Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - CALADRIUS BIOSCIENCES, INC. | Financial_Report.xls |
| EX-10.44 - EXHIBIT 10.44 - CALADRIUS BIOSCIENCES, INC. | exh1045.htm |
| EX-31.2 - EXHIBIT 31.2 - CALADRIUS BIOSCIENCES, INC. | nbs-ex312_20141231xq4.htm |
| EX-21.1 - EXHIBIT 21.1 - CALADRIUS BIOSCIENCES, INC. | exh2111.htm |
| EX-23.1 - EXHIBIT 23.1 - CALADRIUS BIOSCIENCES, INC. | exh2311.htm |
| EX-32.2 - EXHIBIT 32.2 - CALADRIUS BIOSCIENCES, INC. | nbs-ex322_20141231xq4.htm |
| EX-31.1 - EXHIBIT 31.1 - CALADRIUS BIOSCIENCES, INC. | nbs-ex311_20141231xq4.htm |
| EX-32.1 - EXHIBIT 32.1 - CALADRIUS BIOSCIENCES, INC. | nbs-ex321_20141231xq4.htm |
| EX-10.9 - EXHIBIT 10.9 - CALADRIUS BIOSCIENCES, INC. | exhibit109_irvineleases.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL ENDED DECEMBER 31, 2014
OR
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from __________________ to _________________________
Commission File Number 001-33650
NEOSTEM, INC.
(Exact name of registrant as specified in its charter)
DELAWARE | 22-2343568 |
(State or other jurisdiction of | (I.R.S. Employer |
incorporation or organization) | Identification No.) |
420 LEXINGTON AVE, SUITE 350 NEW YORK, NEW YORK | 10170 |
(Address of principal executive offices) | (zip code) |
Registrant’s telephone number, including area code: 212-584-4180
Securities Registered Pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange On Which Registered | |
Common Stock, par value $0.001 per share | NasdaqCM | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this Chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | Accelerated filer x |
Non-accelerated filer o (Do not check if a smaller reporting company) | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes o No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2014 (the last business day of the most recently completed second fiscal quarter) was approximately $227.8 million, computed by reference to the closing sales price of $6.52 for the common stock on the NasdaqCM reported for such date. Shares held by executive officers, directors and persons owning directly or indirectly more than 10% of the outstanding common stock have been excluded from the preceding number because such persons may be deemed to be affiliates of the registrant. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
On February 27, 2015, 38,459,297 shares of the registrant's common stock, par value 0.001 per share, were outstanding.
EXPLANATORY NOTE
The Registrant meets the "accelerated filed" requirements as of the end of its 2013 fiscal year pursuant to Rule 12b-2 of the Securities Exchange Act of 1934, as amended. However, pursuant to Rule 12b-2 and SEC Release No. 33-8876, the Registrant (as a smaller reporting company transitioning to the larger reporting company system based on its public float as of June 30, 2013) is not required to satisfy the larger reporting company requirements until its first quarterly report on Form 10-Q for the 2014 fiscal year and thus is eligible to check the "Smaller Reporting Company" box on the cover of this Form 10-K.
DOCUMENTS INCORPORATED BY REFERENCE
None.
All references in this Annual Report on Form 10-K to “we,” “us,” the “Company” and “NeoStem” mean NeoStem, Inc., including subsidiaries and predecessors, except where it is clear that the term refers only to NeoStem, Inc. This Annual Report on Form 10-K contains forward-looking statements, which involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including those set forth under “Cautionary Note Regarding Forward-Looking Statements” and under “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
Unless otherwise indicated to the contrary, all share numbers and per share prices in this Annual Report on Form 10-K have been retrospectively adjusted, as appropriate, to give effect to the one-for-ten reverse stock split implemented on July 16, 2013.
TABLE OF CONTENTS
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS | ||
ITEM 1. BUSINESS | ||
ITEM 1A. RISK FACTORS | ||
ITEM 1B. UNRESOLVED STAFF COMMENTS | ||
ITEM 2. PROPERTIES | ||
ITEM 3. LEGAL PROCEEDINGS | ||
ITEM 4. MINE SAFTEY DISCLOSURES | ||
PART II | ||
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES | ||
ITEM 6. SELECTED FINANCIAL DATA | ||
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS | ||
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK | ||
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA | ||
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE | ||
ITEM 9A. CONTROLS AND PROCEDURES | ||
ITEM 9B. OTHER INFORMATION | ||
PART III | ||
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE | ||
ITEM 11. EXECUTIVE COMPENSATION | ||
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS | ||
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE | ||
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES | ||
PART IV | ||
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES | ||
2
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K includes “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995, as well as historical information. Such forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements, or industry results, to be materially different from anticipated results, performance or achievements expressed or implied by such forward-looking statements. When used in this Annual Report on Form 10-K, statements that are not statements of current or historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, the words “plan,” “intend,” “may,” “will,” “expect,” “believe,” “could,” “anticipate,” “estimate,” or “continue” or similar expressions or other variations or comparable terminology are intended to identify such forward-looking statements, although some forward-looking statements are expressed differently. We remind readers that forward-looking statements are merely predictions and therefore inherently subject to uncertainties and other factors and involve known and unknown risks that could cause the actual results, performance, levels of activity or our achievements or industry results, to be materially different from any future results, performance levels of activity or our achievements or industry results expressed or implied by such forward-looking statements. Factors that could cause our actual results to differ materially from anticipated results expressed or implied by forward-looking statements include, among others:
• | our ability to obtain sufficient capital or strategic business arrangements to fund our operations and expansion plans, including meeting our financial obligations under various licensing and other strategic arrangements, the funding of our clinical trials for product candidates in our development programs for our Cancer Immunotherapy Program, our Ischemic Repair Program and our Immune Modulation Program, and the commercialization of the relevant technology; |
• | our ability to build and maintain the management and human resources infrastructure necessary to support the growth of our business; |
• | our ability to integrate our acquired businesses successfully and grow such acquired businesses as anticipated, including expanding our PCT business; |
• | whether a large global market is established for our cellular-based products and services and our ability to capture a meaningful share of this market; |
• | scientific and medical developments beyond our control; |
• | our ability to obtain and maintain, as applicable, appropriate governmental licenses, accreditations or certifications or comply with healthcare laws and regulations or any other adverse effect or limitations caused by government regulation of our business; |
• | whether any of our current or future patent applications result in issued patents, the scope of those patents and our ability to obtain and maintain other rights to technology required or desirable for the conduct of our business; and our ability to commercialize products without infringing the claims of third party patents; |
• | whether any potential strategic or financial benefits of various licensing agreements will be realized; |
• | the results of our development activities, including the results of our Intus Phase 3 clinical trial of NBS20, being developed to treat metastatic melanoma, and the results of our PreSERVE Phase 2 clinical trial of NBS10 being developed to treat acute myocardial infarction for which we released results of the primary analysis on November 17, 2014; however it is subject to ongoing analysis, and currently reported results, although promising, there can be no assurance that further analysis may not reveal negative, or less promising, results; |
• | our ability to complete our other planned clinical trials (or initiate other trials) in accordance with our estimated timelines due to delays associated with enrolling patients due to the novelty of the treatment, the size of the patient population and the need of patients to meet the inclusion criteria of the trial or otherwise; and |
• | our ability to satisfy our obligations under our credit facility. |
The factors discussed herein, including those risks described in Item 1A. “Risk Factors” and elsewhere in this Annual Report on Form 10-K and in the Company's other periodic filings with the Securities and Exchange Commission (the “SEC”) which are available for review at www.sec.gov under “Search for Company Filings” could cause actual results and developments to be materially different from those expressed or implied by such statements. All forward-looking statements attributable to us are expressly qualified in their entirety by these and other factors. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. Except as required by law, the Company undertakes no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise.
3
PART I
ITEM 1. BUSINESS.
OVERVIEW
NeoStem, Inc. (“we,” “NeoStem” or the “Company”) is a vertically integrated, clinical-stage biopharmaceutical company that is pursuing the preservation and enhancement of human health through the development of cell based therapeutics that leverage the body’s natural ability to heal and fight disease. Our diversified pipeline and unique capabilities for innovative, cost-effective and efficient in-house development set us apart in this emerging industry as we work to fundamentally change the treatment paradigm for several serious diseases.
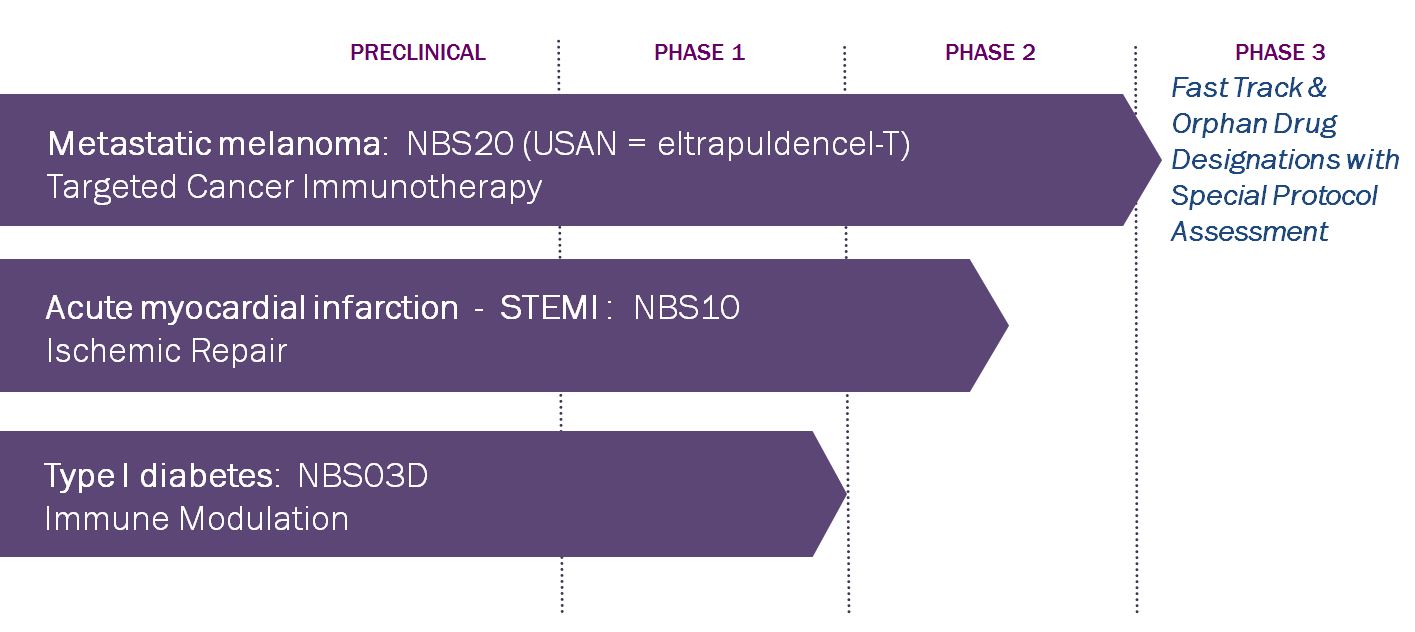
Our most advanced clinical program is based on our dendritic cell/cancer cell technology. It is focused on the development of an innovative cancer treatment that is designed to target the cells responsible for tumor growth and metastasis, known as cancer or tumor initiating cells (CSCs), using purified CSCs from a patient’s own tumor as an antigen source to induce or enhance an anti-tumor immune response in the patient. Our lead product candidate based on this platform technology, NBS20, targets malignant melanoma as an initial indication. NBS20 is being studied in patients with recurrent Stage III or Stage IV metastatic melanoma. The program has been granted Fast Track and Orphan designation by the Food and Drug Administration (FDA) and the protocol for the Phase 3 study, known as the Intus study, is the subject of a Special Protocol Assessment (SPA). Our SPA letter states that our Phase 3 clinical trial is adequately designed to provide the necessary data that, depending on outcome, could support a Biologics License Application (BLA) seeking marketing approval of NBS20. This protocol calls for randomizing 250 patients. Patient screening began in 1Q15 and randomization of the first patient is expected in 2Q15. Interim analysis of the data is targeted for 4Q2017. We are also evaluating other clinical indications for which we may advance this program, including lung, colon, ovarian and liver cancers.
Our company is also developing therapies that are designed to utilize CD34 cells to prevent heart failure and major adverse cardiac events following a severe heart attack, known as an ST-elevation myocardial infarction (STEMI), through the use of CD34 cells to regenerate tissue impacted by ischemia. Ischemia occurs when the supply of oxygenated blood in the body is restricted, causing tissue distress and death. We seek to improve oxygen delivery to tissues through the development and formation of new blood vessels. NBS10 is our most clinically advanced product candidate in our ischemic repair program. At the American Heart Association’s Scientific Sessions in November 2014, we reported data from the primary analysis of our 161 patient PreSERVE (acute myocardial infarction) AMI study. We are also planning the release of our one year data from the phase 2 trial on March 15, 2015 at the Annual Scientific Sessions of the American College of Cardiology. PreSERVE AMI is a randomized, double-blinded, placebo-controlled Phase 2 clinical trial testing NBS10, a “personalized” adult stem cell product being developed for the treatment of patients with left ventricular dysfunction following a STEMI. The primary endpoints are measured by, (i) the change from baseline to six months in myocardial perfusion (RTSS) measured by an imaging technique (SPECT); and (ii) safety of bone marrow procurement and infusion as measured by occurrence of adverse events, serious adverse events (SAEs) and major adverse cardiac events (MACE). There are five secondary efficacy endpoints, two evaluating left ventricular ejection fraction, one evaluating MACE, and two evaluating quality of life. The reported data were based on all treated patients that had received six month follow-up for imaging. The median length follow up for mortality, adverse events, SAEs and MACE in these patients was twelve months. The reported results allowed for important observations about a potential dose-dependent treatment effect that will help guide the next phase of development. These observations about a potential dose dependent treatment effect were based on post hoc analyses of subsets of treated patients based on the number of cells they received. Notably, statistically significant dose-dependent increases in left ventricular function and decreases in serious adverse events were seen in patients who received the highest dose of cells (n=15 patients), though no statistical significance was observed when NBS10 overall was compared to placebo on these measures. With respect to mortality, at one year there were no treatment group deaths while the control group saw a mortality rate of 3.6% (n=3), equating to a statistically significant reduction in mortality. Regarding MACE, while more events occurred with NBS10 overall versus placebo, a non-significant trend toward fewer events was observed in patients who received higher doses of cells, with MACE occurring in 14% of placebo patients, 17% of patients who received less than 14 million cells, 10% of patients who received greater than 14 million cells and 7% of patients who received greater than 20 million cells. Finally, our hypothesis that SPECT used to measure perfusion could be used as a surrogate marker for the current medically relevant and regulatory endpoints was disproven, giving us valuable direction regarding endpoints and analyses for future clinical trials. We expect to complete the PreSERVE AMI study as defined through the final three-year follow-up and, in the meantime, plan to meet with the FDA to discuss our results and our proposal for the next step(s) in development. Finalization of the decision for next steps for NBS10 are expected in the second half of 2015. We also are evaluating other clinical indications that involve ischemia into which we may advance this program, including critical limb ischemia (CLI) and congestive heart failure (CHF).
Another platform technology we are developing is designed to utilize Regulatory T Cells (Tregs) to treat diseases caused by imbalances in an individual's immune system. This novel approach seeks to restore immune balance by enhancing Treg cell
4
number and function. Tregs are a natural part of the human immune system and regulate the activity of T effector cells, the cells that are responsible for protecting the body from viruses and other foreign antigens. When Tregs function properly, only harmful foreign materials are attacked by T effector cells. In autoimmune disease, it is thought that deficient Treg activity permits the T effector cells to attack the body's own tissues. We have received a letter from the FDA stating that we may proceed on a Phase 2 study of NBS03D, a Treg based therapeutic being developed to treat type 1 diabetes mellitus (T1DM) in adolescents, and we plan to initiate the trial in late 2015 or 2016 depending on resource availability. We are evaluating other clinical indications into which we may advance this program, including graft versus host disease, chronic obstructive pulmonary disease (COPD), multiple sclerosis (MS), inflammatory bowel disease (IBD) and steroid resistant asthma.
Finally, we are actively exploring means by which we can take advantage of new regulations in Japan that permit conditional approval for regenerative medicine products that show sufficient safety evidence and signals of efficacy. Potential indications for this unique opportunity include our targeted cancer immunotherapy program in liver cancer and our ischemic repair program in CLI.
We believe that cell-based therapies have the potential to create a paradigm change in the treatment for a variety of diseases and conditions and we are evaluating other programs that we view as holding particular promise, including an aesthetics program for a topical skin application and a very small embryonic like (VSEL TM) stem cell program for the treatment of retinal degeneration, bone restoration and wound healing.
Through our wholly owned subsidiary, Progenitor Cell Therapy, LLC (PCT), we are recognized as a world industry leader in providing high quality innovative and reliable manufacturing capabilities and engineering solutions (e.g. process development) in the development of cell-based therapies. We operate three current Good Manufacturing Practice (cCGMP) facilities in Allendale, NJ, Mountain View, CA and Irvine, CA, respectively, and are poised to expand our facilities internationally. In addition to leveraging this core expertise in the development of our own products, we partner opportunistically with other industry leaders who recognize our unique ability to significantly improve their manufacturing processes and supply clinical and commercial material.
We look forward to further advancement of our cell based therapies to the market and to helping patients suffering from life-threatening medical conditions. Coupling our development expertise with our strong process development and manufacturing capability, we believe the stage is set for us to realize meaningful clinical development or our own proprietary platform technologies and manufacturing advancements, further positioning NeoStem to lead the cell therapy industry.
____________________________________
5
We are a Delaware corporation with our principal executive offices located at 420 Lexington Avenue, Suite 350, New York, New York 10170. Our telephone number is (212) 584-4180 and our corporate website address is www.neostem.com. We include our website address in this Annual Report on Form 10-K only as an inactive textual reference and do not intend it to be an active link to our website. The information on our website is not incorporated by reference in this Annual Report on Form 10-K.
Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports, as well as other documents we file with the U.S. Securities and Exchange Commission ("SEC"), are available free of charge through the Investor Insights section of our website as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. The public can obtain documents that we file with the SEC at www.sec.gov.
This report includes the following trademarks, service marks and trade names owned by us: NeoStem, Inc.®, Amorcyte, LLC®, AthelosTM, Progenitor Cell Therapy LLCTM and VSELTM Technology. These trademarks, service marks and trade names are the property of NeoStem and its affiliates. This report also includes other trademarks, service marks and trade names owned by us or other companies. All trademarks, service marks and traded names included herein are the property of their respective owners.
____________________________________
OVERVIEW OF THE CELL THERAPY FIELD
Regenerative medicine is defined as the process of replacing or regenerating human cells, tissues or organs to restore normal function. Among the categories of therapeutic technology platforms within this field are cell therapy; tissue engineering; tools, devices and diagnostics; and aesthetic medicine. NeoStem's business model is focused on two of these areas. First, cell therapy, in which we introduce cells (adult, donor or patient, stem cell or differentiated) into the body to prevent and treat disease; and second, tools, devices and diagnostics in which we intend to utilize engineering and innovation to automate, integrate or otherwise modify cell therapy manufacturing platforms to improve the deliverability of cellular therapeutics to patients.
All living complex organisms start as a single cell that replicates, differentiates (matures) and perpetuates in an adult organism throughout its lifetime. Cell therapy is the process that uses cells to prevent, treat or cure disease, or regenerate damaged or aged tissue. To date, the most common type of cell therapy has been the replacement of mature, functioning cells through blood and platelet transfusions. Since the 1970s, first bone marrow and then blood and umbilical cord-derived stem cells have been used to restore bone marrow, as well as blood and immune system cells damaged by the chemotherapy and radiation that are used to treat many cancers. These types of cell therapies are standard of practice world-wide and are typically reimbursed by insurance.
Within the field of cell therapy, research and development using stem cells to treat a host of diseases and conditions has greatly expanded. Stem cells (in either embryonic or adult forms) are primitive and undifferentiated cells that have the unique ability to transform into or otherwise affect many different cells, such as white blood cells, nerve cells or heart muscle cells. NeoStem's cell therapy development efforts using stem cells are focused on the use of adult stem cells; these cells are found in the bone marrow, peripheral blood, umbilical cord blood and other body organs.
According to Robin R. Young’s (CEO and Founder of NY Stem Cell Summit) Stem Cell Summit Executive Summary-Analysis and Market Forecasts 2014-2024, the U.S. stem cell therapy market is estimated to grow from an estimated $237 million in 2013 to more than $5.7 billion in 2020.
There are two general classes of cell therapies: Patient Specific Cell Therapies (PSCTs) and Off-the-Shelf Cell Therapies (OSCTs). In PSCTs, cells collected from a person (donor) are transplanted into, or used to develop a treatment for a patient (recipient) with or without modification. In cases where the donor and the recipient are the same individual, these procedures are referred to as “autologous”. In cases in which the donor and the recipient are not the same individual, these procedures are referred to as “allogeneic.” A notable form of autologous PSCT involves the use of autologous cells to create vaccines directed against tumor cells in the body which has been demonstrated to be effective and safe in clinical trials. NeoStem's targeted cancer immunotherapy program, ischemic repair program and immune modulation program also focuses on PSCTs using autologous cells. Autologous cells offer a low likelihood of rejection by the patient and we believe the long-term benefits of these PSCTs can best be achieved with an autologous product. In the case of OSCT, donor cells are expanded many fold in tissue culture, and large banks of cells are frozen in individual aliquots that may result in treatments for as many as 10,000 people from a single donor tissue.
6
Various adult stem cell therapies are in clinical development for an array of human diseases, including autoimmune, oncologic, neurologic and orthopedic diseases, among other indications. NeoStem, as well as other companies, are developing cell therapies that are designed to address cancers, ischemic repair and immune modulation. While no assurances can be given regarding future medical developments, we believe that the field of cell therapy holds the promise to better the human experience and minimize or ameliorate the pain and suffering from many common diseases and/or from the process of aging.
PCT provides us with a unique and fundamental base platform of experience and expertise with a multitude of cell types in development. PCT is strategically helping to position us in a way that allows us to participate in the cell therapy field on multiple levels as the cell therapy industry evolves. Our goal is to nurture our reputation as a premier service provider in the regenerative medicine industry by continuing to leverage the experience and expertise of PCT as a recognized leader of cell therapy manufacturing and development.
CELL THERAPY PRODUCT DEVELOPMENT
NeoStem has a multi-pronged research and clinical development strategy with three therapeutic platforms: targeted cancer immunotherapy (Immuno-Oncology Program), ischemic repair (CD34 Cell Regenerative Medicine Program) and immune modulation (T Regulatory Cell Program). The following chart depicts our robust, diversified pipeline:
Targeted Cancer Immunotherapy (Immuno-Oncology Program)
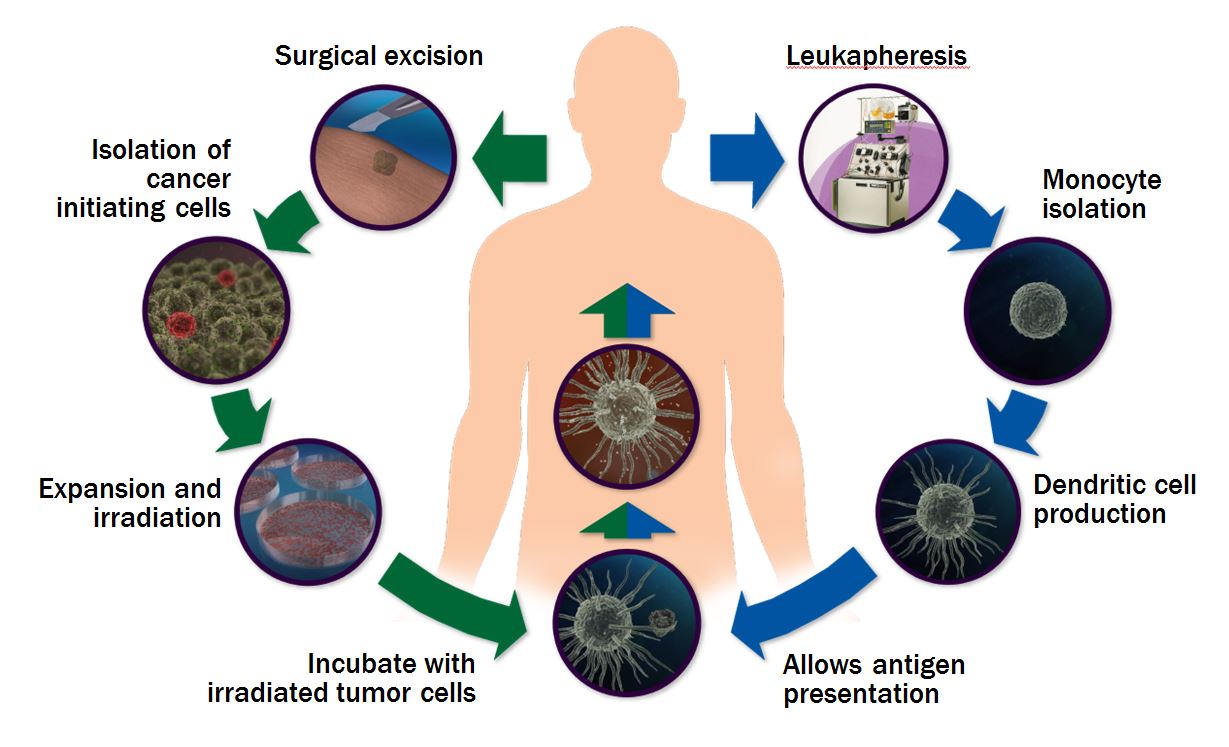
It has been well established that the human immune system can provide a powerful response in the treatment of cancer if the immune system can be properly “educated” to attack cancerous cells while leaving the normal tissue unharmed. It is thought that many recurrences of cancers treated with the standard of care are the result of tumor or cancer initiating cells (commonly referred to as “cancer stem cells”) that evade the initial therapy and initiate tumor re-growth. Targeting tumor or cancer initiating cells after medically induced tumor regression remains a seminal goal of cancer therapeutics, in that eradication of such cells could lead to long-term disease-free survival, better overall survival and potential cures.
With the acquisition of California Stem Cell in May 2014, NeoStem has undertaken the continued development of a patient-specific cancer immunotherapy now known as NBS20 (USAN generic name: eltrapuldencel-T). NBS20, which uses a patient’s own immune cells and tumor or cancer initiating cells to create a therapeutic vaccine, is currently in Phase 3 clinical investigation by NeoStem for the treatment of metastatic melanoma with sites across the country presently screening patients.
NBS20 Represents Novel technology
Traditional preventive vaccines contain weakened or inactivated versions of antigens that cause specific diseases, such as measles, mumps, or flu. When injected into the body, the immune system can recognize and remember the antigen, so that if a person becomes exposed later, the immune system will fight it, preventing illness. Therapeutic cancer vaccines are different in that they aim to treat, not prevent, cancer. The current challenge with most cancer vaccines is that they are based on a limited
7
number, or even a single, antigen, often targeting the most common and well known antigens or peptides linked to a particular cancer. The practical application of this approach is challenging due to temporary expression of the antigen (for example a regulatory protein present only in a certain stage of cell cycle), and extreme variability from patient to patient. In contrast, NeoStem utilizes the advantages of proprietary in vitro manipulations to isolate and substantially expand the number of tumor or cancer initiating cells to be used as the antigen source, or loading agent, for the dendritic cells. The isolation and growth in vitro of these proliferating tumor or cancer initiating cells originating from a patient’s tumor provides a source of highly enriched and patient-specific antigens expressed by the sub-population of cells that is most capable of re-establishing the tumor in vivo and causing disease relapse and death. As such, we believe the antigen profile is highly matched to that of the tumor or cancer initiating cells and has a greater likelihood of achieving its intended effect. Importantly, given that the therapy is autologous (i.e., based on patient’s own cells), and targeted to diseased cells (i.e., the tumor or cancer initiating cells) we believe the level of potential adverse effects is greatly reduced. We believe this platform technology can be applied to any solid tumor, for example, ovarian, lung, colon and liver cancers.
Clinical Development Efforts
With only a 15% historical 5 year survival rate for stage IV metastatic melanoma (AJCC Cancer Staging 2010), there is an unmet medical need to adequately treat patients with metastatic melanoma. NBS20 potentially addresses this unmet need by providing an adjunctive treatment in those patients with metastatic melanoma whose disease has been “controlled” by surgery, radiation therapy, or other systemic therapies. For more than 20 years Dr. Robert O. Dillman (NeoStem’s Vice President of Oncology) and colleagues have investigated the safety and therapeutic potential of patient-specific autologous vaccines derived from autologous cell lines. Prior to the current Phase 3 study, a 42-subject, open-label, randomized Phase 2 trial was conducted wherein 24 subjects received irradiated tumor cells (TC) and 18 subjects received NBS20. The Phase 2 trial was designed to detect survival rates at 2.5 years. Results from that trial were presented and published in 2012 showing a two year survival advantage for patients treated with NBS20 compared to TC (72% vs. 31% (p=.007), respectively). In the Phase 2 trial, common adverse events included infusion site reactions and flu like responses; none of the Grade 3 or higher events that occurred in the trial were deemed treatment related by the investigator.
Our Phase 3, double-blinded, randomized, controlled trial (the Intus trial) began screening patients in 1Q15 and randomization of the first patient is expected in 2Q15. It has been granted Special Protocol Assessment (SPA) from the FDA, and NBS20 has received Fast Track designation and Orphan Drug designation for the treatment of metastatic melanoma. We are targeting approximately 50 sites in the U.S, Canada, Australia and New Zealand and potentially other geographic regions. Eligible patients will have recurrent stage 3 or stage IV melanoma with at least one lesion amenable to surgical resection. After successful establishment of a cell line and referral for treatment, 250 patients with good performance status (ECOG 0-1) will be stratified by whether they have no evidence of disease, non-measurable disease by RECIST, or measurable disease with elevated LDH or without elevated LDH, and then randomized 2:1 to receive either NBS20 or autologous mononuclear cells. Both products will be injected weekly for 3 consecutive weeks, and then monthly for 5 consecutive months. The primary endpoint is overall survival (death from any cause).
Additionally, we are exploring means to take advantage of the efficiencies offered by the Canadian health authority regarding the development and commercialization of regenerative medicine products. Potential indications for this opportunity include our targeted cancer immunotherapy program in melanoma, and ovarian cancer.
8
Timeline
We began screening patients for our Phase 3 melanoma study in 1Q of 2015 and expect to randomize our first patient in 2Q 2015, with enrollment scheduled for completion in 4Q 2016. There is an interim results look at the data scheduled to occur after 99 patients have died, and this is presently estimated to happen in 4Q 2017. If that interim review shows robust efficacy and data warranting early termination of the trial, we could potentially submit a BLA in 2Q 2018 for approval of NBS20. Alternatively, our study would continue until 162 study patient deaths have occurred, which event is presently projected for 2Q 2019 with a potential BLA submission in Q4 2019.
Market Opportunity and Competition
The global melanoma therapeutics market was estimated to be valued at $1.3 billion in 2013 and is expected to expand at a compound annual growth rate of 15.4% to reach $3.6 billion by 2020. The United States has the biggest baseline market size due to its large population, and higher prevalence and treatment rates, and when accounting for the high annual cost of therapy, comprised approximately 78% of the total world market in 2013 (World Cancer Report 2014).
The field of melanoma drug development is highly competitive. According to a 2015 Research and Markets report, there are approximately 274 companies plus partners developing 310 drugs targeting melanoma in development (Research and Markets: Melanoma Drug Pipeline Update 2015). Several recently approved approaches have demonstrated improved clinical benefits in melanoma, including ipilimumab, an anti-CTLA-4 antibody (Bristol-Myers Squibb) and anti-PD-1 antibodies, nivolumab and pembrolizumab (Bristol-Myers Squibb and Merck, respectively). These products represent a new class of “immunotherapeutics” and have novel mechanisms of action targeting immune checkpoints. BRAF enzyme inhibitors include vemurafenib, marketed by Roche and dabrafenib from GlaxoSmithKline. Also recently approved is trametinib, a mitogen-activated protein kinase (MEK) from GlaxoSmithKline. These target critical intracellular protein pathways. In addition to the immune checkpoint modulators, several companies, including Amgen, Northwest Biotherapeutics, Lion Biotechnologies and Argos Therapeutics are developing therapeutic vaccines that act by priming a person’s immune system to recognize and attack cancer cells.
Ischemic Repair (CD34 Cell Program)
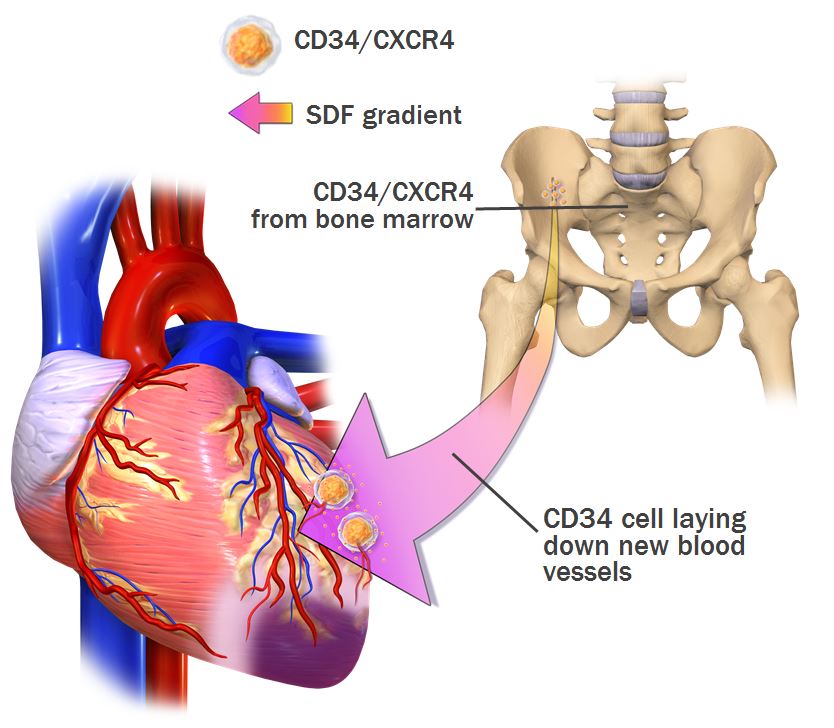
Our CD34 Cell Program develops therapies designed to address diseases and conditions caused by ischemia. Ischemia occurs when the supply of oxygenated blood to tissue is restricted. Through the administration of CD34 cells, we seek to promote the development and formation of new blood vessels and thereby eliminate the ischemic condition. Our most advanced product candidate in this program, NBS10, is a chemotactic hematopoietic stem cell product comprised of autologous bone marrow derived CD34/CXCR4 cells selected to preserve heart muscle function following an acute myocardial infarction (AMI) (acute myocardial infarction or heart attack).
9
The goal of NBS10 is to increase microvascular blood flow in the heart muscle via the development and formation of new blood vessels, thereby reversing the restriction of blood supply caused by a heart attack and rescuing tissue from eventual cell death. The treatment process works as follows:
•A patient's own bone marrow is harvested and a sterile pharmaceutical composition of stem cells found in the bone marrow, enriched for CD34/CXCR4 cells, is prepared using our patented technology. The cell preparation has a 72 hour shelf life.
•The isolated cells are then infused back into the patient via catheter into the infarct-related artery 5 to 11 days following an AMI, which we believe to be the optimal time frame for cellular intervention, after the pro-inflammatory “hot phase” and prior to permanent scar formation, while the heart tissue is naturally and actively attracting CD34/CXCR4 cells.
•The cells are attracted to certain chemicals that are released in higher concentrations in oxygen-starved tissue and when they reach that tissue begin to orchestrate the process of building new blood vessels to restore blood supply and thereby enhance the function of the damaged heart muscle.
Clinical Development Efforts
In light of extensive pre-clinical data showing that CD34 cells can induce the development and formation of new blood vessels over time and prevent heart cell death caused by chronic ischemia, a Phase 1 study of NBS10 treating 31 patients with damaged heart muscle following AMI was conducted, the analysis of which was instructive in the design of our subsequent Phase 2 study.
At the American Heart Association’s Scientific Sessions in November 2014, we reported data from the primary analysis of our 160 patient PreSERVE AMI study. PreSERVE AMI is a randomized, double-blinded, placebo-controlled Phase 2 clinical trial testing NBS10 following a STEMI. The primary endpoints are measured by, (i) the change from baseline to six months in myocardial perfusion (RTSS) measured by an imaging technique (SPECT); and (ii) safety of bone marrow procurement and infusion as measured by occurrence of adverse events, serious adverse events (SAEs) and major adverse cardiac events (MACE). There are five secondary efficacy endpoints, two evaluating left ventricular ejection fraction, one evaluating MACE, and two evaluating quality of life. The reported data were based on all treated patients that had received six month follow-up for imaging. The median length follow up for mortality, adverse events, SAEs and MACE in these patients was twelve months. The reported results allowed for important observations about a potential dose-dependent treatment effect that will help guide the next phase of development. These observations about a potential dose dependent treatment effect were based on post hoc analyses of subsets of treated patients based on the number of cells they received. Notably, statistically significant dose-dependent increases in left ventricular function
10
and decreases in serious adverse events were seen in patients who received the highest dose of cells (n=15 patients), though no statistical significance was observed when NBS10 overall was compared to placebo on these measures. With respect to mortality, at one year there were no treatment group deaths while the control group saw a mortality rate of 3.6% (n=3), equating to a statistically significant reduction in mortality. Regarding MACE, while more events occurred with NBS10 overall versus placebo, a non-significant trend toward fewer events was observed in patients who received higher doses of cells, with MACE occurring in 14% of placebo patients, 17% of patients who received less than 14 million cells, 10% of patients who received greater than 14 million cells and 7% of patients who received greater than 20 million cells. Finally, our hypothesis that SPECT used to measure perfusion could be used as a surrogate marker for the current medically relevant and regulatory endpoints was disproven, giving us valuable direction regarding endpoints and analyses for future clinical trials. We are planning the release of our one year data from the phase 2 trial on March 15, 2015 at the Annual Scientific Sessions of the American College of Cardiology. We expect to complete the PreSERVE AMI study as defined through the final three-year follow-up and, in the meantime, plan to meet with the FDA to discuss our results and our proposal for the next step(s) in development. Finalization of the decision for next steps for NBS10 are expected in the second half of 2015.
Other Conditions
We believe that the CD34 Cell Program may be applicable to other conditions caused by underlying ischemic injury,. Published reports provide preliminary evidence that CD34 cell therapy can exert significant therapeutic effects in patients with critical limb ischemia (CLI), a condition in which the blood flow to the legs is severely impaired, causing pain and non-healing ulcers and, ultimately, potentially resulting in the need for amputation. Prior studies have shown benefits of CD34 cell therapy that included pain relief, ulcer healing and reduced amputation rates. Conditions such as CLI are often difficult to study in large randomized controlled programs and Japan’s new Regenerative Medicine’s Law is designed to advance regenerative medicine therapies such as these. The new regulation supports conditional approval when there is data to show sufficient safety and some preliminary evidence of efficacy. NeoStem is exploring how best to work within the Japanese Regenerative Medicine Law framework to advance this and potentially other programs. NeoStem has available to it in Kobe, Japan space to evaluate a potential CD34 cell therapy program in CLI and a small team of regulatory professionals in Japan who would work with us in all interactions with the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) and Ministry of Health, Labour and Welfare (MHLW).
We are also evaluating the potential of a CD34 product in treating congestive heart failure (CHF). Published reports have provided evidence that CD34 cells administered into the coronary arteries of patients with CHF can improve survival compared to patients treated with standard medical therapy.
Market Opportunity and Competition
A statistical report from Agency for Healthcare Research and Quality in 2011 surveyed the most expensive hospitalization conditions by payer and lists AMIs as the sixth most expensive condition treated in U.S. hospitals, with a national hospital bill of more than $37 billion annually. AMI patients are at significant risk of downstream adverse events including chronic heart failure, re-current AMI, significant arrhythmias, premature death and acute coronary syndrome, and thus AMI is one of the target populations in our CD34 Cell Program. The American Heart Association (AHA) estimates that in the U.S. each year, there are 175,000 to 225,000 patients who suffer a STEMI, the most dangerous type of heart attack, which results from a sudden blockage of one of the arteries that supplies nutrient-rich blood to the heart muscle. We estimate that each year approximately 65,000 of these STEMI patients, successfully stented, later experience heart failure. Treatment of these patients post-heart attack represents a significant financial burden for many managed care programs. We expect that this burden will increase as the “baby boomer” population ages and the annual number of STEMIs likely increases.
The field of cardiovascular cell therapy development is competitive. There are a number of companies that are developing stem cell-based therapies for cardiovascular diseases, including, but not limited to, Cardio3 Biosciences SA, Capricor, Inc., Mesoblast Limited, Athersys, Inc., Pluristem Therapeutics Inc., Vericel Corporation and Cytori Therapeutics, Inc. These companies are utilizing a number of different therapeutic approaches in their development efforts. There are both autologous and allogeneic based competitive therapies that derive cells principally from four sources: fat, peripheral blood, cord blood, and bone marrow. NBS10 is an autologous therapy that derives its cells from bone marrow.
Immune Modulation (T Regulatory Cell Program)
We are collaborating with the University of California, San Francisco (UCSF) on our T Regulatory Cell program, using the technology platform of our majority-owned subsidiary, Athelos Corporation (Athelos) to pursue the development of cell
11
therapies designed to use autologous immune cells as a therapeutic product to treat disorders of the immune system. Many immune-mediated diseases are a result of an imbalance between immune effector and regulatory mechanisms, whereby eventually, pro-inflammatory cells and cytokines go unchecked. Therapy using T Regulatory Cells (Treg) represents a novel approach to restoring immune balance by enhancing regulatory T-cells (Tregs) cell number and function to control pathologic immune responses.
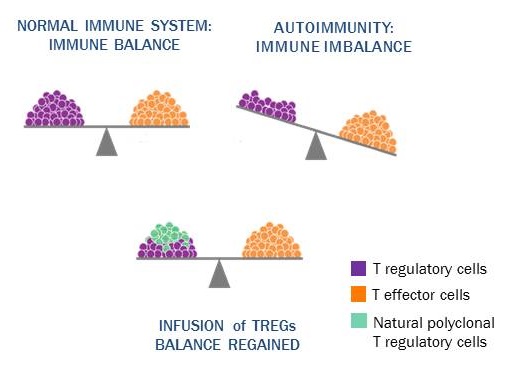
Preclinical and Clinical Development
Through exclusive world-wide licenses to more than 30 issued patents and patent applications, we have secured the rights to a broad patent estate within the Treg field, covering natural or thymic Tregs (Tregs), induced Tregs (iTregs) and methods of treating or preventing certain conditions and/or diseases by use of Tregs. Both types of Tregs have been shown in pre-clinical studies to play a pivotal role in modulating autoimmune and inflammatory diseases. Tregs have been evaluated by others in early phase human clinical trials and shown to be safe with suggestions of clinical benefit in Type 1 diabetes mellitus (T1DM), Crohn’s disease and graft-versus-host disease (GVHD). Both Tregs and iTregs have demonstrated the potential to treat autoimmune related conditions like T1DM, and inflammatory bowel disease. Preclinically, Tregs have also been shown to be beneficial in alloimmune related diseases such as enhancing allogeneic graft tolerance.
T1DM, also known as insulin dependent diabetes or juvenile diabetes, is caused by the autoimmune destruction of insulin-producing beta cells of the pancreas. We have established a collaboration with University of California San Francisco (UCSF) and the laboratories of Drs. Jeffrey Bluestone and Qizhi Tang, world renowned thought leaders in the field of Tregs and immune tolerance, to collaborate on the development of NBS03D, autologous ex-vivo expanded polyclonal Tregs, for the treatment of T1DM. This collaboration, comprised of a data license, patent license and research agreement, is advancing the Company's Treg Program towards a Phase 2 trial, expected to be initiated in late 2015 or 2016, depending on resource availability, o evaluate the efficacy of autologous Tregs in T1DM, effectively advancing this program more quickly than if the Company had developed a program for this clinical indication on its own. In this collaboration, NeoStem will sponsor and manufacture a Treg product consisting of polyclonally expanded Tregs for a planned Phase 2 trial to treat adolescents newly diagnosed with T1DM. The collaboration also includes research efforts to develop the next generation of Treg products for therapeutic use. The Phase 1 open-label uncontrolled dose escalation study of autologous Treg immunotherapy for T1DM was funded by the Juvenile Diabetes Research Foundation and conducted by Dr. Stephen Gitelman at UCSF and Dr. Kevan Harold at Yale University, in collaboration with Dr. Bluestone. Results were reported by Dr. Gitelman at the American Diabetes Association 74th Scientific Sessions in San Francisco in June 2014. The study provided preliminary safety and feasibility data that support developing a novel therapy for the treatment of T1DM with the goal of inducing immune tolerance and preserving pancreatic beta cell function and the Company plans to initiate a Phase 2 study in 2015. The investigators reported that, in the study, 14 patients between 18 and 45 years of age with a mean duration of disease of 10 months all of whom retained a baseline level of insulin production, received a single infusion of autologous Tregs expanded ~500-fold. The majority of adverse events reported were mild. There were three SAEs: two were deemed unrelated by the investigator and a third SAE of grade 3 pre-syncope was deemed unlikely related. Common side effects included mild infections. Infused Tregs peaked in circulation 3 -7 days after infusion and were detectable at up to six months. The average levels of stimulated C-peptide, an indicator of pancreatic islets beta cell function that was measured in the study as a safety biomarker, for some patients remained stable from baseline for as long as two years post treatment. These data suggest that the treatment was manageable and did not adversely affect residual beta cell function. The Tregs were observed to be highly functional and long lived in treated individuals.
12
While the US Phase 1 study was designed to evaluate safety and tolerability in adults who have suffered T1DM for various durations, supportive evidence of the utility of Tregs for T1DM in humans was provided by a study of pediatric patients with new onset T1DM, published in the July 2014 issue of Clinical Immunology. In that open label non-randomized study, Marek-Trzonkowska, et al., conducted in Poland, reported that treatment with expanded autologous Tregs preserved function of pancreatic beta cells and reduced the need for exogenous insulin in the majority of patients. Through 12 months of follow-up, about 66 percent of children treated were in remission according to study specified criteria, compared to only 20 percent of concurrent controls. In addition, two Treg treated children achieved complete insulin independence, while none of the children in the control group achieved this endpoint. Importantly, the study utilized a Treg based product similar to NBS03D, and provided additional information of the safety and feasibility of this approach in new onset children with T1DM. We plan to initiate a Phase 2 study of NBS03D to treat Type 1 diabetes in adolescents in late 2016 or 2016 depending on resource availability. the primary objective of this study is to assess the safety and one year efficacy of the two doses of NBS03D compared with placebo in adolescents with recent onset T1DM. This study has already been submitted to the FDA as part of the Investigational New Drug Application (IND) and can proceed once we are ready to initiate it. We are presently assessing development timelines for this trial as well as partnering opportunities.
We are considering other potential therapeutic targets for Tregs. Asthma, another condition caused by an imbalance in the immune system, occurs when excessive inflammation is triggered in the lungs resulting in constriction of the airways and difficulty breathing. The causes of asthma are complex, through it is known that over-activity of T-helper type 2 (Th2) cells is a common feature. Th2 cells secrete the inflammatory signals that lead to the symptoms of asthma. Existing evidence indicates that Treg cells may regulate Th2 activity and therefore may have a beneficial effect on severe asthma. Additionally, tRegs have been evaluated in early phase human clinical trials and have indicated clinical benefit in graft vs. host disease (GVHD). Allogeneic hematopoietic cell transplantation, multiple sclerosis, chronic obstructive pulmonary disease and inflammatory bowel disease are also other indications in which we believe tRegs may have a meaningful therapeutic effect.
NeoStem's ongoing Treg Program is establishing methods to isolate and expand human Tregs for large scale manufacturing to enable our planned clinical trials. NeoStem’s unique manufacturing expertise has enabled the successful relevant technology transfer of the methods licensed from UCSF, and has combined it, for the first time in this setting, with scale up in the control group quality manufacturing process to meet regulatory expectations, and form a solid foundation for commercial manufacturing.
Market Opportunity and Competition
International Diabetes Foundation Atlas reported an estimated 79,100 children younger than 15 develop T1DM annually worldwide, with annual increase in incidence of about 3%. In the US a SEARCH for Diabetes in Youth study reports an annual incidence of 18,436 in individuals less than 20 years of age. T1DM inflicts a staggering economic cost on the US healthcare system, estimated at $14.4 billion annually, and it is expected that a therapeutic that can modify the course of T1DM will potentially achieve significant cost savings, and thus command high market penetration and premium pricing. The market for T1DM is expected to continue to be dominated by insulin replacement therapies. However, other novel approaches, including immune modulatory agents such as NBS03D, are expected to progressively penetrate the market in the near term. Most recently, the market size for immune modulators was valued at about $45 million in 2017.
Currently, there are no approved therapies. There are many agents in development targeting the modification of the course of the disease. Current approaches can be broadly divided into immune modulatory agents aiming to improve metabolic function by rescuing insulin producing beta cells, or regenerative agents that are aiming to replace beta cells. The pipeline for all novel agents is skewed towards early development, including ultra-low dosing of Interleukin-2, which increases the number of tolerogenic cells and which has been advanced to phase 2 by ILTOO Pharma. Also in early development are beta-cell replacement therapies, such as that of ViaCYTE, Inc., which employ technologies that protect transplanted beta-cells from immune attack.
Other Research and Development
VSELs
Our scientists have been evaluating the therapeutic potential of so-called very small embryonic-like stem cells (adult), which we refer to as "VSELsTM" or "VSELTM stem cells". Preclinical animal models have demonstrated that highly enriched human VSELsTM, when injected in the vitreal or subretinal space can migrate and integrate into areas of damage and have the ability to differentiate and express markers of retinal stem cells, neuronal cells, and photoreceptors and thus, through further studies, may demonstrate VSELsTM potential to treat ocular diseases such as macular degeneration, retinitis pigmentosa, and other retinal degenerative diseases that have no effective treatment options today.
13
Therapeutic uses of VSELsTM are assessed on the basis of unmet medical need, target patient population size, regulatory strategy, and overall commercial market. Through either grant funding or NeoStem's own funding, we are exploring VSELTM treatment development for chronic wounds, retinal repair and bone applications.
Topical Aesthetics
Our aesthetics program, based on expertise in cellular therapy and manufacturing, is developing a topical skin application using growth factors secreted by stem cells. The growth factors are harvested from a proprietary manufacturing process that includes a complex composition of human stem cells of epidermal originating lineages cultured in a controlled microenvironment to promote healthy skin cell biology.
NeoStem has generated in vitro data demonstrating these growth factors can enhance the proliferation and viability of keratinocytes and fibroblasts, cells that play an integral role in maintaining the structural integrity of the epidermis. In vivo and histological data have shown changes in several dermis and basal skin layer components including rete pegs and the expression of filaggrin and aquaporin, which are important for the maintenance and integrity of the skin and its architecture. As a topical application, these growth factors have shown evidence of beneficial effect in a clinical study of skin health. A dermatologist-controlled study was performed in aged, female patients treated twice daily with our product for 12 weeks to determine the efficacy and safety as measured by a standard dermatology scoring system. The dermatologist observed outstanding results as measured by benefit to skin tone, clarity, radiance, decrease in redness, fine lines and deep wrinkles and enhanced skin smoothness. The test formulation was deemed safe and efficacious by the patients, the managing dermatologist, and the clinical histologist. Additionally, toxicology studies indicate no toxicity, pro-inflammatory or allergic effects with the growth factors. Histological analyses of patient skin biopsies were also performed, and we observed statistical significance in some aging measures, and a positive trend in others.
Regulatory and Clinical Affairs Strategy
Our cell therapy regulatory and clinical strategy is to utilize all opportunities to discuss with our development plans with regulators. We will take the opportunity to meet with regulators at various stages in our product development from preclinical to more advanced stages of development. We intend to utilize this strategy with all regulatory agencies as we move forward with the product pipeline.
Intellectual Property
Targeted Cancer Immunotherapy Program
NeoStem owns, through acquisition, the following intellectual property:
▪ | Portfolio of 5 granted and approximately 60 pending patents covering most facets of the dendritic cell vaccine product and manufacture process, including: |
◦ | Stem cell growth media and methods of making and using growth media; |
◦ | Antigen-presenting cancer vaccines, methods of manufacturing vaccines and methods of treating disease using the vaccines; |
◦ | Methods of making individualized high purity carcinoma initiating (stem) cells for target indications |
▪ | The portfolio is international, including filings in the U.S., Europe, Japan, China, Hong Kong, Australia, New Zealand, Israel, Singapore, China, Korea and Canada. |
Ischemic Repair (CD34 Cell) Program
NeoStem’s developed and owned ischemic repair patent portfolio is comprised of the following:
▪ | 6 U.S. patents, 2 EU patents (each filed in 30 individual countries) and 12 other OUS (outside U.S.) composition and methods patents granted (Japan, South Africa, Malaysia, Philippines, Canada, Russia) |
▪ | Claims cover, inter alia, a pharmaceutical composition that contains a therapeutic concentration of non-expanded CD34/CXCR4 stem cells that move in response to SDF-1 or VEGF, together with a stabilizing amount of serum, and that can be delivered parenterally through a catheter to repair an injury caused by vascular insufficiency. |
14
▪ | Issued and pending claims can be applied to broad range of conditions caused by underlying ischemia, including: AMI, chronic myocardial ischemia post-AMI; chronic heart failure; critical limb ischemia; and ischemic brain injury |
▪ | 3 U.S. and 12 OUS patents pending |
T Regulatory Cell Program
NeoStem has created a patent portfolio through exclusive licenses from leaders in the field of Tregs (The University of Pennsylvania/Carl June, Bruce Blazar, et al and the University of California at San Francisco/Jeffrey Bluestone et al) comprised of:
▪ | 13 patents and 10 pending patents |
▪ | Claims covering many facets of Tregs, including: |
◦ | composition claims to engineered antigen presenting cells (APCs); |
◦ | methods of Treg isolation, expansion and activation/stimulation as sourced from peripheral blood and cord blood; |
◦ | methods of treating or preventing certain conditions and/or diseases, including Type 1 diabetes, organ transplant rejection, and GVHD using Tregs. |
▪ | Patents and applications cover international geographies (US, Europe, Japan, China, Australia, Canada) |
▪ | An option on patent licenses to critical reagents employed in Treg therapeutic development |
VSELTM Technology Program
NeoStem continues prosecution of patent applications relating to methods of identifying and purifying its regenerative adult stem cell candidate (VSELs TM ) as well as methods of treating an array of maladies using VSELs, including cardiac repair, bone and cartilage regeneration, ocular disease and cutaneous wound healing.
CELL THERAPY DEVELOPMENT AND MANUFACTURING OPERATIONS
Through our wholly owned subsidiary, Progenitor Cell Therapy, LLC (PCT), we are recognized as a world industry leader in providing high quality manufacturing capabilities and innovative engineering solutions in the development of cell-based therapies. Our strategy is to leverage this core expertise in the development of our own products while we partner opportunistically with other industry leaders who recognize our unique ability to significantly improve their manufacturing processes.
We operate three state-of-the-art, accredited and certified U.S. facilities, one in Allendale, New Jersey, one in Irvine, California and one in Mountain View, California. We are seeking to expand our manufacturing facilities internationally.
Our in-house expertise provides us with know-how and resources to cost-effectively and efficiently develop our own cell therapy products, as well as translate our own research and development efforts, capabilities and proprietary technologies into stable, reproducible, well-characterized cell therapy products. This expertise has provided us with a unique advantage in the advancement of our targeted cancer immunotherapy, ischemic repair and immune modulation program where all manufacturing has been carried out by our in-house team.
Experience and Expertise
Our management team has extensive and unique experience in domestic and internationally regulated cell therapy development, including contract research, development and manufacturing across a broad range of science, technologies, and process operations. Team members are recognized and credentialed experts in all aspects of clinical and product development, characterization, manufacturing, delivery, and use, of cellular products and have extensive experience designing, validating, and operating cGMP/GLP cell therapy manufacturing facilities.
PCT's expertise is focused on advancing product candidates from conception through to commercialization by reducing manufacturing risks, shortening the time to regulatory approval, and lowering the overall costs of a clinical development program. With its established facilities and infrastructure, PCT offers expertise at all stages of the product development lifecycle and cost-effective development and manufacturing services that meet applicable quality standards.
During its more than 15 years in existence, PCT has worked with more than 20 different cell types therapeutics including
15
neuronal and skin based cells for brain and spinal cord repair, myoblast, mesenchymal cells and bone marrow derived cells for heart disease, tumor, dendritic cells and monocytes for cancer treatment, cord blood, peripheral blood, bone marrow CD34 selected cells for transplantation and islet cells for diabetes.
PCT's expertise is in high quality delivery of cell therapy:
• | Manufacturing: Manufacturers of cell therapy-based products face a number of challenges, including limited unit sizes and process scalability, short processing turnaround times and stringent and evolving regulatory requirements. |
• | Engineering and Innovation: We think beyond current practices, and develop long-term solutions to the unique challenges of cell therapy manufacturing. Our team accelerates the use of automation, integration, closed processing and other strategies to address scale up, cost of goods, quality control and robustness of manufacturing process. In order to bolster our unique expertise and further reduce cost of goods sold for products, PCT continually seeks innovation drivers, including new opportunities for automation in its manufacturing operations. |
• | Product and Process Development: PCT develops, optimizes, implements and validates various aspects of cell therapy product and process development. |
• | Cell and Tissue Processing: PCT provides cost-effective cell collection and processing services that meet cGTP standards. |
Over the next several years, we anticipate that the number of companies in the cell therapy field will continue to increase and the relative distribution of stage of development of the therapeutics will begin to skew more heavily towards Phase 2 and Phase 3 trials, and into commercial distribution if regulatory approvals are obtained. As this industry continues to develop and mature, we believe PCT is well positioned to capture a meaningful share of this larger, more profitable market. To meet this coming demand, we are poised to expand internationally.
Improving Deliverability of Cell Therapy Products through PCT's Center for Engineering & Innovation
As the field of regenerative medicine matures, and an increasing number of products are reaching the marketplace, valuable lessons are being learned about the strengths and weaknesses of various business models that may allow for therapies to be delivered to large numbers of patients. Our Center for Engineering & Innovation is working to think beyond current practices to accelerate the use of automation, integration and other engineering strategies to address the important issues of scale up, cost of goods, and improved robustness of manufacturing process in anticipation of commercial production.
We are applying engineering principles to transition cell therapy science to manufacturing, and applying development by design principles, as well as structure development methodology centered on unit operations to increase the chance of successful commercial-scale manufacturing. In addition to building our internal core of engineering and innovation expertise, we are partnering with solutions providers and academic institutions to leverage existing and develop novel closed systems, single-use disposables, automation, and integration as key drivers for innovation. In this way, we believe we will be able to support the manufacture of high quality products at a reasonable cost of goods, to meet product demand in a scalable manner as it grows throughout the commercial life of the therapeutic. For example, we are collaborating with Invetech Pty Ltd, (Invetech) to develop a new closed processing system for cell therapy manufacturing whereby Invetech will provide system design and engineering development and we will develop applications for performing closed cell processing manipulations.
Facilities
With more than 55,000 square feet of development and manufacturing space in its Allendale, NJ, and Mountain View, CA facilities, PCT is a cGMP cell therapy center of excellence with facilities on both the East and the West Coast of the United States. These facilities include 5,500 square feet of controlled environment rooms (CERs) or clean rooms that are unidirectional-flow, negative-pressure, International Organization for Standardization (ISO) designation 7 (ISO7)/Class 10,000 classified and ISO6/Class 1000 which allows samples to be sent to EU for trials, and material pass-throughs. Each CER has controlled access, live facility and equipment monitoring with automated alarm call-out, dedicated HVAC systems, and is on an uninterruptible power supply (UPS) connection, maintained by an external diesel-fueled back-power generator. Each facility also contains cell and tissue cryogenic storage rooms, with controlled access, live facility and equipment monitoring with automated alarm call-out, and UPS connection, to ensure highest level of quality control and risk mitigation for product storage.
Our facilities are accredited by AABB (American Association of Blood Banks) and FACT (Foundation for the Accreditation of Cellular Therapy), hold all requisite licensures, are registered with the FDA as human cells, tissues, and cellular and tissue-based products (HCT/Ps) facilities, and maintain cGMP compliant quality systems. The Allendale facility has been
16
designed to be compliant with FDA and European Medicines Agency (EMA) standards for the manufacture of human cells for therapeutic use.
Our 15,000 square foot Irvine, CA facility also includes six ISO 14644-1 Class 7 (ISO7) positive-pressure clean rooms. Four of the clean rooms are designated for clinical manufacturing, with two designated for research and development. The clean rooms have certified ISO 14644-1 Class 8 (ISO8) gown rooms and are equipped with air-lock pass-through chambers for the transfer of material between clean rooms and outside lab areas. Each room has controlled access, live facility and equipment monitoring with automated alarm call-out, and dedicated HVAC systems. The Irvine facility has been granted a Tissue Bank License by the State of California Department of Public Health, enabling the facility to maintain patient samples and other biological materials indefinitely in its controlled access cryogenic storage room. The cGMP facility is designed to be FDA compliant and has received all requisite licensures for the manufacture of clinical grade products.
Competition
PCT's manufacturing business faces competition from other third party contract manufacturers, as well as more general competition from companies and academic and research institutions that may choose to self-manufacture rather than utilize a contract manufacturer. The two largest third party contract manufacturer competitors in the field of cell therapy are Lonza Group Ltd. and WuXi AppTec. Both of these companies are large, well-established manufacturers with financial, technical, research and development and sales and marketing resources that are significantly greater than those of PCT. In addition, both Lonza and WuXi have international capabilities that we do not currently possess though are pursuing. We also face competition from a number of other manufacturers that are somewhat smaller in size and have fewer resources than PCT.
More generally, we face competition inherent in any third party manufacturer's business: namely, that potential customers may instead choose to invest in their own facilities and infrastructure, affording them greater control over their products and the hope of long-term cost savings compared to a third party contract manufacturer. To be successful, we will need to convince potential customers that PCT's capabilities are more innovative, of higher-quality and more cost-effective than could be achieved through internal manufacturing and that our experience and expertise is unique in the industry. Our ability to achieve this and to successfully compete against other manufacturers will depend, in large part, on our success in developing superior automation technologies that improve both the quality and profitability associated with cell therapy manufacturing.
Cell Processing and Storage
We provide cell therapy processing and storage services in support of stem cell transplant programs at select hospitals throughout the country on a contract basis, where such hospitals do not have their own laboratory and processing services. Such services are provided at the cGMP standards, consistent with applicable national standards.
GOVERNMENT REGULATION
The health care industry is one of the most highly regulated industries in the United States and abroad. Various governmental regulatory authorities, as well as private accreditation organizations, oversee and monitor the activities of individuals and businesses engaged in the development, manufacture and delivery of health care products and services. The following is a general description of certain current laws and regulations that are relevant to our business.
HCT/P Regulations
Manufacturing facilities that produce cellular therapies are subject to extensive regulation by the FDA. In particular, FDA regulations set forth requirements pertaining to establishments that manufacture human cells, tissues, and cellular and tissue-based products (“HCT/Ps”). Title 21, Code of Federal Regulations, Part 1271 provides for a unified registration and listing system, donor-eligibility, current Good Tissue Practices (“cGTP”), and other requirements that are intended to prevent the introduction, transmission, and spread of communicable diseases by HCT/Ps. More specifically, key elements of Part 1271 include:
• | Registration and listing requirements for establishments that manufacture HCT/Ps; |
• | Requirements for determining donor eligibility, including donor screening and testing; |
• | cGTP requirements, which include requirements pertaining to the manufacturer's quality program, personnel, procedures, manufacturing facilities, environmental controls, equipment, supplies and reagents, recovery, processing and process controls, labeling, storage, record-keeping, tracking, complaint files, receipt, pre-distribution shipment, distribution, and donor eligibility determinations, donor screening, and donor testing; |
17
• | Adverse reaction reporting; |
• | Labeling of HCT/Ps; and |
• | FDA inspection, retention, recall, destruction, and cessation of manufacturing operations. |
PCT currently collects, processes, stores and manufactures HCT/Ps, including the manufacture of cellular therapy products. NeoStem Family Storage also collects, processes, and stores HCT/Ps. Therefore, both PCT and NeoStem Family Storage must comply with cGTP and with the current Good Manufacturing Practices ("cGMP") requirements that apply to biological products. Cell and tissue based products may also be subject to the same approval standards, including demonstration of safety and efficacy, as other biologic and drug products if they meet certain criteria such as if the cells or tissues are more than minimally manipulated or if they are intended for a non-homologous use. Management believes that requirements pertaining to premarket approval do not currently apply to PCT or NeoStem Family Storage because those entities are not currently investigating, marketing or selling cellular therapy products. If either PCT or NeoStem Family Storage changes its business operations in the future, the FDA requirements that apply to PCT or NeoStem Family Storage may also change.
State Regulation of Cell Therapy
Certain state and local governments regulate cell-processing facilities by requiring them to obtain other specific licenses. As required under applicable state law, PCT's New Jersey and California facilities are licensed, respectively, as a blood bank in New Jersey and as a biologics manufacturing facility in California. PCT also maintains licenses with respect to states that require licensure of out-of-state facilities that process cell, tissue and/or blood samples of residents of such states (e.g., New York and Maryland). PCT has the relevant state licenses needed for processing and is AABB (American Association of Blood Banks) accredited for this purpose. Management believes that it is in material compliance with currently applicable federal, state, and local laboratory licensure requirements, and intends to continue to comply with new licensing requirements that may become applicable in the future.
Certain states may also have enacted laws and regulations, or may be considering laws and regulations, regarding the use and marketing of stem cells or cell therapy products, such as those derived from human embryos. While these laws and regulations should not directly affect PCT's business, they could affect the business of some of PCT's clients and therefore the amount of business PCT receives from these clients.
Federal Regulation of Clinical Laboratories
The Clinical Laboratory Improvement Amendments (“CLIA”) extends federal oversight to clinical laboratories that examine or conduct testing on materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of disease or for the assessment of the health of human beings. CLIA requirements apply to those laboratories that handle biological matter. CLIA requires that these laboratories be certified by the government, satisfy governmental quality and personnel standards, undergo proficiency testing, be subject to biennial inspections, and remit fees. The sanctions for failure to comply with CLIA include suspension, revocation, or limitation of a laboratory's CLIA certificate necessary to conduct business, fines, or criminal penalties. Additionally, CLIA certification may sometimes be needed when an entity, such as PCT or NeoStem Family Storage, desire to obtain accreditation, certification, or license from non-government entities for cord blood collection, storage, and processing. PCT has obtained CLIA certification for its facilities in New Jersey. We have been advised that, currently, CLIA certification is not required for our PCT facilities in California. However, to the extent that any of the activities of PCT or NeoStem Family Storage (for example, with regard to processing or testing blood and blood products) require CLIA certification, PCT intends to obtain and maintain such certification and/or licensure.
Stem Cell Therapeutic and Research Act of 2005
The Stem Cell Therapeutic and Research Act of 2005 established a national donor bank of cord blood and created a national network for matching cord blood to patients. The National Marrow Donor Program (NMDP) carries out this legislation, which entails acting as the nation's Cord Blood Coordinating Center and actively recruiting parents for cord blood donations. The NMDP also administers the National Cord Blood Inventory (NCBI), which has a goal of collecting 150,000 cord blood units that could be used to treat patients all over the United States. Importantly, the legislation also authorized federal funding to support the legislation's goals for collecting cord blood units.
Pharmaceutical and Biologic Products
Government authorities in the United States, at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising and promotion, distribution, marketing, import and export of biological products such
18
as NBS10, NBS20 and NBS03D. The process of obtaining required regulatory approvals and the subsequent compliance with appropriate statutes and regulations require the expenditure of substantial time and money, and there is no guarantee that we will successfully complete the steps needed to obtain regulatory approval of NBS10, NBS20, NBS03D or any future product candidates. In addition, these regulations may change and our product candidates may be subject to new legislation or regulations.
In the United States, pharmaceutical and biologic products, including cellular therapies, are subject to extensive pre- and post-market regulation by the U.S. FDA. The Federal Food, Drug, and Cosmetic Act (“FD&C Act”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products are approved for marketing under provisions of the Public Health Service Act, or PHS Act. However, because most biological products also meet the definition of “drugs” under the FD&C Act, they are also subject to regulation under FD&C Act provisions. The PHS Act requires the submission of a biologics license application (“BLA”), rather than a New Drug Application ("NDA"), for market authorization. However, the application process and requirements for approval of BLAs are similar to those for NDAs, and biologics are associated with similar approval risks and costs as drugs.
Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical and biologic product development in the U.S. typically involves preclinical laboratory and animal tests, the submission to the FDA of a notice of claimed investigational exemption or an investigational new drug application (“IND”), which must become effective before clinical testing can commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug or biologic for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
Submission of an IND may not result in FDA authorization to initiate a clinical trial if FDA raises concerns or questions about the design of the clinical trial or the preclinical or manufacturing information supporting it, including concerns that human research subjects will be exposed to unreasonable health risks. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations; good clinical practice, or GCP, as set forth in FDA guidance, which is meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND. Sponsors of clinical trials of FDA regulated products, including drugs and biologics, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial is not being conducted in accordance with FDA requirements, or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB's requirements, or may impose other conditions.
Clinical trials to support NDAs or BLAs for marketing approval are typically conducted in four sequential phases, but the phases may overlap.
19
• | Phase 1: Studies are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients, such as cancer patients when the drug or biologic is too toxic to be ethically given to healthy individuals. |
• | Phase 2: Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
• | Phase 3: These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites. In most cases FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible. |
• | Phase 4: In some cases, FDA may condition approval of an NDA or BLA for a product candidate on the sponsor's agreement to conduct additional clinical trials after NDA or BLA approval. In other cases, a sponsor may voluntarily carry out additional trials post approval to gain more information about the drug or biologic. Such post approval trials are typically referred to as Phase 4 studies. |
After completion of the required clinical testing, an NDA or BLA is prepared and submitted to the FDA. FDA approval of the NDA or BLA is required before marketing of the product may begin in the U.S. The NDA or BLA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product's pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA or BLA is substantial. Under federal law, the submission of most NDAs or BLAs is additionally subject to a substantial application user fee, currently exceeding $2,335,000, and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees, currently exceeding $110,000 per product and $569,000 per establishment. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency's threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs and BLAs. Most such applications for standard review drug or biologic products are reviewed within ten to twelve months; most applications for priority review drugs or biologics are reviewed in six to eight months. FDA can extend these reviews by three months. Priority review can be applied to drugs or biologics that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. For biologics, priority review is further limited only for products intended to treat a serious or life-threatening disease relative to the currently approved products.
The FDA may refer applications for novel drug or biologic products, or drug or biologic products which present difficult questions of safety or efficacy, to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
Before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMP - a quality system regulating manufacturing - is satisfactory and the NDA or BLA contains data that provide substantial evidence that the drug or biologic is safe and effective in the indication studied.
After the FDA evaluates the NDA or BLA and the manufacturing facilities, it issues an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in 2 or 6 months depending on the type of information included.
Additional Controls
The PHS Act also provides authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the
20
creation and enforcement of regulations to prevent the introduction or spread of communicable diseases in the U.S. and between states.
Biosimilars
The Affordable Care Act (ACA), signed into law on March 23, 2010, included a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, which created an abbreviated approval pathway for biological products shown to be highly similar to, or interchangeable with, an FDA-licensed reference biological product. This is conceptually similar to the established process for generic drug approval in that it attempts to minimize duplicative testing. Biosimilarity sufficient to reference a prior FDA-approved product requires that there be no differences in conditions of use, route of administration, dosage form, and strength, and that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, which must be shown through analytical studies, animal studies, and a clinical trial or trials, unless the Secretary removes a required element. A biosimilar product may be deemed interchangeable with prior approved product if it meets the higher handle of demonstrating that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. No biosimilar or interchangeable products have been approved under the BPCIA to date. Complexities associated with the larger and often more complex structures of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being evaluated by the FDA.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) eighteen months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) 18 months after the resolution of a lawsuit over the patents of the reference biologics in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar's application has been approved if a patent lawsuit is ongoing within the 42 month period.
Post-Approval Regulation
Once an NDA or BLA is approved, a product will be subject to certain post-approval requirements. For instance, FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. As a condition of NDA or BLA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the product drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. The requirement for a REMS can materially affect the potential market and profitability of the product.
Drugs and biologics may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA supplement or BLA supplement before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements and BLA supplements as it does in reviewing NDAs or BLAs. The FDA has broad enforcement authority under the FDC Act, and failure to abide by these regulations can result in enforcement action, including the issuance of a Warning Letter directing entities to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and federal civil and criminal investigations, prosecutions and penalties. State enforcement actions relating to promotional violations are also becoming more common.
Adverse experiences associated with the use of the drug or biologic must be reported to the FDA and could result in the imposition of market restrictions through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval. The FDA may also require a labeling change if it becomes aware of new safety information that it believes should be included in the labeling of a drug or biologic.
Current Good Manufacturing Practices (cGMP) Standards
21
The FD&C Act and FDA regulations govern the quality control, manufacture, packaging, and labeling procedures of products regulated as a drug or biological product, including cellular therapies comprised of HCT/Ps. These laws and regulations include requirements for cGMP. These requirements are designed to ensure that a facility's processes - and products resulting from those processes - meet defined safety requirements. The cGMP requirements are federal regulations that govern the manufacture, processing, packaging and holding of drug and cell therapy products.
The objective of compliance with cGMP standards is to protect the public health and safety by ensuring that products (i) have the identity, strength, quality and purity that they purport or are represented to possess; (ii) meet their specifications; and (iii) are free of objectionable microorganisms and contamination.
A central focus of the cGMP requirements is to design and build quality into the manufacturing processes and the facilities in which products are produced and to ensure the consistency, product integrity, and reproducibility of results and product characteristics. This is done by implementing quality systems and processes including specifications and documentation.
In addition, drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs. Failure to comply with applicable FDA requirements can result in regulatory inspections and associated observations, warning letters, other requirements of remedial action, and, in the case of failures that are more serious, suspension of manufacturing operations, seizure, injunctions, product recalls, fines, and other penalties. We believe that our facilities are in material compliance with applicable existing FDA requirements.
Additionally, FDA, other regulatory agencies, or the United States Congress may be considering, and may enact laws or regulations regarding the use and marketing of stem cells, cell therapy products, or products derived from human cells or tissue. These laws and regulations can affect us directly or the business of some of PCT's clients and therefore the amount of business PCT receives from these clients.
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or BLAs or supplements to NDAs or BLAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers or deferrals for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The Best Pharmaceuticals for Children Act, or BPCA, provides NDA holders a six-month extension of any exclusivity-patent or non-patent-for a drug if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers. Under the BPCIA, BLA-holders may obtain a six-month extension of non-patent market exclusivity for a biologic if certain conditions are met.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition - generally a disease or condition that affects fewer than 200,000 individuals in the U.S. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA or BLA application user fee.
22
Cosmetics
Cosmetics are not subject to pre-market approval by the FDA, but the products, their ingredients and their label and labeling content are regulated by the FDA, and it is the burden of those who sell cosmetics to ensure that they are safe for use as directed. The FDA prohibits certain ingredients from being contained in cosmetic products that are authorized only for drug use or are deemed adulterated. In addition, the labeling of cosmetic products is subject to the requirements of the FD&C Act, the Fair Packaging Labeling Act and FDA regulations. The FDA limits cosmetic product claims to those of cleansing, beautification and enhancement to the external appearance of the skin. Claims to affect the structure or any function of the body are prohibited for cosmetic products as are disease prevention and treatment claims. It is possible that claims now commonly in use concerning cosmetic reduction in the external appearance of aging, the effect of cosmetic ingredients on fine lines and wrinkles, or on other aspects of appearance may in the future be deemed prohibited, implied disease treatment claims. FDA may initiate enforcement action against a cosmetic for violations of the FD&C Act and regulations, including claims that FDA deems to be drug claims.
Other Health Care Regulations
Health Privacy Laws
The Administrative Simplification provisions of the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH Act”), require health care plans, health care providers and health care clearinghouses, collectively defined under HIPAA as “Covered Entities,” to comply with standards for the use and disclosure of health information within such organizations and with third parties. These include standards for:
• | Common health care transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures; |
• | Unique identifiers for providers, employers, health plans and individuals; and |
• | Security and privacy of health information. |
Although the obligations of HIPAA only apply directly to Covered Entities, any Covered Entity that uses third parties (referred to in HIPAA as “Business Associates”) to perform functions on its behalf involving the creation or use of certain patient health information is required to have a contract with the Business Associate that limits the use and disclosure of such information by the Business Associate.
HIPAA does not preempt, or override, state privacy laws that provide even more protection for individuals' health information. These laws' requirements could further complicate Amorcyte's ability to obtain necessary research data from its collaborators. In addition, certain state privacy and genetic testing laws may directly regulate our research activities, affecting the manner in which we use and disclose individuals' health information, potentially increasing the cost of doing business, and exposing us to liability claims. In addition, patients and research collaborators may have contractual rights that further limit our ability to use and disclose individually identifiable health information. Claims that we violated individuals' privacy rights or breached its contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm the business.
While we believe that the current business operations of PCT or NeoStem Family Storage would not cause either of them to be considered a Covered Entity, there is a risk that due to conflicting interpretations of the regulations, NeoStem Family Storage may be deemed to be a Covered Entity. If NeoStem Family Storage is a Covered Entity, there is a risk of liability that NeoStem Family Storage may not be complying fully with all HIPAA requirements. PCT has signed Business Associate Agreements where requested by PCT's customers who are Covered Entities, which would require compliance with certain privacy and security requirements relating to individually identifiable health information created or used in connection with such relationships. PCT is in substantial compliance with such Business Associate Agreements. However, given the law's complexity and the possibility that the regulations may change and may be subject to changing and even conflicting interpretation, PCT's ability to comply fully with all of the HIPAA requirements and requirements of its Business Associate Agreements is uncertain. Further, as a result of amendments the HITECH Act, PCT's and NeoStem Family Storage's compliance burden has increased and they will be subject to audit and enforcement by the federal government and, in some cases, by state authorities. Further, they are obligated to publicly disclose wrongful disclosures or losses of personal health information.
Fraud and Abuse Laws
23
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal prosecution, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Affordable Care Act
In late March 2010, the Federal government enacted the comprehensive health care reform package, the Affordable Care Act (ACA). Among other provisions, the ACA imposes individual and employer health insurance requirements, provides certain insurance subsidies (e.g., premiums and cost sharing), mandates extensive insurance market reforms, creates new health insurance access points (e.g., State and federal-based health insurance exchanges), expands the Medicaid program, promotes research on comparative clinical effectiveness of different technologies and procedures, and makes a number of changes to how products and services will be reimbursed by the Medicare program. Amendments to the Federal False Claims Act under the ACA have made it easier for private parties to bring “qui tam” (whistleblower) lawsuits against companies, under which the whistleblower may be entitled to receive a percentage of any money paid to the government.
There are a number of provisions in the ACA that may directly impact our customers and, therefore, indirectly affect us. For example, the ACA expands the number of individuals that will be covered by either private or public health insurance, which may, in turn, increase the pool of potential purchasers for our customers' products to the extent they are reimbursable by private or public health insurance. The ACA also requires health insurance issuers in the individual and small group markets to cover certain “essential health benefits,” which include prescription drugs and which may increase coverage for our customers' products. In addition, the Affordable Care Act reduces income and raises costs for our customers through, for instance, the imposition of drug price discounts for Medicare Part D enrollees in the “donut hole” and the imposition of an annual fee on prescription drug and biologic manufacturers. Such provisions may cause our customers to seek to restrain costs in other areas, including the services that we provide. The effective dates of the various provisions within the ACA are staggered over the next several years, with some changes occurring immediately. Much of the interpretation of the ACA has been, and will continue to be, subject to administrative rulemaking, the development of agency guidance, and court interpretation.
Other Applicable Laws
In addition to those described above, other federal and state laws and regulations that could directly or indirectly affect our ability to operate the business and/or financial performance include:
• | state and local licensure, registration and regulation of laboratories, the processing and storage of human cells and tissue, and the development and manufacture of pharmaceuticals and biologics; |
• | other laws and regulations administered by the United States FDA, including the FDC Act and related laws and regulations and the PHS Act and related laws and regulations; |
• | laws and regulations administered by the United States Department of Health and Human Services, including the Office for Human Research Protections; |
• | state laws and regulations governing human subject research; |
• | federal and state coverage and reimbursement laws and regulations, including laws and regulations administered by the Centers for Medicare & Medicaid Services and state Medicaid agencies; |
24
• | the federal Medicare and Medicaid Anti-Kickback Law and similar state laws and regulations; |
• | the federal physician self-referral prohibition commonly known as the Stark Law, and state equivalents of the Stark Law; |
• | Occupational Safety and Health Administration (“OSHA”) requirements; |
• | state and local laws and regulations dealing with the handling and disposal of medical waste; and |
• | the Intermediate Sanctions rules of the IRS providing for potential financial sanctions with respect to “Excess Benefit Transactions” with HUMC or other tax-exempt organizations. |
Other Regulations
The Centers for Medicare & Medicaid Services, or CMS, has issued a final rule that implements a statutory requirement under the ACA that requires applicable manufacturers of drugs, devices, biologicals, or medical supplies that are covered under Medicare, Medicaid, or the Children’s Health Insurance Program, or CHIP, to begin collecting and reporting annually information on payments or transfers of value to physicians and teaching hospitals, as well as investment interests held by physicians and their immediate family members. Manufacturers had to begin collecting information in 2013, with the first reports due in 2014. On September 30, 2014, CMS posted the first round of data in searchable form on a public website. Failure to submit required information may result in civil monetary penalties.
In addition, several states now require prescription drug companies to report expenses relating to the marketing and promotion of drug products and to report gifts and payments to individual physicians in these states. Other states prohibit various other marketing-related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, California, Connecticut, Nevada, and Massachusetts require pharmaceutical companies to implement compliance programs and/or marketing codes. Several additional states are considering similar proposals. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws face civil penalties.
We are also subject to various local, state and federal laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including chemicals, micro-organisms and various radioactive compounds used in connection with our research and development activities. These laws include, but are not limited to, the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, there can be no assurances that accidental contamination or injury to employees and third parties from these materials will not occur. Our insurance program does not include environmental coverage.
Regulation in the European Union
In the European Union, or EU, medicinal products, including advanced therapy medicinal products, are subject to extensive pre- and post-market regulation by regulatory authorities at both the EU and national levels. Advanced therapy medicinal products comprise gene therapy products, somatic cell therapy products and tissue engineered products, which are cells or tissues that have undergone substantial manipulation and that are administered to human beings in order to regenerate, repair or replace a human tissue. We anticipate that our cell therapy products in development, including NBS10 and NBS10, would be regulated as advanced therapy medicinal products in the EU.
Clinical Trials
Clinical trials of medicinal products in the EU must be conducted in accordance with EU and national regulations and the International Conference on Harmonization, or ICH, guidelines on Good Clinical Practices, or GCP. Additional GCP guidelines from the European Commission, focusing in particular on traceability, apply to clinical trials of advanced therapy medicinal products. If the sponsor of the clinical trial is not established within the EU, it must appoint an entity within the EU to act as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU countries the sponsor is liable to provide ‘no fault’ compensation to any study subject injured in the clinical trial.
Prior to commencing a clinical trial, the sponsor must obtain a clinical trial authorization from the competent authority, and a positive opinion from an independent ethics committee. The application for a clinical trial authorization must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. Currently, clinical trial authorization applications must be submitted to the competent authority in each EU member state in which the trial will be conducted. Under the new Regulation on Clinical Trials, which will take effect in May 2016 at the earliest, there will be a centralized application procedure where one national authority takes the lead in reviewing the application and the other national authorities have only a limited involvement.
25
Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be notified to or approved by the relevant competent authorities and ethics committees.
The sponsor of a clinical trial must register the clinical trial in advance, and information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial will be made public as part of the registration. The results of the clinical trial must be submitted to the competent authorities and, with the exception of non-pediatric Phase 1 trials, will be made public at the latest within 12 months after the end of the trial.
During the development of a medicinal product, the European Medicines Agency, or EMA, and national medicines regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Scientific Advice Working Party of the Committee for Medicinal Products for Human Use, or CHMP. A fee is incurred with each scientific advice procedure. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future marketing authorization application of the product concerned. To date, we have not initiated any scientific advice procedures or other discussions with the EMA or any national regulatory authorities in the EU.
Marketing Authorizations
After completion of the required clinical testing, we must obtain a marketing authorization before we may place a medicinal product on the market in the EU. There are various application procedures available, depending on the type of product involved. All application procedures require an application in the common technical document, or CTD, format, which includes the submission of detailed information about the manufacturing and quality of the product, and non-clinical and clinical trial information. There is an increasing trend in the EU towards greater transparency and, while the manufacturing or quality information is currently generally protected as confidential information, the EMA and national regulatory authorities are now liable to disclose much of the non-clinical and clinical information in marketing authorization dossiers, including the full clinical study reports, in response to freedom of information requests after the marketing authorization has been granted. In October 2014, the EMA adopted a policy under which clinical study reports would be posted on the agency’s website following the grant, denial or withdrawal of a marketing authorization application, subject to procedures for limited redactions and protection against unfair commercial use. A similar requirement is contained in the new Regulation on Clinical Trials that will take effect in May 2016 at the earliest.
The centralized procedure gives rise to marketing authorizations that are valid throughout the EU and, by extension (after national implementing decisions), in Norway, Iceland and Liechtenstein, which, together with the EU member states, comprise the European Economic Area, or EEA. Applicants file marketing authorization applications with the EMA, where they are reviewed by a relevant scientific committee, in most cases the CHMP. The EMA forwards CHMP opinions to the European Commission, which uses them as the basis for deciding whether to grant a marketing authorization. The centralized procedure is compulsory for medicinal products that (1) are derived from biotechnology processes, (2) contain a new active substance (not yet approved on 20 November 2005) indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders, viral diseases or autoimmune diseases and other immune dysfunctions, (3) are orphan medicinal products or (4) are advanced therapy medicinal products, such as cell therapy medicines. For medicines that do not fall within these categories, an applicant may voluntarily submit an application for a centralized marketing authorization to the EMA, as long as the CHMP agrees that (i) the medicine concerned contains a new active substance (not yet approved on 20 November 2005), (ii) the medicine is a significant therapeutic, scientific, or technical innovation, or (iii) if its authorization under the centralized procedure would be in the interest of public health.
For those medicinal products for which the centralized procedure is not available, the applicant must submit marketing authorization applications to the national medicines regulators through one of three procedures: (1) a national procedure, which results in a marketing authorization in a single EU member state; (2) the decentralized procedure, in which applications are submitted simultaneously in two or more EU member states; and (3) the mutual recognition procedure, which must be used if the product has already been authorized in at least one other EU member state, and in which the EU member states are required to grant an authorization recognizing the existing authorization in the other EU member state, unless they identify a serious risk to public health. A national procedure is only possible for one member state; as soon as an application is submitted in a second member state the mutual recognition or decentralized procedure will be triggered.
Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria: the seriousness of the disease (e.g. heavy disabling or life-
26
threatening diseases) to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days.
Data Exclusivity
Marketing authorization applications for generic medicinal products do not need to include the results of pre-clinical and clinical trials, but instead can refer to the data included in the marketing authorization of a reference product for which regulatory data exclusivity has expired. If a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic marketing authorization applications referring to the data of that product may not be accepted by the regulatory authorities, and a further two years of market exclusivity, during which such generic products may not be placed on the market. The two-year period may be extended to three years if during the first eight years a new therapeutic indication with significant clinical benefit over existing therapies is approved.
There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials or manufacturing processes. For such products, the results of appropriate pre-clinical or clinical trials must be provided, and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and regulatory experience gained at the time.
Orphan Medicinal Products
The EMA’s Committee for Orphan Medicinal Products, or COMP, may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the EU. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria (for instance because in the meantime a new product was approved for the indication and no convincing data are available to demonstrate a significant benefit over that product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve any similar medicinal product, unless it offers a significant clinical benefit. This period may be reduced to 6 years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. The EMA is currently preparing guidance on assessing similarity of active ingredients for purposes of orphan exclusivity. It is possible that for biological products a narrow interpretation of similarity will be adopted.
Pediatric Development
In the EU, companies developing a new medicinal product must agree to a Pediatric Investigation Plan, or PIP, with the EMA and must conduct pediatric clinical trials in accordance with that PIP, unless a waiver applies, e.g. because the relevant disease or condition occurs only in adults. The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Post-Approval Controls
The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, or QPPV, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
27
All new marketing authorization applications must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. Risk management plans and PSURs are routinely available to third parties requesting access, subject to limited redactions.
All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another.
Manufacturing
Medicinal products may only be manufactured in the EU, or imported into the EU from another country, by the holder of a manufacturing authorization from the competent national authority. The manufacturer or importer must have a qualified person, or QP, who is responsible for certifying that each batch of product has been manufactured in accordance with EU standards of good manufacturing practice, or GMP, before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with GMP.
Human Cells and Tissues
Human cells and tissues that are intended for human applications but that do not fall within the scope of rules governing medicinal products or medical devices are not subject to premarket review and approval, nor do they require extensive preclinical and clinical testing. However, there are EU rules governing the donation, procurement, testing and storage of human cells and tissues intended for human application, whether or not they are advanced therapy medicinal products. These rules also cover the processing, preservation and distribution of human cell and tissues that are not advanced therapy medicinal products. Establishments that conduct such activities must be licensed and are subject to inspection by regulatory authorities. Such establishments must implement appropriate quality systems and maintain appropriate records to ensure that cells and tissues can be traced from the donor to the recipient and vice versa. There are also requirements to report serious adverse events and reactions linked to the quality and safety of cells and tissues. More detailed rules may exist at the national level.
Named Patient Sales
The EU medicines rules allow individual member states to permit the supply of a medicinal product without a marketing authorization to fulfill special needs, where the product is supplied in response to a bona fide unsolicited order, formulated in accordance with the specifications of a healthcare professional and for use by an individual patient under his direct personal responsibility. This may in certain countries also apply to products manufactured in a country outside the EU and imported to treat specific patients or small groups of patients. In addition, advanced therapy medicinal products do not need a marketing authorization if they are prepared on a non-routine basis and are used within the same EU member state in a hospital in accordance with a medical prescription for an individual patient.
These exemptions may allow us to make limited sales of our products before we obtain a marketing authorization in the EU. However, the exemptions could also allow our competitors to make sales without having obtained a marketing authorization and without undergoing the expense of clinical trials, especially if those competitors have cell processing facilities in the relevant EU member state. Similarly, certain hospitals may be able to compete with us on the basis of these rules.
Pricing and Reimbursement
Governments influence the price of medicinal products in the EU through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Regulation in Other Countries
28
We intend to seek to market our products in jurisdictions outside the U.S. and the EU. Most of these jurisdictions have product approval and post-approval regulatory processes that are similar in principle to those in the U.S. or EU. Any such considerations are in the early stages.
EMPLOYEES
As of December 31, 2014, we had 182 full-time employees, including the employees of our subsidiaries. Most of our senior management and professional employees have had prior experience in pharmaceutical or biotechnology companies. None of our employees is covered by collective bargaining agreements. We believe that our relations with our employees are good.
29
ITEM 1A. RISK FACTORS.
Our business, financial condition, operating results and cash flows can be affected by a number of factors, including, but not limited to, those set forth below, any one of which could cause our actual results to vary materially from recent results or from our anticipated future results. The risks described below are not the only ones we face, but those we currently consider to be material. There may be other risks which we now consider immaterial, or which are unknown or unpredictable, with respect to our business, our competition, the regulatory environment or otherwise that could have a material adverse effect on our business.
RISKS RELATED TO OUR FINANCIAL CONDITION AND CAPITAL REQUIREMENTS
We have incurred substantial losses and negative cash flow from operations in the past, and expect to continue to incur losses and negative cash flow for the foreseeable future.
We have a limited operating history, limited capital, and limited sources of revenue. Since our inception in 1980 through December 31, 2014, we have incurred aggregate net losses of approximately $291.2 million. Our net losses attributable to common stockholders for the years ended December 31, 2014 and December 31, 2013 were approximately $54.9 million and $39.0 million, respectively. As of December 31, 2014, our cash and cash equivalents were $19.2 million. The revenues generated in our cell therapy services business have not been, and are not expected in the foreseeable future to be, sufficient to cover costs attributable to that business or to our operations as a whole, including our development activities associated with our product candidates. Ultimately, we may never generate sufficient revenue from our cell therapy services business for us to reach profitability, generate positive cash flow or sustain, on an ongoing basis, our current or projected levels of product development and other operations.
We anticipate that we will need substantial additional financing to continue our operations; if we are unable to raise additional capital, we may be forced to delay, reduce or eliminate one or more of our product development programs, or expansion of our manufacturing operations and our business will be harmed.
Our current operating plan will require significant levels of additional capital to fund, the continued development of our cell therapy product candidates and the operation, enhancement and expansion of our manufacturing operations and our clinical development activities.
Our research and development expenses increased significantly over the past three years as a result of conducting the PreSERVE AMI Phase 2 clinical trial of NBS10 for which we reported primary analysis results in November 2014. We plan to meet with the FDA to determine next steps for development. Research and development expenses also have been increasing with our initiation in Q4 2014 of clinical trial sites for our Intus Phase 3 clinical trial for NBS20 for metastatic melanoma, for which we expect to randomize the first patient in Q2 2015. The cost of this trial is expected to be $25 million. We are also preparing for the initiation in late 2015 or 2016, depending on resource availability, of a Phase 2 clinical trial of NBS03D for diabetes and have other costs relating to that program, particularly due to the licensing of patents, data and collaboration with third parties. The Company's clinical activities are expected to continue to grow as these programs are advanced and they will require significant investment over a period of several years before they could be approved by FDA and commercialized by us, if ever. Even with encouraging results from the Phase 2 PreSERVE clinical trial and encouraging results that we could receive from our other earlier stage clinical trials for NBS10 and other products, we are required us to conduct additional clinical studies of the product, including larger and more expensive pivotal Phase 3 studies. To do so, we will need to raise additional money in the capital markets, enter into collaboration agreements with third parties or undertake some combination thereof. If we are unsuccessful in these efforts, we will likely need to otherwise delay or abandon the trials.
The amount and timing of our future capital requirements also will likely depend on many other factors, including:
• | the scope, progress, results, costs, timing and outcomes of our cell therapy research and development programs and product candidates; |
• | our ability to enter into any collaboration agreements with third parties for our product candidates and the timing and terms of any such agreements; |
• | the costs associated with the consummation of one or more strategic transactions; |
• | the timing of and the costs involved in obtaining regulatory approvals for our product candidates, a process which could be particularly lengthy or complex given the FDA's limited experience with marketing approval for cell therapy products; |
• | the costs of maintaining, expanding and protecting our intellectual property portfolio, including potential litigation costs and liabilities; and |
• | the cost of expansion of our development and manufacturing operations, including but not limited to the costs of expanded facilities, equipment costs, engineering and innovation initiatives and personnel. |
30
To both fund our clinical studies and support our future operations, we would likely seek to raise capital through a variety of different public and/or private financings vehicles. This could include, but not be limited to, use of our common stock purchase agreement with Aspire Capital, as described below, potential warrant exercises, option exercises, issuances of other debt or equity securities in public or private financings, and/or sale of assets. If we raise capital through the sale of equity, or securities convertible into equity, it would result in dilution to our then existing stockholders. Servicing the interest and principal repayment obligations under debt facilities, including our Oxford debt facility, or whether we call it, diverts funds that would otherwise be available to support research and development, clinical or commercialization activities. In addition, debt financing involves covenants that restrict our ability to operate our business. In certain cases, we also may seek funding through collaborative arrangements, that would likely require us to relinquish certain rights to our technology or product candidates and share in the future revenues associated with the partnered product.
Ultimately, we may be unable to raise capital or enter into collaborative relationships on terms that are acceptable to us, if at all. Our inability to obtain necessary capital or financing to fund our future operating needs could adversely affect our business, results of operations and financial condition.
We have never generated any revenue from product sales and our ability to generate revenue from product sales and become profitable depends significantly on our success in a number of factors.
We have no products approved for commercial sale, have not generated any revenue from product sales, and do not anticipate generating any revenue from product sales until some time after we have received regulatory approval for the commercial sale of a product candidate, which may never occur. Our ability to generate revenue and achieve profitability depends significantly on our success in many factors, including:
• | completing research regarding, and nonclinical and clinical development of, our product candidates; |
• | obtaining regulatory approvals and marketing authorizations for product candidates for which we complete clinical studies; |
• | developing a sustainable and scalable manufacturing process for our product candidates, including growing our own manufacturing capabilities and infrastructure; |
• | launching and commercializing product candidates for which we obtain regulatory approvals and marketing authorizations, either directly or with a collaborator or distributor; |
• | obtaining market acceptance of our product candidates as viable treatment options; |
• | addressing any competing technological and market developments; |
• | identifying, assessing, acquiring and/or developing new product candidates; |
• | negotiating favorable terms in any collaboration, licensing, or other arrangements into which we may enter; |
• | maintaining, protecting, and expanding our portfolio of intellectual property rights, including patents, trade secrets, and know-how; and |
• | attracting, hiring, and retaining qualified personnel. |
Even if one or more of the product candidates that we develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our expenses could increase beyond expectations if we are required by the FDA, or other regulatory agencies, domestic or foreign, to change our manufacturing processes or assays, or to perform clinical, nonclinical, or other types of studies in addition to those that we currently anticipate. If we are successful in obtaining regulatory approvals to market one or more of our product candidates, our revenue will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease patients is not as significant as we estimate, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved. If we are not able to generate revenue from the sale of any approved products, we may never become profitable.
Compliance with public company obligations, including the securities laws and regulations, is costly and requires significant management resources, and we may fail to comply. We are an “accelerated filer,” and beginning with our Form 10-Q for the
31
quarter ending March 31, 2014 and no longer qualify to report under smaller reporting company disclosure rules, and as a result are subject to more comprehensive disclosure obligations, with increased compliance costs.
The federal securities laws and regulations, including the corporate governance and other requirements of the Sarbanes-Oxley Act of 2002, impose complex and continually changing regulatory requirements on our operations and reporting. Because the aggregate market value of our public float was in excess of $75 million as of June 30, 2013, we became an “accelerated filer” as of the end of our 2013 fiscal year. As a result, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, our independent registered public accounting firm auditing our financial statements is now required to attest to and report on the effectiveness of our internal controls over financial reporting. The auditor attestation requirement applied to us for the first time with the filing of our Annual Report on Form 10-K for 2013. In addition, beginning with our Form 10-Q for March 31, 2014, we were required to satisfy all of the larger reporting company disclosure requirements. These requirements will increase our legal compliance obligations and costs, which could harm our results of operations and divert management’s attention from business operations.
Relatively speaking, we are a small company with limited resources. There can be no assurances that we will be able to comply with the added “accelerated filer” requirements by applicable deadlines and to maintain compliance in the future. If our independent registered public accounting firm is unable to provide us with an unqualified report as to the effectiveness of our internal control over financial reporting for future year ends, investors could lose confidence in the reliability of our financial reporting.
RISKS RELATED TO OUR CELL THERAPY PRODUCT DEVELOPMENT EFFORTS
Our future success is significantly dependent on the timely and successful development and commercialization of NBS20, our metastatic melanoma product candidate and if we encounter delays or difficulties in the development of this product candidate, as well as NBS10, our post-AMI product candidate, and NBS03D, our T1DM product candidate, that are at earlier stages of development, our business prospects would be significantly harmed.
We are dependent upon the successful development, approval and commercialization of our product candidates. Before we are able to seek regulatory approval of our product candidates, we must conduct and complete extensive clinical trials to demonstrate their safety and efficacy in humans. All of our product candidates are in early stages of development except for NBS20 which is the subject of a Phase 3 clinical trial for stage IV or recurrent stage III metastatic melanoma for which we began initiating clinical sites in Q42014 and expect to randomize the first patient in 2Q2015. We expect to complete enrollment of our Phase 3 Intus study in the fourth quarter of 2016.
For our Phase 2 PreSERVE study, we reported in November 2014 results from the primary analysis that allowed for important observations of a potential dose-dependent treatment effect that will help guide the next phase of development. However, for our primary efficacy endpoint, our hypothesis that an imaging technique (SPECT) used to measure perfusion could be used as a surrogate marker for the current medically relevant and regulatory endpoints was disproven, though it gave us valuable direction regarding endpoints and analyses for future clinical trials. We expect to complete the PreSERVE AMI study as defined through the final three-year follow-up and, in the meantime, plan to meet with the FDA to discuss our results and our proposal for the next step(s) in development. Finalization of the decision for next steps for NBS10 are expected in the second half of 2015. We also plan to initiate in late 2015 or early 2016 a Phase 2 study of NBS03D, a Treg based therapeutic being developed to treat T1DM. We are also actively exploring means by which we can take advantage of the paradigm of conditional approval for regenerative medicine products established by new regulations in Japan for products that show sufficient safety evidence and some evidence of efficacy with CLI and liver cancer being the indications we are considering. Clinical testing is expensive, difficult to design and implement, and can take many years to complete. Importantly, a failure of one or more of these or any other clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to complete our clinical studies, receive regulatory approval or commercialize our cell therapy product candidates, including the following:
• | suspensions, delays or changes in the design, initiation, enrollment, implementation or completion of required clinical trials; |
• | adverse changes in our financial position or significant and unexpected increases in the cost of our clinical development program; |
• | changes or uncertainties in, or additions to, the regulatory approval process that require us to alter our current development strategy; |
• | clinical trial results that are negative, inconclusive or even less than desired as to safety and/or efficacy, which could result in the need for additional clinical studies or the termination of the product's development; |
• | delays in our ability to manufacture the product in quantities or in a form that is suitable for any required clinical trials; |
32
• | intellectual property constraints that prevent us from making, using, or commercializing any of our cell therapy product candidates; |
• | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of these product candidates may be insufficient or inadequate: |
• | inability to generate sufficient preclinical, toxicology, or other in vivo or in vitro data to support the initiation of clinical studies; |
• | delays in reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical study sites; |
• | delays in obtaining required Institutional Review Board, or IRB, approval at each clinical study site; |
• | imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including after review of an IND application or amendment, or equivalent application or amendment; as a result of a new safety finding that presents unreasonable risk to clinical trial participants; a negative finding from an inspection of our clinical study operations or study sites; developments on trials conducted by competitors or approved products post-market for related technology that raises FDA concerns about risk to patients of the technology broadly; or if FDA finds that the investigational protocol or plan is clearly deficient to meet its stated objectives; |
• | difficulty collaborating with patient groups and investigators; |
• | failure by our CROs, other third parties, or us to adhere to clinical study requirements; |
• | failure to perform in accordance with the FDA’s current good clinical practices, or cGCPs, requirements, or applicable regulatory guidelines in other countries; |
• | delays in having patients qualify for or complete participation in a study or return for post-treatment follow-up; |
• | patients dropping out of a study; |
• | occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
• | changes in the standard of care on which a clinical development plan was based, which may require new or additional trials; |
• | transfer of manufacturing processes from our academic collaborators to larger-scale facilities operated by either a contract manufacturing organization, or CMO, or by us, and delays or failure by our CMOs or us to make any necessary changes to such manufacturing process; |
• | delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical studies or the inability to do any of the foregoing; and |
• | FDA may not accept clinical data from trials that are conducted at clinical sites in countries where the standard of care is potentially different from the United States. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
During our Phase 1 and Phase 2 trials of NBS10 for post AMI patients, serious adverse events occurred in subjects treated with NBS10 as well as in the control group, such as cardiogenic shock that resulted in the death of a patient in the Phase 2 trial; although the investigator deemed the event not related to the study product or procedures, we are not able to rule out a relationship. There can be no assurance that similar or other additional events will not occur as we continue to follow patients in the Phase 2 trial or that additional events will not occur. No concerns have been articulated by the Data Safety Monitoring Board ("DSMB"), a group charged with looking at unblinded results during the course of the study which recommended continuing our PreServe AMI Phase 2 clinical trial. In the Phase 2 clinical trial of NBS20 in metastatic melanoma serious adverse events included AMI, seizures, and acute myelogenous leukemia. However, the events in the trial were judged as unrelated to study participation by the investigator.
The Phase 2 study of NBS20 in metastatic melanoma originally was designed to include 200 patients. However, the study was terminated early due to funding issues faced by the prior owner of NBS20; as a result, a final analysis was conducted of 42 patients who had been randomized and received either TC or DC-TC.
33
Creation of NBS20, our cancer vaccine for melanoma, is based on a complex process that involves, among other things, the growing out in culture of cancer initiating cells for each patient until a sufficient number of purified cells have been obtained for the treatment, which is the first step to potential randomization into the Intus Phase 3 clinical trial if we experience problems with the manufacture of NBS20, our Phase 3 clinical trial could be delayed.
Even if we are able to successfully complete our clinical development program for our product candidates, and ultimately receive regulatory approval to market one or more of the products, we may, among other things:
Ÿ | obtain approval for indications that are not as broad as the indications we sought; | |
Ÿ | have the product removed from the market after obtaining marketing approval; | |
Ÿ | encounter issues with respect to the manufacturing of commercial supplies; | |
Ÿ | be subject to additional post-marketing testing requirements; and/or | |
Ÿ | be subject to restrictions on how the product is distributed or used. | |
We may experience delays in enrolling patients in our clinical trials, which could delay or prevent the receipt of necessary regulatory approvals.
We may not be able to initiate or complete as planned any clinical trials if we are unable to identify and enroll a sufficient number of eligible patients to participate in the clinical trials required by the FDA or other regulatory authorities. For example, we had originally expected to complete enrollment for the PreSERVE AMI Phase 2 trial of NBS10 earlier than its December 2013 completion. For our Intus Phase 3 trial of NBS20 in metastatic melanoma, we are targeting to complete enrollment by the fourth quarter of 2016 but challenges in enrolling patients could impede our ability to achieve this goal. We also may be unable to engage a sufficient number of clinical trial sites to conduct our trials. The challenge of enrolling patients is increased as we seek patients to participate in our trials from Europe, Australia and New Zealand or other jurisdictions to increase the likelihood of timely completing enrollment of our trial, which could raise clinical trial costs and could raise regulatory uncertainties, such as the willingness of the FDA to accept clinical data from certain foreign countries. Moreover, because PCT does not currently have manufacturing facilities operating outside of the United States, our ability to conduct trials outside of the U.S. may be constrained by our ability to transport trial materials to foreign destinations within the expiry period of such materials unless and until we commence operation outside of the United States or find another source of supply.
We may face challenges in enrolling patients to participate in our clinical trials due to the novelty of our cell-based therapies, the size of the patient populations and the eligibility criteria for enrollment in the trial. In addition, some patients may have concerns regarding cell therapy that may negatively affect their perception of therapies under development and their decision to enroll in the trials. Furthermore, patients suffering from diseases within target indications may enroll in competing clinical trials, which could negatively affect our ability to complete enrollment of our trials. Enrollment challenges in clinical trials often result in increased development costs for a product candidate, significant delays and potentially the abandonment of the clinical trial.
We may have other delays in completing our clinical trials and we may not complete them at all.
We have not completed the clinical trials necessary to obtain FDA approval to market NBS20, NBS10, NBS03D or any of our other products in development. Other than INTUS, we have not initiated Phase 3 studies for any of our other products in development. Our management lacks significant experience in completing Phase 3 trials and bringing a drug through commercialization. INTUS and clinical trials for other products in development may be delayed or terminated as a result of many factors, including the following:
• | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; |
• | failure by regulators to authorize us to commence a clinical trial; |
• | suspension or termination by regulators of clinical research for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with cGMP requirements; |
• | delays or failure to obtain clinical supply for our products necessary to conduct clinical trials from contract manufacturers, including commercial grade clinical supply for our Phase 3 clinical trials; |
• | treatment candidates demonstrating a lack of efficacy during clinical trials; |
• | inability to continue to fund clinical trials or to find a partner to fund the clinical trials; |
34
• | competition with ongoing clinical trials and scheduling conflicts with participating clinicians; and |
• | delays in completing data collection and analysis for clinical trials. |
Any delay or failure to complete clinical trials and obtain FDA approval for our product candidates could have a material adverse effect on our cost to develop and commercialize, and our ability to generate revenue from, a particular product candidate.
We may be unable to manage multiple late stage clinical trials for a variety of product candidates simultaneously.
As our current clinical trials progress, we may need to manage multiple late stage clinical trials simultaneously in order to continue developing all of our current products. Our management team does not have significant experience in completing late stage clinical trials and the management of late stage clinical trials is more complex and time consuming than early stage trials. Typically, early stage trials involve several hundred patients in no more than 10‑30 clinical sites. Late stage (Phase 3) trials may involve up to several thousand patients in up to several hundred clinical sites and may require facilities in several countries. Therefore, the project management required to supervise and control such an extensive program is substantially larger than early stage programs. As the need for these resources is not known until some months before the trials begin, it is necessary to recruit large numbers of experienced and talented individuals very quickly. If the labor market does not allow this team to be recruited quickly, the sponsor is faced with a decision to delay the program or to initiate it with inadequate management resources. This may result in recruitment of inappropriate patients, inadequate monitoring of clinical investigators and inappropriate handling of data or data analysis. Consequently it is possible that conclusions of efficacy or safety may not be acceptable to permit filing of a BLA for any one of the above reasons or a combination of several.
The development of our cell therapy product candidates are subject to uncertainty because autologous cell therapy is inherently variable.
When manufacturing an autologous cell therapy, the number and the composition of the cell population varies from patient to patient. Such variability in the number and composition of these cells could adversely affect our ability to manufacture autologous cell therapies in a cost-effective or profitable manner and meet acceptable product release specifications for use in a clinical trial or, if approved, for commercial sale. As a consequence, the development and regulatory approval process for autologous cell therapy products could be delayed or may never be completed.
Any disruption to our access to the reagents we are using in the clinical development of our cell therapy product candidates could adversely affect our ability to perform clinical trials and seek future regulatory submissions.
Reagents, including CD3 and CD28 antibody conjugated magnetic beads manufactured by Life Technologies Corporation, as well as, devices, materials and systems that we are using in our clinical trials, that we intend to use in our planned clinical trials and that we may need or use in commercial production, are provided by unaffiliated third parties. Any lack of continued availability of these reagents, devices, materials and systems for any reason would have a material adverse effect on our ability to complete these studies and could adversely impact our ability to achieve commercial manufacture of our planned therapeutic products. Although other available sources for these reagents, devices, materials and systems may exist in the marketplace, we have not evaluated their cost, effectiveness, or intellectual property foundation and therefore cannot guarantee the suitability or availability of such other potential sources.
The initiation of pivotal Phase 3 clinical trials for cell therapy product candidates, including NBS20, require the validation and establishment of manufacturing controls that may delay the products' development timeline.
To conduct pivotal Phase 3 clinical trials, we are required to have certain validated and established manufacturing controls with respect to the safety, purity and potency of our product when administered to patients. For example, in 2014, we began initiating clinical sites for our Intus Phase 3 clinical trial for stage IV or recurrent stage III metastatic melanoma. Although the FDA had questions about our potency the results of assay and placed the trial on clinical hold, we have since addressed those concerns, and the clinical hold was lifted. We believe our Phase 2 PreSERVE clinical trial of NBS10 in post-AMI patients were compelling and warrants further development although we expect to meet with the FDA to discuss our results and our proposal for the next step(s) in development. If we determine that the results of our planned Phase 2 clinical trial in T1DM or the results of any other Phase 2 clinical trial we may conduct support Phase 3 development, we expect to initiate and complete one or more pivotal Phase 3 clinical trials for that program and would need to address any outstanding chemistry, manufacturing and controls, or CMC, issues raised by the FDA prior to initiating such trials. We may not be successful in our efforts to address any CMC issues raised by the FDA. If we cannot initiate, or if we are delayed in initiating, a pivotal Phase 3 clinical program as a result of our failure to satisfy the FDA's CMC concerns or otherwise, the timing of our planned regulatory submission for commercialization
35
of our product candidates would be delayed, or we may be unable to seek regulatory approval to commercialize our products at all.
We may not obtain, or may be unable to maintain, approval for our pivotal Intus Phase 3 clinical trial in countries outside the US.
We intend to expand our Intus Phase 3 clinical trial of NBS20 to include sites in Europe, Australia, New Zealand and possibly elsewhere. In each country in which we intend to have a study site, we will need to obtain a clinical trial authorization from the local regulatory authority and an approval from the appropriate ethics committee. Such local regulatory authorities and ethics committees may refuse to approve our clinical trial, or may significantly delay approval if they raise issues with our application. We may not be successful in addressing any issues raised by the authorities and ethics committees. The authorities and ethics committees may also require amendments to the study protocol or other study documentation as a condition of approval and such changes may make it impractical for us to proceed with the study in that country. Following any approval, the authorities and ethics committees could suspend or terminate the trial if we are unable to comply with the applicable local regulatory requirements or if safety concerns arise during the conduct of the trial. Failure to obtain or maintain approval of our Intus Phase 3 clinical trial in one or more countries could delay the timing of our planned regulatory submission for commercialization of NBS20 or could negatively impact the likelihood of obtaining regulatory approval to commercialize NBS20.
The FDA has reviewed the protocol for the Phase 3 Intus clinical trial of NBS20; however, agreement by the FDA with the protocol under the SPA process does not guarantee that the trial will be successful or that, if successful, NBS20 will receive marketing approval on the basis of a single Phase 3 trial.
The FDA has reviewed, under the SPA process, the protocol for the Phase 3 Intus clinical trial of NBS20. An SPA is an agreement from the FDA that the design of a particular Phase 3 trial, including clinical endpoints, and statistical analyses are acceptable to serve as the primary basis for submission of a BLA. An SPA does not guarantee that NBS20 will receive marketing approval. Although an SPA is generally binding on the FDA, the FDA can choose not to honor an SPA for a number of reasons, including if the FDA identifies a substantial scientific issue essential to determining safety or efficacy after the study begins, public health concerns emerge that were unrecognized at the time of the protocol assessment, the sponsor and FDA agree to the change in writing, or if the study sponsor fails to follow the protocol that was agreed upon with the FDA. While FDA generally follows this practice, the agency has on occasion refused to honor an SPA because it no longer agrees with the study design at the time of the review of the application. Issues related to safety, study conduct, bias, deviation from the protocol, statistical power, patient completion rates, changes in scientific or medical parameters or internal inconsistencies in the data may also affect approvability. In addition, NBS20 may not achieve the primary endpoint of the trial. Even with the SPA agreement, the FDA may still require additional pivotal clinical trials as a condition for approving NBS20. Many companies that have been granted SPAs have ultimately failed to obtain final approval to market their products.
While the FDA requires in most cases two adequate and well-controlled pivotal clinical trials to demonstrate the efficacy of a product candidate, a single pivotal trial with other confirmatory evidence may be sufficient in rare instances where the trial is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible. Even if the primary endpoint in our single pivotal Phase 3 clinical trial is achieved, NBS20 may not be approved.
If the results for the primary endpoint are not robust, are subject to confounding factors, or are not adequately supported by other study endpoints, the FDA may refuse to approve our BLA based upon a single clinical trial. The FDA may also seek the guidance of an outside advisory committee prior to making its final decision.
Products that appear promising in research and development may be delayed or may fail to reach later stages of clinical development.
The successful development of pharmaceutical products is highly uncertain. Product candidates that appear promising in research and development may be delayed or fail to reach later stages of development. Decisions regarding the further development of product candidates must be made with limited and incomplete data, which makes it difficult to ensure or even accurately predict whether the allocation of limited resources and the expenditure of additional capital on specific product candidates will result in desired outcomes. Preclinical and clinical data can be interpreted in different ways, and negative or inconclusive results or adverse events during a clinical trial could delay, limit or prevent the development of a product candidate.
A Fast Track designation by the FDA may not lead to a faster development, regulatory review or approval process.
36
If a product is intended for the treatment of a serious or life-threatening condition and the product demonstrates the potential to address unmet needs for this condition, the treatment sponsor may apply for FDA Fast Track designation. NBS20 has received Fast Track designation. Fast track designation does not ensure that we will experience a faster development, regulatory review or approval process compared to conventional FDA procedures. Additionally, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from the clinical development program.
There can be no assurances that we will realize the desired benefits of the Orphan Drug designation granted to NBS20.
The FDA granted Orphan Drug designation for NBS20 for the treatment of stage IIIb through stage IV metastatic melanoma because it is intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as (i) the drug’s Orphan Designation in the US is revoked; (ii) its marketing approval is withdrawn; (iii) the Orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the Orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity.
Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. In addition to the potential for a period of market exclusivity, we may be eligible for grant funding of up to $400,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption form the FDA application user fee. There can be no assurances that we will realize the desired benefits of the orphan drug designation granted to NBS20. Even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve another drug for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care.
Our clinical trials may fail to demonstrate adequately the safety and efficacy of our product candidates, which would prevent or delay regulatory approval and commercialization.
The clinical trials of our product candidates are, and the manufacturing and marketing of our products will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and market our product candidates. Before obtaining regulatory approvals for the commercial sale of any of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that our product candidates are both safe and effective for use in each target indication. In particular, because our product candidates are subject to regulation as biological drug products, we will need to demonstrate that they are safe, pure, and potent for use in their target indications. Each product candidate must demonstrate an adequate risk versus benefit profile in its intended patient population and for its intended use. The risk/benefit profile required for product licensure will vary depending on these factors and may include not only the ability to show tumor shrinkage (in the case of NBS20), but also adequate duration of response, a delay in the progression of the disease, and/or an improvement in survival. For example, response rates from the use of our product candidates may not be sufficient to obtain regulatory approval unless we can also show an adequate duration of response. In addition, our Intus trial for NBS20 in metastatic melanoma contains a single endpoint - overall survival. While we believe unequivocal robust positive findings on overall survival would support approval, the lack of additional endpoints may make a finding of internally consistent results difficult and could present a risk should there be unexpected safety or efficacy results.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. Exploratory trends and results observed in earlier stage clinical trials, particularly trends and results observed for small subsets that were not prespecified, may not be replicated in later stage clinical trials. As is common with Phase 2 trials, we explored a number of endpoints and analyzed the data from our PreSERVE clinical trial of NBS10 in a number of ways, some of which were not prespecified. For example, the potential dose-dependent treatment effects we observed were seen in post hoc analyses of subsets based on the number of CD34 cells received, with our most encouraging results in a small subset of 15 patients. Product candidates such as NBS10 in Phase 3 clinical trials may fail to demonstrate sufficient efficacy despite having progressed through initial clinical trials, even if certain exploratory subset analyses of primary or secondary endpoints in those early trials showed trends toward efficacy or, in some analyses, nominal statistical significance. The results of studies in one set of patients or line of treatment may not be predictive of those obtained in another.
37
We expect there may be greater variability in results for products processed and administered on a patient-by-patient basis, as anticipated for our product candidates, than for “off-the-shelf” products, like many other drugs. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization.
Data from earlier studies conducted by the third-party research institutions such as Hoag Hospital for NBS20 and UCSF/Yale for NBS03D, should not be relied upon as evidence that later or larger-scale clinical trials will succeed. Some future trials may have different patient populations than current studies and will test our product candidates in different indications, among other differences. In addition, our proposed manufacturing processes for our product candidates include what we believe will be process improvements that are not part of the production processes that were previously used in the earlier conducted clinical trials being conducted by the research institutions. Accordingly, our results with our product candidates may not be consistent with the results of the clinical trials.
In addition, even if such trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. To the extent that the results of the trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates.
We presently lack sufficient manufacturing capabilities to produce our product candidates at commercial scale quantities and do not have an alternate manufacturing supply, which could negatively impact our ability to meet any future demand for the products.
Currently, PCT exclusively provides the cell processing services necessary for clinical production of NBS20 for our Phase 3 melanoma trial and will provide the cell processing services for our planned NBS03D Phase 2 diabetes trial as well as anticipated future trials of NBS10 for STEMI. PCT also provides services and produces materials for clinical trials on behalf of unaffiliated third parties. To date, PCT has not produced any products at commercial scale quantities. We expect that we would need to significantly expand our manufacturing capabilities to meet potential commercial demand for NBS20, NBS10, NBS03D and any other of our product candidates, if approved, as well as any of our other product candidates that might attain regulatory approval. Such expansion would require additional regulatory approvals. Even if we increase our manufacturing capabilities, it is possible that we may still lack sufficient capacity to meet demand. Ultimately, if we are unable to supply our products to meet commercial demand, whether because of processing constraints or other disruptions, delays or difficulties that we experience, sales of the products and their long term commercial prospects could be significantly damaged.
We do not presently have a third-party supply for NBS20, NBS10, NBS03D or any of our other product candidates. If our facilities where these product candidates are being manufactured or equipment were significantly damaged or destroyed, or if there were other disruptions, delays or difficulties affecting manufacturing capacity, our planned and future clinical studies and commercial production for these product candidates would likely be significantly disrupted and delayed. It would be both time consuming and expensive to replace this capacity with third parties, particularly since any new facility would need to comply with the regulatory requirements.
Ultimately, if we are unable to supply our cell therapy product candidates to meet commercial demand, were commercial approval obtained, whether because of processing constraints or other disruptions, delays or difficulties that we experience, our production costs could dramatically increase and sales of the product and its long-term commercial prospects could be significantly damaged.
The commercial potential and profitability of our products are unknown and subject to significant risk and uncertainty.
Even if we successfully develop and obtain regulatory approval for our cell therapy product candidates, the market may not understand or accept the products, which could adversely affect both the timing and level of future sales. Ultimately, the degree of market acceptance of our product candidates (or any of our future product candidates) will depend on a number of factors, including:
38
Ÿ | the clinical effectiveness, safety and convenience of the product particularly in relation to alternative treatments; | |
Ÿ | our ability to distinguish our products (which involve adult cells) from any ethical and political controversies associated with stem cell products derived from human embryonic or fetal tissue; and | |
Ÿ | the cost of the product, the reimbursement policies of government and third-party payors and our ability to obtain sufficient third-party coverage or reimbursement. | |
Even if we are successful in achieving sales of our product candidates, it is not clear to what extent, if any, the products will be profitable. The costs of goods associated with production of cell therapy products are significant. While we are working to improve the speed and efficiency and lower the cost of our manufacturing processes, there can be no assurance that we will be successful in these efforts. In addition, some changes in manufacturing processes or procedures generally require FDA or foreign regulatory authority review and approval prior to implementation. We may need to conduct additional preclinical studies and clinical trials to support approval of any such changes. Furthermore, this review process could be costly and time-consuming and could delay or prevent the commercialization of product candidates.
We may form or seek collaborations or strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements.
We may form or seek strategic alliances, create joint ventures or collaborations, or enter into additional licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute the shares of our existing stockholders, or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy.
Further, collaborations involving our product candidates, such as our collaborations with third-party research institutions, are subject to numerous risks, which may include the following:
• | collaborators have significant discretion in determining the efforts and resources that they will apply to a collaboration; |
• | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; |
• | collaborators may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing; |
• | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates; |
• | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to their marketing and distribution; |
• | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; |
• | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our product candidates, or that result in costly litigation or arbitration that diverts management attention and resources; |
• | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; and |
39
• | collaborators may own or co-own intellectual property covering our products that results from our collaborating with them, and in such cases, we would not have the exclusive right to commercialize such intellectual property. |
As a result, if we enter into collaboration agreements and strategic partnerships or license our products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture, which could delay our timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new collaborations or strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition, and results of operations.
We have limited experience in the development and marketing of cell therapies and may be unsuccessful in our efforts to establish a profitable business.
Over the past four years, we shifted our business plan entirely to focus on capturing a piece of the burgeoning field of cell therapy. Despite being in business for over eight years, we have limited experience in the areas of cell therapy product development and marketing, and in the related regulatory issues and processes. Although we have recruited a team that has experience with designing and conducting clinical trials, as a company, we have limited experience in conducting clinical trials and no experience in conducting clinical trials through to regulatory approval of any product candidate. In part because of this lack of experience, we cannot be certain that ongoing or planned clinical trials will begin or be completed on time, if at all. While PCT historically has provided services in connection with our development activities, we cannot assure you that our management will successfully oversee our clinical development efforts and our plans to capture a piece of the cell therapy market.
Our cell therapy business is based on novel technologies that are inherently expensive, risky and may not be understood by or accepted in the marketplace, which could adversely affect our future value.
The clinical development, commercialization and marketing of cell and tissue-based therapies are at an early-stage, substantially research-oriented, and financially speculative. To date, very few companies have been successful in their efforts to develop and commercialize a cell therapy product. In general, cell-based or tissue-based products may be susceptible to various risks, including undesirable and unintended side effects, unintended immune system responses, inadequate therapeutic efficacy, or other characteristics that may prevent or limit their approval or commercial use. In addition, NBS20 is an immunotherapy candidate that is produced by using a patient’s own dendritic cells loaded with antigens from irradiated tumor stem cells from the patient which are then suspended in an immune stimulant. Regulatory approval of novel product candidates such as NBS20, which is manufactured using novel manufacturing processes, can be more complex and expensive and take longer than other, more well-known or extensively studied pharmaceutical or biopharmaceutical products, due to the FDA’s lack of experience with them. To our knowledge, the FDA has only approved one personalized immunotherapy product to date. This lack of experience may lengthen the regulatory review process, require us to conduct additional studies or clinical trials, which would increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these product candidates or lead to significant post-approval limitations or restrictions. Furthermore, the number of people who may use cell or tissue-based therapies is difficult to forecast with accuracy. Our future success is dependent on the establishment of a large global market for cell- and tissue-based therapies and our ability to capture a share of this market with our product candidates.
If competitors develop and market products that are more effective, safer, or less expensive than our product candidates or offer other advantages, our commercial prospects will be limited.
Our cell therapy development programs now face, and will continue to face, intense competition from pharmaceutical, biopharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies engaged in drug discovery activities or funding, both in the United States and abroad. Some of these competitors are pursuing the development of drugs and other therapies that target the same diseases and conditions that we are targeting with our product candidates.
Because NBS10 generally targets patients without other revascularization options, we do not believe it will compete directly with pharmaceutical therapies being developed to treat less severe stages of our target indications. However, to the extent that therapies are developed that reverse the progression of the ischemic damage or improve blood flow to damaged tissue, they could have the effect of reducing demand for our product. In addition, because NBS10 requires the removal of bone marrow from the patient, potential competing products offering efficacious alternatives through a less invasive procedure may have a competitive advantage in terms of patient appeal. New pharmaceutical agents or devices that improve the repair of cardiac injury after a heart attack, with the result that fewer patients develop ischemic heart failure, would also represent a competitive threat for NBS10.
40
Furthermore, cell-based therapies, such as cardiac derived cells, bone marrow-derived stem cells and adipose cells are being pursued by companies such as Cardio3, Capricor, Mesoblast, Vericel Corporation, Angioblast Systems, Inc., Athersys, Inc., Pluristem Therapeutics, Inc., ReNeuron Group, Stemedica Cell Technologies Inc. and Bioheart, Inc. Some other companies, such as Cytori and Miltenyi, are developing medical devices to facilitate the production of therapeutic cell populations by clinicians for the treatment of NBS10’s target indications. Such devices may be approved by the FDA under a less rigorous regulatory process, and less extensive clinical testing and manufacturing controls than we are required to pursue for NBS10 and thus could reach the market well before NBS10.
With respect to NBS20, the field of melanoma drug development is highly competitive. According to a 2015 Research and Markets report, there are approximately 274 companies plus partners developing 310 drugs targeting melanoma in development. Several recently approved approaches have demonstrated improved clinical benefits in melanoma, including ipilumimab, an anti-CTLA-4 antibody (Bristol-Myers Squibb) and anti-PD-1 antibodies, nivolumab and pembrolizumab (Bristol-Myers Squibb and Merck, respectively). These drugs represent a new class of “immunotherapeutics” and have novel mechanisms of action targeting immune checkpoints. BRAF enzyme inhibitors include vemurafenib, marketed by Roche and dabrafenib from GlaxoSmithKline. Also recently approved is trametinib, a mitogen-activated protein kinase (MEK) from GlaxoSmithKline. These target critical intracellular protein pathways. In addition to the immune checkpoint modulators, several companies are developing therapeutic vaccines that act by priming a person’s immune system to recognize and attack the cancer cells. Amgen presented promising results with its Phase 3 melanoma vaccine, talimogene laherparepvec, T-VEC, which is an engineered virus injected directly into tumor and demonstrated survival. Northwest Biotherapeutics is developing a dendritic cell therapy which uses antigens derived from the lysate from the patient’s surgically resected tumor tissue. Northwest Biotherapeutics is in Phase 3 clinical studies in glioblastoma. Argos Therapeutics uses dendritic cells loaded with RNA amplified from a patient’s tumor to generate tumor specific antigens. A Phase 3 study in renal cell cancer is currently underway and is expected to complete in 2015. Some of these agents are presently on the market and others could reach the market before NBS20, representing competition that could materially impact the commercial success of NBS 20.
Our T regulatory cell therapy product candidate for recent onset Type 1 diabetes (NBS03D) faces competition from other immunomodulatory drugs being developed for other autoimmune diseases as well from other cellular therapies that fall outside of the coverage of our intellectual property. Current approaches include immune modulatory agents aiming to improve metabolic function by rescuing insulin producing beta cells, as well as regenerative agents that are hoping to replace beta cells altogether. There are many novel agents in early development, including ultra-low dose of Interleukin-2, which has been advanced to phase 2 by ILTOO Pharma. Also in early development are beta-cell replacement therapies such as that of ViaCYTE, Inc., which employ technologies that protect transplanted beta-cells from immune attack. If these therapies are easier to manufacture and have similar or better safety and efficacy profiles to NBS03D, the commercial prospects of our T regulatory cell therapy may be limited.
As a general matter, we also face competition from many other companies that are researching and developing cell therapies. Many of these companies have financial and other resources substantially greater than ours. In addition, many of these competitors have significantly greater experience in testing pharmaceutical and other therapeutic products, obtaining FDA and other regulatory approvals, and marketing and selling. If we ultimately obtain regulatory approval for any of our product candidates, we also will be competing with respect to manufacturing efficiency and marketing capabilities, areas in which we have limited or no commercial-scale experience. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in resources being even more concentrated by our competitors. Competition may increase further as a result of advances made in the commercial applicability of our technologies and greater availability of capital for investment in these fields.
Our cell therapy product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
With the enactment of the Biologics Price Competition and Innovation Act of 2009, or BPCIA, an abbreviated pathway for the approval of biosimilar and interchangeable biological products was created. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing reference product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product is approved under a biologics license application, or BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
We believe that if any of our product candidates are approved as a biological product under a BLA it should qualify for the 12-year period of exclusivity. However, there is a risk that the FDA will not consider any of our therapeutic candidates to be
41
reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Additionally, this period of regulatory exclusivity does not apply to companies pursuing regulatory approval via their own traditional BLA, rather than via the abbreviated pathway. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
We may be subject to significant product liability claims and litigation, including potential exposure from the use of our product candidates in human subjects, and our insurance may be inadequate to cover claims that may arise.
Our business exposes us to potential product liability risks inherent in the testing, processing and marketing of cell therapy products. Such liability claims may be expensive to defend and result in large judgments against us. We face an inherent risk of product liability exposure related to the testing of our current and any future product candidates in human clinical trials and will face an even greater risk with respect to any commercial sales of our products should they be approved. No product candidate has been widely used over an extended period of time, and therefore safety data is limited. Cell therapy companies derive the raw materials for manufacturing of product candidates from human cell sources, and therefore the manufacturing process and handling requirements are extensive, which increases the risk of quality failures and subsequent product liability claims. We presently have product liability insurance limited to $5 million per incident and $5 million in annual aggregate.
We will need to increase our insurance coverage when we begin commercializing product candidates, if ever. At that time, we may not be able to obtain or maintain product liability insurance on acceptable terms with adequate coverage or at all, or if claims against us substantially exceed our coverage, then our financial position could be significantly impaired.
Whether or not we are ultimately successful in any product liability litigation that may arise, such litigation could consume substantial amounts of our financial and managerial resources, decreased demand for our products and injure our reputation.
We seek to maintain errors and omissions, directors and officers, workers' compensation and other insurance at levels we believe to be appropriate to our business activities. If, however, we were subject to a claim in excess of this coverage or to a claim not covered by our insurance and the claim succeeded, we would be required to pay the claim from our own limited resources, which could have a material adverse effect on our financial condition, results of operations and business. Additionally, liability or alleged liability could harm our business by diverting the attention and resources of our management and damaging our reputation.
We may be unable to retain key officers or employees or hire new key officers or employees needed to implement our business strategy and develop our products and businesses.
Given the specialized nature of cell therapy and that it is a relatively new field, there is an inherent scarcity of experienced personnel in the field. We are substantially dependent on the skills and efforts of current senior management for their management and operations, as well as for the implementation of our business strategy. In addition, our future success depends upon our ability to attract and retain additional qualified personnel (including medical, scientific, technical, commercial, business and administrative personnel) necessary to support our anticipated growth, develop our business, perform our contractual obligations to third parties and maintain appropriate licensure. There can be no assurance that we will be successful in attracting or retaining personnel required by us to continue to grow our operations. The loss of a key employee, the failure of a key employee to perform in his or her current position or our inability to attract and/or retain skilled employees, as needed, could result in our inability to continue to grow our business or to implement our business strategy, or may have a material adverse effect on our business, financial condition and operating results.
The company's internal computer systems, or those used by its clinical investigators, clinical research organizations or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of development programs for the company's product candidates.
We rely on information technology systems to keep financial records, maintain laboratory and corporate records, communicate with staff and external parties and operate other critical functions. Any significant degradation or failure of these computer systems could cause the combined company to inaccurately calculate or lose its data. Despite the implementation of security measures, these internal computer systems and those used by the combined company's clinical investigators, clinical research organizations, and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. The techniques that could be used by criminal elements or foreign governments to attack these computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. While we have not experienced any such system failure, theft of information, accident or security breach to date, if such an event were to occur and cause interruptions in its operations, it could result in a material disruption of the combined company's clinical development activities. For example, the loss of clinical trial data from historical
42
or future clinical trials could result in delays in regulatory approval efforts and significantly increase costs to recover or reproduce the data. To the extent that any disruption, theft of information, or security breach were to result in a loss of or damage to data or applications, or inappropriate disclosure of confidential or proprietary information, the combined company could incur liability and the clinical development and the future development of its product candidates could be delayed.
RISKS RELATED TO OUR MANUFACTURING BUSINESS
Cell therapy is in its early stages; it is still a developing field and a significant global market for manufacturing services may never emerge.
Cell therapy is in its early stages and is still a developing area of research, with few cell therapy products approved for clinical use. Many of the existing cellular therapy candidates are based on novel cell technologies that are inherently risky and may not be understood or accepted by the marketplace, making difficult their own funding to enable them to continue their business. In addition to providing in-house process development and manufacturing expertise for our own product candidates in development, PCT provides consulting and manufacturing of cell and tissue-based therapeutic products in clinical trials and processing of stem cell products for transplantation programs for third parties. The number of people who may use cell or tissue-based therapies and thus the demand for cell processing services is difficult to forecast. If cell therapies under development by us or by others to treat disease are not proven effective, demonstrate unacceptable risks or side effects or, where required, fail to receive regulatory approval, our manufacturing business will be significantly impaired. While the therapeutic application of cells to treat serious diseases is currently being explored by a number of companies, to date there are only a handful of approved cell therapy products in the United States. Ultimately, our success in deriving revenue from manufacturing depends on the development and growth of a broad and profitable global market for cell- and tissue-based therapies and services and our ability to capture a share of this market through PCT.
PCT's revenues may vary dramatically from period to period making it difficult to forecast future results.
The nature and duration of PCT's contracts with customers often involve regular renegotiation of the scope, level and price of the services we are providing. If our customers reduce the level of their spending on research and development or marketing or are unsuccessful in attaining or retaining product sales due to market conditions, reimbursement issues or other factors, our results of operations may be materially impacted. In addition, other factors, including the rate of enrollment for clinical studies, will directly impact the level and timing of the products and services we deliver. As such, the levels of our revenues and profitability can fluctuate significantly from one period to another and it can be difficult to forecast the level of future revenues with any certainty.
We have a finite manufacturing capacity at PCT, which could inhibit the long-term growth prospects of this business.
We currently provide services and produce materials for clinical trials at our existing manufacturing facilities in Allendale, New Jersey and Mountain View, California. We also produce material for our Phase 3 Intus trial at our Irvine, California facility. These facilities are intended and have been designed to be compliant with FDA cGMP, and cGTP requirements. While we believe these facilities provide us with sufficient capacity to meet our expected near term needs, it is possible that the demand for our services and products could exceed our existing manufacturing capacity. We expect as our own cell therapy development programs progress and demand for cell therapy services in the industry expand, it may become necessary or desirable for us to expand our manufacturing capabilities for cell therapy services and products in the future, which may require us to invest significant amounts of capital and to obtain regulatory approvals. In this regard, we are reviewing opportunities for expansion to both commercial level and international manufacturing capabilities. If we are unable to meet rising demand for products and services on a timely basis or unable to maintain cGMP compliance standards, then it is likely that our clients and potential clients will elect to obtain the products and services from competitors and that the progress of our own programs will be impaired which could materially and adversely affect the level of our revenues, prospects for growth and overall success of our development programs.
Components of therapeutic products approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance with cGMP. Manufacturers of cell-based product candidates such as NBS20, NBS10 and NBS03D also must comply with cGTP. In addition, therapeutic products may be required to modify their manufacturing process from time to time in response to FDA requests. The manufacture of live cellular-based products is complex and subjects companies to significant regulatory burdens that may change over time. We may encounter difficulties in the production of our product candidates due to our limited manufacturing experience.
We will need to improve manufacturing efficiency at PCT in order to establish cost of goods levels that will permit approved products to succeed commercially.
43
PCT is working to improve the efficiency of cell therapy product development through the development of engineering and innovation solutions that think beyond current practices to develop long-term solutions to the unique challenges of cell therapy manufacturing with the ultimate goal of improving scale up, cost of goods quality control and robustness of the manufacturing process. We cannot provide assurances that we will be able to develop process enhancements that are acceptable to the FDA, on a timely basis, on commercially reasonable terms, or at all, or that any expected improvement in profitability will be realized. If we are unsuccessful in our efforts to develop these improvements, we may be unable to develop for ourselves or for our customers commercially viable products, and this inability would impair our ability to continue our operations.
We have a limited marketing staff and budget for our PCT operations, which could limit our ability to grow this business.
The degree of market acceptance of our products and services depends upon a number of factors, including the strength of our sales and marketing support. If our marketing is not effective, our ability to generate revenues could be significantly impaired. The newness of the industry and capital constraints provide challenges to our marketing and sales activities at PCT, and the failure to attract a sufficient base of customers will affect our ability to increase our revenues and operate profitably.
The logistics associated with the distribution of materials produced by PCT are significant, complex and expensive and may negatively impact our ability to generate and meet future demand for our products and improve profitability.
Current cell therapy products and product candidates, including our own, have a limited shelf life, in certain instances limited to less than 12 hours. Thus, it is necessary to minimize the amount of time between when the cell product is extracted from a patient, arrives at one of our facilities for processing, and is returned for infusion in the patient.
To do so, we need our cell therapy facilities to be located in major population centers in which patients are likely to be located and within close proximity of major airports. In the future, it may be necessary to build new facilities, which would require a significant commitment of capital and may not then be available to us. Even if we are able to establish such new facilities, we may experience challenges in ensuring that they are compliant with cGMP standards, FDA requirements, and/or applicable state or local regulations. We cannot be certain that we would be able to recoup the costs of establishing a facility in a given market. Given these risks, we could choose not to expand our cell processing and manufacturing services into new geographic markets which will limit our future growth prospects.
To effectively and efficiently deliver cell therapy product, we also need to establish and maintain cost-effective relationships with reliable and experienced transportation carriers. Most existing transportation carriers are not optimally designed for the transportation of cell therapy products. For example, these carriers generally lack a true point-to-point chain of control, may have non-controlled X-ray and inspection, do not guarantee package orientation, handling or storage conditions and, in many cases, lack a standard, documented and tracked operating procedures. While reliable ground carriers with experience in the transport of blood products exist in major U.S. metropolitan areas, air carriers meeting such needs are limited. If carriers we currently use should cease medical shipping operations or otherwise become unable to properly meet our transportation needs, the lack of access to safe, reliable and effective transportation options could adversely affect our ability to meet our customers' and our own needs.
44
RISKS RELATED TO GOVERNMENT REGULATION
The development and commercialization of our product candidates is subject to extensive regulation by the FDA and other regulatory agencies in the United States and abroad, and the failure to receive regulatory approvals for our cell therapy product candidates would likely have a material and adverse effect on our business and prospects.
To date, we have not received regulatory approval to market any of our product candidates in any jurisdiction. If we seek approval of any of our cell therapy product candidates, we will be required to submit to FDA and European and potentially other regulatory authorities extensive preclinical and clinical data supporting its safety and efficacy, as well as information about the manufacturing process and to undergo inspection of our PCT manufacturing facilities, among other things. The process of obtaining FDA and other regulatory approvals is expensive, generally takes many years and is subject to numerous risks and uncertainties, particularly with complex and/or novel product candidates such as our cell-based product candidates. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application or may make it easier for our competitors to gain regulatory approval to enter the marketplace. Ultimately, the FDA and other regulatory agencies have substantial discretion in the approval process and may refuse to accept any application or may decide that our product candidate data are insufficient for approval without the submission of additional preclinical, clinical or other studies. In addition, varying agency interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
Any of the following factors, among others, could cause regulatory approval for our product candidates to be delayed, limited or denied:
Ÿ | the product candidates require significant clinical testing to demonstrate safety and effectiveness before applications for marketing approval can be submitted to the FDA and other regulatory authorities; | |
Ÿ | data obtained from preclinical and nonclinical animal testing and clinical trials can be interpreted in different ways, and regulatory authorities may not agree with our respective interpretations or may require us to conduct additional testing; | |
Ÿ | negative or inconclusive results or the occurrence of serious or unexpected adverse events during a clinical trial could cause us to delay or terminate development efforts for a product candidate; and/or | |
Ÿ | FDA and other regulatory authorities may require expansion of the size and scope of the clinical trials. | |
Any difficulties or failures that we encounter in securing regulatory approval for our product candidates would likely have a substantial adverse impact on our ability to generate product sales, and could make any search for a collaborative partner more difficult.
We may be unsuccessful in our efforts to comply with applicable federal, state and international laws and regulations, which could result in loss of licensure, certification or accreditation or other government enforcement actions or impact our ability to secure regulatory approval of our product candidates.
Although we seek to conduct our business in compliance with applicable governmental healthcare laws and regulations, these laws and regulations are exceedingly complex and often subject to varying interpretations. The cell therapy industry is the topic of significant government interest, and thus the laws and regulations applicable to our business are subject to frequent change and/or reinterpretation. As such, there can be no assurance that we will be able, or will have the resources, to maintain compliance with all such healthcare laws and regulations. Failure to comply with such healthcare laws and regulations, as well as the costs associated with such compliance or with enforcement of such healthcare laws and regulations, may have a material adverse effect on our operations or may require restructuring of our operations or impair our ability to operate profitably.
Facilities engaged in the recovery, processing, storage, labeling, packaging or distribution of any HCT/Ps, or the screening or testing of a donor, are required to register with the FDA. Any third party retained by us to process our samples must be similarly registered with the FDA and comply with HCT/P regulations. We also are required to comply with FDA's cGTP regulations. If we fail to register or update registration information in a timely way, or fail to comply with cGTP regulations, we will be out of compliance with FDA regulations which could adversely affect our business.
Our manufacture of certain cellular therapy products for ourselves or at PCT on behalf of our customers triggers additional FDA requirements applicable to HCT/Ps, or products comprised of HCT/Ps, which are regulated as a drug, biological product, or medical device. FDA's cGMP regulations govern the manufacture, processing, packaging and holding of cell therapy products
45
regulated as drugs. FDA's Quality System Regulation, or QSR, similarly governs the manufacture, processing, packaging and holding of cell therapy products regulated as medical devices. We must comply with cGMP or QSR requirements including quality control, quality assurance and the maintenance of records and documentation for certain products. We may be unable to comply with these cGMP or QSR requirements and with other FDA, state and foreign regulatory requirements. These requirements may change over time and we or third-party manufacturers may be unable to comply with the revised requirements.
If we are unable to conduct clinical studies in accordance with regulations and accepted standards, we may be delayed in receiving, or may never receive, regulatory approvals of our product candidates from the FDA and other regulatory authorities.
To obtain marketing approvals for our product candidates in the United States and abroad, we must, among other requirements, complete adequate and well-controlled clinical trials sufficient to demonstrate to the FDA and other regulatory bodies that the product candidate is safe and effective for each indication for which approval is sought. If the FDA finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury, due to, among other things, occurrence of a serious adverse event in an ongoing clinical trial, the FDA can place one or more of our clinical trials on hold. If safety concerns develop, we may, or the FDA or an institutional review board may require us to, stop the affected trials before completion. Our Phase 1 trial of NBS10 was subject to a clinical hold following the death of a subject in the study. We presented evidence that the death was the result of ventricular fibrillation attributed to recurrent myocardial infarction from stent thrombosis preceding infusion of NBS10 and the FDA lifted the clinical hold.
The completion of our clinical trials also may be delayed or terminated for a number of other reasons, including if:
Ÿ | third-party clinical investigators do not perform the clinical trials on the anticipated schedule or consistent with the clinical trial protocol, good clinical practices required by the FDA and other regulatory requirements, or other third parties do not perform data collection and analysis in a timely or accurate manner; | |
Ÿ | inspections of clinical trial sites by the FDA or by institutional review boards of research institutions participating in the clinical trials, reveal regulatory violations that require the sponsor of the trial to undertake corrective action, suspend or terminate one or more sites, or prohibit use of some or all of the data in support of marketing applications; or | |
Ÿ | the FDA or one or more institutional review boards suspends or terminates the trial at an investigational site, or precludes enrollment of additional subjects. | |
Our development costs will increase if there are material delays in our clinical trials, or if we are required to modify, suspend, terminate or repeat a clinical trial. If we are unable to conduct our clinical trials properly, we may never receive regulatory approval to market our product candidates.
We will continue to be subject to extensive FDA regulation following any product approvals, and if we fail to comply with these regulations, we may suffer a significant setback in our business.
Even if we are successful in obtaining regulatory approval of our product candidates, we will continue to be subject to the requirements of and review by, the FDA and comparable regulatory authorities in the areas of manufacturing processes, post-approval clinical data, adverse event reporting, labeling, advertising and promotional activities, among other things. In addition, any marketing approval we receive may be limited in terms of the approved product indication or require costly post-marketing testing and surveillance. Discovery after approval of previously unknown problems with a product, manufacturer or manufacturing process, or a failure to comply with regulatory requirements, may result in actions such as:
Ÿ | warning letters or untitled letters or other actions requiring changes in product manufacturing processes or restrictions on product marketing or distribution; | |
Ÿ | product recalls or seizures or the temporary or permanent withdrawal of a product from the market; and | |
Ÿ | fines, restitution or disgorgement of profits or revenue, the imposition of civil penalties or criminal prosecution. | |
The occurrence of any of these actions would likely cause a material adverse effect on our business, financial condition and results of operations.
Health care companies have been the subjects of federal and state investigations, and we could become subject to investigations in the future.
Both federal and state government agencies have heightened civil and criminal enforcement efforts. There are numerous ongoing investigations of health care companies, as well as their executives and managers. In addition, amendments to the Federal False Claims Act, including under Healthcare Reform, have made it easier for private parties to bring “qui tam” (whistleblower)
46
lawsuits against companies under which the whistleblower may be entitled to receive a percentage of any money paid to the government. The Federal False Claims Act provides, in part, that an action can be brought against any person or entity that has knowingly presented, or caused to be presented, a false or fraudulent request for payment from the federal government, or who has made a false statement or used a false record to get a claim approved. The government has taken the position that claims presented in violation of the federal anti-kickback law, Stark Law or other healthcare-related laws, including laws enforced by the FDA, may be considered a violation of the Federal False Claims Act. Penalties include substantial fines for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person or entity and/or exclusion from the Medicare program. In addition, a majority of states have adopted similar state whistleblower and false claims provisions.
We are not aware of any government investigations involving any of our facilities or management. While we believe that we are in material compliance with applicable governmental healthcare laws and regulations, any future investigations of our business or executives could cause us to incur substantial costs, and result in significant liabilities or penalties, as well as damage to our reputation.
It is uncertain to what extent the government, private health insurers and third-party payors will approve coverage or provide reimbursement for the therapies and products to which our services relate. Availability for such reimbursement may be further limited by an increasing uninsured population and reductions in Medicare and Medicaid funding in the United States.
To the extent that health care providers cannot obtain coverage or reimbursement for our therapies and products, they may elect not to provide such therapies and products to their patients and, thus, may not need our services. Further, as cost containment pressures are increasing in the health care industry, government and private payors may adopt strategies designed to limit the amount of reimbursement paid to health care providers.
Similarly, the trend toward managed health care and bundled pricing for health care services in the United States, could significantly influence the purchase of healthcare services and products, resulting in lower prices and reduced demand for our therapeutic products under development.
We may receive a portion of our revenues from services rendered to patients enrolled in federal health care programs, such as Medicare, and we may also directly or indirectly receive revenues from federal health care programs. Federal health care programs are subject to changes in coverage and reimbursement rules and procedures, including retroactive rate adjustments. These contingencies could materially decrease the range of services covered by such programs or the reimbursement rates paid directly or indirectly for our products and services. To the extent that any health care reform favors the reimbursement of other therapies over our therapeutic products under development, such reform could affect our ability to sell our services, which may have a material adverse effect on our revenues.
The limitation on reimbursement available from private and government payors may reduce the demand for, or the price of, our services, which could have a material adverse effect on our revenues. Additional legislation or regulation relating to the health care industry or third-party coverage and reimbursement may be enacted in the future which could adversely affect the revenues generated from the sale of our products and services.
Furthermore, there has been a trend in recent years towards reductions in overall funding for Medicare and Medicaid. There has also been an increase in the number of people who do not have any form of health care coverage in recent years and who are not eligible for or enrolled in Medicare, Medicaid or other governmental programs. The extent to which the reforms brought about under Healthcare Reform may be successful in reducing the number of such uninsured is unclear, and the reduced funding of governmental programs and increase in uninsured populations could have a negative impact on the demand for our services to the extent they relate to products and services which are reimbursed by government and private payors.
Unintended consequences of healthcare reform legislation in the U.S. may adversely affect our business.
The healthcare industry is undergoing fundamental changes resulting from political, economic and regulatory influences. In the U.S., comprehensive programs are under consideration that seek to, among other things, increase access to healthcare for the uninsured and control the escalation of healthcare expenditures within the economy. On March 23, 2010, healthcare reform legislation was approved by Congress and has been signed into law. While we do not believe this legislation will have a direct impact on our business, the legislation requires the adoption of implementing regulations, which may have unintended consequences or indirectly impact our business. For instance, the scope and implications of the amendments pursuant to the Fraud Enforcement and Recovery Act of 2009 (“FERA”), have yet to be fully determined or adjudicated and as a result it is difficult to predict how future enforcement initiatives may impact our business. Also, in some instances our clients may be health insurers that will be subject to limitations on their administrative expenses and federal review of “unreasonable” rate increases that could impact the
47
prices they pay for our services. If the legislation causes such unintended consequences or indirect impact, it could have a material adverse effect on our business, financial condition and results of operations.
Competitor companies or hospitals may be able to take advantage of EU rules permitting sales of unlicensed medicines for individual patients to sell competing products without a marketing authorization.
The EU medicines rules allow individual member states to permit the supply of a medicinal product without a marketing authorization to fulfill special needs, where the product is supplied in response to a bona fide unsolicited order, formulated in accordance with the specifications of a healthcare professional and for use by an individual patient under his direct personal responsibility. This may in certain countries also apply to products manufactured in a country outside the EU and imported to treat specific patients or small groups of patients. In addition, advanced therapy medicinal products do not need a marketing authorization if they are prepared on a non-routine basis and are used within the same EU member state in a hospital in accordance with a medical prescription for an individual patient.
These exemptions could allow our competitors to make sales in the EU without having obtained a marketing authorization and without undergoing the expense of clinical trials, especially if those competitors have cell processing facilities in the relevant EU member state. Similarly, certain hospitals may be able to compete with us on the basis of these rules. Because any such sales would be made without a marketing authorization, there would be no need for the competitor company or hospital to refer to the clinical data in our marketing authorization dossiers, and so any data exclusivity protection that we may obtain for our products would not prevent such competing sales.
A variety of risks associated with operating our business internationally could materially adversely affect our business.
We plan to seek regulatory approval of our product candidates outside of the United States and, accordingly, we expect that we, and any potential collaborators in those jurisdictions, will be subject to additional risks related to operating in foreign countries, including:
• | differing regulatory requirements in foreign countries; |
• | unexpected changes in tariffs, trade barriers, price and exchange controls, and other regulatory requirements; |
• | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
• | compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
• | foreign taxes, including withholding of payroll taxes; |
• | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
• | difficulties staffing and managing foreign operations; |
• | workforce uncertainty in countries where labor unrest is more common than in the United States; |
• | potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign laws; |
• | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
• | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
• | business interruptions resulting from geo-political actions, including war and terrorism. |
These and other risks associated with our planned international operations may materially adversely affect our ability to attain or maintain profitable operations.
48
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
We may be unable to obtain or maintain patent protection for our products and product candidates, which could have a material adverse effect on our business.
Our commercial success will depend, in part, on obtaining and maintaining patent protection for new technologies, product candidates, products and processes and successfully defending such patents against third party challenges. To that end, we file patent applications, and have been issued patents, that are intended to cover certain methods and uses of stem cells, including very small embryonic-like stem cells, as well as compositions and methods relating to T regulatory cells and hematopoietic stem cells, and methods of making and using dendritic cell-based vaccines. These patent applications may never result in the issuance of patents.
The patent positions of biotechnology companies can be highly uncertain and involve complex legal, scientific and factual questions and recent court decisions have introduced significant uncertainty regarding the strength of patents in the industry. Moreover, the legal systems of some foreign countries do not favor the aggressive enforcement of patents and may not protect our intellectual property rights to the same extent as the laws of the United States. Any of the issued patents we own or license may be challenged by third parties and held to be invalid, unenforceable or with a narrower or different scope of coverage that what we currently believe, effectively reducing or eliminating protection we believed we had against competitors with similar products or technologies. If we ultimately engage in and lose any such patent disputes, we could be subject to competition and/or significant liabilities, we could be required to enter into third-party licenses or we could be required to cease using the disputed technology or product. In addition, even if such licenses are available, the terms of any license requested by a third party could be unacceptable or unaffordable to us.
Product development and approval timelines in the biotechnology industry are very lengthy. As such, it is possible that any patents that may cover an approved product may have expired at the time of commercialization or only have a short remaining period of exclusivity, thereby reducing the commercial advantages of the patent. In such case, we would then rely solely on other forms of exclusivity, such as regulatory exclusivity provided by the FDC Act, which may provide less protection to our competitive position.
Litigation relating to intellectual property is expensive, time consuming and uncertain, and we may be unsuccessful in our efforts to protect against infringement by third parties or defend ourselves against claims of infringement.
To protect our intellectual property, we may initiate litigation or other proceedings. In general, intellectual property litigation is costly, time-consuming, diverts the attention of management and technical personnel and could result in substantial uncertainty regarding our future viability, even if we ultimately prevail. Some of our competitors may be able to sustain the costs of such litigation or other proceedings more effectively than can we because of their substantially greater financial resources. The loss or narrowing of our intellectual property protection, the inability to secure or enforce our intellectual property rights or a finding that we have infringed the intellectual property rights of a third party could limit our ability to develop or market our products and services in the future or adversely affect our revenues. Furthermore, any public announcements related to such litigation or regulatory proceedings could adversely affect the price of our common stock.
Third parties may allege that the research, development and commercialization activities we conduct infringe patents or other proprietary rights owned by such parties. While we do not believe any of our current activities infringe the rights of others, we have not conducted an exhaustive search or analysis of third-party patent rights to determine whether our pre-clinical or clinical research and development or activities may infringe or be alleged to infringe any third-party patent rights. If we are found to have infringed the patents of a third party, we may be required to pay substantial damages; we also may be required to seek from such party a license, which may not be available on acceptable terms, if at all, to continue our activities. A judicial finding or infringement or the failure to obtain necessary licenses could prevent us from commercializing our products, which would have a material adverse effect on our business, operating results and financial condition.
If we are unable to maintain our licenses, patents or other intellectual property we could lose important protections that are material to continuing our operations and our future prospects.
To obtain and maintain patent protection and licensing rights under certain of our license agreement, we must, among other things, ensure the timely payment of all applicable filing and maintenance fees. Any failure to do so could result in the loss of some or all of our rights to proprietary technology or the inability to secure or enforce intellectual property protection.
49
Additionally, our license agreements require us to meet certain diligence obligations in the development of the licensed products. Our failure to meet these diligence obligations could result in the loss of some or all of our rights, which could materially and adversely affect our business and future prospects.
If we are unable to protect the confidentiality of trade secrets, our competitive position could be impaired.
A significant amount of our technology, especially regarding manufacturing processes, is unpatented and is maintained as trade secrets. We expend significant energy, resources and know-how in an effort to protect these trade secrets and know-how, including through the use of confidentiality agreement. Even so, improper use or disclosure of our confidential information could occur and in such case adequate remedies may not exist. The disclosure of our trade secrets and trade-secrets could impair our Company's competitive position.
In certain countries, patent holders may be required to grant compulsory licenses, which would likely have a significant and detrimental effect on any future revenues in such country.
Many countries, including some countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, most countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may be limited to monetary relief and may be unable to enjoin infringement, which could materially diminish the value of the patent. Compulsory licensing of life-saving products is also becoming increasingly common in developing countries, either through direct legislation or international initiatives. Such compulsory licenses could be extended to our product candidates, which may limit our potential revenue opportunities, including with respect to any future revenues that may result from our product candidates.
Changes to U.S. Patent Law may have a material adverse effect on our intellectual property rights.
The Leahy-Smith America Invents Act (AIA), which was signed into law on September 16, 2011, significantly changes United States patent law. It may take some time to establish what the law means, since it is just being interpreted by the lower courts, and any lower court decisions have not been reviewed by either the Federal Circuit Court of Appeals or the Supreme Court, a process that will take years. The first major change is that AIA switches the U.S. patent system from a “first to invent” system to a “first to file” system. Now that the first to file system is in effect, there is a risk that another company may independently develop identical or similar patents at approximately the same time, and be awarded the patents instead of us. Further, for the second major change, AIA abolished interference proceedings, and establishes derivation proceedings to replace interference proceedings in all cases in which the time period for instituting an interference proceeding has not lapsed where an inventor named in an earlier application derived the claimed invention from a named inventor. Now that the derivation proceedings are in effect, there is a risk that the inventorship of any pending patent application can be challenged for reasons of derivation. The third major change is that AIA established post-grant opposition proceedings that will apply only to patent applications filed after “first to file” became effective. Post-grant opposition will enable a person who is not the patent owner to initiate proceedings in the Patent office within 9 months after the grant of a patent that can result in cancellation of a patent as invalid. Therefore there is a risk that any of our patents once granted after the effective date of these provisions of the AIA (March 16, 2013) may be subject to post-grant opposition, which will increase uncertainty on the validity of any newly granted patent or may ultimately result in cancellation of the patent.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents, trademarks and proprietary rights of third parties. There have been many lawsuits and other proceedings involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions, and reexamination proceedings before the USPTO and corresponding foreign patent offices and trademark violations. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing products and services. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our products and services may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture, or methods for treatment related to the use or manufacture of our products and services. We have conducted freedom to operate analyses with respect to only certain of our products and services, and therefore we do not know whether there are any third-party patents that would impair our ability to commercialize these products and services. We also cannot guarantee that any of our analyses are complete and thorough, nor can we be sure that we have identified each and every patent and pending application in the United States and abroad that is relevant or necessary to the commercialization of our products and services. Because patent applications can take many
50
years to issue, there may be currently pending patent applications that may later result in issued patents that our products or services may infringe.
In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover aspects of our products or services, the holders of any such patents may be able to block our ability to commercialize such products or services unless we obtained a license under the applicable patents, or until such patents expire or are finally determined to be invalid or unenforceable. Such a license may not be available on commercially reasonable terms or at all.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our products or services. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
RISKS RELATED TO OUR CAPITAL STOCK
Our stock price has been, and will likely continue to be, highly volatile.
The market price of our common stock has been and in the future may continue to be highly volatile. For example, from January 1, 2014 through March 2, 2015 our Common Stock traded as low as $3.08 per share and as high as $8.29 per share; in 2013, our common stock traded as low as $5.00 per share and as high as $9.89 per share.
The market price for our common stock is highly dependent on, among other things, our clinical development efforts the profitability and growth of our cell therapy services business and the growth of our business in general, the amount of our available cash and investments and our level of cash utilization. Future events could increase the volatility seen in our common stock and ultimately cause a significant decline in the price of our common stock and ultimately impact our ability to raise additional capital in the future. These events could include the following, among others:
• | low levels of trading volume for our shares; |
• | capital-raising or other transactions that are, or may in the future be, dilutive to existing stockholders or that involve the issuance of debt securities; |
• | delays in our clinical trials, negative clinical trial results or adverse regulatory decisions relating to our product candidates; |
• | adverse fluctuations in our revenues or operating results or financial results that otherwise fall below the market's expectations; |
• | disappointing developments concerning our cell therapy services clients or other collaborators for our product candidates; and |
• | legal challenges, disputes and/or other adverse developments impacting our patents or other proprietary rights that protect our products. |
In addition, broader external events, such as news concerning economic or market conditions in the general economy or within our industry, the activities of our competitors, changes (or the threat of changes) in U.S. or foreign government regulations impacting the life sciences industry or the movement of capital into or out of our industry, are likely to affect the price of our Common Stock. There can be no assurance that the market price of our common stock will not continue to fluctuate or decline significantly in the future.
In addition to potential dilution associated with future fundraising transactions, we currently have significant numbers of securities outstanding that are exercisable for our Common Stock, which could result in significant additional dilution and downward pressure on our stock price.
As of December 31, 2014, there were 36,783,857 shares of our Common Stock outstanding. In addition, there were outstanding stock options and warrants representing the potential issuance of an additional 7,978,232 shares of our Common Stock. The issuance of these shares in the future would result in significant dilution to our current stockholders and could adversely affect the price of our Common Stock and the terms on which we could raise additional capital. In addition, the issuance and subsequent trading of shares could cause the supply of our Common Stock available for purchase in the market to exceed the purchase demand for our Common Stock. Such supply in excess of demand could cause the market price of our Common Stock to decline.
51
Sales of our Common Stock to Aspire Capital pursuant to our Purchase Agreement may cause substantial dilution to our existing stockholders and the sale of the shares of Common Stock acquired by Aspire Capital could cause the price of our Common Stock to decline.
The Company entered into a Purchase Agreement with Aspire Capital Fund, LLC in March 2014, pursuant to which Aspire Capital committed to the purchase of up to $30 million of shares of the Company’s Common Stock over the term of that Agreement, subject to certain terms and conditions.
Pursuant to the agreement, after Aspire Capital acquires shares under the Purchase Agreement, it may sell all or some of those shares. Sales to Aspire Capital by us pursuant to the Purchase Agreement may result in substantial dilution to the interests of other holders of our common stock. The sale of a substantial number of shares of our common stock to Aspire Capital, or anticipation of such sales, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales. However, we have the right to control the timing and amount of any sales of our shares to Aspire Capital and the Purchase Agreement may be terminated by us at any time at our discretion without any cost to us.
Failure to maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our business and stock price.
During the course of testing our disclosure controls and procedures and internal control over financial reporting, we may identify and disclose material weaknesses or significant deficiencies in internal control over financial reporting that will have to be remedied. Implementing any appropriate changes to our internal control may require specific compliance training of our directors, officers and employees, entail substantial costs to modify our existing accounting systems, and take a significant period of time to complete. Such changes may not, however, be effective in maintaining the adequacy of our internal control over financial reporting, and any failure to maintain that adequacy or inability to produce accurate financial statements on a timely basis could result in our financial statements being unreliable, increase our operating costs and materially impair our ability to operate our business.
Failure to achieve and maintain effective internal control over financial reporting could result in a loss of investor confidence in our financial reports and could have a material adverse effect on our stock price. Additionally, failure to maintain effective internal control over our financial reporting could result in government investigation or sanctions by regulatory authorities.
52
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
ITEM 2. PROPERTIES.
Corporate Headquarters
The Company's corporate headquarters is located in New York City. In January 2014, the Company executed a fourth modification and additional space agreement (the “fourth modification”) modifying to its existing lease in order to (1) obtain additional office space adjacent to its current, third floor executive offices and (2) extend the lease term for both the existing office space and the additional, adjacent space through January 31, 2018. The base monthly rent for the Company’s existing executive offices, which includes storage space and the recent additional, adjacent space, currently, is approximately $34,000 per month.
Cell Therapy Manufacturing Facilities
We presently operate three cell therapy manufacturing facilities, in Allendale, New Jersey, in Mountain View, California, and in Irvine, California. Longer-term plans include the acquisition and development of other such buildings within and outside of the United States, to be developed into replicable and scalable manufacturing facilities, strategically located to best serve our and our clients’ needs. Inherent in the nature of cell therapy today is the biologic shelf life of the cell therapy product itself. This limits the transit times between the time the cell product is extracted from a patient until it arrives at a manufacturing facility and the time that a processed product leaves the manufacturing facility and arrives for re-infusion in the patient. Therefore, it is preferable for cell therapy manufacturing facilities to be located in major population centers and within close proximity of major airport hubs.
The Allendale facility has been developed into a cell manufacturing facility, of which 22,000 square feet of its approximate 30,000 square feet have been developed, allowing for the possibility of future expansion. The Allendale facility is comprised of (a) four ISO Class 7, Class 10,000 manufacturing suites, (b) one ISO Class 6, Class 1,000 manufacturing suite that is designed to meet EU production standards and (c) quality control, research and development laboratories and support facilities. The Allendale facility has been designed to meet the accreditation requirements of the Foundation for the Accreditation of Cellular Therapy (FACT) and to comply with the FDA’s requirements, including applicable cGMP regulations, and to meet the standards of the American Association of Blood Banks (AABB). The Allendale facility is also in compliance with a range of state and federal regulatory and licensing requirements. We recently completed an expansion of the facility in 2014, adding laboratory, clean room suites and support facilities.
The Mountain View facility is also a licensed cell therapy manufacturing facility, encompassing 25,024 square feet within a single building, of which 17,425 square feet is developed. The developed space is presently used for manufacturing client products. Mountain View is equipped with six ISO Class 7, Class 10,000 manufacturing suites and quality control, research and development laboratories and support facilities. The Mountain View facility is subject to a lease agreement, as amended to date, having a current term that extends through June 2017. The base monthly rent is currently $43,000 subject to annual cost of living adjustments provided, however, that each such annual rental adjustment will not be less than 3% or more than 7%.
With our acquisition of California Stem Cell in May 2014, we assumed a facility lease located in Irvine. This facility is approximately 12,000 square feet. It consists of office space and is also a manufacturing facility with six clean rooms. It maintains test methods and proprietary media that enable controlled, current Good Manufacturing Practice (“cGMP”)-compliant production of critical, high-purity cell product(s). The base monthly rent is approximately $25,000. We recently signed an amendment expanding our office space in Irvine by 4,000 square feet. In accordance with the amendment, we plan to occupy the additional space by the second quarter of 2015.
Additional Office Space
We recently entered into an assignment agreement for general office space located in Basking Ridge, New Jersey. The space is approximately 18,467 rentable square feet. Pursuant to the agreement, we are not obligated to make any payments for the space until January 2016. The base monthly rent during the period ending January 31, 2016 is currently $25,000 and the lease term ends July 31, 2020. In addition, there are two (2) five (5) year renewal options. In connection with the assumption of the lease, the third party (a) conveyed its rights in various scheduled furniture and equipment and (b) paid the Company approximately $580,000 which amount will offset the rental payments to be paid by NeoStem. A security deposit of approximately $115,000 payable by NeoStem will offset the amount payable by the third party. With the additional space, the Company believes the total leased space is sufficient for the near future.
53
ITEM 3. LEGAL PROCEEDINGS.
None.
ITEM 4. MINE SAFTEY DISCLOSURES.
Not applicable.
54
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
ITEM 5(a). MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS.
Market For Our Common Equity
Our common stock trades on the NASDAQ Capital Market under the symbol “NBS.” The following table sets forth the high and low sales prices of our common stock for each quarterly period presented, as reported by the NASDAQ. All prices in the tables have been adjusted, as appropriate, to give effect to the one-for-ten reverse stock split of our common stock effected as of July 16, 2013.
2014 | High | Low |
First Quarter | $8.29 | $6.23 |
Second Quarter | $7.39 | $4.56 |
Third Quarter | $6.68 | $5.10 |
Fourth Quarter | $7.22 | $3.08 |
2013 | High | Low |
First Quarter | $7.00 | $5.00 |
Second Quarter | $7.00 | $5.00 |
Third Quarter | $9.89 | $5.20 |
Fourth Quarter | $8.92 | $5.98 |
2012 | High | Low |
First Quarter | $9.00 | $3.70 |
Second Quarter | $6.10 | $3.00 |
Third Quarter | $8.40 | $4.90 |
Fourth Quarter | $7.80 | $5.90 |
55
Performance Graph
Set forth below is a line graph comparing changes in the cumulative total return over the past five years on (i) NeoStem’s common stock, (ii) a broad market index (the NASDAQ Market Index), and (iii) a peer group consisting the following companies in the biotechnology industry: Aastrom Biosciences, Inc., Athersys, Inc., Cytori Therapeutics, Inc., Dendreon Corporation, Immunocellular Therapeutics Ltd., Opexa Therapeutics, Inc., Northwest Biotherapeutics, and Stemcells, Inc. (the “Peer Group”), for the period commencing on December 31, 2009 and ending on December 31, 2014 (1).
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Assumes Initial Investment of $100
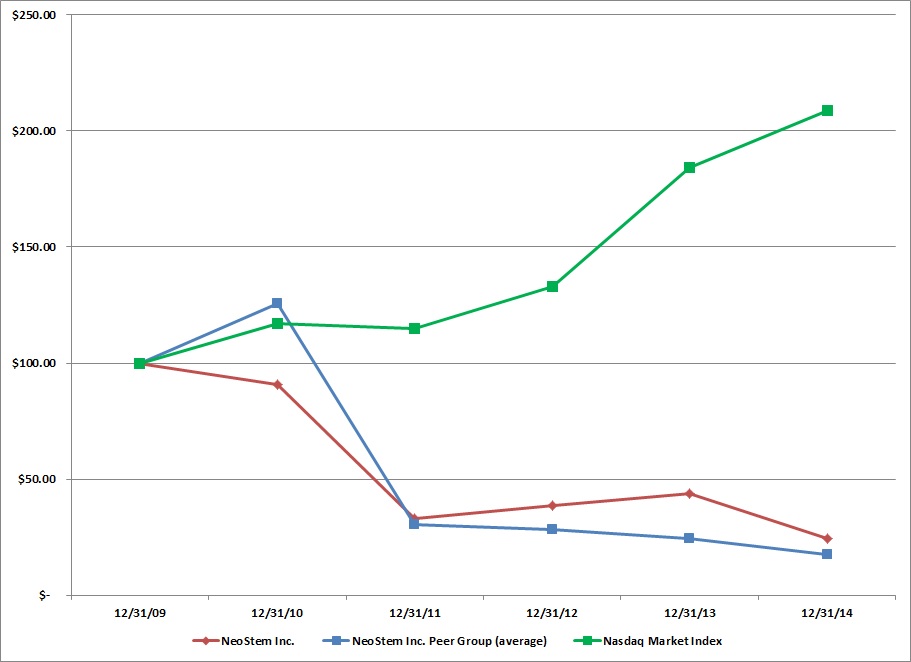
*$100 invested on 12/31/09 in stock or index, including reinvestment of dividends.
NeoStem/Market/Index | 12/31/2009 | 12/31/2010 | 12/31/2011 | 12/31/2012 | 12/31/2013 | 12/31/2014 | ||||||
NeoStem, Inc. | $100.00 | $90.97 | $32.90 | $38.71 | $44.00 | $24.32 | ||||||
NASDAQ Market Index | $100.00 | $116.91 | $114.81 | $133.07 | $184.06 | $208.71 | ||||||
Peer Group | $100.00 | $125.61 | $30.40 | $28.36 | $24.46 | $17.47 | ||||||
_______________
(1) Assumes that $100 was invested on December 31, 2009 in NeoStem’s common stock and each index, and that all dividends were reinvested. No cash dividends have been declared on NeoStem’s common stock. Shareholder returns over the indicated period should not be considered indicative of future shareholder returns.
56
Holders
As of February 27, 2015 , there were approximately 1,311 stockholders of record of our common stock (which does not include beneficial owners for whom Cede & Co. or others act as nominees).
Dividends and Dividend Policy
We have not paid cash dividends on our common stock during the periods set forth in the stock price table that appears above. The holders of our common stock are each entitled to receive dividends when and if declared by the board of directors out of funds legally available therefor, subject to the terms of any outstanding series of preferred stock. We intend to retain any future earnings to fund the development and growth of our business, and therefore we do not anticipate paying any cash dividends on our common stock in the foreseeable future.
Equity Compensation Plan Information
The following table provides information as of December 31, 2014 regarding shares of our common stock that may be issued under our existing equity compensation plans, including our 2003 Stock Option and Incentive Plan (the “2003 Plan”), 2009 Stock Option and Incentive Plan (the “2009 Plan”) and our 2012 Employee Stock Purchase Plan (the “2012 ESPP Plan”).
Equity Compensation Plan Information | ||||
Number of securities to be issued upon exercise of outstanding options (1) | Weighted Average exercise price of outstanding options and rights | Number of securities remaining available for future issuance under equity compensation plan (excluding securities referenced in column (a)) | ||
Equity compensation plans approved by security holders (2) | 4,427,276 | $9.19 | 4,097,111 | (3) |
(1) | Includes stock options only; does not include purchase rights accruing under the 2012 ESPP Plan because the purchase price (and therefore the number of shares to be purchased) will not be determined until the end of the purchase period. |
(2) | Consists of the 2003 Plan, 2009 Plan and 2012 ESPP Plan. |
(3) | Includes shares available for future issuance under the 2009 Plan and the 2012 ESPP Plan. |
Recent Sales of Unregistered Securities
As previously disclosed, and as follows:
The Company has agreed to issue equity to certain consultants for services. Effective November 18, 2014 pursuant to a six month consulting agreement for consulting services in corporate finance, investor communications, investor relations and other specified matters, the Company agreed to issue to a consultant 50,000 shares of the Company's restricted common stock, vesting as to 25,000 shares on execution and as to 25,000 shares on May 18, 2015. Effective November 20, 2014, pursuant to a three month agreement for consulting services in business development, financial and media relations, the Company agreed to issue to consultant 45,000 shares of the Company's restricted common stock vesting ratably through the term of the agreement. Effective November 24, 2014 pursuant to a four month agreement for consulting services in investor relations and other specified matters, the Company agreed to issue to a consultant 24,000 shares of the Company's restricted common stock vesting as to 6,000 shares monthly. On December 30, 2014, the Company entered into a five month extension letter which was effective on February 1, 2015, for consulting services in media, marketing, advertising, and other specified matters, pursuant to which the Company agreed to issue to a consultant 18,000 shares of the Company's restricted common stock, vesting ratably throughout the term of the agreement. Also on December 30, 2014, the Company entered into a three month extension letter with a consultant effective on
57
February 20, 2015, for consulting services in analysis of industry competition, and investor and media relations, whereby the Company agreed to issue to a consultant 45,000 shares of the Company's restricted common stock vesting as to 15,000 shares monthly. Effective January 1, 2015, pursuant to a six month extension for consulting services in information technology and accounting systems, the Company agreed to issue to a consultant, 9,000 shares of the Company's restricted common stock, vesting ratably throughout the term of the agreement on a monthly basis. Also effective on January 1, 2015, pursuant to a six month extension for consulting services in accounting systems and regulatory compliance, the Company agreed to issue to a consultant, 7,500 shares of the Company's common stock vesting ratably throughout the term of the agreement on a monthly basis. Effective January 16, 2015, pursuant to a six month agreement for consulting services in strategic planning and tactical application of those services and other specified matters, the Company agreed to issue to a consultant 34,000 shares of the Company's restricted common stock, vesting ratably over the term of the agreement. Effective February 28, 2015, pursuant to a four month agreement for consulting services in investor relations and other specified matters, the Company agreed to issue to a consultant 24,000 shares of the Company's restricted common stock vesting ratably throughout the term of the agreement.
The offer and sale by the Company of the securities described above were made in reliance upon the exemption from registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”), for transactions by an issuer not involving a public offering. The offer and sale of such securities were made without general solicitation or advertising to “accredited investors” as such term is defined in Rule 501(a) of Regulation D promulgated under the Securities Act and/or pursuant to Regulation D or Regulation S, each promulgated under the Securities Act and may not be resold in the United States or to U.S. persons unless registered under the Securities Act or pursuant to an exemption from registration under the Securities Act.
ITEM 6. SELECTED FINANCIAL DATA.
The selected financial data set forth below for the years ended December 31, 2014, 2013, and 2012 and as of December 31, 2014 and 2013 are derived from and should be read in conjunction with our audited financial statements, including the notes thereto, included elsewhere in this report. The selected financial data for the years ended December 31, 2011 and 2010 and as of December 31, 2012, 2011, and 2010 are derived from our audited financial statements not included in this report.
(in thousands, except per share data) | Year Ended December 31, | ||||||||||||||||||
Consolidated Statement of Income Data: | 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||
Revenues | $ | 17,939 | $ | 14,668 | $ | 14,330 | $ | 10,050 | $ | 181 | |||||||||
Total operating costs and expenses | $ | 75,680 | $ | 51,477 | $ | 44,716 | $ | 44,055 | $ | 26,035 | |||||||||
Net loss from continuing operations | $ | (55,466 | ) | $ | (39,485 | ) | $ | (36,101 | ) | $ | (34,566 | ) | $ | (25,809 | ) | ||||
Net loss from continuing operations attributable to NeoStem, Inc. common stockholders | $ | (54,873 | ) | $ | (38,981 | ) | $ | (35,814 | ) | $ | (34,267 | ) | $ | (25,809 | ) | ||||
Basic and diluted loss from continuing operations per share attributable to NeoStem, Inc. common stockholders | $ | (1.68 | ) | $ | (1.90 | ) | $ | (2.59 | ) | $ | (3.87 | ) | $ | (5.00 | ) | ||||
Weighted average common shares outstanding | 32,756 | 20,496 | 13,842 | 8,860 | 5,163 | ||||||||||||||
As of December 31, | |||||||||||||||||||
Consolidated Balance Sheet Data: | 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||
Cash, cash equivalents, and marketable securities | $ | 26,254 | $ | 46,134 | $ | 13,737 | $ | 3,935 | $ | 8,469 | |||||||||
Assets related to discontinued operations (current and long-term) | $ | — | $ | — | $ | — | $ | 107,938 | $ | 130,408 | |||||||||
Total assets | $ | 126,275 | $ | 89,816 | $ | 54,406 | $ | 155,328 | $ | 143,025 | |||||||||
Long-term debt (current and long-term) | $ | 15,000 | $ | — | $ | — | $ | — | $ | — | |||||||||
Mortgages payable (current and long-term) | $ | — | $ | 3,237 | $ | 3,438 | $ | 3,635 | $ | — | |||||||||
Notes payable (current and long-term) | $ | 1,643 | $ | 912 | $ | 374 | $ | 148 | $ | 117 | |||||||||
Convertible Redeemable Series E Preferred Stock | $ | — | $ | — | $ | — | $ | 4,811 | $ | 6,532 | |||||||||
Liabilities for acquisition-related contingent consideration | $ | 18,260 | $ | 9,450 | $ | 7,550 | $ | 3,130 | $ | — | |||||||||
Liabilities related to discontinued operations (current and long-term) | $ | — | $ | — | $ | — | $ | 54,554 | $ | 45,659 | |||||||||
Total stockholders' equity | $ | 58,074 | $ | 62,026 | $ | 32,820 | $ | 80,133 | $ | 86,488 | |||||||||
58
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under “Cautionary Note Regarding Forward-Looking Statements” and under “Risk Factors” herein.
Overview
NeoStem, Inc. (“we,” “NeoStem” or the “Company”) is a vertically integrated, clinical-stage biopharmaceutical company that is pursuing the preservation and enhancement of human health through the development of cell based therapeutics that leverage the body’s natural ability to heal and fight disease. Our diversified pipeline and unique capabilities for innovative, cost-effective and efficient in-house development set us apart in this emerging industry as we work to fundamentally change the treatment paradigm for several serious diseases.
Our most advanced clinical program is based on our dendritic cell/cancer cell technology. It is focused on the development of an innovative cancer treatment that is designed to target the cells responsible for tumor growth and metastasis, known as cancer or tumor initiating cells (CSCs), using purified CSCs from a patient’s own tumor as an antigen source to induce or enhance an anti-tumor immune response in the patient. Our lead product candidate based on this platform technology, NBS20, targets malignant melanoma as an initial indication. NBS20 is being studied in patients with recurrent Stage III or Stage IV metastatic melanoma. The program has been granted Fast Track and Orphan designation by the Food and Drug Administration (FDA) and the protocol for the Phase 3 study, known as the Intus study, is the subject of a Special Protocol Assessment (SPA). Our SPA letter states that our Phase 3 clinical trial is adequately designed to provide the necessary data that, depending on outcome, could support a Biologics License Application (BLA) seeking marketing approval of NBS20. This protocol calls for randomizing 250 patients. Patient screening began in 1Q15 and randomization of the first patient is expected in 2Q15. Interim analysis of the data is targeted for 4Q2017. We are also evaluating other clinical indications for which we may advance this program, including lung, colon, ovarian and liver cancers.
Our company is also developing therapies that are designed to utilize CD34 cells to prevent heart failure and major adverse cardiac events following a severe heart attack, known as an ST-elevation myocardial infarction (STEMI), through the use of CD34 cells to regenerate tissue impacted by ischemia. Ischemia occurs when the supply of oxygenated blood in the body is restricted, causing tissue distress and death. We seek to improve oxygen delivery to tissues through the development and formation of new blood vessels. NBS10 is our most clinically advanced product candidate in our ischemic repair program. At the American Heart Association’s Scientific Sessions in November 2014, we reported data from the primary analysis of our 161 patient PreSERVE (acute myocardial infarction) AMI study. We are also planning the release of our one year data from the phase 2 trial on March 15, 2015 at the Annual Scientific Sessions of the American College of Cardiology. PreSERVE AMI is a randomized, double-blinded, placebo-controlled Phase 2 clinical trial testing NBS10, a “personalized” adult stem cell product being developed for the treatment of patients with left ventricular dysfunction following a STEMI. The primary endpoints are measured by, (i) the change from baseline to six months in myocardial perfusion (RTSS) measured by an imaging technique (SPECT); and (ii) safety of bone marrow procurement and infusion as measured by occurrence of adverse events, serious adverse events (SAEs) and major adverse cardiac events (MACE). There are five secondary efficacy endpoints, two evaluating left ventricular ejection fraction, one evaluating MACE, and two evaluating quality of life. The reported data were based on all treated patients that had received six month follow-up for imaging. The median length follow up for mortality, adverse events, SAEs and MACE in these patients was twelve months. The reported results allowed for important observations about a potential dose-dependent treatment effect that will help guide the next phase of development. These observations about a potential dose dependent treatment effect were based on post hoc analyses of subsets of treated patients based on the number of cells they received. Notably, statistically significant dose-dependent increases in left ventricular function and decreases in serious adverse events were seen in patients who received the highest dose of cells (n=15 patients), though no statistical significance was observed when NBS10 overall was compared to placebo on these measures. With respect to mortality, at one year there were no treatment group deaths while the control group saw a mortality rate of 3.6% (n=3), equating to a statistically significant reduction in mortality. Regarding MACE, while more events occurred with NBS10 overall versus placebo, a non-significant trend toward fewer events was observed in patients who received higher doses of cells, with MACE occurring in 14% of placebo patients, 17% of patients who received less than 14 million cells, 10% of patients who received greater than 14 million cells and 7% of patients who received greater than 20 million cells. Finally, our hypothesis that SPECT used to measure perfusion could be used as a surrogate marker for the current medically relevant and regulatory endpoints was disproven, giving us valuable direction regarding endpoints and analyses for future clinical trials. We expect to complete the PreSERVE AMI study as defined through the final three-year follow-up and, in the meantime, plan to meet with the FDA to discuss our results and our proposal for the next step(s) in development. Finalization of the decision for next steps for NBS10 are expected in the second half of 2015. We also are evaluating other clinical indications that involve
59
ischemia into which we may advance this program, including critical limb ischemia (CLI) and congestive heart failure (CHF).
Another platform technology we are developing is designed to utilize Regulatory T Cells (Tregs) to treat diseases caused by imbalances in an individual's immune system. This novel approach seeks to restore immune balance by enhancing Treg cell number and function. Tregs are a natural part of the human immune system and regulate the activity of T effector cells, the cells that are responsible for protecting the body from viruses and other foreign antigens. When Tregs function properly, only harmful foreign materials are attacked by T effector cells. In autoimmune disease, it is thought that deficient Treg activity permits the T effector cells to attack the body's own tissues. We have received a letter from the FDA stating that we may proceed on a Phase 2 study of NBS03D, a Treg based therapeutic being developed to treat type 1 diabetes mellitus (T1DM) in adolescents, and we plan to initiate the trial in late 2015 or 2016 depending on resource availability. We are evaluating other clinical indications into which we may advance this program, including graft versus host disease, chronic obstructive pulmonary disease (COPD), multiple sclerosis (MS), inflammatory bowel disease (IBD) and steroid resistant asthma.
Finally, we are actively exploring means by which we can take advantage of new regulations in Japan that permit conditional approval for regenerative medicine products that show sufficient safety evidence and signals of efficacy. Potential indications for this unique opportunity include our targeted cancer immunotherapy program in liver cancer and our ischemic repair program in CLI.
We believe that cell-based therapies have the potential to create a paradigm change in the treatment for a variety of diseases and conditions and we are evaluating other programs that we view as holding particular promise, including an aesthetics program for a topical skin application and a very small embryonic like (VSEL TM) stem cell program for the treatment of retinal degeneration, bone restoration and wound healing.
Through our wholly owned subsidiary, Progenitor Cell Therapy, LLC (PCT), we are recognized as a world industry leader in providing high quality innovative and reliable manufacturing capabilities and engineering solutions (e.g. process development) in the development of cell-based therapies. We operate three current Good Manufacturing Practice (cCGMP) facilities in Allendale, NJ, Mountain View, CA and Irvine, CA, respectively, and are poised to expand our facilities internationally. In addition to leveraging this core expertise in the development of our own products, we partner opportunistically with other industry leaders who recognize our unique ability to significantly improve their manufacturing processes and supply clinical and commercial material.
We look forward to further advancement of our cell based therapies to the market and to helping patients suffering from life-threatening medical conditions. Coupling our development expertise with our strong process development and manufacturing capability, we believe the stage is set for us to realize meaningful clinical development or our own proprietary platform technologies and manufacturing advancements, further positioning NeoStem to lead the cell therapy industry.
Results of Operations
Year Ended December 31, 2014 Compared to Year Ended December 31, 2013
Net loss for the year ended December 31, 2014 was approximately $55.5 million compared to $39.5 million for the year ended December 31, 2013.
Revenues
For the year ended December 31, 2014, total revenues were approximately $17.9 million compared to $14.7 million for the year ended December 31, 2013, representing an increase of $3.3 million, or 22%. Revenues were comprised of the following (in thousands):
Year Ended December 31, | |||||||
2014 | 2013 | ||||||
Clinical Services | $ | 10,442.9 | $ | 9,146.3 | |||
Clinical Services Reimbursables | 3,725.0 | 2,085.4 | |||||
Processing and Storage Services | 3,770.9 | 3,436.8 | |||||
$ | 17,938.8 | $ | 14,668.5 | ||||
60
• | Clinical Services, representing process development and clinical manufacturing services provided at PCT to its various clients, were approximately $10.4 million for the year ended December 31, 2014 compared to $9.1 million for the year ended December 31, 2013, representing an increase of approximately $1.3 million or 14%. The increase was primarily due to $2.0 million of higher process development revenue (such revenue being recognized on a "completed contract" basis) as a result of an increase in the number of new process development agreements with our existing clients, which was partially offset by $0.6 million of lower clinical manufacturing revenue (which is recognized as services are rendered). |
◦ | Process Development Revenue - Process development revenues were approximately $4.0 million for the year ended December 31, 2014, compared to $2.0 million for the year ended December 31, 2013. During the year ended December 31, 2014, the number of process development contracts initiated and completed were higher compared to the prior year period. In accordance with our revenue recognition policy, process development revenue is recognized upon contract completion (i.e., when the services under a particular contract are completed). As of December 31, 2014, approximately $3.9 million process development revenue has been deferred to future periods for contracts that have been initiated but not yet completed. This revenue will be recognized in future periods upon completion of those contracts. Process development revenue will continue to fluctuate from period to period as a result of this revenue recognition policy. |
◦ | Clinical Manufacturing Revenue - Clinical manufacturing revenues were approximately $6.4 million for the year ended December 31, 2014, compared to $7.0 million for the year ended December 31, 2013. The decrease is primarily due to lower enrollment of patients being treated in our customers' clinical trials. |
• | Clinical Services Reimbursables, representing reimbursement of expenses for certain consumables incurred on behalf of our clinical service revenue clients, were approximately $3.7 million for the year ended December 31, 2014 compared to $2.1 million for the year ended December 31, 2013, representing an increase of approximately $1.6 million or 79%. Generally, clinical services reimbursables correlate with clinical services revenues. However, differences in the cost of supplies to be reimbursed can vary greatly from contract to contract based on the cost of supplies needed for each client's manufacturing and development process, and may impact this correlation. In addition, our terms for billing reimbursable expenses do not include a significant mark up in the acquisition cost of such consumables, and as a result, changes in this revenue category have little impact on our gross profit and net loss. |
• | Processing and Storage Services, primarily representing revenues from our oncology stem cell processing, were approximately $3.8 million for the year ended December 31, 2014 compared to $3.4 million for the year ended December 31, 2013, representing an increase of approximately $0.3 million or 10%. The increase is primarily due to increased volume and pricing for the processing services. |
Operating Costs and Expenses of Revenues
For the year ended December 31, 2014, operating expenses totaled $75.7 million compared to $51.5 million for the year ended December 31, 2013, representing an increase of $24.2 million or 47%. Operating expenses were comprised of the following:
• | Cost of revenues were approximately $15.7 million for the year ended December 31, 2014 compared to $12.9 million for the year ended December 31, 2013, representing an increase of $2.7 million or 21%. The increase is primarily due to increased clinical services costs to support our customer's process development and clinical manufacturing efforts, as well as additional investment in our internal facilities and capabilities. Overall, gross profit for the year ended December 31, 2014 was $2.3 million or 13% of 2014 revenues, compared to gross profit for the year ended December 31, 2013 of $1.7 million or 12% of 2013 revenues. Gross profit percentages generally will increase as clinical service revenue increases. However, gross profit percentages will also fluctuate from period to period due to the mix of service and reimbursable revenues and costs. |
• | Research and development expenses were approximately $29.2 million for the year ended December 31, 2014 compared to $16.9 million for the year ended December 31, 2013, representing an increase of approximately $12.4 million, or 73%. Research and development expenses associated with our targeted cancer immunotherapy program, including the initiation of the Intus Phase 3 clinical trial for our lead immunotherapy product candidate NBS20, were $6.9 million for the year ended December 31, 2014. The targeted cancer immunotherapy program was acquired in the CSC merger on May 8, 2014. Research and development expenses related to our ischemic repair program, including expenses associated with the Preserve AMI Phase 2 clinical trial for our product candidate NBS10, increased by approximately $0.2 million for the year ended December 31, 2014 compared to the prior year period. The increase reflects costs related to evaluating additional potential therapeutic indications in the ischemic repair program, which were partially offset by lower expenses |
61
in the Preserve AMI Phase 2 clinical trial which completed patient enrollment in the fourth quarter of 2013. Research and development expenses associated with our immune modulation program increased by approximately $4.3 million, and was primarily due to our efforts to develop Tregs. We continue to focus efforts on initiating a Phase 2 study of NBS03D in type 1 diabetes expected to be initiated in 2015. Other research and development expenses associated with engineering and innovation initiatives at PCT to improve scale up, automation, and integration capabilities increased during year ended December 31, 2014 compared to the prior year. Equity-based compensation included in research and development expenses for the year ended December 31, 2014 and December 31, 2013 were approximately $2.1 million and $0.8 million, respectively.
• | Selling, general and administrative expenses were approximately $30.8 million for the year ended December 31, 2014 compared to $21.6 million for the year ended December 31, 2013, representing an increase of approximately $9.2 million, or 43%. Equity-based compensation included in selling, general and administrative expenses for the year ended December 31, 2014 was approximately $8.7 million, compared to approximately $5.7 million for the year ended December 31, 2013, representing an increase of $3.0 million. The increase in equity-based compensation was due to its broader use during the year ended December 31, 2014, and in particular, equity awards issued as a bonus for the successful completion of the CSC Acquisition. Equity-based compensation expense will continue to fluctuate in future years as equity-linked instruments are used to compensate employees, consultants and other service providers. Non-equity-based general and administrative expenses for the year ended December 31, 2014 were approximately 22.1 million, compared to approximately 15.9 million for the year ended December 31, 2013. The increase was related to higher corporate development activities, expenses associated with the additional CSC operating activities since the acquisition date on May 8, 2014, and increased corporate infrastructure to support our expanded clinical activities. |
Historically, to minimize our use of cash, we have used a variety of equity and equity-linked instruments as compensation to employees, consultants, directors and other service providers. The use of these instruments has resulted in charges to the results of operations, which has been significant in the past.
Other Income (Expense)
Other income, net for the year ended December 31, 2014 totaled approximately $2.9 million, and primarily represented the decrease in the estimated fair value of our contingent consideration liability associated with potential earn out payments on the net sales of our lead product candidate NBS10 (in the event of and following the date of first commercial sale of NBS10), which was partially offset by an increase in the fair value of our contingent consideration for potential future milestone payments related to the CSC acquisition. Other expense, net for the year ended December 31, 2013 totaled approximately $1.6 million, and primarily represented an increase in the estimated fair value of our contingent consideration liability associated with potential earn out payments on the net sales of NBS10.
For the year ended December 31, 2014 interest expense was $0.8 million compared with $0.3 million for the year ended December 31, 2013. The increase is primarily due to the $15.0 million loan from Oxford Finance LLC in September 2014, which bears an annual interest rate of 8.5%, amortization of related debt issuance costs, and accretion of the 8% final payment fee due in September 2018. Interest expense in each period also relates to mortgage payables, which were fully repaid in September 2014.
Provision for Income Taxes
The provision for income taxes for the years ended December 31, 2014 and December 31, 2013 primarily relate to the taxable temporary differences on the goodwill recognized in the PCT acquisition in 2011, which is being amortized over 15 years for tax purposes. A tax provision will continue to be recognized each period over the amortization period, and will only reverse when the goodwill is eliminated through a sale, impairment, or reclassification from an indefinite-lived asset to a finite-lived asset.
Noncontrolling Interests
In March 2011, we acquired rights to use patents under licenses from Becton, Dickinson and Company ("BD") in exchange for a 19.9% interest in our Athelos subsidiary. Pursuant to the Stock Purchase Agreement signed in March 2011, BD's ownership will be diluted based on new investment in Athelos (subject to certain anti-dilution provisions). As of December 31, 2014, BD's ownership interest in Athelos was decreased to 3.8%. For the years ended December 31, 2014 and 2013, BD's minority shareholder’s share of Athelos’ net loss totaled approximately $0.6 million and $0.5 million, respectively.
62
Year Ended December 31, 2013 Compared to Year Ended December 31, 2012
Net loss for the year ended December 31, 2013 was approximately $39.5 million compared to $66.4 million for the year ended December 31, 2012. Our net losses from continuing operations for the year ended December 31, 2013 and 2012 were approximately $39.5 million and $36.1 million, respectively. The loss from discontinued operations - net for the year ended December 31, 2012 was approximately $30.3 million, and represents the operations of our former Regenerative Medicine – China segment which was deconsolidated in the first quarter of 2012, and the operations of our former Pharmaceutical Manufacturing - China segment, which related to the sale of our 51% interest in Suzhou Erye Pharmaceuticals Company Ltd. ("Suzhu Erye"), in the fourth quarter of 2012.
Revenues
For the year ended December 31, 2013, total revenues were approximately $14.7 million compared to $14.3 million for the year ended December 31, 2012, representing an increase of $0.3 million, or 2%. Revenues were comprised of the following (in thousands):
Year Ended December 31, | |||||||
2013 | 2012 | ||||||
Clinical Services | $ | 9,146.3 | $ | 8,034.8 | |||
Clinical Services Reimbursables | 2,085.4 | 3,462.2 | |||||
Processing and Storage Services | 3,436.8 | 2,644.7 | |||||
Other | — | 188.2 | |||||
$ | 14,668.5 | $ | 14,329.9 | ||||
• | Clinical Services, representing process development and clinical manufacturing services provided at PCT to its various clients, were approximately $9.1 million for the year ended December 31, 2013 compared to $8.0 million for the year ended December 31, 2012, representing an increase of approximately $1.1 million or 14%. The increase was primarily due to $2.3 million of higher clinical manufacturing revenue (which is recognized as services are rendered), which was partially offset by $1.2 million lower process development revenue (such revenue being recognized on a "completed contract" basis). Overall, there were approximately 50% more active Clinical Services clients as of December 31, 2013 compared to December 31, 2012. |
◦ | Clinical Manufacturing Revenue - Clinical manufacturing revenues were approximately $7.0 million for the year ended December 31, 2013, compared to $4.7 million for the year ended December 31, 2012. The increase is primarily due to an increase in the number of patients our customers enrolled and were treating in clinical trials being conducted by our customers. |
◦ | Process Development Revenue - Process development revenues were approximately $2.0 million for the year ended December 31, 2013, compared to $3.2 million for the year ended December 31, 2012. The decrease was due to the migration of certain customers from the Process Development phase to the Clinical Manufacturing phase, as well as the impact of revenue recognition associated with existing Process Development clients during the year ended December 31, 2013 compared to the year ended December 31, 2012. In accordance with our revenue recognition policy, process development revenue is recognized upon contract completion (i.e., when the services under a particular contract are completed). As a result, there is no revenue recognized for process development contracts that have yet to be completed, regardless of the amount of progress billing. Process development revenue will continue to fluctuate from period to period as a result of this revenue recognition policy. |
• | Clinical Services Reimbursables, representing reimbursement of expenses for certain consumables incurred on behalf of our clinical service revenue clients, were approximately $2.1 million for the year ended December 31, 2013 compared to $3.5 million for the year ended December 31, 2012, representing a decrease of approximately $1.4 million or 40%. Our reimbursable revenue decrease was partly the result of changes in contractual terms with certain clients that shifted clinical service expense reimbursables to a fully absorbed billing rate which is now reflected in Clinical Manufacturing Revenue. Generally, our terms for billing reimbursable expenses do not include significant mark up in the acquisition cost of such consumables, and as a result the impact of changes in this revenue category has little or no impact on our net loss. |
63
• | Processing and Storage Services, representing revenues from our oncology, cord blood, and adult stem cell processing and banking activities, were approximately $3.4 million for the year ended December 31, 2013 compared to $2.6 million for the year ended December 31, 2012, representing an increase of approximately $0.8 million or 30%. The increase is primarily attributable to increased revenue from our oncology stem cell processing service. |
• | Other Revenue of approximately $0.2 million for the year ended December 31, 2012 represent license fees related to our adult stem cell technology. |
Operating Costs and Expenses of Revenues
For the year ended December 31, 2013 operating expenses totaled $51.5 million compared to $44.7 million for the year ended December 31, 2012, representing an increase of $6.8 million or 15%. Operating expenses were comprised of the following:
• | Cost of revenues were approximately $12.9 million for the year ended December 31, 2013 compared to $11.9 million for the year ended December 31, 2012, representing an increase of $1.0 million or 8%. The increase is primarily due to increased clinical services costs to support our customer's process development and clinical manufacturing efforts. Overall, gross profit for the year ended December 31, 2013 was $1.7 million or 12% of 2013 revenues, compared to gross profit for the year ended December 31, 2012 of $2.4 million or 17% of 2012 revenues. Gross profit percentages generally will increase as clinical service revenue increases. However, gross profit percentages will also fluctuate from period to period due to the mix of service and reimbursable revenues and costs. |
• | Research and development expenses were approximately $16.9 million for the year ended December 31, 2013 compared to $10.5 million for the year ended December 31, 2012, representing an increase of approximately $6.4 million, or 62%. Research and development expenses associated with our ischemic repair program increased by approximately $3.7 million for the year ended December 31, 2013 compared to the prior year period. Research and development expenses associated with our immune modulation program increased by approximately $1.3 million compared to the prior year period. Other research and development associated with our VSELTM Technology Program, patent-related costs, and engineering and innovation initiatives at PCT to improve scale up, automation, and integration capabilities also increased during year ended December 31, 2013. Equity-based compensation included in research and development expenses for the year ended December 31, 2013 and December 31, 2012 were approximately $0.8 million and $0.4 million, respectively. |
• | Selling, general and administrative expenses were approximately $21.6 million for the year ended December 31, 2013 compared to $22.3 million for the year ended December 31, 2012, representing a decrease of approximately $0.7 million, or 3%. Equity-based compensation included in selling, general and administrative expenses for the year ended December 31, 2013 was approximately $5.7 million, compared to approximately $6.1 million for the year ended December 31, 2012, representing a decrease of $0.4 million. Non-equity-based general and administrative expenses for the year ended December 31, 2013 were unchanged at approximately $15.6 million, compared to the prior year period. Selling expenses decreased $0.4 million compared to the prior year period. |
Historically, to minimize our use of cash, we have used a variety of equity and equity-linked instruments to compensate employees, consultants and other service providers. The use of these instruments has resulted in charges to the results of operations, which has been significant in the past. In general, these equity and equity-linked instruments were used to pay for employee and consultant compensation, director fees, marketing services, investor relations and other activities.
Other Income (Expense)
Other expense, net for the year ended December 31, 2013 totaled approximately $1.6 million, and primarily represented the increase in the estimated fair value of our contingent consideration liability associated with potential earn out payments on the net sales of our lead product candidate NBS10 (in the event of and following the date of first commercial sale of NBS10). Other expense, net for the year ended December 31, 2012 totaled approximately $4.3 million, and also primarily represented the increase in the estimated fair value of our contingent consideration liability associated with potential earn out payments on the net sales of our lead product candidate NBS10.
For the year ended December 31, 2013 interest expense was $0.3 million compared with $1.6 million for the year ended December 31, 2012. Interest expense in the prior year period was primarily due to the amortization of debt discount related to the Series E Preferred Stock, which was fully redeemed in October 2012.
Provision for Income Taxes
64
The provision for income taxes for the years ended December 31, 2013 and December 31, 2012 primarily relate to the taxable temporary differences on the goodwill recognized in the PCT acquisition in 2011, which is being amortized over 15 years for tax purposes. A tax provision will continue to be recognized each period over the amortization period, and will only reverse when the goodwill is eliminated through a sale, impairment, or reclassification from an indefinite-lived asset to a finite-lived asset.
Discontinued Operations
Regenerative Medicine - China segment
In the first quarter of 2012, we exited our regenerative medicine business in the People's Republic of China. Accordingly, the operations and cash flows for the Regenerative Medicine - China business in 2012 were reported in discontinued operations. For the year ended December 31, 2012, the loss from discontinued operations was $1.7 million, and included a $1.1 million loss on exit of segment.
Pharmaceutical Manufacturing - China segment
On November 13, 2012, we completed the divestiture of our 51% interest (the “Erye Interest”) in Suzhou Erye Pharmaceuticals Company Ltd., a Sino-foreign equity joint venture with limited liability organized under the laws of the People's Republic of China. Pursuant to the Equity Purchase Agreement, the aggregate purchase price paid to us for the Erye Interest consisted of (i) $12.3 million in cash, (ii) the return to us of 104,000 shares of NeoStem common stock and (iii) the cancellation of 117,000 options and 64,000 warrants to purchase our common stock. This transaction resulted in a loss on exit of segment of $3.4 million, which was recorded in the fourth quarter of 2012. The operations and cash flows of the Pharmaceutical Manufacturing - China business were eliminated from ongoing operations upon the divestiture. Accordingly, the operating results of the Pharmaceutical Manufacturing - China business for the year ended December 31, 2012 were classified as discontinued operations. For the year ended December 31, 2012, the loss from discontinued operations was $28.5 million.
Noncontrolling Interests
In connection with accounting for our 51% interest in Erye, which is reported in discontinued operations, we account for the 49% minority shareholder share of Erye's net income or loss with a charge to Noncontrolling Interests. For the year ended December 31, 2012, Erye's minority shareholders' share of net income totaled approximately $12.3 million. On November 13, 2012, we completed the divestiture of our 51% interest in Erye.
In March 2011, we acquired rights to use patents under licenses from Becton, Dickinson and Company ("BD") in exchange for a 19.9% interest in our Athelos subsidiary. Pursuant to the Stock Purchase Agreement signed in March 2011, BD's ownership will be diluted based on new investment in Athelos (subject to certain anti-dilution provisions). As of December 31, 2013, BD's ownership interest in Athelos was decreased to 11.5%. For the years ended 2013 and 2012, BD's minority shareholder’s share of Athelos’ net loss totaled approximately $0.5 million and $0.3 million, respectively.
Warrant Inducements
To raise capital on terms that we deemed favorable, during the year ended December 31, 2012, the Board authorized certain inducements to warrant holders to exercise outstanding common stock purchase warrants significantly before their expiration dates. We determined in each instance that such inducements were modifications of equity instruments, and an incremental fair value of the inducement was determined using the Black-Scholes option pricing model.
For the year ended December 31, 2012, certain warrant holders were induced to exercised warrants to purchase 0.8 million shares of common stock at prices ranging between $5.10 and $18.50 per share, for gross proceeds to the Company of approximately $5.0 million. The incremental fair value of the inducement recorded in 2012 was $1.0 million.
Analysis of Liquidity and Capital Resources
65
At December 31, 2014 we had cash, cash equivalents, and marketable securities of approximately $26.3 million, working capital of approximately $20.0 million, and stockholders’ equity of approximately $58.5 million.
During the year ended December 31, 2014, we met our immediate cash requirements through revenue generated from our PCT operations, existing cash balances, offerings of our common stock (which raised an aggregate of approximately $16.7 million), warrant exercises (which raised approximately $1.7 million), net loan proceeds from Oxford Finance LLC of approximately $11.3 million (which included the repayment all outstanding amounts due under two loans from TD Bank, N.A.), and the use of equity and equity-linked instruments to pay for services and compensation.
Net cash provided by or used in operating, financing and investing activities from continuing operations were as follows (in thousands):
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net cash used in operating activities - continuing operations | $ | (46,895.2 | ) | $ | (27,101.7 | ) | $ | (18,759.9 | ) | ||
Net cash (used in) provided by investing activities - continuing operations | (10,736.7 | ) | (2,691.5 | ) | 11,748.7 | ||||||
Net cash provided by financing activities - continuing operations | 30,672.2 | 62,189.5 | 17,112.0 | ||||||||
Operating Activities - Continuing Operations
Our cash used in operating activities -continuing operations in the year ended December 31, 2014 totaled approximately $46.9 million, which is the sum of (i) our net loss from continuing operations of $55.5 million, and adjusted for non-cash expenses totaling $10.2 million (which includes adjustments for equity-based compensation, depreciation and amortization, and changes in acquisition-related contingent consideration), and (ii) changes in operating assets and liabilities of approximately $1.7 million.
Our cash used in operating activities -continuing operations in the year ended December 31, 2013 totaled approximately $27.1 million, which is the sum of (i) our net loss from continuing operations of $39.5 million, and adjusted for non-cash expenses totaling $10.8 million (which includes adjustments for equity-based compensation, depreciation and amortization, and changes in acquisition-related contingent consideration), and (ii) changes in operating assets and liabilities of approximately $1.6 million.
Our cash used in operating activities -continuing operations in the year ended December 31, 2012 totaled approximately $18.8 million, which is the sum of (i) our net loss from continuing operations of $36.1 million, and adjusted for non-cash expenses totaling $14.3 million (which includes adjustments for equity-based compensation, depreciation and amortization, and changes in acquisition-related contingent consideration), and (ii) changes in operating assets and liabilities of approximately $3.1 million.
Investing Activities - Continuing Operations
During the year ended December 31, 2014, we spent approximately $3.7 million for property and equipment. In addition, we spent approximately $7.1 million for net purchases of marketable securities available for sale during year ended December 31, 2014.
During the year ended December 31, 2013, we spent approximately $2.7 million for property and equipment.
During the year ended December 31, 2012, we completed the sale of our 51% interest in Erye for approximately 13.4 million in total consideration, including 12.3 million in cash. In addition, we spent approximately $0.5 million for property and equipment.
Financing Activities - Continuing Operations
During the year ended December 31, 2014, our financing activities consisted of the following:
• | We raised gross proceeds of approximately $15.0 million from loan proceeds from Oxford Finance LLC in September 2014. In connection with the loan, we repaid all outstanding amounts due under two loans from TD Bank, N.A. in the amount of approximately $3.1 million. In addition, debt offering/issuance costs of $0.5 million were paid in connection with the loan. |
66
• | We raised gross proceeds of approximately $16.5 million through the issuance of approximately 2.8 million shares of Common Stock under the provisions of our Common Stock Purchase Agreements with Aspire. |
• | We raised approximately $0.3 million from the exercise of 48,987 options. |
• | We raised approximately $1.7 million from the exercise of 333,250 warrants. |
• | We received proceeds of $1.8 million from the issuance of notes payable relating to certain insurance policies and equipment financings, less repayments of $1.1 million. |
During the year ended December 31, 2013, our financing activities consisted of the following:
• | We raised $11.5 million (or $10.5 million in net proceeds after deducting underwriting discounts and commissions and offering expenses) through an underwritten offering of 2.3 million shares of our common stock at a public offering price of $5.00 per share in April 2013. |
• | We raised $40.3 million (or $37.1 million in net proceeds after deducting underwriting discounts and commissions and offering expenses) through an underwritten offering of 5.75 million shares of our common stock at a public offering price of $7.00 per share in October 2013. |
• | We raised gross proceeds of approximately $11.1 million through the issuance of approximately 1.6 million shares of common stock under the provisions of our common stock purchase agreement with Aspire. |
• | We raised approximately $0.2 million from the exercise of 0.03 million options. |
• | We raised approximately $3.0 million from the exercise of 0.6 million warrants. To induce the exercise of certain of these warrants, we provided consideration to the warrant holders in the form of cash. |
During the year ended December 31, 2012, our financing activities consisted of the following:
• | We raised $6.8 million (or $6.0 million in net proceeds after deducting underwriting discounts and offering expenses) through an underwritten offering of 1.7 million units, each unit consisting of one share of common stock and a five year warrant to purchase one share of common stock at an exercise price of $5.10 per share. |
• | We raised an aggregate of approximately $7.1 million in private placements through the issuance of approximately 1.3 million shares of common stock and 0.9 million five year warrants at exercise prices ranging from $5.10 to $7.40. |
• | We raised gross proceeds of approximately $3.3 million through the issuance of 0.5 million shares of common stock under the provisions of our common stock purchase agreement with Aspire. |
• | We raised approximately $6.6 million from the exercise of approximately 0.8 million warrants. To induce the exercise of certain of these warrants, we provided consideration to the warrant holders in the form of either cash, stock or additional warrants. |
• | During 2012, we made cash payment totaling $5.7 million for the repayment of our Series E Preferred Stock and dividends. |
Liquidity and Capital Requirements Outlook
Liquidity
We anticipate requiring additional capital in order to fund the development of cell therapy product candidates, particularly in our Targeted Cancer Immunotherapy Program, Ischemic Repair Program and Immune Modulation Program, as well as to engage in strategic transactions. The most significant funding needs are anticipated to be in connection with the conduct of our Intus Phase 3 clinical trial which is expected to cost approximately $43 million, for which we began activating clinical sites during the fourth quarter of 2014, and other costs related to the targeted cancer immunotherapy operations acquired from CSC in May 2014. In the fourth quarter of 2014 we began activating clinical trial sites for the Intus Phase 3 clinical trial. The acquisition of CSC and the interim results of our PreSERVE Phase 2 clinical trial reported in November 2014 could result in our re-prioritizing the development of certain of our other earlier stage clinical trials. We also anticipate requiring additional capital to grow the PCT business, including implementing additional automation capabilities and pursuing plans to establish commercial capacity, harmonize across locations, strengthen quality systems and expand internationally. We recently completed expansion in the
67
Allendale, New Jersey facility adding laboratory, clean room suites and support facilities, and completed expansion in the Mountain View, California facility adding manufacturing capacity with additional clean rooms, laboratory space and support facilities.
To meet our short and long term liquidity needs, we currently expect to use existing cash balances, our revenue generating activities, and a variety of other means. Those other means include the continued use of a common stock purchase agreement with Aspire (the "Aspire Agreement"). We entered into a $30 million common stock purchase agreement with Aspire in March 2014, of which we had $19.1 million remaining available at December 31, 2014. In addition, in September 2014, we entered into a loan and security agreement with Oxford Finance LLC and to date received $15.0 million of a potential $20.0 million in gross proceeds. In connection with the $15.0 million loan, we repaid all outstanding amounts due under two loans from TD Bank, N.A. in the amount of approximately $3.1 million, and paid debt offering/issuance costs and interim period interest, resulting in net proceeds from the loan of $11.3 million. The additional $5.0 million loan may be obtained from Oxford if we enter into a strategic arrangement with respect to NBS10 and receive an upfront payment of not less than $10.0 million in connection therewith, before September 19, 2015. Other sources of liquidity could include additional potential issuances of debt or equity securities in public or private financings, additional warrant exercises, option exercises, partnerships and/or collaborations, and/or sale of assets. In addition, we will continue to seek as appropriate grants for scientific and clinical studies from the California Institute for Regenerative Medicine, National Institutes of Health, Department of Defense, and other US and other governmental agencies and foundations, but there can be no assurance that we will be successful in qualifying for or obtaining such grants. Our history of operating losses and liquidity challenges, may make it difficult for us to raise capital on acceptable terms or at all. The demand for the equity and debt of biopharmaceutical companies like ours is dependent upon many factors, including the general state of the financial markets. During times of extreme market volatility, capital may not be available on favorable terms, if at all. Our inability to obtain such additional capital could materially and adversely affect our business operations. We believe that our current cash balances and revenue generating activities, along with access to the Aspire Agreement, will be sufficient to fund the business through the next 12 months.
While we continue to seek capital through a number of means, there can be no assurance that additional financing will be available on acceptable terms, if at all, and our negotiating position in capital generating efforts may worsen as existing resources are used. Additional equity financing may be dilutive to our stockholders; debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate as a business; our stock price may not reach levels necessary to induce option or warrant exercises; and asset sales may not be possible on terms we consider acceptable. If we are unable to raise the funds necessary to meet our long-term liquidity needs, we may have to delay or discontinue the acquisition and development of cell therapies, and/or the expansion of our business or raise funds on terms that we currently consider unfavorable.
Commitments and Contingencies
The following table summarizes our obligations to make future payments under current contracts as of December 31, 2014 (in thousands):
Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||||||
Contractual Obligations | |||||||||||||||||||
Notes Payable | $ | 1,642.7 | $ | 816.8 | $ | 825.9 | $ | — | $ | — | |||||||||
Long Term Debt | 16,200.0 | 1,109.6 | 9,775.9 | 5,314.5 | — | ||||||||||||||
Purchase Obligations | 834.0 | 333.6 | 500.4 | — | — | ||||||||||||||
Operating Lease Obligations | 6,247.8 | 1,567.9 | 3,024.2 | 1,027.6 | 628.1 | ||||||||||||||
$ | 24,924.5 | $ | 3,827.9 | $ | 14,126.4 | $ | 6,342.1 | $ | 628.1 | ||||||||||
Other significant commitments and contingencies include the following:
• | Under agreements with external clinical research organizations (“CROs”), we will incur expenses relating to our clinical trials for our therapeutic product candidates in development. The timing and amount of these expenses are based on performance of services rendered and expenses as incurred by the CROs and therefore, we cannot reasonably estimate the timing of these payments. |
• | Under certain license, collaboration, and merger agreements, we are required to pay royalties, milestone and/or other payments upon successful development and commercialization of products. However, successful research and development of pharmaceutical products is high risk, and most products fail to reach the market. Therefore, at this time the amount and timing of the payments, if any, are not known. |
68
• | From time to time, we are subject to legal proceedings and claims, either asserted or unasserted, that arise in the ordinary course of business. While the outcome of pending claims cannot be predicted with certainty, we do not believe that the outcome of any pending claims will have a material adverse effect on our financial condition or operating results. |
SEASONALITY
NeoStem does not believe that its operations are seasonal in nature.
OFF-BALANCE SHEET ARRANGEMENTS
NeoStem does not have any off-balance sheet arrangements.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and judgments that affect the amounts reported in the financial statements. On an ongoing basis, the Company evaluates its estimates and assumptions. The Company bases its estimates on historical experience and other assumptions believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results could differ from these estimates.
An accounting policy is considered to be critical if it is important to the Company’s financial condition and results of operations and if it requires management’s most difficult, subjective and complex judgments in its application. For a summary of all of the Company’s significant accounting policies, see Note 2 to the Company’s Consolidated Financial Statements.
Revenue Recognition
Clinical Services: The Company recognizes revenue for its (i) cell process development and (ii) cell manufacturing services based on the terms of individual contracts.
We recognize revenues for cell development services when all of the following conditions are met:
• | persuasive evidence of an arrangement exists; |
• | delivery has occurred or the services have been rendered; |
• | the fee is fixed or determinable; and |
• | collectability is probable. |
The Company considers signed contracts as evidence of an arrangement. The Company assesses whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the payment terms are subject to refund or adjustment. The Company assesses cash collectability based on a number of factors, including past collection history with the client and the client's creditworthiness. If the Company determines that collectability is not reasonably assured, it defers revenue recognition until collectability becomes reasonably assured, which is generally upon receipt of the cash. The Company's arrangements are generally non-cancellable, though clients typically have the right to terminate their agreement for cause if the Company materially fails to perform.
Revenues associated with cell process development services generally contain multiple stages that do not have stand-alone values and are dependent upon one another, and are recognized as revenue on a completed contract basis. Progress billings collected prior to contract completion are recorded as unearned revenue until such time the contract is completed, which usually requires formal client acceptance.
Cell manufacturing services are generally distinct arrangements whereby the Company is paid for time and materials or for fixed monthly amounts. Revenue is recognized when efforts are expended or contractual terms have been met.
Some client agreements include multiple elements, comprised of cell process development and cell manufacturing services. The Company believes that cell process development and cell manufacturing services each have stand-alone value
69
because these services can be provided separately by other companies. In accordance with ASC Update No. 2009-13, “Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements,” the Company (1) separates deliverables into separate units of accounting when deliverables are sold in a bundled arrangement and (2) allocates the arrangement's consideration to each unit in the arrangement based on its relative selling price.
Clinical Services Reimbursements: The Company separately charges the customers for the expenses associated with certain consumable resources (reimbursable expenses) that are specified in each clinical services contract. On a monthly basis, the Company bills customers for reimbursable expenses and immediately recognizes these billings as revenue, as the revenue is deemed earned as reimbursable expenses are incurred.
Processing and Storage Services: The Company recognizes revenue related to the collection and cryopreservation of cord blood and autologous adult stem cells when the cryopreservation process is completed which is approximately twenty-four hours after cells have been collected. Revenue related to advance payments of storage fees is deferred and recognized ratably over the period covered by the advance payments.
Share-Based Compensation
The Company expenses all share-based payment awards to employees, directors, advisors and consultants, including grants of stock options, warrants, and restricted stock, over the requisite service period based on the grant date fair value of the awards. Advisor and consultant awards are remeasured each reporting period through vesting. For awards with performance-based vesting criteria, the Company estimates the probability of achievement of the performance criteria and recognizes compensation expense related to those awards expected to vest. The Company determines the fair value of option awards using the Black-Scholes option-pricing model which uses both historical and current market data to estimate the fair value. This method incorporates various assumptions such as the risk-free interest rate, expected volatility, expected dividend yield and expected life of the options or warrants. The fair value of the Company’s restricted stock and restricted stock units is based on the closing market price of the Company’s common stock on the date of grant.
Goodwill and Other Intangible Assets
Goodwill is the excess of purchase price over the fair value of identified net assets of businesses acquired. The Company’s intangible assets with an indefinite life are related to in process research and development (IPR&D) for NBS10, the clinical candidate acquired in the Amorcyte acquisition, as the Company expects this research and development to provide the Company with substantial benefit for a period that extends beyond the foreseeable horizon. Intangible assets with indefinite useful lives are measured at their respective fair values as of the acquisition date. The Company does not amortize goodwill and intangible assets with indefinite useful lives. Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated R&D efforts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time.
The Company reviews goodwill and indefinite-lived intangible assets at least annually, or at the time a triggering event is identified for possible impairment. Goodwill and indefinite-lived intangible assets are reviewed for possible impairment between annual tests if an event occurs or circumstances change that would more likely than not reduce the fair value of the reporting unit or the IPR&D below its carrying value. The Company tests its goodwill and indefinite-lived intangible assets each year on December 31. The Company reviews the carrying value of goodwill and indefinite-lived intangible assets utilizing an income approach model, and, where appropriate, a market value approach is also utilized to supplement the discounted cash flow model. The Company makes assumptions regarding estimated future cash flows, discount rates, long-term growth rates and market values to determine each reporting unit’s and IPR&D's estimated fair value. If these estimates or related assumptions change in the future, the Company may be required to record impairment charges. In accordance with its accounting policy, the Company tested goodwill for impairment as of December 31, 2014 and 2013 for its two reporting units as well as its IPR&D, and concluded there was no goodwill and IPR&D impairment.
Amortized intangible assets consist of customer lists, manufacturing technology, and tradename, as well as patents and rights associated primarily with the VSEL™ Technology. These intangible assets are amortized on a straight line basis over their respective useful lives.
Evaluation of Long-lived Assets
70
The Company reviews long-lived assets and finite-lived intangibles assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset exceeds the fair value of the asset. If other events or changes in circumstances indicate that the carrying amount of an asset that the Company expects to hold and use may not be recoverable, the Company will estimate the undiscounted future cash flows expected to result from the use of the asset and/or its eventual disposition, and recognize an impairment loss. The impairment loss, if determined to be necessary, would be measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets. No events were noted in 2014 or 2013.
Recognizing and Measuring Assets Acquired and Liabilities Assumed in Business Combinations at Fair Value
The Company accounts for acquired businesses using the purchase method of accounting, which requires that assets acquired and liabilities assumed be recorded at date of acquisition at their respective fair values. The consolidated financial statements and results of operations reflect an acquired business after the completion of the acquisition. The fair value of the consideration paid, including contingent consideration, is assigned to the underlying net assets of the acquired business based on their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill. Amounts allocated to IPR&D are included on the balance sheet. Intangible assets, including IPR&D assets upon successful completion of the project and approval of the product, are amortized on a straight-line basis to amortization expense over the expected life of the asset. Significant judgments are used in determining the estimated fair values assigned to the assets acquired and liabilities assumed and in determining estimates of useful lives of long-lived assets. Fair value determinations and useful life estimates are based on, among other factors, estimates of expected future net cash flows, estimates of appropriate discount rates used to present value expected from future net cash flow streams, the timing of approvals for IPR&D projects and the timing of related product launch dates, the assessment of each asset’s life cycle, the impact of competitive trends on each asset’s life cycle and other factors. These judgments can materially impact the estimates used to allocate acquisition date fair values to assets acquired and liabilities assumed and the resulting timing and amount charged to, or recognized in current and future operating results. For these and other reasons, actual results may vary significantly from estimated results.
The Company determines the acquisition date fair value of contingent consideration obligations based on a probability-weighted income approach derived from revenue estimates, post-tax gross profit levels and a probability assessment with respect to the likelihood of achieving contingent obligations including contingent payments such as milestone obligations, royalty obligations and contract earn-out criteria, where applicable. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting. The resulting probability-weighted cash flows are discounted using an appropriate effective annual interest rate. At each reporting date, the contingent consideration obligation will be revalued to estimated fair value at that time and changes in fair value will be reflected as income or expense in our consolidated statement of operations. Changes in the fair value of the contingent consideration obligations may result from changes in discount periods and rates, changes in the timing and amount of revenue estimates and changes in probability assumptions with respect to the likelihood of achieving the various contingent payment obligations. Changes in assumptions utilized in our contingent consideration fair value estimates could result in an increase or decrease in our contingent consideration obligation and a corresponding charge to operating loss or gain.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Market risk is the risk of change in fair value of a financial instrument due to changes in interest rates, equity prices, creditworthiness, financing, exchange rates or other factors. Our primary market risk exposure relates to changes in interest rates. Our earnings and cash flows are subject to fluctuations due to changes in interest rates, principally in connection with our investments in marketable securities, which consist primarily of short-term money market funds and municipal debt securities. However, as of December 31, 2014, we do not believe we are materially exposed to changes in interest rates given the short-term duration of the securities. Additionally, our outstanding $15.0 million Long-Term Loan with Oxford Finance LLC, representing our largest component of debt, has a fixed interest rate until 2018, and is not subject to interest rate exposure. As a result, we do not believe we have material exposure to market risk related to interest rate changes as of December 31, 2014.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
The financial statements and notes thereto required to be filed under this Item are presented commencing on page
of this Annual Report on Form 10-K.
71
NeoStem, Inc. and Subsidiaries
Table of Contents
Report of Independent Registered Public Accounting Firm | ||
Financial Statements: | ||
Consolidated Balance Sheets at December 31, 2014 and 2013 | ||
Consolidated Statements of Operations - Years Ended December 31, 2014, 2013, and 2012 | ||
Consolidated Statements of Comprehensive Loss - Years Ended December 31, 2014, 2013, and 2012 | ||
Consolidated Statements of Equity - Years Ended December 31, 2014, 2013, and 2012 | ||
Consolidated Statements of Cash Flows - Years Ended December 31, 2014, 2013, and 2012 | ||
Notes to Consolidated Financial Statements | ||
72
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
NeoStem, Inc.
We have audited the accompanying consolidated balance sheets of NeoStem, Inc. (a Delaware corporation) and subsidiaries (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive loss, equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of NeoStem, Inc. and subsidiaries as of December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2014, based on criteria established in the 2013 Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 2, 2015 expressed an unqualified opinion.
/s/ GRANT THORNTON LLP
New York, New York
March 2, 2015
73
NEOSTEM, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
December 31, 2014 | December 31, 2013 | ||||||
ASSETS | |||||||
Current Assets | |||||||
Cash and cash equivalents | $ | 19,174,061 | $ | 46,133,759 | |||
Marketable securities | 7,080,053 | — | |||||
Accounts receivable trade, net of allowance for doubtful accounts of $385,362 and $391,829, respectively | 3,111,274 | 1,860,835 | |||||
Deferred costs | 2,566,989 | 1,270,223 | |||||
Prepaids and other current assets | 4,349,167 | 1,561,933 | |||||
Total current assets | 36,281,544 | 50,826,750 | |||||
Property, plant and equipment, net | 15,960,731 | 12,844,216 | |||||
Goodwill | 25,209,336 | 11,117,770 | |||||
Intangible assets, net | 47,560,406 | 13,875,617 | |||||
Other assets | 1,263,375 | 1,151,729 | |||||
Total assets | $ | 126,275,392 | $ | 89,816,082 | |||
LIABILITIES AND EQUITY | |||||||
Current Liabilities | |||||||
Accounts payable | $ | 5,661,173 | $ | 3,354,908 | |||
Accrued liabilities | 4,322,901 | 4,018,026 | |||||
Long-term debt, current | 1,109,612 | — | |||||
Notes payable | 816,776 | 381,097 | |||||
Mortgages payable | — | 213,112 | |||||
Derivative liabilities | — | 23,175 | |||||
Unearned revenues | 4,334,120 | 1,816,601 | |||||
Total current liabilities | 16,244,582 | 9,806,919 | |||||
Deferred income taxes | 18,176,190 | 4,379,226 | |||||
Notes payable | 825,897 | 531,164 | |||||
Mortgages payable | — | 3,023,609 | |||||
Long term debt | 13,890,388 | — | |||||
Acquisition-related contingent consideration | 18,260,000 | 9,450,000 | |||||
Other long-term liabilities | 804,546 | 598,729 | |||||
Total liabilities | 68,201,603 | 27,789,647 | |||||
Commitments and Contingencies | |||||||
EQUITY | |||||||
Stockholders' Equity | |||||||
Preferred stock; authorized, 20,000,000 shares Series B convertible redeemable preferred stock liquidation value, 1 share of common stock, $.01 par value; 825,000 shares designated; issued and outstanding, 10,000 shares at December 31, 2014 and December 31, 2013 | 100 | 100 | |||||
Common stock, $.001 par value, authorized 500,000,000 shares; issued and outstanding, 36,783,857 and 27,196,537 shares, at December 31, 2014 and December 31, 2013, respectively | 36,784 | 27,197 | |||||
Additional paid-in capital | 350,428,903 | 299,594,525 | |||||
Treasury stock, at cost | (705,742 | ) | (705,742 | ) | |||
Accumulated deficit | (291,246,538 | ) | (236,373,605 | ) | |||
Accumulated other comprehensive income | 1,329 | — | |||||
Total NeoStem, Inc. stockholders' equity | 58,514,836 | 62,542,475 | |||||
Noncontrolling interests | (441,047 | ) | (516,040 | ) | |||
Total equity | 58,073,789 | 62,026,435 | |||||
$ | 126,275,392 | $ | 89,816,082 | ||||
See accompanying notes to consolidated financial statements.
74
NEOSTEM, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Revenues | $ | 17,938,790 | $ | 14,668,455 | $ | 14,329,889 | |||||
Expenses: | |||||||||||
Cost of revenues | 15,678,475 | 12,947,217 | 11,949,124 | ||||||||
Research and development | 29,194,262 | 16,917,396 | 10,451,070 | ||||||||
Selling, general, and administrative | 30,806,807 | 21,612,793 | 22,315,346 | ||||||||
Operating Expenses | 75,679,544 | 51,477,406 | 44,715,540 | ||||||||
Operating loss | (57,740,754 | ) | (36,808,951 | ) | (30,385,651 | ) | |||||
Other income (expense): | |||||||||||
Other income (expense), net | 2,926,003 | (1,614,858 | ) | (4,314,228 | ) | ||||||
Interest expense | (755,697 | ) | (281,421 | ) | (1,576,975 | ) | |||||
2,170,306 | (1,896,279 | ) | (5,891,203 | ) | |||||||
Loss from operations before (benefit) provision for income taxes | (55,570,448 | ) | (38,705,230 | ) | (36,276,854 | ) | |||||
(Benefit) provision for income taxes | (104,202 | ) | 780,104 | (175,533 | ) | ||||||
Net loss from continuing operations | (55,466,246 | ) | (39,485,334 | ) | (36,101,321 | ) | |||||
Loss from discontinued operations - net | — | — | (30,267,990 | ) | |||||||
Net loss | (55,466,246 | ) | (39,485,334 | ) | (66,369,311 | ) | |||||
Less - loss from continuing operations attributable to noncontrolling interests | (593,313 | ) | (504,090 | ) | (287,181 | ) | |||||
Less - loss from discontinued operations attributable to noncontrolling interests | — | — | (12,312,646 | ) | |||||||
Net loss attributable to NeoStem, Inc. | (54,872,933 | ) | (38,981,244 | ) | (53,769,484 | ) | |||||
Warrant inducements | — | — | (1,012,819 | ) | |||||||
Preferred dividends | — | — | (528,023 | ) | |||||||
Net loss attributable to NeoStem, Inc. common stockholders | $ | (54,872,933 | ) | (38,981,244 | ) | (55,310,326 | ) | ||||
Amounts Attributable to NeoStem, Inc. common stockholders: | |||||||||||
Loss from continuing operations | $ | (54,872,933 | ) | $ | (38,981,244 | ) | $ | (35,814,140 | ) | ||
Loss from discontinued operations - net of taxes | — | — | (17,955,344 | ) | |||||||
Warrant inducements | — | — | (1,012,819 | ) | |||||||
Preferred dividends | — | — | (528,023 | ) | |||||||
Net loss attributable to NeoStem, Inc. common stockholders | $ | (54,872,933 | ) | $ | (38,981,244 | ) | $ | (55,310,326 | ) | ||
Basic and diluted loss per share attributable to NeoStem, Inc. common stockholders: | |||||||||||
Continuing operations | $ | (1.68 | ) | $ | (1.90 | ) | $ | (2.59 | ) | ||
Discontinued operations | $ | — | — | (1.30 | ) | ||||||
NeoStem, Inc. common stockholders | $ | (1.68 | ) | $ | (1.90 | ) | $ | (4.00 | ) | ||
Weighted average common shares outstanding | 32,756,102 | 20,495,771 | 13,841,997 | ||||||||
See accompanying notes to consolidated financial statements.
75
NEOSTEM, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
Year Ended December 31, | ||||||||||||
2014 | 2013 | 2,012 | ||||||||||
Net loss | $ | (55,466,246 | ) | $ | (39,485,334 | ) | $ | (66,369,311 | ) | |||
Other comprehensive income (loss): | ||||||||||||
Available for sale securities - net unrealized gain | 1,329 | — | — | |||||||||
Foreign currency translation elimination on exit of segment | — | — | (169,993 | ) | ||||||||
Foreign currency translation elimination on sale of segment | — | — | (4,387,371 | ) | ||||||||
Foreign currency translation | — | — | 405,021 | |||||||||
Total other comprehensive income (loss) | 1,329 | — | (4,152,343 | ) | ||||||||
Comprehensive loss | (55,464,917 | ) | (39,485,334 | ) | (70,521,654 | ) | ||||||
Noncontrolling interests elimination on sale of segment | — | — | (6,014,981 | ) | ||||||||
Comprehensive loss attributable to noncontrolling interests | (593,313 | ) | (504,090 | ) | (12,448,950 | ) | ||||||
Comprehensive net loss attributable to NeoStem, Inc. common stockholders | $ | (54,871,604 | ) | $ | (38,981,244 | ) | $ | (52,057,723 | ) | |||
See accompanying notes to consolidated financial statements.
76
NEOSTEM, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF EQUITY
Series B Convertible Preferred Stock | Common Stock | Additional Paid in Capital | Accumulated Other Comprehensive Income | Accumulated Deficit | Treasury Stock | Total NeoStem, Inc. Stockholders' Equity | Non- Controlling Interest in Subsidiary | Total Equity | |||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||||
Balance at December 31, 2011 | 10,000 | $ | 100 | 10,932,959 | $ | 10,933 | $ | 200,957,035 | $ | 4,152,343 | $ | (143,094,854 | ) | $ | — | $ | 62,025,557 | $ | 18,106,961 | $ | 80,132,518 | ||||||||||||||||||||
Net loss | — | — | — | — | — | — | (53,769,484 | ) | — | (53,769,484 | ) | (12,599,827 | ) | (66,369,311 | ) | ||||||||||||||||||||||||||
Foreign currency translation | — | — | — | — | — | 235,028 | — | — | 235,028 | 150,877 | 385,905 | ||||||||||||||||||||||||||||||
Share-based compensation | — | — | 336,427 | 336 | 6,712,200 | — | — | — | 6,712,536 | — | 6,712,536 | ||||||||||||||||||||||||||||||
Proceeds from issuance of common stock | — | — | 3,573,229 | 3,573 | 16,425,254 | — | — | — | 16,428,827 | — | 16,428,827 | ||||||||||||||||||||||||||||||
Proceeds from warrant exercises | — | — | 1,107,618 | 1,108 | 6,603,311 | — | — | — | 6,604,419 | — | 6,604,419 | ||||||||||||||||||||||||||||||
Shares, options and warrants received in Erye Sale | — | — | — | — | (452,301 | ) | — | — | (665,600 | ) | (1,117,901 | ) | — | (1,117,901 | ) | ||||||||||||||||||||||||||
Elimination of equity upon Erye Sale | — | — | — | — | — | (4,387,371 | ) | — | — | (4,387,371 | ) | (6,014,981 | ) | (10,402,352 | ) | ||||||||||||||||||||||||||
Repayment of Series E Preferred Principal and Dividends | — | — | 279,238 | 279 | 1,201,938 | — | (528,023 | ) | — | 674,194 | — | 674,194 | |||||||||||||||||||||||||||||
Warrant inducements | — | — | 145,895 | 146 | (228,822 | ) | — | — | — | (228,676 | ) | — | (228,676 | ) | |||||||||||||||||||||||||||
Balance at December 31, 2012 | 10,000 | $ | 100 | 16,375,366 | $ | 16,375 | $ | 231,218,615 | $ | — | $ | (197,392,361 | ) | $ | (665,600 | ) | $ | 33,177,129 | $ | (356,970 | ) | $ | 32,820,159 | ||||||||||||||||||
Net loss | — | — | — | — | — | — | (38,981,244 | ) | — | (38,981,244 | ) | (504,090 | ) | (39,485,334 | ) | ||||||||||||||||||||||||||
Share-based compensation | — | — | 513,912 | 514 | 6,878,187 | — | — | (40,142 | ) | 6,838,559 | — | 6,838,559 | |||||||||||||||||||||||||||||
Net proceeds from issuance of common stock | — | — | 9,712,724 | 9,713 | 58,726,453 | — | — | — | 58,736,166 | — | 58,736,166 | ||||||||||||||||||||||||||||||
Proceeds from option exercises | 31,369 | 31 | 150,627 | 150,658 | 150,658 | ||||||||||||||||||||||||||||||||||||
Proceeds from warrant exercises | 563,167 | 564 | 3,027,677 | 3,028,241 | 3,028,241 | ||||||||||||||||||||||||||||||||||||
Change in Ownership in Subsidiary | (345,020 | ) | (345,020 | ) | 345,020 | — | |||||||||||||||||||||||||||||||||||
Warrant inducements | — | — | — | — | (62,014 | ) | — | — | — | (62,014 | ) | — | (62,014 | ) | |||||||||||||||||||||||||||
Balance at December 31, 2013 | 10,000 | $ | 100 | 27,196,538 | $ | 27,197 | $ | 299,594,525 | $ | — | $ | (236,373,605 | ) | $ | (705,742 | ) | $ | 62,542,475 | $ | (516,040 | ) | $ | 62,026,435 | ||||||||||||||||||
Net loss | — | — | — | — | — | — | (54,872,933 | ) | — | (54,872,933 | ) | (593,313 | ) | (55,466,246 | ) | ||||||||||||||||||||||||||
Unrealized gain/loss on marketable securities | — | — | — | — | — | 1,329 | — | — | 1,329 | — | 1,329 | ||||||||||||||||||||||||||||||
Share-based compensation | — | — | 916,359 | 916 | 11,208,626 | — | — | — | 11,209,542 | — | 11,209,542 | ||||||||||||||||||||||||||||||
Net proceeds from issuance of common stock | — | — | 2,959,214 | 2,959 | 16,707,686 | — | — | — | 16,710,645 | — | 16,710,645 | ||||||||||||||||||||||||||||||
Proceeds from option exercises | 48,987 | 49 | 270,959 | 271,008 | 271,008 | ||||||||||||||||||||||||||||||||||||
Proceeds from warrant exercises | — | — | 333,250 | 333 | 1,720,392 | — | — | — | 1,720,725 | — | 1,720,725 | ||||||||||||||||||||||||||||||
Shares issued in CSC merger | — | — | 5,329,510 | 5,330 | 21,595,021 | — | — | — | 21,600,351 | — | 21,600,351 | ||||||||||||||||||||||||||||||
Change in Ownership in Subsidiary | — | — | — | — | (668,306 | ) | — | — | — | (668,306 | ) | 668,306 | — | ||||||||||||||||||||||||||||
Balance at December 31, 2014 | 10,000 | $ | 100 | 36,783,857 | $ | 36,784 | $ | 350,428,903 | $ | 1,329 | $ | (291,246,538 | ) | $ | (705,742 | ) | $ | 58,514,836 | $ | (441,047 | ) | $ | 58,073,789 | ||||||||||||||||||
See accompanying notes to consolidated financial statements.
77
NEOSTEM, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash flows from operating activities: | |||||||||||
Net loss | $ | (55,466,246 | ) | $ | (39,485,334 | ) | $ | (66,369,311 | ) | ||
Loss from discontinued operations | — | — | 30,267,990 | ||||||||
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||||
Common stock, stock options and warrants issued as payment for compensation, services rendered | 11,209,542 | 6,838,559 | 6,712,536 | ||||||||
Depreciation and amortization | 2,186,949 | 1,605,608 | 1,550,571 | ||||||||
Amortization of preferred stock discount and issuance cost | — | — | 1,609,495 | ||||||||
Changes in fair value of derivative liability | (23,175 | ) | (77,981 | ) | (373,307 | ) | |||||
Changes in acquisition-related contingent consideration | (3,080,000 | ) | 1,900,000 | 4,420,000 | |||||||
Loss on disposal of assets | — | — | 13,653 | ||||||||
Bad debt (recovery) expense | (6,467 | ) | (234,225 | ) | 511,755 | ||||||
Deferred income taxes | (104,202 | ) | 780,104 | (175,533 | ) | ||||||
Amortization/Accretion on Marketable Securities | 51,517 | — | — | ||||||||
Changes in operating assets and liabilities: | |||||||||||
Prepaid expenses and other current assets | (2,768,062 | ) | (758,798 | ) | (178,011 | ) | |||||
Accounts receivable | (1,198,844 | ) | (573,005 | ) | (554,884 | ) | |||||
Deferred costs | (1,296,766 | ) | (157,198 | ) | (465,280 | ) | |||||
Unearned revenues | 2,517,520 | 348,260 | 178,008 | ||||||||
Other assets | 613,175 | (204,422 | ) | 2,414,842 | |||||||
Accounts payable, accrued expenses and other liabilities | 469,821 | 2,916,739 | 1,677,551 | ||||||||
Net cash used in operating activities - continuing operations | (46,895,238 | ) | (27,101,693 | ) | (18,759,925 | ) | |||||
Net cash provided by operating activities - discontinued operations | — | — | 4,907,407 | ||||||||
Net cash used in operating activities | (46,895,238 | ) | (27,101,693 | ) | (13,852,518 | ) | |||||
Cash flows from investing activities: | |||||||||||
Cash received in acquisitions | 50,894 | — | — | ||||||||
Cash received in divestiture | — | — | 12,280,000 | ||||||||
Purchase of short term investments | (8,043,241 | ) | — | — | |||||||
Sales of marketable securities | 913,000 | — | — | ||||||||
Acquisition of property and equipment | (3,657,352 | ) | (2,691,471 | ) | (531,315 | ) | |||||
Net cash (used in) provided by investing activities - continuing operations | (10,736,699 | ) | (2,691,471 | ) | 11,748,685 | ||||||
Net cash used in investing activities - discontinued operations | — | — | (5,660,305 | ) | |||||||
Net cash (used in) provided by investing activities | (10,736,699 | ) | (2,691,471 | ) | 6,088,380 | ||||||
Cash flows from financing activities: | |||||||||||
Proceeds from exercise of options | 271,008 | 150,658 | — | ||||||||
Proceeds from exercise of warrants | 1,720,725 | 3,028,241 | 6,604,418 | ||||||||
Net proceeds from issuance of capital stock | 16,710,645 | 58,736,165 | 16,428,827 | ||||||||
Proceeds from long term debt | 15,000,000 | — | — | ||||||||
Debt issuance costs | (523,830 | ) | — | — | |||||||
Repayment of mortgage loan | (3,236,721 | ) | (201,754 | ) | (196,585 | ) | |||||
Proceeds from notes payable | 1,827,413 | 1,041,347 | 666,501 | ||||||||
78
Repayment of notes payable | (1,097,001 | ) | (503,172 | ) | (440,477 | ) | |||||
Repayment of preferred stock | — | — | (5,394,263 | ) | |||||||
Payment of dividend for preferred stock | — | — | (327,748 | ) | |||||||
Payment for warrant inducement | — | (62,014 | ) | (228,676 | ) | ||||||
Net cash provided by financing activities - continuing operations | 30,672,239 | 62,189,471 | 17,111,997 | ||||||||
Net cash used in provided by financing activities - discontinued operations | — | — | (8,370,228 | ) | |||||||
Net cash provided by financing activities | 30,672,239 | 62,189,471 | 8,741,769 | ||||||||
Impact of changes of foreign exchange rates | — | — | 14,389 | ||||||||
Net (decrease) increase in cash and cash equivalents | (26,959,698 | ) | 32,396,307 | 992,020 | |||||||
Cash and cash equivalents at beginning of year | 46,133,759 | 13,737,452 | 12,745,432 | ||||||||
Cash and cash equivalents at end of year | $ | 19,174,061 | $ | 46,133,759 | $ | 13,737,452 | |||||
Supplemental Disclosure of Cash Flow Information: | |||||||||||
Cash paid during the period for: | |||||||||||
Interest | $ | 232,500 | $ | 274,100 | $ | 1,771,800 | |||||
Taxes | — | — | 2,100,000 | ||||||||
Supplemental Schedule of non-cash investing activities: | |||||||||||
Capitalized interest | — | — | 182,000 | ||||||||
Common stock, warrants and options received upon sale of Erye | — | — | 1,117,901 | ||||||||
Supplemental schedule of non-cash financing activities | |||||||||||
Common stock and contingent consideration issued with the acquisition of CSC | 33,490,351 | — | — | ||||||||
Common stock issued pursuant to the redemption of Convertible Redeemable Series E 7% Preferred Stock | — | — | 1,026,600 | ||||||||
Common stock issued in payment of dividends for the Convertible Redeemable Series E 7% Preferred Stock | — | — | 175,700 | ||||||||
See accompanying notes to consolidated financial statements.
79
NEOSTEM, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 – The Business
Overview
NeoStem, Inc. (“we,” “NeoStem” or the “Company”) is a vertically integrated, clinical-stage biopharmaceutical company that is pursuing the preservation and enhancement of human health through the development of cell based therapeutics that leverage the body’s natural ability to heal and fight disease. Our diversified pipeline and unique capabilities for innovative, cost-effective and efficient in-house development set us apart in this emerging industry as we work to fundamentally change the treatment paradigm for several serious diseases.
Our most advanced clinical program is based on our dendritic cell/cancer cell technology. It is focused on the development of an innovative cancer treatment that is designed to target the cells responsible for tumor growth and metastasis, known as cancer or tumor initiating cells (CSCs), using purified CSCs from a patient’s own tumor as an antigen source to induce or enhance an anti-tumor immune response in the patient. Our lead product candidate based on this platform technology, NBS20, targets malignant melanoma as an initial indication. NBS20 is being studied in patients with recurrent Stage III or Stage IV metastatic melanoma. The program has been granted Fast Track and Orphan designation by the Food and Drug Administration (FDA) and the protocol for the Phase 3 study, known as the Intus study, is the subject of a Special Protocol Assessment (SPA). Our SPA letter states that our Phase 3 clinical trial is adequately designed to provide the necessary data that, depending on outcome, could support a Biologics License Application (BLA) seeking marketing approval of NBS20. This protocol calls for randomizing 250 patients. Patient screening began in 1Q15 and randomization of the first patient is expected in 2Q15. Interim analysis of the data is targeted for 4Q2017. We are also evaluating other clinical indications for which we may advance this program, including lung, colon, ovarian and liver cancers.
Our company is also developing therapies that are designed to utilize CD34 cells to prevent heart failure and major adverse cardiac events following a severe heart attack, known as an ST-elevation myocardial infarction (STEMI), through the use of CD34 cells to regenerate tissue impacted by ischemia. Ischemia occurs when the supply of oxygenated blood in the body is restricted, causing tissue distress and death. We seek to improve oxygen delivery to tissues through the development and formation of new blood vessels. NBS10 is our most clinically advanced product candidate in our ischemic repair program. At the American Heart Association’s Scientific Sessions in November 2014, we reported data from the primary analysis of our 161 patient PreSERVE (acute myocardial infarction) AMI study. We are also planning the release of our one year data from the phase 2 trial on March 15, 2015 at the Annual Scientific Sessions of the American College of Cardiology. PreSERVE AMI is a randomized, double-blinded, placebo-controlled Phase 2 clinical trial testing NBS10, a “personalized” adult stem cell product being developed for the treatment of patients with left ventricular dysfunction following a STEMI. The primary endpoints are measured by, (i) the change from baseline to six months in myocardial perfusion (RTSS) measured by an imaging technique (SPECT); and (ii) safety of bone marrow procurement and infusion as measured by occurrence of adverse events, serious adverse events (SAEs) and major adverse cardiac events (MACE). There are five secondary efficacy endpoints, two evaluating left ventricular ejection fraction, one evaluating MACE, and two evaluating quality of life. The reported data were based on all treated patients that had received six month follow-up for imaging. The median length follow up for mortality, adverse events, SAEs and MACE in these patients was twelve months. The reported results allowed for important observations about a potential dose-dependent treatment effect that will help guide the next phase of development. These observations about a potential dose dependent treatment effect were based on post hoc analyses of subsets of treated patients based on the number of cells they received. Notably, statistically significant dose-dependent increases in left ventricular function and decreases in serious adverse events were seen in patients who received the highest dose of cells (n=15 patients), though no statistical significance was observed when NBS10 overall was compared to placebo on these measures. With respect to mortality, at one year there were no treatment group deaths while the control group saw a mortality rate of 3.6% (n=3), equating to a statistically significant reduction in mortality. Regarding MACE, while more events occurred with NBS10 overall versus placebo, a non-significant trend toward fewer events was observed in patients who received higher doses of cells, with MACE occurring in 14% of placebo patients, 17% of patients who received less than 14 million cells, 10% of patients who received greater than 14 million cells and 7% of patients who received greater than 20 million cells. Finally, our hypothesis that SPECT used to measure perfusion could be used as a surrogate marker for the current medically relevant and regulatory endpoints was disproven, giving us valuable direction regarding endpoints and analyses for future clinical trials. We expect to complete the PreSERVE AMI study as defined through the final three-year follow-up and, in the meantime, plan to meet with the FDA to discuss our results and our proposal for the next step(s) in development. Finalization of the decision for next steps for NBS10 are expected in the second half of 2015. We also are evaluating other clinical indications that involve ischemia into which we may advance this program, including critical limb ischemia (CLI) and congestive heart failure (CHF).
Another platform technology we are developing is designed to utilize Regulatory T Cells (Tregs) to treat diseases caused
80
by imbalances in an individual's immune system. This novel approach seeks to restore immune balance by enhancing Treg cell number and function. Tregs are a natural part of the human immune system and regulate the activity of T effector cells, the cells that are responsible for protecting the body from viruses and other foreign antigens. When Tregs function properly, only harmful foreign materials are attacked by T effector cells. In autoimmune disease, it is thought that deficient Treg activity permits the T effector cells to attack the body's own tissues. We have received a letter from the FDA stating that we may proceed on a Phase 2 study of NBS03D, a Treg based therapeutic being developed to treat type 1 diabetes mellitus (T1DM) in adolescents, and we plan to initiate the trial in late 2015 or 2016 depending on resource availability. We are evaluating other clinical indications into which we may advance this program, including graft versus host disease, chronic obstructive pulmonary disease (COPD), multiple sclerosis (MS), inflammatory bowel disease (IBD) and steroid resistant asthma.
Finally, we are actively exploring means by which we can take advantage of new regulations in Japan that permit conditional approval for regenerative medicine products that show sufficient safety evidence and signals of efficacy. Potential indications for this unique opportunity include our targeted cancer immunotherapy program in liver cancer and our ischemic repair program in CLI.
We believe that cell-based therapies have the potential to create a paradigm change in the treatment for a variety of diseases and conditions and we are evaluating other programs that we view as holding particular promise, including an aesthetics program for a topical skin application and a very small embryonic like (VSEL TM) stem cell program for the treatment of retinal degeneration, bone restoration and wound healing.
Through our wholly owned subsidiary, Progenitor Cell Therapy, LLC (PCT), we are recognized as a world industry leader in providing high quality innovative and reliable manufacturing capabilities and engineering solutions (e.g. process development) in the development of cell-based therapies. We operate three current Good Manufacturing Practice (cCGMP) facilities in Allendale, NJ, Mountain View, CA and Irvine, CA, respectively, and are poised to expand our facilities internationally. In addition to leveraging this core expertise in the development of our own products, we partner opportunistically with other industry leaders who recognize our unique ability to significantly improve their manufacturing processes and supply clinical and commercial material.
We look forward to further advancement of our cell based therapies to the market and to helping patients suffering from life-threatening medical conditions. Coupling our development expertise with our strong process development and manufacturing capability, we believe the stage is set for us to realize meaningful clinical development or our own proprietary platform technologies and manufacturing advancements, further positioning NeoStem to lead the cell therapy industry.
We anticipate requiring additional capital in order to fund the development of cell therapy product candidates and to grow the PCT business. To meet our short and long term liquidity needs, we currently expect to use existing cash, cash equivalents and marketable securities balances, our revenue generating activities, and a variety of other means, including the continued use of a common stock purchase agreement with Aspire Capital. Other sources of liquidity could include additional potential issuances of debt or equity securities in public or private financings, additional warrant exercises, option exercises, partnerships and/or collaborations, and/or sale of assets. In addition, we will continue to seek as appropriate grants for scientific and clinical studies from various governmental agencies and foundations. We believe that our current cash, cash equivalents and marketable securities balances and revenue generating activities, along with access to funds under our agreement with Aspire Capital, will be sufficient to fund the business through the next 12 months. While we continue to seek capital through a number of means, there can be no assurance that additional financing will be available on acceptable terms, if at all. If we are unable to access capital necessary to meet our long-term liquidity needs, we may have to delay or discontinue the acquisition and development of cell therapies, and/or the expansion of our business or raise funds on terms that we currently consider unfavorable.
Basis of Presentation
The accompanying Consolidated Financial Statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“generally accepted accounting principles”) and include the accounts of the Company and its wholly owned and partially owned subsidiaries, the operations of our former Regenerative Medicine - China segment through the deconsolidation date on March 31, 2012 (see Note 15), and the operations of our former Pharmaceutical Manufacturing - China segment through November 13, 2012, the date on which the segment was sold (see Note 15). These former segments are reported in discontinued operations. In the opinion of management, the accompanying Consolidated Financial Statements of the Company and its subsidiaries, include all normal and recurring adjustments considered necessary to present fairly the Company's financial position as of December 31, 2014 and 2013, and the results of its operations and its cash flows for the years ended December 31, 2014, 2013, and 2012.
Use of Estimates
81
The preparation of financial statements in conformity with generally accepted accounting principles in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements. Estimates also affect the reported amounts of revenues and expenses during the reporting period. The Company bases its estimates on historical experience and other assumptions believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. The Company makes critical estimates and assumptions in determining the fair values of goodwill for potential goodwill impairments for our reporting units, fair values of In-Process R&D assets, fair values of acquisition-related contingent considerations, useful lives of our tangible and intangible assets, allowances for doubtful accounts, and stock-based awards values. Accordingly, actual results could differ from those estimates and assumptions.
An accounting policy is considered to be critical if it is important to the Company’s financial condition and results of operations and if it requires management’s most difficult, subjective and complex judgments in its application.
Reclassifications
Certain reclassifications have been made to the Consolidated Financial Statements and Notes to the Consolidated Financial Statements for the year ended December 31, 2012 and 2013 to conform to the presentation for the year ended December 31, 2014.
Principles of Consolidation
The Consolidated Financial Statements include the accounts of NeoStem, Inc. and its wholly owned and partially owned subsidiaries and affiliates as listed below, as well as the operations of our former Regenerative Medicine - China segment through the deconsolidation date on March 31, 2012 (see Note 15), and the operations of our former Pharmaceutical Manufacturing - China segment through November 13, 2012, the date which the segment was sold (see Note 15). These former segments are reported in discontinued operations.
Entity | Percentage of Ownership | Location | ||
NeoStem, Inc. | 100% | United States of America | ||
NeoStem Therapies, Inc. | 100% | United States of America | ||
Stem Cell Technologies, Inc. | 100% | United States of America | ||
Amorcyte, LLC | 100% | United States of America | ||
Progenitor Cell Therapy, LLC (PCT) | 100% | United States of America | ||
NeoStem Family Storage, LLC | 100% | United States of America | ||
Athelos Corporation (1) | 96.2% | United States of America | ||
PCT Allendale, LLC | 100% | United States of America | ||
NeoStem Oncology, LLC (2) | 100% | United States of America | ||
(1) Pursuant to the Stock Purchase Agreement signed in March 2011, our initial ownership in Athelos was 80.1%, and Becton Dickinson's (BD) initial minority ownership was 19.9%. Per the Agreement, BD will be diluted based on new investment in Athelos by us (subject to certain anti-dilution provisions). As of December 31, 2014, BD's ownership interest in Athelos was decreased to 3.8%, and our ownership increased to 96.2%. As a result in the change in ownership, approximately $0.7 million was transferred from additional paid in capital to non-controlling interests in 2014.
(2) On May 8, 2014, NeoStem acquired California Stem Cell, now known as NeoStem Oncology, LLC (see Note 3, Acquisition). Accordingly, the operating results of NeoStem Oncology, LLC prior to May 8, 2014 are not included in the Company's consolidated statements of operations and cash flows.
Note 2 – Summary of Significant Accounting Policies
Cash and Cash Equivalents
Cash and cash equivalents include short-term, highly liquid, investments with maturities of ninety days or less when purchased.
Concentration of Risks
82
We are subject to credit risk from our portfolio of cash and cash equivalents and marketable securities. Under our investment policy, we limit amounts invested in such securities by credit rating, maturity, industry group, investment type and issuer, except for securities issued by the U.S. government. Cash is held at major banks in the United States. Therefore, the Company is not exposed to any significant concentrations of credit risk from these financial instruments. The goals of our investment policy, in order of priority, are as follows: safety and preservation of principal and diversification of risk; liquidity of investments sufficient to meet cash flow requirements; and a competitive after-tax rate of return.
We are also subject to credit risk from our accounts receivable related to our services. The majority of our trade accounts receivable arises from services in the United States.
For the year ended December 31, 2014, four customers represented 16%, 11%, 11%, and 10% respectively, of total revenues recognized. As of December 31, 2014, four customers represented 68% of our accounts receivable.
Marketable Securities
The Company determines the appropriate classification of our marketable securities at the time of purchase and reevaluate such designation at each balance sheet date. All of our marketable securities are considered as available-for-sale and carried at estimated fair values and reported in either cash equivalents or marketable securities. Unrealized gains and losses on available-for-sale securities are excluded from net income and reported in accumulated other comprehensive income (loss) as a separate component of stockholders' equity. Other income (expense), net, includes interest, dividends, amortization of purchase premiums and discounts, realized gains and losses on sales of securities and other-than-temporary declines in the fair value of securities, if any. The cost of securities sold is based on the specific identification method. We regularly review all of our investments for other-than-temporary declines in fair value. Our review includes the consideration of the cause of the impairment, including the creditworthiness of the security issuers, the number of securities in an unrealized loss position, the severity and duration of the unrealized losses, whether we have the intent to sell the securities and whether it is more likely than not that we will be required to sell the securities before the recovery of their amortized cost basis. When we determine that the decline in fair value of an investment is below our accounting basis and this decline is other-than-temporary, we reduce the carrying value of the security we hold and record a loss for the amount of such decline.
Accounts Receivable
Accounts receivable are carried at original invoice amount less an estimate made for doubtful accounts. The Company applies judgment in connection with establishing the allowance for doubtful accounts. Specifically, the Company analyzes the aging of accounts receivable balances, historical bad debts, customer concentration and credit-worthiness, current economic trends and changes in the Company’s customer payment terms. Significant changes in customer concentrations or payment terms, deterioration of customer credit-worthiness or weakening economic trends could have a significant impact on the collectability of the receivables and the Company’s operating results. If the financial condition of the Company’s customers were to deteriorate, resulting in an impairment of their ability to make payments, additional allowances may be required. Management regularly reviews the aging of receivables and changes in payment trends by its customers, and records a reserve when it believes collection of amounts due are at risk.
Deferred Costs
The Company, through its PCT subsidiary, regularly enters into contracts with clients for services that have multiple stages and are dependent on one another to complete the contract and recognize revenue. The Company's deferred costs represents work in process for costs incurred on such projects at PCT that have not been completed. The Company reviews these projects periodically to determine that the value of each project is stated at the lower of cost or market.
Property, Plant, and Equipment
The cost of property, plant and equipment is depreciated over the estimated useful lives of the related assets. Depreciation is computed on the straight-line method. Repairs and maintenance expenditures that do not extend original asset lives are charged to expense as incurred. The estimated useful lives of property, plant and equipment are as follows:
83
Building and improvements | 25-30 years |
Machinery and equipment | 8-12 years |
Lab equipment | 5-7 years |
Furniture and fixtures | 5-12 years |
Software | 3-5 years |
Leasehold improvements | Life of lease |
Goodwill and Indefinite-Lived Intangible Assets
Goodwill is the excess of purchase price over the fair value of identified net assets of businesses acquired. The Company’s intangible assets with an indefinite life are related to in process research and development (IPR&D) for NBS10, the clinical candidate acquired in the Amorcyte acquisition, as the Company expects this research and development to provide the Company with substantial benefit for a period that extends beyond the foreseeable horizon. Intangible assets with indefinite useful lives are measured at their respective fair values as of the acquisition date. The Company does not amortize goodwill and intangible assets with indefinite useful lives. Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated R&D efforts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time.
The Company reviews goodwill and indefinite-lived intangible assets at least annually, or at the time a triggering event is identified for possible impairment. Goodwill and indefinite-lived intangible assets are reviewed for possible impairment between annual tests if an event occurs or circumstances change that would more likely than not reduce the fair value of the reporting unit or the IPR&D below its carrying value. The Company tests its goodwill and indefinite-lived intangible assets each year on December 31. The Company reviews the carrying value of goodwill and indefinite-lived intangible assets utilizing an income approach model, and, where appropriate, a market value approach is also utilized to supplement the discounted cash flow model. The Company makes assumptions regarding estimated future cash flows, discount rates, long-term growth rates and market values to determine each reporting unit’s and IPR&D's estimated fair value. If these estimates or related assumptions change in the future, the Company may be required to record impairment charges. In accordance with its accounting policy, the Company tested goodwill for impairment as of December 31, 2014, 2013, and 2012 for its two reporting units as well as its IPR&D, and concluded there was no goodwill and IPR&D impairment.
Definite-lived Intangible Assets
Definite-lived intangible assets consist of customer lists, manufacturing technology, tradenames, patents and rights. These intangible assets are amortized on a straight line basis over their respective useful lives. The Company reviews definite-lived intangibles assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset exceeds the fair value of the asset. If other events or changes in circumstances indicate that the carrying amount of an asset that the Company expects to hold and use may not be recoverable, the Company will estimate the undiscounted future cash flows expected to result from the use of the asset and/or its eventual disposition, and recognize an impairment loss, if any. The impairment loss, if determined to be necessary, would be measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets. No events were noted in 2014, 2013, and 2012.
Recognizing and Measuring Assets Acquired and Liabilities Assumed in Business Combinations at Fair Value
The Company accounts for acquired businesses using the purchase method of accounting, which requires that assets acquired and liabilities assumed be recorded at date of acquisition at their respective fair values. The consolidated financial statements and results of operations reflect an acquired business after the completion of the acquisition. The fair value of the consideration paid, including contingent consideration, is assigned to the underlying net assets of the acquired business based on their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill. Amounts allocated to IPR&D are included on the balance sheet . Intangible assets, including IPR&D assets upon successful completion of the project and approval of the product, are amortized on a straight-line basis to amortization expense over the expected life of the asset. Significant judgments are used in determining the estimated fair values assigned to the assets acquired and liabilities assumed and in determining estimates of useful lives of long-lived assets. Fair value determinations and useful life estimates are based on, among other factors, estimates of expected future net cash flows, estimates of appropriate discount rates used to present value expected from future net cash flow streams, the timing of approvals for IPR&D projects and the timing of related product launch dates, the assessment of each asset’s life cycle, the impact of competitive trends on each asset’s life cycle and other factors. These judgments can materially impact the estimates used to allocate acquisition date fair values to assets acquired and liabilities
84
assumed and the resulting timing and amount charged to, or recognized in current and future operating results. For these and other reasons, actual results may vary significantly from estimated results.
The Company determines the acquisition date fair value of contingent consideration obligations based on a probability-weighted income approach derived from revenue estimates, post-tax gross profit levels and a probability assessment with respect to the likelihood of achieving contingent obligations including contingent payments such as milestone obligations, royalty obligations and contract earn-out criteria, where applicable. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting. The resulting probability-weighted cash flows are discounted using an appropriate effective annual interest rate. At each reporting date, the contingent consideration obligation will be revalued to estimated fair value at that time and changes in fair value will be reflected as income or expense in our consolidated statement of operations. Changes in the fair value of the contingent consideration obligations may result from changes in discount periods and rates, changes in the timing and amount of revenue estimates and changes in probability assumptions with respect to the likelihood of achieving the various contingent payment obligations. Changes in assumptions utilized in our contingent consideration fair value estimates could result in an increase or decrease in our contingent consideration obligation and a corresponding charge to operating loss or gain.
Share-Based Compensation
The Company expenses all share-based payment awards to employees, directors, consultants, including grants of stock options, warrants, and restricted stock, over the requisite service period based on the grant date fair value of the awards. Consultant awards are remeasured each reporting period through vesting. For awards with performance-based vesting criteria, the Company estimates the probability of achievement of the performance criteria and recognizes compensation expense related to those awards expected to vest. The Company determines the fair value of option awards using the Black-Scholes option-pricing model which uses both historical and current market data to estimate the fair value. This method incorporates various assumptions such as the risk-free interest rate, expected volatility, expected dividend yield and expected life of the options or warrants. The fair value of the Company’s restricted stock and restricted stock units is based on the closing market price of the Company’s common stock on the date of grant.
Loss Per Share
Basic loss per share is based on the weighted effect of all common shares issued and outstanding, and is calculated by dividing net loss attributable to common stockholders by the weighted average shares outstanding during the period. Diluted loss per share, which is calculated by dividing net loss attributable to common stockholders by the weighted average number of common shares used in the basic loss per share calculation plus the number of common shares that would be issued assuming conversion of all potentially dilutive securities outstanding. Diluted loss per share is not presented as such potentially dilutive securities are anti-dilutive in all periods presented due to losses incurred.
Derivatives
Derivative instruments, including derivative instruments embedded in other contracts, are recorded on the balance sheet as either an asset or liability measured at its fair value. Changes in the fair value of derivative instruments are recognized currently in results of operations unless specific hedge accounting criteria are met. The Company has not entered into hedging activities to date. Changes in the derivative value are recorded as other income (expense) on the consolidated statements of operations.
Income Taxes
The Company recognizes (a) the amount of taxes payable or refundable for the current year and (b) deferred tax liabilities and assets for the future tax consequences of events that have been recognized in the Company’s financial statements or tax returns. The Company continues to evaluate the accounting for uncertainty in tax positions at the end of each reporting period. The guidance requires companies to recognize in their financial statements the impact of a tax position if the position is more likely than not of being sustained if the position were to be challenged by a taxing authority. The position ascertained inherently requires judgment and estimates by management. The Company recognizes interest and penalties as a component of income tax expense.
Foreign Currency Translation
Results of the Company’s former Chinese operating segments were translated at the average exchange rates during the period, and assets and liabilities were translated at the closing rate at the end of each reporting period. Cash flows were also translated at average exchange rates for the period, therefore, amounts reported on the consolidated statement of cash flows did not necessarily agree with changes in the corresponding balances on the consolidated balance sheet.
85
Treasury Stock
Treasury stock purchases are accounted for under the cost method whereby the entire cost of the acquired stock is recorded as treasury stock. Gains or losses on the subsequent reissuance of shares are credited or charged to additional paid in capital.
Revenue Recognition
Clinical Services: The Company recognizes revenue for its (i) process development and (ii) clinical manufacturing services based on the terms of individual contracts.
We recognize revenues when all of the following conditions are met:
• | persuasive evidence of an arrangement exists; |
• | delivery has occurred or the services have been rendered; |
• | the fee is fixed or determinable; and |
• | collectability is probable. |
The Company considers signed contracts as evidence of an arrangement. The Company assesses whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the payment terms are subject to refund or adjustment. The Company assesses cash collectability based on a number of factors, including past collection history with the client and the client's creditworthiness. If the Company determines that collectability is not reasonably assured, it defers revenue recognition until collectability becomes reasonably assured, which is generally upon receipt of the cash. The Company's arrangements are generally non-cancellable, though clients typically have the right to terminate their agreement for cause if the Company materially fails to perform.
Revenues associated with process development services generally contain multiple stages that do not have stand-alone values and are dependent upon one another, and are recognized as revenue on a completed contract basis. Progress billings collected prior to contract completion are recorded as unearned revenue until such time the contract is completed, which usually requires formal client acceptance.
Clinical manufacturing services are generally distinct arrangements whereby the Company is paid for time and materials or for fixed monthly amounts. Revenue is recognized when efforts are expended or contractual terms have been met.
Some client agreements include multiple elements, comprised of cell process development and cell manufacturing services. The Company believes that process development and clinical manufacturing services each have stand-alone value because these services can be provided separately by other companies. In accordance with ASC Update No. 2009-13, “Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements,” the Company (1) separates deliverables into separate units of accounting when deliverables are sold in a bundled arrangement and (2) allocates the arrangement's consideration to each unit in the arrangement based on its relative selling price.
Clinical Services Reimbursements: The Company separately charges the customers for the expenses associated with certain consumable resources (reimbursable expenses) that are specified in each clinical services contract. On a monthly basis, the Company bills customers for reimbursable expenses and immediately recognizes these billings as revenue, as the revenue is deemed earned as reimbursable expenses are incurred. For the years ended December 31, 2014, 2013, and 2012, clinical services reimbursements were $3.7 million, $2.1 million, and $3.5 million, respectively.
Processing and Storage Services: The Company recognizes revenue related to the collection and cryopreservation of cord blood and autologous adult stem cells when the cryopreservation process is completed which is approximately twenty-four hours after cells have been collected. Revenue related to advance payments of storage fees is recognized ratably over the period covered by the advance payments.
Research and Development Costs
86
Research and development (“R&D”) expenses include salaries, benefits, and other headcount related costs, clinical trial and related clinical manufacturing costs, contract and other outside service fees including sponsored research agreements, and facilities and overhead costs. The Company expenses the costs associated with research and development activities when incurred.
To further drive the Company’s cell therapy initiatives, the Company will continue targeting key governmental agencies, congressional committees and not-for-profit organizations to contribute funds for the Company’s research and development programs. The Company accounts for such grants as a deduction to the related expense in research and development operating expenses when earned.
New Accounting Pronouncement
In May 2014, the FASB issued ASU 2014-09, "Revenue from Contracts with Customers (Topic 606)." The new revenue recognition standard provides a five-step analysis to determine when and how revenue is recognized. The standard requires that a company recognizes revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. This ASU is effective for annual periods beginning after December 15, 2016 and will be applied retrospectively to each period presented or as a cumulative-effect adjustment as of the date of adoption. The Company is currently evaluating the impact of the pending adoption of ASU 2014-09 on its consolidated financial statements.
In August 2014, FASB issued Accounting Standards Update (ASU) No. 2014-15 Preparation of Financial Statements - Going Concern (Subtopic 205-40), Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. Under generally accepted accounting principles (GAAP), continuation of a reporting entity as a going concern is presumed as the basis for preparing financial statements unless and until the entity's liquidation becomes imminent. Preparation of financial statements under this presumption is commonly referred to as the going concern basis of accounting. If and when an entity's liquidation becomes imminent, financial statements should be prepared under the liquidation basis of accounting in accordance with Subtopic 205-30, Presentation of Financial Statements - Liquidation Basis of Accounting. Even when an entity's liquidation is not imminent, there may be conditions or events that raise substantial doubt about the entity's ability to continue as a going concern. In those situations, financial statements should continue to be prepared under the going concern basis of accounting, but the provisions in this ASU should be followed to determine whether to disclose information about the relevant conditions and events. The ASU is effective for the annual period ending after December 15, 2016, and for annual periods and interim periods thereafter. Early application is permitted. The Company is currently evaluating the adoption of this ASU and its impact on the consolidated financial statements.
Note 3 – Acquisition
On May 8, 2014, NeoStem closed (the “Closing”) its acquisition of CSC (the “CSC Acquisition”), pursuant to the terms of the Agreement and Plan of Merger, dated as of April 11, 2014 (the “Merger Agreement”), by and among NeoStem and its acquisition subsidiaries (collectively, "Subco"), CSC, and Jason Livingston, solely in his capacity as CSC stockholder representative (together with his permitted successors, the “CSC Representative”). At Closing, Fortis Advisors LLC succeeded to the duties of the CSC Representative pursuant to the Merger Agreement. Pursuant to the Merger Agreement, on the Closing Date, Subco was merged with CSC (the “Merger”), with Subco surviving the Merger as a wholly-owned subsidiary of NeoStem. At Closing, Subco changed its legal name to NeoStem Oncology, LLC.
Aggregate Merger Consideration
Pursuant to the terms of the Merger Agreement, all shares of CSC common stock (CSC Common Stock) and CSC preferred stock (“CSC Preferred Stock”, and collectively with the CSC Common Stock, the “CSC Capital Stock”) outstanding immediately prior to the Closing, and all outstanding unexercised options to purchase CSC Common Stock (CSC Options) (treated as if a net exercise had occurred), were canceled and converted into the right to receive, in the aggregate (and giving effect to the liquidation preferences accorded to the CSC Preferred Stock):
(1)An aggregate of 5,329,593 shares of NeoStem common stock (subject to payment of nominal cash in lieu of fractional shares) (the “Closing Merger Consideration”).
(2)if payable after the Closing, certain payments in an amount of up to $90 million in the aggregate, payable in shares of NeoStem Common Stock or cash, in NeoStem’s sole discretion, in the event of the successful completion of certain milestone events in connection with the CSC business being acquired by NeoStem (the “Milestone Payments”, and together with the Closing Merger Consideration, the “Merger Consideration”).
87
The fair value of the net assets acquired in the CSC Acquisition was $19.4 million. The fair value of the Merger Consideration paid by NeoStem was valued at $33.5 million, resulting in the recognition of goodwill in the amount of $14.1 million. The consideration paid was comprised of equity issued and milestone payments. The fair value of the equity issued by NeoStem was valued at $21.6 million. The fair value of the milestone payments was valued at $11.9 million, and is contingent on the achievement of certain milestones associated with the future development of the acquired programs. Such contingent consideration has been classified as a liability and will be subject to remeasurement at the end of each reporting period.
The fair value of assets acquired and liabilities assumed on May 8, 2014 is as follows (in thousands):
Cash and cash equivalents | $ | 51 | |
Accounts receivable trade, net | 45 | ||
Prepaids and other current assets | 19 | ||
Property, plant and equipment, net | 1,041 | ||
Other assets | 201 | ||
Goodwill | 14,092 | ||
In-Process R&D | 34,290 | ||
Accounts payable | (333 | ) | |
Accrued liabilities | (2,014 | ) | |
Deferred tax liability | (13,901 | ) | |
$ | 33,491 | ||
The total cost of the acquisition, which is still preliminary, has been allocated to the assets acquired and the liabilities assumed based on their estimated fair values at the date of the acquisition. The final allocation is expected to be completed during the measurement period which is one year from the date of acquisition. For the period since the CSC Acquisition (May 9, 2014 to December 31, 2014), NeoStem recorded $0.04 million in revenues and a net loss of approximately $7.6 million attributable to CSC.
Pro Forma Financial Information (unaudited)
The following supplemental table presents unaudited consolidated pro forma financial information as if the closing of the acquisition of CSC had occurred on January 1, 2013 (in thousands, except per share amounts):
Twelve Months Ended December 31, 2014 | Twelve Months Ended December 31, 2013 | ||||||||||||||
(As Reported) | (Proforma -Unaudited) | (As Reported) | (Proforma -Unaudited) | ||||||||||||
Revenues | $ | 17,939 | $ | 18,649 | $ | 14,668 | $ | 15,471 | |||||||
Net loss | $ | (55,467 | ) | $ | (57,964 | ) | $ | (39,485 | ) | $ | (44,790 | ) | |||
Net loss attributable to NeoStem | $ | (54,873 | ) | $ | (57,371 | ) | $ | (38,981 | ) | $ | (44,286 | ) | |||
Net loss per share attributable to NeoStem | $ | (1.68 | ) | $ | (1.51 | ) | $ | (1.90 | ) | $ | (1.71 | ) | |||
The unaudited supplemental pro forma financial information should not be considered indicative of the results that would have occurred if the acquisition of CSC had been consummated on January 1, 2013, nor are they indicative of future results.
Note 4 – Available-for-Sale-Securities
The following table is a summary of available-for-sale securities recorded in cash and cash equivalents or marketable securities in our Consolidated Balance Sheets (in thousands):
88
December 31, 2014 | |||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||
Certificate of deposits | $ | 249.0 | $ | — | $ | — | $ | 249.0 | |||||||
Money market funds | 12,791.9 | — | — | 12,791.9 | |||||||||||
Municipal debt securities | 9,317.3 | 1.3 | — | 9,318.6 | |||||||||||
Total | $ | 22,358.2 | $ | 1.3 | $ | — | $ | 22,359.5 | |||||||
Estimated fair values of available-for-sale securities are generally based on prices obtained from commercial pricing services. The following table summarizes the classification of the available-for-sale debt securities on our Consolidated Balance Sheets (in thousands):
December 31, 2014 | |||
Cash and cash equivalents | $ | 15,279.4 | |
Marketable securities | 7,080.1 | ||
Total | $ | 22,359.5 | |
The following table summarizes our portfolio of available-for-sale debt securities by contractual maturity (in thousands):
December 31, 2014 | |||||||
Amortized Cost | Estimated Fair Value | ||||||
Less than one year | $ | 22,358.2 | $ | 22,359.5 | |||
Greater than one year | — | — | |||||
Total | $ | 22,358.2 | $ | 22,359.5 | |||
Note 5 – Deferred Costs
Deferred costs representing work in process for costs incurred on process development contracts that have not been completed, were $2.6 million and $1.3 million as of December 31, 2014 and December 31, 2013, respectively. The Company also has deferred revenue of approximately $3.9 million and $1.5 million of progress billings received as of December 31, 2014 and December 31, 2013, respectively, related to these contracts.
Note 6 – Property, Plant and Equipment
Property, plant, and equipment consisted of the following (in thousands):
December 31, | ||||||||
2014 | 2013 | |||||||
Building and improvements | $ | 11,298.7 | $ | 11,229.9 | ||||
Machinery and equipment | 68.3 | 58.2 | ||||||
Lab equipment | 6,324.7 | 2,743.7 | ||||||
Furniture and fixtures | 1,166.3 | 958.0 | ||||||
Software | 312.4 | 203.1 | ||||||
Leasehold improvements | 2,219.6 | 674.1 | ||||||
Property, plant and equipment, gross | 21,390.0 | 15,867.0 | ||||||
Accumulated depreciation | (5,429.3 | ) | (3,022.8 | ) | ||||
Property, plant and equipment, net | $ | 15,960.7 | $ | 12,844.2 | ||||
The Company’s results included depreciation expense of approximately $1.6 million, $1.0 million and $1.0 million for the years ended December 31, 2014, 2013, and 2012, respectively.
Note 7 – Loss Per Share
89
For the years ended December 31, 2014, 2013, and 2012 the Company incurred net losses and therefore no common stock equivalents were utilized in the calculation of loss per share as they are anti-dilutive in the periods presented. At December 31, 2014, 2013, and 2012 the Company excluded the following potentially dilutive securities:
December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
Stock Options | 4,427,276 | 2,932,191 | 2,168,668 | |||||
Warrants | 3,550,956 | 4,898,266 | 5,528,761 | |||||
Restricted Shares | 280,481 | 78,500 | 34,250 | |||||
Note 8 – Fair Value Measurements
Fair value of financial assets and liabilities that are being measured and reported are defined as the exchange price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants in the principal market at the measurement date (exit price). The Company is required to classify fair value measurements in one of the following categories:
Level 1 inputs are defined as quoted prices (unadjusted) in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date.
Level 2 inputs are defined as inputs other than quoted prices included within Level 1 that are observable for the assets or liabilities, either directly or indirectly.
Level 3 inputs are defined as unobservable inputs for the assets or liabilities. Financial assets and liabilities are classified based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement requires judgment, and may affect the valuation of the fair value of assets and liabilities and their placement within the fair value hierarchy levels.
The Company classifies the fair value of the warrant derivative liabilities as level 3 inputs. These inputs require material subjectivity because value is derived through the use of a lattice model that values the derivatives based on probability weighted discounted cash flows. In May 2014, the warrants expired and the value of the warrant derivative liabilities were written off and recorded in other expenses in our consolidated statement of operations.
The Company classifies the fair value of contingent consideration obligations as level 3 inputs. The Company has recognized contingent consideration obligations related to the following:
• | In October 2011, in connection with the Company's acquisition of Amorcyte, contingent consideration obligations were recognized relating to earn out payments equal to 10% of the net sales of the lead product candidate NBS10 (in the event of and following the date of first commercial sale of NBS10), provided that in the event NeoStem sublicenses NBS10, the applicable earn out payment will be equal to 30% of any sublicensing fees, and provided further that NeoStem will be entitled to recover direct out-of-pocket clinical development costs not previously paid or reimbursed and any costs, expenses, liabilities and settlement amounts arising out of claims of patent infringement or otherwise challenging Amorcyte’s right to use intellectual property, by reducing any earn out payments due by 50% until such costs have been recouped in full (the “Earn Out Payments”). The contingent consideration fair value decreased from $9.5 million as of December 31, 2013 to $5.5 million as of December 31, 2014. The change in estimated fair value is based primarily on the Company's updates to the discounted cash flow model using a probability-weighted income approach subsequent to the reporting of the primary analysis from the Preserve AMI Phase 2 clinical trial in the fourth quarter of 2014, and has been recorded in other expenses in our consolidated statement of operations. |
• | In May 2014, in connection with the Company's acquisition of CSC, contingent consideration obligations were recognized relating to milestone payments of up to $90 million, based on the achievement of certain milestones associated with the future development of the acquired programs. The contingent consideration fair value recognized in the acquisition in May 2014 was $11.9 million. The contingent consideration fair value increased to $12.8 million as of December 31, 2014. The change in estimated fair value is based on changes in assumptions regarding the timing of certain milestone achievements, as well as the time progression to reach those milestones as of December 31, 2014, and has been recorded in other expenses in our consolidated statement of operations. |
90
The fair value of contingent consideration obligations is based on discounted cash flow models using a probability-weighted income approach. The measurements are based upon unobservable inputs supported by little or no market activity based on our own assumptions and experience. The Company bases the timing to complete the development and approval programs on the current development stage of the product and the inherent difficulties and uncertainties in developing a product candidate, such as obtaining U.S. Food and Drug Administration (FDA) and other regulatory approvals. In determining the probability of regulatory approval and commercial success, we utilize data regarding similar milestone events from several sources, including industry studies and our own experience. These fair value measurements represent Level 3 measurements as they are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we record in any given period.
The following table sets forth by level within the fair value hierarchy the Company’s financial assets and liabilities that were accounted for at fair value on a recurring basis as of December 31, 2014, and December 31, 2013 (in thousands):
December 31, 2014 | December 31, 2013 | |||||||||||||||||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | Total | |||||||||||||||||||||||||
Assets: | ||||||||||||||||||||||||||||||||
Marketable securities - available for sale | $ | — | $ | 7,080.0 | $ | — | $ | 7,080.0 | $ | — | $ | — | $ | — | $ | — | ||||||||||||||||
$ | — | $ | 7,080.0 | $ | — | $ | 7,080.0 | $ | — | $ | — | $ | — | $ | — | |||||||||||||||||
Liabilities: | ||||||||||||||||||||||||||||||||
Warrant derivative liabilities | — | — | — | — | — | — | 23.2 | 23.2 | ||||||||||||||||||||||||
Contingent consideration | — | — | 18,260.0 | 18,260.0 | — | — | 9,450.0 | 9,450.0 | ||||||||||||||||||||||||
$ | — | $ | — | $ | 18,260.0 | $ | 18,260.0 | $ | — | $ | — | $ | 9,473.2 | $ | 9,473.2 | |||||||||||||||||
There were no transfers of financial instruments to or from Levels 1, 2 or 3 during the periods presented. For those financial instruments with significant Level 3 inputs, the following table summarizes the activity for the year ended December 31, 2014 by type of instrument (in thousands):
Year Ended | ||||||||||||
December 31, 2014 | ||||||||||||
Warrants | Contingent Consideration | Total | ||||||||||
Beginning liability balance | $ | 23.2 | $ | 9,450.0 | $ | 9,473.2 | ||||||
Amount issued in acquisition | — | 11,890.0 | 11,890.0 | |||||||||
Change in fair value recorded in operations | — | (3,080.0 | ) | (3,080.0 | ) | |||||||
Expiration | (23.2 | ) | — | (23.2 | ) | |||||||
Ending liability balance | $ | — | $ | 18,260.0 | $ | 18,260.0 | ||||||
Some of the Company’s financial instruments are not measured at fair value on a recurring basis, but are recorded at amounts that approximate fair value due to their liquid or short-term nature, such as cash and cash equivalents, accounts receivable, and accounts payable. Our long-term debt and notes payable are carried at cost and approximate fair value due to their variable or fixed interest rates, which are consistent with the interest rates in the market.
Note 9 – Goodwill and Other Intangible Assets
The following table summarizes the changes in the carrying amount of goodwill (in thousands):
91
Total | |||
Balance as of December 31, 2013 | $ | 11,117.8 | |
Goodwill resulting from the acquisition of CSC | 14,091.5 | ||
Balance as of December 31, 2014 | $ | 25,209.3 | |
The Company's intangible assets and related accumulated amortization as of December 31, 2014 and December 31, 2013 consisted of the following (in thousands):
December 31, 2014 | December 31, 2013 | ||||||||||||||||||||||||
Useful Life | Gross | Accumulated Amortization | Net | Gross | Accumulated Amortization | Net | |||||||||||||||||||
Customer list | 10 years | $ | 1,000.0 | $ | (395.1 | ) | $ | 604.9 | $ | 1,000.0 | $ | (295.1 | ) | $ | 704.9 | ||||||||||
Manufacturing technology | 10 years | 3,900.0 | (1,540.9 | ) | 2,359.1 | 3,900.0 | (1,150.9 | ) | 2,749.1 | ||||||||||||||||
Tradename | 10 years | 800.0 | (316.1 | ) | 483.9 | 800.0 | (236.1 | ) | 563.9 | ||||||||||||||||
In process R&D | Indefinite | 43,690.0 | — | 43,690.0 | 9,400.0 | — | 9,400.0 | ||||||||||||||||||
Patent rights | 19 years | 669.0 | (246.5 | ) | 422.5 | 669.0 | (211.3 | ) | 457.7 | ||||||||||||||||
Total Intangible Assets | $ | 50,059.0 | $ | (2,498.6 | ) | $ | 47,560.4 | $ | 15,769.0 | $ | (1,893.4 | ) | $ | 13,875.6 | |||||||||||
Total intangible amortization expense was classified in the operating expense categories for the periods included below as follows (in thousands):
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cost of revenue | $ | 316.8 | $ | 390.0 | $ | 390.0 | |||||
Research and development | 108.4 | 35.2 | 35.2 | ||||||||
Selling, general and administrative | 180.0 | 180.0 | 180.0 | ||||||||
Total | $ | 605.2 | $ | 605.2 | $ | 605.2 | |||||
Estimated intangible amortization expense on an annual basis for the succeeding five years is as follow (in thousands):
2015 | $ | 605.2 | |
2016 | 605.2 | ||
2017 | 605.2 | ||
2018 | 605.2 | ||
2019 | 605.2 | ||
Thereafter | 44,534.4 | ||
$ | 47,560.4 | ||
92
Note 10 – Accrued Liabilities
Accrued liabilities were as follow (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Salaries, employee benefits and related taxes | $ | 2,807.2 | $ | 2,325.8 | |||
Professional fees | 495.4 | 544.8 | |||||
License fees | — | 500.0 | |||||
Other | 1,020.3 | 647.4 | |||||
$ | 4,322.9 | $ | 4,018.0 | ||||
Note 11 – Debt
Notes Payable
As of December 31, 2014 and December 31, 2013, the Company had notes payable of approximately $1.6 million and $0.9 million, respectively. The notes relate to certain insurance policies and equipment financings, require monthly payments, and mature within one to three years.
Long-Term Debt
On September 26, 2014, the Company entered into a loan and security agreement (the “Loan and Security Agreement”) with Oxford Finance LLC (together with its successors and assigns, the “Lender”) pursuant to which the Lender has agreed to lend the Company up to $20.0 million. Upon entering into the Loan and Security Agreement, the Lender disbursed $15.0 million (“Term Loan A”). Under the terms of the Loan and Security Agreement, during the Second Draw Period (as defined below), the Company may, subject to certain conditions, borrow from Lender an additional $5.0 million (“Term Loan B”, together with Term Loan A, the “Term Loans”). The “Second Draw Period” is the period of time: (a) commencing on the date that Lender receives evidence in a form and substance satisfactory to Lender that the Company has entered into a strategic arrangement with respect to the Company’s NBS10 product candidate for ST Elevation Myocardial Infarction and receives an upfront payment of not less than $10.0 million in connection therewith, and (b) ending on the earlier of September 19, 2015 or the occurrence of an event of default under the Term Loans. After repayment of all outstanding amounts due under two loans from TD Bank, N.A. in the amount of approximately $3.1 million, and deductions for debt offering/issuance costs and interim period interest, the net proceeds from Term Loan A were $11.3 million. The debt offering/issuance costs have been recorded as debt issuance costs in other assets in the consolidated balance sheet, and will be amortized to interest expense throughout the life of the Term Loans using the effective interest rate method. The proceeds from the Term Loans may be used to satisfy the Company’s future working capital needs, including the development of its cell therapy product candidates.
The Company will make interest only payments on the outstanding amount of Term Loans on a monthly basis until October 1, 2015 at a rate of 8.50% per annum; provided however, such interest-only period may be extended to April 1, 2016, in the event of either (1) the signing of a partnership for (x) traumatic brain injury indication for the Company's Ischemic Repair Program or for its VSELTM Program or (y) critical limb ischemia indication for its Ischemic Repair Program; or (2) the initiation of the Intus Phase 3 study evaluating the Company's product candidate NBS20 (also referred to as DC/TC) in patients with Stage IV or recurrent Stage III metastatic melanoma. Commencing on the date that principal payments commence, the Company will make consecutive monthly payments of principal and interest based upon a repayment schedule equal to (a) 36 months, if the Term Loans begin amortizing on October 1, 2015, or (b) 30 months, if the Term Loans begin amortizing on April 1, 2016. The Term Loans mature on September 1, 2018. At its option, the Company may prepay all amounts owed under the Loan and Security Agreement (including all accrued and unpaid interest), subject to a prepayment fee that is determined based on the date the loan is prepaid. The Company is also required to pay Lender a final payment fee equal to 8% of the Term Loan A and Term Loan B (if disbursed). The final payment fee will be amortized to interest expense throughout the life of the Term Loans using the effective interest rate method. The Company paid a facility fee in the amount of $100,000 in connection with Term Loan A.
Under the Loan and Security Agreement and a related mortgage, the Company granted to Lender a security interest in all of the Company’s real property and personal property now owned or hereafter acquired, excluding intellectual property, and certain other assets and exemptions. The Company also entered into a Mortgage and Absolute Assignment of Leases and Rents (the "Mortgage"). The Company also granted Lender a security interest in the shares of the Company’s subsidiaries. The Loan and Security Agreement restricts the ability of the Company to: (a) convey, lease, sell, transfer or otherwise dispose of any part
93
of its business or property; and (b) incur any additional indebtedness. The Loan and Security Agreement provides for standard indemnification of Lender and contains representations, warranties and certain covenants of the Company. Upon the occurrence of an event of default by the Company under the Loan and Security Agreement, Lender will have customary acceleration, collection and foreclosure remedies. There are no financial covenants associated with the Loan and Security Agreement. As of December 31, 2014, the Company was in compliance with all covenants under the Loan and Security Agreement.
Estimated future principal payments, interest, and fees due under the Loan and Security Agreement are as follows:
Years Ending December 31, | (in millions) | ||
2015 | $ | 2.4 | |
2016 | 5.7 | ||
2017 | 5.7 | ||
2018 | 5.4 | ||
Total | $ | 19.2 | |
During the year ended December 31, 2014, the Company recognized $0.3 million of interest expense related to the Loan and Security Agreement.
Mortgages Payable
In October 2007, PCT issued a note to borrow $3.1 million (the “First Mortgage”) in connection with its $3.8 million purchase of condominium units in an existing building in Allendale, New Jersey (the “Property”). The First Mortgage was payable in 239 consecutive monthly payments of principal and interest, based on a 20 year amortization schedule; and one final payment of all outstanding principal plus accrued interest then due. The monthly installment was $20,766, which includes interest at an initial rate of 5.00%; the interest rate and monthly installments payments were subject to adjustment on October 1, 2017. The outstanding balance was approximately $2.5 million at December 31, 2013. In connection with the Loan and Security Agreement signed in September 2014, the remaining mortgage obligation was repaid, along with accrued interest and mortgage termination fees.
In December 2010 PCT Allendale, a wholly-owned subsidiary of PCT, entered into a note for a second mortgage in the amount of $1 million (the "Second Mortgage") on the Allendale Property with TD Bank, N.A. The initial guarantors of the Second Mortgage were PCT, DomaniCell (a wholly-owned subsidiary of PCT, now known as NeoStem Family Storage, LLC), Regional Cancer Care Associates LLC and certain of its partners. The Second Mortgage was for124 months at a fixed rate of 6% for the first 64 months. The outstanding balance was approximately $0.8 million at December 31, 2013. In connection with the Loan and Security Agreement signed in September 2014, the remaining mortgage obligation was repaid, along with accrued interest and mortgage termination fees.
Prior to the full repayment of the mortgages in September 2014, the Company modified both the First Mortgage and Second Mortgage with TD Bank, N.A. in December 2013, whereby prior guarantors were released (see Note 16) and replaced with NeoStem, PCT, and NeoStem Family Storage, LLC.
Note 12 – Stockholders' Equity
Reverse Stock Split
On June 28, 2013, pursuant to prior shareholder authorization, the Company’s board of directors unanimously approved a 1-for-10 reverse stock split of the Company’s common stock, which the Company effected on July 16, 2013. All share and per share amounts of common stock, options and warrants in the accompanying financial statements have been restated for all periods to give retroactive effect to the reverse stock split. The shares of common stock retained a par value of $0.001 per share. Accordingly, the stockholders’ deficit reflects the reverse stock split by reclassifying from “common stock” to “Additional paid-in capital” an amount equal to the par value of the decreased shares resulting from the reverse stock split.
Equity Plans
The Company's 2003 Equity Participation Plan (the “2003 Equity Plan”) expired in 2013 and accordingly, equity awards under the 2003 Equity Plan can no longer be issued. The Company's 2009 Equity Compensation Plan (the “2009 Equity Plan”) makes up to 8,995,000 shares of common stock of the Company (as of December 31, 2014) available for issuance to employees,
94
consultants, advisors and directors of the Company and its subsidiaries pursuant to incentive or non-statutory stock options, restricted and unrestricted stock awards and stock appreciation rights.
All stock options under the 2003 Equity Plan were granted and the 2009 Equity Plan are granted at the fair market value of the common stock at the grant date. Stock options vest either on the date of grant, ratably over a period determined at time of grant, or upon the accomplishment of specified business milestones, and generally expire 2, 3, or 10 years from the grant date depending on the status of the recipient as a consultant, employee or director of the Company.
The 2009 Equity Plan was originally adopted by the stockholders of the Company on May 8, 2009. On October 29, 2009, the stockholders of the Company approved an amendment to the 2009 Equity Plan to increase the number of shares of common stock available for issuance thereunder from 380,000 to 975,000. At the 2010 Annual Meeting of Stockholders of the Company held on June 2, 2010, the stockholders approved an amendment to increase this number to 1,375,000. At a Special Meeting of Stockholders of the Company held on January 18, 2011, the stockholders approved an amendment to increase this number to 1,775,000. At the 2011 Annual Meeting of Stockholders of the Company held on October 14, 2011, the stockholders approved an amendment to increase this number to 2,375,000. At the 2012 Annual Meeting of Stockholders of the Company held on October 5, 2012, the stockholders approved an amendment to (i) merge the 570,000 shares reserved for issuance under the Company's 2009 Non-U.S. Based Equity Compensation Plan (the "Non-U.S. Plan") with and into the 2009 Equity Plan, and (ii) increase by 450,000 the aggregate number of shares authorized for issuance under the 2009 Equity Plan (the “2009 Amended & Restated Equity Plan”). At the Company's 2013 Annual Meeting held October 3, 2013, the Company's stockholders approved an amendment to the 2009 Amended & Restated Equity Plan to increase the number of shares authorized for issuance to 5,995,000. At the Company's 2014 Annual Meeting held October 6, 2014, the Company's stockholders approved an amendment to the 2009 Amended & Restated Equity Plan to increase the number of shares authorized for issuance to 8,995,000.
The number of remaining shares authorized to be issued under the various equity plans are as follows:
2003 Equity Plan | 2009 Equity Plan | |||||
Shares Authorized for Issuance | 250,000 | 8,995,000 | ||||
Outstanding Stock Options | (122,395 | ) | (4,304,881 | ) | ||
Exercised Stock Options | (9,250 | ) | (80,856 | ) | ||
Restricted stock or equity grants issued under Equity Plans | (88,993 | ) | (946,711 | ) | ||
Shares Expired | (29,362 | ) | — | |||
Total common shares remaining to be issued under the Equity Plans | — | 3,662,552 | ||||
The Company adopted an employee stock purchase plan effective January 1, 2013, and authorized 500,000 shares under the plan. The plan has two six-month offering periods per year under which eligible employees may contribute up to 15% of their compensation toward the purchase of the Company's common stock per offering period (with a $25,000 cap per calendar year). The employee's purchase price is equal to (i) 85% of the closing price of a share of the Company's common stock on the enrollment date of such offering period or (ii) 85% of the closing price of a share of the Company's Common Stock on the Exercise Date of such Offering Period, whichever is lower. During the year ended December 31, 2014, 65,441 shares were issued under the employee stock purchase plan. At December 31, 2014, the Company had 434,559 shares of the Company's common stock available for future grant in connection with this plan.
Equity Issuances
In September 2011, the Company entered into a common stock Purchase Agreement (the “Initial Purchase Agreement”) with Aspire Capital Fund, LLC, an Illinois limited liability company (“Aspire Capital”), which provided that Aspire Capital was committed to purchase up to an aggregate of $20.0 million worth of shares of the Company’s common stock over the 24-month term. In August, 2012, the Initial Purchase Agreement was extended for an additional 24-month term through September 2015. During the three months ended March 31, 2014, the Company issued 0.8 million shares of Common Stock under the provisions the Initial Purchase Agreement for gross proceeds of approximately $5.6 million. As a result, the full $20.0 million worth of shares of the Company's stock have been issued under the Initial Purchase Agreement.
In March 2014, the Company entered into a new common stock purchase agreement (the “Purchase Agreement”) with Aspire Capital, which provides that, subject to certain terms and conditions, Aspire Capital is committed to purchase up to an aggregate of $30.0 million worth of shares of the Company’s common stock over the 24-month term. At the Company’s discretion, it may present Aspire Capital with purchase notices from time to time to purchase the Company’s common stock, provided certain price
95
and other requirements are met. The purchase price for the shares of stock is based upon one of two formulas set forth in the Purchase Agreement depending on the type of purchase notice the Company submits to Aspire Capital, and is based on market prices of the Company’s common stock (in the case of regular purchases) or a discount of 5% applied to volume weighted average prices (in the case of Volume Weighted Average Price purchases), in each case as determined by parameters defined in the Purchase Agreement. As consideration for entering into the Purchase Agreement, we issued 150,000 shares of our common stock to Aspire Capital. During the year ended December 31, 2014, the Company issued 1.6 million shares of Common Stock under the provisions of the Purchase Agreement with Aspire for gross proceeds of approximately $10.9 million. As of December 31, 2014, the remaining amount available to the Company under the Purchase Agreement was $19.1 million.
Option Exercises
During the year ended December 31, 2014, option holders exercised an aggregate of 48,987 options at exercise prices between $5.20 and $6.20 per share for gross proceeds of approximately $0.3 million.
Warrant Exercises
During the year ended December 31, 2014, warrant holders exercised an aggregate of 333,250 warrants at an exercise price of $5.16 per share for gross proceeds of approximately $1.7 million.
Stock Options and Warrants
The following table summarizes the activity for stock options and warrants for the years ended December 31, 2014 and December 31, 2013:
Stock Options | Warrants | ||||||||||||||||||||||||
Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value (In Thousands) | Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value (In Thousands) | ||||||||||||||||||
Outstanding at December 31, 2013 | 2,932,191 | $ | 11.19 | 6.81 years | $ | 1,658.1 | 4,898,266 | $ | 16.50 | 2.63 | $ | 1,811.0 | |||||||||||||
Changes during the Year: | |||||||||||||||||||||||||
Granted | 2,265,850 | $ | 6.72 | 2,722 | $ | 12.26 | |||||||||||||||||||
Exercised | (48,987 | ) | $ | 5.53 | (333,250 | ) | $ | 5.16 | |||||||||||||||||
Forfeited | (368,704 | ) | $ | 6.78 | (100,108 | ) | $ | 70.00 | |||||||||||||||||
Expired | (353,116 | ) | $ | 12.94 | (916,674 | ) | $ | 23.95 | |||||||||||||||||
Outstanding at December 31, 2014 | 4,427,234 | $ | 9.19 | 6.93 | $ | 28.6 | 3,550,956 | $ | 14.12 | 2.12 | $ | 1.0 | |||||||||||||
Vested at December 31, 2014 or expected to vest in the future | 4,253,465 | $ | 9.28 | 6.84 | $ | 27.0 | 3,550,956 | $ | 14.12 | 2.12 | $ | 1.0 | |||||||||||||
Exercisable at December 31, 2014 | 2,968,379 | $ | 10.22 | 6.11 | $ | 16.6 | 3,543,456 | $ | 14.13 | 2.12 | $ | 1.0 | |||||||||||||
The total intrinsic value of stock options exercised during the years ended December 31, 2014 and December 31, 2013 was $61,641 and $104,360, respectively.
During the years ended December 31, 2014 and 2013, the Company issued warrants for services as follows ($ in thousands, except share data):
Year Ended December 31, | |||||||||
2014 | 2013 | ||||||||
Number of Common Stock Purchase Warrants Issued | — | 40,407 | |||||||
Value of Common Stock Purchase Warrants Issued | $ | — | $ | 149.9 | |||||
96
Restricted Stock
During the years ended December 31, 2014 and 2013, the Company issued restricted stock for services as follows ($ in thousands, except share data):
2014 | 2013 | ||||||||
Number of Restricted Stock Issued | 917,907 | 514,700 | |||||||
Value of Restricted Stock Issued | $ | 4,996.3 | $ | 3,360.0 | |||||
The weighted average estimated fair value of restricted stock issued for services in the years ended December 31, 2014 and 2013 was $5.44 and $6.53 per share, respectively. The fair value of the restricted stock was determined using the Company’s closing stock price on the date of issuance. The vesting terms of restricted stock issuances are generally within one year.
Note 13 – Share-Based Compensation
Share-based Compensation
We utilize share-based compensation in the form of stock options, warrants and restricted stock. The following table summarizes the components of share-based compensation expense for the years ended December 31, 2014 and 2013 ($ in thousands):
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cost of revenues | $ | 494.2 | $ | 314.0 | $ | 195.0 | |||||
Research and development | 2,058.2 | 822.2 | 432.9 | ||||||||
Selling, general and administrative | 8,657.1 | 5,702.5 | 6,084.6 | ||||||||
Total share-based compensation expense | $ | 11,209.5 | $ | 6,838.7 | $ | 6,712.5 | |||||
Total compensation cost related to nonvested awards not yet recognized and the weighted-average periods over which the awards are expected to be recognized at December 31, 2014 were as follows ($ in thousands):
Stock Options | Warrants | Restricted Stock | |||||||||
Unrecognized compensation cost | $ | 4,598.4 | $ | 7.2 | $ | 528.6 | |||||
Expected weighted-average period in years of compensation cost to be recognized | 3.79 | 0.54 | 0.31 | ||||||||
Total fair value of shares vested and the weighted average estimated fair values of shares grant for the year ended December 31, 2014, 2013, and 2012 were as follows ($ in thousands):
Stock Options | Warrants | |||||||||||||||||||||||
Year Ended December 31, | Year Ended December 31, | |||||||||||||||||||||||
2014 | 2013 | 2012 | 2014 | 2013 | 2012 | |||||||||||||||||||
Total fair value of shares vested | $ | 5,387.1 | $ | 3,375.7 | $ | 5,408.0 | $ | 9.6 | $ | 129.0 | $ | 171.6 | ||||||||||||
Weighted average estimated fair value of shares granted | 4.56 | 4.29 | 3.63 | — | 3.71 | 4.10 | ||||||||||||||||||
Valuation Assumptions
The fair value of stock options and warrants at the date of grant was estimated using the Black-Scholes option pricing model. The expected volatility is based upon historical volatility of the Company’s stock. The expected term for the options is based upon observation of actual time elapsed between date of grant and exercise of options for all employees. The expected term for the warrants is based upon the contractual term of the warrants.
97
The range of assumptions made in calculating the fair values of stock options and warrants was as follow:
Stock Options | Warrants | |||||||||||||||||
Year Ended December 31, | Year Ended December 31, | |||||||||||||||||
2014 | 2013 | 2012 | 2014 | 2013 | 2012 | |||||||||||||
Expected term - minimum (in years) | 0 | 1 | 2 | 3 | 2 | 2 | ||||||||||||
Expected term - maximum (in years) | 10 | 10 | 10 | 3 | 5 | 5 | ||||||||||||
Expected volatility - minimum | 62% | 61% | 73% | 66% | 73% | 76% | ||||||||||||
Expected volatility - maximum | 77% | 79% | 84% | 66% | 79% | 83% | ||||||||||||
Weighted Average volatility | 74 | % | 72 | % | 83 | % | 66 | % | 74 | % | 82 | % | ||||||
Expected dividend yield | — | — | — | — | — | — | ||||||||||||
Risk-free interest rate - minimum | 0.12% | 0.13% | 0.28% | 0.79% | 0.32% | 0.27% | ||||||||||||
Risk-free interest rate - maximum | 3.00% | 2.67% | 1.99% | 0.79% | 1.73% | 0.88% | ||||||||||||
Note 14 – Income Taxes
The provision (benefit) for income taxes is based on loss from operations before provision for income taxes and noncontrolling interests as follows ($ in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
United States | $ | (55,570.4 | ) | $ | (38,705.2 | ) | $ | (36,276.9 | ) | ||
$ | (55,570.4 | ) | $ | (38,705.2 | ) | $ | (36,276.9 | ) | |||
The provision (benefit) for income taxes was as follows ($ in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Current | ||||||||||||
US Federal | $ | — | $ | — | $ | — | ||||||
State and local | — | — | — | |||||||||
$ | — | $ | — | $ | — | |||||||
Deferred | ||||||||||||
US Federal | $ | 159.0 | $ | 476.9 | $ | — | ||||||
State and local | (263.2 | ) | 303.2 | (175.5 | ) | |||||||
$ | (104.2 | ) | $ | 780.1 | $ | (175.5 | ) | |||||
Total | ||||||||||||
US Federal | $ | 159.0 | $ | 476.9 | $ | — | ||||||
State and local | (263.2 | ) | 303.2 | (175.5 | ) | |||||||
$ | (104.2 | ) | $ | 780.1 | $ | (175.5 | ) | |||||
The provision (benefit) for income taxes is determined by applying the U.S. Federal statutory rate of 34% to income before income taxes as a result of the following ($ in thousands):
98
Years Ended December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
U.S. Federal benefit at statutory rate | $ | (18,894.0 | ) | $ | (13,159.8 | ) | $ | (12,334.1 | ) | |||
State and local benefit net of U.S. federal tax | (3,435.0 | ) | (3,430.9 | ) | (2,154.1 | ) | ||||||
Permanent non deductible expenses for U.S. taxes | 1,094.6 | 1,798.2 | (2,781.4 | ) | ||||||||
True-up of prior year net operating loss | (25.5 | ) | (91.4 | ) | 321.6 | |||||||
Return to actual | — | (3,822.9 | ) | (384.8 | ) | |||||||
Foreign earnings not permanently reinvested | — | — | (1,810.3 | ) | ||||||||
Effect of change in deferred tax rate | 1,075.7 | (1,094.8 | ) | 525.7 | ||||||||
Valuation allowance for deferred tax assets | 20,080.0 | 20,581.7 | 18,441.9 | |||||||||
Tax provision | $ | (104.2 | ) | $ | 780.1 | $ | (175.5 | ) | ||||
Deferred income taxes at December 31, 2014, 2013 and 2012 consist of the following ($ in thousands):
December 31, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Deferred Tax Assets: | ||||||||||||
Accumulated net operating losses (tax effected) | $ | 69,047.0 | $ | 43,334.8 | $ | 25,727.7 | ||||||
Deferred revenue | — | 10.5 | 23.1 | |||||||||
Contingent accounts payable | (7.8 | ) | 13.6 | 15.2 | ||||||||
Share-based compensation | 9,577.2 | 7,971.9 | 5,466.7 | |||||||||
Intangibles | 715.1 | 704.6 | 287.3 | |||||||||
Accumulated depreciation | — | — | 348.7 | |||||||||
Charitable contributions | 409.8 | 414.9 | 391.8 | |||||||||
Bad debt provision | 296.9 | 304.3 | 239.7 | |||||||||
Capital loss carry-forward | 6,925.1 | 7,036.8 | 6,644.5 | |||||||||
Other | 609.7 | — | — | |||||||||
Deferred tax assets prior to tax credit carryovers | 87,573.0 | 59,791.4 | 39,144.7 | |||||||||
Deferred Tax Liabilities: | ||||||||||||
Accumulated depreciation | $ | (18.8 | ) | $ | (64.8 | ) | $ | — | ||||
Intangible and indefinite lived assets | (18,176.3 | ) | (4,379.2 | ) | (3,599.1 | ) | ||||||
Deferred tax liabilities | (18,195.1 | ) | (4,444.0 | ) | (3,599.1 | ) | ||||||
69,377.9 | 55,347.4 | 35,545.6 | ||||||||||
Valuation reserve | (87,554.1 | ) | (59,726.6 | ) | (39,144.7 | ) | ||||||
Net deferred tax liability | $ | (18,176.2 | ) | $ | (4,379.2 | ) | $ | (3,599.1 | ) | |||
In assessing the realizability of deferred tax assets, including the net operating loss carryforwards (NOLs), the Company assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize its existing deferred tax assets. Based on its assessment, the Company has provided a full valuation allowance against its net deferred tax assets as their future utilization remains uncertain at this time.
As of December 31, 2014 and 2013, the Company had approximately $177.2 million and $110.6 million, respectively of Federal NOLs available to offset future taxable income expiring from 2025 through 2033. In accordance with Section 382 of the Internal Revenue code, the usage of the Company’s NOLs could be limited in the event of a change in ownership. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period when those temporary differences become deductible. If a change of ownership did occur there would be an annual limitation on the usage of the Company’s losses which are available through 2032.
99
The Company applies the FASB’s provisions for uncertain tax positions. The Company utilizes the two step process to determine the amount of recognized tax benefit. For tax positions meeting the more-likely-than-not threshold, the amount recognized in the consolidated financial statements is the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement with the relevant tax authority. The Company recognizes interest and penalties associated with certain tax positions as a component of income tax expense.
As of December 31, 2014, management does not believe the Company has any material uncertain tax positions that would require it to measure and reflect the potential lack of sustainability of a position on audit in its financial statements. The Company will continue to evaluate its uncertain tax positions in future periods to determine if measurement and recognition in its financial statements is necessary. The Company does not believe there will be any material changes in its unrecognized tax positions over the next year.
The Federal tax returns are currently being audited for the years 2012 and 2013. For years prior to 2011 the federal statute of limitations is closed. Most of the remaining states remain open to examination for a period of 3 to 4 years from date of filing. The Company files tax returns in all of the foreign jurisdictions that it has a permanent establishment and the tax filings remain subject to examination for 4 to 5 years.
Note 15 – Discontinued Operations
Regenerative Medicine - China segment
In the first quarter of 2012, the Company exited its regenerative medicine business in the People's Republic of China. As of March 31, 2012, the Company recognized the following loss on exit of the Regenerative Medicine-China business (in thousands):
Cash | $ | 195.1 | |
Prepaid expenses and other current assets | 14.9 | ||
Property, plant and equipment, net | 1,023.7 | ||
Other Assets | 330.5 | ||
Accounts payable | (177.1 | ) | |
Accrued liabilities | (79.2 | ) | |
Accumulated comprehensive income | (169.9 | ) | |
Loss on exit of segment | $ | 1,138.0 | |
The operations and cash flows of the Regenerative Medicine - China business were eliminated from ongoing operations as a result of our exit decision, and the Company will not have continuing involvement in this business going forward. The operating results of the Regenerative Medicine – China business for the year ended December 31, 2012, which are included in discontinued operations, were as follows (in thousands):
Year Ended December 31, | |||
2012 | |||
Revenue | $ | 52.3 | |
Cost of revenues | (30.6 | ) | |
Research and development | (103.3 | ) | |
Selling, general, and administrative | (497.3 | ) | |
Other income (expense) | (6.8 | ) | |
Loss on exit of segment | (1,138.0 | ) | |
Loss from discontinued operations | $ | (1,723.7 | ) |
Pharmaceutical Manufacturing - China segment
100
On November 13, 2012, the Company completed the divestiture of its 51% interest (the “Erye Interest”) in Suzhou Erye Pharmaceuticals Company Ltd., a Sino-foreign equity joint venture with limited liability organized under the laws of the People's Republic of China. Pursuant to the Equity Purchase Agreement, the aggregate purchase price paid to the Company for the Erye Interest consisted of (i) approximately $12.3 million in cash, (ii) the return to the Company of 104,000 shares of NeoStem common stock and (iii) the cancellation of 117,000 options and 64,000 warrants to purchase our common stock. The fair value of the shares was based on the Company's closing price on the date of sale, and was recorded as Treasury Stock in our balance sheet. The fair values of the canceled options and warrants were based on the Black-Scholes values on the date of sale, and were recorded against Additional Paid in Capital in the accompanying balance sheet. The Company recognized the following loss on the date of sale of its 51% interest in Erye (in thousands):
Fair value of consideration received | $ | 13,397.9 | |
Carrying value of segment non-controlling interest | 6,015.0 | ||
Carrying value of segment accumulated comprehensive income | 4,387.4 | ||
$ | 23,800.3 | ||
Less carrying amount of assets and liabilities sold: | |||
Cash | $ | 8,457.5 | |
Restricted Cash | 2,918.1 | ||
Accounts Receivable | 6,130.2 | ||
Inventories | 15,077.7 | ||
Prepaid expenses and other current assets | 957.8 | ||
Property, plant and equipment, net | 38,102.0 | ||
Other assets | 5,946.3 | ||
Accounts payable | (9,604.8 | ) | |
Accrued liabilities | (2,008.8 | ) | |
Bank loans | (15,133.5 | ) | |
Notes payable | (6,599.3 | ) | |
Other liabilities | (9,166.8 | ) | |
Amount due related party | (7,859.7 | ) | |
$ | 27,216.7 | ||
Loss on exit of segment | $ | (3,416.4 | ) |
The operations and cash flows of the Pharmaceutical Manufacturing - China business were eliminated from ongoing operations with the sale of the Company's Erye Interest. The operating results of the Pharmaceutical Manufacturing - China business for the year ended December 31, 2012, which are included in discontinued operations, were as follows (in thousands):
Year Ended December 31, | |||
2012 | |||
Revenue | $ | 61,703.1 | |
Cost of revenues | (40,245.2 | ) | |
Research and development | (1,836.4 | ) | |
Selling, general, and administrative | (10,740.0 | ) | |
Other expense | (1,045.2 | ) | |
Provision for income taxes | (1,794.1 | ) | |
Asset impairments | (31,170.1 | ) | |
Loss on sale of segment | (3,416.4 | ) | |
Loss from discontinued operations | $ | (28,544.3 | ) |
101
Note 16 – Related Party Transactions
On November 13, 2012, we and our subsidiary, CBH, sold our 51% ownership interest in Erye to Fullbright and EET (see Note 15). EET was prior to the sale the holder of the minority 49% ownership interest in Erye, and was a party along with our subsidiary CBH to the Joint Venture Agreement which had governed the ownership of the respective interests in Erye. Fullbright is an affiliate of EET. Mr. Shi Mingsheng (a former member of our Board of Directors, and Chairman of the Board of Erye) and Madam Zhang Jian (the General Manager of Erye, and formerly our Vice President of Pharmaceutical Operations) are the principal equity holders of each of EET and Fullbright. Fullbright assigned all its rights and obligations under the Equity Purchase Agreement (except for its obligations in respect of the return of certain NeoStem securities held by it as part of the purchase price, and its obligations in respect of closing deliverables) to Highacheive Holdings Limited, a limited liability company organized under the laws of the British Virgin Islands and an affiliate of Fullbright (“Highacheive”). As a result of the assignment, the Purchasers of our Erye Interest were EET and Highacheive.
In December 2013, the Company modified both the First Mortgage and Second Mortgage with TD Bank, N.A. (see Note 11). Pursuant to the Loan Modifications, Andrew L. Pecora, M.D., Regional Cancer Care Associates LLC (Dr. Pecora’s medical practice), and certain partners in such practice, including Dr. Pecora, have been released as guarantors of the Second Mortgage Loan, and NeoStem has become a guarantor of the Loans pursuant to a Guaranty of Payment delivered by NeoStem to the Lender. Dr. Pecora, currently currently serves as a NeoStem director, NeoStem’s Chief Visionary Officer, PCT’s Chief Medical Officer and Amorcyte’s Chief Scientific Officer.
Note 17 – Commitments and Contingencies
Lease Commitments
The Company leases offices, of which certain have escalation clauses and renewal options, and also leases equipment under certain noncancelable operating leases that expire from time to time through 2018. In January 2014, the Company signed a new lease for a larger space at its current executive offices at 420 Lexington Avenue, New York, NY 10170. The new lease shall extend through 2018. This property is used as the Company's corporate headquarters. In connection with the CSC Acquisition on May 8, 2014, the Company assumed a facility lease in Irvine, California, with a termination at the end of 2017. We recently signed an amendment expanding our office space in Irvine by 4,000 square feet, and extending the term through 2021. In accordance with the amendment, we plan to occupy the additional space by the second quarter of 2015.
A summary of future minimum rental payments required under operating leases that have initial or remaining terms in excess of one year as of December 31, 2014 are as follows (in thousands):
Years ended | Operating Leases | |||
2015 | $ | 1,567.9 | ||
2016 | 1,656.9 | |||
2017 | 1,367.3 | |||
2018 | 535.2 | |||
2019 | 492.4 | |||
2020 and thereafter | 628.1 | |||
Total minimum lease payments | $ | 6,247.8 | ||
Expense incurred under operating leases were approximately $1.3 million , $1.1 million , and $1.5 million for the years ended December 31, 2014, 2013, and 2012, respectively.
Contingencies
Under license agreements with third parties the Company is typically required to pay maintenance fees, make milestone payments and/or pay other fees and expenses and pay royalties upon commercialization of products. The Company also sponsors research at various academic institutions, which research agreements generally provide us with an option to license new technology discovered during the course of the sponsored research.
102
From time to time, the Company is subject to legal proceedings and claims, either asserted or unasserted, that arise in the ordinary course of business. While the outcome of pending claims cannot be predicted with certainty, the Company does not believe that the outcome of any pending claims will have a material adverse effect on the Company's financial condition or operating results.
Note 18 – Quarterly Financial Data (unaudited)
The tables below summarize the Company's unaudited quarterly operating results for the years ended December 31, 2014 and 2013, respectively.
Three Months Ended | |||||||||||||||
(in thousands, except per share data) | March 31, 2014 | June 30, 2014 | September 30, 2014 | December 31, 2014 | |||||||||||
Revenues | $ | 4,056 | $ | 4,489 | $ | 4,118 | $ | 5,276 | |||||||
Total operating costs and expenses | $ | 17,555 | $ | 16,919 | $ | 20,376 | $ | 20,830 | |||||||
Net loss from continuing operations | $ | (13,830 | ) | $ | (12,769 | ) | $ | (17,177 | ) | $ | (11,691 | ) | |||
Net loss attributable to NeoStem, Inc. common stockholders | $ | (13,682 | ) | $ | (12,605 | ) | $ | (16,974 | ) | $ | (11,612 | ) | |||
Basic and diluted loss per share attributable to NeoStem, Inc. common stockholders | $ | (0.49 | ) | $ | (0.40 | ) | $ | (0.48 | ) | $ | (0.32 | ) | |||
Three Months Ended | |||||||||||||||
(in thousands, except per share data) | March 31, 2013 | June 30, 2013 | September 30, 2013 | December 31, 2013 | |||||||||||
Revenues | $ | 2,524 | $ | 4,359 | $ | 3,707 | $ | 4,078 | |||||||
Total operating costs and expenses | $ | 11,355 | $ | 12,530 | $ | 13,020 | $ | 14,573 | |||||||
Net loss from continuing operations | $ | (8,864 | ) | $ | (8,626 | ) | $ | (9,277 | ) | $ | (12,719 | ) | |||
Net loss attributable to NeoStem, Inc. common stockholders | $ | (8,801 | ) | $ | (8,575 | ) | $ | (9,071 | ) | $ | (12,535 | ) | |||
Basic and diluted loss per share attributable to NeoStem, Inc. common stockholders | $ | (0.53 | ) | $ | (0.46 | ) | $ | (0.45 | ) | $ | (0.47 | ) | |||
Note 19 – Subsequent Events
New Facility Lease
We recently entered into an assignment agreement, effective February 19, 2015, for general office space located in Basking Ridge, New Jersey. The space is approximately 18,467 rentable square feet. Pursuant to the agreement, we are not obligated to make any payments for the space until January 2016.The base monthly rent during the period ending January 31, 2016 is currently $25,000 and the lease term ends July 31, 2020. In addition, there are two (2) five (5) year renewal options. In connection with the assumption of the lease, the third party (a) conveyed its rights in various scheduled furniture and equipment and (b) paid the Company approximately $580,000 which amount will offset the rental payments to be paid by NeoStem. A security deposit of approximately $115,000 payable by NeoStem will offset the amount payable by the third party.
Common Stock Issuances
Pursuant to the Purchase Agreement with Aspire (see Note 12), from January 1, 2015 through March 1, 2015, Aspire has purchased 1.2 million shares of the Company's common stock for an aggregate consideration of approximately $4.4 million.
103
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES.
Disclosure Controls and Procedures
Disclosure controls and procedures are the Company’s controls and other procedures that are designed to ensure that information required to be disclosed in the reports that the Company files or submits under the Securities Exchange Act of 1934 (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934), as amended (the “Exchange Act”) is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports that the Company files under the Exchange Act is accumulated and communicated to management, including the Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. Due to the inherent limitations of control systems, not all misstatements may be detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. Controls and procedures can only provide reasonable, not absolute, assurance that the above objectives have been met.
As of December 31, 2014, the Company carried out an evaluation, with the participation of the Company’s management, including the Company’s Chief Executive Officer and Chief Financial Officer, of the effectiveness of the Company’s disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934. Based on that evaluation, the Company’s Chief Executive Officer and Chief Financial Officer concluded that the Company’s disclosure controls and procedures were effective, at the reasonable assurance level, in ensuring that information required to be disclosed by the Company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to management, including the Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Internal Control Over Financial Reporting
Management’s Annual Report on Internal Control Over Financial Reporting
The management of NeoStem, Inc. and its subsidiaries (the “Company”) is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934.
The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
• | Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; |
• | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and the board of directors of the Company; and |
• | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
104
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to risk that controls may become inadequate because of changes in conditions or because of declines in the degree of compliance with policies or procedures.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2014. In making this assessment, the Company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control-Integrated Framework (2013).
As of December 31, 2014, based on management’s assessment, the Company’s internal control over financial reporting was effective.
Changes in Internal Control over Financial Reporting
There have been no changes in the Company’s internal control over financial reporting that occurred during the quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Attestation Report of the Registered Public Accounting Firm
Our independent registered public accounting firm, Grant Thornton LLP, audited our internal control over financial reporting as of December 31, 2014. Their attestation report, dated March 2, 2015 and which appears below, expressed an unqualified opinion on our internal control over financial reporting.
105
REPORT OF INDEPENDENT REGISTRED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
NeoStem, Inc.
We have audited the internal control over financial reporting of NeoStem, Inc. (a Delaware corporation) and subsidiaries (the “Company”) as of December 31, 2014, based on criteria established in the 2013 Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in the 2013 Internal Control-Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements of the Company as of and for the year ended December 31, 2014, and our report dated March 2, 2015 expressed an unqualified opinion on those financial statements.
/s/ GRANT THORNTON LLP
New York, New York
March 2, 2015
106
ITEM 9B. OTHER INFORMATION.
107
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Pursuant to Paragraph G(3) of the General Instructions to Form 10-K, the information required by Part III (Items 10, 11, 12, 13 and 14) is being incorporated by reference herein from our definitive proxy statement (or an amendment to our Annual Report on Form 10-K) to be filed with the SEC within 120 days of the end of the fiscal year ended December 31, 2014 in connection with our 2015 Annual Meeting of Stockholders.
ITEM 11. EXECUTIVE COMPENSATION
See Item 10.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
See Item 10.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
See Item 10.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
See Item 10.
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
The following documents are being filed as part of this Report:
(a)(1) FINANCIAL STATEMENTS:
Reference is made to the Index to Financial Statements and Financial Statement Schedule on Page .
(a)(2) FINANCIAL STATEMENT SCHEDULE:
Reference is made to the Index to Financial Statements and Financial Statement Schedule on Page .
All other schedules have been omitted because the required information is not present or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the Financial Statements or Notes thereto.
108
NEOSTEM, INC.
FORM 10K
(a)(3) EXHIBITS:
The following is a list of exhibits filed (or furnished, where specified) as part of this Annual Report on Form 10-K. Exhibits that were previously filed are described below and are incorporated by reference. For exhibits incorporated by reference, the location of the exhibit in the previous filing is indicated.
Exhibit | Description |
2.1 | Equity Purchase Agreement, dated as of June 18, 2012, by and among NeoStem, Inc., China Biopharmaceuticals Holdings, Inc., Fullbright Finance Limited, Suzhou Erye Economy & Trading Co., Ltd., and Suzhou Erye Pharmaceutical Co., Ltd. (filed as Exhibit 2.1 to the Company's Current Report on Form 8-K dated June 18, 2012). |
2.2 | Amendment to Equity Purchase Agreement, dated as of August 14, 2012, by and among NeoStem, Inc., China Biopharmaceuticals Holdings, Inc., Highacheive Holdings Limited, Fullbright Finance Limited, Suzhou Erye Economy & Trading Co., Ltd. and Suzhou Erye Pharmaceutical Co., Ltd. (filed as Exhibit 2.1 to the Company's Current Report on Form 8-K dated August 23, 2012). |
2.3 | Agreement and Plan of Merger, dated as of July 13, 2011, by and among NeoStem, Inc., Amo Acquisition Company I, Inc., Amo Acquisition Company II, LLC and Amorcyte, Inc. (filed as Exhibit 2.1 to the Company's Current Report on Form 8-K dated July 11, 2011). |
2.4 | Agreement and Plan of Merger, dated as of September 23, 2010, by and among NeoStem, Inc., NBS Acquisition Company LLC, and Progenitor Cell Therapy, LLC (filed as Exhibit 2.1 to the Company's Current Report on Form 8-K dated September 23, 2010). |
3.1 | Amended and Restated Certificate of Incorporation of NeoStem, Inc., filed with the Secretary of State of the State of Delaware on October 3, 2013 (filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K dated October 3, 2013). |
3.2 | Amended and Restated By-Laws dated January 5, 2015 (filed as Exhibit 3.1 to the Company's Current Report on Form 8-K on January 5, 2015). |
4.1 | Form of Redeemable Service Provider Warrant (filed as Exhibit 4.19 to the Company's Registration Statement on Form S-3/A, File No. 333.173853, filed with the SEC on September 16, 2011). |
4.2 | Form of 2011 Redeemable Service Provider Warrant (filed as Exhibit 4.20 to the Company's Registration Statement on Form S-3/A, File No. 333-173853, filed with the SEC on September 16, 2011). |
4.3 | Form of Redeemable Service Provider Warrant with cashless exercise rights (filed as Exhibit 4.21 to the Company's Registration Statement on Form S-3/A, File No. 333-173853, filed with the SEC on September 16, 2011). |
4.4 | Form of 2010/2011 Redeemable Service Provider Warrant with cashless exercise rights (filed as Exhibit 4.22 to the Company's Registration Statement on Form S-3/A, File No. 333-173853, filed with the SEC on September 16, 2011). |
4.5 | Letter Agreement dated December 18, 2008 between NeoStem, Inc. and RimAsia Capital Partners, L.P. (filed as Exhibit 4.1 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2008 as filed with the SEC on March 31, 2009). |
4.6 | Specimen Certificate for Common Stock (filed as Exhibit 4.1 to the Company's Registration Statement on Form S-3, File No. 333-145988, filed with the SEC on September 11, 2007). |
4.7 | Form of Placement Agent Warrant from June 2010 (filed as Exhibit 4.2 to the Company's Current Report on Form 8-K dated June 25, 2010 and filed with the SEC on June 28, 2010). |
4.8 | Amended and Restated Warrant, dated March 15, 2010, issued to RimAsia Capital Partners, L.P. (filed as Exhibit 4.1 to the Company's Current Report on Form 8-K dated March 15, 2010 and filed with the SEC on March 18, 2010). |
4.9 | Form of Warrant from the November 2010 Common Stock Offering (filed as Exhibit 4.1 to the Company's Current Report on Form 8-K dated and filed with the SEC on November 16, 2010). |
4.10 | Warrant Agreement, dated as of January 19, 2011, between NeoStem, Inc. and Continental Stock Transfer & Trust Company, with the forms of $3.00 Warrant and $5.00 Warrant attached thereto (filed as Exhibit 4.1 to the Company's Current Report on Form 8-K dated January 18, 2011 and filed with the SEC on January 24, 2011). |
109
4.11 | Warrant Agreement, dated as of July 22, 2011, between NeoStem, Inc. and Continental Stock Transfer & Trust Company, with the form of Series NA Warrant attached thereto (filed as Exhibit 4.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 as filed with the SEC on November 10, 2011). |
4.12 | Registration Rights Agreement, dated as of March 10, 2014, by and between NeoStem, Inc. and Aspire Capital Fund, LLC. (Filed as Exhibit 4.18 to the Company's Annual Report on Form 10-K filed with the SEC on March 13, 2014) |
4.13 | Warrant Agreement, dated as of October 17, 2011, between NeoStem, Inc. and Continental Stock Transfer & Trust Company, with the form of Global Series AMO Warrant attached thereto (filed as Exhibit 4.1 to the Company's Current Report on Form 8-K dated October 14, 2011). |
4.14 | Form of Common Stock Purchase Warrant from the March 2012 Underwritten Offering (filed as Exhibit 4.1 to the Company's Current Report on Form 8-K dated March 29, 2012). |
4.15 | Form of Common Stock Purchase Warrant for the May-July 2012 private placement (filed as Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2012 as filed with the SEC on August 14, 2012). |
4.16 | Form of New Warrant from July 2012 (filed as Exhibit 10.6 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2012 as filed with the SEC on August 14, 2012). |
4.17 | Form of Warrant from August 2012 private placement (filed as Exhibit 4.6 to the Company's Registration Statement on Form S-3, File No. 333-183542, filed with the SEC on August 24, 2012). |
4.18 | Form of 2011/2012 Service Provider Warrant (filed as Exhibit 4.10 to the Company's Registration Statement on Form S-3, File No. 333-183542, filed with the SEC on August 24, 2012). |
4.19 | Warrant issued to Aspire Capital Fund, LLC in August 2012 (filed as Exhibit 4.9 to the Company's Registration Statement on Form S-3, File No. 333-183542, filed with the SEC on August 24, 2012). |
4.20 | Form of Warrant for November 2012 Unit private placement (filed as Exhibit 4.4 to the Company's Registration Statement on Form S-3, File No. 333-185346, filed with the SEC on December 7, 2012). |
10.1 | Consulting Agreement, dated as of May 11, 2010 between NeoStem, Inc. and RimAsia Capital Partners, LP (filed as Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2010 as filed with the SEC on August 16, 2010). |
10.2 | Common Stock Purchase Agreement, dated as of March 11, 2014, by and between NeoStem, Inc. and Aspire Capital Fund, LLC. Filed as Exhibit 10.10 to the Company's Annual Report on Form 10-K filed on March 13, 2014) |
10.3 | Underwriting Agreement, dated April 29, 2013, between NeoStem, Inc. and Aegis Capital Corp. (filed as Exhibit 1.1 to the Company’s Current Report on Form 8-K dated April 29, 2013). |
10.4 | Underwriting Agreement, dated October 3, 2013, between NeoStem, Inc. and Aegis Capital Corp. (filed as Exhibit 1.1 to the Company’s Current Report on Form 8-K dated October 3, 2013). |
10.5 | Escrow Agreement, dated as of October 17, 2011, among NeoStem, Inc., Amorcyte, Inc., Paul J. Schmitt, as Amorcyte Representative, and Continental Stock Transfer & Trust Company, as Escrow Agent (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated October 14, 2011). |
10.6 | Lease dated September 1, 2005 between Vanni Business Park, LLC and Progenitor Cell Therapy, LLC, as amended by First Amendment of Lease effective as of July 1, 2006 (filed as Exhibit 10.48 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2010 as filed with the SEC on April 6, 2011). |
10.7 | Second Amendment of Lease, executed July 11, 2011 and effective July 1, 2011, by and between Vanni Business Park, LLC and Progenitor Cell Therapy, LLC (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated July 11, 2011). |
10.8 | Guaranty of Lease, executed July 11, 2011 and effective as of July 1, 2011, by NeoStem, Inc. for the benefit of Vanni Business Park, LLC (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K dated July 11, 2011). |
10.9† | First Amendment to Office Lease dated December 10, 2010, by and between WW VKO Owner, LLC and California Stem Cell, Inc; Second Amendment to Office Lease dated February 1, 2012, by and between CGGL 18301 LLC, and California Stem Cell, Inc. Third Amendment to Office Lease dated February 28, 2014, by and between CGGL 18301 LLC, and California Stem Cell, Inc.; and Fourth Amendment to Office Lease Agreement, executed December 19, 2014, effective April 1, 2015, by and between NeoStem, Inc. and CGGL 18301 LLC. |
10.10 | Stock Purchase and Assignment Agreement dated March 28, 2011, by and among Progenitor Cell Therapy, LLC, Athelos Corporation and Becton Dickinson and Company (filed as Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 as filed with the SEC on May 17, 2011). |
110
10.11 | Stockholders' Agreement dated March 28, 2011, by and among Progenitor Cell Therapy, LLC, Athelos Corporation and Becton Dickinson and Company (filed as Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 as filed with the SEC on May 17, 2011). |
10.12 | NeoStem, Inc. 2003 Equity Participation Plan, as amended (filed as Exhibit 10.2 to the Company's Registration Statement on Form S-1/A, File No. 333-137045, filed with the SEC on November 3, 2006). + |
10.13 | Form of Stock Option Agreement (filed as Exhibit 10.2 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2003 as filed with the SEC on March 30, 2004). + |
10.14 | Form of Option Agreement dated July 20, 2005 (filed as Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2005 as filed with the SEC on August 15, 2005). + |
10.15 | Amended and Restated NeoStem, Inc. 2009 Equity Compensation Plan, as amended (filed as Annex A to the Company's Definitive Proxy Statement on Schedule 14A filed on August 29, 2014). + |
10.16 | Form of Stock Option Grant Agreement under NeoStem, Inc. 2009 Equity Compensation Plan (filed as Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2010 as filed with the SEC on August 16, 2010). + |
10.17 | Description of the NeoStem, Inc. Board of Directors Compensation Plan (incorporated by reference to the first paragraph of Item 5.02 contained within the Company's Current Report on Form 8-K dated January 4, 2012, and the last paragraph appearing under Item 11 of this Annual Report on Form 10-K for the fiscal year ended December 31, 2012). + |
10.18 | NeoStem, Inc. 2012 Employee Stock Purchase Plan (filed as Appendix A to the Company's Definitive Proxy Statement on Schedule 14A for the 2012 Annual Meeting of Stockholders as filed with the SEC on September 7, 2012).+ |
10.19 | Loan and Security Agreement, dated September 26, 2014, by and between NeoStem, Inc., and Oxford Finance LLC. (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated September 26, 2014). |
10.20 | Employment Agreement between Phase III Medical, Inc. and Dr. Robin L. Smith, dated May 26, 2006 (filed as Exhibit 10.4 to the Company's Current Report on Form 8-K dated June 2, 2006). + |
10.21 | January 26, 2007 Amendment to Employment Agreement of Dr. Robin L. Smith (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated January 26, 2007). + |
10.22 | September 27, 2007 Amendment to Employment Agreement of Dr. Robin L. Smith (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated September 27, 2007). + |
10.23 | Letter agreement dated January 9, 2008 with Dr. Robin L. Smith (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated January 9, 2008). + |
10.24 | Amendment dated July 29, 2009 to Employment Agreement dated May 26, 2006 between NeoStem, Inc. and Dr. Robin L. Smith (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated July 29, 2009). + |
10.25 | Amendment dated April 4, 2011 to Employment Agreement dated May 26, 2006 between NeoStem, Inc. and Dr. Robin L. Smith (filed as Exhibit 10.66 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2010 as filed with the SEC on April 6, 2011). + |
10.26 | Amendment dated November 13, 2012 to Employment Agreement dated May 26, 2006 between NeoStem, Inc. and Dr. Robin L. Smith (filed as Exhibit 10.43 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2012 as filed with the SEC on March 8, 2013). + |
10.27 | Letter Agreement dated March 11, 2014 to Employment Agreement dated May 26, 2006 between NeoStem, Inc. and Dr. Robin L. Smith (filed as Exhibit 10.49 to the Company's Annual Report on Form 10-K filed with the SEC on March 13, 2014) + |
10.28 | Amendment, dated as of January 1, 2015, to Employment Agreement by and between NeoStem, Inc. and Robin L. Smith, M.D. dated May 26, 2006 (filed as Exhibit 10.1 on the Company's Current Report on Form 8-K filed with the SEC on January 5, 2015).+ |
10.29 | Amendment, dated as of January16, 2015, to Amendment dated as of January 1, 2015 to Employment Agreement by and between NeoStem, Inc. and Robin L. Smith, M.D. (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K filed with the SEC on January 16, 2015). + |
10.30 | January 26, 2007 Employment Agreement with Catherine M. Vaczy (filed as Exhibit 10.4 to the Company's Current Report on Form 8-K dated January 26, 2007). + |
10.31 | Letter agreement dated January 9, 2008 with Catherine M. Vaczy (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K dated January 9, 2008). + |
10.32 | Letter Agreement dated July 8, 2009 between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K dated July 6, 2009). + |
111
10.33 | Letter Agreement dated July 7, 2010 between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10(a) to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2010 as filed with the SEC on November 12, 2010). + |
10.34 | Letter Agreement dated January 6, 2012 between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.92 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011 as filed with the SEC on March 20, 2012). + |
10.35 | Letter Agreement dated November 13, 2012 between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.57 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2012 as filed with the SEC on March 8, 2013). + |
10.36 | Letter Agreement, dated July 12, 2013, between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated July 12, 2013). + |
10.37 | Letter Agreement, dated March 11, 2014, between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.57 to the Company's Annual Report on Form 10-K filed with the SEC on March 13, 2014).+ |
10.38 | Letter Agreement, dated October 11, 2014, between NeoStem, Inc. and Catherine M. Vaczy, Esq. (filed as Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed with the SEC on October 30, 2014).+ |
10.39 | Employment Agreement, dated as of September 23, 2010 and effective on January 19, 2011, by and between Progenitor Cell Therapy, LLC, NeoStem, Inc. and Andrew L. Pecora (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated January 18, 2011 and filed with the SEC on January 24, 2011). + |
10.40 | Amendment dated August 17, 2011 to Employment Agreement dated September 23, 2010 and effective January 19, 2011 between Progenitor Cell Therapy, LLC, NeoStem, Inc. and Andrew L. Pecora (filed as Exhibit 10.95 to the Company's Registration Statement on Form S-4, File No. 333-176673, filed with the SEC on September 2, 2011). + |
10.41 | Letter Agreement dated April 11, 2012 between NeoStem, Inc. and Andrew Pecora, M.D., F.A.C.P. (filed as Exhibit 10.107 to the Company's Annual Report on Form 10-K/A for the year ended December 31, 2011 as filed with the SEC on April 27, 2012). + |
10.42 | Amendment dated July 31, 2013 and effective August 5, 2013, by and among Andrew L. Pecora, M.D., FACP, NeoStem, Inc., Progenitor Cell Therapy, LLC and Amorcyte, LLC (filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K dated August 5, 2013). + |
10.43 | Employment Agreement, dated as of September 23, 2010 and effective on January 19, 2011, by and between Progenitor Cell Therapy, LLC, NeoStem, Inc. and Robert A. Preti (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K dated January 18, 2011 and filed with the SEC on January 24, 2011). + |
10.44† | Form of Indemnification Agreement for executive officers. |
10.45 | Letter Agreement dated June 28, 2011 between NeoStem, Inc. and Joseph Talamo (filed as Exhibit 10.10 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2011 as filed with the SEC on August 12, 2011). + |
10.46 | Employment Agreement, dated as of July 15, 2013, by and between NeoStem, Inc. and Stephen W. Potter (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated July 15, 2013). + |
10.47 | Employment Agreement, dated as of July 23, 2013 and effective August 5, 2013, by and between NeoStem, Inc. and Douglas W. Losordo, M.D. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated August 5, 2013). + |
10.48 | Employment Agreement, dated as of August 16, 2013 and effective August 19, 2013, by and between NeoStem, Inc. and Robert Dickey IV (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated August 19, 2013). + |
10.49 | Offer Letter dated August 14, 2013 and effective August 19, 2013, by and between NeoStem, Inc. and Larry May (filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K dated August 19, 2013). + |
10.50 | Employment Agreement, dated as of January 5, 2015 and effective on January 5, 2015, by and between NeoStem, Inc. and David J. Mazzo, Ph.D. (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K filed on January 5, 2015).+ |
10.51 | Amendment, dated as of January 16, 2015, to Employment Agreement, dated as of January 5, 2015 and effective on January 5, 2015, by and between NeoStem, Inc. and David J. Mazzo, Ph.D. (filed as Exhibit 10.2 to the Company's Current Report on Form 8-K filed on January 16, 2015). + |
10.52 | Employment Agreement, dated as of January 5, 2015 and effective on January 5, 2015, by and between NeoStem, Inc. and Robert S. Vaters (filed as Exhibit 10.3 to the Company's Current Report on Form 8-K filed on January 5, 2015).+ |
112
10.53 | Amendment, dated as of January16, 2015, to Employment Agreement, dated as of January 5, 2015 and effective on January 5, 2015, by and between NeoStem, Inc. and Robert S. Vaters (filed as Exhibit 10.3 to the Company's Current Report on Form 8-K filed on January 16, 2015).+ |
14.1 | Code of Ethics for Senior Financial Officers (filed as Exhibit 14.1 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2010 as filed with the SEC on April 6, 2011). |
21.1† | Subsidiaries of NeoStem, Inc. |
23.1† | Consent of Grant Thornton LLP |
31.1† | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
31.2† | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
32.1†† | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
32.2†† | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
101.INS | XBRL Instance Document |
101.SCH | XBRL Taxonomy Extension Schema |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase |
101.DEF | XBRL Taxonomy Extension Definition Linkbase |
101.LAB | XBRL Taxonomy Extension Label Linkbase |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase |
_______________
+ | Management contract or compensatory plan, contract or arrangement required to be filed as an exhibit to this Form 10-K pursuant to Item 15(b) of Form 10-K. |
† | Filed herewith. |
†† | Furnished herewith. |
(1) | Certain portions of this exhibit were omitted based upon a request for confidential treatment, and the omitted portions were filed separately with the SEC on a confidential basis. |
113
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of New York, State of New York, on March 2, 2015.
NEOSTEM, INC. | ||
By: /s/ David J. Mazzo, PhD Name: David J. Mazzo Title: Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ David J. Mazzo, PhD. David J. Mazzo, PhD. | Director, and Chief Executive Officer (Principal Executive Officer) | March 2, 2015 | ||
/s/ Robert S. Vaters Robert S. Vaters | Director, President & Chief Financial Officer (Principal Financial Officer) | March 2, 2015 | ||
/s/ Joseph Talamo Joseph Talamo | Vice President, Corporate Controller and Chief Accounting Officer (Principal Accounting Officer) | March 2, 2015 | ||
/s/ Robin L. Smith, M.D. Robin L. Smith | Executive Chair of the Board of Directors | March 2, 2015 | ||
/s/ Richard Berman Richard Berman | Director | March 2, 2015 | ||
/s/ Steven S. Myers Steven S. Myers | Director | March 2, 2015 | ||
/s/ Drew Bernstein Drew Bernstein | Director | March 2, 2015 | ||
/s/ Eric Wei Eric Wei | Director | March 2, 2015 | ||
/s/ Andrew L. Pecora, M.D. Andrew L. Pecora, M.D. | Director | March 2, 2015 | ||
/s/ Martyn D. Greenacre Martyn D. Greenacre | Director | March 2, 2015 | ||
/s/ Steven M. Klosk Steven M. Klosk | Director | March 2, 2015 | ||
/s/ Peter Traber Peter Traber | Director | March 2, 2015 | ||
114