purposes |
53,439 | 61,770 | |||||
Nonqualified stock options |
37,424 | 42,697 | |||||
SERP |
17,141 | 17,478 | |||||
STAP awards |
95,152 | 86,414 | |||||
Other |
27,074 | 29,086 | |||||
| | | | | | | | |
Total deferred tax assets |
230,524 | 239,922 | |||||
Deferred tax liabilities: |
|||||||
Plant and equipment principally due to differences in depreciation |
(26,461 | ) | (30,758 | ) | |||
Other |
(7,995 | ) | (7,854 | ) | |||
| | | | | | | | |
Net deferred tax asset before valuation allowance |
196,068 | 201,310 | |||||
Valuation allowance |
(3,392 | ) | (2,981 | ) | |||
| | | | | | | | |
Net deferred tax assets |
$ | 192,676 | $ | 198,329 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Deferred tax assets are reduced by a valuation allowance when, in our judgment, it is more likely than not that a portion or all of the deferred tax assets will not be realized. In evaluating our ability to realize deferred tax assets, we consider all available positive and negative evidence. Accordingly, we consider past operating results, forecasts of earnings and taxable income, the reversal of temporary differences and any prudent and feasible tax planning strategies. Future increases in the valuation allowance would result in a corresponding charge to earnings in the period such a determination is made. Conversely, future reductions to the valuation allowance would result in the recognition of a tax benefit in the period we conclude a reduction is warranted.
F-36
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
13. Income Taxes (Continued)
A reconciliation of the beginning and ending balances of unrecognized tax benefits for the years indicated is as follows (in thousands):
Unrecognized tax benefits at January 1, 2013 |
$ | 1,511 | ||
Gross increases—tax positions in prior period |
1,325 | |||
| | | | | |
Unrecognized tax benefits at December 31, 2013 |
$ | 2,836 | ||
| | | | | |
| | | | | |
| | | | | |
Unrecognized tax benefits at January 1, 2014 |
$ | 2,836 | ||
Gross increases—tax positions in prior period |
28 | |||
Gross decreases—tax positions in prior period |
(1,419 | ) | ||
| | | | | |
Unrecognized tax benefits at December 31, 2014 |
$ | 1,445 | ||
| | | | | |
| | | | | |
| | | | | |
Unrecognized tax benefits at January 1, 2015 |
$ | 1,445 | ||
Lapse of statute of limitations |
(945 | ) | ||
| | | | | |
Unrecognized tax benefits at December 31, 2015 |
$ | 500 | ||
| | | | | |
| | | | | |
| | | | | |
Included in unrecognized tax benefits at December 31, 2015, 2014 and 2013, is $342,000, $1.0 million, and $2.4 million, respectively, of tax benefits that, if recognized, would impact the effective tax rate. As of December 31, 2015 and 2014, we accrued zero and $28,000, respectively, in interest expense relating to uncertain state tax positions.
We are subject to federal and state taxation in the United States and various foreign jurisdictions. Currently, our 2014, 2013, 2012 and 2011 tax years are subject to examination by the IRS and by state taxing authorities. We are unaware of any positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next twelve months.
14. Employee Benefit Plans
Supplemental Executive Retirement Plan
We maintain the United Therapeutics Corporation Supplemental Executive Retirement Plan (SERP) to provide retirement benefits to certain senior members of our management team.
Participants who retire at age 60 or older are eligible to receive either monthly payments or a lump sum payment based on an average of their total gross base salary over the last 36 months of active employment, subject to certain adjustments. Related benefit payments commence on the first day of the sixth month after retirement. Participants who elect to receive monthly payments will continue payments through the remainder of their life. Alternatively, participants who elect to receive a lump sum distribution will receive a payment equal to the present value of the estimated monthly payments that would have been received upon retirement. As of December 31, 2015 and 2014, all SERP participants had elected to receive a lump sum distribution. Participants who terminate employment for any reason other than death, disability, or change in control prior to age 60 will not be entitled to receive any benefits under the SERP.
We recognize the unfunded balance of the SERP as a liability on our consolidated balance sheets. Since we do not fund the SERP, the liability is equal to the projected benefit obligation as measured at the end of each fiscal year. Expenses related to the SERP are reported under the captions, "Research
F-37
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
14. Employee Benefit Plans (Continued)
and development expense" and "Selling, general and administrative expense" in the accompanying consolidated statements of operations.
A reconciliation of the beginning and ending balances of the projected benefit obligation is presented below (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Projected benefit obligation at the beginning of the year |
$ | 57,955 | $ | 51,034 | |||
Service cost |
3,518 | 5,517 | |||||
Interest cost |
1,836 | 2,367 | |||||
Plan amendments |
— | 3,862 | |||||
Benefits paid |
(7,117 | ) | — | ||||
Actuarial gain |
(1,379 | ) | (4,825 | ) | |||
| | | | | | | | |
Projected benefit obligation at the end of the year |
$ | 54,813 | $ | 57,955 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Fair value of plan assets at the end of the year |
— | — | |||||
| | | | | | | | |
Unfunded at end of the year(1) |
$ | 54,813 | $ | 57,955 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
- (1)
- At December 31, 2015, the aggregate balance of the SERP liability was $54.8 million, of which $14.7 million, representing the benefit obligation due for participants who are currently eligible to retire, has been classified as current liabilities under the caption "Other current liabilities" on our consolidated balance sheets.
The accumulated benefit obligation, a measure that does not consider future increases in participants' salaries, was $42.3 million and $43.5 million at December 31, 2015 and 2014, respectively.
Future estimated benefit payments, based on current assumptions, including election of lump-sum distributions and expected future service, are as follows (in thousands):
Year Ended December 31,
|
|
|||
|---|---|---|---|---|
2016 |
$ | 14,651 | ||
2017 |
— | |||
2018 |
— | |||
2019 |
4,580 | |||
2020 |
— | |||
2021 - 2025 |
51,836 | |||
| | | | | |
Total |
$ | 71,067 | ||
| | | | | |
| | | | | |
| | | | | |
F-38
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
14. Employee Benefit Plans (Continued)
The following weighted-average assumptions were used to measure the SERP obligation:
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | |||||
Discount Rate |
3.82 | % | 3.64 | % | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Salary Increases |
4.00 | % | 5.00 | % | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The components of net periodic pension cost recognized on our consolidated statement of operations consist of the following (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Service cost |
$ | 3,518 | $ | 5,517 | $ | 5,406 | ||||
Interest cost |
1,836 | 2,367 | 1,584 | |||||||
Amortization of prior service cost |
1,234 | 1,234 | 827 | |||||||
Amortization of net actuarial loss |
160 | 210 | 794 | |||||||
| | | | | | | | | | | |
Total |
$ | 6,748 | $ | 9,328 | $ | 8,611 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Reclassification adjustments related to the SERP from accumulated other comprehensive loss to the statement of operations by line item and the tax impact of these reclassifications is presented below (in thousands):
Components Reclassified from Accumulated Other Comprehensive Loss(1) |
As of December 31, 2015 |
As of December 31, 2014 |
|||||
|---|---|---|---|---|---|---|---|
Prior service cost: |
|||||||
Research and development |
$ | 408 | $ | 408 | |||
Selling, general and administrative |
826 | 826 | |||||
| | | | | | | | |
Total |
1,234 | 1,234 | |||||
Amortization of net actuarial loss: |
|||||||
Research and development |
— | 69 | |||||
Selling, general and administrative |
160 | 141 | |||||
| | | | | | | | |
Total |
160 | 210 | |||||
Total prior service cost and amortization of net actuarial loss |
1,394 | 1,444 | |||||
Tax benefit |
(517 | ) | (540 | ) | |||
| | | | | | | | |
Total, net of tax |
$ | 877 | $ | 904 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
- (1)
- Refer to Note 12—Accumulated Other Comprehensive Loss.
F-39
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
14. Employee Benefit Plans (Continued)
Amounts relating to the SERP that have been recognized in other comprehensive gain (loss) are as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Net unrecognized actuarial gain |
$ | 1,539 | $ | 5,035 | $ | 3,956 | ||||
Net unrecognized prior service cost (benefit) |
1,234 | (2,627 | ) | 827 | ||||||
| | | | | | | | | | | |
Total |
2,773 | 2,408 | 4,783 | |||||||
Tax benefit |
(1,070 | ) | (920 | ) | (1,688 | ) | ||||
| | | | | | | | | | | |
Total, net of tax |
$ | 1,703 | $ | 1,488 | $ | 3,095 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The table below presents amounts relating to the SERP included in accumulated other comprehensive loss that have not yet been recognized as a component of net periodic pension cost on our consolidated statements of operations (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2015 | 2014 | 2013 | |||||||
Net unrecognized actuarial loss |
$ | 1,228 | $ | 2,767 | $ | 7,803 | ||||
Net unrecognized prior service cost |
7,093 | 8,326 | 5,698 | |||||||
| | | | | | | | | | | |
Total |
8,321 | 11,093 | 13,501 | |||||||
Tax benefit |
(3,067 | ) | (4,150 | ) | (5,074 | ) | ||||
| | | | | | | | | | | |
Total, net of tax |
$ | 5,254 | $ | 6,943 | $ | 8,427 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Estimated amounts included in accumulated other comprehensive loss as of December 31, 2015 that are expected to be recognized as components of net periodic pension expense on our statement of operations for the year ended December 31, 2016 comprise the following (in thousands):
Amortization of prior service cost |
$ | 1,234 | ||
Amortization of net actuarial loss |
— | |||
| | | | | |
Total |
$ | 1,234 | ||
| | | | | |
| | | | | |
| | | | | |
Employee Retirement Plan
We maintain a Section 401(k) Salary Reduction Plan which is open to all eligible full-time employees. Under the 401(k) Plan, eligible employees can make pre-tax contributions up to statutory limits. Currently, we make discretionary matching contributions to the 401(k) Plan equal to 40 percent of a participant's elected salary deferral. Matching contributions vest immediately for participants who have been employed for three years; otherwise, matching contributions vest annually, in one-third increments over a three-year period until the three-year employment requirement has been met.
F-40
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
15. Assignment and License Agreements
GlaxoSmithKline plc
In 1997, GlaxoSmithKline plc (Glaxo) assigned to us patents and patent applications covering treprostinil for the treatment of PAH and congestive heart failure. Under the agreement, Glaxo was entitled to receive royalties on sales exceeding a specified threshold for a minimum period of ten years (or until expiration of the licensed patents) following the date of the first commercial sale of any initial product containing treprostinil. Pursuant to these terms, our royalty obligation ended in October 2014.
Supernus Pharmaceuticals, Inc.
In June 2006, we entered into an exclusive license agreement with Supernus Pharmaceuticals, Inc. (Supernus) for the use of certain technologies developed by Supernus in our Orenitram tablet. Under this agreement, we paid Supernus certain amounts upon the achievement of specified milestones based on the development and commercial launch of Orenitram for PAH, and we would be obligated to make additional milestone payments if we develop Orenitram for a second indication. Additionally, we will pay a single digit royalty under this agreement, based on net product sales of Orenitram. Royalties will be paid for approximately twelve years commencing with the first commercial sale, subject to adjustments. The royalties commenced in the second quarter of 2014 with the first sale of Orenitram.
Eli Lilly and Company
In November 2008, we acquired from Lilly exclusive rights to develop, market, promote and commercialize Adcirca for the treatment of pulmonary hypertension in the United States. In exchange for these license rights, we agreed to pay Lilly, among other fees, royalties of five percent of our net product sales of Adcirca as a pass through of Lilly's third-party royalty obligations for as long as Lilly is required to make such royalty payments. Pursuant to the terms of our license arrangement, Lilly manufactures Adcirca for us and distributes Adcirca via its wholesaler network in the same manner that it distributes its own pharmaceutical products. We purchase Adcirca from Lilly at a fixed manufacturing cost, which is adjusted by Lilly from time to time. The terms of this licensing arrangement will continue generally until the later of: (1) the expiration or lapse of the last to expire claim within a Lilly patent covering commercialization of Adcirca; or (2) the expiration of any government conferred exclusivity rights to Adcirca. In addition, at Lilly's discretion the license agreement may be terminated in the event that we undergo a change in control.
National Cancer Institute
In July 2010, we entered into a Cooperative Research and Development Agreement (CRADA) with the National Cancer Institute (NCI) of the United States National Institutes for Health (NIH) to collaborate on the late-stage development and regulatory approval process for Unituxin for children with high-risk neuroblastoma and patients with other forms of cancer. In lieu of a royalty payment to the NCI, we have an ongoing obligation to provide the NCI with Unituxin for its studies free of charge.
Toray Industries, Inc.
In 2000, we entered into an agreement with Toray to obtain exclusive rights to develop and market beraprost, a chemically stable oral prostacyclin analogue, in a sustained release formulation in the United States and Canada for the treatment of all cardiovascular indications. In 2007, we amended the agreement to expand our rights to commercialize modified release formulations of beraprost, which
F-41
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
15. Assignment and License Agreements (Continued)
include esuberaprost. As part of the 2007 amendment, we issued 200,000 shares of our common stock (which have since split into 400,000 shares) to Toray with certain put rights. These put rights provide Toray the ability to request at its discretion that we repurchase these shares at a price of $27.21 per share upon 30 days' prior written notice. Accordingly, we classified the value of the shares within temporary equity on our consolidated balance sheets. In the event that Toray requests that we repurchase these shares, we will reclassify the repurchase value of the stock as a liability until settlement. The 2007 amendment also provided for certain milestone payments during the development period and upon receipt of regulatory approval in the United States or the European Union.
In July 2011, we amended our license agreement with Toray. The amendment did not materially change the terms of our license agreement, except for a reduction in royalty rates in exchange for a total of $50.0 million in equal, non-refundable payments to Toray over the five-year period ending in 2015. As of December 31, 2015, we have fulfilled this obligation to Toray.
Pluristem License Agreement
In June 2011, we entered into a license agreement with Pluristem Ltd. (Pluristem) for exclusive worldwide rights to develop and commercialize a cell-based product for the treatment of PAH using Pluristem's proprietary PLX cell technology. The agreement provided for additional milestone payments to Pluristem at various stages, as well as royalties on commercial sales. In December 2015, we terminated our licensing agreement with Pluristem and our development of the PLX program.
Medtronic Inc.
In 2009, we entered into an agreement with Medtronic, Inc. (Medtronic) providing us exclusive rights in the United States, the United Kingdom, Canada, France, Germany, Italy and Japan to develop Medtronic's proprietary intravascular infusion catheter to be used with its SynchroMed® II implantable infusion pump and related infusion system components (together referred to as the Remodulin Implantable System) in order to deliver Remodulin for the treatment of PAH. If this development program is successful, our agreement provides that, upon commercialization, we will purchase infusion pumps and supplies from Medtronic and will also pay a royalty to Medtronic based on net product sales of Remodulin for use in the Remodulin Implantable System within the exclusive territories, subject to certain adjustments specified in the agreement. The Remodulin Implantable System will be exclusive to Remodulin so long as we purchase a minimum percentage of our annual requirement for implantable pump systems from Medtronic. We will be solely responsible for all marketing and promotion of the Remodulin Implantable System for the delivery of Remodulin for the treatment of PAH in the exclusive territories.
DEKA Research & Development Corp.
In December 2014, we entered into an exclusive agreement with DEKA Research & Development Corp. (DEKA) to develop a pre-filled, semi-disposable pump system for subcutaneous delivery of Remodulin. Under the terms of the agreement, we will fund the development costs related to the semi-disposable pump system and will pay product fees and a single-digit royalty to DEKA based on commercial sales of the system and the Remodulin sold for use with the system.
F-42
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
15. Assignment and License Agreements (Continued)
Other
We are party to various other license agreements relating to therapies under development. These license agreements require us to make payments based on a percentage of sales, if we are successful in commercially developing these therapies, and may require other payments upon the achievement of certain milestones.
16. Distribution Agreements
U.S.-Based Specialty Distributors
We are party to separate distribution agreements for Remodulin, Tyvaso and Orenitram with two U.S.-based specialty pharmaceutical distributors. The distribution agreements are similar to one another, and generally have one-year terms that renew automatically for additional one-year periods, unless terminated earlier. The agreements contain contractual responsibilities relating to ordering specifications, inventory requirements and exchange rights. We also have agreements with these distributors to perform certain services for us on a fee-for-service basis. If any of our distribution agreements expire or terminate, we may be required under certain circumstances to repurchase any unsold inventory held by our distributors. In the second quarter of 2015, we entered into an exclusive distribution agreement with ASD Specialty Healthcare, Inc. (ASD), an affiliate of AmerisourceBergen Corporation, to distribute Unituxin in the United States. Under this Agreement, we sell Unituxin to ASD at a transfer price that we establish, and we pay ASD fees for services provided in connection with the distribution and support of Unituxin.
International Distributors
We currently sell Remodulin and Tyvaso internationally through various distributors. The financial terms and conditions relating to these distributor arrangements are structured in a manner substantially similar to those of our U.S. distribution agreements described above.
17. Segment Information
We currently operate as one operating segment. However, our chief operating decision makers regularly review net product sales, cost of product sales and gross profit data as a primary measure of performance for each of our five commercial products. We commenced sales of Orenitram and Unituxin during the second quarter of 2014 and third quarter of 2015, respectively.
F-43
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
17. Segment Information (Continued)
Net product sales, cost of product sales and gross profit for each of our commercial products were as follows (in thousands):
Year Ended December 31, 2015
|
Remodulin | Tyvaso | Adcirca | Orenitram | Unituxin | Total | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Net product sales |
$ | 572,795 | $ | 470,069 | $ | 278,829 | $ | 118,434 | $ | 20,443 | $ | 1,460,570 | |||||||
Cost of product sales |
12,373 | 23,925 | 16,504 | 12,569 | 3,665 | 69,036 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Gross profit |
$ | 560,422 | $ | 446,144 | $ | 262,325 | $ | 105,865 | $ | 16,778 | $ | 1,391,534 | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Year Ended December 31, 2014 |
|||||||||||||||||||
Net product sales |
$ |
553,728 |
$ |
463,067 |
$ |
221,471 |
$ |
41,267 |
$ |
— |
$ |
1,279,533 |
|||||||
Cost of product sales |
47,327 | 57,442 | 13,495 | 7,619 | — | 125,883 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Gross profit |
$ | 506,401 | $ | 405,625 | $ | 207,976 | $ | 33,648 | $ | — | $ | 1,153,650 | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Year Ended December 31, 2013 |
|||||||||||||||||||
Net product sales |
$ |
491,179 |
$ |
438,793 |
$ |
176,972 |
$ |
— |
$ |
— |
$ |
1,106,944 |
|||||||
Cost of product sales |
59,314 | 60,831 | 10,982 | — | — | 131,127 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Gross profit |
$ | 431,865 | $ | 377,962 | $ | 165,990 | $ | — | $ | — | $ | 975,817 | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Geographic revenues are determined based on the country in which our customers (distributors) are located. Total revenues from external customers by geographic area are as follows (in thousands):
Year Ended December 31,
|
2015 | 2014 | 2013 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
United States |
$ | 1,353,002 | $ | 1,180,759 | $ | 1,032,435 | ||||
Rest-of-World(1) |
112,759 | 107,760 | 84,549 | |||||||
| | | | | | | | | | | |
Total(2) |
$ | 1,465,761 | $ | 1,288,519 | $ | 1,116,984 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- Primarily
Europe.
- (2)
- Total includes other revenue of $5.2 million, $9.0 million and $10.0 million for the years ended December 31, 2015, 2014 and 2013, respectively.
For the years ended December 31, 2015, 2014 and 2013, net product sales to Accredo Health Group, Inc. comprised 55 percent, 58 percent and 57 percent, respectively of total revenues.
Long-lived assets (property, plant and equipment) located by geographic area are as follows (in thousands):
Year Ended December 31,
|
2015 | 2014 | |||||
|---|---|---|---|---|---|---|---|
United States |
$ | 481,219 | $ | 462,377 | |||
Rest-of-World(1) |
14,555 | 16,044 | |||||
| | | | | | | | |
Total |
$ | 495,774 | $ | 478,421 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
- (1)
- Facilities principally located in the United Kingdom.
F-44
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
18. Quarterly Financial Information (Unaudited)
Summarized quarterly financial information for each of the years ended December 31, 2015 and 2014 are as follows (in thousands, except per share amounts):
| |
Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
December 31, 2015 |
September 30, 2015 |
June 30, 2015 |
March 31, 2015 |
|||||||||
Total revenues |
$ | 404,875 | $ | 386,221 | $ | 347,161 | $ | 327,504 | |||||
Cost of product sales |
25,309 | 6,891 | 16,058 | 20,778 | |||||||||
Gross profit |
379,566 | 379,330 | 331,103 | 306,726 | |||||||||
Net income (loss)(1)(2) |
104,644 | 464,425 | 99,211 | (16,641 | ) | ||||||||
Net income (loss) per share—basic |
$ | 2.29 | $ | 10.20 | $ | 2.15 | $ | (0.36 | ) | ||||
Net income (loss) per share—diluted |
$ | 2.10 | $ | 9.24 | $ | 1.91 | $ | (0.36 | ) | ||||
| |
Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
December 31, 2014 |
September 30, 2014 |
June 30, 2014 |
March 31, 2014 |
|||||||||
Total revenues |
$ | 346,363 | $ | 329,950 | $ | 322,802 | $ | 289,403 | |||||
Cost of product sales |
15,770 | 40,804 | 38,709 | 30,600 | |||||||||
Gross profit |
330,593 | 289,147 | 284,093 | 258,803 | |||||||||
Net income (loss)(3) |
115,935 | (25,237 | ) | 111,852 | 137,524 | ||||||||
Net income (loss) per share—basic |
$ | 2.44 | $ | (0.53 | ) | $ | 2.35 | $ | 2.73 | ||||
Net income (loss) per share—diluted |
$ | 2.17 | $ | (0.53 | ) | $ | 2.10 | $ | 2.43 | ||||
- (1)
- Operating
results for the quarter ended December 31, 2015, September 30, 2015, June 30, 2015 and March 31, 2015 include
$71.6 million, $(75.7) million, $27.5 million and $145.7 million net of tax expense (benefit) to operating expenses for STAP related share-based compensation expense,
respectively.
- (2)
- Operating
results for the quarter ended September 30, 2015, include a gain on sale of the PPRV we received from the FDA in connection with the
approval of Unituxin, for $350.0 million in cash. The proceeds from the sale of the PPRV were recognized as a gain on the sale of an intangible asset, as the PPRV did not have a carrying value
on our consolidated balance sheet at the time of sale.
- (3)
- Operating results for the quarter ended December 31, 2014, September 30, 2014, June 30, 2014, and March 31, 2014 include $19.9 million, $122.1 million, $(1.4) million and $(39.6) million net of tax expense (benefit) to operating expenses for STAP related share-based compensation expense, respectively.
19. Litigation
Sandoz Inc.
In February 2012, we received a Paragraph IV certification notice letter (the Original Notice Letter) from Sandoz Inc. (Sandoz) advising that Sandoz had submitted an abbreviated new drug application (ANDA) to the FDA requesting approval to market a generic version of the 10 mg/mL strength of Remodulin. In December 2012, we received notice (the Second Notice Letter) that Sandoz
F-45
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
19. Litigation (Continued)
had amended its previously filed ANDA to request additional approval to market generic versions of the 1 mg/mL, 2.5 mg/mL, and 5 mg/mL strengths of Remodulin. In the Original Notice Letter and the Second Notice Letter, Sandoz stated that it intends to market a generic version of Remodulin before the expiration of the following patents relating to Remodulin: U.S. Patent No. 5,153,222, which expired in October 2014; U.S. Patent No. 6,765,117, which expires in October 2017; and U.S. Patent No. 7,999,007, which expires in March 2029. Each of these patents is listed in the FDA's Orange Book, which contains a listing of patents covering a drug or biologic or its method of use, and which have been submitted to the FDA by the filer of a New Drug Application. We responded to the Original Notice Letter by filing a lawsuit in March 2012 against Sandoz in the U.S. District Court for the District of New Jersey alleging patent infringement (the First Action). We responded to the Second Notice Letter by filing an additional lawsuit in January 2013 for patent infringement in the U.S. District Court for the District of New Jersey (the Second Action). Sandoz filed counterclaims in each action alleging that the patents at issue in the litigation are invalid or will not be infringed by the commercial manufacture, use or sale of the proposed product described in Sandoz's ANDA submission. Shortly before trial, Sandoz withdrew its request to market a generic version of Remodulin before the expiration of U.S. Patent No. 5,153,222, but maintained its request to market a generic version of Remodulin before the expiration of the other two patents. The trial for both lawsuits, limited to U.S. Patent Nos. 6,765,117 and 7,999,007, occurred in May and June 2014 and we received the Court's decision in August 2014. In that decision, with respect to U.S. Patent No. 6,765,117 the Court both ruled that the patent is valid and enforceable against Sandoz, and enjoined Sandoz from marketing its generic product until the expiration of that patent in October 2017. With respect to U.S. Patent No. 7,999,007, the Court ruled that the patent is valid, but that it would not be infringed by Sandoz's generic product.
Sandoz appealed the ruling that U.S. Patent No. 6,765,117 is valid and would be infringed, and that U.S. Patent No. 7,999,007 is valid. We filed a cross-appeal challenging the Court's ruling that U.S. Patent No. 7,999,007 would not be infringed by Sandoz's generic version of Remodulin.
In July 2014, we received an additional Paragraph IV certification notice letter (Third Notice Letter) from Sandoz, seeking permission to market and sell its generic version of Remodulin before the expiration of U.S. Patent No. 8,497,393, which expires in December 2028 and is also listed in the Orange Book. We responded to Sandoz's Third Notice Letter by filing a lawsuit in September 2014 in the U.S. District Court for the District of New Jersey for patent infringement with respect to U.S. Patent No. 8,497,393 (the Third Action).
On September 29, 2015, we entered into a Settlement Agreement with Sandoz to settle all ongoing litigation between the parties (all three lawsuits described above, including pending appeals) concerning Remodulin patents. Under the settlement agreement, we granted Sandoz a non-exclusive license to manufacture and commercialize in the United States the generic version of Remodulin described in Sandoz's ANDA filing beginning on June 26, 2018, although Sandoz may be permitted to enter the market earlier under certain circumstances. The settlement agreement does not grant Sandoz a license to manufacture a generic version of any other product, such as Tyvaso or Orenitram, nor does it grant any rights with respect to any technology associated with the Remodulin Implantable System we are developing in conjunction with Medtronic Inc., or the pre-filled semi-disposable pump system we are developing with DEKA Research & Development Corp. The settlement agreement does not grant Sandoz any rights other than those required to launch Sandoz's generic version of Remodulin.
F-46
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
19. Litigation (Continued)
In accordance with the terms of the settlement agreement, the parties have submitted the settlement agreement to the U.S. Federal Trade Commission and the U.S. Department of Justice for review. The parties have terminated the appeal relating to the First Action and the Second Action. The parties' claims in the Third Action have been dismissed with prejudice. Following a motion by the parties, the U.S. District Court vacated the portions of the ruling in the First Action and Second Action regarding whether Sandoz's ANDA product infringed U.S. Patent Nos. 6,765,117 and 7,999,007. The vacatur did not affect the portions of the Court's decisions that these patents are valid.
Teva Pharmaceuticals USA, Inc.
On July 21, 2014, we received a Paragraph IV certification notice letter from Teva Pharmaceuticals USA, Inc. (Teva) advising that Teva had submitted an ANDA to the FDA requesting approval to market a generic version of Remodulin. In its notice letter, Teva states that it intends to market a generic version of Remodulin before the expiration of U.S. Patent Nos. 6,765,117 and 8,497,393, both of which are also the subject of Paragraph IV certifications by Sandoz, as discussed above. Teva's notice letter states that the ANDA contains a Paragraph IV certification alleging that these patents are not valid, not enforceable and/or will not be infringed by the commercial manufacture, use or sale of the proposed product described in Teva's ANDA submission.
We responded to Teva's notice letter by filing a lawsuit in September 2014 against Teva in the U.S. District Court for the District of New Jersey alleging infringement of U.S. Patent Nos. 6,765,117, 7,999,007 and 8,497,393, as well as infringement of U.S. Patent Nos. 8,653,137 and 8,658,694, both of which expire in September 2028.
On January 15, 2016, we entered into a Settlement Agreement with Teva to settle the parties' ongoing litigation concerning Remodulin patents. Under the settlement agreement, we granted Teva a non-exclusive license beginning on December 23, 2018 to manufacture and commercialize in the United States the generic version of Remodulin described in Teva's ANDA filing, although Teva may be permitted to enter the market earlier under certain circumstances. The settlement agreement does not grant Teva a license to manufacture a generic version of any other product, such as Tyvaso or Orenitram, nor does it grant any rights with respect to any technology associated with the Remodulin Implantable System we are developing in conjunction with Medtronic Inc., or the pre-filled semi-disposable pump system we are developing with DEKA Research & Development Corp. The settlement agreement does not grant Teva any rights other than those required to launch Teva's generic version of Remodulin. In accordance with the terms of the settlement agreement, the parties have submitted the settlement agreement to the U.S. Federal Trade Commission and the U.S. Department of Justice for review. The parties have also terminated the outstanding lawsuit.
Watson Laboratories, Inc.
In June 2015, we received a Paragraph IV certification notice letter from Watson Laboratories, Inc. (Watson) indicating that Watson has submitted an ANDA to the FDA to market a generic version of Tyvaso. In its notice letter, Watson states that it intends to market a generic version of Tyvaso before the expiration of U.S. Patent Nos. 6,521,212 and 6,756,033, each of which expires in November 2018; and U.S. Patent No. 8,497,393, which expires in December 2028. Watson's notice letter states that the ANDA contains a Paragraph IV certification alleging that these patents are not valid, not enforceable, and/or will not be infringed by the commercial manufacture, use or sale of the proposed product
F-47
UNITED THERAPEUTICS CORPORATION
Notes to Consolidated Financial Statements (Continued)
19. Litigation (Continued)
described in Watson's ANDA submission. We responded to the Watson notice letter by filing a lawsuit on July 22, 2015 against Watson in the U.S. District Court for the District of New Jersey alleging infringement of U.S. Patent Nos. 6,521,212, 6,756,033, and 8,497,393. Under the Hatch-Waxman Act, the FDA is automatically precluded from approving Watson's ANDA for up to 30 months from receipt of Watson's notice letter or until the issuance of a U.S. District Court decision that is adverse to us, whichever occurs first. On September 1, 2015, Watson filed (1) a motion to dismiss some, but not all, counts of the complaint, and (2) its answer to the complaint as well as certain counterclaims against us. The Court granted Watson's motion to dismiss certain counts of our complaint. On September 25, 2015, we filed our answer to Watson's counterclaims. The parties are currently engaged in discovery, and trial is currently scheduled to take place in September 2017.
We intend to vigorously enforce our intellectual property rights relating to Tyvaso.
SteadyMed Ltd.
On October 1, 2015, SteadyMed Ltd. (SteadyMed) filed a petition with the Patent Trial and Appeal Board (PTAB) of the U.S. Patent and Trademark Office for inter partes review (the IPR Petition) of U.S. Patent No. 8,497,393 (the '393 Patent), which is owned by United Therapeutics. In its IPR Petition, SteadyMed seeks to invalidate '393 Patent, which expires in December 2028 and covers a method of making treprostinil, which is the active pharmaceutical ingredient in our Remodulin, Tyvaso and Orenitram products. The '393 Patent was also the subject of the recently-settled Sandoz and Teva litigation, and remains the subject of our pending litigation with Watson, described above. We filed a response to the IPR Petition in January 2016 and intend to vigorously defend the '393 Patent. SteadyMed has announced that it is developing a product called Trevyent™, which is a single-use, pre-filled pump it plans to seek FDA approval for delivery of a two-day supply of treprostinil subcutaneously using its PatchPump® technology. We expect the PTAB to decide whether to institute the review of the '393 patent requested by SteadyMed by mid-April 2016.
F-48
United Therapeutics Corporation
Schedule II—Valuation and Qualifying Accounts
Years Ended December 31, 2015, 2014, and 2013
(In thousands)
| |
Valuation Allowance on Deferred Tax Assets | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at Beginning of Year |
Additions Charged to Expense |
Deductions | Balance at End of Year |
|||||||||
Year Ended December 31, 2015 |
$ | 2,981 | $ | 411 | $ | — | $ | 3,392 | |||||
Year Ended December 31, 2014 |
$ | 2,507 | $ | 474 | $ | — | $ | 2,981 | |||||
Year Ended December 31, 2013 |
$ | 5,665 | $ | 169 | $ | (3,327 | ) | $ | 2,507 | ||||
| |
Reserve for Inventory Obsolescence | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at Beginning of Year |
Additions Charged to Expense |
Deductions | Balance at End of Year |
|||||||||
Year Ended December 31, 2015 |
$ | 10,537 | $ | 7,913 | $ | (6,286 | ) | $ | 12,164 | ||||
Year Ended December 31, 2014 |
$ | 18,301 | $ | 3,431 | $ | (11,195 | ) | $ | 10,537 | ||||
Year Ended December 31, 2013 |
$ | 16,679 | $ | 3,341 | $ | (1,719 | ) | $ | 18,301 | ||||
F-49
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with participation of our Chairman and Co-Chief Executive Officer, President and Co-Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as of December 31, 2015. Based on that evaluation, our Chairman and Co-Chief Executive Officer, President and Co-Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2015.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended). Our internal control over financial reporting was designed to provide reasonable assurance to our management and Board of Directors regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. All internal controls over financial reporting, no matter how well designed, have inherent limitations. As a result of these inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those internal controls determined to be effective can provide only reasonable assurance with respect to the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2015, based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework (2013). Management's assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of our internal control over financial reporting. Based on this assessment, our management concluded that, as of December 31, 2015, our internal control over financial reporting was effective.
Ernst & Young LLP, an independent registered public accounting firm, has issued an attestation report on our internal control over financial reporting. The report of Ernst & Young LLP is contained in Item 8 of this Annual Report on Form 10-K.
Attestation of Independent Registered Public Accounting Firm
The attestation report of our independent registered public accounting firm regarding internal control over financial reporting is set forth in Item 8 of this Annual Report on Form 10-K under the caption "Report of Independent Registered Public Accounting Firm" and incorporated herein by reference.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
84
Paragraph IV Notice Letter for Orenitram
On February 18, 2016, we received a Paragraph IV Certification Notice Letter (the Notice Letter) from Actavis Laboratories FL, Inc. (Actavis) advising that Actavis has submitted an Abbreviated New Drug Application (ANDA) to the U.S. Food and Drug Administration (FDA) requesting approval to market a generic version of the 2.5 mg strength of Orenitram® (treprostinil) Extended-Release Tablets.
In the Notice Letter, Actavis states that it intends to market a generic version of Orenitram before the expiration of U.S. Patent No. 7,417,070, which expires in July 2026; U.S. Patent No. 7,544,713, which expires in July 2024; U.S. Patent No. 8,252,839, which expires in May 2024; U.S. Patent No. 8,349,892, which expires in January 2031; U.S. Patent No. 8,410,169, which expires in February 2030; U.S. Patent No. 8,497,393, which expires in December 2028; U.S. Patent No. 9,050,311, which expires in May 2024; and U.S. Patent No. 8,747,897, which expires in October 2029. The Notice Letter states that the ANDA contains a Paragraph IV Certification alleging that these patents are not valid, not enforceable, and/or will not be infringed by the commercial manufacture, use or sale of the proposed product described in Actavis ANDA submission.
We intend to vigorously enforce our intellectual property rights relating to Orenitram, including the patents mentioned above, which are listed in FDA's Approved Drug Products with Therapeutic Equivalence Evaluations list (the Orange Book).
We are currently reviewing the Notice Letter, which was directed to eight of the nine Orange Book-listed patents for Orenitram. The only patent not included expires in October 2017. We have 45 days from receipt of the Notice Letter to commence a patent infringement lawsuit against Actavis to trigger a stay precluding FDA from approving Actavis's ANDA for up to 30 months or entry of judgment holding the patents invalid, unenforceable, or not infringed, whichever occurs first.
85
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Information as to the individuals serving on our board of directors is set forth below under the heading Board of Directors. Additional information required by Item 10 regarding nominees and directors appearing under Proposal No. 1: Election of Directors in our definitive proxy statement for our 2016 annual meeting of shareholders scheduled for June 28, 2016 (the 2016 Proxy Statement) is hereby incorporated herein by this reference. Information regarding our executive officers appears in Item 1 of this Annual Report on Form 10-K under the heading Executive Officers of the Registrant. Information regarding the Audit Committee and the Audit Committee's financial expert appearing under the heading Committees of our Board of Directors—Audit Committee in our 2016 Proxy Statement is hereby incorporated herein by this reference.
Information appearing under the heading Section 16(a) Beneficial Ownership Reporting Compliance in our 2016 Proxy Statement is hereby incorporated herein by this reference.
We have a written Code of Conduct and Business Ethics that applies to our co-principal executive officers, principal financial officer and our principal accounting officer and every other director, officer and employee of United Therapeutics. The Code of Conduct and Business Ethics is available on our Internet website at http://ir.unither.com/corporate-governance.cfm. A copy of the Code of Conduct and Business Ethics will be provided free of charge by making a written request and mailing it to our corporate headquarters offices to the attention of the Investor Relations Department. If any amendment to, or a waiver from, a provision of the Code of Conduct and Business Ethics that applies to the principal executive officer, principal financial officer and principal accounting officer is made, such information will be posted on our Internet website within four business days at www.unither.com.
| Christopher Causey, M.B.A. | ||
Principal, Causey Consortium |
||
| Raymond Dwek, F.R.S. | ||
Director of the Glycobiology Institute and Professor Emeritus, University of Oxford |
||
| Richard Giltner | ||
Private Investor |
||
| Roger Jeffs, Ph.D. | ||
President and Co-Chief Executive Officer of United Therapeutics |
||
| Katherine Klein, Ph.D. | ||
Vice-Dean and Professor, The Wharton School of the University of Pennsylvania |
||
| Ray Kurzweil | ||
Director of Engineering, Google Inc. |
||
| Judy D. Olian, Ph.D. | ||
Dean, UCLA Anderson School of Management and John E. Anderson Chair in Management |
||
| Christopher Patusky, J.D., M.G.A. | ||
Founding Principal, Patusky Associates, LLC |
||
| Martine Rothblatt, Ph.D., J.D., M.B.A. | ||
Chairman and Co-Chief Executive Officer of United Therapeutics |
||
| Louis Sullivan, M.D. | ||
Former Secretary, U.S. Department of Health and Human Services |
||
| Tommy Thompson, J.D. | ||
Former Secretary, U.S. Department of Health and Human Services |
||
86
ITEM 11. EXECUTIVE COMPENSATION
Information concerning executive compensation required by Item 11 will appear under the headings Director Compensation, Compensation Discussion and Analysis, Summary Compensation Table and Grants of Plan-Based Awards in 2015, Narratives to Summary Compensation Table and Grants of Plan-Based Awards Table, Summary of Terms of Plan-Based Awards, Supplemental Executive Retirement Plan, Rabbi Trust, Potential Payments Upon Termination or Change in Control, and Director Compensation in our 2016 Proxy Statement and is incorporated herein by reference.
Information concerning the Compensation Committee required by Item 11 will appear under the heading Compensation Committee Report in our 2016 Proxy Statement and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information regarding beneficial ownership of our common stock required by Item 12 will appear under Beneficial Ownership of Common Stock in our 2016 Proxy Statement and is incorporated herein by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table presents information as of December 31, 2015, regarding our securities authorized for issuance under equity compensation plans:
Plan category
|
Number of securities to be issued upon exercise of outstanding options (a) |
Weighted average exercise price of outstanding options |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Equity compensation plan approved by security holders |
3,247,438 | $ | 93.09 | 15,233,766 | ||||||
Equity compensation plans not approved by security holders |
— | 0.00 | N/A | |||||||
| | | | | | | | | | | |
Total |
3,247,438 | $ | 93.09 | 15,233,766 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
All outstanding stock options were issued under our two equity incentive plans approved by security holders in 1999 (the 1999 Plan) and 2015 (the 2015 Plan). Information regarding these plans is contained in Note 11—Stockholders' Equity to the consolidated financial statements included in this Annual Report on Form 10-K. Aside from stock options issued under the 1999 Plan and the 2015 Plan, we do not have any outstanding stock options, warrants or rights that are outstanding or available for issuance as described in Regulation S-K Item 201(d). No further awards will be issued under the 1999 Plan.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Information concerning related party transactions and director independence required by Item 13 will appear under the headings Other Matters—Certain Relationships and Related Party Transactions, Board of Directors, Committees, Corporate Governance—Director Independence and Committees of our Board of Directors in our 2016 Proxy Statement and is incorporated herein by reference.
87
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Information required by Item 14 concerning the principal accounting fees paid by the Registrant and the Audit Committee's pre-approval policies and procedures, will appear under the heading Report of the Audit Committee and Information on our Independent Auditors in our 2016 Proxy Statement and is incorporated herein by reference.
88
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
In reviewing the agreements included or incorporated by reference as exhibits to this Annual Report on Form 10-K, it is important to note that they are included to provide investors with information regarding their terms, and are not intended to provide any other factual or disclosure information about United Therapeutics or the other parties to the agreements. The agreements contain representations and warranties made by each of the parties to the applicable agreement. These representations and warranties have been made solely for the benefit of the other parties to the applicable agreement, and: (1) should not be treated as categorical statements of fact, but rather as a way of allocating risk between the parties; (2) have in some cases been qualified by disclosures that were made to the other party in connection with the negotiation of the applicable agreement, which disclosures are not necessarily reflected in the agreement; (3) may apply standards of materiality in a way that is different from what may be material to investors; and (4) were made only as of the date of the applicable agreement or such other date or dates as may be specified in the agreement and are subject to more recent developments.
Accordingly, these representations and warranties may not describe the actual state of affairs as of the date they were made or at any other time. Additional information about United Therapeutics may be found elsewhere in this Annual Report on Form 10-K and our other public filings, which are available without charge through the SEC's website at http://www.sec.gov.
| (a)(1) | Our financial statements filed as part of this report on Form 10-K are set forth in the Index to Consolidated Financial Statements under Part II, Item 8 of this Form 10-K. | |
(a)(2) |
The Schedule II—Valuation and Qualifying Accounts is filed as part of this Form 10-K. All other schedules are omitted because they are not applicable or not required, or because the required information is included in the consolidated statements or notes thereto. |
|
(a)(3) |
Exhibits filed as a part of this Form 10-K are listed on the Exhibit Index, which is incorporated by reference herein. |
Certain exhibits to this report have been included only with the copies of this report filed with the Securities and Exchange Commission. Copies of individual exhibits will be furnished to shareholders upon written request to United Therapeutics and payment of a reasonable fee (covering the expense of furnishing copies). Shareholders may request exhibit copies by contacting: United Therapeutics Corporation, Attn: Investor Relations, 1040 Spring Street, Silver Spring, Maryland 20910.
89
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereto duly authorized.
|
UNITED THERAPEUTICS CORPORATION | |||
|
By: |
/s/ MARTINE A. ROTHBLATT Martine A. Rothblatt, Ph.D. |
||
February 25, 2016 |
Chairman and Co-Chief Executive Officer | |||
|
By: |
/s/ ROGER A. JEFFS Roger A. Jeffs, Ph.D. |
||
February 25, 2016 |
President and Co-Chief Executive Officer | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signatures
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ MARTINE A. ROTHBLATT Martine A. Rothblatt |
Chairman and Co-Chief Executive Officer (Co-Principal Executive Officer) | February 25, 2016 | ||
/s/ ROGER A. JEFFS Roger A. Jeffs |
President, Co-Chief Executive Officer and Director (Co-Principal Executive Officer) |
February 25, 2016 |
||
/s/ JAMES C. EDGEMOND James C. Edgemond |
Chief Financial Officer and Treasurer (Principal Financial Officer and Principal Accounting Officer) |
February 25, 2016 |
||
/s/ CHRISTOPHER CAUSEY Christopher Causey |
Director |
February 25, 2016 |
||
/s/ RAYMOND DWEK Raymond Dwek |
Director |
February 25, 2016 |
||
/s/ RICHARD GILTNER Richard Giltner |
Director |
February 25, 2016 |
90
Signatures
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ KATHERINE KLEIN Katherine Klein |
Director | February 25, 2016 | ||
/s/ RAYMOND KURZWEIL Raymond Kurzweil |
Director |
February 25, 2016 |
||
/s/ JUDY D. OLIAN Judy D. Olian |
Director |
February 25, 2016 |
||
/s/ CHRISTOPHER PATUSKY Christopher Patusky |
Director |
February 25, 2016 |
||
/s/ LOUIS W. SULLIVAN Louis W. Sullivan |
Director |
February 25, 2016 |
||
/s/ TOMMY THOMPSON Tommy Thompson |
Director |
February 25, 2016 |
91
| Exhibit No. | Description | ||
|---|---|---|---|
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant, incorporated by reference to Exhibit 3.1 of the Registrant's Registration Statement on Form S-1 (Registration No. 333-76409). | ||
| 3.2 | Certificate of Amendment to Amended and Restated Certificate of Incorporation of the Registrant, incorporated by reference to Exhibit 3.1 of the Registrant's Current Report on Form 8-K, filed on June 28, 2010. | ||
| 3.3 | Fourth Amended and Restated By-laws of the Registrant, incorporated by reference to Exhibit 3.1 of the Registrant's Current Report on Form 8-K filed on June 26, 2015. | ||
| 3.4 | Form of Certificate of Designation, Preferences and Rights of Series A Junior Participating Preferred Stock of the Registrant, incorporated by reference to Exhibit A to Exhibit 4 to the Registrant's Current Report on Form 8-K, filed December 18, 2000. | ||
| 4.1 | Reference is made to Exhibits 3.1, 3.2, 3.3 and 3.4. | ||
| 4.2 | First Amended and Restated Rights Agreement, incorporated by reference to Exhibit 4.1 of the Registrant's Current Report on Form 8-K filed on July 3, 2008. | ||
| 4.3 | Indenture, dated as of October 17, 2011, between the Registrant and The Bank of New York Mellon Trust Company, N.A., as trustee (including form of 1.0% Convertible Senior Note due September 15, 2016), incorporated by reference to Exhibit 4.1 of the Registrant's Current Report on Form 8-K filed October 17, 2011. | ||
| 4.4 | Form of 1.0% Convertible Senior Note due September 15, 2016, incorporated by reference to Exhibit 4.2 of the Registrant's Current Report on Form 8-K filed October 17, 2011. | ||
| 10.1 | ** | United Therapeutics Corporation Amended and Restated Equity Incentive Plan, as amended effective as of September 24, 2004, incorporated by reference to Exhibit 10.1 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2004. | |
| 10.2 | ** | Amended and Restated Executive Employment Agreement dated as of January 1, 2009, between the Registrant and Martine A. Rothblatt, incorporated by reference to Exhibit 10.2 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2009. | |
| 10.3 | ** | Employment Agreement dated as of June 16, 2001 between the Registrant and Paul A. Mahon, incorporated by reference to Exhibit 10.4 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2002. | |
| 10.4 | ** | Employment Agreement dated November 29, 2000 between the Registrant and Roger Jeffs, incorporated by reference to Exhibit 10.9 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2002. | |
| 10.5 | Form of Indemnification Agreement between the Registrant and each of its Directors and Executive Officers, incorporated by reference to Exhibit 10.1 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2009. | ||
| 10.6 | ** | Amendment dated December 11, 2002 to Employment Agreement between the Registrant and Roger Jeffs, incorporated by reference to Exhibit 10.40 of the Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2002. | |
92
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.7 | ** | Amendment dated December 11, 2002 to Employment Agreement between the Registrant and Paul Mahon, incorporated by reference to Exhibit 10.43 of the Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2002. | |
| 10.8 | ** | Amendment dated December 29, 2004 to Employment Agreement between Roger Jeffs and the Registrant dated November 29, 2000, as previously amended, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on December 29, 2004. | |
| 10.9 | ** | Amendment dated December 29, 2004 to Employment Agreement between Paul A. Mahon and the Registrant dated June 16, 2001, as previously amended, incorporated by reference to Exhibit 10.4 of the Registrant's Current Report on Form 8-K filed on December 29, 2004. | |
| 10.10 | ** | Form of terms and conditions for awards granted to Employees by the Registrant under the Amended and Restated Equity Incentive Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on December 17, 2004. | |
| 10.11 | ** | Form of Terms and Conditions for Awards granted to Non-Employees by the Registrant under the Amended and Restated Equity Incentive Plan, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on December 17, 2004. | |
| 10.12 | ** | United Therapeutics Corporation Supplemental Executive Retirement Plan, effective as of July 1, 2006, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on May 4, 2006. | |
| 10.13 | ** | Employment Agreement, dated as of August 2, 2006, between John Ferrari and the Registrant, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on August 4, 2006. | |
| 10.14 | ** | Amendment, dated as of July 31, 2006, to amended Employment Agreement, dated November 29, 2000, between Roger Jeffs and the Registrant, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on August 4, 2006. | |
| 10.15 | ** | Amendment, dated as of July 31, 2006, to amended Employment Agreement, dated June 16, 2001, between Paul A. Mahon and the Registrant, incorporated by reference to Exhibit 10.3 of the Registrant's Current Report on Form 8-K filed on August 4, 2006. | |
| 10.16 | ** | Amendment, dated as of December 28, 2006, to Employment Agreement, dated August 2, 2006, between John Ferrari and the Registrant, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on December 29, 2006. | |
| 10.17 | United Therapeutics Corporation Supplemental Executive Retirement Plan Rabbi Trust Document entered into on December 28, 2007, by and between the Registrant and Wilmington Trust Company, as trustee, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on December 28, 2007. | ||
| 10.18 | ** | United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2008. | |
| 10.19 | ** | First Amendment to the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on September 18, 2009. | |
93
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.20 | ** | Second Amendment to the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on February 6, 2012. | |
| 10.21 | ** | Form of terms and conditions for awards granted to non-employees by the Registrant under the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.2 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2008. | |
| 10.22 | ** | Form of terms and conditions for awards granted to employees by the Registrant prior to January 1, 2010, under the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.3 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2008. | |
| 10.23 | ** | Form of terms and conditions for awards granted to employees by the Registrant on or after January 1, 2010, under the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.48 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2009. | |
| 10.24 | ** | Form of terms and conditions for awards granted to employees on or after March 15, 2011 under the United Therapeutics Corporation 2011 Share Tracking Awards Plan and the United Therapeutics Corporation 2008 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.2 of Registrant's Registration Statement on Form S-8 (Registration No. 333-173858) filed on May 2, 2011. | |
| 10.25 | ** | Form of grant letter used by Registrant under the United Therapeutics Corporation Share Tracking Awards Plan, incorporated by reference to Exhibit 10.4 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2008. | |
| 10.26 | ** | United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on March 18, 2011. | |
| 10.27 | ** | First Amendment to the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on February 6, 2012. | |
| 10.28 | ** | Second Amendment to the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012. | |
| 10.29 | ** | Third Amendment to the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on February 4, 2013. | |
| 10.30 | ** | Fourth Amendment to the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on January 31, 2014. | |
| 10.31 | ** | Form of terms and conditions for awards granted to employees by the Registrant on or after March 15, 2011 under the United Therapeutics Corporation Share Tracking Awards Plan or the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on March 18, 2011. | |
94
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.32 | ** | Form of terms and conditions for awards granted to non-employees by the Registrant on or after March 15, 2011 under the United Therapeutics Corporation Share Tracking Awards Plan or the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.3 of the Registrant's Current Report on Form 8-K filed on March 18, 2011. | |
| 10.33 | ** | Form of grant letter used by Registrant under the United Therapeutics Corporation 2011 Share Tracking Awards Plan, incorporated by reference to Exhibit 10.4 of the Registrant's Current Report on Form 8-K filed on March 18, 2011. | |
| 10.34 | ** | United Therapeutics Corporation Employee Stock Purchase Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2012. | |
| 10.35 | * | License Agreement, dated as of November 14, 2008, by and between Eli Lilly and Company and the Registrant, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on December 24, 2008. | |
| 10.36 | * | Manufacturing and Supply Agreement, dated as of November 14, 2008, by and between Eli Lilly and Company, Lilly del Caribe, Inc. and the Registrant incorporated by reference to Exhibit 10.3 of the Registrant's Current Report on Form 8-K filed on December 24, 2008. | |
| 10.37 | ** | Form of Amendment to Employment Agreement between the Registrant and each of Roger Jeffs, Paul Mahon and John Ferrari, each dated as of January 1, 2009, incorporated by reference to Exhibit 10.3 of the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2009. | |
| 10.38 | ** | Form of Amendment to Employment Agreements between the Registrant and each of Roger Jeffs, Paul Mahon and John Ferrari, each dated as of February 22, 2010, incorporated by reference to Exhibit 10.46 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2009. | |
| 10.39 | Distribution Agreement relating to Tyvaso, dated as of August 17, 2009 between the Registrant and Accredo Health Group, Inc., incorporated by reference to Exhibit 10.47 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2009. | ||
| 10.40 | First Amendment to Distribution Agreement relating to Tyvaso, dated as of September 1, 2011, between the Registrant and Accredo Health Group, Inc., incorporated by reference to Exhibit 10.44 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2013. | ||
| 10.41 | Second Amendment to Distribution Agreement relating to Tyvaso, dated as of December 18, 2013, between the Registrant, Accredo Health Group, Inc., CuraScript, Inc. and Priority Healthcare Distribution, Inc., incorporated by reference to Exhibit 10.45 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2013. | ||
| 10.42 | Stipulation of Settlement, dated October 25, 2010, among the parties to a derivative lawsuit against the directors and officers of the Registrant identified therein, incorporated by reference to Exhibit 10.1 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2010. | ||
95
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.43 | *** | Amended and Restated Distribution Agreement relating to Remodulin, dated as of February 21, 2011, between the Registrant and Accredo Health Group, Inc., incorporated by reference to Exhibit 10.38 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2010. | |
| 10.44 | First Amendment to Amended and Restated Distribution Agreement relating to Remodulin, dated as of December 18, 2013, between the Registrant, Accredo Health Group, Inc., CuraScript, Inc. and Priority Healthcare Distribution, Inc., incorporated by reference to Exhibit 10.49 of the Registrant's Annual Report on Form 10-K for the year ended December 31, 2013. | ||
| 10.45 | * | Confirmation, dated October 11, 2011, of a note hedging transaction between the Registrant and Deutsche Bank AG, London Branch, incorporated by reference to Exhibit 10.3 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. | |
| 10.46 | * | Confirmation, dated October 11, 2011, of a warrant transaction between the Registrant and Deutsche Bank AG, London Branch, incorporated by reference to Exhibit 10.2 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. | |
| 10.47 | Credit Agreement dated as of September 26, 2013, by and among the Registrant, the lenders party thereto from time to time, Wells Fargo Bank, National Association, as the Administrative Agent, and a subsidiary of the Registrant, as guarantor, incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed September 27, 2013. | ||
| 10.48 | ** | Amendment to Amended and Restated Executive Employment Agreement between the Registrant and Martine Rothblatt, Ph.D., dated as of January 1, 2015, incorporated by reference to Exhibit 10.1 to Registrant's Current Report on Form 8-K filed December 17, 2014. | |
| 10.49 | ** | Employment Agreement, dated as of June 26, 2006, between the Company and David Zaccardelli, Pharm.D., together with three amendments thereto, dated January 26, 2007, September 23, 2009 and February 24, 2010, respectively, incorporated by reference to Exhibit 10.2 to Registrant's Current Report on Form 8-K filed December 17, 2014. | |
| 10.50 | ** | Change in Control Severance Agreement between the Company and David Zaccardelli, Pharm.D., dated as of February 14, 2012, incorporated by reference to Exhibit 10.3 to Registrant's Current Report on Form 8-K filed December 17, 2014. | |
| 10.51 | Amendment No. 1 to Credit Agreement, dated as of July 24, 2014, by and among the Registrant, the lenders party thereto from time to time, Wells Fargo Bank, National Association, as the Administrative Agent, and a subsidiary of the Registrant, as guarantor, incorporated by reference to Exhibit 10.1 to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2014. | ||
| 10.52 | ** | United Therapeutics Corporation Section 162(m) Bonus Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed June 27, 2014. | |
| 10.53 | * | Third Amendment to Distribution Agreement relating to Tyvaso, dated October 20, 2014, by and among the Registrant, Accredo Health Group, Inc., CuraScript, Inc., and Priority Healthcare Distribution, Inc, incorporated by reference to Exhibit 10.54 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2014. | |
96
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.54 | ** | Employment Agreement, dated as of March 13, 2015, between the Company and James Edgemond, incorporated by reference to Exhibit 10.55 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2014. | |
| 10.55 | ** | Change in Control Severance Agreement between the Company and James Edgemond, dated as of November 12, 2014, incorporated by reference to Exhibit 10.56 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2014. | |
| 10.56 | ** | United Therapeutics Corporation 2015 Stock Incentive Plan, incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on June 29, 2015. | |
| 10.57 | ** | Form of Grant Notice and Standard Terms and Conditions for Non-Qualified Stock Options Granted to Non-Employee Directors under the United Therapeutics Corporation 2015 Stock Incentive Plan, incorporated by reference to Exhibit 10.2 of the Registrant's Current Report on Form 8-K filed on June 29, 2015. | |
| 10.58 | ** | Form of Grant Notice and Standard Terms and Conditions for Non-Qualified Stock Options Granted to Certain Executives under the United Therapeutics Corporation 2015 Stock Incentive Plan, incorporated by reference to Exhibit 10.3 of the Registrant's Current Report on Form 8-K filed on June 29, 2015. | |
| 10.59 | ** | Form of Grant Notice and Standard Terms and Conditions for Non-Qualified Stock Options Granted to Employees under the United Therapeutics Corporation 2015 Stock Incentive Plan, incorporated by reference to Exhibit 10.4 of the Registrant's Current Report on Form 8-K filed on June 29, 2015. | |
| 10.60 | Amendment No. 2 to Credit Agreement, dated as of July 24, 2015, by and among the Registrant, the lenders party thereto from time to time, Wells Fargo Bank, National Association, as the Administrative Agent, and a subsidiary of the Registrant, as guarantor, incorporated by reference to Exhibit 10.6 to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2015. | ||
| 10.61 | ** | First Amendment to the United Therapeutics Corporation Amended and Restated Equity Incentive Plan, effective as of June 2, 2015, incorporated by reference to Exhibit 10.1 to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2015. | |
| 10.62 | Asset Purchase Agreement, dated as of August 18, 2015, by and between the Registrant and AbbVie Ireland Unlimited Company, incorporated by reference to Exhibit 2.1 of the Registrant's Current Report on Form 8-K filed on August 19, 2015. | ||
| 10.63 | * | Settlement Agreement, dated September 29, 2015, between the Registrant and Sandoz Inc., incorporated by reference to Exhibit 10.2 to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015. | |
| 10.64 | Amendment No. 3 to Credit Agreement, dated as of October 12, 2015, by and among the Registrant, the lenders party thereto from time to time, Wells Fargo Bank, National Association, as the Administrative Agent, and a subsidiary of the Registrant, as guarantor, incorporated by reference to Exhibit 10.3 to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2015. | ||
| 10.65 | Credit Agreement, dated as of January 29, 2016, among the Registrant, certain of its subsidiaries party thereto, as guarantors, the lenders referred to therein, and Wells Fargo Bank, National Association, as administrative agent and as a swingline lender, , incorporated by reference to Exhibit 10.1 of the Registrant's Current Report on Form 8-K filed on February 1, 2016. | ||
97
| Exhibit No. | Description | ||
|---|---|---|---|
| 21 | Subsidiaries of the Registrant. | ||
| 23.1 | Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm. | ||
| 31.1 | Certification of Co-Principal Executive Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. | ||
| 31.2 | Certification of Co-Principal Executive Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. | ||
| 31.3 | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. | ||
| 32.1 | Certification of Co-Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| 32.2 | Certification of Co-Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| 32.3 | Certification of Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| 101 | The following financial information from our Annual Report on Form 10-K for the year ended December 31, 2015, filed with the SEC on February 25, 2016, formatted in Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets as of December 31, 2015 and 2014, (ii) Consolidated Statements of Operations for each of three years in the period ended December 31, 2015, (iii) Consolidated Statements of Comprehensive Income for each of the three years in the period ended December 31, 2015, (iv) Consolidated Statements of Stockholders' Equity for each of the three years in the period ended December 31, 2015, (v) Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2015, and (vi) Notes to Consolidated Financial Statements. | ||
- *
- Confidential
treatment has been granted with respect to certain portions of this exhibit pursuant to Rule 406 of the Securities Act of 1933, as amended
or Rule 24b-2 of the Securities Act of 1934, as amended. The omitted portions of this document have been filed with the Securities and Exchange Commission.
- **
- Designates
management contracts and compensation plans.
- ***
- The
Company as has requested an extension of confidential treatment for certain portions of this exhibit pursuant to Rule 24b-2 of the Securities Act
of 1934, as amended. The omitted portions of this document have been filed with the Securities and Exchange Commission.
Note: Except as otherwise noted above, all exhibits incorporated by reference to the Registrant's previously filed reports with the Securities and Exchange Commission are filed under File No. 000-26301.
98