Attached files
| file | filename |
|---|---|
| EX-5.1 - EX-5.1 - Vericel Corp | a13-11022_1ex5d1.htm |
| EX-23.1 - EX-23.1 - Vericel Corp | a13-11022_1ex23d1.htm |
As filed with the Securities and Exchange Commission on August 12, 2013.
Registration No. 333-188186
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 5
TO
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
AASTROM BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
|
Michigan |
|
94-3096597 |
24 Frank Lloyd Wright Drive
Lobby K
Ann Arbor, Michigan 48105
(800) 556-0311
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Dominick C. Colangelo
President and Chief Executive Officer
Aastrom Biosciences, Inc.
Lobby K
Ann Arbor, Michigan 48105
(800) 556-0311
(Name, address, including zip code, and telephone number, including area code, of agent for service)
|
Mitchell S. Bloom |
Yvan-Claude Pierre |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended, check the following box. x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated |
|
Accelerated |
|
Non-accelerated filer o |
|
Smaller reporting |
CALCULATION OF REGISTRATION FEE
|
|
|
|
|
|
| ||
|
|
|
Proposed |
|
|
| ||
|
|
|
Maximum |
|
|
| ||
|
Title of Each Class of |
|
Aggregate |
|
Amount of |
| ||
|
Securities to be Registered(1) |
|
Offering Price (2) (3) |
|
Registration Fee (4) |
| ||
|
Common Stock (no par value) |
|
$ |
10,000,000 |
|
$ |
1,364 |
|
|
Common Stock Purchase Warrants |
|
500,000 |
|
68 |
| ||
|
Shares of Common Stock underlying Common Stock Purchase Warrants |
|
12,500,000 |
|
1,705 |
| ||
|
Total |
|
$ |
23,000,000 |
|
$ |
3,137 |
|
(1) This Registration Statement also relates to the rights to purchase shares of Series A Junior Participating Cumulative Preferred Stock of the Registrant which are attached to all shares of Common Stock outstanding as of, and issued subsequent to, August 15, 2011, pursuant to the terms of the Registrant’s Shareholder Rights Agreement, dated August 11, 2011, as amended on March 9, 2012. Until the occurrence of certain prescribed events, the Rights are not exercisable, are evidenced by the certificates for the Common Stock and will be transferred with and only with such stock.
(2) Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act.
(3) Includes the shares of common stock that the underwriters have an option to purchase to cover over-allotments, if any.
(4) The Registrant previously paid the registration fee of $3,922 in connection with the initial and amended filings of this Registration Statement
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until this Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting offers to buy these securities in any state where the offer or sale is not permitted.
|
|
|
| ||
|
PRELIMINARY PROSPECTUS |
SUBJECT TO COMPLETION |
DATED August 12, 2013 | ||
Shares of Common Stock
|
Warrants to Purchase Shares of Common Stock |

We are offering shares of our common stock and warrants to purchase up to an aggregate shares of our common stock. The warrants will have a per share exercise price of $ [125% of public offering price of the common stock]. The warrants are exercisable immediately and will expire five years from the date of issuance.
Our common stock is traded on The NASDAQ Capital Market under the symbol “ASTM.” On August 9, 2013, the last reported price for our common stock was $0.57 per share. There is no established public trading market for the warrants, and we do not expect a market to develop. In addition, we do not intend to apply for a listing of the warrants on any national securities exchange.
Certain of our affiliates have indicated preliminary interest in purchasing up to 25% of the total number of shares of our common stock and/or warrants to be sold by us in this offering.
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 10 of this prospectus for a discussion of information that should be considered in connection with an investment in our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
|
|
|
Per Share |
|
Per Warrant |
|
Total |
|
|
Public offering price |
|
$ |
|
$ |
|
$ |
|
|
Underwriting discounts (1) |
|
$ |
|
$ |
|
$ |
|
|
Proceeds, before expenses, to us |
|
$ |
|
$ |
|
$ |
|
(1) The underwriters will receive compensation in addition to the underwriting discount. See “Underwriting” beginning on page 45 of this prospectus for a description of compensation payable to the underwriters.
We have granted a 45-day option to the underwriters to purchase up to of additional shares of common stock and/or warrants solely to cover over-allotments, if any.
The underwriters expect to deliver the shares to purchasers in the offering on or about , 2013.
Sole Book-Running Manager
Aegis Capital Corp
, 2013
|
|
Page |
|
|
|
|
1 | |
|
|
|
|
2 | |
|
|
|
|
10 | |
|
|
|
|
23 | |
|
|
|
|
24 | |
|
|
|
|
24 | |
|
|
|
|
25 | |
|
|
|
|
25 | |
|
|
|
|
26 | |
|
|
|
|
27 | |
|
|
|
|
40 | |
|
|
|
|
CERTAIN PROVISIONS OF MICHIGAN LAW AND OF OUR CHARTER AND BYLAWS |
44 |
|
|
|
|
45 | |
|
|
|
|
54 | |
|
|
|
|
54 | |
|
|
|
|
55 | |
|
|
|
|
55 | |
|
|
|
|
56 |
You may rely only on the information provided or incorporated by reference in this prospectus and the documents incorporated herein and therein by reference, or in a prospectus supplement or amendment thereto. We and the underwriters have not authorized anyone to provide you with information different from that contained in or incorporated by reference into this prospectus. We and the underwriters are offering to sell, and seeking offers to buy, common stock only in jurisdictions where offers and sales are permitted. The information contained in this prospectus, any free writing prospectus, or document incorporated by reference is accurate only as of its date, regardless of the time of delivery of this prospectus or of any sale of our common stock.
Information contained on our website is not part of this prospectus. You should read this prospectus together with additional information described under the heading “Where You Can Find More Information” below. In various places in this prospectus, we refer you to sections for additional information by indicating the caption heading of the other sections. All cross-references in this prospectus are to captions contained in this prospectus, unless otherwise indicated.
For investors outside the United States: We have not and the underwriters have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the shares of common stock and the distribution of this prospectus outside the United States.
This summary highlights information contained elsewhere in this prospectus or incorporated by reference in this prospectus. This summary does not contain all of the information that you should consider in making your investment decision. You should read the entire prospectus carefully, especially the discussion regarding the risks of investing in our securities under the heading “Risk Factors” beginning on page of this prospectus and our financial statements and related notes incorporated by reference in this prospectus, before investing in our securities. In this prospectus, “Aastrom,” the “Company,” “we,” “us,” and “our” refer to Aastrom Biosciences, Inc. Please refer to our Glossary at the end of this Prospectus for certain industry-specific and technical definitions.
Aastrom Biosciences, Inc.
Business Overview
We are a clinical-stage biotechnology company focused on developing innovative cell therapies that repair and regenerate damaged tissue for use in the treatment of severe, chronic ischemic cardiovascular diseases. We are developing patient-specific (autologous) multicellular therapies utilizing our proprietary, highly automated and scalable manufacturing system. Our manufacturing technology platform, the Aastrom Replicell System (ARS), enables the expansion of a variety of cell types, including the production of multicellular therapies expanded from an adult patient’s own bone marrow, which can be delivered directly to damaged tissues using conventional syringes and cell injection catheter systems.
Our lead product, ixmyelocel-T, has demonstrated multiple biological activities that promote tissue repair and regeneration by reducing inflammation, promoting angiogenesis and remodeling ischemic tissue. Preclinical and clinical data suggest that ixmyelocel-T is safe and effective in treating patients with severe, chronic ischemic cardiovascular diseases such as advanced heart failure due to dilated cardiomyopathy (DCM), the third leading cause of heart failure, and critical limb ischemia (CLI), the most severe form of peripheral arterial disease (PAD).
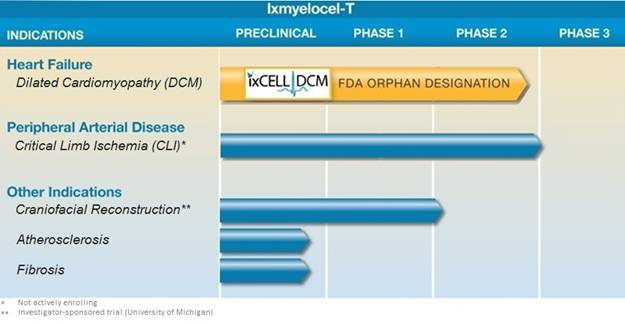
Our lead ixmyelocel-T clinical development program is for the treatment of advanced heart failure due to ischemic DCM. Ixmyelocel-T has been granted a U.S. Orphan Drug designation by the U.S. Food and Drug Administration (FDA) for the treatment of DCM, which we believe provides an efficient and cost-effective path to approval for ixmyelocel-T in this heart failure indication. We are currently enrolling our phase 2b ixCELL-DCM study, which is a randomized, double-blind, placebo-controlled clinical trial for patients with advanced heart failure due to ischemic DCM. The study is designed to enroll 108 patients at approximately 35 sites across the United States and Canada. We also have ongoing ixmyelocel-T clinical programs for the treatment of CLI and craniofacial reconstruction, as well as preclinical research and development programs for the treatment of cardiovascular diseases.
Our Therapy
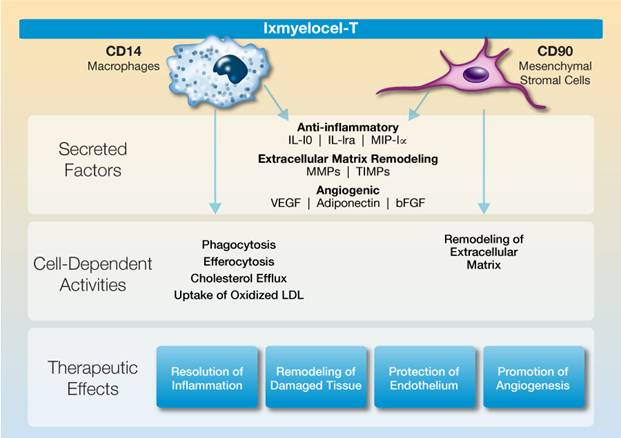
Ixmyelocel-T is a unique multicellular product derived from an adult patient’s own bone marrow. Our proprietary cell manufacturing process significantly expands the mesenchymal stromal cells (MSCs) and M2-like anti-inflammatory macrophages in the patient’s bone marrow mononuclear cells while retaining many of the hematopoietic cells. These cell types are known to regulate the immune response and play a key role in tissue repair and regeneration by resolving pathologic inflammation, promoting angiogenesis, and remodeling ischemic tissue. Ixmyelocel-T is the only multicellular product known to have expanded cell populations of both MSCs and M-2 like anti-inflammatory macrophages.
MSCs and M2-like macrophages have a wide range of biological activities that promote repair and regeneration of damaged tissues through the paracrine effects of their secreted factors, as well as their direct cell activities. These cells produce high levels of potent anti-inflammatory and angiogenic factors, as well as factors involved in extracellular matrix remodeling. These cells also have direct activities such as phagocytosis of cellular debris and apoptotic cells, which control the inflammatory response, uptake of LDL and removal of cholesterol, and
remodeling of extracellular matrix. We believe that, together, these paracrine effects and direct cell activities are responsible for ixmyelocel-T’s demonstrated therapeutic effects of resolving inflammation, promoting angiogenesis, and remodeling and repairing damaged tissue.
The following illustration summarizes the multiple biological activities of ixmyelocel-T that promote repair and regeneration of ischemic tissue:
Ixmyelocel-T has several features that we believe are primarily responsible for success in treating adult patients with severe ischemic cardiovascular diseases such as DCM and critical limb ischemia:
Patient-specific (autologous) — we start with the patient’s own cells, which are accepted by the patient’s immune system, allowing the cells to integrate into existing functional tissues. We believe that this characteristic of our therapy eliminates both the risk of rejection and the need to use immunosuppressive therapy pre- or post-therapy. Our data also suggests that ixmyelocel-T provides the potential for long-term engraftment and tissue repair.
Expanded — we begin with a small amount of bone marrow from the patient (up to 60 ml) and significantly expand the number of certain cell types, primarily MSCs and M2-like anti-inflammatory macrophages, to a substantially greater number than are present in the patient’s own bone marrow (up to 200 times the number of certain cell types compared with the starting bone marrow).
Multicellular — we believe the multiple cell types in ixmyelocel-T, which are normally found in bone marrow but in smaller quantities, possess the key functions required for reducing chronic inflammation and promoting angiogenesis and tissue repair. By reducing inflammation, we believe that ixmyelocel-T provides the ideal conditions to allow for the growth of new tissue and blood vessels.
Minimally invasive — our procedure for collecting bone marrow can be performed in an out-patient setting and takes approximately 15 minutes. Administration of ixmyelocel-T for the treatment of DCM is performed in the cardiac catheterization laboratory using a cell injection catheter system in a one-time procedure. For diseases such as CLI, administration of ixmyelocel-T is performed with a syringe in an outpatient setting in a one-time, approximately 20 minute procedure.
Safe — bone marrow and bone marrow-derived therapies have been used safely and efficaciously in medicine for over three decades. Ixmyelocel-T leverages this body of scientific study and medical experience, and appears well tolerated in over 200 patients treated to date.
Our Technology Platform
Our patient-specific multicellular therapies are manufactured using the Company’s proprietary Aastrom Repicell System (ARS) cell manufacturing system. Our manufacturing process is conducted in a highly-automated, fully-closed and rigorously controlled system. Our system is highly scalable and reproducible and located in a 5,000-square-foot centralized manufacturing facility in Ann Arbor, Michigan. Production is conducted under current Good Manufacturing Practices (cGMP) guidelines required by the FDA with current annual capacity to treat up to 3,000 patients.

Our Strategy
Our objective is to become the leading global biotechnology company in the development, manufacture, and commercialization of autologous multicellular therapies for the treatment of severe ischemic cardiovascular diseases. To achieve this objective, we intend to:
· Complete our phase 2b ixCELL-DCM clinical study for the treatment of advanced heart failure due to ischemic DCM and, if successful, progress ixmyelocel-T into pivotal phase 3 clinical studies for this orphan indication.
· Complete patient follow-up in the REVIVE-CLI study to evaluate safety and efficacy endpoints, and pursue opportunities through investigator-sponsored studies and strategic relationships to continue to develop ixmyelocel-T as a stand-alone and/or adjunct therapy for the treatment of critical limb ischemia.
· Conduct additional preclinical and clinical studies of ixmyelocel-T to pursue additional high-value indications for the treatment of severe ischemic cardiovascular diseases.
· Utilize our proprietary ARS cell-expansion manufacturing platform to expand our product portfolio of cell therapies for the treatment of immune/inflammatory, cardiovascular and fibrovascular diseases.
· Leverage our leading proprietary cell manufacturing platform and expertise to provide manufacturing services and capabilities to other development and commercial-stage biopharmaceutical companies.
· Prepare to commercialize ixmyelocel-T through continued development of our internal commercialization capabilities and/or strategic partnerships for North America, Europe and Asia.
Our Clinical Development Programs
Our clinical development programs are focused on addressing areas of high unmet medical need in severe, chronic ischemic cardiovascular diseases. We have completed our Phase 1/2 clinical trials in DCM and we are currently enrolling our phase 2b ixCELL-DCM study, which is a randomized, double-blind, placebo-controlled clinical trial for patients with advanced heart failure due to ischemic DCM. Ixmyelocel-T has been granted a U.S. Orphan Drug designation by the FDA for the treatment of DCM. We also have ongoing ixmyelocel-T clinical programs for the treatment of CLI and craniofacial reconstruction.
The following summarizes the status of our clinical programs:
Heart Failure Due to Dilated Cardiomyopathy
Heart failure represents a significant unmet medical need and a growing public health problem. The American Heart Association reports that there are approximately 6 million patients currently suffering from heart failure in the United States and an estimated 650,000 new cases in the U.S. each year. Current medical costs to treat these patients exceed $25 billion and this is expected to more than triple to nearly $80 billion by 2030 as a result of a growing patient population and the high cost of the limited treatment alternatives for advanced heart failure patients, as described below.
DCM is the third leading cause of heart failure and the leading cause of heart transplantation in the United States. DCM is a disease characterized by weakening of the heart muscle, thinning of the heart walls, enlargement of the heart chambers, and the inability to sufficiently pump blood throughout the body. Patients with DCM typically present with symptoms of congestive heart failure, including limitations in physical activity and shortness of breath. Ischemic DCM is associated with atherosclerotic cardiovascular disease and prior heart attacks and is the most common form of dilated cardiomyopathy, representing an estimated 60% of all DCM patients. Patient prognosis depends on the stage and cause of the disease, but is typically characterized by a very poor quality of life and a high mortality rate.
Current treatments for ischemic DCM patients that are refractory to further medical therapy such as prescription drugs, devices, and/or further revascularization procedures including bypass surgery and angioplasty, are limited to heart transplantation and placement of left ventricular assist devices (LVADs). There are less than 2,500 heart transplantations in the United States each year. Many refractory DCM patients are not eligible for heart transplantation and transplants are extremely expensive at an estimated cost of over $1 million. LVADs are also expensive at an estimated cost of over $175,000 and have a mortality rate of 50% at two years.
A majority of advanced heart failure patients that are refractory to medical therapy have DCM, and we believe that the refractory ischemic DCM market represents a substantial market opportunity for ixmyelocel-T. These refractory ischemic DCM patients are currently the target patient population for our clinical development of ixmyelocel-T, with approximately 175,000 patients in the United States alone. Ixmyelocel-T has been granted a U.S. Orphan Drug designation by the FDA for the treatment of DCM, which we believe provides an efficient and cost-effective path to approval for ixmyelocel-T in this heart failure indication.
We have conducted two phase 2a multicenter, randomized, open-label clinical studies in patients with ischemic DCM and nonischemic DCM investigating surgical (IMPACT-DCM) and catheter-based (Catheter-DCM) delivery of ixmyelocel-T. We reported 12-month data for the surgical IMPACT-DCM study at the Heart Failure Society of America meeting in September 2011 and final 12-month results from the Catheter-DCM study at the Society for Cardiovascular Angiography and Interventions (SCAI) 2012 Scientific Sessions. Results from these studies demonstrated that ixmyelocel-T was well-tolerated in patients with DCM. In the Catheter-DCM study and post-surgery in the IMPACT-DCM study, the incidence of adverse events was comparable between the ixmyelocel-T groups and the control groups. Cardiac failure was reported more frequently in the control group relative to ixmyelocel-T in both studies.
While these exploratory phase 2a studies were not powered for determining differences in efficacy between treatment groups, there were consistent trends of clinically meaningful improvement in clinical endpoints observed in the ischemic DCM groups in both studies. In the combined ischemic DCM groups across both studies, major adverse cardiovascular events (MACE) were experienced by a lower percentage of ixmyelocel T-treated patients compared to control patients, representing a 45% reduction in the number of patients having a MACE event. Likewise, patients in the combined ischemic DCM groups that were treated with ixmyelocel-T had a lower average number of MACE events at 12 months compared to those in the control group, representing a 61% reduction in the average number of MACE events per patient. MACE is the recommended endpoint (mortality and cardiovascular hospitalizations) in Phase 3 heart failure studies as stated in the FDA 2009 Somatic Cell Therapy for Cardiac Diseases Draft Guidance. Consistent positive trends also were observed in several secondary efficacy measures in the ischemic DCM groups. The majority of ixmyelocel T-treated patients, but not placebo-treated patients (both IDCM and NIDCM), had improvement in NYHA Class over the 12 months following treatment. Improvement in NYHA Class is considered clinically meaningful. There was also a trend toward improved function, with a higher percentage of ixmyelocel T-treated IDCM patients showing an improvement in ejection fraction and increased 6 minute walk performance compared to the IDCM control patients.
We are currently enrolling patients in the Phase 2b ixCELL-DCM clinical study, which is a multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of ixmyelocel-T in patients with advanced heart failure due to ischemic DCM. The study is designed to enroll 108 patients at approximately 30 sites in the U.S. and Canada. Patients will be followed for 12 months for the primary efficacy endpoint of MACE events, defined as all-cause deaths, all-cause hospitalizations, and unplanned outpatient or emergency department visits for IV treatment of acute worsening heart failure. Secondary endpoints include clinical, functional, structural, symptomatic, quality of life, and biomarker measures at 3, 6 and 9 months. Patients will be followed for an additional 12 months for safety. We expect to complete enrollment of the ixCELL-DCM study by the end of the first quarter of 2014, and have top-line efficacy results in the second quarter of 2015.
Critical Limb Ischemia
CLI is the most serious and advanced stage of PAD resulting from chronic inflammation and lipid accumulation. PAD is a chronic atherosclerotic disease that progressively restricts blood flow in the limbs and can lead to serious medical complications. This disease is often associated with other serious clinical conditions including hypertension, cardiovascular disease, dyslipidemia, diabetes, obesity and stroke. CLI is used to describe
patients with chronic ischemia-induced pain (even at rest) or tissue loss (ulcers or gangrene) in the limbs, often leading to amputation and death. Many CLI patients are considered unsuitable for revascularization as they have exhausted all other reasonable treatment options and will likely require amputation. The one-year and four-year mortality rates for CLI patients that are unsuitable for revascularization that progress to amputation are approximately 25% and 70%, respectively. Currently, there are an estimated 250,000 CLI patients that are unsuitable for revascularization in the United States.
Ixmyelocel-T has shown significant promise in the treatment of CLI patients with existing tissue loss that are unsuitable for revascularization. Our U.S. Phase 2b RESTORE-CLI program was a multi-center, randomized, double-blind, placebo-controlled clinical trial designed to evaluate the safety and efficacy of ixmyelocel-T in the treatment of patients with CLI that are unsuitable for revascularization. It was the largest multi-center, randomized, double-blind, placebo-controlled cellular therapy study ever conducted in CLI patients. We completed enrollment of this trial in February 2010 with a total of 86 patients at 18 sites across the United States.
Final results of the Phase 2b RESTORE-CLI clinical trial were presented at the American Heart Association Scientific Sessions in November 2011 and published in the peer-reviewed journal Molecular Therapy in April 2012. Patients in the treatment arm showed a 62% reduction in risk relative to placebo in the primary efficacy endpoint of time to first occurrence of treatment failure (p=0.0032). While the study was not powered to show statistical significance in the secondary endpoint of amputation free survival, results from a subgroup of 45 patients with wounds at baseline (the approximate profile of the Phase 3 patient population) showed a 61% reduction in risk (21% ixmyelocel-T treated versus 44% control event rate; p=0.0802). The study also met the primary safety endpoint with no meaningful differences between the treated and control groups.
We initiated the Phase 3 REVIVE-CLI clinical study, a multicenter, randomized, double-blind, placebo controlled study to evaluate the efficacy and safety of ixmyelocel-T in patients with CLI, in 2012. We had previously received Fast Track Designation from the FDA for use of ixmyelocel-T for the treatment of CLI and reached agreement with the FDA on a Special Protocol Assessment. Patients were randomized 1:1 and were to be followed for 12 months for the primary efficacy endpoint of amputation-free survival. On March 27, 2013 we announced that we were stopping enrollment in the study for strategic business reasons. This study has been amended and is ongoing for the 41 patients that are enrolled in the study, and we plan to continue following these patients for 12 months to evaluate safety and certain efficacy measures. We expect to have results from this study in the second quarter of 2014.
Risks Associated with Our Business
An investment in our common stock involves a high degree of risk. You should carefully consider the risks summarized below. The risks are discussed more fully in the “Risk Factors” section of this prospectus beginning on page 10 of this prospectus. These risks include, but are not limited to, the following:
· We currently depend heavily on the success of ixmyelocel-T, our sole product candidate. Any failure to commercialize ixmyelocel-T, or significant delays in doing so, will have a material adverse effect on our business, operating results and financial condition.
· Our product development programs are based on novel technologies and are inherently risky.
· We may not be able to raise the required capital to conduct our operations and develop and commercialize our products.
· If we do not keep pace with our competitors and with technological and market changes, our products will become obsolete and our business may suffer.
· If our patents and proprietary rights do not provide substantial protection, then our business and competitive position will suffer.
· Our past losses and expected future losses cast doubt on our ability to continue as a going concern and operate profitably.
Company Information
We were incorporated under the laws of the State of Michigan on March 24, 1989. Our principal executive offices are located at 24 Frank Lloyd Wright Drive, Lobby K, Ann Arbor, Michigan 48105 and our telephone number is (800) 556-0311. Our website address is www.aastrom.com. The reference to our website is intended to be an inactive textual reference and, except for the documents incorporated by reference as noted above, the information on, or accessible through, our website is not part of this prospectus.
|
Issuer |
|
Aastrom Biosciences, Inc. |
|
|
|
|
|
Securities offered by us |
|
of shares of common stock and warrants to purchase up to an aggregate of shares of common stock ( shares and warrants if the underwriters exercise their over-allotment option in full). |
|
|
|
|
|
Common stock outstanding before this offering |
|
45,664,079 shares of common stock. |
|
Common stock to be outstanding after this offering |
|
shares ( if the warrants are exercised in full). If the underwriters’ over-allotment option is exercised in full, the total number of shares of common stock outstanding immediately after this offering would be ( if the warrants are exercised in full). |
|
|
|
|
|
Description of warrants |
|
The warrants will have a per share exercise price equal to $ [125% of the public offering price of common stock]. The warrants are exercisable immediately and expire five years from the date of issuance. |
|
Use of proceeds |
|
We expect to use the net proceeds from this offering to fund the development of ixmyelocel-T through our ixCELL-DCM Phase 2b clinical trial, fund development costs associated with preclinical studies and to further develop our manufacturing platform, as well as for working capital and general corporate purposes, including funding the costs of operating as a public company. See "Use of Proceeds" for a more complete description of our intended use of the net proceeds from this offering. |
|
Risk Factors |
|
You should carefully read “Risk Factors” in this prospectus for a discussion of factors that you should consider before deciding to invest in our common stock. |
|
|
|
|
|
NASDAQ Capital Market Symbol |
|
ASTM |
The number of shares of our common stock that will be outstanding immediately after this offering is based on 45,664,079 shares of common stock outstanding as of June 30, 2013 and excludes:
· 7,253,866 shares of common stock issuable upon the exercise of stock options outstanding as of June 30, 2013 at a weighted-average exercise price of $2.05 per share;
· 4,834,078 shares of common stock issuable upon the exercise of warrants outstanding as of June 30, 2013 at exercise prices of $2.44 per share (January 2010 - 4,525,978 shares) and $1.25 per share (December 2010 - 308,100 shares) in each case, before any adjustment to this offering, which warrants are exercisable to purchase common stock; and
· 12,308,000 shares of common stock issuable upon the conversion of preferred stock outstanding as of June 30, 2013.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below and in the documents incorporated by reference in this prospectus and any prospectus supplement, as well as other information we include or incorporate by reference into this prospectus and any applicable prospectus supplement, before making an investment decision. Our business, financial condition or results of operations could be materially adversely affected by the materialization of any of these risks. The trading price of our securities could decline due to the materialization of any of these risks, and you may lose all or part of your investment. This prospectus and the documents incorporated herein by reference also contain forward-looking statements that involve risks and uncertainties. Actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks described below and in the documents incorporated herein by reference, including (i) our Annual Report on Form 10-K for the year ended December 31, 2012 and (ii) other documents we file with the SEC that are deemed incorporated by reference into this prospectus.
Risks Related to Our Business
Our past losses and expected future losses cast doubt on our ability to continue as a going concern and operate profitably.
As of June 30, 2013, we had $4,494,000 of cash and cash equivalents. This is not sufficient to sustain our operations for one year. In light of our financial position, we are evaluating strategic and financial opportunities in the short-term in order to maintain adequate liquidity through December 31, 2013 and beyond. Additionally, we could sell shares through an At-the-Market Sales Agreement (ATM) in order to raise additional capital, though there are certain factors, such as volume of trading in our common stock, our stock price and the ability to terminate the agreement with notice, which could limit the amount we could raise in a short period of time. On a longer term basis, we will need to raise additional funds in order to complete product development programs and complete clinical trials needed to market and commercialize our products. We cannot be certain that such funding will be available on favorable terms, if at all. Some of the factors that will impact our ability to raise additional capital and our overall success include: the rate and degree of progress for our product development, the rate of regulatory approval to proceed with clinical trial programs, the level of success achieved in clinical trials, the requirements for marketing authorization from regulatory bodies in the United States and other countries, the liquidity and market volatility of our equity securities, regulatory and manufacturing requirements and uncertainties, technological developments by competitors, and other factors. If we cannot raise such funds, we will not be able to develop or enhance products, take advantage of future opportunities, or respond to competitive pressures or unanticipated requirements, which would have a material adverse impact on our business, financial condition and results of operations. As a result of the need to raise additional capital and a net capital deficiency, there is uncertainty regarding our ability to maintain liquidity sufficient to operate our business effectively over at least the next twelve months, which raises substantial doubt as to our ability to continue as a going concern. The consolidated financial statements incorporated by reference in this prospectus do not include any adjustments that might result from the outcome of this uncertainty.
We were incorporated in 1989 and have experienced substantial operating losses since inception. As of June 30, 2013, we had incurred a cumulative net loss attributable to common shareholders totaling approximately $287,011,000 and we have continued to incur losses since that date. These losses have resulted principally from costs incurred in the research and development (including clinical trials) of our cell culture technologies and our cell manufacturing system, general and administrative expenses, and the prosecution of patent applications. We expect to continue to incur significant operating losses over the next several years and at least until, and probably after, product sales increase, primarily owing to our research and development programs, including preclinical studies and clinical trials, and the establishment of marketing and distribution capabilities necessary to support commercialization efforts for our products. We cannot predict with any certainty the amount of future losses. Our ability to achieve profitability will depend, among other things, on successfully completing the development of our product candidates, timely initiation and completion of clinical trials, obtaining regulatory approvals, establishing manufacturing, sales and marketing arrangements with third parties, maintaining supplies of key manufacturing components, acquisition and development of complementary activities and raising sufficient cash to fund our operating activities. Therefore, we may not be able to achieve or sustain profitability.
We may not be able to raise the required capital to conduct our operations and develop and commercialize our products.
We will require substantial additional capital resources in order to conduct our operations, complete our product development programs, complete our clinical trials needed to market our products (including a Phase 2b clinical trial for DCM), and commercialize these products and cell manufacturing facilities. In order to grow and expand our business, to introduce our new product candidates into the marketplace and to acquire or develop complementary business activities, we will need to raise a significant amount of additional funds. We will also need significant additional funds or a collaborative partner, or both, to finance the research and development activities of our cell product candidates for additional indications. Accordingly, we are continuing to pursue additional sources of financing.
Our future capital requirements will depend upon many factors, including:
|
· |
continued scientific progress in our research, clinical and development programs; |
|
|
|
|
· |
costs and timing of conducting clinical trials and seeking regulatory approvals; |
|
|
|
|
· |
competing technological and market developments; |
|
|
|
|
· |
avoiding infringement and misappropriation of third-party intellectual property; |
|
|
|
|
· |
obtaining valid and enforceable patents that give us a competitive advantage; |
|
|
|
|
· |
our ability to establish additional collaborative relationships; |
|
|
|
|
· |
our ability to effectively launch a commercial product; |
|
|
|
|
· |
the effect of commercialization activities and facility expansions, if and as required; and |
|
|
|
|
· |
complementary business acquisition or development opportunities. |
We entered into an ATM on June 16, 2011, which allows us to raise approximately $20,000,000 through sales of our common stock from time to time. However, there are certain factors, such as volume of trading in our common stock, our stock price and the ability to terminate the agreement with notice, which limit the amount that can be raised in a short period of time through the ATM. Additionally, we have temporarily suspended the ATM in connection with this offering. Regardless of the usage of the ATM, we will need to raise additional capital in order to fund the clinical trials of ixmyelocel-T for DCM, complete our product development programs, complete clinical trials needed to market our products and commercialize these products.
We will need to raise additional funds in order to complete our product development programs, complete clinical trials needed to market our products (including clinical trials for our DCM program), and commercialize these products. Because of our long-term funding requirements, we may try to access the public or private equity markets if conditions are favorable to complete a financing, even if we do not have an immediate need for additional capital at that time, or whenever we require additional operating capital. In addition, we may seek collaborative relationships, incur debt and access other available funding sources. This additional funding may not be available to us on reasonable terms, or at all. Some of the factors that will impact our ability to raise additional capital and our overall success include:
|
· |
the rate and degree of progress for our product development; |
|
|
|
|
· |
the rate of regulatory approval to proceed with clinical trial programs; |
|
|
|
|
· |
the level of success achieved in clinical trials; |
|
· |
the requirements for marketing authorization from regulatory bodies in the United States and other |
|
|
countries; |
|
|
|
|
· |
the liquidity and market volatility of our equity securities; and |
|
|
|
|
· |
regulatory and manufacturing requirements and uncertainties, and technological developments by competitors. |
If adequate funds are not available in the future, we may not be able to develop or enhance our products, take advantage of future opportunities, or respond to competitive pressures or unanticipated requirements and we may be required to delay or terminate research and development programs, curtail capital expenditures, and reduce business development and other operating activities. Should the financing we require to sustain our working capital needs be unavailable or prohibitively expensive when we require it, the consequences could have a material adverse effect on our business, operating results, financial condition and prospects.
Failure to obtain and maintain required regulatory approvals would severely limit our ability to sell our products.
We must obtain the approval of the FDA before commercial sales of our cell product candidates may commence in the United States, which we believe will ultimately be the largest market for our products. We will also be required to obtain additional approvals from various foreign regulatory authorities to initiate sales activities of cell products in those jurisdictions. If we cannot demonstrate the safety, purity and potency of our product candidates, including our cell product candidates, produced in our production system, the FDA or other regulatory authorities could delay or withhold regulatory approval of our product candidates.
Finally, even if we obtain regulatory approval of a product, that approval may be subject to limitations on the indicated uses for which it may be marketed. Even after granting regulatory approval, the FDA and regulatory agencies in other countries continue to review and inspect marketed products, manufacturers and manufacturing facilities, which may create additional regulatory burdens. Later discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions on the product or manufacturer, including a withdrawal of the product from the market. Further, regulatory agencies may establish additional regulations that could prevent or delay regulatory approval of our products.
We currently depend heavily on the success of ixmyelocel-T, our sole product candidate. Any failure to commercialize ixmyelocel-T, or significant delays in doing so, will have a material adverse effect on our business, operating results and financial condition.
We have invested a significant portion of our efforts and financial resources in the development of ixmyelocel-T. Our ability to generate future product revenue depends heavily on the successful development and commercialization of ixmyelocel-T. The successful commercialization of ixmyelocel-T will depend on several factors, including the following:
|
· |
obtaining marketing approvals from the FDA and other foreign regulatory authorities; |
|
|
|
|
· |
successful enrollment of patients in our ongoing clinical studies of ixmyelocel-T; |
|
|
|
|
· |
successful completion of our ongoing clinical studies of ixmyelocel-T; |
|
|
|
|
· |
the successful audit of our facilities by additional regulatory authorities; |
|
|
|
|
· |
maintaining the cGMP and cGTP compliance of our manufacturing facility; |
|
|
|
|
· |
maintaining current manufacturing arrangements with third parties and establishing new manufacturing arrangements; |
|
|
|
|
· |
our development of a successful sales and marketing organization for ixmyelocel-T; |
|
· |
an acceptable safety and efficacy profile of our product candidates following approval; |
|
|
|
|
· |
the availability of reimbursement to patients from healthcare payers for our drug products, if approved; and |
|
|
|
|
· |
other risks described in this “Risk Factors” section. |
Any failure to commercialize ixmyelocel-T or significant delays in doing so will have a material adverse effect on our business, results of operations and financial condition.
Our sole product candidate, ixmyelocel-T, is still in clinical development. If we do not successfully continue or complete the clinical development of ixmyelocel-T, our likelihood of success as a company and our ability to finance our operations will be substantially harmed.
Our near-term prospects substantially depend upon our ability to successfully continue and complete clinical trials of our lead product candidate, ixmyelocel-T, and to demonstrate its safety and efficacy, as well as its superiority over existing therapies and standards of care, if any. We are currently enrolling patients with ischemic DCM for the ixCELL-DCM trial, a Phase 2b clinical trial, and have recently treated the first patients in the trial. All of our other potential product candidates are in preclinical research or early clinical development. Our ability to finance our company and to generate revenues will depend heavily on our ability to obtain favorable results in the ongoing and planned clinical trials of ixmyelocel-T, including the ongoing ixCELL-DCM Phase 2b clinical trial, and to successfully develop and commercialize ixmyelocel-T. Ixmyelocel-T could be unsuccessful if it:
|
· |
does not demonstrate acceptable safety and efficacy in clinical trials, or otherwise does not meet applicable regulatory standards for approval; |
|
|
|
|
· |
does not offer sufficient, clinically meaningful therapeutic or other improvements over existing or future drugs used to treat the DCM indications for which it is being tested; |
|
|
|
|
· |
is not capable of being produced in commercial quantities at acceptable costs; or |
|
|
|
|
· |
is not accepted as safe, efficacious, cost-effective, less costly and preferable to current therapies in the medical community and by third-party payers. |
If we are not successful in developing and commercializing ixmyelocel-T or are significantly delayed in doing so, our financial condition and future prospects may be adversely affected and we may experience difficulties in raising the substantial additional capital required to fund our business.
Our product development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of our therapeutics creates significant challenges in regard to product development and optimization, manufacturing, government regulation, third-party reimbursement and market acceptance. For example, if regulatory agencies have limited experience in approving cellular therapies for commercialization, the development and commercialization pathway for our therapies may be subject to increased uncertainty, as compared to the pathway for new conventional drugs.
Any changes in the governmental regulatory classifications of our products could prevent, limit or delay our ability to market or develop our products.
The FDA establishes regulatory requirements based on the classification of a product. Because our product development programs are designed to satisfy the standards applicable to biological licensure for our cellular products, any change in the regulatory classification or designation would affect our ability to obtain FDA approval of our products. Each of these cell products is, under current regulations, regulated as a biologic, which requires a BLA.
Our inability to complete our product development activities successfully would severely limit our ability to operate or finance operations.
In order to commercialize our cell product candidates in the United States, we must complete substantial clinical trials and obtain sufficient safety, purity and potency results to support required registration approval and market acceptance of our cell product candidates. We may not be able to successfully complete the development of our product candidates, or successfully market our technologies or product candidates. We, and any of our potential collaborators, may encounter problems and delays relating to research and development, regulatory approval and intellectual property rights of our technologies and product candidates. Our research and development programs may not be successful, and our cell culture technologies and product candidates may not facilitate the production of cells outside the human body with the expected results. Our technologies and cell product candidates may not prove to be safe and efficacious in clinical trials, and we may not obtain the requisite regulatory approvals for our technologies or product candidates and the cells produced in such products. If any of these events occur, we may not have adequate resources to continue operations for the period required to resolve any issues delaying commercialization and we may not be able to raise capital to finance our continued operation during the period required for resolution of any such issues.
We must successfully complete our clinical trials to be able to market certain of our products.
To be able to market therapeutic cell products in the United States, we must demonstrate, through extensive preclinical studies and clinical trials, the safety and efficacy of our processes and product candidates. If our clinical trials are not successful, our products may not be marketable. The results of early stage clinical trials do not ensure success in later clinical trials, and interim results are not necessarily predictive of final results.
Our ability to complete our clinical trials in a timely manner depends on many factors, including the rate of patient enrollment. Patient enrollment can vary with the size of the patient population, the proximity of suitable patients to clinical sites, perceptions of the utility of cell therapy for the treatment of certain diseases, and the eligibility criteria for the study. For example, patients enrolling in our studies need to provide an adequate amount of bone marrow to process and expand for injection and some patients may not be able to provide sufficient starting material despite our study inclusion and exclusion criteria designed to prevent this. Bone marrow is an inherently variable starting material. We have experienced delays in patient accrual in our previous clinical trials. On March 27, 2013, we announced that we were stopping enrollment in the Phase 3 REVIVE clinical trial due to the slow patient accrual rate for the study and to optimize the use of our financial resources. If we experience similar delays in patient enrollment for other clinical trials, we could experience increased costs and delays associated with these trials, which would impair our product development programs and our ability to market our products.
Furthermore, the FDA monitors the progress of clinical trials and it may suspend or terminate clinical trials at any time due to patient safety or other considerations.
Our research programs are currently directed at improving product functionality for certain clinical indications, improving product shelf life, and decreasing the cost of manufacturing our products. These production process changes may alter the functionality of our cells and require various additional levels of experimental and clinical testing and evaluation. Any such testing could lengthen the time before these products would be commercially available.
Even if successful clinical results are reported for a product from a completed clinical trial, this does not mean that the results will be sustained over time, or will be sufficient for a marketable or regulatory approvable product.
We may rely on third parties to conduct some of our clinical trials, and their failure to perform their obligations in a timely or competent manner may delay development and commercialization of our product candidates.
We may use clinical research organizations (CROs) to assist in conduct of our clinical trials. There are numerous alternative sources to provide these services. However, we may face delays outside of our control if these parties do not perform their obligations in a timely or competent fashion, or if we are forced to change service providers. Any third party that we hire to conduct clinical trials may also provide services to our competitors, which could compromise the performance of their obligations to us. If we experience significant delays in the progress of our clinical trials, the commercial prospects for product candidates could be harmed and our ability to generate product
revenue would be delayed or prevented. In addition, we and any provider that we retain will be subject to Good Clinical Practice, (GCP) requirements. If GCP and other regulatory requirements are not adhered to by us or our third-party providers, the development and commercialization of our product candidates could be delayed.
Any failure of such CRO to successfully accomplish clinical trial monitoring, data collection, safety monitoring and data management and the other services it provides for us in a timely manner and in compliance with regulatory requirements could have a material adverse effect on our ability to complete clinical development of our products and obtain regulatory approval. Problems with the timeliness or quality of the work of a CRO may lead us to seek to terminate the relationship and use an alternate service provider. However, making such changes may be costly and may delay our trials, and contractual restrictions may make such a change difficult or impossible. Additionally, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
Failure of third parties, including Vention Medical, to manufacture or supply certain components, equipment, disposable devices and other materials used in our cell manufacturing process would impair our cell product development.
We rely on third parties, including Vention Medical (Vention), to manufacture and/or supply certain of our devices/manufacturing equipment and to manufacture and/or supply certain components, equipment, disposable devices and other materials used in our cell manufacturing process to develop our cell products. Vention is our sole supplier of cell cassettes for which it would be difficult to obtain alternate sources of supply on a short-term basis. If any of our manufacturers or suppliers fails to perform its respective obligations, or if our supply of certain components, equipment, disposable devices and other materials is limited or interrupted, it could impair our ability to manufacture our products, which would delay our ability to conduct our clinical trials or market our product candidates on a timely and cost-competitive basis, if at all.
In addition, we may not be able to continue our present arrangements with our suppliers, supplement existing relationships, establish new relationships or be able to identify and obtain the ancillary materials that are necessary to develop our product candidates in the future. Our dependence upon third parties for the supply and manufacture of these items could adversely affect our ability to develop and deliver commercially feasible products on a timely and competitive basis.
Manufacturing of our cell products in centralized facilities may increase the risk that we will not have adequate quantities of our cell products for clinical programs.
We are subject to regulatory compliance and quality assurance requirements at our production site in Ann Arbor, Michigan. This site could be subject to ongoing, periodic, unannounced inspection by regulatory agencies to ensure strict compliance with GMP regulations and other governmental regulations. We do not have redundant cell manufacturing sites. In the event our cell production facility is damaged or destroyed or is subject to regulatory restrictions, our clinical trial programs and other business prospects would be adversely affected.
Even if we obtain regulatory approvals to sell our products, lack of commercial acceptance could impair our business.
We will be seeking to obtain regulatory approvals to market our cell products for tissue repair treatments. Even if we obtain all required regulatory approvals, we cannot be certain that our products and processes will be accepted in the marketplace at a level that would allow us to operate profitably. Our products may be unable to achieve commercial acceptance for a number of reasons, such as the availability of alternatives that are less expensive, more effective, or easier to use; the perception of a low cost-benefit ratio for the product amongst physicians and hospitals; or an inadequate level of product support from ourselves or a commercial partner. Our technologies or product candidates may not be employed in all potential applications being investigated, and any reduction in applications would limit the market acceptance of our technologies and product candidates, and our potential revenues.
The market for our products will be heavily dependent on third-party reimbursement policies.
Our ability to successfully commercialize our product candidates will depend on the extent to which government
healthcare programs, such as Medicare and Medicaid, as well as private health insurers, health maintenance organizations and other third-party payers will pay for our products and related treatments.
Reimbursement by third-party payers depends on a number of factors, including the payer’s determination that use of the product is safe and effective, not experimental or investigational, medically necessary, appropriate for the specific patient and cost-effective. Reimbursement in the United States or foreign countries may not be available or maintained for any of our product candidates. If we do not obtain approvals for adequate third-party reimbursements, we may not be able to establish or maintain price levels sufficient to realize an appropriate return on our investment in product development. Any limits on reimbursement from third-party payers may reduce the demand for, or negatively affect the price of, our products. For example, in the past, published studies suggested that stem cell transplantation for breast cancer, which constituted a significant portion of the overall stem cell therapy market at the time, may have limited clinical benefit. The lack of reimbursement for these procedures by insurance payers has negatively affected the market for our products in this indication in the past.
Managing and reducing health care costs has been a general concern of federal and state governments in the United States and of foreign governments. In addition, third-party payers are increasingly challenging the price and cost-effectiveness of medical products and services, and many limit reimbursement for newly approved health care products. In particular, third-party payers may limit the indications for which they will reimburse patients who use any products that we may develop. Cost control initiatives could decrease the price for products that we may develop, which would result in lower product revenues to us.
Use of animal-derived materials could harm our product development and commercialization efforts.
Some of the manufacturing materials and/or components that we use in, and which are critical to, implementation of our technology involve the use of animal-derived products, including fetal bovine serum. Suppliers or regulatory changes may limit or restrict the availability of such materials for clinical and commercial use. We currently purchase all of our fetal bovine sera from protected herds in Australia and New Zealand. These sources are considered to be the safest and raise the least amount of concern from the global regulatory agencies. If, for example, the so-called “mad cow disease” occurs in New Zealand or in Australia, it may lead to a restricted supply of the serum currently required for our product manufacturing processes. Any restrictions on these materials would impose a potential competitive disadvantage for our products or prevent our ability to manufacture our cell products. The FDA has issued draft regulations for controls over bovine materials. These proposed regulations do not appear to affect our ability to purchase the manufacturing materials we currently use. However, the FDA may issue final regulations that could affect our operations. Our inability to develop or obtain alternative compounds would harm our product development and commercialization efforts. There are certain limitations in the supply of certain animal-derived materials, which may lead to delays in our ability to complete clinical trials or eventually to meet the anticipated market demand for our cell products.
Given our limited internal manufacturing, sales, marketing and distribution capabilities, we need to develop increased internal capability or collaborative relationships to manufacture, sell, market and distribute our products.
We have only limited internal manufacturing, sales, marketing and distribution capabilities. As market needs develop, we intend to establish and operate commercial-scale manufacturing facilities, which will need to comply with all applicable regulatory requirements. We will also need to develop new configurations of our cell manufacturing system for these facilities to enable processes and cost efficiencies associated with large-scale manufacturing. Establishing these facilities will require significant capital and expertise. We may need to make such expenditures when there are significant uncertainties as to the market opportunity. Any delay in establishing, or difficulties in operating, these facilities will limit our ability to meet the anticipated market demand for our cell products. We intend to get assistance to market some of our future cell products through collaborative relationships with companies with established sales, marketing and distribution capabilities. Our inability to develop and maintain those relationships would limit our ability to market, sell and distribute our products. Our inability to enter into successful, long-term relationships could require us to develop alternate arrangements at a time when we need sales, marketing or distribution capabilities to meet existing demand. We may market one or more of our products through our own sales force. Our inability to develop and retain a qualified sales force could limit our ability to market, sell and distribute our cell products.
If we do not keep pace with our competitors and with technological and market changes, our products will become obsolete and our business may suffer.
The markets for our products are very competitive, subject to rapid technological changes, and vary for different candidates and processes that directly compete with our products. Our competitors may have developed, or could in the future develop, new technologies that compete with our products or even render our products obsolete. As an example, in the past, published studies have suggested that hematopoietic stem cell therapy use for bone marrow transplantation, following marrow ablation due to chemotherapy, may have limited clinical benefit in the treatment of breast cancer, which was a significant portion of the overall hematopoietic stem cell transplant market. This resulted in the practical elimination of this market for our cell-based product for this application.
Our cell manufacturing system is designed to improve and automate the processes for producing cells used in therapeutic procedures. Even if we are able to demonstrate improved or equivalent results, the cost or process of treatment and other factors may cause researchers and practitioners to not use our products and we could suffer a competitive disadvantage. Finally, to the extent that others develop new technologies that address the targeted application for our products, our business will suffer.
The current credit and financial market conditions may exacerbate certain risks affecting our business.
We rely upon third parties for certain aspects of our business, including collaboration partners, wholesale distributors, contract clinical trial providers, contract manufacturers and third-party suppliers. Because of the recent tightening of global credit and the volatility in the financial markets, there may be a delay or disruption in the performance or satisfaction of commitments to us by these third parties, which could adversely affect our business.
If we cannot attract and retain key personnel, our business may suffer.
Our success depends in large part upon our ability to attract and retain highly qualified scientific and management personnel. We face competition for such personnel from other companies, research and academic institutions and other entities. Further, in an effort to conserve financial resources, we have implemented reductions in our work force on four previous occasions, most recently in the first quarter of 2013. As a result of these and other factors, we may not be successful in hiring or retaining key personnel. Our inability to replace any key employee could harm our operations.
Risks Related to Intellectual Property
If our patents and proprietary rights do not provide substantial protection, then our business and competitive position will suffer.
Our success depends in large part on our ability to develop or license intellectual property rights to protect our proprietary products and technologies. This involves complex legal, scientific, and factual questions and uncertainties. We rely upon patent, trade secret, copyright and contract laws to protect proprietary technology and trademark law to protect brand identities. However, we cannot assure you that any patent applications filed by, assigned to, or licensed to us will be granted, and that the scope of any of our issued or licensed patents will be sufficiently broad to offer meaningful protection. In addition, our issued patents or patents licensed to us could be successfully challenged, invalidated, held to be unenforceable, or circumvented so that our patent rights would not create an effective competitive barrier. We also cannot assure you that the inventors of the patents and applications that we own or license were the first to invent or the first to file on the inventions, or that a third party will not claim ownership in one of our patents or patent applications. We cannot assure you that a third party does not have or will not obtain patents that dominate the patents we own or license now or in the future. Furthermore, we rely on exclusive, world-wide licenses relating to the production of human cells granted to us by the University of Michigan. If we materially breach such agreements or otherwise fail to materially comply with such agreements, or if such agreements expire or are otherwise terminated by us, we may lose our rights under the patents held by the University of Michigan. At the latest, each of these licenses will terminate when the patent underlying the license expires, with the last to expire during the third quarter of 2014. Once the patents expire, third parties may be able to practice the inventions covered by those patents and thus compete with us.
Patent law relating to the scope of claims in the biotechnology field is evolving and our patent rights in this country and abroad are subject to this uncertainty.
We also rely on trade secrets and un-patentable know-how that we seek to protect, in part, by confidentiality agreements with our employees, consultants, suppliers and licensees. These agreements may be breached, and we might not have adequate remedies for any breach. Our competitors may also independently develop technologies substantially equivalent or superior to ours. If this were to occur, our business and competitive position would suffer.
Intellectual property litigation could harm our business.
Our success will also depend in part on our ability to develop commercially viable products without infringing the proprietary rights of others. Our cell processing system and cell compositions utilize a wide variety of technologies and we can give no assurance that we have identified or can identify all inventions and patents that may be infringed by development and manufacture of our cell compositions. Although we have not been subject to any filed infringement claims, patents could exist or could be filed which would prohibit or limit our ability to market our products or maintain our competitive position. In the event of an intellectual property dispute, we may be forced to litigate. Such litigation is typically protracted and the results are unpredictable. Intellectual property litigation would divert management’s attention from developing our products and would force us to incur substantial costs regardless of whether we are successful. An adverse outcome could subject us to significant liabilities to third parties including treble damages and the opposing party’s attorney fees, and force us to pay significant license fees and royalties or cease the development and sale of our products and processes.
We have hired and will continue to hire individuals who have experience in cell culture and cell based therapeutics and may have confidential trade secret or proprietary information of third parties. We caution these individuals not to use or reveal this third-party information, but we cannot assure you that these individuals will not use or reveal this third-party information. Thus, we could be sued for misappropriation of proprietary information and trade secrets. Such claims are expensive to defend and could divert our attention and could result in substantial damage awards and injunctions that could have a material adverse effect on our business, financial condition or results of operations.
We may need to initiate lawsuits to protect or enforce our patents or other proprietary rights, which would be expensive and, if unsuccessful, may cause us to lose some of our intellectual property rights.
To protect or enforce our patent rights, it may be necessary for us to initiate patent litigation proceedings against third parties, such as infringement suits or interference proceedings. These lawsuits would be expensive, take significant time and would divert management’s attention from other business concerns. These lawsuits could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly, and our patent applications at risk of not being issued. Further, these lawsuits may provoke the defendants to assert claims against us. The patent position of biotechnology firms is highly uncertain, involves complex legal and factual questions and recently has been the subject of much litigation. We cannot assure you that we will prevail in any of such suits or proceedings or that the damages or other remedies awarded to us, if any, will be commercially valuable.
The government maintains certain rights in technology that we develop using government grant money and we may lose the revenues from such technology if we do not commercialize and utilize the technology pursuant to established government guidelines.
Certain of our and our licensors’ research have been or are being funded in part by government grants. As a result of such funding, the United States government has established guidelines and has certain rights in the technology developed with the grant. These rights include a non-exclusive, fully paid-up, worldwide license under such inventions for any governmental purpose. In addition, the United States government has the right to require us to grant an exclusive license under any of such inventions to a third party if the United States government determines that: (i) adequate steps have not been taken to commercialize such inventions; (ii) such action is necessary to meet public health or safety needs; or (iii) such action is necessary to meet requirements for public use under federal regulations. Additionally, under the federal Bayh Dole Act, a party which acquires an exclusive license for an invention that was partially funded by a federal research grant is subject to the following government rights: (x) products using the
invention which are sold in the United States are to be manufactured substantially in the United States, unless a waiver is obtained; (y) the government may force the granting of a license to a third party who will make and sell the needed product if the licensee does not pursue reasonable commercialization of a needed product using the invention; and (z) the United States government may use the invention for its own needs. If we fail to meet these guidelines, we would lose our exclusive rights to these products, and we would lose potential revenue derived from the sale of these products.
Potential product liability claims could affect our earnings and financial condition.
We face an inherent business risk of exposure to product liability claims in the event that the manufacture and/or use of our products during clinical trials, or after commercialization, results in adverse events. As a result, we may incur significant product liability exposure, which could exceed existing insurance coverage. We may not be able to maintain adequate levels of insurance at reasonable cost and/or on reasonable terms. Excessive insurance costs or uninsured claims would increase our operating loss and adversely affect our financial condition.
Risks Related to Our Common Stock and Warrants
We may be unable to continue as a going concern in which case our securities will have little or no value.
We have incurred substantial losses since inception and have a net capital deficiency. This raises substantial doubt about our ability to continue as a going concern. In the event we are not able to continue operations you will likely suffer a complete loss of your investment in our securities.
Our common stock price has been volatile and future sales of shares of common stock could have an adverse effect on the market price of such shares.
The market price of shares of our common stock has been volatile, ranging in closing price between $0.40 and $1.41 during the six months ended June 30, 2013. The price of our common stock may continue to fluctuate in response to a number of events and factors, such as:
|
· |
clinical trial results; |
|
|
|
|
· |
the amount of our cash resources and our ability to obtain additional funding; |
|
|
|
|
· |
announcements of research activities, business developments, technological innovations or new products by us or our competitors; |
|
|
|
|
· |
entering into or terminating strategic relationships; |
|
|
|
|
· |
regulatory developments in both the United States and abroad; |
|
|
|
|
· |
disputes concerning patents or proprietary rights; |
|
|
|
|
· |
changes in our revenues or expense levels; |
|
|
|
|
· |
public concern regarding the safety, efficacy or other aspects of the products or methodologies we are developing; |
|
|
|
|
· |
news or reports from other stem cell, cell therapy or regenerative medicine companies; |
|
|
|
|
· |
reports by securities analysts; |
|
|
|
|
· |
status of the investment markets; |
|
|
|
|
· |
concerns related to management transitions; and |
· delisting from The NASDAQ Capital Market.
Any of these events may cause the price of our shares to fall, which may adversely affect our business and financing opportunities. In addition, the stock market in general and the market prices for biotechnology companies in particular have experienced significant volatility recently that often has been unrelated to the operating performance or financial conditions of such companies. These broad market and industry fluctuations may adversely affect the trading price of our common stock, regardless of our operating performance or prospects.
Our common stock may be delisted from The NASDAQ Capital Market, which could affect its market price and liquidity.
Currently, our common stock trades on The NASDAQ Capital Market. Continued listing of a security on The NASDAQ Capital Market is conditioned upon compliance with various continued listing standards, which require, among other things, that for 30 consecutive trading days the closing minimum bid price for our listed securities not be lower than $1.00 per share and for ten consecutive trading days the market value of listed securities for our common stock close at or above $35 million. We received notice from The NASDAQ Capital Market on May 9, 2013 that our common stock bid price has fallen below $1.00 per share, and on May 20, 2013 that we have not maintained a market value of at least $35 million. As a result, our common stock is in jeopardy of being delisted. The NASDAQ Capital Market has informed us that we have until November 5, 2013, to meet the minimum bid price threshold and until November 18, 2013, to meet the market value threshold to maintain the listing of our common stock on The NASDAQ Capital Market.
While we are exercising diligent efforts to maintain the listing of our common stock on The NASDAQ Capital Market, there can be no assurance that we will be able to meet these thresholds within the required timeframes. With respect to the minimum bid price, we may receive a second 180 day grace period if certain conditions are met. Additionally, The NASDAQ Capital Market rules permit us to appeal to a NASDAQ Hearings Panel. We intend to regain compliance prior to the expiration of the notice periods by exploring a number of options, such as effecting a reverse stock split on our outstanding common stock. However, even if the reverse stock split achieves the requisite increase in the market price of our common stock, we cannot assure you that we will be able to continue to comply with the minimum bid price thresholds or the other continued listing standards of The NASDAQ Capital Market. Additionally, a reverse stock split may decrease the liquidity of the shares of our common stock, and the resulting market price of our common stock following such a split may not attract new investors. Consequently, the trading liquidity of our common stock may not improve.
While we have filed an Amended and Restated Certificate of Designations, Preferences and Rights of the Company classifying and designating the Series B-1 Non-Voting Convertible Preferred Stock and the Series B-2 Voting Convertible Preferred Stock that we believe will result in a shareholders’ deficit of $3,345,000 as of June 30, 2013, we cannot assure you that we will continue to comply with the other continued listing standards of The NASDAQ Capital Market.
If we are unable to maintain or regain compliance in a timely manner and our common stock is delisted, it could be more difficult to buy or sell our common stock and obtain accurate quotations, and the price of our common stock could suffer a material decline. Delisting may also impair our ability to raise capital. Moreover, if we were delisted we would be subject to rules that impose additional sales practice requirements on broker-dealers who sell our securities. The additional burdens imposed upon broker-dealers by these requirements could discourage broker-dealers from effecting transactions in our common stock, which could severely limit the market liquidity of the common stock and your ability to sell our securities in the secondary market.
You will experience immediate and substantial dilution in the net tangible book value per share of the common stock you purchase in this offering.
Because the public offering price per share of our common stock is higher than the net tangible book value per share of our common stock, you will suffer dilution in the net tangible book value of the common stock you purchase in this offering. Based on an assumed public offering price of $0.57 per share, if you purchase shares of common stock in this offering, you will suffer immediate and substantial dilution of approximately $1.08 per share in the net tangible book value of the common stock. See the section entitled “Dilution” in this prospectus for a more detailed discussion of the dilution you will incur if you purchase common stock in this offering.
The sale of our common stock through future equity offerings may cause dilution and could cause the price of our common stock to decline.
We have registered $100,000,000 of securities for public sale pursuant to our registration statement on Form S-3 declared effective in July 2011. In addition, we registered $75,000,000 of securities for public sale pursuant to our registration statement on Form S-3 filed in November 2010. In December 2010, we offered 10,000,000 shares of common stock and warrants to purchase up to 10,000,000 shares of common stock under such registration statement and pursuant to a prospectus supplement first made available on December 10, 2010. Additionally, we entered into an ATM on June 16, 2011, which has a remaining capacity of approximately $17,300,000 through sales of our common stock from time to time under such registration statement. However, there are certain factors, such as volume of trading in our common stock, our stock price and the ability to terminate the agreement with notice, which limit the amount that can be raised in a short period of time through the ATM. Additionally, we have temporarily suspended the ATM in connection with this offering.
Sales of our common stock offered through future equity offerings may result in substantial dilution to the interests of other holders of our common stock. The sale of a substantial number of shares of our common stock to investors, or anticipation of such sales, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales.
The warrants are speculative in nature.
The warrants do not confer any rights of common stock ownership on its holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price for a limited period of time. Specifically, commencing on the date of issuance, holders of the warrants may exercise their right to acquire the common stock and pay an exercise price of $ per share [125% of public offering price of the common stock], prior to five years from the date of issuance, after which date any unexercised warrants will expire and have no further value. Moreover, following this offering, the market value of the warrants is uncertain and there can be no assurance that the market value of the warrants will equal or exceed their public offering price. There can be no assurance that the market price of the common stock will ever equal or exceed the exercise price of the warrants, and consequently, whether it will ever be profitable for holders of the warrants to exercise the warrants.
Our outstanding warrants include anti-dilution protection for any issuance of securities lower than the exercise price of such warrants such as is contemplated by this offering if such lower issuance occurs prior to the exercise or during the exercise period of the warrants. This anti-dilution protection could result in dilution to the shareholders and may contribute to downward pressure on the trading price of our common stock.
We currently have outstanding Class A warrants to purchase 4,525,978 shares of common stock issued in January 2010 and warrants to purchase 308,100 shares of common stock issued December 2010, with current exercise prices of $2.44 per common share and $1.25 per common share before any adjustment related to this offering, respectively. These warrants contain anti-dilution provisions that reduce the exercise price of the warrants if we issue or sell, or are deemed to have issued or sold, any shares of its common stock or securities exercisable or convertible into shares of common stock for no consideration or for a consideration per share less than the applicable exercise price in effect immediately prior to the time of such issue or sale, as is contemplated by this offering. The exercise of the warrants at prices below the market price of our common stock could adversely affect the price of shares of our common stock. In addition, sales of the shares of our common stock issuable upon exercise of the warrants could have a depressive effect on the price of our common stock, particularly if there is not a coinciding increase in demand by purchasers of our common stock.
Eastern Capital Limited holds a large percentage of our outstanding capital stock and has significant influence over the outcome of corporate actions requiring shareholder approval; and such shareholder’s priorities for our business may be different from other shareholders’.
Eastern Capital Limited (Eastern Capital) has not entered into a lock-up agreement in connection with this offering. All of the accumulated dividends in Series B-1 non-voting preferred stock and outstanding Series B-2 voting preferred stock, representing a significant amount of our outstanding capital stock on a fully-converted basis, are held by Eastern Capital. The accumulated dividends in our Series B-1 non-voting preferred stock are exchangeable for shares of Series B-2 voting preferred stock and, in March 2017, are convertible into shares of our common stock. Based solely on the number of shares of Series B-2 preferred stock that Eastern Capital held as of June 30, 2013, Eastern Capital has beneficial ownership of approximately twenty-one percent (21%) (calculated on an as converted to common stock basis and excluding any shares that will accrue as a dividend on the shares of Series B-2 preferred) of our voting securities based on the approximately 58,000,000 shares of common stock and Series B-2 preferred stock outstanding as of August 9, 2013. Furthermore, in connection with the March 2012 financing, we amended our Shareholder Rights Plan described below under “Description of Capital Stock” to allow Eastern Capital to acquire beneficial ownership of up to 49.9% of the Company’s outstanding securities without being deemed an “Acquiring Person” for purposes of our Shareholder Rights Plan. As a result of their current ownership and their ability to acquire more of our securities, they will be able to significantly influence the outcome of any financing transaction or other matter submitted to our shareholders for approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. The interests of Eastern Capital may differ from the interests of our other shareholders. For example, Eastern Capital could delay or prevent a change of control of the Company even if such a change of control would benefit our other shareholders. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to our investors’ perception that conflicts of interest may exist or arise.
In addition, the shares of Series B-1 preferred stock and the shares of Series B-2 preferred stock which may be issued upon exchange of the shares of Series B-1 preferred stock have certain rights, preferences and privileges that rank senior to the shares of our common stock. For example, the shares of Series B-1 preferred stock and Series B-2 preferred stock are entitled to receive a liquidation preference prior to any payment being made to holders of common stock upon a voluntary or involuntary liquidation, dissolution or winding up of the Company, or, in certain cases, if we experience a change of control. Furthermore, if the shares of Series B-1 preferred stock are never exchanged for shares of Series B-2 preferred stock and/or converted into shares of our common stock, at any time after March 2017, we may be required to redeem the then outstanding shares of Series B-1 preferred stock and any dividend shares accrued thereon at a price equal to the greater of (A) $3,250 (subject to adjustments for stock splits and similar events) plus all accrued dividends and (B) the then fair market value of a share of common stock multiplied by the number of shares of common stock into which such share of Series B-1 preferred stock is then convertible. Such redemption would be completed in three annual installments beginning not more than 120 days after we receive a request for redemption. The requirement for us to redeem Eastern Capital’s shares of Series B-1 preferred stock in cash could diminish our working capital, the consequences of which could have a material adverse effect on our business, operating results, financial condition and prospects.
Our corporate documents and Michigan law contain provisions that may make it more difficult for us to be acquired.
Our Board of Directors (Board) has the authority, without shareholder approval, to issue additional shares of preferred stock and to fix the rights, preferences, privileges and restrictions of these shares without any further vote or action by our shareholders. Michigan law contains a provision that makes it more difficult for a 10% shareholder, or its officers, to acquire a company. This authority, together with certain provisions of our charter documents, may have the effect of making it more difficult for a third party to acquire, or of discouraging a third-party from attempting to acquire, control of our company. This effect could occur even if our shareholders consider the change in control to be in their best interest. We have adopted a shareholder rights plan, the purpose of which is, among other things, to enhance our Board’s ability to protect shareholder interests and to ensure that shareholders receive fair treatment in the event any coercive takeover attempt of our company is made in the future. The shareholder rights plan could make it more difficult for a third party to acquire, or could discourage a third party from acquiring, our company or a large block of our company’s common stock.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This prospectus, including the documents that we incorporate by reference, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (Securities Act) and Section 21E of the Securities Exchange Act of 1934, as amended (Exchange Act). Any statements about our expectations, beliefs, plans, objectives, assumptions or future events or performance are not historical facts and may be forward-looking. These statements are often, but are not always, made through the use of words or phrases such as “anticipates,” “estimates,” “plans,” “projects,” “trends,” “opportunity,” “comfortable,” “current,” “intention,” “position,” “assume,” “potential,” “outlook,” “remain,” “continue,” “maintain,” “sustain,” “seek,” “achieve,” “continuing,” “ongoing,” “expects,” “believe,” “intend” and similar words or phrases, or future or conditional verbs such as “will,” “would,” “should,” “could,” “may,” or similar expressions. Accordingly, these statements involve estimates, assumptions and uncertainties which could cause actual results to differ materially from those expressed in them. Any forward-looking statements are qualified in their entirety by reference to the factors discussed throughout this report, and in particular those factors referenced in the section “Risk Factors.”
Because the factors referred to in the preceding paragraph could cause actual results or outcomes to differ materially from those expressed in any forward-looking statements we make, you should not place undue reliance on any such forward-looking statements. Further, any forward-looking statement speaks only as of the date on which it is made, and we undertake no obligation to update any forward-looking statement or statements to reflect events or circumstances after the date on which such statement is made or to reflect the occurrence of unanticipated events. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. These forward-looking statements include, but are not limited to, statements regarding:
· potential strategic collaborations with others;
· future capital needs;
· adequacy of existing capital to support operations for a specified time;
· product development and marketing plans;
· features and successes of our cellular therapies;
· manufacturing and facility capabilities;
· clinical trial plans and anticipated results, including the publication thereof;
· anticipation of future losses;
· replacement of manufacturing sources;
· commercialization plans;
· regaining and maintaining our compliance with the continued listing standards of The NASDAQ Stock Market; or
· revenue expectations and operating results.
We have never declared dividends on our common stock, and currently do not plan to declare dividends on shares of our common stock in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our Board and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our Board.
We estimate the net proceeds from this offering will be approximately $8,790,000 after deducting underwriting discounts and commissions and estimated transaction expenses payable by us of approximately $1,210,000 (or net proceeds of approximately $10,185,000 if the underwriters’ over-allotment option is exercised in full), in each case assuming a public offering price of $0.57 per share, which was the last reported sale price of our common stock on The NASDAQ Capital Market on August 9, 2013.
The principal purposes of this offering are to obtain additional capital to support our operations and continued development of ixmyelocel-T for severe, chronic ischemic cardiovascular diseases.
We estimate that we will use the net proceeds from this offering as follows:
· approximately $6.0 million to fund the internal and external clinical development costs associated with the ixCELL-DCM Phase 2b clinical trial, the REVIVE-CLI study, and investigator-sponsored trials;
· approximately $1.0 million to fund development costs associated with preclinical studies and to further develop our manufacturing platform; and
· the remainder for general corporate purposes, including internal development costs, working capital, general administrative costs and the prosecution and maintenance of our intellectual property.
We will be required to raise substantial additional capital to continue to fund the clinical development of our cell therapy applications. We may raise additional capital through additional public or private financings, as well as collaborative relationships, incurring debt and other available sources. Please see the discussion of the risks associated with our liquidity in the section “Risk Factors.”
MARKET PRICE OF OUR COMMON STOCK
Our common stock is traded on The NASDAQ Capital Market under the symbol “ASTM.” Our common stock has, from time to time, traded on a limited, sporadic and volatile basis. The table below shows the high and low closing prices for our common stock for the periods indicated, as reported by NASDAQ.
|
|
|
Price Ranges |
| ||||
|
|
|
High |
|
Low |
| ||
|
Fiscal Year Ending December 31, 2013 |
|
|
|
|
| ||
|
First Quarter |
|
$ |
1.41 |
|
$ |
0.70 |
|
|
Second Quarter |
|
0.80 |
|
0.40 |
| ||
|
Third Quarter — through August 9, 2013 |
|
0.77 |
|
0.35 |
| ||
|
|
|
|
|
|
| ||
|
Fiscal Year Ended December 31, 2012 |
|
|
|
|
| ||
|
First Quarter |
|
$ |
2.20 |
|
$ |
1.78 |
|
|
Second Quarter |
|
2.64 |
|
1.94 |
| ||
|
Third Quarter |
|
2.18 |
|
1.57 |
| ||
|
Fourth Quarter |
|
1.63 |
|
1.15 |
| ||
|
|
|
|
|
|
| ||
|
Fiscal Year Ended December 31, 2011 |
|
|
|
|
| ||
|
First Quarter |
|
$ |
3.25 |
|
$ |
2.06 |
|
|
Second Quarter |
|
3.27 |
|
2.48 |
| ||
|
Third Quarter |
|
2.89 |
|
2.01 |
| ||
|
Fourth Quarter |
|
2.75 |
|
1.79 |
| ||
The following table sets forth our capitalization as of June 30, 2013:
· on an actual basis; and
· on a pro forma as adjusted basis to give further effect to our issuance and sale of $10,000,000 of shares of our common stock and warrants in this offering at an assumed public offering price of $0.57 per share, which was the last reported sale price of our common stock on The NASDAQ Capital Market on August 9, 2013, and $ per warrant, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
|
|
|
June 30, 2013 |
| ||||
|
|
|
Actual |
|
Pro forma |
| ||
|
|
|
(In thousands) |
| ||||
|
Cash and cash equivalents |
|
$ |
4,494 |
|
$ |
13,284 |
|
|
|
|
|
|
|
| ||
|
Series B-1 non-voting convertible preferred stock, no par value; shares authorized and reserved — 39 actual, shares issued and outstanding — zero, pro forma |
|
$ |
5,186 |
|
$ |
5,186 |
|
|
Series B-2 voting convertible preferred stock, no par value; shares authorized and reserved — 39 actual, shares issued and outstanding — 12, pro forma |
|
37,690 |
|
37,690 |
| ||
|
Total convertible preferred stock |
|
42,876 |
|
42,876 |
| ||
|
|
|
|
|
|
| ||
|
SHAREHOLDERS’ DEFICIT: |
|
|
|
|
| ||
|
Common stock, no par value; shares authorized — 150,000, shares issued and outstanding — 45,664, actual; 63,208 shares issued and outstanding, pro forma |
|
245,976 |
|
254,766 |
| ||
|
Accumulated deficit |
|
(287,011 |
) |
(287,011 |
) | ||
|
Total shareholders’ deficit |
|
(41,035 |
) |
(32,245 |
) | ||
|
|
|
|
|
|
| ||
|
Total capitalization |
|
$ |
1,841 |
|
$ |
10,631 |
|
The table above does not include:
· 7,253,866 shares of common stock issuable upon the exercise of stock options outstanding as of June 30, 2013 at a weighted-average exercise price of $2.05 per share;
· 4,834,078 shares of common stock issuable upon the exercise of warrants outstanding as of June 30, 2013 at exercise prices of $2.44 per share (January 2010 - 4,525,978 shares) and $1.25 per share (December 2010 - 308,100 shares) in each case, before any adjustment related to this offering, which warrants are exercisable to purchase common stock; and
· 12,308 shares of preferred stock outstanding as of June 30, 2013.
Each $0.25 increase (decrease) in the assumed public offering price of $0.57 per share, which was the last reported sale price of our common stock on The NASDAQ Capital Market on August 9, 2013, would increase (decrease) the as adjusted amount of cash and cash equivalents, working capital, total assets and total shareholders’ equity by approximately $4,079,000, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase of 1,000,000 shares in the number of shares offered by us would increase the as adjusted amount of cash and cash equivalents, and total shareholders’ equity by approximately $530,000, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, each decrease of 1,000,000 shares in the number of shares offered by us would decrease the as adjusted amount of cash and cash equivalents, and total shareholders’ equity by approximately $530,000, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The as adjusted information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
If you invest in our common stock in this offering, your interest will be diluted to the extent of the difference between the public offering price per share of our common stock and the pro forma net tangible book value per share of our common stock immediately after this offering.
The net tangible book value of our common stock as of June 30, 2013 was a deficit of $41,035,000, or $(0.90) per share. Net tangible book value per share represents our total tangible assets less our total tangible liabilities, divided by the number of shares of common stock before giving effect to the conversion of all outstanding shares of preferred stock into shares of common stock upon the completion of this offering. After giving effect to the conversion of all outstanding shares of preferred stock into shares of common stock upon completion of this offering, the pro forma net tangible book value of our common stock as of June 30, 2013 was a deficit of $32,245,000, or a deficit of $(0.51) per share.
Net tangible book value dilution per share to new investors represents the difference between the amount per share paid by purchasers in this offering and the pro forma net tangible book value per share of our common stock immediately after the completion of this offering. After giving effect to our issuance and sale of $10,000,000 shares of common stock and warrants in this offering at an assumed public offering price of $0.57 per share, which was the last reported sale price of our common stock on The NASDAQ Capital Market on August 9, 2013, and $ per warrant, and after deducting estimated underwriting discounts and commissions and estimated offering expenses, our pro forma net tangible book value as of June 30, 2013 would have been $(0.51) per share. This represents an immediate increase in net tangible book value of $0.39 per share to existing shareholders and an immediate dilution in net tangible book value of $1.08 per share to purchasers of common stock in this offering, as illustrated in the following table:
|
Assumed public offering price per share |
|
|
|
$ |
0.57 |
| |
|
Net tangible book value per share as of June 30, 2013 |
|
$ |
(0.90 |
) |
|
| |
|
Increase per share attributable to new investors |
|
$ |
0.39 |
|
|
| |
|
Pro forma net tangible book value per share at June 30, 2013 after giving effect to the offering |
|
|
|
$ |
(0.51 |
) | |
|
Dilution per share to new investors |
|
|
|
$ |
1.08 |
| |
Business Overview
We were incorporated in 1989 and are a biotechnology company focused on the development of innovative cell therapies to repair or regenerate damaged or diseased tissues. We are developing patient-specific, expanded multicellular therapies for use in the treatment of severe, chronic ischemic cardiovascular diseases. We believe ixmyelocel-T (the generic name for our multicellular therapy) is a disease modifying therapy with multi-functional properties including: tissue remodeling, immunomodulation and the promotion of angiogenesis. Our proprietary cell-manufacturing technology enables the manufacture of multicellular therapies, expanded from an adult patient’s own bone marrow, to be delivered directly to damaged tissues. Preclinical and clinical data suggest that ixmyelocel-T may be safe and effective in treating patients with severe, chronic ischemic cardiovascular diseases such as dilated cardiomyopathy (DCM), the third leading cause of heart failure, and critical limb ischemia (CLI), the most severe form of peripheral arterial disease (PAD). Over 400 patients have been safely treated since our inception, with over 200 of those using ixmyelocel-T. In November 2011, we released positive Phase 2b data from our RESTORE-CLI clinical trial and launched our pivotal Phase 3 REVIVE trial in CLI in February 2012. During the fourth quarter of 2012, we launched a randomized, placebo-controlled, double-blinded Phase 2b clinical trial (ixCELL-DCM) for patients with advance heart failure due to ischemic DCM.
On March 27, 2013, we announced a strategic change in our research and development programs to focus on the clinical development of ixmyelocel-T for the treatment of advanced heart failure due to ischemic DCM. We believe heart failure represents a significant unmet medical need and a growing public health problem. DCM is the third leading cause of heart failure and the leading cause of heart transplantation in the United States. A majority of advanced heart failure patients that are refractory to medical therapy have DCM, which leads us to believe that the refractory ischemic DCM market represents a substantial market opportunity for ixmyelocel-T. The DCM program has received a U.S. Orphan Drug designation, and we believe that this orphan designation will allow us to pursue this heart failure indication with a more cost-effective path to approval for ixmyelocel-T.
As a result of the strategic change, we stopped enrollment of patients in the Phase 3 REVIVE clinical trial in patients with CLI. In addition, we are executing a corporate restructuring that we expect will reduce staff and ongoing operating cash needs by approximately 50 percent. As a result, we recorded a one-time restructuring charge of approximately $400,000 in the first quarter of 2013, primarily representing cash payments for severance and other personnel-related expenses. Severance payments were paid out during the second quarter of 2013 and will continue into the fourth quarter of 2013.
Our Therapy
Ixmyelocel-T is a unique multicellular product derived from an adult patient’s own bone marrow. Our proprietary cell manufacturing process significantly expands the mesenchymal stromal cells (MSCs) and M2-like anti-inflammatory macrophages in the patient’s bone marrow mononuclear cells while retaining many of the hematopoietic cells. These cell types are known to regulate the immune response and play a key role in tissue repair and regeneration by resolving pathologic inflammation, promoting angiogenesis, and remodeling ischemic tissue. Ixmyelocel-T is the only multicellular product known to have expanded cell populations of both MSCs and M-2 like anti-inflammatory macrophages.
MSCs and M2-like macrophages have a wide range of biological activities that promote repair and regeneration of damaged tissues through the paracrine effects of their secreted factors, as well as their direct cell activities. These cells produce high levels of potent anti-inflammatory and angiogenic factors, as well as factors involved in extracellular matrix remodeling. These cells also have direct activities such as phagocytosis of cellular debris and apoptotic cells, which control the inflammatory response, uptake of LDL and removal of cholesterol, and remodeling of extracellular matrix. We believe that, together, these paracrine effects and direct cell activities are responsible for ixmyelocel-T’s demonstrated therapeutic effects of resolving inflammation, promoting angiogenesis, and remodeling and repairing damaged tissue.
The following illustration summarizes the multiple biological activities of ixmyelocel-T that promote repair and regeneration of ischemic tissue:
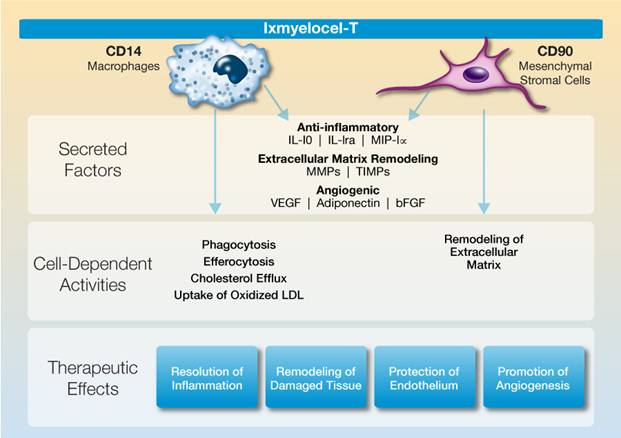
Ixmyelocel-T has several features that we believe are primarily responsible for success in treating adult patients with severe ischemic cardiovascular diseases such as DCM and critical limb ischemia:
Patient-specific (autologous) — we start with the patient’s own cells, which are accepted by the patient’s immune system, allowing the cells to integrate into existing functional tissues. We believe that this characteristic of our therapy eliminates both the risk of rejection and the need to use immunosuppressive therapy pre- or post-therapy. Our data also suggests that ixmyelocel-T provides the potential for long-term engraftment and tissue repair.
Expanded — we begin with a small amount of bone marrow from the patient (up to 60 ml) and significantly expand the number of certain cell types, primarily MSCs and M2-like anti-inflammatory macrophages, to a substantially greater number than are present in the patient’s own bone marrow (up to 200 times the number of certain cell types compared with the starting bone marrow).
Multicellular — we believe the multiple cell types in ixmyelocel-T, which are normally found in bone marrow but in smaller quantities, possess the key functions required for reducing chronic inflammation and promoting angiogenesis and tissue repair. By reducing inflammation, we believe that ixmyelocel-T provides the ideal conditions to allow for the growth of new tissue and blood vessels.
Minimally invasive — our procedure for collecting bone marrow can be performed in an out-patient setting and takes approximately 15 minutes. Administration of ixmyelocel-T for the treatment of DCM is performed in the cardiac catheterization laboratory using a cell injection catheter system in a one-time procedure. For diseases such as CLI, administration of ixmyelocel-T is performed with a syringe in an outpatient setting in a one-time, approximately 20 minute procedure.
Safe — bone marrow and bone marrow-derived therapies have been used safely and efficaciously in medicine for over three decades. Ixmyelocel-T leverages this body of scientific study and medical experience, and appears well tolerated in over 200 patients treated to date.
Our Technology Platform
Our patient-specific multicellular therapies are manufactured using the Company’s proprietary Aastrom Repicell System (ARS) cell manufacturing system. Our manufacturing process is conducted in a highly-automated, fully-closed and rigorously controlled system. Our system is highly scalable and reproducible and located in a 5,000-square-foot centralized manufacturing facility in Ann Arbor, Michigan. Production is conducted under current Good Manufacturing Practices (cGMP) guidelines required by the FDA with current annual capacity to treat up to 3,000 patients.

Our Strategy
Our objective is to become the leading global biotechnology company in the development, manufacture, and commercialization of autologous multicellular therapies for the treatment of severe ischemic cardiovascular diseases. To achieve this objective, we intend to:
· Complete our phase 2b ixCELL-DCM clinical study for the treatment of advanced heart failure due to ischemic DCM and, if successful, progress ixmyelocel-T into pivotal phase 3 clinical studies for this orphan indication.
· Complete patient follow-up in the REVIVE-CLI study to evaluate safety and efficacy endpoints, and pursue opportunities through investigator-sponsored studies and strategic relationships to continue to develop ixmyelocel-T as a stand-alone and/or adjunct therapy for the treatment of critical limb ischemia.
· Conduct additional preclinical and clinical studies of ixmyelocel-T to pursue additional high-value indications for the treatment of severe ischemic cardiovascular diseases.
· Utilize our proprietary ARS cell-expansion manufacturing platform to expand our product portfolio of cell therapies for the treatment of immune/inflammatory, cardiovascular and fibrovascular diseases.
· Leverage our leading proprietary cell manufacturing platform and expertise to provide manufacturing services and capabilities to other development and commercial-stage biopharmaceutical companies.
· Prepare to commercialize ixmyelocel-T through continued development of our internal commercialization capabilities and/or strategic partnerships for North America, Europe and Asia.
Clinical Development Programs
Our clinical development programs are focused on addressing areas of high unmet medical need in severe, chronic ischemic cardiovascular diseases. We have completed our Phase 1/2 clinical trials in DCM and we are currently enrolling our phase 2b ixCELL-DCM study, which is a randomized, double-blind, placebo-controlled clinical trial for patients with advanced heart failure due to ischemic DCM. Ixmyelocel-T has been granted a U.S.Orphan Drug designation by the FDA for the treatment of DCM. We also have ongoing ixmyelocel-T clinical programs for the treatment of CLI and craniofacial reconstruction.
The following summarizes the status of our clinical programs:

* Not actively enrolling
** Investigator-sponsored trial (University of Michigan)
Results to date in our clinical trials may not be indicative of results obtained from subsequent patients enrolled in those trials or from future clinical trials. Further, our future clinical trials may not be successful or we may not be able to obtain the required Biologic License Application (BLA) approval to commercialize our products in the United States in a timely fashion, or at all. See “Risk Factors.”
Dilated Cardiomyopathy
Background
DCM is a severe, chronic cardiovascular disease that leads to weakening of the heart muscle, enlargement of the heart, and reduction of the pumping function of the heart to the point that blood circulation is impaired. Patients with DCM typically present with symptoms of congestive heart failure, including limitations in physical activity and shortness of breath. DCM is now the third leading cause of heart failure and the leading cause of heart transplantation in the United States. There are two types of DCM: ischemic and non-ischemic. Ischemic DCM, the most common form representing an estimated 60% of all DCM patients, is associated with atherosclerotic cardiovascular disease. These refractory ischemic DCM patients are currently the target patient population for our clinical development of ixmyelocel-T, with approximately 175,000 patients in the United States alone. Patient prognosis depends on the stage and cause of the disease, but is typically characterized by a very poor quality of life and a high mortality rate.
Current treatments for refractory ischemic DCM patients are limited to heart transplantation and placement of left ventricular assist devices (LVADs). There are less than 2,500 heart transplantations in the United States each year. Many refractory DCM patients are not eligible for heart transplantation and transplants are extremely expensive at an estimated cost of over $1 million. LVADs are also expensive at an estimated cost of over $175,000 and have a mortality rate of 50% at two years.
In February 2007, the FDA granted Orphan Drug designation to ixmyelocel-T for the treatment of DCM. Our DCM development program is currently in Phase 2b clinical development. We recently completed follow up on two U.S. Phase 1/2 trials investigating surgical and catheter-based delivery for our product in the treatment of DCM. The final results from these Phase 1/2 clinical trials were presented at the Society for Cardiovascular Angiography and Interventions (SCAI) meeting on May 10, 2012.
Surgical Trial Program — DCM
We completed enrollment of 40 ischemic and non-ischemic DCM patients in the IMPACT-DCM clinical trial in January 2010 and the final patient was treated in March 2010. Participants in the IMPACT-DCM clinical trial were required to have New York Heart Association (NYHA) functional class III or IV heart failure, a left ventricular ejection fraction (LVEF) of less than or equal to 30% (55-75% is typical for a healthy person), and meet other eligibility criteria, including optimized medical therapy. Patients were randomized in an approximate 2:1 ratio of treatment to control group. Patients in the treatment group received our therapy through direct injection into the heart muscle during minimally invasive-surgery (involving a chest incision of approximately two inches). The primary objective of this study was to assess the safety of ixmyelocel-T in patients with DCM. Efficacy measures included cardiac dimensions and tissue mass, cardiac function (e.g., cardiac output, LVEF, cardiopulmonary exercise testing parameters), cardiac perfusion and viability, as well as other efficacy endpoints. NYHA functional class and quality of life were also assessed. Patients were followed for 12 months after treatment with an additional two year follow-up phone call recently completed for all patients.
Patients in the IDCM group who were treated with ixmyelocel-T experienced a lower percentage of major adverse cardiac events (MACE) compared to control subjects. The majority of ixmyelocel-T treated subjects (both IDCM and NIDCM) had improved NYHA Class over 12 months. There was also a trend toward improved function with a higher percentage of ixmyelocel-T treated IDCM subjects showing improved performance in the 6-minute walk as compared to IDCM control subjects. Following the week of surgery, adverse events were comparable between the treatment and control groups.
Catheter Trial Program — DCM
The Catheter-DCM clinical trial was designed to explore catheter-based direct injection delivery of ixmyelocel-T to treat DCM patients. This multi-center, randomized, controlled, prospective, open-label, Phase 2 study enrolled DCM patients at clinical sites across the United States.
We reported final 12-month results from the Catheter-DCM Phase 2 trial at the SCAI 2012 Scientific Sessions on May 10, 2012. The trial included 22 ischemic DCM (IDCM) and non-ischemic DCM (NIDCM) patients with a NYHA heart failure class of III or IV, or moderate to severe heart failure, and a left ventricular ejection fraction of 30 percent or less, which is a measure of how much blood leaves the heart with each pump. Patients were randomized 2:1 to receive an injection of the treatment into their heart muscles or to a control group, and were followed at three, six and 12 months. After 12 months, no procedural complications were reported and adverse events were comparable among patients who received the treatment and the control group. IDCM patients who received ixmyelocel-T had a lower mean number of major adverse cardiac events (MACE) (0.22 compared to 1.67 in the control group). IDCM patients who received the treatment were more likely to see improvement in NYHA class, six-minute walking distance and ejection fraction, compared to those in the control group. No consistent trends were noted in NIDCM patients.
Phase 2b Clinical Program — ixCELL-DCM
In February 2013, several sites began screening patients with ischemic DCM in the ixCELL-DCM trial, which is a randomized, double-blind, placebo-controlled clinical trial. The first patient was randomized in April 2013. The Phase 2b ixCELL-DCM trial will enroll 108 ischemic DCM patients. To be eligible, patients must be between the ages of 30 and 85, not be a candidate for reasonable revascularization procedures, have a LVEF less than or equal to 30%, and have NYHA class III or IV heart failure. Patients will be randomized 1:1 and followed for 12 months for the primary efficacy endpoint of major adverse cardiac events (MACE), defined as all-cause deaths, all-cause hospitalizations, and unplanned outpatient or emergency department visits for IV treatment of acute worsening heart failure. Secondary endpoints include clinical, functional, structural, symptomatic, quality of life, and biomarker measures at three, six and nine months. Patients will be followed for an additional 12 months for safety. We anticipate that enrollment will occur at approximately 35 sites across the United States and Canada and be completed by the end of the first quarter of 2014, with top-line data in the second quarter of 2015.
Critical Limb Ischemia
Background
CLI is the most serious and advanced stage of peripheral arterial disease (PAD) resulting from chronic inflammation and lipid accumulation. PAD is a chronic atherosclerotic disease that progressively restricts blood flow in the limbs and can lead to serious medical complications. This disease is often associated with other serious clinical conditions including hypertension, cardiovascular disease, dyslipidemia, diabetes, obesity and stroke. CLI is
used to describe patients with chronic ischemia-induced pain (even at rest) or tissue loss (ulcers or gangrene) in the limbs, often leading to amputation and death. Many CLI patients are considered “unsuitable for revascularization” patients as they have exhausted all other reasonable treatment options and will likely require amputation. The one-year and four-year mortality rates for CLI patients that are unsuitable for revascularization that progress to amputation are approximately 25% and 70%, respectively. Ixmyelocel-T, our disease modifying therapy with multiple functions, has shown significant promise in the treatment of CLI patients with existing tissue loss that are unsuitable for revascularization. Currently, there are an estimated 250,000 CLI patients that are unsuitable for revascularization in the United States.
Phase 2b Clinical Program — RESTORE CLI
Our U.S. Phase 2b RESTORE-CLI program was a multi-center, randomized, double-blind, placebo-controlled clinical trial. This clinical trial was designed to evaluate the safety and efficacy of ixmyelocel-T in the treatment of patients with CLI that are unsuitable for revascularization. It was the largest multi-center, randomized, double-blind, placebo-controlled cellular therapy study ever conducted in CLI patients. We completed enrollment of this trial in February 2010 with a total of 86 patients at 18 sites across the United States, with the last patient treated in March 2010. These patients were followed for a period of 12 months after treatment. In addition to assessing the safety of our product, efficacy endpoints included time to first occurrence of treatment failure — the trial’s primary efficacy endpoint — (defined as major amputation, all-cause mortality, doubling in wound surface area and de novo gangrene), amputation-free survival (defined as major amputation and all-cause mortality), major amputation rates, level of amputation, wound healing, patient quality of life and pain scores. The primary purpose of the trial was to assess performance of our therapy and, if positive, prepare for a Phase 3 program.
Final results of the Phase 2b RESTORE-CLI clinical trial were presented at the American Heart Association Scientific Sessions in November 2011 and published in the peer-reviewed journal Molecular Therapy in April 2012. Patients in the treatment arm showed a 62% reduction in risk relative to placebo in the primary efficacy endpoint of time to first occurrence of treatment failure (p=0.0032). While the study was not powered to show statistical significance in the secondary endpoint of amputation free survival, results from a subgroup of 45 patients with wounds at baseline (the approximate profile of the Phase 3 patient population) showed a 61% reduction in risk (21% ixmyelocel-T treated versus 44% control event rate; p=0.0802). The study also met the primary safety endpoint with no meaningful differences between the treated and control groups.
Phase 3 Clinical Program — REVIVE
In February 2012, several principal investigators participating in the pivotal Phase 3 REVIVE clinical trial for patients with CLI that are unsuitable for revascularization began screening patients. The first patient was randomized and aspirated in May 2012. We had previously received Fast Track Designation from the FDA for use of ixmyelocel-T for CLI in October 2010 and reached agreement with the FDA on a Special Protocol Assessment (SPA) in July 2011. Patients were randomized 1:1 and were to be followed for 12 months for the primary efficacy endpoint of amputation-free survival. On March 27, 2013, we announced that we were stopping enrollment in the Phase 3 REVIVE clinical trial. We had enrolled approximately 40 patients through that date and amended the protocol to continue following the patients for 12 months for safety and certain efficacy measures. We expect to have results from this study in the second quarter of 2014.
Production
Cell Manufacturing and Cell Production Components
We operate a centralized cell manufacturing facility in Ann Arbor, Michigan. The facility supports the current United States clinical trial and has sufficient capacity, with minor modifications, to supply our early commercialization requirements. We may establish and operate larger commercial-scale cell manufacturing facilities for the United States market in the future to accommodate potential market growth. We have reached agreement with the FDA on Chemistry, Manufacturing and Control (CMC) which was completed as part of the Special Protocol Assessment (SPA) process with the FDA for the Phase 3 REVIVE clinical trial.
We have established relationships with manufacturers that are registered with the FDA as suppliers of medical products to produce various components of our patented cell manufacturing system.
We have established relationships with various third parties who manufacture and/or supply certain components, equipment, disposable devices and other materials used in our cell manufacturing process to develop our cell products, as well as our final assemblies, component parts, subassemblies and associated spare parts used in the instrumentation platform of our cell production system.
There can be no assurance that we will be able to continue our present arrangements with our manufacturers and/or suppliers, supplement existing relationships or establish new relationships, or that we will be able to identify and obtain certain components, equipment, disposable devices, other materials, including ancillary materials that are necessary to develop our product candidates or that are used in our cell manufacturing and cell production components processes. Our dependence upon third parties for the supply and manufacture of such items could adversely affect our ability to develop and deliver commercially feasible cell products on a timely and competitive basis. See “Risk Factors.”
Our Arrangement with Vention Medical
In October 2010, we entered into a contract manufacturing and supply agreement (Supply Agreement) with ATEK Medical, LLC (ATEK) for the manufacture of our proprietary cell cassette for use in our manufacturing process. In November 2011, ATEK was purchased by Vention Medical (Vention) and currently operates as a division of Vention. There have been no changes to the terms of the Supply Agreement as a result of this purchase.
Pursuant to the terms of the Supply Agreement, we have granted Vention the exclusive right to manufacture our proprietary cell cassette, which includes assembly, labeling, packaging and sterilization. Vention will be responsible for obtaining all of our approved components pertaining to the cassettes and we are obligated to order and purchase the cassettes from Vention on an agreed upon schedule and in agreed upon quantities. In addition, we will provide Vention with reasonable engineering support to initiate and ramp up manufacturing of the cassettes and will supply all manufacturing equipment.
The Supply Agreement has an initial term of four years and will terminate automatically without notice unless prior to that time the term is extended by mutual written consent delivered at least six months prior to the termination date. The minimum term extension is generally to be no less than two years.
The Supply Agreement provides that we may discontinue the manufacture of the cassettes at our sole discretion. In such event, we agree to use commercially best efforts to notify Vention at least 120 days prior to our intention to discontinue manufacture of the cassettes. Failure to provide such notice will not be a breach of the Supply Agreement, but without such notice, we agree to purchase from Vention (i) certain finished goods that are in usable condition and (ii) certain components or raw materials inventory or work in process in each case to the extent convertible into finished cassettes.
We or Vention may terminate the Supply Agreement if the other party materially defaults in the performance of any provision of the Supply Agreement and, should any such default occur, then the non-defaulting party may give written notice to the defaulting party that if the default is not cured within 45 days, the Supply Agreement will be terminated. If the non-defaulting party gives such notice and the default is not cured during the 45 day period, then the Supply Agreement shall automatically terminate at the end of such period unless an extension is mutually agreed to by Vention and us. In addition to other remedies, either party may terminate the Supply Agreement at any time if either of us breaches our respective confidentiality obligations under the Supply Agreement, in which case termination shall be effective immediately upon receipt of notice from the non-breaching party of the breach and of termination. Either party may immediately terminate the Supply Agreement by written notice if the other party is or becomes insolvent, appoints or has appointed a receiver for all or substantially all of its assets, or makes an assignment for the benefit of its creditors. In addition, either party may terminate the Supply Agreement by written notice if the other party files a voluntary petition, or has filed against it an involuntary petition for bankruptcy and such petition is not dismissed within 90 days.
Upon termination of the Supply Agreement, Vention agrees to provide reasonable technical support at Vention’s published engineering rates for the transfer of manufacturing technology to an alternative manufacturer chosen by us to conduct final manufacture, package and test of the cassettes in the event that Vention, for a period of 150 days
from the date of receipt of the associated purchase order, is unable to manufacture all of our orders for any reason, or if Vention fails or refuses to meet our orders for cassettes pursuant to the terms of the Supply Agreement.
There can be no assurance that we will be able to continue our present arrangement with Vention. Our dependence upon our arrangement with Vention for the supply and manufacture of our proprietary cell cassette could adversely affect our ability to develop and deliver commercially feasible cell products on a timely and competitive basis. See “Risk Factors.”
Research & Development
Our therapy is produced from the patient’s bone marrow using Aastrom’s proprietary manufacturing system. The product is composed of a mixture of cell types normally found in bone marrow but at different quantities. For example, the mesenchymal stromal cells, identified with the CD90+ cell surface marker, as well as monocytes and activated macrophages, identified with CD14 marker, are expanded approximately 50 and 200 fold, respectively, while other CD45+ mononuclear cells from the bone marrow remain during the manufacturing process. We have demonstrated in the laboratory that the cells in our therapy are capable of multiple biological activities thought to play a critical role in repairing diseased and damaged tissues. These activities include aspects of tissue remodeling, promotion of angiogenesis and resolution of inflammation. In addition to these properties demonstrated in vitro, we have also shown that the therapy increases blood perfusion in both rat and mouse models of critical limb ischemia. In addition to these initial preclinical observations, we have ongoing preclinical studies designed to further characterize the mechanism of action of our product in the treatment of cardiovascular diseases. This data supports our current clinical-stage research where we are exploring the use of our therapy to regenerate tissue in patients with DCM and CLI.
In addition, our proprietary cell manufacturing system has demonstrated the capability to produce other types of cells. In the future, we may continue to explore the application of our manufacturing technology for the production of other cell types where there are potential opportunities to collaborate in the development of new cell therapies.
Patents and Proprietary Rights
Our success depends in part on our ability, and the ability of our licensors, to obtain patent protection for our products and processes. We have exclusive rights to approximately 19 unexpired issued United States patents. Eleven of these patents are material patents that protect our cellular therapy. We own ten of these patents and one has been licensed exclusively from the University of Michigan. These patents present various claims relating to (i) the composition of our cellular therapy, (ii) methods to manufacture or administer the cellular therapy, and (iii) the bioreactor device (the Aastrom Replicell System) that is used to make our product. The number of United States patents of each type with expiration range is listed in the table below.
|
Patent Type |
|
Number |
|
Expiry (Years) |
|
|
Composition of Matter |
|
2 |
|
1 and 16 |
|
|
Methods |
|
2 |
|
14 |
|
|
Bioreactor Device |
|
7 |
|
1 - 2 |
|
Certain patent equivalents to the United States patents have also been issued in other jurisdictions including Australia, Japan, and Canada, and under the European Patent Convention. In addition, we have filed applications for patents in the United States and equivalent applications in certain other countries claiming other aspects of our cell products and manufacturing processes. Our most significant patent that protects the composition of the cellular therapy directly, “Mixed cell populations for tissue repair and separation technique for cell processing” (US Patent 7,871,605), was issued in January 2011 and will expire in 2029. A divisional application of 7,871,605 for administration of this composition to patients was allowed by the USPTO in January 2012 and was issued in the April 2012 and will expire in 2027. A second divisional application of 7,871,605 directed to the methods of manufacture of our cell compositions was issued in March 2013 and will expire in 2027. Patents that protect our automated bioreactor device and culture system expire in 2015, but we will continue to rely on trade secrets and un-patentable know-how.
The validity and breadth of claims in medical technology patents involve complex legal and factual questions and, therefore, may be highly uncertain. No assurance can be given that any patents based on pending patent applications or any future patent applications by us, or our licensors, will be issued, that the scope of any patent protection will exclude competitors or provide competitive advantages to us, that any of the patents that have been or may be issued to us or our licensors will be held valid if subsequently challenged or that others will not claim rights in or ownership of the patents and other proprietary rights held or licensed by us. Furthermore, there can be no assurance that others have not developed or will not develop similar products, duplicate any of our products or design around any patents that have been or may be issued to us or our licensors. Since patent applications in the United States are maintained in secrecy until they are published 18 months after filing, we also cannot be certain that others did not first file applications for inventions covered by our and our licensors’ pending patent applications, nor can we be certain that we will not infringe any patents that may be issued to others on such applications.
We rely on certain licenses granted by the University of Michigan for certain patent rights. If we breach such agreements or otherwise fail to comply with such agreements, or if such agreements expire or are otherwise terminated, we may lose our rights in such patents.
We also rely on trade secrets and un-patentable know-how that we seek to protect, in part, by confidentiality agreements. It is our policy to require our employees, consultants, contractors, manufacturers, outside scientific collaborators and sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific limited circumstances. We also require signed confidentiality or material transfer agreements from any company that is to receive our confidential information. In the case of employees, consultants and contractors, the agreements generally provide that all inventions conceived by the individual while rendering services to us shall be assigned to us as the exclusive property of Aastrom. There can be no assurance, however, that these agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets or un-patentable know-how will not otherwise become known or be independently developed by competitors.
Our success will also depend in part on our ability to develop commercially viable products without infringing the proprietary rights of others. We do not believe any of our currently contemplated products or processes infringe any existing valid issued patent. However, the results of patent litigation are unpredictable, and no assurance can be given that patents do not exist or could not be filed which would have an adverse effect on our ability to market our products or maintain our competitive position with respect to our products. If our technology components, designs, products, processes or other subject matter are claimed under other existing United States or foreign patents, or are otherwise protected by third-party proprietary rights, we may be subject to infringement actions. In such event, we may challenge the validity of such patents or other proprietary rights or we may be required to obtain licenses from such companies in order to develop, manufacture or market our products. There can be no assurances that we would be able to obtain such licenses or that such licenses, if available, could be obtained on commercially reasonable terms. Furthermore, the failure either to develop a commercially viable alternative or obtain such licenses could result in delays in marketing our proposed products or the inability to proceed with the development, manufacture or sale of products requiring such licenses, which could have a material adverse effect on our business, financial condition and results of operations. If we are required to defend ourselves against charges of patent infringement or to protect our proprietary rights against third parties, substantial costs will be incurred regardless of whether we are successful. Such proceedings are typically protracted with no certainty of success. An adverse outcome could subject us to significant liabilities to third parties and force us to curtail or cease our development and sale of our products and processes.
Certain of our and our licensors’ research has been or is being funded in part by the Department of Commerce and by a Small Business Innovation Research Grant obtained from the Department of Health and Human Services. As a result of such funding, the United States government has certain rights in the technology developed with such funding. These rights include a non-exclusive, fully paid-up, worldwide license under such inventions for any governmental purpose. In addition, the United States government has the right to require us to grant an exclusive license under any of such inventions to a third party if the United States government determines that: (i) adequate steps have not been taken to commercialize such inventions; (ii) such action is necessary to meet public health or safety needs; or (iii) such action is necessary to meet requirements for public use under federal regulations. Additionally, under the federal Bayh
Dole Act, a party which acquires an exclusive license for an invention that was partially funded by a federal research grant is subject to the following government rights: (i) products using the invention which are sold in the United States are to be manufactured substantially in the United States, unless a waiver is obtained; (ii) the government may force the granting of a license to a third party who will make and sell the needed product if the licensee does not pursue reasonable commercialization of a needed product using the invention; and (iii) the United States government may use the invention for its own needs.
Sales and Marketing
We currently do not have the sales or marketing resources required to fully commercialize our therapeutic products. We intend to advance our programs to a point where we can evaluate the options to seek a development and/or commercialization partnership, or to make the investment to complete development and commercialize a product alone. We may also choose to undertake some pilot level of sales and marketing activity while seeking a commercial partnership.
Government Regulation
Our research and development activities and the manufacturing and marketing of our products are subject to the laws and regulations of governmental authorities in the United States and other countries in which our products will be marketed. Specifically, in the United States, the FDA, among other activities, regulates new product approvals to establish safety and efficacy of these products. Governments in other countries have similar requirements for testing and marketing. In the United States, in addition to meeting FDA regulations, we are also subject to other federal laws, such as the Occupational Safety and Health Act and the Environmental Protection Act, as well as certain state laws.
Our cell products will be regulated as somatic cell therapies/biologics/pharmaceuticals. With this classification, commercial production of our products will need to occur in registered/licensed facilities in compliance with Good Manufacturing Practice (GMP) for biologics (cellular products) or drugs.
Regulatory Process
Our products are subject to regulation as biological products under the Public Health Service Act and the Food, Drug and Cosmetic Act. Different regulatory requirements may apply to our products depending on how they are categorized by the FDA under these laws. The FDA has indicated that it intends to regulate products based on our technology as licensed biologics through the Center for Biologics Evaluation and Research. As current regulations exist, the FDA will require regulatory approval for certain human cellular- or tissue-based products, including our cell products, through a BLA submission.
Approval of new biological products is a lengthy procedure leading from development of a new product through preclinical and clinical testing. This process takes a number of years and the expenditure of significant resources. There can be no assurance that our product candidates will ultimately receive regulatory approval.
Regardless of how our product candidates are regulated, the Federal Food, Drug, and Cosmetic Act and other Federal and State statutes and regulations govern or influence the research, testing, manufacture, safety, labeling, storage, record-keeping, approval, distribution, use, product reporting, advertising and promotion of such products. Noncompliance with applicable requirements can result in civil penalties, recall, injunction or seizure of products, refusal of the government to approve or clear product approval applications or to allow us to enter into government supply contracts, withdrawal of previously approved applications and criminal prosecution.
Product Approval
In order to obtain FDA approval of a new medical product, sponsors must submit proof of safety and efficacy. In most cases, such proof entails extensive preclinical studies and clinical trials. The testing, preparation of necessary applications and processing of those applications by the FDA is expensive and may take several years to complete. There can be no assurance that the FDA will act favorably or in a timely manner in reviewing submitted applications, and we may encounter significant difficulties or costs in our efforts to obtain FDA approvals, in turn, which could delay or preclude us from marketing any products we may develop. The FDA may also require post-
marketing testing and surveillance of approved products, or place other conditions on the approvals. These requirements could cause it to be more difficult or expensive to sell the products, and could therefore restrict the commercial applications of such products. Product approvals may be withdrawn if compliance with applicable regulations is not maintained or if problems occur following commercialization. For patented technologies, delays imposed by the governmental approval process may materially reduce the period during which we will have the exclusive right to exploit such technologies.
If clinical trials of a proposed medical product are required, the manufacturer or distributor of a drug or biologic will have to submit an IND application with the FDA prior to commencing human clinical trials. The submission must be supported by data, typically including the results of preclinical and laboratory testing. Following submission of the IND, the FDA has 30 days to review the application and raise safety and other clinical trial issues. If we are not notified of objections within that period, clinical trials may be initiated, and human clinical trials may commence at a specified number of investigational sites with the number of patients approved by the FDA. We have submitted several INDs for our cell products, and we have conducted clinical trials under these INDs.
Our products will be regulated by the FDA as a licensed biologic, although there can be no assurance that the FDA will not choose to regulate this product in a different manner in the future. The FDA categorizes human cell- or tissue-based products as either minimally manipulated or more than minimally manipulated, and has determined that more than minimally manipulated products require clinical trials to demonstrate product safety and efficacy and the submission of a BLA for marketing authorization. For products that may be regulated as biologics, the FDA requires: (i) preclinical laboratory and animal testing; (ii) submission to the FDA of an IND application, which must be approved prior to the initiation of human clinical trials; (iii) adequate and well-controlled clinical trials to establish the safety and efficacy of the product for its intended use; (iv) submission to the FDA of a BLA; and (v) review and approval of the BLA as well as inspections of the manufacturing facility by the FDA prior to commercial marketing of the product.
We conduct preclinical testing for internal use and as support for submissions to the FDA. Preclinical testing generally includes various types of in-vitro laboratory evaluations of our products as well as animal studies to assess the safety and the functionality of the product. Clinical trials are identified by phases (i.e., Phase 1, Phase 2, Phase 3, etc.). Depending on the type of preclinical and/or clinical data available, the trial sponsor will submit a request to the FDA to initiate a specific phase study (e.g., a Phase 1 trial represents an initial study in a small group of patients to test for safety and other relevant factors; a Phase 2 trial represents a study in a larger number of patients to assess the safety and efficacy of a product; and, Phase 3 trials are initiated to establish safety and efficacy in an expanded patient population at multiple clinical trial sites).
The results of the preclinical tests and clinical trials are submitted to the FDA in the form of a BLA for marketing approval. The testing, clinical trials and approval process are likely to require substantial time and effort, and there can be no assurance that any approval will be granted on a timely basis, if at all. Additional animal studies or clinical trials may be requested during the FDA review period that may delay marketing approval. After FDA approval for the initial indications, further clinical trials may be necessary to gain approval for the use of the product for additional indications. The FDA requires that adverse effects be reported to the FDA and may also require post-marketing testing to monitor for adverse events, which can involve significant expense.
Under current requirements, facilities manufacturing biological products for commercial distribution must be licensed. To accomplish this, an establishment registration must be filed with the FDA. In addition to the preclinical studies and clinical trials, the BLA includes a description of the facilities, equipment and personnel involved in the manufacturing process. An establishment registration/license is granted on the basis of inspections of the applicant’s facilities in which the primary focus is on compliance with GMPs and the ability to consistently manufacture the product in the facility in accordance with the BLA. If the FDA finds the results of the inspection unsatisfactory, it may decline to approve the BLA, resulting in a delay in production of products.
As part of the approval process for human biological products, each manufacturing facility must be registered and inspected by the FDA prior to marketing approval. In addition, state agency inspections and approvals may also be required for a biological product to be shipped out of state.
Commercial Strategy
We are currently focused on utilizing our technology to produce expanded, patient specific multicellular products for use in severe, chronic ischemic cardiovascular indications. At such time as we satisfy applicable regulatory approval requirements, we expect the sales of our cell-based products to constitute nearly all of our product sales revenues.
We do not expect to generate positive cash flows from our consolidated operations for at least the next several years and then only if we achieve significant product sales. Until that time, we expect that our revenue sources from our current activities will consist of only minor sales of our cell products and manufacturing supplies to our academic collaborators, grant revenue, research funding and potential licensing fees or other financial support from potential future corporate collaborators.
We expect that we will need to raise significant additional funds or pursue strategic transactions or other strategic alternatives in order to complete our product development programs, complete clinical trials needed to market our products, and commercialize our products. To date, we have financed our operations primarily through public and private sales of our equity securities, and we expect to continue to seek to obtain the required capital in a similar manner. As a development stage company, we have never been profitable and do not anticipate having net income unless and until significant product sales commence. With respect to our current activities, this is not likely to occur until we obtain significant additional funding, complete the required clinical trials for regulatory approvals, and receive the necessary approvals to market our products. If we cannot raise such funds, we will not be able to develop or enhance products, take advantage of future opportunities, or respond to competitive pressures or unanticipated requirements, which would have a material adverse impact on our business, financial condition and results of operations. As a result of the need to raise additional capital and a net capital deficiency, there is uncertainty regarding our ability to maintain liquidity sufficient to operate our business effectively over at least the next twelve months, which raises substantial doubt as to our ability to continue as a going concern. Through June 30, 2013, we have accumulated a net loss attributable to common shareholders of approximately $287,011,000. We cannot provide any assurance that we will be able to achieve profitability on a sustained basis, if at all, obtain the required funding, obtain the required regulatory approvals, or complete additional corporate partnering or acquisition transactions.
Competitive Environment
The biotechnology and medical device industries are characterized by rapidly evolving technology and intense competition. Our competitors include major multinational medical device companies, pharmaceutical companies, biotechnology companies and stem cell companies operating in the fields of tissue engineering, regenerative medicine, cardiac, vascular, orthopedics and neural medicine. Many of these companies are well-established and possess technical, research and development, financial, and sales and marketing resources significantly greater than ours. In addition, many of our smaller potential competitors have formed strategic collaborations, partnerships and other types of joint ventures with larger, well established industry competitors that afford these companies potential research and development and commercialization advantages in the technology and therapeutic areas currently being pursued by us. Academic institutions, governmental agencies and other public and private research organizations are also conducting and financing research activities which may produce products directly competitive to those being commercialized by us. Moreover, many of these competitors may be able to obtain patent protection, obtain FDA and other regulatory approvals and begin commercial sales of their products before us.
Our potential commercial products address a broad range of existing and emerging therapeutic markets, in which cell-based therapy is a new and as of yet, unproven, commercial strategy. In a large part, we face primary competition from existing medical devices and drug products. Some of our competitors have longer operating histories and substantially greater resources. These include companies such as Baxter International, Inc. (Baxter), Biomet, Inc., Johnson & Johnson, Inc., Medtronic, Inc. (Medtronic), and others.
In the general area of cell-based therapies, we potentially compete with a variety of companies, most of whom are specialty medical products or biotechnology companies. Some of these, such as Baxter, Johnson & Johnson, Medtronic and Miltenyi Biotec are well-established and have substantial technical and financial resources compared to ours. However, as cell-based products are only just emerging as viable medical therapies, many of our most direct competitors are smaller biotechnology and specialty medical products companies. These include Advanced
Cell Technology, Inc., Cytomedix, Inc. (formerly Aldagen, Inc.), Arteriocyte Medical Systems, Inc., Athersys, Inc., Bioheart, Inc., Cytori Therapeutics, Inc., International Stem Cell Corporation, Neostem, Inc., Terumo Medical Corporation (formerly Harvest Technologies Corporation), Mesoblast, Osiris Therapeutics, Inc., Pluristem, Inc. Stem Cells, Inc., Tengion, Inc., and others.
Employees
As of August 1, 2013, we employed approximately 40 individuals on a full-time equivalent basis. A significant number of our management and professional employees have had prior experience with pharmaceutical, biotechnology or medical product companies. None of our employees are covered by collective bargaining agreements, and management considers relations with our employees to be good.
Executive Officers
|
Name |
|
Position |
|
Age |
|
Executive |
|
|
Dominick C. Colangelo |
|
President and Chief Executive Officer |
|
49 |
|
2013 |
|
|
Daniel R. Orlando |
|
Chief Commercial Officer |
|
48 |
|
2012 |
|
|
Ronnda L. Bartel, Ph.D. |
|
Chief Scientific Officer |
|
54 |
|
2010 |
|
|
Brian D. Gibson |
|
Vice President of Finance |
|
34 |
|
2011 |
|
Dominick C. Colangelo — Mr. Colangelo joined Aastrom in 2013 with more than twenty years of executive management and corporate development experience in the biopharmaceutical industry, including nearly a decade with Eli Lilly and Company. Most recently, he was President and Chief Executive Officer of Promedior, Inc. During his career, he has held a variety of executive positions of increasing responsibility in product development, pharmaceutical operations, sales and marketing, and corporate development. He has extensive experience in the acquisition, development and commercialization of therapies to treat fibrovascular, metabolic and cardiovascular diseases. During his tenure at Eli Lilly and Company, he held positions as Director of Strategy and Business Development for Lilly’s Diabetes Product Group and also served as a founding Managing Director of Lilly Ventures. Mr. Colangelo received his B.S.B.A. in Accounting, Magna Cum Laude, from the State University of New York at Buffalo and a J.D. degree, with Honors, from the Duke University School of Law.
Daniel R. Orlando — Mr. Orlando joined Aastrom as Chief Commercial Officer in August of 2012. Mr. Orlando served as interim Chief Executive Officer of Aastrom from December 2012 to March 2013. He has more than 20 years of commercial product preparation and launch experience including leadership roles in sales, marketing and most recently as a vice president of business development for North and South America at Takeda Pharmaceuticals. As an early employee at Takeda North America, he served as the original brand director for Actos, which became the #1 branded anti-diabetic agent in the United States. Mr. Orlando’s initial pharmaceutical experience came in progressively expanding roles in sales and marketing at Abbott Laboratories. He holds an MBA from Florida Atlantic University and a BA in economics with Honors from the University of Florida.
Ronnda L. Bartel, Ph.D. — Dr. Bartel joined Aastrom in 2006 and is responsible for research, development, quality, IT, manufacturing and engineering operations. Dr. Bartel has more than 20 years of research and product development experience and most recently was Executive Director, Biological Research at MicroIslet and Vice President, Scientific Development at StemCells, Inc. Earlier in her career, she was Senior Principal Scientist, Cell Biology at Advanced Tissue Sciences and was involved in the development and approval of two of the first three cell based products approved by the FDA. She has also worked as Senior Director, Science and Technology at SRS Capital, LLC evaluating life science investments and has also held positions in clinical development, drug delivery, business development and manufacturing. Dr. Bartel holds a Ph.D. in Biochemistry from the University of Kansas, completed postdoctoral work at the University of Michigan and received a B.A. in Chemistry and Biology from Tabor College.
Brian D. Gibson — Mr. Gibson joined Aastrom in July 2010 and is Vice President of Finance. He brings more than 12 years of finance and accounting experience to Aastrom. Prior to joining Aastrom, Mr. Gibson was a senior manager at PricewaterhouseCoopers, with broad experience in multiple industries, including life sciences and healthcare. Mr. Gibson holds a B.A. in accounting with high honor from the Eli Broad College of Business at Michigan State University and is a certified public accountant.
NASDAQ Delisting Notification
We received notice from NASDAQ on May 9, 2013 that our common stock bid price has fallen below $1.00 per share, and on May 20, 2013 that we have not maintained a market value of at least $35 million. Continued listing of a security on NASDAQ is conditioned upon compliance with various continued listing standards, which require, among other things, that for 30 consecutive trading days the closing minimum bid price for our listed securities not be lower than $1.00 per share and for ten consecutive trading days the market value of listed securities for our common stock close at or above $35 million. As a result, our common stock is in jeopardy of being delisted. NASDAQ has informed us that we have until November 5, 2013, to meet the minimum bid price threshold and until November 18, 2013 to meet the market value threshold to maintain the listing of our common stock on The NASDAQ Capital Market.
While we are exercising diligent efforts to maintain the listing of our common stock on NASDAQ, such as the amendment and restatement of the Certificate of Designations, Preferences and Rights of Series B-1 Non-Voting Preferred Stock and Series B-2 Voting Preferred Stock (the Certificate of Designations), there can be no assurance that we will be able to meet these thresholds within the required timeframes. With respect to the minimum bid price, we may receive a second 180 day grace period if certain conditions are met. Additionally, NASDAQ rules permit us to appeal to a NASDAQ Hearings Panel. We intend to regain compliance prior to the expiration of the notice periods by exploring a number of options, such as effecting a reverse stock split on our outstanding common stock.
Recent Business Developments
On August 12, 2013, we amended and restated the Certificate of Designations with the consent of Eastern Capital. The amendment removes the redemption feature from the Series B-2 Voting Preferred Stock and increases the redemption value assigned to the Series B-1 Non-Voting Preferred Stock in the event of a change of control, subject to certain terms and limitations. As a result of the amendment, we believe that the Series B-2 Voting Preferred Stock will be reclassified from mezzanine equity to shareholders’ deficit on our balance sheet, reducing our shareholders’ deficit by $37.7 million.
Corporate Information
Aastrom is incorporated under the laws of the State of Michigan. Our principal executive offices are located at 24 Frank Lloyd Wright Drive, Lobby K, Ann Arbor, Michigan 48105. Our telephone number is (800) 556-0311. The address of our website is www.aastrom.com. The reference to our website is intended to be an inactive textual reference and, except for the documents incorporated by reference as noted above, the information on, or accessible through, our website is not intended to be part of this prospectus.
The following briefly summarizes the general terms and provisions of our shares of common preferred stock and the warrants. You should read the provisions of our articles of incorporation, as amended (Charter), our amended and restated bylaws (Bylaws) and other relevant instruments and agreements relating to our securities before you make an investment decision with respect to our shares of common and preferred stock.
The following description of our common and preferred stock and certain provisions of our Charter, and our amended and restated Bylaws, is a summary and is qualified in its entirety by the provisions of our Charter and Bylaws.
Our authorized capital stock consists of 150,000,000 shares of common stock, no par value per share, and 5,000,000 shares of preferred stock, no par value per share. Please see “Certain Provisions of Michigan Law and of Our Charter and Bylaws” for a description of those provisions in our Charter and Bylaws that would have an effect of delaying, deferring or preventing a change in control of the Company and that would operate only with respect to an extraordinary corporate transaction involving us or our subsidiaries.
Common Stock
Holders of our common stock are entitled to one vote for each share held of record on all matters submitted to a vote of shareholders. We do not have a classified Board and shareholders do not have cumulative voting rights. Holders of common stock have no preemptive, redemption or conversion rights and are not subject to future calls or assessments. No sinking fund provisions apply to our common stock. All outstanding shares are fully-paid and non-assessable. In the event of our liquidation, dissolution or winding up, holders of common stock are entitled to share ratably in assets available for distribution, subject to any prior distribution rights of any preferred stock then outstanding. Holders of common stock are entitled to receive proportionately any such dividends declared by our Board, out of legally available funds for dividends, subject to any preferences that may be applicable to any shares of preferred stock that may be outstanding at that time. The rights, preferences and privileges of holders of common stock are set forth in our Charter, which may be amended by the holders of a majority of the outstanding shares of
common stock. We have adopted a shareholder rights plan, which could make it more difficult for a third party to acquire, or could discourage a third party from acquiring, our company or a large block of our common stock. Please see the description above in “Certain Provisions of Michigan Law and of our Charter and Bylaws; Transfer Agent and Registrar.”
Preferred Stock
Our Board may issue preferred stock in one or more series without shareholder approval. Our Board may determine the rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, of each series of preferred stock. The issuance of preferred stock, while providing desirable flexibility in connection with possible acquisitions and other corporate purposes, could make it more difficult for a third party to acquire, or could discourage a third party from acquiring, a majority of our outstanding voting stock. The rights of holders of our common stock described above, will be subject to, and may be adversely affected by, the rights of any preferred stock that we may designate and issue in the future.
Shareholder Rights Agreement - Series A Junior Participating Cumulative Preferred Stock
On August 11, 2011, our Board adopted a shareholder rights agreement (Rights Agreement), the purpose of which is, among other things, to enhance the Board’s ability to protect shareholder interests and to ensure that shareholders receive fair treatment in the event any coercive takeover attempt of Aastrom is made in the future. The Rights Agreement could make it more difficult for a third-party to acquire, or could discourage a third party from acquiring, us or a large block of our common stock. The following summary description of the Rights Agreement should be read in conjunction with the Rights Agreement, which was filed with the SEC as an exhibit to a Registration Statement on Form 8-A on August 12, 2011 and amended in March 2012 to allow Eastern Capital to acquire beneficial ownership of up to 49.9% of the Company’s outstanding securities without being deemed an “acquiring person” for purposes of our Rights Agreement.
In connection with the adoption of the Rights Agreement, the Board declared a dividend distribution of one preferred stock purchase right (Right) for each outstanding share of common stock to shareholders of record as of the close of business on August 15, 2011. In addition, one Right will automatically attach to each share of common stock issued between August 15, 2011 and the distribution date. The Rights currently are not exercisable and are attached to and trade with the outstanding shares of common stock. Under the Rights Agreement, the Rights become exercisable if a person or group becomes an “acquiring person” by acquiring 15% or more of the outstanding shares of common stock or if a person or group commences a tender offer that would result in that person owning 15% or more of the common stock. If a person or group becomes an “acquiring person,” each holder of a Right (other than the acquiring person and its affiliates, associates and transferees) would be entitled to purchase, at the then-current exercise price, such number of shares of our preferred stock which are equivalent to shares of common stock having a value of twice the exercise price of the Right. If we are is acquired in a merger or other business combination transaction after any such event, each holder of a Right would then be entitled to purchase, at the then-current exercise price, shares of the acquiring company’s common stock having a value of twice the exercise price of the Right.
Each share of preferred stock is entitled to payment of a quarterly dividend, an increased vote multiple, and a liquidation preference. In addition, each share of preferred stock is granted the exclusive right to vote for two additional members of the Board whose positions are created upon the vesting of such rights upon holders of preferred stock. Once purchased, said shares are not redeemable by the Company.
The Rights may be redeemed in whole, but not in part, at a price of $0.001 per Right (payable in cash, common stock or other consideration deemed appropriate by the Board) by the Board only until the earlier of (i) the time at which any person becomes an “acquiring person” or (ii) the expiration date of the Rights Agreement. Immediately upon the action of the Board ordering redemption of the Rights, the Right will terminate and thereafter the only right of the holders of Rights will be to receive the redemption price. The Rights will expire at the close of business on August 15, 2021, unless previously redeemed or exchanged by us as described above.
Series B Convertible Preferred Stock
On March 9, 2012, the Company completed the sale of 12,308 shares of Series B-1 non-voting convertible preferred stock (Series B-1 preferred stock) at an offering price of $3,250 per share. The Company received $37,620,000 in net proceeds from the sale of the shares of Series B-1 preferred stock, after offering expenses. In addition to the Series B-1 preferred stock, which was issued at the closing, the Company also authorized Series B-2 Voting Convertible Preferred Stock (Series B-2 preferred stock). The Series B-1 preferred stock and Series B-2 preferred stock collectively are referred to as the Series B preferred stock. On August 12, 2013, we amended the terms of the Series B-1 preferred stock and Series B-2 preferred stock with the consent of the holder of the Series B preferred stock, to modify the redemption feature such that in the event of a change of control, the Series B-1 preferred stock is redeemable at a greater price per share, subject to certain terms and limitations, but the Series B-2 preferred stock is not redeemable. The Series B-1 preferred stock is not entitled to vote on matters on which the common shareholders are generally entitled to vote. The Series B-2 preferred stock is entitled to vote with the holders of the common stock as a single class, with each share of Series B-2 preferred stock having the number of votes equal to the number of shares of common stock issuable upon conversion of such Series B-2 preferred stock. On May 3, 2012, shareholder approval was obtained in accordance with Nasdaq Marketplace Rule 5635(b), which allowed the holder of Series B-1 preferred stock to exchange all of the then outstanding shares for shares of Series B-2 preferred stock on a one-for-one basis. The Series B preferred stock will, with respect to dividend rights and rights on liquidation, winding-up and dissolution, rank on parity with any other class or series of the Company capital stock that the Company may issue in the future which is designated as being on parity with the Series B preferred stock, and rank senior to our common stock and Series A preferred stock. The Series B preferred stock is convertible, at the option of the holder thereof at any time after the five year anniversary of the closing of the offering, into shares of common stock at a conversion price of $3.25 per share of common stock. At any time after the five year anniversary of issuance, the Company may elect to convert any or all outstanding shares of Series B preferred stock into shares of our common stock, subject to certain limitations. Dividends on the Series B preferred stock will be cumulative and compound daily, at a rate of 11.5% per annum, payable upon conversion, liquidation, redemption or other similar events, and payable in cash or Series B-1 preferred stock until the five year anniversary of issuance. Following the five year anniversary of issuance and until the earlier of the tenth anniversary of the issuance and the date no Series B preferred stock remain outstanding, dividends will accrue at a rate of 8% per annum and will be payable in cash or Series B-1 preferred stock, at our option. The Series B-1 preferred stock is redeemable at any time after the fifth anniversary of issuance or upon a change of control, in each case, subject to certain terms and limitations.
Warrants
The following summary of certain terms and provisions of the warrants offered hereby is not complete and is subject to, and qualified in its entirety by the provisions of the form of the warrant, which is filed as an exhibit to the registration statement of which this prospectus is a part of. Prospective investors should carefully review the terms and provisions set forth in the form of warrant.
Exercisability
The warrants are exercisable immediately upon issuance and at any time up to the date that is five years from the date of issuance. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). Unless otherwise specified in the warrant, the holder will not have the right to exercise any portion of the warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% (which the holder may increase to 9.99% upon 61 days’ notice to us) of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the warrants.
Exercise Price
The initial exercise price per share of common stock purchasable upon exercise of the warrants is $ per share [125% of the public offering price of the common stock]. The exercise price and the number of shares issuable upon exercise are subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock.
Cashless Exercise
In the event that a registration statement covering shares of common stock underlying the warrants, or an exemption from registration, is not available for the resale of such shares of common stock underlying the warrants, the holder may, in its sole discretion, exercise the warrant in whole or in part and, in lieu of making cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, elect instead to receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the warrant. In no event shall we be required to make any cash payments or net settlement to the registered holder in lieu of issuance of common stock underlying the warrants.
Transferability
Subject to applicable laws, the warrants may be transferred at the option of the holders upon surrender of the warrants to us together with the appropriate instruments of transfer.
Rights as a Shareholder
Except as otherwise provided in the warrants or by virtue of such holder’s ownership of shares of our common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant.
Transfer Agent
The transfer agent of our common stock is Continental Stock Transfer & Trust Company.
CERTAIN PROVISIONS OF MICHIGAN LAW AND OF OUR CHARTER AND
BYLAWS
We are subject to certain anti-takeover provisions of the Michigan Business Corporation Act (MBCA) that could delay or make more difficult a merger or tender offer involving us. Chapter 7A of the MBCA prevents, in general, an “interested shareholder” (defined generally as a person owning 10% or more of a corporation’s outstanding voting shares) from engaging in a “business combination” (as defined therein) with a Michigan corporation unless: (a) the board of directors issues an advisory statement, holders of 90% of the shares of each class of stock entitled to vote approve the transaction, and holders of two-thirds of the “disinterested” shares of each class of stock approve the transaction; (b) the interested shareholder has been an interested shareholder for at least five years and has not acquired beneficial ownership of any additional shares of the corporation subsequent to the transaction which resulted in such shareholder being classified as an interested shareholder, and meets certain requirements, including provisions relating to the fairness of the price and the form of consideration paid; or (c) the board of directors, by resolution, exempts a particular interested shareholder from these provisions prior to the interested shareholder becoming an interested shareholder. The MBCA also contains certain other provisions that could have anti-takeover effects.
Our Charter does not provide shareholders with the right to act without a meeting and does not provide for cumulative voting in the election of directors. The amendment of any of these provisions would require approval by holders of at least a majority of the shares of our outstanding common stock.
These and other provisions of our Charter or Bylaws, as well as our Rights Agreement described above under “Description of Capital Stock,” could have the effect of deterring certain takeovers or delaying or preventing certain changes in control or changes in our management, including transactions in which shareholders might otherwise receive a premium for their shares over then-current market prices.
Aegis Capital Corp. is acting as the sole book-running manager of the offering and as representative of the underwriters, or the Representative. We have entered into an underwriting agreement, dated , 2013, with the Representative. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to each underwriter named below and each underwriter named below has severally and not jointly agreed to purchase from us, at the public offering price per share less the underwriting discounts set forth on the cover page of this prospectus, the number of shares of common stock and warrants listed next to its name in the following table:
|
Name of Underwriter |
|
Number of Shares |
|
Number of Warrants |
|
|
Aegis Capital Corp |
|
|
|
|
|
|
Total |
|
|
|
|
|
The underwriters are committed to purchase all the shares of common stock and warrants offered by us other than those covered by the option to purchase additional shares and/or warrants described below, if they purchase any shares and warrants. The obligations of the underwriters may be terminated upon the occurrence of certain events specified in the underwriting agreement. Furthermore, pursuant to the underwriting agreement, the underwriters’ obligations are subject to customary conditions, representations and warranties contained in the underwriting agreement, such as receipt by the underwriters of officers’ certificates and legal opinions.
We have agreed to indemnify the underwriters against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect thereof.
The underwriters are offering the shares and warrants, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel and other conditions specified in the underwriting agreement. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 45 days after the date of this prospectus, permits the underwriters to purchase a maximum of additional shares and/or additional warrants (15% of the shares sold in this offering) from us to cover over-allotments, if any. If the underwriters exercise all or part of this option, they will purchase shares and/or warrants covered by the option at the public offering price that appears on the cover page of this prospectus, less the underwriting
discount. If this option is exercised in full, the total price to the public will be $ and the total net proceeds, before expenses, to us will be .
Discount. The following table shows the public offering price, underwriting discount and proceeds, before expenses, to us. The information assumes either no exercise or full exercise by the underwriters of their over-allotment option.
|
|
|
Per |
|
Per |
|
Total Without Over- |
|
Total With Over- |
| ||||
|
Public offering price |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
Underwriting discount (7%)(1) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
Non-accountable expense allowance (1%)(2) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
Proceeds, before expenses, to us |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
(1) The underwriting discount of 7% is not payable with respect to the shares and/or warrants issued to Eastern Capital Limited.
(2) The expense allowance of 1% is not payable with respect to the shares and/or warrants issued to Eastern Capital Limited or the shares and/or warrants sold upon exercise of the underwriters’ over-allotment option.
The underwriters propose to offer the shares and/or warrants offered by us to the public at the public offering price set forth on the cover of this prospectus. In addition, the underwriters may offer some of the shares and/or warrants to other securities dealers at such price less a concession of $ . After the initial offering, the public offering price and concession to dealers may be changed.
We have paid an expense deposit of $25,000 to the Representative, which will be applied against the accountable expenses that will be paid by us to the Representative in connection with this offering. The underwriting agreement provides that in the event the offering is terminated, the $25,000 expense deposit paid to the Representative will be returned to us to the extent that offering expenses are not actually incurred by the Representative.
We have agreed to pay the Representative an accountable expense allowance of $75,000, which shall cover the following accountable expenses in full: (a) all fees, expenses and disbursements relating to background checks of our officers and directors; (b) the cost associated with the use of Ipreo’s book building, prospectus tracking and compliance software for the offering; (c) the Representative’s “road show” expenses for the offering; (d) the costs associated with post-closing advertising of the offering in the national editions of the Wall Street Journal and New York Times, subject to the our approval; (e) the costs associated with bound volumes of the public offering materials as well as commemorative mementos and lucite tombstones in an amount not to exceed $5,000; (f) the fees and expenses of the underwriters’ legal counsel and other agents and representatives; and (g) such other expenses of the Representative not described above.
We estimate that the total expenses of the offering payable by us, excluding underwriting discounts and commissions, will be approximately $ .
Discretionary Accounts. The underwriters do not intend to confirm sales of the securities offered hereby to any accounts over which they have discretionary authority.
Lock-Up Agreements. Pursuant to certain “lock-up” agreements, we, our executive officers and directors, and holders of 5% or more of our outstanding shares of common stock (other than Eastern Capital) have agreed, subject to certain exceptions, not to offer, sell, assign, transfer, pledge, contract to sell, or otherwise dispose of or announce the intention to otherwise dispose of directly or indirectly, or enter into any swap, hedge or similar agreement or arrangement that transfers, in whole or in part, the economic risk of ownership of any common stock or securities convertible into or exchangeable or exercisable for any common stock, whether currently owned or subsequently acquired, without the prior written consent of the Representative, for a period of ninety (90) days from the date of effectiveness of the offering.
Right of First Refusal. Subject to certain limited exceptions, until twelve (12) months after the date of effectiveness of the registration statement of which this prospectus is a part, the Representative has a right of first refusal to purchase for its account or to sell for our account, or any subsidiary or successor, any securities of our company or any such subsidiary or successor which we or any subsidiary or successor may seek to sell in public or private equity and public debt offerings during such twelve (12)-month period.
Listing. Our common stock is presently listed on The NASDAQ Capital Market under the symbol “ASTM.”
Electronic Offer, Sale and Distribution of Shares. A prospectus in electronic format may be made available on the websites maintained by one or more of the underwriters or selling group members, if any, participating in this offering, and one or more of the underwriters participating in this offering may distribute prospectuses electronically. The Representative may agree to allocate a number of shares to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations. Other than the prospectus in electronic format, the information on these websites is not part of, nor incorporated by reference into, this prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or any underwriter in its capacity as underwriter, and should not be relied upon by investors.
Stabilization. In connection with this offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate-covering transactions, penalty bids and purchases to cover positions created by short sales.
· Stabilizing transactions permit bids to purchase shares so long as the stabilizing bids do not exceed a specified maximum, and are engaged in for the purpose of preventing or retarding a decline in the market price of the shares while the offering is in progress.
· Over-allotment transactions involve sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing shares in the open market.
· Syndicate covering transactions involve purchases of shares in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared with the price at which they may purchase shares through exercise of the over-allotment option. If the underwriters sell more shares than could be covered by exercise of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could be downward pressure on the price of the shares in the open market that could adversely affect investors who purchase in the offering.
· Penalty bids permit the Representative to reclaim a selling concession from a syndicate member when the shares originally sold by that syndicate member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions.
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our shares or common stock or preventing or retarding a decline in the market price of our shares or common stock. As a result, the price of our common stock in the open market may be higher than it would otherwise be in the absence of these transactions. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The NASDAQ Capital Market or otherwise and, if commenced, may be discontinued at any time.
Passive market making. In connection with this offering, underwriters and selling group members may engage in passive market making transactions in our common stock on The NASDAQ Capital Market in accordance with Rule 103 of Regulation M under the Exchange Act, during a period before the commencement of offers or sales of the shares and extending through the completion of the distribution. A passive market maker must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered below the passive market maker’s bid, then that bid must then be lowered when specified purchase limits are exceeded.
Other Relationships. Certain of the underwriters and their affiliates have provided, and may in the future provide, various investment banking, commercial banking and other financial services for us and our affiliates for which they have received, and may in the future receive, customary fees. However, except as disclosed in this prospectus, we have no present arrangements with any of the underwriters for any further services.
Offer Restrictions Outside the United States
Other than in the United States, no action has been taken by us or the underwriters that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to the offering and the distribution of this prospectus. This
prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter 6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer for the offeree under this prospectus.
China
The information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s Republic of China (PRC) (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly to “qualified domestic institutional investors.”
European Economic Area—Belgium, Germany, Luxembourg and Netherlands
The information in this document has been prepared on the basis that all offers of securities will be made pursuant to an exemption under the Directive 2003/71/EC (“Prospectus Directive”), as implemented in Member States of the European Economic Area (each, a “Relevant Member State”), from the requirement to produce a prospectus for offers of securities.
An offer to the public of securities has not been made, and may not be made, in a Relevant Member State except pursuant to one of the following exemptions under the Prospectus Directive as implemented in that Relevant Member State:
(a) to legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity that has two or more of (i) an average of at least 250 employees during its last fiscal year; (ii) a total balance sheet of more than €43,000,000 (as shown on its last annual unconsolidated or consolidated financial statements) and (iii) an annual net turnover of more than €50,000,000 (as shown on its last annual unconsolidated or consolidated financial statements);
(c) to fewer than 100 natural or legal persons (other than qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive) subject to obtaining the prior consent of the Company or any underwriter for any such offer; or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall result in a requirement for the publication by the Company of a prospectus pursuant to Article 3 of the Prospectus Directive.
France
This document is not being distributed in the context of a public offering of financial securities (offre au public de titres financiers) in France within the meaning of Article L.411-1 of the French Monetary and Financial Code (Code monétaire et financier) and Articles 211-1 et seq. of the General Regulation of the French Autorité des marchés financiers (“AMF”). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France.
This document and any other offering material relating to the securities have not been, and will not be, submitted to the AMF for approval in France and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in France.
Such offers, sales and distributions have been and shall only be made in France to (i) qualified investors (investisseurs qualifiés) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-1 to D.411-3, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation and/or (ii) a restricted number of non-qualified investors (cercle restreint d’investisseurs) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-4, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation.
Pursuant to Article 211-3 of the General Regulation of the AMF, investors in France are informed that the securities cannot be distributed (directly or indirectly) to the public by the investors otherwise than in accordance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 to L.621-8-3 of the French Monetary and Financial Code.
Ireland
The information in this document does not constitute a prospectus under any Irish laws or regulations and this document has not been filed with or approved by any Irish regulatory authority as the information has not been prepared in the context of a public offering of securities in Ireland within the meaning of the Irish Prospectus (Directive 2003/71/EC) Regulations 2005 (the “Prospectus Regulations”). The securities have not been offered or sold, and will not be offered, sold or delivered directly or indirectly in Ireland by way of a public offering, except to (i) qualified investors as defined in Regulation 2(l) of the Prospectus Regulations and (ii) fewer than 100 natural or legal persons who are not qualified investors.
Israel
The securities offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority, or the ISA, nor have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with the offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Italy
The offering of the securities in the Republic of Italy has not been authorized by the Italian Securities and Exchange Commission (Commissione Nazionale per le Società e la Borsa, “CONSOB”) pursuant to the Italian securities legislation and, accordingly, no offering material relating to the securities may be distributed in Italy and such securities may not be offered or sold in Italy in a public offer within the meaning of Article 1.1(t) of Legislative Decree No. 58 of 24 February 1998 (“Decree No. 58”), other than:
· to Italian qualified investors, as defined in Article 100 of Decree no. 58 by reference to Article 34-ter of CONSOB Regulation no. 11971 of 14 May 1999 (“Regulation no. 1197l”) as amended (“Qualified Investors”); and
· in other circumstances that are exempt from the rules on public offer pursuant to Article 100 of Decree No. 58 and Article 34-ter of Regulation No. 11971 as amended.
Any offer, sale or delivery of the securities or distribution of any offer document relating to the securities in Italy (excluding placements where a Qualified Investor solicits an offer from the issuer) under the paragraphs above must be:
· made by investment firms, banks or financial intermediaries permitted to conduct such activities in Italy in accordance with Legislative Decree No. 385 of 1 September 1993 (as amended), Decree No. 58, CONSOB Regulation No. 16190 of 29 October 2007 and any other applicable laws; and
· in compliance with all relevant Italian securities, tax and exchange controls and any other applicable laws.
Any subsequent distribution of the securities in Italy must be made in compliance with the public offer and prospectus requirement rules provided under Decree No. 58 and the Regulation No. 11971 as amended, unless an exception from those rules applies. Failure to comply with such rules may result in the sale of such securities being declared null and void and in the liability of the entity transferring the securities for any damages suffered by the investors.
Japan
The securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an exemption from the registration requirements applicable to a private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly, in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition by any such person of securities is conditional upon the execution of an agreement to that effect.
Portugal
This document is not being distributed in the context of a public offer of financial securities (oferta pública de valores mobiliários) in Portugal, within the meaning of Article 109 of the Portuguese Securities Code (Código dos Valores Mobiliários). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in Portugal. This document and any other offering material relating to the securities have not been, and will not be, submitted to the Portuguese Securities Market Commission (Comissão do Mercado de Valores Mobiliários) for approval in Portugal and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in Portugal, other than under circumstances that are deemed not to qualify as a public offer under the Portuguese Securities Code. Such offers, sales and distributions of securities in Portugal are limited to persons who are “qualified investors” (as defined in the Portuguese Securities Code). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Sweden
This document has not been, and will not be, registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority). Accordingly, this document may not be made available, nor may the securities be offered for sale in Sweden, other than under circumstances that are deemed not to require a prospectus under the Swedish Financial Instruments Trading Act (1991:980) (Sw. lag (1991:980) om handel med finansiella instrument). Any offering of securities in Sweden is limited to persons who are “qualified investors” (as defined in the Financial Instruments Trading Act). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority.
This document is personal to the recipient only and not for general circulation in Switzerland.
United Arab Emirates
Neither this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates, nor has the Company received authorization or licensing from the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to
market or sell the securities within the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. No services relating to the securities, including the receipt of applications and/or the allotment or redemption of such shares, may be rendered within the United Arab Emirates by the Company.
No offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
United Kingdom
Neither the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”)) has been published or is intended to be published in respect of the securities. This document is issued on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document, except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA.
This document should not be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in the United Kingdom.
Any invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply to us.
In the United Kingdom, this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005 (“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”). The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
Certain legal matters, including the legality of the securities offered, will be passed upon for us by Dykema Gossett PLLC, Ann Arbor, Michigan, acting as special counsel to the Company. In connection with the offering, other legal matters will be passed upon for us by Goodwin Procter LLP, Boston, Massachusetts. Certain legal matters in connection with this offering will be passed upon for the underwriters by Reed Smith LLP, New York, New York.
The consolidated financial statements and management’s assessment of the effectiveness of internal control over financial reporting (which is included in Management’s Report on Internal Control over Financial Reporting) incorporated in this Prospectus by reference to the Annual Report on Form 10-K for the year ended December 31, 2012 have been so incorporated in reliance on the report (which contains an explanatory paragraph relating to the Company’s ability to continue as a going concern as described in Note 1 to the consolidated financial statements) of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the Securities and Exchange Commission (SEC) a registration statement on Form S-1 under the Securities Act with respect to the securities offered by this prospectus. This prospectus, which forms a part of the registration statement, does not contain all the information set forth in the registration statement, as permitted by the rules and regulations of the SEC. For further information with respect to us and the securities offered by this prospectus, reference is made to the registration statement.
Statements contained in this prospectus as to the contents of any contract or other document that we have filed as an exhibit to the registration statement are qualified in their entirety by reference to the exhibits for a complete statement of their terms and conditions.
We are subject to the information requirements of the Exchange Act and, in accordance therewith, file annual, quarterly and special reports, proxy statements and other information with the SEC. You may read and copy any document we file, including the registration statement, at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. You may call the SEC at 1-800-SEC-0330 for further information on the operation of the Public Reference Room. These documents also may be accessed through the SEC’s electronic data gathering, analysis and retrieval system, or EDGAR, via electronic means, including the SEC’s home page on the Internet (www.sec.gov). You may also inspect the registration statement on this website.
This prospectus incorporates by reference important business and financial information that we file with the SEC and that we are not including in or delivering with this prospectus. As the SEC allows, incorporated documents are considered part of this prospectus, and we can disclose important information to you by referring you to those documents. We incorporate by reference the documents listed below:
· our annual report on Form 10-K for the period ended December 31, 2012, filed with the SEC on March 18, 2013;
· our quarterly reports on Form 10-Q for the quarters ended March 31, 2013, filed with the SEC on May 8, 2013, and June 30, 2013, filed with the SEC on August 7, 2013;
· our current reports on Form 8-K, filed with the SEC on March 8, 2013, March 29, 2013, April 9, 2013, April 23, 2013 (including exhibit 99.1 thereto), May 3, 2013, May 13, 2013, May 24, 2013, and August 12, 2013, respectively (excluding any information furnished in such reports under Item 2.02, Item 7.01 or Item 9.01);
· our definitive Proxy Statement on Schedule 14A for the Annual Meeting of Shareholders, filed with the SEC on March 22, 2013;
· the description of the rights to purchase shares of our Series A Junior Participating Cumulative Preferred Stock contained in the Registration Statement on Form 8-A, filed with the SEC on August 12, 2011, including any amendment or report for the purpose of updating such description; and
· the description of our common stock contained in our registration statement on Form S-1, filed with the SEC on November 1, 1996, including any amendment or report filed for the purpose of updating such description.
Pursuant to Rule 412 under the Securities Act, any statement contained in a document incorporated or deemed to be incorporated by reference into this prospectus will be deemed to be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus or any other subsequently filed document that is deemed to be incorporated by reference into this prospectus modifies or supersedes the statement. Any statement so modified or superseded will not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
You may request a copy of any or all of these filings, at no cost, by writing to us at: Aastrom Biosciences, Inc., 24 Frank Lloyd Wright Drive, Lobby K, Ann Arbor, Michigan 48105, Attention: Investor Relations, or by telephoning us at (800) 556-0311. These filings may also be obtained through our website located at http://www.aastrom.com. The reference to our website is intended to be an inactive textual reference and, except for the documents incorporated by reference as noted above, the information on, or accessible through, our website is not intended to be part of this prospectus.
You should rely only on the information incorporated by reference or provided in this prospectus or any supplement. We have not authorized anyone else to provide you with different information. You should not assume that information in this prospectus or any supplement is accurate as of any date other than the date on the front of these documents.
We advise that there have been no material changes in our affairs that have occurred since the end of the latest fiscal period for which audited financial statements were included in the latest Form 10-K and that have not been described in a Form 8-K filed under the Exchange Act.
|
TERM |
|
DEFINITION |
|
Adverse Event |
|
Any adverse change in health or “side-effect” that occurs in a person participating in a clinical trial, from the time they consent to joining the trial until a pre-specified period of time after their treatment has been completed. |
|
Autologous (Patient Specific) |
|
Originating from the patient receiving treatment. (Aastrom uses only autologous cells) |
|
BLA — Biologics License Application |
|
An application containing product safety, efficacy and manufacturing information required by the FDA to market biologics products in the United States. |
|
CLI — Critical Limb Ischemia |
|
A vascular disease characterized by insufficient blood flow in the lower extremities that causes severe pain, tissue loss or both. |
|
Chemistry, Manufacturing, and Control |
|
The composition, manufacture, and control of the drug substance and the drug product. It is information on the identification, quality, purity, and strength of the investigational product. |
|
Controlled Clinical Trial |
|
A clinical study that compares patients receiving a specific treatment to patients receiving an alternate treatment for the condition of interest. The alternate treatment may be another active treatment, standard of care for the condition and/or a placebo (inactive) treatment. |
|
DCM — Dilated Cardiomyopathy |
|
A chronic cardiac disease where expansion of the patient’s heart reduces the pumping function to a point that the normal circulation of blood cannot be maintained. |
|
Double-Blind Clinical Trial |
|
Clinical trials in which neither the patient nor the physician know if the patient received the experimental treatment or a control/placebo. |
|
FDA — Food & Drug Administration |
|
The U.S. FDA ensures that medicines, medical devices, and radiation-emitting consumer products are safe and effective. Authorized by Congress to enforce the Federal Food, Drug, and Cosmetic Act and several other public health laws, the agency monitors the manufacture, import, transport, storage, and sale of $1 trillion worth of goods annually. |
|
GMP — Good Manufacturing Practice |
|
GMP regulations require that manufacturers, processors, and packagers of drugs, medical devices, some food, and blood take proactive steps to ensure that their products are safe, pure, and effective. GMP regulations require a quality approach to manufacturing, enabling companies to minimize or eliminate instances of contamination, mix-ups, and errors. |
|
Hematopoietic Stem Cells |
|
Stem cells that give rise to all the blood cell types including myeloid (monocytes and macrophages, neutrophils, basophils, eosinophils, erythrocytes, megakaryocytes/platelets, dendritic cells), and lymphoid lineages (T-cells, B-cells, NK-cells). |
|
IMPACT-DCM |
|
Our U.S. Phase 2 dilated cardiomyopathy clinical trial. |
|
IND — Investigational New Drug |
|
An application submitted to the FDA for a new drug or biologic that, if allowed, will be used in a clinical trial. |
|
Ischemia |
|
A shortage or inadequate flow of blood to a body part (commonly an organ or tissue) caused by a constriction or obstruction of the blood vessels supplying it. |
|
LVEF — Left Ventricular Ejection Fraction |
|
The fraction of blood pumped out of the left ventricle with each heart beat. |
|
Mesenchymal stromal cells |
|
Connective tissue cells that, in the case of bone marrow derived MSC, function to support blood forming cells and secrete anti-inflammatory factors. |
|
TERM |
|
DEFINITION |
|
M2 anti-inflammatory macrophages |
|
Specialized blood cells that remove damaged tissue and bacteria and secrete anti-inflammatory factors. |
|
Open-label Clinical Trial |
|
A trial in which both the treating physician and the patient know whether they are receiving the experimental treatment or control/placebo treatment. |
|
|
|
|
|
Orphan Drug Designation |
|
“Orphan drug” refers to a drug or biologic that is intended for use in the treatment of a rare disease or condition. Orphan drug designation from the U.S. Food and Drug Association (FDA) qualifies the sponsor to receive certain benefits from the Government in exchange for developing the drug for a rare disease or condition. The drug must then go through the FDA marketing approval process like any other drug or biologic which evaluates for safety and efficacy. Usually a sponsor receives a quicker review time and lower application fees for an orphan product. |
|
Phase 1 Clinical Trial |
|
A Phase 1 trial represents an initial study in a small group of patients to test for safety and other relevant factors. |
|
Phase 2 Clinical Trial |
|
A Phase 2 trial represents a study in a moderate number of patients to assess the safety and efficacy of a product. |
|
Phase 2b Clinical Trial |
|
A Phase 2b trial is a moderately-sized Phase 2 trial that is more specifically designed assess the efficacy of a product than a Phase 2a trial. |
|
Phase 3 Clinical Trial |
|
Phase 3 studies are initiated to establish safety and efficacy in an expanded patient population at multiple clinical trial sites and are generally larger than trials in earlier phases of development. |
|
Prospective Clinical Trial |
|
A clinical trial in which participants are identified and then followed throughout the study going forward in time. |
|
Randomized Clinical Trial |
|
A clinical trial in which the participants are assigned randomly to different treatment groups. |
|
Somatic Cell |
|
Any of the cells responsible for forming the body of an organism such as internal organs, bones, skin, connective tissues and blood. |
|
Stem Cell |
|
Unspecialized (undifferentiated) cells that retain the ability to divide throughout a lifetime and give rise to more specialized (differentiated) cells which take the place of cells that die or are lost. In culture, these undifferentiated cells possess the ability to divide for indefinite periods in culture and may give rise to highly specialized cells. |
Shares of Common Stock
Warrants to Purchase Shares of Common Stock

Prospectus
Sole Book-Running Manager
Aegis Capital Corp
, 2013
Part II—INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The expenses payable by Aastrom Biosciences, Inc. (the “Registrant” or the “Company”) in connection with the issuance and distribution of the securities being registered (other than underwriting discounts and commissions, if any) are set forth below. Each item listed is estimated, except for the Securities and Exchange Commission (the “SEC”) registration fee and FINRA filing fee.
|
Securities and Exchange Commission registration fee |
|
$ |
3,137 |
|
|
FINRA filing fee |
|
|
3,950 |
|
|
NASDAQ listing fee |
|
65,000 |
| |
|
Legal fees and expenses |
|
150,000 |
| |
|
Accounting fees and expenses |
|
43,000 |
| |
|
Printing fees and expenses |
|
10,000 |
| |
|
Transfer agent and registrar fees |
|
5,000 |
| |
|
Accountable expense allowance |
|
75,000 |
| |
|
Non-accountable expense allowance |
|
150,000 |
| |
|
Miscellaneous fees and expenses |
|
4,913 |
| |
|
|
|
|
| |
|
Total |
|
$ |
510,000 |
|
Item 14. Indemnification of Directors and Officers
Sections 1561 through 1571 of the Michigan Business Corporation Act (the “MBCA”) authorize a corporation to grant or a court to award, indemnity to directors, officers, employees and agents in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities (including reimbursement for expenses incurred) arising under the Securities Act.
The Company’s amended and restated bylaws (the “Bylaws”) provide that the Company shall, to the fullest extent authorized or permitted by the MBCA, or other applicable law, indemnify a director or officer who was or is a party or is threatened to be made a party to any proceeding by or in the right of the Company to procure a judgment in its favor by reason of the fact that such person is or was a director, officer, employee or agent of the Company, against expenses, including actual and reasonable attorneys’ fees, and amounts paid in settlement incurred in connection with the action or suit, if the indemnitee acted in good faith and in a manner the person reasonably believed to be in, or not opposed to, the best interests of the Company or its shareholders. This section also requires the Company to advance expenses incurred by any agent of the Company in defending any proceeding prior to the final disposition of such proceeding upon receipt of an undertaking by or on behalf of the agent to repay such amount unless it shall be determined ultimately that the agent is entitled to be indemnified.
The Bylaws also authorize the Company to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the Company against any liability asserted against or incurred by such person in such capacity or arising out of such person’s status as such, regardless of whether the Company would have the power to indemnify such person against such liability under the provisions of the MBCA.
The Company has entered into indemnification agreements with certain individuals which contain provisions that may in some respects be broader than the specific indemnification provisions contained under applicable law. The indemnification agreement may require the Company, among other things, to indemnify such directors, officers and key personnel against certain liabilities that may arise by reason of their status or service as directors, officers or employees of the Company, to advance the expenses incurred by such parties as a result of any threatened claims or proceedings brought against them as to which they could be indemnified, and to the maximum extent that insurance coverage of such directors, officers and key employees under the Company’s directors’ and officers’ liability insurance policies is maintained.
Section 1209 of the MBCA permits a Michigan corporation to include in its articles of incorporation a provision eliminating or limiting a director’s liability to a corporation or its shareholders for monetary damages for breaches of
fiduciary duty. The enabling statute provides, however, that liability for breaches of the duty of loyalty, acts or omissions not in good faith or involving intentional misconduct or knowing violations of the law, or the receipt of improper personal benefits cannot be eliminated or limited in this manner. The Company’s Restated Articles of Incorporation (as amended, the “Charter”) includes a provision which eliminates, to the fullest extent permitted by the MBCA, director liability for monetary damages for breaches of fiduciary duty.
Item 15. Recent Sales of Unregistered Securities
The following is a summary of all securities that we have sold within the past three years without registration under the Securities Act of 1933, as amended.
On March 9, 2012, the Company entered into a Securities Purchase Agreement with Eastern Capital Limited, a Cayman exempted company (“ECL”), to sell 12,308 shares of Series B-1 Non-Voting Preferred Stock in a private placement to ECL, an “accredited investor” (as defined in Regulation D) under the Securities Act, at a price of $3,250.00 per share. The Series B-1 Shares were exchanged on a one-for-one basis for shares of the Series B-2 Voting Preferred Stock of the Company. The sales of the shares of Series B preferred stock were made only to a select number of accredited investors in reliance upon the exemptions from registration afforded by Rule 506 of Regulation D as promulgated by the SEC under the Securities Act and/or Section 4(2) of the Securities Act.
On June 27, 2012, the Company entered into separate warrant exchange agreements with each of certain holders of the Company’s outstanding warrants to purchase the Company’s common stock, issued in connection with the Company’s December 2010 public offering, with an exercise price of $3.22 and an expiration date of December 15, 2015. Pursuant to such warrant exchange agreements, on June 27, 2012, the Company issued an aggregate of 3,833,334 shares of Common Stock to Great Point Partners and its affiliated investment funds, Heights Capital Management and its affiliated investment funds, Deerfield Capital and its affiliated investment funds, and Millenium Management and its affiliated investment funds in exchange for the surrender of an aggregate of 7,666,666 warrants.
On July 30, 2012, the Company announced the results of its previously announced offer to exchange (the “Exchange Offer”) any warrant to purchase shares of common stock, no par value per share, of the Company issued in connection with the Company’s December 2010 public offering, that was tendered and accepted, for shares of the Company’s common stock. Such Exchange Offer was made upon the terms and subject to the conditions set forth in the Company’s offer to exchange, dated June 28, 2012, and in the related Exchange Offer materials filed as exhibits to the Tender Offer Statement on Schedule TO originally filed with the Securities and Exchange Commission on June 28, 2012, as amended. The Exchange Offer expired at 5:00 p.m., Eastern Standard Time, on Friday, July 27, 2012.
The issuance of shares of Common Stock in the warrant exchanges was made pursuant to the exemption from the registration requirements of the Securities Act of 1933, as amended, provided by Section 3(a)(9) of the Securities Act. No proceeds were received and no commissions were paid by the Company in connection with the Exchange Offer.
Item 16. Exhibits and Financial Statements Schedules
(a) Exhibits
A list of exhibits filed with this registration statement on Form S-1 is set forth on the Exhibit Index and is incorporated herein by reference.
(b) Financial Statement Schedules
All schedules have been omitted because either they are not required, are not applicable or the information is otherwise set forth in the financial statements and related notes thereto.
Item 17. Undertakings.
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement;
(2) To include any prospectus required by Section 10(a)(3) of the Securities Act;
(3) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement; and
(4) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(b) That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(c) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(d) The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, as amended, the information omitted from a form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933, as amended, shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, as amended, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, or the Act, may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant has duly caused this amendment no. 5 to registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Ann Arbor, state of Michigan on August 12, 2013.
|
|
AASTROM BIOSCIENCES, INC. | |
|
|
|
|
|
|
By: |
/s/ Dominick C. Colangelo |
|
|
|
Dominick C. Colangelo |
|
|
|
|
|
|
|
President and Chief Executive Officer |
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Dominick C. Colangelo |
|
President, Chief Executive Officer and Director |
|
August 12, 2013 |
|
Dominick C. Colangelo |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/s/ Brian D. Gibson |
|
Vice President of Finance, Chief Accounting Officer and Treasurer |
|
August 12, 2013 |
|
Brian D. Gibson |
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
|
|
* |
|
Chairman of the Board of Directors |
|
August 12, 2013 |
|
Robert L. Zerbe, M.D. |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
August 12, 2013 |
|
Ronald M. Creswell, Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
August 12, 2013 |
|
Alan L. Rubino |
|
|
|
|
|
|
|
|
|
|
|
* |
|
Director |
|
August 12, 2013 |
|
Nelson M. Sims |
|
|
|
|
|
|
|
|
|
|
|
*/s/ Dominick C. Colangelo |
|
|
|
August 12, 2013 |
|
Dominick C. Colangelo, Attorney-in-fact |
|
|
|
|
EXHIBIT INDEX
|
Exhibit |
|
Description |
|
1.1 # |
|
Form of Underwriting Agreement. |
|
|
|
|
|
3.1 |
|
Restated Articles of Incorporation of the Company, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on December 17, 2009 and incorporated herein by reference (File No. 000-22025). |
|
|
|
|
|
3.2 |
|
Certificate of Amendment to Restated Articles of Incorporation of the Company dated February 9, 2010, filed as Exhibit 3.2 to the Company’s Post-Effective Amendment No. 1 to Form S-1 filed on March 31, 2010 and incorporated herein by reference (File No. 333-160044). |
|
|
|
|
|
3.3 |
|
Certificate of Amendment to Restated Articles of Incorporation of the Company dated March 22, 2011, filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed on March 25, 2011 and incorporated herein by reference (File No. 000-22025). |
|
|
|
|
|
3.4 |
|
Certificate of Designation, Preferences and Rights, of the Company classifying and designating the Series A Junior Participating Cumulative Preferred Stock, attached as Exhibit 3.1 to the Company’s Current Report on Form 8-A filed on August 12, 2011, incorporated herein by reference. |
|
|
|
|
|
3.5 |
|
Amended and Restated Certificate of Designations, Preferences and Rights, of the Company classifying and designating the Series B-1 Non-Voting Convertible Preferred Stock and the Series B-2 Voting Convertible Preferred Stock, attached as Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on August 12, 2013, incorporated herein by reference. |
|
|
|
|
|
3.6 |
|
Amended and Restated Bylaws, filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on November 12, 2010 and incorporated herein by reference. |
|
|
|
|
|
4.1 |
|
Specimen Common Stock Certificate, filed as Exhibit 4.1 to Amendment No. 2 to the Company’s Registration Statement on Form S-1/A filed on December 20, 1996 and incorporated herein by reference. |
|
|
|
|
|
4.2 |
|
Shareholder Rights Agreement, dated as of August 11, 2011, between the Company and Continental Stock Transfer & Trust Company, as Rights Agent, attached as Exhibit 4.1 to the Company’s Current Report on Form 8-A filed on August 12, 2011, incorporated herein by reference. |
|
|
|
|
|
4.3 |
|
Amendment to Shareholder Rights Agreement, dated as of March 9, 2012, between the Company and Continental Stock Transfer & Trust Company, as Rights Agent, attached as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on March 9, 2012, incorporated herein by reference. |
|
|
|
|
|
4.4 * |
|
Form of Warrant. |
|
|
|
|
|
5.1 # |
|
Opinion of Dykema Gossett PLLC. |
|
|
|
|
|
10.1 |
|
Form of Indemnification Agreement, attached as Exhibit 10.1 to the Company’s Registration Statement on Form S-1 (No. 333-15415), filed on November 1, 1996, incorporated herein by reference. |
|
|
|
|
|
10.2 |
|
Amended and Restated 1992 Incentive and Non-Qualified Stock Option Plan and forms of agreements thereunder, attached as Exhibit 10.5 to the Company’s Registration Statement on Form S-1 (No. 333-15415), filed on November 1, 1996, incorporated herein by reference. |
|
10.3 |
|
Form of Employment Agreement, attached as Exhibit 10.8 to the Company’s Registration Statement on Form S-1 (No. 333-15415), filed on November 1, 1996, incorporated herein by reference. |
|
|
|
|
|
10.4 |
|
License Agreement, dated March 13, 1992, between the Company and the University of Michigan and amendments thereto dated March 13, 1992, October 8, 1993 and June 21, 1995, attached as Exhibit 10.17 to the Company’s Registration Statement on Form S-1 (No. 333-15415), filed on November 1, 1996, incorporated herein by reference. |
|
|
|
|
|
10.5 |
|
2001 Stock Option Plan, attached as Exhibit 10.72 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2002, incorporated herein by reference. |
|
|
|
|
|
10.6 |
|
2004 Equity Incentive Plan, attached as Exhibit 10.82 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q/A for the quarter ended September 30, 2004, incorporated herein by reference. |
|
|
|
|
|
10.7 |
|
Form of Option and Restricted Stock Award Agreements for Grants under 2004 Equity Incentive Plan, attached as Exhibit 10.84 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2005, incorporated herein by reference. |
|
|
|
|
|
10.8 |
|
Amendment dated December 5, 2002 to License Agreement with the University of Michigan, attached as Exhibit 10.87 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2005, incorporated herein by reference. |
|
|
|
|
|
10.9 |
|
2004 Equity Incentive Plan, as amended, attached as Exhibit 99.1 to the Company’s Current Report on Form 8-K filed on November 8, 2006, incorporated herein by reference. |
|
|
|
|
|
10.10 |
|
Forms of Grant Notice and Stock Option Agreement for Grants under 2004 Equity Incentive Plan, as amended, attached as Exhibit 99.2 to the Company’s Current Report on Form 8-K filed on November 8, 2006, incorporated herein by reference. |
|
|
|
|
|
10.11 |
|
Form of Purchase Agreement, attached as Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on October 16, 2007, incorporated herein by reference. |
|
|
|
|
|
10.12 |
|
Form of Warrant, attached as Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on October 16, 2007, incorporated herein by reference. |
|
|
|
|
|
10.13 |
|
Lease agreement between Domino’s Farms Office Park, LLC and the Company, as amended., attached as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 9, 2013, incorporated herein by reference. |
|
|
|
|
|
10.14 |
|
2009 Omnibus Incentive Plan, attached as Appendix II to the Company’s Proxy Statement filed on October 9, 2009, incorporated herein by reference. |
|
|
|
|
|
10.15 |
|
Class A Warrant Agreement, dated as of January 21, 2010, by and between the Company and Continental Stock Transfer & Trust Company (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed with the SEC on January 27, 2010). |
|
|
|
|
|
10.16 |
|
Class B Warrant Agreement, dated as of January 21, 2010, by and between the Company and Continental Stock Transfer & Trust Company (incorporated herein by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K filed with the SEC on January 27, 2010). |
|
|
|
|
|
10.17 |
|
Underwriting Agreement, dated as of January 15, 2010, and between the Company and Oppenheimer & Co. Inc. (incorporated herein by reference to Exhibit 1.1 to the Company’s Current Report on Form 8-K filed |
|
|
|
on January 15, 2010). |
|
|
|
|
|
10.18 |
|
Form of indemnification agreement entered into between the Company and each of its directors, attached as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 31, 2010, incorporated herein by reference. |
|
|
|
|
|
10.19 |
|
Amended Code of Business Conduct and Ethics, attached as Exhibit 14.1 to the Company’s Current Report on Form 8-K filed on August 31, 2010, incorporated herein by reference. |
|
|
|
|
|
10.20 |
|
Contract Manufacturing and Supply Agreement, dated as of November 1, 2010, by and between Vention Medical (formerly ATEK Medical, LLC) and the Company (incorporated herein by reference to Exhibit 10.30 to the Company’s Annual Report on Form 10-KT for the year ended December 31, 2010). |
|
|
|
|
|
10.21 |
|
Warrant Agreement, dated as of December 15, 2010, by and between the Company and Continental Stock Transfer & Trust Company (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on December 16, 2010). |
|
|
|
|
|
10.22 |
|
Underwriting Agreement, dated as of December 10, 2010, and between the Company and Stifel, Nicolaus & Company, Incorporated, Needham & Company, LLC and Roth Capital Partners (incorporated herein by reference to Exhibit 1.1 to the Company’s Current Report on Form 8-K filed on December 10, 2010). |
|
|
|
|
|
10.23 |
|
Amendment to the 2009 Omnibus Incentive Plan, dated March 21, 2011 (incorporated herein by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K, filed on March 25, 2011). |
|
|
|
|
|
10.24 |
|
Employment Agreement with Ronnda L. Bartel, PhD, dated March 22, 2011 (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on March 25, 2011). |
|
|
|
|
|
10.25 |
|
Senior Executive Incentive Bonus Plan (incorporated herein by reference to Exhibit 10.3 to the Company’s current Report on Form 8-K, filed on March 25, 2011). |
|
|
|
|
|
10.26 |
|
At Market Issuance Sales Agreement, dated June 16, 2011, by and among the Company and McNicoll, Lewis & Vlak LLC (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 16, 2011). |
|
|
|
|
|
10.27 |
|
Master Services Agreement by and between the Company and PPD, made and entered into as of September 23, 2011 (the “Master Services Agreement”) (incorporated herein by reference to Exhibit 10.28 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2012). |
|
|
|
|
|
10.28 |
|
Project Addendum to the Master Services Agreement, dated as of November 16, 2011 (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed November 22, 2011). |
|
|
|
|
|
10.29 |
|
Registration Rights Agreement, dated March 9, 2012, between the Company and Eastern Capital Limited, attached as Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on March 9, 2012, incorporated herein by reference. |
|
|
|
|
|
10.30 |
|
Securities Purchase Agreement, dated as of March 9, 2012, by and between the Company and Eastern Capital Limited (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on March 9, 2012). |
|
|
|
|
|
10.31 |
|
Employment Agreement, dated as of April 3, 2013, by and between the Company and Daniel R. Orlando (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on |
|
|
|
April 9, 2013). |
|
|
|
|
|
10.32 |
|
Employment Agreement, dated as of October 26, 2012, by and between the Company and Brian Gibson (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 26, 2012). |
|
|
|
|
|
10.33 |
|
Amendment to the 2009 Omnibus Incentive Plan, dated May 3, 2012 (incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q, filed on August 7, 2012). |
|
|
|
|
|
10.34 |
|
Form of Warrant Exchange Agreement, dated June 27, 2012 (incorporated herein by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed on June 27, 2012). |
|
|
|
|
|
10.35 |
|
Executive Resignation Agreement, executed on December 12, 2012 and effective December 14, 2012, by and between the Company and Tim M. Mayleben (incorporated herein by reference to Exhibit 10.30 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2012). |
|
|
|
|
|
10.36 |
|
Executive Employment Agreement, executed March 4, 2013 and effective March 1, 2013, by and between the Company and Dominick C. Colangelo (incorporated herein by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed on March 8, 2013). |
|
23.1# |
|
Consent of PricewaterhouseCoopers LLP. |
|
|
|
|
|
23.2# |
|
Consent of Dykema Gossett PLLC (included in Exhibit 5.1 hereto). |
|
|
|
|
|
24.1* |
|
Power of Attorney (included in signature pages to this Registration Statement). |
# Filed herewith.
* Previously filed.