Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - Assertio Therapeutics, Inc | a2223144zex-32_2.htm |
| EX-21 - EX-21 - Assertio Therapeutics, Inc | a2223144zex-21.htm |
| EX-31.1 - EX-31.1 - Assertio Therapeutics, Inc | a2223144zex-31_1.htm |
| EX-12.1 - EX-12.1 - Assertio Therapeutics, Inc | a2223144zex-12_1.htm |
| EX-32.1 - EX-32.1 - Assertio Therapeutics, Inc | a2223144zex-32_1.htm |
| EX-31.2 - EX-31.2 - Assertio Therapeutics, Inc | a2223144zex-31_2.htm |
| EX-10.15 - EX-10.15 - Assertio Therapeutics, Inc | a2223144zex-10_15.htm |
| EX-10.22 - EX-10.22 - Assertio Therapeutics, Inc | a2223144zex-10_22.htm |
| EX-10.16 - EX-10.16 - Assertio Therapeutics, Inc | a2223144zex-10_16.htm |
| EXCEL - IDEA: XBRL DOCUMENT - Assertio Therapeutics, Inc | Financial_Report.xls |
| EX-23.1 - EX-23.1 - Assertio Therapeutics, Inc | a2223144zex-23_1.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
PART IV
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark one) | ||
ý |
Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
|
For the fiscal year ended December 31, 2014 |
||
OR |
||
o |
Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
|
For the transition period from: to |
||
Commission File Number: 001-13111
DEPOMED, INC.
(Exact Name of Registrant as Specified in its Charter)
| California (State or other jurisdiction of incorporation or organization) |
94-3229046 (I.R.S. Employer Identification No.) |
|
7999 Gateway Boulevard, Suite 300, Newark, California (Address of principal executive offices) |
94560 (Zip Code) |
Registrant's telephone number, including area code: (510) 744-8000
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class: | Name of each exchange on which registered: | |
|---|---|---|
| Common Stock, no par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer, as defined in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ý | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant based upon the closing price of Common Stock on the Nasdaq Stock Market on June 30, 2014 was approximately $808,470,496. Shares of Common Stock held by each officer and director and by each person who owned 10% or more of the outstanding Common Stock as of June 30, 2014 have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares outstanding of the registrant's Common Stock, no par value, as of February 25, 2015 was 59,558,855
Documents Incorporated by Reference
Portions of the registrant's Proxy Statement, which will be filed with the Securities and Exchange Commission (SEC) pursuant to Regulation 14A in connection with the registrant's 2015 Annual Meeting of Shareholders, expected to be held on or about May 12, 2015, are incorporated by reference in Part III of this Form 10-K.
DEPOMED, INC.
FORM 10-K FOR THE FISCAL YEAR ENDED DECEMBER 31, 2014
TABLE OF CONTENTS
2
NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements made in this Annual Report on Form 10-K that are not statements of historical fact are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the Securities Act), and Section 21E of the Securities Exchange Act of 1934, as amended (the Exchange Act). We have based these forward-looking statements on our current expectations and projections about future events. Our actual results could differ materially from those discussed in, or implied by, these forward-looking statements. Forward-looking statements are identified by words such as "believe," "anticipate," "expect," "intend," "plan," "will," "may" and other similar expressions. In addition, any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements. Forward-looking statements include, but are not necessarily limited to, those relating to:
- •
- the commercial success and market acceptance of Gralise® (gabapentin), our once-daily product for the management of
postherpetic neuralgia, CAMBIA® (diclofenac potassium for oral solution), our non-steroidal anti-inflammatory drug for the acute treatment
of migraine attacks, Zipsor® (diclofenac potassium) liquid filled capsules, our non-steroidal anti-inflammatory drug for the treatment of
mild to moderate pain in adults, and Lazanda® (fentanyl) nasal spray, our product for the management of breakthrough cancer pain in adult,
opioid-tolerant cancer patients;
- •
- our ability to consummate our previously announced acquisition of the rights to the NUCYNTA® franchise of pharmaceutical
products in the United States as further described below, and, if consummated, the commercial success of NUCYNTA®;
- •
- the results of our ongoing litigation against the filer of an Abbreviated New Drug Application (ANDA) to market generic versions of
Zipsor® in the United States;
- •
- any additional patent infringement or other litigation or proceeding that may be instituted related to Gralise®,
CAMBIA®, Zipsor®,
Lazanda® or any other of our products, product candidates or products we may acquire, including NUCYNTA®;
- •
- our and our collaborative partners' compliance or non-compliance with legal and regulatory requirements related to the promotion of
pharmaceutical products in the United States;
- •
- the outcome of our ongoing patent infringement litigation against Purdue Pharma L.P. (Purdue) and Endo
Pharmaceuticals Inc. (Endo);
- •
- our plans to acquire, in-license or co-promote other products;
- •
- the results of our research and development efforts;
- •
- submission, acceptance and approval of regulatory filings;
- •
- our ability to raise additional capital; and
- •
- our collaborative partners' compliance or non-compliance with obligations under our collaboration agreements.
Factors that could cause actual results or conditions to differ from those anticipated by these and other forward-looking statements include those more fully described in the "ITEM 1A. RISK FACTORS" section and elsewhere in this Annual Report on Form 10-K. Except as required by law, we assume no obligation to update any forward-looking statement publicly, or to revise any forward-looking statement to reflect events or developments occurring after the date of this Annual Report on Form 10-K, even if new information becomes available in the future. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in any such forward-looking statement.
3
CORPORATE INFORMATION
The address of our Internet website is http://www.depomed.com. We make available, free of charge through our website or upon written request, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other periodic SEC reports, along with amendments to all of those reports, as soon as reasonably practicable after we file the reports with the SEC.
Unless the context indicates otherwise, "Depomed," "the Company," "we," "our" and "us" refer to Depomed, Inc. Depomed was incorporated in the State of California on August 7, 1995. Our principal executive offices are located at 7999 Gateway Boulevard, Suite 300, Newark, California, 94560 and our telephone number is (510) 744-8000.
Depomed®, Gralise®, CAMBIA®, Zipsor®,Lazanda® and Acuform® are registered trademarks of Depomed. Glumetza® is a registered trademark of Valeant International (Barbados) SRL exclusively licensed in the United States (U.S.) to Depomed. All other trademarks and trade names referenced in this Annual Report on Form 10-K are the property of their respective owners.
4
COMPANY OVERVIEW
Depomed is a specialty pharmaceutical company focused on pain and other central nervous system (CNS) conditions. The products that comprise our current specialty pharmaceutical business are Gralise® (gabapentin), a once-daily product for the management of postherpetic neuralgia (PHN) that we launched in October 2011, CAMBIA® (diclofenac potassium for oral solution), a non-steroidal anti-inflammatory drug for the acute treatment of migraine attacks that we acquired in December 2013, Zipsor® (diclofenac potassium) liquid filled capsules, a non-steroidal anti-inflammatory drug for the treatment of mild to moderate acute pain that we acquired in June 2012, and Lazanda® (fentanyl) nasal spray, a product for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain, that we acquired in July 2013. We actively seek to expand our product portfolio through in-licensing, acquiring or obtaining co-promotion rights to commercially available products or late-stage product candidates that could be marketed and sold effectively with our existing products through our sales and marketing capability.
We also have a portfolio of royalty and milestone producing license agreements based on our proprietary Acuform® gastroretentive drug delivery technology with Mallinckrodt Inc. (Mallinckrodt), Ironwood Pharmaceuticals, Inc. (Ironwood) and Janssen Pharmaceuticals, Inc. (Janssen Pharma).
In October 2013, we sold our interests in royalty and milestone payments under our license agreements in the Type 2 diabetes therapeutic area to PDL BioPharma, Inc. (PDL) for $240.5 million (PDL Transaction). The interests sold include royalty and milestone payments accruing from and after October 1, 2013 from: (a) Salix Pharmaceuticals, Inc. (Salix) with respect to sales of Glumetza® (metformin HCL extended-release tablets) in the United States; (b) Merck & Co. Inc. (Merck) with respect to sales of Janumet® XR (sitagliptin and metformin HCL extended-release); (c) Janssen Pharmaceutica N.V. and Janssen Pharma (collectively, Janssen) with respect to potential future development milestones and sales of Janssen's investigational fixed-dose combination of Invokana® (canagliflozin) and extended-release metformin; (d) Boehringer Ingelheim International GMBH (Boehringer Ingelheim) with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to our license agreement with Boehringer Ingelheim; and (e) LG Life Sciences Ltd. (LG) and Valeant International Bermuda SRL (Valeant SRL) for sales of extended-release metformin in Korea and Canada, respectively.
On January 15, 2015, we entered into an Asset Purchase Agreement (Asset Purchase Agreement) with Janssen Pharma, pursuant to which we will acquire from Janssen Pharma and its affiliates the rights to the NUCYNTA® franchise of pharmaceutical products in the United States as well as certain related assets for $1.05 billion in cash (NUCYNTA® Acquisition). The NUCYNTA® franchise includes NUCYNTA® ER (tapentadol) extended release tablets indicated for the management of pain, including neuropathic pain associated with diabetic peripheral neuropathy (DPN), severe enough to require daily, around-the-clock, long-term opioid treatment, NUCYNTA® (tapentadol), an immediate release version of tapentadol, for management of moderate to severe acute pain in adults, and NUCYNTA® (tapentadol) oral solution, an approved oral form of tapentadol that has not been commercialized. Upon execution of the Asset Purchase Agreement, we delivered a cash deposit in the amount of $500.0 million to JPMorgan Chase Bank, N.A., (Escrow Agent) in accordance with an Escrow Agreement, dated January 15, 2015, by and among the Company, Janssen Pharma and the Escrow Agent. The cash deposit will be credited against the total purchase price payable to Janssen Pharma upon the consummation of the NUCYNTA® Acquisition. The consummation of the NUCYNTA® Acquisition, which we expect to occur in the second quarter of 2015, is subject to the satisfaction of a number of conditions which we cannot be certain will be satisfied. See "Note 16—Subsequent Events"
5
of the Notes to Consolidated Financial Statements for further information on the Asset Purchase Agreement.
SIGNIFICANT DEVELOPMENTS
Among the significant developments in our business during 2014 were the following:
- •
- Total revenues for the year ended December 31, 2014 were $390.4 million, including product revenues of
$114.2 million. Total revenue includes approximately $243.0 million of non-cash PDL royalty revenue.
- •
- Cash, cash equivalents and marketable securities were $566.4 million as of December 31, 2014, of which
$500.0 million was delivered to the Escrow Agent on January 15, 2015 as a deposit to be credited against the purchase price payable to Janssen Pharma upon the consummation of the
NUCYNTA® Acquisition.
- •
- In March 2014, the FDA approved Mallinckrodt's New Drug Applications (NDA) for XARTEMIS™ XR. The approval of the NDA
triggered a $10.0 million milestone payment to us, which we recognized as revenue in the first quarter of 2014 and received in April 2014.
- •
- In May 2014, the FDA accepted for filing the NDA for MNK-155. The acceptance for filing of the NDA triggered a $5.0 million
milestone payment to us which we recognized in the second quarter of 2014 and received in June 2014.
- •
- In September 2014, we issued $345.0 million aggregate principal amount of convertible senior notes in a public offering.
- •
- In August 2014, the U.S. district court for the district of New Jersey ruled in our favor in our patent infringement lawsuit against
Actavis Elizabeth LLC and Actavis Inc., upholding the validity of all seven of our patents asserted in the litigation and providing expected market exclusivity for Gralise®
until 2024.
- •
- In September 2014, the federal district court for the District of Columbia ruled in our favor in our lawsuit against the U.S. Food and Drug Administration (FDA), resulting in an order requiring the FDA to grant Gralise® seven years of orphan drug exclusivity for the management of PHN.
Commercialized Products
The following table summarizes our and our partners' commercialized products and product candidate development pipeline:
Depomed Commercialized Products
Product
|
Indication | Status | ||
|---|---|---|---|---|
Gralise® |
Management of postherpetic neuralgia | Currently sold in the United States. Launched in October 2011 |
||
CAMBIA® |
Acute treatment of migraine attacks in adults 18 years of age or older | Currently sold in the United States. Acquired in December 2013 |
||
Zipsor® |
Mild to moderate acute pain | Currently sold in the United States. Acquired in June 2012 |
||
Lazanda® |
Breakthrough pain in cancer patients 18 years of age and older who are already receiving and who are tolerant to continuous opioid therapy for their underlying persistent cancer pain | Currently sold in the United States. Acquired in July 2013 |
6
Partner Commercialized Products and Product Candidates
Product / Product Candidate
|
Indication | Partner | Status | |||
|---|---|---|---|---|---|---|
XARTEMIS™ XR (oxycodone hydrochloride and acetaminophen) |
Management of acute pain severe enough to require opioid treatment and in patients for whom alternative treatment options are ineffective, not tolerated or would otherwise be inadequate | Mallinckrodt | Approved by the FDA and launched in March 2014 | |||
MNK-155 |
Pain | Mallinckrodt | NDA accepted for filing by the FDA in May 2014 | |||
|
Foreign regulatory filings in process | |||||
NUCYNTA® ER |
Moderate to severe chronic pain; neuropathic pain associated with diabetic peripheral neuropathy (DPN) | Janssen Pharma | License covers sales of NUCYNTA® ER in the United States, Canada and Japan | |||
IW-3718 Refractory gastroesophageal reflux disease (GERD) program using Acuform® |
Refractory GERD | Ironwood | In clinical development |
OUR BUSINESS OPERATIONS
As of December 31, 2014, our revenues are generated primarily from commercialized products and license and development arrangements.
Commercialized Products
Gralise® (Gabapentin) Tablets for the Management of PHN
Gralise® is our proprietary, once-daily formulation of gabapentin for the management of PHN. We made Gralise commercially available in October 2011, following its FDA approval in January 2011 and our reacquisition of the product in March 2011 from Abbott Products, Inc. (Abbott Products), our former licensee.
Gralise® product sales were $60.4 million for the year ended December 31, 2014, $36.2 million for the year ended December 31, 2013 and $17.3 million for the year ended December 31, 2012.
Postherpetic Neuralgia. PHN is a persistent pain condition caused by nerve damage during a shingles, or herpes zoster, viral infection. The Centers for Disease Control and Prevention Advisory Committee on Immunization Practices recommends that adults 60 years of age and older be vaccinated with a shingles vaccine. While the shingles vaccine is not a treatment for PHN, it could impact the future market for therapies for PHN, including Gralise®.
Orphan Drug Designation. In November 2010, the FDA granted Gralise® Orphan Drug designation for the management of PHN, but did not recognize Orphan drug exclusivity for Gralise® in
7
January 2011 when Gralise® was approved for marketing in the United States. In September 2012, we filed an action in federal district court for the District of Columbia against the FDA seeking an order requiring the FDA to grant Gralise® Orphan Drug exclusivity for the management of PHN. In September 2014, the court issued an order granting our request for summary judgment, and ordering the FDA to grant Orphan Drug exclusivity for Gralise® for the management of PHN, which the FDA formally granted in October 2014.
CAMBIA® (Diclofenac Potassium for Oral Solution) for the Acute Treatment of Migraine Attacks in Adults 18 Years of Age or Older
CAMBIA® is a non-steroidal anti-inflammatory drug (NSAID) indicated for the acute treatment of migraine attacks with or without aura in adults 18 years of age or older. We acquired CAMBIA® and related product inventory on December 17, 2013 from Nautilus Neurosciences, Inc. (Nautilus). We also assumed certain annual third party royalty obligations totaling not more than 11% of CAMBIA® net sales.
We began shipping and recognizing product sales on CAMBIA® in December 2013. We began commercial promotion of CAMBIA® in February 2014. Our CAMBIA® product sales were $21.7 million for the year ended December 31, 2014 and $0.6 million for the year ended December 31, 2013, which includes approximately two weeks of sales.
Zipsor® (Diclofenac Potassium) Liquid-Filled Capsules for Treatment of Mild to Moderate Acute Pain
Zipsor® is an NSAID indicated for relief of mild to moderate acute pain in adults. Zipsor® uses proprietary ProSorb® delivery technology to deliver a finely dispersed, rapidly absorbed formulation of diclofenac. We acquired Zipsor® on June 21, 2012 from Xanodyne Pharmaceuticals, Inc. (Xanodyne).
We began shipping and recognizing product sales on Zipsor® at the end of June 2012. We began commercial promotion of Zipsor® in July 2012. Our Zipsor® product sales were $25.2 million for the year ended December 31, 2014, $20.3 million for the year ended December 31, 2013 and $9.8 million for the year ended December 31, 2012.
Lazanda® (Fentanyl) Nasal Spray for the Management of Breakthrough Pain in Cancer Patients, 18 Years of Age and Older, who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain
Lazanda® nasal spray is an intranasal fentanyl drug used to manage breakthrough pain in adults (18 years of age or older) who are already routinely taking other opioid pain medicines around-the-clock for cancer pain. We acquired Lazanda® and certain related product inventory on July 29, 2013, from Archimedes Pharma US Inc., Archimedes Pharma Ltd., and Archimedes Development Ltd. (collectively, Archimedes).
We began shipping and recognizing product sales on Lazanda® in August 2013. We began commercial promotion of Lazanda® in October 2013. Our Lazanda® product sales were $6.9 million for the year ended December 31, 2014 and $1.2 million for the year ended December 31, 2013.
Segment and Customer Information
The Company operates in one operating segment and has operations solely in the United States. To date, all of the Company's revenues from product sales are related to sales in the United States. The Company has recognized license and royalty revenue from license agreements in the territories of the United States, Canada and Korea.
Three wholesale distributors represented 26%, 27% and 35% of product shipments for the year ended December 31, 2014. These three customers individually comprised 33%, 32% and 35%, respectively, of product sales-related accounts receivable as of December 31, 2014. Three wholesale distributors represented 35%, 37% and 21% of product shipments for the year ended December 31,
8
2013. These three customers individually comprised 23%, 35% and 34%, respectively, of product sales-related accounts receivable as of December 31, 2013.
License and Development Arrangements
Janssen Pharmaceuticals, Inc.—NUCYNTA® ER
In August 2012, we entered into a license agreement with Janssen Pharma that grants Janssen Pharma a non-exclusive license to certain patents and other intellectual property rights to our Acuform drug delivery technology for the development and commercialization of tapentadol extended release products, including NUCYNTA® ER (tapentadol extended-release tablets). We received a $10.0 million upfront license fee and receive low single digit royalties on net sales of NUCYNTA® ER in the United States, Canada, and Japan from and after July 2, 2012 through December 31, 2021. We will not receive any royalties from Janssen Pharma on net sales of NUCYNTA® ER in the United States for any period after the consummation of the NUCYNTA® Acquisition.
Mallinckrodt (Formerly Covidien)—Acetaminophen/Opiate Combination Products
In November 2008, we entered into a license agreement related to acetaminophen/opiate combination products with Mallinckrodt. The license agreement grants Mallinckrodt worldwide rights to utilize our Acuform technology for the exclusive development of up to four products containing acetaminophen in combination with opiates, two of which Mallinckrodt has elected to develop.
We have received $27.5 million in upfront fees and milestones under the agreement. The upfront fees included a $4.0 million upfront license fee and a $1.5 million advance payment for formulation work we performed under the agreement. The milestone payments include four $0.5 million clinical development milestones, a $5.0 million milestone following the FDA's July 2013 acceptance for filing of the NDA for XARTEMIS™ XR (oxycodone hydrochloride and acetaminophen) Extended-Release Tablets (CII), previously known as MNK-795, a $10.0 million milestone on FDA approval of XARTEMIS™ XR, and a $5.0 million milestone following the FDA's May 2014 acceptance for filing of the NDA for MNK-155.
In March 2014, the FDA approved XARTEMIS™ XR for the management of acute pain severe enough to require opioid treatment and in patients for whom alternative treatment options (e.g., non-opioid analgesics) are ineffective, not tolerated or would otherwise be inadequate. The approval of the NDA triggered a $10.0 million milestone payment to us, which we recognized in first quarter 2014 and received in April 2014. In May 2014, the FDA accepted for filing the NDA for MNK-155. The acceptance for filing of the NDA triggered a $5.0 million milestone payment to us which we recognized in the second quarter of 2014 and received in June 2014. We receive high single digit royalties on net sales of XARTEMIS™ XR, which was launched in March 2014, and we will receive the same high single digit royalty on net sales of MNK-155 if it is approved.
Ironwood Pharmaceuticals, Inc.—IW-3718 for Refractory GERD
In July 2011, we entered into a collaboration and license agreement with Ironwood granting Ironwood a license for worldwide rights to certain patents and other intellectual property rights to our Acuform drug delivery technology for IW-3718, an Ironwood product candidate under evaluation for refractory GERD. We have received $3.4 million under the agreement, which includes an upfront payment, reimbursement of initial product formulation work and three milestones payments, including a milestone payment of $1.0 million in March 2014 as a result of the initiation of clinical trials relating to IW-3718 by Ironwood.
Licensing and Development Agreement Royalties Sold to PDL in October 2013
In October 2013, we sold to PDL our milestone and royalty interests in our license agreements in the type 2 diabetes therapeutic area (and any replacements for the agreements) for $240.5 million. The material agreements included in the sale are described below. From and after October 1, 2013, PDL
9
will receive all royalty and milestone payments due under the agreements until PDL has received payments equal to $481 million, after which we and PDL will share evenly all net payments received.
Salix Pharmaceuticals, Inc. (formerly Santarus, Inc.)—Glumetza®
Glumetza is a once-daily extended release metformin product approved in the United States for type 2 diabetes that we have licensed to Salix. In August 2011, we entered into a commercialization agreement with Salix granting Salix exclusive rights to manufacture and commercialize Glumetza in the United States. The commercialization agreement supersedes the previous promotion agreement between the parties originally entered into in July 2008. Under the commercialization agreement, we granted Salix exclusive rights to manufacture and commercialize Glumetza in the United States in return for a royalty on Glumetza net sales. We recognized $42.8 million in royalty revenue for the year ended December 31, 2012. We recognized $42.1 million royalty revenue for the year ended December 31, 2013, all of which was recognized in the nine months ended September 30, 2013. The 2013 amount does not include royalties we sold to PDL.
Salix pays royalties on Glumetza net product sales in the United States as follows: 26.5% in 2011; 29.5% in 2012; 32.0% in 2013 and 2014; and 34.5% in 2015 and beyond, prior to a generic entry of a Glumetza product. In the event of a generic entry of a Glumetza product in the United States, the parties will thereafter equally share Glumetza proceeds based on a gross margin split.
Merck—Janumet® XR
We have received $12.5 million in upfront and milestone payments and we receive royalties on Merck's net sales of Janumet® XR in the United States and other licensed territories through the expiration of the licensed patents under a July 2009 license agreement with Merck. The non-exclusive license agreement grants Merck a license as well as other rights to certain of our patents directed to metformin extended release technology for Janumet® XR, Merck's fixed-dose combination product for type 2 diabetes containing sitagliptin and extended release metformin that was approved by the FDA in February 2012. Merck began selling Janumet® XR during the first quarter of 2012.
Janssen—Canaglifozin/Metformin XR Combination Products
We have received $10.0 million in upfront and milestone payments, and are eligible for additional milestone payments and royalties under an August 2010 non-exclusive license agreement between us and Janssen related to fixed dose combinations of extended release metformin and Janssen's type 2 diabetes product candidate canagliflozin.
Under the agreement, we granted Janssen a license to certain patents related to our Acuform drug delivery technology to be used in developing the combination products. We also granted Janssen a right to reference the Glumetza NDA in Janssen's regulatory filings covering the products. In February 2013, we completed a project for Janssen related to this program and recognized $2.2 million in revenue during the first quarter of 2013.
Boehringer Ingelheim—Undisclosed Compounds/Metformin XR Combination Products
We have received $12.5 million in upfront and milestone payments and may receive additional development milestones, as well as royalties, pursuant to a March 2011 license and service agreement with Boehringer Ingelheim related to fixed dose combinations of extended release metformin and proprietary Boehringer Ingelheim compounds in development for type 2 diabetes. Under the agreement, we granted Boehringer Ingelheim a license to certain patents related to our Acuform drug delivery technology to be used in developing the combination products. Boehringer Ingelheim was also granted a right to reference the Glumetza NDA in regulatory submissions for the products.
We received a $10.0 million upfront license payment and, in March 2012, we received an additional $2.5 million milestone payment upon delivery of experimental batches of prototype formulations that met agreed-upon specifications. The agreement provides for additional milestone payments based on regulatory filings and approval events, as well as royalties on worldwide net sales of products.
10
OUR DRUG DELIVERY TECHNOLOGY
The Acuform technology is based on our proprietary oral drug delivery technologies and is designed to include formulations of drug-containing polymeric tablets that allow multi-hour delivery of an incorporated drug. Although our formulations are proprietary, the polymers utilized in the Acuform technology are commonly used in the food and drug industries and are included in the list of inert substances approved by the FDA for use in oral pharmaceuticals. By using different formulations of the polymers, we believe that the Acuform technology is able to provide continuous, controlled delivery of drugs of varying molecular complexity and solubility. With the use of different polymers and polymers of varying molecular weight, our Acuform tablet technology can deliver drugs by diffusion, tablet erosion, or from a bi-layer matrix. In addition, our technology allows for the delivery of more than one drug from a single tablet. If taken with a meal, these polymeric tablets remain in the stomach for an extended period of time to provide continuous, controlled delivery of an incorporated drug.
The Acuform technology's design is based in part on principles of human gastric emptying and gastrointestinal transit. Following a meal, liquids and small particles flow continuously from the stomach into the intestine, leaving behind the larger undigested particles until the digestive process is complete. As a result, drugs in liquid or dissolved form or those consisting of small particles tend to empty rapidly from the stomach and continue into the small intestine and on into the large intestine, often before the drug has time to act locally or to be absorbed in the stomach and/or upper small intestine. The drug-containing polymeric tablets of the Acuform technology are formulated into easily swallowed shapes and are designed to swell upon ingestion. The tablets attain a size after ingestion sufficient to be retained in the stomach for multiple hours during the digestive process while delivering the drug content at a controlled rate. After drug delivery is complete, the polymeric tablet dissolves and becomes a watery gel, which is safely eliminated through the intestine sight unseen.
The Acuform technology is designed to address certain limitations of drug delivery and to provide for orally-administered, conveniently-dosed, cost-effective drug therapy that provides continuous, controlled-delivery of a drug over a multi-hour period. We believe that the Acuform technology can provide one or more of the following advantages over conventional methods of drug administration:
- •
- Greater Patient and Caregiver Convenience. We believe
that the Acuform technology may offer once-daily or reduced frequency dosing for certain drugs that are currently required to be administered several times daily.
- •
- Enhanced Safety and Efficacy through Controlled
Delivery. We believe that the Acuform technology may improve the ratio of therapeutic effect to toxicity by decreasing the initial peak
concentrations of a drug associated with toxicity, while maintaining levels of the drug at therapeutic, subtoxic concentrations for an extended period of time.
- •
- More Efficient Gastrointestinal Drug Absorption. We
believe that the Acuform technology can be used for improved oral administration of drugs that are inadequately absorbed when delivered as conventional tablets or capsules. Many drugs are primarily
absorbed in the stomach, duodenum or upper small intestine regions, through which drugs administered in conventional oral dosage forms transit quickly. In contrast, the Acuform technology is designed
to be retained in the stomach, allowing for multi-hour flow of drugs to these regions of the gastrointestinal tract.
- •
- Rational Drug Combinations. We believe that the Acuform technology may allow for rational combinations of drugs with different biological half-lives. Physicians frequently prescribe multiple drugs for treatment of a single medical condition. By appropriately incorporating different drugs into an Acuform technology we believe that we can provide for the release of each incorporated drug continuously at a rate and duration (dose) appropriately adjusted for the specific biological half-lives of the drugs.
11
Gralise® uses our Acuform technology. CAMBIA®, Zipsor® and Lazanda® do not use the technology.
RESEARCH AND DEVELOPMENT EXPENSES
Our research and development expenses were $7.1 million in 2014, $8.1 million in 2013 and $15.5 million in 2012. We expect research and development expense in 2015 to increase from 2014 levels, primarily as a result of pediatric studies relating to CAMBIA® and Zipsor® that we intend to undertake in 2015. If we consummate the NUCYNTA® Acquisition, we will assume responsibility for certain post marketing regulatory requirements and pediatric studies relating to the products, which may increase our research and development expenses for future periods.
PATENTS AND PROPRIETARY RIGHTS
The material issued in the United States patents we own or have in-licensed, and the products they cover, are as follows:
Product
|
U.S. Patent Nos. (Exp. Dates) | |
|---|---|---|
| Gralise® | 7,438,927 (February 26, 2024) | |
7,731,989 (October 25, 2022) |
||
8,192,756 (October 25, 2022) |
||
8,252,332(October 25, 2022) |
||
8,333,992 (October 25, 2022) |
||
6,723,340 (October 25, 2021) |
||
6,488,962 (June 20, 2020) |
||
6,340,475 and 6,635,280 (September 19, 2016) |
||
Zipsor® |
7,662,858 (February 24, 2029) |
|
7,884,095 (February 24, 2029) |
||
7,939,518 (February 24, 2029) |
||
8,110,606 (February 24, 2029) |
||
8,623,920 (February 24, 2029) |
||
6,365,180 (July 15, 2019) |
||
6,287,594 (January 15, 2019) |
||
CAMBIA® |
7,759,394*, 8,097,651* and 8,927,604* (June 16, 2026) |
|
6,974,595* and 7,482,377* (May 15, 2017) |
||
Lazanda® |
8,216,604 (October 3, 2024) |
|
6,432,440 (April 20, 2018) |
||
8,889,176 (January 16, 2024) |
- *
- Patent rights are exclusively in-licensed by the Company.
12
Our success will depend in part on our ability to obtain and maintain patent protection for our products and technologies. Our policy is to seek to protect our proprietary rights, by among other methods, filing patent applications in the United States and foreign jurisdictions to cover certain aspects of our technology. In addition to those patents noted on the above table, we have 19 patent applications pending in the United States. Our pending patent applications may lack priority over other applications or may not result in the issuance of patents. Even if issued, our patents may not be sufficiently broad to provide protection against competitors with similar technologies and may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products or may not provide us with competitive advantages against competing products. We also rely on trade secrets and proprietary know-how, which are difficult to protect. We seek to protect such information, in part, through entering into confidentiality agreements with employees, consultants, collaborative partners and others before such persons or entities have access to our proprietary trade secrets and know-how. These confidentiality agreements may not be effective in certain cases. In addition, our trade secrets may otherwise become known or be independently developed by competitors.
Our ability to develop our technologies and to make commercial sales of products using our technologies also depends on not infringing other patents or intellectual property rights. We are not aware of any intellectual property claims against us. However, the pharmaceutical industry has experienced extensive litigation regarding patents and other intellectual property rights. If claims concerning any of our products were to arise and it is determined that these products infringe a third party's proprietary rights, we could be subject to substantial damages for past infringement or be forced to stop or delay our activities with respect to any infringing product, unless we can obtain a license, or we may have to redesign our product so that it does not infringe upon such third party's patent rights, which may not be possible or could require substantial funds or time. Such a license may not be available on acceptable terms, or at all. Even if we, our collaborators or our licensors were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property.
From time to time, we may become aware of activities by third parties that may infringe our patents. We may need to engage in litigation to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights, such as litigation described in "LEGAL PROCEEDINGS". Our issued or licensed patents may not be held valid by a court of competent jurisdiction. Whether or not the outcome of litigation is favorable to us, defending a lawsuit takes significant time, may be expensive and may divert management attention from other business concerns. Adverse determinations in litigation or interference proceedings could require us to seek licenses which may not be available on commercially reasonable terms, or at all, or subject us to significant liabilities to third parties. If we need but cannot obtain a license, we may be prevented from marketing the affected product.
MARKETING AND SALES
We have developed capabilities in various aspects of our commercial organization through our commercialization of Gralise®, CAMBIA®, Zipsor® and Lazanda®, including sales, marketing, manufacturing, quality assurance, wholesale distribution, medical affairs, managed market contracting, government price reporting, compliance, maintenance of the product NDA and review, and submission of promotional materials. Members of our commercial organization are also engaged in the commercial and marketing assessments of other potential product candidates.
Our sales organization includes 188 full-time sales representatives. If we consummate the NUCYNTA® Acquisition, we expect to significantly increase the number of sales representatives. Our sales force primarily calls on pain specialists, neurologists and primary care physicians throughout most of the United States. Our marketing organization is comprised of professionals who have developed a
13
variety of marketing techniques and programs to promote our products, including promotional materials, speaker programs, industry publications, advertising and other media.
MANUFACTURING
Our facility is used for office and research and development (R&D) purposes. No commercial manufacturing takes place at our facility. The R&D work includes preclinical development of pharmaceutical formulations, their characterization, and the development of pharmaceutical processes for external commercial manufacturing. The total laboratory area includes the following individual labs: Analytical Development Lab, Formulation Dry Lab, Process Lab, and Quality Lab.
We are responsible for the supply and distribution of our marketed products. For Gralise®, we have entered into a manufacturing agreement with Patheon, as our sole commercial supplier. MiPharm, S.p.A. (MiPharm) is our sole supplier for CAMBIA® pursuant to a manufacturing and supply agreement that we assumed in connection with our December 2013 acquisition of CAMBIA®. Accucaps Industries Limited (Accucaps) is our sole supplier for Zipsor® pursuant to a manufacturing agreement we assumed in connection with our acquisition of Zipsor® from Xanodyne in June 2012. DPT Lakewood, Inc. (DPT) is our sole supplier for Lazanda® pursuant to a manufacturing and supply agreement that we assumed in connection with our July 2013 acquisition of Lazanda®. If we consumate the NUCYNTA® Acquisition, Janssen Pharma and its affiliates will be our sole supplier of NUCYNTA® and NUCYNTA® ER.
We have one qualified supplier for the active pharmaceutical ingredient in each of Gralise®, CAMBIA®, Zipsor® and Lazanda®. If we consummate the NUCYNTA® Acquisition, Janssen Pharma and its affiliates will be our sole supplier for the active pharmaceutical ingredient in NUCYNTA®. We have a supply agreement with the suppliers of the active pharmaceutical ingredient for Gralise®, CAMBIA® and Zipsor®. We also obtain polyethylene oxide, one of the excipients common to Gralise® and products under development by our partners, on a purchase order basis from Dow Chemical, our sole source for polyethylene oxide. We currently have no long-term supply arrangement with respect to polyethylene oxide.
Applicable current Good Manufacturing Practices (cGMP) requirements and other rules and regulations prescribed by foreign regulatory authorities apply to the manufacture of our products, including products using the Acuform technology. We depend on the manufacturers of our products to comply with cGMP and applicable foreign standards. Any failure by a manufacturer to maintain cGMP or comply with applicable foreign standards could delay or prevent the initial or continued commercial sale of our products and the products being sold or developed by parties with whom we have license or development agreements.
COMPETITION
General. We believe that we compete favorably in the markets described above on the basis of the safety and efficacy of our products, and in some cases on the basis of the price of our products. However, competition in pharmaceutical products and drug delivery technologies is intense, and we expect competition to increase. There may be other companies developing products competitive with ours of which we are unaware. Competing product or technologies developed in the future may prove superior to our products or technologies, either generally or in particular market segments. These developments could make our products or technologies noncompetitive or obsolete.
Most of our principal competitors have substantially greater financial, sales, marketing, personnel and research and development resources than we do. In addition, many of our potential collaborative partners have devoted, and continue to devote, significant resources to the development of their own products and drug delivery technologies.
14
Gralise® for Postherpetic Neuralgia. Gabapentin is currently marketed by Pfizer Inc. (Pfizer) as Neurontin and by several generic manufacturers for adjunctive therapy for epileptic seizures and for postherpetic pain. In addition, Pfizer's product Lyrica® (pregabalin) has been approved for marketing in the United States and the European Union for the management of PHN, diabetic nerve pain, spinal cord injury nerve pain, fibromyalgia, and for therapy in partial onset seizures. In December 2014, Pfizer announced positive Phase 3 clinical trial results for its controlled release formulation of Lyrica® as a treatment for PHN. Gralise® competes against these products and other neuropathic pain treatments, such as anti-depressants, anti-convulsants, local anesthetics used as regional nerve blockers, anti-arrythmics and opioids.
CAMBIA® for the Acute Treatment of Migraine Attacks. Diclofenac, the active pharmaceutical ingredient in CAMBIA®, is a NSAID approved in the United States for the acute treatment of migraine in adults. CAMBIA® competes with a number of triptans which are used to treat migraine and certain other headaches. Currently, seven triptans are available and sold in the United States (almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan and zolmitriptan), as well as a fixed-dose combination product containing sumatriptan plus naproxen. There are other products prescribed for or under development for the treatment of migraines which are now or may become competitive with CAMBIA®.
Zipsor® for Mild to Moderate Pain. Diclofenac, the active pharmaceutical ingredient in Zipsor®, is a NSAID that is approved in the United States for the treatment of mild to moderate pain and inflammation, including the symptoms of arthritis. Both branded and generic versions of diclofenac are marketed in the United States. Zipsor® competes against other drugs that are widely used to treat mild to moderate acute pain. In addition, a number of other companies are developing NSAIDs in a variety of dosage forms for the treatment of mild to moderate pain and related indications. Other drugs are in clinical development to treat acute pain.
Lazanda® for the Management of Breakthrough Pain in Cancer Patients. Lazanda® (fentanyl) nasal spray is an intranasal fentanyl drug used to manage breakthrough pain in adults (18 years of age or older) who are already routinely taking other opioid pain medicines around-the-clock for cancer pain. Fentanyl, an opioid analgesic, is currently sold by a number of companies for the treatment of breakthrough pain in opioid-tolerant cancer patients. Branded fentanyl products against which Lazanda® currently competes include Subsys®, which is sold by Insys Therapeutics, Inc. (Insys), Fentora® and Actiq®, which are sold by Cephalon, Inc. (Cephalon), Abstral®, which is sold by Galena Biopharma, Inc. (Galena) and Onsolis®, which is sold by BioDelivery Sciences International, Inc. (BDSI). Generic fentanyl products against which Lazanda® currently competes are sold by Mallinckrodt, Par Pharmaceutical Companies, Inc. (Par) and Actavis, Inc. (Actavis).
Drug Delivery Technologies. Other companies that have oral drug delivery technologies competitive with the Acuform technology include Alkermes plc, Bristol-Myers Squibb, Teva Pharmaceutical Industries Ltd., Johnson & Johnson, SkyePharma plc, Valeant, Flamel Technologies S.A., Ranbaxy Laboratories, Ltd., and Intec Pharma, all of which develop oral tablet products designed to release the incorporated drugs over time. Each of these companies has patented technologies with attributes different from ours, and in some cases with different sites of delivery to the gastrointestinal tract.
GOVERNMENT REGULATION
Product Development
Numerous governmental authorities in the United States and other countries regulate our research and development activities and those of our collaborative partners. Governmental approval is required of all potential pharmaceutical products using the Acuform technology and the manufacture and marketing of products using the Acuform technology prior to the commercial use of those products.
15
The regulatory process takes several years and requires substantial funds. If new products using the Acuform technology do not receive the required regulatory approvals or if such approvals are delayed, our business would be materially adversely affected. We cannot be certain that the requisite regulatory approvals will be obtained without lengthy delays, if at all.
In the United States, the FDA rigorously regulates pharmaceutical products, including any drugs using the Acuform technology. If a company fails to comply with applicable requirements, the FDA or the courts may impose sanctions. These sanctions may include civil penalties, criminal prosecution of the company or its officers and employees, injunctions, product seizure or detention, product recalls, and total or partial suspension of production. The FDA may withdraw approved applications or refuse to approve pending new drug applications, premarket approval applications, or supplements to approved applications.
We may be required to conduct preclinical testing on laboratory animals of new pharmaceutical products prior to commencement of clinical studies involving human beings. These studies evaluate the potential efficacy and safety of the product. If preclinical testing is required, we must submit the results of the studies to the FDA as part of an Investigational New Drug Application, which must become effective before beginning clinical testing in humans.
The products we develop generally are or will be submitted for approval under Section 505(b)(2) of the FDCA which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of a NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. For instance, the NDA for Gralise® relies on the FDA's prior approval of Neurontin® (gabapentin), the immediate release formulation of gabapentin initially approved by the FDA.
Typically, human clinical evaluation involves a time-consuming and costly three-phase process:
- •
- In Phase 1, we conduct clinical trials with a small number of subjects to determine a drug's early safety profile and its
pharmacokinetic pattern.
- •
- In Phase 2, we conduct limited clinical trials with groups of patients afflicted with a specific disease in order to determine
preliminary efficacy, optimal dosages and further evidence of safety.
- •
- In Phase 3, we conduct large-scale, multi-center, comparative trials with patients afflicted with a target disease in order to provide enough data to statistically evaluate the efficacy and safety of the product candidate, as required by the FDA.
The FDA closely monitors the progress of each phase of clinical testing. The FDA may, at its discretion, re-evaluate, alter, suspend or terminate testing based upon the data accumulated to that point and the FDA's assessment of the risk/benefit ratio to patients. The FDA may also require additional clinical trials after approvals, which are known as Phase 4 trials.
The results of preclinical and clinical testing are submitted to the FDA in the form of an NDA, for approval prior to commercialization. An NDA requires that our products are compliant with cGMP. Failure to achieve or maintain cGMP standards for our products would adversely impact their marketability. In responding to an NDA, the FDA may grant marketing approval, request additional information or deny the application. Failure to receive approval for any products using the Acuform technology would have a material adverse effect on us.
Foreign regulatory approval of a product must also be obtained prior to marketing the product internationally. Foreign approval procedures vary from country to country. The time required for approval may delay or prevent marketing in certain countries. In certain instances we or our collaborative partners may seek approval to market and sell certain products outside of the United
16
States before submitting an application for United States approval to the FDA. The clinical testing requirements and the time required to obtain foreign regulatory approvals may differ from that required for FDA approval. Although there is now a centralized European Union (EU) approval mechanism in place, each EU country may nonetheless impose its own procedures and requirements. Many of these procedures and requirements are time-consuming and expensive. Some EU countries require price approval as part of the regulatory process. These constraints can cause substantial delays in obtaining required approval from both the FDA and foreign regulatory authorities after the relevant applications are filed, and approval in any single country may not meaningfully indicate that another country will approve the product.
Reimbursement
Sales of pharmaceutical products in the United States depend in significant part on the extent of coverage and reimbursement from government programs, including Medicare and Medicaid, as well as other third party payers. Third party payers are undertaking efforts to control the cost of pharmaceutical products, including by implementing cost containment measures to control, restrict access to, or influence the purchase of drugs, and other health care products and services.
Government programs may regulate reimbursement, pricing, and coverage of products in order to control costs or to affect levels of use of certain products. Private health insurance plans may restrict coverage of some products, such as by using payer formularies under which only selected drugs are covered, variable co-payments that make drugs that are not preferred by the payer more expensive for patients, and by employing utilization management controls, such as requirements for prior authorization or prior failure on another type of treatment.
Fraud and Abuse
Pharmaceutical companies that participate in federal healthcare programs are subject to various U.S. federal and state laws pertaining to healthcare "fraud and abuse," including anti-kickback and false claims laws. Violations of U.S. federal and state fraud and abuse laws may be punishable by criminal or civil sanctions, including fines, civil monetary penalties and exclusion from federal healthcare programs (including Medicare and Medicaid).
Federal statutes that apply to us include the federal Anti-Kickback Statute, which prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration in exchange for or to generate business, including the purchase or prescription of a drug, that is reimbursable by a federal healthcare program such as Medicare and Medicaid, and the Federal False Claims Act (FCA), which generally prohibits knowingly and willingly presenting, or causing to be presented, for payment to the federal government any false, fraudulent or medically unnecessary claims for reimbursed drugs or services. Government enforcement agencies and private whistleblowers have asserted liability under the FCA for claims submitted involving inadequate care, kickbacks, improper promotion of off-label uses and misreporting of drug prices to federal agencies.
Similar state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payers, including private insurers. These state laws may be broader in scope than their federal analogues, such as state false claims laws that apply where a claim is submitted to any third-party payer, regardless of whether the payer is a private health insurer or a government healthcare program, and state laws that require pharmaceutical companies to certify compliance with the pharmaceutical industry's voluntary compliance guidelines.
Federal and state authorities have increased enforcement of fraud and abuse laws within the pharmaceutical industry, and private individuals have been active in alleging violations of the law and bringing suits on behalf of the government under the FCA. These laws are broad in scope and there may not be regulations, guidance, or court decisions that definitively interpret these laws and apply them to particular industry practices. In addition, these laws and their interpretations are subject to change.
17
Other U.S. Healthcare Laws
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the ACA) contains provisions that have or could potentially impact our business, including (a) an increase in the minimum Medicaid rebate to states participating in the Medicaid program on branded prescription drugs; (b) the extension of the Medicaid rebate to managed care organizations that dispense drugs to Medicaid beneficiaries; and (c) the expansion of the 340B Public Health Service Act drug pricing program, which provides outpatient drugs at reduced rates, to include certain children's hospitals, free standing cancer hospitals, critical access hospitals and rural referral centers.
Additionally, the federal Physician Payments Sunshine Act ("sunshine") provisions, enacted in 2010 as part of ACA, require pharmaceutical manufacturers, among others, to disclose annually to the federal government (for re-disclosure to the public) certain payments made to physicians and certain other healthcare practitioners or to teaching hospitals. State laws may also require disclosure of pharmaceutical pricing information and marketing expenditures and impose penalties for failures to disclose. Many of these laws and regulations contain ambiguous requirements. As a result of the ambiguity in certain of these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent federal and state laws and regulations.
Our operations and business are subject to a number of other laws and regulations, including those relating to the workplace, privacy, laboratory practices and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances as well as controlled substances. In addition, state laws may also govern the privacy and security of health information in some circumstances and may contain different or broader privacy protections than the federal provisions.
EMPLOYEES
As of December 31, 2014, we had 324 full-time employees. None of our employees are represented by a collective bargaining agreement, nor have we experienced any work stoppage. We believe that our relations with our employees are good.
In addition to other information in this report, the following factors should be considered carefully in evaluating an investment in our securities. If any of the risks or uncertainties described in this Form 10-K actually occurs, our business, results of operations or financial condition would be materially and adversely affected. The risks and uncertainties described in this Form 10-K are not the only ones facing us. Additional risks and uncertainties of which we are unaware or that we currently deem immaterial may also become important factors that may harm our business, results of operations and financial condition.
If we do not successfully commercialize Gralise®, CAMBIA®, Zipsor®, Lazanda® and NUCYNTA® (if we consummate the NUCYNTA® Acquisition) our business will suffer.
In October 2011, we began commercial sales of Gralise®. In June 2012, we acquired Zipsor and began commercial promotion of Zipsor® in July 2012. In July 2013, we acquired Lazanda® and began commercial promotion of Lazanda® in October 2013. In December 2013, we acquired CAMBIA® and began commercial promotion of CAMBIA® in February 2014. In January 2015, we entered into an agreement with Janssen Pharma to acquire the United States rights to the NUCYNTA® franchise of pharmaceutical products. As a Company, we have a limited history of selling and marketing pharmaceutical products. In addition to the risks discussed elsewhere in this section, our ability to
18
successfully commercialize and generate revenues from Gralise®, CAMBIA®, Zipsor®, Lazanda® and NUCYNTA® (if we consummate the NUCYNTA® Acquisition) depend on a number of factors, including, but not limited to, our ability to:
- •
- develop and execute our sales and marketing strategies for our products;
- •
- achieve market acceptance of our products;
- •
- obtain and maintain adequate coverage, reimbursement and pricing from managed care, government and other third-party payers;
- •
- maintain, manage or scale the necessary sales, marketing, manufacturing, managed markets, and other capabilities and infrastructure
that are required to successfully commercialize our products;
- •
- maintain intellectual property protection for our products; and
- •
- comply with applicable legal and regulatory requirements.
If we are unable to successfully achieve or perform these functions, we will not be able to maintain or increase our revenues from Gralise®, CAMBIA®, Zipsor®, Lazanda® and NUCYNTA® (if we consummate the NUCYNTA® Acquisition) our business will suffer.
If generic manufacturers use litigation and regulatory means to obtain approval for generic versions of our products, our business will suffer.
Under the Federal Food, Drug and Cosmetics Act (FDCA), the FDA can approve an Abbreviated New Drug Application (ANDA) for a generic version of a branded drug without the ANDA applicant undertaking the clinical testing necessary to obtain approval to market a new drug. In place of such clinical studies, an ANDA applicant usually needs only to submit data demonstrating that its product has the same active ingredient(s) and is bioequivalent to the branded product, in addition to any data necessary to establish that any difference in strength, dosage, form, inactive ingredients or delivery mechanism does not result in different safety or efficacy profiles, as compared to the reference drug.
The FDCA requires an applicant for a drug that relies, at least in part, on the patent of one of our branded drugs to notify us of their application and potential infringement of our patent rights. Upon receipt of this notice we have 45 days to bring a patent infringement suit in federal district court against the company seeking approval of a product covered by one of our patents. The discovery, trial and appeals process in such suits can take several years. If such a suit is commenced, the FDCA provides a 30-month stay on the FDA's approval of the competitor's application. Such litigation is often time-consuming and quite costly and may result in generic competition if the patents at issue are not upheld or if the generic competitor is found not to infringe such patents. If the litigation is resolved in favor of the applicant or the challenged patent expires during the 30-month stay period, the stay is lifted and the FDA may thereafter approve the application based on the standards for approval of ANDAs.
As described in greater detail under "LEGAL PROCEEDINGS" below, in August 2014, we received a favorable ruling in our patent litigation against Actavis. The lawsuit was filed in March 2012 against Actavis for infringement of certain U.S. patents listed in the Patent and Exclusivity Information Addendum of FDA's publication, Approved Drug Products with Therapeutic Equivalence Evaluations (commonly known as the Orange Book) for Gralise®. A bench trial was completed on May 20, 2014 and in August 2014 the court ruled in our favor, finding that Actavis infringed all asserted claims of all seven patents asserted in trial and upholding the validity of the patents, which expire between September 2016 and February 2024. On September 15, 2014, Actavis filed a notice appealing the decision to the United States Court of Appeals for the Federal Circuit. On February 2, 2015, Actavis filed its opening brief with the United States Court of Appeals for the Federal Circuit. A successful
19
appeal of the trial court's ruling by Actavis would harm our business, financial condition and results of operations.
On June 28, 2013, we received from Banner Pharmacaps Inc. (Banner) a Notice of Certification for U.S. Patent Nos. 6,365,180; 7,662,858; 7,884,095; 7,939,518 and 8,110,606 under 21 U.S.C. § 355 (j)(2)(A)(vii)(IV) (Zipsor® Paragraph IV Letter) certifying that Banner has submitted and the FDA has accepted for filing an ANDA for 25mg diclofenac potassium capsules, (Banner ANDA Product). Banner has granted exclusive rights to the Banner ANDA Product to Watson Laboratories Inc., a subsidiary of Actavis plc. The letter states that the Banner ANDA Product contains the required bioavailability or bioequivalence data to Zipsor® and certifies that Banner intends to obtain FDA approval to engage in commercial manufacture, use or sale of Banner's ANDA product before the expiration of the above identified patents, which are listed for Zipsor® in the Orange Book. We commenced the lawsuit within the 45 days required to automatically bar the FDA from approving the Banner ANDA Product for 30 months or until a district court decision that is adverse to the asserted patents, whichever may occur earlier. Absent a court order, the 30-month stay is expected to expire in December 2015.
Several ANDAs have been filed with respect to NUCYNTA®. Janssen Pharma and Grünenthal have timely commenced lawsuits against each ANDA filer for infringing certain patents licensed by Janssen Pharma from Grünenthal which are listed for NUCYNTA® in the Orange Book. We will assume responsibility for these lawsuits if we consummate the NUCYNTA® Acquisition.
Any introduction of one or more products generic to Gralise®, CAMBIA®, Zipsor® or Lazanda®, and if we consummate the NUCYNTA® Acquisition, to NUCYNTA® , whether as a result of an ANDA or otherwise, would harm our business, financial condition and results of operations. The filing of the ANDAs described above, or any other ANDA or similar application in respect to any of our products, or the successful appeal of the favorable ruling received in the Gralise® litigation, could have an adverse impact on our stock price. Moreover, if the patents covering our products were not upheld in litigation or if a generic competitor is found not to infringe these patents, the resulting generic competition would have a material adverse effect on our business, financial condition, and results of operations.
If we are unable to negotiate acceptable pricing or obtain adequate reimbursement for our products from third-party payers, our business will suffer.
Sales of our products will depend in part on the availability of acceptable pricing and adequate reimbursement from third-party payers such as:
- •
- government health administration authorities;
- •
- private health insurers;
- •
- health maintenance organizations;
- •
- managed care organizations;
- •
- pharmacy benefit management companies; and
- •
- other healthcare-related organizations.
If reimbursement is not available for our products or product candidates, demand for these products may be limited. Further, any delay in receiving approval for reimbursement from third-party payers could have an adverse effect on our future revenues. Third-party payers are increasingly challenging the price and cost-effectiveness of medical products and services, including pharmaceuticals. Significant uncertainty exists as to the reimbursement status of pharmaceutical products. Our products may not be considered cost effective, and adequate third-party reimbursement may be unavailable to
20
enable us to maintain price levels sufficient to realize an acceptable return on our investment. Any third-party payer decision not to approve pricing for, or provide adequate coverage and reimbursement of, our products, including by limiting or denying reimbursement for new products or excluding products that were previously eligible for reimbursement, would limit the market acceptance and commercial prospects of our products and harm our business, financial condition, and results of operations.
Federal and state governments in the United States continue to propose and pass new legislation, such as the ACA, which is designed to contain or reduce the cost of healthcare. Cost control initiatives could decrease the price that we receive for our products and any product that we may develop or acquire.
We may be unable to compete successfully in the pharmaceutical industry.
Gabapentin is currently sold by Pfizer Inc. as Neurontin® for adjunctive therapy for epileptic seizures and for PHN. Pfizer's basic U.S. patents relating to Neurontin® have expired, and numerous companies have received approval to market generic versions of the immediate release product. Pfizer has also developed Lyrica® (pregabalin), which has been approved for marketing in the United States for post herpetic pain, fibromyalgia, diabetic nerve pain, adjunctive therapy, epileptic seizures, and nerve pain associated with spinal cord injury and has captured a significant portion of the market. In December 2014, Pfizer announced positive Phase 3 clinical trial results for its controlled release formulation of Lyrica® as a treatment for PHN. In June 2012, GlaxoSmithKline and Xenoport, Inc. received approval to market Horizant™ (gabapentin enacarbil extended-release tablets) for the management of PHN. There are other products prescribed for or under development for PHN which are now or may become competitive with Gralise®.
Diclofenac, the active pharmaceutical ingredient in Zipsor®, is an NSAID that is approved in the United States for the treatment of mild to moderate pain in adults, including the symptoms of arthritis. Both branded and generic versions of diclofenac are marketed in the U.S. Zipsor® competes against other drugs that are widely used to treat mild to moderate pain in the acute setting. In addition, a number of other companies are developing NSAIDs in a variety of dosage forms for the treatment of mild to moderate pain and related indications. Other drugs are in clinical development to treat acute pain.
An alternate formulation of diclofenac is the active ingredient in CAMBIA® that is approved in the United States for the acute treatment of migraine in adults. CAMBIA® competes with a number of triptans which are used to treat migraine and certain other headaches. Currently, seven triptans are available and sold in the United States (almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan and zolmitriptan), as well as a fixed-dose combination product containing sumatriptan plus naproxen. There are other products prescribed for or under development for the treatment of migraines which are now or may become competitive with CAMBIA®.
Fentanyl, an opioid analgesic, is currently sold by a number of companies for the treatment of breakthrough pain in opioid-tolerant cancer patients. Branded fentanyl products against which Lazanda® currently competes include Subsys®, which is sold by Insys Therapeutics, Inc., Fentora® and Actiq®, which are sold by Cephalon Inc., Abstral®, which is sold by Galena Biophama Inc., and Onsolis®, which is sold by BioDelivery Sciences International, Inc. (BDSI). Generic fentanyl products against which Lazanda® currently competes are sold by Mallinckrodt, Par and Actavis.
Competition in the pharmaceutical industry is intense and we expect competition to increase. If we consummate the NUCYNTA® Acquisition, we will face significant competition from a number of pharmaceutical products competitive with NUCYNTA®. Competing products currently under development or developed in the future may prove superior to our products and achieve greater
21
commercial acceptance. Most of our principal competitors have substantially greater financial, sales, marketing, personnel and research and development resources than we do.
We may incur significant liability if it is determined that we are promoting or have in the past promoted the "off-label" use of drugs.
Companies may not promote drugs for "off-label" use—that is, uses that are not described in the product's labeling and that differ from those approved by the FDA. Physicians may prescribe drug products for off-label uses, and such off-label uses are common across some medical specialties. Although the FDA and other regulatory agencies do not regulate a physician's choice of treatments, the FDCA and FDA regulations restrict communications on the subject of off-label uses of drug products by pharmaceutical companies. The Office of Inspector General of the Department of Health and Human Services (OIG), the FDA, and the Department of Justice (DOJ) all actively enforce laws and regulations prohibiting promotion of off-label use and the promotion of products for which marketing clearance has not been obtained. If the OIG or the FDA takes the position that we are or may be out of compliance with the requirements and restrictions described above, and we are investigated for or found to have improperly promoted off-label use, we may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions. In addition, management's attention could be diverted from our business operations and our reputation could be damaged.
Pharmaceutical marketing is subject to substantial regulation in the United States and any failure by us or our collaborative partners to comply with applicable statutes or regulations could adversely affect our business.
All marketing activities associated with Gralise®, Zipsor®, Lazanda® and CAMBIA®, as well as marketing activities related to any other products which we may acquire, such as NUCYNTA® , or for which we obtain regulatory approval, will be subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products. The FDA regulates post-approval promotional labeling and advertising to ensure that they conform to statutory and regulatory requirements. In addition to FDA restrictions, the marketing of prescription drugs is subject to laws and regulations prohibiting fraud and abuse under government healthcare programs. For example, the federal healthcare program anti-kickback statute prohibits giving things of value to induce the prescribing or purchase of products that are reimbursed by federal healthcare programs, such as Medicare and Medicaid. In addition, federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government. Under this law, in recent years, the federal government has brought claims against drug manufacturers alleging that certain marketing activities caused false claims for prescription drugs to be submitted to federal programs. Many states have similar statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, and, in some states, such statutes or regulations apply regardless of the payer. If we, or our collaborative partners, fail to comply with applicable FDA regulations or other laws or regulations relating to the marketing of our products, we could be subject to criminal prosecution, civil penalties, seizure of products, injunctions, and exclusion of our products from reimbursement under government programs, as well as other regulatory actions against our product candidates, our collaborative partners or us.
Acquisition of new and complementary businesses, products, and technologies is a key element of our corporate strategy. If we are unable to successfully identify and acquire such businesses, products or technologies, our business and prospects will be limited.
Since June 2012, we have acquired Zipsor®, Lazanda® and CAMBIA®. In January 2015, we entered into an agreement with Janssen Pharma to acquire the United States rights to the NUCYNTA® franchise of pharmaceutical products. An important element of our business strategy is to
22
actively seek to acquire products or companies and to in-license or seek co-promotion rights to products that could be sold by our sales force. We cannot be certain that we will be able to successfully pursue and complete any further acquisitions, including the NUCYNTA® Acquisition, or whether we would be able to successfully integrate any acquired business, product or technology or retain any key employees. Integrating any business, product or technology we may acquire, including NUCYNTA®, could be expensive and time consuming, disrupt our ongoing business, and distract our management. If we are unable to enhance and broaden our product offerings, unable to effectively integrate any acquired businesses, products or technologies, or achieve the anticipated benefits of any acquired business, product or technology, our business and prospects will be limited. In addition, any amortization or charges resulting from the costs of such acquisitions will adversely affect our results of operations.
If we engage in strategic transactions that fail to achieve the anticipated results and synergies, our business will suffer.
We may seek to engage in strategic transactions with third parties, such as company acquisitions, strategic partnerships, joint ventures, divestitures or business combinations. We may face significant competition in seeking potential strategic partners and transactions, and the negotiation process for acquiring any product or engaging in strategic transactions can be time-consuming and complex. Engaging in strategic transactions, including the NUCYNTA® Acquisition, may require us to incur non-recurring and other charges, increase our near and long-term expenditures, pose integration challenges and fail to achieve the anticipated results or synergies or distract our management and business, which may harm our business.
As part of an effort to acquire a product or company or to enter into other strategic transactions, we conduct business, legal and financial due diligence with the goal of identifying, evaluating and assessing material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining, evaluating and accurately assessing all such risks and, as a result, might not realize the intended advantages of the transaction. We may also assume liabilities and legal risks in connection with a transaction, including those relating to activities of the seller prior to the consummation of the transaction and contracts that we assume. Failure to realize the expected benefits from acquisitions or strategic transactions that we may consummate, including the NUCYNTA® Acquisition, whether as a result of identified or unidentified risks, integration difficulties, regulatory setbacks, governmental investigations, litigation or other events, could adversely affect our business, results of operations and financial condition.
We depend on third parties that are single source suppliers to manufacture our products. If these suppliers are unable to manufacture and supply our products, our business will suffer.
Patheon Puerto Rico Inc. is our sole supplier for Gralise® pursuant to a manufacturing and supply agreement we entered into with Patheon in September 2011. Accucaps Industries Limited is our sole supplier for Zipsor® pursuant to a manufacturing agreement we assumed in connection with our acquisition of Zipsor® in June 2012. DPT Lakewood Inc. is our sole supplier for Lazanda® pursuant to a manufacturing and supply agreement that we assumed in connection with our acquisition of Lazanda® in July 2013. MiPharm, S.p.A is our sole supplier for CAMBIA® pursuant to a manufacturing and supply agreement that we assumed in connection with our acquisition of CAMBIA® in December 2013. We have one qualified supplier for the active pharmaceutical ingredient in each of Gralise®, CAMBIA®, Zipsor® and Lazanda®. If we consummate the NUCYNTA® Acquisition, Janssen Pharma and its affiliates will be our sole supplier of both NUCYNTA® and tapentadol, the active pharmaceutical ingredient in NUCYNTA®. We do not have, and we do not intend to establish in the foreseeable future, internal commercial scale manufacturing capabilities. Rather, we intend to use the facilities of third parties to manufacture products for clinical trials and commercialization. Our
23
dependence on third parties for the manufacture of our products and our product candidates may adversely affect our ability to deliver such products on a timely or competitive basis, if at all. Any failure to obtain Gralise®, CAMBIA®, Zipsor®, Lazanda® or NUCYNTA® (if we consummate the NUCYNTA® Acquisition), or active pharmaceutical ingredients, excipients or components from our suppliers could adversely affect our business, results of operations and financial condition.
The manufacturing process for pharmaceutical products is highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations. We, our third-party manufacturers and our suppliers are subject to numerous regulations, including current FDA regulations governing manufacturing processes, stability testing, record keeping and quality standards. Similar regulations are in effect in other countries. Our third-party manufacturers and suppliers are independent entities who are subject to their own unique operational and financial risks which are out of our control. If we or any of these third-party manufacturers or suppliers fail to perform as required or fail to comply with the regulations of the FDA and other applicable governmental authorities, our ability to deliver our products on a timely basis or receive royalties or continue our clinical trials could be adversely affected. The manufacturing processes of our third party manufacturers and suppliers may also be found to violate the proprietary rights of others. To the extent these risks materialize and adversely affect such third-party manufacturers' performance obligations to us, and we are unable to contract for a sufficient supply of required products on acceptable terms, or if we encounter delays and difficulties in our relationships with manufacturers or suppliers, our business, results of operation and financial condition could be adversely affected.
We may be unable to protect our intellectual property and may be liable for infringing the intellectual property of others.
Our success will depend in part on our ability to obtain and maintain patent protection for our products and technologies, and to preserve our trade secrets. Our policy is to seek to protect our proprietary rights, by, among other methods, filing patent applications in the United States and foreign jurisdictions to cover certain aspects of our technology. We hold issued United States patents and have patent applications pending in the United States. In addition, we are pursuing patent applications relating to our technologies in the United States and abroad. We have also applied for patents in numerous foreign countries. Some of those countries have granted our applications and other applications are still pending. Our pending patent applications may lack priority over other applications or may not result in the issuance of patents. Even if issued, our patents may not be sufficiently broad to provide protection against competitors with similar technologies and may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products or may not provide us with competitive advantages against competing products. We also rely on trade secrets and proprietary know-how, which are difficult to protect. We seek to protect such information, in part, through entering into confidentiality agreements with employees, consultants, collaborative partners and others before such persons or entities have access to our proprietary trade secrets and know-how. These confidentiality agreements may not be effective in certain cases, due to, among other things, the lack of an adequate remedy for breach of an agreement or a finding that an agreement is unenforceable. In addition, our trade secrets may otherwise become known or be independently developed by competitors.
Our ability to develop our technologies and to make commercial sales of products using our technologies also depends on not infringing other patents or intellectual property rights. We are not aware of any such intellectual property claims against us. However, the pharmaceutical industry has experienced extensive litigation regarding patents and other intellectual property rights. Patents issued to third parties relating to sustained release drug formulations or particular pharmaceutical compounds could in the future be asserted against us, although we believe that we do not infringe any valid claim of any patents. If claims concerning any of our products were to arise and it was determined that these
24
products infringe a third party's proprietary rights, we could be subject to substantial damages for past infringement or be forced to stop or delay our activities with respect to any infringing product, unless we can obtain a license, or we may have to redesign our product so that it does not infringe upon such third party's patent rights, which may not be possible or could require substantial funds or time. Such a license may not be available on acceptable terms, or at all. Even if we, our collaborators or our licensors were able to obtain a license, the rights may be nonexclusive, which could give our competitors access to the same intellectual property. In addition, any public announcements related to litigation or interference proceedings initiated or threatened against us, even if such claims are without merit, could cause our stock price to decline.
From time to time, we may become aware of activities by third parties that may infringe our patents. Infringement by others of our patents may reduce our market shares (if a related product is approved) and, consequently, our potential future revenues and adversely affect our patent rights if we do not take appropriate enforcement action. We may need to engage in litigation to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights. For instance, we are engaged in litigation against one Zipsor® ANDA filer. If we consummate the NUCYNTA® Acquisition, we will assume responsibility for the pending litigation against the NUCYNTA® ANDA filers. Also, in January 2013 and April 2013, we filed lawsuits against Purdue and Endo, respectively, for infringement of certain of our Acuform drug delivery technology patents. In response to our lawsuits, Purdue and Endo are challenging the validity of the patents we asserted in inter partes review proceedings before the United States Patent Trial and Appeal Board (PTAB) at the United States Patent and Trademark Office. In these or other proceedings, our issued or licensed patents may not be held valid by a court of competent jurisdiction or the PTAB. Whether or not the outcome of litigation or the PTAB proceeding is favorable to us, the litigation and the proceedings takes significant time, may be expensive, and may divert management attention from other business concerns. We may also be required to participate in derivation proceedings or other post-grant proceedings declared by the United States Patent and Trademark Office for the purposes of, respectively, determining the priority of inventions in connection with our patent applications or determining validity of claims in our issued patents. Adverse determinations in litigation or proceedings at the United States Patent and Trademark Office would adversely affect our business, results of operations and financial condition and could require us to seek licenses which may not be available on commercially reasonable terms, or at all, or subject us to significant liabilities to third parties. If we need but cannot obtain a license, we may be prevented from marketing the affected product.
Health care reform could increase our expenses and adversely affect the commercial success of our products.
The ACA includes numerous provisions that affect pharmaceutical companies, some of which became effective immediately upon President Obama signing the law, and others of which are scheduled to take effect over the next several years. For example, the ACA seeks to expand healthcare coverage to the uninsured through private health insurance reforms and an expansion of Medicaid. The ACA also imposes substantial costs on pharmaceutical manufacturers, such as an increase in liability for rebates paid to Medicaid, new drug discounts that must be offered to certain enrollees in the Medicare prescription drug benefit and an annual fee imposed on all manufacturers of brand prescription drugs in the U.S. The ACA also requires increased disclosure obligations and an expansion of an existing program requiring pharmaceutical discounts to certain types of hospitals and federally subsidized clinics and contains cost-containment measures that could reduce reimbursement levels for pharmaceutical products. The ACA also includes provisions known as the Physician Payments Sunshine Act, which require manufacturers of drugs, biologics, devices and medical supplies covered under Medicare and Medicaid to record any transfers of value to physicians and teaching hospitals and to report this data to the Centers for Medicare and Medicaid Services for subsequent public disclosure. Similar reporting requirements have also been enacted on the state level domestically, and an increasing number of countries worldwide either have adopted or are considering similar laws requiring transparency of
25
interactions with health care professionals. Failure to report appropriate data may result in civil or criminal fines and/or penalties. These and other aspects of the ACA, including the regulations that may be imposed in connection with the implementation of the ACA, could increase our expenses and adversely affect our ability to successfully commercialize our products and product candidates.
Changes in laws and regulations may adversely affect our business.
The manufacture, marketing, sale, promotion and distribution of our products are subject to comprehensive government regulation. Changes in laws and regulations applicable to the pharmaceutical industry could potentially affect our business. For example, federal, state and local governments have recently given increased attention to the public health issue of opioid abuse. At the federal level, the White House Office of National Drug Control Policy continues to coordinate efforts between the FDA, United States Drug Enforcement Agency (DEA) and other agencies to address this issue. The DEA continues to increase its efforts to hold manufacturers, distributors, prescribers and pharmacies accountable through various enforcement actions as well as the implementation of compliance practices for controlled substances. In addition, many state legislatures are considering various bills intended to reduce opioid abuse, for example by establishing prescription drug monitoring programs and mandating prescriber education. These and other changes in laws and regulations could adversely affect our business, financial condition and results of operations.
If we are unable to obtain or maintain regulatory approval, we will be limited in our ability to commercialize our products, and our business will suffer.
The regulatory process is expensive and time consuming. Even after investing significant time and expenditures on clinical trials, we may not obtain regulatory approval of our product candidates. Data obtained from clinical trials are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval, and the FDA may not agree with our methods of clinical data analysis or our conclusions regarding safety and/or efficacy. Significant clinical trial delays could impair our ability to commercialize our products and could allow our competitors to bring products to market before we do. In addition, changes in regulatory policy for product approval during the period of product development and regulatory agency review of each submitted new application may cause delays or rejections. Even if we receive regulatory approval, this approval may entail limitations on the indicated uses for which we can market a product.
Further, with respect to our approved products, once regulatory approval is obtained, a marketed product and its manufacturer are subject to continual review. The discovery of previously unknown problems with a product or manufacturer may result in restrictions on the product, manufacturer or manufacturing facility, including withdrawal of the product from the market. Manufacturers of approved products are also subject to ongoing regulation and inspection, including compliance with FDA regulations governing current Good Manufacturing Practices (cGMP) or Quality System Regulation (QSR). The FDCA, the Controlled Substances Act of 1970 (CSA) and other federal and foreign statutes and regulations govern and influence the testing, manufacturing, packing, labeling, storing, record keeping, safety, approval, advertising, promotion, sale and distribution of our products. In addition, with respect to Lazanda®, we and our partners are also subject to ongoing DEA regulatory obligations, including annual registration renewal, security, recordkeeping, theft and loss reporting, periodic inspection and annual quota allotments for the raw material for commercial production of our products. The failure to comply with these regulations could result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, non-renewal of marketing applications or authorizations or criminal prosecution, which could adversely affect our business, results of operations and financial condition.
We are also required to report adverse events associated with our products to the FDA and other regulatory authorities. Unexpected or serious health or safety concerns could result in labeling changes,
26
recalls, market withdrawals or other regulatory actions. Recalls may be issued at our discretion or at the discretion of the FDA or other empowered regulatory agencies. For example, in June 2010, we instituted a voluntary class 2 recall of 52 lots of our 500mg Glumetza product after chemical traces of 2,4,6-tribromoanisole (TBA) were found in the product bottle.
We are subject to risks associated with NDAs submitted under Section 505(b)(2) of the Food, Drug and Cosmetic Act.
The products we develop generally are or will be submitted for approval under Section 505(b)(2) of the FDCA, which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of a NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. For instance, the NDA for Gralise® relies on the FDA's prior approval of Neurontin, the immediate release formulation of gabapentin initially approved by the FDA.
For NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch-Waxman Act, such NDAs may be required to include certifications, known as "Paragraph IV certifications," that certify any patents listed in the FDA's Orange Book publication in respect to any product referenced in the 505(b)(2) application are invalid, unenforceable, and/or will not be infringed by the manufacture, use, or sale of the product that is the subject of the 505(b)(2) application. Under the Hatch-Waxman Act, the holder of the NDA which the 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the Paragraph IV certification. Filing of a patent infringement lawsuit triggers a one-time automatic 30-month stay of the FDA's ability to approve the 505(b)(2) application. Accordingly, we may invest a significant amount of time and expense in the development of one or more products only to be subject to significant delay and patent litigation before such products may be commercialized, if at all. A Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. The FDA may also require us to perform one or more additional clinical studies or measurements to support the change from the approved product. The FDA may then approve the new formulation for all or only some of the indications sought by us. The FDA may also reject our future Section 505(b)(2) submissions and require us to file such submissions under Section 501(b)(1) of the FDCA, which could be considerably more expensive and time consuming. These factors, among others, may limit our ability to successfully commercialize our product candidates.
If a product liability claim against us is successful, our business will suffer.
Our business involves exposure to potential product liability risks that are inherent in the development production and commercialization of pharmaceutical products. Side effects, manufacturing defects, misuse or abuse of our products could result in patient injury or death. For instance, Lazanda® is a self-administered, opioid analgesic that contains fentanyl, a Schedule II "controlled substance" under the CSA. A patient's failure to follow instructions on the use and administration of, or the abuse of Lazanda® could result in injury or death. In addition, patients using Lazanda® have been diagnosed with cancer, an often fatal disease. Patient injury or death can result in product liability claims being brought against us, even if our products did not cause an injury or death. Product liability claims may be brought against us by consumers, health care providers, pharmaceutical companies or others who come into contact with our products.
27
We have obtained product liability insurance for clinical trials currently underway and forecasted 2015 sales of our products, but:
- •
- we may be unable to maintain product liability insurance on acceptable terms;
- •
- we may be unable to obtain product liability insurance for future trials;
- •
- we may be unable to obtain product liability insurance for future products;
- •
- we may be unable to secure increased coverage as the commercialization of our Acuform gastric retentive technology expands; or
- •
- our insurance may not provide adequate protection against potential liabilities.
Our inability to obtain or maintain adequate insurance coverage at an acceptable cost could prevent or inhibit the commercialization of our products. Defending a law suit could be costly and significantly divert management's attention from conducting our business. If third parties were to bring a successful product liability claim or series of claims against us for uninsured liabilities or in excess of insured liability limits, our business, results of operations and financial condition could be adversely affected.
If we consummate the NUCYNTA® Acquisition, the foregoing risks will also apply to tapentadol, a Schedule II substance and the active pharmaceutical ingredient in NUCYNTA®.
Our collaborative arrangements may give rise to disputes over commercial terms, contract interpretation and ownership or protection of our intellectual property and may adversely affect the commercial success of our products.
We currently have collaboration or license arrangements with a number of companies, including Mallinckrodt, Janssen Pharma, Salix and Ironwood. In addition, we have in the past and may in the future enter into other collaborative arrangements, some of which have been based on less definitive agreements, such as memoranda of understanding, material transfer agreements, options or feasibility agreements. We may not execute definitive agreements formalizing these arrangements.
Collaborative relationships are generally complex and may give rise to disputes regarding the relative rights, obligations and revenues of the parties, including the ownership of intellectual property and associated rights and obligations, especially when the applicable collaborative provisions have not been fully negotiated and documented. Such disputes can delay collaborative research, development or commercialization of potential products, and can lead to lengthy, expensive litigation or arbitration. The terms of collaborative arrangements may also limit or preclude us from developing products or technologies developed pursuant to such collaborations. Additionally, the collaborative partners under these arrangements might breach the terms of their respective agreements or fail to maintain, protect or prevent infringement of the licensed patents or our other intellectual property rights by third parties. Moreover, negotiating collaborative arrangements often takes considerably longer to conclude than the parties initially anticipate, which could cause us to enter into less favorable agreement terms that delay or defer recovery of our development costs and reduce the funding available to support key programs. Any failure by our collaborative partners to abide by the terms of their respective agreements with us, including their failure to accurately calculate, report or pay any royalties payable to us, may adversely affect our results of operations.
We may be unable to enter into future collaborative arrangements on acceptable terms, which could harm our ability to develop and commercialize our current and potential future products and technologies. Other factors relating to collaborations that may adversely affect the commercial success of our products include:
- •
- any parallel development by a collaborative partner of competitive technologies or products;
28
- •
- arrangements with collaborative partners that limit or preclude us from developing products or technologies;
- •
- premature termination of a collaboration agreement; or
- •
- failure by a collaborative partner to devote sufficient resources to the development and commercial sales of products using our current and potential future products and technologies.
Our collaborative arrangements do not necessarily restrict our collaborative partners from competing with us or restrict their ability to market or sell competitive products. Our current and any future collaborative partners may pursue existing or other development-stage products or alternative technologies in preference to those being developed in collaboration with us. Our collaborative partners may also terminate their collaborative relationships with us or otherwise decide not to proceed with development and commercialization of our products.
Any failure by us or our partners to comply with applicable statutes or regulations relating to controlled substances could adversely affect our business.
Lazanda® is an opioid analgesic that contains fentanyl, a regulated "controlled substance" under the CSA. The CSA establishes, among other things, certain registration, production quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. The DEA regulates controlled substances as Schedule I, II, III, IV or V substances, with Schedule II substances being those that present the highest risk of abuse. Fentanyl is listed by the DEA as a Schedule II substance under the CSA. The manufacture, shipment, storage, sale and use, among other things, of controlled substances that are pharmaceutical products are subject to a high degree of regulation. For example, generally all Schedule II substance prescriptions, such as prescriptions for fentanyl, must be written and signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription.
The DEA also conducts periodic inspections of certain registered establishments that handle controlled substances. Facilities that conduct research, manufacture, distribute, import or export controlled substances must be registered to perform these activities and have the security, control and inventory mechanisms required by the DEA to prevent drug loss and diversion. Failure to maintain compliance, particularly non-compliance resulting in loss or diversion, can result in regulatory action that could adversely affect our business, results of operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to restrict, suspend or revoke those registrations and in certain circumstances, violations could lead to criminal proceedings against us or our manufacturing and distribution partners, and our respective employees, officers and directors.
In addition to federal regulations, many individual states also have controlled substances laws. Although state controlled substances laws generally mirror federal law, because the states are separate jurisdictions they may separately schedule our products. Any failure by us or our partners to obtain separate state registrations, permits, or licenses in order to be able to obtain, handle, and distribute fentanyl or to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law and would adversely affect our business, results of operations and financial condition.
If we consummate the NUCYNTA® Acquisition, the foregoing risks will apply to tapentadol, which is a Schedule II substance under the CSA and the active pharmaceutical ingredient in NUCYNTA®.
29
Limitations on the production of Schedule II substances in the United States could limit our ability to successfully commercialize Lazanda® and NUCYNTA® (if we consummate the NUCYNTA® Acquisition).
The availability and production of all Schedule II substances, including fentanyl, is limited by the DEA through a quota system that includes a national aggregate quota, production quotas for individual manufacturers and procurement quotas that authorize the procurement of specific quantities of Schedule II controlled substances for use in drug manufacturing. The DEA annually establishes an aggregate quota for total fentanyl production in the United States based on the DEA's estimate of the quantity needed to meet commercial and scientific need. The aggregate quota of fentanyl that the DEA allows to be produced in the United States annually is allocated among individual fentanyl drug manufacturers, which must submit applications annually to the DEA for individual production quotas. In turn, the manufacturer of Lazanda® has to obtain a procurement quota to source fentanyl for the production of Lazanda®. The DEA requires substantial evidence and documentation of expected legitimate medical and scientific needs before assigning quotas for these activities. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Based on a variety of factors, including public policy considerations, the DEA may set the aggregate fentanyl quota lower than the total amount requested by individual manufacturers. Although through our manufacturing partner we are permitted to ask the DEA to increase our manufacturer's procurement quota after it is initially established, we cannot be certain that the DEA would act favorably upon such a request. In addition, our manufacturer obtains a procurement quota for fentanyl for all fentanyl products manufactured at their facility, which is allocated to Lazanda® at the manufacturer's discretion. If the available quota of fentanyl is insufficient to meet our commercial demand or clinical needs, our business, results of operations and financial condition could be adversely affected. In addition, any delay or refusal by the DEA or our manufacturer in establishing the production or procurement quota or any reduction by the DEA or our manufacturer in the allocated quota for fentanyl could adversely affect our business, results of operations and financial condition.
If we consummate the NUCYNTA® Acquisition, the foregoing risks will also apply to tapentadol, a Schedule II substance and the active pharmaceutical ingredient in NUCYNTA®.
The FDA-mandated Risk Evaluation and Mitigation Strategy program may limit the commercial success of Lazanda®.
Lazanda® is subject to a FDA-mandated Risk Evaluation and Mitigation Strategy (REMS) protocol that requires enrollment and participation in the REMS program to prescribe, dispense or distribute Transmucosal Immediate Release Fentanyl (TIRF) medicines, including Lazanda®, for outpatient use. Many physicians, health care practitioners and pharmacies are unwilling to enroll and participate in the TIRF REMS program. As a result, there are relatively few prescribers and dispensers of TIRF products. If we are not able to successfully promote Lazanda® to participants in the TIRF REMS program, our business, results of operations and financial condition could be adversely affected.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays outside of our control.
We rely on clinical investigators and clinical sites to enroll patients and other third parties to manage our trials and to perform related data collection and analysis. However, we may be unable to control the amount and timing of resources that the clinical sites that conduct the clinical testing may devote to our clinical trials. If our clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to enroll them on our planned schedule, we will be unable to complete these trials or to complete them as planned, which could delay or prevent us from obtaining regulatory approvals for our product candidates.
30
Our agreements with clinical investigators and clinical sites for clinical testing and for trial management services place substantial responsibilities on these parties, which could result in delays in, or termination of, our clinical trials if these parties fail to perform as expected. For example, if any of our clinical trial sites fail to comply with FDA-approved good clinical practices, we may be unable to use the data gathered at those sites. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our product candidates.
The development of drug candidates is inherently difficult and uncertain and we cannot be certain that any of our product candidates or those of our collaborative partners will be approved for marketing or, if approved, will achieve market acceptance.
Clinical development is a long, expensive and uncertain process and is subject to delays and failures. Our own product candidates and those of our collaborative partners are subject to the risk that any or all of them may be found to be ineffective or unsafe, or otherwise may fail to receive necessary regulatory clearances. Positive or encouraging results of prior clinical trials are not necessarily indicative of the results obtained in later clinical trials, as was the case with the Phase 3 trial for Gralise® for the management of PHN that we completed in 2007, and with the Phase 3 trials evaluating Sefelsa, our prior product candidate, for menopausal hot flashes, the last of which we completed in October 2011. In addition, data obtained from pivotal clinical trials are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval.
Other factors could delay or result in termination of our clinical trials, including:
- •
- negative or inconclusive results;
- •
- patient noncompliance with the protocol;
- •
- adverse medical events or side effects among patients during the clinical trials;
- •
- FDA inspections of our clinical operations; and
- •
- actual or perceived lack of efficacy or safety of the product candidate.
We are unable to predict whether any of our product candidates or those of our collaborative partners will receive regulatory clearances or be successfully manufactured or marketed. Further, due to the extended testing and regulatory review process required before marketing clearance can be obtained, the time frame for commercializing a product is long and uncertain. Even if these other product candidates receive regulatory clearance, our products may not achieve or maintain market acceptance. Also, DM-1992 uses the Acuform technology. If it is discovered that the Acuform technology could have adverse effects or other characteristics that indicate it is unlikely to be effective as a delivery system for drugs or therapeutics, our product development efforts and our business could be significantly harmed.
Even assuming our products obtain regulatory approval, successful commercialization requires:
- •
- market acceptance;
- •
- cost-effective commercial scale production; and
- •
- reimbursement under private or governmental health plans.
Any material delay or failure in the governmental approval process and/or the successful commercialization of our potential products or those of our collaborative partners could adversely impact our financial position and liquidity.
31
The market price of our common stock historically has been volatile. Our results of operations may fluctuate and affect our stock price.
The trading price of our common stock has been, and is likely to continue to be, volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. From June 30, 2013 through December 31, 2014, our stock price has ranged from $5.63 to $16.64 per share.
Factors affecting our operating results and that could adversely affect our stock price include:
- •
- the degree of commercial success and market acceptance of Gralise®,
CAMBIA®, Zipsor® and
Lazanda®;
- •
- our ability to consummate the NUCYNTA® Acquisition, and if consummated, the degree of commercial success of
NUCYNTA® and the outcome of the pending patent litigation against the filers of ANDAs for NUCYNTA®.
- •
- filings and other regulatory or governmental actions or proceedings related to our products and product candidates and those of our
collaborative partners;
- •
- the outcome of our patent infringement litigation against the filer of an ANDA for
Zipsor®;
- •
- the reversal or any appeal of the district court's favorable ruling in our patent infringement litigation against the filer of an ANDA
for Gralise®;
- •
- developments concerning proprietary rights, including patents, infringement allegations, inter party review proceedings and litigation
matters;
- •
- our collaborative partners' compliance or non-compliance with legal and regulatory requirements and with obligations under our
collaborative agreements;
- •
- our plans to acquire, in-license or co-promote other products, or acquire or combine with other companies, and our degree of success
in realizing the intended advantages of, and mitigating any risks associated with, any such transaction;
- •
- adverse events related to our products, including recalls;
- •
- interruptions of manufacturing or supply, or other manufacture or supply difficulties;
- •
- variations in revenues obtained from collaborative agreements, including milestone payments, royalties, license fees and other
contract revenues;
- •
- adoption of new technologies by us or our competitors;
- •
- the outcome of our patent infringement litigation against Purdue and Endo;
- •
- decisions by collaborative partners to proceed or not to proceed with subsequent phases of a collaboration or program;
- •
- sales of large blocks of our common stock or the dilutive effect of our 2021 Notes; and
- •
- variations in our operating results, earnings per share, cash flows from operating activities, deferred revenue, and other financial metrics and non-financial metrics, and how those results compare to analyst expectations.
As a result of these factors, our stock price may continue to be volatile and investors may be unable to sell their shares at a price equal to, or above, the price paid. Any significant drops in our stock price could give rise to shareholder lawsuits, which are costly and time consuming to defend against and which may adversely affect our ability to raise capital while the suits are pending, even if the suits are ultimately resolved in our favor.
32
In addition, if the market for pharmaceutical stocks or the stock market in general experiences uneven investor confidence, the market price of our common stock could decline for reasons unrelated to our business, operating results or financial condition. The market price of our common stock might also decline in reaction to events that affect other companies within, or outside, our industry even if these events do not directly affect us.
We have incurred operating losses in the past and may incur operating losses in the future.
To date, we have recorded revenues from license fees, product sales, royalties, collaborative research and development arrangements and feasibility studies. For 2014 and 2013, we recognized net income of $131.8 million and $43.3 million, respectively. Although we have achieved profitability in recent periods, we have incurred operating losses in the past and may incur operating losses in 2015 and in future years. Any such losses may have an adverse impact on our total assets, shareholders' equity and working capital.
Our existing capital resources are not sufficient to fund the NUCYNTA® Acquisition and may not be sufficient to fund our future operations or product acquisitions and strategic transactions which we may pursue.
We fund our operations primarily through revenues from product sales and do not have any committed sources of capital. To the extent that our existing capital resources and revenues from ongoing operations are insufficient to fund our future operations, or product acquisitions and strategic transactions which we may pursue, such as the NUCYNTA® Acquisition, we will have to raise additional funds through the sale of our equity securities, through additional debt financing, from development and licensing arrangements, or the sale of assets. We may be unable to raise such additional capital on favorable terms, or at all. If we raise additional capital by selling our equity or convertible debt securities, the issuance of such securities could result in dilution of our shareholders' equity positions.
Our success is dependent in large part upon the continued services of our Chief Executive Officer and senior management with whom we do not have employment agreements.
Our success is dependent in large part upon the continued services of our President and Chief Executive Officer, James A. Schoeneck, and other members of our executive management team, and on our ability to attract and retain key management and operating personnel. We do not have agreements with Mr. Schoeneck or any of our other executive officers that provide for their continued employment with us. We may have difficulty filling open senior commercial, scientific and financial positions. Management, scientific and operating personnel are in high demand in our industry and are often subject to competing offers. The loss of the services of one or more members of management or key employees or the inability to hire additional personnel as needed could result in delays in the research, development, and commercialization of our products and potential product candidates.
Provisions in our restated articles of incorporation and bylaws and California law might discourage, delay or prevent a change of control of our company or changes in our management and, therefore, depress the market price of our common stock.
Certain provisions of our articles of incorporation and the California General Corporation Law could discourage a third party from acquiring, or make it more difficult for a third party to acquire, control of our company without approval of our board of directors. These provisions could also limit the price that certain investors might be willing to pay in the future for shares of our common stock. Certain provisions allow the board of directors to authorize the issuance of preferred stock with rights superior to those of the common stock. We are also subject to the provisions of Section 1203 of the California General Corporation Law which requires a fairness opinion to be provided to our
33
shareholders in connection with their consideration of any proposed "interested party" reorganization transaction.
We have adopted a shareholder rights plan, commonly known as a "poison pill," which contains provisions that may discourage, delay or prevent a third party from acquiring us. These provisions could also discourage proxy contests and make it more difficult for shareholders to elect directors of their choosing and to cause us to take other corporate actions they desire, any of which, under certain circumstances, could depress the market price of our common stock.
If we are unable to satisfy regulatory requirements relating to internal controls, our stock price could suffer.
Section 404 of the Sarbanes-Oxley Act of 2002 requires companies to conduct a comprehensive evaluation of the effectiveness of their internal control over financial reporting. At the end of each fiscal year, we must perform an evaluation of our internal control over financial reporting, include in our annual report the results of the evaluation and have our external auditors also publicly attest to the effectiveness of our internal control over financial reporting. If material weaknesses are found in our internal controls in the future, if we fail to complete future evaluations on time or if our external auditors cannot attest to the effectiveness of our internal control over financial reporting, we could fail to meet our regulatory reporting requirements and be subject to regulatory scrutiny and a loss of public confidence in our internal controls, which could have an adverse effect on our stock price.
Changes in fair value of contingent consideration and/or the liability for the unfavorable contract assumed as part of our acquisitions could adversely affect our results of operations.
Contingent consideration obligations arise from the Zipsor®, CAMBIA®, and Lazanda® acquisitions and relate to the potential future milestone payments and royalties payable under the respective agreements. The liability for the unfavorable contract arose from the acquisition of CAMBIA® and represents the milestone payable to the vendor as well as the value of the amounts by which the contract terms are unfavorable compared to current market pricing. The contingent consideration and the liability for the unfavorable contract is initially recognized at its fair value on the acquisition date and is re-measured to fair value at each reporting date until the contingency is resolved with changes in fair value recognized in earnings. The estimates of fair values for the contingent consideration and the unfavorable manufacturing contract contain uncertainties as it involves assumptions about the probability assigned to the potential milestones and royalties being achieved and the discount rate. Significant judgment is employed in determining these assumptions as of the acquisition date and for each subsequent period. Updates to assumptions could have a significant impact on our results of operations in any given period.
The value of our deferred tax assets could become impaired, which could adversely affect our results of operations.
As of December 31, 2014, we had a significant amount of deferred tax assets, exclusive of a deferred tax liability for the convertible debt issuance. These deferred tax assets are principally comprised of state net operating loss carryovers and temporary differences related to intangible assets and other temporary differences that are expected to reverse in the future. We assess on a quarterly basis the probability of the realization of deferred tax assets, using significant judgments and estimates with respect to, among other things, the deferred tax liability as a source of income, historical operating results, expectations of future earnings and significant risks and uncertainties related to our business. If we determine in the future that there is not sufficient positive evidence to support the valuation of these assets, due to the risk factors described herein or other factors, we may be required to further adjust the valuation allowance to reduce our deferred tax assets. Such a reduction could result in material non-cash expenses in the period in which the valuation allowance is adjusted and could have an adverse effect on our results of operations.
34
The investment of our cash is subject to risks, which may cause losses or adversely affect the liquidity of these investments and our results of operations, liquidity and financial condition.
As of December 31, 2014, we had $488.7 million in cash and cash equivalents and $77.7 million in investments, of which $500.0 million was delivered to the Escrow Agent on January 15, 2015 as a deposit to be credited against the purchase price payable to Janssen Pharma upon the consummation of the NUCYNTA® Acquisition. These investments are subject to general credit, liquidity, market and interest rate risks, which have been and may, in the future, be exacerbated by a U.S. and/or global financial crisis. We may realize losses in the fair value of certain of our investments or a complete loss of these investments if the credit markets tighten, which would have an adverse effect on our results of operations, liquidity and financial condition.
Our failure to generate sufficient cash flow from our business to make payments on our debt would adversely affect our business, financial condition and results of operations.
We incurred significant indebtedness in the aggregate principal amount of $345.0 million under our 2021 Notes. We will incur significant additional indebtedness in connection with obtaining the additional funds necessary to consummate the NUCYNTA® Acquisition. Our ability to make scheduled payments of the principal of, to pay interest on, or to refinance, the 2021 Notes and any additional debt obligations we may incur, depends on our future performance, which is subject to economic, financial, competitive and other factors that may be beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt and to make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on commercially reasonable or acceptable terms, which could result in a default on our obligations, including the 2021 Notes.
In addition, our significant indebtedness, combined with our other financial obligations and contractual commitments, could have other important consequences to our business. For example, it could:
- •
- make us more vulnerable to adverse changes in general economic, industry and competitive conditions and adverse changes in government
regulation;
- •
- limit our flexibility in planning for, or reacting to, changes in our business and our industry;
- •
- put us at a disadvantage compared to our competitors who have less debt; and
- •
- limit our ability to borrow additional amounts for working capital and other general corporate purposes, including funding possible acquisitions of, or investments in, additional products, technologies, and companies.
Any of these factors could adversely affect our business, financial condition and results of operations. In addition, if we incur additional indebtedness, the risks related to our business and our ability to service or repay our indebtedness would increase.
We may not have the ability to raise the funds necessary to settle conversions of the 2021 Notes in cash or to repurchase the 2021 Notes upon a fundamental change.
Holders of the 2021 Notes will have the right to require us to repurchase all or a portion of their 2021 Notes upon the occurrence of certain events deemed to be a "fundamental change" at a repurchase price equal to 100% of the principal amount of the 2021 Notes to be repurchased, plus accrued and unpaid interest, if any. In addition, upon conversion of the 2021 Notes, unless we elect to
35
deliver solely shares of our common stock to settle such conversion (other than paying cash in lieu of delivering any fractional share), we will be required to make cash payments in respect of the 2021 Notes being converted. However, we may not have enough available cash or be able to obtain financing at the time we are required to make repurchases of 2021 Notes surrendered therefore or pay cash with respect to 2021 Notes being converted. In addition, our ability to repurchase or to pay cash upon conversion of the 2021 Notes may be limited by law, regulatory authority or agreements governing our future indebtedness. Our failure to repurchase 2021 Notes at a time when the repurchase is required by the indenture or to pay cash upon conversion of the 2021 Notes as required by the indenture would constitute a default under the indenture. A default under the indenture or the fundamental change itself could also lead to a default under agreements governing our future indebtedness. Moreover, the occurrence of a fundamental change under the indenture could constitute an event of default under any such agreements. If the payment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and repurchase the 2021 Notes or to pay cash upon conversion of the 2021 Notes.
The conditional conversion feature of the 2021 Notes, if triggered, may adversely affect our financial condition and operating results.
In the event the conditional conversion feature of the 2021 Notes is triggered, holders of 2021 Notes will be entitled to convert the 2021 Notes at any time during specified periods at their option. If one or more holders elect to convert their 2021 Notes, unless we elect to satisfy our conversion obligation by delivering solely shares of our common stock (other than paying cash in lieu of delivering any fractional share), we would be required to settle a portion or all of our conversion obligation in cash, which could adversely affect our liquidity. In addition, even if holders do not elect to convert their 2021 Notes, we could be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the 2021 Notes as a current rather than long-term liability, which would result in a material reduction of our net working capital.
The accounting method for convertible debt securities that may be settled in cash, such as the 2021 Notes could have a material effect on our reported financial results.
In May 2008, FASB issued FASB Staff Position No. APB 14-1, Accounting for Convertible Debt Instruments That May Be Settled in Cash upon Conversion (Including Partial Cash Settlement), which has subsequently been codified as Accounting Standards Codification 470-20, Debt with Conversion and Other Options (ASC 470-20). Under ASC 470-20, an entity must separately account for the liability and equity components of the convertible debt instruments (such as the 2021 Notes) that may be settled entirely or partially in cash upon conversion in a manner that reflects the issuer's economic interest cost. The effect of ASC 470-20 on the accounting for the 2021 Notes is that the equity component is required to be included in the additional paid-in capital within shareholders' equity on our consolidated balance sheet at the issuance date and the value of the equity component would be treated as debt discount for purposes of accounting for the debt component of the 2021 Notes. As a result, we will be required to record a greater amount of non-cash interest expense as a result of the accretion of the discounted carrying value of the 2021 Notes to their face amount over the term of the notes. We will report lower net income (or larger net losses) in our financial results because ASC 470-20 will require interest to include both the accretion of the debt discount and the instrument's non-convertible coupon interest rate, which could adversely affect our reported or future financial results, the trading price of our common stock.
In addition, if the 2021 Notes become convertible, we would be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the 2021 Notes as a current rather than a long-term liability, which would result in a material reduction of our net working capital. Finally, we use the if-converted method to compute diluted earnings per share with respect to
36
our convertible debt, which could more dilutive than assuming the debt would be settled in cash as opposed to shares.
Any of these factors could cause a decrease in the market price of our common stock.
We do not intend to pay dividends on our common stock so any returns on shares of our common stock will be limited to changes in the value of our common stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, our ability to pay cash dividends on our common stock may be prohibited or limited by the terms of any future debt financing arrangement. Any return to shareholders will therefore be limited to the increase, if any, of our stock price.
Business interruptions could limit our ability to operate our business.
Our operations and infrastructure, and those of our partners, third party suppliers and vendors are vulnerable to damage or interruption from cyber attacks and security breaches, human error, natural disasters, fire, flood, power loss, telecommunications failures, equipment failures, intentional acts of theft, vandalism, terrorism and similar events. In particular, our corporate headquarters are located in the San Francisco Bay area, which has a history of seismic activity. We have not established a formal disaster recovery plan, and our back-up operations and our business interruption insurance may not be adequate to compensate us for losses that occur. A significant business interruption could result in losses or damages incurred by us and require us to cease or curtail our operations.
Our product revenues have historically been lower in the first quarter of the year as compared to the fourth quarter of the preceding year, which may cause our stock price to decline.
Our product revenues have historically been lower in the first quarter of the year as compared to the fourth quarter of the preceding year. We believe this arises primarily as a result of the reduction by our wholesalers of inventory of our products in the first quarter and annual changes in health insurance plans that occur at the beginning of the calendar year.
In 2012 and 2013, our wholesalers ended the calendar year with higher levels of inventory of our products than at the end of the first quarter of the following year. As a result, in the first quarter of 2013 and 2014, product shipments were lower than prescription demand and net sales decreased as a result of the reduction of product inventory at our wholesalers. Any material reduction by our wholesalers of their inventory of our products in the first quarter of any calendar year as compared to the fourth quarter of the preceding calendar year, could adversely affect our operating results and may cause our stock price to decline.
Many health insurance plans and government programs reset annual limits on deductibles and out-of-pocket costs at the beginning of each calendar year and require participants to pay for substantially all of the costs of medical services and prescription drug products until such deductibles and annual out-of-pocket cost limits are met. In addition, enrollment in high-deductible health insurance plans has increased significantly in recent years. As a result of these factors, patients may delay filling or refilling prescriptions for our products or substitute less expensive generic products until such deductibles and annual out-of-pocket cost limits are met. Any reduction in the demand for our products, including as a result of the foregoing factors, could adversely affect our business, operating results and financial condition.
37
The consummation of the NUCYNTA® Acquisition is subject to a number of conditions, including certain conditions that may not be satisfied on a timely basis, if at all. Failure to consummate the NUCYNTA® Acquisition could negatively impact our stock price and our future business and financial results.
The completion of the NUCYNTA® Acquisition is subject to a number of conditions, including (i) our obtaining financing, through one or more transactions, in an aggregate amount sufficient to pay the purchase price (minus the $500.0 cash deposit) and any related fees and expenses and (ii) the expiration or termination of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended. The completion of the NUCYNTA® Acquisition is not certain and is subject to risks, including the risk that we are unable to obtain financing for the remainder of the purchase price, the approval of governmental agencies is not obtained or that other closing conditions are not satisfied.
If the NUCYNTA® Acquisition is not consummated, our ongoing business may be adversely affected and, without realizing any of the benefits of having consummated the NUCYNTA® Acquisition, we will be subject to a number of risks, including the following:
- •
- if the acquisition agreement is terminated under specified circumstances, we may be required to pay to Janssen Pharma a termination
fee equal to $73.5 million;
- •
- the current price of our common stock may reflect a market assumption that the NUCYNTA® Acquisition will occur, meaning
that a failure to complete the acquisition could result in a decline in the price of our common stock; and
- •
- matters relating to the acquisition have required and will continue to require substantial commitments of time and resources by management, which could otherwise have been devoted to other opportunities that may have been beneficial to us.
We also could be subject to litigation related to any failure to consummate the NUCYNTA® Acquisition or to perform our obligations under the Asset Purchase Agreement, or related to any enforcement proceeding commenced against us. If the NUCYNTA® Acquisition is not consummated, these risks may materialize and may adversely affect our business, financial results and stock price.
Additional financing we intend to obtain to consummate the NUCYNTA® Acquisition will cause us to incur a significant amount of additional indebtedness and may negatively impact our shareholders' equity position and subject us to restrictive operating and financial covenants.
In order to consummate the NUCYNTA® Acquisition, we expect to raise at least $550.0 million of capital (net of fees) through the sale of our equity or equity-linked securities or through debt financing or a combination of debt and equity financing. The issuance of such securities may be highly dilutive, or otherwise disadvantageous, to existing shareholders or subject us to restrictive operating and financial covenants. Such financing may not be available on commercially reasonable or acceptable terms, if at all. Our failure to raise capital when needed and on acceptable terms may require us to reduce our costs and expenses and would limit our ability to respond to competitive pressures or unanticipated circumstances and to continue operations. Any one of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
We will incur substantial transaction-related costs in connection with the NUCYNTA® Acquisition.
We expect to incur substantial expenses in connection with consummating the NUCYNTA® Acquisition and integrating the businesses and operations related to NUCYNTA®. There are a number of factors beyond our control that could affect the total amount or the timing of integration expenses. Many of the expenses that will be incurred, by their nature, are currently difficult to estimate accurately. Due to these factors, if we are unable to consummate the NUCYNTA® Acquisition and realize the benefits thereof, the transaction and integration expenses associated with the NUCYNTA® Acquisition could have a material adverse effect on our business, financial condition and results of operations.
38
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
In April 2012, we entered into an office and laboratory lease agreement with BMR-Pacific Research Center LP (BMR) to lease approximately 52,500 rentable square feet in Newark, California commencing on December 1, 2012. The initial term of the lease is approximately ten years. We will pay approximately $12.2 million in aggregate as rent over the term of the lease to the landlord. As part of the lease, BMR agreed to provide various financial allowances so that we can build initial and future laboratories, offices and other improvements, subject to customary terms and conditions relating to landlord-funded tenant improvements. As part of the lease, we are obligated to lease approximately 8,000 additional rentable square feet commencing no later than December 1, 2015. The lease will expire on November 30, 2022. However, we have the right to renew the lease for one additional five year term, provided that written notice is made to BMR no later than 12 months prior to the lease expiration. We will have the one-time right to terminate the lease in its entirety effective as of November 30, 2017 by delivering written notice to the landlord on or before December 1, 2016. In the event of such termination, we will pay BMR the unamortized portion of the tenant improvement allowance, specified additional allowances, waived base rent and leasing commissions, in each case amortized at 8% interest.
The property subject to the office and laboratory lease is the only property utilized by us. We believe our office and laboratory space is adequate to meet our current and future needs.
Depomed v. Gralise® ANDA Filers
Between March 2012 and May 2012, we filed lawsuits in the United States District Court for the District of New Jersey in response to six Abbreviated New Drug Applicants (ANDAs) filed by companies seeking to market generic versions of 300mg and 600mg dosage strengths of Gralise® prior to the expiration of our patents listed in the Orange Book for Gralise®. The lawsuits have been consolidated for purposes of all pretrial proceedings. Our lawsuits against two of the six Gralise® ANDA filers, Impax Laboratories and Watson Laboratories, have been dismissed as a result of the withdrawal of the ANDAs from consideration by the FDA. Our lawsuit against another ANDA filer, Par Pharmaceuticals Inc., has been dismissed because the ANDA filer no longer seeks approval of its Gralise ANDA prior to the expiration of our Gralise® Orange Book-listed patents. In April 2014, we entered settlement agreements with Incepta Pharmaceuticals and Abon Pharmaceuticals LLC (collectively, Incepta) and with Zydus Pharmaceuticals USA Inc. and Cadila Healthcare Limited (collectively, Zydus) pursuant to which Incepta and Zydus may begin selling generic versions of Gralise® on January 1, 2024, or earlier under certain circumstances.
A bench trial involving defendants Actavis Elizabeth LLC and Actavis Inc. (collectively, Actavis) was completed on May 20, 2014 as to U.S. Patent Nos. 6,635,280; 6,488,962; 7,438,927; 7,731,989; 8,192,756; 8,252,332; and 8,333,992, which expire between September 2016 and February 2024. In August 2014, the court ruled in our favor, finding that Actavis infringed all patent claims we asserted and upholding the validity of the patents. On September 15, 2014, Actavis filed a notice appealing the decision to the United States Court of Appeals for the Federal Circuit. On February 2, 2015, Actavis filed its opening brief with the United States Court of Appeals for the Federal Circuit.
39
Depomed v. FDA
In November 2010, the FDA granted Gralise® Orphan Drug designation for the management of PHN, but did not recognize Orphan drug exclusivity for Gralise® in January 2011 when Gralise® was approved for marketing in the United States. In September 2012, we filed an action in federal district court for the District of Columbia against the FDA seeking an order requiring the FDA to grant Gralise® Orphan Drug exclusivity for the management of PHN. Briefing in the case was completed in March 2013 and a hearing on our summary judgment motion was held in August 2013. In September 2014, the court issued an order granting our request for summary judgment, and ordering the FDA to grant Orphan Drug exclusivity for Gralise® for the management of PHN, which the FDA did formally in October 2014. On November 3, 2014, the FDA filed a notice appealing the order to the United States Court of Appeals for the Federal Circuit. On November 5, 2014, the government dismissed its appeal.
Depomed v. Purdue and Depomed v. Endo Pharmaceuticals—Patent Infringement Litigation and Related Inter Partes Review Proceedings
We have sued Purdue Pharma and Endo Pharmaceuticals for patent infringement in separate lawsuits filed in the United States District Court for the District of New Jersey. The lawsuits arise from Purdue's commercialization of reformulated OxyContin® (oxycodone hydrochloride controlled-release) in the United States and Endo's commercialization of OPANA® ER (oxymorphone hydrochloride extended-release) in the United States. We sued Purdue in January 2013 for infringement of U.S. Patent Nos. 6,340,475 (the '475 Patent) and 6,635,280 (the '280 Patent), which expire in September 2016. We sued Endo in April 2013 for infringement of the '475 Patent, the '280 Patent and U.S. Patent No. 6,723,340 (the '340 Patent), which expires in October 2021. The Purdue lawsuit has been stayed pending completion of the inter partes reviews described below. The District Court has not yet ruled on Endo's request to stay the Endo litigation.
In response to two petitions filed by Purdue and six petitions filed by Endo, the United States Patent and Trademark Office Patent Trial and Appeal Board (PTAB) has instituted inter partes reviews (each, an IPR) of certain of the claims asserted in our lawsuits against Purdue and Endo. An IPR is a proceeding that became available in September 2012 in accordance with the America Invents Act (the AIA). In an IPR, a petitioner may request that the PTAB reconsider the validity of issued patent claims on the basis of prior art in the form of printed publications and other patents. Any patent claim the PTAB determines to be unpatentable is stricken from the challenged patent. Any party may appeal final written decisions of the PTAB to the United States Court of Appeals for the Federal Circuit. But, the PTAB's decisions denying institution of an IPR are non-appealable. Accordingly, if the PTAB finds a challenged claim patentable, or declines to institute an IPR as to a challenged claim, the IPR petitioner is estopped from asserting in a patent infringement lawsuit that those claims are invalid on any ground the petitioner raised or reasonably could have raised in the IPR.
In the Purdue IPRs, the PTAB declined to institute an IPR as to two claims of the '475 patent and two claims of the '280 Patent. The PTAB instituted an IPR as to the other 15 claims of the '475 Patent and as to the other 10 claims of the '280 Patent asserted against Purdue.
Endo filed two IPR petitions for each of the '475 Patent, the '280 Patent and the '340 Patent. The PTAB declined to institute an IPR as to three of Endo's petitions. The PTAB also declined to institute an IPR as to five claims of the '475 Patent, three claims of the '280 Patent and one claim of the '340 Patent in the Endo IPRs. The PTAB instituted an IPR as to the other 13 claims of the '475 Patent, as to the other ten claims of the '280 Patent and as to the other eight claims of the '340 patent asserted against Endo. The PTAB also declined to institute an IPR as to a number of Endo's requested grounds. On December 22, 2014, Depomed filed patent owner responses opposing the instituted IPRs.
40
Discovery, briefing and oral argument is scheduled to be complete in the Purdue IPRs in March 2015 and in the Endo IPRs in June 2015. In accordance with the requirements of the AIA, we expect final decisions from the PTAB not later than one year after the PTAB's decisions to institute the IPRs, or not later than July 10, 2015 in the Purdue IPRs and not later than September 29, 2015 in the Endo IPRs.
Depomed v. Banner Pharmacaps
On June 28, 2013, we received from Banner a Notice of Certification for U.S. Patent Nos. 6,365,180; 7,662,858; 7,884,095; 7,939,518; and 8,110,606 under 21 U.S.C. § 355 (j)(2)(A)(vii)(IV) (Zipsor® Paragraph IV Letter) certifying that Banner has submitted and the FDA has accepted for filing an ANDA for diclofenac potassium capsules, 25mg. The letter states that the Banner ANDA product contains the required bioavailability or bioequivalence data to Zipsor® and certifies that Banner intends to obtain FDA approval to engage in commercial manufacture, use or sale of Banner's ANDA product before the expiration of the above identified patents, which are listed for Zipsor® in the Orange Book. U.S. Patent No. 6,365,180 expires in 2019 and U.S. Patent Nos. 7,662,858; 7,884,095; 7,939,518; and 8,110,606 expire in 2029. The Zipsor® Paragraph IV letter indicates Banner has granted to Watson Laboratories Inc. (Watson) exclusive rights to Banner's proposed generic Zipsor® product.
On July 26, 2013, we filed a lawsuit in the United States District Court for District of New Jersey against Banner and Watson for infringement of the patents identified above. The lawsuit was commenced within the 45 days required to automatically stay, or bar, the FDA from approving Banner's ANDA for 25 mg diclofenac for 30 months or until a district court decision that is adverse to Depomed, whichever may occur earlier. Absent a court order, the 30-month stay would be expected to expire in December 2015.
On April 2, 2014, we filed an amended complaint to include infringement of U.S. Patent Nos. 6,287,594 and 8,623,920, which were recently added to the Orange Book listing for Zipsor® and expire in 2019 and 2029, respectively. Fact discovery and claim construction in the case are ongoing and no trial date has been set.
General
We cannot reasonably predict the outcome of the legal proceedings described above, nor can we estimate the amount of loss, range of loss or other adverse consequence, if any, that may result from these proceedings. As such we are not currently able to estimate the impact of the above litigation on our financial position or results of operations.
We may from time to time become party to actions, claims, suits, investigations or proceedings arising from the ordinary course of our business, including actions with respect to intellectual property claims, breach of contract claims, labor and employment claims and other matters. Although actions, claims, suits, investigations and proceedings are inherently uncertain and their results cannot be predicted with certainty, other than the matters set forth above, we are not currently involved in any matters that we believe may have a material adverse effect on our business, results of operations or financial condition. However, regardless of the outcome, litigation can have an adverse impact on us because of associated cost and diversion of management time.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
41
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on the NASDAQ Global Market (NASDAQ) under the symbol "DEPO." The following table sets forth, for the periods indicated, the intraday high and low prices of our common stock as reported by the NASDAQ from January 1, 2013 to December 31, 2014.
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2014 |
|||||||
First Quarter |
$ | 15.39 | $ | 10.20 | |||
Second Quarter |
$ | 14.85 | $ | 10.29 | |||
Third Quarter |
$ | 15.51 | $ | 9.85 | |||
Fourth Quarter |
$ | 16.64 | $ | 13.55 | |||
2013 |
|||||||
First Quarter |
$ | 7.15 | $ | 5.12 | |||
Second Quarter |
$ | 6.19 | $ | 4.99 | |||
Third Quarter |
$ | 7.70 | $ | 5.63 | |||
Fourth Quarter |
$ | 10.77 | $ | 6.95 | |||
On February 25, 2015, the closing price of our common stock was $22.52. As of February 25, 2015, there were approximately 23 shareholders of record of our common stock, one of which is Cede & Co., a nominee for Depository Trust Company, or DTC. All of the shares of common stock held by brokerage firms, banks and other financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and are therefore considered to be held of record by Cede & Co. as one shareholder.
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain any future earnings to finance the growth and development of our business.
Issuer Purchases of Securities
None.
Unregistered Sales of Securities
None.
Equity Compensation Plan Information
The information under the principal heading "Equity Compensation Plan Information" in our definitive Proxy Statement for the Annual Meeting of Stockholders to be held on or about May 12, 2015, to be filed with the SEC, is incorporated herein by reference.
Stock Price Performance Graph
The following graph compares total shareholder returns of the Company for the past five years to two indices: the NASDAQ Composite Index and the NASDAQ Pharmaceutical Index. The total return for our common stock and for each index assumes the reinvestment of all dividends, although cash dividends have never been declared on our common stock.
42
The performance graph and related information shall not be deemed to be "soliciting material" or to be "filed" with the SEC, and shall not be deemed to be incorporated by reference into any of our filings under the Securities Act or Exchange Act
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Depomed, Inc., The NASDAQ Composite Index
And The NASDAQ Pharmaceutical Index
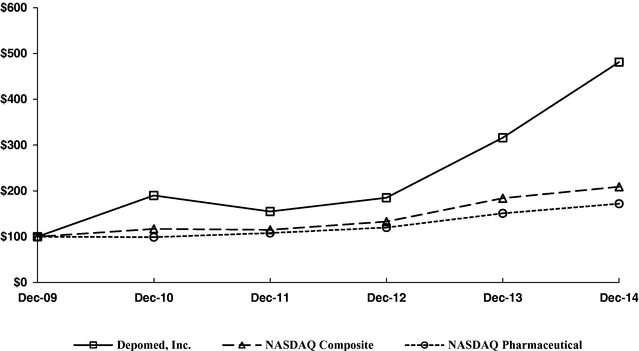
- *
- $100
invested on 12/31/08 in stock or index, including reinvestment of dividends.
Fiscal year ending December 31.
43
ITEM 6. SELECTED FINANCIAL DATA
The data set forth below is not necessarily indicative of the results of future operations and should be read in conjunction with the financial statements and the notes included elsewhere in this annual report on Form 10-K and also with "ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS."
| |
2014 | 2013 | 2012 | 2011(1) | 2010 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Consolidated Statement of Operations Data (in thousands): |
||||||||||||||||
Revenues: |
||||||||||||||||
Product sales |
$ | 114,219 | $ | 58,302 | $ | 27,483 | $ | 41,178 | $ | 45,637 | ||||||
Royalties |
1,821 | 45,003 | 44,535 | 9,997 | 306 | |||||||||||
License and other revenue(2) |
31,515 | 12,796 | 18,798 | 81,798 | 34,821 | |||||||||||
Non-cash PDL royalty revenue(2) |
242,808 | 18,104 | — | — | — | |||||||||||
| | | | | | | | | | | | | | | | | |
Total revenues |
390,363 | 134,205 | 90,816 | 132,973 | 80,764 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total costs and expenses |
153,549 | 124,888 | 121,169 | 102,275 | 77,139 | |||||||||||
Gain on termination of Abbott agreement |
— | — | — | 40,000 | — | |||||||||||
Income (loss) from operations |
236,814 | 9,317 | (30,353 | ) | 70,698 | 3,625 | ||||||||||
Net income (loss) before income taxes |
213,108 | 4,580 | (29,872 | ) | 71,122 | 3,892 | ||||||||||
(Provision for) benefit from income taxes |
(81,346 | ) | 38,733 | 91 | (396 | ) | 4 | |||||||||
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | $ | 70,726 | $ | 3,896 | |||||
Basic net income (loss) per share |
$ | 2.26 | $ | 0.76 | $ | (0.53 | ) | $ | 1.30 | $ | 0.07 | |||||
Diluted net income (loss) per share |
$ | 2.05 | $ | 0.75 | $ | (0.53 | ) | $ | 1.26 | $ | 0.07 | |||||
Shares used in computing basic net income (loss) per share |
58,292,633 | 56,736,009 | 55,892,563 | 54,562,820 | 52,533,256 | |||||||||||
Shares used in computing diluted net income (loss) per share |
66,307,364 | 57,543,979 | 55,892,563 | 56,089,796 | 53,463,749 | |||||||||||
| |
2014 | 2013 | 2012 | 2011(1) | 2010 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Consolidated Balance Sheet Data |
||||||||||||||||
Cash, cash equivalents and marketable securities |
$ | 566,402 | $ | 276,017 | $ | 77,892 | $ | 139,793 | $ | 76,888 | ||||||
Total assets |
711,065 | 508,653 | 141,653 | 164,372 | 87,031 | |||||||||||
Total current liabilities(2),(3) |
57,499 | 156,857 | 36,681 | 39,840 | 32,984 | |||||||||||
Deferred revenue, non-current portion(2) |
— | 12,475 | 15,516 | 17,932 | 30,926 | |||||||||||
Liability related to the sale of future royalties and milestones, less current portion(2) |
— | 177,624 | — | — | — | |||||||||||
Contingent consideration liability, non-current |
14,252 | 11,264 | 1,342 | — | — | |||||||||||
Other long-term liabilities |
12,387 | 13,017 | 4,178 | — | 15 | |||||||||||
Accumulated earnings (deficit) |
47,714 | (84,048 | ) | (127,361 | ) | (97,580 | ) | (168,306 | ) | |||||||
Total shareholders' equity |
364,447 | 137,416 | 83,936 | 105,918 | 23,106 | |||||||||||
- (1)
- Total revenues, income from operations, net income before income taxes, net income and net income per share in 2011 include a one-time $48.0 million milestone received from Abbott Laboratories for the FDA approval of Gralise®.
44
Income from operations, net income before income taxes, net income and net income per share in 2011 include a $40.0 million gain on termination of our agreement with Abbott related to Gralise®.
- (2)
- Effective
October 1, 2014, the Company amended its agreements with Salix and Valeant, which eliminated any and all continuing obligations on the part
of the Company in the manufacture and supply of 1000mg Glumetza tablets. As a result, the unamortized deferred revenue balance as of October 1, 2014 of $13.2 million was recognized as
license and other revenue during 2014. The Company also recognized the entire remaining balance of the liability related to sale of future royalties and milestones of approximately
$147.0 million as non-cash PDL royalty revenue during 2014.
- (3)
- Total current liabilities as of December 31, 2013 included income taxes payable of $61.9 million and liability related to sale of future royalties of $49.5 million.
45
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
Depomed is a specialty pharmaceutical company focused on pain and other central nervous system (CNS) conditions. The products that comprise our current specialty pharmaceutical business are Gralise® (gabapentin), a once-daily product for the management of postherpetic neuralgia (PHN) that we launched in October 2011, CAMBIA® (diclofenac potassium for oral solution), a non-steroidal anti-inflammatory drug for the acute treatment of migraine attacks that we acquired in December 2013, Zipsor® (diclofenac potassium) liquid filled capsules, a non-steroidal anti-inflammatory drug for the treatment of mild to moderate acute pain that we acquired in June 2012, and Lazanda® (fentanyl nasal spray), a product for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain, that we acquired in July 2013. We actively seek to expand our product portfolio through in-licensing, acquiring or obtaining co-promotion rights to commercially available products or late-stage product candidates that could be marketed and sold effectively with our existing products through our sales and marketing capability.
We also have a portfolio of royalty and milestone producing license agreements based on our proprietary Acuform® gastroretentive drug delivery technology with Mallinckrodt Inc. (Mallinckrodt), Ironwood Pharmaceuticals, Inc. (Ironwood) and Janssen Pharmaceuticals, Inc. (Janssen Pharma).
In October 2013, we sold our interests in royalty and milestone payments under our license agreements in the Type 2 diabetes therapeutic area to PDL BioPharma, Inc. (PDL) for $240.5 million (PDL Transaction). The interests sold include royalty and milestone payments accruing from and after October 1, 2013 from: (a) Salix Pharmaceuticals, Inc. (Salix) with respect to sales of Glumetza® (metformin HCL extended-release tablets) in the United States; (b) Merck & Co. Inc. (Merck) with respect to sales of Janumet® XR (sitagliptin and metformin HCL extended-release); (c) Janssen Pharmaceutica N.V. and Janssen Pharma (collectively, Janssen) with respect to potential future development milestones and sales of Janssen's investigational fixed-dose combination of Invokana® (canagliflozin) and extended-release metformin; (d) Boehringer Ingelheim with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to our license agreement with Boehringer Ingelheim International GMBH (Boehringer Ingelheim); and (e) LG Life Sciences Ltd. (LG) and Valeant International Bermuda SRL (Valeant SRL) for sales of extended-release metformin in Korea and Canada, respectively.
As of December 31, 2014, we have one product candidate under clinical development, DM-1992 for Parkinson's disease. DM-1992 completed a Phase 2 trial for Parkinson's disease, and we announced a summary of the results of that trial in November 2012. We continue to evaluate clinical and regulatory strategies and commercial prospects for DM-1992.
On January 15, 2015, we entered into an Asset Purchase Agreement with Janssen Pharma, pursuant to which we will in the United States acquire from Janssen and its affiliates the rights to the NUCYNTA® franchise of pharmaceutical products as well as certain related assets for $1.05 billion in cash (NUCYNTA® Acquisition). The NUCYNTA® franchise includes NUCYNTA® ER (tapentadol extended release tablets) indicated for the management of pain, including neuropathic pain associated with diabetic peripheral neuropathy (DPN), severe enough to require daily, around-the-clock, long-term opioid treatment, NUCYNTA® (tapentadol), an immediate release version of tapentadol, for management of moderate to severe acute pain in adults, and NUCYNTA® (tapentadol oral solution), an approved oral form of tapentadol that has not been commercialized. Upon execution of the Asset Purchase Agreement, we delivered a cash deposit in the amount of $500.0 million to JPMorgan Chase Bank, N.A., (Escrow Agent) in accordance with an Escrow Agreement, dated January 15, 2015, by and
46
among the Company, Janssen Pharma and the Escrow Agent. The cash deposit will be credited against the total cash payable upon the consummation of the NUCYNTA® Acquisition. We need to raise approximately $550.0 million (net of fees) in capital to consummate the NUCYNTA® Acquisition. The consummation of the NUCYNTA® Acquisition is subject to the satisfaction of a number of customary conditions. See "Note 16—Subsequent Events" of the Note to Consolidated Financial Statements for further information on the Asset Purchase Agreement. We recorded $0.3 million of NUCYNTA® Acquisition related costs in 2014.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
A detailed discussion of our significant accounting policies can be found in Note 1 of the Notes to Financial Statements, and the impact and risks associated with our accounting policies are discussed throughout this Annual Report on Form 10-K and in the footnotes to the financial statements. Critical accounting policies are those that require significant judgment and/or estimates by management at the time that financial statements are prepared such that materially different results might have been reported if other assumptions had been made. We consider certain accounting policies related to revenue recognition, accrued liabilities, and use of estimates to be critical policies. These estimates form the basis for making judgments about the carrying values of assets and liabilities. We base our estimates and judgments on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results could differ materially from these estimates.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
We recognize revenue from the sale of our products, and from license fees, milestones and royalties earned on license agreements and collaborative arrangements. Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred and title has passed, the price is fixed or determinable and we are reasonably assured of collecting the resulting receivable. Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items.
Product Sales
We sell our commercial products to wholesale distributors and retail pharmacies. Products sales revenue is recognized when title has transferred to the customer and the customer has assumed the risks and rewards of ownership, which typically occurs on delivery to the customer.
Product Sales Allowances
We recognize product sales allowances as a reduction of product sales in the same period the related revenue is recognized. Product sales allowances are based on amounts owed or to be claimed on the related sales. These estimates take into consideration the terms of our agreements with customers, historical product returns, rebates or discounts taken, estimated levels of inventory in the distribution channel, the shelf life of the product, and specific known market events, such as competitive pricing and new product introductions. If actual future results vary from our estimates, we may need to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustment. Our product sales allowances include:
- •
- Product Returns—We allow customers to return product for credit on returned product that is within six months before and up to 12 months after its product expiration date. We estimate
47
- •
- Wholesaler and Retail Pharmacy Discounts—We offer contractually determined discounts to certain wholesale distributors and
retail pharmacies that purchase directly from us. These discounts are either taken off-invoice at the time of shipment or paid to the customer on a quarterly basis one to two months after the quarter
in which product was shipped to the customer.
- •
- Prompt Pay Discounts—We offer cash discounts to our customers, generally 2% of the sales price, as an incentive for prompt
payment. Based on our experience, we expect our customers to comply with the payment terms to earn the cash discount.
- •
- Patient Discount Programs—We offer patient discount co-pay assistance programs in which patients receive certain discounts
off their prescription at participating retail pharmacies. The discounts are reimbursed by us approximately one month after the prescriptions subject to the discount are filled.
- •
- Medicaid Rebates—We participate in Medicaid rebate programs, which provide assistance to certain low-income patients based
on each individual state's guidelines regarding eligibility and services. Under the Medicaid rebate programs, we pay a rebate to each participating state, generally two to three months after the
quarter in which prescriptions subject to the rebate are filled.
- •
- Chargebacks—We provide discounts to authorized users of the Federal Supply Schedule (FSS) of the General Services Administration under an FSS contract with the Department of Veterans Affairs. These federal entities purchase products from the wholesale distributors at a discounted price, and the wholesale distributors then charge back to us the difference between the current retail price and the price the federal entity paid for the product.
product returns on Gralise®, CAMBIA®, Zipsor®, and Lazanda®. Under the terms of the CAMBIA® Asset Purchase Agreement, we also assumed financial responsibility for returns of CAMBIA® product previously sold by Nautilus. We also estimate returns on sales of Glumetza made by us through August 2011, as we are financially responsible for return credits on Glumetza product we shipped as part of the our commercialization with Santarus in August 2011. Under the terms of the Zipsor® Asset Purchase Agreement, we also assumed financial responsibility for returns of Zipsor® product previously sold by Xanodyne. We did not assume financial responsibility for returns of Lazanda® product previously sold by Archimedes. See Note 15 of the Notes to Financial Information for further information on the acquisition of Zipsor®, CAMBIA® and Lazanda®.
The shelf life of Gralise® is 24 to 36 months from the date of tablet manufacture. The shelf life of CAMBIA® is 24 to 48 months from the manufacture date. The shelf life of Zipsor® is 36 months from the date of tablet manufacture. The shelf life of Lazanda® is 24 to 36 months from the manufacture date. The shelf life of the 500mg Glumetza is 48 months from the date of tablet manufacture and the shelf life of the 1000mg Glumetza is 24 to 36 months from the date of tablet manufacture. We monitor actual return history on an individual product lot basis since product launch, which provides us with a basis to reasonably estimate future product returns, taking into consideration the shelf life of product at the time of shipment, shipment and prescription trends, estimated distribution channel inventory levels and consideration of the introduction of competitive products.
Because of the shelf life of our products and our return policy of issuing credits on returned product that is within six months before and up to 12 months after its product expiration date, there may be a significant period of time between when the product is shipped and when we issue credits on returned product. Accordingly, we may have to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustments.
48
- •
- Managed Care Rebates—We offer discounts under contracts with certain managed care providers who do not purchase directly
from us. We generally pay managed care rebates one to two months after the quarter in which prescriptions subject to the rebate are filled.
- •
- Medicare Part D Coverage Gap Rebates—We participate in the Medicare Part D Coverage Gap Discount Program under which we provide rebates on prescriptions that fall within the "donut hole" coverage gap. We generally pay Medicare Part D Coverage Gap rebates two to three months after the quarter in which prescriptions subject to the rebate are filled.
We believe our estimates related to gross-to-net sales adjustments for wholesaler and retail pharmacy fees and discounts, prompt payment discounts, patient discount programs, launch discounts, managed care rebates and other government chargebacks do not have a high degree of estimation complexity or uncertainty as the related amounts are settled within a relatively short period of time. We believe that our estimated product return allowances require a high degree of judgment and are subject to change based on our experience and certain quantitative and qualitative factors.
Our product sales allowances and related accruals are evaluated each reporting period and adjusted when trends or significant events indicate that a change in estimate is appropriate. Such changes in estimate could affect our results of operations of financial position.
A rollforward of our product sales allowances for the three years ended December 31, 2014 is as follows: -
| (in thousands) |
Contract Discounts(1) |
Product Returns(2) |
Cash Discounts |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance at December 31, 2011 |
$ | 2,626 | $ | 9,842 | $ | 90 | $ | 12,558 | |||||
Revenue Allowances: |
|||||||||||||
Acquisition of Zipsor® |
— | 1,812 | — | 1,812 | |||||||||
Provision related to current period sales(2) |
7,624 | 3,506 | 784 | 11,914 | |||||||||
Changes in estimates related to sales made in prior years |
— | (853 | ) | — | (853 | ) | |||||||
Recorded to balance sheet(2) |
(464 | ) | — | (147 | ) | (611 | ) | ||||||
Payments and credits related to sales made in current period |
(2,910 | ) | — | (558 | ) | (3,468 | ) | ||||||
Payments and credits related to sales made in prior periods |
(2,626 | ) | (3,476 | ) | (91 | ) | (6,193 | ) | |||||
| | | | | | | | | | | | | | |
Balance at December 31, 2012 |
$ | 4,250 | $ | 10,831 | $ | 78 | $ | 15,159 | |||||
Revenue Allowances: |
|||||||||||||
Acquisition of CAMBIA® |
— | 930 | — | 930 | |||||||||
Provision related to current period sales(2) |
20,419 | 5,709 | 1,719 | 27,847 | |||||||||
Changes in estimates related to sales made in prior years |
— | (34 | ) | — | (34 | ) | |||||||
Payments and credits related to sales made in current period |
(12,132 | ) | — | (1,484 | ) | (13,616 | ) | ||||||
Payments and credits related to sales made in prior periods |
(4,250 | ) | (6,227 | ) | (79 | ) | (10,556 | ) | |||||
| | | | | | | | | | | | | | |
Balance at December 31, 2013 |
$ | 8,287 | $ | 11,209 | $ | 234 | $ | 19,730 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Revenue Allowances: |
|||||||||||||
Provision related to current period sales(2) |
60,701 | 8,668 | 3,748 | 73,117 | |||||||||
Changes in estimates related to sales made in prior years |
— | 781 | — | 781 | |||||||||
Payments and credits related to sales made in current period |
(40,007 | ) | — | (3,180 | ) | (43,187 | ) | ||||||
Payments and credits related to sales made in prior periods |
(8,286 | ) | (5,643 | ) | (235 | ) | (14,164 | ) | |||||
| | | | | | | | | | | | | | |
Balance at December 31, 2014 |
$ | 20,695 | $ | 15,015 | $ | 567 | $ | 36,277 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Includes wholesaler fees and retail discounts, launch discounts, patient support programs, managed care rebates, and government chargebacks and rebates.
49
- (2)
- Beginning in the fourth quarter of 2012, we began recognizing Gralise® product sales at the time title transfers to our customer, and began providing for an estimate of future product returns at that time. In June 2012, we acquired Zipsor® and assumed financial responsibility on returns of Zipsor® previously sold by Xanodyne. In December 2013, we acquired CAMBIA® and assumed financial responsibility on returns of CAMBIA® previously sold by Nautilus.
License and Collaborative Arrangements
Revenue from license and collaborative arrangements, including license fees creditable against future royalty obligations (if any), of the licensee, is recognized when an arrangement is entered into if we have substantially completed our obligations under the terms of the arrangement and our remaining involvement is inconsequential and perfunctory. If we have significant continuing involvement under such an arrangement, license fees are deferred and recognized over the estimated performance period. License fee and collaborative payments received in excess of amounts earned are classified as deferred revenue until earned.
We recognize milestone payments upon the achievement of specified milestones if (1) the milestone is substantive in nature, and the achievement of the milestone was not reasonably assured at the inception of the agreement, (2) the achievement relates to past performance and (3) the fees are nonrefundable. Milestone payments received in excess of amounts earned are classified as deferred revenue until earned.
Non-Cash Interest Expense on PDL Liability
In October 2013, we sold our interests in royalty and milestone payments under our license agreements in the Type 2 diabetes therapeutic area to PDL for $240.5 million. We had significant continuing involvement in the PDL transaction until September 30, 2014, primarily due to our obligation to act as the intermediary for the supply of 1000 mg Glumetza to Salix, the licensee of Glumetza. Under the relevant accounting guidance, because of our significant continuing involvement, the $240.5 million payment received from PDL was accounted for as debt until September 30, 2014, and we were required to amortize the debt using the interest method over the life of the arrangement. In order to determine the amortization of this debt, we were required to estimate the total amount of future royalty payments to be received by PDL and payments we were required to make to PDL, if any, over the life of the agreement. The sum of these amounts less the $240.5 million proceeds we received was recorded as interest expense over the life of the royalty obligation. Consequently, we imputed interest on the transaction and recorded interest expense using an estimated interest rate to reflect an arms-length debt transaction. Our estimate of the interest rate under the agreement was based on the amount of royalty and milestone payments expected to be received by PDL over the life of the arrangement. Our estimate of this total interest expense resulted in an effective annual interest rate of approximately 10%. The non-cash royalty revenues and non-cash interest expense was within our consolidated statement of operations over the term of the PDL agreement until September 30, 2014.
Effective October 1, 2014, the Company, Valeant, Salix and PDL executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the manufacture and supply of 1000mg Glumetza tablets. Consequently, the entire outstanding balance of the liability related to the sale of future royalties and milestones of approximately $147.0 million was recognized within "Non-cash PDL royalty revenue" in the accompanying Consolidated Statement of Operations. As a result, we will no longer report any amounts relating to the non-cash royalty revenue or non-cash interest expense relating to the PDL transaction in future periods.
50
Research and Development Expense and Accruals
Research and development expenses include related salaries, clinical trial costs, consultant fees, supplies, manufacturing costs for research and development programs and allocations of corporate costs. All such costs are charged to research and development expense as incurred. These expenses result from our independent research and development efforts as well as efforts associated with collaborations. Our expense accruals for clinical trials are based on estimates of the services received from clinical trial centers and clinical research organizations. If possible, we obtain information regarding unbilled services directly from service providers. However, we may be required to estimate these services based on information available to our product development or administrative staff. If we underestimate or overestimate the activity associated with a study or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Historically, our estimated accrued liabilities have approximated actual expense incurred.
Stock-Based Compensation
We estimate the fair value of stock options and Employee Stock Purchase Plan (ESPP) shares using the Black-Scholes valuation model. The Black-Scholes model requires the input of highly subjective assumptions. The most significant assumptions are our estimates of the expected volatility and the expected term of the award.
Risk-Free Interest Rate. The risk-free interest rate is based on the implied yield available on U.S. Treasury zero-coupon issues with a remaining term equal to the expected term of the option.
Expected Volatility. The volatility assumption is based on the historical volatility of our common stock over the expected term of the options.
Expected Life of Options. We use historical option exercise data to estimate the expected life of the options.
Expected Dividend Yield. We have never paid any dividends and do not intend to in the near future.
As required, we review our valuation assumptions at each grant date and, as a result, we are likely to change our valuation assumptions used to value employee stock-based awards granted in future periods. Employee and director stock-based compensation costs are to be recognized over the vesting period of the award, and we have elected to use the straight-line attribution method. Forfeitures are to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimate forfeitures based on historical experience.
Restricted stock units (RSUs) are measured at the fair value of our common stock on the date of grant and expensed over the period of vesting using the straight-line attribution approach.
Recognizing and Measuring Assets Acquired and Liabilities Assumed in Business Combinations at Fair Value
We account for acquired businesses using the acquisition method of accounting, which requires that assets acquired and liabilities assumed be recorded at date of acquisition at their respective fair values. The fair value of the consideration paid, including contingent consideration, is assigned to the underlying net assets of the acquired business based on their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill or bargain purchase, as applicable.
Significant judgments are used in determining the estimated fair values assigned to the assets acquired and liabilities assumed and in determining estimates of useful lives of long-lived assets. Fair value determinations and useful life estimates are based on, among other factors, estimates of expected future net cash flows, estimates of appropriate discount rates used to present value expected future net cash flow streams, the assessment of each asset's life cycle, the impact of competitive trends on each
51
asset's life cycle and other factors. These judgments can materially impact the estimates used to allocate acquisition date fair values to assets acquired and liabilities assumed and the resulting timing and amounts charged to, or recognized in current and future operating results. For these and other reasons, actual results may vary significantly from estimated results.
Any changes in the fair value of contingent consideration resulting from a change in the underlying inputs is recognized in operating expenses until the contingent consideration arrangement is settled. Changes in the fair value of contingent consideration resulting from the passage of time are recorded within interest expense until the contingent consideration is settled.
Intangible Assets
Intangible assets consist of purchased developed technology and trademarks. We determine the fair values of acquired intangible assets as of the acquisition date. Discounted cash flow models are typically used in these valuations, which require the use of significant estimates and assumptions, including but not limited to, developing appropriate discount rates and estimating future cash flows from product sales and related expenses. We evaluate purchased intangibles for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Estimating future cash flows related to an intangible asset involves significant estimates and assumptions. If our assumptions are not correct, there could be an impairment loss or, in the case of a change in the estimated useful life of the asset, a change in amortization expense.
Our intangible assets relate to CAMBIA®, Zipsor® and Lazanda® product rights of $51.4 million, $27.3 million and $10.5 million, respectively, and have been recorded as intangible assets on the accompanying Consolidated Balance Sheet, and are being amortized ratably over the estimated useful life of the asset through December 2023, July 2019 and August 2022, respectively. As of December 31, 2014 the carrying values of the intangible assets for CAMBIA®, Zipsor® and Lazanda®, were $46.0 million, $17.5 million and $8.8 million, respectively.
Income Taxes
The Company's income tax policy is to record the estimated future tax effects of temporary differences between the tax bases of assets and liabilities and amounts reported in the Company's accompanying consolidated balance sheets, as well as operating loss and tax credit carryforwards. The Company follows the guidelines set forth in the applicable accounting guidance regarding the recoverability of any tax assets recorded on the consolidated balance Sheet and provides any necessary allowances as required. Determining necessary allowances requires the Company to make assessments about the timing of future events, including the probability of expected future taxable income and available tax planning opportunities.
The Company is subject to examination of its income tax returns by various tax authorities on a periodic basis. The Company regularly assesses the likelihood of adverse outcomes resulting from such examinations to determine the adequacy of its provision for income taxes. The Company has applied the provisions of the applicable accounting guidance on accounting for uncertainty in income taxes, which requires application of a more-likely-than-not threshold to the recognition and de-recognition of uncertain tax positions. If the recognition threshold is met, the applicable accounting guidance permits the Company to recognize a tax benefit measured at the largest amount of tax benefit that, in the Company's judgment, is more than 50 percent likely to be realized upon settlement. It further requires that a change in judgment related to the expected ultimate resolution of uncertain tax positions be recognized in earnings in the period of such change.
52
Convertible Debt
On September 9, 2014, we issued and sold $345.0 million aggregate principal amount of convertible senior notes in a public offering (2021 Notes). The convertible debt offering resulted in net proceeds of $334.2 million after deducting the underwriting discount and offering expenses of $10.4 million and $0.4 million, respectively. The 2021 Notes are accounted for in accordance with ASC Subtopic 470-20, Debt with Conversion and Other Options. Under ASC Subtopic 470-20, issuers of certain convertible debt instruments that have a net settlement feature and may be settled in cash upon conversion, including partial cash settlement, are required to separately account for the liability (debt) and equity (conversion option) components of the instrument. The carrying amount of the liability component of any outstanding debt instrument is computed by estimating the fair value of a similar liability without the conversion option. The amount of the equity component is then calculated by deducting the fair value of the liability component from the principal amount of the convertible debt instrument. See "Note 8—Debt" of the Notes to Consolidated Financial Statements for further information regarding the 2021 Notes.
RESULTS OF OPERATIONS
Our results of operations in 2014 differ significantly from our reported results for 2013 and 2012. In October 2013, we sold our interests in royalties and milestone payments related to certain license agreements in the Type 2 diabetes area to PDL for $240.5 million. In 2013, we reflect nine months of royalty revenue for Glumetza and Janumet XR and three months of non-cash royalty revenue and non-cash interest expense related to the sale of future royalties to PDL. In 2014, we reflect nine months of non-cash PDL royalty revenue and non-cash interest expense on PDL liability. Effective October 1, 2014, the Company, Valeant, Salix and PDL executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the supply of 1000mg Glumetza tablets. Consequently, the entire outstanding balance of the liability related to the sale of future royalties and milestones of approximately $147.0 million was recognized within "Non-cash PDL royalty revenue" in the accompanying Consolidated Statement of Operations.
In addition, we acquired Zipsor® in June 2012, Lazanda® in June 2013 and CAMBIA® in December 2013. Zipsor® revenue and expense is reflected in our results of operations for an entire year in 2013 and 2014 but only for the second half of 2012. Lazanda® revenue and expense is reflected in our results of operations for an entire year in 2014 but only for the second half of 2013. CAMBIA® revenue and expense is reflected in our results of operations for an entire year in 2014 but only for a partial month in December 2013.
53
Revenues
Total revenues are summarized in the following table (in thousands):–
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Product sales: |
||||||||||
Gralise |
$ | 60,411 | $ | 36,188 | $ | 17,288 | ||||
Zipsor |
25,155 | 20,341 | 9,835 | |||||||
Cambia |
21,681 | 555 | — | |||||||
Lazanda |
6,972 | 1,218 | — | |||||||
Proquin XR |
— | — | 360 | |||||||
| | | | | | | | | | | |
Total product sales |
114,219 | 58,302 | 27,483 | |||||||
Royalties: |
||||||||||
Glumetza US |
— | 42,060 | 42,792 | |||||||
Others |
1,821 | 2,943 | 1,743 | |||||||
| | | | | | | | | | | |
Total royalty revenue |
1,821 | 45,003 | 44,535 | |||||||
Non-cash PDL royalty revenue |
$ |
242,808 |
$ |
18,104 |
$ |
— |
||||
License and Other revenue: |
||||||||||
Glumetza |
$ | 15,515 | $ | 3,041 | $ | 4,926 | ||||
Boehringer Ingelheim |
— | — | 2,617 | |||||||
Mallinckrodt |
15,000 | 5,000 | — | |||||||
Janssen |
— | 3,554 | 10,005 | |||||||
Other |
1,000 | 1,201 | 1,250 | |||||||
| | | | | | | | | | | |
Total license and other revenue: |
31,515 | 12,796 | 18,798 | |||||||
| | | | | | | | | | | |
Total revenues |
$ | 390,363 | $ | 134,205 | $ | 90,816 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Product sales
Gralise®. In October 2011, we announced the commercial availability of Gralise® and began distributing Gralise® to wholesalers and retail pharmacies. Until the fourth quarter of 2012, we deferred recognition of revenue on product shipments of Gralise® until the right of return no longer existed, which occurred at the earlier of (a) the time Gralise® units were dispensed through patient prescriptions or (b) expiration of the right of return. In the fourth quarter of 2012, we concluded that we had the information needed to reasonably estimate product returns thereby changing our revenue recognition policy for Gralise® and began recognizing revenue upon delivery to our customers. The increase in Gralise® product sales in 2014 is primarily a result of higher prescription demand and, to a lesser extent, price increases. We expect Gralise® product sales and prescriptions to increase in 2015.
CAMBIA®. We began shipping and recognizing product sales on CAMBIA® in December 2013. We began commercial promotion of CAMBIA® in February 2014. We expect CAMBIA® product sales and prescriptions to increase in 2015.
Zipsor®. We began shipping and recognizing product sales on Zipsor® at the end of June 2012. We began commercial promotion of Zipsor® in July 2012. The increase in Zipsor® product sales for the year ended December 31, 2014 compared to the year ended December 31, 2013 is primarily the result of price increases. We expect Zipsor® product sales to increase in 2015.
Lazanda®. We began shipping and recognizing product sales on Lazanda® in August 2013. We began commercial promotion of Lazanda® in October 2013. We expect Lazanda® product sales and prescriptions to increase in 2015.
54
Royalties
Glumetza US. Until October 1, 2013, we received royalties from Salix based on net sales of Glumetza in the United States. Royalty revenue from Salix for the year ended December 31, 2013 was $42.1 million which represents a 32.0% royalty on net sales of Glumetza for the nine months ended September 30, 2013. Royalty revenue from Salix for the year ended December 31, 2012 was $42.8 million which represents a 29.5% royalty on net sales of Glumetza. In October 2013, we sold our interest in the Glumetza royalties to PDL, as discussed below.
Other Royalties. In October 2013, we sold our interest in Janumet XR, Valeant and LG Life Sciences royalties to PDL as discussed below. In January 2012, Merck received FDA approval to market Janumet XR in the United States, and Merck began selling Janumet XR during the first quarter of 2012. We received very low single digit royalties on net product sales of Janumet XR. As such, we began recognizing royalty revenue in the first quarter of 2012. Other royalties also include royalties we received from Valeant on net sales of Glumetza in Canada and from LG Life Sciences on net sales of LG's version of Glumetza, Novamet GR, in Korea.
In August 2012, we entered into a license agreement with Janssen Pharma relating to NUCYNTA® ER and currently receive a low single digit royalty on net sales of NUCYNTA® ER in the United States, Canada and Japan, which began in the third quarter of 2012. We will not receive any royalties from Janssen Pharma on net sales of NUCYNTA® ER in the United States for any period after the consummation of the NUCYNTA® Acquisition.
We receive high single digit royalties on net sales of XARTEMIS™ XR, which was launched in March 2014, and we will receive the same high single digit royalty on net sales of MNK-155 if it is approved.
Non-Cash PDL Royalty Revenue. In October 2013, as noted above, we sold our interests in royalty and milestone payments under our license agreements in the Type 2 diabetes therapeutic area to PDL for $240.5 million. Until September 30, 2014, this transaction was accounted for as debt that was amortized using the interest method over the life of the arrangement. As a result of the debt accounting, even though we did not retain the related royalties and milestones under the transaction (as the amounts are remitted to PDL), we continued to record revenue related to these royalties and milestones until September 30, 2014.
Effective October 1, 2014, the Company, Valeant, Salix and PDL executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the supply of 1000mg Glumetza tablets. Consequently, the entire outstanding balance of the liability related to the sale of future royalties and milestones of approximately $147.0 million was recognized within "Non-cash PDL royalty revenue" in the accompanying Consolidated Statement of Operations.
We recognized $242.8 million and $18.1 million of non-cash revenue associated with the PDL Transaction for the years ended December 31, 2014 and 2013, respectively.
License and other revenue
Mallinckrodt (formerly Covidien). In November 2008, we entered into a license agreement related to acetaminophen/opiate combination products with Mallinckrodt. The license agreement grants Mallinckrodt worldwide rights to utilize our Acuform technology for the exclusive development of up to four products containing acetaminophen in combination with opiates, two of which Mallinckrodt has elected to develop.
Since the inception of the contract, we have received $27.5 million in upfront fees and milestones under the agreement. The upfront fees included a $4.0 million upfront license fee and a $1.5 million advance payment for formulation work we performed under the agreement. The milestone payments include four $0.5 million clinical development milestones and $5.0 million following the FDA's July
55
2013 acceptance for filing of the NDA for XARTEMIS™ XR (oxycodone hydrochloride and acetaminophen) Extended-Release Tablets (CII), previously known as MNK-795.
In March 2014, the FDA approved XARTEMIS™ XR for the management of acute pain severe enough to require opioid treatment and in patients for whom alternative treatment options (e.g., non-opioid analgesics) are ineffective, not tolerated or would otherwise be inadequate. The approval of the NDA triggered a $10.0 million contingent payment to us, which we received in April 2014. This $10.0 million contingent payment was recognized as revenue during the three months ended March 31, 2014. In May 2014, the FDA accepted for filing the NDA for MNK-155, and this acceptance triggered a $5.0 million contingent payment to us, which we received in June 2014. This $5.0 million contingent payment was recognized as revenue during the three months ended June 30, 2014. If MNK-155 is approved by the FDA, we will receive a $10.0 million contingent payment. We receive high single digit royalties on net sales of XARTEMIS™ XR, which was launched in March 2014, and will receive the same high single digit royalties on net sales of MNK-155 if that product is approved.
Janssen Pharmaceuticals, Inc. In August 2012, we entered into a license agreement with Janssen Pharma that grants Janssen Pharma a non-exclusive license to certain patents and other intellectual property rights to its Acuform drug delivery technology for the development and commercialization of tapentadol extended release products, including NUCYNTA® ER (tapentadol extended-release tablets). We received a $10.0 million upfront license fee, which was recognized as revenue in 2012, and receive low single digit royalties on net sales of NUCYNTA® ER in the United States, Canada and Japan from and after July 2, 2012 through December 31, 2021. We will not receive any royalties from Janssen Pharma on net sales of NUCYNTA® ER in the United States for any period after the consummation of the NUCYNTA® Acquisition.
Ironwood Pharmaceuticals, Inc. In July 2011, we entered into a collaboration and license agreement with Ironwood granting Ironwood a license for worldwide rights to certain patents and other intellectual property rights to our Acuform drug delivery technology for IW-3718, an Ironwood product candidate under evaluation for refractory GERD.
Since the inception of the contract, we received $3.4 million under the agreement, which includes an upfront payment, reimbursement of initial product formulation work and three milestones payments. We recognized as revenue a non-refundable payment of $1.0 million in March 2014 as a result of the initiation of clinical trials relating to IW-3718 by Ironwood. As we had no continuing involvement in this arrangement, we recognized the $1.0 million as revenue in March 2014.
Salix Pharmaceuticals, Inc. (formerly Santarus, Inc.) In August 2011, we entered into a commercialization agreement with Santarus, Inc., which was acquired by Salix Pharmaceuticals, Inc. (Salix) in January 2014, granting Salix exclusive rights to manufacture and commercialize Glumetza in the United States. The commercialization agreement supersedes the promotion agreement between the parties previously entered into in July 2008. Under the commercialization agreement, we granted Salix exclusive rights to manufacture and commercialize Glumetza in the United States in return for a royalty on Glumetza net sales.
Under the commercialization agreement, Salix is also required to pay us royalties on net product sales of Glumetza in the United States of 26.5% in 2011; 29.5% in 2012; 32.0% in 2013 and 2014; and 34.5% in 2015 and beyond, prior to generic entry of a Glumetza product. In the event of generic entry of a Glumetza product in the United States, the parties were to share proceeds equally based on a gross margin split.
Pursuant to the original promotion agreement, Salix paid us a $12.0 million upfront fee in July 2008. The upfront payment received was originally being amortized as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the promotion agreement related to manufacturing Glumetza and paying Salix promotion fees on gross margin of Glumetza. The commercialization agreement in August 2011 superseded the promotion agreement and
56
removed our promotion fee obligations and contemplated removal of its manufacturing obligations. The commercialization agreement included obligations with respect to manufacturing and regulatory transition to Salix and managing the patent infringement lawsuits against Sun Pharmaceutical Industries, Inc. (Sun) and Lupin Limited (Lupin). At the time of the commercialization agreement, all of these obligations were estimated to be completed in December 2013. During the fourth quarter of 2012, events occurred related to the transfer of manufacturing with one of the contract manufacturers of Glumetza that extended the estimated completion date of our manufacturing obligations with respect to 1000mg Glumetza to February 2016, which was the estimated date we expected our obligations would be completed under the commercialization agreement.
Effective October 1, 2014, the Company, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the supply of 1000mg Glumetza tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, we recognized the remaining unamortized deferred revenue of $1.9 million as of October 1, 2014 in the accompanying Consolidated Statements of Operations.
We recognized approximately $3.0 million, $1.4 million, and $3.3 million of revenue associated with this upfront license fee during 2014, 2013, and 2012, respectively.
Valeant Pharmaceuticals International, Inc. (formerly Biovail Laboratories, Inc.) In May 2002, we entered into a development and license agreement granting Valeant Pharmaceuticals International, Inc. (Valeant) an exclusive license in the United States and Canada to manufacture and market Glumetza. Under the terms of the agreement, we were responsible for completing the clinical development program in support of the 500mg Glumetza. In July 2005, Valeant received FDA approval to market Glumetza in the United States. In accordance with the license agreement, Valeant paid a $25.0 million license fee payment to Depomed.
Until September 30, 2014, we were recognizing the $25.0 million license fee payment as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the arrangement related to royalties that we were obligated to pay Valeant on net sales of the 500mg Glumetza in the United States and to use Valeant as the sole supplier of the 1000mg Glumetza. Effective October 1, 2014, the Company, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the manufacture and supply of 1000mg Glumetza tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, we recognized the remaining unamortized deferred revenue of $11.3 million as of October 1, 2014 in the accompanying Consolidated Statement of Operations. We recognized approximately $12.5 million, $1.6 million and $1.6 million of revenue associated with this upfront license fee during 2014, 2013 and 2012, respectively.
Licensing and Development Agreements Sold to PDL in October 2013
In October 2013, we sold to PDL our milestone and royalty interests in our license agreements in the type 2 diabetes therapeutic area (and any replacements for the agreements) for $240.5 million. The interests sold include royalty and milestone payments accruing from and after October 1, 2013 from: (a) Salix with respect to sales of Glumetza® (metformin HCL extended-release tablets) in the United States; (b) Merck with respect to sales of Janumet® XR (sitagliptin and metformin HCL extended-release); (c) Janssen with respect to potential future development milestones and sales of Janssen's investigational fixed-dose combination of Invokana® (canagliflozin) and extended-release metformin; (d) Boehringer Ingelheim with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to our license agreement with Boehringer Ingelheim; and (e) LG and Valeant SRL for sales of extended-release metformin in Korea and Canada, respectively. From and after October 1, 2013, PDL will receive all royalty and milestone payments due under the agreements until PDL has received payments equal to $481 million, after which we and PDL will share evenly all net payments received.
57
Cost of Sales
Cost of sales consists of costs of the active pharmaceutical ingredient, contract manufacturing and packaging costs, inventory write-downs, product quality testing, internal employee costs related to the manufacturing process, distribution costs and shipping costs related to our product sales. Total cost of sales for 2014, 2013 and 2012 was as follows (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cost of Sales |
$ | 15,146 | $ | 7,091 | $ | 6,039 | ||||
Dollar change from prior year |
8,055 | 1,052 | ||||||||
Percentage change from prior year |
113.6 | % | 17.4 | % | ||||||
We expect cost of sales to increase in 2015 as we expect product sales to increase from current levels.
Cost of sales increased in 2014 as a result of increased sales of Gralise® and the acquisition of CAMBIA® and Lazanda® products in 2013. We began selling CAMBIA® in December 2013. The fair value of inventories acquired included a step-up in the value of CAMBIA® inventories of $3.7 million which was being amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of CAMBIA® was $3.5 million and $0.2 million in 2014 and 2013, respectively. We began selling Lazanda® in August 2013. The fair value of inventories acquired included a step-up in the value of Lazanda® inventories of $0.6 million which is being amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of Lazanda® was $0.3 million and $0.1 million in 2014 and 2013, respectively. The fair value of inventories acquired included a step-up in the value of Zipsor® inventories of $1.9 million, of which $0.7 million was amortized to cost of sales in 2013 and $1.2 million was amortized to cost of sales in 2012.
Cost of sales for 2012 includes a $0.7 million charge related to slow-moving Gralise® starter pack inventory that is not expected to be sold prior to expiry. Cost of sales increased in 2013 as compared to 2012 as a result of increased product sales.
Research and Development Expense
Our research and development expenses currently include salaries, clinical trial costs, consultant fees, supplies, manufacturing costs for research and development programs and allocations of corporate costs. The scope and magnitude of future research and development expenses cannot be predicted at this time for our product candidate in research and development, as it is not possible to determine the nature, timing and extent of clinical trials and studies, the FDA's requirements for a particular drug and the requirements and level of participation, if any, by potential partners. As potential products proceed through the development process, each step is typically more extensive, and therefore more expensive, than the previous step. Therefore, success in development generally results in increasing expenditures until actual product approval. Total research and development expense for 2014, 2013, and 2012 was as follows (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Research and development expense |
$ | 7,116 | $ | 8,073 | $ | 15,462 | ||||
Dollar change from prior year |
(957 | ) | (7,389 | ) | ||||||
Percentage change from prior year |
–11.9 | % | –47.8 | % | ||||||
The decrease in research and development expense in 2014 as compared to 2013 was primarily driven by lower costs associated with our Sefelsa program, which ceased in the first quarter of 2013.
The decrease in research and development expense in 2013 as compared to 2012 was primarily due to reduced costs related to Sefelsa and the Phase 2 clinical trial for our DM-1992 program, which was completed in 2012.
58
We expect research and development expense in 2015 to increase from 2014 levels, primarily as a result of pediatric studies relating to CAMBIA® and Zipsor® that we intend to undertake in 2015. If we consummate the NUCYNTA® Acquisition, we will assume responsibility for certain post marketing regulatory requirements and pediatric studies, which may increase our research and development expenses for future periods.
Selling, General and Administrative Expense
Selling, general and administrative expenses primarily consist of personnel, contract personnel, marketing and promotion expenses associated with our commercial products, personnel expenses to support our administrative and operating activities, facility costs, and professional expenses, such as legal fees. Total selling, general and administrative expenses were as follows (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Selling, general and administrative expense |
$ | 121,126 | $ | 105,176 | $ | 97,646 | ||||
Dollar change from prior year |
15,950 | 7,530 | ||||||||
Percentage change from prior year |
15.2 | % | 7.7 | % | ||||||
The increase in selling, general and administrative expense in 2014 as compared to the same period in 2013 was primarily due to sales and marketing expense related to Lazanda® and CAMBIA® which we acquired in July 2013 and December 2013, respectively, and higher legal expenses related to our ongoing patent litigation.
The increase in selling, general and administrative expense in 2013 as compared to 2012 was primarily due to the build out of our commercial infrastructure and sales and marketing expense related to Zipsor® and Lazanda®, which we acquired in June 2012 and July 2013 respectively.
We expect selling, general and administrative expenses to increase in 2015 over 2014 levels, primarily as a result of continued build out of our commercial infrastructure. If we consummate the NUCYNTA® Acquisition, selling, general and administrative expenses will increase substantially as we intend to hire additional field sales persons along with additional personnel to support the increased sales force.
Amortization of Intangible Assets
| (In thousands) |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Amortization of intangible assets—Zipsor |
$ | 3,858 | $ | 3,853 | $ | 2,022 | ||||
Amortization of intangible assets—Lazanda |
1,167 | 484 | — | |||||||
Amortization of intangible assets—CAMBIA |
5,136 | 211 | — | |||||||
| | | | | | | | | | | |
|
$ | 10,161 | $ | 4,548 | $ | 2,022 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The Zipsor® product rights of $27.1 million have been recorded as intangible assets on the accompanying consolidated balance sheet and are being amortized over the estimated useful life of the asset on a straight-line basis through July 2019. Total amortization expense for 2014 was approximately $3.9 million. The estimated amortization expense for each of the four succeeding fiscal years is expected to be $3.9 million and $2.1 million for 2019.
59
The Lazanda® product rights of $10.5 million have been recorded as intangible assets on the accompanying consolidated balance sheet and are being amortized over the estimated useful life of the asset on a straight-line basis through August 2022. Amortization commenced on July 29, 2013, the date we acquired Lazanda®. Total amortization expense for 2014 was approximately $1.2 million. The estimated amortization expense for each of the five succeeding fiscal years is expected to be $1.2 million.
The CAMBIA® product rights of $51.4 million have been recorded as intangible assets on the accompanying consolidated balance sheet and are being amortized over the estimated useful life of the asset on a straight-line basis through December 2023. Amortization commenced on December 17, 2013, the date we acquired CAMBIA®. Total amortization expense for 2014 was approximately $5.1 million. The estimated amortization expense for each of the five succeeding fiscal years is expected to be $5.1 million.
Interest Income and Expense
| (In thousands) |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Interest and other income |
$ | 215 | $ | 662 | $ | 520 | ||||
Non-cash interest expense on PDL liability |
(14,646 | ) | (4,488 | ) | — | |||||
Interest expense |
(9,275 | ) | (911 | ) | (39 | ) | ||||
| | | | | | | | | | | |
Net interest (expense) income |
$ | (23,706 | ) | $ | (4,737 | ) | $ | 481 | ||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The decrease in interest and other income for the year ended December 31, 2014 compared to the year ended December 31, 2013 is attributable a $0.5 million gain from a bargain purchase relating to the CAMBIA® acquisition recorded in 2013, partially offset by higher interest income. Interest and other income for 2012 includes $0.1 million in respect of the gain from a bargain purchase relating to the Zipsor® acquisition.
The increase in non-cash interest expense on liability related to sale of future royalties for the year ended December 31, 2014 compared to the year ended December 31, 2013 is attributable to the royalty sale transaction that we completed in October 2013. The non-cash interest expense for the year ended December 31, 2014 includes the imputed interest expense until September 30, 2014, the date through which we are required to account for the royalty sale transaction as debt.
Interest expense primarily relates to the $345.0 million aggregate principal amount of the 2021 Notes issued in September 2014. The offering resulted in net proceeds of $334.2 million after deducting the underwriting discount and offering expenses of $10.4 million and $0.4 million, respectively. The interest rate for the 2021 Notes is fixed at 2.50% per annum and is payable semi-annually in arrears on March 1 and September 1 of each year, commencing on March 1, 2015. In 2014, we recognized $2.7 million of accrued coupon interest expense related to the 2021 Notes. Interest expense also includes $2.4 million recorded in 2014 for the change in the fair value of the contingent consideration obligations.
In accordance with accounting guidance on embedded conversion features, we valued and bifurcated the conversion option associated with the 2021 Notes from the respective host debt instrument and initially recorded the conversion option of $115.3 million for the 2021 Notes in "Shareholders' equity" on our consolidated balance sheets. The resulting debt discounts on the 2021 Notes are being amortized to interest expense at an effective interest rate of 9.34% over the contractual term of the notes. In 2014, we recognized $4.2 million of interest expense related to the amortization of these debt discounts. We expect interest expense to increase in future periods as the 2014 expense reflects accrued interest and amortization of the debt discount and debt issuance costs only since September 9, 2014.
60
Income Tax Provision (Benefit)
During 2014, we provided income tax expense of approximately $81.3 million that represents an effective tax rate of 38.2% on income from continuing operations. The difference between income tax expense of $81.3 million and the tax at the statutory rate of 35% on current year operations is principally due to state income tax, non-deductible stock and other individually immaterial non-deductible tax expenses.
During 2013, we recognized an income tax benefit of approximately $38.7 million which resulted primarily from our reversal of a valuation allowance on all of our U.S. federal deferred tax assets and most of our state deferred tax assets. Our 2013 effective tax rate from continuing operations was (846)%. The tax benefit represents a reversal of a valuation allowance on a significant portion of our U.S. federal and state deferred assets resulting in a deferred tax benefit of $103.2 million, offset by a current income tax provision of $64.5 million.
Our tax benefit for the year ended December 31, 2012 was due to Federal and state refundable credits offset by foreign taxes withheld on royalty revenue related to the Company's agreement with LG by the Republic of Korea.
Non-GAAP Financial Measures
To supplement our financial results presented on a U.S. generally accepted accounting principles, or GAAP, basis, we have included information about non-GAAP adjusted earnings and non-GAAP adjusted earnings per share, non-GAAP financial measures, as useful operating metrics for 2014, 2013 and 2012. We believe that the presentation of these non-GAAP financial measures, when viewed with our results under GAAP and the accompanying reconciliation, provides supplementary information to investors. We use these non-GAAP measures in connection with our own planning and forecasting purposes and for measuring our performance. These non-GAAP financial measures should be considered in addition to, and not a substitute for, or superior to, net income or other financial measures calculated in accordance with GAAP. Non-GAAP adjusted earnings and non-GAAP adjusted earnings per share for 2014, 2013 and 2012 are not based on any standardized methodology prescribed by GAAP and represent GAAP net income and GAAP earnings per share adjusted to exclude (1) non-cash PDL royalty revenue, net of related costs, (2) non-cash interest expense on PDL liability, (3) amortization related to product acquisitions, (4) stock-based compensation expense, (5) non-cash interest expense related to convertible debt, and to adjust (6) the income tax provision to reflect the estimated amounts payable in cash. Non-GAAP financial measures used by us may be calculated differently from, and therefore may not be comparable to, non-GAAP measures used by other companies.
The following table reconciles the Company's GAAP net income to non-GAAP adjusted income for 2014, 2013 and 2012:
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
GAAP net income |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
Non-cash PDL royalties, net of related costs |
(241,714 | ) | (17,909 | ) | — | |||||
Non-cash interest expense on PDL liability |
14,646 | 4,488 | — | |||||||
Non-cash interest expense on convertible debt |
4,200 | — | — | |||||||
Amortization related to product acquisitions |
16,853 | 6,352 | 4,243 | |||||||
Stock based compensation |
8,930 | 6,108 | 5,070 | |||||||
Non-cash income tax adjustment |
81,345 | (38,733 | ) | (91 | ) | |||||
| | | | | | | | | | | |
Non-GAAP adjusted earnings |
$ | 16,022 | $ | 3,619 | $ | (20,559 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Non-GAAP adjusted earnings per share |
$ | 0.26 | $ | 0.06 | $ | (0.37 | ) | |||
61
LIQUIDITY AND CAPITAL RESOURCES
| |
As of December 31, | ||||||
|---|---|---|---|---|---|---|---|
| (In thousands) |
2014 | 2013 | |||||
Cash, cash equivalents and marketable securities |
$ | 566,402 | $ | 276,017 | |||
The increase in cash, cash equivalents and marketable securities during 2014 is primarily attributable to $334.2 million of net proceeds received from the issuance of the 2021 Notes, partially offset by net income tax payments totaling approximately $58.3 million related to the year ended December 31, 2014.
Since inception through December 31, 2014, we have financed our product development efforts and operations primarily from private and public sales of equity securities, including convertible debt securities, the sale of rights to future royalties and milestones to PDL, upfront license, milestone and termination fees from collaborative and license partners, and product sales. In September 2014, we issued and sold $345.0 million of convertible senior notes due 2021.
We may incur operating losses in future years. We believe that our existing cash balances and cash we expect to generate from operations will be sufficient to fund our operations, and to meet our existing obligations for the foreseeable future, including our obligations under the 2021 notes. We base this expectation on our current operating plan, which may change as a result of many factors.
Our cash needs may vary materially from our current expectations because of numerous factors, including:
- •
- acquisitions or licenses of complementary businesses, products, technologies or companies;
- •
- sales of our marketed products;
- •
- expenditures related to our commercialization of Gralise®,
CAMBIA®, Zipsor®,
Lazanda® and NUCYNTA® (if we consummate the NUCYNTA® Acquisition)
- •
- milestone and royalty revenue we receive under our collaborative development and commercialization arrangements;
- •
- interest and principal payments on our 2021 Notes and any other indebtedness we may incur, including in connection with the
consummation of the NUCYNTA® Acquisition;
- •
- financial terms of definitive license agreements or other commercial agreements we may enter into;
- •
- results of research and development efforts;
- •
- changes in the focus and direction of our business strategy and/or research and development programs; and
- •
- results of clinical testing requirements of the FDA and comparable foreign regulatory agencies.
We fund our operations primarily through revenues from product sales and do not have any committed sources of capital. In connection with the signing of the Asset Purchase Agreement relating to the NUCYNTA® Acquisition, on January 15, 2015 we delivered $500.0 million into an escrow account to be credited against the total purchase price payable to Janssen Pharma upon the consummation of the NUCYNTA® Acquisition. In order to close the NUCYNTA® Acquisition we need to raise $550.0 million of capital (net of fees), and we will have to raise additional funds through the sale of our equity or equity-linked securities or through debt financing. We may be unable to raise such additional capital on favorable terms or at all in which case we could be liable to Janssen Pharma for a fee of $73.5 million that would be deducted from the amount in escrow and $426.5 million will be
62
returned to the Company. If we raise additional capital by selling our equity or equity-linked securities, the issuance of such securities could result in dilution of our shareholders' equity positions.
The inability to raise any additional capital that may be required to fund our future operations or product acquisitions and strategic transactions which we may pursue could have a material adverse effect on our company.
The following table summarizes our cash flow activities (in thousands):
| |
As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (In thousands) |
2014 | 2013 | 2012 | |||||||
Cash (used in) provided by operating activities |
$ | (55,917 | ) | $ | 9,754 | $ | (30,985 | ) | ||
Cash (used in) provided by investing activities |
(47,307 | ) | (38,036 | ) | 33,332 | |||||
Cash provided by financing activities |
347,218 | 243,880 | 2,686 | |||||||
Cash Flows from Operating Activities
Cash used in operating activities during 2014 was approximately $55.9 million, compared to cash provided by operating activities of $9.8 million during 2013. The difference was primarily due to net income for each respective period adjusted for income tax provision, non-cash interest expense on PDL liability, depreciation and amortization expense and stock-based compensation expense, partially offset by non-cash PDL royalty revenue of $242.8 million, movements in working capital and net income tax payments totaling approximately $58.3 million related to the year ended December 31, 2014. Cash provided by operating activities during 2013 was approximately $9.8 million, compared to cash used in operating activities of $31.0 million during 2012. The difference was primarily due to net income/loss for each respective period adjusted for movements in working capital, stock-based compensation, depreciation expense and income tax benefit. Cash used in operating activities during 2012 was primarily due to our net loss adjusted for movements in working capital, stock-based compensation and depreciation expense.
Cash Flows from Investing Activities
Net cash used in investing activities during 2014 was approximately $47.3 million primarily due to higher purchases of marketable securities relative to maturities of marketable securities. Net cash used in investing activities during 2013 was approximately $38.0 million, which was primarily due to cash used in the Lazanda® and CAMBIA® acquisitions offset by higher proceeds from maturities of marketable securities relative to purchases of marketable securities. Net cash provided by investing activities during 2012 was approximately $33.3 million and consisted of sales and maturities of marketable securities offset by purchases of marketable securities and approximately $26.4 million in cash paid for the acquisition of Zipsor® in June 2012.
Cash Flows from Financing Activities
Cash provided by financing activities during 2014 was $347.2 million, primarily due to $334.2 million of net proceeds received from the issuance of the 2021 Notes and $9.8 million of proceeds received from employee option exercises. Cash provided by financing activities during 2013 was approximately $243.9 million, which was primarily due to the sale of our interests in royalty and milestone payments to PDL for $240.5 million, with the remaining $3.7 million consisting of proceeds from employee option exercises. Cash provided by financing activities during 2012 was approximately $2.7 million and consisted of proceeds from employee option exercises.
63
Contractual Obligations
As of December 31, 2014, our contractual obligations are shown in the following table (in thousands):
| |
1 Year | 2 - 3 Years | 4 - 5 Years | More than 5 Years |
Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Convertible senior notes due 2021—principal |
$ | — | $ | — | $ | — | $ | 345,000 | $ | 345,000 | ||||||
Convertible senior notes due 2021—interest |
8,433 | 17,250 | 17,250 | 17,250 | 60,183 | |||||||||||
Operating lease(1) |
3,164 | 5,278 | 3,106 | 4,865 | 16,413 | |||||||||||
Purchase commitments |
3,267 | — | — | — | 3,267 | |||||||||||
| | | | | | | | | | | | | | | | | |
|
$ | 14,864 | $ | 22,528 | $ | 20,356 | $ | 367,115 | $ | 424,863 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
- (1)
- Amounts represent payments under a noncancelable office and laboratory lease and under an operating lease for vehicles used by our sales force.
At December 31, 2014, we had non-cancelable purchase orders and minimum purchase obligations of approximately $3.3 million under our manufacturing agreements related to Gralise®, CAMBIA®, Zipsor® and Lazanda®. The amounts disclosed only represent minimum purchase requirements. Actual purchases are expected to exceed these amounts.
In April 2012, we entered into an office and laboratory lease agreement to lease approximately 52,500 rentable square feet in Newark, California commencing on December 1, 2012 and an additional 8,000 rentable square feet commencing no later than December 1, 2015. The Newark lease included free rent for the first five months of the lease. Lease payments began in May 2013. We have the one-time right to terminate the lease in its entirety effective as of November 30, 2017 by delivering written notice to the landlord on or before December 1, 2016. In the event of such termination, we will pay the landlord the unamortized portion of the tenant improvement allowance, specified additional allowances made by the landlord, waived base rent and leasing commissions, in each case amortized at 8% interest. Our previous lease in Menlo Park, California ended in January 2013.
OFF-BALANCE SHEET ARRANGEMENTS
None.
RECENTLY ISSUED ACCOUNTING PRONOUNCEMENTS
In August 2014, the Financial Accounting Standards Board (FASB) issued guidance which requires management to assess an entity's ability to continue as a going concern and to provide related disclosures in certain circumstances. Under the new guidance, disclosures are required when conditions give rise to substantial doubt about an entity's ability to continue as a going concern within one year from the financial statement issuance date. The guidance is effective for annual periods ending after December 15, 2016, and all annual and interim periods thereafter. Early application is permitted. The adoption of this guidance will not have any impact on the Company's financial position and results of operations and, as this time, the Company does not expect any impact on its disclosures.
In June 2014, the FASB issued guidance which requires that a performance target that affects vesting, and that could be achieved after the requisite service period, be treated as a performance condition. As such, the performance target should not be reflected in estimating the grant date fair value of the award. This update further clarifies that compensation cost should be recognized in the period in which it becomes probable that the performance target will be achieved and should represent the compensation cost attributable to the period(s) for which the requisite service has already been rendered. The guidance is effective for annual periods beginning after December 15, 2015, and all
64
annual and interim periods thereafter. The Company does not anticipate that the adoption of this standard will have a material impact on its consolidated financial statements.
In May 2014, the FASB issued guidance which outlines a new, single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. This new revenue recognition model provides a five-step analysis in determining when and how revenue is recognized. The new model will require revenue recognition to depict the transfer of promised goods or services to customers in an amount that reflects the consideration a company expects to receive in exchange for those goods or services. The guidance is effective for annual periods beginning after December 15, 2016, and all annual and interim periods thereafter. The Company is currently assessing the impact that adopting this new accounting guidance will have on its consolidated financial statements and footnote disclosures.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
We had cash and cash equivalents totaling $488.7 million as of December 31, 2014. A significant portion of our cash and cash equivalents were invested in corporate debt securities and money market funds. Cash and cash equivalents are held for working capital purposes. We do not enter into investments for trading or speculative purposes. We believe that we do not have any material exposure to changes in the fair value as a result of changes in interest rates due to the short term nature of our cash equivalents.
As of December 31, 2014, we had $345.0 million aggregate principal amount of convertible senior notes outstanding, which are fixed rate instruments. Therefore, our results of operations are not subject to fluctuations in interest rates.
Foreign Currency Risk
We have not had any significant transactions in foreign currencies, nor did we have any significant balances that were due or payable in foreign currencies at December 31, 2014. Accordingly, significant changes in foreign currency rates would not have a material impact on our financial position and results of operations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required by this item are set forth beginning on page 69 of this report and are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
65
ITEM 9A. CONTROLS AND PROCEDURES
- (a)
- Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
At the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer, principal financial officer and principal accounting officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended (the Exchange Act). Based on this evaluation, our principal executive officer, our principal financial officer and principal accounting officer concluded that our disclosure controls and procedures were effective as of December 31, 2014 to ensure that information to be disclosed by us in this Annual Report on Form 10-K was recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission's rules and Form 10-K.
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission's rules and forms and that such information is accumulated and communicated to our management, including our chief executive officer, principal financial officer and principal accounting officer, as appropriate, to allow for timely decisions regarding required disclosure.
We intend to review and evaluate the design and effectiveness of our disclosure controls and procedures on an ongoing basis and to correct any material deficiencies that we may discover. Our goal is to ensure that our management has timely access to material information that could affect our business. While we believe the present design of our disclosure controls and procedures is effective to achieve our goal, future events affecting our business may cause us to modify our disclosure controls and procedures. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
- (b)
- Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our management, including our principal executive officer, principal financial officer and principal accounting officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2014. Ernst & Young LLP, our independent registered public accounting firm, has attested to and issued a report on the effectiveness of our internal control over financial reporting, which is included herein.
66
Report of Independent Registered Public Accounting Firm
The
Board of Directors and Shareholders of
Depomed, Inc.
We have audited Depomed, Inc.'s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Depomed, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Depomed, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Depomed, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive income, shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2014 of Depomed, Inc. and our report dated February 26, 2015 expressed an unqualified opinion thereon.
| /s/Ernst & Young LLP |
Redwood
City, California
February 26, 2015
67
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item with respect to executive officers, directors and corporate governance matters is incorporated by reference to the information set forth under the captions "Executive Officers and Senior Management" and "Election of Directors" in the company's Proxy Statement for the 2015 Annual Meeting of Shareholders.
The section entitled "Compliance Under Section 16(a) of the Securities Exchange Act of 1934" appearing in the Proxy Statement for the 2015 Annual Meeting of Shareholders sets forth the information concerning compliance by officers, directors and 10% shareholders of the company with Section 16 of the Exchange Act of 1934 and is incorporated herein by reference.
The Board has adopted a Code of Business Conduct and Ethics that applies to all of the Company's employees, officers and directors, including its principal executive officer and its principal financial officer. A copy of the code is available on the Company's website at: http://www.depomed.com and any amendments to or waivers of the code will posted to such website.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Executive Compensation" in the Proxy Statement for the 2015 Annual Meeting of Shareholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters" in the Proxy Statement for the 2015 Annual Meeting of Shareholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item is incorporated herein by reference to the information set forth under the captions "Directors" and "Certain Relationships and Related Transactions" in the Proxy Statement for the 2015 Annual Meeting of Shareholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item is incorporated herein by reference to the information set forth under the caption "Principal Accountant Fees and Services" in the Proxy Statement for the 2015 Annual Meeting of Shareholders.
68
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)
1. Financial Statements
2. Financial Statement Schedules
Schedule II is included on page 122 of this report. All other schedules are omitted because they are not required or the required information is included in the financial statements or notes thereto.
69
3. Exhibits:
| Exhibit | Footnote | Description of Document | ||||
|---|---|---|---|---|---|---|
| 3.1 | (1 | ) | Amended and Restated Articles of Incorporation | |||
| 3.2 | (2 | ) | Certificate of Amendment to Amended and Restated Articles of Incorporation | |||
| 3.3 | (3 | ) | Bylaws, as amended | |||
| 3.4 | (5 | ) | Certificate of Determination of Series RP Preferred Stock of the Company | |||
| 4.1 | (4 | ) | Rights Agreement, dated as of April 21, 2005, between the Company and Continental Stock Transfer and Trust Company as Rights Agent | |||
| 4.2 | (17 | ) | Senior Indenture dated as of September 9, 2014 between the Company and The Bank of New York Mellon Trust Company, N.A., as trustee | |||
| 4.3 | (17 | ) | First Supplemental Indenture dated as of September 9, 2014 between the Company and The Bank of New York Mellon Trust Company, N.A., as trustee, supplementing the Senior Indenture dated as of September 9, 2014 | |||
| 10.1 | (6 | ) | Offer Letter, dated June 14, 2006, between the Company and Matthew M. Gosling | |||
| 10.2 | (7 | ) | Form of Indemnification Agreement between the Company and its directors and executive officers | |||
| 10.3 | (10 | ) | 2004 Equity Incentive Plan, as amended | |||
| 10.4 | (10 | ) | Form of Restricted Stock Unit Award Agreement under the 2004 Equity Incentive Plan | |||
| 10.5 | (15 | ) | 2004 Employee Stock Purchase Plan, as amended | |||
| 10.6 | (8 | ) | Offer Letter, dated April 3, 2011, between the Company and James A. Schoeneck | |||
| †10.7 | (9 | ) | Commercial Manufacturing Services Agreement dated June 1, 2011 between the Company and Patheon Puerto Rico, Inc. | |||
| †10.8 | (9 | ) | Commercialization Agreement dated August 22, 2011 between the Company and Santarus, Inc. | |||
| 10.9 | (11 | ) | Offer Letter dated January 13, 2012 between the Company and August J. Moretti | |||
| 10.10 | (12 | ) | Lease dated April 4, 2012 between the Company and BMR-Pacific Research Center LP | |||
| 10.11 | (13 | ) | Asset Purchase Agreement dated June 21, 2012 between the Company and Xanodyne Pharmaceuticals, Inc. | |||
| †10.12 | (14 | ) | Asset Purchase Agreement, dated July 29, 2013, among the Company, Archimedes Pharma US Inc., Archimedes Pharma Ltd. and Archimedes Development Ltd. | |||
| †10.13 | (15 | ) | Royalty Purchase and Sale Agreement dated October 18, 2013, among the Company, Depo DR Sub, LLC and PDL BioPharma, Inc. | |||
| †10.14 | (15 | ) | Asset Purchase Agreement dated December 17, 2013 between the Company and Nautilus Pharmaceuticals, Inc. | |||
| 10.15 | (* | ) | 2014 Omnibus Incentive Plan and Forms of Award Documents | |||
70
| Exhibit | Footnote | Description of Document | ||||
|---|---|---|---|---|---|---|
| 10.16 | (* | ) | Depomed, Inc. Annual Bonus Plan, as adopted on February 5, 2015 | |||
| 10.17 | (16 | ) | Non-Employee Director Compensation and Grant Policy | |||
| 10.18 | (16 | ) | Form of Management Continuity Agreement between the Company and its executive officers | |||
| 10.19 | (18 | ) | Offer Letter dated as of July 14, 2014 between the Company and Srinivas G. Rao, M.D., Ph.D. | |||
| 10.20 | (18 | ) | Offer Letter dated as of July 31, 2014 between the Company and Richard Scott Shively | |||
| 10.21 | (17 | ) | Underwriting Agreement dated as of September 3, 2014 between the Company and Morgan Stanley & Co. LLC and RBC Capital Markets, LLC., as representatives of the several underwriters named therein | |||
| +10.22 | (* | ) | Asset Purchase Agreement dated January 15, 2015 between the Company and Janssen Pharmaceuticals, Inc. | |||
| 12.1 | (* | ) | Ratio of Earnings to Fixed Charges | |||
| 21 | (* | ) | List of Subsidiaries | |||
| 23.1 | (* | ) | Consent of Independent Registered Public Accounting Firm | |||
| 24.1 | (* | ) | Power of Attorney (included on signature page hereto) | |||
| 31.1 | (* | ) | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934 of James A. Schoeneck | |||
| 31.2 | (* | ) | Certification pursuant to Rule 13a-14(a) under the Securities Exchange Act of August J. Moretti | |||
| 32.1 | (* | ) | Certification pursuant to 18 U.S.C. Section 1350 of James A. Schoeneck | |||
| 32.2 | (* | ) | Certification pursuant to 18 U.S.C. Section 1350 of August J. Moretti | |||
| 101.INS | XBRL Instance Document | |||||
| 101.SCH | XBRL Taxonomy Extension Schema Document | |||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |||||
| 101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |||||
- (1)
- Incorporated
by reference to the Company's registration statement on Form SB-2 (File No. 333-25445)
- (2)
- Incorporated
by reference to the Company's Form 10-K filed on March 31, 2003
- (3)
- Incorporated
by reference to the Company's Form 8-K filed on April 19, 2005
- (4)
- Incorporated
by reference to the Company's Form 8-A filed on April 22, 2005
- (5)
- Incorporated
by reference to the Company's Form 10-Q filed on May 10, 2005
- (6)
- Incorporated
by reference to the Company's Form 8-K filed on June 30, 2006
- (7)
- Incorporated by reference to the Company's Form 10-Q filed on November 9, 2006
71
- (8)
- Incorporated
by reference to the Company's Form 10-Q filed on May 6, 2011
- (9)
- Incorporated
by reference to the Company's Form 10-Q filed on November 7, 2011
- (10)
- Incorporated
by reference to the Company's Form 8-K filed on January 17, 2012
- (11)
- Incorporated
by reference to the Company's Form 10-K filed on March 8, 2012
- (12)
- Incorporated
by reference to the Company's Form 10-Q filed on May 8, 2012
- (13)
- Incorporated
by reference to the Company's Form 10-Q filed on August 3, 2012
- (14)
- Incorporated
by reference to the Company's Form 10-Q filed on November 7, 2013
- (15)
- Incorporated
by reference to the Company's Form 10-K filed on March 17, 2014
- (16)
- Incorporated
by reference to the Company's Form 8-K filed on May 23, 2014
- (17)
- Incorporated
by reference to the Company's Form 8-K filed on September 9, 2014
- (18)
- Incorporated
by reference to the Company's Form 10-Q filed on November 6, 2014
- †
- Confidential
treatment granted
- +
- Confidential
treatment requested
- *
- Filed herewith
72
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the issuer, a corporation organized and existing under the laws of the State of California, has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized, in the City of Newark, State of California, on the 26th day of February 2015.
| Depomed, Inc. | ||||
By |
/s/ JAMES A. SCHOENECK James A. Schoeneck President and Chief Executive Officer |
|||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints James A. Schoeneck and August J. Moretti, and each of them acting individually, as his true and lawful attorneys-in-fact and agents, each with full power of substitution, for him in any and all capacities, to sign any and all amendments to this report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, with full power of each to act alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or his or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed by the following persons in the capacities and on the dates indicated.
Signature
|
|
|
||
|---|---|---|---|---|
| /s/ JAMES A. SCHOENECK James A. Schoeneck |
President and Chief Executive Officer (Principal Executive Officer) | February 26, 2015 | ||
/s/ AUGUST J. MORETTI August J. Moretti |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
February 26, 2015 |
||
/s/ PETER D. STAPLE Peter D. Staple |
Chairman of the Board of Directors |
February 26, 2015 |
||
/s/ VICENTE ANIDO JR., PH.D. Vicente Anido, Jr., Ph.D. |
Director |
February 26, 2015 |
73
Signature
|
|
|
||
|---|---|---|---|---|
| /s/ KAREN A. DAWES Karen A. Dawes |
Director | February 26, 2015 | ||
/s/ LOUIS J. LAVIGNE JR. Louis J. Lavigne Jr. |
Director |
February 26, 2015 |
||
/s/ SAMUEL R. SAKS, M.D. Samuel R. Saks, M.D. |
Director |
February 26, 2015 |
||
/s/ DAVID B. ZENOFF, D.B.A. David B. Zenoff, D.B.A. |
Director |
February 26, 2015 |
74
DEPOMED, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
DEPOMED, INC. CONSOLIDATED FINANCIAL STATEMENTS
75
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Shareholders of
Depomed, Inc.
We have audited the accompanying consolidated balance sheets of Depomed, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive income (loss), shareholders' equity, and cash flows for each of the three years in the period ended December 31, 2014. Our audits also included the financial statement schedule listed in the Index at Item 15(a)(2). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Depomed, Inc. at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Depomed, Inc.'s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework) and our report dated February 26, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood
City California
February 26, 2015
76
DEPOMED, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share amounts)
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
ASSETS |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 488,668 | $ | 244,674 | |||
Marketable securities |
70,773 | 27,263 | |||||
Accounts receivable, net |
27,008 | 11,451 | |||||
Receivables from collaborative partners |
1,070 | 10,824 | |||||
Inventories |
8,456 | 10,145 | |||||
Income taxes receivable |
4,030 | — | |||||
Deferred tax assets, net |
9,601 | 26,860 | |||||
Prepaid and other current assets |
8,014 | 5,828 | |||||
| | | | | | | | |
Total current assets |
617,620 | 337,045 | |||||
Marketable securities, long-term |
6,961 | 4,080 | |||||
Property and equipment, net |
7,055 | 8,340 | |||||
Intangible assets, net |
72,361 | 82,521 | |||||
Deferred tax assets, net, non-current |
— | 76,342 | |||||
Other assets |
7,068 | 325 | |||||
| | | | | | | | |
|
$ | 711,065 | $ | 508,653 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
LIABILITIES AND SHAREHOLDERS' EQUITY |
|||||||
Current liabilities: |
|||||||
Accounts payable and accrued liabilities |
$ | 52,686 | $ | 34,935 | |||
Income taxes payable |
— | 61,875 | |||||
Deferred license revenue |
— | 3,041 | |||||
Liability related to the sale of future royalties and milestones |
— | 56,357 | |||||
Other current liabilities |
4,813 | 649 | |||||
| | | | | | | | |
Total current liabilities |
57,499 | 156,857 | |||||
Deferred license revenue, non-current portion |
— | 12,475 | |||||
Contingent consideration liability |
14,252 | 11,264 | |||||
Liability related to the sale of future royalties and milestones, less current portion |
— | 177,624 | |||||
Deferred tax liabilities, net, non-current |
32,589 | — | |||||
Convertible debt |
229,891 | — | |||||
Other long-term liabilities |
12,387 | 13,017 | |||||
Commitments |
|||||||
Shareholders' equity: |
|||||||
Preferred stock, no par value, 5,000,000 shares authorized; Series A convertible preferred stock, 25,000 shares designated, 18,158 shares issued and surrendered, and zero shares outstanding at December 31, 2014 and December 31, 2013 |
— | — | |||||
Common stock, no par value, 100,000,000 shares authorized; 59,293,428 and 57,369,683 shares issued and outstanding at December 31, 2014 and December 31, 2013, respectively |
239,961 | 221,124 | |||||
Additional paid-in capital |
76,809 | 347 | |||||
Accumulated earnings (deficit) |
47,714 | (84,048 | ) | ||||
Accumulated other comprehensive loss, net of tax |
(37 | ) | (7 | ) | |||
| | | | | | | | |
Total shareholders' equity |
364,447 | 137,416 | |||||
| | | | | | | | |
|
$ | 711,065 | $ | 508,653 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
77
DEPOMED, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except share and per share amounts)
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Revenues: |
||||||||||
Product sales |
$ | 114,219 | $ | 58,302 | $ | 27,483 | ||||
Royalties |
1,821 | 45,003 | 44,535 | |||||||
License and other revenue |
31,515 | 12,796 | 18,798 | |||||||
Non-cash PDL royalty revenue |
242,808 | 18,104 | — | |||||||
| | | | | | | | | | | |
Total revenues |
390,363 | 134,205 | 90,816 | |||||||
Costs and expenses: |
||||||||||
Cost of sales |
15,146 | 7,091 | 6,039 | |||||||
Research and development expense |
7,116 | 8,073 | 15,462 | |||||||
Selling, general and administrative expense |
121,126 | 105,176 | 97,646 | |||||||
Amortization of intangible assets |
10,161 | 4,548 | 2,022 | |||||||
| | | | | | | | | | | |
Total costs and expenses |
153,549 | 124,888 | 121,169 | |||||||
| | | | | | | | | | | |
Income (loss) from operations |
236,814 | 9,317 | (30,353 | ) | ||||||
Other (expense) income: |
||||||||||
Interest and other income |
215 | 662 | 520 | |||||||
Interest expense |
(9,275 | ) | (911 | ) | (39 | ) | ||||
Non-cash interest expense on PDL liability |
(14,646 | ) | (4,488 | ) | — | |||||
| | | | | | | | | | | |
Total other (expense) income |
(23,706 | ) | (4,737 | ) | 481 | |||||
Net income (loss) before income taxes |
213,108 | 4,580 | (29,872 | ) | ||||||
(Provision for) benefit from income taxes |
(81,346 | ) | 38,733 | 91 | ||||||
| | | | | | | | | | | |
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Basic net income (loss) per share |
$ | 2.26 | $ | 0.76 | $ | (0.53 | ) | |||
Diluted net income (loss) per share |
$ | 2.05 | $ | 0.75 | $ | (0.53 | ) | |||
Shares used in computing basic net income (loss) per share |
58,292,633 | 56,736,009 | 55,892,563 | |||||||
Shares used in computing diluted net income (loss) per share |
66,307,364 | 57,543,979 | 55,892,563 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
78
DEPOMED, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(in thousands)
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
Unrealized gains (losses) on available-for-sale securities: |
||||||||||
Unrealized (losses) gains during period, net of income taxes |
(30 | ) | (37 | ) | 58 | |||||
Less: Reclassification adjustments for gains included to net income (loss), net of taxes |
— | 1 | 14 | |||||||
| | | | | | | | | | | |
Net unrealized (losses) gains on available-for-sale securities |
(30 | ) | (38 | ) | 44 | |||||
| | | | | | | | | | | |
Comprehensive income (loss) |
$ | 131,732 | $ | 43,275 | $ | (29,737 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
79
DEPOMED, INC.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS' EQUITY
(in thousands, except share amounts)
| |
Common Stock | |
Accumulated Other Comprehensive Loss |
|
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-In Capital |
Accumulated Earnings (Deficit) |
Shareholders' Equity |
||||||||||||||||
| |
Shares | Amount | |||||||||||||||||
Balances at Dec. 31, 2011 |
55,506,120 | $ | 203,511 | $ | — | $ | (13 | ) | $ | (97,580 | ) | $ | 105,918 | ||||||
Issuance of common stock upon exercise of options |
628,394 | 1,835 | — | — | — | 1,835 | |||||||||||||
Issuance of common stock under employee stock purchase plan |
203,389 | 851 | — | — | — | 851 | |||||||||||||
Issuance of common stock in conjunction with vesting of restricted stock units |
45,810 | 278 | — | — | — | 278 | |||||||||||||
Stock-based compensation |
— | 4,791 | — | — | — | 4,791 | |||||||||||||
Net income (loss) |
— | — | — | — | (29,781 | ) | (29,781 | ) | |||||||||||
Unrealized gain (loss) on available-for-sale securities |
— | — | — | 44 | — | 44 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Balances at Dec. 31, 2012 |
56,383,713 | $ | 211,266 | $ | — | $ | 31 | $ | (127,361 | ) | $ | 83,936 | |||||||
Issuance of common stock upon exercise of options |
621,090 | 2,782 | — | — | — | 2,782 | |||||||||||||
Issuance of common stock under employee stock purchase plan |
222,062 | 966 | — | — | — | 966 | |||||||||||||
Issuance of common stock in conjunction with vesting of restricted stock units |
142,818 | 765 | — | — | — | 765 | |||||||||||||
Stock-based compensation |
— | 5,345 | — | — | — | 5,345 | |||||||||||||
Windfall tax benefit |
— | — | 347 | — | — | 347 | |||||||||||||
Net income (loss) |
— | — | — | — | 43,313 | 43,313 | |||||||||||||
Unrealized gain (loss) on available-for-sale securities |
— | — | — | (38 | ) | — | (38 | ) | |||||||||||
| | | | | | | | | | | | | | | | | | | | |
Balances at Dec. 31, 2013 |
57,369,683 | $ | 221,124 | $ | 347 | $ | (7 | ) | $ | (84,048 | ) | $ | 137,416 | ||||||
Issuance of common stock upon exercise of options |
1,515,023 | 8,370 | — | — | — | 8,370 | |||||||||||||
Issuance of common stock under employee stock purchase plan |
177,036 | 1,536 | — | — | — | 1,536 | |||||||||||||
Issuance of common stock in conjunction with vesting of restricted stock units |
231,686 | 1,799 | — | — | — | 1,799 | |||||||||||||
Stock-based compensation |
— | 7,132 | — | — | — | 7,132 | |||||||||||||
Equity component of convertible debt issued, net of tax |
— | — | 73,272 | — | — | 73,272 | |||||||||||||
Windfall tax benefit |
— | — | 3,190 | — | — | 3,190 | |||||||||||||
Net income (loss) |
— | — | — | — | 131,762 | 131,762 | |||||||||||||
Unrealized gain (loss) on available-for-sale securities |
— | — | — | (30 | ) | — | (30 | ) | |||||||||||
| | | | | | | | | | | | | | | | | | | | |
Balances at Dec. 31, 2014 |
59,293,428 | $ | 239,961 | $ | 76,809 | $ | (37 | ) | $ | 47,714 | $ | 364,447 | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
80
DEPOMED, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Operating Activities |
||||||||||
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
Adjustments for non-cash items: |
||||||||||
Non-cash interest expense on PDL liability |
14,646 | 4,488 | — | |||||||
Non-cash PDL royalty revenue |
(242,808 | ) | (18,104 | ) | — | |||||
Depreciation and amortization |
12,025 | 6,161 | 2,561 | |||||||
Amortization of investments |
288 | 783 | 331 | |||||||
Gain on bargain purchase |
— | (484 | ) | (92 | ) | |||||
Provision for inventory obsolescence |
— | 347 | 584 | |||||||
Loss on disposal of property and equipment |
19 | — | 28 | |||||||
Stock-based compensation |
8,930 | 6,109 | 5,070 | |||||||
Change in fair value of contingent consideration and unfavorable contract |
2,791 | 909 | — | |||||||
Accretion of debt discount |
4,200 | — | — | |||||||
Deferred income tax provision (benefit) |
87,378 | (103,202 | ) | — | ||||||
Excess tax benefit from stock-based compensation |
(3,190 | ) | 347 | — | ||||||
Changes in assets and liabilities: |
||||||||||
Accounts receivable |
(15,453 | ) | (7,838 | ) | 807 | |||||
Receivables from collaborative partners |
10,170 | (726 | ) | (1,943 | ) | |||||
Inventories |
1,689 | 4,266 | (2,346 | ) | ||||||
Prepaid and other assets |
(2,190 | ) | 1,540 | (1,509 | ) | |||||
Income taxes receivable |
(4,030 | ) | — | — | ||||||
Accounts payable and other accrued liabilities |
15,076 | 10,833 | 5,661 | |||||||
Accrued compensation |
171 | 2,063 | 1,780 | |||||||
Income taxes payable |
(61,875 | ) | 62,222 | — | ||||||
Deferred revenue |
(15,516 | ) | (3,273 | ) | (12,136 | ) | ||||
| | | | | | | | | | | |
Net cash (used in) provided by operating activities |
(55,917 | ) | 9,754 | (30,985 | ) | |||||
| | | | | | | | | | | |
Investing Activities |
||||||||||
Purchases of property and equipment |
(599 | ) | (1,812 | ) | (6,880 | ) | ||||
Acquisition of business |
— | (52,725 | ) | (26,435 | ) | |||||
Acquisition of patents |
— | (150 | ) | — | ||||||
Purchases of marketable securities |
(73,754 | ) | (37,746 | ) | (38,462 | ) | ||||
Maturities of marketable securities |
27,046 | 53,056 | 61,405 | |||||||
Sales of marketable securities |
— | 1,341 | 43,704 | |||||||
| | | | | | | | | | | |
Net cash (used in) provided by investing activities |
(47,307 | ) | (38,036 | ) | 33,332 | |||||
| | | | | | | | | | | |
Financing Activities |
||||||||||
Proceeds from issuance of convertible debt |
345,000 | — | — | |||||||
Convertible debt issuance costs |
(10,775 | ) | — | — | ||||||
Proceeds from sale of future royalties and milestones to PDL |
— | 240,500 | — | |||||||
Proceeds from issuance of common stock |
9,803 | 3,727 | 2,686 | |||||||
Excess tax benefit from stock-based compensation |
3,190 | (347 | ) | — | ||||||
| | | | | | | | | | | |
Net cash provided by financing activities |
347,218 | 243,880 | 2,686 | |||||||
| | | | | | | | | | | |
Net increase in cash and cash equivalents |
243,994 | 215,598 | 5,033 | |||||||
Cash and cash equivalents at beginning of year |
244,674 | 29,076 | 24,043 | |||||||
| | | | | | | | | | | |
Cash and cash equivalents at end of year |
$ | 488,668 | $ | 244,674 | $ | 29,076 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplemental Disclosure of Cash Flow Information |
||||||||||
Cash paid during the period for: |
||||||||||
| | | | | | | | | | | |
Taxes, net of refunds |
$ | 58,318 | $ | 45 | $ | 144 | ||||
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
81
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization
Depomed, Inc. (Depomed or the Company) is a specialty pharmaceutical company focused on pain and other central nervous sytem (CNS) conditions. The products that comprise the Company's current specialty pharmaceutical business are Gralise® (gabapentin), a once-daily product for the management of postherpetic neuralgia (PHN) that we launched in October 2011, CAMBIA® (diclofenac potassium for oral solution), a product for the acute treatment of migraine attacks that we acquired in December 2013, Zipsor® (diclofenac potassium) liquid filled capsules, a product for the treatment of mild to moderate acute pain that we acquired in June 2012, and Lazanda® (fentanyl) nasal spray, a product for the management of breakthrough pain in cancer patients that we acquired in July 2013.
The Company also has a portfolio of royalty and milestone producing license agreements based on its proprietary Acuform® gastroretentive drug delivery technology with Mallinckrodt Inc. (Mallinckrodt), Ironwood Pharmaceuticals, Inc. (Ironwood) and Janssen Pharmaceuticals, Inc. (Janssen Pharma).
On October 18, 2013, the Company sold its interests in royalty and milestone payments under its license agreements in the Type 2 diabetes therapeutic area to PDL BioPharma, Inc. (PDL) for $240.5 million (PDL Transaction). The interests sold include royalty and milestone payments accruing from and after October 1, 2013: (a) from Salix Pharmaceuticals, Inc. (Salix) with respect to sales of Glumetza® (metformin HCL extended-release tablets) in the United States; (b) from Merck & Co., Inc. (Merck) with respect to sales of Janumet® XR (sitagliptin and metformin HCL extended-release); (c) from Janssen Pharmaceutica N.V. and Janssen Pharma (collectively, Janssen) with respect to potential future development milestones and sales of Janssen's investigational fixed-dose combination of Invokana® (canagliflozin) and extended-release metformin; (d) from Boehringer Ingelheim International GMBH (Boehringer Ingelheim) with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to the Company's license agreement with Boehringer Ingelheim; and (e) from LG Life Sciences Ltd. (LG) and Valeant International Bermuda SRL (Valeant SRL) for sales of extended-release metformin in Korea and Canada, respectively.
The Company has one product candidate under clinical development, DM-1992 for Parkinson's disease. DM-1992 completed a Phase 2 trial for Parkinson's disease, and the Company announced a summary of the results of that trial in November 2012. The Company continues to evaluate clinical and regulatory strategies and commercial prospects for DM-1992.
On January 15, 2015, the company entered into an Asset Purchase Agreement with Janssen Pharma, pursuant to which the Company will in the United States acquire from Janssen and its affiliates the rights to the NUCYNTA® franchise of pharmaceutical products as well as certain related assets for $1.05 billion in cash (NUCYNTA® Acquisition). The NUCYNTA® franchise includes NUCYNTA® ER (tapentadol extended release tablets) indicated for the management of pain, including neuropathic pain associated with diabetic peripheral neuropathy (DPN), severe enough to require daily, around-the-clock, long term opioid treatment, NUCYNTA® (tapentadol), an immediate release version of tapentadol, for management of moderate to severe acute pain in adults, and NUCYNTA® (tapentadol oral solution), an approved oral form of tapentadol that has not been commercialized. Upon execution of the Asset Purchase Agreement, the Company delivered a cash deposit in the amount of $500.0 million to JP Morgan Chase Bank, N.A., (Escrow Agent) in accordance with an Escrow Agreement, dated January 15, 2015, by and among the Company, Janssen Pharma and the Escrow Agent. The cash deposit will be credited against the total cash payable upon the consummation of the NUCYNTA® Acquisition. The Company needs to raise approximately $550.0 million (net of fees) in capital to consummate the NUCYNTA® Acquisition. The consummation of the NUCYNTA®
82
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Acquisition is subject to the satisfaction of a number of customary conditions. See "Note 16—Subsequent Events" for further information on the Asset Purchase Agreement.
Basis of Preparation
The Company's consolidated financial statements are prepared in accordance with the Financial Accounting Standards Board Accounting Standards Codification, or the Codification, which is the single source for all authoritative U.S. generally accepted accounting principles, or U.S. GAAP.
Reclassification
The Company has reclassified a royalty payable to PDL of $6.9 million from "Accounts payable and accrued liabilities" to the current portion of "Liability related to the sale of future royalties" in the accompanying consolidated balance sheet as of December 31, 2013 to conform to the current period presentation.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary, Depo DR Sub LLC (Depo DR Sub). All intercompany accounts and transactions have been eliminated on consolidation.
Depo DR Sub was formed in October 2013 for the sole purpose of facilitating the PDL Transaction. The Company contributed to Depo DR Sub all of its right, title and interest in each of the license agreements to receive royalty and milestone payments. Immediately following the transaction, Depo DR Sub sold to PDL, among other things, such right to receive royalty and milestone payments, for an upfront cash purchase price of $240.5 million.
The Company and Depo DR Sub continue to retain the duties and obligations under the specified license agreements. These include the collection of the royalty and milestone amounts due and enforcement of related provisions under the specified license agreements, among others. In addition, the Company and Depo DR Sub must prepare a quarterly distribution report relating to the specified license agreements, containing, among other items, the amount of royalty payments received by the Company, reimbursable expenses and set-offs. The Company and Depo DR Sub must also provide PDL with notice of certain communications, events or actions with respect to the specified license agreements and infringement of any underlying intellectual property.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Although management believes these estimates are based upon reasonable assumptions within the bounds of its knowledge of the Company's business and operations, actual results could differ materially from those estimates.
Recognizing and Measuring Assets Acquired and Liabilities Assumed in Business Combinations at Fair Value
The Company accounts for acquired businesses using the acquisition method of accounting, which requires that assets acquired and liabilities assumed be recorded at date of acquisition at their respective fair values. The fair value of the consideration paid, including contingent consideration, is assigned to the underlying net assets of the acquired business based on their respective fair values. Any
83
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
excess of the purchase price over the estimated fair values of the net assets acquired is recorded as goodwill or bargain purchase, as applicable.
Significant judgments are used in determining the estimated fair values assigned to the assets acquired and liabilities assumed and in determining estimates of useful lives of long-lived assets. Fair value determinations and useful life estimates are based on, among other factors, estimates of expected future net cash flows, estimates of appropriate discount rates used to present value expected future net cash flow streams, the assessment of each asset's life cycle, the impact of competitive trends on each asset's life cycle and other factors. These judgments can materially impact the estimates used to allocate acquisition date fair values to assets acquired and liabilities assumed and the resulting timing and amounts charged to, or recognized in current and future operating results. For these and other reasons, actual results may vary significantly from estimated results.
Any changes in the fair value of contingent consideration resulting from a change in the underlying inputs is recognized in operating expenses until the contingent consideration arrangement is settled. Changes in the fair value of contingent consideration resulting from the passage of time are recorded within interest expense until the contingent consideration is settled.
Revenue Recognition
The Company recognizes revenue from the sale of its products, royalties earned, and payments received and services performed under contractual arrangements.
Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred and title has passed, the price is fixed or determinable and the Company is reasonably assured of collecting the resulting receivable. Revenue arrangements with multiple elements are evaluated to determine whether the multiple elements meet certain criteria for dividing the arrangement into separate units of accounting, including whether the delivered element(s) have stand-alone value to the Company's customer or licensee. Where there are multiple deliverables combined as a single unit of accounting, revenues are deferred and recognized over the period that the Company remains obligated to perform services.
- •
- Product Sales—The Company sells commercial products to wholesale distributors and retail pharmacies. Products sales
revenue is recognized when title has transferred to the customer and the customer has assumed the risks and rewards of ownership, which typically occurs on delivery to the customer.
- •
- Product Sales Allowances—The Company recognizes product sales allowances as a reduction of product sales in the same
period the related revenue is recognized. Product sales allowances are based on amounts owed or to be claimed on the related sales. These estimates take into consideration the terms of the Company's
agreements with customers, historical product returns, rebates or discounts taken, estimated levels of inventory in the distribution channel, the shelf life of the product and specific known market
events, such as competitive pricing and new product introductions. If actual future results vary from the Company's estimates, the Company may need to adjust these estimates, which could have an
effect on product sales and earnings in the period of adjustment. The Company's sales allowances include:
- •
- Product Returns—The Company allows customers to return product for credit with respect to product that is within six months before and up to 12 months after its product expiration date. The Company estimates product returns on Gralise®, CAMBIA®, Zipsor® and Lazanda®. The Company also estimates returns on sales of Glumetza made by the Company through August
84
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
- •
- Wholesaler and Retail Pharmacy Discounts—The Company offers contractually determined discounts to certain wholesale
distributors and retail pharmacies that purchase directly from it. These discounts are either taken off-invoice at the time of shipment or paid to the customer on a quarterly basis one to two months
after the quarter in which product was shipped to the customer.
- •
- Prompt Pay Discounts—The Company offers cash discounts to its customers, (generally 2% of the sales price), as an
incentive for prompt payment. Based on the Company's experience, the Company expects its customers to comply with the payment terms to earn the cash discount.
- •
- Patient Discount Programs—The Company offers patient discount co-pay assistance programs in which patients receive certain
discounts off their prescriptions at participating retail pharmacies. The discounts are reimbursed by the Company approximately one month after the prescriptions subject to the discount are filled.
- •
- Medicaid Rebates—The Company participates in Medicaid rebate programs, which provide assistance to certain low-income
patients based on each individual state's guidelines regarding eligibility and services. Under the Medicaid rebate programs, the Company pays a rebate to each participating state, generally two to
three months after the quarter in which prescriptions subject to the rebate are filled.
- •
- Chargebacks—The Company provides discounts to authorized users of the Federal Supply Schedule (FSS) of the General Services Administration under an FSS contract with the
2011,
as the Company is financially responsible for return credits on Glumetza product the Company shipped as part of its commercialization agreement with Salix in August 2011. Under the terms of the
Zipsor® Asset Purchase Agreement, the Company assumed financial responsibility for returns of
Zipsor® product previously sold by Xanodyne Pharmaceuticals, Inc. (Xanodyne). Under the terms of the
CAMBIA® Asset Purchase Agreement, the Company also assumed financial responsibility for returns of
CAMBIA® product previously sold by Nautilus. The Company did not assume financial responsibility for returns of
Lazanda® product previously sold by Archimedes Pharma US Inc. See Note 15 for further information on the acquisition of
Zipsor®, CAMBIA® and
Lazanda®.
The shelf life of Gralise® is 24 to 36 months from the date of tablet manufacture. The shelf life of CAMBIA® is 24 to 48 months from the manufacture date. The
shelf life of Zipsor® is 36 months from the date of tablet manufacture. The shelf life of Lazanda® is 24 to 36 months from the manufacture date. The shelf life of
the 500mg Glumetza is 48 months from the date of tablet manufacture and the shelf life of the 1000mg Glumetza is 24 to 36 months from the date of tablet manufacture. The Company monitors
actual return history on an individual product lot basis since product launch, which provides it with a basis to reasonably estimate future product returns, taking into consideration the shelf life of
product at the time of shipment, shipment and prescription trends, estimated distribution channel inventory levels and consideration of the introduction of competitive products.
Because of the shelf life of the Company's products and its return policy of issuing credits with respect to product that is returned within six months before and up to 12 months after its
product expiration date, there may be a significant period of time between when the product is shipped and when the Company issues credit on a returned product. Accordingly, the Company may have to
adjust these estimates, which could have an effect on product sales and earnings in the period of adjustments.
85
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
- •
- Managed Care Rebates—The Company offers discounts under contracts with certain managed care providers. The Company
generally pays managed care rebates one to three months after the quarter in which prescriptions subject to the rebate are filled.
- •
- Medicare Part D Coverage Gap Rebates—The Company participates in the Medicare Part D Coverage Gap Discount
Program under which it provides rebates on prescriptions that fall within the "donut hole" coverage gap. The Company generally pays Medicare Part D Coverage Gap rebates two to three months
after the quarter in which prescriptions subject to the rebate are filled.
- •
- Royalties—Royalties are recognized as earned in accordance with the contract terms when royalties from licensees can be reliably measured and collectability is reasonably assured.
Department of Veterans Affairs. These federal entities purchase products from the wholesale distributors at a discounted price, and the wholesale distributors then charge back to the Company the difference between the current retail price and the price the federal entity paid for the product.
- •
- License and Collaborative Arrangements—Revenue from license and collaborative arrangements is recognized when the Company has substantially completed its obligations under the terms of the arrangement and the Company's remaining involvement is inconsequential and perfunctory. If the Company has significant continuing involvement under such an arrangement, license and collaborative fees are recognized over the estimated performance period. The Company
Royalties received from Mallinckrodt on sales of XARTEMIS XR™ and from Janssen Pharma on sales of NUCYNTA® ER are recognized in the period earned as the royalty amounts can be estimated and collectability is reasonably assured.
Until October 1, 2013, the Company received royalties from Salix Pharmaceuticals, Inc. (Salix) based on net sales of Glumetza and from Merck based on net sales of Janumet® XR. The royalties were recognized in the period earned as the royalty amounts could be estimated and collectability was reasonably assured.
In October 2013, the Company sold its interests in royalty and milestone payments under its license agreements in the Type 2 diabetes therapeutic area, including the Glumetza royalty and the Janumet® XR royalty, to PDL for $240.5 million. We had significant continuing involvement in the PDL transaction until September 30, 2014, primarily due to our obligation to act as the intermediary for the supply of 1000 mg Glumetza to Santarus, the licensee of Glumetza. Under the relevant accounting guidance, because of our significant continuing involvement, the $240.5 million payment received from PDL was accounted for as debt until September 30, 2014. As a result of debt accounting, even though the Company did not retain the related royalties and milestones under the transaction, the Company was required to record the revenue related to these royalties and milestones in its Consolidated Statement of Operations until September 30, 2014.
Effective October 1, 2014, the Company, Valeant, Salix and PDL executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the manufacture and supply of 1000mg Glumetza tablets. Consequently, the entire outstanding balance of the liability related to the sale of future royalties and milestones of approximately $147.0 million was recognized within "Non-cash PDL royalty revenue" in the accompanying Consolidated Statement of Operations.
86
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
recognizes milestone payments for its research and development collaborations upon the achievement of specified milestones if (1) the milestone is substantive in nature, and the achievement of the milestone was not reasonably assured at the inception of the agreement, (2) consideration earned relates to past performance and (3) the milestone payment is nonrefundable. A milestone is considered substantive if the consideration earned from the achievement of the milestone is consistent with the Company's performance required to achieve the milestone or consistent with the increase in value to the collaboration resulting from the Company's performance; the consideration earned relates solely to past performance; and the consideration earned is reasonable relative to all of the other deliverables and payments within the arrangement. License, milestones and collaborative fee payments received in excess of amounts earned are classified as deferred revenue until earned.
Stock-Based Compensation
Compensation expense for stock-based compensation is based on the single-option approach, includes an estimate for forfeitures and is recognized over the vesting term of the options using the straight-line method. The Company estimates forfeitures based on historical experience. The Company uses historical option exercise data to estimate the expected life of the options.
Research and Development Expense and Accruals
Research and development expenses include salaries, clinical trial costs, consultant fees, supplies, manufacturing costs for research and development programs and allocations of corporate costs. All such costs are charged to research and development expense as incurred. These expenses result from the Company's independent research and development efforts as well as efforts associated with collaborations. The Company reviews and accrues clinical trial expenses based on work performed, which relies on estimates of total costs incurred based on patient enrollment, completion of patient studies and other events. The Company follows this method since reasonably dependable estimates of the costs applicable to various stages of a research agreement or clinical trial can be made. Accrued clinical costs are subject to revisions as trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known.
Shipping and Handling Costs
Shipping and handling costs incurred for inventory purchases and product shipments are recorded in cost of sales in the Statements of Operations.
Advertising Costs
Costs associated with advertising are expensed as incurred. Advertising expense for the years ended December 31, 2014, 2013 and 2012 were $1.8 million, $2.4 million and $1.5 million, respectively.
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net income (loss) and other comprehensive income (loss). Other comprehensive income (loss) includes certain changes in equity of the Company that are excluded from net income (loss). Unrealized gains and losses on the Company's available-for-sale securities are reported separately in shareholders' equity and included in accumulated other comprehensive loss. Comprehensive income (loss) for the years ended December 31, 2014, 2013 and 2012 has been reflected in the consolidated statements of comprehensive income (loss).
87
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Cash, Cash Equivalents and Marketable Securities
The Company considers all highly liquid investments with an original maturity (at date of purchase) of three months or less to be cash equivalents. Cash and cash equivalents consist of cash on deposit with banks, money market instruments and commercial paper. The Company places its cash, cash equivalents and marketable securities with high quality U.S. government and financial institutions and to date has not experienced material losses on any of its balances. The Company records cash and cash equivalents at amortized cost, which approximates the fair value. All marketable securities are classified as available-for-sale since these instruments are readily marketable. These securities are carried at fair value, which is based on readily available market information, with unrealized gains and losses included in accumulated other comprehensive loss within shareholders' equity.
The Company uses the specific identification method to determine the amount of realized gains or losses on sales of marketable securities. We regularly review all of our investments for other-than-temporary declines in fair value. Our review includes the consideration of the cause of the impairment including the creditworthiness of the security issuers, the number of securities in an unrealized loss position and the severity and duration of the unrealized losses. When we determine that the decline in fair value of an investment is below our accounting basis and this decline is other-than-temporary, we reduce the carrying value of the security we hold and record a loss in the amount of such decline. Realized gains or losses have been insignificant and are included in interest and other income in the consolidated statements of operations.
Accounts Receivable
Trade accounts receivable are recorded net of allowances for cash discounts for prompt payment. To date the Company has not recorded a bad debt allowance due to the fact that the majority of its product revenue comes from sales to a limited number of financially sound companies who have historically paid their balances timely. The need for bad debt allowance is evaluated each reporting period based on our assessment of the credit worthiness of our customers or any other potential circumstances that could result in bad debt.
Receivables from collaborative partners represent amounts due from Salix, Merck and Janssen.
Inventories
Inventories are stated at the lower of cost or market with cost determined by specific manufactured lot. Inventories consist of costs of the active pharmaceutical ingredient, contract manufacturing and packaging costs. The Company writes-off the value of inventory for potentially excess, dated or obsolete inventories based on an analysis of inventory on hand and projected demand.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization (See Note 5 of the Notes to the Consolidated Financial Statements). Depreciation is calculated using the straight-line method over the estimated useful lives of the respective assets, as follows:
Furniture and office equipment |
3 - 5 years | |
Laboratory equipment |
3 - 5 years | |
Leasehold improvements |
Shorter of estimated useful life or lease term |
88
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Intangible Assets
Intangible assets consist of purchased developed technology and trademarks. We determine the fair values of acquired intangible assets as of the acquisition date. Discounted cash flow models are typically used in these valuations, which require the use of significant estimates and assumptions, including but not limited to, developing appropriate discount rates and estimating future cash flows from product sales and related expenses. We evaluate purchased intangibles for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Estimating future cash flows related to an intangible asset involves significant estimates and assumptions. If our assumptions are not correct, there could be an impairment loss or, in the case of a change in the estimated useful life of the asset, a change in amortization expense.
Our intangible assets relate to CAMBIA®, Zipsor® and Lazanda® product rights of $51.4 million, $27.3 million and $10.5 million, respectively, and have been recorded as intangible assets on the accompanying balance sheet, and are being amortized ratably over the estimated useful life of the asset through December 2023, July 2019 and August 2022, respectively. As of December 31, 2014 the carrying values of the intangible assets for CAMBIA®, Zipsor® and Lazanda®, were $46.0 million, $17.5 million and $8.9 million, respectively. Accumulated amortization relating to our intangible assets are $16.8 million and $6.7 million as of December 31, 2014 and 2013, respectively. Estimated amortization expense for 2015 through 2019 is $10.2 million, $10.2 million, $10.2 million, $10.2 million and $8.2 million, respectively. Estimated amortization expense for 2020 and thereafter is $23.4 million.
Net Income (Loss) Per Share
Basic net income (loss) per share is calculated by dividing the net income by the weighted-average number of shares of common stock outstanding during the period. Diluted net income (loss) per share is calculated by dividing the net income by the weighted-average number of shares of common stock outstanding during the period, plus potentially dilutive common shares, consisting of stock options and convertible debt. The Company uses the treasury-stock method to compute diluted earnings per share with respect to its stock options and equivalents. The Company uses the if-converted method to compute diluted earnings per share with respect to its convertible debt. For purposes of this calculation, options to purchase stock are considered to be potential common shares and are only
89
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
included in the calculation of diluted net income (loss) per share when their effect is dilutive. Basic and diluted earnings per common share are calculated as follows:
| (in thousands, except for per share amounts) |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Basic income (loss) per share |
||||||||||
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
Denominator |
58,293 | 56,736 | 55,893 | |||||||
| | | | | | | | | | | |
Basic net income (loss) per share |
$ | 2.26 | $ | 0.76 | $ | (0.53 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Diluted net income (loss) per share |
||||||||||
Numerator: |
||||||||||
Net income (loss) |
$ | 131,762 | $ | 43,313 | $ | (29,781 | ) | |||
Add interest expense on convertible debt, net of tax |
4,256 | — | — | |||||||
| | | | | | | | | | | |
|
$ | 136,018 | $ | 43,313 | $ | (29,781 | ) | |||
| | | | | | | | | | | |
Denominator: |
||||||||||
Denominator for basic income (loss) per share |
58,293 | 56,736 | 55,893 | |||||||
| | | | | | | | | | | |
Add effect of dilutive securities: |
||||||||||
Stock options and equivalents |
2,463 | 808 | — | |||||||
Convertible debt |
5,551 | — | — | |||||||
| | | | | | | | | | | |
Denominator for diluted net income (loss) per share: |
66,307 | 57,544 | 55,893 | |||||||
| | | | | | | | | | | |
Diluted net income (loss) per share |
$ | 2.05 | $ | 0.75 | $ | (0.53 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The following table sets forth outstanding potential shares of common stock that are not included in the computation of diluted net income (loss) per share because, to do so would be anti-dilutive:
| (in thousands) |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Convertible debt |
— | — | — | |||||||
Stock options and equivalents |
1,578 | 4,824 | 6,000 | |||||||
| | | | | | | | | | | |
Total potentially dilutive shares |
1,578 | 4,824 | 6,000 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Income Taxes
The Company's income tax policy is to record the estimated future tax effects of temporary differences between the tax bases of assets and liabilities and amounts reported in the Company's accompanying consolidated balance sheets, as well as operating loss and tax credit carryforwards. The Company follows the guidelines set forth in the applicable accounting guidance regarding the recoverability of any tax assets recorded on the consolidated balance Sheet and provides any necessary allowances as required. Determining necessary allowances requires the Company to make assessments about the timing of future events, including the probability of expected future taxable income and available tax planning opportunities.
The Company is subject to examination of its income tax returns by various tax authorities on a periodic basis. The Company regularly assesses the likelihood of adverse outcomes resulting from such examinations to determine the adequacy of its provision for income taxes. The Company has applied
90
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
the provisions of the applicable accounting guidance on accounting for uncertainty in income taxes, which requires application of a more-likely-than-not threshold to the recognition and de-recognition of uncertain tax positions. If the recognition threshold is met, the applicable accounting guidance permits the Company to recognize a tax benefit measured at the largest amount of tax benefit that, in the Company's judgment, is more than 50 percent likely to be realized upon settlement. It further requires that a change in judgment related to the expected ultimate resolution of uncertain tax positions be recognized in earnings in the period of such change.
Segment Information
The Company operates in one operating segment and has operations solely in the United States. To date, all of the Company's revenues from product sales are related to sales in the United States. The Company has recognized license and royalty revenue from license agreements in the territories of the United States, Canada and Korea.
Concentration of Risk
The Company invests cash that is currently not being used for operational purposes in accordance with its investment policy in low-risk debt securities of the U.S. Treasury, U.S. government sponsored agencies and very highly rated banks and corporations. The Company is exposed to credit risk in the event of a default by the institutions holding the cash equivalents and available-for sale securities to the extent recorded on the consolidated balance sheet.
The Company is subject to credit risk from its accounts receivable related to product sales and royalties. The majority of the Company's trade accounts receivable arises from product sales in the United States. Three wholesale distributors represented 26%, 27% and 35% of product shipments for the year ended December 31, 2014. These three customers individually comprised 33%, 32% and 35%, respectively, of product sales-related accounts receivable as of December 31, 2014. Three wholesale distributors represented 35%, 37% and 21% of product shipments for the year ended December 31, 2013. These three customers individually comprised 23%, 35% and 34%, respectively, of product sales-related accounts receivable as of December 31, 2013. Three wholesale distributors represented 39%, 40% and 14% of product shipments for the year ended December 31, 2012. These three customers individually comprised 46%, 42% and 5%, respectively, of product sales-related accounts receivable as of December 31, 2012. Accounts receivable balances related to product sales were $27.0 million, $11.4 million and $3.6 million for the years ended December 31, 2014, 2013 and 2012, respectively. The Company relies on a single third-party contract manufacturer organization in Puerto Rico to manufacture Gralise® and one third-party supplier for the supply of gabapentin, the active pharmaceutical ingredient in Gralise®. The Company also relies on single third-party contract suppliers: MiPharm, S.p.A., Accucaps and DPT Lakewood, Inc. for supply of CAMBIA®, Zipsor® and Lazanda® respectively.
Accounts receivable related to royalties was $0.5 million for the year ended December 31, 2014. Accounts receivable related to royalties was $7.2 million for the year ended December 31, 2013, of which $6.4 million was a receivable from Salix.
To date, the Company has not experienced any losses with respect to the collection of its accounts receivable and believes that its entire accounts receivable balances are collectible.
91
NOTE 1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Recent Accounting Pronouncements
In August 2014, the Financial Accounting Standards Board (FASB) issued guidance which requires management to assess an entity's ability to continue as a going concern and to provide related disclosures in certain circumstances. Under the new guidance, disclosures are required when conditions give rise to substantial doubt about an entity's ability to continue as a going concern within one year from the financial statement issuance date. The guidance is effective for annual periods ending after December 15, 2016, and all annual and interim periods thereafter. Early application is permitted. The adoption of this guidance will not have any impact on the Company's financial position and results of operations and, as this time, the Company does not expect any impact on its disclosures.
In June 2014, the FASB issued guidance which requires that a performance target that affects vesting, and that could be achieved after the requisite service period, be treated as a performance condition. As such, the performance target should not be reflected in estimating the grant date fair value of the award. This update further clarifies that compensation cost should be recognized in the period in which it becomes probable that the performance target will be achieved and should represent the compensation cost attributable to the period(s) for which the requisite service has already been rendered. The guidance is effective for annual periods beginning after December 15, 2015, and all annual and interim periods thereafter. The Company does not anticipate that the adoption of this standard will have a material impact on its consolidated financial statements.
In May 2014, the FASB issued guidance which outlines a new, single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. This new revenue recognition model provides a five-step analysis in determining when and how revenue is recognized. The new model will require revenue recognition to depict the transfer of promised goods or services to customers in an amount that reflects the consideration a company expects to receive in exchange for those goods or services. The guidance is effective for annual periods beginning after December 15, 2016, and all annual and interim periods thereafter. The Company is currently assessing the impact that adopting this new accounting guidance will have on its consolidated financial statements and footnote disclosures.
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS
Mallinckrodt Inc. (formerly Covidien, Ltd.)
In November 2008, the Company entered into a license agreement related to acetaminophen/opiate combination products with Mallinckrodt. The license agreement grants Mallinckrodt worldwide rights to utilize Depomed's Acuform technology for the exclusive development of up to four products containing acetaminophen in combination with opiates, two of which Mallinckrodt has elected to develop.
Since the inception of the contract, the Company has received $27.5 million in upfront fees and milestones under the agreement. The upfront fees included a $4.0 million upfront license fee and a $1.5 million advance payment for formulation work the Company performed under the agreement. The milestone payments include four $0.5 million clinical development milestones and $5.0 million following the FDA's July 2013 acceptance for filing of the NDA for XARTEMIS™ XR (oxycodone hydrochloride and acetaminophen) Extended-Release Tablets (CII), previously known as MNK-795. In March 2014, the FDA approved XARTEMIS™ XR for the management of acute pain severe enough to require opioid treatment and in patients for whom alternative treatment options (e.g., non-opioid analgesics)
92
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
are ineffective, not tolerated or would otherwise be inadequate. The approval of the NDA triggered a $10.0 million milestone payment to the Company, which the Company received in April 2014. This $10.0 million milestone payment was recognized as revenue during the three months ended March 31, 2014. In May 2014, the FDA accepted for filing the NDA for MNK-155, and this acceptance triggered a $5.0 million milestone payment to the Company, which the Company received in June 2014. This $5.0 million milestone payment was recognized as revenue during the three months ended June 30, 2014. If MNK-155 is approved by the FDA, the Company will receive a $10.0 million milestone payment. The Company receives high single digit royalties on net sales of XARTEMIS™ XR, which was launched in March 2014, and will receive the same high single digit royalties on net sales of MNK-155 if that product is approved.
Janssen Pharmaceuticals, Inc.
In August 2012, the Company entered into a license agreement with Janssen Pharma that grants Janssen Pharma a non-exclusive license to certain patents and other intellectual property rights to its Acuform drug delivery technology for the development and commercialization of tapentadol extended release products, including NUCYNTA® ER (tapentadol extended-release tablets). The Company received a $10.0 million upfront license fee, which was recognized as revenue in 2012, and receives low single digit royalties on net sales of NUCYNTA® ER in the U.S., Canada and Japan from and after July 2, 2012 through December 31, 2021.
Janssen Pharmaceutica N.V
In August 2010, the Company entered into a license agreement with Janssen that grants Janssen a non-exclusive license to certain patents related to Depomed's Acuform drug delivery technology to be used in developing fixed dose combinations of extended release metformin and Janssen's type 2 diabetes product candidate canagliflozin. In 2010, the Company received $10.0 million in upfront and milestone payments which was recognized as revenue. The Company also granted Janssen a right to reference the Glumetza NDA in Janssen's regulatory filings covering the products. In February 2013 and December 2013, the Company completed two projects for Janssen related to this program and recognized $2.2 million in revenue during the first quarter of 2013 and $1.4 million during the fourth quarter of 2013.
In October 2013, the Company sold all of its rights to future payments under the license agreement relating to fixed dose combinations of metformin and canaglifozin to PDL.
Ironwood Pharmaceuticals, Inc.
In July 2011, the Company entered into a collaboration and license agreement with Ironwood granting Ironwood a license for worldwide rights to certain patents and other intellectual property rights to Depomed's Acuform drug delivery technology for IW-3718, an Ironwood product candidate under evaluation for refractory GERD.
Since the inception of the contract, the Company has received $3.4 million under the agreement, which includes an upfront payment, reimbursement of initial product formulation work and three milestones payments. The Company recognized a non-refundable milestone payment of $1.0 million in March 2014 as a result of the initiation of clinical trials relating to IW-3718 by Ironwood. As the non-refundable milestone was both substantive in nature and related to past performance, the Company recognized the $1.0 million as revenue in March 2014.
93
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
Salix Pharmaceuticals, Inc. (formerly Santarus, Inc.)
In August 2011, the Company entered into a commercialization agreement with Santarus, Inc., which was acquired by Salix in January 2014, granting Salix exclusive rights to manufacture and commercialize Glumetza in the United States. The commercialization agreement supersedes the promotion agreement between the parties previously entered into in July 2008. Under the commercialization agreement, we granted Salix exclusive rights to manufacture and commercialize Glumetza in the United States in return for a royalty on Glumetza net sales.
Under the commercialization agreement, Salix is also required to pay the Company royalties on net product sales of Glumetza in the United States of 26.5% in 2011; 29.5% in 2012; 32.0% in 2013 and 2014; and 34.5% in 2015 and beyond, prior to generic entry of a Glumetza product. In the event of generic entry of a Glumetza product in the United States, the parties were to share proceeds equally based on a gross margin split. Royalty revenue from Salix for the years ended December 31, 2013 and 2012 was $42.1 million and $42.8 million, respectively. In October 2013, the Company sold its interest in the Glumetza royalties to PDL.
Pursuant to the original promotion agreement, Salix paid us a $12.0 million upfront fee in July 2008. The upfront payment received was originally being amortized as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the promotion agreement related to manufacturing Glumetza and paying Salix promotion fees on gross margin of Glumetza. The commercialization agreement in August 2011 superseded the promotion agreement and removed our promotion fee obligations and contemplated removal of its manufacturing obligations. The commercialization agreement included obligations with respect to manufacturing and regulatory transition to Salix and managing the patent infringement lawsuits against Sun Pharmaceutical Industries, Inc. (Sun) and Lupin Limited (Lupin). At the time of the commercialization agreement, all of these obligations were estimated to be completed in December 2013. During the fourth quarter of 2012, events occurred related to the transfer of manufacturing with one of the contract manufacturers of Glumetza that extended the estimated completion date of our manufacturing obligations with respect to 1000 mg Glumetza to February 2016, which is the estimated date we expected our obligations would be completed under the commercialization agreement. Effective October 1, 2014, Depomed, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of Depomed in the supply of Glumetza 1000mg tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, the Company recognized the remaining unamortized deferred revenue of $1.9 million as of October 1, 2014 in the accompanying Consolidated Statement of Operations.
We recognized approximately $3.0 million, $1.4 million, and $3.3 million of revenue associated with this upfront license fee during 2014, 2013, and 2012, respectively.
Valeant Pharmaceuticals International, Inc. (formerly Biovail Laboratories, Inc.)
In May 2002, we entered into a development and license agreement granting Valeant Pharmaceuticals International, Inc. (Valeant) an exclusive license in the United States and Canada to manufacture and market Glumetza. Under the terms of the agreement, we were responsible for completing the clinical development program in support of the 500mg Glumetza. In July 2005, Valeant received FDA approval to market Glumetza in the United States. In accordance with the license agreement, Valeant paid a $25.0 million license fee payment to Depomed.
94
NOTE 2. LICENSE AND COLLABORATIVE ARRANGEMENTS (Continued)
Until September 30, 2014, we were recognizing the $25.0 million license fee payment as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the arrangement related to royalties that we were obligated to pay Valeant on net sales of the 500mg Glumetza in the United States and to use Valeant as the sole supplier of the 1000mg Glumetza. Effective October 1, 2014, Depomed, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of Depomed in the manufacture and supply of 1000mg Glumetza tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, the Company recognized the remaining unamortized deferred revenue of $11.3 million as of October 1, 2014 in the accompanying Consolidated Statement of Operations. We recognized approximately $12.5 million, $1.6 million and $1.6 million of revenue associated with this upfront license fee during 2014, 2013 and 2012, respectively.
Patheon Puerto Rico, Inc.
In September 2011, the Company entered into a manufacturing agreement with Patheon Puerto Rico, Inc. (Patheon), pursuant to which Patheon manufactures, packages and supplies commercial quantities of Gralise®.
Under the agreement, the Company provides rolling forecasts to Patheon of its requirements for the product, a portion of which will be considered a firm purchase order. At December 31, 2014, the Company had non-cancelable purchase orders and minimum purchase obligations of approximately $1.3 million under the manufacturing agreement with Patheon for the manufacture of Gralise®. The Company may obtain a portion of its product requirements from a second manufacturing source. The Company is responsible for providing Patheon with the active pharmaceutical ingredient in Gralise®. The agreement will expire on May 31, 2016, subject to early termination under certain circumstances.
NOTE 3. MARKETABLE SECURITIES
Securities classified as cash and cash equivalents and available-for-sale marketable securities as of December 31, 2014 and 2013 are summarized below (in thousands). Estimated fair value is based on quoted market prices for these investments.
December 31, 2014
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cash and cash equivalents: |
|||||||||||||
Cash |
$ | 22,452 | $ | — | $ | — | $ | 22,452 | |||||
Money market funds |
179,923 | — | — | 179,923 | |||||||||
Corporate debt securities |
286,292 | 3 | (2 | ) | 286,293 | ||||||||
| | | | | | | | | | | | | | |
Total cash and cash equivalents |
$ | 488,667 | $ | 3 | $ | (2 | ) | $ | 488,668 | ||||
Available-for-sale securities: |
|||||||||||||
Total maturing within 1 year and included in marketable securities: |
|||||||||||||
Corporate debt securities |
$ | 70,777 | 1 | (5 | ) | $ | 70,773 | ||||||
Total maturing between 1 and 2 years and included in marketable securities: |
|||||||||||||
Corporate debt securities |
6,974 | — | (13 | ) | 6,961 | ||||||||
| | | | | | | | | | | | | | |
Total available-for-sale securities |
$ | 77,751 | $ | 1 | $ | (18 | ) | $ | 77,734 | ||||
| | | | | | | | | | | | | | |
Total cash, cash equivalents and marketable securities |
$ | 566,418 | $ | 4 | $ | (20 | ) | $ | 566,402 | ||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
95
NOTE 3. MARKETABLE SECURITIES (Continued)
December 31, 2013
|
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cash and cash equivalents: |
|||||||||||||
Cash |
$ | 26,728 | $ | — | $ | — | $ | 26,728 | |||||
Money market funds |
217,946 | — | — | 217,946 | |||||||||
| | | | | | | | | | | | | | |
Total cash and cash equivalents |
$ | 244,674 | $ | — | $ | — | $ | 244,674 | |||||
Available-for-sale securities: |
|||||||||||||
Total maturing within 1 year and included in marketable securities: |
|||||||||||||
Corporate debt securities |
$ | 12,440 | $ | 8 | $ | (2 | ) | $ | 12,446 | ||||
Government agency debt securities |
14,814 | 3 | — | 14,817 | |||||||||
Total maturing between 1 and 2 years and included in marketable securities: |
|||||||||||||
Corporate debt securities |
4,075 | 5 | — | 4,080 | |||||||||
| | | | | | | | | | | | | | |
Total available-for-sale securities |
$ | 31,329 | $ | 16 | $ | (2 | ) | $ | 31,343 | ||||
| | | | | | | | | | | | | | |
Total cash, cash equivalents and marketable securities |
$ | 276,003 | $ | 16 | $ | (2 | ) | $ | 276,017 | ||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The Company considers all highly liquid investments with a maturity at date of purchase of three months or less to be cash equivalents. Cash and cash equivalents consist of cash on deposit with banks, money market instruments and corporate debt securities. The Company invests its cash in marketable securities with U.S. Treasury and government agency securities, and high quality securities of financial and commercial institutions. To date, the Company has not experienced material losses on any of its balances. These securities are carried at fair value, which is based on readily available market information, with unrealized gains and losses included in "accumulated other comprehensive loss" within shareholders' equity on the consolidated balance sheets. The Company uses the specific identification method to determine the amount of realized gains or losses on sales of marketable securities. Realized gains or losses have been insignificant and are included in "interest and other income" in the consolidated statement of operations.
At December 31, 2014, the Company had 45 securities in an unrealized loss position. The following table shows the gross unrealized losses and fair value of the Company's investments with unrealized losses that are not deemed to be other-than-temporarily impaired, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2014 (in thousands):
| |
Less than 12 months | 12 months or greater | Total | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Fair Value | Gross Unrealized Losses |
Fair Value | Gross Unrealized Losses |
Fair Value | Gross Unrealized Losses |
|||||||||||||
Corporate debt securities |
$ | 80,854 | $ | (20 | ) | $ | — | $ | — | $ | 80,854 | $ | (20 | ) | |||||
| | | | | | | | | | | | | | | | | | | | |
Total available-for-sale |
$ | 80,854 | $ | (20 | ) | $ | — | $ | — | $ | 80,854 | $ | (20 | ) | |||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
The gross unrealized losses above were caused by interest rate increases. No significant facts or circumstances have arisen to indicate that there has been any deterioration in the creditworthiness of the issuers of the securities held by the Company. Based on the Company's review of these securities, including the assessment of the duration and severity of the unrealized losses and the Company's ability and intent to hold the investments until maturity, there were no material other-than-temporary impairments for these securities at December 31, 2014.
96
NOTE 3. MARKETABLE SECURITIES (Continued)
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs.
- •
- Level 1: Quoted prices in active markets for identical assets or liabilities.
- •
- Level 2: Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar
assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets
or liabilities.
- •
- Level 3: Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The following table represents the Company's fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2014 (in thousands):
| |
Level 1 | Level 2 | Level 3 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Money market funds |
$ | 179,923 | $ | — | $ | — | $ | 179,923 | |||||
Commercial Paper |
— | 253,837 | — | 253,837 | |||||||||
Corporate debt securities |
110,190 | — | — | 110,190 | |||||||||
| | | | | | | | | | | | | | |
Total |
$ | 290,113 | $ | 253,837 | $ | — | $ | 543,950 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Liabilities: |
|||||||||||||
Contingent consideration—Zipsor |
$ | — | $ | — | $ | 1,800 | $ | 1,800 | |||||
Contingent consideration—Lazanda |
— | — | 11,209 | 11,209 | |||||||||
Contingent consideration—CAMBIA |
— | — | 1,243 | 1,243 | |||||||||
Unfavorable contract assumed |
— | — | 3,343 | 3,343 | |||||||||
| | | | | | | | | | | | | | |
|
$ | — | $ | — | $ | 17,595 | $ | 17,595 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The fair value measurement of the contingent consideration obligations arises from the Zipsor®, CAMBIA® and Lazanda® acquisitions and relates to fair value of the potential future milestone payments and royalties payable under the respective agreements which are determined using Level 3 inputs. The key assumptions in determining the fair value are the discount rate and the probability assigned to the potential milestones and royalties being achieved. At each reporting date, the Company re-measures the contingent consideration obligation arising from the above acquisitions to their estimated fair values. Any changes in the fair value of contingent consideration resulting from a change in the underlying is recognized in operating expenses until the contingent consideration arrangement is settled. Changes in the fair value of contingent consideration resulting from the passage of time are recorded within interest expense until the contingent consideration is settled. The table below provides a summary of the changes in fair value recorded in interest expense and selling, general and administrative expense for the year ended December 31, 2014. Changes in fair value included within interest expense in the accompanying Consolidated Statement of Operations was $0.9 million and $0.1 million for the years ended December 31, 2013 and 2012, respectively.
The liability for the unfavorable contract assumed represents an obligation for the Company to make certain payments to a vendor upon the achievement of certain milestones by such vendor. This contract was entered into by Nautilus Neurosciences, Inc. (Nautilus) as part of a legal settlement unrelated to the CAMBIA® acquisition. The liability of $3.3 million recorded above, as of December 31
97
NOTE 3. MARKETABLE SECURITIES (Continued)
2014, represents the fair value of the amounts by which the contract terms are unfavorable compared to the current market pricing and a probability—weighted assessment of the likelihood that the stipulated milestones will be achieved by the third party. The contract may be terminated if the third party fails to achieve these milestones, in which case the fair value of the liability as of the date of the termination will be reversed on the consolidated balance sheet and reflected in the consolidated statement of operations. Any changes in the fair value of this liability resulting from a change in the underlying inputs are recognized in operating expenses until the contract is settled. Changes in the fair value of the liability resulting from the passage of time are recorded within interest expense until the contract is settled.
The table below provides a summary of the changes in fair value of all financial liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for the year ended December 31, 2014 (in thousands):
| |
Balance at December 31, 2013 |
Changes in fair value recorded in interest expense |
Changes in fair value recorded in selling, general and administrative expense |
Balance at December 31, 2014 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Liabilities: |
|||||||||||||
Contingent consideration obligations—Zipsor® |
$ | 1,638 | $ | 162 | $ | — | $ | 1,800 | |||||
Contingent consideration obligations—Lazanda® |
8,616 | 1,533 | 1,060 | 11,209 | |||||||||
Contingent consideration obligations—CAMBIA® |
1,010 | 233 | — | 1,243 | |||||||||
Unfavorable contract assumed |
3,540 | 445 | (642 | ) | 3,343 | ||||||||
| | | | | | | | | | | | | | |
Total |
$ | 14,804 | $ | 2,373 | $ | 418 | $ | 17,595 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The estimated fair value of the 2.50% Convertible Senior Notes Due 2021, which the Company issued on September 9, 2014 (the 2021 Notes), is based on a market approach. The estimated fair value was approximately $375.2 million (par value $345.0 million) as of December 31, 2014 and represents a Level 2 valuation. When determining the estimated fair value of the Company's long-term debt, the Company used a commonly accepted valuation methodology and market-based risk measurements that are indirectly observable, such as credit risk.
The following table represents the Company's fair value hierarchy for its financial assets measured at fair value on a recurring basis as of December 31, 2013 (in thousands):
| |
Level 1 | Level 2 | Level 3 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Money market funds |
$ | 217,946 | $ | — | $ | — | $ | 217,946 | |||||
Corporate debt securities |
— | 16,526 | — | 16,526 | |||||||||
Government agency debt securities |
— | 14,817 | — | 14,817 | |||||||||
| | | | | | | | | | | | | | |
Total |
$ | 217,946 | $ | 31,343 | $ | — | $ | 249,289 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Liabilities: |
|||||||||||||
Contingent consideration—Zipsor |
$ | — | $ | — | $ | 1,638 | $ | 1,638 | |||||
Contingent consideration—Lazanda |
— | — | 8,616 | 8,616 | |||||||||
Contingent consideration—CAMBIA |
— | — | 1,010 | 1,010 | |||||||||
Unfavorable contract assumed |
— | — | 3,540 | 3,540 | |||||||||
| | | | | | | | | | | | | | |
|
$ | — | $ | — | $ | 14,804 | $ | 14,804 | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
98
NOTE 4. INVENTORIES
Inventories consist of finished goods, raw materials and work in process and are stated at the lower of cost or market and consist of the following (in thousands):
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
Raw materials |
$ | 2,141 | $ | 1,951 | |||
Work-in-process |
1,348 | 181 | |||||
Finished goods |
4,984 | 9,056 | |||||
Less: allowance for obsolescence |
(17 | ) | (1,043 | ) | |||
| | | | | | | | |
Total |
$ | 8,456 | $ | 10,145 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The fair value of inventories acquired in 2013 included a step-up in the value of CAMBIA® and Lazanda® inventories of $3.7 million and $0.6 million, respectively. The step-up in the value of CAMBIA® and Lazanda® inventories are being amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of CAMBIA® was $3.5 million and $0.2 million in 2014 and 2013, respectively. The cost of sales related to the step-up value of Lazanda® was $0.3 million and $0.1 million in 2014 and 2013, respectively.
As of December 31, 2014, the unamortized portion of step-up related purely to Lazanda® inventories of $0.2 million. As of December 31, 2013, the unamortized portion of step-up related to CAMBIA® and Lazanda® inventories were $3.5 million and $0.5 million, respectively.
NOTE 5. PROPERTY AND EQUIPMENT
Property and equipment consists of the following (in thousands):
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
Furniture and office equipment |
$ | 4,097 | $ | 3,547 | |||
Laboratory equipment |
5,156 | 5,151 | |||||
Leasehold improvements |
6,045 | 6,045 | |||||
| | | | | | | | |
|
15,298 | 14,743 | |||||
Less: Accumulated depreciation and amortization |
(8,243 | ) | (6,403 | ) | |||
| | | | | | | | |
Property and equipment, net |
$ | 7,055 | $ | 8,340 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
There was no property and equipment included under capitalized leases as of December 31, 2014 or December 31, 2013. Depreciation expense was $1.8 million, $1.6 million and $2.6 million for the years ended December 31, 2014, 2013 and 2012, respectively.
99
NOTE 6. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
Accounts payable and accrued liabilities consist of the following (in thousands):
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
Accounts payable |
$ | 1,278 | $ | 2,232 | |||
Accrued compensation |
7,248 | 7,077 | |||||
Accrued rebates and sales discounts |
20,695 | 8,594 | |||||
Allowance for product returns |
15,015 | 10,278 | |||||
Accrued contract sales organization fees |
— | 962 | |||||
Inventory and other contract manufacturing accruals |
360 | 87 | |||||
Other accrued liabilities |
8,090 | 5,705 | |||||
| | | | | | | | |
Total accounts payable and accrued liabilities |
$ | 52,686 | $ | 34,935 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
NOTE 7. DEFERRED REVENUE
Deferred revenue consists of the following (in thousands):
| |
December 31, 2014 |
December 31, 2013 |
|||||
|---|---|---|---|---|---|---|---|
Deferred revenue, current portion: |
|||||||
Deferred product sales |
|||||||
Deferred license revenue, current portion: |
|||||||
Valeant |
$ | — | $ | 1,598 | |||
Salix |
— | 1,443 | |||||
| | | | | | | | |
Deferred license revenue, current portion |
— | 3,041 | |||||
| | | | | | | | |
Deferred revenue, current portion |
$ | — | $ | 3,041 | |||
| | | | | | | | |
Deferred license revenue, non-current portion: |
|||||||
Valeant |
— | 10,905 | |||||
Salix |
— | 1,570 | |||||
| | | | | | | | |
Deferred license revenue, non-current portion |
— | 12,475 | |||||
| | | | | | | | |
Total deferred revenue |
$ | — | $ | 15,516 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Deferred license revenue relates to upfront payments received by the Company under license and collaborative agreements with its partners. At December 31, 2013, deferred license revenue consisted of upfront license fee payments received from Salix and Valeant.
In December 2004, the Company received a $25.0 million license fee payment under its agreements with Valeant. Until September 30, 2014, we were recognizing the $25.0 million license fee payment as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the arrangement related to royalties that we were obligated to pay Valeant on net sales of the 500mg Glumetza in the United States and to use Valeant as the sole supplier of the 1000mg Glumetza. Effective October 1, 2014, the Company, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the manufacture and supply of 1000mg Glumetza tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, the Company recognized the remaining unamortized deferred revenue of $11.3 million as of October 1, 2014 in the accompanying Consolidated
100
NOTE 7. DEFERRED REVENUE (Continued)
Statement of Operations. The Company recognized approximately $12.5 million, $1.6 million and $1.6 million of revenue associated with this upfront license fee during 2014, 2013 and 2012, respectively.
In July 2008, the Company received a $12.0 million upfront payment under its promotion agreement with Salix. The upfront payment received was originally being amortized as revenue ratably until October 2021, which represented the estimated length of time our obligations existed under the promotion agreement related to manufacturing Glumetza and paying Salix promotion fees on gross margin of Glumetza. The commercialization agreement in August 2011 superseded the promotion agreement and removed our promotion fee obligations and contemplated removal of its manufacturing obligations. The commercialization agreement included obligations with respect to manufacturing and regulatory transition to Salix and managing the patent infringement lawsuits against Sun Pharmaceutical Industries, Inc. (Sun) and Lupin Limited (Lupin). At the time of the commercialization agreement, all of these obligations were estimated to be completed in December 2013. During the fourth quarter of 2012, events occurred related to the transfer of manufacturing with one of the contract manufacturers of Glumetza that extended the estimated completion date of our manufacturing obligations with respect to 1000 mg Glumetza to February 2016, which is the estimated date we expected our obligations would be completed under the commercialization agreement. Effective October 1, 2014, the Company, Valeant and Salix executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the supply of 1000mg Glumetza tablets. The execution of that agreement represented the completion of the final deliverable and, consequently, the Company recognized the remaining unamortized deferred revenue of $1.9 million as of October 1, 2014 in the accompanying Consolidated Statement of Operations
We recognized approximately $3.0 million, $1.4 million, and $3.3 million of revenue associated with this upfront license fee during 2014, 2013, and 2012, respectively.
In July 2011, Ironwood paid the Company a $0.9 million upfront license fee associated with the collaboration and license agreement. The $0.9 million was amortized ratably through June 2012, which is the estimated length of time the Company was obligated to perform formulation work under the agreement.
NOTE 8. DEBT
On September 9, 2014, the Company issued $345.0 million aggregate principal amount of the 2021 Notes in a public offering. The convertible debt offering resulted in net proceeds of $334.2 million after deducting the underwriting discount and offering expenses of $10.4 million and $0.4 million, respectively.
The 2021 Notes were issued pursuant to an indenture, as supplemented by a supplemental indenture between the Company and The Bank of New York Mellon Trust Company, N.A., as trustee (the Trustee), and mature on September 1, 2021, unless earlier converted, redeemed or repurchased. The 2021 Notes bear interest at the rate of 2.50% per annum, payable semi-annually in arrears on March 1 and September 1 of each year, beginning March 1, 2015.
Upon the occurrence of certain circumstances, holders may convert their 2021 Notes prior to the close of business on the business day immediately preceding March 1, 2021. On or after March 1, 2021, until the close of business on the second trading day immediately preceding the maturity date, holders may surrender their 2021 Notes for conversion at any time. Upon conversion, the Company will pay or deliver, at its option, cash, shares of its common stock or a combination of cash and shares of its common stock. The initial conversion rate of 51.9852 shares of common stock per $1,000 principal
101
NOTE 8. DEBT (Continued)
amount of 2021 Notes is equivalent to a conversion price of approximately $19.24 per share of common stock. The conversion rate is subject to adjustment upon the occurrence of certain events.
In addition, upon the occurrence of certain events defined in the indenture as a fundamental change, holders of the 2021 Notes may require us to purchase for cash all or any portion of their 2021 Notes at a purchase price equal to 100% of the principal amount of the Notes to be purchased plus accrued and unpaid interest, if any, to, but excluding, the fundamental change purchase date.
The 2021 Notes are accounted for in accordance with ASC Subtopic 470-20, Debt with Conversion and Other Options. Pursuant to ASC Subtopic 470-20, since the 2021 Notes can be settled in cash, shares of common stock or a combination of cash and shares of common stock at the Company's option, the Company is required to separately account for the liability (debt) and equity (conversion option) components of the instrument. The carrying amount of the liability component of any outstanding debt instrument is computed by estimating the fair value of a similar liability without the conversion option. The amount of the equity component is then calculated by deducting the fair value of the liability component from the principal amount of the convertible debt instrument. The effective interest rate used in determining the liability component of the 2021 Notes was 9.34%. This resulted in the recognition of $230.0 million as the liability component net of a $115.1 million debt discount with a corresponding increase to paid-in capital representing the equity component of the 2021 Notes. The underwriting discount of $10.4 million and offering expenses of $0.4 million were allocated between debt issuance costs and equity issuance costs in proportion to the allocation of the proceeds. Debt issuance costs of $7.1 million are included in "Other assets" on the consolidated balance sheets as of the issuance date. Equity issuance costs of $3.7 million related to the convertible debt offering were recorded as an offset to additional paid-in capital.
The Company recognized a net deferred tax liability of $41.9 million created by the book-tax basis difference for the liability component of our convertible debt offering. The deferred tax liability has been offset within the applicable "Deferred tax assets" sections of the consolidated balance sheets and, under the applicable accounting guidance, a corresponding off-set was recorded to additional paid-in capital on the accompanying consolidated balance sheets. The net deferred tax liability will be reduced and a deferred tax benefit will be recognized as the debt discount is amortized over the life of the instrument.
The following is a summary of the liability component of the 2021 Notes as of December 31, 2014 (in thousands):
| |
2014 | |||
|---|---|---|---|---|
Net carrying amount of the liability component |
$ | 229,891 | ||
Unamortized discount of the liability component |
115,109 | |||
| | | | | |
Principal amount of the 2021 Notes |
$ | 345,000 | ||
| | | | | |
| | | | | |
| | | | | |
The debt discount and debt issuance costs will be amortized as interest expense through September 2021. The following is a summary of interest expense for the 2021 Notes at December 31, 2014 (in thousands):
| |
2014 | |||
|---|---|---|---|---|
Stated coupon interest |
$ | 2,683 | ||
Amortization of debt discount and debt issuance costs |
4,200 | |||
| | | | | |
Total interest expense |
$ | 6,883 | ||
| | | | | |
| | | | | |
| | | | | |
102
NOTE 8. DEBT (Continued)
The balance of unamortized debt discount and debt issuance costs was $121.8 million of which $115.1 million is included in "Senior Convertible Notes" and $6.7 million is included within "Other assets" as of December 31, 2014 on the accompanying consolidated balance sheets.
NOTE 9. LIABILITY RELATED TO SALE OF FUTURE ROYALTIES
In October 2013, as noted above, we sold our interests in royalty and milestone payments under our license agreements in the Type 2 diabetes therapeutic area to PDL for $240.5 million. Until September 30, 2014, this transaction was accounted for as debt that was amortized using the interest method over the life of the arrangement. In order to determine the amortization of the debt, the Company is required to estimate the total amount of future royalty payments to be received by PDL and payments the Company is required to make to PDL, if any, over the life of the arrangement. The sum of these amounts less the $240.5 million proceeds the Company received will be recorded as interest expense over the life of the debt. Consequently, the Company imputes interest on the unamortized portion of the debt and records interest expense using an estimated interest rate for an arms-length debt transaction. Our estimate of the interest rate under the arrangement is based on the amount of royalty and milestone payments expected to be received by PDL over the life of the arrangement. Our estimate of this total interest expense resulted in an effective annual interest rate of approximately 10%.
As a result of the debt accounting, even though we did not retain the related royalties and milestones under the transaction (as the amounts are remitted to PDL), we continued to record revenue related to these royalties and milestones until September 30, 2014. We recognized $242.8 million and $18.1 million of non-cash revenue associated with the PDL Transaction for the years ended December 31, 2014 and December 31, 2013, respectively.
Effective October 1, 2014, the Company, Valeant, Salix and PDL executed an amended agreement which eliminated any and all continuing obligations on the part of the Company in the supply of 1000mg Glumetza tablets. Consequently, the entire outstanding balance of the liability related to the sale of future royalties and milestones of approximately $147.0 million was recognized within "Non-cash PDL royalty revenue" in the accompanying Consolidated Statement of Operations.
The following table shows the activity within the liability account during the year ended December 31, 2014 (in thousands):
Liability related to sale of future royalties as of December 31, 2013 |
$ | 227,079 | ||
Non-cash interest expense recognized until September 30, 2014 |
14,646 | |||
Amounts collected and remitted to PDL until September 30, 2014 |
(75,314 | ) | ||
| | | | | |
Total liability related to sale of future royalties as of September 30, 2014 |
166,411 | |||
Amounts collected and remitted to PDL in Q4, 2014 relating to Q3, 2014 |
(19,455 | ) | ||
Income recognized in Q4, 2014 |
(146,956 | ) | ||
| | | | | |
Total liability related to sale of future royalties as of December 31, 2014 |
$ | — | ||
| | | | | |
| | | | | |
| | | | | |
NOTE 10. COMMITMENTS AND CONTINGENCIES
Leases
In April 2012, the Company entered into an office and laboratory lease agreement to lease approximately 52,500 rentable square feet in Newark, California commencing on December 1, 2012.
103
NOTE 10. COMMITMENTS AND CONTINGENCIES (Continued)
The Company was obligated to lease approximately 8,000 additional rentable square feet commencing no later than December 1, 2015. The Lease will expire on November 30, 2022. However, the Company has the right to renew the lease for one additional five year term, provided that written notice is made to the landlord no later than 12 months prior to the lease expiration. The Company will have the one-time right to terminate the lease in its entirety effective as of November 30, 2017 by delivering written notice to the landlord on or before December 1, 2016. In the event of such termination, the Company will pay the landlord the unamortized portion of the tenant improvement allowance, specified additional allowances made by the landlord, waived base rent and leasing commissions, in each case amortized at 8% interest. Our previous lease in Menlo Park, California ended in January 2013.
The Company was allowed to control physical access to the premises upon signing the lease. Therefore, in accordance with the applicable accounting guidance, the lease term was deemed to have commenced in April 2012. Accordingly, the rent free periods and the escalating rent payments contained within the lease are being recognized on a straight-line basis from April 2012. The Company will pay approximately $12.1 million in aggregate rent over the remaining term of the lease for the above premises. Deferred rent for the years ended December 31, 2014 and 2013 was approximately $1.7 million and $1.5 million, respectively.
In December 2013, the Company entered into an operating lease agreement with Enterprise FM Trust (Enterprise) for the lease of vehicles to be used by the Company's sales force. The Company began receiving vehicles in the second quarter of 2014, with the lease terms ranging from 18 to 36 months. The Company will pay approximately $4.3 million in aggregate rent over the remaining term of the lease for the vehicles.
Rent expense relating to the office and laboratory lease agreement for 2014, 2013, and 2012 was $0.7 million, $1.0 million and $2.3 million, respectively. Rent expense relating to the lease of cars for 2014 was $1.1 million.
As of December 31, 2014 future minimum payments under operating leases for facilities and vehicles were as follows (in thousands):
2015 |
$ | 3,164 | ||
2016 |
3,209 | |||
2017 |
2,069 | |||
2018 |
1,530 | |||
thereafter |
6,441 | |||
| | | | | |
Total |
$ | 16,413 | ||
| | | | | |
| | | | | |
| | | | | |
Landlord Contributions to Leasehold Improvements
In conjunction with entering into leases for office space, the Company received contributions from the landlord toward leasehold improvements which are included in the Deferred Rent and Other Non-Current Liabilities line item of the Company's consolidated statements of financial condition. These contributions are amortized as a reduction to rent expense over the non-cancelable lease terms to which they pertain. The unamortized contributions from the landlord toward leasehold improvements were $5.1 million and $5.8 million for the years ended December 31, 2014 and 2013, respectively. For the years ended December 31, 2013 and 2012, cash contributions from landlords were $5.0 million and $1.3 million.
104
NOTE 10. COMMITMENTS AND CONTINGENCIES (Continued)
Legal matters
Depomed v. Gralise® ANDA Filers
Between March 2012 and May 2012, we filed lawsuits in the United States District Court for the District of New Jersey in response to six Abbreviated New Drug Applicants (ANDAs) filed by companies seeking to market generic versions of 300mg and 600mg dosage strengths of Gralise® prior to the expiration of our patents listed in the Orange Book for Gralise®. The lawsuits have been consolidated for purposes of all pretrial proceedings. Our lawsuits against two of the six Gralise® ANDA filers, Impax Laboratories and Watson Laboratories, have been dismissed as a result of the withdrawal of the ANDAs from consideration by the FDA. Our lawsuit against another ANDA filer, Par Pharmaceuticals Inc., has been dismissed because the ANDA filer no longer seeks approval of its Gralise ANDA prior to the expiration of our Gralise® Orange Book-listed patents. In April 2014, we entered settlement agreements with Incepta Pharmaceuticals and Abon Pharmaceuticals LLC (collectively, Incepta) and with Zydus Pharmaceuticals USA Inc. and Cadila Healthcare Limited (collectively, Zydus) pursuant to which Incepta and Zydus may begin selling generic versions of Gralise® on January 1, 2024, or earlier under certain circumstances.
A bench trial involving defendants Actavis Elizabeth LLC and Actavis Inc. (collectively, Actavis) was completed on May 20, 2014 as to U.S. Patent Nos. 6,635,280; 6,488,962; 7,438,927; 7,731,989; 8,192,756; 8,252,332; and 8,333,992, which expire between September 2016 and February 2024. In August 2014, the court ruled in our favor, finding that Actavis infringed all patent claims we asserted and upholding the validity of the patents. On September 15, 2014, Actavis filed a notice appealing the decision to the United States Court of Appeals for the Federal Circuit. On February 2, 2015, Actavis filed its opening brief with the United States Court of Appeals for the Federal Circuit.
Depomed v. FDA
In November 2010, the FDA granted Gralise® Orphan Drug designation for the management of PHN, but did not recognize Orphan drug exclusivity for Gralise® in January 2011 when Gralise® was approved for marketing in the United States. In September 2012, we filed an action in federal district court for the District of Columbia against the FDA seeking an order requiring the FDA to grant Gralise® Orphan Drug exclusivity for the management of PHN. Briefing in the case was completed in March 2013 and a hearing on our summary judgment motion was held in August 2013. In September 2014, the court issued an order granting our request for summary judgment, and ordering the FDA to grant Orphan Drug exclusivity for Gralise® for the management of PHN, which the FDA did formally in October 2014. On November 3, 2014, the FDA filed a notice appealing the order to the United States Court of Appeals for the Federal Circuit. On November 5, 2014, the government dismissed its appeal.
Depomed v. Purdue and Depomed v. Endo Pharmaceuticals—Patent Infringement Litigation and Related Inter Partes Review Proceedings
We have sued Purdue Pharma and Endo Pharmaceuticals for patent infringement in separate lawsuits filed in the United States District Court for the District of New Jersey. The lawsuits arise from Purdue's commercialization of reformulated OxyContin® (oxycodone hydrochloride controlled-release) in the United States and Endo's commercialization of OPANA® ER (oxymorphone hydrochloride extended-release) in the United States. We sued Purdue in January 2013 for infringement of U.S. Patent Nos. 6,340,475 (the '475 Patent) and 6,635,280 (the '280 Patent), which expire in September 2016. We sued Endo in April 2013 for infringement of the '475 Patent, the '280 Patent and U.S. Patent No. 6,723,340 (the '340 Patent), which expires in October 2021. The Purdue lawsuit has been stayed
105
NOTE 10. COMMITMENTS AND CONTINGENCIES (Continued)
pending completion of the inter partes reviews described below. The District Court has not yet ruled on Endo's request to stay the Endo litigation.
In response to two petitions filed by Purdue and six petitions filed by Endo, the United States Patent and Trademark Office Patent Trial and Appeal Board (PTAB) has instituted inter partes reviews (each, an IPR) of certain of the claims asserted in our lawsuits against Purdue and Endo. An IPR is a proceeding that became available in September 2012 in accordance with the America Invents Act (the AIA). In an IPR, a petitioner may request that the PTAB reconsider the validity of issued patent claims on the basis of prior art in the form of printed publications and other patents. Any patent claim the PTAB determines to be unpatentable is stricken from the challenged patent. Any party may appeal final written decisions of the PTAB to the United States Court of Appeals for the Federal Circuit. But, the PTAB's decisions denying institution of an IPR are non-appealable. Accordingly, if the PTAB finds a challenged claim patentable, or declines to institute an IPR as to a challenged claim, the IPR petitioner is estopped from asserting in a patent infringement lawsuit that those claims are invalid on any ground the petitioner raised or reasonably could have raised in the IPR.
In the Purdue IPRs, the PTAB declined to institute an IPR as to two claims of the '475 patent and two claims of the '280 Patent. The PTAB instituted an IPR as to the other 15 claims of the '475 Patent and as to the other 10 claims of the '280 Patent asserted against Purdue.
Endo filed two IPR petitions for each of the '475 Patent, the '280 Patent and the '340 Patent. The PTAB declined to institute an IPR as to three of Endo's petitions. The PTAB also declined to institute an IPR as to five claims of the '475 Patent, three claims of the '280 Patent and one claim of the '340 Patent in the Endo IPRs. The PTAB instituted an IPR as to the other 13 claims of the '475 Patent, as to the other ten claims of the '280 Patent and as to the other eight claims of the '340 patent asserted against Endo. The PTAB also declined to institute an IPR as to a number of Endo's requested grounds. On December 22, 2014, Depomed filed patent owner responses opposing the instituted IPRs.
Discovery, briefing and oral argument is scheduled to be complete in the Purdue IPRs in March 2015 and in the Endo IPRs in June 2015. In accordance with the requirements of the AIA, we expect final decisions from the PTAB not later than one year after the PTAB's decisions to institute the IPRs, or not later than July 10, 2015 in the Purdue IPRs and not later than September 29, 2015 in the Endo IPRs.
Depomed v. Banner Pharmacaps
On June 28, 2013, we received from Banner a Notice of Certification for U.S. Patent Nos. 6,365,180; 7,662,858; 7,884,095; 7,939,518; and 8,110,606 under 21 U.S.C. § 355 (j)(2)(A)(vii)(IV) (Zipsor® Paragraph IV Letter) certifying that Banner has submitted and the FDA has accepted for filing an ANDA for diclofenac potassium capsules, 25mg. The letter states that the Banner ANDA product contains the required bioavailability or bioequivalence data to Zipsor® and certifies that Banner intends to obtain FDA approval to engage in commercial manufacture, use or sale of Banner's ANDA product before the expiration of the above identified patents, which are listed for Zipsor® in the Orange Book. U.S. Patent No. 6,365,180 expires in 2019 and U.S. Patent Nos. 7,662,858; 7,884,095; 7,939,518; and 8,110,606 expire in 2029. The Zipsor® Paragraph IV letter indicates Banner has granted to Watson Laboratories Inc. (Watson) exclusive rights to Banner's proposed generic Zipsor® product.
On July 26, 2013, we filed a lawsuit in the United States District Court for District of New Jersey against Banner and Watson for infringement of the patents identified above. The lawsuit was commenced within the 45 days required to automatically stay, or bar, the FDA from approving Banner's ANDA for 25 mg diclofenac for 30 months or until a district court decision that is adverse to
106
NOTE 10. COMMITMENTS AND CONTINGENCIES (Continued)
the Company, whichever may occur earlier. Absent a court order, the 30-month stay would be expected to expire in December 2015.
On April 2, 2014, we filed an amended complaint to include infringement of U.S. Patent Nos. 6,287,594 and 8,623,920, which were recently added to the Orange Book listing for Zipsor® and expire in 2019 and 2029, respectively. Fact discovery and claim construction in the case are ongoing and no trial date has been set.
General
We cannot reasonably predict the outcome of the legal proceedings described above, nor can we estimate the amount of loss, range of loss or other adverse consequence, if any, that may result from these proceedings. As such we are not currently able to estimate the impact of the above litigation on our financial position or results of operations.
We may from time to time become party to actions, claims, suits, investigations or proceedings arising from the ordinary course of our business, including actions with respect to intellectual property claims, breach of contract claims, labor and employment claims and other matters. Although actions, claims, suits, investigations and proceedings are inherently uncertain and their results cannot be predicted with certainty, other than the matters set forth above, we are not currently involved in any matters that we believe may have a material adverse effect on our business, results of operations or financial condition. However, regardless of the outcome, litigation can have an adverse impact on us because of associated cost and diversion of management time.
NOTE 11. STOCK-BASED COMPENSATION
The Company uses the Black-Scholes option valuation model to determine the fair value of stock options and employee stock purchase plan (ESPP) shares. The determination of the fair value of stock-based payment awards on the date of grant using an option valuation model is affected by the Company's stock price as well as assumptions, which include the Company's expected term of the award, the expected stock price volatility, risk-free interest rate and expected dividends over the expected term of the award.
The Company uses historical option exercise data to estimate the expected life of the options. The Company estimates the volatility of its common stock price by using the historical volatility over the expected term of the options. The Company bases the risk-free interest rate on U.S. Treasury zero-coupon issues with terms similar to the expected term of the options as of the date of grant. The Company does not anticipate paying any cash dividends in the foreseeable future and therefore uses an expected dividend yield of zero in the option valuation model.
The Company used the following assumptions to calculate the fair value of option grants for the years ended December 31, 2014, 2013 and 2012
| |
2014 | 2013 | 2012 | |||
|---|---|---|---|---|---|---|
Employee and Director Stock Options |
||||||
Risk-free interest rate |
1.26 - 1.51% | 0.67 - 1.38% | 0.51 - 0.78% | |||
Dividend yield |
None | None | None | |||
Expected option term (in years) |
4.33 - 4.41 | 4.48 - 4.51 | 4.50 - 4.54 | |||
Expected stock price volatility |
42.6 - 49.0% | 49.2 - 63.5% | 64.1 - 67.5% |
107
NOTE 11. STOCK-BASED COMPENSATION (Continued)
The Company used the following assumptions to calculate the fair value of stock purchase rights granted under the ESPP for the years ended December 31, 2014, 2013 and 2012:
| |
2014 | 2013 | 2012 | |||
|---|---|---|---|---|---|---|
Employee and Director Stock Purchase Rights |
||||||
Risk-free interest rate |
0.06 - 0.08% | 0.08 - 0.10% | 0.12 - 0.16% | |||
Dividend yield |
None | None | None | |||
Expected option term (in years) |
0.5 | 0.5 | 0.5 | |||
Expected stock price volatility |
44.8 - 61.4% | 28.8 - 35% | 35.6 - 44.3% |
The following table presents stock-based compensation expense recognized for stock options, restricted stock units and the ESPP in the Company's Statements of Operations (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cost of sales |
$ | 14 | $ | 42 | $ | 41 | ||||
Research and development expense |
258 | 364 | 568 | |||||||
Selling, general and administrative expense |
8,658 | 5,702 | 4,461 | |||||||
| | | | | | | | | | | |
Total |
$ | 8,930 | $ | 6,108 | $ | 5,070 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The weighted-average grant date fair value of options granted during the years ended December 31, 2014, 2013 and 2012 was $4.83, $3.35 and $3.09, respectively. The weighted-average grant date fair value of stock purchase rights granted under the ESPP during the years ended December 31, 2014, 2013 and 2012 was $4.05, $1.87 and $1.44, respectively. The total intrinsic value of options exercised during the years ended December 31, 2014, 2013 and 2012 was $12.1 million, $1.7 million and $1.8 million, respectively. The total fair value of options that vested during the years ended December 31, 2014, 2013 and 2012 was $6.2 million, $5.1 million and $4.1 million, respectively. At December 31, 2014, the Company had $9.6 million of total unrecognized compensation expense, net of estimated forfeitures, related to stock option plans that will be recognized over an average vesting period of 1.9 years. Cash received from stock option exercises was $8.4 million, $2.8 million and $1.8 million for the years ended December 31, 2014, 2013 and 2012, respectively. There is no stock-based compensation recorded within inventory in any of the years presented.
2004 Equity Incentive Plan
The Company's 2004 Equity Incentive Plan (2004 Plan) was adopted by the Board of Directors and approved by the shareholders in May 2004. The 2004 Plan provides for the grant to employees of the Company, including officers, of incentive stock options, and for the grant of nonstatutory stock options to employees, directors and consultants of the Company. The number of shares authorized under the 2004 Plan was 14,450,000 shares and there are no more shares available for future issuance at December 31, 2014.
Generally, the exercise price of all incentive stock options and nonstatutory stock options granted under the 2004 Plan must be at least 100% and 85%, respectively, of the fair value of the common stock of the Company on the grant date. The term of incentive and nonstatutory stock options may not exceed 10 years from the date of grant. An option shall be exercisable on or after each vesting date in accordance with the terms set forth in the option agreement. The right to exercise an option generally vests over four years at the rate of at least 25% by the end of the first year and then ratably in monthly installments over the remaining vesting period of the option.
108
NOTE 11. STOCK-BASED COMPENSATION (Continued)
The following tables summarize the activity for the year ended December 31, 2014 under the 2004 Plan:
| |
Shares | Weighted- Average Exercise Price |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2013 |
6,518,971 | $ | 6.14 | ||||
Options granted |
1,605,385 | 12.64 | |||||
Options exercised |
(1,515,023 | ) | 5.52 | ||||
Options expired |
(606,999 | ) | 8.30 | ||||
| | | | | | | | |
Options outstanding at December 31, 2014 |
6,002,334 | $ | 7.84 | ||||
Options vested and expected to vest at December 31, 2014 |
5,581,055 | $ | 7.63 | ||||
Options exercisable at December 31, 2014 |
3,491,018 | $ | 6.76 | ||||
| |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in thousands) |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2014 |
7.12 | $ | 49,613 | ||||
Options exercisable and expected to become exercisable at December 31, 2014 |
7.02 | $ | 47,317 | ||||
Options exercisable at December 31, 2014 |
6.43 | $ | 32,648 | ||||
The total intrinsic value of options exercised during 2014, 2013 and 2012 was $12.1 million, $1.6 million and $1.7 million, respectively
Information regarding the stock options outstanding at December 31, 2014 under the 2004 Plan is summarized below:
Range of Exercise Prices
|
Number Outstanding |
Weighted- Average Remaining Contractual Term (Years) |
Weighted- Average Exercise Price (Outstanding) |
Number Exercisable |
Weighted- Average Exercise Price (Exercisable) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
$1.49 - $5.35 |
860,494 | 5.07 | $ | 3.95 | 759,278 | $ | 3.77 | |||||||||
$5.56 - $5.91 |
690,578 | 7.35 | $ | 5.78 | 441,736 | $ | 5.75 | |||||||||
$5.94 - $6.11 |
702,771 | 6.84 | $ | 6.06 | 472,510 | $ | 6.06 | |||||||||
$6.14 - $6.29 |
145,741 | 4.68 | $ | 6.21 | 101,679 | $ | 6.24 | |||||||||
$6.77 - $6.77 |
893,078 | 7.76 | $ | 6.77 | 380,299 | $ | 6.77 | |||||||||
$7.02 - $8.36 |
764,825 | 6.62 | $ | 7.88 | 631,493 | $ | 7.95 | |||||||||
$8.45 - $8.71 |
518,058 | 6.26 | $ | 8.53 | 438,430 | $ | 8.52 | |||||||||
$9.02 - $9.02 |
2,187 | 6.22 | $ | 9.02 | 1,875 | $ | 9.02 | |||||||||
$12.05 - $12.05 |
97,000 | 8.90 | $ | 12.05 | 1,146 | $ | 12.05 | |||||||||
$12.69 - $12.69 |
1,327,602 | 8.81 | $ | 12.69 | 262,572 | $ | 12.69 | |||||||||
| | | | | | | | | | | | | | | | | |
|
6,002,334 | 7.12 | $ | 7.84 | 3,491,018 | $ | 6.76 | |||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
109
NOTE 11. STOCK-BASED COMPENSATION (Continued)
Restricted stock units generally vest over four years, with 25% of each award vesting annually.
| |
Number of Shares |
Weighted Average Grant Date Fair Value Per Share |
Weighted Average Remaining Contractual Term (In years) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Non-vested restricted stock units at December 31, 2013 |
382,619 | $ | 6.61 | |||||||
Granted |
438,582 | $ | 12.69 | |||||||
Vested |
(231,686 | ) | $ | 12.69 | ||||||
Forfeited |
(68,229 | ) | $ | 9.32 | ||||||
| | | | | | | | | | | |
Non-vested restricted stock units at December 31, 2014 |
521,286 | $ | 8.67 | 1.67 | ||||||
The total fair value of restricted stock vested during 2014 was $2.9 million.
2014 Omnibus Incentive Plan
The Company's 2014 Omnibus Incentive Plan (2014 Plan) was adopted by the Board of Directors and approved by the shareholders in May 2014. The 2014 Plan provides for the grant of stock options, stock appreciation rights, stock awards, cash awards and performance award to the employees, non-employee directors and consultants of the Company. The number of shares authorized under the 2014 Plan is 6,150,000 shares, of which 5,368,400 were available for future issuance at December 31, 2014.
Generally, the exercise price of all incentive stock options and nonstatutory stock options granted under the 2014 Plan must be the fair value of the common stock of the Company on the grant date. The term of incentive and nonstatutory stock options may not exceed 10 years from the date of grant. An option shall be exercisable on or after each vesting date in accordance with the terms set forth in the option agreement. The right to exercise an option generally vests over four years at the rate of at least 25% by the end of the first year and then ratably in monthly installments over the remaining vesting period of the option.
The following table summarize the activity for the year ended December 31, 2014 under the 2014 Plan:
| |
Shares | Weighted Average Exercise Price |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2013 |
— | $ | — | ||||
Options granted |
753,672 | 13.40 | |||||
Options expired |
(8,000 | ) | 11.90 | ||||
| | | | | | | | |
Options outstanding at December 31, 2014 |
745,672 | $ | 13.42 | ||||
Options vested and expected to vest at December 31, 2014 |
526,914 | $ | 13.40 | ||||
Options exercisable at December 31, 2014 |
10,167 | $ | 11.80 | ||||
110
NOTE 11. STOCK-BASED COMPENSATION (Continued)
| |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in thousands) |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2014 |
9.61 | $ | 2,008 | ||||
Options exercisable and expected to become exercisable at December 31, 2014 |
9.61 | $ | 1,429 | ||||
Options exercisable at December 31, 2014 |
9.43 | $ | 44 | ||||
Information regarding the stock options outstanding at December 31, 2014 under the 2014 Plan is summarized below:
Range of Exercise Prices
|
Number Outstanding |
Weighted- Average Remaining Contractual Term (Years) |
Weighted- Average Exercise Price (Outstanding) |
Number Exercisable |
Weighted- Average Exercise Price (Exercisable) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
$10.80 - $10.80 |
82,000 | 9.58 | $ | 10.80 | 0 | $ | 0 | |||||||||
$10.86 - $10.86 |
107,960 | 9.21 | $ | 10.86 | 0 | $ | 0 | |||||||||
$11.11 - $11.11 |
10,000 | 9.39 | $ | 11.11 | 8,333 | $ | 11.11 | |||||||||
$11.83 - $11.83 |
150,000 | 9.54 | $ | 11.83 | 0 | $ | 0 | |||||||||
$11.84 - $11.84 |
7,000 | 9.41 | $ | 11.84 | 0 | $ | 0 | |||||||||
$14.94 - $14.94 |
22,000 | 9.63 | $ | 14.94 | 1,834 | $ | 14.94 | |||||||||
$15.02 - $15.02 |
63,300 | 9.73 | $ | 15.02 | 0 | $ | 0 | |||||||||
$15.31 - $15.31 |
150,000 | 9.67 | $ | 15.31 | 0 | $ | 0 | |||||||||
$15.53 - $15.53 |
59,112 | 9.92 | $ | 15.53 | — | $ | 0 | |||||||||
$15.74 - $15.74 |
94,300 | 9.90 | $ | 15.74 | 0 | $ | 0 | |||||||||
| | | | | | | | | | | | | | | | | |
|
745,672 | 9.61 | $ | 13.42 | 10,167 | $ | 11.80 | |||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Restricted stock units generally vest over four years, with 25% of each award vesting annually
| |
Number of Shares |
Weighted Average Grant Date Fair Value Per Share |
Weighted Average Remaining Contractual Term (In years) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Non-vested restricted stock units at December 31, 2013 |
— | $ | — | |||||||
Granted |
23,178 | $ | 15.53 | |||||||
| | | | | | | | | | | |
Non-vested restricted stock units at December 31, 2014 |
23,178 | $ | 15.53 | 0.92 | ||||||
NOTE 12. SHAREHOLDERS' EQUITY
Employee Stock Purchase Plan
In May 2004, the ESPP was approved by the shareholders. The ESPP is qualified under Section 423 of the Internal Revenue Code. The ESPP is designed to allow eligible employees to purchase shares of the Company's common stock through periodic payroll deductions. The price of the common stock purchased under the ESPP must be equal to at least 85% of the lower of the fair market value of the common stock on the commencement date of each offering period or the specified purchase date. The number of shares authorized for issuance under the ESPP as of December 31, 2014 was 2,500,000, of which 536,556 shares were available for future issuance.
111
NOTE 12. SHAREHOLDERS' EQUITY (Continued)
In 2014, the Company sold 177,036 shares of its common stock under the ESPP. The shares were purchased at a weighted-average purchase price of $8.68 with proceeds of approximately $1.5 million. In 2013, the Company sold 222,062 shares of its common stock under the ESPP. The shares were purchased at a weighted-average purchase price of $4.35 with proceeds of approximately $1.0 million.
Option Exercises
Employees exercised options to purchase 1,515,023 shares of the Company's common stock with net proceeds to the Company of approximately $8.4 million during 2014. Employees exercised options to purchase 621,090 shares of the Company's common stock with net proceeds to the Company of approximately $2.8 million during 2013.
Shareholder Rights Plan
On April 21, 2005, the Company adopted a shareholder rights plan, (Rights Plan). Under the Rights Plan, the Company distributed one preferred share purchase right for each share of common stock outstanding at the close of business on May 5, 2005. If a person or group acquires 20% or more of the Company's common stock in a transaction not pre-approved by the Company's Board of Directors, each right will entitle its holder, other than the acquirer, to buy additional shares of the Company's common stock at 50% of its market value, as defined in the Rights Plan. In addition, if an unapproved party acquires more than 20% of the Company's common stock, and the Company is later acquired by the unapproved party or in a transaction in which all shareholders are not treated alike, shareholders with unexercised rights, other than the unapproved party, will be entitled to receive upon exercise of the rights, common stock of the merger party or asset buyer with a value of twice the exercise price of the rights. Each right also becomes exercisable for one one-thousandth of a share of the Company's Series RP preferred stock at the right's then current exercise price ten days after an unapproved third party makes, or announces an intention to make, a tender offer or exchange offer that, if completed, would result in the unapproved party acquiring 20% or more of the Company's common stock. The Board of Directors may redeem the rights for a nominal amount before an event that causes the rights to become exercisable. The rights will expire on April 21, 2015.
NOTE 13. RELATED PARTY TRANSACTIONS
There were no related party transactions in 2014.
In 2013, Burrill Securities acted as a financial advisor to PDL in connection with the PDL transaction. Burrill Securities is the merchant banking division of Burrill & Company. G. Steven Burrill, a former director of the Company, was at the time of the PDL transaction a director of the Company and the Chief Executive Officer and sole shareholder of Burrill & Company. The Board of Directors was aware of Burrill & Company's interest in the transaction and Mr. Burrill recused himself from all deliberations and actions taken by the Board of Directors with respect to the transaction. Burrill Securities' engagement with PDL in the transaction was led by Fredrick Frank, the Chairman of Burrill Securities and a former member of the Board of Directors of PDL. The Company was informed that Burrill Securities received a fee of up to $500,000 from PDL in connection with the transaction.
112
NOTE 14. INCOME TAXES
The provision for (benefit from) income taxes consists of the following (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Current: |
||||||||||
Federal |
$ | (2,853 | ) | $ | 60,874 | $ | (19 | ) | ||
State |
11 | 3,593 | (75 | ) | ||||||
Foreign |
— | 3 | 3 | |||||||
| | | | | | | | | | | |
|
(2,842 | ) | 64,470 | (91 | ) | |||||
| | | | | | | | | | | |
Deferred: |
||||||||||
Federal |
$ | 80,293 | $ | (97,690 | ) | $ | — | |||
State |
3,895 | (5,513 | ) | — | ||||||
Foreign |
— | — | — | |||||||
| | | | | | | | | | | |
|
84,188 | (103,203 | ) | — | ||||||
| | | | | | | | | | | |
Total provision (benefit) for income taxes |
$ | 81,346 | $ | (38,733 | ) | $ | (91 | ) | ||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
A reconciliation of income taxes at the statutory federal income tax rate to the actual tax rate included in the statements of operations is as follows (in thousands):
| |
2014 | 2013 | 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Tax at federal statutory rate |
$ | 74,588 | $ | 1,603 | $ | (10,454 | ) | |||
State tax, net of federal benefit |
3,903 | (3,177 | ) | (48 | ) | |||||
Foreign tax |
— | 3 | 3 | |||||||
Research Credit |
— | (258 | ) | — | ||||||
Net Operating Losses not benefited (benefited) |
— | (17,510 | ) | 9,094 | ||||||
Stock Based Compensation |
366 | 923 | 1,038 | |||||||
Non deductible meals and entertainment |
440 | 442 | 131 | |||||||
Non-deductible other expense |
2,049 | (133 | ) | 145 | ||||||
Change in Valuation Allowance |
— | (20,626 | ) | — | ||||||
| | | | | | | | | | | |
Total |
$ | 81,346 | $ | (38,733 | ) | $ | (91 | ) | ||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
During 2014, the Company provided for income tax expense of approximately $81.3 million which primarily related to the recognition for financial statement purposes the deferred revenue related to the 2013 PDL transaction.
During 2013, we recognized an income tax benefit of approximately $38.7 million which resulted primarily from our reversal of a valuation allowance on all of our U.S. federal deferred tax assets and most of our state deferred tax assets. Our 2013 effective tax rate from continuing operations was (846)%. The tax benefit represents a reversal of a valuation allowance on a significant portion of our U.S. federal and state deferred assets resulting in a deferred tax benefit of $103.2 million, offset by a current income tax provision of $64.5 million.
The Company's tax benefits for the year ended December 31, 2012 was due to Federal and state refundable credits offset by foreign taxes withheld on royalty revenue related to the Company's agreement with LG by the Republic of Korea.
On January 2, 2013, the enactment of the American Taxpayer Relief Act of 2013 extended retroactively through the end of calendar 2013 the U.S. federal research and development credit which
113
NOTE 14. INCOME TAXES (Continued)
had expired on December 31, 2011. As a result, an income tax benefit for the year ended December 2013 includes the tax benefit for the reinstatement of the 2012 federal research tax credit.
As of December 31, 2014, the Company had net operating loss carry forwards for federal income tax purposes of approximately $11.0 million, which expire in the years 2021 through 2032. Net operating loss carryforwards for state income tax purposes were approximately $106.0 million, which expire in the years 2017 through 2033 and state research and development tax credits were approximately $1.4 million which have no expiration date.
Utilization of the Company's net operating loss and credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations provided by the Internal Revenue Code of 1986 and similar state provisions. The annual limitation may result in the expiration of net operating losses and credits before utilization.
Deferred income taxes reflect the temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company's deferred tax assets are as follows (in thousands):
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Deferred Tax Assets: |
|||||||
Net operating loss carryforwards |
$ | 5,390 | $ | 4,237 | |||
Tax carryforwards |
80 | 133 | |||||
In-process research and development |
330 | 665 | |||||
Deferred revenue |
— | 88,248 | |||||
Intangibles |
3,282 | 1,363 | |||||
Other, net |
14,180 | 11,226 | |||||
| | | | | | | | |
Total deferred tax assets |
23,262 | 105,872 | |||||
Convertible Debt |
(41,940 | ) | — | ||||
Valuation allowance for deferred tax assets |
(4,310 | ) | (2,670 | ) | |||
| | | | | | | | |
Deferred tax (liabilities) assets, net |
$ | (22,988 | ) | $ | 103,202 | ||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Management regularly assesses the ability to realize deferred tax assets based on the weight of all available evidence, including such factors as the history of recent earnings and expected future taxable income on a jurisdiction by jurisdiction basis. Management determined that a valuation allowance was no longer needed for a substantial portion of the deferred tax assets based on an assessment of the relative impact of all positive and negative evidence as of December 31, 2014, including an evaluation of cumulative income in recent years, future sources of taxable income exclusive of reversing temporary differences, and significant risks and uncertainties related to our business. The valuation allowance increased by $1.6 million, decreased by $39.8 million and increased by $7.8 million during the years ended December 31, 2014, 2013 and 2012 respectively.
At December 31, 2014, the portion of the federal and state net operating loss carryforwards related to stock option deductions is approximately $11 million for federal and $13.9 million for state, which is not included in the Company's gross or net deferred tax assets. Pursuant to ASC 718-740-25-10, the tax effect of the stock option benefit of approximately $4.7 million will be recorded to equity when they reduce cash taxes payable in the future.
The Company files income tax returns in the United States federal jurisdiction and in various states, and the tax returns filed for the years 1996 through 2013 and the applicable statutes of
114
NOTE 14. INCOME TAXES (Continued)
limitation have not expired with respect to those returns. Because of net operating loss carryovers, substantially all of the Company's tax years remain open to examination.
Interest and penalties, if any, related to unrecognized tax benefits would be recognized as income tax expense by the Company. At December 31, 2014, the Company had approximately $0.3 million and $0.1 million of accrued interest and penalties associated with any unrecognized tax benefits.
The following table summarizes the activity related to our unrecognized tax benefits for the three years ended December 31, 2014 (in thousands):
Unrecognized tax benefits—January 1, 2012 |
$ | 3,573 | ||
Gross increases—current year tax positions |
21 | |||
| | | | | |
Unrecognized tax benefits—December 31, 2012 |
$ | 3,594 | ||
Gross increases—current year tax positions |
174 | |||
Gross increases—prior year tax positions |
111 | |||
| | | | | |
Unrecognized tax benefits—December 31, 2013 |
$ | 3,879 | ||
Gross increases—current year tax positions |
27 | |||
Gross increases—prior year tax positions |
1,273 | |||
| | | | | |
Unrecognized tax benefits—December 31, 2014 |
$ | 5,179 | ||
| | | | | |
| | | | | |
| | | | | |
The total amount of unrecognized tax benefit that would affect the effective tax rate is approximately $5.2 million at December 31, 2014.
Though our unrecognized tax benefits may change during the next year for items that arise in the ordinary course of business, we do not expect any such change to be significant. As of December 31, 2014, the Company anticipates that the balance of gross unrecognized tax benefits will decrease by $0.1 million due to the expiration of the applicable statute of limitation over the next 12 months.
NOTE 15. BUSINESS COMBINATIONS
The CAMBIA® Acquisition
On December 17, 2013, the Company entered into an Asset Purchase Agreement (CAMBIA® Asset Purchase Agreement) with Nautilus Neurosciences, Inc., a Delaware corporation (Nautilus), pursuant to which the Company acquired from Nautilus all of the rights to CAMBIA® (diclofenac potassium for oral solution), including related product inventory, and assumed from Nautilus certain liabilities relating to CAMBIA®, for an initial payment of $48.7 million in cash.
Pursuant to the CAMBIA® Asset Purchase Agreement, $7.5 million of the Initial Payment will be held in escrow for 24 months and applied towards the indemnification obligations of Nautilus as set forth in the CAMBIA® Asset Purchase Agreement.
In addition to the initial payment, the Company agreed to pay one-time, contingent cash payments upon the achievement of certain CAMBIA® net sales milestones. Up to $5.0 million in sales milestones are payable to Nautilus, and up to $10.0 million in sales milestones are payable to third parties pursuant to contracts assigned to the Company. The net sales thresholds triggering such milestone payments to Nautilus range up to $100 million in calendar year net sales. The Company also assumed certain third party royalty obligations totaling not more than 11% of CAMBIA® net sales.
115
NOTE 15. BUSINESS COMBINATIONS (Continued)
In accordance with the authoritative guidance for business combinations, the transaction with Nautilus was determined to be a business combination and was accounted for using the acquisition method of accounting.
The following table presents a summary of the purchase price consideration for the CAMBIA® acquisition (in thousands):
Cash for CAMBIA and related inventories |
$ | 48,725 | ||
Fair value of contingent consideration |
1,010 | |||
| | | | | |
Purchase price |
$ | 49,735 | ||
| | | | | |
| | | | | |
| | | | | |
The contingent consideration was recognized and measured at fair value as of the acquisition date. The Company determined the acquisition date fair value of the contingent consideration obligation based on an income approach derived from CAMBIA® revenue estimates and a probability assessment with respect to the likelihood of achieving the level of net sales that would trigger the contingent payment. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting. The key assumptions in determining the fair value are the discount rate and the probability assigned to the potential milestones being achieved. At each reporting date, the Company will re-measure the contingent consideration obligation to estimated fair value.
The following table summarizes the fair values of the tangible and identifiable intangible assets acquired and liabilities assumed at the acquisition date (in thousands):
Intangible asset—CAMBIA product rights |
$ | 51,360 | ||
Inventories |
3,837 | |||
Other assets |
409 | |||
Sales reserve liabilities |
(1,847 | ) | ||
Unfavorable contract assumed |
(3,540 | ) | ||
Bargain purchase |
(484 | ) | ||
| | | | | |
|
$ | 49,735 | ||
| | | | | |
| | | | | |
| | | | | |
The CAMBIA® product rights of $51.4 million have been recorded as intangible assets on the accompanying consolidated balance sheet and are being amortized over the estimated useful life of the assets on a ratable basis through December 2023 as no other method could be reliably estimated. Total amortization expense for 2014 and 2013 was approximately $5.1 million and $0.2 million, respectively. The Company incurred an aggregate of $0.1 million in acquisition-related costs during 2013. These expenses are included in selling, general and administrative expenses in the Company's consolidated statement of operations.
The liability for the unfavorable contract assumed represents an obligation for the Company to make certain payments to a vendor upon the achievement of certain milestones by such vendor. This contract was entered into by Nautilus as part of a legal settlement unrelated to the CAMBIA® acquisition. The liability of $3.5 million recorded above represents the fair value of the amounts by which the contract terms are unfavorable compared to the current market pricing and a probability—weighted assessment of the likelihood that the stipulated milestones will be achieved by the third party. The contract may be terminated if the third party fails to achieve these milestones, in which case the fair value of the liability as of the date of the termination will be reversed on the consolidated balance
116
NOTE 15. BUSINESS COMBINATIONS (Continued)
sheet and reflected in the consolidated statement of operations as a credit within interest and other income. The Company determines the fair value of this liability at each reporting period.
The fair value of inventories acquired included a step-up in the value of CAMBIA® inventories of $3.7 million that will be amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of CAMBIA® inventories was $3.5 million and $0.2 million in 2014 and 2013, respectively. The bargain purchase amount has been recorded within interest and other income during 2013.
The Lazanda® Acquisition
On July 29, 2013, the Company entered into an Asset Purchase Agreement (Lazanda® Asset Purchase Agreement) with each of Archimedes Pharma US Inc., a Delaware corporation, Archimedes Pharma Ltd., a corporation registered under the laws of England and Wales, and Archimedes Development Ltd., a company registered under the laws of England and Wales (collectively, Archimedes), pursuant to which the Company acquired all of the U.S. and Canadian rights to Archimedes' product Lazanda® (fentanyl) nasal spray and related inventory for an initial payment of $4.0 million in cash. The Company also assumed certain liabilities related to Lazanda®.
Pursuant to the Lazanda® Asset Purchase Agreement, $1.0 million of the Initial Payment will be held in escrow for 18 months and applied towards the indemnification obligations of Archimedes as set forth in the Lazanda® Asset Purchase Agreement.
In addition to the initial payment, the Company will also pay royalties on its net sales of Lazanda®. In 2013 and 2014, the Company will not pay royalties to Archimedes, and third party royalties assumed by the Company in connection with the acquisition will be less than 5% of the Company's net sales of Lazanda®. Thereafter, the Company will pay royalties to Archimedes and third parties totaling 13% to 15% of the Company's net sales of Lazanda®. In addition to the initial payment and royalties, the Company will pay to Archimedes the following one-time, cash contingent payments upon the achievement by the Company's net sales of Lazanda® equal to or in excess of the following net sales milestones: (i) $1.0 million at the end of the first calendar year in which net sales of Lazanda® are $20.0 million; (ii) $2.5 million at the end of the first calendar year in which net sales of Lazanda® are $45.0 million; (iii) $5.0 million at the end of the first calendar year in which net sales of Lazanda® are $75.0 million; and (iv) $7.5 million at the end of the first calendar year in which net sales of Lazanda® are $100.0 million.
In accordance with the authoritative guidance for business combinations, the Asset Purchase Agreement with Archimedes was determined to be a business combination and was accounted for using the acquisition method of accounting. Pursuant to a letter dated August 21, 2013 (Letter) from the staff of the Division of Corporate Finance (Division) of the Securities and Exchange Commission, the Division stated that it would waive the requirement to provide a pro forma statement of operations if the use of forward-looking information is necessary to meaningfully present the effects of the acquisition of Lazanda® by the Company. The Company's expense structure and commercialization infrastructure related to Lazanda® are anticipated to differ significantly from the expense structure and commercialization infrastructure maintained by Archimedes with regard to Lazanda®. As a result, the Company has concluded that the use of forward-looking information is necessary to meaningfully present the effects of the acquisition. Based on the guidance provided by the Division in the Letter, the Company has not presented a pro forma statement of operations.
117
NOTE 15. BUSINESS COMBINATIONS (Continued)
The following table presents a summary of the purchase price consideration for the Lazanda® acquisition (in thousands):
Cash for Lazanda, related property, inventories, and other assets |
$ | 4,000 | ||
Fair Value of contingent consideration |
8,004 | |||
| | | | | |
Purchase Price |
$ | 12,004 | ||
| | | | | |
| | | | | |
| | | | | |
The contingent consideration was recognized and measured at fair value as of the acquisition date. The Company determined the acquisition date fair value of the contingent consideration obligation based on an income approach derived from Lazanda® revenue estimates and a probability assessment with respect to the likelihood of achieving the level of net sales that would trigger the contingent payment. The contingent consideration also includes royalties payable to Archimedes based on net sales where increase in the royalty rate is tied to a reduction in cost of goods sold. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting. The key assumptions in determining the fair value are the discount rate and the probability assigned to the potential milestones being achieved. At each reporting date, the Company will re-measure the contingent consideration obligation to estimated fair value.
The following table summarizes the fair values of the tangible and identifiable intangible assets acquired and liabilities assumed at the acquisition date (in thousands):
Intangible asset—Lazanda product rights |
$ | 10,450 | ||
Inventories |
1,334 | |||
Property, plant and equipment |
356 | |||
Other assets |
116 | |||
Current liabilities |
(283 | ) | ||
Goodwill |
31 | |||
| | | | | |
|
$ | 12,004 | ||
| | | | | |
| | | | | |
| | | | | |
The Lazanda® product rights of $10.5 million have been recorded as intangible assets on the accompanying condensed balance sheet and are being amortized over the estimated useful life of the asset on a ratable basis through August 2022. Total amortization expense for 2014 and 2013 was approximately $1.2 million and $0.5 million, respectively. The Company incurred an aggregate of $0.1 million in acquisition-related costs in 2013. These expenses are included in selling, general and administrative expenses in the Company's consolidated statement of operations.
The fair value of inventories acquired included a step-up in the value of Lazanda® inventories of $0.6 million which will be amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of Lazanda® inventories was $0.3 million and $0.1 million in 2014 and 2013, respectively.
The Zipsor® Acquisition
On June 21, 2012, the Company entered into an Asset Purchase Agreement (Zipsor® Asset Purchase Agreement) with Xanodyne, pursuant to which the Company acquired Xanodyne's product Zipsor®and related inventory for $26.4 million in cash, and assumed certain product related liabilities relating to Zipsor®. In addition, the Company will make a one-time contingent payment to Xanodyne of $2.0 million in cash at the end of the first calendar year in which the Company's net sales of Zipsor®
118
NOTE 15. BUSINESS COMBINATIONS (Continued)
products exceed $30.0 million and an additional, one-time contingent payment to Xanodyne of $3.0 million in cash at the end of the first year in which the Company's net sales of Zipsor® products exceed $60.0 million.
In accordance with the authoritative guidance for business combinations, the Zipsor® Asset Purchase Agreement with Xanodyne was determined to be a business combination and was accounted for using the acquisition method of accounting. Neither separate financial statements nor pro forma results of operations have been presented because the acquisition transaction does not meet the qualitative or quantitative materiality tests under SEC Regulation S-X.
Pursuant to the Zipsor® Asset Purchase Agreement, we have $1.0 million in escrow as of December 31, 2014.
The following table presents a summary of the purchase price consideration for the Zipsor® acquisition (in thousands):
Cash for Zipsor and related inventories |
$ | 26,436 | ||
Fair Value of contingent consideration |
1,303 | |||
| | | | | |
Purchase Price |
$ | 27,739 | ||
| | | | | |
| | | | | |
| | | | | |
The contingent consideration was recognized and measured at fair value as of the acquisition date and is included within other long-term liabilities in the accompanying balance sheet. The Company determined the acquisition date fair value of the contingent consideration obligation based on an income approach derived from Zipsor® revenue estimates and a probability assessment with respect to the likelihood of achieving the level of net sales that would trigger the contingent payment. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting. The key assumptions in determining the fair value are the discount rate and the probability assigned to the potential milestones being achieved. At each reporting date, the Company will re-measure the contingent consideration obligation to estimated fair value.
The following table summarizes the fair values of the tangible and identifiable intangible assets acquired and liabilities assumed at the acquisition date (in thousands):
Intangible asset—Zipsor product rights |
$ | 27,100 | ||
Inventories |
2,428 | |||
Other assets |
100 | |||
Property, plant and equipment |
43 | |||
Current liabilities |
(1,840 | ) | ||
Bargain purchase |
(92 | ) | ||
| | | | | |
|
$ | 27,739 | ||
| | | | | |
| | | | | |
| | | | | |
The Zipsor® product rights of $27.1 million have been recorded as intangible assets on the accompanying condensed balance sheet and are being amortized over the estimated useful life of the asset on a ratable basis through July 2019. Total amortization expense for 2014, 2013 and 2012 was approximately $3.9 million, $3.8 million and $2.0 million, respectively.
The fair value of inventories acquired included a step-up in the value of Zipsor® inventories of $1.9 million which is being amortized to cost of sales as the acquired inventories are sold. The cost of sales related to the step-up value of Zipsor® for 2013 and 2012 was $0.7 million and $1.2 million,
119
NOTE 15. BUSINESS COMBINATIONS (Continued)
respectively. The bargain purchase amount has been recorded within Interest and other income during 2012.
NOTE 16. SUBSEQUENT EVENTS
On January 15, 2015, the Company entered into an Asset Purchase Agreement with Janssen Pharmaceuticals, Inc., a Pennsylvania corporation (Janssen), pursuant to which the Company will acquire from Janssen and its affiliates the U.S. rights to the NUCYNTA® franchise of pharmaceutical products (Products) as well as certain related assets (Transaction) for $1.05 billion in cash (Cash Payment).
The NUCYNTA® franchise includes NUCYNTA® ER (tapentadol) extended release tablets indicated for the management of pain, including neuropathic pain associated with diabetic peripheral neuropathy (DPN), severe enough to require daily, around-the-clock, long-term opioid treatment, NUCYNTA®(tapentadol), an immediate release version of tapentadol, for management of moderate to severe acute pain in adults, and NUCYNTA® (tapentadol) oral solution, an approved oral form of tapentadol that has not been commercialized.
Upon the consummation of the Transaction, the Company will acquire (i) rights to commercialize the Products in the United States including the investigational new drug applications (INDs) and the new drug applications (NDAs) for the Products in the United States, and (ii) certain other assets relating to the Products, including finished goods product inventory and certain manufacturing equipment. In addition, Janssen will assign to the Company all of its rights and obligations under the License Agreement (U.S.) (License Agreement) by and among Janssen, Janssen Research & Development, LLC and Grünenthal GmbH (Grünenthal) pursuant to which Janssen has a royalty-bearing license to certain Grünenthal patents and other intellectual property rights covering the commercialization of the Products in the United States.
In connection with the Transaction, the Company will assume certain liabilities relating to the Products, including responsibility for the ongoing legal proceedings relating to certain of the Grünenthal patents licensed under the License Agreement and Janssen's clinical obligations relating to the Products. Other than as set forth in the Asset Purchase Agreement, Janssen will retain all liabilities relating to the Products associated with Janssen's commercialization of the Products prior to the consummation of the Transaction.
In connection with the Transaction, the Company, Janssen and certain affiliates of Janssen will enter into (i) supply agreements pursuant to which Janssen will manufacture and supply the Products to the Company until the Company, or its contract manufacturer, begins commercial production of the Products, following which the Company will manufacture and supply Janssen for its requirements for NUCYNTA® outside of the United States and (ii) a supply agreement pursuant to which an affiliate of Janssen will manufacture and supply the Company with the active pharmaceutical ingredient contained in the Products.
Upon execution of the Asset Purchase Agreement, the Company delivered a cash deposit in the amount of $500.0 million (Cash Deposit) to JPMorgan Chase Bank, N.A., (Escrow Agent) in accordance with an Escrow Agreement, dated January 15, 2015 (Escrow Agreement), by and among the Company, Janssen and the Escrow Agent. The Cash Deposit will be credited against the total cash payable to Janssen upon the consummation of the Transaction.
The Asset Purchase Agreement contains customary representations and warranties, covenants and indemnification provisions. The consummation of the Transaction is subject to the satisfaction of a number of conditions, including, but not limited to, (i) the Company obtaining financing, through one
120
NOTE 16. SUBSEQUENT EVENTS (Continued)
or more transactions, in an aggregate amount sufficient to pay the total cash payable (minus the Cash Deposit) and any related fees and expenses applicable to the Transaction (Financing Condition) and (ii) the expiration or termination of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended.
The Asset Purchase Agreement provides for certain termination rights of the parties. If Janssen terminates the Asset Purchase Agreement due to the Company's failure to satisfy the Financing Condition and Janssen has materially complied with its obligations under the Asset Purchase Agreement, the Cash Deposit less $73.5 million (Reverse Termination Fee) will be returned to the Company, and the Reverse Termination Fee will be paid to Janssen in accordance with the Asset Purchase Agreement and the Escrow Agreement. If the Asset Purchase Agreement is terminated under certain other circumstances, the entire amount of the Cash Deposit will be returned to the Company in accordance with the Asset Purchase Agreement and the Escrow Agreement.
NOTE 17. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
The following tables set forth certain unaudited quarterly financial data for each of the eight quarters beginning with the quarter ended March 31, 2013 through the quarter ended December 31, 2014 (in thousands). This quarterly financial data is unaudited, but has been prepared on the same basis as the annual financial statements and, in the opinion of management, reflects all adjustments, consisting only of normal recurring adjustments necessary for a fair representation of the information for the periods presented. Operating results for any quarter are not necessarily indicative of results for any future period.
| |
2014 Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (in thousands) |
March 31 | June 30 | September 30 | December 31 | |||||||||
Product sales |
$ | 21,506 | $ | 28,245 | $ | 30,584 | $ | 33,884 | |||||
Total revenues |
76,544 | 67,732 | 51,485 | 194,601 | |||||||||
Gross margin on product sales |
17,804 | 23,570 | 27,061 | 30,638 | |||||||||
Income from operations |
35,744 | 26,545 | 16,700 | 157,822 | |||||||||
Net income |
17,939 | 12,746 | 6,454 | 94,623 | |||||||||
Basic net income per share |
$ | 0.31 | $ | 0.22 | $ | 0.11 | $ | 1.61 | |||||
Diluted net income per share |
$ | 0.30 | $ | 0.21 | $ | 0.11 | $ | 1.23 | |||||
| |
2013 Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31 | June 30 | September 30 | December 31 | |||||||||
Product sales |
$ | 9,129 | $ | 14,106 | $ | 16,278 | $ | 18,789 | |||||
Total revenues |
26,174 | 29,963 | 37,460 | 40,608 | |||||||||
Gross margin on product sales |
7,645 | 12,418 | 14,527 | 16,621 | |||||||||
Income (loss) from operations |
(5,533 | ) | 532 | 6,838 | 7,480 | ||||||||
Net income (loss) |
(5,480 | ) | 478 | 6,514 | 41,801 | ||||||||
Basic net income (loss) per share |
$ | (0.10 | ) | $ | 0.01 | $ | 0.11 | $ | 0.73 | ||||
Diluted net income (loss) per share |
$ | (0.10 | ) | $ | 0.01 | $ | 0.11 | $ | 0.72 | ||||
121
SCHEDULE II: VALUATION AND QUALIFYING ACCOUNTS
(in thousands)
| |
|
Additions | |
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Description
|
Balance at Beginning of Year |
Charged as a Reduction to Revenue(1) |
Change in Deferred Revenue(1) |
Deductions(2) | Balance at End of Year |
|||||||||||
Sales & return allowances, discounts, chargebacks and rebates: |
||||||||||||||||
Year ended December 31, 2014 |
$ | 19,730 | $ | 73,898 | $ | — | $ | (57,351 | ) | $ | 36,277 | |||||
Year ended December 31, 2013 |
$ | 15,159 | $ | 28,743 | $ | — | $ | (24,172 | ) | $ | 19,730 | |||||
Year ended December 31, 2012 |
$ | 12,559 | $ | 12,872 | $ | (611 | ) | $ | (9,661 | ) | $ | 15,159 | ||||
| |
|
Additions | |
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Balance at Beginning of Year |
Additions charged to costs and expenses |
Other Additions |
Deductions | Balance at End of Year |
|||||||||||
Deferred tax asset valuation allowance: |
||||||||||||||||
Year ended December 31, 2014(4) |
$ | 2,670 | $ | — | $ | — | $ | 1,640 | $ | 4,310 | ||||||
Year ended December 31, 2013(3) |
$ | 42,500 | $ | — | $ | — | $ | (39,830 | ) | $ | 2,670 | |||||
Year ended December 31, 2012 |
$ | 34,700 | $ | 7,800 | $ | — | $ | — | $ | 42,500 | ||||||
- (1)
- Additions
to sales discounts and allowances are recorded as a reduction of deferred revenue until such time revenue is recognized.
- (2)
- Deductions
to sales discounts and allowances relate to discounts or allowances actually taken or paid.
- (3)
- The
Company reversed a valuation allowance of $39.8 million during 2013.
- (4)
- The Company recorded a valuation allowance of $1.6 million during 2014.
122