Attached files
| file | filename |
|---|---|
| EX-99.1 - EX-99.1 - ZIOPHARM ONCOLOGY INC | d861898dex991.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
Date of report (Date of earliest event reported): February 2, 2015
ZIOPHARM Oncology, Inc.
(Exact Name of Registrant as Specified in Charter)
| Delaware | 001-33038 | 84-1475672 | ||
| (State or Other Jurisdiction of Incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) |
| One First Avenue, Parris Building 34, Navy Yard Plaza Boston, Massachusetts |
02129 | |
| (Address of Principal Executive Offices) | (Zip Code) |
(617) 259-1970
(Registrant’s telephone number, including area code)
Not applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425). |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12). |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)). |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)). |
| Item 2.02 | Results of Operations and Financial Condition |
Based upon preliminary estimates, ZIOPHARM Oncology, Inc., or the Company, expects to have approximately $43 million in cash and cash equivalents as of December 31, 2014. We have not yet completed our year-end financial close process for the year ended December 31, 2014. This estimate of our cash and cash equivalents as of December 31, 2014 is based on preliminary estimates of our financial results that we expect to report for the period. These estimates are subject to completion of our financial closing procedures. Our independent registered public accounting firm, McGladrey LLP, has not audited, reviewed or compiled these estimates. These estimates are not a comprehensive statement of our financial results for the year ended December 31, 2014 and our actual results may differ materially from these estimates as a result of the completion of our financial closing procedures, final adjustments and other developments arising between now and the time that our financial results for this period are finalized.
| Item 7.01 | Regulation FD Disclosure |
Public Offering Announcement
On February 2, 2015, the Company issued a press release announcing that it intends to commence an underwritten public offering of $75,000,000 of shares of its common stock. J.P. Morgan Securities LLC is acting as sole book-running manager for the offering and BMO Capital Markets Corp. is acting as senior lead manager. ZIOPHARM intends to grant the underwriters a 30-day option to purchase up to $11,250,000 of additional shares of common stock. The offering is subject to market and other conditions, and there can be no assurance as to whether or when the offering may be completed, or as to the actual size or terms of the offering.
A copy of the above referenced press release is furnished as Exhibit 99.1 to this Current Report on Form 8-K. This information, including the information contained in the press release furnished as Exhibit 99.1, shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and is not incorporated by reference into any of the Company’s filings, whether made before or after the date hereof, regardless of any general incorporation language in any such filing.
| Item 8.01 | Other Events |
Updated Business Description and Risk Factors
On February 2, 2015, the Company filed with the Securities and Exchange Commission a Registration Statement on Form S-3 (File No. 333-201826) and a prospectus supplement thereto, which included an updated business description and the new and/or revised risk factors set forth below.
All references below to “ZIOPHARM Oncology,” “ZIOPHARM,” the “Company,” “we,” “us,” “our” and similar references refer to ZIOPHARM Oncology, Inc., except where the context otherwise requires or as otherwise indicated.
Description of Business
Company overview
ZIOPHARM Oncology, Inc. is a biopharmaceutical company that seeks to acquire, develop and commercialize, on its own or with commercial partners, a diverse portfolio of cancer therapies that can address unmet medical needs through synthetic biology. Pursuant to an exclusive channel agreement with Intrexon Corporation, or Intrexon, we obtained rights to Intrexon’s synthetic biology platform for use in the field of oncology, which included a clinical stage product candidate, Ad-RTS-IL-12 used with the oral activator veledimex. The synthetic biology platform is an industrialized engineering approach for molecular and cell biology and gene control. It employs an inducible gene-delivery system that enables controlled in vivo expression of genes that produce therapeutic proteins to treat cancer. Ad-RTS-IL-12 + veledimex uses this gene delivery system to produce Interleukin-12, or IL-12, a potent, naturally occurring anti-cancer protein. We have completed two Phase 2 studies evaluating Ad-RTS-IL-12 + veledimex, the first for the treatment of metastatic melanoma, and the second for the treatment of metastatic breast cancer; data from these Phase 2 studies was presented in December 2014. We are continuing to pursue intratumoral injection of Ad-RTS-IL-12 + veledimex in breast cancer and brain cancer.
In addition to our synthetic biology programs, we recently obtained an exclusive, worldwide license to certain immuno-oncology technologies owned and licensed by The University of Texas M.D. Anderson Cancer Center, or MD Anderson, including technologies relating to novel chimeric antigen receptors, or CARs, natural killer, or NK cells and T cell receptors, or TCRs. Combining these technologies with Intrexon’s technology suite and clinically tested RheoSwitch Therapeutic System®, or RTS®, IL-12 modules, we plan to develop CAR-T and other immune cells that will target and kill cancer cells. We plan to leverage the synergy between the platforms to accelerate a promising synthetic immuno-oncology pipeline, with up to five CAR-T therapies expected to enter the clinic in 2015 and programs for the development of allogeneic CAR-T therapies that can be used off-the-shelf expected to be initiated in 2016.
We plan to continue to combine Intrexon’s technology suite with our capabilities to translate science to the patient to identify and develop additional products to stimulate or inhibit or stimulate key pathways, including those used by the body’s immune system, to treat cancer.”
We also have a portfolio of small molecule drug candidates, which are no longer a strategic focus of our development activities and for some of which we are seeking partners to pursue further development and potential commercialization.
Enabling technologies
Synthetic biology
Synthetic biology entails the application of engineering principles to biological systems for the purpose of designing and constructing new biological systems or redesigning/modifying existing biological systems. Biological systems are governed by DNA, the building block of gene programs, which control cellular processes by coding for the production of proteins and other molecules that have a functional purpose and by regulating the activities of these molecules. This regulation occurs via complex biochemical and cellular reactions working through intricate cell signaling pathways, and control over these molecules modifies the output of biological systems. Synthetic biology has been enabled by the application of information technology and advanced statistical analysis, also known as bioinformatics, to genetic engineering, as well as by improvements in DNA synthesis. Synthetic biology aims to engineer gene-based programs or codes to modify cellular function to achieve a desired biological outcome. Its application is intended to allow more precise control of drug concentration and dose, thereby improving the therapeutic index associated with the resulting drug.
On January 6, 2011, we entered into an Exclusive Channel Partner Agreement with Intrexon, which we refer to as the Channel Agreement, to develop and commercialize novel DNA-based therapeutics in the field of cancer treatment by combining Intrexon’s synthetic biology platform with our capabilities to translate science to the patient. As a result, our DNA synthetic biology platform employs an inducible gene-delivery system that enables regulated and controlled delivery of genes that produce therapeutic proteins to treat cancer. The first example of this regulated controlled delivery is achieved by producing IL-12, a potent, naturally occurring anti-cancer protein, under the control of Intrexon’s proprietary biological “switch” to turn on and off (and on and off repeatedly) the therapeutic protein expression at the tumor site. We and Intrexon refer to this “switch” as the RheoSwitch Therapeutic System® or RTS® platform. Our initial drug candidate being developed using the synthetic biology platform is Ad-RTS-IL-12 + veledimex.
More detailed descriptions of our clinical development for each are set forth in this report under the caption “—Product candidates” below.
Immuno-oncology
Immuno-oncology, which utilizes a patient’s own immune system to treat cancer, is one of the most actively pursued areas of research by biotechnology and pharmaceutical companies today. Cancer cells contain mutated proteins and may overexpress other proteins usually found in the body at low levels. The immune system typically recognizes unusual or aberrant cell protein expression and eliminates these cells in a highly efficient process known as immune surveillance. A central player in immune surveillance is a type of white blood cell known as the T cell. In healthy individuals, T cells identify and kill infected or abnormal cells, including cancer cells. Cancer cells develop the ability to evade immune surveillance, which is a key factor in their growth, spread, and persistence. In the last five years, there has been substantial scientific progress in countering these evasion mechanisms using immunotherapies, or therapies that activate the immune system.
On January 13, 2015, we, together with Intrexon, entered into a license agreement with MD Anderson, which we refer to as the MD Anderson License. Pursuant to the MD Anderson License, we and Intrexon hold an exclusive, worldwide license to certain technologies owned and licensed by MD Anderson including technologies relating to novel CAR-T cell therapies arising from the laboratory of Laurence Cooper, M.D., Ph.D., professor of pediatrics at MD Anderson, as well as either co-exclusive or non-exclusive licenses under certain related technologies.
Combining the non-viral genetic engineering technologies we licensed from MD Anderson together with Intrexon’s industrialized approach to gene engineering and cell control, we believe we can rapidly and efficiently reprogram T cells to express a particular CAR or TCR construct that will enable the T cell to recognize and target cancer cells. CAR-T cells target cell surface tumor antigens, such as CD-19, that exist on cancer cells and that are independent of human leukocyte antigens, or HLAs, and which we refer to as “public” antigens. TCR+ cells target tumor antigens that are dependent on HLAs and which we refer to as “private” antigens. Natural killer cells target tumors with loss of HLAs, or tumors with no antigens. Most CAR-T cell and TCR products currently being developed by competitors are autologous, or derived from the patient’s own blood, and gene engineered with viral technology. As a result, the patient’s blood must be harvested, shipped to a manufacturing facility where it is modified using a retrovirus to express the CAR or TCR, and then shipped back to be infused into the patient. The process can take several weeks to a month and is very labor intensive and costly. Currently, this complex technique can only be done in very sophisticated laboratories. We believe we will be
able to manufacture our CAR-T cells and TCRs using non-viral methods, which we expect will enable a simpler process requiring only days or hours and result in a lower cost of manufacturing. Our non-viral methods could also potentially enable autologous point of care treatment, where a patient’s own T cells would be modified at or near the point of care, for example, utilizing a local blood bank, to express the CAR-T or TCR construct and then infused back into the patient, potentially during the same visit. In addition, we intend to use our non-viral methods to develop allogeneic treatments that can be used off-the-shelf. An allogeneic off-the-shelf treatment would enable a patient to be treated with a CAR-T or TCR construct that is created from a separate healthy donor, personalized for that patient, and then distributed to the point of care. Our non-viral methods, which we believe are nimble, fast and less costly than other approaches, together with our industrialized, scalable engineering approach are expected to enable highly efficient and less costly manufacturing approaches to gene engineered cell-based therapy. In addition, our proprietary RheoSwitch Therapeutic System® may give us the ability to control in vivo gene expression (on-off-on-off etc.) in CAR-T or TCR cells, which we believe could result in significantly lower toxicity compared to other products currently in development.
Product candidates
The following chart identifies our current synthetic biology product candidates and their stage of development, each of which are described in more detail below.
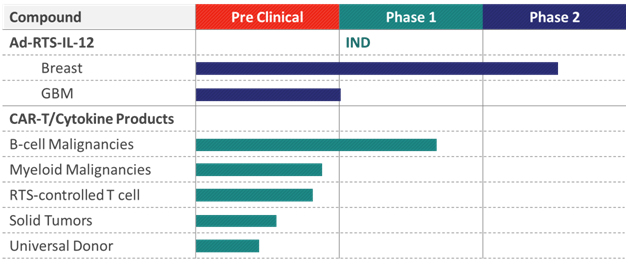
Synthetic biology programs
Ad-RTS-IL-12 + veledimex
Ad-RTS-IL-12 + veledimex has been evaluated in two Phase 2 studies, the first for the treatment of metastatic melanoma, and the second for the treatment of unresectable recurrent or metastatic breast cancer. Ad-RTS-IL-12 + veledimex is our lead product candidate, which uses our gene delivery system to produce IL-12, a potent, naturally occurring anti-cancer protein.
More specifically, IL-12 is a potent immunostimulatory cytokine which activates and recruits dendritic cells that facilitate the cross-priming of tumor antigen-specific T cells. Intratumoral administration of Ad-RTS-IL-12 + veledimex, which allows for adjustment of IL-12 gene expression upon varying the dose of veledimex, is designed to reduce the toxicity elicited by systemic delivery of IL-12, and increase efficacy through high intratumoral expression.
We reported the controlled local expression of IL-12 as an immunotherapeutic treatment of glioma (brain cancer) in animal models through the use of the RTS® at the October 2013 AACR-NCI-EORTC conference. Veledimex brain penetration was demonstrated in normal mice and monkeys with intact blood brain barriers. Treatment with Ad-RTS-IL-12 + veledimex and DC-RTS-IL-12 + veledimex both demonstrated dose-related increase in survival in the mouse GL-261 glioma model with no adverse clinical signs observed. In December 2013, we announced the unanimous approval of the Recombinant DNA Advisory Committee of the National Institutes of Health, or the RAC/NIH, for the initiation of a Phase 1 study of Ad-RTS-IL-12 + veledimex, in subjects with recurrent or progressive high grade gliomas. The U.S Food and Drug Administration, or FDA, has requested additional nonclinical information to support the Phase 1 study and this data has been generated. Subject to reaching agreement with the FDA, we anticipate initiation of the Phase 1 study during the first half of 2015. Glioblastoma is by far the most frequent malignant brain tumor and is associated with a particularly aggressive course and dismal prognosis. The current standard of care is based on surgical resection to the maximum feasible extent, followed by radiotherapy and concomitant adjuvant temozolomide. Such aggressive treatment, however, is associated with only modest improvements in survival resulting in a very high unmet medical need.
At the American Association for Cancer Research, or AACR, 2014 Annual Meeting, in April 2014, we presented data from a preclinical study conducted jointly by us and Intrexon demonstrating the anti-tumor effects and tolerability of Ad-RTS-mIL-12 in a glioblastoma murine model. Veledimex was found to effectively cross the blood brain barrier, with dose-related increases in plasma and brain tissue exposure, and no accumulation in brain tissue following repeat dosing. The study data demonstrated that administration of Ad-RTS-mIL-12 + veledimex resulted in dose-related increases in survival of four- to five-fold, without exhibiting an adverse safety profile, when compared to median survival in vehicle control groups.
At the 17th Annual Meeting of the American Society of Gene and Cell Therapy, or ASGCT, in May 2014, we presented results demonstrating the potent anti-tumor and anti-cancer stem cell effects of Ad-RTS-IL-12 in a preclinical glioma model. Results from human and laboratory studies of Ad-RTS-IL-12 demonstrated that precise control of IL-12 gene expression levels can be achieved using Intrexon’s RTS®. Rapid, tight modulation of in vivo expression of IL-12 using the activator ligand, veledimex, was demonstrated across these studies. When IL-12 expression was “switched on” it rapidly led to expression and an immune response. This immune response was characterized by an increase in tumor infiltrating lymphocytes with system wide immune activation. The data presented in May 2014 further demonstrated that Ad-RTS-IL-12 has potent anti-cancer effects in a glioma model, showing both a reduction in tumor mass and prolonged survival when compared to existing treatment standards. The data also showed a significant reduction in cancer stem cells, as measured by dramatically reduced nestin levels. Cancer stem cells are thought to play a critical role in recurrence and metastasis.
At the AACR 2014 Immunology and Immunotherapy Meeting in December 2014, together with Intrexon, we presented clinical results from the Ad-RTS-hIL-12 + veledimex studies in patients with advanced breast cancer and melanoma demonstrating local and systemic IL-12-mediated anti-cancer activity, as well as safety and control of both immune- and IL-12-mediated toxicity with use of the RTS® gene switch. In two open-label Phase 2 clinical studies, twelve patients with metastatic advanced stage breast cancer and twenty-six patients with metastatic melanoma were administered Ad-RTS-hIL-12. Following intra-tumoral injection of Ad-RTS-hIL-12, expression of IL-12 within patients was controlled by the RTS® gene switch using the oral activator ligand, veledimex, at doses ranging from 5mg to 160mg. All subjects had heavy tumor burden and disease progression at the time of enrollment, with mean number of prior therapies at 14 and 10 for breast cancer and melanoma patients, respectively. Treatment with Ad-RTS-hIL-12 + veledimex resulted in an increase in the immune cytokine IL-12 and downstream cytokines, IFN-g, IP-10 and IL-10, resulting in a significant increase in the number of CD8+ T-cells. Among seven evaluable subjects in the Phase 2 clinical study of Ad-RTS-IL-12 + veledimex in patients with recurrent or metastatic breast cancer, three had stable disease, including one triple negative breast cancer subject who crossed the primary endpoint of 16 week progression free survival, for a disease control rate (stable disease or better) of 43%. Target lesions and tumor burden were significantly reduced in approximately 40% of patients. In the Phase 1/2 study of Ad-RTS-hIL-12 + veledimex in subjects with unresectable stage III/IV melanoma, of eighteen evaluable subjects, one had a partial response and six had stable disease, for a disease control rate of 39%. In melanoma patients for whom a response was observed, there was evidence of local and systemic anti-cancer activity. The adverse event profile of Ad-RTS-hIL-12 + veledimex in both melanoma and breast cancer was predictable, reversible and characteristic of immune activation. The most common ³ Grade 3 treatment emergent adverse events, or TEAEs, in breast cancer and melanoma included neutropenia and electrolyte abnormalities (21%) each, LFTs increased (16%), leukopenia (13%) and pyrexia, hypotension, lymphopenia, anemia, and cytokine release syndrome (11%) each. Importantly, all TEAEs and SAEs ³ Grade 3 reversed rapidly upon discontinuation of veledimex oral dosing.
Also at the AACR 2014 Immunology and Immunotherapy Meeting in December 2014, together with Intrexon, we presented preclinical data supporting the potential for cytolytic activity against solid tumor targets with allogeneic, genetically-modified stem cells enabled for controlled release of cell-linking moieties, or CLMs, within the tumor micro-environment and preclinical data describing the development of a novel, high-throughput screening technology for rapidly identifying bi-specific antibodies capable of inducing targeted immunologic activity through the activation of T-cells or other immune cells against tumors. CLMs are small bi-specific antibody fragments capable of directing potent T-cell mediated tumor lysis by bridging the immunologic synapses of T-cells and surface targets on tumor cells. Previous studies have shown that the systemic distribution and pharmacokinetic profile of bi-specific antibodies limit their utility for many target/effector combinations. In two preclinical studies, Intrexon and Ziopharm researchers interrogated a large number of CLM-based effectors for their ability to activate white blood cells from peripheral blood and lyse receptor target-positive tumor cells. Allogeneic, tumor targeting stem cells were then genetically modified to express CLMs within the tumor microenvironment using the RTS® platform as a mechanism for providing spatial and temporal control. The first study demonstrated the ability of Intrexon’s proprietary image-based screening systems and rapid DNA assembly to screen a large number of EGFR and HER2 receptor-targeted CLM variants for their ability to recruit CD3+ T-cells and mediate selective cell killing against target positive cells in peripheral blood co-cultures. The image-based screening platform allowed for real time target cell killing information to be obtained, as well as kinetic cell morphologic analyses to understand the dynamics of killing activity, thereby shortening the developmental timeline to lead candidate selection. The second study validated these CLM candidates in scalable, allogeneic endometrial regenerative cells, or ERCs, genetically modified to express an anti-CD3-anti-EGFR CLM under RTS® ligand inducible control. Expression of CLMs under the RTS® inducible promoter provided effective control of CLM secretion and modulation of killing activity, with veledimex-dependent cytotoxicity of greater than 80% against an EGFR+ KRAS mutant lung cancer cell model. CLM-expressing ERCs were found to be effective in co-culture killing assays at cellular doses as low as 1% of target cells. These data supported the feasibility of localized cytolytic activity of CLM-secreting allogeneic cell therapy products against EGFR+ KRAS mutant solid tumor malignancies.
We have completed the Phase 2 monotherapy studies in melanoma and breast cancer using Ad-RTS-IL-12 + veledimex. Additionally, we expect a future trial with IL-12 in combination therapies with standard of care for breast cancer. As the treatment of advanced melanoma has undergone and continues to undergo a rapid evolution with the introduction and approval of highly promising new single and combination agents, the standard of care in this indication has become uncertain, resulting in a much more competitive and commercially unpredictable environment. As a result, we are pursuing intratumoral injection of Ad-RTS-IL-12 + veledimex in brain cancer and breast cancer, and will pause further development of Ad-RTS-IL-12 + veledimex in melanoma with intratumoral injection. However, through current strategic initiatives, we expect to utilize RTS-IL-12 + veledimex in cell based immunotherapy of melanoma and other cancers. We plan to initiate a Phase 1 trial to evaluate Ad-RTS-IL-12 + veledimex as a single agent in the treatment of patients with brain cancer in the first half of 2015.
CAR-T/cytokine programs
We are actively pursuing non-viral, genetic engineering technologies to develop novel CAR-T, NK and TCR cells. Combining this technology with Intrexon’s industrialized synthetic biologic engineering and clinically tested and validated RTS IL-12 modules, represents a differentiated approach to genetically modified CAR-T cell and other immune cells. Employing novel cell engineering techniques and multigenic gene programs, we expect to implement next-generation non-viral adoptive cellular therapies based on designer cytokines and CARs under control of RTS® technology targeting both hematologic malignancies and solid tumors. We plan to leverage the synergy between the platforms to accelerate a promising synthetic immuno-oncology pipeline, with up to five CAR-T therapies expected to enter the clinic in 2015 and programs for the development of allogeneic CAR-T therapies that can be used off-the-shelf expected to be initiated in 2016.
Research suggests that T cells can be re-programmed to have a very strong anti-cancer therapeutic effect through the expression of CARs to redirect specificity to tumors without HLA restrictions. The signature event within this field has resulted from the infusion of T cells expressing CARs into patients with B cells leukemias and lymphomas. Many of these patients have responded to these new therapies with a durable and dramatic anti-tumor effect after infusing CD19-specific T cells. Despite the highly promising results that have been demonstrated by early researchers in the field, current technologies and approaches have shown a number of serious drawbacks, including toxicity, manufacturing complexity and expense. A particular problem is that infusions of T cells into patients with large amounts of disease have invariably led to significant issues of toxicity for recipient patients. These toxicities primarily involve three major, potentially catastrophic side-effects:
| 1. | The rapid killing of tumor cells releases a large number intracellular constituents that are very toxic to various organs and is called “tumor lysis syndrome” that can be fatal, |
| 2. | the supra-physiologic release of cytokines (“cytokine storm”) that causes fever, instability of blood pressure, mental status changes and on occasion, death, and |
| 3. | on-target, and off-tissue toxicity represented by the concomitant damage of normal B cells and loss of humoral (antibody) immunity. |
We expect to be able to tightly control expansion and activation of CAR-T cells in the body, which has the potential to alleviate or abrogate these toxicities.
MD Anderson’s platform, which uses the exclusive Sleeping Beauty system, or SB system, generates and characterizes new CAR-T designs, which enables a high throughput approach to evaluate the CAR-T. This “EZ-CAR-T” non-viral system is used to fashion immuno-receptors that differ in specificity and ability to activate T cells. These CAR-T molecules are evaluated in a “go/no go” system based on serial killing and protection of T cells from activation-induced cell death. Non-viral gene transfer using the SB system is unique in the field of oncology. Examples of cell engineering techniques that we expect to employ with the SB system are induced pluripotent stem cell (iPSC) processing technologies combined with Laser-enabled Analysis and Processing, or LEAP®, which consists of computerized image-based selection and laser processing for very rapid cell identification and purification as well as AttSite® Recombinases, which involves stable, targeted gene integration and expression with proprietary serine recombinases. We believe the advanced DNA vectors derived from SB can be used to avoid the expense and manufacturing difficulty associated with creating CAR-T cells using viral vectors. After electroporation, the transposon/transposase improves the efficiency of integration of plasmids used to express CAR and other transgenes in T cells. Propagation of genetically modified T cells on activating and propagating cells, or AaPC, provide a competitive advantage over other non-viral methods of modification. The SB system combined with artificial antigen-presenting cells can selectively propagate and thus retrieve CAR+ T cells suitable for human application. T cells can also be genetically modified using these technologies to target a panel (several) cancer antigen targets. We are on the verge of implementing technology to manufacture “minimally-manipulated” T cells within days of gene transfer by electroporation.
We expect this platform will rapidly integrate with Intrexon’s RTS® and multigenic control gene programs. The programs are also designed and built for rapid transition to universal donor products or minimally manipulated point of care products. Dr. Laurence Cooper and colleagues at MD Anderson recently published research which
demonstrated that transformed, primary, and pluripotent stem cells can be permanently modified to eliminate HLA-A expression, demonstrating how to generate a priori cells from one allogeneic donor for infusion into multiple recipients representing a significant step towards our goal of on-demand therapy that can be pre-deployed at multiple sites and infused when needed. The primary factor limiting the development of a universal donor product is the existence of graft-vs-host response, or GVHD. GVHD occurs because the newly transplanted cells regard the recipient’s body as foreign. When this happens, the newly transplanted cells attack the recipient’s body. Additional research from Dr. Cooper and colleagues at MD Anderson suggests that “universal” allogeneic T cells generated from one donor could be administered to multiple recipients. This is achieved by genetically editing CD19-specific CAR+ T cells to eliminate expression of the endogenous ab TCR, the gene responsible for triggering GVHD, without compromising CAR-dependent effector functions. Genetically modified T cells are generated using the SB system to stably introduce the CD19-specific CAR with subsequent permanent deletion of a or b TCR chains with nucleases. The translation of the SB system and AaPC for use in clinical trials highlights how a nimble and cost-effective approach to developing genetically modified T cells can be used to implement clinical trials infusing next-generation T cells with improved therapeutic potential. We are expanding our initial trials targeting CD19 and planning to conduct additional trials with re-designed CAR-Ts expanding beyond CD19+ tumor cells.
Anticipated Milestones
We expect the following milestones to occur in 2015 and 2016:
| • | Intra-tumoral IL-12 RheoSwitch® programs: |
| • | Early data is expected in the fourth quarter of 2015 for our Phase 1/2 study in Breast Cancer with standard of care. |
| • | Early data is expected in the fourth quarter of 2015 for our Phase 1 study of Glioblastoma multiforme (GBM). |
| • | CAR-T programs: |
| • | We expect to initiate two Phase 1 studies of next-generation CD19 CARs in the second quarter of 2015. |
| • | We expect to initiate a Phase 1 study of next-generation CAR with an inducible cytokine in the fourth quarter of 2015. |
| • | We expect to initiate a novel CAR for myeloid malignancies in the fourth quarter of 2015. |
| • | We expect to receive interim data on two Phase 1 CARs studies in advanced leukemia and lymphomas in the fourth quarter of 2015. |
| • | We expect to initiate other leukemia and solid tumor CAR-T cell studies in 2016. |
| • | We expect to initiate allogeneic, off-the-shelf T-cell studies in 2016. |
Data from all programs is expected in 2015 and 2016. We are also evaluating additional potential preclinical candidates and continuing discovery efforts aimed at identifying other potential product candidates under our Channel Agreement with Intrexon. In addition, we may seek to enhance our pipeline in synthetic biology through focused strategic transactions, which may include acquisitions, partnerships and in-licensing activities. We are actively seeking to out-license some or all of our small molecule programs to further support our synthetic biology efforts.
Small molecule programs
In addition to our synthetic biology programs discussed above, we have certain rights to three small molecule programs, palifosfamide (or isophosphoramide mustard), darinaparsin and indibulin, all of which we are no longer actively pursuing. With respect to palifosfamide, in March 2013, we announced that the pivotal Phase 3 study, PICASSO 3, did not meet its primary endpoint of progression-free survival, and that we would terminate our development program in metastatic soft tissue sarcoma. In addition, we recently received the overall survival endpoint data from our study of palifosfamide in combination with carboplatin and etoposide chemotherapy versus carboplatin and etoposide alone in chemotherapy naïve patients with metastatic small cell lung cancer, which we refer to as MATISSE, which data will be submitted for presentation at a scientific forum during the first half of 2015.
We are seeking transactions with third parties for the possible out-license of palifosfamide. With respect to darinaparsin, we have entered into an amended and restated global licensing agreement with Solasia Pharma K.K., or Solasia, on July 31, 2014 granting Solasia an exclusive worldwide license to develop and commercialize darinaparsin, and related organoarsenic molecules, in both intravenous and oral forms in all indications for human use. In exchange, we will be eligible to receive from Solasia development-and sales-based milestones, a royalty on net sales of darinaparsin, once commercialized, and a percentage of any sublicense revenues generated by Solasia. During 2014, we determined to no longer pursue clinical development of indibulin.
Recent developments
Expected cash as of December 31, 2014
Based upon preliminary estimates, we expect to have approximately $43 million in cash and cash equivalents as of December 31, 2014. We have not yet completed our year-end financial close process for the year ended December 31, 2014. This estimate of our cash and cash equivalents as of December 31, 2014 is based on preliminary estimates of our financial results that we expect to report for the period. These estimates are subject to completion of our financial closing procedures. Our independent registered public accounting firm, McGladrey LLP, has not audited, reviewed or compiled these estimates. These estimates are not a comprehensive statement of our financial results for the year ended December 31, 2014 and our actual results may differ materially from these estimates as a result of the completion of our financial closing procedures, final adjustments and other developments arising between now and the time that our financial results for this period are finalized.
Development plans
Our current plan is to raise additional capital to support further development activities for our strategic product candidates. Based upon our current plans and without taking into account the net proceeds of this offering, we anticipate that our cash resources will be sufficient to fund our operations into the third quarter of 2015. This forecast of cash resources is forward-looking information that involves risks and uncertainties, and the actual amount of our expenses could vary materially and adversely as a result of a number of factors, including the factors discussed in the “Risk Factors” section of this prospectus supplement and the uncertainties applicable to our forecast for the overall sufficiency of our capital resources. We have based our estimates on assumptions that may prove to be wrong, and our expenses could prove to be significantly higher than we currently anticipate. In particular, pursuant to the MD Anderson License, MD Anderson agreed to transfer to us certain existing research programs described in the MD Anderson License and we, together with Intrexon, agreed to enter into a research and development agreement pursuant to which we will provide funding for certain research and development activities of MD Anderson for a period of three years from the date of the MD Anderson License, in an amount between $15 and $20 million per year. In addition, we also expect to enter into additional collaboration and technology transfer agreements with MD Anderson and Intrexon to accelerate technology and clinical development of these product candidates. We expect to increase the level of our overall research and development expenses significantly going forward as a result of each of these items. Further, in light of our entry into the MD Anderson License, we expect to establish operations in Houston, Texas that will enable us to join and collaborate with the MD Anderson academic and medical community, which may require that we add headcount in the future, and which could add to our general and administrative expenses going forward. Although our forecasts for expenses and the sufficiency of our capital resources takes into account our plans to develop the technology licensed from MD Anderson and our obligations under the MD Anderson License, the MD Anderson License was entered into on January 13, 2015 and is only beginning to be implemented, therefore the actual costs associated therewith may be significantly in excess of forecasted amounts.
Risk Factors
Risks related to our business
Our plans to develop and commercialize, through the MD Anderson License with MD Anderson and Intrexon and our Channel Agreement with Intrexon, nonviral adoptive cellular therapies based on designer cytokines and novel chimeric antigen receptor (CAR) T-cell therapies are new approaches to cancer treatment that present significant challenges in a competitive landscape and the success of our efforts depends in large part on our owned and licensed intellectual property, and our efforts may be affected by litigation and developments in intellectual property law outside of our control.
We intend to employ technologies licensed from MD Anderson pursuant to the MD Anderson License described above in “Summary—Recent developments”, and from Intrexon, pursuant to our existing Channel Agreement with Intrexon, to pursue the development and commercialization of nonviral adoptive cellular therapies based on cytokines and CARs under control of the RTS® technology targeting both hematologic and solid tumor malignancies. Because this is a new approach to cancer immunotherapy and cancer treatment generally, developing and commercializing product candidates subjects us to a number of challenges, including:
| • | obtaining regulatory approval from the FDA and other regulatory authorities that have very limited experience with the commercial development of genetically modified T-cell therapies for cancer; |
| • | developing and deploying consistent and reliable processes for engineering a patient’s T-cells ex vivo and infusing the engineered T-cells back into the patient; |
| • | possibly conditioning patients with chemotherapy in conjunction with delivering each of the potential products, which may increase the risk of adverse side effects of the potential products; |
| • | educating medical personnel regarding the potential side effect profile of each of the potential products, such as the potential adverse side effects related to cytokine release; |
| • | developing processes for the safe administration of these potential products, including long-term follow-up for all patients who receive the potential products; |
| • | sourcing additional clinical and, if approved, commercial supplies for the materials used to manufacture and process the potential products; |
| • | developing a manufacturing process and distribution network with a cost of goods that allows for an attractive return on investment; |
| • | establishing sales and marketing capabilities after obtaining any regulatory approval to gain market acceptance; |
| • | developing therapies for types of cancers beyond those addressed by the current potential products; and |
| • | not infringing the intellectual property rights, in particular, the patent rights, of third parties, including competitors developing alternative CAR-T cell therapies. |
We cannot be sure that T-cell immunotherapy technologies that we intend to develop in partnership with MD Anderson and Intrexon will yield satisfactory products that are safe and effective, scalable, or profitable. Moreover, public perception of therapy safety issues, including adoption of new therapeutics or novel approaches to treatment, may adversely influence the willingness of subjects to participate in clinical trials, or if approved, of physicians to subscribe to the novel treatment mechanics. Physicians, hospitals and third-party payors often are slow to adopt new products, technologies and treatment practices that require additional upfront costs and training. Physicians may not be willing to undergo training to adopt this novel and personalized therapy, may decide the therapy is too complex to adopt without appropriate training and may choose not to administer the therapy. Based on these and other factors, hospitals and payors may decide that the benefits of this new therapy do not or will not outweigh its costs.
Our CAR-T product candidates are supported by limited clinical data, all of which has been generated through trials conducted by MD Anderson, not by us. We plan to assume control of the overall clinical and regulatory development of our CAR-T product candidates, and any failure to obtain, or delays in obtaining, sponsorship of investigational new drug applications, or INDs, or in filing new INDs sponsored by us for these or any other product candidates we determine to advance could negatively affect the timing of our potential future clinical trials. Such an impact on timing could increase research and development costs and could delay or prevent obtaining regulatory approval for our product candidates, either of which could have a material adverse effect on our business. Further, we did not control the design or conduct of the previous trials. It is possible that the FDA will not accept these previous trials as providing adequate support for future clinical trials, whether controlled by us or third parties, for any of one or more reasons, including the safety, purity, and potency of the product candidate, the degree of product characterization, elements of the design or execution of the previous trials or safety concerns, or other trial results. We may also be subject to liabilities arising from any treatment-related injuries or adverse effects in patients enrolled in these previous trials. As a result, we may be subject to unforeseen third-party claims and delays in our potential future clinical trials. We may also be required to repeat in whole or in part clinical trials previously conducted by MD Anderson, which will be expensive and delay the submission and licensure or other regulatory approvals with respect to any of our product candidates.
In addition, the results of the limited clinical trials conduct by MD Anderson to date may not be replicated in future clinical trials. Our CAR-T product candidates may fail to show the desired safety and efficacy in clinical development and we cannot assure you that the results of any future trials will demonstrate the value and efficacy of our product candidates. Moreover, there are a number of regulatory requirements that we must satisfy before we can continue clinical trials of CAR-T or other cellular therapy product candidates in the United States. Satisfaction of these requirements will entail substantial time, effort and financial resources. Any time, effort and financial resources we expend on our CAR-T and other early-stage product candidate development programs may adversely affect our ability to continue development and commercialization of our synthetic biology product candidates. Moreover, public perception of therapy safety issues, including adoption of new therapeutics or novel approaches to treatment, may adversely influence the willingness of subjects to participate in clinical trials, or if approved, of physicians to subscribe to the novel treatment mechanics. Physicians, hospitals and third-party payors often are slow to adopt new products, technologies and treatment practices that require additional upfront costs and training. Physicians may not be willing to undergo training to adopt this novel and personalized therapy, may decide the therapy is too complex to adopt without appropriate training and may choose not to administer the therapy. Based on these and other factors, hospitals and payors may decide that the benefits of this new therapy do not or will not outweigh its costs.
The biopharmaceutical industry, and the rapidly evolving market for developing genetically engineered T-cells in particular, is characterized by intense competition and rapid innovation. Genetically engineering T-cells faces significant competition in the CAR technology space from multiple companies and their collaborators, such as Novartis/University of Pennsylvania, Bluebird bio/Celgene/Baylor College of Medicine, Kite Pharma/National Cancer Institute, Juno Therapeutics/Fred Hutchinson Cancer Research Center/Memorial Sloan-Kettering Cancer Center/Seattle Children’s Research Institute, Cellectis/Pfizer and Adaptimmune/GSK. We face competition from non-cell based treatments offered by other companies such as Amgen, AstraZeneca, Bristol-Myers, Incyte, Merck, and Roche. Even if we obtain regulatory approval of potential products, we may not be the first to market and that may affect the price or demand for our potential products. Additionally, the availability and price of our competitors’ products could limit the demand and the price we are able to charge for our potential products. We may not be able to implement our business plan if the acceptance of our potential products is inhibited by price competition or the reluctance of physicians to switch from existing methods of treatment to our potential products, or if physicians switch to other new drug or biologic products or choose to reserve our potential products. Additionally, a competitor could obtain orphan product exclusivity from the FDA with respect to such competitor’s product. If such competitor product is determined to be the same product as one of our potential products, that may prevent us from obtaining approval from the FDA for such potential products for the same indication for seven years, except in limited circumstances.
We are dependent on patents, know-how, and proprietary technology that are licensed from others, particularly MD Anderson and Intrexon. Any termination of these licenses could result in the loss of significant rights and could harm our ability to commercialize our product candidates. Disputes may also arise between us and these licensors regarding intellectual property subject to a license agreement, including those relating to:
| • | the scope of rights granted under the applicable license agreement and other interpretation-related issues; |
| • | whether and the extent to which our technology and processes, and the technology and processes of Intrexon, MD Anderson and our other licensors, infringe on intellectual property of the licensor that is not subject to the applicable license agreement; |
| • | our right to sublicense patent and other rights to third parties pursuant to our relationships with our licensors and partners; |
| • | whether we and/or Intrexon are complying with our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our potential products under the MD Anderson License; and |
| • | the allocation of ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and by us. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements, particularly with MD Anderson, on acceptable terms, we may be unable to successfully develop and commercialize the affected potential products. We are generally also subject to all of the same risks with respect to protection of intellectual property that we license as we are for intellectual property that we own. If we or our licensors fail to adequately protect this intellectual property, our ability to commercialize potential products under our applicable licenses could suffer.
There is a substantial amount of litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference, derivation, and reexamination proceedings before the United States Patent and Trademark Office, or U.S. PTO, or oppositions and other comparable proceedings in foreign jurisdictions. Recently, due to changes in U.S. law referred to as patent reform, new procedures including inter partes review and post-grant review have been implemented, which adds uncertainty to the possibility of challenge to our or our licensors’ patents in the future.
We will require additional financial resources in order to continue ongoing development of our product candidates; if we are unable to obtain these additional resources, we may be forced to delay or discontinue clinical testing of our product candidates.
We have not generated significant revenue and have incurred significant net losses in each year since our inception. For the nine months ended September 30, 2014, we had a net loss of $21.4 million, and, as of September 30, 2014, we had incurred approximately $362.2 million of cumulative net losses since our inception in 2003. We expect to continue to incur significant operating expenditures and net losses. Further development of our product candidates, including product candidates that we may develop under our Channel Agreement with Intrexon and/or pursuant to the MD Anderson License, will likely require substantial increases in our expenses as we:
| • | continue to undertake clinical trials for product candidates; |
| • | scale-up the formulation and manufacturing of our product candidates; |
| • | seek regulatory approvals for product candidates; |
| • | work with regulatory authorities to identify and address program related inquiries; |
| • | implement additional internal systems and infrastructure; |
| • | hire additional personnel; |
| • | begin to advance candidates pursuant to the MD Anderson License; and |
| • | commence providing funding for certain research and development activities of MD Anderson pursuant to the terms of the MD Anderson License. |
We continue to seek additional financial resources to fund the further development of our product candidates. If we are unable to obtain sufficient additional capital, one or more of these programs could be placed on hold. Because we are currently devoting a significant portion of our resources to the development of synthetic biology, further progress with the development of our other candidates may be significantly delayed and may depend on the licensing of those compounds to third parties.
We anticipate that our cash resources, without giving effect to the proceeds of this offering, will be sufficient to fund our operations into the third quarter of 2015, and we have no current committed sources of additional capital. We do not know whether additional financing will be available on terms favorable or acceptable to us when needed, if at all. Our business is highly cash-intensive and our ability to continue operations after our current cash resources are exhausted depends on our ability to obtain additional financing and/or achieve profitable operations, as to which no assurances can be given. If adequate additional funds are not available when required, or if we are unsuccessful in entering into partnership agreements for the further development of our products, we will be required to delay, reduce or eliminate planned preclinical and clinical trials and may be forced to terminate the approval process for our product candidates from the FDA or other regulatory authorities. In addition, we could be forced to discontinue product development, forego attractive business opportunities or pursue merger or divestiture strategies. In the event we are unable to obtain additional financing, we may be forced to cease operations altogether.
We need to raise additional capital to fund our operations. The manner in which we raise any additional funds may affect the value of your investment in our common stock.
We expect to have approximately $43 million of cash and cash equivalents as of December 31, 2014. We anticipate that our cash resources, without giving effect to the proceeds of this offering, will be sufficient to fund our operations into the third quarter of 2015. However, changes may occur that would consume our existing capital prior to the third quarter of 2015, including expansion of the scope of, and/or slower than expected progress of, our research and development efforts and changes in governmental regulation. Actual costs may ultimately vary from our current expectations, which could materially impact our use of capital and our forecast of the period of time through which our financial resources will be adequate to support our operations. We have estimated the sufficiency of our cash resources based in part on the discontinuation of the PICASSO 3 pivotal trial for first-line metastatic STS and our adaptive Phase 3 trial for first-line SCLC for IV palifosfamide. Also our estimates include the advancement of our synthetic biology product candidates in the clinic under our Channel Agreement with Intrexon and our increased expenses as we begin to advance candidates pursuant to the MD Anderson License and commence providing funding for certain research and development activities of MD Anderson pursuant to the terms of the MD Anderson License, and we expect that the costs associated with these and additional product candidates will increase the level of our overall research and development expenses significantly going forward.
In addition to above factors, our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, changes in the focus and direction of our development programs, competitive and technical advances, costs associated with the development of our product candidates, our ability to secure partnering arrangements, and costs of filing, prosecuting, defending and enforcing our intellectual property rights. If we exhaust our capital reserves
more quickly than anticipated, regardless of the reason, and we are unable to obtain additional financing on terms acceptable to us or at all, we will be unable to proceed with development of some or all of our product candidates on expected timelines and will be forced to prioritize among them.
The unpredictability of the capital markets may severely hinder our ability to raise capital within the time periods needed or on terms we consider acceptable, if at all. Moreover, if we fail to advance one or more of our current product candidates to later-stage clinical trials, successfully commercialize one or more of our product candidates, or acquire new product candidates for development, we may have difficulty attracting investors that might otherwise be a source of additional financing.
Our need for additional capital and limited capital resources may force us to accept financing terms that could be significantly dilutive to existing stockholders. To the extent that we raise additional capital by issuing equity securities, our stockholders may experience dilution. In addition, we may grant future investors rights superior to those of our existing stockholders. If we raise additional funds through collaborations and licensing arrangements, it may be necessary to relinquish some rights to our technologies, product candidates or products, or grant licenses on terms that are not favorable to us. If we raise additional funds by incurring debt, we could incur significant interest expense and become subject to covenants in the related transaction documentation that could affect the manner in which we conduct our business.
Clinical trials are very expensive, time-consuming, and difficult to design, initiate and implement.
Human clinical trials are very expensive and difficult to design, initiate and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial start up and process itself is also time-consuming and results are inherently uncertain. We estimate that clinical trials of our product candidates will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to delay the start of, abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
| • | additional nonclinical data requests by regulatory agencies; |
| • | unforeseen safety issues; |
| • | determination of dosing issues; |
| • | lack of effectiveness during clinical trials; |
| • | slower than expected rates of patient recruitment and enrollment; |
| • | inability to monitor patients adequately during or after treatment; |
| • | inability or unwillingness of medical investigators to follow our clinical protocols; and |
| • | regulatory determinations to temporarily or permanently cease enrollment for other reasons not related to patient safety. |
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. For example, despite positive findings in earlier clinical trials, our product candidate palifosfamide failed to meet the primary endpoint of the Phase 3 PICASSO 3 trial. In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our Investigational New Drug, or IND, submission or in the conduct of these trials.
See also “—Risks relating to the clinical testing, regulatory approval and manufacturing of our product candidates—Our product candidates are in various stages of clinical trials, which are very expensive and time-consuming. We cannot be certain when we will be able to submit an NDA or BLA, to the FDA and any failure or delay in completing clinical trials for our product candidates could harm our business.”
Ethical, legal and social concerns about synthetic biologically engineered products could limit or prevent the use of our product candidates.
Our products candidates use a synthetic biology platform. Public perception about the safety and environmental hazards of, and ethical concerns over, genetically engineered products could influence public acceptance of our product candidates. If we and our collaborators are not able to overcome the ethical, legal and social concerns relating to synthetic biological engineering, our product candidates may not be accepted. These concerns could result in increased expenses, regulatory scrutiny, delays or other impediments to the public acceptance and commercialization of our product candidates. Our ability to develop and commercialize products could be limited by public attitudes and governmental regulation.
The subject of genetically modified organisms has received negative publicity, which has aroused public debate. This adverse publicity could lead to greater regulation and trade restrictions on the development and commercialization of genetically altered products. Further, there is a risk that our product candidates could cause adverse health effects or other adverse events, which could also lead to negative publicity.
The synthetic biological platform that we use may have significantly enhanced characteristics compared to those found in naturally occurring organisms, enzymes or microbes. While we believe we produce synthetic biological technologies only for use in a controlled laboratory and industrial environment, the release of such synthetic biological technologies into uncontrolled environments could have unintended consequences. Any adverse effect resulting from such a release could have a material adverse effect on our business and financial condition, and we may have exposure to liability for any resulting harm.
The technology on which our Channel Agreement with Intrexon Corporation is based in part on early stage technology in the field of human oncology therapeutics.
Our Channel Agreement with Intrexon contemplates our using Intrexon’s advanced transgene engineering platform for the controlled and precise cellular production of anti-cancer effectors. The synthetic biology effector platform in which we have acquired rights represents early-stage technology in the field of human oncology biotherapeutic, with Ad-RTS-IL-12 + veledimex having completed two Phase 2 studies, in melanoma and breast cancer. The Company is continuing to pursue intratumoral injection of Ad-RTS-IL-12 + veledimex in brain cancer and breast cancer. Although we plan to leverage Intrexon’s synthetic biology platform for additional products targeting key pathways used by cancers to grow and metastasize, we may not be successful in developing and commercializing these products for a variety of reasons. The risk factors set forth herein that apply to our small molecule drug candidates, which are in various stages of development, also apply to product candidates that we seek to develop under our Channel Agreement with Intrexon.
We will incur additional expenses in connection with our Channel Agreement with Intrexon Corporation.
The synthetic biology platform, in which we have acquired rights for cancer indications from Intrexon, includes two existing product candidates, Ad-RTS-IL-12+ veledimex and DC-RTS-IL-12 + veledimex. Upon entry into the Channel Agreement with Intrexon, we assumed responsibility for the clinical development of these product candidates, which we expect will increase the level of our overall research and development expenses significantly going forward. Although all human clinical trials are expensive and difficult to design and implement, we believe that due to complexity, costs associated with clinical trials for synthetic biology products are greater than the corresponding costs associated with clinical trials for small molecule candidates. In addition to increased research and development costs, prior to the adoption of our April 2013 workforce reduction plan, we added headcount in part to support our Channel Agreement endeavors, and we may need to do so again in the future which would add to our general and administrative expenses going forward.
Although our forecasts for expenses and the sufficiency of our capital resources takes into account our plans to develop the Intrexon products, the actual costs associated therewith may be significantly in excess of forecasted amounts. In addition to the amount and timing of expenses related to the clinical trials, our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, changes in the focus and direction of our development programs, competitive and technical advances, costs associated with the development of our product candidates and costs of filing, prosecuting, defending and enforcing our intellectual property rights. If we exhaust our capital reserves more quickly than anticipated, regardless of the reason, and we are unable to obtain additional financing on terms acceptable to us or at all, we will be unable to proceed with development of some or all of our product candidates on expected timelines and will be forced to prioritize among them.
We may not be able to retain the exclusive rights licensed to us by Intrexon Corporation to develop and commercialize products involving DNA administered to humans for expression of anti-cancer effectors for the purpose of treatment or prophylaxis of cancer.
Under the Channel Agreement, we use Intrexon’s technology directed towards in vivo expression of effectors in connection with the development of Ad-RTS-IL-12+ veledimex and generally to research, develop and commercialize products, in each case in which DNA is administered to humans for expression of anti-cancer effectors for the purpose of treatment or prophylaxis of cancer, which we collectively refer to as the Cancer Program. The Channel Agreement grants us a worldwide license to use patents and other intellectual property of Intrexon in connection with the research, development, use, importing, manufacture, sale, and offer for sale of products involving DNA administered to humans for expression of anti-cancer effectors for the purpose of treatment or prophylaxis of cancer, which we refer to collectively as the ZIOPHARM Products. Such license is exclusive with respect to any clinical development, selling, offering for sale or other commercialization of ZIOPHARM Products, and otherwise is non-exclusive. Subject to limited exceptions, we may not sublicense the rights described without Intrexon’s written consent. Under the Channel Agreement, and subject to certain exceptions, we are responsible for, among other things, the performance of the Cancer Program, including development, commercialization and certain aspects of manufacturing of ZIOPHARM Products.
Intrexon may terminate the Channel Agreement if we fail to use diligent efforts to develop and commercialize ZIOPHARM Products or if we elect not to pursue the development of a Cancer Program identified by Intrexon that is a “Superior Therapy” as defined in the Channel Agreement. We may voluntarily terminate the Channel Agreement upon 90 days written notice to Intrexon. Upon termination of the Channel Agreement, we may continue to develop and commercialize any ZIOPHARM Product that, at the time of termination:
| • | is being commercialized by us; |
| • | has received regulatory approval; |
| • | is a subject of an application for regulatory approval that is pending before the applicable regulatory authority; or |
| • | is the subject of at least an ongoing Phase 2 clinical trial (in the case of a termination by Intrexon due to an uncured breach or a voluntary termination by us), or an ongoing Phase 1 clinical trial in the field (in the case of a termination by us due to an uncured breach or a termination by Intrexon following an unconsented assignment by us or our election not to pursue development of a Superior Therapy). |
Our obligation to pay 50% of net profits or revenue as described further in our Annual Report on Form 10-K under the heading “Business—License Agreements, Intellectual Property and Other Agreements—Exclusive Channel Partner Agreement with Intrexon Corporation” with respect to these “retained” products will survive termination of the Channel Agreement.
There can be no assurance that we will be able to successfully perform under the Channel Agreement and if the Channel Agreement is terminated it may prevent us from achieving our business objectives.
We will incur additional expenses in connection with our License Agreement with The University of Texas M.D. Anderson Cancer Center
Pursuant to the MD Anderson License, we, together with Intrexon, obtained an exclusive, worldwide license to certain technologies owned and licensed by MD Anderson including technologies relating to novel CAR-T cell, T-NK and TCR cell therapies arising from the laboratory of Laurence Cooper, M.D., Ph.D., professor of pediatrics at MD Anderson, as well as either co-exclusive or non-exclusive licenses under certain related technologies. Pursuant to the MD Anderson License, MD Anderson agreed to transfer to us certain existing research programs described in the MD Anderson License and we, together with Intrexon, agreed to enter into a research and development agreement
pursuant to which we will provide funding for certain research and development activities of MD Anderson for a period of three years from the date of the MD Anderson License, in an amount between $15 and $20 million per year. In addition, we also expect to enter into additional collaboration and technology transfer agreements with MD Anderson and Intrexon to accelerate technology and clinical development of these product candidates. We expect to increase the level of our overall research and development expenses significantly going forward as a result of each of these items.
In addition, in light of our entry into the MD Anderson License, we expect to build a base of operations in Houston, Texas to join and collaborate with the MD Anderson academic and medical community, which may require that we add headcount in the future, and which could add to our general and administrative expenses going forward.
Although our forecasts for expenses and the sufficiency of our capital resources takes into account our plans to develop the technology licensed from MD Anderson and our obligations under the MD Anderson License, the MD Anderson License was entered into on January 13, 2015 and is only beginning to be implemented, therefore the actual costs associated therewith may be significantly in excess of forecasted amounts. In addition to the amount and timing of expenses related to our relationship with MD Anderson, our actual cash requirements may vary materially from our current expectations for a number of other factors that may include, but are not limited to, the final terms and conditions of the research and development agreement contemplated by the MD Anderson License, costs associated with opening a new operational facility in Houston, Texas, changes in the focus and direction of our development programs, competitive and technical advances, costs associated with the development of our product candidates and costs of filing, prosecuting, defending and enforcing our intellectual property rights. If we exhaust our capital reserves more quickly than anticipated, regardless of the reason, and we are unable to obtain additional financing on terms acceptable to us or at all, we will be unable to proceed with development of some or all of our product candidates on expected timelines and will be forced to prioritize among them.
We may not be able to retain the rights licensed to us and Intrexon by The University of Texas M.D. Anderson Cancer Center to technologies relating to novel chimeric antigen receptor (CAR) T-cell therapies and other related technologies.
Under the MD Anderson License, we, together with Intrexon, received an exclusive, worldwide license to certain technologies owned and licensed by MD Anderson including technologies relating to novel CAR-T cell, T-NK and TCR cell therapies arising from the laboratory of Laurence Cooper, M.D., Ph.D., professor of pediatrics at MD Anderson, as well as either co-exclusive or non-exclusive licenses under certain related technologies. When combined with Intrexon’s technology suite and ZIOPHARM’s clinically tested RheoSwitch Therapeutic System® interleukin-12 modules, the resulting proprietary methods and technologies may help realize the promise of genetically modified CAR-T cell and other immune cells by tightly controlling cell expansion and activation in the body, minimizing off-target and unwanted on-target effects and toxicity while maximizing therapeutic efficacy. The term of the MD Anderson License expires on the last to occur of (a) the expiration of all patents licensed thereunder, or (b) the twentieth anniversary of the date of the MD Anderson License; provided, however, that following the expiration of the term, the Company and Intrexon shall then have a fully-paid up, royalty free, perpetual, irrevocable and sublicensable license to use the licensed intellectual property thereunder.
After 10 years from the date of the MD Anderson License and subject to a 90 day cure period, MD Anderson will have the right to convert the MD Anderson License into a non-exclusive license if we and Intrexon are not using commercially reasonable efforts to commercialize the licensed intellectual property on a case-by-case basis. After five years from the date of the MD Anderson License and subject to a 180-day cure period, MD Anderson will have the right to terminate the MD Anderson License with respect to specific technology(ies) funded by the government or subject to a third party contract if we and Intrexon are not meeting the diligence requirements in such funding agreement or contract, as applicable. Subject to a 30-day cure period, MD Anderson has the right to terminate the MD Anderson License if we and Intrexon fail to timely deliver the shares due in consideration for the MD Anderson License. MD Anderson may also terminate the agreement with written notice upon material breach by us or Intrexon, if such breach has not been cured within 60 days of receiving such notice. In addition, the MD Anderson License will terminate upon the occurrence of certain insolvency events for both us or Intrexon and may be terminated by the mutual written agreement of us, Intrexon and MD Anderson.
There can be no assurance that we will be able to successfully perform under the MD Anderson License and if the MD Anderson License is terminated it may prevent us from achieving our business objectives.
We have a limited operating history upon which to base an investment decision.
We have not demonstrated an ability to perform the functions necessary for the successful commercialization of any product candidates. The successful commercialization of any product candidates will require us to perform a variety of functions, including:
| • | continuing to undertake preclinical development and clinical trials; |
| • | participating in regulatory approval processes; |
| • | formulating and manufacturing products; and |
| • | conducting sales and marketing activities. |
Our operations have been limited to organizing and staffing our company, acquiring, developing and securing our proprietary product candidates, and undertaking preclinical and clinical trials of our product candidates. These operations provide a limited basis for you to assess our ability to commercialize our product candidates and the advisability of investing in our securities.
We rely on key executive officers and scientific and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace.
We are highly dependent on Dr. Jonathan Lewis, our Chief Executive Officer, Caesar J. Belbel, our Executive Vice President and Chief Legal Officer and our principal scientific, regulatory, and medical advisors. Mr. Belbel’s employment is governed by a written employment agreement, while our previously effective written employment agreement with Dr. Lewis expired in accordance with its terms in January 2015. Dr. Lewis and Mr. Belbel may terminate their employment with us at any time, subject, however, to certain non-compete and non-solicitation covenants which, in Dr. Lewis’s case, survive the expiration of his written employment agreement in certain circumstances. The loss of the technical knowledge and management and industry expertise of Dr. Lewis and Mr. Belbel, or any of our other key personnel, could result in delays in product development, loss of customers and sales, and diversion of management resources, which could adversely affect our operating results. We do not carry “key person” life insurance policies on any of our officers or key employees.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products, if approved. Even a successful defense would require significant financial and management resources. Regardless of the merit or eventual outcome, liability claims may result in:
| • | decreased demand for our product candidates; |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants; |
| • | withdrawal of prior governmental approvals; |
| • | costs of related litigation; |
| • | substantial monetary awards to patients; |
| • | product recalls; |
| • | loss of revenue; and |
| • | the inability to commercialize our product candidates. |
We currently carry clinical trial insurance and product liability insurance. However, an inability to renew our policies or to obtain sufficient insurance at an acceptable cost could prevent or inhibit the commercialization of pharmaceutical products that we develop, alone or with collaborators.
Risks related to the clinical testing, regulatory approval and manufacturing of our product candidates
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any potential marketing approval.
As with many pharmaceutical and biological products, treatment with our product candidates may produce undesirable side effects or adverse reactions or events. If our product candidates or similar products or product candidates under development by third parties demonstrate unacceptable adverse events, we may be required to halt or delay further clinical development of our product candidates. The FDA, the EMA or other foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The product-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Our synthetic biology product candidates are based on a novel technology, which makes it difficult to predict the time and cost of product candidate development and subsequently obtaining regulatory approval. Currently, no gene therapy products have been approved in the United States and only one product has been approved in Europe.
We have recently focused our product research and development efforts on our synthetic biology product candidates under our Channel Agreement with Intrexon. These products, including Ad-RTS-IL-12+ veledimex, are based on gene therapy technology. Due to the novelty of this medical technology, there can be no assurance that any development problems we experience in the future related to our synthetic biology platform will not cause significant delays or unanticipated costs, or that such development problems can be solved. We may also experience unanticipated problems or delays in expanding our manufacturing capacity or transferring our manufacturing process to commercial partners, which may prevent us from completing our clinical studies or commercializing our synthetic biology product candidates on a timely or profitable basis, if at all.
In addition, the clinical study requirements of the FDA, the EMA and other regulatory agencies and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential products. The regulatory approval process for novel product candidates such as ours can be more expensive and take longer than for other, better known or extensively studied pharmaceutical or other product candidates. Currently, only one gene therapy product, UniQure’s Glybera, which received marketing authorization from the EMA in 2012, has been approved in Europe but has not yet been launched for commercial sale, which makes it difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates in either the United States or Europe. Approvals by the EMA may not be indicative of what the FDA may require for approval.
Regulatory requirements governing gene and cell therapy products have changed frequently and may continue to change in the future. For example, the FDA has established the Office of Cellular, Tissue and Gene Therapies within its Center for Biologics Evaluation and Research, or CBER, to consolidate the review of gene therapy and related products, and the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its review. Gene therapy clinical studies conducted at institutions that receive funding for recombinant DNA research from the U.S. National Institutes of Health, or the NIH, are also subject to review by the NIH Office of Biotechnology Activities’ Recombinant DNA Advisory Committee, or the RAC. Although the FDA decides whether individual gene therapy protocols may proceed, the RAC review process can impede the initiation of a clinical trial, even if the FDA has reviewed the trial and approved its initiation. Conversely, the FDA can put an IND on clinical hold even if the RAC has provided a favorable review. For example, our planned Phase 1 clinical trial of Ad-RTS-IL-12 +
veledimex, in subjects with recurrent or progressive high grade gliomas (brain cancer) received approval from the NIH RAC in December 2013, but the FDA requested additional nonclinical information prior to permitting clinical study initiation, which we are currently generating. Also, before a clinical trial can begin at an NIH-funded institution, that institution’s institutional review board, or IRB, and its Institutional Biosafety Committee will have to review the proposed clinical trial to assess the safety of the trial. In addition, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA or other regulatory bodies to change the requirements for approval of any of our product candidates.
These regulatory review committees and advisory groups and the new guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these treatment candidates or lead to significant post-approval limitations or restrictions. As we advance our synthetic biology product candidates, we will be required to consult with these regulatory and advisory groups, and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of our product candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected for oncology product candidates. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue to maintain our business.
Our reliance on third parties to formulate and manufacture our product candidates exposes us to a number of risks that may delay the development, regulatory approval and commercialization of our products or result in higher product costs.
We do not have experience in drug formulation or manufacturing of drugs or biologics and do not intend to establish our own manufacturing facilities. Although we will work closely with and rely upon Intrexon on the manufacturing and scale-up of Intrexon product candidates, we lack the resources and expertise to formulate or manufacture our own product candidates. We currently are contracting for the manufacture of our product candidates. We intend to contract with one or more manufacturers to manufacture, supply, store, and distribute drug supplies for our clinical trials. If a product candidate we develop or acquire in the future receives FDA approval, we will rely on one or more third-party contractors or Intrexon to manufacture our products. Our anticipated future reliance on a limited number of third-party manufacturers exposes us to the following risks:
| • | We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any. |
| • | Our third-party manufacturers might be unable to formulate and manufacture our products in the volume and of the quality required to meet our clinical needs and commercial needs, if any. |
| • | Our future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store, and distribute our products. |
| • | Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Administration and corresponding state and foreign agencies to ensure strict compliance with current good manufacturing practices, or cGMP, and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards. |
| • | If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation. |
| • | Our third-party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party |
| manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of drug candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products. |
Each of these risks could delay our clinical trials, the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates or result in higher costs or deprive us of potential product revenues.
Any drug candidate for which we obtain marketing approval could be subject to post-marketing restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products, when and if any of them are approved.
Any drug candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include, among other things, submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a drug candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, including the requirement to implement a risk evaluation and mitigation strategy, or REMS, which could include requirements for a restricted distribution system. If any of our drug candidates receives marketing approval, the accompanying label may limit the approved use of our drug, which could limit sales of the product.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of our approved products. The FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use and if we market our products outside of their approved indications, we may be subject to enforcement action for off-label marketing. Violations of the FDCA relating to the promotion of prescription drugs may lead to investigations alleging violations of federal and state health care fraud and abuse laws, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
| • | litigation involving patients taking our drug; |
| • | restrictions on such products, manufacturers or manufacturing processes; |
| • | restrictions on the labeling or marketing of a product; |
| • | restrictions on product distribution or use; |
| • | requirements to conduct post-marketing studies or clinical trials; |
| • | warning letters; |
| • | withdrawal of the products from the market; |
| • | refusal to approve pending applications or supplements to approved applications that we submit; |
| • | recall of products; |
| • | fines, restitution or disgorgement of profits or revenues; |
| • | suspension or withdrawal of marketing approvals; |
| • | damage to relationships with existing and potential collaborators; |
| • | unfavorable press coverage and damage to our reputation; |
| • | refusal to permit the import or export of our products; |
| • | product seizure; or |
| • | injunctions or the imposition of civil or criminal penalties. |
Noncompliance with similar European Union requirements regarding safety monitoring or pharmacovigilance can also result in significant financial penalties. Similarly, failure to comply with U.S. and foreign regulatory requirements regarding the development of products for pediatric populations and the protection of personal health information can also lead to significant penalties and sanctions.
Risks related to our ability to commercialize our product candidates
If physicians and patients do not accept and use our product candidates, our ability to generate revenue from sales of our products will be materially impaired.
Even if the FDA and/or foreign equivalents thereof approve our product candidates, physicians and patients may not accept and use them. Acceptance and use of our products will depend upon a number of factors including:
| • | perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our drugs; |
| • | pharmacological benefit and cost-effectiveness of our products relative to competing products; |
| • | availability of coverage and adequate reimbursement for our products from government or other healthcare payors; |
| • | effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any; and |
| • | the price at which we sell our products. |
Because we expect sales of our current product candidates, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of a drug to find market acceptance would harm our business and could require us to seek additional financing in order to fund the development of future product candidates.
Our ability to generate product revenues will be diminished if our drugs do not obtain coverage or adequate reimbursement from payors.
Our ability to commercialize our drugs, alone or with collaborators, will depend in part on the extent to which coverage and reimbursement will be available from government authorities, private managed care organizations and health insurers and other third-party payors.
Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Even if we obtain coverage for our product candidates, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. Patients are unlikely to use our product candidates unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our product candidates.
In addition, the market for our product candidates for which we may receive regulatory approval will depend significantly on access to third-party payors’ drug formularies, or lists of medications for which third-party payors provide coverage and reimbursement, which might not include all of the FDA-approved
drugs for a particular indication. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payors may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that requires us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that approval will be obtained. If we are unable to obtain coverage of and adequate payment levels for our product candidates from third-party payors, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition, and future success.
In addition, in many foreign countries, particularly the countries of the European Union, the pricing of prescription drugs is subject to government control. In some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. We may face competition for our product candidates from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, there may be importation of foreign products that compete with our own products, which could negatively impact our profitability.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory enactments in recent years that change the healthcare system in ways that could impact our future ability to sell our product candidates profitably.
Furthermore, there have been and continue to be a number of initiatives at the federal and state level that seek to reduce healthcare costs. Most significantly, in March 2010, President Obama signed into law the Patient Protection and Affordable Health Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the PPACA, which includes measures that significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the PPACA of importance to the pharmaceutical industry are the following:
| • | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| • | increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | an extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | new methodologies by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and for drugs that are line extensions; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals with income at or below 133% of the Federal Poverty Level, thereby potentially increasing both the volume of sales and manufacturers’ Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| • | a new requirement to annually report drug samples that certain manufacturers and authorized distributors provide to practitioners; |
| • | expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| • | a licensure framework for follow-on biologic products; |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| • | creation of the Independent Payment Advisory Board which has authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs and those recommendations could have the effect of law even if Congress does not act on the recommendations; and |
| • | establishment of a Center for Medicare & Medicaid Innovation at the Centers for Medicare & Medicaid Services, or CMS, to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. |
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, in August 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee on Deficit Reduction did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions include aggregate reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and, due to subsequent legislative amendments, will stay in effect through 2024 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. The full impact of these new laws, as well as laws and other reform and cost containment measures that may be proposed and adopted in the future, remains uncertain, but may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our future customers and accordingly, our ability to generate revenue, attain profitability, or commercialize our products.
If we fail to comply with federal and state healthcare laws, including fraud and abuse and health information privacy and security laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. For example, we could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include, among others:
| • | the federal Anti-Kickback Statute, which constrains our business activities, including our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities, by prohibiting, among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual or the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| • | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| • | requirements to report annually to CMS certain financial arrangements with physicians and teaching hospitals, as defined in the PPACA and its implementing regulations, including reporting any “transfer of value” made or distributed to teaching hospitals, prescribers, and other healthcare providers and reporting any ownership and investment interests held by physicians and their immediate family members and applicable group purchasing organizations during the preceding calendar year; and |
| • | state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government that otherwise restricts certain payments that may be made to healthcare providers and entities; state laws that require drug manufacturers to report information related to payments and other transfer of value to physicians and other healthcare providers and entities; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent health care reform legislation has further strengthened these laws. For example, the PPACA, among other things, amends the intent requirement of the federal anti-kickback and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. Moreover, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
To the extent that any of our product candidates is ultimately sold in a foreign country, we may be subject to similar foreign laws and regulations.
If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, exclusion from participation in United States federal or state health care programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, any of which could materially adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
Our ability to use net operating loss carryforwards and research tax credits to reduce future tax payments may be limited or restricted.
We have generated significant net operating loss carryforwards, or NOLs, and research and development tax credits, or R&D credits, as a result of our incurrence of losses and our conduct of research activities since inception. We generally are able to carry NOLs and R&D credits forward to reduce our tax liability in future years. However, our ability to utilize the NOLs and R&D credits is subject to the rules of Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the “Code”), respectively. Those sections generally restrict the use of NOLs and R&D credits after an “ownership change.” An ownership change occurs if, among other things, the stockholders (or specified groups of stockholders) who own or have owned, directly or indirectly, 5% or more of a corporation’s common stock or are otherwise treated as 5% stockholders under Section 382 of the Code and the United States Treasury Department regulations promulgated thereunder increase their aggregate percentage ownership of that corporation’s stock by more than 50 percentage points over the lowest percentage of the stock owned by these stockholders over the applicable testing period. In the event of an ownership change, Section 382 imposes an annual limitation on the amount of taxable income a corporation may offset with NOL carry forwards and Section 383 imposes an annual limitation on the amount of tax a corporation may offset with business credit (including the R&D credit) carry forwards. Any unused annual limitation may be carried over to later years until the applicable expiration date for the respective NOL or R&D credit carry forwards. We may have experienced an “ownership change” within the meaning of Section 382 in the past and there can be no assurance that we have not experienced additional ownership changes, or that we will not experience an ownership change as a result of this offering. As a result, our NOLs and business (including the R&D credit) may be subject to limitations and we may be required to pay taxes earlier and in larger amounts than would be the case if our NOLs or R&D credits were freely usable.
Our synthetic biology product candidates may face competition in the future from follow-on biologics.
With the enactment of the Biologics Price Competition and Innovation Act of 2009, or BPCIA, as part of the Patient Protection and Affordable Care Act, an abbreviated pathway for the approval of follow-on biological products was created. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” with an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. This data exclusivity does not prevent another company from developing a product that is highly similar to the original branded product, generating its own data and seeking approval. Data exclusivity only assures that another company cannot rely upon the data within the innovator’s application to support the biosimilar product’s approval.
In his proposed budget for fiscal year 2014, President Obama proposed to cut this 12-year period of exclusivity down to seven years. He also proposed to prohibit additional periods of exclusivity due to minor changes in product formulations, a practice often referred to as “evergreening.” It is possible that Congress may take these or other measures to reduce or eliminate periods of exclusivity.
The new law is complex and is only beginning to be interpreted and implemented by the FDA. As a result, its ultimate impact is subject to uncertainty, and could have a material adverse effect on the future commercial prospects for our biological products. Since enactment of the BCPIA, the FDA has issued several draft guidance documents discussing the biosimilar pathway, but to date, no biosimilar or interchangeable product has been approved. Although it is unclear what final implementation of the BCPIA will entail, such FDA implementation could have a material adverse effect on the future commercial prospects for our product candidates.
Risks related to our intellectual property
If we or our licensors fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish and our ability to successfully commercialize our products may be impaired.
Our success, competitive position, and future revenues will depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights, and to operate without infringing the proprietary rights of third parties.
To date, we have exclusive rights to certain United States and foreign intellectual property with respect to our small molecule product candidates, with respect to the Intrexon technology, including the existing Intrexon product candidates, and with respect to CAR-T cell, NK and TCR cell therapies arising from the laboratory of Laurence Cooper, M.D., Ph.D., professor of pediatrics at MD Anderson. Under our Channel Agreement with Intrexon, Intrexon has the sole right to conduct and control the filings, prosecution and maintenance of the patents and patent applications licensed to us. Although under the agreement Intrexon has agreed to consider in good faith and consult with us regarding any comments we may have regarding these patents and patent applications, we cannot guarantee that our comments will be solicited or followed. Under the MD Anderson License, future filings and applications require the agreement of each of MD Anderson, Intrexon and us, and MD Anderson has the right to control the preparation and filing of additional patent applications unless the parties agree that we or Intrexon may prosecute the application directly. Although under the agreement MD Anderson has agreed to review and incorporate any reasonable comments that we or Intrexon may have regarding these patents and patent applications, we cannot guarantee that our comments will be solicited or followed. Without direct control of the channel program patents and patent applications, we are dependent on Intrexon or MD Anderson, as applicable, to keep us advised of prosecution, particularly in foreign jurisdictions where prosecution information may not be publicly available. We anticipate that we, Intrexon and MD Anderson will file additional patent applications both in the United States and in other countries. However, we cannot predict or guarantee:
| • | the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent our patents; |
| • | if and when patents will be issued; |
| • | whether or not others will obtain patents claiming subject matter related to or relevant to our product candidates; or |
| • | whether we will need to initiate litigation or administrative proceedings that may be costly whether we win or lose. |
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost, in a timely manner, or in all jurisdictions. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from third parties. We may also require the cooperation of our licensors in order to enforce the licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States and we may fail to seek or obtain patent protection in all major markets. For example, European patent law restricts the patentability of methods of treatment of the human body more than United States law does. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all.
Changes in patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. In September 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law, resulting in a number of significant changes to United States patent law. These changes include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. In addition, the United States Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the value of patents, once obtained, and with regard to our ability to obtain patents in the future. As the U.S. PTO continues to implement the Leahy-Smith Act, and as the federal courts have the opportunity to interpret the Leahy-Smith Act, the laws and regulations governing patents, and the rules regarding patent procurement could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Certain technologies utilized in our research and development programs are already in the public domain. Moreover, a number of our competitors have developed technologies, filed patent applications or obtained patents on technologies, compositions and methods of use that are related to our business and may cover or conflict with our owned or licensed patent applications, technologies or product candidates. Such conflicts could limit the scope of the patents that we may be able to obtain or may result in the rejection of claims in our patent applications. Because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that others have not filed or maintained patent applications for technology used by us or covered by our pending patent applications without our being aware of these applications. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned patents or pending patent applications, or that we were the first to file for patent protection of such inventions, nor can we know whether those from whom we license patents were the first to make the inventions claimed or were the first to file. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection. In addition, our own earlier filed patents and applications or those of Intrexon may limit the scope of later patents we obtain or may result in the rejection of claims in our later filed patent applications. If third parties filed patent applications or obtained patents on technologies, compositions and methods of use that are related to our business and that cover or conflict with our owned or licensed patent applications, technologies or product candidates, we may be required to challenge such protection, terminate or modify our programs impacted by such protection or obtain licenses from such third parties, which might not be available on acceptable terms, or at all.
Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
If we breach any of the agreements under which we license rights to products or technology from others, we could lose license rights that are material to our business or be subject to claims by our licensors.
We license rights to products and technology that are important to our business, and we expect to enter into additional licenses in the future. For instance, we have exclusively licensed patents and patent applications under our Channel Agreement with Intrexon and under the MD Anderson License. Under these agreements, we are subject to a range of commercialization and development, sublicensing, royalty, patent prosecution and maintenance, insurance and other obligations.
Any failure by us to comply with any of these obligations or any other breach by us of our license agreements could give the licensor the right to terminate the license in whole, terminate the exclusive nature of the license or bring a claim against us for damages. Any such termination or claim could have a material adverse effect on our financial condition, results of operations, liquidity or business. Even if we contest any such termination or claim and are ultimately successful, such dispute could lead to delays in the development or commercialization of potential products and result in time-consuming and expensive litigation or arbitration. On termination we may be required to license to the licensor any related intellectual property that we developed.
In addition, in certain cases, the rights licensed to us are rights of a third party licensed to our licensor. In such instances, if our licensors do not comply with their obligations under such licenses, our rights under our license agreements with our licensor may be adversely affected.
Other risks related to our company
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Provisions of our amended and restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders. These provisions authorize the issuance of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares and hinder a takeover attempt, and limit who may call a special meeting of stockholders. In addition, Section 203 of the Delaware General Corporation Law generally prohibits a publicly-held Delaware corporation from engaging in a business combination with a party that owns at least 15% of its common stock unless the business combination is approved by the company’s board of directors before the person acquires the 15% ownership stake or later by its board of directors and two-thirds of its stockholders. Section 203 could have the effect of delaying, deferring or preventing a change in control that our stockholders might consider to be in their best interests.
In connection with our January 2011 issuance of shares of common stock to Intrexon in a private placement transaction, our board of directors waived the Section 203 prohibition with respect to a future business combination with Intrexon.
| Item 9.01 | Financial Statements and Exhibits |
| (d) | Exhibits. |
| Exhibit |
Description | |
| 99.1 | Press Release, dated February 2, 2015 | |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| ZIOPHARM Oncology, Inc. | ||||||||
| By: | /s/ Kevin G. Lafond | |||||||
| Date: February 2, 2015 | Name: | Kevin G. Lafond | ||||||
| Title: | Vice President, Chief Accounting Officer and Treasurer | |||||||
INDEX OF EXHIBITS
| Exhibit |
Description | |
| 99.1 | Press Release, dated February 2, 2015 | |