Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - INSITE VISION INC | Financial_Report.xls |
| EX-31.1 - EX-31.1 - INSITE VISION INC | d841402dex311.htm |
| EX-23.1 - EX-23.1 - INSITE VISION INC | d841402dex231.htm |
| EX-31.2 - EX-31.2 - INSITE VISION INC | d841402dex312.htm |
| EX-21.1 - EX-21.1 - INSITE VISION INC | d841402dex211.htm |
| EX-32.1 - EX-32.1 - INSITE VISION INC | d841402dex321.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 0-22332
INSITE VISION INCORPORATED
(Exact name of registrant as specified in its charter)
| Delaware | 94-3015807 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 965 Atlantic Avenue, Alameda, CA | 94501 | |
| (Address of principal executive offices) | (Zip Code) |
(510)-865-8800
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.01 par value per share | OTC Bulletin Board |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer ¨ | |
| Non-accelerated filer ¨ |
Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of registrant’s Common Stock, $0.01 par value, held by non-affiliates of the Registrant as of June 30, 2014 was approximately $12,932,000 (based upon the closing sale price of the Common Stock on the last business day of the registrant’s most recently completed second fiscal quarter). Shares of Common Stock held by each officer and director and by each person who owns 5% or more of the Common Stock have been excluded from such calculation as such persons may be deemed affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
Number of shares of Common Stock, $0.01 par value, outstanding as of February 12, 2015: 131,951,033
Portions of the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission in connection with its 2015 annual meeting of stockholders are incorporated by reference into Part III hereof.
Table of Contents
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2014
Table of Contents
Except for the historical information contained herein, the discussion in this Annual Report on Form 10-K contains certain forward-looking statements that involve risks and uncertainties, such as statements of our plans, beliefs, objectives, expectations and intentions. Our actual results could differ materially from those discussed herein. Factors that could cause or contribute to such differences include those discussed below under “Risk Factors,” and elsewhere herein. The cautionary statements made in this document should be read as applicable to all related forward-looking statements wherever they appear in this document. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. We undertake no obligation to update any forward-looking statements to reflect events or circumstances after the date hereof or to reflect the occurrence of unanticipated events. References in this Annual Report on Form 10-K to the “Company,” “InSite,” “we,” “our” and “us” refer to InSite Vision, Incorporated and its consolidated subsidiaries, unless we state, or the context indicates, otherwise.
PART I
| Item 1. | Business |
THE COMPANY
We are an ophthalmic product development company advancing ophthalmic pharmaceutical products to address unmet eye care needs. Our current portfolio of products is based on our proprietary DuraSite® sustained drug delivery technology. Our near term strategy is to primarily focus on filing New Drug Applications (NDAs) for BromSite and DexaSite with the United States Food and Drug Administration (FDA).
We face significant challenges related to our lack of financial resources. We expect that our cash on hand, anticipated cash flow from operations and the net proceeds from our existing debt financing arrangements (for further discussion, see Note 1 and 8 to the consolidated financial statements included in this annual report) will only enable us to continue our operations as currently planned until approximately May 2015. Our independent auditors included an explanatory paragraph in their audit report related to our consolidated financial statements for the year ended December 31, 2014, referring to our recurring losses from operations, available cash balance and accumulated deficit and a substantial doubt about our ability to continue as a going concern.
Our DuraSite sustained drug delivery technology is a proven synthetic polymer-based formulation designed to extend the residence time of a drug relative to conventional topical therapies. It enables topical delivery of a drug as a solution, gel or suspension and can be customized for delivering a wide variety of drug candidates. We have focused our research and development and commercial support efforts on the following topical products formulated with our DuraSite drug delivery technology.
| • | AzaSite® (azithromycin ophthalmic solution) 1% is a DuraSite formulation of azithromycin, a broad spectrum ocular antibiotic approved by the FDA in April 2007 to treat bacterial conjunctivitis (pink eye). It is commercialized in the United States by Akorn, Inc. (Akorn). We received a 25% royalty on net sales of AzaSite in North America through March 31, 2014, which was required to service the outstanding, non-recourse promissory notes due 2019 of our wholly-owned subsidiary, Azithromycin Royalty Sub, LLC (AzaSite Notes). On June 10, 2014, we entered into the Third Amendment to License Agreement (Amendment) with Akorn to amend the North American license (Akorn License) of AzaSite to Akorn. Under the Amendment, Akorn paid us an amendment fee of $6.0 million, which was used to repurchase and cancel the AzaSite Notes, in return for a lower royalty on net sales of AzaSite in North America. Effective April 1, 2014, for annual net sales of AzaSite, the royalty is (1) 8.0% on net sales less than $20.0 million, subject to increase to 9.0% in the event that Akorn enters into certain settlements with third parties seeking to launch generic versions of AzaSite, (2) 12.5% on net sales greater than or equal to $20.0 million and less than or equal to $50.0 million and (3) 15.0% on net sales in excess of $50.0 million. |
1
Table of Contents
| • | Besivance® (besifloxacin ophthalmic suspension) 0.6% is a DuraSite formulation of besifloxacin, a broad spectrum ocular antibiotic approved by the FDA in May 2009 to treat bacterial conjunctivitis (pink eye). Besivance was developed by Bausch + Lomb Incorporated (Bausch & Lomb) (wholly-owned by Valeant Pharmaceuticals International, Inc. (Valeant) as of August 2013) and was launched in the United States in the second half of 2009. In 2011, Besivance was launched internationally in select countries. Until we sold the Besivance royalty rights in April 2013, we received a middle single-digit royalty on net sales of Besivance globally. In April 2013, we sold the rights to receive royalty payments on sales of Besivance, beginning on January 1, 2013, for $15 million at closing, with an additional $1 million paid in February 2014 after certain 2013 Besivance sales targets were met. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021. |
| • | BromSiteTM is a DuraSite formulation of bromfenac in development for the treatment of post-operative inflammation and prevention of post-operative eye pain. We initiated a Phase 1/2 clinical trial for this product candidate in August 2010 and we received positive top-line results from this study in the first quarter of 2011, which demonstrated the efficacy and safety of BromSite. In the third quarter of 2011, we completed an additional Phase 2 clinical trial to investigate the pharmacokinetics (PK) of BromSite in humans. We received positive top-line results that showed that the mean concentration of bromfenac in the aqueous humor of patients using BromSite was more than double compared to the currently available bromfenac eye product. In July 2012, we initiated a Phase 3 clinical trial for this product candidate and completed patient enrollment in November 2012. The BromSite Phase 3 clinical trial enrolled 268 patients undergoing cataract surgery in a two-arm trial designed to evaluate the efficacy and safety of BromSite against the DuraSite vehicle alone. In March 2013, we received positive top-line results from this Phase 3 clinical trial demonstrating statistically significant superiority compared to vehicle (p < 0.001) in alleviating ocular pain and inflammation among patients following cataract surgery. In May 2013, we initiated the second Phase 3 clinical trial for this product candidate, which was completed in November 2013 with 248 patients enrolled. We received positive top-line results from this confirmatory Phase 3 clinical trial in December 2013. We plan to file an NDA with the FDA in the first quarter of 2015 for this product candidate. In May 2014, the Swedish Medical Products Agency (MPA) concluded that the existing Phase 3 clinical data for BromSite was likely sufficient to support the filing of a Marketing Authorization Application (MAA) with the MPA. In July 2014, the U.S. Patent and Trademark Office (USPTO) issued a patent on BromSite. The patent provides protection for bromfenac formulations in DuraSite, including ISV-101, to August 8 2029. |
| • | DexaSiteTM is a DuraSite formulation of dexamethasone in development for the treatment of ocular inflammation. In November 2011, we initiated a Phase 3 clinical trial for this product candidate in blepharitis and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. While DexaSite did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis, it demonstrated statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. After ongoing meetings with the FDA, in July 2014, the FDA agreed that the results of the Phase 3 clinical trial could support marketing approval for DexaSite for the treatment of blepharitis. During the fourth quarter of 2014, we executed post-Phase 3 regulatory meetings with European Health Authorities and the FDA to outline our proposed filings. We currently plan to file an NDA with the FDA in 2016 for this product in this indication. As a next step toward this filing, we will be executing additional regulatory meetings with European Health Authorities and the FDA during the first and second quarters of 2015 to attempt to harmonize the chemistry, manufacturing and controls (CMC) section in support of DexaSite NDA and MAA filings for this indication. In addition, in November 2013, we submitted a protocol to the FDA for Phase 3 clinical trials with respect to the use of DexaSite for the treatment of inflammation and prevention of eye pain in ocular surgery. |
| • | AzaSite PlusTM is a fixed combination of azithromycin and dexamethasone in DuraSite for the treatment of ocular inflammation and infection (blepharitis and/or blepharoconjunctivitis) for which |
2
Table of Contents
| there is no FDA approved indicated treatment. In November 2011, we initiated a Phase 3 clinical trial for this product candidate in blepharitis and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. AzaSite Plus did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis. However, AzaSite Plus did demonstrate statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. We continue to meet with the FDA to review the overall results of the clinical trial and determine the next steps in the development of this product candidate. |
| • | DuraSite® 2 is our next-generation enhanced drug delivery system, which is designed to provide a broad platform for developing superior ophthalmic therapeutics. DuraSite 2 is based on the original DuraSite technology, and incorporates a cationic polymer to achieve sustained and enhanced ocular delivery of drugs. DuraSite 2 is designed to increase the tissue penetration for topically delivered ocular drugs with the aim of improved efficacy and dosing convenience. In August 2013, the USPTO issued a patent on DuraSite 2. The patent provides protection to March 5, 2029 for both the delivery system and the drugs that are formulated with DuraSite 2. In November 2013, we obtained preclinical data from a comparative study that demonstrated superior drug retention and tissue penetration compared to DuraSite. We plan to utilize the DuraSite 2 platform in the development of all future pipeline products. We plan to initiate a broad licensing program that provides access to industry partners through both exclusive and non-exclusive licensing and/or commercialization agreements. |
| • | ISV-101 is a DuraSite formulation with a low concentration of bromfenac for the treatment of dry eye disease. We filed an Investigational New Drug Application (IND) with the FDA for this product candidate in the first quarter of 2011. We plan to initiate a Phase 1/2 clinical trial for this product candidate, but no time frame has been set. |
Business Strategy.
Assuming we can raise sufficient funding to continue our operations, our business strategy consists of the following:
1. Develop our pipeline of ocular product candidates. We seek to identify new product candidates from proven drugs that can be improved by formulation in DuraSite 2, which can substantially reduce the clinical risk involved in these product candidates. As appropriate, we plan to conduct preclinical and clinical testing of our product candidates.
2. Partner our product candidates. When we deem it appropriate, we seek to partner with larger pharmaceutical companies to manufacture and market our products. Partnering agreements generally include upfront and milestone payments, as well as on-going royalty payments upon commercialization, payable to us.
Corporate Information. Our principal executive offices are located at 965 Atlantic Avenue, Alameda, California 94501. Our telephone number is (510) 865-8800. We were incorporated in 1986 as a California corporation and reincorporated in Delaware in 1987. We make our periodic and current reports available, free of charge, through our website (http:///www.insitevision.com) under “Investor Relations – SEC Filings” as soon as reasonably practicable after such material is electronically filed with the Securities and Exchange Commission (SEC). Additionally, copies of materials filed by us with the SEC may be accessed at the SEC’s Public Reference Room at 100 F Street NE, Washington D.C. or at the SEC’s website at http://www.sec.gov. For information about the SEC’s Public Reference Room, the public may contact 1-800-SEC-0330.
Ophthalmic Anti-Infective Market
The ocular anti-infective market is estimated to exceed $2.0 billion annually. Eye infections are routinely treated with topical antibiotics or antibiotic/corticosteroid fixed-dose combination products. We are developing topical products to treat eye and eye-lid infections and/or inflammations. Some of these infections and/or inflammations are either under-treated or do not have an FDA-approved product indication. These infections and/
3
Table of Contents
or inflammations can be both acute and chronic. Our goal is to provide highly effective, safe and differentiated therapeutics for the treatment of acute and chronic ocular infection and inflammation. There are two general areas where our topical ocular anti-infective products have been utilized by eye-care physicians, or where we believe our product candidates are well-suited to improving patient care:
| • | Acute Eye Infections. |
| • | Acute bacterial conjunctivitis (pink eye) is a common ophthalmic condition is experienced by most people at some point in their lives, and is especially prevalent among children. Due to its common occurrence, and contagiousness, conjunctivitis is a global economic burden. Approximately four million cases of bacterial conjunctivitis are seen annually in the United States in children and adults, with direct and indirect costs estimated to be as much as $857 million annually. Bacterial conjunctivitis afflicts the conjunctiva or the transparent lining on the inside of the eyelids and the white part of the eye. In bacterial conjunctivitis, bacteria infect these parts of the eye and the white part of the eye may look pink from the inflammation. As it is an extremely contagious condition, immediate treatment is recommended. Our developed ocular antibiotics, AzaSite and Besivance, are designed to treat this disease with significantly lower dosing than competing products. |
| • | Chronic and Acute Eye-lid Infections. |
| • | Blepharitis (also known as lid margin disease) is a chronic inflammation of the eyelids, particularly the eyelid margins where the eyelashes grow. It is a common disorder that may be caused by bacterial growth, viral infection, allergies, environmental conditions or systemic disease. In a survey conducted by researchers at George Washington University, ophthalmologists and optometrists reported that blepharitis is commonly seen in 37% and 47% of their patients, respectively. In spite of that frequency, blepharitis is often misdiagnosed or under-diagnosed. |
Blepharitis is a chronic condition with periodic acute episodes that are difficult to treat. An eyelid with blepharitis may become itchy and appear red and swollen with scaly, dandruff-like or greasy debris along the lid margin. There are no FDA-approved product for the treatment of blepharitis. Patients are typically advised to use lid scrubs, hot compresses, lid massage, antibiotics, corticosteroids and fixed-combination products. We have developed two novel ophthalmic products that utilize our DuraSite drug delivery technology to alleviate the signs and symptoms of blepharitis; AzaSite Plus, a topical anti-bacterial and anti-inflammatory combination product; and DexaSite, a corticosteroid anti-inflammatory agent. In July 2013, we completed a Phase 3 clinical trial for DexaSite and AzaSite Plus in blepharitis, enrolled 907 patients and announced top-line results. While DexaSite and AzaSite Plus did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis, they demonstrated statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. After ongoing meetings with the FDA, in July 2014, the FDA agreed that the results of the Phase 3 clinical trial could support marketing approval for DexaSite for the treatment of blepharitis. We currently plan to file an NDA with the FDA in 2016 for DexaSite for the treatment of blepharitis.
| • | Blepharoconjunctivitis occurs when conjunctivitis accompanies blepharitis, a condition that occurs frequently. A unilateral or bilateral conjunctivitis that persists for four or more weeks is diagnosed as chronic. There is a considerable overlap of symptoms of all types of blepharitis and it often leads to associated ocular surface inflammation, including conjunctivitis, function tear deficiency and keratitis (an inflammation of the cornea which can develop into corneal ulcers). There is no approved drug therapy for the chronic symptoms of blepharoconjunctivitis. Typical treatment includes eye hygiene using lid scrubs, topical and/or systemic antibiotics and topical corticosteroids. |
4
Table of Contents
Ocular Inflammation and Pain Market
We are developing novel ophthalmic therapeutics for the treatment of conditions associated with ocular inflammation. Our product development programs are initially focused on two indications, post-operative inflammation and pain, and dry eye disease.
| • | Post-Operative Ocular Inflammation and Pain is typically treated using non-steroidal anti-inflammatory drugs (NSAIDs) and/or topical ophthalmic steroids. Ophthalmic surgeons deploy topical NSAIDs before and after cataract and other procedures to address patient pain. Topical ophthalmic steroids are prescribed post-surgery to address inflammation that can cause significant discomfort. Frequently utilized together, NSAIDs and corticosteroids prevent complications of inflammation, such as cystoid macular edema. Cystoid macular edema (CME) is a serious post-surgical complication that occurs when inflammation and swelling develop in the center of the retina. CME is the most common cause of decreased vision or even blindness following cataract surgery. Cataract surgery is currently the most frequent ophthalmic procedure conducted in the United States, with more than 3 million cataract surgeries performed annually. With the growing elderly population in the U.S., the demand for cataract surgeries continues to rise. People can have an age-related cataract in their 40s and 50s, but it is after age 60 that most cataracts impact the patient’s vision. By age 80, more than half of all Americans will either have a cataract or have had cataract surgery. More than 20 million Americans have at least one cataract. By 2020, the incidence is expected to increase to 30 million. In 2011, the ocular anti-inflammatory products (corticosteroids, NSAIDs, and anti-inflammatory/anti-infective combinations) market was valued at over $1.7 billion. We completed two Phase 3 clinical studies of our BromSite product candidate, which is intended to relieve post-operative ocular inflammation and pain while providing superior tissue penetration of drug to the eye that may reduce the incidences of CME with convenient twice-daily dosing. We plan to file an NDA in the first quarter of 2015 for BromSite. In November 2013, the FDA approved the protocol for Phase 3 clinical trials in the use of DexaSite for the prevention of inflammation and eye pain in ocular surgery. We are also ready to start Phase 3 clinical studies of DexaSite, our topical ophthalmic steroid product candidate, in this indication but no time frame has been set. |
| • | Dry Eye Disease occurs when the ocular surface and/or tear film is compromised. While causes of dry eye may vary, it is frequently associated with inflammation of the surface of the eye, the lacrimal gland, or the conjunctiva. A chronic condition that can occur at any age, dry eye disease is most prevalent among the elderly. Symptoms of dry eye disease include a scratchy irritation as if something is in the eye, stinging or burning of the eye; episodes of excess tearing that follow periods of very dry sensation; stringy discharge from the eye and pain or redness. According to the National Eye Institute, dry eye disease is estimated to affect five million people age 50 and older in the U.S. alone, with tens of millions more experiencing less severe symptoms. We have developed a low-dose topical NSAID, known as ISV-101, for the treatment of dry eye disease. We plan to initiate a Phase 1/2 clinical trial for ISV-101, but no time frame has been set. |
5
Table of Contents
Products and Product Candidates
The following table summarizes the current status of our principal products and product candidates in our development pipeline. A more detailed description of each product and product candidate follows the table.
Principal DuraSite Products and Product Candidates
Active Programs
| Product |
Indications |
Anticipated Benefits |
Status | |||
| AzaSite |
Bacterial conjunctivitis (pink eye) | Broad-spectrum macrolide antibiotic with reduced dosing frequency | *Approved and launched in U.S. *Approved in Canada | |||
| Besivance |
Bacterial conjunctivitis (pink eye) | Broad-spectrum fluoroquinolone antibiotic with reduced dosing frequency | *Approved and launched in the U.S. and select countries internationally | |||
| BromSite |
Post-operative inflammation and eye pain | A non-steroidal anti-inflammatory to treat pain and inflammation | NDA expected to be filed in the first quarter of 2015 | |||
| DexaSite |
Blepharitis
Post-operative inflammation and eye pain |
A potent corticosteroid with reduced dosing frequency to treat inflammation | NDA expected to be filed in 2016. | |||
| AzaSite Plus |
Blepharitis | Broad-spectrum antibiotic combined with a potent corticosteroid with reduced dosing frequency to treat both inflammation and infection | Phase 3 clinical trial demonstrated statistically significant improvements in clinical signs and symptoms but did not meet the primary endpoint | |||
| ISV-101 |
Dry eye disease | A low dose non-steroidal anti-inflammatory to treat dry eye disease | Filed an IND with the FDA
Phase 1/2 clinical trial planned | |||
The DuraSite Product Family of Topical Anti-infectives and Product Candidates
AzaSite: Launched commercially in the United States by Inspire (acquired by Merck in May 2011 and Akorn in November 2013) in August 2007 for Bacterial Conjunctivitis (pink eye)
We developed a topical formulation of the antibiotic azithromycin to treat bacterial conjunctivitis and other infections of the eye. Bacterial conjunctivitis is a common ocular surface disease characterized by inflammation of the delicate skin and mucosa on the inside of the eyelids and the white part of the eye. These bacterial infections are contagious and are generally accompanied by irritation, itching, foreign body sensation, watering, mucus discharge and redness. The bacterial form of the disease is generally more common in children than adults.
6
Table of Contents
Azithromycin has a broad spectrum of antibiotic activity and is widely used to treat respiratory and other infections in its oral and parenteral forms. AzaSite is an eye drop of 1% azithromycin formulated to deliver sufficient tissue concentrations over a seven-day dosing period using our proprietary DuraSite technology. The eye drop is designed to enable superior bactericidal activity against common ocular pathogens and even difficult bacteria such as pseudomonas. We believe the key advantages of AzaSite include its once-a-day dosing after the first two days of twice-a-day dosing and the high and persistent levels of azithromycin achieved in the tissues of the eye. Clinical studies have shown that AzaSite is well tolerated and effective. AzaSite was approved by the FDA in April 2007. In August 2007, Inspire Pharmaceuticals commercially launched AzaSite in the United States pursuant to its license from InSite. In May 2011, Merck acquired Inspire and Inspire became a wholly-owned subsidiary of Merck. In November 2013, Akorn acquired Inspire from Merck and Inspire became a wholly-owned subsidiary of Akorn. AzaSite is commercialized by Akorn in the United States.
Besivance: Launched commercially in the United States by Bausch & Lomb in the second half of 2009 for Bacterial Conjunctivitis (pink eye) and available in select countries internationally
Besivance (besifloxacin ophthalmic suspension) 0.6% is indicated for the treatment of bacterial conjunctivitis in patients one year or older and is marketed by Bausch & Lomb (wholly-owned by Valeant as of August 2013).
Besivance is the first fluoroquinolone specifically developed for ophthalmic use and is the first and only ophthalmic fluoroquinolone with no previous systemic use. It offers broad-spectrum antibacterial activity, including activity against the strains that are the most common causes of bacterial conjunctivitis.
In clinical trials, investigators found that Besivance treatment resulted in a greater proportion of patients experiencing clinical resolution and microbial eradication when compared to its vehicle.
Besivance was approved by the FDA in May 2009. The product was launched commercially in the second half of 2009 in the United States. In 2011, Besivance was launched internationally in select countries.
In April 2013, we sold the rights to receive royalty payments from Bausch & Lomb on sales of Besivance. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021.
BromSite: Phase 3 clinical trial for post-operative inflammation and eye pain completed
We developed a topical formulation of the non-steroidal, anti-inflammatory bromfenac to treat post-operative inflammation and eye pain in patients who have undergone cataract surgery. In the first half of 2010, we completed our preclinical development of this product candidate and filed an IND with the FDA. In the second half of 2010, we initiated and completed enrollment for the Phase 1/2 clinical trial for this product candidate. In the first quarter of 2011, we received positive top-line results from this study, which demonstrated the efficacy and safety of BromSite. In the third quarter of 2011, we completed an additional Phase 2 clinical trial to investigate the PK of BromSite in humans. We received positive top-line results that showed that the mean concentration of bromfenac in the aqueous humor of patients using BromSite was more than double compared to the currently available bromfenac eye product. We discussed the design of the Phase 3 clinical trial with the FDA. The BromSite Phase 3 clinical study was a two-arm, double-blind, placebo-controlled clinical trial where the placebo arm was the DuraSite vehicle. Patients undergoing cataract surgery were randomized and then dosed twice-a-day beginning the day before surgery, continuing the day of surgery and 14 days post-surgery. The primary study endpoint was the reduction of pain and inflammation after surgery. In July 2012, we initiated a Phase 3 clinical trial for this product candidate and completed patient enrollment in November 2012 with 268 patients enrolled. In March 2013, we received positive top-line results that demonstrated a reduction of inflammation and pain after cataract surgery at a lower drug concentration compared to the current market leader.
7
Table of Contents
In May 2013, we initiated the second Phase 3 clinical trial for this product candidate, which was completed in November 2013 with 248 patients enrolled. We received positive top-line results from this confirmatory Phase 3 clinical trial in December 2013. We plan to file an NDA in the first quarter of 2015 for this product candidate for the treatment of post-operative inflammation and prevention of eye pain in patients who have undergone cataract surgery.
DexaSite: A second candidate for blepharitis in Phase 3 development and a new Phase 3 candidate for the prevention of inflammation and pain in ocular surgery.
We developed a topical formulation of the corticosteroid dexamethasone to treat eye inflammation caused by infections, injury, surgery or other conditions. In 2007, we completed our preclinical development of this product candidate. This is a second product candidate originating from the IND filed for AzaSite Plus.
In 2008, the data from our Phase 3 clinical trial indicated that DexaSite was efficacious and well tolerated with no serious adverse events. Treatment-related ocular adverse events were minimal in frequency and equivalent between all groups. There were no significant differences in intraocular pressure between the DexaSite group and the other group containing dexamethasone after 14 days of treatment.
In 2010, we discussed a development pathway for this product candidate with the FDA. DexaSite was included in the Phase 3 clinical trial SPA for AzaSite Plus, which allowed us to simultaneously evaluate both agents in a single Phase 3 clinical trial for the treatment of blepharitis. Per the SPA-approved protocol, the efficacy and safety of DexaSite was measured against the DuraSite vehicle for the primary clinical endpoint of resolution of clinical signs and symptoms of blepharitis at the end of the dosing period. DexaSite was also evaluated for the secondary clinical endpoint of clinical improvement of signs and symptoms of blepharitis. In November 2011, we initiated a Phase 3 clinical trial for this product candidate and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. DexaSite did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis. However, DexaSite did demonstrate statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. After ongoing meetings with the FDA, in July 2014, the FDA agreed that the results of the Phase 3 clinical trial could support marketing approval for blepharitis. We currently plan to file an NDA with the FDA in 2016 for DexaSite for the treatment of blepharitis.
In November 2013, we submitted a protocol to the FDA for Phase 3 clinical trials with respect to the use of DexaSite for the treatment of inflammation and prevention of eye pain in ocular surgery.
AzaSite Plus: Phase 3 Clinical Trials for Blepharitis/Blepharoconjunctivitis
Expansion of our AzaSite product into a larger franchise includes a fixed combination of azithromycin with dexamethasone for the treatment of blepharitis/blepharoconjunctivitis, an infection of the eyelid and the conjunctiva, as well as other ophthalmic infections. In 2006, we completed our preclinical development of this combination product candidate, filed an IND with the FDA and conducted a Phase 1 clinical trial.
In February 2007, we announced that the preliminary safety data from our Phase 1 clinical trial indicated that AzaSite Plus was well tolerated and no serious adverse events were reported. Treatment-related ocular adverse events were minimal in frequency and equivalent between the treatment and placebo groups. There were no significant differences in intraocular pressure between the AzaSite Plus group and placebo group after 14 days of treatment.
In the fall of 2007, we conducted a pilot study to evaluate endpoints and time points for use in the Phase 3 trial for AzaSite Plus. There were 32 patients with blepharoconjunctivitis who completed the double-masked and randomized trial and received eye drops two times a day for 14 days. The results led to the selection of endpoints for the first Phase 3 trial, which included lid margin redness, lid swelling, conjunctival redness, ocular discharge and lid irritation in at least one eye.
8
Table of Contents
The Phase 3 trial tested a total of 417 patients with blepharoconjunctivitis. The dosing regimen consisted of one drop in the eye and one on the eyelid, two times a day for 14 days. The trial design included three treatment arms with the objective of demonstrating the superiority of AzaSite Plus in treating blepharoconjunctivitis over AzaSite alone or DexaSite alone.
Results from the Phase 3 trial indicated that AzaSite Plus improved clinical outcomes as compared to treatment with AzaSite alone in the reduction of inflammatory signs and symptoms and dexamethasone alone in bacterial eradication. AzaSite Plus was very well tolerated. However, the trial did not achieve its primary clinical endpoint as the reduction of inflammatory signs and symptoms between AzaSite Plus and DexaSite was statistically equivalent.
In April 2009, we discussed the results of this trial with the FDA and, based on this meeting, we developed a protocol for the treatment of blepharitis that would seek to demonstrate AzaSite Plus’s ability to delay exacerbation and/or recurrence of acute episodes of blepharitis. This study would serve as a basis for revisions to the pivotal Phase 3 clinical trial protocols.
In 2010, we discussed a development pathway for this product candidate with the FDA. In May 2011, we reached an agreement with the FDA on a Special Protocol Assessment (SPA) for the design of a Phase 3 clinical trial of AzaSite Plus in patients with blepharitis. The trial design includes four study arms to receive AzaSite Plus, DexaSite, AzaSite or the DuraSite vehicle twice-daily for a period of 14 days. Under the SPA-approved trial protocol, AzaSite Plus was evaluated for safety and efficacy against AzaSite for the primary clinical endpoint of resolution of the clinical signs and symptoms of blepharitis and against DexaSite to compare the length of time to recurrence or exacerbation of symptoms following the treatment period. AzaSite Plus was also evaluated for the secondary clinical endpoint of clinical improvement of signs and symptoms of blepharitis against AzaSite. In November 2011, we initiated a Phase 3 clinical trial for this product candidate and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. AzaSite Plus did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis. However, AzaSite Plus did demonstrate statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. We continue to meet with the FDA to review the overall results of the clinical trial and determine the next steps in the development of this product candidate.
ISV-101: Filed an IND for this product candidate
We developed a topical formulation of the non-steroidal anti-inflammatory bromfenac to treat dry eye disease. In January 2011, we filed an IND with the FDA. We plan to initiate a Phase 1/2 clinical trial for this product candidate, but no time frame has been set.
DuraSite Sustained Delivery Technology
At the core of our AzaSite franchise is our proprietary DuraSite drug delivery technology. Our DuraSite sustained drug delivery technology is a synthetic polymer-based formulation designed to extend the residence time of a drug relative to conventional topical therapies. It enables topical delivery of a drug as a solution, gel, or suspension and can be customized for delivering a wide variety of potential drug candidates.
The combination of DuraSite and proven drug products is designed to result in differentiated products that have increased efficacy and improved compliance through a reduced dosing frequency that yields better outcomes, lowers the development risk by using the proven DuraSite technology with a proven drug product and lowers development costs. In addition to its formulation with azithromycin in our AzaSite family of products, our DuraSite technology may be used in the formulation of new ocular product candidates using either non-proprietary drugs or compounds developed by others for non-ophthalmic indications.
9
Table of Contents
Physical Properties. DuraSite is composed of a cross-linked polyacrylic acid polymer, water and salts. We have developed considerable knowledge of how to formulate DuraSite for topical applications that have a range of viscosities and physical forms including gels, suspensions and solutions. The size of the dry polymer particle averages 5 microns. The molecular weight of the polymer exceeds 3x107 Daltons. Upon the addition of water, DuraSite swells to ~100x its original weight.
The polymer entraps water and the active drug product in a bioadhesive matrix. The viscosity of the matrix is controlled by pH. The bioavailability and release characteristics of the drug can be adjusted by altering the chemical environment. The resulting drug delivery system is bioadhesive, demonstrates sustained release and is compatible with both water soluble and water insoluble molecules.
Regulatory Status. The ingredients in the DuraSite sustained release technology are classified by the FDA as Category 1 GRAS (generally regarded as safe). It has been approved by many pharmacopeias, which helps to facilitate worldwide approvals of drugs that contain DuraSite. DuraSite has been used commercially in AquaSite, an ophthalmic product for dry eye syndrome; AzaSite, a topical anti-infective product for the treatment of bacterial conjunctivitis; and Besivance, besifloxacin ophthalmic suspension, 0.6%.
DuraSite 2 is our next-generation enhanced drug delivery system, which is designed to provide a broad platform for developing superior ophthalmic therapeutics. DuraSite 2 is based on the original DuraSite technology, and incorporates a cationic polymer to achieve sustained and enhanced ocular delivery of drugs. DuraSite 2 is designed to increase the tissue penetration for topically delivered ocular drugs with the aim of improved efficacy and dosing convenience. We have preclinical data from a comparative study that demonstrated superior drug retention and tissue penetration compared to DuraSite. In August 2013, the U.S. Patent and Trademark Office (USPTO) issued a patent on DuraSite 2. The patent provides protection to 2029 for both the delivery system and the drugs that are formulated with DuraSite 2. In November 2013, we obtained preclinical data from a comparative study that demonstrated superior drug retention and tissue penetration compared to DuraSite. We plan to utilize the DuraSite 2 platform in the development of all future pipeline products.
Additional Research and Development Opportunities
In addition to products leveraging our DuraSite technology, we occasionally seek to in-license or acquire promising product candidates and technologies from other companies and universities and research institutions and to apply our expertise to create novel differentiated ophthalmic products.
Collaborative, Licensing and Service Agreements
As part of our business strategy, we have entered into, and will continue to pursue additional licensing agreements, corporate collaborations and service agreements. There can be no assurance that we will be able to negotiate acceptable collaborative, licensing or service agreements, or that our existing arrangements will be successful or renewed or that they will not be terminated.
Pfizer and Pfizer Products, Inc. In February 2007, we entered into a worldwide, exclusive, royalty-bearing licensing agreement with Pfizer (Pfizer License) under Pfizer’s patent family titled “Method of Treating Eye Infections with Azithromycin” for ocular anti-infective product candidates known as AzaSite and AzaSite Plus. Under the Pfizer License, we are required to pay Pfizer a low single-digit royalty based on net sales of the licensed products and to use reasonable commercial efforts to seek regulatory approval for and market licensed products. We have the right to grant sublicenses, subject to Pfizer’s prior approval which shall not be unreasonably withheld. The Pfizer License expires worldwide in November 2018 upon expiration of the underlying Pfizer patent. The Pfizer License may be terminated by Pfizer or us upon material breach of the agreement and following notice and failure to cure, or upon Pfizer or us becoming bankrupt or insolvent.
10
Table of Contents
Akorn. In February 2007, we entered into a license agreement with Inspire. In May 2011, Merck acquired Inspire and Inspire became a wholly-owned subsidiary of Merck. In November 2013, Akorn acquired Inspire from Merck and Inspire is currently a wholly-owned subsidiary of Akorn. Under the license agreement (Akorn License), we licensed exclusive development and commercialization rights under our AzaSite patent rights and certain know-how for topical anti-infective products containing azithromycin as the sole active ingredient for human ocular or ophthalmic indications in the United States and Canada and their respective territories. The Akorn License also provides for nonexclusive licenses under our DuraSite patent rights, container patent rights, Columbia Laboratories, Inc. polymer technology patent rights and certain know-how in the same field of use as described above. We also granted Akorn an exclusive sublicense under the Pfizer patent rights that we have licensed under the Pfizer License discussed above. Akorn has the right to grant sublicenses under the terms of the Akorn License.
Under the Akorn License, Inspire paid us license fees and a milestone payment totaling $32 million through FDA approval in April 2007. Akorn also pays us a royalty on net sales of AzaSite in North America. Akorn, and previously Merck prior to November 2013, was obligated to pay us 25% royalties under the Akorn License for the longer of (i) eleven years from the launch of the first product (August 13, 2007) or (ii) the period during which a valid claim under a patent licensed from us covers a licensed product. For five years after the first year of commercial sale, Merck was required to pay us the greater of the royalty discussed above or certain tiered minimum royalties, which minimum royalties terminated in September 2013. On June 10, 2014, we entered into an amendment to the Akorn License (the “Amendment”). Under the Amendment, Akorn paid us an amendment fee of $6.0 million in return for a lower royalty on net sales of AzaSite in North America. The $6.0 million was used by us to repurchase and cancel the AzaSite Notes (for further discussion, see Note 8). Effective April 1, 2014, for annual net sales of AzaSite, the royalty is (1) 8.0% on net sales less than $20.0 million, subject to increase to 9.0% in the event that Akorn enters into certain settlements with third parties seeking to launch generic versions of AzaSite, (2) 12.5% on net sales greater than or equal to $20.0 million and less than or equal to $50.0 million and (3) 15.0% on net sales in excess of $50.0 million. For annual net sales of AzaSite Xtra, the royalty would be 12.5% on net sales less than $30.0 million and 15.0% on net sales greater than or equal to $30.0 million. We waived any right to consent to any settlements regarding AzaSite (for further discussion, see Note 13) and agreed to cooperate to effectuate any such settlement. The royalties under the Akorn License are subject to certain reductions in the event of patent invalidity, third party licenses, generic competition and uncured material breach.
After obtaining regulatory approval in the United States and Canada, we transferred all regulatory documentation regarding AzaSite to Inspire. Akorn, through its ownership of Inspire, is currently responsible for all regulatory obligations and strategies relating to the further development and commercialization of products in the United States and Canada. Akorn is also responsible for commercialization in both the U.S. and Canada. Akorn focuses its marketing and sales efforts on ophthalmologists rather than pediatricians and primary care physicians, which could limit the effectiveness its AzaSite sales efforts.
We also entered into a trademark license agreement with Inspire in February 2007 under which we granted an exclusive license to the AzaSite trademark and domain name and a nonexclusive license to the DuraSite trademark in connection with the commercialization of products in the United States and Canada under the terms of the Akorn License.
We also entered into a supply agreement (Supply Agreement) with Inspire in February 2007 for azithromycin. We had previously entered into a third-party supply agreement for the production of azithromycin. The Supply Agreement was terminated in July 2012.
During the years ended December 31, 2014, 2013 and 2012, Merck/Akorn royalties and other revenues represented approximately 98%, 48% and 90%, respectively, of our total revenues.
11
Table of Contents
Catalent Pharma Solutions, formerly Cardinal Health PTS, L.L.C. The AzaSite New Drug Application (NDA) was transferred to Inspire and manufacturing responsibilities for AzaSite were transferred to Inspire for sales in the United States and Canada. We continue to have a relationship with Catalent for the manufacture of AzaSite for sales outside the U.S. and Canada and for research and development purposes, as well as for other products in our pipeline.
Bausch and Lomb Incorporated. In December 2003, we completed the sale of a drug candidate for the treatment of ocular infections to Bausch & Lomb (wholly-owned by Valeant as of August 2013) pursuant to a purchase agreement and a license agreement (License Agreement) (collectively, the Asset Sale). Under the terms of the Asset Sale, Bausch & Lomb has assumed all future Besivance development and commercialization expenses and is responsible for all development activities. The drug candidate, Besivance, was developed by Bausch & Lomb. In May 2009, the FDA approved Besivance to treat bacterial conjunctivitis (pink eye). Besivance was launched in the United States by Bausch & Lomb in 2009. In 2011, Besivance was launched internationally in select countries. In April 2013, we sold the rights to receive royalty payments on sales of Besivance. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021.
Until we sold the rights to receive royalty payments in 2013, we were entitled to a middle single-digit royalty on Besivance net product sales, globally, ending upon the later of the expiration of the patent rights underlying Besivance or ten years from the date of the first Besivance product sale by Bausch & Lomb.
The License Agreement provides Bausch & Lomb a license to certain of our patents related to our DuraSite delivery system for use with Besivance and to other non-patented intellectual property used in Besivance. The License Agreement provides for Bausch & Lomb to complete development of the SS734 fluoroquinolone products that combine certain compounds we licensed from SSP Co., Ltd. (SSP) with the DuraSite delivery system and to commercialize any such products. The patent license is exclusive in the particular field of developing, testing, manufacturing, obtaining regulatory approval of, marketing, selling and otherwise disposing of such products. The license of non-patented intellectual property granted to Bausch & Lomb is nonexclusive.
Other. We continually pursue agreements with other companies, universities and research institutions concerning additional therapeutic agents and drug delivery technologies to complement and expand our family of proprietary ophthalmic products as well as collaborative agreements for the further development and marketing of our current products and product candidates. We intend to continue exploring licensing and collaborative opportunities, although there is no certainty that we can successfully enter into, or maintain, any such agreements.
Patents and Proprietary Rights
Patents and other proprietary rights are important to our business. Our policy is to file patent applications seeking to protect technology, inventions and improvements to our inventions that we consider valuable. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. Our DuraSite drug delivery products are made under patents and applications, and we have filed a number of patent applications in the United States relating to our DuraSite technology with delivery tips and drug compounds. Of these applications, six U.S. patents have been issued. We have four U.S. patents on our retinal drug delivery device that have been issued. Six U.S. patents have been issued related to our antibiotic programs with one application pending. At least eight other patent applications by us relating to the foregoing and other aspects of our business and potential business are also pending. Foreign counterparts of our patents have also been filed or issued in many countries.
United States Patent 7,795,231 titled “Concentrated Aqueous Azalide Formulations” is owned by InSite Vision and will expire in October 2027. The product AzaSite Xtra includes inventions under this patent. InSite Vision also owns pending foreign patent applications which claim priority to US Patent 7,795,231.
12
Table of Contents
InSite Vision’s United States Patents 6,239,113, 6,569,443, 7,056,893, 7,732,415 and 7,749,970 titled “Topical Treatment or Prevention of Ocular Infections” are based on a 1999 application (US 09/282,165) and will expire in March 2019. The products AzaSite, AzaSite Plus and AzaSite Xtra include inventions under these patents. InSite Vision also owns foreign patents stemming from the 1999 application in the following countries: Australia, Austria, Belgium, Canada, Cyprus, Denmark, Finland, France, Germany, Great Britain, Greece, Hong Kong, Ireland, Italy, Korea, Japan, Luxembourg, Monaco, Mexico, Netherlands, New Zealand, Portugal, Spain, Sweden, and Switzerland. The foreign patents will expire in March 2020.
InSite Vision has licensed from Pfizer United States Patent 6,861,411, titled “Method of Treating Eye Infections with Azithromycin”. The products AzaSite, AzaSite Plus and AzaSite Xtra include inventions covered by some or all of these patents. Foreign patents based on US 6,861,411 and licensed to InSite Vision have been issued in the following countries: Austria, Belgium, Canada, Cyprus, Denmark, Finland, France, Germany, Great Britain, Greece, Ireland, Italy, Korea, Luxembourg, Mexico, Netherlands, Portugal, Spain, Sweden, and Switzerland. All of these patents will expire in November 2018.
United States Patent 8,778,999 titled “Non-Steroidal Anti-Inflammatory Ophthalmic Compositions” is owned by InSite Vision and will expire in March 2029. The product BromSite includes inventions under this patent. InSite Vision also owns pending foreign patent applications which claim priority to US Patent 8,778,999.
The patent positions of pharmaceutical companies, including ours, are uncertain and involve complex legal and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before a patent is issued. Consequently, we do not know whether any of our pending patent applications will result in the issuance of patents or if any of our patents will provide significant proprietary protection. Since patent applications are maintained in secrecy until they are published, we cannot be certain that we or any licensor was the first to file patent applications for such inventions or that patents issued to our competitors will not block or limit our ability to exploit our technology. Moreover, we might have to participate in administrative proceedings to determine our entitlement to patent rights, which could result in substantial cost to us, even if the eventual outcome is favorable. There can be no assurance that our patents will be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents. See Item 3. “Legal Proceedings.”
A number of pharmaceutical companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. These conflicts, whether actual or perceived, could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our patent applications. In addition, if patents that cover our activities have been or are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost, or at all, or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in or be precluded altogether from introducing products to the market.
In addition to patent protection, we also rely upon trade secret protection for our confidential and proprietary information. There can be no assurance that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets, that such trade secrets will not be disclosed or that we can effectively protect our rights to unpatented trade secrets.
We believe our drug delivery technology may expand the ophthalmic pharmaceutical market by permitting the novel use of drugs for ophthalmic indications that are currently used or being developed for non-ophthalmic indications. However, we may be required to obtain licenses from third parties that have rights to these compounds in order to conduct research, to develop or to market products that contain such compounds. There can be no assurance that such licenses will be available on commercially reasonable terms, if at all.
13
Table of Contents
Research and Development
On December 31, 2014, our research and development staff numbered 25 people, of whom five have Ph.Ds. Our research and development expenses for the years ended December 31, 2014, 2013 and 2012 were as follows:
Research and Development Cost by Program
(in millions)
| Program |
2014 | 2013 | 2012 | |||||||||
| BromSite |
$ | 3.2 | $ | 4.2 | $ | 2.7 | ||||||
| AzaSite Plus/DexaSite |
0.2 | 1.2 | 6.6 | |||||||||
| New products and other |
— | 0.2 | 0.4 | |||||||||
| Programs—non-specific |
5.7 | 6.0 | 5.8 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 9.1 | $ | 11.6 | $ | 15.5 | ||||||
|
|
|
|
|
|
|
|||||||
For 2014, our BromSite program expenses were primarily related to costs to prepare to file an NDA with the FDA, which is currently expected to occur in the first quarter of 2015. Non-specific program costs, which comprised facility, internal personnel and stock-based compensation costs, are not allocated to a specific development program. If we are able to raise sufficient additional funding, we expect to incur additional R&D expense to file an NDA for BromSite in the first quarter of 2015, an NDA for DexaSite in 2016 and develop new product candidates.
In 2013, our BromSite program expenses were primarily related to costs of our Phase 3 clinical trials. For the first Phase 3 clinical trial, we completed patient enrollment in November 2012 with 268 patients and received positive top-line results in March 2013. In May 2013, we initiated the second Phase 3 clinical trial for BromSite and completed patient enrollment in September 2013 with 248 patients enrolled and received positive top-line results in December 2013. Our AzaSite Plus/DexaSite program expenses were primarily related to costs for our Phase 3 clinical trial. We completed patient enrollment for the AzaSite Plus/DexaSite clinical trials in September 2012 and enrolled 907 patients. In July 2013, we announced top-line results for the AzaSite Plus and DexaSite Phase 3 clinical trial.
In 2012, program expenses consisted of our AzaSite Plus/DexaSite program primarily related to costs for our Phase 3 clinical trial. We completed patient enrollment for the AzaSite Plus/DexaSite clinical trial in September 2012 and enrolled 907 patients. Our BromSite program expenses primarily related to the costs for our Phase 3 clinical trial. We completed patient enrollment for the BromSite clinical trial in November 2012 and enrolled 268 patients. Non-specific program costs, which comprised facility, internal personnel and stock-based compensation costs that are not allocated to a specific development program, increased primarily due to an increase in headcount as a direct result of the Phase 3 clinical trials.
Manufacturing
We have no experience or facilities for the manufacture of products for commercial purposes and we currently have no intention of developing such experience or building such facilities. We have a pilot facility, licensed by the State of California, to produce potential products for Phase 1 and some of our Phase 2 clinical trials. However, we rely on third parties for supplies and materials necessary for our Phase 3 clinical trials and commercial needs. If we should encounter delays or difficulties in establishing and maintaining our relationship with qualified manufacturers to produce, package and distribute our finished products, then clinical trials, regulatory filings, market introduction and subsequent sales of such products would be harmed.
Under the Akorn License, Akorn is responsible for the manufacture of AzaSite for the United States and Canada. The AzaSite NDA was transferred to Inspire (currently owned by Akorn) and manufacturing
14
Table of Contents
responsibilities for AzaSite were transferred to Inspire for sales in the United States and Canada. We have a relationship with Catalent for the manufacture of AzaSite for sales outside the U.S. and Canada and for research and development purposes, as well as for other product candidates in our pipeline.
Marketing and Sales
The cost to develop and maintain a marketing organization and sales force is significant and would result in the reallocation of our limited resources needed for the development of our product candidates. We do not currently plan on establishing a dedicated sales force or a marketing organization for our AzaSite, AzaSite Plus or other product candidates.
We have entered into corporate collaborations, and we may continue to pursue additional collaborations with one or more additional pharmaceutical companies in the United States and abroad, to market our products.
Competition
The pharmaceutical industry is highly competitive and requires an ongoing commitment to the pursuit of technological innovation. Such commitment requires significant investment in the resources necessary to discover, develop, test and obtain regulatory approvals for products. It also involves the need to effectively commercialize, market and promote approved products, including communicating the effectiveness, safety and value of products to customers and medical professionals.
The global ophthalmic market is anticipated to grow and will become even more competitive going forward as the prevalence of eye disease increases, leading to increased demand for new and novel ophthalmic products. The market segments that treat diseases and conditions of the eye are subject to ongoing technological change and evolution.
Many companies are engaged in activities similar to our own. Many of these companies have substantially greater financial, technical, marketing and human resources available to them, which may allow them to succeed in developing technologies and products at a faster rate, thereby gaining greater market acceptance than the therapies that we are developing or have developed with our more limited resources. By being first to the market, these competitors may also succeed in obtaining cost advantages or intellectual property rights that would limit our ability to develop and commercialize our own product opportunities. Consequently, they might obtain a more timely and effective regulatory approval for the commercialization of their products in comparison to our timeline.
The global ophthalmic pharmaceutical market is currently comprised of a number of large and well-established companies, including Novartis/Alcon Laboratories, Inc., Allergan, Inc., Santen Pharmaceutical Co., Ltd., Bausch + Lomb, Johnson & Johnson, and Pfizer. While there are many other large- and medium-sized companies participating in the ophthalmic market, smaller companies such as our own find it challenging to successfully develop and market products without entering into collaborations.
Certain segments of the greater ophthalmic market, such as those for glaucoma, anti-infective and anti-inflammatory agents, already have well-established competing products currently available as well as many in development by prominent competitors. Therefore, in order to penetrate these competitive and mature markets, new products must exhibit improved efficacy and safety profiles, be supported by strong marketing and sales initiatives, and have the support of industry thought leaders.
Competition in our industry depends on a variety of factors including the ability to develop enhanced and innovative products, maintain a proprietary technology position, obtain required government approvals for products on a timely basis, attract and retain key personnel, effective and timely marketing of approved products and enter into effective collaborations for the manufacture, commercial marketing and distribution of products in key worldwide markets.
15
Table of Contents
Government Regulation
The manufacturing and marketing of our products and our research and development activities are subject to regulation by numerous governmental authorities in the United States and other countries. In the United States, drugs are subject to rigorous FDA regulation. The Federal Food, Drug and Cosmetic Act and regulations promulgated thereunder govern the testing, manufacture, labeling, storage, record keeping, approval, advertising and promotion in the United States of our products. In addition to FDA regulations, we are also subject to other federal and state regulations such as the Occupational Safety and Health Act and the Environmental Protection Act. Product development and approval within this regulatory framework takes a number of years and involves the expenditure of substantial resources.
The steps required before a pharmaceutical agent may be marketed in the United States include:
| • | preclinical laboratory testing; |
| • | submission to the FDA of an IND; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug; |
| • | the submission of an NDA or Biological License Application (BLA) to the FDA; and |
| • | the FDA approval of the NDA or BLA, prior to any commercial sale or shipment of the drug. |
In addition to obtaining FDA approval for each product, each domestic drug manufacturer and facility must be registered with, and approved by, the FDA. Drug product manufacturing establishments located in California also must be licensed by the State of California in compliance with separate regulatory requirements.
Preclinical tests include laboratory evaluation of product chemistry and animal studies to assess the potential safety and efficacy of the product and its formulation. The results of the preclinical tests are submitted to the FDA as part of an IND and, unless the FDA objects, the IND will become effective 30 days following its receipt by the FDA.
Clinical trials involve the administration of the drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Before any clinical trial can commence, each protocol is submitted to the FDA as part of the IND. Each clinical study is conducted under the auspices of an independent Institutional Review Board that considers, among other things, ethical factors and the rights, welfare and safety of human subjects.
Clinical trials are typically conducted in three sequential phases, but the phases may involve multiple studies and may overlap. In Phase 1, the initial introduction of the drug into human subjects, the drug is tested for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase 2 involves studies in a limited patient population to (i) determine the efficacy of the drug for specific targeted indications, (ii) determine dosage tolerance and optimal dosage and (iii) identify possible adverse effects and safety risks. When a compound is found to be effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 clinical trials are undertaken to further evaluate clinical efficacy and to further test for safety within an expanded patient population at multiple clinical study sites. The FDA reviews both the clinical plans and the results of the trials and may discontinue the trials at any time if there are significant safety issues.
The results of the preclinical studies and clinical studies are submitted to the FDA in the form of an NDA or BLA for marketing approval. The testing and approval process requires substantial time and effort and there can be no assurance that any approval will be granted on a timely basis, if at all. Additional animal studies or clinical trials may be requested during the FDA review period and may delay marketing approval. After FDA approval for the initial indications, further clinical trials are necessary to gain approval for the use of the product for additional indications. The FDA may also require post-marketing testing to monitor for adverse effects, which can involve significant expense.
16
Table of Contents
Among the conditions for manufacture of clinical drug supplies and for NDA or BLA approval is the requirement that the prospective manufacturer’s quality control and manufacturing procedures conform to current Good Manufacturing Practice, or cGMP. Prior to approval, manufacturing facilities are subject to FDA and/or other regulatory agency inspection to ensure compliance with cGMP. Manufacturing facilities are subject to periodic regulatory inspection to ensure ongoing compliance.
For marketing outside the United States, we or our licensees are also subject to foreign regulatory requirements governing human clinical trials and marketing approval for drugs. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary widely from country to country and in some cases are even more rigorous than in the United States.
Scientific and Business Advisors
We have access to a number of academic and industry advisors with expertise in clinical ophthalmology and pharmaceutical development, marketing and sales. Our advisors meet with our management and key scientific employees on an ad hoc basis to provide advice in their respective areas of expertise and further assist us by periodically reviewing with management our preclinical, clinical and marketing activities. We established the Scientific Advisory Board (SAB) to help guide and shape our research programs in the development of novel ophthalmic medicines. Members of the SAB represent leaders in ophthalmic research, treatment and clinical drug development, including Golman Peyman, M.D., Richard Lindstrom, M.D., Gary Foulks, M.D., and Kelly Nichols, O.D., M.P.H., Ph.D. The SAB is led by our Chief Medical Officer, Kamran Hosseini, M.D., Ph.D., and Brian Levy, O.D. M.Sc., a member of our Board of Directors. Our SAB members possess deep insight into the etiology of important ocular diseases, which will be instrumental in advancing our therapeutic programs. Our SAB members have already made significant contributions to our current clinical development programs, providing input on trial protocols and endpoint design.
We plan to add additional advisors as appropriate. Although we expect to receive guidance from our advisors, all of our advisors are employed on a full-time basis by other entities, or are primarily engaged in outside business activities, and may have other commitments to, or consulting or advisory contracts with, other entities that may conflict or compete with their obligations to us.
Executive Officers of the Company
As of February 18, 2015, our executive officers were as follows:
| Name |
Title |
Age | ||
| Timothy Ruane |
Chief Executive Officer and member of the Board | 50 | ||
| Louis Drapeau |
Vice President and Chief Financial Officer | 70 | ||
| Lyle M. Bowman, Ph.D. |
Vice President, Development | 66 | ||
| Kamran Hosseini, M.D., Ph.D. |
Vice President, Clinical & Regulatory Affairs and Chief Medical Officer | 50 | ||
| Surendra Patel |
Vice President, Operations & Quality | 60 |
Timothy Ruane joined us on December 1, 2010 as Chief Executive Officer and was appointed as a member of the Board. Previously, Mr. Ruane served as President and Chief Executive Officer of Tekmira Pharmaceuticals and INEX Pharmaceuticals (which spun-out to Tekmira in 2007), a biopharmaceutical company, from 2005 to 2008. From 2004 to 2005, he served as the Senior Vice President of Corporate Development of INEX. From 2002 to 2004, Mr. Ruane was the Senior Vice President of Business Management of ILEX Oncology. Mr. Ruane has more than 24 years of experience with pharmaceutical and biotechnology companies in various management positions. Mr. Ruane has a Bachelor of Science degree in business finance from Wake Forest University and a Masters in Business Administration from the University of Washington.
17
Table of Contents
Louis Drapeau joined us on October 1, 2007 as Vice President and Chief Financial Officer and served as our interim Chief Executive Officer from October 2008 through November 2010. Mr. Drapeau served as Senior Vice President, Finance and Chief Financial Officer of Nektar Therapeutics, a biopharmaceutical company, from January 2006 until September 2007. From August 2002 to August 2005, Mr. Drapeau was Senior Vice President and Chief Financial Officer of BioMarin Pharmaceutical, a fully integrated biopharmaceutical company. From August 2004 to May 2005, Mr. Drapeau also held the position of Acting Chief Executive Officer of BioMarin. Prior to that, Mr. Drapeau spent over 30 years with Arthur Andersen including 19 years as an Audit Partner in Arthur Andersen’s Northern California Audit and Business Consulting practice, including 12 years as Managing Partner. Mr. Drapeau currently serves on the boards of Bio-Rad Laboratories, Inc. and Ampliphi Biosciences Corporation. He holds an undergraduate degree in mechanical engineering and a Masters in Business Administration from Stanford University.
Lyle M. Bowman joined us in October 1988 as Director of Drug Delivery Systems. Previously, Dr. Bowman had worked at Syntex Ophthalmics as Manager/Director of Analytical Polymer Characterization working on contact lenses and solutions from 1979 through September 1988. From 1989 to 1991, Dr. Bowman served as Vice President, Science and Technology. From 1991 to 1995, he served as Vice President, Development and from 1995 to 2008, he served as Vice President Development and Operations. Dr. Bowman currently is Vice President Development, holds a Ph.D. in Physical Chemistry from the University of Utah and has considerable experience in material science as applied to ophthalmic products.
Kamran Hosseini joined us in February 2008 as Vice President, Clinical & Regulatory Affairs and Chief Medical Officer. From November 2007 to February 2008, Dr. Hosseini served as the ophthalmic expert at JGB BioPharma consulting for R&D, preclinical, clinical, and business development projects. From May 2005 to October 2007, he was the director of ophthalmology drug delivery programs at Alza Corporation, a member of the Johnson and Johnson Family of Companies, where he provided ophthalmology and visual science expertise for new technology assessment activities aimed at enhancing the drug/device unit pipeline. From November 2003 to May 2005, he was a post-doctoral fellow in retinal degenerative diseases at the University of California, San Francisco. Dr. Hosseini received his M.D. from the University of Groningen Faculty of Medicine, The Netherlands, and his Ph.D. as part of a joint program at the University of Texas, Medical Branch in Galveston and the University of Maastricht, The Netherlands.
Surendra Patel joined us in April 2008 as Vice President, Operations & Quality. From 2002 to 2008, Mr. Patel served as Senior Director, Manufacturing Operations at Nektar Therapeutics where he managed clinical and commercial manufacturing operations and played a strategic role in the selection of domestic and international contract manufacturing sites. Mr. Patel has more than 30 years of development and operational experience in various management positions at pharmaceutical and biotechnology companies, including Syntex, Roche Bioscience, Oread Inc., and DrugAbuse Sciences. Mr. Patel has a Bachelor of Science degree in pharmaceutical formulation from De Montford University, Leicester, United Kingdom.
Executive officers are appointed to serve at the discretion of the Board until their successors are appointed. There are no family relationships between any members of the Board and our executive officers.
Employees
As of December 31, 2014, we had 34 employees, 29 of whom were full time. None of our employees are covered by a collective bargaining agreement. We believe we have good employee relations. We also utilize independent consultants to provide services in certain areas of our scientific and business operations.
18
Table of Contents
| Item 1A. | Risk Factors |
Risks Relating to Our Business
Our cash on hand, anticipated cash flow from operations and the net proceeds from our existing debt financing arrangements will only fund our business until approximately May 2015. We need to enter into a strategic transaction or raise additional funds in the near future, which funding could be difficult to obtain or could significantly dilute the value and rights of our common stock, and if not obtained in satisfactory amounts, may prevent us from developing our products, conducting clinical trials or otherwise taking advantage of future opportunities or growing our business, any of which could harm our business
Our independent auditors included an explanatory paragraph in their audit report to our consolidated financial statements for the year ended December 31, 2014, included in this report, referring to our recurring losses from operations, available cash balance and accumulated deficit and a substantial doubt about our ability to continue as a going concern.
We expect that our cash on hand, anticipated cash flow from operations and the net proceeds from our existing debt financing arrangements will only enable us to continue operations until approximately May 2015. At that point, or earlier if circumstances change from our current expectations, we will either have to enter into a strategic transaction or we will require substantial additional funding from the issuance of debt or equity securities, payments under existing or future collaboration agreements, asset sales or other sources. We cannot assure you that any strategic transaction or additional funding will be available on a timely basis, or on reasonable terms, or at all.
The current worldwide financing environment is challenging, particularly for smaller capitalized businesses such as ours, which combined with the negative results of our AzaSite Plus/DexaSite Phase 3 trial, challenges in our past relationship with Merck, uncertainties regarding our current and future relationship with Akorn and uncertainties regarding our ability to commercialize additional products could make it more difficult for us to raise funds on reasonable terms, or at all. If we issue additional equity securities, our stockholders will experience dilution, which could be substantial, and the new equity securities may have rights, preferences or privileges senior to those of existing holders of common stock. If we raise funds through debt, we will have to pay interest and may be subject to restrictive covenants, which would restrict operating flexibility and could harm our business. In addition, the existence of the explanatory paragraph in the audit report may make it more difficult for us to raise additional financing, may adversely affect the terms of such financing and could prevent investors from purchasing our shares in the open market as certain investors may be restricted or precluded from investing in companies that have received this explanatory paragraph in an audit report. Further, the factors leading to the explanatory paragraph in the audit report may harm our ability to obtain additional funding and could make the terms of any such funding, if available, less favorable than might otherwise be the case. We sometimes receive inquiries related to strategic transactions and have and will continue to actively consider potential strategic transactions. If we are unable to secure additional financing or consummate a strategic transaction, we would have to cease operations and liquidate our assets.
It is difficult to evaluate our business because we are in an early stage of commercializing our products, our product candidates are still in clinical trials and successful development of pharmaceutical products is highly uncertain and requires significant expenditures, risk and time
We are still in an early stage of commercializing our products. AzaSite received regulatory approval in the U.S. in April 2007 and commercial sales of AzaSite began in the third quarter of 2007. Besivance received regulatory approval in May 2009 and commercial sales of Besivance began in the second half of 2009. We must receive approval in other countries prior to marketing AzaSite in such countries. Before regulatory authorities grant us marketing approval for additional products, we need to conduct significant additional research and development and preclinical and clinical testing and submit New Drug Applications, or NDAs. Successful
19
Table of Contents
development of pharmaceutical products is highly uncertain. Products that appear promising in research or development may be delayed or fail to reach later stages of development or the market for several reasons, including:
| • | preclinical tests may show the product to be toxic or lack efficacy in animal models; |
| • | failure to receive the necessary U.S. or international regulatory approvals or a delay in receiving such approvals due to, among other things, slow enrollment in clinical studies, failure to achieve study endpoints within the time period prescribed by the study, or at all, additional time requirements for data analysis or BLA or NDA preparation, discussions with the FDA, FDA requests for additional preclinical or clinical data, analyses or changes to study design; or safety, efficacy or manufacturing issues; |
| • | clinical trial results may show the product to be less effective than desired or to have harmful or problematic side effects; |
| • | difficulties in formulating the product, scaling the manufacturing process, or getting necessary manufacturing approvals; |
| • | even if safe and effective, manufacturing costs, pricing, reimbursement issues or other factors may make the product uneconomical; |
| • | proprietary rights of others and their competing products and technologies may prevent the product from being developed or commercialized; or |
| • | inability to compete with superior, equivalent, more cost-effective or more effectively promoted products offered by competitors. |
Therefore, our research and development activities may not result in any commercially viable products.
We have a history of operating losses and we expect to continue to have losses in the future
We have incurred significant operating losses since our inception in 1986 and have pursued numerous drug development candidates that failed to achieve clinical end points or did not prove to have commercial potential. We expect to incur net losses for the foreseeable future or until we are able to achieve significant royalties or other revenues from sales of our products. Attaining significant revenue or profitability depends upon our ability, alone or with third parties, to develop our potential products successfully, conduct clinical trials, obtain required regulatory approvals and manufacture and market our products successfully. We may not ever achieve or be able to maintain significant revenue or profitability, including with respect to AzaSite, our lead product which has experienced declining sales in the United States, and has continued to decline further following Akorn’s acquisition of Inspire from Merck in November 2013. In addition, our right to receive minimum royalties from Merck terminated in September 2013. AzaSite has not been commercially launched outside the United States.
Clinical trials are expensive, time-consuming and difficult to design, enroll and implement and there can never be any assurance that the results of such clinical trials will be favorable
Human clinical trials for our product candidates are very expensive and difficult to design, enroll and implement, in part because they are subject to rigorous regulatory requirements. A significant portion of our operating expenses in the past three years were incurred on our AzaSite Plus/DexaSite Phase 3 clinical trial and BromSite Phase 3 clinical trial. Despite the significant time, expense and resources devoted to the trial, the AzaSite Plus/DexaSite Phase 3 clinical trial did not meet its clinical endpoints as required by the FDA and the future development of these product candidates is uncertain. The clinical trial process is also time-consuming. We may experience difficulties or delays in enrolling our clinical trials, which can delay the trials and our ability to obtain ultimate approval of our product candidates. In addition, we require various clinical materials to conduct our clinical trials and any unavailability or delay in our ability to obtain these materials may delay our
20
Table of Contents
trials, cause them to be more expensive or preclude us from completing these trials, which would harm our ability to obtain approval for our product candidates and therefore harm our business. We estimate that any particular clinical trial may take over a year to complete and will be very expensive. Furthermore, we could encounter problems that might cause us to abandon or repeat clinical trials resulting in additional expense, further delays and potentially preventing the completion of such trials. The commencement and completion of clinical trials may be delayed or terminated due to several factors, including, among others:
| • | unforeseen safety issues; |
| • | lack of effectiveness during clinical trials, including failure to meet required clinical endpoints; |
| • | difficulty in determining dosing and trial protocols; |
| • | slower than expected rates of patient recruitment; |
| • | difficulties in obtaining clinical materials or participants necessary for the conduct of our clinical trials; |
| • | inability to monitor patients adequately during or after treatment; and |
| • | inability or unwillingness of clinical investigators to follow our clinical protocols. |
In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our submissions or the conduct of these trials. In any such case, we may not be able to obtain regulatory approval for our product candidates, in which case we would not obtain any benefit from our substantial investment in developing the product and conducting clinical trials for such products.
The results of our clinical trials may not support our product candidate claims
Even if our clinical trials are completed as planned, we cannot be certain that the results will support our product candidate claims. Even if pre-clinical testing and clinical trials for a product candidate are successful, this does not ensure that later clinical trials will be successful and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing, meet our expectations or defined clinical endpoints, or satisfy the FDA or other regulatory bodies. The clinical trial process may fail to demonstrate that our product candidates are safe for humans or effective for indicated uses. In addition, our clinical trials involve relatively small patient populations. Because of the small sample size, the results of these clinical trials may not be indicative of future results. Any such failure would likely cause us to abandon the product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay or preclude the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues.
For example, results from our 2008 Phase 3 clinical trial of AzaSite Plus for the treatment of blepharoconjunctivitis showed improved clinical outcomes as compared to treatment with a corticosteroid or antibiotic alone in the reduction of inflammatory signs and symptoms and bacterial eradication, respectively. However, the trial did not achieve its primary clinical endpoint as defined by the protocol. In 2013, our AzaSite Plus/DexaSite Phase 3 clinical trial also failed to meet its primary endpoint. The future development of these product candidates is unclear and we may not receive any return on our significant investments in AzaSite Plus or DexaSite. Moreover, we cannot assure you that we will be able to file an NDA for BromSite, in a timely manner or at all, or that we will ever achieve FDA approval for the commercialization of AzaSite Plus, DexaSite, or BromSite. Our lack of cash resources may cause us to have to delay filing NDAs for BromSite or DexaSite, delaying any FDA approvals and commercialization of either product.
21
Table of Contents
Our strategy for commercialization of our products requires us to enter into successful arrangements with corporate collaborators
We generally intend to enter into partnering and collaborative arrangements with respect to the commercialization of our product candidates. However, we cannot assure you that we will be able to enter into such arrangements or that they will be beneficial to us. The success of our partnering and collaboration arrangements will depend upon many factors, including, among others:
| • | the progress and results of our preclinical and clinical testing and research and development programs; |
| • | the time and cost involved in obtaining regulatory approvals; |
| • | the market, or perceived market, for our product candidates; |
| • | our financial condition, and ability to perform our obligations to our potential partners; |
| • | our ability to negotiate favorable terms with potential collaborators; |
| • | the efforts and success of our collaborators in further developing or marketing the product; |
| • | our ability to prosecute, defend and enforce patent claims and other intellectual property rights; |
| • | the outcome of possible future legal actions; and |
| • | competing technological and market developments. |
We may not be able to enter into arrangements with third parties to support the commercialization of our products on acceptable terms, or at all, and may not be able to maintain any arrangement that we do enter into. If we pursue a partnership for BromSite or our other product candidates prior to successfully filing an NDA or completing Phase 3 trials, we will likely receive less favorable economic terms than if we successfully filed an NDA or completed Phase 3 trials.
The commercial success of our products is dependent on the diligent efforts of our corporate collaborators
Because we generally rely on third parties for the marketing and sale of our products, revenues that we receive will be highly dependent on the efforts and success of these third parties. In November 2013, Akorn acquired Inspire from Merck and Akorn became responsible for the commercialization of AzaSite in the United States. Prior to Akorn’s acquisition of Inspire, the monthly prescriptions and our earned royalty revenues on net sales of AzaSite by Merck had decreased significantly, a trend that has continued subsequent to Akorn’s acquisition of Inspire in November 2013. In June 2014, we amended the Akorn License to, among other things, reduce the royalty rate we receive to a tiered royalty percentage ranging from 8.0% to 15.0% of AzaSite net sales, depending on the level of such sales. We believe that our AzaSite royalties will likely decline further in future periods due to the reduced royalty rate and declining prescriptions for AzaSite. In addition, our partners, including Akorn, may terminate their relationships with us and/or may not diligently or successfully market or sell our products. Akorn can terminate the Akorn License at any time at its discretion. Akorn or other partners may also emphasize sales and marketing of their own or other products or pursue alternative or competing technologies or develop alternative products either on their own or in collaboration with others, including our competitors. In addition, marketing consultants and contract sales organizations that we use for our products may market products that compete with our products and we must rely on their efforts and ability to market and sell our products effectively.
If we fail to enter into future collaborations or our current collaborations are terminated, we will need to enter into new collaborations or establish our own sales and marketing organization; our royalties under the Akorn license are subject to reduction, termination and suspension
We may not be able to enter into or maintain collaborative arrangements with third parties, including Akorn. If we are not successful in entering into future collaborations or maintaining our existing collaborations, we may be required to find new corporate collaborators or establish our own sales and marketing organization.
22
Table of Contents
We do not have a long-standing relationship with Akorn, which acquired Inspire from Merck in November 2013. Prior to Akorn’s acquisition of Inspire, and continuing since Akorn’s acquisition, the number of monthly prescriptions and sales of AzaSite have significantly declined. While the minimum royalty payments from Merck had previously made up for this decline, Merck’s minimum royalty payment obligations terminated in September 2013 and Akorn has no such obligations. Royalties under the Akorn License are subject to a cumulative reduction or offset in the event of patent invalidity, generic competition, uncured material breaches by us or in the event that Akorn is required to pay royalties, milestone payments or license fees to third parties for the continued use of AzaSite. In addition, the applicable royalty rate under the Akorn License is subject to reduction by up to 50% in any country during any period in which AzaSite does not have patent protection and Akorn’s obligation to pay royalties may be suspended entirely upon the occurrence of certain events, such as a requirement by the FDA or other governmental agency to suspend the marketing of AzaSite or withdraw it from the market in the United States or if we are unable to obtain commercial quantities of AzaSite for Akorn to market and sell. Despite Akorn’s acquisition of Inspire from Merck, we believe that our AzaSite royalties will likely decline further in future periods due to the reduced royalty rate and declining prescriptions for AzaSite.
If the Akorn License is terminated, we will have to find a new marketing partner or market AzaSite ourselves. There can be no assurance that any new partnership would be possible or on similar terms as the Akorn License or that it would be successful. If the Akorn License is terminated, our efforts to find a new third-party collaborator or pursue direct commercialization efforts ourselves will divert the attention of senior management from our current business operations, which could delay the development or licensing of our other product candidates. If we elect to commercialize AzaSite ourselves, we would have to expend significant resources as we currently have no sales, marketing or distribution capabilities or experience, and have no current plans to establish any such resources. Our limited cash resources would likely preclude us from establishing any marketing or sales capabilities. Accordingly, our efforts to commercialize AzaSite ourselves, if undertaken, may not be successful and could harm our financial condition and results of operation. If we are unable to maintain existing collaborations, enter into additional collaborations or successfully market our products ourselves, our revenues and financial results would be significantly harmed.
Our future success depends on our ability to engage third parties to assist us with the development of new products, new indications for existing products and the conduct of our clinical trials to achieve regulatory approval for commercialization and any failure or delay by those parties to fulfill their obligations could adversely affect our development and commercialization plans
For our business model to succeed, we must continually develop new products or discover new indications for our existing products. As part of that process, we rely on third parties such as clinical research organizations, clinical investigators and outside testing labs for development activities, such as Phase 2 and/or Phase 3 clinical testing, and to assist us in obtaining regulatory approvals for our product candidates. We rely heavily on these parties for successful execution of their responsibilities but have no control over how these parties manage their businesses and cannot assure you that such parties will diligently or effectively perform their activities. For example, the clinical investigators that conducted our clinical trials, including our second BromSite Phase 3 clinical trial, were not our employees and we anticipate that any future clinical trials for any of our product candidates will also be conducted by third parties. We are responsible for ensuring that each of our clinical trials is conducted in accordance with applicable protocols, rules and regulations or in accordance with the general investigational plan and protocols for the trial as well as the various rules and regulations governing clinical trials in the United States and abroad. Any failure by those parties to perform their duties effectively, in compliance with clinical trial protocols, or on a timely basis, could delay or cause cancellation of our clinical trials, cause us to have to repeat the clinical trials, thereby increasing our expenses, harm our ability to develop and commercialize new products, subject us to potential liabilities and harm our business.
23
Table of Contents
Physicians and patients may not accept or use our products
Even if the FDA approves our product candidates, physicians and patients may not accept or use them. Acceptance and use of our products will depend upon a number of factors including:
| • | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our products; |
| • | effectiveness of marketing and distribution efforts by us and our licensees and distributors; |
| • | cost-effectiveness of our products relative to competing products or treatments; |
| • | actual or perceived benefits of competing products or treatments; |
| • | physicians’ comfort level and prior experience with and use of competing products or treatments; and |
| • | availability of reimbursement for our products from government or other healthcare payers. |
We may require additional licenses or be subject to expensive and uncertain patent litigation in order to sell our products
A number of pharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. As is common in the pharmaceutical and biotech industry, from time to time we receive notices from third parties alleging various challenges to our patent rights. Such conflicts, if proven, could invalidate our issued patents, limit the scope of the patents, if any, that we may be able to obtain, result in the denial of our patent applications or block our rights to exploit our technology. If the USPTO or foreign patent agencies have issued or in the future issue patents to other companies that cover our activities, we may not be able to obtain licenses to these patents at a reasonable cost, or at all, or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in or be precluded altogether from introducing products to the market. If we are required to obtain additional licenses from third parties for the sale of AzaSite in the United States and Canada, we will be required to pay for such additional licenses from our existing cash or royalties received from Akorn.
In addition, we may need to litigate in order to defend against claims of infringement by others, to enforce patents issued to us or to protect trade secrets or know-how owned or licensed by us. Litigation could result in substantial cost to and diversion of effort by us, which may harm our business, prospects, financial condition and results of operations, particularly given our limited and declining cash resources. We have also agreed to indemnify our licensees against infringement claims by third parties related to our technology, which could result in additional litigation costs and liability for us. In addition, our efforts to protect or defend our proprietary rights may not be successful or, even if successful, may result in substantial cost to us, thereby utilizing our very limited resources for purposes other than product development and commercialization.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
| • | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| • | redesign our products or processes to avoid infringement; |
| • | stop using the subject matter claimed in the patents held by others, which could preclude us from commercializing our products; |
| • | pay damages; or |
| • | defend litigation or administrative proceedings, which may be costly whether we win or lose, and which could result in a substantial diversion of our valuable management resources. |
24
Table of Contents
Our business depends upon our proprietary rights and we may not be able to protect, enforce, or secure our intellectual property rights adequately
Our success depends in large part on our ability to obtain patents, protect trade secrets, obtain and maintain rights to technology developed by others, and operate without infringing upon the proprietary rights of others. A substantial number of patents in the field of ophthalmology and genetics have been issued to pharmaceutical, biotechnology and biopharmaceutical companies. Moreover, competitors may have filed patent applications, may have been issued patents or may obtain additional patents and proprietary rights relating to products or processes competitive with ours. We cannot assure you that patents will be granted on a timely basis, or at all, on any of our patent applications or that the scope of any of our issued patents will be sufficiently broad to offer meaningful protection. We may not be able to develop additional proprietary products that are patentable. Even if we receive patent issuances, those issued patents may not provide us with adequate protection for our inventions or may be challenged by others.
In April 2011, we received a letter (Notice Letter) from Sandoz, Inc. (Sandoz) providing notice that Sandoz has filed an Abbreviated New Drug Application (ANDA) with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution (Sandoz Product) prior to the expiration of the five U.S. patents listed in the Orange Book for AzaSite which include four of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Sandoz ANDA filing, Sandoz alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed by the Sandoz Product. On May 26, 2011, we, Merck and Pfizer filed a patent infringement lawsuit against Sandoz and related entities. The plaintiff companies agreed that Merck will take the lead in prosecuting this lawsuit. On November 15, 2013, Akorn acquired Inspire from Merck and, as such, acquired the rights to AzaSite in North America. Akorn has taken the lead in prosecuting this lawsuit. Before the trial, the patents involved in the litigation were limited to the one Pfizer patent and three of our patents. On October 4, 2013, the United States District Court for the District of New Jersey entered a Final Judgment in favor of us and the other plaintiffs finding all the asserted claims of the patents in the litigation valid and infringed by Sandoz and related entities. The Court Order specified that the effective date of any FDA approval of a Sandoz ANDA for generic 1% azithromycin ophthalmic solution products would be no earlier than the expiration date of the patents in the litigation. On November 4, 2013, Sandoz filed an appeal of this decision to the United States Court of Appeals for the Federal Circuit. We and the other plaintiffs intend to vigorously contest any Sandoz assertions that these patents should have been found not infringed, invalid and/or unenforceable. An adverse decision on the issues of validity or enforceability on any such appeal would significantly harm our royalty revenue stream from AzaSite.
In May 2013, we received a Notice Letter that Mylan Pharmaceuticals, Inc. or Mylan, has filed an ANDA with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution, or the Mylan Product, prior to the expiration of the U.S. patents listed in the Orange Book for AzaSite, which include three of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Mylan ANDA filing, Mylan alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed upon by the Mylan Product. On June 14, 2013, we, Merck and Pfizer filed a patent infringement lawsuit against Mylan and a related entity. On November 15, 2013, Akorn acquired Inspire from Merck and, as such, acquired the rights to AzaSite in North America. The plaintiff companies have agreed that Akorn will take the lead in prosecuting this lawsuit. The filing of this lawsuit triggered an automatic stay, or bar, of the FDA’s approval of the ANDA for up to 30 months or until a final district court decision of the infringement lawsuit, whichever comes first. We and the other plaintiffs intend to vigorously enforce our patent rights relating to AzaSite and vigorously contest any Mylan assertions that these patents are invalid and/or unenforceable.
Furthermore, the patents of others may impair our ability to commercialize our products. The patent positions of firms in the pharmaceutical and biotechnology industries generally are highly uncertain, involve complex legal and factual questions, and have recently been the subject of significant litigation. The USPTO and the courts have not developed, formulated, or presented a consistent policy regarding the breadth of claims
25
Table of Contents
allowed or the degree of protection afforded under pharmaceutical and genetic patents. Despite our efforts to protect our proprietary rights, others may independently develop similar products, duplicate any of our products or design around any of our patents. In addition, third parties from whom we have licensed or otherwise obtained technology may attempt to terminate or scale back our rights.
We also depend upon unpatented trade secrets to maintain our competitive position. Others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets. Our trade secrets may also be disclosed, and we may not be able to protect our rights to unpatented trade secrets effectively. To the extent that we, our consultants or our research collaborators use intellectual property owned by others, disputes also may arise as to the rights in related or resulting know-how and inventions.
The failure to successfully market and commercialize AzaSite has continued to harm sales of AzaSite, which has made it unlikely that we will receive significant future revenue from sales of AzaSite
The success of the commercialization of AzaSite and our receipt of royalties under the Akorn License will depend on a number of factors, including, among others:
| • | the scope of Akorn’s marketing of AzaSite in the United States; |
| • | Akorn’s commitment to continuing the Akorn License with us; |
| • | Akorn’s deployment of resources to market and sell AzaSite or lack thereof; |
| • | Akorn’s marketing efforts outside of ophthalmologists; |
| • | Akorn’s pricing decisions regarding AzaSite; |
| • | Akorn’s marketing and selling of any current or future competing products; |
| • | the effectiveness and extent of Akorn’s promotional, sales and marketing efforts; |
| • | Akorn’s ability to build, train and retain an effective sales force; |
| • | Akorn’s marketing efforts and success outside of eye care professionals, such as to pediatricians and primary care physicians; |
| • | Akorn’s ability to successfully market AzaSite to physicians and patients; |
| • | Akorn’s ability to compete against larger and more experienced competitors; |
| • | the discovery of any side effects or negative efficacy findings for AzaSite; |
| • | product recalls or product liability claims relating to AzaSite; |
| • | the introduction of generic competition; |
| • | the extent to which competing products for the treatment of bacterial conjunctivitis obtain more favorable formulary status than AzaSite; and |
| • | the relevant parties’ ability to adequately maintain or enforce the intellectual property rights relevant to AzaSite. |
Pediatricians and primary care physicians write more than 67% of prescriptions for ophthalmic antibiotics. However, Akorn has no experience calling on pediatricians and primary care physicians. Akorn’s focus on eye care professionals rather than pediatricians and primary care providers could result in lower AzaSite sales and therefore lower royalties paid to us. A large number of pharmaceutical companies, including those with competing products, much larger sales forces and much greater financial resources, and those with products for indications that are completely unrelated to AzaSite, compete for the time and attention of eye care professionals, pediatricians and primary care physicians. We have no control over how Akorn manages and operates its sales force, how effective Akorn’s sales efforts will be or Akorn’s pricing decisions regarding AzaSite.
26
Table of Contents
Akorn could experience financial or other difficulties unrelated to AzaSite that could adversely affect the marketing or sale of AzaSite. Moreover, Akorn could change its commercial strategy and deemphasize or sell or sublicense its rights to AzaSite. We cannot prevent Akorn from developing or licensing a product that competes with AzaSite or limiting or withdrawing its support of AzaSite.
We rely on a sole source for the supply of the active pharmaceutical ingredient for AzaSite
We currently have a single supplier for azithromycin, the active drug incorporated into AzaSite, as well as AzaSite Plus and AzaSite Xtra. The supplier of azithromycin has a drug master file on the compound with the FDA and is subject to the FDA’s review and oversight. The supplier’s manufacturing facility is subject to potential natural disasters, including earthquakes, hurricanes, tornadoes, floods, fires or explosions, and other interruptions in operation due to factors including labor unrest or strikes, failures of utility services or microbial or other contamination. If the supplier were to fail or refuse to continue to supply us or Akorn, if the FDA were to identify issues in the production of azithromycin that the supplier was unable to resolve quickly and cost-effectively, or if other issues were to arise that impact production, the manufacture and commercialization of AzaSite could be interrupted which could prevent us from receiving future revenue from AzaSite. Additional suppliers for azithromycin exist, but qualification of an alternative source would be required and could be time consuming and expensive and, during such qualification process, any shortage of azithromycin would negatively impact the sales of AzaSite and could delay the development timeline of AzaSite Plus and AzaSite Xtra.
In addition, certain of the raw materials that we use in formulating DuraSite, the drug delivery system used in AzaSite and our other products, are available only from Lubrizol Advanced Materials, Inc., or Lubrizol. Although we do not have a current supply agreement with Lubrizol, we have not encountered any difficulties obtaining necessary materials from Lubrizol. Any significant interruption in the supply of these raw materials could delay sales of AzaSite, which could prevent us from receiving future revenue from AzaSite.
We compete in highly competitive markets and our competitors’ financial, technical, marketing, manufacturing and human resources may surpass ours and limit our ability to develop and/or market our products and technologies
Our success depends upon developing and maintaining a competitive advantage in the development of products and technologies in our areas of focus. We have many competitors in the United States and abroad, including pharmaceutical, biotechnology and other companies with varying resources and degrees of concentration in the ophthalmic market. Our competitors may have existing products or products under development which may be technically superior to ours or which may be less costly or more acceptable to the market. Our competitors may obtain cost advantages, patent protection or other intellectual property rights that would block or limit our ability to develop our potential products. Competition from these companies is intense and is expected to increase as new products enter the market and new technologies become available. Many of our competitors have substantially greater financial, technical, marketing, manufacturing and human resources than we do, particularly in light of our current financial condition. In addition, they may succeed in developing technologies and products that are more effective, safer, less expensive or otherwise more commercially acceptable than any that we have or will develop. Our competitors may also obtain regulatory approval for commercialization of their products more effectively or rapidly than we will. If we decide to manufacture and market our products by ourselves, we will be competing in areas in which we have limited or no experience such as manufacturing efficiency and marketing capabilities.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer
The market for our product candidates is characterized by intense competition and rapid technological advances. If our product candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing
27
Table of Contents
products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will be harmed.
We compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have products competitive with AzaSite already approved or in development, including Zymar and Ocuflox by Allergan, Vigamox and Ciloxan by Alcon, and Quixin by Johnson & Johnson. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do, as well as significantly greater experience in:
| • | developing drugs; |
| • | undertaking pre-clinical testing and human clinical trials; |
| • | obtaining FDA and other regulatory approvals of drugs; |
| • | formulating and manufacturing drugs; |
| • | launching, marketing and selling drugs; and |
| • | attracting qualified personnel, parties for acquisitions, joint ventures or other collaborations. |
We have to attract and retain key employees to be successful
A critical factor to our success will be retaining our personnel or recruiting replacement personnel. Competition for skilled individuals in the biotechnology business, particularly in the San Francisco Bay Area, is highly competitive, and we may not be able to continue to attract and retain personnel necessary for the development of our business, particularly in light of our financial condition. Our ability to attract and retain such individuals may be harmed by our current financial situation, uncertainties regarding our ability to raise additional funds or continue our operations and the other challenges we face. Our employees may leave us to pursue other opportunities due to uncertainties regarding our financial condition or announcements or rumors regarding potential strategic transactions. The loss of key personnel, the failure to recruit replacement personnel or to develop needed expertise would harm our business.
Our products are subject to government regulations and approvals which may delay or prevent the marketing of potential products and impose costly procedures upon our activities
The FDA and comparable agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon preclinical and clinical testing, manufacturing and marketing of pharmaceutical products. Lengthy and detailed preclinical and clinical testing, validation of manufacturing and quality control processes, and other costly and time-consuming procedures are required. Satisfaction of these requirements typically takes several years and the time needed to satisfy them may vary substantially, based on the type, complexity and novelty of the pharmaceutical product. The effect of government regulation may be to delay or to prevent marketing of potential products for a considerable period of time and to impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approval on a timely basis, or at all, for any products we develop. Success in preclinical or early stage clinical trials does not assure success in later stage clinical trials. Data obtained from preclinical and clinical activities are susceptible to varying interpretations that could delay, limit or prevent regulatory approval. If regulatory approval of a product is granted, such approval may impose limitations on the indicated uses for which a product may be marketed. Further, even after we have obtained regulatory approval, later discovery of previously unknown problems with a product may result in restrictions on the product, including withdrawal of the product from the market. Moreover, the FDA has recently reduced previous restrictions on the marketing, sale and prescription of products for indications other than those specifically approved by the FDA. Accordingly, even if we receive FDA approval of a product for certain
28
Table of Contents
indicated uses, our competitors, including our collaborators, could market products for such indications even if such products have not been specifically approved for such indications. If the FDA determines regulatory approval is required, any delay in obtaining or failure to obtain regulatory approvals would make it difficult or impossible to market our products and would harm our business, prospects, financial condition, and results of operations.
The FDA’s policies may change and additional government regulations may be promulgated which could prevent or delay regulatory approval of our potential products. Moreover, increased attention to the containment of health care costs in the United States could result in new government regulations that could harm our business. Adverse governmental regulation might arise from future legislative or administrative action, either in the United States or abroad. See “Uncertainties regarding healthcare reform and third-party reimbursement may impair our ability to raise capital, form collaborations and sell our products.”
We have no experience in commercial manufacturing and if contract manufacturing is not available to us or does not satisfy regulatory requirements, we will have to establish our own regulatory compliant manufacturing capability and may not have the financial resources to do so
We have no experience manufacturing products for Phase 3 clinical trials and commercial purposes at our own facility. We have a pilot facility licensed by the State of California to manufacture a number of our products for Phase 1 and Phase 2 clinical trials but not for late stage clinical trials or commercial purposes. Therefore, we rely on a single contract manufacturer for a substantial portion of our manufacturing requirements. Any delays or difficulties that we may encounter in establishing and maintaining a relationship with qualified manufacturers to produce, package and distribute our products may harm our clinical trials, regulatory filings, market introduction and subsequent sales of our products.
Contract manufacturers must adhere to cGMP regulations that are strictly enforced by the FDA on an ongoing basis through the FDA’s facilities inspection program, as well as by foreign governmental associations outside the United States. Contract manufacturing facilities must pass a pre-approval plant inspection before the FDA will approve an NDA. Some of the material manufacturing changes that occur after approval are also subject to FDA review and clearance or approval. While the FDA has approved the AzaSite manufacturing process and facility, the FDA or other regulatory agencies may not approve the process or the facilities by which any of our other products may be manufactured or could rescind their approval of the AzaSite manufacturing process or facility. Our dependence on third parties to manufacture our products may harm our ability to develop and deliver products on a timely and competitive basis. To the extent that we change manufacturers or engage additional manufacturers in the United States or abroad, we may experience delays, increased costs, quality-control issues and other issues that could harm our ability to conduct clinical trials and market and sell our products. Should we be required to manufacture products ourselves, we will:
| • | be required to expend significant amounts of capital to install a manufacturing capability; |
| • | be subject to the regulatory requirements described above; |
| • | be subject to similar risks regarding delays or difficulties encountered in manufacturing any such products; and |
| • | require substantially more additional capital than we otherwise may require. |
Therefore, we may not be able to manufacture any products successfully or in a cost-effective manner.
Uncertainties regarding healthcare reform and third-party reimbursement may impair our ability to raise capital, form collaborations and sell our products
The continuing efforts of governmental and third-party payers to contain or reduce the costs of healthcare through various means may harm our business. For example, in some foreign markets, the pricing or profitability of healthcare products is subject to government control. In the United States, there have been, and we expect
29
Table of Contents
there will continue to be, a number of federal and state proposals to implement similar government control, which could lead to lower reimbursement rates for our products or no reimbursement at all. The implementation or even the announcement of any of these legislative or regulatory proposals or reforms could harm our business by reducing the prices we or our partners are able to charge for our products, impeding our ability to achieve profitability, raise capital or form collaborations. In addition, the availability of reimbursement from third-party payers determines, in large part, the demand for healthcare products in the United States and elsewhere. Examples of such third-party payers are government and private insurance plans. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products and third-party payers are increasingly challenging the prices charged for medical products and services. If we or our partners succeed in bringing one or more products to the market, reimbursement from third-party payers may not be available or may not be sufficient to allow the sale of these products on a competitive or profitable basis.
Our insurance coverage may not adequately cover our potential product liability exposure
We are exposed to potential product liability risks inherent in the development, testing, manufacturing, marketing and sale of human therapeutic products. Product liability insurance for the pharmaceutical industry is expensive. Although we believe our current insurance coverage is adequate to cover likely claims we may encounter given our current stage of development and activities, our present product liability insurance coverage may not be adequate to cover all potential claims we may encounter, particularly if AzaSite is commercialized outside the United States and Canada. If AzaSite is commercialized in other countries, we may have to increase our coverage, which will be expensive, and we may not be able to obtain or afford adequate insurance coverage against potential claims in sufficient amounts or at a reasonable cost.
Our use of hazardous materials may pose environmental risks and liabilities which may cause us to incur significant costs
Our research, development and manufacturing processes involve the controlled use of small amounts of hazardous solvents used in pharmaceutical development and manufacturing, including acetic acid, acetone, acrylic acid, calcium chloride, chloroform, dimethyl sulfoxide, ethyl alcohol, hydrogen chloride, nitric acid, phosphoric acid and other similar solvents. We retain a licensed outside contractor that specializes in the disposal of hazardous materials used in the biotechnology industry to properly dispose of these materials, but we cannot completely eliminate the risk of accidental contamination or injury from these materials. Our cost for the disposal services rendered by our outside contractor was not material for the years ended 2014, 2013, or 2012, respectively. In the event of an accident involving these materials, we could be held liable for any damages that result and any such liability could exceed our resources. Moreover, as our business develops we may be required to incur significant costs to comply with federal, state and local environmental laws, regulations and policies, especially to the extent that we manufacture our own products.
Management and principal stockholders may be able to exert significant control on matters requiring approval by our stockholders
As of January 31, 2015, our management and principal stockholders (those owning more than 5% of our outstanding shares) together beneficially owned approximately 47% of our shares of common stock. As a result, our management and principal stockholders, acting together or individually, may be able to exert significant control on matters requiring approval by our stockholders, including the election of all or at least a majority of our Board of Directors, the approval of amendments to our charter, and the approval of business combinations and certain financing transactions. In September 2008, a group of our stockholders prevailed in a proxy contest that resulted in the replacement of all members of our Board of Directors.
30
Table of Contents
The market prices for securities of biopharmaceutical and biotechnology companies such as ours have been and are likely to continue to be highly volatile due to reasons that are related and unrelated to our operating performance and progress; registration of the common stock underlying the warrants issued, or to be issued, pursuant to the Purchase Agreement we entered into in connection with our October 2014 debt financing, or the subsequent sale of such common stock, may cause our stock price to decline; we have not paid dividends in the past and do not anticipate doing so in the future
The market prices for securities of biopharmaceutical and biotechnology companies, including ours, have been highly volatile. The market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. In addition, future announcements and circumstances such as our current financial condition and need for substantial additional funding to continue our operations beyond May 2015, the audit report included in this annual report on Form 10-K for the year ended December 31, 2014 that included an explanatory paragraph referring to our recurring losses from operations, available cash balance and accumulated deficit and a substantial doubt about our ability to continue as a going concern, our ability to obtain new financing, any announcements or rumors regarding strategic alternatives we pursue or that terminate, our likelihood of raising additional funding, the status of our relationships or proposed relationships with third-party collaborators, including Akorn, the results of testing and clinical trials, future sales of equity or debt securities by us, the exercise of outstanding options and warrants that could result in dilution to our current holders of common stock, developments in our patents or other proprietary rights or those of our competitors, our failure to meet analyst expectations, any litigation regarding the same, technological innovations or new therapeutic products, governmental regulation, or public concern as to the safety of products developed by us or others and general market conditions concerning us, our competitors or other biopharmaceutical companies may have a significant effect on the market price of our common stock. For example, in the twelve months ended December 31, 2014, our closing stock price fluctuated from a high of $0.36 to a low of $0.12. Such fluctuations can lead to securities class action litigation and make it difficult to obtain financing. Securities litigation against us could result in substantial costs and a diversion of our management’s attention and resources, which could have an adverse effect on our business.
We have filed one registration statement with the Securities and Exchange Commission, or SEC, to register the resale of the common stock issuable upon exercise of the warrants issued pursuant to the Purchase Agreement we entered into in connection with our October 2014 debt financing. To the extent that we draw down additional amounts under the debt financing, we will be obligated to file one or more additional registration statements with the SEC to register the resale of the common stock issuable upon exercise of the warrants issued in connection with such additional borrowings. Filing of a registration statement, or the subsequent sale of the common stock covered by such registration statement, may cause our stock price to decline.
We have not paid any cash dividends on our common stock and we do not anticipate paying any dividends on our common stock in the foreseeable future.
Our common stock trades on the OTCBB
Our common stock currently trades on the over-the-counter bulletin board (OTCBB) market, although there are no assurances that it will continue to trade on this market. Over-the-counter (OTC) transactions involve risks in addition to those associated with transactions on a stock exchange. Listing on the OTC rather than a stock exchange could harm the trading volume and liquidity of our common stock and, as a result, the market price for our common stock might become more volatile. Listing on the OTC rather than on a major stock exchange could also cause a reduction in the number of investors willing or able to hold or acquire our common stock, transactions in our common stock could be delayed and securities analysts’ and news media coverage of us may be reduced. These factors could result in lower prices and larger spreads in the bid and ask prices for shares of common stock. Listing on the OTC rather than on a major stock exchange could also make our common stock substantially less attractive as collateral for loans, for investment by potential financing sources under their internal policies or state and federal securities laws or as consideration in future capital raising transactions.
31
Table of Contents
Furthermore, the listing on the OTC rather than a stock exchange may have other negative implications, including the potential loss of confidence by suppliers, partners and employees. Our OTC status may also make it more difficult and expensive for us to comply with state and federal securities laws in connection with future financings, acquisitions or equity issuances to employees and other service providers, thereby making it more difficult and expensive for us to raise capital, acquire other businesses using our stock and compensate our employees using equity.
We have adopted and are subject to anti-takeover provisions that could delay or prevent an acquisition of our Company and could prevent or make it more difficult to replace or remove current management
Provisions of our certificate of incorporation and bylaws may constrain or discourage a third party from acquiring or attempting to acquire control of us. Such provisions could limit the price that investors might be willing to pay in the future for shares of our common stock. In addition, such provisions could also prevent or make it more difficult for our stockholders to replace or remove current management and could adversely affect the price of our common stock if they are viewed as discouraging takeover attempts, business combinations or management changes that stockholders consider in their best interest. Our Board of Directors has the authority to issue up to 5,000,000 shares of our preferred stock (Preferred Stock). Our Board of Directors has the authority to determine the price, rights, preferences, privileges and restrictions, including voting rights, of the unissued shares of Preferred Stock without any further vote or action by the stockholders. The rights of the holders of common stock will be subject to, and may be harmed by, the rights of the holders of any Preferred Stock that may be issued in the future. The issuance of Preferred Stock, while providing desirable flexibility in connection with possible financings, acquisitions and other corporate purposes, could have the effect of making it more difficult for a third party to acquire a majority of our outstanding voting stock, even if the transaction might be desired by our stockholders. Provisions of Delaware law applicable to us could also delay or make more difficult a merger, tender offer or proxy contest involving us, including Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years unless conditions set forth in the Delaware General Corporation Law are met. The issuance of Preferred Stock or Section 203 of the Delaware General Corporation Law could also be deemed to benefit incumbent management to the extent that these provisions deter offers by persons who would wish to make changes in management or exercise control over management. Other provisions of our certificate of incorporation and bylaws may also have the effect of delaying, deterring or preventing a takeover attempt or management changes that our stockholders might consider in their best interest. For example, our bylaws limit the ability of stockholders to remove directors and fill vacancies on our Board of Directors. Our bylaws also impose advance notice requirements for stockholder proposals and nominations of directors and prohibit stockholders from calling special meetings or acting by written consent.
If earthquakes and other catastrophic events strike, our business may be negatively affected
Our corporate headquarters, including our research and development and pilot plant operations, are located in the San Francisco Bay Area, a region known for seismic activity. A significant natural disaster such as an earthquake would have a material adverse impact on our business, results of operations, and financial condition. If we were able to use the equipment at our contract manufacturing site we could conduct our pilot plant operations although we would incur significant additional costs and delays in our product development timelines.
We face the risk of a decrease in our cash balances and losses in our investment portfolio
Our investment policy is structured to limit credit risk and manage interest rate risk. By policy, we only invest in what we view as very high quality debt securities, such as U.S. government securities. However, the recent uncertainties in the credit markets, including the recent downgrade by Standard & Poor’s of the U.S. debt rating, could negatively affect our ability to liquidate our positions in securities that we currently believe constitute high quality investments and could also result in the loss of some or all of our principal if the issuer of such securities defaults on its credit obligations. The ability to achieve our investment objectives is affected by
32
Table of Contents
many factors, some of which are beyond our control. Our interest income will be affected by changes in interest rates, which are highly sensitive to many factors, including governmental monetary policies and domestic and international economic and political conditions. The outlook for our investment income is dependent on the future direction of interest rates and the amount of cash flows from operations, if any, that are available for investment. Any significant decline in our investment income or the value of our investments as a result of falling interest rates, deterioration in the credit of the securities in which we have invested or general market conditions, could harm our ability to liquidate our investments, our cash position and our income.
We are subject to risks related to our information technology systems and the information gathered in our clinical trials
We rely on information technology systems in order to conduct business, including internal and external communications, ordering materials for our operations, storing operational information and maintaining and reporting our results. These systems are vulnerable to interruption or failure due to the age of certain of our systems, viruses, malware, security breaches, fire, power loss, system malfunction and other events, which may be beyond our control. Systems interruptions or failures could reduce our ability to develop our products or continue our business, which could have a material adverse effect on our operations and financial performance.
Additionally, federal and state laws governing our ability to obtain and, in some cases, to use and disclose data we need to conduct research activities, including our clinical trials, could increase our costs of doing business. These laws’ requirements could further complicate our ability to obtain necessary research data from our collaborators. In the event that our systems are breached and certain clinical data is compromised, we could become subject to costs arising from failure to maintain the privacy of protected health information. Claims that we have violated individuals’ privacy rights or breached our privacy obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We currently lease approximately 39,123 square feet of research laboratory and office space located in Alameda, California. The facility includes laboratories for formulation, analytical, microbiology, pharmacology, quality control and development as well as a pilot manufacturing plant. The lease expires on December 31, 2020, and may be renewed by us for an additional 5-year term. In October 2010, we subleased approximately 11,640 square feet of office space at this facility. The sublease expired on January 31, 2015. On January 1, 2015, the existing leased space was expanded by approximately 5,000 square feet. We believe our existing facilities will be suitable and adequate to meet our needs for the immediate future.
| Item 3. | Legal Proceedings |
The Company is subject to various claims and legal actions during the ordinary course of its business.
In April 2011, the Company received a Notice Letter that Sandoz, Inc. or Sandoz, has filed an Abbreviated New Drug Application, or ANDA, with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution, or the Sandoz Product, prior to the expiration of the five U.S. patents listed in the Orange Book for AzaSite, which include four of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Sandoz ANDA filing, Sandoz alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed upon by the Sandoz Product. On May 26, 2011, we, Merck and Pfizer filed a patent infringement lawsuit against Sandoz and related entities. The plaintiff
33
Table of Contents
companies agreed that Merck would take the lead in prosecuting this lawsuit. Before the trial, the patents involved in the litigation were limited to the one Pfizer patent and three of our patents. On October 4, 2013, the United States District Court for the District of New Jersey entered a Final Judgment in favor of us and the other plaintiffs finding all the asserted claims of the patents in the litigation valid and infringed by Sandoz and related entities. The Court Order specified that the effective date of any FDA approval of a Sandoz ANDA for generic 1% azithromycin ophthalmic solution products would be no earlier than the expiration date of the patents in the litigation. On November 4, 2013, Sandoz filed an appeal of this decision to the United States Court of Appeals for the Federal Circuit. On November 15, 2013, Akorn acquired Inspire from Merck, and as such, acquired the rights to AzaSite in North America. Akorn has taken the lead in prosecuting this lawsuit. We and the other plaintiffs intend to vigorously contest any Sandoz assertions that these patents should have been found not infringed, invalid and/or unenforceable.
On January 3, 2013, certain plaintiffs filed a complaint in circuit court in Fayette County, Kentucky against Bausch & Lomb and the Company alleging that they were injured when treated with the Bausch & Lomb product Besivance following a photorefractive keratectomy. In February 2013, Bausch & Lomb removed the case to the United States District Court for the Eastern District of Kentucky. We moved to dismiss for lack of jurisdiction and in January 2014, the plaintiffs filed a response conceding to our motion to dismiss for lack of personal jurisdiction. In March 2014, the matter was taken off docket by the District Court. We subsequently received a complete release of all claims.
In May 2013, the Company received a Notice Letter that Mylan Pharmaceuticals, Inc. or Mylan, has filed an ANDA with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution, or the Mylan Product, prior to the expiration of the U.S. patents listed in the Orange Book for AzaSite, which include three of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Mylan ANDA filing, Mylan alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed upon by the Mylan Product. On June 14, 2013, we, Merck and Pfizer filed a patent infringement lawsuit against Mylan and a related entity. On November 15, 2013, Akorn acquired Inspire from Merck, and as such, acquired the rights to AzaSite in North America. The plaintiff companies have agreed that Akorn will take the lead in prosecuting this lawsuit. The filing of this lawsuit triggered an automatic stay, or bar, of the FDA’s approval of the ANDA for up to 30 months or until a final district court decision of the infringement lawsuit, whichever comes first. We and the other plaintiffs intend to vigorously enforce our patent rights relating to AzaSite and vigorously contest any Mylan assertions that these patents are invalid and/or unenforceable.
On July 24, 2014, Karin Stiens filed a complaint against Bausch & Lomb, Dr. Lance Ferguson and the Company (Fayette (Kentucky) Circuit Court Civil Action No. 14-CI-2829) alleging that she suffered ophthalmological damage when Dr. Ferguson engaged in off-label use of Besivance in a photorefractive keratectomy procedure as a prophylactic antibiotic. The plaintiff alleges that Bausch & Lomb and the Company negligently tested, marketed and distributed Besivance, and negligently informed medical providers and consumers about Besivance. The complaint contains no facts supporting these general allegations, no facts supporting the plaintiff’s claim of permanent injuries and no allegations asserting Kentucky jurisdiction over the Company. The plaintiff has not pleaded any specific amount in damages nor made any demand. The Company is investigating the plaintiff’s allegations. The Company filed its answer on August 18, 2014.
There are currently no other claims or legal actions that would have a material adverse impact on our financial position, operations or potential performance.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
34
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock is traded on the OTC Bulletin Board under the symbol “INSV.” Over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions. The following table sets forth the high and low closing sales prices for our common stock as reported by the OTC Bulletin Board for the periods indicated. These prices do not include retail mark-ups, mark-downs or commissions.
| 2014 |
High | Low | ||||||
| First Quarter |
0.32 | 0.20 | ||||||
| Second Quarter |
0.20 | 0.12 | ||||||
| Third Quarter |
0.36 | 0.16 | ||||||
| Fourth Quarter |
0.35 | 0.17 | ||||||
| 2013 |
High | Low | ||||||
| First Quarter |
0.36 | 0.28 | ||||||
| Second Quarter |
0.34 | 0.30 | ||||||
| Third Quarter |
0.34 | 0.17 | ||||||
| Fourth Quarter |
0.30 | 0.19 | ||||||
Dividends
We have never declared or paid dividends on our common stock and do not anticipate paying any cash dividends in the foreseeable future. It is the present policy of our Board of Directors to retain our earnings, if any, for the development of our business.
Other Information
As of February 12, 2015, we had approximately 149 registered stockholders of record of our Common Stock. On February 12, 2015, the last sale price reported on the OTCBB for our common stock was $0.20 per share.
35
Table of Contents
Stock Performance Graph
The following graph compares the percentage change in (i) the cumulative total stockholder return on our common stock from December 31, 2009 through December 31, 2014 with (ii) the cumulative total return on (a) the American Stock Exchange (U.S. Index) and (b) the American Stock Exchange (biotech) index. The comparison assumes (i) an investment of $100 on December 31, 2009 in each of the foregoing indices and (ii) reinvestment of dividends, if any.
The stock price performance shown on the graph below represents historical price performance and is not necessarily indicative of any future stock price performance.
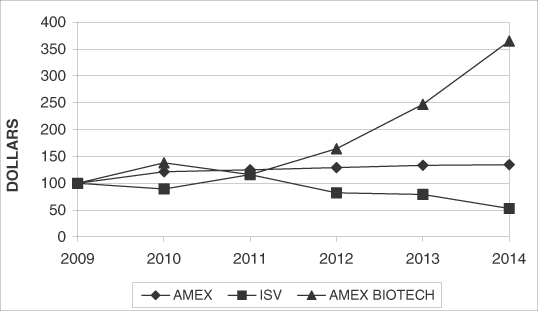
| AMEX | ISV | AMEX BIOTECH |
||||||||||
| 12/31/09 |
100 | 100 | 100 | |||||||||
| 12/31/10 |
121 | 89 | 138 | |||||||||
| 12/31/11 |
125 | 116 | 116 | |||||||||
| 12/31/12 |
129 | 82 | 164 | |||||||||
| 12/31/13 |
133 | 79 | 247 | |||||||||
| 12/31/14 |
134 | 53 | 365 | |||||||||
Notwithstanding anything to the contrary set forth in any of our previous filings under the Securities Act or the Securities Exchange Act of 1934, as amended (Exchange Act) which might incorporate any of our future filings made under those statutes, the preceding Stock Performance Graph will not be incorporated by reference into any of those prior filings, nor will such graph be incorporated by reference into any of our future filings made under those statues.
Recent Sales of Unregistered Securities
Please see “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Major Developments” for a discussion of the 12% Senior Secured Notes and related warrants we sold in the fourth quarter of 2014.
Issuer Purchases of Securities
None.
36
Table of Contents
| Item 6. | Selected Financial Data |
The comparability of the following selected financial data is affected by a variety of factors, and this data is qualified by reference to and should be read in conjunction with the audited financial statements and notes thereto and the Management’s Discussion and Analysis of Financial Condition and Results of Operations contained elsewhere in this Annual Report on Form 10-K. The following table sets forth selected financial data for us for the five years ended December 31, 2014 (in thousands except per share amounts):
| Year Ended December 31, | ||||||||||||||||||||
| (in thousands, except per share data) |
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
| Statement of Operations Data |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Royalties |
$ | 2,005 | $ | 14,787 | $ | 21,641 | $ | 15,138 | $ | 11,120 | ||||||||||
| Sale of royalty rights, net |
— | 15,490 | — | — | — | |||||||||||||||
| Licensing fee, milestone amortization and other |
6,209 | 545 | — | 785 | 747 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total revenues |
8,214 | 30,822 | 21,641 | 15,923 | 11,867 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Expenses: |
||||||||||||||||||||
| Research and development |
9,134 | 11,578 | 15,479 | 7,337 | 4,974 | |||||||||||||||
| General and administrative |
5,721 | 5,754 | 5,781 | 5,645 | 4,511 | |||||||||||||||
| Cost of revenues, principally royalties to third parties |
578 | 385 | 1,062 | 1,917 | 1,727 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total expenses |
15,433 | 17,717 | 22,322 | 14,899 | 11,212 | |||||||||||||||
| Gain on extinguishment of debt |
35,999 | — | — | — | — | |||||||||||||||
| Interest expense and other, net |
(3,516 | ) | (7,896 | ) | (9,494 | ) | (10,167 | ) | (10,248 | ) | ||||||||||
| Change in fair value of warrant liability |
1,497 | 572 | 1,898 | 2,201 | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) |
$26,761 | $5,781 | $(8,277) | $(6,942) | $(9,593) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) per share: |
||||||||||||||||||||
| Income (loss) per share—basic |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | $ | (0.06 | ) | $ | (0.10 | ) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Income (loss) per share—diluted |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | $ | (0.06 | ) | $ | (0.10 | ) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average shares used in per share calculation: |
||||||||||||||||||||
| —Basic |
131,951 | 131,951 | 131,951 | 111,769 | 94,774 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| —Diluted |
132,385 | 132,433 | 131,951 | 111,769 | 94,774 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| As of December 31, | ||||||||||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 1,656 | $ | 8,251 | $ | 9,322 | $ | 26,395 | $ | 16,468 | ||||||||||
| Working capital, exclusive of deferred revenues |
(7,171 | ) | (35,656 | ) | (3,424 | ) | 14,303 | 14,104 | ||||||||||||
| Total assets |
4,905 | 14,165 | 17,759 | 32,401 | 23,586 | |||||||||||||||
| Secured notes payable |
5,199 | 41,281 | 51,883 | 58,558 | 60,000 | |||||||||||||||
| Accumulated deficit |
(175,262 | ) | (202,023 | ) | (207,804 | ) | (199,527 | ) | (192,585 | ) | ||||||||||
| Total stockholders’ deficit |
$ | (7,502 | ) | $ | (35,154 | ) | $ | (41,869 | ) | $ | (34,539 | ) | $ | (42,220 | ) | |||||
No cash dividends have been declared or paid by us since our inception.
37
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion should be read in conjunction with the financial statements and notes thereto included in Item 8 of this Annual Report on Form 10-K.
Overview
We are an ophthalmic product development company advancing ophthalmic pharmaceutical products to address unmet eye care needs. Our current portfolio of products is based on our proprietary DuraSite® sustained drug delivery technology. Our near term strategy is to primarily focus on filing New Drug Applications (NDAs) for BromSite and DexaSite with the United States Food and Drug Administration (FDA).
We face significant challenges related to our lack of financial resources. We expect that our cash on hand, anticipated cash flow from operations and the net proceeds from our existing debt financing arrangements (for further discussion, see Note 1 and 8 to the consolidated financial statements included in this annual report) will only enable us to continue our operations as currently planned until approximately May 2015. Our independent auditors included an explanatory paragraph in their audit report related to our consolidated financial statements for the year ended December 31, 2014, referring to our recurring losses from operations, available cash balance and accumulated deficit and a substantial doubt about our ability to continue as a going concern.
Our DuraSite sustained drug delivery technology is a proven synthetic polymer-based formulation designed to extend the residence time of a drug relative to conventional topical therapies. It enables topical delivery of a drug as a solution, gel or suspension and can be customized for delivering a wide variety of drug candidates. We have focused our research and development and commercial support efforts on the following topical products formulated with our DuraSite drug delivery technology.
| • | AzaSite® (azithromycin ophthalmic solution) 1% is a DuraSite formulation of azithromycin, a broad spectrum ocular antibiotic approved by the FDA in April 2007 to treat bacterial conjunctivitis (pink eye). It is commercialized in the United States by Akorn, Inc. (Akorn). We received a 25% royalty on net sales of AzaSite in North America through March 31, 2014, which was required to service the outstanding, non-recourse promissory notes due 2019 of our wholly-owned subsidiary, Azithromycin Royalty Sub, LLC (AzaSite Notes). On June 10, 2014, we entered into the Third Amendment to License Agreement (Amendment) with Akorn to amend the North American license (Akorn License) of AzaSite to Akorn. Under the Amendment, Akorn paid us an amendment fee of $6.0 million, which was used to repurchase and cancel the AzaSite Notes, in return for a lower royalty on net sales of AzaSite in North America. Effective April 1, 2014, for annual net sales of AzaSite, the royalty is (1) 8.0% on net sales less than $20.0 million, subject to increase to 9.0% in the event that Akorn enters into certain settlements with third parties seeking to launch generic versions of AzaSite, (2) 12.5% on net sales greater than or equal to $20.0 million and less than or equal to $50.0 million and (3) 15.0% on net sales in excess of $50.0 million. |
| • | Besivance® (besifloxacin ophthalmic suspension) 0.6% is a DuraSite formulation of besifloxacin, a broad spectrum ocular antibiotic approved by the FDA in May 2009 to treat bacterial conjunctivitis (pink eye). Besivance was developed by Bausch + Lomb Incorporated (Bausch & Lomb) (wholly-owned by Valeant Pharmaceuticals International, Inc. (Valeant) as of August 2013) and was launched in the United States in the second half of 2009. In 2011, Besivance was launched internationally in select countries. Until we sold the Besivance royalty rights in April 2013, we received a middle single-digit royalty on net sales of Besivance globally. In April 2013, we sold the rights to receive royalty payments on sales of Besivance, beginning on January 1, 2013, for $15 million at closing, with an additional $1 million paid in February 2014 after certain 2013 Besivance sales targets were met. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021. |
38
Table of Contents
| • | BromSiteTM is a DuraSite formulation of bromfenac in development for the treatment of post-operative inflammation and prevention of post-operative eye pain. We initiated a Phase 1/2 clinical trial for this product candidate in August 2010 and we received positive top-line results from this study in the first quarter of 2011, which demonstrated the efficacy and safety of BromSite. In the third quarter of 2011, we completed an additional Phase 2 clinical trial to investigate the pharmacokinetics (PK) of BromSite in humans. We received positive top-line results that showed that the mean concentration of bromfenac in the aqueous humor of patients using BromSite was more than double compared to the currently available bromfenac eye product. In July 2012, we initiated a Phase 3 clinical trial for this product candidate and completed patient enrollment in November 2012. The BromSite Phase 3 clinical trial enrolled 268 patients undergoing cataract surgery in a two-arm trial designed to evaluate the efficacy and safety of BromSite against the DuraSite vehicle alone. In March 2013, we received positive top-line results from this Phase 3 clinical trial demonstrating statistically significant superiority compared to vehicle (p < 0.001) in alleviating ocular pain and inflammation among patients following cataract surgery. In May 2013, we initiated the second Phase 3 clinical trial for this product candidate, which was completed in November 2013 with 248 patients enrolled. We received positive top-line results from this confirmatory Phase 3 clinical trial in December 2013. We plan to file an NDA with the FDA in the first quarter of 2015 for this product candidate. In May 2014, the Swedish Medical Products Agency (MPA) concluded that the existing Phase 3 clinical data for BromSite was likely sufficient to support the filing of a Marketing Authorization Application (MAA) with the MPA. In July 2014, the U.S. Patent and Trademark Office (USPTO) issued a patent on BromSite. The patent provides protection for bromfenac formulations in DuraSite, including ISV-101, to August 8, 2029. |
| • | DexaSiteTM is a DuraSite formulation of dexamethasone in development for the treatment of ocular inflammation. In November 2011, we initiated a Phase 3 clinical trial for this product candidate in blepharitis and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. While DexaSite did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis, it demonstrated statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. After ongoing meetings with the FDA, in July 2014, the FDA agreed that the results of the Phase 3 clinical trial could support marketing approval for DexaSite for the treatment of blepharitis. During the fourth quarter of 2014, we executed post-Phase 3 regulatory meetings with European Health Authorities and the FDA to outline our proposed filings. We currently plan to file an NDA with the FDA in 2016 for this product in this indication. As a next step toward this filing, we will be executing additional regulatory meetings with European Health Authorities and the FDA during the first and second quarters of 2015 to attempt to harmonize the chemistry, manufacturing and controls (CMC) section in support of DexaSite NDA and MAA filings for this indication. In addition, in November 2013, we submitted a protocol to the FDA for Phase 3 clinical trials with respect to the use of DexaSite for the treatment of inflammation and prevention of eye pain in ocular surgery. |
| • | AzaSite PlusTM is a fixed combination of azithromycin and dexamethasone in DuraSite for the treatment of ocular inflammation and infection (blepharitis and/or blepharoconjunctivitis) for which there is no FDA approved indicated treatment. In November 2011, we initiated a Phase 3 clinical trial for this product candidate in blepharitis and completed patient enrollment in the clinical trial in September 2012. This study enrolled 907 patients and we announced top-line results in July 2013. AzaSite Plus did not meet the primary endpoint of complete resolution of all clinical signs and symptoms of blepharitis. However, AzaSite Plus did demonstrate statistically significant improvements in the disease’s clinical signs and symptoms at the end of treatment on Day 15. We continue to meet with the FDA to review the overall results of the clinical trial and determine the next steps in the development of this product candidate. |
| • | DuraSite® 2 is our next-generation enhanced drug delivery system, which is designed to provide a broad platform for developing superior ophthalmic therapeutics. DuraSite 2 is based on the original DuraSite technology, and incorporates a cationic polymer to achieve sustained and enhanced ocular delivery of drugs. DuraSite 2 is designed to increase the tissue penetration for topically delivered |
39
Table of Contents
| ocular drugs with the aim of improved efficacy and dosing convenience. In August 2013, the USPTO issued a patent on DuraSite 2. The patent provides protection to March 5, 2029 for both the delivery system and the drugs that are formulated with DuraSite 2. In November 2013, we obtained preclinical data from a comparative study that demonstrated superior drug retention and tissue penetration compared to DuraSite. We plan to utilize the DuraSite 2 platform in the development of all future pipeline products. We plan to initiate a broad licensing program that provides access to industry partners through both exclusive and non-exclusive licensing and/or commercialization agreements. |
| • | ISV-101 is a DuraSite formulation with a low concentration of bromfenac for the treatment of dry eye disease. We filed an Investigational New Drug Application (IND) with the FDA for this product candidate in the first quarter of 2011. We plan to initiate a Phase 1/2 clinical trial for this product candidate, but no time frame has been set. |
Major Developments
Our major developments and events in 2014 included:
| • | In May 2014, the MPA concluded that the existing Phase 3 clinical data for BromSite was likely sufficient to support the filing of a MAA with the MPA. |
| • | In June 2014, we amended the Akorn License. Akorn paid an amendment fee of $6.0 million in return for a lower royalty on net sales of AzaSite in North America. Effective April 1, 2014, for annual net sales of AzaSite, the royalty is (1) 8.0% on net sales less than $20.0 million, subject to increase to 9.0% in the event that Akorn enters into certain settlements with third parties seeking to launch generic versions of AzaSite, (2) 12.5% on net sales greater than or equal to $20.0 million and less than or equal to $50.0 million and (3) 15.0% on net sales in excess of $50.0 million. For annual net sales of AzaSite Xtra, the royalty would be 12.5% on net sales less than $30.0 million and 15.0% on net sales greater than or equal to $30.0 million. |
| • | In June 2014, we repurchased and cancelled the AzaSite Notes for a single payment of $6.0 million. As a result of the cancellation, the remaining principal on the AzaSite Notes of $41.3 million and accrued interest of $2.8 million was extinguished. |
| • | In July 2014, the USPTO issued a patent on BromSite. The patent provides protection for bromfenac formulations in DuraSite, including ISV-101, to 2029. |
| • | In July 2014, the FDA agreed that the results of the DexaSite Phase 3 clinical trial could support marketing approval for DexaSite for the treatment of blepharitis. We plan to file an NDA for DexaSite with the FDA in 2016. |
| • | In October 2014, we entered into a Securities Purchase Agreement (Purchase Agreement) with certain purchasers (Purchasers) pursuant to which we agreed to sell 12% Senior Secured Notes (Notes) in an aggregate principal amount of up to $15 million and issue warrants to purchase shares of our common stock. In the last quarter of 2014, we sold and issued Notes to the Purchasers having an aggregate principal amount equal to $5.2 million and issued to the Purchasers warrants to purchase an aggregate 2,053,169 shares, 200,620 shares and 2,824,281 shares of common stock that are exercisable at $0.33, $0.31 and $0.26, respectively per share. The total loan commitment by the Purchasers pursuant to the Purchase Agreement is $7.8 million. Accordingly, we have the option to sell additional Notes in an aggregate principal amount of $2.6 million to the Purchasers. Our right to sell and issue additional Notes and to issue additional warrants in connection with such Notes expires on October 9, 2016. |
40
Table of Contents
Business Strategy
Assuming we can raise sufficient funding to continue our operations, our business strategy consists of the following:
| 1. | Develop our pipeline of ocular product candidates. We seek to identify new product candidates from proven drugs that can be improved by formulation in DuraSite 2, which can substantially reduce the clinical risk involved in these product candidates. As appropriate, we plan to conduct preclinical and clinical testing of our product candidates. |
| 2. | Partner our product candidates. When we deem it appropriate, we seek to partner with larger pharmaceutical companies to manufacture and market our products. Partnering agreements generally include upfront and milestone payments, as well as on-going royalty payments upon commercialization, payable to us. |
Critical Accounting Policies and Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States requires us to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make significant estimates, assumptions and judgments about matters that are uncertain:
Revenue Recognition. We recognize revenue when four basic criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. We have arrangements with multiple revenue-generating elements. We analyze our multiple element arrangements to determine whether the elements can be separated and accounted for individually as separate units of accounting. An item can generally be considered a separate unit of accounting if both of the following criteria are met: the delivered item(s) has value to the customer on a stand-alone basis and if the arrangement includes a general right of return and delivery or performance of the undelivered item(s) is considered probable and substantially in our control. Items that cannot be divided into separate units are combined with other units of accounting, as appropriate. Consideration received is allocated among the separate units based on the unit’s estimated selling price and is recognized in full when the criteria are met. We deem service to be rendered if no continuing obligation exists on our part.
Our revenues are primarily related to royalties on product sales and licensing agreements, and such agreements may provide for various types of payments, including upfront payments, research funding and related fees during the terms of the agreements, milestone payments based on the achievement of established development objectives and licensing fees.
We receive royalties from licensees based on third-party sales. The royalties are recorded as earned in accordance with the contract terms when third-party results are reliably measured and collectability is reasonably assured.
Revenue associated with non-refundable up-front license fees under arrangements where the license fees cannot be accounted for as separate units of accounting is deferred and recognized as revenue on a straight-line basis over the expected term of our continued involvement. Revenues from the achievement of milestones are recognized as revenue when the milestones are achieved and the milestone payments are due and collectible.
The Company allocates revenue in multiple-deliverable revenue arrangements using estimated selling prices of the delivered goods and services based on the relative selling price method.
41
Table of Contents
Income Taxes. We account for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain. We record a valuation allowance against deferred tax assets to reduce their carrying value to an amount that is more likely than not to be realized. When we establish or reduce the valuation allowance related to the deferred tax assets, our provision for income taxes will increase or decrease, respectively, in the period such determination is made.
We utilize a two-step approach to recognize and measure uncertain tax positions, if any. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained upon tax authority examination, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon ultimate settlement.
Stock-Based Compensation. We granted stock-based awards to eligible employees and directors to purchase shares of our common stock under our stock compensation plan approved in 1994 (1994 Plan) and its successor the 2007 Performance Incentive Plan (2007 Plan). In addition, we have a qualified employee stock purchase plan (Purchase Plan) in which eligible employees may elect to withhold up to 15% of their compensation to purchase shares of our common stock on a quarterly basis at a discounted price equal to 85% of the lower of the employee’s offering price or the closing price of the stock on the date of purchase. In August 2009, the Purchase Plan was suspended. The benefits provided by these plans qualify as stock-based compensation which requires us to recognize compensation expense based on their estimated fair values determined on the date of grant for all stock-based awards granted, modified or cancelled.
We estimate the fair value of share-based awards on the date of grant using the Black-Scholes option-pricing method (Black-Scholes method). The determination of fair value of share-based awards using an option-pricing model requires the use of certain estimates and assumptions that affect the reported amount of share-based compensation cost recognized in our Consolidated Statements of Operations. These include estimates of the expected term of share-based awards, expected volatility of our stock price, expected dividends and the risk-free interest rate. These estimates and assumptions are highly subjective and may result in materially different amounts should circumstances change and we employ different assumptions in future periods.
For stock-based awards issued, we estimated the expected term by considering various factors including the vesting period of options granted and employees’ historical exercise and post-employment termination behavior. Our estimated volatility was derived using our historical stock price volatility. We have never declared or paid any cash dividends on our common stock and currently do not anticipate paying such cash dividends. We currently anticipate that we will retain all of our future earnings for use in the development and expansion of our business and for general corporate purposes. Any determination to pay dividends in the future will be at the discretion of our Board of Directors and will depend upon our results of operations, financial condition, financial covenants, tax laws and other factors as the Board of Directors, in its discretion, deems relevant. The risk-free interest rate is based upon U.S. Treasury securities with remaining terms similar to the expected term of the stock-based awards.
42
Table of Contents
Results of Operations
Revenues.
Our revenues for the years ended December 31, 2014, 2013 and 2012 were, as follows (in millions):
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| AzaSite royalties |
$ | 2.0 | $ | 14.3 | $ | 19.5 | ||||||
| Sale of royalty rights, net |
— | 15.5 | — | |||||||||
| Licensing fee |
6.0 | — | — | |||||||||
| Besivance royalties |
— | 0.5 | 2.1 | |||||||||
| Other |
0.2 | 0.5 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 8.2 | $ | 30.8 | $ | 21.6 | ||||||
|
|
|
|
|
|
|
|||||||
For 2014, AzaSite royalties included $2.0 million of earned royalties based on net sales of AzaSite. Earned AzaSite royalties declined by 48% compared to 2013 due to a reduction of the royalty rate from 25% to 8%, effective on April 1, 2014, in connection with entry into the Amendment. In June 2014, we received a $6.0 million license fee for the amendment to the Akorn License. Revenues in 2014 also included $0.2 million from manufacturing equipment rental and the sale of bulk drug, azithromycin, to international partners.
For 2013, AzaSite royalties included $3.8 million of royalties based on net sales of AzaSite and an additional $10.5 million minimum royalty true-up payment by Merck. Earned royalties on AzaSite declined by 49% compared to 2012, which resulted from ceased sales promotion of AzaSite in August 2013 by Merck and a scheduled production suite upgrade at the manufacturing plant of AzaSite in the fourth quarter of 2013 which resulted in a supply shortage. The required minimum royalty for the fiscal year ended September 30, 2013, the measurement period pursuant to the terms of the Akorn License, was $19 million. Merck’s obligation to make minimum royalty payments terminated in September 2013 and Akorn’s obligation to pay earned royalties may be suspended upon the occurrence of certain events, such as a requirement by the FDA or other governmental agency to suspend the marketing of AzaSite or withdraw it from the market in the United States. See “Risk Factors,” “Liquidity and Capital Resources,” and Note 1 and 8 to our audited consolidated financial statements included in this Annual Report on Form 10-K.
For 2013, we also recorded $15.5 million from the sale of the rights to receive royalty payments on sales of Besivance. In April 2013, we sold the rights to receive royalty payments on sales of Besivance, beginning on January 1, 2013, for $15 million at closing, with an additional $1 million paid in February 2014 after certain 2013 Besivance sales targets were met. $0.5 million of previously recorded Besivance royalties in 2013 were netted against the sales price. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021. Revenues in 2013 also included $0.5 million from manufacturing equipment rental and the sale of bulk drug, azithromycin, to Merck.
For 2012, AzaSite royalties included $7.6 million of royalties based on net sales and an additional $11.9 million minimum royalty true-up payment by Merck, compared to $10.0 million of earned royalties and an additional $3.9 million minimum royalty true-up payment from Merck in 2011. The increase in royalties was driven by an increase in the required minimum royalty to $17 million for the fiscal year ended September 30, 2012, the measurement period pursuant to the terms of the Akorn License, and the pro-rata share of $19 million for the fiscal year ended September 30, 2013. In addition, a decline in net sales of AzaSite by Merck further increased the minimum royalty true-up payment. Earned AzaSite royalties declined by 24% compared to the same period in 2011. The increase in Besivance royalties in 2012 was driven by an increase in net sales of Besivance by Bausch & Lomb.
Research and development.
Our research and development activities can be separated into two major segments, research and clinical development. Research includes activities involved in evaluating a potential product, related preclinical testing
43
Table of Contents
and manufacturing. Clinical development includes activities related to filings with the FDA and the related human clinical testing required to obtain marketing approval for a potential product. The following represents the approximate cost of these activities for 2014, 2013 and 2012 (in thousands):
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Research |
$ | 5,040 | $ | 4,977 | $ | 5,047 | ||||||
| Clinical development |
4,094 | 6,601 | 10,432 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development |
$ | 9,134 | $ | 11,578 | $ | 15,479 | ||||||
|
|
|
|
|
|
|
|||||||
Our research and development expenses by program for 2014, 2013 and 2012 (in millions) were:
| Program |
2014 | 2013 | 2012 | |||||||||
| BromSite |
$ | 3.2 | $ | 4.2 | $ | 2.7 | ||||||
| AzaSite Plus/DexaSite |
0.2 | 1.2 | 6.6 | |||||||||
| New products and other |
— | 0.2 | 0.4 | |||||||||
| Programs—non-specific |
5.7 | 6.0 | 5.8 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 9.1 | $ | 11.6 | $ | 15.5 | ||||||
|
|
|
|
|
|
|
|||||||
Research and development expenses were $9.1 million in 2014. For 2014, our BromSite program expenses were primarily related to costs to prepare to file an NDA with the FDA in the first quarter of 2015. Non-specific program costs, which comprised facility, internal personnel and stock-based compensation costs, are not allocated to a specific development program. If we are able to raise sufficient additional funding, we expect to incur additional R&D expense to file an NDA for BromSite in the first quarter of 2015, an NDA for DexaSite in 2016 and develop new product candidates.
Research and development expenses were $11.6 million in 2013. In 2013, our BromSite program expenses were primarily related to costs of our Phase 3 clinical trials. For the first Phase 3 clinical trial, we completed patient enrollment in November 2012 with 268 patients and received positive top-line results in March 2013. In May 2013, we initiated the second Phase 3 clinical trial for BromSite and completed patient enrollment in September 2013 with 248 patients enrolled and received positive top-line results in December 2013. Our AzaSite Plus/DexaSite program expenses were primarily related to costs for our Phase 3 clinical trial. We completed patient enrollment for the AzaSite Plus/DexaSite clinical trials in September 2012 and enrolled 907 patients. In July 2013, we announced top-line results for the AzaSite Plus and DexaSite Phase 3 clinical trial.
Research and development expenses were $15.5 million in 2012. In 2012, program expenses consisted of our AzaSite Plus/DexaSite program primarily related to costs for our Phase 3 clinical trial. We completed patient enrollment for the AzaSite Plus/DexaSite clinical trial in September 2012 and enrolled 907 patients. Our BromSite program expenses primarily related to the costs for our Phase 3 clinical trial. We completed patient enrollment for the BromSite clinical trial in November 2012 and enrolled 268 patients.
Our future research and development expenses will depend on our ability to fund our business and the results and magnitude or scope of our clinical, preclinical and research activities and requirements imposed by regulatory agencies. Accordingly, our research and development expenses may fluctuate significantly from period to period. In addition, if we in-license or out-license rights to product candidates, our research and development expenses may fluctuate significantly.
General and administrative.
General and administrative expenses were $5.7 million, $5.8 million and $5.8 million in 2014, 2013 and 2012, respectively. In 2014, we incurred additional legal expenses from our on-going partnering and financing efforts. In 2013, we incurred higher legal and professional fees related to the sale of the Besivance royalty payment and international license agreements. In 2012, we incurred higher costs related to financial reporting services and investor relations.
44
Table of Contents
Cost of revenues.
Our cost of revenues was $0.6 million, $0.4 million and $1.1 million for 2014, 2013 and 2012, respectively. Cost of revenues was primarily comprised of royalties accrued for third parties, including Pfizer, based on AzaSite net sales by Merck or Akorn.
Gain on extinguishment of debt.
Gain on extinguishment of debt was $36.0 million for the year ended December 31, 2014. On June 10, 2014, our Subsidiary and the noteholders entered into a Note Purchase Agreement in which the Subsidiary repurchased and cancelled the AzaSite Notes for a single payment of $6.0 million. As a result of the cancellation, the remaining principal on the AzaSite Notes of $41.3 million and accrued interest of $2.8 million was extinguished. In addition, the Subsidiary wrote-off the remaining balance of $2.1 million in debt issuance costs and incurred less than $0.1 million of legal and professional fees. For the year ended December 31, 2014, the gain on the extinguishment of debt represented basic and diluted net income per share of $0.27.
Interest expense and other, net.
Interest expense and other, net, was an expense of $3.5 million, $7.9 million and $9.5 million for 2014, 2013 and 2012, respectively. Interest expense was primarily due to the interest expense on the non-recourse AzaSite Notes issued in February 2008 and related amortization of the debt issuance costs incurred from our subsidiary’s issuance of the AzaSite Notes. Interest expense was lower in 2014 as a result of payments of principal in 2013 and the repurchase and cancellation of the AzaSite Notes in June 2014. 2014 also included $0.3 million of interest expense and related amortization of debt issuance costs related to the issuance of the Notes pursuant to the Purchase Agreement in the last quarter of 2014. Interest expense decreased in 2013 as a result of payments of principal on the AzaSite Notes.
Change in fair value of warrant liability.
Change in fair value of warrant liability was income of $1.5 million, $0.6 million and $1.9 million for 2014, 2013 and 2012, respectively. The income resulted from a decrease in the fair value of our warrant liability that was initially valued in July 2011 or the last quarter of 2014, and revalued as of December 31, 2014, 2013 and 2012. The decrease in fair value was primarily driven by a decrease in our stock price.
Liquidity and Capital Resources
In recent years, we have financed our operations primarily through private placements of equity securities, debt financings and payments from corporate collaborations. At December 31, 2014 and 2013, our cash, cash equivalents and short-term investments were $1.7 million and $8.3 million, respectively. It is our policy to invest our cash and cash equivalents in highly liquid securities, such as interest-bearing money market funds, treasury and federal agency notes. The current uncertain credit markets may affect the liquidity of such money market funds or other cash investments.
On October 9, 2014, we entered into the Purchase Agreement with the Purchasers pursuant to which we agreed to sell Notes in an aggregate principal amount of up to $15 million and issue warrants to purchase shares of our common stock. We sold and issued Notes to the Purchasers having an aggregate principal amount equal to $5.2 million and issued to the Purchasers warrants to purchase an aggregate of 2,053,169 shares, 200,620 shares and 2,824,281 shares of common stock that are exercisable at $0.33, $0.31 and $0.26, respectively. The total loan commitment by the Purchasers pursuant to the Purchase Agreement is $7.8 million. Accordingly, we have the option to sell additional Notes in an aggregate principal amount of $2.6 million to the Purchasers. Our right to sell and issue additional Notes and to issue additional warrants in connection with such Notes expires on October 9, 2016.
45
Table of Contents
We have incurred significant losses since inception. As of December 31, 2014, our accumulated deficit was $175.3 million and cash and cash equivalents were $1.7 million. Further, we anticipate that our existing cash and short-term investment balances together with cash flows from operations will only be adequate to fund our cash requirements through May 2015. Our ability to fund our operations is dependent primarily upon our ability to raise additional funding, as well as our ability to execute on our business plan, including generating sufficient cash inflows from operating activities.
Our auditors have included an explanatory paragraph in their audit report referring to our recurring losses from operations, available cash balance and accumulated deficit and a substantial doubt about our ability to continue as a going concern. Absent additional funding from private or public equity or debt financings, collaborative or other partnering arrangements, including under the Akorn License, asset sales, or other sources, we expect that our cash on hand, anticipated cash flow from operations and current cash commitments to us will only be adequate to fund our operations until approximately May 2015. If we are unable to secure sufficient additional funding prior to that time, we will need to enter into a strategic transaction or else cease operations and liquidate our assets. Our financial statements were prepared on the assumption that we will continue as a going concern and do not include any adjustments that might result should we be unable to continue as a going concern.
Even if we are able to obtain additional financing in order to continue long-term operations beyond approximately May 2015, we will require and will seek additional funding through collaborative or other partnering arrangements, public or private equity or debt financings and from other sources. However, there can be no assurance that we will obtain interim or longer-term financing or that such funding, if obtained, will be sufficient to continue our operations as currently conducted or in a manner necessary for the continued development of our products or the long-term success of our company. If we raise funds through the issuance of debt securities, such debt will likely be secured by a security interest or pledge of all of our assets, will require us to make principal and interest payments, would likely include the issuance of warrants and may subject us to restrictive covenants. If we raise funds through one or more equity offerings, our stockholders may suffer substantial dilution.
Cash used in operating activities was $5.0 million and $10.3 million for 2014 and 2012. Cash provided by operating activities was $9.6 million for 2013. In 2014, we incurred approximately $3.4 million in direct costs related to our Phase 3 clinical trials and costs to prepare to file an NDA for BromSite and $0.5 million of additional legal expenses from our on-going partnering and financing efforts. In addition, we received a $6.0 million license fee for the amendment of the Akorn License. In 2013, we incurred approximately $5.4 million in direct costs related to our Phase 3 clinical trials for AzaSite/DexaSite and BromSite. In addition, in April 2013, we sold the rights to receive royalty payments on sales of Besivance, beginning on January 1, 2013, for $15 million at closing, with an additional $1 million paid in February 2014 after certain 2013 Besivance sales targets were met. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to us in whole or in part. Patent protection for Besivance in the United States expires in 2021. In 2012, we incurred approximately $9.3 million, respectively, in direct costs related to our Phase 3 clinical trials for AzaSite/DexaSite and BromSite.
Cash provided by investing activities was $4.7 million, $2.9 million and $16.4 million for 2014, 2013 and 2012, respectively. In 2014, 2013 and 2012, we converted $5.0 million, $3.0 million and $16.5 million, respectively, from short-term investments to cash and cash equivalents.
Cash used in financing activities was $1.3 million, $10.6 million and $6.7 million for 2014, 2013 and 2012, respectively. In 2014, 2013 and 2012, we made principal payments of $6.0 million, $10.6 million and $6.7 million, respectively, on the AzaSite Notes. In the last quarter of 2014, we completed private placement debt financing transactions in which we issued secured notes and issued warrants to purchase shares of common stock, which resulted in approximately $4.7 million in net proceeds after deducting placement agent and legal fees associated with the transactions. The Company incurred $0.5 million in placement agent and legal fees and $1.0 million (non-cash) for the initial valuation of the warrants issued with the Notes. These amounts are recorded as debt issuance costs and are amortized over the expected life of the Notes.
46
Table of Contents
The tables below set forth the amount of cash that we raised for fiscal years 2012 through 2014 from debt financings.
Cash Received from Private Placements of Notes
| Date |
Net Proceeds | Type of Notes | Interest Rate | Maturity Date | ||||||||
| October 2014 |
$ | 2.1 million | Short-term Secured Notes | 12 | % | October 2015* | ||||||
| November 2014 |
$ | 0.2 million | Short-term Secured Notes | 12 | % | November 2015* | ||||||
| December 2014 |
$ | 2.4 million | Short-term Secured Notes | 12 | % | December 2015* | ||||||
| * | The Notes have a maturity date of one year from the date of issuance, which may be extended for one year at our option. In the event we extend the maturity date, the interest rate increases to 14% per annum. |
Our future capital expenditures and requirements will depend on numerous factors, including our ability to raise additional financing by May 2015 when we expect our cash resources to be depleted, the progress of our clinical testing, research and development programs and preclinical testing, the time and costs involved in obtaining regulatory approvals, our ability to successfully commercialize any products that we may launch in the future, our ability to establish collaborative arrangements, the cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights, competing technological and market developments, changes in our existing collaborative and licensing relationships, acquisition of new businesses, products and technologies, the completion of commercialization activities and arrangements, and the purchase of additional property and equipment.
We anticipate no material capital expenditures to be incurred for environmental compliance in fiscal year 2014. Based on our environmental compliance record to date, and our belief that we are current in compliance with applicable environmental laws and regulations, environmental compliance is not expected to materially harm our operations.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements that have or are reasonably likely to have a current or future material effect.
Contractual Obligations
The following table summarizes our significant contractual obligations as of December 31, 2014 and the effect such obligations are expected to have on our liquidity and cash flows in future periods. Some of these amounts are based on management’s estimates and assumptions about these obligations including their duration, the possibility of renewal and other factors. Because these estimates are necessarily subjective, our actual payments in the future may vary from those listed in this table.
| Payments due by period (in thousands) |
||||||||||||||||||||
| Total | Less than 1 year |
1-3 years |
3-5 Years |
More than 5 years |
||||||||||||||||
| Facilities lease obligations(1) |
4,623 | 696 | 1,498 | 1,603 | 826 | |||||||||||||||
| Licensing agreement obligations(2) |
1,800 | 450 | 900 | 450 | — | |||||||||||||||
| Secured notes(3) |
5,199 | 5,199 | — | — | — | |||||||||||||||
| Interest expense on secured notes(4) |
539 | 539 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total commitments |
$ | 12,161 | $ | 6,884 | $ | 2,398 | $ | 2,053 | $ | 826 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | We lease our facilities under a non-cancelable operating lease that expires in 2020. |
| (2) | We have entered into certain license agreements that require us to make royalty payments for the life of the licensed patents. The estimated royalties due under certain of these agreements are as noted for 2014 through 2018 based on future expected net sales of AzaSite. |
| (3) | Principal repayments are based on estimated payment dates. |
| (4) | Interest expense represents an estimate based on expected principal balance. |
47
Table of Contents
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers. The FASB and the IASB initiated a joint project to clarify the principles for recognizing revenue and developed a common revenue recognition standard for GAAP and IFRS. Under the guidance, an entity should recognize revenue when the entity satisfies a performance obligation within a contract at a determined transaction price. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is not permitted. The Company does not expect the adoption of this standard will materially impact the Company’s consolidated statement of financial position or results of operations.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements – Going Concern. This standard includes guidance about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern within one year after the financial statements are issued. If conditions or events raise substantial doubt, the entity must disclose the conditions or events that raise substantial doubt about the entity’s ability to continue as a going concern, management’s evaluation of those conditions or events, and management’s plans to mitigate the conditions or events. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is permitted. The Company does not expect the adoption of this standard will materially impact the Company’s consolidated statement of financial position or results of operations.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
The following discusses our exposure to market risk related to changes in interest rates.
We have debt in the form of secured notes issued with fixed interest rates. As a result, our exposure to market risk caused by fluctuations in interest rates is minimal. We had $5.2 million in the Notes outstanding as of December 31, 2014, with a fixed interest rate of 12%. If the market interest rates were to increase by 10% from the December 31, 2014 levels, it would not result in an increase in interest expense. At December 31, 2014, our debt was reflected in the accompanying consolidated financial statements at face value.
The securities in our investment portfolio are not leveraged and are subject to minimal interest rate risk. Due to their original maturities of twelve months or less, the securities are classified as cash and cash equivalents or short-term investments. They are classified as trading securities principally bought and held for the purpose of selling them in the near term, with unrealized gains and losses included in earnings. Because of the short-term maturities of our investments, we do not believe that a change in market rates would have a significant negative impact on the value of our investment portfolio. While a hypothetical decrease in market interest rates by 10% from the December 31, 2014 levels would cause a decrease in interest income, it would not likely result in loss of principal.
The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. To achieve this objective in the current economic environment, we maintain our portfolio in cash equivalents or short-term investments, including obligations of U.S. government-sponsored enterprises and money market funds. These securities are classified as cash and cash equivalents or short-term investments and consequently are recorded on the balance sheet at fair value. We do not utilize derivative financial instruments to manage our interest rate risks.
48
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
The following Consolidated Financial Statements and Report of Independent Registered Public Accounting Firm are included on the pages that follow:
49
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
InSite Vision Incorporated
We have audited the accompanying consolidated balance sheets of InSite Vision Incorporated and its subsidiaries (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, stockholders’ deficit, and cash flows for each of the three years in the period ended December 31, 2014. InSite Vision Incorporated’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of InSite Vision Incorporated and its subsidiaries as of December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company’s recurring losses from operations, available cash balance and accumulated deficit raise substantial doubt about its ability to continue as a going concern. Management’s plans regarding those matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Burr Pilger Mayer, Inc.
E. Palo Alto, California
February 18, 2015
50
Table of Contents
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||
| (in thousands, except share data) |
2014 | 2013 | ||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 1,656 | $ | 3,251 | ||||
| Short-term investments |
— | 5,000 | ||||||
| Accounts receivable |
349 | 1,026 | ||||||
| Other receivables |
— | 1,114 | ||||||
| Prepaid expenses and other current assets |
36 | 95 | ||||||
| Debt issuance costs, net |
1,267 | 2,248 | ||||||
|
|
|
|
|
|||||
| Total current assets |
3,308 | 12,734 | ||||||
| Property and equipment, net |
1,597 | 1,431 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 4,905 | $ | 14,165 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 611 | $ | 394 | ||||
| Accrued liabilities |
660 | 2,184 | ||||||
| Accrued compensation and related expense |
1,655 | 1,090 | ||||||
| Accrued royalties |
892 | 776 | ||||||
| Accrued interest |
85 | 826 | ||||||
| Lease incentive, current |
186 | 154 | ||||||
| Secured notes payable |
5,199 | 41,281 | ||||||
| Warrant liability |
1,191 | 1,685 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
10,479 | 48,390 | ||||||
| Deferred revenues |
1,000 | 7 | ||||||
| Lease incentive |
928 | 922 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
12,407 | 49,319 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 9) |
||||||||
| Stockholders’ deficit: |
||||||||
| Preferred stock, $0.01 par value, 5,000,000 shares authorized, none issued and outstanding |
— | — | ||||||
| Common stock, $0.01 par value, 240,000,000 shares authorized; 131,951,033 shares issued and outstanding at December 31, 2014 and 2013 |
1,320 | 1,320 | ||||||
| Additional paid-in capital |
166,440 | 165,549 | ||||||
| Accumulated deficit |
(175,262 | ) | (202,023 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ deficit |
(7,502 | ) | (35,154 | ) | ||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ deficit |
$ | 4,905 | $ | 14,165 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
51
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
| Year Ended December 31, | ||||||||||||
| (in thousands, except per share data) |
2014 | 2013 | 2012 | |||||||||
| Revenues: |
||||||||||||
| Royalties |
$ | 2,005 | $ | 14,787 | $ | 21,641 | ||||||
| Sale of royalty rights, net |
— | 15,490 | — | |||||||||
| Licensing fee |
6,000 | — | — | |||||||||
| Other product and service revenues |
209 | 545 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
8,214 | 30,822 | 21,641 | |||||||||
|
|
|
|
|
|
|
|||||||
| Expenses: |
||||||||||||
| Research and development |
9,134 | 11,578 | 15,479 | |||||||||
| General and administrative |
5,721 | 5,754 | 5,781 | |||||||||
| Cost of revenues, principally royalties to third parties |
578 | 385 | 1,062 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total expenses |
15,433 | 17,717 | 22,322 | |||||||||
|
|
|
|
|
|
|
|||||||
| Income (loss) from operations |
(7,219 | ) | 13,105 | (681 | ) | |||||||
| Gain on extinguishment of debt |
35,999 | — | — | |||||||||
| Interest expense and other, net |
(3,516 | ) | (7,896 | ) | (9,494 | ) | ||||||
| Change in fair value of warrant liability |
1,497 | 572 | 1,898 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) |
$ | 26,761 | $ | 5,781 | $ | (8,277 | ) | |||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) per share: |
||||||||||||
| Income (loss) per share—basic |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | |||||
|
|
|
|
|
|
|
|||||||
| Income (loss) per share—diluted |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | |||||
|
|
|
|
|
|
|
|||||||
| Weighted average shares used in per share calculation: |
||||||||||||
| —Basic |
131,951 | 131,951 | 131,951 | |||||||||
|
|
|
|
|
|
|
|||||||
| —Diluted |
132,385 | 132,433 | 131,951 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
52
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
| Additional Paid-in-Capital |
Accumulated Deficit |
Total Stockholders’ Deficit |
||||||||||||||||||
| Common Stock | ||||||||||||||||||||
| (in thousands, except share data) |
Shares | Amount | ||||||||||||||||||
| Balances, December 31, 2011 |
131,951,033 | 1,320 | 163,668 | (199,527 | ) | (34,539 | ) | |||||||||||||
| Stock-based compensation |
— | — | 947 | — | 947 | |||||||||||||||
| Net loss |
— | — | — | (8,277 | ) | (8,277 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balances, December 31, 2012 |
131,951,033 | 1,320 | 164,615 | (207,804 | ) | (41,869 | ) | |||||||||||||
| Stock-based compensation |
— | — | 934 | — | 934 | |||||||||||||||
| Net income |
— | — | — | 5,781 | 5,781 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balances, December 31, 2013 |
131,951,033 | 1,320 | 165,549 | (202,023 | ) | (35,154 | ) | |||||||||||||
| Stock-based compensation |
— | — | 891 | — | 891 | |||||||||||||||
| Net income |
— | — | — | 26,761 | 26,761 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balances, December 31, 2014 |
131,951,033 | $ | 1,320 | $ | 166,440 | $ | (175,262 | ) | $ | (7,502 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
See accompanying notes consolidated financial statements.
53
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| OPERATING ACTIVITIES: |
||||||||||||
| Net income (loss) |
$ | 26,761 | $ | 5,781 | $ | (8,277 | ) | |||||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation and amortization |
335 | 107 | 95 | |||||||||
| Amortization of debt issuance costs |
405 | 418 | 419 | |||||||||
| Amortization of lease incentive |
(186 | ) | — | — | ||||||||
| Gain on extinguishment of debt |
(35,999 | ) | — | — | ||||||||
| Stock-based compensation |
891 | 934 | 947 | |||||||||
| Change in fair value of warrant liability |
(1,497 | ) | (572 | ) | (1,898 | ) | ||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
677 | 4,224 | (2,686 | ) | ||||||||
| Other receivables |
1,114 | (1,114 | ) | — | ||||||||
| Prepaid expenses and other current assets |
59 | 49 | (132 | ) | ||||||||
| Accounts payable |
169 | (283 | ) | (26 | ) | |||||||
| Accrued liabilities |
(1,524 | ) | 656 | 1,124 | ||||||||
| Accrued compensation and related expense |
565 | (44 | ) | 156 | ||||||||
| Accrued royalties |
116 | (328 | ) | 140 | ||||||||
| Accrued interest |
2,088 | (212 | ) | (133 | ) | |||||||
| Deferred revenues |
993 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) operating activities |
(5,033 | ) | 9,616 | (10,271 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| INVESTING ACTIVITIES: |
||||||||||||
| Purchase of property and equipment, net |
(277 | ) | (85 | ) | (127 | ) | ||||||
| Decrease in short-term investments |
5,000 | 2,999 | 16,496 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by investing activities |
4,723 | 2,914 | 16,369 | |||||||||
|
|
|
|
|
|
|
|||||||
| FINANCING ACTIVITIES: |
||||||||||||
| Proceeds from secured notes payable, net of issuance costs |
4,715 | — | — | |||||||||
| Payment of secured notes payable |
(6,000 | ) | (10,602 | ) | (6,675 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in financing activities |
(1,285 | ) | (10,602 | ) | (6,675 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
(1,595 | ) | 1,928 | (577 | ) | |||||||
| Cash and cash equivalents at beginning of year |
3,251 | 1,323 | 1,900 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of year |
$ | 1,656 | $ | 3,251 | $ | 1,323 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Interest received |
$ | 1 | $ | 5 | $ | 17 | ||||||
|
|
|
|
|
|
|
|||||||
| Interest paid |
$ | 1,335 | $ | 7,694 | $ | 9,224 | ||||||
|
|
|
|
|
|
|
|||||||
| Income taxes received |
$ | 22 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Income taxes paid |
$ | 1 | $ | 110 | $ | 1 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing activities—Lease incentives |
$ | 224 | $ | 1,076 | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash financing activities—Debt extinguishment |
$ | 36,047 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash financing activities—Warrant issuance |
$ | 1,003 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
54
Table of Contents
Notes to Consolidated Financial Statements
For the years ended December 31, 2014, 2013 and 2012
1. Business and Summary of Significant Accounting Policies
InSite Vision Incorporated (“InSite,” the “Company,” “we,” or “our”) is an ophthalmic product development company advancing ophthalmic pharmaceutical products to address unmet eye care needs. The Company’s current portfolio of products is based on its proprietary DuraSite® drug delivery technology. The Company’s DuraSite sustained drug delivery technology is a proven synthetic polymer-based formulation designed to extend the residence time of a drug relative to conventional topical therapies. It enables topical delivery of a drug as a solution, gel or suspension and can be customized for delivering a wide variety of drug candidates.
DuraSite® 2 is the Company’s next-generation enhanced drug delivery system, which is designed to provide a broad platform for developing superior ophthalmic therapeutics. DuraSite 2 is based on the original DuraSite technology, and incorporates a cationic polymer to achieve sustained and enhanced ocular delivery of drugs. The Company plans to focus its research and development and commercial support efforts on topical products formulated with the DuraSite 2 drug delivery technology.
Historically, the Company has incurred substantial cumulative losses and negative cash flows from operations. Clinical trials and costs to prepare a New Drug Application (“NDA”) for the Company’s product candidates with the U.S. Food and Drug Administration (“FDA”) are very expensive and difficult, in part because they are subject to rigorous regulatory requirements. As of December 31, 2014, the Company’s accumulated deficit was $175.3 million and its cash and cash equivalents were $1.7 million. Further, the Company anticipates that its existing cash and cash equivalent balances, together with cash flows from operations, and the net proceeds from existing debt financing arrangements (See Note 8, “Secured Notes Payable” for further discussion) will only be adequate to fund its cash requirements through May 2015. Management’s plans include exploring strategic alternatives or raising additional financing. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The financial statements do not include any adjustments to reflect the possible future effects relating to the recoverability and classification of the recorded asset amounts or amounts and classification of liabilities that might result from the outcome of this uncertainty.
Principles of Consolidation. The consolidated financial statements include the accounts of InSite Vision Incorporated as well as its wholly-owned subsidiaries. All intercompany transactions have been eliminated in consolidation.
Industry Segment and Geographic Information. The Company operates in one segment and is focused on developing drugs and drug delivery systems principally for ophthalmic indications. The Company had limited foreign-based operations for the years ended December 31, 2014, 2013 and 2012. All long-lived assets are located in the United States.
Use of Estimates. The preparation of financial statements in conformity with generally accepted accounting principles in the United States of America requires the Company to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates.
Reclassifications. Certain amounts in prior years’ financial statements have been reclassified to conform to the current presentation. These reclassifications had no impact on previously reported results of operations or stockholders’ deficit.
55
Table of Contents
Cash and cash equivalents. The Company considers all highly liquid investments with maturities of 90 days or less from the date of purchase to be cash equivalents.
Short-term investments. The Company considers all investments with original maturities of 12 months or less from the date of purchase to be short-term investments. They are classified as trading securities principally bought and held for the purpose of selling them in the near term, with unrealized gains and losses included in earnings.
Property and Equipment. Property and equipment is stated at cost, less accumulated depreciation and amortization. Depreciation of property and equipment is provided over the estimated useful lives of the respective assets, which range from three to five years, using the straight-line method. Leasehold improvements and property acquired under capital leases are amortized over the lives of the related leases or their estimated useful lives, whichever is shorter, using the straight-line method. Depreciation and amortization expense for the years ended December 31, 2014, 2013 and 2012 was $335,000, $107,000 and $95,000, respectively. The costs of repairs and maintenance are expensed as incurred.
Impairment of Long-Lived Assets. The Company periodically assesses the recoverability of its long-lived assets for which an indicator of impairment exists by determining whether the carrying value of such assets can be recovered through undiscounted future operating cash flows. If the Company concludes that the carrying value will not be recovered, the Company measures the amount of such impairment by comparing the fair value to the carrying value. For the years ended December 31, 2014, 2013 and 2012, no impairment of property and equipment was recorded.
Patents. As a result of the Company’s research and development efforts, the Company has obtained, or is applying for, a number of patents to protect proprietary technology and inventions. All costs associated with patents for product candidates under development are expensed as incurred. As of December 31, 2014 and 2013, the Company had no capitalized patent costs.
Debt Issuance Costs. Debt issuance costs paid to third parties are capitalized and amortized over the life of the underlying debt, using the effective interest method. Amortization of debt issuance costs for the years ended December 31, 2014, 2013 and 2012 were $405,000, $418,000 and $419,000, respectively, and are included in interest expense and other, net in the Consolidated Statements of Operations. See Note 8, “Secured Notes Payable” for further discussion of the underlying debt.
Warrant Liability. The Company issued warrants to purchase shares of the Company’s common stock in connection with a private placement equity financing transaction in July 2011 and private placement debt financing transactions in the fourth quarter of 2014. The Company accounted for these warrants as a liability measured at fair value due to a provision included in the warrant agreements that provides the warrant holders with an option to require the Company (or its successor) to purchase their warrants for cash in the event of a “Fundamental Transaction” (as defined in the warrant agreements). The actual amount of cash required if the option is exercised would be determined using the Black-Scholes Option Pricing Model (the “Black-Scholes Model”) as determined in accordance with the terms of the warrant agreements. The fair value of the warrant liability is estimated using the Black-Scholes Model, which requires inputs such as the remaining term of the warrants, share price volatility and risk-free interest rate. These assumptions are reviewed on a monthly basis and changes in the estimated fair value of the outstanding warrants are recognized each reporting period in the Consolidated Statements of Operations under “Change in fair value of warrant liability.”
Fair Value Measurements. Fair value is the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value is estimated by applying the following hierarchy, which prioritizes the inputs used to measure fair value into three levels and bases the categorization within the hierarchy upon the lowest level of input that is available and significant to the fair value measurement:
| Level 1: | Quoted prices in active markets for identical assets or liabilities. |
56
Table of Contents
| Level 2: | Observable inputs other than quoted prices in active markets for identical assets and liabilities, quoted prices for identical or similar assets or liabilities in inactive markets, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| Level 3: | Inputs that are generally unobservable and typically reflect management’s estimate of assumptions that market participants would use in pricing the asset or liability. |
As of December 31, 2014 and 2013, less than $0.1 million and $8.2 million, respectively, of the Company’s cash, cash equivalents and short-term investments consisted of Level 1 Treasury-backed government securities or money market funds that are measured at fair value on a recurring basis.
The Company’s financial instruments consist mainly of cash and cash equivalents, short-term investments, accounts receivable, other receivables, accounts payable, accrued liabilities and debt obligations. Accounts receivable, other receivables, and accounts payable are reflected in the accompanying consolidated financial statements at cost, which approximates fair value due to the short-term nature of these instruments. While the Company believes its valuation methodologies are appropriate and consistent with those of other market participants, the use of different methodologies or assumptions to determine the fair value of certain financial instruments could result in a different estimate of fair value at the reporting date.
As discussed above, the fair value of the warrant liability, determined using Level 3 criteria, was initially recorded on the grant date and remeasured at December 31, 2014 and 2013 using the Black-Scholes Model, which requires inputs such as the remaining term of the warrants, share price volatility and risk-free interest rate. These inputs are subjective and generally require significant analysis and judgment to develop.
The fair value of the warrant liability was estimated using the following weighted-average assumptions, as determined in accordance with the terms of the warrant agreements, at December 31, 2014 and 2013:
| December 31, 2014 |
December 31, 2013 |
|||||||
| Risk-free interest rate |
0.9 | % | 0.8 | % | ||||
| Remaining term (years) |
2.4 | 2.5 | ||||||
| Expected dividends |
0.0 | % | 0.0 | % | ||||
| Volatility |
101.2 | % | 100.0 | % | ||||
The expected dividend yield is set at zero because the Company has never paid cash dividends and has no present intention to pay cash dividends. Per the terms of the warrant agreements, expected volatility is based on the historical volatility of the Company’s common stock and is equal to the greater of 100% or the 30-day volatility rate. The risk-free interest rates are taken from the Daily Federal Yield Curve Rates as published by the Federal Reserve and represent the yields on actively-traded U.S. Treasury securities for a term equal to the remaining term of the warrants.
The following table provides a summary of changes in the fair value of the Company’s Level 3 financial liabilities for the year ended December 31, 2014 and 2013 (in thousands):
| Year Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Beginning balance |
$ |
1,685 |
|
$ | 2,257 | |||
| Initial fair value of warrants as of issuance |
1,003 | — | ||||||
| Net decrease in fair value of warrant liability on remeasurement |
(1,497 | ) | (572 | ) | ||||
|
|
|
|
|
|||||
| Ending balance |
$ | 1,191 | $ | 1,685 | ||||
|
|
|
|
|
|||||
57
Table of Contents
The initial value of the warrants issued in 2014 were recorded as “Debt issuance costs, net” in the Consolidated Balance Sheets. The net decrease in the estimated fair value of the warrant liability was recognized as income under “Change in fair value of warrant liability” in the Consolidated Statements of Operations.
Revenue Recognition. The Company recognizes revenue when four basic criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. The Company has arrangements with multiple revenue-generating elements. The Company analyzes its multiple element arrangements to determine whether the elements can be separated and accounted for individually as separate units of accounting. An item can generally be considered a separate unit of accounting if both of the following criteria are met: (i) the delivered item(s) has value to the customer on a stand-alone basis and (ii) the arrangement includes a general right of return and delivery or performance of the undelivered item(s) is considered probable and substantially in the control of the Company. Items that cannot be divided into separate units are combined with other units of accounting, as appropriate. Consideration received is allocated among the separate units based on the unit’s selling price and is recognized in full when the criteria are met. The Company deems service to be rendered if no continuing obligation exists on the part of the Company.
The Company’s revenues are primarily derived from royalties on product sales and licensing agreements, and such agreements may provide for various types of payments, including upfront payments, research funding and related fees during the terms of the agreements, milestone payments based on the achievement of established development objectives and licensing fees.
The Company receives royalties from licensees based on third-party sales. The royalties are recorded as earned in accordance with the contract terms when third-party results are reliably measured and collectability is reasonably assured.
Revenues associated with non-refundable up-front license fees under arrangements where the license fees cannot be accounted for as separate units of accounting are deferred and recognized as revenues on a straight-line basis over the expected term of the Company’s continued involvement. Revenues from the achievement of milestones are recognized as revenues when the milestones are achieved and the milestone payments are due and collectible.
The Company allocates revenue in multiple-deliverable revenue arrangements using estimated selling prices of the delivered goods and services based on the relative selling price method.
Research and Development Expenses. Research and development expenses include salaries, benefits, facility costs, services provided by outside consultants and contractors, administrative costs and materials for the Company’s research and development activities. The Company expenses these research and development activities as they are incurred.
The Company accrues and expenses the costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations and clinical trial sites. The Company determines the estimates through discussions with internal clinical personnel and external service providers as to progress or stage of completion of trials or services and the agreed upon fee to be paid for such services. Costs of setting up clinical trial sites for participation in the trials are expensed immediately as research and development expenses. Clinical trial site costs related to patient enrollment are accrued as patients are entered into the trial.
General and Administrative Expenses. General and administrative expenses include salaries, benefits, facility costs, services provided by outside consultants and contractors, legal services, advertising and marketing, investor relations, financial reporting, materials and other expenses related to general corporate and sales and marketing activities. The Company recognizes such costs as they are incurred.
58
Table of Contents
Cost of Revenues. The Company recognizes royalties to third parties and the cost of inventory shipped related to the sale of the Company’s products when they are incurred.
Stock-Based Compensation. The Company’s stock-based compensation programs consist of stock options granted to employees as well as our employee stock purchase plan, based on the grant date fair value of those awards.
The grant date fair value of the award is recognized as expense over the requisite service period. The Company uses the Black-Sholes option pricing model to compute the estimated fair value of stock option awards. Using this model, fair value is calculated based on assumptions with respect to: expected volatility of our common stock price: the periods of time over which employees and members of our board are expected to hold their options prior to exercise; expected dividend yield on our common stock; and risk-free interest rates. Stock-based compensation expense also includes an estimate, which is made at the time of grant, of the number of awards that are expected to be forfeited. The estimate is revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
See Note 12, “Stock-Based Compensation” for further discussion of employee stock-based compensation.
The Company occasionally issues stock options and warrants to consultants of the Company in exchange for services. The Company has valued these options and warrants using the Black-Scholes option pricing model, at each reporting period and has recorded charges to operations over the vesting periods of the individual stock options or warrants. Such charges amounted to approximately $1,000, $8,000 and $56,000 during the years ended December 31, 2014, 2013 and 2012, respectively.
Net Income (Loss) per Share. Basic net income (loss) per share has been computed using the weighted-average number of common shares outstanding during the period. Dilutive net income (loss) per share is computed using the sum of the weighted-average number of common shares outstanding and the potential number of dilutive common shares outstanding during the period. Potential common shares consist of the shares issuable upon exercise of stock options and warrants. Potentially dilutive securities have been excluded from the computation of diluted net income (loss) per share in 2014, 2013 and 2012 as their inclusion would be anti-dilutive.
The following table sets forth the computation of basic and diluted net income (loss) per share:
| Year Ended December 31, | ||||||||||||
| (in thousands, except per share data) |
2014 | 2013 | 2012 | |||||||||
| Numerator: |
||||||||||||
| Net income (loss) |
$ | 26,761 | $ | 5,781 | $ | (8,277 | ) | |||||
|
|
|
|
|
|
|
|||||||
| Denominator: |
||||||||||||
| Weighted-average shares outstanding |
131,951 | 131,951 | 131,951 | |||||||||
| Effect of dilutive securities: |
||||||||||||
| Stock options |
434 | 482 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Weighted-average shares outstanding for diluted loss |
132,385 | 132,433 | 131,951 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) per share: |
||||||||||||
| Basic |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | |||||
|
|
|
|
|
|
|
|||||||
| Diluted |
$ | 0.20 | $ | 0.04 | $ | (0.06 | ) | |||||
|
|
|
|
|
|
|
|||||||
For the year ended December 31, 2012, due to the loss applicable to common stockholders, loss per share is based on the weighted average number of common shares only, as the effect of including equivalent shares from stock options and warrants would be anti-dilutive. At December 31, 2014, 2013 and 2012, 40,179,706, 31,407,604 and 28,156,898 options and warrants, respectively, were excluded from the calculation of diluted earnings per share because the effect was anti-dilutive.
59
Table of Contents
Comprehensive Income (Loss). Comprehensive income (loss) is the change in equity from transactions and other events and circumstances other than those resulting from investments by owners and distributions to owners. The consolidated comprehensive income (loss) for the Company was equal to the net income (loss) attributable to the Company for the years ended December 31, 2014, 2013 and 2012.
Key Suppliers. The Company is dependent on single or limited source suppliers for certain materials used in its research and development and commercial activities. The Company has generally been able to obtain adequate supplies of these components. However, an extended interruption in the supply of these components currently obtained from single or limited source suppliers could adversely affect the Company’s research and development and commercial efforts.
Income Taxes. The Company accounts for income taxes under the liability method; under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain.
The Company utilizes a two-step approach to recognize and measure uncertain tax positions, if any. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained upon tax authority examination, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon ultimate settlement.
Significant Customers and Risk. All revenues recognized and/or deferred were primarily from AzaSite licensees. The Company is entitled to receive royalty revenues from net sales of AzaSite under the terms of its agreement with Inspire. Inspire was acquired by Merck in May 2011 and subsequently acquired by Akorn in November 2013. Accordingly, all trade receivables are concentrated with these parties during the years ended December 31, 2014, 2013 and 2012. Under the terms of the license with Inspire, Inspire, through its applicable parent company, has significant influence over the commercial success of AzaSite. Revenues from Akorn/Merck represented approximately 98%, 48% and 90% of total revenues for the years ended December 31, 2014, 2013 and 2012, respectively. In addition, in April 2013, the Company sold its rights to receive royalty payments relating to sales of Besivance and the amounts received in this sale represented 50% of the Company’s revenues for the year ended December 31, 2013.
Credit Risk. Financial instruments that potentially subject the Company to significant concentrations of credit risk consist principally of cash, cash equivalents and short-term investments. The Company’s cash, cash equivalents and short-term investments are primarily deposited in demand accounts with two financial institutions.
Risks from Third Party Manufacturing Concentration. The Company relies on a single source manufacturer for each of its product candidates and on a single source manufacturer for the active pharmaceutical ingredient in its product candidates. Accordingly, delays in the manufacture of the Company’s product candidates or the active pharmaceutical ingredients could adversely impact the development of the Company’s product candidates. Furthermore, the Company has no control over the manufacture and the overall product supply chain of products for which it is entitled to receive revenue.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09 (“ASU 2014-09”), Revenue from Contracts with Customers. The FASB and the International Accounting Standards Board (“IASB”) initiated a joint project to clarify the principles for recognizing revenue and developed a common revenue recognition standard for U.S. generally accepted accounting principles
60
Table of Contents
(“GAAP’) and International Financial Reporting Standards (“IFRS”). Under the guidance, an entity should recognize revenue when the entity satisfies a performance obligation within a contract at a determined transaction price. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is not permitted. The Company does not expect that the adoption of this standard will materially impact the Company’s consolidated statement of financial position or results of operations.
In August 2014, the FASB issued Accounting Standards Update No. 2014-15 (“ASU 2014-15”), Presentation of Financial Statements – Going Concern. This standard includes guidance about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern within one year after the financial statements are issued. If conditions or events raise substantial doubt, the entity must disclose the conditions or events that raise substantial doubt about the entity’s ability to continue as a going concern, management’s evaluation of those conditions or events, and management’s plans to mitigate the conditions or events. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is permitted. The Company does not expect that the adoption of this standard will materially impact the Company’s consolidated statement of financial position or results of operations.
2. License Agreements
In December 2003, the Company completed the sale of its drug candidate for the treatment of ocular infections to Bausch & Lomb Incorporated (“Bausch & Lomb”), wholly-owned by Valeant Pharmaceuticals International, Inc. (“Valeant”) as of August 2013, pursuant to a Purchase Agreement and a License Agreement. Accordingly, Besivance was developed by Bausch & Lomb. In May 2009, the United States Food and Drug Administration (“FDA”) approved Besivance to treat bacterial conjunctivitis (pink eye). Besivance was launched in the United States by Bausch & Lomb in the last half of 2009. In 2011, Besivance was launched internationally in select countries. During the years ended December 31, 2012, the Company recognized $2,126,000 of royalties related to net sales of Besivance by Bausch & Lomb. In April 2013, the Company sold the rights to receive royalty payments on sales of Besivance, beginning on January 1, 2013, for $15 million at closing, with an additional $1 million paid in February 2014 after certain 2013 Besivance sales targets were met. $0.5 million of Besivance royalties previously recorded in 2013 were netted against the sales price. Under the terms of the agreement, if the purchasers receive specified levels of total cash from the Besivance royalty, the royalty would be returned to the Company in whole or in part. Patent protection for Besivance in the United States expires in 2021.
On February 15, 2007, the Company entered into a license agreement for AzaSite® (the “Akorn License”) with Inspire Pharmaceuticals, Inc. (“Inspire”). In May 2011, Merck & Co. (“Merck”) acquired Inspire and Inspire became a wholly-owned subsidiary of Merck. In November 2013, Akorn, Inc. (“Akorn”) acquired Inspire from Merck and Inspire became a wholly-owned subsidiary of Akorn. Under the Akorn License, the Company licensed to Inspire exclusive development and commercialization rights in the United States and Canada for topical anti-infective products containing azithromycin as the sole active ingredient for human ocular or ophthalmic indications. The Company also granted Inspire an exclusive sublicense under the Pfizer patent rights the Company has licensed under the Pfizer License discussed below. Inspire has the right to grant sublicenses under the terms of the Akorn License.
Akorn, due to its acquisition of Inspire, pays the Company a royalty on net sales of AzaSite in the United States and Canada. Prior to the June 2014 amendment discussed below, the royalty rate was 25% of net sales of AzaSite in North America. Akorn is obligated to pay the Company royalties under the Akorn License for the longer of (i) eleven years from the launch of the first product or (ii) the period during which a valid claim under a patent exists. Until September 30, 2013, Merck paid the Company certain minimum royalties. The royalties are subject to certain reductions in the event of patent invalidity, generic competition, uncured material breach under the Akorn License or in the event that Akorn is required to pay license fees to third parties for the continued use of AzaSite. Akorn may also terminate the Akorn License at any time upon 6 months’ notice.
61
Table of Contents
The Company also entered into a supply agreement (the “Supply Agreement”) with Inspire on February 15, 2007 for the active pharmaceutical ingredient azithromycin. The Company had previously entered into a third-party supply agreement for the production of this active ingredient. The Supply Agreement was terminated in July 2012.
On August 9, 2012, the Company and Merck amended the payment terms of the Akorn License. Under the amended terms, on a quarterly basis, Merck paid the Company the higher of the pro-rata annual minimum royalty or the earned royalty for 2012 and 2013. In addition, in August 2012, Merck paid the Company a $7.3 million catch-up payment for the difference between the earned royalties already paid for the fourth quarter of 2011 and the first and second quarters of 2012, and the pro-rata annual minimum royalties for those quarters. For the fiscal years ended September 30, 2013, 2012 and 2011, the measurement period pursuant to the terms of the Akorn License, the Company received $19 million, $17 million and $15 million, respectively, in minimum royalties from Merck. The obligation to make minimum royalty payments terminated in September 2013 and Akorn’s obligation to pay earned royalties may be suspended upon the occurrence of certain events, such as a requirement by the FDA or other governmental agency to suspend the marketing of AzaSite or withdraw it from the market in the United States.
On June 20, 2013, the Company and Merck entered into an amendment to the Akorn License. The amendment granted back to the Company, the right to develop and commercialize AzaSite Xtra™ in the United States and Canada. The amendment also granted Akorn the exclusive option, but not the obligation, to re-acquire the exclusive right to develop and commercialize AzaSite Xtra in the United States and Canada. The option can be exercised during the period commencing on the effective date of the amendment and ending on the date that is ten (10) business days following the first regulatory approval of AzaSite Xtra in the United States, unless the Akorn License is earlier terminated or has expired in accordance with its terms. Upon exercise of the option, Akorn is required to pay the Company a one-time payment. In addition, in the event that Akorn exercises the option, Akorn will be obligated to pay the Company royalties on net sales of AzaSite Xtra.
On June 10, 2014, the Company entered into an amendment to the Akorn License (the “Amendment”). Under the Amendment, Akorn paid the Company an amendment fee of $6.0 million in return for a lower royalty on net sales of AzaSite in North America. The $6.0 million was used by the Company to repurchase and cancel the AzaSite Notes (see Note 8, “Secured Notes Payable” for further discussion). Pursuant to the Amendment, effective April 1, 2014, for annual net sales of AzaSite, the royalty is (1) 8.0% on net sales less than $20.0 million, subject to increase to 9.0% in the event that Akorn enters into certain settlements with third parties seeking to launch generic versions of AzaSite, (2) 12.5% on net sales greater than or equal to $20.0 million and less than or equal to $50.0 million and (3) 15.0% on net sales in excess of $50.0 million. For annual net sales of AzaSite Xtra, the royalty would be 12.5% on net sales less than $30.0 million and 15.0% on net sales greater than or equal to $30.0 million. The Company waived any right to consent to any settlements regarding AzaSite (see Note 13, “Legal Proceedings” for further discussion) and agreed to cooperate to effectuate any such settlement.
The Amendment further provides that in the event AzaSite Xtra has received regulatory approval and has been commercially launched by Akorn, as to any calendar quarter, Akorn would not be obligated to pay any royalties as to a licensed product in a given country where sales of a generic equivalent of such licensed product occur in such country. Akorn may, at Akorn’s expense, act to acquire and maintain on the Company’s behalf, registrations for certain Company trademarks in North America. In addition, Akorn may, but is not obligated to, pay any maintenance or other fees related to the maintenance of certain patents licensed by the Company to Akorn in the event the Company fails to pay any such fee.
During the years ended December 31, 2014, 2013 and 2012, the Company recognized $2.0 million, $14.3 million and $19.5 million, respectively, of royalties related to the Akorn License. During the years ended December 31, 2014 and 2013, the Company recognized $0.1 million and $0.5 million, respectively, of revenue from Akorn for manufacturing equipment rental and the sale of the active ingredient, azithromycin.
62
Table of Contents
On February 15, 2007, the Company entered into a worldwide, exclusive, royalty-bearing license agreement with Pfizer Inc. (“Pfizer”) under Pfizer’s patent family titled “Method of Treating Eye Infections with Azithromycin” for ocular anti-infective product candidates known as AzaSite and AzaSite Plus (the “Pfizer License”). Under the Pfizer License, the Company is required to pay Pfizer a low single digit royalty based on net sales of the licensed products and to use reasonable commercial efforts to seek regulatory approval for and market licensed products. The Pfizer License provides the Company the right to grant sublicenses thereunder, subject to Pfizer’s prior approval, which approval shall not be unreasonably withheld. Pfizer approved the sublicense granted to Akorn, and previously to Merck prior to November 2013. Based on the royalty reports provided by Merck and Akorn, for the years ended December 31, 2014, 2013 and 2012, the Company recorded third-party royalties of $0.5 million, $0.4 million and $1.1 million, respectively, due primarily under the Pfizer License.
The Company has entered into, and will continue to pursue additional licensing agreements, corporate collaborations and service contracts. There can be no assurance that the Company will be able to negotiate acceptable collaborative, licensing or service agreements, or that the existing arrangements will be successful or renewed or will not be terminated.
3. Short-term Investments
As of December 31, 2014, the Company had no short-term investments. As of December 31, 2013, the Company had $5.0 million in short-term investments. The Company’s investment policy is to limit the risk of principal loss and to ensure safety of invested funds by generally attempting to limit market risk. Accordingly, the Company’s short-term investments were invested in U.S. Treasury securities with original maturities of 12 months or less. They are classified as trading securities principally bought and held for the purpose of selling them in the near term, with unrealized gains and losses included in earnings. At December 31, 2013, the unrealized gains on these short-term investments were insignificant.
4. Accounts Receivable
Accounts receivable represent amounts due to the Company from its licensees or former licenses, including Akorn and other third parties. At December 31, 2014, accounts receivable included $0.3 million due from Akorn for earned royalties on net sales of AzaSite and manufacturing equipment rental. At December 31, 2013, accounts receivable included $1.0 million for reaching certain 2013 Besivance sales targets related to the sale of the rights to receive Besivance royalty payments. At December 31, 2014 and 2013, the Company did not record a bad debt allowance related to any accounts receivable as all amounts were reasonably expected to be collected. The need for a bad debt allowance is evaluated each reporting period based on the Company’s assessment of the collectability of such amounts.
5. Property and Equipment, net
Property and equipment, net consisted of the following (in thousands):
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Laboratory and other equipment |
$ | 2,408 | $ | 1,445 | ||||
| Leasehold improvements |
663 | 45 | ||||||
| Furniture and fixtures |
49 | 14 | ||||||
| Construction in Progress |
— | 1,115 | ||||||
|
|
|
|
|
|||||
| 3,120 | 2,619 | |||||||
| Accumulated depreciation and amortization |
(1,523 | ) | (1,188 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 1,597 | $ | 1,431 | ||||
|
|
|
|
|
|||||
63
Table of Contents
6. Warrant Liability
On July 18, 2011, the Company completed a private placement financing transaction in which it sold shares of its common stock and warrants to purchase shares of its common stock. The Company sold a total of 36,978,440 shares of common stock, at a price of $0.60 per share, and issued warrants to purchase up to 14,791,376 shares of common stock. The warrants are exercisable at $0.75 per share and expire five years from the date of issuance. The private placement resulted in $22.2 million in gross proceeds and approximately $20.4 million in net proceeds to the Company after deducting placement agent fees, legal, accounting and other costs associated with the transaction. The Company has used the net proceeds of the transaction to fund clinical trials and for general corporate purposes, including working capital.
In the fourth quarter of 2014, the Company completed private placement debt financing transactions, see Note 8, “Secured Notes Payable” for further discussion. In connection with the debt financings, the Company issued warrants to purchase up to 2,053,169 shares, 200,620 shares and 2,824,281 shares of common stock that are exercisable at $0.33, $0.31 and $0.26, respectively, per share and expire five years from the date of issuance.
As discussed in Note 1, “Business and Summary of Significant Accounting Policies”, the warrants issued in 2011 and 2014 included a provision that provides the warrant holders with an option to require the Company (or its successor) to purchase the warrants for cash in an amount equal to the Black-Scholes value in the event of a “Fundamental Transaction” (as defined in the warrant agreements). Accordingly, the fair value of the warrants at the issuance date was estimated using the Black-Scholes Model, as determined in accordance with the terms of the warrant agreements, and the Company recorded an initial warrant liability valuation of $6.4 million in 2011 and $1.0 million in 2014. The Company remeasured the warrant liability at December 31, 2014 and 2013, and recorded a decrease to the warrant liability of approximately $1.5 million and $0.6 million, respectively, which was recognized as income in the Company’s Consolidated Statement of Operations for the years ended December 31, 2014 and 2013. Additional disclosures regarding assumptions used in calculating the fair value of the warrant liability are included in Note 1, “Business and Summary of Significant Accounting Policies.”
7. Lease Incentive
On August 1, 2013, the Company and Legacy Partners I Alameda, LLC entered into the fifth amendment to the Company’s lease, dated September 1, 1996, which covers the Company’s space at 965 Atlantic Avenue and 2020 Challenger Drive, in Alameda, California. Under the terms of the amendment, the term of the Company’s lease was extended until December 2020 and the Company was eligible to receive up to $1.3 million from Legacy Partners for leasehold improvements for both buildings.
For the years ended December 31, 2014 and 2013, the Company incurred $0.2 million and $1.1 million, respectively, for leasehold improvements under the terms of the amendment for both buildings. The lease incentives were recognized as reductions of rental expense on a straight-line basis over the term of the lease. As of December 31, 2013, the leasehold improvements had not placed in service and were recorded as Construction in Progress, see Note 5, “Property and Equipment, net.” The leasehold improvements were placed in service during the first quarter of 2014 and amortized over the life of the lease or their estimated useful lives, whichever is shorter, using the straight-line method. As of December 31, 2014, $0.9 million of the lease incentives were classified as long-term and $0.2 million were classified as current in the Company’s Consolidated Balance Sheets. As of December 31, 2013, the reimbursement of $1.1 million from Legacy Partners was recorded as Other receivables in the Company’s Consolidated Balance Sheets. As of December 31, 2014, there was no remaining reimbursement receivable from Legacy Partners.
8. Secured Notes Payable
In February 2008, the Company’s wholly-owned subsidiary, Azithromycin Royalty Sub, LLC, (the “Subsidiary”) completed a private placement of $60.0 million in aggregate principal amount of the AzaSite Notes, which were non-convertible, non-recourse promissory notes due in 2019. Net proceeds from the financing
64
Table of Contents
were approximately $55.3 million after transaction costs of approximately $4.7 million. The annual interest rate on the notes is 16% with interest payable quarterly in arrears. The notes were secured by royalties to be paid to the Company by Akorn from net sales of AzaSite in the United States. The secured notes were non-recourse to InSite Vision Incorporated.
For the quarter ended December 31, 2013, the Subsidiary received insufficient royalties to make the interest payment in full that was due on February 15, 2014. The Company had the ability to make-up this shortfall with its own cash resources. This shortfall in the interest payment was not an event of default. However, if the Company did not pay in full the shortfall (plus interest thereon) by May 15, 2014, the Subsidiary would trigger an event of default under the indenture. The Company had no intention to pay this shortfall. Accordingly, an event of default was likely on May 15, 2014. To the extent that an event of default occurred, the bondholders could seek available remedies, which included foreclosure on the Subsidiary, which would mean the Company would lose all interest in AzaSite Royalty Sub and lose its right to receive AzaSite royalties in North America. The Company’s ability to receive future revenue from sales of AzaSite was dependent on the Company’s subsidiary repaying the AzaSite Notes and interest in a timely fashion. If the Company’s subsidiary did not cure the expected event of default, the Company was highly unlikely to receive future revenue from AzaSite. Based on earned royalty levels, the earned royalties would not cover future required interest payments. As such, as of December 31, 2013, $41.3 million of secured notes was classified as current along with the unamortized debt issuance costs.
For the quarter ended March 31, 2014, the Subsidiary received insufficient royalties to make the interest payments in full that were due on the notes. Accordingly, an event of default occurred on May 15, 2014.
On June 10, 2014, the Subsidiary and the noteholders entered into a Note Purchase Agreement under which the Subsidiary repurchased and cancelled the AzaSite Notes for a single payment of $6.0 million. The $6.0 million payment to the noteholders was funded by the $6.0 million amendment fee paid by Akorn pursuant to the Amendment. As a result of the cancellation, the remaining principal on the AzaSite Notes of $41.3 million and accrued interest of $2.8 million was extinguished. In addition, the Subsidiary wrote-off the remaining balance of $2.1 million in debt issuance costs and incurred less than $0.1 million of legal and professional fees. In the Consolidated Statement of Operations for the year ended December 31, 2014, the unpaid principal of $35.3 million and unpaid interest of $2.8 million, less the write-off of debt issuance costs of $2.1 million and legal fees, totaling $36.0 million, are included under “Gain on Extinguishment of Debt” and represented basic and diluted net income per share of $0.27. In the Consolidated Statement of Cash Flows for the year ended December 31, 2014, $36.0 million is included under “Non-cash financing activities – Debt extinguishment.”
In the fourth quarter of 2014, the Company entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain purchasers (the “Purchasers”) pursuant to which the Company agreed to sell 12% Senior Secured Notes (the “Notes”) in an aggregate principal amount of up to $15 million and issue warrants to purchase shares of the Company’s common stock. The Company sold and issued Notes to the Purchasers and issued to the Purchasers warrants to purchase shares of the Company’s common stock, as follows:
| Date Issued |
Aggregate Principal Amount |
Warrant Shares |
Warrant Exercise Price |
|||||||||
| October 9, 2014 |
$ | 2.4 million | 2,053,169 | $ | 0.33 | |||||||
| November 21, 2014 |
$ | 0.2 million | 200,620 | $ | 0.31 | |||||||
| December 10, 2014 |
$ | 2.6 million | 2,824,281 | $ | 0.26 | |||||||
|
|
|
|
|
|||||||||
| Total |
$ | 5.2 million | 5,078,070 | |||||||||
|
|
|
|
|
|||||||||
The total loan commitment by the Purchasers pursuant to the Purchase Agreement is $7.8 million. Accordingly, the Company has the option to sell additional Notes in an aggregate principal amount of $2.6 million to the Purchasers. The Company’s right to sell and issue additional Notes and to issue additional warrants in connection with such Notes expires on October 9, 2016.
65
Table of Contents
The Notes are senior secured obligations of the Company and are secured by substantially all of the assets of the Company, including its intellectual property. The Notes have a maturity date of one year from the date of issuance, which may be extended for one year at the option of the Company. The Notes bear interest at a rate of 12% per annum. In the event that the Company extends the maturity date, the interest rate increases to 14% per annum.
Riverbank Capital Securities acted as the placement agent (the “Placement Agent”) for the offering of Notes and warrants pursuant to the Purchase Agreement and received $311,800, a 6% cash commission on the gross proceeds from the sale of the Notes and issuance of the warrants on October 9, 2014. Timothy McInerney, the Chairman of the Board of the Company and a member of the Company’s Board of Directors, is a principal of the Placement Agent and is a Purchaser in the offering of Notes and warrants under the same terms as the other Purchasers.
The Company incurred $0.5 million in placement agent and legal fees and $1.0 million (non-cash) for the initial valuation of the warrants issued with the Notes. These amounts are recorded as debt issuance costs and are amortized using the effective interest rate method.
9. Commitments and Contingencies
The Company has entered into certain license agreements that require it to make royalty payments for the life of the licensed patents. The estimated royalty payments related to the net sales of AzaSite in North America are approximately $1.8 million for the period 2015 through 2018. These contractual obligations are reflected in the Company’s financial statements once the related obligation becomes due.
The Company conducts its operations from leased facilities in Alameda, California under non-cancelable operating lease agreements that expire in December 2020. Lease payments include rent and the Company’s pro-rata share of operation expenses. The Company subleases a portion of the facility under a lease agreement that expired on January 31, 2015. Lease income includes rent and a pro-rata share of operation expenses. For accounting purposes, the Company is amortizing all rent payments, receipts and lease incentives ratably over the life of the lease. Rent expense for the years ended December 31, 2014, 2013 and 2012, was $314,000, $583,000, and $697,000, respectively. Future minimum lease payments under the operating lease and future cash receipts from the sublease, are as follows (in thousands):
| Year Ending December 31, |
Operating Lease Cash Payments |
Operating Sublease Cash Receipts |
Operating Lease Cash Payments, net |
|||||||||
| 2015 |
$ | 709 | $ | 13 | $ | 696 | ||||||
| 2016 |
736 | — | 736 | |||||||||
| 2017 |
762 | — | 762 | |||||||||
| 2018 |
788 | — | 788 | |||||||||
| 2019 |
815 | — | 815 | |||||||||
| 2020 and thereafter |
826 | — | 826 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 4,636 | $ | 13 | $ | 4,623 | |||||||
|
|
|
|
|
|
|
|||||||
66
Table of Contents
10. Income Taxes
Provision for Income Taxes
There was no provision for income taxes for the years ended December 31, 2014, 2013 and 2012 due to the Company’s net operating losses.
Income tax provision related to continuing operations differ from the amounts computed by applying the statutory income tax rate of 34% to pretax loss as follows (in thousands):
| 2014 | 2013 | 2012 | ||||||||||
| Tax provision at federal statutory rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State taxes, net of federal benefit |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Warrant liability |
-1.9 | % | -3.4 | % | 7.8 | % | ||||||
| Other permanent differences |
0.0 | % | 0.1 | % | 0.0 | % | ||||||
| Credits |
-4.1 | % | -11.9 | % | 0.0 | % | ||||||
| Credit Reserves |
4.4 | % | 0.0 | % | 0.0 | % | ||||||
| Expiring net operating losses |
0.0 | % | 0.0 | % | -15.0 | % | ||||||
| True up of deferred tax assets |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Valuation allowance |
-32.4 | % | -18.8 | % | -26.8 | % | ||||||
|
|
|
|
|
|
|
|||||||
| Effective tax rate |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
|
|
|
|
|
|
|
|||||||
Deferred Tax Assets and Liabilities
Deferred income taxes reflect the net tax effects of loss and credit carryforwards and temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets for federal and state income taxes as of December 31, 2014 and 2013 were as follows (in thousands):
| 2014 | 2013 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 31,342 | $ | 39,555 | ||||
| Tax credit carryforwards |
7,937 | 8,936 | ||||||
| Capitalized research and development |
9,244 | 11,746 | ||||||
| Depreciation |
162 | 165 | ||||||
| Other |
2,114 | 1,251 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
50,799 | 61,653 | ||||||
| Valuation allowance |
(50,799 | ) | (61,653 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
Realization of the Company’s deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Because of the Company’s lack of earnings history, the net deferred tax assets have been fully offset by a valuation allowance. During the years ended December 31, 2014 and 2013, the valuation allowance decreased by $10.9 million and $1.0 million, respectively.
At December 31, 2014, the Company had net operating loss carryforwards for federal income tax purposes of approximately $80.4 million, which expire in the years 2022 through 2034 and federal tax credits of approximately $5.2 million, which expire in the years 2018 through 2034. At December 31, 2014, the Company also had net operating loss carryforwards for state income tax purposes of approximately $66.7 million, which expire in the years 2016 through 2034, and state research and development tax credits of approximately
67
Table of Contents
$5.4 million, which carry forward indefinitely. The reduction of net operating loss carryforwards from the prior year is due primarily to debt forgiveness that was excluded from taxable income but resulted in a reduction of net operating loss carryforwards for federal and state purposes and expiring net operating losses.
The future utilization of the Company’s net operating loss carryforwards to offset future taxable income is subject to an annual limitation as a result of ownership changes that have occurred previously, and may be further impacted by future ownership changes. As necessary, the deferred tax assets have been reduced by any carryforwards that expire prior to utilization as a result of such limitations, with a corresponding reduction of the valuation allowance. These carryforwards may be further reduced if the Company has any additional ownership changes in the future.
The valuation allowance includes amounts of benefit at both December 31, 2014 and 2013 related to stock-based compensation and exercises, prior to the implementation of Accounting Standards Codification 515 and 718 that will be credited to additional paid-in capital when realized.
Unrecognized Tax Benefits
The Company has incurred net operating losses since inception and does not have any unrecognized tax benefits. The Company’s policy is to include interest and penalties related to unrecognized tax benefits, if any, within the provision for taxes in the consolidated statements of operations. If the Company is eventually able to recognize its uncertain positions, its effective tax rate would be reduced. The Company currently has a full valuation allowance against its net deferred tax assets which would impact the timing of the effective tax rate benefit should any uncertain tax positions be favorably settled in the future. Any adjustments to the Company’s uncertain tax positions would result in an adjustment of its net operating loss or tax credit carry forwards.
The Company files income tax returns in the U.S. federal and California jurisdictions. The Company is no longer subject to tax examinations for years before 2011 for federal returns and 2010 for California returns, except to the extent that it utilizes net operating losses or tax credit carryforwards that originated before those years. The Company is not currently under audit by any major tax jurisdiction nor has it been in the past.
The Company had the following activity relating to unrecognized tax benefits (in thousands):
| 2014 | 2013 | 2012 | ||||||||||
| Beginning Balance |
— | — | — | |||||||||
| Tax positions related to current year: |
||||||||||||
| Additions: |
||||||||||||
| Federal |
110 | — | — | |||||||||
| State |
108 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
218 | — | — | |||||||||
| Reductions |
— | — | — | |||||||||
| Tax positions related to prior years: |
||||||||||||
| Additions |
||||||||||||
| Federal |
1,179 | — | — | |||||||||
| State |
1,249 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
2,428 | — | — | |||||||||
| Reductions |
— | — | — | |||||||||
| Settlements |
— | — | — | |||||||||
| Lapses in Statutes of Limitations |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Ending Balance |
2,646 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
68
Table of Contents
Although it is reasonably possible that certain unrecognized tax benefits may increase or decrease within the next twelve months due to tax examination changes, settlement activities, expirations of statute of limitations, or the impact on recognition and measurement considerations related to the results of published tax cases or other similar activities, the Company does not anticipate any significant changes to unrecognized tax benefits over the next 12 months. During the years ended December 31, 2014 and 2013, no interest or penalties were required to be recognized relating to unrecognized tax benefits.
11. Common Stock Warrants
The following table shows outstanding warrants as of December 31, 2014, issued in the July 2011 private placement financing transaction and in private placement debt financing transactions that closed in the fourth quarter of 2014. All of the outstanding warrants have cashless exercise provisions in the event the registration statement registering the resale of the shares of common stock issuable upon exercise of the warrants is not effective or the prospectus forming a part of the registration statement is not current. All warrants are exercisable for common stock.
| Date Issued |
Warrant Shares |
Exercise Price |
Expiration Date | Cash if Exercised |
||||||||||||
| July 18, 2011 |
14,791,376 | $ | 0.75 | July 18, 2016 | $ | 11,093,532 | ||||||||||
| October 9, 2014 |
2,053,169 | $ | 0.33 | October 9, 2019 | $ | 677,546 | ||||||||||
| November 21, 2014 |
200,620 | $ | 0.31 | November 21, 2019 | $ | 62,192 | ||||||||||
| December 10, 2014 |
2,824,281 | $ | 0.26 | December 10, 2019 | $ | 734,313 | ||||||||||
|
|
|
|
|
|||||||||||||
| Total |
19,869,446 | $ | 12,567,583 | |||||||||||||
|
|
|
|
|
|||||||||||||
12. Stock-Based Compensation
Equity Incentive Program
In 2007, the Company’s stockholders approved the Company’s 2007 Performance Incentive Plan (the “2007 Plan”), which provides for grants of options and other equity-based awards to the Company’s employees, directors and consultants. Options granted under this plan, and its predecessor plan, expire 10 years after the date of grant and become exercisable at such times and under such conditions as determined by the Company’s Board of Directors (generally, with 25% vesting after one year and the balance vesting on a daily basis over the next three years of service). Upon termination of the optionee’s service, unvested options terminate and vested options generally expire three months thereafter, if not exercised. Only nonqualified stock options have been granted under this plan to date. On January 1 of each calendar year during the term of the 2007 Plan, the shares of common stock available for issuance under the 2007 Plan will be increased by the lesser of (i) 2% of the total outstanding shares of common stock on December 31 of the preceding calendar year and (ii) 3,000,000 shares. The Board of Directors of the Company delayed the effectiveness of the January 1, 2015 increase in the number of shares available for issuance under the 2007 Plan until such time as (i) the Board determines otherwise, or (ii) the Company’s stockholders approve an increase in the authorized shares of common stock under the Company’s Certificate of Incorporation (as amended).
Employee Stock Purchase Plans
The Company maintained an employee stock purchase plan, adopted in 1994 and amended thereafter (the “Purchase Plan”), until August 2009. In August 2009, the Purchase Plan was suspended. No new offering period will commence and no additional shares will be added to the Purchase Plan under its evergreen provision unless and until approved by the Company’s Board of Directors. The Purchase Plan operated in 24-month “offering periods” that are each divided into four six-month “purchase periods.” The Purchase Plan allowed eligible employees to purchase Common Stock at 85% of the lower of the fair market value of the Common Stock on the first day of the applicable offering period or the fair market value of the Common Stock on the last day of the
69
Table of Contents
applicable purchase period. Purchases were limited to 10% of each employee’s eligible compensation, subject to certain Internal Revenue Service restrictions. All of the Company’s employees were eligible to participate in the Purchase Plan after certain service periods were met. The number of shares available for issuance under the Purchase Plan was automatically increased on the first trading day in January each calendar year, by an amount equal to 0.5% of the total number of shares of Common Stock outstanding on the last trading day in December in the immediately preceding calendar year, but in no event will any such annual increase exceed 125,000 shares. No shares have been issued under the Purchase Plan since 2009. As of December 31, 2014, there was no remaining unrecorded deferred stock-based compensation expense related to the Purchase Plan. As of December 31, 2014, 515,183 shares were reserved for issuance under the Purchase Plan.
Stock-based Compensation
Stock-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as expense over the requisite service period. All of the Company’s stock compensation is accounted for as an equity instrument.
The effect of recording stock-based compensation for the years ended December 31, 2014, 2013 and 2012 was as follows (in thousands):
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Stock-based compensation expense by type of award: |
||||||||||||
| Employee stock options |
$ | 890 | $ | 926 | $ | 891 | ||||||
| Non-employee stock options Scientific Advisory Board stock options |
1 | 8 | 56 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation |
$ | 891 | $ | 934 | $ | 947 | ||||||
|
|
|
|
|
|
|
|||||||
Stock-based compensation included in expense line items in the Consolidated Statements of Operations for the year ended December 31, 2014, 2013 and 2012 was as follows (in thousands):
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Research and development |
$ | 351 | $ | 331 | $ | 271 | ||||||
| General and administrative |
540 | 603 | 676 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 891 | $ | 934 | $ | 947 | |||||||
|
|
|
|
|
|
|
|||||||
During the years ended December 31, 2014 and 2013, respectively, the Company granted options to purchase 3,939,500 and 4,124,374 shares of common stock with an estimated total grant date fair value of $0.7 million and $0.9 million. Based on the Company’s historical experience of option pre-vesting cancellations and estimates of future forfeiture rates, the Company has assumed an annualized forfeiture rate of 10% for its options for all periods disclosed. Accordingly, for the years ended December 31, 2014 and 2013, the Company estimated that the stock-based compensation for the awards not expected to vest was $0.2 million and $0.3 million, respectively.
As of December 31, 2014 and 2013, the unrecorded deferred stock-based compensation balances related to stock options were $1.0 million and $1.2 million, respectively, and will be recognized over an estimated weighted-average amortization period of 2.4 years.
70
Table of Contents
Fair Value Assumptions
The fair value of each option grant is estimated using the Black-Scholes valuation model on the date of grant and the graded-vesting method with the following weighted-average assumptions:
| Year ended December 31, | ||||||||||||
| Stock Options |
2014 | 2013 | 2012 | |||||||||
| Risk-free interest rate |
1.7 | % | 0.9 | % | 0.7 | % | ||||||
| Expected term (years) |
5 | 5 | 5 | |||||||||
| Expected dividends |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Volatility |
88.1 | % | 89.9 | % | 90.9 | % | ||||||
The expected dividend yield was set at zero because the Company has never paid cash dividends and has no present intention to pay cash dividends. Expected volatility was based on the historical volatility of the Company’s common stock and the expected moderation in future volatility over the period commensurate with the expected life of the options and other factors. The risk-free interest rates were taken from the Daily Federal Yield Curve Rates as of the grant dates as published by the Federal Reserve and represent the yields on actively traded U.S. Treasury securities for terms equal to the expected term of the options. The expected term calculation was based on the terms utilized by the Company’s peers, observed historical option exercise behavior and forfeitures of options by the Company’s employees.
The following is a summary of activity under the Company’s stock option plans for the indicated periods:
| Number of shares |
Weighted- |
Weighted-Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value (in thousands) |
|||||||||||
| Outstanding at December 31, 2011 |
11,003,963 | $0.41 | ||||||||||||
|
|
|
|||||||||||||
| Granted |
4,047,500 | 0.42 | ||||||||||||
| Exercised |
— | 0.00 | ||||||||||||
| Forfeited |
(784,223 | ) | 0.37 | |||||||||||
| Expired |
(901,718 | ) | 0.38 | |||||||||||
|
|
|
|||||||||||||
| Outstanding at December 31, 2012 |
13,365,522 | 0.42 | 7.69 | $ | 156 | |||||||||
|
|
|
|||||||||||||
| Granted |
4,124,374 | 0.31 | ||||||||||||
| Exercised |
— | 0.00 | ||||||||||||
| Forfeited |
(323,479 | ) | 0.33 | |||||||||||
| Expired |
(68,433 | ) | 0.62 | |||||||||||
|
|
|
|||||||||||||
| Outstanding at December 31, 2013 |
17,097,984 | 0.39 | 7.22 | 195 | ||||||||||
|
|
|
|||||||||||||
| Granted |
3,939,500 | 0.27 | ||||||||||||
| Exercised |
— | 0.00 | ||||||||||||
| Forfeited |
(108,190 | ) | 0.33 | |||||||||||
| Expired |
(184,653 | ) | 0.61 | |||||||||||
|
|
|
|||||||||||||
| Outstanding at December 31, 2014 |
20,744,641 | $0.37 | 6.82 | $ | — | |||||||||
|
|
|
|||||||||||||
| Options vested and expected to vest at December 31, 2014 |
20,211,174 | $0.37 | 6.76 | $ | — | |||||||||
| Options exercisable at December 31, 2014 |
14,750,731 | $0.39 | 6.04 | $ | — | |||||||||
At December 31, 2014, the Company had 3,513,717 shares of common stock available for grant under its 2007 Plan. The weighted average grant date fair value of options granted during the years ended December 31, 2014, 2013 and 2012 was $0.19, $0.21 and $0.29, respectively. No options were exercised during the year ended December 31, 2014, 2013 and 2012.
71
Table of Contents
At December 31, 2013 and 2012, options to purchase 11,094,139 and 7,752,944 shares of common stock were exercisable at weighted-average exercise prices of $0.41 and $0.42, per share, respectively.
13. Legal Proceedings
The Company is subject to various claims and legal actions during the ordinary course of its business.
In April 2011, the Company received a Notice Letter that Sandoz, Inc. or Sandoz, has filed an Abbreviated New Drug Application, or ANDA, with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution, or the Sandoz Product, prior to the expiration of the five U.S. patents listed in the Orange Book for AzaSite, which include four of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Sandoz ANDA filing, Sandoz alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed upon by the Sandoz Product. On May 26, 2011, we, Merck and Pfizer filed a patent infringement lawsuit against Sandoz and related entities. The plaintiff companies agreed that Merck would take the lead in prosecuting this lawsuit. Before the trial, the patents involved in the litigation were limited to the one Pfizer patent and three of our patents. On October 4, 2013, the United States District Court for the District of New Jersey entered a Final Judgment in favor of us and the other plaintiffs finding all the asserted claims of the patents in the litigation valid and infringed by Sandoz and related entities. The Court Order specified that the effective date of any FDA approval of a Sandoz ANDA for generic 1% azithromycin ophthalmic solution products would be no earlier than the expiration date of the patents in the litigation. On November 4, 2013, Sandoz filed an appeal of this decision to the United States Court of Appeals for the Federal Circuit. On November 15, 2013, Akorn acquired Inspire from Merck, and as such, acquired the rights to AzaSite in North America. Akorn has taken the lead in prosecuting this lawsuit. We and the other plaintiffs intend to vigorously contest any Sandoz assertions that these patents should have been found not infringed, invalid and/or unenforceable.
On January 3, 2013, certain plaintiffs filed a complaint in circuit court in Fayette County, Kentucky against Bausch & Lomb and the Company alleging that they were injured when treated with the Bausch & Lomb product Besivance following a photorefractive keratectomy. In February 2013, Bausch & Lomb removed the case to the United States District Court for the Eastern District of Kentucky. The Company moved to dismiss for lack of jurisdiction and in January 2014, the plaintiffs filed a response conceding to the Company’s motion to dismiss for lack of personal jurisdiction. On March 7, 2014, the matter was taken off docket by the District Court. The Company subsequently received a complete release of all claims.
In May 2013, the Company received a Notice Letter that Mylan Pharmaceuticals, Inc. or Mylan, has filed an ANDA with the FDA seeking marketing approval for a 1% azithromycin ophthalmic solution, or the Mylan Product, prior to the expiration of the U.S. patents listed in the Orange Book for AzaSite, which include three of our patents and one patent licensed to us by Pfizer. In the paragraph IV Certification accompanying the Mylan ANDA filing, Mylan alleges that the claims of the Orange Book listed patents are invalid, unenforceable and/or will not be infringed upon by the Mylan Product. On June 14, 2013, we, Merck and Pfizer filed a patent infringement lawsuit against Mylan and a related entity. On November 15, 2013, Akorn acquired Inspire from Merck, and as such, acquired the rights to AzaSite in North America. The plaintiff companies have agreed that Akorn will take the lead in prosecuting this lawsuit. The filing of this lawsuit triggered an automatic stay, or bar, of the FDA’s approval of the ANDA for up to 30 months or until a final district court decision of the infringement lawsuit, whichever comes first. We and the other plaintiffs intend to vigorously enforce our patent rights relating to AzaSite and vigorously contest any Mylan assertions that these patents are invalid and/or unenforceable.
On July 24, 2014, Karin Stiens filed a complaint against Bausch & Lomb, Dr. Lance Ferguson and the Company (Fayette (Kentucky) Circuit Court Civil Action No. 14-CI-2829) alleging that she suffered ophthalmological damage when Dr. Ferguson engaged in off-label use of Besivance in a photorefractive keratectomy procedure as a prophylactic antibiotic. The plaintiff alleges that Bausch & Lomb and the Company
72
Table of Contents
negligently tested, marketed and distributed Besivance, and negligently informed medical providers and consumers about Besivance. The complaint contains no facts supporting these general allegations, no facts supporting the plaintiff’s claim of permanent injuries and no allegations asserting Kentucky jurisdiction over the Company. The plaintiff has not pleaded any specific amount in damages nor made any demand. The Company is investigating the plaintiff’s allegations. The Company filed its answer on August 18, 2014.
There are currently no other claims or legal actions that would have a material adverse impact on our financial position, operations or potential performance.
14. Quarterly Results (Unaudited)
The following table is a summary of the quarterly results of operations for the years ended December 31, 2014 and 2013. The Company believes that the following information reflects all normal recurring adjustments necessary for a fair presentation of the information for the periods presented. The Company’s operating results for any quarter are not necessarily indicative of results for any future period.
| 2014 | ||||||||||||||||
| (In thousands, except per share amounts) |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||||
| Revenues |
$ | 1,153 | $ | 6,349 | $ | 360 | $ | 352 | ||||||||
| Cost of revenues |
138 | 158 | 159 | 123 | ||||||||||||
| Gross profit |
1,015 | 6,191 | 201 | 229 | ||||||||||||
| Income (loss) from operations |
(3,472 | ) | 2,256 | (2,915 | ) | (3,088 | ) | |||||||||
| Net income (loss) |
(4,679 | ) | 37,149 | (3,280 | ) | (2,429 | ) | |||||||||
| —basic |
$ | (0.04 | ) | $ | 0.28 | $ | (0.02 | ) | $ | (0.02 | ) | |||||
| —diluted |
$ | (0.04 | ) | $ | 0.28 | $ | (0.02 | ) | $ | (0.02 | ) | |||||
| 2013 | ||||||||||||||||
| (In thousands, except per share amounts) |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||||
| Revenues |
$ | 5,260 | $ | 19,240 | $ | 5,254 | $ | 1,068 | ||||||||
| Cost of revenues |
85 | 167 | 130 | 3 | ||||||||||||
| Gross profit |
5,175 | 19,073 | 5,124 | 1,065 | ||||||||||||
| Income (loss) from operations |
355 | 13,972 | 1,319 | (2,541 | ) | |||||||||||
| Net income (loss) |
(1,921 | ) | 12,116 | 579 | (4,993 | ) | ||||||||||
| —basic |
$ | (0.01 | ) | $ | 0.09 | $ | 0.00 | $ | (0.04 | ) | ||||||
| —diluted |
$ | (0.01 | ) | $ | 0.09 | $ | 0.00 | $ | (0.04 | ) | ||||||
73
Table of Contents
15. Subsequent Events
On January 29, 2015, the Company entered into a license agreement with Nicox S.A. (“Nicox”), a France-based publicly traded company, for the development and commercialization of AzaSite (1% azithromycin), AzaSite Xtra (2% azithromycin) and BromSite (0.075% bromfenac) in Europe, Middle East and Africa (105 total countries). Under the terms of the license, the Company received an upfront payment of $3.0 million and will potentially receive $13.75 million in milestone payments. It will also receive tiered, mid-single digit to double-digit royalties on the net sales of these three products.
Under the license agreement, Nicox can request up to twelve full-time equivalent (“FTE”) employees from the Company to assist with presenting data to regulatory authorities in the European Union, obtaining European Union regulatory approvals and dealing with the approved product manufacturer in the United States. Nicox has agreed to reimburse the Company for the use of its employees. Should the twelve FTE employees be needed for a full year, the reimbursement to the Company would be approximately $3.6 million.
A joint collaboration and development committee will oversee further product development of the licensed products for the European Union including AzaSite Xtra and any additional indications for BromSite. The Company has the right to utilize the jointly developed data for regulatory approval outside of the Nicox territory including in the United States. Each company will bear 50% of the development cost.
The Company evaluated subsequent events through the date on which the financial statements were issued, and has determined that there are no additional subsequent events that require adjustments or the addition of disclosure to the financial statements for the year ended December 31, 2014.
74
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures. Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of the end of the period covered by this report (Evaluation Date). Based upon the evaluation, our principal executive officer and principal financial officer concluded as of the Evaluation Date that our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act (i) is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms, and (ii) is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Disclosure controls are controls and procedures designed to reasonably ensure that information required to be disclosed in our reports filed under the Exchange Act, such as this report, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls include controls and procedures designed to reasonably ensure that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Our quarterly evaluation of disclosure controls includes an evaluation of some components of our internal control over financial reporting, and internal control over financial reporting is also separately evaluated on an annual basis for purposes of providing the management report which is set forth below.
Report of Management on Internal Control Over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
There are inherent limitations in the effectiveness of any system of internal control, including the possibility of human error and the circumvention or overriding of controls. Accordingly, even effective internal controls can provide only reasonable assurances with respect to financial statement preparation. Further, because of changes in conditions, the effectiveness of internal control may vary over time.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations (COSO) of the Treadway Commission in Internal Control—2013 Integrated Framework. Based on its assessment using those criteria, our management concluded that, as of December 31, 2014, our internal control over financial reporting was effective.
Changes in Internal Control Over Financial Reporting. There were no changes in our internal control over financial reporting (as defined in Exchange act Rule 13a-15(f)) during the quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
75
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
(a) Information regarding our executive officers appears under the heading “Executive Officers of the Company” in Item 1 of Part I of this Annual Report on Form 10-K.
(b) The remaining information required by this Item will appear under the headings labeled “Proposal One—Election of Directors” and “Section 16(a) Beneficial Ownership Reporting Compliance” of our Proxy Statement and such required information is incorporated herein by reference.
| Item 11. | Executive Compensation |
The information required by this Item will appear under the headings labeled “Director Compensation – Fiscal 2014,” “Compensation Discussion and Analysis,” “2014 Summary Compensation Table,” “2014 Grants of Plan Based Awards,” “Outstanding Equity Awards at 2014 Fiscal Year End,” “ 2014 Option Exercises and Stock Vested,” “Potential Payments Upon Termination or Change in Control,” “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report” of our Proxy Statement and such required information is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item will appear under the headings labeled “Equity Compensation Plan Information” and “Security Ownership of Certain Beneficial Owners and Management” of our Proxy Statement and such required information is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item will appear under the headings labeled “Certain Relationships and Related Transactions” and “Family Relationships and Director Independence” of our Proxy Statement and such required information is incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
Independent Auditor Fees
The information required by this Item will appear under the headings labeled “Proposal Two – Ratification of Independent Registered Public Accounting Firm” of our Proxy Statement and such required information is incorporated herein by reference.
76
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a)(1) Financial Statements
The Financial Statements and Report of Independent Auditors are included in Item 8 of Part II of this Annual Report on Form 10-K. See index to consolidated financial statements at Item 8 of Part II of this Annual Report on Form 10-K.
(2) Financial Statement Schedules
The information required under this Item appears in the Financial Statements or notes thereto included in Item 8 of Part II of this Annual Report on Form 10-K. See index to consolidated financial statements at Item 8 of this Annual Report on Form 10-K.
(3) Exhibits
The information required under this Item appears under the heading “Exhibit Index” of this Annual Report on Form 10-K.
77
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: February 18, 2015
| INSITE VISION INCORPORATED | ||
| By: | /S/ LOUIS DRAPEAU | |
| Louis Drapeau | ||
| Chief Financial Officer | ||
POWER OF ATTORNEY
KNOW ALL PEOPLE BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Louis C. Drapeau, his attorney in fact and agent, with the power of substitution and resubstition, for him and in his name, place and stead, in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney in fact, or his substitutes or agents, each acting alone, may do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Name |
Capacity |
Date | ||
|
/s/ TIMOTHY RUANE Timothy Ruane |
Chief Executive Officer and Director (Principal Executive Officer) |
February 18, 2015 | ||
| /s/ LOUIS DRAPEAU Louis Drapeau |
Chief Financial Officer (Principal Financial and Accounting Officer) |
February 18, 2015 | ||
| /s/ BRIAN LEVY Brian Levy, O.D. M.Sc. |
Director | February 18, 2015 | ||
|
/s/ TIMOTHY MCINERNEY Timothy McInerney |
Chairman of Board, Director | February 18, 2015 | ||
| /s/ ROBERT O’HOLLA Robert O’Holla |
Director | February 18, 2015 | ||
| /s/ CRAIG A. TOOMAN Craig A. Tooman |
Director | February 18, 2015 | ||
|
/s/ ANTHONY J. YOST Anthony J. Yost |
Director | February 18, 2015 | ||
78
Table of Contents
| Exhibit |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 3.1 | Restated Certificate of Incorporation as filed with the Delaware Secretary of State on October 25, 1993. | 10-K | 000-22332 | 3.2 | (1) | |||||||
| 3.2 | Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock as filed with the Delaware Secretary of State on September 11, 1997. | S-3 | 333-36673 | 4.1 | 9-29-97 | |||||||
| 3.3 | Certificate of Correction of the Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock as filed with the Delaware Secretary of State on September 26, 1997. | S-3 | 333-36673 | 4.2 | 9-29-97 | |||||||
| 3.4 | Certificate of Designations, Preferences and Rights of Series A-1 Preferred Stock as filed with the Delaware Secretary of State on July 3, 2002. |
10-Q | 001-14207 | 4.5 | 11-14-02 | |||||||
| 3.5 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on June 3, 1994. | S-3 | 333-126084 | 4.2 | 6-23-05 | |||||||
| 3.6 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on July 20, 2000. | 10-K | 001-14207 | 3.7 | 3-15-07 | |||||||
| 3.7 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on June 1, 2004. | 10-K | 001-14207 | 3.8 | 3-15-07 | |||||||
| 3.8 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on October 23, 2006. |
10-Q | 001-14207 | 3.7 | 11-14-06 | |||||||
| 3.9 | Amended Bylaws. | 8-K | 000-22332 | 3.1 | 12-16-11 | |||||||
| 4.1 | Reference is made to Exhibits 3.1 through 3.9. |
|||||||||||
| 4.2 | Securities Purchase Agreement, entered into on October 9, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | 8-K | 000-22332 | 10.1 | 10-16-14 | |||||||
| 4.3 | Amendment No. 1 to Securities Purchase Agreement, entered into on November 21, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | S-1 | 333-201052 | 10.33 | 12-18-2014 | |||||||
79
Table of Contents
| Exhibit |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 4.4 | Form of Warrant to Purchase Common Stock issued pursuant to the Securities Purchase Agreement entered into on October 9, 2014, as amended. | 8-K | 000-22332 | 4.1 | 10-16-14 | |||||||
| 10.1* | InSite Vision Incorporated Amended and Restated Employee Stock Purchase Plan adopted October 15, 2007. | 8-K | 001-14207 | 10.4 | 10-19-07 | |||||||
| 10.2* | InSite Vision Incorporated 1994 Stock Option Plan (Amended and Restated as of December 15, 1997). | S-8 | 333-60057 | 99.1 | 7-28-98 | |||||||
| 10.3* | InSite Vision Incorporated 2007 Performance Incentive Plan. | 8-K | 001-14207 | 10.1 | 10-19-07 | |||||||
| 10.4* | Form of Nonqualified Stock Option Agreement (2007). | 8-K | 001-14207 | 10.2 | 10-19-07 | |||||||
| 10.5* | Form of Incentive Stock Option Agreement (2007). | 8-K | 001-14207 | 10.3 | 10-19-07 | |||||||
| 10.6* | Form of Indemnification Agreement between the Company and its directors and officers. | 8-K | 000-22332 | 10.1 | 7-31-14 | |||||||
| 10.7* | Form of Employee’s Proprietary Information and Inventions Agreement. | S-1 | 333-68024 | 10.7 | 8-27-93 | |||||||
| 10.8 | Facilities Lease, dated September 1, 1996, between the Registrant and Alameda Real Estate Investments. | 10-K | 000-22332 | 10.17 | 3-5-97 | |||||||
| 10.9 | Amendment No. 1 to Marina Village Office Tech Lease, dated July 20, 2001 and effective January 1, 2002. |
10-Q | 001-14207 | 10.40 | 11-14-01 | |||||||
| 10.10 | Amendment No. 3 to Marina Village Office Tech Lease, dated November 28, 2006. | 10-K | 001-14207 | 10.50 | 3-15-07 | |||||||
| 10.11§ | Exclusive License Agreement, dated as of February 15, 2007, by and between the Company and Pfizer, Inc. and Pfizer Products, Inc. | 10-Q | 001-14207 | 10.2 | 5-10-07 | |||||||
| 10.12§ | License Agreement, dated as of February 15, 2007, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 001-14207 | 10.1 | 5-10-07 | |||||||
| 10.13§ | Trademark License Agreement, dated as of February 15, 2007, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 001-14207 | 10.5 | 5-10-07 | |||||||
| 10.14§ | First Amendment to License Agreement, dated as of August 9, 2012, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 000-22332 | 10.1 | 8-14-12 | |||||||
80
Table of Contents
| Exhibit |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 10.15 | Purchase and Sale Agreement, dated as of February 21, 2008, by and between Azithromycin Royalty Sub LLC and the Company. | 10-Q | 001-14207 | 10.1 | 5-12-08 | |||||||
| 10.16 | Note Purchase Agreement, dated as of February 21, 2008, by and among Azithromycin Royalty Sub LLC, the Company and the purchasers named therein. | 10-Q | 001-14207 | 10.2 | 5-12-08 | |||||||
| 10.17 | Indenture, dated as of February 21, 2008, by and between Azithromycin Royalty Sub LLC and U.S. Bank National Association. | 10-Q | 001-14207 | 10.3 | 5-12-08 | |||||||
| 10.18 | Pledge and Security Agreement made by the Company to U.S. Bank National Association, as Trustee, dated February 21, 2008. | 10-Q | 001-14207 | 10.4 | 5-12-08 | |||||||
| 10.19 | Residual License Agreement by and between Azithromycin Royalty Sub LLC and the Company dated February 21, 2008. | 10-Q | 001-14207 | 10.5 | 5-12-08 | |||||||
| 10.20* | InSite Vision Incorporated Annual Bonus Plan. | 10-Q | 001-14207 | 10.1 | 8-11-08 | |||||||
| 10.21* | InSite Vision Incorporated Severance Plan. | 8-K | 001-14207 | 10.1 | 4-29-09 | |||||||
| 10.22* | Offer letter, by and between the Company and Louis Drapeau, dated October 29, 2008. | 10-K/A | 001-14207 | 10.37 | 4-30-09 | |||||||
| 10.23* | Offer letter, by and between the Company and Timothy Ruane, dated December 1, 2010. | 8-K | 000-22332 | 10.1 | 11-30-10 | |||||||
| 10.24 | Form of Securities Purchase Agreement, dated July 12, 2011. | 8-K | 000-22332 | 10.1 | 7-18-11 | |||||||
| 10.25 | Form of Common Stock Warrant issued pursuant to Securities Purchase Agreement, dated July 12, 2011. | 8-K | 000-22332 | 4.1 | 7-18-11 | |||||||
| 10.26* | Option Cancellation Agreement, by and between the Company and Timothy Ruane, dated December 27, 2012. | 8-K | 000-22332 | 10.1 | 12-31-12 | |||||||
| 10.27§ | Royalty Purchase Agreement, by and among SWK Funding LLC and Bess Royalty, L.P. and InSite Vision Incorporated, dated as of April 2, 2013. | 10-Q | 000-22332 | 10.1 | 5-9-13 | |||||||
| 10.28§ | Second Amendment to License Agreement, entered into on June 20, 2013, by and between InSite Vision Incorporated and Inspire Pharmaceuticals. | 10-Q/A | 000-22332 | 10.1 | 10-7-13 | |||||||
| 10.29 | Amendment No. 5 to Marina Village Office Tech Lease, dated August 1, 2013. | 10-Q | 000-22332 | 10.1 | 11-13-13 | |||||||
81
Table of Contents
| Exhibit |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 10.30 | Note Purchase Agreement, entered into on June 10, 2014, between Azithromycin Royalty Sub LLC and the noteholders party thereto. | 8-K | 000-22332 | 10.1 | 6-13-14 | |||||||
| 10.31 | Third Amendment to License Agreement, entered into on June 10, 2014, between InSite Vision Incorporated, Akorn, Inc. and Inspire Pharmaceuticals. | 8-K | 000-22332 | 10.2 | 6-13-14 | |||||||
| 10.32 | Securities Purchase Agreement, entered into on October 9, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | 8-K | 000-22332 | 10.1 | 10-16-14 | |||||||
| 10.33 | Amendment No. 1 to Securities Purchase Agreement, entered into on November 21, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | S-1 | 333-201052 | 10.33 | 12-18-2014 | |||||||
| 10.34 | Security Agreement, entered into on October 9, 2014, between InSite Vision Incorporated and U.S. Bank National Association. | 8-K | 000-22332 | 10.2 | 10-16-14 | |||||||
| 10.35 | Form of 12% Senior Secured Note. | 8-K | 000-22332 | 10.3 | 10-16-14 | |||||||
| 10.36 | Form of Warrant to Purchase Common Stock issued pursuant to the Securities Purchase Agreement entered into on October 9, 2014, as amended. | 8-K | 000-22332 | 4.1 | 10-16-14 | |||||||
| 21.1 | Subsidiaries of the Company. | X | ||||||||||
| 23.1 | Consent of Burr Pilger Mayer, Inc., Independent Registered Public Accounting Firm. | X | ||||||||||
| 24.1 | Reference is hereby made to the Power of Attorney included on the signature page to this Annual Report on Form 10-K. | X | ||||||||||
| 31.1 | Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | X | ||||||||||
| 31.2 | Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | X | ||||||||||
| 32.1 | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||
82
Table of Contents
| Exhibit |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 101 | The following materials from Insite Vision, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (eXtensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Stockholders’ Deficit, (iv) the Consolidated Statements of Cash Flows, and (v) the Notes to the Consolidated Financial Statements. | X | ||||||||||
| (1) | Filed with the Company’s Annual Report on Form 10-K for the year ended December 31, 1993. |
| § | Confidential treatment has been granted with respect to certain portions of this agreement. |
| * | Management contract or compensatory plan. |
83
Table of Contents
| Exhibit Number |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 3.1 | Restated Certificate of Incorporation as filed with the Delaware Secretary of State on October 25, 1993. | 10-K | 000-22332 | 3.2 | (1) | |||||||
| 3.2 | Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock as filed with the Delaware Secretary of State on September 11, 1997. | S-3 | 333-36673 | 4.1 | 9-29-97 | |||||||
| 3.3 | Certificate of Correction of the Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock as filed with the Delaware Secretary of State on September 26, 1997. | S-3 | 333-36673 | 4.2 | 9-29-97 | |||||||
| 3.4 | Certificate of Designations, Preferences and Rights of Series A-1 Preferred Stock as filed with the Delaware Secretary of State on July 3, 2002. |
10-Q | 001-14207 | 4.5 | 11-14-02 | |||||||
| 3.5 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on June 3, 1994. | S-3 | 333-126084 | 4.2 | 6-23-05 | |||||||
| 3.6 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on July 20, 2000. | 10-K | 001-14207 | 3.7 | 3-15-07 | |||||||
| 3.7 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on June 1, 2004. | 10-K | 001-14207 | 3.8 | 3-15-07 | |||||||
| 3.8 | Certificate of Amendment to Restated Certificate of Incorporation as filed with the Delaware Secretary of State on October 23, 2006. | 10-Q | 001-14207 | 3.7 | 11-14-06 | |||||||
| 3.9 | Amended Bylaws. | 8-K | 000-22332 | 3.1 | 12-16-11 | |||||||
| 4.1 | Reference is made to Exhibits 3.1 through 3.9. |
|||||||||||
| 4.2 | Securities Purchase Agreement, entered into on October 9, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | 8-K | 000-22332 | 10.1 | 10-16-14 | |||||||
| 4.3 | Amendment No. 1 to Securities Purchase Agreement, entered into on November 21, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | S-1 | 333-201052 | 10.33 | 12-18-2014 | |||||||
84
Table of Contents
| Exhibit Number |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 4.4 | Form of Warrant to Purchase Common Stock issued pursuant to the Securities Purchase Agreement entered into on October 9, 2014, as amended. | 8-K | 000-22332 | 4.1 | 10-16-14 | |||||||
| 10.1* | InSite Vision Incorporated Amended and Restated Employee Stock Purchase Plan adopted October 15, 2007. | 8-K | 001-14207 | 10.4 | 10-19-07 | |||||||
| 10.2* | InSite Vision Incorporated 1994 Stock Option Plan (Amended and Restated as of December 15, 1997). | S-8 | 333-60057 | 99.1 | 7-28-98 | |||||||
| 10.3* | InSite Vision Incorporated 2007 Performance Incentive Plan. | 8-K | 001-14207 | 10.1 | 10-19-07 | |||||||
| 10.4* | Form of Nonqualified Stock Option Agreement (2007). | 8-K | 001-14207 | 10.2 | 10-19-07 | |||||||
| 10.5* | Form of Incentive Stock Option Agreement (2007). | 8-K | 001-14207 | 10.3 | 10-19-07 | |||||||
| 10.6* | Form of Indemnification Agreement between the Company and its directors and officers. | 8-K | 000-22332 | 10.1 | 7-31-14 | |||||||
| 10.7* | Form of Employee’s Proprietary Information and Inventions Agreement. | S-1 | 333-68024 | 10.7 | 8-27-93 | |||||||
| 10.8 | Facilities Lease, dated September 1, 1996, between the Registrant and Alameda Real Estate Investments. | 10-K | 000-22332 | 10.17 | 3-5-97 | |||||||
| 10.9 | Amendment No. 1 to Marina Village Office Tech Lease, dated July 20, 2001 and effective January 1, 2002. | 10-Q | 001-14207 | 10.40 | 11-14-01 | |||||||
| 10.10 | Amendment No. 3 to Marina Village Office Tech Lease, dated November 28, 2006. | 10-K | 001-14207 | 10.50 | 3-15-07 | |||||||
| 10.11§ | Exclusive License Agreement, dated as of February 15, 2007, by and between the Company and Pfizer, Inc. and Pfizer Products, Inc. | 10-Q | 001-14207 | 10.2 | 5-10-07 | |||||||
| 10.12§ | License Agreement, dated as of February 15, 2007, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 001-14207 | 10.1 | 5-10-07 | |||||||
| 10.13§ | Trademark License Agreement, dated as of February 15, 2007, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 001-14207 | 10.5 | 5-10-07 | |||||||
| 10.14§ | First Amendment to License Agreement, dated as of August 9, 2012, by and between the Company and Inspire Pharmaceuticals, Inc. | 10-Q | 000-22332 | 10.1 | 8-14-12 | |||||||
85
Table of Contents
| Exhibit Number |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 10.15 | Purchase and Sale Agreement, dated as of February 21, 2008, by and between Azithromycin Royalty Sub LLC and the Company. | 10-Q | 001-14207 | 10.1 | 5-12-08 | |||||||
| 10.16 | Note Purchase Agreement, dated as of February 21, 2008, by and among Azithromycin Royalty Sub LLC, the Company and the purchasers named therein. | 10-Q | 001-14207 | 10.2 | 5-12-08 | |||||||
| 10.17 | Indenture, dated as of February 21, 2008, by and between Azithromycin Royalty Sub LLC and U.S. Bank National Association. | 10-Q | 001-14207 | 10.3 | 5-12-08 | |||||||
| 10.18 | Pledge and Security Agreement made by the Company to U.S. Bank National Association, as Trustee, dated February 21, 2008. | 10-Q | 001-14207 | 10.4 | 5-12-08 | |||||||
| 10.19 | Residual License Agreement by and between Azithromycin Royalty Sub LLC and the Company dated February 21, 2008. | 10-Q | 001-14207 | 10.5 | 5-12-08 | |||||||
| 10.20* | InSite Vision Incorporated Annual Bonus Plan. | 10-Q | 001-14207 | 10.1 | 8-11-08 | |||||||
| 10.21* | InSite Vision Incorporated Severance Plan. | 8-K | 001-14207 | 10.1 | 4-29-09 | |||||||
| 10.22* | Offer letter, by and between the Company and Louis Drapeau, dated October 29, 2008. | 10-K/A | 001-14207 | 10.37 | 4-30-09 | |||||||
| 10.23* | Offer letter, by and between the Company and Timothy Ruane, dated December 1, 2010. | 8-K | 000-22332 | 10.1 | 11-30-10 | |||||||
| 10.24 | Form of Securities Purchase Agreement, dated July 12, 2011. | 8-K | 000-22332 | 10.1 | 7-18-11 | |||||||
| 10.25 | Form of Common Stock Warrant issued pursuant to Securities Purchase Agreement, dated July 12, 2011. | 8-K | 000-22332 | 4.1 | 7-18-11 | |||||||
| 10.26* | Option Cancellation Agreement, by and between the Company and Timothy Ruane, dated December 27, 2012. | 8-K | 000-22332 | 10.1 | 12-31-12 | |||||||
| 10.27§ | Royalty Purchase Agreement, by and among SWK Funding LLC and Bess Royalty, L.P. and InSite Vision Incorporated, dated as of April 2, 2013. | 10-Q | 000-22332 | 10.1 | 5-9-13 | |||||||
| 10.28§ | Second Amendment to License Agreement, entered into on June 20, 2013, by and between InSite Vision Incorporated and Inspire Pharmaceuticals. | 10-Q/A | 000-22332 | 10.1 | 10-7-13 | |||||||
86
Table of Contents
| Exhibit Number |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 10.29 | Amendment No. 5 to Marina Village Office Tech Lease, dated August 1, 2013. | 10-Q | 000-22332 | 10.1 | 11-13-13 | |||||||
| 10.30 | Note Purchase Agreement, entered into on June 10, 2014, between Azithromycin Royalty Sub LLC and the noteholders party thereto. | 8-K | 000-22332 | 10.1 | 6-13-14 | |||||||
| 10.31 | Third Amendment to License Agreement, entered into on June 10, 2014, between InSite Vision Incorporated, Akorn, Inc. and Inspire Pharmaceuticals. | 8-K | 000-22332 | 10.2 | 6-13-14 | |||||||
| 10.32 | Securities Purchase Agreement, entered into on October 9, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | 8-K | 000-22332 | 10.1 | 10-16-14 | |||||||
| 10.33 | Amendment No. 1 to Securities Purchase Agreement, entered into on November 21, 2014, between InSite Vision Incorporated, Riverbank Capital Securities, Inc., and the purchasers party thereto. | S-1 | 333-201052 | 10.33 | 12-18-2014 | |||||||
| 10.34 | Security Agreement, entered into on October 9, 2014, between InSite Vision Incorporated and U.S. Bank National Association. | 8-K | 000-22332 | 10.2 | 10-16-14 | |||||||
| 10.35 | Form of 12% Senior Secured Note. | 8-K | 000-22332 | 10.3 | 10-16-14 | |||||||
| 10.36 | Form of Warrant to Purchase Common Stock issued pursuant to the Securities Purchase Agreement entered into on October 9, 2014, as amended. | 8-K | 000-22332 | 4.1 | 10-16-14 | |||||||
| 21.1 | Subsidiaries of the Company. | X | ||||||||||
| 23.1 | Consent of Burr Pilger Mayer, Inc., Independent Registered Public Accounting Firm. | X | ||||||||||
| 24.1 | Reference is hereby made to the Power of Attorney included on the signature page to this Annual Report on Form 10-K. | X | ||||||||||
| 31.1 | Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | X | ||||||||||
| 31.2 | Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | X | ||||||||||
87
Table of Contents
| Exhibit Number |
Incorporated by Reference | Filed Herewith | ||||||||||
| Exhibit Description |
Form | File No. | Exhibit | Filing Date | ||||||||
| 32.1 | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||
| 101 | The following materials from Insite Vision, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Stockholders’ Deficit, (iv) the Consolidated Statements of Cash Flows, and (v)the Notes to the Consolidated Financial Statements. | X | ||||||||||
| (1) | Filed with the Company’s Annual Report on Form 10-K for the year ended December 31, 1993. |
| § | Confidential treatment has been granted with respect to certain portions of this agreement. |
| * | Management contract or compensatory plan. |
88