Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
x | Annual report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2014
or
¨ | Transition report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission file number: 0-27756
ALEXION PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
Delaware | 13-3648318 |
(State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) |
352 Knotter Drive, Cheshire Connecticut 06410
(Address of Principal Executive Offices) (Zip Code)
203-272-2596
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: | Common Stock, par value $0.0001 | |
Rights to Purchase Junior Participating Cumulative Preferred Stock, par value $0.0001 | ||
Name of each exchange on which registered: The NASDAQ Stock Market LLC | ||
Securities registered pursuant to Section 12(g) of the Act: None | ||
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check One:
Large accelerated filer x Accelerated filer ¨ Non-accelerated filer ¨ (Do not check if a smaller reporting company)
Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the Common Stock held by non-affiliates of the registrant, based upon the last sale price of the Common Stock reported on The NASDAQ Stock Market LLC on June 30, 2014, was $30,706,252,188.(1)
The number of shares of Common Stock outstanding as of February 3, 2015 was 202,148,509.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Definitive Proxy Statement to be used in connection with its Annual Meeting of Stockholders to be held on May 6, 2015, are incorporated by reference into Part III of this report.
1
(1) Excludes 1,378,571 shares of common stock held by directors and executive officers at June 30, 2014. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, directly or indirectly, to direct or cause the direction of the management or policies of the registrant, or that such person is controlled by or under common control with the registrant.
2
Alexion Pharmaceuticals, Inc.
Table of Contents
PART I | Page | |
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
Item 2. | ||
Item 3. | ||
Item 4. | ||
PART II | ||
Item 5. | ||
Item 6. | ||
Item 7. | ||
Item 7.A | ||
Item 8. | ||
Item 9. | ||
Item 9A. | ||
Item 9A(T). | ||
Item 9B. | ||
PART III | ||
Item 10. | ||
Item 11. | ||
Item 12. | ||
Item 13. | ||
Item 14. | ||
PART IV | ||
Item 15. | ||
3
PART I
Unless the context requires otherwise, references in this report to "Alexion", the “Company”, "we", "our" or "us" refer to Alexion Pharmaceuticals, Inc. and its subsidiaries.
Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements that have been made pursuant to the provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements are based on current expectations, estimates and projections about our industry, management's beliefs, and certain assumptions made by our management, and may include, but are not limited to, statements regarding the potential benefits and commercial potential of Soliris® (eculizumab) for its approved indications and any expanded uses, timing and effect of sales of Soliris in various markets worldwide, pricing for Soliris, level of insurance coverage and reimbursement for Soliris, level of future Soliris sales and collections, timing regarding development and regulatory approvals for additional indications or in additional territories for Soliris, the medical and commercial potential of additional indications for Soliris, failure to satisfactorily address the issues raised by the U.S. Food and Drug Administration (FDA) in the March 2013 Warning Letter and Form 483 issued by the FDA in August 2014, costs, expenses and capital requirements, cash outflows, cash from operations, status of reimbursement, price approval and funding processes in various countries worldwide, progress in developing commercial infrastructure and interest about Soliris and our drug candidates in the patient, physician and payer communities, the safety and efficacy of Soliris and our product candidates, estimates of the potential markets and estimated commercialization dates for Soliris and our drug candidates around the world, sales and marketing plans, any changes in the current or anticipated market demand or medical need for Soliris or our drug candidates, status of our ongoing clinical trials for eculizumab, asfotase alfa and our other product candidates, commencement dates for new clinical trials, clinical trial results, evaluation of our clinical trial results by regulatory agencies, the adequacy of our pharmacovigilance and drug safety reporting processes, prospects for regulatory approval of asfotase alfa and our other product candidates, need for additional research and testing, the uncertainties involved in the drug development process and manufacturing, performance and reliance on third party service providers, our future research and development activities, plans for acquired programs, our ability to develop and commercialize products with our collaborators, assessment of competitors and potential competitors, the outcome of challenges and opposition proceedings to our intellectual property, assertion or potential assertion by third parties that the manufacture, use or sale of Soliris infringes their intellectual property, estimates of the capacity of manufacturing and other service facilities to support Soliris and our product candidates, potential costs resulting from product liability or other third party claims, the sufficiency of our existing capital resources and projected cash needs, the possibility that expected tax benefits will not be realized, assessment of impact of recent accounting pronouncements, declines in sovereign credit ratings or sovereign defaults in countries where we sell Soliris, delay of collection or reduction in reimbursement due to adverse economic conditions or changes in government and private insurer regulations and approaches to reimbursement, the short and long term effects of other government healthcare measures, and the effect of shifting foreign exchange rates. Words such as “anticipates,” “expects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. These statements are not guarantees of future performance and are subject to certain risks, uncertainties, and assumptions that are difficult to predict; therefore, actual results may differ materially from those expressed or forecasted in any such forward-looking statements. Such risks and uncertainties include, but are not limited to, those discussed later in this report under the section entitled “Risk Factors”. Unless required by law, we undertake no obligation to update publicly any forward-looking statements, whether because of new information, future events or otherwise. However, readers should carefully review the risk factors set forth in this and other reports or documents we file from time to time with the Securities and Exchange Commission.
Item 1. | BUSINESS. |
(dollars and shares in thousands)
Overview
We are a biopharmaceutical company focused on serving patients with severe and ultra-rare disorders through the innovation, development and commercialization of life-transforming therapeutic products. Our marketed product Soliris is the first and only therapeutic approved for patients with either of two severe and ultra-rare disorders resulting from chronic uncontrolled activation of the complement component of the immune system: paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening and ultra-rare genetic blood disorder, and atypical hemolytic uremic syndrome (aHUS), a life-threatening and ultra-rare genetic disease. We are also evaluating additional potential indications for Soliris in severe and devastating diseases in which we believe that uncontrolled complement activation is the underlying mechanism, and we are progressing in various
4
stages of development with additional biotechnology product candidates as treatments for patients with severe and life-threatening ultra-rare disorders. We were incorporated in 1992 and began commercial sale of Soliris in 2007.
Soliris is designed to inhibit a specific aspect of the complement component of the immune system and thereby treat inflammation associated with chronic disorders in several therapeutic areas, including hematology, nephrology, transplant rejection and neurology. Soliris is a humanized monoclonal antibody that effectively blocks terminal complement activity at the doses currently prescribed. The initial indication for which we received approval for Soliris is PNH. PNH is a debilitating and life-threatening, ultra-rare genetic blood disorder defined by chronic uncontrolled complement activation leading to the destruction of red blood cells (hemolysis). The chronic hemolysis in patients with PNH may be associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breath and intermittent episodes of dark-colored urine (hemoglobinuria).
Soliris was approved for the treatment of PNH by the U.S. Food and Drug Administration (FDA) and the European Commission (EC) in 2007 and by Japan’s Ministry of Health, Labour and Welfare (MHLW) in 2010, and has been approved in several other territories. Additionally, Soliris has been granted orphan drug designation for the treatment of PNH in the United States, Europe, Japan and several other territories.
In September and November 2011, Soliris was approved by the FDA and EC, respectively, for the treatment of pediatric and adult patients with aHUS in the United States and Europe. In September 2013, the MHLW approved Soliris for the treatment of pediatric and adult patients with aHUS in Japan. aHUS is a severe and life-threatening genetic ultra-rare disease characterized by chronic uncontrolled complement activation and thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, causing a reduction in platelet count (thrombocytopenia) and life-threatening damage to the kidney, brain, heart and other vital organs. In addition, the FDA and EC have granted Soliris orphan drug designation for the treatment of patients with aHUS.
Products and Development Programs
The human immune system defends the body from attack or invasion by infectious agents or pathogens. This defense is accomplished through a complex system of proteins and cells, primarily complement proteins, antibodies and white blood cells, each with a specialized function. Under normal circumstances, complement proteins, together with antibodies and white blood cells, act to protect the body by removing:
• | harmful micro-organisms; |
• | cells containing foreign proteins known as antigens; and |
• | potential disease-causing combinations of antigens and antibodies known as immune complexes. |
When activated by certain stimuli, the immune system triggers a series of enzymatic and biochemical reactions called the complement cascade that results in a pro-inflammatory, pro-thrombotic and cytolytic (cell lysis) response. This set of responses is one of the immune system’s weapons against foreign pathogens or otherwise diseased tissue. Given this role of the complement cascade, it must be tightly regulated so that damage to healthy cells, tissues and organs does not occur. However, in certain settings, the complement cascade is subject to uncontrolled excessive or inappropriate activation, or as well an individual may be deficient in naturally occurring complement inhibitors (regulatory proteins). Any of these circumstances may result in acute and chronic inflammatory conditions and damage to healthy tissues and organs.
We focus our product development programs on life-transforming therapeutics for severe and life-threatening ultra-rare diseases for which we believe current treatments are either non-existent or inadequate. Eculizumab is a humanized antibody known as a C5 terminal complement inhibitor (C5 Inhibitor), which is designed to selectively block the cleavage of C5 and hence the production of the pro-inflammatory, pro-thrombotic and cytolytic proteins of the terminal complement cascade. In addition to PNH and aHUS, for which the use of eculizumab has been approved in the United States, Europe, Japan, and other countries, we believe that C5 Inhibitors may be useful in the treatment of a variety of other serious diseases and conditions resulting from uncontrolled complement activation.
5
Marketed Products
Our marketed products include the following:
Product | Development Area | Description | Development Stage | |||
Soliris (eculizumab) | Hematology | Paroxysmal Nocturnal Hemoglobinuria (PNH) | Commercial | |||
PNH Registry | Phase IV | |||||
Hematology/Nephrology | Atypical Hemolytic Uremic Syndrome (aHUS) | Commercial | ||||
aHUS Registry | Phase IV | |||||
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Soliris is the first and only therapy approved for the treatment of patients with PNH, a debilitating and life-threatening ultra-rare blood disorder in which an acquired genetic deficiency causes uncontrolled complement activation which leads to life-threatening complications. We continue to work with researchers to expand the base of knowledge in PNH and the utility of Soliris to treat patients with PNH. In 2013, the EC extended the Soliris label to include pediatric patients with PNH. The Committee for Medicinal Products for Human Use (CHMP) recommends that the renewal be granted with unlimited validity. Additionally, we are sponsoring a multinational registry to gather information regarding the natural history of patients with PNH and the longer term outcomes during Soliris treatment.
Atypical Hemolytic Uremic Syndrome (aHUS)
aHUS is a chronic and life-threatening ultra-rare genetic disease in which uncontrolled complement activation causes blood clots in small blood vessels throughout the body, or TMA, leading to kidney failure, stroke, heart attack and death. Soliris is the first and only therapy approved for the treatment of pediatric and adult patients with aHUS. Pursuant to a post marketing requirement imposed by the FDA, we have now completed enrollment in a prospective open-label trial in adults with aHUS and, separately, enrollment has been completed in a prospective trial of pediatric patients with aHUS. In May 2014, based on data from these trials, the FDA approved conversion of Soliris accelerated approval in aHUS to regular approval for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA.
6
Clinical Development Program
Our programs, including investigator sponsored clinical programs, include the following:
Product | Development Area | Description | Development Stage | |||
Soliris (eculizumab) | Transplant | Delayed Kidney Transplant Graft Function | Phase III | |||
Antibody Mediated Rejection (AMR) Presensitized Renal Transplant - Living Donor | Phase II | |||||
Antibody Mediated Rejection (AMR) Presensitized Renal Transplant - Deceased Donor | Phase II | |||||
Treatment of Antibody Mediated Rejection (AMR) Following Renal Transplantation* | Phase II | |||||
Neurology | Neuromyelitis Optica (NMO) | Phase III | ||||
Myasthenia Gravis (MG) | Phase III | |||||
Asfotase alfa | Metabolic Disorders | Hypophosphatasia (HPP) | Phase II | |||
cPMP (ALXN 1101) | Metabolic Disorders | MoCD Type A | Phase II | |||
ALXN 1007 | Inflammatory Disorders | GI Graft versus Host Disease | Phase II | |||
Anti-phospholipid Syndrome | Phase II | |||||
ALXN 1210 | Next Generation | Phase I | ||||
ALXN 5500 | Next Generation | Phase I | ||||
* Investigator Initiated Trial
Soliris (eculizumab)
Transplant
Delayed Kidney Transplant Graft Function
Delayed graft function (DGF) is the term used to describe the failure of a kidney or other organs to function immediately after transplantation due to ischemia-reperfusion and immunological injury. Enrollment has been completed in this Phase III registration trial study of eculizumab in patients at elevated risk for DGF following kidney transplant. Eculizumab has been granted orphan drug designation for DGF by the FDA and, in the first quarter of 2014, the EC granted orphan drug designation to eculizumab for prevention of DGF after solid organ transplantation. In August 2014, we announced the initiation of dosing in a single, multinational, placebo-controlled DGF registration trial based on positive discussions with regulators in the U.S. and EU.
Antibody Mediated Rejection (AMR) in Presensitized Kidney Transplant Patients
AMR is the term used to describe a type of transplant rejection that occurs when the recipient has antibodies to the donor organ. Enrollment in a multi-national, multi-site controlled clinical trial of eculizumab in presensitized kidney transplant patients at elevated risk for AMR who received kidneys from deceased organ donors was completed in March 2013. The study was re-opened in October 2013 to enroll additional patients at the request of participating investigators. Enrollment and dosing in this expanded trial has been completed and patient follow-up in the trial is continuing. In September 2013, researchers presented positive preliminary data from the eculizumab deceased-donor AMR kidney transplant study at the European Society of Organ Transplant in Vienna, Austria.
Enrollment in a multi-national, multi-site randomized controlled clinical trial of eculizumab in presensitized kidney transplant patients at elevated risk for AMR who received kidneys from living donors has been completed and patient follow-up in the trial is ongoing. In January 2015, we reported results from a randomized, open-label, multicenter Phase II clinical trial to determine the safety and efficacy of eculizumab in the prevention of AMR in living-donor kidney transplant recipients requiring desensitization. The primary composite endpoint of the trial did not reach statistical significance. Data analyses are
7
ongoing and based on discussions with regulators, we are developing plans to commence a clinical trial with eculizumab as a treatment for patients with AMR.
In April 2014, the EC granted orphan drug designation to eculizumab for the prevention of graft rejection following solid organ transplantation.
Neurology
Neuromyelitis Optica (NMO)
NMO is a severe and ultra-rare autoimmune disease of the central nervous system (CNS) that primarily affects the optic nerves and spinal cord. In an investigator-initiated Phase II clinical trial of eculizumab in severe and relapsing NMO, eculizumab reduced the median number of NMO attacks at 12 months with a high degree of statistical significance. In the first half of 2014, we commenced a Phase III pivotal trial to evaluate eculizumab as as treatment for patients with relapsing NMO. The FDA , EC, and MHLW have each granted orphan designation for eculizumab as a treatment for patients with NMO.
Myasthenia Gravis (MG)
MG is an ultra-rare autoimmune syndrome characterized by complement activation leading to the failure of neuromuscular transmission. Data from a Phase II trial evaluating the safety and efficacy of eculizumab in patients with refractory generalized MG indicated improvement in clinical measures. In the second quarter of 2014, we commenced a Phase III pivotal trial to evaluate eculizumab as a treatment for patients with refractory generalized MG. In addition, the FDA, EC and MHLW have granted orphan drug designation for eculizumab as a treatment for patients with MG.
Asfotase Alfa
Hypophosphatasia (HPP)
HPP is an ultra-rare, genetic, and life-threatening metabolic disease characterized by impaired phosphate and calcium regulation, leading to progressive damage to multiple vital organs including destruction and deformity of bones, profound muscle weakness, seizures, impaired renal function, and respiratory failure.
Asfotase alfa, a targeted enzyme replacement therapy in Phase II clinical trials for patients with HPP, is designed to directly address the morbidities and mortality of HPP by targeting alkaline phosphatase directly to the deficient tissue. In this way, asfotase alfa is designed to normalize the genetically defective metabolic process and prevent or reverse its severe and life-threatening complications in patients with HPP. Studies with asfotase alfa in HPP patients indicate that the treatment significantly decreases the levels of targeted metabolic substrates. In 2013, asfotase alfa received Breakthrough Therapy Designation from the FDA. In September 2014, the MHLW granted orphan drug designation to asfotase alfa for the treatment of patients with HPP.
Interim results from a separate multinational Phase II open-label study of infants and children with HPP were presented at the European Society of Pediatric Endocrinology meeting held in September 2013. Results of 15 enrolled and treated patients representing a range of HPP characteristics were summarized, showing that the primary efficacy endpoint was achieved with a high degree of clinical and statistical significance and several key secondary endpoints were also achieved. The study continues to enroll and dose patients.
We have completed two natural history studies in infantile-onset patients with HPP and juveniles with HPP. We have completed our initial analysis for the studies. We have commenced and completed a rolling submission of our U.S. Biologics License Application (BLA) for asfotase alfa, which allows completed portions of the application to be submitted and reviewed by the FDA on an ongoing basis. In July 2014, we announced that the European Medicines Agency (EMA) informed us that it had validated our Marketing Authorization Application (MAA) for asfotase alfa for the treatment of HPP. We believe the analysis of our clinical data supports our regulatory filings in the U.S., EU, and Japan.
In September 2014, results of several HPP trials were presented at the 2014 Annual Meeting of the American Society of Bone and Mineral Research (ASBMR). Results of these trials indicate that in HPP patients at high risk of death, overall survival rates significantly greater in asfotase alfa patients compared with survival in historical control patients. In addition, patients receiving asfotase alfa significantly improved ventilator-free survival.
8
cPMP (ALXN 1101)
Molybdenum Cofactor Deficiency (MoCD) Disease Type A (MoCD Type A)
MoCD Type A is an ultra-rare metabolic disorder characterized by severe and rapidly progressive neurologic damage and death in newborns. MoCD Type A results from a genetic deficiency in cyclic Pyranopterin Monophosphate (cPMP), a molecule that enables production of certain enzymes, the absence of which allows neurotoxic sulfite to accumulate in the brain. To date, there is no approved therapy available for MoCD Type A. There has been some early clinical experience with the cPMP replacement therapy in a small number of children with MoCD Type A, and we have initiated a natural history study in patients with MoCD Type A. In October 2013, cPMP received Breakthrough Therapy Designation from the FDA for the treatment of patients with MoCD Type A. Evaluation of our synthetic cPMP replacement therapy in a Phase I healthy volunteer study is complete. As a result, we have initiated a multi-center, multinational, open-label clinical trial of synthetic cPMP in patients with MoCD Type A being treated with recombinant cPMP.
ALXN 1007
ALXN 1007 is a novel humanized antibody designed to target rare and severe inflammatory disorders and is a product of our proprietary antibody discovery technologies. We have completed enrollment in both a Phase I single-dose, dose escalating safety and pharmacology study in healthy volunteers, as well as in a multi-dose, dose escalating safety and pharmacology study in healthy volunteers. As a result of meetings with the FDA, we commenced dosing in the second quarter of 2014 a Phase II proof-of-concept study in patients with anti-phospholipid syndrome (APS). APS is an ultra-rare autoimmune, hypercoagulable state caused by antiphospholipid antibodies. A second proof-of-concept study in patients with another ultra-rare disorder, gastrointestinal graft versus host disease (GI-GVHD) was initiated in September 2014. Patients with GI-GVHD following bone marrow or hematopoietic stem cell transplant experience engrafted hematopoietic cells that attack host gastrointestinal tissues in the first 100 days post-transplant causing damage to the GI tract, liver and skin.
Manufacturing
We currently rely on two manufacturing facilities, Alexion's Rhode Island manufacturing facility (ARIMF) and one facility operated by Lonza Group AG and its affiliates (Lonza), to produce commercial and clinical bulk quantities of Soliris, and we rely on a facility operated by Lonza for clinical quantities of asfotase alfa. We produce our clinical and preclinical quantities of our other product candidates at ARIMF. We have entered into an agreement with Lonza to manufacture commercial and clinical supplies of Soliris and asfotase alfa at an additional site. We also depend on a limited number of third party providers for other services with respect to our clinical and commercial requirements, including manufacturing services, product finishing, packaging, filling and labeling.
We have various agreements with Lonza through 2026, with remaining total non-cancellable commitments of approximately $383,500 through 2018. Our agreements with Lonza also include potential payments totaling up to $5,000 that will become payable if and when certain milestones are achieved. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of Soliris manufactured at ARIMF and a payment with respect to sales of Soliris manufactured at Lonza facilities.
In March 2013, we received a Warning Letter (Warning Letter)from the FDA regarding compliance with current Good Manufacturing Practices (cGMP) at ARIMF. The Warning Letter followed an FDA inspection which concluded in August 2012. At the conclusion of that inspection, the FDA issued a Form 483 Inspectional Observations, to which we responded in August 2012 and provided additional information to the FDA in September and December 2012. The observations relate to commercial and clinical manufacture of Soliris at ARIMF. We responded to the Warning Letter in a letter to the FDA dated in April 2013. At the conclusion of another inspection of ARIMF in August 2014, the FDA issued a Form 483 with three inspectional observations, none of which were designated as a repeat observation to the Warning Letter. The observations are inspectional and do not represent a final FDA determination of compliance. We continue to manufacture products, including Soliris, at ARIMF. While the resolution of the issues raised in this Warning Letter is difficult to predict, we do not currently believe a loss related to this matter is probable or that the potential magnitude of such loss or range of loss, if any, can be reasonably estimated. To the extent that circumstances related to this matter change, the impact could have a material adverse effect on our financial operations.
The EMA inspected ARIMF in January 2013 and issued a cGMP certificate in May 2013.
Unrelated to the Warning Letter, we initiated voluntary recalls and replacements of certain lots of Soliris in 2013 and 2014 due to the presence of visible particles detected in a limited number of vials in these lots. These recalls did not interrupt
9
the supply of Soliris to patients. Following investigation, we believe that we have identified the fill/finish process step at our third party provider that resulted in the presence of the visible particles and we have implemented the changes necessary to modify the process step. During the fourth quarter of 2013, we recorded expense of $14,277 in costs of sales resulting from the disposal of inventory in 2014. Expenses associated with recalls were not material in 2014.
In April 2014, we purchased a fill/finish facility in Athlone, Ireland. Following refurbishment of the facility, and after successful completion of the appropriate validation processes and regulatory approvals, the facility will become our first company-owned fill/finish and packaging facility for Soliris and other clinical and commercial products. Our plans for future expansion in Ireland also include the construction of office and laboratory facilities on property in Dublin, Ireland, which we purchased in April 2014.
Sales and Marketing
We have established a commercial organization to support current and future sales of Soliris in the United States, Europe, Japan, Asia Pacific countries, and other territories. Our sales force for Soliris is small compared to that of other drugs with similar revenues; however, we believe that a relatively smaller sales force is appropriate to effectively market Soliris due to the incidence and prevalence of PNH and aHUS. If we receive regulatory approval in new territories or for new products or indications, we may expand our own commercial organizations in such territories and market and sell Soliris through our own sales force in these territories. However, we evaluate each jurisdiction on a country-by-country basis, and, in certain territories, we promote Soliris in collaboration with marketing partners or rely on relationships with one or more companies with established distribution systems and direct sales forces in certain countries.
Customers
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other health care providers. In some cases, we may also sell Soliris to governments and government agencies.
During 2014, sales to our largest customer accounted for 18% of our Soliris net product sales. During 2013, sales to our largest customer accounted for 20% of our Soliris net product sales.
Because of factors such as the pricing of Soliris, the limited number of patients, the short period from product sale to patient infusion and the lack of contractual return rights, Soliris customers often carry limited inventory. We also monitor inventory within our sales channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms and financial strength of distributors.
Please also see "Management’s Discussion and Analysis – Net Product Sales," and Note 17 of the Consolidated Financial Statements included in this Annual Report on Form 10-K, for financial information about geographic areas.
Intellectual Property Rights and Market Exclusivity
Patents and other intellectual property rights are important to our business. We own or license a number of patents in the U.S. and foreign countries that cover our products and investigational compounds; also we file and prosecute patent applications covering new technologies and inventions that are meaningful to our business. In addition to patents, we rely on trade secrets, know-how, trademarks, regulatory exclusivity and other forms of intellectual property. Our intellectual property rights have material value and we act to protect them.
In the biopharmaceutical industry, two forms of intellectual property generally determine the period of a product’s market exclusivity: patent rights and regulatory forms of exclusivity. It is during the period of market exclusivity that an innovative product generally realizes most of its commercial value.
Patents provide the owner with a right to exclude others from practicing an invention. Patents may cover the active ingredients, uses, formulations, doses, administrations, delivery mechanisms, manufacturing processes and other aspects of a product. The period of patent protection for any given product may depend on the expiration date of various patents and may differ from country to country according to the type of patents, the scope of coverage and the remedies for infringement available in a country.
Most of our products and investigational compounds are protected by patents with varying terms that depend on the type of patent and its filing date. However, a significant portion of a product's patent life can elapse during the time it takes to develop and obtain regulatory approval of the product. As compensation, certain developed countries will extend a patent’s term, subject to a number of factors and caps.
10
With respect to Soliris, we own an issued U.S. patent that covers the product and will expire in 2021, taking into account patent term extension. We also own a corresponding issued European patent that covers Soliris and will expire in 2015, though in certain European countries where we filed for supplementary protection certificates we expect exclusivity to extend into 2020. In Japan and other countries where we own patents covering Soliris the patents will expire between 2015 and 2020. We also own U.S. and foreign patents and patent applications that protect our investigational compounds and product candidates. At present, it is not known whether any such investigational compound or product candidate will be approved for human use and sale.
Regulatory forms of exclusivity are another source of valuable rights that can contribute toward market exclusivity for an innovative biopharmaceutical product such as Soliris. Many developed countries provide such non-patent incentives to develop medicines. In the U.S., Europe and Japan, for instance, regulatory intellectual property rights provide incentives to develop medicines for rare diseases, or orphan drugs, and medicines for pediatric patients. Those countries and others also provide data protection for a period of time after the approval of a new drug, during which regulatory agencies may not rely on the innovator’s data to approve a biosimilar or generic copy. Regulatory forms of exclusivity can work in conjunction with patents to strengthen market exclusivity, and in countries where patent protection has expired or does not exist, regulatory forms of exclusivity can extend a product’s market exclusivity period.
With respect to Soliris, we rely on regulatory forms of exclusivity such as data protection and orphan drug protection to support the product’s market exclusivity. Specific aspects of the laws governing regulatory exclusivity vary by country, but most forms of regulatory exclusivity do not prevent competitive products from gaining regulatory approval on the basis of the competitor’s own safety and efficacy data, even when the competitive product is a biosimilar or generic copy. In certain countries, however, orphan drugs can obtain a period of exclusivity during which no competitive product containing the same drug may be approved for the same orphan indication.
Our success will depend in part on our ability to obtain and maintain patent and regulatory protections for our products and investigational compounds, to preserve our trade secrets and other proprietary rights, to operate without infringing the proprietary rights of third parties, and to prevent third parties from circumventing our rights. Due to the time and expense of bringing new products through development and regulatory approval to the marketplace, there is particular importance in obtaining patent and trade secret protection for significant new technologies, products and processes. As of December 31, 2014, we owned or in-licensed patents and patent applications that relate to C5 inhibitors, high throughput screening, biologic manufacturing processes, vectors, cancer, recombinant antibodies, bone delivery conjugates, nucleic acid-based therapies, natriuretic peptides, human molybdenum cofactor deficiency, targeted complement inhibitors, and other technologies.
Significant legal questions exist concerning the extent and scope of patent protection for biopharmaceutical products and processes in the United States and elsewhere. Accordingly, there is no certainty that patent applications owned or licensed by us will issue as patents, or that our issued patents will afford meaningful protection against competitors. Once issued, patents are subject to challenge through both administrative and judicial proceedings in the U.S. and other countries. Such proceedings include re-examinations, inter partes reviews, post-grant reviews and interference proceedings before the U.S. Patent and Trademark Office, as well as opposition proceedings before the European Patent Office. Litigation may be required to enforce, defend or obtain our patent and other intellectual property rights. Any administrative proceeding or litigation could require a significant commitment of our resources and, depending on outcome, could adversely affect the scope, validity or enforceability of certain of our patent or other proprietary rights.
In regard to third party intellectual property, we have in the past received, and may in the future receive, notices claiming infringement of their patents. We are aware of other patents owned by third parties that the owners might claim to be infringed by the development and commercialization of Soliris or some of our investigational compounds. We have obtained licenses to some of those patents and may obtain licenses to others. In other instances, we have determined in our judgment that:
• | our products and investigational compounds do not infringe the patents; |
• | the patents are not valid or enforceable; or |
• | we have identified and are testing various alternatives that should not infringe the patents and which should permit continued development and commercialization of our products and investigational compounds. |
If a patent holder successfully challenges our judgment that our products do not infringe their patents or that their patents are invalid, we could be required to pay costly damages or to obtain a license to sell or develop our products. A required license may be costly or may not be available on acceptable terms, if at all. A costly license or inability to obtain a necessary license could materially and adversely affect our ability to commercialize our products, including Soliris.
11
On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims, we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adjustment to our operating results.
The market exclusivity of our products may be impacted by competitive products that are either innovative or biosimilar or generic copies. In our industry, the potential for biosimilar challenges has been an increasing risk to product market exclusivity. U.S. law enacted in 2010 created a new approval pathway for biosimilar versions of innovative biological products. Prior to that time, innovative biologics had essentially unlimited regulatory exclusivity. Under the new pathway, the FDA may approve products that are similar to (but not generic copies of) innovative biologics on the basis of less extensive data than is required by a full biologic license application. After an innovator has marketed its product for four years, other manufacturers may apply for approval of a biosimilar version of the innovator product. However, qualified innovative biological products will receive 12 years of regulatory exclusivity, meaning that the FDA may not actually approve a biosimilar version until 12 years after the innovative product received its approval. The law also provides a mechanism for innovators to enforce their patents that protect their products and for biosimilar applicants to challenge the patents. Such litigation may begin as early as four years after the innovative biological product is first approved by the FDA. Pathways for biosimilar products also exist in many other markets also, including Europe and Japan.
We estimate the market exclusivity period for our products solely for business planning purposes. The actual length of market exclusivity for any product is impossible to predict with certainty due to the complex interaction between patent and regulatory factors and the inherent uncertainties of litigation.
License and Collaboration Agreements
In January 2015, we entered into a license agreement with a third party to obtain certain intellectual property rights and technology related to specific therapeutic compounds. The agreement provides an exclusive research, development and commercial license for products to be developed using such compounds. Pursuant to the terms of the agreement, we made an upfront payment of $50,000 during the first quarter 2015. We could be required to pay up to an additional $213,000 in development and regulatory milestones related to a product developed under the agreement for a single disease indication. An additional $437,000 in milestone payments could be due if certain development and regulatory milestones are achieved for additional disease indications. The agreement also provides for royalty payments and potential milestone payments of up to $180,000 on commercial sales of products developed under the agreement.
In December 2014, we entered into an agreement with X-Chem Pharmaceuticals (X-Chem) that allows us to identify novel drug candidates from X-Chem's proprietary drug discovery engine. Alexion will have the exclusive worldwide rights to develop and commercialize up to three targets arising from the collaboration. Due to the early stage of these assets, we recorded expense for an upfront payment of $8,000. In addition, for each drug target, to a maximum of three targets, we could be required to make additional payments upon the achievement of specified research, development and regulatory milestones up to $75,000, as well as royalties on commercial sales.
In January 2014, we entered into an agreement with Moderna Therapeutics, Inc. (Moderna) that provides the option to purchase drug products for clinical development and commercialization of Moderna's messenger RNA (mRNA) therapeutics to treat rare diseases. Due to the early stage of these assets, we recorded expense for an upfront payment of $100,000 in 2014. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of ten targets, would could be required to make an option exercise payment of $15,000 and to pay up to an additional $120,000 with respect to a rare disease product and $400,000 with respect to a non-rare disease product in development and sales milestones if the specific milestones are met over time as well as royalties on commercial sales.
In July 2013, we entered into a license and collaboration agreement with Ensemble Therapeutics Corporation for the identification, development and commercialization of therapeutic candidates based on specific drug targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $11,500 during the third quarter of 2013. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of four targets, we could be required to pay up to an additional $90,750 in development milestones as the specific milestones are met over time. The agreement also provides for royalty payments on commercial sales of each product developed under the agreement.
12
In January 2013, we entered into a license agreement for a technology, which provides an exclusive research license and an option for an exclusive commercial license for specific targets and products to be developed. Due to the early stage of this asset, we recorded expense for an upfront payment of $3,000 during the first quarter of 2013. We will also be required to pay annual maintenance fees during the term of the arrangement. In addition, for each target, up to a maximum of six targets we develop, we could be required to pay up to an additional $70,500 in license fees, development and sales milestones as the specific milestones are met over time.
In October 2012, we entered into a settlement and non-exclusive license agreement with a third party. Under the terms of the agreement, we made an upfront payment and will pay royalties on sales of Soliris through 2018 in accordance with the terms of the agreement.
In March 1996, we entered into a license agreement with the Medical Research Council (MRC) whereby MRC granted to us worldwide non-exclusive rights to certain patents related to the humanization and production of monoclonal antibodies. The license agreement requires us to pay MRC royalties on a quarterly basis with respect to sales of Soliris in the United States and Canada, as well as foreign sales of Soliris for product manufactured and vialed in the U.S. The royalty is payable until the expiration of the last patent covered by the license agreement, which is expected to be in 2015, except that royalties for sales in Canada will continue until January 2017. MRC may terminate the license if we file for bankruptcy or become insolvent, or if we fail to perform our obligations under the agreement and such failure is not remedied within three months after delivery of notice. Under the agreement, we agreed to (a) make royalty payments with respect to sales of licensed products, (b) promote the sale of Soliris of good marketable quality, and (c) use reasonable endeavors to meet market demand for licensed products.
Government Regulation
Drug Development and Approval in the U.S.
The preclinical studies and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, export, and marketing, among other things, of our products and product candidates, including Soliris, are subject to extensive regulation by governmental authorities in the United States, the European Union and other territories. In the United States, pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. Soliris is regulated by the FDA as a biologic. Biologics require the submission of a Biologics License Application (BLA) and approval by the FDA prior to being marketed in the United States. Manufacturers of biologics may also be subject to state regulation. Failure to comply with FDA requirements, both before and after product approval, may subject us and/or our partners, contract manufacturers, and suppliers to administrative or judicial sanctions, including FDA refusal to approve applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, fines and/or criminal prosecution.
The process for obtaining regulatory approval to market a biologic is expensive, often takes many years, and can vary substantially based on the type, complexity, and novelty of the product candidates involved. The steps required before a biologic may be approved for marketing of an indication in the United States generally include:
(1) preclinical laboratory tests and animal tests;
(2) submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may commence;
(3) adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended use;
(4) submission to the FDA of a BLA or supplemental BLA;
(5) FDA pre-approval inspection of the manufacturing sites identified in the BLA; and
(6) FDA review and approval of the BLA or supplemental BLA. Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests intended for submission to FDA must be conducted in compliance with FDA’s Good Laboratory Practice (GLP) regulations and the United States Department of Agriculture’s Animal Welfare Act. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND which must become effective before human clinical trials may be commenced. The investigational new drug (IND) will automatically become effective 30 days after receipt by the FDA, unless the FDA before that time raises concerns about the drug candidate or the conduct of the trials as outlined in the IND. The IND sponsor and the FDA must resolve any outstanding concerns
13
before clinical trials can proceed. We cannot assure you that submission of an IND will result in FDA authorization to commence clinical trials or that once commenced, other concerns will not arise. FDA may stop the clinical trials by placing them on “clinical hold” because of concerns about the safety of the product being tested, or for other reasons.
Clinical trials involve the administration of the investigational product to healthy volunteers or to patients, under the supervision of qualified principal investigators. The conduct of clinical trials is subject to extensive regulation, including compliance with the FDA’s bioresearch monitoring regulations and Good Clinical Practice (GCP) requirements, which establish standards for conducting, recording data from, and reporting the results of clinical trials, and are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected. Clinical trials must be conducted in accordance with protocols that detail the objectives of the study, the criteria for determining subject eligibility, the dosing plan, patient monitoring requirements, timely reporting of adverse events, and other elements necessary to ensure patient safety, and any efficacy criteria to be evaluated. Each protocol must be submitted to FDA as part of the IND; further, each clinical study at each clinical site must be reviewed and approved by an independent institutional review board, prior to the recruitment of subjects. The institutional review board’s role is to protect the rights and welfare of human subjects involved in clinical studies by evaluating, among other things, the potential risks and benefits to subjects, processes for obtaining informed consent, monitoring of data to ensure subject safety, and provisions to protect the subjects’ privacy. Foreign studies conducted under an IND application must meet the same requirements that apply to studies being conducted in the United States. Data from a foreign study not conducted under an IND may be submitted in support of a BLA if the study was conducted in accordance with GCP and FDA is able to validate the data.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap and different trials may be initiated with the same drug candidate within the same phase of development in similar or differing patient populations. Phase I studies may be conducted in a limited number of patients, but are usually conducted in healthy volunteer subjects. The drug is usually tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmaco-dynamics and pharmaco-kinetics. Phase II usually involves studies in a larger, but still limited patient population to evaluate preliminarily the efficacy of the drug candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible short-term adverse effects and safety risks.
Phase III trials are undertaken to gather additional information to evaluate the product’s overall risk-benefit profile, and to provide a basis for physician labeling. Phase III trials evaluate clinical efficacy of a specific endpoint and test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase I, Phase II or Phase III testing might not be completed successfully within any specific time period, if at all, with respect to any of our product candidates. Results from one trial are not necessarily predictive of results from later trials. Furthermore, the FDA, sponsor or institutional review board may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
We must register each controlled clinical trial, other than Phase I trials, on a website administered by National Institutes of Health (NIH) (http://clinicaltrials.gov). Registration must occur not later than 21 days after the first patient is enrolled, and the submission must include descriptive information (e.g., a summary in lay terms of the study design, type and desired outcome), recruitment information (e.g., target number of participants and whether healthy volunteers are accepted), location and contact information, and other administrative data (e.g., FDA identification numbers). Within one year of a trial’s completion, information about the trial including characteristics of the patient sample, primary and secondary outcomes, trial results written in lay and technical terms, and the full trial protocol must be submitted to the FDA. The results information is posted to the website unless the drug has not yet been approved, in which case the FDA posts the information shortly after approval. A BLA, BLA supplement, and certain other submissions to the FDA require certification of compliance with the FDAAA clinical trials database requirements. There are proposals to expand these registration requirements to additional studies.
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to the FDA as part of a BLA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act, as amended, the fees payable to the FDA for reviewing a BLA, as well as annual fees for commercial manufacturing establishments and for approved products, can be substantial. The BLA review fee alone can exceed $2,000 subject to certain limited deferrals, waivers and reductions that may be available. Each BLA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within 60 days following submission of the application. If the FDA finds the BLA sufficiently complete, the FDA will “file” the BLA, thus triggering a full review of the application. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission. FDA performance goals provide for action on an application
14
within 12 months of submission. The FDA, however, may not approve a drug within these established goals and its review goals are subject to change from time to time because the review process is often significantly extended by FDA requests for additional information or clarification. As part of its review, the FDA may refer the BLA to an advisory committee composed of outside experts for evaluation and a recommendation as to whether the application should be approved. Although the FDA is not bound by the recommendation of an advisory committee, the agency usually has followed such recommendations.
Further, the outcome of the review, even if generally favorable, may not be an actual approval but instead a “complete response letter” communicating the FDA's decision not to approve the application, outlining the deficiencies in the BLA, and identifying what information and/or data (including additional pre-clinical or clinical data) is required before the application can be approved. Even if such additional information and data are submitted, the FDA may decide that the BLA still does not meet the standards for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we do.
Before approving a BLA, the FDA typically will inspect the facilities at which the product is manufactured and will not approve the product unless cGMP compliance is satisfactory. The FDA may deny approval of a BLA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, and may require that additional studies be conducted following approval as a condition of the approval. FDA also may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a Risk Evaluation Mitigation Strategies (REMS), or otherwise limit the scope of any approval. A REMS may include various elements, ranging from a medication guide to limitations on who may prescribe or dispense the drug, depending on what the FDA considers necessary for the safe use of the drug. To market a product for other indicated uses, or to make certain manufacturing or other changes requires FDA review and approval of a BLA Supplement or new BLA. Further post-marketing testing and surveillance to monitor the safety or efficacy of a product may be required. In addition, new government requirements may be established that could delay or prevent regulatory approval of our product candidates under development.
In 2010, the Biologics Price Competition and Innovation Act (BPCI) was enacted, creating a statutory pathway for licensure, or approval, of biological products that are biosimilar to, and possibly interchangeable with, reference biological products licensed under the Public Health Service Act. Under the BPCI, innovator manufacturers of original reference biological products are granted 12 years of exclusive use before biosimilar versions of such products can be licensed for marketing in the United States. This means that the FDA may not approve an application for a biosimilar version of a reference biological product until 12 years after the date of approval of the reference biological product (with a potential six-month extension of exclusivity if certain pediatric studies are conducted and the results reported to FDA), although a biosimilar application may be submitted four years after the date of licensure of the reference biological product. Additionally, the BPCI establishes procedures by which the biosimilar applicant must provide information about its application and product to the reference product sponsor, and by which information about potentially relevant patents is shared and litigation over patents may proceed in advance of approval. The BPCI also provides a period of exclusivity for the first biosimilar to be determined by the FDA to be interchangeable with the reference product.
The objectives of the BPCI are conceptually similar to those of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the "Hatch-Waxman Act", which established abbreviated pathways for the approval of small molecule drug products. The FDA is currently in the process of establishing the procedures and standards it will apply in implementing the abbreviated approval pathway for biological products created by the BPCI. We anticipate that contours of the BPCI will be defined as the statute is implemented over a period of years. This likely will be accomplished by a variety of means, including FDA issuance of guidance documents, proposed regulations, and decisions in the course of considering specific applications. FDA has released guidance documents interpreting the BPCI in each of the last three years. These guidance documents, among other things, elaborate on the definition of a biosimilar as a biological product that is highly similar to an already approved biological product, notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biosimilar and the approved biological product in terms of the safety, purity, and potency. Under this proposed approval pathway, biological products are approved based on demonstrating they are biosimilar to, or interchangeable with, a biological product that is already approved by the FDA, which is called a reference product. The approval of a biologic product biosimilar to one of our products could have a material impact on our business because it may be significantly less costly to bring to market and may be priced significantly lower than our products.
Both before and after the FDA approves a product, the manufacturer and the holder or holders of the BLA for the product are subject to comprehensive regulatory oversight. If ongoing regulatory requirements are not satisfied or if safety problems occur after the product reaches the market, the FDA may at any time withdraw its product approval or take actions that would
15
suspend marketing. For example, quality control and manufacturing procedures must conform, on an ongoing basis, to cGMP requirements, and the FDA periodically subjects manufacturing facilities to unannounced inspections to assess compliance with cGMP. Failure to comply with applicable cGMP requirements and other conditions of product approval may lead the FDA to seek sanctions, including fines, recalls, civil penalties, injunctions, suspension of manufacturing operations, operating restrictions, withdrawal of FDA approval, seizure or recall of products, and criminal prosecution. Accordingly, manufacturers must continue to spend time, money and effort to maintain cGMP compliance.
The FDA and other federal regulatory agencies also closely regulate the promotion of drugs and biologics through, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities, and promotional activities involving the Internet and social media. A product cannot be commercially promoted before it is approved. After approval, product promotion can include only those claims relating to safety and effectiveness that are consistent with the labeling approved by the FDA. Healthcare providers are permitted to prescribe drugs and biologics for “off-label” uses - that is, uses not approved by the FDA and therefore not described in the product's labeling - because the FDA does not regulate the practice of medicine. However, FDA regulations impose stringent restrictions on manufacturers' communications regarding off-label uses. Broadly speaking, a manufacturer may not promote a drug or biologic for off-label use, but may engage in non-promotional, balanced communication regarding off-label use under certain conditions. Failure to comply with applicable FDA requirements and restrictions in this area may subject a company to adverse publicity and enforcement action by the FDA, the Department of Justice, or the Office of the Inspector General of the Department of Health and Human Services, as well as state authorities. Noncompliance could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug or biologic products.
Orphan Drug Designation in the United States, the European Union and Other Foreign Jurisdictions
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs and biological products intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting a BLA or supplemental BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are publicly disclosed by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for that drug or biologic for the indication for which it has such designation, the product is entitled to an orphan exclusivity period, in which the FDA may not approve any other applications to market the same drug or biologic for the same indication for seven years, except in limited circumstances, such as where the sponsor of different version of the product is able to demonstrate that its product is clinically superior to the approved orphan drug product. This exclusivity does not prevent a competitor from obtaining approval to market a different product that treats the same disease or condition or the same product to treat a different disease or condition. The FDA can revoke a product’s orphan drug exclusivity under certain circumstances, including when the holder of the approved orphan drug application is unable to assure the availability of sufficient quantities of the drug to meet patient needs. A sponsor of a product application that has received an orphan drug designation is also granted tax incentives for clinical research undertaken to support the application. In addition, the FDA will typically coordinate with the sponsor on research study design for an orphan drug and may exercise its discretion to grant marketing approval on the basis of more limited product safety and efficacy data than would ordinarily be required.
Medicinal products: (a) that are used to treat life-threatening or chronically debilitating conditions; (b) that affect no more than five in 10,000 people in the European Union; (c) that, for economic reasons, would be unlikely to be developed without incentives; and (d) where no satisfactory method of diagnosis, prevention or treatment of the condition concerned exists, or, if such a method exists, the medicinal product must be of significant benefit to those affected by the condition, may be granted an orphan designation in the European Union. The application for orphan designation is submitted to the EMA before an application is made for marketing authorization. Once authorized, orphan medicinal products are entitled to ten years of market exclusivity. During this ten year period, with a limited number of exceptions, neither the competent authorities of the European Union member states, the EMA, or the European Commission are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. However, marketing authorization may be granted to a similar medicinal product with the same orphan indication during the ten year period with the consent of the marketing authorization holder for the original orphan medicinal product or if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if this product is safer, more effective or otherwise clinically superior to the original orphan medicinal product. The period of market exclusivity may, in addition, be reduced to six years if it can be demonstrated on the
16
basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity.
Soliris has received orphan drug designation for the treatment of PNH and aHUS in the United States, the European Union, and in several other territories, for the prevention of delayed graft function in renal transplant patients in the United States, for the treatment of patients with myasthenia gravis in the United States, Japan, and the European Union, and for prevention of graft rejection and delayed graft rejection following solid organ transplantation in the European Union. Orphan drug designation provides certain regulatory and filing fee advantages, including market exclusivity, except in limited circumstances, for several years after approval. In 2008, asfotase alfa received orphan drug designation for the treatment of patients with hypophosphatasia in the United States and the European Union, and in Japan in November 2014.
Breakthrough Designation in the United States
With the passage of the Food and Drug Administration Safety Act (FDASIA) of 2012, Congress created the Breakthrough Therapy designation program. FDA may grant Breakthrough Therapy status to a drug intended for the treatment of a serious condition when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint over existing therapies. The Breakthrough Therapy designation, which may be requested by a sponsor when filing or amending an IND, is intended to facilitate and expedite the development and FDA review of a product candidate. Specifically, the Breakthrough Therapy designation may entitle the sponsor to more frequent meetings with FDA during drug development, intensive guidance on clinical trial design, and expedited FDA review by a cross-disciplinary team comprised of senior managers. The designation does not guarantee a faster development or review time as compared to other drugs, however, nor does it assure that the drug will obtain ultimate marketing approval by the FDA. Once granted, the FDA may withdraw this designation at any time. We have received Breakthrough Therapy designations for asfotase alfa, intended to treat hypophosphatasia in perinatal-, infant-, and juvenile-onset patients, and cyclic Pyranopterin Monophosphate, intended to treat Molybdenum Cofactor Deficiency Type A. Because the Breakthrough Therapy designation program is relatively new, it is difficult for us to predict the impact that these designations will have on the development and FDA review of our products.
Foreign Regulation of Drug Development and Approval
In addition to regulations in the United States, we are subject to a variety of foreign regulatory requirements governing human clinical trials, marketing approval, and post-marketing regulation for drugs. The foreign regulatory approval process includes all of the risks associated with FDA approval set forth above, as well as additional country-specific regulations. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. The approval process varies from country to country, can involve additional testing beyond that required by FDA, and may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country.
Under the European Union regulatory system, we may submit applications for marketing authorizations either under a centralized, decentralized, or mutual recognition marketing authorization procedure. The centralized procedure provides for the grant of a single marketing authorization for a medicinal product by the European Commission on the basis of an opinion by the EMA. A centralized marketing authorization is valid for all European Union member states and three of the four EFTA States (Iceland, Liechtenstein and Norway). The decentralized procedure and the mutual recognition procedure apply between European Union member states. The decentralized marketing authorization procedure involves the submission of an application for marketing authorization to the competent authority of all European Union member states in which the product is to be marketed. One national competent authority, selected by the applicant, assesses the application for marketing authorization. The competent authorities of the other European Union member states are subsequently required to grant marketing authorization for their territory on the basis of this assessment, except where grounds of potential serious risk to public health require this authorization to be refused. The mutual recognition procedure provides for mutual recognition of marketing authorizations delivered by the national competent authorities of European Union member states by the approval authorities of other European Union member states. The holder of a national marketing authorization may submit an application to the competent authority of a European Union member state requesting that this authority recognize the marketing authorization delivered by the competent authority of another European Union member state.
Similarly to the U.S., both marketing authorization holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA and the competent authorities of the individual European Union member states both before and after grant of the manufacturing and marketing authorizations. This includes European Union cGMP rules,
17
which govern quality control of the manufacturing process and require documentation policies and procedures. We and our third party manufacturers are required to ensure that all of our processes, methods, and equipment are compliant with cGMP.
Failure by us or by any of our third party partners, including suppliers, manufacturers, and distributors to comply with European Union laws and the related national laws of individual European Union member states governing the conduct of clinical trials, manufacturing approval, marketing authorization of medicinal products and marketing of such products, both before and after grant of marketing authorization, may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal, or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing, or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
The European Union has had an established regulatory pathway for biosimilars since 2005 and has approved several biosimilar products. The approval of a biosimilar of one of our products marketed in the European Union could have a material impact on our business. The biosimilar may be significantly less costly to bring to market and may be priced significantly lower than our products.
Pharmaceutical Pricing and Reimbursement
Sales of pharmaceutical products depend in significant part on the extent of coverage and reimbursement from government programs, including Medicare and Medicaid in the United States, and other third party payers. Third party payers are sensitive to the cost of drugs and are increasingly seeking to implement cost containment measures to control, restrict access to, or influence the purchase of drugs, biologicals, and other health care products and services. Governments may regulate reimbursement, pricing, and coverage of products in order to control costs or to affect levels of use of certain products. Private health insurance plans may restrict coverage of some products, such as by using payer formularies under which only selected drugs are covered, variable co-payments that make drugs that are not preferred by the payer more expensive for patients, and by employing utilization management controls, such as requirements for prior authorization or prior failure on another type of treatment. Payers may especially impose these obstacles to coverage for higher-priced drugs such as Soliris. Consequently, Soliris may be subject to payer-driven restrictions, rendering patients responsible for a higher percentage of the total cost of drugs in the outpatient setting. This can lower the demand for our products if the increased patient cost-sharing obligations are more than they can afford.
Medicare is a U.S. federal government insurance program that covers individuals aged 65 years or older, as well as individuals of any age with certain disabilities, and individuals with End-Stage Renal Disease. The primary Medicare programs that may affect reimbursement for Soliris are Medicare Part B, which covers physician services and outpatient care, and Medicare Part D, which provides a voluntary outpatient prescription drug benefit. Medicare Part B provides limited coverage of certain outpatient drugs and biologicals that are reasonable and necessary for diagnosis or treatment of an illness or injury. Under Part B, reimbursement for most drugs is based on a fixed percentage above the applicable product’s average sales price (ASP). Manufacturers calculate ASP based on a statutory formula and must report ASP information to the Centers for Medicare and Medicaid Services (CMS), the federal agency that administers Medicare and the Medicaid Drug Rebate Program, on a quarterly basis. For 2015, the reimbursement rate for drugs and biologicals in both the hospital outpatient department setting and the physician office setting is ASP + 4.3%. The rate for the physician clinic setting is set by statute, but CMS has the authority to adjust the rate for the hospital outpatient setting on an annual basis. This reimbursement rate may decrease in the future. In both settings, the amount of reimbursement is updated quarterly based on the manufacturer’s submission of new ASP information.
Medicare Part D coverage is available through private plans, and the list of prescription drugs covered by Part D plans varies by plan. However, drug lists maintained by individual plans are required by statute to cover certain therapeutic categories and classes of drugs or biologicals and to have at least two drugs in each unique therapeutic category or class, with certain exceptions.
Medicare Part A covers inpatient hospital benefits. Hospitals typically receive a single payment for an inpatient stay depending on the Medicare Severity Diagnosis Related Group (MS-DRG) to which the inpatient stay is assigned. The MS-DRG for a hospital inpatient stay varies based on the patient’s condition. Hospitals generally do not receive separate payment for drugs and biologicals administered to patients during an inpatient hospital stay. As a result, hospitals may not have a financial incentive to utilize Soliris for inpatients.
Beginning April 1, 2013, the Budget Control Act of 2011, Pub. L. No. 112-25, as amended by the American Taxpayer Relief Act of 2012, Pub. L. 112-240, required Medicare payments for all items and services, including drugs and biologicals, to
18
be reduced by 2% under the sequestration (i.e., automatic spending reductions). This 2% reduction was extended to 2023 by the Bipartisan Budget Act of 2013, Pub. L. No. 113-67. This 2% reduction in Medicare payments affects all Parts of the Medicare program and could impact sales of Soliris.
Medicaid is a government health insurance program for low-income children, families, pregnant women, and people with disabilities. It is jointly funded by the federal and state governments, and it is administered by individual states within parameters established by the federal government. Coverage and reimbursement for drugs and biologicals thus varies by state. Drugs and biologicals may be covered under the medical or pharmacy benefit. State Medicaid programs may impose utilization management controls, such as prior authorization, step therapy, or quantity limits on drugs and biologicals. Medicaid also includes the Drug Rebate Program, under which we are required to pay a rebate to each state Medicaid program for quantities of Soliris that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for Soliris under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS. These data include the average manufacturer price and the best price for Soliris. As further described below under “U.S. Healthcare Reform and Other U.S. Healthcare Laws,” the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the PPACA), made significant changes to the Medicaid Drug Rebate Program that could negatively impact our results of operations.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. Changes to the definition of average manufacturer price and the Medicaid rebate amount under PPACA and CMS’s issuance of final regulations implementing those changes also could affect our 340B ceiling price calculation for Soliris and could negatively impact our results of operations. As descried below under “U.S. Healthcare Reform and Other U.S. Healthcare Laws,” PPACA expanded the 340B program to include additional types of covered entities but exempts “orphan drugs”-those designated under section 526 of the FDCA, such as Soliris-from the ceiling price requirements for these newly-eligible entities.
In order to be eligible to have our products paid for with federal funds under the Medicaid program and purchased by certain federal agencies, we participate in the Department of Veterans Affairs Federal Supply Schedule, or FSS, pricing program, established by Section 603 of the Veterans Health Care Act of 1992. Under this program, we are obligated to make our product available for procurement on an FSS contract and charge a price to four federal agencies, Department of Veterans Affairs, Department of Defense, Public Health Service and Coast Guard that is no higher than the statutory Federal Ceiling Price, or FCP. The FCP is based on the non-federal average manufacturer price, or Non-FAMP, which we calculate and report to the Department of Veterans Affairs on a quarterly and annual basis. We also participate in the Tricare Retail Pharmacy program, established by Section 703 of the National Defense Authorization Act for FY 2008 and related regulations, under which we pay quarterly rebates on utilization of innovator products that are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries. The rebates are calculated as the difference between Annual Non-FAMP and FCP.
Payors also are increasingly considering new metrics as the basis for reimbursement rates, such as ASP, average manufacturer price, and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS has begun posting drafts of this retail survey price information on at least a monthly basis in the form of draft National Average Drug Acquisition Cost, or NADAC, files, which reflect retail community pharmacy invoice costs, and National Average Retail Price, or NARP, files, which reflect retail community pharmacy prices to consumers. In July 2013, CMS suspended the publication of draft NARP data, pending funding decisions. In November 2013, CMS moved to publishing final rather than draft NADAC data and has since made updated NADAC data publicly available on a weekly basis. Therefore, it may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payors to cover Soliris.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, in the European Union the sole legal instrument at the European Union level governing the pricing and reimbursement of medicinal products is Council Directive 89/105/EEC (the Price Transparency Directive). The aim of the Price Transparency Directive is to ensure that pricing and reimbursement mechanisms established in European Union member states are transparent and objective, do not
19
hinder the free movement and trade of medicinal products in the European Union and do not hinder, prevent or distort competition on the market. The Price Transparency Directive does not, however, provide any guidance concerning the specific criteria on the basis of which pricing and reimbursement decisions are to be made in individual European Union member states. Neither does it have any direct consequence for pricing or levels of reimbursement in individual European Union member states. The national authorities of the individual European Union member states are free to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices and/or reimbursement of medicinal products for human use. Individual European Union member states adopt policies according to which a specific price or level of reimbursement is approved for the medicinal product. Other European Union member states adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market, including volume-based arrangements and reference pricing mechanisms.
Health Technology Assessment (HTA) of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some European Union member states. These countries include the United Kingdom, France, Germany and Sweden. The HTA process in the European Union member states is governed by the national laws of these countries. HTA is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of the use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well as their potential implications for the healthcare system. Those elements of medicinal products are compared with other treatment options available on the market.
The outcome of HTA will often influence the pricing and reimbursement status for specific medicinal products within individual European Union member states. The extent to which pricing and reimbursement decisions are influenced by the HTA of a specific medicinal product vary between the European Union member states.
In 2011, Directive 2011/24/EU was adopted at the European Union level. This Directive concerns the application of patients' rights in cross-border healthcare. The Directive is intended to establish rules for facilitating access to safe and high-quality cross-border healthcare in the European Union. It also provides for the establishment of a voluntary network of national authorities or bodies responsible for HTA in the individual European Union member states. The purpose of the network is to facilitate and support the exchange of scientific information concerning HTAs. This could lead to harmonization of the criteria taken into account in the conduct of HTA and their impact on pricing and reimbursement decisions between European Union member states.
On a continuous basis, we engage with appropriate authorities on the operational, reimbursement, price approval and funding processes that are separately required in each country.
Fraud and Abuse
Pharmaceutical companies participating in federal healthcare programs like Medicare or Medicaid are subject to various U.S. federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback and false claims laws. Violations of U.S. federal and state fraud and abuse laws may be punishable by criminal, civil and administrative sanctions, including fines, damages, civil monetary penalties and exclusion from federal healthcare programs (including Medicare and Medicaid). Applicable U.S. statutes, include, but are not limited to, the following:
• | The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully soliciting, offering, receiving, or paying any remuneration, directly or indirectly, in cash or in kind, to induce or reward purchasing, ordering or arranging for or recommending the purchase or order of any item or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. In addition, PPACA amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. A conviction for violation of the Anti-kickback Statute requires mandatory exclusion from participation in federal health care programs. |
• | The federal civil False Claims Act (“FCA”) prohibits, among other things, knowingly presenting, or causing to be presented claims for payment of government funds that are false or fraudulent, or knowingly making, using or causing to be made or used a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. This statute permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. Government enforcement |
20
agencies and private whistleblowers have asserted liability under the FCA for, among others, claims submitted involving inadequate or medically unnecessary care or items or services, kickbacks, promotion of off-label uses, and misreporting of drug prices to federal agencies.
• | The federal False Statements Statute prohibits knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry, in connection with the delivery of or payment for healthcare benefits, items, or services. |
• | The federal Civil Monetary Penalties Law, which authorizes the imposition of substantial civil monetary penalties against an entity, such as a pharmaceutical manufacturer, that engages in activities including, among others (1) knowingly presenting, or causing to be presented, a claim for services not provided as claimed or that is otherwise false or fraudulent in any way; (2) arranging for or contracting with an individual or entity that is excluded from participation in federal health care programs to provide items or services reimbursable by a federal health care program; (3) violations of the federal Anti-Kickback Statute; or (4) failing to report and return a known overpayment. |
• | Analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payers, including private insurers. Some of these state laws may be broader in scope than their federal analogues, such as state false claims laws that apply where a claim is submitted to any third-party payer, regardless of whether the payer is a private health insurer or a government healthcare program, state laws that prohibit certain marketing-related activities including the provision of gifts, meals or other items to certain health care providers, and state laws that require pharmaceutical companies to implement compliance programs or codes of conduct governing their sales and marketing activities. |
Federal and state authorities are continuing to devote significant attention and resources to enforcement of fraud and abuse laws within the pharmaceutical industry, and private individuals have been active in alleging violations of the law and bringing suits on behalf of the government under the FCA. These laws are broad in scope and there may not be regulations, guidance, or court decisions that definitively interpret these laws and apply them to particular industry practices. In addition, these laws and their interpretations are subject to change.
U.S. Healthcare Reform and Other U.S. Healthcare Laws
PPACA was adopted in the United States in March 2010. This law substantially changes the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that are expected to impact our business and operations. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under the health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, and fraud and abuse and enforcement. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
PPACA contains several provisions that have or could potentially impact our business. PPACA made significant changes to the Medicaid Drug Rebate Program. Effective March 23, 2010, rebate liability expanded from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well. With regard to the amount of the rebates owed, PPACA increased the minimum Medicaid rebate from 15.1% to 23.1% of the average manufacturer price for most innovator products; changed the calculation of the rebate for certain innovator products that qualify as line extensions of existing drugs; and capped the total rebate amount for innovator drugs at 100% of the average manufacturer price. In addition, PPACA and subsequent legislation changed the definition of average manufacturer price. A final regulation regarding these changes to the Medicaid Drug Rebate Program currently is expected in 2015. Finally, PPACA requires pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government beginning in 2011. Each individual pharmaceutical manufacturer pays a prorated share of the branded prescription drug fee of $3.0 billion in 2014 (and set to increase in ensuing years), based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. Sales of “orphan drugs” those designated under section 526 of the FDCA, such as Soliris, are excluded from this fee to the extent that no non-orphan indications have been approved for the orphan drug.
Additional provisions of PPACA, some of which became effective in 2011, may negatively affect manufacturer's revenues in the future. For example, as part of PPACA’s provisions closing a coverage gap that currently exists in the Medicare Part D
21
prescription drug program (commonly known as the “donut hole”), manufacturers are required to provide a 50% discount on branded prescription drugs dispensed to beneficiaries within this donut hole.
PPACA also expanded the Public Health Service’s 340B drug pricing discount program. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. PPACA expanded the 340B program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by PPACA. PPACA exempts “orphan drugs”-those designated under section 526 of the FDCA, such as Soliris-from the ceiling price requirements for these newly-eligible entities. On July 21, 2014, the Health Resources and Services Administration, or HRSA, which administers the 340B program, issued an interpretive rule to implement the orphan drug exception which interprets the orphan drug exception narrowly. It exempts orphan drugs from the ceiling price requirements for the newly-eligible entities only when the orphan drug is used for its orphan indication. The newly-eligible entities are entitled to purchase orphan drugs at the ceiling price when the orphan drug is not used for its orphan indication. A manufacturer trade group has filed a lawsuit challenging the interpretive rule as inconsistent with the statutory language. That challenge remains ongoing. The uncertainty regarding how the statutory orphan drug exception will be applied will increase the complexity of compliance, will make compliance more time-consuming, and could negatively impact our results of operations. If HRSA’s narrow interpretation of the scope of the orphan drug exemption prevails, it could potentially negatively impact the price we are paid for Soliris by certain entities for some uses and increase the complexity of compliance with the 340B program. Additionally, PPACA enacted the federal “Physician Payment Sunshine Act, being implemented as the Open Payments program, that requires pharmaceutical manufacturers, among others, with products for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program, to track and report annually to the federal government (for disclosure to the public) certain payments and other transfers of value made to physicians and teaching hospitals. State laws may also require disclosure of pharmaceutical pricing information and marketing expenditures and impose penalties for failures to disclose. Many of these laws and regulations contain ambiguous requirements that government officials have not yet clarified. Given the lack of clarity in the laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent federal and state laws and regulations.
Finally, numerous federal and state laws, including state security breach notification laws, state health information privacy laws, and federal and state consumer protection laws govern the collection, use, and disclosure of personal information. In addition, most healthcare providers who prescribe and dispense our products and research institutions with whom we collaborate for our sponsored clinical trials are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and its implementing regulations. Although we are neither a “covered entity” nor a “business associate” under HIPAA, and these privacy and security requirements do not apply to us, the regulations may affect our interactions with health care providers, health plans, and research institutions from whom we obtain patient health information. Further, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting the violation of HIPAA.
Other Regulations
We are also subject to the United States Foreign Corrupt Practices Act (FCPA), the U.K. Bribery Act (U.K. Bribery Act), and other anti-corruption laws and regulations pertaining to our financial relationships with foreign government officials. The FCPA prohibits U.S. companies and their representatives from paying, offering to pay, promising, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate to obtain or retain business or to otherwise seek favorable treatment. In many countries in which we operate or sell Soliris, the health care professionals with whom we interact may be deemed to be foreign government officials for purposes of the FCPA. The U.K. Bribery Act, which applies to any company incorporated or doing business in the UK, prohibits giving, offering, or promising bribes in the public and private sectors, bribing a foreign public official or private person, and failing to have adequate procedures to prevent bribery amongst employees and other agents. Penalties under the Bribery Act include potentially unlimited fines for companies and criminal sanctions for corporate officers under certain circumstances. Liability in relation to breaches of the Bribery Act is strict. This means that it is not necessary to demonstrate elements of a corrupt state of mind. However, a defense of having in place adequate procedures designed to prevent bribery is available.
Our present and future business has been and will continue to be subject to various other laws and regulations. Laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances,
22
including radioactive compounds, used in connection with our research work are or may be applicable to our activities. We cannot predict the impact of government regulation, which may result from future legislation or administrative action, on our business.
Competition
There are currently no approved drugs other than Soliris for the treatment of PNH and aHUS. However, many companies, including major pharmaceutical and chemical companies as well as specialized biotechnology companies, are engaged in activities similar to our activities. Universities, governmental agencies and other public and private research organizations also conduct research and may market commercial products on their own or through joint ventures. Some of these entities may have:
• | substantially greater financial and other resources; |
• | larger research and development staffs; |
• | lower labor costs; and/or |
• | more extensive marketing and manufacturing organizations. |
Many of these companies and organizations have significant experience in preclinical testing, human clinical trials, product manufacturing, marketing, sales and distribution and other regulatory approval and commercial procedures. They may also have a greater number of significant patents and greater legal resources to seek remedies for cases of alleged infringement of their patents by us to block, delay or compromise our own drug development process.
We compete with large pharmaceutical companies that produce and market synthetic compounds and with specialized biotechnology firms in the United States, Europe and in other countries and regions, as well as a growing number of large pharmaceutical companies that are developing biotechnology products. A number of biotechnology and pharmaceutical companies are developing new products for the treatment of the same diseases being targeted by us; in some instances, these products have already entered clinical trials or are already being marketed. Other companies are engaged in research and development based on complement proteins.
Several companies have either publicly announced their intentions to develop drugs which target the inflammatory effects of complement in the immune system or have had programs to develop complement inhibitor therapies. We believe that Soliris differs substantially from compounds of our potential competitors because Soliris has demonstrated to be safe and effective in two clinical indications by regulators in many jurisdictions around the world.
Employees
As of December 31, 2014, we had 2,273 full-time, world-wide employees, of which 914 were engaged in research, product development, manufacturing, and clinical development, 880 in sales and marketing, and 479 in administration, human resources, information technology and finance. Our U.S. employees are not represented by any collective bargaining unit, and we regard the relationships with all our employees as satisfactory.
23
EXECUTIVE OFFICERS OF THE COMPANY
The executive officers of the Company and their respective ages and positions as of February 3, 2015 are as follows:
Name | Age | Position with Alexion | |
Leonard Bell, M.D. | 56 | Chairman and Chief Executive Officer | |
David L. Hallal | 48 | Chief Operating Officer and CEO-elect | |
Clare Carmichael | 55 | Executive Vice President and Chief Human Resources Officer | |
Martin Mackay | 58 | Executive Vice President and Global Head of Research and Development | |
John B. Moriarty, J.D. | 47 | Executive Vice President and General Counsel | |
Julie O'Neill | 48 | Executive Vice President of Global Operations | |
Vikas Sinha, M.B.A., C.A., C.P.A. | 51 | Executive Vice President and Chief Financial Officer | |
Saqib Islam | 45 | Senior Vice President and Chief Strategy and Portfolio Officer | |
Edward Miller | 50 | Senior Vice President and Global Chief Compliance Officer | |
Dominique Monnet | 54 | Senior Vice President and Chief Marketing Officer | |
Carsten Thiel, Ph.D. | 51 | Senior Vice President EMEA and Asia Pacific | |
Leonard Bell, M.D. is the principal founder of Alexion and has been a director of Alexion since February 1992 and the Company’s Chief Executive Officer since January 1992 and Chairman since October 2014. From 1991 to 1992, Dr. Bell was an Assistant Professor of Medicine and Pathology and co-Director of the program in Vascular Biology at the Yale University School of Medicine. From 1990 to 1992, Dr. Bell was an attending physician at the Yale-New Haven Hospital and an Assistant Professor in the Department of Internal Medicine at the Yale University School of Medicine. Dr. Bell was a recipient of the Physician Scientist Award from the National Institutes of Health and Grant-in-Aid from the American Heart Association as well as various honors and awards from academic and professional organizations. His work has resulted in more than 20 scientific publications and 9 patent applications. Dr. Bell received his A.B. from Brown University and M.D. from Yale University School of Medicine. Dr. Bell is currently an Adjunct Assistant Professor of Medicine and Pathology at the Yale University School of Medicine.
David L. Hallal has been with Alexion since June 2006 and has served as Chief Operating Officer since September 2014. On January 29, 2015, Alexion announced that Mr. Hallal was appointed Chief Executive Officer (CEO) and Mr. Hallal will become CEO of Alexion effective April 1, 2015. Mr. Hallal has also been a member of the Board of Directors since September 2014. Since joining Alexion, Mr. Hallal has served in senior commercial positions, including Senior Vice President, US Commercial Operations from June 2006 until November 2008, Senior Vice President, Commercial Operations Americas from November 2008 to May 2010, Senior Vice President, Global Commercial Operations from May 2010 until October 2012 and then Executive Vice President and Chief Commercial Officer from October 2012 to September 2014. Prior to joining Alexion, Mr. Hallal served as Vice President, Sales at OSI Eyetech from April 2004 until June 2006, where he led the U.S. launch of a first-in-class anti-VEGF therapy for age-related macular degeneration. Prior to OSI Eyetech, from 1992 until 2004, Mr. Hallal held various sales and marketing leadership positions at Amgen and Biogen Idec, where he was involved in multiple product launches in the areas of hematology, oncology, nephrology and immunology. Mr. Hallal received a B.A. in Psychology from the University of New Hampshire.
Clare Carmichael has been with Alexion since August 2011 and has served as Executive Vice President and Chief Human Resources Officer since September 2014. From August 2011 to September 2014, Ms. Carmichael served as Senior Vice President and Chief Human Resources Officer. From August 2008 to March 2011, Ms. Carmichael served as Senior Vice President, Global Human Resources at Watson Pharmaceuticals, Inc., where she established and executed global HR strategies. From December 2005 to August 2008, Ms. Carmichael held various human resources positions of increasing responsibility at Schering-Plough Corporation, including Vice President of Global Human Resources at the Schering-Plough Research Institute. From December 2003 to December 2005, Ms. Carmichael was Vice President of Human Resources at Eyetech Pharmaceuticals, Inc. Prior to Eyetech, she held various positions of increasing responsibility in human resources at Pharmacia Corporation. Ms. Carmichael received a B.A. in Psychology from Rider University.
Martin Mackay has been Executive Vice President, Global Head of Research & Development since joining Alexion in May 2013. Prior to joining Alexion, Dr. Mackay served as President, Research and Development at AstraZeneca from June 2010 to February 2012, where he led all R&D functions worldwide, including discovery research, clinical development, regulatory affairs and key related R&D functions. From April 1995 to May 2010, he held various positions of increasing responsibility at Pfizer,
24
including President, Head of Pfizer Pharmatherapeutics, R&D, where he oversaw all aspects of small molecule discovery and development across multiple therapeutic areas. Dr. Mackay has also worked in the CIBA organization, now Novartis, and held positions within academia. Dr. Mackay received a Microbiology First Class Honors Degree from Heriot-Watt University, Scotland, and a Ph.D. in Molecular Genetics from the University of Edinburgh, Scotland.
John B. Moriarty, J.D. has been with Alexion since December 2012 and has served as Executive Vice President and General Counsel since September 2014. From December 2012 to September 2014, Mr. Moriarty served as Senior Vice President and General Counsel. From December 2010 to December 2012, Mr. Moriarty served as General Counsel and Chief Legal Officer at Elan Corporation plc, an Irish public limited company traded on the New York and Irish Stock Exchanges, and also served as a member of Elan's Executive Management team. Prior to assuming the role of General Counsel, Mr. Moriarty served as Senior Vice President of Law, Litigation and Commercial Operations at Elan from December 2008 to December 2010. From 2002 to 2008, Mr. Moriarty held various positions with Amgen, Inc., including Executive Director and Associate General Counsel, Global Commercial Operations - Amgen Oncology and Senior Counsel, Complex Litigation, Products Liability and Government Investigations. Between 1994 and 2002, Mr. Moriarty served in various capacities in private practice focused on healthcare and as a healthcare fraud prosecutor in the U.S. Attorney's Office and the Virginia Attorney General's Office. Mr. Moriarty received his J.D., cum laude, from the University of Georgia School of Law and his B.A., with distinction, from the University of Virginia
Julie O'Neill has been with Alexion since February 2014 and has served as Executive Vice President of Global Operations since January 2015. From January 2014 to January 2015, Ms. O'Neill was Senior Vice President Global Manufacturing Operations and General Manager of Alexion Pharma International Trading. Prior to joining Alexion, Ms. O'Neill served in various leadership positions at Gilead Sciences from February 1997 to February 2014 including Vice President of Operations and General Manager of Ireland from 2011 to 2014. Prior to Gilead Sciences, Ms. O'Neill held leadership positions in operations, manufacturing and quality functions at Burnil Pharmacies and Helsinn Birex Pharmaceuticals. She is the Chairperson for the National Standards Authority of Ireland and is a member of the Governing Body of University College Cork. Ms. O'Neill received a Bachelor's of Science in Pharmacy from University of Dublin, Trinity College and a Masters of Business Administration from University College Dublin (Smurfit School of Business).
Vikas Sinha, M.B.A., C.A., C.P.A. has been with Alexion since September 2005 and has served as Alexion's Executive Vice President and Chief Financial Officer since October 2012. From September 2005 to October 2012, Mr. Sinha was Senior Vice President and Chief Financial Officer. Prior to joining Alexion, Mr. Sinha held various positions with Bayer AG in the United States, Japan, Germany, and Canada, including Vice President and Chief Financial Officer of Bayer Pharmaceuticals Corporation, USA, Vice President and Chief Financial Officer of Bayer Yakuhin Ltd., in Japan, and Manager, Mergers and Acquisitions with Bayer AG in Germany. He also was a member of the Pharmaceutical Management Committee for North America. Prior to Bayer, Mr. Sinha held several positions of increasing responsibilities with ANZ Bank and Citibank in South Asia. Mr. Sinha holds a Masters of Business Administration from the Asian Institute of Management which included an exchange program with the University of Western Ontario (Richard Ivey School of Business). He is also a qualified Chartered Accountant from the Institute of Chartered Accountants of India and a Certified Public Accountant in the United States.
Saqib Islam has been Senior Vice President, Chief Strategy and Portfolio Officer since joining Alexion in April 2013. Prior to joining Alexion, Mr. Islam worked for 18 years in international business management with a focus on business development, strategic decision-making and planning, and capital markets, and most recently as Managing Director, Head of Healthcare and Diversified Industrials Capital Markets at Credit Suisse Securities from November 2009 until April 2013. Prior to Credit Suisse, Mr. Islam held various positions of increasing responsibility in the investment banking divisions of Merrill Lynch and Morgan Stanley and provided strategic analysis and advice to client firms across diverse industry segments for The Boston Consulting Group. Mr. Islam received a Bachelor of Commerce from McGill University, where he was a Faculty and University Scholar, and a J.D. from Columbia Law School, where he was a Harlan Fiske Stone Scholar.
Edward Miller has been Senior Vice President and Global Chief Compliance Officer since joining Alexion in September 2014. Prior to joining Alexion, Mr. Miller served in various compliance and legal leadership positions at Boehringer Ingelheim from 2000 to August 2014, including Vice President, Associate General Counsel, Global Head of Litigation and Government Investigations; Vice President and Acting Global Compliance Officer and Vice President, Chief Compliance Officer and Head of Litigation. Prior to Boehringer Ingelheim, Mr. Miller was a Senior Trial Attorney at the U. S. Department of Justice in Washington, D.C. Mr. Miller received a Bachelors Degree from Princeton University and his J.D. from Rutgers University School of Law.
Dominique Monnet has been Senior Vice President and Chief Marketing Officer since joining Alexion in May 2014. Prior to joining Alexion, Mr. Monnet served in various marketing leadership positions at Amgen, Inc from 2002 to 2013, including Vice President and General Manager, Inflammation Business Unit, Vice President and Head of Global Marketing and Commercial Development and Vice President International Marketing and Business Operations. Prior to Amgen, Mr. Monnet
25
held positions of increasing responsibility at Schering-Plough, including General Manager for the company’s UK and Ireland entity. Mr. Monnet earned his undergraduate business degree from EDHEC Business School in Lille, France, and his MBA from INSEAD in Fontainebleau, France.
Carsten Thiel, Ph.D. has been with Alexion since September 2014 and has served as Senior Vice President EMEA and Asia Pacific since January 2015. From September 2014 to January 2015, Mr. Thiel was Senior Vice President EMEA and Australasia-Canada. Prior to joining Alexion, Mr. Thiel served in various senior leadership positions at Amgen from 2002 to 2014, including Vice President, Head of Europe, General Manager, Germany, General Manager, CEE and Head of the Oncology Franchise in Europe. Prior to Amgen, Mr. Thiel held several sales and marketing leadership roles across Europe at Roche. Mr. Thiel has a Ph.D. in Molecular Biology and Biochemistry from the Max Planck Institute, Germany, and a Master’s Degree in Biochemistry from the University of Marburg, Germany.
Available Information
Our internet website address is http://www.alexion.com. Through our website, we make available, free of charge, our Annual Reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, any amendments to those reports, proxy and registration statements, and all of our insider Section 16 reports, as soon as reasonably practicable after such material is electronically filed with, or furnished to, the Securities and Exchange Commission (SEC). These SEC reports can be accessed through the “Investors” section of our website. The information found on our website is not part of this or any other report we file with, or furnish to, the SEC. Paper copies of our SEC reports are available free of charge upon request in writing to Investor Relations, Alexion Pharmaceuticals, Inc., 352 Knotter Drive, Cheshire, CT 06410. In addition, any document we file may be inspected, without charge, at the SEC’s public reference room at 100 F Street NE, Washington, DC 20549, or at the SEC’s internet address at http://www.sec.gov. (This website address is not intended to function as a hyperlink, and the information contained in the SEC’s website is not intended to be a part of this filing). Information related to the operation of the SEC’s public reference room may be obtained by calling the SEC at 800-SEC-0330 (800-732-0330).
26
Item 1A. | Risk Factors. |
(amounts in thousands, except percentages)
You should carefully consider the following risk factors before you decide to invest in Alexion and our business because these risk factors may have a significant impact on our business, operating results, financial condition, and cash flows. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations. If any of the following risks actually occurs, our business, financial condition and results of operations could be materially and adversely affected.
Risks Related to Our Products
We depend heavily on the success of our lead product, Soliris. If we are unable to increase sales of Soliris, or obtain approval or commercialize Soliris in new territories for the treatment of PNH, aHUS or for additional indications, or if we are significantly delayed or limited in doing so, our business may be materially harmed.
Our ability to generate revenues will continue to depend on commercial success of Soliris and whether physicians, patients and health care payers view Soliris as therapeutically effective and safe relative to cost. Since we launched Soliris in the United States in April 2007, essentially all of our revenue has been attributed to sales of Soliris, and we expect that Soliris product sales will continue to contribute to a significant percentage or almost all of our total revenue over the next several years.
In September and November 2011, we obtained marketing approval in the United States and the European Union, respectively, for Soliris for the treatment of a second indication, aHUS. In September 2013, the MHLW approved Soliris for the treatment of patients with aHUS in Japan.
We dedicate significant resources to the worldwide commercialization of Soliris. We have established sales and marketing capabilities in the United States and in many countries throughout the world. We cannot guarantee that any marketing application for Soliris for the treatment of PNH, aHUS or any other indication, will be approved or maintained in any country where we seek marketing authorization to sell Soliris. In certain countries, we continue discussions with authorities to finalize operational, reimbursement, price approval and funding processes so that we may, upon conclusion of such discussions, commence commercial sales of Soliris for the treatment of PNH in those countries. We have had and will continue to have similar discussions with authorities to facilitate the commercialization of Soliris for the treatment of aHUS in certain countries in the European Union. Our ability to complete such processes successfully is subject to the risks and uncertainties described in this Annual Report on Form 10-K. We cannot guarantee that we will be able to obtain reimbursement for Soliris or successfully commercialize Soliris in any additional countries, or that we will be able to maintain coverage or reimbursement at anticipated levels in any country in which we have already received marketing approval, including the U.S., certain European countries, or Japan. As a result, sales in certain countries may be delayed or never occur, or may be subsequently reduced.
The commercial success of Soliris and our ability to generate and increase revenues will depend on several factors, including the following:
• | receipt of marketing approvals for Soliris for the treatment of PNH and aHUS in new territories, and the maintenance of marketing approvals in the United States, the European Union, Japan and other territories; |
• | our ability to obtain sufficient coverage or reimbursement by government or third-party payers and our ability to maintain coverage or reimbursement at anticipated levels; |
• | establishment and maintenance of commercial manufacturing capabilities ourselves or through third-party manufacturers; |
• | the number of patients with PNH and aHUS, and the number of those patients who are diagnosed with PNH and aHUS and identified to us; |
• | the number of patients with PNH and aHUS that may be treated with Soliris; |
• | successful continuation of commercial sales in the United States, Japan and in European countries where we are already selling Soliris for the treatment of PNH and aHUS, and successful launch in countries where we have not yet obtained, or only recently obtained, marketing approval or commenced sales; |
27
• | acceptance of Soliris and maintenance of safety and efficacy in the medical community; and |
• | our ability to develop, register and commercialize Soliris for indications other than PNH and aHUS. |
If we are not successful in increasing sales of Soliris in the United States, Europe and Japan and commercializing in the rest of the world, or are significantly delayed or limited in doing so, we may experience surplus inventory, our business may be materially harmed and we may need to significantly curtail operations.
If we are unable to obtain, or maintain at anticipated levels, reimbursement for Soliris from government health administration authorities, private health insurers and other organizations, our pricing may be affected or our product sales, results of operations or financial condition could be harmed.
We may not be able to sell Soliris on a profitable basis or our profitability may be reduced if we are required to sell our product at lower than anticipated prices or reimbursement is unavailable or limited in scope or amount. Soliris is significantly more expensive than traditional drug treatments and almost all patients require some form of third party coverage to afford its cost. Our future revenues and profitability will be adversely affected if we cannot depend on governmental payers, such as Medicare and Medicaid in the United States or country specific governmental organizations in foreign countries, and private third-party payers to defray the cost of Soliris to patients. These entities may refuse to provide coverage and reimbursement with respect to Soliris, determine to provide a lower level of coverage and reimbursement than anticipated, or reduce previously approved levels of coverage and reimbursement, including in the form of higher mandatory rebates or modified pricing terms. In any such case, our pricing or reimbursement for Soliris may be affected and our product sales, results of operations or financial condition could be harmed.
In certain countries where we sell or are seeking or may seek to commercialize Soliris, including certain countries where we both sell Soliris for the treatment of PNH and sell or seek to commercialize Soliris for the treatment of aHUS, if approved by the appropriate regulatory authority, pricing, coverage and level of reimbursement of prescription drugs are subject to governmental control. We may be unable to timely or successfully negotiate coverage, pricing, and reimbursement on terms that are favorable to us, or such coverage, pricing, and reimbursement may differ in separate regions in the same country. In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country, and we cannot guarantee that we will have the capabilities or resources to successfully conclude the necessary processes and commercialize Soliris in every, or even most countries in which we seek to sell Soliris.
Reimbursement sources are different in each country and in each country may include a combination of distinct potential payers, including private insurance and governmental payers. For example, the European Union member states' authorities may restrict the range of medicinal products for which their national health insurance systems provide reimbursement and adopt additional measures to control the prices of medicinal products for human use. This includes the use of reference pricing and Health Technology Assessment (HTA). HTA is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of the use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well as their potential implications for the healthcare system. These elements of medicinal products are compared with other treatment options available on the market. The national authorities of some European Union member states may from time to time approve a specific price for the medicinal product. Others may adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the national market. Some countries have and others may seek to impose limits on the aggregate reimbursement for Soliris or for the use of Soliris for certain indications. In such cases, our commercial operations in such countries and our results of operations and our business are and may be adversely affected. Our results of operations may suffer if we are unable to successfully and timely conclude reimbursement, price approval or funding processes and market Soliris in such foreign countries or if coverage and reimbursement for Soliris is limited or reduced. If we are not able to obtain coverage, pricing or reimbursement on terms acceptable to us or at all, or if such terms should change in any foreign countries, we may not be able to or we may determine not to sell Soliris for one or more indications in such countries, or we could decide to sell Soliris at a lower than anticipated price in such countries, and our revenues may be adversely affected as a result.
The potential increase in the number of patients receiving Soliris may cause third-party payers to modify or limit coverage or reimbursement for Soliris for the treatment of PNH, aHUS, or both indications.
Changes in pricing or the amount of reimbursement in countries where we currently commercialize Soliris may also reduce our profitability and worsen our financial condition. In the United States, the European Union member states, and elsewhere, there have been, and we expect there will continue to be, efforts to control and reduce health care costs. Third party payers decide which drugs they will pay for and establish reimbursement and co-payment levels. Government and other third-party payers in the United States and the European Union member states are increasingly challenging the prices charged for health care products, examining the cost effectiveness of drugs in addition to their safety and efficacy, and limiting or attempting to limit both coverage and the level of reimbursement for prescription drugs.
28
A significant reduction in the amount of reimbursement or pricing for Soliris in one or more countries may have a material adverse effect on our business. See additional discussion below under the headings "Changes in healthcare law and implementing regulations, including those based on recently enacted legislation, as well as changes in healthcare policy and Government initiatives that affect coverage and reimbursement of drug products may impact our business in ways that we cannot currently predict and these changes could adversely affect our business and financial condition" and "The credit and financial market conditions may aggravate certain risks affecting our business." In addition, certain countries establish pricing and reimbursement amounts by reference to the price of the same or similar products in other countries. If coverage or the level of reimbursement is limited in one or more countries, we may be unable to obtain or maintain anticipated pricing or reimbursement in current or new territories.
Many third-party payers cover only selected drugs, making drugs that are not preferred by such payer more expensive for patients, and require prior authorization or failure on another type of treatment before covering a particular drug. Third-party payers may be especially likely to impose these obstacles to coverage for higher-priced drugs such as Soliris.
Payers in the U.S. also are increasingly considering new metrics as the basis for reimbursement rates, such as ASP, average manufacturer price, and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. The Centers for Medicare and Medicaid Services (CMS), the federal agency that administers Medicare and the Medicaid Drug Rebate Program, has begun posting drafts of this retail survey price information on at least a monthly basis in the form of draft National Average Drug Acquisition Cost, or NADAC, files, which reflect retail community pharmacy invoice costs, and National Average Retail Price, or NARP, files, which reflect retail community pharmacy prices to consumers. In July 2013, CMS suspended the publication of draft NARP data, pending funding decisions. In November 2013, CMS moved to publishing final rather than draft NADAC data and has since made updated NADAC data publicly available on a weekly basis. Therefore, it may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payers to cover Soliris.
Even in countries where patients have access to insurance, their insurance co-payment amounts or annual or lifetime caps on reimbursements may represent a barrier to obtaining or continuing Soliris. We have financially supported non-profit organizations which assist patients in accessing treatment for PNH and aHUS, including Soliris. Such organizations assist patients whose insurance coverage leaves them with prohibitive co-payment amounts or other expensive financial obligations. Such organizations' ability to provide assistance to patients is dependent on funding from external sources, and we cannot guarantee that such funding will be provided at adequate levels, if at all. We have also provided Soliris without charge to patients who have no insurance coverage for drugs through related charitable purposes. We are not able to predict the financial impact of the support we may provide for these and other charitable purposes; however, substantial support could have a material adverse effect on our profitability in the future.
We are also focusing development efforts on the use of eculizumab for the treatment of additional diseases. The success of these programs depends on many factors, including those described in this report. As Soliris is approved by regulatory agencies for indications other than PNH and aHUS, the potential increase in the number of patients receiving Soliris may cause third-party payers to refuse coverage or reimbursement for Soliris for the treatment of PNH, aHUS or for any other approved indication, or provide a lower level of coverage or reimbursement than anticipated or currently in effect.
We may not be able to maintain market acceptance of Soliris among the medical community or patients, or gain market acceptance of our products in the future, which could prevent us from maintaining profitability or growth.
We cannot be certain that Soliris will maintain market acceptance in a particular country among physicians, patients, health care payers, and others. Although we have received regulatory approval for Soliris in certain territories, including the United States, Japan and the European Union, such approvals do not guarantee future revenue. We cannot predict whether physicians, other health care providers, government agencies or private insurers will determine or continue to accept that Soliris is safe and therapeutically effective relative to its cost. Physicians' willingness to prescribe, and patients' willingness to accept, our products, such as Soliris, depends on many factors, including prevalence and severity of adverse side effects in both clinical trials and commercial use, the timing of the market introduction of competitive drugs, lower demonstrated clinical safety and efficacy compared to other drugs, perceived lack of cost-effectiveness, pricing and lack of availability of reimbursement from third-party payers, convenience and ease of administration, effectiveness of our marketing strategy, publicity concerning the product, our other product candidates or competing products, and availability of alternative treatments, including bone marrow transplant as an alternative treatment for PNH. The likelihood of physicians to prescribe Soliris for patients with aHUS may also depend on how quickly Soliris can be delivered to the hospital or clinic and our distribution methods may not be sufficient to satisfy this need. In addition, we are aware that medical doctors have determined not to continue Soliris treatment for some patients with aHUS.
Health insurance programs may restrict coverage of some products by using payer formularies under which only selected drugs are covered, variable co-payments that make drugs that are not preferred by the payer more expensive for patients, and by using utilization management controls, such as requirements for prior authorization or failure on another type of treatment.
29
Payers may especially impose these obstacles to coverage for higher-priced drugs, and consequently our drug products may be subject to payer-driven restrictions. In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, European Union member states may restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices and/or reimbursement of medicinal products for human use. A European Union member state may approve a specific price or level of reimbursement for the medicinal product, or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. The reimbursement or budget identified by a government or non-government payer for our products, including Soliris in a new indication, if obtained, may be adversely affected by the reimbursement or budget for Soliris in previously approved indications and/or adversely affect the reimbursement or budget for Soliris in such previously approved indication by that payer.
If Soliris fails to achieve or maintain market acceptance among the medical community or patients in a particular country, we may not be able to market and sell it successfully in such country, which would limit our ability to generate revenue and could harm our overall business.
If we or any third party manufacturer or provider fails to provide sufficient quantities of Soliris or our product candidates, including Soliris for new indications, we could experience product shortages, our commercialization of Soliris may be stopped or delayed, our clinical trials could be disrupted or regulatory approvals could be delayed.
Soliris is manufactured by Alexion at ARIMF and by Lonza. We depend on a very limited number of third party providers for the manufacture and supply of Soliris and our product candidates. The manufacture of Soliris and our product candidates is difficult, requiring a multi-step controlled process and even minor problems or deviations could result in defects or failures. Manufacture of our products, including Soliris, is highly technical, and only a small number of companies have the ability and capacity to manufacture our products for our development and commercialization needs. Due to the highly technical requirements of manufacturing our products and the strict quality and control specifications, we and our third party providers may be unable to manufacture or supply our products despite our and their efforts. In addition, we cannot be certain that any third party will be able or willing to honor the terms of its agreement, including any obligations to manufacture our products in accordance with regulatory requirements and to our quality specifications and volume requirements.
We cannot be certain that we, Lonza or our other third party providers will be able to perform uninterrupted supply chain services. The failure to manufacture appropriate supplies of Soliris, on a timely basis, or at all, may prevent or interrupt the commercialization of Soliris. If we, Lonza or our other third party providers were unable to manufacture Soliris for any period for any reason, including due to the loss of approvals, or if we, Lonza or our other third party providers do not obtain approval for the manufacturing of Soliris in the respective facility by the applicable regulatory agencies, we may incur substantial loss of sales. See also our Risk Factor "If we or our contract manufacturers fail to comply with continuing United States and foreign regulations, we could lose our approvals to market Soliris or our manufacturers could lose their approvals to manufacture Soliris or our product candidates, and our business would be seriously harmed." We may also lose any redundancy in our manufacturing capabilities if we are no longer able to perform operations at ARIMF or any other facility. The failure to manufacture appropriate supplies of our product candidates, on a timely basis, or at all, may prevent or interrupt clinical development of our products, including Soliris for new indications. If we are forced to find an alternative supplier or other third party providers, in addition to loss of sales and disruption to patients, we may also incur significant costs and experience significant delay in establishing a new arrangement.
We are authorized to sell Soliris that is manufactured by Lonza and at ARIMF in the United States, the European Union, Japan and certain other territories. However, manufacturing Soliris for commercial sale in certain other territories may only be performed at a single facility until such time as we have received the required regulatory approval for an additional facility, if ever. We will continue to depend entirely on one facility to manufacture Soliris for commercial sale in such other territories until that time.
We have obtained marketing approval for Soliris for the treatment of patients with aHUS in the United States, the European Union, Japan and other territories. We expect that the demand for Soliris will increase. We may underestimate demand, or experience product interruptions at ARIMF, Lonza or a facility of a third party provider, including as a result of risks and uncertainties described in this report. If we, Lonza or our other third party providers do not manufacture sufficient quantities of Soliris to satisfy demand, our business will be materially harmed.
We depend on a very limited number of third party providers for other services with respect to our clinical and commercial requirements, including product filling, finishing, packaging, and labeling. We have changed or added third party fill/finish providers in the past in order to support uninterrupted supply, and may do so in the future. We currently rely on three third party fill/finish providers to support our commercial requirements in the United States and the European Union, and two to support requirements in Japan. No guarantee can be made that regulators will approve additional third party fill/finish providers in a timely manner or at all, or that any third party fill/finish providers will be able to perform such services for sufficient product volumes for any country or territory. We do not have control over any third party provider's compliance with
30
our internal or external specifications or the rules and regulations of the FDA, EMA, competent authorities of the European Union member states, MHLW or any other applicable regulations or standards. In the past, we have had to write off and incur other charges and expenses for production that failed to meet requirements, including with respect to recalls initiated in 2013 and 2014.
Any difficulties or delays in our third party manufacturing of Soliris, or any failure of our third party providers to comply with our internal and external specifications or any applicable rules, regulations and standards could increase our costs, constrain our ability to satisfy demand for Soliris from customers, cause us to lose revenue or incur penalties for failure to deliver product, make us postpone or cancel clinical trials, or cause our products to be recalled or withdrawn, such as the voluntary recalls, that we initiated in 2013 and 2014 due to the presence of visible particles in a limited number of vials in specific lots. Even if we are able to find alternatives they may ultimately be insufficient for our needs.
Due to the nature of the current market for third-party commercial manufacturing, many arrangements require substantial penalty payments by the customer for failure to use the manufacturing capacity for which it contracted. Penalty payments under these agreements typically decrease over the life of the agreement, and may be substantial initially and de minimis or non-existent in the final period. The payment of a substantial penalty could harm our financial condition.
In April 2014, we acquired a fill/finish facility in Ireland to support global distribution of Soliris and Alexion's other clinical and commercial products. To date, we have relied entirely on third party fill/finish providers and have never operated our own fill/finish facility. We cannot guarantee that we will be able to successfully complete the appropriate validation processes or obtain the necessary regulatory approvals, or that we will be able to perform fill/finish services at this facility to support our product requirements.
Many additional factors could cause production interruptions at ARIMF or at the facilities of Lonza or our third party providers, including natural disasters, labor disputes, acts of terrorism or war, human error, equipment malfunctions, contamination, or raw material shortages. The occurrence of any such event could adversely affect our ability to satisfy demand for Soliris, which could materially and adversely affect our operating results.
If we or our contract manufacturers fail to comply with continuing United States and foreign regulations, we or our manufacturers could lose our approvals to market Soliris or our product candidates, and our business would be seriously harmed.
We cannot guarantee that we will be able to maintain our regulatory approvals for Soliris. If we do not maintain our regulatory approvals for Soliris, the value of our company and our results of operations will be materially harmed. We and our current and future partners, contract manufacturers and suppliers are subject to rigorous and extensive regulation by governmental authorities around the world, including the FDA, EMA, the competent authorities of the European Union member states, and MHLW. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us. For example, in March 2013, we received a Warning Letter from the FDA relating to compliance with cGMP at ARIMF. In August 2014 we announced that we received a Form 483 with three observations following an FDA inspection at ARIMF. If we do not resolve outstanding concerns expressed by the FDA in the Warning Letter and the August 2014 Form 483 to the satisfaction of the FDA, EMA or any other regulatory agency, or we or our third-party providers, including our product fill/finish providers, packagers and labelers, fail to comply fully with applicable regulations then we may be required to initiate a recall or withdrawal of our products.
The safety profile of any product continues to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. Regulations continue to apply after product approval, and cover, among other things, testing, manufacturing, quality control, finishing, filling, labeling, advertising, promotion, risk mitigation, adverse event reporting requirements, and export of biologics. For example, the risk management program established in 2007 upon the FDA's approval of Soliris for the treatment of PNH was replaced with a Risk Evaluation and Mitigation Strategy (REMS) program, approved by the FDA in 2010. The REMS program requires mandatory physician certification in the United States. Each physician must certify that the physician is aware of the potential risks associated with the administration of Soliris and that the physician will inform each patient of these risks using educational material approved by the FDA. In November 2014, we met with the FDA Drug Safety and Risk Management Advisory Committee to discuss adjustments to the REMS with elements to assure safe use (ETASU). A majority of the Committee favored revising the REMS and made suggestions for streamlining prescriber assessments and broadening the program’s educational outreach. Changes to the Soliris REMS could be costly and burdensome to implement.
As a condition of approval for marketing Soliris, governmental authorities may require us to conduct additional studies. For example, in connection with the approval of Soliris in the United States, European Union and Japan, for the treatment of PNH, we agreed to establish a PNH Registry, monitor immunogenicity, monitor compliance with vaccination requirements, and determine the effects of anticoagulant withdrawal among PNH patients receiving eculizumab, and, specifically in Japan, we
31
agreed to conduct a trial in a limited number of Japanese PNH patients to evaluate the safety of a meningococcal vaccine. Further, in connection with the approval of Soliris in the United States for the treatment of aHUS, we agreed to establish an aHUS Registry and complete additional human clinical studies in adult and pediatric patients. In the United States, for example, the FDA can propose to withdraw approval for a product if it determines that such additional studies are inadequate or if new clinical data or information shows that a product is not safe for use in an approved indication. We are required to report any serious and unexpected adverse experiences and certain quality problems with Soliris to the FDA, the EMA, the competent authorities of the European Union member states, MHLW, and certain other health agencies. We or any health agency may have to notify health care providers of any such developments.
The discovery of any previously unknown problems with Soliris, a manufacturer or a facility may result in restrictions on Soliris, a manufacturer or a facility, including withdrawal of Soliris from the market, batch failures, or interruption of production or a product recall such as the recalls we announced and voluntarily initiated in 2013 and 2014. Certain changes to an approved product, including the way it is manufactured or promoted, often require prior regulatory approval before the product as modified may be marketed. Our manufacturing and other facilities and those of any third parties manufacturing Soliris will be subject to inspection prior to grant of marketing approval by each regulatory authority where we seek marketing approval and subject to continued review and periodic inspections by the regulatory authorities, such as the inspections that resulted in issuance of the Warning Letter. We and any third party we would use to manufacture Soliris for sale, including Lonza, must also be licensed by applicable regulatory authorities.
The FDA requires reporting of certain information on side effects and adverse events reported during clinical studies and after marketing approval. Non-compliance with safety reporting requirements could result in regulatory action that may include civil action or criminal penalties.
Failure to comply with the laws and requirements, including statutes and regulations, administered by the FDA, the EMA, the competent authorities of the European Union member states, the MHLW or other agencies, including without limitation, failures or delays in resolving the concerns raised by the FDA in the Warning Letter, could result in:
• | a product recall; |
• | a product withdrawal; |
• | significant administrative and judicial sanctions, including, warning letters or untitled letters; |
• | significant fines and other civil penalties; |
• | suspension, variation or withdrawal of a previously granted approval for Soliris; |
• | interruption of production; |
• | operating restrictions, such as a shutdown of production facilities or production lines, or new manufacturing requirements; |
• | suspension of ongoing clinical trials; |
• | delays in approving or refusal to approve our products including pending BLAs or BLA supplements for Soliris or asfotase alfa, or a facility that manufactures our products; |
• | seizing or detaining product; |
• | requiring us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
• | injunctions; and/or |
• | criminal prosecution. |
If the use of Soliris harms people, or is perceived to harm patients even when such harm is unrelated to Soliris, our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The testing, manufacturing, marketing and sale of drugs for use in humans exposes us to product liability risks. Side effects and other problems from using Soliris could (1) lessen the frequency with which physicians decide to prescribe Soliris, (2) encourage physicians to stop prescribing Soliris to their patients who previously had been prescribed Soliris, (3) cause serious adverse events and give rise to product liability claims against us, and (4) result in our need to withdraw or recall Soliris from the marketplace. Some of these risks are unknown at this time.
We tested Soliris in only a small number of patients. The FDA marketing approval for the treatment of patients with aHUS was based on two prospective studies in a total of 37 adult and adolescent patients, together with a retrospective study
32
that included 19 pediatric patients. PNH and aHUS are ultra-rare diseases. As more patients use Soliris, including more children and adolescents, new risks and side effects may be discovered, the rate of known risks or side effects may increase, and risks previously viewed as less significant could be determined to be significant. Previously unknown risks and adverse effects of Soliris may also be discovered in connection with unapproved uses of Soliris, which may include administration of Soliris under acute emergency conditions, such as the Enterohemorrhagic E. coli health crisis in Europe, primarily Germany, that began in May 2011. We do not promote, or in any way support or encourage the promotion of Soliris for unapproved uses in violation of applicable law, but physicians are permitted to use products for unapproved purposes and we are aware of such uses of Soliris. In addition, we are studying and expect to continue to study Soliris in diseases other than PNH and aHUS in controlled clinical settings, and independent investigators are doing so as well. In the event of any new risks or adverse effects discovered as new patients are treated for approved indications and as Soliris is studied in or used by patients for other indications, regulatory authorities may delay or revoke their approvals, we may be required to conduct additional clinical trials and safety studies, make changes in labeling of Soliris, reformulate Soliris or make changes and obtain new approvals for our and our suppliers' manufacturing facilities. We may also experience a significant drop in the potential sales of Soliris, experience harm to our reputation and the reputation of Soliris in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales of Soliris or substantially increase the costs and expenses of commercializing and marketing Soliris.
We may be sued by people who use Soliris, whether as a prescribed therapy, during a clinical trial, during an investigator initiated study, or otherwise. Many patients who use Soliris are already very ill. Any informed consents or waivers obtained from people who enroll in our trials or use Soliris may not protect us from liability or litigation. Our product liability insurance may not cover all potential types of liabilities or may not cover certain liabilities completely. Moreover, we may not be able to maintain our insurance on acceptable terms. In addition, negative publicity relating to the use of Soliris or a product candidate, or to a product liability claim, may make it more difficult, or impossible, for us to market and sell Soliris. As a result of these factors, a product liability claim, even if successfully defended, could have a material adverse effect on our business, financial condition or results of operations.
Patients who use Soliris already often have severe and advanced stages of disease and known as well as unknown significant pre-existing and potentially life-threatening health risks, including, for example, bone marrow failure, kidney failure and thrombosis. During the course of treatment, patients may suffer adverse events, including death, for reasons that may or may not be related to Soliris. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market Soliris, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to Soliris, the investigation into the circumstance may be time consuming or inconclusive. These investigations may interrupt our sales efforts, delay our regulatory approval process in other countries, or impact and limit the type of regulatory approvals Soliris receives or maintains.
Some patients treated with Soliris for PNH and other diseases, including patients who have participated in our clinical trials, have died or suffered potentially life-threatening diseases either during or after ending their Soliris treatments. In particular, use of C5 Inhibitors, such as Soliris, is associated with an increased risk for certain types of infection, including meningococcal infection. Serious cases of meningococcal infection can result in severe illness, including but not limited to brain damage, loss of limbs or parts of limbs, kidney failure, or death. Under controlled settings, patients in our eculizumab trials all receive vaccination against meningococcal infection prior to first administration of Soliris and patients who are prescribed Soliris in most countries are required by prescribing guidelines to be vaccinated prior to receiving their first dose. A physician may not have the opportunity to timely vaccinate a patient in the event of an acute emergency episode, such as in a patient presenting with aHUS or during the health crisis that began in May 2011 in Europe, principally in Germany, due to the epidemic of infections from Enterohemorrhagic E. coli. Vaccination does not, however, eliminate all risk of meningococcal infection. Additionally, in some countries there may not be any vaccine approved for general use or approved for use in infants and children. Some patients treated with Soliris who had been vaccinated have nonetheless experienced meningococcal infection, including patients who have suffered serious illness or death. Each such incident is required to be reported to appropriate regulatory agencies in accordance with relevant regulations.
We are also aware of a potential risk for PNH patients who delay a dose of Soliris or discontinue their treatment of Soliris. Treatment with Soliris blocks complement and allows complement-sensitive PNH red blood cells to increase in number. If treatment with Soliris is thereafter delayed or discontinued, a greater number of red blood cells therefore would become susceptible to destruction when the patient's complement system is no longer blocked. The rapid destruction of a larger number of a patient's red blood cells may lead to numerous complications, including death. Several PNH patients in our studies of Soliris have received delayed doses or discontinued their treatment. In none of those circumstances were significant complications shown to be due to rapid destruction of a larger number of PNH red blood cells; however, we have not studied the delay or termination of treatment in enough patients to determine that such complications in the future are unlikely to occur. Additionally, such delays or discontinuations may be associated with significant complications without evidence of such rapid cell destruction.
33
We are aware of a risk for aHUS patients who delay or miss a dose of Soliris or discontinue their treatment of Soliris. Treatment with Soliris blocks complement and inhibits complement-mediated TMA. After missing a dose or discontinuing Soliris, blood clots may form in small blood vessels throughout the body, causing a reduction in platelet count. The reduction in platelet count may lead to numerous complications, including changes in mental status, seizures, angina, thrombosis, renal failure or even death. In our aHUS clinical studies, such TMA complications were observed in some patients who missed a dose.
Clinical evaluations of outcomes in the post-marketing setting are required to be reported to appropriate regulatory agencies in accordance with relevant regulations. Determination of significant complications associated with the delay or discontinuation of Soliris could have a material adverse effect on our ability to sell Soliris.
If we are unable to establish and maintain effective sales, marketing and distribution capabilities, or to enter into agreements with third parties to do so, we will be unable to successfully commercialize Soliris.
We are marketing and selling Soliris ourselves in the United States, Europe, Japan and several other territories. If we are unable to establish and/or expand our capabilities to sell, market and distribute Soliris for the treatment of PNH, aHUS or, if approved by the necessary regulatory agencies, other future indications, either through our own capabilities or by entering into agreements with others, or to maintain such capabilities in countries where we have already commenced commercial sales, we will not be able to successfully sell Soliris. In that event, we will not be able to generate significant revenues. We cannot guarantee that we will be able to establish and maintain our own capabilities or enter into and maintain any marketing or distribution agreements with third-party providers on acceptable terms, if at all. Even if we hire the qualified sales and marketing personnel we need to support our objectives, or enter into marketing and distribution agreements with third parties on acceptable terms, we may not do so in an efficient manner or on a timely basis. We may not be able to correctly judge the size and experience of the sales and marketing force and the scale of distribution capabilities necessary to successfully market and sell Soliris. Establishing and maintaining sales, marketing and distribution capabilities are expensive and time-consuming. Our expenses associated with building up and maintaining the sales force and distribution capabilities around the world may be disproportionate compared to the revenues we may be able to generate on sales of Soliris. We cannot guarantee that we will be successful in commercializing Soliris.
If we market Soliris in a manner that violates health care fraud and abuse laws and other laws regulating marketing and promotion, we may be subject to investigations and civil or criminal penalties.
In addition to FDA and related regulatory requirements, we are subject to health care "fraud and abuse" laws, such as the federal False Claims Act, the anti-kickback provisions of the federal Social Security Act, and other state and federal laws and regulations. The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, in cash or in kind to induce, or reward the purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid, or other federal health care programs. This statute has been interpreted to apply broadly to arrangements between pharmaceutical manufacturers on the one hand and prescribers, patients, purchasers and formulary managers on the other. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. In addition, PPACA amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act (FCA). A conviction for violation of the Anti-kickback Statute requires mandatory exclusion from participation in federal health care programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration to those who prescribe, purchase, or recommend pharmaceutical products, including certain discounts, education and research grants, purchase of speaking or consulting services, and patient assistance programs, may be subject to scrutiny or penalty if they do not qualify for an exemption or safe harbor. We seek to comply with the anti-kickback laws and with the available statutory exemptions and safe harbors. However, our practices may not in all cases fit within the safe harbors, and our practices may therefore be subject to case-by-case scrutiny.
The FCA prohibits any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim. Pharmaceutical companies have been investigated and have reached substantial financial settlements with the Federal government under the FCA for a variety of alleged promotional and marketing activities, such as allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees and other benefits to physicians to induce them to prescribe products; reporting inflated prices to private publications that were then used by federal programs to set reimbursement rates; engaging in promotion for uses that the FDA has not approved, or "off-label" uses that caused claims to be submitted to Federal programs for non-covered off-label uses; and submitting inflated best price information to the Medicaid Rebate Program.
34
The majority of states also have statutes similar to the federal anti-kickback law and false claims laws, that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. In addition, several U.S. states and localities have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state or make periodic public disclosures on sales, marketing, pricing, clinical trials, and other activities. Some state laws prohibit certain marketing-related activities including the provision of gifts, meals or other items to certain health care providers. Similar legislation is being considered in other states. Additionally, PPACA enacted the Physician Payment Sunshine Act, being implemented as the Open Payments program, that requires manufacturers to track and report to the federal government, for public dissemination, payments and other transfers of value made to physicians and teaching hospitals. Many of these requirements are new and there is limited guidance on many aspects of how they will be interpreted, implemented and enforced. , Nonetheless, if we are found not to be in full compliance with these laws, we could face enforcement action and fines and other penalties, and could receive adverse publicity.
Sanctions under these federal and state fraud and abuse laws may include civil monetary penalties, exclusion of a manufacturer's products from reimbursement under government programs, monetary damages, criminal fines, and imprisonment. Efforts to ensure that our business arrangements continue to comply with applicable healthcare laws and regulations could be costly. Because of the breadth of these laws and the narrowness of the safe harbors and because government scrutiny in this area is high, it is possible that some of our business activities could come under that scrutiny. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could also harm our financial condition. Responding to government investigations or whistleblower lawsuits, defending any claims raised, and any resulting fines, damages, penalties, settlement payments or administrative actions, as well as any related actions brought by stockholders or other third parties, could have a material impact on our reputation, business and financial condition and divert the attention of our management from operating our business.
Although physicians are permitted to, based on their medical judgment, prescribe products for indications other than those cleared or approved by the FDA, manufacturers are prohibited from promoting their products for such off-label uses. In the United States, we market Soliris for PNH and aHUS and provide promotional materials and training programs to physicians regarding the use of Soliris for PNH and aHUS. Although we believe our marketing materials and training programs for physicians do not constitute off-label promotion of Soliris, the FDA, the U.S. Justice Department, or other federal or state government agencies may disagree. If the FDA or other government agencies determine that our promotional materials, training or other activities constitute off-label promotion of Soliris, it could request that we modify our training or promotional materials or other activities or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal or state enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false or fraudulent claims for payment of government funds. Even if it is later determined we are not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our position and have to divert significant management resources from other matters.
Similar strict restrictions are imposed on the promotion and marketing of drug products in the European Union, where a large portion of our non-U.S. business is conducted, and other territories. Laws in the European Union, including in the individual European Union member states, require promotional materials and advertising for drug products to comply with the product's Summary of Product Characteristics (SmPC), which is approved by the competent authorities. Promotion of a medicinal product which does not comply with the SmPC is considered to constitute off-label promotion. The off-label promotion of medicinal products is prohibited in the European Union and in other territories. The promotion of medicinal products that are not subject to a marketing authorization is also prohibited in the European Union. Laws in the European Union, including in the individual European Union member states, also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the European Union and in other territories could be penalized by administrative measures, fines and imprisonment.
Interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry self-regulation codes of conduct and physicians' codes of professional conduct in the individual European Union member states. The provision of any inducements to physicians to prescribe, recommend, endorse, order, purchase, supply, use or administer a medicinal product is prohibited. A number of European Union member states have introduced additional rules requiring pharmaceutical companies to publicly disclose their interactions with physicians and to obtain approval from employers, professional organizations and/or competent authorities before entering into agreements with physicians. These rules have been supplemented by provisions of related industry codes. Additional countries may consider or implement similar laws and regulations. Violations of these rules could lead to reputational risk, public reprimands, and/or the imposition of fines or imprisonment.
We are also subject to the United States Foreign Corrupt Practices Act (FCPA), the U.K. Bribery Act, and other anti-corruption laws and regulations that generally prohibit companies and their intermediaries from making improper payments to
35
government officials and/or other persons for the purpose of obtaining or retaining business. Worldwide regulators are increasing their regulatory and enforcement efforts in this area. For example, the Bribery Act in the United Kingdom, effective as of July 2011 applies to any company incorporated in or "carrying on business" in the United Kingdom, regardless of the country in which the alleged bribery activity occurs and even if the inappropriate activity is undertaken by our international distribution partners.
Recent years have seen a substantial increase in anti-bribery law enforcement activity by U.S. regulators, with more frequent and aggressive investigations and enforcement proceedings by both the Department of Justice ("DOJ") and the U.S. Securities and Exchange Commission ("SEC"), increased enforcement activity by non-U.S. regulators, and increases in criminal and civil proceedings brought against companies and individuals. Our policies mandate compliance with these anti-bribery laws. We may operate in many parts of the world that are recognized as having a greater potential for governmental and commercial corruption. We cannot assure that our policies and procedures will always protect us from reckless or criminal acts committed by our employees or third-party intermediaries. From time-to-time, we may conduct internal investigations and compliance reviews, the findings of which could negatively impact our business. Any determination that our operations or activities are not, or were not, in compliance with existing United States or foreign laws or regulations could result in the imposition of substantial fines, interruptions of business, loss of supplier, vendor or other third-party relationships, termination of necessary licenses and permits, and other legal or equitable sanctions. Other internal or government investigations or legal or regulatory proceedings, including lawsuits brought by private litigants, may also follow as a consequence. Violations of these laws may result in criminal or civil sanctions, which could disrupt our business and result in a material adverse effect on our reputation, business, results of operations or financial condition. Increasing regulatory scrutiny of the promotional activities of pharmaceutical companies also has been observed in a number of European Union member states.
Laws, including those governing promotion, marketing and anti-kickback/anti-bribery provisions, and industry regulations are often strictly enforced. In the United States, additional governmental resources are being added to enforce these laws and to prosecute companies and individuals believed to be violating them. For example, PPACA included a number of provisions aimed at strengthening the government’s ability to pursue anti-kickback and false claims cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers for government authorities, and amendments to the civil False Claims Act that make it easier for the government and whistleblowers to pursue cases for alleged kickback and false claim violations. We anticipate that government scrutiny of pharmaceutical sales and marketing practices will continue for the foreseeable future and subject us to the risk of government investigations and whistleblower lawsuits. Responding to a government investigation or whistleblower lawsuit would be expensive and time-consuming, and could have a material adverse effect on our business and financial condition and growth prospects.
None of our product candidates except for Soliris has received regulatory approvals. Soliris has not been approved for any indication other than for the treatment of patients with PNH and aHUS. If we are unable to obtain regulatory approvals to market one or more of our product candidates, including asfotase alfa and Soliris for other indications, our business may be adversely affected.
All of our product candidates except Soliris and asfotase alfa are in early stages of development, and we do not expect our early stage product candidates to be commercially available for several years, if at all. Although we are preparing for a commercial launch of asfotase alfa for the treatment of hypophosphatasia, we do not know when or if asfotase alfa will be approved by the FDA, EMA or any other regulatory agency. We completed a rolling submission of our BLA for asfotase alfa in the U.S., which allowed completed portions of the application to be submitted and reviewed by the FDA on an ongoing basis. While we believe a rolling submission will allow us to expedite the review of the application, we cannot predict how long the approval process will take or when we will receive approval, if at all. We do not know when or if our other product candidates will be approved. Unfavorable clinical trial results, failure to comply with regulatory requirements, resolve pending concerns described in the Warning Letter, and inadequate manufacturing processes are examples of problems that could prevent approval. In addition, we may encounter delays or rejections due to additional government regulation from future legislation, administrative action or changes in the FDA policy. Even if the FDA approves a product, the approval will be limited to those indications covered in the approval.
Outside the United States, our ability to market any of our potential products is dependent upon receiving marketing approvals from the appropriate regulatory authorities. These foreign regulatory approval processes include all of the risks associated with the FDA approval process described above. If we are unable to receive regulatory approvals, we will be unable to commercialize our product candidates, and our business may be adversely affected.
Completion of preclinical studies or clinical trials does not guarantee advancement to the next phase of development.
Completion of preclinical studies or clinical trials does not guarantee that we will initiate additional studies or trials for our product candidates, that if further studies or trials are initiated what the scope and phase of the trial will be or that they will be completed, or that if these further studies or trials are completed, that the design or results will provide a sufficient basis to apply for or receive regulatory approvals or to commercialize products. Results of clinical trials could be inconclusive,
36
requiring additional or repeat trials. Data obtained from preclinical studies and clinical trials are subject to varying interpretations that could delay, limit or prevent regulatory approval. Data that we believe is highly clinically significant, including the results of our HPP trials, could be interpreted differently by the FDA or other regulatory agencies. The results generated in clinical studies of asfotase alfa which we believe to be positive, do not ensure that the product will be approved and the FDA or other regulatory agency could require additional preclinical or clinical data. If the design or results achieved in our clinical trials are insufficient to proceed to further trials or to regulatory approval of our product candidates, our company could be materially adversely affected. Failure of a clinical trial to achieve its pre-specified primary endpoint, such as the Phase 2 Soliris trial for AMR that we announced in January 2015, generally increases the likelihood that additional studies or trials will be required if we determine to continue development of the product candidate, reduces the likelihood of timely development of and regulatory approval to market the product candidate, and may decrease the chances for successfully achieving the primary endpoint in scientifically similar indications.
There are many reasons why drug testing could be delayed or terminated.
For human trials, patients must be recruited and each product candidate must be tested at various doses and formulations for each clinical indication. In addition, to ensure safety and effectiveness, the effect of drugs often must be studied over a long period of time, especially for the chronic diseases that we are studying. Many of our programs focus on diseases with small patient populations and insufficient patient enrollment in our clinical trials could delay or cause us to abandon a product development program. We may decide to abandon development of a product candidate at any time due to unfavorable results or other reasons, or we may have to spend considerable resources repeating clinical trials or conducting additional trials, either of which would increase costs and delay any revenue from those product candidates, if any.
Additional factors that can cause delay, impairment or termination of our clinical trials or our product development efforts include:
• | delay or failure in obtaining institutional review board (IRB), approval or the approval of other reviewing entities to conduct a clinical trial at each site; |
• | delay or failure in reaching agreement on acceptable terms with prospective contract research organizations(CROs), and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
• | withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials; |
• | clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial; |
• | slow patient enrollment, including, for example, due to the rarity of the disease being studied; |
• | delay or failure in having patients complete a trial or return for post-treatment follow-up; |
• | long treatment time required to demonstrate effectiveness; |
• | lack of sufficient supplies of the product candidate; |
• | disruption of operations at the clinical trial sites; |
• | adverse medical events or side effects in treated patients, and the threat of legal claims and litigation alleging injuries; |
• | failure of patients taking the placebo to continue to participate in our clinical trials; |
• | insufficient clinical trial data to support effectiveness of the product candidates; |
• | lack of effectiveness or safety of the product candidate being tested; |
• | lack of sufficient funds; |
• | inability to meet required specifications or to manufacture sufficient quantities of the product candidate for development or commercialization activities in a timely and cost-efficient manner; |
• | decisions by regulatory authorities, the IRB, ethics committee, or us, or recommendation by a data safety monitoring board, to suspend or terminate clinical trials at any time for safety issues or for any other reason; |
• | failure to obtain the necessary regulatory approvals for the product candidate or the approvals for the facilities in which such product candidate is manufactured; and |
• | decisions by competent authorities, IRBs or ethics committees to demand variations in protocols or conduct of clinical trials. |
37
The regulatory approval process is costly and lengthy and we may not be able to successfully obtain all required regulatory approvals.
In April 2014, Alexion initiated a BLA with the FDA for asfotase alfa as a treatment for patients with hypophosphatasia (HPP). In July 2014, the Marketing Authorization Application (MAA) for asfotase alfa was validated by the European Medicines Agency (EMA). In October 2014, Alexion submitted a New Drug Application for asfotase alfa to Japan’s MHLW.
The preclinical development, clinical trials, manufacturing, marketing and labeling of pharmaceuticals are all subject to extensive regulation by numerous governmental authorities and agencies in the United States, the European Union and other territories. We must obtain regulatory approval for each of our product candidates, such as asfotase alfa, before marketing or selling any of them. It is not possible to predict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any of our product candidates will take or whether any such approvals ultimately will be granted. For example, the EMA transitioned the MAA for asfotase alfa from an accelerated assessment to a regular assessment. The FDA and foreign regulatory agencies have substantial discretion in the drug approval process, and positive results in preclinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. The approval process varies from country to country and the requirements governing the conduct of clinical trials, product manufacturing, product licensing, pricing and reimbursement vary greatly from country to country. Generally, preclinical and clinical testing of product candidates can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. If we encounter significant delays in the regulatory process, this may prevent us from continuing to develop our product candidates due to excessive costs or otherwise. Any delay in obtaining, or failure to obtain, approvals could adversely affect the marketing of our products and our ability to generate product revenue. The risks associated with the approval process include:
• | failure of our product candidates to meet a regulatory agency's requirements for safety, efficacy and quality; |
• | disagreement over interpretation of data from preclinical studies or clinical trials; |
• | restricted distribution or limitation on the indicated uses for which a product may be marketed; |
• | unforeseen safety issues or side effects and potential requirements to establish REMS or post-marketing obligations; |
• | disapproval of the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
• | governmental or regulatory delays and changes in regulatory requirements and guidelines. |
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data, to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that is not desirable for the successful commercialization of that product candidate. In addition, if our product candidate produces undesirable side effects or safety issues, the FDA may require the establishment of Risk Evaluation Mitigation Strategies, or REMS, or a comparable foreign regulatory authority may require the establishment of a similar strategy, that may, for instance, restrict distribution of our products and impose burdensome implementation requirements on us. Any of the foregoing scenarios could materially harm the commercial prospects of our product candidates.
Risks Related to Intellectual Property
If we cannot obtain new patents, maintain our existing patents and protect the confidentiality and proprietary nature of our trade secrets and other intellectual property, our business and competitive position will be harmed.
In order to protect our drugs and technology more effectively, we need to obtain and maintain patents covering the drugs and technologies we develop. We have and may in the future obtain patents or the right to practice patents through ownership or license. Our patent applications may not result in the issue of patents in the United States or other countries. Our patents may not afford adequate protection for our products. Third parties may challenge our patents, and have challenged our patents in the past. If any of our patents are narrowed, invalidated or become unenforceable, competitors may develop and market products similar to ours that do not conflict with or infringe our patents rights, which could have a material adverse effect on our financial condition. We may also finance and collaborate in research conducted by government organizations, hospitals, universities or other educational or research institutions. Such research partners may be unwilling to grant us exclusive rights to technology or products developed through such collaborations. There is also a risk that disputes may arise as to the rights to technology or products developed in collaboration with other parties. Soliris and our drug candidates are expensive and time-consuming to test and develop. Even if we obtain and maintain patents, our business may be significantly harmed if the patents are not broad enough to protect our drugs from copycat products.
38
In addition, our business requires using sensitive technology, techniques and proprietary compounds that we protect as trade secrets. However, we may also rely heavily on collaboration with, or discuss the potential for collaboration with, suppliers, outside scientists and other drug companies. Collaboration and discussion of potential collaboration present a strong risk of exposing our trade secrets. If our trade secrets were exposed, it would help our competitors and adversely affect our business prospects.
If we are found to be infringing on patents owned by others, we may be forced to pay damages to the patent owner and/or obtain a license to continue the manufacture, sale or development of our drugs. If we cannot obtain a license, we may be prevented from the manufacture, sale or development of our drugs, including Soliris, which would adversely affect our business.
Parts of our technology, techniques and proprietary compounds and potential drug candidates, including those which are or may be in-licensed, may be found to infringe patents owned by or granted to others. We previously reported that Novartis and other third parties have filed civil lawsuits against us claiming infringement of their intellectual property rights. Each of these matters has been resolved, however, additional third parties may claim that the manufacture, use or sale of Soliris or other drugs under development infringes patents owned or granted to such third parties. In addition to the civil actions referenced above, we have in the past received, and may in the future receive, notices from third parties claiming that their patents may be infringed by the development, manufacture or sale of Soliris or some of our drug candidates. We are aware of patents owned by third parties that might be claimed by such third parties to be infringed by the development and commercialization of Soliris and some of our drug candidates. In respect to some of these patents, we have obtained licenses, or expect to obtain licenses. However, with regard to such other patents, we have determined in our judgment that:
• | Soliris and our product candidates do not infringe the patents; |
• | the patents are not valid; or |
• | we have identified and tested or are testing various modifications that we believe should not infringe the patents and which should permit commercialization of our product candidates. |
Any holder of these patents or other patents covering similar technology could sue us for damages and seek to prevent us from manufacturing, selling or developing our drugs. Legal disputes can be costly and time consuming to defend. If we cannot successfully defend against any future actions or conflicts, if they arise, we may incur substantial legal costs and may be liable for damages, be required to obtain costly licenses or need to stop manufacturing, using or selling Soliris, which would adversely affect our business. We may seek to obtain a license prior to or during legal actions in order to reduce further costs and the risk of a court determination that our product infringes the third party's patents. A required license may be costly or may not be available on acceptable terms, if at all. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our business.
There can be no assurance that we would prevail in a patent infringement action or that we would be able to obtain a license to any third-party patent on commercially reasonable terms or any terms at all; successfully develop non-infringing alternatives on a timely basis; or license alternative non-infringing technology, if any exists, on commercially reasonable terms. Any impediment to our ability to manufacture, use or sell approved forms of Soliris or our product candidates could have a material adverse effect on our business and prospects.
It is possible that we could lose market exclusivity for a product earlier than expected, which would harm our competitive position.
In our industry, much of an innovative product’s commercial value is realized while it has market exclusivity. When market exclusivity expires and biosimilar or generic versions of the product are approved and marketed, there can be substantial decline in the innovative product’s sales.
Market exclusivity for Soliris is based upon patent rights and certain regulatory forms of exclusivity. The scope of Soliris patent rights vary from country to country and are dependent on the availability of meaningful legal remedies in each country. The failure to obtain patent and other intellectual property rights, or limitations on the use, or loss of such rights, could be material to our business. In some countries, patent protections for Soliris may not exist because certain countries did not historically offer the right to obtain specific types of patents or we did not file patents in those markets. Also, the patent environment is unpredictable and the validity and enforceability of patents cannot be predicted with certainty. Absent relevant patent protection for a product, once regulatory exclusivity periods expire, biosimilar or generic versions of the product can be approved and marketed. Even prior to the expiration of regulatory exclusivity, a competitor could seek to obtain marketing approval by submitting its own clinical trial data.
39
Risks Related to Our Operations
We cannot guarantee that we will achieve our financial goals, including our ability to maintain profitability on a quarterly or annual basis in the future.
Until the quarter ended June 30, 2008, we had never been profitable since we were incorporated in January 1992. We have maintained profitability on a quarterly basis since the quarter ended June 30, 2008 and on an annual basis beginning with the year ended December 31, 2008. We believe that we formulate our annual operating budgets with reasonable assumptions and targets, however we cannot guarantee that we will be able to generate sufficient revenues or control expenses to achieve our financial goals, including continued profitability. Even if we do achieve profitability in any subsequent quarters, we may not be able to sustain or increase profitability on a quarterly or annual basis. You should not consider our revenue growth in recent periods as indicative of our future performance. Our revenue in future periods could decline. We may make errors in predicting and reacting to relevant business trends or our business may be subject to factors beyond our control, which could harm our operations. Since we began our business, we have focused on research and development of product candidates. We cannot guarantee that we will be successful in marketing and selling Soliris on a continued basis in countries or regions where we have obtained marketing approval, including the United States, Europe and Japan, and we do not know when we will have Soliris available for sale in territories where we have applied or will apply for marketing approval, if ever. We will have substantial expenses as we continue our research and development efforts, continue to conduct clinical trials and continue to develop manufacturing, sales, marketing and distribution capabilities in the United States and abroad. The achievement of our financial goals, including the extent of our future profitability, depends on many factors, including our ability to successfully market Soliris in the United States, the European Union and Japan and other territories, our ability to obtain regulatory, pricing, coverage, and reimbursement approvals of our drug candidates, such as asfotase alfa, and for Soliris in additional territories and other indications, our ability to successfully market Soliris in additional territories, our ability to successfully manufacture and commercialize our drug candidates and our ability to successfully bring our other product candidates to the major commercial markets throughout the world.
If our competitors get to the marketplace before we do, or with better or less expensive drugs, it may not be profitable to continue to produce Soliris and our product candidates.
The FDA, EC and the MHLW granted orphan drug designation for Soliris in the treatment of PNH and the FDA and EC granted orphan drug designation for aHUS. Orphan drug status entitles Soliris to market exclusivity for a total of seven years in the United States and for ten years in the European Union and Japan. However, if a competitive product that is the same as or similar to Soliris, as defined under the applicable regulations, is shown to be clinically superior to Soliris in the treatment of PNH or aHUS, or if a competitive product is different from Soliris, as defined under the applicable regulations, the orphan drug exclusivity we have obtained may not block the approval of such competitive product. Several biotechnology and pharmaceutical companies throughout the world have programs to develop complement inhibitor therapies or have publicly announced their intentions to develop drugs which target the inflammatory effects of complement in the immune system. Pharmaceutical companies have publicly announced intentions to establish or develop rare disease programs and these companies may introduce products that are competitive with ours. These and other companies, many of which have significantly greater resources than us, may develop, manufacture, and market better or cheaper drugs than Soliris or our product candidates. They may establish themselves in the marketplace before us for Soliris for other indications or for any of our other product candidates. Other pharmaceutical companies also compete with us to attract academic research institutions as drug development partners, including for licensing these institutions' proprietary technology. If our competitors successfully enter into such arrangements with academic institutions, we will be precluded from pursuing those unique opportunities and may not be able to find equivalent opportunities elsewhere.
If we fail to recruit and retain personnel, we may not be able to implement our business strategy.
We are highly dependent upon the efforts of our executive officers, and other key personnel in our commercial and technical organizations. There is intense competition in the biopharmaceutical industry for qualified commercial and technical personnel. Our business is specialized and global and we must attract and retain highly qualified individuals across many geographies. We may not be able to continue to attract and retain the qualified personnel necessary for developing, manufacturing and commercializing our products and product candidates.
We are subject to environmental laws and potential exposure to environmental liabilities.
We are subject to various federal, state and local environmental laws and regulations that govern our operations, including our manufacturing operations at ARIMF and in Ireland, the handling and disposal of non-hazardous and hazardous wastes, such as medical and biological wastes, and emissions and discharges into the environment, such as air, soils and water sources. Failure to comply with such laws and regulations could result in costs for corrective action, penalties or the imposition
40
of other liabilities. We also are subject to laws and regulations that impose liability and clean-up responsibility for releases of hazardous substances into the environment. Under certain of these laws and regulations, a current or previous owner or operator of property may be liable for the costs of remediating its property or locations to which wastes were sent from its facilities, without regard to whether the owner or operator knew of, or necessarily caused, the contamination. Such obligations and liabilities, which to date have not been material, could have a material impact on our business and financial condition.
We are seeking to expand our business through acquisitions and we may not realize the benefits of such acquisitions.
Our business strategy includes expanding our products and capabilities. We may seek additional acquisitions or in-licensing of businesses or products to expand our products and capabilities. Acquisitions of new businesses or products and in-licensing of new products may involve numerous risks, including:
• | substantial cash expenditures; |
• | potentially dilutive issuance of equity securities; |
• | incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; |
• | difficulties in assimilating the operations of the acquired companies; |
• | diverting our management's attention away from other business concerns; |
• | risks of entering markets in which we have limited or no direct experience; |
• | the potential loss of our key employees or key employees of the acquired companies; and |
• | failure of any acquired businesses or products or in-licensed products to achieve the scientific, medical, commercial or other results anticipated. |
A substantial portion of our strategic efforts are focused on opportunities for rare disorders and life-saving therapies. The availability of such development opportunities is limited. We may not be able to identify opportunities that are acceptable to us or our shareholders. Several companies have publicly announced intentions to establish or develop rare disease programs. For these and other reasons, we may not be able to acquire the rights to additional product candidates and approved products on terms that we or our shareholders find acceptable, or at all. The development or expansion of our business, any acquired business or any acquired or in-licensed products may require a substantial capital investment by us. We may not have these necessary funds or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our capital stock, which could dilute current stockholders' ownership interest in our company, or securities convertible into our capital stock, which could dilute current stockholders' ownership interest in our company upon conversion.
Even if we are able to successfully identify and complete acquisitions and other strategic transactions, we may not be able to integrate them or take full advantage of them. An acquisition or other strategic transaction may not result in short-term or long-term benefits to us. We may also incorrectly judge the value or worth of an acquired company or business or an acquired or in-licensed product.
To effectively manage our current and future potential growth, we must continue to effectively grow and manage our global employee base, and enhance our operational and financial processes. Supporting our growth strategy will require significant capital expenditures and management resources, including investments in research and development, sales and marketing, manufacturing and other areas of our operations. If we do not successfully manage our current growth and do not successfully execute our strategy, then our business and financial results may be adversely affected and we may incur asset impairment or restructuring charges.
Our business could be affected by litigation, government investigations and enforcement actions.
We operate in many jurisdictions in a highly regulated industry and we could be subject to litigation, government investigation and enforcement actions on a variety of matters in the United States or foreign jurisdictions, including, without limitation, intellectual property, regulatory, product liability, environmental, whistleblower, Qui Tam, false claims, privacy, anti-kickback, anti-bribery, securities, commercial, employment, and other claims and legal proceedings which may arise from conducting our business. Legal proceedings, government investigations and enforcement actions can be expensive and time consuming. An adverse outcome could result in significant damages awards, fines, penalties, exclusion from the federal healthcare programs, healthcare debarment, injunctive relief, product recalls, reputational damage and modifications of our business practices, which could have a material adverse effect on our business and results of operations.
41
The intended efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate and we may have exposure to additional tax liabilities or our effective tax rate could change, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the United States and various foreign jurisdictions. Significant judgment is required in determining our worldwide tax liabilities. Although we believe our estimates are reasonable, the ultimate outcome with respect to the taxes we owe may differ from the amounts recorded in our financial statements. If the Internal Revenue Service, or other taxing authority, disagrees with the positions we take, we could have additional tax liability, and this could have a material impact on our results of operations and financial position. Our effective tax rate could be adversely affected by changes in the mix of earnings in countries with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in tax laws and regulations, changes in interpretations of tax laws, including pending tax law changes, changes in our manufacturing activities and changes in our future levels of research and development spending.
We have designed our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our affiliates in a way that is intended to enhance our operational and financial efficiency and increase our overall profitability. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing. If tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase and harm our financial position and results of operations.
In addition, the United States government and other governments are considering and may adopt tax reform measures that significantly increase our worldwide tax liabilities. The U.S. Congress, the Organization for Economic Co-operation and Development and other government agencies in countries where we and our affiliates operate have focused on issues related to the taxation of multinational corporation, including , for example, in the area of “base erosion and profit shifting,” where payments are made between affiliates from a jurisdiction with high tax rates to a jurisdiction with lower tax rates. We established operations in Ireland in 2013 and recently, Ireland tax authorities announced changes to the treatment of non-resident Irish entities. The changes are not expected to impact existing non-resident Irish entities, such as ours, until after December 31, 2020. These changes, and other prospective changes in the United States and other countries in which we and our affiliates operate could increase our effective tax rate, and harm our financial position and results of operations.
Our sales and operations are subject to the economic, political, legal and business conditions in the countries in which we do business, and our failure to operate successfully or adapt to changes in these conditions could cause our sales and operations to be limited or disrupted.
Since 2007, we have significantly expanded our operations and expect to continue to do so in the future. Our operations in foreign countries subject us to the following additional risks:
• | fluctuations in currency exchange rates; |
• | political or economic determinations that adversely impact pricing or reimbursement policies; |
• | economic problems or political instability that disrupt health care payment systems; |
• | difficulties or inability to obtain financing in markets; |
• | unexpected changes in tariffs, trade barriers and regulatory requirements; |
• | difficulties enforcing contractual and intellectual property rights; |
• | changes in laws, regulations or enforcement practices with respect to our business, including without limitation laws relating to reimbursement, competition, pricing and sales and marketing of our products; |
• | trade restrictions and restrictions on direct investments by foreign entities; |
• | compliance with tax, employment and labor laws; |
• | costs and difficulties in recruiting and retaining qualified managers and employees to manage and operate the business in local jurisdictions; |
• | costs and difficulties in managing and monitoring international operations; and |
• | longer payment cycles. |
Our business and marketing methods are also subject to regulation by the governments of the countries in which we operate. The FCPA and similar anti-bribery laws in other countries prohibit companies and their representatives from offering,
42
promising, authorizing or making payments to foreign officials for the purpose of obtaining or retaining business. We have policies and procedures designed to help ensure that we and our representatives, including our employees, comply with such laws, however we cannot guarantee that these policies and procedures will protect us against liability under the FCPA or other anti-bribery laws for actions taken by our representatives. Failure to comply with the laws and regulations of the countries in which we operate could materially harm our business.
We conduct, or anticipate that we will conduct, a substantial portion of our business in currencies other than the U.S. dollar and we are exposed to fluctuations in foreign currency exchange rates in the normal course of our business. See also Risk Factor "Currency fluctuations and changes in exchange rates could adversely affect our revenue growth, increase our costs and negatively affect our profitability."
The credit and financial market conditions may aggravate certain risks affecting our business.
Sales of Soliris and other products are or will be dependent, in large part, on reimbursement from government health administration organizations and private and governmental third-party payers, and also co-payments from individual patients in certain situations. As a result of adverse credit and financial market conditions, and the overall financial climate, these governmental organizations and payers, and/or individuals, may reduce or delay initiation of treatment, may be unable to satisfy their reimbursement obligations, may delay payment or may seek to reduce reimbursement for our products, including Soliris, in the future, which could have a material adverse effect on our business and results of operations. Soliris is approved for the treatment of patients with PNH and aHUS in the United States, the European Union and Japan and for the treatment of PNH in several other territories. If Soliris is approved in additional territories for PNH, aHUS, or for additional indications that are under clinical development, the reimbursement risks and uncertainties associated with adverse credit and financial market conditions may be exacerbated due to increases in the number of patients receiving Soliris that require reimbursement. Payment defaults by a government payer could require us to expense previously recorded revenue as uncollectible, and might cause us to end or restrict sales to patients in that country. Further, the risk of payment default by a government payer could require us to revise our revenue recognition policies in regard to that payer, causing revenue to be recorded only on a cash basis, and we may be required to end or restrict sales to patients in that country.
We continue to monitor economic conditions, including volatility associated with U.S. and international economies, associated impacts on the financial markets and our business, and the sovereign debt issues in Europe.
We may not be able to successfully mitigate or prevent our exposures to volatile economic and financial conditions and our failure to operate successfully or adapt to changes in these conditions could cause our sales and operations to be limited or disrupted or otherwise harm our business.
Additionally, we rely upon third-parties for certain parts of our business, including Lonza, licensees, wholesale distributors of Soliris, contract clinical trial providers, contract manufacturers and other third-party suppliers and financial institutions. Because of the volatility in the financial markets, there may be a disruption or delay in the performance or satisfaction of commitments to us by these third parties which could have a material adverse effect on our business and results of operations.
Currency fluctuations and changes in exchange rates could adversely affect our revenue growth, increase our costs and negatively affect our profitability.
We conduct, or anticipate that we will conduct, a substantial portion of our business in currencies other than the U.S. dollar. We are exposed to fluctuations in foreign currency exchange rates in the normal course of our business and we expect these exposures to increase during 2015 due to the strengthening of the U.S. dollar. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, including the Euro, Japanese Yen, British Pound, Swiss Franc, and Russian Ruble. We manage our foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. We enter into foreign exchange forward contracts, with durations of up to 60 months, to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. The purpose of the hedges of revenue is to reduce the volatility of exchange rate fluctuations on our operating results and to increase the visibility of the foreign exchange impact on forecasted revenues. Further, we enter into foreign exchange forward contracts, with durations of approximately 30 days, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. While we attempt to hedge certain currency risks, currency fluctuations between the U.S. dollar and the currencies in which we do business have, in the past, caused foreign currency transaction gains and losses and have also impacted the amounts of revenues and expenses calculated in U.S. dollars and will likely do so in the future. Likewise, past currency fluctuations have at times resulted in foreign currency transaction gains, and there can be no assurance that these gains can be reproduced.
43
Changes in healthcare law and implementing regulations, including those based on recently enacted legislation, as well as changes in healthcare policy and coverage and reimbursement of drug products may impact our business in ways that we cannot currently predict and these changes could adversely affect our business and financial condition.
Governments in countries where we operate have adopted or have shown significant interest in pursuing legislative initiatives to reduce costs of health care. Any such government-adopted health care measures could adversely impact the pricing of Soliris or the amount of coverage and reimbursement available for Soliris from governmental agencies or other third-party payers.
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the PPACA), was adopted in the United States in March 2010. This law substantially changes the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, and fraud and abuse enforcement. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
PPACA contains several provisions that have or could potentially impact our business. PPACA made significant changes to the Medicaid Drug Rebate Program. Effective March 23, 2010, rebate liability expanded from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well. With regard to the amount of the rebates owed, PPACA increased the minimum Medicaid rebate from 15.1% to 23.1% of the average manufacturer price for most innovator products; changed the calculation of the rebate for certain innovator products that qualify as line extensions of existing drugs; and capped the total rebate amount for innovator drugs at 100% of the average manufacturer price. In addition, PPACA and subsequent legislation changed the definition of average manufacturer price. Finally, PPACA requires pharmaceutical manufacturers of branded prescription drugs, such as Soliris, to pay a branded prescription drug fee to the federal government beginning in 2011. Each individual pharmaceutical manufacturer pays a prorated share of the branded prescription drug fee of $3.0 billion in 2014 (and set to increase in ensuing years), based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. Sales of "orphan drugs"-those designated under section 526 of the FDCA, like Soliris-are excluded from this fee as long as no non-orphan indications have been approved for the orphan drug.
In 2012, CMS issued proposed regulations to implement the changes to the Medicaid Drug Rebate Program under PPACA but has not yet issued final regulations. CMS is currently expected to release the final regulations in 2015. Moreover, in the future, Congress could enact legislation that further increases Medicaid drug rebates or other costs and charges associated with participating in the Medicaid Drug Rebate Program. The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program has and will continue to increase our costs and the complexity of compliance, has been and will be time-consuming, and could have a material adverse effect on our results of operations.
Additional provisions of PPACA, some of which became effective in 2011, may negatively affect our revenues in the future. For example, as part of PPACA’s provisions closing a coverage gap that currently exists in the Medicare Part D prescription drug program (commonly known as the "donut hole"), we are required to provide a 50% discount on branded prescription drugs dispensed to beneficiaries within this donut hole.
PPACA also expanded the Public Health Service’s 340B drug pricing discount program. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B "ceiling price" for the manufacturer’s covered outpatient drugs. PPACA expanded the 340B program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by PPACA. PPACA exempts "orphan drugs"-those designated under section 526 of the FDCA, such as Soliris-from the ceiling price requirements for these newly-eligible entities. On July 21, 2014, the Health Resources and Services Administration, or HRSA, which administers the 340B program, issued an interpretive rule to implement the orphan drug exception which interprets the orphan drug exception narrowly. It exempts orphan drugs from the ceiling price requirements for the newly-eligible entities only when the orphan drug is used for its orphan indication. The newly-eligible entities are entitled to purchase orphan drugs at the ceiling price when the orphan drug is not used for its orphan indication. A manufacturer trade group has filed a lawsuit challenging the interpretive rule as inconsistent with the statutory language. That challenge remains ongoing. The uncertainty regarding how the statutory orphan drug exception will be applied will increase the complexity of compliance, will make compliance more time-consuming, and could negatively impact our results of operations. If HRSA’s narrow interpretation of the scope of the orphan drug exemption prevails, it could potentially negatively
44
impact the price we are paid for Soliris by certain entities for some uses and increase the complexity of compliance with the 340B program.
In addition, our industry may be affected by broader legislation addressing federal spending, including, for example, a sequester required by the Budget Control Act of 2011, Pub. L. No. 112-25, as amended by the American Taxpayer Relief Act of 2012, Pub. L. 112-240, that took effect in April 2013 and was expended by the Bipartisan Budget Act of 2013, Pub. L. No. 113-67. Under the sequestration, Medicare payments for all items and services, including drugs and biologicals, have been reduced by 2%. This 2% reduction in Medicare payments affects all Parts of the Medicare program and could impact sales of Soliris. As another example, the governments of Germany and Spain each approved increases to mandatory rebates on the sales of pharmaceutical products.
We expect that the implementation of current laws and policies, the amendment of those laws and policies in the future, as well as the adoption of new laws and policies, could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or successfully commercialize our product candidates, or could limit or eliminate our future spending on development projects. In many cases, these government initiatives, even if enacted into law, are subject to future rulemaking by regulatory agencies. Although we have evaluated these government initiatives and the impact on our business, we cannot know with certainty whether any such law, rule or regulation will adversely affect coverage and reimbursement of Soliris, or to what extent, until such laws, rules and regulations are promulgated, implemented and enforced. The announcement or adoption of regulatory or legislative proposals could delay or prevent our entry into new markets, affect our reimbursement or sales in the markets where we are already selling Soliris and materially harm our business, financial condition and results of operations.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program, Medicare, or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Medicare is a U.S. federal government insurance program that covers individuals aged 65 years or older, certain younger individuals with certain disabilities, and individuals with End-Stage Renal Disease. The primary Medicare programs that may affect reimbursement for Soliris are Medicare Part B, which covers physician services and outpatient care, and Medicare Part D, which provides a voluntary outpatient prescription drug benefit. Medicare Part B provides limited coverage of certain outpatient drugs and biologicals that are reasonable and necessary for diagnosis or treatment of an illness or injury. Under Part B, reimbursement is based on a fixed percentage of the applicable product’s average sales price (ASP). Manufacturers calculate ASP based on a statutory formula and must report ASP information to the Centers for Medicare and Medicaid Services (CMS), the federal agency that administers Medicare and the Medicaid Drug Rebate Program, on a quarterly basis.
Medicaid is a government health insurance program for low-income children, families, pregnant women, and people with disabilities. It is jointly funded by the federal and state governments, and it is administered by individual states within parameters established by the federal government. Coverage and reimbursement for drugs and biologicals thus varies by state. Drugs and biologicals may be covered under the medical or pharmacy benefit. State Medicaid programs may impose utilization management controls, such as prior authorization, step therapy, or quantity limits on drugs and biologicals. Medicaid also includes the Medicaid Drug Rebate Program, under which we are required to pay a rebate to each state Medicaid program for quantities of Soliris that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for Soliris under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS. These data include the average manufacturer price and the best price for Soliris.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B "ceiling price" for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program.
Pricing and rebate calculations vary among products and programs. The calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current average manufacturer price and best price for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the
45
Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities, such as safety-net providers, under the 340B drug discount program.
We are liable for errors associated with our submission of pricing data. In addition to retroactive rebates and the potential for 340B program refunds, if we are found to have knowingly submitted false average manufacturer price, ASP, or best price information to the government, we may be liable for civil monetary penalties in the amount of $100,000 per item of false information. If we are found to have made a misrepresentation in the reporting of our average sales price, the Medicare statute provides for civil monetary penalties of up to $10,000 for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit monthly/quarterly average manufacturer price, ASP, and best price data on a timely basis could result in a civil monetary penalty of $10,000 per day for each day the information is late beyond the due date. Such failure also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs.
In September 2010, CMS and the Office of Inspector General (OIG) indicated that they intend to pursue more aggressively those companies who fail to report these data to the government in a timely manner. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. We cannot assure you that our submissions will not be found by CMS to be incomplete or incorrect.
Federal law requires that for a company to be eligible to have its products paid for with federal funds under the Medicaid program as well as to be purchased by certain federal agencies and grantees, it also must participate in the Department of Veterans Affairs, or VA, Federal Supply Schedule, or FSS, pricing program. To participate, we are required to enter into an FSS contract with the VA, under which we must make our innovator "covered drugs" available to the "Big Four" federal agencies - the VA, the Department of Defense, or DoD, the Public Health Service, and the Coast Guard - at pricing that is capped pursuant to a statutory federal ceiling price, or FCP, formula set forth in Section 603 of the Veterans Health Care Act of 1992, or VHCA. The FCP is based on a weighted average non-federal average manufacturer price, or Non-FAMP, which manufacturers are required to report on a quarterly and annual basis to the VA. If a company misstates Non-FAMPs or FCPs it must restate these figures. Pursuant to the VHCA, knowing provision of false information in connection with a Non-FAMP filing can subject a manufacturer to penalties of $100,000 for each item of false information.
FSS contracts are federal procurement contracts that include standard government terms and conditions, separate pricing for each product, and extensive disclosure and certification requirements. All items on FSS contracts are subject to a standard FSS contract clause that requires FSS contract price reductions under certain circumstances where pricing is reduced to an agreed "tracking customer." Further, in addition to the "Big Four" agencies, all other federal agencies and some non-federal entities are authorized to access FSS contracts. FSS contractors are permitted to charge FSS purchasers other than the Big Four agencies "negotiated pricing" for covered drugs that is not capped by the FCP; instead, such pricing is negotiated based on a mandatory disclosure of the contractor’s commercial "most favored customer" pricing. We offer dual pricing on our FSS contract.
In addition, pursuant to regulations issued by the DoD TRICARE Management Activity, now the Defense Health Agency, to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, each of our covered drugs is listed on a Section 703 Agreement under which we have agreed to pay rebates on covered drug prescriptions dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. Companies are required to list their innovator products on Section 703 Agreements in order for those products to be eligible for DoD formulary inclusion. The formula for determining the rebate is established in the regulations and our Section 703 Agreement and is based on the difference between the annual Non-FAMP and the FCP (as described above, these price points are required to be calculated by us under the VHCA).
If we overcharge the government in connection with our FSS contract or Section 703 Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S., some of the laws that may
46
apply include state security breach notification laws, state health information privacy laws and federal and state consumer protections laws which impose requirements for the collection, use, disclosure and transmission of personal information. Each of these laws may be subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations we could be subject to penalties or sanctions. Accordingly, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting the violation of HIPAA.
In addition, the receipt of personal health information in connection with our clinical trial initiatives is subject to state and federal human subject protection laws. These laws could create liability for us if one of our research collaborators were to use or disclose research subject information without consent and in violation of applicable laws.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. European Union member states and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. For example, the EU Data Protection Directive, as implemented into national laws by the EU member states, imposes strict obligations and restrictions on the ability to collect, analyze, and transfer personal data, including health data from clinical trials and adverse event reporting. Data protection authorities from different EU member states may interpret the EU Data Protection Directive and national laws differently, which adds to the complexity of processing personal data in the EU, and guidance on implementation and compliance practices are often updated or otherwise revised. The EU Data Protection Directive prohibits the transfer of personal data to countries outside of the EU member states that are not considered by the European Commission to provide an adequate level of data protection. These countries include the United States. Any failure to comply with the rules arising from the EU Data Protection Directive and related national laws of European Union member states could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results
A proposal for an EU Data Protection Regulation, intended to replace the current EU Data Protection Directive, is currently under consideration. The EU Data Protection Regulation is expected to introduce new data protection requirements in the EU and substantial fines for breaches of the data protection rules. If the draft EU Data Protection Regulation is adopted in its current form, it may increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
Security breaches, cyber-attacks, or other disruptions could expose us to liability and affect our business and reputation.
We collect, store, and transmit sensitive information including intellectual property, proprietary business information and personal information in connection with business operations. We have implemented information security measures to protect patients’ personal information against the risk of inappropriate and unauthorized external use and disclosure. However, despite these measures, and due to the ever changing information cyber-threat landscape, we may be subject to data breaches through cyber-attacks perpetrated by individuals that attempt to compromise our security controls. If our systems were to fail or be disrupted for an extended period of time we could lose product sales and our revenue and reputation would suffer. In the event our systems were to be breached by an unauthorized third-party, they could potentially access confidential personal information, which could cause us to suffer reputational damage and loss of customer confidence. Such incidents would result in notification obligations to affected individuals and government agencies, legal claims or proceedings, and liability under federal and state laws that protect the privacy and security of personal information. Any one of these events could cause our business to be materially harmed and our results of operations would be adversely impacted.
47
Risks Related to Our Common Stock
If the trading price of our common stock continues to fluctuate in a wide range, our stockholders will have uncertainty with respect to an investment in our common stock.
The trading price of our common stock has been volatile and may continue to be volatile in the future. Factors such as announcements of fluctuations in our or our competitors' operating results or clinical or scientific results, fluctuations in the trading prices or business prospects of our competitors and collaborators, changes in our prospects, particularly with respect to sales of Soliris, failure to resolve, delays in resolving or other developments with respect to the issues raised in the Warning Letter, and market conditions for biopharmaceutical stocks in general could have a significant impact on the future trading prices of our common stock. In particular, the trading price of the common stock of many biopharmaceutical companies, including ours, has experienced extreme price and volume fluctuations, which have at times been unrelated to the operating performance of the companies whose stocks were affected. This is due to several factors, including general market conditions, sales of Soliris, the announcement of the results of our clinical trials or product development and the results of our efforts to obtain regulatory approval for our products. While we cannot predict our future performance, if our stock price continues to fluctuate in a wide range, an investment in our common stock may result in considerable uncertainty for an investor.
Anti-takeover provisions of Delaware law, provisions in our charter and bylaws and our stockholders' rights plan, or poison pill, could make a third-party acquisition of us difficult and may frustrate any attempt to remove or replace our current management.
Because we are a Delaware corporation, the anti-takeover provisions of Delaware law could make it more difficult for a third party to acquire control of us, even if the change in control would be beneficial to stockholders. We are subject to the provisions of Section 203 of the Delaware General Laws, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our corporate charter and by-law provisions and stockholder rights plan may discourage certain types of transactions involving an actual or potential change of control that might be beneficial to us or our stockholders. Our bylaws provide that special meetings of our stockholders may be called only by the Chairman of the Board, the President, the Secretary, or a majority of the Board of Directors, or upon the written request of stockholders who together own of record 50% of the outstanding stock of all classes entitled to vote at such meeting. Our bylaws also specify that the authorized number of directors may be changed only by resolution of the board of directors. Our charter does not include a provision for cumulative voting for directors, which may have enabled a minority stockholder holding a sufficient percentage of a class of shares to elect one or more directors. Under our charter, our board of directors has the authority, without further action by stockholders, to designate up to 5,000 shares of preferred stock in one or more series. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of the holders of any class or series of preferred stock that may be issued in the future.
Pursuant to our stockholder rights plan, each share of common stock has an associated preferred stock purchase right. The rights will not trade separately from the common stock until, and are exercisable only upon, the acquisition or the potential acquisition through tender offer by a person or group of 20% or more of the outstanding common stock. The rights are designed to make it more likely that all of our stockholders receive fair and equal treatment in the event of any proposed takeover of us and to guard against the use of partial tender offers or other coercive tactics to gain control of us. These provisions could delay or discourage transactions involving an actual or potential change in control of us or our management, including transactions in which stockholders might otherwise receive a premium for their shares over then current prices. These provisions could also limit the ability of stockholders to remove current management or approve transactions that stockholders may deem to be in their best interests and could adversely affect the price of our common stock.
Item 1B. | UNRESOLVED STAFF COMMENTS. |
None.
48
Item 2. | PROPERTIES. |
We conduct our primary operations at the owned and leased facilities described below.
Location | Operations Conducted | Approximate Square Feet | Lease Expiration Dates | ||||
Cheshire, Connecticut | Corporate headquarters and executive, sales, research and development offices | 254,000 | 2016 and 2020 | ||||
Smithfield, Rhode Island | Commercial, research and development manufacturing | 67,000 | N/A | ||||
Lausanne, Switzerland | Regional executive and sales offices | 48,000 | 2019 | ||||
Dublin, Ireland | Global supply chain and distribution | 15,800 | 2023 | ||||
We believe that our administrative office space is adequate to meet our needs for the foreseeable future. We also believe that our research and development facilities and our manufacturing facility, together with third party manufacturing facilities, will be adequate for our on-going activities. In addition to the locations above, we also lease space in other U.S. locations and foreign countries to support our operations as a global organization.
In November 2012, we entered into a new lease agreement for approximately 328,000 square feet of office and laboratory space to be constructed in New Haven, Connecticut. We amended the lease in July 2013 to expand the building and increase the leased space to a total of approximately 408,000 square feet. The construction of the facility began in June 2013 and is expected to be completed in 2015. Upon completion of the new facility, we will relocate our headquarters and Cheshire operations to New Haven.
In April 2014, we purchased a fill/finish facility in Athlone, Ireland. Following refurbishment of the facility, and after successful completion of the appropriate validation processes and regulatory approvals, the facility will become our first company-owned fill/finish and packaging facility for Soliris and other clinical and commercial products. Our plans for future expansion in Ireland also include the construction of office and laboratory facilities on property in Dublin, Ireland, which we purchased in April 2014.
In January 2015, we entered into a new lease agreement for approximately 44,000 square feet of office space in Zurich, Switzerland to support the relocation of the European headquarters. The term of the new lease is estimated to commence in the second quarter of 2015 and will expire 10 years later, with a minimum renewal option of 5 years and a maximum renewal option of 10 years.
Item 3. | LEGAL PROCEEDINGS. |
From time to time, we are party to legal proceedings in the course of our business. We do not expect any such current legal proceedings to have a material adverse impact on our business or financial condition.
Item 4. | MINE SAFETY DISCLOSURES. |
Not applicable.
49
PART II
Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Our common stock is quoted on The NASDAQ Stock Market, LLC under the symbol “ALXN.” The following table sets forth the range of high and low sales prices for our common stock on The NASDAQ Stock Market, LLC for the periods indicated since January 1, 2013.
Fiscal 2013 | High | Low | |||||
First Quarter | |||||||
(January 1, 2013 to March 31, 2013) | $ | 103.20 | $ | 81.82 | |||
Second Quarter | |||||||
(April 1, 2013 to June 30, 2013) | $ | 108.13 | $ | 87.01 | |||
Third Quarter | |||||||
(July 1, 2013 to September 30, 2013) | $ | 125.65 | $ | 93.34 | |||
Fourth Quarter | |||||||
(October 1, 2013 to December 31, 2013) | $ | 133.75 | $ | 100.89 | |||
Fiscal 2014 | |||||||
First Quarter | |||||||
(January 1, 2014 to March 31, 2014) | $ | 185.43 | $ | 126.76 | |||
Second Quarter | |||||||
(April 1, 2014 to June 30, 2014) | $ | 172.50 | $ | 136.37 | |||
Third Quarter | |||||||
(July 1, 2014 to September 30, 2014) | $ | 173.70 | $ | 154.38 | |||
Fourth Quarter | |||||||
(October 1, 2014 to December 31, 2014) | $ | 203.30 | $ | 155.01 | |||
As of January 28, 2015, we had approximately 57 stockholders of record of our common stock and an estimated 146,830 beneficial owners. The closing sale price of our common stock on January 28, 2015 was $177.78 per share.
DIVIDEND POLICY
We have never paid cash dividends. We do not expect to declare or pay any cash dividends on our common stock in the near future. We intend to retain all earnings, if any, to invest in our operations. The payment of future dividends is within the discretion of our board of directors and will depend upon our future earnings, if any, our capital requirements, financial condition and other relevant factors.
ISSUER PURCHASES OF EQUITY SECURITIES (amounts in thousands except per share amounts)
The following table summarizes our common stock repurchase activity during the fourth quarter of 2014:
Period | Total Number of Shares Purchased | Average Price Paid per Share | Total Number of Shares Purchased as Part of Publicly Announced Programs | Maximum Dollar Value of Shares that May Yet Be Purchased Under the Programs | |||||||
October 1-31, 2014 | 100 | 169.58 | 100 | 22,273 | |||||||
November 1-30, 2014 | — | — | — | 22,273 | |||||||
December 1-31, 2014 | 14 | 178.88 | 14 | 519,712 | |||||||
Total | 114 | 170.72 | 114 | ||||||||
On November 8, 2012, we announced that our Board of Directors authorized the repurchase of up to $400,000 of our common stock. On December 15, 2014 we announced that our Board of Directors authorized the repurchase of up to an additional $500,000 of our common stock. The repurchase program does not have an expiration date. As of December 31, 2014, the maximum dollar value of shares remaining for purchase under the program was $519,712.
50
EQUITY COMPENSATION PLAN INFORMATION (amounts in thousands except per share amounts)
Plan Category | Number of shares of common stock to be issued upon exercise of outstanding options (2) | Weighted- average exercise price of outstanding options | Weighted- average term to expiration of options outstanding | Number of shares of common stock remaining available for future issuance under equity compensation plans | ||||||||
Equity compensation plans approved by stockholders (1) | 6,420 | $ | 85.65 | 6.98 | 12,043 | |||||||
Equity compensation plans not approved by stockholders | — | $ | — | — | — | |||||||
(1) | Reflects number of shares of common stock to be issued upon exercise of outstanding options under all our equity compensation plans, including our Amended and Restated 2004 Incentive Plan. All 12,043 shares of common stock remaining available for future issuance are available under the Amended and Restated 2004 Incentive Plan. |
(2) | Does not include 1,808 restricted shares outstanding that were issued under the Amended and Restated 2004 Incentive Plan. |
The outstanding options and restricted shares are not transferable for consideration and do not have dividend equivalent rights attached.
51
THE COMPANY’S STOCK PERFORMANCE
The following graph compares cumulative total return of the Company’s Common Stock with the cumulative total return of (i) the NASDAQ Stock Market-United States, and (ii) the NASDAQ Biotechnology Index. The graph assumes (a) $100 was invested on December 31, 2009 in each of the Company’s Common Stock, the stocks comprising the NASDAQ Stock Market-United States and the stocks comprising the NASDAQ Biotechnology Index, and (b) the reinvestment of dividends. The comparisons shown in the graph are based on historical data and the stock price performance shown in the graph is not necessarily indicative of, or intended to forecast, future performance of our stock.
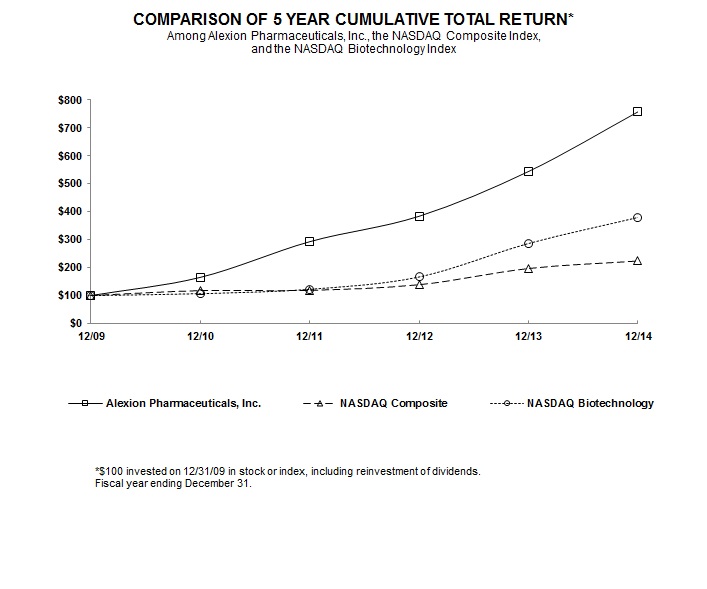
CUMULATIVE TOTAL RETURN
12/09 | 12/10 | 12/11 | 12/12 | 12/13 | 12/14 | ||||||||||||
Alexion Pharmaceuticals, Inc. | 100.00 | 164.99 | 292.91 | 384.02 | 544.38 | 758.01 | |||||||||||
NASDAQ Composite | 100.00 | 117.61 | 118.70 | 139.00 | 196.83 | 223.74 | |||||||||||
NASDAQ Biotechnology | 100.00 | 106.73 | 122.40 | 166.72 | 286.55 | 379.71 | |||||||||||
52
Item 6. | SELECTED FINANCIAL DATA. |
The following selected financial data is derived from, and should be read in conjunction with, the financial statements, including the notes thereto, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K.
(amounts in thousands, except per share amounts)
Consolidated Statements of Operations Data: | |||||||||||||||||||
Year Ended December 31, | |||||||||||||||||||
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
Net product sales | $ | 2,233,733 | $ | 1,551,346 | $ | 1,134,114 | $ | 783,431 | $ | 540,957 | |||||||||
Cost of sales: | |||||||||||||||||||
Cost of sales | 173,862 | 168,375 | 126,214 | 93,140 | 64,437 | ||||||||||||||
Change in contingent liability from intellectual property settlements | — | 9,181 | (53,377 | ) | — | — | |||||||||||||
Total cost of sales | 173,862 | 177,556 | 72,837 | 93,140 | 64,437 | ||||||||||||||
Operating expenses: | |||||||||||||||||||
Research and development | 513,782 | 317,093 | 222,732 | 137,421 | 98,394 | ||||||||||||||
Selling, general and administrative | 630,209 | 489,720 | 384,678 | 308,176 | 226,766 | ||||||||||||||
Acquisition-related costs | 20,295 | 5,029 | 22,812 | 13,486 | 722 | ||||||||||||||
Impairment of intangible assets | 11,514 | 33,521 | 26,300 | — | — | ||||||||||||||
Restructuring expenses | 15,365 | — | — | — | — | ||||||||||||||
Amortization of purchased intangible assets | — | 417 | 417 | 382 | — | ||||||||||||||
Total operating expenses | 1,191,165 | 845,780 | 656,939 | 459,465 | 325,882 | ||||||||||||||
Operating income | 868,706 | 528,010 | 404,338 | 230,826 | 150,638 | ||||||||||||||
Other income (expense) | 3,401 | (1,741 | ) | (6,772 | ) | (1,158 | ) | (1,627 | ) | ||||||||||
Income before income taxes | 872,107 | 526,269 | 397,566 | 229,668 | 149,011 | ||||||||||||||
Income tax provision | 215,195 | 273,374 | 142,744 | 54,353 | 51,981 | ||||||||||||||
Net income | $ | 656,912 | $ | 252,895 | $ | 254,822 | $ | 175,315 | $ | 97,030 | |||||||||
Earnings per common share | |||||||||||||||||||
Basic | $ | 3.32 | $ | 1.29 | $ | 1.34 | $ | 0.96 | $ | 0.54 | |||||||||
Diluted | $ | 3.26 | $ | 1.27 | $ | 1.28 | $ | 0.91 | $ | 0.52 | |||||||||
Shares used in computing earnings per common share | |||||||||||||||||||
Basic | 198,103 | 195,532 | 190,461 | 183,220 | 178,542 | ||||||||||||||
Diluted | 201,623 | 199,712 | 198,501 | 191,806 | 186,074 | ||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||||
As of December 31, | |||||||||||||||||||
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
Cash, cash equivalents and marketable securities | $ | 1,961,566 | $ | 1,514,851 | $ | 989,501 | $ | 540,865 | $ | 361,605 | |||||||||
Total assets | 4,201,962 | 3,317,696 | 2,613,560 | 1,394,751 | 1,012,037 | ||||||||||||||
Long-term debt and convertible notes (current and noncurrent) | 57,500 | 113,000 | 149,000 | — | 3,718 | ||||||||||||||
Contingent consideration (current and noncurrent) | 162,971 | 142,676 | 141,670 | 18,120 | — | ||||||||||||||
Facility lease obligation | 107,099 | 32,230 | — | — | — | ||||||||||||||
Total stockholders’ equity | 3,302,018 | 2,382,079 | 1,970,850 | 1,134,492 | 859,736 | ||||||||||||||
53
Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. (amounts in thousands, except percentages and per share data) |
In addition to historical information, this report contains forward-looking statements that involve risks and uncertainties which may cause our actual results to differ materially from plans and results discussed in forward-looking statements. We encourage you to review the risks and uncertainties, discussed in the section entitled item 1A “Risk Factors”, and the “Note Regarding Forward-Looking Statements”, included at the beginning of this Annual Report on Form 10-K. The risks and uncertainties can cause actual results to differ significantly from those forecast in forward-looking statements or implied in historical results and trends.
The following discussion should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K.
Overview
We are a biopharmaceutical company focused on serving patients with severe and ultra-rare disorders through the innovation, development and commercialization of life-transforming therapeutic products. Our marketed product Soliris is the first and only therapeutic approved for patients with either of two severe and ultra-rare disorders resulting from chronic uncontrolled activation of the complement component of the immune system: paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening and ultra-rare genetic blood disorder, and atypical hemolytic uremic syndrome (aHUS), a life-threatening and ultra-rare genetic disease. We are also evaluating additional potential indications for Soliris in severe and devastating diseases in which we believe that uncontrolled complement activation is the underlying mechanism, and we are progressing in various stages of development with additional biotechnology product candidates as treatments for patients with severe and life-threatening ultra-rare disorders. We were incorporated in 1992 and began commercial sale of Soliris in 2007.
Soliris is designed to inhibit a specific aspect of the complement component of the immune system and thereby treat inflammation associated with chronic disorders in several therapeutic areas, including hematology, nephrology, transplant rejection and neurology. Soliris is a humanized monoclonal antibody that effectively blocks terminal complement activity at the doses currently prescribed. The initial indication for which we received approval for Soliris is PNH. PNH is a debilitating and life-threatening, ultra-rare genetic blood disorder defined by chronic uncontrolled complement activation leading to the destruction of red blood cells (hemolysis). The chronic hemolysis in patients with PNH may be associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breath and intermittent episodes of dark-colored urine (hemoglobinuria).
Soliris was approved for the treatment of PNH by the U.S. Food and Drug Administration (FDA) and the European Commission (EC) in 2007 and by Japan’s Ministry of Health, Labour and Welfare (MHLW) in 2010, and has been approved in several other territories. Additionally, Soliris has been granted orphan drug designation for the treatment of PNH in the United States, Europe, Japan and several other territories.
In September and November 2011, Soliris was approved by the FDA and EC, respectively, for the treatment of pediatric and adult patients with aHUS in the United States and Europe. In September 2013, the MHLW approved Soliris for the treatment of pediatric and adult patients with aHUS in Japan. aHUS is a severe and life-threatening genetic ultra-rare disease characterized by chronic uncontrolled complement activation and thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, causing a reduction in platelet count (thrombocytopenia) and life-threatening damage to the kidney, brain, heart and other vital organs. In addition, the FDA and EC have granted Soliris orphan drug designation for the treatment of patients with aHUS.
In addition to PNH and aHUS, we believe that Soliris may be useful in the treatment of a variety of other serious diseases and conditions resulting from uncontrolled complement activation. We are currently evaluating additional potential indications for Soliris in severe and ultra-rare diseases in which uncontrolled complement activation is the underlying mechanism. We are also progressing in various stages of development with additional biotechnology product candidates that target severe and life-threatening ultra-rare diseases for which we believe current treatments are either non-existent or inadequate. These therapeutics focus on metabolic and inflammatory diseases. We are also involved in the research associated with the identification and development of new therapeutics pursuant to ongoing license and collaboration agreements.
Critical Accounting Policies and the Use of Estimates
The significant accounting policies and basis of preparation of our consolidated financial statements are described in Note 1, “Business Overview and Summary of Significant Accounting Policies” of the Consolidated Financial Statements included in
54
this Annual Report on Form 10-K. Under accounting principles generally accepted in the United States, we are required to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and disclosure of contingent assets and liabilities in our financial statements. Actual results could differ from those estimates.
We believe the judgments, estimates and assumptions associated with the following critical accounting policies have the greatest potential impact on our consolidated financial statements:
• | Revenue recognition; |
• | Contingent liabilities; |
• | Inventories; |
• | Share-based compensation; |
• | Valuation of goodwill, acquired intangible assets and in-process research and development (IPR&D); |
• | Valuation of contingent consideration; and |
• | Income taxes. |
Revenue Recognition
Net Product Sales
Our principal source of revenue is product sales. We recognize revenue from product sales when persuasive evidence of an arrangement exists, title to product and associated risk of loss has passed to the customer, the price is fixed or determinable, collection from the customer is reasonably assured, and we have no further performance obligations. Depending on these criteria, revenue is usually recorded upon receipt of the product by the end customer, which is typically a hospital, physician’s office, private or government pharmacy or other health care facility. On a regular basis, we review revenue arrangements, such as distributor relationships, to determine whether changes in these criteria have an impact on revenue recognition. Amounts collected from customers and remitted to governmental authorities, such as value-added taxes (VAT) in foreign jurisdictions, are presented on a net basis in our consolidated statements of operations and do not impact net product sales.
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other health care providers. In some cases, we may also sell Soliris to governments and government agencies.
Because of factors such as the pricing of Soliris, the limited number of patients, the short period from product sale to patient infusion and the lack of contractual return rights, Soliris customers often carry limited inventory. We also monitor inventory within our sales channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms and financial strength of distributors. In certain countries, exact quantities of inventory in the channel are not precisely known, requiring us to estimate these amounts. If actual amounts of inventory differ from these estimates, these adjustments could have an impact in the period in which these estimates change.
In addition to sales in countries where Soliris is commercially available, we have also recorded revenue on sales for patients receiving Soliris treatment through named-patient programs. The relevant authorities or institutions in those countries have agreed to reimburse for product sold on a named-patient basis where Soliris has not received final approval for commercial sale.
We record estimated rebates payable under governmental programs, including Medicaid in the United States and other programs outside the United States, as a reduction of revenue at the time of product sale. Our calculations related to these rebate accruals require analysis of historical claim patterns and estimates of customer mix to determine which sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions each period and record any necessary adjustments, which may have an impact on revenue in the period in which the adjustment is made. Generally, the length of time between product sale and the processing and reporting of the rebates is three to six months.
We have entered into volume-based arrangements with governments in certain countries in which reimbursement is limited to a contractual amount. Under this type of arrangement, amounts billed in excess of the contractual limitation are repaid to these governments as a rebate. We estimate incremental discounts resulting from these contractual limitations, based on estimated sales during the limitation period, and we apply the discount percentage to product shipments as a reduction of revenue. Our calculations related to these arrangements require estimation of sales during the limitation period, and adjustments in these estimates may have an impact in the period in which these estimates change.
55
We have provided balances and activity in the rebates payable account for the years ended December 31, 2014, 2013 and 2012 as follows:
Rebates Payable | |||
Balance at December 31, 2011 | $ | 21,746 | |
Current provisions relating to sales in current year | 80,131 | ||
Adjustments relating to prior years | (2,566 | ) | |
Payments/credits relating to sales in current year | (22,634 | ) | |
Payments/credits relating to sales in prior years | (14,343 | ) | |
Balance at December 31, 2012 | $ | 62,334 | |
Current provisions relating to sales in current year | 149,247 | ||
Adjustments relating to prior years | (2,180 | ) | |
Payments/credits relating to sales in current year | (29,574 | ) | |
Payments/credits relating to sales in prior years | (55,530 | ) | |
Balance at December 31, 2013 | $ | 124,297 | |
Current provisions relating to sales in current year | 62,478 | ||
Adjustments relating to prior years | (87,004 | ) | |
Payments/credits relating to sales in current year | (33,922 | ) | |
Payments/credits relating to sales in prior years | (29,022 | ) | |
Balance at December 31, 2014 | $ | 36,827 | |
In March 2014, we entered into an agreement with the French government which positively impacts prospective reimbursement of Soliris and also provides for reimbursement for shipments in years prior to January 1, 2014. As a result of this agreement, in the first quarter 2014, we reduced the rebate payable and recognized $87,830 of net product sales from Soliris in France relating to years prior to January 1, 2014. In addition, our current provisions relating to sales in the current year decreased by $86,769 during 2014 primarily due to this agreement.
In 2013 compared to 2012, current provisions relating to sales in the current year increased by $69,116 primarily due to estimated rebates payable in France during 2013. The increase in rebates payable in France of approximately $57,900 in 2013 was due to increased unit volumes and contractual reimbursement limitations. The remaining increase in current provisions related to increased unit volumes in the United States and Europe which were subject to rebates.
We record distribution and other fees paid to our customers as a reduction of revenue, unless we receive an identifiable and separate benefit for the consideration and we can reasonably estimate the fair value of the benefit received. If both conditions are met, we record the consideration paid to the customer as an operating expense. These costs are typically known at the time of sale, resulting in minimal adjustments subsequent to the period of sale.
We enter into foreign exchange forward contracts to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. These hedges are designated as cash flow hedges upon inception. We record the effective portion of these cash flow hedges to revenue in the period in which the sale is made to an unrelated third party and the derivative contract is settled.
We evaluate the creditworthiness of customers on a regular basis. In certain European countries, sales by us are subject to payment terms that are statutorily determined. This is primarily the case in countries where the payer is government-owned or government-funded, which we consider to be creditworthy. The length of time from sale to receipt of payment in certain countries exceeds our credit terms. In countries in which collections from customers extend beyond normal payment terms, we seek to collect interest. We record interest on customer receivables as interest income when collected. For non-interest bearing receivables with an estimated payment beyond one year, we discount the accounts receivable to present value at the date of sale, with a corresponding adjustment to revenue. Subsequent adjustments for further declines in credit rating are recorded as bad debt expense as a component of selling, general and administrative expense. We also use judgments as to our ability to collect outstanding receivables and provide allowances for the portion of receivables if and when collection becomes doubtful, and we also assess on an ongoing basis whether collectibility is reasonably assured at the time of sale.
We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. For additional information related to our concentration of credit
56
risk associated with certain international accounts receivable balances, refer to the "Financial Condition, Liquidity and Capital Resources" section below.
Contingent liabilities
We are currently involved in various claims, lawsuits and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims, we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adjustment to our operating results.
Inventories
Inventories are stated at the lower of cost or estimated realizable value. We determine the cost of inventory using the weighted-average cost method.
We capitalize inventory produced for commercial sale, which may include costs incurred for certain products awaiting regulatory approval. We capitalize inventory produced in preparation of product launches sufficient to support estimated initial market demand. Capitalization of such inventory begins when we have (i) obtained positive results in clinical trials that we believe are necessary to support regulatory approval, (ii) concluded that uncertainties regarding regulatory approval have been sufficiently reduced, and (iii) determined that the inventory has probable future economic benefit. In evaluating whether these conditions have been met, we consider clinical trial results for the underlying product candidate, results from meetings with regulatory authorities, and the compilation of the regulatory application. If we are aware of any material risks or contingencies outside of the standard regulatory review and approval process, or if there are any specific negative issues identified relating to the safety, efficacy, manufacturing, marketing or labeling of the product that would have a significant negative impact on its future economic benefits, the related inventory would not be capitalized.
Products that have been approved by the FDA or other regulatory authorities, such as Soliris, are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of Soliris utilized for both commercial and clinical programs is identical and, as a result, the inventory has an "alternative future use" as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to research and development expense when the product enters the research and development process and no longer can be used for commercial purposes and, therefore, does not have an "alternative future use".
For products which are under development and have not yet been approved by regulatory authorities, purchased drug product is charged to research and development expense when the inventory passes quality inspection and ownership transfers to us. Nonrefundable advance payments for research and development activities, including production of purchased drug product, are deferred and capitalized until the goods are delivered. We also recognize expense for raw materials purchased when the raw materials pass quality inspection, and we have an obligation to pay for the materials.
We analyze our inventory levels to identify inventory that may expire prior to sale, inventory that has a cost basis in excess of its estimated realizable value, or inventory in excess of expected sales requirements. Although the manufacturing of our product is subject to strict quality control, certain batches or units of product may no longer meet quality specifications or may expire, which would require adjustments to our inventory values. We also apply judgment related to the results of quality tests that we perform throughout the production process, as well as our understanding of regulatory guidelines, to determine if it is probable that inventory will be saleable. These quality tests are performed throughout the pre- and post-production process, and we continually gather information regarding product quality for periods after the manufacturing date. Soliris currently has a maximum estimated life of 48 months and, based on our sales forecasts, we expect to realize the carrying value of the Soliris inventory. In the future, reduced demand, quality issues or excess supply beyond those anticipated by management may result in a material adjustment to inventory levels, which would be recorded as an increase to cost of sales.
The determination of whether or not inventory costs will be realizable requires estimates by our management. A critical input in this determination is future expected inventory requirements based on internal sales forecasts. We then compare these requirements to the expiry dates of inventory on hand. For inventories that are capitalized in preparation of product launch, we also consider the expected approval date in assessing realizability. To the extent that inventory is expected to expire prior to
57
being sold, we will write down the value of inventory. If actual results differ from those estimates, additional inventory write-offs may be required.
Share-Based Compensation
We have one share-based compensation plan known as the Amended and Restated 2004 Incentive Plan. Under this plan, restricted stock, restricted stock units, stock options and other stock-related awards may be granted to our directors, officers, employees and consultants or advisors of the Company or any subsidiary. To date, share-based compensation issued under the plan consists of incentive and non-qualified stock options, restricted stock and restricted stock units, including restricted stock units with market and non-market performance conditions. Stock-related awards are also outstanding under other share-based compensation plans, but we have not granted awards under these plans since 2004.
Compensation expense for our share-based awards is recognized based on the estimated fair value of the awards on the grant date. Compensation expense reflects an estimate of the number of awards expected to vest and is primarily recognized on a straight-line basis over the requisite service period of the individual grants, which typically equals the vesting period. Compensation expense for awards with performance conditions is recognized using the graded-vesting method.
Our estimates of employee stock option values rely on estimates of factors we input into the Black-Scholes model. The key factors involve an estimate of future uncertain events. Significant assumptions include the use of historical volatility to determine the expected stock price volatility. We also estimate expected term until exercise and the reduction in the expense from expected forfeitures. We currently use historical exercise and cancellation patterns as our best estimate of future estimated life. Actual volatility and lives of options may be significantly different from our estimates.
For our non-market performance-based awards, we estimate the anticipated achievement of the performance targets, including forecasting the achievement of future financial targets. These estimates are revised periodically based on the probability of achieving the performance targets and adjustments are made throughout the performance period as necessary. We use payout simulation models to estimate the grant date fair value of market performance-based awards. The payout simulation models assume volatility of our common stock and the common stock of a comparator group of companies, as well as correlations of returns of the price of our common stock and the common stock prices of the comparator group.
If factors change or we employ different assumptions to value our stock-based awards, the share-based compensation expense that we record in future periods may differ materially from our prior recorded amounts.
Valuation of Goodwill, Acquired Intangible Assets and In-Process Research and Development (IPR&D)
We have recorded goodwill, acquired intangible assets and IPR&D related to our business combinations. When identifiable intangible assets, including IPR&D, are acquired, we determine the fair values of the assets as of the acquisition date. Discounted cash flow models are typically used in these valuations if quoted market prices are not available, and the models require the use of significant estimates and assumptions including but not limited to:
•timing and costs to complete the in-process projects;
•timing and probability of success of clinical events or regulatory approvals;
• | estimated future cash flows from product sales resulting from completed products and in-process projects; and |
•discount rates.
We may also utilize a cost approach, which estimates the costs that would be incurred to replace the assets being purchased. Significant inputs into the cost approach include estimated rates of return on historical costs that a market participant would expect to pay for these assets.
Intangible assets related to IPR&D are treated as indefinite-lived intangible assets and are not amortized until the product is approved for sale by regulatory authorities in specified markets. At that time, we will determine the useful life of the asset, reclassify the asset out of IPR&D and begin amortization. Impairment testing is performed at least annually or when a triggering event occurs that could indicate a potential impairment. In 2014, 2013, and 2012, we recognized impairment charges of $11,514, $29,736, and $26,300, respectively, associated with early stage indefinite-lived intangible assets acquired in connection with the purchases of Taligen and Orphatec. As of December 31, 2014, the remaining carrying value of our IPR&D was not impaired.
Intangible assets with definite useful lives are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events occur. In 2013, we also recognized an impairment charge of $3,785 associated with a purchased technology asset acquired in connection with the Taligen acquisition.
58
If projects are not successfully developed, our sales and profitability may be adversely affected in future periods. Additionally, the value of the acquired intangible assets, including IPR&D, may become impaired if the underlying projects do not progress as we initially estimated. We believe that the assumptions used in developing our estimates of intangible asset values were reasonable at the time of the respective acquisitions. However, the underlying assumptions used to estimate expected project sales, development costs, profitability, or the events associated with such projects, such as clinical results, may not occur as we estimated at the acquisition date.
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We are organized and operate as a single reporting unit and therefore the goodwill impairment test is performed using our overall market value, as determined by our traded share price, compared to our book value of net assets. We completed our annual impairment test as of December 31, 2014 and determined the carrying value of goodwill was not impaired.
Valuation of Contingent Consideration
We record contingent consideration resulting from a business combination at its fair value on the acquisition date. We determine the fair value of the contingent consideration based primarily on the following factors:
•timing and probability of success of clinical events or regulatory approvals;
• | timing and probability of success of meeting commercial milestones, such as estimated future sales levels of a specific compound; and |
•discount rates.
Our contingent consideration liabilities arose in connection with our business combinations. On a quarterly basis, we revalue these obligations and record increases or decreases in their fair value as an adjustment to operating earnings. Changes to contingent consideration obligations can result from adjustments to discount rates, accretion of the discount rates due to the passage of time, changes in our estimates of the likelihood or timing of achieving development or commercial milestones, changes in the probability of certain clinical events or changes in the assumed probability associated with regulatory approval.
The assumptions related to determining the value of contingent consideration include a significant amount of judgment, and any changes in the underlying estimates could have a material impact on the amount of contingent consideration expense recorded in any given period.
Income Taxes
We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax basis of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. We provide a valuation allowance when it is more likely than not that deferred tax assets will not be realized. We recognize the benefit of an uncertain tax position that has been taken or we expect to take on income tax returns if such tax position is more likely than not to be sustained.
We follow the authoritative guidance regarding accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. These unrecognized tax benefits relate primarily to issues common among multinational corporations in our industry. We apply a variety of methodologies in making these estimates which include studies performed by independent economists, advice from industry and subject experts, evaluation of public actions taken by the Internal Revenue Service and other taxing authorities, as well as our own industry experience. We provide estimates for unrecognized tax benefits which may be subject to material adjustments until matters are resolved with taxing authorities or statutes expire. If our estimates are not representative of actual outcomes, our results of operations could be materially impacted.
We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized. We consider many factors when assessing the likelihood of future realization of deferred tax assets, including our recent cumulative earnings experience by taxing jurisdiction, expectations of future taxable income, carryforward periods available to us for tax reporting purposes, various income tax strategies and other relevant factors. Significant judgment is required in making this assessment and, to the extent future expectations change, we would assess the recoverability of our deferred tax assets at that time. If we determine that the
59
deferred tax assets are not realizable in a future period, we would record material adjustments to income tax expense in that period.
New Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. We are currently assessing the method of adoption and the expected impact the new standard has on our financial position and results of operations.
Results of Operations
The following table sets forth consolidated statements of operations data for the periods indicated. This information has been derived from the consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net product sales | $ | 2,233,733 | $ | 1,551,346 | $ | 1,134,114 | |||||
Cost of sales: | |||||||||||
Cost of sales | 173,862 | 168,375 | 126,214 | ||||||||
Change in contingent liability from intellectual property settlements | — | 9,181 | (53,377 | ) | |||||||
Total cost of sales | 173,862 | 177,556 | 72,837 | ||||||||
Operating expenses: | |||||||||||
Research and development | 513,782 | 317,093 | 222,732 | ||||||||
Selling, general and administrative | 630,209 | 489,720 | 384,678 | ||||||||
Acquisition-related costs | 20,295 | 5,029 | 22,812 | ||||||||
Impairment of intangible assets | 11,514 | 33,521 | 26,300 | ||||||||
Restructuring expenses | 15,365 | — | — | ||||||||
Amortization of purchased intangible assets | — | 417 | 417 | ||||||||
Total operating expenses | 1,191,165 | 845,780 | 656,939 | ||||||||
Operating income | 868,706 | 528,010 | 404,338 | ||||||||
Other income and expense | 3,401 | (1,741 | ) | (6,772 | ) | ||||||
Income before income taxes | 872,107 | 526,269 | 397,566 | ||||||||
Income tax provision | 215,195 | 273,374 | 142,744 | ||||||||
Net income | $ | 656,912 | $ | 252,895 | $ | 254,822 | |||||
Earnings per common share: | |||||||||||
Basic | $ | 3.32 | $ | 1.29 | $ | 1.34 | |||||
Diluted | $ | 3.26 | $ | 1.27 | $ | 1.28 | |||||
60
Comparison of the Year Ended December 31, 2014 to the Year Ended December 31, 2013
Net Product Sales
Net product sales by significant geographic region are as follows:
Year Ended December 31, | ||||||||||
2014 | 2013 | % Change | ||||||||
Net product sales: | ||||||||||
United States | $ | 730,089 | $ | 561,405 | 30 | % | ||||
Europe (1) | 836,134 | 514,987 | 62 | % | ||||||
Asia Pacific | 244,059 | 203,538 | 20 | % | ||||||
Other | 423,451 | 271,416 | 56 | % | ||||||
$ | 2,233,733 | $ | 1,551,346 | 44 | % | |||||
(1) In March 2014, we entered into an agreement with the French government which positively impacts prospective reimbursement of Soliris and also provides for reimbursement for shipments made in years prior to January 1, 2014. As a result of the agreement, in the first quarter of 2014, we recognized $87,830 of net product sales from Soliris in France relating to years prior to January 1, 2014. | ||||||||||
The components of the increase in net product sales for the year ended December 31, 2014, exclusive of the $87,830 recognized related to prior years, are as follows:
Year Ended December 31, | ||
2014 | ||
Components of change: | ||
Price | 6 | % |
Volume | 34 | % |
Foreign exchange | (2 | )% |
Total change in net product sales | 38 | % |
The increase in net product sales for fiscal year 2014 as compared to the same period in 2013, was primarily due to an increase in unit volumes of 34% due to increased physician demand globally for Soliris therapy for patients with PNH or aHUS during the respective periods.
Price had a positive impact on net product sales of 6%,for the year ended December 31, 2014, as compared to the same period in 2013. The positive price impact was primarily due to the agreement with the French government and a reduction in estimated rebates in Germany.
The positive impacts of volume and price on net product sales were offset by the negative impact on foreign exchange of 2%, for the year ended December 31, 2014, as compared to the same period in 2013. The negative impact on foreign exchange of $27,993, or 2%, was due to changes in foreign currency exchange rates (inclusive of hedging activity) versus the U.S. dollar for the year ended December 31, 2013. The negative impact was primarily due to the weakening of the Japanese Yen, Russian Ruble and the Canadian Dollar, partly offset by the positive impacts of the British Pound during the same respective period. We recorded a gain in revenue of $18,873 and $20,569 related to our foreign currency cash flow hedging program, for the years ended December 31, 2014 and 2013, respectively. We expect the strong dollar compared to other currencies, especially the Euro, Japanese Yen and Russian Ruble, to continue to have a negative impact on revenue in 2015 compared to 2014.
Cost of Sales
Cost of sales includes manufacturing costs as well as actual and estimated royalty expenses associated with sales of Soliris.
61
The following table summarizes cost of sales for the year ended December 31, 2014 and 2013:
Year Ended December 31, | |||||||||||
2014 | 2013 | % Change | |||||||||
Cost of sales | $ | 173,862 | $ | 168,375 | $ | 5,487 | |||||
Cost of sales as a percentage of net product sales | 8 | % | 11 | % | (3 | )% | |||||
The decrease in cost of sales as a percentage of net product sales for the year ended December 31, 2014 was partially due to a $14,277 of voluntary recall expense recognized in 2013. Additionally, in the first quarter of 2014, we entered into a settlement agreement with a third party related to the calculation of royalties payable to such third party under a pre-existing license agreement. Based on this settlement agreement, the Company recorded a reversal of accrued royalties of $5,124 as a reduction of cost of sales.
In the first quarter of 2014, we also recorded an incremental impact in cost of sales of $2,055 for additional royalties related to the $87,830 of net product sales from prior year shipments.
The remaining decrease in cost of sales for the years ended December 31, 2014 as a percentage of net product sales resulted from a decrease in royalties paid on sales of Soliris.
In October 2013, we entered into a settlement agreement and dismissal with Novartis Vaccines and Diagnostics, Inc. pursuant to which Alexion was granted a nonexclusive, fully paid license and the case was dismissed with prejudice. As a result, we recorded expense of $9,181 in cost of sales in the third quarter 2013 related to our change in contingent liabilities resulting from this litigation settlement agreement.
Research and Development Expense
Our research and development expense includes personnel, facility and external costs associated with the research and development of our product candidates, as well as product development costs. We group our research and development expenses into two major categories: external direct expenses and all other research and development (R&D) expenses.
External direct expenses are comprised of costs paid to outside parties for clinical development, product development and discovery research, as well as costs associated with strategic licensing agreements we have entered into with third parties. Clinical development costs are comprised of costs to conduct and manage clinical trials related to eculizumab and other product candidates. Product development costs are those incurred in performing duties related to manufacturing development and regulatory functions, including manufacturing of material for clinical and research activities. Discovery research costs are incurred in conducting laboratory studies and performing preclinical research for other uses of eculizumab and other product candidates. Licensing agreement costs include upfront and milestone payments made in connection with strategic licensing arrangements we have entered into with third parties. Clinical development costs have been accumulated and allocated to each of our programs, while product development and discovery research costs have not been allocated.
All other R&D expenses consist of costs to compensate personnel, to maintain our facility, equipment and overhead and similar costs of our research and development efforts. These costs relate to efforts on our clinical and preclinical products, our product development and our discovery research efforts. These costs have not been allocated directly to each program.
62
The following table provides information regarding research and development expenses:
Year Ended December 31, 2014 | Year Ended December 31, 2013 | $ Change | % Change | |||||||||||
Clinical development | $ | 111,435 | $ | 72,281 | $ | 39,154 | 54 | % | ||||||
Product development | 63,235 | 62,832 | 403 | 1 | % | |||||||||
Licensing agreements | 109,925 | 14,500 | 95,425 | 658 | % | |||||||||
Discovery research | 13,403 | 5,546 | 7,857 | 142 | % | |||||||||
Total external direct expenses | 297,998 | 155,159 | 142,839 | 92 | % | |||||||||
Payroll and benefits | 190,669 | 144,034 | 46,635 | 32 | % | |||||||||
Operating and occupancy | 11,050 | 7,765 | 3,285 | 42 | % | |||||||||
Depreciation and amortization | 14,065 | 10,135 | 3,930 | 39 | % | |||||||||
Total other R&D expenses | 215,784 | 161,934 | 53,850 | 33 | % | |||||||||
Research and development expense | $ | 513,782 | $ | 317,093 | $ | 196,689 | 62 | % | ||||||
During the year ended December 31, 2014, we incurred research and development expenses of $513,782, an increase of $196,689, or 62%, versus the $317,093 incurred during the year ended December 31, 2013. The increase was primarily related to the following:
• | Increase of $39,154 in external clinical development expenses related primarily to an expansion of studies for eculizumab and asfotase alfa (see table below). |
• | Increase of $95,425 in licensing agreement costs primarily due to the upfront payment of $100,000 on the option agreement entered into with Moderna Therapeutics, Inc. in the first quarter of 2014. |
• | Increase of $7,857 in discovery research expenses primarily related to increases in external research expenses associated with our Moderna agreement and other external research expenses. |
• | Increase of $46,635 in R&D payroll and benefit expense related primarily to the continued global expansion of staff supporting our increasing number of clinical and development programs. |
• | Increases of $3,285 and $3,930 in R&D operating and occupancy and depreciation and amortization expenses. respectively, related primarily to the continued expansion of global supply chain facilities and support services. |
The following table summarizes external direct expenses related to our clinical development programs. Please refer to Item 1, "Business", for a description of each of these programs:
Year Ended December 31, 2014 | Year Ended December 31, 2013 | Accumulated Expenditures (Non-Approved Products) | |||||||||
External direct expenses | |||||||||||
Eculizumab | $ | 67,224 | $ | 44,577 | (a) | ||||||
Asfotase alfa | 25,034 | 13,677 | $ | 43,511 | |||||||
cPMP | 7,802 | 6,408 | 16,354 | ||||||||
Other programs | 5,947 | 5,546 | 19,037 | ||||||||
Unallocated | 5,428 | 2,073 | (b) | ||||||||
$ | 111,435 | $ | 72,281 | $ | 78,902 | ||||||
(a) From 1992 through 2006, substantially all research and development expenses were related to two products, eculizumab and pexelizumab. We obtained approval in the U.S. for eculizumab for PNH in 2007 and for aHUS in 2010, and we ceased development of pexelizumab in 2006. | |||||||||||
(b) External costs shared across various development programs. | |||||||||||
The successful development of our drug candidates is uncertain and subject to a number of risks. We cannot guarantee that results of clinical trials will be favorable or sufficient to support regulatory approvals for our other programs. We could decide to
63
abandon development or be required to spend considerable resources not otherwise contemplated. For additional discussion regarding the risks and uncertainties regarding our development programs, please refer to Item 1A "Risk Factors" in this Annual Report on Form 10-K.
We expect our research and development expenses to increase in 2015 due to clinical development and manufacturing costs related to our expanding development programs. For additional information on these programs, please refer to “Product and Development Programs” in Item I "Business" of this Annual Report on Form 10-K.
Selling, General and Administrative Expense
Our selling, general and administrative expense includes commercial and administrative personnel, corporate facility and external costs required to support the marketing and sales of our commercialized products. These selling, general and administrative costs include: corporate facility operating expenses and depreciation; marketing and sales operations in support of Soliris; human resources; finance, legal, information technology and support personnel expenses; and other corporate costs such as telecommunications, insurance, audit, government affairs and our global corporate compliance program.
The table below provides information regarding selling, general and administrative expense:
Year Ended December 31, 2014 | Year Ended December 31, 2013 | $ Change | |||||||||
Salary, benefits and other labor expense | $ | 388,738 | $ | 292,881 | $ | 95,857 | |||||
External selling, general and administrative expense | 241,471 | 196,839 | 44,632 | ||||||||
Total selling, general and administrative expense | $ | 630,209 | $ | 489,720 | $ | 140,489 | |||||
During the year ended December 31, 2014, we incurred selling, general and administrative expenses of $630,209, an increase of $140,489, or 29%, versus the $489,720 incurred during the year ended December 31, 2013. The increase was primarily related to the following:
• | Increase in salary, benefits and other labor expenses of $95,857. The increase was a result of increased headcount related to commercial development activities, including increases in payroll and benefits costs of $49,023 related to our global commercial staff to support global expansion. This increase was also due to increases in payroll and benefits of $46,835 within our general and administrative functions to support our infrastructure growth as a global commercial entity. |
• | Increase in external selling, general and administrative expenses of $44,632. This increase was primarily due to an increase in marketing costs to support the continued growth in global sales of Soliris, as well as an increase in other administrative costs to support our infrastructure growth. |
We expect our selling, general and administrative expenses to increase, at a lower rate than our revenue, in 2015, reflecting our continued growth as a commercial organization throughout the world.
Acquisition-related Costs
For the years ended December 31, 2014 and 2013, acquisition-related costs associated with our business combinations included the following:
Year Ended December 31, 2014 | Year Ended December 31, 2013 | ||||||
Separately-identifiable employee costs | $ | — | $ | 248 | |||
Professional fees | — | 775 | |||||
Changes in fair value of contingent consideration | 20,295 | 4,006 | |||||
$ | 20,295 | $ | 5,029 | ||||
64
The increase in expense associated with changes in the fair value of contingent consideration for the year ended December 31, 2014 as compared the prior year resulted primarily from increases in the likelihood of payments for contingent consideration and related to a decrease in discount rates.
Restructuring Expenses
In the fourth quarter of 2014 we announced plans to move the European headquarters from Lausanne, Switzerland to Zurich, Switzerland resulting in restructuring expenses of $15,365. The relocation of the European headquarters will support our growing operational needs based on current business forecasts. We expect to incur approximately $10,000 to $15,000 of additional restructuring related charges in 2015 related to additional employee costs and contract terminations costs. We expect to pay all accrued amounts related to this restructuring activity in 2015.
Impairment of Intangible Asset
During the fourth quarter of 2014, we reviewed for impairment the value of the early stage, Phase II indefinite-lived intangible asset related to the Orphatec acquisition. We initiated such review as part of our annual impairment testing and increased costs associated with clinical trial studies. Although we will continue to develop this asset, the estimated fair value that can be obtained from a market participant in an arm's length transaction was determined to be de minimis as of December 31, 2014. As a result, in the fourth quarter 2014, we recognized an impairment charge of $8,050 to write-down these assets to fair value.
During the first quarter of 2014 and the fourth quarter of 2013, we reviewed for impairment the value of an early stage, Phase I indefinite-lived intangible asset related to our acquisition of Taligen Therapeutics, Inc. We initiated such review based on a reassessment of scientific findings associated with this acquired asset. In the fourth quarter 2013, we also reviewed for impairment the value of purchased technology associated with the Taligen acquisition. As a result, we recognized impairment charges of $3,464 and $33,521 for the years ended December 31, 2014 and 2013 to adjust these assets to fair value, which was determined to be de minimis.
Other Income and Expense
The following table provides information regarding other income and expense:
Year Ended December 31, 2014 | Year Ended December 31, 2013 | $ Change | |||||||||
Investment income | $ | 8,373 | $ | 3,346 | $ | 5,027 | |||||
Interest expense | (2,982 | ) | (4,112 | ) | 1,130 | ||||||
Foreign currency loss | (1,990 | ) | (975 | ) | (1,015 | ) | |||||
Total other income (expense) | $ | 3,401 | $ | (1,741 | ) | $ | 5,142 | ||||
Income Taxes
During the year ended December 31, 2014, we recorded an income tax provision of $215,195 and an effective tax rate of 24.7%, compared to an income tax provision of $273,374 and an effective tax rate of 51.9% for the year ended December 31, 2013. The reduction in the effective tax rate is primarily attributable to the centralization of our global supply chain and technical operations in Ireland.
The income tax provision for 2014 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations. Additionally, included for the year ended December 31, 2014 is $2,128 of tax attributable to our agreement with the French government that provided reimbursement for shipments of Soliris made prior to January 1, 2014.
The income tax provision for 2013 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations, as well as the tax expense of resulting from the centralization of our global supply chain and technical operations in Ireland undertaken in the fourth quarter of 2013 in the amount of approximately $95,800. We were also impacted by a tax benefit of $2,719 attributable to the 2012 U.S. Federal tax credit for research and experimentation due to retroactive extension of this credit signed into law in January 2013.
We were granted an incentive tax holiday in the Canton of Vaud in Switzerland effective January 1, 2010. This tax holiday had exempted us from most local corporate income taxes in Switzerland through the end of 2014 and was renewable for an
65
additional 5 years with final expiration in 2019. During 2013, we undertook a restructuring which significantly changed our business model in Switzerland and we converted from a principal company to a distribution and service company. As a result of the significant change to our business activities in Switzerland, the Canton of Vaud in Switzerland provided final notification to us in December 2014 that our structure no longer complied with the conditions of the incentive tax holiday. In the fourth quarter of 2014, we made a payment of $22,817 in satisfaction of the clawback of previously exempted cantonal income taxes for tax years 2010 through 2013. This amount was fully accrued on our balance sheet as of December 31, 2013. Prospectively, our federal and cantonal tax will be based on the current enacted tax rates in Switzerland.
The U.S. Federal tax credit for research and experimentation expenses expired December 31, 2013. In connection with this expiration, our 2014 tax expense for the first three quarters of the year did not include any benefit from the U.S. Federal tax credit for research and experimentation. In December 2014, the Tax Increase Prevention Act of 2014, which retroactively extended the tax credit for research and experimentation back to January 1, 2014 through the end of 2014, was signed into law. The effects of a change in tax law is recognized in the period that includes the date of enactment and, therefore, our tax benefit attributable to the 2014 U.S. Federal tax credit of $3,222 for research and experimentation was recorded in the fourth quarter of 2014.
We continue to maintain a valuation allowance against certain other deferred tax assets where the realization is not certain. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized.
Comparison of the Year Ended December 31, 2013 to the Year Ended December 31, 2012
Net Product Sales
Net product sales by significant geographic region are as follows:
Year Ended December 31, | ||||||||||
Net product sales: | 2013 | 2012 | % Change | |||||||
United States | $ | 561,405 | $ | 400,483 | 40 | % | ||||
Europe | 514,987 | 418,321 | 23 | % | ||||||
Asia Pacific | 203,538 | 161,480 | 26 | % | ||||||
Other | 271,416 | 153,830 | 76 | % | ||||||
$ | 1,551,346 | $ | 1,134,114 | 37 | % | |||||
The increase in revenue for fiscal year 2013 versus 2012 was primarily due to an increased volume of unit shipments, partially offset by a negative impact of price and foreign exchange.
The increase in revenue of 37% for the year ended December 31, 2013 was due to an increase in unit volumes of 40%, offset by a negative price impact of 2%, and a negative impact on foreign exchange of 1%. The increase in volume was largely due to physicians globally requesting Soliris therapy for additional patients. The negative price impact of 2% for the year ended December 31, 2013 was primarily due to increased rebates in certain countries in Europe, offset by a price increase in the United States.
The negative impact on foreign exchange of $15,876, or 1%, for the year ended December 31, 2013 was due to changes in foreign currency exchange rates (inclusive of hedging activity) versus the U.S. dollar for the year ended December 31, 2012. The negative impact was primarily due to the weakening of the Japanese Yen. We recorded a gain in revenue of $20,569 and $12,869 related to our foreign currency cash flow hedging program, which is included in revenue from outside the United States, for the years ended December 31, 2013 and 2012, respectively.
Cost of Sales
In October 2013, we entered into a settlement agreement and dismissal with Novartis pursuant to which Alexion was granted a non-exclusive, fully paid license and the case was dismissed with prejudice. As a result, we recorded expense of $9,181 in cost of sales in the third quarter 2013 related to this litigation settlement agreement.
In the third quarter of 2012, we reduced our estimate for probable contingent liabilities as of September 30, 2012 due to the execution of a settlement and non-exclusive license agreement in October 2012 with a third party related to the third party's intellectual property. The adjustment reflected the actual, negotiated royalty rate set forth in the agreement. This change in estimate resulted in a positive impact in cost of sales of $53,377 during the third quarter 2012.
66
Exclusive of the changes in estimates of contingent liabilities for the settlements noted above, cost of sales were $168,375 and $126,214, or 11% of product revenue, for the years ended December 31, 2013 and 2012, respectively. Cost of sales includes manufacturing costs as well as actual and estimated royalty expenses associated with sales of Soliris. Included in cost of sales for the year ended December 31, 2013, was $14,277 or 1% of product sales related to the expected disposal of inventory in 2014 associated with our voluntary recall announced in November 2013. Offsetting this increase in cost of sales was a decrease in our ongoing royalty expense as a result of the settlement and non-exclusive license agreement we entered into in October 2012.
Research and Development Expense
The following table provides information regarding research and development expenses:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | $ Change | % Change | |||||||||||
Clinical development | $ | 72,281 | $ | 46,711 | $ | 25,570 | 55 | % | ||||||
Product development | 62,832 | 57,028 | 5,804 | 10 | % | |||||||||
Discovery research | 20,046 | 8,271 | 11,775 | 142 | % | |||||||||
Total external direct expenses | 155,159 | 112,010 | 43,149 | 39 | % | |||||||||
Payroll and benefits | 144,034 | 95,609 | 48,425 | 51 | % | |||||||||
Operating and occupancy | 7,765 | 7,958 | (193 | ) | (2 | )% | ||||||||
Depreciation and amortization | 10,135 | 7,155 | 2,980 | 42 | % | |||||||||
Total other R&D expenses | 161,934 | 110,722 | 51,212 | 46 | % | |||||||||
Research and development expense | $ | 317,093 | $ | 222,732 | $ | 94,361 | 42 | % | ||||||
During the year ended December 31, 2013, we incurred research and development expenses of $317,093, an increase of $94,361, or 42%, versus the $222,732 incurred during the year ended December 31, 2012. The increase was primarily related to the following:
• | Increase of $25,570 in external clinical development expenses related primarily to an expansion of studies of eculizumab, asfotase alfa and cPMP programs (see table below). |
• | Increase of $5,804 in external product development expenses related primarily to costs associated with the preparation of regulatory filings for asfotase alfa and an increase in manufacturing costs related to our other product development programs, offset by a decrease in costs associated with the production of asfotase alfa for clinical studies. |
• | Increase of $11,775 in discovery research expenses primarily related to the upfront payment of $14,500 on the license agreements entered into in 2013. |
• | Increase of $48,425 in R&D payroll and benefit expense related primarily to global expansion of staff supporting our increasing number of clinical and development programs. |
67
The following table summarizes external direct expenses related to our clinical development programs. Please refer to Item 1, "Business", for a description of each of these programs:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | ||||||
External direct expenses | |||||||
Eculizumab | $ | 44,577 | $ | 35,732 | |||
Asfotase alfa | 13,677 | 4,800 | |||||
cPMP | 6,408 | 2,144 | |||||
Other programs | 5,546 | 3,396 | |||||
Unallocated | 2,073 | 639 | |||||
$ | 72,281 | $ | 46,711 | ||||
(a) From 1992 through 2006, substantially all research and development expenses were related to two products, eculizumab and pexelizumab. We obtained approval in the U.S. for eculizumab for PNH in 2007 and for aHUS in 2010, and we ceased development of pexelizumab in 2006. | |||||||
Selling, General and Administrative Expense
The table below provides information regarding selling, general and administrative expense:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | $ Change | |||||||||
Salary, benefits and other labor expense | $ | 292,881 | $ | 223,053 | $ | 69,828 | |||||
External selling, general and administrative expense | 196,839 | 161,625 | 35,214 | ||||||||
Total selling, general and administrative expense | $ | 489,720 | $ | 384,678 | $ | 105,042 | |||||
During the year ended December 31, 2013, we incurred selling, general and administrative expenses of $489,720, an increase of $105,042, or 27%, versus the $384,678 incurred during the year ended December 31, 2012. The increase was primarily related to the following:
• | Increase in salary, benefits and other labor expenses of $69,828. The increase was a result of increased headcount related to commercial development activities, including increases in payroll and benefits costs of $51,700 related to our global commercial staff to support global expansion. This increase was also due to increases in payroll and benefits of $18,100 within our general and administrative functions to support our infrastructure growth as a global commercial entity. |
• | Increase in external selling, general and administrative expenses of $35,214. This increase was primarily due to an increase in legal costs associated with the Novartis litigation, an increase in consulting fees related to our global supply chain expansion in Ireland, an increase in marketing costs to support the continued growth in global sales, as well as an increase in general administrative expenses due to infrastructure growth. |
Acquisition-related Costs
For the years ended December 31, 2013 and 2012, acquisition-related costs associated with our business combinations included the following:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | ||||||
Separately-identifiable employee costs | $ | 248 | $ | 3,669 | |||
Professional fees | 775 | 12,593 | |||||
Changes in fair value of contingent consideration | 4,006 | 6,550 | |||||
$ | 5,029 | $ | 22,812 | ||||
68
The following table provides information for acquisition-related costs for each business combination:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | ||||||
Enobia Pharma Corp. | $ | 9,625 | $ | 23,673 | |||
Taligen Therapeutics, Inc. | (5,777 | ) | (2,948 | ) | |||
Orphatec Pharmaceuticals GmbH | 1,181 | 2,087 | |||||
$ | 5,029 | $ | 22,812 | ||||
Included in the acquisition-related costs for Taligen for the year ended December 31, 2013 and 2012 is a gain of $5,973 and $4,331, respectively, related to the decrease in the fair value of the contingent consideration related to this acquisition. The decrease in fair value was a result of a decreased likelihood of payments for contingent consideration due to a reassessment of scientific findings.
Impairment of Intangible Asset
During the fourth quarter of 2013, we reviewed for impairment the value of an early stage, Phase I indefinite-lived intangible asset related to the Taligen acquisition. We initiated such review as part of our annual impairment testing and based our evaluation on preliminary scientific findings of a Phase I clinical trial which led us to reassess the development of this acquired asset. Based on these factors, the estimated value that can be obtained from a market participant in an arm's length transaction of $3,464 was lower than the carrying amount. We also reviewed for impairment the value of purchased technology associated with the Taligen acquisition and determined the estimated fair value to be de minimis. As a result, in the fourth quarter 2013, we recognized an impairment charge of $33,521 to write-down these assets to fair value.
During the year ended December 31, 2012, we reviewed for impairment the value of an early stage, preclinical indefinite-lived intangible asset related to the Taligen acquisition. We initiated such review based on our evaluation of negative scientific findings associated with our development of a different asset for the treatment of age-related macular degeneration, the likelihood of success for ophthalmic use and the value that can be obtained from a market participant in an arm's length transaction. These developments led us to deprioritize the development of this acquired asset. As a result, in the third quarter 2012, we recognized an impairment charge of $26,300 to write-down this asset to fair value, which was determined to be de minimis.
Other Income and Expense
The following table provides information regarding other income and expense:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | $ Change | |||||||||
Investment income | $ | 3,346 | $ | 1,838 | $ | 1,508 | |||||
Interest expense | (4,112 | ) | (7,402 | ) | 3,290 | ||||||
Foreign currency loss | (975 | ) | (1,208 | ) | 233 | ||||||
Total other income (expense) | $ | (1,741 | ) | $ | (6,772 | ) | $ | 5,031 | |||
We recognize investment income primarily from our portfolio of cash equivalents. During the year ended December 31, 2013, investment income increased $1,508, or 82%, to $3,346.
We incur interest on our term notes and revolving credit facility. During the year ended December 31, 2013, interest expense decreased $3,290 to $4,112 due to a decrease in amounts outstanding under our credit facility.
Foreign currency transaction gains and losses relate to changes in the fair value of monetary assets and liabilities denominated in foreign currencies. The foreign currency transaction losses totaled $975 and $1,208 for the years ended December 31, 2013 and December 31, 2012, respectively. The amounts recorded in these periods were a result of the costs of hedging our exposures, as well as the fluctuation in exchange rates on the portion of our monetary assets and liabilities that were not fully hedged as part of our hedging programs.
69
Income Taxes
During the year ended December 31, 2013, we recorded an income tax provision of $273,374 and an effective tax rate of 51.9%, compared to an income tax provision of $142,744 and an effective tax rate of 35.9% for the year ended December 31, 2012.
The income tax provision for 2013 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations, as well as the tax expense of resulting from the centralization of our global supply chain and technical operations in Ireland undertaken in the fourth quarter of 2013 in the amount of approximately $95,800. We were also impacted by a tax benefit of $2,719 attributable to the 2012 U.S. Federal tax credit for research and experimentation due to retroactive extension of this credit signed into law in January 2013.
The tax provision for 2012 is principally attributable to the U.S. federal, state, and foreign income taxes in our profitable operations, as well as the tax expense of $21,812 associated with the structuring of the Enobia business.
We were granted an incentive tax holiday in the Canton of Vaud in Switzerland effective January 1, 2010. This tax holiday will exempt us from most local corporate income taxes in Switzerland through the end of 2014 and is renewable for an additional 5 years with final expiration in 2019. The impact of this tax holiday decreased foreign tax expense by $4,351 in 2013 and $3,173 in 2012.
We continue to maintain a valuation allowance against certain other deferred tax assets where the realization is not certain. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized.
Financial Condition, Liquidity and Capital Resources
The following table summarizes the components of our financial condition as of December 31, 2014 and 2013:
December 31, 2014 | December 31, 2013 | $ Change | |||||||||
Cash and cash equivalents | $ | 943,999 | $ | 529,857 | $ | 414,142 | |||||
Marketable securities | 1,017,567 | 984,994 | 32,573 | ||||||||
Long-term debt (includes current portion) | 57,500 | 113,000 | (55,500 | ) | |||||||
Current assets | $ | 2,796,029 | $ | 2,186,857 | $ | 609,172 | |||||
Current liabilities | 606,740 | 582,429 | 24,311 | ||||||||
Working capital | $ | 2,189,289 | $ | 1,604,428 | $ | 584,861 | |||||
The increase in cash and cash equivalents was primarily attributable to cash generated from operations, proceeds from the maturity or sale of available-for-sale securities, and net proceeds from the exercise of stock options. Offsetting these increases in cash were purchases of marketable securities, payments on our outstanding term loan, purchases of property, plant and equipment, and the repurchase of common stock. We also paid an upfront fee of $100,000 during the first quarter of 2014 related to an option agreement we entered into with Moderna Therapeutics, Inc. and an additional $37,500 for the purchase of Moderna LLC preferred equity during 2014.
We expect continued growth in our expenditures, particularly those related to research and product development, clinical trials, regulatory approvals, international expansion, commercialization of products and capital investment. However, we anticipate that cash generated from operations and our existing available cash, cash equivalents and marketable securities should provide us adequate resources to fund our operations as currently planned.
We have financed our operations and capital expenditures primarily through positive cash flows from operations. We expect to continue to be able to fund our operations, including principal and interest payments on our credit facility and contingent payments from our acquisitions principally through our cash flows from operations. We may, from time to time, also seek additional funding through a combination of equity or debt financings or from other sources, if necessary for future acquisitions or other strategic purposes.
Financial Instruments
Until required for use in the business, we may invest our cash reserves in money market funds or high-quality marketable securities in accordance with our investment policy. The stated objectives of our investment policy is to preserve capital, provide
70
liquidity consistent with forecasted cash flow requirements, maintain appropriate diversification and generate returns relative to these investment objectives and prevailing market conditions.
Financial instruments that potentially expose us to concentrations of credit risk are cash equivalents, marketable securities, accounts receivable and our foreign exchange derivative contracts. At December 31, 2014, four individual customers accounted for 58% of the accounts receivable balance, with individual customers accounting for 10% to 23% of the accounts receivable balance. At December 31, 2013, two individual customers accounted for 30% of the accounts receivable balance, with individual customers accounting for 10% and 20% of the accounts receivable balance. For the years ended December 31, 2014 and 2013, one customer accounted for 18% and 20%, respectively, of our product sales.
We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. Substantially all of our accounts receivable due from these countries are due from or backed by sovereign or local governments, and the amount of non-sovereign accounts receivable is not material. Although collection of our accounts receivables from certain countries may extend beyond our standard credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operations.
We manage our foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. As of December 31, 2014, we have foreign exchange forward contracts with notional amounts totaling $1,748,931. These outstanding foreign exchange forward contracts had a net fair value of $135,166, of which an unrealized gain of $136,046 is included in other assets, offset by an unrealized loss of $880 included in other liabilities. The counterparties to these foreign exchange forward contracts are large multinational commercial banks, and we believe the risk of nonperformance is not material.
At December 31, 2014, our financial assets and liabilities were recorded at fair value. We have classified our financial assets and liabilities as Level 1, 2 or 3 within the fair value hierarchy. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Our Level 1 assets consist of mutual fund investments. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, but substantially the full term of the financial instrument. Our Level 2 assets consist primarily of institutional money market funds, commercial paper, municipal bonds, U.S. and foreign government-related debt, corporate debt securities, certificates of deposit and foreign exchange forward contracts. Our Level 2 liabilities consist also of foreign exchange forward contracts. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value. Our Level 3 liabilities consist of contingent consideration related to acquisitions.
Business Combinations and Contingent Consideration Obligations
The purchase agreements for our business combinations include contingent payments totaling up to $876,000 that will become payable if and when certain development and commercial milestones are achieved. Of these milestone amounts, $561,000 and $315,000 of the contingent payments relate to development and commercial milestones, respectively. We do not expect these amounts to have an impact on our liquidity in the near-term, and, during the next 12 months, we expect to make milestone payments totaling approximately $50,000. As additional future payments become probable, we will evaluate methods of funding payments, which could be made from available cash and marketable securities, cash generated from operations or proceeds from other financing.
Financing Lease Obligation
In November 2012, we entered into a lease agreement for office and laboratory space to be constructed in New Haven, Connecticut. Although we will not legally own the premises, we are deemed to be the owner of the building during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord's costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. As of December 31, 2014, we recorded a construction-in-process asset of $126,566, inclusive of the landlord's costs as well as costs incurred by Alexion, and an offsetting facility lease obligation of $107,099, associated with the new facility.
License Agreements
In January 2015, we entered into a license agreement with a third party to obtain certain intellectual property rights and technology related to specific therapeutic compounds. The agreement provides an exclusive research, development and commercial license for products to be developed using such compounds. Pursuant to the terms of the agreement, we made an upfront payment of $50,000 during the first quarter 2015. We could be required to pay up to an additional $213,000 in development and regulatory milestones related to a product developed under the agreement for a single disease indication. An
71
additional $437,000 in milestone payments could be due if certain development and regulatory milestones are achieved for additional disease indications. The agreement also provides for royalty payments and potential milestone payments of up to $180,000 on commercial sales of products developed under the agreement.
In December 2014, we entered into an agreement with X-Chem Pharmaceuticals (X-Chem) that allows us to identify novel drug candidates from X-Chem's proprietary drug discovery engine. Alexion will have the exclusive worldwide rights to develop and commercialize products arising from the collaboration in up to three program targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $8,000. In addition, for each program target, for a maximum of three targets, we could be required to make additional payments upon the achievement of specified research, development and regulatory milestones up to $75,000, as well as royalties on commercial sales.
In January 2014, we entered into an agreement with Moderna Therapeutics, Inc. (Moderna) that allows us to purchase ten product options to develop and commercialize treatments for rare diseases with Moderna's messenger RNA (mRNA) therapeutics platform. Alexion will lead the discovery, development and commercialization of the treatments produced through this broad, long-term strategic agreement, while Moderna will retain responsibility for the design and manufacture of the messenger RNA against selected targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $100,000. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of ten targets, we could be required to make an option exercise payment of $15,000 and to pay up to an additional $120,000 with respect to a rare disease product and $400,000 with respect to a non-rare disease product in development and sales milestones if the specific milestones are met over time as well as royalties on commercial sales.
Long-term Debt
In February 2012, we entered into a Credit Agreement (Credit Agreement) with a syndicate of lenders and other parties named in the Credit Agreement that provides for a $240,000 senior secured term loan facility payable in equal quarterly installments of $12,000 starting June 30, 2012 and a $200,000 senior secured revolving credit facility, which includes up to a $60,000 sublimit for letters of credit and a $10,000 sublimit for swingline loans. We may also use the facilities for working capital requirements, acquisitions and other general corporate purposes. Any of Alexion's wholly-owned foreign subsidiaries may borrow funds under the facilities upon satisfaction of certain conditions described in the Credit Agreement. As of December 31, 2014, we had $57,500 outstanding on the term loan of which $12,000 was paid in January 2015. As of December 31, 2014, we had open letters of credit of $10,284, and our borrowing availability under the revolving facility was $189,716 at December 31, 2014. We expect that cash generated from operations will be sufficient to meet debt service obligations.
Lonza Agreement
We have supply agreements with Lonza through 2026 relating to the manufacture of eculizumab and asfotase alfa, which requires payments to Lonza at the inception of contract and upon the initiation and completion of product manufactured. On an ongoing basis, we evaluate our plans for future levels of manufacturing by Lonza, which depends upon our commercial requirements, the progress of our clinical development programs and the production levels of ARIMF.
We have various agreements with Lonza, with remaining total non-cancellable commitments of approximately $383,500 through 2018. Our agreements with Lonza also include potential payments totaling up to $5,000 that will become payable if and when certain milestones are achieved. Such commitments may be canceled only in limited circumstances. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of Soliris manufactured at ARIMF and a payment with respect to sales of Soliris manufactured at Lonza facilities.
Taxes
We do not record U.S. tax expense on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries as these earnings are intended to be permanently reinvested offshore. At December 31, 2014, the cumulative amount of these earnings was approximately $359,000. During the fourth quarter of 2013, in connection with the centralization of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner in a foreign partnership subsidiary. To the extent that our U.S. parent company receives its allocation of partnership taxable income, the amounts will be taxable in the U.S. and therefore the permanent reinvestment assertion will no longer apply.
We do not have any present or anticipated future need for cash held by our CFCs, as cash generated in the U.S., as well as borrowings, are expected to be sufficient to meet U.S. liquidity needs for the foreseeable future. At December 31, 2014, approximately $674,000 of our cash and cash equivalents was held by foreign subsidiaries, a significant portion of which is required for liquidity needs of our foreign subsidiaries. Due to the liability position of our foreign subsidiaries, these subsidiaries
72
will repay any outstanding intercompany debt, prior to having excess cash available which could be used to repatriate to our entities in the United States. While our expectation is that all future undistributed earnings of our CFCs will be permanently reinvested, there could be certain unforeseen future events that could impact our permanent reinvestment assertion. Such events include acquisitions, corporate restructurings or tax law changes not currently contemplated.
Common Stock Repurchase Program
In November 2012 and December 2014, we announced that our Board of Directors authorized the repurchase of up to $400,000 and $500,000 of our common stock. The repurchase program does not have an expiration date, and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at the Company's discretion. We expect that cash generated from operations and our existing available cash and cash equivalents are sufficient to fund any share repurchases.
Under the program, we repurchased 1,903 and 758 shares of our common stock at a cost of $302,599 and $66,136 during the years ended December 31, 2014 and 2013, respectively. At December 31, 2014, there is a total of $519,712 remaining for repurchases under these repurchase program.
Subsequent to December 31, 2014, we repurchased 87 shares of our common stock under our repurchase program at a cost of $15,531.
Cash Flows
The following summarizes our net change in cash and cash equivalents:
Year Ended December 31, | |||||||||||
2014 | 2013 | $ Change | |||||||||
Net cash provided by operating activities | $ | 640,075 | $ | 497,349 | $ | 142,726 | |||||
Net cash used in investing activities | (222,869 | ) | (1,027,141 | ) | 804,272 | ||||||
Net cash provided by financing activities | 7,126 | 71,639 | (64,513 | ) | |||||||
Effect of exchange rate changes on cash | (10,190 | ) | (1,491 | ) | (8,699 | ) | |||||
Net change in cash and cash equivalents | $ | 414,142 | $ | (459,644 | ) | $ | 873,786 | ||||
The increase in cash and cash equivalents was primarily attributable to cash generated from operations, proceeds from the maturity or sale of available-for-sale securities, net proceeds from the exercise of stock options and a reduction of income taxes payable due to excess tax benefits from stock options. Offsetting these increases in cash were purchases of marketable securities, payments on our outstanding term loan, purchases of property, plant and equipment, and the repurchase of common stock. We also paid an upfront fee of $100,000 during the first quarter of 2014 related to an option agreement we entered into with Moderna Therapeutics, Inc. and an additional $37,500 for the purchase of Moderna LLC preferred equity during 2014.
Operating Activities
The components of cash flows from operating activities, as reported in our Consolidated Statements of Cash Flows, are as follows:
• | Our reported net income was $656,912 and $252,895 for the years ended December 31, 2014 and 2013, respectively. During the first quarter of 2014, we recorded expense of $100,000 for an upfront payment related to an option agreement we entered into with Moderna Therapeutics, Inc. |
• | Non-cash items included depreciation and amortization, impairment of intangible assets, change in fair value of contingent consideration, share-based compensation expense, premium amortization of available-for-sale securities, and deferred taxes, and were increases to reconcile net income to net cash flows from operating activities of $76,869 and $240,529 for the years ended December 31, 2014 and 2013, respectively. |
• | Non-cash items also included $251,136 and $105,714 of windfall tax benefits for the years ended December 31, 2014 and 2013, respectively. The amount of the windfall tax benefit was significantly higher during the year ended December 31, 2014 due to an increased stock price and increased level of stock option exercises. |
73
• | Net cash inflows due to changes in operating assets and liabilities was $157,430 and $109,639 for the years ended December 31, 2014 and 2013, respectively. The $157,430 change in operating assets and liabilities primarily relates to: |
• | Increase in accounts receivable of $28,137 due primarily to increasing revenue. |
• | Increase of $66,812 in inventory related to increased production of inventory to support commercial growth and the capitalization of $22,005 of inventory produced for commercial sale for products awaiting regulatory approval. |
• | Increase of $18,392 in prepaid expenses and other assets related to an increase in prepaid manufacturing costs. |
• | Increase of $264,572 in accounts payable, accrued expenses and other liabilities primarily related to an increase in trade accounts payable, accrued manufacturing, accruals for purchases of property, plant and equipment, clinical costs, accrued compensation and a change in accrued income taxes. These increases were offset by a decrease in accruals for rebates of approximately $87,800 resulting from the agreement with the French government for reimbursement of prior year shipments and a decrease in accruals for royalties of approximately $20,100 resulting from the settlement agreement we entered into with a third party related to the calculation of royalties under a pre-existing license agreement. |
In 2015, we expect increases in cash flow from operations which will be highly dependent on sales levels, and the related cash collections from Soliris.
Investing Activities
The components of cash flows from investing activities primarily consisted of the following:
• | Purchases of available-for-sale marketable securities of $664,228 and $1,048,429 for the year ended December 31, 2014 and 2013, respectively, offset by proceeds from the maturity or sale of available-for-sale marketable securities of $619,447 and $60,917 during the same periods. |
• | Purchase of $37,500 of preferred equity of Moderna LLC during the year ended December 31, 2014. |
• | Additions to property, plant and equipment of $136,650 and $29,329 for the years ended December 31, 2014 and 2013, respectively. |
We expect to increase spending on property, plant and equipment in 2015 due to the capital projects for our New Haven headquarters and our two facilities in Ireland.
Financing Activities
Net cash flows from financing activities reflected proceeds from the exercise of stock options of $114,350 and $71,281 for the years ended December 31, 2014 and 2013, respectively. Net cash flows from financing activities for the years ended December 31, 2014 and 2013 also include $251,136 and $105,714, respectively, of excess tax benefits from stock options attributable to the utilization of the excess tax benefit portion of federal and state net operating losses and tax credits.
During the years ended December 31, 2014 and 2013, we made payments of $55,500 and $36,000 against the term loan facility. As of December 31, 2014, the facility had $57,500 remaining outstanding.
During the years ended December 31, 2014 and 2013, we repurchased $302,599 and $66,136 worth of shares of our common stock under a repurchase program that was approved by our Board of Directors in November 2012. In December 2014, our Board of Directors approved an additional $500,000 to repurchase shares. As of December 31, 2014, there is a total of $519,712 remaining for repurchases under these repurchase program.
74
Contractual Obligations
The following table summarizes our contractual obligations at December 31, 2014 and the effect such obligations and commercial commitments are expected to have on our liquidity and cash flow in future fiscal years. These do not include potential milestone payments and assume non-termination of agreements.
These obligations, commitments and supporting arrangements represent payments based on current operating forecasts, which are subject to change:
Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||||||
Contractual obligations: | |||||||||||||||||||
Long-term debt | $ | 57,500 | $ | 10,000 | $ | 47,500 | $ | — | $ | — | |||||||||
Interest expense (1) | 1,308 | 807 | 501 | — | — | ||||||||||||||
Pension obligations | 12,342 | 1,985 | 2,961 | 2,474 | 4,922 | ||||||||||||||
Facility Lease Obligation (2) | 149,529 | 1,941 | 23,302 | 23,915 | 100,371 | ||||||||||||||
Operating leases | 72,561 | 21,481 | 30,952 | 13,868 | 6,260 | ||||||||||||||
Total contractual obligations | $ | 293,240 | $ | 36,214 | $ | 105,216 | $ | 40,257 | $ | 111,553 | |||||||||
Commercial commitments: | |||||||||||||||||||
Clinical and manufacturing development (3) | $ | 383,500 | $ | 81,810 | $ | 198,710 | $ | 102,980 | $ | — | |||||||||
Licenses (4) | 17,080 | 8,964 | 2,878 | 4,228 | 1,010 | ||||||||||||||
Total commercial commitments | $ | 400,580 | $ | 90,774 | $ | 201,588 | $ | 107,208 | $ | 1,010 | |||||||||
(1) Interest on variable rate debt calculated based on interest rates at December 31, 2014. | |||||||||||||||||||
(2) Facility lease obligation includes the lease agreement signed in November 2012, for office and laboratory space to be constructed in New Haven, Connecticut. Although we will not legally own the premises, we are deemed to be the owner of the building during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord's costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. Construction of the new facility began in June 2013 and commitments included within this schedule assume an estimated November 2015 occupancy date. | |||||||||||||||||||
(3) Clinical and manufacturing development commitments include only non-cancellable commitments at December 31, 2014. | |||||||||||||||||||
(4) License commitments do not include the $50,000 upfront payment associated with the license agreement we entered into in January 2015. | |||||||||||||||||||
The contractual obligations table above does not include contingent royalties and other contingent contractual payments we may owe to third parties in the future because such payments are contingent on future sales of our products and the existence and scope of third party intellectual property rights and other factors described in Item 1A "Risk Factors" and Note 9 "Commitments and Contingencies" of the Consolidated Financial Statements included in the Annual Report on Form 10-K.
The table above also does not include a liability for unrecognized tax benefits related to various federal, state and foreign income tax matters of $28,675 at December 31, 2014. The timing of the settlement of these amounts was not reasonably estimable at December 31, 2014. We do not expect payment of amounts related to the unrecognized tax benefits within the next twelve months.
We also did not include contingent payments related to business acquisitions completed in prior years, as the timing of payment for these amounts was not reasonably estimable at December 31, 2014. Contingent payments associated with these business combinations total up to $876,000 which will become payable if and when certain development and commercial milestones are achieved. During the next 12 months, we expect to make milestone payments totaling approximately $50,000.
75
Credit Facilities
In February 2012, we entered into a credit agreement, as amended (the Credit Agreement) with a syndicate of lenders and other parties named in the Credit Agreement that provides for a $240,000 senior secured term loan facility payable in equal quarterly installments of $12,000 beginning on June 30, 2012 and a $200,000 senior secured revolving credit facility through February 7, 2017.
We may elect that the loans under the Credit Agreement bear interest at a rate per annum equal to (i) LIBOR plus 1.25% to 2.00% depending on our consolidated leverage ratio (as calculated in accordance with the Credit Agreement), or (ii) in the case of loans denominated in U.S. dollars, a Base Rate equal to the higher of the (A) Prime Rate then in effect, (B) Federal Funds Rate then in effect plus 0.50%, and (C) Eurodollar Rate then in effect plus 1.00%, plus in each case of (A), (B) or (C), 0.25% to 1.00% depending on our consolidated leverage ratio of our cash to liabilities (as calculated in accordance with the Credit Agreement). Interest is payable quarterly for Base Rate loans and, in the case of LIBOR-based loans, at the end of the applicable interest period, with the principal due on February 7, 2017, the maturity date.
Our obligations under the credit facilities are unconditionally guaranteed, jointly and severally, by certain of our existing domestic subsidiaries and are required to be guaranteed by certain of our future domestic subsidiaries. The obligations of our non-U.S. subsidiaries under the credit facilities are unconditionally guaranteed, jointly and severally, by us, certain of our existing domestic subsidiaries, and certain of our foreign subsidiaries, and are required to be guaranteed by certain of our future subsidiaries. All obligations of each borrower under the credit facilities, and the guarantees of those obligations, are secured, subject to certain exceptions, by substantially all of each borrower's assets and the assets of certain guarantors, including the pledge of the equity interests of certain of our subsidiaries and real estate located in Smithfield, Rhode Island, but excluding intellectual property and assets of certain foreign subsidiaries.
The Credit Agreement requires us to comply with certain financial covenants on a quarterly basis. Further, the Credit Agreement includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, engage in certain investment, acquisition and disposition transactions, pay dividends, repurchase capital stock and enter into transactions with affiliates. The Credit Agreement also contains customary representations and warranties, affirmative covenants and events of default, including payment defaults, breach of representations and warranties, covenant defaults and cross defaults. If an event of default occurs, the interest rate would increase and the administrative agent would be entitled to take various actions, including the acceleration of amounts due under the loan.
Operating Leases
Our operating leases are principally for facilities and equipment. We currently lease office and laboratory space at our headquarters and research and development facility in Cheshire, Connecticut, as well as office space at our regional executive and sales offices in Lausanne, Switzerland. We also lease space at our global supply chain and distribution headquarters in Dublin, Ireland. In addition to the locations above, we also lease space in other U.S. states and foreign countries to support our operations as a global organization.
In January 2015, we entered into a new lease agreement of office space in Zurich, Switzerland to support the relocation of the European headquarters currently located in Lausanne, Switzerland. The term of the lease is 10 years with a minimum renewal option of 5 years and a maximum renewal option of 10 years. Annual lease payments of approximately $2,600 will commence in the second quarter of 2015.
We believe that our administrative office space is adequate to meet our needs for the foreseeable future. We also believe that our research and development facilities and our manufacturing facility, together with third party manufacturing facilities, will be adequate for our on-going activities.
Commercial Commitments
Our commercial commitments consist of research and development, license, operational, clinical development, and manufacturing cost commitments, along with anticipated supporting arrangements, subject to certain limitations and cancellation clauses. The timing and level of our commercial scale manufacturing costs, which may or may not be realized, are contingent upon the progress of our clinical development programs and our commercialization plans. Our commercial commitments are represented principally by our supply agreement with Lonza described above.
76
Item 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
(amounts in thousands, except percentages)
Interest Rate Risk
As of December 31, 2014, we invested our cash in a variety of financial instruments, principally money market funds, corporate bonds, municipal bonds, commercial paper and government-related obligations. Most of our interest-bearing securities are subject to interest rate risk and could decline in value if interest rates fluctuate. Our investment portfolio is comprised of marketable securities of highly rated financial institutions and investment-grade debt instruments, and we have guidelines to limit the term-to-maturity of our investments. Based on the type of securities we hold, we do not believe a change in interest rates would have a material impact on our financial statements. If interest rates were to increase or decrease by 1%, the fair value of our investment portfolio would (decrease) increase by approximately $(11,195) and $8,455, respectively.
In February 2012, we entered into the Credit Agreement with a floating rate of interest based on LIBOR, Prime Rate, Federal Funds Rate or Eurodollar Rate, at our election, plus an applicable credit spread. We do not expect changes in interest rates related to the Credit Agreement to have a material effect on our financial statements. As of December 31, 2014, we had approximately $57,500 of variable rate debt outstanding. If interest rates were to increase or decrease by 1% for the year, annual interest expense would increase or decrease by approximately $575.
Foreign Exchange Market Risk
Our operations include activities in many countries outside the United States, including, countries in Europe, Latin America and Asia Pacific. As a result, our financial results are impacted by factors such as changes in foreign currency exchange rates or weak economic conditions in the foreign markets where we operate. We have exposure to movements in foreign currency exchange rates, the most significant of which are the Euro and Japanese Yen, against the U.S. dollar. We are a net receiver of many foreign currencies, and our consolidated financial results benefit from a weaker U.S. Dollar and are adversely impacted by a stronger U.S. Dollar relative to foreign currencies in which we sell our product.
Our monetary exposures on our balance sheet arise primarily from cash, accounts receivable, intercompany receivables and payables denominated in foreign currencies. Approximately 60% of our product sales were denominated in foreign currencies during 2014, and our revenues are also exposed to fluctuations in the foreign currency exchange rates over time. In certain foreign countries, we may sell in U.S. Dollar, but our customers may be impacted adversely in fluctuations in foreign currency exchange rates which may also impact us in the future.
Both positive and negative impacts to our international product sales from movements in foreign currency exchange rates are only partially mitigated by the natural, opposite impact that foreign currency exchange rates have on our international operating expenses. Additionally, we have operations based in Switzerland, and accordingly, our expenses are impacted by fluctuations in the value of the Swiss Franc against the U.S. dollar.
We currently have a derivative program in place to achieve the following: 1) limit the foreign currency exposure of our monetary assets and liabilities on our balance sheet, using contracts with durations of up to 30 days and 2) hedge a portion of our forecasted product sales (in some currencies), including intercompany sales, using contracts with durations of up to 60 months. The objectives of this program are to reduce the volatility of our operating results due to fluctuation of foreign exchange and to increase the visibility of the foreign exchange impact on forecasted revenues. This program utilizes foreign exchange forward contracts intended to reduce, not eliminate, the volatility of operating results due to fluctuations in foreign exchange rates.
As of December 31, 2014 and 2013, we held foreign exchange forward contracts with notional amounts totaling $1,748,931 and $1,222,464, respectively. The increase in outstanding foreign exchange forward contracts resulted primarily from increases in forecasted revenues and, for certain currencies, extended duration of hedges. As of December 31, 2014 and 2013, our outstanding foreign exchange forward contracts had a net fair value of $135,166 and $(3,438), respectively. The increase in the net fair value of outstanding foreign exchange forward contracts is primarily due to the strengthening of the U.S. dollar in 2014.
We do not use derivative financial instruments for speculative trading purposes. The counterparties to these foreign exchange forward contracts are multinational commercial banks. We believe the risk of counterparty nonperformance is not material.
Based on our foreign currency exchange rate exposures at December 31, 2014, a hypothetical 10% adverse fluctuation in exchange rates would decrease the fair value of our foreign exchange forward contracts that are designated as cash flow hedges by approximately $136,500 at December 31, 2014. The resulting loss on these forward contracts would be offset by the gain on the underlying transactions and therefore would have minimal impact on future anticipated earnings and cash flows. Similarly,
77
adverse fluctuations in exchange rates that would decrease the fair value of our foreign exchange forward contracts that are not designated as hedge instruments would be offset by a positive impact of the underlying monetary assets and liabilities.
Credit Risk
As a result of our foreign operations, we are exposed to changes in the general economic conditions in the countries in which we conduct business. Substantially all of our accounts receivable due from these countries are due from or backed by sovereign or local governments, and the amount of non-sovereign accounts receivable is not material. We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. Although collection of our accounts receivables from certain countries may extend beyond our standard credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operations.
Item 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The consolidated financial statements and supplementary data of the Company required in this item are set forth beginning on page F-1.
Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
Item 9A. | CONTROLS AND PROCEDURES. |
Disclosure Controls and Procedures.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act,) as of December 31, 2014. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2014, our disclosure controls and procedures were effective to provide reasonable assurance that information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure, and ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms.
Management's Report on Internal Control over Financial Reporting.
Management of Alexion Pharmaceuticals, Inc. is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management utilized the criteria set forth in “Internal Control-Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission to conduct an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2014. Based on the assessment, management has concluded that, as of December 31, 2014, our internal control over financial reporting is effective.
The effectiveness of our internal control over financial reporting as of December 31, 2014 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Changes in Internal Control over Financial Reporting.
There has been no change in our internal control over financial reporting that occurred during the quarter ended December 31, 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
78
Item 9A(T). | CONTROLS AND PROCEDURES. |
Not applicable
Item 9B. | OTHER INFORMATION. |
None.
PART III
Item 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
The information required by this item with respect to our executive officers is provided under the caption entitled “Executive Officers of the Company” in Part I of this Annual Report on Form 10-K and is incorporated by reference herein. The information required by this item with respect to our directors and our audit committee and audit committee financial expert will be set forth in our definitive Proxy Statement under the captions “General Information About the Board of Directors” and “Election of Directors”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
SECTION 16(a) BENEFICIAL OWNERSHIP REPORTING COMPLIANCE
The information regarding compliance with Section 16(a) of the Securities Exchange Act of 1934 required by this Item will be set forth in our definitive Proxy Statement under the caption “Section 16(a) Beneficial Ownership Reporting Compliance”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
CODE OF ETHICS
We have adopted the Alexion Pharmaceuticals, Inc. Code of Conduct, or code of ethics, that applies to directors, officers and employees of Alexion and its subsidiaries and complies with the requirements of Item 406 of Regulation S-K and the listing standards of the NASDAQ Global Select Market. Our code of ethics is located on our website (http://ir.alexionpharm.com/governance.cfm). We amended the code of ethics in April 2011 and any future amendments or waivers to our code of ethics will be promptly disclosed on our website and as required by applicable laws, rules and regulations of the SEC and NASDAQ.
Item 11. | EXECUTIVE COMPENSATION. |
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
Item 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES. |
The information required by this Item will be set forth in our definitive Proxy Statement under the caption “Independent Registered Public Accounting Firm”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
80
PART IV
Item 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES. |
Item 15(a) |
(1) | Financial Statements |
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this report.
(2) | Financial Statement Schedules |
Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in the financial statements or notes thereto beginning on page F-1 of this report.
(3) | Exhibits: |
2.1 | Agreement and Plan of Merger by and among Alexion, TPCA Corporation, Taligen Therapeutics, Inc., each stockholder of Taligen that signed the Agreement as a seller of Series Bl Call Rights, and, only for the limited purposes described therein as Stockholders’ Representatives (and not in their individual capacities), Nick Galakatos, Ed Hurwitz and Timothy Mills, dated as of January 28, 2011.(1)+ | |
2.2 | Agreement and Plan of Merger by and among Alexion, EMRD Corporation, Enobia Pharma Corp., and the Stockholder Representatives named therein, dated as of December 28, 2011.(2)+ | |
2.3 | Amendment No. 1 to the Agreement and Plan of Merger, dated December 28, 2011, by and among Alexion, EMRD Corporation, Enobia Pharma Corp., and the Stockholder Representatives named therein, dated February 1, 2012.(3) | |
3.1 | Certificate of Incorporation, as amended.(4) | |
3.2 | Certificate of Amendment of the Certificate of Incorporation.(5) | |
3.3 | Bylaws, as amended.(6) | |
4.1 | Specimen Common Stock Certificate.(7) | |
4.2 | Rights Agreement between Alexion and Continental Stock Transfer & Trust Company, Rights Agent, dated as of February 14, 1997.(8) | |
4.3 | Amendment No. 1 to Rights Agreement, dated as of September 18, 2000, between Alexion and Continental Stock Transfer and Trust Company.(9) | |
4.4 | Amendment No. 2 to Rights Agreement, dated as of December 12, 2001, between Alexion and Continental Stock Transfer and Trust Company, which includes as Exhibit B the form of Right Certificate.(10) | |
4.5 | Amendment No. 3 to Rights Agreement, dated as of November 16, 2004, between Alexion and Continental Stock Transfer and Trust Company.(11) | |
4.6 | Amendment No. 4 to Rights Agreement, dated February 23, 2007, between Alexion and Continental Stock Transfer and Trust Company.(12) | |
10.1 | Employment Agreement, dated as of February 14, 2006, between Alexion and Dr. Leonard Bell.(13)** | |
10.2 | Amendment No. 1 to the Employment Agreement, dated as of December 23, 2009, between Alexion and Dr. Leonard Bell.(14)** | |
10.3 | Employment Agreement, dated as of February 14, 2006, between Alexion and Dr. Stephen P. Squinto.(13)** | |
10.4 | Amendment No. 1 to the Employment Agreement, dated as of December 23, 2009, between Alexion and Dr. Stephen P. Squinto.(14)** | |
10.5 | Employment Agreement, dated as of February 14, 2006, between Alexion and Vikas Sinha.(13)** | |
10.6 | Amendment No. 1 to the Employment Agreement, dated as of December 23, 2009, between Alexion and Vikas Sinha.(14)** | |
10.7 | Form of Employment Agreement (Senior Vice Presidents).(13)** | |
10.8 | Form of Amendment No. 1 to Employment Agreements (Senior Vice Presidents). (14)** | |
10.9 | Form of Indemnification Agreement for Officers and Directors. (15) | |
81
10.10 | Agreement of Lease, dated May 9, 2000, between Alexion and WE Knotter L.L.C.(16)+ | |
10.11 | Lease, dated November 15, 2012, between Alexion and WE Route 34, LLC.(17) | |
10.12 | Alexion’s 2000 Stock Option Plan, as amended.(18)** | |
10.13 | Alexion’s 1992 Outside Directors Stock Option Plan, as amended.(19)** | |
10.14 | Alexion’s Amended and Restated 2004 Incentive Plan.** | |
10.15 | License Agreement dated March 27, 1996 between Alexion and Medical Research Council.(20)+ | |
10.16 | Master Manufacturing and Supply Agreement, dated December 16, 2014 between Alexion Pharma International Trading, Alexion Pharmaceuticals, Inc,, Lonza Group AG, Lonza Biologics Tuas PTE LTD and Lonza Sales AG* | |
10.17 | Form of Stock Option Agreement for Directors.(22)** | |
10.18 | Form of Stock Option Agreement for Executive Officers (Form A).(23)** | |
10.19 | Form of Stock Option Agreement for Executive Officers (Form B).(23)** | |
10.20 | Form of Restricted Stock Award Agreement for Executive Officers (Form A).(24)** | |
10.21 | Form of Stock Option Agreement (Incentive Stock Options).(21) | |
10.22 | Form of Stock Option Agreement (Nonqualified Stock Options).(21) | |
10.23 | Form of Restricted Stock Award Agreement.(21) | |
10.24 | Form of Restricted Stock Unit Award Agreement.(25) | |
10.25 | Form of Stock Option Agreement for Participants in France.(21)** | |
10.26 | Form of Restricted Stock Unit Agreement for Participants in France.(21)** | |
10.27 | Credit Agreement by and among Alexion, certain subsidiaries of Alexion, the lenders party hereto, Bank of America, N.A., as Administrative Agent, Merrill Lynch, Pierce, Fenner & Smith Incorporated and J.P. Morgan Securities LLC, as joint lead arrangers and joint book managers. (3) | |
10.28 | First Amendment to Credit Agreement dated November 14, 2012 by and among Alexion, certain subsidiaries of Alexion, the lenders party thereto and Bank of America, N.A., as Administrative Agent. | |
10.29 | Consent and Second Amendment to Credit Agreement and First Amendment to Administrative Borrower Guaranty, Domestic Subsidiary Guaranty and Foreign Subsidiary Guaranty dated December 17, 2013 by and among Alexion, certain subsidiaries of Alexion, the lenders party thereto and Bank of America, N.A., as Administrative Agent. | |
21.1 | Subsidiaries of Alexion Pharmaceuticals, Inc. | |
23.1 | Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm | |
31.1 | Certificate of Chief Executive Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 Sarbanes Oxley Act of 2002. | |
31.2 | Certificate of Chief Financial Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 of Sarbanes Oxley Act of 2002. | |
32.1 | Certificate of Chief Executive Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. | |
32.2 | Certificate of Chief Financial Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. | |
101 | The following materials from the Alexion Pharmaceuticals, Inc. Annual Report on Form 10-K for the year ended December 31, 2014 formatted in eXtensible Business Reporting Language (XBRL): (i) the Consolidated Statements of Operations, (ii) the Consolidated Statements of Comprehensive Income, (iii) the Consolidated Balance Sheets, (iv) the Consolidated Statements of Changes in Stockholders' Equity, (v) the Consolidated Statements of Cash Flows and (vi) related notes, tagged as blocks of text. | |
_____________________
(1) | Incorporated by reference to our Report on Form 8-K, filed on February 3, 2011. |
(2) | Incorporated by reference to our Report on Form 8-K, filed on January 4, 2012. |
(3) | Incorporated by reference to our Report on Form 8-K, filed on February 7, 2012. |
(4) | Incorporated by reference to our Registration Statement on Form S-3 (Reg. No. 333-128085), filed on September 2, 2005. |
82
(5) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2011. |
(6) | Incorporated by reference to our Report on Form 10-Q, filed on October 25, 2013. |
(7) | Incorporated by reference to our Registration Statement on Form S-1 (Reg. No. 333-00202). |
(8) | Incorporated by reference to our Registration Statement on Form 8-A (Reg. No. 000-27756), filed on February 21, 1997. |
(9) | Incorporated by reference to Amendment No. 1 to our Registration Statement on Form 8-A (Reg. No. 000-27756), filed on October 6, 2000. |
(10) | Incorporated by reference to Amendment No. 2 to our Registration Statement on Form 8-A (Reg. No. 000-27756), filed on February 12, 2002. |
(11) | Incorporated by reference to Amendment No. 3 to our Registration Statement on Form 8-A (Reg. No. 000-27756), filed on November 17, 2004. |
(12) | Incorporated by reference to our Annual Report on Form 10-K for the year ended December 31, 2006. |
(13) | Incorporated by reference to our Report on Form 8-K filed on February 16, 2006. |
(14) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2009. |
(15) | Incorporated by reference to our Report on Form 8-K, filed on September 17, 2010. |
(16) Incorporated by reference to our Registration Statement on Form S-3 (Reg. No. 333-36738) filed on May 10, 2000.
(17) Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2013.
(18) Incorporated by reference to our quarterly report on Form 10-Q for the quarter ended January 31, 2004.
(19) Incorporated by reference to our Registration Statement on Form S-8 (Reg. No. 333-71879) filed on February 5, 1999.(20) Incorporated by reference to our Annual Report on Form 10-K/A for the fiscal year ended July 31, 1996.
(21) Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2008.
(22) Incorporated by reference to our report on Form 8-K, filed on December 16, 2004.
(23) Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended January 31, 2005.
(24) Incorporated by reference to our report on Form 8-K, filed on March 14, 2005.
(25) Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2010.
+ | Confidential treatment was granted for portions of such exhibit. |
* | Confidential treatment requested under 17 C.F.R. §§200.80(b)(4) and 24b-2. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the SEC pursuant to the confidential treatment request. |
** Indicates a management contract or compensatory plan or arrangement required to be filed pursuant to Item 15(b) of Form 10-K.
Item 15(b) Exhibits
See (a) (3) above.
Item 15(c) Financial Statement Schedules
See (a) (2) above.
83
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ALEXION PHARMACEUTICALS, INC. | |
By: | /s/ Leonard Bell |
Leonard Bell, M.D. Chairman and Chief Executive Officer Dated: February 6, 2015 | |
By: | /s/ Vikas Sinha |
Vikas Sinha, M.B.A., C.A. Executive Vice President and Chief Financial Officer Dated: February 6, 2015 | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
/s/ Leonard Bell | Chairman and Chief Executive Officer, (principal executive officer) | February 6, 2015 | |
Leonard Bell, M.D. | |||
/s/ Vikas Sinha | Executive Vice President and Chief Financial Officer (principal financial officer) | February 6, 2015 | |
Vikas Sinha, M.B.A., C.A., C.P.A. | |||
/s/ Scott Phillips | Vice President, Corporate Controller and Chief Accounting Officer (principal accounting officer) | February 6, 2015 | |
Scott Phillips, C.P.A. | |||
/s/ David R. Brennan | Director | February 6, 2015 | |
David R. Brennan | |||
/s/ M. Michele Burns | Director | February 6, 2015 | |
M. Michele Burns | |||
/s/ Christopher J. Coughlin | Director | February 6, 2015 | |
Christopher J. Coughlin | |||
/s/ David Hallal | Director, Chief Operating Officer and CEO-elect | February 6, 2015 | |
David Hallal | |||
/s/ William Keller | Director | February 6, 2015 | |
William Keller | |||
/s/ John T. Mollen | Director | February 6, 2015 | |
John T. Mollen | |||
/s/ R. Douglas Norby | Director | February 6, 2015 | |
R. Douglas Norby | |||
/s/ Alvin S. Parven | Director | February 6, 2015 | |
Alvin S. Parven | |||
/s/ Andreas Rummelt | Director | February 6, 2015 | |
Andreas Rummelt, Ph.D. | |||
/s/ Ann Veneman | Director | February 6, 2015 | |
Ann Veneman | |||
Alexion Pharmaceuticals, Inc.
Contents
For the Years Ended December 31, 2014, 2013 and 2012
Page(s) | |
Consolidated Financial Statements | |
F-1
Report of Independent Registered Public Accounting Firm
To Board of Directors and Stockholders
of Alexion Pharmaceuticals, Inc.:
In our opinion, the consolidated financial statements listed in the accompanying index present fairly, in all material respects, the financial position of Alexion Pharmaceuticals, Inc. and its subsidiaries at December 31, 2014 and December 31, 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Hartford, Connecticut
February 6, 2015
F-2
Alexion Pharmaceuticals, Inc.
Consolidated Balance Sheets
(amounts in thousands, except per share amounts)
December 31, | |||||||
2014 | 2013 | ||||||
Assets | |||||||
Current Assets: | |||||||
Cash and cash equivalents | $ | 943,999 | $ | 529,857 | |||
Marketable securities | 1,017,567 | 984,994 | |||||
Trade accounts receivable, net | 432,888 | 421,752 | |||||
Inventories | 176,441 | 102,602 | |||||
Deferred tax assets | 35,733 | 41,432 | |||||
Prepaid expenses and other current assets | 189,401 | 106,220 | |||||
Total current assets | 2,796,029 | 2,186,857 | |||||
Property, plant and equipment, net | 392,248 | 201,109 | |||||
Intangible assets, net | 587,046 | 609,719 | |||||
Goodwill | 254,073 | 254,073 | |||||
Deferred tax assets | 34,939 | 3,394 | |||||
Other assets | 137,627 | 62,544 | |||||
Total assets | $ | 4,201,962 | $ | 3,317,696 | |||
Liabilities and Stockholders' Equity | |||||||
Current Liabilities: | |||||||
Accounts payable | $ | 44,016 | $ | 21,596 | |||
Accrued expenses | 395,232 | 402,344 | |||||
Deferred revenue | 58,837 | 53,801 | |||||
Current portion of long-term debt | 48,000 | 48,000 | |||||
Other current liabilities | 60,655 | 56,688 | |||||
Total current liabilities | 606,740 | 582,429 | |||||
Long-term debt, less current portion | 9,500 | 65,000 | |||||
Contingent consideration | 116,425 | 106,744 | |||||
Facility lease obligation | 107,099 | 32,230 | |||||
Deferred tax liabilities | 7,046 | 101,241 | |||||
Other liabilities | 53,134 | 47,973 | |||||
Total liabilities | 899,944 | 935,617 | |||||
Commitments and contingencies (Note 9) | |||||||
Stockholders' Equity: | |||||||
Preferred stock, $.0001 par value; 5,000 shares authorized, no shares issued or outstanding | — | — | |||||
Common stock, $.0001 par value; 290,000 shares authorized; 201,944 and 197,941 shares issued at December 31, 2014 and 2013, respectively | 20 | 20 | |||||
Additional paid-in capital | 2,592,167 | 2,106,183 | |||||
Treasury stock, at cost, 2,888 and 985 shares at December 31, 2014 and 2013, respectively | (382,964 | ) | (80,365 | ) | |||
Accumulated other comprehensive income (loss) | 56,785 | (22,857 | ) | ||||
Retained earnings | 1,036,010 | 379,098 | |||||
Total stockholders' equity | 3,302,018 | 2,382,079 | |||||
Total liabilities and stockholders' equity | $ | 4,201,962 | $ | 3,317,696 | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Operations
(amounts in thousands, except per share amounts)
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net product sales | $ | 2,233,733 | $ | 1,551,346 | $ | 1,134,114 | |||||
Cost of sales: | |||||||||||
Cost of sales | 173,862 | 168,375 | 126,214 | ||||||||
Change in contingent liability from intellectual property settlements | — | 9,181 | (53,377 | ) | |||||||
Total cost of sales | 173,862 | 177,556 | 72,837 | ||||||||
Operating expenses: | |||||||||||
Research and development | 513,782 | 317,093 | 222,732 | ||||||||
Selling, general and administrative | 630,209 | 489,720 | 384,678 | ||||||||
Acquisition-related costs | 20,295 | 5,029 | 22,812 | ||||||||
Impairment of intangible assets | 11,514 | 33,521 | 26,300 | ||||||||
Restructuring expenses | 15,365 | — | — | ||||||||
Amortization of purchased intangible assets | — | 417 | 417 | ||||||||
Total operating expenses | 1,191,165 | 845,780 | 656,939 | ||||||||
Operating income | 868,706 | 528,010 | 404,338 | ||||||||
Other income and expense: | |||||||||||
Investment income | 8,373 | 3,346 | 1,838 | ||||||||
Interest expense | (2,982 | ) | (4,112 | ) | (7,402 | ) | |||||
Foreign currency loss | (1,990 | ) | (975 | ) | (1,208 | ) | |||||
Income before income taxes | 872,107 | 526,269 | 397,566 | ||||||||
Income tax provision | 215,195 | 273,374 | 142,744 | ||||||||
Net income | $ | 656,912 | $ | 252,895 | $ | 254,822 | |||||
Earnings per common share | |||||||||||
Basic | $ | 3.32 | $ | 1.29 | $ | 1.34 | |||||
Diluted | $ | 3.26 | $ | 1.27 | $ | 1.28 | |||||
Shares used in computing earnings per common share | |||||||||||
Basic | 198,103 | 195,532 | 190,461 | ||||||||
Diluted | 201,623 | 199,712 | 198,501 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Comprehensive Income
(amounts in thousands)
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net income | $ | 656,912 | $ | 252,895 | $ | 254,822 | |||||
Other comprehensive income (loss), net of tax: | |||||||||||
Foreign currency translation | (6,337 | ) | (4,573 | ) | 150 | ||||||
Unrealized losses on marketable securities, net of tax of $99, $(75) and $0, respectively | (88 | ) | (146 | ) | — | ||||||
Unrealized losses on pension obligation, net of tax of $(1,574), $(547) and $(143), respectively | (5,068 | ) | (5,790 | ) | (1,529 | ) | |||||
Unrealized gains (losses) on hedging activities, net of tax of $45,448, $(871) and $232, respectively | 91,135 | (18,983 | ) | 3,835 | |||||||
Other comprehensive income (loss), net of tax | 79,642 | (29,492 | ) | 2,456 | |||||||
Comprehensive income | $ | 736,554 | $ | 223,403 | $ | 257,278 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Changes in Stockholders’ Equity
(amounts in thousands)
Common Stock | Additional Paid-In Capital | Treasury Stock at Cost | Accumulated Other Comprehensive Income (Loss) | Retained Earnings (Deficit) | Total Stockholders Equity | ||||||||||||||||||||||||
Shares Issued | Amount | Shares | Amount | ||||||||||||||||||||||||||
Balances, December 31, 2011 | 185,616 | $ | 19 | $ | 1,261,589 | 97 | $ | (2,676 | ) | $ | 4,179 | $ | (128,619 | ) | $ | 1,134,492 | |||||||||||||
Issuance of common stock, net of issuance costs of $207 | 5,000 | 1 | 462,212 | — | — | — | — | 462,213 | |||||||||||||||||||||
Conversion of convertible notes to common stock | 91 | — | 718 | — | — | — | — | 718 | |||||||||||||||||||||
Repurchase of common stock | — | — | — | 130 | (11,553 | ) | — | — | (11,553 | ) | |||||||||||||||||||
Issuance of common stock from exercise of options | 3,918 | — | 66,438 | — | — | — | — | 66,438 | |||||||||||||||||||||
Issuance of restricted common stock | 293 | — | — | — | — | — | — | — | |||||||||||||||||||||
Excess tax benefit from stock options | — | — | 7,228 | — | — | — | — | 7,228 | |||||||||||||||||||||
Share-based compensation expense | — | — | 54,036 | — | — | — | — | 54,036 | |||||||||||||||||||||
Net income | — | — | — | — | — | — | 254,822 | 254,822 | |||||||||||||||||||||
Other comprehensive income | — | — | $ | — | — | — | 2,456 | — | $ | 2,456 | |||||||||||||||||||
Balances, December 31, 2012 | 194,918 | $ | 20 | $ | 1,852,221 | 227 | $ | (14,229 | ) | $ | 6,635 | $ | 126,203 | $ | 1,970,850 | ||||||||||||||
Repurchase of common stock | — | — | — | 758 | (66,136 | ) | — | — | (66,136 | ) | |||||||||||||||||||
Issuance of common stock from exercise of options | 2,481 | — | 71,281 | — | — | — | — | 71,281 | |||||||||||||||||||||
Issuance of restricted common stock | 542 | — | — | — | — | — | — | — | |||||||||||||||||||||
Excess tax benefit from stock options | — | — | 105,714 | — | — | — | — | 105,714 | |||||||||||||||||||||
Share-based compensation expense | — | — | 76,967 | — | — | — | — | 76,967 | |||||||||||||||||||||
Net income | — | — | — | — | — | — | 252,895 | 252,895 | |||||||||||||||||||||
Other comprehensive loss | — | — | — | — | — | (29,492 | ) | — | (29,492 | ) | |||||||||||||||||||
Balances, December 31, 2013 | 197,941 | $ | 20 | $ | 2,106,183 | 985 | $ | (80,365 | ) | $ | (22,857 | ) | $ | 379,098 | $ | 2,382,079 | |||||||||||||
Repurchase of common stock | — | — | — | 1,903 | (302,599 | ) | — | — | (302,599 | ) | |||||||||||||||||||
Issuance of common stock from exercise of options | 3,408 | — | 114,350 | — | — | — | — | 114,350 | |||||||||||||||||||||
Issuance of restricted common stock | 595 | — | — | — | — | — | — | — | |||||||||||||||||||||
Excess tax benefit from stock options | — | — | 251,136 | — | — | — | — | 251,136 | |||||||||||||||||||||
Share-based compensation expense | — | — | 120,498 | — | — | — | — | 120,498 | |||||||||||||||||||||
Net income | — | — | — | — | — | — | 656,912 | 656,912 | |||||||||||||||||||||
Other comprehensive loss | — | — | — | — | — | 79,642 | — | 79,642 | |||||||||||||||||||||
Balances, December 31, 2014 | 201,944 | $ | 20 | $ | 2,592,167 | 2,888 | $ | (382,964 | ) | $ | 56,785 | $ | 1,036,010 | $ | 3,302,018 | ||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(amounts in thousands)
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash flows from operating activities: | |||||||||||
Net income | $ | 656,912 | $ | 252,895 | $ | 254,822 | |||||
Adjustments to reconcile net income to net cash flows from operating activities: | |||||||||||
Depreciation and amortization | 46,939 | 28,693 | 23,887 | ||||||||
Impairment of intangible assets | 11,514 | 33,521 | 26,300 | ||||||||
Change in fair value of contingent consideration | 20,295 | 4,006 | 6,550 | ||||||||
Share-based compensation expense | 114,461 | 76,203 | 54,013 | ||||||||
Premium amortization of available-for-sale securities | 15,519 | 3,235 | — | ||||||||
Deferred taxes | (153,905 | ) | 92,831 | 71,155 | |||||||
Excess tax benefit from stock options | (251,136 | ) | (105,714 | ) | (7,228 | ) | |||||
Other | 22,046 | 2,040 | (4,833 | ) | |||||||
Changes in operating assets and liabilities, excluding the effect of acquisitions: | |||||||||||
Accounts receivable | (28,137 | ) | (116,439 | ) | (72,870 | ) | |||||
Inventories | (66,812 | ) | 126 | (6,265 | ) | ||||||
Prepaid expenses and other assets | (18,392 | ) | (39,879 | ) | (17,041 | ) | |||||
Accounts payable, accrued expenses and other liabilities | 264,572 | 242,355 | 68,951 | ||||||||
Deferred revenue | 6,199 | 23,476 | 13,172 | ||||||||
Net cash provided by operating activities | 640,075 | 497,349 | 410,613 | ||||||||
Cash flows from investing activities: | |||||||||||
Purchases of available-for-sale securities | (664,228 | ) | (1,048,429 | ) | — | ||||||
Proceeds from maturity or sale of available-for-sale securities | 619,447 | 60,917 | — | ||||||||
Purchases of trading securities | (3,431 | ) | (985 | ) | — | ||||||
Purchases of property, plant and equipment | (136,650 | ) | (29,329 | ) | (21,846 | ) | |||||
Purchases of other investments | (37,500 | ) | — | — | |||||||
Payments for acquisitions of businesses, net of cash acquired | — | — | (605,735 | ) | |||||||
Other | (507 | ) | (9,315 | ) | (4 | ) | |||||
Net cash used in investing activities | (222,869 | ) | (1,027,141 | ) | (627,585 | ) | |||||
Cash flows from financing activities: | |||||||||||
Debt issuance costs | — | — | (6,184 | ) | |||||||
Proceeds from revolving credit facility | — | — | 115,000 | ||||||||
Payments on revolving credit facility | — | — | (115,000 | ) | |||||||
Proceeds from term loan | — | — | 240,000 | ||||||||
Payments on term loan | (55,500 | ) | (36,000 | ) | (91,000 | ) | |||||
Excess tax benefit from stock options | 251,136 | 105,714 | 7,228 | ||||||||
Repurchase of common stock | (302,599 | ) | (66,136 | ) | (11,553 | ) | |||||
Net proceeds from issuance of common stock | — | — | 462,212 | ||||||||
Net proceeds from the exercise of stock options | 114,350 | 71,281 | 66,438 | ||||||||
Payment of contingent consideration | (3,000 | ) | — | ||||||||
Other | (261 | ) | (220 | ) | (765 | ) | |||||
Net cash provided by financing activities | 7,126 | 71,639 | 666,376 | ||||||||
Effect of exchange rate changes on cash | (10,190 | ) | (1,491 | ) | (768 | ) | |||||
Net change in cash and cash equivalents | 414,142 | (459,644 | ) | 448,636 | |||||||
Cash and cash equivalents at beginning of period | 529,857 | 989,501 | 540,865 | ||||||||
Cash and cash equivalents at end of period | $ | 943,999 | $ | 529,857 | $ | 989,501 | |||||
Supplemental cash flow disclosures: | |||||||||||
Cash paid for interest (net of amounts capitalized) | $ | 1,910 | $ | 2,831 | $ | 4,475 | |||||
Cash paid for income taxes | $ | 91,195 | $ | 76,165 | $ | 18,272 | |||||
Supplemental cash flow disclosures from investing and financing activities: | |||||||||||
Contingent consideration issued in acquisitions | $ | — | $ | — | $ | 117,000 | |||||
Construction in process related to facility lease obligation | $ | 74,869 | $ | 32,230 | $ | — | |||||
Increase in accrued expenses for purchases of property, plant and equipment | $ | 17,092 | $ | — | $ | — | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
1. | Business Overview and Summary of Significant Accounting Policies |
Business
Alexion Pharmaceuticals, Inc. (Alexion, the Company, we, our or us) is a biopharmaceutical company focused on serving patients with severe and ultra-rare disorders through the innovation, development and commercialization of life-transforming therapeutic products. Our marketed product Soliris is the first and only therapeutic approved for patients with either of two severe and ultra-rare disorders resulting from chronic uncontrolled activation of the complement component of the immune system: paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening and ultra-rare genetic blood disorder, and atypical hemolytic uremic syndrome (aHUS), a life-threatening and ultra-rare genetic disease. We are also evaluating additional potential indications for Soliris in other severe and devastating diseases in which uncontrolled complement activation is the underlying mechanism, and we are progressing in various stages of development with additional product candidates as potential treatments for patients with severe and life-threatening ultra-rare disorders. We were incorporated in 1992 and began commercial sale of Soliris in 2007.
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Alexion and its wholly-owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation. For each of our business combinations, all of the assets acquired and liabilities assumed were recorded at their respective fair values as of the date of acquisition, and their results of operations are included in the consolidated financial statements from the date of acquisition.
Dividend Policy
We have never paid a cash dividend on shares of our stock. We currently intend to retain our earnings to finance future operations and do not anticipate paying any cash dividends on our stock in the foreseeable future.
Critical Accounting Estimates
The preparation of our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, revenues, expenses and related disclosure of contingent assets and liabilities in our financial statements. We believe the most complex judgments result primarily from the need to make estimates about the effects of matters that are inherently uncertain and are significant to our consolidated financial statements. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. We evaluate our estimates, judgments and assumptions on an ongoing basis. Actual results may differ from these estimates under different assumptions or conditions.
The most significant areas involving estimates, judgments and assumptions used in the preparation of our consolidated financial statements are as follows:
• | Revenue recognition; |
• | Contingent liabilities; |
• | Inventories; |
• | Research and development expenses; |
• | Share-based compensation; |
• | Valuation of goodwill, acquired intangible assets and in-process research and development (IPR&D); |
• | Valuation of contingent consideration; and |
• | Income taxes. |
Foreign Currency Translation
The financial statements of our subsidiaries with functional currencies other than the U.S. dollar are translated into U.S. dollars using period-end exchange rates for assets and liabilities, historical exchange rates for stockholders’ equity and weighted average exchange rates for operating results. Translation gains and losses are included in accumulated other
F-8
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
comprehensive income (loss), net of tax, in stockholders’ equity. Foreign currency transaction gains and losses are included in the results of operations in other income and expense.
Cash and Cash Equivalents
Cash and cash equivalents are stated at cost plus accrued interest, which approximates fair value, and include short-term highly liquid investments with original maturities of three months or less.
Fair Value of Financial Instruments
The carrying amounts reflected in the consolidated balance sheets for cash and cash equivalents, accounts receivable, other assets, accounts payable, accrued expenses and other liabilities approximate fair value due to their short-term maturities. Our marketable securities are valued based upon pricing of securities with similar investment characteristics and holdings. Our derivative financial instruments are measured at fair value using observable market inputs such as forward rates, interest rates, our own credit risk and our counterparties’ credit risks. Our debt obligations are carried at historical cost, which approximates fair value. Our contingent consideration liabilities related to our acquisitions are valued based on various estimates, including probability of success, estimated revenues, discount rates and amount of time until the conditions of the milestone payments are met.
Marketable Securities
We invest our excess cash balances in marketable securities of highly rated financial institutions and investment-grade debt instruments. We seek to diversify our investments and limit the amount of investment concentrations for individual institutions, maturities and investment types. We classify these marketable securities as available-for-sale and, accordingly, record such securities at fair value. We classify these marketable securities as current assets as these investments are intended to be available to the Company for use in funding current operations.
Unrealized gains and losses that are deemed temporary are included in accumulated other comprehensive income (loss) as a separate component of stockholders' equity. If any adjustment to fair value reflects a significant decline in the value of the security, we evaluate the extent to which the decline is determined to be other-than-temporary and would mark the security to market through a charge to our consolidated statement of operations. Credit losses are identified when we do not expect to receive cash flows sufficient to recover the amortized cost basis of a security. In the event of a credit loss, only the amount associated with the credit loss is recognized in operating results, with the amount of loss relating to other factors recorded in accumulated other comprehensive income (loss).
We sponsor a nonqualified deferred compensation plan which allows certain highly-compensated employees to elect to defer income to future periods. Participants in the plan earn a return on their deferrals based on several investments options, which mirror returns on underlying mutual fund investments. We choose to invest in the underlying mutual fund investments to offset the liability associated with our nonqualified deferred compensation plan. These securities are classified as trading securities and are carried at fair value with gains and losses included in investment income. The changes in the underlying liability to the employee are recorded in operating expenses.
Accounts Receivable
Our standard credit terms vary based on the country of sale and range from 30 to 120 days. Our consolidated average days’ sales outstanding ranges from 60 to 70 days. We evaluate the creditworthiness of customers on a regular basis. In certain European countries, sales by us are subject to payment terms that are statutorily determined. This is primarily the case in countries where the payer is government-owned or government-funded, which we consider to be creditworthy. The length of time from sale to receipt of payment in certain countries exceeds our credit terms. In countries in which collections from customers extend beyond normal payment terms, we seek to collect interest. We record interest on customer receivables as interest income when collected. For non-interest bearing receivables with an estimated payment beyond one year, we discount the accounts receivable to present value at the date of sale, with a corresponding adjustment to revenue. Subsequent adjustments for further declines in credit rating are recorded as bad debt expense as a component of selling, general and administrative expense. We also use judgments as to our ability to collect outstanding receivables and provide allowances for the portion of receivables if and when collection becomes doubtful, and we also assess on an ongoing basis whether collectibility is reasonably assured at the time of sale.
F-9
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Concentration of Credit Risk
Financial instruments that potentially expose the Company to concentrations of credit risk are limited to cash equivalents, marketable securities, accounts receivable and our foreign exchange derivative contracts. We invest our cash reserves in money market funds or high-quality marketable securities in accordance with our investment policy. The stated objectives of our investment policy is to preserve capital, provide liquidity consistent with forecasted cash flow requirements, maintain appropriate diversification and generate returns relative to these investment objectives and prevailing market conditions.
At December 31, 2014, four individual customers accounted for 58% of the accounts receivable balance, with individual customers ranging from 10% to 23% of the accounts receivable balance. At December 31, 2013, two individual customers accounted for 30% of the accounts receivable balance, with individual customers accounting for 10% and 20%. For the years ended December 31, 2014 and 2013, one customer accounted for 18% and 20%, respectively, of our product sales. No other customers accounted for more than 10% of net product sales or accounts receivable.
As a result of our foreign operations, we are exposed to changes in the general economic conditions in the countries in which we conduct business. Substantially all of our accounts receivable due from these countries are due from or backed by sovereign or local governments, and the amount of non-sovereign accounts receivable is not material. We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. The adverse credit and economic conditions in Italy and Spain, among other members of the European Union, have improved in recent periods and, although collection of our accounts receivables due from these countries may extend beyond our standard credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operations.
Inventories
Inventories are stated at the lower of cost or estimated realizable value. We determine the cost of inventory using the weighted-average cost method.
The components of inventory are as follows:
December 31, | |||||||
2014 | 2013 | ||||||
Raw materials | $ | 14,570 | $ | 12,170 | |||
Work-in-process | 107,170 | 62,192 | |||||
Finished goods | 54,701 | 28,240 | |||||
$ | 176,441 | $ | 102,602 | ||||
Capitalization of Inventory Costs
We capitalize inventory produced for commercial sale, which may include costs incurred for certain products awaiting regulatory approval. We capitalize inventory produced in preparation of product launches sufficient to support estimated initial market demand. Capitalization of such inventory begins when we have (i) obtained positive results in clinical trials that we believe are necessary to support regulatory approval, (ii) concluded that uncertainties regarding regulatory approval have been sufficiently reduced, and (iii) determined that the inventory has probable future economic benefit. In evaluating whether these conditions have been met, we consider clinical trial results for the underlying product candidate, results from meetings with regulatory authorities, and the compilation of the regulatory application. If we are aware of any material risks or contingencies outside of the standard regulatory review and approval process, or if there are any specific negative issues identified relating to the safety, efficacy, manufacturing, marketing or labeling of the product that would have a significant negative impact on its future economic benefits, the related inventory would not be capitalized. At December 31, 2014, we capitalized $22,005 of inventory produced for commercial sale for products awaiting regulatory approval. At December 31, 2013, we did not capitalize any inventory associated with such products.
Products that have been approved by the U.S. Food and Drug Administration (FDA) or other regulatory authorities, such as Soliris, are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of Soliris utilized for both commercial and clinical programs is identical and, as a result, the inventory has an "alternative future use" as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to
F-10
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
research and development expense when the product enters the research and development process and no longer can be used for commercial purposes and, therefore, does not have an "alternative future use".
For products which are under development and have not yet been approved by regulatory authorities, purchased drug product is charged to research and development expense upon delivery. Delivery occurs when the inventory passes quality inspection and ownership transfers to us. Nonrefundable advance payments for research and development activities, including production of purchased drug product, are deferred and capitalized until the goods are delivered. We also recognize expense for raw materials purchased for developmental purposes when the raw materials pass quality inspection and we have an obligation to pay for the materials.
Inventory Write-Offs
We analyze our inventory levels to identify inventory that may expire prior to sale, inventory that has a cost basis in excess of its estimated realizable value, or inventory in excess of expected sales requirements. Although the manufacturing of our product is subject to strict quality control, certain batches or units of product may no longer meet quality specifications or may expire, which requires adjustments to our inventory values. We also apply judgment related to the results of quality tests that we perform throughout the production process, as well as our understanding of regulatory guidelines, to determine if it is probable that inventory will be saleable. These quality tests are performed throughout the pre-and post-production process, and we continually gather additional information regarding product quality for periods after the manufacture date. Soliris currently has a maximum estimated life of 48 months and, based on our sales forecasts, we expect to realize the carrying value of the Soliris inventory. In the future, reduced demand, quality issues or excess supply beyond those anticipated by management may result in a material adjustment to inventory levels, which would be recorded as an increase to cost of sales.
The determination of whether or not inventory costs will be realizable requires estimates by our management. A critical input in this determination is future expected inventory requirements based on internal sales forecasts. We then compare these requirements to the expiry dates of inventory on hand. For inventories that are capitalized in preparation of product launch, we also consider the expected approval date in assessing realizability. To the extent that inventory is expected to expire prior to being sold, we will write down the value of inventory.
Derivative Instruments
We record the fair value of derivative instruments as either assets or liabilities on the balance sheet. The accounting for gains and losses resulting from changes in fair value is dependent on the use of the derivative and whether it is designated and qualifies for hedge accounting.
All qualifying hedging activities are documented at the inception of the hedge and must meet the definition of highly effective in offsetting changes to future cash. The effectiveness of the qualifying hedge contract is assessed quarterly. We record the fair value of the qualifying hedges in other current assets, other assets, other current liabilities and other liabilities. Gains or losses resulting from changes in the fair value of qualifying hedges are recorded in other comprehensive income (loss) until the forecasted transaction occurs. When the forecasted transaction occurs, this amount is reclassified into revenue. Any non-qualifying portion of the gains or losses resulting from changes in fair value, if any, is reported in other income and expense.
Property, Plant and Equipment
Property, plant and equipment are stated at cost and are depreciated on a straight-line basis over the estimated useful lives of the assets. We estimate economic lives as follows:
• | Building and improvements—five to thirty years |
• | Machinery and laboratory equipment—three to ten years |
• | Computer hardware and software—three to five years |
• | Furniture and office equipment—three years |
Leasehold improvements and assets under capital lease arrangements are amortized over the lesser of the asset's estimated useful life or the term of the respective lease. Maintenance costs are expensed as incurred.
Construction-in-progress reflects amounts incurred for property, plant, or equipment construction or improvements that have not been placed in service.
F-11
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Manufacturing Facilities
We capitalize costs incurred for the construction of facilities which support commercial manufacturing. We also capitalize costs related to validation activities which are directly attributable to preparing the facility for its intended use, including engineering runs and inventory production necessary to obtain approval of the facility from government regulators for the production of a commercially approved drug. When the facility is substantially complete and ready for its intended use and regulatory approval for commercial production has been received, we will place the asset in service.
The production of inventory for preparing the facility for its intended use requires two types of production: engineering runs which are used for testing purposes only and do not result in saleable inventory, and validation runs which are used for validating equipment and may result in saleable inventory. The costs associated with inventory produced during engineering runs and normal production losses during validation runs are capitalized to fixed assets and depreciated over the asset's useful life. Saleable inventory produced during the validation process is initially treated as a fixed asset; however, upon regulatory approval, this inventory is reclassified to inventory and expensed in cost of goods sold as product is sold, or in research and development expenses as product is utilized in R&D activities. Abnormal production costs incurred during the validation process are expensed as incurred.
Acquisitions
Business combinations are accounted for using the acquisition method of accounting. Under the acquisition method of accounting, the tangible and intangible assets acquired and the liabilities assumed are recorded as of the acquisition date at their respective fair values. We evaluate a business as an integrated set of activities and assets that is capable of being managed for the purpose of providing a return in the form of dividends, lower costs or other economic benefits and consists of inputs and processes that provide or have the ability to provide outputs. In an acquisition of a business, the excess of the fair value of the consideration transferred over the fair value of the net assets acquired is recorded as goodwill. In an acquisition of net assets that does not constitute a business, no goodwill is recognized.
Our consolidated financial statements include the results of operations of an acquired business after the completion of the acquisition.
Intangible Assets
Our intangible assets consist of licenses, patents, purchased technology and acquired in-process research and development (IPR&D). Intangible assets with definite lives are amortized over their estimated useful lives and reviewed periodically for impairment.
Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment. If and when development is complete, which generally occurs when regulatory approval to market a product is obtained, the associated assets are deemed finite-lived and are amortized based on their estimated useful lives at that point in time.
Goodwill
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We are organized and operate as a single reporting unit and therefore the goodwill impairment test is performed using our overall market value, as determined by our traded share price, compared to our book value of net assets. We completed our annual impairment test as of December 31, 2014 and determined the carrying value of goodwill was not impaired.
Impairment of Long-Lived Assets
Our long-lived assets are primarily comprised of intangible assets and property, plant and equipment. We evaluate our finite-lived intangible assets and property, plant and equipment, for impairment whenever events or changes in circumstances indicate the carrying value of an asset or group of assets is not recoverable. If these circumstances exist, recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset group to future undiscounted net cash flows expected to be generated by the asset group. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the fair value of the assets.
F-12
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
In addition, indefinite-lived intangible assets, comprised of IPR&D, are reviewed for impairment annually and whenever events or changes in circumstances indicate that it is more likely than not that the asset is impaired by comparing the fair value to the carrying value of the asset.
We recognized impairment charges associated with early stage indefinite-lived intangible assets acquired in connection with the purchase of Taligen and Orphatec of $11,514 in 2014. We recognized impairment charges associated with an early stage indefinite-lived intangible asset and a purchased technology asset acquired in connection with the purchase of Taligen of $33,521 in 2013, and we recognized impairment charges associated with an early stage indefinite-lived intangible asset acquired in connection with the purchase of Taligen of $26,300 in 2012. We did not recognize any other impairment loss for long-lived assets during the years ended December 31, 2014, 2013 and 2012.
Contingent Consideration
We record contingent consideration resulting from a business combination at its fair value on the acquisition date. On a quarterly basis, we revalue these obligations and record increases or decreases in their fair value as an adjustment to operating earnings. Changes to contingent consideration obligations can result from adjustments to discount rates, accretion of the liability due to the passage of time, changes in our estimates of the likelihood or timing of achieving development or commercial milestones, changes in the probability of certain clinical events or changes in the assumed probability associated with regulatory approval.
Contingent Liabilities
We are currently involved in various claims and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims, we may reassess the potential liability related to these matters and may revise these estimates.
Treasury Stock
Treasury stock is accounted for using the cost method, with the purchase price of the common stock recorded separately as a deduction from stockholders' equity.
Revenue Recognition
Our principal source of revenue is product sales. We recognize revenue from product sales when persuasive evidence of an arrangement exists, title to product and associated risk of loss has passed to the customer, the price is fixed or determinable, collection from the customer is reasonably assured, and we have no further performance obligations. Depending on these criteria, revenue is usually recorded upon receipt of the product by the end customer, which is typically a hospital, physician’s office, private or government pharmacy or other health care facility. On a regular basis, we review revenue arrangements, such as distributor relationships, to determine whether changes in these criteria have an impact on revenue recognition. Amounts collected from customers and remitted to governmental authorities, such as value-added taxes (VAT) in foreign jurisdictions, are presented on a net basis in our consolidated statements of operations and do not impact net product sales.
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other health care providers. In some cases, we may also sell Soliris to governments and government agencies.
Because of factors such as the pricing of Soliris, the limited number of patients, the short period from product sale to patient infusion and the lack of contractual return rights, Soliris customers often carry limited inventory. We also monitor inventory within our sales channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms and financial strength of distributors. In some cases, exact quantities of inventory in the channel are not precisely known, requiring us to estimate these amounts. If actual amounts of inventory differ from these estimates, these adjustments could have an impact in the period in which these estimates change.
In addition to sales in countries where Soliris is commercially available, we have also recorded revenue on sales for patients receiving Soliris treatment through named-patient programs. The relevant authorities or institutions in those countries
F-13
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
have agreed to reimburse for product sold on a named-patient basis where Soliris has not received final approval for commercial sale.
We record estimated rebates payable under governmental programs, including Medicaid in the United States and other programs outside the United States, as a reduction of revenue at the time of product sale. Our calculations related to these rebate accruals require analysis of historical claim patterns and estimates of customer mix to determine which sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions each period and record any necessary adjustments, which may have an impact on revenue in the period in which the adjustment is made. Generally, the length of time between product sale and the processing and reporting of the rebates is three to six months.
We have entered into volume-based arrangements with governments in certain countries in which reimbursement is limited to a contractual amount. Under this type of arrangement, amounts billed in excess of the contractual limitation are repaid to these governments as a rebate. We estimate incremental discounts resulting from these contractual limitations, based on estimated sales during the limitation period, and we apply the discount percentage to product shipments as a reduction of revenue. Our calculations related to these arrangements require estimation of sales during the limitation period, and adjustments in these estimates may have a material impact in the period in which these estimates change.
We record distribution and other fees paid to our customers as a reduction of revenue, unless we receive an identifiable and separate benefit for the consideration and we can reasonably estimate the fair value of the benefit received. If both conditions are met, we record the consideration paid to the customer as an operating expense. These costs are typically known at the time of sale, resulting in minimal adjustments subsequent to the period of sale.
We enter into foreign exchange forward contracts to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. These hedges are designated as cash flow hedges upon inception. We record the effective portion of these cash flow hedges to revenue in the period in which the sale is made to an unrelated third party and the derivative contract is settled.
Research and Development Expenses
Research and development expenses are comprised of costs incurred in performing research and development activities including payroll and benefits, pre-clinical, clinical trial and related clinical manufacturing costs, manufacturing development and scale-up costs, product development and regulatory costs, contract services and other outside contractor costs, research license fees, depreciation and amortization of lab facilities, and lab supplies. These costs are expensed as incurred. We accrue costs for clinical trial activities based upon estimates of the services received and related expenses incurred that have yet to be invoiced by the contract research organizations, clinical study sites, laboratories, consultants, or other clinical trial vendors that perform the activities.
Share-Based Compensation
We have one share-based compensation plan known as the Amended and Restated 2004 Incentive Plan. Under this plan, restricted stock, restricted stock units, stock options and other stock-related awards may be granted to our directors, officers, employees and consultants or advisors of the Company or any subsidiary. To date, share-based compensation issued under the plan consists of incentive and non-qualified stock options, restricted stock and restricted stock units, including restricted stock units with market and non-market performance conditions.
Compensation expense for our share-based awards is recognized based on the estimated fair value of the awards on the grant date. Compensation expense reflects an estimate of the number of awards expected to vest and is primarily recognized on a straight-line basis over the requisite service period of the individual grants, which typically equals the vesting period. Compensation expense for awards with performance conditions is recognized using the graded-vesting method.
Our estimates of employee stock option values rely on estimates of factors we input into the Black-Scholes model. The key factors involve an estimate of future uncertain events. Significant assumptions include the use of historical volatility to determine the expected stock price volatility. We also estimate expected term until exercise and the reduction in the expense from expected forfeitures. We currently use historical exercise and cancellation patterns as our best estimate of future estimated life.
For our non-market performance-based awards, we estimate the anticipated achievement of the performance targets, including forecasting the achievement of future financial targets. These estimates are revised periodically based on the probability of achieving the performance targets and adjustments are made throughout the performance period as necessary. We use payout simulation models to estimate the grant date fair value of market performance-based awards. The payout
F-14
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
simulation models assume volatility of our common stock and the common stock of a comparator group of companies, as well as correlations of returns of the price of our common stock and the common stock prices of the comparator group.
Earnings Per Common Share
Basic earnings per common share (EPS) are computed by dividing net income by the weighted-average number of shares of common stock outstanding. For purposes of calculating diluted EPS, the denominator reflects the potential dilution that could occur if stock options, unvested restricted stock, unvested restricted stock units or other contracts to issue common stock were exercised or converted into common stock, using the treasury stock method.
The following table summarizes the calculation of basic and diluted EPS for years ended December 31, 2014, 2013 and 2012:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Net income used for basic and diluted calculation | $ | 656,912 | $ | 252,895 | $ | 254,822 | |||||
Shares used in computing earnings per common share—basic | 198,103 | 195,532 | 190,461 | ||||||||
Weighted-average effect of dilutive securities: | |||||||||||
Shares issuable upon the assumed conversion of our 1.375% convertible senior notes | — | — | 8 | ||||||||
Stock awards | 3,520 | 4,180 | 8,032 | ||||||||
Dilutive potential common shares | 3,520 | 4,180 | 8,040 | ||||||||
Shares used in computing earnings per common share—diluted | 201,623 | 199,712 | 198,501 | ||||||||
Earnings per common share: | |||||||||||
Basic | $ | 3.32 | $ | 1.29 | $ | 1.34 | |||||
Diluted | $ | 3.26 | $ | 1.27 | $ | 1.28 | |||||
We exclude from EPS the weighted-average number of securities whose effect is anti-dilutive. Excluded from the calculation of EPS for the years ended December 31, 2014, 2013 and 2012 were 1,099, 2,243, and 1,899 shares of common stock, respectively, because their effect is anti-dilutive.
Income Taxes
We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax basis of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance when it is more likely than not that deferred tax assets will not be realized.
We recognize the benefit of an uncertain tax position that has been taken or we expect to take on income tax returns if such tax position is more likely than not to be sustained. The tax benefit recognized in the financial statements for a particular tax position is based on the largest benefit that is more likely than not to be realized. The amount of unrecognized tax benefits is adjusted, as appropriate, for changes in facts and circumstances, such as significant amendments to existing tax law, new regulations or interpretations by the taxing authorities, or new information obtained during a tax examination or resolution of an examination. We also accrued for potential interest and penalties related to unrecognized tax benefits as a component of tax expense.
Comprehensive Income
Comprehensive income is comprised of net income and other comprehensive income (loss). Other comprehensive income (loss) includes changes in equity that are excluded from net income, such as changes in pension liabilities, unrealized gains and losses on marketable securities, unrealized gains and losses on hedge contracts and foreign currency translation adjustments. Certain of these changes in equity are reflected net of tax.
F-15
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Other Investments
We invest in companies with securities that are not publicly traded and where fair value is not readily available. Other investments include an investment in the preferred stock of the non-public entity Moderna LLC. During 2014, we purchased $37,500 of preferred equity of Moderna LLC. We recorded our investment at cost within other assets in our condensed consolidated balance sheets. We regularly monitor these investments to evaluate whether there has been an other-than-temporary decline in its fair value, based on the implied value of recent company financings, public market prices of comparable companies, and general market conditions. The carrying value of these investments was not impaired as of December 31, 2014.
New Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. We are currently assessing the method of adoption and the expected impact the new standard has on our financial position and results of operations.
2. | Acquisitions |
Acquisition of Enobia Pharma Corp.
In February 2012, we acquired Enobia Pharma Corp. (Enobia), a privately held clinical-stage biotechnology company based in Montreal, Canada and Cambridge, Massachusetts, in a transaction accounted for under the acquisition method of accounting for business combinations. Under the acquisition method of accounting, the assets acquired and liabilities assumed of Enobia were recorded as of the acquisition date at their respective fair values. The reported consolidated financial condition after completion of the acquisition reflects these fair values. Enobia's results of operations are included in the consolidated financial statements from the date of acquisition. The acquisition was intended to further our objective to develop and commercialize therapies for patients with severe, ultra-rare and life-threatening disorders. Enobia's lead product candidate, asfotase alfa, is a human recombinant targeted alkaline phosphatase enzyme-replacement therapy for patients suffering with hypophosphatasia (HPP), an ultra-rare, life-threatening, genetic metabolic disease for which there are no approved treatments.
We made an upfront cash payment of $623,876 for 100% of Enobia's capital stock. Additional contingent payments of up to an aggregate of $470,000 would be due upon reaching various regulatory and sales milestones. We financed the acquisition with existing cash and proceeds from our new credit facility.
A reconciliation of upfront payments in accordance with the purchase agreement to the total purchase price is presented below:
Enobia | |||
Base payment per agreement | $ | 610,000 | |
Cash acquired | 18,141 | ||
Working capital adjustment | (4,265 | ) | |
Upfront payment in accordance with agreement | 623,876 | ||
Estimated fair value of contingent consideration | 117,000 | ||
Total purchase price | $ | 740,876 | |
The initial estimate of fair value of contingent consideration was $117,000, which was recorded as a noncurrent liability. We determined the fair value of these obligations to pay additional milestone payments using various estimates, including probabilities of success, discount rates and amount of time until the conditions of the milestone payments are met. This fair value measurement is based on significant inputs not observable in the market, representing a Level 3 measurement within the fair value hierarchy. The resulting probability-weighted cash flows were discounted using a cost of debt rate of 5.2% for developmental milestones and a weighted average cost of capital rate of 13.0% for commercial milestones. These rates were
F-16
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
representative of market participant assumptions. The range of estimated milestone payments is from zero if no clinical milestones are achieved for any product to $470,000 if various regulatory and sales milestones are achieved.
Subsequent to the acquisition date, we have adjusted the contingent consideration to fair value with changes in fair value recognized in operating earnings. Changes in fair values reflect new information about the probability and timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of contingent consideration related to the passage of time as development work progresses towards the achievement of the milestones. At December 31, 2014, the fair value of the contingent consideration for Enobia was $155,299. Changes in fair value of the consideration for Enobia were $22,286 and $8,602 for the years ended December 31, 2014 and 2013, respectively.
The following table summarizes the estimated fair values of assets acquired and liabilities assumed:
Enobia | |||
Cash and cash equivalents | $ | 18,141 | |
Current assets | 5,536 | ||
In-process research and development | 587,000 | ||
Other noncurrent assets | 910 | ||
Assets acquired | 611,587 | ||
Deferred tax liability | (31,471 | ) | |
Other liabilities assumed | (13,674 | ) | |
Liabilities assumed | (45,145 | ) | |
Goodwill | 174,434 | ||
Net assets acquired | $ | 740,876 | |
Asset categories acquired in the Enobia acquisition included working capital, fixed assets, deferred tax assets and IPR&D. The fair value of working capital was determined to approximate book values.
Intangible assets associated with IPR&D projects relate to Enobia's lead product candidate, asfotase alfa. The estimated fair value of $587,000 was determined using the multi-period excess earnings method, a variation of the income approach. The multi-period excess earnings method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset. The fair value using the multi-period excess earnings method was dependent on an estimated weighted average cost of capital for Enobia of 13.0%, which represents a rate of return that a market participant would expect for these assets.
The excess of purchase price over the fair value amounts of the assets acquired and liabilities assumed represents the goodwill amount resulting from the acquisition. We do not expect any portion of this goodwill to be deductible for tax purposes. The goodwill attributable to our acquisition of Enobia has been recorded as a noncurrent asset and is not amortized, but is subject to an annual review for impairment. The factors that contributed to the recognition of goodwill included the synergies that are specific to our business and not available to market participants, including our unique ability to commercialize therapies for rare diseases, our skills and relationships related to biologics manufacturing, our existing relationships with specialty physicians who can identify patients with HPP and a global distribution network to facilitate immediate drug delivery.
We recorded a net deferred tax liability of $31,471. This amount was primarily comprised of $78,527 related to IPR&D, offset by acquired net operating losses and research credit carryovers totaling $47,056.
For the year ended December 31, 2012, we recorded $6,794 of expenses associated with the operations of Enobia from February 7, 2012 through March 31, 2012 in our condensed consolidated statement of comprehensive income. Effective April 1, 2012, the operations of Enobia were integrated into our operations.
Pro forma financial information (unaudited)
The following unaudited pro forma information presents the combined results of operations for the years ended December 31, 2012, as if the acquisition of Enobia had been completed on January 1, 2012. The pro forma results do not reflect operating efficiencies or potential cost savings which may result from the consolidation of operations. The pro forma results have been adjusted to remove costs associated with changes in the fair value of Enobia's preferred stock. Included in the pro forma net income for the year ended December 31, 2012, are approximately $23,673 and $7,900 of Alexion and Enobia acquisition-related costs, respectively, which are not expected to have an ongoing impact.
F-17
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Year Ended December 31, | |||
2012 | |||
Revenues | $ | 1,134,114 | |
Net income | 236,407 | ||
Earnings per common share | |||
Basic | $ | 1.24 | |
Diluted | $ | 1.19 | |
Other Acquisitions
Orphatec Pharmaceuticals GmbH
In February 2011, we acquired certain patents and assets from Orphatec Pharmaceuticals GmbH (Orphatec) related to an investigational therapy for patients with molybdenum cofactor deficiency (MoCD) Type A, an ultra-rare genetic disorder characterized by severe brain damage and rapid death in newborns. We made initial payments of $3,050 in cash and may make additional future payments of up to $42,000 in contingent milestone payments upon various development, regulatory and commercial milestones. The range of estimated milestone payments is from zero if no products gain market approval to $42,000 if all indications for up to two products gain both U.S. and European marketing approval and reach applicable sales levels.
The initial estimate of fair value of contingent consideration was $5,086. Subsequent to the acquisition date, we have measured the contingent consideration arrangement at fair value with changes in fair value recognized in operating earnings. As of December 31, 2014 we made a milestone payment of $3,000 related to this acquisition. At December 31, 2014, the fair value of the contingent consideration for Orphatec was $5,936. Changes in fair value of the consideration for Orphatec were $232, $1,181 and $2,087 for the years ended December 31, 2014, 2013 and 2012, respectively.
Taligen Therapeutics, Inc.
In January 2011, we acquired all of the outstanding capital stock of Taligen Therapeutics, Inc. (Taligen) in a transaction accounted for under the acquisition method of accounting for business combinations. We made initial payments of $111,773 in cash and may make additional future payments of up to $367,000 in contingent milestone payments upon achievement of various development and commercial milestones. The range of estimated milestone payments is from zero if no clinical milestones are achieved for any product to $367,000 if six products gain both U.S. and European marketing approval.
The initial estimate of fair value of contingent consideration was $11,634. Subsequent to the acquisition date, we have adjusted the contingent consideration to fair value with changes in fair value recognized in operating earnings. At December 31, 2014, the fair value of the contingent consideration for Taligen was $1,736. Changes in fair value of the consideration for Taligen were $(2,223), $(5,777) and $(2,948) for the years ended December 31, 2014, 2013 and 2012, respectively. Included in the change in fair value for the years ended December 31, 2014 and 2013 is a benefit of $2,458 and $5,973, respectively, related to the decrease in the fair value of the contingent consideration related to this acquisition. The decrease in fair value was a result of a decreased likelihood of payments for contingent consideration due to a reassessment of scientific findings.
Acquisition-Related Costs
Acquisition-related costs associated with our business combinations for the years ended December 31, 2014, 2013 and 2012 include the following:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Separately-identifiable employee costs | $ | — | $ | 248 | $ | 3,669 | |||||
Professional fees | — | 775 | 12,593 | ||||||||
Changes in fair value of contingent consideration | 20,295 | 4,006 | 6,550 | ||||||||
$ | 20,295 | $ | 5,029 | $ | 22,812 | ||||||
During the years ended December 31, 2014, 2013 and 2012, we incurred costs of approximately $22,286, $9,625 and $23,673, respectively, related to the Enobia acquisition, which are included in the amounts above.
F-18
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
3. | Property, Plant and Equipment, Net |
A summary of property, plant and equipment is as follows:
December 31, 2014 | December 31, 2013 | ||||||
Land | $ | 9,130 | $ | 692 | |||
Buildings and improvements | 170,355 | 154,996 | |||||
Machinery and laboratory equipment | 65,079 | 56,130 | |||||
Computer hardware and software | 59,927 | 41,704 | |||||
Furniture and office equipment | 11,371 | 10,119 | |||||
Construction-in-progress | 214,041 | 41,573 | |||||
529,903 | 305,214 | ||||||
Less: Accumulated depreciation and amortization | (137,655 | ) | (104,105 | ) | |||
$ | 392,248 | $ | 201,109 | ||||
Included in construction-in-progress at December 31, 2014 and 2013 was $126,566 and $32,230, respectively of costs associated with the construction of a new facility in New Haven, Connecticut. In November 2012, we entered into a lease agreement for office and laboratory space to be constructed in New Haven. Although we will not legally own the premises, we are deemed to be the owner of the building during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord's costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. Construction of the new facility began in June 2013 and is expected to be completed in 2015.
Depreciation and amortization of property, plant and equipment was approximately $34,901, $19,084 and $15,192 for the years ended December 31, 2014, 2013 and 2012, respectively.
At December 31, 2014 and 2013, computer software costs included in property, plant and equipment were $16,292 and $9,691, respectively. Depreciation and amortization expense for capitalized computer software costs was $7,016, $4,503 and $4,228 for the years ended December 31, 2014, 2013 and 2012, respectively.
4. | Intangible Assets and Goodwill |
Intangible assets and goodwill, net of accumulated amortization, are as follows:
December 31, 2014 | December 31, 2013 | ||||||||||||||||||||||||
Estimated Life (months) | Cost | Accumulated Amortization | Net | Cost | Accumulated Amortization | Net | |||||||||||||||||||
Licenses | 72-96 | $ | 28,507 | $ | (28,461 | ) | $ | 46 | $ | 28,507 | $ | (18,719 | ) | $ | 9,788 | ||||||||||
Patents | 84 | 10,517 | (10,517 | ) | — | 10,517 | (9,100 | ) | 1,417 | ||||||||||||||||
Purchased technology | 144 | — | — | — | — | — | — | ||||||||||||||||||
Acquired IPR&D | Indefinite | 587,000 | — | 587,000 | 598,514 | — | 598,514 | ||||||||||||||||||
Total | $ | 626,024 | $ | (38,978 | ) | $ | 587,046 | $ | 637,538 | $ | (27,819 | ) | $ | 609,719 | |||||||||||
Goodwill | Indefinite | $ | 256,974 | $ | (2,901 | ) | $ | 254,073 | $ | 256,974 | $ | (2,901 | ) | $ | 254,073 | ||||||||||
Amortization of our intangible assets was approximately $11,159, $8,257 and $5,660 for the years ended December 31, 2014, 2013 and 2012, respectively. Assuming no changes in the gross cost basis of intangible assets, amortization of $46 will occur in 2015.
As of December 31, 2014, we have recorded indefinite-lived intangible assets of $587,000 of purchased IPR&D from prior business acquisitions. As of December 31, 2014, except as noted below, there have been no significant changes that would impact the carrying value of IPR&D since the date of acquisition.
During the fourth quarter 2014, we reviewed for impairment the value of the early stage, Phase II indefinite-lived intangible asset related to the Orphatec acquisition. We initiated such review as part of our annual impairment testing and increased costs associated with clinical trial studies. The estimated value that can be obtained from a market participant in an arm's length transaction was determined to be de minimis as of December 31, 2014. As a result, in the fourth quarter 2014, we
F-19
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
recognized an impairment charge of $8,050 to write-down these assets to fair value. In addition, during the first quarter of 2014, we reviewed for impairment the value of an early stage, Phase I indefinite-lived intangible asset related to our acquisition of Taligen Therapeutics, Inc. We initiated such review based on a reassessment of scientific findings associated with this acquired asset. As a result, we recognized an impairment of $3,464 for the year ended December 31, 2014 to adjust this asset to fair value, which was determined to be de minimis.
During 2013, we reviewed for impairment the value of an early stage, Phase I indefinite-lived intangible asset related to the Taligen acquisition. We initiated such review as part of our annual impairment testing and based our evaluation on preliminary scientific findings of a Phase I clinical trial which led us to reassess the development of this acquired asset. The fair value of this IPR&D asset was determined using the income approach, which used significant unobservable (Level 3) inputs. These unobservable inputs included, among other things, risk-adjusted forecast future cash flows to be generated by this asset, contributory asset charges for other assets employed in this IPR&D project and the determination of an appropriate discount rate based on a weighted average cost of capital of 21.5% to be applied in calculating the present value of future cash flows. Based on these factors, the estimated value that can be obtained from a market participant in an arm's length transaction of $3,464 was lower than the carrying amount. We also reviewed for impairment the value of purchased technology associated with the Taligen acquisition and determined the estimated value to be de minimis. As a result, we recognized an impairment charge of $33,521 to write-down these assets to fair value, which was recorded in operating expenses in our consolidated statement of operations for the year ended December 31, 2013.
During 2012, we reviewed for impairment the value of an early stage, preclinical indefinite-lived intangible asset related to the Taligen acquisition. We initiated such review based on our evaluation of negative scientific findings associated with our development of a different asset for the treatment of age-related macular degeneration, the likelihood of success for ophthalmic use and the value that can be obtained from a market participant in an arm's length transaction. These developments led us to deprioritize the development of this acquired asset. As a result, we recognized an impairment charge of $26,300 to write-down this asset to fair value, which was determined to be de minimis based on the value of the asset to a market participant in an arm's length transaction. The fair value of this IPR&D asset was determined using the income approach, which used significant unobservable (Level 3) inputs. These unobservable inputs included, among other things, risk-adjusted forecast future cash flows to be generated by this asset, contributory asset charges for other assets employed in this IPR&D project and the determination of an appropriate discount rate based on a weighted average cost of capital of 22.0% to be applied in calculating the present value of future cash flows. The impairment charge was recorded in operating expenses in our consolidated statement of operations for the year ended December 31, 2012.
The following table summarizes the changes in the carrying amount of goodwill:
Balance at December 31, 2012 | $ | 253,645 | |
Change in goodwill associated with prior acquisition | 428 | ||
Balance at December 31, 2013 and 2014 | $ | 254,073 | |
F-20
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
5. | Marketable Securities |
The amortized cost, gross unrealized holding gains, gross unrealized holding losses and estimated fair value of available-for-sale investments by type of security at December 31, 2014 and December 31, 2013 were as follows:
December 31, 2014 | ||||||||||||||||
Amortized Cost | Gross Unrealized Holding Gains | Gross Unrealized Holding Losses | Estimated Fair Value | |||||||||||||
Commercial paper | $ | 142,495 | $ | — | $ | — | $ | 142,495 | ||||||||
Corporate bonds | 494,032 | 415 | (581 | ) | 493,866 | |||||||||||
Municipal bonds | 174,759 | 132 | (46 | ) | 174,845 | |||||||||||
Other government related obligations: | ||||||||||||||||
U.S. | 99,668 | 14 | (71 | ) | 99,611 | |||||||||||
Foreign | 193,439 | 100 | (174 | ) | 193,365 | |||||||||||
Bank certificates of deposit | 77,000 | — | — | 77,000 | ||||||||||||
$ | 1,181,393 | $ | 661 | $ | (872 | ) | $ | 1,181,182 | ||||||||
December 31, 2013 | ||||||||||||||||
Amortized Cost Basis | Gross Unrealized Holding Gains | Gross Unrealized Holding Losses | Aggregate Fair Value | |||||||||||||
Commercial paper | $ | 112,679 | $ | — | $ | — | $ | 112,679 | ||||||||
Corporate bonds | 476,459 | 487 | (588 | ) | 476,358 | |||||||||||
Municipal bonds | 202,396 | 47 | (40 | ) | 202,403 | |||||||||||
Other government related obligations: | ||||||||||||||||
U.S. | 46,466 | 30 | (7 | ) | 46,489 | |||||||||||
Foreign | 156,974 | 54 | (204 | ) | 156,824 | |||||||||||
Bank certificates of deposit | 33,004 | — | — | 33,004 | ||||||||||||
$ | 1,027,978 | $ | 618 | $ | (839 | ) | $ | 1,027,757 | ||||||||
The aggregate fair value of available-for-sale securities in an unrealized loss position as of December 31, 2014 and December 31, 2013 was $472,241 and $461,634. Investments that have been in a continuous unrealized loss position for more than 12 months were not material. As of December 31, 2014 we believe that the cost basis of our available-for-sale investments is recoverable.
The fair values of available-for-sale securities by classification in the consolidated balance sheet were as follows:
December 31, 2014 | December 31, 2013 | ||||||
Cash and cash equivalents | $ | 167,892 | $ | 43,780 | |||
Marketable securities | 1,013,290 | 983,977 | |||||
$ | 1,181,182 | $ | 1,027,757 | ||||
The fair values of available-for-sale debt securities at December 31, 2014, by contractual maturity, are summarized as follows:
December 31, 2014 | |||
Due in one year or less | $ | 641,052 | |
Due after one year through three years | 540,130 | ||
Due after three years through five years | — | ||
$ | 1,181,182 | ||
As of December 31, 2014 and December 31, 2013, the fair value of our trading securities was $4,277 and $1,017.
F-21
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
We utilize the specific identification method in computing realized gains and losses. Realized gains and losses on our available-for-sale and trading securities were not material for the year ended December 31, 2014 and 2013.
6. | Derivative Instruments and Hedging Activities |
We operate internationally and, in the normal course of business, are exposed to fluctuations in foreign currency exchange rates. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, primarily the Euro and Japanese Yen. We manage our foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes.
We enter into foreign exchange forward contracts, with durations of up to 60 months, to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. The purpose of the hedges of revenue is to reduce the volatility of exchange rate fluctuations on our operating results and to increase the visibility of the foreign exchange impact on forecasted revenues. These hedges are designated as cash flow hedges upon contract inception. At December 31, 2014, we had open contracts with notional amounts totaling $1,524,521 that qualified for hedge accounting.
The impact on accumulated other comprehensive income (AOCI) and earnings from foreign exchange contracts that qualified as cash flow hedges, for the years ended December 31, 2014 and 2013 were as follows:
Year Ended December 31, | |||||||
2014 | 2013 | ||||||
Gain (loss) recognized in AOCI, net of tax | $ | 110,088 | $ | (1,000 | ) | ||
Gain reclassified from AOCI to net product sales (effective portion), net of tax | $ | 16,514 | $ | 18,820 | |||
Gain (loss) reclassified from AOCI to other income and expense (ineffective portion), net of tax | $ | 2,439 | $ | (837 | ) | ||
Assuming no change in foreign exchange rates from market rates at December 31, 2014, $74,142 of gains recognized in AOCI will be reclassified to revenue over the next 12 months.
We enter into foreign exchange forward contracts, with durations of approximately 30 days, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Hedge accounting is not applied to these derivative instruments as gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. As of December 31, 2014, the notional amount of foreign exchange contracts where hedge accounting is not applied was $224,410.
We recognized a gain of $26,295, $8,306 and $3,518, in other income and expense, for the years ended December 31, 2014, 2013 and 2012, respectively, associated with the foreign exchange contracts not designated as hedging instruments. These amounts were largely offset by gains or losses in monetary assets and liabilities.
The following tables summarize the fair value of outstanding derivatives at December 31, 2014 and 2013:
December 31, 2014 | |||||||||||
Asset Derivatives | Liability Derivatives | ||||||||||
Balance Sheet Location | Fair Value | Balance Sheet Location | Fair Value | ||||||||
Derivatives designated as hedging instruments: | |||||||||||
Foreign exchange forward contracts | Other current assets | $ | 77,348 | Other current liabilities | $ | 794 | |||||
Foreign exchange forward contracts | Other non-current assets | 58,698 | Other non-current liabilities | 86 | |||||||
Total fair value of derivative instruments | $ | 136,046 | $ | 880 | |||||||
F-22
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
December 31, 2013 | |||||||||||
Asset Derivatives | Liability Derivatives | ||||||||||
Balance Sheet Location | Fair Value | Balance Sheet Location | Fair Value | ||||||||
Derivatives designated as hedging instruments: | |||||||||||
Foreign exchange forward contracts | Other current assets | $ | 21,815 | Other current liabilities | $ | 20,228 | |||||
Foreign exchange forward contracts | Other non-current assets | 9,839 | Other non-current liabilities | 14,864 | |||||||
Total fair value of derivative instruments | $ | 31,654 | $ | 35,092 | |||||||
The fair value of our foreign exchange forward contracts that are not designated as hedging instruments was zero as of December 31, 2014 and December 31, 2013.
Although we do not offset derivative assets and liabilities within our condensed consolidated balance sheets, our International Swap and Derivatives Association (ISDA) agreements provide for net settlement of transactions that are due to or from the same counterparty upon early termination of the agreement due to an event of default or other termination event. The following tables summarize the potential effect on our condensed consolidated balance sheets of offsetting our foreign exchange forward contracts subject to such provisions:
December 31, 2014 | ||||||||||||||||||||||||
Gross Amounts Not Offset in the Consolidated Balance Sheet | ||||||||||||||||||||||||
Description | Gross Amounts of Recognized Assets/Liabilities | Gross Amounts Offset in the Consolidated Balance Sheet | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | Derivative Financial Instruments | Cash Collateral Received (Pledged) | Net Amount | ||||||||||||||||||
Derivative assets | $ | 136,046 | $ | — | $ | 136,046 | $ | (880 | ) | $ | — | $ | 135,166 | |||||||||||
Derivative liabilities | (880 | ) | — | (880 | ) | 880 | — | — | ||||||||||||||||
December 31, 2013 | ||||||||||||||||||||||||
Gross Amounts Not Offset in the Consolidated Balance Sheet | ||||||||||||||||||||||||
Description | Gross Amounts of Recognized Assets/Liabilities | Gross Amounts Offset in the Consolidated Balance Sheet | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | Derivative Financial Instruments | Cash Collateral Received (Pledged) | Net Amount | ||||||||||||||||||
Derivative assets | $ | 31,654 | $ | — | $ | 31,654 | $ | (27,256 | ) | $ | — | $ | 4,398 | |||||||||||
Derivative liabilities | (35,092 | ) | — | (35,092 | ) | 27,256 | — | (7,836 | ) | |||||||||||||||
F-23
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
7. | Accrued Expenses |
Accrued expenses consist of the following:
December 31, 2014 | December 31, 2013 | ||||||
Royalties | $ | 25,863 | $ | 40,945 | |||
Payroll and employee benefits | 88,467 | 64,950 | |||||
Taxes payable | 94,823 | 108,907 | |||||
Rebates payable | 36,827 | 124,297 | |||||
Clinical | 30,123 | 17,818 | |||||
Manufacturing | 42,631 | 7,250 | |||||
Other | 76,498 | 38,177 | |||||
$ | 395,232 | $ | 402,344 | ||||
8. | Debt |
In February 2012, we entered into a credit agreement, as amended (the Credit Agreement) with a syndicate of banks, that provides for a $240,000 senior secured term loan facility payable in equal quarterly installments of $12,000 starting June 30, 2012 and a $200,000 senior secured revolving credit facility through February 7, 2017. In addition to borrowings upon prior notice, the revolving credit facility includes borrowing capacity in the form of letters of credit up to $60,000 and borrowings on same-day notice, referred to as swingline loans, of up to $10,000. Borrowings can be used for working capital requirements, acquisitions and other general corporate purposes. With the consent of the lenders and the administrative agent and subject to satisfaction of certain conditions, we may increase the term loan facility and/or the revolving credit facility by an aggregate amount not to exceed $150,000.
We may elect that the loans under the Credit Agreement bear interest at a rate per annum equal to (i) LIBOR plus 1.25% to 2.00% depending on our consolidated leverage ratio (as calculated in accordance with the Credit Agreement), or (ii) in the case of loans denominated in U.S. dollars, a Base Rate equal to the higher of the (A) Prime Rate then in effect, (B) Federal Funds Rate then in effect plus 0.50%, and (C) Eurodollar Rate then in effect plus 1.00%, plus in each case of (A), (B) or (C), 0.25% to 1.00% depending on our consolidated leverage ratio (as calculated in accordance with the Credit Agreement). Interest is payable quarterly for Base Rate loans and, in the case of LIBOR-based loans, at the end of the applicable interest period, with the principal due on February 7, 2017, the maturity date. At December 31, 2014 and 2013, the interest rate on our outstanding loans under the Credit Agreement was 1.41%.
Our obligations under the credit facilities are unconditionally guaranteed, jointly and severally, by certain of our existing domestic subsidiaries and are required to be guaranteed by certain of our future domestic subsidiaries. The obligations of our non-U.S. subsidiaries under the credit facilities are unconditionally guaranteed, jointly and severally, by us, certain of our existing domestic subsidiaries, and certain of our foreign subsidiaries, and are required to be guaranteed by certain of our future subsidiaries. All obligations of each borrower under the credit facilities, and the guarantees of those obligations, are backed, subject to certain exceptions, by substantially all of each borrower's assets and the assets of certain guarantors, including the pledge of the equity interests of certain of our subsidiaries and real estate located in Smithfield, Rhode Island, but excluding intellectual property and assets of certain foreign subsidiaries.
The Credit Agreement requires us to comply with certain financial covenants on a quarterly basis. Further, the Credit Agreement includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, engage in certain investment, acquisition and disposition transactions, pay dividends, repurchase capital stock and enter into transactions with affiliates. The Credit Agreement also contains customary representations and warranties, affirmative covenants and events of default, including payment defaults, breach of representations and warranties, covenant defaults and cross defaults. If an event of default occurs, the interest rate would increase and the administrative agent would be entitled to take various actions, including the acceleration of amounts due under the loan.
For the years ended December 31, 2014, 2013 and 2012 we made payments of $55,500, $36,000, and $91,000, against the term loan, respectively. As of December 31, 2014, we had $57,500 outstanding on the term loan. As of December 31, 2014, we had open letters of credit of $10,284, and our borrowing availability under the revolving facility was $189,716.
The fair value of our long term debt, which is measured using Level 2 inputs, approximates book value.
F-24
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The contractual maturities of our long-term debt obligations due subsequent to December 31, 2014 are $10,000 in 2015, $38,000 in 2016, and $9,500 in 2017. As of December 31, 2014, we recorded $48,000 due under our term loan in current liabilities based on our intent and ability to make payments in this amount during 2015. In January 2015, we made a payment of $12,000.
9. | Commitments and Contingencies |
Commitments
License Agreements
In December 2014, we entered into an agreement with X-Chem Pharmaceuticals (X-Chem) that allows us to identify novel drug candidates from X-Chem's proprietary drug discovery engine. Alexion will have the exclusive worldwide rights to develop and commercialize products arising from the collaboration in up to three program targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $8,000. In addition, for each program target, for a maximum of three targets, we could be required to make additional payments upon the achievement of specified research, development and regulatory milestones up to $75,000, as well as royalties on commercial sales.
In January 2014, we entered into an agreement with Moderna Therapeutics, Inc. (Moderna) that allows us to purchase ten product options to develop and commercialize treatments for rare diseases with Moderna's messenger RNA (mRNA) therapeutics platform. Alexion will lead the discovery, development and commercialization of the treatments produced through this broad, long-term strategic agreement, while Moderna will retain responsibility for the design and manufacture of the messenger RNA against selected targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $100,000. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of ten targets, we could be required to make an option exercise payment of $15,000 and to pay up to an additional $120,000 with respect to a rare disease product and $400,000 with respect to a non-rare disease product in development and sales milestones if the specific milestones are met over time as well as royalties on commercial sales.
In July 2013, we entered into a license and collaboration agreement with Ensemble Therapeutics Corporation for the identification, development and commercialization of therapeutic candidates based on specific drug targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $11,500 during the third quarter of 2013. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of four targets, we could be required to pay up to an additional $90,750 in development milestones as the specific milestones are met over time. The agreement also provides for royalty payments on commercial sales of each product developed under the agreement.
In January 2013, we entered into a license agreement for a technology, which provides an exclusive research license and an option for an exclusive commercial license for specific targets and products to be developed. Due to the early stage of this asset, we recorded expense for an upfront payment of $3,000 during the first quarter of 2013. We will also be required to pay annual maintenance fees during the term of the arrangement. In addition, for each target up to a maximum of six targets we develop, we could be required to pay up to an additional $70,500 in license fees, development and sales milestones as the specific milestones are met over time.
Lonza Agreement
We rely on Lonza Group AG and its affiliates (Lonza), a third party manufacturer, to produce a portion of commercial and clinical quantities of Soliris and for clinical quantities of asfotase alfa, and we have contracted and expect to continue contracting for product finishing, filling and packaging through third parties. We have various agreements with Lonza, with remaining total non-cancellable future commitments of approximately $383,500. Our agreements with Lonza also include potential payments totaling up to $5,000 that will become payable if and when certain milestones are achieved. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of Soliris manufactured at Alexion Rhode Island Manufacturing Facility (ARIMF) and a payment with respect to sales of Soliris manufactured at Lonza facilities.
Contingent Liabilities
We are currently involved in various claims, lawsuits and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available
F-25
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims, we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adverse adjustment to our operating results.
We have in the past received, and may in the future receive, notices from third parties claiming that their patents may be infringed by the development, manufacture or sale of Soliris. Under the guidance of ASC 450, Contingencies, we record a royalty accrual based on our best estimate of the fair value percent of net sales of Soliris that we could be required to pay the owners of patents for technology used in the manufacture and sale of Soliris. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our financial results.
In January 2011, Novartis Vaccines and Diagnostics, Inc. (Novartis) filed an action against us and other biopharmaceutical companies in the U.S. District Court for the District of Delaware claiming willful infringement by us of U.S. Patent No. 5,688,688 (688 Patent). During the third quarter of 2013, the parties engaged in discussions to resolve the matter. In October 2013, we and Novartis agreed to resolve all claims asserted by Novartis in the action. In October 2013, the parties entered into a settlement agreement and dismissal pursuant to which Novartis granted Alexion a non-exclusive, fully paid license to the 688 Patent for our products and dismissed its case with prejudice. As a result, we recorded expense of $9,181 in cost of sales in the third quarter 2013 related to this litigation settlement agreement.
As previously reported, in the third quarter of 2012 we reduced our estimate for probable contingent liabilities due to the execution of a settlement and non-exclusive license agreement in October 2012 with a third party related to the third party's intellectual property. We adjusted the liability to reflect the actual, negotiated royalty rate set forth in the agreement. This change in estimate resulted in a positive impact in cost of sales of $53,377 during the third quarter 2012.
In March 2013, we received a Warning Letter (Warning Letter) from the U.S. Food and Drug Administration (FDA) regarding compliance with current Good Manufacturing Practices (cGMP) at ARIMF. The Warning Letter followed an FDA inspection which concluded in August 2012. At the conclusion of that inspection, the FDA issued a Form 483 Inspectional Observations, to which we responded in August 2012 and provided additional information to the FDA in September and December 2012. The observations relate to commercial and clinical manufacture of Soliris at ARIMF. We responded to the Warning Letter in a letter to the FDA dated in April 2013. At the conclusion of another inspection of ARIMF in August 2014, the FDA issued a Form 483 with three inspectional observations, none of which was designated as a repeat observation to the Warning Letter. The observations are inspectional and do not represent a final FDA determination of compliance. We continue to manufacture products, including Soliris, at ARIMF. While the resolution of the issues raised in the Warning Letter is difficult to predict, we do not currently believe a loss related to this matter is probable or that the potential magnitude of such loss or range of loss, if any, can be reasonably estimated. To the extent that circumstances related to this matter change, the impact could have a material adverse effect on our financial operations.
Unrelated to the Warning Letter, we initiated voluntary recalls and replacements of certain lots of Soliris in 2013 and 2014 due to the presence of visible particles detected in a limited number of vials in these lots. These recalls did not interrupt the supply of Soliris to patients. Following investigation, we believe that we have identified the filling process step at our third party fill/finish provider that resulted in the presence of the visible particles and we have implemented the changes necessary to modify the process step. During the fourth quarter of 2013, we recorded expense of $14,277 in cost of sales resulting from the expected disposal of inventory in 2014. Expenses associated with recalls were not material in 2014.
Operating Leases
As of December 31, 2014, we lease our headquarters and primary research and development facilities in Cheshire, Connecticut. The leases are set to expire in 2016 and 2020. Monthly fixed rent started at approximately $315, increasing to approximately $324 over the term of this lease. We also lease space for our regional executive and sales offices in Lausanne, Switzerland, and our global supply chain and distribution headquarters in Dublin, Ireland, as well as in other U.S. locations and foreign countries to support our operations as a global organization.
Aggregate lease expense was $22,738, $19,094 and $16,758 for the years ended December 31, 2014, 2013 and 2012, respectively. Lease expense is being recorded on a straight-line basis over the applicable lease terms.
F-26
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Aggregate future minimum annual rental payments, for the next five years and thereafter under non-cancellable operating leases (including facilities and equipment) as of December 31, 2014 are:
Year | |||
2015 | $ | 21,481 | |
2016 | 18,878 | ||
2017 | 12,074 | ||
2018 | 8,371 | ||
2019 | 5,497 | ||
Thereafter | 6,260 | ||
In January 2015, we entered into a new lease agreement for office space in Zurich, Switzerland to support the relocation of the European headquarters. The new lease is estimated to commence in the second quarter of 2015 and will expire 10 years later, with a minimum renewal option of 5 years and a maximum renewal option of 10 years. The lease provides for annual payments of $2,600 over the term of the lease.
Facility Lease Obligation
In November 2012, we entered into a new lease agreement for office and laboratory space to be constructed in New Haven, Connecticut. The term of the new lease will commence upon the landlord's substantial completion of the building and will expire 12 years later, with a minimum renewal option of 7 years and a maximum renewal option of 20 years, provided that we expand our lease to include all rentable space in the building. Although we will not legally own the premises, we are deemed to be the owner of the building during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord's costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. As of December 31, 2014, we recorded a construction-in-process asset of $126,566, inclusive of the landlord's costs as well as costs incurred by Alexion, and an offsetting facility lease obligation of $107,099 associated with the new facility.
We also entered into an agreement with the State of Connecticut Department of Economic and Community Development which provides for a forgivable loan and grants totaling $26,000 and tax credits of up to $25,000 related to the lease agreement in New Haven, Connecticut. The program requires that we meet certain criteria in order to prevent forfeiture or repayment of the loan, grants and credits, which include (i) maintaining corporate headquarters in Connecticut for the next 10 years; and (ii) achieving and maintaining up to 668 full-time employment positions in the State of Connecticut over the next 6 years. Proceeds from the forgivable loan and grants will reduce our basis in the cost of the building. As of December 31, 2014, we have not received any grant funds or tax credits associated with our agreement with the State of Connecticut.
Aggregate future minimum non-cancellable commitments under this facility lease obligation, assuming a substantial completion date of November 2015, as of December 31, 2014 are as follows:
Year | ||
2015 | 1,941 | |
2016 | 11,651 | |
2017 | 11,651 | |
2018 | 11,738 | |
2019 | 12,177 | |
Thereafter | 100,371 | |
License and Research and Development Agreements
We have entered into a number of license, research and development and manufacturing development agreements since our inception. These agreements have been made with various research institutions, universities, contractors, collaborators, and government agencies in order to advance and obtain technologies and services related to our business.
License agreements generally provide for us to pay an initial fee followed by annual minimum royalty payments. Additionally, certain agreements call for future payments upon the attainment of agreed upon milestones, such as, but not
F-27
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
limited to, Investigational New Drug (IND) application or approval of Biologics License Application. These agreements require minimum royalty payments based on sales of products developed from the applicable technologies, if any.
Clinical and manufacturing development agreements generally provide for us to fund manufacturing development and on-going clinical trials. Clinical trial and development agreements include contract services and outside contractor services including contracted clinical site services related to patient enrollment for our clinical trials. Manufacturing development agreements include clinical manufacturing and manufacturing development and scale-up. We have executed a large-scale product supply agreement with Lonza Sales AG for the long-term commercial manufacture of Soliris.
The minimum fixed payments (assuming non-termination of the above agreements) as of December 31, 2014, for each of the next five years are as follows:
Year | License Agreements | Clinical and Manufacturing Development Agreements | |||||
2015 | $ | 8,964 | $ | 81,810 | |||
2016 | 2,214 | 95,730 | |||||
2017 | 664 | 102,980 | |||||
2018 | 3,164 | 102,980 | |||||
2019 | 1,064 | — | |||||
10. | Income Taxes |
The income tax provision is based on income before income taxes as follows:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
U.S. | $ | 222,088 | $ | 376,067 | $ | 294,794 | |||||
Non-U.S. | 650,019 | 150,202 | 102,772 | ||||||||
$ | 872,107 | $ | 526,269 | $ | 397,566 | ||||||
The components of the income tax provision are as follows:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Domestic | |||||||||||
Current | $ | 285,624 | $ | 141,051 | $ | (2,094 | ) | ||||
Deferred | (111,890 | ) | 92,040 | 114,807 | |||||||
173,734 | 233,091 | 112,713 | |||||||||
Foreign | |||||||||||
Current | 81,810 | 34,975 | 73,287 | ||||||||
Deferred | (40,349 | ) | 5,308 | (43,256 | ) | ||||||
41,461 | 40,283 | 30,031 | |||||||||
Total | |||||||||||
Current | 367,434 | 176,026 | 71,193 | ||||||||
Deferred | (152,239 | ) | 97,348 | 71,551 | |||||||
$ | 215,195 | $ | 273,374 | $ | 142,744 | ||||||
We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain.
We continue to pay cash taxes in U.S. Federal, various U.S. states, and foreign jurisdictions where we have operations and have utilized all of our net operating losses.
F-28
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
At December 31, 2014, we have federal and state net operating loss carryforwards of $7,718 and $12,377, respectively. Included in the NOL’s are state NOL’s of $6,593 attributable to excess tax benefits from the exercise of non-qualified stock options and vests of restricted stock. The tax benefits attributable to these NOL’s will be credited directly to additional paid in capital when utilized to offset taxes payable. Our NOL’s expire between 2022 and 2033. We also have federal and state income tax credit carryforwards of $117,109 and $2,609, respectively. These income tax credits expire between 2015 and 2034. All of these U.S. federal and state income tax credit carryforwards are attributable to excess tax benefits from the exercise of non-qualified stock options and vests of restricted stock.
Certain stock option exercises and restricted stock vestings resulted in tax deductions in excess of previously recorded benefits based on the value at the time of grant. Although these additional tax benefits or “windfalls” are reflected in U.S. state net operating loss carryforwards and U.S. federal and state income tax credit carryforwards, pursuant to authoritative guidance, the additional tax benefit associated with the windfall is not recognized until the deduction reduces taxes payable. Accordingly, since the tax benefit does not reduce our current taxes payable due to net operating loss carryforwards and credit carryforwards, these “windfall” tax benefits are not reflected in our net operating losses and credit carryforwards in deferred tax assets for all periods presented.
We were granted an incentive tax holiday in the Canton of Vaud in Switzerland effective January 1, 2010. This tax holiday had exempted us from most local corporate income taxes in Switzerland through the end of 2014 and was renewable for an additional 5 years with final expiration in 2019. During 2013, we undertook a restructuring which significantly changed our business model in Switzerland and we converted from a principal company to a distribution and service company. As a result of the significant change to our business activities in Switzerland, the Canton of Vaud in Switzerland provided final notification to us in December 2014 that our structure no longer complied with the conditions of the incentive tax holiday. In the fourth quarter of 2014, we made a payment of $22,817 in satisfaction of the clawback of previously exempted cantonal income taxes for tax years 2010 through 2013. This amount was fully accrued on our balance sheet as of December 31, 2013. Prospectively, our federal and cantonal tax will be based on the current enacted tax rates in Switzerland.
The Tax Reform Act of 1986 contains certain provisions that can limit a taxpayer's ability to utilize net operating loss and tax credit carryforwards in any given year resulting from cumulative changes in ownership interests in excess of 50% over a three-year period. We have determined that these limiting provisions were triggered during a prior year. However, we believe that such limitations are not expected to result in the expiration or loss of any significant amount of our federal NOL’s and income tax credit carryforwards.
The provision (benefit) for income taxes differs from the U.S. federal statutory tax rate. The reconciliation of the statutory U.S. federal income tax rate to our effective income tax rate is as follows:
Year Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
U.S. federal statutory tax rate | 35.0 | % | 35.0 | % | 35.0 | % | ||
State and local income taxes | 0.9 | % | 3.3 | % | 1.7 | % | ||
Foreign income tax rate differential | (19.7 | )% | (14.1 | )% | (6.7 | )% | ||
Income tax credits | (3.7 | )% | (3.5 | )% | (0.3 | )% | ||
Foreign income tax credits | (4.8 | )% | (20.5 | )% | (0.1 | )% | ||
Foreign income subject to U.S. taxation | 15.8 | % | 10.2 | % | 0.2 | % | ||
Stock option compensation | 0.1 | % | 1.1 | % | 0.7 | % | ||
State tax incentives | — | % | — | % | (1.1 | )% | ||
Structuring related costs | — | % | — | % | 4.8 | % | ||
Non-deductible acquisition related costs | — | % | — | % | 0.3 | % | ||
U.S. deferred taxes on foreign earnings | — | % | 27.2 | % | — | % | ||
Other nondeductible and permanent differences | 1.1 | % | 13.2 | % | 1.8 | % | ||
Provision (benefit) attributable to valuation allowances | — | % | — | % | (0.4 | )% | ||
Effective income tax rate | 24.7 | % | 51.9 | % | 35.9 | % | ||
The U.S. Federal tax credit for research and experimentation expenses expired December 31, 2013. In connection with this expiration, our 2014 tax expense, for the first three quarters of the year, did not include any benefit from the U.S. Federal
F-29
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
tax credit for research and experimentation. In December 2014, the Tax Increase Prevention Act of 2014, which retroactively extended the tax credit for research and experimentation back to January 1, 2014 through the end of 2014, was signed into law. The effects of a change in tax law is recognized in the period that includes the date of enactment and, therefore, our tax benefit attributable to the 2014 U.S. Federal tax credit of $3,222 for research and experimentation was recorded in the fourth quarter of 2014.
The U.S. Federal tax credit for research and experimentation expenses had previously expired December 31, 2011. In connection with this expiration, our 2012 tax expense did not include any benefit from the U.S. Federal tax credit for research and experimentation. In January 2013, the American Taxpayer Relief Act of 2012, which retroactively extended the tax credit for research and experimentation back to January 1, 2012 through the end of 2013, was signed into law. The effects of a change in tax law is recognized in the period that includes the date of enactment and, therefore, our tax benefit attributable to the 2012 U.S. Federal tax credit of $2,719 for research and experimentation was recorded in the first quarter of 2013.
In 2012, as a result of structuring the Enobia business, we recorded income tax expense of $21,812 in our income statement. The structuring also required us to make a cash payment of $47,200 in early 2013, and this amount was fully accrued on our balance sheet as of December 31, 2012.
Provisions have been made for deferred taxes based on the differences between the basis of the assets and liabilities for financial statement purposes and the basis of the assets and liabilities for tax purposes using currently enacted tax rates and regulations that will be in effect when the differences are expected to be recovered or settled. The components of the deferred tax assets and liabilities, which exclude "windfall" tax benefits, are as follows:
December 31, | December 31, | ||||||
2014 | 2013 | ||||||
Deferred tax assets: | |||||||
Net operating losses | $ | 3,401 | $ | 5,398 | |||
Income tax credits | 1,706 | 4,863 | |||||
Stock compensation | 49,090 | 33,539 | |||||
Accruals and allowances | 29,072 | 26,092 | |||||
Research and development expenses | 129,995 | 3,418 | |||||
Accrued royalties | 41,201 | 14,346 | |||||
254,465 | 87,656 | ||||||
Valuation allowance | (1,117 | ) | (1,934 | ) | |||
Total deferred tax assets | 253,348 | 85,722 | |||||
Deferred tax liabilities: | |||||||
Depreciable assets | (40,648 | ) | (41,281 | ) | |||
Unrealized gains | (45,191 | ) | (134 | ) | |||
Investment in foreign partnership | (116,359 | ) | (100,746 | ) | |||
Total deferred tax liabilities | (202,198 | ) | (142,161 | ) | |||
Net deferred tax asset (liability) | $ | 51,150 | $ | (56,439 | ) | ||
We follow authoritative guidance regarding accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The interpretation also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosures, and transition.
F-30
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The beginning and ending amounts of unrecognized tax benefits reconciles as follows:
2014 | 2013 | 2012 | |||||||||
Beginning of period balance | $ | 46,389 | $ | 12,393 | $ | 9,773 | |||||
Increases for tax positions taken during a prior period | 899 | 2,571 | 99 | ||||||||
Decreases for tax positions taken during a prior period | (2,468 | ) | (812 | ) | (1,931 | ) | |||||
Increases for tax positions taken during the current period | 9,063 | 33,056 | 4,651 | ||||||||
Decreases for tax positions related to settlements | (24,812 | ) | (419 | ) | (199 | ) | |||||
Decreases for tax positions related to lapse of statute | (396 | ) | (400 | ) | — | ||||||
$ | 28,675 | $ | 46,389 | $ | 12,393 | ||||||
The total amount of accrued interest and penalties was not significant as of December 31, 2014. The total amount of tax benefit recorded during 2014 which related to unrecognized tax benefits was $17,012. Expense recognized during 2013 was $7,897 and 2012 was not material. Unless related to excess tax benefits from stock options, all of our unrecognized tax benefits, if recognized, would have a favorable impact on the effective tax rate.
We expect none of our total unrecognized tax benefits to reverse within the next twelve months. We file federal and state income tax returns in the U.S. and in numerous foreign jurisdictions. The U.S. and foreign jurisdictions have statute of limitations ranging from 3 to 5 years. However, the statute of limitations could be extended due to our NOL carryforward position in a number of our jurisdictions. The tax authorities generally have the ability to review income tax returns for periods where the statute of limitation has previously expired and can subsequently adjust the NOL carryforward or tax credit amounts. Accordingly, we do not expect to reverse any significant portion of the unrecognized tax benefits.
The Internal Revenue Service (IRS) commenced an examination of our U.S. income tax returns for 2008 and 2009 during the second quarter 2011. This examination was completed during the fourth quarter of 2013. As a result of this audit, there was not a material change in the liability for unrecognized tax benefits.
We do not record U.S. tax expense on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries as these earnings are intended to be permanently reinvested offshore. At December 31, 2014, the cumulative amount of these earnings was approximately $359,000. During the fourth quarter of 2013, in connection with the centralization of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner in a captive foreign partnership. To the extent that our U.S. parent company receives its allocation of partnership income, the amounts will be taxable in the U.S. each year and therefore the permanent reinvestment assertion will no longer apply to such earnings. The recognition of deferred tax liabilities associated with the aforementioned partnership resulted in tax expense of approximately $95,800 during the fourth quarter of 2013. We also distributed the majority of earnings and profits of our non U.S. subsidiaries via a dividend in the amount of $152,000 during the fourth quarter of 2013. This dividend did not give rise to any U.S. cash tax liability. This resulted in repatriation of a significant portion of our remitted earnings at December 31, 2013.
We do not have any present or anticipated future need for cash held by our CFCs, as cash generated in the U.S., as well as borrowings, are expected to be sufficient to meet U.S. liquidity needs for the foreseeable future.
It is not practicable to estimate the amount of additional taxes which might be payable on our CFCs’ undistributed earnings due to a variety of factors, including the timing, extent and nature of any repatriation. While our expectation is that all foreign undistributed earnings, other than our U.S. parent company's share of the foreign partnership profits, are permanently invested, there could be certain unforeseen future events that could impact our permanent reinvestment assertion. Such events include acquisitions, corporate restructuring or tax law changes not currently contemplated.
11. | Share-based Compensation |
At December 31, 2014, we have one stock option plan, the Amended and Restated 2004 Incentive Plan (“2004 Plan”). Under the 2004 Plan, restricted stock and restricted stock units (collectively referred to as Restricted Stock), incentive and non-qualified stock options, and other stock-related awards, may be granted for up to a maximum of 47,874 shares to our directors, officers, key employees and consultants. Stock options granted under the 2004 Plan have a maximum contractual term of ten years from the date of grant, have an exercise price not less than the fair value of the stock on the grant date and generally vest over four years. Restricted stock awards also generally vest over four years, with performance-based restricted stock units having a three-year vesting period.
F-31
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The following table summarizes the components of share-based compensation expense in the consolidated statements of operations:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cost of sales | $ | 4,174 | $ | 3,214 | $ | 2,815 | |||||
Research and development | 36,203 | 23,905 | 13,839 | ||||||||
Selling, general and administrative | 74,084 | 49,084 | 37,359 | ||||||||
Total share-based compensation expense | 114,461 | 76,203 | 54,013 | ||||||||
Income tax effect | (42,082 | ) | (28,652 | ) | (20,188 | ) | |||||
Total share-based compensation expense, net of tax | $ | 72,379 | $ | 47,551 | $ | 33,825 | |||||
Share-based compensation expense capitalized to inventory during the years ended December 31, 2014, 2013 and 2012 was $10,211, $3,978, and $2,838, respectively.
As of December 31, 2014, there was $249,511 of total unrecognized share-based compensation expense related to non-vested share-based compensation arrangements granted under the Plan. The expense is expected to be recognized over a weighted-average period of 2.70 years.
Stock Options
A summary of the status of our stock option plans at December 31, 2014, and changes during the year then ended is presented in the table and narrative below:
Number of shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value | |||||||||
Outstanding at December 31, 2013 | 8,619 | $ | 50.69 | |||||||||
Granted | 1,472 | 173.04 | ||||||||||
Exercised | (3,408 | ) | 33.64 | |||||||||
Forfeited and canceled | (263 | ) | 102.99 | |||||||||
Outstanding at December 31, 2014 | 6,420 | $ | 85.65 | 6.98 | $ | 638,145 | ||||||
Vested and unvested expected to vest at December 31, 2014 | 6,320 | $ | 84.85 | 6.96 | $ | 633,345 | ||||||
Exercisable at December 31, 2014 | 3,440 | $ | 48.59 | 5.72 | $ | 469,383 | ||||||
Total intrinsic value of stock options exercised during the years ended December 31, 2014, 2013 and 2012 was $459,940, $204,470 and $308,009, respectively. We primarily utilize newly issued shares to satisfy the exercise of stock options. The total fair value of options vested during the years ended December 31, 2014, 2013 and 2012 was $35,859, $32,249 and $27,301, respectively.
The fair value of options at the date of grant was estimated using the Black-Scholes model with the following ranges of weighted average assumptions:
December 31, | December 31, | December 31, | |||
2014 | 2013 | 2012 | |||
Expected life in years | 3.64 - 5.30 | 3.30 - 5.37 | 3.30 - 4.19 | ||
Interest rate | .97% - 1.74% | 0.30% - 1.21% | 0.45% - 0.78% | ||
Volatility | 32.15% - 34.87% | 29.81% - 36.93% | 29.82% - 38.57% | ||
Dividend yield | — | — | — | ||
The expected stock price volatility rates are based on historical volatilities of our common stock. The risk-free interest rates are based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option. The average expected life represents the weighted average period of time that options granted are expected to be
F-32
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
outstanding. We have evaluated three distinct employee groups in determining the expected life assumptions, and we estimate the expected life of stock options based on historical experience of exercises, cancellations and forfeitures of our stock options.
The weighted average fair value at the date of grant for options granted during the years ended December 31, 2014, 2013 and 2012 was $51.22, $23.99 and $24.04 per option, respectively.
Restricted Stock
A summary of the status of our nonvested Restricted Stock and changes during the period then ended is as follows:
Number of Shares | Weighted Average Grant Date Fair Value | |||||
Nonvested Restricted Stock at December 31, 2013 | 1,789 | $ | 78.63 | |||
Shares granted | 855 | 174.22 | ||||
Shares forfeited | (186 | ) | 99.88 | |||
Shares vested | (650 | ) | 63.53 | |||
Nonvested Restricted Stock at December 31, 2014 | 1,808 | $ | 127.08 | |||
Restricted stock awards granted in 2014 and 2013 include 129 and 81 restricted stock units granted to senior management, which have both non-market performance-based and service-based vesting conditions. The weighted average grant date fair value of these awards granted in 2014 and 2013 was $176.04 and $97.07 respectively. The number of non-market performance-based restricted stock units granted represents the number of shares earned during the performance period, which ended on December 31, 2014 and 2013, respectively, based on specific pre-established performance goals. These awards will vest over a three year period, subject to the employees' continued employment with the Company.
The fair value of restricted stock at the date of grant is based on the fair market value of the shares of common stock underlying the awards on the date of grant. The weighted average fair value at the date of grant for restricted stock awards granted during the years ended December 31, 2014, 2013 and 2012, including restricted stock units with non-market performance conditions, was $174.22, $95.06 and $82.13 per share, respectively. The total fair value of restricted stock vested during the years ended December 31, 2014, 2013 and 2012 was $41,304, $26,679 and $18,573, respectively.
During 2014, we granted market-based performance awards to senior management which provides the recipient the right to receive restricted stock at the end of a three year performance period, based on pre-established market-based performance goals. We used payout simulation models to estimate the grant date fair value of the awards and recognized expense of $301 during 2014.
12. | Stockholders' Equity |
Preferred Stock
In February 1997, our Board of Directors declared a dividend of one preferred stock purchase right for each outstanding share of common stock (including all future issuances of common stock). Under certain conditions, each right may be exercised to purchase one hundredth of a share of a new series of preferred stock at an exercise price of $75.00, subject to adjustment (see below). The rights may be exercised only after a public announcement that a party acquired 20% or more of our common stock or after commencement or public announcement to make a tender offer for 20% or more of our common stock. The rights, which do not have voting rights, expire on March 6, 2017, and may be redeemed by us at a price of $0.01 per right at any time prior to their expiration or the acquisition of 20% or more of our stock. The preferred stock purchasable upon exercise of the rights will have a minimum preferential dividend of $10.00 per year, but will be entitled to receive, in the aggregate, a dividend of 100 times the dividend declared on a share of Common Stock. In the event of liquidation, the holders of the shares of preferred stock will be entitled to receive a minimum liquidation payment of $100 per share, but will be entitled to receive an aggregate liquidation payment equal to 100 times the payment to be made per share of Common Stock.
On February 23, 2007, our Board of Directors amended the purchase price under the preferred stock purchase rights. Further, as a result of the two-for-one stock split of the Company's outstanding shares of common stock effected on August 22, 2008, the number of shares of preferred stock purchasable upon proper exercise of each preferred stock purchase right automatically adjusted from one hundredth of a share of preferred stock to two hundredths of a share of preferred stock. Therefore, the purchase price, for each two hundredths of a share of preferred stock to be issued upon the exercise of each
F-33
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
preferred stock purchase right is $300.00. Except for the increase in the purchase price, the terms and conditions of the rights remain unchanged.
In the event that we are acquired in a merger, other business combination transaction, or 50% or more of our assets, cash flow, or earning power are sold, proper provision shall be made so that each holder of a right shall have the right to receive, upon exercise thereof at the then current exercise price, that number of shares of common stock of the surviving company which at the time of such transaction would have a market value of two times the exercise price of the right.
Common Stock
In May 2012, in conjunction with our addition into the S&P 500 Index, we completed the sale of 5,000 shares of our common stock in a public offering. The net proceeds from the sale of shares in the offering were $462,212.
Share Repurchases
In November 2012, we announced that our Board of Directors authorized the repurchase of up to $400,000 of our common stock. In December 2014, our Board of Directors authorized the repurchase of up to an additional $500,000 of our common stock. The repurchase program does not have an expiration date and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at the Company's discretion. Under the program, we repurchased 1,903 and 758 shares of our common stock at a cost of $302,599 and $66,136 during the years ended December 31, 2014 and 2013, respectively. At December 31, 2014, there is a total of $519,712 remaining for repurchases under these repurchase program.
13. | Other Comprehensive Income and Accumulated Other Comprehensive Income |
The following table summarizes the changes in AOCI, by component, for the years ended December 31, 2014, 2013 and 2012:
Defined Benefit Pension Plans | Unrealized Gains (Losses) from Marketable Securities | Unrealized Gains (Losses) from Hedging Activities | Foreign Currency Translation Adjustment | Total Accumulated Other Comprehensive Income (Loss) | |||||||||||||||
Balances, December 31, 2011 | $ | (4,183 | ) | $ | — | $ | 11,321 | $ | (2,959 | ) | $ | 4,179 | |||||||
Other comprehensive income before reclassifications | (1,807 | ) | — | 14,856 | 150 | 13,199 | |||||||||||||
Amounts reclassified from other comprehensive income | 278 | — | (11,021 | ) | — | (10,743 | ) | ||||||||||||
Net other comprehensive income (loss) | (1,529 | ) | — | 3,835 | 150 | 2,456 | |||||||||||||
Balances, December 31, 2012 | $ | (5,712 | ) | $ | — | $ | 15,156 | $ | (2,809 | ) | $ | 6,635 | |||||||
Other comprehensive income before reclassifications | (6,175 | ) | (197 | ) | (1,000 | ) | (4,573 | ) | (11,945 | ) | |||||||||
Amounts reclassified from other comprehensive income | 385 | 51 | (17,983 | ) | — | (17,547 | ) | ||||||||||||
Net other comprehensive income (loss) | (5,790 | ) | (146 | ) | (18,983 | ) | (4,573 | ) | (29,492 | ) | |||||||||
Balances, December 31, 2013 | $ | (11,502 | ) | $ | (146 | ) | $ | (3,827 | ) | $ | (7,382 | ) | $ | (22,857 | ) | ||||
Other comprehensive income before reclassifications | (5,732 | ) | (63 | ) | 110,088 | (6,337 | ) | 97,956 | |||||||||||
Amounts reclassified from other comprehensive income | 664 | (25 | ) | (18,953 | ) | — | (18,314 | ) | |||||||||||
Net other comprehensive income (loss) | (5,068 | ) | (88 | ) | 91,135 | (6,337 | ) | 79,642 | |||||||||||
Balances, December 31, 2014 | $ | (16,570 | ) | $ | (234 | ) | $ | 87,308 | $ | (13,719 | ) | $ | 56,785 | ||||||
F-34
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The table below provides details regarding significant reclassifications from AOCI during the years ended December 31, 2014, 2013 and 2012:
Details about Accumulated Other Comprehensive Income Components | Amount Reclassified From Accumulated Other Comprehensive Income during the year ended December 31, | Affected Line Item in the Consolidated Statements of Operations | ||||||||||
2014 | 2013 | 2012 | ||||||||||
Unrealized Gains (Losses) on Hedging Activity | ||||||||||||
Effective portion of foreign exchange contracts | $ | 18,874 | $ | 20,569 | $ | 12,869 | Net product sales | |||||
Ineffective portion of foreign exchange contracts | 2,787 | (915 | ) | (824 | ) | Foreign currency loss | ||||||
21,661 | 19,654 | 12,045 | ||||||||||
(2,708 | ) | (1,671 | ) | (1,024 | ) | Income tax provision | ||||||
$ | 18,953 | $ | 17,983 | $ | 11,021 | |||||||
Unrealized Gains (Losses) from Marketable Securities | ||||||||||||
Realized gains (losses) on sale of securities | $ | 40 | $ | (81 | ) | $ | — | Investment income | ||||
40 | (81 | ) | — | |||||||||
(15 | ) | 30 | — | Income tax provision | ||||||||
$ | 25 | $ | (51 | ) | $ | — | ||||||
Defined Benefit Pension Items | ||||||||||||
Amortization of prior service costs and actuarial losses | $ | (865 | ) | $ | (421 | ) | $ | (304 | ) | (a) | ||
(865 | ) | (421 | ) | (304 | ) | |||||||
201 | 36 | 26 | Income tax provision | |||||||||
$ | (664 | ) | $ | (385 | ) | $ | (278 | ) | ||||
(a) This AOCI component is included in the computation of net periodic pension benefit cost (see Note 15 for additional details).
14. | Fair Value Measurement |
Authoritative guidance establishes a valuation hierarchy for disclosure of the inputs to the valuation used to measure fair value. This hierarchy prioritizes the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value.
F-35
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The following tables present information about our assets and liabilities that are measured at fair value on a recurring basis as of December 31, 2014 and 2013, and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value.
Fair Value Measurement at December 31, 2014 | ||||||||||||||||
Balance Sheet Classification | Type of Instrument | Total | Level 1 | Level 2 | Level 3 | |||||||||||
Cash equivalents | Institutional money market funds | $ | 176,331 | $ | — | $ | 176,331 | $ | — | |||||||
Cash equivalents | Commercial paper | $ | 117,529 | $ | — | $ | 117,529 | $ | — | |||||||
Cash equivalents | Corporate bonds | $ | 9,315 | $ | — | $ | 9,315 | $ | — | |||||||
Cash equivalents | Municipal bonds | $ | 12,050 | $ | — | $ | 12,050 | $ | — | |||||||
Cash equivalents | Other government-related obligations | $ | 23,998 | $ | — | $ | 23,998 | $ | — | |||||||
Cash equivalents | Bank certificates of deposit | $ | 5,000 | $ | — | $ | 5,000 | $ | — | |||||||
Marketable securities | Mutual funds | $ | 4,277 | $ | 4,277 | $ | — | $ | — | |||||||
Marketable securities | Commercial paper | $ | 24,966 | $ | — | $ | 24,966 | $ | — | |||||||
Marketable securities | Corporate bonds | $ | 484,551 | $ | — | $ | 484,551 | $ | — | |||||||
Marketable securities | Municipal bonds | $ | 162,795 | $ | — | $ | 162,795 | $ | — | |||||||
Marketable securities | Other government-related obligations | $ | 268,978 | $ | — | $ | 268,978 | $ | — | |||||||
Marketable securities | Bank certificates of deposit | $ | 72,000 | $ | — | $ | 72,000 | $ | — | |||||||
Other current assets | Foreign exchange forward contracts | $ | 77,348 | $ | — | $ | 77,348 | $ | — | |||||||
Other assets | Foreign exchange forward contracts | $ | 58,698 | $ | — | $ | 58,698 | $ | — | |||||||
Other current liabilities | Foreign exchange forward contracts | $ | 794 | $ | — | $ | 794 | $ | — | |||||||
Other liabilities | Foreign exchange forward contracts | $ | 86 | $ | — | $ | 86 | $ | — | |||||||
Other current liabilities | Acquisition-related contingent consideration | $ | 46,546 | $ | — | $ | — | $ | 46,546 | |||||||
Contingent consideration | Acquisition-related contingent consideration | $ | 116,425 | $ | — | $ | — | $ | 116,425 | |||||||
F-36
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Fair Value Measurement at December 31, 2013 | ||||||||||||||||
Balance Sheet Classification | Type of Instrument | Total | Level 1 | Level 2 | Level 3 | |||||||||||
Cash equivalents | Institutional money market funds | $ | 234,212 | $ | — | $ | 234,212 | $ | — | |||||||
Cash equivalents | Commercial paper | $ | 6,298 | $ | — | $ | 6,298 | $ | — | |||||||
Cash equivalents | Corporate bonds | $ | 15,255 | $ | — | $ | 15,255 | $ | — | |||||||
Cash equivalents | Municipal bonds | $ | 2,225 | $ | — | $ | 2,225 | $ | — | |||||||
Cash equivalents | Bank certificates of deposit | $ | 20,003 | $ | — | $ | 20,003 | $ | — | |||||||
Marketable securities | Mutual funds | $ | 1,017 | $ | 1,017 | $ | — | $ | — | |||||||
Marketable securities | Commercial paper | $ | 106,381 | $ | — | $ | 106,381 | $ | — | |||||||
Marketable securities | Corporate bonds | $ | 461,103 | $ | — | $ | 461,103 | $ | — | |||||||
Marketable securities | Municipal bonds | $ | 200,178 | $ | — | $ | 200,178 | $ | — | |||||||
Marketable securities | Other government-related obligations | $ | 203,313 | $ | — | $ | 203,313 | $ | — | |||||||
Marketable securities | Bank certificates of deposit | $ | 13,001 | $ | — | $ | 13,001 | $ | — | |||||||
Other current assets | Foreign exchange forward contracts | $ | 21,815 | $ | — | $ | 21,815 | $ | — | |||||||
Other assets | Foreign exchange forward contracts | $ | 9,839 | $ | — | $ | 9,839 | $ | — | |||||||
Other current liabilities | Foreign exchange forward contracts | $ | 20,228 | $ | — | $ | 20,228 | $ | — | |||||||
Other liabilities | Foreign exchange forward contracts | $ | 14,864 | $ | — | $ | 14,864 | $ | — | |||||||
Other current liabilities | Acquisition-related contingent consideration | $ | 35,932 | $ | — | $ | — | $ | 35,932 | |||||||
Contingent consideration | Acquisition-related contingent consideration | $ | 106,744 | $ | — | $ | — | $ | 106,744 | |||||||
There were no securities transferred between Level 1, 2 and 3 for the year ended December 31, 2014.
Valuation Techniques
We classify mutual fund investments, which are valued based on quoted market prices in active markets with no valuation adjustment, as Level 1 assets within the fair value hierarchy.
Cash equivalents and marketable securities classified as Level 2 within the valuation hierarchy consist of institutional money market funds, commercial paper, municipal bonds, U.S. and foreign government-related debt, corporate debt securities and certificates of deposit. We estimate the fair values of these marketable securities by taking into consideration valuations obtained from third-party pricing sources. These pricing sources utilize industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include market pricing based on real-time trade data for the same or similar securities, issuer credit spreads, benchmark yields, and other observable inputs. We validate the prices provided by our third-party pricing sources by understanding the models used, obtaining market values from other pricing sources and analyzing pricing data in certain instances.
F-37
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Our derivative assets and liabilities include foreign exchange derivatives that are measured at fair value using observable market inputs such as forward rates, interest rates, our own credit risk as well as an evaluation of our counterparties’ credit risks. Based on these inputs, the derivative assets and liabilities are classified within Level 2 of the valuation hierarchy.
Contingent consideration liabilities related to acquisitions are classified as Level 3 within the valuation hierarchy and are valued based on various estimates, including probability of success, discount rates and amount of time until the conditions of the milestone payments are met.
As of December 31, 2014, there has not been any impact to the fair value of our derivative liabilities due to our own credit risk. Similarly, there has not been any significant adverse impact to our derivative assets based on our evaluation of our counterparties’ credit risks.
Contingent Consideration
In connection with prior acquisitions, we may be required to pay future consideration that is contingent upon the achievement of specified development, regulatory approval or sales-based milestone events. We determine the fair value of these obligations on the acquisition date using various estimates that are not observable in the market and represent a Level 3 measurement within the fair value hierarchy. The resulting probability-weighted cash flows were discounted using a cost of debt of 4.8% for developmental milestones and a weighted average cost of capital ranging from 12% to 21% for sales-based milestones.
Each reporting period, we adjust the contingent consideration to fair value with changes in fair value recognized in operating earnings. Changes in fair values reflect new information about the probability and timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of contingent consideration related to the passage of time as development work progresses towards the achievement of the milestones.
Estimated future contingent milestone payments related to prior business combinations range from zero if no milestone events are achieved, to a maximum of $876,000 if all development, regulatory and sales-based milestones are reached. As of December 31, 2014, the fair value of acquisition-related contingent consideration was $162,971. The following table represents a roll-forward of our acquisition-related contingent consideration:
December 31, 2014 | |||
Balance at beginning of period | $ | (142,676 | ) |
Change in fair value | (20,295 | ) | |
Balance at end of period | $ | (162,971 | ) |
15. | Employee Benefit Plans |
Defined Contribution Plan
We have one qualified 401(k) plan covering all eligible employees. Under the plan, employees may contribute up to the statutory allowable amount for any calendar year. We make matching contributions equal to $1.00 for each dollar contributed up to the first 6% of an individual's base salary and incentive cash bonus up to the annual IRS maximum. For the years ended December 31, 2014, 2013 and 2012, we recorded matching contributions of approximately $8,782, $6,360, and $3,700 respectively.
Defined Benefit Plans
We maintain defined benefit plans for employees in certain countries outside the United States, including retirement benefit plans required by applicable local law. The plans are valued by independent actuaries using the projected unit credit method. The liabilities correspond to the projected benefit obligations of which the discounted net present value is calculated based on years of employment, expected salary increases, and pension adjustments.
F-38
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
The following table sets forth the funded status and the amounts recognized for defined benefit plans:
December 31, | |||||||
2014 | 2013 | ||||||
Change in benefit obligation: | |||||||
Projected benefit obligation, beginning of year | $ | 38,166 | $ | 24,484 | |||
Prior service cost | — | — | |||||
Service cost | 8,136 | 5,413 | |||||
Interest cost | 780 | 504 | |||||
Change in assumptions | 5,571 | 2,643 | |||||
Recognized actuarial net loss | 1,350 | 3,701 | |||||
Foreign currency exchange rate changes | (3,055 | ) | 573 | ||||
Net transfers (from) to plan | (247 | ) | 848 | ||||
Projected benefit obligation, end of year | $ | 50,701 | $ | 38,166 | |||
Accumulated benefit obligation, end of year | $ | 43,141 | $ | 30,655 | |||
December 31, | |||||||
2014 | 2013 | ||||||
Change in plan assets: | |||||||
Fair value of plan assets, beginning of year | $ | 23,327 | $ | 16,006 | |||
Return on plan assets | 393 | 244 | |||||
Employer contributions | 4,417 | 3,811 | |||||
Plan participants' contributions | 1,741 | 1,523 | |||||
Foreign currency exchange rate changes | (2,855 | ) | 895 | ||||
Net transfers (from) to plan | (247 | ) | 848 | ||||
Fair value of plan assets, end of year | $ | 26,776 | $ | 23,327 | |||
Funded status at end of year | $ | (23,925 | ) | $ | (14,839 | ) | |
The Company measures the fair value of plan assets based on the prices that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The following table presents total plan assets by investment category as of December 31, 2014 and the classification of each investment category within the fair value hierarchy with respect to the inputs used to measure fair value:
December 31, 2014 | December 31, 2013 | ||||||||||||
Fair Value (Level 2) | as % of total plan assets | Fair Value (Level 2) | as % of total plan assets | ||||||||||
Cash and cash equivalents | $ | 1,794 | 7 | % | $ | 1,470 | 6 | % | |||||
Equity security funds | 10,791 | 40 | % | 9,237 | 40 | % | |||||||
Debt security funds | 11,246 | 42 | % | 9,704 | 42 | % | |||||||
Real estate funds | 2,945 | 11 | % | 2,916 | 12 | % | |||||||
$ | 26,776 | 100 | % | $ | 23,327 | 100 | % | ||||||
All plan asset investments are classified as Level 2 within the fair value hierarchy and are valued utilizing observable prices for similar instruments and quoted prices for identical or similar instruments in markets that are not active. The investment objective is to maximize the overall return from investment income and capital appreciation consistent with the preservation of capital considering investment strategies and asset allocation limits as determined by pension law. The targeted allocation for these funds (if any) is as follows:
F-39
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Target Allocation Ranges in % | |
Cash and notes receivable issued by banks or insurance companies | 0-10% |
Equity securities | 30-60% |
Debt securities | 16-45% |
Real estate | 10-20% |
Other | 0-12% |
At December 31, 2014, we have recorded a liability of $23,925 in other non-current liabilities and a charge to accumulated other comprehensive income, net of tax, of $16,570 related to an additional minimum liability.
The following table provides the weighted average assumptions used to calculate net periodic benefit cost and the actuarial present value of projected benefit obligations:
December 31, | |||||
2014 | 2013 | ||||
Weighted average assumptions - Net Periodic Benefit Cost: | |||||
Discount rate | 2.0 | % | 2.0 | % | |
Long term rate of return on assets | 4.0 | % | 4.0 | % | |
Rate of compensation increase | 1.6 | % | 1.6 | % | |
Weighted average assumptions - Projected Benefit Obligation: | |||||
Discount Rate | 1.4 | % | 2.1 | % | |
Rate of compensation increase | 1.6 | % | 1.6 | % | |
The discount rates used to determine the net periodic benefit cost and projected benefit obligation represent the yield on high quality AA-rated corporate bonds for periods that match the duration of the benefit obligations.
The expected long-term rate of return on plan assets represents a weighted average of expected returns per asset category. The rate of return considers historical and estimated future risk free rates of return as well as risk premiums for the relevant investment categories.
The components of net periodic benefit cost are as follows:
Year Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Service cost | $ | 8,136 | $ | 5,413 | $ | 4,733 | |||||
Interest cost | 780 | 504 | 464 | ||||||||
Expected return on plan assets | (900 | ) | (633 | ) | (515 | ) | |||||
Employee contributions | (1,741 | ) | (1,523 | ) | (1,163 | ) | |||||
Amortization of prior service costs | 9 | 9 | 9 | ||||||||
Amortization and deferral of actuarial gain | 846 | 410 | 217 | ||||||||
Total net periodic benefit cost | $ | 7,130 | $ | 4,180 | $ | 3,745 | |||||
F-40
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Other changes in plan assets and benefit obligations recognized in AOCI are as follows:
Amount included in AOCI - December 31, 2012 | $ | (5,712 | ) |
Prior service cost | 9 | ||
Net loss arising during the period | (3,710 | ) | |
Change in assumptions | (2,657 | ) | |
Amortization of net gain | 412 | ||
Plan assets losses | (391 | ) | |
Taxes | 547 | ||
Amount included in AOCI - December 31, 2013 | $ | (11,502 | ) |
Prior service cost | 9 | ||
Net loss arising during the period | (1,354 | ) | |
Change in assumptions | (5,640 | ) | |
Amortization of net gain | 856 | ||
Plan assets losses | (513 | ) | |
Taxes | 1,574 | ||
Amount included in AOCI - December 31, 2014 | $ | (16,570 | ) |
The amount in accumulated other comprehensive income as of December 31, 2014 that is expected to be recognized as a component of the net periodic pension costs in 2015 is $1,216.
We estimate that we will pay employer contributions of approximately $4,238 in 2014. The expected future cash flows to be paid in respect of the pension plans as of December 31, 2014 were as follows:
Year | ||
2015 | 1,985 | |
2016 | 1,511 | |
2017 | 1,450 | |
2018 | 1,247 | |
2019 | 1,227 | |
2020 to 2024 | 4,922 | |
16. | Restructuring |
In the fourth quarter 2014, we announced plans to move the European headquarters from Lausanne to Zurich, Switzerland. The relocation of the European headquarters will support our growing operational needs based on current business forecasts. The activities at our Lausanne site will be relocated to our Zurich, Cheshire, Connecticut, and Dublin, Ireland locations. As a result of this action, we recorded restructuring expenses of $15,365 related to employee costs in the fourth quarter of 2014. We expect to incur approximately $10,000 to $15,000 of additional restructuring related charges in 2015 related to contract termination and additional employee costs. We expect to pay all accrued amounts related to this restructuring activity by the end of fiscal year 2015. The restructuring reserve of $15,365 is included within accrued expenses liabilities on the Company's consolidated balance sheet as of December 31, 2014.
17. | Segment Information |
We operate as one business segment, which is the innovation, development and commercialization of life-transforming therapeutic products. Therefore, our chief operating decision-maker manages our operations as a single operating segment.
F-41
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
Revenues and tangible long-lived assets by significant geographic region are as follows:
Year Ended December 31, | |||||||||||
Revenues: | 2014 | 2013 | 2012 | ||||||||
United States | $ | 730,089 | $ | 561,405 | $ | 400,483 | |||||
Europe (1) | 836,134 | 514,987 | 418,321 | ||||||||
Asia Pacific | 244,059 | 203,538 | 161,480 | ||||||||
Other | 423,451 | 271,416 | 153,830 | ||||||||
$ | 2,233,733 | $ | 1,551,346 | $ | 1,134,114 | ||||||
(1) As described in Note 19 "Quarterly Financial Information (unaudited)", included within the Europe revenues for 2014 is a reimbursement of $87,830 for shipments made in years prior to January 1, 2014 as a result of an agreement with the French government. | |||||||||||
December 31, | |||||||||||
Long-lived assets (2): | 2014 | 2013 | |||||||||
United States | $ | 298,122 | $ | 190,791 | |||||||
Europe | 88,543 | 5,413 | |||||||||
Other | 5,583 | 4,905 | |||||||||
$ | 392,248 | $ | 201,109 | ||||||||
(2) | Long-lived assets consist of property, plant and equipment. |
18. | Subsequent Events |
In January 2015, we entered into a license agreement with a third party to obtain certain intellectual property rights and technology related to specific therapeutic compounds. The agreement provides an exclusive research, development and commercial license for products to be developed using such compounds. Pursuant to the terms of the agreement, we made an upfront payment of $50,000 during the first quarter 2015. We could be required to pay up to an additional $213,000 in development and regulatory milestones related to a product developed under the agreement for a single disease indication. An additional $437,000 in milestone payments could be due if certain development and regulatory milestones are achieved for additional disease indications. The agreement also provides for royalty payments and potential milestone payments of up to $180,000 on commercial sales of products developed under the agreement.
In January 2015, we entered into a new lease agreement for approximately 44,000 square feet of office space in Zurich, Switzerland to support the relocation of the European headquarters. The lease is estimated to commence in the second quarter of 2015 and will expire 10 years later, with a minimum renewal option of 5 years and a maximum renewal option of 10 years. The lease provides for annual payments of $2,600 over the term of the lease.
F-42
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2014, 2013 and 2012
(amounts in thousands except per share amounts)
19. | Quarterly Financial Information (unaudited) |
The following condensed quarterly financial information is for the years ended December 31, 2014 and 2013:
March 31 | June 30 | September 30 | December 31 | |||||||||||||
2014: | ||||||||||||||||
Revenues | $ | 566,616 | (1) | $ | 512,495 | $ | 555,146 | $ | 599,476 | |||||||
Cost of sales | 32,939 | (1) | 39,626 | 51,858 | 49,439 | |||||||||||
Operating expenses | 324,174 | 254,020 | 266,629 | 346,342 | (2) | |||||||||||
Operating income | 209,503 | 218,849 | 236,659 | 203,695 | ||||||||||||
Net income (loss) | $ | 159,354 | $ | 166,495 | $ | 177,731 | $ | 153,332 | ||||||||
Earnings (loss) per common share | ||||||||||||||||
Basic | $ | 0.81 | $ | 0.84 | $ | 0.90 | $ | 0.77 | ||||||||
Diluted | $ | 0.79 | $ | 0.83 | $ | 0.88 | $ | 0.76 | ||||||||
March 31 | June 30 | September 30 | December 31 | |||||||||||||
2013: | ||||||||||||||||
Revenues | $ | 338,941 | $ | 370,091 | $ | 400,405 | $ | 441,909 | ||||||||
Cost of sales | 35,269 | 39,377 | 51,358 | 51,552 | ||||||||||||
Operating expenses | 186,700 | 193,023 | 213,772 | 252,285 | (3) | |||||||||||
Operating income | 116,972 | 137,691 | 135,275 | 138,072 | ||||||||||||
Net income | $ | 82,217 | $ | 95,885 | $ | 93,785 | $ | (18,992 | ) | |||||||
Earnings per common share | ||||||||||||||||
Basic | $ | 0.42 | $ | 0.49 | $ | 0.48 | $ | (0.10 | ) | |||||||
Diluted | $ | 0.41 | $ | 0.48 | $ | 0.47 | $ | (0.10 | ) | |||||||
(1) Included within revenues for the first quarter of 2014 is a reimbursement for shipments made in years prior to January 1, 2014 as a result of an agreement with the French government which positively impacted reimbursement for Soliris. As a result of the agreement, in the first quarter of 2014, we recognized $87,830 of net product sales from Soliris in France relating to years prior to January 1, 2014. Also, included within cost of sales for the first quarter of 2014 is the incremental impact in cost of sales of $2,055 for additional royalties related to the $87,830 of net product sales from prior year shipments.
(2) Included within operating expenses for the fourth quarter of 2014 is $15,365 for restructuring expenses recognized in connection with the relocation of the European headquarters.
(3) Included within operating expenses for the fourth quarter of 2013 is an impairment charge of $33,521 to write-down the value of an early stage, Phase I indefinite-lived intangible asset and purchased technology asset related to the Taligen acquisition.
F-43