Attached files
| file | filename |
|---|---|
| EX-23.1 - CONSENT - Ocata Therapeutics, Inc. | ocata_ex2301.htm |
| EX-21.1 - SUBSIDIARIES OF THE REGISTRANT - Ocata Therapeutics, Inc. | ocata_ex2101.htm |
| EXCEL - IDEA: XBRL DOCUMENT - Ocata Therapeutics, Inc. | Financial_Report.xls |
| EX-31.1 - CERTIFICATION - Ocata Therapeutics, Inc. | ocata_ex3101.htm |
| EX-31.2 - CERTIFICATION - Ocata Therapeutics, Inc. | ocata_ex3102.htm |
| EX-23.1 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - Ocata Therapeutics, Inc. | ocata_ex2302.htm |
| EX-32.1 - CERTIFICATION - Ocata Therapeutics, Inc. | ocata_ex3201.htm |
| EX-32.2 - CERTIFICATION - Ocata Therapeutics, Inc. | ocata_ex3202.htm |
| EX-10.30 - 2014 STOCK OPTION AND INCENTIVE PLAN AND FORMS OF OPTION AGREEMENTS THEREUNDER - Ocata Therapeutics, Inc. | ocata_ex1030.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2014
OR
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from _____________ to _____________
Commission file number 0-50295
OCATA THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 87-0656515 |
| (STATE OR OTHER JURISDICTION OF | (I.R.S. EMPLOYER IDENTIFICATION NO.) |
| INCORPORATION OR ORGANIZATION) |
33 Locke Drive, Marlborough, Massachusetts
01752
(508) 756-1212
(Address and telephone number, including area code, of registrant’s principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| Common Stock, $0.001 par value per share | The Nasdaq Stock Market |
| (Title of each class) | Name of each exchange on which registered |
Securities registered pursuant to Section
12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer x |
| Non-accelerated filer o (Do not check if a smaller reporting company) | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act.) Yes o No x
The aggregate market value of the registrant’s Common Stock held by non-affiliates of the registrant (based upon the closing price of $6.70 for the registrant’s Common Stock as of June 30, 2014) was approximately $222.6 million (based on 33,071,871 shares of common stock outstanding and held by non-affiliates on such date all on an 100:1 reverse split-adjusted basis). Shares of the registrant’s Common Stock held by each executive officer and director and by each entity or person that, to the registrant’s knowledge, owned 10% or more of the registrant’s outstanding Common Stock as of June 30, 2014 have been excluded in that such persons may be deemed to be affiliates of the registrant. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of outstanding shares of the registrant’s Common Stock, $0.001 par value, was 35,042,363 shares as of March 1, 2015.
OCATA THERAPEUTICS, INC.
2014 ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| Page | ||
| PART I | 1 | |
| Item 1. | Business | 1 |
| Item 1A. | Risk Factors | 13 |
| Item 1B. | Unresolved Staff Comments | 34 |
| Item 2. | Properties | 34 |
| Item 3. | Legal Proceedings | 34 |
| Item 4. | Mine Safety Disclosures | 34 |
| PART II | 35 | |
| Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 35 |
| Item 6. | Selected Financial Data | 36 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 37 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 44 |
| Item 8. | Financial Statements and Supplementary Data | 44 |
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 44 |
| Item 9A. | Controls and Procedures | 44 |
| Item 9B. | Other Information | 47 |
| PART III | 47 | |
| Item 10. | Directors, Executive Officers and Corporate Governance | 47 |
| Item 11. | Executive Compensation | 47 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 47 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 47 |
| Item 14. | Principal Accountant Fees and Services | 47 |
| Item 15. | Exhibits and Financial Statement Schedules | 47 |
CAUTIONARY STATEMENT RELATING TO FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and the information incorporated by reference includes “forward-looking statements” All statements regarding our expected financial position and operating results, our business strategy, our financing plans and the outcome of any contingencies are forward-looking statements. Any such forward-looking statements are based on current expectations, estimates, and projections about our industry and our business. Words such as “anticipates,” “expects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” or variations of those words and similar expressions are intended to identify such forward-looking statements. Forward-looking statements are subject to risks and uncertainties that could cause actual results to differ materially from those stated in or implied by any forward-looking statements.
Forward-looking statements are only current predictions and are subject to known and unknown risks, uncertainties, and other factors that may cause our or our industry’s actual results, levels of activity, performance, or achievements to be materially different from those anticipated by such statements. These factors include, among other things, the fact that we have no product revenue and no products approved for marketing; our limited operating history; the need for and limited sources of future capital; potential failures or delays in obtaining regulatory approval of products; risks inherent in the development and commercialization of potential products; reliance on new and unproven technology in the development of products; the need to protect our intellectual property; the challenges associated with conducting and enrolling clinical trials; the risk that the results of clinical trials may not support our product candidate claims; even if approved, the risk that physicians and patients may not accept or use our products; our reliance on third parties to conduct its clinical trials and to formulate and manufacture its product candidates; economic conditions generally; as well as those factors listed under “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K and elsewhere in this Annual Report on Form 10-K. These and other factors could cause results to differ materially from those expressed in these forward-looking statements.
Although we believe that the expectations reflected in the forward-looking statements contained in this Annual Report on Form 10-K are reasonable, we cannot guarantee future results, performance, or achievements. Except as required by law, we are under no duty to update or revise any of such forward-looking statements, whether as a result of new information, future events or otherwise, after the date of this Annual Report on Form 10-K.
Unless otherwise indicated, information contained in this Annual Report on Form 10-K concerning our clinical trials, therapeutic candidates, number of patients that may benefit from these therapeutic candidates and the potential commercial opportunity for our therapeutic candidates, is based on information from independent industry analysts and third-party sources (including industry publications, surveys, and forecasts), our internal research, and management estimates. Management estimates are derived from publicly available information released by independent industry analysts and third-party sources, as well as data from our internal research, and based on assumptions made by us based on such data and our knowledge of such industry, which we believe to be reasonable. None of the sources cited in this Annual Report on Form 10-K has consented to the inclusion of any data from its reports, nor have we sought their consent. Our internal research has not been verified by any independent source, and we have not independently verified any third-party information. While we believe that such information included in this Annual Report on Form 10-K is generally reliable, such information is inherently imprecise. In addition, projections, assumptions, and estimates of our future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K and elsewhere in this Annual Report on Form 10-K. These and other factors could cause results to differ materially from those expressed in the estimates made by the independent parties and by us.
PART I
Item 1. Business.
Overview
Ocata Therapeutics, Inc., a Delaware corporation formerly known as Advanced Cell Technology, Inc. (the “Company”, “Ocata”, “we”, “us”, or “our”) is a clinical stage biotechnology company focused on the development and commercialization of Regenerative Ophthalmology therapeutics. Ocata’s most advanced products are in clinical trials for the treatment of Stargardt’s macular degeneration, dry age-related macular degeneration, and myopic macular degeneration. We are also developing several pre-clinical terminally differentiated-cell therapies for the treatment of other ocular disorders. Additionally, we have a number of pre-clinical stage assets in disease areas outside the field of ophthalmology, including autoimmune, inflammatory and wound healing-related disorders. Our intellectual property portfolio includes pluripotent human embryonic stem cell, or hESC; induced pluripotent stem cell, or iPSC, platforms; and other cell therapy technologies. We have no therapeutic products currently available for sale and do not expect to have any therapeutic products commercially available for sale for a period of years, if at all. These factors indicate that our ability to continue research and development activities is dependent upon the ability of management to obtain additional financing as required.
We pursue a number of approaches to generating transplantable tissues both in-house and through collaborations with other researchers who have particular interests in, and skills related to, cellular differentiation. Our research in this area includes projects focusing on the development of many different cell types that may be used to treat a range of diseases within ophthalmology and other therapeutic areas. Control of cellular differentiation and the culture and growth of stem and differentiated cells are important areas of research and development for us.
Key Management Updates
During 2014 there were a number of key changes to our management team. In July 2014, Paul Wotton joined us as President and Chief Executive Officer and a member of our Board of Directors. In October 2014, LeRoux Jooste, joined our company as Senior Vice President of Business Development and Chief Commercial Officer.
An Overview of Regenerative Medicine
Our business focus is the development of new therapies in the field of regenerative medicine. Regenerative medicine is defined as the process of replacing or "regenerating" human cells, tissues or organs to restore or establish normal function and has been called the "next evolution of medical treatments" and “the vanguard of 21st century healthcare” by the U.S. Department of Health and Human Services.
Cell therapies, such as those we are developing, have the potential to offer a complete solution for complex physiologic processes in ways, or through mechanisms, which are not expected to be attainable using traditional small-molecule or protein therapeutics. This field holds the potential to therapeutically address damaged tissues and organs in the body by replacing damaged tissue or by stimulating the body's own repair mechanisms to heal tissues or organs. By altering the course of disease, regenerative medicine may make it possible to eliminate the need for daily therapies, reduce hospitalizations and avoid expensive medical procedures, thus enabling patients to lead healthier and more productive lives. Regenerative medicine, such as those therapies we are developing, could provide more effective solutions or potential cures for a broad range of conditions, and provide meaningful advances for a range of chronic, orphan, and aging-related conditions that traditional medicine has to date been unable to treat and that represent a quality-of-life and economic burden on society.
We believe that regenerative medicine, including Regenerative Ophthalmology ™ will become a highly relevant area of medicine as our population continues to age. Chronic diseases and impairments, which are among the leading causes of disability in older people, can negatively impact quality of life, lead to a decline in independent living, and impose an economic burden on patients and the healthcare system. About 80 percent of seniors in developed countries have at least one chronic health condition and 50 percent have at least two. According to the Centers for Disease Control, of the roughly 150,000 people who die each day, about two-thirds die of age-related causes. In industrialized nations, the proportion is much higher, reaching 90%. Concern is growing that medical advances leading to longevity will in turn lead to an older population who have a higher incidence of functional and cognitive impairment.
| 1 |
Figure 1 shows the projected growth of two groups of older people – those aged 65-79 and those 80 years and older.

By 2050, the number of people over the age of 65 living in developed countries is projected to exceed 325 million; when including developing countries, this number rises to 1.5 billion – a near tripling of the over 65 population as compared to today. In the United States, as an example, the number of Americans aged 65 and older is projected to be 88.5 million in 2050, more than double the population of 40.2 million in 2010. The U.S. population is projected to grow to 439 million by 2050, an increase of 42 percent relative to the 2010 census numbers of 310 million. The population is also expected to become much older. Those over the age of 85 accounted for 5.8 million Americans in the 2010 census, and are expected to reach 8.7 million by 2030 and then 19 million by 2050.
The majority of treatments available today for chronic and/or life-threatening diseases are palliative, meaning they merely treat the symptoms rather than cure the underlying cause. Others delay disease progression and the onset of complications associated with the underlying illness. Only a very limited number of these available therapies are capable of curing or significantly changing the course of disease. The result is a healthcare system burdened by costly treatments for an aging, increasingly ailing population, with few solutions for containing rising costs.
Regenerative medicine has the potential to change the thinking about disease and aging, as well as to help potentially reduce continuously growing health care costs. The best way to address the escalating economics of healthcare includes developing more effective treatments and cures for the most burdensome diseases (such as diabetes, neurodegenerative disorders, stroke and cardiovascular disease) which may help to facilitate longer, healthier and more productive lives.
We believe our therapeutic programs, such as our macular degeneration programs, will contribute to the medical community’s response to this growing problem. Our regenerative cell therapies currently in development may have both therapeutic benefit and provide meaningful reduction to the otherwise-predicted increase in healthcare costs resulting from aging. We believe those factors may help us obtain favorable pricing and reimbursement considerations for our therapies.
Pluripotent Stem Cell Platforms
There are two broad categories of stem cells: adult stem cells and pluripotent stem cells. The term “stem cells” is used to describe those cell types that can give rise to the different cells found in tissues. A common feature to all stem cells is the ability to both replicate (propagate) as well as differentiate into two or more different mature cell types. There are however, differences between adult and pluripotent stem cells. Adult stem cells are derived from various tissues in the human body and are typically limited in the diversity of other cell types they can become, usually only able to produce two or three different types of mature cells. Adult stem cells also are often limited in their ability to divide and renew in culture before ceasing to grow. In contrast, pluripotent stem cells are often termed “true” stem cells because they have the potential to differentiate into almost any cell in the body, and have a near infinite capacity to replicate. Pluripotent stem cells can potentially provide a renewable source of healthy cells and tissues to treat a wide array of diseases, making pluripotent stem cells a central aspect to our strategy in the development of effective cellular therapies that can be used in a commercially scalable manner.
From a single master stem cell bank the manufacturing of the therapeutic doses to be used can be readily controlled for consistency, lack of infectious agents and cleared by regulatory agencies to be used in an entire patient population. The pluripotent stem cell approach also permits the use of cell culturing and manufacturing techniques that we believe will prove to be less costly and intrinsically more scalable than the high-touch process that otherwise characterize the majority of “autologous” cell therapies.
Pluripotent stem cells presently include two distinct cell types: (1) embryonic stem, or ESCs, and (2) induced pluripotency stem cells, or iPSCs, which have ESC-like properties.
| 2 |
Embryonic Stem Cell Platform
A human ESC, or hESC, line represents a potentially inexhaustible supply of pluripotent cells. Derived from a single cell, the replicative capacity of an hESC line could be very significant. Embryonic stem cells have specific properties that make them particularly useful for cell-based therapies. Because they are able to differentiate into all of the more than 200 types of cells in the human adult body, they may offer substantial therapeutic potential. Our primary focus has been in the development of therapies that are terminally differentiated into the cells of interest, with hESCs representing the starting raw material for our therapeutic products.
iPSC Platform
iPSCs are adult cells that have been genetically or environmentally reprogrammed to an embryonic stem cell-like state by being genetically manipulated to express genes and factors important for maintaining the defining properties of embryonic stem cells. The reprogramming of adult cells into embryonic stem cell-like cells enables the generation of patient-specific stem cells and thus has potential for the treatment of degenerative diseases. Given that iPSCs can be made in a patient-specific manner, the ultimate goal for iPSC-derived tissues and differentiated cells is that these can be transplanted back into the same patient without rejection, and so might be used in treatment settings where donor-recipient matching would otherwise be necessary to prevent rejection of the transplanted cells.
Our Cell Therapy Research Programs
Our product development programs are based on our ability to induce hESCs to become a specific cell type of interest and to become this type of cell permanently. This is known as terminal differentiation and we believe it is a core competency of our research and development programs. We have demonstrated our ability to terminally differentiate hESCs into a wide variety of cell types. We have also demonstrated that many of these terminally differentiated cells may be safe, and, in some cases, potentially effective in treating human disease as in the case of our lead programs for macular degeneration. Our research with these terminally differentiated cells includes projects using many different cell types that may be used to treat a range of diseases across several therapeutic categories. We are pursuing a variety of approaches to generate transplantable tissues both through in-house and external collaborations with other researchers who have particular interests in, and skills related to, cellular differentiation. Control of differentiation and the culture and growth of stem and differentiated cells are also important current areas of research for us.
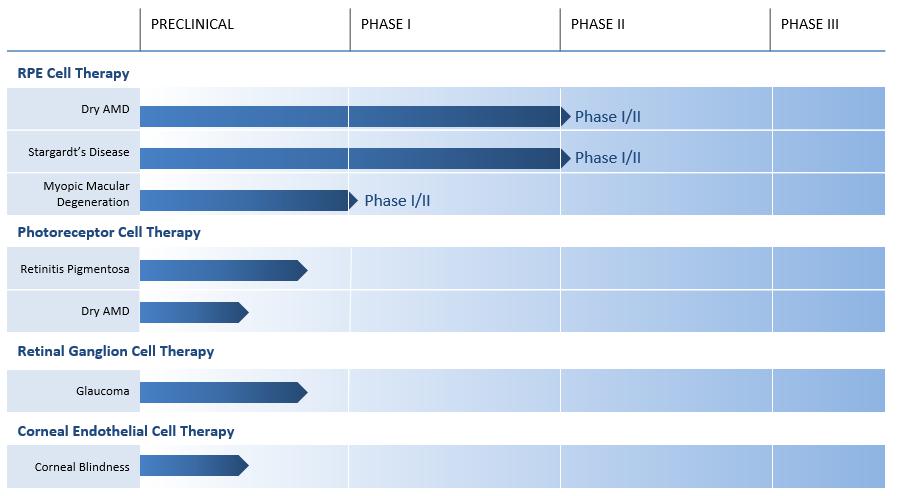
Based on the success to date of our Phase 1 clinical trials and what we see as favorable market dynamics of the ophthalmology sector, we have shifted our strategic priorities to focus primarily on the development, and ultimate commercialization of ophthalmology therapies. We believe that the eye is well suited for cellular transplantation for a number of reasons. First, the eye has relative immune privilege, which means that the body’s immune system is less likely to target a foreign substance, such as allogeneic cells, for rejection. Second, the eye is a compact structure and therefore the doses are relatively small (approximately 200,000-300,000 cells), which may enable relative ease of manufacturing scalability. Third, there are many well-known and validated tools that are accepted for measurement of clinical outcomes. Finally, the community of surgeons who will ultimately develop our back-of-the-eye therapies is small and highly concentrated. There are approximately 2,000 retinal surgeons in the United States, we believe we can access the majority of macular degeneration patients with a relatively small sales force to call on this concentrated physician base. Our current ophthalmology development pipeline is presented below:
| 3 |
Ophthalmology Programs
We are developing a pipeline of stem cell derived therapeutics which may have use as treatment for degenerative diseases of the eye. In some instances, stem cell derived therapies may repair and replace damaged tissue in the eye, permitting restoration of otherwise lost vision or prevention of further vision loss. As our understanding of the underlying pathophysiology of ocular disease increases, we believe we will have additional opportunities to develop other therapeutic products for the ophthalmology market.
Chronic diseases of the eye are common globally, particularly in aging populations. The recent success of palliative therapies in the wet age-related macular degeneration market highlights the therapeutic and commercial potential of this sector. This is underscored by large pharmaceutical and global biotech companies that have either recently repositioned themselves through further emphasis in funding and/or acquiring ophthalmology programs, or entering ophthalmology for the first time, as the industry comes to appreciate the potential size of this market and its projected growth rate, largely as a consequence of an aging population. These companies include, as examples, Bayer Healthcare, GlaxoSmithKline, Pfizer, Novartis and Roche. Despite this growing interest, several disease areas of ophthalmology remain underserved by prescription pharmaceuticals.
A significant unmet medical need relates to diseases affecting the back of the eye, such as age-related macular degeneration as well as other forms of macular degeneration, diabetic retinopathy and retinitis pigmentosa. Inflammatory diseases such as uveitis, and vision loss from photoreceptor and other neurosensory retinal damage due to glaucoma, also represent significant patient populations for which effective therapies have remained elusive. These conditions have been under-served primarily because of their pathophysiological complexity, which the development of new drugs – traditional small molecule and biologics – has been unable to solve. We are developing a pipeline of cell-based therapeutics which may have use as treatment for degenerative diseases of the eye. In some instances, stem cell derived therapies may repair and replace damaged tissue in the eye, permitting restoration of otherwise lost vision. As our understanding of the underlying pathophysiology of ocular disease increases, we believe we will have additional opportunities to develop other therapeutic products for the ophthalmology market.
Macular Degeneration Programs
The largest indication involving macular degeneration is “age-related macular degeneration”, or AMD. AMD is the leading cause of blindness and visual impairment in adults over fifty years of age. It is estimated that the clinically detectable AMD patient population in North America and Europe includes about 25-30 million people across the range of disease, from early-stage to late-stage, or legal blindness. Furthermore, it is estimated that there are nearly two million new diagnoses of AMD per year in the United States. AMD represents one of the largest unmet medical needs in medicine today in terms of the lack of useful therapeutics.
Retinal pigment epithelium, or RPE, is a single-cell-thick layer of pigmented cells that form part of the blood-ocular barrier. The presence and integrity of the RPE layer is required for normal vision. The RPE layer is positioned between the photoreceptor cell layer of the retina and the Bruch's membrane and choroid, a layer filled with blood vessels. Because the photoreceptors see no direct blood supply, it is the role of the RPE layer to transport nutrients and oxygen to the photoreceptor cells, as well as to supply, recycle, and detoxify products involved with the phototransduction process — the process by which the photoreceptors turn light into a signal to be propagated along the optic nerve to the brain. In particular, the RPE layer serves as the transport layer that maintains the structure of the photoreceptor environment by acting as an intermediary between the nerve layer and blood vessels, supplying small molecules, transporting ions and water from the blood vessels to the photoreceptor layer. The RPE cells take up nutrients such as glucose, retinol (Vitamin A), and fatty acids from the blood and deliver these nutrients to photoreceptors. The RPE layer also prevents the buildup of toxic metabolites around the nerve cells by transporting the metabolites to the blood. In addition, the RPE cells are able to secrete a variety of growth and survival factors helping to maintain the structural integrity and organization of the photoreceptors.
| 4 |
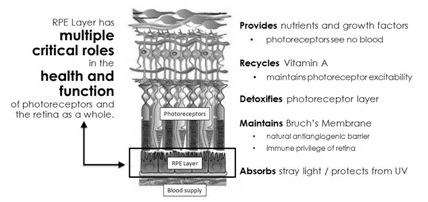
Maintenance of the Bruch’s membrane, which serves as a natural anti-angiogenic barrier that prevents the capillary bed of the choroid from invading and disrupting the photoreceptor and nerve microarchitecture of the retina, is also an important function of the RPE layer. RPE cells also recycle proteins and other components involved in a process is known as the visual cycle of retinal, which isomerizes all trans-retinol to 11-cis retinal – the latter of which is required by photoreceptors for vision. A failure of any one of these functions of the RPE layer can lead to degeneration of the retina, loss of visual function, and/or blindness. Dysfunction and degeneration of the RPE layer is in fact implicated in many disease processes, the most prominent being various forms of macular degeneration.
As the name implies, age-related macular degeneration usually affects older adults, with loss of central vision required for reading, driving and other important activities of daily living due to chronic damage of the central retina. It occurs in "dry" (aka "atrophic" or "geographic") and "wet" (aka "neovascular" or "exudative") forms. In the case of dry AMD, the disease process appears to begin with loss (death) of RPE cells followed by some period of photoreceptor atrophy and inactivity, and eventually, photoreceptor death. For most dry AMD patients, gradual loss of central vision occurs first. Wet AMD is often an end-stage manifestation seen in approximately 10% - 15% of dry AMD patients, with the loss of the RPE layer and its ability to maintain the Bruch's membrane function as a barrier resulting in failure of the membrane's integrity and abnormal blood vessels penetrating into the subretinal space with ensuing rapid loss of vision. In addition to AMD, other forms of macular degenerative diseases exist which, even if the underlying causes are different, appear to follow a similar course of RPE cell loss followed by atrophy, inactivity and ultimately death of the photoreceptor. These include, for example, an inherited juvenile onset form of macular degeneration called Stargardt's Macular Degeneration, or SMD.
Our research has indicated that RPE cells generated from pluripotent stem cell sources, such as our hESC lines, may potentially solve the sourcing of transplantable RPE cells for treating macular degenerative conditions. It is possible that the area in which the RPE layer exists will maintain its relative immune-privilege in dry AMD patients. If so, the need for donor matching is not likely to be a significant limitation and a single and scalable allogeneic source of RPE cells, one that can be manufactured in culture, might provide a therapeutic solution for the millions of patients affected by this disease. We have created GMP-compliant hESC master stem cell banks and a GMP protocol for manufacturing of human RPE cells from our hESC master banks. Extensive animal testing of the human RPE cells generated in culture has been conducted and has established that when injected into the eyes of test animals as a suspension of cells, the human RPE cells were able to home to areas of damage in the RPE layer, with engraftment and recapitulation of the correct anatomical structure in the back of the eye of the animals. As published in 2006 the journal Cloning and Stem Cells , we have also demonstrated that in animal models of macular degeneration, not only did the human RPE cells reform the correct structure, but also that the injection of the cells resulted in preservation of the photoreceptor layer and its function. That is, the injected human RPE cells repaired and restored the function of the RPE layer in animal models of disease.
This data, along with safety data we collected on the human RPE cells, or our RPE Program, formed the basis of several Investigational New Drug applications filed with the U.S. Food and Drug Administration and an Investigational Medicinal Product Dossier, or IMPD, relied upon by the U.K. Medicines and Healthcare Products Regulatory Agency. It also served as the basis of our clinical trials using RPE cells in patients with SMD and dry AMD.
| 5 |
We believe that the results from the SMD and dry AMD clinical trials are promising. Preliminary results for the first dry AMD and first SMD patient were published in January 2012 in The Lancet. This publication followed two patients who were treated with our RPE therapy for at least three months of post-transplant follow up and the study reported that there were no serious adverse events due to the injected RPE cells, which is the primary endpoint of the trials. The trial sites have provided regular follow-up on all of the patients, and have been able to include data relating to the engraftment and persistence of the injected cells as well the impact on visual acuity. The preliminary data suggest that the injected cells are well tolerated and appear generally to be capable of engrafting at the site of injection, forming the appropriate anatomical monolayer structure around the injected area. Visual acuity improvement has been observed to varying degrees in several of these very late-stage patients, a result that was not anticipated in the original design of these studies.
In October 2014, we announced that Phase 1/2 clinical data published online in The Lancet demonstrate positive long-term safety results using our RPE cells for the treatment of SMD and AMD (nine patients from each study). The publication features data from 18 U.S.-based patients with at least six months of post-transplant follow-up. These two studies provide the first evidence of the mid- to long-term safety, survival, and potential biologic activity of pluripotent stem cell progeny into humans with any disease. In addition to showing no adverse safety issues related to the transplanted tissue, anatomic evidence confirmed successful engraftment of the RPE cells, which included increased pigmentation at the level of the RPE layer after transplantation in 13 of 18 patients. The publication also noted that ten of the 18 patients experienced clinically significant (greater than 15 letters on the standard EDTRS charts) improvements in best corrected visual acuity. We have subsequently updated the visual acuity data, as patients have continued their post-transplant follow up measurements, and have noted that visual acuity gains have persisted in several patients for more than 2 years.
Based on the data published in The Lancet in October 2014, and the continued safety profile observed in all 38 patients that have been treated, to date, with our experimental therapy, we plan to advance our programs to Phase 2 clinical proof-of-concept trials. To this end, we have met with regulatory agencies, including the US Food and Drug Administration (FDA), the European Medicines Agency (EMA) and the United Kingdom Medicines Healthcare Products Regulatory Agency (“MHRA”), to obtain feedback on our Phase 2 study designs. Based on this feedback and in consultation with our clinical investigators and other external consultants, we believe we have developed a set of clinical studies that will achieve our development objectives over the next several years.
We plan to initiate our Phase 2 trial for dry AMD in the second quarter of this year. The purpose of the dry AMD trial is to evaluate the safety and exploratory efficacy of our hESC-derived RPE cells. This Phase 2 study will include three cohorts of 20 subjects each, 15 who will be treated with our RPE therapy and 5 untreated control subjects. Each of the three cohorts will be treated with different immune suppression regimens. We intend to complete the first cohort during 2015 and the second two cohorts by the end of 2016.
The purpose of our pivotal SMD trial will be to evaluate the safety and efficacy of our hESC-derived RPE cells. This trial will include 100 subjects, 50 who will be treated with our RPE therapy and 50 untreated control subjects.
To support these plans we intend to expand our clinical operations capabilities. We currently work with four clinical sites in the US and two in the UK. In addition, we are expanding the network of contract service providers and also plan to expand our internal workforce. These expansions and the increased spending that will result from these expanded capabilities is consistent with our previously stated plans to transition into a late-stage clinical development company.
In February 2013, we announced that our clinical partner, the Jules Stein Eye Institute at the University of California, Los Angeles had received approval of its Investigator IND Application to initiate a Phase 1/2 study using our RPE cells to treat myopic macular degeneration, or MMD, a form of macular degeneration that can occur in association with severe forms of myopia. Myopia, or nearsightedness, is the most common eye disorder in the world, and is a significant global public health concern. Myopic macular degeneration seems to be associated with stress on the RPE layer as a consequence of pathological elongation of the eye in myopic patients. The apparent stress can induce fissures in the RPE layer, leading to RPE cell death and ultimately macular dystrophy and degeneration. It is an important public health issue lacking safe and effective treatments. Overall, MMD is reported to be the seventh-ranking cause of legal blindness in the United States, the fourth-ranking cause in Hong Kong and the second in parts of China and Japan. In June 2014, we announced that Jules Stein Eye Institute has initiated the trial. The actual enrollment of patients in this trial has been delayed by technical considerations to maximize patient safety.
As we continue to manage our clinical trials and expand the indications for which our RPE cell therapy is being investigated, we have also begun to take the steps to define our final product formulation, as well as to lay the early ground work to support appropriate pricing, coverage and reimbursement programs. We believe that our RPE therapy provides pricing justification across all categories of consideration by Medicare, Medicaid, National Health Service (UK) and private payers. Our program related to RPE cell therapy for the treatment of SMD has been granted Orphan Drug status in both the U.S. and Europe, which could accordingly lead to provide regulatory market exclusivity and potential FDA grant opportunities.
| 6 |
Photoreceptor Progenitor Program
Photoreceptors mediate the first step in vision, capturing light which, in turn, is converted into nerve signals to the brain. Photoreceptor atrophy, and subsequent cell death and permanent loss of photoreceptors, is seen as a consequence of degenerative diseases including AMD, and SMD as well as various inherited retinopathies. Loss of photoreceptors is also a consequence of acquired conditions such as diabetes. We recognize the potential value of being able to repair the retina with replacement photoreceptor cells derived from pluripotent stem cell sources such as iPSCs and hESCs. We believe those therapies can provide the basis for new approaches for treating a wide variety of retinal degenerations in diseases where photoreceptors malfunction and/or die, either alone or in combination with our RPE therapy.
We have developed a human photoreceptor progenitor cell which we believe is unique with respect to both the markers they express as well as their plasticity, meaning that they can differentiate into both rods and cones, and therefore provide a viable source of new photoreceptors for retinal repair. In addition, our photoreceptor progenitors appear to secrete neuroprotective factors, and have the ability to phagocytose (digest) such materials as the drusen deposits that build up in the eyes of dry AMD patients, and so may provide additional benefits beyond forming new photoreceptors when injected into the subretinal space in the eyes of patients. We will continue our pre-clinical investigation in animal models, establish appropriate correlation between integration of the transplanted cells and visual function in the animals, and then consider preparation of an IND and/or IMPD application to commence clinical studies with these cells. As part of our evaluation of this program, we will consider, among other things, the data generated from pre-clinical studies, our understanding of the potential market opportunity, and our ability to allocate capital resources to adequately fund the program to the next milestone.
Retinal Ganglion Cell Progenitor Program
In the United States alone, approximately 100,000 people are legally blind from glaucoma. Proven treatments include drug therapy or surgery to lower intraocular pressure; however, many patients lose vision despite receiving these treatments. In glaucoma, retinal ganglion cells degenerate before photoreceptors are lost. We are currently conducting pre-clinical research and development activities regarding differentiation of stem cells into retinal ganglion cells and demonstration of the ability of those cells to protect against elevated intraocular pressure in glaucoma models. We have succeeded in generating a unique human ganglion progenitor cell which, when injected in animal models of glaucoma, appear to protect against damage. We will continue our pre-clinical investigation in animal models, establish appropriate correlation between integration and visual function in the animals, and then consider preparation of an IND and/or IMPD application to commence clinical studies with these cells. As part of our evaluation of this program, we will consider, among other things, the data generated from pre-clinical studies, our understanding of the potential market opportunity, and our ability to allocate capital resources to adequately fund the program to the next milestone.
Corneal Endothelial Program
Diseases and injuries affecting the cornea are another major cause of blindness worldwide. Although the cornea is clear and seems to lack substance, it is actually a highly organized group of cells and proteins. To see well, all layers of the cornea must be free of any cloudy or opaque areas which can be caused by, among other reasons, swelling of the cornea due loss of the corneal endothelial cells (CECs). The CECs are single layer of cells required to maintain the health and clarity of the cornea and are not know to regenerate spontaneously. In instances where the cornea is damaged or scarred, such as due to chemical injury or infection or thinning as a consequence of aging or an inherited disorder, the current standard of care is a cornea transplant, also referred to as a keratoplasty or corneal graft. The graft replaces damaged corneal tissue with healthy corneal tissue donated from an eye bank. In the past, full thickness corneal transplants were used as part of the procedure. However, a newer version of corneal transplant, known as Descemet's Stripping Endothelial Keratoplasty, (DSEK), is gaining prominence as the surgical method for visual rehabilitation of secondary to corneal pathology [gReg1] . DSEK utilizes the innermost layers, i.e., the endothelial layer and Descemet membrane, for transplant. While corneal transplants are performed routinely (more than 40,000 corneal transplants are performed in the U.S. each year), there is still a pressing need for transplantable corneal tissue. The cadaveric source of donor eyes in the eye banks in the U.S. is not sufficient to meet the demand for the number of patients in need of the surgery, and the corneal tissue being used is often from older donors and hence not as dense or robust as might be desired. We believe that corneal blindness is a significant unmet medical need.
We have been able to generate sheets of corneal endothelial cells, with Descemet membrane, from hESCs. These endothelial sheets, which resemble fetal cornea in cell density, and thickness and durability of the tissue graft, could serve as the transplanted tissue in DSEK. In culture, our corneal endothelial cells have all the hallmarks, both marker expression and morphology, of native human corneal endothelium. We have tested these cells in several animal models of corneal diseases. As part of our evaluation of this program, we will consider, among other things, the data generated from pre-clinical studies, our understanding of the potential market opportunity, and our ability to allocate capital resources to adequately fund the program to the next milestone.
Other Programs
In addition to our ophthalmology programs, we are investing a limited portion of our resources to advance other programs where we feel that we can leverage our expertise in cellular and developmental biology to generate therapies that have the potential to improve health care in other prevalent degenerative diseases and diseases of aging. At the core of our pipeline planning are approaches intended to address large unmet medical needs with allogeneic stem cell-derived therapeutics. The criteria for prioritizing and allocating resources to these programs include stem cell capability, competitive landscape within the therapeutic area, severity/prevalence of the therapeutic need and our perceived ability to out-license these non-core programs to, or otherwise collaborate with third parties that may be able to help advance these programs more efficiently than we can on our own. We utilize a proof-of-concept approach in our product development process, testing our candidate therapies in relevant animal models of human disease in order to assess the likelihood of success when it comes time to try those therapies in human patients.
| 7 |
Hemangio-derived Mesenchymal Cells™
Pluripotent stem-cell derived mesenchymal stem cells (MSCs) regulate immune and inflammatory responses, making them an attractive tool for the treatment of autoimmunity and inflammation. Their underlying molecular mechanisms of action together with their clinical benefit — for example, in autoimmunity — are, in our opinion, being revealed by an increasing number of clinical trials and pre-clinical studies of MSCs. The immunosuppressive/ immunomodulatory activity of these cells allows MSCs to be transplanted nearly universally, i.e., as an allogeneic cell therapy, without matching between donors and recipients. This versatility, along with the ability to manufacture and store these cells, presents a unique opportunity to produce an "off-the-shelf" cellular therapy ready for treatment of diseases in both acute and chronic settings.
Current competitive MSC products for therapeutic applications are isolated from donor-dependent primary tissue sources such as umbilical cord, bone marrow, and adipose (fat) tissue. To achieve commercial scale quantities, MSCs isolated from these tissues need to be expanded in culture, yet the process of expansion compromises the therapeutic potency of these cells. Accordingly, the number of doses of MSCs that can be generated from each donor must be limited in order to preserve potency. Multiple donors must therefore be used to achieve large scale manufacturing required for an off-the-shelf therapy. The use of multiple donors introduces variability in the final MSC product and also drives up costs as pathogen screening must be performed for each new donor.
We believe we have succeeded at creating a unique cellular product, which we refer to as Hemangio-derived Mesenchymal Cells™ (“HMC’s) with attributes similar to MSCs but that circumvent the issues encountered with donor-dependent sources, Our HMCs are produced from a single, pluripotent stem cell source, the renewable nature of which, permits us to manufacture large scale quantities of HMCs without the need for extensive in vitro culture, thus preserving their potency. The stem-cell-sourced manufacturing process may be scalable for global commercialization and therefore may prove to be less costly (particularly at commercial scale) than the primary tissue-sourced MSC products in development by other companies.
Pre-clinical testing of our stem cell-derived HMCs has demonstrated their therapeutic efficacy in various autoimmune disease models. During the course of this testing, we noted another differentiating feature of our cells: the HMCs generated using our proprietary manufacturing approach appear to be more potent with respect to suppressing autoimmune disease than equivalent doses of bone marrow MSCs. This potency was dependent on the number of passages in culture, with the earlier passage HMCs retaining the greatest potency. This correlates with reports in the scientific literature which suggest that not only does the time in culture affect MSC potency but the age of the tissue source affects its potency as well. Evidence in the field suggests that MSCs from young tissue sources, such as embryonic, may be more potent than those from adult (e.g., bone marrow or adipose) tissue sources. Being derived from embryonic stem cells, the early-passage HMCs we are testing for potential therapeutic use are both youthful and replenishable, representing the earliest and most potent stage of biological development.
Our goal is to conduct a limited number of pre-clinical proof-of-concept studies, and based on those results, advance certain of these HMC discovery programs into IND-enabling pre-clinical studies and perhaps file IND applications, as circumstances dictate. In parallel, we are evaluating opportunities for strategic partnering relationships, out-licensing or other commercial transactions with outside parties, such as large pharmaceutical and biotech companies, with the objective of obtaining external funding for these programs so they may continue to be advanced with minimal costs to our company.
Neuroprotective Biologics
In the course of our work with various progenitor cells for treating ocular degenerative diseases, we have discovered that certain progenitor cells not only have the ability to participate directly in the formation of new tissue in the eye, but also were able to exert a neuroprotective effect that reduces the rate of degeneration of native photoreceptors in the animals’ eyes, for example, in animal models of macular degeneration. These cells appeared to also be a source of neuroprotective paracrine factors; biological agents which may themselves be useful as drugs. Further, we observed that these protective effects were uniquely produced by particular progenitor cell sub-types. The restriction of this protective activity to only a certain progenitor cell type permits us to examine which factors are differentially produced by these cells as compared with other closely related progenitor cells which do not seem to secrete any protective agents. We anticipate that the neuroprotective agent(s) that we may ultimately develop as drug candidates may be useful not only in retinal diseases and dystrophies, but may have broader applications in central nervous system and peripheral nervous system diseases and disorders, including diseases causing cognitive function impairment, movement disorders such as Parkinson’s Disease, and ischemic events such as caused by stroke.
| 8 |
Platelets
Platelets are key elements in maintaining blood vessel integrity, or hemostasis, and are therefore central to wound healing and tissue regeneration after injury or surgery. Platelets are a mainstay in treating trauma, and are increasingly being used to promote healing from a wide range of surgeries. When platelet levels decrease and result in thrombocytopenia, such as when bone marrow is destroyed or suppressed, the decrease in platelet function is often a leading cause of morbidity.
Platelets are the most difficult blood product to maintain. They cannot be frozen or refrigerated. Instead, they must be stored at room temperature which limits the shelf life of platelets to five to seven days both because of loss of activity and risk of bacterial contamination during storage. Accordingly, it is our belief that the practical use of platelets is limited by availability. Our estimates are that, but for limitations on donated platelet supplies, there would be a demand for a substantial number of additional units of platelets each year beyond the current platelet usage, particularly for expanded use in surgical settings such as joint replacement or to prevent scarring.
We have developed a manufacturing process for generating megakaryocytes, proplatelet forming cells and ultimately platelets using either hESCs or iPSCs as the starting materials. This process can be carried out under GMP conditions, and we are approaching the ability to produce clinical doses of platelets. We have also solved an important problem in this process, in that we have developed a feeder-free process for making platelets from start to finish. This means our process may be portable into a continuous flow bioreactor to permit large-scale manufacturing.
Overall, we observe that the platelets made by our stem cell process have ultrastructural and morphological features that are indistinguishable from normal blood platelets. We believe our platelets function appropriately as well, both in vitro and in vivo. They respond to thrombin stimulation, form micro-aggregates, and facilitate clot formation and retraction. In animal models of injury, our stem cell derived platelets contribute to developing thrombi at sites of vascular injury.
While the early data that we have generated are encouraging, we believe that our limited resources are best allocated to our other programs, at this time. In the future we may look to partner this program, pursue government funding or permanently cancel the program.
Our Intellectual Property
Our research and development is supported by a robust intellectual property portfolio, including pending patent applications and issued patents. As of February 11, 2015, we have 58 issued patents and 180 pending patent applications filed worldwide, of which 13 issued patents and 82 pending patent applications pertain to our active product development programs. Any patents that may issue from our pending patent applications and which are directed to our current clinical and pre-clinical therapeutic programs would expire between 2025 and 2035, excluding any patent term extension. These patents and patent applications disclose compositions of matter, pharmaceutical compositions, methods of use and manufacturing methods. For instance, we already have issued patents with broad reaching claims in the U.S. and other major markets around our RPE clinical programs, and continue to actively improve our existing portfolio with filings around the improvements we make as we have translated the use of RPE cells from the bench to a regulated manufacturing and human therapy program. To illustrate, over the past few years, the United States Patent and Trademark Office, and other patent offices in major market countries, have granted several of our patents covering the methods we use to derive and produce our RPE cell therapy, as well as patents that cover the use of the RPE cells for formulating pharmaceutical preparations for use in human patients and for treating various macular degenerative diseases such as dry AMD and SMD.
With respect to our therapeutic programs generally, we have filed a number of patent applications, including broad omnibus patent applications, intended to cover the generation of transplantable cells and tissues from any pluripotent stem cell source including hESC, iPSC and other pluripotent stem cell sources as may be identified. Our patent strategy has been to protect the method of manufacturing these transplantable cells and tissues, as well as pharmaceutical preparations of the cells/tissues and the use of those pharmaceutical preparations in patient treatment settings. Our patent strategy includes very broad claims, as well as claims more narrowly directed to our actual processes and formulations. In the case of our RPE Program, to illustrate, we have pursued layers of various independent and dependent claims that range from very broad methods and formulations, to narrower claims which further define and protect the methods and compositions we actually use; as for example, the particular steps in our derivation process, defining the resulting RPE cells by marker and/or functional characteristics, the format of the RPE cells in the final formulation (cell suspension, sheet of cells, cells on a matrix support), etc. In the course of pursuing broad claims with the intention of covering not only our business but creating a barrier to entry to potential competitors who wish to use similar though not identical technology, we have focused on both literal claim scope as well as claims intended to provide additional coverage under the doctrine of equivalents.
Our success will likely depend upon our ability to preserve our proprietary technologies as well as operate without infringing the proprietary rights of other parties. However, we may also need to rely on certain proprietary technologies and know-how that are not patentable. With regard to our own proprietary information, we seek to protect such information, in part, by the use of confidentiality agreements with our employees, consultants and certain of our contractors.
| 9 |
We maintain a strategic patent policy and, when appropriate, seek patent protection for inventions in our core technologies and in ancillary technologies that support our core technologies, or which we otherwise believe will provide us with a competitive advantage. We pursue this strategy by filing patent applications for discoveries we make, either alone or in collaboration with scientific collaborators and strategic partners. Typically, although not always, we file patent applications both in the United States and in select international markets. In addition, we sometimes obtain licenses or options, if available, to acquire licenses to patent filings from other individuals and organizations that we anticipate could be useful in advancing our research, development and commercialization initiatives and our strategic business interests.
Our patents do have a finite life with respect to enforcement against third parties and will eventually expire. The fundamental consequence of patent expiration is that the invention covered by that patent will enter the public domain. However, the expiration of patent protection, or anticipated patent protection, for the bulk of our portfolio is not scheduled to begin for approximately ten to fifteen years. In some instances, we believe that patent term extensions and adjustments, or other forms of exclusivity dependent on our patent rights, may be available in particular instances, such as by operation of patent and/or regulatory laws and regulations. As we make improvements to formulations and dosage amounts, find new combinations of cells and combinations of cells and other therapeutics, refine manufacturing, and elucidate new indications for which our therapies can be used, we expect that we will continue to file additional patent applications covering these new inventions in the future. Any actual products that we develop are expected to be supported by intellectual property covered by granted patents or current patent applications that, if granted, would not expire for 20 years from the date first filed. For example, the granted United States patents covering our RPE cell therapy do not begin to expire until 2025 at the earliest, and then only if no patent term extensions and adjustments are provided. As we have made improvements to our RPE program, particularly arising from the translation of the cell therapy into a human patient treatment setting, we have diligently filed on those improvements. These additional patent filings may prove to be significant barriers to entry for third parties wishing to compete, and would extend the patent portfolio well into the 2030's.
Research and License Agreements
Stem Cell & Regenerative Medicine International
On December 1, 2008, we formed an international joint venture with CHA Biotech. The new company, SCRMI, was tasked with developing human blood cells and other clinical therapies based on our hemangioblast program, one of our core technologies. Under the terms of this agreement, we received a 33% interest in the joint venture upfront, and another 7% interest upon fulfilling certain obligations under the agreement over a period of 3 years. Our contribution included (a) the uninterrupted use of a portion of our leased facility at our expense, (b) the uninterrupted use of certain equipment in the leased facility, and (c) the release of certain of our research and science personnel who were subsequently employed by the joint venture. In return, for a 60% interest, CHA Biotech contributed $150,000 cash upfront and funded operational costs thereafter. Additionally, SCRMI paid us a fee of $500,000 for an exclusive, worldwide license to the hemangioblast program. In parallel, SCRMI granted an exclusive license to CHA Biotech for the commercialization of products arising from the hemangioblast in the territory of South Korea. As of December 31, 2014, we hold a 40% interest in the joint venture and CHA Biotech owns a 60% interest.
In July 2011, we entered into a binding term sheet with CHA Biotech in which SCRMI was realigned around both product development rights and research responsibilities. Under the terms of the binding term sheet, SCRMI exclusively licensed the rights to the hemangioblast program to us for United States and Canada and expanded the jurisdictional scope of the license to CHA Biotech to include Japan (in addition to South Korea, which was already exclusively licensed to CHA Biotech). As part of the agreement, the scientists at SCRMI involved in the hemangioblast program were transferred to us, and SCRMI discontinued its research activity and became solely a licensing entity. In order to maintain our exclusive license, we are obligated to satisfy certain diligence requirements relating to licensed products, defined in the license agreement as “any therapeutic, diagnostic, bioinformatics or other human or veterinarian health care product and/or service and or research reagent utilizing or derived in any manner whatsoever from the Technology”. By filing the investigational new animal drug application on September 12, 2013, with the U.S. FDA, we met the commitment required to maintain its exclusive license. Intellectual property rights created by us in the course of our research are subject to a non-exclusive license to CHA Biotech for Japan and South Korea, and to SCRMI to be sub-licensable under certain circumstances for countries other than the United States, Canada, Japan and South Korea.
CHA Biotech
On March 31, 2009, we entered into a licensing agreement under which we have licensed our RPE technology, for the treatment of diseases of the eye, to CHA Biotech for development and commercialization exclusively in Korea. We are eligible to receive up to $1.9 million in fees based upon achieving certain milestones, including us making an IND submission to the U.S. FDA to commence clinical trials in humans using the technology, which we completed during the second half of 2009. We received an up-front fee of $250,000 and additional consideration under the agreement in the amount of $850,000. Under the terms of the agreement, CHA Biotech will incur all of the cost associated with RPE clinical trials in Korea.
On May 21, 2009, we entered into a licensing agreement under which we licensed our proprietary single blastomere technology, which has the potential to generate stable cell lines, including RPE cells for the treatment of diseases of the eye, to CHA Biotech for development and commercialization exclusively in Korea. We received a $300,000 up-front license fee, and received an additional $300,000 in December 2009.
| 10 |
Embryome Sciences, Inc.
In 2008, we entered into three license agreements whereby we licensed to Embryome Sciences certain cell processing technologies, including technology licensed from Kirin Beer. We received up-front payments of $470,000 and will receive royalties from future sales, if any, of product that utilizes the technologies from the licenses.
Regulations
Our research and development activities and the future manufacturing and marketing of our potential therapeutic products are, and will continue to be, subject to regulation for safety and efficacy by numerous governmental authorities in the United States and other countries.
In the United States, pharmaceuticals, biologicals and medical devices are subject to rigorous regulation by the FDA. The Federal Food, Drug and Cosmetic Act, the Public Health Service Act, applicable FDA regulations, and other federal and state statutes and regulations govern, among other things, the testing, manufacture, labeling, storage, export, record keeping, approval, marketing, advertising, and promotion of our potential products. Product development and approval within this regulatory framework takes a number of years and involves significant uncertainty combined with the expenditure of substantial resources.
The steps required before our potential therapeutic products may be marketed in the United States include: pre-clinical laboratory and animal tests; submission and acceptance of an IND application; safe and efficacious human clinical trials; submission of a Biologics Licensing Application; and Regulatory Approval.
The testing and approval process will require substantial time, effort and expense. The time for approval is affected by a number of factors, including relative risks and benefits demonstrated in clinical trials, the availability of alternative treatments and the severity of the disease. Additional animal studies or clinical trials may be requested during the FDA review period, which might add to that time. FDA approval of the application(s) is required prior to any commercial sale or shipment of the therapeutic product. Biologic product manufacturing facilities located in certain states also may be subject to separate regulatory and licensing requirements.
In addition, the FDA may require post-marketing studies. After receiving FDA marketing approval for a product for an initial indication, further clinical trials may be required to gain approval for the use of the product for additional indications. The FDA may also require post-marketing testing and surveillance to monitor for adverse effects, which could involve significant expense, or the FDA may elect to grant only conditional approvals subject to collection of post-marketing data.
Among the conditions for product licensure is the requirement that the prospective manufacturer’s quality control and manufacturing procedures conform to the FDA’s current good manufacturing practice (GMP) requirements. Even after a product’s licensure approval, its manufacturer must comply with GMP on a continuing basis, and what constitutes GMP may change as the state of the art of manufacturing changes. Domestic manufacturing facilities are subject to regular FDA inspections for GMP compliance, which are normally held at least every two years. Foreign manufacturing facilities are subject to periodic FDA inspections or inspections by the foreign regulatory authorities. Domestic manufacturing facilities may also be subject to inspection by foreign authorities.
In addition to safety regulations enforced by the FDA, we are also subject to regulations under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act and other present and potential future and federal, state, local, and foreign regulations.
Outside the United States, we will be subject to regulations that govern the import of drug products from the United States or other manufacturing sites and foreign regulatory requirements governing human clinical trials and marketing approval for our products. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursements vary widely from country to country.
The United States Congress, several states and foreign countries have considered legislation banning or restricting human application of ESC-based and nuclear transfer based technologies. No assurance can be given regarding future restrictions or prohibitions that might affect our technology and business. In addition, we cannot assure you that future judicial rulings with respect to nuclear transfer technology or hESCs will not have the effect of delaying, limiting or preventing the use of nuclear transfer technology or ESC-based technology or delaying, limiting or preventing the sale, manufacture or use of products or services derived from nuclear transfer technology or ESC-derived material. Any such legislative or judicial development would harm our ability to generate revenues and operate profitably.
For additional information about governmental regulations that will affect our planned and intended business operations, see the section entitled “Risk Factors” beginning below.
| 11 |
Competition
We are engaged in activities in the biotechnology field, which is characterized by extensive research efforts and rapid technological progress. If we fail to anticipate or respond adequately to technological developments, our ability to operate profitably could suffer. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, products that are more effective or less costly than any product candidate that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization and market penetration than us. Additionally, technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing our product candidates against competitors. We cannot assure you that research and discoveries by other biotechnology, agricultural, pharmaceutical or other companies will not render our technologies or potential products or services uneconomical or result in products superior to those we develop or that any technologies, products or services we develop will be preferred to any existing or newly-developed technologies, products or services.
We are aware that several companies and non-profit entities are working on various RPE formulations for treating macular degeneration. For example, Pfizer, Regenerative Patch Technologies and the Riken Center for Developmental Biology (Japan) have publicly stated that each is working towards clinical trials of RPE patches (sheets of cells) for treating wet AMD, and have also stated that they believe their formulations of RPE cells could potentially be used for treating dry AMD. Cell Cure Neurosciences Ltd. (Israel) has previously announced that it is developing RPE cell formulations for dry AMD.
Other cell types are also being developed for subretinal use in treating various forms of macular degeneration. StemCells Inc. recently commenced treating dry AMD patients with purified human neural stem cells. Bioheart, Inc. sponsors an active clinical trial for treating dry AMD with adipose stem cell (ASC), while the University of California Davis and Retinal Associates of South Florida are the sponsors of FDA approved pilot studies to determine whether it would be safe and feasible to inject CD34+ stem cells from bone marrow into the eye as treatment for patients who are irreversibly blind from various retinal conditions including dry AMD. Neurotech, Inc. recently completed a phase I study testing the safety of injecting encapsulated cells that express CNTF in dry AMD patients, while Janssen Research & Development, LLC suspended temporarily its safety study of umbilical cord stem cells administered subretinally in dry AMD patients.
Research and Development Expenditures
We spent the following amounts on company-sponsored research and development activities during each of the last three fiscal years:
| Fiscal Year | Research and Development Expenditures | |||
| 2014 | $10,529,321 | |||
| 2013 | $11,564,768 | |||
| 2012 | $14,158,936 |
Employees
As of March 1, 2015, we had 37 full-time employees, of whom 10 hold Ph.D. or M.D. degrees. Twenty-six employees are directly involved in research and development activities and 11 are engaged in business development and administration. We also use the services of numerous outside consultants in business and scientific matters. We believe that we have good relations with our employees and consultants.
Corporate Information
We were incorporated in Nevada under the name Two Moon Kachinas Corp. on May 18, 2000. On December 30, 2004, we changed our corporate name to A.C.T. Holdings, Inc. On January 31, 2005, we completed the acquisition of Advanced Cell Technology, Inc., a Delaware corporation, or ACT, pursuant to the terms of an Agreement and Plan of Merger dated December 31, 2004. On June 17, 2005, we changed our corporate name to Advanced Cell Technology, Inc. On November 18, 2005, we consummated a merger with and into our wholly-owned subsidiary ACT. As a result of the reincorporation, we became a Delaware corporation. On November 12, 2014, we changed our corporate name to Ocata Therapeutics, Inc.
| 12 |
Item 1A. Risk Factors.
An investment in our common stock involves a high degree of risk. You should carefully consider the risks described below, including information in the section of this document entitled “Forward Looking Statements.” The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our common stock could decline, and you may lose all or part of your investment.
Risks Relating to our Early Stage of Development and Capital Resources
We have a history of operating losses and we may not achieve future revenues or operating profits.
We have generated modest revenue to date from our operations. Historically, we have had net operating losses each year since our inception. As of December 31, 2014, we have an accumulated deficit of $349,134,225 and a stockholders’ deficit of $2,735,545. We incurred net losses of $34,748,945, $31,022,248, and $34,584,115 for the years ended December 31, 2014, 2013, and 2012, respectively. We have limited current potential sources of income from licensing fees and we do not generate significant revenue from any other source. We have no therapeutic products currently available for sale and do not expect to have any therapeutic products commercially available for sale for a period of years, if at all. Additionally, even if we are able to commercialize our technologies or any products or services related to our technologies if approved, it is not certain that they will result in revenue or profitability.
Our business is at an early stage of development and we may not develop therapeutic products that can be commercialized.
Our most advanced product candidates are being prepared for use in Phase 2 clinical trials and we do not have any products that are currently in the marketplace. Though we have recently released clinical data from our Phase I/II clinical trial regarding the safety and tolerability of sub-retinal transplantation of hESC-derived RPE cells transplanted into patients with SMD and dry AMD, our potential therapeutic products will require additional extensive pre-clinical and clinical testing prior to any possible regulatory approval in the United States and other countries and may additionally require post-authorization outcome studies. We may not be able to obtain regulatory approvals for any of our products (see the subsection entitled “Regulatory Risks” below), or commence or continue clinical trials for any of our products, or commercialize any products. Any of our therapeutic and product candidates may prove to have undesirable and unintended side effects or other characteristics that could cause adverse effects on patient safety, efficacy or cost-effectiveness that could prevent or limit their therapeutic use, commercialization or acceptance in the medical community. Clinical trial results that we view as positive or proof of safety and/or efficacy may not be viewed in the same manner by regulators or potential collaborators. Any product using any of our technologies may fail to provide the intended therapeutic benefits, or even achieve therapeutic benefits equal to or better than the standard of treatment at the time of testing or production, or may not be safe for use in humans. In addition, we will need to determine whether any of our potential products can be manufactured in commercial quantities or at an acceptable cost, with or without third-party support. Our efforts may not result in a product that can be or will be marketed successfully. Physicians may not prescribe our products, patients may not use our products, or third-party payors may not cover or provide adequate reimbursement for our products. For these reasons and others, we may not be able to generate product revenues.
We have never generated any revenue from product sales and may never be profitable.
We have limited clinical testing, regulatory, manufacturing, marketing, distribution and sales experience capabilities, which may limit our ability to generate revenues. Due to the early stage of our therapeutic products, including regenerative medical therapies and stem cell therapy-based programs, we have not yet invested significantly in regulatory, marketing, distribution or product sales resources. We cannot assure you that we will be able to invest or develop any of these resources successfully or as expediently as necessary, either alone or with strategic partners, to generate revenue.
Our ability to become profitable depends upon our ability to generate revenue. We do not anticipate generating revenues from product sales for the foreseeable future, if ever. Our ability to generate future revenues from product sales depends heavily on our success in:
| · | continuing and completing research and pre-clinical and clinical development of our therapeutic candidates, including Phase 2 trials of our RPE cell therapies for the treatment of SMD and dry AMD; |
| · | seeking and obtaining regulatory and marketing approvals for therapeutic candidates for which we complete clinical studies; |
| · | developing a sustainable, scalable, reproducible, and transferable manufacturing process for our therapeutic candidates; |
| · | establishing and maintaining supply and manufacturing relationships with third parties that can provide adequate (in amount and quality) products and services to support clinical development and the market demand for our therapeutic candidates, if approved; |
| 13 |
| · | commercializing therapeutic candidates for which we obtain regulatory and marketing approval, either by collaborating with a partner or, if launched independently, by establishing a sales force, marketing and distribution infrastructure; |
| · | obtaining market acceptance of our therapeutic candidates as a viable treatment option; |
| · | adequately addressing any competing technological and market developments; |
| · | implementing additional internal systems and infrastructure, as needed; |
| · | identifying and validating new therapeutic candidates; |
| · | negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter; |
| · | maintaining, protecting and expanding our portfolio of intellectual property rights, including patents, trade secrets and know-how, to cover our existing and future products; |
| · | obtaining and maintaining coverage and adequate reimbursement from third-party payors; |
| · | having appropriate “freedom to operate” necessary to manufacture, use and sell our existing and future products; and |
| · | attracting, hiring and retaining qualified personnel. |
Even if one or more of the therapeutic candidates that we may develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate, including costs related to additional clinical studies, and such costs may exceed our estimates. Even if we are able to generate revenues from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations. The inability to do so will inhibit or harm our ability to generate revenues or operate profitably.
Our ability to generate revenues depends on obtaining regulatory approvals for our products.
In order to successfully commercialize a product, we or our potential partners will be required to conduct tests and successfully complete clinical trials needed in order to meet regulatory requirements and to obtain applicable regulatory approvals. The costs of developing and obtaining regulatory approvals for therapeutics can be substantial. Our ability to commercialize any of our products in development is dependent upon the success of development efforts in clinical studies. If these clinical trials fail to produce satisfactory results, or if we are unable to maintain the financial and operational capability to complete these development efforts, we may be unable to generate revenues. Even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize products successfully. For example, if the approval process takes too long, we may miss market opportunities, have diminished or exhausted relevant patent rights, and enable other companies to develop competing products or establish market dominance. Additionally, the terms of any approvals may not have the scope or breadth needed for us to commercialize products successfully.
We have a limited operating history on which investors may evaluate our operations and prospects for profitable operations.
If we continue to suffer losses as we have in the past, investors may not receive any return on their investment and may perhaps lose their entire investment. Our prospects must be considered speculative in light of the risks, expenses and difficulties frequently encountered by companies in their early stages of development, particularly in light of the uncertainties relating to the new, competitive and rapidly evolving markets in which we anticipate we will operate. A substantial risk is involved in investing in us because, as an early stage company, we have fewer resources than an established company, our management may be more likely to make mistakes at such an early stage, and we may be more vulnerable operationally and financially to any mistakes that may be made, as well as to external factors beyond our control. We also have no experience bringing therapeutics candidates through the regulatory approval process to commercialization, and we operate with little budgetary margin for error. To attempt to address these risks, we must, among other things, further develop our technologies, products and services, successfully implement our research, development, marketing and commercialization strategies, respond to competitive developments, update our organizational structure to meet new business or market demands, and attract, retain and motivate qualified personnel. Any failure to achieve any of the forgoing would result in an inability to achieve profitability.
| 14 |
We will need to expand our organization and we may experience difficulties in managing this growth, which could disrupt our operations.
As of December 31, 2014, we had 37 full-time employees. As we mature and undertake the activities required to further develop and commercialize our therapeutic candidates, we may expand our full-time employee base and hire more consultants and contractors. Our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenues could be reduced, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
We have determined that material weaknesses existed in our system of internal control over financial reporting, which have in the past, and could in the future, result in a material misstatement of our annual and interim financial statements, which could have a material impact on our business and share price.
Our ability to implement our business plan and comply with regulations requires an effective planning and management process. We expect that we will need to improve existing operational and financial systems, procedures and controls, and implement new ones, to manage our future business effectively. Any implementation delays, or disruption in the transition to new or enhanced systems, procedures or controls, could harm our ability to forecast sales, manage our supply chain, and record and report financial and management information on a timely and accurate basis.
Furthermore we are required to maintain internal control over financial reporting adequate to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our consolidated financial statements in accordance with generally accepted accounting principles. In connection with the restatement of certain of our financial statements for fiscal years December 31, 2009, 2010, 2011, and 2012, for each quarter in our fiscal years ended December 31, 2011 and December 31, 2012, and for the first three quarters of the fiscal year ended December 31, 2013, we determined that we have a material weakness as of December 31, 2013, namely that our controls over the evaluation and review of complex and non-routine transactions were not effective. During the year ended December 31, 2014, management has remediated the aforementioned material weakness through a combination of hiring and training of employees.
We do not expect that our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. Over time, controls may become inadequate because changes in conditions or deterioration in the degree of compliance with policies or procedures may occur. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. As a result, we cannot assure you that significant deficiencies or material weaknesses in our internal control over financial reporting will not be identified in the future. A material weakness means a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the registrant’s annual or interim financial statements will not be prevented or detected on a timely basis.
Any failure to maintain or implement required new or improved controls, or any difficulties that we encounter in their implementation, could result in additional significant deficiencies or material weaknesses, cause us to fail to timely meet our periodic reporting obligations, or result in material misstatements in our consolidated financial statements. Any such failure could adversely affect the results of periodic management evaluations and annual auditor attestation reports regarding disclosure controls and the effectiveness of our internal control over financial reporting.
Our primary source of liquidity is our financing arrangement with Lincoln Park, and changes in our share price directly affect our ability to fund our operations.
We currently rely on our share purchase arrangement with Lincoln Park to fund our ongoing operations. Pursuant to the 2014 Purchase Agreement with Lincoln Park, the purchase price of such common stock sold to Lincoln Park is based on the prevailing market price of our common stock immediately preceding the time of sales; we control the timing and amount of any future sales, if any, of common stock. There are no upper limits to the price Lincoln Park may pay to purchase our common stock. The purchase price in most cases is directly derived from the prevailing market price of our common stock on OTCBB. Though the purchase price cannot be less than $1.00, subject to adjustment as set forth in the 2014 Purchase Agreement, this arrangement means that our prevailing share price directly affects the number of shares we need to issue to Lincoln Park at any given time to fund short-term operations. The number of shares issuable under our Certificate of Incorporation and the number of shares sellable to Lincoln Park are both limited, and a share price that falls and stays too low would make it difficult or impossible to fund our operations through sales of shares to Lincoln Park due to these limitations. As of December 31, 2014 $18,619,451 in proceeds remained available to us under the 2014 Purchase Agreement with Lincoln Park.
| 15 |
We will require substantial additional resources to fund our operations and to develop our product candidates. If we cannot find additional capital resources, we will have difficulty in operating as a going concern and growing our business.
The biotechnology industry in general, and research and development efforts in particular, is capital-intensive. Our future capital requirements will depend on many factors, including the:
| · | Progress, interim results and costs of pre-clinical development and laboratory testing and clinical trials; |
| · | time and costs involved in obtaining regulatory approvals; |
| · | number of product candidates we pursue; |
| · | costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; and |
| · | the costs associated with commercializing our product candidates if they receive regulatory approval, including the cost and timing of developing sales and marketing capabilities, or entering into strategic collaboration with others relating to the commercialization of our product candidates. |
Other than our arrangement with Lincoln Park, we have no sources of debt or equity capital committed for funding. Recent attempts to raise capital in the public equity markets have proven unsuccessful, and we can provide no assurance that we will be successful in any future funding effort. The timing and degree of any future capital requirements will depend on many factors, including:
| · | our ability to establish, enforce and maintain strategic arrangements for research, development, clinical testing, manufacturing and marketing; |
| · | the accuracy of the assumptions underlying our estimates of our short- and long-term capital needs, including capital needed to fund the clinical studies of our therapy candidates; |
| · | scientific progress in our research and development programs; |
| · | the magnitude and scope of our research and development programs; |
| · | our progress with pre-clinical development and clinical trials; |
| · | the time and costs involved in obtaining regulatory approvals; |
| · | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; and |
| · | the number and type of therapeutic candidates that we pursue. |
Our ability to execute our business strategy and sustain our infrastructure at our currently planned levels will be impacted by whether or not we have sufficient funds. Depending on market conditions and our ability to maintain financial stability, we may not have access to additional funds on reasonable terms or at all. Any inability to obtain additional funds when needed would have an adverse effect on our business and on our ability to operate on an ongoing basis.
Our independent auditor’s report for the fiscal year ended December 31, 2014 includes an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern.
Due to the uncertainty of our ability to meet our current operating and capital expenses, in their report on our audited annual financial statements as of and for the year ended December 31, 2014, our independent auditors included an explanatory paragraph regarding concerns about our ability to continue as a going concern. Recurring losses from operations raise substantial doubt about our ability to continue as a going concern. If we are unable to continue as a going concern, we might have to liquidate our assets and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements. In addition, the inclusion of an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern and our lack of cash resources may materially adversely affect our share price and our ability to raise new capital or to enter into critical contractual relations with third parties.
| 16 |
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights.
We may seek additional capital through a combination of private and public equity offerings, debt financings and collaboration, strategic and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted (with such dilution potentially being immediate and substantial), and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder.
Additional debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends, or requiring us to devote specific amounts of revenue or other capital to debt service or deleveraging payments. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. Any or all of the above may adversely affect the value of your investment in us.
Risks Relating to Technology
We are dependent on new and unproven technologies.
Our risks as an early stage company are compounded by our heavy dependence on unproven technologies. If these technologies do not produce satisfactory results in the clinical trial setting and/or are unable to gain regulatory approval, our business may be harmed. We have not shown an ability to bring any therapeutic candidate through the regulatory process to marketing approval. Given the unproven nature of our technology and potential product candidates, the FDA or other regulatory agencies may require additional clinical data or manufacturing practices than that required of conventional therapies or may otherwise require additional regulatory processes and reviews prior to any potential approval. Additionally, some of our technologies and potential revenue sources involve ethically sensitive and controversial issues which could become the subject of legislation or regulations materially restricting our development programs, future sales and marketing and other operations and, therefore, harm our financial condition and operating results.
We may not be able to commercially develop our technologies and proposed product lines, which, in turn, would significantly harm our ability to earn revenues and result in a loss of investment.
Our ability to commercially develop our technologies may be limited in part by a number of factors including, but not limited to, general economic conditions, the success of our research and pre-clinical and field testing, the availability of collaborative partners willing and able to finance our work in pursuing applications of cell therapy technologies, and technological or other developments in the biomedical field which may render our technologies obsolete or competitively unattractive. We may not pursue one or more commercialization strategies at all if we cannot locate a collaborative partner or entity willing to fund research and development or if we cannot agree to acceptable terms governing a potential development or marketing collaboration. Our decisions regarding the ultimate products and/or services we pursue could have a significant adverse effect on our ability to earn revenue if we misinterpret trends, underestimate development costs and/or pursue wrong products or services. Any of these factors either alone or in concert could materially harm our ability to earn revenues or could result in a loss of any investment in us.
State and/or country regulations relating to use and sale of products derived from hESCs may limit the markets we can penetrate.
Our product candidates consist of cells derived from a renewable hESC line. Certain states have, or have shown interest in, restricting or even banning embryonic stem cell research. Similarly, there are certain countries that restrict or ban hESC research. The laws and regulations surrounding hESC research continue to evolve and change, and this may affect our ability to conduct research, development or commercialization activities these markets.
We may not be able to maintain our hESC line that is used for the manufacture of our product candidates reliably and cost-effectively, and we may not be able to grow those cells at sufficient scale to continue development or commercialize our product candidates.
Operations with human cells, even with a stable, renewable hESC line, can be subject to conditions and influences that we may not be able to control. Cells could be lost due to contamination, equipment failure or improper installation or operation of equipment or laboratory technician error. Storage facilities could also be affected by labor shortages, natural disasters, power failures and numerous other factors that could result in the loss of all or a portion of our cell banks. It is also possible that the cells will simply cease to function, or that materials we use in manufacturing the cells could contain viruses or other pathogens, which would result in contamination of the cells. While we take precautions to prevent the cells from ceasing to function or becoming contaminated, long-term maintenance of hESC for our purpose has not been demonstrated and we could encounter unforeseen complications, including contamination, which could result in infection in patients or other adverse effects. In addition, if not all the cells respond to the directed differentiation cues as expected, it is possible that residual hESC could result in the potential for formation of hESC-derived teratoma, a tumor comprised of tissues not normally present at the site that might arise due to uncontrolled, off-target growth of the cells.
As we increase production to support future development and if approved, commercialization, we may experience significant quality and cost control issues and our business could be materially harmed.
| 17 |
Risks Related to Intellectual Property
Certain aspects of our business are dependent upon maintaining licenses with respect to key technology; if we are unable to obtain or protect intellectual property rights related to our product candidates, we may not be able to compete effectively in our markets.
Several of the patents we utilize are licensed to us by third parties. These licenses are subject to termination under certain circumstances (including, for example, our failure to make minimum royalty payments or to timely achieve spending, development and commercialization benchmarks). The loss of any of such licenses, or the conversion of such licenses to non-exclusive licenses, could harm our operations and/or enhance the prospects of our competitors. Certain of these licenses also contain restrictions, such as limitations on our ability to grant sublicenses that could materially interfere with our ability to generate revenue through the licensing or sale to third parties of important and valuable technologies that we have, for strategic reasons, elected not to pursue directly. The possibility exists that in the future we will require further licenses to complete and/or commercialize our proposed products. We cannot assure you that we will be able to acquire any such licenses on a commercially viable basis.
Certain parts of our technology are not protectable by patent.
Certain parts of our know-how and technology are not patentable. To protect our proprietary position in such know-how and technology, we require all employees, consultants, advisors and collaborators to enter into confidentiality and invention ownership agreements with us. We cannot assure you, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, in the absence of patent protection, competitors who independently develop substantially equivalent technology may harm our business.
Patent litigation presents an ongoing threat to our business with respect to both outcomes and costs.
Companies in the life science industry typically obtain patents and frequently engage in substantial intellectual property litigation. Our products and technologies could infringe on the rights of others. If a third party successfully asserts a claim for infringement against us, we may be liable for substantial damages, be unable to sell products using that technology, or have to seek a license (which may or may not be on favorable terms) or redesign the related product. These alternatives may be uneconomical or impossible. Intellectual property litigation could be costly, result in product development delays and divert the efforts and attention of management from our business. We may also be unable to obtain licenses under economically viable terms needed to develop its technology or for certain intellectual property needed to develop and commercialize its products.
In addition to our ability to avoid infringing the proprietary rights of others, our success will also depend, in part, on our ability to maintain protection for our products and technologies under the patent laws of the United States and other countries. Our patent rights could be challenged by others, or if issued, could later be deemed invalid or unenforceable. Patent prosecution, related proceedings, and litigation in the U.S. and in other countries may be expensive, time consuming and ultimately unsuccessful. In addition, patents issued by foreign countries may afford less protection than is available under U.S. patent law and may not adequately protect our proprietary information. We have previously been involved in patent interference litigation, and it is possible that further litigation or patent office proceedings (such as oppositions, observations and/or reexaminations) over one or more of our own patent filings could arise. We could incur substantial litigation costs or costs associated with patent office proceedings in defending ourselves against suits or other actions brought against us or in suits in which we may assert our patents against others. If the outcome of any such litigation or patent office proceeding is unfavorable, our business could be materially adversely affected. The expiration of patents on which we rely for protection of key products could diminish our competitive advantage and adversely affect our business and prospects. Our competitors may independently develop proprietary technologies and processes that design around the coverage of our patents.
Without additional capital, we may not have the resources to adequately defend or pursue such litigation or patent office proceedings. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have an adverse effect on the price of our common stock.
We may not be able to protect our proprietary technology and/or the practice of our technology may infringe other third party patents, which could harm our ability to operate profitably.
The biotechnology and pharmaceutical industries place considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Our success will depend, to a substantial degree, on our ability to obtain and enforce patent protection for our products, preserve any trade secrets and operate without infringing the proprietary rights of others. We cannot assure you that:
| · | we were the first to file patent applications for these inventions; |
| · | we were the first to make the inventions covered by each of our pending patent applications; |
| · | any patents issued to us will cover our products as ultimately developed; |
| 18 |
| · | the term of any patents issued to us can be extended under the provisions of patent term extension afforded by U.S. law or similar provisions in foreign countries, where available; |
| · | there is no prior art of which we are not aware that may affect the validity or enforceability of a patent claim; |
| · | there is no prior art of which we are aware, which we do not believe affects the validity or enforceability of a patent claim, but which, nonetheless, ultimately may be found to affect the validity or enforceability of a patent claim; |
| · | we will succeed in obtaining any patents in a timely manner or at all, or that the breadth or degree of protection of any such patents will protect our interests, or that such patents would even be enforceable; |
| · | the use of our technology will not infringe on the proprietary rights of others; |
| · | patent applications relating to our potential products or technologies will result in the issuance of any patents or that, if issued, such patents will afford adequate protection to us or not be challenged invalidated or infringed; |
| · | if issued, patents might not be declared as unenforceable or invalid by operation of law; |
| · | patents will not issue to other parties, which may be infringed by our potential products or technologies; and |
| · | we will continue to have the financial resources necessary to prosecute our existing patent applications, pay maintenance fees on patents and patent applications, or file patent applications on new inventions. |
We are aware of certain patents that have been granted to others and certain patent applications that have been filed by others with respect to iPSCs, ESCs, and other stem cell technologies. The fields in which we operate have been characterized by significant efforts by competitors to establish dominant or blocking patent rights to gain a competitive advantage, and by considerable differences of opinion as to the value and legal legitimacy of competitors’ purported patent rights and the technologies they actually utilize in their businesses.
Patents obtained by other persons may result in infringement claims against us that are costly to defend and which may limit our ability to use the disputed technologies and prevent us from pursuing research and development or commercialization of potential products.
A number of other pharmaceutical, biotechnology and other companies, universities and research institutions have patents or patent applications potentially relevant to or required in the manufacturing, storage, sale or use of our expected products. In the case of pending patent application, we cannot predict which, if any, of such applications will issue as patents or the claims that might be allowed. We are aware that a number of companies have filed patent applications, which in some cases have resulted in issued patents, relating to the generation, formulation and uses of various stem cells, as well as RPE cells, photoreceptor progenitor cells, and mesenchymal stem cells.
If third party patents or patent applications contain claims infringed by us or any strategic partner or other licensee of our products, such as for the manufacturing, storage, sale or use of our expected products, and such patent claims are ultimately determined to be valid and enforceable against us or our licensees, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost, if at all, or be able to develop or obtain alternative technology. If we are unable to obtain such licenses at a reasonable cost, we may not be able to develop some products commercially. We may be required to defend ourselves in court against allegations of infringement of third party patents. Patent litigation is very expensive and could consume substantial resources and create significant uncertainties. An adverse outcome in such a suit could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties, or require us or our licensees to cease using such technology.
Changes in U.S. patent law and in patent law in other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and therefore is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings and rulings from the European Patent Office Board of Appeals have narrowed the scope of patent protection available in certain circumstances and weakened and potentially eliminated the rights of patent owners in certain situations, including potentially relating to the patentability of certain cells and tissues, including those generated from hESC lines, and their uses as therapeutics. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, and equivalents bodies in other major markets, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
| 19 |
We may not be able to adequately defend against piracy of intellectual property in foreign jurisdictions.
Considerable research in the areas of stem cells, cell therapeutics and regenerative medicine is being performed in countries outside of the United States, and a number of potential competitors are located in these countries. The laws protecting intellectual property in some of those countries may not provide adequate protection to prevent our competitors from misappropriating our intellectual property. Several of these potential competitors may be further along in the process of product development and also operate large, company-funded research and development programs. As a result, our competitors may develop more competitive or affordable products, or achieve earlier patent protection or product commercialization than we are able to achieve. Competitive products may render any products or product candidates that we develop obsolete.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In this event, competitors might be able to enter the market earlier than would otherwise have been the case.
Risks Related to Clinical Development
The risk of failure of clinical development is high. Clinical studies are expensive, time-consuming, uncertain and susceptible to change, delay or termination. Any delay or failure in obtaining required approvals could have a material adverse effect on our business.
We do not currently have marketing approval for any products. Before obtaining marketing approval from regulatory authorities for sale of our products, we must conduct extensive clinical studies to demonstrate the safety and efficacy of such product in humans for the desired indications. We are planning to initiate Phase 2 clinical studies for some of our product candidates, initially pursuing treatment of several forms of macular degeneration, such as SMD, dry-AMD and MMD. However, clinical testing is expensive and can take many years to complete, its outcome is inherently uncertain and it will take years to obtain approval, if at all. It is impossible to predict when or if any of our product candidates for which we may seek marketing approval will be deemed by the FDA to be safe and effective enough to receive regulatory approval for the desired indications.
Despite promising results in earlier clinical studies, including the trials for age-related macular degeneration and Stargardt’s macular dystrophy, the results from our clinical studies may neither demonstrate adequate safety and effectiveness to the satisfaction of the FDA nor be predictive of the outcome of later stage trials.
Clinical trial results are subject to varying interpretation. Even if we conclude that the results from our clinical studies demonstrate the safety and effectiveness of our product candidates, the FDA may not agree with us or could also conclude that the clinical study results are not clinically meaningful, that there were human errors in the conduct of the clinical study, or that for some other reason the clinical study results are inadequate to support approval. Numerous other factors could affect the timing, cost or outcome of our development efforts, including the following:
| · | conditions imposed on us by regulatory authorities regarding the scope or design of our clinical studies; |
| · | delays in reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical study sites; |
| · | failure to obtain, or delays in obtaining, the required regulatory approvals for the facilities or the processes used to manufacture our product candidates or any changes to such processes; |
| · | insufficient or inadequate supply or quality of product candidates being tested or other necessary materials necessary to conduct our clinical studies; |
| · | delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites; |
| · | problems in engaging institutional review boards, or IRBs, to oversee trials or problems in obtaining or maintaining IRB approval of studies at each clinical study site; |
| 20 |
| · | delays in recruiting and enrolling suitable patients to participate in our clinical studies in conformity with required protocols or projected timelines; |
| · | imposition of a clinical hold on our studies by regulatory agencies; |
| · | failure by our CROs, other third parties or us to adhere to clinical study requirements, comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| · | failure to perform in accordance with the FDA’s good clinical practices, or GCPs, or applicable regulatory guidelines in other countries; |
| · | delays in having patients complete participation in a study or return for post-treatment follow-up; |
| · | clinical study sites or patients dropping out of a study; |
| · | occurrence of undesirable toxicities, side effects or other unexpected adverse results; |
| · | occurrence of serious adverse events associated with the product being tested that are viewed to outweigh its potential benefits; |
| · | negative or inconclusive results from our clinical studies or the clinical studies of others for similar product being tested or inability to generate statistically significant data confirming the efficacy of the product being tested; |
| · | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; |
| · | changes in the regulatory environment, including pricing, coverage and reimbursement, that make development no longer desirable; |
| · | delays in filing or acceptance of a supplement to our Biologics License Application; or |
| · | failure or delays in obtaining pricing, coverage and reimbursement approvals. |
Any inability to successfully complete pre-clinical and clinical development could result in additional costs to us or impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, clinical study delays could also shorten any periods during which we may have the exclusive right to commercialize our products or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our products and may harm our business and results of operations.
If side effects are identified during the time our products candidates are in development or after they are approved and on the market, we may either choose to or be required to perform lengthy additional clinical trials, discontinue development of the affected product candidate, change the labeling of any such products, or withdraw any such products from the market, any of which would hinder or preclude our ability to generate revenues.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. The side effects could affect patient recruitment or the ability of enrolled patients to complete any ongoing clinical trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly. Even if any of our product candidates receives marketing approval, as greater numbers of patients use a product following its approval, an increase in the incidence of side effects or the incidence of other post-approval problems that were not seen or anticipated during pre-approval clinical trials could result in a number of potentially significant negative consequences, including:
| · | regulatory authorities may withdraw their approval of the product; |
| · | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| · | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| · | we could be sued and held liable for harm caused to patients; and |
| · | our reputation with patients and healthcare providers may suffer. |
Any of these events could substantially increase the costs and expenses of developing, commercializing and marketing any such product candidates or could harm or prevent sales of any approved products.
We rely on third parties to conduct, supervise and monitor our clinical studies, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies.
We do not independently conduct our own clinical studies but rather rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to perform this function. Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it could delay our product development activities.
Our reliance on these third parties for clinical development activities reduces our control over these activities but does not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our pre-clinical and clinical studies are conducted in accordance with the study plan and protocols and legal, regulatory, and scientific standards. We and our third party contractors are required to comply with requirements for Good Clinical Practices (GCPs), which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP requirements. In addition, our clinical trials must be conducted with product produced under current Good Manufacturing Practices (GMPs) regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
| 21 |
The performance of these third parties may be adversely affected by various issues, including less advanced medical infrastructure, lack of familiarity with conducting clinical studies using U.S. standards or using our research materials, insufficient training of personnel and communication difficulties. We remain responsible for ensuring that each of our clinical studies is conducted in accordance with the general investigational plan and protocols for the study. Moreover, the FDA requires us to comply with GCP, for conducting, recording and reporting the results of clinical studies to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of patients in clinical studies are protected.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical studies in accordance with regulatory requirements or our stated study plans and protocols, we will not be able to obtain, or may be delayed in obtaining, the necessary regulatory approvals and will not be able to, or may be delayed in our efforts to, successfully commercialize our products.
If our clinical studies fail to demonstrate safety and effectiveness to the satisfaction of the FDA or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
If the results of our clinical studies are inconclusive or if there are safety concerns or adverse events associated with the product being tested, we may:
| · | be delayed in obtaining marketing approval for such product, if at all; |
| · | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| · | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| · | be subject to changes with the way such product is administered; |
| · | be required to perform additional clinical studies to support approval or be subject to additional post-marketing testing requirements; |
| · | have regulatory authorities withdraw their approval of such product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation strategy; |
| · | be subject to the addition of labeling statements, such as warnings or contraindications; |
| · | be sued; or |
| · | otherwise experience damage to our reputation. |
Even if the results of our clinical studies are favorable, the time required to obtain FDA and other approvals is unpredictable, and often can take years following the commencement of clinical studies. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all.
The FDA or any other regulatory authority may deny or delay an approval because it is not satisfied with the structure or conduct of clinical studies or due to its assessment of the data we supply. A regulatory authority, for instance, may not believe that we have adequately addressed negative safety issues. Clinical data is subject to varied interpretations, and regulatory authorities may disagree with our assessments of data. In any such case, a regulatory authority could insist that we provide additional data, which could substantially delay or even prevent commercialization efforts, particularly if we are required to conduct additional pre-approval clinical studies.
Clinical studies also require the review and oversight of IRBs, which approve and continually review clinical investigations and protect the rights and welfare of human subjects. An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical studies, and the FDA may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB review and approval.
Any of the foregoing could have a material adverse effect on our business, results of operations and financial condition.
If we fail to obtain or maintain orphan exclusivity for our products, our competitors may sell products to treat the same conditions, and our revenues will be reduced.
The FDA has designated MA09-hRPE cells as an orphan drug for the treatment of Stargardt’s Macular Dystrophy. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is intended to treat a rare disease or condition, defined as a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. Orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer is unable to assure sufficient product quantity.
| 22 |
Even if orphan drug designation has been granted, we may not be the first to obtain marketing approval for this indication due to the uncertainties associated with developing biological products. Further, even if we obtain orphan drug exclusivity, that exclusivity may not effectively protect our product from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve the same product for the same condition if the FDA concludes that the later product is safer, more effective, or makes a major contribution to patient care. Orphan exclusivity neither shortens the development time or regulatory review time, nor gives the product any advantage in the regulatory review or approval process. Orphan exclusive marketing rights may be lost if the FDA later determines that the request for designation was materially defective, if the manufacturer is unable to assure sufficient quantity of the product, or if a second applicant demonstrates its product is “clinically superior” to the original orphan drug.
Finally, as a result of the expiration or successful challenge of our patent rights, we could face more litigation with respect to the validity and/or scope of patents relating to our competitors’ products. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize.
We may find it difficult to enroll patients in our clinical studies, which could delay or prevent our clinical studies.
Identifying and qualifying patients to participate in our clinical studies is critical to our success. The timing of our clinical studies depends on the speed at which we can recruit patients to participate. If we have difficulty enrolling a sufficient number of patients to conduct our clinical studies as planned, we may need to delay, limit or terminate ongoing or planned clinical studies, any of which would have an adverse effect on our business.
Patient enrollment is affected by a number of factors including:
| · | severity of the disease under investigation; |
| · | design of the study protocol; |
| · | estimated and actual size of the patient population; |
| · | eligibility criteria for the study in question; |
| · | perceived risks and benefits of the product being tested; |
| · | proximity and availability of clinical study sites for prospective patients; |
| · | availability of competing therapies and clinical studies; |
| · | efforts to facilitate timely enrollment in clinical studies; |
| · | patient referral practices of physicians; and |
| · | ability to monitor patients adequately during and after treatment. |
We may experience difficulties with patient enrollment for our clinical studies due to the small patient populations for our proposed products, and the process of finding and diagnosing patients may prove costly. Additionally, patients may be unwilling to participate in our studies due to negative publicity from adverse events in the biotechnology or other industries or for other reasons, such as concerns about the requirement of surgery with its attendant risks including infection, bleeding, loss of vision, loss of eye, death due to complication, increased risk of infection or malignancy due to immunosuppression, need for randomization of the eye to be treated and ethical concerns regarding embryonic stem cell based therapy. We may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a study, to complete our clinical studies in a timely manner.
Our employees, consultants and commercial partners may engage in misconduct or other improper activities, including insider trading and non-compliance with regulatory standards and requirements.
We are exposed to the risk that our employees, consultants and commercial partners may engage in fraudulent or illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates the regulations of the FDA and non-U.S. regulators, including those laws requiring the reporting of true, complete and accurate information to such regulators, manufacturing standards, healthcare fraud and abuse laws and regulations in the United States and abroad or laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment of operations, any of which could adversely affect our ability to operate our business and our results of operations. Whether or not we are successful in defending against such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these claims or investigations.
| 23 |
We are subject to federal and state fraud and abuse laws, health information privacy and security laws, which, if violated, could subject us to substantial penalties. Additionally, any challenge to or investigation into our practices under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
There are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback, false claims and physician transparency laws. Our relationships with providers and hospitals are subject to scrutiny under these laws. We may also be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| · | the Federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual or furnishing or arranging for a good or service, for which payment may be made, in whole or in part, under federal healthcare programs, such as Medicare and Medicaid; |
| · | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| · | HIPAA, which created federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| · | the Physician Payment Sunshine Act under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively referred to as the PPACA, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians, which is defined broadly to include other healthcare providers, and teaching hospitals and ownership and investment interests held by physicians and their immediate family members; |
| · | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; |
| · | state laws that require manufacturers to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and |
| · | state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. |
These laws, among other things, constrain our sales, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with hospitals, physicians or other potential purchasers of our products. We have a variety of arrangements with our customers that could implicate these laws. Due to the breadth of these laws, the narrowness of statutory exceptions and safe harbors available, and the range of interpretations to which they are subject to, it is possible that some of our current or future practices might be challenged under one or more of these laws. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity, and be costly to respond to, and thus could harm our business, financial condition and results of operations.
In addition, recent healthcare reform legislation has strengthened these laws. For example, the PPACA, among other things, amends the intent requirement of the Federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them to have committed a violation. Moreover, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the Federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring of our operations, any of which could negatively impact our ability to operate our business and our results of operations.
| 24 |
Healthcare policy changes, including legislation reforming the United States healthcare system, may have a material adverse effect on our financial condition and results of operations.
The PPACA makes changes that are expected to significantly impact the pharmaceutical, biological product and medical device industries. The PPACA, among other things, established annual fees and taxes on manufacturers of certain branded prescription drugs and biological products and included coordination and promotion of research on comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. In addition, the PPACA established an Independent Payment Advisory Board, or IPAB, to reduce the per capita rate of growth in Medicare spending. The IPAB has broad discretion to propose policies to reduce health care expenditures, which may have a negative impact on payment rates for services, including our products. The IPAB proposals may impact payments for hospital services beginning in 2020.
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation's automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will remain in effect until 2024 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012, or the ATRA, was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
The full impact on our business of the PPACA and the other new laws is uncertain. Nor is it clear whether other legislative changes will be adopted or how such changes would affect our industry generally or our ability to successfully commercialize any of our product candidates.
Regulatory Risks
Our products may not receive regulatory approval, which would prevent us from commercially marketing our products and producing revenues.
The FDA and comparable government agencies in foreign countries impose substantial regulations on the manufacture and marketing of pharmaceutical products through lengthy and detailed laboratory, pre-clinical and clinical testing procedures, sampling activities and other costly and time-consuming procedures. Satisfaction of these regulations typically takes several years or more and varies substantially based upon the type, complexity and novelty of the proposed product. We cannot assure you that FDA approvals for any products developed by us will be granted on a timely basis, if at all. We may encounter significant delays or excessive costs in our efforts to secure necessary approvals or licenses. Even if we obtain FDA regulatory approvals, the FDA extensively regulates manufacturing, labeling, distributing, marketing, promotion and advertising after product approval. Moreover, several of our product development areas involve relatively new technologies and have not been the subject of extensive laboratory testing and clinical studies. The regulatory requirements governing these products and related clinical procedures remain uncertain and the products themselves may be subject to substantial review by the FDA and other foreign governmental regulatory authorities that could prevent or delay approval in the United States and any other foreign country. Regulatory requirements ultimately imposed on our products could limit our ability to test, manufacture and, ultimately, commercialize our products and thereby could adversely affect our financial condition and results of operations. Any inability or delay in obtaining, or failure to obtain, such approvals could have a material adverse effect on the marketing of our products and our ability to generate product revenue.
We cannot market our product candidates until we receive regulatory approval; even if we complete the necessary pre-clinical and clinical studies, we cannot predict when or if we will obtain regulatory approval to commercialize a product candidate or the approval may be for a more narrow indication than we expect.
Development of our products is subject to extensive regulation by governmental authorities in the United States and other countries. The process of obtaining regulatory approval is lengthy, expensive and uncertain. In the United States, the FDA imposes substantial requirements on the introduction of biological products and many medical devices through lengthy and detailed laboratory and clinical testing procedures, sampling activities and other costly and time-consuming procedures. Satisfaction of these requirements typically takes several years and the time required to do so may vary substantially based upon the type and complexity of the biological product or medical device.
| 25 |
Product candidates that we believe should be classified as medical devices for purposes of the FDA regulatory pathway may be determined by the FDA to be biologic products subject to the satisfaction of significantly more stringent requirements for FDA approval. Any difficulties that we encounter in obtaining regulatory approval may have a substantial adverse impact on our business and cause our stock price to significantly decline.
If we fail to obtain regulatory approval of any of our product candidates for at least one indication, we will not be permitted to market our product candidates and may be forced to cease our operations. Even if our product candidates demonstrate safety and efficacy in clinical studies, the regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval at all. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on any approved indications. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical studies and the review process. Regulatory agencies also may approve a treatment candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our treatment candidates.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory scrutiny.
Even if we obtain regulatory approval in a jurisdiction, the regulatory authority may still impose significant restrictions on the indicated uses or marketing of our product candidates, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. Conditions of approval, such as limiting the category of patients who can use the product, may significantly impact our ability to commercialize the product and may make it difficult or impossible for us to market a product profitably. In addition, product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with good manufacturing practices, or GMP, and adherence to commitments made to the FDA in the approval process. If we or a regulatory agency discovers previously unknown problems with a product such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured or manufacturing issues, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
FDA approval of our products may also entail ongoing requirements for post-marketing studies, or limit how or to whom we can sell its products. Even if we obtain regulatory approval, labeling, promotional and manufacturing activities are subject to continual scrutiny by the FDA, state regulatory agencies and, in some circumstances, the Federal Trade Commission. In addition, FDA enforcement policy prohibits the marketing of approved products for unapproved, or off-label, uses. These regulations and the FDA’s and other third-party payers’ interpretation of them could materially increase our expenses, impair its ability to effectively market its products, and limit our revenue.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. In addition, if our product candidates are approved by the FDA or other regulatory authorities for the treatment of any indications, regulatory labeling may specify that our product candidates may be used in conjunction with other therapies. The occurrence of any of these events or penalties may inhibit our ability to commercialize our product candidates and generate revenues.
Once obtained, regulatory approvals may be withdrawn and can be expensive to maintain.
Regulatory approval may be withdrawn for a number of reasons, including the later discovery of previously unknown problems with the product. Regulatory approval may also require costly post-marketing follow-up studies, and failure of our product candidates to demonstrate sufficient efficacy and safety in these studies may result in either withdrawal of marketing approval or severe limitations on permitted product usage. In addition, numerous additional regulatory requirements relating to, among other things, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record-keeping will also apply. Furthermore, regulatory agencies subject a marketed product, its manufacturer and the manufacturer’s facilities to continual review and periodic inspections. Compliance with these regulatory requirements is time-consuming and requires the expenditure of substantial resources.
If any of our product candidates is approved, we will be required to report certain adverse events involving our products to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning the advertisement and promotional labeling of our products. As a result, even if we obtain necessary regulatory approvals to market our product candidates for any indication, any adverse results, circumstances or events that are subsequently discovered could require that we cease marketing the product for that indication or expend additional money, time and effort to ensure full compliance, which could have an adverse effect on our business or results of operations.
| 26 |
If our products do not comply with applicable laws and regulations our business will be harmed.
Any failure by us or by any third parties that may manufacture or market our products, to comply with the law, including statutes and regulations administered by the FDA or other U.S. or foreign regulatory authorities, could result in, among other things, warning letters, fines and other civil penalties, suspension of regulatory approvals and the resulting requirement that we suspend sales of our products, refusal to approve pending applications or supplements to approved applications, export or import restrictions, interruption of production, operating restrictions, closure of the facilities used by us or third parties to manufacture our product candidates, injunctions or criminal prosecution. Any of the foregoing actions could have an adverse effect on our business.
Restrictions on the use of hESCs, the ethical, legal and social implications of stem cell research, and negative public opinion about stem cell therapy may damage public perception of our therapeutic candidates and could prevent us from developing or gaining acceptance for commercially viable products.
Some of our most important programs involve the use of stem cells that are derived from human embryos. The use of hESCs gives rise to ethical, legal and social issues regarding the appropriate derivation of these cells. In the event that our research related to hESCs becomes the subject of adverse commentary or publicity or increased scrutiny by governmental or regulatory organizations, our business could be harmed or otherwise substantially impaired, and the market price for our common stock could be significantly harmed. Some political and religious groups have voiced opposition to our technology and practices. We use stem cells derived from human embryos that have been created for in vitro fertilization procedures but are no longer desired or suitable for that use and are donated with appropriate informed consent for research use. Also, many research institutions, including some of our scientific collaborators, have adopted policies regarding the ethical use of human embryonic tissue. These policies may have the effect of limiting the scope of research conducted using hESCs, thereby impairing our ability to conduct research in this field.
Governmental regulations and laws could change.
There can be no assurance that our operations will not be restricted by any future legislative or administrative efforts by politicians or groups opposed to the development of hESC technology or nuclear transfer technology. Additionally, the scope of the Dickey–Wicker Amendment, a 16-year-old ban on U.S. federal funding for activity related to the harm or destruction of an embryo, was recently under review by the federal courts and while it was determined not to preclude funding of hESC research by the federal government, there can be no assurance that it will not be challenged again or the language modified by Congress so as to restrict government funding of hESC research. Judicial review of this or other U.S. federal or state laws, the occurrence and results of which are difficult to predict with any certainty, could result in a more restrictive interpretation of those laws than is previously the case, and may limit or require us to terminate certain of our research and therapeutic programs.
We may not be able to obtain required approvals in countries other than the United States.
The requirements governing the conduct of clinical trials and cell culturing as well as the marketing approval process for our product candidates outside the United States vary widely from country to country. Foreign approvals may take longer to obtain than FDA approvals and can require, among other things, additional testing and different clinical trial designs. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval processes. Some foreign regulatory agencies also must approve prices of the products. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. We may not be able to file for regulatory approvals and may not receive necessary approvals to market our product candidates in any foreign country. If we fail to comply with these regulatory requirements or fail to obtain and maintain required approvals in any foreign country, we will not be able to sell our product candidates in that country and our ability to generate revenue will be adversely affected.
Licensure of our products may not be available even if safety and efficacy is demonstrated in clinical trials.
Regulatory requirements for cell therapies, including manufacturing operations, can be different from one jurisdiction to the next in which are or intend to carry out clinical trials. The implementation or interpretation of the rules and regulations relating to manufacturing processes and source materials may occasionally be changed by the regulators, or modified or replaced by other governmental action. Our ability to obtain market authorization or the equivalent in any given jurisdiction may be dependent on our ability to demonstrate compliance with the rules and regulations covering source materials and manufacturing requirements in those jurisdictions, which we may not be able to meet.
MA09-hRPE cells for the treatment of Stargardt’s Macular Dystrophy, or any other product candidate for which we seek approval as a biologic, may face competition sooner than anticipated.
With the enactment of the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, as part of the Patient Protection and Affordable Care Act, an abbreviated pathway for the approval of biosimilar and interchangeable biological products was created. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing reference product. Under the BPCI Act, an application for a biosimilar product cannot be approved by the FDA until twelve years after the original reference product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement the BPCI Act may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
| 27 |
While products approved under a BLA should qualify for the twelve-year period of exclusivity, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider the approved BLA product to be a reference product for competing products, potentially creating the opportunity for competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our potential reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Our products may be used by physicians for indications that are not approved by the FDA. If the FDA finds that we marketed our products in a manner that promoted off-label use, we may be subject to civil or criminal penalties.
We plan to pursue FDA approval of product candidates for other indications other than age-related macular degeneration and Stargardt’s macular dystrophy, including: myopic macular degeneration; retinal degenerative conditions; diabetic retinopathy; retinal vascular disorders; macular dysfunction, glaucoma, non-infectious uveitis and inflammatory diseases of the retina and choroid Under the Federal Food, Drug, and Cosmetic Act and other laws, we are prohibited from promoting any products for which we may obtain approval for off-label uses. This means that we may not make claims about the safety or effectiveness of a product or product candidate candidates outside of the approved indication and we may not proactively discuss or provide information on the off-label uses of products or product candidates with very specific and limited exceptions. Physicians, however, may lawfully choose prescribing products for off-label uses in the practice of medicine.
A company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil and administrative remedies, and even criminal sanctions. There can be no assurance that any of our clinical trials will generate data necessary to support approval of any of our product candidates. Should the FDA determine, however, that our activities constitute the promotion of off-label use, the FDA could bring action to prevent us from distributing products for the off-label use and could impose fines and penalties on us and our executives. In addition, failure to follow FDA rules and guidelines relating to promotion and advertising can result in, among other things, the FDA's refusal to approve a product, the suspension or withdrawal of an approved product from the market, product recalls, fines, disgorgement of money, operating restrictions, injunctions or criminal prosecutions.
If the Office of Inspector General within the Department of Health and Human Services, the Department of Justice, or DOJ, or another federal or state agency determines that we have promoted off-label use of our products, we may be subject to various penalties, including civil or criminal penalties, and the off-label use of our products may result in injuries that lead to product liability suits, which could be costly to our business.
In addition to the FDA restrictions on marketing of approved products, several other types of state and federal healthcare laws have been applied by DOJ and state attorneys general to restrict certain marketing practices in the pharmaceutical industry. While physicians may prescribe products for off-label uses and indications other than the approved indications for use, if other federal or state regulatory authorities determine that we have engaged in off-label promotion through remuneration, kickbacks or other monetary benefits to prescribers, we may be subject to civil or criminal penalties and could be prohibited from participating in government healthcare programs such as Medicaid and Medicare. In addition, government agencies or departments could conclude that we have engaged in off-label promotion and, potentially, caused the submission of false claims. Even if we are successful in resolving such matters without incurring penalties, responding to investigations or prosecutions will likely result in substantial costs and could significantly and adversely impact our reputation and divert management's attention and resources, which could have a material adverse effect on our business, operating results, financial condition and ability to finance our operations. In addition, the off-label use of our products may increase the risk of injury to patients, and, in turn, the risk of product liability claims. Product liability claims are expensive to defend and could divert our management's attention and result in substantial damage awards against us.
Risks Related to Competition
We face intense competition and rapid technological change and the possibility that our competitors may develop therapies that are more advanced or effective than ours, which may adversely affect our financial condition and our ability to successfully commercialize our product candidates.
We are engaged in activities in the biotechnology field, and particularly in the development of products for treating ophthalmological diseases and disorders, which is characterized by extensive research efforts and rapid technological progress. If we fail to anticipate or respond adequately to technological developments, our ability to operate profitably could suffer. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. The biotechnology industries are characterized by rapidly evolving technology and intense competition. Our competitors include major multinational pharmaceutical companies, specialty biotechnology companies and chemical and medical products companies operating in the fields of regenerative medicine, cell therapy, tissue engineering and tissue regeneration. Development of drug candidates for use in ophthalmology, in particular, has received a greater degree of interest and financing from global pharmaceutical companies to start-up companies. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, products that are more effective or less costly than any product candidate that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization and market penetration than us. Additionally, technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing our product candidates against competitors. We cannot assure you that research and discoveries by other life science, pharmaceutical or other companies will not render our technologies or potential products or services uneconomical or result in products superior to those we develop or that any technologies, products or services we develop will be preferred to any existing or newly-developed technologies, products or services.
| 28 |
We believe potential competitors may be developing a variety of technologies and products for treating the same ophthalmological indications for which we are developing products, or indications which are similar and provide potential bases for eventually pursuing the same indications as us. In particular, there are several companies and non-profit entities working on various RPE formulations for treating macular degeneration, including Pfizer, Regenerative Patch Technologies, the Riken Center for Developmental Biology (Japan) and Cell Cure Neurosciences Ltd. (Israel). Other cell types are also being developed for subretinal use in treating various forms of macular degeneration, including by StemCells Inc. (purified human neural stem cells), Bioheart, Inc. (adipose stem cell), the University of California Davis and Retinal Associates of South Florida (CD34+ stem cells from bone marrow), Neurotech, Inc. (encapsulated cells that express CNTF) and Janssen Research & Development, LLC (umbilical cord stem cells).
In addition to cell-based therapies, there are a number of gene therapy, biologics and small molecule approaches (non-cell therapies) that range in development status from pre-clinical to phase III trials. Examples of companies involved in the development of gene therapy treatments for macular degeneration which may compete with our RPE programs include Oxford Biomedica, Spark Therapeutics, Avalanche Therapeutics and Genzyme. With respect to small molecules and biological agents (proteins) which may be competitive with our RPE programs, such products in development include, to illustrate, drugs that decrease oxidative stress (AREDS2, OT-551); Visual cycle modulators (emixustat, Fenretinide, ALK-001); Neuroprotectants (CNTF, NT-501, Brimonidine tartrate, Tandospirone); Drugs that reduce toxic by-products (RN6G, GSK933776); Drugs that suppress inflammation (fluocinolone acetonide, compastatin, eculizumab, lampalizumab, LFG316, ARC1905); and Vascular enhancers (MC-1101) .
Many of these companies are well-established and possess technical, research and development, financial and sales and marketing resources significantly greater than ours. In addition, certain smaller biotech companies have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies’ potential research and development and commercialization advantages. Academic institutions, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those we are developing. Moreover, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before we do.
There are currently no effective long-term therapies for the diseases and medical conditions we are targeting. Nevertheless, we expect that our technologies and products will compete with a variety of therapeutic products and procedures offered by major pharmaceutical companies. Many pharmaceutical and biotechnology companies are investigating new drugs and therapeutic approaches for the same purposes, which may achieve new efficacy profiles, extend the therapeutic window for such products, alter the prognosis of these diseases or prevent their onset.
We are aware of ongoing clinical trials and pre-clinical development efforts directed to cell therapies as well as small molecules and biologics for treating many of the same indications for which we pursue treatments. In the case of macular degeneration, these include antibody therapies, small molecules, gene therapies and cell therapies. Some of these third party drug development programs have already reached Phase 3 in clinical trials, some with positive results for patients treated in Phase 1 or 2 of those trials. For example, Roche/Genentech is currently in Phase 3 development of a monoclonal antibody, lampalizumab, for AMD on the strength of positive Phase 2 data. We believe that our products, when and if successfully developed, will compete with these products principally on the basis of improved and extended efficacy and safety and their overall economic benefit to the health care system. Competition for any stem cell products that we may develop may be in the form of existing and new drugs, other forms of cell transplantation, ablative and stimulative procedures and gene therapy. We believe that some of our competitors are also trying to develop stem and progenitor cell-based technologies. We expect that all of these products will compete with our potential stem cell products based on efficacy, safety, cost and intellectual property positions. We may also face competition from companies that have filed patent applications relating to the use of genetically modified cells to treat disease, disorder or injury. In the event our therapies should require the use of such genetically modified cells, we may be required to seek licenses from these competitors in order to commercialize certain of our proposed products, and such licenses may not be granted.
If we develop products that receive regulatory approval, they would then have to compete for market acceptance and market share. For certain of our potential products, an important success factor will be the timing of market introduction of competitive products. This timing will be a function of the relative speed with which we and our competitors can develop products, complete the clinical testing and approval processes and supply commercial quantities of a product to market. These competitive products may also impact the timing of clinical testing and approval processes by limiting the number of clinical investigators and patients available to test our potential products.
| 29 |
Many of our competitors have significantly greater experience than we have in the development, pre-clinical testing and human clinical trials of biotechnology and pharmaceutical products, in obtaining FDA and other regulatory approvals of such products and in manufacturing and marketing such products.
Accordingly our competitors may succeed in obtaining FDA approval for products more rapidly or effectively than we can. Our competitors may also be the first to discover and obtain a valid patent to a particular stem cell technology which may effectively block all others from doing so. It will be important for us or our collaborators to be the first to discover any stem cell technology that we are seeking to discover. Failure to be the first could prevent us from commercializing all of our research and development affected by that discovery. Additionally, if we commence commercial sales of any products, we will also be competing with respect to manufacturing efficiency and sales and marketing capabilities: areas in which we have no experience.
General Risks Relating to Our Business
We are subject to litigation that will be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us. That litigation is likely to be expensive and may require a significant amount of management’s time and attention, at the expense of other aspects of our business. The outcome of litigation is always uncertain, and in some cases could include judgments against us that require us to pay damages, enjoin us from certain activities or otherwise affect our legal or contractual rights, which could have a significant adverse effect on our business.
For example, in mid-April of 2014 we received a notice of default from CAMOFI Master LDC and CAMHZN Master LDC stating that a failure by us to deliver shares of common stock issuable to this investors under convertible debentures held by these investors event resulted in an event of default under the debentures. In late April 2014, these investors delivered a notice to us stating that all amounts payable under the debentures, subject to adjustment as set forth therein, were immediately due and payable in accordance with their terms. The investors also demanded damages to which we believe they were not entitled and that they did not incur. On May 2, 2014, we paid these holders an amount that we believe satisfies all of our obligations under the debentures. However, there can be no assurance that CAMOFI Master LDC and CAMHZN Master LDC agree with our position that we have satisfied all of our obligations under the debentures and not pursue additional monetary or other damages.
A significant adverse determination in any claim against us could adversely affect our operating results or financial condition. The amount we may be required to pay, in cash or in stock, in connection with any Claim may prove to exceed our estimated reserves and, in the case of payment in the form of stock, may prove to be highly dilutive to our stockholders. Should any judgment or settlement occur that exceeds our estimate, or a new claim arise, or if we become aware of additional information that requires us to adjust our estimation of potential exposure, we may need to adjust our overall reserve and, depending on the amount, such adjustment could be material and adversely affect our operating results or financial condition.
Form 4 filing delays by our former Chief Executive Officer gave rise to an investigation by the Securities and Exchange Commission into the delays and our Section 16 compliance procedures, and this investigation resulted in penalties and sanctions against us.
As previously disclosed by us, in April 2013, it was determined that Gary Rabin, our then-Chief Executive Officer, failed to report 27 transactions in which Mr. Rabin sold shares of our common stock that took place between February 7, 2011 and January 10, 2013. Mr. Rabin filed a Form 4 under Section 16 of the Exchange Act on April 15, 2013 reporting the previously unreported sale transactions and correcting the total number of shares of our common stock that Mr. Rabin owned as of the date of filing of the Form 4. Our board of directors initiated an investigation into this matter upon becoming aware of it. In September 2014, we settled the SEC action arising from the SEC’s investigation. Under the terms of the settlement accepted by the SEC, we consented to the entry of order under which we neither admit nor deny liability and has agreed to pay a civil penalty of $375,000, which has been previously accrued for, by July 2015. In addition, the settlement requires us to engage an independent Section 16 compliance consultant, provide Section 16(a) training to each Section 16(a) reporting person, and provide a certification of compliance that each of the preceding requirements were completed. The settlement also requires us to cease and desist from committing or causing any violations and any future violations of Section 17(a)(2) of the Securities Act, Sections 13(a) and 14(a) of the Exchange Act, and Rules 12b-20, 13a-1, and 14a-9 thereunder. The terms of this settlement require us to allocate financial and management resources to complying with the settlement’s terms, which may have adverse effect on our business. Also, if the SEC deems us to not have complied with any portion of the settlement, it may issue additional fines or sanctions against us which may limit our ability to issue securities or otherwise conduct our business as currently conducted.
| 30 |
The commercial success of any current or future product candidate will depend upon the degree of market acceptance by physicians, patients, third-party payors and others in the medical community, and our products may not be accepted in the marketplace.
If we are successful in obtaining regulatory approval for any of our product candidates, the degree of market acceptance of those products will depend on many factors, including:
| · | our ability to provide acceptable evidence of, and the perception of patients and the healthcare community, including third party payors, of, the potential advantages of our product candidates relative to existing treatment methods; |
| · | the incidence and severity of any adverse side effects of our product candidates; |
| · | the availability, pricing and efficacy of alternative treatments; |
| · | the labeling requirements imposed by the FDA and foreign regulatory agencies on our products and related marketing materials, including the scope of approved indications and any safety warnings; |
| · | our ability to obtain sufficient third party insurance coverage or reimbursement for our product candidates; |
| · | the inclusion of our products on insurance company coverage policies; |
| · | the willingness and ability of patients and the healthcare community to adopt new technologies or therapeutics; |
| · | public opinion and acceptance of stem cell therapy in general, including media coverage and activism by religious, social or political groups; |
| · | The ease of administration and procedure time associated with the use of our product candidates, including time between and frequency of dosage; |
| · | our ability to manufacture or obtain from third party manufacturers sufficient quantities of our product candidates with acceptable quality and at an acceptable cost to meet demand; and |
| · | internal or external marketing and distribution support for our products. |
We cannot predict or guarantee that physicians, patients, healthcare insurers, third party payors or health maintenance organizations, or the healthcare community in general, will accept or utilize any of our product candidates. Failure to achieve market acceptance would limit our ability to generate revenue and would have a material adverse effect on our business. In addition, if any of our product candidates achieve market acceptance, we may not be able to maintain that market acceptance over time if competing products or technologies are introduced that are received more favorably or are more cost-effective.
We may not be able to obtain third-party payer reimbursement or favorable product pricing, which would reduce our ability to operate profitably.
Our ability to successfully commercialize some of our proposed products in the human therapeutic field may depend on a significant degree on obtaining coverage and adequate reimbursement of the costs of such products and related treatments at acceptable levels from government authorities, private health insurers and other third-party payers. We cannot assure you that coverage and adequate reimbursement in the United States or foreign countries will be available for any products we may develop or, if available, will not be decreased in the future, or that reimbursement amounts will not reduce the demand for, or the price of, our products with a consequent harm to our business. We cannot predict what additional regulation or legislation relating to the health care industry or third-party coverage and reimbursement may be enacted in the future or what effect such regulation or legislation may have on our business. If additional regulations are overly onerous or expensive, or if health care related legislation makes our business more expensive or burdensome than originally anticipated, we may be forced to significantly downsize our business plans or completely abandon our business model.
| 31 |
Our products are likely to be expensive to manufacture, and they may not be profitable if we are unable to control the costs to manufacture them.
Our products are likely to be significantly more expensive to manufacture than most other biologics currently on the market today. Our present manufacturing processes produce modest quantities of product intended for use in our ongoing research activities, and we have not developed processes, procedures and capability to produce commercial volumes of product. We hope to substantially reduce manufacturing costs through process improvements, development of new science, increases in manufacturing scale and outsourcing to experienced manufacturers. If we are not able to make these or other improvements, and depending on the pricing of the product, our profit margins may be significantly less than that of most biologics on the market today. In addition, we may not be able to charge a high enough price for any cell therapy product we develop, even if they are safe and effective, to make a profit. If we are unable to realize significant profits from our potential product candidates, our business would be materially harmed.
Our current source of revenues depends on the stability and performance of our sub-licensees.
Our ability to collect royalties on product sales from our sub-licensees will depend on the financial and operational success of the companies operating under a sublicense. Revenues from those licensees will depend upon the financial and operational success of those third parties. We cannot assure you that these licensees will be successful in obtaining requisite financing or in developing and successfully marketing their products. These licensees may experience unanticipated obstacles including regulatory hurdles, and scientific or technical challenges, which could have the effect of reducing their ability to generate revenues and pay us royalties.
We depend on key personnel for our continued operations and future success, and a loss of certain key personnel could significantly hinder our ability to move forward with our business plan.
Because of the specialized nature of our business, we are highly dependent on our ability to identify, hire, train and retain highly qualified scientific and technical personnel for the research and development activities we conduct or sponsor. The loss of one or more certain key executive officers, or scientific officers, would be significantly detrimental to us. In addition, recruiting and retaining qualified scientific personnel to perform research and development work is critical to our success. Our anticipated growth and expansion into areas and activities requiring additional expertise, such as clinical testing, regulatory compliance, manufacturing and marketing, will require the addition of new management personnel and the development of additional expertise by existing management personnel. There is intense competition for qualified personnel in the areas of our present and planned activities, and there can be no assurance that we will be able to continue to attract and retain the qualified personnel necessary for the development of our business. The failure to attract and retain such personnel or to develop such expertise would adversely affect our business.
Our insurance policies may be inadequate and potentially expose us to unrecoverable risks.
Any significant insurance claims would have a material adverse effect on our business, financial condition and results of operations. Insurance availability, coverage terms and pricing continue to vary with market conditions. We endeavor to obtain appropriate insurance coverage for insurable risks that we identify, however, we may fail to correctly anticipate or quantify insurable risks, we may not be able to obtain appropriate insurance coverage and insurers may not respond as we intend to cover insurable events that may occur. We have observed rapidly changing conditions in the insurance markets relating to nearly all areas of traditional corporate insurance. Such conditions have resulted in higher premium costs, higher policy deductibles, and lower coverage limits. For some risks, we may not have or maintain insurance coverage because of cost or availability.
Attacks perpetrated against our information systems could result in loss of assets and critical information and exposes us to remediation costs and reputational damage.
We rely on information technology in virtually all aspects of our business. A significant disruption or failure of our information technology systems could result in service interruptions, safety failures, security violations, regulatory compliance failures, an inability to protect corporate information assets against intruders, and other operational difficulties.
Although we have taken steps intended to mitigate these risks, including business continuity planning, disaster recovery planning and business impact analysis, a significant disruption or cyber intrusion could lead to misappropriation of assets or data corruption and could adversely affect our results of operations, financial condition or liquidity. Additionally, if we are unable to acquire or implement new technology, we may suffer a competitive disadvantage, which could also have an adverse effect on our results of operations, financial condition or liquidity.
Cyber attacks could further adversely affect our ability to operate facilities, information technology and business systems, or compromise confidential clinical and employee information. Furthermore, instability in the financial markets as a result of cyber terrorism, as well as sustained or significant cyber attacks, could also materially adversely affect our and our subsidiaries’ ability to raise capital.
| 32 |
We have limited product liability insurance, which may leave us vulnerable to future claims we will be unable to satisfy.
The testing, manufacturing, marketing and sale of human therapeutic products entail an inherent risk of product liability claims, and we cannot assure you that substantial product liability claims will not be asserted against us. We have limited product liability insurance. In the event we are forced to expend significant funds on defending product liability actions, and in the event those funds come from operating capital, we will be required to reduce our business activities, which could lead to significant losses. We cannot assure you that adequate insurance coverage will be available in the future on acceptable terms, if at all, or that, if available, we will be able to maintain any such insurance at sufficient levels of coverage or that any such insurance will provide adequate protection against potential liabilities. Whether or not a product liability insurance policy is maintained in the future, any product liability claim could harm our business or financial condition.
Risks Relating to Our Common Stock
Stock prices for biotechnology companies have historically tended to be very volatile.
Stock prices and trading volumes for many biotechnology companies fluctuate widely for a number of reasons, including but not limited to the following factors (some of which may be unrelated to their businesses or results of operations):
| · | clinical trial results; |
| · | the amount of cash resources and ability to obtain additional funding; |
| · | announcements of financing transactions, research activities, business developments, technological innovations or new products by companies or their competitors; |
| · | entering into or terminating strategic relationships; |
| · | changes in government regulation; |
| · | disputes concerning patents or proprietary rights; |
| · | changes in revenues or expense levels; |
| · | public concern regarding the safety, efficacy or other aspects of the products or methodologies being developed, |
| · | reports by securities analysts; |
| · | activities of various interest groups or organizations; |
| · | media coverage; and |
| · | status of the investment markets. |
This market volatility, as well as general domestic or international economic, market and political conditions, could materially and adversely affect the market price of our common stock and the return on your investment.
A significant number of shares of our common stock have become available for sale and their sale could depress the price of our common stock.
Substantially all of our common stock is freely tradable in the equity markets.
We may also sell a substantial number of additional shares of our common stock in connection with a private placement or public offering of shares of our common stock (or other series or class of capital stock to be designated in the future). The terms of any such transactions would likely require us to register the resale of any shares of capital stock issued or issuable in the transaction. For example, we currently have the ability to issue $18,619,451 worth of our common stock to Lincoln Park under the 2014 Purchase Agreement, which amount would equal 3,057,381 shares of our common stock based on the closing price of our common stock of $6.09 as of December 31, 2014.
Sales of a substantial number of shares of our common stock under any of the circumstances described above could adversely affect the market price for our common stock and make it more difficult for you to sell shares of our common stock at times and prices that you feel are appropriate.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
Any payment of cash dividends will depend upon our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. We do not anticipate paying cash dividends on our common stock in the foreseeable future. Furthermore, we may incur additional indebtedness that may severely restrict or prohibit the payment of dividends.
| 33 |
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties.
Our principal executive offices are located in Marlborough, Massachusetts, where we lease approximately 30,000 square foot of office and laboratory facilities, pursuant to two separate leases, in different suites of the same building. The first lease agreement relates to 12,257 square feet of office and laboratory facilities, requires a monthly rent payment of $14,771 and continues until July 31, 2015. The second lease agreement relates to 17,696 of office and laboratory facilities, requires a monthly rent payment of $21,383 started in April 2013 (with scheduled increases thereafter) and continues until March 31, 2018 with an option to extend the lease for an additional five year period. We also lease approximately 1,568 square feet of corporate office space in Santa Monica, California for $6,272 per month (with schedule increases thereafter), which lease continues until June 30, 2018. We have closed our Santa Monica office and we are in the process of consolidating all of our operations in our Marlborough, Massachusetts facility. Beginning in March 2015, we have sub-leased our Santa Monica office to a third party.
Item 3. Legal Proceedings.
In the ordinary course of business, we are from time to time involved in lawsuits, claims, investigations, proceedings, and threats of litigation relating to intellectual property, commercial arrangements and other matters. While the outcome of these proceedings and claims cannot be predicted with certainty, as of December 31, 2014, we were not party to any legal or arbitration proceedings that may have, or have had in the recent past, significant effects on our financial position or profitability. No governmental proceedings are pending or, to our knowledge, contemplated against us. We are not a party to any material proceedings in which any director, member of senior management or affiliate of ours is either a party adverse to us or our subsidiaries or has a material interest adverse to us or our subsidiaries.
Item 4. Mine Safety Disclosures.
Not applicable.
| 34 |
PART II
Item 5. Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock is quoted on the NASDAQ Global Market under the symbol “OCAT.” For the periods indicated, the following table sets forth the high and low bid prices per share of our common stock. These prices represent inter-dealer quotations without retail markup, markdown or commission and may not necessarily represent actual transactions, and take into account for all periods the effect of the 100-to-1 reverse split of our common stock effected on August 28, 2014.
| High | Low | |||||||
| Fiscal Year 2014 | Bid | Bid | ||||||
| First Quarter | $ | 10.30 | $ | 5.70 | ||||
| Second Quarter | $ | 8.30 | $ | 4.95 | ||||
| Third Quarter | $ | 12.73 | $ | 6.01 | ||||
| Fourth Quarter | $ | 9.45 | $ | 5.85 | ||||
| High | Low | |||||||
| Fiscal Year 2013 | Bid | Bid | ||||||
| First Quarter | $ | 9.81 | $ | 5.57 | ||||
| Second Quarter | $ | 9.40 | $ | 6.21 | ||||
| Third Quarter | $ | 8.00 | $ | 5.55 | ||||
| Fourth Quarter | $ | 7.30 | $ | 5.30 | ||||
Stock Price Performance Graph
A five-year comparison of the performance of our common stock with a broad equity market index and a peer group is set forth below. The broad equity market index used is the Nasdaq Composite Index and the peer group is the Dow Jones U.S. Biotechnology Index. The below comparison assumes $100 was invested on December 31, 2009 and dividends are reinvested for all years ending December 31.
| 35 |
Holders
As of March 1, 2015, there were approximately 223 stockholders of record of our common stock.
Dividends
We have never paid cash dividends and have no plans to do so in the foreseeable future. Our future dividend policy will be determined by our board of directors and will depend upon a number of factors, including our financial condition and performance, our cash needs and expansion plans, income tax consequences, and the restrictions that applicable laws, our current preferred stock instruments, and our future credit arrangements may then impose.
Recent Sales of Unregistered Securities
On December 31, 2014, we issued various board members 7,500 shares of common stock valued at $45,675 as compensation for board services.
We relied on the exemption from registration provided by Section 4(a)(2) of the Securities Act, as amended, or the Securities Act, with respect to each of the issuances of unregistered securities set forth above.
Item 6. Selected Financial Data.
The following tables set forth selected historical consolidated financial and operating data for the periods indicated. The statement of operations and balance sheet data is derived from audited financial statements for the years 2014, 2013, 2012, 2011 and 2010. The Company’s financial statements as of December 31, 2014 and 2013, and for each of the three years in the period ended December 31, 2014 are included in Item 8, Financial Statements and Supplementary Data, in Part II of this Form 10-K.
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| Revenue | $ | 157,875 | $ | 224,985 | $ | 466,487 | $ | 506,419 | $ | 725,044 | ||||||||||
| Cost of revenue | 62,436 | 82,436 | 117,436 | 343,950 | 216,600 | |||||||||||||||
| Gross profit | 95,439 | 142,549 | 349,051 | 162,469 | 508,444 | |||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 10,529,321 | 11,564,768 | 14,158,936 | 9,753,759 | 8,598,383 | |||||||||||||||
| Grant reimbursements | – | – | – | – | (977,917 | ) | ||||||||||||||
| General and administrative expenses | 11,052,123 | 12,057,067 | 11,432,866 | 7,435,709 | 14,551,042 | |||||||||||||||
| Change in estimate of accrued liabilities | – | – | – | – | (1,263,009 | ) | ||||||||||||||
| Change in estimate of loss on settlement of litigation | – | 6,228,621 | – | – | – | |||||||||||||||
| Loss on settlement of litigation | 13,468,547 | – | – | 294,144 | 11,132,467 | |||||||||||||||
| Total operating expenses | 35,049,991 | 29,850,456 | 25,591,802 | 17,483,612 | 32,040,966 | |||||||||||||||
| Loss from operations | (34,954,552 | ) | (29,707,907 | ) | (25,242,751 | ) | (17,321,143 | ) | (31,532,522 | ) | ||||||||||
| Non-operating income (expense): | ||||||||||||||||||||
| Interest income | 55,840 | 165,918 | 15,581 | 35,114 | 16,724 | |||||||||||||||
| Interest expense and late fees | (429,573 | ) | (1,437,584 | ) | (1,104,602 | ) | (1,510,693 | ) | (11,726,120 | ) | ||||||||||
| Other gain (loss) | (172,656 | ) | (867,065 | ) | (10,515,470 | ) | (54,984,170 | ) | (4,332,277 | ) | ||||||||||
| Gain (loss) on disposal of fixed assets | (10,922 | ) | – | (17,138 | ) | – | 9,500 | |||||||||||||
| Gain on extinguishment of debt | – | 438,587 | – | – | 197,370 | |||||||||||||||
| Loss attributable to equity method investments | – | – | – | (820,000 | ) | – | ||||||||||||||
| Adjustments to fair value of unsettled warrant obligation | 18,959 | (107,438 | ) | 1,390,382 | 7,963,101 | (7,331,109 | ) | |||||||||||||
| Adjustments to fair value of derivatives | 743,959 | 493,241 | 889,883 | 11,444,988 | (6,209,898 | ) | ||||||||||||||
| Total non-operating income (expense) | 205,607 | (1,314,341 | ) | (9,341,364 | ) | (37,871,660 | ) | (29,375,810 | ) | |||||||||||
| Loss before provision for income tax | (34,748,945 | ) | (31,022,248 | ) | (34,584,115 | ) | (55,192,803 | ) | (60,908,332 | ) | ||||||||||
| Provision for income tax | – | – | – | – | – | |||||||||||||||
| Net loss | $ | (34,748,945 | ) | $ | (31,022,248 | ) | $ | (34,584,115 | ) | $ | (55,192,803 | ) | $ | (60,908,332 | ) | |||||
| Preferred stock dividend | 1,889,192 | 2,364,947 | 2,048,007 | 1,432,661 | 196,986 | |||||||||||||||
| Net loss applicable to common stock (1) | $ | (36,638,137 | ) | $ | (33,387,195 | ) | $ | (36,632,122 | ) | $ | (56,625,464 | ) | $ | (61,105,318 | ) | |||||
| Net loss applicable to common share: | ||||||||||||||||||||
| Basic and diluted | $ | (1.17 | ) | $ | (1.34 | ) | $ | (1.76 | ) | $ | (3.58 | ) | $ | (5.02 | ) | |||||
| Weighted average shares outstanding: | ||||||||||||||||||||
| Basic and diluted | 31,270,460 | 24,918,727 | 20,866,197 | 15,820,951 | 12,181,910 | |||||||||||||||
| 36 |
| As of December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| Balance Sheet Data: | ||||||||||||||||||||
| Cash and cash equivalents | $ | 4,424,374 | $ | 1,743,485 | $ | 7,241,852 | $ | 13,103,007 | $ | 15,889,409 | ||||||||||
| Current assets | 4,749,035 | 2,849,424 | 7,552,756 | 13,406,690 | 15,981,007 | |||||||||||||||
| Total assets | 5,737,484 | 3,907,919 | 8,496,542 | 15,185,326 | 19,054,152 | |||||||||||||||
| Current liabilities | 5,676,074 | 22,916,789 | 23,490,235 | 58,181,785 | 24,490,141 | |||||||||||||||
| Total liabilities | 8,473,029 | 26,441,529 | 30,042,223 | 62,312,262 | 54,580,237 | |||||||||||||||
| Series A-1 redeemable preferred stock | – | – | 1,598,533 | 1,429,126 | 1,272,441 | |||||||||||||||
| Total stockholders' deficit | 2,735,545 | 22,533,610 | 23,144,214 | 48,556,062 | 36,798,526 | |||||||||||||||
| (1) | Due to the impact the reverse stock split executed in 2014 had on net loss per share, the Company has changed the presentation of certain prior periods to enhance the disclosure surrounding net loss applicable to common stock. |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Certain statements in this annual report on Form 10-K that are not historical in fact constitute “forward-looking statements.” Words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “intend,” “plan,” and similar expressions are intended to identify forward-looking statements. Such forward-looking statements involve known and unknown risks, uncertainties and other factors based on the Company’s estimates and expectations concerning future events that may cause the actual results of the Company to be materially different from historical results or from any results expressed or implied by such forward-looking statements. These risks and uncertainties, as well as the Company’s critical accounting policies, are discussed in more detail under “Management’s Discussion and Analysis—Critical Accounting Policies” and in periodic filings with the Securities and Exchange Commission. You should review carefully the factors identified in this report in Item 1A, “Risk Factors”. We disclaim any intent to update or announce revisions to any forward-looking statements to reflect actual events or developments, except as required by law. Except as otherwise indicated herein, all dates referred to in this report represent periods or dates fixed with reference to our fiscal year ended December 31. The Company undertakes no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise.
You should read the following discussion of our financial condition and results of operations together with the audited financial statements and the notes to the audited financial statements included in this annual report. This discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
We are a clinical stage biotechnology company focused on the development and commercialization of Regenerative Ophthalmology therapeutics. Ocata’s most advanced products are in clinical trials for the treatment of Stargardt’s macular degeneration, dry age-related macular degeneration, and myopic macular degeneration. We are also developing several pre-clinical terminally differentiated-cell therapies for the treatment of other ocular disorders. Additionally, we have a number of pre-clinical stage assets in disease areas outside the field of ophthalmology, including autoimmune, inflammatory and wound healing-related disorders. Our intellectual property portfolio includes pluripotent human embryonic stem cell, or hESC; induced pluripotent stem cell, or iPSC, platforms; and other cell therapy technologies. We have no therapeutic products currently available for sale and do not expect to have any therapeutic products commercially available for sale for a period of years, if at all. These factors indicate that our ability to continue research and development activities is dependent upon the ability of management to obtain additional financing as required.
| 37 |
We pursue a number of approaches to generating transplantable tissues both in-house and through collaborations with other researchers who have particular interests in, and skills related to, cellular differentiation. Our research in this area includes projects focusing on the development of many different cell types that may be used to treat a range of diseases within ophthalmology and other therapeutic areas. Control of cellular differentiation and the culture and growth of stem and differentiated cells are important areas of research and development for us.
Comparison of Years Ended December 31, 2014 and 2013
| Dollar | Percentage | |||||||||||||||
| 2014 | 2013 | Change | Change | |||||||||||||
| Revenue | $ | 157,875 | $ | 224,985 | $ | (67,110 | ) | (29.8 | )% | |||||||
| Cost of Revenue | 62,436 | 82,436 | (20,000 | ) | (24.3 | )% | ||||||||||
| Gross Profit | 95,439 | 142,549 | (47,110 | ) | (33.0 | )% | ||||||||||
| Research and Development expenses: | ||||||||||||||||
| -R&D expenses, excluding non-cash, stock compensation | 9,625,486 | 10,171,842 | (546,356 | ) | (5.4 | )% | ||||||||||
| -R&D stock compensation | 903,835 | 1,392,926 | (489,091 | ) | (35.1 | )% | ||||||||||
| Total Research and Development | 10,529,321 | 11,564,768 | (1,035,447 | ) | (9.0 | )% | ||||||||||
| General and administrative expenses | ||||||||||||||||
| -G&A expenses, excluding non-cash, stock compensation | 9,165,847 | 9,569,934 | (404,087 | ) | (4.2 | )% | ||||||||||
| -G&A stock compensation | 1,886,276 | 2,487,133 | (600,857 | ) | (24.2 | )% | ||||||||||
| Total General and Administrative | 11,052,123 | 12,057,067 | (1,004,944 | ) | (8.3 | )% | ||||||||||
| Litigation settlement contingency | – | 6,228,621 | (6,228,621 | ) | (100 | )% | ||||||||||
| Loss on settlement of litigation | 13,468,547 | – | 13,468,547 | 100 | % | |||||||||||
| Non-operating income (expense) | 205,607 | (1,314,341 | ) | 1,519,948 | (115.6 | )% | ||||||||||
| Net Loss | $ | (34,748,945 | ) | $ | (31,022,248 | ) | $ | (3,726,697 | ) | 12.0 | % | |||||
Revenue
Revenue was $157,875 for 2014, which was a decrease of $67,110 or 29.8% compared to 2013. The decrease is due to license agreements that expired in 2013. Deferred revenue of $1,749,699, as of December 31, 2014, will be amortized and recorded to revenue over approximately 11 years. We currently have no therapeutic products available for sale and do not expect to have any commercially available for sale for a period of years, if at all.
Research and Development Expenses
Our research and development expenses, consist mainly of payroll and payroll related expenses for our scientific, manufacturing, clinical and regulatory staff, services attained in connection with our ongoing clinical trials and pre-clinical programs, our R&D and manufacturing facilities, and research supplies and materials. Our primary focus is on the development of novel therapies based on terminally differentiated cells. These expenses represent both pre-clinical and clinical development costs and costs associated with support activities such as manufacturing, quality control and regulatory processes. The cost of our research and development personnel is the most significant category of R&D expense; however, we also incur expenses with third parties, including license agreements, sponsored research programs and consulting expenses.
R&D expenditures, excluding non-cash, stock compensation expense, decreased from $10,171,842 for 2013 to $9,625,486 for 2014, for a decrease of $546,356 or 5.4%. The decrease in R&D expenditures was primarily due to a decrease in clinical costs of approximately $742,000, primarily due to lower patient enrollment in the period. Other decreases included: approximately $339,000 in license costs due to the expiration of certain licenses in 2013: approximately $214,000 related to lower lab supply costs; and approximately $269,000 related to a reversal of various accruals which we deemed we had been legally released from in 2014. These decreases were partially offset by the increase in payroll and other compensation costs of approximately $607,000 and an increase in consulting expenses of approximately $429,000. These increases in payroll and consulting were driven primarily by our planned expansion of our clinical capabilities as we continue to prepare for the commencement of our Phase 2 clinical trials in AMD and SMD.
R&D expenses related to non-cash, stock compensation decreased from $1,392,926 for 2013 to $903,835 for 2014, for a decrease of $489,091, or 35.1%. This decrease is related to the change in vesting schedules of new 2014 grants. In 2013 new grants vested 20% upfront and subsequently over a 24 month period, whereas new grants in 2014 had no upfront vesting and typically vest over a 48 month period, resulting in higher stock compensation expense for 2013 as compared to 2014.
| 38 |
We expect that R&D expenses will increase from period to period, for the foreseeable future. This planned increase will be driven primarily by our expansion of our clinical operations capabilities as we initiate and scale our phase 2 programs for SMD and AMD. Spending will continue to increase throughout 2015 as our trials are initiated and patients are being enrolled in the trials. We currently work with four clinical sites in the US and 2 in the UK. We plan to expand the number of sites in both the US and in Europe. In addition, we are expanding the network of consultants and service providers we contract and we also plan to expand our internal workforce. These expansions and the increased spend that will result from these expanded capabilities is consistent with our previously stated plans to transition to become a product development company. Our spending is impacted by the timing of enrollment and treatment of clinical trial patients along with interim results of our many pre-clinical programs. The amount and timing of these fluctuations can be difficult to predict due to the uncertainty inherent in the timing and extent of progress in our research programs, initiation of new clinical trials and rate of progression of existing clinical trials. In addition, the results from our basic research and pre-clinical trials, as well as the results of trials of similar therapeutics under development by others, will influence the number, size and duration of future trials. As our research efforts mature, we will continue to review the direction of our research based on an assessment of the value of possible commercial applications emerging from these efforts. Based on this continuing review, we expect to establish discrete research programs and evaluate the cost and potential for cash inflows from commercializing products, partnering with others in the biotechnology or pharmaceutical industry, or licensing the technologies associated with these programs to third parties.
We believe that it is not possible at this stage to provide a meaningful estimate of the total cost to complete our ongoing projects and bring any proposed products to market. The use of human embryonic stem cells as a therapy is an emerging area of medicine, and it is not known what clinical trials will be required by the FDA in order to gain marketing approval. Costs to complete could vary substantially depending upon the projects selected for development, the number of clinical trials required and the number of patients needed for each study. It is possible that the completion of these studies could be delayed for a variety of reasons, including difficulties in enrolling patients, delays in manufacturing, incomplete or inconsistent data from the pre-clinical or clinical trials, and difficulties evaluating the trial results. Any delay in completion of a trial would increase the cost of that trial, which would harm our results of operations. Due to these uncertainties, we cannot reasonably estimate the size, nature nor timing of the costs to complete, or the amount or timing of the net cash inflows from our current activities. Until we obtain further relevant pre-clinical and clinical data, we will not be able to estimate our future expenses related to these programs or when, if ever, and to what extent we will receive cash inflows from resulting products.
General and Administrative Expenses
General and administrative, or G&A, consist mainly of payroll and payroll related expenses, legal costs relating to corporate matters and litigation, and fees for consultants, service providers and other administrative costs. G&A expenditures, excluding non-cash, stock compensation expense, decreased from $9,569,934 for 2013 to $9,165,847 for 2014, for a decrease of $404,087 or 4.2%. The decrease in G&A expenditures was primarily due to a reduction in legal costs of approximately $1,011,000 and decreased investor relation costs of approximately $277,000. G&A expenses were also decreased, by approximately $566,000, as a result of a reversal of various accruals which we deemed we had been legally released from in 2014. These decreases were partially offset by increases in payroll and other compensation costs of approximately $673,000. Payroll costs were higher due to a full year of costs for a Chief Financial Officer in 2014 versus six months during 2013 and the hiring of a Chief Commercial Officer during the last quarter of 2014. Other increases related to costs for audit and professional and consulting fees associated with financial restatements and the remediation of other historical matters of approximately $659,000 and an increase in recruiting fees of approximately $183,000, mainly related to the recruitment of the CEO.
G&A expenses related to non-cash, stock compensation decreased from $2,487,133 for 2013 to $1,886,276 for 2014, for a decrease of $600,857, or 24.2%. This decrease is related to the change in vesting schedules of new 2014 grants. In 2013 new grants vested 20% upfront and subsequently over a 24 month period, whereas new grants in 2014 had no upfront vesting and vest over a 48 month period, resulting in higher stock compensation expense for 2013 as compared to 2014.
We expect G&A expenses to decrease in 2015, relative to 2014. While we’ve increased our staffing in G&A, we believe that we’ve resolved all legacy legal matters and therefore we don’t expect to incur legal costs related to such matters.
Loss on Settlement of Litigation
The loss on settlement of litigation relates to the settlement in June 2014, of the warrant holder litigation. The total value of the litigation settlement was recorded at $23,577,600 based on a 3,840,000 share settlement valued at $6.14 per share. Partially offsetting this charge was the reversal of previously recorded accruals related to the unsettled warrant obligation and loss contingency.
| 39 |
Other Income (Expense)
Other, non-operating income (expense) consisted of the following:
| 2014 | 2013 | $ Change | % Change | |||||||||||||
| Interest income | $ | 55,840 | $ | 165,918 | $ | (110,078 | ) | (66.3 | )% | |||||||
| Interest expense | (429,573 | ) | (1,437,584 | ) | 1,008,011 | (70.1 | )% | |||||||||
| Other loss | (172,656 | ) | (867,065 | ) | 694,409 | (80.1 | )% | |||||||||
| Adjustments to fair value of unsettled warrant obligation (expense) | 18,959 | (107,438 | ) | 126,397 | (117.6 | )% | ||||||||||
| Loss on disposal of fixed assets | (10,922 | ) | – | (10,922 | ) | (100 | )% | |||||||||
| Gain on the extinguishment of debt | – | 438,587 | (438,587 | ) | (100 | )% | ||||||||||
| Adjustments to fair value of derivatives | 743,959 | 493,241 | 250,718 | 50.8 | % | |||||||||||
| Total non-operating expense | $ | 205,607 | $ | (1,314,341 | ) | $ | 1,519,948 | (115.6 | )% | |||||||
Interest expense for 2014 compared to 2013 decreased by $1,008,011 to $429,573. The decrease is due to the extinguishment of debt and related interest expense on financing from JMJ Financial and Violation Capital in 2013 and a lower principal balance on the debt held by CAMOFI Master LDC and CAMHZN Master LDC, or the CAMOFI Notes, in 2014 as compared to 2013.
The decrease in other loss during 2014, compared to 2013, relates primarily to items included as part of the accrued loss contingency that were deemed no longer to be likely to be settled as of December 31, 2014.
Adjustments to fair value of unsettled warrant obligation was a gain of $18,959 for 2014 as compared to a loss of $107,438 for 2013 resulting in an increase of $126,397. The fair value account adjusts the 6.3 million shares which are contractually obligated by the change in the stock price for each period. The increase in other income resulted from the stock price decreasing from approximately $6.17 at December 31, 2013 to $6.14 at June 4, 2014 when the obligation was settled, whereas in 2013, during the period of January 1, 2013 to December 31, 2013, the stock price increased.
Adjustment to fair value of derivatives was a gain of $743,959 for 2014 compared to a gain of $493,241 for 2013. The 2014 balance is mainly due to the valuation of the derivative related to the CAMOFI debentures being revalued to $0 with the retirement of the debentures, resulting in a gain of approximately $663,000 for 2014.
Comparison of Years Ended December 31, 2013 and 2012
| Dollar | Percentage | |||||||||||||||
| 2013 | 2012 | Change | Change | |||||||||||||
| Revenue | $ | 224,985 | $ | 466,487 | $ | (241,502 | ) | (51.8 | )% | |||||||
| Cost of Revenue | 82,436 | 117,436 | (35,000 | ) | (29.8 | )% | ||||||||||
| Gross Profit | 142,549 | 349,051 | (206,502 | ) | (59.2 | )% | ||||||||||
| Research and Development expenses: | ||||||||||||||||
| -R&D expenses, excluding non-cash, stock compensation | 10,171,842 | 10,366,542 | (194,700 | ) | (1.9 | )% | ||||||||||
| -R&D stock compensation | 1,392,926 | 3,792,394 | (2,399,468 | ) | (63.3 | )% | ||||||||||
| Total Research and Development | 11,564,768 | 14,158,936 | (2,594,168 | ) | (18.3 | )% | ||||||||||
| General and administrative expenses | ||||||||||||||||
| -G&A expenses, excluding non-cash, stock compensation | 9,569,934 | 7,429,375 | 2,140,559 | 28.8 | % | |||||||||||
| -G&A stock compensation | 2,487,133 | 4,003,491 | (1,516,358 | ) | (37.9 | )% | ||||||||||
| Total General and Administrative | 12,057,067 | 11,432,866 | 624,201 | 5.5 | % | |||||||||||
| Litigation settlement contingency | 6,228,621 | – | 6,228,621 | 100 | % | |||||||||||
| Non-operating expense | (1,314,341 | ) | (9,341,364 | ) | 8,027,023 | 85.9 | % | |||||||||
| Net Loss | $ | (31,022,248 | ) | $ | (34,584,115 | ) | $ | 3,561,867 | 10.3 | % | ||||||
Revenue
Revenue relates to license fees and royalties collected that are being amortized over the period of the license granted, and are therefore typically consistent between periods. The decrease of 51.8% is related to the termination of our license agreement with International Stem Cell Corporation in early 2013. Deferred revenue of $1,907,574 as of December 31, 2013 will be amortized to revenue over approximately 12 years. We currently have no therapeutic products available for sale and do not expect to have any commercially available for sale for a period of years, if at all.
| 40 |
Research and Development Expenses
R&D expenses, excluding non-cash, stock compensation expense, decreased from $10,366,542 for the year ended December 31, 2012 to $10,171,842 for the year ended December 31, 2013, for a decrease of $194,700 or 1.9%. In the beginning of 2013, a number of our employees who are located in our Massachusetts R&D and manufacturing facility changed their roles and responsibilities consistent with our scale up of the general and administrative function within the company. This shift in roles and responsibilities in early 2013 resulted in a decrease in R&D payroll related expenses of approximately $1,144,000. Aside from this shift in responsibilities of these employees, and the related shift in their cost allocation, R&D increased from 2012 to 2013. The primary driver of this increased spending was our planned expansion of our clinical trial activities which increased by approximately $639,000 during the year, as we expanded our clinical sites and increased the activities to screen, enroll and treat patients in our AMD and SMD clinical trials, in the US and in the UK. Also contributing to the increase in R&D spending for the year was approximately $557,000 of increased collaboration and licensing costs incurred, in support of our pre-clinical programs and an increase of approximately $317,000 of additional occupancy costs due to the additional lab and manufacturing space rented in our Marlborough facility. Other items of increased spending were recruiting costs of approximately $94,000; and depreciation expenses of approximately $51,000.
Research and development expenses related to non-cash, stock compensation was $1,392,926 for the year ended December 31, 2013 and $3,792,394, as restated, for the year ended December 31, 2012. This decrease of $2,399,468, or 63.3%, is due to the revaluation of stock option expense, pursuant to the adjustment of the stock option pool from liability to equity classification.
General and Administrative Expenses
G&A expenses, excluding non-cash, stock compensation increased from $7,429,374 for the year ended December 31, 2012, to $9,569,934 for the year ended December 31, 2013, for an increase of $2,140,559 or 28.8%. The increase in G&A spending primarily related to increases in salaries and wages of approximately $1,347,000. In the beginning of 2013, a number of our employees who are located in our Massachusetts R&D and manufacturing facility changed their roles and responsibilities consistent with our scale up of the general and administrative function within the company. This further emphasis on the G&A function is consistent with our plans to relocate the majority of our G&A function to our Massachusetts facility. We also realized an increase in legal expenses of approximately $1,235,000 in 2013, as compared to 2012. These increases were partially offset by a reduction in the amounts incurred for outside services and professional fees of approximately $162,000.
General and administrative expenses related to non-cash, stock compensation was $2,487,133 for the year ended December 31, 2013, and $4,003,491, as restated, for the year ended December 31, 2012, for a decrease of $1,516,358 or 37.9%. This decrease is due to the revaluation of stock option expense, pursuant to the adjustment of the stock option pool from liability to equity classification.
Other Income (Expense)
Other, non-operating income (expense) consisted of the following:
| 2013 | 2012 | $ Change | % Change | |||||||||||||
| Interest income | $ | 165,918 | $ | 15,581 | $ | 150,337 | 964.9 | % | ||||||||
| Interest expense | (1,437,584 | ) | (1,104,602 | ) | (332,982 | ) | (30.1 | )% | ||||||||
| Other (loss) | (867,065 | ) | (10,515,470 | ) | 9,648,405 | 91.8 | % | |||||||||
| Adjustments to fair value of unsettled warrant obligation (expense) | (107,438 | ) | 1,390,382 | (1,497,820 | ) | (107.7 | )% | |||||||||
| Loss on disposal of fixed assets | – | (17,138 | ) | 17,138 | 100 | % | ||||||||||
| Gain on the extinguishment of debt | 438,587 | – | 438,587 | 100 | % | |||||||||||
| Adjustments to fair value of derivatives | 493,241 | 889,883 | (396,642 | ) | (44.6 | )% | ||||||||||
| Total non-operating expense | $ | (1,314,341 | ) | $ | (9,341,364 | ) | $ | 8,027,023 | 85.9 | % | ||||||
Interest expense for the year ended December 31, 2013 compared to the year ended December 31, 2012 increased by $332,982, or 30.1%. The increase is due to interest on the CAMOFI Notes which were effective in December 2012 as part of a settlement agreement. The principal amount of the CAMOFI Notes at issuance was $6,000,000 and the debentures accrue interest at a rate of 8%. This interest expense increase was partially offset by the discontinuation of interest from the debt issued to JMJ Financial and Volation. Convertible promissory notes originally issued to JMJ Financial in 2010 were retired in May 2013, when the Company entered into a Mutual Release and Waiver Agreement with JMJ Financial. In 2009, the Company entered into a purchase agreement with Volation and issued Series A-1 redeemable convertible preferred stock which paid dividends at a rate of 10% and was recorded as interest expense. In April 2013, the Company entered into an exchange agreement whereby the Series A-1 preferred stock was exchanged for shares of common stock of the Company.
Other gain (loss) for the year ended December 31, 2013, changed by $9,648,405, from a loss of $10,515,470 in 2012 to a loss of $867,065 in 2013. Finance charges for the year ended December 31, 2012 consist of $3,586,000 related to the final settlement with Alpha Capital concerning a dispute over convertible notes and warrants they held plus an additional $2,887,000 related to the final settlement with CAMOFI and an increase in the estimate of various additional potential settlement claims of $542,470. Fines and penalties for the year ended December 31, 2013 were $962,227. Approximately $587,000 of the balance was due to us being named as a defendant in a civil action brought by the SEC, alleging that we violated the Securities Act because certain sales of shares to outside organizations completed in 2008 and 2009 were neither registered under the Securities act nor subject to an exemption from registration. This amount was in addition to the $3,500,000 we expensed in 2012. The SEC civil suit was settled in December 2013, for approximately $4,087,000, which includes the $3,500,000 and approximately $587,000 of pre-judgment interest. The remaining balance in 2013 of approximately $375,000 relates to an SEC investigation of our previous CEO’s failure to report transactions for shares of common stock sold between February 7, 2011 and January 10, 2013.
| 41 |
Adjustments to fair value of unsettled warrant obligation for the year ended December 31, 2013 was a loss of $107,438 compared to a gain of $1,390,382 for the year ended December 31, 2012. The fair value account adjusts the 63.2 million shares, on a pre-reverse split basis, which are contractually obligated by the change in the stock price for each period. In 2013 the stock price was relatively flat and on average increased slightly, leading to expense for the year. In 2012 the stock price decreased leading to a gain for the year.
The gain on the extinguishment of debt for the year ended December 31, 2013 relates to the settlement with JMJ Financial.
The adjustment to fair value of derivatives changed to a gain of $493,241 during the year ended December 31, 2013, from a gain of $889,883 during the year ended December 31, 2012. The decrease in gain of $396,642 is due to the revaluation of the embedded derivative related to the CAMOFI debentures and lower expected volatility relating to the decreased time to maturity offset by a slightly higher stock price at December 31, 2013 than at December 31, 2012.
Liquidity and Capital Resources
Cash Flows
The following table sets forth a summary of our cash flows for the periods indicated below:
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Net cash used in operating activities | $ | (21,447,451 | ) | $ | (21,965,199 | ) | $ | (14,606,357 | ) | |||
| Net cash used in investing activities | (263,375 | ) | (710,772 | ) | (111,350 | ) | ||||||
| Net cash provided by financing activities | 24,391,715 | 17,177,604 | 8,856,552 | |||||||||
| Net increase (decrease) in cash and cash equivalents | 2,680,889 | (5,498,367 | ) | (5,861,155 | ) | |||||||
| Cash and cash equivalents at the end of the period | $ | 4,424,374 | $ | 1,743,485 | $ | 7,241,852 | ||||||
Cash Used in Operating Activities
Our net cash used in operating activities during the years ended December 31, 2014, 2013 and 2012 was $21,447,451, $21,965,199, and $14,606,357, respectively. Net cash used in operating activities for 2014 includes the net loss of $34,748,945, partially offset by non-cash expenses such as the loss on settlement of litigation of $13,468,547. Partially offsetting this decrease in cash used in operating activities was the amount of loss that related to stock compensation. For 2014 the Company incurred $2,546,126 in stock compensation expense, as compared to $3,880,058 for 2013. During 2014, there was approximately $2,408,000 of cash used from changes in operating assets and liabilities, whereas during 2013 there was approximately $2,965,000 of cash used from changes in operating assets and liabilities, for a difference of approximately $557,000.
Cash Used in Investing Activities
Cash used in investing activities during the years ended December 31, 2014, 2013 and 2012 was $263,375, $710,772 and $111,350, respectively. Our cash used in investing activities decreased during the year ended December 31, 2014 over the comparable amount from December 31, 2013 mainly due to the decrease in the purchase of fixed assets year over year. The majority of the fixed asset purchases in 2013 related to leasehold improvements for additional leased space in Marlborough, MA. Our purchases for this build-out of additional space were substantially complete as of the end of 2013 so our investing levels relating to the purchase of fixed assets have decreased in 2014.
Cash Flows from Financing Activities
Cash flows provided by financing activities during the years ended December 31, 2014, 2013 and 2012 was $24,391,715, $17,177,604, and $8,856,552, respectively. On September 19, 2012, we entered into a purchase agreement, or the 2012 Purchase Agreement, with Lincoln Park Capital Fund, LLC, or Lincoln Park. Pursuant to the 2012 Purchase Agreement, we had the right to sell to Lincoln Park up to $35,000,000 in shares of our common stock. In June 2014 we had sold Lincoln Park the maximum amount of $35,000,000 under the Purchase Agreement.
On June 27, 2014, we entered into a new purchase agreement, or the 2014 Purchase Agreement with Lincoln Park pursuant to which we have the right to sell to Lincoln Park up to $30,000,000 in shares of its common stock, subject to certain limitations set forth in the 2014 Purchase Agreement.
| 42 |
Upon the satisfaction of the conditions set forth in the 2014 Purchase Agreement, we obtained the right over a 36-month period to sell up to $30,000,000 worth of shares of our common stock to Lincoln Park based upon the terms set forth in the 2014 Purchase Agreement. Pursuant to the 2014 Purchase Agreement, the purchase price of such common stock will be based on the prevailing market price of our common stock immediately preceding the time of sales, with our controlling the timing and amount of any future sales, if any, of common stock to Lincoln Park. There are no upper limits to the price Lincoln Park may pay to purchase our common stock. Lincoln Park shall not have the right or the obligation to purchase any shares of common stock on any business day that the closing price of our common stock is below a floor price of $1.00, as adjusted under the 2014 Purchase Agreement due to the 100-to-1 reverse split of our common stock effected in August 2014.
The cash provided by financing activities during the year ended December 31, 2014 included $25,661,844 from the sale of common stock to Lincoln Park. 2,269,750 shares of common stock were sold to Lincoln Park pursuant to the purchase agreement executed in 2012, for total proceeds of $14,281,295. 1,633,309 shares (including 106,008 shares issued as a commitment fee) of common stock were sold to Lincoln Park pursuant to the 2014 Purchase Agreement for total proceeds of $11,380,549. During the same period in 2013, cash provided by financing activities included $17,777,604 of proceeds from the sale of common stock to Lincoln Park.
| · | During the year ended December 31, 2014, we repaid $1,200,000 on the CAMOFI Notes. |
| · | During the year ended December 31, 2014, we redeemed 1,000 shares of Series B preferred stock which resulted in a cash use of $70,129. |
| · | We plan to fund our operations for the foreseeable future from the following sources: | |
| o | As of December 31, 2014, we have approximately $4,424,374 in cash. |
| o | As of December 31, 2014, $18,619,451 is available to us through the Lincoln Park financing arrangement. On various dates from January 1, 2015 through March 16, 2015, Lincoln Park purchased 932,182 shares of common stock for cash proceeds to the Company of $5,872,996. As of March 16, 2015, the Company has $12,746,455 available to us through the Lincoln Park financing arrangement. |
On a long-term basis, we have no expectation of generating any meaningful revenues from our product candidates for a substantial period of time and must rely on raising funds in capital transactions to finance our research and development programs. Our future cash requirements will depend on many factors, including the pace and scope of our research and development programs, the costs involved in filing, prosecuting and enforcing patents, and other costs associated with commercializing our potential products.
We believe that our current cash balance, and the $18,619,451 available to us under the Lincoln Park financing arrangement, will be sufficient to fund our operations into early 2016. This belief is based on the assumption that our stock price does not realize any significant or prolonged decreases. Our ability to fund our operations through the Lincoln Park arrangement is highly dependent on our stock price. A significant decline in our share price could force us to curtail our operations in part, or entirely. We are continually in discussions with potential investors and collaborators to explore alternative sources of funding which may or may not result in immediate and substantial dilution to our stockholders, so that we may either extend our current cash runway beyond early 2016 or accelerate the rate of investment in our many clinical and pre-clinical programs.
We cannot assure you that public or private financing or grants will be available on acceptable terms, if at all. Several factors will affect our ability to raise additional funding, including, but not limited to, the volatility of our common stock and the broader public equity market. If we are unable to raise additional funds, we will be forced to either scale back our business efforts or curtail our business activities entirely. As of December 31, 2014, the Company has an accumulated deficit of $349.1 million, recurring losses from operations, and negative working capital which raise substantial doubt about the ability of the Company to continue as a going concern. Furthermore, an emphasis of matter paragraph related to an uncertainty as to the Company’s ability to continue as a going concern has been included in the auditor’s opinion.
Contractual Obligations
Our significant contractual obligations are as follows:
| Less than | One to | Three to | More Than | |||||||||||||||||
| One Year | Three Years | Five Years | Five Years | Total | ||||||||||||||||
| Operating lease obligations | $ | 433,104 | $ | 706,462 | $ | 110,927 | $ | – | $ | 1,250,493 | ||||||||||
| Other Liability - SEC settlements | 1,731,202 | – | – | – | 1,731,202 | |||||||||||||||
| Total | $ | 2,164,306 | $ | 706,462 | $ | 110,927 | $ | – | $ | 2,955,854 | ||||||||||
Off-Balance Sheet Arrangements
We do not maintain any off-balance sheet arrangements, transactions, obligations or other relationships with unconsolidated entities that would be expected to have a material current or future effect upon our financial condition or results of operations.
| 43 |
Shelf Registration Statement
On October 14, 2014, we filed with the SEC a universal shelf registration statement on Form S-3 (Registration No. 333-199311), which provides for the offer, from time to time, of an indeterminate amount (up to $100,000,000) of: common stock; preferred stock; debt securities; warrants to purchase any of these securities; and any combination of these securities, individually or as units. In addition, if we identify any security holder(s) in a prospectus supplement, they may also offer identified securities under this registration statement although we will not receive any of the proceeds from the sale of securities by any of these selling security holders. This universal shelf registration statement was declared effective by the SEC on November 14, 2014. The addition of any newly issued securities into the market may be dilutive to existing stockholders and new issuances by us or sales by our selling security holders could have an adverse effect on the price of our securities.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Our exposure to market risk is limited primarily to interest income sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because a significant portion of our investments are in short-term debt securities issued by the U.S. government and institutional money market funds. The primary objective of our investment activities is to preserve principal. Due to the nature of our marketable securities, we believe that we are not exposed to any material market risk. We do not have any derivative financial instruments or foreign currency instruments. If interest rates had varied by 10% in the year ended December 31, 2014, it would not have had a material effect on our results of operations or cash flows for that period.
Item 8. Financial Statements and Supplementary Data.
The information required by this item is set forth beginning on page F-1 of this Annual Report on Form 10-K.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures.
Our principal executive and financial officer after evaluating the effectiveness of our "disclosure controls and procedures" (as defined in the Exchange Act Rule 13a- 15(e) or Rule 15d-15(e)), with the participation of our management has concluded that as of the end of the period covered by this Annual Report on Form 10-K our disclosure controls and procedures were effective and were designed to ensure that information we are required to disclose in the reports that we file or submit under the Securities Exchange Act of 1934, as amended, is accumulated and communicated to management including our principal executive and financial officer as appropriate to allow timely decisions regarding required disclosure and is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission rules and forms. It should be noted that any system of controls is designed to provide reasonable but not absolute assurances that the system will achieve its stated goals under all reasonably foreseeable circumstances. Our principal executive and financial officer has concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective at a level that provides such reasonable assurances.
We carried out an evaluation, under the supervision and with the participation of our management, including our chief executive officer and principal financial officer, of the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2014. Based upon that evaluation, the Principal Executive Officer and Principal Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting for our company. Internal control over financial reporting is defined in Exchange Act Rules 13a-15(f) and 15(d) -15(f) as a process designed by, or under the supervision of, our Chief Executive and Chief Financial Officer and effected by our board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| · | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and disposition of our assets; |
| · | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles; |
| · | provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
| · | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. |
| 44 |
Because of inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), in the 2013 Internal Control-Integrated Framework. Based on our assessment and those criteria, our management believes that, as of December 31, 2014, our internal controls over financial reporting were effective based on those criteria.
As of December 31, 2013, management conducted the same evaluation as described above and identified a material weakness in internal control over financial reporting as of that date. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the annual or interim financial statement will not be prevented or detected on a timely basis. Based upon management’s evaluation in the prior year, we concluded that we did not maintain adequate and effective internal control in the area of technical accounting relating to the application of applicable accounting literature related to accounting for derivatives. Specifically, the control deficiency related to our interpretation of the accounting for an embedded derivative instrument with an anti-dilution ratchet provision and the subsequent unsettled share obligation. It was discovered during the preparation of our year-end financial statements for fiscal year 2013 that certain prior technical accounting judgments and conclusions related to the recording of the embedded derivative liability were not supportable, leading management to conclude that the execution of certain internal control activities had not been adequate. Specifically, we believe that, in the context of the small size of our business, we did not have sufficient staffing and technical expertise in this area of technical accounting to provide adequate review and control with respect to accounting for certain types of embedded derivatives. The material weakness contributed to the restatement of prior period financial statements, which were reflected in the financial statements for the three years ended December 31, 2013.
BDO USA LLP, our independent registered public accounting firm, has audited our consolidated financial statements and the effectiveness of our internal control over financial reporting as of December 31, 2014. This report appears below.
Changes in Internal Controls Over Financial Reporting.
As previously described in Item 9A “Controls and Procedures” in our Annual Report on Form 10-K filed for the year ended December 31, 2013, we identified a material weakness in internal controls over financial reporting relating our accounting of derivatives, specifically embedded derivatives relating to anti-dilution ratchet provisions. This material weakness contributed to material post-closing adjustments and restatement of prior period financial statements, which were reflected in the financial statements for the three years ended December 31, 2013.
In connection with the review, we identified a control deficiency relating to the application of applicable accounting literature related to accounting for derivatives. Specifically, the control deficiency related to our interpretation of the FASB Accounting Standards Codification for derivatives (ASC 815) in determining the proper accounting treatment for an embedded derivative with anti-dilution ratchet provisions. The warrant agreement was entered into in 2005 and the applicable ratchet provision extended through January 2009. The resulting unissued share obligation remained an issue through the third quarter of 2013.
Management, with the input, oversight, and support of the Audit Committee has identified and taken the following steps, which management believes assisted us in the identification and resolution of the technical accounting matters which resulted in the remediation of the material weakness described above as of December 31, 2014:
| · | in June 2013, we hired a new Chief Financial Officer, who has extensive experience leading the accounting and finance functions at publicly traded companies and adds accounting expertise to our staff. For several years we had outsourced many accounting duties and the previous CEO acted as the company CFO as well; |
| · | in November 2013, we hired a new Corporate Controller, who has extensive experience leading the accounting and finance functions at publicly traded companies and adds accounting expertise to our staff; and |
| · | in December 2013, we engaged external advisors knowledgeable in many technical accounting matters, including derivatives to assist us in the interpretation of key technical accounting standards and associated interpretations and the determination of how to adequately apply such standards; |
| · | in August 2014, we hired an Accounting Manager who has more than five years of progressive experience at PricewaterhouseCooper LLP. He brings additional capabilities to our finance and accounting organization and will assist us in identifying complex accounting positions and in determining the appropriate treatment. |
Additionally, we have enhanced our training programs to ensure that our accounting personnel have the competence and the on-going accounting and financial reporting training necessary for their assigned duties, including specific technical training courses related to derivative accounting and other complex accounting issues.
Other than the continued implementation of the remediation efforts described above, there were no changes in our internal control over financial reporting that occurred during the fourth quarter of 2014 that have materially affected or are reasonably likely to materially affect our internal control over financial reporting.
| 45 |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Ocata Therapeutics, Inc. and Subsidiary
Marlborough, Massachusetts
We have audited Ocata Therapeutics, Inc. and Subsidiary's (the “Company”) internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Ocata Therapeutics, Inc. and Subsidiary's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Item 9A, Management Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Ocata Therapeutics, Inc. and Subsidiary maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet of Ocata Therapeutics, Inc. and Subsidiary as of December 31, 2014, and the related consolidated statements of operations, stockholders' deficit, and cash flows for the year then ended and our report dated March 16, 2015 expressed an unqualified opinion thereon and included an emphasis of a matter paragraph relating to an uncertainty as to the Company’s ability to continue as a going concern.
/s/ BDO USA, LLP
Boston, Massachusetts
March 16, 2015
| 46 |
Item 9B. Other Information.
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, to be filed with the SEC not later than 120 days after the close of our year ended December 31, 2014.
Code of Ethics
Our Board of Directors has adopted a code of business conduct and ethics that applies to our directors, officers and employees. There have been no material modifications to, or waivers from, the provisions of such code. This code is available on the corporate governance section of our website (which is a subsection of the investor relations section of our website) at the following address: www.advancedcell.com. Any waivers from or amendments to the code will be filed with the SEC on Form 8-K. You may also request a printed copy of the code, without charge, by writing to us at Corporate Secretary, Ocata Therapeutics, Inc., 33 Locke Drive, Marlborough, Massachusetts.
Item 11. Executive Compensation
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, to be filed with the SEC not later than 120 days after the close of our year ended December 31, 2014.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, to be filed with the SEC not later than 120 days after the close of our year ended December 31, 2014.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, to be filed with the SEC not later than 120 days after the close of our year ended December 31, 2014.
Item 14. Principal Accountant Fees and Services
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, to be filed with the SEC not later than 120 days after the close of our year ended December 31, 2014.
Item 15. Exhibits and Financial Statement Schedules
(a)(1) Financial Statements
The following is a list of the Financial Statements included in Item 8 of Part II of this Report.
| Page | |
| Report of Independent Registered Public Accounting Firms | F-1 |
| Consolidated Balance Sheets as of December 31, 2014 and December 31, 2013 | F-2 |
| Consolidated Statements of Operations for the Years Ended December 31, 2014, 2013 and 2012 | F-3 |
| Consolidated Statements of Stockholders’ Deficit for the Years Ended December 31, 2014, 2013 and 2012 | F-4 |
| Consolidated Statements of Cash Flows for the Years Ended December 31, 2014, 2013 and 2012 | F-5 |
| Notes to Financial Statements | F-6 |
| 47 |
(a)(2) Financial Statement Schedules
Schedules not included herein are omitted because they are inapplicable or not required or because the required information is given in the financial statements and notes thereto.
(b)
The exhibits required by this item and included in this report or incorporated herein by reference are as follows:
| Exhibit No. | Document Description | Incorporation by Reference | ||
| 3.1 | Certificate of Incorporation of the Registrant dated November 17, 2005 | Previously filed as Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on November 21, 2005 and incorporated by reference herein. | ||
| 3.2 | Certificate of Amendment to Certificate of Incorporation dated October 13, 2006 | Previously filed as Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on October 13, 2006 and incorporated by reference herein. | ||
| 3.3 | Certificate of the Powers, Designations, Preferences and Rights of the Series A-1 Convertible Preferred Stock dated March 5, 2009 | Previously filed as Exhibit 4.1 to the Registrant’s Quarterly Report on Form 10-Q filed on July 20, 2009 and incorporated by reference herein. | ||
| 3.4 | Certificate of Amendment to Certificate of Incorporation dated September 15, 2009 | Previously filed as Exhibit 3.15 to Registrant’s Registration Statement on Form S-1 filed November 18, 2009 and herein incorporated by reference. | ||
| 3.5 | Certificate of Designations of Preferences, Rights and Limitations of Series B Preferred Stock dated November 3, 2009 | Previously filed as Exhibit 3.1 to the Registrant's Current Report on Form 8-K filed on November 13, 2009 and incorporated by reference herein. | ||
| 3.6 | Certificate of Designations of Preferences, Rights and Limitations of Series C Preferred Stock dated December 30, 2010 | Previously filed as Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on January 3, 2011 and incorporated herein by reference. | ||
| 3.7 | Certificate of Amendment to Certificate of Incorporation dated January 24, 2012 | Previously filed as Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on January 30, 2012 and incorporated herein by reference. | ||
| 3.8 | Certificate of Amendment to Certificate of Incorporation dated October 24, 2013 | Previously filed as Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on October 24, 2013 and incorporated herein by reference. | ||
| 3.9 | Certificate of Amendment to Certificate of Incorporation dated August 27, 2014 | Previously filed as Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on August 27, 2014 and incorporated herein by reference. | ||
| 3.10 | Bylaws of the Registrant | Previously filed as Exhibit 3.2 to the Registrant's Current Report on Form 8-K filed on November 21, 2005 and incorporated by reference herein. | ||
| 3.11 | Amendment No. 1 to Bylaws of the Registrant | Previously filed as Exhibit 3.2 to the Registrant’s Current Report on Form 8-K filed on November 30, 2007 and incorporated by reference herein | ||
| 4.1 | Form of Subscription Agreement to Purchase Series A Convertible Preferred Stock of the Registrant | Previously filed as Exhibit 4.8 to the Registrant's Quarterly Report on Form 10-QSB filed on May 23, 2005 and incorporated by reference herein. | ||
| 10.1 | A.C.T. Holdings, Inc. 2005 Stock Option Plan* | Previously filed as Appendix A to the Registrant's preliminary proxy statement on Form PRE-14A filed on May 10, 2005 and incorporated by reference herein. | ||
| 10.2 | Form of Incentive Stock Option Agreement under A.C.T. Holdings, Inc. 2005 Stock Option Plan * | Previously filed as Exhibit 10.36 to the Registrant's Quarterly Report on Form 10-QSB filed on May 23, 2005 and incorporated by reference herein. | ||
| 10.3 | Form of Nonqualified Stock Option Agreement under A.C.T. Holdings, Inc. 2005 Stock Option Plan * | Previously filed as Exhibit 10.37 to the Registrant's Quarterly Report on Form 10- QSB filed on May 23, 2005 and incorporated by reference herein. | ||
| 10.4 | Confidentiality and Nondisclosure Agreement dated February 3, 1999 by and between the Registrant and Robert Lanza, M.D.* | Previously filed as Exhibit 10.54 to the Registrant's Quarterly Report on Form 10-QSB filed on May 23, 2005 and incorporated by reference herein. | ||
| 10.5 | Agreement dated September 15, 2005 by and among ACT, Advanced Cell, Inc. and A.C.T. Group, Inc. | Previously filed as Exhibit 10.9 to the Registrant's Current Report on Form 8-K filed on September 19, 2005 and incorporated by reference herein. | ||
| 10.6 | Agreement and Plan of Merger dated July 31, 2007 by and among the Registrant, ACT Acquisition Sub, Inc., Mytogen, Inc. and certain shareholders of Mytogen, Inc. | Previously filed as exhibit 10.101 to the Amendment No. 1 to the Registrant’s 10-KSB for the year ended December 31, 2007 filed with the SEC on June 30, 2008 and incorporated by reference herein. |
| 48 |
| 10.7 | Form of Additional Investment Right | Previously filed as Exhibit 10.132 to the Registrant’s Registration Statement on Form S-1 filed on November 19, 2009 and incorporated herein by reference. | ||
| 10.8 | Employment Agreement dated October 1, 2009 by and between the Registrant and Robert P. Lanza* | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on November 17, 2009 and incorporated herein by reference. | ||
| 10.9 | Amended and Restated Employment Agreement dated July 1, 2011 by and between the Registrant and Robert P. Lanza* | Previously filed as Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on August 17, 2011 and incorporated herein by reference. | ||
| 10.10 | Settlement Agreement and Mutual Release Form used between the Registrant and several counter parties | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on December 12, 2011 and incorporated herein by reference. | ||
| 10.11 | Settlement Agreement and Mutual Release dated December 31, 2012 by and between the Registrant and CAMOFI Master LDC, and CAMHZN Master LDC | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on January 17, 2013 and incorporated herein by reference. | ||
| 10.12 | Form of Amortizing Senior Secured Convertible Debenture Issued to CAMOFI Master LDC | Previously filed as Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on January 17, 2013 and incorporated herein by reference. | ||
| 10.13 | Form of Amortizing Senior Secured Convertible Debenture Issued to CAMHZN Master LDC | Previously filed as Exhibit 10.3 to the Registrant’s Current Report on Form 8-K filed on January 17, 2013 and incorporated herein by reference. | ||
| 10.14 | Form of Registration Rights Agreement between the Registrant and each of the holders signatory thereto | Previously filed as Exhibit 10.4 to the Registrant’s Current Report on Form 8-K filed on January 17, 2013 and incorporated herein by reference. | ||
| 10.15 | Office Lease Agreement dated January 11, 2013 by and between the Registrant and Wendy Jolles and Linda Olstein, Trustees of The Janelon Trust under Declaration of Trust dated January 28, 1975 and recorded with the Suffolk County Registry of Deeds in Book 8766, Page 558, as amended by instrument dated January 7, 1988 and recorded in Book 14432, Page 267 | Previously filed as Exhibit 10.5 to the Registrant’s Current Report on Form 8-K filed on January 17, 2013 and incorporated herein by reference. | ||
| 10.16 | Executive Employment Agreement dated as of May 20, 2013, by and between the Registrant and Edward Myles | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on May 24, 2013 and incorporated herein by reference. | ||
| 10.17 | Mutual Release and Waiver Agreement, dated May 31, 2013 by and between the Registrant and JMJ Financial. | Previously filed as Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q filed on August 7, 2013 and incorporated herein by reference. | ||
| 10.18 | Share Exchange Agreement dated April 25, 2013 between the Registrant and Volation Capital Partners, LLC | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on May1, 2013 and incorporated herein by reference. | ||
| 10.19 | Separation Agreement, dated as of January 21, 2014 by and between the Registrant and Gary Rabin. | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on January 22, 2014 and incorporated herein by reference. | ||
| 10.20 | Employment Agreement dated December 13, 2013 by and between the Registrant and Eddy Anglade | Previously filed as Exhibit 10.47 to the Registrant’s Annual Report on Form 10-K filed on April 1, 2014 and incorporated herein by reference. | ||
| 10.21 | Executive Employment Agreement dated as of May 22, 2014, by and between the Company and Edward Myles | Previously filed as Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q filed on August 11, 2014 and incorporated d herein by reference. | ||
| 10.22 | Settlement Agreement, dated as of June 3, 2014 by and among Advanced Cell Technology, Inc. and Gary D. Aronson, John S. Gorton, individually and as trustee of the John S. Gorton Separate Property Trust dated March 3, 1993, herronlaw apc, Miller and Steele LLP, and Michael A. Bourke | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on June 6, 2014 and incorporated herein by reference. |
| 49 |
| 10.23 | Executive Employment Agreement, dated as of June 18, 2014 by and between the Company and Paul K. Wotton, Ph.D.* | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on June 24, 2014 and incorporated herein by reference. | ||
| 10.24 | Purchase Agreement, dated as of June 27, 2014, by and between the Company and Lincoln Park Capital Fund, LLC (incorporated herein by reference to, File No. 000-50295). | Previously filed as Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on July 3, 2014 and incorporated herein by reference. | ||
| 10.25 | Registration Rights Agreement, dated as of June 27, 2014, by and between the Company and Lincoln Park Capital Fund, LLC | Previously filed as Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on July 3, 2014 and incorporated herein by reference. | ||
| 10.26 | Stock Repurchase and Release Agreement dated as of August 7, 2014, by and between the Company and Socius CGII, Ltd. | Previously filed as Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q filed on August 11, 2014 and incorporated herein by reference. | ||
| 10.27 | Stock Repurchase and Release Agreement dated as of August 7, 2014, by and between the Company and Optimus Life Sciences Capital Partners, LLC | Previously filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q filed on August 11, 2014 and incorporated herein by reference. | ||
| 10.28 | Amended and Restated Executive Employment Agreement, dated as of October 1, 2014 by and between the Company and Robert Lanza | Previously filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q filed on November 10, 2014 and incorporated herein by reference. | ||
| 10.29 | Executive Employment Agreement, dated as of October 14, 2014 by and between the Company and H. LeRoux Jooste* | Previously filed as Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q filed on November 10, 2014 and incorporated herein by reference. | ||
| 10.30 | 2014 Stock Option and Incentive Plan and forms of option agreements thereunder* | Filed herewith. | ||
| 21.1 | Subsidiaries of the Registrant | Filed herewith. | ||
| 23.1 | Consent of Singer Lewak LLP, Independent Registered Public Accounting Firm | Filed herewith. | ||
| 23.2 | Consent of BDO USA LLP, Independent Registered Public Accounting Firm | Filed herewith. | ||
| 24.1 | Power of Attorney | Included on the signature page(s) hereto | ||
| 31.1 | Certification of the Principal Executive Officer Pursuant to Rule 13a-14 of the Securities Exchange Act of 1934 | Filed herewith. | ||
| 31.2 | Certification of the Principal Financial Officer Pursuant to Rule 13a-14 of the Securities Exchange Act of 1934 | Filed herewith. | ||
| 32.1 | Certification of Principal Executive Officer Pursuant to Section 1350 | Filed herewith. | ||
| 32.2 | Certification of Principal Financial Officer Pursuant to Section 1350 | Filed herewith. |
| 101.INS | XBRL Instance Document** |
| 101.SCH | XBRL Schema Document** |
| 101.CAL | XBRL Calculation Linkbase Document** |
| 101.DEF | XBRL Definition Linkbase Document** |
| 101.LAB | XBRL Label Linkbase Document** |
| 101.PRE | XBRL Presentation Linkbase Document** |
* Management contract or compensatory plan or arrangement
** Pursuant to Rule 406T of Regulation S-T, these interactive data files are not deemed filed or part of a registration statement or prospectus for purposes of Section 11 or 12 of the Securities Act or Section 18 of the Securities Exchange Act and otherwise not subject to liability.
| 50 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| OCATA THERAPEUTICS, INC. | ||
| Dated: March 16, 2015 | By: | /s/ Paul Wotton |
| Paul Wotton | ||
| President and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| OCATA THERAPEUTICS, INC. | ||
| Dated: March 16, 2015 | By: | /s/ Edward Myles |
| Edward Myles | ||
| Chief Operating Officer and Chief Financial Officer | ||
| (Principal Financial Officer and Principal Accounting Officer) | ||
We, the undersigned officers and directors of Ocata Therapeutics, Inc., hereby severally constitute and appoint Paul Wotton and Edward Myles, and each of them singly, our true and lawful attorneys, with full power to them and each of them singly, to sign for us in our names in the capacities indicated below, all amendments to this report, and generally to do all things in our names and on our behalf in such capacities to enable Ocata Therapeutics, Inc. to comply with the provisions of the Securities Exchange Act of 1934, as amended, and all requirements of the Securities and Exchange Commission.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| /s/ Paul Wotton | March 16, 2015 | |
| Paul Wotton | ||
| President and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| /s/ Edward Myles | March 16, 2015 | |
| Edward Myles | ||
| Chief Operating Officer and Chief Financial Officer | ||
| (Principal Financial Officer and Principal Accounting Officer) | ||
| /s/ Robert Langer | March 16, 2015 | |
| Robert Langer | ||
| Director | ||
| /s/ Alan Shapiro | March 16, 2015 | |
| Alan Shapiro | ||
| Director | ||
| /s/ Gregory D. Perry | March 16, 2015 | |
| Gregory D. Perry | ||
| Director | ||
| /s/ Zohar Loshitzer | March 16, 2015 | |
| Zohar Loshitzer | ||
| Director | ||
| /s/ Michael Heffernan | March 16, 2015 | |
| Michael Heffernan | ||
| Director and Chairman of the Board |
| 51 |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Ocata Therapeutics, Inc. and Subsidiary
Marlborough, Massachusetts
We have audited the accompanying consolidated balance sheet of Ocata Therapeutics, Inc. and Subsidiary (the “Company”) as of December 31, 2014, and the related consolidated statements of operations, stockholders' deficit, and cash flows for the year then ended. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Ocata Therapeutics, Inc. and Subsidiary as of December 31, 2014, and the results of their operations and their cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As described in Note 2 to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Ocata Therapeutics, Inc. and Subsidiary’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated March 16, 2015 expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
Boston, Massachusetts
March 16, 2015
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Ocata Therapeutics, Inc. and subsidiary
We have audited the accompanying consolidated balance sheet of Ocata Therapeutics, Inc. and subsidiary, formerly Advanced Cell
Technology, Inc. (collectively, the “Company”) as of December 31, 2013, and the related consolidated statements of
operations, stockholders' deficit and cash flows for each of the two years in the period ended December 31, 2013. These financial
statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements
based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management,
as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our
opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial
position of the Company as of December 31, 2013, and the results of their operations and their cash flows for each of the two years
in the period ended December 31, 2013, in conformity with U.S. generally accepted accounting principles.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern.
/s/ SingerLewak LLP
Los Angeles, California
April 1, 2014, except for the reclassification paragraph in Note 2 as to which the date is March 16, 2015
| F-2 |
OCATA THERAPETUICS, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
AS OF DECEMBER 31, 2014 AND 2013
| December 31, | December 31, | |||||||
| 2014 | 2013 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | 4,424,374 | $ | 1,743,485 | ||||
| Other receivable | 16,217 | 209,198 | ||||||
| Prepaid expenses and other current assets | 308,444 | 896,741 | ||||||
| Total current assets | 4,749,035 | 2,849,424 | ||||||
| Property and equipment, net | 832,963 | 753,576 | ||||||
| Deferred royalty fees | 107,779 | 170,215 | ||||||
| Other assets | 47,707 | 68,801 | ||||||
| Deferred costs | – | 65,903 | ||||||
| TOTAL ASSETS | $ | 5,737,484 | $ | 3,907,919 | ||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable | $ | 1,271,325 | $ | 2,285,331 | ||||
| Accrued expenses | 2,515,674 | 3,545,713 | ||||||
| Accrued settlement | 1,731,202 | 4,086,619 | ||||||
| Senior secured convertible promissory notes, current portion, net of discount of $225,324 at December 31, 2013 | – | 2,174,676 | ||||||
| Embedded conversion option liabilities, current portion | – | 335,208 | ||||||
| Unsettled warrant obligation | – | 3,899,391 | ||||||
| Loss contingency accrual | – | 6,431,979 | ||||||
| Deferred revenue, current portion | 157,873 | 157,872 | ||||||
| Total current liabilities | 5,676,074 | 22,916,789 | ||||||
| Senior secured convertible promissory notes, less current portion, net of discount of $37,553 at December 31, 2013 | – | 1,162,447 | ||||||
| Embedded conversion option liabilities, less current portion | – | 327,792 | ||||||
| Warrant and option derivative liabilities | 16,255 | 284,799 | ||||||
| Other liabilities | 1,188,874 | – | ||||||
| Deferred revenue, less current portion | 1,591,826 | 1,749,702 | ||||||
| Total liabilities | 8,473,029 | 26,441,529 | ||||||
| Commitments and contingencies (Note 14) | ||||||||
| STOCKHOLDERS' DEFICIT: | ||||||||
| Preferred stock, Series B; $0.001 par value; 50,000,000 shares authorized, 0 and 1,000 shares issued and outstanding at December 31, 2014 and 2013, respectively | – | 1 | ||||||
| Preferred stock, Series C; $0.001 par value; 50,000,000 shares authorized, 0 and 1,750 shares issued and outstanding at December 31, 2014 and 2013, respectively | – | 2 | ||||||
| Common stock, $0.001 par value; 60,000,000 shares authorized, 34,620,218 and 26,402,650 shares issued and outstanding at December 31, 2014 and 2013, respectively | 34,620 | 26,403 | ||||||
| Additional paid-in capital | 346,364,060 | 325,297,736 | ||||||
| Promissory notes receivable, net of discount of $2,018,321 at December 31, 2013 | – | (34,013,395 | ) | |||||
| Accumulated deficit | (349,134,225 | ) | (313,844,357 | ) | ||||
| Total stockholders' deficit | (2,735,545 | ) | (22,533,610 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS' DEFICIT | $ | 5,737,484 | $ | 3,907,919 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
OCATA THERAPEUTICS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2014, 2013 AND 2012
| 2014 | 2013 | 2012 | ||||||||||
| Revenue (License fees and royalties) | $ | 157,875 | $ | 224,985 | $ | 466,487 | ||||||
| Cost of Revenue | 62,436 | 82,436 | 117,436 | |||||||||
| Gross profit | 95,439 | 142,549 | 349,051 | |||||||||
| Operating expenses: | ||||||||||||
| Research and development | 10,529,321 | 11,564,768 | 14,158,936 | |||||||||
| General and administrative | 11,052,123 | 12,057,067 | 11,432,866 | |||||||||
| Litigation settlement contingency | – | 6,228,621 | – | |||||||||
| Loss on settlement of litigation | 13,468,547 | – | – | |||||||||
| Total operating expenses | 35,049,991 | 29,850,456 | 25,591,802 | |||||||||
| Loss from operations | (34,954,552 | ) | (29,707,907 | ) | (25,242,751 | ) | ||||||
| Non-operating income (expense): | ||||||||||||
| Interest income | 55,840 | 165,918 | 15,581 | |||||||||
| Interest expense | (429,573 | ) | (1,437,584 | ) | (1,104,602 | ) | ||||||
| Other loss | (172,656 | ) | (867,065 | ) | (10,515,470 | ) | ||||||
| Adjustment to fair value of unsettled warrant obligation | 18,959 | (107,438 | ) | 1,390,382 | ||||||||
| Loss on disposal of fixed assets | (10,922 | ) | – | (17,138 | ) | |||||||
| Gain on extinguishment of debt | – | 438,587 | – | |||||||||
| Adjustments to fair value of derivatives | 743,959 | 493,241 | 889,883 | |||||||||
| Total non-operating income (expense) | 205,607 | (1,314,341 | ) | (9,341,364 | ) | |||||||
| Loss before provision for income tax | (34,748,945 | ) | (31,022,248 | ) | (34,584,115 | ) | ||||||
| Provision for income tax | – | – | – | |||||||||
| Net loss | $ | (34,748,945 | ) | $ | (31,022,248 | ) | $ | (34,584,115 | ) | |||
| Preferred stock dividend | 1,889,192 | 2,364,947 | 2,048,007 | |||||||||
| Net loss applicable to common stock | $ | (36,638,137 | ) | $ | (33,387,195 | ) | $ | (36,632,122 | ) | |||
| Weighted average shares outstanding : | ||||||||||||
| Basic and diluted | 31,270,460 | 24,918,727 | 20,866,197 | |||||||||
| Net loss applicable to common share: | ||||||||||||
| Basic and diluted | $ | (1.17 | ) | $ | (1.34 | ) | $ | (1.76 | ) | |||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
OCATA THERAPEUTICS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' DEFICIT
FOR THE YEARS ENDED DECEMBER 31, 2014, 2013 AND 2012
| Series B Preferred Stock | Series C Preferred Stock | Common Stock | Additional Paid-in | Promissory
Notes Receivables, | Accumulated | Total Stockholders' | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | net | Deficit | Deficit | |||||||||||||||||||||||||||||||
| Balance – December 31, 2011 | 1,000 | $ | 1 | 1,150 | $ | 1 | 17,435,693 | $ | 17,436 | $ | 223,091,744 | $ | (23,381,185 | ) | $ | (248,284,059 | ) | $ | (48,556,062 | ) | ||||||||||||||||||||
| Shares issued for settlements | – | – | – | – | 3,306,910 | 3,307 | 38,423,706 | – | – | 38,427,013 | ||||||||||||||||||||||||||||||
| Shares issued for services | – | – | – | – | 140,779 | 141 | 1,816,489 | – | – | 1,816,630 | ||||||||||||||||||||||||||||||
| Accrued dividends on Series B and C Preferred Stock | – | – | – | – | – | – | 2,048,007 | – | (2,048,007 | ) | – | |||||||||||||||||||||||||||||
| Accretion of note receivable discount on Series B and C Preferred Stock | – | – | – | – | – | – | – | (2,068,320 | ) | 2,068,320 | – | |||||||||||||||||||||||||||||
| Stock based compensation | – | – | – | – | – | – | 7,795,885 | – | – | 7,795,885 | ||||||||||||||||||||||||||||||
| Issuance of Series C preferred stock | – | – | 600 | 1 | – | – | 5,999,999 | – | – | 6,000,000 | ||||||||||||||||||||||||||||||
| Issuance of Common Stock to Series C Preferred Stock holder for note receivable | – | – | – | – | 738,172 | 738 | 5,143,588 | (5,144,326 | ) | – | – | |||||||||||||||||||||||||||||
| Common stock issued upon exercise of Series C Preferred Stock warrants and issuance of note receivable | – | – | – | – | 147,634 | 148 | 1,028,717 | (1,028,865 | ) | – | – | |||||||||||||||||||||||||||||
| Issuance of 558,020 shares of common stock in financing arrangement (net of issuance costs of $700,000) | – | – | – | – | 558,020 | 557 | 2,855,995 | – | – | 2,856,552 | ||||||||||||||||||||||||||||||
| Stock option reclass from liability to equity | – | – | – | – | – | – | 3,099,883 | – | – | 3,099,883 | ||||||||||||||||||||||||||||||
| Net loss for year ended December 31, 2012(1) | – | – | – | – | – | – | – | – | (34,584,115 | ) | (34,584,115 | ) | ||||||||||||||||||||||||||||
| Balance December 31, 2012 | 1,000 | $ | 1 | 1,750 | $ | 2 | 22,327,208 | $ | 22,327 | $ | 291,304,013 | $ | (31,622,696 | ) | $ | (282,847,861 | ) | $ | (23,144,214 | ) | ||||||||||||||||||||
| Shares issued for settlements | – | – | – | – | 1,121,646 | 1,122 | 6,298,878 | – | – | 6,300,000 | ||||||||||||||||||||||||||||||
| Shares issued for services | – | – | – | – | 145,968 | 146 | 1,762,207 | – | – | 1,762,353 | ||||||||||||||||||||||||||||||
| Accrued dividends on Series B and C Preferred Stock | – | – | – | – | – | – | 2,364,947 | – | (2,364,947 | ) | – | |||||||||||||||||||||||||||||
| Accretion of note receivable discount on Series B and C Preferred Stock | – | – | – | – | – | – | – | (2,390,699 | ) | 2,390,699 | – | |||||||||||||||||||||||||||||
| Stock based compensation | – | – | – | – | – | – | 3,880,058 | – | – | 3,880,058 | ||||||||||||||||||||||||||||||
| Redemption of Series A Preferred Stock | – | – | – | – | 275,228 | 275 | 1,912,562 | – | – | 1,912,837 | ||||||||||||||||||||||||||||||
| Issuance of 2,532,600 shares of common stock in financing arrangement | – | – | – | – | 2,532,600 | 2,533 | 17,775,071 | – | – | 17,777,604 | ||||||||||||||||||||||||||||||
| Net loss for the year ended December 31, 2013 | – | – | – | – | – | – | – | – | (31,022,248 | ) | (31,022,248 | ) | ||||||||||||||||||||||||||||
| Balance December 31, 2013 | 1,000 | $ | 1 | 1,750 | $ | 2 | 26,402,650 | $ | 26,403 | $ | 325,297,736 | $ | (34,013,395 | ) | $ | (313,844,357 | ) | $ | (22,533,610 | ) | ||||||||||||||||||||
| Shares issued for settlements | – | – | – | – | 4,273,737 | 4,274 | 25,973,326 | – | – | 25,977,600 | ||||||||||||||||||||||||||||||
| Shares issued for services | – | – | – | – | 34,729 | 34 | 243,950 | – | – | 243,984 | ||||||||||||||||||||||||||||||
| Accrued dividends on Series B and C Preferred Stock | – | – | – | – | – | – | 1,442,082 | – | (1,442,082 | ) | – | |||||||||||||||||||||||||||||
| Accretion of note receivable discount on Series B and C Preferred Stock | – | – | – | – | – | – | (1,348,269 | ) | 1,348,269 | – | ||||||||||||||||||||||||||||||
| Redemption of Series B and C Preferred Stock | (1,000 | ) | (1 | ) | (1,750 | ) | (2 | ) | – | – | (34,984,680 | ) | 35,361,664 | (447,110 | ) | (70,129 | ) | |||||||||||||||||||||||
| Stock based compensation | – | – | – | – | – | – | 2,546,126 | – | – | 2,546,126 | ||||||||||||||||||||||||||||||
| Issuance of 3,903,060 shares of common stock in financing arrangement (net of issuance costs of $698,926) | – | – | – | – | 3,903,060 | 3,903 | 25,657,941 | – | – | 25,661,844 | ||||||||||||||||||||||||||||||
| Adjustment for 1/100 reverse stock split | – | – | – | – | 6,042 | 6 | (6 | ) | – | – | – | |||||||||||||||||||||||||||||
| Stock option reclass from liability to equity | – | – | – | – | – | – | 187,585 | – | – | 187,585 | ||||||||||||||||||||||||||||||
| Net loss for the year ended December 31, 2014 | – | – | – | – | – | – | – | – | (34,748,945 | ) | (34,748,945 | ) | ||||||||||||||||||||||||||||
| Balance December 31, 2014 | – | $ | – | – | $ | – | 34,620,218 | $ | 34,620 | $ | 346,364,060 | $ | – | $ | (349,134,225 | ) | $ | (2,735,545 | ) | |||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
OCATA THERAPETUICS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2014, 2013 AND 2012
| 2014 | 2013 | 2012 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Net loss | $ | (34,748,945 | ) | $ | (31,022,248 | ) | $ | (34,584,115 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation | 173,065 | 93,507 | 58,637 | |||||||||
| Amortization of deferred charges | 62,436 | 112,436 | 117,436 | |||||||||
| Amortization of deferred revenue | (157,875 | ) | (224,935 | ) | (466,487 | ) | ||||||
| Stock based compensation | 2,546,126 | 3,880,058 | 7,795,885 | |||||||||
| Amortization of deferred issuance costs | 65,903 | 502,555 | 807,989 | |||||||||
| Amortization of discounts on senior secured convertible debentures | 262,877 | 502,022 | 161,379 | |||||||||
| Changes in the fair value of warrant obligation | (18,959 | ) | 107,438 | (1,390,382 | ) | |||||||
| Changes in the fair value of derivatives | (743,959 | ) | (493,241 | ) | (889,883 | ) | ||||||
| Shares of common stock issued for services | 243,984 | 1,762,353 | 1,816,630 | |||||||||
| Non-cash financing (gain) loss | (203,358 | ) | 6,133,459 | 7,015,470 | ||||||||
| Loss on settlement of litigation | 13,468,547 | – | – | |||||||||
| Gain on extinguishment of debt | – | (438,587 | ) | – | ||||||||
| Loss on disposal of fixed assets | 10,923 | – | 17,138 | |||||||||
| Warrant and options issued for consulting services | – | 41,460 | 60,388 | |||||||||
| Other | – | 43,873 | 135,235 | |||||||||
| Changes in operating assets and liabilities | ||||||||||||
| Grants receivable | – | (112,773 | ) | (96,425 | ) | |||||||
| Prepaid expenses and other current assets | 802,372 | (794,697 | ) | 34,204 | ||||||||
| Accounts payable and other current liabilities | (3,210,588 | ) | (2,057,879 | ) | 4,800,544 | |||||||
| Net cash used in operating activities | (21,447,451 | ) | (21,965,199 | ) | (14,606,357 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||||||
| Purchases of property and equipment | (263,375 | ) | (671,827 | ) | (96,260 | ) | ||||||
| Payment of lease deposits | – | (38,945 | ) | (15,090 | ) | |||||||
| Net cash used in investing activities | (263,375 | ) | (710,772 | ) | (111,350 | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Principal repayment of senior secured convertible debentures | (1,200,000 | ) | (600,000 | ) | – | |||||||
| Proceeds from issuance of preferred stock | – | – | 6,000,000 | |||||||||
| Net proceeds from issuance of common stock | 25,661,844 | 17,777,604 | 2,941,102 | |||||||||
| Costs associated with issuance of common stock | – | – | (84,550 | ) | ||||||||
| Redemption of Series B Preferred Stock, net | (70,129 | ) | – | – | ||||||||
| Net cash provided by financing activities | 24,391,715 | 17,177,604 | 8,856,552 | |||||||||
| NET DECREASE IN CASH AND CASH EQUIVALENTS | 2,680,889 | (5,498,367 | ) | (5,861,155 | ) | |||||||
| CASH AND CASH EQUIVALENTS, BEGINNING BALANCE | 1,743,485 | 7,241,852 | 13,103,007 | |||||||||
| CASH AND CASH EQUIVALENTS, ENDING BALANCE | $ | 4,424,374 | $ | 1,743,485 | $ | 7,241,852 | ||||||
| CASH PAID FOR: | ||||||||||||
| Interest | $ | 100,793 | $ | 389,133 | $ | – | ||||||
| Income taxes | $ | – | $ | – | $ | – | ||||||
| SUPPLEMENTAL DISCLOSURE OF NON-CASH FINANCING ACTIVITIES: | ||||||||||||
| Issuance of note receivable on issuance of shares and exercise of warrants for 0, 0, and 885,807 shares of common stock | $ | – | $ | – | $ | 7,200,000 | ||||||
| Record note receivable discount related to Series C preferred stock | $ | – | $ | – | $ | (1,026,809 | ) | |||||
| Accrued dividends on Series B and C Preferred Stock | $ | 1,442,082 | $ | 2,364,947 | $ | 2,048,007 | ||||||
| Accretion of note receivable discount on Series B and C Preferred Stock | $ | 1,348,270 | $ | 2,390,699 | $ | 2,068,320 | ||||||
| Issuance of 4,273,737, 1,121,646, and 3,306,910 shares of common stock for accrued settlement | $ | 25,977,600 | $ | 6,300,000 | $ | 38,427,013 | ||||||
| Issuance of 106,008, 0, and 87,500 shares of common stock as commitment financing fee | $ | 698,926 | $ | – | $ | 700,000 | ||||||
| Conversion of Series A Preferred stock for 275,229 shares of common stock | $ | – | $ | 1,912,837 | $ | – | ||||||
| Issuance of senior secured convertible promissory notes for settlement | $ | – | $ | – | $ | 6,000,000 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
OCATA THERAPEUTICS, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. ORGANIZATIONAL MATTERS
Organization and Nature of Business
Ocata Therapeutics, Inc., and Subsidiary (formerly Advanced Cell Technology, and collectively, the “Company”) is a biotechnology company incorporated in the state of Delaware, are focused on the development of novel cell-based therapies. The Company’s therapeutic area of focus is ophthalmology and the Company’s most advanced products are in clinical trials for the treatment of dry age-related macular degeneration, Stargardt’s macular degeneration and myopic macular degeneration. The Company is also developing several pre-clinical cell therapies for the treatment of other ocular disorders. Additionally, the Company also has a number of pre-clinical stage assets in disease areas outside the field of ophthalmology, including autoimmune, inflammatory and wound healing-related disorders. The Company’s intellectual property portfolio includes pluripotent human embryonic stem cell, or hESC; induced pluripotent stem cell, or iPSC, platforms; and other cell therapy research programs. The corporate headquarters and principal laboratory and manufacturing facilities are located in Marlborough, Massachusetts.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Management’s Plans — The Company follows accounting standards set by the Financial Accounting Standards Board, (“FASB”). The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, GAAP. References to GAAP issued by the FASB in these footnotes are to the FASB Accounting Standards Codification,™ sometimes referred to as the Codification or ASC.
The accompanying consolidated financial statements have been prepared in conformity with GAAP which contemplate continuation of the Company as a going concern. However, as of December 31, 2014, the Company has an accumulated deficit of $349.1 million, recurring losses from operations, and negative working capital which raise substantial doubt about the ability of the Company to continue as a going concern. The ability to continue as a going concern is dependent upon many factors, including the Company’s ability to raise additional capital in a timely manner. On a long-term basis, we have no expectation of generating any meaningful revenues from our product candidates for a substantial period of time and must rely on raising funds in capital transactions to finance our research and development programs. Our future cash requirements will depend on many factors, including the pace and scope of our research and development programs, the costs involved in filing, prosecuting and enforcing patents, and other costs associated with commercializing our potential products. Accordingly, management’s plans to continue as a going concern contemplate raising additional capital including the execution of an agreement for a $30 million equity line in late June 2014, of which approximately $18.6 million remains available as of December 31, 2014. There can be no assurances that management can raise the necessary additional capital on favorable terms or at all. The accompanying financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
On August 28, 2014, the Company effected a 100-to-1 reverse stock split of its common stock. Unless otherwise noted, all references in these financial statements to number of shares, price per share and weighted average number of shares outstanding of common stock have been adjusted to reflect the reverse stock split on a retroactive basis. The split was also applied to any outstanding equity-based awards.
Principles of Consolidation — The accounts of the Company and its wholly-owned subsidiary Mytogen, Inc. are included in the accompanying consolidated financial statements. All intercompany balances and transactions were eliminated in consolidation.
Segment Reporting — ASC 280, Segment Reporting requires use of the “management approach” model for segment reporting. The management approach model is based on the way a company’s management organizes segments within the company for making operating decisions and assessing performance. The Company determined it has one operating segment.
Use of Estimates — These consolidated financial statements have been prepared in accordance with GAAP and, accordingly, require management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Specifically, the Company’s management has estimated loss contingencies related to outstanding litigation. In addition, management has estimated variables used to calculate the Black-Scholes option pricing model used to value derivative instruments and the Company estimates the fair value of the embedded conversion option associated with the senior secured convertible debentures using a binomial lattice model as discussed below under “Fair Value Measurements”. Also, management has estimated the expected economic life and value of the Company’s licensed technology, the Company’s net operating loss for tax purposes, share-based payments for compensation to employees, directors, consultants and investment banks, and the useful lives of the Company’s fixed assets. Actual results could differ from those estimates.
Reclassifications — Certain prior period financial statement balances have been reclassified to conform to the current period presentation. Items include the reclassification of deferred costs from current assets to long-term assets and the presentation of net loss applicable to common stock and net loss applicable to common share on the face of the statement of operations.
On August 28, 2014, the Company effected a 100-to-1 reverse stock split of its common stock. As such, the Company determined that current year presentation of net loss applicable to common stock and net loss applicable to common share was appropriate. The Company retroactively adjusted the presentation for net loss applicable to common stock and net loss applicable to common share for prior periods on the statement of operations to conform to the current year’s presentation.
Cash and Cash Equivalents — Cash equivalents are comprised of certain highly liquid investments with maturities of three months or less when purchased. The Company maintains its cash in bank deposit accounts, which at times, may exceed federally insured limits. The Company has not experienced any losses related to this concentration of risk. As of December 31, 2014 and December 31, 2013, the Company had deposits in excess of federally-insured limits totaling $4,325,886 and $1,668,232, respectively.
| F-7 |
Commitments and Contingencies — The Company is subject to various claims and contingencies related to lawsuits as well as commitments under contractual and other obligations. The Company recognizes liabilities for contingencies and commitments when a loss is probable and can be reasonably estimated.
Relating to loss contingencies the Company accrues the best estimate of a loss within a range. If no estimate in a range is better than any other, the minimum amount is accrued. The Company discloses a reasonably possible loss in excess of the amount accrued, if applicable. For reasonably possible loss contingencies, the Company discloses the nature of the loss contingency and provides a range of the estimate of possible loss or state that an estimate cannot be made.
Included in the accounts payable balance as of December 31, 2013 is approximately $1,200,000 primarily related to the acquisition of Mytogen which the Company expected to settle during 2014. This balance was reclassified to the other liabilities balance as of December 31, 2014 as the Company does not expect to settle within the coming year.
Grant Received — From time to time the Company participates in research grants both as an initiator of grants as well as a sub-recipient of grant funds. The Company incurs costs for the grant and is subsequently reimbursed for these expenses by grant receipts. The Company records such receipts as a reduction in research and development costs. For the years ended December 31, 2014, 2013 and 2012 , the Company recorded as a reduction in research and development costs, $0, $160,054, and $320,112, respectively.
Grants Receivable — The Company periodically assesses its grants receivable for collectability on a specific identification basis. If collectability of an account becomes unlikely, the Company records an allowance for that doubtful account. Once the Company has exhausted efforts to collect, management writes off the grants receivable against the allowance it has already created.
Property and Equipment — The Company records its property and equipment at historical cost. The Company expenses maintenance and repairs as incurred. Upon disposition of property and equipment, the gross cost and accumulated depreciation are written off and the difference between the proceeds and the net book value is recorded as a gain or loss on sale of assets. In the case of certain assets acquired under capital leases, the assets are recorded net of imputed interest, based upon the net present value of future payments. Assets under capital lease are pledged as collateral for the related lease.
The Company provides for depreciation over the assets’ estimated useful lives as follows:
| Machinery & equipment | 4 years | |
| Computer equipment | 3 years | |
| Office furniture | 4 years | |
| Leasehold improvements | Lesser of remaining lease term or economic life |
Patents — The Company follows ASC 350-30, General Intangibles Other than Goodwill, (“ASC 350-30”) in accounting for its patents. ASC 350-30 provides that costs of internally developing, maintaining, or restoring intangible assets that are not specifically identifiable, that have indeterminate lives, or that are inherent in a continuing business and related to an entity as a whole, shall be recognized as an expense when incurred. The Company has expensed as research and development expense all costs associated with developing its patents.
Equity Method Investment — The Company follows ASC 323, Investments-Equity Method and Joint Ventures, in accounting for its investment in the joint venture. In the event the Company’s share of the joint venture’s net losses reduces the Company’s investment to zero, the Company will discontinue applying the equity method and will not provide for additional losses unless the Company has guaranteed obligations of the joint venture or is otherwise committed to provide further financial support for the joint venture. If the joint venture subsequently reports net income, the Company will resume applying the equity method only after its share of that net income equals the share of net losses not recognized during the period the equity method was suspended.
Deferred Costs — Consist of the following:
(a) Payments, either in cash or share-based, made in connection with the sale of debentures which are amortized using the effective interest method over the lives of the related debentures. These deferred issuance costs are charged to interest expense when and if the related debt instrument is retired or converted early. The weighted average amortization period for deferred debt issuance costs is 48 months.
(b) Payments made to secure commitments under certain financing arrangements. These amounts are recognized in interest expense ratably over the period of the financing arrangements, and are recognized in financing costs immediately if the arrangement is cancelled, forfeited or the utility of the arrangement to the company is otherwise compromised.
(c) Payments made to financial institutions in order to provide financing related services. These costs are being amortized to interest expense over the terms of the related agreements.
| F-8 |
Long-Lived Assets — The Company follows ASC 360-10, Property, Plant, and Equipment, which established a “primary asset” approach to determine the cash flow estimation period for a group of assets and liabilities that represents the unit of accounting for a long-lived asset to be held and used. Long-lived assets to be held and used are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The carrying amount of a long-lived asset is not recoverable if it exceeds the sum of the undiscounted cash flows expected to result from the use and eventual disposition of the asset. Long-lived assets to be disposed of are reported at the lower of carrying amount or fair value less cost to sell. Through December 31, 2014, the Company had not experienced impairment losses on its long-lived assets.
Fair Value Measurements — The Company applies the provisions of ASC 820-10, Fair Value Measurements and Disclosures. ASC 820-10 defines fair value, and establishes a three-level valuation hierarchy for disclosures of fair value measurement that enhances disclosure requirements for fair value measures. For certain financial instruments, including cash and cash equivalents, grants receivable, prepaid expenses, accounts payable and accrued expenses, the carrying amounts approximate fair value due to their relatively short maturities. The carrying amount of senior secured convertible debentures approximates fair value as the interest rate charged on the debentures is based on the prevailing rate. The three levels of valuation hierarchy are defined as follows:
| · | Level 1 inputs to the valuation methodology are quoted prices for identical assets or liabilities in active markets. |
| · | Level 2 inputs to the valuation methodology include quoted prices for similar assets and liabilities in active markets, and inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the financial instrument. |
| · | Level 3 inputs to the valuation methodology are unobservable and significant to the fair value measurement. |
The Company analyzes all financial instruments with features of both liabilities and equity under ASC 480, Distinguishing Liabilities From Equity, and ASC 815, Derivatives and Hedging. Derivative liabilities are adjusted to reflect fair value at each period end, with any increase or decrease in the fair value being recorded in results of operations as adjustments to fair value of derivatives. The effects of interactions between embedded derivatives are calculated and accounted for in arriving at the overall fair value of the financial instruments. In addition, the fair values of freestanding derivative instruments such as warrant and option derivatives are valued using the Black-Scholes model.
The Company uses Level 2 inputs for its valuation methodology for certain warrant derivative liabilities. The Company’s derivative liabilities are adjusted to reflect fair value at each period end, with any increase or decrease in the fair value being recorded in results of operations as adjustments to fair value of derivatives.
The Company uses Level 3 inputs for its valuation methodology for the fair value of certain embedded conversion options and warrant and option derivative liabilities.
The Company estimates the fair value of the embedded conversion option associated with its 8% convertible debentures using a binomial lattice, which estimates and compares the present value of the principal and interest payments to the as converted value to determine whether the holder of the notes should convert the notes into the Company’s common stock or continue to receive principal and interest payments. The Company uses this methodology to determine the beneficial conversion features because there are no observable inputs available with respect to the fair value.
The binomial lattice relies on the following Level 3 inputs: (1) expected volatility of the Company’s common stock; (2) potential discount for illiquidity of large blocks of the Company’s common stock, and (3) discount rate for contractual debt principal and interest payments. The fair value of the embedded beneficial conversion feature is estimated as the difference between the fair value of the notes with and without the conversion feature. The fair value of the notes without the conversion feature is determined using one Level 3 input, the discount rate for contractual debt interest and principal payments.
| · | The expected volatility of the Company’s common stock is estimated from the historical volatility of daily returns in the Company’s common stock price. The Company monitors the volatility of its common stock on a quarterly basis to observe trends that may impact the fair value of the notes. |
| · | The discount for illiquidity is measured using an average-strike option that calculates the discount as the opportunity cost for not being able to sell a large block of the Company’s common stock immediately at prevailing observable market prices. Inputs to the average-strike option model include the expected volatility of the Company’s common stock and time to sell a large block of the Company’s stock as Level 3 inputs and other observable inputs. The time to sell the stock is estimated considering the historical daily trading volume of our common stock and market maker estimates of the amount of shares that can be offered for sale above the normal the daily trading volume without depressing the price of the Company’s common stock. |
| F-9 |
At December 31, 2014, the Company identified the following assets and liabilities that are required to be presented on the balance sheet at fair value:
| Fair Value | Fair Value Measurements at | |||||||||||||||
| As of | December 31, 2014 | |||||||||||||||
| Description | December 31, 2014 | Using Fair Value Hierarchy | ||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
| Warrant and option liabilities | $ | 16,255 | $ | – | $ | – | $ | 16,255 | ||||||||
| Total | $ | 16,255 | $ | – | $ | – | $ | 16,255 | ||||||||
At December 31, 2013, the Company identified the following assets and liabilities that are required to be presented on the balance sheet at fair value:
| Fair Value | Fair Value Measurements at | |||||||||||||||
| As of | December 31, 2013 | |||||||||||||||
| Description | December 31, 2013 | Using Fair Value Hierarchy | ||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
| Warrant and option derivative liabilities | $ | 284,799 | $ | – | $ | – | $ | 284,799 | ||||||||
| Embedded conversion option liabilities | 663,000 | – | – | 663,000 | ||||||||||||
| Unsettled warrant obligation | 3,899,391 | 3,899,391 | – | – | ||||||||||||
| Total | $ | 4,847,190 | $ | 3,899,391 | $ | – | $ | 947,799 | ||||||||
The following tables reconcile the change in fair value for measurements categorized within Level 3 of the fair value hierarchy:
| Embedded | ||||
| Conversion | ||||
| Option | ||||
| Liabilities | ||||
| Balance at December 31, 2013 | $ | 663,000 | ||
| Total (gains) or losses for the period included in earnings | (663,000 | ) | ||
| Balance at December 31, 2014 | $ | – | ||
| Warrant and Option Derivative Liabilities | ||||
| Balance at December 31, 2013 | $ | 284,799 | ||
| Reclass of options from liability to equity | (187,585 | ) | ||
| Total (gains) or losses for the period included in earnings | (80,959 | ) | ||
| Balance at December 31, 2014 | $ | 16,255 | ||
Gains and losses included in earnings for the twelve months ended December 31, 2014 are reported as follows:
| Embedded | ||||
| Conversion | ||||
| Option Liabilities | ||||
| Total gain included in earnings | $ | 663,000 | ||
| Warrant and Option Derivative Liabilities | ||||
| Total gain included in earnings | $ | 80,959 | ||
| F-10 |
The following table provides quantitative information about measurements categorized within Level 3 of the fair value hierarchy:
| Fair Value at | ||||||||
| December 31, | Valuation | |||||||
| Description | 2014 | Technique | Unobservable Input | Value | ||||
| Warrant and Option derivative liabilities | $16,255 | Black Scholes Model | Expected volatility of the Company's common stock | 70% - 75% |
For the years ended December 31, 2014, 2013 and 2012 the Company recognized a gain of $743,959, $493,241, and $889,883, respectively, for the changes in the valuation of derivative liabilities.
The Company did not identify any non-recurring assets and liabilities that were recorded at fair value during the periods presented.
Revenue Recognition and Deferred Revenue — The Company’s revenues are primarily generated from license and research agreements with collaborators. Licensing revenue is recognized on a straight-line basis over the shorter of the life of the license or the estimated economic life of the patents related to the license.
License fee revenue begins to be recognized in the first full month following the effective date of the license agreement. Deferred revenue represents the portion of the license and other payments received that has not been earned. Costs associated with license revenue are deferred and recognized over the same term as the revenue. Reimbursements of research expense pursuant to grants are recorded in the period during which collection of the reimbursement becomes assured, because the reimbursements are subject to approval.
In some cases, the Company is entitled to receive royalty payments from licensees. In such cases, the Company recognizes the royalties when they are earned and collectability of those royalty payments is reasonably assured.
In connection with its license agreements, the Company recorded $157,875, $224,985, and $466,487 in license fee revenue for the years ended December 31, 2014, 2013 and 2012, respectively, in its consolidated statements of operations. The remainder of the license fees are reflected in deferred revenue at December 31, 2014 and December 31, 2013, respectively.
Research and Development Costs — Costs incurred in connection with research and development activities are expensed as incurred. Research and development expenses consist of (i) employee-related expenses, including salaries, benefits, travel and stock compensation expense; (ii) external research and development expenses incurred under arrangements with third parties, such as contract research organizations, investigational sites and consultants; (iii) the cost of acquiring, developing and manufacturing clinical study materials; (iv) facilities and other expenses, which include direct and allocated expenses for rent and maintenance of facilities and laboratory and other supplies; and (v) costs associated with pre-clinical and clinical activities and regulatory operations.
The Company enters into consulting, research and other agreements with commercial firms, researchers, universities and others for the provision of goods and services. Under such agreements, the Company may pay for services on a monthly, quarterly, project or other basis. Such arrangements are generally cancellable upon reasonable notice and payment of costs incurred. Costs are considered incurred based on an evaluation of the progress to completion of specific tasks under each contract using information and data provided to us by the Company’s clinical sites and vendors. These costs consist of direct and indirect costs associated with specific projects, as well as fees paid to various entities that perform certain research on behalf of the Company. Reimbursements of research expense pursuant to grants are recorded in the period during which collection of the reimbursement becomes assured, because the reimbursements are subject to approval.
In certain circumstances, the Company is required to make advance payments to vendors for goods or services that will be received in the future for use in research and development activities. In such circumstances, the advance payments are deferred and are expensed when the activity has been performed or when the goods have been received.
Share Compensation — The Company records stock compensation in accordance with ASC 718, Compensation – Stock Compensation. ASC 718 requires companies to measure compensation cost for stock employee compensation at fair value at the grant date and recognize the expense over the employee’s requisite service period. The Company recognizes in the statement of operations the grant-date fair value of stock options and other equity-based compensation issued to employees and non-employees.
Income Taxes — Deferred income taxes are provided using the liability method whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carryforwards, and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of the changes in tax laws and rates at the date of enactment.
| F-11 |
When tax returns are filed, it is highly certain that some positions taken would be sustained upon examination by the taxing authorities, while others are subject to uncertainty about the merits of the position taken or the amount of the position that would be ultimately sustained. The benefit of a tax position is recognized in the financial statements in the period during which, based on the weight of available evidence, management believes it is more likely than not that the position will be sustained upon examination, including the resolution of appeals or litigation processes, if any. Tax positions taken are not offset or aggregated with other positions. Tax positions that meet the more-likely-than-not recognition threshold are measured as the largest amount of tax benefit that is more than 50 percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefits associated with tax positions taken that exceeds the amount measured as described above is reflected as a liability for unrecognized tax benefits in the balance sheets along with any associated interest and penalties that would be payable to the taxing authorities upon examination.
Applicable interest and penalties associated with unrecognized tax benefits are classified as additional income taxes in the statements of operations.
Net Loss Per Share — Basic net loss per share applicable to common stockholders is calculated by dividing net loss applicable to common stockholders by the weighted average shares outstanding during the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by adjusting weighted average shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock method and the if-converted method. For purposes of the diluted net loss per share calculation, convertible preferred stock, stock options, unvested restricted stock, and warrants are considered to be common stock equivalents but have been excluded from the calculation of diluted net loss per share, as their effect would be anti-dilutive for all periods presented. Therefore, basic and diluted net loss per share was the same for all periods presented. Additionally, dividends associated with the extinguished preferred stock has been added back in order to calculate the net loss applicable to common stockholders.
At December 31, 2014, 2013 and 2012, approximately 2,784,287, 1,975,323, and 2,359,890 potentially dilutive shares, respectively, were excluded from the shares used to calculate diluted earnings per share as their inclusion would be anti-dilutive.
Recent Accounting Pronouncements
In July 2013, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update No. 2013-11, Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists (ASU 2013-11). ASU 2013-11 clarifies guidance and eliminates diversity in practice on the presentation of unrecognized tax benefits when a net operating loss carryforward, a similar tax loss, or a tax credit carryforward exists at the reporting date. This new guidance is effective on a prospective basis for fiscal years and interim reporting periods within those years, beginning after December 15, 2013. The adoption of ASU 2013-11 did not have a material impact on consolidated results of operations, financial condition, or liquidity.
In May 2014, the Financial Accounting Standards Board (FASB) issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606), which supersedes all existing revenue recognition requirements, including most industry-specific guidance. The new standard requires a company to recognize revenue when it transfers goods or services to customers in an amount that reflects the consideration that the company expects to receive for those goods or services. The new standard will be effective for the Company on January 1, 2017. The Company is currently evaluating the potential impact that Topic 606 may have on its financial position and results of operations and its method of adoption.
In August 2014, the Financial Accounting Standards Board (FASB) issued ASU No. 2014-15, Presentation of Financial Statement-Going Concern (Subtopic 205-40) –Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The new standard requires a company to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern or to provide related footnote disclosures. The new standard will be effective for the Company on January 1, 2017. The Company is currently evaluating the potential impact that the new standard may have on its financial statements and related disclosures.
3. INVESTMENT IN JOINT VENTURE
On December 1, 2008, the Company and CHA Bio & Diostech Co., Ltd. (“CHA”), formed an international joint venture. The new company, Stem Cell & Regenerative Medicine International, Inc. (“SCRMI”), will work towards developing human blood cells and other clinical therapies based on the Company’s hemangioblast program. Under the terms of the agreement, the Company purchased upfront a 33% interest in the joint venture, and has received another 7% interest upon fulfilling certain obligations under the agreement over a period of 3 years. The Company’s contribution includes (a) the uninterrupted use of a portion of its leased facility at the Company’s expense, (b) the uninterrupted use of certain equipment in the leased facility, and (c) the release of certain of the Company’s research and science personnel to be employed by the joint venture. In return, for a 60% interest, CHA has agreed to contribute $150,000 cash and to fund all operational costs in order to conduct the hemangioblast program. Effective May 1, 2010, the Company was no longer obligated to provide laboratory space to SCRMI. As of December 31, 2014, the Company holds a 40% interest in the joint venture and CHA owns a 60% interest. The two partners to the joint venture are in negotiations on further funding of the joint venture, but there can be no assurances that an agreement will be reached. Any financial statement impact at this time is unclear should an agreement not be reached.
| F-12 |
The Company has agreed to collaborate with the joint venture in securing grants to further research and development of its technology. Additionally, SCRMI has agreed to pay the Company a fee of $500,000 for an exclusive, worldwide license to the hemangioblast program. The Company recorded $29,412, $29,412 and $29,412 in license fee revenue for the years ended December 31, 2014, 2013 and 2012, respectively, in its accompanying consolidated statements of operations, and the balance of unamortized license fee of $322,304 and $351,715, of which $29,412 and $29,412 is current, is included in deferred revenue in the accompanying consolidated balance sheets at December 31, 2014 and 2013, respectively.
On July 15, 2011, the Company and CHA entered into a binding term sheet, with the expectation of entering into a future definitive agreement, in which the joint venture was realigned around both product development rights and research responsibilities. Under the terms of the binding term sheet, SCRMI exclusively licensed the rights to the hemangioblast program to the Company for United States and Canada and expanded the jurisdictional scope of the license to CHA to include Japan (in addition to South Korea, which was already exclusively licensed to CHA). As part of the agreement, the scientists at SCRMI involved in the hemangioblast program were transferred to the Company, and SCRMI discontinued its research activity and became solely a licensing entity. The Company fulfilled its obligation to meet a minimal research spending requirement of $6.75 million by July 31, 2014 in order to maintain its exclusive license, up to the point of filing an investigational new drug for a therapeutic product. Intellectual property rights created by the Company in the course of its research are subject to a non-exclusive license to CHA for Japan and South Korea, and to SCRMI to be sub-licensable under certain circumstances for countries other than the United States, Canada, Japan and South Korea. Pursuant to the agreement, the Company paid $820,000 to SCRMI which is recorded as “losses attributable to equity method investments.” By filing the investigational new animal drug application on September 12, 2013 with the Federal Drug Administration, the Company has met the commitment required to maintain its exclusive license.
4. PROPERTY AND EQUIPMENT
Property and equipment consisted of the following at December 31, 2014 and 2013:
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Machinery & equipment (1) | $ | 1,186,232 | $ | 1,086,800 | ||||
| Computer equipment | 104,857 | 49,707 | ||||||
| Office furniture | 54,226 | 38,783 | ||||||
| Leasehold improvements (1) | 620,252 | 559,969 | ||||||
| 1,965,567 | 1,735,259 | |||||||
| Accumulated depreciation | (1,132,604 | ) | (981,683 | ) | ||||
| Property and equipment, net | $ | 832,963 | $ | 753,576 | ||||
| (1) | The 2013 balances includes approximately $125,000 in machinery & equipment and $339,000 in leasehold improvements that were not yet placed in service at December 31, 2013 and therefore had not started being depreciated as of that date. |
Depreciation expense for the years ended December 31, 2014, 2013 and 2012 was $173,065, $93,507, and $58,637, respectively.
5. ACCRUED SETTLEMENT
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| SEC Civil Action | $ | 1,356,202 | $ | 4,086,619 | ||||
| SEC Section 16 Investigation | 375,000 | – | ||||||
| Total | $ | 1,731,202 | $ | 4,086,619 | ||||
| F-13 |
Securities and Exchange Commission – Civil Action
In May 2012, the Company was named as a defendant in a civil action brought by the Securities and Exchange Commission (the “SEC”) related to transactions involving the sale and issuance of the Company’s securities. The Securities and Exchange Commission alleges that Company violated Section 5(a) and 5(c) of the Securities Act of 1933 (the “Securities Act”) because certain sales of shares to outside organizations, completed in late 2008 and early 2009 under the Company’s former management, resulted in $3.5 million in proceeds to the Company, were neither registered under the Securities Act nor subject to an exemption from registration under Section 3(a)(10) of the Securities Act, as amended. In addition, the Company is alleged to have violated Section 13(a) of the Exchange Act of 1934 because the Company did not disclose the sale and issuance of the shares to the SEC on a timely basis.
In December 2013, the Company settled the civil action. Under the terms of the settlement accepted by the SEC, the Company consented to entry of judgment under which it neither admits nor denies liability and agreed to the disgorgement of $3.5 million in proceeds from the transactions in question. In addition, the Company will pay approximately $587,000 in pre-judgment interest. The total amount due, approximately $4.1 million, will be paid over six equal quarterly installments. In addition, the settlement permanently restrains and enjoins the Company from violations of Sections 5(a) and 5(c) of the Securities Act, Section 13(a) of the Exchange Act and Rule 13a-11 under the Exchange Act.
SEC Section 16 Investigation
In April 2013, it was determined that Gary Rabin, the Company’s former Chief Executive Officer, failed to report 27 transactions in a timely manner on Form 4 under Section 16 of the Exchange Act in which Mr. Rabin sold shares of the Company’s common stock that took place between February 7, 2011 and January 10, 2013. The SEC then investigated the unreported transactions involving sales of shares of the Company’s common stock. In September 2014, the Company settled the SEC action arising from the SEC’s investigation. Under the terms of the settlement accepted by the SEC, the Company consented to the entry of order under which it neither admits nor denies liability and has agreed to pay a civil penalty of $375,000, which has been previously accrued for, by July 2015. In addition, the settlement requires the Company to engage an independent Section 16 compliance consultant, provide Section 16(a) training to each Section 16(a) reporting person, and provide a certification of compliance that each of the preceding requirements were completed. The settlement also requires the Company to cease and desist from committing or causing any violations and any future violations of Section 17(a)(2) of the Securities Act, Sections 13(a) and 14(a) of the Exchange Act, and Rules 12b-20, 13a-1, and 14a-9 thereunder. The terms of this settlement require the Company to allocate financial and management resources to complying with the settlement’s terms, which may have adverse effect on our business. Also, if the SEC deems the Company to not have complied with any portion of the settlement, it may issue additional fines or sanctions against us which may limit our ability to issue securities or otherwise conduct our business as currently conducted.
6. LOSS CONTINGENCY ACCRUAL
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Warrant holder litigation | $ | – | $ | 6,228,621 | ||||
| Miscellaneous settlements | – | 203,358 | ||||||
| Total | $ | – | $ | 6,431,979 | ||||
Warrant holder litigation
On June 4, 2014, the Company entered into a settlement agreement (the “June 2014 Agreement”) with each of Gary D. Aronson (“Aronson”), John S. Gorton, individually and as trustee of the John S. Gorton Separate Property Trust dated March 3, 1993 (“Gorton”), herronlaw apc, attorneys for Aronson (“Herron”), Miller and Steele LLP, attorneys for Gorton (“Miller/Steele”) and Michael A. Bourke, attorney for both Aronson and Gorton (“Bourke”). The June 2014 Agreement relates to previously disclosed lawsuits filed against the Company by each of Aronson and Gorton in August 2011 in the United States District Court for the District of Massachusetts claiming that the Company breached an anti-dilution provision contained in warrants held by each of Aronson and Gorton as a result of certain transactions between the Company and other third-party investors.
Pursuant to the June 2014 Agreement, in exchange for dismissal of the lawsuit by the warrant holders, the Company issued 3,840,000 shares of its common stock. On the date of the execution of the June 2014 Agreement, the shares were valued at $6.14 per share for a total value of $23,577,600. As a result the Company recorded a loss on settlement of litigation of $13,468,547 for the year-ended December 31, 2014.
At December 31, 2013, the Company had determined that a loss was probable and the amount of loss was reasonably estimable, based on the facts and circumstances surrounding the litigation with Aronson and Gorton during the last quarter of 2013. The loss contingency represented the estimated number of shares to settle above a determined share amount necessary to settle the warrant share obligation plus an additional amount for potential interest charges. Refer to footnote 8 for further details of the liabilities recorded in 2013 related to this matter.
Miscellaneous settlements
The Company was not able to reach settlement agreements on potential litigation with all of holders of convertible promissory notes and warrants that were issued between 2005 and 2010. As of December 31, 2014, the Company believes the probability of a future settlement to be remote and therefore has removed the loss contingency related to these settlements.
| F-14 |
7. CONVERTIBLE PROMISSORY NOTES
2010 JMJ Convertible Promissory Notes
During 2010, the Company issued three convertible promissory notes to JMJ Financial, for a total of $3,000,000 available to receive in cash, for a principal sum of $3,850,000, which included an original issue discount of $850,000. The notes bear a one-time interest charge of 10% on the principal sum. The holder may at its election convert all or part of these notes into shares of the Company's common stock at the conversion rate of the lesser of: (a) $0.10 per share, or (b) 85% of the average of the three lowest trade prices in the 20 trading days prior to the conversion. During 2010, the Company received the entire $3,000,000 on these notes. Of the $3,850,000 borrowed, the Company converted $3,562,215 into 76,465,706 shares of common stock during 2010.
On May 31, 2013, the Company and JMJ entered into a Mutual Release and Waiver Agreement (“Waiver Agreement”) whereby JMJ released and discharged the Company from any and all claims connected with the JMJ convertible promissory notes. At the date of the Waiver Agreement, the convertible promissory note balance was $287,785 and the conversion option liability associated with the convertible promissory note had a fair value of $150,802. The value was determined using the Black-Scholes model with the following assumptions: (1) dividend yield of 0%; (2) expected volatility of 80%, (3) risk-free interest rate of 0.04%, and (4) expected life of 0.001 years. The Company recorded a gain on extinguishment of debt of $438,587 during the year ended December 31, 2013.
As of both December 31, 2014 and December 31, 2013 the outstanding balance of the JMJ Convertible Promissory Notes was $0.
The fair value of the embedded conversion option was $0 for both December 31, 2014 and 2013, respectively. The decrease in the fair value of this liability was $0, $28,134 and $104,878 during the years ended December 31, 2014, 2013 and 2012, respectively, which was recorded through the statements of operations as an adjustment to fair value of derivatives.
Interest expense from amortization of debt discounts related to the JMJ Convertible Promissory Notes for the years ended December 31, 2014, 2013 and 2012 was $0, $30,935 and $127,207, respectively.
CAMOFI Senior Secured Convertible Debentures
On January 11, 2013, the Company entered into a settlement agreement and mutual release (“the Settlement Agreement”) with CAMOFI Master LDC (“CAMOFI”) and CAMHZN Master LDC (“CAMHZN” and together with CAMOFI, the “CAMOFI Parties”), relating to the lawsuit between the CAMOFI Parties, as plaintiffs, and the Company, as defendant, in the Supreme Court of New York. Pursuant to the Settlement Agreement the Company issued Debentures in the principal amount of $4,732,781 and $1,267,219 to CAMOFI and CAMHZN, respectively (together the “Debentures”).
The Debentures had an effective date of December 31, 2012, accrue interest at the rate of 8% per annum and mature on June 30, 2015. The Company may pre-pay all or a portion of the amounts due under the Debentures prior to maturity without penalty. Both of the Debentures are convertible at the option of the holder at a price per share of common stock equal to 80% of the volume weighted average price (“VWAP”) of the ten consecutive trading days prior to the conversion date. The Company must make quarterly payments under the Debentures on the last day of each calendar quarter commencing on March 31, 2013 in the amount of $600,000. The quarterly payments may, at the option of the Company and subject to the satisfaction of certain conditions, be paid in shares of Common Stock. In such case, the conversion price for such payment will be based on the lesser of (i) the conversion price as defined in the agreement or (ii) 80% of the average of the 10 closing prices immediately prior to the date the quarterly payment is due. To secure its obligations under the Debentures, the Company granted a security interest in substantially all of the Company’s assets, including its intellectual property, to the CAMOFI Parties. The Debentures contain certain covenants customary for debt instruments of their kind.
On April 29, 2014, the Company received an Acceleration Notice from the CAMOFI Parties of a declaration that the aggregate principal amount remaining under the Debentures subject to adjustment as set forth therein, together with any other amounts owed under the Debentures were immediately due and payable in accordance with their terms. The Acceleration Notice followed the delivery of a notice to the Company on April 15, 2014 stating that, due to the Company’s failure to deliver shares of common stock issuable to the CAMOFI Parties within three days of a conversion event, an “Event of Default” had occurred and the CAMOFI Parties were reserving all rights held by them arising from such Event of Default. At the time of the conversion event, the Company determined not to deliver the shares to the CAMOFI Parties in order to comply with applicable securities laws. The Company later delivered the shares to the CAMOFI Parties in compliance with applicable securities laws, prior to the delivery of the Default Notice.
| F-15 |
On May 2, 2014, the Company paid to the CAMOFI Parties an aggregate amount of approximately $1,616,000 calculated in accordance with the payment acceleration provisions of the Debentures and satisfying the Company’s obligations under the Debentures. The payment represented the remaining $1,200,000 in principal amount due and an additional amount of approximately $416,000, which represented penalties, interest and legal cost reimbursement to the CAMOFI Parties. With the payment, the Company satisfied in full its obligations under the Debentures and the terms of the Settlement Agreement and Mutual Release dated as of December 31, 2012 pursuant to which the Debentures were issued in January 2013. The Company received correspondence from the CAMOFI Parties stating that the CAMOFI Parties believe the aggregate amount due to be different than the amount the Company paid. The Company believes that the Company’s interpretation of the Debentures is accurate and complete.
The Debentures had contained an embedded beneficial conversion feature as the Debentures are convertible at a price per share of common stock equal to 80% of VWAP of the ten consecutive trading days prior to the conversion date. The embedded beneficial conversion feature was modeled using a binomial lattice model, and had a calculated value at December 31, 2013 of $663,000. The Company recorded a gain of $663,000 for the change in the fair value of the embedded conversion option liability for the year-ended December 31, 2014 as the derivative was recorded at $0 at December 31, 2014 with the retirement of the remaining debentures.
The Company recorded a debt discount of $725,000, which was to be amortized over the life of the Debentures using the effective interest rate of 22.9%. The unamortized discount balance of $108,229 was written off to interest expense with the retirement of the remaining outstanding Debentures.
8. Unsettled Warrant Obligation
The Company determined that it had an unsettled warrant obligation related to two warrant agreements entered into in 2005. The warrant agreements contained “full ratchet” anti-dilution provisions which the Company determined led to a contractual obligation, which became fixed on January 15, 2009, to issue approximately 63.2 million common shares on a pre-reverse stock split basis. The Company further determined that those common shares represent a liability which should be recorded at fair value at each accounting period with changes to that fair value being recorded in earnings. Fair value is based on the share obligation multiplied by the stock price at the end of each reporting period, with a liability “floor” established, at $0.06 per share on a pre-reverse stock split basis, based on the stock price at the time the anti-dilution provision was triggered. At December 31, 2013, the liability had been recorded at $3,899,391.
In June 2014, the warrant holder litigation with Aronson and Gorton was settled as the Company entered into a settlement agreement with the parties. The Company conducted a final valuation of the 63.2 million shares, on a pre-reverse stock split basis, of unsettled warrant obligation on the day of settlement, June 4, 2014. For the year-ended December 31, 2014, the Company recorded a gain of $18,959 through earnings related to the matter.
9. SERIES B PREFERRED STOCK
On November 2, 2009 (“Effective Date”), the Company entered into a preferred stock purchase agreement with Optimus Life Sciences Capital Partners, LLC (“Optimus”). Pursuant to the purchase agreement, the Company agreed to sell, and Optimus agreed to purchase, in one or more purchases from time to time at the Company’s sole discretion (each, a “Series B Tranche”), (i) up to 1,000 shares of Series B preferred stock at a purchase price of $10,000 per share, for an aggregate purchase price of up to $10,000,000, and (ii) five-year warrants to purchase shares of the Company’s common stock with an aggregate exercise price equal to 135% of the purchase price paid by the Investor.
Dividends
Commencing on the date of the issuance of any shares of Series B preferred stock, Holders of Series B preferred stock will be entitled to receive dividends on each outstanding share of Series B preferred stock, which will accrue in shares of Series B preferred stock at a rate equal to 10% per annum from the issuance date compounded annually. Accrued dividends will be payable upon redemption of the Series B preferred stock. Accrued dividends were $0 and $3,587,748 at December 31, 2014 and 2013, respectively.
Related Secured Promissory Notes Receivable:
In accordance with the terms of the Series B preferred stock agreement, Optimus issued to the Company a secured promissory note in consideration for receiving and exercising warrants under each tranche. The value of each secured promissory note equals the value of the warrants that Optimus received. Interest on the notes accrues at 2% per year, compounding annually if the interest remains unpaid at the end of each year. The note is secured by freely tradable marketable securities belonging to Optimus. Each promissory note matures on the fourth anniversary of its issuance.
In the event the Company redeems all or a portion of any shares of Series B preferred stock held by Optimus, the Company will be permitted to offset the full amount of such proceeds against amounts outstanding under the promissory notes. Accordingly, the Company included the discounted value of the secured promissory notes as a separate component of stockholders’ deficit at December 31, 2013.
| F-16 |
June 2014 Redemption
In June 2014 the Company redeemed 250 shares of the Series B preferred stock, all of which were upon or after the fourth anniversary of their initial issuance date, representing three separate tranches. As part of the redemption, the promissory notes receivable balances for these tranches were netted against the accrued dividends payable, with the net dividend payable balance of $70,129 being paid out in cash and a charge being recorded to stockholders equity.
August 2014 Redemption
On August 7, 2014, the Company redeemed the remaining 750 shares of its Series B Preferred Stock and the associated outstanding dividends payable from Optimus in exchange for (i) cancellation of the secured promissory notes and associated accrued interest of $10,874,257 issued to the Company by Optimus and (ii) $25,000 cash. The difference on the redemption of $112,978 was recorded as a credit to accumulated deficit.
During the years ended December 31, 2014, 2013 and 2012 the Company accreted interest on the promissory notes in the amount of $578,543 $1,233,050 and $1,120,623 respectively, which was recorded in accumulated deficit during the periods then ended. The Company recorded dividends on its Series B preferred stock during the years ended December 31, 2013, 2012 and 2011 of $735,508, $1,235,427 and $1,122,783 respectively. The accrued dividends are offset by the accretion of the note receivable discount.
Following this repurchase, we have zero shares of Series B Preferred Stock outstanding as of December 31, 2014. As of December 31, 2013, 1,000 shares of Series B preferred stock were outstanding.
10. SERIES C PREFERRED STOCK
On December 30, 2010 (the “Series C Effective Date”), the Company entered into a securities purchase agreement (the “Series C Purchase Agreement”) with Socius CG II, Ltd., a Bermuda exempted company (“Socius”). Pursuant to the Series C Purchase Agreement, the Company agreed to sell, and Socius agreed to purchase, in one or more purchases from time to time (each such purchase, a “Series C Tranche”) in the Company’s sole discretion (subject to the conditions set forth therein), (i) up to 2,500 shares of Series C preferred stock at a purchase price of $10,000 per share, for an aggregate purchase price of up to $25,000,000, and (ii) a two-year warrant (the “Socius Warrant”) obligating Socius to purchase shares of the Company’s common stock with an aggregate exercise price equal to 20% of the purchase price paid by Socius for the Series C preferred stock sold in each Series C Tranche, at an exercise price per share equal to the closing bid price of the Company’s common stock on the date the Company provides notice of such Series C Tranche (the “Series C Tranche Notice”).
As of December 31, 2013, the Company had drawn $17,500,000 of the $25,000,000 commitment.
Dividends
Commencing on the date of the issuance of any shares of Series C preferred stock, holders of Series C preferred stock will be entitled to receive dividends on each outstanding share of Series C preferred stock, which will accrue in shares of Series C preferred stock at a rate equal to 6% per annum from the issuance date compounded annually. Accrued dividends will be payable upon redemption of the Series C preferred stock. Accrued dividends were $0 and $2,454,853 at December 31, 2014 and 2013, respectively.
Related Secured Promissory Notes Receivable:
Interest on the notes accrues at 2% per year, compounding annually if the interest remains unpaid at the end of each year. The notes are secured by freely tradable marketable securities belonging to Socius. Each promissory note matures on the fourth anniversary of its issuance.
In the event the Company redeems all or a portion of any shares of Series C preferred stock held by Socius, the Company will be permitted to offset the full amount of such proceeds against amounts outstanding under the promissory notes. Accordingly, the Company included the discounted value of the secured promissory notes as a separate component of stockholders’ deficit at December 31, 2013.
At December 31, 2014 and 2013, the value of the secured promissory notes in the consolidated balance sheet was $0 and $20,451,788, net of discounts of $1,363,762 and accrued interest of $815,549, reflecting a face value of $21,000,000.
The Company determined that a 6% discount is appropriate, in order to consistently reflect the Company’s cost of borrowing under the terms of the underlying Series C preferred stock that permits offset.
| F-17 |
August 2014 Redemption
On August 7, 2014, the Company redeemed the 1,750 shares of its Series C Preferred Stock and the associated outstanding dividends payable from Socius in exchange for (i) cancellation of the secured promissory notes and associated accrued interest of $22,186,899, issued to the Company by Socius and (ii) $25,000 cash. Following this repurchase, the Company has zero shares of Series C Preferred Stock outstanding. The difference on the redemption of $560,088 was recorded as a debit to accumulated deficit.
During the years ended December 31, 2014, 2013, and 2012, the Company accreted interest on the promissory note in the amount of $769,726, $1,157,649, and $947,696, respectively, which was recorded in accumulated deficit during the periods then ended. The Company recorded dividends on its Series C preferred stock during the years ended December 31, 2014, 2013, and 2012 of $706,574, $1,129,520, and $925,222, respectively. The accrued dividends are offset by the accretion of the note receivable discount.
The Company has classified the Series C preferred stock in the equity section in its consolidated balance sheets. As of December 31, 2014 and December 31, 2013, 0 and 1,750 shares of Series C preferred stock were outstanding, respectively.
11. WARRANT SUMMARY
Warrant Activity
A summary of warrant activity for the year ended December 31, 2014 is as follows:
| Weighted | ||||||||||||||||
| Weighted | Average | Aggregate | ||||||||||||||
| Average | Remaining | Intrinsic | ||||||||||||||
| Number of | Exercise | Contractual | Value | |||||||||||||
| Warrants | Price $ | Life (in years) | (000) $ | |||||||||||||
| Outstanding, December 31, 2013 | 78,299 | 28.00 | 1.82 | – | ||||||||||||
| Granted | – | – | – | – | ||||||||||||
| Exercised | – | – | – | – | ||||||||||||
| Forfeited/Canceled | (32,393 | ) | 10.00 | – | – | |||||||||||
| Outstanding, December 31, 2014 | 45,906 | 40.46 | 1.45 | – | ||||||||||||
| Exercisable, December 31, 2014 | 45,906 | 40.46 | 1.45 | – | ||||||||||||
The aggregate intrinsic value in the table above is before applicable income taxes and is calculated based on the difference between the exercise price of the warrants and the quoted price of the Company’s common stock as of the reporting date.
12. STOCKHOLDERS’ DEFICIT TRANSACTIONS
On September 19, 2012 the Company entered into a purchase agreement with Lincoln Park Capital, LLC (“Lincoln Park”). Pursuant to the purchase agreement, the Company had the right to sell to Lincoln Park up to $35,000,000 in shares of its common stock. On June 18, 2014, the Company completed the last sale of common stock to Lincoln Park under this agreement.
On June 27, 2014, the Company entered into a similar purchase agreement with Lincoln Park pursuant to which the Company has the right to sell to Lincoln Park up to $30,000,000 in shares of its common stock, subject to certain limitations set forth in the purchase agreement.
On June 27, 2014, the Company and Lincoln Park also entered into a registration rights agreement, pursuant to which the Company is required to file a registration statement with the SEC to register the resale of the shares of common stock issued and issuable to Lincoln Park on a best efforts basis, pursuant to the purchase agreement.
Upon the satisfaction of the conditions set forth in the purchase agreement, including the registration statement being declared effective by the SEC, the Company has the right over a 36-month period commencing in June 2014 to sell up to $30,000,000 million worth of shares of its Common Stock to Lincoln Park, upon the terms set forth in the purchase agreement. Pursuant to the purchase agreement, the purchase price of such common stock will be based on the prevailing market price of the Company’s common stock immediately preceding the time of sales, with the Company controlling the timing and amount of any future sales, if any, of common stock to Lincoln Park. There are no upper limits to the price Lincoln Park may pay to purchase the Company’s common stock. Lincoln Park shall not have the right or the obligation to purchase any shares of common stock on any business day that the closing price of the Company’s common stock is below a floor price as provided in the purchase agreement. The purchase price means, with respect to any regular purchase, the lower of: (i) the lowest sale price on the applicable purchase date and (ii) the arithmetic average of the three (3) lowest closing sale prices for the common stock during the ten (10) consecutive business days ending on the business day immediately preceding such purchase date (in each case, to be appropriately adjusted for any reorganization, recapitalization, non-cash dividend, stock split or other similar transaction that occurs on or after the execution of the purchase agreement).Due to the 100-to-1 reverse stock split of the Company’s common stock effected in August 2014, as of December 31, 2014, the purchase price cannot be below $1.00.
The purchase agreement contains customary representations, warranties, covenants, closing conditions and indemnification and termination provisions by, among and for the benefit of the parties. Lincoln Park has covenanted not to cause or engage in any manner whatsoever, any direct or indirect short selling or hedging of the Company’s common stock. The purchase agreement may be terminated by the Company at any time at its discretion without any cost to the Company.
| F-18 |
In consideration for entering into the purchase agreement, the Company issued to Lincoln Park 106,008 shares of its common stock as a commitment fee. At the time of issuance, the Company’s common stock had a fair value of $6.59 per share, for a total commitment fee of $698,926. The shares related to the commitment fee have been issued in reliance on an exemption from registration under the Securities Act of 1933, as amended pursuant to Section 4(a)(2) thereof and Regulation D promulgated thereunder, and will be registered for resale on the registration statement that the Company must file pursuant to the purchase agreement and the registration rights agreement. The shares related to the commitment fee are fully earned as of the date of the agreement. There were no other considerations given to Lincoln Park for entering into this agreement with the Company.
During the twelve months ended December 31, 2014, Lincoln Park purchased 3,903,059 shares of common stock for cash proceeds of $25,661,844. 2,269,750 shares of common stock were sold to Lincoln Park pursuant to the 2012 Lincoln Park purchase agreement, for total proceeds of $14,281,295. 1,633,309 shares (including 106,008 shares issued as a commitment fee) of common stock were sold to Lincoln Park pursuant to the 2014 purchase agreement for total proceeds of $11,380,549.
During the twelve months ended December 31, 2014, the Company issued an aggregate of 4,273,737 shares in settlements of disputes, including 3,840,000 shares in settlement of the Aronson-Gorton warrant holder litigation valued at $23,577,600 and 433,737 shares in settlement of $2,400,000 Debentures to the CAMOFI Parties as required by the Settlement Agreement.
During the twelve months ended December 31, 2014, the Company issued various board members 34,729 shares of common stock valued at $243,984 as compensation for board services.
On August 28, 2014, the Company completed a 100-to-1 reverse stock split of its common stock. Unless otherwise noted, all references in these financial statements to number of shares, price per share and weighted average number of shares outstanding of common stock have been adjusted to reflect the reverse stock split on a retroactive basis. The split was also applied to any outstanding equity-based awards.
At the 2014 Annual Meeting, the Company’s stockholders approved a Certificates of Amendment to the Company’s Certificate of Incorporation. The Certificate of Amendment provided for an increase in the number of authorized shares of the Company’s common stock, par value $0.001 per share, from 37,500,000 to 60,000,000. The Certificate of Amendment became effective upon its filing with the Secretary of State of the State of Delaware on November 12, 2014.
13. STOCK COMPENSATION
The Company determined that in certain periods in 2009 and again in Q4 2011 it did not have the required amount of authorized, unissued common shares to deliver to option holders under its stock plans. As a result, under ASC 718, Compensation - Stock Compensation, certain equity balances were adjusted and accounted for as liabilities and marked-to-market until shareholder approval for additional shares to cover outstanding stock options occurred. In 2011 the lack of required authorized, unissued shares occurred on November 2, 2011 and shareholder approval for necessary shares to cover the outstanding option pool took place on January 24, 2012. As a result at that time in 2012 the stock options were considered equity classified share compensation.
At the 2014 Annual Meeting, which was held on November 12, 2014, the Company’s stockholders approved the Company’s 2014 Stock Option and Incentive Plan (the “2014 Stock Option Plan”). The 2014 Stock Option Plan previously had been approved, subject to stockholder approval, by the Company’s Board of Directors. The Company’s executive officers and directors are eligible to receive awards under the 2014 Stock Option Plan, including stock options and restricted stock units, in accordance with the terms and conditions of the 2014 Stock Option Plan.
Stock Plans
| Options/Shares | ||||||||||||||||
| Options/Shares | Options | Available | Total | |||||||||||||
| Stock Plan | Issued | Outstanding | For Grant | Authorized | ||||||||||||
| 2005 Stock Plan | 2,756,356 | 2,170,381 | 3,180,007 | 5,818,388 | ||||||||||||
| 2014 Stock Plan | 100,000 | 75,000 | 150,000 | 250,000 | ||||||||||||
| 2,856,356 | 2,245,381 | 3,330,007 | 6,068,388 | |||||||||||||
| F-19 |
Stock Option Activity
A summary of option activity for the year ended December 31, 2014:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | Aggregate | ||||||||||||||
| Number of | Exercise | Contractual | Intrinsic | |||||||||||||
| Options | Price | Life (in years) | Value | |||||||||||||
| Outstanding, December 31, 2013 | 1,182,795 | $ | 19.22 | 6.95 | $ | 10,319 | ||||||||||
| Granted | 1,190,250 | 7.80 | ||||||||||||||
| Exercised | – | – | ||||||||||||||
| Forfeited/canceled | (127,664 | ) | 39.06 | |||||||||||||
| Outstanding, December 31, 2014 | 2,245,381 | $ | 12.03 | 8.12 | $ | 12,110 | ||||||||||
| Vested and expected to vest at December 31, 2014 | 2,099,889 | $ | 12.33 | 8.02 | $ | 11,586 | ||||||||||
| Exercisable, December 31, 2014 | 1,126,215 | $ | 16.28 | 6.70 | $ | 8,081 | ||||||||||
The aggregate intrinsic value in the table above is before applicable income taxes and is calculated based on the difference between the exercise price of the options and the quoted price of the Company’s common stock as of the reporting date.
The assumptions used in calculating the fair value of options granted using the Black-Scholes option- pricing model for options granted during the three years ended December 31, 2014 are as follows:
| December 31, | December 31, | December 31, | |||||||
| 2014 | 2013 | 2012 | |||||||
| Risk-free interest rate | 0.02% – 2.29% | 0.66% – 2.17% | 0.66% – .92% | ||||||
| Expected life of the options | 5.00 – 6.25 years | 3.90 - 6.02 years | 3.90 – 5.45 years | ||||||
| Expected volatility | 112% – 148% | 134% –158% | 149% – 158% | ||||||
| Expected dividend yield | 0% | 0% | 0% | ||||||
The weighted average grant-date fair value for the options granted during the years ended December 31, 2014, 2013 and 2012 was $7.49, $7.00, and $9.00, respectively.
The compensation expense related to the unvested options as of December 31, 2014, was $8,276,199 which will be recognized over the weighted average vesting period of 2.44 years.
Restricted Stock Units Activity
A summary of the restricted stock unit activity for the year ended December 31, 2014:
| Number of Shares Underlying Restricted Units |
Weighted Average Grant-Date Fair Value per Share |
Weighted
Average Remaining Recognition Period (in years) |
||||||||||
| Outstanding, December 31, 2013 | – | $ | – | – | ||||||||
| Granted | 493,000 | 7.28 | ||||||||||
| Outstanding, December 31, 2014 | 493,000 | 7.28 | 2.6 | |||||||||
| Unearned stock-based compensation expense of outstanding restricted units | $ | 3,196,969 | ||||||||||
Stock compensation expense to employees and non-employees for the years ended December 31, 2014, 2013 and 2012 are as follows:
| December 31, | December 31, | December 31, | |||||||||
| 2014 | 2013 | 2012 | |||||||||
| R&D | $ | 903,835 | $ | 1,392,926 | $ | 3,792,394 | |||||
| G&A | 1,642,291 | 2,487,133 | 4,003,491 | ||||||||
| $ | 2,546,126 | $ | 3,880,059 | $ | 7,795,885 | ||||||
| F-20 |
14. COMMITMENTS AND CONTINGENCIES
Employment Contracts
The Company has entered into employment contracts with certain executives and research personnel. The contracts provide for salaries, bonuses and stock option grants, along with other employee benefits.
Leases
On January 29, 2010, the Company signed a lease to move from its Worcester facility to a new 10,607 square-foot facility in Marlborough, Massachusetts. The lease term is from April 1, 2010 through June 30, 2015. The Company amended the lease effective March 1, 2011 adding an additional 1,650 square feet. On January 11, 2013 the Company added an additional 17,696 square feet at its Marlborough location with a lease term from January 2013 through March 2018.
During 2011, the Company renewed its site in Los Angeles, California through February 28, 2013 with a monthly base rent of $2,170. In November 2012, the Company entered into a new lease agreement that became effective July 1, 2013 and terminates on June 30, 2018. Beginning in March 2015, the Company has sub-leased their Santa Monica office to a third party. Any future sub-lease income is deemed to be immaterial.
Annual minimum lease payments are as follows:
| 2015 | $ | 433,104 | ||
| 2016 | 349,803 | |||
| 2017 | 356,659 | |||
| 2018 | 110,927 | |||
| $ | 1,250,493 |
Rent expense recorded in the financial statements for the years ended December 31, 2014, 2013 and 2012 was approximately $513,820, $419,347, and $187,000 respectively.
15. INCOME TAXES
The items accounting for the difference between income taxes computed at the federal statutory rate and the provision (benefit) for income taxes were as follows:
| 2014 | 2013 | 2012 | ||||||||||
| Statutory federal income tax rate | (34 | )% | (34 | )% | (34 | )% | ||||||
| State income taxes, net of federal taxes | (1 | )% | (6 | )% | (6 | )% | ||||||
| Loss on settlement of litigation | 13 | % | 0 | % | 0 | % | ||||||
| Provision to return differences | (3 | )% | 0 | % | 0 | % | ||||||
| Non-includable items | 1 | % | 17 | % | 11 | % | ||||||
| Increase in valuation allowance | 24 | % | 23 | % | 29 | % | ||||||
| Effective income tax rate | – | – | – | |||||||||
Deferred tax assets consist of the following at December 31, 2014 and 2013:
| 2014 | 2013 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 54,573,453 | $ | 54,916,539 | ||||
| Depreciation | 57,083 | 155,651 | ||||||
| Capitalized R&D expenses | 9,435,513 | 4,142,634 | ||||||
| Deferred revenue | 687,282 | (167,791 | ) | |||||
| Stock compensation | 3,131,186 | – | ||||||
| Accrued expenses | 475,894 | 47,558 | ||||||
| Reversal of unpaid liabilities | 466,990 | 1,238,538 | ||||||
| Other | 21,000 | 332,304 | ||||||
| Valuation allowance | (68,848,401 | ) | (60,665,433 | ) | ||||
| Net deferred tax asset | $ | – | $ | – | ||||
The Company files income tax returns in the U.S. federal jurisdiction, and various state jurisdictions. With few exceptions, the Company is no longer subject to U.S. federal, state and local income tax examinations by tax authorities for years before 2005.
| F-21 |
At December 31, 2014, the Company had federal and state net operating loss carry forwards available to offset future taxable income of approximately $148 million and $79 million respectively. These carry forwards will begin to expire in the years ending December 31, 2020 and December 31, 2015, respectively. Utilization of the net operating loss carryforwards may be subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986 due to ownership changes that have occurred previously or that could occur in the future. These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50% over a three-year period. The Company has not conducted a study to assess whether a change of control has occurred or whether there have been multiple changes of control since inception due to the significant complexity and cost associated with such a study. If the Company has experienced a change of control, as defined by Section 382, at any time since inception, utilization of the net operating loss carryforwards would be subject to an annual limitation under Section 382, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term tax-exempt rate, and then could be subject to additional adjustments, as required. Any limitation may result in expiration of a portion of the net operating loss carryforwards before utilization. Net operating loss carryforwards may be reduced to limitations imposed by Section 382 and due to exit of activity in certain states.
The Company periodically evaluates the likelihood of the realization of deferred tax assets, and adjusts the carrying amount of the deferred tax assets by the valuation allowance to the extent the future realization of the deferred tax assets is not judged to be more likely than not. The Company considers many factors when assessing the likelihood of future realization of its deferred tax assets, including its recent cumulative earnings experience by taxing jurisdiction, expectations of future taxable income or loss, the carryforward periods available to the Company for tax reporting purposes, and other relevant factors.
Management has considered the weight of all available evidence in determining whether a valuation allowance remains to be required against its deferred tax assets at December 31, 2014. Given the significant losses incurred in 2014 and the overall cumulative loss history, the Company has determined that it is more likely than not that the net deferred tax assets will not be realized. The Company has a $68.8 million valuation allowance associated with its deferred tax assets as of December 31, 2014. The amount of the deferred tax asset considered realizable is subject to change based on future events, including generating taxable income in future periods. The Company continues to assess the need for the valuation allowance at each balance sheet date based on all available evidence.
The components of income tax expense are as follows:
| 2014 | 2013 | 2012 | ||||||||||
| Current federal income tax | $ | – | $ | – | $ | – | ||||||
| Current state income tax | – | – | – | |||||||||
| Deferred taxes | 8,182,968 | 7,968,310 | 2,021,705 | |||||||||
| Valuation allowance | (8,182,968 | ) | (7,968,310 | ) | (2,021,705 | ) | ||||||
| $ | – | $ | – | $ | – | |||||||
The Company notes no uncertain tax positions and estimates that the unrecognized tax benefit will not change significantly within the next twelve months. The Company will continue to classify income tax penalties and interest as part of general and administrative expense in its consolidated statements of operations. There were no interest or penalties accrued as of December 31, 2014, 2013 or 2012.
The following table summarizes the open tax years for each major jurisdiction:
| Jurisdiction | Open Tax Years | |
| Federal | 2005 - 2013 | |
| States | 2005 - 2013 |
16. SUBSEQUENT EVENTS
Lincoln Park
On various dates from January 1, 2015 through March 16, 2015, Lincoln Park purchased 932,182 shares of common stock for cash proceeds to the Company of $5,872,996. As of March 16, 2015, the Company has $12,746,455 available to us through the Lincoln Park financing arrangement.
| F-22 |
17. SUMMARIZED QUARTERLY UNAUDITED FINANCIAL DATA
The Company’s unaudited quarterly operating results are summarized below for 2014 and 2013:
| For the Quarter Ended | ||||||||||||||||
| March 31, 2014 | June 30, 2014 | September 30, 2014 | December 31, 2014 | |||||||||||||
| Revenue | $ | 39,468 | $ | 39,469 | $ | 39,467 | $ | 39,471 | ||||||||
| Loss from operations | (7,698,086 | ) | (16,437,522 | ) | (3,980,179 | ) | (6,838,765 | ) | ||||||||
| Net loss | (8,689,774 | ) | (15,517,976 | ) | (3,716,460 | ) | (6,824,735 | ) | ||||||||
| Net loss applicable to common share: | ||||||||||||||||
| Basic and diluted | $ | (0.33 | ) | $ | (0.55 | ) | $ | (0.13 | ) | $ | (0.16 | ) | ||||
| For the Quarter Ended | ||||||||||||||||
| March 31, 2013 | June 30, 2013 | September 30, 2013 | December 31, 2013 | |||||||||||||
| Revenue | $ | 87,781 | $ | 58,268 | $ | 39,468 | $ | 39,468 | ||||||||
| Loss from operations | (5,879,006 | ) | (5,586,704 | ) | (6,257,907 | ) | (11,984,290 | ) | ||||||||
| Net loss | (6,510,637 | ) | (6,894,056 | ) | (5,745,735 | ) | (11,871,820 | ) | ||||||||
| Net loss applicable to common share: | ||||||||||||||||
| Basic and diluted | $ | (0.31 | ) | $ | (0.31 | ) | $ | (0.25 | ) | $ | (0.48 | ) | ||||
| F-23 |