Attached files
| file | filename |
|---|---|
| EX-10.32 - SERVIER TERMINATION LETTER - PHARMACYCLICS INC | exhibit1032servierterminat.htm |
| EXCEL - IDEA: XBRL DOCUMENT - PHARMACYCLICS INC | Financial_Report.xls |
| EX-10.34 - CCO OFFER LETTER - PHARMACYCLICS INC | exhibit1034cooofferletter.htm |
| EX-10.33 - SERVIER TERMINATION COMMITMENT - PHARMACYCLICS INC | exhibit1033servierterminat.htm |
| EX-23.1 - CONSENT - PHARMACYCLICS INC | exhibit231consent201410-k1.htm |
| EX-31.1 - CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER - PHARMACYCLICS INC | pcyc20141231exhibit3111.htm |
| EX-31.2 - CERTIFICATION OF CHIEF FINANCIAL OFFICER - PHARMACYCLICS INC | pcyc20141231exhibit3121.htm |
| EX-21 - SUBSIDIARY LISTING - PHARMACYCLICS INC | exhibit21-subsidiarylistin.htm |
| EX-10.9 - FORM OF OPTION AGREEMENT - PHARMACYCLICS INC | exhibit109formofoptionagr.htm |
| EX-32.1 - CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER AND PRINCIPAL FINANCIAL OFFICER - PHARMACYCLICS INC | pcyc20141231exhibit3211.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
____________________________________
FORM 10-K
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
OR
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from __________ to __________
Commission File Number: 000-26658
Pharmacyclics, Inc.
(Exact name of Registrant as specified in its charter)
Delaware | 94-3148201 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
995 E. Arques Avenue, Sunnyvale, CA | 94085-4521 |
(Address of principal executive offices) | (Zip code) |
Registrant’s telephone number, including area code: (408) 774-0330
____________________________________
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange On Which Registered | |
Common Stock, $.0001 Par Value | Nasdaq Capital Market | |
Securities registered pursuant to Section 12(g) of the Act: None
(Title of Class)
____________________________________
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý
No o
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§229.405 of this chapter)
during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files) Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. o
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check one:
Large accelerated filer ý Accelerated filer o Non-accelerated filer o Smaller reporting company o
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
As of June 30, 2014, the aggregate market value of the voting and non-voting stock held by non-affiliates of the Registrant was $4,026,965,098 based on the closing sale price of the Registrant's common stock on The NASDAQ Stock Market LLC on that date. Shares of the Registrant's common stock beneficially owned by each executive officer and director of the Registrant and by each person known by the Registrant to beneficially own 10% or more of its outstanding common stock have been excluded, in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The number of outstanding shares of the Registrant’s common stock as of February 12, 2015 was 76,016,912.
____________________________________
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the following document are incorporated by reference into Part III of this Form 10-K: the Definitive Proxy Statement for the Registrant’s 2015 Annual Meeting of Stockholders which will be filed with the Securities and Exchange Commission within 120 days after the end of the Registrant’s fiscal year ended December 31, 2014.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2014
TABLE OF CONTENTS
Page | ||||||
PART I | ||||||
Item | 1 | Business | ||||
Item | 1A. | Risk Factors | ||||
Item | 1B. | Unresolved Staff Comments | ||||
Item | 2 | Properties | ||||
Item | 3 | Legal Proceedings | ||||
Item | 4 | Mine Safety Disclosures | ||||
PART II | ||||||
Item | 5 | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | ||||
Item | 6 | Selected Financial Data | ||||
Item | 7 | Management’s Discussion and Analysis of Financial Condition and Results of Operations | ||||
Item | 7A. | Quantitative and Qualitative Disclosures About Market Risk | ||||
Item | 8 | Financial Statements and Supplementary Data | ||||
Item | 9 | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | ||||
Item | 9A. | Controls and Procedures | ||||
Item | 9B. | Other Information | ||||
PART III | ||||||
Item | 10 | Directors, Executive Officers and Corporate Governance | ||||
Item | 11 | Executive Compensation | ||||
Item | 12 | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | ||||
Item | 13 | Certain Relationships and Related Transactions and Director Independence | ||||
Item | 14 | Principal Accountant Fees and Services | ||||
PART IV | ||||||
Item | 15 | Exhibits and Financial Statement Schedules | ||||
Signatures | ||||||
Exhibits Index | ||||||
Part I
Important Factors Regarding Forward-Looking Statements
This report contains forward-looking statements. These statements relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “goal,” “intend,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “should” or “will” or the negative of such terms or other comparable terminology. In particular, forward-looking statements include:
• | statements about our future capital requirements and the sufficiency of our cash, cash equivalents, marketable securities and other financing proceeds to meet these requirements; |
• | information concerning possible or assumed future results of operations, trends in financial results and business plans; |
• | statements about our product development schedule; |
• | statements about our expectations for and timing of regulatory approvals for any of our product candidates; |
• | statements about the level of our expected revenues, costs and expenses; |
• | statements about the potential results of ongoing or future clinical trials; |
• | other statements about our plans, objectives, expectations and intentions; and |
• | other statements that are not historical fact. |
From time to time, we also may provide oral or written forward-looking statements in other materials we release to the public. Forward-looking statements are only predictions that provide our current expectations or forecasts of future events. Any or all of our forward-looking statements in this report and in any other public statements are subject to unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, performance or achievements. You should not place undue reliance on these forward-looking statements.
We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosures we make on related subjects in our Quarterly Reports on Form 10-Q and Current Reports on Form 8-K. Also note that we provide a cautionary discussion of risks, uncertainties, assumptions and other factors relevant to our business under the caption Risk Factors and elsewhere in this report. These are risks that we think could cause our actual results to differ materially from expected or historical results.
Item 1. Business
Company Overview
We are a fully integrated biopharmaceutical company focused on developing and commercializing novel therapies for the treatment of cancer and immune-mediated diseases. We are currently an approximately 630-person company with in-house research and development, commercial and third-party contracted manufacturing capabilities and a growing U.S. footprint and global presence. Our goal is to make available therapies intended to improve quality of life, increase duration of life, and resolve serious unmet medical needs for patients. We will do this by identifying and controlling promising product candidates based on our scientific development and administrational expertise, developing our products in a rapid, cost-efficient manner, and by pursuing commercialization and/or development partners when and where appropriate. To that end, Pharmacyclics is at the forefront at transforming the speed by which innovative, high-quality medicines can advance from bench to bedside. Our first commercial product, IMBRUVICA® (ibrutinib), was developed and commercialized in 4.5 years from the start of its first clinical trial in 2009.
IMBRUVICA is a first-in-class, oral, once-daily, single-agent therapy which has demonstrated a survival advantage over an approved, standard-of-care therapy in a difficult-to-treat blood cancer. IMBRUVICA inhibits a protein called Bruton's tyrosine kinase (BTK), a key signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B-cells. IMBRUVICA blocks signals that tell malignant B-cells to multiply and spread uncontrollably.
We market IMBRUVICA in the United States (U.S.) for our four FDA-approved indications for the treatment of patients with: chronic lymphocytic leukemia (CLL) who have received at least one prior therapy; all lines of CLL with deletion of the short arm of chromosome 17 (del 17p CLL); mantle cell lymphoma (MCL) who have received at least one prior therapy; and all lines of Waldenström's macroglobulinemia (WM).
Accelerated approval was granted for the MCL indication based on overall response rate (ORR). Improvements in survival or disease symptoms have not been established. Continued approval for the MCL indication may be contingent upon verification of clinical benefit in confirmatory trials. IMBRUVICA is the only medicine approved to treat patients with del 17p CLL and WM.
IMBRUVICA was one of the first medicines to receive FDA approval via the new Breakthrough Therapy Designation pathway, and is the only product to have received three Breakthrough Therapy Designations. In the U.S., IMBRUVICA received its first four FDA approvals in a period of less than fifteen months, ranging from November 2013 through January 2015, echoing the same speed by which the product was developed (see IMBRUVICA Regulatory Updates). IMBRUVICA currently is approved for use in approximately 40 countries including the U.S., Canada, and the 28 countries which comprise the European Union (EU).
We believe that IMBRUVICA is helping to transform the management of blood cancers, and we are encouraged by the high clinical adoption rate we have seen in our approved indications. In fact, IMBRUVICA currently occupies the position of one of the most successful oncology drug launches in history based on launch revenue trajectories.
In commercial use and in the clinical trial setting, IMBRUVICA has demonstrated -- and continues to demonstrate -- a favorable efficacy, safety, toxicity, and durability of response profile. To date, over 5,600 patients have been treated in Company-sponsored IMBRUVICA trials conducted in over 35 countries involving more than 800 investigators. We are continuing to investigate how IMBRUVICA may benefit a broader group of patients in the future. Our development program has several clinical trials underway studying IMBRUVICA alone and in combination with other therapies in several blood cancers including in CLL, which is a clinical development program that currently includes seven Phase III trials and covers all lines of therapy and various combinations of treatments. Additional blood cancers we are investigating in which we believe IMBRUVICA may have a benefit include: small lymphocytic leukemia (SLL), MCL, WM, diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), multiple myeloma (MM), and other forms of cancer. A clinical trial is also underway studying IMBRUVICA in Graft versus host disease (GvHD). In addition, we are beginning the exploration of IMBRUVICA in select solid tumor types. As of December 31, 2014, 13 Phase III trials have been initiated with IMBRUVICA and approximately 58 trials are registered on www.clinicaltrials.gov. Information found on this website is not incorporated by reference into this report.
We are conducting this research together with our partner Janssen Biotech Inc. and its affiliates (Janssen), one of the Janssen Pharmaceutical companies of Johnson & Johnson, under our 2011 worldwide collaboration and license agreement (the Agreement). However, we recently have formed a number of strategic collaborations with other world-class companies including Amgen Inc., AstraZeneca, Bristol-Myers Squibb Co., Celgene Corp., and F. Hoffmann-La
1
Roche Ltd. (Roche) in order to explore the potential of IMBRUVICA as a combination agent and a backbone of therapy for certain blood cancers and solid tumors.
Under the Agreement, we and our partner Janssen are jointly commercializing IMBRUVICA in the U.S. Janssen is commercializing IMBRUVICA outside the U.S.
In addition to IMBRUVICA, we have other product candidates in clinical development and several pre-clinical molecules in lead optimization. We will continue to pioneer research into the development of next-generation BTK inhibitors. Our pre-clinical molecules include BTK inhibitors for autoimmune disorders, such as rheumatoid arthritis. We remain committed to high standards of ethics, scientific rigor, and operational efficiency throughout our development efforts.
We are headquartered in Sunnyvale, California and are listed on NASDAQ under the symbol PCYC. To learn more about how Pharmacyclics advances science to improve human healthcare visit us at http://www.pharmacyclics.com. Information found on our website is not incorporated by reference into this report.
Our Business Strategy
The five key elements of our business strategy are to:
• | Extend IMBRUVICA’s reach and enhance our leadership position across hematology. We intend to continue to grow our IMBRUVICA business, further strengthen our market leadership within our approved indications, and expand the use of IMBRUVICA. We have established fully integrated commercial and manufacturing capabilities to address key areas for the successful ongoing commercialization of our product in the U.S., as well as to support the commercial needs of our partner outside of the U.S. In addition, we are developing future indications for IMBRUVICA across a variety of other blood cancers through a mix of the development efforts we undertake ourselves and with other world-class companies through strategic clinical and drug supply collaborations. We intend to expand the use of our key product through ongoing label expansion efforts in areas of need that will allow us to maintain leadership positions in those markets. |
• | Establish IMBRUVICA in multiple solid tumor indications. Based on early data, IMBRUVICA demonstrates promise in its ability to inhibit growth of breast cancer and non-small cell lung cancer cell lines due to the inhibition of HER-2 and epidermal growth factor receptor (EGFR) inhibitor. In addition, pre-clinical experiments suggest that the combination of IMBRUVICA and an antibody targeting PD-L1 causes improved inhibition of tumor growth. This potentially synergistic combination effect, along with the provocative single-agent activity, has provided the basis for our further exploration of the utility of IMBRUVICA in solid tumors in several trials, one of which is being conducted through a strategic clinical and drug supply collaboration with world-class companies. |
• | Develop next-generation BTK inhibitors as a new standard-of-care for immune-mediated diseases. Based on our expertise in chemistry, biology and clinical development that has led to the success of IMBRUVICA and the acknowledgment of the importance of BTK and BCR pathway signals, we are continuing to create novel, patentable, differentiated drug candidates that address areas of unmet medical need for the treatment of immune-mediated diseases. Proof-of-concept data in immune-mediated diseases demonstrates compelling evidence of the potential of our candidate to modify the activity of destructive cells involved in these two conditions. |
• | Leverage our core capabilities to ensure accelerated innovation and long-term growth. With our particular expertise in BTK and kinase inhibition, we are making strategic investments to sustain ongoing innovation and fuel our long-term growth, namely in the area of strategic collaborations with like-minded companies who also want to lead in innovation. Moreover, we enjoy a broad intellectual property (IP) portfolio to protect our pre-clinical insights. |
• | Focus on profitability while investing to expand and sustain our business for the future. We have a record of steady revenue growth and a solid cash position with a goal of maintaining profitability while investing wisely in IMBRUVICA and product candidate pipeline. |
BTK Inhibitor Program
We are pioneers in the development of orally bioavailable inhibitors of BTK, a signaling protein that is critically important for the activity of B-cells (immune cells that can develop into antibody producing cells). B-cell lymphomas
2
and leukemias, which are common blood cancers, result from mutations acquired during B-cell development that lead to uncontrolled B-cell proliferation. Additionally, when B-cells are overactive, the immune system can produce antibodies that begin to attack the body's own tissue, leading to autoimmune diseases. Both autoimmune diseases and B-cell malignancies are thought to be driven by overactive signaling and activation of the B-cell antigen receptor (BCR), a process that is dependent on BTK.
To date, we have developed and commercialized IMBRUVICA in four indications, for the treatment of patients with: CLL who have received at least one prior therapy; all lines of del 17p CLL; MCL who have received one prior therapy; and all lines of WM. We continue to develop IMBRUVICA, which has demonstrated novel activity in pre-clinical studies across a variety of B-cell malignancies and solid tumors. We are pursuing multiple indications as part of our clinical development plan for IMBRUVICA, which include the following B-cell malignancies: CLL, SLL, MCL, FL, DLBCL, MM, WM, marginal zone lymphoma (MZL), acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML) and other cancer types. We are also pursuing the development of IMBRUVICA for GvHD. In addition, we have a pre-clinical testing program underway to develop novel inhibitors of BTK for inflammatory and autoimmune diseases.
IMBRUVICA Mechanism of Action
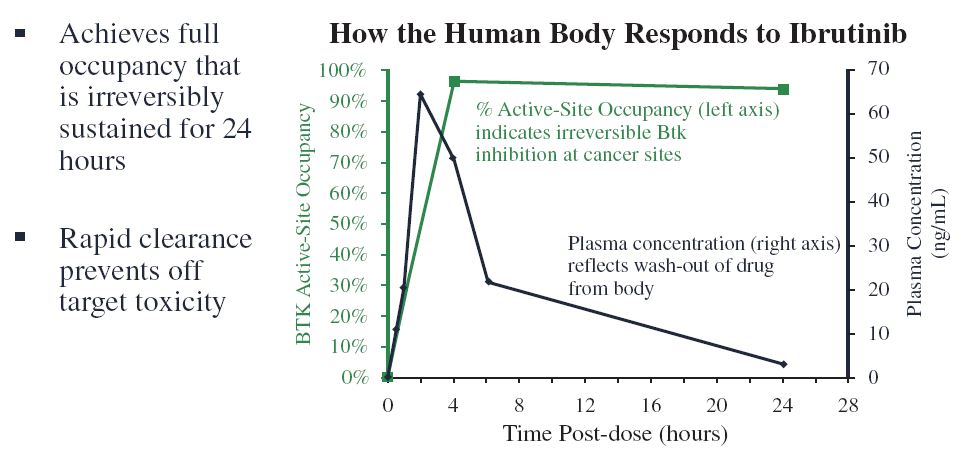
IMBRUVICA is a potent and selective small molecule inhibitor of BTK, a signaling kinase expressed in B-cells. BTK is an enzyme that functions downstream of the BCR, which is present on the surface of B-cells. When engaged, the BCR signaling pathway causes the B-cell to grow and develop. In a study funded entirely by Pharmacyclics, we found that selective inhibition of BTK with IMBRUVICA blocks BCR signaling and prevents B-cell activation (Honigberg et al., Proc Natl Acad Sci USA, 2010; 107: 13075-80). IMBRUVICA binds covalently to an unpaired cysteine residue (cysteine-481) in the ATP-binding site of BTK, thereby inhibiting the activity of BTK (IC50 of 0.5 nM). With few exceptions, IMBRUVICA does not appear to bind to other cellular proteins as strongly or as rapidly as it does to BTK in vivo. Importantly, new evidence has demonstrated IMBRUVICA inhibits B-cell lymphoma adhesion in vitro (Chang et al, Blood 2013; de Rooij et al; Blood, 2012), which may be related to the rapid lymphocytosis that is observed clinically following administration of the drug (Advani et al., JCO; 2012). In humans, the levels of IMBRUVICA in the blood are reduced by half within 2 to 4 hours of peak exposure. With the combination of irreversible “on-target” kinase inhibition and rapid elimination from the blood, we achieve 24-hour BTK inhibition with once-daily dosing while reducing the duration of reversible inhibition of many “off-target” kinases.
In CLL, multiple studies have documented evidence of enhanced BCR signaling, especially in patients with immunoglobulin variable heavy chain (IgVH) unmutated disease or those with increased ZAP-70 expression, which are predictors of poor prognosis to cytotoxic chemotherapy. It has been published that IMBRUVICA promotes apoptosis, inhibits proliferation, and prevents CLL cells from responding to survival stimuli provided by the microenvironment (Herman et al, Blood, 2011; 117:6287-6296). In this study, treatment of activated CLL cells with IMBRUVICA inhibited the phosphorylation activity of BTK and effectively abrogated BTK-dependent downstream survival pathways including those involving ERK1/2, PI3K and NF-κB. Additionally, IMBRUVICA inhibited activation-induced proliferation of CLL cells in vitro, effectively blocking survival signals provided externally to CLL cells by components of the microenvironment including soluble factors (CD40L, BAFF, IL-6, IL-4 and TNF-α), fibronectin engagement, and stromal cell contact.
3
Several lines of evidence suggest that signaling through the BCR pathway is necessary to sustain the viability of B-cell lymphomas, and BTK was identified in a conventionally-designed small interfering RNA compounds (siRNA) screen as an essential kinase for survival in a subset of DLBCLs driven by activated BCR. In these cells, chronic active BCR signaling drives constitutive NF-κB signaling blocking apoptosis; blocking BTK with IMBRUVICA was shown to promote apoptosis in these cells (Davis et al., Nature, 2010; 463: 88-94).
IMBRUVICA Resistance
Clinically effective cancer therapies are typically associated with escape mechanisms and resistance. Pharmacyclics is committed to understanding the underlying reason for patients who initially respond to IMBRUVICA, who subsequently regress. To this end, we have studied (1) tumor cell lines that have been selected to become resistant to IMBRUVICA and blood samples from the small number of CLL patients who have acquired resistance to IMBRUVICA over time.
As reported at the International Workshop on CLL (IWCLL) meeting in September 2013, at a median follow up of 22.1 months, the estimated median progression-free survival (PFS) rates in our PCYC-1102-CA study were: 96% for treatment-naive CLL patients (n=31), 92% for relapsed/refractory CLL patients excluding high-risk patients with del 17p and del 11q (n=29), and 74% for relapsed/refractory patients including high risk patients (n=85) at 26 months. In patients with CLL, the development of therapeutic resistance has been uncommon so far. Nevertheless, early research has revealed the acquisition of specific point mutations in B-cell receptor pathway genes, including BTK and its downstream mediator PLCgamma, in several patients with acquired IMBRUVICA resistance (Woyach NEJM 2014). These mutations may help to explain the therapeutic resistance and have validated BTK as a relevant target of IMBRUVICA in CLL.
As reported at the European Hematology Association (EHA) meeting in June 2013, our PCYC-1104-CA study of IMBRUVICA in MCL patients (n=111) had an estimated median progression-free survival of 13.9 months. In addition, as reported in an abstract at the American Society of Hematology (ASH) annual meeting in December 2012, our PCYC-1106 study of IMBRUVICA in DLBCL patients (n=70) had an estimated median progression-free survival of 1.6 months. The rate of acquired resistance is higher in MCL and DLBCL patients compared to CLL patients. Research using MCL lines has revealed several potential mechanisms of resistance, including the activation of alternate B-cell signaling pathways inside the cell. Samples from MCL patients have also been evaluated and genes such as PIM1 and Erb4 have been noted to be mutated more frequent in patients with progressive disease compared with those who were not resistant to therapy (ASH 2014). Further, data presented at ASH 2014 and the EHA 2013 has implicated CARD11, MYD88, and Bcl-2 as possible resistance-associated genes in DLBCL.
We do not yet understand the full scope of these mechanisms in cancer patients who relapse, nor do we understand the full breadth of mutations that may underlie acquired resistance to IMBRUVICA in the different B-cell malignancies. We intend to actively study this beginning in 2015.
BTK Inhibitor Market Opportunity
There are significant and distinct areas of unmet medical needs across the B-cell malignancies. Within the indolent lymphomas, we believe a need still exists for active therapies that avoid the toxicities typically seen with conventional chemotherapies. Such active therapies are needed as part of effective combinations early in the course of treatment, and as effective single-agent treatments later in the course of disease progression. In particular, drugs are needed that are well tolerated and which do not limit subsequent treatment options because of bone marrow or other organ toxicity. In the aggressive lymphomas, it is our belief that the need exists for agents that can combine with standard therapies to improve cure rates and that are effective in patients who fail potentially curative therapy. While advances have been made in the treatment of MM, patients still need effective and tolerable therapies as there currently is no cure. To that end, we are actively evaluating IMBRUVICA in combination with established MM therapies.
Our comprehensive clinical development program addresses a broad range of B-cell malignancies as well as select solid tumors.
Commercialization of IMBRUVICA
IMBRUVICA is available in approximately 40 countries including the U.S., Canada, and the 28 countries which comprise the EU. In the U.S., IMBRUVICA is approved by the U.S. Food and Drug Administration (FDA) as an oral single agent drug for four indications for the treatment of patients with: CLL who have received at least one prior therapy; all lines of del 17p CLL; MCL who have received at least one prior therapy; and all lines of WM. In the 28 member countries of the EU, IMBRUVICA is approved for the treatment of adult patients with relapsed or refractory
4
(R/R) MCL, or patients with CLL who have received at least one prior therapy, or in first line CLL patients in the presence of a del 17p or TP53 mutation in patients unsuitable for chemotherapy.
Pharmacyclics leads the U.S. commercial activities together with Janssen. IMBRUVICA is being marketed by a Commercial team which has extensive and relevant expertise in the promotion, distribution, and reimbursement of oncology/hematology therapies. The Commercial team covers sales, brand marketing, strategy and analytics, reimbursement and distribution, training, business analytics, forecasting, and operations, as well as medical affairs. Pharmacyclics and Janssen participate equally and collaboratively in the U.S. sales activities and we believe that the size of our U.S. sales organization, which is supplemented by Janssen’s oncology sales organization who is involved in co-promoting IMBRUVICA, is appropriate to effectively reach our target audience. Our Marketing team develops strategic positioning and messaging that communicate the efficacy and safety of IMBRUVICA, and has developed creative campaigns for each indication to support the necessary promotional and educational activities.
Sales of pharmaceutical products depend, in significant part, on the coverage and reimbursement policies of government programs, including Medicare and Medicaid in the U.S., and other third-party payers. All third-party payers are sensitive to the cost of drugs and have taken efforts to control those costs and will continue to do so in the future. Private health insurance plans may restrict coverage of some products by using payer formularies under which only selected drugs are covered, variable co-payments that make drugs that are not preferred by the payer more expensive for patients, and by using utilization management controls, such as requirements for prior authorization or prior failure on another type of treatment. Payers may especially impose these obstacles with respect to coverage for higher priced drugs, and consequently IMBRUVICA may be subject to payer-driven restrictions. Therefore, our Market Access team covers Medicare, Medicaid, Commercial Health Plans, Federal Markets, Trade, Pricing, Policy, Contracting and Patient Access Services. Our Market Access team created several effective and comprehensive access and affordability programs which are dedicated to helping appropriate patients receive access to IMBRUVICA.
Our Medical Affairs group is well established at Pharmacyclics and consists of teams focused on Medical Science, Medical Communications and Medical Information. In 2015, we are operationalizing a team of Clinical Educators with advanced degrees and clinical experience who will be focused on the community setting to further educate healthcare providers on IMBRUVICA, connect community physicians to Medical Affairs resources, and to launch our informCLL™ registry.
MCL, CLL and WM are considered orphan diseases. Today, it is estimated that there are approximately 13,300 MCL patients, 121,200 CLL patients and 12,000 WM patients living with these diseases in the U.S. (Note: this information is an estimate derived from the use of information under license from the following IMS Health Incorporated information service: IMS Oncology Tracking Reports for the period July 2013 to June 2014. IMS expressly reserves all rights, including rights of copying, distribution and republication). Many of these patients cycle through multiple lines of treatments often requiring combination therapy with extensive supportive care. During September 2014, we increased the price of IMBRUVICA by 7%. For patients with MCL, the approved dose is four 140 mg pills of IMBRUVICA once daily for an initial cost of approximately $11,699 for a 30-day supply. For patients with CLL (including del 17p CLL) and WM, the approved dose is three 140 mg pills of IMBRUVICA once daily for an initial cost of approximately $8,774 for a 30-day supply. We have a well established and comprehensive Patient Access Support program to ensure access for patients in need of IMBRUVICA.
IMBRUVICA IMPORTANT SAFETY INFORMATION
In accordance with U.S. federal law, when information is presented about a prescription drug product’s intended use and makes claims about its benefit for a disease or medical condition, a company is required to provide a fair balance of information about the drug’s risk compared to its benefits. This means that the drug’s most important risks must be appropriately similar in prominence to the information being presented about its benefits. This information, which is referred to as Important Safety Information, is derived directly from the IMBRUVICA product label and should not be altered. It is intended for use by physicians who rely on this information to help inform and guide patient treatment and care.
Warnings and Precautions
Hemorrhage - Fatal bleeding events have occurred in patients treated with IMBRUVICA. Grade 3 or higher bleeding events (subdural hematoma, gastrointestinal bleeding, hematuria, and post-procedural hemorrhage) have occurred in up to 6% of patients. Bleeding events of any grade, including bruising and petechiae, occurred in approximately half of patients treated with IMBRUVICA.
5
The mechanism for the bleeding events is not well understood. IMBRUVICA may increase the risk of hemorrhage in patients receiving antiplatelet or anticoagulant therapies. Consider the benefit-risk of withholding IMBRUVICA for at least 3 to 7 days pre and post-surgery depending upon the type of surgery and the risk of bleeding.
Infections - Fatal and non-fatal infections have occurred with IMBRUVICA therapy. Grade 3 or greater infections occurred in 14% to 26% of patients. Cases of progressive multifocal leukoencephalopathy (PML) have occurred in patients treated with IMBRUVICA. Monitor patients for fever and infections and evaluate promptly.
Cytopenias - Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (range, 19 to 29%), thrombocytopenia (range, 5 to 17%), and anemia (range, 0 to 9%) occurred in patients treated with IMBRUVICA. Monitor complete blood counts monthly.
Atrial Fibrillation - Atrial fibrillation and atrial flutter (range, 6 to 9%) have occurred in patients treated with IMBRUVICA, particularly in patients with cardiac risk factors, acute infections, and a previous history of atrial fibrillation. Periodically monitor patients clinically for atrial fibrillation. Patients who develop arrhythmic symptoms (e.g. palpitations, lightheadedness) or new-onset dyspnea should have an ECG performed. If atrial fibrillation persists, consider the risks and benefits of IMBRUVICA treatment and dose modification.
Second Primary Malignancies - Other malignancies (range, 5 to 14%) including non-skin carcinomas (range, 1 to 3%) have occurred in patients treated with IMBRUVICA. The most frequent second primary malignancy was non-melanoma skin cancer (range, 4 to 11%).
Tumor Lysis Syndrome - Tumor lysis syndrome has been reported with IMBRUVICA therapy. Monitor patients closely and take appropriate precautions in patients at risk for tumor lysis syndrome (e.g. high tumor burden).
Embryo-Fetal Toxicity - Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. Advise women to avoid becoming pregnant while taking IMBRUVICA. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus.
Adverse Reactions
The most common adverse reactions (≥25%) in patients with B-cell malignancies (MCL, CLL, WM) were thrombocytopenia, neutropenia, diarrhea, anemia, fatigue, musculoskeletal pain, bruising, nausea, upper respiratory tract infection, and rash. Seven percent of patients receiving IMBRUVICA discontinued treatment due to adverse events.
Drug Interactions
CYP3A Inhibitors - Avoid co-administration with strong and moderate CYP3A inhibitors. If a moderate CYP3A inhibitor must be used, reduce the IMBRUVICA dose.
CYP3A Inducers - Avoid co-administration with strong CYP3A inducers.
Specific Populations
Hepatic Impairment - Avoid use in patients with moderate or severe baseline hepatic impairment. In patients with mild impairment, reduce IMBRUVICA dose.
For additional important safety information, please see Full Prescribing Information at http://www.imbruvica.com/downloads/Prescribing_Information.pdf.
Patient Access to IMBRUVICA
Patients who are prescribed IMBRUVICA can receive access support through several distinct programs:
• | The YOU&i Start™ program enables eligible patients who are experiencing insurance coverage delays to access free product for a limited time. |
• | The YOU&i Access™ Instant Savings Program helps commercially insured, eligible patients who have difficulties with out-of-pocket expenses for IMBRUVICA. Eligible patients may receive support to reduce their monthly out-of-pocket costs to $10 per month*. |
- *Month refers to a 30-day supply. Subject to a maximum benefit, 12 months after activation or 12 monthly fills (1-year supply), whichever comes first, unless the maximum dollar benefit has been
6
reach.
- Not valid for patients enrolled in Medicare or Medicaid.
• | The YOU&i Access Service Center assists patients with all access-related administration issues. |
• | Pharmacyclics makes donations to the Johnson & Johnson Patient Assistance Foundation (JJPAF), an independent, non-profit organization that provides assistance to patients who need access to IMBRUVICA who are eligible based on financial need and if they are uninsured. |
• | Pharmacyclics is supporting third-party foundations, organizations and other efforts to help patients in need get access to appropriate care. |
Customers
We sell IMBRUVICA directly to some customers who have in-house dispensing capabilities, specialty pharmacies that sell to individual patients, specialty distributors that sell to hospital pharmacies and other organizations that we have contracted with. For details on our customers, refer to Notes 2 and 3 to the consolidated financial statements.
Competition
There are many companies focused on the development of small molecules and antibodies for treating B-cell malignancies. Companies such as AbbVie Inc., Celgene Corp., Gilead Sciences Inc., Roche and others are known to be active in the development and/or commercialization of therapies for B-cell malignancies.
Our potential competitors include major pharmaceutical and biotechnology companies, clinical reference laboratories and government agencies, as well as academic research institutions that are pursuing research activities similar to ours. Many of our potential competitors have significantly more financial and other resources, larger research and development staffs, lower labor costs, and/or more extensive marketing and manufacturing organizations and other resources than we do, which may allow them to have a competitive advantage.
Many of these companies and organizations have significant experience in pre-clinical testing, human clinical trials, product manufacturing, marketing, sales and distribution and other regulatory approval and commercial procedures. They may also have a greater number of significant patents and greater legal resources to seek remedies for cases of alleged infringement of their patents by us to block, delay or compromise our own drug development process.
The markets for which we have and intend to pursue regulatory approval of IMBRUVICA are highly competitive. We are aware of products in research or development by our competitors that are intended to treat the B-cell malignancies we are targeting, and any of these products may compete with IMBRUVICA and/or be combined with IMBRUVICA to improve patient outcomes. Our competitors may succeed in obtaining approvals for their products more rapidly than we do from the FDA or from regulatory agencies in other regions of the world, or developing products that are more effective than IMBRUVICA. These products or technologies might render our technology obsolete or noncompetitive. There may also be drug candidates of which we are not aware at earlier stages of development that may compete with IMBRUVICA. In addition, IMBRUVICA competes with existing therapies that have long histories of use, such as chemotherapy used in cancer indications.
Our Pipeline
Our current clinical development program examines product candidates that are small-molecule enzyme inhibitors designed to target key biochemical pathways involved in life-threatening human diseases. IMBRUVICA, our first product to market, is indicated for the treatment of specific subsets of CLL, MCL and WM patients. We are continuing the development of IMBRUVICA across a variety of B-cell malignancies as well as in solid tumors and certain immune-mediated diseases. In addition to IMBRUVICA, we currently have other product candidates in clinical development and several pre-clinical molecules in lead optimization; including a BTK inhibitor currently in a Phase I study in rheumatoid arthritis (RA) patients, an inhibitor of Factor VIIa (PCI-27483) and a HDAC inhibitor, abexinostat (formerly known as PCI-24781). Our BTK inhibitor program, consisting of several novel BTK inhibitors, has been established to continue our clinical development and commercial success in oncology and immune-mediated diseases and support long-term growth.
Status of Products Currently in Pre-Clinical and Clinical Development
The table below summarizes our pre-clinical programs and clinical product candidates and their stage of development:
7
Product Candidates/Programs | Disease Indication | Development Status(1) | ||
IMBRUVICA BTK Inhibitor | • Chronic lymphocytic leukemia (CLL) | Multiple trials (Phase I, II, III) in | ||
• Small lymphocytic lymphoma (SLL) | treatment naive and in relapsed/ | |||
• Mantle cell lymphoma (MCL) | refractory patients | |||
• Diffuse large B-cell lymphoma (DLBCL) | ||||
• Follicular lymphoma (FL) | ||||
• Multiple myeloma (MM) | ||||
• Waldenström's macroglobulinemia (WM) | ||||
• Marginal zone lymphoma (MZL) | ||||
• Graft versus host disease (GvHD) | ||||
• Acute Lymphoblastic Leukemia (ALL) | ||||
• Acute Myeloid Leukemia (AML) | ||||
• Solid tumors and others | ||||
BTK Inhibitor Program | Autoimmune | Pre-clinical testing, Phase I | ||
Abexinostat HDAC Inhibitor (PCI-24781) | Relapsed/refractory lymphomas and solid tumors | Multiple trials (Phase I, II) | ||
Factor VIIa Inhibitor (PCI-27483) | Cancer | Phase II complete/program under review | ||
(1) "Phase I" means initial human clinical trials designed to establish the safety, dose tolerance, pharmacokinetics (i.e., absorption, metabolism, excretion) and pharmacodynamics (i.e. biological markers for activity) of a compound. "Phase II" means human clinical trials designed to establish safety, optimal dosage and preliminary activity of a compound in a patient population. "Phase III" means human clinical trials designed to establish the safety and efficacy of a compound. These are the most important trials required by the FDA and are done to rigorously establish the clinical benefit and safety profile of a drug in a particular patient population. "Pre-clinical" means the stage of drug development prior to human clinical trials in which a molecule is optimized for "drug like" properties and evaluated for efficacy, pharmacokinetics, pharmacodynamics and safety.
IMBRUVICA for Solid Tumors
Based on early data, IMBRUVICA demonstrates promise in its ability to inhibit growth of HER-2 breast cancer cells as a potent epidermal growth factor receptor (EGFR) inhibitor. In addition, early data suggest that in combination with PD-L1, IMBRUVICA enhances the checkpoint inhibitor’s ability to attack tumors and allow the immune response to fight the cancer. The enhanced effects observed with the combination, along with the provocative single-agent activity, has provided the basis for our further exploration of the utility of IMBRUVICA in solid tumors in several trials, one of which is being conducted through a strategic clinical and drug supply collaborations with world-class companies.
BTK Inhibitor for Autoimmune Diseases
In animal models of RA, we have observed that once-daily oral administration of our proprietary BTK inhibitors lead to regression of established disease. Based on data from a study funded entirely by Pharmacyclics, we reported that our BTK inhibitors reduce cytokine releases from human monocytes in cell culture and reduced inflammatory synovitis, pannus formation, synovial fluid cytokines, cartilage damage and bone erosion in mice with collagen-induced arthritis (Chang et al., ACR Annual Meeting Abstracts, 2010). In 2014, we continued working on a series of BTK inhibitors which were optimized pre-clinically for eventual treatment of patients with anti-inflammatory and autoimmune diseases, including RA. In January 2014, we filed an investigational new drug application (IND) for our lead molecule for anti-inflammatory and autoimmune diseases. This IND was deemed safe to proceed by the FDA in mid-February 2014.
IMBRUVICA Recent Clinical Development Updates
During the year ended December 31, 2014, we provided updates on several of our clinical programs at various scientific conferences including the 56th ASH Annual Meeting in San Francisco from December 5-9, 2014 and the
8
50th Annual Meeting of the American Society of Clinical Oncology (ASCO) in Chicago, IL from May 30-June 3, 2014.
The following are abstracts presented at ASH and ASCO during 2014 that were made available in our press releases and filings with the SEC:
Selected Abstracts from the ASH 2014 Annual Meeting:
Efficacy and Safety of Ibrutinib in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Leukemia with 17p Deletion: Results from the Phase II RESONATE-17 Trial (Oral Presentation - ASH Abstract # 327)
Results were presented from the Phase II open-label, single-arm, multi-center RESONATE-17 (PCYC-1117) trial in which 144 del 17p patients (95% with CLL, 5% with SLL) received single-agent IMBRUVICA once-daily until progression. The primary endpoint was ORR, as determined by an independent review committee (IRC). Key secondary endpoints were duration of response (DOR), PFS and safety. The investigator-assessed ORR was 83% with 17% of these patients achieving a PR-L including 1% who achieved a CR. The IRC-assessed ORR was 65%. Neither the median PFS nor DOR had been reached with a median follow-up of 11.5 months. Seventy-nine percent of patients were alive and had not progressed at 11.5 months, with an overall survival (OS) rate of 84%.
At the time of the data cut, the median time on study (treatment duration) was 11.5 months, and 101 of 144 patients (70%) continued to take IMBRUVICA.
The most common Grade 3 or 4 AEs in the RESONATE-17 trial (occurring in ≥5% of IMBRUVICA patients) were neutropenia (14%), anemia (8%), and hypertension (8%). The most frequently reported AEs of any grade were: diarrhea (36%); fatigue (31% ); cough (24%); and, arthralgia (22%). Atrial fibrillation of any grade was reported in 11 patients (8%). Major bleeding was reported in seven patients taking IMBRUVICA (5%).
Ibrutinib, Single Agent or in Combination with Dexamethasone, in Patients with Relapsed or Relapsed/Refractory Multiple Myeloma (MM): Preliminary Phase II Results (Oral Presentation - ASH Abstract # 31)
Data from an open-label, Phase II dose escalation trial evaluated potential IMBRUVICA dosing regimens either as a monotherapy or in combination with dexamethasone 40 mg in the treatment of 69 heavily pre-treated (relapsed or relapsed/refractory) patients. Efficacy and safety were assessed at four-week intervals using the International Myeloma Working Group (IMWG) response criteria for efficacy results and Common Terminology Criteria for AEs (CTCAE) to evaluate safety.
Heavily pre-treated patients (median of 4.5 prior lines of therapy) who received IMBRUVICA 840 mg daily in combination with dexamethasone 40 mg weekly (n=20) experienced the highest clinical benefit rate of 25%, including one partial response (PR) and four minor responses (MR). Also, an additional five patients (25%) showed sustained stable disease (SD; >4 cycles). IMBRUVICA-treatment in combination with dexamethasone resulted in positive responses and disease stabilization which led to a median progression-free survival of 5.6 months. As a result of this trend toward improved efficacy and manageable toxicities, investigators expanded the treatment group per protocol design. Twenty-three additional patients are currently enrolled in this cohort; follow-up is ongoing. Based on these encouraging data, IMBRUVICA is currently being evaluated as a combination agent to treat relapsed/refractory multiple myeloma with agents such as carfilzomib.
The safety profile of IMBRUVICA was tolerable, with similar AE rates across dosing cohorts. Across all cohorts, 57% of patients experienced Grade 3 or greater adverse events (AEs). The most commonly reported non-hematologic AEs of any grade were: diarrhea (51%); fatigue (41%); nausea (35%); dizziness (25%); and muscle spasms (23%). Myelosuppression had an overall incidence of any grade anemia (29%), thrombocytopenia (23%), and neutropenia (7%), with 16%, 9% and 4% being Grade 3, respectively. Notably, there were no clinically meaningful differences among dose levels. Thirty-three percent of patients experienced a treatment-emergent serious AE. At the time of data cut-off, 4 patients remained on treatment; the most common reason for treatment discontinuation in 47% was progressive disease.
Ibrutinib and Rituximab are an Efficacious and Safe Combination in Relapsed Mantle Cell Lymphoma: Preliminary Results from a Phase II Clinical Trial (Oral Presentation - ASH Abstract # 627)
IMBRUVICA was combined with rituximab in a single-center, Phase II trial in 50 relapsed/refractory MCL patients. After a median follow-up of 11 months (range 4-16 months) among 34 evaluable patients with lower levels (< 50) of the Ki-67 protein, a known marker associated with cell growth, the ORR was 100% (56% CRs and 44% partial
9
responses [PRs]). Among 12 evaluable patients with higher Ki-67 (≥ 50%) protein levels, the ORR was 50% (8% CRs, 42% PRs).
The median duration of response and progression-free survival have not yet been reached.
Ten patients discontinued treatment during the study due to progressive MCL, all of whom had Ki67 levels greater than 60% (range 50-100%). There were no deaths due to toxicity. Grade 1 hematologic toxicity events included anemia (30%) and thrombocytopenia (25%). The most common treatment-emergent, non-hematologic adverse events (occurring in > 15% of patients treated with IMBRUVICA plus rituximab) included fatigue, diarrhea, myalgia and dyspnea. This combination has been well tolerated.
Addition of Rituximab Abrogates Ibrutinib-Induced Lymphocytosis and Promotes More Rapid Decrease in Absolute Lymphocyte Counts in Patients with Relapsed Chronic Lymphocytic Leukemia (Poster Presentation - ASH Abstract # 1998)
Data from a multi-center, Phase Ib/II trial also suggest that when IMBRUVICA and rituximab are combined in patients with relapsed CLL, short-term lymphocytosis, which sometimes is associated with IMBRUVICA treatment, decreased and patients experienced faster clearing of leukemia cells from the bloodstream versus patients who received IMBRUVICA alone. These data were presented in a poster presentation on Saturday, December 6th by Ekaterina Kim, M.S. from The University of Texas MD Anderson Cancer Center.
The median absolute lymphocyte count (ALC) during pre-treatment was similar in the IMBRUVICA single-agent and combination treatment arms (25 and 27.6 K/µl, respectively). In the IMBRUVICA single-agent arm, ALC levels rose immediately after treatment was started and began to stabilize. After four months of follow-up, patients in the combination therapy arm experienced a more rapid decrease in ALC levels, suggesting the combination decreased lymphocytosis over this period. Plasma levels of chemokines CCL3 and CCL4 are known to reflect the activation status of CLL cells. Concentration of both chemokines was significantly reduced after a week of treatment in both arms. Further analysis of the CCL3 and CCL4 patterns is still in progress. No additional safety findings were reported.
Updated Efficacy Including Genetic and Clinical Subgroup Analysis and Overall Safety in the Phase III RESONATE™ Trial of Ibrutinib Versus Ofatumumab in Previously Treated Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Poster Presentation - ASH Abstract # 3331)
Researchers presented median 16-month follow-up data from the pivotal Phase III RESONATE (PCYC-1112) trial studying IMBRUVICA versus ofatumumab (n=391; 195 IMBRUVICA, 196 ofatumumab). The patient population was heavily pretreated (a median of three prior therapies in the IMBRUVICA patients vs. a median of two prior therapies in the ofatumumab patients) and largely elderly, often with baseline comorbidities. At 12 months, 84% of IMBRUVICA-treated patients were progression free versus 18% of patients receiving ofatumumab (hazard ratio = 0.106; 95% confidence interval: 0.073, 0.153; P<0.001). The median overall survival in patients taking IMBRUVICA has not been reached with 168 of 195 patients (86%) alive. One hundred twenty-two patients (62%) randomized to ofatumumab crossed over to IMBRUVICA; the best ORR for single-agent IMBRUVICA was 90% versus 25% for ofatumumab, with 74% of IMBRUVICA patients achieving a partial response (PR), 8% partial responses with lymphocytosis (PR-L) and 4% complete responses (CR).
Patients who had received only one prior therapy before receiving IMBRUVICA had higher PFS and ORR rates than those who had received two or more prior therapies. At 12 months, 94% of IMBRUVICA patients who received one prior therapy, 84% who received two prior therapies and 80% who received three or more prior therapies, were progression free (P=0.0001). The ORR for patients who had received one prior therapy was 100%, while the ORR for patients who received two and three or more prior therapies was 79% and 78% (P=0.0001).
The most frequent Grade 3 or 4 adverse events (AEs) in the RESONATE trial analysis occurring in IMBRUVICA patients were: neutropenia (18%); pneumonia (9%); thrombocytopenia (6%); anemia (6%); and, hypertension (6%). Overall, at a median follow-up of 16 months 47 patients (24%) discontinued IMBRUVICA: 17 due to progressive disease (9%), 13 for AEs (7%) and 10 due to death (5%).
Hematologic and Immunologic Function and Patient Well-Being for the Phase III RESONATE™ Study of Ibrutinib vs. Ofatumumab in Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphoma (Poster Presentation - ASH Abstract # 4696)
IMBRUVICA’s impact on patient well-being also was presented from a sub-analysis of the Phase III RESONATE trial showing that IMBRUVICA was associated with improvements in hematologic function and disease burden in previously treated CLL/SLL patients versus those treated with ofatumumab. The data suggest the survival benefit
10
afforded by IMBRUVICA, combined with sustained improvements in key endpoints of the trial including patient-reported outcomes, may enhance patient quality of life while also prolonging survival. More patients taking IMBRUVICA experienced clinically meaningful improvement in EORTC QLQ-C30* global health scores than patients taking ofatumumab (47%, n=117 vs. 40%, n=87, respectively). At week 24, IMBRUVICA was associated with a greater improvement (≥ 3 points) in FACiT-F (FACiT fatigue; a measure of therapy-fatigue in patients with chronic illnesses1) than patients taking ofatumumab (56% vs. 43%). An IRC-assessment also observed a ≥50% reduction in lymph node size in 92% of the evaluable IMBRUVICA patients, as compared with a 14% reduction for patients in the ofatumumab-treatment arm.
Baseline disease-related symptoms were similar between the IMBRUVICA and ofatumumab groups; however, IMBRUVICA was associated with improvements over ofatumumab in weight loss (100% vs. 87%); fatigue (79% vs. 64%); night sweats (89% vs. 77%); abdominal pain/discomfort (96% vs. 75%); and, anorexia (100% vs. 64%). While rates of overall medical resource use was comparable for growth factors, IMBRUVICA patients experienced longer median treatment duration than ofatumumab patients (16 months vs. 5 months). Hospitalizations in the first 30 days occurred less frequently with IMBRUVICA than with ofatumumab (0.087 vs. 0.184 events/patient).
Single-Agent Ibrutinib Demonstrates Safety and Durability of Response at 2 Years Follow-up in Patients with Relapsed or Refractory Mantle Cell Lymphoma: Updated Results of an International, Multi-center, Open-Label Phase II Study (Poster Presentation - ASH Abstract # 4453)
In the Phase II, multi-center, single-arm, open-label trial (PCYC-1104) in patients with relapsed/refractory mantle cell lymphoma, patients received IMBRUVICA once daily until disease progression or unacceptable toxicity (n=111). Patients were allowed to continue treatment through a long-term extension trial. While the median treatment duration in the trial was 8.3 months, 46% of patients received treatment for more than one year and 20% continued on treatment in the extension trial for more than two years. At 24 months, approximately one-third of patients (31%) remained progression-free and almost half (47%) remained alive. The median overall survival was 22.5 months and the median progression-free survival was 13 months. Investigators observed a 67% overall response rate (ORR), which was the primary endpoint of the trial, and 23% of patients experienced a complete response. The median response time was less than two months (1.9 months) and the median duration of response was 17.5 months.
Data from the follow-up analysis were consistent with earlier results from the trial. With an estimated median follow up of 26.7 months, the most common Grade 3 or greater AEs in the trial were infection (28%), diarrhea (5%) and bleeding (6%). Serious adverse events (SAEs (≥ 2), regardless of attribution, included: disease progression (10%); pneumonia (7%); atrial fibrillation (6%); and, urinary tract infection (4%). SAEs occurred in 21% of patients and generally both grade ≥ 3 and SAE infections decreased over time.
Efficacy and Safety of Single-Agent Ibrutinib in Patients with Mantle Cell Lymphoma Who Progressed after Bortezomib Therapy (Poster Presentation - ASH Abstract # 4471)
Data was presented highlighting the results from a Phase II, multi-center, single-arm trial (MCL2001), which investigated once-daily IMBRUVICA in patients with relapsed/refractory MCL who previously had received a rituximab-containing treatment regimen and had progressed after at least two cycles of bortezomib (n=120). An Independent Review Committee (IRC) found the ORR, which was the primary endpoint of the trial, was 63% after a median follow-up of 14.9 months and 21% of patients achieved a complete response. Secondary endpoints included DOR, PFS, OS and safety. The median DOR based on IRC assessment was 14.9 months and the median time to first response was 2.1 months. The median PFS was 10.5 months, with 47% of patients remaining progression-free at one year. The median PFS has not yet been reached. The OS rate at 18 months was 61%.
The most frequently reported AEs of any grade were fatigue (43%) and diarrhea (43%). Diarrhea, when observed generally occurred early after initial treatment, but resolved quickly and was not treatment limiting. The majority of AEs were grade 1 and 2. The most common AEs ≥ grade 3 were neutropenia (21%), thrombocytopenia (13%) and pneumonia (13%). Atrial fibrillation was reported in 13 patients (11%); six patients (5%) experienced Grade 3 or 4 atrial fibrillation which resolved in 1 to 4 days. Five of these six patients had a history of atrial fibrillation.
Selected Abstracts from the ASCO 2014 Annual Meeting:
Randomized Comparison of Ibrutinib Versus Ofatumumab in Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: Results From the Phase III RESONATETM Trial (Oral Presentation - ASCO Abstract #LBA7008)
11
IMBRUVICA significantly prolonged progression-free survival in patients with relapsed/refractory (R/R) CLL in comparison to those randomized to receive ofatumumab. Patients treated with IMBRUVICA experienced a 78% reduction in risk of progression or death versus patients receiving ofatumumab (HR 0.215, 95% CI, 0.146 to 0.317, p<0.0001) per Independent Review Committee (IRC) evaluation. The median PFS for IMBRUVICA was not reached with a median time on study of 9.4 months. The median PFS for ofatumumab was 8.1 months. IMBRUVICA significantly prolonged overall survival compared with ofatumumab. IMBRUVICA reduced the risk of death by 57% (HR=0.434, 95% CI, 0.238 to 0.789; p<0.005) compared to the ofatumumab arm. This was observed despite a total of 57 patients who were initially randomized to ofatumumab crossing over to receive IMBRUVICA prior to the analysis. Patient characteristics were well balanced between arms: patients receiving IMBRUVICA had a median of 3 prior therapies with 32% del 17p, a mutation typically associated with poor prognosis, and 56% RAI stage III/IV (indicative of high risk CLL patients) versus patients receiving ofatumumab who had a median of 2 prior therapies with 33% del 17p and 58% RAI stage III/IV.
ORR was significantly higher in patients receiving IMBRUVICA by both investigator and IRC evaluations. Investigator assessed ORR (including complete responses (CR), partial responses (PR) and partial responses with lymphocytosis (PR+L)) was 85 % for IMBRUVICA and 23% for patients receiving ofatumumab, evaluated by the investigators based on the International Workshop on CLL (IWCLL) response criteria. The RESONATE study also had the IRC evaluate sequential CT scans (performed per protocol, approximately 12 weeks apart) to assess and confirm response. With this analysis, 63% of IMBRUVICA patients achieved a partial response (PR or a PR+L) compared to only 4% of patients receiving ofatumumab (p<0.0001). Significant benefit measured by PFS, OS and response rates were observed in the IMBRUVICA arm consistently across all subgroups by baseline disease risk factors, including those patients with del 17p or those whose disease was considered refractory to purine analogue therapy. Only 3% of the patients receiving IMBRUVICA experienced progressive disease as a best response versus 10% receiving ofatumumab as evaluated by the IRC.
The most commonly occurring adverse events (AE) independent of Grade (AEs in 20% or more of patients) were diarrhea (48% vs. 18%), fatigue (28% vs. 30%), pyrexia (fever; 24% vs. 15%), nausea (26% vs.18%), anemia (23% vs. 17%) and neutropenia (22% vs. 15%). Hematologic AEs that were Grade 3 or 4 in the RESONATE trial were neutropenia (decreased amount of white blood cells; 16% in the IMBRUVICA arm vs. 14% in the ofatumumab arm), thrombocytopenia (decrease in platelets in the blood; 6% vs. 4%), and anemia (5% vs. 8%). Atrial fibrillation of any Grade was noted more frequently in patients receiving IMBRUVICA (5% versus 0.5% with ofatumumab), these events were manageable and lead to discontinuation for only 1 patient. There was no difference in major bleeding between the IMBRUVICA or ofatumumab arms (reported in 2 patients randomized to IMBRUVICA and 3 patients receiving ofatumumab). The incidence of Grade 3 or higher infection was 24% for the IMBRUVICA arm and 22% for the ofatumumab arm. Richter’s Transformation (RT) occurred in 2 patients in the IMBRUVICA arm and in 2 patients in the ofatumumab arm.
With a median follow-up of 9.6 months, patients receiving IMBRUVICA showed a discontinuation rate due to progressive disease, adverse events or death of 13%, with 86% of patients continuing on therapy. Patients receiving ofatumumab showed a discontinuation rate due to progressive disease, adverse events or death of 28%.
Independent Evaluation of Ibrutinib Efficacy 3 Years Post-Initiation of Monotherapy in Patients With Chronic Lymphocytic Leukemia/Small Lymphocytic Leukemia Including Deletion 17p Disease (Oral Presentation - ASCO Abstract #7014)
In the treatment naïve group, 31 CLL patients, aged 65 years or older, received single agent IMBRUVICA once daily and were followed in the study for a median time of 32.1 months. The ORR, as assessed by the Investigator, was 87% (CR 13%, nodular PR (nPR) 3%, PR 65%, PR+L 6%). Only one patient had progressive disease during the median follow up of 32.1 months and the PFS at 30 months was 96%. The OS for these patients at 30 months was 97%.
In the R/R group, 101 patients with CLL/SLL (median of 4 prior therapies, 57% Rai stage III or IV (indicative of high risk CLL patients), 69% of patients had the high-risk characteristics of deletion of short arm of chromosome 17 or 11 (del 17p or del 11q)) received single-agent IMBRUVICA once daily and were followed in the study for a median time of 26.6 months. Overall 90% of patients achieved a best response of PR+L or higher. For patients without del 17p or del 11q, the estimated PFS was 89% at 30 months. The median PFS for del 17p patients was 28.1 months and for patients with del 11q the median PFS had not been reached and was estimated to be 74% at 30 months. The estimated OS for these R/R patients was 79.9%.
12
Following all the patients in this study for up to 3 years, the percentage of patients with AEs as a primary reason leading to discontinuation decreased over time, with 8% in the first year, 2% in the second year and 3% in the third year. Similarly, the percentage of patients who experienced a death on the study decreased over time, with 5% in the first year, 4% in the second year and 3% in the third year.
A Phase Ib/II study evaluating activity and tolerability of the BTK inhibitor ibrutinib in combination with ofatumumab in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and related diseases (Poster Presentation - ASCO Abstract #7009)
The study design consisted of three dosing cohorts, group 1 received one month of IMBRUVICA monotherapy followed by IMBRUVICA and ofatumumab in combination thereafter, group 2 received IMBRUVICA and ofatumumab in combination from the outset, and group 3 received two months of ofatumumab monotherapy followed by IMBRUVICA and ofatumumab thereafter. Seventy-one (71) patients with R/R CLL/SLL/Prolymphocytic Leukemia (PLL) and RT, with a median of 3 prior therapies, were enrolled in the study (27, 20 and 24 in groups 1, 2 and 3 respectively). Sixty-one percent (61%) of patients had RAI stage III/IV and were considered high risk, 44% had del 17p, and 31% had del11q, genetic mutations typically associated with poor prognosis.
The overall response (including CR, PR and PR+L) in CLL/SLL/PLL patients was 100%, 84%, and 75% in groups 1, 2 and 3 respectively, which were achieved within a median of 4.6 months during the study. At the study end, 90% (52 of 58) of responders were progression free with a follow up of 16, 12 and 11 months for groups 1, 2 and 3, respectively. At 12 months, the PFS was 89%, 85%, and 75% in groups 1, 2 and 3, respectively.
The AEs for all 3 cohorts combined were mostly Grade 1 or 2. The most frequent events reported in 15% or more of patients were diarrhea (68%, 5 cases of Grade ≥ 3), infusion related reaction (45%, 1 case of Grade ≥ 3), peripheral sensory neuropathy (42%, 2 cases of Grade ≥ 3), and stomatitis (37%, 2 cases of Grade ≥ 3). The Grade 3 or higher AE, reported in 15% or more of patients, was neutropenia (22.5%, 16 cases). Of all 71 patients, 6 patients had AEs leading to IMBRUVICA discontinuation.
IMBRUVICA Selected Clinical Trials
Chronic Lymphocytic Leukemia/ Small Lymphocytic Lymphoma (CLL/SLL)
• | RESONATE™ (PCYC-1112): Phase III study of IMBRUVICA versus ofatumumab in patients with relapsed/refractory (R/R) CLL/SLL was initiated in the first quarter of 2012. This was a randomized, multi-center, open-label Phase III trial of IMBRUVICA administered as monotherapy. This 391 patient study met its primary end point of PFS as well as a key secondary endpoint of OS at the pre-planned interim analysis in January 2014. This study confirmed IMBRUVICA’s clinical benefit in CLL patients who have received one prior therapy, resulting in regular (full) FDA approval for this patient population on July 28, 2014. |
• | RESONATE™-17 (PCYC-1117): Open-label, single-arm, Phase II study of IMBRUVICA as a single agent in patients with CLL who have deletion of chromosome 17p and who did not respond to or relapsed after at least one prior treatment (a high unmet need population) was initiated in the first quarter of 2013. The primary endpoint of the study is ORR. This study completed enrollment of 111 patients worldwide in the third quarter of 2013. Data was presented at the 56th ASH Annual Meeting on December 9, 2014. |
• | RESONATE™-2 (PCYC-1115): Phase III study of IMBRUVICA versus chlorambucil in newly diagnosed elderly CLL/SLL patients was initiated in the first quarter of 2013. This is a randomized, multi-center, open-label trial of IMBRUVICA as a monotherapy versus chlorambucil in patients 65 years or older with treatment naïve CLL/SLL. The study design was agreed upon with the FDA under a Special Protocol Assessment (SPA). The primary objective of the study is to demonstrate a clinically significant improvement in PFS when compared to chlorambucil. This study completed enrollment of 273 patients worldwide in the first quarter of 2014. |
• | ILLUMINATE (PCYC-1130): Phase III study of IMBRUVICA in combination with GAZYVA® versus chlorambucil in combination with GAZYVA in newly diagnosed CLL/SLL patients was initiated in the fourth quarter of 2014. This is a randomized, multi-center, open-label trial in patients 18 years or older with treatment naïve CLL/SLL. The primary objective of the study is to demonstrate a clinically significant improvement in PFS when compared to chlorambucil plus GAZYVA. The enrollment target of this study is 212 patients. |
13
• | HELIOS (CLL3001): Phase III study of IMBRUVICA in combination with bendamustine and rituximab in patients with R/R CLL/SLL was initiated in the third quarter of 2012. This is a randomized, multi-center, double-blinded, placebo-controlled trial of IMBRUVICA in combination with bendamustine and rituximab versus placebo in combination with bendamustine and rituximab (BR) in R/R CLL/SLL patients who have received at least one line of prior therapy. The primary objective of the study is to demonstrate a clinically significant improvement in PFS when compared to bendamustine and rituximab. This study completed enrollment of 578 patients worldwide in the first quarter of 2014. |
• | BRILLIANCE (CLL3002): Phase III study of IMBRUVICA versus rituximab in patients with R/R CLL/SLL was initiated in the fourth quarter of 2013. This is a randomized, open-label, multi-center study to evaluate the efficacy and safety of IMBRUVICA versus rituximab in adult Asia Pacific region patients with R/R CLL or SLL with active disease requiring treatment, who have failed at least one prior line of therapy and are not considered appropriate candidates for treatment or retreatment with purine analog-based therapy or combination chemoimmunotherapy. The primary objective of the study is to demonstrate a clinically significant improvement in PFS. The enrollment target of this study is 150 patients. |
• | Third-party sponsored: Phase III study of IMBRUVICA versus IMBRUVICA + rituximab versus bendamustine + rituximab in frontline newly diagnosed elderly (≥ 65 Years of Age) CLL/SLL patients (Alliance A041202) was initiated by the National Cancer Institute in the fourth quarter of 2013. This is a randomized, multi-center study designed to evaluate the improvement in PFS of IMBRUVICA with or without rituximab vs bendamustine and rituximab. Secondary outcome measures include OS and duration of response. The enrollment target of this multi-center study is 523 patients. |
• | Third-party sponsored: Phase III study in treatment-naive, young fit patients with CLL, comparing the combination of IMBRUVICA and Rituxan to chemoimmunotherapy of FCR (fludarabine, cyclophosphamide, and rituximab), (ECOG1912), was initiated by the Eastern Cooperative Oncology Group in the first quarter of 2014. This is a randomized study designed to evaluate the improvement in PFS of IMBRUVICA with rituximab vs FCR. Secondary outcome measures include OS and adverse events. The enrollment target of this multi-center study is 519 patients. |
• | Third-party sponsored: Phase III study in untreated, intermediate and high-risk patients with CLL, comparing monotherapy IMBRUVICA to placebo or no therapy, (CLL12), was initiated by the German Study Group in the first quarter of 2014. This is a randomized study designed to evaluate the improvement in event-free survival (EFS) of IMBRUVICA vs. watch and waiting. Secondary outcome measures include ORR and PFS. The enrollment target of this multi-center study is 302 patients. |
Mantle Cell Lymphoma (MCL)
• | RAY (MCL3001): Phase III study of IMBRUVICA versus temsirolimus in R/R MCL patients was initiated in the fourth quarter of 2012. This is a randomized, multi-center, open-label trial of IMBRUVICA as a monotherapy versus temsirolimus in R/R MCL patients who received at least one prior rituximab-containing chemotherapy regimen. The primary endpoint of the study is PFS. This ex-U.S. study completed enrollment of 280 patients in the fourth quarter of 2013. |
• | SHINE (MCL3002): Phase III study of IMBRUVICA in combination with BR in elderly patients with newly diagnosed MCL was initiated in the second quarter of 2013. This is a randomized, multi-center, double-blinded, placebo-controlled trial of IMBRUVICA plus BR versus placebo plus BR in patients 65 years or older with newly diagnosed MCL. The primary endpoint of the study is PFS. The enrollment target of this global study is 520 patients. |
Waldenström's Macroglobulinemia (WM)
• | INNOVATE (PCYC-1127): Phase III study of IMBRUVICA or placebo in combination with rituximab in patients with previously treated WM was initiated in the second quarter of 2014. This is a randomized, multi-center, double-blinded, placebo-controlled trial of IMBRUVICA. The primary outcome measure of this study is PFS. The secondary outcome measures include ORR, time to next treatment, OS and the number of participants with AEs as a measure of safety and tolerability within each treatment arm. The enrollment target of this study is 180 patients. |
Diffuse Large B-cell Lymphoma (DLBCL)
14
• | PHOENIX (DBL3001): Phase III study of IMBRUVICA in combination with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in patients with newly diagnosed non-GCB subtype of DLBCL was initiated in the third quarter of 2013. This is a randomized, multi-center, double-blinded, controlled trial of IMBRUVICA plus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) versus R-CHOP in patients with newly diagnosed non-GCB subtype DLBCL. The primary endpoint of the study is to demonstrate a clinically significant improvement in EFS when compared to R-CHOP. The enrollment target of this global study is 800 patients. |
• | PCYC-1123: Phase Ib/II randomized, multi-center, open-label, study of IMBRUVICA, in combination with lenalidomide with or without rituximab in relapsed or refractory patients with diffuse large b-cell lymphoma was initiated in the first quarter of 2014. The primary endpoint of the Phase IIb portion of this study is maximum tolerated dose of the investigational combination regimen and the primary endpoint of the Phase II portion is ORR. The enrollment target of this study is 130 patients. |
• | PCYC-1124: Phase Ib/II randomized, multi-center, open-label, study of IMBRUVICA, in combination with dose adjusted EPOCH-R in relapsed or refractory patients with diffuse large b-cell lymphoma was initiated in the second quarter of 2014. The primary endpoint of the Phase IIb portion of this study is maximum tolerated dose of the investigational combination regimen and the primary endpoint of the Phase II portion is ORR. The enrollment target of this study is 56 patients. |
Follicular Lymphoma (FL)
• | PCYC-1125: Phase II multi-center, open-label, study of IMBRUVICA, in combination with rituximab in previously untreated patients with follicular lymphoma was initiated in the fourth quarter of 2013. The primary endpoint of this study is ORR. The enrollment target of this study is 80 patients. |
• | DAWN (FLR2002): Phase II study of IMBRUVICA in patients with R/R FL was initiated in the second quarter of 2013. This is a multi-center, open-label, single-arm, global trial of IMBRUVICA in patients with chemoimmunotherapy-resistant FL, whose disease has relapsed from at least two prior lines of therapy, including at least one rituximab combination chemotherapy regimen. The primary endpoint of this study is ORR. This study completed enrollment of 111 patients worldwide in the second quarter of 2014. |
• | SELENE (FLR3001): Phase III study of IMBRUVICA in patients with R/R FL and marginal zone lymphoma was initiated in the first quarter of 2014. This is a randomized, multi-center, placebo-controlled trial in combination with either BR or R-CHOP in patients with previously treated indolent Non-Hodgkin Lymphoma (iNHL). The primary endpoint of this study is PFS. The enrollment target of this global study is 400 patients. |
Marginal Zone Lymphoma (MZL)
• | PCYC-1121: Phase II study of IMBRUVICA in patients with R/R marginal zone lymphoma was initiated in the fourth quarter of 2013. This is a multi-center, open-label, monotherapy study to evaluate the safety and efficacy of IMBRUVICA in patients with R/R marginal zone lymphoma. The primary endpoint of this study is ORR and the enrollment target of this study is 60 patients. |
Multiple Myeloma (MM)
• | PCYC-1111: Phase II study of IMBRUVICA in patients with R/R multiple myeloma was initiated in the first quarter of 2012. This is a Phase II, multi-center, open-label trial designed to assess the safety and efficacy of IMBRUVICA as a single agent and in combination with dexamethasone in patients with R/R MM. Data was presented at the 56th ASH Annual Meeting on December 6, 2014. The clinical development focus of this indication is with IMBRUVICA-based combination therapies. |
• | PCYC-1119: Phase I/IIb study of IMBRUVICA in combination with carfilzomib in patients with R/R MM was initiated in the third quarter of 2013. The Phase I portion of this study is a dose escalation study designed to assess the safety and recommended Phase IIb dose of IMBRUVICA and carfilzomib. The Phase IIb portion will be a randomized, double-blind, placebo-controlled study to evaluate the efficacy of IMBRUVICA and carfilzomib versus carfilzomib and placebo. The primary endpoint of the Phase IIb portion of the study is PFS. The enrollment target of this study is 176 patients. |
Graft versus Host Disease (GvHD)
15
• | PCYC-1129: Phase Ib/II study of IMBRUVICA in patients with steroid dependent or refractory GvHD was initiated in the third quarter of 2014. This is a multi-center, open-label trial designed to assess the safety and efficacy of IMBRUVICA as a single agent. The Phase I portion of the study identified 420 mg as the recommended Phase II dose. The Phase II portion of the study, which is currently enrolling, has a primary outcome measure of overall GvHD response rate. The enrollment target of this study is 39 patients. |
Acute Lymphoblastic Leukemia (ALL)
• | Third-party sponsored: Phase II study of IMBRUVICA in patients with relapsed or refractory ALL was initiated by the National Cancer Institute in the second quarter of 2014. This is an open-label trial designed to assess the efficacy of IMBRUVICA as a single agent. The primary endpoints of this study are ORR and OS. The enrollment target of this study is 20 patients. |
Acute Myeloid Leukemia (AML)
• | PCYC-1131: Phase II study of IMBRUVICA in patients with AML who have failed standard treatment, or subjects without prior therapy who refuse standard chemotherapy with or without low dose cytarabine (LD-AraC) was initiated in the first quarter of 2015. This is a multi-center, open-label trial designed to assess the safety and efficacy of IMBRUVICA as a single agent and in combination. The primary endpoints of this study are CR or incomplete blood count recovery (CRi) and safety with secondary endpoints being relapse-free survival, EFS, OS and clinical benefit rate. The enrollment target of this study is 67 patients. |
Collaborations and Other Agreements
Worldwide Collaboration and License Agreement with Janssen Biotech, Inc.
In December 2011, we entered into the worldwide collaboration and license agreement (the Agreement) with Janssen for the joint development and commercialization of IMBRUVICA, and certain compounds structurally related to IMBRUVICA, for oncology and other indications, excluding all immune- and inflammatory-mediated diseases or conditions and all psychiatric or psychological diseases or conditions, in the U.S. and outside the U.S.
The collaboration provides Janssen with an exclusive license to exploit the underlying technology outside of the U.S. (the License Territory) and co-exclusively with Pharmacyclics in the U.S. Both parties are responsible for the development, manufacturing and marketing of any products resulting from the Agreement. We continue to work with Janssen on protocols and the design, schedules and timing of trials.
The collaboration has no fixed duration or expiration date and provided for payments by Janssen to us of a $150.0 million non-refundable upfront payment upon execution, as well as potential future milestone payments of up to $825.0 million based upon continued development progress ($250.0 million), regulatory progress ($225.0 million) and approval of the product in both the U.S. and the License Territory ($350.0 million). As of December 31, 2014, we have earned $605.0 million in milestone payments under the Agreement, of which $230.0 million represented total approval milestones earned, $200.0 million represented total development milestones earned and $175.0 million represented total regulatory milestones earned. As of December 31, 2014, we may receive up to an additional $220.0 million in development, regulatory and approval milestone payments.
However, clinical development entails risks and we have no assurance as to whether or when the milestone targets might be achieved.
The Agreement includes a cost sharing arrangement for associated collaboration activities. Except in certain cases, in general Janssen is responsible for approximately 60% of collaboration development costs and we are responsible for the remaining 40% of collaboration development costs. Generally, costs associated with commercialization will be included in determining pre-tax commercial profits or pre-tax commercial losses, which are to be shared 50% by us and 50% by Janssen.
The collaboration with Janssen provides us with an annual cap of our share of IMBRUVICA related research and development and commercial-related expenses, offset by pre-tax commercial profits for each calendar year. In the event that our share of aggregate development costs in any given calendar year, together with any other amounts that become due from us, plus our share of pre-tax commercial losses for any calendar quarter in such calendar year, less our share of pre-tax commercial profits for any calendar quarter in such calendar year, exceeds $50.0 million, then amounts that are in excess of $50.0 million (the Excess Amounts) are funded by Janssen. Under the Agreement, the total Excess Amounts plus interest may not exceed $225.0 million at any given time. Interest shall be accrued on the outstanding balance with interest calculated at the average annual European Interbank Offered Rate (EURIBOR) for
16
the EURO or average annual London Interbank Offered Rate (LIBOR) for U.S. Dollars as reported in the Wall Street Journal, plus 2%, calculated on the number of days from the date on which our payment would be due to Janssen. The interest rate on outstanding Excess Amounts shall not exceed 5% per annum, and the cumulative interest on Excess Amounts shall not in the aggregate exceed $25.0 million.
In the event the Excess Amounts plus interest reaches a maximum of $225.0 million, we shall be responsible for our share of development costs, together with any other amounts that become due from us, plus our share of any pre-tax commercial loss beyond such maximum.
For all calendar quarters following the third profitable calendar quarter for the collaboration, as determined in the Agreement, we can no longer add to Excess Amounts and shall be responsible for our own share of development costs along with our share of pre-tax commercial losses incurred in such quarters. Janssen may only recoup the Excess Amounts, together with interest from our share of pre-tax commercial profits in calendar quarters after the third profitable calendar quarter for the collaboration from our share of pre-tax commercial profits, commencing in the fourth quarter of profitability under the Agreement until the Excess Amounts and applicable interest has been fully paid. As per the Agreement, a profitable quarter shall mean a full calendar quarter beginning after the first commercial sale of IMBRUVICA in which the combined pre-tax commercial profit or loss for the U.S. and the License Territory is positive and exceeds the development costs for such calendar quarter.
As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements for additional information about the Agreement).
We recognize Excess Amounts as a reduction to costs and expenses as our payment of Excess Amounts to Janssen is contingent and would become payable from our share of pre-tax commercial profits only after the third profitable calendar quarter for the collaboration.
The Agreement also provides that any net profits from the commercialization of products resulting from the collaboration will be shared 50% by Pharmacyclics and 50% by Janssen. Janssen has sole responsibility and exclusive rights to commercialize the products in the License Territory. The parties hold joint responsibility and co-exclusive rights to commercialize the products in the U.S., and Pharmacyclics is serving as the lead party in such effort. Janssen is commercializing IMBRUVICA outside the U.S.
Other Collaborations
Clinical Collaboration Agreements with AstraZeneca
In September and October 2014, we entered into three separate clinical collaboration agreements with AstraZeneca.
In one of the collaborations, which is a three-party arrangement with Janssen Research & Development LLC, we will explore the combination of IMBRUVICA and MEDI4736 as a treatment for patients with hematologic cancers including DLBCL and FL, which are investigational uses for both compounds. MEDI4736 blocks the signals that help tumors avoid detection by the immune system, countering the tumor's immune-evading tactics. IMBRUVICA blocks signals that tell malignant B cells (white blood cells that produce antibodies) to multiply and spread uncontrollably. Pre-clinical evidence suggests that the combination of these two agents may lead to an enhanced anti-tumor immune response. The Phase I part of the trial is expected to establish a recommended dose regimen for the combination of MEDI4736 and IMBRUVICA, and the Phase II part of the study will assess the safety and efficacy of the investigational combination. Under the terms of the agreement, the trial will be conducted by Pharmacyclics.
In a second collaboration, we will evaluate IMBRUVICA and MEDI4736 with AstraZeneca as a novel combination therapy targeting solid tumors. Pre-clinical evidence suggests that the combination of IMBRUVICA with this investigational medicine may enhance their effects. Under the terms of the agreement, we will collaborate with AstraZeneca on a non-exclusive basis and multiple studies may be considered and conducted. These studies will be led by us.
In the third collaboration, two different AstraZeneca investigational PI3 kinase pathway inhibitors in combination with IMBRUVICA for the treatment of patients with relapsed or refractory DLBCL will be investigated. Pre-clinical evidence suggests that the combination of IMBRUVICA with these investigational medicines may enhance their
17
effects. Under the terms of the agreement, we will collaborate with AstraZeneca on a non-exclusive basis and multiple studies may be considered and conducted. The studies will be led by AstraZeneca.
The Phase I element of each study is expected to establish a recommended safe and tolerable dose and schedule for the combination, and the Phase IIa element will assess its safety and efficacy in an expanded patient population.
Clinical Drug Supply Agreement with Roche
In July 2014, we entered into a master clinical drug supply agreement with Roche to evaluate the safety, tolerability and preliminary efficacy of IMBRUVICA in combination with GAZYVA® (obinutuzumab), a new CD20-directed antibody that attacks targeted cells both directly and together with the body's immune system, in patients with non-Hodgkin Lymphoma (NHL) and CLL/SLL. The agreement allows for multiple studies to be considered and conducted. We currently have a Phase III study, PCYC-1130, investigating the combination of IMBRUVICA and Gazyva versus Chlorambucil and Gazyva in previously untreated CLL/SLL patients. Plans to evaluate the combination for NHL currently are in development.
Clinical Drug Supply Agreement with Celgene
In March 2014, we entered into a clinical drug supply agreement with Celgene Corp. to explore the combination therapy potential of IMBRUVICA with REVLIMID® (lenalidomide) to address several histologies, including CLL/SLL and DLBCL. We currently have two phase II studies investigating the combination of IMBRUVICA and Revlimid. PCYC-1123 is studying the impact of adding Rituxan to the combination of IMBRUVICA and Revlimid in R/R DLBCL patients who are transplant ineligible. PCYC-1124 is studying the combination of IMBRUVICA and DA-EPOCH-R 2 in relapsed/refractory DLBCL patients who are transplant eligible.
Clinical Drug Supply Agreement with Onyx/Amgen
In July 2013, we entered into a master clinical drug supply agreement with Onyx Pharmaceuticals, now part of Amgen Inc., to evaluate the potential of IMBRUVICA with KRYPOLIS® (carfilzomib) as a potential combination therapy for patients with MCL and MM. We currently have a phase I/IIb study, PCYC-1119, investigating the combination of IMBRUVICA and Kyprolis in relapsed/refractory multiple myeloma patients.
Clinical Trial Collaboration Agreement with Bristol-Myers Squibb Company
In October 2014, we entered into a clinical trial collaboration agreement with Bristol-Myers Squibb Company and Janssen Research & Development, LLC to evaluate the safety, tolerability and preliminary efficacy of Bristol-Myers Squibb’s investigational PD-1 immune checkpoint inhibitor nivolumab in combination with IMBRUVICA. The initial Phase I/II study will focus on evaluating the safety and anti-tumor activity of combining nivolumab and IMBRUVICA as a potential treatment option for patients with NHL, including relapsed DLBCL, relapsed FL and relapsed CLL. Nivolumab is part of a class of cancer treatments known as immunotherapies, which are designed to harness the body’s own immune system in fighting cancer by targeting distinct regulatory components of the immune system. While each agent individually has shown activity against various malignancies in clinical trials; the combination of the two agents shows high levels of synergy in pre-clinical studies.
Collaboration and License Agreement with Servier
In September 2014, we concluded our 2009 collaboration and license agreement with Servier, entered into in April 2009, pertaining to the pan-HDAC inhibitor, abexinostat, an orally active, novel, small molecule inhibitor of pan-HDAC enzymes. Under the terms of that agreement, Servier had acquired the exclusive right to develop and commercialize the pan-HDAC inhibitor product worldwide except for the U.S. and its possessions. We continued to own all rights within the U.S. We and Servier terminated the collaboration and license agreement effective November 23, 2014. Upon the termination of the agreement, Servier’s rights to ex-U.S. development and commercialization of our pan-HDAC inhibitor compounds were returned to Pharmacyclics, giving us full global development and commercialization rights.
License Agreement with Novo Nordisk A/S
In October 2012, we entered into a license agreement with Novo Nordisk A/S (Novo Nordisk). Under the terms of the agreement, Novo Nordisk acquired the exclusive worldwide rights for our small molecule Factor VIIa inhibitor, PCI-27483, as an excipient in a product containing a Novo Nordisk active pharmaceutical ingredient for a restricted disease indication outside of oncology. Novo Nordisk will utilize PCI-27483 as an excipient in a product within Novo
18
Nordisk's biopharmaceutical unit. Novo Nordisk is solely responsible for all further research and development activities within the restricted disease indication outside of oncology.
In connection with entering into the license agreement with Novo Nordisk, we received an upfront payment of $5.0 million in October 2012. In addition, we may receive up to $55.0 million based on the achievement of certain development, regulatory and sales milestones. Upon commercialization, we will also receive low, single-digit, tiered royalties on Novo Nordisk's net sales of biopharmaceutical formulations utilizing the addition of PCI-27483.
On June 28, 2013, we entered into an amended and restated license agreement with Novo Nordisk to expand the scope of the license granted by us to Novo Nordisk in October 2012. Under the amended and restated license agreement with Novo Nordisk, we have no additional obligations related to the delivery of the license.
Cooperative Research and Development Agreement (CRADA) with the National Cancer Institute (NCI)
In August 2011, we entered into a five-year CRADA with the NCI to collaborate on the development of IMBRUVICA. Under the Agreement, the NCI's Division of Cancer Treatment and Diagnosis is sponsoring multiple trials of IMBRUVICA in various hematologic malignancies. In addition, we are participating in several other investigator-sponsored trials.
IMBRUVICA Regulatory Updates
In June 2013, a New Drug Application was submitted to the U.S. FDA for two R/R indications: MCL and CLL. IMBRUVICA was first approved as a single-agent treatment by the U.S. FDA on November 13, 2013, for its first indication for the treatment of patients with MCL who have received at least one prior therapy. This accelerated approval was based on ORR. An improvement in survival or disease-related symptoms has not been established. Per the limitations of an accelerated approval, continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials. FDA Breakthrough Therapy Designation was granted for this indication in February 2013.
The FDA granted accelerated approval for single-agent IMBRUVICA on February 12, 2014 for the treatment of patients with CLL who have received at least one prior therapy. This second indication also was based on ORR. In April 2014, Phase III data from the RESONATE™ trial in patients with CLL was submitted to the FDA in order to convert the accelerated approval into a regular (full) approval for this indication. Subsequently, on July 28, 2014, IMBRUVICA received regular (full) FDA approval for the treatment of patients with CLL who have received at least one prior therapy. This was the first full FDA approval for IMBRUVICA, and was granted only five and a half months after the original accelerated approval for this indication, which received FDA Breakthrough Designation in March 2013.
In addition, on that same day, IMBRUVICA was granted full approval for its third indication for the treatment of CLL patients with del 17p, including treatment naïve (frontline) and previously treated del 17p CLL patients. The approval of IMBRUVICA for the treatment of del 17p CLL patients made it the first approved treatment for this difficult-to-treat patient population.
In October 2014, a supplemental New Drug Application was submitted to the FDA based on data evaluating the use of IMBRUVICA in patients with WM, a rare type of B-cell lymphoma. On January 29, 2015, single-agent IMBRUVICA received regular (full) FDA approval as the first and only treatment for patients with WM, and it is approved in all lines of therapy. This is the fourth indication for IMBRUVICA, which received FDA Breakthrough Therapy Designation for this indication in February 2013.
The European Commission (EC) granted marketing authorization (approval) for IMBRUVICA throughout the 28 member states of the EU on October 21, 2014 for the treatment of patients with R/R MCL, patients with CLL who have received at least one prior therapy, or in first line in the presence of 17p deletion or TP53 mutation in patients unsuitable for chemoimmunotherapy. Subsequently, the European Medicines Agency (EMA) accepted a Type II variation application for IMBRUVICA for a potential label expansion for IMBRUVICA in the EU for the treatment for patients with WM. If approved, IMBRUVICA would be the first medicinal product authorized to treat WM in all 28 member states.
IMBRUVICA Regulatory Designations: Breakthrough Therapy, Fast Track and Orphan Drug
The U.S. FDA granted Orphan Drug Designation to IMBRUVICA for the following orphan disease indications: CLL on April 6, 2012; MCL on December 3, 2012; MM on May 16, 2013; SLL on May 30, 2013; WM on October 15, 2013; DLBCL on October 23, 2013; FL on September 8, 2014; splenic MZL on February 5, 2015; and nodal MZL on
19
February 5, 2015. Orphan Drug Designation provides the company with several benefits and incentives, including a seven-year period of orphan drug exclusivity following the marketing approval during which time the FDA will not grant marketing approval to another drug of the same type for the same indication if the orphan drug is the first of its type approved for the specified indication.
The EC also has designated IMBRUVICA as an orphan medicinal product for the following indications: CLL/SLL on April 26, 2012; MCL on March 12, 2013; DLBCL on November 13, 2013; FL on January 16, 2014; and lymphoplasmacytic lymphoma/WM on April 29, 2014. An EU Orphan Drug Designation provides the drug developer with similar benefits and incentives related to the orphan drug as offered by the U.S. FDA, most importantly including a ten-year period of market exclusivity during which time the EMA will not grant marketing authorization to another drug of the same type for the same indication if the orphan drug is the first of its type approved for the specified indication.
The FDA also granted IMBRUVICA with Fast Track Designation for the following indications: CLL/SLL on October 29, 2012; MCL on December 18, 2012; FL on August 7, 2014, and; DLBCL on October 24, 2014. Fast Track Designation is a process designed to facilitate the development and expedite the review of drugs to treat serious and life-threatening conditions and address unmet medical needs for the condition.
As part of the 2012 FDA Safety and Innovation Act, the designation of a drug as a Breakthrough Therapy was enacted to expedite the development and review of a potential new drug for serious or life-threatening diseases whereby “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.”
IMBRUVICA became one of the first drugs to receive Breakthrough Therapy Designation and is the only drug to have received three separate Breakthrough Therapy Designations. The FDA granted Breakthrough Therapy Designation to IMBRUVICA as a monotherapy for the treatment of patients with relapsed or refractory MCL and as a monotherapy for the treatment of patients with WM on February 8, 2013, and granted Breakthrough Therapy Designation to IMBRUVICA as a monotherapy for the treatment of CLL/SLL patients with del 17p on March 18, 2013.
FACTOR VIIa INHIBITOR PROGRAM
Tissue Factor (TF) up-regulation is associated with increased tumor invasiveness and progression, worsened prognosis and increased thromboembolism (VTE). Factor VII is an enzyme that becomes activated (FVIIa) by binding to the cell surface protein tissue factor (TF), a protein found in the body that helps to trigger the process of blood clotting in response to injury. TF is over expressed in many cancers including gastric, breast, colon, lung, prostate, ovarian and pancreatic cancers. Activation of protease activated receptors by TF:FVIIa complex leads to increases in IL-8, VEGF and other invasiveness promoting factors. Our Factor VIIa inhibitor PCI-27483 is a novel first-in-human small molecule inhibitor that selectively targets FVIIa. As an inhibitor of FVIIa, PCI-27483 has two potential mechanisms of action: 1) inhibition of intracellular signaling involved in tumor growth and metastases and 2) inhibition of early coagulation processes associated with thromboembolism. Novo Nordisk is currently developing PCI-27483 for use as an excipient. This clinical development program for PCI-27483 is being evaluated at this time and there are no active clinical trials underway.
PCI-27483, claimed as a composition of matter, is covered by granted patents in the U.S., Canada, Japan, China and India. We have pending patent applications covering PCI-24783, as compositions of matter, in the U.S., and Europe. As of December 31, 2014, the duration of granted patents in Canada, Japan, China and India that claim PCI-27483 as compositions of matter is through December 2023 and in the U.S. through June 2025, subject to any patent term extensions that may be obtained in certain territories. The projected duration of any patent that may grant on any of the pending patent applications covering PCI-24783 as compositions of matter in Europe is through December 2023 and in the U.S. is through June 2025, subject to any patent term extensions that may be obtained in those territories.
ABEXINOSTAT (PCI-24781) PAN-HDAC INHIBITOR
Histone deacetylases (HDACs) are well-validated drug targets in a number of disease areas including cancer. These enzymes control several vital cellular processes, such as transcription, cell cycle progression, protein transport and degradation etc, and their activity is often dysregulated in cancer. Classically, the major function of these enzymes is controlling the expression of genes, i.e. whether genes are turned "on" or "off" via epigenetic mechanisms. In cancer, HDACs are often differentially expressed from normal cells, resulting in gene expression changes that favor a
20
tumor's ability to multiply, to avoid apoptosis (i.e. programmed cell death) or to become resistant to chemotherapy. Treatment with HDAC inhibitors reverses these changes, resulting in cancer cell death in vitro (i.e. in cultured cells) and tumor growth inhibition in vivo (i.e. in animals) at non-toxic concentrations.
Abexinostat is an orally dosed, broad spectrum, hydroxamic acid-based small molecule HDAC inhibitor that has been evaluated in Phase I and II clinical trials for refractory solid tumors and lymphoma. Abexinostat has shown promising anti-tumor activity in vitro and in vivo (Buggy et al, Mol Cancer Ther 2006; 5: 1309-17).
Abexinostat treatment leads to synergistic efficacy in tumor cells in combination with many cancer therapeutics, such as bortezomib, as well as DNA-damaging agents such as radiation (Banuelos et al Clin Cancer Res 2007 13:6816-26) and chemotherapy agents such as doxorubicin (Lopez et al, Clin Cancer Res 2009 15:3472-83, Yang et al, Anticancer Res. 2011 31:1115-23). In lymphoma cells, abexinostat together with bortezomib greatly enhances proteasome and NF-κB inhibition, increases oxidative stress, causes cell cycle arrest and results in increased cell death (Bhalla et al Clin Cancer Res 2009 15:3354-65). In solid tumor cells, we have shown that abexinostat inhibits DNA repair following damage by radiation or chemotheraputic agents, thereby enhancing the efficacy of these anti-cancer agents. The mechanism of the synergy may involve inhibition of the homologous recombination pathway, a major double-strand break (DSB) repair pathway. In a study funded entirely by us, we showed that abexinostat also effectively synergizes with inhibitors of single-strand break repair such as poly ADP ribose polymerase inhibitors (PARP is a protein important for repairing single-strand breaks in DNA) (Adimoolam et al 2007). Furthermore, abexinostat demonstrated highly synergistic growth inhibition of chemotherapy-resistant tumors in combination with chloroquine (an inhibitor of autophagy, a protective mechanism in cells under stress), particularly in certain subtypes of sarcoma (Lopez et al. Cancer Res 2010 71:185-96). In a model of epigenetic regulation of cancers, abexinostat showed synergistic cell death in glioblastoma cells when combined with a demethylase inhibitor (Singh et al 2011 13:894-903). Other recent pre-clinical publications have also demonstrated activity of abexinostat in a mouse model of gallbladder carcinoma (Kitamura et al J Hepatology 2012 57:84-91) and in combination with radiation on breast cancer stem cells (Al-Assar et al Cancer Biol & Ther 11: 1028-36, 2011).
Abexinostat has been tested in several clinical trials in the U.S. by us and internationally by Servier. In the U.S., we have completed two Phase I studies using abexinostat as a single agent in patients with advanced solid tumors, a Phase I/II trial testing abexinostat single agent in patients with relapsed or refractory NHL and a Phase I trial in soft-tissue sarcoma patients (in combination with doxorubicin, an anti-tumor agent) co-sponsored by the Massachusetts General Hospital and Dana-Farber Cancer Institute. Results from this trial were presented at the annual meeting of the Connective Tissue Oncology Society in November 2012 in Prague, Czech Republic and updated at the AACR Annual Meeting in April 2013. A Phase II study was undertaken in R/R FL and MCL. The results from this study were presented in poster at the 12th International Conference on Malignant Lymphoma (IMCL) in Lugano, Switzerland (June 19-22, 2013). Servier developed a new formulation of our HDAC inhibitor PCI-24781 covered by a U.S. patent application we own, and we are actively considering additional clinical development opportunities. We are also continuing to investigate combination strategies for PCI-24781 through nonclinical studies.
Abexinostat and pan-HDAC inhibitors, claimed as compositions of matter, are covered by granted patents in the U.S., Australia, Europe, Canada, Mexico, Japan, Korea, New Zealand, Singapore, South Africa, Taiwan, Ukraine, and India. We have pending applications covering abexinostat and pan-HDAC inhibitors, as compositions of matter, in China, Israel, Brazil, and Norway. As of December 31, 2014, the duration of the granted patents covering the composition of matter in Australia, Europe, Canada, Mexico, Japan, Korea, New Zealand, Singapore, South Africa, Taiwan, Ukraine, and India is through 2024, and in the U.S. through 2025 due to a Patent Term Adjustment. The projected duration of any patent that may grant on any of the pending patent applications in China, Israel, Brazil and Norway is through 2024. In addition, the projected duration of any patent that may grant on the U.S. patent application, or any foreign counterpart patent application, that we own covering the new formulation of PCI-24781 developed by Servier is through 2034. The durations of the granted patents and the projected duration of any patent that may be granted on any of the pending patent applications are subject to any patent term extensions that may be obtained in certain territories.
Acquired Products
Celera Corporation
In April 2006, we acquired multiple small molecule drug candidates for the treatment of cancer and other diseases from Celera Genomics, an Applera Corporation business (now Celera Corporation - a subsidiary of Quest Diagnostics Incorporated). Future milestone payments under the agreement, as amended, could total as much as approximately $97.0 million, although we currently cannot predict if or when any of the milestones will be achieved. Approximately
21
two-thirds of the milestone payments relate to our HDAC inhibitor program and approximately one-third relates to our Factor VIIa inhibitor program. Approximately 90% of the potential future milestone payments would be paid to Celera after obtaining regulatory approval in various countries. To date, there has been no milestone payments triggered related to our HDAC inhibitor or Factor VIIa programs.
In addition to the milestone payments, we are required to make single-digit royalty payments based on annual sales of IMBRUVICA and would be required to make single-digit royalty payments based on annual sales of drugs commercialized from our HDAC inhibitor, Factor VIIa inhibitor and certain other BTK inhibitor programs. We commercially launched IMBRUVICA for MCL on November 13, 2013. For the year ended December 31, 2014 and year ended December 31, 2013, we recognized royalty expense under the Celera agreement of approximately $27.4 million and $1.0 million, respectively, on net product sales of IMBRUVICA. Royalty expense related to net product sales of IMBRUVICA is included within cost of goods sold in the consolidated statement of operations. No royalty expense was incurred under the Celera agreement prior to the year ended December 31, 2013 as there were no product sales.
For any BTK inhibitor product or Factor VIIa inhibitor product obtained from Celera, the agreement with Celera expires on a product-by-product and country-by-country basis. The term of the agreement for a given BTK inhibitor product shall expire in a given country upon the expiration of the last-to-expire Celera patent assigned to us that covers the manufacture, use, sale, offer for sale, or importation of such product in such country. For any HDAC inhibitor product obtained from Celera, the agreement with Celera expires on a product-by-product and country-by-country basis. The term of the agreement for a given HDAC inhibitor product shall expire in a given country upon the expiration of the last-to-expire Celera patent assigned to us that covers the sale of such product in such country.
We may terminate the agreement with Celera in its entirety, or with respect to one or more of the three classes of products (BTK inhibitor products, HDAC inhibitor products and Factor VIIa inhibitor products) obtained from Celera, at any time by giving Celera at least 60 days' prior written notice. If we terminate the agreement with respect to a particular class of products, ownership of the Celera intellectual property assigned to us relating to the products in the terminated product class will revert to Celera. If we terminate the agreement in its entirety, ownership of all of the Celera intellectual property assigned to us will revert to Celera.
The agreement with Celera may be terminated effective immediately upon a party's written notice to the other party for a breach by the other party that remains uncured for 90 days after notice of the breach is given to the breaching party. If we breach the agreement only with respect to one or two of the three classes of products obtained from Celera, but not with respect to all three classes of products, and if our breach remains uncured for 90 days after we have received notice of breach from Celera, Celera may terminate the agreement solely with respect to the class or classes of products affected by our breach, but may not terminate the agreement with respect to the class or classes of products unaffected by our breach.
Patents and Proprietary Technology
We believe our success depends in part on our ability to protect and defend our proprietary technology and product candidates through patents and trade secret protection. We, therefore, aggressively pursue and prosecute, patent applications and protect our trade secrets. While we intend to defend against threats to our intellectual property, these patents and trade secrets may not adequately protect our intellectual property.
We have a number of patents and patent applications related to our compounds, but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will issue as patents. The evaluation of the patentability of U.S. and foreign patent applications can take years to complete and can involve considerable expense. Even if patents are issued and maintained, these patents may not be of adequate scope to benefit us, or may be held invalid and unenforceable against third parties. Third parties could obtain patents that may require us to negotiate licenses to conduct our business, and the required licenses may not be available on reasonable terms or at all.
Our patents, patent applications, and licensed patent rights cover various compounds, pharmaceutical formulations and methods of use. As of December 31, 2014, we own or hold licenses to:
•77 issued U.S. patents; and
•81 other pending U.S. patent applications, including 8 allowed U.S. patent applications.
These issued U.S. patents expire at various times depending on product programs (see above program sections for certain specific compounds). In addition, we own or hold licenses to approximately 243 issued foreign patents, 21
22
Patent Cooperation Treaty (PCT) patent applications, and more than 350 pending non-U.S. patent applications filed with the European Patent Office, and nationally in Canada, Japan, China, India, Australia and other non-U.S. international territories.
Some of these issued patents would be subject to potential patent term extensions in the U.S. and certain non-U.S. territories.
Furthermore, we own patents which claim the IMBRUVICA compound and related BTK inhibitor compounds as compositions of matter in the U.S., Europe, Japan, China, Canada, Mexico, South Korea, Singapore, Hong Kong, South Africa, Russia, New Zealand, India, and Australia. As of December 31, 2014, the duration of the granted patents covering the IMBRUVICA compound and related BTK inhibitor compounds as compositions of matter in the countries noted above is through December 2026, subject to any patent term extensions that may be obtained in certain territories.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We depend on confidentiality and assignment-of-invention agreements with certain of our employees, consultants and other parties, to protect in part trade secrets and other proprietary rights. We cannot be certain that these agreements will not be breached, that we will have adequate remedies for any breach, that others will not independently develop substantially equivalent proprietary information, or that third parties will not otherwise gain access to our trade secrets or proprietary information.
Manufacturing
Overview
Generally the raw materials required for the production of our product are available from several suppliers, yet in some cases the raw materials are only available through one supplier. We have adopted policies to attempt, to the extent feasible, to minimize our raw material supply risks, including maintenance of greater levels of raw materials inventory and implementation of multiple raw materials sourcing strategies, especially for critical raw materials. Although to date we have not experienced any significant delays in obtaining any raw materials from our suppliers, we cannot provide assurance that we will not face shortages from one or more of them in the future.
We contract with third parties to manufacture the raw materials and the active pharmaceutical ingredient (API) of IMBRUVICA for clinical and commercial uses. We currently do not have internal manufacturing facilities for clinical or commercial production of IMBRUVICA. In addition, we expect in the foreseeable future to continue to rely on third parties for the manufacture of the raw materials and API for IMBRUVICA.
Raw materials required for the production of the API are generally sourced from several third-party suppliers. Contract manufacturers in Asia convert these raw materials into API for clinical and commercial purposes. We use a third-party facility in North America to manufacture drug product. A dedicated facility was constructed at this site to provide drug product manufacturing. Another third-party is used to package and label the finished product for commercial purposes. We use a third-party logistics provider to handle shipping and warehousing of our commercial supply of IMBRUVICA and a range of specialty pharmacies and distributors to dispense IMBRUVICA to patients in fulfillment of prescriptions.
Our third-party manufacturers are independent entities, under contract with us, who are subject to their own unique operational and financial risks which are out of our control. If we or any of our third-party manufacturers fail to perform as required, this could impair our ability to deliver IMBRUVICA on a timely basis or cause delays in supplies for our clinical trials and commercial activities. To the extent these risks materialize, our financial results may be adversely affected.
The processes used to manufacture our products are proprietary. For products manufactured by our third-party contract manufacturers, we have licensed the necessary knowhow of these processes which are proprietary to us to enable them to manufacture the products for us. We have agreements with these third-party manufacturers to restrict these manufacturers from using or revealing our processes, but we cannot be certain that these third-party manufacturers will comply with these restrictions.
To establish a new third-party manufacturer to produce our products may take several years. While there are third parties potentially capable of providing most of the materials and services we need to manufacture and distribute IMBRUVICA, it is very expensive, time consuming, and we may be subject to other implementation execution risks should we be required to replace any member of our existing supply chain. Should any third-party manufacturer in
23
our supply chain fail to perform for any reason, it may have significant consequences on our ability to deliver the commercial and clinical supplies of IMBRUVICA.
We believe that our current arrangement for the supply of starting materials, intermediates and active ingredients, will be adequate to satisfy our currently forecasted commercial requirements of IMBRUVICA and our current and planned clinical studies. Due to the significant lead times involved in our supply chain for IMBRUVICA, we have less flexibility to adjust our supply in response to changes in demand than if we had shorter lead times.
Manufacturing Agreements
Agreement with Lonza Sales Ltd. (Lonza)
On February 5, 2013, we entered into a Commercial Manufacturing Agreement (the Lonza Agreement) with Lonza dated as of February 5, 2013 (the Lonza Effective Date), pursuant to which Lonza will manufacture for us a human pharmaceutical oncology agent (the Lonza Product), as identified in the Lonza Agreement, for the sale and distribution by Pharmacyclics. Since then, Lonza and us have entered into additional amendments to the Lonza Agreement (the Lonza Amendment), which amend certain pricing, delivery terms and other matters originally included in the Lonza Agreement.
Under the Lonza Agreement, Lonza has agreed to supply the Lonza Product and we have agreed to purchase Lonza Product, subject to Lonza’s continued material compliance with the terms of the Lonza Agreement. We are obligated to purchase a minimum amount of Lonza Product, to be calculated based on a percentage of our global requirements of Lonza Product in each calendar year. In the event that the minimum amount of the Lonza Product is not purchased by us, Lonza may invoice us for the amount equal to the shortfall between the minimum purchase requirement and the number of units of Lonza Product actually ordered during such year. The Lonza Agreement commenced on the Lonza Effective Date and will continue for a certain specified number of years (the Lonza Agreement Term), unless terminated earlier as provided in the Lonza Agreement. Upon expiration of the Lonza Agreement Term or any extension thereof, the Lonza Agreement will automatically extend for an additional set period, as set forth in the Lonza Agreement.
Agreement with Catalent CTS, LLC (Catalent)
On May 1, 2013, we entered into a Build-Out and Commercial Supply Agreement (the Catalent Agreement) with Catalent dated as of May 1, 2013 (the Catalent Effective Date), pursuant to which Catalent will manufacture and supply us fully formulated bulk pharmaceutical product (the Catalent Product).
Under the Catalent Agreement, we will purchase from Catalent the Catalent Product during the term of the Catalent Agreement for commercial use in certain specified territories that we and Catalent mutually agree to in writing (the Territory). The Catalent Agreement commenced on the Catalent Effective Date and will continue until the earlier of the end of a specified anniversary of the completion of the building improvements in accordance with the terms of the Catalent Agreement (the Completion Date) or July 1, 2014, whichever is earlier unless earlier terminated in accordance with the Catalent Agreement. A contract year means each consecutive 12 month period beginning on the earlier of the Completion Date or July 1, 2014, and each anniversary thereof, as applicable. The term of the Catalent Agreement will be automatically extended for successive five year periods, with a total maximum extension of a specified number of years, as indicated in the Catalent Agreement.
We will be obligated to purchase from Catalent a certain minimum amount with respect to the Catalent Product. This amount will be determined in accordance with the requirements of Catalent Product for countries in the Territory in which Pharmacyclics has the right to sell, have sold, or grant a license to sell the Catalent Product. We will pay Catalent a non-refundable annual facility fee, which includes the cost of a designated number of batches of Catalent Product to be produced. Any additional batches of Catalent Product ordered in a contract year will be paid to Catalent in accordance with the terms of the Catalent Agreement.
Research and Development
The majority of our costs and expenses to date have been related to research and development (R&D). Our R&D expenses consist of independent R&D costs and costs associated with collaborative R&D.
We are pursuing multiple indications as part of our clinical development plan for IMBRUVICA, which include the following B-cell malignancies: CLL, SLL, MCL, FL, DLBCL, MM, WM, MZL, ALL, AML and other cancer types. We are also pursuing the development of IMBRUVICA for GvHD.
24
Under our worldwide collaboration and license agreement with Janssen, we are jointly developing IMBRUVICA for oncology and other indications, excluding all immune- and inflammatory-mediated diseases or conditions and all psychiatric or psychological diseases or conditions, in the U.S. and outside the U.S. In addition, we recently have formed a number of strategic collaborations with other world-class companies including Amgen Inc., AstraZeneca, Bristol-Myers Squibb Co., Celgene Corp., and Roche in order to explore the potential of IMBRUVICA as a combination agent and a backbone of therapy for certain blood cancers.
R&D expenses were $172.5 million for the year ended December 31, 2014 (net of Janssen's share of costs under the cost sharing arrangement of $40.1 million). R&D expenses were $85.1 million for the year ended December 31, 2013 (net of Janssen's share of costs under the cost sharing arrangement of $48.2 million and Excess Amounts of $85.7 million which were recorded as reductions to R&D expense). R&D expenses were $29.3 million for the six months ended December 31, 2012 (net of Janssen's share of costs under the cost sharing arrangement of $17.3 million and Excess Amounts of $17.3 million which were recorded as reductions to R&D expense). R&D expenses were $54.5 million for the year ended June 30, 2012 (net of Janssen's share of costs under the cost sharing arrangement of $18.4 million). For further information regarding the terms and conditions underlying our cost sharing arrangement with Janssen, see "Collaboration and License Agreement with Janssen Biotech, Inc." above.
Government Regulation and Product Approval
The FDA and applicable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our product candidates. Failure to comply with FDA requirements, both before and after product approval, may subject us to administrative or judicial sanctions, including but not limited to, FDA refusal to approve pending applications, warning letters, product recalls, product seizures, or total or partial suspension of production or distribution, fines, injunctions, or civil or criminal penalties.
The process required by the FDA before our products may be marketed or receive a label extension in the U.S. generally involves the following:
• | completion of pre-clinical laboratory and animal tests; |
• | submission of an Investigational New Drug (IND) application, which must become effective before clinical trials may begin; |
• | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy for each intended use; |
• | submission to the FDA of a New Drug Application (NDA) or Supplemental NDA; and |
• | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the product candidate is made to assess compliance with the FDA’s current good manufacturing practice (cGMP) regulations. |
The testing and approval process requires substantial time, effort, and financial resources; and we cannot be certain that any approval will be granted on a timely basis, if at all.
Pre-clinical tests include laboratory evaluation of the product, its chemistry, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product. We then submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we may begin human clinical trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the trials as outlined in the IND, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. Our submission of an IND may not result in FDA authorization to commence clinical trials. Further, an independent Institutional Review Board/Ethics Committee responsible for the medical center proposing to conduct the clinical trials must review and approve any clinical study.
Human clinical trials are typically conducted in three sequential phases which may overlap:
25
• | Phase I: The drug is initially introduced into healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. |
• | Phase II: Involves studies in a limited patient population to identify possible adverse effects and safety risks, to evaluate preliminarily the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage/schedule. |
• | Phase III: When Phase II evaluations demonstrate that a dosage range of the product may be effective and has an acceptable safety profile, Phase lll trials are undertaken to evaluate clinical efficacy and safety in the patient population intended for registration at geographically dispersed clinical study sites. |
In the case of products for severe or life-threatening diseases such as cancer, the initial human testing is often conducted in patients rather than in healthy volunteers. Since these patients already have the target disease, these studies may provide initial evidence of efficacy traditionally obtained in Phase II trials and thus these trials are frequently referred to as Phase I/II trials. We cannot be certain that we will successfully complete Phase I, Phase II or Phase III testing of our product candidates within any specific time period, if at all. Furthermore, the FDA/applicable regulatory agencies, the relevant Institutional Review Board/Ethics Committee or the sponsor may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of product development, preclinical studies and clinical studies are submitted to the FDA as part of a NDA, for approval of the product allowing commercial shipment and marketing of the product. The FDA may not accept the NDA for review if the applicable regulatory criteria are not satisfied or may require additional data. Even if such data are accepted for filing, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. In addition, before approving an NDA, the FDA will inspect the facilities at which the product is manufactured to ensure compliance with all cGMP regulations. Once issued, the FDA/applicable regulatory agencies may withdraw product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. In addition, the FDA/applicable regulatory agency may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the agency has the authority to prevent or limit further marketing of a product based on the results of these post-marketing studies or evaluations.
Satisfaction of the above FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially, based upon the type, complexity and novelty of the pharmaceutical product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon our activities. We cannot be certain that the FDA or any other regulatory agency will grant approval for any of our products under development on a timely basis, if at all. Success in preclinical or early stage clinical trials does not assure success in later stage clinical trials. Data obtained from pre-clinical and clinical activities is not always conclusive and may be susceptible to varying interpretations which could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business. Marketing our products abroad will require similar regulatory approvals and is subject to similar risks. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA (announced or unannounced) and certain state agencies, and are subject to periodic inspections by the FDA and certain state agencies for compliance with Good Manufacturing Practice regulations, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the current Good Manufacturing Practice, or cGMP, regulations and other FDA regulatory requirements.
The FDA regulates drug labeling and promotion activities. The FDA has actively enforced regulations prohibiting the marketing of products for unapproved uses. Pharmacyclics, our partners, and our products are also subject to a variety of federal, state and foreign laws and regulations in those states or localities where our products are or will be marketed. Any applicable state or local regulations may hinder our ability to market our products in those states or
26
localities. We are also subject to numerous federal, state, local and foreign laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
The FDA's policies may change and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. Moreover, increased attention to the containment of health care costs in the U.S. and in foreign markets could result in new government regulations that could have a material adverse effect on our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
Employees
As of December 31, 2014, we had 607 employees, all of whom were full-time employees. 331 of our employees are engaged in research, development, preclinical and clinical testing, manufacturing, quality assurance and quality control and regulatory affairs and 276 are in sales and marketing, medical affairs, legal and compliance, human resources, procurement, finance and administration. Sixty-one of our employees have an M.D. or Ph.D. Our future performance depends in significant part upon the continued service of our key scientific, technical and senior management personnel, none of whom is bound by an employment agreement requiring service for any defined period of time. None of our employees are represented by a labor union.
Change in Fiscal Year End
On November 14, 2012, the Board of Directors approved a change in the fiscal year end from June 30 to December 31, effective December 31, 2012. All references to "fiscal years", unless otherwise noted, refer to the twelve-month fiscal year, which prior to July 1, 2012, ended on June 30, and beginning on January 1, 2013, ends on December 31, of each year.
Available Information
We were incorporated in Delaware in 1991 and commenced operations in 1992.
We file electronically with the Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly interim reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We maintain a site on the worldwide web at www.pharmacyclics.com; however, information found on our website is not incorporated by reference into this report. We make our SEC filings available free of charge on or through our website, including our annual report on Form 10-K, quarterly interim reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act of 1934 as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Further, a copy of this annual report on Form 10-K is located at the Securities and Exchange Commission’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the Securities and Exchange Commission at 1-800-SEC-0330. The Securities and Exchange Commission maintains a website that contains reports, proxy and information statements and other information regarding our filings at www.sec.gov.
On May 21, 2013, we adopted our current Code of Business Conduct and Ethics (the Code) that applies to our officers, directors and employees, including our principal executive officer, principal financial officer and principal accounting officer. We have posted the text of the Code on our website at www.pharmacyclics.com in connection with “Investor” materials. In addition, we intend to promptly disclose (1) the nature of any amendment to the Code that applies to our principal executive officer, principal financial officer, principal accounting officer, or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of the Code that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
Item 1A. Risk Factors
An investment in our securities involves a high degree of risk. Anyone who is making an investment decision regarding our securities should carefully consider the following risk factors, as well as the other information contained or incorporated by reference in this report. The risks and uncertainties described below are those that we currently believe may materially affect our company or your investment. Other risks and uncertainties that we do not presently consider to be material, or of which we
27
are not presently aware, may become important factors that adversely affect our security holders or us in the future. If any of the risks discussed below actually materialize, then our business, financial condition, operating results, cash flows and future prospects, or your investment in our securities, could be materially and adversely affected, resulting in a loss of all or part of your investment.
Risks Relating to Pharmacyclics
IMBRUVICA® (ibrutinib) is our only approved drug.
Our prospects are largely dependent on (a) successful commercialization of IMBRUVICA in the U.S. and outside the U.S. to treat the approved patient populations, and (b) obtaining regulatory approval of, and successfully commercializing, IMBRUVICA both in and outside the U.S. to treat other indications. If we are unsuccessful in achieving any one or more of these critical business objectives, our ability to generate significant revenue or achieve profitability will be adversely affected and our business may fail.
The commercialization of IMBRUVICA for the treatment of current patient populations or any patient populations for which IMBRUVICA may be subsequently approved may not be successful for a number of reasons, including:
• | we and our collaborative partner, Janssen, may face unexpected challenges from competitors with potential new therapeutic options and also in overcoming inertia in the adoption of upcoming novel therapies such as IMBRUVICA; |
• | new safety issues or concerns with respect to IMBRUVICA being reported that may impact or narrow the approved indications; |
• | our level of experience in marketing IMBRUVICA is limited to the time during which IMBRUVICA has been commercially available for any patient population; |
• | reimbursement and coverage policies of government and private payers such as Medicare, Medicaid, insurance companies, health maintenance organizations and other plan administrators could change unexpectedly; |
• | government price controls and reimbursement policies in foreign countries; |
• | the relative price of IMBRUVICA as compared to alternative treatment options; |
• | changed or increased legal or regulatory restrictions and our ability to comply with such restrictions; |
• | changes to the label for IMBRUVICA that further restrict how we and Janssen market IMBRUVICA, arising from the results of any other on-going or future studies, including post-marketing studies; and |
• | we and Janssen may not be able to obtain adequate commercial supplies of IMBRUVICA to meet demand or at an acceptable cost because of manufacturing or other issues, including a potential recall of IMBRUVICA. |
If the commercialization of IMBRUVICA is unsuccessful, our ability to generate revenue from product sales and achieve profitability will be adversely affected.
Our success will depend on our ability to effectively and profitably commercialize IMBRUVICA.
Our success will depend on our ability to effectively and profitably commercialize IMBRUVICA, which will include our ability to:
• | create continued market demand for IMBRUVICA through education, marketing and sales activities; |
• | achieve market acceptance and generate product sales; |
• | receive continued reimbursement from third-party payers, such as federal government payers and private insurance programs; |
• | comply with post-marketing requirements established by the U.S. Food and Drug Administration, or FDA, and applicable foreign regulatory agencies, for IMBRUVICA, and any other requirements established by the FDA or foreign regulatory agencies in the future; |
• | comply with the regulations and guidelines of the FDA, and applicable foreign regulatory agencies, surrounding promotional activities for IMBRUVICA; |
• | conduct the post-marketing studies required by the FDA; |
• | comply with other healthcare regulatory requirements; |
• | ensure that the active pharmaceutical ingredient, or API, for IMBRUVICA and the finished product are manufactured in sufficient quantities and in compliance with requirements of the FDA and similar foreign regulatory agencies and with an acceptable quality and pricing level in order to meet commercial demand; and |
28
• | ensure that the entire supply chain for IMBRUVICA - from API to finished product - efficiently and consistently delivers IMBRUVICA to our customers. |
Prior to our commercialization of IMBRUVICA, we did not have any commercial products. We believe our Sales and Marketing organizations have the technical expertise and experience that will be required for the successful marketing of IMBRUVICA in the U.S. However, we still cannot be certain that our commercialization efforts will continue to be successful for a number of reasons, including the fact that our level of experience in marketing IMBRUVICA is limited to the time during which IMBRUVICA has been commercially available for any patient population. If we are unable to continue to successfully commercialize IMBRUVICA, our ability to generate product sales will be severely limited, which will have a material adverse impact on our business, financial condition, and results of operations.
Acceptance of our products in the marketplace is uncertain, and failure to achieve market acceptance may harm our business.
Although approved for marketing for the treatment of certain patient populations, IMBRUVICA may not achieve robust market acceptance. The degree of market acceptance for IMBRUVICA and our other product candidates will depend upon a number of factors, including:
• | the receipt of regulatory approvals for additional indications of IMBRUVICA and the desired indications for our other product candidates, and the acceptance by physicians and patients of the clinical benefits that IMBRUVICA and our other product candidates may offer; |
• | the establishment and demonstration in the medical community of the safety, clinical efficacy and cost-effectiveness of IMBRUVICA and our other product candidates and their potential advantages over existing therapeutic products; |
• | marketing and distribution support; |
• | the introduction, market penetration, and pricing of competing and future products; and |
• | the general willingness of physicians, patients, payers or the medical community to accept, purchase, utilize or recommend IMBRUVICA or any of our other product candidates. |
If we are unable to obtain additional marketing approvals for IMBRUVICA, or if we are significantly delayed or limited in doing so, our results of operations and business will be materially and adversely affected and our stock price would likely decline.
Despite the marketing approvals that we have received to date, we do not know nor can we predict with any certainty that we will obtain additional marketing approvals for IMBRUVICA from the FDA or foreign regulatory authorities. In addition, we cannot predict the timeframe for any additional marketing approvals that we may receive. The review and potential approval of IMBRUVICA carries many risks and uncertainties, and our regulatory submissions outside of the U.S. may not be satisfactory to the applicable regulatory authorities, including with regard to demonstrating adequate safety and efficacy for regulatory approval. We have made, and expect to make in the future, assumptions, estimations, calculations and decisions as part of our analyses of data and regulatory submissions, and the applicable regulatory authorities may not accept or agree with our assumptions, estimations, calculations, decisions or analyses or may interpret or weigh the importance of data differently.
Furthermore, as was the case with approvals that we received from the FDA and European Commission (EC), other regulatory approvals, even if obtained, may be limited to specific indications, limit the type of patients in which the drug may be used, or otherwise require specific warning or labeling language, any of which might reduce the commercial potential of IMBRUVICA. As with the FDA’s and EC's approval of IMBRUVICA, regulatory authorities in other territories may condition IMBRUVICA marketing approval on the conduct of specific post-marketing studies to further evaluate safety and efficacy in particular and/or general patient populations. The results of these studies, discovery of previously unknown issues involving safety or efficacy or failure to comply with post-approval regulatory requirements, including requirements with respect to manufacturing practices, reporting of adverse events, advertising, promotion and marketing, may result in restrictions on the marketing of IMBRUVICA or the withdrawal of IMBRUVICA from the market.
We are subject to certain healthcare laws, regulations and enforcement that may impact the commercialization of IMBRUVICA and our product candidates. Failure to comply with such evolving laws, regulations and enforcement could subject us to significant fines and penalties and result in a material adverse effect on our results of operations and financial condition.
We are subject to statutory and administrative regulation of healthcare enforced by the federal government and the states, and applicable foreign equivalents, in which we conduct our business. These laws may impact, among other things, the viability of the corporation as a whole, and more specifically, the sales, marketing and education programs for IMBRUVICA or any of
29
our potential future product candidates that may be approved for commercial sale. Among the most significant healthcare laws, regulations and enforcement areas are the following:
• | the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (collectively, HIPAA), as amended by the Health Information for Economic and Clinical Health Act of 2009 (HITECH) which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; |
• | the federal healthcare programs’ Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
• | federal False Claims Act which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent; |
• | the federal Food, Drug, and Cosmetic Act, which, among other things, strictly regulates drug product marketing, prohibits manufacturers from marketing drug products for off-label use, and regulates the distribution of drug samples; |
• | the Foreign Corrupt Practices Act, which among other things, prohibits bribery of non-U.S. government (i.e., foreign) officials; |
• | the PhRMA Code on Interactions with Healthcare Professionals (codified into law in California), which among other things, governs appropriate interactions between pharmaceutical manufacturers and healthcare professionals; |
• | the federal Physician Payment Sunshine Act, which requires manufacturers to report payments and other "transfers of value" provided to physicians and teaching hospitals to the Centers for Medicare and Medicaid Services; |
• | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
• | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including federal government payers and commercial insurers; and |
• | international law equivalents of each of the above federal laws, where applicable, such as EFPIA Code, UK Bribery Act, or international transparency and reporting requirements. |
Additionally, the compliance environment is changing, with more states, such as California and Massachusetts, mandating implementation of compliance programs, compliance with industry ethics codes, and spending limits, and other states, such as Vermont, District of Columbia, and Minnesota limiting the provision of and/or requiring reporting to state governments of gifts, compensation, and other remuneration to healthcare professionals. Moreover, Section 6002 of the Patient Protection and Affordable Care Act, or PPACA, includes new requirements for pharmaceuticals manufacturers, among others, to report certain financial arrangements with physicians and teaching hospitals, as defined in PPACA, including reporting any payment or “transfer of value” made or distributed to teaching hospitals, prescribers, and other healthcare providers and reporting any ownership and investment interests held by physicians and their immediate family members and applicable group purchasing organizations during the preceding calendar year. Section 6002 of PPACA includes in its reporting requirements a broad range of transfers of value (such as, for example, consulting fees, charitable contributions, payments for research, and grants), but excludes from the reporting obligation any transfer of value which is less than $10, unless the aggregate amount of such transfers of value to a recipient exceeds $100 annually. On February 8, 2013, the Centers for Medicare and Medicaid Services, or CMS, released the final rule implementing Section 6002 of PPACA. Under the rule, drug manufacturers are required to track payments or transfers of value to recipients beginning on August 1, 2013, and annually report payments, transfers for value, and investments interests beginning on March 31, 2014. Under the rule, with our product approval on November 13, 2013 for MCL, we commenced data capture from May 12, 2014, 6 months from product registration, for reporting by March 31, 2015. Several states currently have similar laws and more states may enact similar legislation. Reporting and potential public disclosure of these expenses may make it more difficult to recruit physicians for assistance with activities that would be helpful to our business. Tracking and reporting the required expenses may result in considerable expense and additional resources.
While we have developed and implemented a corporate compliance program, which adheres to: (1) the requirements set forth by the OIG in OIG’s Compliance Program Guidance for Pharmaceutical Manufacturers, as codified into law in California, (2) the PhRMA Code on Interactions with Healthcare Professionals as codified into law in California, and (3) other applicable healthcare compliance laws and regulations, and is largely based on what we believe are the current pharmaceutical industry best practices, we cannot provide any assurance that governmental authorities will find that our business practices comply with current or future administrative or judicial interpretations of potentially applicable laws and regulations.
30
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Current health care laws and regulations, including the recently enacted health care reform, as well as future legislative or regulatory changes to the healthcare system, may affect our ability to sell our products profitably.
In the U.S., there has been recent legislation, as well as legislative and regulatory proposals, changing the healthcare system in ways that may affect our future revenue and profitability, and the future revenue and profitability of our potential customers, suppliers and collaborative partners, and the availability of capital. Federal and state lawmakers regularly propose and, at times, enact legislation that would result in significant changes to the healthcare system and, in particular, that are intended to contain or reduce the costs of medical products and services.
The most significant recent health care legislation is the PPACA, as amended by the Health Care and Education Reconciliation Act, or the “Healthcare Reform Act”, which President Obama signed into law in March 2010. This law substantially changes how health care is funded by the state and federal government as well as private insurers, and significantly impacts the pharmaceutical industry. Though the full effect of the Healthcare Reform Act on pharmaceutical companies has yet to be seen, the changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, new governmental programs, and fraud and abuse enforcements. The Healthcare Reform Act takes effect in stages through 2018.
Certain aspects of the Health Care Reform Act are likely to adversely affect pharmaceutical manufacturers in particular. For example, in 2011, the Healthcare Reform Act imposed non-deductible annual flat fees on pharmaceutical manufacturers and importers based upon relative market share. Furthermore, as part of the Healthcare Reform Act closing a funding gap in the Medicare Part D prescription drug program, certain pharmaceutical manufacturers will be required to provide a 50% discount on drugs dispensed to beneficiaries within this funding gap.
There also have been and likely will continue to be legislative and regulatory proposals at the state and federal levels that could bring about significant changes to the Medicaid drug rebate program and other federal pharmaceutical pricing and rebate programs. Given these and other recent federal and state government initiatives directed at lowering the total cost of health care, federal and state lawmakers will likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid programs. These efforts could adversely affect our business by, among other possibilities, limiting the prices that can be charged for drugs we develop or the amount of reimbursement available for these products from governmental agencies or third-party payers, limiting the profits that pharmaceutical companies may earn on certain sales, increasing the tax obligations on pharmaceutical companies, increasing our rebate liability, or limiting our commercial opportunity. We cannot predict the impact on our business of any legislation or regulations that may be adopted in the future. Any cost containment measures and other healthcare system reforms that are adopted could have a material adverse effect on our ability to operate profitably.
Price controls imposed in foreign markets may adversely affect our future profitability.
In some countries, particularly member states of the EU, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Drug pricing may be made against a reference price set by the healthcare providers as a measure for healthcare cost containment. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. In some countries, we or our collaborative partners may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our therapeutic candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. If reimbursement of any product candidate approved for marketing is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, any one of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
Even though we have obtained approval to market IMBRUVICA for MCL in the U.S., we are subject to ongoing regulatory obligations and review, including post-approval requirements that could result in the removal of the MCL indication from the approval of IMBRUVICA.
IMBRUVICA was approved for treating patients with MCL, who have received at least one prior therapy, under the FDA’s priority review program which provides for an expedited review for drugs that may offer significant improvement in treatment or provide treatment where no satisfactory alternative therapy exists. Even though we have obtained approval to market
31
IMBRUVICA for treating patients with MCL in the U.S., we are subject to extensive ongoing obligations and continued regulatory review from the FDA and other applicable regulatory agencies, such as continued adverse event reporting requirements, the requirement to have our promotional materials relating to MCL pre-cleared by the FDA, and the requirement to conduct and complete a Phase III clinical study that confirms the safety and efficacy of IMBRUVICA in patients with MCL who have received at least one prior therapy. Although we are conducting two Phase III clinical studies designed to confirm the safety and efficacy of IMBRUVICA in patients with MCL who have received at least one prior therapy, we cannot guarantee that we will successfully complete either or both of these studies or that the results of either or both of these studies will be adequate to fulfill the post-approval requirement. Failure to fulfill the post-approval requirement for a confirmatory Phase III clinical study could result in the removal of the MCL indication from the approval of IMBRUVICA in the U.S. There may also be additional FDA post-marketing obligations, all of which may result in significant expense and limit our ability to commercialize IMBRUVICA in the U.S. or potentially other jurisdictions.
Failure to comply with applicable FDA or other foreign and domestic regulatory requirements and guidelines may subject us to administrative or judicially imposed sanctions. If we fail to comply with foreign or domestic regulatory requirements or guidelines, we may face sanctions, including:
• | issuance of Form FDA 483 or Warning Letters by the FDA or notices of noncompliance from other regulatory agencies; |
• | imposition of fines and other civil penalties; |
• | criminal prosecutions; |
• | injunctions, suspensions or revocations of regulatory approvals; |
• | suspension of any ongoing clinical trials; |
• | total or partial suspension of manufacturing; |
• | refusal by the FDA to approve pending applications or supplements to approved applications filed by us; |
• | refusals to permit drugs to be imported into or exported from the U.S.; |
• | restrictions on operations, including costly new manufacturing requirements; and |
• | product recalls or seizures. |
The policies of the FDA and other regulatory agencies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of IMBRUVICA in other indications or further restrict or regulate post-approval activities. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are not able to maintain regulatory compliance, we or Janssen might not be permitted to market IMBRUVICA and our business would suffer.
Our failure to continue to manage and maintain a distribution network could compromise the distribution of IMBRUVICA.
We have contracted with a third-party logistics company to warehouse IMBRUVICA and distribute it to wholesalers, distributors, pharmacies, hospitals, and other drug suppliers that ultimately distribute IMBRUVICA directly to patients. Our third-party logistics company will also provide billing, collection and returns services. This distribution network requires significant coordination with our market access, finance, quality and manufacturing teams.
Failure to maintain our contracts with our third-party logistics company, wholesalers, distributors, pharmacies, hospitals or other drug suppliers, or the inability or failure of any of them to adequately perform under the contracts, could negatively impact the distribution of IMBRUVICA. Failure to coordinate financial systems could also negatively impact our ability to accurately report and forecast product revenue. If we are unable to effectively establish and manage the distribution process, sales of IMBRUVICA could be severely compromised and our business, financial condition and results of operations would be harmed.
We may need substantial additional financing and we may have difficulty raising needed capital in the future.
We have expended and will continue to expend substantial funds for the commercialization, manufacturing, marketing and sales of IMBRUVICA and to complete the research, development and clinical testing of investigational uses of IMBRUVICA and additional products. We may be unable to entirely fund these efforts with our current financial resources. We may also raise from time to time additional funds through the public or private sale of securities, bank debt, collaborations or otherwise. If we are unable to secure additional funds, whether through additional partnership collaborations, bank debt financings, or sale of our securities, we may have to delay, reduce the scope of or discontinue one or more of our product development programs. Based upon the current status of our product development plans, we believe that our cash, cash equivalents and marketable
32
securities will be adequate to satisfy our capital needs for at least the next 12 months. We may, however, choose to raise additional funds before then. Our actual capital requirements will depend on many factors, including:
• | the level of sales of IMBRUVICA; |
• | the timing of marketing authorization of any of our additional products granted by the FDA, EC or other regulatory authority; |
• | the timing of market launch of any of our additional products after grant of marketing authorization by the FDA, EC or other regulatory agency; |
• | the speed of market uptake and the timing and extent of demand for any of our additional products after grant of marketing authorization by the FDA, EC or other regulatory agency; |
• | continued progress of our research and development programs; |
• | progress with preclinical studies and clinical trials; |
• | the time and costs involved in obtaining regulatory approval; |
• | our ability to establish and maintain collaborative arrangements with third parties; |
• | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims; |
• | the amount and timing of capital equipment purchases; and |
• | competing technological and market developments. |
We also expect to raise any necessary additional funds through the public or private sale of securities, bank debt financings, collaborative arrangements with corporate partners or other sources that may be highly dilutive, or otherwise disadvantageous, to existing stockholders or subject us to restrictive covenants. In addition, in the event that additional funds are obtained through arrangements with collaborative partners or other sources, such arrangements may require us to relinquish rights to some of our technologies, product candidates or products under development that we would otherwise seek to develop or commercialize ourselves. Additional funds may not be available on acceptable terms, if at all. Our failure to raise capital when needed and on acceptable terms may require us to reduce our costs and expenses, delay or reduce the scope of or eliminate one or more of our research or development programs and would limit our ability to respond to competitive pressures or unanticipated requirements and to continue operations. Any one of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
If we are unable to successfully develop and test our drug candidates, we may not be successful.
The success of our business depends primarily upon our ability, and the ability of our collaborators, if any, to develop and commercialize our drug candidates, including IMBRUVICA for indications other than those approved by the FDA, and outside of the U.S., successfully. The time frame necessary to achieve these goals for any individual product or indication is long and uncertain. We cannot be certain that we will be permitted to begin or continue our planned clinical trials for our potential products.
The completion rate of our clinical trials depends upon, among other factors, the rate of patient enrollment, the adequacy of patient follow-up and the completion of required clinical evaluations. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, competing clinical trials and new drugs or procedures used for the indications we are investigating. Other companies are conducting clinical trials and have announced plans for future trials that are seeking or are likely to seek patients with the same diseases that we are studying. We may experience delays in reaching planned levels of patient enrollment in our clinical trials. Delays in planned patient enrollment may result in increased costs, and delays or termination of clinical trials, which could have a material adverse effect on us. Many factors can affect the adequacy of patient follow-up and completion of required clinical evaluations, including failure of patients to return for scheduled visits or failure of clinical sites to complete necessary documentation. Delays in or failure to obtain required clinical follow-up and completion of clinical evaluations could also have a material adverse effect on the timing and outcome of our clinical trials and product approvals.
Additionally, clinical trials require substantial administration and monitoring. We may fail to effectively oversee and monitor the various trials we have underway at any particular time, which would result in increased costs or delays of our clinical trials or compromise our ability to obtain product approvals.
Our drug candidates are in various stages of development and must satisfy rigorous standards of safety and efficacy before they can be approved by the FDA or comparable foreign regulatory authorities for sale. To satisfy these standards, we and/or our collaborators must allocate resources among our various development programs and must engage in expensive and lengthy testing of our drug candidates. Despite our efforts, our drug candidates may:
33
• | cause, or may appear to cause serious adverse events or potentially dangerous drug interactions; |
• | have dose limiting toxicities; |
• | not show signs of efficacy in any disease as a single agent or in combination, or may only work in combination with other drugs; |
• | cause resistance in patients that may diminish the clinical benefit; |
• | not offer therapeutic or other improvement over existing competitive drugs; or |
• | not be proven adequately safe and effective in clinical trials. |
The failure to adequately demonstrate the safety and effectiveness of a product under development could limit or prevent regulatory approval of the potential product and would materially harm our business. Our clinical trials may not demonstrate the sufficient levels of safety and efficacy necessary to obtain the requisite regulatory approval or may not result in marketable products.
The strength of our pipeline of drug candidates and potential drug candidates will depend in part upon the outcomes of our ongoing and planned Phase I, Phase II and Phase III clinical trials. Findings, including toxicity, tolerability, efficacy and/or safety findings, in nonclinical studies conducted concurrently with clinical trials as well as results of our clinical trials could lead to certain changes in our development activities, including the possible cessation of development activities associated with a particular drug candidate or program, including development of our next-generation BTK inhibitor drug candidate for rheumatoid arthritis and/or other autoimmune indications, which may be discontinued at any time due to unacceptable toxicity, tolerability, efficacy and/or safety issues discovered in our ongoing Phase I clinical trial and/or nonclinical studies of this drug candidate, and including development of IMBRUVICA for indications other than those approved by the FDA and outside of the U.S. Furthermore, results from our clinical trials may not meet the level of statistical significance required by the FDA or other regulatory authorities for approval of a drug candidate.
Data already obtained from preclinical studies and clinical trials of our products under development do not necessarily predict the results that will be obtained from later preclinical studies and clinical trials. As such, positive results in preclinical studies and early clinical trials may not be replicated in later clinical trials. Further, data from clinical trials we are conducting are susceptible to varying interpretations that could delay, limit or prevent regulatory approval. We and many other companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in earlier-stage clinical trials. In addition, from time to time we report interim data from our clinical trials, including our BTK program and its effects on various types of cancers. Interim data are subject to change, and there can be no assurances that interim data will be confirmed upon the analysis of final data.
Serious adverse events may force us to limit or discontinue any of our drug development and commercialization programs.
Our ability to develop and commercialize any of our drug candidates or products may be adversely impacted by any serious adverse events that may arise. If significant safety issues arise when patients are treated with our marketed product or product candidates, our future sales may be reduced, which would adversely affect our results of operations.
The data supporting the marketing approvals for our products and forming the basis for the safety warnings in our IMBRUVICA product label were obtained from a limited number of patients in a limited number of clinical studies. As our product is used over longer periods of time and by many patients outside of the more exclusive and controlled environment of a clinical study, including patients with underlying health problems, taking numerous medications, we expect to continue to find new safety signals and gather new information, which may require us to provide additional warnings to our label, which could reduce the market acceptance of our product. Further, if serious safety, resistance or drug interaction issues arise with our product, sales could be limited or halted by us or by regulatory authorities and our results of operations would be adversely affected. For further details of safety and efficacy information, please refer to the full U.S. Prescribing Information (USPI).
We may fail to adequately protect or enforce our intellectual property rights or secure rights to third-party patents.
Our success depends in part upon our ability to protect and defend our proprietary technology and product candidates through patents and trade secret protection. We, therefore, aggressively pursue, prosecute, protect and defend patent applications, issued patents, trade secrets, and licensed patent and trade secret rights covering certain aspects of our technology. The evaluation of the patentability of U.S. and foreign patent applications can take years to complete and can involve considerable expense and uncertainty.
We have a number of patents and patent applications related to our compounds but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will issue as patents. Even if patents are issued and maintained, these patents may not be of adequate scope to benefit us, or may be held invalid and unenforceable against third parties. In addition, if we breach our agreements with our licensors and do not cure such breaches, we may lose
34
rights under patents and other intellectual property licensed to us. Moreover, if our agreement with Celera is terminated by Celera under certain circumstances, our rights in our intellectual property acquired from Celera, including that related to IMBRUVICA, would revert to Celera and we would lose our right to commercialize such intellectual property.
We face risks and uncertainties related to our intellectual property rights. For example:
• | we may be unable to obtain or maintain patent or other intellectual property protection for any products or processes that we may develop; |
• | third parties may obtain patents covering the manufacture, use or sale of these products, which may prevent us from commercializing IMBRUVICA or any of our products under development globally or in certain regions; and |
• | any future patents that we may obtain may not prevent other companies from competing with us if such companies can design their products or conduct their activities so as to avoid the coverage of our patents. |
The actual protection afforded by a patent varies depending on the product candidate and country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patents under existing and future laws. Our ability to maintain or enhance our proprietary position for our product candidates will depend on our success in obtaining effective patent claims and enforcing those patent claims once granted. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar products. Due to the extensive amount of time required for the development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. Although we take steps to protect our proprietary rights and information, including the use of confidentiality and other agreements with our employees and consultants, and in our academic and commercial relationships, these steps may be inadequate, these agreements may be violated, or there may be no adequate remedy available for a violation. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Furthermore, our trade secrets may become known to our competitors even in the absence of any violation of our rights. Our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology. We may be unable to meaningfully protect our rights in unpatented proprietary technology.
Our success depends in large part on our ability to operate without infringing upon the patents or other proprietary rights of third parties.
If we infringe the patents of others, we may be prevented from commercializing future products or may be required to obtain licenses from these third parties. We may not be able to obtain alternative technologies or any required license on reasonable terms or at all. If we fail to obtain these licenses or alternative technologies, we may be unable to develop or commercialize some or all of our products. We own patents that claim the active pharmaceutical ingredient of IMBRUVICA as a chemical entity. However, the existence of issued patents does not guarantee our right to practice the patented technology or commercialize the patented product. Third parties may have or obtain rights to patents which they may use to prevent or attempt to prevent us from commercializing IMBRUVICA or any of our patented product candidates. If these other parties are successful in obtaining valid and enforceable patents, and establishing our infringement of those patents, we could be prevented from selling IMBRUVICA or any of our other products unless we were able to obtain a license under such patents. If any license is needed it may not be available on commercially reasonable terms or at all.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these or other claims. If we fail in defending such claims, in addition to paying monetary damages, we may face injunctions that restrict or preclude our access to important markets, intellectual property, or personnel. Any restriction or loss of access to markets, intellectual property, research personnel or work product that are key to our operations could hamper or negate our ability to commercialize IMBRUVICA or certain potential drugs, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
35
We are dependent on our collaboration agreement with Janssen to further develop and commercialize IMBRUVICA outside of the U.S. The failure to maintain this agreement or the failure of Janssen to perform its obligations under this agreement, could negatively impact our business.
Pursuant to the terms of our collaboration and licensing agreement with Janssen, we granted Janssen a license to co-develop (with us) IMBRUVICA, our BTK Inhibitor, globally, to co-commercialize it (with us) in the U.S., and to exclusively commercialize it outside of the U.S., in each case for all non-immunology related indications. Integral to the success of our collaboration with Janssen is our ability to timely achieve certain milestones and obtain regulatory approvals. Under a global development plan, each party will be responsible for conducting certain clinical trials. The collaboration and license agreement includes a cost sharing arrangement for associated development activities. Except in certain cases, in general Janssen is responsible for approximately 60% of development costs and we are responsible for the remaining 40% of development costs. The agreement also provides that pre-tax commercial profits or pre-tax commercial losses from the commercialization of any products resulting from the collaboration are shared 50% by us and 50% by Janssen.
The collaboration with Janssen provides us with an annual cap of our share of collaboration costs and pre-tax commercial losses for each calendar year until the third profitable calendar quarter for the collaboration, as determined in the agreement. In the event that our share of aggregate development costs in any given calendar year, together with any other amounts that become due from us, plus our share of pre-tax commercial losses for any calendar quarter in such calendar year, less our share of pre-tax commercial profits for any calendar quarter in such calendar year, exceeds $50.0 million, then amounts that are in excess of $50.0 million (Excess Amounts) are funded by Janssen. Under the Agreement, total Excess Amounts plus interest may not exceed $225.0 million. Janssen may recoup the Excess Amounts, together with interest, from our share of pre-tax commercial profits after the third profitable calendar quarter for the collaboration until the Excess Amounts and applicable interest has been fully paid. As of December 31, 2014, total Excess Amounts were $138.3 million, which was comprised of the cumulative amount funded by Janssen to-date of $134.3 million and interest of $4.0 million. As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements for additional information about the Agreement).
We have limited control over the development or commercialization costs incurred by Janssen through yearly budget approvals, and limited control over the execution of development and commercial activities performed by them. Our costs and revenue are therefore tied to efforts made by ourselves and Janssen in developing and marketing our product. We have limited control over the amount of time and effort Janssen will devote to the development, manufacturing and commercialization of IMBRUVICA, and very limited control over the manner in which Janssen conducts its business with regard to obtaining additional regulatory and other approvals and commercializing the product, especially outside the U.S. Accordingly, our revenue and financial position may be adversely affected if Janssen does not dedicate sufficient time to the development and commercialization of IMBRUVICA, fails to obtain additional regulatory approvals, or otherwise fails to comply with its obligations under the agreement.
We are subject to a number of other risks associated with our dependence on our collaboration and license agreement with Janssen, including:
• | We and Janssen could disagree as to future development plans and Janssen may delay future clinical trials or stop a future clinical trial; |
• | There may be disputes between us and Janssen, including disagreements regarding the collaboration and license agreement, that may result in (1) the delay in or loss of additional milestone or profit share payments due to any failure to achieve regulatory and commercial objectives, (2) the delay or termination of any future development or commercialization of IMBRUVICA, and/or (3) costly litigation or arbitration that diverts our management's attention and resources; |
• | Janssen may not provide us with timely and accurate information regarding supply forecasts, which could adversely impact our ability to comply with our supply obligations to Janssen and manage our own inventory of IMBRUVICA, as well as our ability to generate accurate financial forecasts; |
• | Business combinations or significant changes in Janssen's business strategy may adversely affect Janssen's ability or willingness to perform its obligations under our collaboration agreement; |
• | If Janssen is unsuccessful in performing clinical trials, or in obtaining additional regulatory approvals for or commercializing IMBRUVICA outside the U.S., we may not receive certain corresponding milestone payments or profit payments related to international sales under the collaboration and license agreement and our business prospects and financial results may be materially harmed; |
36
• | Our co-promotion and licensing arrangement with our partner Janssen for the marketing of IMBRUVICA in the U.S. may fail to increase the commercial success of IMBRUVICA; and |
• | Janssen may not comply with applicable regulatory requirements or guidelines with respect to developing or commercializing IMBRUVICA outside the U.S., which could adversely impact future development or sales of IMBRUVICA outside of the U.S. |
The collaboration and license agreement is subject to early termination, including through Janssen's right to terminate without cause upon advance notice to us. Termination of the collaboration and license agreement would hinder our efforts to effectively develop and commercialize IMBRUVICA. Such termination could result in a delay in getting our product to market. Such delay could likely result in higher costs for us and could adversely affect any progress we have made in clinical trials. If Janssen terminates its relationship with us, we may decide to enter into a new co-promotion or other licensing arrangement with another large pharmaceutical or biotechnology firm in order to commercialize IMBRUVICA and may not be successful. We may have difficulty finding another collaboration partner on favorable terms if Janssen terminates our agreement. We may be unable to pursue continued development and commercialization of IMBRUVICA or raise capital on our own.
We rely heavily on third parties for research, pre-clinical development and clinical development of our products.
We currently depend heavily and will continue to depend heavily in the future on third parties for support in research, pre-clinical development and clinical development of our products. The termination of a significant number of our existing collaborative arrangements, or our inability to establish and maintain collaborative arrangements could have a material adverse effect on our ability to complete clinical development of our products. Given our limited resources, it may be necessary to establish collaborations with other pharmaceutical companies that have greater financial and technical resources in order to successfully develop and commercialize our products. Although we entered into a global strategic alliance with Servier related to the research, development, and commercialization of abexinostat (formerly known as PCI-24781), we and Servier agreed to terminate the alliance effective November 23, 2014 (see Note 5 to the consolidated financial statements). There is no assurance that any additional collaborations can be entered into, and even if entered into, that any such collaborations will be successful and will not require us to relinquish product rights to the detriment of the financial success of these products.
We engage clinical investigators and medical institutions to enroll subjects in our clinical trials and contract clinical research organizations, or CROs, for various aspects of our clinical development activities including clinical trial monitoring, data collection, safety monitoring and data management. As a result, we have had and continue to have less control over the conduct of clinical trials, the timing and completion of the trials, the required reporting of adverse events and the management of data developed through the trial than would be the case if we were relying entirely upon our own staff. Although we rely on CROs to conduct some of our clinical trials, we are responsible for confirming that each of our clinical trials is conducted in accordance with the investigational plan and protocol. Moreover, the FDA, the Office of the Inspector General of the Department of Health and Human Services (OIG) and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. We could be held accountable, from a regulatory or compliance standpoint, for any erroneous acts or omissions of these third parties, as they are acting on behalf of Pharmacyclics and as our agents.
Outside parties may have staffing difficulties, may undergo changes in priorities, may become financially distressed, or may cease their operations, adversely affecting their willingness or ability to conduct our trials. We may experience unexpected cost increases that are beyond our control. Any failure of such CROs to successfully accomplish clinical trial monitoring, data collection, safety monitoring and data management and the other services they provide for us in a timely manner and in compliance with regulatory requirements could have a material adverse effect on our ability to complete clinical development of our products and obtain regulatory approval. Problems with the timeliness or quality of the work of a CRO or the cessation of a CRO's business may lead us to seek to terminate the relationship and use an alternate service provider. However, making such changes may be costly and may delay our trials, and contractual restrictions or other business obstacles may make such a change difficult or impossible. Additionally, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
We rely upon contract manufacturers to manufacture pharmaceuticals for clinical development and for commercial sale.
We currently rely on third parties to perform activities related to the manufacture and storage of pharmaceuticals for our clinical trials and commercial sales. Our manufacturing strategy presents the following risks:
• | we currently rely on a single manufacturer and supply chain for IMBRUVICA and expect to continue to do so for the foreseeable future. |
37
• | delays in scale-up to quantities needed for multiple clinical trials, or failure to manufacture such quantities to our specifications, or deliver such quantities on the dates we require, could cause delay or suspension of clinical trials, regulatory submissions and commercialization of our products; |
• | there is no guarantee that the supply of clinical materials can be maintained during the clinical development of our product candidates; |
• | our current and future manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding foreign regulatory agencies for compliance with strictly enforced GMP and similar standards in other countries. Failure to achieve an acceptable inspection outcome could have a material adverse effect on our ability to produce our products to support our operations; |
• | our reliance on third parties for manufacturing and storage does not relieve us of responsibilities and requirements of the FDA, OIG, and corresponding foreign regulatory agencies. We could be held accountable from a regulatory or compliance standpoint, for any erroneous acts or omissions of these third parties, as they are acting on behalf of Pharmacyclics and as our agents; |
• | we may be unsuccessful in maintaining our current supply agreements and if we are required to seek new supply agreements, there is no guarantee that we will be able to obtain such agreements on favorable terms or at all. These contractors must achieve an acceptable inspection outcome by the FDA and comparable foreign regulators prior to our use; |
• | our current manufacturer might not be able to fulfill our commercial needs, which would require us to seek new manufacturing arrangements and may result in substantial delays in meeting market demand; |
• | any disruption of the ability of our manufacturing contractors to supply necessary quantities of our products could have a material adverse effect on our ability to support our operations; and |
• | our current manufacturer may have insufficient capacity to support our requirements in the event of an increase in market demand. |
Any of these factors could delay clinical trials or commercialization of our products under development and entail higher costs.
Our manufacturing process may require the use of highly flammable solvents and agents, and may result in hazardous situations in our contract manufacturing facilities.
The manufacturing processes for IMBRUVICA involve the use of hazardous, volatile and flammable materials capable of igniting and/or exploding if not handled properly. Although our manufacturing processes incorporate carefully designed safety procedures to prevent the creation of conditions under which the materials could ignite and/or explode, we cannot be certain that such safety procedures will be followed or if followed will be adequate to mitigate or eliminate the risk of harm caused by fire and/or explosion during the manufacturing of IMBRUVICA. Any such event may incapacitate the manufacturing capability of any of our contract manufacturing organizations (CMOs). We have no assurance that we will avoid liability for harm caused by any such event. If we are found liable for damages or agree to pay damages in settlement of a claim against us for any harm caused by any such event, our insurance coverage may be inadequate or may not cover part or all of such damages. In addition, we do not have an alternate or back-up supply chain for the manufacturing and supply of IMBRUVICA. If such an event causes us to lose the operational capacity of any manufacturing element in the supply chain for IMBRUVICA, our ability to manufacture and supply the product will be substantially impaired or prevented, and we may be unable to supply enough product to support our development and/or commercialization programs, which may force us to curtail or discontinue our business operations.
We may have exposure to greater than anticipated tax liabilities.
Our income tax obligations are based on our corporate operating structure, including the manner in which we develop, value, and use our intellectual property and the scope of our international operations. The tax laws applicable to our international business activities, including the laws of the U.S. and other jurisdictions, are subject to interpretation. The taxing authorities of the jurisdictions in which we operate may challenge our methodologies for valuing developed technology or intercompany arrangements and our positions that defer or avoid the recognition of revenue from and taxation of intercompany technology transfer transactions, which could increase our worldwide effective tax rate and harm our financial position and results of operations. In addition, our future income taxes could be adversely affected by earnings being lower than anticipated in jurisdictions that have lower statutory tax rates and higher than anticipated in jurisdictions that have higher statutory tax rates, by changes in the valuation of our deferred tax assets and liabilities, or by changes in tax laws, regulations, or accounting principles. We are subject to regular review and audit by both U.S. federal and state and foreign tax authorities. Any adverse outcome of such a review or audit could have a negative effect on our financial position and results of operations. In addition, the determination of our worldwide provision for income taxes and other tax liabilities requires significant judgment by
38
management, and there are many transactions where the ultimate tax determination is uncertain. Although we believe that our estimates are reasonable, the ultimate tax outcome may differ from the amounts recorded in our financial statements and may materially affect our financial results in the period or periods for which such determination is made.
The enactment of legislation implementing changes in the U.S. taxation of international business activities or the adoption of other U.S. or foreign tax reform policies could materially affect our financial position, cash flows and results of operations.
The current administration has made public statements indicating that it has made international tax reform a priority, and key members of the U.S. Congress have conducted hearings and proposed a wide variety of potential changes. Certain changes to U.S. tax laws, including limitations on the ability to defer U.S. taxation on earnings outside of the U.S. until those earnings are repatriated to the U.S., could affect the tax treatment of our foreign earnings, as well as cash and cash equivalent balances we currently maintain outside of the U.S.
Furthermore, changes to foreign rules may also materially affect us. Due to the expanding scale of our international business activities, any changes in the U.S. or foreign taxation of such activities may increase our worldwide effective tax rate and harm our financial position and results of operations. The Organisation for Economic Co-operation and Development (OECD) has published an action plan to address base erosion and profit shifting (BEPS) impacting its member countries and other jurisdictions. It is possible that jurisdictions in which we do business could react to the BEPS initiative or their own concerns by enacting tax legislation that could adversely affect us or our shareholders.
A number of multilateral organizations, including the EU and the OECD have, in recent years, expressed concern about some countries not participating in adequate tax information exchange arrangements and have threatened those that do not agree to cooperate with punitive sanctions by member countries. It is as yet unclear what all of these sanctions might be, which countries might adopt them, and when or if they might be imposed. We cannot assure, however, that the Tax Information Exchange Agreements (TIEAs) that have been or will be entered into by Switzerland and Cayman will be sufficient to preclude all of the sanctions described above, which, if ultimately adopted, could adversely affect us or our shareholders.
We are exposed to fluctuations in foreign currency exchange rates, and an adverse change in foreign currency exchange rates could have a material adverse impact on our business, financial condition, cash flows and results of operations.
All of our revenues and the majority of our expenses are denominated in U.S. dollars and as a result, we have not experienced significant foreign currency transaction gains and losses to date. We conduct some transactions in foreign currencies, primarily related to ex-U.S. clinical trial and manufacturing activities, and we expect to continue to do so. We have not entered into any agreements or transactions for currency exchange to hedge the risk associated with potential fluctuations in currencies; accordingly, we are subject to foreign currency exchange risk related to these ex-U.S. clinical trial and manufacturing activities. While we may enter into hedge or other agreements in the future to actively manage this risk, we do not believe this risk is material to our financial statements.
Risks Related to Our Common Stock
If our results do not meet the financial guidance we provide to investors in any period, analysts' forecasts or the expectations of our investors, our stock price could decline.
Analysts who cover our business and operations provide valuations regarding our stock price and make recommendations whether to buy, hold or sell our stock. Our stock price may be dependent upon such valuations and recommendations. Analysts' valuations and recommendations are based primarily on our reported results and our and their forecasts and expectations concerning our future results regarding, for example, expenses, revenues, clinical trials, regulatory marketing approvals and competition. Our future results are subject to substantial uncertainty, and we may fail to meet or exceed the financial guidance we provide to investors in any period or analysts' forecasts or investors' expectations as a result of a number of factors, including those discussed under the section entitled “Risks Related to Pharmacyclics” above. If our results do not meet the financial guidance that we provide to investors in any period or analysts' forecasts or investors' expectations, our stock price could decline as a result of analysts lowering their valuations and recommendations or otherwise.
Future equity issuances or a sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
Because we may need to raise additional capital in the future to continue to expand our business and our research and development activities, among other things, we may conduct additional equity offerings. If we or our stockholders sell substantial amounts of our common stock (including shares issued upon the exercise of options and warrants) in the public
39
market, the market price of our common stock could fall. A decline in the market price of our common stock could make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem appropriate.
Anti-takeover provisions in our charter documents and Delaware law could prevent or delay a change in control.
Our certificate of incorporation and bylaws include provisions that may discourage, delay or prevent a merger or acquisition that a stockholder may consider favorable. In addition, provisions of the Delaware General Corporation Law also restrict certain business combinations with interested stockholders. These provisions are intended to encourage potential acquirers to negotiate with us and allow our board of directors the opportunity to consider alternative proposals in the interest of maximizing stockholder value. However, these provisions may also discourage acquisition proposals or delay or prevent a change in control, which could harm our stock price.
The price of our common stock is volatile.
The market prices for securities of biotechnology companies, including ours, have historically been highly volatile. Our stock, like that of many other companies, has from time to time experienced significant price and volume fluctuations sometimes unrelated to operating performance. For example, during the three year period beginning January 1, 2012 and ending December 31, 2014, the sales price for one share of our common stock reached a high closing price of $151.61 per share and a low closing price of $14.99 per share. The market price of our common stock may fluctuate significantly due to a variety of factors, including:
• | the commercial success of IMBRUVICA and the revenues we generate; |
• | the progress and results of our preclinical testing, clinical trials, product development and partnering activities; |
• | quarterly fluctuations in our financial results; |
• | our actual results of operations may deviate materially from projected results; |
• | the development of technological innovations or new therapeutic products by us, our competitors or others; |
• | changes in governmental regulation; |
• | the success or failure of our efforts to obtain marketing authorization for any of our products from the FDA or other regulatory authority; |
• | our ability to satisfy regulatory requirements for the maintenance of any marketing authorization granted by FDA or other regulatory authority for any of our products; |
• | the emergence of drug safety issues that require us to add restrictions or warnings to the label or withdraw from the market any of our products after approval by the FDA or other regulatory authority; |
• | developments in patent or other proprietary rights by us, our competitors or others; |
• | developments and/or announcements by us, our competitors or others; |
• | litigation; |
• | public concern as to the safety of products developed by us, our competitors or others; |
• | departure of key personnel; |
• | ability to manufacture our products to commercial standards; |
• | changes in the structure of healthcare payment systems and the coverage and reimbursement policies of governmental and other third-party payers; |
• | our ability to successfully commercialize our products if they are approved; |
• | comments by securities analysts; and |
• | general market conditions in our industry. |
In addition, if any of the risks described in this section entitled “Risk Factors” actually occur, there could be a dramatic and material adverse impact on the market price of our common stock.
Risks Related to Our Industry
Our business exposes us to product liability claims.
The testing, manufacture, marketing and sale of our products involve an inherent risk that product liability claims will be asserted against us. We face the risk that the use of our products when sold to patients and when used in human clinical trials will result in adverse effects. IMBRUVICA is currently marketed, and if we complete clinical testing for our additional products and receive regulatory approval to market such additional products, we will market our additional products, with
40
warnings that identify the known potential adverse effects and the patients who should not receive our product. We cannot ensure that physicians and patients will comply with these warnings. In addition, unexpected adverse effects may occur even with use of our products that have received approval for commercial sale. Although we are insured against such risks in connection with clinical trials and commercial activities, our present product liability insurance may be inadequate. A successful product liability claim in excess of our insurance coverage could have a material adverse effect on our business, financial condition and results of operations. Any successful product liability claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable or reasonable terms. In addition, product liability coverage may cease to be available in sufficient amounts or at an acceptable cost. An inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our pharmaceutical products. A product liability claim or recall would have a material adverse effect on our reputation, business, financial condition and results of operations.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
As of December 31, 2014, we lease a total of 132,176 square feet for our corporate headquarters in Sunnyvale, California. Of this total space, 79,776 square feet are leased under an operating lease that expires in November 2017, with an option to extend the lease term for an additional five years. The remaining 52,400 square feet are leased under an operating lease which expires in February 2023, which provides for an option to early terminate the lease in 2018 and an option to extend the lease term for an additional five years. In addition, we lease approximately 8,000 square feet for our Pharmacyclics Switzerland GmbH office under an operating lease that expires in May 2015 and lease 7,000 square feet of office space in South San Francisco, California under an operating lease that expires in March 2015. In January 2015, we entered into a lease agreement for additional office space for our corporate headquarters in Sunnyvale, California for approximately 36,000 square feet under an operating lease that expires in May 2016. In addition, in February 2015, we entered into a lease agreement for approximately 10,000 square feet of office space in South San Francisco, California, expiring in February 2017.
We believe our existing facilities are adequate to meet our current needs and our capital resources are sufficient to lease additional spaces as needed.
Item 3. Legal Proceedings
We may be involved, from time to time, in legal proceedings and claims arising in the ordinary course of our business. Such matters are subject to many uncertainties and outcomes are not predictable with assurance. We accrue amounts, to the extent they can be reasonably estimated, that we believe are adequate to address any liabilities related to legal proceedings and other loss contingencies that we believe will result in a probable loss. While there can be no assurances as to the ultimate outcome of any legal proceeding or other loss contingency involving us, management does not believe any pending matter will be resolved in a manner that would have a material adverse effect on our consolidated financial position, results of operations or cash flows.
Item 4. Mine Safety Disclosures
None.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock trades on the NASDAQ Stock Market under the symbol "PCYC." The following table sets forth the range of high and low closing prices for our common stock for the periods indicated.
41
HIGH | LOW | |||||||
FISCAL YEAR ENDED December 31, 2014 | ||||||||
First Quarter | $ | 151.61 | $ | 100.22 | ||||
Second Quarter | 107.42 | 85.85 | ||||||
Third Quarter | 127.81 | 89.58 | ||||||
Fourth Quarter | 141.55 | 103.79 | ||||||
FISCAL YEAR ENDED December 31, 2013 | ||||||||
First Quarter | $ | 94.20 | $ | 61.43 | ||||
Second Quarter | 91.64 | 72.51 | ||||||
Third Quarter | 138.26 | 84.50 | ||||||
Fourth Quarter | 139.89 | 99.28 | ||||||
As of February 12, 2015, there were 70 holders of record of our common stock. We have not paid cash dividends on our common stock since our inception and we expect to retain earnings primarily for use in the operation and expansion of our business, and therefore, do not anticipate paying any cash dividends in the foreseeable future.
Performance Graph (1)
The following graph compares our total stockholder returns for the past 5 years to two indices: the Nasdaq Composite Index and the Nasdaq Biotechnology Index. The total return for each index assumes the reinvestment of all dividends, if any, paid by companies included in these indices and are calculated as of December 31 of each year.
The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder return.
Comparison of Cumulative Total Return on Investment for the Past 5 Years (2)
(1) | This section is not "soliciting material," is not deemed "filed" with the SEC and is not to be incorporated by reference in any of our filings under the Securities Act or the Exchange Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
42
(2) | Shows the cumulative return on investment assuming an investment of $100 in our common stock, the Nasdaq Composite Index and the Nasdaq Biotechnology Index at December 31, 2009. |
Sales of Unregistered Securities
Not Applicable.
Stock Repurchases during the three months ended December 31, 2014
Not Applicable.
Securities Authorized for Issuance Under Equity Compensation Plans
See Item 12, "Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters" for information with respect to our compensation plans under which equity securities are authorized for issuance.
Item 6. Selected Financial Data
On November 14, 2012, the Board of Directors approved a change in the fiscal year end from June 30 to December 31, effective December 31, 2012. All references to "fiscal years", unless otherwise noted, refer to the twelve-month fiscal year, which prior to July 1, 2012, ended on June 30, and beginning on January 1, 2013, ends on December 31, of each year.
The following table sets forth selected historical financial data for the years ended December 31, 2014 and December 31, 2013, the six-month transition period ended December 31, 2012 and the previous three fiscal years. The financial data presented below is derived from our audited consolidated financial statements and should be read in conjunction with Item 7, "Management’s Discussion and Analysis of Financial Condition and Results of Operations" and the consolidated financial statements and related notes included elsewhere herein.
43
Selected Consolidated Financial Data | ||||||||||||||||||||||||
(in thousands, except share and per share data) | ||||||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||||||
Years Ended December 31, | Six Months Ended December 31, | Years Ended June 30, | ||||||||||||||||||||||
2014 | 2013 | 2012 | 2012 | 2011 | 2010 | |||||||||||||||||||
Revenue: | ||||||||||||||||||||||||
Product revenue, net | $ | 492,418 | $ | 13,573 | $ | — | $ | — | $ | — | $ | — | ||||||||||||
License and milestone revenue (1) | 220,000 | 235,000 | 155,000 | 77,605 | 4,355 | 6,645 | ||||||||||||||||||
Collaboration services revenue (1) | 17,311 | 11,596 | 5,658 | 4,385 | 3,878 | 2,662 | ||||||||||||||||||
Total revenue | 729,729 | 260,169 | 160,658 | 81,990 | 8,233 | 9,307 | ||||||||||||||||||
Costs and expenses (2): | ||||||||||||||||||||||||
Cost of goods sold | 40,410 | 3,501 | — | — | — | — | ||||||||||||||||||
Research and development | 172,459 | 170,865 | 46,639 | 54,537 | 34,482 | 17,358 | ||||||||||||||||||
Less: Excess Amounts related to Research and development (3) | — | (85,732 | ) | (17,306 | ) | — | — | — | ||||||||||||||||
Research and development, net | 172,459 | 85,133 | 29,333 | 54,537 | 34,482 | 17,358 | ||||||||||||||||||
Selling, general and administrative | 167,955 | 104,016 | 12,093 | 15,575 | 9,125 | 7,561 | ||||||||||||||||||
Less: Excess Amounts related to Selling, general and administrative (3) | — | (30,405 | ) | (819 | ) | — | — | — | ||||||||||||||||
Selling, general and administrative, net | 167,955 | 73,611 | 11,274 | 15,575 | 9,125 | 7,561 | ||||||||||||||||||
Costs of collaboration | 226,004 | — | — | — | — | |||||||||||||||||||
Amortization of intangible assets | 520 | — | — | — | — | — | ||||||||||||||||||
Total costs and expenses | 607,348 | 162,245 | 40,607 | 70,112 | 43,607 | 24,919 | ||||||||||||||||||
Income (loss) from operations | 122,381 | 97,924 | 120,051 | 11,878 | (35,374 | ) | (15,612 | ) | ||||||||||||||||
Interest income | 440 | 341 | 137 | 178 | 169 | 81 | ||||||||||||||||||
Other income (expense), net | (374 | ) | (129 | ) | (8 | ) | (31 | ) | 2 | (43 | ) | |||||||||||||
Income (loss) before income taxes | 122,447 | 98,136 | 120,180 | 12,025 | (35,203 | ) | (15,574 | ) | ||||||||||||||||
Income tax provision (benefit) | 36,323 | 31,126 | 2,647 | 39 | — | (550 | ) | |||||||||||||||||
Net income (loss) | $ | 86,124 | $ | 67,010 | $ | 117,533 | $ | 11,986 | $ | (35,203 | ) | $ | (15,024 | ) | ||||||||||
Net income (loss) per share (4): | ||||||||||||||||||||||||
Basic | $ | 1.14 | $ | 0.92 | $ | 1.69 | $ | 0.17 | $ | (0.59 | ) | $ | (0.31 | ) | ||||||||||
Diluted | $ | 1.10 | $ | 0.87 | $ | 1.58 | $ | 0.17 | $ | (0.59 | ) | $ | (0.31 | ) | ||||||||||
Weighted average shares used to compute net income (loss) per share: | ||||||||||||||||||||||||
Basic | 75,247 | 72,777 | 69,676 | 68,728 | 59,973 | 48,344 | ||||||||||||||||||
Diluted | 78,151 | 77,083 | 74,408 | 72,617 | 59,973 | 48,344 | ||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||||||
As of December 31, | As of June 30, | |||||||||||||||||||||||
2014 | 2013 | 2012 | 2012 | 2011 | 2010 | |||||||||||||||||||
Cash, cash equivalents and marketable securities | $ | 856,987 | $ | 635,628 | $ | 317,144 | $ | 203,607 | $ | 112,329 | $ | 74,149 | ||||||||||||
Total assets | 1,060,107 | 768,751 | 355,129 | 219,120 | 116,352 | 76,820 | ||||||||||||||||||
Deferred revenue | 53,658 | 59,606 | 70,701 | 75,378 | 7,000 | 6,099 | ||||||||||||||||||
Total liabilities | 230,942 | 141,128 | 92,603 | 86,997 | 14,678 | 10,059 | ||||||||||||||||||
Accumulated deficit | (130,472 | ) | (216,596 | ) | (283,606 | ) | (401,139 | ) | (413,125 | ) | (377,922 | ) | ||||||||||||
Total stockholders' equity | 829,165 | 627,623 | 262,526 | 132,123 | 101,674 | 66,761 | ||||||||||||||||||
44
(1) See Note 5 to the consolidated financial statements for a discussion of our collaboration and license agreements. | |||
(2) See Note 9 to the consolidated financial statements for a description of stock-based compensation included in costs and expenses for the years ended December 31, 2014 and December 31, 2013, the six months ended December 31, 2012 and for the fiscal year ended June 30, 2012. | |||
(3) See Note 5 to the consolidated financial statements for a description of Excess Amounts which are included as a reduction to costs and expenses for the year ended December 31, 2013 and the six months ended December 31, 2012. | |||
(4) See Note 13 to the consolidated financial statements for a description of the computation of basic and diluted net income (loss) per share. | |||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
In addition to historical information, this report contains predictions, estimates, assumptions and other forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended and Section 21E of the Securities Exchange Act of 1934, as amended. Actual results could differ materially from any future performance suggested in this report as a result of the risks, uncertainties and other factors described herein and elsewhere in this report, including those discussed in “Risk Factors.” The following discussion should be read in conjunction with the consolidated financial statements and notes thereto included elsewhere in this Annual Report on Form 10-K and with Part I, Item 1A, "Risk Factors."
Company Overview
We are a fully integrated biopharmaceutical company focused on developing and commercializing novel therapies for the treatment of cancer and immune-mediated diseases. We are currently an approximately 630-person company with in-house research and development (R&D), commercial and third-party contracted manufacturing capabilities and a growing U.S. footprint and global presence. Our goal is to make available therapies intended to improve quality of life, increase duration of life, and resolve serious unmet medical needs for patients. We will do this by identifying and controlling promising product candidates based on our scientific development and administrational expertise, developing our products in a rapid, cost-efficient manner, and by pursuing commercialization and/or development partners when and where appropriate. To that end, Pharmacyclics is at the forefront at transforming the speed by which innovative, high-quality medicines can advance from bench to bedside. Our first commercial product, IMBRUVICA® (ibrutinib), was developed and commercialized in 4.5 years from the start of its first clinical trial in 2009.
IMBRUVICA is a first-in-class, oral, once-daily, single-agent therapy which has demonstrated a survival advantage over an approved, standard-of-care therapy in a difficult-to-treat blood cancer. IMBRUVICA inhibits a protein called Bruton's tyrosine kinase (BTK), a key signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B-cells. IMBRUVICA blocks signals that tell malignant B-cells to multiply and spread uncontrollably.
We market IMBRUVICA in the United States (U.S.) for our four FDA-approved indications for the treatment of patients with: chronic lymphocytic leukemia (CLL) who have received at least one prior therapy; all lines of CLL with deletion of the short arm of chromosome 17 (del 17p CLL); mantle cell lymphoma (MCL) who have received at least one prior therapy; and all lines of Waldenström's macroglobulinemia (WM).
Accelerated approval was granted for the MCL indication based on overall response rate (ORR). Improvements in survival or disease symptoms have not been established. Continued approval for the MCL indication may be contingent upon verification of clinical benefit in confirmatory trials. IMBRUVICA is the only medicine approved to treat patients with del 17p CLL and WM.
IMBRUVICA was one of the first medicines to receive FDA approval via the new Breakthrough Therapy Designation pathway, and is the only product to have received three Breakthrough Therapy Designations. In the U.S., IMBRUVICA received its first four FDA approvals in a period of less than fifteen months, ranging from November 2013 through January 2015, echoing the same speed by which the product was developed (see Item 1 - IMBRUVICA Regulatory Updates). IMBRUVICA currently is approved for use in approximately 40 countries including the U.S., Canada, and the 28 countries which comprise the European Union (EU).
We believe that IMBRUVICA is helping to transform the management of blood cancers, and we are encouraged by the high clinical adoption rate we have seen in our approved indications. In fact, IMBRUVICA currently occupies the position of one of the most successful oncology drug launches in history based on launch revenue trajectories.
45
In commercial use and in the clinical trial setting, IMBRUVICA has demonstrated -- and continues to demonstrate -- a favorable efficacy, safety, toxicity, and durability of response profile. To date, over 5,600 patients have been treated in Company-sponsored IMBRUVICA trials conducted in over 35 countries involving more than 800 investigators. We are continuing to investigate how IMBRUVICA may benefit a broader group of patients in the future. Our development program has several clinical trials underway studying IMBRUVICA alone and in combination with other therapies in several blood cancers including in CLL, which is a clinical development program that currently includes seven Phase III trials and covers all lines of therapy and various combinations of treatments. Additional blood cancers we are investigating in which we believe IMBRUVICA may have a benefit include: small lymphocytic leukemia (SLL), MCL, WM, diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), multiple myeloma (MM), and other forms of cancer. A clinical trial is also underway studying IMBRUVICA in Graft versus host disease (GvHD). In addition, we are beginning the exploration of IMBRUVICA in select solid tumor types. As of December 31, 2014, 13 Phase III trials have been initiated with IMBRUVICA and approximately 58 trials are registered on www.clinicaltrials.gov. Information found on this website is not incorporated by reference into this report.
We are conducting this research together with our partner Janssen Biotech Inc. and its affiliates (Janssen), one of the Janssen Pharmaceutical companies of Johnson & Johnson, under our 2011 worldwide collaboration and license agreement (the Agreement). However, we recently have formed a number of strategic collaborations with other world-class companies including Amgen Inc., AstraZeneca, Bristol-Myers Squibb Co., Celgene Corp., and F. Hoffmann-La Roche Ltd. (Roche) in order to explore the potential of IMBRUVICA as a combination agent and a backbone of therapy for certain blood cancers and solid tumors.
Under the Agreement, we and our partner Janssen are jointly commercializing IMBRUVICA in the U.S. Janssen is commercializing IMBRUVICA outside the U.S.
In addition to IMBRUVICA, we have other product candidates in clinical development and several pre-clinical molecules in lead optimization. We will continue to pioneer research into the development of next-generation BTK inhibitors. Our pre-clinical molecules include BTK inhibitors for autoimmune disorders, such as rheumatoid arthritis. We remain committed to high standards of ethics, scientific rigor, and operational efficiency throughout our development efforts.
Change in Fiscal Year End
On November 14, 2012, the Board of Directors approved a change in the fiscal year end from June 30 to December 31, effective December 31, 2012. All references to "fiscal years", unless otherwise noted, refer to the twelve-month fiscal year, which prior to July 1, 2012, ended on June 30, and beginning on January 1, 2013, ends on December 31, of each year.
Critical Accounting Policies, Estimates and Judgments
This discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses, and related disclosures. On an on-going basis, we evaluate our estimates, including those related to revenue recognition and income taxes. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results, however, may differ significantly from these estimates under different assumptions or conditions and may adversely affect the financial statements.
We believe the following critical accounting policies reflect the more significant judgments and estimates used in the preparation of our consolidated financial statements and accompanying notes.
Revenue Recognition
Product revenue, net is recognized in accordance with the Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 605, Revenue Recognition, when the following criteria have been met: (1) persuasive evidence of an arrangement exists, (2) delivery has occurred or services have been rendered, (3) the price is fixed or determinable and (4) collectability is reasonably assured. Revenues are deferred for fees received before earned or until no further obligations exist. We exercise judgment in determining that collectability is reasonably assured or that services have been delivered in accordance with the arrangement. We assess whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the sales price is subject to refund or adjustment. We assess collectability based primarily on the customer's payment history and on the creditworthiness of the customer.
46
Product Revenue, Net
Product revenue, net consists of U.S. sales of IMBRUVICA and is recognized once all four revenue recognition criteria described above have been met. We sell IMBRUVICA directly to some customers who have in-house dispensing capabilities, specialty pharmacies (SP) that sell to individual patients, specialty distributors (SD) that sell to hospital pharmacies and other organizations that we have contracted with. We recognize revenue from sales of IMBRUVICA when the product’s title and risk of loss transfers to the customer. We determined that we have the ability to make reasonable estimates of product returns in order to recognize revenue at the time that title and risk of loss transfers to the customer based on the following factors: (1) we believe that we have sufficient insight into the distribution channel at the SP's and SD's in order to ascertain their inventory level and dispense data, (2) due to the price of our product and limited patient population, our SP and SD customers have not built up significant levels of inventory, nor do we expect they will do so for the foreseeable future, (3) inventory on hand at our SP customers was approximately two weeks as of December 31, 2014, (4) there have been no product returns to-date since our commercial launch of IMBRUVICA on November 13, 2013 and (5) we believe there is limited risk of return of inventory in the channel because there is significant remaining shelf-life at the point of sale.
We recognize product revenue net of adjustments for customer credits, including estimated government rebates and charge-backs, returns, prompt payment discounts, U.S. Department of Veteran's Affairs (VA) negotiated discounts and administrative service fees related to our patient assistance program which includes co-payment and deductible assistance, and Medicare Part D coverage gap reimbursements. Each of the above adjustments is recorded at the time of revenue recognition, resulting in a reduction in product revenue, net and an increase in accrued expenses or a reduction in accounts receivable, net. The above adjustments require significant estimates, judgment and information obtained from external sources. If management's estimates differ from actual results, we will record adjustments that would affect product revenue, net in the period of adjustment.
The following table summarizes the provisions, and credits/payments, for these adjustments (in thousands):
Total | ||||
Balance as of December 31, 2012 | $ | — | ||
Provision related to current period sales | 1,395 | |||
Credits/payments | (189 | ) | ||
Balance as of December 31, 2013 | 1,206 | |||
Provision related to current period sales | 60,725 | |||
Credits/payments | (35,196 | ) | ||
Balance as of December 31, 2014 | $ | 26,735 | ||
Gross-to-net sales adjustments
Rebates
We record an allowance for rebates including mandated discounts under the Medicaid Drug Rebate Program, discounts provided under the TRICARE Retail Pharmacy Refunds Program (TRICARE) and to members of organizations with whom we have contracted with. The allowance for rebates is based upon our contractual agreements and/or legal requirements and public sector benefit providers, including Medicaid and TRICARE. For estimated amounts owed to public sector benefit providers, including Medicaid and TRICARE, the allowance for rebates is based on the estimated rebate percentage of forecasted eligible sales. The estimated rebate percentage is based on statutory discount rates and expected utilization. The forecasted eligible Medicaid and TRICARE sales represent those sales made by us that will ultimately be consumed by patients covered by Medicaid and TRICARE. To estimate the allowance for rebates, we use the estimated patient mix information which is provided by our SP customers, as well as third-party sources. For organizations that we have contracted with, the rebate is based upon contracted volume discount amounts. In addition, we incur administrative fees in exchange for administrative services provided that are also accrued at the time of sale. Rebates for public sector benefit providers and organizational discounts are generally invoiced and paid in arrears. As such, the allowance for rebates consists of an estimate of the amount expected to be incurred for the current quarter's shipments to patients, plus an accrual balance for estimated unpaid rebates from prior periods. The allowance for rebates is recorded within accrued liabilities in the consolidated balance sheets.
Charge-backs
Charge-backs are discounts that result from the difference between the prices at which we make IMBRUVICA available to wholesalers for purchase by discount customers under pricing agreements we have with the discount customers and the sales price paid to us by the wholesalers who service the discount customers. Such discount customers, which primarily consist of
47
the U.S. Department of Defense (DOD), VA, Public Health Services (PHS), and other Federal Government institutions, purchase products through wholesalers at a lower price provided for in pricing contracts and the wholesalers then charge us the difference between the wholesale acquisition cost and the lower price paid by the discount customer. These reductions are settled through charge-backs from our wholesalers. Charge-backs are recorded as a reduction to accounts receivable, net in the consolidated balance sheets.
Product Returns
Consistent with industry practice, we generally offer customers a limited right to return. We generally allow for the return of product that is a few months prior to and up to a few months after the product expiration date. Additionally, we consider several other factors in the estimation process including the expiration dates of product shipped, third-party data in monitoring channel inventory levels, shelf life of the product, prescription trends and other relevant factors. Provisions for estimated product returns are recorded within accrued liabilities in the consolidated balance sheets.
Medicare Part D coverage gap
Medicare Part D, also known as the Medicare prescription drug benefit, is a federal program to subsidize the costs of prescription drugs for Medicare beneficiaries in the United States. The Medicare Part D prescription drug benefit mandates that drug manufacturers fund 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Funding of the Medicare Part D gap is invoiced and paid in arrears. As such, the allowance for Medicare Part D consists of an estimate of the amount expected to be incurred for the current quarter's shipments to patients, plus an accrual balance for estimated shipments remaining in the channel at period end which are estimated to ship to Medicare Part D patients. The allowance for rebates is recorded within accrued liabilities in the consolidated balance sheets.
Prompt payment discounts
We generally offer cash discounts to our customers, generally a 2% discount applied to the invoice amount, as an incentive for prompt payment. We expect that all of our customers to whom we offer cash discounts for prompt payment to take advantage of the full amount of the 2% discount. We record the prompt-payment discount as a reduction to accounts receivable, net in the consolidated balance sheets.
Co-payment assistance
Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. We accrue for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators. The allowance for co-payment assistance is recorded within accrued liabilities in the consolidated balance sheets.
Collaboration Revenues
Revenue under our license and collaboration arrangements is recognized based on the performance requirements of the contract. Determinations of whether persuasive evidence of an arrangement exists and whether delivery has occurred or services have been rendered are based on management’s judgments regarding the fixed nature of the fees charged for deliverables and the collectability of those fees. Should changes in conditions cause management to determine that these criteria are not met for any new or modified transactions, revenue recognized could be adversely affected.
We recognize revenue related to collaboration and license arrangements in accordance with the provisions of Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 605-25, “Revenue Recognition – Multiple-Element Arrangements,” or ASC Topic 605-25. Additionally, we adopted, effective July 1, 2010, Accounting Standards Update, or ASU, No. 2009-13, “Multiple Deliverable Revenue Arrangements,” or ASU 2009-13, which amended ASC Topic 605-25 and:
• | provided guidance on how deliverables in an arrangement should be separated and how the arrangement consideration should be allocated to the separate units of accounting; |
• | required an entity to determine the selling price of a separate deliverable using a hierarchy of (i) vendor-specific objective evidence, or VSOE, (ii) third-party evidence, or TPE, or (iii) best estimate of selling price, or BESP; and |
• | required the allocation of the arrangement consideration, at the inception of the arrangement, to the separate units of accounting based on relative fair value. |
We evaluate all deliverables within an arrangement to determine whether or not they provide value on a stand-alone basis. Based on this evaluation, the deliverables are separated into units of accounting. The arrangement consideration that is fixed or
48
determinable at the inception of the arrangement is allocated to the separate units of accounting based on their relative selling prices. We may exercise significant judgment in determining whether a deliverable is a separate unit of accounting, as well as in estimating the selling prices of such unit of accounting.
To determine the selling price of a separate deliverable, we use the hierarchy as prescribed in ASC Topic 605-25 based on VSOE, TPE or BESP. VSOE is based on the price charged when the element is sold separately and is the price actually charged for that deliverable. TPE is determined based on third-party evidence for a similar deliverable when sold separately and BESP is the price at which we would transact a sale if the elements of collaboration and license arrangements were sold on a stand-alone basis. We may not be able to establish VSOE or TPE for the deliverables within collaboration and license arrangements, as we do not have a history of entering into such arrangements or selling the individual deliverables within such arrangements separately. In addition, there may be significant differentiation in these arrangements, which indicates that comparable third-party pricing may not be available. We may determine that the selling price for the deliverables within collaboration and license arrangements should be determined using BESP. The process for determining BESP involves significant judgment on our part and includes consideration of multiple factors such as estimated direct expenses and other costs, and available data.
For collaborations entered into after July 1, 2010, we have determined its best estimate of selling prices for the license unit of accounting based on the income approach as defined in ASC 820-10-35-32. This measurement is based on the value indicated by current estimates about those future amounts and reflects management determined estimates and assumptions. These estimates and assumptions include, but are not limited to, how a market participant would use the license, estimated market opportunity and expected market share and assumed royalty rates that would be paid for sales resulting from products developed using the license, similar arrangements entered into by third parties and entity-specific factors such as the terms of our previous collaborative agreement, our pricing practices and pricing objectives, the likelihood that clinical trials will be successful, the likelihood that regulatory approval will be received and that the products will become commercialized and the markets served. We have also determined BESP for services-related deliverables based on the nature of the services to be performed and estimates of the associated effort as well as estimated market rates for similar services.
For each unit of accounting identified within an arrangement, we determine the period over which the performance obligation occurs. Revenue is then recognized using either a proportional performance or straight-line method. We recognize revenue using the proportional performance method when the level of effort to complete our performance obligations under an arrangement can be reasonably estimated. Direct labor hours or full time equivalents are typically used as the measurement of performance.
Effective July 1, 2010, we adopted ASU No. 2010-17, “Milestone Method of Revenue Recognition,” or ASU 2010-17, which provides guidance on revenue recognition using the milestone method. Under the milestone method, a payment that is contingent upon the achievement of a substantive milestone is recognized in the period in which the milestone is achieved. The determination that a milestone is substantive is subject to considerable judgment.
Inventory valuation and related reserves
Inventories are stated at the lower of cost or market value with the approximate cost being determined on a first-in, first-out basis. Costs capitalized as inventories include third-party manufacturing costs and labor costs for personnel involved in the manufacturing process.
During the year ended December 31, 2013, we began capitalizing costs associated with manufacturing inventory as a result of the FDA approval for IMBRUVICA. Until September 30, 2013, all inventory costs incurred were recorded as R&D expense in the consolidated statements of operations.
Products that have been approved by the FDA or other regulatory agencies, such as IMBRUVICA, are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of IMBRUVICA (i.e., raw materials and API) utilized for both commercial and clinical programs is identical and, as a result, the inventory has an "alternative future use" as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to R&D expense when the product enters the R&D process and can no longer be used for commercial purposes and, therefore, does not have "alternative future use."
We evaluate inventory levels quarterly to determine if the inventory has a cost basis in excess of its expected net realizable value, inventory has expired, inventory on-hand is in excess of expected sales forecasts, and/or inventory that does not meet its quality specifications. Our raw materials and API has no expiration date and can be revalidated for use after testing. Our finished goods inventory currently has an estimated life of 24 months and, based on our sales forecasts, we expect to realize the carrying value of the IMBRUVICA inventory. If any impairment of inventory is identified, the related inventory is written
49
down with a corresponding charge to cost of goods sold in the consolidated statements of operations in the period that the inventory impairment is first identified.
Research and Development, net
R&D expenses include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. R&D costs are expensed as incurred. Nonrefundable advance payments for goods or services that will be used or rendered for future R&D activities are deferred and amortized over the period that the goods are delivered or the related services are performed, subject to an assessment of recoverability.
Clinical development costs are a significant component of R&D expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the ongoing development of our product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow. We accrue and expense costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations and clinical trial sites. We determine our estimates through discussions with internal clinical personnel and outside service providers as to the progress or stage of completion of trials or services and the agreed upon fee to be paid for such services.
Stock-based Compensation
Stock-based compensation cost for employee and director stock options with time-based vesting and employee stock plan purchase rights is generally measured and recognized based on the fair value of the award at the grant date. Stock-based compensation cost for restricted stock units (RSUs) with time-based vesting is measured based on the closing fair market value of our common stock on the date of the grant, multiplied by the number of RSUs granted. The accounting grant date for employee stock options and restricted stock units with performance obligations is the date on which the performance goals have been defined and a mutual understanding of the terms has been reached. Generally, our time-based stock option and RSU grants vest over a four year period. Stock options and RSUs with performance obligations generally vest over a four year period, with the goals set and agreed upon annually. Stock-based compensation for non-employee stock options is re-estimated at each period-end through the vesting date. We estimate a forfeiture rate to calculate the stock-based compensation cost for its awards primarily based on an analysis of its historical pre-vesting forfeitures.
The fair value of each stock option is estimated using the Black Scholes option-pricing model. Expected volatility is based on historical volatility data of our stock. The expected term of stock options granted represents the period of time that stock options are expected to be outstanding. We generally do not expect substantially different exercise or post-vesting termination behavior among our employee or non-employee population. As such, for the majority of stock options granted and our Employee Stock Purchase Plan, we generally calculate and apply an overall expected term assumption based on historical data. In certain cases, we use a shorter expected term for performance-based stock options based on a combination of historical data and management's estimates of the period of time that options will be outstanding. The risk-free interest rate is based on a zero-coupon U.S. Treasury bond whose maturity period equals the expected term of our options.
Options and RSUs vest upon the passage of time or a combination of time and the achievement of certain performance obligations. Vesting of performance-based options and RSUs for executive officers depends on their attainment of key corporate and departmental goals. The Compensation Committee of the Board of Directors will determine if the performance conditions have been met. Stock-based compensation expense for the options and RSUs with performance obligations is recorded when we believe that the vesting of these options and RSUs is probable (see Note 9 to the consolidated financial statements).
Income Taxes
We are subject to income taxes in both the U.S. and foreign jurisdictions, and we use estimates in determining our provisions for income taxes. We provide for income taxes using the asset and liability method, whereby deferred tax assets or liability account balances are calculated at the balance sheet date using current tax laws and rates in effect for the year in which the differences are expected to affect taxable income.
Recognition of deferred tax assets is appropriate when realization of such assets is more likely than not. We recognize a valuation allowance against our net deferred tax assets if it is more likely than not that some portion of the deferred tax assets will not be fully realizable. This assessment requires judgment as to the likelihood and amounts of future taxable income by tax jurisdiction.
50
We apply the provisions of FASB's guidance on accounting for uncertainty in income taxes. We assess all material positions taken in any income tax return, including all significant uncertain positions, in all tax years that are still subject to assessment or challenge by relevant taxing authorities. Assessing an uncertain tax position begins with the initial determination of the position’s sustainability and the tax benefit to be recognized is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. As of each balance sheet date, unresolved uncertain tax positions must be reassessed, and we will determine whether (i) the factors underlying the sustainability assertion have changed and (ii) the amount of the recognized tax benefit is still appropriate. The recognition and measurement of tax benefits requires significant judgment. Judgments concerning the recognition and measurement of a tax benefit might change as new information becomes available.
Recent Accounting Pronouncements
See Note 2, Significant Accounting Policies, in Notes to the Consolidated Financial Statements in Item 8 of Part II of this Annual Report on Form 10-K, for a full description of recent accounting pronouncements, including the expected dates of adoption and estimated effects on financial condition and results of operations, which is incorporated herein by reference.
Financial Data for the Year Ended December 31, 2014 and 2013:
Total Revenue (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Product revenue, net | $ | 492,418 | $ | 13,573 | $ | 478,845 | ||||||
License and milestone revenue | 220,000 | 235,000 | (15,000 | ) | ||||||||
Collaboration services revenue | 17,311 | 11,596 | 5,715 | |||||||||
Total revenue | $ | 729,729 | $ | 260,169 | $ | 469,560 | ||||||
Total revenue increased $469.6 million during the year ended December 31, 2014 compared to the year ended December 31, 2013 primarily due to a $478.8 million increase in product revenue, net from sales of IMBRUVICA, and a $5.7 million increase in collaboration services revenue primarily related to the Agreement, partially offset by a $15.0 million decrease in milestone revenue earned under the Agreement.
Product revenue, net
Product revenue, net consists of revenue recorded on sales of IMBRUVICA. During the year ended December 31, 2014, IMBRUVICA was marketed in the U.S. for the treatment of patients with: CLL who have received at least one prior therapy; all lines of del 17p CLL; and MCL who have received at least one prior therapy.
During the year ended December 31, 2013, IMBRUVICA was approved by the FDA and marketed in the U.S. for the treatment of patients with MCL who have received at least one prior therapy. IMBRUVICA first came to market on November 13, 2013 when it was approved by the FDA on November 13, 2013 for this indication. The year ended December 31, 2014 represented our first full year of IMBRUVICA sales.
We recognize revenue from sale of IMBRUVICA when the product’s title and risk of loss transfers to the customer. We derive product revenue, net based on net sales to our customers, less estimated government rebates, charge-backs, returns reserve and prompt payment discounts.
Product revenue, net was calculated as follows (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Product revenue, gross | $ | 553,143 | $ | 14,968 | $ | 538,175 | ||||||
Government rebates, return reserve and other | (34,502 | ) | (1,024 | ) | (33,478 | ) | ||||||
Chargebacks and cash discounts | (26,223 | ) | (371 | ) | (25,852 | ) | ||||||
Product revenue, net | $ | 492,418 | $ | 13,573 | $ | 478,845 | ||||||
License and milestone revenue
51
License and milestone revenue of $220.0 million for the year ended December 31, 2014 was comprised of the following milestone payments that were earned under the Agreement:
• | Two approval milestones of $60.0 million and $30.0 million, respectively, were earned upon the FDA approval of IMBRUVICA as a single agent for the treatment of patients with CLL who have received at least one prior therapy and for del 17p CLL patients. |
• | One regulatory milestone of $30.0 million earned upon the FDA's acceptance of our new drug application filed for IMBRUVICA for the treatment of patients with del 17p CLL. |
• | Two approval milestones totaling $80.0 million were earned upon the EU approvals of IMBRUVICA for the treatment of adult patients with relapsed or refractory MCL, or adult patients with CLL who have received at least one prior therapy, or in first line in the presence of 17p deletion or TP53 mutation in patients unsuitable for chemoimmunotherapy. |
• | One regulatory milestone of $20.0 million was earned upon the European Medicines Agency (EMA) acceptance of a Type II variation application of IMBRUVICA for the treatment of adult patients with WM. |
In comparison, for the year ended December 31, 2013, license and milestone revenue included $235.0 million in connection with our achievement of four regulatory, one development, and one approval milestones earned during the period under the Agreement.
Collaboration services revenue
Collaboration services revenue for the years ended December 31, 2014 included revenue related to commercial and development services under the Agreement and net income from the sale of our product to Janssen. For the year ended December 2013, collaboration services revenue included revenue related to commercial and development services under the Agreement.
For the year ended December 31, 2014, collaboration services revenue increased by $5.7 million compared to the year ended December 31, 2013 primarily related to sales of our product to Janssen. For the year ended December 31, 2014, total collaboration services revenue from Janssen of $17.2 million was comprised of $12.1 million of amortization of development services related to the Agreement, $0.9 million of amortization of committee services related to the Agreement and $4.2 million of net income from the sale of the Company's product to Janssen. For the year ended December 31, 2013, total collaboration services revenue related to the Agreement was $11.0 million which was comprised of $10.1 million of amortization of development services and $0.9 million of amortization of committee services. The increase in collaboration services revenue recognized under the Agreement was due to an increase in development efforts under the collaboration with Janssen.
In December 2011, we entered into the Agreement which provided for a $150.0 million non-refundable upfront payment upon execution (see Note 5 to the consolidated financial statements). The revenue related to the upfront payment was allocated $70.6 million to the licenses, $15.0 million to the committee services and $64.4 million to the development services. Since inception, the $15.0 million and $64.4 million allocated to committee and development services, respectively, is being recognized as revenue as the related services are provided over the estimated service periods of 17 years and 9 years, which are equivalent to the estimated remaining life of the underlying technology and the estimated remaining development period, respectively.
As of December 31, 2014, approximately $46.5 million was included in deferred revenue related to the committee and development services, of which $34.9 million was included in deferred revenue non-current.
Cost of goods sold (in thousands):
Years Ended December 31, | |||||||||||||
2014 | 2013 | Change | |||||||||||
Cost of goods sold | $ | 40,410 | $ | 3,501 | $ | 36,909 | |||||||
Cost of goods sold includes third-party manufacturing cost of products sold, fixed manufacturing overhead, royalty fees, and other indirect costs such as employee compensation. Cost of goods sold increased $36.9 million during the year ended December 31, 2014 compared to the year ended December 31, 2013 primarily due to a $26.5 million increase in royalty fees incurred under the Celera agreement (see Note 5 to the consolidated financial statements), a $7.0 million increase in the cost of manufacturing primarily as a result of 2014 being the first full year of IMBRUVICA sales and a $3.4 million increase in facility fee.
52
We began capitalizing inventory during the year ended December 31, 2013 in connection with the FDA’s approval of IMBRUVICA, as the related costs were expected to be recoverable through the commercialization of the product. As of December 31, 2013, inventory related costs of $16.1 million incurred prior to FDA approval were recorded as R&D expenses in our statements of operations, of which $1.6 million is remaining on hand as of December 31, 2014. We expect to sell the remaining pre-commercialization inventory on hand over the next 6 to 9 months. Subsequent to the utilization of all our pre-commercialization inventory, we estimate cost of goods sold as a percentage of product revenue, net will be in the range of high single digit to low double digit percentage. The estimated cost of goods sold percentage of product revenue, net includes third-party manufacturing costs of products sold, fixed manufacturing overhead, royalty fees, and other indirect costs such as employee compensation. The range is impacted by our estimate of materials costs from our suppliers as well the level of our fixed overhead costs estimated in relation to our future sales levels.
Research and development, net (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Research and development | $ | 172,459 | $ | 170,865 | $ | 1,594 | ||||||
Less: Excess Amounts related to Research and development | — | (85,732 | ) | 85,732 | ||||||||
Research and development, net | $ | 172,459 | $ | 85,133 | $ | 87,326 | ||||||
R&D, net for the year ended December 31, 2014 increased by $87.3 million compared to the year ended December 31, 2013. The increase was primarily due to the $85.7 million of Excess Amounts recorded as a reduction to R&D expense in the year ended December 31, 2013 (see Note 5 to the consolidated financial statements). In addition, there was an increase of $15.9 million in clinical and research related expenses, an increase in employee related expenses of $9.4 million, and a $8.1 million increase in cost sharing expenses under the Agreement, partially offset by a decrease of $29.6 million in inventory related costs, which prior to FDA approval were recorded as R&D and since FDA approval on November 13, 2013 are being capitalized, and a decrease of $2.0 million in stock-based compensation expense.
R&D headcount was 331 at December 31, 2014, compared to 269 at December 31, 2013. In the near term, we expect to hire additional R&D employees, as well as incur costs under our collaboration agreements as we continue to invest in the development of our products.
Excess Amounts, when funded by Janssen, are recorded as reductions to costs and expenses. Under the Agreement, Excess Amounts would become payable only after the third profitable quarter for the collaboration from our share of pre-tax commercial profits, commencing in the fourth quarter of profitability under the Agreement until the Excess Amounts and applicable interest has been fully paid. As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements).
R&D costs are identified as either directly attributed to one of our R&D programs or as an indirect cost, with only direct costs being tracked by specific program. Direct costs consist of personnel costs directly associated with a program, preclinical study costs, clinical trial costs, and related clinical drug and manufacturing costs, drug formulation costs, contract services and other research expenditures. Indirect costs consist of personnel costs not directly associated with a program, overhead and facility costs and other support service expenses.
Direct costs by program and indirect costs were as follows (in thousands):
53
Years Ended December 31, | ||||||||||||
Program | Description | Phase of Development | 2014 | 2013 | ||||||||
BTK Inhibitors | Cancer & Autoimmune | Phase I/II/III, Pre-clinical | $ | 120,115 | $ | 120,850 | ||||||
HDAC Inhibitors | Cancer | Phase I/II | 1,738 | 1,232 | ||||||||
Factor VIIa Inhibitor | Cancer | Phase II | 42 | 410 | ||||||||
Total direct costs | 121,895 | 122,492 | ||||||||||
Indirect costs | 50,564 | 48,373 | ||||||||||
Less: Excess Amounts related to Research and development | — | (85,732 | ) | |||||||||
Research and development, net | $ | 172,459 | $ | 85,133 | ||||||||
Selling, general and administrative, net (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Selling, general and administrative | $ | 167,955 | $ | 104,016 | $ | 63,939 | ||||||
Less: Excess Amounts related to Selling, general and administrative | — | (30,405 | ) | 30,405 | ||||||||
Selling, general and administrative, net | $ | 167,955 | $ | 73,611 | $ | 94,344 | ||||||
Selling, general and administrative (SG&A), net for the year ended December 31, 2014 increased by $94.3 million compared to the year ended December 31, 2013. The increase was primarily due to a $26.8 million increase in employee and related expenses, a $15.6 million increase in donations to third-party patient assistance foundations, a $12.4 million increase in consulting and outside service expenses, a $3.8 million increase in stock-based compensation expense, a $2.9 million increase in legal expenses and a $2.5 million in medical affairs expenses. The increase in expense was also due to $30.4 million in Excess Amounts recorded as a reduction to SG&A expense in the year ended December 31, 2013 (see Note 5 to the consolidated financial statements).
SG&A headcount was 276 at December 31, 2014 compared to 215 at December 31, 2013. In the near term, we expect to hire additional SG&A employees, as well as incur costs under our collaboration agreements as we continue to invest in the commercialization of our product and build support functions.
Excess Amounts, when funded by Janssen, are recorded as reductions to costs and expenses. Under the Agreement, Excess Amounts would become payable only after the third profitable quarter for the collaboration from our share of pre-tax commercial profits, commencing in the fourth quarter of profitability under the Agreement until the Excess Amounts and applicable interest has been fully paid. As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements).
Costs of collaboration (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Costs of collaboration | $ | 226,004 | $ | — | $ | 226,004 | ||||||
Costs of collaboration is derived from Janssen's 50% share of U.S. net product revenue less cost of goods sold of IMBRUVICA. Costs of collaboration for the year ended December 31, 2014 increased by $226 million compared to the year ended December 31, 2013. The increase was primarily due to increase in our net product revenue during fiscal year 2014. We started the commercial launch of IMBRUVICA on November 13, 2013. For the year ended December 31, 2013, Janssen's 50% share of U.S. product revenue, net less cost of goods sold of IMBRUVICA of $5.0 million was fully absorbed by Excess Amounts funded by Janssen that were recorded as a reduction to SG&A, net.
54
Income tax provision (in thousands):
Years Ended December 31, | ||||||||||||
2014 | 2013 | Change | ||||||||||
Income tax provision | $ | 36,323 | $ | 31,126 | $ | 5,197 | ||||||
For the year ended December 31, 2014, we recorded an income tax provision of $36.3 million compared to an income tax provision of $31.1 million for the year ended December 31, 2013. The estimated annual effective tax rate for the year ended December 31, 2014 was 29.66%.
In December 2014, Congress passed the Tax Increase Prevention Act which included a retroactive renewal of the Federal R&D credit back to January 1, 2014, which had previously expired. The impact of this retroactive tax legislation will not affect our fiscal 2014 effective tax rate, but will reduce future income tax payments.
We have established and are expanding our international operations and staff to better support our future expansion into international markets which have relatively low statutory tax rates when compared with the U.S. statutory rate. In 2011, we formed two entities, Pharmacyclics Switzerland GmbH, a wholly-owned subsidiary of Pharmacyclics, Inc., and Pharmacyclics Cayman Ltd., a wholly-owned subsidiary of Pharmacyclics Switzerland GmbH. In 2013, we formed Pharmacyclics (Shanghai) Management Consulting Services Ltd., a wholly-owned subsidiary of Pharmacyclics Switzerland GmbH. On December 8, 2011, Pharmacyclics, Inc. transferred certain intellectual property rights to Pharmacyclics Cayman Ltd.
We intend to continue maintaining a full valuation allowance on our deferred tax assets until there is sufficient evidence to support the reversal of all or some portion of these allowances. However, should there be greater market demand for or greater market acceptance of IMBRUVICA than management has currently forecasted, there is a reasonable possibility that within the next year sufficient positive evidence may become available to reach a conclusion that a significant portion of the valuation allowance will no longer be needed. As such, we may release a significant portion of our valuation allowance against our deferred tax assets within the next 12 months. This release would result in the recognition of certain deferred tax assets and a decrease to income tax expense for the period such release is recorded.
Financial Data for the Years Ended December 31, 2013 and 2012 (unaudited)
Total Revenue (in thousands):
Years Ended December 31, | ||||||||||||
2013 | 2012 | Change | ||||||||||
(unaudited) | ||||||||||||
License and milestone revenue | $ | 235,000 | $ | 155,000 | $ | 80,000 | ||||||
Collaboration services revenue | 11,596 | 9,708 | 1,888 | |||||||||
Product revenue, net | 13,573 | — | 13,573 | |||||||||
Total revenue | $ | 260,169 | $ | 164,708 | $ | 95,461 | ||||||
Total revenue increased by $95.5 million for the year ended December 31, 2013 compared to the year ended December 31, 2012 primarily due to an $80 million increase in milestone revenue earned under the worldwide collaboration and license agreement with Janssen (Agreement), a $13.6 million increase in product revenue, net due to product sales which began during the period due to the FDA approval of IMBRUVICA on November 13, 2013 and a $1.9 million increase in collaboration services revenue primarily related to the Janssen agreement.
License and milestone revenue
License and milestone revenue of $235.0 million for the year ended December 31, 2013 was comprised of the following milestone payments that were earned under the Agreement:
• | Approval milestone of $60.0 million earned on November 13, 2013 upon the FDA approval of IMBRUVICA as a single agent for the treatment of patients with MCL who have received at least one prior therapy, |
• | Regulatory milestone of $75.0 million earned upon the FDA's acceptance for filing of the new drug application upon the FDA's acceptance for filing of the new drug application for IMBRUVICA, for previously treated MCL and previously treated CLL/small lymphocytic lymphoma (CLL/SLL), |
55
• | Regulatory milestone of $50.0 million earned upon the European Medicines Agency's confirmation that Janssen's Marketing Authorization Application (MAA) for IMBRUVICA for the treatment of adult patients with relapsed or refractory CLL/SLL and relapsed or refractory MCL was valid, and |
• | Development milestone of $50.0 million earned as a result of the enrollment of a fifth patient in our Phase III clinical trial of IMBRUVICA in a monotherapy trial using IMBRUVICA versus chlorambucil in elderly patients with newly diagnosed chronic lymphocytic leukemia/small lymphocytic leukemia (CLL/SLL). |
In comparison, for the year ended December 31, 2012, license and milestone revenue included $150.0 million in connection with our achievement of three development milestones earned during the period under the Agreement and $5.0 million of license revenue related to our license agreement with Novo Nordisk (A/S).
Collaboration services revenue
Collaboration services revenue for the years ended December 31, 2013 and 2012 consists primarily of amounts earned under the Agreement. In December 2011, we entered into the Agreement which provided for a $150.0 million non-refundable upfront payment upon execution (see Note 5 to the consolidated financial statements). The revenue related to the upfront payment was allocated $70.6 million to the licenses, $15.0 million to the committee services and $64.4 million to the development services. The $15.0 million and $64.4 million allocated to committee and development services, respectively, is being recognized as revenue as the related services are provided over the estimated service periods of 17 years and 9 years, which are equivalent to the estimated remaining life of the underlying technology and the estimated remaining development period, respectively.
For the year ended December 31, 2013, total collaboration services revenue related to the Agreement was $11.0 million which was comprised of $10.1 million of amortization of development services and $0.9 million of amortization of committee services. For the year ended December 31, 2012, total collaboration services revenue related to the Agreement was $8.7 million which was comprised of $7.8 million of amortization of development services and $0.9 million of amortization of committee services. The increase in collaboration services revenue recognized under the Agreement was due to an increase in development efforts under the collaboration with Janssen.
As of December 31, 2013, approximately $59.5 million was included in deferred revenue related to the committee and development services under the Agreement, of which $52.0 million was included in deferred revenue non-current.
Product revenue, net
Product revenue, net consists of revenue recorded on sales of IMBRUVICA. IMBRUVICA was approved by the FDA and commercially launched on November 13, 2013 for MCL. IMBRUVICA is currently marketed in the United States as a single agent for the treatment of patients with MCL and since February 12, 2014 also for the treatment of patients with CLL, who in each case have received at least one prior therapy. We recognize revenue from sale of IMBRUVICA when the product’s title and risk of loss transfers to the customer. We derive product revenue, net based on net sales to our specialty pharmacy and specialty distributor customers, less estimated government rebates, charge-backs, returns reserve and prompt payment discounts.
Product revenue, net was calculated as follows (in thousands):
Year Ended | ||||
December 31, 2013 | ||||
Product revenue, gross | $ | 14,968 | ||
Government rebates, charge-backs and other | (1,018 | ) | ||
Returns reserve and cash discounts | (377 | ) | ||
Product revenue, net | $ | 13,573 | ||
Cost of goods sold (in thousands):
Year Ended | ||||
December 31, 2013 | ||||
Cost of goods sold | $ | 3,501 | ||
56
Cost of goods sold includes third-party manufacturing costs of products sold, royalty fees, and other indirect costs such as employee compensation.
Costs incurred prior to September 30, 2013 were recorded as R&D expenses, net in the consolidated statements of operations. This zero cost inventory primarily consisted of raw materials for which the shelf life has not yet started. As a result, as of December 31, 2013, we had approximately $16.1 million of zero cost inventory on hand which had been previously charged to R&D expenses (as those costs were incurred prior to FDA approval).
Research and development, net (in thousands):
Years Ended December 31, | ||||||||||||
2013 | 2012 | Change | ||||||||||
(unaudited) | ||||||||||||
Research and development | $ | 170,865 | $ | 77,852 | $ | 93,013 | ||||||
Less: Excess Amounts related to Research and development | (85,732 | ) | (17,306 | ) | (68,426 | ) | ||||||
Research and development, net | $ | 85,133 | $ | 60,546 | $ | 24,587 | ||||||
R&D expense for the year ended December 31, 2013 increased by $24.6 million, or 41%, compared to the year ended December 31, 2012. The increase was primarily attributable to a $28.2 million increase in payroll and related expenses primarily due to an increase in headcount, a $20.2 million increase in drug manufacturing costs, a $16.5 million increase in costs related to our clinical trials, $15.6 million increase in stock-based compensation expense primarily due to the timing and accounting of grants for performance-based stock options, an increase in our stock price which resulted in a higher fair value for options granted and growth in our employee base and other increases related to an increase in expense for research activities, consulting expenses, outside services and recruiting and relocation expenses. Partially offsetting these increases in expense was a $82 million increase in the reduction to R&D expense related to the Agreement, of which $13.6 million represented an increase in Janssen's share of expenses incurred under the cost sharing arrangement and $68.4 million represented an increase in Excess Amounts. See Note 5 to the consolidated financial statements for a description of the Agreement.
Average R&D headcount was 222 for the year ended December 31, 2013, compared to 133 for the year ended December 31, 2012. In the near term, we expect to hire additional R&D employees, as well as incur costs under our collaboration agreements as we continue to invest in the development of our products.
Excess Amounts, which are recorded as a reduction to costs and expenses, reduced R&D expense by $85.7 million and $17.3 million for the years ended December 31, 2013 and 2012, respectively. In the event that we achieve a third profitable calendar quarter from sales of IMBRUVICA, which was commercially launched during the year ended December 31, 2013, or once we fully utilize the maximum Excess Amounts provided for in the Agreement, we will no longer receive Excess Amounts and our R&D expense will increase (see Note 5 to the consolidated financial statements).
R&D costs are identified as either directly attributed to one of our R&D programs or as an indirect cost, with only direct costs being tracked by specific program. Direct costs consist of personnel costs directly associated with a program, preclinical study costs, clinical trial costs, and related clinical drug and manufacturing costs, drug formulation costs, contract services and other research expenditures. Indirect costs consist of personnel costs not directly associated with a program, overhead and facility costs and other support service expenses.
The following table summarizes our principal product development initiatives, including the related stages of development for each product, the direct costs attributable to each product and total indirect costs for each respective period.
For a discussion of the risks and uncertainties associated with the timing and cost of completing a product development phase, see the Risk Factors discussed in this Annual Report.
Direct costs by program and indirect costs were as follows (in thousands):
57
Years Ended December 31, | ||||||||||||
Product | Description | Phase of Development | 2013 | 2012 | ||||||||
(unaudited) | ||||||||||||
BTK Inhibitors | Cancer/autoimmune | Phase I/II/III, Pre-clinical | $ | 120,850 | $ | 56,945 | ||||||
HDAC Inhibitors | Cancer | Phase I/II | 1,232 | 1,928 | ||||||||
Factor VIIa Inhibitor | Cancer | Phase II | 410 | 1,664 | ||||||||
Total direct costs | 122,492 | 60,537 | ||||||||||
Indirect costs | 48,373 | 17,315 | ||||||||||
Less: Excess Amounts related to Research and development | (85,732 | ) | (17,306 | ) | ||||||||
Research and development, net | $ | 85,133 | $ | 60,546 | ||||||||
Selling, general and administrative, net (in thousands):
Years Ended December 31, | ||||||||||||
2013 | 2012 | Change | ||||||||||
(unaudited) | ||||||||||||
Selling, general and administrative | $ | 104,016 | $ | 20,374 | $ | 83,642 | ||||||
Less: Excess Amounts related to Selling, general and administrative | (30,405 | ) | (819 | ) | (29,586 | ) | ||||||
Selling, general and administrative, net | $ | 73,611 | $ | 19,555 | $ | 54,056 | ||||||
SG&A expense for the year ended December 31, 2013 increased by $54.1 million, or 276%, compared to the year ended December 31, 2012. The increase was primarily attributable to a $22 million increase in stock-based compensation expense primarily due to the timing and accounting of grants for performance-based stock options, an increase in our stock price which resulted in a higher fair value for options granted and growth in our employee base, a $21.1 million increase in payroll and related expenses primarily due to the increase in headcount, a $10.4 million increase in cost sharing expenses under the Janssen arrangement, $10.3 million increase in marketing related expenses due to activities related to the launch of IMBRUVICA, a $4.1 million increase in medical affairs expenses and increases in other expenses including patent and legal costs, consulting expenses, travel related expense and recruitment fees and expenses. Partially offsetting these increases was a $29.6 million increase in Excess Amounts which were recorded as a reduction to SG&A (see Note 5 to the consolidated financial statements).
Average SG&A headcount was 133 for the year ended December 31, 2013 compared to 31 for the year ended December 31, 2012. In the near term, we expect to hire additional SG&A employees, as well as incur costs under our collaboration agreements as we continue to invest in the development of our products.
Excess Amounts, which are recorded as a reduction to costs and expenses, reduced SG&A expense by $30.4 million and $0.8 million for the years ended December 31, 2013 and 2012, respectively. In the event that we achieve a third profitable calendar quarter from sales of IMBRUVICA, which was commercially launched during the year ended December 31, 2013, or once we fully utilize the maximum Excess Amounts provided for in the Agreement, we will no longer receive Excess Amounts and our SG&A expense will increase (see Note 5 to the consolidated financial statements).
Income tax provision (benefit) (in thousands):
Years Ended December 31, | |||||||||||||
2013 | 2012 | Change | |||||||||||
(unaudited) | |||||||||||||
Income tax provision (benefit) | $ | 31,126 | $ | (2,965 | ) | $ | 34,091 | ||||||
For the year ended December 31, 2013, we recorded an income tax provision of $31.1 million, compared to an income tax benefit of $3.0 million for the year ended December 31, 2012. The estimated annual effective tax rate for the year ended December 31, 2013 was 31.72%. The income tax provision for the year ended December 31, 2013 and the income tax benefit for the year ended December 31, 2012 was calculated based upon tax provision amounts for respective years spanning multiple
58
tax return fiscal years, which for the year ended December 31, 2012 was comprised of tax provision amounts for the last six month period underlying the tax return year ended June 30, 2012 and first six months for the tax year ended June 30, 2013 and, which for the year ended December 31, 2013 was comprised of tax provision amounts for the last six months period underlying the tax return year ended June 30, 2013 and for the short tax return year ended December 31, 2013.
We have established and are expanding our international operations and staff to better support our future expansion into international markets which have relatively low statutory tax rates when compared with the U.S. statutory rate. In 2011, we formed two entities, Pharmacyclics Switzerland GmbH, a wholly-owned subsidiary of Pharmacyclics, Inc. and Pharmacyclics Cayman Ltd., a wholly-owned subsidiary of Pharmacyclics Switzerland GmbH. On December 8, 2011, Pharmacyclics, Inc. transferred certain intellectual property rights to Pharmacyclics Cayman Ltd.
Through June 30, 2013, we had a tax year end that is different from our fiscal year end and we applied for a change in tax year to align the tax return period with the fiscal year.
Liquidity and Capital Resources
Our principal sources of liquidity were cash provided by operating activities and our cash and cash equivalents and marketable securities, the majority of which were held in the U.S. Cash, cash equivalents and marketable securities at December 31, 2014 and 2013 were as follows (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Cash and cash equivalents | $ | 845,035 | $ | 623,956 | |||
Marketable securities | 11,952 | 11,672 | |||||
Total cash, cash equivalents and marketable securities | $ | 856,987 | $ | 635,628 | |||
Our primary cash inflows and outflows for the years ended December 31, 2014 and 2013 were as follows (in thousands):
Years Ended | |||||||
December 31, | |||||||
2014 | 2013 | ||||||
Net cash flow provided by (used in): | |||||||
Operating activities | $ | 183,767 | $ | 88,772 | |||
Investing activities | (23,449 | ) | (20,313 | ) | |||
Financing activities | 60,761 | 248,064 | |||||
Net increase in cash and cash equivalents | $ | 221,079 | $ | 316,523 | |||
Net cash provided by operating activities of $183.8 million for the year ended December 31, 2014 resulted primarily from our net income of $86.1 million, adjusted by $53.7 million for stock-based compensation expense, a $25.0 million decrease in receivable from collaboration partners, a $20.1 million increase in accrued liabilities primarily due to increases in accrued discounts and rebates and higher accrued royalties, a $76.4 million increase in the payable to collaboration partner primarily due to an increase in costs of collaboration from IMBRUVICA sales, an $8.0 million decrease in advances to manufacturers due to the timing of inventory receipts. These increases in cash flows provided by operating activities were partially offset by an increase in trade receivables of $53.3 million due to the increase in sales of IMBRUVICA during the year ended December 31, 2014, a $21.0 million increase in inventory to meet customer demand, and a $12.2 million increase in prepaid expenses and other assets primarily due to prepaid income taxes and prepaid contract research.
Net cash provided by operating activities of $88.8 million for the year ended December 31, 2013 resulted primarily from our net income of $67.0 million, adjusted by $50.2 million for stock-based compensation expense, a $55.8 million increase in accrued liabilities due to higher accrued payroll and related expenses due to the increase in headcount and a new bonus plan for employees, higher accrued clinical trial costs due to the expansion of our BTK program, an increase in accrued property and equipment purchases due to our capital expansion and an increase in value added taxes. These increases in cash flows provided by operating activities were partially offset by decreases of $25.3 million due to the increase in the receivable from collaboration partners which was driven by an increase in receivables from Janssen related to Excess Amounts under the Agreement, $20.3 million from the increase in advances to manufacturers, $12.4 million from the increase inventory as a result of the capitalization of inventory, $11.1 million from the decrease in deferred revenue due to higher collaboration services
59
revenue recognized under the Agreement and $11.0 million from the increase in accounts receivable, net due to product sales of IMBRUVICA.
Net cash used in investing activities of $23.4 million for the year ended December 31, 2014 consisted primarily of $13.9 million used for the purchase of property and equipment, $9.3 million used to purchase intangible assets, and $13.6 million used for purchases of marketable securities, partially offset by $13.4 million of proceeds from the maturities of marketable securities.
Net cash used in investing activities of $20.3 million for the year ended December 31, 2013 consisted primarily of $18.3 million used for the purchase of property and equipment and $14.8 million used for purchases of marketable securities, partially offset by $0.5 million of proceeds from the maturities of marketable securities.
Net cash provided by financing activities of $60.8 million for the year ended December 31, 2014 consisted primarily of $25.9 million of proceeds from the issuance of common stock under our employee stock plan and the excess tax benefit from stock-based compensation arrangements of $34.9 million.
Net cash provided by financing activities of $248.1 million for the year ended December 31, 2013 consisted primarily of net proceeds of $201.0 million from the issuance of shares in a public offering at $94.20 per share after deducting expenses of the offering, $17.0 million of proceeds from the issuance of common stock under our employee stock plans and the excess tax benefit from stock-based compensation arrangements of $30.0 million.
In December 2011, we received a $150.0 million upfront payment under the Agreement. The Agreement provided us with the potential to receive future milestone payments of up to $825.0 million. As of December 31, 2014, $605.0 million in milestone payments have been earned by us under the Agreement and we may receive up to an additional $220.0 million in development, regulatory and approval milestone payments. However, clinical development entails risks and we have no assurance as to whether or when the milestone targets might be achieved (See Note 5 to the consolidated financial statements for additional information).
We anticipate that our capital requirements will increase in 2015 as we continue to incur expenses to further develop IMBRUVICA and our other product candidates, further commercialize IMBRUVICA, and to manufacture additional commercial products.
Under the Agreement, the payment of Excess Amounts is contingent and would become payable, with interest, to Janssen from our quarterly share of pre-tax commercial profits, commencing after the third profitable quarter for the collaboration until the total Excess Amounts have been paid. As of December 31, 2014, total Excess Amounts were $138.3 million, which was comprised of the cumulative amount funded by Janssen to date of $134.3 million and interest of $4.0 million. As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements).
Based upon the current status of our product development plans, we believe that our existing cash, cash equivalents and marketable securities will be adequate to satisfy our capital needs through at least the next twelve months. Our actual capital requirements will depend on many factors, including the following:
• | our continued ability to successfully market IMBRUVICA; |
• | the amount of sales of IMBRUVICA and any other products that we may commercialize; |
• | the timing of achieving a third profitable calendar quarter for the product, under the Agreement, which would trigger the payment of Excess Amounts; |
• | the costs of obtaining clinical and commercial supplies of IMBRUVICA; |
• | progress with preclinical studies and clinical trials; |
• | the time and costs involved in obtaining regulatory approval; |
• | continued progress of our R&D programs; |
• | our ability to maintain and establish collaborative arrangements with third parties; |
• | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims; |
• | the amount and timing of capital equipment purchases; and |
• | competing technological and market developments. |
60
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially. The factors described above will impact our future capital requirements and the adequacy of our available funds. If we are required to raise additional funds, we cannot be certain that such additional funding will be available on terms favorable to us, or at all. Furthermore, any additional equity financing may be highly dilutive, or otherwise disadvantageous, to existing stockholders and debt financing, if available, may involve restrictive covenants. Collaborative arrangements, if necessary to raise additional funds, may require us to relinquish rights to certain of our technologies, products or marketing territories. Our failure to raise capital when needed and on acceptable terms, would require us to reduce our costs and expenses and would limit our ability to respond to competitive pressures or unanticipated requirements to develop our product candidates and to continue operations, any of which would have a material adverse effect on our business, financial condition and results of operations.
Contractual Obligations
The following table summarizes our primary non-cancelable contractual obligations as of December 31, 2014 (in thousands):
Payments due by period | ||||||||||||||||||||||||||||
Contractual Obligations(1)(2) | Total | 2015 | 2016 | 2017 | 2018 | 2019 | Thereafter | |||||||||||||||||||||
Operating lease obligations | $ | 11,128 | $ | 2,226 | $ | 2,161 | $ | 2,112 | $ | 857 | $ | 882 | $ | 2,890 | ||||||||||||||
Purchase commitments (3) | 85,871 | 21,046 | 7,945 | 7,643 | 7,575 | 7,575 | 34,087 | |||||||||||||||||||||
Total | $ | 96,999 | $ | 23,272 | $ | 10,106 | $ | 9,755 | $ | 8,432 | $ | 8,457 | $ | 36,977 | ||||||||||||||
(1) | Purchase commitments primarily consist of non-cancelable fees related to our long-term agreement with a contract manufacturer for the use of a dedicated drug manufacturing facility and non-cancelable orders for purchases from our contract manufacturers. |
(2) | Due to the uncertainty with respect to the timing of future cash flows associated with our unrecognized tax benefits as of December 31, 2014, we are unable to make reasonably reliable estimates of the period of cash settlement with the respective taxing authority. Therefore, $26.9 million of unrecognized tax benefits have been excluded from the contractual obligations table above (see Note 11 to the consolidated financial statements). |
(3) | Excluded from the above table are Excess Amounts under the collaboration and license agreement with Janssen. Under the Agreement, the payment of Excess Amounts is contingent and would become payable, with interest, to Janssen from our quarterly share of pre-tax commercial profits, commencing after the third profitable quarter for the collaboration until the total Excess Amounts have been paid. As of December 31, 2014, total Excess Amounts were $138.3 million, which was comprised of the cumulative amount funded by Janssen to date of $134.3 million and interest of $4.0 million. As of December 31, 2014, we have achieved two profitable quarters for the collaboration and expect the three months ending March 31, 2015 to represent the third profitable quarter for the collaboration. Accordingly, we will begin to pay Excess Amounts to Janssen from our quarterly share of pre-tax commercial profits beginning in the fourth profitable quarter for the collaboration and until Excess Amounts have been fully paid (see Note 5 to the consolidated financial statements for additional information about the Agreement). |
Off-Balance Sheet Arrangements
None.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Risk
Our exposure to interest rate risk relates primarily to our investment portfolio. The fair market value of fixed rate securities may be adversely impacted due to fluctuations in interest rates, while floating rate securities may produce less income than expected if interest rates fall. Due in part to these factors, our future investment income may fall short of expectations due to changes in interest rates or we may suffer losses in principal if forced to sell securities which have declined in market value due to changes in interest rates. The primary objective of our investment activities is to preserve principal while at the same time improving yields without significantly increasing risk. To achieve this objective, our cash and cash equivalents are primarily held in cash deposits and money market funds. As of December 31, 2014, our marketable securities consist of FDIC-insured certificates of deposit which generally had maturities of one year or less and we believe we do not have any material exposure
61
to changes in the fair value of our investment portfolio as a result of changes in interest rates. Assuming a hypothetical increase in interest rates of one percentage point, the fair value of our total investment portfolio as of December 31, 2014 and 2013, would have potentially declined by approximately $0.1 million and $0.1 million, respectively.
The table below presents the fair value of our marketable securities at December 31, 2014 and weighted-average interest rates by year of stated maturity for our investment portfolio (in thousands, except interest rates):
Matures in Fiscal Year 2015 | |||
Marketable Securities | $ | 11,952 | |
Weighted-average interest rate | 0.31 | % | |
Foreign Currency Risk
Our functional currency is the U.S. dollar. All of our revenues and the majority of our expenses are denominated in U.S. dollars and as a result, we have not experienced significant foreign currency transaction gains and losses to date. We conduct some transactions in foreign currencies, primarily related to ex-U.S. clinical trial activities, and we expect to continue to do so. We have not entered into any agreements or transactions to hedge the risk associated with potential fluctuations in currencies; accordingly, we are subject to foreign currency exchange risk related to these ex-U.S. clinical trial activities. While we may enter into hedge or other agreements in the future to actively manage this risk, we do not believe this risk is material to our financial statements.
62
Item 8. Financial Statements and Supplementary Data
INDEX TO FINANCIAL STATEMENTS
Page | |
Report of Independent Registered Public Accounting Firm | |
Consolidated Balance Sheets | |
Consolidated Statements of Operations | |
Consolidated Statements of Comprehensive Income | |
Consolidated Statements of Stockholders' Equity | |
Consolidated Statements of Cash Flows | |
Notes to Consolidated Financial Statements | |
63
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Pharmacyclics, Inc.:
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, comprehensive income, stockholders' equity and cash flows present fairly, in all material respects, the financial position of Pharmacyclics, Inc. and its subsidiaries (the "Company") at December 31, 2014 and December 31, 2013, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2014, the six month period ended December 31, 2012, and the year ended June 30, 2012 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedules listed in the accompanying index present fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements and financial statement schedules, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Annual Report on Internal Control over Financial Reporting, appearing under Item 9A(b). Our responsibility is to express opinions on these financial statements, on the financial statement schedules, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
San Jose, California
February 18, 2015
64
PHARMACYCLICS, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
December 31, 2014 | December 31, 2013 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 845,035 | $ | 623,956 | |||
Marketable securities | 11,952 | 11,672 | |||||
Accounts receivable, net | 64,297 | 11,044 | |||||
Receivable from collaboration partners | 26,970 | 51,957 | |||||
Inventory | 34,446 | 12,603 | |||||
Advances to manufacturers | 12,288 | 20,316 | |||||
Prepaid expenses and other current assets | 20,571 | 9,253 | |||||
Total current assets | 1,015,559 | 740,801 | |||||
Property and equipment, net | 32,476 | 25,471 | |||||
Intangible assets, net | 8,730 | — | |||||
Other assets | 3,342 | 2,479 | |||||
Total assets | $ | 1,060,107 | $ | 768,751 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 7,315 | $ | 4,026 | |||
Accrued liabilities | 88,279 | 71,188 | |||||
Payable to collaboration partner | 79,799 | 3,388 | |||||
Income taxes payable | 70 | 1,448 | |||||
Deferred revenue - current portion | 18,773 | 7,581 | |||||
Total current liabilities | 194,236 | 87,631 | |||||
Deferred revenue - non-current portion | 34,885 | 52,025 | |||||
Other long-term liabilities | 1,821 | 1,472 | |||||
Total liabilities | 230,942 | 141,128 | |||||
Commitments and contingencies (Notes 5 and 12) | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $0.0001 par value; 1,000,000 shares authorized; none issued and outstanding | — | — | |||||
Common stock, $0.0001 par value; 150,000,000 shares authorized at December 31, 2014 and December 31, 2013, respectively; shares issued and outstanding 75,933,661 and 74,167,169 at December 31, 2014 and December 31, 2013, respectively | 8 | 7 | |||||
Additional paid-in capital | 959,637 | 844,220 | |||||
Accumulated other comprehensive loss | (8 | ) | (8 | ) | |||
Accumulated deficit | (130,472 | ) | (216,596 | ) | |||
Total stockholders’ equity | 829,165 | 627,623 | |||||
Total liabilities and stockholders’ equity | $ | 1,060,107 | $ | 768,751 | |||
The accompanying notes are an integral part of these consolidated financial statements.
65
PHARMACYCLICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Revenue: | |||||||||||||||
Product revenue, net | $ | 492,418 | $ | 13,573 | $ | — | $ | — | |||||||
License and milestone revenue | 220,000 | 235,000 | 155,000 | 77,605 | |||||||||||
Collaboration services revenue | 17,311 | 11,596 | 5,658 | 4,385 | |||||||||||
Total revenue | 729,729 | 260,169 | 160,658 | 81,990 | |||||||||||
Costs and expenses: | |||||||||||||||
Cost of goods sold | 40,410 | 3,501 | — | — | |||||||||||
Research and development | 172,459 | 170,865 | 46,639 | 54,537 | |||||||||||
Less: Excess Amounts related to Research and development (See Note 5) | — | (85,732 | ) | (17,306 | ) | — | |||||||||
Research and development, net | 172,459 | 85,133 | 29,333 | 54,537 | |||||||||||
Selling, general and administrative | 167,955 | 104,016 | 12,093 | 15,575 | |||||||||||
Less: Excess Amounts related to Selling, general and administrative (See Note 5) | — | (30,405 | ) | (819 | ) | — | |||||||||
Selling, general and administrative, net | 167,955 | 73,611 | 11,274 | 15,575 | |||||||||||
Costs of collaboration (see Note 5) | 226,004 | — | — | — | |||||||||||
Amortization of intangible assets (see Note 8) | 520 | — | — | — | |||||||||||
Total costs and expenses | 607,348 | 162,245 | 40,607 | 70,112 | |||||||||||
Income from operations | 122,381 | 97,924 | 120,051 | 11,878 | |||||||||||
Interest income | 440 | 341 | 137 | 178 | |||||||||||
Other expense, net | (374 | ) | (129 | ) | (8 | ) | (31 | ) | |||||||
Income before income taxes | 122,447 | 98,136 | 120,180 | 12,025 | |||||||||||
Income tax provision | 36,323 | 31,126 | 2,647 | 39 | |||||||||||
Net income | $ | 86,124 | $ | 67,010 | $ | 117,533 | $ | 11,986 | |||||||
Net income per share: | |||||||||||||||
Basic | $ | 1.14 | $ | 0.92 | $ | 1.69 | $ | 0.17 | |||||||
Diluted | $ | 1.10 | $ | 0.87 | $ | 1.58 | $ | 0.17 | |||||||
Weighted average shares used to compute net income per share: | |||||||||||||||
Basic | 75,247 | 72,777 | 69,676 | 68,728 | |||||||||||
Diluted | 78,151 | 77,083 | 74,408 | 72,617 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
66
PHARMACYCLICS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(in thousands)
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Net income | $ | 86,124 | $ | 67,010 | $ | 117,533 | $ | 11,986 | |||||||
Change in unrealized gain (loss) on marketable securities | — | (4 | ) | 5 | 12 | ||||||||||
Comprehensive income, net of tax | $ | 86,124 | $ | 67,006 | $ | 117,538 | $ | 11,998 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
67
PHARMACYCLICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands, except share and per share amounts)
Common Stock | ||||||||||||||||||||||
Shares | Amount | Additional Paid-in Capital | Accumulated Other Comprehensive Loss | Accumulated Deficit | Total | |||||||||||||||||
Balance at June 30, 2011 | 67,915,865 | $ | 7 | $ | 514,813 | $ | (21 | ) | $ | (413,125 | ) | $ | 101,674 | |||||||||
Issuance of common stock upon under employee stock plans | 1,401,792 | — | 8,578 | — | — | 8,578 | ||||||||||||||||
Stock-based compensation expense | — | — | 9,873 | — | — | 9,873 | ||||||||||||||||
Change in unrealized gain on marketable securities | — | — | — | 12 | — | 12 | ||||||||||||||||
Net income | — | — | — | — | 11,986 | 11,986 | ||||||||||||||||
Balance at June 30, 2012 | 69,317,657 | 7 | 533,264 | (9 | ) | (401,139 | ) | 132,123 | ||||||||||||||
Issuance of common stock upon under employee stock plans | 898,729 | — | 5,429 | — | — | 5,429 | ||||||||||||||||
Stock-based compensation expense | — | — | 7,436 | — | — | 7,436 | ||||||||||||||||
Change in unrealized gain on marketable securities | — | — | — | 5 | — | 5 | ||||||||||||||||
Net income | — | — | — | — | 117,533 | 117,533 | ||||||||||||||||
Balance at December 31, 2012 | 70,216,386 | 7 | 546,129 | (4 | ) | (283,606 | ) | 262,526 | ||||||||||||||
Issuance of common stock in a public offering at $94.20 per share, net of issuance costs | 2,200,000 | — | 201,023 | — | — | 201,023 | ||||||||||||||||
Issuance of common stock under employee stock plans | 1,750,783 | — | 16,640 | — | — | 16,640 | ||||||||||||||||
Stock-based compensation expense | — | — | 50,395 | — | — | 50,395 | ||||||||||||||||
Excess tax benefit from stock-based compensation arrangements | — | — | 30,033 | — | — | 30,033 | ||||||||||||||||
Change in unrealized loss on marketable securities | — | — | — | (4 | ) | — | (4 | ) | ||||||||||||||
Net income | — | — | — | — | 67,010 | 67,010 | ||||||||||||||||
Balance at December 31, 2013 | 74,167,169 | 7 | 844,220 | (8 | ) | (216,596 | ) | 627,623 | ||||||||||||||
Issuance of common stock under employee stock plans | 1,766,492 | 1 | 25,909 | — | — | 25,910 | ||||||||||||||||
Stock-based compensation expense | — | — | 54,631 | — | — | 54,631 | ||||||||||||||||
Excess tax benefit from stock-based compensation arrangements | — | — | 34,877 | — | — | 34,877 | ||||||||||||||||
Net income | — | — | — | — | 86,124 | 86,124 | ||||||||||||||||
Balance at December 31, 2014 | 75,933,661 | $ | 8 | $ | 959,637 | $ | (8 | ) | $ | (130,472 | ) | $ | 829,165 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
68
PHARMACYCLICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | ||||||||||||||
2014 | 2013 | 2012 | 2012 | |||||||||||||
Cash flows from operating activities: | ||||||||||||||||
Net income | $ | 86,124 | $ | 67,010 | $ | 117,533 | $ | 11,986 | ||||||||
Adjustments to reconcile net income to net cash provided by operating activities: | ||||||||||||||||
Depreciation and amortization | 4,411 | 2,219 | 595 | 563 | ||||||||||||
Amortization of premium on marketable securities, net | — | — | — | 89 | ||||||||||||
Stock-based compensation expense | 53,741 | 50,210 | 7,436 | 9,873 | ||||||||||||
Excess tax benefit from stock-based compensation arrangements | (34,877 | ) | (30,033 | ) | — | — | ||||||||||
Income tax benefit from stock-based compensation arrangements | 34,877 | 30,033 | — | — | ||||||||||||
Loss on property and equipment | — | 188 | 9 | 25 | ||||||||||||
Provision for bad debts | 14 | — | — | — | ||||||||||||
Changes in assets and liabilities: | ||||||||||||||||
Accounts receivable | (53,267 | ) | (11,044 | ) | — | — | ||||||||||
Receivable from collaboration partners | 24,987 | (25,260 | ) | (20,773 | ) | (5,877 | ) | |||||||||
Inventory | (20,953 | ) | (12,419 | ) | — | — | ||||||||||
Advances to manufacturers | 8,028 | (20,316 | ) | — | — | |||||||||||
Prepaid expenses and other assets | (12,155 | ) | (7,184 | ) | 1,033 | (3,307 | ) | |||||||||
Accounts payable | 3,289 | (37 | ) | 1,557 | 2,206 | |||||||||||
Accrued liabilities | 20,114 | 52,365 | 6,495 | 1,874 | ||||||||||||
Payable to collaboration partner | 76,411 | 3,388 | — | — | ||||||||||||
Income taxes payable | (1,378 | ) | 59 | 1,389 | — | |||||||||||
Deferred revenue | (5,948 | ) | (11,095 | ) | (4,677 | ) | 68,378 | |||||||||
Other long-term liabilities | 349 | 688 | 97 | 277 | ||||||||||||
Net cash provided by operating activities | 183,767 | 88,772 | 110,694 | 86,087 | ||||||||||||
Cash flows from investing activities: | ||||||||||||||||
Purchase of property and equipment | (13,919 | ) | (18,318 | ) | (2,421 | ) | (2,975 | ) | ||||||||
Purchase of marketable securities | (13,640 | ) | (14,800 | ) | (5,645 | ) | (5,720 | ) | ||||||||
Proceeds from sales of marketable securities | — | 480 | — | — | ||||||||||||
Purchase of intangible assets | (9,250 | ) | — | — | — | |||||||||||
Proceeds from maturities of marketable securities | 13,360 | 12,325 | 1,680 | 24,504 | ||||||||||||
Net cash (used in) provided by investing activities | (23,449 | ) | (20,313 | ) | (6,386 | ) | 15,809 | |||||||||
Cash flows from financing activities: | ||||||||||||||||
Issuance of common stock, net of issuance costs | — | 201,023 | — | (559 | ) | |||||||||||
Proceeds from issuance of common stock under employee stock plan | 25,884 | 17,008 | 5,229 | 8,802 | ||||||||||||
Excess tax benefit from stock-based compensation arrangements | 34,877 | 30,033 | — | — | ||||||||||||
Net cash provided by financing activities | 60,761 | 248,064 | 5,229 | 8,243 | ||||||||||||
Increase in cash and cash equivalents | 221,079 | 316,523 | 109,537 | 110,139 | ||||||||||||
Cash and cash equivalents at beginning of period | 623,956 | 307,433 | 197,896 | 87,757 | ||||||||||||
Cash and cash equivalents at end of period | $ | 845,035 | $ | 623,956 | $ | 307,433 | $ | 197,896 | ||||||||
Supplemental disclosure of cash flow information: | ||||||||||||||||
Cash paid for income taxes | $ | 10,368 | $ | 820 | $ | 980 | $ | 310 | ||||||||
Supplemental disclosure of non-cash investing and financing activities: | ||||||||||||||||
Receivable for stock option exercises | $ | 36 | $ | — | $ | 368 | $ | 167 | ||||||||
Property and equipment purchases included in accounts payable and accrued liabilities | 1,016 | 4,039 | 883 | 143 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
69
PHARMACYCLICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 — DESCRIPTION OF THE COMPANY AND BASIS OF PRESENTATION
Description of the Company
Pharmacyclics, Inc. (Pharmacyclics or the Company) is a biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Pharmacyclics markets IMBRUVICA® (ibrutinib) and has other product candidates in clinical development and several preclinical molecules in lead optimization.
IMBRUVICA is a first-in-class, oral, once-daily therapy that inhibits a protein called Bruton's tyrosine kinase (BTK). IMBRUVICA is being jointly developed and commercialized by Pharmacyclics and Janssen Biotech, Inc. and its affiliates (Janssen), one of the Janssen Pharmaceutical companies of Johnson & Johnson.
IMBRUVICA currently is approved for use in approximately 40 countries including the U.S., Canada, and the 28 countries within the European Union (EU).
IMBRUVICA first came to market on November 13, 2013, when it was approved by the U.S. Food and Drug Administration (FDA) under accelerated approval as a single agent for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. Improvements in survival or disease symptoms have not been established. On February 12, 2014, the FDA approved IMBRUVICA under accelerated approval as a single agent for the treatment of patients with chronic lymphocytic leukemia (CLL) who have received at least one prior therapy. On July 28, 2014, IMBRUVICA received regular (full) FDA approval for IMBRUVICA for the treatment of patients with CLL who have received at least one prior therapy, and for the treatment of CLL patients with deletion of the short arm of chromosome 17 (del 17p CLL), including treatment naive and previously treated del 17p patients. On October 17, 2014, the European Commission (EC) granted marketing approval for IMBRUVICA in the EU for the treatment of adult patients with relapsed or refractory MCL, or adult patients with CLL who have received at least one prior therapy, or in first line in the presence of 17p deletion or TP53 mutation in patients unsuitable for chemoimmunotherapy. On January 29, 2015, single-agent IMBRUVICA received regular (full) FDA approval for patients with Waldenström's macroglobulinemia (WM), and it is approved in all lines of therapy.
Change in Fiscal Year End
On November 14, 2012, the Board of Directors approved a change in the fiscal year end from June 30 to December 31, effective December 31, 2012. All references to "fiscal years", unless otherwise noted, refer to the twelve-month fiscal year, which prior to July 1, 2012, ended on June 30, and beginning on January 1, 2013, ends on December 31, of each year.
Basis of presentation
The accompanying consolidated financial statements include the accounts of Pharmacyclics, Inc. and its wholly-owned subsidiaries, Pharmacyclics (Europe) Limited, Pharmacyclics Switzerland GmbH, Pharmacyclics Cayman Ltd. and Pharmacyclics (Shanghai) Management Consulting Service Limited. All intercompany accounts and transactions have been eliminated. The U.S. dollar is the functional currency for all of the Company's consolidated operations.
Reclassification
Certain prior year balances were reclassified to conform to the current year's presentation. None of these reclassifications had an impact on reported financial position or cash flows for any of the periods presented.
NOTE 2 — SIGNIFICANT ACCOUNTING POLICIES
Management's use of estimates and assumptions
The preparation of financial statements in accordance with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reported period. Actual results could differ from those estimates.
Basic and diluted net income per share
70
Basic net income per share is computed using the weighted average number of common shares outstanding during the period. Diluted net income per share is computed using the weighted average number of shares of common stock outstanding during the period increased to include the number of additional shares of common stock that would have been outstanding if the potentially dilutive securities had been issued. Potentially dilutive securities include outstanding stock options, including performance-based stock options and restricted stock units for which performance criteria have been achieved, restricted stock units with time-based vesting and shares to be purchased under the employee stock purchase plan. The dilutive effect of potentially dilutive securities is reflected in diluted net income per common share by application of the treasury stock method. Under the treasury stock method, an increase in the fair market value of the Company's common stock can result in a greater dilutive effect from potentially dilutive securities.
Cash and cash equivalents
All highly liquid investments purchased with an original maturity date of three months or less that are readily convertible into cash and have an insignificant interest rate risk are considered to be cash equivalents.
Marketable securities
The Company's marketable securities are classified as “available-for-sale”. The Company includes these investments in current assets and carry them at fair value. Unrealized gains and losses on available-for-sale securities are included in accumulated other comprehensive loss. The amortized cost of debt securities is adjusted for the amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income. Gains and losses on securities sold are recorded based on the specific identification method and are included in other expense, net in the consolidated statement of operations.
Management assesses whether declines in the fair value of marketable securities are other than temporary. If the decline is judged to be other than temporary, the cost basis of the individual security is written down to fair value and the amount of the write down is included in the statement of operations within other expense, net. In determining whether a decline is other than temporary, management considers various factors including the length of time and the extent to which the market value has been less than cost, the financial condition and near-term prospects of the issuer and the Company's intent and ability to retain its investment in the issuer for a period of time sufficient to allow for any anticipated recovery in market value. To date, the Company has not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value.
Fair value measurements
The fair value of the Company's financial assets is determined by using three levels of input which are defined as follows:
Level 1 - Quoted prices in active markets for identical assets. At December 31, 2014, the Company's Level 1 assets were comprised of money market funds. At December 31, 2013, the Company's Level 1 assets were comprised of U.S. treasury securities and money market funds.
Level 2 - Observable inputs other than Level 1 prices such as quoted prices for similar assets, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets. The Company's short-term investments primarily utilize broker quotes in markets with infrequent transactions for valuation of these securities. At December 31, 2014 and December 31, 2013, the Company's Level 2 assets were comprised of FDIC insured certificates of deposit.
Level 3 - Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets. At December 31, 2014 and December 31, 2013, the Company did not hold any Level 3 assets.
The Company utilizes the market approach to measure fair value for its financial assets. The market approach uses prices and other relevant information generated by market transactions involving identical or comparable assets.
Fair value of financial instruments
Cash and cash equivalents and marketable securities are carried at fair value. Accounts receivable, net, receivables from collaboration partners, accounts payable and accrued liabilities are valued at their carrying amounts, which approximate fair value due to their short-term nature. As of December 31, 2014 and December 31, 2013, the Company had no liabilities measured at fair value.
Restricted investments
71
Under one of its facilities lease agreements, the Company is required to maintain a $0.3 million letter of credit as security for performance under the lease. The letter of credit is secured by a $0.3 million certificate of deposit which is included in Other assets at December 31, 2014 and December 31, 2013.
Accounts Receivable, net
Accounts receivable, net represents amounts due to the Company from sales of IMBRUVICA. The Accounts receivable balance is recorded net of allowances which generally include wholesaler chargebacks from third-party payers including government and other programs, cash discounts for prompt payment and doubtful accounts. Estimates for wholesaler chargebacks, cash discounts and prompt payment discounts are based on contractual payment terms and the Company's expectations about utilization rates for these programs. The Company's estimate of the allowance for doubtful accounts is based on the creditworthiness of its customers, historical payment patterns, the aging of accounts receivable balances and general economic conditions. Accounts receivables balances are written off against the allowance when it is probable that the receivable will not be collected (see Note 4).
Inventory valuation and related reserves
Inventories are stated at the lower of cost or market value with the approximate cost being determined on a first-in, first-out basis. Costs capitalized as inventories include third-party manufacturing costs and labor costs for personnel involved in the manufacturing process.
During the year ended December 31, 2013, the Company began capitalizing costs associated with manufacturing inventory as a result of the FDA approval for IMBRUVICA. Until September 30, 2013, all inventory costs incurred were recorded as research and development expense in the consolidated statements of operations.
Products that have been approved by the FDA or other regulatory agencies, such as IMBRUVICA, are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of IMBRUVICA (i.e., raw materials and active pharmaceutical ingredient (API)) utilized for both commercial and clinical programs is identical and, as a result, the inventory has an "alternative future use" as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to research and development expense when the product enters the research and development process and can no longer be used for commercial purposes and, therefore, does not have "alternative future use."
The Company evaluates inventory levels quarterly to determine if the inventory has a cost basis in excess of its expected net realizable value, inventory has expired, inventory on-hand is in excess of expected sales forecasts, and/or inventory that does not meet its quality specifications. The Company's raw materials and API has no expiration date and can be revalidated for use after testing. The Company's finished goods inventory currently has an estimated life of 24 months and, based on the Company's sales forecasts, it expects to realize the carrying value of the IMBRUVICA inventory. If any impairment of inventory is identified, the related inventory is written down with a corresponding charge to cost of goods sold in the period that the inventory impairment is first identified. The Company's inventory valuation reserve balance was zero as of December 31, 2014 and December 31, 2013, respectively.
Inventories consist of raw materials, work-in-process and finished goods related to the production of IMBRUVICA. Raw materials include IMBRUVICA API. Work-in-process includes third-party manufacturing and associated labor costs relating to the Company’s personnel involved in the production process. Included in inventories are raw materials and work-in-process that may be used as clinical products, which are charged to research and development expense when the product enters the research and development process.
The components of inventory are as follows (in thousands):
As of December 31, | |||||||
2014 | 2013 | ||||||
Raw materials | $ | — | $ | 8,007 | |||
Work-in-process | 27,591 | 3,489 | |||||
Finished goods | 6,855 | 1,107 | |||||
$ | 34,446 | $ | 12,603 | ||||
Advances to manufacturers
72
The Company records cash advances that it pays prior to the receipt of inventory as advances to manufacturers in the consolidated balance sheets. The cash advances may be forfeited if the Company terminates the scheduled production. The Company expects the carrying value of the advances to manufacturers to be fully realized.
Concentration of credit risk and other risks and uncertainties
Financial instruments that potentially subject the Company to credit risk consist principally of cash, cash equivalents, marketable securities, accounts receivable, net and receivables from collaboration partners. The Company places its cash and cash equivalents with high-credit quality financial institutions and invests in debt instruments of financial institutions, corporations and government entities with strong credit ratings. The Company's management believes it has established guidelines relative to credit quality, diversification and maturities that maintain safety and liquidity.
The Company sells IMBRUVICA through a limited number of specialty pharmacies and specialty distributors. Therefore, the Company's accounts receivable balance is comprised of amounts due from a small number of customers. The Company continuously monitors the creditworthiness of its customers and has internal policies regarding customer credit limits. The Company's policy is to estimate the allowance for doubtful accounts based on the credit worthiness of its customers, historical payment patterns, the aging of accounts receivable balances and general economic conditions. For the year ended December 31, 2014, four individual customers accounted for 25%, 22%, 19% and 15% of accounts receivable, net, respectively. For the year ended December 31, 2013, three individual customers accounted for 26%, 26%, and 16% of accounts receivable, net, respectively.
As of December 31, 2014, Company had $27.0 million receivable from Janssen, which consisted primarily of a milestone receivable of $20.0 million, $3.9 million related to material and product sales and $1.7 million related to value added taxes (see Note 5). As of December 31, 2013, the Company's receivable from collaboration partner balance of $52.0 million was comprised primarily of $50.2 million due from Janssen under the collaboration agreement. To date, the Company has not experienced credit losses from its receivables from collaborative partners, including Janssen. The amounts receivable from Janssen are included within Receivable from collaboration partners on the consolidated balance sheets.
The Company's products require approvals from the FDA and international regulatory agencies prior to commercial sales. Although the FDA has approved IMBRUVICA as a single agent for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy, the Company's prospects are largely dependent on (a) successful commercialization of IMBRUVICA in the U.S. to treat patients with MCL who have received at least one prior therapy, (b) obtaining regulatory approval of, and successfully commercializing, IMBRUVICA outside the U.S. to treat patients with MCL who have received at least one prior therapy, and (c) obtaining regulatory approval of, and successfully commercializing, IMBRUVICA both in and outside the U.S. to treat other indications. If the Company is unsuccessful in achieving any one or more of these critical business objectives, its ability to generate significant revenue or achieve profitability will be adversely affected and its business may fail. Further, there can be no assurance that the Company's other product candidates will receive required approvals. If the Company was denied such approvals or such approvals were delayed, it could have a materially adverse impact on the Company and the execution of its business strategy.
The Company has expended and will continue to expend substantial funds for the commercialization, manufacturing, marketing and sales of IMBRUVICA and to complete the research, development and clinical testing of investigational uses of IMBRUVICA and additional products. The Company may be unable to entirely fund these efforts with its current financial resources and may require additional funds to continue to develop and commercialize its products. Additional funds may not be available on acceptable terms, if at all. If adequate funds are unavailable on a timely basis from operations or additional sources of financing, the Company may have to delay, reduce the scope of or eliminate one or more of its research or development programs which would materially and adversely affect its business, financial condition and operations.
Property and equipment
Property and equipment are stated at cost. Equipment is depreciated by the straight-line method over the estimated useful lives of the assets, generally 2 to 5 years. Furniture and fixtures are depreciated by the straight-line method over the estimated useful lives of the assets, generally 5 years. Leasehold improvements are generally amortized by the straight-line method over the shorter of the life of the related asset or the term of the underlying lease. Assets not yet placed in use are not depreciated. Substantially all of the Company's property and equipment is located within the U.S.
Long-lived assets
The Company evaluates long-lived assets (including finite-lived intangible assets) for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset group may not be recoverable. In accordance with Accounting Standards Codification (ASC) ASC 350-10, Goodwill and Other Intangible Assets, intangible assets with estimable
73
useful lives are amortized over their respective estimated useful lives, and reviewed for impairment in accordance with ASC 360-10, "Accounting for the Impairment or Disposal of Long-Lived Assets." The Company reviews long-lived assets, such as acquired intangibles and property and equipment, for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company measures recoverability of assets to be held and used by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, the Company recognizes an impairment charge at the amount by which the carrying amount of the asset exceeds the fair value of the asset. The Company's impairment review requires significant judgment with respect to future revenue and expense growth rates, selection of appropriate discount rate and other assumptions and estimates. No significant impairment losses have been recorded to date with respect to the Company's long-lived assets (see Note 8).
Donations to Third-Party Patient Assistance Foundations
The Company provides cash and product donations to independent third-party patient assistance foundations that, in turn, make grants to help qualifying patients meet their out-of-pocket costs (including co-pay payments or co-insurance obligations) in connection with their use of the Company's products as well as those of other pharmaceutical and biotechnology companies. These independent patient assistance foundations maintain their own patient eligibility criteria and independently determine what type of assistance patients may qualify for. Given the non-reciprocal nature of these transactions with parties who are not considered direct or indirect customers, and for which the Company does not receive any consideration, and the fact that the donation of cash does not have any conditions attached to the donation, the Company accounts for the cash donations as a component of selling, general and administrative expenses in the consolidated statements of operations.
Revenue Recognition
Product revenue, net is recognized in accordance with the Financial Accounting Standards Board (FASB) ASC 605, Revenue Recognition, when the following criteria have been met: (1) persuasive evidence of an arrangement exists, (2) delivery has occurred or services have been rendered, (3) the price is fixed or determinable and (4) collectability is reasonably assured. Revenues are deferred for fees received before earned or until no further obligations exist. We exercise judgment in determining that collectability is reasonably assured or that services have been delivered in accordance with the arrangement. We assess whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the sales price is subject to refund or adjustment. We assess collectability based primarily on the customer's payment history and on the creditworthiness of the customer.
Product Revenue, Net
Product revenue, net consists of U.S. sales of IMBRUVICA and is recognized once all four revenue recognition criteria described above have been met. The Company sells IMBRUVICA directly to some customers who have in-house dispensing capabilities, specialty pharmacies (SP) that sell to individual patients, specialty distributors (SD) that sell to hospital pharmacies and other organizations that the Company has contracted with. The Company recognizes revenue from sales of IMBRUVICA when the product’s title and risk of loss transfers to the customer. The Company determined that it has the ability to make reasonable estimates of product returns in order to recognize revenue at the time that title and risk of loss transfers to the customer based on the following factors: (1) the Company believes that it has sufficient insight into the distribution channel at the SP's and SD's in order to ascertain their inventory level and dispense data, (2) due to the price of the Company's product and limited patient population, its SP and SD customers have not built up significant levels of inventory, nor does the Company expect they will do so for the foreseeable future, (3) inventory on hand at the Company's SP customers was approximately two weeks as of December 31, 2014, (4) there have been no product returns to-date since the Company's commercial launch of IMBRUVICA on November 13, 2013 and (5) the Company believes there is limited risk of return of inventory in the channel because there is significant remaining shelf life at the point of sale.
The Company recognizes product revenue net of adjustments for customer credits, including estimated government rebates and charge-backs, returns, prompt payment discounts, U.S. Department of Veteran's Affairs (VA) negotiated discounts and administrative service fees related to its patient assistance program which includes co-payment and deductible assistance, and Medicare Part D coverage gap reimbursements. Each of the above adjustments is recorded at the time of revenue recognition, resulting in a reduction in product revenue, net and an increase in accrued expenses or a reduction in accounts receivable, net. The above adjustments require significant estimates, judgment and information obtained from external sources. If management's estimates differ from actual results, the Company will record adjustments that would affect product revenue, net in the period of adjustment.
Gross-to-net sales adjustments
Rebates
74
The Company records an allowance for rebates including mandated discounts under the Medicaid Drug Rebate Program, discounts provided under the TRICARE Retail Pharmacy Refunds Program (TRICARE) and to members of organizations with whom the Company has contracted with. The allowance for rebates is based upon the Company's contractual agreements and/or legal requirements and public sector benefit providers, including Medicaid and TRICARE. For estimated amounts owed to public sector benefit providers, including Medicaid and TRICARE, the allowance for rebates is based on the estimated rebate percentage of forecasted eligible sales. The estimated rebate percentage is based on statutory discount rates and expected utilization. The forecasted eligible Medicaid and TRICARE sales represent those sales made by the Company that will ultimately be consumed by patients covered by Medicaid and TRICARE. To estimate the allowance for rebates, the Company uses the estimated patient mix information which is provided by its SP customers, as well as third party sources. For organizations that the Company has contracted with, the rebate is based upon contracted volume discount amounts. In addition, the Company incurs administrative fees in exchange for administrative services provided that are also accrued at the time of sale. Rebates for public sector benefit providers and organizational discounts are generally invoiced and paid in arrears. As such, the allowance for rebates consists of an estimate of the amount expected to be incurred for the current quarter's shipments to patients, plus an accrual balance for estimated unpaid rebates from prior periods. The allowance for rebates is recorded within accrued liabilities in the consolidated balance sheets.
Charge-backs
Charge-backs are discounts that result from the difference between the prices at which the Company makes IMBRUVICA available to wholesalers for purchase by discount customers under pricing agreements the Company has with the discount customers and the sales price paid to the Company by the wholesalers who service the discount customers. Such discount customers, which primarily consist of the U.S. Department of Defense (DOD), VA, Public Health Services (PHS), and other Federal Government institutions, purchase products through wholesalers at a lower price provided for in pricing contracts and the wholesalers then charge the Company the difference between the wholesale acquisition cost and the lower price paid by the discount customer. These reductions are settled through charge-backs from the Company's wholesalers. Charge-backs are recorded as a reduction to accounts receivable, net in the consolidated balance sheets.
Product Returns
Consistent with industry practice, the Company generally offers its customers a limited right to return. The Company generally allows for the return of product that is a few months prior to and up to a few months after the product expiration date. Additionally, the Company considers several other factors in the estimation process including the expiration dates of product shipped, third party data in monitoring channel inventory levels, shelf life of the product, prescription trends and other relevant factors. Provisions for estimated product returns are recorded within accrued liabilities in the consolidated balance sheets.
Medicare Part D coverage gap
Medicare Part D, also known as the Medicare prescription drug benefit, is a federal program to subsidize the costs of prescription drugs for Medicare beneficiaries in the United States. The Medicare Part D prescription drug benefit mandates that drug manufacturers fund 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Funding of the Medicare Part D gap is invoiced and paid in arrears. As such, the allowance for Medicare Part D consists of an estimate of the amount expected to be incurred for the current quarter's shipments to patients, plus an accrual balance for estimated shipments remaining in the channel at period end which are estimated to ship to Medicare Part D patients. The allowance for rebates is recorded within accrued liabilities in the consolidated balance sheets.
Prompt payment discounts
The Company generally offers cash discounts to its customers, generally a 2% discount applied to the invoice amount, as an incentive for prompt payment. The Company expects that all of its customers to whom it offers cash discounts for prompt payment to take advantage of the full amount of the 2% discount. The Company records the prompt-payment discount as a reduction to accounts receivable, net in the consolidated balance sheets.
Co-payment assistance
Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. The Company accrues for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators. The allowance for co-payment assistance is recorded within accrued liabilities in the consolidated balance sheets.
Collaboration Revenues
75
Revenue under the Company's license and collaboration arrangements is recognized based on the performance requirements of the contract. Determinations of whether persuasive evidence of an arrangement exists and whether delivery has occurred or services have been rendered are based on management’s judgments regarding the fixed nature of the fees charged for deliverables and the collectability of those fees. Should changes in conditions cause management to determine that these criteria are not met for any new or modified transactions, revenue recognized could be adversely affected.
The Company recognizes revenue related to collaboration and license arrangements in accordance with the provisions of Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 605-25, “Revenue Recognition – Multiple-Element Arrangements,” or ASC Topic 605-25. Additionally, the Company adopted, effective July 1, 2010, Accounting Standards Update, or ASU, No. 2009-13, “Multiple Deliverable Revenue Arrangements,” or ASU 2009-13, which amended ASC Topic 605-25 and:
• | provided guidance on how deliverables in an arrangement should be separated and how the arrangement consideration should be allocated to the separate units of accounting; |
• | required an entity to determine the selling price of a separate deliverable using a hierarchy of (i) vendor-specific objective evidence, or VSOE, (ii) third-party evidence, or TPE, or (iii) best estimate of selling price, or BESP; and |
• | required the allocation of the arrangement consideration, at the inception of the arrangement, to the separate units of accounting based on relative fair value. |
The Company evaluates all deliverables within an arrangement to determine whether or not they provide value on a stand-alone basis. Based on this evaluation, the deliverables are separated into units of accounting. The arrangement consideration that is fixed or determinable at the inception of the arrangement is allocated to the separate units of accounting based on their relative selling prices. The Company may exercise significant judgment in determining whether a deliverable is a separate unit of accounting, as well as in estimating the selling prices of such unit of accounting.
To determine the selling price of a separate deliverable, the Company uses the hierarchy as prescribed in ASC Topic 605-25 based on VSOE, TPE or BESP. VSOE is based on the price charged when the element is sold separately and is the price actually charged for that deliverable. TPE is determined based on third-party evidence for a similar deliverable when sold separately and BESP is the price at which the Company would transact a sale if the elements of collaboration and license arrangements were sold on a stand-alone basis. The Company may not be able to establish VSOE or TPE for the deliverables within collaboration and license arrangements, as the Company does not have a history of entering into such arrangements or selling the individual deliverables within such arrangements separately. In addition, there may be significant differentiation in these arrangements, which indicates that comparable third-party pricing may not be available. The Company may determine that the selling price for the deliverables within collaboration and license arrangements should be determined using BESP. The process for determining BESP involves significant judgment on the Company's part and includes consideration of multiple factors such as estimated direct expenses and other costs, and available data.
For collaborations entered into after July 1, 2010, the Company has determined its best estimate of selling prices for the license unit of accounting based on the income approach as defined in ASC 820-10-35-32. This measurement is based on the value indicated by current estimates about those future amounts and reflects management determined estimates and assumptions. These estimates and assumptions include, but are not limited to, how a market participant would use the license, estimated market opportunity and expected market share and assumed royalty rates that would be paid for sales resulting from products developed using the license, similar arrangements entered into by third parties and entity-specific factors such as the terms of the Company's previous collaborative agreement, the Company's pricing practices and pricing objectives, the likelihood that clinical trials will be successful, the likelihood that regulatory approval will be received and that the products will become commercialized and the markets served. The Company has also determined BESP for services-related deliverables based on the nature of the services to be performed and estimates of the associated effort as well as estimated market rates for similar services.
For each unit of accounting identified within an arrangement, the Company determines the period over which the performance obligation occurs. Revenue is then recognized using either a proportional performance or straight-line method. The Company recognizes revenue using the proportional performance method when the level of effort to complete our performance obligations under an arrangement can be reasonably estimated. Direct labor hours or full time equivalents are typically used as the measurement of performance.
Effective July 1, 2010, the Company adopted ASU No. 2010-17, “Milestone Method of Revenue Recognition,” or ASU 2010-17, which provides guidance on revenue recognition using the milestone method. Under the milestone method, a payment that is contingent upon the achievement of a substantive milestone is recognized in the period in which the milestone is achieved. The determination that a milestone is substantive is subject to considerable judgment.
76
Cost of goods sold
Cost of goods sold includes third-party manufacturing costs of products sold, fixed manufacturing overhead, royalty fees, and other indirect costs such as personnel compensation.
During the fiscal year 2013, the Company began capitalizing costs associated with manufacturing inventory as a result of the FDA approval of IMBRUVICA for MCL on November 13, 2013. Until September 30, 2013, all inventory costs incurred were recorded as research and development expense in the consolidated statements of operations. This zero cost inventory primarily consisted of raw materials for which the shelf-life has not yet started.
Research and development expense
Research and development expenses include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and amortized over the period that the goods are delivered or the related services are performed, subject to an assessment of recoverability.
Clinical development costs are a significant component of research and development expenses. The Company has a history of contracting with third parties that perform various clinical trial activities on its behalf in the ongoing development of its product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow. The Company accrues and expenses costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations and clinical trial sites. The Company determines its estimates through discussions with internal clinical personnel and outside service providers as to the progress or stage of completion of trials or services and the agreed upon fee to be paid for such services.
The Company's worldwide collaboration and license agreement with Janssen (the Agreement) includes a cost sharing arrangement for certain collaboration activities. Except in certain cases, in general Janssen is responsible for approximately 60% of collaboration development costs and the Company is responsible for the remaining 40% of collaboration development costs. Further, the Agreement provides the Company with a $50.0 million annual cap of its share of collaboration costs and pre-tax commercialization losses for each calendar year until after the third profitable calendar quarter for the product and any amounts in excess of the annual cap (Excess Amounts) are funded by Janssen. Under the Agreement, total Excess Amounts plus interest may not exceed $225.0 million.
The Company's policy is to account for cost-sharing payments to Janssen related to development services as a component of research and development expense and reimbursements for development services under the cost-sharing arrangement as an offset to research and development expense, upon delivery of the related services when expenses have been incurred and reimbursements have been earned. During the year ended December 31, 2013 and the six months ended December 31, 2012, the Company recognized Excess Amounts related to development services as a reduction to research and development expenses. The Company recognizes Excess Amounts as a reduction to Costs and expenses as the Company's payment of Excess Amounts to Janssen is contingent and would become payable only after the third profitable calendar quarter for the product. Further, Excess Amounts shall be reimbursable only from the Company's share of pre-tax profits after the third profitable calendar quarter for the product (see Note 5).
Selling, general and administrative expense
The Company expenses the cost of selling, general and administrative activities as incurred. Selling, general and administrative expenses consist primarily of personnel and facility-related expenses, outside contracted services and other costs not associated with the research and development activities of the Company. In connection with the Agreement, the Company also classifies certain commercial-related collaboration costs within Selling, general and administrative expense in the consolidated statements of operations. Costs under the Agreement that are recorded within selling, general and administrative expense are generally shared 50% by the Company and 50% by Janssen and include marketing costs, patent costs and the Company's share of pre-tax commercial losses on sales of IMBRUVICA.
During the year ended December 31, 2013 and the six months ended December 31, 2012, the Company recognized Excess Amounts related to certain collaboration costs as a reduction to selling, general and administrative expenses (see Note 5).
Income taxes
The Company is subject to income taxes in both the U.S. and foreign jurisdictions, and it uses estimates in determining its provisions for income taxes. The Company provides for income taxes using the asset and liability method, whereby deferred
77
tax assets or liability account balances are calculated at the balance sheet date using current tax laws and rates in effect for the year in which the differences are expected to affect taxable income. Recognition of deferred tax assets is appropriate when realization of such assets is more likely than not. The Company recognizes a valuation allowance against its net deferred tax assets if it is more likely than not that some portion of the deferred tax assets will not be fully realizable. This assessment requires judgment as to the likelihood and amounts of future taxable income by tax jurisdiction.
The Company applies the provisions of FASB's guidance on accounting for uncertainty in income taxes. The Company assesses all material positions taken in any income tax return, including all significant uncertain positions, in all tax years that are still subject to assessment or challenge by relevant taxing authorities. Assessing an uncertain tax position begins with the initial determination of the position’s sustainability and the tax benefit to be recognized is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. As of each balance sheet date, unresolved uncertain tax positions must be reassessed, and the Company will determine whether (i) the factors underlying the sustainability assertion have changed and (ii) the amount of the recognized tax benefit is still appropriate. The recognition and measurement of tax benefits requires significant judgment. Judgments concerning the recognition and measurement of a tax benefit might change as new information becomes available.
Stock-based compensation
Stock-based compensation cost for employee and director stock options with time-based vesting and employee stock plan purchase rights is generally measured and recognized based on the fair value of the award at the grant date. Stock-based compensation cost for restricted stock units (RSUs) with time-based vesting is measured based on the closing fair market value of the Company's common stock on the date of the grant, multiplied by the number of RSUs granted. The accounting grant date for employee stock options and restricted stock units with performance obligations is the date on which the performance goals have been defined and a mutual understanding of the terms has been reached. Generally, the Company's time-based stock option and RSU grants vest over a four year period. Stock options and RSUs with performance obligations generally vest over a four year period, with the goals set and agreed upon annually. Stock-based compensation for non-employee stock options is re-estimated at each period-end through the vesting date. The Company estimates a forfeiture rate to calculate the stock-based compensation cost for its awards primarily based on an analysis of its historical pre-vesting forfeitures.
The fair value of each stock option is estimated using the Black Scholes option-pricing model. Expected volatility is based on historical volatility data of the Company's stock. The expected term of stock options granted represents the period of time that stock options are expected to be outstanding. The Company generally does not expect substantially different exercise or post-vesting termination behavior among its employee or non-employee population. As such, for the majority of stock options granted and the Company's Employee Stock Purchase Plan, the Company generally calculates and applies an overall expected term assumption based on historical data. In certain cases, the Company uses a shorter expected term for performance-based stock options based on a combination of historical data and management's estimates of the period of time that options will be outstanding. The risk-free interest rate is based on a zero-coupon U.S. Treasury bond whose maturity period equals the expected term of the Company's options.
Options and RSUs vest upon the passage of time or a combination of time and the achievement of certain performance obligations. Vesting of performance-based options and RSUs for executive officers depends on their attainment of key corporate and departmental goals. The Compensation Committee of the Board of Directors will determine if the performance conditions have been met. Stock-based compensation expense for the options and RSUs with performance obligations is recorded when the Company believes that the vesting of these options and RSUs is probable.
Shipping and handling costs
Shipping and handling costs incurred for inventory purchases and product shipments are recorded in Selling, general and administrative, net in the consolidated statements of income. Shipping and handling costs were not material for the years ended December 31, 2014 and December 31, 2013.
Advertising expenses
The Company expenses the costs of advertising, including promotional expenses, as incurred. The Company began incurring advertising expenses during the year ended December 31, 2014 due to the commercialization of IMBRUVICA on November 13, 2013. Advertising expenses were $4.4 million for the year ended December 31, 2014 and $0.3 million for the year ended December 31, 2013.
Segment reporting
78
Operating segments are defined as components of an enterprise that engage in business activities for which separate financial information is available and evaluated by the chief operating decision maker in deciding how to allocate resources and assessing performance. The Company’s chief operating decision maker is its Chief Executive Officer (CEO). The Company operates as a single reportable segment, which primarily focuses on developing and commercializing novel therapies for the treatment of cancer and immune-mediated diseases. Product revenue, net is attributed to geography based upon the country in which the product is delivered. Long-lived assets are attributed to geography based on the country where the assets are located.
Recent Accounting Pronouncements
In April 2014, the FASB issued ASU 2014-08, "Presentation of Financial Statements (Topic 205) and Property, Plant, and Equipment (Topic 360): Reporting Discontinued Operations and Disclosures of Disposals of Components of an Entity", or ASU 2014-08. Under ASU 2014-08, only disposals representing a strategic shift in operations should be presented as discontinued operations. Those strategic shifts should have a major effect on the organization’s operations and financial results. Additionally, ASU 2014-08 requires expanded disclosures about discontinued operations that will provide financial statement users with more information about the assets, liabilities, income, and expenses of discontinued operations. ASU 2014-08 is effective for fiscal and interim periods beginning on or after December 15, 2014, with early adoption permitted. The Company will adopt the provisions of ASU 2014-08 beginning with the Company’s fiscal 2015.
In May 2014, the FASB issued ASU 2014-09, "Revenue from Contracts with Customers (Topic 606)," which provides guidance for revenue recognition. This ASU will supersede the revenue recognition requirements in Topic 605, Revenue Recognition, and most industry-specific guidance. The standard’s core principle is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and make more estimates than under today’s guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. This standard is effective for annual and interim reporting periods beginning after December 15, 2016. Early adoption is not permitted. The Company is currently evaluating the impact of the adoption of this accounting standard on its consolidated financial statements.
In June 2014, the FASB issued ASU 2014-12, "Accounting for Stock-based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period." ASU 2014-12 requires that a performance target that affects vesting and could be achieved after the requisite service period be treated as a performance condition. This new guidance is effective for fiscal years and interim periods within those years beginning after December 15, 2015. Early adoption is permitted. This standard is not expected to have any impact on current disclosures in the consolidated financial statements.
In August 2014, the FASB issued ASU 2014-15, "Presentation of Financial Statements - Going Concern, which requires management to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and provide related footnote disclosures." ASU 2014-15 is effective for annual and interim reporting periods beginning on or after December 15, 2016. Early adoption is permitted for financial statements that have not been previously issued. The standard allows for either a full retrospective or modified retrospective transition method. The Company does not expect this standard to have a any impact on the Company’s consolidated financial statements upon adoption.
In November 2014, the FASB issued ASU 2014-17, "Business Combinations (Topic 805): Pushdown Accounting (a consensus of the FASB Emerging Issues Task Force)." ASU 2014-17 provides an acquired entity with an option to apply pushdown accounting in its separate financial statements upon occurrence of an event in which an acquirer obtains control of the acquired entity. An acquired entity may elect the option to apply pushdown accounting in the reporting period in which the change-in-control event occurs. This new standard is effective on November 18, 2014. Early application is prohibited. After the effective date, an acquired entity can make an election to apply the guidance to future change-in-control events or to its most recent change-in-control event. However, if the financial statements for the period in which the most recent change-in-control event occurred already have been issued or made available to be issued, the application of this guidance would be a change in accounting principle. The Company has adopted this new guidance and there's no impact on its consolidated financial statements.
NOTE 3 — PRODUCT REVENUE, NET
Product revenue, net consists of revenue recorded on the sale of IMBRUVICA which was approved by the FDA and commercially launched by the Company on November 13, 2013 for MCL. All product revenue, net for the years ended December 31, 2014 and December 31, 2013 was from sales to U.S. customers.
79
The Company recorded no product revenue, net for the six months ended December 31, 2012 and the year ended June 30, 2012 as these periods were prior to FDA approval for IMBRUVICA.
For the years ended December 31, 2014 and December 31, 2013, product revenue, net was calculated as follows (in thousands):
Years Ended December 31, | ||||||||
2014 | 2013 | |||||||
Product revenue, gross | $ | 553,143 | $ | 14,968 | ||||
Government rebates, return reserve and other | (34,502 | ) | (1,024 | ) | ||||
Chargebacks and cash discounts | (26,223 | ) | (371 | ) | ||||
Product revenue, net | $ | 492,418 | $ | 13,573 | ||||
The following table summarizes the provisions, and credits/payments, for product revenue, net adjustments for the years ended December 31, 2014 and December 31, 2013 (in thousands):
Total | ||||
2013 | ||||
Beginning Balance | $ | — | ||
Provision related to current period sales | 1,395 | |||
Credits/payments | (189 | ) | ||
Ending Balance | $ | 1,206 | ||
2014 | ||||
Beginning Balance | $ | 1,206 | ||
Provision related to current period sales | 60,725 | |||
Credits/payments | (35,196 | ) | ||
Ending Balance | $ | 26,735 | ||
The following table sets forth customers who represented 10% or more of the Company's product revenue, net for the years ended December 31, 2014 and December 31, 2013.
Years Ended December 31, | ||||||
Customers: | 2014 | 2013 | ||||
A | 30 | % | 24 | % | ||
B | 20 | % | 15 | % | ||
C | 15 | % | 12 | % | ||
D | 14 | % | 20 | % | ||
E | 11 | % | 12 | % | ||
80
NOTE 4 — ACCOUNTS RECEIVABLE, NET
Accounts receivable, net includes (in thousands):
As of December 31, | |||||||
2014 | 2013 | ||||||
Accounts receivable, gross | $ | 72,921 | $ | 11,345 | |||
Allowance for doubtful accounts | (14 | ) | — | ||||
Allowance for chargebacks related to government and other programs | (7,105 | ) | (72 | ) | |||
Allowance for cash discounts | (1,505 | ) | (229 | ) | |||
Accounts receivable, net | $ | 64,297 | $ | 11,044 | |||
NOTE 5 — COLLABORATIONS AND OTHER AGREEMENTS
The Company recognized revenue related to its collaboration and license arrangements as follows (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Janssen | $ | 237,164 | $ | 245,970 | $ | 154,878 | $ | 74,622 | |||||||
Les Laboratoires Servier (Servier) | 139 | 159 | 93 | 7,157 | |||||||||||
Novo Nordisk A/S (Novo Nordisk) | 8 | 454 | 5,631 | — | |||||||||||
Other | — | 13 | 56 | 211 | |||||||||||
Total | $ | 237,311 | $ | 246,596 | $ | 160,658 | $ | 81,990 | |||||||
Worldwide Collaboration and License Agreement with Janssen Biotech, Inc.
Background
In December 2011, the Company entered into a worldwide collaboration and license agreement (the Agreement) with Janssen for the joint development and commercialization of IMBRUVICA, a novel, orally active, selective covalent inhibitor of Bruton’s Tyrosine Kinase (BTK), and certain compounds structurally related to IMBRUVICA, for oncology and other indications, excluding all immune and inflammatory mediated diseases or conditions and all psychiatric or psychological diseases or conditions, in the U.S. and outside the U.S.
The collaboration provides Janssen with an exclusive license to exploit the underlying technology outside of the U.S. (the License Territory) and co-exclusively with Pharmacyclics in the U.S. Both parties are responsible for the development, manufacturing and marketing of any products resulting from the Agreement. The Company continues to work with Janssen on protocols and the design, schedules and timing of trials.
The collaboration has no fixed duration or expiration date and provided for payments by Janssen to the Company of a $150.0 million non-refundable upfront payment upon execution, as well as potential future milestone payments of up to $825.0 million, based upon continued development progress ($250.0 million), regulatory progress ($225.0 million) and approval of the product in both the U.S. and the License Territory ($350.0 million). As of December 31, 2014, $605.0 million in milestone payments had been earned by the Company under the Agreement and it may receive up to an additional $220.0 million in development, regulatory and approval milestone payments. However, clinical developments entails risks and the Company has no assurance as to whether or when the milestone targets might be achieved.
The development, regulatory and approval milestones represents non-refundable amounts that would be paid by Janssen to the Company if certain milestones are achieved in the future. The Company has elected to apply the guidance in ASC 605-28 to the milestones. These milestones, if achieved, are substantive as they relate solely to past performance, are commensurate with estimated enhancement of value associated with the achievement of each milestone as a result of the Company's performance,
81
which are reasonable relative to the other deliverables and terms of the arrangement, and are unrelated to the delivery of any further elements under the arrangement.
The Agreement includes a cost sharing arrangement for associated collaboration activities. Except in certain cases, in general, Janssen is responsible for approximately 60% of collaboration development costs and the Company is responsible for the remaining 40% of collaboration development costs. Generally, costs associated with commercialization will be included in determining pre-tax commercial profits or pre-tax commercial losses, which are to be shared 50% by the Company and 50% by Janssen.
The collaboration with Janssen provides the Company with an annual cap of its share of IMBRUVICA related research and development and commercial-related expenses, offset by pre-tax commercial profits for each calendar year. In the event that the Company's share of aggregate development costs in any given calendar year, together with any other amounts that become due from the Company, plus the Company's share of pre-tax commercial losses for any calendar quarter in such calendar year, less the Company's share of pre-tax commercial profits for any calendar quarter in such calendar year, exceeds $50.0 million, then Excess Amounts are funded by Janssen. Under the Agreement, total Excess Amounts plus interest may not exceed $225.0 million at any given time. Interest shall be accrued on the outstanding balance with interest calculated at the average annual European Interbank Offered Rate (EURIBOR) for the EURO or average annual London Interbank Offered Rate (LIBOR) for U.S. Dollars as reported in the Wall Street Journal, plus 2%, calculated on the number of days from the date on which the Company's payment would be due to Janssen. The interest rate on outstanding Excess Amounts shall not exceed 5% per annum, and the cumulative interest on Excess Amounts shall not in the aggregate exceed $25.0 million.
In the event the Excess Amounts plus interest reaches a maximum of $225.0 million, the Company shall be responsible for its share of development costs, together with any other amounts that become due from the Company, plus its share of any pre-tax commercial loss beyond such maximum. For all calendar quarters following the Company's third profitable calendar quarter for the collaboration, as determined in the Agreement, the Company can no longer add to Excess Amounts and shall be responsible for its own share of development costs along with its share of pre-tax commercial losses incurred in such quarters. Janssen may only recoup the Excess Amounts, together with interest from the Company's share of pre-tax commercial profits in calendar quarters subsequent to its third profitable calendar quarter for the collaboration until the Excess Amounts and applicable interest has been fully paid.
As per the Agreement, a profitable quarter shall mean a full calendar quarter beginning after the first commercial sale of IMBRUVICA in which the combined pre-tax profit or loss for the U.S. and the License Territory is positive and exceeds the development costs for such calendar quarter.
The Company recognizes Excess Amounts as a reduction to costs and expenses as the Company's payment of Excess Amounts to Janssen is contingent and would become payable from the Company's share of pre-tax commercial profits only after the third profitable calendar quarter for the collaboration. As of December 31, 2014, total Excess Amounts were $138.3 million, which was comprised of the cumulative amount funded by Janssen to date of $134.3 million and interest of $4.0 million. As of December 31, 2014, the Company has achieved two profitable quarters for the collaboration.
The Agreement also provides that any net profits from the commercialization of products resulting from the collaboration will be shared 50% by the Company and 50% by Janssen. Janssen has sole responsibility and exclusive rights to commercialize the products in the License Territory. The parties hold joint responsibility and co-exclusive rights to commercialize the products in the U.S., and Pharmacyclics will serve as the lead party in such effort. Janssen is commercializing IMBRUVICA outside the U.S.
In accordance with ASU No. 2009-13 (and as incorporated into ASC Topic 605-25), the Company identified all of the deliverables at the inception of the Agreement. The significant deliverables were determined to be the license, committee services, development services and commercialization services. The commercialization services represent a contingent deliverable for which there is not a significant incremental discount.
The Company has determined that the license represents a separate unit of accounting as the license, which includes rights to the underlying technologies for IMBRUVICA, has standalone value apart from the committee and development services because the development, manufacturing and commercialization rights conveyed would permit Janssen to perform all efforts necessary to bring the compound to commercialization and begin selling the drug upon regulatory approval. The Company has also determined that the committee and development services each represent individual units of accounting as they have standalone value from each other. The Company has determined its best estimate of selling prices for the license unit of accounting based on the income approach as defined in ASC 820-10-35-32. This measurement is based on the value indicated by current estimates about those future amounts and reflects management determined estimates and assumptions. These estimates and assumptions include, but are not limited to, how a market participant would use the license, estimated market opportunity and expected market share and assumed royalty rates that would be paid for sales resulting from products
82
developed using the license, similar arrangements entered into by third parties and entity-specific factors such as the terms of the Company's previous collaborative agreement, the Company's pricing practices and pricing objectives, the likelihood that clinical trials will be successful, the likelihood that regulatory approval will be received and that the products will become commercialized and the markets served. These estimates and assumptions led to an expected future cash flow which was discounted based on estimated weighted average cost of capital of 12% and royalty rates ranging from 30% to 40%. The Company has also determined its best estimate of selling prices for the committee and development services, based on the nature of the services to be performed and estimates of the associated effort as well as estimated market rates for similar services. The arrangement consideration of $150.0 million was allocated to the units of accounting based on the relative selling price method.
Of the $150.0 million upfront payment received, $70.6 million was allocated to the licenses, $15.0 million to the committee services and $64.4 million to the development services. The Company has recognized license revenue upon execution of the arrangement as the associated unit of accounting had been delivered pursuant to the terms of the Agreement. Since inception, the $15.0 million and $64.4 million allocated to committee and development services, respectively, is being recognized as revenue as the related services are provided over the estimated service periods of 17 years and 9 years, which are equivalent to the estimated remaining life of the underlying technology and the estimated remaining development period, respectively.
Janssen Collaboration Revenue
Total revenue recognized with respect to the Agreement consisted of the following (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
License and milestone revenue | $ | 220,000 | $ | 235,000 | $ | 150,000 | $ | 70,605 | |||||||
Collaboration services revenue | 17,164 | 10,970 | 4,878 | 4,017 | |||||||||||
Total | $ | 237,164 | $ | 245,970 | $ | 154,878 | $ | 74,622 | |||||||
For the year ended December 31, 2014, total collaboration services revenue from Janssen included $4.2 million of net income from the sale of the Company's product to Janssen.
As of December 31, 2014, total deferred revenue related to committee and development services under the Agreement with Janssen was $46.5 million, of which $34.9 million was included in deferred revenue non-current portion.
License and milestone revenue recognized with respect to the Agreement with Janssen consisted of the following (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
License revenue | $ | — | $ | — | $ | — | $ | 70,605 | |||||||
Approval milestone revenue | 170,000 | 60,000 | — | — | |||||||||||
Regulatory milestone revenue | 50,000 | 125,000 | — | — | |||||||||||
Development milestone revenue | — | 50,000 | 150,000 | — | |||||||||||
Total | $ | 220,000 | $ | 235,000 | $ | 150,000 | $ | 70,605 | |||||||
Janssen Collaboration Cost Sharing and Excess Amounts
The Company recognized development costs under the collaboration as a component of Research and development expense, net in the consolidated statements of operations. The Company also recognized certain selling, general and administrative expenses under the collaboration, including marketing costs, patent costs and the Company's share of pre-tax commercial losses on sales of IMBRUVICA outside of the U.S. as a component of Selling, general and administrative, net in the consolidated statements of operations. Expenses which were charged to the collaboration are as follows (in thousands):
83
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Collaboration expenses, unadjusted | $ | 155,027 | $ | 175,567 | $ | 57,690 | $ | 43,272 | |||||||
Increase (decrease) for cost sharing | (40,121 | ) | (48,223 | ) | (17,339 | ) | (18,381 | ) | |||||||
Excess Amounts | — | (85,732 | ) | (17,306 | ) | — | |||||||||
Research and development, net | $ | 114,906 | $ | 41,612 | $ | 23,045 | $ | 24,891 | |||||||
Collaboration expenses, unadjusted | $ | 88,110 | $ | 28,118 | $ | 2,426 | $ | 1,070 | |||||||
Increase (decrease) for cost sharing | 5,775 | 10,675 | 247 | (4 | ) | ||||||||||
Excess Amounts | — | (30,405 | ) | (819 | ) | — | |||||||||
Selling, general and administrative, net | $ | 93,885 | $ | 8,388 | $ | 1,854 | $ | 1,066 | |||||||
Under the Agreement, Excess Amounts would become payable only after the third profitable quarter for the collaboration from the Company's share of pre-tax commercial profits, commencing in the fourth quarter of profitability under the Agreement until the Excess Amounts and applicable interest has been fully paid. The quarters ended September 30, 2014 and December 31, 2014 represented the first and second profitable quarters for the collaboration, respectively. The Company's net profit (loss) under the Agreement for its first and second profitable quarters for the collaboration was calculated as follows (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
50% of Pharmacyclics' U.S. product revenue, net | $ | 246,209 | $ | — | $ | — | $ | — | |||||||
Less: 50% of Pharmacyclics' U.S. cost of goods sold | (20,205 | ) | — | — | — | ||||||||||
Pharmacyclics' 50% share of U.S. net product revenue, less cost of goods sold | 226,004 | — | — | — | |||||||||||
Less: Pharmacyclics' share of U.S. commercial expenses under the Agreement | (70,597 | ) | (8,006 | ) | (1,073 | ) | (1,058 | ) | |||||||
Pharmacyclics' share of U.S. pre-tax profits from the commercialization of IMBRUVICA under the Agreement | 155,407 | (8,006 | ) | (1,073 | ) | (1,058 | ) | ||||||||
Less: Pharmacyclics' share of outside-U.S. pre-tax commercial loss under the Agreement | (23,288 | ) | (382 | ) | (781 | ) | (8 | ) | |||||||
Pharmacyclics' share of worldwide pre-tax profits from the commercialization of IMBRUVICA under the Agreement | 132,119 | (8,388 | ) | (1,854 | ) | (1,066 | ) | ||||||||
Less: Pharmacyclics' share of world-wide research and development expenses under the Agreement | (114,906 | ) | (41,612 | ) | (23,045 | ) | (24,891 | ) | |||||||
Pharmacyclics' share of IMBRUVICA related pre-tax net profit under the Agreement | $ | 17,213 | $ | (50,000 | ) | $ | (24,899 | ) | $ | (25,957 | ) | ||||
For the year ended December 31, 2013, Janssen's 50% share of U.S. product revenue, net less cost of goods sold of IMBRUVICA of $5.0 million was fully absorbed by Excess Amounts funded by Janssen that were recorded as a reduction to selling, general and administrative, net. For the year ended December 31, 2014, the Company determined Janssen's 50% share of U.S. net product revenue less cost of goods sold of IMBRUVICA, which was included in costs of collaboration in the Company's consolidated statement of operations as follows (in thousands):
84
Year Ended December 31, | |||
2014 | |||
Product revenue, net | $ | 492,418 | |
Less: Cost of goods sold | 40,410 | ||
452,008 | |||
Janssen's share of U.S. pre-tax commercial profits | 50 | % | |
Total costs of collaboration (Janssen's 50% share under the Agreement) | $ | 226,004 | |
Pharmacyclics' 50% share of U.S. net product revenue less cost of goods sold | $ | 226,004 | |
Under the Agreement, the Company recorded Excess Amounts as follows:
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | ||||||||||||||
2014 | 2013 | 2012 | 2012 | |||||||||||||
Research and development | $ | — | $ | 85,732 | $ | 17,306 | $ | — | ||||||||
General and administrative | — | 30,405 | 819 | — | ||||||||||||
Total Excess Amounts | $ | — | $ | 116,137 | $ | 18,125 | $ | — | ||||||||
As of December 31, 2014, the Company's had $27.0 million receivable from Janssen, which consisted primarily of a milestone of $20.0 million, $3.9 million related to material and product sales and $1.7 million related to value added taxes. As of December 31, 2013, the Company's receivable from collaboration partner balance of $52.0 million was comprised primarily of $50.2 million due from Janssen under the collaboration agreement. The receivable from Janssen is included within Receivable from collaboration partners on the consolidated balance sheets.
As of December 31, 2014, the Company had $79.8 million payable to Janssen, of which $76.7 million was related to Janssen's share of pre-tax commercial profits, $2.8 million was related to cost-sharing under the Agreement and $0.3 million was related to value added taxes.
As of December 31, 2014, total Excess Amounts of $138.3 million (which comprises the cumulative amount funded by Janssen to-date of $134.3 million and interest of $4.0 million) would become payable once the Company reaches a third profitable calendar quarter for the product. Further, Excess Amounts shall be reimbursable only from the Company's share of pre-tax profits after the third profitable calendar quarter for the product.
Collaboration and License Agreement with Servier
In April 2009, the Company entered into a collaboration and license agreement with Servier to research, develop and commercialize abexinostat (PCI-24781), an orally active, novel, small molecule inhibitor of pan-HDAC enzymes. Under the terms of the agreement, Servier acquired the exclusive right to develop and commercialize the pan-HDAC inhibitor product worldwide except for the U.S. The Company and Servier terminated the collaboration and license agreement effective November 23, 2014. Upon the termination of the alliance, Servier’s rights to ex-U.S. development and commercialization of the Company's pan-HDAC inhibitor compounds will be returned to the Company, giving the Company full global development and commercialization rights. As of December 31, 2014, the Company's estimated costs associated with the termination of the Servier agreement of $0.8 million was recorded within accrued liabilities on the consolidated balance sheet.
Total revenue recognized with respect to the Company's collaboration and license agreement with Servier consisted of the following (in thousands):
85
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
License and milestone revenue | $ | — | $ | — | $ | — | $ | 7,000 | |||||||
Collaboration services revenue | 139 | 159 | 93 | 157 | |||||||||||
Total | $ | 139 | $ | 159 | $ | 93 | $ | 7,157 | |||||||
License Agreement with Novo Nordisk A/S
In October 2012, the Company entered into a license agreement with Novo Nordisk A/S (Novo Nordisk). Under the terms of the agreement, Novo Nordisk acquired the exclusive worldwide rights for the Company's small molecule Factor VIIa inhibitor, PCI-27483, as an excipient in a product containing a Novo Nordisk active pharmaceutical ingredient for a restricted disease indication outside of oncology. Novo Nordisk will utilize PCI-27483 as an excipient in a product within Novo Nordisk's biopharmaceutical unit. Novo Nordisk is solely responsible for all further research and development activities within the restricted disease indication outside of oncology.
In connection with entering into the license agreement with Novo Nordisk, the Company received an upfront payment of $5.0 million in October 2012. In addition, the Company may receive up to $55.0 million based on the achievement of certain development, regulatory and sales milestones. Upon commercialization, the Company will also receive low single digit tiered royalties on Novo Nordisk's net sales of biopharmaceutical formulations utilizing the addition of PCI-27483.
On June 28, 2013, the Company entered into an amended and restated license agreement with Novo Nordisk to expand the scope of the license granted by the Company to Novo Nordisk in October 2012. Under the amended and restated license agreement with Novo Nordisk, the Company has no additional obligations related to the delivery of the license.
Total revenue recognized with respect to the Company's license agreement with Novo Nordisk consisted of the following (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
License and milestone revenue | $ | — | $ | — | $ | 5,000 | $ | — | |||||||
Collaboration services revenue | 8 | 454 | 631 | — | |||||||||||
Total | $ | 8 | $ | 454 | $ | 5,631 | $ | — | |||||||
Acquired Products
Celera Corporation
Background
In April 2006, the Company acquired multiple small molecule drug candidates for the treatment of cancer and other diseases from Celera Genomics, an Applera Corporation business (now Celera Corporation - a subsidiary of Quest Diagnostics Incorporated). Future milestone payments under the agreement, as amended, could total as much as approximately $97.0 million although the Company currently cannot predict if or when any of the milestones will be achieved. Approximately two-thirds of the milestone payments relate to the Company's HDAC inhibitor program and approximately one-third relates to the Company's Factor VIIa inhibitor program. Approximately 90% of the potential future milestone payments would be paid to Celera after obtaining regulatory approval in various countries. To date, no milestone payments have been triggered related to the Company's HDAC inhibitor or Factor VIIa programs.
In addition to the milestone payments, the Company is required to make single-digit royalty payments based on annual sales of IMBRUVICA and would be required to make single-digit royalty payments based on annual sales of drugs commercialized from the Company's HDAC inhibitor, Factor VIIa inhibitor and certain other BTK inhibitor programs. For the year ended December 31, 2014 and year ended December 31, 2013, the Company recognized royalty expense under the Celera agreement of approximately $27.4 million and $1.0 million, respectively, on net product sales of IMBRUVICA. Royalty
86
expense related to net product sales of IMBRUVICA is included within cost of goods sold in the consolidated statement of operations. No royalty expense was incurred under the Celera agreement prior to the year ended December 31, 2013.
For any BTK inhibitor product or Factor VIIa inhibitor product obtained from Celera, the agreement with Celera expires on a product-by-product and country-by-country basis. The term of the agreement for a given BTK inhibitor product or Factor VIIa inhibitor product shall expire in a given country upon the expiration of the last-to-expire Celera patent assigned to the Company that covers the manufacture, use, sale, offer for sale, or importation of such product in such country. For any HDAC inhibitor product obtained from Celera, the agreement with Celera expires on a product-by-product and country-by-country basis. The term of the agreement for a given HDAC inhibitor product shall expire in a given country upon the expiration of the last-to-expire Celera patent assigned to the Company that covers the sale of such product in such country.
The Company may terminate the agreement with Celera in its entirety, or with respect to one or more of the three classes of products (BTK inhibitor products, HDAC inhibitor products and Factor VIIa inhibitor products) obtained from Celera, at any time by giving Celera at least 60 days' prior written notice. If the Company terminates the agreement with respect to a particular class of products, ownership of the Celera intellectual property assigned to the Company relating to the products in the terminated product class will revert to Celera. If the Company terminates the agreement in its entirety, ownership of all of the Celera intellectual property assigned to the Company will revert to Celera.
The agreement with Celera may be terminated effective immediately upon a party's written notice to the other party for a breach by the other party that remains uncured for 90 days after notice of the breach is given to the breaching party. If the Company breaches the agreement only with respect to one or two of the three classes of products obtained from Celera, but not with respect to all three classes of products, and if the Company's breach remains uncured for 90 days after the Company has received notice of breach from Celera, Celera may terminate the agreement solely with respect to the class or classes of products affected by the Company's breach, but may not terminate the agreement with respect to the class or classes of products unaffected by the Company's breach.
NOTE 6 — CASH, CASH EQUIVALENTS AND MARKETABLE SECURITIES
The following tables set forth the Company's cash, cash equivalents and marketable securities (in thousands):
As of December 31, | |||||||
2014 | 2013 | ||||||
Cash - Demand deposits | $ | 471,916 | $ | 225,035 | |||
Cash equivalents - money market funds | 373,119 | 129,193 | |||||
Cash equivalents - U.S. treasury bills | — | 269,488 | |||||
Cash equivalents - Certificates of deposit - FDIC insured | — | 240 | |||||
Cash and cash equivalents | $ | 845,035 | $ | 623,956 | |||
As of December 31, | |||||||
2014 | 2013 | ||||||
Certificates of deposit - FDIC insured | $ | 11,952 | $ | 11,672 | |||
Marketable securities | $ | 11,952 | $ | 11,672 | |||
To date, the Company has not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value. In determining whether a decline is other than temporary, the Company considers various factors including the length of time and extent to which the market value has been less than cost, the financial condition and near-term prospects of the issuer and the Company's intent and ability to retain its investment in the issuer for a period of time sufficient to allow for any anticipated recovery in market value.
For the year ended December 31, 2014 and year ended December 31, 2013, gross realized losses and gains on the sale of available-for-sale securities were not material.
The following is a summary of the Company's available-for-sale securities measured a amortized costs, gross unrealized gains, gross unrealized losses and fair value (in thousands):
87
As of December 31, 2014 | Amortized Cost | Gross Unrealized Gain | Gross Unrealized Loss | Fair Value | ||||||||||||
Certificates of deposit – FDIC insured | $ | 11,960 | $ | — | $ | (8 | ) | $ | 11,952 | |||||||
Marketable Securities | $ | 11,960 | $ | — | $ | (8 | ) | $ | 11,952 | |||||||
As of December 31, 2013 | Amortized Cost | Gross Unrealized Gain | Gross Unrealized Loss | Fair Value | ||||||||||||
Certificates of deposit – FDIC insured | $ | 11,680 | $ | — | $ | (8 | ) | $ | 11,672 | |||||||
Marketable Securities | $ | 11,680 | $ | — | $ | (8 | ) | $ | 11,672 | |||||||
As of December 31, 2014, the Company's marketable securities had the following remaining contractual maturities (in thousands):
Amortized Cost | Fair Value | ||||||
Less than one year | $ | 11,960 | $ | 11,952 | |||
The following table sets forth the basis of fair value measurements for the Company's cash equivalents and available-for-sale securities (in thousands):
As of December 31, 2014 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Fair Value | ||||||||||||
Cash equivalents: | |||||||||||||||
Money market funds | $ | 373,119 | $ | — | $ | — | $ | 373,119 | |||||||
Marketable securities: | |||||||||||||||
Certificates of deposit- FDIC insured | — | 11,952 | — | 11,952 | |||||||||||
$ | 373,119 | $ | 11,952 | $ | — | $ | 385,071 | ||||||||
As of December 31, 2013 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Fair value | ||||||||||||
Cash equivalents: | |||||||||||||||
U.S. treasury bills | $ | 269,488 | $ | — | $ | — | $ | 269,488 | |||||||
Money market funds | 129,193 | — | — | 129,193 | |||||||||||
Certificates of deposit - FDIC insured | — | 240 | — | 240 | |||||||||||
Marketable securities: | |||||||||||||||
Certificates of deposit - FDIC insured | — | 11,672 | — | 11,672 | |||||||||||
$ | 398,681 | $ | 11,912 | $ | — | $ | 410,593 | ||||||||
There were no transfers between levels of the fair value hierarchy during fiscal year 2014.
NOTE 7 — BALANCE SHEET COMPONENTS
Property and equipment, net consists of the following (in thousands):
88
As of December 31, | ||||||||
2014 | 2013 | |||||||
Equipment | $ | 13,363 | $ | 11,827 | ||||
Leasehold improvements | 9,951 | 5,493 | ||||||
Furniture and fixtures | 1,278 | 1,278 | ||||||
Construction in progress | 19,461 | 14,592 | ||||||
44,053 | 33,190 | |||||||
Less: Accumulated depreciation and amortization | (11,577 | ) | (7,719 | ) | ||||
$ | 32,476 | $ | 25,471 | |||||
Depreciation and amortization of property and equipment was $3.9 million for the year ended December 31, 2014, $2.2 million for the year ended December 31, 2013, $0.6 million for the six months ended December 31, 2012 and $0.6 million for the year ended June 30, 2012.
Accrued liabilities consist of the following (in thousands):
As of December 31, | ||||||||
2014 | 2013 | |||||||
Accrued payroll and employee related expenses | $ | 19,645 | $ | 20,500 | ||||
Accrued discounts and rebates | 18,125 | 906 | ||||||
Accrued clinical related | 14,806 | 17,062 | ||||||
Accrued royalty to Celera (see Note 5) | 10,646 | 949 | ||||||
Accrued value added taxes | 7,395 | 7,041 | ||||||
Accrued outside services | 5,114 | 5,077 | ||||||
Accrued contract manufacturing | 2,380 | 1,556 | ||||||
Accrued marketing | 1,288 | — | ||||||
Accrued property and equipment purchases | 934 | 4,039 | ||||||
Accrued inventory | 101 | 7,592 | ||||||
Accrued other | 7,845 | 6,466 | ||||||
$ | 88,279 | $ | 71,188 | |||||
Deferred revenue consists of the following (in thousands):
Current portion: | As of December 31, | |||||||
2014 | 2013 | |||||||
Deferred revenue related to the Agreement (see Note 5) | $ | 11,656 | $ | 7,505 | ||||
Deferred revenue from sales to collaboration partner (see Note 5) | 7,117 | 76 | ||||||
$ | 18,773 | $ | 7,581 | |||||
Non-current portion: | As of December 31, | |||||||
2014 | 2013 | |||||||
Deferred revenue related to the Agreement (see Note 5) | $ | 34,885 | $ | 52,025 | ||||
89
Note 8 – Intangible Assets
On April 15, 2014, the Company entered into an agreement with a third party to acquire technology assets in the amount of $9.3 million in cash. The Company expects to amortize the technology assets over 13 years which reflects the estimated remaining useful life of the technology assets. Amortization expense was $0.5 million for the year ended December 31, 2014. Accumulated amortization as of December 31, 2014 was $0.5 million.
As of December 31, 2014, the future annual amortization expense for the Company's intangible assets is expected to be as follows (in thousands):
Fiscal Year | Future Amortization of Intangible Assets | |||
2015 | $ | 728 | ||
2016 | 730 | |||
2017 | 728 | |||
2018 | 728 | |||
2019 | 728 | |||
Thereafter | 5,088 | |||
Total | $ | 8,730 | ||
NOTE 9 — STOCKHOLDERS' EQUITY
Common stock
Public Offering
In March 2013, the Company sold 2.2 million shares of its common stock in an underwritten public offering at $94.20 per share for net proceeds of $201.0 million after deducting expenses of the offering. The closing of the offering took place on March 13, 2013.
Preferred stock
As amended, the Company's Certificate of Incorporation authorizes 1.0 million shares of preferred stock, par value $0.0001 per share. The Board of Directors is authorized to issue the preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions thereof, including dividend rights, dividend rates, conversion rights, voting rights, terms of redemption, redemption prices, liquidation preferences and the number of shares constituting any series or the designation of such series, without further vote or action by the stockholders. As of December 31, 2014 and 2013, no preferred stock was issued or outstanding.
The ability of the Company's Board of Directors to issue shares of preferred stock without stockholder approval may have certain anti-takeover effects. The Company is also subject to provisions of the Delaware General Corporation Law, which may make certain business combinations more difficult.
Stock plans
Equity Incentive Award Plans
The Company maintains three stock-based compensation plans: the 2014 Equity Incentive Award Plan (2014 Plan), which was approved during the Company's annual meeting of stockholders on May 8, 2014, the 2004 Equity Incentive Award Plan (2004 Plan) and the Employee Stock Purchase Plan (ESPP). The 2014 Plan serves as the successor to the 2004 Plan and provides for the issuance of various forms of awards including among others, incentive and non-qualified stock options and restricted stock units (RSUs). The exercise price of all stock options granted under the 2014 Plan may not be less than the fair market value of the Company's common stock on the date of grant and no stock option will be exercisable more than ten years after the date it is granted. Stock options and restricted stock units granted to employees generally vest over four years. The Company settles employee stock option exercises, ESPP purchases, and the vesting of RSUs with newly issued common shares.
90
The following table summarizes stock option activity related to shares of common stock under the Company's stock option plans (in thousands, except per share amounts and years data):
Options Outstanding | |||||||||||||
Number of Options | Weighted Average Exercise Price per Share | Weighted Average Remaining Contractual Term (in Years) | Aggregate Intrinsic Value | ||||||||||
Balance at December 31, 2013 | 5,111 | $ | 23.54 | ||||||||||
Exercised | (1,682 | ) | 11.93 | ||||||||||
Granted | 1,142 | 91.68 | |||||||||||
Forfeited or expired | (371 | ) | 59.84 | ||||||||||
Balance at December 31, 2014 | 4,200 | 43.50 | 5.7 | $ | 332,507 | ||||||||
Vested and expected to vest at December 31, 2014 | 4,073 | $ | 41.72 | 6.6 | $ | 328,684 | |||||||
The above table excludes approximately 0.8 million options which comprise the portion of performance options granted for which the performance criteria had not been established as of December 31, 2014.
The following table summarizes restricted stock unit activity under the Company's 2014 Plan (in thousands, except per share amounts and years data):
Number of Options | Weighted Average Grant Date Fair Value | Weighted Average Remaining Contractual Term (in Years) | Aggregate Intrinsic Value | ||||||||||
Outstanding at December 31, 2013 | — | $ | — | ||||||||||
Granted | 211 | 114.98 | |||||||||||
Vested and released | (13 | ) | 121.89 | ||||||||||
Forfeited | (10 | ) | 100.09 | ||||||||||
Outstanding and unvested at December 31, 2014 | 188 | $ | 115.32 | 2.0 | $ | 23,052 | |||||||
Expected to vest at December 31, 2014 | 163 | $ | 115.61 | 1.9 | $ | 19,960 | |||||||
The above table excludes approximately 0.1 million restricted stock units which comprise the portion of performance RSUs granted for which the performance criteria had not been established as of December 31, 2014.
The components of stock-based compensation recognized in the Company's consolidated statements of operations were as follows (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | ||||||||||||||
2014 | 2013 | 2012 | 2012 | |||||||||||||
Cost of goods sold | $ | 1,453 | $ | 436 | $ | — | $ | — | ||||||||
Research and development | 22,710 | 24,041 | 5,357 | 6,947 | ||||||||||||
Selling, general and administrative | 29,578 | 25,733 | 2,079 | 2,926 | ||||||||||||
Total stock-based compensation | $ | 53,741 | $ | 50,210 | $ | 7,436 | $ | 9,873 | ||||||||
During the year ended December 31, 2014 and the year ended December 31, 2013, the Company capitalized stock-based compensation of approximately $0.9 million and $0.2 million, respectively. The Company capitalized no stock-based compensation during the six months ended December 31, 2012 and the year ended June 30, 2012, as these periods were prior to the commercialization of its product.
The fair value of each stock option is estimated using the Black Scholes option-pricing model. The expected term of stock options granted represents the period of time that stock options are expected to be outstanding. The Company generally does
91
not expect substantially different exercise or post-vesting termination behavior among its employee or non-employee population. As such, for the majority of stock options granted and the Company's Employee Stock Purchase Plan, the Company generally calculates and applies an overall expected term assumption based on historical data. In certain cases, the Company uses a shorter expected term for performance-based stock options based on a combination of historical data and management's estimates of the period of time that options will be outstanding. The risk-free interest rate is based on a zero-coupon U.S. Treasury bond whose maturity period equals the expected term of the Company's options.
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||
2014 | 2013 | 2012 | 2012 | ||||||||
Employee stock options (1): | |||||||||||
Expected dividend yield | — | — | — | — | |||||||
Expected stock price volatility (2) | 46.9% - 67.4% | 50.9% - 84.8% | 51.5% - 85.5% | 88 | % | ||||||
Risk free interest rate (2) | 0.7% - 1.8% | 0.5% - 1.5% | 0.3% - 0.8% | 1.00 | % | ||||||
Expected life (years) (2) | 2.52 - 5.04 | 2.52 - 5.20 | 2.60 - 5.20 | 5.00 | |||||||
Non-employee stock options: | |||||||||||
Expected dividend yield | — | — | — | — | |||||||
Expected stock price volatility | 79 | % | 79 | % | 89 | % | 84% - 89% | ||||
Risk free interest rate | 2.6 | % | 1.9 | % | 3.89 | % | 2.00% - 3.89% | ||||
Expected life (years) | 10.00 | 10.00 | 8.00 | 8.10 – 10.00 | |||||||
(1) The above table includes assumptions used in the valuation of performance-based stock option grants for which the Company set performance criteria during the period.
(2) Amounts for the year ended June 30, 2012 are presented on a weighted average basis.
The weighted average grant date fair value for employee options granted under the Company's stock option plans during the year ended December 31, 2014, the year ended December 31, 2013, the six months ended December 31, 2012, and the year ended June 30, 2012 was $59.28, $55.32, $43.55 and $11.44, respectively.
The total pre-tax intrinsic value of stock options exercised during the year ended December 31, 2014, the year ended December 31, 2013, the six months ended December 31, 2012, and the year ended June 30, 2012 was $196.6 million, $145.5 million, $45.8 million and $17.3 million, respectively.
For the year ended December 31, 2014 and year ended December 31, 2013, the Company recognized a tax benefit from stock-based compensation of $34.9 million and $30.0 million, respectively, as a decrease to income taxes payable, with a corresponding increase to additional paid-in capital. No income tax benefits were recognized during the six months ended December 31, 2012 and the year ended June 30, 2012.
As of December 31, 2014, there were approximately 2.0 million shares reserved for issuance and available for grant under the 2014 Plan.
As of December 31, 2014, $87.6 million of total unrecognized compensation costs related to non vested employee options are scheduled to be recognized over a weighted average period of 2.6 years.
The total fair value of shares vested for the year ended December 31, 2014, the year ended December 31, 2013, the six months ended December 31, 2012 and the year ended June 30, 2012 was $43.3 million, $37.4 million, $3.7 million and $5.6 million, respectively.
A summary of outstanding, exercisable and vested stock options as of December 31, 2014 is as follows (in thousands, except per share amounts):
92
Options Outstanding | Exercisable | Exercisable and Vested | |||||||||||||||||||||||||||||||||||
Range of Exercise Prices | Number of Shares | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price Per Share | Aggregate Intrinsic Value | Number of Shares | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price Per Share | Aggregate Intrinsic Value | Number of Shares | Weighted Average Exercise Price Per Share | Aggregate Intrinsic Value | ||||||||||||||||||||||||||
$0.75 - $6.49 | 837 | 4.28 | $ | 2.57 | 837 | 4.28 | $ | 2.57 | 822 | $ | 2.50 | ||||||||||||||||||||||||||
$6.52 - $12.00 | 851 | 5.41 | $ | 7.73 | 831 | 5.43 | $ | 7.67 | 815 | $ | 7.61 | ||||||||||||||||||||||||||
$12.10 - $32.38 | 863 | 6.78 | $ | 20.28 | 796 | 6.76 | $ | 20.28 | 489 | $ | 18.62 | ||||||||||||||||||||||||||
$36.73 - $86.78 | 924 | 7.72 | $ | 68.11 | 924 | 7.72 | $ | 68.11 | 437 | $ | 66.52 | ||||||||||||||||||||||||||
$87.19 - $102.22 | 1,013 | 9.11 | $ | 96.07 | 91 | 8.58 | $ | 90.62 | 91 | $ | 90.62 | ||||||||||||||||||||||||||
$102.77 - $151.48 | 521 | 9.26 | $ | 119.51 | 44 | 8.69 | $ | 113.87 | 43 | $ | 113.87 | ||||||||||||||||||||||||||
5,009 | 7.03 | $ | 49.67 | $ | 364,377 | 3,523 | 6.18 | $ | 28.62 | $ | 329,982 | 2,697 | $ | 22.11 | $ | 270,222 | |||||||||||||||||||||
Employee Stock Purchase Plan
The Company adopted an Employee Stock Purchase Plan (the "Purchase Plan") in August 1995. Qualified employees may elect to have a certain percentage of their salary withheld to purchase shares of the Company's common stock under the Purchase Plan. The purchase price per share is equal to 85% of the fair market value of the stock on specified dates.
The number of shares purchased under the Purchase Plan in the year ended December 31, 2014, the year ended December 31, 2013, the six months ended December 31, 2012, and the year ended June 30, 2012 were approximately 0.1 million, 0.2 million, 0.1 million, and 0.2 million shares of common stock at an average price of $80.55, $21.65, $13.06, and $5.19 per share, respectively. In May 2013, stockholders approved an increase of 0.3 million shares available for issuance under the Purchase Plan. As of December 31, 2014, approximately 0.5 million shares were available for future purchase under the Purchase Plan.
Compensation cost is estimated using the Black Scholes option-pricing model using the weighted average assumptions noted in the following table:
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||
2014 | 2013 | 2012 | 2012 | ||||||||
Expected dividend yield | — | — | — | — | |||||||
Stock price volatility | 49 | % | 48 | % | 50 | % | 54 | % | |||
Risk free interest rate | 0.30 | % | 0.16 | % | 0.16 | % | 0.15 | % | |||
Expected life (years) | 1.59 | 1.10 | 0.68 | 1.14 | |||||||
The weighted average estimated grant date fair value of purchase awards under the Company's employee stock purchase plan during the year ended December 31, 2014, the year ended December 31, 2013, six months ended December 31, 2012, and the year ended June 30, 2012 was $36.79, $41.19, $33.32 and $6.38 per share, respectively.
As of December 31, 2014, $4.4 million of total unrecognized compensation costs related to purchase awards under the Company's employee stock purchase plan were scheduled to be recognized over a weighted average period of 1 year.
NOTE 10 — EMPLOYEE BENEFIT PLAN
The Company maintains a defined contribution plan covering substantially all U.S. employees under Section 401(k) of the Internal Revenue Code and a defined contribution plan for non-U.S. employees. The Company's matching contribution to the plan was $1.9 million, $1.0 million, $0.1 million, $0.2 million for the year ended December 31, 2014, the year ended December 31, 2013, the six months ended December 31, 2012, and the year ended June 30, 2012, respectively.
NOTE 11 — INCOME TAXES
The components of the provision for income taxes are as follows (in thousands):
93
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Current: | |||||||||||||||
Federal | $ | 31,938 | $ | 30,879 | $ | 2,047 | $ | 26 | |||||||
State | 4,294 | 202 | 599 | — | |||||||||||
Foreign | 91 | 45 | 1 | 13 | |||||||||||
Total provision for income taxes | 36,323 | 31,126 | 2,647 | 39 | |||||||||||
Income before taxes | $ | 122,447 | $ | 98,136 | $ | 120,180 | $ | 12,025 | |||||||
Tax rate | 29.66 | % | 31.72 | % | 2.20 | % | 0.33 | % | |||||||
The following is a geographical breakdown of consolidated income before income taxes by income tax jurisdiction (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
United States | $ | 100,270 | $ | 70,254 | $ | 69,563 | $ | (4,596 | ) | ||||||
Foreign | 22,177 | 27,882 | 50,617 | 16,621 | |||||||||||
Income before taxes | $ | 122,447 | $ | 98,136 | $ | 120,180 | $ | 12,025 | |||||||
Net deferred tax assets (liabilities) are summarized as follows (in thousands):
As of December 31, | ||||||||
2014 | 2013 | |||||||
Deferred tax assets: | ||||||||
Net operating loss carryforwards | $ | 4,546 | $ | 3,552 | ||||
Tax credit carryforwards | 25,978 | 26,110 | ||||||
Capitalized research and development costs | 200 | 230 | ||||||
Depreciation and amortization | 775 | 916 | ||||||
Partnership investment | 4,922 | — | ||||||
Stock-based compensation | 16,320 | 9,712 | ||||||
Reserves and accruals | 18,039 | 18,557 | ||||||
Gross deferred tax assets | 70,780 | 59,077 | ||||||
Less: valuation allowance | (70,350 | ) | (58,679 | ) | ||||
Net deferred tax assets | 430 | 398 | ||||||
Deferred tax liabilities: | ||||||||
Depreciation | (430 | ) | (398 | ) | ||||
Net Deferred tax assets | $ | — | $ | — | ||||
A full valuation allowance has been established for the Company's deferred tax assets as of December 31, 2014 and December 31, 2013, since realization of such assets through the generation of future taxable income is uncertain. The increases in the valuation allowance were $11.7 million, and $26.8 million for year ended December 31, 2014 and the year ended December 31, 2013, respectively.
The Company intends to continue maintaining a full valuation allowance on its deferred tax assets until there is sufficient evidence to support the reversal of all or some portion of these allowances. However, should there be greater market demand for or greater market acceptance of IMBRUVICA than management has currently forecasted, there is a reasonable possibility
94
that within the next year sufficient positive evidence may become available to reach a conclusion that a significant portion of the valuation allowance will no longer be needed. As such, the Company may release a significant portion of its valuation allowance against its deferred tax assets within the next 12 months. This release would result in the recognition of certain deferred tax assets and a decrease to income tax expense for the period such release is recorded.
The provision for income taxes differs from the amount determined by applying the U.S. statutory income tax rate of 35% for the year ended December 31, 2014, year ended December 31, 2013, the six months ended December 31, 2012, and year ended June 30, 2012 to the income before income taxes as summarized below (in thousands):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Tax provision (benefit) at statutory rate | $ | 42,857 | $ | 34,347 | $ | 42,064 | $ | 4,209 | |||||||
State Tax, net of federal benefit | 3,156 | 202 | 389 | — | |||||||||||
Research and development and orphan drug credits | (16,944 | ) | (21,774 | ) | — | (529 | ) | ||||||||
Deferred tax assets benefited (not benefited) | 7,151 | 28,376 | (35,181 | ) | (5,727 | ) | |||||||||
Stock-based compensation | 4,532 | 5,261 | 1,056 | 2,009 | |||||||||||
Other | 3,238 | 744 | (629 | ) | 38 | ||||||||||
Federal – alternative minimum tax | — | — | 2,047 | 26 | |||||||||||
Foreign taxes greater than (less than) U.S. rates | (7,667 | ) | (16,030 | ) | (7,099 | ) | 13 | ||||||||
$ | 36,323 | $ | 31,126 | $ | 2,647 | $ | 39 | ||||||||
At December 31, 2014 and December 31, 2013, the Company had federal net operating loss carryforwards of approximately $9.4 million and $10.1 million, respectively. At December 31, 2014 and December 31, 2013, the Company had state net operating loss carryforwards of approximately $58.0 million and $37.3 million, respectively. The state operating losses are attributable to stock option benefits which will be recorded to equity when they reduce cash taxes payable. The federal and state net operating loss carryforwards will begin to expire in 2016. At December 31, 2014 and December 31, 2013, federal tax research credit carryforwards of $9.1 million and $6.9 million, respectively and state tax research credit carryforwards of $8.9 million and $5.5 million, respectively, are available to offset future taxable income. At December 31, 2014 and December 31, 2013, federal orphan drug credits of $71.1 million and $48.6 million, respectively, are also available to offset future taxable income. The federal tax research and orphan drug credits can be carried forward 20 years and will begin to expire in 2032. State tax research credits can be carried forward indefinitely. At December 31, 2014 and December 31, 2013, the Company also had federal and state alternative minimum tax credits of $1.5 million and $2.4 million, respectively, that can be used in the future that have no expiration date.
The Company is tracking the portion of its net operating losses attributable to stock option benefits in a separate memo account pursuant to ASC 718-740. Therefore, these amounts are no longer included in the Company's gross or net deferred tax assets. Pursuant to ASC 718-740-25-10, the stock option benefits of approximately $2.1 million will be only recorded to equity when they reduce cash taxes payable.
The Company reviewed whether the utilization of its net operating losses and research credits are subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Utilization of these carryforwards is restricted and results in some amount of these carryforwards expiring prior to benefiting the Company. The deferred tax assets shown above have been adjusted to reflect these expiring carryforwards.
The following table summarizes the activity related to the Company's gross amounts of unrecognized tax benefits (in thousands):
95
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Beginning balance | $ | 15,976 | $ | 2,845 | $ | 2,307 | $ | 1,726 | |||||||
Additions based on tax positions related to current year | 11,520 | 10,075 | 538 | 581 | |||||||||||
Additions (reduction) for tax positions of prior years | (623 | ) | 3,056 | — | — | ||||||||||
Ending balance | $ | 26,873 | $ | 15,976 | $ | 2,845 | $ | 2,307 | |||||||
The total amount of the unrecognized tax benefits if recognized would be an adjustment to the amount of deferred tax assets reported. The Company may from time to time be assessed interest or penalties by major tax jurisdictions, although there have been no such assessments historically. In the event the Company receives an assessment for interest and/or penalties, it would be classified in the financial statements as income tax expense. As of December 31, 2014, all tax years in the U.S. remain open due to the taxing authorities' ability to adjust operating loss carry forwards. The Company does not expect any material changes to the unrecognized tax benefits reported above during the next twelve months.
U.S. income taxes were not provided on approximately $65.9 million of undistributed earnings of certain non-U.S. subsidiaries. The amount of the unrecognized deferred tax liability for temporary differences relates to investments in these non-U.S. subsidiaries that are essentially permanent in duration and the Company anticipates that, if ever distributed, such amounts would not be taxed at greater than 40%. The Company has not provided U.S. income taxes and foreign withholding taxes on the undistributed earnings of foreign subsidiaries as of December 31, 2014, because the Company intends to permanently reinvest such earnings outside the U.S. As of December 31, 2014, the amount of potential U.S. income tax of a future hypothetical distribution would not trigger a significant U.S. tax liability because the Company has tax attribute carryovers available that it can use to reduce its liability.
During the year ended December 31, 2013, the Company was notified by the Internal Revenue Service (IRS) that it will be audited for the tax years ended June 30, 2012 and 2011. In the third quarter of 2014, the IRS issued an Agreed Tax Change (Form 4549) accepting a proposed adjustment to reduce the Company's R&D credit forward by $0.3 million for the tax year ended June 30, 2011. As of December 31, 2014, no additional adjustments have been proposed by the IRS.
In December 2014, Congress passed the Tax Increase Prevention Act which included a retroactive renewal of the Federal R&D credit back to January 1, 2014, which had previously expired. The impact of this retroactive tax legislation will not affect the Company's fiscal 2014 effective tax rate, but will reduce future income tax payments.
NOTE 12 — COMMITMENTS AND CONTINGENCIES
Facilities Lease
As of December 31, 2014, the Company leases a total of 132,176 square feet for its corporate headquarters in Sunnyvale, California. Of this total space, 79,776 square feet are leased under an operating lease that expires in November 2017, with an option to extend the lease term for an additional five years. The remaining 52,400 square feet are leased under an operating lease that expires in February 2023, which provides for an option to early terminate the lease in 2018 and has an option to extend the lease term for an additional five years. In addition, the Company leases approximately 8,000 square feet for its Pharmacyclics Switzerland GmbH office under an agreement that expires in May 2015 and leases 7,000 square feet of office space in South San Francisco, California under an operating lease which expires in March 2015. In January 2015, the Company entered into a lease agreement for additional office space for its corporate headquarters in Sunnyvale, California for approximately 36,000 square feet with annual rental expense of $0.5 million under an operating lease that expires in May 2016. In addition, in February 2015, the Company entered into a lease agreement for approximately 10,000 square feet of office space in South San Francisco, California, with annual rental expense of $0.3 million under an operating lease, expiring in February 2017.
The Company recognizes rental expense under the lease on a straight line basis over the lease term. Differences between the straight line rent expense and rent payments are classified as deferred rent liability on the balance sheet. As of December 31, 2014, future minimum lease payments under non-cancelable operating leases are as follows (in thousands):
96
Operating Lease Commitments | |||
2015 | $ | 2,226 | |
2016 | 2,161 | ||
2017 | 2,112 | ||
2018 | 857 | ||
2019 | 882 | ||
Thereafter | 2,890 | ||
Total | $ | 11,128 | |
Rent expense for the year ended December 31, 2014, year ended December 31, 2013, six months ended December 31, 2012, and the year ended June 30, 2012 was $2.5 million, $1.8 million, $0.6 million, and $1.0 million, respectively.
Purchase Commitments
The Company had non-cancelable purchase obligations for approximately $85.9 million, and $34.8 million as of December 31, 2014 and December 31, 2013, respectively.
Excess amounts under collaboration and license agreement with Janssen
The Company's worldwide collaboration and license agreement with Janssen provides the Company with an annual cap of its share of development costs and pre-tax commercial losses for each calendar year until the third profitable calendar quarter for the collaboration, as determined in the agreement, and any Excess Amounts are funded by Janssen. Janssen may recoup the Excess Amounts, together with interest, from the Company's share of pre-tax commercial profits after the third profitable calendar quarter for the collaboration until the Excess Amounts and applicable interest has been fully paid. As of December 31, 2014, total Excess Amounts were $138.3 million, which was comprised of the cumulative amount funded by Janssen to-date of $134.3 million and interest of $4.0 million. As of December 31, 2014, we have achieved two profitable quarters for the collaboration (see Note 5).
Legal Proceedings
The Company may be involved, from time to time, in legal proceedings and claims arising in the ordinary course of its business. Such matters are subject to many uncertainties and outcomes are not predictable with assurance. The Company accrues amounts, to the extent they can be reasonably estimated, that it believes are adequate to address any liabilities related to legal proceedings and other loss contingencies that the Company believe will result in a probable loss. While there can be no assurances as to the ultimate outcome of any legal proceeding or other loss contingency involving the Company, management does not believe any pending matter will be resolved in a manner that would have a material adverse effect on the Company's consolidated financial position, results of operations or cash flows.
NOTE 13 – BASIC AND DILUTED NET INCOME PER SHARE
97
The computations of basic and diluted net income per share are as follows (in thousands, except per share amounts):
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
Numerator: | |||||||||||||||
Net income | $ | 86,124 | $ | 67,010 | $ | 117,533 | $ | 11,986 | |||||||
Denominator: | |||||||||||||||
Weighted average common shares-basic | 75,247 | 72,777 | 69,676 | 68,728 | |||||||||||
Effect of dilutive securities: | |||||||||||||||
Employee stock options | 2,873 | 4,237 | 4,607 | 3,781 | |||||||||||
Employee stock purchase plan | 16 | 69 | 125 | 108 | |||||||||||
Restricted stock units | 15 | — | — | — | |||||||||||
Weighted average common shares - diluted | 78,151 | 77,083 | 74,408 | 72,617 | |||||||||||
Net income per share: | |||||||||||||||
Basic | $ | 1.14 | $ | 0.92 | $ | 1.69 | $ | 0.17 | |||||||
Diluted | $ | 1.10 | $ | 0.87 | $ | 1.58 | $ | 0.17 | |||||||
Potentially dilutive securities excluded from net income per share - diluted because their effect is anti-dilutive | 1,243 | 848 | 445 | 441 | |||||||||||
NOTE 14 – SEGMENT INFORMATION
The Company operates as a single reportable segment, which primarily focuses on developing and commercializing novel therapies for the treatment of cancer and immune-mediated diseases (see Note 2). The following table summarizes total revenue from external customers by geographic region, which consists of product revenue, net. IMBRUVICA first came to market on November 13, 2013.
Years Ended December 31, | Six Months Ended December 31, | Year Ended June 30, | |||||||||||||
2014 | 2013 | 2012 | 2012 | ||||||||||||
United States | $ | 492,418 | $ | 13,573 | $ | — | $ | — | |||||||
Other | — | — | — | — | |||||||||||
Total | $ | 492,418 | $ | 13,573 | $ | — | $ | — | |||||||
As of December 31, 2014, substantially all of the Company's long-lived assets were located in the U.S.
Note 15 — QUARTERLY RESULTS (UNAUDITED)
The following table is in thousands, except per share amounts:
98
Quarters Ended | |||||||||||||||
March 31, | June 30, | September 30, | December 31, | ||||||||||||
2014 | 2014 | 2014 | 2014 | ||||||||||||
Total revenue | $ | 119,377 | $ | 113,020 | $ | 207,140 | $ | 290,192 | |||||||
Total costs and expenses | $ | 101,152 | $ | 150,224 | $ | 158,771 | $ | 197,201 | |||||||
Net income (loss) from operations | $ | 18,225 | $ | (37,204 | ) | $ | 48,369 | $ | 92,991 | ||||||
Net income (loss) | $ | 18,275 | $ | (37,057 | ) | $ | 41,422 | $ | 63,484 | ||||||
Net income (loss) per share: (1) | |||||||||||||||
Basic | $ | 0.24 | $ | (0.49 | ) | $ | 0.55 | $ | 0.84 | ||||||
Diluted | $ | 0.23 | $ | (0.49 | ) | $ | 0.53 | $ | 0.81 | ||||||
Shares used in computation of net income (loss) per share: | |||||||||||||||
Basic | 74,754 | 75,139 | 75,392 | 75,692 | |||||||||||
Diluted | 78,064 | 75,139 | 78,025 | 78,471 | |||||||||||
Quarters Ended | |||||||||||||||
March 31, | June 30, | September 30, | December 31, | ||||||||||||
2013 | 2013 | 2013 | 2013 | ||||||||||||
Total revenue | $ | 2,846 | $ | 54,684 | $ | 79,088 | $ | 123,551 | |||||||
Total costs and expenses | $ | 55,810 | $ | 42,653 | $ | 25,662 | $ | 38,120 | |||||||
Net income (loss) from operations | $ | (52,964 | ) | $ | 12,031 | $ | 53,426 | $ | 85,431 | ||||||
Net income (loss) | $ | (51,911 | ) | $ | 12,351 | $ | 42,333 | $ | 64,237 | ||||||
Net income (loss) per share: (1) | |||||||||||||||
Basic | $ | (0.73 | ) | $ | 0.17 | $ | 0.58 | $ | 0.87 | ||||||
Diluted | $ | (0.73 | ) | $ | 0.16 | $ | 0.55 | $ | 0.82 | ||||||
Shares used in computation of net income (loss) per share: | |||||||||||||||
Basic | 70,933 | 72,953 | 73,316 | 73,868 | |||||||||||
Diluted | 70,933 | 77,194 | 77,659 | 77,917 | |||||||||||
(1) Basic and diluted net income (loss) per share amounts are computed independently for each of the quarters presented. Therefore, the sum of quarterly basic and diluted net income (loss) per share information may not equal annual basic and diluted net income (loss) per share.
NOTE 16 – Transition Period Comparative Data (unaudited)
99
(in thousands, except per share data) | |||||||
Six Months Ended December 31, | |||||||
2012 | 2011 | ||||||
(unaudited) | |||||||
Statement of Operations Data: | |||||||
Revenue: | |||||||
License and milestone revenue | $ | 155,000 | $ | 77,605 | |||
Collaboration services revenue | 5,658 | 335 | |||||
Total revenue | 160,658 | 77,940 | |||||
Costs and expenses: | |||||||
Research and development | 46,639 | 23,324 | |||||
Less: Excess Amounts related to Research and development (See Note 5) | (17,306 | ) | — | ||||
Research and development, net | 29,333 | 23,324 | |||||
Selling, general and administrative | 12,093 | 7,294 | |||||
Less: Excess Amounts related to Selling, general and administrative (See Note 5) | (819 | ) | — | ||||
Selling, general and administrative, net | 11,274 | 7,294 | |||||
Total costs and expenses | 40,607 | 30,618 | |||||
Income from operations | 120,051 | 47,322 | |||||
Interest income | 137 | 68 | |||||
Other expense, net | (8 | ) | (24 | ) | |||
Income before income taxes | 120,180 | 47,366 | |||||
Income tax provision | (2,647 | ) | (5,651 | ) | |||
Net income | $ | 117,533 | $ | 41,715 | |||
Net income per share: | |||||||
Basic | $ | 1.69 | $ | 0.61 | |||
Diluted | $ | 1.58 | $ | 0.58 | |||
Weighted average shares used to compute net income (loss) per share: | |||||||
Basic | 69,676 | 68,491 | |||||
Diluted | 74,408 | 71,312 | |||||
Statement of Cash Flows Data: | |||||||
Net cash provided by (used in) operating activities | $ | 110,694 | $ | 123,904 | |||
Net cash (used in) provided by investing activities | (6,386 | ) | 13,063 | ||||
Net cash provided by financing activities | 5,229 | 5,490 | |||||
Increase in cash and cash equivalents | $ | 109,537 | $ | 142,457 | |||
NOTE 17 – RELATED PARTY TRANSACTIONS
Kenneth A. Clark, a director of the Company since November 2012, is a member of the law firm of Wilson Sonsini Goodrich & Rosati (WSGR). The Company retains WSGR as legal counsel for various matters, primarily consisting of intellectual property matters. During the years ended December 31, 2014 and 2013, the Company made payments to WSGR of $3.7 million and $2.9 million, respectively. In addition, from the time that Mr. Clark was elected as a director on November 9, 2012 until December 31, 2012, the Company made payments to WSGR of approximately $0.4 million. As of December 31, 2014 and 2013, $1.3 million and $0.9 million, respectively, was included in accrued liabilities in the consolidated bal
100
ance sheet for amounts due to WSGR.
During the year ended June 30, 2012, the Company paid Dr. Gwen Fyfe, a former member of its Board of Directors, approximately $0.1 million for consulting services under a Consulting Agreement entered into prior to Dr. Fyfe joining the Company's Board in December 2010. The Company made no payments to Dr. Fyfe during the year ended December 31, 2014, the year ended December 31, 2013 or the six months ended December 31, 2012. In November 2011, the Company entered into an amendment (the “Amendment”) to its Consulting Agreement with Dr. Fyfe. The Amendment provided that Dr. Fyfe would receive a lump sum of $0.1 million and that she will continue to provide limited consulting services to the Company for a period of two years. Payment of the $0.1 million lump sum occurred in November 2011. In addition, the options to purchase 330,000 shares of the Company's common stock previously granted to Dr. Fyfe in connection with her consulting services continued to vest through November 30, 2011 and were to remain exercisable for a period of two years following the date of the Amendment. Dr. Fyfe did not stand for reelection at the Company's December 15, 2011 Annual Meeting of Stockholders. Options granted to Dr. Fyfe upon her initial election to the Board continued to vest through December 15, 2011; all such vested options and all additional options received by Dr. Fyfe in connection with her Board service were to remain exercisable for a period of three years from this date. During the year ended December 31, 2014, Dr. Fyfe exercised all remaining stock options.
NOTE 18 – SUBSEQUENT EVENT
In January 2015, the Company entered into a six month consulting services agreement with Dr. Robert Booth, a member of its Board of Directors (the Consulting Agreement). In addition, on January 22, 2015, Dr. Booth was granted 10,000 RSUs which vest over four years. The grant date fair value of the RSUs is $1.6 million based on the Company’s closing stock price on January 22, 2015. The Consulting Agreement provides for maximum payments to Dr. Booth of $0.1 million over the six month term and for the reimbursement of documented, authorized travel and out-of-pocket expenses.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not Applicable.
Item 9A. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures:
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As of the end of the period covered by this Form 10-K, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
(b) Management’s Annual Report on Internal Control Over Financial Reporting:
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2014 based on the framework in Internal Control—Integrated Framework 2013 issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2014.
The independent registered public accounting firm, PricewaterhouseCoopers LLP, has issued an attestation report on our internal control over financial reporting. The report on the audit of internal control over financial reporting is included in Item 8 in this Form 10-K.
101
(c) Changes in Internal Control Over Financial Reporting:
There has been no change in our internal control over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
On February 17, 2015, the Compensation Committee of the Board of Directors of Pharmacyclics, Inc. (the “Company”) approved 2014 year-end cash bonuses for certain officers of the Company, including $227,027 for Mahkam Zanganeh, the Company’s Chief Operating Officer, $183,838 for Manmeet Soni, the Company’s Chief Financial Officer, and $107,915 for Heow Tan, the Company’s Chief of Quality and Technical Operations. Separate mid-year bonuses for certain officers of the Company were previously paid and disclosed in August 2014.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Certain information required by this Item 10 is hereby incorporated by reference to the information under the captions, (i) “Election of Directors,” (ii) “Audit Committee,” (iii) “Code of Business Conduct and Ethics” and (iv) “Section 16(a) Beneficial Ownership Reporting Compliance,” contained in our Definitive Proxy Statement to be filed with the Securities and Exchange Commission no later than 120 days from the end of our fiscal year ended December 31, 2014.
Item 11. Executive Compensation
The information required by this Item 11 is incorporated by reference to the information under the caption “Executive Compensation and Other Information” in the Definitive Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item 12 with respect to stock ownership of certain beneficial owners and management and securities authorized for issuance under equity compensation plans are incorporated by reference to the information under the captions “Stock Ownership of Management and Certain Beneficial Owners” and “Securities Authorized For Issuance Under Equity Compensation Plans” in the Definitive Proxy Statement.
Item 13. Certain Relationships and Related Transactions and Director Independence
The information required by this Item 13 is incorporated by reference to the information under the caption “Certain Relationships and Related Transactions” in the Definitive Proxy Statement.
Item 14. Principal Accountant Fees and Services
The information required by this Item 14 is incorporated by reference to the information in the Definitive Proxy Statement.
102
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) 1. | Financial Statements | |
See Index to Financial Statements under Item 8.
(a) 2. | Financial Statement Schedules | |
The following financial statement schedules are filed as part of this Annual Report on Form 10-K:
Schedule II - Valuation and Qualifying Accounts and Reserves
Schedule II: Valuation and Qualifying Accounts and Reserves | ||||||||||||||||
(in thousands) | ||||||||||||||||
Balance at Beginning of Period | Additions | Write-offs/Adjustments | Balance at End of Period | |||||||||||||
Income Tax Valuation Allowance: | ||||||||||||||||
Fiscal year ended December 31, 2014 | $ | 58,679 | $ | 11,671 | $ | — | $ | 70,350 | ||||||||
Fiscal year ended December 31, 2013 | $ | 31,915 | $ | 26,764 | $ | — | $ | 58,679 | ||||||||
Six months ended December 31, 2012 | $ | 70,001 | $ | — | $ | (38,086 | ) | $ | 31,915 | |||||||
Fiscal year ended June 30, 2012 | $ | 79,161 | $ | — | $ | (9,160 | ) | $ | 70,001 | |||||||
Allowance for Cash Discounts and Other Programs: | ||||||||||||||||
Fiscal year ended December 31, 2014 | $ | 301 | $ | 26,641 | $ | (18,332 | ) | $ | 8,610 | |||||||
Fiscal year ended December 31, 2013 | $ | — | $ | 371 | $ | (70 | ) | $ | 301 | |||||||
Six months ended December 31, 2012 | $ | — | $ | — | $ | — | $ | — | ||||||||
Fiscal year ended June 30, 2012 | $ | — | $ | — | $ | — | $ | — | ||||||||
Allowance for Doubtful Accounts: | ||||||||||||||||
Fiscal year ended December 31, 2014 | $ | — | $ | 14 | $ | — | $ | 14 | ||||||||
Fiscal year ended December 31, 2013 | $ | — | $ | — | $ | — | $ | — | ||||||||
Six months ended December 31, 2012 | $ | — | $ | — | $ | — | $ | — | ||||||||
Fiscal year ended June 30, 2012 | $ | — | $ | — | $ | — | $ | — | ||||||||
All other schedules have been omitted because they are not applicable or are not required or the information required to be set forth therein is included in the financial statements or notes thereto. |
(a) 3. | Exhibits | |
See Index to Exhibits.
103
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, (the “Exchange Act”) the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
PHARMACYCLICS, INC. | |||
Dated: February 18, 2015 | By: | /s/ ROBERT W. DUGGAN | |
Robert W. Duggan | |||
Chairman of the Board & Chief Executive Officer (Principal Executive Officer) | |||
104
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints jointly and severally, Robert W. Duggan and Manmeet S. Soni, or either of them as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated.
Pursuant to the requirements of the Exchange Act, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ ROBERT W. DUGGAN | ||||
Robert W. Duggan | Chairman of the Board and Chief Executive Officer (Principal Executive Officer) | February 18, 2015 | ||
/s/ MANMEET S. SONI | ||||
Manmeet S. Soni | Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) | February 18, 2015 | ||
/s/ ROBERT F. BOOTH, Ph.D. | ||||
Robert F. Booth, Ph.D. | Director | February 18, 2015 | ||
/s/ KENNETH A. CLARK | ||||
Kenneth A. Clark | Director | February 18, 2015 | ||
/s/ ERIC H. HALVORSON | ||||
Eric H. Halvorson | Director | February 18, 2015 | ||
/s/ MINESH P. MEHTA, M.D. | ||||
Minesh P. Mehta, M.D | Director | February 18, 2015 | ||
/s/ DAVID D. SMITH, Ph.D. | ||||
David D. Smith, Ph.D. | Director | February 18, 2015 | ||
/s/ RICHARD A. VAN DEN BROEK | ||||
Richard A. van den Broek | Director | February 18, 2015 | ||
105
EXHIBITS INDEX
Exhibit Number | Description |
3.1 | Amended and Restated Certificate of Incorporation of the Company (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 16, 2008). |
3.2 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of the Company (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 19, 2011). |
3.3 | Amended and Restated Bylaws of the Company (incorporated by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 19, 2011). |
4.1 | Specimen Certificate of the Company's Common Stock (incorporated by reference to Exhibit 4.2 to the Company's Registration Statement on Form S-1, Commission File No. 33-96048). |
10.1+ | The Company's Employee Stock Purchase Plan as amended and restated on October 25, 2011 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 19, 2011). |
10.2+ | Form of Employee Stock Purchase Plan Enrollment/Change Form (incorporated by reference to Exhibit 99.12 to the Company's Registration Statement on Form S-8, Commission File No. 33-98514). |
10.3 | Lease and Lease Termination Agreement dated June 14, 2000 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.49 to the Annual Report on Form 10-K for the year ended June 30, 2001). |
10.4 | First Amendment to New Lease dated April 10, 2001 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.50 to the Annual Report on Form 10-K for the year ended June 30, 2001). |
10.5 | Second Amendment to New Lease dated June 29, 2001 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.51 to the Annual Report on Form 10-K for the year ended June 30, 2001). |
10.6 | Third Amendment to New Lease dated February 5, 2003 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2003). |
10.7+ | The Company's 2004 Equity Incentive Award Plan (the "2004 Plan") as amended and restated on October 25, 2011 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 19, 2011). |
10.8+ | The Company’s 2014 Equity Incentive Award Plan (the “2014 Plan”) (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on May 14, 2014). |
10.9+ | Form of Option Agreement. |
10.10* | Assignment Agreement By and Between Pharmacyclics, Inc. and Applera Corporation dated April 7, 2006 (incorporated by reference to Exhibit 10.64 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2006). |
10.11 | Fourth Amendment to New Lease dated August 14, 2006 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed with the Securities and Exchange Commission on August 17, 2006). |
10.12 | Fifth Amendment to New Lease dated July 11, 2008 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.6 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2008). |
10.13* | Amendment No. 1 to Assignment Agreement by and between Pharmacyclics, Inc. and Applera Corporation dated May 12, 2008 (incorporated by reference to Exhibit 10.68 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2008). |
10.14* | Amendment No. 2 to Assignment Agreement by and between Pharmacyclics, Inc. and Celera Corporation dated March 2, 2009 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009). |
10.15* | Amendment No. 3 to Assignment Agreement by and between Pharmacyclics, Inc. and Celera Corporation dated March 30, 2009 (incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2009). |
10.16* | Collaboration Agreement by and between Pharmacyclics, Inc. and Les Laboratoires Servier and Institut de Recherches Internationales Servier dated April 9, 2009 (incorporated by reference to Exhibit 10.83 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2009). |
106
10.17* | Drug Supply Agreement by and between Pharmacyclics, Inc. and Les Laboratoires Servier dated December 18, 2009 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2009). |
10.18 | Sixth Amendment to New Lease dated January 20, 2011 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended March 31, 2011). |
10.19* | Collaboration and License Agreement by and between the Company and Janssen Biotech, Inc. dated as of December 8, 2011 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q for the quarter ended December 31, 2011). |
10.20 | Seventh Amendment to New Lease dated February 14, 2012 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2012). |
10.21 | Amendment No. 1 to the Collaboration Agreement by and between the Company and Les Laboratoires Servier and Institut De Recherches Internationales Servier dated as of January 5, 2012 (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2012). |
10.22* | Amended and Restated License Agreement by and between the Company and Novo Nordisk A/S, dated as of June 28, 2013 (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013). |
10.23* | Manufacturing Supply Agreement by and between the Company and Lonza Sales Ltd., dated February 5, 2013 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K/A filed with the Securities and Exchange Commission on August 14, 2014). |
10.24* | Amendment One to Manufacturing Supply Agreement by and between the Company and Lonza Sales Ltd., dated October 2, 2013 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K/A filed with the Securities and Exchange Commission on August 14, 2014). |
10.25* | Build Out and Commercial Supply Agreement by and between the Company and Catalent CTS, LLC dated May 1, 2013 (incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K/A filed with the Securities and Exchange Commission on August 14, 2014). |
10.26 | First Amendment to the Company's Employee Stock Purchase Plan, as amended January 21, 2014 (incorporated by reference to Exhibit 10.38 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2013). |
10.27 | Second Amendment to the Company's Employee Stock Purchase Plan, as amended January 21, 2014 (incorporated by reference to Exhibit 10.39 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2013). |
10.28 | Eighth Amendment to New Lease dated February 14, 2012 by and between the Company and Metropolitan Life Insurance Company (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2012). |
10.29 | Commercial Lease Agreement by and between the Company and John G. and Theresa L. Hessler as Trustees of the Hessler 2007 Revocable Trust; Mark Dellamano, John Dellamano and Joseph Dellamano, as Tenants in Common (incorporated by reference to Exhibit 10.41 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2013). |
10.30 | Binding Notice to Landlord to Exercise Expansion Option pursuant to Section 1.6 of the Commercial Lease between the Company and John G. and Theresa L. Hessler as Trustees of the Hessler 2007 Revocable Trust; Mark Dellamano, John Dellamano and Joseph Dellamano, as Tenants in Common (incorporated by reference to Exhibit 10.42 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2013). |
10.31 | Form of Restricted Stock Unit Agreement (incorporated by reference to Exhibit 10.43 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2013). |
10.32 | Termination Letter dated September 24, 2014, by and between the Company and Les Laboratoires Servier and Institut de Recherches Internationales Servier. |
10.33** | Letter re: Servier’s Termination Commitments dated September 24, 2014, by and between the Company and Les Laboratoires Servier and Institut de Recherches Internationales Servier. |
10.34 | Offer Letter effective as of August 18, 2014 by and between the Company and Shawn Tomasello. |
21.1 | Subsidiaries of the Company. |
23.1 | Consent of Independent Registered Public Accounting Firm. |
24.1 | Power of Attorney (included on the signature page of this Annual Report on Form 10-K). |
31.1 | Certification of Principal Executive Officer. |
31.2 | Certification of Principal Financial Officer. |
107
32.1 | Certification of Principal Executive Officer and Principal Financial Officer. |
101.INS | XBRL Instance Document |
101.SCH | XBRL Taxonomy Extension Schema Document |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB | XBRL Taxonomy Extension Label Linkbase Document |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document |
________________
* Confidential treatment has been granted as to certain portions of this agreement.
** Confidential treatment has been requested as to certain portions of this agreement.
+ Indicates a management contract or compensatory plan or arrangement.
108