Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT 31.1 - NephroGenex, Inc. | nrx10-k2014exhibit311.htm |
| EX-31.2 - EXHIBIT 31.2 - NephroGenex, Inc. | nrx10-k2014exhibit312.htm |
| EX-23.1 - EXHIBIT 23.1 - NephroGenex, Inc. | nrx10-k2014exhibit231conse.htm |
| EXCEL - IDEA: XBRL DOCUMENT - NephroGenex, Inc. | Financial_Report.xls |
| EX-32 - EXHIBIT 32 - NephroGenex, Inc. | nrx10-kexhibit32.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One) | |
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014 | |
OR | |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to | |
Commission file number: 001‑36303
NephroGenex, Inc.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 20‑1295171 (I.R.S. Employer Identification No.) |
3200 Beechleaf Court Suite 900 Raleigh, NC (Address of principal executive offices) | 27604 (Zip Code) |
(609) 986‑1780
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Exchange Act:
Title of each class | Name of each exchange on which registered |
Common Stock, $0.001 Par Value Per Share | NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S‑T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S‑K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10‑K or any amendment to this Form 10‑K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b‑2 of the Exchange Act. (Check one):
Large accelerated filer o | Accelerated filer o | |
Non-accelerated filer o | Smaller reporting company x | |
(Do not check if a smaller reporting company) | ||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Exchange Act). Yes No x
The aggregate market value of the registrant’s voting and non‑voting common stock held by non‑affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) computed by reference to the price at which the common stock was last sold as of June 30, 2014, the last business day of the registrant's most recently completed second fiscal quarter, was $32,889,304.
As of March 24, 2015 the registrant had 8,863,614 shares of common stock outstanding.
TABLE OF CONTENTS
Page | ||
1
Forward‑Looking Statements
This Annual Report on Form 10‑K contains forward‑looking statements. All statements other than statements of historical facts contained in this Annual Report on Form 10‑K, including statements regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth are forward‑looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward‑looking statements.
The words “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward‑looking statements, although not all forward‑looking statements contain these identifying words. These forward‑looking statements include, among other things, statements about:
• | our ability to obtain additional financing; |
• | the accuracy of our estimates regarding expenses, future revenues and capital requirements; |
• | the success and timing of our preclinical studies and clinical trials; |
• | our ability to obtain and maintain regulatory approval of Pyridorin and any other product candidates we may develop, and the labeling under any approval we may obtain; |
• | regulatory developments in the United States and other countries; |
• | the performance of third‑party manufacturers; |
• | our plans to develop and commercialize our product candidates; |
• | our ability to obtain and maintain intellectual property protection for our product candidates; |
• | the successful development of our sales and marketing capabilities; |
• | the potential markets for our product candidates and our ability to serve those markets; |
• | the rate and degree of market acceptance of any future products; |
• | the success of competing drugs that are or become available; and |
• | the loss of key scientific or management personnel. |
These forward‑looking statements are only predictions and we may not actually achieve the plans, intentions or expectations disclosed in our forward‑looking statements, so you should not place undue reliance on our forward‑looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward‑looking statements we make. We have based these forward‑looking statements largely on our current expectations and projections about future events and trends that we believe may affect our business, financial condition and operating results. We have included important factors in the cautionary statements included in this Annual Report on Form 10‑K, particularly in Item 1.A. Risk Factors, that could cause actual future results or events to differ materially from the forward‑looking statements that we make. Our forward‑looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make.
You should read this Annual Report on Form 10‑K and the documents that we have filed as exhibits to the Annual Report on Form 10‑K with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward‑looking statements whether as a result of new information, future events or otherwise, except as required by applicable law.
2
PART 1
Item 1. BUSINESS
All brand names or trademarks appearing in this report are the property of their respective holders. Unless the context requires otherwise, references in this report to “NephroGenex,” the “Company,” “we,” “us,” and “our” refer to NephroGenex, Inc.
Overview
We are a pharmaceutical company focused on the development of therapeutics to treat kidney disease, an area of significant unmet medical need. Since our inception, we have collaborated with the world’s leading experts in kidney disease and leveraged our knowledge of pathogenic oxidative chemistries to build a strong portfolio of intellectual property and to advance the development of our drug candidates. We believe that our comprehensive effort to develop a new generation of therapeutics that target kidney disease provides us with a leadership position in this large and attractive market.
Pathogenic oxidative chemistries are collectively a group of oxygen‑based chemical reactions that occur in the body during stress, injury, or disease, to form compounds that can induce pathological changes in tissues that effect normal physiological function. These include (i) advanced glycation end‑products (AGE’s), which are oxidative end products of glucose‑modified biomolecules which adversely affect their function; (ii) reactive oxygen species (ROS), which are chemically reactive molecules containing oxygen such as oxygen ions and peroxides that when elevated in the body can induce pathology; and (iii) toxic carbonyls which are reactive compounds that can modify biomolecules and affect their function. These chemistries are generally agreed to be involved in the etiology of diabetic nephropathy, a common complication of diabetes, and in cases of acute kidney injury (AKI). We are developing Pyridorin™ (“Pyridorin”), a small molecule drug that is a unique and broadly acting inhibitor of the pathogenic oxidative chemistries which are elevated in diabetic patients.
We licensed patents covering methods of use and synthesis of Pyridorin from BioStratum, Inc. in May of 2006. We subsequently acquired Pyridorin‑related patents from BioStratum through a Series A financing completed in May of 2007. At the time of acquisition, BioStratum, through its contracted investigators, contract research organizations, and collaborators had completed 5 preclinical efficacy studies, 36 preclinical safety studies, 4 Phase 1 studies and 5 Phase 2 studies with Pyridorin. After the acquisition, we conducted a multi‑center, randomized, placebo‑controlled Phase 2b study, namely PYR‑210 and recently completed the Phase 1 QT/QTc (TQT) cardiac safety study. In addition, we worked with the FDA to establish a new regulatory pathway for Pyridorin approval, as well as received support from the European Medicines Agency (EMA) regarding the pivotal Phase 3 program with Pyridorin in diabetic nephropathy.
Pyridorin has demonstrated preliminary evidence of efficacy in slowing the progression of diabetic nephropathy in relevant patient populations in three Phase 2 clinical studies. Based on these results, Pyridorin entered into a Phase 3 program in 2014 termed the PIONEER trial which was agreed to by the U.S. Food and Drug Administration (FDA), with fast track designation, under a Special Protocol Assessment (SPA). This Phase 3 program is using a novel , events‑based endpoint based on end stage renal disease (ESRD) or a 50% increase in serum creatinine (SCr). We believe this change will significantly reduce the cost and time for completion of our Phase 3 program compared to the traditional endpoint used in previous pivotal trials for diabetic nephropathy which is a 100% increase in SCr from baseline or end stage renal disease (ESRD). Based on an analysis of the Irbesartan Type II Diabetic Nephropathy Trial (IDNT) used for the approval of the drug irbesartan, the follow‑up time required to reach the new endpoint of a 50% SCr increase would be approximately 50% less than the follow‑up time required to reach the traditional endpoint in a similar patient population. We believe that we are the first company to use this novel endpoint in a Phase 3 trial.
We are also studying the application of an intravenous formulation of Pyridorin to specific types of AKI in patients at increased risk and where pathogenic oxidative chemistries have been identified as a possible contributing factor to the severity of this condition. Our preclinical program has shown encouraging results in animal models of ischemia-reperfusion AKI including an observed treatment effect on post injury fibrosis.
Corporate Objectives
There is a large medical need and market opportunity for treatments that can (1) slow the progression of renal disease and thus delay or avoid the onset of ESRD; or (2) reduce the severity of AKI and its associated potential treatment costs and long term complications.
3
Our principal corporate objective is the maximization of shareholder value by advancing Pyridorin through Phase 3 development and approval. In order to maximize the market potential of Pyridorin, we intend to consider entering into a partnership for the launch and marketing of the product at the end of Phase 3 or possibly earlier, based on interim clinical data. We also intend to consider acquisitions and the development of other clinical candidates as we see appropriate.
We acquired commercial rights to Pyridorin in 2007 and, since then, have been investigating the safety and efficacy of Pyridorin therapy for diseases in which pathogenic oxidative chemistries are an established and/or causative and contributing factor in kidney disease. These include diabetic nephropathy and AKI.
We anticipate seeking corporate partners to aid us in commercialization and market entry.
Our Strategy
We are committed to applying our leadership position in the field of kidney disease to transform the lives of patients with debilitating, costly diseases or conditions. Each of our ongoing and planned development projects addresses kidney diseases or conditions with high unmet medical need that presents a significant market opportunity. The core elements of our strategy include:
• | advancing Pyridorin through Phase 3 development for the treatment of diabetic nephropathy in patients with type 2 diabetes; |
• | submission and approval of a new drug application (NDA) in the United States and a Market Authorization Application (MAA) in Europe; |
• | commercializing Pyridorin using a highly‑targeted sales force in the United States and the rest of the world; |
• | continued development of an intravenous formulation of Pyridorin for AKI, with an investigational new drug application (IND) filing and launch of the initial clinical study during the second half of 2015; and |
• | deploying capital strategically to develop our portfolio of product candidates and create shareholder value. |
Rationale for Development of Pyridorin
Diabetic microvascular complications arise in tissues that are not under direct insulin control and are thus exposed to elevated levels of glucose in hyperglycemic conditions. This exposure leads to a perturbation or deviation of many metabolic pathways and the emergence of non‑enzymatic oxidative chemistries that form pathogenic reactive compounds including: (1) reactive oxygen species; (2) reactive carbonyl intermediates (which are reactive compounds containing a carbonyl function group that can react with biomolecules and modify their function, a process collectively referred to as carbonyl stress); and (3) glycated protein amino groups and their subsequent AGEs.
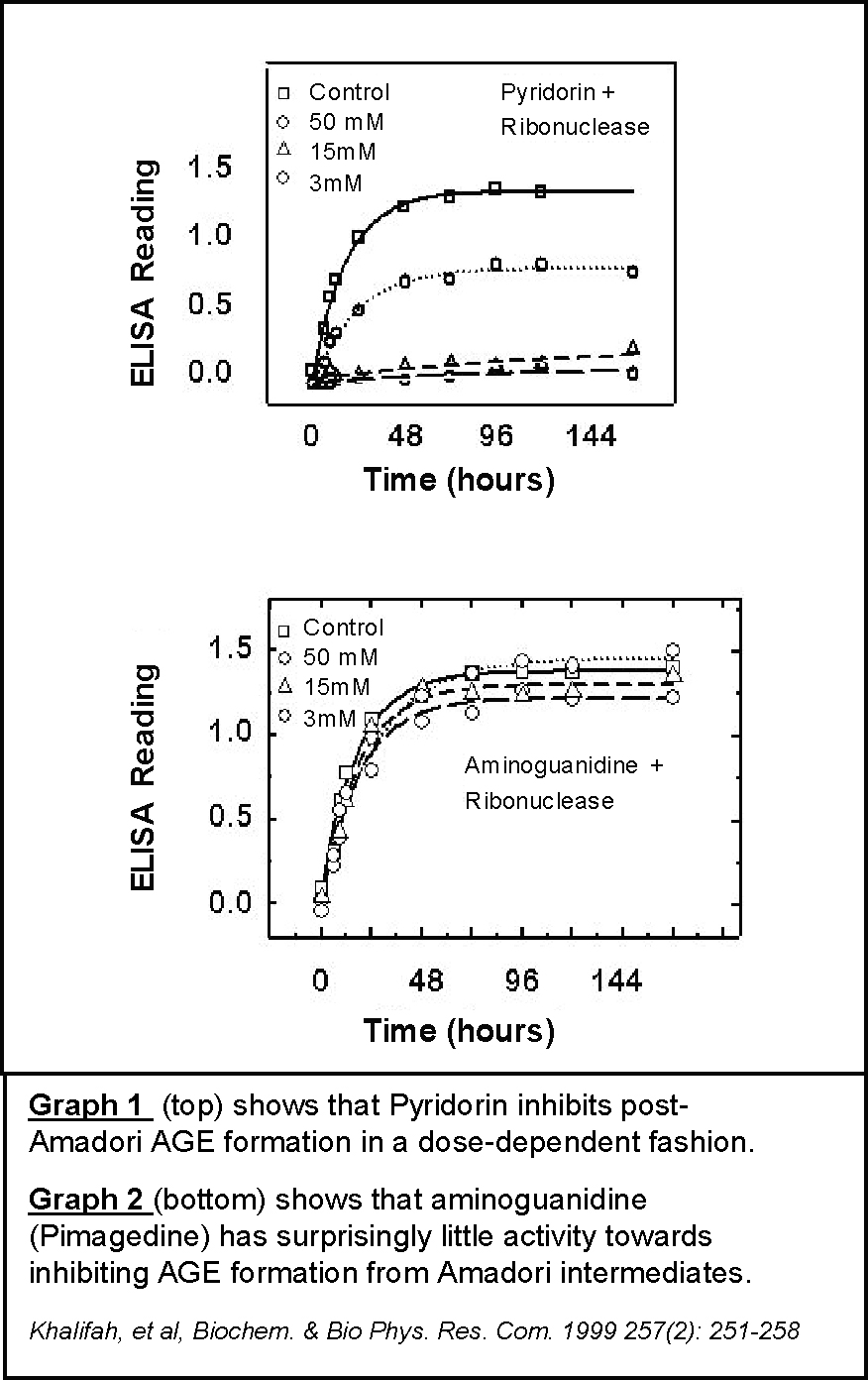
One pathway of particular interest is the post‑Amadori pathway of AGE formation. The study of this pathway led to the discovery of Pyridorin as a promising drug candidate for diabetic nephropathy. Scientists first isolated protein‑Amadori intermediates and utilized them to search for compounds that could specifically block the degradation of protein‑Amadori intermediates into AGEs. They examined many previously studied AGE inhibitors in this screening assay, including aminoguanidine (pimagedine). The majority of such AGE inhibitors, including aminoguanidine (Graph 2), did not exhibit inhibitory activity towards formation of the AGE carboxymethlylysine (CML) under these conditions. However, Pyridorin uniquely exhibited potent post‑Amadori inhibitory activity (Graph 1). Due to the possible importance of this AGE pathway, this inhibitory activity may form the basis for the activity of Pyridorin in inhibiting the progression of diabetic nephropathy, as evidenced in nonclinical studies and as summarized below.
4
Chronic hyperglycemia is directly associated with end‑organ damage in patients with diabetes. The major target organs affected, namely the kidney, peripheral nerves, retina, and the vasculature, are all exposed to glucose fluctuations since they are not under insulin regulation. This hyperglycemia damage may be initiated by direct chemical reaction of glucose (an aldehyde) with protein amino groups, leading to the formation of harmful products collectively designated as AGEs. It has been established that circulating and tissue levels of AGEs are elevated in patients with poorly controlled diabetes and increase dramatically when the glomerular filtration rate (GFR) declines. GFR is the calculation of the flow rate of filtered fluid through the glomerulus that determines how well the kidney is filtering the blood.
In extensive in‑vitro studies, Pyridorin has been shown to inhibit AGE formation and scavenge ROS and toxic carbonyl compounds. For example, Pyridorin has been shown to:
5
• | inhibit the degradation of glycated proteins to AGEs; |
• | inhibit lipoxidation (lipid oxidation) by trapping lipoxidation intermediates, (reactive lipid compounds that form during the oxidation of lipids that normally proceed to lipid oxidation end‑products), particularly 1,4‑dicarbonyls; |
• | scavenge glycoaldehyde and dicarbonyls intermediates of carbonyl stress such as glyoxal and methylglyoxal; |
• | trap the hydroxyl radical (which is a highly reactive and short‑lived neutral form of the hydroxide ion (HO−); and |
• | bind redox transition metal ions (such as Cu2+, Mn2+, and Fe 2+),which interfere with their catalytic role in oxidative reactions (redox chemical reactions are common physiological chemical reactions involving the transfer of electrons). |
All of the above processes and reactive compounds have been implicated directly or indirectly in the development of diabetic microvascular disease, the basis of diabetic complications.
Pyridorin Targets Specific
Pathogenic Oxidative Chemistries
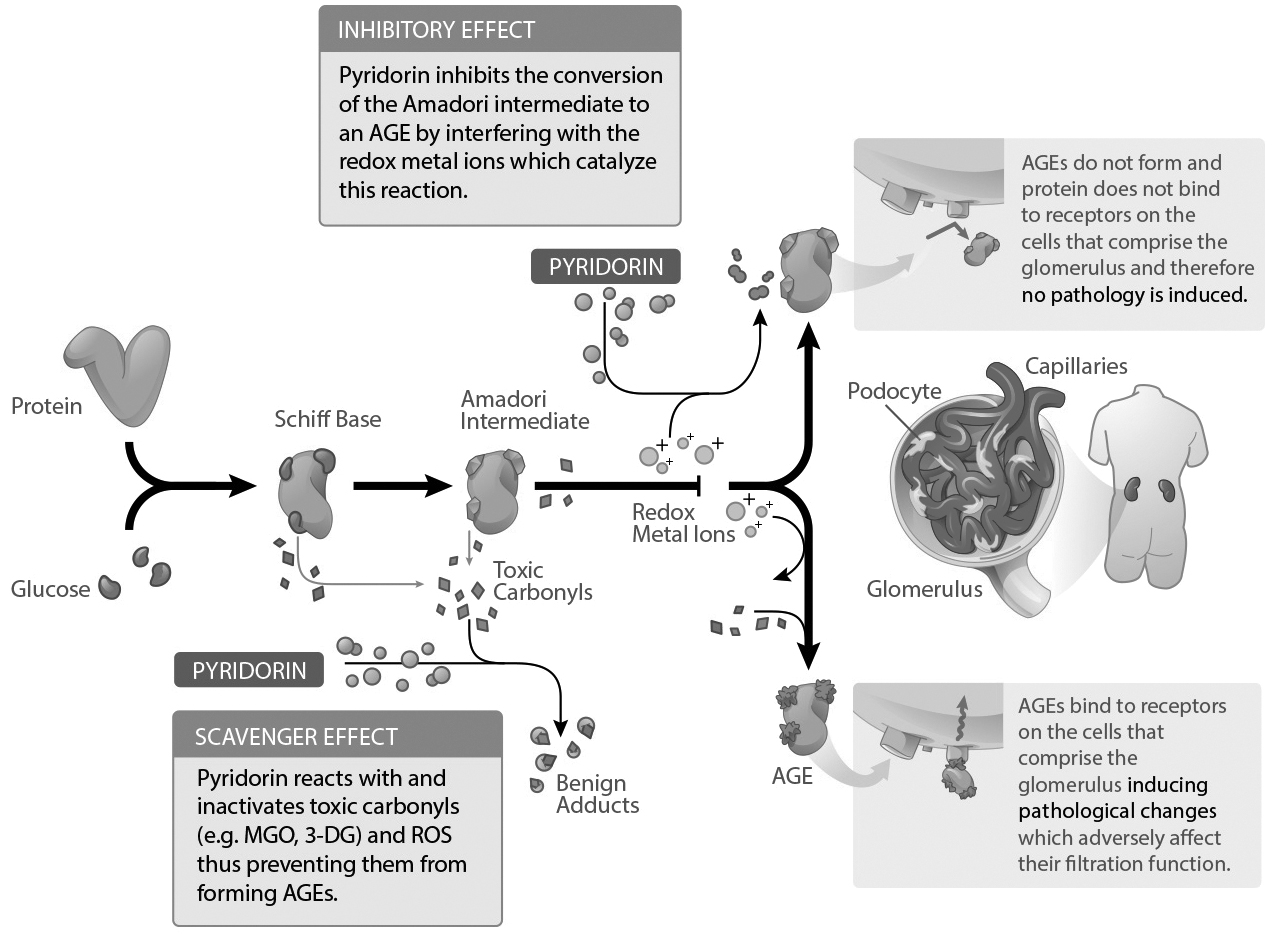
The above graphic is for illustrative purposes only, complete details of the mechanism of action are unknown at this time.
6
Preclinical Efficacy Results
The ability of Pyridorin to slow the progression of diabetic nephropathy in animals has been examined in several preventative and interventional preclinical studies. These include a “proof‑of‑principle” rat model of AGE‑albumin induced nephropathy (Khalifah, et al, J. Am. Soc. Nephrol. 1997 Sep; 8:641A), an STZ‑treated rat classical model of type 1 diabetic nephropathy (Degenhardt, et al, Kidney Int. 2002; 61:939‑950), a db/db mouse spontaneous model of type 2 diabetic nephropathy Zheng, et al, Kidney Int. 2006; 70: 507‑514), the Zucker fa/fa rat model of non‑diabetic, hyperlipidemic nephropathy (Alderson, et al, Kidney Int. 2003; 63:2123‑2133), and the type 2 diabetic KK‑Ay/Ta mouse (Tanimoto, et al, Metabolism. 56:160‑7, 2007).
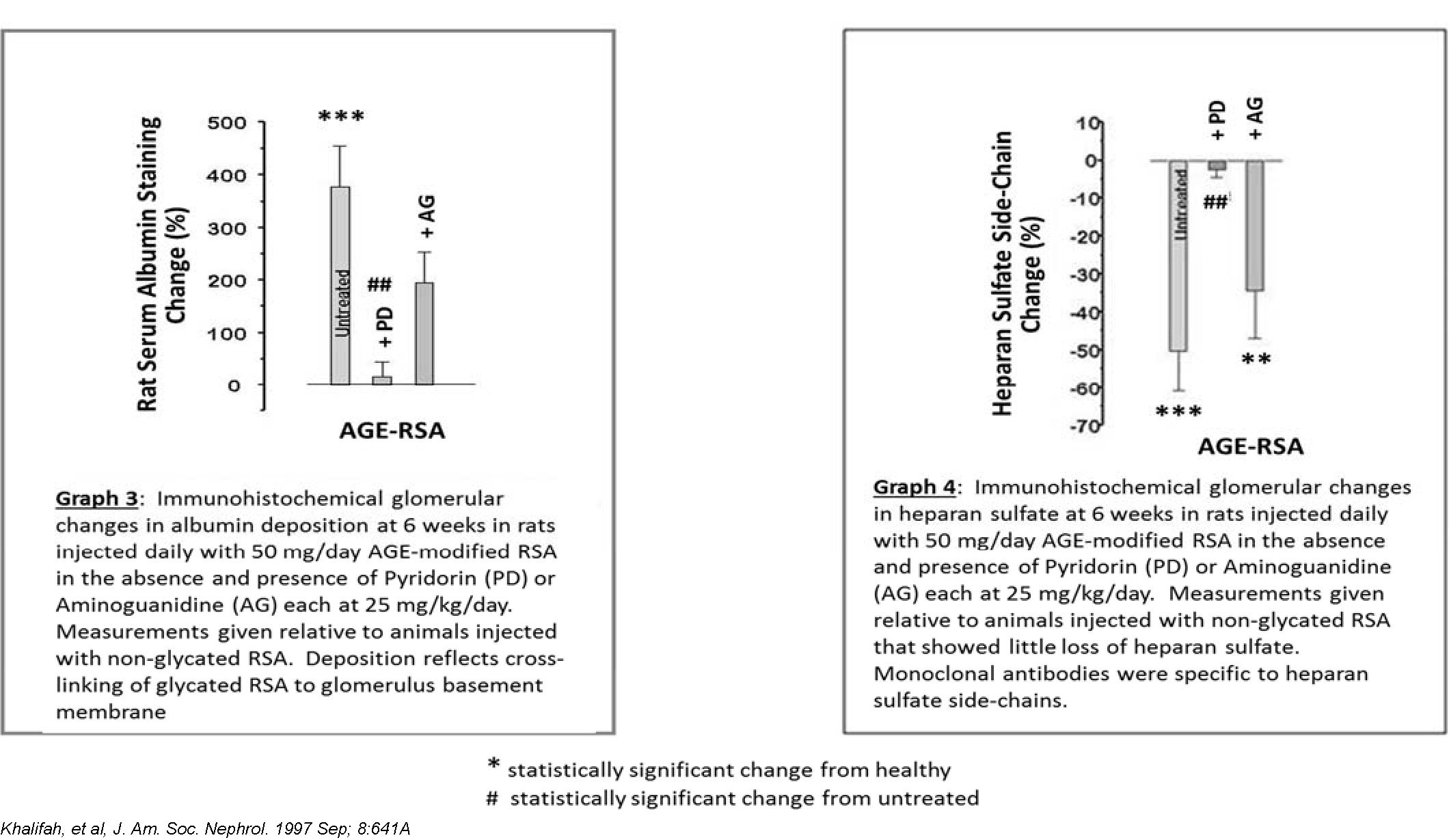
In the first model, AGE‑modified rat serum albumin (RSA), which is the most abundant protein in rat blood plasma, was injected daily for 6 weeks into normoglycemic rats to mimic damage from circulating AGE‑modified plasma proteins. These normoglycemic rats were given daily tail vein injections of AGE‑modified RSA at 50 mg/kg/day with and without concomitant treatment with 25 mg/kg/day Pyridorin in the drinking water. Another AGE inhibitor, aminoguanidine (pimagedine) was also evaluated in this model for comparative purposes. At the time of this study, aminoguanidine was being developed by Alteon for the treatment of diabetic nephropathy. Previous studies have demonstrated that such daily injections of AGE‑modified RSA induce pathological changes in the kidney consistent with the onset of diabetic nephropathy. As expected, overt nephropathy did not develop during this short‑term study. However, statistically significant early diabetic‑like morphological changes were observed in the glomerulus, such as an increase in glomerular volume, an increase in albumin deposition (Graph 3), and a decrease in heparin sulfate, a component of the kidney anionic filtration barrier (Graph 4).
Treatment with Pyridorin protected the animals from the damaging effects of AGE‑albumin with regard to all three parameters mentioned above. All of the results were statistically significant when compared to untreated animals. Treatment with similar amounts of aminoguanidine did not lead to significant amelioration except for a partial reduction in albumin deposition.
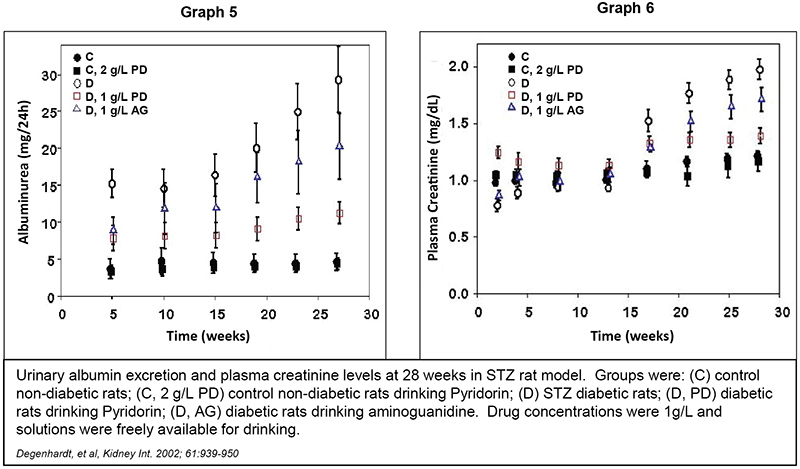
Results from an STZ‑treated rat model of type 1 diabetic nephropathy are shown in Graphs 5 and 6 below. Pyridorin inhibited the development of albuminuria compared to untreated animals (p = 0.0001 at 27 weeks). It also inhibited the increase in plasma creatinine levels compared to untreated animals (p = 0.0001 at 28 weeks). Increases in albuminuria and plasma creatinine levels are indications of decreasing kidney function. Additionally, at equal doses, Pyridorin exhibited an improvement over aminoguanidine in preventing increases in plasma creatinine (p = 0.021 at 28 weeks) and albuminuria.
7
In addition to these results on kidney function, this study demonstrated that Pyridorin significantly inhibited AGE formation in skin collagen, as measured by standard methods of quantifying AGE levels (i.e. pepsin digestibility, AGE fluorescence, and carboxymethyllysine AGE content).
In a second STZ study similar in design to the above, treatment with Pyridorin at 1 g/L drinking water was compared to treatment with the ACE inhibitor enalapril (the standard of care treatment for diabetic nephropathy) dosed at 50 mg/L drinking water (Alderson, et al, Diabetologia 2004; 47:1385‑1395). At 28 weeks, Pyridorin significantly inhibited the development of albuminuria relative to both untreated diabetic controls (43 mg/24 hr versus 12mg/24 hr) and diabetic animals treated with enalapril (26 mg/24 hr versus 12 mg/24 hr). The differences were statistically significant. Pyridorin also significantly reduced the increases in plasma creatinine relative to both untreated diabetic controls (110 ìmol/L versus 45 ìmol/L) and diabetic animals treated with enalapril (70 ìmol/L versus 45 ìmol/L). The differences were statistically significant.
Pyridorin has also been evaluated in a standard model of type 2 diabetic nephropathy. The db/db mouse is a commonly used mouse model of type 2 diabetes and develops histologic changes in the kidney which are very similar to those observed in humans with diabetic nephropathy. The study was designed to evaluate the effects of Pyridorin in established diabetic nephropathy. In mice with biopsy‑proven diabetic nephropathy, Pyridorin orally administered at 250 mg/kg/day for 2 months resulted in a 43% reduction in the urinary albumin/creatinine ratio. In contrast, the placebo group albumin/creatinine ratio increased 215% (p<0.05). The ACE inhibitor treated group increased 40%. Microscopic lesions of glomerulosclerosis in the kidney were also reduced in the Pyridorin group when compared with control animals (p<0.05).
A second db/db mouse study of 16‑week treatment duration was conducted to assess the combination of Pyridorin plus the ACE inhibitor enalapril versus enalapril alone. As in the initial study, there were significant effects on urinary albumin/creatinine ratio. In the placebo group albumin/creatinine ratio increased approximately 350% over 16 weeks. The enalapril treated group increased approximately 220%. The Pyridorin plus enalapril group increased approximately 50% (p<0.05 compared to control). There was also a reduction in glomerular lesions in the Pyridorin plus ACE inhibitor group (p<0.05 compared to control). In addition, Pyridorin plus enalapril significantly improved survival versus the control or enalapril alone (p<0.05).
8
Pyridorin has also been studied in a non‑diabetic, “syndrome X‑like” model to assess its effects on the development of nephropathy in the absence of diabetes. In this study, the development of nephropathy and dyslipidemia in treated and untreated obese fa/fa rats was compared to those in lean Fa/fa littermates. Pyridorin, administered at 1 g/L in the drinking water, markedly inhibited the development of dyslipidemia and nephropathy in the fa/fa rats. A 10‑fold increase in albuminurea was observed in the untreated obese fa/fa rats over 32 weeks as well as an increase in plasma creatinine from 0.9 mg/dL to 1.5 mg/dL. Pyridorin provided nearly complete protection against increases in both of these parameters (p<0.0001). Pyridorin also inhibited the thickening of the aortic and coronary vasculature observed in the untreated obese fa/fa rats by approximately 90% (p<0.05). Furthermore, Pyridorin significantly reduced AGE levels in the rat skin collagen when compared to the untreated fa/fa group (p<0.05).
Pyridorin was also studied in the type 2 diabetic KK‑Ay/Ta mouse. KK‑Ay/Ta mice were given Pyridorin (200 or 400 mg/kg per day) starting at 8 weeks of age for 12 weeks. Pyridorin therapy, especially at 400 mg/kg per day, prevented an increase in albuminuria relative to untreated controls (increase of 6.4 mg/L versus 43.5 mg/L, p<0.05). Accumulations or Carboxymethyllysine (an AGE) and nitrotyrosine in the kidney were also decreased (p<0.05). TGF‑β1 and laminin‑β1 messenger RNA expressions in kidneys were significantly lower than those in the controls (p<0.05).
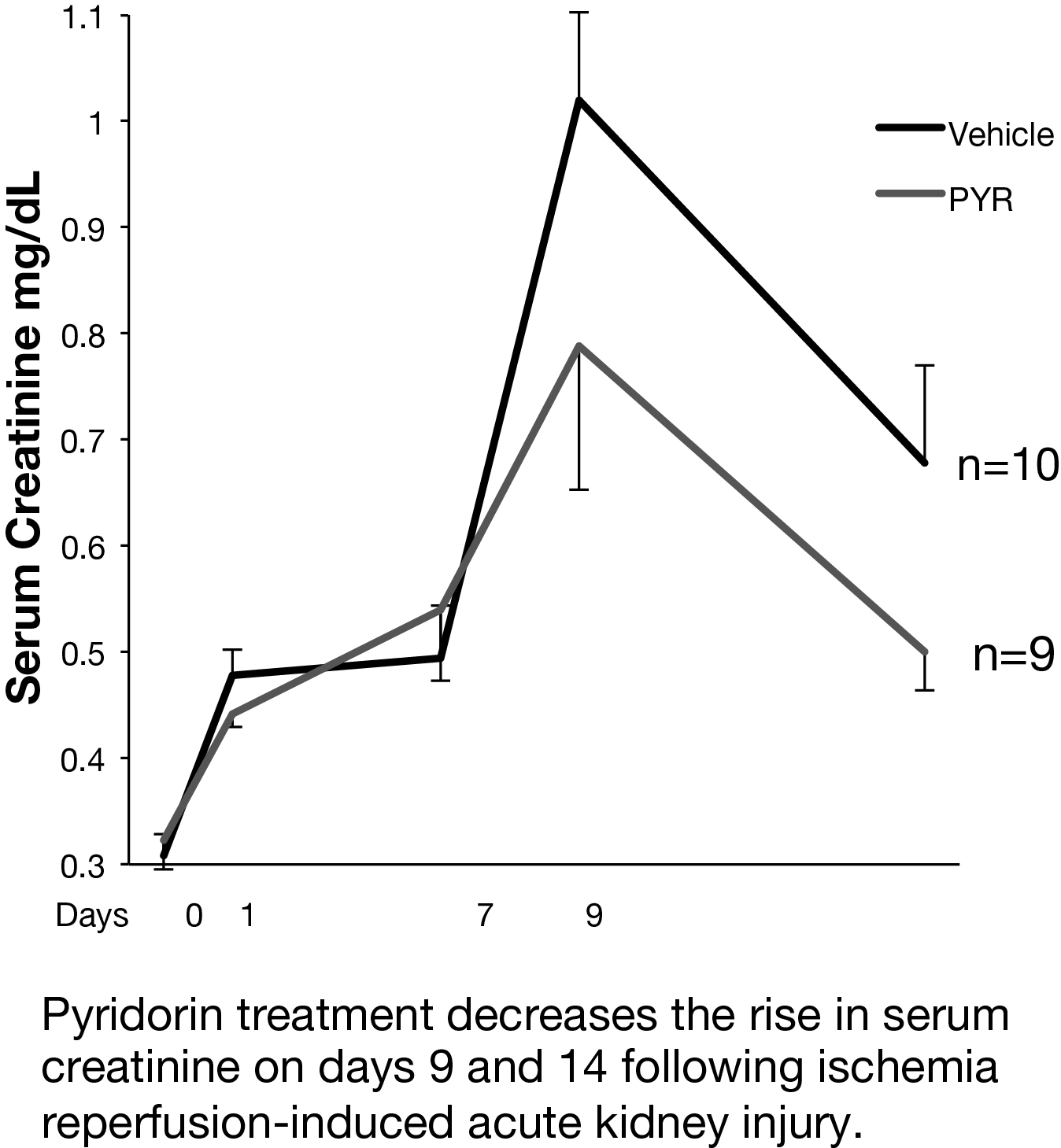
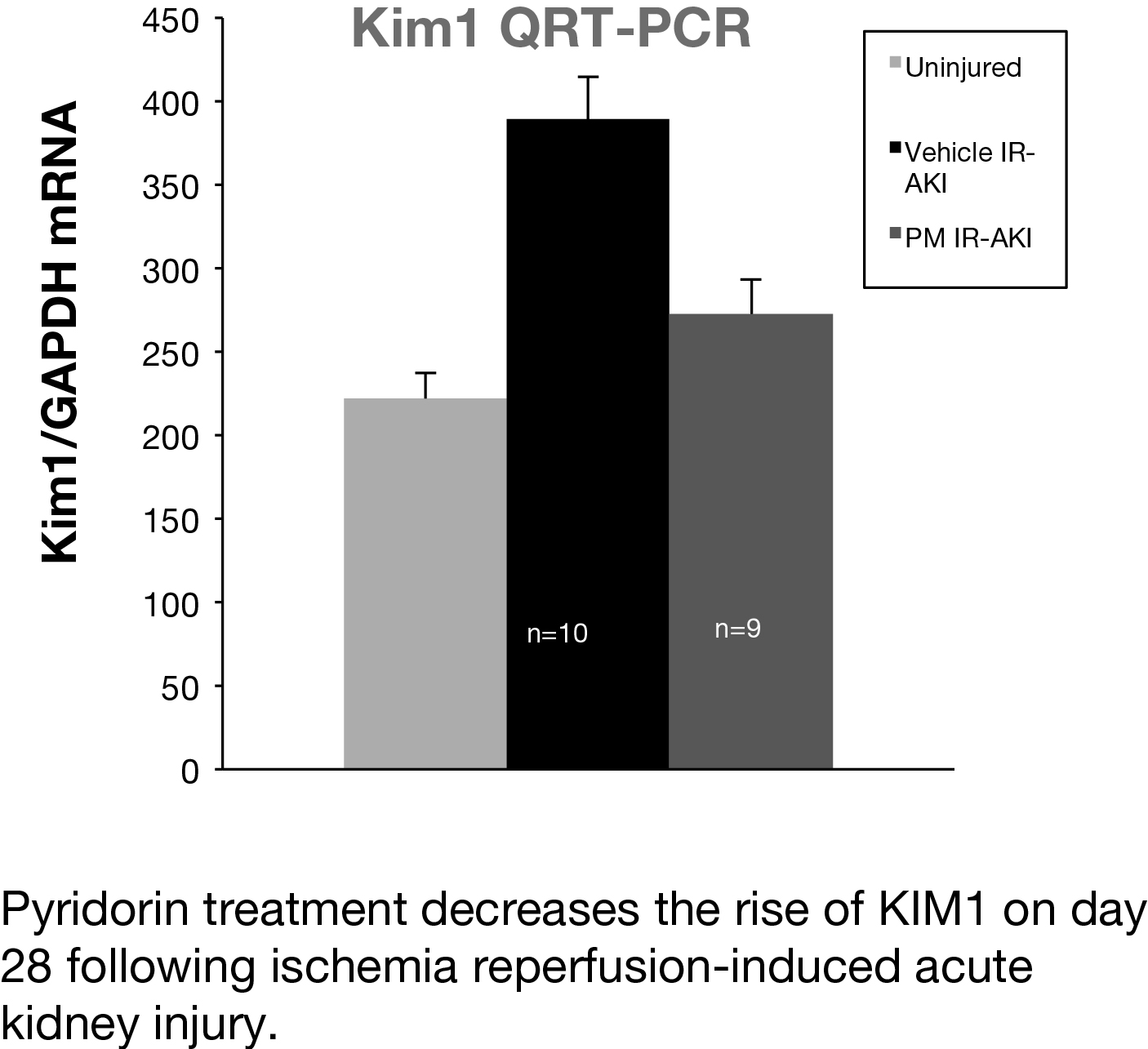
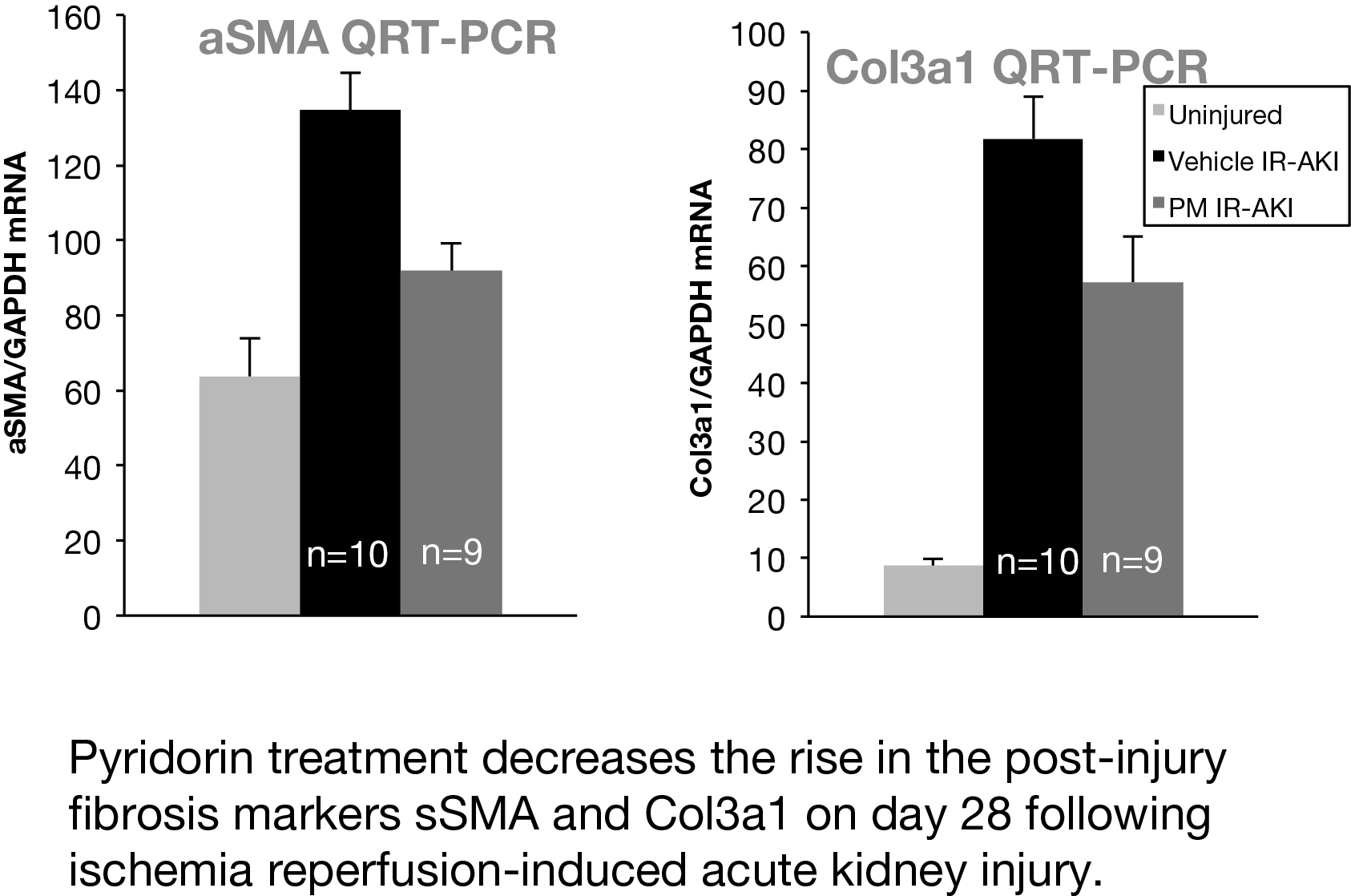
In a preclinical mouse model of AKI, we studied whether Pyridorin therapy could reduce injury and prevent long-term fibrosis following ischemia-reperfusion acute kidney injury (IR-AKI). Two mouse IR-AKI models were studied: moderate ischemia-reperfusion AKI (unilateral ischemia time 26 min and simultaneous contralateral nephrectomy) and severe ischemia-reperfusion AKI (unilateral ischemia time 31 min and delayed contralateral nephrectomy on day eight). Pyridorin was provided to mice in drinking water at 500 mg/kg BW/day starting 72 hours prior to injury and continued until sacrificed, except for mice with moderate IR-AKI where Pyridorin was administered one day after injury induction. Pyridorin was given to mice via gavage feeding twice a day at 200 mg/kg for 72 hours after AKI induction to ensure proper post-operative dosing. Renal function was assessed by serum creatinine, renal tubular injury with urinary Kim1 (days one and three), and post-injury fibrosis with qRT-PCR for renal fibrosis markers (aSMA, Col1a1, Col3a1) on day 28 after injury. Pyridorin ameliorated the increase in serum creatinine on days nine and 14 after injury, in urinary Kim1 expression on days one and three, and in post-injury fibrosis markers on day 28 after severe I/R-AKI. Pyridorin had no effect on serum creatinine (day three, five and seven) in moderate I/R-AKI when treatment was delayed 24 hours after injury. Pyridorin ameliorated injury and reduces post-injury fibrosis in severe IR-AKI when administered three days before injury, but had no effect on functional recovery or renal injury when administered 24 hours after moderate injury. These data suggest that pre-treatment with Pyridorin may ameliorate injury and prevent progression to chronic kidney disease in patients with AKI.
9
Preclinical Safety Summary
Pyridorin was studied in acute and chronic rat, rabbit and dog studies for up to one year. Acute and chronic toxicology studies were conducted by Quintiles Preclinical Services. Developmental & reproductive toxicology studies were conducted by Charles River Laboratories Inc. All of these studies were sponsored by BioStratum, Inc. There were no observable side effects seen at blood levels as high as 100x over therapeutic blood levels in humans. In a full battery of genotoxicity tests, no mutagenicity or clastogenicity was observed. These studies were conducted by Bioreliance Labs, Quintiles Toxicology/Pathology Services, and Sequani Ltd and sponsored by BioStratum, Inc. Human hepatic cytochrome P450 enzymes are involved in the metabolism and elimination of many widely used drugs. Any induction or inhibition of these enzymes can potentially lead to drug‑drug interactions. In human hepatic cell assays, Pyridorin had no effect on cytochrome P450 enzymes.
10
Thus, the potential for Pyridorin to interact with the metabolism of other drugs in‑vivo is unlikely. The P450 enzyme studies were conducted by RTI International and sponsored by BioStratum, Inc.
Clinical Safety Summary
An IND was submitted for Pyridorin by BioStratum, Inc. on July 30, 1999. The sponsorship of the IND was transferred to NephroGenex on July 10, 2007.
The safety, tolerability, and pharmacokinetics of Pyridorin has been investigated in five Phase 1 studies conducted in healthy volunteers. A summary of these studies is provided in the table below:
Protocol # | 440‑01 (PO) | 440‑01 (IV) | 440‑02 | PYR‑103 | PYR‑110 |
Conducted | Sep 99 ‑ Nov 99 | Sep 99 ‑ Nov 99 | Nov 99 ‑ Dec 99 | Mar 2001 | May 14, 2014 |
CRO/Sponsor | MDS Harris/BioStratum | MDS Harris/BioStratum | MDS Harris/BioStratum | PPD Development/BioStratum | Parexel/Nephrogenex |
Location(s) | Lincoln, NE | Lincoln, NE | N. Ireland | Morrisville, NC | Baltimore, MD |
Active/Placebo | 16/8 | 4/2 | 18/6 | 6/0 | N=43 randomized/treated |
Type of Subject M/F | Healthy 24/0 | Healthy 6/0 | Healthy 24/0 | Healthy 6/0 | Healthy 23/20 |
Age range | 19 ‑ 41 yrs | 19 ‑ 41 yrs | 18 ‑ 45 yrs | 19 ‑ 50 yrs | 20-55 yrs |
Study Design | Ascending Single dose Randomized Double Blind Placebo control | Single dose Randomized Double Blind | Ascending Multiple dose Randomized Double Blind Placebo control | Single dose High fat meal vs fasted 2‑way crossover | Single dose 4-Period, randomized, Cross-over Study, Partially blind (blinded for Pyridorin and Placebo, open label for moxifloxacin [control] |
Route of administration | Oral | I.V. | Oral | Oral | Oral |
Dose | 3 mg/kg 10 mg/kg 30 mg/kg 50 mg/kg | 10 mg/kg | 5mg/kg BID 15 mg/kg BID 25 mg/kg BID | 500 mg | 300 mg Pyridorin; 1200 mg Pyridorin; 400 mg Control; Placebo |
Duration | Single dose | Single dose | 7 days | Single dose | 4 clinic confinement periods, each 2 days, separated by 5-7 day washout period |
Results | No safety signal | No safety signal | No safety signal | No safety signal | No safety signal |
In all five of these studies, Pyridorin was well tolerated with no drug‑ related toxicity observed in any research subject. Based on its benign profile in healthy volunteers (studies: 440-01 (PO), 440-01 (IV), 440-020 and PYR-103), the decision was made by BioStratum to advance Pyridorin into Phase 2 testing in patients with diabetic nephropathy.
In December 2014, we completed a QT/QTc (TQT) cardiac safety study (PYR-110) on Pyridorin. A TQT study assesses a drug’s risk of QT prolongation and its proarrhythmic potential, and is a standard component of all clinical development programs for new molecular entities. The QT/QTc interval is a measure of the time between the start of the Q wave and the end of the T wave in the heart’s electrical cycle. In general, the QT interval represents electrical depolarization and repolarization of the left and right ventricles. A lengthened QT interval is a biomarker for ventricular tachyarrhythmias and a risk factor for sudden death. Fridericia’s and Bazett’s formulae are two different correction methods commonly used to correct for heart rate differences when calculating the QT interval. Pyridorin showed no effect on the QT/QTc interval at the expected therapeutic dose of 300 mg and at a higher supratherapeutic dose of 1200 mg. In all previously conducted Phase 1 and Phase 2 studies, Pyridorin has shown no effect on the QT/QTc interval.
11
The safety, tolerability, and pharmacokinetics of Pyridorin was investigated by BioStratum in a Phase 2 study conducted in patients with Type 1 diabetic nephropathy. In addition, the safety, tolerability and biological activity of Pyridorin was investigated in another Phase 2 study conducted in Type 2 diabetic patients with microalbuminuria (ACR≤ 300 mg/g). This study was conducted in Japan under the sponsorship and management of Kowa Company Ltd.
A summary of these two studies is provided in the table below:
Protocol # | PYR‑202 | K‑163‑04 | |
Conducted | Nov 2000 ‑ Mar 2001 | 2005 ‑ 2006 | |
CRO/Sponsor | PPD Development/BioStratum | Kowa | |
Location(s) | USA (5 sites) | Japan | |
Active/Placebo | 9/3 | 68/67 | |
Type of Subject M/F | Type 1 Diabetic nephropathy 8/4 | Type 2 Diabetes w/microalbuminurea 107/28 | |
Age range | 28 ‑ 54 yrs | 20 ‑ 70 yrs | |
Study Design | Multiple dose Randomized Escalating dose Double Blind Placebo control | Multiple dose Randomized Double Blind Placebo control | |
Route of administration | Oral | Oral | |
Dose | 50 mg BID for 7 days then 250 mg BID for 7 days then 500 mg BID for 28 days | 300 mg BID | |
Duration | 6 weeks | 26 weeks | |
Results | No safety signal | No safety signal No effect on microalbuminuria | |
In both of these studies, Pyridorin was well tolerated with no drug‑related toxicity observed in any patients. Based on its benign profile in diabetic nephropathy patients, the decision was made by BioStratum to continue evaluation of the safety, tolerability and biological activity of Pyridorin in type 1 and type 2 diabetic nephropathy patients with macroalbuminuria (ACR >300 mg/g).
In two randomized, placebo‑controlled, Phase 2 studies of 24‑week treatment duration, patients with nephropathy due to either type 1 or type 2 diabetes showed no consistent across‑study differences between Pyridorin and placebo groups in the type or incidence of adverse event reporting or in vital signs, weight, blood pressure, electrocardiograms (ECGs), general chemistry, urinalysis, hematology or special laboratories (coagulation and thyroid function tests). In the first study, the adverse events defined as definitely, probably, or possibly related to the study drug as determined by the investigator, were reported in 26.2% and 33.3% Pyridorin and Placebo patients, respectively. In the second study, the adverse events defined as definitely, probably, or possibly related to the study drug as determined by the investigator, were reported in 35.1% and 44.4% Pyridorin and Placebo patients, respectively. The types of serious adverse events (SAEs) observed were quite varied and very similar to what is typically observed in diabetic nephropathy patients. Cardiac related events were the most common, followed by infections. While a numerical imbalance in SAE reporting was seen, the lack of a specific type of SAE reported in patients receiving Pyridorin, the similarity to the types of SAEs reported in other diabetic nephropathy studies, and the significant baseline medical conditions in these patients suggest that the SAEs were related to the underlying medical conditions, not an effect attributable to Pyridorin. In a retrospective ECG analysis using pooled data from the two 24‑week studies, there was no evidence for an effect of Pyridorin on the QT/QTc interval, either at the group level or at the individual patient level (using Fridericia’s and Bazett’s formulae).
In a 12‑month Phase 2 study treatment with Pyridorin, up to 300 mg twice daily (BID) was generally well tolerated. Most of the AEs were mild or moderate in severity and there was a slight increase in the incidence of diarrhea and constipation in the 300 mg BID group relative to placebo. The pattern and occurrence of AEs were consistent with the patient population being studied. The overall incidence of AEs and AEs deemed drug‑related was similar among the treatment groups. The types of serious adverse events (SAEs) observed were quite varied and very similar to what is observed in diabetic nephropathy patients. Cardiac related events were the most common, followed by infections. There were no meaningful differences in SAEs
12
between the placebo group and the Pyridorin group. The observed SAEs were attributed to underlying baseline medical conditions in these patients and not attributed to Pyridorin therapy.
Phase 2 Efficacy Results
PYR‑206
PYR‑206 was a Phase 2, multi‑center, placebo‑controlled, randomized, double‑blind study which evaluated the safety and tolerability of Pyridorin administered orally via 50 mg capsules BID for 24 weeks to patients with nephropathy due to type 1 or type 2 diabetes. This study was conducted by BioStratum Inc. which utilized the services of the contract research organization Pharmaceutical Product Development (PPD). The study was conducted from October 2001 to January 2003 in the United States.
Although PYR‑206 was designed as a safety and tolerability study, post‑hoc analyses were performed on various efficacy parameters, including serum creatinine (SCr), urinary creatinine clearance, and TGF‑β1. Creatinine is a breakdown product of creatine. Its level in serum reflects the efficiency of the kidney to remove waste products from the blood. Serum creatinine is the most commonly used indicator of renal function. The SCr change from baseline was analyzed for all patients and for the patient subgroups listed in Table 1 below using a repeated measures mixed model with baseline SCr as a fixed covariate.
Treatment with Pyridorin reduced the change in SCr concentration from baseline by 27% for all patients (65 Pyridorin and 63 placebo). While the treatment was not statistically significant in the Intent to Treat (ITT) patient population, which included all patients that received at least one dose of study drug, this effect was statistically significant for a subgroup of patients with type 2 diabetes and a starting baseline SCr ≥ 1.3 mg/dL (Table 1 and Figure 1).
Table 1: PYR‑206-Serum Creatinine Change from Baseline Analysis
Patient Population | Treatment Group | N | Baseline SCr(1) | SCr Change from Baseline(2) | Treatment Effect(3) | |
All Patients | Pyridorin | 65 | 1.27 ± 0.34 | 0.12 ± 0.40 | −27% | |
Placebo | 63 | 1.33 ± 0.38 | 0.16 ± 0.28 | |||
Type 2 Diabetes | Pyridorin | 40 | 1.28 ± 0.34 | 0.08 ± 0.29 | −53% | |
Placebo | 40 | 1.30 ± 0.36 | 0.17 ± 0.30 | |||
Baseline SCr ≥ 1.3 mg/dL | Pyridorin | 34 | 1.54 ± 0.21 | 0.13 ± 0.53 | −50% | |
Placebo | 30 | 1.65 ± 0.28 | 0.26 ± 0.33 | |||
Type 2, Baseline SCr ≥ 1.3 mg/dL | Pyridorin | 22 | 1.53 ± 0.20 | 0.06 ± 0.37 | −79%** | |
Placebo | 19 | 1.59 ± 0.73 | 0.29 ± 0.35 | |||
(1) | Mean ± SD in mg/dL |
(2) | Unadjusted mean within group change from baseline in mg/dL |
(3) | Difference relative to placebo in unadjusted mean change from baseline where a negative value indicates a lesser change from baseline in Pyridorin patients (i.e. reno‑protection) |
** | Statistically significant, p<0.01 |
13
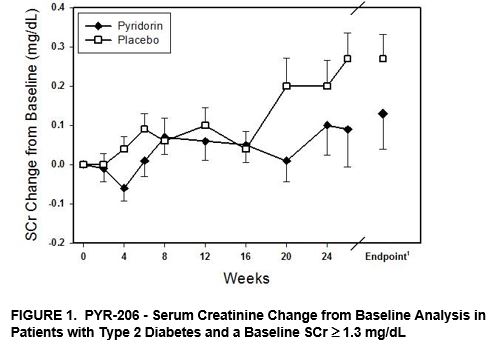
________________________
(1) Mean ± SEM; P= 0.0074 (Repeated measures mixed model analysis with baseline serum creatinine as a fixed covariate)
In the total patient population, Pyridorin also reduced the rate of rise in SCr levels by 23% relative to placebo. The rise in SCr was 0.161 mg/dL/yr and 0.210 mg/dL/yr in the Pyridorin (n=65) and placebo (n=63) groups, respectively. In the sub‑population of patients with more substantial renal impairment as evidenced by a baseline SCr level of ≥ 1.3 mg/dL, the ability of Pyridorin to preserve renal function was more pronounced with a 59% reduction in the rate of rise in SCr relative to placebo. In this sub‑population of patients, the rise in SCr was 0.183 mg/dL/yr and 0.445 mg/dL/yr in the Pyridorin (n=34) and placebo (n=31) groups, respectively. This result suggests Pyridorin therapy may be slowing the progression of kidney disease in diabetic patients with more substantial renal impairment exhibiting a larger increase in SCr over the treatment period. However, it is part of a post‑hoc analysis, and this effect may not be observed in a subsequent study.
Urinary creatinine clearance findings were consistent with the beneficial effects of Pyridorin on slowing the decline of renal function with an 18% reduction in the decline of creatinine clearance in the Pyridorin group relative to patients treated with placebo in the total patient population.
Urinary excretion of TGF‑β1, a factor implicated in the pathogenesis of chronic renal failure in diabetic nephropathy, was also assessed. The mean change from baseline to endpoint in urinary TGF‑β1 levels was −9.34 and 14.38 pg/mg creatinine in the Pyridorin and placebo patients respectively, with a relative change from baseline of −24.7% and 41.8%, respectively, in the total patient population. As in the case of the observed changes in SCr and urinary creatinine clearance, these results on urinary TGF‑β1 are part of a post‑hoc analysis, and they may not repeat in a subsequent clinical study.
PYR‑205/207
PYR‑205 and PYR‑207 were identical in design, with the exception of the patient entrance criteria for SCr (≤ 2.0 mg/dL and > 2.0 mg/dL but ≤ 3.5 mg/dL, respectively). The data were merged, as prespecified in the Statistical Analysis Plan, and analyzed as a single study. PYR‑205 and 207 were Phase 2, international, multi‑center, randomized, double‑blind, placebo‑controlled, escalating dose studies to evaluate the safety, tolerability, and biologic activity of Pyridorin given orally in a sequential fashion to patients with diabetic nephropathy due to type 1 or type 2 diabetes at:
• | 50 mg BID for two weeks, |
• | 100 mg BID for two weeks, and |
• | 250 mg BID for 20 weeks. |
14
This study was conducted by BioStratum Inc. which utilized the services of the contract research organizations Pharmaceutical Product Development (PPD), Cato Research, and PharmaNet. The study was conducted from July 2002 to September 2003 in the United States, Belgium, the United Kingdom, Canada and South Africa.
In PYR‑205/207, baseline renal function was more impaired than patients studied in PYR‑206. In PYR‑205/207, Pyridorin reduced the change from baseline SCr in either a statistically significant fashion or trending toward a significant p‑value close to 0.05 in all prospectively defined patient sub‑groups. The reno‑protective effect of Pyridorin as compared to placebo was seen to an equal degree across all patient groups with an approximate 70% reduction relative to placebo in the increase of baseline SCr (Table 2 and Figure 2).
Table 2: PYR‑205/207-Serum Creatinine Change from Baseline Analysis
Patient Population | Treatment Group | N | Baseline SCr(1) | SCr Change from Baseline(2) | Treatment Effect(3) | |
All Patients | Pyridorin | 57 | 1.75 ± 0.64 | 0.11 ± 0.26 | −68%* | |
Placebo | 27 | 1.96 ± 0.86 | 0.34 ± 0.92 | |||
Type 2 Diabetes | Pyridorin | 45 | 1.74 ± 0.67 | 0.12 ± 0.27 | −68%* | |
Placebo | 22 | 1.94 ± 0.92 | 0.38 ± 1.02 | |||
Baseline SCr ≥ 1.3 mg/dL | Pyridorin | 42 | 2.00 ± 0.55 | 0.12 ± 0.30 | −74%* | |
Placebo | 19 | 2.37 ± 0.67 | 0.47 ± 1.09 | |||
Type 2, Baseline SCr ≥ 1.3 mg/dL | Pyridorin | 33 | 2.00 ± 0.58 | 0.14 ± 0.31 | −75% | |
Placebo | 15 | 2.40 ± 0.73 | 0.55 ± 1.22 | |||
(1) | Mean ± SD in mg/dL |
(2) | Unadjusted mean within group change from baseline in mg/dL |
(3) | Difference relative to placebo in unadjusted mean change from baseline, where a negative value indicates a lesser change from baseline in Pyridorin patients (i.e., reno‑protection) |
(4) | Determined using repeated measures mixed model analysis with baseline SCr as a fixed covariate and treatment effect being the difference relative to placebo in change from baseline measured in mg/dL. |
* | Statistically significant, p<0.05 |
15
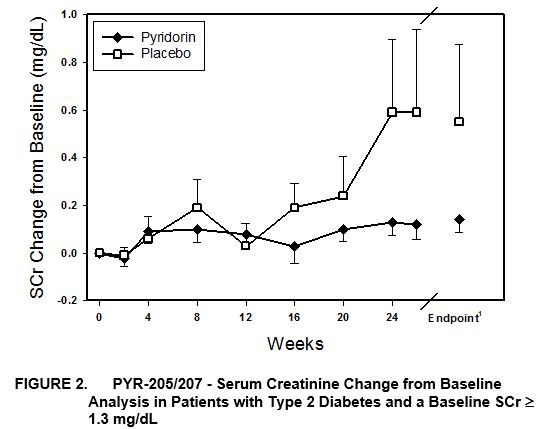
____________
(1) | Mean ± SEM; P= 0.058 (Repeated measures mixed model analysis with baseline serum creatinine as a fixed covariate) |
Relative to placebo, Pyridorin treatment also slowed the rate of SCr increase (slope analysis) by approximately 70% in all populations analyzed. The rise in SCr was 0.177 mg/dL/yr in Pyridorin group (n=57) and 0.629 mg/dL/yr in the placebo group (n=27), with a P value of 0.062.
No significant between‑group differences were observed in urinary albumin excretion. Short term effects on proteinuria are usually only seen with anti‑hypertensive drugs that improve renal hemodynamics. Pyridorin treatment did not affect blood pressure.
AGE measurements were performed in plasma of patients with more advanced renal disease (all PYR‑207 patients) using gas chromatography‑mass spectrometry. Whereas carboxymethyllysine (CML) and carboxyethyllysine (CEL) levels increased from baseline by 0.02 and 0.015 mmol/mol Lys, respectively, in the placebo group, CML and CEL levels were decreased from baseline by 0.04 and 0.01 mmol/mol Lys in the Pyridorin‑treated group. These data suggest that Pyridorin‑induced inhibition of AGE formation occurs concomitantly with the beneficial effects of Pyridorin on renal function, thus lending support to the hypothesis that Pyridorin exerts beneficial effects on renal function via an AGE‑dependent mechanism.
The mean change from baseline to endpoint in urinary TGF‑β1 levels was −9.7 pg/mg creatinine in Pyridorin patients and +14.2 pg/mg creatinine in placebo patients with a relative change from baseline of −13.1% and 55.7% in the Pyridorin and placebo groups, respectively. These relative differences in TGF‑β1 levels could represent one of the mechanisms by which Pyridorin could potentially slow the progressive decline in renal function.
PYR‑210
PYR‑210 was a randomized, double‑blind, placebo‑controlled study of Pyridorin at doses of 150 mg BID, 300 mg twice daily (BID) or placebo for 12 months. PYR‑210 was designed to further study the efficacy and safety of Pyridorin in patients with overt nephropathy due to type 2 diabetes and to identify the appropriate dose and patient population for Phase 3 pivotal trials.
We conducted the study and utilized the services of the contract research organization Medpace. The study was conducted from August 2008 to August 2010 in the United States, Australia and Israel.
16
The population selected had macroalbuminuria and impaired renal function. Although previous pivotal trials for diabetic nephropathy (notably, the IDNT study of the drug Irbesartan and the RENAAL study of the drug Losartan) have excluded patients with baseline SCr values ≥ 3.0 mg/dL, patients with higher bSCr values (up to 3.7 mg/dL) were included in the PYR‑210 study in order to evaluate Pyridorin safety in more advanced renal disease patients. Pre‑specified efficacy analyses according to starting baseline SCr levels were included in the statistical analysis plan. Patients were required to be on an established diabetic nephropathy standard of care (SOC) at screening. Specifically, patients must have received a renin‑aldosterone‑angiotensin‑system (RAAS) inhibitor (ACE‑I) or an ARB for at least 3 months prior to screening where the dose of the ACE‑I or the ARB was considered appropriate for that patient and had been stable for at least 2 months. Patients were also required to be on stable blood pressure medications (other than an ACE‑I or ARB) for 2 months prior to screening.
Patients not on an established, stable regimen of SOC were allowed to enter a screening phase (designated the “run‑in period”) during which ACE‑I/ARB or blood pressure dosing was initiated or adjusted to establish SOC. This was followed by a run‑in period of at least 2 months at these same doses before patients could be randomized. These patients were required to meet the other entry criteria at the screening visit. Because changes in ACE‑I/ARB or blood pressure medications are known to affect baseline SCr values, a pre‑specified analysis of patients on an established standard of care at screening, excluding run‑in patients, was included in the statistical analysis plan.
Eligible patients also had:
• | a history of overt diabetic nephropathy defined by a SCr measurement of 1.3 mg/dl to 3.3 mg/dl (women) or 1.5 mg/dl to 3.5 mg/dl (men), inclusive, and |
• | a 24‑hour urine collection Protein to Creatinine Ratio (PCR) > 1200 mg/g. |
The trial did not reach its primary endpoint on the intent to treat (ITT) population. In the overall patient population, Pyridorin did not demonstrate a significant treatment effect on the progressive increase in serum creatinine concentration that these patients experienced over one year. However, results from the pre‑specified analysis of patients on established SOC at screening showed a treatment effect of 45% for Pyridorin 300 mg BID and 21% for Pyridorin 150 mg BID treatment as compared to placebo treatment. This analysis included patients with a baseline SCr ≥ 3.0 mg/dL, which is higher than the baseline SCr used in the precedent IDNT and RENAAL clinical studies and represents patients who are not appropriate for a pivotal trial in diabetic nephropathy due to their baseline instability and advanced stage of renal insufficiency. Nonetheless, these patients were included in PYR‑210 for the purposes of a broad safety assessment. When patients with a baseline SCr < 3.0 mg/dL (the patient population studied in the RENAAL trial of Losartan) that were on established SOC at screening were analyzed, a statistically significant treatment effect of 57% for the Pyridorin 300 mg dose (p=0.0094) and 45% for the Pyridorin 150 mg dose (p=0.0414) was observed. The more robust treatment effect observed in the Pyridorin 300 mg BID group over the Pyridorin 150 mg BID group suggests a potential dose response in this patient population. This subgroup is the patient population that is being studied in the Phase 3 trial. Our subgroup analysis carries the inherent risk that the results may not be repeatable in a subsequent trial. It is possible that the treatment effect observed in this subgroup of PYR‑210 may not be repeated in the Phase 3 trials.
A summary of these results is shown in Table 3.
Table 3: Change in Serum Creatinine (mg/dl) From Baseline to Endpoint in Various Subgroups from PYR‑210
Patient Population | Treatment Group | N | Baseline SCr | SCr Change from Baseline | Treatment Effect | |
ITT Population | Pyridorin 300mg | 105 | 2.17 ± 0.57 | 0.36 ± 0.57 | N/A | |
Pyridorin 150mg | 99 | 2.22 ± 0.55 | 0.42 ± 0.72 | N/A | ||
Placebo | 103 | 2.20 ± 0.56 | 0.36 ± 0.70 | |||
Patients requiring a run‑in period(1) | Pyridorin 300mg | 36 | 2.32 ± 0.59 | 0.62 ± 0.75 | N/A | |
Pyridorin 150mg | 30 | 2.33 ± 0.56 | 0.73 ± 0.90 | N/A | ||
Placebo | 34 | 2.34 ± 0.67 | 0.31 ± 0.68 | |||
Patients on SOC @ screening in the RENAAL population (bSCr < 3.0)(1) (FDA approved patient population for Phase 3) | Pyridorin 300mg | 64 | 2.01 ± 0.49 | 0.18 ± 0.34 | −57%** | |
Pyridorin 150mg | 60 | 2.03 ± 0.40 | 0.23 ± 0.45 | −45%* | ||
Placebo | 63 | 2.04 ± 0.40 | 0.42 ± 0.70 | |||
17
(1) | A separate analysis of this group was pre‑specified in the statistical analysis plan. |
(2) | The patient population used in the RENAAL clinical trial of Losartan is considered to be the established population used for pivotal trials in diabetic nephropathy. |
* | Statistically significant, p<0.05 |
** | Statistically significant, p<0.01 |
Patients who were not on a stable regimen of SOC at screening, and required a run‑in period, are also shown in Table 3. These patients did not show a Pyridorin treatment effect. The analysis of the ITT patient population also showed no Pyridorin treatment effect. Since the patients on SOC did show a Pyridorin treatment effect, it is possible that inclusion of patients requiring a run‑in period confounded the analysis of the ITT population. It is generally accepted that the initiation or change in ACEi/ARB or blood pressure medication dosing in overt diabetic nephropathy patients with established renal insufficiency can result in an increase in SCr levels (or a decrease in GFR). A recently published post‑hoc analysis of the RENAAL study showed that patients assigned to Losartan (an ARB marketed by Merck & Co. Inc.) had a greater acute fall in eGFR during the first three months compared to patients assigned to placebo. A post‑hoc analysis of the database of the IDNT study indicates that this effect of a blood pressure medication can persist for up to 6 months. Since the run‑in period in PYR‑210 only required stable doses of ACEi/ARB or blood pressure medications for 2 months prior to randomization, it is likely that some run‑in patients had not reached a stable SCr baseline value prior to randomization. In addition, there was an increased number of post‑randomization blood pressure medication changes in the run‑in patients as compared to patients on established SOC at screening. For future Pyridorin studies, the FDA has agreed that all patients will need to be on stable SOC for at least 6 months prior to screening.
When the subgroup of patients that will be studied in the Phase 3 trials was examined (the RENAAL patient population with bSCr < 3.0 mg/dL on stable SOC @ screening) a dose dependent statistically significant treatment effect of 57% at 300 mg BID was observed.
In addition to the primary efficacy endpoint of change from baseline in SCr, the changes in serum cystatin C were also measured based on the demonstration of a 50% reduction in serum cystatin C by Pyridorin relative to placebo in all patients in Study PYR‑205/207. The cystatin C results in PYR‑210 followed similar trends to what was observed in the subgroups analyzed for SCr changes. A 26% treatment effect was observed in both treated arms (300 mg BID and 150 mg BID) of patients on SOC at screening in the RENAAL population (bSCr < 3.0 mg/dL).
Changes in urinary TGF‑β1 were measured based on the demonstration of a reduction in TGF‑β1 in PYR 206 and PYR 205/207. The mean change from baseline to endpoint in urinary TGF‑β1 levels was −5.8 pg/mg for the Pyridorin 300 mg BID group, +21.4 pg/mg for the Pyridorin 150 mg BID group and +264 pg/mg for the placebo group. Although a dose dependent trend of decreasing TGF‑β1 was observed in treated patients, the differences did not reach statistical significance.
Changes in 24 hour urinary protein creatinine ratio (PCR) were also measured. The mean change from baseline to endpoint in urinary PCR was −118 mg/g for the Pyridorin 300 mg BID group, +182 mg/g for the Pyridorin 150 mg BID group and +179 mg/g for the placebo group. Although there was evidence of a possible reduction in the 300 mg BID group relative to the placebo group, the difference was not statistically significant. The average baseline PCR was extremely high in this patient population (~3000 mg/gm) making the likelihood of observing significant effects within one year very low. It is possible that Pyridorin would further reduce urinary PCR with exposures longer than those in the PYR‑210 study. Shorter term effects on proteinuria are usually only seen with anti‑hypertensive drugs that improve renal hemodynamics. Pyridorin treatment did not affect blood pressure.
In summary, treatment with Pyridorin up to 300 mg BID was well tolerated. No safety signals were observed in this study. Treatment with Pyridorin for one year demonstrated a statistically significant treatment effect of 57% for the Pyridorin 300 mg dose (p=0.0094) and 45% for the Pyridorin 150 mg dose (p=0.0414) in the subgroup of patients with a baseline SCr < 3.0 that were on established SOC at screening. The more robust treatment effect observed in the Pyridorin 300 mg BID group over the Pyridorin 150 mg BID group indicates evidence for a dose response in this patient population. Pyridorin also demonstrated evidence of a reduction in serum cystatin C and urinary TGF‑β1.
The efficacy data from PYR‑210 was consistent with the previous Phase 2 trials PYR‑206 and PYR‑205/207. These results support the use of the 300 mg BID dose for pivotal studies, as all doses were well tolerated and there was a suggestion of a better treatment effect with the highest dose.
18
In 2013, we reached agreement with the FDA in a Special Protocol Assessment (SPA) on the patient population to be studied in the pivotal Phase 3 studies: type 2 diabetic patients with overt nephropathy and a bSCr < 3.0 mg/dL that are on an established and stable SOC regimen at screening. In this specific patient population, Pyridorin dosed at 300 mg BID demonstrated a 57% treatment effect in PYR‑210 in the endpoint of SCr change from baseline relative to placebo.
We also received supportive Scientific Advice from the European Medicines Agency (EMA) regarding the pivotal Phase 3 program with Pyridorin in diabetic nephropathy that has been accepted by the FDA under a SPA referenced above. The EMA indicated that the current Phase 3 program could be adequate to support a Marketing Authorization Application for full market approval in Europe.
Clinical Development Strategy
The clinical development path for a drug to treat diabetic nephropathy has traditionally been very long and associated with significant risk. In the past few years there have been four drug candidates for diabetic nephropathy that failed in Phase 3 clinical trials: Pimagedine, Sulonex, Avosantan and Bardoxalone. These drug candidates all looked promising in their respective Phase 2 studies, but all four failed in pivotal trials. A close examination of these clinical development programs reveals that in each case the Phase 3 studies were conducted in a different patient population using a different endpoint than was studied in their respective Phase 2 programs. This unusual circumstance arose because of the very challenging regulatory pathway that previously existed in this field. The long term endpoint that the FDA previously required in Phase 3 (time to SCr doubling or ESRD) made it nearly impossible to evaluate the drug against a similar endpoint in a Phase 2 trial. For example, the recruitment and patient follow‑up time for the IDNT study totaled 60 months or 5 years. Bearing in mind trial costs and patent lifetime, this is very long and expensive for a Phase 2 study. Companies chose to use Phase 2 trials to study surrogate endpoints. They also chose patient populations where a treatment effect on the surrogate endpoint would be the most pronounced. Since the FDA did not accept these surrogate endpoints and narrow patient populations for the Phase 3 program, the transition to a Phase 3 trial was quite risky. All four companies ended up evaluating a significant number of types of patients in Phase 3 that they had never evaluated before, using an endpoint for which they had relatively little data.
We took a different approach in our clinical development strategy for Pyridorin. Specifically, during the Phase 2 program, working closely with the FDA, we examined broader patient populations under different conditions of standard of care to identify those patients most appropriate for the Phase 3 program. The pre‑specified subgroup analyses of the Phase 2b study indicate that the appropriate diabetic nephropathy patient population to study in Phase 3 is patients on long term establish standard of care at screening with a baseline SCr >1.3 and < 3.0 mg/dL. In this patient population, Pyridorin therapy produced a greater than a 50% treatment effect that was statistically significant (P = 0.009) at the 300 mg bid dose. The Phase 2b study also indicated that patients that would not be appropriate to include in the Phase 3 pivotal study are those not on a stable regimen of standard of care at screening. These patients did not demonstrate a Pyridorin treatment effect and very likely did not reach a stable blood pressure and stable SCr baseline prior to the start of the study which would confound the treatment effect analysis.
We also used a SCr increase‑based endpoint that would correlate with a potentially approvable endpoint. Simultaneously, we provided the FDA with analyses from previously completed Phase 3 clinical studies in diabetic nephropathy that supported a new, lower SCr increase‑based endpoint. As a result, we potentially significantly reduced the cost of the Phase 3 trials and made our Phase 2b endpoint even closer to the Phase 3 endpoint.
As agreed to in the SPA referenced above, the Pyridorin Phase 3 study is being conducted in the specific patient population where Pyridorin has previously shown greater than a 50% treatment effect on a year‑1 SCr endpoint (PYR‑210).
Phase 3 Development Plan
Based on these clinical results and the SPA agreement with the FDA, the first of two Pyridorin Phase 3 diabetic nephropathy clinical trials (PYR-311) commenced in June 2014. We intend to commence the second of the Phase 3 trials (PYR‑312) after we receive the interim analysis on PYR-311, which we estimate will be in 2016, or earlier if we’re able to find a collaborator. These two clinical trials (PYR‑311 and PYR‑312), if successful, will serve as the basis for the product registration application.
PYR‑311 and PYR‑312 will be identical Phase 3 randomized, double‑ blind, placebo‑controlled, international multi‑center studies to evaluate the efficacy of Pyridorin 300 mg twice daily (BID) compared to placebo in reducing the rate of progression of renal disease due to type 2 diabetes. Each study will provide approximately 90% power to detect a 28% treatment effect. This progression rate will be estimated by the time to the composite endpoint consisting of the earliest event amongst:
19
• | A SCr increase of ≥ 50% from baseline that occurs during follow‑up; or |
• | End Stage Renal Disease. |
The FDA has agreed to the SCr increase of ≥ 50% from baseline endpoint as indicated in our SPA agreement with the FDA which covers the design of the Pyridorin Phase 3 program and the endpoint to be used for drug approval. This endpoint was previously validated by an FDA‑NKF (National Kidney Foundation) Workshop held in December of 2012 that included leading nephrology clinical investigators and extensive analyses of completed kidney disease clinical studies demonstrating a highly significant correlation between time to a 50% SCr increase and time to ESRD.
The key secondary objective of the studies is to determine the safety of Pyridorin compared to placebo, as assessed by adverse events, 12‑lead ECGs, vital signs, physical examination, clinical chemistries, glycosylated hemoglobin (HbA1c), and hematology.
Each study will enroll approximately 600 patients with a history of overt diabetic nephropathy defined by a SCr measurement of ≥ 1.3 mg/dL for female patients or ≥ 1.5 mg/dL for male patients, < 3.0 mg/dL for all patients, and a urine PCR ≥ 1200 mg/g at screening. Patients must be on stable standard of care (SOC) regimen which is defined as an ACE‑I or ARB at a constant dose for at least 26 weeks prior to randomization.
PYR‑311 will include one interim analysis that will be conducted approximately six months following the randomization of 600 patients in the study. At that time, an independent biostatistician will perform an analysis of its effect on the rate of SCr progression. If the independent biostatistician determines that Pyridorin is not safe or that it is futile to continue the trial because of lack of efficacy, the trial will be terminated. If the independent biostatistician determines Pyridorin is safe and it is not futile to continue the study, the study will be continued until the necessary number of events have accrued per the study design. An independent Data Safety Monitoring Board will assess the general safety of Pyridorin throughout the progression of the trial.
Acute Kidney Injury (AKI)
Pyridorin targets specific pathogenic oxidative chemistries that emerge in diabetes. These same pathogenic oxidative chemistries emerge with the onset of AKI and are believed to contribute to the severity of the AKI. An intravenous formulation of Pyridorin could provide significant benefit in this acute setting. Because of its benign safety profile, Pyridorin could also be used as a treatment for patients at increased risk of developing AKI.
We believe AKI constitutes a very significant market opportunity for Pyridorin. Since this would be an intravenous product used in an acute setting, it would not compete with an oral Pyridorin product used for the chronic treatment of diabetic nephropathy.
Acute kidney injury is characterized by a rapid reduction in kidney function resulting in a failure to maintain fluid, electrolyte and acid‑base homeostasis. It covers a wide spectrum of disease ranging from less severe forms of injury to more advanced injury when acute kidney failure may require renal replacement therapy (RRT). The incidence of AKI varies from 20% to 40% in critical care patients. In the United States, it is estimated that up to 7% of all patients who visit the hospital will experience AKI. Patients with uncomplicated AKI have a mortality rate of up to 10%. If RRT is required, the mortality rate rises to as high as 80%.
The most common causes of AKI include:
• | Sepsis |
• | Cardiovascular surgery |
• | Ischemic reperfusion injury |
• | Contrast dye induced AKI |
• | Chemotherapy induced AKI |
• | Trauma |
• | Serious Burns |
20
Severe AKI is characterized by a surge in pathogenic oxidative chemistries. These oxidative chemistries can lead to further damage to the kidneys and ultimately result in acute renal failure (ARF). Even if ARF does not occur, there is evidence that patients who experience AKI have a much higher incidence of subsequent chronic kidney disease.
Biomarkers have been identified that allow for earlier detection of AKI. One such biomarker is neutrophil gelatinase‑associated lipocalin (NGAL). Kidney injury molecule-1 (KIM-1) is another promising biomarker of AKI based on animal and early human studies. Early detection of AKI would allow therapeutic intervention with an agent such as Pyridorin that could inhibit these pathogenic oxidative chemistries and prevent further damage to the kidneys. Because of its benign safety profile, Pyridorin is an attractive candidate for early intervention (e.g. elevated KIM-1). Pyridorin may have application in, among other areas, the treatment of patients at increased risk of developing cardiac surgery associated AKI, and the treatment of patients receiving contrast dye or as treatment in patients among the other most common causes of AKI.
We will conduct additional preclinical studies to identify those indications where Pyridorin would be most effective. This will form the basis for our clinical development plan.
Commercialization
Given our stage of development, we have not yet established a commercial organization or distribution capabilities. Pyridorin, if approved, is intended to be prescribed to patients with diabetic nephropathy. These patients are normally under the care of a nephrologist, an endocrinologist, and/or a primary care physician (PCP). All of these specialties prescribe therapy for diabetic nephropathy, with the endocrinologist or the PCP typically treating patients in the earlier stage of the disease and the nephrologist typically treating patients in the later stages of the disease (overt diabetic nephropathy). Our current plan is to evaluate a possible partnership to commercialize Pyridorin, if approved, for the treatment of diabetic nephropathy in patients with type 2 diabetes in the United States and Europe. We may also build our own commercial infrastructure or utilize contract reimbursement specialists, sales people and medical education specialists, and take other steps to establish the necessary commercial infrastructure at such time as we believe that Pyridorin is approaching marketing approval. Outside of the United States and Europe, subject to obtaining necessary marketing approvals, we will likely seek to commercialize Pyridorin through distribution or other collaboration arrangements for kidney disease in patients with type 2 diabetes. As a result of our ongoing clinical work, we have been engaged in dialogue with specialists who treat patients with kidney disease. We believe that these activities have provided us with a growing knowledge of the physicians we plan to target for commercial launch of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, subject to marketing approval in the United States and Europe.
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Although we believe that Pyridorin is one of the few drug candidates in advanced clinical trials for diabetic kidney disease that targets an underlying cause of the disease, our competitors may be able to develop other compounds or drugs that are able to achieve similar or better results. Our potential competitors include major multinational pharmaceutical companies, established biotechnology
21
companies, specialty pharmaceutical companies and universities and other research institutions. Smaller or early‑stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.
Diabetic Nephropathy
As of 2010, the Center for Disease Control and U.S. Census data estimate the prevalence of diabetic nephropathy across all stages of disease to be approximately 6 million patients in the United States and this population is expected to grow. According to a 2010 study commissioned by us, approximately 2.8 million diabetic patients have overt nephropathy, approximately 3.5 million patients have early stage diabetic nephropathy and approximately 3.6 million patients are at high risk of progressing to diabetic nephropathy.
While the market opportunity for drugs to treat diabetic nephropathy is large and growing, the availability of drugs to treat this condition is very limited. There are two classes of drugs currently approved to slow the progression of diabetic nephropathy: ACE‑Inhibitors and ARBs. These agents target the renin‑angiotensin system. Approved initially as anti‑hypertension drugs, these agents are now considered standard of care for patients with diabetic nephropathy. Pyridorin is intended to be given in conjunction with these therapies; therefore, actual competition will not come from drugs targeting the renin‑angiotensin system. Instead, it may come from companies seeking to treat diabetic nephropathy through some other mechanism of action. The table below summarizes the competitive landscape.
COMPANIES WITH CLINICAL PROGRAMS IN DIABETIC NEPHROPATHY
Company | Agent | Phase | Program Status | |
AbbVie | Endothelin receptor antagonist | 3 | Active | |
Janssen Pharmaceuticals | INVOKANA SGLT2 Inhibitor | 3 | Active | |
Pfizer | Chemokine CCR2/5 Receptor Antagonist | 2 | Completed | |
Phosphodiesterase type 5 inhibitor | 2 | Completed | ||
Eli Lilly | Transforming Growth Factor B-Monoclonal Antibody (IV) | 2 | Terminated | |
MR Antagonist | 2 | Active | ||
Bayer Healthcare | Mineralcorticoid Receptor Antagonist | 2 | Completed | |
BMS | BMS-813160 CCCR2 antagonist | 2 | Active | |
Eli Lilly & Incyte Corp. | Janus Kinase 1 Inhibitor, TKI | 2 | Completed | |
Gilead Sciences | GS-4997 Mitogen-activated Protein Kinase Inhibitor | 2 | Active | |
La Jolla Pharmaceuticals Co. | GCS-100 (injection) Angiogenesis Inhibitor, Apoptosis Stimulant | 2 | Planned | |
Concert Pharmaceuticals | CTP-499 Unidentified pharmacological activity | 2 | Completed | |
ChemoCentryx | Chemokine CCR2 Receptor Antagonist | 2 | Completed | |
Genkyotex Innovation SAS | NOX 1 Inhibitor | 2 | Active | |
Vascular Pharmaceuticals, Inc. | VPI-2690B injection targets Insulin-like growth factor 1 | 2 | Active | |
Sanwa Kagaku Kenkyusho Co., Ltd | Topiroxostat Xanthine oxidase inhibitor | 2 | Planned | |
Yuhan Corporation | 5-hydroxytryptamine 2A Receptor Antagonist | 2 | Completed | |
Daiichi Sankyo, Inc. | Mineralcorticoid-receptor antagonist | 2 | Active | |
Kyowa Hakko Kirin Co. Ltd | RTA-402 Bardoxolone Methy Activator of Nrf2 | 2 | Active | |
Korea Otsuka Pharmaceutical Co., Ltd | Probucol Cholesterol inhibitor reducing agent | 2 | Completed | |
Yuhan Corporation | Anplag (Sarpogrelate) 5-hydroxytryptamine 2A receptor antagonist | 2 | Completed | |
Dong Wha Pharmaceutical Co., Ltd | DW1029 Botantical extract | 2 | Completed | |
Competition for Phase 3 Recruitment
We believe AbbVie’s Phase 3 trial is actively recruiting over 6,200 patients worldwide and Janssen’s Phase 3 trial is actively recruiting over 3,700 patients worldwide. While the eligible patient population is not identical, it is similar enough to
22
impact enrollment goals set by our Pyridorin Phase 3 program. Accordingly, we have increased our planned spending on investigator and CRO costs to minimize any enrollment impact.
Acute Kidney Injury (AKI)
In the United States, the incidence of AKI varies from 20% to 40% in critical care patients. It is estimated that up to 7% of all patients who visit the hospital will experience AKI. Patients with uncomplicated AKI have a mortality rate of up to 10%. If RRT is required, the mortality rate rises to as high as 80%.
The current treatment for AKI is mainly supportive in nature; no therapeutic modalities to date have shown efficacy in treating the condition.
We believe the market opportunity for effective treatments for AKI is large. There are a small number of industry drug trials in later stage development. Companies with an active AKI agent or program in Phase 2 or beyond include AM-Pharma, Baxter, Complexa, Kringle Pharma, NephroGuard, Stealth, Tenax Therapeutics, and Thrasos Innovation.
Sales of Pyridoxamine as a Dietary Supplement
Following the publication of the initial Phase 2 results that evaluated pyridoxamine therapy in diabetic nephropathy patients, a number of dietary supplement companies began selling pyridoxamine over the internet.
In January 2009, in response to a citizen petition filed on behalf of a pharmaceutical company, the FDA determined that products containing pyridoxamine are not dietary supplements and may not be marketed as such. A significant decline in product availability occurred after the determination.
In the case of Pyridorin, we believe that illegal sales of pyridoxamine will have little if any effect on Pyridorin sales for the following reasons:
1. The FDA has a track record of enforcing the regulations against dietary supplement companies that attempt to sell the active ingredient of an FDA approved drug. Since pyridoxamine would be approved for diabetic patients with substantial kidney disease, it is likely the FDA will continue this policy.
2. NephroGenex has issued patents covering pyridoxamine as an agent to treat diabetic nephropathy patients and other diabetic complications, and also as an agent to inhibit pathogenic oxidative chemistries that emerge in diabetes. This intellectual property makes it difficult to effectively market pyridoxamine as a dietary supplement without infringing on these issued patents.
3. A significant investment in pyridoxamine production capacity would be required by the dietary supplement industry just to impact a small percentage of Pyridorin drug sales. Furthermore, a non‑oxidative method of pyridoxamine production would have to be developed, since the commonly used oxidative method cannot be scaled up due to safety and environmental concerns. We have already developed and patented a non‑oxidative method of pyridoxamine production (used in the Phase 2b study), thus making the task of developing a new, non‑infringing, non‑oxidative method of pyridoxamine production that much more difficult and expensive.
Food and dietary supplements in Europe are regulated by Directive 2002/46/EC, European Commission, Health and Consumers Directorate‑General. Those approved are listed in Annex I and II of this directive. Pyridoxamine is not included on either list, and therefore the sale of pyridoxamine in foods and supplements in Europe is not permitted. We have kept the European Commission Health and Consumers Protection Directorate‑General up to date on the clinical status of Pyridorin, and plans for Phase 3 trials.
This office has indicated to NephroGenex as recently as April of 2014, that no applications for pyridoxamine have been received and that any new product intended for preventing, curing or treating diseases, would fall under the scope of medicinal products and not dietary supplements products.
Intellectual Property
The proprietary nature of, and protection for, our product candidates and our discovery programs, processes and know‑how are important to our business. We have sought patent protection in the United States and internationally for Pyridorin and our discovery programs, and any other inventions to which we have rights, where available and when appropriate. Our policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third
23
parties, and to protect the technology, inventions and improvements that are commercially important to the development of our business. We also rely on trade secrets that may be important to the development of our business. However, we do not have composition of matter patent protection for Pyridorin which may result in competitors being able to offer and sell products including pyridoxamine so long as these competitors do not infringe any other patents that we or third parties hold, including synthesis and method of use patents.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third‑party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our product candidates, discovery programs and processes. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors-Risks Relating to Our Intellectual Property.”
Patents and Proprietary Rights Covering Our Drug Candidates
We strive to protect our product candidates and exclusivity rights, as well as both maintain and fortify our position in the field of kidney disease therapeutics. We believe our intellectual property portfolio consists of early and broad filings in the area. We have focused on patents and patent applications covering, where possible, use of our products in disease treatment. We have sought and continue to seek the strongest possible intellectual property protection available to us in order to prevent others from directly competing with us, as well as to exclude competition around our products where possible, their manufacture, and methods for use of the products in disease treatment. Our intellectual property portfolio contains 28 issued patents and at least four pending patent applications in the United States and worldwide of both in‑licensed and NephroGenex‑owned inventions. This portfolio includes patents and proprietary rights around:
(i) Methods for using Pyridorin (pyridoxamine dihydrochoride) as a therapeutic agent to treat diabetic nephropathy;
(ii) Methods for manufacture of Pyridorin;
(iii) Methods for using Pyridorin as a therapeutic agent to treat a variety of other kidney diseases and other disorders; and
(iv) Pyridorin analog drug candidates, and their use for treating kidney disease.
We own patents covering methods for using Pyridorin to treat diabetic nephropathy in patients with type 2 diabetes and elevated levels of SCr, and thus closely track the anticipated drug label for an approved Pyridorin drug. These patents consist of an issued U.S. patent (U.S. Patent 8067444) and corresponding issued patents in Canada and Europe, which will expire in 2024 absent any extension to the patent term. As discussed in more detail herein, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products.
We also have worldwide, exclusive licenses from Kansas University Medical Center, the University of South Carolina, and Vanderbilt University to patents covering methods for using Pyridorin to treat a variety of other disorders. These patents include patents for treating urinary stone disease (US Patent 6521645), proteinuria (U.S. Patent 6472400), retinopathy (U.S. Patent 6750209), neurodegenerative disease (U.S. Patent 6750209), diabetic neuropathy (U.S. Patent 7030146), oxidative protein modification (U.S. Patent No. 6730686), oxidative stress‑related disorders (U.S. Patent No. 6716858), diabetes-associated hypercholesterolemia (U.S. Patent No. 6740668), and some corresponding foreign patents. The term of these patents will expire at various times, but all would expire by 2021. These patents further include a pending application in the United States for treating symptoms of kidney disorders; if a patent is granted on this application, it would expire in 2026.
We also own patents covering Methods for manufacture of Pyridorin; these patents consist of two issued U.S. patents (U.S. Patents 7214799 and 8431712), which will expire in 2025.
We also have worldwide, exclusive licenses from Kansas University Medical Center, the University of South Carolina, and Vanderbilt University to patents covering methods for using Pyridorin to treat a variety of other disorders. These patents include patents for treating urinary stone disease (US Patent 6521645), proteinuria (U.S. Patent 6472400), retinopathy (U.S. Patent 6750209), neuropathy (U.S. Patents 6750209 and 7030146), oxidative protein modification (U.S. Patent No. 6730686),
24
oxidative stress‑related disorders (U.S. Patent No. 6716858), hypercholesterolemia (U.S. Patent No. 6740668), and some corresponding foreign patents. The term of these patents will expire at various times, but all would expire by 2021. These patents further include pending applications in the United States for treating symptoms of kidney disorders, and inflammatory disorders. If granted, patents issuing from these patent applications would expire at different times, but all would expire by 2032.
We own pending patent applications in the United States and Europe covering Pyridorin analogs, and uses of such analogs as therapeutics to treat a variety of disorders, including kidney disorders such as nephropathy. Patent protection, to the extent it issues, would be expected to extend to 2027.
Intellectual Property Strategy
We continually assess our intellectual property strategy in order to fortify our position in our market space. To that end, we are prepared to file additional patent applications in any of the above families should our intellectual property strategy require such filings and/or where we seek to adapt to competition or seize business opportunities. Further, we are prepared to file patent applications relating to the other products in our pipeline soon after the experimental data necessary for a strong application become available and our cost‑benefit analyses justify filing such applications. In addition to filing and prosecuting patent applications in the United States, we typically file counterpart patent applications in Europe and additional countries where we think such foreign filing is likely to be beneficial.
We do not know if patents will be issued for all of the patent applications in our portfolio. Furthermore, for patent claims now issued and for claims to be issued in the future, we do not know if such claims will provide significant proprietary protection to our drug candidates and proprietary technologies or if they will be challenged, circumvented, or invalidated. Our success will in part depend on our ability to obtain and maintain patents protecting our drug candidates, technologies and inventions, to operate without infringing the proprietary rights of third parties, and to enforce and defend our patents and ensure others do not infringe on our proprietary rights.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing for a non‑ provisional patent application to which the patent claims priority. In the United States, a patent’s term may be shortened if a patent is terminally disclaimed over another patent, and a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent.
The patent term of a patent that covers an FDA‑approved drug or biologic may also be eligible for patent term extension, as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch‑Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug or biologic is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug or biologic may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug or biologic. In the future, if and when our pharmaceutical products receive FDA approval we expect to apply for patent term extensions on patents covering those products. We anticipate that some of our issued patents may be eligible for patent term extensions. For more information regarding U.S. patent laws, see “Business-Government Regulation.”
In addition to the patent term extension rights described above, some of our product candidates that receive FDA approval may also be eligible for market exclusivity protection under the Federal Food, Drug and Cosmetic Act or the Biologics Price Competition and Innovation Act of 2009. For more information regarding market exclusivity laws, see “Business-Government Regulation.”
Many pharmaceutical companies, biotechnology companies and academic institutions are competing with us in the field of diabetic nephropathy and filing patent applications potentially relevant to our business. In order to contend with the inevitable possibility of third party intellectual property conflicts, from time to time, we review and assess the third‑party intellectual property landscape for competitive and other developments that may inform or impact our intellectual property development and commercialization strategies. From time to time, we may find it necessary or prudent to obtain licenses from third party intellectual property holders. Where licenses are readily available at reasonable cost, such licenses are considered a normal cost of doing business. In other instances, however, where a third party holds relevant intellectual property and is a direct competitor, a license might not be available on commercially reasonable terms or available at all. Accordingly, we attempt to manage the risk that such third party intellectual property may pose by conducting, among other measures, freedom‑to‑operate studies to guide our early‑stage research away from areas where we are likely to encounter obstacles in the
25
form of third party intellectual property. As our programs advance, we continue to monitor the intellectual property landscape in an effort to assess the advisability of licensing third party intellectual property or taking other appropriate steps to address such freedom‑to‑operate or development issues in the manner we deem in the best interests of the Company.
With respect to third party intellectual property, it is impossible to establish with certainty that our product candidates will be free of claims by third party intellectual property holders or whether we will require licenses from such third parties. Even with modern databases and on‑line search engines, literature searches are imperfect and may fail to identify relevant patents and published applications. Even when a third party patent is identified, we may conclude upon a thorough analysis, that we do not infringe the patent or that the patent is invalid. If the third party patent owner disagrees with our conclusion and we continue with the business activity in question, we might have patent litigation thrust upon us. Alternatively, we might decide to initiate litigation in an attempt to have a court declare the third party patent invalid or not infringed by our activity. In either scenario, patent litigation typically is costly and time‑consuming, and the outcome is uncertain. The outcome of patent litigation is subject to uncertainties that cannot be quantified in advance, for example, the credibility of expert witnesses who may disagree on technical interpretation of scientific data. Ultimately, in the case of an adverse outcome in litigation, we could be prevented from commercializing a product or using certain aspects of our discovery platform as a result of patent infringement claims asserted against us. This could have a material adverse effect on our business.
To protect our competitive position, it may be necessary to enforce our patent rights through litigation against infringing third parties. Litigation to enforce our own patent rights is subject to the same uncertainties discussed above. In addition, however, litigation involving our patents carries the risk that one or more of our patents will be held invalid (in whole or in part, on a claim‑by‑claim basis) or held unenforceable. Such an adverse court ruling could allow third parties to commercialize our products, and then compete directly with us, without payment to us.
Trade Secrets
In addition to patents, we rely on trade secrets and know‑how to develop and maintain our competitive position. Trade secrets and know‑how can be difficult to protect. We seek to protect our proprietary processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information. We also seek to preserve the integrity and confidentiality of our data, trade secrets and know‑how by maintaining physical security of our premises and physical and electronic security of our information technology systems.
License Agreements
26
Licensing Payments
Set forth below is a summary chart outlining various potential license payments due under our license agreements referenced below:
Indication | Diabetic Nephropathy Phase III | Acute Kidney Injury, Chemotherapy Protection, or Radiation Damage Pre‑clinical AKI | Diabetic Neuropathy or Hyperlipedemia Not in current pipeline | |
Institution | Kansas University Medical Center | Vanderbilt University | South Carolina Research Foundation | |
FDA approval of SPA for designated indication | $25,000 | — | — | |
Submission of IND | — | $75,000 | — | |
Commencement of first Phase 1 | — | $100,000 | — | |
Commencement of first Phase 2 | — | $150,000 | $325,000 | |
Commencement of first Phase 3 | — | $250,000 | $500,000 | |
Submit NDA or foreign equivalent | — | — | $750,000 | |
FDA approval of NDA | $200,000 | $500,000 ($250,000 credited against royalty) | $2,000,000 | |
First commercial sale | — | — | $2,500,000 | |
Royalty on net sales | — | 5% (minus $250,000 credit) | — | |
Licensing fee | — | — | $120,000 per year with $30,000 payable quarterly (credited against milestone payments & upfront sublicense fees) | |
Upon execution of a sublicense | — | 25% of any sublicense fees or milestone payments | $35,000 plus 25% of upfront sublicense fees | |
License Agreements
Kansas University Medical Center (KUMC) Exclusive License Agreement
In May 2007, we entered into an amended license agreement with KUMC. Under the agreement, KUMC grants us an exclusive, royalty‑free, worldwide license, with a right to grant sublicenses, to make, have made, use, distribute, sell, have sold, have distributed, offer to sell, market, import, have imported or otherwise dispose of licensed products for diagnostic testing and palliative, prophylactic and therapeutic treatments which incorporate the use of the technology relating to the licensed patents and improvements. The patents licensed from KUMC include claims reciting methods for using Pyridorin to: (a) treat diabetic nephropathy (expires by 2016 absent any extension); (b) treat proteinuria or albuminuria associated with elevated blood sugar levels (expires by 2016 absent any extension); (c) treat retinopathy or neurodegenerative disease (expires by 2016 absent any extension); (d) inhibiting oxidative modification of proteins or treating atherosclerosis in a non‑hyperglycemic mammal (expires by 2016 in the United States and 2019 outside the United States absent any extension); (e) treat a condition associated with oxidative stress in a hyperglycemic mammal (expires by 2016 absent any extension); (f) treat diabetes‑associated increases in hypercholesterolemia or hypertriglyceridemia in a diabetic mammal; (expires by 2016 in the United States and 2019 outside the United States absent any extension); (g) treat diabetic neuropathy (expires by 2016 in the United States and 2019 outside the United States absent any extension); (h) decrease dialysis‑related amyloidosis or dialysis‑related increases in permeability of the peritoneal membrane in a dialysis patient (expires by 2016 absent any extension); and (i) urinary stone disease (expires by 2021 absent any extension).
The patents licensed from KUMC also include patents with claims reciting novel Pyridorin analogues, and methods for using them to treat AGE‑related pathologies, diabetic nephropathy, proteinuria, albuminuria; diabetes‑associated increases in hypercholesterolemia or hypertriglyceridemia in a diabetic mammal; and for inhibiting oxidative modification of proteins or treating atherosclerosis in a non‑hyperglycemic mammal (expire by 2016 in the United States and 2019 outside the United States absent any extension). The granted license is subject to certain rights and license granted to the United States and to foreign governments pursuant to U.S. government patent laws and regulations.
27
We must pay KUMC milestone payments related to milestones met in the FDA regulatory approval process. These milestone payments include $25,000 upon receipt of FDA approval of our SPA for our first licensed product and $200,000 upon receipt of FDA approval of our submitted NDA for our first licensed product in respect to the first primary indication. We must exercise commercially reasonable efforts to seek regulatory approval for the marketing of a licensed product for at least one primary indication, effect the introduction of a licensed product for at least one primary indication into the commercial market and to maximize these sales. Primary indications are the diagnosis, treatment, palliation or prophylaxis of diabetic nephropathy, diabetic retinopathy and diabetic neuropathy.
The agreement survives until expiration of the last to expire licensed patent, or in November 2018, whichever occurs last. We may terminate the license for any reason upon 90 days written notice. If either we or KUMC breach a material obligation under the agreement the non‑breaching party may terminate the agreement upon an additional written notice.
The South Carolina Research Foundation (SCRF) Exclusive License Agreement
In April 2012, we entered into an amended license agreement with SCRF. Under the agreement, SCRF grants us an exclusive, royalty‑free, worldwide license, under certain patent rights and related technology (including know‑how) with a right to sub‑license to utilize the patent rights and the technology during the term of the agreement and to practice under the patent rights to make, have made, use, sell, have sold, offer to sell, market, import, lease, or otherwise dispose of licensed products for all uses covered under the patent rights. The licensed product is Pyridorin or any other pharmaceutical compound labeled for an FDA‑approved indication that would infringe a valid claim of the patent rights in the absence of the license.
The patents licensed from SCRF include claims reciting methods for using Pyridorin to: (a) inhibit oxidative modification of proteins or treating atherosclerosis in a non‑hyperglycemic mammal (expires by 2016 in the United States and 2019 outside the United States absent any extension); (b) treat diabetes‑associated increases in hypercholesterolemia or hypertriglyceridemia in a diabetic mammal; (expires by 2016 in the United States and 2019 outside the United States absent any extension); and (c) treat diabetic neuropathy (expires by 2016 in the United States and 2019 outside the United States absent any extension). The patents licensed from SCRF also include patents with claims reciting novel Pyridorin analogues, and methods for using them to treat diabetes‑associated increases in hypercholesterolemia or hypertriglyceridemia in a diabetic mammal, and for inhibiting oxidative modification of proteins or treating atherosclerosis in a non‑hyperglycemic mammal; (expire in 2016 in the United States and 2019 outside the United States absent any extension).
Under the license, SCRF retains the right to practice under the patents in the field solely for non‑profit, educational, research, and academic purposes. The license also is subject to any U.S. government rights in the patent rights, if the technology or patent rights were developed with the support of the U.S. government or an agency thereof.
We must exercise commercially reasonable efforts to develop and commercialize one or more licensed products. If we fail to comply with our diligence obligations with respect to at least one licensed product, then SCRF may terminate the license. If we develop Pyridorin for the treatment of hyperlipidemia or diabetic neuropathy, we must pay SCRF milestone payments related to milestones met in the FDA regulatory approval process in the aggregate amount of $6,075,000. We must pay SCRF an annual license fee each year that we are actively marketing Pyridorin or have an active sublicense for Pyridorin for the treatment of hyperlipidemia or diabetic neuropathy, which are creditable only against Licensed Product Sublicense upfront fees and milestone payments earned and payable in the same calendar year. We must pay SCRF an annual fee of $120,000 for 2015 and the years thereafter. We must pay SCRF a one‑time fee of $35,000 upon execution of a sub‑license between NephroGenex and a third party, and must pay to SCRF 25% of any non‑royalty sublicense payments made by such sub‑licensee to NephroGenex. The planned phase 3 program for Pyridorin is for the treatment of diabetic nephropathy. Hyperlipidemia and diabetic neuropathy are not being evaluated in the current trial.
The agreement survives until the expiration or other disposition of the licensed patent rights. We may terminate the license at any time on three months prior written notice to SCRF. If we breach a material obligation under the agreement, and such obligation is not cured within 90 days after we receive written notice of the breach, then SCRF may terminate the agreement upon an additional written notice. SCRF may also terminate the license if (i) we cease operations and have not assigned the license to a third party; (ii) we become insolvent or make a general assignment of substantially all of our assets for the benefit of creditors, or if a petition of bankruptcy or any reorganization shall be commenced by, against, or in respect of us; or (iii) we fail to make a payment due under the license and the default is not cured within 30 days after written notice of such default, and SCRF has provided additional written notice.
28
Vanderbilt University (VU) Exclusive License Agreement
In connection with our additional pipeline opportunities for specific types of acute kidney injury, in July 2012, we entered into a license agreement with VU, which was amended on November 6, 2013. Under the agreement, VU grants us an exclusive, royalty‑bearing, worldwide license, under certain patent rights, and a corresponding nonexclusive license under related know‑how, with a right to sub‑license, to make, have made, use, offer to sell, sell, and import licensed products incorporating the technology embodied in the licensed VU patent rights for use of pyridoxamine in the field of use, which is defined as treatment of acute renal failure or acute renal injury, use for radiation protection, and use for chemotherapy protection. The patent applications licensed from VU include claims reciting methods for using Pyridorin to: (a) ameliorate at least one symptom of a kidney disorder associated with oxidative stress, carbonyl stress, or combinations thereof (if issued, would expire by 2026); and (b) treat or prevent acute renal injury or acute renal failure (if issued, would expire by 2026).
The patent applications licensed from VU also include claims reciting intravenous formulations of Pyridorin (if issued, would expire by 2026). Federal government rights in the licensed patents are reserved, as are VU’s right to use the subject matter of the licensed patents for academic research or other not‑for‑profit scholarly purposes, and to grant to other academic, governmental, or not‑for‑profit organizations a non‑exclusive right, non‑transferable, non‑sublicensable right to practice the licensed patent rights for academic research or other not‑for‑profit scholarly research purposes, expressly excluding any human use.
We must pay VU milestone payments related to milestones met in the FDA regulatory approval process in the aggregate amount of $1,075,000. We must also pay VU a 5% royalty on net sales of licensed products in the field of use. We must also pay VU 25% of non‑royalty sublicense payments to us such as milestone payments we recoup from sub licensees. We must exercise commercially reasonable efforts to develop and commercialize a licensed product for at least one indication. Our diligence obligations include a series of patent prosecution and clinical trial milestones. If we fail to comply with our diligence obligations with respect to at least one licensed product, then VU may terminate the license.
The agreement survives until the last to expire of the licensed patent rights. We may terminate the agreement upon 60 days written notice to VU. If either we or VU breach a material obligation under the agreement, and such obligation, then the non‑breaching party may terminate the agreement upon an additional written notice. VU may also terminate the license if we become insolvent or suspend business, or file a voluntary petition or an answer admitting the jurisdiction of the court, or consent to an involuntary petition pursuant to any reorganization or insolvency law of any jurisdiction, or make an assignment for the benefit of creditors, or apply for or consent to the appointment of a receiver or trustee of a substantial part of our property.
BioStratum, Inc. (BioStratum) Grant Back License Agreement
In May 2007, we entered into a grant‑back license agreement with BioStratum as part of our acquisition of certain of BioStratum’s assets, including certain patent rights. The licensed patent rights include all patents and patent applications licensed by NephroGenex from BioStratum under an earlier, terminated license agreement between the parties. These rights include all patents owned or licensed by us with the exception of the patent applications that we license from VU. Under this agreement, we grant BioStratum an exclusive, sublicensable license and sublicense under those patent rights to make, have made, use, sell, offer for sale and import licensed products solely in Japan, Taiwan, Korea and China. The licensed products are Pyridorin or AGE inhibitor products that are covered by the licensed patents. As this license has been fully paid, there are no milestone payments under this agreement. In this agreement, we also agreed not to modify the Kansas or USC license agreements in a manner that would adversely affect BioStratum’s rights.
The license grant to BioStratum was made solely to enable BioStratum to exercise its rights and perform its obligations pursuant to a license agreement with Kowa Company, Ltd. (Kowa) pursuant to which BioStratum granted Kowa an exclusive license (the Kowa Agreement) to manufacture and use licensed products in Japan, Taiwan, Korea, and China. The Kowa Agreement was terminated by Kowa on December 5, 2007.
After termination of the BioStratum grant‑back license agreement for any reason other than assignment or transfer of the Kowa Agreement to NephroGenex, we are required to obtain the written consent of BioStratum to grant a license to any third party to develop, make, have made, use, sell, offer for sale, or import Licensed Products in Japan, Taiwan, Korea or China.
29
Manufacturing
We do not own or operate manufacturing facilities for the production of any of our product candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently rely on third‑party contract manufacturers for all of our required raw materials, active pharmaceutical ingredient (API) and finished product for our preclinical research and clinical trials, including the Phase 3 trials for Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes. We are currently negotiating a new manufacturing agreement to manufacture pyridoxamine dihydrochloride, the API in Pyridorin. At our direction, Patheon manufactures clinical trial drug supply of pyridoxamine dihydrochloride capsules and placebo for our clinical supply. We do not have any current contractual relationships for the manufacture of commercial supplies of any of our product candidates. If any of our products are approved by any regulatory agency, we intend to enter into agreements with a third‑party contract manufacturer and one or more back‑up manufacturers for the commercial production of those products. Development and commercial quantities of any products that we develop will need to be manufactured in facilities, and by processes, that comply with the current Good Manufacturing Practice regulations, or cGMPs, and other requirements of the FDA and the regulatory agencies of other jurisdictions in which we are seeking approval. We currently employ internal resources to manage our manufacturing contractors.
The typical route for the chemical synthesis of Pyridorin (pyridoxamine) uses oxidative methods where the starting material is the readily and economically available pyridoxine (vitamin B6). Although such oxidative manufacturing methods are usable at a small scale, oxidative methods are not viable for large‑scale production and commercialization. For example, the first step in the metabolism of pyridoxine is an enzymatic oxidation of the alcohol group to an aldehyde, thus converting pyridoxine to pyridoxal. The oxidative chemical synthetic parallels this by utilizing oxidizing agents such as manganese dioxide to convert pyridoxine to pyridoxal. However, the oxidation of pyridoxine is problematic at the scale required for commercial manufacturing for several reasons, including the need to rapidly remove large amounts of solid oxidants to minimize the potential for continuing oxidation reactions. Such overoxidation not only can convert pyridoxal to pyridoxic acid but can also lead to non‑selective oxidation of the second hydroxymethyl group at the 5‑position. Other difficulties can be encountered subsequent to the formation of pyridoxal. For example, in order to form the desired amine, pyridoxal is conveniently reacted with hydroxylamine to form an intermediate oxime that must be subsequently reduced. Hydroxylamine is a dangerous reagent to handle on an industrial scale due to its instability, its high reactivity and its toxicity. Reduction of the oxime is known and can be performed by methods such as using zinc. However, this is also an unfavorable reagent for large scale manufacturing. Reduction with hydrogen catalysts such as platinum or palladium is possible, but this route is expensive, difficult to control, and difficult to scale up. Over‑reduction can lead to the generation of deoxy impurities that may be toxic anti‑metabolites contaminating the API.
To overcome this barrier to commercialization, we have developed and patented a non‑oxidative method for the synthesis of pyridoxamine and all of its intermediate compounds and salts. This method provides for large scale synthesis at a fraction of the price required using traditional oxidative methods. It also eliminates the safety and environmental hazards associated with these oxidative methods.
Government Regulation and Product Approval
Governmental authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States and by the EMA through the MAA process before they may be legally marketed in most countries in Europe. Our product candidates will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
NDA Approval Processes
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (the FDCA) and implementing regulations. Failure to comply with the applicable U.S. requirements at any time during the product development process or approval process, or after approval, may subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
• | refusal to approve pending applications; |
30
• | withdrawal of an approval; |
• | imposition of a clinical hold; |
• | warning letters; |
• | product seizures; |
• | total or partial suspension of production or distribution; or |
• | injunctions, fines, disgorgement of profits, or civil or criminal penalties. |
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
• | completion of nonclinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory Practices (GLPs) or other applicable regulations; |
• | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
• | performance of adequate and well‑controlled human clinical trials according to Good Clinical Practices (GCPs) to establish the safety and efficacy of the proposed drug for its intended use; |
• | submission to the FDA of a NDA; |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (cGMPs) to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
• | FDA review and approval of the NDA. |
Once a pharmaceutical candidate is identified for development, it enters the preclinical or nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the nonclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some nonclinical testing may continue even after the IND is submitted. In addition to including the results of the nonclinical studies, the IND will also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30‑day time period, places the IND on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND, and may affect one or more specific studies or all studies conducted under the IND.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCPs. They must be conducted under protocols detailing, among other things, the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors also must timely report to FDA serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigation brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug. An institutional review board, or IRB, at each institution participating in the clinical trial must review and approve the protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each research subject or the subject’s legal representative, monitor the study until completed and otherwise comply with IRB regulations.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some products for severe or life‑threatening
31
diseases, such as cancer, especially when the product may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
Phase 2. Clinical trials are performed on a limited patient population intended to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk‑benefit ratio of the product and provide an adequate basis for product labeling.
Human clinical trials are inherently uncertain and Phase 1, Phase 2 and Phase 3 testing may not be successfully completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to the submission of an IND, at the end of Phase 2 and before a NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trials that they believe will support the approval of the new drug. If a Phase 2 clinical trial is the subject of discussion at the end of Phase 2 meeting with the FDA, a sponsor may be able to request a Special Protocol Assessment, or SPA, the purpose of which is to reach agreement with the FDA on the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
According to published guidance on the SPA process, a sponsor that meets the prerequisites may make a specific request for a SPA and provide information regarding the design and size of the proposed clinical trial. The FDA is supposed to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. A SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies and also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing commercial quantities of the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug and the manufacturer must develop methods for testing the quality, purity and potency of the drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its proposed shelf‑life.
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of a NDA requesting approval to market the product. The submission of a NDA is subject to the payment of user fees, but a waiver of such fees may be obtained under specified circumstances. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may request additional information rather than accept a NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in‑depth review. NDAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. The FDA may refuse to approve a NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA reviews a NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP‑compliant. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not
32
bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving a NDA, the FDA will inspect the facility or facilities where the product is manufactured and tested.
Expedited Review and Approval
The FDA has various programs, including Fast Track, priority review, and accelerated approval, which are intended to expedite or simplify the process for reviewing drugs, and/or provide for the approval of a drug on the basis of a surrogate endpoint. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. Generally, drugs that are eligible for these programs are those for serious or life‑threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious or life‑threatening diseases or conditions and fill unmet medical needs. Priority review is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of ten months.
Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated drug and expedite review of the application for a drug designated for priority review. Accelerated approval, which is described in Subpart H of 21 CFR Part 314, provides for an earlier approval for a new drug that is intended to treat a serious or life‑threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. A surrogate endpoint is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. As a condition of approval, the FDA may require that a sponsor of a product candidate receiving accelerated approval perform post‑marketing clinical trials.
In the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was signed into law in July 2012, Congress encouraged the FDA to utilize innovative and flexible approaches to the assessment of products under accelerated approval. The law required the FDA to issue related draft guidance within a year after the law’s enactment and also promulgate confirming regulatory changes. In June 2013, the FDA published a draft Guidance for Industry entitled, “Expedited Programs for Serious Conditions-Drugs and Biologics” which provides guidance on FDA programs that are intended to facilitate and expedite development and review of new drugs as well as threshold criteria generally applicable to concluding that a drug is a candidate for these expedited development and review programs. In addition to the Fast Track, accelerated approval and priority review programs discussed above, the FDA also provided guidance on a new program for Breakthrough Therapy designation. A request for Breakthrough Therapy designation should be submitted concurrently with, or as an amendment to an IND. FDA has already granted this designation to over 70 new drugs and has approved almost 20 Breakthrough Therapy designated drug.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch‑Waxman Act. The Hatch‑Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one‑half the time between the effective date of an IND, and the submission date of a NDA, plus the time between the submission date of a NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for extension must be made prior to expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five‑year period of non‑patent marketing exclusivity within the United States to the first applicant to gain approval of a NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non‑infringement. The FDCA also provides three years of marketing exclusivity for a NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were
33
conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three‑year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five‑year and three‑year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well‑controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric Exclusivity and Pediatric Use
Under the Best Pharmaceuticals for Children Act (BPCA) certain drugs may obtain an additional six months of exclusivity, if the sponsor submits information requested in writing by the FDA (a Written Request) relating to the use of the active moiety of the drug in children. The FDA may not issue a Written Request for studies on unapproved or approved indications or where it determines that information relating to the use of a drug in a pediatric population, or part of the pediatric population, may not produce health benefits in that population.
We have not received a Written Request for such pediatric studies, although we may ask the FDA to issue a Written Request for such studies in the future. To receive the six‑month pediatric market exclusivity, we would have to receive a Written Request from the FDA, conduct the requested studies in accordance with a written agreement with the FDA or, if there is no written agreement, in accordance with commonly accepted scientific principles, and submit reports of the studies. A Written Request may include studies for indications that are not currently in the labeling if the FDA determines that such information will benefit the public health. The FDA will accept the reports upon its determination that the studies were conducted in accordance with and are responsive to the original Written Request or commonly accepted scientific principles, as appropriate, and that the reports comply with the FDA’s filing requirements.
In addition, the Pediatric Research Equity Act (PREA) requires all applications (or supplements to an application) submitted under section 505 of the FDCA (21 U.S.C. Section 355) for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration to contain a pediatric assessment unless the applicant has obtained a waiver or deferral. It also authorizes the FDA to require holders of approved NDAs for marketed drugs to conduct pediatric studies under certain circumstances. In general, PREA applies only to those drugs developed for diseases and/or conditions that occur in both the adult and pediatric populations. Products intended for pediatric‑specific indications will be subject to the requirements of PREA only if they are initially developed for a subset of the relevant pediatric population.
As part of the FDASIA, Congress reauthorized both BPCA and PREA, which were slated to expire on September 30, 2012, and made both laws permanent.
Post‑approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post‑marketing programs.
Any drug products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
• | record‑keeping requirements; |
• | reporting of adverse experiences with the drug; |
• | providing the FDA with updated safety and efficacy information; |
• | drug sampling and distribution requirements; |
• | notifying the FDA and gaining its approval of specified manufacturing or labeling changes; and |
• | complying with FDA promotion and advertising requirements. |
34
Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMP and other laws.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Regulation Outside of the United States
In addition to regulations in the United States, we will be subject to regulations of other countries governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the United States before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology or those medicines intended to treat AIDS, cancer, neurodegenerative disorders or diabetes and optional for those medicines which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessments report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Reimbursement
Sales of our products will depend, in part, on the extent to which the costs of our products will be covered by third‑party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third‑party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost‑containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost‑containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third‑party payors do not consider our products to be cost‑effective compared to other therapies, they may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the MMA) imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand‑alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in
35
setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non‑governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third‑party payors do not consider our products to be cost‑effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the ACA), enacted in March 2010, is expected to have a significant impact on the health care industry. ACA is expected to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the impact of ACA on pharmaceutical companies, as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred. In addition, although the United States Supreme Court upheld the constitutionality of much of the law, some members of the U.S. Congress continue to try to overturn at least portions of the legislation, and we expect they will continue to review and assess this legislation and alternative health care reform proposals. Any legal challenges to ACA, as well as Congressional efforts to repeal ACA, add to the uncertainty of the legislative changes enacted as part of ACA.
In addition, in some non‑U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
Corporate Information
We are subject to the information requirements of the Exchange Act. Therefore, we file public reports, proxy statements and other information with the Securities and Exchange Commission (SEC), which may be obtained by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, DC 20549 or by calling the SEC at 1-(800)-SEC-0330. The SEC also maintains a website (www.sec.gov) that contains reports, proxy information statements, and other information that issuers file electronically.
In addition, we maintain a website at www.nephrogenex.com and make available free of charge through this website our Annual Reports on Form 10-K, our Quarterly Reports on Form 10-Q, our Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. We also similarly make available, free of charge on our website, the reports filed with the SEC by our executive officers, directors and 10% stockholders pursuant to Section 16 under the Exchange Act as soon as reasonably practicable after copies of those filings are provided to us by those persons. We are not including the information contained at www.nephrogenex.com, or at any other Internet address as part of, or incorporating it by reference into, this Annual Report on Form 10-K. We also make available on our website (i) the charters for the committees of our Board of Directors, including the Audit Committee, Compensation Committee and Nominating and Governance Committee, and (ii) our Corporate Code of Conduct and Ethics and Whisleblower Policy governing our directors, officers and employees. We intend to disclose on our website any amendments to, or waivers from, our Code of Business Conduct and Ethics that are required to be disclosed pursuant to the rules of the SEC.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) December 31, 2019; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer
36
under the rules of the SEC. We refer to the Jumpstart Our Business Startups Act of 2012 herein as the “JOBS Act,” and references herein to “emerging growth company” shall have the meaning associated with it in the JOBS Act.
Employees
As of March 24, 2015, we had eleven full-time employees, of which all are involved in our drug development operations or in general and administrative functions.
Item 1A. RISK FACTORS
Except for the historical information contained herein or incorporated by reference, this report and the information incorporated by reference contains forward‑looking statements that involve risks and uncertainties. These statements include projections about our accounting and finances, plans and objectives for the future, future operating and economic performance and other statements regarding future performance. These statements are not guarantees of future performance or events. Our actual results could differ materially from those discussed in this report. Factors that could cause or contribute to these differences include, but are not limited to, those discussed in the following section, as well as those discussed in Part II, Item 7 entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere throughout this report and in any documents incorporated in this report by reference.
You should consider carefully the following risk factors, together with all of the other information included or incorporated in this report. If any of the following risks, either alone or taken together, or other risks not presently known to us or that we currently believe to not be significant, develop into actual events, then our business, financial condition, results of operations or prospects could be materially adversely affected. If that happens, the market price of our common stock could decline, and stockholders may lose all or part of their investment.
Risks Relating to Our Financial Position and Need for Additional Capital
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We are currently advancing Pyridorin through clinical development for diabetic nephropathy and an intravenous formulation of Pyridorin for AKI through preclinical development. Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We will require substantial additional future capital in order to complete clinical development and commercialize Pyridorin. If the FDA or EMA requires that we perform additional nonclinical studies or clinical trials, our expenses would further increase beyond what we currently expect and the anticipated timing of any potential NDA or MAA would likely be delayed. Further, there can be no assurance that the costs to obtain regulatory approval of Pyridorin as a treatment for diabetic nephropathy in patients with type 2 diabetes or as a treatment for AKI will not increase.
We will continue to require substantial additional capital to continue our clinical development and commercialization activities. Because successful development of our product candidates is uncertain, we are unable to estimate the actual funds we will require to complete research and development and commercialize our products under development.
The amount and timing of our future funding requirements will depend on many factors, including but not limited to:
• | the progress, costs, results of and timing of our Phase 3 Pyridorin PIONEER program for the treatment of diabetic nephropathy in patients with type 2 diabetes, and the preclinical and clinical development of an intravenous formulation of Pyridorin for AKI |
• | the outcome, costs and timing of seeking and obtaining FDA, EMA and any other regulatory approvals; |
• | the number and characteristics of product candidates that we pursue; |
• | the ability of our product candidates to progress through clinical development successfully; |
• | our need to expand our research and development activities; |
• | the costs associated with securing and establishing commercialization and manufacturing capabilities; |
• | market acceptance of our product candidates; |
37
• | the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies; |
• | our ability to maintain, expand and enforce the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
• | our need and ability to hire additional management and scientific and medical personnel; |
• | the effect of competing drug candidates and new product approvals; |
• | our need to implement additional internal systems and infrastructure, including financial and reporting systems; and |
• | the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future. |
Some of these factors are outside of our control. Based upon our currently expected level of operating expenditures, we believe that we will be able to fund our operations into early 2016. This period could be shortened if there are any significant increases in planned spending on development programs or more rapid progress of development programs than anticipated. We do not expect our existing capital resources to be sufficient to enable us to complete the commercialization of Pyridorin, if approved, or to initiate any clinical trials or additional development work needed for any other product candidates, other than as described above. Accordingly, we expect that we will need to raise additional funds in the future.
We may seek additional funding through a combination of equity offerings, debt financings, government or other third‑party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. Additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. In addition, the issuance of additional shares by us, or the possibility of such issuance, may cause the market price of our shares to decline.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our research or development programs. We also could be required to seek funds through arrangements with collaborative partners or otherwise that may require us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us.
We have never been profitable. Currently, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to reduce our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have not yet submitted any product candidates for approval by regulatory authorities in the United States or elsewhere for our lead indication, the treatment of diabetic nephropathy in patients with type 2 diabetes, or any other indication. We have incurred net losses in each year since our inception, including net losses of $16.8 million and $6.3 million for the years ended December 31, 2014 and 2013, respectively. We had an accumulated deficit of approximately $57.8 million as of December 31, 2014.
To date, we have devoted most of our financial resources to our corporate overhead and research and development, including our drug discovery research, preclinical development activities and clinical trials. We have not generated any revenues from product sales. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for Pyridorin, prepare for and begin the commercialization of any approved products, and add infrastructure and personnel to support our continuing product development efforts. We anticipate that any such losses could be significant for the next several years as we continue our Phase 3 clinical program of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, which we call the PIONEER program, and related activities required for regulatory approval of Pyridorin and pursuing an intravenous formulation of Pyridorin for AKI in clinical trials. If Pyridorin or any of our other product candidates fails in clinical trials or does not gain regulatory approval, or if our product candidates do not achieve market acceptance, we may never become profitable. As a result of the foregoing, we expect to continue to experience net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital.
38
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. In addition, our expenses could increase if we are required by the FDA or the EMA, to perform studies or trials in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our product candidates. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues.
Borrowings under our credit facility may not be available to us to operate our business and successfully develop and commercialize our primary product candidate.
On November 20, 2014, we entered into a Loan and Security Agreement with East West Bank (East West) for a term loan (the Initial Term Loan) with an aggregate principal amount of $7.0 million and, subject to the terms and conditions set forth in the agreement, a second term loan (the Second Term Loan) with an aggregate principal amount of $5.0 million. As security for our obligations under the Loan Agreement, we granted East West a lien in substantially all of our assets, including owned and licensed intellectual property. At the Company’s option, the Company may borrow the Second Term Loan on or before May 29, 2015, if the Company has met certain clinical milestones. As of the date hereof, the Company does not believe that it will meet the clinical milestones for the Second Term Loan. However, the Company has made a proposal to East West to amend the clinical milestones necessary for incurrence of the Second Term Loan.
Our loan agreement contains customary affirmative and negative covenants, indemnification provisions and events of default. The affirmative covenants include, among others, covenants requiring us to maintain our legal existence and governmental approvals, deliver certain financial reports and maintain certain intellectual property rights. The negative covenants include, among others, restrictions on transferring or licensing our assets, changing our business, incurring additional indebtedness, engaging in mergers or acquisitions, paying dividends or making other distributions, and creating other liens on our assets, in each case subject to customary exceptions. If we default under the Loan Agreement, East West may accelerate all of our repayment obligations and take control of our pledged assets, potentially requiring us to renegotiate the Loan Agreement on terms less favorable to us or to immediately cease operations. Further, if we are liquidated, East West has the right to repayment would be senior to the rights of the holders of our common shares to receive any proceeds from the liquidation. East West could declare a default under the Loan Agreement upon the occurrence of any event that East West interprets as a material adverse change as defined under the Loan Agreement, thereby requiring us to repay the loan immediately or to attempt to reverse the declaration of default through negotiation or litigation. Any declaration by our lender of an event of default could significantly harm our business and prospects and could cause the price of our common shares to decline. If we raise any additional debt financing, the terms of such additional debt could further restrict our operating and financial flexibility.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make it difficult to predict our future performance.
We are a development stage pharmaceutical company with a limited operating history. Our operations to date have been limited to developing our technology and undertaking preclinical studies and clinical trials of our product candidates. We have not yet obtained regulatory approvals for any of our product candidates. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or approved products on the market. Our financial condition and operating results have varied significantly in the past and are expected to continue to significantly fluctuate from quarter‑to‑quarter or year‑to‑year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include:
• | any delays in regulatory review and approval of our product candidates in clinical development, including our ability to receive approval from the FDA and the EMA for Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes based on our Phase 3 Pyridorin program, and our other completed and planned clinical trials and nonclinical studies and other work, as the basis for review and approval of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes; |
• | delays in the commencement, enrollment and timing of clinical trials; |
• | difficulties in identifying and randomizing patients suffering from our target indications, and kidney disease in patients with type 2 diabetes in particular; |
• | the success of our clinical trials through all phases of clinical development, including our Phase 3 trial of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes; |
39
• | potential side effects of our product candidate that could delay or prevent approval or cause an approved drug to be taken off the market; |
• | our ability to obtain additional funding to develop product candidates; |
• | our ability to identify and develop additional product candidates; |
• | market acceptance of our product candidates; |
• | our ability to establish an effective sales and marketing infrastructure directly or through collaborations with third parties; |
• | competition from existing products or new products that continue to emerge; |
• | the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products; |
• | our ability to adhere to clinical trial requirements directly or with third parties such as contract research organizations (CROs); |
• | our dependency on third‑party manufacturers to manufacture our products and key ingredients; |
• | our ability to establish or maintain collaborations, licensing or other arrangements; |
• | the costs to us, and our ability and our third‑party collaborators’ ability to obtain, maintain and protect our intellectual property rights; |
• | costs related to and outcomes of potential intellectual property litigation; |
• | our ability to adequately support future growth; |
• | our ability to attract and retain key personnel to manage our business effectively; and |
• | potential product liability claims. |
Accordingly, the results of any quarterly or annual periods should not be relied upon as indications of future operating performance.
Our recurring losses from operations may raise substantial doubt regarding our ability to continue as a going concern.
Our recurring losses from operations may raise substantial doubt about our ability to continue as a going concern. There is no assurance that sufficient financing will be available when needed to allow us to continue as a going concern. The perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations.
Risks Relating to Regulatory Review and Approval of Our Product Candidates
We cannot be certain that Pyridorin will receive regulatory approval, and without regulatory approval we will not be able to market Pyridorin.
Our business currently depends entirely on the successful development and commercialization of Pyridorin. Our ability to generate revenue related to product sales, if ever, will depend on the successful development and regulatory approval of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes or an intravenous formulation of Pyridorin for AKI.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the United States, the EMA in Europe and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the United States or Europe until we receive
40
approval of a NDA from the FDA or a MAA from the EMA, respectively. We have not submitted any marketing applications for any of our product candidates.
NDAs and MAAs must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. NDAs and MAAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a NDA or a MAA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA and the EMA review processes can take years to complete and approval is never guaranteed. If we submit a NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators in other jurisdictions, such as the EMA, have their own procedures for approval of product candidates. Even if a product is approved, the FDA or the EMA, as the case may be, may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time‑consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and Europe also have requirements for approval of drug candidates with which we must comply with prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. In addition, delays in approvals or rejections of marketing applications in the United States, Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding our product candidates or other products. Also, regulatory approval for any of our product candidates may be withdrawn.
We have completed three Phase 2 trials for Pyridorin and are enrolling patients for our Phase 3 PIONEER trial. In addition, we have successfully completed a QT/QTc (TQT) cardiac safety study. Before we submit a NDA to the FDA or a MAA to the EMA for Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, we must successfully conduct two Phase 3 trials. In addition, we must complete other nonclinical studies and clinical trials, such as two nonclinical carcinogenicity studies and a nonclinical cardiac safety study. We cannot predict whether our future trials and studies will be successful or whether regulators will agree with our conclusions regarding the preclinical studies and clinical trials we have conducted to date.
If we are unable to obtain approval from the FDA, the EMA or other regulatory agencies for Pyridorin and our other product candidates, or if, subsequent to approval, we are unable to successfully commercialize Pyridorin or our other product candidates, we will not be able to generate sufficient revenue to become profitable or to continue our operations.
Any statements in this document indicating that Pyridorin has demonstrated preliminary evidence of efficacy are our own and are not based on the FDA’s or any other comparable governmental agency’s assessment of Pyridorin and do not indicate that Pyridorin will achieve favorable efficacy results in any later stage trials or that the FDA or any comparable agency will ultimately determine that Pyridorin is effective for purposes of granting marketing approval.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for Pyridorin and our other product candidates.
Delays in the commencement, enrollment and completion of clinical trials could increase our product development costs or limit the regulatory approval of our product candidates. We do not know whether any future trials or studies of our other product candidates will begin on time or will be completed on schedule, if at all. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, including delays or shortages in available drug product, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, that include the age and condition of the patients and the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments and/or availability of investigational treatment options for the relevant disease.
A product candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for product candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may
41
suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
• | inability to obtain sufficient funds required for a clinical trial; |
• | inability to reach agreements on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
• | negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; |
• | serious and unexpected drug‑related side effects experienced by subjects in our clinical trials or by individuals using drugs similar to our product candidates; |
• | inability to obtain approval from institutional review boards (IRBs), to conduct a clinical trial at their respective sites; |
• | inability to obtain approval from regulatory authorities outside the United States to conduct a clinical trial in their respective country; |
• | conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
• | delays in enrolling research subjects in clinical trials; |
• | high drop‑out rates of research subjects; |
• | high screen fail rates of research subjects; |
• | inadequate supply or quality of product candidate components or materials or other supplies necessary for the conduct of our clinical trials; |
• | greater than anticipated clinical trial costs; |
• | poor effectiveness of our product candidates during clinical trials; |
• | unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or vendor; |
• | failure of our third‑party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all; |
• | delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or |
• | varying interpretations of data by the FDA and similar foreign regulatory agencies. |
Although the FDA has agreed to our endpoint for approval for the pivotal Phase 3 PIONEER program, other regulatory agencies outside the United States may not agree to our proposed endpoint for approval of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, in which case we would need to complete one or more additional clinical trials in order to seek approval outside the United States.
Regulatory authorities in other countries in which we may seek approval for and market Pyridorin may require additional nonclinical studies and/or clinical trials prior to granting approval. It may be expensive and time consuming to conduct and complete additional nonclinical studies and clinical trials that other regulatory authorities may require us to perform. As such, any requirement by other regulatory authorities that we conduct additional nonclinical studies or clinical trials could materially and adversely affect our business, financial condition and results of operations. Furthermore, even if we
42
receive regulatory approval of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, the labeling for Pyridorin in the United States, Europe or other countries in which we seek approval may include limitations that could impact the commercial success of Pyridorin.
Clinical failure can occur at any stage of clinical development and we have never conducted a Phase 3 trial or submitted a NDA or MAA before. The results of earlier clinical trials are not necessarily predictive of future results and any product candidate we or our potential future collaborators advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of our clinical development. Clinical trials may produce negative or inconclusive results, and we or our collaborators may decide, or regulators may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Success in preclinical studies and early clinical trials does not ensure that subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in Phase 3 clinical trials, even after seeing promising results in earlier clinical trials.
Pyridorin did not reach its primary endpoint in the intent to treat (ITT) population in the Phase 2b trial (PYR‑210). However, in a prespecified subgroup of patients on stable long term standard of care, Pyridorin showed a dose-dependent treatment effect of approximately 50%. This subgroup is the patient population that will be studied in the Phase 3 program. Subgroup analysis carries the inherent risk that the results may not be repeatable in a subsequent trial. It is possible that the treatment effect observed in this subgroup of PYR‑210 may not repeat in our Phase 3 trials.
Pyridorin has demonstrated a promising treatment effect in Phase 2 clinical trials using a rate of change in SCr endpoint. The Phase 3 PIONEER trial is utilizing a new ≥50% SCr increase event endpoint or ESRD. While there is a strong correlation between the rate of change of SCr and the 50% SCr increase event endpoint, no clinical trials have been conducted using this new endpoint. We cannot assure you that our PIONEER Pyridorin program will achieve positive results using this new endpoint.
In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well‑advanced. We may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts.
If Pyridorin is found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for it and our business would be harmed. For example, if the results of our Phase 3 Pyridorin program do not achieve the primary efficacy endpoints or demonstrate expected safety, the prospects for approval of Pyridorin would be materially and adversely affected.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether any Phase 2, Phase 3 or other clinical trials we or any of our potential future collaborators may conduct will demonstrate the consistent or adequate efficacy and safety that would be required to obtain regulatory approval and market Pyridorin. If we are unable to bring Pyridorin to market, or to acquire other products that are on the market or can be developed, our ability to create long‑term stockholder value will be limited.
Our product candidates may have undesirable side effects which may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Pyridorin targets a broad range of pathogenic oxidative chemistries, including advanced glycation end‑products, toxic carbonyls, and reactive oxygen species that develop in patients with diabetes and are considered a principal causative factor in the development and progression of diabetic microvascular disease. Unforeseen side effects from any of our product candidates could arise either during clinical development or, if approved, after the approved product has been marketed. The most common side effects observed in clinical trials of Pyridorin were a slight increase in diarrhea and constipation. No patients were withdrawn from the study for these side effects. Additional or unforeseen side effects from these or any of our other product candidates could arise either during clinical development or, if approved, after the approved product has been marketed.
43
The range and potential severity of possible side effects from systemic therapies is significant. The results of future clinical trials may show that Pyridorin causes undesirable or unacceptable side effects, which could interrupt, delay or halt clinical trials, and result in delay of, or failure to obtain, marketing approval from the FDA and other regulatory authorities, or result in marketing approval from the FDA and other regulatory authorities with restrictive label warnings.
If any of our product candidates receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by such products:
• | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies; |
• | we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
• | we may be subject to limitations on how we may promote the product; |
• | sales of the product may decrease significantly; |
• | regulatory authorities may require us to take our approved product off the market; |
• | we may be subject to litigation or product liability claims; and |
• | our reputation may suffer. |
Any of these events could prevent us or our potential future collaborators from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
Reimbursement decisions by third‑party payors may have an adverse effect on pricing and market acceptance. If there is not sufficient reimbursement for our products, it is less likely that they will be widely used.
Market acceptance and sales of Pyridorin or any other product candidates that we develop, if approved, will depend on reimbursement policies and may be affected, among other things, by future healthcare reform measures. Government authorities and third‑party payors, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for Pyridorin or any other product candidates that we develop. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize Pyridorin or any other product candidates that we develop.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA) changed the way Medicare covers and pays for pharmaceutical products. The legislation established Medicare Part D, which expanded Medicare coverage for outpatient prescription drug purchases by the elderly but provided authority for limiting the number of drugs that will be covered in any therapeutic class. The MMA also introduced a new reimbursement methodology based on average sales prices for physician‑ administered drugs. Any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain in the United States. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non‑governmental payors.
The United States and several other jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. We expect to experience pricing pressures in connection with the sale of Pyridorin and any other products that we develop, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals.
44
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, ACA) became law in the United States. The goal of ACA is to reduce the cost of health care and substantially change the way health care is financed by both governmental and private insurers. While we cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically, the ACA may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of Pyridorin or any future product candidates. In addition, some members of the U.S. Congress have been seeking to overturn at least portions of the legislation and we expect they will continue to review and assess this legislation and alternative health care reform proposals. We cannot predict whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
In the European Union (EU), prescription drug pricing and reimbursement is subject to governmental control and reimbursement mechanisms used by private and public health insurers in the EU vary by member state. For the public systems, reimbursement is determined by guidelines established by the legislator or responsible national authority. As elsewhere, inclusion in reimbursement catalogues focuses on the medical usefulness, need, quality and economic benefits to patients and the health care system. Acceptance for reimbursement comes with cost, use and often volume restrictions, which can vary by member state. In those member states that impose price controls, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some member states, we or our partners may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies.
Some EU member states require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some member states, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we or our partners might obtain marketing approval for a product in a particular member state, but then be subject to price regulations that delay commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues that are generated from the sale of the product in that country. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, or if there is competition from lower priced cross-border sales, our profitability may be negatively affected.
If we do not obtain protection under the Hatch‑Waxman Act and similar legislation outside of the United States by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of Pyridorin and our other product candidates, if any, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch‑Waxman Act. The Hatch‑Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product will be shortened and our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially. In the event that we are unable to obtain any patent term extensions, the issued patents for methods of using Pyridorin are expected to expire in June 2024 assuming they withstand any challenge.
If we market products in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare laws, commonly referred to as “fraud and abuse” laws, have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. Other jurisdictions such as Europe have similar laws. These laws include false claims and anti‑kickback statutes. If we market our products and our products are paid for by governmental programs, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. The
45
federal healthcare program anti‑kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service covered by Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers or formulary managers on the other. Although there are several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Most states also have statutes or regulations similar to the federal anti‑kickback law and federal false claims laws, which apply to items and services covered by Medicaid and other state programs, or, in several states, apply regardless of the payor. Administrative, civil and criminal sanctions may be imposed under these federal and state laws.
Over the past few years, a number of pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of promotional and marketing activities, such as: providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off‑label promotion; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates.
If the FDA and EMA and other regulatory agencies do not find the manufacturing facilities of our future contract manufacturers acceptable for commercial production, we may not be able to commercialize any of our product candidates.
We do not intend to manufacture the pharmaceutical products that we plan to sell. We currently have agreements with and are negotiating additional agreements with contract manufacturers for the production of the active pharmaceutical ingredients and the formulation of drug product for our Phase 3 trial of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes and the other trials and nonclinical studies that we believe we will need to conduct prior to seeking regulatory approval. However, we do not have agreements for commercial supplies of Pyridorin or any of our other product candidates and we may not be able to reach agreements with these or other contract manufacturers for sufficient supplies to commercialize Pyridorin if it is approved. Additionally, the facilities used by any contract manufacturer to manufacture Pyridorin or any of our other product candidates must be the subject of a satisfactory inspection before the FDA or the regulators in other jurisdictions approve the product candidate manufactured at that facility. We are completely dependent on these third‑party manufacturers for compliance with the requirements of U.S. and non‑U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture material that conform to our specifications and cGMP and other requirements of any governmental agency whose jurisdiction to which we are subject, our product candidates will not be approved or, if already approved, may be subject to recalls. Reliance on third‑party manufacturers entails risks to which we would not be subject if we manufactured the product candidates, including:
• | the possibility that we are unable to enter into a manufacturing agreement with a third party to manufacture our product candidates; |
• | the possible breach of the manufacturing agreements by the third parties because of factors beyond our control; and |
• | the possibility of termination or nonrenewal of the agreements by the third parties before we are able to arrange for a qualified replacement third‑party manufacturer. |
Any of these factors could cause the delay of approval or commercialization of our product candidates, cause us to incur higher costs or prevent us from commercializing our product candidates successfully. Furthermore, if any of our product candidates are approved and contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis and at commercially reasonable prices and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the government agencies that regulate our products.
Even if our product candidates receive regulatory approval, we may still face future development and regulatory difficulties.
Our product candidates, if approved, will also be subject to ongoing regulatory requirements for labeling, packaging, storage, advertising, promotion, record‑keeping and submission of safety and other post‑market information. In addition, approved products, manufacturers and manufacturers’ facilities are required to comply with extensive FDA and EMA
46
requirements and requirements of other similar agencies, including ensuring that quality control and manufacturing procedures conform to cGMPs. As such, we and our contract manufacturers are subject to continual review and periodic inspections to assess compliance with cGMPs. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control. We will also be required to report certain adverse reactions and production problems, if any, to the FDA and EMA and other similar agencies and to comply with certain requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. Accordingly, we may not promote our approved products, if any, for indications or uses for which they are not approved.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of a product, it may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
• | issue warning letters; |
• | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
• | require us or our potential future collaborators to enter into a consent decree or permanent injunction, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
• | impose other administrative or judicial civil or criminal penalties; |
• | withdraw regulatory approval; |
• | refuse to approve pending applications or supplements to approved applications filed by us or our potential future collaborators; |
• | impose restrictions on operations, including costly new manufacturing requirements; or |
• | seize or detain products. |
Risks Relating to the Commercialization of Our Products
Even if approved, our product candidates may not achieve broad market acceptance among physicians, patients and healthcare payors, and as a result our revenues generated from their sales may be limited.
The commercial success of Pyridorin, if approved, will depend upon its acceptance among the medical community, including physicians, health care payors and patients. The degree of market acceptance of Pyridorin or future product candidates will depend on a number of factors, including:
• | limitations or warnings contained in our product candidates’ FDA‑approved labeling; |
• | changes in the standard of care or availability of alternative therapies at similar or lower costs for the targeted indications for any of our product candidates; |
• | limitations in the approved clinical indications for our product candidates; |
• | demonstrated clinical safety and efficacy compared to other products; |
• | lack of significant adverse side effects; |
• | sales, marketing and distribution support; |
• | availability of reimbursement from managed care plans and other third‑party payors; |
47
• | timing of market introduction and perceived effectiveness of competitive products; |
• | the degree of cost‑effectiveness; |
• | availability of alternative therapies at similar or lower cost, including generics and over‑the‑counter products; |
• | enforcement by the FDA and EMA of laws and rulings that prohibit the sale of pyridoxamine as a dietary supplement; |
• | the extent to which our product candidates are approved for inclusion on formularies of hospitals and managed care organizations; |
• | whether our product candidates are designated under physician treatment guidelines for the treatment of the indications for which we have received regulatory approval; |
• | adverse publicity about our product candidates or favorable publicity about competitive products; |
• | convenience and ease of administration of our product candidates; |
• | potential product liability claims; and |
• | countries accepting the EMA and FDA approvals without study conduct in their respective countries or among a patient population representative of their respective country. |
If our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, patients, the medical community and healthcare payors, sufficient revenue may not be generated from these products and we may not become or remain profitable. In addition, efforts to educate the medical community and third‑party payors on the benefits of our product candidates may require significant resources and may never be successful.
We have no sales, marketing or distribution experience and we will have to invest significant resources to develop those capabilities or enter into acceptable third‑party sales and marketing arrangements.
We have no sales, marketing or distribution experience. To develop sales, distribution and marketing capabilities, we will have to invest significant amounts of financial and management resources, some of which will be committed prior to any confirmation that Pyridorin or any of our other product candidates will be approved. For product candidates where we decide to perform sales, marketing and distribution functions ourselves or through third parties, we could face a number of additional risks, including:
• | we or our third‑party sales collaborators may not be able to attract and build an effective marketing or sales force; |
• | the cost of securing or establishing a marketing or sales force may exceed the revenues generated by any products; and |
• | our direct sales and marketing efforts may not be successful. |
We may have limited or no control over the sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties.
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop certain of our product candidates and our financial condition and operating results.
Because developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products are expensive, we may seek to enter into collaborations with companies that have more experience. Additionally, if any of our product candidates receives marketing approval, we may enter into sales and marketing arrangements with third parties with respect to our unlicensed territories. If we are unable to enter into arrangements on acceptable terms, if at all, we may be unable to effectively market and sell our products in our target markets. We expect to face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement and they may require substantial resources to maintain. We may not be
48
successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of our product candidates.
When we collaborate with a third party for development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the future success of that product candidate to the third party. For example, we may relinquish the rights to Pyridorin in jurisdictions outside of the United States. Our collaboration partner may not devote sufficient resources to the commercialization of our product candidates or may otherwise fail in their commercialization. The terms of any collaboration or other arrangement that we establish may not be favorable to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of our product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a product candidate or research program under a collaboration arrangement, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development. If we are unable to reach agreements with suitable collaborators for our product candidates, we would face increased costs, we may be forced to limit the number of our product candidates we can commercially develop or the territories in which we commercialize them and we might fail to commercialize products or programs for which a suitable collaborator cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition will be materially and adversely affected.
The success of the company depends greatly on the success of Pyridorin’s development in diabetic nephropathy, and the company’s pipeline of product candidates beyond this lead indication is limited.
We are evaluating the application of an intravenous formulation of Pyridorin to specific types of acute renal injury in which pathogenic oxidative chemistries have been identified as likely causative factors in the onset, severity and progression of this condition. These include ischemia‑reperfusion and contrast‑dye‑induced acute renal injury, which can arise in cardiac and vascular surgeries. However, the intravenous formulation of Pyridorin has never been evaluated in a clinical setting and there is no clinical evidence that the therapy will be effective in additional indications. Moreover, the completion of development, securing of approval and commercialization of an intravenous formulation of Pyridorin for additional indications will require substantial additional funding and is prone to the risks of failure inherent in drug development. We cannot provide you any assurance that we will be able to successfully advance any of these indications through the development process. Even if we receive FDA approval to market an intravenous formulation of Pyridorin for additional indications, we cannot provide assurance that this will be successfully commercialized, widely accepted in the marketplace or more effective than other commercially available alternatives.
If serious adverse events or other undesirable side effects are identified during the development of Pyridorin for one indication, we may need to abandon our development of Pyridorin for other indications.
Product candidates in clinical stages of development have a high risk of failure. We cannot predict when or if Pyridorin will prove effective or safe in humans or will receive regulatory approval. To date, the most common side effects observed in clinical trials of Pyridorin were a slight increase in diarrhea and constipation. New side effects could, however, be identified as we expand the size of our clinical trials and apply Pyridorin to other indications. If new side effects are found during the development of Pyridorin for any indication, if known side effects are shown to be more severe than previously observed or if Pyridorin is found to have other unexpected characteristics, we may need to abandon our development of Pyridorin for kidney disease in patients with type 2 diabetes and other potential indications. Additional or more severe adverse side effects with respect to Pyridorin may develop in future clinical trials, which could delay or preclude regulatory approval of Pyridorin or limit its commercial use.
Risks Relating to Our Business and Strategy
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have competitors in the United States, Europe and other jurisdictions, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies and universities and other research institutions. Many of our competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to
49
in‑license novel compounds that could make the product candidates that we develop obsolete. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing drugs for the diseases that we are targeting before we do. Smaller or early‑stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Some of the pharmaceutical and biotechnology companies we expect to compete with include AbbVie Inc., Bayer Corporation, Bristol-Meyers Squibb, Thrasos Therapeutics, Inc., Genkyotex S.A., Janssen Pharmaceutical, Inc., Pfizer Inc., Chemocentryx, Inc., Eli Lilly and Company, and Mitsubishi Tanabe Pharma. In addition, many universities and private and public research institutes may become active in our target disease areas. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, technologies and drug products that are more effective or less costly than Pyridorin or any other product candidates that we are currently developing or that we may develop, which could render our products obsolete and noncompetitive.
We believe that our ability to successfully compete will depend on, among other things:
• | the results of our and our potential strategic collaborators’ clinical trials and preclinical studies; |
• | our ability to recruit and randomize patients for our clinical trials; |
• | the efficacy, safety and reliability of our product candidates; |
• | the speed at which we develop our product candidates; |
• | our ability to design and successfully execute appropriate clinical trials; |
• | our ability to maintain a good relationship with regulatory authorities; |
• | the timing and scope of regulatory approvals, if any; |
• | our ability to commercialize and market any of our product candidates that receive regulatory approval; |
• | the price of our products; |
• | adequate levels of reimbursement under private and governmental health insurance plans, including Medicare; |
• | our ability to protect intellectual property rights related to our products; |
• | our ability to manufacture and sell commercial quantities of any approved products to the market; and |
• | acceptance of our product candidates by physicians and other health care providers. |
If our competitors market products that are more effective, safer or less expensive or that reach the market sooner than our future products, if any, we may not achieve commercial success. In addition, the biopharmaceutical industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
We depend on third‑party contractors for a substantial portion of our operations and may not be able to control their work as effectively as if we performed these functions ourselves.
We outsource substantial portions of our operations to third‑party service providers, including the conduct of preclinical studies and clinical trials, collection and analysis of data, and manufacturing. Our agreements with third‑party service providers and CROs are on a study‑by‑study and project‑by‑project basis. Typically, we may terminate the agreements with notice and are responsible for the supplier’s previously incurred costs. In addition, any CRO that we retain will be subject to the FDA’s and EMA’s regulatory requirements and similar standards outside of the United States and Europe and we do not have control over compliance with these regulations by these providers. Consequently, if these providers do not adhere to applicable governing practices and standards, the development and commercialization of our product candidates could be delayed or stopped, which could severely harm our business and financial condition.
50
Because we have relied on third parties, our internal capacity to perform these functions is limited to management oversight. Outsourcing these functions involves the risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. Although we have not experienced any significant difficulties with our third‑party contractors, it is possible that we could experience difficulties in the future. In addition, the use of third‑party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. There are a limited number of third‑party service providers that specialize or have the expertise required to achieve our business objectives. Identifying, qualifying and managing performance of third‑party service providers can be difficult, time consuming and cause delays in our development programs. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor third‑party service providers. To the extent we are unable to identify, retain and successfully manage the performance of third‑party service providers in the future, our business may be adversely affected, and we may be subject to the imposition of civil or criminal penalties if their conduct of clinical trials violates applicable law.
A variety of risks associated with our possible international business relationships could materially adversely affect our business.
We may enter into agreements with other third parties for the development and commercialization of Pyridorin or our other product candidates in international markets. International business relationships subject us to additional risks that may materially adversely affect our ability to attain or sustain profitable operations, including:
• | differing regulatory requirements for drug approvals internationally; |
• | potentially reduced protection for intellectual property rights; |
• | potential third‑party patent rights in countries outside of the United States; |
• | the potential for so‑called “parallel importing,” which is what occurs when a local seller, faced with relatively high local prices, opts to import goods from another jurisdiction with relatively low prices, rather than buying them locally; |
• | unexpected changes in tariffs, trade barriers and regulatory requirements; |
• | economic weakness, including inflation, or political instability, particularly in non‑U.S. economies and markets, including several countries in Europe; |
• | compliance with tax, employment, immigration and labor laws for employees traveling abroad; |
• | taxes in other countries; |
• | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
• | workforce uncertainty in countries where labor unrest is more common than in the United States; |
• | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
• | business interruptions resulting from geo‑political actions, including war and terrorism, or natural disasters, including earthquakes, volcanoes, typhoons, floods, hurricanes and fires. |
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As of March 24, 2015, we had eleven employees. As we increase the number of ongoing product development programs and advance our product candidates through preclinical studies and clinical trials, we will need to increase our product development, scientific and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we may need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
51
• | successfully attract and recruit new employees or consultants with the expertise and experience we will require; |
• | manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites, among multiple vendors and countries; |
• | develop a marketing and sales infrastructure; and |
• | continue to improve our operational, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel and consultants due to the intense competition for qualified personnel and consultants among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel and consultants to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our industry has experienced a high rate of turnover of management personnel in recent years. We are highly dependent on the development, regulatory, commercialization and business development expertise of Pierre Legault, our chief executive officer; John P. Hamill, our chief financial officer; J. Wesley Fox, our president and chief scientific officer; and our other key employees and consultants. If we lose one or more of our executive officers or key employees or consultants, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers or key employees or consultants may terminate their employment at any time. Replacing executive officers, key employees and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire and retain employees and consultants from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel and consultants. Our failure to retain key personnel or consultants could materially harm our business.
In addition, we have scientific and clinical advisors and consultants who assist us in formulating our research, development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us and typically they will not enter into non‑compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
Failure to continue improving our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with the Sarbanes‑Oxley Act of 2002, and the related rules and regulations of the SEC, expanded disclosure requirements, accelerated reporting requirements and more complex accounting rules. Company responsibilities required by the Sarbanes‑Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
We have implemented a system of internal controls over financial reporting and preparing the documentation necessary to perform the evaluation needed to comply with Section 404(a) of the Sarbanes‑Oxley Act. We may need to retain additional finance capabilities and build our financial infrastructure as a public company, including complying with the requirements of Section 404 of the Sarbanes‑Oxley Act. We plan to continue improving our financial infrastructure with the enhancement of internal controls and additional training for our financial and accounting staff.
Section 404(a) of the Sarbanes‑Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC. However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we have and intend to continue to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that
52
are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes‑Oxley Act. We may continue to take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) December 31, 2019; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with the regulations of the FDA and non‑U.S. regulators, provide accurate information to the FDA and non‑U.S. regulators, comply with health care fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self‑dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or our potential future collaborators by participants enrolled in our clinical trials, patients, health care providers or others using, administering or selling our products. If we cannot successfully defend ourselves against any such claims, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
• | withdrawal of clinical trial participants; |
• | termination of clinical trial sites or entire trial programs; |
• | costs of related litigation; |
• | substantial monetary awards to patients or other claimants; |
• | decreased demand for our product candidates and loss of revenues; |
• | impairment of our business reputation; |
• | diversion of management and scientific resources from our business operations; and |
• | the inability to commercialize our product candidates. |
We currently maintain products liability insurance ($20 million coverage) which covers our clinical trials liability. Our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any
53
products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash resources and adversely affect our business.
We purchase commercially available insurance at limits suggested by our insurance broker based on our business operations. Our insurance policies do not cover all of our business exposures thus leaving us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability ($2 million coverage), umbrella liability ($2 million coverage), employment practices liability, property, auto, workers’ compensation, and directors’ and officers’ insurance. We currently maintain products liability insurance ($20 million coverage) which covers our clinical trials liability. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
If we engage in an acquisition, reorganization or business combination, we will incur a variety of risks that could adversely affect our business operations or our stockholders.
From time to time we have considered, and we will continue to consider in the future, strategic business initiatives intended to further the expansion and development of our business. These initiatives may include acquiring businesses, technologies or products or entering into a business combination with another company. If we pursue such a strategy, we could, among other things:
• | issue equity securities that would dilute our current stockholders’ percentage ownership; |
• | incur substantial debt that may place strains on our operations; |
• | spend substantial operational, financial and management resources to integrate new businesses, technologies and products; |
• | assume substantial actual or contingent liabilities; |
• | reprioritize our development programs and even cease development and commercialization of our product candidates; or |
• | merge with, or otherwise enter into a business combination with, another company in which our stockholders would receive cash and/or shares of the other company on terms that certain of our stockholders may not deem desirable. |
Although we intend to evaluate and consider acquisitions, reorganizations and business combinations in the future, we have no agreements or understandings with respect to any acquisition, reorganization or business combination at this time.
Risks Relating to Our Intellectual Property
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If our patent position does not adequately protect our product candidates, others could compete against us more directly, which would harm our business, possibly materially.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third‑party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our product candidates, discovery programs and processes.
54
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved.
No consistent policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the United States or in many jurisdictions outside of the United States. Changes in either the patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that may be issued from the applications we currently or, may in the future, own or license from third parties. Further, if any patents we obtain or license are deemed invalid and unenforceable, our ability to commercialize or license our technology could be adversely affected.
In the future others may file patent applications covering products and technologies that are similar, identical or competitive to ours or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed or in‑licensed by us, or that we or our licensors will not be involved in interference, opposition or invalidity proceedings before U.S. or non‑U.S. patent offices.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
• | others may be able to develop a platform similar to, or better than, ours in a way that is not covered by the claims of our patents; |
• | others may be able to make compounds that are similar to our product candidates but that are not covered by the claims of our patents; |
• | we might not have been the first to make the inventions covered by our pending patent applications; |
• | we might not have been the first to file patent applications for these inventions; |
• | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
• | any patents that we obtain may not provide us with any competitive advantages; |
• | we may not develop additional proprietary technologies that are patentable; or |
• | the patents of others may have an adverse effect on our business. |
As of December 31, 2014, we were the owner of record or the licensee of 28 issued or granted U.S. and non‑U.S. patents relating to Pyridorin with claims directed to methods of making Pyridorin, and methods of using Pyridorin in various indications. We were also the owner of record or licensee of three pending U.S. and non‑U.S. patent applications relating to Pyridorin in these areas. In addition, as of December 31, 2014, we were the owner of record of two pending U.S. and non‑U.S. applications relating to our product candidates other than Pyridorin, with claims directed to pharmaceutical compounds, pharmaceutical compositions and methods of using these compounds in various indications.
Patents covering methods of using Pyridorin expire in 2024 if the appropriate maintenance fee renewal, annuity, or other government fees are paid, unless a patent term extension based on regulatory delay is obtained. We expect that expiration in 2016 of some of our method‑of‑use patents, or their foreign equivalents, covering use of Pyridorin for treating diabetic nephropathy will have a limited impact on our ability to protect our intellectual property in the United States, Europe, and Canada, where we have additional issued patents covering this use that extend until 2024. In other countries, our patent protection covering use of Pyridorin for treating diabetic nephropathy will expire in 2016. We will attempt to mitigate the effect of patent expiration by seeking data exclusivity, or the foreign equivalent thereof, in conjunction with product approval, as well as by filing additional patent applications covering improvements in our intellectual property.
We expect that the other patents and patent applications for the Pyridorin portfolio, if issued, and if the appropriate maintenance, renewal, annuity or other governmental fees are paid, would expire from 2016 to 2035. We own pending applications in the United States and Europe covering Pyridorin analogs, and uses of such analogs as therapeutics to treat a variety of disorders, including kidney disorders such as nephropathy. Patent protection, to the extent it issues, would be expected to extend to 2027, unless a patent term extension based on regulatory delay is obtained.
55
Due to the patent laws of a country, or the decisions of a patent examiner in a country, or our own filing strategies, we may not obtain patent coverage for all of our product candidates or methods involving these candidates in the parent patent application. We plan to pursue divisional patent applications or continuation patent applications in the United States and other countries to obtain claim coverage for inventions which were disclosed but not claimed in the parent patent application.
We may also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or feasible. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know‑how.
Pyridorin does not have composition of matter patent protection.
Although we own and exclusively license patents and patent applications with claims directed to the methods of use of Pyridorin (pyridoxamine) to treat diabetic nephropathy and other conditions, and methods for its synthesis, we are unaware of any composition of matter patent protection for Pyridorin in the United States or elsewhere. As a result, competitors may be able to offer and sell products including pyridoxamine so long as these competitors do not infringe any other patents that we or third parties hold, including synthesis and method of use patents. However, method of use patents, in particular, are more difficult to enforce than composition of matter patents because of the risk of off‑label sale or use of the subject compounds. Physicians are permitted to prescribe an approved product for uses that are not described in the product’s labeling. Although off‑label prescriptions may infringe our method of use patents, the practice is common across medical specialties and such infringement is difficult to prevent or prosecute. Off‑label sales would limit our ability to generate revenue from the sale of Pyridorin, if approved for commercial sale.
In addition, other third parties have obtained patents in the United States and elsewhere relating to methods of use of pyridoxamine for the treatment of certain diseases. As a result, it is possible that we could face competition from third party products that have pyridoxamine as the active pharmaceutical ingredient. If a third party were to obtain FDA approval in the United States for the use of pyridoxamine, or regulatory approval in another jurisdiction, for an indication before we did, such third party would be first to market and could establish the price for pyridoxamine in these jurisdictions. This could adversely impact our ability to implement our pricing strategy for the product and may limit our ability to maximize the commercial potential of Pyridorin in the United States and elsewhere. The presence of a lower priced competitive product with the same active pharmaceutical ingredients as our product could lead to use of the competitive product for our diabetic nephropathy indication. This could lead to pricing pressure for Pyridorin, which would adversely affect our ability to generate revenue from the sale of Pyridorin for treating diabetic nephropathy. This would also limit the length of data exclusivity and patent term extension available if we later obtain approval to market Pyridorin for treating diabetic nephropathy.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
If we choose to go to court to stop another party from using the inventions claimed in any patents we obtain, that individual or company has the right to ask the court to rule that such patents are invalid or should not be enforced against that third party. These lawsuits are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to such patents. In addition, in recent years the U.S. Supreme Court modified some tests used by the U.S. Patent and Trademark Office (USPTO) in granting patents over the past 20 years, which may decrease the likelihood that we will be able to obtain patents and increase the likelihood of challenge of any patents we obtain or license.
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidates.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. We cannot guarantee that our products, or manufacture or use of our product candidates, will not infringe third‑party patents. Furthermore, a third party may claim that we or our manufacturing or commercialization collaborators are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities,
56
including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. There is a risk that a court would decide that we or our commercialization collaborators are infringing the third party’s patents and would order us or our collaborators to stop the activities covered by the patents. In that event, we or our commercialization collaborators may not have a viable way around the patent and may need to halt commercialization of the relevant product. In addition, there is a risk that a court will order us or our collaborators to pay the other party damages for having violated the other party’s patents. In the future, we may agree to indemnify our commercial collaborators against certain intellectual property infringement claims brought by third parties. The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and divert management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, which may not be available, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non‑infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may incur substantial monetary damages, encounter significant delays in bringing our product candidates to market and be precluded from manufacturing or selling our product candidates.
We cannot be certain that others have not filed patent applications for technology covered by our pending applications, or that we were the first to invent the technology, because:
• | some patent applications in the United States may be maintained in secrecy until the patents are issued; |
• | patent applications in the United States are typically not published until 18 months after the priority date; and |
• | publications in the scientific literature often lag behind actual discoveries. |
Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent application may have priority over our patent applications, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions. Other countries have similar laws that permit secrecy of patent applications, and may be entitled to priority over our applications in such jurisdictions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non‑compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We employ an outside firm and rely on our outside counsel to pay these fees due to non‑U.S. patent agencies and this outside firm has systems in place to ensure compliance on payment of fees. The USPTO and various non‑U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
57
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers. If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and time‑consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Failure to secure trademark registrations could adversely affect our business.
If we seek to register additional trademarks, our trademark applications may not be allowed for registration or our registered trademarks may not be maintained or enforced. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many other jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
If the FDA, EMA or other regulatory agencies fail to monitor and enforce the illegal sale of pyridoxamine as a dietary supplement, the commercial success of Pyridorin may be limited.
Following the publication of the initial Phase 2 studies that evaluated pyridoxamine therapy in diabetic nephropathy patients, a number of dietary supplement companies began selling pyridoxamine over the internet. In January 2009, the FDA ruled that pyridoxamine is an investigational drug candidate not eligible for sale as a dietary supplement. A significant decline in product availability occurred after the issuance of the above mentioned FDA ruling. However, approximately 5 sites on the internet can be found that continue to illegally sell pyridoxamine. In at least one example, the FDA has taken action against a dietary supplement company and prohibited such company from selling an FDA approved active drug ingredient in a dietary supplement. However, there is no guarantee that the FDA will take action against other companies that illegally sell pyridoxamine after its approval. Food and dietary supplements in Europe are regulated by Directive 2002/46/EC, European Commission, Health and Consumers Directorate‑General. Those approved are listed in Annex I and II of Directive 2002/46/EC. Pyridoxamine is not included on either list, and therefore the sale of pyridoxamine in foods and supplements in Europe is not permitted. The European Commission, Health and Consumers Directorate‑General has indicated to us in April of this year that no applications for pyridoxamine have been received and that any new product intended for preventing, curing or treating diseases, would fall under the scope of medicinal products and not dietary supplements products. We are not aware of any direct action that this agency has taken against a company illegally selling an EMA approved drug for preventing, curing or treating disease, in the European Union. It is possible that this agency would not be successful in prohibiting such sales. We will rely on the FDA, EMA and other regulatory agencies to enforce laws and rulings that prohibit the illegal sale of pyridoxamine as a dietary supplement. If these agencies fail to enforce such laws and rulings, the commercial success of Pyridorin may be limited.
58
Risks Relating to Owning Our Common Stock
The trading market in our common stock has been extremely limited and substantially less liquid than the average trading market for a stock quoted on the NASDAQ Capital Market.
Since our initial listing on the NASDAQ Capital Market on February 11, 2014, the trading market in our common stock has been limited. The quotation of our common stock on the NASDAQ Capital Market does not assure that a meaningful, consistent and liquid trading market currently exists. We cannot predict whether a more active market for our common stock will develop in the future. An absence of an active trading market could adversely affect our stockholders’ ability to sell our common stock at current market prices in short time periods, or possibly at all. Additionally, market visibility for our common stock may be limited and such lack of visibility may have a depressive effect on the market price for our common stock. As of March 24, 2015, 77.7% of our outstanding shares of common stock were held by our officers, directors, beneficial owners of 5% or more of our securities and their respective affiliates, which adversely affects the liquidity of the trading market for our common stock, in as much as federal securities laws restrict sales of our shares by these stockholders. If our affiliates continue to hold their shares of common stock, there will be limited trading volume in our common stock, which may make it more difficult for investors to sell their shares or increase the volatility of our stock price.
Our share price may be volatile, which could subject us to securities class action litigation and result in substantial losses to our stockholders.
The trading price of our common stock is highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. Our stock price is likely to remain volatile. The stock market in general and the market for pharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the price at which it was purchased. The market price for our common stock may be influenced by many factors, including:
• | results of our clinical trials; |
• | results of clinical trials of our competitors’ products; |
• | regulatory actions with respect to our products or our competitors’ products; |
• | actual or anticipated fluctuations in our financial condition and operating results; |
• | actual or anticipated changes in our growth rate relative to our competitors; |
• | actual or anticipated fluctuations in our competitors’ operating results or changes in their growth rate; |
• | competition from existing products or new products that may emerge; |
• | announcements by us, our potential future collaborators or our competitors of significant acquisitions, strategic collaborations, joint ventures, or capital commitments; |
• | issuance of new or updated research or reports by securities analysts; |
• | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
• | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
• | additions or departures of key management or scientific personnel; |
• | disputes or other developments related to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
• | announcement or expectation of additional financing efforts; |
• | sales of our common stock by us, our insiders or our other stockholders; |
59
• | market conditions for biopharmaceutical stocks in general; and |
• | general economic and market conditions. |
Furthermore, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of shares of our common stock, regardless of our actual operating performance. In addition, such fluctuations could subject us to securities class action litigation, which could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business. As a result of this volatility, our stockholders could incur substantial losses.
We have a significant stockholder, which will limit your ability to influence corporate matters and may give rise to conflicts of interest.
Care Capital III LLC, together with its affiliates (collectively, Care Capital) is our largest stockholder. As of March 24, 2015, Care Capital beneficially owned 4,241,097 shares of our common stock. The shares of common stock beneficially owned by Care Capital represent approximately 47.8% of our outstanding shares of common stock. Accordingly, Care Capital exerts significant influence over us and any action requiring the approval of the holders of our common stock, including the election of directors and approval of significant corporate transactions. This concentration of voting power makes it less likely that any other holder of common stock or directors of our business will be able to affect the way we are managed and could delay or prevent an acquisition of us on terms that other stockholders may desire. In addition, if Care Capital obtains a majority of our common stock, Care Capital would be able to control all matters submitted to our stockholders for approval, as well as our management and affairs. For example, Care Capital would be able to control the election of directors, amendments to our organizational documents and approval of any merger, consolidation, sale of all or substantially all of our assets or other business combination or reorganization. In addition, if Care Capital obtains a majority of our common stock, we would be deemed a “controlled company” for purposes of NASDAQ listing requirements. Under NASDAQ rules, a “controlled company” may elect not to comply with certain NASDAQ corporate governance requirements, including (i) the requirement that a majority of our board of directors consist of independent directors, (ii) the requirement that the compensation of our officers be determined or recommended to the board by a majority of independent directors or a compensation committee that is composed entirely of independent directors, and (iii) the requirement that director nominees be selected or recommended to the board by a majority of independent directors or a nominating committee that is composed of entirely independent directors.
Furthermore, the interests of Care Capital may not always coincide with your interests or the interests of other stockholders and Care Capital may act in a manner that advances its best interests and not necessarily those of other stockholders, including seeking a premium value for its common stock, and might affect the prevailing market price for our common stock. Our board of directors, which currently consists of six directors, including two designated by Care Capital, has the power to set the number of directors on our board from time to time. Richard J. Markham and Robert R. Seltzer, partners at Care Capital, are members of our board of directors and some of its committees.
Being a public company has increased our expenses and administrative burden.
As a public company, we are incurring, and will continue to incur significant legal, insurance, accounting and other expenses. In addition, we are required to bear all of the internal and external costs of preparing and distributing periodic public reports in compliance with our obligations under the securities laws.
In addition, laws, regulations and standards applicable to public companies relating to corporate governance and public disclosure, including the Sarbanes‑Oxley Act and related regulations implemented by the SEC and the NASDAQ Stock Market, are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment will result in increased general and administrative expenses and may divert management’s time and attention from product development activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed. In connection with our initial public offering, we increased our directors’ and officers’ insurance coverage, which increased our insurance cost. In
60
the future, it will be more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.
We are an “emerging growth company” and we will continue to avail ourselves of the reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the JOBS Act) and we have and intend to continue to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes‑Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we have and may continue to rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) December 31, 2019; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
The Sarbanes‑Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. Commencing with our annual report on Form 10‑K for the year ending December 31, 2015, we will be required, under Section 404 of the Sarbanes‑Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes‑Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company, as defined in the JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the independent registered public accounting firm’s requirement to attest to the effectiveness of our internal controls over financial reporting.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge, and compile the system and process documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begin its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
61
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision‑making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosure due to error or fraud may occur and not be detected.
Future sales of our common stock, or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing well.
Sales of substantial amounts of our common stock, or the perception that these sales may occur, could materially and adversely affect the price of our common stock and could impair our ability to raise capital through the sale of additional equity securities.
We had outstanding 8,862,114 shares of common stock as of December 31, 2014, 4,248,097 of which are restricted securities that may be sold only in accordance with the resale restrictions under Rule 144 of the Securities Act of 1933, as amended. In addition, as of December 31, 2014, we had outstanding options to purchase 1,272,581 shares of our common stock, 17,000 shares of common stock were issuable upon the settlement of outstanding restricted stock units and we had outstanding warrants to purchase 118,603 shares of our common stock. Shares issued upon the exercise of stock options or upon the settlement of outstanding restricted stock units generally will be eligible for sale in the public market, except that affiliates will continue to be subject to volume limitations and other requirements of Rule 144 under the Securities Act. The issuance or sale of such shares could depress the market price of our common stock.
In the future, we also may issue our securities if we need to raise additional capital. The number of new shares of our common stock issued in connection with raising additional capital could constitute a material portion of the then-outstanding shares of our common stock. We are unable to predict the effect that transactions on our stock may have on the prevailing market price of our common stock.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our common stock will depend on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will continue to cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our share price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our share price or trading volume to decline.
NASDAQ may delist our securities from its exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
If we fail to maintain the listing of our common stock on the NASDAQ Capital Market, the liquidity for our common stock would be significantly impaired, which may substantially decrease the trading price of our common stock. We cannot assure you that, in the future, our securities will meet the continued listing requirements to be listed on NASDAQ. If NASDAQ delists our common stock from trading on its exchange, we could face significant material adverse consequences, including:
• | a limited availability of market quotations for our securities; |
• | a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly resulting in a reduced level of trading activity in the secondary trading market for our common stock; |
62
• | a limited amount of news and analyst coverage for our company; and |
• | a decreased ability to issue additional securities or obtain additional financing in the future. |
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker‑dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not obtain or retain a listing on The NASDAQ Capital Market and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker‑dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker‑dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser’s written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks; and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
Some provisions of our charter documents and Delaware law may have anti‑takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders. These provisions include:
• | authorizing the issuance of “blank check” convertible preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
• | limiting the removal of directors by the stockholders; |
• | creating a staggered board of directors; |
• | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
• | eliminating the ability of stockholders to call a special meeting of stockholders; |
• | permitting our board of directors to accelerate the vesting of outstanding equity awards upon certain transactions that result in a change of control; and |
• | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
These provisions may also frustrate or prevent any attempts by our stockholders to replace or remove our current management or members of our board of directors. In addition, we are subject to Section 203 of the Delaware General Corporation Law (DGCL), which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by our board of directors. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders. Further, other provisions of Delaware law may also discourage, delay or prevent someone from acquiring us or merging with us.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful stockholder claims against us and may reduce the amount of money available to us.
As permitted by Section 102(b)(7) of the DGCL, our restated certificate of incorporation limits the liability of our directors to the fullest extent permitted by law. In addition, as permitted by Section 145 of the DGCL, our restated certificate of
63
incorporation and restated bylaws provide that we shall indemnify, to the fullest extent authorized by the DGCL, each person who is involved in any litigation or other proceeding because such person is or was a director or officer of our company or is or was serving as an officer or director of another entity at our request, against all expense, loss or liability reasonably incurred or suffered in connection therewith. Our restated certificate of incorporation provides that the right to indemnification includes the right to be paid expenses incurred in defending any proceeding in advance of its final disposition, provided, however, that such advance payment will only be made upon delivery to us of an undertaking, by or on behalf of the director or officer, to repay all amounts so advanced if it is ultimately determined that such director is not entitled to indemnification. If we do not pay a proper claim for indemnification in full within 60 days after we receive a written claim for such indemnification, except in the case of a claim for an advancement of expenses, in which case such period is 20 days, our restated certificate of incorporation and our restated bylaws authorize the claimant to bring an action against us and prescribe what constitutes a defense to such action.
Section 145 of the DGCL permits a corporation to indemnify any director or officer of the corporation against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reason to believe his or her conduct was unlawful. In a derivative action, (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought shall determine that the defendant is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
The rights conferred in the restated certificate of incorporation and the restated bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons. We have entered into or plan to enter into indemnification agreements with each of our officers and directors.
The above limitations on liability and our indemnification obligations limit the personal liability of our directors and officers for monetary damages for breach of their fiduciary duty as directors by shifting the burden of such losses and expenses to us. Although we have increased the coverage under our directors’ and officers’ liability insurance, certain liabilities or expenses covered by our indemnification obligations may not be covered by such insurance or the coverage limitation amounts may be exceeded. As a result, we may need to use a significant amount of our funds to satisfy our indemnification obligations, which could severely harm our business and financial condition and limit the funds available to stockholders who may choose to bring a claim against our company.
We do not anticipate paying cash dividends, and accordingly, stockholders must rely on stock appreciation for any return on their investment.
We do not anticipate paying cash dividends in the future. As a result, only appreciation of the market price of our common stock, which may never occur, will provide a return to stockholders. Investors seeking cash dividends should not invest in our common stock.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2014, we had federal net operating loss carryforwards (NOLs) of $31.8 million million which expire from 2024 through 2034. Our ability to utilize our NOLs may be limited under Section 382 of the Internal Revenue Code. The limitations apply if an ownership change, as defined by Section 382, occurs. Generally, an ownership change occurs when certain shareholders increase their aggregate ownership by more than 50 percentage points over their lowest ownership percentage in a testing period (typically three years). Although we have not undergone a Section 382 analysis, it is possible that the utilization of the NOLs, could be substantially limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be utilized against future taxes. As a result, we may not be able to take full advantage of these carryforwards for federal and state tax purposes. Future changes in stock ownership may also trigger an ownership change and, consequently, a Section 382 limitation.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
64
Item 2. PROPERTIES
Facilities
Since November 24, 2014, our corporate headquarters and clinical development operations have been located in 5,514 square feet of office space located at 3200 Beechleaf Court, Raleigh, North Carolina, pursuant to a lease agreement that commenced on December 1, 2014 and terminates on May 31, 2020. The lease agreement gives us the right and option to extend the lease term for an additional 36 months contingent upon certain conditions set forth in the lease agreement.
We believe that our facility is suitable and adequate for our current needs.
Item 3. LEGAL PROEEDINGS
We are not a party to any legal proceedings and we are not aware of any claims or actions pending or threatened against us. In the future, we might from time to time become involved in litigation relating to claims arising from our ordinary course of business.
Item 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
Item 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock began trading on the NASDAQ Capital Market on February 11, 2014, under the symbol “NRX”. Prior to that there was no public market for our common stock. Shares sold in our initial public offering on February 10, 2014, were priced at $12.00 per share.
The following table sets forth the high and low sales prices for the common stock, as reported on the NASDAQ Capital Market since our common stock commenced public trading on February 11, 2014:
Price Range | ||||||
2014 | High | Low | ||||
1st Quarter (beginning February 11, 2014) | $ | 13.00 | $ | 7.26 | ||
2nd Quarter | 8.98 | 5.00 | ||||
3rd Quarter | 6.09 | 3.96 | ||||
4th Quarter | 17.98 | 4.00 | ||||
2015 | ||||||
1st Quarter (through March 13, 2015) | $ | 12.94 | $ | 6.26 | ||
On March 13, 2015, the closing price for our common stock as reported on the NASDAQ Capital Market was $7.70.
Comparative Stock Performance Graph
The following graph illustrates a comparison of the total cumulative stockholder return on our common stock for the period February 11, 2014 to December 31, 2014, to two indices: the NASDAQ Composite Index and the NASDAQ Pharmaceutical Index. The graph assumes an initial investment of $100 on February 11, 2014, in our common stock, the stocks comprising the NASDAQ Composite Index, and the stocks comprising the NASDAQ Pharmaceutical Index. Historical stockholder return is not necessarily indicative of the performance to be expected for any future periods.
65
Holders of Record
As of March 16, 2015, there were approximately 24 stockholders of record of the 8,863,614 outstanding shares of our common stock, which excludes stockholders whose shares were held in nominee or street name by brokers.
Dividends
We have not paid dividends to our stockholders since our inception and we do not plan to pay cash dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance the growth of our Company.
Securities Authorized for issuance under Equity Compensation Plans
The information required by Item 5 of Form 10‑K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report on Form 10-K.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
We did not purchase any of our registered equity securities during the period covered by this Annual Report on Form 10‑K.
Use of Proceeds from Registered Securities
None.
66
Item 6. SELECTED FINANCIAL DATA
Not required as we are a smaller reporting company.
Item 2. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in the forward-looking statements as a result of certain factors. We discuss factors that we believe could cause or contribute to these differences below and elsewhere in this report including those set forth under Item 1.A. "Risk Factors" in this Annual Report on Form 10-K.
Overview
We are a pharmaceutical company focused on the development of therapeutics to treat kidney disease, an area of significant unmet medical need. Since our inception, we have collaborated with the world’s leading experts in kidney disease and leveraged our knowledge of pathogenic oxidative chemistries to build a strong portfolio of intellectual property and to advance the development of our drug candidates. We believe that our comprehensive effort to develop a new generation of therapeutics that target kidney disease provides us with a leadership position in this large and attractive market.
We have devoted substantially all of our resources to development efforts relating to our product candidate, including conducting clinical trials of our product candidate, providing general and administrative support for these operations and protecting our intellectual property. We do not have any products approved for sale and have not generated any revenue from product sales. We have funded our operations primarily through proceeds from our initial public offering, or IPO, and the private placement of preferred stock, common stock, convertible notes and a term loan. In February 2014, we completed our IPO pursuant to a registration statement on Form S-1, and raised approximately $33.4 million in net proceeds, after deducting underwriting discounts, commissions and offering expenses.
We have incurred net losses in each year since our inception in 2004. Our net losses for the years ended December 31, 2014 and 2013 were $16.8 million and $6.3 million, respectively. As of December 31, 2014, we had an accumulated deficit of approximately $57.8 million. Our net losses have resulted primarily from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations and from changes in the value of our preferred stock warrant liability which was settled in February 2014 upon completion of our IPO.
We expect to continue to incur significant expenses and have increasing operating losses for at least the next several years. We anticipate that our expenses will increase substantially as we:
• | continue the development of our lead product candidate, Pyridorin, for the treatment of diabetic nephropathy in patients with type 2 diabetes including the completion of Phase 3 clinical trial activities ; |
• | complete the development of an intravenous formulation of Pyridorin for the treatment of AKI; |
• | seek to obtain regulatory approvals for Pyridorin; |
• | outsource the commercial manufacturing of Pyridorin for any indications for which we receive regulatory approval; |
• | contract with third parties for the sales, marketing and distribution of Pyridorin for any indications for which we receive regulatory approval; |
• | maintain, expand and protect our intellectual property portfolio; |
• | continue our research and development efforts; |
67
• | add operational, financial and management information systems and personnel, including personnel to support our product development and commercialization efforts; and |
• | continue to operate as a public company. |
We do not expect to generate revenue from product sales unless and until we successfully complete development and obtain marketing approval for one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Accordingly, we will need to raise additional capital prior to the commercialization of Pyridorin or any other product candidate. Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our operating activities through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. However, we may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all. Our failure to raise capital or enter into such other arrangements as and when needed would have a negative impact on our financial condition and our ability to develop our product candidates.
Financial Overview
Revenue
We have not generated any revenue since our inception on May 25, 2004. Our ability to generate revenue in the future will depend almost entirely on our ability to successfully develop, obtain regulatory approval for and commercialize Pyridorin in the United States.
Research and Development Expenses
Our research and development activities have included conducting nonclinical studies and clinical trials, manufacturing development efforts and activities related to regulatory filings for Pyridorin. We recognize research and development expenses as they are incurred. Our research and development expenses consist primarily of:
• | salaries and related overhead expenses for personnel in research and development functions, including costs related to stock options or other stock-based compensation; |
• | fees paid to consultants and CROs for our nonclinical and clinical trials, and other related clinical trial fees, including investigator grants, laboratory work, clinical trial database management, clinical trial material management and statistical compilation and analysis; |
• | costs related to acquiring and manufacturing clinical trial materials; and |
• | costs related to compliance with regulatory requirements. |
We plan to increase our research and development expenses for the foreseeable future as we continue the development of Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes, AKI and other indications, subject to the availability of additional funding.
The table below summarizes our direct research and development expenses for Pyridorin for the periods indicated. Our direct research and development expenses consist principally of costs paid to third-party service providers, including fees paid to CROs, investigative sites, consultants, central laboratories and other vendors in connection with our clinical trials, and costs related to acquiring and manufacturing clinical trial materials. We do not allocate personnel related expenses including salaries and stock-based compensation or other indirect costs related to our research and development function to specific product candidates.
68
Year Ended December 31, | ||||||
(in thousands) | 2014 | 2013 | ||||
Direct research and development expense | $ | 8,417 | $ | — | ||
Personnel costs | 1,676 | 941 | ||||
Indirect research and development expense | 1,171 | 539 | ||||
Total research and development expense | $ | 11,264 | $ | 1,480 | ||
The successful development of our clinical and preclinical product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing or costs of the efforts that will be necessary to complete the remainder of the development of any of our clinical or preclinical product candidates or the period, if any, in which material net cash inflows from these product candidates may commence. This is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
• | the scope, rate of progress and expense of our ongoing, as well as any additional, clinical trials and other research and development activities; |
• | future clinical trial results; and |
• | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of a product candidate could result in a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
Pyridorin
Our research and development resources are primarily focused on the Phase 3 Pyridorin program and our other planned clinical and nonclinical studies and other work needed to submit Pyridorin for AKI, as well as the treatment of diabetic nephropathy in patients with type 2 diabetes for regulatory approval in the United States and Europe. We have incurred and expect to continue to incur expense in connection with these efforts, including:
• | working with our CROs to complete our Phase 3 clinical program; |
• | working with third-party service providers to produce sufficient clinical trial supply for our Phase 3 clinical program and other contemplated trials; |
• | working with our clinical nephrology academic research organization that provides scientific and clinical oversight on the conduct of the Pyridorin Phase 3 program. |
In addition, we are evaluating the application of an intravenous formulation of Pyridorin to specific types of acute renal failure in which pathogenic oxidative chemistries have been identified as likely causative factors in the onset, severity and progression of this condition. These include contrast-dye and drug-induced acute renal injury, and ischemia-reperfusion acute renal injury, which can arise in cardiac and vascular surgeries. In connection with these efforts, we have incurred and expect to incur significant expenses relating to:
• | working with research institutions with expertise using animal models of various types of acute renal injury to conduct studies to determine where Pyridorin would have the most beneficial effect in ameliorating the severity and progression of the induced acute renal injury; and |
69
• | working with a third-party drug formulator to produce intravenous Pyridorin solutions for preclinical and clinical studies. |
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs for employees in executive, and finance functions. Other significant general and administrative expenses include facilities costs, insurance, accounting and legal services and other consulting services related to our corporate governance activities.
We expect that our general and administrative expenses may increase in the future as we expand our operating activities, maintain and expand our patent portfolio, and incur additional costs associated with public company support, including legal and accounting fees and director and officers' liability insurance.
Other Income (expense)
Other income consists of interest income earned on our cash and cash equivalents. Other expense includes interest expense accrued for our convertible notes, term loan and the change in value of our preferred stock warrant liability.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which we have prepared in accordance with generally accepted accounting principles in the United States (GAAP). The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. We evaluate these estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our financial statements appearing elsewhere in this Annual Report on Form 10-K, we believe that the following accounting policies are the most critical for fully understanding and evaluating our financial condition and results of operations.
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with selected service providers and make adjustments, if necessary. To date, we have not adjusted our estimate at any particular balance sheet date by any material amount. Examples of estimated accrued expenses include:
• | fees paid to CROs for management of our clinical trial activities |
• | fess paid to investigative sites in connection with clinical trials |
• | fess paid to contract manufacturers in connection with the production of clinical trial supplies; and |
• | professional services and fees. |
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the
70
level of effort to be expended in each period. If we do not accurately identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
Fair Value Measurements
The carrying amounts of certain of our financial instruments, including cash and cash equivalents and short-term investments are stated at fair value. We account for the fair value of our financial instruments in accordance with the provisions of the Fair Value Measurement topic of the Financial Accounting Standards Board Codification (the Codification).
Fair value is the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. We apply the market approach valuation technique for fair value measurements on a recurring basis and attempt to maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. All of our cash equivalents and short-term investments are measured using inputs classified at Level 1 or Level 2 within the fair value hierarchy. Level 1 inputs are quoted prices in active markets for identical assets. Level 2 inputs are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant inputs are observable in the market or can be corroborated by observable market data for substantially the full term of the assets. Level 3 inputs are unobservable inputs that are supported by little or no market activity and are significant to the fair value of the assets or liabilities. Where applicable, these models project future cash flows and discount the future amounts to a present value using market-based observable inputs obtained from various third-party data providers, including but not limited to, benchmark yields, interest rate curves, reported trades, broker/dealer quotes and market reference data.
Stock-Based Compensation
The provisions of the Compensation - Stock Compensation topic of the Codification establish accounting for stock-based awards exchanged for employee services. In accordance with this topic stock-based compensation cost is measured on the grant date, based on the fair value of the award, and is recognized as expense over the requisite employee service period.
We estimate the fair value of stock options and stock purchase rights using a Black-Scholes valuation model which require the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. We have opted to use the simplified method for estimating the expected term as provided by the SEC's Staff Accounting Bulletin No.107. The simplified method calculates the expected term as the average time-to-vesting and the contractual life of the options. The expected stock price volatility assumption was determined by examining the historical volatilities of a group of industry peers. The fair value of each option grant is estimated on the date of grant using the Black-Scholes option valuation model, and the resulting charge is expensed using the straight-line attribution method over the vesting period. Restricted stock units are measured at the fair value of our common stock on the date of grant and expensed over the period of vesting using the straight-line attribution approach. The Black-Scholes option-pricing model was developed for use in estimating the fair value of short-lived, exchange-traded options that have no vesting restrictions and are fully transferable.
Research and Development Expenses
Research and development expenses consist of costs associated with external research and development expenses incurred (i) under agreements with third-party investigative sites, where a substantial portion of our preclinical studies and all of our clinical trials are conducted, (ii) under the agreements with third-party manufacturing organizations, where a substantial portion of our clinical supplies are produced, and (iii) related to consultants and employee-related expenses.
JOBS Act
On April 5, 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (the Securities Act), for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates.
71
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including without limitation, (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (ii) complying with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) December 31, 2019; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Results of Operations
Comparison of the Year Ended December 31, 2014 and the Year Ended December 31, 2013
The following table summarizes our results of operations for each of the years ended December 31, 2014 and 2013, together with the changes in those items in dollars and as a percentage:
Years Ended December 31, | $ | |||||||||||||
(in thousands) | 2014 | 2013 | Change | % Change | ||||||||||
Expenses: | ||||||||||||||
Research and development | $ | 11,264 | $ | 1,480 | $ | 9,784 | 661.1 | % | ||||||
General and administrative | 5,323 | 1,026 | 4,297 | 418.8 | % | |||||||||
Loss from operations | (16,587 | ) | (2,506 | ) | 14,081 | (561.9 | )% | |||||||
Other income (expense): | ||||||||||||||
Change in value of preferred stock warrants | (140 | ) | (3,417 | ) | 3,277 | (95.9 | )% | |||||||
Interest expense | (140 | ) | (383 | ) | 243 | (63.4 | )% | |||||||
Interest income | 47 | 1 | 46 | — | % | |||||||||
Net loss | $ | (16,820 | ) | $ | (6,305 | ) | $ | 10,515 | (166.8 | )% | ||||
Research and Development Expenses
Research and development expenses were approximately $11.3 million and $1.5 million for the years ended December 31, 2014 and 2013, respectively. The increase in research and development expense of $9.8 million, or 661%, is primarily due to our Phase 3 clinical development activities for Pyridorin which began in 2014 and an increase in personnel-related expenses as a result of an increase in headcount.
General and Administrative Expenses
General and administrative expenses were approximately $5.3 million and $1.0 million for the years ended December 31, 2014 and 2013, respectively. The increase in general and administrative expenses of $4.3 million, or 419%, was primarily a result of an increase in personnel-related expenses, including non-cash stock based compensation expense and an increase in our corporate governance expenses, including our director and officer liability insurance and other professional fees incurred for operating as a public company.
Other Income (Expense)
Interest income for the year ended December 31, 2014 was approximately $47,000 from interest received on our cash, cash equivalents and investments. Interest expense for the year ended December 31, 2014 was for interest on our convertible notes payable and term loan. Interest expense for the year ended December 31, 2013 was for interest accrued on our convertible promissory notes, which were converted into common stock upon the closing of the IPO in February 2014. The change in fair value of our preferred stock warrant liability for the year ended December 31, 2013 was $3.4 million. The preferred stock warrant liability was settled upon the closing of the IPO.
72
Liquidity and Capital Resources
Sources of Liquidity
We have incurred losses and cumulative negative cash flows from operations since inception and as of December 31, 2014, we had an accumulated deficit of $57.8 million. We anticipate that we will continue to incur losses for at least the next several years. We expect that our research and development and general and administrative expenses will continue to increase and, as a result, we will need additional capital to fund our operations, which we may seek to obtain through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements.
We have funded our operations principally from the sale of common stock, preferred stock and convertible notes and debt. As of December 31, 2014, we had cash and cash equivalents and short-term investments of approximately $28.7 million. Cash in excess of immediate requirements is invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation. Currently, our funds are held in cash, money market bank accounts, and certificates of deposit held at various banks that do not exceed the Federal Deposit Insurance Corporation insurance limit.
On February 14, 2014, we completed our IPO and sold 3,100,000 shares of common stock at a price of $12.00 per share for total gross proceeds of $37.2 million, less underwriting discounts, commissions and offering expenses totaling $3.8 million.
Cash Flows
The following table sets forth the significant sources and uses of cash for the periods set forth below:
Years Ended December 31, | |||||||
(in thousands) | 2014 | 2013 | |||||
Net cash provided by (used in): | |||||||
Operating activities | $ | (13,638 | ) | $ | (2,712 | ) | |
Investing activities | (14,809 | ) | (12 | ) | |||
Financing activities | 40,293 | 4,532 | |||||
Net increase in cash and cash equivalents | $ | 11,846 | $ | 1,808 | |||
Operating Activities.
Net cash used in operating activities for the year ended December 31, 2014 was primarily related to our net loss from the operation of our business of $16.8 million, including expenses incurred for the development of Pyridorin, partially offset by changes in working capital including an increase in our accrued liabilities, and non-cash charges, including non-cash interest expense for our convertible notes, stock based compensation expense and changes in our preferred stock warrant liability. Cash used in operating activities for the year ended December 31, 2013 was primarily related to our net loss from the operation of our business of $6.3 million during the period, changes in working capital, including a decrease in our accrued liabilities, partially offset by non-cash charges, including interest for our convertible notes, stock based compensation expense and changes in our preferred stock warrant liability.
Investing Activities. Net cash used in investing activities during the year ended December 31, 2014 was primarily related to the purchase of available for sale investments. Net cash used in investing activities during the year ended December 31, 2013 was primarily related to purchases of equipment.
Financing Activities. Net cash provided by financing activities for the year ended December 31, 2014 consisted of approximately $33.4 million in net proceeds received from the issuance of common stock in our IPO and $6.8 million, net of issuance costs from borrowing under our term loan. Net cash provided by financing activities for the year ended December 31, 2013 consisted of approximately $4.6 million of net proceeds from the sale of convertible notes.
Credit Facilities
On November 20, 2014, we entered into a Loan Agreement with East West , pursuant to which East West agreed to extend the Initial Term Loan to us with an aggregate principal amount of $7.0 million and, subject to the terms and conditions
73
set forth in the Loan Agreement, a Second Term Loan with an aggregate principal amount of $5.0 million. Each term loan shall accrue interest at a rate of 2.25% per annum plus the greater of 3.25% or the current prime rate. As of December 31, 2014, the interest rate on the loan was 5.5%. As security for our obligations under the Loan Agreement, we granted East West a lien on substantially all of our assets, including owned and licensed intellectual property.
On November 20, 2014, East West funded the Initial Term Loan which provided us with approximately $6.9 million of net loan proceeds. The Loan Facility matures on October 1, 2018. Interest only payments are due during the first twelve months of the Initial Term Loan and beginning on November 1, 2015, we are required to make 36 equal monthly payments of principal and interest. The Interest Only Term may be extended under the Loan Agreement if certain conditions are met. Upon payment of the final monthly installment under the Loan Agreement, or the remaining balance in the case of a prepayment, we would pay an end-of-term fee of approximately $60,000.
As of December 31, 2014, $7.0 million of principal remains outstanding on the loan.
At our option, we may borrow the Second Term Loan on or before May 29, 2015, if we have met certain milestones for enrollment and recruitment of Phase 3 Pyridorin trial patients and achieved positive TQT cardiac safety study results. We may prepay each term loan in full with no prepayment penalty .As of the date hereof, the Company does not believe that it will meet the clinical milestones for the Second Term Loan. However, the Company has made a proposal to East West to amend the clinical milestones necessary for incurrence of the Second Term Loan.
The Loan Agreement contains customary representations and warranties and customary affirmative and negative covenants, including, among others, covenants that limit or restrict the our ability to incur indebtedness, grant liens, merge or consolidate, dispose of assets, make investments, make acquisitions, enter into certain transactions with affiliates, pay dividends or make distributions, or repurchase stock, in each case subject to customary exceptions for a loan facility of this size and type. In addition, the Loan Agreement contains customary events of default that entitle East West to cause any or all of our indebtedness under the Loan Agreement to become immediately due and payable. The events of default include, among others, non-payment, inaccuracy of representations and warranties, covenant defaults, the occurrence of a material adverse effect (as defined in the Loan Agreement), cross-default to material agreements, cross-default to material indebtedness, bankruptcy and insolvency, material judgment defaults, discontinuation of the Phase 3 Pyridorin trial and defaults related to certain actions taken against the us by the FDA or other equivalent governmental authority.
As of December 31, 2014, we were in compliance with all covenants under the Loan Agreement.
Pursuant to the terms of the Loan Agreement, we issued to East West warrants to purchase up to 56,603 shares of our common stock at an exercise price equal to $4.24 per share. The warrants are immediately exercisable and expire on November 20, 2021.
Future Funding Requirements
To date, we have not generated any revenue. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize Pyridorin or any of our other product candidates. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, our product candidates. We expect to continue to incur costs associated with operating as a public company. In addition, subject to obtaining regulatory approval of any of our product candidates, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate that we will need substantial additional funding in connection with our continuing operations.
Based upon our current operating plan and approximately $33.4 million of net proceeds received from our IPO completed in February 2014 and the $6.8 million received from our term loan, we believe that our existing cash, cash equivalents and short-term investments, will enable us to fund our operating expenses and capital expenditure requirements into early 2016. We intend to devote our cash to fund our Phase 3 Pyridorin program and our planned clinical trials and nonclinical studies and other work needed to submit applications for Pyridorin for the treatment of diabetic nephropathy in patients with type 2 diabetes for regulatory approval in the United States and Europe; to fund the pre-IND work for the program on an intravenous formulation of Pyridorin for AKI; and for general corporate purposes, general and administrative expenses, capital expenditures, working capital and prosecution and maintenance of our intellectual property. We have based our estimates on assumptions that may prove to be wrong, and we may use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our product
74
candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures necessary to complete the development of our product candidates.
Our future capital requirements will depend on many factors, including:
• | the progress, costs, results of and timing of our Phase 3 Pyridorin program for the treatment of diabetic nephropathy in patients with type 2 diabetes, and the clinical development of an intravenous formulation of Pyridorin for AKI; |
• | the willingness of the EMA or other regulatory agencies outside the United States to accept our Phase 3 Pyridorin program, as well as our other completed and planned clinical and nonclinical studies and other work, as the basis for review and approval of Pyridorin in the European Union for the treatment of diabetic nephropathy in patients with type 2 diabetes; |
• | the outcome, costs and timing of seeking and obtaining FDA, EMA and any other regulatory approvals; |
• | the number and characteristics of product candidates that we pursue, including our product candidates in preclinical development; |
• | the ability of our product candidates to progress through clinical development successfully; |
• | our need to expand our research and development activities; |
• | the costs associated with securing and establishing commercialization and manufacturing capabilities; |
• | market acceptance of our product candidates; |
• | the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies; |
• | our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
• | our need and ability to hire additional management and scientific and medical personnel; |
• | the effect of competing technological and market developments; |
• | our need to implement additional internal systems and infrastructure, including financial and reporting systems; and |
• | the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future. |
Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our cash needs through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our common stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, commercialization, marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have
75
to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
Contractual Commitments and Obligations
We are a party to license agreements with universities and other third parties, as well as patent assignment agreements, under which we have obtained rights to patents, patent applications and know‑how. These license agreements are subject to various milestone payments related to milestones met in the FDA regulatory approval process. The commitments under our licensing agreement with The South Carolina Research Foundation (USCRF) are payable quarterly until the expiration of certain patent rights and related technology. The Company can terminate the license at any time upon three months prior written notice to USCRF.
On September 12, 2014, we entered into an office lease for approximately 5,514 square feet of office space located at 3200 Beechleaf Court, Raleigh, North Carolina. These premises will serve as our new corporate headquarters. Under the terms of the lease agreement, the lease term is 66 months, commencing on December 1, 2014 and terminating on May 31, 2020. Our monthly base rent, commencing on February 1, 2015, is approximately $9,500 per month and will increase at a rate of approximately 3.0% per year during the term of the lease.
We have employment agreements with certain employees which require the funding of specific levels of payments, if certain events, such as a change in control or termination without cause, occur. We enter into contracts in the normal course of business with CROs for clinical trials and clinical supply manufacturing and with vendors for preclinical research studies and other services and products for operating purposes, which generally provide for termination within 30 days of notice or less, and therefore are cancelable contracts and not included as contractual obligations and commitments.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements as defined under the rules of the SEC.
Recent Accounting Pronouncements
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-15, “Presentation of Financial Statements - Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern” (“ASU 2014-15”). ASU 2014-15 is intended to define management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Specifically, ASU 2014-15 provides a definition of the term substantial doubt and requires an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). It also requires certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans and requires an express statement and other disclosures when substantial doubt is not alleviated. The new standard will be effective for reporting periods beginning after December 15, 2016, with early adoption permitted. Management is currently evaluating the impact of the adoption of ASU 2014-14 on our financial statements and disclosures.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Market Risk
Our cash and cash equivalents and short-term investments as of December 31, 2014, consisted primarily of cash, cash equivalents, money market funds and certificates of deposits. Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of United States interest rates. However, because of the short-term nature of the instruments in our portfolio, a sudden change in market interest rates would not be expected to have a material impact on our financial condition and/or results of operations.
Item 8. Financial Statements and Supplementary Data
76

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
NephroGenex, Inc.
We have audited the accompanying balance sheets of NephroGenex, Inc., a Development Stage Company, (the “Company”) as of December 31, 2014 and 2013, and the related statements of comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the years in the two‑year period ended December 31, 2014. The financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of NephroGenex, Inc. as of December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the years in the two‑year period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
/s/ EisnerAmper LLP
Jenkintown, PA
March 24, 2015
77
NephroGenex, Inc.
Balance Sheets
(in thousands except share and per share information)
December 31. | |||||||
2014 | 2013 | ||||||
Assets | |||||||
Current assets | |||||||
Cash and cash equivalents | $ | 13,978 | $ | 2,132 | |||
Short-term investments | 14,698 | — | |||||
Prepaid expenses and other assets | 309 | 12 | |||||
Total current assets | 28,985 | 2,144 | |||||
Property and equipment, net | 36 | 11 | |||||
Deferred initial public offering costs | — | 461 | |||||
Other assets | 210 | 4 | |||||
Total assets | $ | 29,231 | $ | 2,620 | |||
Liabilities and Stockholders’ Equity (Deficit) | |||||||
Current liabilities | |||||||
Accounts payable | $ | 1,750 | $ | 48 | |||
Accrued and other liabilities | 1,405 | 1,208 | |||||
Current portion of note payable | 293 | — | |||||
Preferred stock warrant liability | — | 6,983 | |||||
Convertible notes payable | — | 8,567 | |||||
Total current liabilities | 3,448 | 16,806 | |||||
Note payable, less current portion | 6,442 | — | |||||
Other long-term liabilities | 10 | — | |||||
Total liabilities | 9,900 | 16,806 | |||||
Commitments and Contingencies (note 12) | 0 | 0 | |||||
Stockholders’ equity (deficit) | |||||||
Series A preferred stock: $.001 par value; 32,690,676 shares authorized; 0 and 23,688,396 shares issued and outstanding as of December 31, 2014 and 2013, respectively | — | 24 | |||||
Preferred stock; $.001 par value; 5,000,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock; $.001 par value; 100,000,000 shares authorized; 8,862,114 and 319,882 shares issued and outstanding as of December 31, 2014 and 2013, respectively | 9 | — | |||||
Additional paid-in capital | 77,149 | 26,789 | |||||
Accumulated other comprehensive loss | (8 | ) | — | ||||
Accumulated deficit | (57,819 | ) | (40,999 | ) | |||
Total stockholders’ equity (deficit) | 19,331 | (14,186 | ) | ||||
Total liabilities and stockholders’ equity (deficit) | $ | 29,231 | $ | 2,620 | |||
(See accompanying Notes to Financial Statements)
78
NephroGenex, Inc.
Statements of Comprehensive Loss
(in thousands except share and per share information)
Year Ended | |||||||||
December 31, | |||||||||
2014 | 2013 | ||||||||
Expenses: | |||||||||
Research and development | $ | 11,264 | $ | 1,480 | |||||
General and administrative | 5,323 | 1,026 | |||||||
Total expenses | 16,587 | 2,506 | |||||||
Loss from operations | (16,587 | ) | (2,506 | ) | |||||
Other income (expense): | |||||||||
Change in value of preferred stock warrants | (140 | ) | (3,417 | ) | |||||
Interest expense | (140 | ) | (383 | ) | |||||
Interest income | 47 | 1 | |||||||
Net loss | $ | (16,820 | ) | $ | (6,305 | ) | |||
Net loss per share – basic and diluted | $ | (2.15 | ) | $ | (19.71 | ) | |||
Weighted average shares outstanding – basic and diluted | 7,827,519 | 319,882 | |||||||
Other comprehensive loss: | |||||||||
Net loss | $ | (16,820 | ) | $ | (6,305 | ) | |||
Unrealized loss on short-term investments | (8 | ) | — | ||||||
Comprehensive loss | $ | (16,828 | ) | $ | (6,305 | ) | |||
(See accompanying Notes to Financial Statements)
79
NephroGenex, Inc.
Statement of Stockholders’ Equity (Deficit)
(in thousands except share information)
Series A Convertible Preferred Stock | Common Stock | Additional | Accumulated | ||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Paid-In Capital | Other Comprehensive Loss | Accumulated Deficit | Total | ||||||||||||||||||||||||||||
Balance at December 31, 2012 | 23,688,396 | — | $ | 24 | — | 319,882 | — | $ | — | — | $ | 26,701 | — | $ | — | — | $ | (34,694 | ) | $ | (7,969 | ) | |||||||||||||
Stock based compensation | — | — | — | — | 88 | — | — | 88 | |||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | (6,305 | ) | (6,305 | ) | |||||||||||||||||||||||||
Balance at December 31, 2013 | 23,688,396 | 24 | 319,882 | — | 26,789 | — | (40,999 | ) | (14,186 | ) | |||||||||||||||||||||||||
Issuance of common stock at IPO, net of expenses of $3,767 | — | — | 3,100,000 | 3 | 33,430 | — | — | 33,433 | |||||||||||||||||||||||||||
Issuance of common stock for preferred stock warrant | — | — | 593,589 | 1 | 7,123 | — | — | 7,124 | |||||||||||||||||||||||||||
Issuance of common stock for convertible notes and accrued interest | — | — | 1,197,289 | 1 | 8,644 | — | — | 8,645 | |||||||||||||||||||||||||||
Issuance of common stock for preferred stock | (23,688,396 | ) | (24 | ) | 3,644,354 | 4 | 20 | — | — | — | |||||||||||||||||||||||||
Issuance of common stock for restricted stock units | — | — | 7,000 | — | — | — | — | — | |||||||||||||||||||||||||||
Issuance of warrants with term loan | — | — | — | — | 192 | — | — | 192 | |||||||||||||||||||||||||||
Stock based compensation | — | — | — | — | 951 | — | — | 951 | |||||||||||||||||||||||||||
Other comprehensive loss | — | — | — | — | — | (8 | ) | — | (8 | ) | |||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | (16,820 | ) | (16,820 | ) | |||||||||||||||||||||||||
Balance at December 31, 2014 | — | $ | — | 8,862,114 | $ | 9 | $ | 77,149 | $ | (8 | ) | $ | (57,819 | ) | $ | 19,331 | |||||||||||||||||||
(See accompanying Notes to Financial Statements)
80
NephroGenex, Inc.
Statements of Cash Flows
(in thousands)
Year Ended | ||||||||
December 31, | ||||||||
2014 | 2013 | |||||||
Operating activities | ||||||||
Net loss | $ | (16,820 | ) | $ | (6,305 | ) | ||
Adjustments to reconcile net loss to net cash and cash equivalents used in operating activities | ||||||||
Depreciation and amortization | 4 | 4 | ||||||
Change in fair value of preferred stock warrants | 140 | 3,417 | ||||||
Non-cash interest expense | 95 | 383 | ||||||
Accretion of premium on investment activities | 71 | — | ||||||
Accrued interest receivable | 4 | — | ||||||
Stock based compensation expense | 951 | 88 | ||||||
Changes in operating assets and liabilities | ||||||||
Prepaid expenses and other assets | (455 | ) | 22 | |||||
Accounts payable, accrued and other liabilities | 2,372 | (321 | ) | |||||
Net cash and cash equivalents used in operating activities | (13,638 | ) | (2,712 | ) | ||||
Investing activities | ||||||||
Purchases of investments | (26,383 | ) | — | |||||
Sales of investments | 11,603 | — | ||||||
Property and equipment purchases | (29 | ) | (12 | ) | ||||
Net cash and cash equivalents used in investing activities | (14,809 | ) | (12 | ) | ||||
Financing activities | ||||||||
Proceeds from issuance of convertible notes payable | — | 4,562 | ||||||
Proceeds from issuance of note payable | 6,880 | — | ||||||
Payment of debt issuance costs | (50 | ) | — | |||||
Payment of initial public offering costs | (3,737 | ) | (30 | ) | ||||
Proceeds from issuance of common stock | 37,200 | — | ||||||
Net cash and cash equivalents provided by financing activities | 40,293 | 4,532 | ||||||
Net increase in cash and cash equivalents | 11,846 | 1,808 | ||||||
Cash and cash equivalents at beginning of year | 2,132 | 324 | ||||||
Cash and cash equivalents at end of year | $ | 13,978 | $ | 2,132 | ||||
Supplemental disclosure of cash flow information | ||||||||
Cash paid for interest | $ | 12 | $ | — | ||||
Supplemental disclosure of noncash financing activities | ||||||||
Conversion of convertible notes payable, accrued interest, preferred stock and warrants into common stock | $ | 15,793 | $ | — | ||||
Initial public offering costs included in accrued and other liabilities | $ | — | $ | 431 | ||||
(See accompanying Notes to Financial Statements)
81
NephroGenex, Inc.
Notes to Financial Statements
1. Description of Business and Basis of Presentation
Description of Business
NephroGenex, Inc. (the “Company”) was incorporated in Delaware on May 25, 2004. The Company is a drug development company focused on developing novel therapies for kidney disease. The Company acquired commercial rights to Pyridorin™ and has initiated a Phase 3 clinical study in patients with diabetic nephropathy.
The Company’s primary efforts to date have been devoted to raising capital, recruiting senior management and staff and conducting research and development activities. The Company has experienced net losses since its inception and, as of December 31, 2014, has an accumulated deficit of $57.8 million.
The Company currently has no commercially approved products and has recognized no revenue since its inception in 2004. We do not expect to generate revenue from product sales unless and until we successfully complete development and obtain marketing approval for one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Developing and commercializing a product requires significant time and capital and is subject to regulatory review and approval. There can be no assurance that the Company’s current products in development, if approved, will be successfully commercialized due to a variety of factors, including competition from other biotechnology and pharmaceutical companies.
Basis of Presentation
The financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
Reclassification
Certain amounts in the 2013 financial statements have been reclassified to conform to the 2014 presentation. The reclassifications had no effect on net loss or stockholders’ equity as previously reported.
Liquidity
The Company's financial statements have been prepared on a basis which assumes that the Company will continue as a going concern and which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since its inception, expects to incur additional costs and requires additional capital to continue as a going concern. As a result, the Company will require additional funds and will continue to seek private or public equity, debt financing, research funding and revenue or expense sharing from collaborative agreements to meet its capital requirements. Even if the Company does not have an immediate need for additional cash, it may seek access to the private or public equity markets if and when conditions are favorable. If such funds are not available, management may need to reassess its business plans. There is no assurance that such additional funds will be available for the Company to finance its operations on acceptable terms, if at all.
2. Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Reverse Stock Split
On February 6, 2014, the Company effected a 1-for-6.5 reverse stock split of its issued and outstanding shares of common stock and a proportional adjustment to the conversion ratio for the Company’s outstanding Series A Preferred Stock. Accordingly, all share and per share amounts for all periods presented in these financial statements and notes thereto have been
82
NephroGenex, Inc.
Notes to Financial Statements
adjusted retroactively, where applicable, to reflect the reverse stock split and adjustment of the preferred share conversion ratios.
Initial Public Offering
On February 14, 2014, the Company completed its initial public offering of common stock (the “IPO”) pursuant to a registration statement that was declared effective on February 10, 2014. The Company sold 3,100,000 shares of its common stock, at a price of $12.00. The Company received a total of $33.4 million in net proceeds after deducting underwriting discounts and commissions and offering expenses of approximately $3.8 million. As of December 31, 2013, costs directly associated with the IPO were capitalized and recorded as deferred costs. Upon closing of the IPO, these costs were recorded as a reduction of the proceeds received in arriving at the amount to be recorded as additional paid-in capital.
In connection with the IPO, 3,644,354 shares of common stock were issued for the conversion of all outstanding shares of Series A Preferred stock, 1,197,289 shares of common stock were issued for the conversion of outstanding convertible notes and accrued interest and 593,589 aggregate shares of common stock were issued in connection with the settlement of the Company’s outstanding preferred stock warrant liability.
Warrant Liability
Certain warrants to purchase the Company’s capital stock had historically been classified as liabilities and were recorded at estimated fair value. At each reporting period, any change in fair value of the freestanding warrants was recorded as other (expense) income. The preferred stock warrant liability was settled upon the closing of the IPO.
Segment Reporting
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The Company views its operations and manages its business in one operating segment.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less to be cash equivalents.
Investments
The Company invests in money market funds and certificates of deposits and considers all investments purchased with original maturity dates greater than three months and less than one year to be short-term investments. Those investments with original maturity dates greater than one year at each balance sheet date are considered to be long-term investments. As of December 31, 2014, all investments were classified as available-for-sale and had original maturity dates less than one year. These investments are carried at estimated fair value with unrealized gains and losses included in stockholders' equity (deficit). The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity, which is included in interest income.
Concentration of Credit Risk
The Company invests its available cash balances in bank deposits, money market funds and certificates of deposit. The Company maintains deposits in federally insured financial institutions in excess of federally insured limits. Management believes that the Company is not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held. Additionally, the Company has established guidelines regarding approved investments and maturities of investments, which are designed to maintain safety and liquidity.
Property and Equipment
Property and equipment, including leasehold improvements, are stated at cost. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets or the shorter of the lease term or the estimated useful life for leasehold improvements. Useful lives generally range from three to seven years.
83
NephroGenex, Inc.
Notes to Financial Statements
Fair Value of Financial Instruments
As of December 31, 2014, financial instruments consist of cash and cash equivalents, short-term investments, a note payable, accounts receivable and accounts payable.
The Company defines fair value (“FV”) as the price that would be received to sell an asset or paid to transfer a liability ("the exit price") in an orderly transaction between market participants at the measurement date. The FV hierarchy for inputs maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. The Company uses the following hierarchy of inputs to measure FV:
•Level 1: Quoted prices in active markets for identical assets or liabilities;
• | Level 2: Inputs, other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities in active markets; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; and |
• | Level 3: Unobservable inputs that are supported by little or no market activity, which require the reporting entity to develop its own assumptions. |
The Company values investments using the most observable inputs available that are current as of the measurement date and classifies them according to the lowest level of inputs used. Observable inputs are inputs that market participants would use in pricing the asset or liability developed from market data obtained from independent sources. Unobservable inputs are those which reflect the Company’s judgment concerning the assumptions that market participants would use in pricing the asset or liability developed from the best information available under the circumstances.
The Company targets investments principally in Level 1 and Level 2 cash equivalents and financial instruments and records them at FV. The Company did not rely on Level 3 inputs for the valuation of any investments at December 31, 2014. The Company expects that the carrying values of cash equivalents will approximate FV because of their short maturities.
The Company classifies as Level 2 investments in certificates of deposits and values them using the market approach based on significant other observable inputs including quoted prices in active markets for instruments that are similar or quoted prices in markets that are not traded on a daily basis for identical or similar instruments.
The following table sets forth our financial instruments carried at FV within the ASC 820 hierarchy and using the lowest level of input as of December 31, 2014:
Quoted Prices | ||||||||||||
in Active | Significant | |||||||||||
(in thousands) | Markets | Other | Significant | |||||||||
Balance | For Identical Assets | Observable Inputs | Unobservable Inputs | |||||||||
Assets: | December 31, 2014 | Level 1 | Level 2 | Level 3 | ||||||||
Money Market Funds | $ | 4,807 | $ | 4,807 | $ | — | $ | — | ||||
Certificates of Deposit | 16,765 | — | 16,765 | — | ||||||||
Total Assets | $ | 21,572 | $ | 4,807 | $ | 16,765 | $ | — | ||||
84
NephroGenex, Inc.
Notes to Financial Statements
Assets and liabilities measured at fair value on a recurring basis as of December 31, 2013 were as follows:
Quoted Prices | ||||||||||||
in Active | Significant | |||||||||||
(in thousands) | Markets | Other | Significant | |||||||||
Balance | For Identical Assets | Observable Inputs | Unobservable Inputs | |||||||||
Assets: | December 31, 2013 | Level 1 | Level 2 | Level 3 | ||||||||
Money Market Funds | $ | 2,119 | $ | 2,119 | $ | — | $ | — | ||||
Total Assets | $ | 2,119 | $ | 2,119 | $ | — | $ | — | ||||
Liabilities: | ||||||||||||
Preferred stock warrant liability | $ | 6,983 | $ | — | $ | — | $ | 6,983 | ||||
Total liabilities | $ | 6,983 | $ | — | $ | — | $ | 6,983 | ||||
At December 31, 2013, certain warrants to purchase the Company’s capital stock were classified as liabilities and were recorded at estimated FV. The Company measured its warrant liability using significant unobservable inputs that were based on little or no verifiable market data, which is Level 3 in the FV hierarchy. At each reporting period, any change in fair value of the freestanding warrants was recorded as other (expense) income. The Company recorded a $140,000 and $3.4 million as other expense as as a result of the change in fair value of the preferred stock warrant liability for the years ended December 31, 2014 and 2013, respectively. The preferred stock warrant liability was settled upon closing of the IPO. As of December 31, 2014, the Company has no financial instruments that are valued using Level 3 inputs.
Debt Issuance Costs
Debt issuance costs represent legal and other direct costs related to the Company's outstanding loan. These costs are recorded as an asset on the accompanying balance sheets and are being amortized to interest expense utilizing the effective interest method through the earliest date at which the Company can be required to repay the notes. At December 31, 2014, the balance of debt issuance costs was $48,000.
Research and Development Costs
Research and development costs are expensed as incurred. Research and development expenses include personnel costs associated with research and development activities including non-cash share-based compensation, costs for third-party contractors to perform research, conduct clinical trials and manufacture drug supplies and materials. The Company accrues for costs incurred by external service providers, including contract research organizations and clinical investigators, based on its estimates of service performed and costs incurred. These estimates include the level of services performed by the third parties, patient enrollment in clinical trials, administrative costs incurred by the third parties, and other indicators of the services completed. Based on the timing of amounts invoiced by service providers, the Company may also record payments made to those providers as prepaid expenses that will be recognized as expense in future periods as the related services are rendered.
Stock-Based Compensation
The Company estimates the FV of stock options and stock purchase rights using a Black-Scholes option valuation model which requires the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. The Company uses the simplified method for estimating the expected term as provided by the Securities and Exchange Commission's Staff Accounting Bulletin No. 107. The simplified method calculates the expected term as the average time-to-vesting and the contractual life of the options. The expected stock price volatility assumption was determined by examining the historical volatilities of a group of industry peers. The FV of each option grant is estimated on the date of grant using the Black-Scholes option valuation model, and the resulting FV is expensed using the straight-line attribution method over the vesting period, which is the same as the requisite service period. Restricted stock units are measured at the FV of the Company's common stock on the date of grant and expensed over the period of vesting, which is the same as the requisite service period using the straight-line attribution method.
85
NephroGenex, Inc.
Notes to Financial Statements
Recent Accounting Pronouncements
Occasionally, new accounting standards are issued or proposed by the Financial Accounting Standards Board (the “FASB”), or other standards-setting bodies that the Company adopts by the effective date specified within the standard. Unless otherwise discussed, standards that do not require adoption until a future date are not expected to have a material impact on the Company’s financial statements upon adoption.
In July 2013, the FASB issued Accounting Standards Update, or ASU, No. 2013-11, Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists. ASU 2013-11 provides explicit guidance on the financial statement presentation of an unrecognized tax benefit when a net operating loss carryforward, a similar tax loss, or a tax credit carryforward exists. The guidance is effective prospectively for fiscal years, and interim periods within those years, beginning after December 15, 2013, with an option for early adoption. The Company adopted this guidance on January 1, 2014. The adoption did not impact the Company’s financial position or results of operations.
In July 2014, the FASB issued ASU No. 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation. The amendments in this ASU remove all incremental financial reporting requirements from U.S. GAAP for development stage entities, including the removal of Topic 915, Development Stage Entities, from the FASB Accounting Standards Codification™. A development stage entity is one that devotes substantially all of its efforts to establishing a new business and for which: (a) planned principal operations have not commenced; or (b) planned principal operations have commenced, but have produced no significant revenue. Current U.S. GAAP requires a development stage entity to present the same basic financial statements and apply the same recognition and measurement rules as established companies. In addition, U.S. GAAP requires a development stage entity to present inception-to-date information about income statement line items, cash flows, and equity transactions. For public business entities, the presentation and disclosure requirements in Topic 915 will no longer be required for the first annual period beginning after December 15, 2014. The revised consolidation standards are effective one year later, in annual periods beginning after December 15, 2015. Early adoption is permitted. The Company adopted the guidance for the quarterly periods ended June 30, 2014. The adoption did not impact the Company’s financial position or results of operations.
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-15, “Presentation of Financial Statements - Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern” (“ASU 2014-15”). ASU 2014-15 is intended to define management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Specifically, ASU 2014-15 provides a definition of the term substantial doubt and requires an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). It also requires certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans and requires an express statement and other disclosures when substantial doubt is not alleviated. The new standard will be effective for reporting periods beginning after December 15, 2016, with early adoption permitted. Management is currently evaluating the impact of the adoption of ASU 2014-14 on the Company's financial statements and disclosures.
3. Earnings Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted average number of shares of the Company’s common stock outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is computed by dividing the net loss by the weighted average number of common share equivalents outstanding for the period determined using the treasury-stock method. Under the treasury-stock method earnings per share data is computed as if the common share equivalents were outstanding at the beginning of the period (or at the time of issuance, if later) and as if the funds obtained from exercise of the common stock equivalents were used to purchase common stock at the average market price during the period. If there is little or no market for the common stock, a reasonable estimate of FV shall be used.
For purposes of this calculation, preferred stock, stock options, restricted stock units and warrants to purchase capital stock are considered to be common stock equivalents and are only included in the calculation of diluted net loss per share when their effect is dilutive.
86
NephroGenex, Inc.
Notes to Financial Statements
The following table sets forth the computation of basic and diluted net loss per share in thousands, except share and per share data:
Year Ended December 31, | ||||||
2014 | 2013 | |||||
Numerator: | ||||||
Net loss | $ | (16,820 | ) | $ | (6,305 | ) |
Denominator: | ||||||
Weighted average common shares outstanding | 7,827,519 | 319,882 | ||||
Net loss per share-basic and diluted | $ | (2.15 | ) | $ | (19.71 | ) |
Potentially dilutive securities not included in the calculation of diluted net loss per common share because to do so would be anti-dilutive are as follows (in common equivalent shares on a weighted-average basis):
2014 | 2013 | |||
Common stock options | 848,025 | 533,199 | ||
Restricted stock units | 24,000 | 3,616 | ||
Common stock warrants | 61,039 | — | ||
In addition to the potentially dilutive securities noted above, the Company has excluded from the table above 3,644,354 shares of common stock that were issued for the conversion of all outstanding shares of Series A Preferred stock, 1,197,289 shares of common stock that were issued for convertible notes and accrued interest and 593,589 aggregate shares of common stock that were issued in connection with the settlement of the Company’s outstanding warrant obligations upon closing of the IPO.
4. Balance Sheet Items
Investments
The following table summarizes the Company's available for sale investments as of December 31, 2014 (in thousands):
Maturity | Amortized | Unrealized | Unrealized | Estimated | |||||||||
Short-term Investments | (in years) | Cost | Gains | Loss | Fair Value | ||||||||
Money Market | 1 or less | $ | — | $ | — | $ | — | $ | — | ||||
Certificates of Deposit | 1 or less | 14,706 | — | (8 | ) | 14,698 | |||||||
Total Investments | $ | 14,706 | $ | — | $ | (8 | ) | $ | 14,698 | ||||
At each reporting date, the Company performs an evaluation of impairment to determine if the unrealized losses are other-than-temporary. For debt securities, management determines whether it intends to sell the impaired securities, and if there is no intent or expected requirement to sell, management considers whether it is likely that the amortized cost will be recovered. The Company does not consider unrealized losses on its debt investment securities to be credit-related. These unrealized losses relate to changes in interest rates and market spreads subsequent to purchase. The Company has not made a decision to sell securities with unrealized losses and believes it is more likely than not it would not be required to sell such securities before recovery of its amortized cost. There have been no other than temporary losses recognized in earnings.
87
NephroGenex, Inc.
Notes to Financial Statements
Property and Equipment
As of December 31, 2014 and 2013 property and equipment were as follows (in thousands):
Useful Life | 2014 | 2013 | |||||
Computer equipment | 5 years | $ | 54 | $ | 51 | ||
Furniture and fixtures | 7 years | 66 | 66 | ||||
Leasehold improvements | 5.5 years | 18 | — | ||||
138 | 117 | ||||||
Less accumulated depreciation | (102 | ) | (106 | ) | |||
Property and equipment, net | $ | 36 | $ | 11 | |||
For the years ended December 31, 2014 and 2013, depreciation and amortization expense was approximately $3,600 and $4,100, respectively.
Accrued Liabilities
As of December 31, 2014 and 2013 accrued liabilities were as follows (in thousands):
2014 | 2013 | |||||
Accrued clinical trial expenses | $ | 393 | $ | 209 | ||
Accrued compensation | 909 | 365 | ||||
Other accruals | 103 | 634 | ||||
Total | $ | 1,405 | $ | 1,208 | ||
5. License Agreements
The University of South Carolina Research Foundation, Corp.
During 2007, the Company licensed certain technology from the University of South Carolina Research Foundation, Corp. (“USCRF”) to the Company. The license gives the Company worldwide rights to use the technology as defined in the agreement. The agreement was amended in August 2013. The Company paid an annual licensing fee of $30,000 through 2008, $60,000 from 2009 through 2010, $62,000 from 2011 through 2012 and $122,000 in 2013. The Company is obligated to pay an annual licensing fee of $120,000 thereafter. Upon the achievement of certain defined product development milestones for diabetic neuropathy or hyperlipidemia, the Company would be obligated to make up to $6.1 million of payments to USCRF. The Company will be obligated to pay USCRF a one-time fee of $35,000 upon execution of a sublicense and would pay to USCRF 25% of any non-royalty sublicense payments received from a sub-licensee. The term of the agreement expires on the expiration of the underlying USCRF patents. The Company can terminate the license at any time upon three months prior written notice to USCRF. As of December 31, 2014, no development milestones have been paid or accrued nor does the Company expect to achieve any development milestones during the next few years. The Company paid $120,000 and $122,000 for the years ended December 31, 2014 and 2013, respectively, for annual licensing fees due under this agreement.
Vanderbilt University
During 2006, the Company entered into a licensing agreement with Vanderbilt University (“Vanderbilt”) for the rights to use certain technology. The agreement, as amended, requires the Company to make milestone payments totaling approximately $1.1 million in the event certain defined events occur. Should the Company successfully develop a product using the licensed technology, Vanderbilt will be due royalties based on net sales at a rate of 5%. The Company must also pay Vanderbilt 25% of non‑royalty sub‑licensee payments received from a sub‑licensee. Annual minimum royalties due under the licensing agreement are $10,000 and will increase to $25,000 when a claim in the licensed patent rights is issued in a major market country, as defined. The licensing agreement expires when the underlying patents to the licensed technology expire. The Company may terminate the agreement upon sixty days written notice to Vanderbilt.
88
NephroGenex, Inc.
Notes to Financial Statements
Certain milestones can be paid in stock or are creditable against future royalties due based on net sales. As of December 31, 2014, no milestone or royalty payments have been paid or accrued.
The University of Kansas Medical Center Research Institute, Inc.:
During 2007, the Company received rights to certain technology licensed from the University of Kansas Medical Center Research Institute, Inc. ("KUMC") to the Company. The license gives the Company worldwide royalty-free rights to use certain technology. Upon the achievement of certain defined product development milestones, the Company would be obligated to make up to $225,000 of payments to KUMC. As of December 31, 2014, no milestones have been paid or accrued. The term of the agreement expires on the expiration of the underlying KUMC patents or November 2018, whichever occurs last. The Company can terminate the agreement with 90 days notice.
6. Convertible Notes Payable
On February 14, 2014, in connection with the closing of the Company’s IPO, $7.9 million of convertible promissory notes and $728,000 of accrued interest were converted into 1,197,289 shares of common stock.
The Company had accrued interest of approximately $650,000 as of December 31, 2013 which is included in the carrying value of convertible notes payable. Interest expense for the years ended December 31, 2014 and 2013 relating to the notes was approximately $78,000 and $383,000, respectively.
7. Term Loan
On November 20, 2014, the Company entered into a Loan and Security Agreement with a bank for a term loan (the “Initial Term Loan”) with an aggregate principal amount of $7.0 million and, subject to the terms and conditions set forth in the agreement, a second term loan (the “Second Term Loan”) with an aggregate principal amount of $5.0 million. Each term loan shall accrue interest at a rate of 2.25% per annum plus the greater of 3.25% or the current prime rate. As of December 31, 2014 the interest rate on the loan was 5.5%. As security for its obligations under the Loan Agreement, the Company granted the bank a lien on substantially all of its assets, including owned and licensed intellectual property.
On November 20, 2014, the bank funded the Initial Term Loan, which matures on October 1, 2018. Interest only payments are due during the first twelve months of the Initial Term Loan (the “Interest Only Term”) and beginning on November 1, 2015, the Company is required to make thirty-six (36) equal monthly payments of principal and interest. The Interest Only Term may be extended under the Loan Agreement if certain conditions are met. The Company paid a $120,000 facility fee which was recorded as a debt discount to be amortized as interest expense over the term of the loan using the effective interest rate method.
At the Company’s option, the Company may borrow the Second Term Loan on or before May 29, 2015, if the Company has met certain clinical milestones. The Company may prepay each term loan in full with no prepayment penalty. Upon payment of the final monthly installment of the loan, or the remaining balance in the case of a prepayment, the Company would pay an end-of-term fee of approximately $60,000.
In connection with the Initial Term Loan, the Company issued warrants to purchase an aggregate of 56,603 shares of the Company's common stock at an exercise price of $4.24 per share. The warrants are immediately exercisable and will expire on November 20, 2021. The Company determined the fair value of the warrants to be $192,450 using the Black-Scholes pricing model and recorded the warrants as a debt discount to be amortized as interest expense over the term of the Notes using the effective interest rate method. The Company also paid $50,000 in debt issuance costs, which were capitalized as a deferred asset and are being amortized over the expected remaining life of the loan using the effective interest method.
The Company recognized $62,000 in interest expense related to the term loan, including the amortization of the warrants, for the year ended December 31, 2014.
89
NephroGenex, Inc.
Notes to Financial Statements
The following represents the outstanding principal balances, carrying amounts and maturities of notes payable as of December 31, 2014 (in thousands):
December 31, 2014 | ||||||||||||
Amortization of | Accrued | Carrying Value | ||||||||||
Principal Value | Debt Discount | Interest | of Note Payable | |||||||||
2015 | $ | 389 | (129 | ) | 33 | 293 | ||||||
2016 | 2,333 | (99 | ) | — | 2,234 | |||||||
2017 | 2,334 | (56 | ) | 2,278 | ||||||||
2018 | 1,944 | (14 | ) | — | 1,930 | |||||||
$ | 7,000 | $ | (298 | ) | $ | 33 | $ | 6,735 | ||||
Less current portion | 293 | |||||||||||
Long-term note payable, net of discount | $ | 6,442 | ||||||||||
8. Stockholders' equity (deficit)
Series A Preferred Stock
In connection with the completion of the IPO, 3,644,354 shares of common stock were issued for the conversion of all outstanding shares of the Company’s Series A Preferred stock.
Warrants
On January 16, 2014, an agreement was reached among the Company’s significant shareholders to cancel warrants held by its majority shareholder, Care Capital Investments III, LP, together with its affiliates (collectively, Care Capital), and by funds affiliated with Rho Venture Partners (Rho). Pursuant to this agreement, an aggregate of 593,589 shares of the Company’s common stock were issued to Care Capital and Rho concurrently with the completion of the Company’s IPO in return for cancelling the warrants. In connection with the cancellation of the warrants, the Company settled the preferred stock warrant liability on its balance sheet.
On February 10, 2014, the Company, in connection with the IPO, issued the underwriter warrants to purchase up to 62,000 shares of common stock. The warrants are exercisable at any time commencing one year from the effective date of the Company’s IPO. The warrants are exercisable at a price of $15.00 per share and expire on February 10, 2018.
On November 20, 2014, the Company, in connection with the issuance of a term loan, issued warrants to a lender to purchase up to an aggregate of 56,603 shares of the Company's common stock at an exercise price of $4.24 per share. The warrants are immediately exercisable and will expire on November 20, 2021.
As of December 31, 2014, the following warrants to purchase common stock were outstanding:
Exercise | ||||||
Issuance Date | Shares | Price | Expiration | |||
2/10/2014 | 62,000 | $ | 15.00 | 2/10/2018 | ||
11/20/2014 | 56,603 | $ | 4.24 | 11/20/2021 | ||
Total warrants outstanding | 118,603 | |||||
Common Stock
On February 14, 2014, the Company filed an Amended and Restated Certificate of Incorporation which authorizes the issuance of 100,000,000 shares of common stock, and 5,000,000 shares of undesignated preferred stock.
90
NephroGenex, Inc.
Notes to Financial Statements
During 2014, the Company issued 7,000 shares of common stock to its chief executive officer for restricted stock units that vested during the year.
Shares Reserved for Future Issuance
As of December 31, 2014, the Company had 8,862,114 shares of common stock outstanding. The Company has reserved shares of common stock for future issuance as of December 31, 2014 as follows:
Stock options outstanding | 1,272,581 | |
Shares available for grant under stock option plans | 169 | |
Restricted stock units | 17,000 | |
Common stock warrants | 118,603 | |
Total shares reserved for future issuance | 1,408,353 | |
Stock Based Compensation
In 2005, the Company adopted the NephroGenex, Inc. 2005 Stock Option Plan. On May 15, 2014, the 2005 Stock Option Plan, was amended and restated to the 2007 Equity Incentive Plan (the “Plan”). The amendment authorized an increase of 673,923 shares and provided for the granting of up to 1,283,226 shares of common stock to employees and consultants of the Company in the form of incentive and nonqualified stock options and shares of restricted stock. As of December 31, 2014, there were 169 shares available for issuance from the Plan.
The table below summarizes stock option activity for the years ended December 31, 2014 and 2013.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (years) | |||||
Outstanding as of December 31, 2012 | 473,805 | $ | 1.11 | ||||
Granted | 91,261 | 2.02 | |||||
Exercised | — | — | |||||
Forfeitures | (1,613 | ) | 32.50 | ||||
Outstanding as of December 31, 2013 | 563,453 | 1.18 | |||||
Granted | 712,204 | 6.60 | |||||
Exercised | — | — | |||||
Forfeitures | (3,076 | ) | 11.90 | ||||
Outstanding as of December 31, 2014 | 1,272,581 | $ | 4.19 | 7.5 | |||
Exercisable as of December 31, 2014 | 580,217 | $ | 2.08 | 5 | |||
During the years ended December 31, 2014 and 2013, the Company’s Board of Directors granted stock options of 712,204 and 91,261, respectively to employees and Directors of the Company with a weighted average fair value of $5.40 and $1.38 per share, respectively.
The weighted-average assumptions used in the Black-Scholes valuation model for equity awards granted during the years ended December 31, 2014 and 2013 are shown in the table below.
2014 | 2013 | |||
Expected volatility | 88.3 | % | 60.0 | % |
Expected dividends | — | — | ||
Expected life in years | 6.5 | 7 | ||
Risk-free interest rate | 1.98 | % | 2.00 | % |
The Company determines the options’ life based upon the use of the simplified method. As a newly public company, sufficient history to estimate the volatility and dividend yield of our common stock is not available. The Company uses a pool
91
NephroGenex, Inc.
Notes to Financial Statements
of comparable companies as a basis for the expected volatility assumption and dividend yield. The Company intends to continue to consistently apply this process using the comparable companies until sufficient amount of historical information becomes available. The risk free interest rate is based upon the yield of an applicable Treasury instrument.
At December 31, 2014, the aggregate intrinsic value of options outstanding was $11.8 million. The aggregate intrinsic value of options outstanding as of December 31, 2014 represents the pretax value (the Company's closing market price of$13.35 per share on December 31, 2014, less the exercise price per share, times the number of in-the-money options) that would have been received by all option holders had they exercised their options at the end of the period. No options were exercised for the years ended December 31, 2014 or 2013.
As of December 31, 2014, there was $3.1 million of unrecognized compensation expense related to unvested stock options, which is expected to be recognized over a weighted average period of 1.67 years.
In November 2013, the Company issued 24,000 Restricted Stock Units (RSU) to its CEO in connection with his employment agreement. The RSU represent the right to receive shares of common stock, subject to the terms and conditions of a restricted stock unit agreement and grant notice and were not issued under the Plan. The RSU’s are subject to time based vesting with 25% of the RSU’s vesting on October 21, 2014 and the remaining 75% are vesting in equal monthly installments on the 1st day of each calendar month beginning November 1, 2014. As of December 31, 2014, the Company had issued 7,000 shares of common stock for RSU's that vested during the year.
The Company recognized non-cash share-based compensation expense in its research and development and general and administrative expenses as follows:
Year Ended December 31, | ||||||
(in thousands) | 2014 | 2013 | ||||
Research and development | $ | 130 | $ | 5 | ||
General and administrative | 821 | 83 | ||||
Total | $ | 951 | $ | 88 | ||
9. Retirement Savings Plan
The Company provides a qualified 401(k) savings plan for its employees. All employees are eligible to participate, provided they meet the requirements of the plan. The Company provides a contribution on the first 3% of an employee's eligible salary subject to statutory limitations as prescribed by law. For the year ended December 31, 2014, the Company recorded $48,000 of expense for 401(k) contributions. No contributions were made for the year ended December 31, 2013.
10. Related Party Transactions
Prior to June 30, 2014, the Company reimbursed Care Capital, LLC (“Care”), an affiliate of the majority shareholder of the Company, for services of a Care employee and reimbursed Care for such personnel services incurred by Care on behalf of the Company. Total expense recognized in operating results for the years ended December 31, 2014 and 2013 in connection with services provided by Care was $70,000 and $124,000, respectively.
11. Income Taxes
The Company recognized deferred tax liabilities and assets for the expected future tax consequences of events that have been recognized differently between the financial statements and tax returns. Under this method, deferred tax liabilities and assets are determined based on the difference between the financial statement carrying amounts and tax basis of liabilities and assets using enacted tax rates and laws in effect in the years in which the differences are expected to reverse. Deferred tax assets are evaluated for realization based on a more‑likely‑than‑not criteria in determining if a valuation allowance should be provided.
There was no income tax provision for the years ended December 31, 2014 and 2013.
92
NephroGenex, Inc.
Notes to Financial Statements
The components of the Company’s deferred tax assets at December 31, 2014 and 2013 are as follows:
(in thousands) | 2014 | 2013 | ||||
Net operating loss carry forwards | $ | 11,044 | $ | 9,294 | ||
Stock based compensation | 357 | 57 | ||||
Tax credits | 1,516 | 1,071 | ||||
Depreciation | 1 | 5 | ||||
Amortization | 6,392 | 3,400 | ||||
Accrued bonus | — | 139 | ||||
Accrued expenses | 4 | 122 | ||||
Accrued interest | 12 | 33 | ||||
Deferred tax assets | 19,326 | 14,121 | ||||
Less: valuation allowance | (19,326 | ) | (14,121 | ) | ||
Net deferred tax asset | $ | — | $ | — | ||
The Company’s valuation allowance increased by $5.2 million and $1.2 million during the years ended December 31, 2014 and 2013, respectively. The reconciliation between the Company’s effective tax rate and the federal statutory rate for the years ended December 31, 2014 and 2013 are as follows:
2014 | 2013 | |||
Federal statutory rate | (34.00 | )% | (34.00 | )% |
State income taxes | 0.73 | % | 19.13 | % |
Valuation allowance | 33.27 | % | 14.87 | % |
Effective tax rate | — | % | — | % |
As of December 31, 2014, the Company had approximately $31.8 million of Federal net operating losses that will begin to expire in 2024 and approximately $7.2 million of State net operating losses that will begin to expire in 2030. As of December 31, 2014, the Company has research and development credit carryovers for Federal and New Jersey of approximately $1.3 million and $244,000, respectively; these will begin to expire in 2024 for federal and 2015 for New Jersey tax purposes. The Internal Revenue Code (“IRC”) limits the amounts of net operating loss carryforwards that a company may use in any one year in the event of certain cumulative changes in ownership over a three‑year period as described in Section 382 of the IRC. The Company has not performed a detailed analysis to determine whether an ownership change has occurred. Such a change of ownership could limit the utilization of the net operating losses, and could be triggered by subsequent sales of securities by the Company or its stockholders.
The Company did not have a liability related to unrecognized tax benefits as of December 31, 2014 or 2013.
The Company records interest accrued and penalties related to unrecognized tax benefits within the income tax expense. The Company had not accrued any interest or penalties related to unrecognized benefits. The Company is no longer subject to federal income tax assessment for years before 2011 and for years before 2010 for New Jersey income tax purposes. However, since the Company has incurred net operating losses in every year since inception, all of its income tax returns are subject to examination and adjustments by the Internal Revenue Service for at least three years following the year in which the tax attributes are utilized. The Company does not believe that there will be a material change in its unrecognized tax positions over the next twelve months. There is no amount of unrecognized tax benefit that, if recognized, would affect the effective tax rate.
12. Commitments
Lease
On September 12, 2014, the Company entered into an agreement to lease office space at 3200 Beechleaf Court, Raleigh, North Carolina for the period December 1, 2014 through May 31, 2020. These premises will serve as the Company's corporate headquarters. The lease provides for abatement of rent during certain periods and escalating rent payments during the lease term. The Company records rent expense on a straight-line basis over the life of the lease.
93
NephroGenex, Inc.
Notes to Financial Statements
The following is a schedule of future non-cancellable minimum lease payments for operating leases at December 31, 2014 (in thousands):
Year | |||
2015 | $ | 105 | |
2016 | 118 | ||
2017 | 122 | ||
2018 | 125 | ||
2019 | 129 | ||
Thereafter | 55 | ||
$ | 654 | ||
Rent expense was approximately $62,000 and $52,000 for the years ended December 31, 2014 and 2013, respectively.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures
Management’s Evaluation of our Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial and accounting officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) as of December 31, 2014. Based on that evaluation, our principal executive officer and principal financial and accounting officer concluded that our disclosure controls and procedures as of December 31, 2014 are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated to our management, including our principal executive officer and principal financial and accounting officer, as appropriate to allow timely decisions regarding disclosures. A controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
Changes in Internal Control Over Financial Reporting
During the year ended December 31, 2014, there have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15(d)-15(f) promulgated under the Securities Exchange Act of 1934, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. Management has used the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), or the COSO framework, to evaluate the effectiveness of internal control over financial reporting. Management believes that the COSO framework is a suitable framework for its evaluation of financial reporting because it is free from bias, permits reasonably consistent qualitative and quantitative measurements of our internal control over financial reporting, is sufficiently complete so that those relevant factors that would alter a conclusion about the effectiveness of our internal control over financial reporting are not omitted and is relevant to an evaluation of internal control over financial reporting. Management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2014 and has concluded that such internal control over financial reporting was effective.
An attestation report of control over financial reporting by our registered public accounting firm is not required for the year ended December 31, 2014.
94
Item 9B. Other Information
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Our restated certificate of incorporation and restated bylaws provide that our business is to be managed by or under the direction of our Board of Directors. Our Board of Directors is divided into three classes for purposes of election. One class is elected at each annual meeting of stockholders to serve for a three-year term. Our Board of Directors currently consists of six members, classified into three classes as follows: (1) James Mitchum and Robert Seltzer constitute a class with a term ending at the 2015 annual meeting; (2) Richard Markham and Pierre Legault constitute a class with a term ending at the 2016 annual meeting and (3) Eugen Steiner, M.D., Ph.D. and Marco Taglietti, M.D. constitute a class with a term ending at the 2017 annual meeting.
Set forth below are our directors as of March 24, 2015, their ages, their offices in the Company, if any, their principal occupations or employment for at least the past five years, the length of their tenure as directors and the names of other public companies in which such persons hold or have held directorships during the past five years. Additionally, information about the specific experience, qualifications, attributes or skills that led to our Board of Directors' conclusion at the time of filing of this Annual Report on Form 10-K that each person listed below should serve as a director is set forth below:
Name | Age | Position with the Company | |
Richard J, Markham(2)(3) | 64 | Chairman of the Board | |
Pierre Legault | 54 | Chief Executive Officer | |
James Mitchum(1)(2) | 62 | Director | |
Robert R. Seltzer(3) | 39 | Director | |
Eugen Steiner, M.D., Ph.D(1) | 60 | Director | |
Marco Taglietti, M.D. (1)(2) | 55 | Director | |
(1) Member of our Audit Committee.
(2) Member of our Compensation Committee.
(3) Member of our Nominating and Governance Committee.
Our Board of Directors has reviewed the materiality of any relationship that each of our directors has with us, either directly or indirectly. Based upon this review, our Board of Directors has determined that the following members of the Board of Directors are "independent directors" as defined by The NASDAQ Stock Market: Richard Markham, James Mitchum, Robert R. Seltzer, Eugen Steiner, M.D. Ph.D., and Marco Taglietti, M.D.
Richard J. Markham has served as a member of our Board of Directors since 2007 and as the chairman of our Board of Directors since October 2013. Mr. Markham has been a partner in the venture capital firm Care Capital, LLC, an affiliate of one of our principal stockholders, since November 2004 and continues in that role. Prior to joining Care Capital, he was the Vice Chairman of the Management Board and COO of Aventis. Previously, he was the CEO of Aventis Pharma and Hoechst Marion Roussel and the President and COO of Marion Merrell Dow, Inc. and a member of its board of directors. From 1973 to 1993, Mr. Markham was associated with Merck & Co., Inc., culminating in his position as President and COO. Prior to this role Richard held a number of positions, starting as a professional representative and then becoming district manager, product manager and director, executive director and then Vice President Marketing for the Merck Sharp & Dohme Division. He later was responsible for Merck's European pharmaceutical business before being named senior vice president of Merck & Co. and president of the Merck Human Health Division, responsible for worldwide marketing and sales of Merck's pharmaceutical products. Mr. Markham received a B.S. in Pharmacy and Pharmacal Sciences from Purdue University and has served as a member of the Dean's Advisory Council of the university. He has also been awarded an honorary Doctor of Science degree, the university's highest honor for achievement. Mr. Markham previously served as a member of the board of directors of Acura Pharmaceuticals, Inc. and Anacor Pharmaceuticals, Inc. In addition, Mr. Markham has been a member of the board of directors and executive committee of the Pharmaceutical Research and Manufacturers Association, a member of the Board of Trustees of the HealthCare Institute of New Jersey and a member of the board of directors of Aventis Pasteur and of Commerce Bank of Kansas City.
95
We believe that Mr. Markham's extensive experience within the life sciences industry, the experience he brings as a Board member of life sciences companies, his knowledge of finance and transactions, and his historic knowledge of our company and our product candidates qualify him to serve as a member of our Board of Directors.
Pierre Legault was named our Chief Executive Officer on October 18, 2013 and has been a member of our Board of Directors since November 2012. From April 2012 until October 2013, Mr. Legault was the Chief Executive Officer of Stone Management LLC, a consulting company. From January 2009 to April 2012, Mr. Legault, was the Chief Executive Officer of Prosidion Ltd., a U.K. mid-size biotechnology firm discovering, developing and commercializing products in the therapeutic areas of diabetes and obesity. From January 2009 to September 2010, he served as Executive VP, Chief Financial Officer and Treasurer with OSI Pharmaceuticals, a mid-size biotechnology company focused on oncology. He was also Senior Executive VP and Chief Administrative Officer of Rite Aid Corporation, a fortune 500 pharmaceutical retail company, from July 2007 to December 2008. From January 2006 to July 2007, Mr. Legault served as Executive VP of The Jean Coutu Group (PJC) Inc. and President of the Eckerd group, with overall management responsibilities for the Brooks Eckerd operations in the US.
Previously Mr. Legault held several senior positions for a period of 15 years with Sanofi-Aventis and predecessor companies, last serving as Worldwide President of Sanofi-Aventis Dermatology/Dermik (2003 to 2005). Prior positions included the Senior VP and Chief Financial Officer of Aventis Pharmaceuticals Inc. (2000 to 2003), Global Senior VP Finance and Treasury of Hoechst Marion Roussel, Inc. (1998 to 2000), VP and Chief Financial Officer, North America Finance, IT and Administration of Marion Merrell Dow, Inc. (1997 to 1998), and VP and Chief Financial Officer of Marion Merrell Dow Pharmaceutical Canada (1989 to 1996). Mr. Legault has served on several public, private and nonprofit company boards and audit committees, as well as on several advisory boards, including the following: Cyclacel Pharmaceutical Inc., a publicly traded biotech company (2006-2008), Forest Laboratories, Inc. (2012-2014), NPS Pharmaceuticals, Inc. (2014-2015) and Regado Biosciences, Inc. (2013-2015), Tobira Biosciences (2015 to present). Mr. Legault also belongs to several professional associations and he studied at McGill University, University of Montreal (HEC) and the Harvard Business School. He has a Six Sigma Green Belt, a BAA, MBA, CA and CPA diploma.
We believe that Mr. Legault's perspective and the experience he brings as our chief executive officer, together with his historic knowledge of our company and our product candidates, operational expertise and continuity to our Board of Directors, and his experience in managing and investing in companies within the life sciences industry, qualify him to serve as a member of our Board of Directors.
James Mitchum has served as a member of our Board of Directors since February 2014. Mr. Mitchum is currently the Chief Executive Officer of Heart to Heart International a non-profit humanitarian organization. From 2009 to July 2012, Mr. Mitchum served as President of the Americas for EUSA Pharma (USA), Inc., where he oversaw the streamlining of that business as well as the development, FDA approval and successful launch of a pediatric oncology drug in 2011. From 2005 to 2008, Mr. Mitchum served as President and Chief Executive Officer of Enturia, Inc., a privately owned drug-device company, based in Kansas City, Missouri. From 2004 to 2005, Mr. Mitchum served as the President and Chief Executive Officer of Sanofi-Aventis Group Japan. Mr. Mitchum has also served as a director on numerous private company and organization boards. Mr. Mitchum earned an MBA in Business from the University of Tennessee in Knoxville, Tennessee and a Bachelor of Science degree in Business and Math from Milligan College in Johnson City, Tennessee.
We believe that Mr. Mitchum's experience in managing companies in the life sciences industry, as well as his financial and operational expertise, qualify him to serve on our Board of Directors.
Robert R. Seltzer has served as a member of our Board of Directors since October 2013. Mr. Seltzer is a Partner at Care Capital, LLC, a life sciences venture capital firm and an affiliate of one of our principal stockholders, which he joined in July 2005. He was previously a management consultant at the Boston Consulting Group (1997 to 2000 and 2004 to 2005), and he was the Co-Founder and President of Trenza Corp (2000 to 2001). He has served on the board of directors of Minerva Neurosciences, Inc. and a number of private biopharmaceutical and drug development companies. Mr. Seltzer received his MBA from The Wharton School, a Master of Biotechnology from the University of Pennsylvania, and a B.S. in Molecular Biophysics and Biochemistry from Yale University.
We believe that Mr. Seltzer's perspective and the experience he brings as a Board member of life sciences companies, his knowledge of finance and transactions, and his historic knowledge of our company and our product candidates qualify him to serve as a member of our Board of Directors.
Eugen Steiner, M.D., Ph.D., has served as a member of our Board of Directors since 2007. Dr. Steiner is a venture partner of HealthCap, a group of multistage venture capital funds, investing globally in the life sciences. He is currently the Chief Executive Officer of Glionova AB, an early-stage Swedish biopharmaceutical company a portfolio company of HealthCap. He has more than 25 years of executive management experience, and since 1997 has served as CEO of certain companies in which HealthCap has invested, leading these companies mostly in start-up and early stages of development. He has been CEO of Affibody AB, Biostratum Inc., Calab Medical AB, Creative Peptides AB, Eurona Medical AB, Melacure Therapeutics AB, Nordic Vision Clincs AS, PyroSequencing AB and Visual Bioinformatics AB. Dr. Steiner has served on
96
several public, private and nonprofit company boards, including Alba Therapeutics, APL, Biolipox (chairman), BioPhausia, Biostratum (chairman), Biotage, Praktikertjänst, and Stockholm School of Entrepreneurship, and also belongs to several professional medical, industry and investor associations. He studied medicine and earned his MD as well as PhD degrees at the Karolinska Institute. Until 1987 Dr. Steiner practiced medicine and was active in medical research at the Karolinska Hospital, Stockholm, Sweden.
We believe that Dr. Steiner's experience, together with his historic knowledge of our company and our product candidates, and continuity to our Board of directors, and his experience in managing and investing in companies within the life sciences industry, qualify him to serve as a member of our Board of directors.
Marco Taglietti, M.D. has served on our Board of Directors since October 2014. Dr. Taglietti is the Chief Executive Officer of Scynexis, Inc., effective as of April 1, 2015. Dr. Taglietti also serves as a member of the Board of Directors of Delcath Systems, Inc. Prior to its recent acquisition, Dr. Taglietti served as Executive Vice President, Research and Development, and Chief Medical Officer of Forest Laboratories, Inc. He also served as President, Chief Medical Officer of the Forest Research Institute. Prior to joining Forest Labs in 2007, Dr. Taglietti held the position of Senior Vice President, Head of Global Research and Development, at Stiefel Laboratories, Inc. for three years. He joined Stiefel after 12 years at Schering-Plough Corporation where he last held the position of Vice President, Worldwide Clinical Research for Anti-Infectives, Oncology, CNS, Endocrinology and Dermatology. Dr. Taglietti began his career at Marion Merrell Dow Research Institute. Over the course of his career, he has brought to market 35 different products in the U.S. and internationally. He received his medical degree and board certifications from the University of Pavia in Italy.
We believe that Dr. Taglietti’s experience in managing companies in the life sciences industry, as well as his financial and operational expertise, qualify him to serve on our Board of Directors.
There are no family relationships between or among any of our directors or nominees. The principal occupation and employment during the past five years of each of our directors and nominees was carried on, in each case except as specifically identified above, with a corporation or organization that is not a parent, subsidiary or other affiliate of us. There is no arrangement or understanding between any of our directors or nominees and any other person or persons pursuant to which he or she is to be selected as a director or nominee.
There are no legal proceedings to which any of our directors is a party adverse to us or any of our subsidiaries or in which any such person has a material interest adverse to us or any of our subsidiaries.
Director Independence
Based upon information requested from and provided by each director concerning their background, employment and affiliations, including family relationships, our Board of Directors has determined that Messrs. Markham, Mitchum and Seltzer and Drs. Steiner and Taglietti are independent under the applicable rules and regulations of the NASDAQ Stock Market. Our Board of Directors also determined that Messrs. Markham and Mitchum and Dr. Taglietti, who comprise our Compensation Committee; and Messrs. Seltzer and Markham, who comprise our Nominating and Governance Committee, all satisfy the independence standards for such committees established by the SEC and the NASDAQ Marketplace Rules, as applicable. With respect to our Audit Committee, our Board of Directors has determined that Mr. Mitchum and Drs. Steiner and Taglietti satisfy the independence standards for such committee established by Rule 10A-3 under the Exchange Act, the Securities and Exchange Commission and the NASDAQ Marketplace Rules, as applicable. In making such determinations, our Board of Directors considered the relationships that each such non-employee director has with our company and all other facts and circumstances the Board of Directors deemed relevant in determining their independence.
Committees of the Board of Directors and Meetings
The following table sets forth the number of meetings held during calendar year 2014 by the Board of Directors and by each committee thereof. Each of the directors, who were serving on our Board of Directors during 2014, attended at least 75% of the total number of meetings of the Board of Directors and of the committees of which he was a member during the time each such individual was a member of the Board of Directors.
Number of Meetings Held | ||
Board of Directors | 10 | |
Audit Committee | 6 | |
Compensation Committee | 6 | |
Nominating and Corporate Governance | 3 | |
Audit Committee. Our Audit Committee has three members: James Mitchum, Eugen Steiner, M.D., Ph.D. and Marco Taglietti, M.D. Mr. Mitchum is the chairperson of the committee. Our Board of Directors has determined that Mr. Mitchum is
97
an audit committee financial expert, as defined by the rules of the SEC, and satisfies the financial sophistication requirements of applicable NASDAQ rules.
Our Board of Directors has determined that each of Mr. Mitchum and Drs. Steiner and Taglietti are independent directors under the NASDAQ Marketplace Rules and Rule 10A-3 of the Exchange Act.
Our Audit Committee is authorized to:
• | approve and retain the independent auditors to conduct the annual audit of our financial statements; |
• | review the proposed scope and results of the audit; |
• | review and pre-approve audit and non-audit fees and services; |
• | review accounting and financial controls with the independent auditors and our financial and accounting staff; |
• | review and approve transactions between us and our directors, officers and affiliates; |
• | recognize and prevent prohibited non-audit services; |
• | establish procedures for complaints received by us regarding accounting matters; |
• | oversee internal audit functions, if any; and |
• | prepare the report of the Audit Committee that the rules of the SEC require to be included in our annual meeting proxy statement. |
A copy of the Audit Committee's written charter is publicly available in the "Investors" section on our website at www.nephrogenex.com.
Compensation Committee. Our Compensation Committee has three members: Richard J. Markham, James Mitchum, and Marco Taglietti, M.D. Mr. Markham is the chairperson of the committee. Our Compensation Committee's role and responsibilities are set forth in the Compensation Committee's written charter and includes reviewing, approving and making recommendations regarding our compensation policies, practices and procedures to ensure that legal and fiduciary responsibilities of the Board of Directors are carried out and that such policies, practices and procedures contribute to our success. Our Compensation Committee also administers the Stock Plan. The Compensation Committee is responsible for the determination of the compensation of our chief executive officer, and shall conduct its decision making process with respect to that issue without the chief executive officer present. All members of the Compensation Committee qualify as independent under the definition promulgated by The NASDAQ Stock Market.
Our Compensation Committee's role and responsibilities are set forth in the Compensation Committee's written charter and include:
• | reviewing and recommending the compensation arrangements with management, including the compensation of our chief executive officer |
• | establishing and reviewing general compensation policies with the objective of aligning, where appropriate, the long-term interests of executive officers and other key employees with those of our stockholders and otherwise encouraging the achievement of superior results over an extended time period; and |
• | overseeing any of our equity incentive plans, as applicable. |
The Compensation Committee’s independent compensation consultant during fiscal year 2014 was the Hay Group (“Hay Group”). Hay Group is engaged by, and reports directly to, the Compensation Committee, which has the sole authority to hire or fire them and to approve fee arrangements for work performed. Hay Group assists the Compensation Committee in fulfilling its responsibilities under its charter, including advising on proposed compensation packages for executive officers, compensation program design and market practices generally. The Compensation Committee has authorized Hay Group to interact with management on behalf of the Compensation Committee, as needed in connection with advising the Compensation Committee, and Hay Group is included in discussions with management and, when applicable, the Compensation Committee’s outside legal counsel on matters being brought to the Compensation Committee for consideration.
98
The Compensation Committee has assessed the independence of Hay Group pursuant to SEC rules and concluded that Hay Group’s work for the Compensation Committee does not raise any conflict of interest.
A copy of the Compensation Committee's written charter is publicly available in the "Investors" section on our website at www.nephrogenex.com.
Nominating and Governance Committee. Our Nominating and Governance Committee is comprised of Richard J. Markham and Robert R. Seltzer. Mr. Seltzer is the chairperson of the committee. The Nominating and Governance Committee's role and responsibilities are set forth in the Nominating and Governance Committee's written charter and include evaluating and making recommendations to the full Board as to the size and composition of the Board of Directors and its committees, evaluating and making recommendations as to potential candidates, and evaluating the performance of current members of the Board of Directors. All members of the Nominating and Governance Committee qualify as independent under the definition promulgated by The NASDAQ Stock Market.
If a stockholder wishes to nominate a candidate for director who is not to be included in our proxy statement, it must follow the procedures described in our bylaws.
In addition, under our current corporate governance policies, the Nominating and Governance Committee may consider candidates recommended by stockholders as well as from other sources such as other directors or officers, third party search firms or other appropriate sources. For all potential candidates, the Nominating and Governance Committee may consider all factors it deems relevant, such as a candidate's personal integrity and sound judgment, business and professional skills and experience, independence, knowledge of the industry in which we operate, possible conflicts of interest, diversity, the extent to which the candidate would fill a present need on the Board of Directors, and concern for the long-term interests of the stockholders. In general, persons recommended by stockholders will be considered on the same basis as candidates from other sources. If a stockholder wishes to propose a candidate for consideration as a nominee by the Nominating and Governance Committee under our corporate governance policies, it should submit such recommendation in writing c/o Secretary, NephroGenex, Inc., 3200 Beechleaf Court, Suite 900, Raleigh, North Carolina, 27604.
Our Nominating and Governance committee has not adopted a formal diversity policy in connection with the consideration of director nominations or the selection of nominees. However, the Nominating and Governance Committee will consider issues of diversity among its members in identifying and considering nominees for director, and will strive where appropriate to achieve a diverse balance of backgrounds, perspectives, experience, age, gender, ethnicity and energy industry experience on our Board of Directors and its committees.
A copy of the Nominating and Governance Committee's written charter is publicly available on the Company's website at www.nephrogenex.com.
Board Leadership Structure and Role in Risk Oversight
The positions of chairman of the board and chief executive officer are presently separated at our company. We believe that separating these positions allows our chief executive officer to focus on our day-to-day business, while allowing our chairman of the board to lead the Board of Directors in its fundamental role of providing advice to, and independent oversight of, management. Our Board of Directors recognizes the time, effort and energy that the chief executive officer is required to devote to his position in the current business environment, as well as the commitment required to serve as our chairman, particularly as the Board of Directors' oversight responsibilities continue to grow. Our Board of Directors also believes that this structure ensures a greater role for the independent directors in the oversight of our company and active participation of the independent directors in setting agendas and establishing priorities and procedures for the work of our Board of Directors. This leadership structure also is preferred by a significant number of our stockholders. Our Board of Directors believes its administration of its risk oversight function has not affected its leadership structure.
While our restated bylaws and corporate governance guidelines do not require that our chairman and chief executive officer positions be separate, our Board of Directors believes that having separate positions is the appropriate leadership structure for us at this time and demonstrates our commitment to good corporate governance.
Risk is inherent with every business, and how well a business manages risk can ultimately determine its success. We face a number of risks, including risks relating to product candidate development, having no commercial manufacturing experience, marketing or sales capability or experience and dependence on key personnel, as more fully discussed under Item 1.A. "Risk Factors" in our annual report on this Annual Report on Form 10-K. Management is responsible for the day-to-day management of risks we face, while our Board of Directors, as a whole and through its committees, has responsibility for the
99
oversight of risk management. In its risk oversight role, our Board of Directors has the responsibility to satisfy itself that the risk management processes designed and implemented by management are adequate and functioning as designed.
Our Board of Directors is actively involved in oversight of risks that could affect us. This oversight is conducted primarily through committees of the Board of Directors, but the full board of directors has retained responsibility for general oversight of risks. Our Board of Directors satisfies this responsibility through full reports by each committee chair regarding the committee's considerations and actions, as well as through regular reports directly from officers responsible for oversight of particular risks within our company as our Board of Directors believes that full and open communication between management and the Board of Directors is essential for effective risk management and oversight.
Stockholder Communications to the Board
Generally, stockholders who have questions or concerns should contact us at http://investors.nephrogenex.com/contactus.cfm. However, any stockholders who wish to address questions regarding our business directly with the Board of Directors, or any individual director, should direct his or her questions in writing to our Secretary, John P. Hamill, NephroGenex, Inc., 3200 Beechleaf Court, Suite 900, Raleigh, North Carolina, 27604. Communications will be distributed to the Board, or to any individual director or directors as appropriate, depending on the facts and circumstances outlined in the communications. Items that are unrelated to the duties and responsibilities of the Board of Directors may be excluded, such as:
• junk mail and mass mailings
• resumes and other forms of job inquiries
• surveys
• solicitations or advertisements.
In addition, any material that is unduly hostile, threatening, or illegal in nature may be excluded, provided that any communication that is filtered out will be made available to any outside director upon request.
Executive Officers
The following table sets forth certain information regarding our executive officers who are not also directors.
Name | Age | Position | |
J. Wesley Fox, Ph.D | 63 | President and Chief Scientific Officer | |
John P. Hamill | 51 | Chief Financial Officer, Treasurer and Secretary | |
We have employment agreements with all of our executive officers.
J. Wesley Fox, Ph.D. is a co-founder of the Company and has served as our President and Chief Scientific Officer since October 2013. From 2005 until October 2013, Dr. Fox served as our President and Chief Executive Officer. Dr. Fox has over 30 years of experience in the organization, funding and management of early and developmental stage biotechnology companies. Prior to co-founding the Company, Dr. Fox was co-founder and Chief Scientific Officer of BioStratum, Inc. where he established and directed research and development operations that identified and advanced Pyridorin, recombinant laminins for tissue regeneration (now being sold and applied to stem cells by BioLamina AB), and licensed inhibitors of GPBP kinase with applications to autoimmune disease, fibrosis, and cancer (now being developed by FibroStatin SL). Dr. Fox is also an advisor for BioLamina AB and FibroStatin SL. Prior to these entrepreneurial activities, Dr. Fox held research and development positions with Abbott Laboratories and Idexx Laboratories, Inc. Dr. Fox received a B.A. in Chemistry from Washington and Jefferson College in Washington, Pennsylvania. Dr. Fox received his Ph.D. in Biochemistry from University of Kansas Medical School in Kansas City, Kansas.
John P. Hamill has served as our Chief Financial Officer since January 2014. From June 2013 until January 2014, Mr. Hamill served as Co-President and Chief Financial Officer of Savient Pharmaceuticals, Inc. ("Savient") and as Senior Vice President and Chief Financial Officer of Savient since September 2012. Savient filed for bankruptcy on October 14, 2013, while Mr. Hamill was Savient's Co-President and Chief Financial Officer and, shortly thereafter, Savient sold its assets in bankruptcy. From 2010 to 2012, Mr. Hamill served as a financial consultant for various private companies. From 2001 until 2009, Mr. Hamill worked for PharmaNet Development Group, Inc., where he served as Executive Vice President and Chief Financial Officer from 2006 until 2009. During the period in which Mr. Hamill served as Executive Vice President and Chief Financial Officer, he also maintained responsibilities as the Chief Financial Officer of PharmaNet Development Group, Inc.'s wholly-owned subsidiary, PharmaNet, Inc. Mr. Hamill earned his B.S. with a dual major in Accounting/Business and Computer Science from DeSales University (formerly Allentown College of St. Francis de Sales) in 1986. Mr. Hamill is a Certified Public Accountant and is a member of the Pennsylvania Institute of Certified Public Accountants and the American Institute of Certified Public Accountants.
100
Item 11. Executive Compensation
Summary Compensation Table
The following table sets forth the compensation paid or accrued during the last two fiscal years to our named executive officers.
Salary | Bonus (1) | Stock Awards (2) | Option Awards (3) | All Other Compensation (4) | Total Compensation | ||||||||
Name and Principal Position | $ | $ | $ | $ | $ | $ | |||||||
Pierre Legault(5) | 2014 | 395,000 | 409,090 | 30,395 | 1,932,321 | 40,935 | 2,807,741 | ||||||
Chief Executive Officer | 2013 | 214,516 | 75,000 | 496,080 | 125,996 | 3,098 | 914,690 | ||||||
J. Wesley Fox(6) | 2014 | 352,504 | 147,170 | — | 392,693 | 43,972 | 936,339 | ||||||
President and Chief Scientific Officer | 2013 | 340,680 | 200,638 | — | — | 16,987 | 558,305 | ||||||
John P. Hamill | 2014 | 291,875 | 140,100 | — | 512,728 | 40,793 | 985,496 | ||||||
Chief Financial Officer | 2013 | — | — | — | — | — | — | ||||||
___________________________________
(1) | Bonus payments to our named executive officer are subject to the discretion of the Board of Directors and Compensation Committee. Amounts represent cash bonuses earned in 2014, which were paid during 2015, and cash bonuses earned in 2013, which were paid in 2014, based on achievement of performance goals and other factors deemed relevant by our Board of Directors and the Compensation Committee. The corporate performance goals for 2013 and 2014 included key strategic and financial goals that related to development programs, obtaining approval from the FDA for Pyridorin, securing new sources of capital/financing and achieving the budgeted financial targets. In addition, Mr. Legault's 2014 bonus included $112,840 for cash compensation earned under a restricted stock unit agreement. |
(2) | The amount for 2014 represents the fair market value of common stock issued to Mr. Legault under a restricted stock agreement.These amounts represent the aggregate grant date fair value of restricted stock awards granted to Mr. Legault in the year ended December 31, 2013 computed in accordance with FASB ASC Topic 718. A discussion of the assumptions used in determined grant date fair value may be found in Note 2 to our Financial Statements, included in our Annual Report on Form 10-K. |
(3) | These amounts represent the aggregate grant date fair value of options granted in fiscal year ended December 31, 2014 and 2013, repectively computed in accordance with FASB ASC Topic 718. A discussion of the assumptions used in determining grant date fair value may be found in Note 2 to our Financial Statements, included in our Annual Report on Form 10-K. |
(4) | Amount represents 401(k) match, medical, dental, vision, life and long-term disability benefits paid by us on behalf of each employee. In addition, amounts include payment for unused vacation for Mr. Legault, Mr. Hamill and Dr. Fox. |
(5) | Mr. Legault was appointed as our Chief Executive Officer on October 18, 2013. Prior to this date, Mr. Legault served as the chairman of our Board of Directors. Compensation information presented for 2013 reflects compensation paid to Mr. Legault as a consultant for part of the year and as our Chief Executive Officer for part of the year. |
(6) | Dr. Fox was appointed as our President and Chief Scientific Officer on October 18, 2013. Prior to this date, Dr. Fox served as our President and Chief Executive Officer. |
Employment Arrangements with Our Named Executive Officers
Pierre Legault. On November 7, 2013, we entered into an employment agreement with Pierre Legault to reflect his role and responsibilities as Chief Executive Officer. Mr. Legault's service as Chief Executive Officer commenced on October 18, 2013 (the "Commencement Date"). The employment agreement will continue until Mr. Legault's employment is terminated by either party pursuant to the terms and provisions of the employment agreement. The employment agreement provides Mr. Legault an annual base salary of $400,000. Mr. Legault’s annual base salary was increased by our Board of Directors on December 12, 2014 to $500,000, effective as of January 1, 2015. For each completed fiscal year during Mr. Legault's service to us, Mr. Legault is eligible to earn a bonus based on achievement of reasonable individual and corporate performance objectives established by our Board of Directors and communicated to Mr. Legault. The target amount of Mr. Legault's annual bonus for each fiscal year will be 50% of the base salary paid or payable to Mr. Legault for his service in that year. To receive any annual bonus otherwise earned for a given year, Mr. Legault must remain employed with us through the last business day of that year.
101
Pursuant to his employment agreement, Mr. Legault received a grant of restricted stock units which represent the right to receive 24,000 shares of our common stock, subject to the terms and conditions of a restricted stock unit agreement and grant notice connected therewith (the "RSU Award"). 25% of the restricted stock units granted to Mr. Legault vested on October 21, 2014, and the remaining 75% of the restricted stock units granted to Mr. Legault vest in equal monthly installments, on the first day of each calendar month, beginning on November 1, 2014 and continuing for 36 months thereafter, provided that Mr. Legault remains in service with us. We will also make special cash bonus payments to Mr. Legault on each date that restricted stock units are delivered to Mr. Legault in an amount equal to the product of the number of shares of common stock underlying the restricted stock units delivered on such date and $16.12, less applicable taxes and withholdings; but in no event shall this bonus amount exceed $387,000 in the aggregate, before adjustment for applicable taxes and withholdings. The per share bonus amount shall be equitably adjusted in the event of any capitalization adjustment of the company.
The employment agreement further provides that during Mr. Legault's continuing service to us, if the number of issued and outstanding shares of our capital stock increases, without limitation, in connection with an initial public offering of our common stock or a stock dividend with respect to our preferred stock, but not including the conversion of convertible debt or convertible promissory notes issued prior to the one year anniversary of the Commencement Date (the "Additional Shares"), we shall grant Mr. Legault an option under the Stock Plan or any successor plan of ours (the "True Up Option") covering the number of shares of common stock equal to three ninety-sevenths of the Additional Shares, rounded to the nearest whole share. In connection with the completion of our initial public offering, on February 14, 2014, we granted Mr. Legault, a True Up Option to purchase 114,234 shares of our common stock with an exercise price of $12.00 per share. With respect to 76,156 of the shares subject to the True Up Option, 1/48th of the shares vest when Mr. Legault completes each month of continuous service to us, with 23,911 shares vested as of the date of grant. With respect to the 38,078 remaining shares subject to the True Up Option, 25% of the shares vested on October 21, 2014 and 75% of the shares vest in equal monthly installments on the first day of each calendar month for 36 months, beginning November 1, 2014, provided however, that Mr. Legault must remain in service to us on each applicable vesting date.
If, while any portion of the True Up Option or RSU Award is outstanding, any of our convertible debt issued prior to the execution of the employment agreement is actually converted into shares of our stock, Mr. Legault will be entitled to an additional option to purchase shares of our common stock in an amount equal to three ninety-sevenths of the shares into which such debt is converted (the "Additional Option"). The exercise price for each such Additional Option shall be $2.02 per share (subject to adjustment for stock splits, reverse splits, mergers, reorganizations, recapitalizations and similar events or transactions). Any Additional Option may only be exercised, to the extent vested, during the period beginning on the earliest of the following to occur after the grant of such Additional Option: (i) Mr. Legault's "separation from service" (as defined by Treasury Regulation § 1.409A-1(h), or, if Treasury Regulation § 1.409A-3(i)(2) applies, six months and one day following Mr. Legault's separation from service), (ii) the time immediately preceding a change in control, but only if such change in control also constitutes a "change in control event" (as that term is defined in Treasury Regulation Section 1.409A-3(i)(5)(i)), or (iii) the first day of the calendar year in which the expiration date of the employment agreement occurs. Once exercisable, each such Additional Option will continue to be exercisable until (x) the last day of the calendar year in which the option became exercisable (if such Additional Option became exercisable pursuant to Mr. Legault's separation from service, as defined above), (y) the closing of a change in control (if such Additional Option became exercisable pursuant to a change in control), or (z) the tenth anniversary of the date of the RSU Award (if such Additional Option became exercisable pursuant to the first day of the tenth year after the date of the RSU Award). On February 14, 2014, in connection with the completion of our initial public offering and the conversion of all of our convertible notes into common stock, we granted Mr. Legault an Additional Option to purchase 37,029 shares of our common stock with an exercise price of $2.02 per share and subject to the terms discussed above in order for such option to comply with Section 409A of the Internal Revenue Code . With respect to 24,686 of the shares subject to Additional Option, 1/48th of the shares vest when Mr. Legault completes each month of continuous service to us, with 7,714 shares vested as of the date of grant. With respect to the 12,343 remaining shares subject to the Additional Option, 25% of the shares vested on October 21, 2014; and 75% of the shares vest in equal monthly installments on the first day of each calendar month for 36 months, beginning November 1, 2014, provided however, that Mr. Legault must remain in service to us on each applicable vesting date.
Mr. Legault also participates in the employee benefit plans, policies or arrangements maintained by us for our management-level employees. Further, we have agreed to pay directly, or reimburse, Mr. Legault for travel and business expenses in accordance with our generally applicable policies relating to such expenses.
Upon the termination of Mr. Legault's employment, he will receive payment for any accrued but unpaid wages, accrued but unused vacation and for any incurred but unreimbursed expenses, subject to our policies for expense reimbursements. If Mr. Legault's employment is terminated by us upon 53 days prior written notice or is terminated by Mr. Legault for "Good Reason," we will (a) make a cash lump sum payment to Mr. Legault equal to 150% of his base salary (at the rate in effect immediately prior to such termination), less applicable taxes and withholdings, and (b) for a period of twelve (12) months (or by lump sum covering twelve (12) months if allowed by applicable law) pay Mr. Legault the monthly benefit stipend equal to Mr. Legault's premiums for continuation of medical and dental benefits pursuant to Mr. Legault's COBRA
102
election, including payments to account for applicable taxes and withholdings. Such payments will be conditioned on Mr. Legault's execution of a general release of claims against us and our affiliates (excluding Mr. Legault's rights as a stockholder, rights with respect to equity incentive awards and rights to indemnification for acts performed in Mr. Legault's capacity as an director, officer or employee) in a mutually acceptable form and on such release becoming effective no later than 53 days following such termination. Notwithstanding the foregoing, for the twenty-four (24) month period following any change in control: we will (a) make a cash lump sum payment to Mr. Legault equal to 225% of his base salary (at the rate in effect immediately prior to such termination) and (b) continue to pay Mr. Legault the monthly benefit stipend described above for eighteen (18) months following such termination.
"Good Reason" is defined under the employment agreement as (i) any adverse change in Mr. Legault's title, authority or duties (including, without limitation, the assignment to Mr. Legault of duties materially inconsistent with his position) or (ii) any other material breach by us of any term or condition of the employment agreement.
Receipt of the severance benefits described above may be modified by us to comply with Section 409A of the Internal Revenue Code of 1986, as amended, or the Code.
J. Wesley Fox, Ph.D. On April 30, 2007, we entered into an employment agreement with Dr. Fox who is currently our president and chief executive officer. The employment agreement provides for an annual base salary at a gross annual rate of not less than $250,000. Dr. Fox’s annual base salary was increased by our Board of Directors on December 12, 2014 to $367,425, effective as of January 1, 2015. In addition, the employment agreement granted Dr. Fox options to purchase 102,992 shares of our common stock pursuant to the terms and conditions of the Stock Plan, with an exercise price being the fair market value of the common stock underlying such options on that date of grant. One quarter of the common stock underlying the options vested after Dr. Fox’s first year of service to us under the employment agreement, and the remainder of shares underlying the options vested on a quarterly basis over the next three years. All of these options are vested.
Under the employment agreement, Dr. Fox is eligible to be considered for an annual incentive bonus which will require approval by our Board of Directors. If we terminate Dr. Fox for any reason other than for cause or permanent disability, he shall be entitled to his base salary at the rate currently in effect at the time of termination for a period of twelve months following such termination, in addition to reimbursement for health care premiums under COBRA for Dr. Fox and his dependents until the earliest of (i) twelve months following termination, (ii) the expiration of Dr. Fox's continuation coverage under COBRA, or (iii) the date Dr. Fox receives substantially equivalent health insurance coverage in connection with new employment or self-employment.
Under the employment agreement, "Cause" is defined as: (i) an unauthorized use or disclosure by Dr. Fox of our confidential information or trade secrets, which use or disclosure causes us material harm; (ii) a material breach by Dr. Fox of any agreement between Dr. Fox and us; (iii) a material failure by Dr. Fox to comply with our written policies or rules; (iv) Dr. Fox's conviction of, or plea of "guilty" or "no contest" to, a felony under the laws of the United States or any state thereof; (v) Dr. Fox's gross negligence or willful misconduct; (vi) a continuing failure by Dr. Fox to perform assigned duties after receiving written notification of such failure from our Board of Directors; (vii) a failure by Dr. Fox to cooperate in good faith with a governmental or internal investigation of us or our directors, officers or employees after request for such cooperation by us; or (viii) Dr. Fox's debarment, or conviction of a crime that could lead to disbarment, under any governmental payment program.
Either we or Dr. Fox shall be entitled to terminate Dr. Fox's employment at any time and for any reasons, with or without cause. We may terminate Dr. Fox's employment with us by giving Dr. Fox notice of such termination in writing. Dr. Fox may terminate his employment with us by providing us with 30 days' advance notice in writing.
Under the employment agreement, Dr. Fox has agreed to refrain from directly or indirectly competing with us or soliciting our employees or independent contractors during his employment and for a two-year period thereafter. In connection with the employment agreement, Dr. Fox entered into our standard proprietary information and inventions agreement.
John P. Hamill. On December 12, 2013, we entered into an employment agreement with John P. Hamill to reflect his role and responsibilities as Chief Financial Officer. Mr. Hamill’s service as Chief Financial Officer commenced on January 21, 2014. The employment agreement will continue until Mr. Hamill’s employment is terminated by either party pursuant to the terms and provisions of the employment agreement. The employment agreement provides Mr. Hamill an annual base salary of $300,000. Mr. Hamill’s annual base salary was increased by our Board of Directors on December 12, 2014 to $331,200, effective as of January 1, 2015. For each completed fiscal year during Mr. Hamill’s service to us, Mr. Hamill is eligible to earn a bonus based on achievement of reasonable individual and corporate performance objectives established by our Board of Directors and communicated to Mr. Hamill. The target amount of Mr. Hamill's annual bonus for each fiscal year will be 40% of the base salary paid or payable to Mr. Hamill for his service in that year. To receive any annual bonus otherwise earned for a given year, Mr. Hamill must remain employed with us through the last business day of that year.
Mr. Hamill also participates in the employee benefit plans, policies or arrangements maintained by us for our management-level employees, subject to the terms and conditions of such plans, policies or arrangements. Further, we have
103
agreed to pay directly, or reimburse, Mr. Hamill for travel and business expenses in accordance with our generally applicable policies relating to such expenses.
Upon the termination of Mr. Hamill’s employment, he will receive payment for any accrued but unpaid wages, accrued but unused vacation and for any incurred but unreimbursed expenses, subject to our policies for expense reimbursements. If Mr. Hamill’s employment is terminated by us upon 30 days prior written notice or is terminated by Mr. Hamill for “Good Reason,” we will (a) make a cash lump sum payment to Mr. Hamill equal to 140% of his base salary (at the rate in effect immediately prior to such termination), less applicable taxes and withholdings, and (b) for a period of twelve (12) months (or by lump sum covering twelve (12) months if allowed by applicable law) pay Mr. Hamill the monthly benefit stipend equal to Mr. Hamill’s premiums for continuation of medical and dental benefits pursuant to Mr. Hamill’s COBRA election, including payments to account for applicable taxes and withholdings. Such payments will be conditioned on Mr. Hamill’s execution of a general release of claims against us and our affiliates (excluding Mr. Hamill’s rights as a stockholder, rights with respect to equity incentive awards and rights to indemnification for acts performed in Mr. Hamill’s capacity as an director, officer or employee) in a mutually acceptable form and on such release becoming effective no later than 60 days following such termination. Notwithstanding the foregoing, for the twenty-four (24) month period following any change in control we will make a cash lump sum payment to Mr. Hamill equal to 210% of his base salary (at the rate in effect immediately prior to
such termination).
“Good Reason” is defined under the employment agreement as (i) any material adverse change in Mr. Hamill’s title, authority or duties (including, without limitation, the assignment to Mr. Hamill of duties materially inconsistent with his position) or (ii) any other material breach by us of any term or condition of the employment agreement.
Receipt of the severance benefits described above may be modified by us to comply with Section 409A of the
Internal Revenue Code of 1986, as amended, or the Code.
Pension Benefits
None of our named executive officers participates in or has account balances in qualified or non-qualified defined benefit plans sponsored by us.
Outstanding Equity Awards at 2014 Fiscal Year-End
The following table shows grants of stock options and grants of restricted stock unit awards outstanding on the last day of the fiscal year ended December 31, 2014, including both awards subject to performance conditions and non-performance-based awards, to each of the executive officers named in the Summary Compensation Table.
104
Option Awards | Restricted Stock Units | |||||||||||||
Name | Number of Securities Underlying Underlying Unexercised Options Exercisable | Number of Securities Underlying Underlying Unexercised Options Unexercisable | Option Exercise Price | Option Expiration Date | Number of Unvested Securities | Market Value of Units that are Unvested | ||||||||
Pierre Legault | 47,531 | 43,730 | (1) | 2.02 | 5/1/2023 | 17,000 | (2) | 226,950 | ||||||
12,857 | 11,829 | (1) | 2.02 | 11/6/2023 | — | — | ||||||||
3,599 | 8,744 | (3) | 2.02 | 5/1/2023 | — | — | ||||||||
39,664 | 36,492 | (1) | 12.00 | 11/6/2023 | — | — | ||||||||
11,105 | 26,973 | (3) | 12.00 | 5/1/2023 | — | — | ||||||||
— | 85,652 | (5) | 4.67 | 12/12/2024 | — | — | ||||||||
— | 64,348 | (5) | 4.67 | 12/12/2024 | — | — | ||||||||
J. Wesley Fox, Ph.D. | 769 | — | 32.50 | 2/1/2015 | — | — | ||||||||
27,227 | — | 0.39 | 8/13/2017 | — | — | |||||||||
27,502 | — | 0.39 | 4/18/2018 | — | — | |||||||||
75,765 | — | 0.39 | 4/18/2018 | — | — | |||||||||
32,538 | — | 1.95 | 6/5/2019 | — | — | |||||||||
20,812 | — | 1.95 | 12/11/2019 | — | — | |||||||||
38,769 | — | 1.82 | 12/8/2021 | — | — | |||||||||
10,961 | 3,654 | (4) | 1.82 | 12/8/2021 | — | — | ||||||||
— | 15,384 | (5) | 11.90 | 2/14/2024 | — | — | ||||||||
— | 28,000 | (5) | 4.49 | 8/14/2024 | — | — | ||||||||
— | 45,000 | (5) | 4.67 | 12/12/2024 | — | — | ||||||||
John. P. Hamill | — | 30,769 | (5) | 11.90 | 2/14/2024 | — | — | |||||||
— | 24,000 | (5) | 4.49 | 8/14/2024 | — | — | ||||||||
— | 45,000 | (5) | 4.67 | 12/12/2024 | — | — | ||||||||
_______________________________
(1 | ) | 1/48th of the option covered by these grants vest monthly commencing November 26, 2012. |
(2 | ) | 25% of the restricted stock units granted to Mr. Legault vested on October 21, 2014, and the remaining 75% of the restricted stock units granted to Mr. Legault vest in 36 equal monthly installments, on the first day of each calendar month, beginning on November 1, 2014. |
(3 | ) | 25% of the options covered by these grants vested on October 21, 2014 and the remaining 75% vest in 36 equal monthly installments beginning November 1, 2014. |
(4 | ) | Options vest monthly through December 14, 2015. |
(5 | ) | The options covered by these grants vest and become exercisable at a rate of 25% of the total grant on the first anniversary of the vesting start date and the remaining 75% vesting in 36 equal monthly installments over the first 36 months following the first anniversary of the vesting start date. |
105
Director Compensation
2014 Non-Employee Director Compensation Policy
Set forth below is our 2014 Non-Employee Director Compensation Policy which became effective on October 1, 2014.
Annual Amount | |||
Annual Retainer for each Board Member: | $ | 40,000 | |
Additional Retainer for the Chairman of the Board: | 25,000 | ||
Audit Committee Chair: | 15,000 | ||
Compensation Committee Chair: | 10,000 | ||
Nominating and Corporate Governance Committee Chair: | 7,500 | ||
In addition, each member will receive the value of an annual stock option award of $37,500 with the number of stock options to be determined based on a Black-Scholes calculation on the date of the grant. All options will vest on the first anniversary of the grant date. Newly elected or appointed directors will receive two times the regular annual option grant or $75,000 of fair value, at the time of their election. It is contemplated that non-employee directors grants will be made annually at the time of the Registrant’s annual meeting. At that time, consideration may also be given to adjust the compensation of the most recently elected director to reflect the new policy.
2014 Non-Employee Director Compensation
The following table presents information regarding the compensation of our non-employee directors in 2014.
Fees Earned or | ||||||
Name | Paid in Cash ($)(1) | Option Awards ($)(2) | Total ($) | |||
Richard J. Markham (3) | 67,750 | 26,782 | 94,532 | |||
James Mitchum | 47,750 | 26,782 | 74,532 | |||
Robert R. Seltzer (3) | 43,250 | 26,782 | 70,032 | |||
Eugen Steiner, M.D., Ph.D. (4) | 37,750 | 26,782 | 64,532 | |||
Marco Taglietti, M.D. | 10,000 | 74,998 | 84,998 | |||
_______________________________
(1) | Amounts in this column represent fees earned under all 2014 Non-Employee Director Compensation Policies. |
(2) | Amounts in this column represent the grant date fair value of option awards granted to non-employee directors during 2014, computed in accordance with FASB ASC Topic 718. These amounts do not necessarily correspond to the actual value that may be realized by non-employee directors. The assumptions made in valuing the option awards reported in this column are discussed in the Company's audited financial statements (Note 2), Summary of Significant Accounting Policies under subsection "Stock-Based Compensation," in this Annual Report on Form 10-K. |
(3) | All directors fees paid to Messrs. Markham and Seltzer are remitted by Messrs. Markham and Seltzer to Care Capital. In addition, any proceeds received upon the exercise of option awards will be remitted by Messrs. Markham and Seltzer to Care Capital. |
(4) | Pursuant to an agreement between Dr. Steiner and the Company, all of these director fees were paid directly to Setraco EHF. |
The aggregate number of shares subject to outstanding option awards held by our non-employee directors as of December 31, 2014 was as follows:
Number of Options | ||
Outstanding at December 31, 2014 | ||
Richard J. Markham | 3,076 | |
James Mitchum | 3,076 | |
Robert R. Seltzer | 3,076 | |
Eugen Steiner, M.D., Ph.D. | 3,076 | |
Marco Taglietti, M.D. | 22,656 | |
106
107
EQUITY COMPENSATION PLAN INFORMATION
The following table provides certain aggregate information with respect to all of the Company's equity compensation plans in effect as of December 31, 2014.
a | b | c | |||||
Number of securities | |||||||
remaining available for | |||||||
Number of securities to be | Weighted-average | future issuance under equity | |||||
issued upon exercise of | exercise price of | compensation plans | |||||
outstanding options, | outstanding options, | (excluding securities | |||||
Plan category | warrants and rights | warrants and rights | reflected in column (a)) | ||||
Equity compensation plans approved by security holders(1) | 1,289,581 | $ | 4.35 | 169 | |||
Equity compensation plans not approved by security holders | — | — | — | ||||
Total | 1,289,581 | $ | 4.35 | 169 | |||
__________________________________
(1) Consists of options to purchase 1,272,581 shares of our common stock granted under the 2007 Equity Incentive Plan. Also consists of unvested restricted stock units for up to 17,000 shares granted to Mr. Legault in November 2013, outside of the Stock Plan but with the approval of the stockholders.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of March 15, 2015, for (a) the executive officers named in Item 11 of this Annual Report on Form 10-K, (b) each of our directors and director nominees, (c) all of our current directors and executive officers as a group and (d) each stockholder known by us to own beneficially more than 5% of our common stock. Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. We deem shares of common stock that may be acquired by an individual or group within 60 days of March 15, 2015 pursuant to the exercise of options or warrants to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them based on information provided to us by these stockholders. Percentage of ownership is based on 8,863,614 shares of common stock outstanding on March 15, 2015.
Unless otherwise indicated, the address for each director and executive officer listed is: c/o NephroGenex, Inc., 3200 Beechleaf Ct., Suite 900, Raleigh, NC 27604.
108
Shares Beneficially Owned | ||||
Name and Address | Number | Percent | ||
Directors and Executive Officers | ||||
Pierre Legault (1) | 145,517 | 1.64 | % | |
J. Wesley Fox, Ph.D. (2) | 239,903 | 2.71 | % | |
John P. Hamill (3) | 9,615 | * | ||
Richard J. Markham (4) | 961 | * | ||
James Mitchum (5) | 961 | * | ||
Robert R. Seltzer (6) | 961 | * | ||
Eugen Steiner M.D., Ph.D. (7) | 961 | * | ||
Marco Taglietti, M.D. | — | * | ||
All directors and current executive officers as a group (8 persons) (8) | 398,879 | 4.5 | % | |
Five Percent Stockholders | ||||
Funds affiliated with Care Capital, LLC (9) | 4,241,097 | 47.8 | % | |
Funds affiliated with Rho Ventures (10) | 1,052,966 | 11.9 | % | |
Funds affiliated with Visium Asset Management (11) | 658,969 | 7.4 | % | |
BioStratum, Incorporated (12) | 538,002 | 6.1 | % | |
* | Represents beneficial ownership of less than 1% of the shares of Common Stock. |
109
(1) | Includes (a) options to purchase 136,017 shares of common stock that are exercisable within 60 days of March 15, 2015; (b) 1,000 shares of common stock issuable under a Restricted Stock Unit Grant within 60 days of March 15, 2015 and (c) 8,500 shares of common stock. | |
(2 | ) | Consists of options to purchase 239,903 shares of common stock that are exercisable within 60 days of March 15, 2015. |
(3 | ) | Consists of options to purchase 961 shares of common stock that are exercisable within 60 days of March 15, 2015. |
(4 | ) | Consists of options to purchase 961 shares of common stock that are exercisable within 60 days of March 15, 2015. Mr. Markham is one of four managing members at Care Capital III, LLC. Care Capital III, LLC is the general partner of Care Capital Investments III, LP and Care Capital Offshore Investments III, LP (collectively referred to herein as “Care Capital”). Mr. Markham disclaims beneficial ownership of the shares held by Care Capital. |
(5 | ) | Consists of options to purchase 961 shares of common stock that are exercisable within 60 days of March 15, 2015. |
(6 | ) | Consists of options to purchase 961 shares of common stock that are exercisable within 60 days of March 15, 2015. |
(7 | ) | Consists of options to purchase 961 shares of common stock that are exercisable within 60 days of March 15, 2015. |
(8 | ) | Consists of options to purchase 389,379 shares of common stock beneficially owned by our officers and directors that are exercisable within 60 days of March 15, 2015 and 1,000 share of commons stock issuable to Mr. Legault under a Restricted Stock Unit Grant. |
(9 | ) | This information is based on the Schedule 13D filed with the SEC on February 24, 2014, by Care Capital III LLC, a Delaware limited liability company (“Care Capital III LLC”), Care Capital Investments III LP, a Delaware limited partnership (“Care Capital Investments III LP”), and Care Capital Offshore Investments III LP, a Cayman Islands exempted limited partnership (“Care Capital Offshore Investments III LP”). Mr. Markham, Jan Leschly, Jerry N. Karabelas and David R. Ramsay are the four managing members at Care Capital III, LLC, and in their capacity as such, may be deemed to exercise shared voting and investment power over the shares held by Care Capital. The address of Care Capital is 47 Hulfish Street, Princeton, New Jersey 08542. |
(10 | ) | This information is based on the Schedule 13G/A filed with the SEC on February 13, 2015 by Visium Balanced Master Fund, Ltd. (“VBMF”), Visium Asset Management, LP (“VAM”), JG Asset, LLC (“JG Asset”), and Jacob Gottlieb (the “Visium 13G/A”). VAM, JG Asset and Mr. Gottlieb disclaim beneficial ownership of the securities, except to the extent of his or its pecuniary interest therein. The mailing address of the beneficial owner is 888 Seventh Avenue, New York, NY 10019. |
(11 | ) | This information is based on the Schedule 13D filed with the SEC on January 7, 2015, by Rho Ventures V, L.P. (“RV V”), Rho Ventures V Affiliates, L.L.C. (“RV V Affiliates”), RMV V, L.L.C. (“RMV”) and Rho Capital Partners LLC (“RCP,” and together with RV V, RV V Affiliates, RMV, collectively, the “Rho Entities”) (the “Rho 13D”). The address for the Rho Ventures group is Carnegie Hall Tower, 152 West 57th Street, 23rd Floor, New York, New York 10019. |
(12 | ) | This information is based on the Schedule 13G filed with the SEC on May 22, 2014, by Biostratum, Incorporated (“Biostratum”). The board of directors of BioStratum, consisting of Dr. Steiner, Per Samuelsson, Eggert Dagbjartson, Birgir Haraldsson and Dr. John Mazur, has sole voting and investment power with respect to the shares held by BioStratum, and each disclaims beneficial ownership of shares held by BioStratum, except to the extent of his pecuniary interest therein. The address of BioStratum is c/o Eugen Steiner, Timmermansgatan 2A, SE 11825 Stockholm, Sweden. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
In addition to the director and executive officer compensation arrangements discussed above in "Executive and Director Compensation," during the past two years, we have been a party to the following transactions in which the amount involved exceeded $120,000 and in which any director, executive officer or holder of more than 5% of our voting securities, whom we refer to as our principal stockholders, or affiliates or immediate family members of our directors, executive officers and principal stockholders had or will have a material interest. We believe that all of these transactions were on terms as favorable as could have been obtained from unrelated third parties.
Issuance of Convertible Notes
In 2013, we sold convertible notes in an aggregate principal amount of approximately $4.6 million. Each convertible note bore a simple interest rate of 8% per annum. We did not pay any accrued interest on the convertible notes. Each of the noteholders converted their notes into common stock upon the completion of our initial public offering.
The following table sets forth the loan amounts provided by our directors, executive officers and principal stockholders, or affiliates or immediate family members of our directors, executive officers and principal stockholders in 2013.
110
Name | Loan Amount | ||
Entities affiliated with Care Capital III LLC(1) | $ | 3,817,259 | |
Entities affiliated with Rho Capital Partners, LP(2) | 351,663 | ||
(1) | Consists of (a) loans in an aggregate principal amount of $3,754,557 provided by Care Capital Investments III LP and (b) loans in an aggregate principal amount of $62,702 provided by Care Capital Offshore Investments III LP. |
(2) | Consists of (a) loans in an aggregate principal amount of $323,279 provided by Rho Ventures V, L.P. and (b) loans in an aggregate principal amount of $28,384 provided by Rho Ventures V Affiliates, L.L.C. |
Director and Executive Officer Compensation
Please see "Executive and Director Compensation" for information regarding compensation of directors and executive officers.
Payments for Services
During 2013 and 2014 we paid Care Capital, LLC, an affiliate of our largest shareholder $124,000 and $70,000, respectively, for accounting services provided to us by a Care Capital, LLC employee.
Participation in our Initial Public Offering
Entities affiliated with Care Capital, LLC purchased an aggregate of 790,000 shares of our common stock in our initial public offering at the initial public offering price for an aggregate purchase price of approximately $9.5 million.
Indemnification Agreements
We enter into indemnification agreements with each of our directors and officers. The indemnification agreements and our restated certificate of incorporation and restated bylaws require us to indemnify our directors and officers to the fullest extent permitted by Delaware law.
Policy for Approval of Related Person Transactions
Pursuant to the written charter of our Audit Committee, the Audit Committee is responsible for reviewing and approving, prior to our entry into any such transaction, all transactions in which we are a participant and in which any parties related to us, including our executive officers, our directors, beneficial owners of more than 5% of our securities, immediate family members of the foregoing persons and any other persons whom our Board of Directors determines may be considered related parties under Item 404 of Regulation S-K, has or will have a direct or indirect material interest.
In reviewing and approving such transactions, the Audit Committee shall obtain, or shall direct our management to obtain on its behalf, all information that the committee believes to be relevant and important to a review of the transaction prior to its approval. Following receipt of the necessary information, a discussion shall be held of the relevant factors if deemed to be necessary by the committee prior to approval. If a discussion is not deemed to be necessary, approval may be given by written consent of the committee. This approval authority may also be delegated to the chair of the Audit Committee in some circumstances. No related party transaction shall be entered into prior to the completion of these procedures.
The Audit Committee or its chair, as the case may be, shall approve only those related party transactions that are determined to be in, or not inconsistent with, the best interests of us and our stockholders, taking into account all available facts and circumstances as the committee or the chair determines in good faith to be necessary in accordance with principles of Delaware law generally applicable to directors of a Delaware corporation. These facts and circumstances will typically include, but not be limited to, the benefits of the transaction to us; the impact on a director's independence in the event the related party is a director, an immediate family member of a director or an entity in which a director is a partner, stockholder or executive officer; the availability of other sources for comparable products or services; the terms of the transaction; and the terms of comparable transactions that would be available to unrelated third parties or to employees generally. No member of the Audit Committee shall participate in any review, consideration or approval of any related party transaction with respect to which the member or any of his or her immediate family members has an interest.
Item 14. Principal Accounting Fees and Services
The Audit Committee has appointed EisnerAmper LLP ("EisnerAmper"), as our independent registered public accounting firm, to audit our financial statements for the fiscal year ending December 31, 2014. Accordingly, EisnerAmper audited our financial statements for the fiscal year ended December 31, 2014.
111
In deciding to appoint EisnerAmper, the Audit Committee reviewed auditor independence issues and existing commercial relationships with EisnerAmper and concluded that EisnerAmper had no commercial relationship with the Company that would impair its independence for the fiscal year ending December 31, 2014.
Audit Fees
The following table presents fees for professional audit services and fees billed for other services rendered by EisnerAmper for the audit of the Company's annual financial statements for the years ended December 31, 2014 and 2013.
2014 | 2013 | |||||
Audit fees | $ | 275,875 | $ | 125,675 | ||
Audit related fees | — | — | ||||
Tax fees | 8,800 | — | ||||
All other fees | — | — | ||||
Total | $ | 284,675 | $ | 125,675 | ||
Audit Fees. Audit fees for 2014 consisted of fees for audit and audit and tax services related to the review of our registration statement on Form S-1 and fees associated with our annual audit and the review of our quarterly reports on Form 10-Q and other regulatory filings. Audit fees for 2013 consisted of audit work performed in the preparation of our 2013 and 2012 financial statements and audit and tax services and fees related to the review of our registration statement on Form S-1.
Audit-Related Fees. There were no fees for the category “Audit-Related Fees” in 2014 and 2013.
Tax Fees. Tax fees for 2014 were for the preparation of our corporate income tax returns. There were no fees for the category “Tax Fees” in 2013.
All Other Fees. There were no fees for the category “All Other Services” in 2014 and 2013.
Pre-Approval Policies and Procedures
Our Audit Committee has established a policy that requires it to pre-approve all services provided by the Company’s independent registered public accounting firm and the fees for such services. The prior approval of our Audit Committee was obtained for all services provided by EisnerAmper after completing the initial public offering in February 2014 and the fees for such services. Audit fees prior to the IPO, consisted of audit work performed in the preparation of our 2013 financial statements as well as work generally only the independent registered public accounting firm can reasonably be expected to provide. Audit fees for 2013 and 2014 also consisted of audit and tax services and other fees related to the review of our registration statement on Form S-1.
112
PART IV
Item 15. Exhibits
Exhibits required by Item 601 of Regulation S-K.
EXHIBIT INDEX
Exhibit | Incorporated by | Filing | SEC | ||
Number | Exhibit Description | Reference | Date | File/Reg. Number | |
3.1 | Restated Certificate of Incorporation of the Registrant. | Form 8-K (Exhibit 3.1) | 2/14/2014 | 001-36303 | |
3.2 | Restated Bylaws of the Registrant. | Form 8-K (Exhibit 3.2) | 2/14/2014 | 001-36303 | |
4.1 | Form of Common Stock Certificate | Form S-1 (Exhibit 4.1) | 1/10/2014 | 333-193023 | |
4.2 | Form of Representative's Warrant. | Form S-1 (Exhibit 4.2) | 1/29/2014 | 333-193023 | |
4.3 | Amended and Restated Investors' Rights Agreement, dated February 28, 2008, as amended February 14, 2014 | Form 10-K (Exhibit 4.3) | 3/31/2014 | 001-36303 | |
10.1* | Executive Employment Agreement by and between the Registrant and Pierre Legault, dated November 7, 2013. | Form S-1 (Exhibit 10.1) | 12/23/2013 | 333-193023 | |
10.1.1* | Restricted Stock Unit Grant Notice and Agreement by and between the Registrant and Pierre Legault, dated November 7, 2013. | Form 10-K (Exhibit 10.1.1) | 9/15/2014 | 001-36303 | |
10.2* | Offer of Employment Letter by and between the Registrant and Bob Peterson, dated August 8, 2009. | Form S-1 (Exhibit 10.2) | 12/23/2013 | 333-193023 | |
10.3* | Employment Agreement by and between J. Wesley Fox and the Registrant, dated April 30, 2007. | Form S-1 (Exhibit 10.3) | 12/23/2013 | 333-193023 | |
10.4* | Form of Indemnification Agreement by and between the Registrant and its directors and officers. | Form S-1 (Exhibit 10.4) | 12/23/2013 | 333-193023 | |
10.5 | Office Lease, dated September 12, 2014 by and between NephroGenex, Inc. and Highwoods Realty Limited Partnership. | Form 8-K (Exhibit 10.2) | 9/15/2014 | 001-36303 | |
10.6.1 | Amended and Restated License Agreement between University of Kansas Medical Center Research Institute, Inc. and BioStratum Incorporated (assigned to the Registrant), effective as of November 19, 1998. | Form S-1 (Exhibit 10.6.1) | 12/23/2013 | 333-193023 | |
10.6.2 | First Amendment to Amended and Restated License Agreement between University of Kansas Medical Center Research Institute, Inc. and the Registrant, effective as of May 4, 2007. | Form S-1 (Exhibit 10.6.2) | 12/23/2013 | 333-193023 | |
10.6.3 | Second Amendment to Amended and Restated License Agreement between University of Kansas Medical Center Research Institute, Inc. and the Registrant, effective as of June 25, 2008. | Form S-1 (Exhibit 10.6.3) | 12/23/2013 | 333-193023 | |
10.7.1 | License Agreement between the University of South Carolina Research Foundation and BioStratum Incorporated (assigned to the Registrant), dated August 27, 2004. | Form S-1 (Exhibit 10.7.1) | 12/23/2013 | 333-193023 | |
10.7.2 | Amendment to License Agreement between The South Carolina Research Foundation and the Registrant, effective as of June 20, 2011. | Form S-1 (Exhibit 10.7.2) | 12/23/2013 | 333-193023 | |
10.7.3 | Second Amendment to License Agreement between The South Carolina Research Foundation and the Registrant, effective as of April 2, 2012. | Form S-1 (Exhibit 10.7.3) | 12/23/2013 | 333-193023 | |
10.7.4 | Third Amendment to License Agreement between The South Carolina Research Foundation and the Registrant, effective as of August 9, 2013. | Form S-1 (Exhibit 10.7.4) | 12/23/2013 | 333-193023 | |
113
10.7.5 | Fourth Amendment to License Agreement between The University of South Carolina Research Foundation and the Registrant, effective as of January 14, 2014. | Form S-1 (Exhibit 10.7.5) | 01/17/2014 | 333-193023 | |
10.8.1 | License Agreement between Vanderbilt University and the Registrant, effective as of January 11, 2006. | Form S-1 (Exhibit 10.8.1) | 12/23/2013 | 333-193023 | |
10.8.2 | First Amendment to License Agreement between Vanderbilt University and the Registrant, effective as of April 30, 2007. | Form S-1 (Exhibit 10.8.2) | 12/23/2013 | 333-193023 | |
10.8.3 | Restated and Amended License Agreement between Vanderbilt University and the Registrant, effective as of July 1, 2012. | Form S-1 (Exhibit 10.8.3) | 12/23/2013 | 333-193023 | |
10.8.4 | First Amendment to Restated and Amended License Agreement between Vanderbilt University and the Registrant, effective as of November 6, 2013. | Form S-1 (Exhibit 10.8.4) | 12/23/2013 | 333-193023 | |
10.9.1 | License Agreement between BioStratum, Incorporated and the Registrant, effective as of May 8, 2006. | Form S-1 (Exhibit 10.9.1) | 12/23/2013 | 333-193023 | |
10.9.2 | Amendment to License Agreement between BioStratum, Incorporated and the Registrant, effective September 13, 2006. | Form S-1 (Exhibit 10.9.2) | 12/23/2013 | 333-193023 | |
10.9.3 | Grant Back License Agreement by and between the Registrant and BioStratum, Incorporated, dated May 4, 2007. | Form S-1 (Exhibit 10.9.3) | 12/23/2013 | 333-193023 | |
10.10* | NephroGenex, Inc. Amended and Restated 2007 Equity Incentive Plan. | Form 8-K (Exhibit 10.1) | 05/15/2014 | 001-36303 | |
10.10.1* | Amendment 2015-1 to the NephroGenex, Inc. Amended and Restated 2007 Equity Incentive Plan | Form 8-K (Exhibit 3.2) | 02/05/2015 | 001-36303 | |
10.11* | Executive Employment Agreement between the Registrant and John P. Hamill, dated December 12, 2013. | Form 8-K (Exhibit 10.11) | 12/23/2013 | 333-193023 | |
10.12 | Form of Omnibus Agreement and Consent among the Registrant, Care Capital Investments III, LP, Care Capital Offshore Investments III, LP, Rho Ventures V, L.P., Rho Ventures V Affiliates, L.L.C., Biostratum, Incorporated, Vanderbilt University, Vanderbilt University Medical Center, Vanderbilt University, by and through its Medical Center and John B. Mazur. | Form 8-K (Exhibit 10.12) | 01/10/2014 | 333-193023 | |
10.13 | Loan and Securities Agreement, dated November 20, 2014, by and between the Company and East West Bank. | Form 8-K (Exhibit 10.1) | 11/20/2014 | 001-36303 | |
10.14 | Intellectual Property Security Agreement, dated November 20, 2014, by and between the Company and East West Bank. | Form 8-K (Exhibit 10.2) | 11/20/2014 | 001-36303 | |
10.15 | Warrants, dated November 20, 2014, issued by the Company to East West Bank. | Form 8-K (Exhibit 10.3) | 11/20/2014 | 001-36303 | |
23.1** | Consent of Independent Auditor | ||||
31.1** | Certification of the Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||||
31.2** | Certification of the Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||||
32** | Certifications of the Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||||
101*** | Financials in XBRL | ||||
* Management contract or compensatory plan
** Filed herewith
*** Furnished herewith
114
Attached as Exhibits 101 to this report are the following financial statements from our Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (eXtensible Business Reporting Language): (i) the Balance Sheets, (ii) the Statements of Comprehensive Loss, (iii) the Statements of Cash Flows and (iv) and related notes to these financial statements.
The XBRL related information in Exhibits 101 to this Annual Report on Form 10-K shall not be deemed “filed” or a part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended (“Securities Act”) and is not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (“Exchange Act”), or otherwise subject to the liabilities of those sections.
115
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
NEPHROGENEX, INC. | ||
(Registrant) | ||
Date: March 24, 2015 | By: | /s/ PIERRE LEGAULT |
Pierre Legault Chief Executive Officer (Principal Executive Officer) | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated below and on the dates indicated.
Signature | Title | Date |
/s/ PIERRE LEGAULT | Chief Executive Officer and Director (Principal Executive Officer) | March 24, 2015 |
Pierre Legault | ||
/s/ JOHN P. HAMILL | Chief Financial Officer (Principal Financial and Accounting Officer) | March 24, 2015 |
John P. Hamill | ||
/s/ RICHARD MARKHAM | Director | March 24, 2015 |
Richard Markham | ||
/s/ JAMES MITCHUM | Director | March 24, 2015 |
James Mitchum | ||
/s/ ROBERT R. SELTZER | Director | March 24, 2015 |
Robert R. Seltzer | ||
/s/ EUGEN STEINER | Director | March 24, 2015 |
Eugene Steiner, M.D., PhD. | ||
/s/ MARCO TAGLIETTI | Director | March 24, 2015 |
Marco Taglietti, M.D. | ||
116