Attached files
| file | filename |
|---|---|
| EX-1.1 - EX-1.1 - Eiger BioPharmaceuticals, Inc. | d762637dex11.htm |
| EX-5.1 - EX-5.1 - Eiger BioPharmaceuticals, Inc. | d762637dex51.htm |
| EX-23.1 - EX-23.1 - Eiger BioPharmaceuticals, Inc. | d762637dex231.htm |
Table of Contents
As filed with the Securities and Exchange Commission on August 7, 2014
Registration No. 333-197720
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1
TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Celladon Corporation
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 2836 | 33-0971591 | ||
| (State or Other Jurisdiction of Incorporation or Organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
11988 El Camino Real, Suite 650
San Diego, California 92130
(858) 366-4288
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant’s Principal Executive Offices)
Paul Cleveland
President and Chief Financial Officer
Celladon Corporation
11988 El Camino Real, Suite 650
San Diego, California 92130
(858) 366-4288
(Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent for Service)
Copies to:
| Jason L. Kent, Esq. Cooley LLP 4401 Eastgate Mall San Diego, California 92121 (858) 550-6000 |
Elizabeth Reed Vice President, General Counsel and Secretary Celladon Corporation 11988 El Camino Real, Suite 650 San Diego, California 92130 (858) 366-4288 |
Cheston J. Larson, Esq. Michael E. Sullivan, Esq. Latham & Watkins LLP 12670 High Bluff Drive San Diego, California 92130 (858) 523-5400 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended (the “Securities Act”), check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | x | |||
CALCULATION OF REGISTRATION FEE
|
| ||||
| Title of each class of securities to be registered |
Proposed maximum aggregate offering price(1) |
Amount of registration fee | ||
| Common Stock, $0.001 par value per share |
$63,250,000 | $8,147(2) | ||
|
| ||||
|
| ||||
| (1) | Estimated solely for the purpose of calculating the amount of the registration fee in accordance with Rule 457(o) under the Securities Act. Includes the offering price of additional shares that the underwriters have the option to purchase. |
| (2) | Previously paid. |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment that specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and we are not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
Preliminary Prospectus
SUBJECT TO COMPLETION, DATED AUGUST 7, 2014
4,000,000 Shares

Common Stock
We are offering 4,000,000 shares of our common stock.
Our common stock is listed on The NASDAQ Global Market under the symbol “CLDN.” The closing price of our common stock on The NASDAQ Global Market on August 6, 2014, was $10.54 per share.
We are an emerging growth company as that term is used in the Jumpstart Our Business Startups Act of 2012 and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
The underwriters have a thirty-day option to purchase a maximum of 600,000 additional shares of our common stock.
Investing in our common stock involves risks. See “Risk Factors” beginning on page 12 of this prospectus.
| Price to Public |
Underwriting Discounts and |
Proceeds to Us, Before | ||||
| Per Share |
$ | $ | $ | |||
| Total |
$ | $ | $ |
| (1) | We have agreed to reimburse the underwriters for certain FINRA-related expenses. See “Underwriting.” |
Delivery of the shares of common stock will be made on or about , 2014.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
Joint Book-Running Managers
| Credit Suisse | Jefferies |
Co-Managers
| Stifel | Wedbush PacGrow Life Sciences |
The date of this prospectus is , 2014
Table of Contents
| Page | ||||
| 1 | ||||
| 12 | ||||
| 14 | ||||
| 16 | ||||
| 17 | ||||
| 17 | ||||
| 18 | ||||
| 19 | ||||
| 22 | ||||
| 24 | ||||
| 70 | ||||
| 79 | ||||
| 85 | ||||
| 89 | ||||
| 94 | ||||
| MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF OUR COMMON STOCK |
96 | |||
| 100 | ||||
| 106 | ||||
| 106 | ||||
| 106 | ||||
| 106 | ||||
Neither we nor any of the underwriters has authorized anyone to provide you with information different from, or in addition to, that contained in or incorporated by reference into this prospectus or any free writing prospectus prepared by or on behalf of us or to which we may have referred you in connection with this offering. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. Neither we nor any of the underwriters is making an offer to sell or seeking offers to buy these securities in any jurisdiction where, or to any person to whom, the offer or sale is not permitted. The information contained in or incorporated by reference into this prospectus is accurate only as of its date, regardless of the time of delivery of this prospectus or of any sale of shares of our common stock, and the information in any free writing prospectus that we may provide you in connection with this offering is accurate only as of the date of that free writing prospectus. Our business, financial condition, results of operations and future growth prospects may have changed since those dates.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data.
For investors outside the United States: neither we nor any of the underwriters have done anything that would permit this offering or possession or distribution of this prospectus or any free writing prospectus we may provide to you in connection with this offering in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about, and to observe any restrictions relating to, this offering and the distribution of this prospectus and any free writing prospectus outside of the United States.
Table of Contents
This summary highlights information contained in other parts of this prospectus. Because it is only a summary, it does not contain all of the information that you should consider before investing in shares of our common stock and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere or incorporated by reference into this prospectus. You should read the entire prospectus and the information incorporated herein carefully, especially “Risk Factors” and our consolidated financial statements and the related notes incorporated by reference into this prospectus, before deciding to buy shares of our common stock. Unless the context requires otherwise, references in this prospectus to “Celladon,” “we,” “us” and “our” refer to Celladon Corporation.
Overview
We are a clinical-stage biotechnology company applying our leadership position in the field of gene therapy and calcium dysregulation to develop novel therapies for diseases with tremendous unmet medical needs. Our lead programs target sarco/endoplasmic reticulum Ca2+ -ATPase, or SERCA, enzymes, which are a family of enzymes that play an integral part in the regulation of intra-cellular calcium in all human cells. Calcium dysregulation is implicated in a number of important and complex medical conditions and diseases, such as heart failure, which is a clinical syndrome characterized by poor heart function resulting in inadequate blood flow to meet the body’s metabolic needs, as well as blood vessel health, diabetes and neurodegenerative diseases. SERCA2a, an enzyme that becomes deficient in patients with heart failure, was scientifically validated as a molecular target for heart failure in the 1990s and became a focus of internal discovery efforts for many large pharmaceutical companies. However, to date, no other company has been successful in targeting SERCA2a using traditional discovery methods.
Our therapeutic portfolio includes both gene therapies and small molecule compounds targeting diseases characterized by SERCA enzyme deficiency. MYDICAR, our most advanced product candidate, uses gene therapy to target SERCA2a. We believe that our gene therapy approach to modulating SERCA2a overcomes the issues encountered by previous efforts and has the potential to provide transformative disease-modifying effects with long-term benefits in patients with heart failure. In addition, we have recently in-licensed worldwide rights to patents covering an additional gene therapy product opportunity, the membrane-bound form of Stem Cell Factor, or mSCF, for the treatment of cardiac ischemic damage. We have also identified a number of potential first-in-class compounds addressing novel targets in diabetes and neurodegenerative diseases with our small molecule platform of SERCA2b modulators.
We are the first company to enter clinical development with a product candidate, MYDICAR, that selectively targets SERCA2a. We refer to our Phase 1 trial and Phase 2a trial of MYDICAR together as our CUPID 1 trial. In Phase 2a of our CUPID 1 trial, 39 patients with systolic heart failure, which is caused by the inability of the heart to pump blood efficiently due to weakening and enlargement of the ventricles, were enrolled in a randomized, double-blind, placebo-controlled trial. MYDICAR was safe and well-tolerated, reduced heart failure-related hospitalizations, improved patients’ symptoms, quality of life and serum biomarkers and improved key markers of cardiac function predictive of survival, such as end systolic volume. Based on these results, as well as our previous preclinical studies and clinical trials, we advanced MYDICAR to a 250-patient randomized, double-blind, placebo-controlled international Phase 2b trial in patients with systolic heart failure, which we refer to as CUPID 2. We completed enrollment of CUPID 2 in February 2014 and expect to announce results in April 2015. If successful, these results, along with other studies, will form the basis for regulatory submissions for approval with the United States Food and Drug Administration, or FDA, and European Medicines Agency, or EMA. In 2012, we obtained a Special Protocol Assessment, or SPA, whereby the FDA agreed to use time-to-multiple heart failure-related hospitalizations as the primary endpoint for a MYDICAR Phase 3 pivotal trial. Our ongoing CUPID 2 trial uses a similar clinical protocol with identical endpoints as agreed to in the SPA. In
1
Table of Contents
November 2013, the EMA indicated that if MYDICAR demonstrates a substantial and highly significant treatment effect in the advanced heart failure population, and no untoward effects attributable to MYDICAR are observed, a safety database of approximately 205 to 230 MYDICAR-treated subjects may be sufficient for a safety assessment to allow for acceptance of a Marketing Authorization Application, or MAA, for MYDICAR for the treatment of systolic heart failure. We therefore believe that, if the above conditions are met, a Phase 3 trial may not be required for marketing approval in Europe.
In April 2014, the FDA’s Center for Biologics Evaluation and Research, or CBER, granted Breakthrough Therapy designation to MYDICAR for reducing hospitalizations for heart failure in patients who test negative for adeno-associated viral vector 1, or AAV1, neutralizing antibodies, are class III or IV heart failure patients under the New York Heart Association, or NYHA, classification system, and are not in immediate need of a left-ventricular assist device, or LVAD, or heart transplant. The Breakthrough Therapy program is intended to expedite drug development and review of innovative new drugs that are intended to treat serious or life-threatening diseases and for which preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on a clinically significant endpoint. MYDICAR was the third product candidate to receive this designation from CBER, and the designation indicates that the FDA has determined that the CUPID 1 trial provided preliminary clinical evidence that MYDICAR may demonstrate substantial improvement over available therapies in heart failure patients for which the designation was granted.
We are initially developing MYDICAR to treat patients with systolic heart failure. We are also developing MYDICAR for additional indications, including arteriovenous fistula, or AVF, maturation failure, and for the treatment of patients with advanced heart failure who are on an LVAD. In addition, we expect to initiate a clinical trial in 2015 for the treatment of diastolic heart failure, a condition caused by the inability of the heart to relax normally between contractions, if data from our preclinical research warrants.
2
Table of Contents
Our Product Pipeline
The following chart depicts key information regarding our development programs, their indications, and their current stage of development:
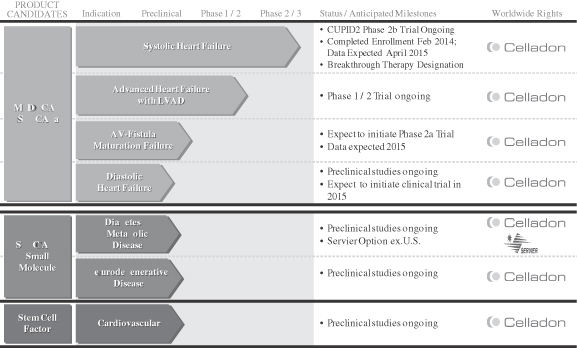
MYDICAR: Genetic Enzyme Replacement Therapy of SERCA2a Deficiency
Our lead product candidate, MYDICAR, uses genetic enzyme replacement therapy to correct the SERCA2a enzyme deficiency in heart failure patients that results in inadequate pumping of the heart. MYDICAR is delivered directly to the heart in a routine outpatient procedure, similar to an angiogram, in a cardiac catheterization laboratory. MYDICAR has the potential to provide transformative disease-modifying effects with long-term benefits in heart failure patients with a single administration.
MYDICAR utilizes a recombinant adeno-associated viral vector 1, or AAV1 serotype, which is a group of adeno-associated viruses, or AAVs, sharing specific antigens, to deliver the gene for the SERCA2a enzyme. We believe AAV1 serotype vectors are particularly well suited for administration to the heart muscle because AAV vectors are safe and are less immunogenic than other viral vectors commonly used in gene therapy. Most people are exposed to wild type AAV (serotype 2) during childhood, without experiencing any symptoms, because AAV causes no disease. In addition, local delivery of AAV1 to the heart requires extremely small quantities to achieve therapeutic effect, which has contributed to the low incidence of side effects in clinical trials to date. We have developed a companion diagnostic to identify the patients who are AAV1 neutralizing antibody, or NAb, negative and therefore eligible for MYDICAR treatment. We believe approximately 40% of patients in the United States are AAV1 NAb negative and estimate that there are over 350,000 systolic heart failure patients in the United States alone who will be eligible for MYDICAR therapy upon launch. In an effort to expand the population of heart failure patients with systolic dysfunction that may be eligible for MYDICAR treatment, we are currently exploring whether plasma exchange can be used to remove AAV1 neutralizing antibodies from the circulation in advance of MYDICAR administration. If we are able to successfully utilize plasma exchange on
3
Table of Contents
patients to reduce AAV1 neutralizing antibodies in advance of MYDICAR administration, we estimate that this number could be increased by approximately 175,000 patients. In late 2014, we plan to initiate a pilot, 24 patient, Phase 1/2 study of MYDICAR in advanced heart failure patients with systolic dysfunction and pre-existing levels of neutralizing antibodies against the AAV1 vector, who will undergo plasma exchange prior to administration of MYDICAR. Initial results from this study are expected in 2015.
We hold worldwide rights to MYDICAR in all indications and markets. We plan to commercialize MYDICAR for all approved heart failure indications using a targeted sales force in the United States focused on selected cardiologists and heart failure specialists who treat the majority of heart failure patients. We believe we can maximize the value of our company by retaining substantial commercialization rights to our product candidates and, where appropriate, entering into partnerships for specific therapeutic indications and/or geographic territories.
MYDICAR for Systolic Heart Failure
Heart failure caused by systolic dysfunction is characterized by a decreased contraction of the heart muscle. In 2013, the American Heart Association estimated that there are nearly six million patients currently diagnosed with heart failure in the United States. Despite optimal guideline-directed therapies employing a wide range of pharmacologic, device, and surgical options, many heart failure patients deteriorate over time. The long-term prognosis associated with heart failure is worse than that associated with the majority of cancers, with a mortality rate of approximately 50% at five years following initial diagnosis. There are one million primary heart failure-related hospitalizations and over 280,000 heart failure-related deaths annually in the United States. The estimated direct cost of heart failure in the United States in 2012 was $60.2 billion, half of which was related to repeated hospitalizations. The one- and six-month readmission rates after heart failure-related hospitalization are close to 25% and 50%, respectively, and there is growing pressure on hospitals to reduce readmissions for heart failure.
MYDICAR was initially evaluated in Phase 1 of our CUPID 1 trial, which was an open-label, dose-escalation trial in which patients with heart failure received a single intracoronary infusion of MYDICAR on top of maximal optimized heart failure therapy. Of the 12 patients who received MYDICAR, several demonstrated improvements from baseline to month six across a number of parameters important in heart failure. Based on these results, we advanced MYDICAR to Phase 2a of our CUPID 1 trial. In this 39-patient trial, MYDICAR was found to be safe and well-tolerated, reduced heart failure-related hospitalizations, improved patients’ symptoms and quality of life, and improved key markers of cardiac function predictive of survival, such as elevated levels of natriuretic peptides and end systolic volume. This trial included a single intracoronary infusion of MYDICAR followed by an on-study observation period of 12 months, plus a two-year long-term follow-up period. High-dose MYDICAR (1 x 1013 DNase resistant particles) met the primary endpoint versus placebo at six months, and all positive trends were confirmed at 12 months. The hazard ratio at 12 months for the high-dose MYDICAR group versus placebo for recurrent adjudicated clinical events was 0.12 (p=0.003, where p-value is the statistical probability of a result due to chance alone) representing a risk reduction of 88% with MYDICAR versus placebo. Benefit in preventing clinical events such as hospitalizations has been confirmed at three years as well as a trend in improved survival. The hazard ratio at 36 months for the high-dose MYDICAR group versus placebo for recurrent adjudicated clinical events was 0.18 (p=0.048) representing a risk reduction of 82% with high-dose MYDICAR versus placebo.
Following the completion of our CUPID 1 trial, we received Fast Track designation from the FDA in December 2011 for MYDICAR for the treatment of systolic heart failure in New York Heart Association Class III/IV heart failure patients. Subsequently, we held an End-of-Phase 2 meeting with the FDA, as a result of which the FDA has indicated that: data supported proceeding to a Phase 3 clinical trial with high-dose MYDICAR; our proposed safety database, which will include approximately 610 patients (one-half treated), may be acceptable if the safety profile is similar to CUPID 1; time-to-recurrent heart failure-related hospitalizations, in the presence of
4
Table of Contents
terminal events (all-cause death, LVAD implantation, and heart transplant), is acceptable as the primary endpoint, pending details of the statistical analysis plan and further discussion with agency statisticians; and a single clinical trial may be acceptable for a biologics license application, or BLA, submission assuming statistically significant primary outcome and strong concordance of primary and secondary endpoint analyses. We have also held a Type A meeting with the FDA, as a result of which the FDA approved a 572-patient Phase 3 trial protocol under the SPA guidance and agreed that the design and planned analyses of this trial would be sufficient to provide data that, depending on outcome, could support a BLA submission. Pursuant to the SPA, we also obtained an agreement from the FDA that the primary efficacy endpoint of time-to-recurrent heart failure-related hospitalizations in the presence of terminal events would be acceptable for a pivotal trial of MYDICAR. This endpoint counts multiple heart failure-related hospitalizations per patient, and “corrects” for the occurrence of terminal events. Based on published FDA guidance, we believe that the FDA may not require us to complete a Phase 3 trial if the results of our CUPID 2 trial meet the requirements necessary to support a BLA submission based on a single trial as outlined by the FDA.
Based on the CUPID 1 results and following discussions with the FDA, we advanced MYDICAR to our CUPID 2 Phase 2b trial. The primary objective of our ongoing CUPID 2 trial is to determine the efficacy of a single intracoronary infusion of high-dose MYDICAR compared to placebo, in conjunction with maximal optimized heart failure therapy, in reducing the frequency of and/or delaying heart failure-related hospitalizations in patients with systolic heart failure (having an ejection fraction less than 35%) who are at increased risk of terminal events based on elevated levels of natriuretic peptides or a recent heart failure-related hospitalization. Ejection fraction, or EF, is the measurement used to describe the contractility of the heart. The dose being used in this trial is equivalent to the high-dose used in CUPID 1. Patients were randomized in parallel to high-dose MYDICAR or placebo in a 1:1 ratio. We completed enrollment of this trial in February 2014. Approximately 250 patients were enrolled to obtain at least 186 adjudicated heart failure-related hospitalizations. The primary efficacy endpoint is time-to-recurrent heart failure-related hospitalizations in the presence of terminal events at the time of primary analysis data cutoff. We expect to announce results from this trial in April 2015.
Upon completion of our ongoing CUPID 2 trial, we plan to discuss results with the FDA and the EMA with the possibility that MYDICAR could qualify for expedited approval if the trial outcome demonstrates substantial reduction in recurrent heart failure-related hospitalizations and concordant trends in reduction in and/or delay of terminal events overall, and death in particular. However, if the FDA requires another trial, we have an SPA in place for a 572-patient Phase 3 pivotal trial using the same endpoint as in our CUPID 2 trial. We believe the results of one or both of these trials could support submission of a BLA for MYDICAR for the treatment of systolic heart failure. In November 2013, the EMA indicated that if MYDICAR demonstrates a substantial and highly significant treatment effect in the advanced heart failure population, and no untoward effects attributable to MYDICAR are observed, a safety database of approximately 205-230 subjects may be sufficient for a safety assessment to allow for acceptance of a MAA for MYDICAR for the treatment of systolic heart failure. We therefore believe that, if the above conditions are met, a Phase 3 trial may not be required for marketing approval in Europe.
In addition to the ongoing CUPID 2 trial, we are planning two other studies for MYDICAR to support our BLA filing, an AAV1 NAb positive trial called CELL-005, and a viral shedding trial called CELL-006. MYDICAR will also be evaluated in an investigator-initiated trial called AGENT-HF. The primary objective of the AAV1 NAb positive trial is to determine the safety of a single intracoronary infusion of high-dose MYDICAR in patients who test positive for NAbs who would otherwise be ineligible for treatment with MYDICAR. The FDA has required this approximately 70 patient safety trial, as a condition to the submission of a BLA, to cover the possibility that MYDICAR may be used in NAb positive patients. The viral shedding trial is required as part of the environmental risk assessment that must be included in a marketing application to regulatory authorities, both in the United States and in Europe. In this open-label trial, ten patients with heart failure will be treated with high-dose MYDICAR and will be followed until they have two consecutive bodily
5
Table of Contents
fluid samples that are negative for presence of the SERCA2a gene. The patients would continue to be followed for safety for up to two years to add to the overall MYDICAR safety database. We expect to initiate the AAV1 NAb positive and viral shedding trials in 2014. The primary objective of the AGENT-HF trial is to determine whether treatment with MYDICAR leads to a reversal in the decline of left-ventricular function of the heart. This trial will enroll approximately 44 heart failure patients in France with half receiving MYDICAR and the other half placebo. The primary endpoint at six months will be change, compared to baseline, in left ventricular end systolic volume as measured by cardiac computed tomography.
MYDICAR in Additional Indications
Beyond our proposed lead indication of systolic heart failure, we are also developing MYDICAR for additional indications including enhancement of AVF maturation, diastolic heart failure and treatment of patients with advanced heart failure who are on an LVAD. Each of these conditions is characterized by a SERCA2a deficiency, and MYDICAR has demonstrated disease-modifying capability in preclinical models of these diseases. We are currently engaged in preclinical research regarding MYDICAR for the treatment of diastolic heart failure, and plan to initiate clinical trials in this indication in 2015 if data warrants. The broad potential of MYDICAR in multiple indications presents opportunities to maximize the value of our development programs for indications that are poorly managed by existing treatment options.
Recent Developments
Regulatory and Clinical Trial Update
| • | We completed enrollment of CUPID 2 in February 2014 and expect to announce results in April 2015. |
| • | In April 2014, the FDA’s Center for Biologics Evaluation and Research granted Breakthrough Therapy designation to MYDICAR for reducing hospitalizations for heart failure in patients who test negative for adeno-associated viral vector 1 neutralizing antibodies, are NYHA class III or IV heart failure patients and are not in immediate need of an LVAD or heart transplant. |
Business Development Update
| • | In February 2014, we entered into a material transfer and exclusivity agreement with Les Laboratories Servier, or Servier, for the purpose of enabling Servier to conduct an evaluation of our small molecule compounds that modulate the SERCA2b enzyme. As part of this agreement, we granted Servier an option to enter into a license and research collaboration agreement for the joint collaboration, research and development of these compounds for the treatment of type 2 diabetes and other metabolic diseases, pursuant to which Servier may obtain an exclusive, royalty-bearing license to commercialize one or more of these compounds and any related products in the field of type 2 diabetes and other metabolic diseases outside of the United States. |
| • | In July 2014, we in-licensed worldwide rights to gene therapy applications for the membrane bound form of Stem Cell Factor, or mSCF, for treatment of cardiac ischemia from Enterprise Partners Management, LLC. Our approach with mSCF gene therapy is to recruit and expand resident stem cells, thereby harnessing advances in gene therapy technologies and also expanding the application to those in which cardiac stem cells have shown promise in clinical and preclinical testing. Our initial focus will be to generate clinically acceptable gene therapy vectors in support of potentially conducting a future clinical trial in patients who have suffered cardiac damage, as well as exploration of other potential applications. mSCF induces c-kit+ stem/progenitor cell expansion in situ, as well as cardiomyocyte proliferation, which may represent a new therapeutic strategy to reverse adverse remodeling after cardiac injury. In a preclinical setting, mSCF has demonstrated potential improvements in cardiac function and survival |
6
Table of Contents
| following a myocardial infarction. Specifically, these data suggest mSCF gene therapy promoted a regenerative response characterized by an enhancement in cardiac hemodynamic function; an improvement in survival; a reduction in fibrosis, infarct size and apoptosis; an increase in cardiac c-kit+ progenitor cells recruitment to the injured area; an increase in cardiomyocyte cell-cycle activation; and Wnt/ß-catenin pathway induction. |
| • | In July 2014, we entered into a Loan and Security Agreement with Hercules Technology III, L.P. and Hercules Technology Growth Capital, Inc. under which we may borrow up to $25.0 million in two tranches. We borrowed the first tranche of $10.0 million on August 1, 2014. We plan to use the proceeds of the Loan and Security Agreement to provide additional funding for the development of MYDICAR, for other development programs in our pipeline and for general corporate purposes. The second tranche of up to $15.0 million can be drawn through May 31, 2015, but only if data from our ongoing CUPID 2 trial supports the continued development of MYDICAR for its Breakthrough Therapy designation to either a Phase 3 clinical trial or for registration for approval. |
Strategy
We are committed to applying our first-mover scientific leadership position in the field of SERCA2 enzymes to transform the lives of patients with debilitating, life-threatening diseases or conditions. Each of our ongoing and planned development projects addresses diseases or conditions with high unmet medical need that are characterized by an underlying SERCA2 enzyme deficiency. The core elements of our strategy include:
| • | successfully develop MYDICAR as a novel, first-in-class therapy for patients with heart failure due to systolic dysfunction; |
| • | advance MYDICAR through an expedited development and approval process as a Breakthrough Therapy product candidate; |
| • | maximize the value of our MYDICAR franchise by expanding into additional indications; |
| • | commercialize MYDICAR using a highly-targeted cardiology-focused sales force in the United States; |
| • | advance our additional preclinical assets including mSCF gene therapy and our small molecule platform targeting SERCA2 enzymes; and |
| • | deploy capital strategically to develop our portfolio of product candidates and create shareholder value. |
Risks Associated With Our Business
Our business and our ability to implement our business strategy are subject to numerous risks, as more fully described in the section entitled “Risk Factors” immediately following this prospectus summary and in our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014. You should read these risks before you invest in our common stock. We may be unable, for many reasons, including those that are beyond our control, to implement our business strategy. In particular, risks associated with our business include:
| • | We have incurred significant losses since our inception, which we anticipate will continue for the foreseeable future. We have never generated revenue from product sales and may never be profitable. |
| • | Failure to obtain additional funding when needed may force us to delay, limit or terminate our product development efforts or other operations. |
| • | MYDICAR is based on a novel technology, which makes it difficult to predict the time and cost of product candidate development and, subsequently, for obtaining regulatory approval. |
| • | We are highly dependent on the success of MYDICAR and we may not be able to successfully obtain regulatory or marketing approval for, or successfully commercialize, this product candidate. |
7
Table of Contents
| • | We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates. |
| • | Failure to successfully validate, commercialize and obtain regulatory approval for our companion diagnostic could delay or prevent commercialization of MYDICAR. |
| • | If our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates. |
| • | We rely on third parties to conduct some or all aspects of our current vector production, product manufacturing, companion diagnostic testing, reagent manufacturing, protocol development, research, and preclinical and clinical testing. If they fail to meet deadlines or perform in an unsatisfactory manner, our business could be harmed. |
| • | The commercial success of any current or future product candidate will depend upon the degree of market acceptance by physicians, patients, third-party payors and others in the medical community. |
| • | Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel. |
| • | If we are unable to obtain or protect intellectual property rights related to our product candidates, we may not be able to compete effectively in our markets. |
Corporate Information
We were originally incorporated in California in December 2000. In April 2012, we reincorporated in Delaware. Our principal executive offices are located at 11988 El Camino Real, Suite 650, San Diego, California 92130, and our telephone number is (858) 366-4288. Our corporate website address is www.celladon.com. Information contained on or accessible through our website is not a part of this prospectus, and the inclusion of our website address in this prospectus is an inactive textual reference only.
We have obtained a registered trademark for MYDICAR® in the United States. This prospectus contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this prospectus, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other company.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of our initial public offering, (b) in which we have total annual gross revenue of at least $1.0 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. We refer to the Jumpstart Our Business Startups Act of 2012 in this prospectus as the “JOBS Act,” and references in this prospectus to “emerging growth company” have the meaning associated with it in the JOBS Act.
8
Table of Contents
The Offering
| Common stock offered by us |
4,000,000 shares |
| Common stock to be outstanding after this offering |
22,534,480 shares |
| Option to purchase additional shares |
We have granted to the underwriters the option, exercisable for 30 days from the date of this prospectus, to purchase up to 600,000 additional shares of common stock. |
| Use of proceeds |
We estimate that we will receive net proceeds of approximately $39.2 million (or approximately $45.1 million if the underwriters’ option to purchase 600,000 additional shares is exercised in full) from the sale of the shares of common stock offered by us in this offering, based on an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014), and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We intend to use the net proceeds from this offering to fund (1) research and development activities related to seeking regulatory approval for MYDICAR and our companion diagnostic for the treatment of systolic heart failure and other indications, (2) development of manufacturing capabilities for the commercial production of MYDICAR and preparation activities for the potential commercial launch of MYDICAR for the treatment of systolic heart failure in the United States and Europe, (3) clinical development of a potential plasma exchange procedure designed to remove AAVI neutralizing antibodies in advanced heart failure patients to enable their treatment with MYDICAR, (4) research and, if supported by pre-clinical data, clinical development of MYDICAR for the treatment of diastolic heart failure, and (5) working capital and general corporate purposes. See “Use of Proceeds.” |
| Risk factors |
You should read the “Risk Factors” section of this prospectus for a discussion of certain of the factors to consider carefully before deciding to purchase any shares of our common stock. |
| NASDAQ Global Market symbol |
CLDN |
The number of shares of our common stock to be outstanding after this offering is based on 18,534,480 shares of common stock outstanding as of June 30, 2014, and excludes:
| • | 2,510,828 shares of common stock issuable upon the exercise of outstanding stock options as of June 30, 2014, at a weighted-average exercise price of $6.06 per share; |
| • | 206,340 shares of common stock issuable upon the exercise of outstanding warrants as of June 30, 2014, each at an exercise price of $5.61 per share; |
| • | 156,748 shares of common stock reserved for future issuance under our 2013 employee stock purchase plan, or the ESPP, as of June 30, 2014; and |
| • | 532,871 shares of common stock reserved for future issuance under our 2013 equity incentive plan, or the 2013 plan, as of June 30, 2014. |
Unless otherwise indicated, all information contained in this prospectus assumes no exercise by the underwriters of their option to purchase up to an additional 600,000 shares of our common stock.
9
Table of Contents
SUMMARY FINANCIAL DATA
The following table summarizes certain of our financial data. We changed our fiscal year end from June 30 to December 31, effective for the fiscal period ended December 31, 2011. We derived the summary statement of operations data for the fiscal year ended June 30, 2011, the six months ended December 31, 2011, and the years ended December 31, 2012 and 2013 from our audited consolidated financial statements incorporated by reference into this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013. The summary statement of operations data for the six months ended June 30, 2013 and 2014 and the period from December 21, 2000 (inception) to June 30, 2014 and the summary balance sheet data as of June 30, 2014 were derived from our unaudited financial statements incorporated by reference into this prospectus from our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014. Our historical results are not necessarily indicative of the results that may be expected in the future and results of interim periods are not necessarily indicative of the results for the entire year. The summary financial data should be read together with our financial statements and related notes, “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” appearing elsewhere or incorporated by reference in this prospectus.
| Year Ended June 30, 2011 |
Six Months Ended December 31, 2011 |
Year Ended |
Six Months Ended June 30, 2014 |
Period From December 21, 2000 (inception) to June 30, 2014 |
||||||||||||||||||||
| 2012 | 2013 | |||||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Research and development |
$ | 4,193 | $ | 1,252 | $ | 13,314 | $ | 16,927 | $ | 10,199 | $ | 102,243 | ||||||||||||
| General and administrative |
1,832 | 920 | 2,631 | 3,037 | 3,730 | 23,252 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total operating expenses |
6,025 | 2,172 | 15,945 | 19,964 | 13,929 | 125,495 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Loss from operations |
(6,025 | ) | (2,172 | ) | (15,945 | ) | (19,964 | ) | (13,929 | ) | (125,495 | ) | ||||||||||||
| Other income (expense) |
(965 | ) | (689 | ) | 74 | (127 | ) | (225 | ) | (1,495 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Consolidated net loss |
(6,990 | ) | (2,861 | ) | (15,871 | ) | (20,091 | ) | (14,154 | ) | (126,990 | ) | ||||||||||||
| Net loss attributable to noncontrolling interest |
— | — | 154 | 96 | — | 250 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss attributable to Celladon Corporation |
(6,990 | ) | (2,861 | ) | (15,717 | ) | (19,995 | ) | (14,154 | ) | (126,740 | ) | ||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | (343 | ) | — | — | (343 | ) | ||||||||||||||||
| Change in fair value of noncontrolling interest |
— | — | (154 | ) | (3,105 | ) | — | (3,259 | ) | |||||||||||||||
| Deemed dividend |
— | — | — | (856 | ) | — | (856 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss attributable to common stockholders |
$ | (6,990 | ) | $ | (2,861 | ) | $ | (16,214 | ) | $ | (23,956 | ) | $ | (14,154 | ) | $ | (131,198 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Other comprehensive loss: |
||||||||||||||||||||||||
| Unrealized gain (loss) on investments |
— | — | 9 | (7 | ) | 16 | 18 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Comprehensive loss |
$ | (6,990 | ) | $ | (2,861 | ) | $ | (15,862 | ) | $ | (20,098 | ) | $ | (14,138 | ) | $ | (126,972 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (2,729.66 | ) | $ | (1,022.52 | ) | $ | (19.74 | ) | $ | (27.09 | ) | $ | (0.94 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Weighted-average shares outstanding, basic and diluted(1) |
2,561 | 2,798 | 821,568 | 884,179 | 15,092,098 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| (1) | See Note 1 to our consolidated audited financial statements incorporated by reference in this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013 for an explanation of the method used to calculate historical and pro forma basic and diluted net loss per common share attributable to common stockholders and the number of shares used in the computation of the per share amounts. |
10
Table of Contents
The unaudited pro forma balance sheet data set forth below give effect to our issuance and sale of 4,000,000 shares of our common stock in this offering at an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014) after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
| As of June 30, 2014 | ||||||||
| Actual | Pro Forma(1) | |||||||
| (unaudited, in thousands) | ||||||||
| Consolidated Balance Sheet Data: |
||||||||
| Cash, cash equivalents and investments |
$ | 51,172 | $ | 90,364 | ||||
| Working capital |
48,094 | 87,286 | ||||||
| Total assets |
52,278 | 91,470 | ||||||
| Deficit accumulated during the development stage |
(126,740 | ) | (126,740 | ) | ||||
| Total stockholders’ equity |
48,564 | 87,756 | ||||||
| (1) | A $1.00 increase (decrease) in the assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014) would increase (decrease) each of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $3.8 million, assuming the number of shares offered by us, as set forth on the cover of this prospectus, remains unchanged and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, a one million share increase (decrease) in the number of shares offered by us, as set forth on the cover of this prospectus, would increase (decrease) each of cash and cash equivalents, working capital, total assets and total stockholders’ equity by $9.9 million, assuming the assumed public offering price of $10.54 per share remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The pro forma information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
11
Table of Contents
An investment in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included or incorporated by reference in this prospectus, including the risks and uncertainties discussed under “Risk Factors” in our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 which are incorporated by reference in this prospectus in their entirety, before deciding whether to invest in our common stock. The occurrence of any of the risks described below or incorporated by reference in this prospectus could have a material adverse effect on our business, financial condition, results of operations and future growth prospects. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to this Offering
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Our executive officers, directors, 5% stockholders and their affiliates beneficially owned approximately 74.9% of our voting stock as of June 30, 2014. Based upon the assumed number of shares to be sold in this offering as set forth on the cover page of this prospectus, upon the closing of this offering, that same group will beneficially own approximately 62.2% of our outstanding voting stock. Therefore, even after this offering these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders, acting together, may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may believe are in your best interest as one of our stockholders.
If you purchase our common stock in this offering, you will incur immediate and substantial dilution in the book value of your shares.
Investors purchasing common stock in this offering will pay a price per share that substantially exceeds the pro forma net tangible book value per share as of June 30, 2014. Net tangible book value is our tangible assets after subtracting our liabilities. As a result, investors purchasing common stock in this offering will incur immediate dilution of $6.65 per share, based on an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014), and our pro forma net tangible book value as of June 30, 2014. For more information on the dilution you may suffer as a result of investing in this offering, see “Dilution.”
This dilution is due to the substantially lower price paid by our investors who purchased shares prior to this offering as compared to the price offered to the public in this offering, and the exercise of stock options granted to our employees and warrants issued to our existing investors. As of June 30, 2014, options to purchase 2,510,828 shares of our common stock at a weighted-average exercise price of $6.06 per share, and warrants to purchase 206,340 shares of our common stock each at an exercise price of $5.61 per share, were outstanding. The exercise of any of these options or warrants would result in additional dilution. As a result of the dilution to investors purchasing shares in this offering, investors may receive significantly less than the purchase price paid in this offering, if anything, in the event of our liquidation.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our common stock.
12
Table of Contents
We, along with our directors, executive management team and the entities affiliated with our directors, as well as certain of our existing stockholders, have agreed that for a period of 90 days after the date of this prospectus, subject to specified exceptions, not to offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any of our common stock. Subject to certain limitations, including sales volume limitations with respect to shares held by our affiliates, approximately 1,281,478 shares of our common stock will become eligible for sale upon expiration of the lock-up period, as calculated and described in more detail in the section entitled “Shares Eligible for Future Sale.” In addition, shares issued or issuable upon exercise of options and warrants vested as of the expiration of the lock-up period will also be eligible for sale at that time. Sales of stock by these stockholders upon expiration of the lock-up period could have a material adverse effect on the trading price of our common stock.
Certain holders of our securities are entitled to rights with respect to the registration of their shares under the Securities Act of 1933, as amended, or the Securities Act, subject to the 90-day lock-up arrangement described above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares held by our affiliates as defined in Rule 144 under the Securities Act. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds, including for any of the purposes described in “Use of Proceeds,” and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. Because of the number and variability of factors that will determine our use of the net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. The failure by our management to apply these funds effectively could harm our business. If we do not invest or apply the net proceeds from this offering in ways that enhance stockholder value, we may fail to achieve expected financial results, which could cause our stock price to decline.
13
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference contain forward-looking statements. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business” in this prospectus or the documents incorporated by reference. We may, in some cases, use words such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” or the negative of those terms, and similar expressions that convey uncertainty of future events or outcomes to identify these forward-looking statements. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. Forward-looking statements in this prospectus include, but are not limited to, statements about:
| • | the success, cost and timing of our product development activities and clinical trials; |
| • | our ability to obtain and maintain regulatory approval for MYDICAR, our companion diagnostic, and any of our future product candidates, and any related restrictions, limitations, and/or warnings in the label of an approved product candidate; |
| • | our ability to obtain funding for our operations, including funding necessary to complete all clinical trials that may potentially be required to file a biologics license application, or BLA, and a Marketing Authorization Application, or MAA, for MYDICAR for the treatment of systolic heart failure; |
| • | the commercialization of our product candidates and companion diagnostic, if approved; |
| • | our plans to research, develop and commercialize our product candidates and companion diagnostic; |
| • | our ability to attract collaborators with development, regulatory and commercialization expertise; |
| • | our plans and expectations with respect to future commercial scale-up activities, including our expectation regarding the building of a commercial manufacturing facility for the production of MYDICAR; |
| • | future agreements with Lonza Houston, Inc., or Lonza, and other third parties in connection with the commercialization of MYDICAR, our companion diagnostic and any other approved product; |
| • | the size and growth potential of the markets for our product candidates, and our ability to serve those markets; |
| • | the rate and degree of market acceptance of our product candidates and companion diagnostic; |
| • | regulatory developments in the United States and foreign countries; |
| • | the performance of our third-party suppliers and manufacturers; |
| • | the success of competing therapies that are or may become available; |
| • | our ability to attract and retain key scientific or management personnel; |
| • | the accuracy of our estimates regarding expenses, future revenues, capital requirements and needs for additional financing; |
| • | our expectations regarding the period during which we qualify as an emerging growth company under the JOBS Act |
| • | our use of the proceeds from this offering; and |
| • | our expectations regarding our ability to obtain and maintain intellectual property protection for our product candidates. |
These forward-looking statements reflect our management’s beliefs and views with respect to future events and are based on estimates and assumptions as of the date of this prospectus and are subject to risks and
14
Table of Contents
uncertainties. We discuss many of these risks in greater detail under “Risk Factors” and in the risk factors incorporated by reference in this prospectus. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements by these cautionary statements. Except as required by law, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise.
15
Table of Contents
We estimate that we will receive net proceeds of approximately $39.2 million (or approximately $45.1 million if the underwriters’ option to purchase additional shares is exercised in full) from the sale of the shares of common stock offered by us in this offering, based upon an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014) and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
A $1.00 increase (decrease) in the assumed public offering price of $10.54 per share would increase (decrease) the net proceeds to us from this offering by approximately $3.8 million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
Similarly, a one million share increase (decrease) in the number of shares offered by us, as set forth on the cover of this prospectus, would increase (decrease) the net proceeds to us by $9.9 million, assuming the assumed public offering price of $10.54 per share remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We currently intend to use the net proceeds from this offering to fund: research and development activities, including internal salaries and external costs, related to seeking regulatory approval for MYDICAR and our companion diagnostic for the treatment of systolic heart failure; development of manufacturing capabilities for the commercial production of MYDICAR; early preparation for the potential commercial launch of MYDICAR for the treatment of systolic heart failure in the United States and Europe; research and development activities related to seeking regulatory approval for MYDICAR for the treatment of patients with end-stage renal disease on hemodialysis undergoing surgery for arteriovenous fistula creation; clinical development of a potential plasma exchange procedure designed to remove AAVI neutralizing antibodies from advanced heart failure patients to enable their treatment with MYDICAR; research and, if supported by pre-clinical data, clinical development of MYDICAR for the treatment of diastolic heart failure; and general and administrative expenses, potential future development programs, early-stage research and development activities and general corporate purposes. We may also use a portion of the remaining net proceeds to in-license, acquire, or invest in complementary businesses, technologies, products or assets. However we have no current commitments or obligations to do so. Our expected use of the net proceeds from this offering represents our current intentions based on our present business plans and business condition. We cannot currently allocate specific percentages of the net proceeds that we may use for the purposes specified above, and we cannot predict with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering, or the amounts that we will actually spend on the uses set forth above. The amounts and timing of our actual use of the net proceeds will vary depending on numerous factors, including our ability to obtain additional financing, the relative success and cost of our research, preclinical and clinical development programs and whether we are able to enter into future licensing arrangements. As a result, our management will have broad discretion in the application of the net proceeds, and investors will be relying on our judgment regarding the application of the net proceeds of this offering. In addition, we might decide to postpone or not pursue clinical trials or preclinical activities if the net proceeds from this offering and our other sources of cash are less than expected. Pending their use, we plan to invest the net proceeds from this offering in short- and intermediate-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the U.S. government. We anticipate that we will need to secure additional funding for the further development, regulatory approval and commercial launch of MYDICAR.
16
Table of Contents
PRICE RANGE OF OUR COMMON STOCK
Our common stock has been listed on The NASDAQ Global Market since January 30, 2014 under the symbol “CLDN.” Prior to that date, there was no public market for our common stock. Shares sold in our initial public offering on January 29, 2014 were priced at $8.00 per share.
On August 6, 2014, the closing price for our common stock as reported on The NASDAQ Global Market was $10.54 per share. The following table sets forth the ranges of high and low sales prices per share of our common stock as reported on The NASDAQ Global Market for the periods indicated. Such quotations represent inter-dealer prices without retail markup, markdown or commission and may not necessarily represent actual transactions.
| Year Ending December 31, 2014 |
High | Low | ||||||
| First Quarter (from January 30, 2014) |
$ | 17.16 | $ | 7.45 | ||||
| Second Quarter |
$ | 16.47 | $ | 7.82 | ||||
| Third Quarter (through August 6, 2014) |
$ | 16.72 | $ | 10.30 | ||||
As of June 30, 2014, there were 37 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
17
Table of Contents
The following table sets forth our cash, cash equivalents and investments, and our capitalization as of June 30, 2014:
| • | on an actual basis; and |
| • | on a pro forma basis, giving effect to the sale by us of 4,000,000 shares of our common stock in this offering at an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014), after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
The pro forma information below is illustrative only and our capitalization following the closing of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. You should read this table together with “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes appearing elsewhere or incorporated by reference in this prospectus.
| As of June 30, 2014 | ||||||||
| Actual | Pro Forma(1) | |||||||
| (unaudited) | ||||||||
| (in thousands, except per share data) | ||||||||
| Cash, cash equivalents and investments |
$ | 51,172 | $ | 90,364 | ||||
|
|
|
|
|
|||||
| Capitalization: |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized and no shares issued or outstanding, actual and pro forma |
$ | — | $ | — | ||||
| Common stock, $0.001 par value; 200,000,000 shares authorized and 18,534,480 shares issued and outstanding, actual; 200,000,000 shares authorized and 22,534,480 issued and outstanding, pro forma |
18 | 22 | ||||||
| Additional paid-in capital |
175,268 | 214,456 | ||||||
| Accumulated other comprehensive income |
18 | 18 | ||||||
| Deficit accumulated during the development stage |
(126,740 | ) | (126,740 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
48,564 | 87,756 | ||||||
|
|
|
|
|
|||||
| Total liabilities, preferred stock and stockholders’ deficit |
$ | 52,278 | $ | 91,470 | ||||
|
|
|
|
|
|||||
| (1) | A $1.00 increase (decrease) in the assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014) would increase (decrease) the amount of cash and cash equivalents, additional paid-in capital and total capitalization by approximately $3.8 million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering costs payable by us. Similarly, a one million share increase (decrease) in the number of shares offered by us, as set forth on the cover page of this prospectus, would increase (decrease) each of cash and cash equivalents and total stockholders’ equity (deficit) and total capitalization by $9.9 million, assuming the assumed public offering price of $10.54 per share remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
The number of common shares shown in the table above is based on the number of shares of our common stock outstanding as of June 30, 2014, and excludes:
| • | 2,510,828 shares of common stock issuable upon the exercise of outstanding stock options as of June 30, 2014, at a weighted-average exercise price of $6.06 per share; |
| • | 206,340 shares of common stock issuable upon the exercise of outstanding warrants issued after June 30, 2014, each at an exercise price of $5.61 per share; |
| • | 156,748 shares of common stock reserved for future issuance under the ESPP as of June 30, 2014; and |
| • | 532,871 shares of common stock reserved for future issuance under the 2013 plan as of June 30, 2014. |
18
Table of Contents
If you invest in our common stock in this offering, your ownership interest will be immediately diluted to the extent of the difference between the public offering price per share of our common stock and the pro forma net tangible book value per share of our common stock after this offering.
Our historical net tangible book value per share is determined by dividing our total tangible assets less our total liabilities by the actual number of outstanding shares of our common stock. Our historical net tangible book value as of June 30, 2014 was approximately $48.6 million, or $2.62 per share of our common stock.
After giving pro forma effect to the sale of shares of our common stock in this offering at an assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014), and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, our net tangible book value as of June 30, 2014 would have been approximately $87.8 million, or $3.89 per share of common stock. This represents an immediate increase in pro forma net tangible book value of $1.27 per share to our existing stockholders, and an immediate dilution of $6.65 per share to new investors purchasing shares of common stock in this offering at the assumed public offering price.
The following table illustrates this dilution on a per share basis:
| Assumed public offering price per share |
$ | 10.54 | ||||||
| Historical net tangible book value per share as of June 30, 2014 |
$ | 2.62 | ||||||
|
|
|
|||||||
| Increase in net tangible book value per share attributable to investors participating in this offering |
$ | 1.27 | ||||||
|
|
|
|||||||
| Pro forma net tangible book value per share after this offering |
$ | 3.89 | ||||||
|
|
|
|||||||
| Pro forma dilution per share to investors participating in this offering |
$ | 6.65 | ||||||
|
|
|
A $1.00 increase (decrease) in the assumed public offering price of $10.54 per share would increase (decrease) the pro forma net tangible book value per share after this offering by approximately $0.17 (0.16) per share and the dilution per share to investors participating in this offering by approximately $0.83 (0.84) per share, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. An increase of one million shares in the number of shares offered by us would increase the net tangible book value by approximately $0.26 per share and decrease the dilution per share to investors in this offering by $0.26 per share, assuming that the assumed public offering price of $10.54 per share remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
Similarly, a decrease of one million shares in the number of shares offered by us, as set forth on the cover of this prospectus, would decrease the pro forma net tangible book value per share after this offering by approximately $0.27 per share and increase the dilution per share to investors participating in this offering by approximately $0.27 per share, assuming the assumed public offering price of $10.54 per share remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
If the underwriters exercise in full their option to purchase 600,000 additional shares of our common stock in this offering, the pro forma net tangible book value will increase to $4.05 per share, representing an immediate increase in pro forma net tangible book value to existing stockholders of $1.43 per share and immediate dilution of $6.49 per share to new investors participating in this offering.
19
Table of Contents
The following table summarizes, on a pro forma basis as of June 30, 2014, the number of shares purchased or to be purchased from us, the total consideration paid or to be paid to us, and the average price per share paid or to be paid to us by existing stockholders and investors participating in this offering at an assumed public offering price of $10.54 per share, before deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. As the table below shows, investors participating in this offering will pay an average price per share substantially higher than our existing stockholders paid.
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||||
| Number | Percent | Amount | Percent | |||||||||||||||||
| Existing stockholders before this offering |
18,534,480 | 82.2 | % | $ | 175,581,000 | 80.6 | % | $ | 9.47 | |||||||||||
| Investors participating in this offering |
4,000,000 | 17.8 | % | 42,160,000 | 19.4 | % | 10.54 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
22,534,480 | 100 | % | $ | 217,741,000 | 100 | % | $ | 9.66 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
A $1.00 increase (decrease) in the assumed public offering price of $10.54 per share (the last reported sale price of our common stock on The NASDAQ Global Market on August 6, 2014) would increase (decrease) the total consideration paid by investors participating in this offering, total consideration paid by all stockholders and the average price per share paid by all stockholders by approximately $4.0 million, $4.0 million and $0.18, respectively, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and before deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
An increase of one million shares in the number of shares offered by us, as set forth on the cover of this prospectus, would increase the total consideration paid by investors participating in this offering, increase total consideration paid by all stockholders and increase the average price per share paid by all stockholders by approximately $10.5 million, $10.5 million and $0.04 per share, respectively, assuming that the assumed public offering price of $10.54 per share remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, a decrease of one million shares in the number of shares offered by us, as set forth on the cover of this prospectus, would decrease the total consideration paid by investors participating in this offering, decrease total consideration paid by all stockholders and decrease the average price per share paid by all stockholders by approximately $10.5 million, $10.5 million and $0.04 per share, respectively, assuming the assumed public offering price of $10.54 per share remains the same, and before deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
If the underwriters exercise in full their option to purchase 600,000 additional shares of our common stock in this offering, the number of shares of common stock held by existing stockholders will be reduced to 80.1% of the total number of shares of common stock to be outstanding after this offering, and the number of shares of common stock held by investors participating in this offering will be further increased to 4,600,000, or 19.9% of the total number of shares of common stock to be outstanding after this offering.
The foregoing discussion and tables are based on 18,534,480 shares of common stock outstanding as of June 30, 2014 and excludes:
| • | 2,510,828 shares of common stock issuable upon the exercise of outstanding stock options as of June 30, 2014, at a weighted-average exercise price of $6.06 per share; |
| • | 206,340 shares of common stock issuable upon the exercise of outstanding warrants issued after June 30, 2014, each at an exercise price of $5.61 per share; |
| • | 156,748 shares of common stock reserved for future issuance under the ESPP as of June 30, 2014; and |
| • | 532,871 shares of common stock reserved for future issuance under the 2013 plan as of June 30, 2014. |
20
Table of Contents
Furthermore, we may choose to raise additional capital through the sale of equity or convertible debt securities due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent we issue additional shares of common stock or other equity or convertible debt securities in the future, there will be further dilution to investors participating in this offering.
21
Table of Contents
The following selected financial data should be read together with our consolidated financial statements and related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” incorporated by reference into this prospectus. The selected financial data in this section are not intended to replace our consolidated financial statements and the related notes. Our historical results are not necessarily indicative of the results that may be expected in the future and results of interim periods are not necessarily indicative of the results for the entire year.
We changed our fiscal year end from June 30 to December 31, effective for the fiscal period ended December 31, 2011. The selected statement of operations data for the fiscal year ended June 30, 2011, the six months ended December 31, 2011 and the years ended December 31, 2012 and 2013, and the selected balance sheet data as of December 31, 2012 and 2013 are derived from our audited consolidated financial statements incorporated by reference into this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013. The selected statement of operations data for the period from December 21, 2000 (inception) to June 30, 2014 and for the six months ended June 30, 2013 and 2014, and the selected balance sheet data as of June 30, 2014 are derived from our unaudited consolidated financial statements incorporated by reference into this prospectus from our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014.
| Year Ended June 30, 2011 |
Six Months Ended December 31, 2011 |
Year Ended |
Six Months Ended June 30, 2014 |
Period From December 21, 2000 (inception) to June 30, 2014 |
||||||||||||||||||||
| 2012 | 2013 | |||||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Research and development |
$ | 4,193 | $ | 1,252 | $ | 13,314 | $ | 16,927 | $ | 10,199 | $ | 102,243 | ||||||||||||
| General and administrative |
1,832 | 920 | 2,631 | 3,037 | 3,730 | 23,252 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total operating expenses |
6,025 | 2,172 | 15,945 | 19,964 | 13,929 | 125,495 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Loss from operations |
(6,025 | ) | (2,172 | ) | (15,945 | ) | (19,964 | ) | (13,929 | ) | (125,495 | ) | ||||||||||||
| Other income (expense) |
(965 | ) | (689 | ) | 74 | (127 | ) | (225 | ) | (1,495 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Consolidated net loss |
(6,990 | ) | (2,861 | ) | (15,871 | ) | (20,091 | ) | (14,154 | ) | (126,990 | ) | ||||||||||||
| Net loss attributable to noncontrolling interest |
— | — | 154 | 96 | — | 250 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss attributable to Celladon Corporation |
(6,990 | ) | (2,861 | ) | (15,717 | ) | (19,995 | ) | (14,154 | ) | (126,740 | ) | ||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | (343 | ) | — | — | (343 | ) | ||||||||||||||||
| Change in fair value of noncontrolling interest |
— | — | (154 | ) | (3,105 | ) | — | (3,259 | ) | |||||||||||||||
| Deemed dividend |
— | — | — | (856 | ) | — | (856 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss attributable to common stockholders |
$ | (6,990 | ) | $ | (2,861 | ) | $ | (16,214 | ) | $ | (23,956 | ) | $ | (14,154 | ) | $ | (131,198 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Other comprehensive loss: |
||||||||||||||||||||||||
| Unrealized gain (loss) on investments |
— | — | 9 | (7 | ) | 16 | 18 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Comprehensive loss |
$ | (6,990 | ) | $ | (2,861 | ) | $ | (15,862 | ) | $ | (20,098 | ) | $ | (14,138 | ) | $ | (126,972 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (2,729.66 | ) | $ | (1,022.52 | ) | $ | (19.74 | ) | $ | (27.09 | ) | $ | (0.94 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Weighted-average shares outstanding, basic and diluted(1) |
2,561 | 2,798 | 821,568 | 884,179 | 15,092,098 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| (1) | See Note 1 to our consolidated financial statements incorporated by reference into this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013 for an explanation of the methods used to calculate historical and pro forma basic and diluted net loss per common share attributable to common stockholders and the number of shares used in the computation of the per share amounts. |
22
Table of Contents
| As of December 31, | As of June 30, 2014 |
|||||||||||
| 2012 | 2013 | |||||||||||
| (unaudited) | ||||||||||||
| (in thousands) | ||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||
| Cash, cash equivalents and investments |
$ | 35,511 | $ | 18,370 | $ | 51,172 | ||||||
| Working capital |
31,159 | 11,990 | 48,094 | |||||||||
| Total assets |
35,929 | 21,154 | 52,278 | |||||||||
| Redeemable non-controlling interest |
4,814 | — | — | |||||||||
| Redeemable convertible preferred stock |
52,274 | 60,098 | — | |||||||||
| Junior preferred stock |
5,450 | 5,450 | — | |||||||||
| Deficit accumulated during the development stage |
(92,591 | ) | (112,586 | ) | (126,740 | ) | ||||||
| Total stockholders’ (deficit) equity |
(28,416 | ) | (50,991 | ) | 48,564 | |||||||
23
Table of Contents
Overview
We are a clinical-stage biotechnology company applying our leadership position in the field of gene therapy and calcium dysregulation to develop novel therapies for diseases with tremendous unmet medical needs. Our lead programs target sarco/endoplasmic reticulum Ca2+ -ATPase, or SERCA, enzymes, which are a family of enzymes that play an integral part in the regulation of intra-cellular calcium in all human cells. Calcium dysregulation is implicated in a number of important and complex medical conditions and diseases, such as heart failure, which is a clinical syndrome characterized by poor heart function resulting in inadequate blood flow to meet the body’s metabolic needs, as well as blood vessel health, diabetes and neurodegenerative diseases. SERCA2a, an enzyme that becomes deficient in patients with heart failure, was scientifically validated as a molecular target for heart failure in the 1990s and became a focus of internal discovery efforts for many large pharmaceutical companies. However, to date, no other company has been successful in targeting SERCA2a using traditional discovery methods.
Our therapeutic portfolio includes both gene therapies and small molecule compounds targeting diseases characterized by SERCA enzyme deficiency. MYDICAR, our most advanced product candidate, uses gene therapy to target SERCA2a. We believe that our gene therapy approach to modulating SERCA2a overcomes the issues encountered by previous efforts and has the potential to provide transformative disease-modifying effects with long-term benefits in patients with heart failure. In addition, we have recently in-licensed worldwide rights to patents covering an additional gene therapy product opportunity, the membrane-bound form of Stem Cell Factor, or mSCF, for the treatment of cardiac ischemic damage. We have also identified a number of potential first-in-class compounds addressing novel targets in diabetes and neurodegenerative diseases with our small molecule platform of SERCA2b modulators.
We are the first company to enter clinical development with a product candidate, MYDICAR, that selectively targets SERCA2a. We refer to our Phase 1 trial and Phase 2a trial of MYDICAR together as our CUPID 1 trial. In Phase 2a of our CUPID 1 trial, 39 patients with systolic heart failure, which is caused by the inability of the heart to pump blood efficiently due to weakening and enlargement of the ventricles, were enrolled in a randomized, double-blind, placebo-controlled trial. MYDICAR was safe and well-tolerated, reduced heart failure-related hospitalizations, improved patients’ symptoms, quality of life and serum biomarkers and improved key markers of cardiac function predictive of survival, such as end systolic volume. Based on these results, as well as our previous preclinical studies and clinical trials, we advanced MYDICAR to a 250-patient randomized, double-blind, placebo-controlled international Phase 2b trial in patients with systolic heart failure, which we refer to as CUPID 2. We completed enrollment of CUPID 2 in February 2014 and expect to announce results in April 2015. If successful, these results, along with other studies, will form the basis for regulatory submissions for approval with the United States Food and Drug Administration, or FDA, and European Medicines Agency, or EMA. In 2012, we obtained a Special Protocol Assessment, or SPA, whereby the FDA agreed to use time-to-multiple heart failure-related hospitalizations as the primary endpoint for a MYDICAR Phase 3 pivotal trial. Our ongoing CUPID 2 trial uses a similar clinical protocol with identical endpoints as agreed to in the SPA. In May 1998, the FDA published “Guidance for Industry—Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products” outlining the conditions in which a single trial might be sufficient to support a BLA submission. We believe that the FDA may not require us to complete additional trials of MYDICAR for the treatment of systolic heart failure if the results of our CUPID 2 trial meet the requirements for a single trial set forth in this guidance. In November 2013, the EMA indicated that if MYDICAR demonstrates a substantial and highly significant treatment effect in the advanced heart failure population, and no untoward effects attributable to MYDICAR are observed, a safety database of approximately 205 to 230 MYDICAR-treated subjects may be sufficient for a safety assessment to allow for acceptance of a Marketing Authorization Application, or MAA, for MYDICAR for the treatment of systolic heart failure. We therefore believe that, if the above conditions are met, a Phase 3 trial may not be required for marketing approval in Europe.
24
Table of Contents
In April 2014, the FDA’s Center for Biologics Evaluation and Research, or CBER, granted Breakthrough Therapy designation to MYDICAR for reducing hospitalizations for heart failure in patients who test negative for adeno-associated viral vector 1, or AAV1, neutralizing antibodies, are class III or IV heart failure patients under the New York Heart Association, or NYHA, classification system, and are not in immediate need of a left-ventricular assist device, or LVAD, or heart transplant. The Breakthrough Therapy program is intended to expedite drug development and review of innovative new drugs that are intended to treat serious or life-threatening diseases and for which preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on a clinically significant endpoint. MYDICAR was the third product candidate to receive this designation from CBER, and the designation indicates that the FDA has determined that the CUPID 1 trial provided preliminary clinical evidence that MYDICAR may demonstrate substantial improvement over available therapies in heart failure patients for which the designation was granted.
MYDICAR utilizes a recombinant adeno-associated viral vector 1, or AAV1 serotype, which is a group of adeno-associated viruses, or AAVs, sharing specific antigens, to deliver the gene for the SERCA2a enzyme. We believe AAV1 serotype vectors are particularly well suited for administration to the heart muscle because AAV vectors are safe and are less immunogenic than other viral vectors commonly used in gene therapy. Most people are exposed to wild type AAV (serotype 2) during childhood, without experiencing any symptoms, because AAV causes no disease. In addition, local delivery of AAV1 to the heart requires extremely small quantities to achieve therapeutic effect, which has contributed to the low incidence of side effects in clinical trials to date. We have developed a companion diagnostic to identify the patients who are AAV1 neutralizing antibody, or NAb, negative and therefore eligible for MYDICAR treatment. We believe approximately 40% of patients in the United States are AAV1 NAb negative. In an effort to expand the population of heart failure patients with systolic dysfunction that may be eligible for MYDICAR treatment, we are currently exploring whether plasma exchange can be used to remove AAV1 neutralizing antibodies from the circulation in advance of MYDICAR administration. In late 2014, we plan to initiate a pilot, 24 patient, Phase 1/2 study of MYDICAR in advanced heart failure patients with systolic dysfunction and pre-existing levels of neutralizing antibodies against the AAV1 vector, who will undergo plasma exchange prior to administration of MYDICAR. Initial results from this study are expected in 2015.
In 2013, the American Heart Association estimated that there are nearly six million patients currently diagnosed with heart failure in the United States. Despite optimal guideline-directed therapies employing a wide range of pharmacologic, device, and surgical options, many heart failure patients deteriorate over time. The long-term prognosis associated with heart failure is worse than that associated with the majority of cancers, with a mortality rate of approximately 50% at five years following initial diagnosis. There are one million primary heart failure-related hospitalizations and over 280,000 heart failure-related deaths annually in the United States. The estimated direct cost of heart failure in the United States in 2012 was $60.2 billion, half of which was related to repeated hospitalizations. The one- and six-month readmission rates after heart failure-related hospitalization are close to 25% and 50%, respectively, and there is growing pressure on hospitals to reduce readmissions for heart failure.
We are initially developing MYDICAR to treat patients with systolic heart failure. Heart failure caused by systolic dysfunction is characterized by a decreased contraction of the heart muscle. We are also developing MYDICAR for additional indications, including arteriovenous fistula, or AVF, maturation failure, and for the treatment of patients with advanced heart failure who are on a left-ventricular assist device, or LVAD. In addition, we expect to initiate a clinical trial in 2015 for the treatment of diastolic heart failure, a condition caused by the inability of the heart to relax normally between contractions, if data from our preclinical research warrants. MYDICAR has demonstrated activity in preclinical models of this condition.
We hold worldwide rights to MYDICAR in all indications and markets. We plan to commercialize MYDICAR for any approved heart failure indications using a targeted sales force in the United States focused on selected cardiologists and heart failure specialists who treat the majority of heart failure patients. We believe we
25
Table of Contents
can maximize the value of our company by retaining substantial commercialization rights to our product candidates and, where appropriate, entering into partnerships for specific therapeutic indications and/or geographic territories.
We are also investigating MYDICAR for enhancing the rate of AVF maturation and have initiated discussions with the FDA to determine what additional preclinical work will be necessary to support an IND for this new indication. Pending completion of additional preclinical work and agreement by the FDA, we intend to conduct a 100-patient Phase 2a trial with MYDICAR in end-stage renal disease patients undergoing surgery for AVF creation, and expect to have initial clinical data from such study in 2015. Over 500,000 Americans have end-stage renal disease requiring dialysis and approximately 100,000 fistulae are placed yearly. An AVF, which is a surgically created connection between an artery and a vein, is placed in the arm to provide access for hemodialysis. The access that is created is routinely used for hemodialysis two to five times per week. The AVF has proven to be the most durable, least complicated, and therefore preferred mode of vascular access for hemodialysis. The clinical problem that has resulted from this practice is that following surgery to create the fistula, approximately 50% of fistulae fail to mature to a usable state for hemodialysis. Furthermore, as many as 25% of hospital admissions in the dialysis population have been attributed to vascular access problems, including fistula malfunction and thrombosis. The biology of SERCA2a in both vascular smooth muscle cells, or VSMC, and endothelial cells provides a unique opportunity to potentially positively impact the pathological processes driving fistula failure. The majority of AVF maturation failures have been attributed to rapid proliferation of VSMC, resulting in vascular blockage or occlusion. In preclinical studies, SERCA2a enzyme deficiency has been associated with VSMC proliferation, and increasing SERCA2a activity has been shown to prevent VSMC proliferation and stenosis of injured blood vessels. In addition to stenosis, maturation of AVF requires that the blood vessels dilate to support the increased blood flow during dialysis sessions. MYDICAR increases blood flow in treated vessels, and therefore these effects may aid AVF maturation.
In February 2014, we entered into a material transfer and exclusivity agreement with Les Laboratories Servier, or Servier, for the purpose of enabling Servier to conduct an evaluation of our small molecule compounds that modulate the SERCA2b enzyme. As part of this agreement, we granted Servier an option to enter into a license and research collaboration agreement for the joint collaboration, research and development of these compounds for the treatment of type 2 diabetes and other metabolic diseases, pursuant to which Servier may obtain an exclusive, royalty-bearing license to commercialize one or more of these compounds and any related products in the field of type 2 diabetes and other metabolic diseases outside of the United States.
In July 2014, we in-licensed world-wide rights to gene therapy applications for mSCF for treatment of cardiac ischemia from Enterprise Partners Management, LLC. Our approach with mSCF gene therapy is to recruit and expand resident stem cells, thereby harnessing advances in gene therapy technologies and also expanding the application to those in which cardiac stem cells have shown promise in clinical and preclinical testing. Our initial focus will be to generate clinically acceptable gene therapy vectors in support of potentially conducting a future clinical trial in patients who have suffered cardiac damage, as well as exploration of potential other applications. We believe mSCF has applications in a number of disease areas, particularly cardiovascular conditions and diseases. mSCF induces c-kit+ stem/progenitor cell expansion in situ, as well as cardiomyocyte proliferation, which may represent a new therapeutic strategy to reverse adverse remodeling after cardiac injury. In a preclinical setting, mSCF has demonstrated potential improvements in cardiac function and survival following a myocardial infarction. Specifically, these data suggest mSCF gene therapy promoted a regenerative response characterized by an enhancement in cardiac hemodynamic function; an improvement in survival; a reduction in fibrosis, infarct size and apoptosis; an increase in cardiac c-kit+ progenitor cells recruitment to the injured area; an increase in cardiomyocyte cell-cycle activation; and Wnt/ß-catenin pathway induction.
In July 2014, we entered into a Loan and Security Agreement with Hercules Technology III, L.P. and Hercules Technology Growth Capital, Inc. under which we may borrow up to $25.0 million in two tranches. We borrowed the first tranche of $10.0 million on August 1, 2014. We plan to use the proceeds of the Loan and Security Agreement to provide additional funding for the development of MYDICAR, for other development
26
Table of Contents
programs in our pipeline and for general corporate purposes. The second tranche of up to $15.0 million can be drawn through May 31, 2015, but only if data from our ongoing CUPID 2 trial supports the continued development of MYDICAR for its Breakthrough Therapy designation to either a Phase 3 clinical trial or for registration for approval.
Strategy
We are committed to apply our first-mover scientific leadership position in the field of gene therapy and SERCA2 enzymes to transform the lives of patients with debilitating, life-threatening diseases or conditions. Each of our ongoing and planned development projects addresses diseases or conditions with high unmet medical need that are characterized by an underlying SERCA2 enzyme deficiency. The core elements of our strategy include:
| • | Successfully develop MYDICAR as a novel, first-in-class therapy for patients with heart failure due to systolic dysfunction. Based on positive results from our CUPID 1 trial for MYDICAR, we are conducting our CUPID 2 trial to evaluate the safety and efficacy of MYDICAR to reduce heart failure-related hospitalizations in patients with systolic heart failure. We completed enrollment of this trial in February 2014 and expect to announce results in April 2015. In the United States alone, several hundreds of thousands of patients with heart failure due to systolic dysfunction currently have a poor prognosis and limited treatment options. We believe MYDICAR, if approved, will become a valuable treatment option for these patients. |
| • | Advance MYDICAR through an expedited development and review process as a designated Breakthrough Therapy product candidate. In 2012, we obtained an SPA in the context of a Phase 3 clinical trial protocol whereby the FDA agreed to the use of time-to-multiple heart failure-related hospitalizations as the primary endpoint for a potential pivotal trial of MYDICAR. Our ongoing CUPID 2 trial uses a similar clinical protocol with identical endpoints as agreed to in the SPA. Following the completion of our ongoing CUPID 2 trial, we anticipate that we will have meetings with the FDA and the EMA to discuss whether any remaining clinical trials will be required for approval of MYDICAR. If the FDA allows us to pursue an expedited approval process, we anticipate that we will seek registration for MYDICAR upon completion of our CUPID 2 trial and would not conduct the Phase 3 trial outlined in the SPA. In April 2014, the FDA granted Breakthrough Therapy designation to MYDICAR for reducing hospitalizations for heart failure in patients who test negative for adeno-associated viral vector 1 neutralizing antibodies, are NYHA class III or IV heart failure patients and are not in immediate need of an LVAD or heart transplant. Following the completion of our ongoing CUPID 2 trial, we anticipate that we will have meetings with the FDA and the EMA to discuss expeditious pathways towards potential approval of MYDICAR in each jurisdiction. |
| • | Maximize the value of our MYDICAR franchise by expanding into additional indications. The broad therapeutic potential of MYDICAR in multiple indications presents opportunities to maximize the value of our MYDICAR franchise. Beyond our lead proposed indication of systolic heart failure, we are also developing MYDICAR for additional indications including as treatment of AVF maturation failure and for the treatment of patients with advanced heart failure who are on an LVAD. We plan to initiate a development program in diastolic heart failure. This condition is characterized by a SERCA2a deficiency. We may selectively form collaborative alliances to expand and accelerate our development capabilities and product offerings for indications that are poorly managed by existing treatment options. |
| • | Commercialize MYDICAR using a highly-targeted cardiology-focused sales force in the United States. Heart failure patients are largely treated at leading hospitals and medical centers of excellence by a select group of high-prescribing cardiologists and heart failure specialists. We plan to commercialize MYDICAR for all potential heart failure indications using a targeted sales force focused on these treating physicians. We believe cardiologists, heart failure specialists and interventional cardiologists are typically early adopters of innovative products, devices and technologies, in part because the rate of innovation in this sector has been sustained, and in part because of the large unmet need that their patients exhibit. We believe that MYDICAR would be adopted first by certain cardiologists and heart failure specialists at high-volume, key-opinion-leading hospitals and medical centers, and progressively by a broader segment of the market. |
27
Table of Contents
| • | Advance our additional preclinical assets including mSCF gene therapy and our small molecule platform targeting SERCA2 enzymes. We intend to leverage our leading position and proprietary scientific expertise in gene therapy and SERCA2 high throughput screening assays to identify SERCA2 small molecule product candidates and advance mSCF gene therapy towards clinical testing. We have established early preclinical proof-of-concept results in the fields of heart failure, diabetes and neurodegenerative diseases. We plan to continue to advance these programs in certain diseases by ourselves or through a partnering strategy. |
| • | Deploy capital strategically to develop our portfolio of product candidates and create stockholder value. We intend to deploy most of our capital resources to further support the manufacture and clinical development of our lead product candidate, MYDICAR. We strive to leverage new clinical design principles and regulatory approval paths to advance our product candidates towards key value inflection points in a capital efficient manner. We believe we can maximize the value of our company by retaining substantial commercial rights to our product candidates and, where appropriate, entering into partnerships for certain indications and/or geographic territories. We believe this combination of independent development and targeted commercialization, together with selective partnering activities, will allow us to capture substantial value of our product candidates while reducing our need for human and capital resources. |
Our Platform
We are applying our leading expertise in the field of gene therapies and SERCA2 biology towards the development of therapeutics for significant un-met diseases. For our SERCA2 program, we are targeting a specific class of proteins, or enzymes, that control calcium levels inside all cells. We believe that SERCA enzymes function as “master switches” that are critical to keeping cells of the body healthy through regulation of calcium levels. SERCA2 enzyme levels are deficient in many disease states, such as heart failure, AVF maturation failure, pulmonary arterial hypertension, or PAH, which is characterized by a SERCA2a deficiency in vascular smooth muscle cells, diabetes and neurodegenerative diseases. We believe that the involvement of SERCA2 deficiencies in multiple diseases and conditions creates “franchise” opportunities for our first-in-class gene therapy and small molecule product candidates.
We have acquired leading AAV gene vector technology and developed proprietary delivery methods which form the basis of our MYDICAR platform. In addition, using our proprietary, patented SERCA2 screening assay, we have developed a broad platform of novel, first-in-class, small molecule modulators of the SERCA2b enzyme, creating development opportunities for product candidates targeting diseases associated with endoplasmic reticulum, or ER, stress-related pathways, such as diabetes and neurodegenerative diseases.
28
Table of Contents
The following figure illustrates the opportunities and approach we are taking to target SERCA2 deficiency states:
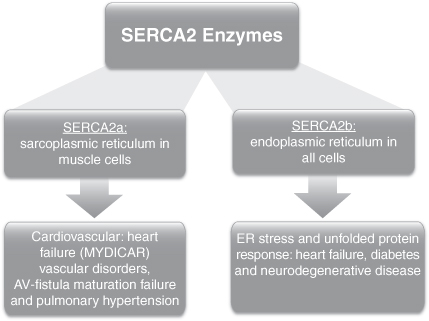
Our lead development program targets calcium dysregulation in the heart. Of the ions involved in the intricate workings of the heart, calcium is considered perhaps the most important. It enables the chambers of the heart to pump, or contract and relax, which causes blood to be propelled in and out of the heart. Calcium directly activates the myofilaments, which are threadlike structures in muscle fibers which cause contraction. Dysregulation of calcium is a central cause of heart failure due to both contractile (systolic) dysfunction, and relaxation (diastolic) dysfunction. One of the central causes of calcium dysregulation in heart failure is a deficiency in the level of SERCA2a enzymes in heart muscle cells. SERCA2a deficiencies are not limited to heart muscle cells, but are also present in blood vessel disorders such as AVF maturation failure and PAH.
Another focus of our research program relates to a different form of the SERCA2 enzyme, SERCA2b. Specifically, these enzymes control calcium movement in the ER in all human cells. SERCA2b enzyme levels become deficient when cells are stressed, and accumulate unfolded proteins in the ER, known as ER stress. There has been a proliferation of publications in scientific medical literature supporting the important role of ER stress in many diseases and conditions, including heart failure, diabetes and neurodegenerative diseases. We believe we are the industry leader in isolating small molecule modulators of the SERCA2b enzyme, which can correct underlying calcium dysregulation and ER stress. Our proprietary, novel, first-in-class, compounds have demonstrated activity in multiple preclinical models of diseases and conditions.
29
Table of Contents
Our Product Pipeline
The following chart depicts key information regarding our development programs, their indications, and their current stage of development:
MYDICAR for Heart Failure
The Heart Failure Epidemic
Heart failure constitutes an important medical, social, and economic problem. Heart failure is a clinical condition in which the output of blood from the heart is insufficient to meet the metabolic demands of the body. In 2013, the American Heart Association estimated that there are nearly six million patients currently diagnosed with heart failure in the United States. The prevalence of heart failure is progressively increasing due to an aging population and increasing prevalence of major cardiovascular risk factors, including obesity and diabetes. Additional risk factors for heart failure include coronary heart disease, hypertension, alcoholism, drug abuse, exposure to toxins and infectious agents, pregnancy and congenital mutations. It is estimated that one in five adults at age 40 will develop heart failure during their remaining lifetime, and that approximately 250,000 to 500,000 patients in the United States are currently in the terminal phase of heart failure and have symptoms that cannot be effectively managed by existing optimized medical therapy. These patients suffer from disabling symptoms and often need hospitalization. The long-term prognosis associated with heart failure is worse than that associated with the majority of cancers, with approximate 50% mortality at five years following initial diagnosis. With over 280,000 heart failure-related deaths annually, we believe MYDICAR will provide a much needed therapeutic alternative for heart failure patients. We estimate that there are over 350,000 systolic heart failure patients in the United States alone who will be eligible for MYDICAR treatment upon launch. If we are able to successfully utilize plasma exchange on patients to reduce AAV1 neutralizing antibodies in advance of MYDICAR administration, we estimate that this number could be increased by approximately 175,000 patients.
Hospitalizations for heart failure are expensive and are particularly problematic, as the risk of death is increased with each recurrent heart failure-related hospitalization. There are one million primary heart failure-
30
Table of Contents
related hospitalizations annually in the United States alone. The estimated direct cost of heart failure in the United States in 2012 was $60.2 billion, half of which was related to repeated hospitalizations. By 2030, the total cost of heart failure in the United States is projected to increase to $70 billion. The one- and six-month readmission rates after heart failure-related hospitalization are close to 25% and 50%, respectively. The Affordable Care Act recently established the “Hospital Readmissions Reduction Program,” which requires Centers for Medicare & Medicaid Services to reduce payments to hospitals with excessive heart failure readmissions. As such, there is a growing pressure on hospitals to reduce readmissions for heart failure.
The pathologies resulting from heart failure are devastating. During heart failure progression, the heart steadily loses its ability to respond to increased metabolic demand, such as during intense physical activity. Patients suffer from increased shortness of breath in a progressive manner, and mild exercise soon exceeds the capacity of the heart to react to the increase in metabolic demand. Towards the end stage of the disease, the heart cannot pump enough blood to meet what the body needs even at rest. At this stage, fluids accumulate in the extremities or in the lungs, making the patient bedridden and unable to perform activities of daily living. In addition to constant shortness of breath, even minor deviation from a physical activity and diet restricted lifestyle can cause acute exacerbations, during which patients experience a drowning sensation and must be urgently hospitalized in intensive care or cardiac care units. Heart failure is classified in relation to the severity of the symptoms experienced by the patient. The most commonly used classification system, established by the New York Heart Association, or NYHA, is as follows:
| • | Class I (mild): patients experience no or very mild symptoms with ordinary physical activity |
| • | Class II (mild): patients experience fatigue and shortness of breath during moderate physical activity |
| • | Class III (moderate): patients experience shortness of breath during even light physical activity |
| • | Class IV (severe): patients are exhausted even at rest |
The survival rate in each of these classes of heart failure is a function of the severity of the disease with the more advanced patients having poorer survival prognosis. Guideline-directed medical therapy for heart failure emphasizes angiotensin-converting enzyme, or ACE, inhibitors, angiotensin-2 receptor blockers if the patient is ACE intolerant, and beta blockers. There is recommendation for cardiac resynchronization therapy in certain patients. Implantable cardioverter-defibrillators, or ICDs, are used in patients at risk for sudden cardiac death. Despite these optimal guideline-directed therapies employing a wide range of pharmacologic, device, and surgical options, many patients deteriorate over time and develop advanced heart failure symptoms that cannot be effectively managed by existing optimized medical therapy. At the end stage of heart failure disease, current treatment options include heart transplant surgery or implantation of an LVAD. LVADs are battery operated mechanical circulatory devices used to partially or completely replace the function of the left ventricle of the heart for patients awaiting a heart transplant, or as a destination therapy for patients with NYHA Class IV heart failure who will never receive a heart transplant. Both of these end-stage treatment options require invasive open-chest surgery, include a host of complications such as lifetime immunosuppressive therapy in the case of transplant and risk of thrombosis and infection in the case of LVADs, and can cost in excess of $150,000. An estimated 1,500 patients per year in the United States have an LVAD implanted and an estimated 2,300 patients per year in the United States undergo heart transplant surgery.
Role of SERCA2a in Heart Failure
SERCA2a’s role in heart failure was scientifically validated in the 1990s and immediately became a focus of pharmaceutical industry discovery efforts. However, due in part to ineffective screening technologies, SERCA2a proved to be an elusive target and to date no other company has been successful in targeting SERCA2a using traditional discovery methods.
31
Table of Contents
Heart failure is characterized by abnormalities in the various steps of the heart muscle pumping process. Intracellular calcium movements in the heart are tightly regulated at various levels within the heart’s cells. An organelle called the sarcoplasmic reticulum, or SR, plays an important role in orchestrating the movement of calcium during each contraction and relaxation. The cardiac cycle is illustrated in the figure below.
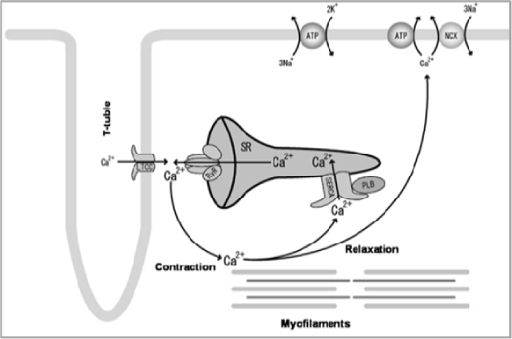
During contraction, calcium is released from the SR, activating the myofilaments leading to muscle contraction. During relaxation, the majority of calcium is sequestered back into the SR by the SERCA2a enzyme leading to muscle relaxation. It is modulated through normal physiology via a protein known as phospholamban (PLB in the figure above), increasing activity when we exercise and decreasing activity when we rest. In advanced heart failure, SERCA2a enzyme levels are abnormally low, so patients cannot effectively modulate SERCA2a activity and increase their cardiac output even upon mild physical activity, such as walking or climbing stairs.
The figure below depicts in vitro studies of the contraction and relaxation and calcium cycle in normal human heart cells, in cells from patients with heart failure, and after correction of the SERCA2a deficiency in heart failure cells.
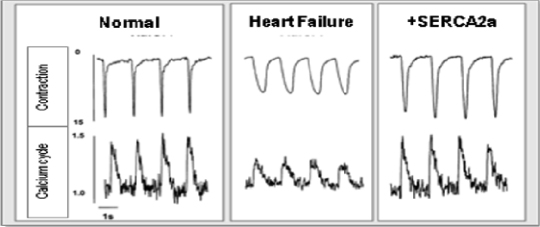
32
Table of Contents
Even in end-stage human heart failure cardiac cells, correction of the SERCA2a deficiency is able to restore normal contractility, relaxation, and calcium cycling. This demonstrates the central importance of SERCA2a deficiency in heart failure, and the ability to reverse the abnormality in contraction and relaxation driving the pathogenesis of this serious medical condition.
Heart failure can be caused by a problem with cardiac contraction, relaxation, or both. Ejection fraction, or EF, is the measurement used to describe the contractility of the heart. Approximately half of heart failure patients suffer from contractility abnormalities (systolic heart failure, EF less than 35%) and the other half suffer from relaxation abnormalities (diastolic heart failure, or heart failure with preserved EF). Both forms represent a significant unmet medical need and we are also developing MYDICAR to target the diastolic form of the disease. Diastolic heart failure is characterized by a “stiff” ventricle, which impairs relaxation of the heart between contractions. We believe MYDICAR can effectively treat diastolic heart failure by correcting the SERCA2a deficiency and improving the ability of the heart to relax between contractions. Based on the Framingham Heart Study conducted by the National Heart, Lung and Blood Institute and Boston University, the five-year mortality rate for patients with diastolic heart failure is 45–60%, which demonstrates the significant unmet need for effective treatments for this condition.
MYDICAR: Genetic Enzyme Replacement Therapy of SERCA2a Deficiency
Our lead product candidate, MYDICAR, uses genetic enzyme replacement therapy to correct the SERCA2a enzyme deficiency in heart failure patients that results in inadequate pumping of the heart. MYDICAR is delivered directly to the heart in a routine outpatient procedure, similar to an angiogram, in a cardiac catheterization laboratory. MYDICAR has the potential to provide transformative disease-modifying effects with long-term benefits in heart failure patients with a single administration. We filed an investigational new drug application, or IND, in December 2006 for MYDICAR for the treatment of heart failure.
Gene therapy alters a person’s deficient genetic material (encoded by deoxyribonucleic acid, or DNA). The altered genes, in turn, through a process called gene expression, can then produce the correct proteins and/or enzymes that were otherwise being produced improperly, or in the case of SERCA2 deficiency, at abnormally low levels. Gene therapy is accomplished through a process known as gene transfer, whereby a functional gene is delivered and incorporated into a patient’s cells through a delivery system called a vector, which are most commonly based on naturally-occurring viruses that have been modified to take advantage of the virus’ natural ability to introduce genes into cells. However, unlike naturally-occurring viruses, which replicate following infection of a target cell and have the capacity to infect new cells, viral vectors are modified to be non-replicating by deleting that portion of the viral genome responsible for replication. We believe that the growing body of gene therapy-based clinical data and the establishment of regulatory guidelines to govern the development and approval of gene therapy products suggest that gene therapy is positioned to emerge as an important new therapeutic modality for patients with significant unmet medical need.
MYDICAR, or AAV1/SERCA2a, utilizes AAV1 to deliver the gene for the enzyme SERCA2a. AAV1/SERCA2a consists of an outer protein shell, called a capsid, and inner DNA genome that contains a gene for SERCA2a. In a treated patient, the capsid delivers the genome to the target cell, where the DNA will direct expression of the SERCA2a protein. Different strains of AAV, called serotypes, have slightly different capsids, which target the vector to different cell types. AAV vectors are particularly well suited for the treatment of heart failure because:
| • | AAV vectors are safe; most people are exposed to wild type AAV (serotype 2) during childhood, without developing any symptoms because AAV causes no disease. Regulatory authorities consider AAV vectors lower risk than other vectors commonly used in gene therapy, such as retroviruses or lentiviruses, because they present a low risk for inserting genetic material into the patient’s chromosomes, which is known as insertional mutagenesis and may lead to cancer. This is because AAV DNA exists in the cell as a circle, or plasmid, outside the main chromosomal DNA. |
33
Table of Contents
| • | AAV vectors are less immunogenic than other viral vectors commonly used in gene therapy, which have caused inflammatory reactions in some patients. |
| • | AAV1 results in a highly efficient delivery of genes into muscle cells so extremely small quantities can be administered directly to the heart to achieve a therapeutic effect; approximately 1/10,000 of a gram of AAV1 capsid protein is contained in a therapeutic dose. We have not observed any toxicities in our preclinical studies or clinical trials. |
| • | AAV particles are small particles and pass freely through the blood vessel wall, bathing the heart muscle and providing broad distribution in the heart without the requirement for invasive or risky procedures. It is delivered directly to the heart over ten minutes in a simple outpatient procedure in a cardiac catheterization laboratory. Patients are awake under mild sedation, and outside of a small puncture in the groin or arm, feel no sensation as a catheter is advanced to the heart. Catheterization procedures like this are routine and are performed thousands of times a day around the globe for imaging the heart. |
| • | Our AAV1 production and manufacturing technology has been developed with a focus on commercialization, and we believe we will be able to produce MYDICAR in large quantities to support our target markets. |
After the AAV1/SERCA2a is infused in the arteries that feed the heart muscle, the AAV1 particle is taken up by the cells and results in expression of the normal SERCA2a human protein in the heart. This results in improved contractility, improved symptoms, and reductions in heart failure-related hospitalizations as demonstrated in our CUPID 1 trial.
Antibodies against AAV1 can block entry of MYDICAR into the target cells, and we have therefore developed a companion diagnostic to identify which patients do not have pre-existing NAbs against the AAV1 capsid proteins, and hence which patients are eligible for MYDICAR treatment. Even though the majority of the population has been exposed to wild type AAV (serotype 2), we believe approximately 40% of heart failure patients in the United States are AAV1 NAb negative and hence are eligible for MYDICAR treatment. In an effort to expand the population of heart failure patients with systolic dysfunction who may be eligible for MYDICAR treatment, we are currently exploring whether plasma exchange can be used to reduce the levels of AAV1 neutralizing antibodies from the circulation prior to administration of MYDICAR. In late 2014, we plan to initiate a pilot, 24 patient, Phase 1/2 study of MYDICAR in advanced heart failure patients with systolic dysfunction and pre-existing levels of neutralizing antibodies against the AAV1 vector who will undergo plasma exchange prior to administration of MYDICAR. Initial results from this study are expected in 2015.
MYDICAR is initially being developed to treat patients with systolic heart failure. Heart failure caused by systolic dysfunction is characterized by a decreased contraction (EF less than 35%). We also plan to develop MYDICAR for additional indications, such as AVF maturation failure, and for the treatment of patients with advanced heart failure who are on an LVAD. We expect to initiate a clinical trial in 2015 for the treatment of diastolic heart failure, a condition caused by the inability of the heart to relax normally between contractions.
We estimate that there are over 350,000 systolic heart failure patients in the United States alone who will be eligible for MYDICAR therapy upon launch. If we are able to successfully utilize plasma exchange on patients to reduce the levels of AAV1 neutralizing antibodies in advance of MYDICAR administration, we estimate that this number could be increased by approximately 175,000 patients.
MYDICAR Previous Human Experience in Systolic Heart Failure
We are the first company to enter clinical development with agents that selectively target this well-validated, key enzyme deficiency. In Phase 2a of the CUPID 1 trial, 39 patients with systolic heart failure were enrolled in a randomized-double-blind, placebo-controlled trial in which MYDICAR compared to placebo was found to be safe, reduced heart failure-related hospitalizations, improved patients’ symptoms, quality of life and
34
Table of Contents
serum biomarkers, and improved key markers of cardiac function predictive of survival, such as end systolic volume, or ESV. The CUPID 1 trial included a single dose of MYDICAR with an on-study observation period of 12 months, plus a two-year long-term follow-up. Details are provided below, but an overall summary is as follows:
| • | MYDICAR was associated with benefit in clinical outcomes such as worsening heart failure, heart failure-related hospitalizations, need for LVAD implantation or heart transplant, or death. |
| • | Benefit in clinical outcomes was supported by improvement in patients’ heart failure symptoms, exercise tolerance, serum biomarkers, and cardiac function. |
| • | High-dose MYDICAR (1 x 1013 DNase resistant particles) met the primary endpoint versus placebo at six months, and all positive trends were confirmed at 12 months. |
| • | Benefit in preventing clinical events such as hospitalizations was confirmed at three years as well as a trend in improved survival. We expect to present the full three-year follow-up data at an upcoming medical conference. |
| • | MYDICAR demonstrated an excellent safety profile. |
CUPID 1, Phase 1 (CELL-001)
A total of 12 patients with heart failure (NYHA class III/IV) received a single intracoronary infusion of MYDICAR in an open-label dose-escalation trial in the United States. Administration of MYDICAR was on top of maximal optimized heart failure therapy. Doses administered ranged from 1.4 x 1011 to 1 x1013 DNase resistant particles, or DRP, per patient. The mode of administration was a ten-minute infusion into the coronary artery. MYDICAR demonstrated an excellent safety profile in this heart failure population, with no treatment related toxicities observed. Of the 12 patients who received MYDICAR, several demonstrated improvements from baseline to month six across a number of parameters important in heart failure, including symptomatic (NYHA and Minnesota Living with Heart Failure Questionnaire, five patients), functional (six-minute walk test and peak maximum oxygen consumption, five patients), biomarker (N-terminal prohormone brain natriuretic peptide, or NT-ProBNP, two patients), and left-ventricular, or LV, function/remodeling (EF and ESV, six patients). Quantitative evidence of biological activity across a number of parameters important for assessing heart failure status could be detected in several patients without pre-existing NAbs in this open-label trial.
CUPID 1, Phase 2a (CELL-001)
The Phase 2a design was a randomized, double-blind, placebo-controlled trial in 39 patients who received one of three different doses of MYDICAR or placebo. Twenty-five patients received MYDICAR and 14 received placebo. The mode of administration was a ten-minute infusion into the coronary arteries. All subjects had systolic heart failure (NYHA class III/IV). Treatment with either MYDICAR or placebo was on top of maximal optimized heart failure therapy. Seven efficacy parameters were assessed in four domains: symptoms (NYHA class and Minnesota Living With Heart Failure Questionnaire), functional status (six-minute walk test and peak maximum oxygen consumption), biomarker (NT-ProBNP), and LV function/remodeling (EF and ESV), plus clinical outcomes. The high-dose MYDICAR group versus placebo met the primary endpoint, which was demonstration of improvement across multiple domains without significant worsening in any domain. This combination of requirements resulted in a probability of success by chance alone (false-positive effect) of approximately 3%. The trial met the primary endpoint at six months (confirmed at 12 months) and demonstrated improvement or stabilization in the four efficacy domains.
The characteristics of recurrent clinical events and terminal events over the 12 months of the active observation period of the trial for Phase 2a portion of our CUPID 1 trial are illustrated in the figure below. Each line represents a single subject. Clinical events are depicted by symbols; a star at the beginning of a line represents subjects who were NAb positive at dosing. Patients who were AAV1 NAb positive at dosing had
35
Table of Contents
developed AAV1 NAbs during the period between their initial selection for participation in the trial and dosing, which in some cases, was as long as six months. We expect to use our companion diagnostic to screen out AAV1 NAb positive subjects going forward, as they are not expected to respond to MYDICAR therapy.
As can be seen from the figure below, despite maximal optimized background therapy, the clinical events (worsening heart failure, or WHF, myocardial infarction, or MI, LVAD implantation, use of chronic intravenous inotrope, heart transplant, or all-cause death) in the placebo group were substantial, underscoring the significant unmet need in this population, while in the high-dose MYDICAR group clinical events were limited. WHF was defined as signs and symptoms of heart failure requiring either hospitalization or treatment with intravenous diuretics, vasodilators or positive inotropes; mechanical fluid removal; or intra-aortic balloon pump.
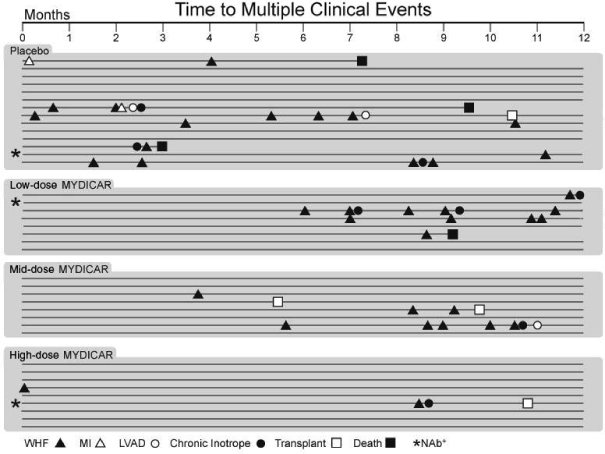
36
Table of Contents
Clinical Events in CELL-001 Phase 2a
In the low-dose (6 × 1011 DNase resistant particles) and mid-dose (3 × 1012 DNase resistant particles) groups, there was a delay to the onset of clinical events, and in the high-dose group, a significant reduction: the relative risk reductions, or hazard ratios, at 12 months for the high-dose MYDICAR group versus placebo for recurrent adjudicated clinical events was 0.12, p=0.003 (where the p-value is the statistical probability of a result due to chance alone), representing a risk reduction of 88% for these important events with high-dose MYDICAR. At 36 months, the high-dose MYDICAR group versus placebo for recurrent adjudicated clinical events was 0.18, p=0.048, representing a risk reduction of 82% for these important events with high-dose MYDICAR. The hazard ratios for recurrent clinical events at 12 months are summarized by treatment group in the table below.
| Time to Multiple Clinical Events Analysis at 12 Months |
||||||
| MYDICAR Dose vs. Placebo |
Hazard Ratio (CI) for Recurrent
Clinical |
Risk Reduction |
||||
| Low-dose |
0.40 (0.13, 1.21), p=0.11 | 60 | % | |||
| Mid-dose |
0.44 (0.16, 1.24), p=0.12 | 54 | % | |||
| High-dose |
0.12 (0.03, 0.49), p=0.003 | 88 | % | |||
| (1) | Recurrent clinical events include WHF and MI. |
In the low- and mid-dose groups, there was a delay to the onset of clinical events, and in the high-dose group a significant reduction. In the low- and mid-dose groups, we believe the dose was not sufficient to insert the SERCA2a gene in enough cells of the myocardium to generate a long-lasting improvement in contractility. We have confirmed this in biopsy samples (see “CUPID 1 (CELL-001) Long-term Follow-up” below), since MYDICAR vector DNA was only detected at long time points in cardiac biopsies in the high-dose patients, but not in biopsies from any other group. MYDICAR increases the presence of an enzyme called nitric oxide synthase in endothelial cells and this enables blood vessels to relax, thereby resulting in short-term increased blood flow. Our hypothesis for why the low- and mid-dose groups demonstrate a delay of the onset of clinical events which is not durable relates to the short-term increase in blood flow to the heart after MYDICAR therapy; higher doses are required to insert the gene deep into the cardiac muscle cells.
The frequency of cardiovascular-related events (WHF and MI), are shown in the figure below.
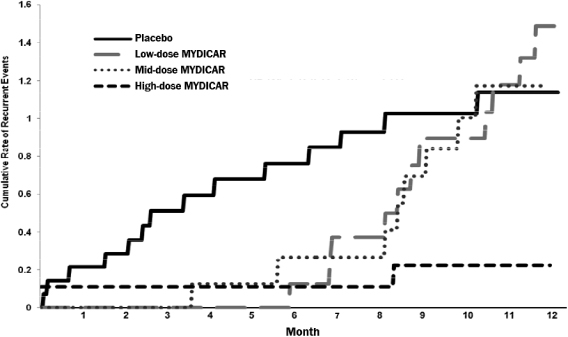
37
Table of Contents
In addition to reducing the frequency of hospitalizations, the mean duration of heart failure-related hospitalizations over 12 months was substantially decreased (0.4 versus 4.5 days; p = 0.05) on high-dose treatment versus placebo. Finally, there were no adverse safety findings.
CUPID 1 (CELL-001) Long-term Follow-up
The patients in the Phase 1 and Phase 2a portions of the CUPID 1 trial were followed for a total of three years. The following clinical events were tracked in all groups: WHF, LVAD implantation, heart transplantation, MI and all-cause death. At three years post-administration, there were 13 deaths: six in the placebo group, three in the low-dose group, three in the mid-dose group and one in the high-dose group (see the figure below for high-dose MYDICAR versus placebo). We expect that data from this trial for the full three year follow-up will be presented at an upcoming conference.
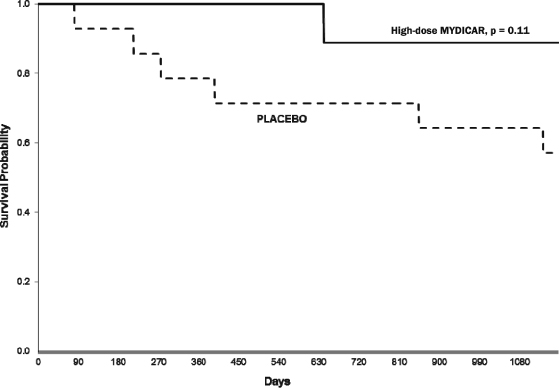
38
Table of Contents
Throughout the three years of follow-up, the number of clinical events was high in the placebo group and high but delayed in the low- and mid-dose groups. Few events occurred in the high-dose group where we found evidence of gene expression (the risk of pre-specified recurrent clinical events over three years of follow-up was reduced by 82% in the high-dose group compared to the placebo group, p=0.048). The figure below depicts cumulative clinical event rates over the three years of follow-up.
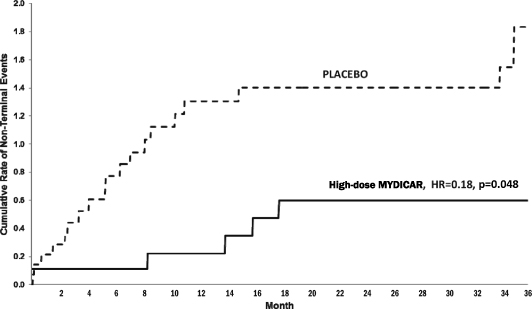
Finally, persistence of the AAV1/SERCA2a vector DNA in biopsy samples of the heart, in cases where heart tissue was made available, was demonstrated by a positive signal from quantitative polymerase chain reaction, or qPCR, testing in high-dose patients. We were only able to obtain heart tissue samples from patients who received an LVAD, cardiac transplant or who died in the hospital. The qPCR assay for AAV1/SERCA2a DNA has demonstrated persistence of the SERCA2a gene out to month 31 in the target tissue of one high-dose patient and to month 22 in another. A third high-dose patient demonstrated presence of vector DNA at month 23. All three patients with qPCR positive vector DNA results showing persistence of the AAV1/SERCA2a vector were in the high-dose group. The qPCR testing of available biopsy samples in patients from the placebo, low- and mid-dose groups did not demonstrate persistence of the AAV1/SERCA2a vector DNA.
Our CUPID 1 trial results demonstrated a favorable safety profile of MYDICAR. No increases in adverse events, disease-related events or laboratory abnormalities were observed in any of the MYDICAR-treated subjects compared to those receiving placebo over the three-year period. There was no indication of an increase in any new occurrences or exacerbation of pre-existing clinical conditions or prior disorders during long-term follow-up including malignancies, neurologic disorders, rheumatologic or other autoimmune disorders, hematologic disorders or other unexpected illnesses associated with MYDICAR administration.
Current and Future Clinical Development of MYDICAR for Systolic Heart Failure
The impact of high-dose MYDICAR on reduction of heart failure-related hospitalizations was an important finding from our CUPID 1 trial and current and future studies are designed to confirm these results and serve as the basis for potential regulatory approvals in the United States. Following completion of our CUPID 1 trial, we held an End-of-Phase 2 meeting with the FDA, as a result of which the FDA indicated that:
| • | data supported proceeding to a Phase 3 clinical trial with high-dose MYDICAR; |
39
Table of Contents
| • | our proposed safety database, which will include approximately 610 patients (one-half treated), may be acceptable if the safety profile is similar to CUPID 1; |
| • | time-to-recurrent heart failure-related hospitalizations, in the presence of terminal events, is acceptable as the primary endpoint, pending details of the statistical analysis plan and further discussion with agency statisticians; and |
| • | a single trial may be acceptable for a BLA submission assuming statistically significant primary outcome and strong concordance of primary and secondary endpoint analyses. |
In November 2013, the EMA indicated that if MYDICAR demonstrates a substantial and highly significant treatment effect in the advanced heart failure population, and no untoward effects attributable to MYDICAR are observed, a safety database of approximately 205-230 MYDICAR-treated subjects may be sufficient for a safety assessment to allow for acceptance of an MAA for MYDICAR for the treatment of systolic heart failure. We therefore believe that, if the above conditions are met, a Phase 3 trial may not be required for marketing approval in Europe. We have also held a Type A SPA meeting with the FDA, as a result of which the FDA approved a 572-patient Phase 3 trial protocol under the SPA guidance and agreed that the design and planned analyses of this trial would be sufficient to provide data that, depending on outcome, could support a license application submission. Pursuant to the SPA, we also obtained an agreement from the FDA that the primary efficacy endpoint of time-to-recurrent heart failure-related hospitalizations in the presence of terminal events would be acceptable for a pivotal trial of MYDICAR. This endpoint counts multiple heart failure-related hospitalizations per patient, and “corrects” for the occurrence of terminal events. Other elements of the Phase 3 SPA protocol may be changed if agreed to in writing by both the FDA and us, including sample size. In April 2014, the FDA granted Breakthrough Therapy designation to MYDICAR for reducing hospitalizations for heart failure in patients who test negative for adeno-associated viral vector 1 neutralizing antibodies, are class III or IV heart failure patients under the New York Heart Association, or NYHA, classification system, and are not in immediate need of a left-ventricular assist device, or LVAD, or heart transplant. We are currently in discussions with the FDA regarding the use of the joint frailty statistical model as a method of analysis for the primary endpoint. Our extensive simulation studies have demonstrated that when recurrent heart failure-related hospitalizations and terminal events are correlated, the joint frailty model provides both high power to detect a treatment effect and strong control of false-positive rate. The FDA is currently performing additional simulations using our proprietary software to validate that the false-positive rate is acceptable for a pivotal trial using the joint frailty model.
The design of our CUPID 2 trial is substantially similar in design to the Phase 3 SPA protocol. Our CUPID 2 trial uses the identical primary efficacy endpoint, which is important as we have obtained an agreement on this endpoint with the FDA for use in a pivotal trial.
In May 2012, we participated in European Scientific Advice Meetings with local authorities at the Paul Ehrlich Institute in Germany and the College ter Beoordeling van Geneesmiddelen, Medicines Evaluation Board in the Netherlands. Advice from these meetings was incorporated into the Clinical Trial Application for the CUPID 2 clinical trial. In November 2013 we met with the Scientific Advice Working Part of the EMA to obtain scientific advice regarding the overall development program and most expeditious approval route for MYDICAR and advice from this meeting was incorporated into the development program.
MYDICAR for Systolic Heart Failure
CUPID 2 Trial (CELL-004)
The primary objective of our ongoing CUPID 2 trial is to determine the efficacy of a single intracoronary infusion of high-dose MYDICAR compared to placebo, in conjunction with maximal optimized heart failure therapy, in reducing the frequency of and/or delaying heart failure-related hospitalizations in patients with systolic heart failure (EF less than 35%) who are at increased risk of terminal events based on elevated levels of NT ProBNP or a recent heart failure-related hospitalization.
40
Table of Contents
The population is adult patients, 18 to 80 years of age, with NYHA class III/IV symptoms of heart failure due to ischemic or non-ischemic cardiomyopathy, and who, despite maximal optimized heart failure therapy regimens, are at high risk of heart failure-related hospitalizations. A total of 250 patients (N= ~125 per treatment arm) were randomized for the purpose of obtaining at least 186 adjudicated heart failure-related hospitalizations.
Patients were randomized in parallel to high-dose MYDICAR or placebo in a 1:1 ratio. The trial is being conducted at approximately 53 sites in the United States, Denmark, Sweden, Germany, Poland, Belgium, the Netherlands, the United Kingdom, Israel and Hungary, with randomization stratified by country.
Potential trial participants were prescreened for the presence of NAbs against AAV1 using our companion diagnostic. Those who tested negative for AAV1 NAbs underwent further screening tests and procedures to determine eligibility prior to randomization and enrollment into the trial. Those who tested positive for AAV1 NAbs were excluded from the trial. Data analyses will be performed when all patients have completed the full 12-month active observation period and at least 186 adjudicated heart failure-related hospitalizations have occurred.
In CUPID 2, we enrolled advanced heart failure patients, at high-risk for serious adverse events and death. An independent data monitoring committee, or DMC, responsible for monitoring safety of the trial, has met three times. In all three meetings the DMC recommended that CUPID 2 proceed with its trial protocol as planned. Because CUPID 2 is an event-driven trial, all clinical events are reviewed by both the unblinded DMC and by an independent blinded Clinical Endpoint Committee, or CEC. The primary endpoint will be assessed at one year and all patients will be followed for a total of five years.
In CUPID 2 the endpoints were chosen to capture disease burden fully and to gain efficiency by including all terminal events (e.g. all-cause death, heart transplants and LVAD implantation) in the analyses. There are many statistical methods for the analysis of recurrent events; however, the joint frailty model addresses the limitations of other approaches, as it accounts for the correlation between the recurrent event process and the terminal event process (informative censoring).
The primary efficacy endpoint is time-to-recurrent advanced heart failure-related hospitalizations in the presence of terminal events at the time of primary analysis data cutoff. In the primary endpoint analysis the treatment effect estimate (hazard ratio for recurrent heart failure-related hospitalizations for MYDICAR versus placebo adjusted for correlated terminal events), will be calculated using the joint frailty model. The secondary endpoint is time-to-first terminal event (all-cause death, heart transplant or LVAD implantation). Additional endpoints include Kansas City Cardiomyopathy Questionnaire (quality of life) and six-minute walk test (exercise capacity). NYHA class will be descriptively summarized by time point for each treatment group.
The sample size for our CUPID 2 trial is based on Monte Carlo simulations so that approximately 250 patients with an estimated total of 186 heart failure-related hospitalizations, should provide at least 83% power at the 0.05 two-sided significance level to detect at least a 45% risk reduction (hazard ratio of 0.55) based on time-to-recurrent heart failure-related hospitalizations in the presence of the terminal events. The assumed magnitude of treatment effect is based on the data from published studies in heart failure patients and a conservative estimate of the anticipated magnitude of effect of MYDICAR based on 12-month results from CUPID 1 that showed an 88% reduction in recurrent clinical events adjusted for correlated terminal events with high-dose MYDICAR compared to placebo. We completed enrollment of this trial in February 2014 and expect to announce results in April 2015.
Upon completion of our CUPID 2 trial, the results will be discussed with the FDA and the EMA with the possibility that MYDICAR could potentially qualify for approval if the trial outcome demonstrates substantial reduction in recurrent heart failure-related hospitalizations and concordant trends in reduction in and/or delay of terminal events overall, and death in particular. We believe the results of this trial and a subsequent Phase 3 trial, if required, could support submission of a BLA for MYDICAR for the treatment of systolic heart failure. In
41
Table of Contents
November 2013, the EMA indicated that if MYDICAR demonstrates a substantial and highly significant treatment effect in the advanced heart failure population, and no untoward effects attributable to MYDICAR are observed, a safety database of approximately 205-230 MYDICAR- treated subjects may be sufficient for a safety assessment to allow for acceptance of an MAA for MYDICAR for the treatment of systolic heart failure. We therefore believe that, if the above conditions are met, a Phase 3 trial may not be required for marketing approval in Europe. However, there can be no assurance that regulatory agencies will not require one or more additional clinical trials prior to granting regulatory approval.
AGENT-HF Trial (AAV1-CMV-SERCA2a Gene Therapy Trial in Heart Failure)
This trial is an investigator initiated clinical trial which commenced screening in December 2013. The trial is partially funded by the French government and sponsored by Assistance Publique – Hôpitaux de Paris. We are providing investigational medicinal product and some financial support. This trial is not required by any regulatory authorities for systolic heart failure indications.
The primary objective of the AGENT-HF Trial is to determine whether treatment with MYDICAR leads to reverse remodeling of the heart. In patients with heart failure, the size, shape, structure and physiology of their heart changes over time, and these changes that lead to a progressive decline in left ventricular function are referred to as remodeling. In reverse remodeling, there would be changes back to the more normal, healthier state of the heart along with an improvement in the functioning of the heart. This trial is expected to enroll approximately 44 heart failure patients in France with half receiving MYDICAR and the other half placebo. The primary endpoint at six months will be change, compared to baseline, in left ventricular end systolic volume as measured by cardiac computed tomography.
CELL-005 AAV1 NAb Positive Trial
The primary objective of the AAV1 NAb positive trial is to determine the safety of a single intracoronary infusion of high-dose MYDICAR in patients who test positive for AAV1 NAbs. The FDA has required this safety study as a condition to the submission of a BLA, to cover the possibility that MYDICAR may be used off-label in AAV1 NAb positive patients. In addition, the trial would explore the potential level of activity of MYDICAR in AAV1 NAb positive patients, although the trial would not be of sufficient size to detect statistical differences in the response in patients who test positive for AAV1 NAbs versus those who test negative. The patient population would be similar to the target patient population in our CUPID 2 trial and would be approximately 60 patients. The study design would be a Phase 2, randomized, double-blinded, parallel study. Patients would be stratified by baseline AAV1 NAb titer – either negative/equivocal or positive (³1:2) – and randomized in parallel, in a 2:1 ratio, to either MYDICAR or placebo. The primary endpoint after all subjects had been followed for at least six months would be safety as measured by the incidence and severity of adverse events, including all-cause mortality and heart failure-related hospitalizations. The percentage of subjects experiencing an event would be calculated for survivors and for all patients enrolled. Frequency, type and duration of cardiovascular hospitalizations would also be analyzed. The CEC would classify all deaths and hospitalizations, distinguishing between the primary cause and immediate underlying cause of death or hospitalization. The following activity/efficacy variables would be summarized descriptively by treatment group as the trial is not powered to detect a statistical significance in any of the variables: left ventricular end systolic volume, distance walked during the six-minute walk test, NT-proBNP levels, NYHA classification, and quality of life assessed by the Kansas City Cardiomyopathy Questionnaire. We currently plan to conduct this trial in 2015.
CELL-006 Viral Shedding Trial
The viral shedding trial is required as part of the environmental risk assessment that must be included in a marketing application to regulatory authorities, both in the United States and in Europe. In this open-label trial, approximately 10 to 20 patients with heart failure (the same target patient population as our CUPID 2 trial and
42
Table of Contents
our AAV1 NAb positive trial) would be treated with high-dose MYDICAR and followed until they have two consecutive bodily fluid samples that are negative for presence of the SERCA2a gene, as assessed by qPCR. The patients would continue to be followed for safety for up to two years to add to the overall MYDICAR safety database. With the information from this trial, the marketing application would have information on how long treated patients would be excreting MYDICAR into the environment, thereby potentially spreading the virus to family members, health care workers and the public. We currently plan to conduct this trial in 2015.
CELL-008 Plasma Exchange Pilot Study
We are currently making arrangements to initiate a pilot, 24 patient, Phase 1/2 study of MYDICAR in advanced heart failure patients with systolic dysfunction who have been previously excluded from MYDICAR studies in this indication due to pre-existing levels of neutralizing antibodies against the AAV1 vector. We expect to initiate this trial in late 2014 and data are expected in 2015.
Preclinical Studies of MYDICAR in Systolic Heart Failure
Preclinical studies have shown that, after administration of an AAV vector, the plasmids containing the vector DNA are cleared from the blood and tissues via the mononuclear phagocyte system in liver, spleen and lymph nodes, and lungs. After intracoronary delivery, AAV particles which are not taken up in cardiac tissues are first passed through to the lung via the coronary sinus, making this the first pass organ. Stable, long-term presence of viral DNA, SERCA2a protein, and vector-derived SERCA2a mRNA have been demonstrated in cardiac tissue of normal rats for up to one year following a single administration of MYDICAR.
Gene transfer of SERCA2a is associated with improved cardiac function in various rodent models of heart failure. Improved heart function and enhanced expression of SERCA2 have also been demonstrated in an ovine (sheep) pacing-induced heart failure model with MYDICAR. SERCA2 gene transfer has also been associated with restoration of SERCA2a expression and improved heart function in both a dog-pacing heart failure model and in a chronic myocardial ischemia-induced heart failure model in mini-pigs. Beyond the effects on enhancing contractility, SERCA2a gene transfer has been shown in preclinical studies to restore the energetic state of the heart (both in terms of energy supply and utilization), to decrease arrhythmias, and enhance blood flow to the heart through expression in endothelial cells.
Several studies we have sponsored have established pharmacologic activity for MYDICAR gene transfer in animals with heart failure, with data demonstrating restored SERCA2a expression and stabilization/improvement in heart function. The pharmacology study was conducted in the porcine (pig) mitral regurgitation, or MR, heart failure model. MR induces reduced myocardial contractility, elevated B-type natriuretic peptide, or BNP, levels and other signs and markers which are virtually identical to those associated with the human disease, including a decrease in SERCA2a expression. MYDICAR-treated animals demonstrated significant improvements in the heart’s ability to contract and relax and improved ventricular volumes. In these studies, there was an absolute increase of 16% in median EF in MYDICAR-treated animals as compared to control animals. ESV increased in the control group by a median of 16 milliliters, or a median relative increase of 35%, an indication of decreased contractility and cardiac enlargement, compared with the MYDICAR group, which showed a tendency to decrease LV ESV by a median of 9.9 milliliters (a median decrease of 14%). In humans, a reduction in ESV of 10% signifies clinically relevant reverse remodeling, which is a strong predictor of lower long-term mortality and heart failure clinical events. Treated animals also had lower BNP levels post-dosing.
We have also sponsored two safety toxicology and biodistribution studies, both in normal mini-pigs. Both were three-month studies simulating the clinical administration procedure for MYDICAR or placebo with 5, 30 and 90 day sacrifice time points. Doses of up to three times the human dose on a weight-adjusted basis were administered. No mortalities were observed in either study and treatment with MYDICAR was not associated with any signs of toxicity or effects on body weight, sperm motility, clinical pathology, gross pathology, clinical chemistry parameters, organ weights or histopathology. No significant effects were observed on cardiovascular
43
Table of Contents
parameters, including electrocardiographic intervals. There were no test article-related observations during the necropsies. Mild increases in troponin I were observed in eight out of a total of 36 MYDICAR-treated animals in the first study, barely above upper limits of normal for humans. These increases were not considered to be related to MYDICAR or biologically significant and were not observed in the second study. No treatment related changes in troponin I values were observed across the other large animal pharmacology studies.
MYDICAR in Additional Indications
Beyond our proposed lead indication of systolic heart failure, we are also developing MYDICAR for additional indications including enhancement of AVF maturation, diastolic heart failure and treatment of patients with advanced heart failure who are on an LVAD. Each of these conditions is characterized by a SERCA2a deficiency, and MYDICAR has demonstrated disease-modifying capability in preclinical models of these diseases. We are currently engaged in preclinical research regarding MYDICAR for the treatment of diastolic heart failure, and plan to initiate human clinical trials in this indication in 2015 if data warrants. The broad potential of MYDICAR in multiple indications presents opportunities to maximize the value of our development programs for indications that are poorly managed by existing treatment options.
MYDICAR in Arteriovenous Fistula Maturation Failure (SERCA2a-AVF)
Currently, over 500,000 Americans have end-stage renal disease requiring dialysis. An arteriovenous fistula, or AVF, which is a surgically created connection between an artery and a vein in the arm of the patient, has proven to be the most durable, least complicated, and therefore preferred mode of vascular access for hemodialysis. The access that is created is routinely used for hemodialysis two to five times per week. Approximately 100,000 fistulae are placed yearly in the United States. However, a clinical problem that has resulted from this practice is that, following surgery to create the fistula, approximately 50% of the fistulae fail to mature to a usable state, and as many as 25% of hospital admissions in the dialysis population have been attributed to vascular access problems, including fistula malfunction and thrombosis.
Role of SERCA2a in Arteriovenous Fistula Maturation Failure
We believe MYDICAR has the ability to provide patients with end-stage renal disease a reliable and durable vascular access site for hemodialysis. The role of SERCA2a in normal and diseased blood vessel biology has been extensively studied. Maturation failure of an AVF has been attributed to rapid proliferation of vascular smooth muscle cells, or VSMC, resulting in vascular occlusion. The histological lesion that appears to be associated with early AVF failure is referred to as neointimal hyperplasia, comprising VSMC, myofibroblasts and endothelial cells within microvessels. In the setting of early AVF failure, both aggressive neointimal hyperplasia and adverse vascular remodeling (vasoconstriction or an inability to dilate adequately) plays a role. In particular, the combination of early and aggressive neointimal hyperplasia together with adverse vascular remodeling results in aggressive early stenosis. The biology of SERCA2a in both VSMC and endothelial cells provides a unique opportunity to potentially positively impact these pathological processes:
| • | Proliferation of VSMC is associated in the rat, rabbit, and human with loss of SERCA2a expression and is thought to be the dominant cell type driving neointimal hyperplasia. SERCA2a gene transfer inhibits in vitro VSMC proliferation and prevents neointimal thickening in a rat carotid-injury model and prevented in-stent restenosis using an ex vivo model of human left internal mammary artery intimal thickening. |
| • | In endothelial cells, SERCA2a modulates endothelial nitric oxide synthase, or eNOS, expression and activity. This enzyme produces nitric oxide, which dilates blood vessels. In a swine model of heart failure, coronary artery blood flow was decreased significantly, and MYDICAR rescued blood flow to levels observed in normal animals. In human artery endothelial cells, SERCA2a overexpression increased eNOS expression, phosphorylation, promoter activity and cellular Ca2+ storage capacity. Thus, SERCA2a gene transfer increases eNOS expression and activity by modulating calcium homeostasis, resulting in dilated blood vessels and improved blood flow. |
44
Table of Contents
| • | MYDICAR was tested in a pharmacology safety study in a swine model of vascular injury. MYDICAR-treated animals demonstrated reduced neointimal hyperplasia and less stenosis as compared to the control animals. |
The purpose of the SERCA2a AVF trial is to determine if MYDICAR, when applied to a limited segment of blood vessel during surgery to create an AVF, is safe, dilates the blood vessel, helps keep vessels open and improves the long-term function of the AVF. Discussions are underway with the FDA to determine what additional preclinical work will be required to support an IND for this potential new indication.
MYDICAR—LVAD Trial Investigation of the Safety and Feasibility of AAV1/SERCA2a Gene Transfer in Patients with Heart Failure and an LVAD
This trial is partially funded by the British Heart Foundation, sponsored by Imperial College London, and was recently initiated. We are providing investigational medicinal product and some financial support. It is not a required trial by any regulatory authorities; however, it could potentially serve as a proof-of-concept trial to support the use of MYDICAR to wean patients off of an LVAD. The use of these devices present a host of risk factors for the patient, such as increased risk of thrombosis and infections, and these devices do not last for long periods of time. Given that the circulatory system of a patient with an LVAD is dependent on these devices, device failure usually translates to a catastrophic event for the patient. The primary objectives of the SERCA2a-LVAD trial are to determine (1) the safety and feasibility of using MYDICAR to treat heart failure patients who have an LVAD, (2) how well MYDICAR delivers the gene for SERCA2a to heart cells and (3) what impact circulating NAbs to AAV1 have on the ability of MYDICAR to deliver the SERCA2a gene to heart muscle cells. This trial is expected to enroll approximately 24 patients in the United Kingdom with varying levels of circulating NAbs to AAV1, 16 of whom will be treated with MYDICAR and eight with placebo. Six months post-treatment, all patients will undergo a heart biopsy for collection of tissue to determine the presence of the SERCA2a gene. In addition, safety data and changes in LV function will be collected and analyzed.
MYDICAR—HF/pEF MYDICAR for Heart Failure with Preserved Ejection Fraction (Diastolic Heart Failure)
As in systolic heart failure, a consistent finding in diastolic heart failure is a decrease in the expression of SERCA2a—a change that is seen in most animal models of heart failure and in human hearts with diastolic dysfunction. In preclinical studies, overexpressing SERCA2a using gene therapy in streptozotocin-treated transgenic mice demonstrated that increasing SERCA2a could improve diastolic function. In human cardiomyocytes isolated from the left ventricle of patients with end-stage heart failure, SERCA2a levels were correlated with improved diastolic function. We have also evaluated MYDICAR in another preclinical study in a rat model for spontaneous non-insulin-dependent type 2 diabetes mellitus, which is characterized by diastolic dysfunction and associated with abnormal calcium levels and decrease in SERCA2a expression. In this study, SERCA2a gene transfer restored diastolic function to normal. These data showed that SERCA2a overexpression may be used as a therapeutic strategy for the treatment of this disease.
SERCA2a gene transfer has also been demonstrated to improve diastolic cardiac function in aged animals. In preclinical studies, cardiac SERCA2a protein and ATPase activity were significantly decreased in elderly rat hearts compared with adult rats and were restored to adult levels after SERCA2a gene transfer. Diastolic function parameters, which were adversely affected in elderly rat hearts, were restored by overexpression of SERCA2a, supporting the hypothesis that decreased SERCA2a contributes to the functional abnormalities observed in elderly hearts and demonstrating that targeting SERCA2a in the elderly heart may lead to improved diastolic cardiac function.
The MYDICAR- HF/pEF trial would be our pilot clinical trial for the treatment of diastolic heart failure, which comprises approximately half of all heart failure cases. We anticipate that the existing data we have generated for our proposed systolic heart failure indication would allow us to launch directly into a Phase 1/2 trial. We expect to initiate further development for this proposed indication in 2014 and initiate clinical studies in 2015.
45
Table of Contents
Small Molecule Program
We have initiated several pre-clinical studies with our novel, first in class, small molecule modulators of SERCA enzymes including for the treatment of diabetes and neurodegenerative diseases. We plan to commence additional pre-clinical studies for heart failure and PAH in 2014. We believe these compounds may correct underlying calcium dysregulation and ER stress which are implicated in many disease states.
Membrane-bound form of Stem Cell Factor (mSCF)
Following our recent acquisition in July 2014 of worldwide rights to gene therapy applications for the membrane-bound form of Stem Cell Factor for treatment of cardiac ischemia, we are pursuing an additional gene therapy product opportunity in mSCF for the treatment of cardiac ischemic damage. Stem cell research to date has demonstrated potential to treat heart failure, pulmonary disease, type 1 diabetes mellitus, Parkinson’s disease, Huntington’s disease, Celiac Disease, muscle damage, along with many others. mSCF is a powerful growth signal for c-kit+ stem cells, and is the ligand for the tyrosine kinase receptor c-kit. mSCF induces c-kit+ stem/progenitor cell expansion in situ, as well as cardiomyocyte proliferation, which may represent a new therapeutic strategy to reverse adverse remodeling after cardiac injury. In a preclinical setting, mSCF has demonstrated potential improvements in cardiac function and survival following a myocardial infarction. Specifically, these data suggest mSCF gene therapy promoted a regenerative response characterized by an enhancement in cardiac hemodynamic function; an improvement in survival; a reduction in fibrosis, infarct size and apoptosis; an increase in cardiac c-kit+ progenitor cells recruitment to the injured area; an increase in cardiomyocyte cell-cycle activation; and Wnt/ß-catenin pathway induction. To date, however, cell therapy for tissue repair has been hampered by the complexities of using cells as products from a delivery, manufacturing, and regulatory perspective.
Our approach with mSCF gene therapy is to recruit and expand resident stem cells, thereby harnessing advances in gene therapy technologies and also expanding the application to those in which cardiac stem cells have shown promise in clinical and preclinical testing. Our initial focus will be to generate clinically acceptable gene therapy vectors in support of potentially conducting a future clinical trial in patients who have suffered cardiac damage, as well as exploration of other potential applications.
Sales and Marketing
We currently have full worldwide commercial rights to all of our development programs. We believe we can maximize the value of our company by retaining substantial commercialization rights to our product candidates and, where appropriate, entering into partnerships for specific therapeutic indications and/or geographic territories.
Our current strategy is to market MYDICAR for all potential heart failure indications using a dedicated direct sales model focused on selected cardiologists and heart failure specialists. These physicians are typically affiliated with leading hospitals and medical centers and we believe that they tend to have well-established referral networks with supporting interventional cardiologists and cardiac catheterization laboratories. We believe they represent a concentrated customer base suitable to a specialist care sales model. We believe that MYDICAR would be adopted first by high-volume key-opinion-leader hospitals and medical centers, and progressively by a broader segment of the market. We believe that therapy adoption generally occurs much faster in the United States compared to Europe or the rest of the world. Cardiologists, heart failure specialists, and interventional cardiologists, have a history of early adoption of innovative products and technologies, in part because the rate of innovation in this sector has been sustained, and in part because of the large unmet need that their patients exhibit.
We therefore believe that a commercial strategy involving a progressive build out of commercial infrastructure in the United States covering key prescribers and centers of excellence is one that we can realistically pursue. Our commercialization strategy for MYDICAR in different geographies and indications beyond heart failure will continue to be evaluated and may involve strategic partners.
46
Table of Contents
Manufacturing of MYDICAR (AAV1/SERCA2a)
AAV has many characteristics that facilitate large scale manufacturing and distribution, when exploited effectively. We believe that our significant investment in AAV1/SERCA2a process development and analytical characterization has paid off in an inherently scalable, proprietary manufacturing process that is capable of supplying a global market as large as heart failure with a gene therapy product.
The technology includes a coordinated design of the AAV1/SERCA2a vector genome (the vector DNA) and the production system. AAV vectors are made “gutless,” meaning that they do not contain viral genes. Only the two small non-coding elements from the parent virus are needed for replicating and packaging the vector DNA during production, which can be provided separately. The genome was also designed to be very close to the size of the parent AAV genome, to optimally fit within the AAV capsid.
Our state of the art manufacturing process for AAV1/SERCA2a was developed based on proven industrial cell culture methodologies. Like many of the manufacturers of recombinant monoclonal antibodies or proteins, we use cell-suspension based culturing techniques and intend to use stirred tank bioreactors for large scale cell culture and production. Our envisioned commercial production scale is 2,000 liters, which is one-tenth the volume of the largest industrial production vessels, so our anticipated production scale is far from the limits of the technology. We selected stirred tank production bioreactor technology as our production system because it has been the workhorse for recombinant protein production for more than 20 years. For purification of AAV1/SERCA2a, we use industrial chromatography columns and resins, and filtration technology common to the biopharmaceutical industry. We believe these materials and equipment are common for manufacturing of FDA approved biological products.
Our Approach for Producing AAV1/SERCA2a
By specifically creating a cell line for the manufacture of AAV1/SERCA2a that has the necessary components stably integrated into the cell line, we have created a production process similar to other industrial processes used to treat large market disease indications.
We use standard cell culture techniques and standard equipment in production and purification found in industrial cell culture drug manufacturing. All media used for cell growth and production are free of animal serum and of high risk animal-derived components. To induce production of AAV1/SERCA2a, the cells are infected with a highly characterized batch of adenovirus. AAV viruses in nature and AAV vectors are not capable of replicating on their own and require a helper virus, such as adenovirus, to initiate replication. The purification process was designed to yield a high purity AAV1/SERCA2a product. Special attention was placed on the inactivation and removal of adenovirus and its free components, clearance of DNA and protein impurities, and even intact host cells.
MYDICAR drug product is produced by an FDA registered contract manufacturer. The manufacturing process is relatively simple: drug product is diluted to a specified concentration, filter-sterilized, and vials are aseptically filled into single-use standard pharmaceutical grade vials and stoppered using an automated filling machine. The final drug product is stored frozen or refrigerated until use.
Our Plans for Scale-Up and Our Approach to Commercial Manufacturing
Our production process has already been successfully scaled up from lab scale to the 250-liter clinical scale. Of the limited number of batches produced at 250 liters, two batches were successfully produced at Targeted Genetics Corporation (now AmpliPhi Biosciences Corporation) in Seattle, Washington. We expect risk for scale-up to the 2,000-liter commercial scale to be minimal, based on our knowledge and experience, and the proven track record of the stirred tank bioreactor technology and industrial chromatography. We have selected Lonza, a worldwide leader of biological product manufacturing with extensive experience in viral manufacturing, as our
47
Table of Contents
contract manufacturing organization for the production of AAV1/SERCA2a. We have worked with Lonza for nearly two years and have successfully transferred key analytical methods, manufacturing processes for critical reagents, and the AAV1/SERCA2a drug substance process to their laboratories. Our experienced technical staff continues to work closely with Lonza staff on all activities related to drug substance process development and manufacturing, and related ancillary activities. We are now preparing a demonstration batch for scale-up to commercial scale of production (2,000-liter production bioreactor), which we expect to begin later this year.
Our plan for commercial manufacturing is to establish commercial supply agreements with Lonza and/or other contract manufacturing organizations for product launch and commercial supply. We plan for the AAV1/SERCA2a manufacturing process to be designed and operated using standard off-the-shelf equipment, including a 2,000-liter disposable bioreactor platform, within a simple modular cleanroom. The concept is to have a production train that can be replicated in standardized fashion to ensure that from facility to facility the manufacturing process is operated exactly the same using identical equipment, material and supplies. We anticipate that one production train will meet our global product requirements for our expected first indication, systolic heart failure. However, if actual product demand is greater than anticipated or additional indications gain approval, we believe that the standardized approach will allow for an easy and quick start-up of additional production trains. Our approach is designed to minimize capital costs and provide nimbleness and expandability of the production process.
MYDICAR Clinical and Commercial Supply
We have enough MYDICAR clinical supplies (drug product) to complete the CUPID 2, MYDICAR-LVAD and AGENT-HF trials. We recently manufactured another batch of drug product which will provide clinical supplies for all of our currently planned clinical trials, including our plasma exchange trial, AAV NAb positive trial, viral shedding trial, and MYDICAR-AVF maturation trial, if commenced. An additional batch of drug substance and drug product supply would be required to conduct a Phase 3 clinical trial of MYDICAR, if required, or a diastolic heart failure trial, if commenced.
We have engaged Lonza for the manufacture of AAV1/SERCA2a, drug substance for MYDICAR, for use in our clinical trials, We are currently evaluating commercial supply arrangements with contract manufacturing organizations, including Lonza. Under such an arrangement, we currently envision construction of a stand-alone commercial manufacturing facility dedicated to MYDICAR production with capability up to 2,000-liter bioreactor capacity. We anticipate that MYDICAR will be launched from such a purpose-built commercial facility and production levels from the facility will be sufficient to meet our initial projected global commercial demand for MYDICAR.
Companion Diagnostic
The presence of pre-existing NAbs against the proteins that encapsulate the AAV1 gene therapy agent can block entry of the gene therapy agents into their target cells. Preclinical and limited clinical results with AAV1 NAb positive animals or patients, as well as in vitro neutralization experiments, have demonstrated that the detection of AAV1 NAbs is important prior to treatment with MYDICAR. Our experience in our CUPID 1 and CUPID 2 trials indicates that approximately 40% of the heart failure patients in the United States are AAV1 NAb negative and hence eligible for MYDICAR therapy. In other countries, such as Poland, the prevalence of pre-existing AAV1 NAbs is significantly higher.
We have developed a companion diagnostic AAV1 NAb assay for use in combination with MYDICAR in order to qualify subjects for treatment in clinical trials and for commercial use. The AAV1 NAb assay is intended to measure the loss of infectivity of AAV1/GFP (green fluorescent protein), an AAV1 recombinant particle with a reporter gene, following treatment with subject’s serum (i.e., neutralization). Diluted samples of a subject’s serum are incubated with AAV1/GFP, and then the mixture is tested for vector activity/infectivity in vitro on a permissive cell line (testing the relative gene expression (fluorescence) as a measure of vector neutralization).
48
Table of Contents
To date, our tests to measure a potential clinical trial participant’s level of pre-existing NAbs have been performed for us by Laboratory Corporation of America Holdings. We expect that the commercial assay, if approved, would be automated and similarly run by a strategic partner in several locations worldwide. It is not expected that the assay will be provided to the laboratories as a stand-alone kit but that approved laboratories would purchase the cells, controls and critical reagent, AAV1/GFP, from qualified suppliers. We intend that Quality System regulation set forth in 21 CFR Part 820 would be followed for the manufacture of AAV1/GFP and for the performance of the assay.
Companion diagnostics are subject to regulation by the FDA, the EMA and other foreign regulatory authorities as medical devices and require separate regulatory clearance or approval prior to commercial use. We anticipate that our companion diagnostic will require approval under a pre-market approval application, or PMA, submitted to the FDA’s Center for Devices and Radiological Health, or CDRH, prior to commercialization. We further anticipate that regulatory approval of our companion diagnostic will be a prerequisite to our ability to market MYDICAR. Representatives of CDRH have participated in our meetings with the Center for Biologics Evaluation and Research, or CBER, regarding MYDICAR to discuss the potential use of our companion diagnostic, and we anticipate that future meetings will include representatives from both CBER and CDRH to ensure that the BLA submission (for MYDICAR) and PMA submission (for the companion diagnostic) are coordinated and subject to parallel review by these respective FDA centers. Accordingly, our objective is to align the development programs such that the companion diagnostic will be developed and approved contemporaneously with MYDICAR.
MYDICAR Administration Devices
MYDICAR is administered in an outpatient cardiac catheterization laboratory by a qualified interventional cardiologist as a single dose intracoronary infusion using a legally marketed syringe pump and off-the-shelf components typically used for minimally invasive interventional procedures, including a 60 mL syringe, tubing, stopcocks and appropriate percutaneous catheter. MYDICAR and the syringe pump and catheters are regulated by the FDA as a biologic-device combination product. Cross-labeling may be required for MYDICAR and the administration devices at the time of a marketing approval of the combination product.
Competition
The biotechnology and pharmaceutical industries in which we operate are subject to rapid change and are characterized by intense competition to develop new technologies and proprietary products. We face potential competition from many different sources, including larger and better-funded pharmaceutical companies. While we believe that MYDICAR’s unique mechanism of action provides us with competitive advantages, particularly given that MYDICAR is designed to be administered in conjunction with other pharmacological agents and devices (except LVADs), we have identified several companies which are active in the advancement of gene therapy products in the heart failure arena as of the date of this prospectus. Not only must we compete with other companies that are focused on gene therapy treatments, any products that we may commercialize will have to compete with existing therapies and new therapies that may become available in the future.
Some of the pharmaceutical and biotechnology companies we expect to potentially compete with include Renova Therapeutics, NanoCor Therapeutics, Juventas Therapeutics, VentriNova and Beat BioTherapeutics. Renova, Beat BioTherapeutics and Juventas are in the clinical stages of development with their gene therapy products targeting moderate to advanced heart failure. Renova is using adenovirus serotype 5 encoding human adenylyl cyclase type 6 in a Phase 1/2 trial, while Juventas is enrolling a Phase 2 trial with its product candidate JVS100, which is a non-viral plasmid that encodes for stromal cell-derived factor-1 (SDF-1). NanoCor (BNP delivery of I1), VentriNova (cyclin A2), and Beat BioTherapeutics (AAV/R1R2) are in the preclinical testing of their gene therapy product candidates for the treatment of heart failure. These companies also compete with us in recruiting human capital and securing licenses to complementary technologies that may be critical to the success
49
Table of Contents
of our business. They also compete with us for potential funding from the biotechnology and pharmaceutical industries. Our potential competitors also include academic institutions, government agencies and research institutions. In addition, as the presence of pre-existing NAbs against the proteins that encapsulate the AAV1 gene therapy agent can block entry of the AAV1 gene therapy agents into their target cells, previous patient exposure to other AAV1-based gene therapies, irrespective of the condition or disease they aim to treat, would render a patient ineligible for MYDICAR therapy and could therefore be considered competitive to MYDICAR.
We believe that the key competitive factors that will affect the development and commercial success of MYDICAR and any other product candidates that we develop are efficacy, safety and tolerability profile, convenience in dosing, product labeling, value, price and the availability of reimbursement from the government and other third-parties. Our commercial opportunity could be reduced or eliminated if our competitors have products which are better in one or more of these categories.
Intellectual Property
We strive to protect and enhance the proprietary technologies that we believe are important to our business, and seek to obtain and maintain patents for any patentable aspects of our products or product candidates, including our companion diagnostic, their methods of use and any other inventions that are important to the development of our business. Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, maintain our licenses to use intellectual property owned by third parties, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and other proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen, and maintain our proprietary position in the fields targeted by our product candidates.
We are the owner or licensee of a portfolio of patents and patent applications and possess substantial know-how and trade secrets which protect various aspects of our business. The patent families comprising our patent portfolio are primarily focused on MYDICAR for the treatment of heart failure and are generally directed to certain genes, AAV vectors and methods of delivering such AAV vectors to cells, methods of delivery to myocardial cells and processes to manufacture our product candidates. We intend to leverage the intellectual property surrounding MYDICAR, together with the 12 years of available regulatory exclusivity that we expect to receive under the Biologics Price Competition and Innovation Act, as an important component of our business strategy.
Patent Protection for MYDICAR
Our portfolio of patents and patent applications related to MYDICAR generally relates to three aspects of MYDICAR: use of the SERCA2a gene for the treatment of heart failure; use and delivery of AAV vectors as a therapy; and manufacture of AAV vectors. The patent families which we believe are important for the protection of MYDICAR after its expected approval are summarized below. See also “Business—License Agreements.”
| • | Delivery of AAV Vectors to the Heart as a Therapy. We are the sole owner of two patent families related to a method of treating cardiovascular disease by infusion of a therapeutic nucleic acid, such as MYDICAR, into the coronary circulation over a specified period of time, either alone or optionally with a vasodilating substance such as nitroglycerine. Two patents have issued from these families (U.S. Patent Nos. 8,221,738 and 8,636,998), which includes claims to the use of a vasodilator in conjunction with MYDICAR. These patents are expected to expire in July 2030 and October 2028, respectively. We are currently prosecuting another method of use applications, and we expect that an additional patent or patents will issue from this family. If issued, these patents would expire between 2027 and 2028, excluding any potential additional term that may be available as a result of patent term adjustments, or if we elect to seek patent term extensions, or PTEs, that may be available under the Hatch-Waxman Act. In addition to the United States, corresponding patents have issued in Europe (EP 2044199), and Israel |
50
Table of Contents
| (IL 196541), and applications are pending in Australia, Europe, Hong Kong, India, and Japan. These patents and any patents issuing from the pending applications are expected to expire in July 2027 or October 2028. |
| • | Composition of MYDICAR. MYDICAR utilizes a hybrid AAV vector, where the various components of the AAV vectors (capsid proteins and/or genetic material) are from different AAV serotypes. We in-licensed two patent families containing patent applications related to recombinant hybrid AAV vectors, the first via a sublicense from the University of Pennsylvania, or UPenn, under our exclusive license agreement with AmpliPhi (formerly Targeted Genetics), and the second under our non-exclusive license agreement with AskBio LLC, or AskBio. We expect that these patent families (U.S. Patent Nos. 6,759,237, 7,186,552 and 7,172,893) will expire in November 2019 and February 2021, and we expect to pay a royalty to UPenn and AskBio upon commercialization of MYDICAR. Foreign patents corresponding to U.S. Patent Nos. 6,759,237 and 7,186,522 have issued in Australia (AU 768729 and AU 2004201463), Canada (CA 2,349,838), Europe (EP 1127150) and Japan (JP 2000/58122700), all of which are expected to expire in November 2019. Foreign patents corresponding to U.S. Patent No. 7,172,893 include issued patents in Australia (AU 780231), Canada (CA 2348382) and Europe (EP 1135468), all of which are expected to expire in November 2019. In addition, U.S. Patent No. 8,637,255, a family member of the patents sublicensed from UPenn, recently issued. This patent is directed to methods of assaying for the presence of neutralizing antibodies specific against a recombinant AAV virion. This patent is expected to expire in December 2019. |
| • | Manufacture of AAV Vectors. The manufacture and purification of the AAV vector used in MYDICAR is complicated and requires technical know-how. Our manufacturing process technology is protected by patents, trade secrets and proprietary know-how. We have obtained an exclusive license from AmpliPhi for certain aspects of the AAV manufacturing technology related to MYDICAR. This includes licenses to several patent families covering products and methods of manufacturing AAV vectors, including patent families related to stably transfected host cells for production of AAV vectors, and methods for commercial scale manufacturing and purification of recombinant AAV vectors. Taken in conjunction with our proprietary know-how, these patents are expected to offer additional protection by restricting competitors’ access to AAV manufacturing methods used to make MYDICAR or competing AAV-based products. In the United States, these patents (U.S. Patent Nos. 6,566,118, 6,989,264, 6,995,006 and 6,475,769) are expected to expire in September 2018. Corresponding foreign patents have issued in Australia (AU 758708, AU 772921, AU 2003204921), Canada (CA 2302992, CA 2342849), Europe (EP 1009808, EP 1109892), and Japan (JP 4472182), all of which are expected to expire in September of 2018 or 2019. Our exclusive license with AmpliPhi includes a patent family related to improved methods for purification of recombinant AAV vectors (WO 2010/148143) is pending in Brazil, Canada, China, Europe, Hong Kong, Israel, India, Japan, South Korea, Mexico, Russia, Singapore and the United States, and any resulting patents are expected to expire in June of 2030. |
| • | Use of SERCA2a for the Treatment of Heart Failure. We are developing MYDICAR for the treatment or prevention of heart failure through the use of AAV vectors to deliver the SERCA2a gene to improve cardiac function. We have licensed certain patent rights from The Regents of the University of California (including U.S. Patent No. 7,745,416) related to gene therapy for the purpose of increasing SERCA2a expression in the treatment of heart failure, which have been important in the development of MYDICAR, but these patent rights are expected to expire in the U.S. in 2015 prior to our anticipated approval of MYDICAR. Corresponding patents have issued in Australia (AU 2004204815) and Israel (IL169663), with expected expiration in January 2024, and Canada (CA 2217967) and Europe (EP 0820310, EP 1977767) which are expected to expire in April 2016. An application is pending in Canada. |
International Patent Protection for MYDICAR
We are the owner or licensee of numerous patents and patent applications in jurisdictions outside the United States. As noted above, most of the patent families discussed above have issued or are pending in foreign
51
Table of Contents
jurisdictions. Depending on the applicable national laws, these patents and patent applications (if applicable) covering MYDICAR may also benefit from extensions of patent term in individual countries.
Trade Secret Protection for MYDICAR
We exclusively in-license certain trade secret technology and know-how for manufacturing the AAV vector used in MYDICAR under our 2012 agreement with AmpliPhi. We believe that the expertise and materials licensed to us provide us with a commercial advantage over competitors attempting to utilize an AAV vector in their products.
U.S. Regulatory Protection for MYDICAR
In addition to patent and trade secret protection, we expect to receive a 12-year period of regulatory exclusivity from the FDA upon approval of MYDICAR pursuant to the Biologics Price Competition and Innovation Act. The exclusivity period, if granted, will run from the time of FDA approval. This exclusivity period, if granted, will supplement the intellectual property protection discussed above, providing an additional barrier to entry of any competitor seeking approval for a biosimilar version of MYDICAR.
In addition, it is possible to extend the patent term of one patent covering MYDICAR following FDA approval. This PTE is intended to compensate a patent owner for the loss of patent term during the FDA approval process. If eligible, we may use a PTE to extend the term of one of the patents discussed above beyond the expected expiration date, providing additional protection for MYDICAR.
Patent Protection of Pipeline Products
While the majority of our patent portfolio is related to MYDICAR and its use for treating heart failure, we are the owner or licensee of several additional patent families which relate to other technology which we are developing, including our small molecule program and our stem cell factor program. This includes treatments for additional indications using SERCA enzymes and MYDICAR, and new drugs for treating other SERCA-related diseases.
| • | Methods of Treating Stenosis. We in-license a patent family from The General Hospital Corporation related to using SERCA2a genes, including delivery by AAV vectors, to reduce stenosis, which is the narrowing of a blood vessel, or restenosis, which is the repeated narrowing in blood vessels. We expect that these patents (U.S. Patent Nos. 7,291,604 and 8,133,878) will expire no earlier than September 2024. |
| • | Methods of Treating Pulmonary Arterial Hypertension. We are the co-owner with the Mount Sinai School of Medicine of New York University, or Mount Sinai, of a patent family containing patent applications (U.S. Patent Pub. 2011/0256101) related to the use of genes, including SERCA, to treat pulmonary arterial hypertension, a type of high blood pressure that affects the arteries in the lungs and the right side of the heart. These applications are currently in prosecution, and we expect that any patents that may issue from this family of patent applications will expire no earlier than April 2031. We are the exclusive licensee of Mount Sinai’s joint ownership interest in this patent family pursuant to a license agreement. |
| • | Methods of Treating Heart Arrhythmia. We in-license a patent family from The General Hospital Corporation containing patent applications (U.S. Patent Pub. 2009/0239940) which disclose methods and materials for treating heart disease, including heart arrhythmia, using SERCA2a and AAV vectors. We expect that any patents which issue from this family of patent applications will expire no earlier than July 2018. |
| • | Activation of SERCA2a using Zinc Finger Technology. We are the sole owner of a patent family containing a patent application (U.S. Patent Pub. 2011/0172144) related to the use of a class of proteins known as zinc finger proteins to augment the expression of SERCA2a in cardiac muscle. Filed in January of 2011, we expect that any patent which issues from this application will expire no earlier than January of 2031. |
52
Table of Contents
| • | High-throughput Screening for SERCA Modulators and Their Use. We are the co-owner, with The Regents of the University of Minnesota, or UMinn, of a patent family (U.S. Patent No. 8,431,356) that relates to high-throughput screening methods used to identify small molecule compounds that modulate SERCA activity, as well as their use in treating SERCA-related disease. We are the exclusive licensee of UMinn’s joint ownership interest in these patents pursuant to a license agreement and we are solely responsible for the prosecution of these patents. We plan to use this technology to help identify product candidates which can be used to increase SERCA activity in muscle tissue, including the heart, to build a pipeline of SERCA-related therapies. We expect patents that may issue from these patent families to expire no earlier than January 2030. The current issued patent will expire in January 2030. |
| • | Methods of Treating Ischemic Diseases Using Stem Cell Factor Coding Sequences. We have received assignment of certain patent rights from Enterprise Partners Management, LLC, or Enterprise, related to certain gene therapy applications of mSCF for treatment of cardiac ischemia. Included within these rights is U.S. Patent No. 8,404,653, which has a projected expiration date of April 2029. Similar applications are pending in Europe (Publication No. EP1948246A2) and Hong Kong (Application No. 09100868.0). |
Trademarks
We have registered the trademark “MYDICAR” in the United States for use in connection with a biological product, namely, a gene transfer product composed of a recombinant AAV vector for medical use. We intend to pursue additional registrations in markets outside the United States where we plan to sell MYDICAR.
Patent Term
The term of individual patents and patent applications listed in previous sections will depend upon the legal term of the patents in the countries in which they are obtained. In most countries, the patent term is 20 years from the date of filing of the patent application (or parent application, if applicable). For example, if an international Patent Cooperation Treaty, or PCT, application is filed, any patent issuing from the PCT application in a specific country expires 20 years from the filing date of the PCT application. In the United States, however, if a patent was in force on June 8, 1995, or issued on an application that was filed before June 8, 1995, that patent will have a term that is the greater of 20 years from the filing date, or 17 years from the date of issue.
Under the Hatch-Waxman Act, the term of a patent that covers an FDA-approved drug or biological product may also be eligible for PTE. PTE permits restoration of a portion of the patent term of a U.S. patent as compensation for the patent term lost during product development and the FDA regulatory review process if approval of the application for the product is the first permitted commercial marketing of a drug or biological product containing the active ingredient. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application. The Hatch-Waxman Act permits a PTE for only one patent applicable to an approved drug, and the maximum period of restoration is five years beyond the expiration of the patent. A PTE cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and a patent can only be extended once, and thus, even if a single patent is applicable to multiple products, it can only be extended based on one product. Similar provisions may be available in Europe and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of a BLA, we expect to apply for PTEs for patents covering our product candidates and their methods of use.
For additional information on PTE, see “Business—Government Regulation.”
Proprietary Rights and Processes
We may rely, in some circumstances, on proprietary technology and processes (including trade secrets) to protect our technology. However, these can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with those who have access to our confidential
53
Table of Contents
information, including our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our proprietary technology and processes by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our proprietary technology and processes may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, scientific advisors, contractors, or any future collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For this and more comprehensive risks related to our proprietary technology and processes, please see “Risk Factors—Risks related to our intellectual property,” incorporated by reference from our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014.
License Agreements
License Agreement with The Regents of the University of California
In February 2001, we entered into a license agreement with The Regents of the University of California, or UC, under which we obtained an exclusive, worldwide license to UC’s patent rights in certain inventions, or the UC Patent Rights, related to the use of gene therapy vectors to deliver the SERCA2a gene to improve cardiac function, including certain patents related to MYDICAR. The agreement was amended twice, once in March 2001 to modify certain financial terms and once in January 2005 to make further amendments to the financial terms, with the second amendment also adding additional patents. We paid to UC an amendment fee of $114,455 and reimbursed UC for approximately $86,000 of previously incurred patent costs relating to the UC Patent Rights in connection with the second amendment of the agreement in January 2005.
Under the agreement, we are permitted to research, develop, manufacture and commercialize products utilizing the UC Patent Rights for gene therapy for the treatment or prevention of heart failure by the delivery of a gene or a synthetic equivalent, including SERCA2a, and to sublicense such rights. UC retained the right, on behalf of itself and other non-profit institutions, to use the UC Patent Rights for educational and research purposes and to publish information about the inventions covered by the UC Patent Rights.
In consideration for the rights granted to us under the agreement, we issued an aggregate of 83 shares of our common stock to UC upon the achievement of certain developmental milestones. We are required to issue to UC an additional 55 shares of our common stock and pay to UC up to an aggregate of approximately $1.6 million upon the achievement of certain developmental and regulatory milestones. In addition, upon commercialization of any product utilizing the UC Patent Rights, we will be required to pay to UC a low single-digit royalty on net sales of such product sold by us or our affiliates subject to minimum annual royalty payments and other adjustments in certain circumstances. However, we do not expect to commercialize MYDICAR prior to the expiration of the UC Patent Rights applicable to MYDICAR in the United States and Europe. Our obligation to pay milestones and royalties to UC terminates upon the expiration of the applicable UC Patent Rights.
In the event we sublicense a UC Patent Right, we are obligated to pay to UC a fee based on a percentage of sublicense fees received by us, which percentage ranges from the low-teens to mid-twenties depending on the country of origin of such UC Patent Right and is subject to adjustment in certain circumstances. In addition, we will also be required to pay to UC a low single-digit percentage sublicense royalty on net sales of products sold by our sublicensees that utilize the sublicensed UC Patent Right, but in no event will we be required to pay more than 50% of the royalties we receive from such sublicensees.
The agreement requires that we diligently develop, manufacture and commercialize products that are covered by the UC Patent Rights, and we have agreed to meet certain developmental and commercial milestones. UC may, at its option, either terminate the agreement or change the license granted from an exclusive license to a non-exclusive license if we fail to meet such milestones. We are currently in compliance with these milestone requirements.
54
Table of Contents
We may unilaterally terminate the agreement for any reason upon 90 days’ written notice to UC. UC may terminate the agreement in the event of our nonperformance or breach of the agreement if such nonperformance or breach remains uncured for 60 days following our receipt of written notice of such nonperformance or breach. Absent early termination, the agreement will continue until the expiration date of the longest-lived patent right included in the UC Patent Rights, which is expected to occur in 2024.
Exclusive License Agreement with Dr. Martin J. Kaplitt
In June 2006, we entered into an exclusive license agreement with Dr. Martin J. Kaplitt pursuant to which Dr. Kaplitt granted to us an exclusive, worldwide license under Dr. Kaplitt’s interest in certain patents related to the use of AAV vectors to deliver genes to cardiac muscles and delivery methods of AAV vectors to heart cells for the development, manufacture, use and sale of MYDICAR. The license granted to us under the agreement automatically became non-exclusive on the fourth anniversary of the effective date of the agreement. We have the right to grant sublicenses to third parties under the agreement.
In consideration for the rights granted to us under the agreement, we paid an upfront fee to Dr. Kaplitt of $25,000. We are also obligated to pay to Dr. Kaplitt an annual license maintenance fee of $6,000 during the term of the agreement. In addition, we are required to pay to Dr. Kaplitt a very low single-digit percentage royalty on net sales of products sold by us, our affiliates and our sublicensees that are covered by the licensed patents. Our royalty obligations continue on a product-by-product and country-by-country basis until the expiration of the last-to-expire valid claim in the licensed patents covering a licensed product in such country. Finally, we are obligated to pay to Dr. Kaplitt up to an aggregate of $200,000 upon the achievement of certain regulatory milestones.
We may unilaterally terminate the agreement upon 60 days’ written notice to Dr. Kaplitt. Dr. Kaplitt may terminate the agreement in the event of our material breach of the agreement if such breach remains uncured for 60 days following our receipt of written notice of such breach. Absent early termination, the agreement will automatically terminate upon the expiration of the last-to-expire of the licensed patents containing a valid claim, which is expected to occur in 2015, prior to the projected launch of our product candidates.
Sublicense Agreement and Amended and Restated License Agreement with AmpliPhi
Sublicense Agreement
In June 2012, we entered into a sublicense agreement with AmpliPhi, or the AmpliPhi Sublicense, pursuant to which AmpliPhi sublicensed to us certain rights under a separate agreement, the UPenn Agreement, which AmpliPhi entered into in 2009 with the Trustees of UPenn. Under the terms of the agreement, we obtained an exclusive, worldwide sublicense from AmpliPhi under certain UPenn patents related to AAV1 vectors for the development, manufacture, use and sale of companion diagnostics to MYDICAR. We have the right to grant sublicenses to our affiliates and third-party collaborators under the agreement solely for research, development or other non-commercial purposes, or as reasonably necessary, to our manufacturers or distributors, provided that we remain primarily liable and such downstream sublicenses are consistent with the terms of our agreement with AmpliPhi and prohibit further sublicensing. In addition, we are required to use commercially reasonable efforts to meet certain developmental, regulatory and commercial milestones with respect to companion diagnostics under the agreement. We are currently in compliance with these milestone requirements. While we have sole control over the development and commercialization of companion diagnostics under the agreement, AmpliPhi has the first right to prosecute and maintain the licensed patents, subject to our right to consult with AmpliPhi with regard to such prosecution and maintenance upon our reasonable request.
In consideration for the sublicense granted to us under the agreement, we paid to AmpliPhi a sublicense initiation fee of $310,000, and we are obligated to pay to AmpliPhi an annual sublicense maintenance fee of $310,000. We are also required to pay to AmpliPhi a low single-digit percentage royalty based on net sales of any companion diagnostic covered by a licensed patent sold by us, our affiliates or our sublicensees. Our royalty
55
Table of Contents
obligations continue on a companion diagnostic-by-companion diagnostic and country-by-country basis until the expiration of the last-to-expire valid claim in a licensed patent covering the applicable companion diagnostic in such country. Finally, we are obligated to pay to AmpliPhi all royalty and milestone payments that become due and payable by AmpliPhi to UPenn under the UPenn Agreement as a result of our exercise of the sublicense granted under our agreement with AmpliPhi, including a low single-digit tiered percentage royalty on net sales of any companion diagnostic sold by us, our affiliates or our sublicensees, which royalty is separate from and in addition to the royalty payable to AmpliPhi described above, and up to an aggregate of $850,000 in potential milestone payments per product covered by the licensed patents.
We may unilaterally terminate the agreement upon 30 days’ written notice to AmpliPhi. Absent early termination, the agreement will automatically terminate upon the expiration of the last-to-expire licensed patent, which is expected to occur in 2019.
Amended and Restated License Agreement
We entered into an amended and restated license agreement with AmpliPhi concurrently with the AmpliPhi Sublicense that both amended the terms of the license agreement which we entered into with AmpliPhi in 2009 and terminated our manufacturing agreement with AmpliPhi which we entered into in 2009. Under the agreement, we obtained an exclusive, worldwide license under certain patents and know-how related to AmpliPhi’s AAV vector and manufacturing technology for the development, manufacture, use and sale of MYDICAR. We have the right to grant sublicenses to our affiliates and third-party collaborators under the agreement for research, development or other non-commercial purposes, or as reasonably necessary, to our manufacturers or distributors, provided that we remain primarily liable and such sublicenses comply with the terms of our agreement with AmpliPhi and prohibit further sublicensing. In addition, we have agreed to use commercially reasonable efforts to meet certain diligence milestones with respect to the development and commercialization of at least one product covered by the UPenn patent rights licensed to AmpliPhi by UPenn under the UPenn Agreement. We are currently in compliance with these milestone requirements. While we have sole control over development and commercialization of products covered by the licensed patents, AmpliPhi has the first right to prosecute and maintain the licensed patents, subject to our right to consult with AmpliPhi with regard to such prosecution and maintenance upon our reasonable request.
During the term of the agreement, we are obligated to pay to AmpliPhi all royalty and milestone payments that become due and payable by AmpliPhi to UPenn under the UPenn Agreement as a result of our exercise of the sublicense granted under our agreement with AmpliPhi. This includes a low single-digit tiered percentage royalty on net sales of MYDICAR and any other product covered by the licensed patents sold by us, our affiliates or our sublicensees, and up to $850,000 in milestone payments upon the achievement of certain developmental and regulatory milestones related to MYDICAR and any other product covered by the licensed patents.
The agreement does not provide either party with termination rights and does not have a provision for expiration or automatic termination.
License Agreement with AdVec
In February 2009, we entered into a license agreement with AdVec, Inc., or AdVec, under which we obtained a non-exclusive, worldwide license to use and acquire from AdVec’s distributor certain human embryo kidney cells transformed by Adenovirus 5 DNA, or 293 Cells, and certain AdVec know-how related to 293 Cells for use in testing of MYDICAR for lot release. In consideration for the rights granted to us under the agreement, we are obligated to pay to AdVec an annual license maintenance fee of $5,000.
Either party may terminate the agreement upon written notice of the other party’s insolvency or bankruptcy or upon the other party’s breach of the agreement if such breach remains uncured after 60 days of receipt of written notice of such breach. Absent early termination, the agreement will remain in effect until the tenth
56
Table of Contents
anniversary of the effective date. Thereafter, the agreement will automatically renew for successive five-year terms unless either party notifies the other party in writing at least 90 days prior to the end of any such five-year term of its election not to renew the agreement.
Non-Exclusive License Agreement with Virovek
In November 2010, we entered into a non-exclusive license agreement with Virovek Incorporation, or Virovek, under which we obtained a non-exclusive, worldwide license under certain patent rights and trade secrets related to Virovek’s AAV baculovirus technology to develop, manufacture, use and sell AAV1/GFP vector reagents as part of a companion diagnostic. Under the terms of the agreement, we have the right to grant sublicenses to third parties, and we are required to use commercially reasonable efforts to develop and commercialize a companion diagnostic to MYDICAR. We are currently in compliance with this requirement.
In consideration for the rights granted to us under the agreement, we paid to Virovek an up-front license fee of $15,000, and we are obligated to pay to Virovek an annual maintenance fee of $20,000, which fee is creditable against royalties due under the agreement. We are also required to pay to Virovek a percentage royalty in the mid-teen range based on upfront, annual, milestone, royalty and other payments received by us as a result of the performance of companion diagnostics by us, our affiliates and our sublicensees, subject to adjustment in certain circumstances. Our royalty obligations continue on a companion diagnostic-by-companion diagnostic and country-by-country basis until the expiration of the last-to-expire valid claim in a licensed patent covering the companion diagnostic in such country, which is expected to occur in 2027, or 10 years from the date of first commercial sale in such country if the companion diagnostic is covered only by licensed trade secrets.
We may unilaterally terminate the agreement upon 60 days’ notice to Virovek. Either party may terminate the agreement for the other party’s material breach of the agreement if such breach remains uncured after 90 days of receiving written notice of such breach. Absent early termination, the agreement will automatically terminate upon the expiration of our royalty payment obligations.
Non-Exclusive License Agreement with AskBio
In January 2008, we entered into a non-exclusive license agreement with AskBio, a wholly owned subsidiary of Asklepios Biopharmaceutical Inc., under which we obtained a non-exclusive, worldwide license under certain patents related to recombinant AAV vectors to develop, manufacture, use and sell MYDICAR. We have the right to grant sublicenses to third parties under the agreement provided that such sublicenses are entered into pursuant to a written sublicense agreement containing terms consistent with our agreement with AskBio.
Under the terms of the agreement, we granted to AskBio an option to obtain a non-exclusive, worldwide license under certain of our patent rights related to infusion of AAV in the arteries of the heart to develop, manufacture, use and sell products for the treatment of cardiac diseases. This option includes our currently pending patent application related to a method of treating cardiovascular disease by infusion of a therapeutic nucleic acid into the coronary circulation over a specified period of time. It does not include our issued patent in this family, which includes claims to the concurrent use of a vasodilating substance such as nitroglycerine. If AskBio timely exercises its option to obtain the license under the agreement on or before the earlier of January 15, 2015 and within 60 days following notice that a patent has issued from the patent applications included within the patent rights subject to the option, we will enter into a separate license agreement with AskBio with respect to such license with previously agreed upon payment terms. Although the scope of the license granted to AskBio upon exercise of the option would enable AskBio to develop and commercialize a competing product with respect to the patent rights to which the option applies, we believe that the exclusion of our issued patent from that license, and the scope of our anticipated regulatory approvals, will prevent AskBio from being able to launch any product that is able to compete directly with MYDICAR.
In consideration for the rights granted to us under the agreement, we paid to AskBio license fee payments of $150,000 in the aggregate. In addition, we are obligated to pay to AskBio an annual maintenance fee of
57
Table of Contents
$100,000. Upon commercialization of any product utilizing the licensed patents, we will also be required to pay to AskBio a low single-digit percentage royalty on net sales of such products, including MYDICAR. Our royalty obligations continue on a product-by-product and country-by-country basis until the expiration of the last-to-expire valid claim in a licensed patent covering the applicable product in such country, which is expected to be in 2021. We are also obligated to reimburse AskBio for up to an aggregate of $355,000 per licensed product upon the achievement of certain clinical, regulatory and sales milestones that may become due and payable by AskBio under a separate agreement between AskBio and the University of North Carolina at Chapel Hill from 2003.
We may unilaterally terminate the agreement upon 180 days’ written notice to AskBio. Either party may terminate the agreement for the other party’s material breach of the agreement if such breach is not cured after 30 days of receiving written notice of such breach. Absent early termination, the agreement will continue in effect until the expiration of our royalty payment obligations under the agreement.
Exclusive Patent License with the Regents of the University of Minnesota
We are joint owners with UMinn of the rights in a certain patent related to screening technology for isolation of small molecule modulators of SERCA enzymes (fluorescence resonance energy transfer, or FRET, assays). In May 2009, we entered into an exclusive patent license agreement with UMinn under which we obtained an exclusive license to UMinn’s joint ownership interest in the patent application that led to the current issued patent. We have the right to grant sublicenses to third parties under the agreement, and UMinn retained the right to use the licensed technology for non-commercial research and educational purposes.
We have agreed to meet certain performance milestones under the agreement, the deadline for which may be extended at our request provided that we have used commercially reasonable efforts to achieve such milestones by the applicable deadlines. We are currently in compliance with these milestone requirements. We have the first right to prosecute and maintain the applicable patent family.
In consideration for the rights granted to us under the agreement, we made an upfront payment to UMinn of $120,000. In addition, we are obligated to pay to UMinn an annual license fee of $120,000. The annual license fee will increase to $325,000 if we (1) undergo a change of control, (2) assign the agreement, any of our rights or obligations under the agreement or our joint ownership interest in the licensed technology, (3) receive a certain amount in license and sublicense revenues under the agreement, (4) file an IND, new drug application, or NDA, BLA or orphan drug application (or a foreign equivalent of any such application) for a product covered by the licensed technology, or (5) enter into any agreement with a third party to market or use the licensed technology, subject to certain exceptions.
We may unilaterally terminate the agreement upon 90 days’ written notice to UMinn. UMinn may terminate the agreement upon 10 days’ written notice to us upon our insolvency or for our breach of the agreement if such breach remains uncured for 90 days after we receive notice of such breach, or 30 days in the case of a non-payment breach. Absent early termination, the agreement will automatically terminate upon the expiration of all active claims in any licensed patent or patent application, which is expected to occur no earlier than January 2030.
Material Transfer and Exclusivity Agreement with Les Laboratoires Servier
In February 2014, we and Servier entered into a material transfer and exclusivity agreement, pursuant to which we agreed to transfer to Servier samples of certain proprietary compounds from our small molecule SERCA2b modulator program and granted to Servier a non-exclusive, non-sublicensable, royalty-free license to conduct certain studies of the samples for the purpose of evaluating Servier’s interest in negotiating a potential license and research collaboration agreement with us relating to small molecule SERCA2b modulators, or Compounds, for the treatment of type 2 diabetes and other metabolic diseases.
58
Table of Contents
Subject to earlier termination of the agreement as described below, the term of Servier’s license to conduct the evaluation, or the evaluation period, will expire six months after Servier’s initial receipt from us of the samples, provided that Servier may extend the evaluation period for up to an additional two months.
Under the terms of the agreement, we also granted to Servier the exclusive right to negotiate for an exclusive, royalty-bearing license to develop and commercialize Compounds, and products containing Compounds, in the field of type 2 diabetes and other metabolic diseases, solely outside of the United States and its territories and possessions on the terms and conditions set forth in the agreement and other commercially reasonable terms to be negotiated in good faith by the parties and set forth in a definitive license and research collaboration agreement.
Exclusive Patent License with Enterprise Partners
On July 18, 2014, we and Enterprise entered into an Assignment and License Agreement, pursuant to which Enterprise granted to us an exclusive, worldwide license and the assignment of patents held by Enterprise relating to certain gene therapy applications of mSCF for the treatment of cardiac ischemia. We have the right to grant sublicenses to third parties under the agreement.
In consideration for the rights granted to us under the agreement, we paid an upfront fee to Enterprise of $160,000. We are also obligated to pay to Enterprise a milestone payment in the amount of $1,000,000 upon the grant to us, or an affiliate or sublicensee of ours, of the first regulatory approval in the United States of a product that is covered by the licensed patents. In addition, we are required to pay to Enterprise a 2% royalty on net sales of products sold by us or by our affiliates or sublicensees that are covered by the licensed patents. Our royalty obligations continue on a product-by-product and country-by-country basis until the expiration of the last-to-expire valid claim in the licensed patents covering a licensed product in such country.
We may unilaterally terminate the agreement upon written notice to Enterprise. Enterprise may terminate the agreement in the event of our material breach of the agreement if such breach remains uncured for 90 days following receipt of written notice of such breach. Absent early termination, the agreement will automatically terminate upon the expiration of the last-to-expire of the licensed patents containing a valid claim.
Manufacturing
Manufacturing Services Agreement with Lonza
In August 2012, we entered into a manufacturing services agreement with Lonza, which we subsequently amended and restated in August 2013. Under the terms of the agreement, Lonza provides manufacturing services to produce MYDICAR at a scale sufficient for our clinical trials to date. We pay for manufacturing services performed by Lonza under the agreement pursuant to statements of work entered into from time to time.
We may unilaterally terminate the agreement upon six months’ written notice to Lonza. Lonza may terminate the agreement upon written notice to us, provided that such termination by Lonza will not be effective until the earlier of one year after the date we receive such written notice or our qualification of an alternative supplier and completion of certain technology transfer assistance services to establish manufacturing capabilities at the alternative supplier’s facilities. Either party may terminate the agreement in the event of the other party’s insolvency or for the other party’s material breach of the agreement if such breach remains uncured after 30 days of receiving written notice of such breach or after 180 days of receiving written notice of such breach if such breach is not a non-payment related breach, is not capable of being cured within 30 days and the breaching party is making diligent efforts to cure such breach. In addition, either party may terminate the agreement, by providing two months’ written notice to the other party if it receives notice that the production of MYDICAR under the agreement or clinical trials for which MYDICAR is being produced has been or will be suspended or terminated by the FDA or EMA due to product failure. Absent early termination, the agreement will continue until the fifth anniversary of the effective date of the original agreement.
59
Table of Contents
Government Regulation
Biological products, including gene therapy products, are subject to regulation under the Federal Food, Drug, and Cosmetic Act, or FD&C Act, and the Public Health Service Act, or PHS Act, and other federal, state, local and foreign statutes and regulations. Both the FD&C Act and the PHS Act and their corresponding regulations govern, among other things, the testing, manufacturing, safety, purity, potency, efficacy, labeling, packaging, storage, record keeping, distribution, reporting, advertising and other promotional practices involving biological products. FDA approval must be obtained before clinical testing of a biological product begins, and each clinical trial protocol for a gene therapy product is reviewed by the FDA and, in some instances, the U.S. National Institutes of Health, or NIH, through its Recombinant DNA Advisory Committee, or RAC. FDA approval also must be obtained before marketing of biological products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources and we may not be able to obtain the required regulatory approvals. To date, the FDA has never approved a gene therapy product for commercial sale. Within the FDA, the Center for Biologics Evaluation and Research, or CBER, regulates gene therapy products, CDRH regulates companion diagnostics, and the Office of Combination Products, or OCP, issues classification and jurisdiction assignments for medical products. Specifically, OCP determines how combination products, such as biologic/medical device combination products, will be regulated and which FDA Center or Lead Center (e.g., CBER or CDRH) will regulate the product.
CBER works closely with the NIH and its RAC, which makes recommendations to the NIH on gene therapy issues and engages in a public discussion of scientific, safety, ethical and societal issues related to proposed and ongoing gene therapy protocols. The FDA and the NIH have published guidance documents with respect to the development and submission of gene therapy protocols. The FDA also has published guidance documents related to, among other things, gene therapy products in general, their preclinical assessment, observing subjects involved in gene therapy studies for delayed adverse events, potency testing and chemistry, manufacturing and control information in gene therapy INDs.
Ethical, social and legal concerns about gene therapy, genetic testing and genetic research could result in additional regulations restricting or prohibiting the processes we may use. Federal and state agencies, congressional committees and foreign governments have expressed interest in further regulating biotechnology. More restrictive regulations or claims that our products are unsafe or pose a hazard could prevent us from commercializing any products. New government requirements may be established that could delay or prevent regulatory approval of our product candidates under development. It is impossible to predict whether legislative changes will be enacted, regulations, policies or guidance changed, or interpretations by agencies or courts changed, or what the impact of such changes, if any, may be.
U.S. Biological Products Development Process
The process required by the FDA before a biological product may be marketed in the United States generally involves the following:
| • | completion of nonclinical laboratory tests and animal studies according to good laboratory practices, or GLPs, and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| • | submission to the FDA of an IND application, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations, commonly referred to as good clinical practices, or GCPs, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed biological product for its intended use; |
60
Table of Contents
| • | submission to the FDA of a BLA for marketing approval that includes substantive evidence of safety, efficacy, purity and potency from results of nonclinical testing and clinical trials; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with GMP, to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity; |
| • | potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and |
| • | FDA review and approval, or licensure, of the BLA. |
Before testing any biological product candidate, including a gene therapy product, in humans, the product candidate enters the preclinical testing stage. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs.
Where a gene therapy trial is conducted at, or sponsored by, institutions receiving NIH funding for recombinant DNA research, prior to the submission of an IND to the FDA, a protocol and related documentation is submitted to and the trial is registered with the NIH Office of Biotechnology Activities, or OBA, pursuant to the NIH Guidelines for Research Involving Recombinant DNA Molecules, or NIH Guidelines. Compliance with the NIH Guidelines is mandatory for investigators at institutions receiving NIH funds for research involving recombinant DNA, however many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them. The NIH is responsible for convening the RAC, a federal advisory committee, which discusses protocols that raise novel or particularly important scientific, safety or ethical considerations at one of its quarterly public meetings. The OBA will notify the FDA of the RAC’s decision regarding the necessity for full public review of a gene therapy protocol. RAC proceedings and reports are posted to the OBA website and may be accessed by the public.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. With gene therapy protocols, if the FDA allows the IND to proceed, but the RAC decides that full public review of the protocol is warranted, the FDA will request at the completion of its IND review that sponsors delay initiation of the protocol until after completion of the RAC review process. The FDA may also impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials involve the administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or
61
Table of Contents
servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Clinical trials also must be reviewed by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees basic and clinical research conducted at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1. The biological product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase 2. The biological product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. The FDA recommends that sponsors observe subjects for potential gene therapy-related delayed adverse events for a 15-year period, including a minimum of five years of annual examinations followed by ten years of annual queries, either in person or by questionnaire, of trial subjects.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, the NIH and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk, including risks inferred from other unrelated gene therapy trials. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
Human gene therapy products are a new category of therapeutics. Because this is a relatively new and expanding area of novel therapeutic interventions, there can be no assurance as to the length of the trial period, the number of patients the FDA will require to be enrolled in the trials in order to establish the safety, efficacy, purity and potency of human gene therapy products, or that the data generated in these trials will be acceptable to
62
Table of Contents
the FDA to support marketing approval. The NIH and the FDA have a publicly accessible database, the Genetic Modification Clinical Research Information System which includes information on gene transfer studies and serves as an electronic tool to facilitate the reporting and analysis of adverse events on these studies.
Concurrently with clinical trials, companies usually complete additional animal studies and must also develop additional information about the physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with GMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHS Act emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
After the completion of clinical trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA must include results of product development, laboratory and animal studies, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. The FDA may grant deferrals for submission of data or full or partial waivers. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual product fee for biological products and an annual establishment fee on facilities used to manufacture prescription biological products. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe and potent, or effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with GMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the biological product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the biological product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve a BLA without a REMS, if required.
Before approving a BLA, the FDA may inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in
63
Table of Contents
compliance with GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure GMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
The FDA has agreed to certain review goals under PDUFA, and aims to complete its review of 90% of standard BLAs within ten months from filing and 90% of priority BLAs within six months from filing. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests, or the BLA sponsor otherwise provides, additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Fast Track Designation, Accelerated Approval, Priority Review and Breakthrough Therapy Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biological product may request the FDA to designate the drug or biological product as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Other types of FDA programs intended to expedite development and review, such as priority review, accelerated approval and Breakthrough Therapy designation, also exist. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may
64
Table of Contents
be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials to confirm the effect of the endpoint. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
A product may also be eligible for receipt of a Breakthrough Therapy designation. The Breakthrough Therapy designation is intended to expedite the FDA’s review of a potential new drug for serious or life-threatening diseases or conditions where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy provides the same benefits as are available under the Fast Track program, as well as intensive FDA guidance on the product’s development program. Fast Track designation, priority review, accelerated approval and Breakthrough Therapy designation do not change the standards for approval, but may expedite the development or approval process.
Combination Products
Combination products include products where two or more separate products are packaged together (e.g., drug and device products); or a product packaged separately but intended for use only with an approved, individually specified product, where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product, the labeling of the individually specified product would need to be changed (e.g., to reflect a change in intended use). We believe MYDICAR and certain delivery device components will be regulated as a combination product.
Regulation of Companion Diagnostics
In the United States, the FD&C Act and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Companion diagnostic tests are classified as medical devices under the FD&C Act. Unless an exemption applies, diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution. The two primary types of FDA marketing authorization applicable to a medical device are premarket notification, also called 510(k) clearance, and PMA approval. We anticipate that the companion diagnostic tests we are developing will be subject to the PMA approval process.
PMA applications must be supported by valid scientific evidence, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. For diagnostic tests, a PMA application typically includes data regarding analytical and clinical validation studies. As part of its review of the PMA, the FDA will conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the Quality System Regulation, or QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures. FDA review of an initial PMA application is required by statute to take between six to ten months, although the process typically takes longer, and may require several years to complete. If the FDA evaluations of
65
Table of Contents
both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an approvable letter, which usually contains a number of conditions that must be met in order to secure the final approval of the PMA. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the PMA approvable. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the PMA. Once granted, PMA approval may be withdrawn by the FDA if compliance with post approval requirements, conditions of approval or other regulatory standards is not maintained or problems are identified following initial marketing.
We and our third-party collaborators who may develop our companion diagnostics will work cooperatively to generate the data required for submission with the PMA application, and will remain in close contact with the CDRH to ensure that any changes in requirements are incorporated into the development plans. We anticipate that, as was the case in our meetings to date, future meetings with the FDA with regard to MYDICAR and its companion diagnostic product candidate will include representatives from both CBER and CDRH to ensure that the BLA and PMA submissions are coordinated to enable the FDA to conduct a parallel review of both submissions. On August 6, 2014, the FDA issued a guidance document addressing the development and approval process for “In Vitro Companion Diagnostic Devices.” According to the guidance, for novel products such as MYDICAR, the PMA for a companion diagnostic device should be developed and approved contemporaneously with the biological product. We believe our programs for the development of our companion diagnostics are consistent with the guidance. On April 23, 2014, the FDA issued for comment a draft guidance document proposing a new, voluntary program for certain medical devices that demonstrate the potential to address unmet medical needs for life threatening or irreversibly debilitating diseases or conditions that are subject to PMA applications, referred to as the Expedited Access PMA or EAP program. The draft guidance includes references to companion diagnostics as potentially being eligible for the EAP program if the stated criteria are satisfied. The proposed program is designed to help patients have more timely access to these medical devices by expediting their development, assessment and review, while preserving the statutory standard of reasonable assurance of safety and effectiveness for premarket approval. For example, as part of the proposed EAP program, on a case-by-case basis, the FDA may, where appropriate, allow a sponsor to provide less manufacturing information in their PMA application. While this draft guidance is not yet finalized, we believe that the review and approval of our companion diagnostic may qualify for and benefit from elements of the EAP program if the program is ultimately implemented as proposed.
Post-approval Requirements
Maintaining substantial compliance with applicable federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect to GMP. We will rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Manufacturers of our products are required to comply with applicable requirements in the GMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biological products include reporting of GMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency and effectiveness of biological products.
66
Table of Contents
We also must comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Biological product manufacturers and other entities involved in the manufacture and distribution of approved biological products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMPs and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain GMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved BLA, including withdrawal of the product from the market. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Coverage and Reimbursement
Sales of our products, when and if approved for marketing, will depend, in part, on the extent to which our products will be covered and reimbursed by third-party payors, such as federal, state and foreign government healthcare programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of our products once approved and have a material adverse effect on our sales, results of operations and financial condition.
Other Healthcare Laws
Although we currently do not have any products on the market, we may be subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation, state and federal anti-kickback, false claims, privacy and security and physician sunshine laws and regulations. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
67
Table of Contents
Additional Regulation
In addition to the foregoing, state and federal laws regarding environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy and Recovery Act and the Toxic Substances Control Act, affect our business. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in, and wastes generated by, our operations. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and that continued compliance therewith will not have a material adverse effect on our business. We cannot predict, however, how changes in these laws may affect our future operations.
U.S. Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act, to which we are subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity.
Government Regulation Outside of the United States
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. Because biologically sourced raw materials are subject to unique contamination risks, their use may be restricted in some countries.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a CTA must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and the IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational biological product under European Union regulatory systems, we must submit a marketing authorization application. The application used to file the BLA in the United States is similar to that required in the European Union, with the exception of, among other things, country-specific document requirements. The European Union also provides opportunities for market exclusivity. For example, in the European Union, upon receiving marketing authorization, new chemical entities generally receive eight years of data exclusivity and an additional two years of market exclusivity. If granted, data exclusivity prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application. During the additional two-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic product can be marketed until the expiration of the market exclusivity. However, there is no guarantee that a product will be considered by the European Union’s regulatory authorities to be a new chemical entity, and products may not qualify for data exclusivity.
68
Table of Contents
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Employees
As of July 30, 2014, we had 22 full-time employees, consisting of research, process development, manufacturing, finance, legal, administration and business development personnel. We also regularly use independent contractors across the organization to augment our regular staff. None of our employees are covered by collective bargaining agreements and we consider relations with our employees to be good. We believe that our future success will depend in part on our continued ability to attract, hire and retain qualified personnel.
Legal Proceedings
From time to time, we are involved in various legal proceedings arising in the ordinary course of our business. We are not presently a party to any legal proceedings the outcome of which, if determined adversely to us, would individually or in the aggregate have a material adverse effect on our business, operating results or financial condition.
Incorporation/Facilities
We were originally incorporated in California in December 2000. In April 2012, we reincorporated in Delaware. Our principal executive offices are located at 11988 El Camino Real, Suite 650, San Diego, California 92130 in a facility we lease encompassing approximately 10,900 square feet of office space. The lease for this facility expires in September 2021.
69
Table of Contents
Executive Officers and Directors
The following table sets forth certain information regarding our current executive officers and directors:
| Name |
Age |
Position(s) | ||||
| Executive Officers |
||||||
| Krisztina M. Zsebo, Ph.D. |
58 | Chief Executive Officer and Director | ||||
| Paul Cleveland. |
57 | President and Chief Financial Officer | ||||
| Rebecque J. Laba |
52 | Vice President, Finance and Administration | ||||
| Elizabeth Reed |
43 | Vice President, General Counsel and Secretary | ||||
| Jeffrey J. Rudy |
52 | Vice President, Clinical Operations | ||||
| Ryan K. Takeya |
45 | Vice President, Manufacturing | ||||
| Fredrik Wiklund |
43 | Vice President, Corporate Development and Investor Relations | ||||
| Non-Employee Directors |
||||||
| Michael Narachi(1)(2) |
54 | Chairman of the Board of Directors | ||||
| Gregg Alton(1)(3) |
48 | Director | ||||
| Graham Cooper(2)(3) |
44 | Director | ||||
| Joshua Funder, Ph.D.(2) |
42 | Director | ||||
| Peter K. Honig, M.D., M.P.H(3). |
58 | Director | ||||
| Patrick Y. Yang, Ph.D(1). |
66 | Director | ||||
| (1) | Member of the compensation committee |
| (2) | Member of the audit committee |
| (3) | Member of the nominating and corporate governance committee |
Executive Officers
Krisztina M. Zsebo, Ph.D. Dr. Zsebo has served as our Chief Executive Officer and a member of our Board of Directors since 2004 and served as our President from 2004 until June 2014. From March 2004 until October 2007, Dr. Zsebo was a venture partner at Enterprise Partners Venture Capital, a venture capital firm. Prior to joining Enterprise Partners, Dr. Zsebo held executive positions at Remedyne Corporation, a biotechnology company, Connetics Corporation, a specialty pharmaceutical company, ALZA Corporation, a pharmaceutical and medical systems company, Cell Genesys, Inc., a biotechnology company, and Amgen Inc., a biotechnology company. Dr. Zsebo received a B.S. in Biochemistry from the University of Maryland, an M.S. in Biochemistry and Biophysics from Oregon State University and a Ph.D. in Comparative Biochemistry from the University of California, Berkeley. Our Nominating and Corporate Governance Committee believes that Dr. Zsebo’s 29 years of experience in the pharmaceutical industry, experience with drug development and service as our President and Chief Executive Officer qualify her to serve on our Board of Directors.
Paul Cleveland. Mr. Cleveland has served as our President and Chief Financial Officer since June 2014. From February 2013 to August 2013, Mr. Cleveland served as Executive Vice President, Corporate Strategy and Chief Financial Officer of Aragon Pharmaceuticals, Inc., a private biotechnology company focused on the development of small-molecule drugs for the treatment of hormone-dependent cancers. From April 2011 to February 2013, Mr. Cleveland served as General Partner and Chief Operating Officer of Mohr Davidow Ventures. From January 2006 to February 2011, Mr. Cleveland served as Executive Vice President, Corporate Development and Chief Financial Officer of Affymax, Inc., a biopharmaceutical company. From April 2004 to December 2005, he served as a managing director at Integrated Finance, Ltd., an investment bank. From September 1996 to April 2003, Mr. Cleveland served as a managing director at investment banks J.P. Morgan Chase and Co. and a predecessor firm, Hambrecht & Quist. From January 1993 to September 1996, Mr. Cleveland was a partner at Cooley LLP, from December 1988 to December 1992, he was a corporate
70
Table of Contents
attorney at Sidley Austin LLP and from September 1981 to November 1988, he was a corporate attorney at Davis Polk & Wardwell LLP. He currently serves on the Board of Directors of Sangamo BioSciences, Inc. Mr. Cleveland received an A.B. from Washington University in St. Louis and a J.D. from Northwestern University School of Law.
Rebecque J. Laba. Ms. Laba has served as our Vice President, Finance and Administration since September 2007, and before that, served as a consultant to us on finance and administrative matters since October 2005. From 1999 to 2005, Ms. Laba served in various financial and operational roles at Idun Pharmaceuticals, Inc. until Idun was acquired by Pfizer Inc., a pharmaceutical company, in 2005. From 1997 to 1999, Ms. Laba worked at Asset Management Group, where she served in various financial and operational roles.
Elizabeth Reed. Ms. Reed has served as our Vice President, General Counsel and Secretary since June 2014. From 2013 to June 2014, Ms. Reed served as a legal consultant for several companies in the life sciences industry. From 2001 to 2012, Ms. Reed led the legal function at Anadys Pharmaceuticals, Inc., a publicly traded biopharmaceutical company, most recently serving as Senior Vice President, Legal Affairs, General Counsel and Corporate Secretary until Anadys’ acquisition by Roche. Prior to Anadys, Ms. Reed was an attorney with the law firms Cooley LLP and Brobeck, Phleger & Harrison LLP. Ms. Reed is a member of the State Bar of California and received her B.S. in Business Administration from the Haas School of Business at the University of California, Berkeley and holds a J.D., cum laude, from Harvard Law School.
Jeffrey J. Rudy. Mr. Rudy has served as our Vice President, Clinical Operations since joining us in 2006. From 1997 to 2006, Mr. Rudy worked at Agouron Pharmaceuticals (prior to its acquisition by the Warner-Lambert Company, which was subsequently acquired by Pfizer) where he served in roles of increasing responsibility within its clinical research operations, including portfolio manager of the ophthalmology franchise and director of development operations. From 1995 to 1997, Mr. Rudy was at Gilead Sciences, Inc., a biopharmaceutical company, where he was clinical program manager in the clinical research department overseeing a number of antiviral compounds in early development. From 1991 to 1994, Mr. Rudy was at Amgen, where he worked in clinical affairs on a number of antiviral programs. Mr. Rudy received his B.S. in Microbiology from Ohio State University.
Ryan K. Takeya. Mr. Takeya has served as our Vice President, Manufacturing since April 2012. From August 1996 to December 2009, Mr. Takeya served in the Manufacturing Group at Targeted Genetics Corporation, a biotechnology company, where he oversaw in-house and contract manufacturing of clinical gene therapy products, including clinical supplies used in the MYDICAR clinical program. From 1993 to 1996, Mr. Takeya held various process development and process transfer roles at Immunex Corporation, a biotechnology company. In 2011, Mr. Takeya was at Dendreon Corporation, a biotechnology company, where he was involved with the transfer of the PROVENGE antigen manufacturing process to a secondary commercial manufacturing site. Mr. Takeya received his B.A. in Chemistry from the University of Washington.
Fredrik Wiklund. Mr. Wiklund has served as our Vice President, Corporate Development and Investor Relations since August 2013 and as our Vice President, Corporate Development from June 2013 to August 2013. Before that, he served as our Senior Director, Corporate Development from April 2012 to June 2013. From September 2009 to April 2012, Mr. Wiklund served as a consultant to us on business development matters. From November 2003 to November 2008, Mr. Wiklund was head of corporate development and investor relations at Tercica, Inc., a biopharmaceutical company, until its acquisition by the Ipsen Group, a biotechnology company, in 2008. From January 2001 to June 2003, Mr. Wiklund was at Lehman Brothers, Inc., a global financial services firm, where he served in the Investment Banking Health Care Group. From 1996 to 2000, Mr. Wiklund served as an antiviral specialist at Gilead Sciences. Mr. Wiklund received his M.B.A. from the University of Southern California and his B.A. in International Relations from the University of San Diego.
Non-Employee Directors
Michael A. Narachi. Mr. Narachi has served as our Chairman of the Board of Directors since October 2013. Since March 2009, Mr. Narachi has served as the President and Chief Executive Officer and a member of the
71
Table of Contents
Board of Directors of Orexigen Therapeutics, Inc., a biopharmaceutical company focused on the treatment of obesity. Previously, Mr. Narachi served as Chairman, Chief Executive Officer and President of Ren Pharmaceuticals, Inc., a private biotechnology company, from November 2006 to March 2009. From August 2002 to January 2008, Mr. Narachi served as Chairman of the Board of Directors of Naryx Pharma, Inc., a private pharmaceutical company. In 2004, Mr. Narachi retired as an officer and Vice President of Amgen Inc., a leading therapeutics company, where he served as General Manager of Amgen’s Anemia Business from 1999 to 2003. Mr. Narachi joined Amgen in 1984 and held various positions throughout the organization including: Product Development Team Leader for NEUPOGEN; Director of Clinical Operations in Thousand Oaks, CA and Cambridge, UK; Vice President of Development and Representative Director for Amgen Japan; Head of Corporate Strategic Planning; Chief Operations Officer of Amgen BioPharma; and Vice President, Licensing and Business Development. He currently serves as Chairman of the Board of Directors of AMAG Pharmaceuticals, Inc. and serves on the Board of Directors of the Pharmaceutical Research and Manufacturers of America and the Biotechnology Industry Organization. Mr. Narachi received a B.S. in Biology and an M.A. degree in Biology and Genetics from the University of California at Davis. He received an M.B.A. from the Anderson Graduate School of Management at University of California, Los Angeles. Our Nominating and Corporate Governance Committee believes that Mr. Narachi’s business, leadership and management experience, as well as his experience in the biotechnology industry, qualifies him to serve on our Board of Directors.
Gregg Alton. Mr. Alton has served on our Board of Directors since August 2013. Since August 2009, Mr. Alton has served as executive vice president of corporate and medical affairs and chief compliance officer at Gilead Sciences. In this role, Mr. Alton oversees legal affairs, public affairs, government affairs, emerging markets and medical affairs. From January 2008 to March 2013, Mr. Alton served as a director of Oculus Innovative Sciences, Inc., a global healthcare company. From March 2000 to August 2009, Mr. Alton served as general counsel at Gilead and from October 1999 to March 2000, served as associate general counsel at the same company. Mr. Alton was a corporate attorney at the law firm of Cooley LLP from November 1993 to December 1996 and from July 1998 to October 1999, and at the law firm Mintz Levin Cohn Ferris Glovsky and Popeo, P.C. from January 1997 to July 1998. Mr. Alton received a B.A. from the University of California, Berkeley and a J.D. from Stanford Law School. Our Nominating and Corporate Governance Committee believes that Mr. Alton’s expertise and experience in the biotechnology industry qualifies him to serve on our Board of Directors.
Graham Cooper. Mr. Cooper has served on our Board of Directors since September 2013. Since February 2013, Mr. Cooper has served as chief financial officer at Receptos, Inc., a publicly held biopharmaceutical company focused on therapeutics for immune disorders. From January 2012 to December 2012, Mr. Cooper served as chief financial officer at Geron Corporation, a biopharmaceutical company focused on cancer therapies. From May 2006 to March 2011, Mr. Cooper served as chief financial officer of Orexigen Therapeutics, Inc., a biotechnology company focused on obesity therapeutics. From 1999 to 2006, Mr. Cooper held positions of increasing responsibility, including director, health care investment banking, at Deutsche Bank Securities, a global investment bank, where he was responsible for executing and managing a wide variety of financing and merger and acquisition transactions in the life sciences field. From August 1992 to January 1995, Mr. Cooper worked as an accountant at Deloitte & Touche LLP, an independent registered public accounting firm, and was previously a C.P.A. Mr. Cooper holds a B.A. in economics from the University of California, Berkeley and an M.B.A. from the Stanford Graduate School of Business. Our Nominating and Corporate Governance Committee believes that Mr. Cooper’s expertise and experience in the biotechnology industry and his financial expertise qualifies him to serve on our Board of Directors.
Joshua Funder, Ph.D. Dr. Funder has served on our Board of Directors since January 2012. Dr. Funder has been a partner with GBS Venture Partners, a venture capital group since April 2004. From January 2003 to March 2004, Dr. Funder was senior manager, corporate strategy and development at Infinity Pharmaceuticals, Inc., a drug discovery company. From June 2004 to December 2004, Dr. Funder served as interim chief executive officer of Proacta Inc., a biopharmaceutical company. Dr. Funder also serves as a member of the Board of Directors of OPAL Inc., Spinifex Pty Ltd and Pathway Therapeutics Ltd. Dr. Funder received a B.S. and a Bachelor of Laws from Melbourne University, and a Master of Laws from the London School of Economics. He
72
Table of Contents
also holds a D.Phil. in intellectual property for biotechnology from Oxford University. Our Nominating and Corporate Governance Committee believes that Dr. Funder’s expertise and experience in the biotechnology industry qualifies him to serve on our Board of Directors.
Peter K. Honig, M.D., M.P.H. Dr. Honig has served on our Board of Directors since March 2014. Dr. Honig currently serves as Senior Vice President of Worldwide Regulatory Affairs at Pfizer Inc. and before that was Head of Global Regulatory Affairs, Patient Safety and Quality Assurance at AstraZeneca, Inc., a global biopharmaceutical company specializing in the discovery, development, manufacturing and marketing of prescription medicines. Dr. Honig also serves as a director of Orexigen Therapeutics, Inc., a biopharmaceutical company focused on the treatment of obesity. From January 2003 through December 2009, Dr. Honig served as Senior Vice President, Worldwide Regulatory Affairs and Product Safety at Merck & Co., Inc., a global healthcare company. From March 2002 to January 2003, Dr. Honig was Merck’s Vice President, Worldwide Product Safety and Quality Assurance. Prior to Merck, from 1993 to 2002, Dr. Honig held various positions at the FDA, including Director of the Office of Drug Safety in the FDA’s Center for Drug Evaluation and Research. Dr. Honig received his B.A. in History from Columbia College of Columbia University, his M.D. from the Columbia College of Physicians & Surgeons and his M.P.H from Columbia University School of Public Health. Our Nominating and Corporate Governance Committee believes that Dr. Honig’s expertise and experience in the pharmaceutical industry qualifies him to serve on our Board of Directors.
Patrick Y. Yang, Ph.D. Dr. Yang has served on our Board of Directors since March 2014. Dr. Yang recently retired from F. Hoffman-La Roche AG, a global pharmaceutical and diagnostics company, where he served as Executive Vice President and Global Head of Pharmaceutical Technical Operations from January 2010 until March 2013. From December 2003 through December 2009, Dr. Yang worked for Genentech, Inc., a biotechnology company, where his most recent position was Executive Vice President of Product Operations. Prior to Genentech, Dr. Yang held several leadership roles at Merck, including Vice President of Asia/Pacific Operations and Vice President of Supply Chain Management. He also previously worked at General Electric Co. and Life Systems, Inc. in research, development, and manufacturing operations. Dr. Yang currently works as a biotech industry consultant. He serves on the board of directors of Tesoro Corporation, an independent refiner and marketer of petroleum products, the board of directors of Codexis, Inc., a company in the development and production of custom industrial enzymes for use in the pharmaceutical, chemical and biofuel production; and on the board of directors of PharmaEssentia Corporation (Taiwan), a biotechnology company. Dr. Yang holds a Ph.D. in engineering from Ohio State University. Our Nominating and Corporate Governance Committee believes that Dr. Yang’s expertise and experience in the pharmaceutical and biotechnology industries qualifies him to serve on our Board of Directors.
Board Composition
Our business and affairs are organized under the direction of our board of directors, which currently consists of seven members. The primary responsibilities of our board of directors are to provide oversight, strategic guidance, counseling and direction to our management. Our board of directors meets on a regular basis and additionally as required.
Our board of directors has determined that all of our directors, except Dr. Zsebo, are independent directors, as defined by Rule 5605(a)(2) of the NASDAQ Listing Rules.
In accordance with the terms of our amended and restated certificate of incorporation and bylaws, our board of directors is divided into three classes, class I, class II and class III, with members of each class serving staggered three-year terms.
The authorized number of directors may be changed only by resolution by a majority of the board of directors. This classification of the board of directors may have the effect of delaying or preventing changes in our control or management. Our directors may be removed for cause by the affirmative vote of the holders of at least 66 2⁄3% of our voting stock.
73
Table of Contents
Board Leadership Structure
Our board of directors is currently chaired by Mr. Narachi. As a general policy, our board of directors believes that separation of the positions of Chairman and Chief Executive Officer reinforces the independence of the board of directors from management, creates an environment that encourages objective oversight of management’s performance and enhances the effectiveness of the board of directors as a whole. As such, Dr. Zsebo serves as our Chief Executive Officer while Mr. Narachi serves as our Chairman of the Board of Directors but is not an officer. We expect and intend the positions of Chairman of the Board of Directors and Chief Executive Officer to continue to be held by two individuals in the future.
Role of the Board in Risk Oversight
One of the key functions of our board of directors is informed oversight of our risk management process. The board of directors does not have a standing risk management committee, but rather administers this oversight function directly through the board of directors as a whole, as well as through various standing committees of our board of directors that address risks inherent in their respective areas of oversight. In particular, our board of directors is responsible for monitoring and assessing strategic risk exposure and our audit committee has the responsibility to consider and discuss our major financial risk exposures and the steps our management has taken to monitor and control these exposures, including guidelines and policies to govern the process by which risk assessment and management is undertaken. The audit committee also monitors compliance with legal and regulatory requirements. Our nominating and corporate governance committee monitors the effectiveness of our corporate governance practices, including whether they are successful in preventing illegal or improper liability-creating conduct. Our compensation committee assesses and monitors whether any of our compensation policies and programs has the potential to encourage excessive risk-taking.
Board Committees
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee.
Audit Committee
Our audit committee consists of Mr. Cooper, Dr. Funder and Mr. Narachi. Our board of directors has determined that each of the members of our audit committee satisfies the NASDAQ Stock Market and SEC independence requirements.
Mr. Cooper serves as the chair of our audit committee. Our board of directors has determined that Mr. Cooper qualifies as an audit committee financial expert within the meaning of SEC regulations and meets the financial sophistication requirements of the NASDAQ Listing Rules. In making this determination, our board has considered Mr. Cooper’s formal education and previous and current experience in financial roles. Both our independent registered public accounting firm and management periodically meet privately with our audit committee.
The functions of this committee include, among other things:
| • | evaluating the performance, independence and qualifications of our independent auditors and determining whether to retain our existing independent auditors or engage new independent auditors; |
| • | reviewing and approving the engagement of our independent auditors to perform audit services and any permissible non-audit services; |
| • | monitoring the rotation of partners of our independent auditors on our engagement team as required by law; |
| • | prior to engagement of any independent auditor, and at least annually thereafter, reviewing relationships that may reasonably be thought to bear on their independence, and assessing and otherwise taking the appropriate action to oversee the independence of our independent auditor; |
74
Table of Contents
| • | reviewing our annual and quarterly financial statements and reports, including the disclosures contained under the caption “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and discussing the statements and reports with our independent auditors and management; |
| • | reviewing with our independent auditors and management significant issues that arise regarding accounting principles and financial statement presentation and matters concerning the scope, adequacy and effectiveness of our financial controls; |
| • | reviewing with management and our auditors any earnings announcements and other public announcements regarding material developments; |
| • | establishing procedures for the receipt, retention and treatment of complaints received by us regarding financial controls, accounting or auditing matters and other matters; |
| • | preparing the report that the SEC requires in our annual proxy statement; |
| • | reviewing and providing oversight of any related-person transactions in accordance with our related-person transaction policy and reviewing and monitoring compliance with legal and regulatory responsibilities, including our code of business conduct and ethics; |
| • | reviewing our major financial risk exposures, including the guidelines and policies to govern the process by which risk assessment and risk management is implemented; |
| • | reviewing on a periodic basis our investment policy; and |
| • | reviewing and evaluating on an annual basis the performance of the audit committee, including compliance of the audit committee with its charter. |
We believe that the composition and functioning of our audit committee complies with all applicable requirements of the Sarbanes-Oxley Act, and all applicable SEC and NASDAQ rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
Compensation Committee
Our compensation committee consists of Mr. Alton, Mr. Narachi and Dr. Yang. Mr. Narachi serves as the chair of our compensation committee. Our board of directors has determined that each of the members of our compensation committee is a non-employee director, as defined in Rule 16b-3 promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act, is an outside director, as defined pursuant to Section 162(m) of the Code, and satisfies the NASDAQ Stock Market independence requirements. The functions of this committee include, among other things:
| • | reviewing, modifying and approving (or if it deems appropriate, making recommendations to the full board of directors regarding) our overall compensation strategy and policies; |
| • | reviewing, modifying and approving (or if it deems appropriate, making recommendations to the full board of directors regarding) the compensation and other terms of employment of our executive officers; |
| • | reviewing, modifying and approving (or if it deems appropriate, making recommendations to the full board of directors regarding) performance goals and objectives relevant to the compensation of our executive officers and assessing their performance against these goals and objectives; |
| • | reviewing and approving (or if it deems it appropriate, making recommendations to the full board of directors regarding) the equity incentive plans, compensation plans and similar programs advisable for us, as well as modifying, amending or terminating existing plans and programs; |
| • | evaluating risks associated with our compensation policies and practices and assessing whether risks arising from our compensation policies and practices for our employees are reasonably likely to have a material adverse effect on us; |
75
Table of Contents
| • | reviewing and making recommendations to the full board of directors regarding the type and amount of compensation to be paid or awarded to our non-employee board members; |
| • | establishing policies with respect to votes by our stockholders to approve executive compensation to the extent required by Section 14A of the Exchange Act and, if applicable, determining our recommendations regarding the frequency of advisory votes on executive compensation; |
| • | reviewing and assessing the independence of compensation consultants, legal counsel and other advisors as required by Section 10C of the Exchange Act; |
| • | administering our equity incentive plans; |
| • | establishing policies with respect to equity compensation arrangements; |
| • | reviewing the competitiveness of our executive compensation programs and evaluating the effectiveness of our compensation policy and strategy in achieving expected benefits to us; |
| • | reviewing and making recommendations to the full board of directors regarding the terms of any employment agreements, severance arrangements, change of control protections and any other compensatory arrangements for our executive officers; |
| • | reviewing the adequacy of its charter on a periodic basis; |
| • | reviewing with management and approving our disclosures under the caption “Compensation Discussion and Analysis” in our periodic reports or proxy statements to be filed with the SEC, to the extent such caption is included in any such report or proxy statement; |
| • | preparing the report that the SEC requires in our annual proxy statement; and |
| • | reviewing and assessing on an annual basis the performance of the compensation committee. |
We believe that the composition and functioning of our compensation committee complies with all applicable requirements of the Sarbanes-Oxley Act of 2002, and all applicable SEC and NASDAQ rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Mr. Alton, Mr. Cooper and Dr. Honig. Our board of directors has determined that each of the members of this committee satisfies the NASDAQ Stock Market independence requirements. Mr. Alton serves as the chair of our nominating and corporate governance committee. The functions of this committee include, among other things:
| • | identifying, reviewing and evaluating candidates to serve on our board of directors consistent with criteria approved by our board of directors; |
| • | determining the minimum qualifications for service on our board of directors; |
| • | evaluating director performance on the board and applicable committees of the board and determining whether continued service on our board is appropriate; |
| • | evaluating, nominating and recommending individuals for membership on our board of directors; |
| • | evaluating nominations by stockholders of candidates for election to our board of directors; |
| • | considering and assessing the independence of members of our board of directors; |
| • | developing a set of corporate governance policies and principles, including a code of business conduct and ethics, periodically reviewing and assessing these policies and principles and their application and recommending to our board of directors any changes to such policies and principles; |
| • | considering questions of possible conflicts of interest of directors as such questions arise; |
76
Table of Contents
| • | reviewing the adequacy of its charter on an annual basis; and |
| • | annually evaluating the performance of the nominating and corporate governance committee. |
We believe that the composition and functioning of our nominating and corporate governance committee complies with all applicable requirements of the Sarbanes-Oxley Act of 2002, and all applicable SEC and NASDAQ rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
Compensation Committee Interlocks and Insider Participation
As stated above, the Compensation Committee currently consists of Mr. Narachi (Chair), Mr. Alton and Dr. Yang. No member of the Compensation Committee has ever been an officer or employee of Celladon. None of our executive officers currently serves, or has served during the last completed fiscal year, on the compensation committee or board of directors of any other entity that has one or more executive officers serving as a member of our Board of Directors or Compensation Committee.
Limitation on Liability and Indemnification of Directors and Officers
Our amended and restated certificate of incorporation and bylaws limit our directors’ and officers’ liability to the fullest extent permitted under Delaware corporate law. Delaware corporate law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability:
| • | for any transaction from which the director derives an improper personal benefit; |
| • | for any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | under Section 174 of the Delaware General Corporation Law (unlawful payment of dividends or redemption of shares); or |
| • | for any breach of a director’s duty of loyalty to the corporation or its stockholders. |
If the Delaware General Corporation Law is amended to authorize corporate action further eliminating or limiting the personal liability of directors or officers, then the liability of our directors or officers shall be eliminated or limited to the fullest extent permitted by the Delaware General Corporation Law, as so amended.
Delaware law and our amended and restated bylaws provide that we will, in certain situations, indemnify any person made or threatened to be made a party to a proceeding by reason of that person’s former or present official capacity with us against judgments, penalties, fines, settlements and reasonable expenses. Any person is also entitled, subject to certain limitations, to payment or reimbursement of reasonable expenses (including attorneys’ fees and disbursements) in advance of the final disposition of the proceeding.
In addition, we have entered, and intend to continue to enter, into separate indemnification agreements with our directors and executive officers. These agreements, among other things, require us to indemnify our directors and executive officers for certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of their services as one of our directors or executive officers or as a director or executive officer of any other company or enterprise to which the person provides services at our request.
We believe that these provisions in our amended and restated certificate of incorporation and amended bylaws and these indemnification agreements are necessary to attract and retain qualified persons as directors and officers. We also maintain a directors’ and officers’ insurance policy pursuant to which our directors and officers are insured against liability for actions taken in their capacities as directors and officers.
77
Table of Contents
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or control persons, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
78
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
The following includes a summary of transactions since January 1, 2010 to which we have been a party, in which the amount involved in the transaction exceeded the lesser of $120,000 or one percent of our average total assets at year end for the last two completed fiscal years, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change of control and other arrangements which are described in our filings with the SEC.
Prior Loan Arrangements
In 2010 and 2011, we entered into various loan arrangements with beneficial owners of more than 5% of our capital stock, pursuant to which we issued secured convertible promissory notes and unsecured convertible promissory notes. The notes carried interest at 12.0% per annum. In January 2012, these notes were cancelled and the aggregate amount of outstanding principal and unpaid accrued interest thereon was exchanged for shares of our Series A-1 preferred stock, Junior preferred stock and common stock, with the balance paid in cash, as described below under the caption “Preferred Stock Financing and Convertible Note and Warrant Financing.” Below is a summary of certain information relating to such notes as of and for the years ended December 31, 2012, 2011 and 2010:
| Years Ended December 31, | ||||||||||||
| 2012 | 2011 | 2010 | ||||||||||
| (in thousands) | ||||||||||||
| Principal amount of promissory notes issued |
$ | — | $ | 12,350 | $ | 9,000 | ||||||
| Largest aggregate principal amount outstanding |
12,350 | 12,350 | 9,000 | |||||||||
| Aggregate interest expense accrued on notes payable |
2,191 | 2,083 | 787 | |||||||||
| Principal and interest repaid |
— | — | — | |||||||||
| Principal and interest converted to equity |
14,429 | — | — | |||||||||
The participants in these loan arrangements included the following holders of more than 5% of our capital stock or entities affiliated with them. The following table presents the aggregate principal amount of secured convertible promissory notes and unsecured convertible promissory notes issued to these related parties in these loan arrangements:
| Participants |
Aggregate Principal Amount of Notes (in thousands) |
|||
| Enterprise Partners and affiliated entities(1) |
$ | 5,839 | ||
| Johnson & Johnson Development Corporation |
$ | 3,702 | ||
| Venrock Partners and affiliated entities(2) |
$ | 2,809 | ||
| (1) | Consists of $2.7 million aggregate principal amount of notes issued to Enterprise Partners V, L.P., $3.0 million aggregate principal amount of notes issued to Enterprise Partners VI, L.P., and $0.1 million aggregate principal amount of notes issued to Enterprise Management, LLC. |
| (2) | Consists of $0.5 million aggregate principal amount of notes issued to Venrock Partners, L.P.; $2.3 million aggregate principal amount of notes issued to Venrock Associates IV, L.P., and $0.1 million aggregate principal amount of notes issued to Venrock Entrepreneurs Fund IV, L.P. |
In January 2012, the noteholders waived their right to receive payment of unpaid accrued interest under these notes in exchange for an aggregate of 849,949 shares of our common stock. See “Preferred Stock Financing” below for further information relating to the outstanding principal amounts under these notes.
79
Table of Contents
Preferred Stock Financing and Convertible Note and Warrant Financing
In January 2012, we issued and sold to investors an aggregate of 27,616,923 shares of our Series A-1 preferred stock and 12,138,080 shares of our Junior preferred stock, at a purchase price of $0.449 per share, for aggregate consideration of $17.8 million. Of this amount, $12.4 million was paid for by cancellation of principal indebtedness under the promissory notes described above under the caption “Loan Arrangements” and the balance was paid for in cash.
In March 2012, we issued and sold to investors an aggregate of 1,913,987 shares of Series A-1 preferred stock for aggregate cash consideration of $0.9 million. In April 2012, we issued and sold to Coôperatief LSP IV UA, or LSP, share capital in our Netherlands-based subsidiary, Celladon Europe B.V., or Celladon Europe, for aggregate cash consideration of $0.8 million, which share capital was immediately exchangeable for 1,683,327 shares of our Series A-1 preferred stock at the investor’s election. In June 2012, in exchange for aggregate cash consideration of $43.1 million, we issued and sold to investors an additional 86,893,215 shares of our Series A-1 preferred stock, at a purchase price of $0.449 per share, and to LSP share capital in Celladon Europe exchangeable for 9,033,078 shares of our Series A-1 preferred stock. In June 2013, LSP exercised its option to exchange its share capital of Celladon Europe and we issued 10,716,405 shares of our Series A-1 preferred stock to LSP for no additional consideration.
The participants in this preferred stock financing included the following holders of more than 5% of our capital stock or entities affiliated with them. The following table presents the number of shares issued to these related parties in this financing:
| Participants |
Junior Preferred Stock |
Series A-1 Preferred Stock |
||||||
| Coôperatief LSP IV UA |
— | 10,716,405 | ||||||
| Enterprise Partners and affiliated entities(1) |
5,741,267 | 11,573,520 | ||||||
| Johnson & Johnson Development Corporation |
3,655,435 | 8,243,822 | ||||||
| GBS Bioventures IV |
— | 11,788,047 | ||||||
| H&Q Healthcare Investors and affiliated entities(2) |
— | 10,723,875 | ||||||
| Lundbeckfond Invest A/S |
— | 19,289,531 | ||||||
| MPM Capital and affiliated entities(3) |
— | 11,788,047 | ||||||
| Novartis Bioventures Ltd. |
— | 17,146,250 | ||||||
| Pfizer Inc. |
— | 19,289,531 | ||||||
| Venrock Partners and affiliated entities(4) |
2,741,378 | 6,182,653 | ||||||
| (1) | Consists of 2,704,061 shares of Junior preferred stock and 5,070,613 shares of Series A-1 preferred stock issued to Enterprise Partners V, L.P.; 2,914,744 shares of Junior preferred stock and 6,370,333 shares of Series A-1 preferred stock issued to Enterprise Partners VI, L.P.; and 122,462 shares of Junior preferred stock and 132,574 shares of Series A-1 preferred stock issued to Enterprise Partners Management, LLC. |
| (2) | Consists of 7,399,474 shares of Series A-1 preferred stock issued to H&Q Healthcare Investors and 3,324,401 shares of Series A-1 preferred stock issued to H&Q Life Sciences Investors. |
| (3) | Consists of 11,048,241 shares of Series A-1 preferred stock issued to MPM BioVentures IV-QP, L.P.; 425,642 shares of Series A-1 preferred stock issued to MPM BioVentures IV GmbH & Co. Beteiligungs KG; and 314,164 shares of Series A-1 preferred stock issued to MPM Asset Management Investors BV4 LLC. |
| (4) | Consists of 455,069 shares of Junior preferred stock and 1,026,321 shares of Series A-1 preferred stock issued to Venrock Partners, L.P.; 2,231,483 shares of Junior preferred stock and 5,032,681 shares of Series A-1 preferred stock issued to Venrock Associates IV, L.P.; and 54,826 shares of Junior preferred stock and 123,651 shares of Series A-1 preferred stock issued to Venrock Entrepreneurs Fund IV, L.P. |
In October 2013, we entered into a convertible note and warrant purchase agreement with each of our greater than 5% stockholders, including entities affiliated with certain members of our board of directors,
80
Table of Contents
pursuant to which we issued $1,097,017 aggregate principal amount of convertible notes, or the 2013 notes, and warrants exercisable for shares of our Series A-1 preferred stock, or the 2013 warrants. The 2013 notes accrued interest at a rate of 6% per annum, compounded annually, and had a maturity date of the earlier of (1) March 31, 2014 or (2) the occurrence of a deemed liquidation event as defined in our amended and restated certificate of incorporation, subject in each case to their earlier conversion in the event we complete a qualified initial public offering or private placement of our equity securities. In connection with the closing of our initial public offering in February 2014, the 2013 notes (including accrued interest thereon) automatically converted into 139,665 shares of common stock at a conversion price of $8.00 per share. The 2013 warrants were initially exercisable for an aggregate of 2,895,570 shares of Series A-1 preferred stock at an exercise price of $0.449 per share. In connection with the closing of our initial public offering, the 2013 warrants became exercisable for an aggregate of 231,821 shares of common stock, at an exercise price of $5.61 per share. 25,481 shares of common stock have been issued pursuant to the exercise of 2013 warrants as of the date of this prospectus. The 2013 warrants will expire in October 2018.
The following table sets forth the aggregate amount of securities acquired by the listed holders of more than 5% of our capital stock, or their affiliates, in the convertible note and warrant financing.
| Participants |
Aggregate Principal Amount of 2013 Notes (in thousands) |
Shares of Series A-1 Preferred Stock Underlying 2013 Warrants |
||||||
| Coôperatief LSP IV UA |
81 | 198,916 | ||||||
| Enterprise Partners and affiliated entities(1) |
106 | 260,859 | ||||||
| Johnson & Johnson Development Corporation |
43 | — | ||||||
| GBS Bioventures IV |
182 | 672,060 | ||||||
| H&Q Healthcare Investors and affiliated entities(2) |
81 | 199,055 | ||||||
| Lundbeckfond Invest A/S |
146 | 358,049 | ||||||
| MPM Capital and affiliated entities(3) |
89 | 218,806 | ||||||
| Novartis Bioventures Ltd. and affiliated entities(4) |
130 | 318,266 | ||||||
| Pfizer Inc. |
182 | 532,818 | ||||||
| Venrock Partners and affiliated entities(5) |
56 | 136,741 | ||||||
| (1) | Consists of $43,752.19 principal amount of 2013 notes and 2013 warrants to purchase 107,187 shares of Series A-1 preferred stock issued to Enterprise Partners V, L.P.; $43,752.19 principal amount of 2013 notes and 2013 warrants to purchase 107,187 shares of Series A-1 preferred stock issued to Enterprise Partners VI, L.P.; and $18,974.54 principal amount of 2013 notes and 2013 warrants to purchase 46,485 shares of Series A-1 preferred stock issued to Enterprise Partners Management, LLC. |
| (2) | Consists of $56,062.96 principal amount of 2013 notes and 2013 warrants to purchase 137,348 shares of Series A-1 preferred stock issued to H & Q Healthcare Investors and $25,187.70 principal amount of 2013 notes and 2013 warrants to purchase 61,707 shares of Series A-1 preferred stock issued to H & Q Life Sciences Investors. |
| (3) | Consists of $83,708.25 principal amount of 2013 notes and 2013 warrants to purchase 205,075 shares of Series A-1 preferred stock issued to MPM BioVentures IV-QP, L.P.; $3,224.93 principal amount of 2013 notes and 2013 warrants to purchase 7,900 shares of Series A-1 preferred stock issued to MPM BioVentures IV GmbH & Co. Beteiligungs KG; and $2,380.30 principal amount of 2013 notes and 2013 warrants to purchase 5,831 shares of Series A-1 preferred stock issued to MPM Asset Management Investors BV4 LLC. |
| (4) | Consists of $129,910.51 principal amount of 2013 notes and 2013 warrants to purchase 318,266 shares of Series A-1 preferred stock issued to Novartis International Pharmaceutical Investment Ltd. |
| (5) | Consists of $9,265.45 principal amount of 2013 notes and 2013 warrants to purchase 22,699 shares of Series A-1 preferred stock issued to Venrock Partners, L.P.; $45,434.20 principal amount of 2013 notes and 2013 warrants to purchase 111,308 shares of Series A-1 preferred stock issued to Venrock Associates IV, L.P.; and $1,116.30 principal amount of 2013 notes and 2013 warrants to purchase 2,734 shares of Series A-1 preferred stock issued to Venrock Entrepreneurs Fund IV, L.P. |
81
Table of Contents
Immediately prior to the closing of our initial public offering in February 2014, all of our outstanding shares of preferred stock were converted into shares of our common stock at a conversion ratio of 12.49-to-1.
Below is a summary of certain information relating to such notes as of and for the year ended December 31, 2013:
| Year Ended December 31, 2013 |
||||
| (in thousands) | ||||
| Principal amount of promissory notes issued |
$ | 1,097 | ||
| Largest aggregate principal amount outstanding |
1,097 | |||
| Aggregate interest expense accrued on notes payable |
14 | |||
| Principal and interest repaid |
— | |||
| Principal and interest converted to equity |
— | |||
License Agreement with Enterprise Partners
On July 18, 2014, we and Enterprise entered into an Assignment and License Agreement, pursuant to which Enterprise granted to us an exclusive, worldwide license and the assignment of patents held by Enterprise relating to certain gene therapy applications of mSCF for the treatment of cardiac ischemia. We have the right to grant sublicenses to third parties under the agreement.
In consideration for the rights granted to us under the agreement, we paid an upfront fee to Enterprise of $160,000. We are also obligated to pay to Enterprise a milestone payment in the amount of $1,000,000 upon the grant to us, or an affiliate or sublicensee of ours, of the first regulatory approval in the United States of a product that is covered by the licensed patents. In addition, we are required to pay to Enterprise a 2% royalty on net sales of products sold by us or by our affiliates or sublicensees that are covered by the licensed patents. Our royalty obligations continue on a product-by-product and country-by-country basis until the expiration of the last-to-expire valid claim in the licensed patents covering a licensed product in such country.
Participation in our Initial Public Offering
Certain of our existing stockholders, officers and directors purchased an aggregate of 1,453,651 shares of our common stock in our initial public offering at a price of $8.00 per share, or $11.6 million in the aggregate.
| Purchaser |
Initial Public Offering Shares |
|||
| Pfizer, Inc. |
227,261 | |||
| H&Q Healthcare Investors |
70,072 | |||
| H&Q Life Sciences Investors |
31,485 | |||
| GBS Bioventure IV |
227,261 | |||
| MPM BioVentures IV-QP, LP |
104,603 | |||
| MPM BioVentures IV BMBH & Co |
4,030 | |||
| MPM Asset Management Investors |
2,975 | |||
| Lundbeckfond Invest A/S |
182,681 | |||
| Cooperatief LSP IV UA |
101,488 | |||
| Novartis Bioventures Ltd. |
162,386 | |||
| Enterprise Liquidating Trust V |
57,229 | |||
82
Table of Contents
| Purchaser |
Initial Public Offering Shares |
|||
| Enterprise Liquidating Trust VI |
57,229 | |||
| Enterprise Partners Management |
18,625 | |||
| Venrock Partners, L.P. |
11,574 | |||
| Venrock Associates IV, L.P. |
56,784 | |||
| Venrock Entrepreneurs Fund IV, L.P. |
1,385 | |||
| Johnson & Johnson Development Corp |
54,083 | |||
| Michael Narachi |
62,500 | |||
| B. Fredrik Wiklund |
20,000 | |||
Certain of our current and former directors have affiliations with the investors that participated in the loan arrangements, preferred stock financing, convertible note and warrant financing and initial public offering described above, as indicated in the table below:
| Director |
Principal Stockholder | |
| Fouad Azzam, Ph.D.* |
Coôperatief LSP IV UA | |
| Barbara Dalton, Ph.D.* |
Pfizer Inc. | |
| Todd Foley* |
MPM Capital and affiliated entities | |
| Joshua Funder, Ph.D. |
GBS Bioventures IV | |
| Johan Kôrdel, Ph.D.* |
Lundbeckfond Invest A/S | |
| Daniel Omstead, Ph.D.* |
H&Q Healthcare Investors and affiliated entities | |
| Andrew E. Senyei, M.D.* |
Enterprise Partners V, L.P. and affiliated entities | |
| Lauren Silverman, Ph.D.* |
Novartis Bioventures Ltd. and affiliated entities |
| * | Former director |
Investor Agreements
In connection with our preferred stock financings, we entered into amended and restated investor rights, voting and right of first refusal and co-sale agreements containing voting rights, information rights, rights of first refusal and registration rights, among other things, with certain holders of our preferred stock and certain holders of our common stock, including all of the holders of more than 5% of our capital stock or entities affiliated with them. These stockholder agreements terminated upon the closing of our initial public offering, except for the amended and restated investor rights agreement which terminates seven years after the closing of our initial public offering, and contains certain registration rights as more fully described below under the heading “Description of Capital Stock—Registration Rights.”
Employment Arrangements
We currently have written employment agreements with our executive officers, as more fully described in our filings with the SEC.
Stock Options Granted to Executive Officers and Directors
We have granted stock options to our executive officers and directors, as more fully described in our filings with the SEC.
Indemnification Agreements
We have entered, and intend to continue to enter, into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated bylaws. These agreements, among other things, require us to indemnify our directors and executive officers for
83
Table of Contents
certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of their services as one of our directors or executive officers or as a director or executive officer of any other company or enterprise to which the person provides services at our request. For more information regarding these indemnification arrangements, see “Management—Limitation on Liability and Indemnification of Directors and Officers.” We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might benefit us and our stockholders. A stockholder’s investment may decline in value to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions
Policies and Procedures for Transactions with Related Persons
We have adopted a written related-person transactions policy that sets forth our policies and procedures regarding the identification, review, consideration and oversight of “related-person transactions.” For purposes of our policy only, a “related-person transaction” is a transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any “related person” are participants involving an amount that exceeds $120,000.
Transactions involving compensation for services provided to us as an employee, consultant or director are not considered related-person transactions under this policy. A related person is any executive officer, director or a holder of more than 5% of our common stock, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, where a transaction has been identified as a related-person transaction, management must present information regarding the proposed related-person transaction to our audit committee (or, where review by our audit committee would be inappropriate, to another independent body of our board of directors) for review. The presentation must include a description of, among other things, the material facts, the direct and indirect interests of the related persons, the benefits of the transaction to us and whether any alternative transactions are available. To identify related-person transactions in advance, we rely on information supplied by our executive officers, directors and certain significant stockholders. In considering related-person transactions, our audit committee or another independent body of our board of directors takes into account the relevant available facts and circumstances including, but not limited to:
| • | the risks, costs and benefits to us; |
| • | the impact on a director’s independence in the event the related person is a director, immediate family member of a director or an entity with which a director is affiliated; |
| • | the terms of the transaction; |
| • | the availability of other sources for comparable services or products; and |
| • | the terms available to or from, as the case may be, unrelated third parties or to or from our employees generally. |
In the event a director has an interest in the proposed transaction, the director must recuse himself or herself from the deliberations and approval.
84
Table of Contents
The following table sets forth information regarding beneficial ownership of our capital stock by:
| • | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our common stock; |
| • | each of our directors; |
| • | each of our named executive officers; and |
| • | all of our current executive officers and directors as a group. |
The percentage ownership information before the offering is based on 18,534,480 shares of common stock outstanding as of June 30, 2014. The percentage ownership information after the offering assumes the sale of 4,000,000 shares in this offering.
The following table is based upon information supplied by officers, directors and principal stockholders and Schedules 13G filed with the SEC. We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of our common stock issuable pursuant to the exercise of stock options or warrants that are either immediately exercisable or exercisable on or before August 29, 2014, which is 60 days after June 30, 2014. These shares are deemed to be outstanding and beneficially owned by the person holding those options or warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
Except as otherwise noted below, the address for each person or entity listed in the table is c/o Celladon Corporation, 11988 El Camino Real, Suite 650, San Diego, California 92130.
| Name and Address of Beneficial Owner |
Number of shares beneficially owned |
Percentage of shares beneficially owned | ||||||||||
| Before offering | After offering | |||||||||||
| Greater than 5% stockholders |
||||||||||||
| Entities affiliated with Enterprise Partners(1). |
1,971,067 | 10.6 | % | 8.7 | % | |||||||
| 2223 Avenida de la Playa, Suite 205 La Jolla, CA 92037 |
||||||||||||
| Pfizer Inc.(2) |
1,837,462 | 9.9 | % | 8.1 | % | |||||||
| c/o Pfizer Venture Investments 235 E. 42nd Street New York, NY 10017 |
||||||||||||
| Lundbeckfond Invest A/S(3) |
1,774,349 | 9.6 | % | 7.9 | % | |||||||
| Vestagervej 17 DK-2900 Hellerup Denmark |
||||||||||||
| Entities affiliated with Novartis Bioventures Ltd.(4) |
1,577,202 | 8.5 | % | 7.0 | % | |||||||
| 131 Front Street Hamilton, HM 12 Bermuda |
||||||||||||
85
Table of Contents
| Name and Address of Beneficial Owner |
Number of shares beneficially owned |
Percentage of shares beneficially owned | ||||||||||
| Before offering | After offering | |||||||||||
| Johnson & Johnson Development Corporation |
1,276,724 | 6.9 | % | 5.7 | % | |||||||
| 410 George Street New Brunswick, NJ 08901 |
||||||||||||
| GBS Bioventures IV(5) |
1,248,011 | 6.7 | % | 5.5 | % | |||||||
| Level 5, 71 Collins Street Melbourne, Vic 3000 Australia |
||||||||||||
| Entities affiliated with MPM Capital(6) |
1,084,292 | 5.8 | % | 4.8 | % | |||||||
| The John Hancock Tower 200 Clarendon Street, 54th Floor Boston, MA 02116 |
||||||||||||
| Entities affiliated with Venrock Partners(7) |
1,002,236 | 5.4 | % | 4.4 | % | |||||||
| 3340 Hillview Ave. Palo Alto, CA 94304 |
||||||||||||
| H&Q Healthcare Investors and H&Q Life Sciences Investors(8) |
986,431 | 5.3 | % | 4.4 | % | |||||||
| 2 Liberty Square, 9th Floor Boston, MA 02109 |
||||||||||||
| Coôperatief LSP IV UA(9) |
985,748 | 5.3 | % | 4.4 | % | |||||||
| Johannes Vermeerplein 9 1071 DV Amsterdam The Netherlands |
||||||||||||
| Directors and Named Executive Officers |
||||||||||||
| Krisztina M. Zsebo, Ph.D.(10) |
464,819 | 2.4 | % | 2.0 | % | |||||||
| Rebecque J. Laba(11) |
110,570 | * | * | |||||||||
| Jeffrey J. Rudy(12) |
109,252 | * | * | |||||||||
| Fredrik Wiklund(13) |
104,404 | * | * | |||||||||
| Gregg Alton(14) |
17,332 | * | * | |||||||||
| Graham Cooper(15). |
17,332 | * | * | |||||||||
| Joshua Funder, Ph.D.(16) |
1,254,399 | 6.7 | % | 5.5 | % | |||||||
| Michael Narachi(17) |
87,554 | * | * | |||||||||
| Peter K. Honig, M.D., M.P.H.(18) |
6,388 | * | * | |||||||||
| Patrick Y. Yang, Ph.D.(19) |
6,388 | * | * | |||||||||
| All current executive officers and directors as a group (13 persons)(20) |
2,254,628 | 11.6 | % | 9.6 | % | |||||||
| * | Represents beneficial ownership of less than one percent |
| (1) | Consists of (a) 883,674 shares of common stock and 8,581 shares issuable upon the exercise of 2013 warrants held by Enterprise Partners Liquidating Trust V, (b) 1,016,477 shares of common stock and 8,581 shares issuable upon the exercise of 2013 warrants held by Enterprise Partners Liquidating Trust VI, and (c) 50,033 shares of common stock and 3,721 shares issuable upon the exercise of 2013 warrants held by Enterprise Partners Management, LLC. |
| (2) | Consists of 1,794,803 shares of common stock and 42,659 shares issuable upon the exercise of warrants. |
| (3) | Consists of 1,745,683 shares of common stock and 28,666 shares issuable upon the exercise of warrants. |
| (4) | Consists of 1,551,721 shares of common stock held by Novartis Bioventures Ltd. and 25,481 shares of common stock held by Novartis International Pharmaceutical Investment Ltd. Novartis Bioventures Ltd. and Novartis International Pharmaceutical Investment Ltd. are indirect wholly-owned subsidiaries of, and controlled by, Novartis AG. |
86
Table of Contents
| (5) | Consists of 1,194,204 shares of common stock and 53,807 shares issuable upon the exercise of warrants. Joshua Funder, Ph.D., one of our directors, shares voting and investment power with respect to the shares held by GBS Bioventures IV. Dr. Funder disclaims beneficial ownership of such shares except to the extent of his pecuniary interest therein. |
| (6) | Consists of (a) 999,825 shares of common stock and 16,419 shares issuable upon the exercise of warrants held by MPM BioVentures IV-QP, L.P., (b) 38,518 shares of common stock and 632 shares issuable upon the exercise of warrants held by MPM BioVentures IV GmbH & Co. Beteiligungs KG, and (c) 28,431 shares of common stock and 466 shares issuable upon the exercise of 2013 warrants held by MPM Asset Management Investors BV4 LLC. MPM BioVentures IV LLC is the Managing Member of MPM BioVentures IV GP LLC, which is the General Partner of MPM BioVentures IV-QP, L.P. and the Managing Limited Partner of MPM BioVentures IV GmbH & Co. Beteiligungs KG. MPM BioVentures IV LLC is the Manager of MPM Asset Management Investors BV4 LLC. Todd Foley is a Member of MPM BioVentures IV LLC and shares the power to vote, hold and dispose of the shares held by MPM BioVentures IV-QP, L.P., MPM Bio BioVentures IV GmbH & Co. Beteiligungs KG and MPM Asset Management Investors BV4 LLC. Mr. Foley and each such other Member of MPM BioVentures IV LLC disclaims beneficial ownership of the securities reported herein except to the extent of his respective pecuniary interest therein. |
| (7) | Consists of (a) 806,926 shares of common stock and 8,911 shares issuable upon the exercise of warrants held by Venrock Associates IV, L.P., (b) 19,814 shares of common stock and 218 shares issuable upon the exercise of warrants held by Venrock Entrepreneurs Fund IV, L.P., and (c) 164,550 shares of common stock and 1,817 shares issuable upon the exercise of warrants held by Venrock Partners, L.P. The sole general partner of Venrock Associates IV, L.P. is Venrock Management IV, LLC. The sole general partner of Venrock Entrepreneurs Fund IV, L.P. is VEF Management IV, LLC. The sole general partner of Venrock Partners, L.P. is Venrock Partners Management, LLC. Venrock Management IV, LLC, VEF Management IV, LLC and Venrock Partners Management, LLC disclaim beneficial ownership over all shares held by Venrock Associates IV, L.P., Venrock Entrepreneurs Fund IV, L.P. and Venrock Partners, L.P., except to the extent of their pecuniary interests therein. Anthony B. Evnin, Ph.D. is a member of Venrock Management IV, LLC, VEF Management IV, LLC and Venrock Partners Management, LLC and as such, he may be deemed to have voting and investment power with respect to these shares. Dr. Evnin disclaims beneficial ownership of these shares except to the extent of his indirect pecuniary interest therein. |
| (8) | Consists of (a) 669,639 shares of common stock and 10,996 shares issuable upon the exercise of warrants held by H&Q Healthcare Investors and (b) 300,856 shares of common stock and 4,940 shares issuable upon the exercise of warrants held by H&Q Life Sciences Investors (together with H&Q Healthcare Investors, the “H&Q Funds”). Tekla Capital Management LLC (“TCM”), the investment adviser to the H&Q Funds, and Daniel Omstead, Ph.D., the controlling member of TCM, have investment power with respect to the foregoing shares and share voting power with respect to the foregoing shares with the H&Q Funds. |
| (9) | Consists of 969,822 shares of common stock and 15,926 shares issuable upon the exercise of warrants. As the sole director of LSP IV, LSP IV Management may be deemed to beneficially own these securities. As managing directors of LSP IV Management, each of Martijn Kleijwegt, Rene Kuijten and Joachim Rothe may also be deemed to beneficially own these securities. |
| (10) | Includes 463,288 shares that Dr. Zsebo has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (11) | Includes 109,879 shares that Ms. Laba has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (12) | Includes 108,838 shares that Mr. Rudy has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (13) | Includes 82,843 shares that Mr. Wiklund has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (14) | Consists of 17,332 shares that Mr. Alton has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (15) | Consists of 17,332 shares that Mr. Cooper has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
87
Table of Contents
| (16) | Includes the securities described in footnote (5) above and 6,388 shares that Dr. Funder has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (17) | Includes 25,054 shares that Mr. Narachi has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (18) | Consists of 6,388 shares that Dr. Honig has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (19) | Consists of 6,388 shares that Dr. Yang has the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
| (20) | Consists of 1,281,478 shares of common stock, 53,807 shares issuable upon exercise of a warrant, and 973,150 shares that all executive officers and directors as a group have the right to acquire from us within 60 days of June 30, 2014 pursuant to the exercise of stock options. |
88
Table of Contents
Our amended and restated certificate of incorporation authorizes us to issue up to 200,000,000 shares of common stock, par value $0.001 per share, and 10,000,000 shares of preferred stock, par value $0.001 per share.
As of June 30, 2014, there were outstanding:
| • | 18,534,480 shares of common stock; |
| • | zero shares of preferred stock; |
| • | options exercisable for up to 2,510,828 shares of common stock; and |
| • | warrants exercisable for up to 206,340 shares of common stock. |
As of June 30, 2014, we had 37 holders of record of our common stock. The actual number of stockholders is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
The following description of our capital stock is not complete and is subject to and qualified in its entirety by our amended and restated certificate of incorporation and amended and restated bylaw, each filed as an exhibit to our Current Report on Form 8-K filed with the SEC on February 10, 2014, and by the relevant provisions of the Delaware General Corporation Law.
Common Stock
Voting
Our common stock is entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, including the election of directors, and does not have cumulative voting rights. Accordingly, the holders of a majority of the shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election.
Dividends
Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Rights and Preferences
Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
89
Table of Contents
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued in this offering will be, fully paid and nonassessable.
Preferred Stock
Our Board of Directors has the authority, without further action by the stockholders, to issue up to 10,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in our control that may otherwise benefit holders of our common stock and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock. As of June 30, 2014, there were no shares of preferred stock outstanding and we have no current plans to issue any shares of preferred stock.
Stock Options
As of June 30, 2014, there were 2,510,828 shares of common stock issuable upon the exercise of outstanding stock options, at a weighted-average exercise price of $6.06 per share.
Warrants
As of June 30, 2014, there were 206,340 shares of common stock issuable upon the exercise of outstanding warrants at an exercise price of $5.61 per share.
These warrants provide for cashless exercise at the option of the holder, and also contain provisions for the adjustment of the number of shares issuable upon the exercise of the warrant in the event of stock splits, recapitalizations, reclassifications and consolidations. Upon closing of this offering, these warrants will be automatically cancelled if not previously exercised. The warrants will expire in October 2018.
Registration Rights
Certain holders of our common stock, or their transferees, are entitled to the registration rights set forth below with respect to registration of the resale of such shares under the Securities Act pursuant to an amended and restated investors’ rights agreement by and among us and certain of our stockholders.
Demand Registration Rights
Upon the written request from the holders of 25% of the registrable securities (excluding registrable securities derived from our Junior preferred stock) then outstanding that we file a registration statement under the Securities Act with an anticipated aggregate price to the public of at least $5.0 million, we will be obligated to notify all holders of registrable securities of such request and to use our reasonable best efforts to register the sale of all registrable securities that holders may request to be registered. We are not required to effect more than two registration statements which are declared or ordered effective, subject to certain exceptions. We may postpone the filing of a registration statement for up to 90 days once in any 12-month period if in the good faith judgment of our board of directors such registration would be detrimental to us.
90
Table of Contents
Form S-3 Registration Rights
If we are eligible to file a registration statement on Form S-3, holders of registrable securities have the right to demand that we file a registration statement on Form S-3 so long as the aggregate amount of securities to be sold under the registration statement on Form S-3 is at least $3.0 million, subject to specified exceptions, conditions and limitations.
“Piggyback” Registration Rights
If we register any securities for public sale, holders of registration rights will have the right to include their shares in the registration statement. The underwriters of any underwritten offering will have the right to limit the number of shares having registration rights to be included in the registration statement, but not below 33% of the total number of shares included in the registration statement, except this offering in which the holders have waived any and all rights to have their shares included.
Expenses of Registration
Generally, we are required to bear all registration and selling expenses incurred in connection with the demand, piggyback and Form S-3 registrations described above, other than underwriting discounts and commissions.
Expiration of Registration Rights
The demand, piggyback and Form S-3 registration rights discussed above will terminate seven years following the closing of our initial public offering or, (i) as to a given holder of registrable securities, at such earlier time as the holder’s registrable securities, taken together with any registrable securities held by such holder’s affiliates, constitute less than 1% of our outstanding common stock and such holder is able to sell of such holder’s registrable securities in a single 90-day period under Rule 144 of the Securities Act, or (ii) as to any securities otherwise registrable pursuant to the exercise of the foregoing registration rights, at such time as the securities are sold (a) to the public, either through a registration statement or under Rule 144 of the Securities Act, or (b) in a private transaction in which the registration rights are not also transferred.
Anti-Takeover Effects of Provisions of Our Amended and Restated Certificate of Incorporation, Our Bylaws and Delaware Law
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law, or Section 203. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
| • | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
| • | the interested stockholder owned at least 85% of the voting stock of the corporation outstanding upon consummation of the transaction, excluding for purposes of determining the number of shares outstanding (1) shares owned by persons who are directors and also officers and (2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| • | on or subsequent to the consummation of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2⁄3% of the outstanding voting stock which is not owned by the interested stockholder. |
91
Table of Contents
Section 203 defines a business combination to include:
| • | any merger or consolidation involving the corporation and the interested stockholder; |
| • | any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; |
| • | subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; |
| • | subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; and |
| • | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Provisions of our amended and restated certificate of incorporation and amended and restated bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our amended and restated certificate of incorporation and amended and restated bylaws:
| • | permit our board of directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in our control); |
| • | provide that the authorized number of directors may be changed only by resolution adopted by a majority of the board of directors; |
| • | provide that the board of directors or any individual director may only be removed with cause and the affirmative vote of the holders of at least 66 2⁄3% of the voting power of all of our then outstanding common stock; |
| • | provide that all vacancies, including newly created directorships, may, except as otherwise required by law or subject to the rights of holders of preferred stock as designated from time to time, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| • | divide our board of directors into three classes; |
| • | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent; |
| • | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice; |
| • | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); |
92
Table of Contents
| • | provide that special meetings of our stockholders may be called only by the chairman of the board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors (whether or not there exists any vacancies); and |
| • | provide that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (1) any derivative action or proceeding brought on our behalf, (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors or officers to us or our stockholders, (3) any action asserting a claim against the us arising pursuant to any provision of the DGCL or our certificate of incorporation or bylaws or (4) any action asserting a claim against us governed by the internal affairs doctrine. |
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require the affirmative vote of the holders of at least 662/3% of the voting power of all of our then outstanding common stock.
NASDAQ Global Market Listing
Our common stock is listed on The NASDAQ Global Market under the symbol “CLDN.”
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company, LLC. The transfer agent and registrar’s address is 6201 15th Avenue, Brooklyn, New York 11219.
93
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Prior to our initial public offering in January 2014, there was no public market for our common stock. Future sales of substantial amounts of common stock in the public market, including shares issued upon exercise of outstanding options and warrants, or the anticipation of these sales, could adversely affect prevailing market prices from time to time and could impair our ability to raise equity capital in the future.
Based on the number of shares of common stock outstanding as of June 30, 2014, upon the completion of this offering we will have 22,534,480 shares of common stock outstanding, assuming (1) no exercise of the underwriters’ option to purchase additional shares of common stock and (2) no exercise of outstanding options or warrants. Of those shares, all of the shares sold in this offering and all 6,325,000 shares sold in our initial public offering will be freely tradable, except that any shares held by our “affiliates,” as that term is defined in Rule 144 under the Securities Act, or Rule 144, may only be sold in compliance with the limitations described below.
Rule 144
In general, under Rule 144 as currently in effect, any person who is not an affiliate of ours and has held their shares for at least six months, including the holding period of any prior owner other than one of our affiliates, may sell shares without restriction, provided current public information about us is available. In addition, under Rule 144, any person who is not an affiliate of ours and has held their shares for at least one year, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares without regard to whether current public information about us is available.
A person who is an affiliate of ours and who has beneficially owned restricted securities for at least six months, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted shares within any three-month period that does not exceed the greater of:
| • | 1% of the number of shares of our common stock then outstanding, which will equal approximately 225,345 shares immediately after this offering; or |
| • | the average weekly trading volume of our common stock on The NASDAQ Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale. |
Sales of restricted shares under Rule 144 held by our affiliates are also subject to requirements regarding the manner of sale, notice and the availability of current public information about us. Rule 144 also provides that affiliates relying on Rule 144 to sell shares of our common stock that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement.
Rule 701
In general, under Rule 701 of the Securities Act, any of our stockholders who purchased shares from us in connection with a qualified compensatory stock plan or other written agreement before we became subject to the reporting requirements of Section 13 or 15(d) of the Exchange Act is eligible to resell those shares in reliance on Rule 144. An affiliate of the issuer can resell shares in reliance on Rule 144 without having to comply with the holding period requirements of Rule 144, and a non-affiliate of the issuer can resell shares in reliance on Rule 144 without having to comply with the holding period requirements of Rule 144 and without regard to the volume of such sales or the availability of public information about the issuer.
As of June 30, 2014, options to purchase a total of 2,510,828 shares of common stock were outstanding, of which 876,334 were vested. Of the total number of shares of our common stock issuable under these options, 1,213,633 are subject to contractual lock-up agreements with the underwriters described below under “Underwriting” and will become eligible for sale at the expiration of those agreements.
94
Table of Contents
Lock-Up Agreements
We, along with our directors, executive management team and the entities affiliated with our directors, as well as certain of our existing stockholders, have agreed with the underwriters that for a period of 90 days (the restricted period) after the date of this prospectus, except with the prior written consent of Credit Suisse Securities (USA) LLC and Jefferies LLC and subject to specified exceptions, not to offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend or otherwise transfer or dispose of, directly or indirectly, any shares of common stock or any securities convertible into or exercisable or exchangeable for shares of common stock, or enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of the common stock. Credit Suisse Securities (USA) LLC and Jefferies LLC have advised us that they have no current intent or arrangement to release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period. Upon expiration of the restricted period, certain of our stockholders and warrantholders will have the right to require us to register their shares under the Securities Act. See “—Registration Rights” below and “Description of Capital Stock—Registration Rights.”
Certain of our employees, including our executive officers and/or directors, have entered into, and may in the future enter into, written trading plans that are intended to comply with Rule 10b5-1 under the Exchange Act. Sales under existing trading plans are exempt from the restrictions of the lock-up agreements relating to the offering described above. Sales under any trading plan entered into in the future, if any, would not be permitted until the expiration of the lock-up agreements relating to the offering described above.
Registration Rights
The holders of 11,119,262 shares of our common stock are entitled to rights with respect to the registration of their shares under the Securities Act, subject to the lock-up agreements described under “—Lock-Up Agreements” above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock. See “Description of Capital Stock—Registration Rights.”
Equity Incentive Plans
Shares of our common stock issued under our 2001 Stock Option Plan, 2012 Equity Incentive Plan, 2013 plan and the ESPP are available for sale in the open market, subject to Rule 144 volume limitations and the lock-up agreements described above, if applicable.
95
Table of Contents
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF OUR COMMON STOCK
The following discussion describes the material U.S. federal income tax consequences of the acquisition, ownership and disposition of our common stock acquired in this offering by Non-U.S. Holders (as defined below). This discussion does not address all aspects of U.S. federal income taxes that may be relevant to Non-U.S. Holders in light of their particular circumstances, nor does it address any U.S. federal estate or gift tax, any state, local or non-U.S. tax consequences. Rules different from those described below may apply to certain Non-U.S. Holders that are subject to special treatment under the Code such as financial institutions, insurance companies, tax-exempt organizations, tax-qualified retirement plans, broker-dealers and traders in securities, commodities or currencies, U.S. expatriates, “controlled foreign corporations,” “passive foreign investment companies,” corporations that accumulate earnings to avoid U.S. federal income tax, persons that hold our common stock as part of a “straddle,” “conversion transaction,” or other risk reduction strategy, holders deemed to sell our common stock under the constructive sale provisions of the Code, holders who hold or receive our common stock pursuant to the exercise of employee stock options or otherwise as compensation, holders who are subject to the alternative minimum tax or Medicare contribution tax, partnerships and other pass-through entities, and investors in such pass-through entities or entities that are treated as disregarded entities for U.S. federal income tax purposes (regardless of their places of organization or formation). Such Non-U.S. Holders are urged to consult their own tax advisors to determine the U.S. federal, state, local and other tax consequences that may be relevant to them. Furthermore, the discussion below is based upon the provisions of the Code and Treasury regulations, published administrative pronouncements, rulings and judicial decisions thereunder as of the date hereof. Such authorities may be repealed, revoked or modified, perhaps retroactively, so as to result in U.S. federal income tax consequences different from those discussed below. We have not requested a ruling from the U.S. Internal Revenue Service, or the IRS, with respect to the statements made and the conclusions reached in the following summary. This discussion assumes that the Non-U.S. Holder holds our common stock as a “capital asset” within the meaning of Section 1221 of the Code (generally, property held for investment).
The following discussion is for general information only and is not tax advice for any Non-U.S. Holder under its particular circumstances. Persons considering the purchase of our common stock pursuant to this offering should consult their own tax advisors concerning the U.S. federal income and estate tax consequences of acquiring, owning and disposing of our common stock in light of their particular situations as well as any consequences arising under the laws of any other taxing jurisdiction, including any state, local and non-U.S. tax consequences and any U.S. federal non-income tax consequences.
For the purposes of this discussion, a “Non-U.S. Holder” is, for U.S. federal income tax purposes, a beneficial owner of common stock that is not a U.S. Holder. A “U.S. Holder” means a beneficial owner of our common stock that is for U.S. federal income tax purposes (a) an individual who is a citizen or resident of the United States, (b) a corporation or other entity treated as a corporation created or organized in or under the laws of the United States, any state thereof or the District of Columbia, (c) an estate the income of which is subject to U.S. federal income taxation regardless of its source or (d) a trust if it (1) is subject to the primary supervision of a court within the United States and one or more U.S. persons have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. Also, partnerships, or other entities that are treated as partnerships for U.S. federal income tax purposes (regardless of their place of organization or formation) and entities that are treated as disregarded entities for U.S. federal income tax purposes (regardless of their place of organization or formation) are not addressed by this discussion and are, therefore, not considered to be Non-U.S. Holders for the purposes of this discussion.
Distributions on Our Common Stock
Subject to the discussion below regarding backup withholding and foreign accounts, distributions, if any, made on our common stock to a Non-U.S. Holder of our common stock generally will constitute dividends for U.S. tax purposes to the extent made out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles) and will be subject to withholding tax at a 30% rate or such lower rate as may
96
Table of Contents
be specified by an applicable income tax treaty. To obtain a reduced rate of withholding under a treaty, a Non-U.S. Holder generally will be required to provide us with a properly executed IRS Form W-8BEN or W-8BEN-E, or other appropriate form, certifying the Non-U.S. Holder’s entitlement to benefits under that treaty. In the case of a Non-U.S. Holder that is an entity, Treasury regulations and the relevant tax treaty provide rules to determine whether, for purposes of determining the applicability of a tax treaty, dividends will be treated as paid to the entity or to those holding an interest in that entity. If a Non-U.S. Holder holds stock through a financial institution or other agent acting on the holder’s behalf, the holder will be required to provide appropriate documentation to such agent. The holder’s agent will then be required to provide certification to the applicable withholding agent, either directly or through other intermediaries. If you are eligible for a reduced rate of U.S. federal withholding tax under an income tax treaty, you should consult with your own tax advisor to determine if you are able to obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim for a refund with the IRS.
We generally are not required to withhold tax on dividends paid to a Non-U.S. Holder that are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States (and, if required by an applicable income tax treaty, are attributable to a permanent establishment that such holder maintains in the United States) if a properly executed IRS Form W-8ECI, stating that the dividends are so connected, is furnished to us (or, if stock is held through a financial institution or other agent, to such agent). In general, such effectively connected dividends will be subject to U.S. federal income tax, on a net income basis at the regular graduated rates, unless a specific treaty exemption applies. A corporate Non-U.S. Holder receiving effectively connected dividends may also be subject to an additional “branch profits tax,” which is imposed, under certain circumstances, at a rate of 30% (or such lower rate as may be specified by an applicable treaty) on the corporate Non-U.S. Holder’s effectively connected earnings and profits, subject to certain adjustments.
To the extent distributions on our common stock, if any, exceed our current and accumulated earnings and profits, they will first reduce your basis in our common stock as a non-taxable return of capital, but not below zero, and then any excess will be treated as gain and taxed in the same manner as gain realized from a sale or other disposition of common stock as described in the next section.
Gain on Disposition of Our Common Stock
Subject to the discussion below regarding backup withholding and foreign accounts, a Non-U.S. Holder generally will not be subject to U.S. federal income tax with respect to gain realized on a sale or other disposition of our common stock unless (a) the gain is effectively connected with a trade or business of such holder in the United States (and, if required by an applicable income tax treaty, is attributable to a permanent establishment that such holder maintains in the United States), (b) the Non-U.S. Holder is a nonresident alien individual and is present in the United States for 183 or more days in the taxable year of the disposition and certain other conditions are met, or (c) we are or have been a “United States real property holding corporation,” or a USRPHC, within the meaning of Code Section 897(c)(2) at any time within the shorter of the five-year period preceding such disposition or such holder’s holding period.
If you are a Non-U.S. Holder described in (a) above, you will be required to pay tax on the net gain derived from the sale at regular graduated U.S. federal income tax rates, unless a specific treaty exemption applies, and corporate Non-U.S. Holders described in (a) above may be subject to the additional branch profits tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty. If you are an individual Non-U.S. Holder described in (b) above, you will be required to pay a flat 30% tax on the gain derived from the sale, which gain may be offset by U.S. source capital losses (even though you are not considered a resident of the United States). With respect to (c) above, in general, we would be a USRPHC if interests in U.S. real estate constituted (by fair market value) at least half of our assets. We believe that we are not, and do not anticipate becoming, a USRPHC, however, there can be no assurance that we will not become a USRPHC in the future. Even if we are treated as a USRPHC, gain realized by a Non-U.S. Holder on a disposition of our common stock will not be subject to U.S. federal income tax so long as (1) the Non-U.S. Holder owned, directly, indirectly and
97
Table of Contents
constructively, no more than 5% of our common stock at all times within the shorter of (a) the five-year period preceding the disposition or (b) the holder’s holding period and (2) our common stock is regularly traded on an established securities market. There can be no assurance that our common stock will continue to qualify as regularly traded on an established securities market.
Information Reporting Requirements and Backup Withholding
Generally, we or certain financial middlemen must report information to the IRS with respect to any dividends we pay on our common stock including the amount of any such dividends, the name and address of the recipient, and the amount, if any, of tax withheld. A similar report is sent to the holder to whom any such dividends are paid. Pursuant to tax treaties or certain other agreements, the IRS may make its reports available to tax authorities in the recipient’s country of residence.
Dividends paid by us (or our paying agents) to a Non-U.S. Holder may also be subject to U.S. backup withholding. U.S. backup withholding generally will not apply to a Non-U.S. Holder who provides a properly executed IRS Form W-8BEN or W-8BEN-E or otherwise establishes an exemption.
Under current U.S. federal income tax law, U.S. information reporting and backup withholding requirements generally will apply to the proceeds of a disposition of our common stock effected by or through a U.S. office of any broker, U.S. or non-U.S., unless the holder provides a properly executed IRS Form W-8BEN or W-8BEN-E or otherwise establishes an exemption. Generally, U.S. information reporting and backup withholding requirements will not apply to a payment of disposition proceeds to a Non-U.S. Holder where the transaction is effected outside the United States through a non-U.S. office of a non-U.S. broker. For information reporting purposes, certain brokers with substantial U.S. ownership or operations will generally be treated in a manner similar to U.S. brokers.
If backup withholding is applied to you, you should consult with your own tax advisor to determine if you are able to obtain a tax refund or credit with respect to the amount withheld.
Foreign Accounts
A U.S. federal withholding tax of 30% may apply to dividends and the gross proceeds of a disposition of our common stock paid to a foreign financial institution (as specifically defined by applicable rules), including when the foreign financial institution holds our common stock on behalf of a Non-U.S. Holder, unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which may include certain equity holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the United States governing these withholding and reporting requirements may be subject to different rules. This U.S. federal withholding tax of 30% may also apply to dividends and the gross proceeds of a disposition of our common stock paid to a non-financial foreign entity unless such entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners or provides information regarding direct and indirect U.S. owners of the entity. The withholding tax described above will not apply if the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from the rules. Under certain circumstances, a Non-U.S. Holder might be eligible for refunds or credits of such tax. Holders are encouraged to consult with their own tax advisors regarding the possible implications of this withholding on their investment in our common stock.
The withholding provisions described above generally apply to payments of dividends on our common stock and will apply to payments of gross proceeds from a sale or other disposition of common stock on or after January 1, 2017.
98
Table of Contents
EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS OWN TAX ADVISOR REGARDING THE TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON STOCK, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAW, AS WELL AS TAX CONSEQUENCES ARISING UNDER ANY STATE, LOCAL, NON-U.S. OR U.S. FEDERAL NON-INCOME TAX LAWS.
99
Table of Contents
Under the terms and subject to the conditions contained in an underwriting agreement dated , 2014, we have agreed to sell to the underwriters named below, for whom Credit Suisse Securities (USA) LLC and Jefferies LLC are acting as representatives, the following respective numbers of shares of common stock:
| Underwriter |
Number of Shares | |
| Credit Suisse Securities (USA) LLC |
||
| Jefferies LLC |
||
| Stifel, Nicolaus & Company, Incorporated |
||
| Wedbush Securities Inc |
||
|
| ||
| Total |
||
|
|
The underwriting agreement provides that the underwriters are obligated to purchase all the shares of common stock in the offering if any are purchased, other than those shares covered by the option to purchase additional shares described below. The underwriting agreement also provides that if an underwriter defaults, the purchase commitments of the non-defaulting underwriters may be increased or the offering may be terminated.
We have granted to the underwriters a 30-day option to purchase on a pro rata basis up to an aggregate of additional shares at the public offering price less the underwriting discounts and commissions.
The underwriters propose to offer the shares of common stock initially at the public offering price on the cover page of this prospectus and to selling group members at that price less a selling concession of up to $ per share. After the public offering the representatives may change the public offering price and selling concession.
The following table summarizes the underwriting discounts and commissions and estimated expenses we will pay. The information assumes either no exercise or full exercise by the underwriters of their option to purchase additional shares.
| Per Share | Total | |||||||||||||||
| Without Option to Purchase Additional Shares |
With Option to Purchase Additional Shares |
Without Option to Purchase Additional Shares |
With Option to Purchase Additional Shares |
|||||||||||||
| Underwriting discounts and commissions paid by us |
$ | $ | $ | $ | ||||||||||||
| Expenses payable by us |
$ | $ | $ | $ | ||||||||||||
The representatives have informed us that they do not expect sales to accounts over which the underwriters have discretionary authority to exceed 5% of the shares of common stock being offered.
We have agreed that we will not (i) offer, sell, issue, contract to sell, pledge or otherwise dispose of, (ii) offer, sell, issue, contract to sell, contract to purchase or grant any option, right or warrant to purchase, (iii) enter into any swap, hedge or any other agreement that transfers, in whole or in part, the economic consequences of ownership, (iv) establish or increase a put equivalent position or liquidate or decrease a call equivalent position or (iv) file with the Securities and Exchange Commission a registration statement under the Securities Act relating to, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, or publicly disclose the intention to take any such action, without the prior written consent of Credit Suisse Securities (USA) LLC and Jefferies LLC, for 90 days after the date of this prospectus, subject to certain exceptions.
The restrictions in the foregoing paragraph do not apply to (i) the sale of shares of common stock to the underwriters pursuant to the underwriting agreement, (ii) the issuance by us of shares of common stock pursuant to the exercise of options outstanding on the date of this prospectus, (iii) the grant of stock options, restricted
100
Table of Contents
stock or other equity-based compensation awards (or the issuance of common stock upon exercise thereof) to eligible participants pursuant to our employee benefit or equity incentive plans described in this prospectus, (iv) the establishment by a director, officer or stockholder of a trading plan pursuant to Rule 10b5-1 under the Exchange Act for the transfer of shares of our common stock; provided that such plan does not provide for the transfer of shares of our common stock during the restricted period and no public announcement or filing under the Exchange Act regarding the establishment of such plan shall be required or shall be voluntarily made by or on behalf of such director, officer or stockholder or us during the restricted period, (v) transactions by an officer relating to shares of our common stock executed under a trading plan pursuant to Rule 10b5-1 under the Exchange Act in existence as of the date of this prospectus providing for the transfer of shares of our common stock; provided that any filing under Section 16(a) of the Exchange Act that is made in connection with any such transaction during the restricted period shall state that such transaction has been executed under a trading plan pursuant to Rule 10b5-1 under the Exchange Act, and shall also state the date such trading plan was adopted, and (vi) the filing of a registration statement on Form S-8 with respect to the registration of securities to be offered under our employee benefit or equity incentive plans described in this prospectus.
Our executive officers, our directors and certain of our principal stockholders have agreed that they will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, enter into a transaction that would have the same effect, or enter into any swap, hedge or other arrangement that transfers, in whole or in part, any of the economic consequences of ownership of our common stock, whether any of these transactions are to be settled by delivery of our common stock or other securities, in cash or otherwise, or publicly disclose the intention to make any offer, sale, pledge or disposition, or to enter into any transaction, swap, hedge or other arrangement, without, in each case, the prior written consent of Credit Suisse Securities (USA) LLC and Jefferies LLC for a period of 90 days after the date of this prospectus, subject to certain exceptions.
We have agreed to indemnify the underwriters against liabilities under the Securities Act, or contribute to payments that the underwriters may be required to make in that respect. We have also agreed to reimburse the underwriters for up to $20,000 of expenses related to the review of this offering by the Financial Industry Regulatory Authority, Inc.
Our common stock is listed on The NASDAQ Global Market under the symbol “CLDN.”
The underwriters and their respective affiliates are full-service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriters and their respective affiliates may in the future provide financial advisory or investment banking services to us from time to time for which they expect to receive customary compensation and expense reimbursement.
In addition, in the ordinary course of their business activities, the underwriters and their affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers. Such investments and securities activities may involve securities and/or instruments of ours or our affiliates. The underwriters and their affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or financial instruments and may hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
In connection with the offering the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Exchange Act.
| • | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. |
101
Table of Contents
| • | Over-allotment involves sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the option to purchase additional shares. In a naked short position, the number of shares involved is greater than the number of shares in the option to purchase additional shares. The underwriters may close out any covered short position by either exercising their option to purchase additional shares and/or purchasing shares in the open market. |
| • | Syndicate covering transactions involve purchases of our common stock in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the option to purchase additional shares. If the underwriters sell more shares than could be covered by the option to purchase additional shares, a naked short position, the position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. |
| • | Penalty bids permit the representatives to reclaim a selling concession from a syndicate member when the common stock originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. These transactions may be effected on The NASDAQ Global Market or otherwise and, if commenced, may be discontinued at any time.
A prospectus in electronic format may be made available on the web sites maintained by one or more of the underwriters, or selling group members, if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses electronically. The representatives may agree to allocate a number of shares to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations.
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive, which we refer to as a Relevant Member State, an offer to the public of any shares of our common stock may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any shares of our common stock may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
| (a) | to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
| (b) | to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or |
| (c) | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares of our common stock shall result in a requirement for the publication by us or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive. |
102
Table of Contents
For the purposes of this provision, the expression an “offer to the public” in relation to any shares of our common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares of our common stock to be offered so as to enable an investor to decide to purchase any shares of our common stock, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
Our common stock may not be offered or sold and will not be offered or sold to any persons in the United Kingdom other than persons whose ordinary activities involve them in acquiring, holding, managing or disposing of investments (as principal or as agent) for the purposes of their businesses and in compliance with all applicable provisions of the Financial Services and Markets Act 2000 (“FSMA”) with respect to anything done in relation to our common stock in, from or otherwise involving the United Kingdom. Each underwriter has represented and agreed that:
| (a) | it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the FSMA) received by it in connection with the issue or sale of the shares of our common stock in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
| (b) | it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares of our common stock in, from or otherwise involving the United Kingdom. |
Hong Kong
The common shares may not be offered or sold in Hong Kong by means of any document other than (a) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (b) to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (c) in other circumstances which do not result in the document being a “prospectus” within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to common shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the common shares may not be circulated or distributed, nor may the common shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (a) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore, or the SFA, (b) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (c) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
103
Table of Contents
Where the common shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
| (a) | a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
| (b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, shares, debentures and units of shares and debentures of that corporation or the beneficiaries’ rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the common shares pursuant to an offer made under Section 275 of the SFA except: |
| i. | to an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in Section 275(2) of the SFA, or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and interest in that trust are acquired at a consideration of not less than S$200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA; |
| ii. | where no consideration is or will be given for the transfer; or |
| iii. | where the transfer is by operation of law. |
Switzerland
The common shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange, or the SIX, or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the common shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the common shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of common shares will not be supervised by, the Swiss Financial Market Supervisory Authority, or FINMA, and the offer of common shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes, or CISA. Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of common shares.
United Arab Emirates
This offering has not been approved or licensed by the Central Bank of the United Arab Emirates, or the UAE, Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the Dubai Financial Services Authority, or DFSA, a regulatory authority of the Dubai International Financial Centre, or DIFC. The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No 8 of 1984 (as amended), DFSA Offered Securities Rules and
104
Table of Contents
NASDAQ Dubai Listing Rules, accordingly, or otherwise. The common shares may not be offered to the public in the UAE and/or any of the free zones.
The common shares may be offered and issued only to a limited number of investors in the UAE or any of its free zones who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned.
France
This prospectus (including any amendment, supplement or replacement thereto) is not being distributed in the context of a public offering in France within the meaning of Article L. 411-1 of the French Monetary and Financial Code (Code monétaire et financier).
This prospectus has not been and will not be submitted to the French Autorité des marchés financiers, or the AMF, for approval in France and accordingly may not and will not be distributed to the public in France.
Pursuant to Article 211-3 of the AMF General Regulation, French residents are hereby informed that:
| (a) | the transaction does not require a prospectus to be submitted for approval to the AMF; |
| (b) | persons or entities referred to in Point 2°, Section II of Article L.411-2 of the Monetary and Financial Code may take part in the transaction solely for their own account, as provided in Articles D. 411-1, D. 734-1, D. 744-1, D. 754-1 and D. 764-1 of the Monetary and Financial Code; and |
| (c) | the financial instruments thus acquired cannot be distributed directly or indirectly to the public otherwise than in accordance with Articles L. 411-1, L. 411-2, L. 412-1 and L. 621-8 to L. 621-8-3 of the Monetary and Financial Code. |
This prospectus is not to be further distributed or reproduced (in whole or in part) in France by the recipients of this prospectus. This prospectus has been distributed on the understanding that such recipients will only participate in the issue or sale of our common stock for their own account and undertake not to transfer, directly or indirectly, our common stock to the public in France, other than in compliance with all applicable laws and regulations and in particular with Articles L. 411-1 and L. 411-2 of the French Monetary and Financial Code.
105
Table of Contents
The validity of the shares of common stock being offered by this prospectus will be passed upon for us by Cooley LLP, San Diego, California. As of the date of this prospectus, Cooley LLP beneficially owned less than one percent of the outstanding shares of our common stock. The underwriters are being represented by Latham & Watkins LLP, San Diego, California.
Ernst & Young LLP, an independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2013, as set forth in their report, which is incorporated by reference into this prospectus. Our financial statements are incorporated by reference in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1, including exhibits and schedules, under the Securities Act, with respect to the shares of common stock being offered by this prospectus. This prospectus, which constitutes part of the registration statement, does not contain all of the information in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.
You can read our SEC filings, including the registration statement, over the Internet at the SEC’s website at www.sec.gov. You may also read and copy any document we file with the SEC at its public reference facilities at 100 F Street, NE, Washington, D.C. 20549. You may also obtain copies of these documents at prescribed rates by writing to the Public Reference Section of the SEC at 100 F Street, NE, Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference facilities. You may also request a copy of these filings, at no cost, by writing us at 11988 El Camino Real, Suite 650, San Diego, California or telephoning us at (858) 366-4288.
We are subject to the information reporting requirements of the Exchange Act and file reports, proxy statements and other information with the SEC. These reports, proxy statements and other information are available for inspection and copying at the public reference room and web site of the SEC referred to above. We also maintain a website at www.celladon.com, at which you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Information contained on or accessible through our website is not a part of this prospectus, and the inclusion of our website address in this prospectus is an inactive textual reference only.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to “incorporate by reference” information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus. Information in this prospectus supersedes information incorporated by reference that we filed with the SEC prior to the date of this prospectus.
106
Table of Contents
We incorporate by reference into this prospectus and the registrations statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC (Commission File No. 001-36183):
| • | our Annual Report on Form 10-K for the year ended December 31, 2013, filed with the SEC on March 31, 2014; |
| • | our Definitive Proxy Statement on Schedule 14A, filed with the SEC on April 21, 2014 (other than the portions thereof which are furnished and not filed); |
| • | our Quarterly Reports on Form 10-Q for the quarters ended March 31, 2014 and June 30, 2014, filed with the SEC on May 13, 2014 and August 7, 2014; and |
| • | our Current Reports on Form 8-K filed with the SEC on February 10, 2014, February 24, 2014, March 3, 2014, March 25, 2014, April 10, 2014, May 21, 2014, June 2, 2014, June 3, 2014, July 21, 2014, July 30, 2014 and August 5, 2014. |
We will furnish without charge to you, on written or oral request, a copy of any or all of the documents incorporated by reference, including exhibits to these documents. You should direct any requests for documents by writing us at 11988 El Camino Real, Suite 650, San Diego, California 92130 or telephoning us at (858) 366-4288.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus will be deemed modified, superseded or replaced for purposes of this prospectus to the extent that a statement contained in this prospectus modifies, supersedes or replaces such statement.
107
Table of Contents
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth all costs and expenses, other than estimated underwriting discounts and commissions, payable by Celladon Corporation (the “Registrant”) in connection with the sale of the common stock being registered. All amounts shown are estimates except for the Securities and Exchange Commission (“SEC”) registration fee, the Financial Industry Regulatory Authority, Inc. (“FINRA”) filing fee and The NASDAQ Global Market listing fee.
| Amount paid or to be paid |
||||
| SEC registration fee |
$ | 8,147 | ||
| FINRA filing fee |
9,988 | |||
| Blue sky qualification fees and expenses |
7,500 | |||
| Printing and engraving expenses |
60,000 | |||
| Legal fees and expenses |
200,000 | |||
| Accounting fees and expenses |
100,000 | |||
| Transfer agent and registrar fees and expenses |
5,000 | |||
| Miscellaneous expenses |
48,000 | |||
|
|
|
|||
| Total |
$ | 438,635 | ||
|
|
|
|||
Item 14. Indemnification of Directors and Officers.
The Registrant is incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law provides that a Delaware corporation may indemnify any persons who were, are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation, or is or was serving at the request of such corporation as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who were, are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses (including attorneys’ fees) actually and reasonably incurred.
The Registrant’s amended and restated certificate of incorporation and amended and restated bylaws provide for the indemnification of its directors and officers to the fullest extent permitted under the Delaware General Corporation Law.
II-1
Table of Contents
Section 102(b)(7) of the Delaware General Corporation Law permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| • | transaction from which the director derives an improper personal benefit; |
| • | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | unlawful payment of dividends or redemption of shares; or |
| • | breach of a director’s duty of loyalty to the corporation or its stockholders. |
The Registrant’s amended and restated certificate of incorporation includes such a provision. Expenses incurred by any officer or director in defending any such action, suit or proceeding in advance of its final disposition shall be paid by the Registrant upon delivery to it of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by the Registrant.
Section 174 of the Delaware General Corporation Law provides, among other things, that a director who willfully or negligently approves of an unlawful payment of dividends or an unlawful stock purchase or redemption, may be held liable for such actions. A director who was either absent when the unlawful actions were approved or dissented at the time may avoid liability by causing his or her dissent to such actions to be entered in the books containing minutes of the meetings of the board of directors at the time such action occurred or immediately after such absent director receives notice of the unlawful acts.
As permitted by the Delaware General Corporation Law, the Registrant has entered into indemnity agreements with each of its directors and executive officers, that require the Registrant to indemnify such persons against any and all costs and expenses (including attorneys’, witness or other professional fees) actually and reasonably incurred by such persons in connection with any action, suit or proceeding (including derivative actions), whether actual or threatened, to which any such person may be made a party by reason of the fact that such person is or was a director or officer or is or was acting or serving as an officer, director, employee or agent of the Registrant or any of its affiliated enterprises. Under these agreements, the Registrant is not required to provided indemnification for certain matters, including:
| • | indemnification beyond that permitted by the Delaware General Corporation Law; |
| • | indemnification for any proceeding with respect to the unlawful payment of remuneration to the director or officer; |
| • | indemnification for certain proceedings involving a final judgment that the director or officer is required to disgorge profits from the purchase or sale of the Registrant’s stock; |
| • | indemnification for proceedings involving a final judgment that the director’s or officer’s conduct was in bad faith, knowingly fraudulent or deliberately dishonest or constituted willful misconduct or a breach of his or her duty of loyalty, but only to the extent of such specific determination; |
| • | indemnification for proceedings or claims brought by an officer or director against us or any of the Registrant’s directors, officers, employees or agents, except for (1) claims to establish a right of indemnification or proceedings, (2) claims approved by the Registrant’s board of directors, (3) claims required by law, (4) when there has been a change of control as defined in the indemnification agreement with each director or officer, or (5) by the Registrant in its sole discretion pursuant to the powers vested to the Registrant under Delaware law; |
| • | indemnification for settlements the director or officer enters into without the Registrant’s consent; or |
| • | indemnification in violation of any undertaking required by the Securities Act or in any registration statement filed by the Registrant. |
II-2
Table of Contents
The indemnification agreements also set forth certain procedures that will apply in the event of a claim for indemnification thereunder.
Except as otherwise disclosed under the heading “Legal Proceedings” in the “Business” section of the prospectus included in this registration statement, there is at present no pending litigation or proceeding involving any of the Registrant’s directors or executive officers as to which indemnification is required or permitted, and the Registrant is not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
The Registrant has an insurance policy in place that covers its officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act of 1933, as amended (the “Securities Act”) or otherwise.
The Registrant plans to enter into an underwriting agreement which provides that the underwriters are obligated, under some circumstances, to indemnify the Registrant’s directors, officers and controlling persons against specified liabilities, including liabilities under the Securities Act.
Item 15. Recent Sales of Unregistered Securities.
The following sets forth information regarding all unregistered securities sold by the Registrant since January 1, 2010:
| (1) | From May 2010 to December 2011, the Registrant issued and sold to investors convertible promissory notes in the aggregate principal amount of $8.9 million, which notes bore interest at the rate of 12% per annum. |
| (2) | In January 2012, the Registrant issued and sold to investors an aggregate of 27,616,923 shares of its Series A-1 preferred stock and 12,138,080 shares of its Junior preferred stock, at a purchase price of $0.449 per share, for aggregate consideration of $17.8 million. Of this amount, $12.4 million was paid for by cancellation of principal indebtedness under the promissory notes described in paragraph (1) above and the balance was paid for in cash. |
| (3) | In January 2012, the Registrant issued an aggregate of 849,949 shares of its common stock to the holders of the convertible promissory notes described in paragraph (1) above in exchange for the waiver of such holders’ rights to receive payment of unpaid accrued interest under such notes. The foregoing share numbers reflect the effect of the 1-for-12.49 reverse stock split of the Registrant’s common stock effected on October 25, 2013. |
| (4) | From March 2012 to June 2012, the Registrant issued and sold to investors an aggregate of 88,807,202 shares of its Series A-1 preferred stock, at a purchase price of $0.449 per share, for aggregate cash consideration of $39.9 million. In addition, from April 2012 to June 2012, the Registrant issued securities exchangeable for 10,716,405 shares of its Series A-1 preferred stock, at a purchase price of $0.449 per share on an as-exchanged basis, for aggregate cash consideration of $4.8 million. |
| (5) | In June 2013, the Registrant issued 10,716,405 shares of its Series A-1 preferred stock to Coôperatief LSP IV UA in consideration for the exchange by Coôperatief LSP IV UA of share capital it held in the Registrant’s Netherlands-based subsidiary, Celladon Europe B.V. No additional consideration was provided for this issuance. |
| (6) | From January 14, 2010 to June 10, 2010, the Registrant granted stock options under its 2001 Stock Option Plan to purchase an aggregate of 1,770 shares of common stock to its employees, directors and consultants, having exercise prices ranging from $224.82 to $349.72 per share. The foregoing share numbers and per share exercise prices reflect the effect of the 1-for-100 reverse split of the Registrant’s capital stock that occurred in January 2012 and the 1-for-12.49 reverse stock split of the Registrant’s common stock effected on October 25, 2013. |
II-3
Table of Contents
| (7) | From June 15, 2012 to October 16, 2013, the Registrant granted stock options under its 2012 Equity Incentive Plan to purchase up to an aggregate of 1,563,355 shares of its common stock to its employees, directors and consultants, at an exercise price of $1.12 per share. The foregoing share numbers and per share exercise prices reflect the effect of the 1-for-12.49 reverse stock split of the Registrant’s common stock effected on October 25, 2013. |
| (8) | In October 2013, the Registrant issued and sold to investors convertible promissory notes in the aggregate principal amount of $1,097,017 and warrants exercisable for an aggregate of 2,895,570 shares of its Series A-1 preferred stock at an exercise price of $0.449 per share. The aggregate consideration paid to the Registrant for these convertible notes and warrants was $1,097,307. In connection with the completion of the Registrant’s initial public offering, the principal amount of the convertible notes and accrued interest automatically converted into 139,665 shares of the Registrant’s common stock at a conversion price of $8.00 per share. Following the completion of the Registrant’s initial public offering, the warrants were initially exercisable for an aggregate of 231,821 shares of the Registrant’s common stock at an exercise price of $5.61 per share. A total of 25,481 of these warrants were exercised for cash as of June 30, 2014. The foregoing share numbers and per share exercise prices reflect the effect of the 1-for-12.49 reverse stock split of the Registrant’s common stock effected on October 25, 2013. |
The offers, sales and issuances of the securities described in paragraphs (1) through (5) and (8) above were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act and Rule 506 promulgated under Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited investor within the meaning of Rule 501 of Regulation D under the Securities Act and had adequate access, through employment, business or other relationships, to information about us. No underwriters were involved in these transactions.
The offers, sales and issuances of the securities described in paragraphs (6) and (7) above were deemed to be exempt from registration under the Securities Act in reliance on Rule 701 in that the transactions were under compensatory benefit plans and contracts relating to compensation as provided under Rule 701. The recipients of such securities were our employees, directors or bona fide consultants and received the securities under the Registrant’s 2001 Stock Option Plan and/or the Registrant’s 2012 Equity Incentive Plan. Appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions had adequate access, through employment, business or other relationships, to information about us.
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits.
| Exhibit |
Description | |||
| 1.1 | Form of Underwriting Agreement. | |||
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed with the SEC on February 10, 2014). | |||
| 3.2 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 to the Registrant’s Current Report on Form 8-K, filed with the SEC on February 10, 2014). | |||
| 4.1 | Reference is made to Exhibits 3.1 and 3.2. | |||
II-4
Table of Contents
| Exhibit |
Description | |||
| 4.2 | Form of Common Stock Certificate of the Registrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 4.3 | Amended and Restated Investor Rights Agreement by and among the Registrant and certain of its stockholders, dated February 4, 2014 (incorporated by reference to Exhibit 4.2 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 4.4 | Form of Warrant to Purchase Common Stock issued to participants in the Registrant’s Convertible Debt and Warrant financing, dated October 15, 2013 (incorporated by reference to Exhibit 4.3 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 5.1 | Opinion of Cooley LLP. | |||
| 10.1+ | Form of Indemnity Agreement by and between the Registrant and its directors and officer (incorporated by reference to Exhibit 10.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.2+ | Celladon Corporation 2001 Stock Option Plan and Form of Notice of Grant of Stock Option, Stock Option Agreement and Stock Option Exercise Notice thereunder (incorporated by reference to Exhibit 10.2 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.3+ | Celladon Corporation 2012 Equity Incentive Plan and Form of Stock Option Grant Notice, Option Agreement and Notice of Exercise thereunder (incorporated by reference to Exhibit 10.3 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.4+ | Celladon Corporation 2013 Equity Incentive Plan and Form of Stock Option Grant Notice, Option Agreement and Notice of Exercise thereunder (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.5+ | Celladon Corporation 2013 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.6+ | Celladon Corporation Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.6 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.7+ | Employment Agreement by and between the Registrant and Jeffrey J. Rudy, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.7 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.8+ | Employment Agreement by and between the Registrant and Rebecque Laba, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.8 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.9+ | Employment Agreement by and between the Registrant and Ryan K. Takeya, dated September 2, 2013, as amended (incorporated by reference to Exhibit 10.9 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
II-5
Table of Contents
| Exhibit |
Description | |||
| 10.10+ | Employment Agreement by and between the Registrant and Fredrik Wiklund, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.10 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.11+ | Employment Agreement by and between the Registrant and Krisztina M. Zsebo, Ph.D., dated August 30, 2013, as amended (incorporated by reference to Exhibit 10.11 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.12+ | Letter Agreement by and between the Registrant and Gregg Huber Alton, dated August 30, 2013 (incorporated by reference to Exhibit 10.12 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.13+ | Letter Agreement by and between the Registrant and Graham Cooper, dated September 2, 2013 (incorporated by reference to Exhibit 10.13 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.14 | Office Lease by and between the Registrant and Arden Realty, Inc., dated March 6, 2012, as amended (incorporated by reference to Exhibit 10.14 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.15* | License Agreement by and between the Registrant and the Regents of the University of California, dated February 10, 2001, as amended (incorporated by reference to Exhibit 10.15 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.16* | Exclusive License Agreement by and between the Registrant and Martin J. Kaplitt, M.D., dated June 7, 2006 (incorporated by reference to Exhibit 10.16 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.17* | Non-Exclusive License Agreement by and between the Registrant and AskBio, LLC, dated January 15, 2008 (incorporated by reference to Exhibit 10.17 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.18 | License Agreement by and between the Registrant and AdVec Inc., dated February 24, 2009 (incorporated by reference to Exhibit 10.18 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.19* | Exclusive Patent License Agreement by and between the Registrant and the Regents of the University of Minnesota, dated May 11, 2009 (incorporated by reference to Exhibit 10.19 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.20* | Non-Exclusive License Agreement by and between the Registrant and Virovek Incorporation, dated November 4, 2010 (incorporated by reference to Exhibit 10.20 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.21* | Amended and Restated License Agreement by and between the Registrant and AmpliPhi Biosciences Corporation, dated June 27, 2012 (incorporated by reference to Exhibit 10.21 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.22* | Sublicense Agreement by and between the Registrant and AmpliPhi Biosciences Corporation, dated June 27, 2012 (incorporated by reference to Exhibit 10.22 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
II-6
Table of Contents
| Exhibit |
Description | |||
| 10.23* | Amended and Restated Manufacturing Services Agreement by and between the Registrant and Lonza Houston, Inc., dated August 26, 2013 (incorporated by reference to Exhibit 10.23 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.24+ | Letter Agreement by and between the Registrant and Michael Narachi, dated October 16, 2013 (incorporated by reference to Exhibit 10.24 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.25* | Material Transfer and Exclusivity Agreement by and between the Registrant and Les Laboratoires Servier, dated February 20, 2014 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q filed with the SEC on May 13, 2014). | |||
| 10.26(1) | Sublease Agreement by and between the Registrant and Brandes Investment Partners, L.P. dated May 28, 2014. | |||
| 10.27(1) | Assignment and License Agreement by and between the Registrant and Enterprise Management Partners, LLC dated July 18, 2014 (incorporated by reference to Exhibit 99.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on July 21, 2014). | |||
| 10.28+ | (1) | Employment Agreement by and between the Registrant and Paul Cleveland, dated May 28, 2014. | ||
| 10.29+ | (1) | Employment Agreement by and between the Registrant and Elizabeth Reed, dated May 30, 2014. | ||
| 10.30 | Loan and Security Agreement by and between the Registrant and Hercules Technology III, L.P. and Hercules Technology Growth Capital, Inc., dated as of July 31, 2014 (incorporated by reference to Exhibit 99.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 5, 2014). | |||
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |||
| 23.2 | Consent of Cooley LLP. See Exhibit 5.1. | |||
| 24.1 | Power of Attorney. Reference is made to the signature page hereto. | |||
| (1) | Previously filed. |
| + | Indicates management contract or compensatory plan. |
| * | The Registrant has obtained confidential treatment with respect to certain portions of this exhibit. |
(b) Financial Statement Schedules.
Not applicable.
Item 17. Undertakings.
The undersigned Registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer, or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-7
Table of Contents
The undersigned Registrant hereby undertakes that:
| (a) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (b) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-8
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of San Diego, State of California, on the 6th day of August, 2014.
| CELLADON CORPORATION |
| /s/ Paul B. Cleveland |
| Paul B. Cleveland |
| President and Chief Financial Officer |
Pursuant to the requirements of the Securities Act, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Krisztina M. Zsebo, Ph.D. Krisztina M. Zsebo, Ph.D. |
Chief Executive Officer and Member of the (Principal Executive Officer) |
August 6, 2014 | ||
| /s/ Paul B. Cleveland Paul B. Cleveland |
President and Chief Financial Officer (Principal Financial Officer) |
August 6, 2014 | ||
| /s/ Rebecque J. Laba Rebecque J. Laba |
Vice President, Finance and Administration (Principal Accounting Officer) |
August 6, 2014 | ||
| /s/ Michael Narachi* Michael Narachi |
Chairman of the Board of Directors | August 6, 2014 | ||
|
Gregg Alton |
Member of the Board of Directors | August , 2014 | ||
| /s/ Graham Cooper* Graham Cooper |
Member of the Board of Directors | August 6, 2014 | ||
| /s/ Joshua Funder, Ph.D.* Joshua Funder, Ph.D. |
Member of the Board of Directors | August 6, 2014 | ||
| /s/ Peter K. Honig, M.D., M.P.H* Peter K. Honig, M.D., M.P.H |
Member of the Board of Directors | August 6, 2014 | ||
| /s/ Patrick Y. Yang, Ph.D.* Patrick Y. Yang, Ph.D. |
Member of the Board of Directors | August 6, 2014 | ||
* Pursuant to Power of Attorney
| By: |
/s/ Paul B. Cleveland | |
| Paul B. Cleveland | ||
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Description | |||
| 1.1 | Form of Underwriting Agreement. | |||
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed with the SEC on February 10, 2014). | |||
| 3.2 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 to the Registrant’s Current Report on Form 8-K, filed with the SEC on February 10, 2014). | |||
| 4.1 | Reference is made to Exhibits 3.1 and 3.2. | |||
| 4.2 | Form of Common Stock Certificate of the Registrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 4.3 | Amended and Restated Investor Rights Agreement by and among the Registrant and certain of its stockholders, dated February 4, 2014 (incorporated by reference to Exhibit 4.2 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 4.4 | Form of Warrant to Purchase Common Stock issued to participants in the Registrant’s Convertible Debt and Warrant financing, dated October 15, 2013 (incorporated by reference to Exhibit 4.3 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 5.1 | Opinion of Cooley LLP. | |||
| 10.1+ | Form of Indemnity Agreement by and between the Registrant and its directors and officer (incorporated by reference to Exhibit 10.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.2+ | Celladon Corporation 2001 Stock Option Plan and Form of Notice of Grant of Stock Option, Stock Option Agreement and Stock Option Exercise Notice thereunder (incorporated by reference to Exhibit 10.2 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.3+ | Celladon Corporation 2012 Equity Incentive Plan and Form of Stock Option Grant Notice, Option Agreement and Notice of Exercise thereunder (incorporated by reference to Exhibit 10.3 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.4+ | Celladon Corporation 2013 Equity Incentive Plan and Form of Stock Option Grant Notice, Option Agreement and Notice of Exercise thereunder (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.5+ | Celladon Corporation 2013 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.6+ | Celladon Corporation Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.6 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.7+ | Employment Agreement by and between the Registrant and Jeffrey J. Rudy, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.7 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
Table of Contents
| Exhibit Number |
Description | |||
| 10.8+ | Employment Agreement by and between the Registrant and Rebecque Laba, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.8 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.9+ | Employment Agreement by and between the Registrant and Ryan K. Takeya, dated September 2, 2013, as amended (incorporated by reference to Exhibit 10.9 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.10+ | Employment Agreement by and between the Registrant and Fredrik Wiklund, dated September 3, 2013, as amended (incorporated by reference to Exhibit 10.10 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.11+ | Employment Agreement by and between the Registrant and Krisztina M. Zsebo, Ph.D., dated August 30, 2013, as amended (incorporated by reference to Exhibit 10.11 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.12+ | Letter Agreement by and between the Registrant and Gregg Huber Alton, dated August 30, 2013 (incorporated by reference to Exhibit 10.12 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.13+ | Letter Agreement by and between the Registrant and Graham Cooper, dated September 2, 2013 (incorporated by reference to Exhibit 10.13 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.14 | Office Lease by and between the Registrant and Arden Realty, Inc., dated March 6, 2012, as amended (incorporated by reference to Exhibit 10.14 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.15* | License Agreement by and between the Registrant and the Regents of the University of California, dated February 10, 2001, as amended (incorporated by reference to Exhibit 10.15 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.16* | Exclusive License Agreement by and between the Registrant and Martin J. Kaplitt, M.D., dated June 7, 2006 (incorporated by reference to Exhibit 10.16 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.17* | Non-Exclusive License Agreement by and between the Registrant and AskBio, LLC, dated January 15, 2008 (incorporated by reference to Exhibit 10.17 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.18 | License Agreement by and between the Registrant and AdVec Inc., dated February 24, 2009 (incorporated by reference to Exhibit 10.18 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.19* | Exclusive Patent License Agreement by and between the Registrant and the Regents of the University of Minnesota, dated May 11, 2009 (incorporated by reference to Exhibit 10.19 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.20* | Non-Exclusive License Agreement by and between the Registrant and Virovek Incorporation, dated November 4, 2010 (incorporated by reference to Exhibit 10.20 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
Table of Contents
| Exhibit Number |
Description | |||
| 10.21* | Amended and Restated License Agreement by and between the Registrant and AmpliPhi Biosciences Corporation, dated June 27, 2012 (incorporated by reference to Exhibit 10.21 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.22* | Sublicense Agreement by and between the Registrant and AmpliPhi Biosciences Corporation, dated June 27, 2012 (incorporated by reference to Exhibit 10.22 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.23* | Amended and Restated Manufacturing Services Agreement by and between the Registrant and Lonza Houston, Inc., dated August 26, 2013 (incorporated by reference to Exhibit 10.23 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.24+ | Letter Agreement by and between the Registrant and Michael Narachi, dated October 16, 2013 (incorporated by reference to Exhibit 10.24 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-191688), originally filed with the SEC on October 11, 2013). | |||
| 10.25* | Material Transfer and Exclusivity Agreement by and between the Registrant and Les Laboratoires Servier, dated February 20, 2014 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q filed with the SEC on May 13, 2014). | |||
| 10.26(1) | Sublease Agreement by and between the Registrant and Brandes Investment Partners, L.P. dated May 28, 2014. | |||
| 10.27(1) | Assignment and License Agreement by and between the Registrant and Enterprise Management Partners, LLC dated July 18, 2014 (incorporated by reference to Exhibit 99.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on July 21, 2014). | |||
| 10.28+(1) | Employment Agreement by and between the Registrant and Paul Cleveland, dated May 28, 2014. | |||
| 10.29+(1) | Employment Agreement by and between the Registrant and Elizabeth Reed, dated May 30, 2014. | |||
| 10.30 | Loan and Security Agreement by and between the Registrant and Hercules Technology III, L.P. and Hercules Technology Growth Capital, Inc., dated as of July 31, 2014 (incorporated by reference to Exhibit 99.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 5, 2014). | |||
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |||
| 23.2 | Consent of Cooley LLP. See Exhibit 5.1. | |||
| 24.1 | Power of Attorney. Reference is made to the signature page hereto. | |||
| (1) | Previously filed. |
| + | Indicates management contract or compensatory plan. |
| * | The Registrant has obtained confidential treatment with respect to certain portions of this exhibit. |