Attached files
| file | filename |
|---|---|
| EX-3.2 - FORM OF THIRD AMENDED AND RESTATED CERTIFICATE OF INCORPORATION - Vyant Bio, Inc. | d254016dex32.htm |
| EX-23.1 - CONSENT OF MCGLADREY LLP - Vyant Bio, Inc. | d254016dex231.htm |
| EX-10.61 - AMENDMENT NO. 2 TO AFFILIATION AGREEMENT - Vyant Bio, Inc. | d254016dex1061.htm |
| EX-10.62 - DESCRIPTION OF AMENDMENT TO LETTER AGREEMENT - Vyant Bio, Inc. | d254016dex1062.htm |
Table of Contents
As filed with the Securities and Exchange Commission on January 8, 2013
Registration No. 333-178836
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 11 to
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
CANCER GENETICS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 8071 | 04-3462475 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
201 Route 17 North 2nd Floor
Rutherford, NJ 07070
(201) 528-9200
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Panna L. Sharma
Chief Executive Officer
Cancer Genetics, Inc.
201 Route 17 North 2nd Floor
Rutherford, NJ 07070
(201) 528-9200
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Alan Wovsaniker Meredith Prithviraj Andrea Schreiber Lowenstein Sandler LLP 65 Livingston Avenue Roseland, NJ 07068 (973) 597-2564 |
Yvan-Claude Pierre Daniel I. Goldberg Reed Smith LLP 599 Lexington Ave New York, NY 10022 (212) 521-5400 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities, nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS | SUBJECT TO COMPLETION | DATED JANUARY 8, 2013 |
5,000,000 Shares
Common Stock

This is an initial public offering of 5,000,000 shares of common stock by Cancer Genetics, Inc. No public market currently exists for our common stock. We anticipate that the initial public offering price per share of our common stock will be between $6.00 and $8.00 per share.
We expect to effect a 1-for-2 reverse stock split of our outstanding common stock just prior to the date of this prospectus. We have applied to have our shares of common stock approved for quotation on the NASDAQ Capital Market under the symbol “CGIX.”
We are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”) and, as such, may elect to comply with certain reduced public company reporting requirements for future filings.
Investing in our common stock involves risk. See “Risk Factors” beginning on page 10 of this prospectus for a discussion of information that should be considered in connection with an investment in our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Discounts and commissions to underwriters(1) |
$ | $ | ||||||
| Offering proceeds to us, before expenses |
$ | $ | ||||||
| (1) | The underwriters will receive compensation in addition to the underwriting discount. See “Underwriting” beginning on page 166 of this prospectus for a description of compensation payable to the underwriters. |
We have granted a 45-day option to the representative of the underwriters to purchase up to 750,000 additional shares of common stock solely to cover over-allotments, if any.
The underwriters expect to deliver the shares against payment therefor on or about , 2013.
| Aegis Capital Corp | Feltl and Company |
, 2013
Table of Contents

Table of Contents
| Page | ||||
| 1 | ||||
| 8 | ||||
| 10 | ||||
| 40 | ||||
| 42 | ||||
| 44 | ||||
| 45 | ||||
| 47 | ||||
| 49 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
51 | |||
| 78 | ||||
| 118 | ||||
| 126 | ||||
| 136 | ||||
| 142 | ||||
| 144 | ||||
| 151 | ||||
| MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF OUR COMMON STOCK |
154 | |||
| 158 | ||||
| 166 | ||||
| 166 | ||||
| 166 | ||||
| 167 | ||||
| F-1 | ||||
You should rely only on the information contained in this prospectus. We have not, and the underwriters have not, authorized anyone to provide you with different information. We are not making an offer of these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information contained in this prospectus is accurate as of the date on the front of this prospectus only. Our business, financial condition, results of operations and prospects may have changed since that date.
Information contained in our website does not constitute part of this prospectus.
We use MatBA®, UroGenRA™, UGenRA™, LeukA™, FHACT™, FReCAD™, Expand DX™, Select One™, Summation™ Report and the Cancer Genetics logo as trademarks in the United States and elsewhere. All other trademarks or trade names referred to in this prospectus are the property of their respective owners.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that they have gathered their information from sources they believe to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe that these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data.
Table of Contents
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should read this entire prospectus carefully, especially the “Risk Factors” section of this prospectus and the consolidated financial statements and related notes appearing at the end of this prospectus before making an investment decision.
Unless the context provides otherwise, all references in this prospectus to “Cancer Genetics,” “CGI,” “we,” “us,” “our,” the “Company,” or similar terms, refer to Cancer Genetics, Inc. and its wholly owned subsidiary, Cancer Genetics Italia, S.r.L.
Unless otherwise indicated, all share amounts and per share amounts in this prospectus have been presented on a pro forma basis as if a reverse stock split of our outstanding shares of common stock at a ratio of 1-for-2, that we expect to effect just prior to the date of this prospectus, has occurred.
Our Company
We are an early-stage diagnostics company focused on developing and commercializing proprietary genomic tests and services to improve and personalize the diagnosis, prognosis and response to treatment (theranosis) of cancer. The proprietary tests we are developing target cancers that are difficult to prognose and predict treatment outcomes by using currently available mainstream techniques. These cancers include hematological, urogenital and HPV-associated cancers. We recently have begun to provide our proprietary tests and services along with a comprehensive range of non-proprietary oncology-focused tests and laboratory services that we have provided historically to oncologists and pathologists at hospitals, cancer centers, and physician offices. We are currently offering our tests and laboratory services in our 17,936 square foot state-of-the-art laboratory located in Rutherford, New Jersey, which has been accredited under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) to perform high complexity testing.
Our proprietary tests are based principally on our expertise in specific cancer types, test development methodologies and proprietary algorithms correlating genetic events with disease specific information. During the first quarter of 2011, we received CLIA approval for, and commercially launched, MatBA®-CLL, our first proprietary microarray test for chronic lymphocytic leukemia (“CLL”) for use in our CLIA-accredited clinical laboratory. In January 2012, we received CLIA approval for MatBA®-SLL, our proprietary microarray for risk stratification in small lymphocytic lymphoma (“SLL”), and we are currently offering MatBA®-SLL in our laboratory. In addition, we are developing a series of other proprietary genomic tests in our core oncology markets.
We have established collaborative relationships with key thought leaders in oncology, which enable us to develop and validate the effectiveness and utility of our tests in a clinical setting and which provide us access to clinically robust patient data. For example, we agreed to form a joint venture in March 2013 with Mayo Foundation for Medical Education and Research (“Mayo”) focused on developing oncology diagnostic services and tests utilizing next-generation sequencing. Additionally, we agreed to a research collaboration with the Cleveland Clinic to validate our renal-cancer microarray UroGenRA™ Renal.
We believe that we can be successful by offering cancer professionals a fully-integrated menu of oncology-focused proprietary and non-proprietary tests and customized laboratory services. Based on our discussions with leading researchers in the oncology field and our interactions with our collaborators, as well as information we learn through performing the nonproprietary genetic diagnostic testing services which are focused on the specific oncology categories where we are developing our proprietary tests, we provide to our
-1-
Table of Contents
customers, we believe that our proprietary tests provide superior diagnostic and prognostic values than currently available tests and services. We believe our ability to rapidly translate research insights about the genetics and molecular mechanisms of cancer into the clinical setting will improve patient treatment and management and that this approach will become a key component in the standard of care for personalized cancer treatment.
Market Overview
Despite many advances in the treatment of cancer, it remains one of the greatest areas of unmet medical need. The World Health Organization attributed 7.6 million deaths worldwide to cancer-related causes in 2008. In addition to the human toll, the financial cost of cancer is overwhelming. An independent study published in 2010 and conducted jointly by the American Cancer Society and LIVESTRONG ranked cancer as the most economically devastating cause of death in the world—estimated to be as high as $895 billion globally in 2008.
Cancer constitutes a heterogeneous class of diseases characterized by uncontrollable cell growth and results from a combination of environmental and hereditary risk factors. It has only been in recent years that technology has sufficiently advanced to enable researchers to understand many cancers at a molecular level and attribute specific cancers to genetic mechanisms.
Limitations of Traditional Cancer Diagnostics
Cancer is difficult to diagnose due to its varying morphology and genetic complexity. Traditional methods of diagnosis, routinely used as the initial step in cancer detection, involve a pathologist examining a thin slice of potentially cancerous tissue under a microscope. A relatively new tissue sample must be used along with chemical staining techniques to view the biopsy. Through visual inspection, the pathologist determines whether the biopsy contains normal or cancerous cells. Cells that are deemed cancerous are graded on a level of progression of disease and aggressiveness.
Use of Genomic-Based Analysis in Cancer Diagnosis and Treatment
Molecular diagnostic tests for cancer aim to remove subjectivity from the diagnostic phase, and add prognostic information, thereby enabling personalized treatments based on cancer analysis at its most basic genetic level. These tests both define the cancer subtype and help determine the best course of treatment by detecting genetic mutations, gene fusions and DNA copy number changes, all of which are possible causes of or precursors to malignant growth. An important method of measuring changes in the genomic profile of cancer cells is copy number variation. This method measures the gain or loss of DNA within specific regions of chromosomes and is commonly performed using DNA microarrays and probes.
Our Proprietary Genomic Tests and Services
Our clinical laboratory is accredited under CLIA to perform our first proprietary test, MatBA®-CLL, which is also, to our knowledge based on our informal communications with New York State Department of Health personnel, the first oncology microarray to be approved by the New York State Department of Health, one of the only state governmental agencies that reviews the clinical utility of new laboratory developed tests (“LDTs”). The test has been validated by us in a clinical study using over 320 CLL specimens in conjunction with a leading CLL thought leader, Dr. Kanti Rai at Long Island Jewish / North Shore Hospital. Another data set of over 200 DLBCL specimens is being analyzed for additional biomarkers in conjunction with Dr. Julie Teruya-Feldstein at Memorial Sloan-Kettering Cancer Center. There are approximately 14,500 new cases of CLL diagnosed in the United States each year, and these cases require risk stratification and guidance on patient management and treatment issues at multiple points during the course of the disease. Prior to the introduction of MatBA®-CLL, clinicians had to rely on diagnostic tests that provided limited information on the genetic
-2-
Table of Contents
abnormalities associated with CLL. In contrast, MatBA®-CLL identifies a much broader range of genomic markers associated with CLL, providing improved diagnostic and prognostic value and critical information for clinicians to consider in planning patient treatment. The MatBA® platform was developed by us under the guidance of Dr. Raju Chaganti, our Chairman and one of our founders. Dr. Chaganti founded one of the earliest comprehensive clinical cytogenetic laboratories focused on cancer in the United States at Memorial Sloan-Kettering Cancer Center, where he is on the faculty of the Department of Medicine and Cell Biology Program and the incumbent of the William E. Snee Chair.
In collaboration with Memorial Sloan-Kettering Cancer Center and Long Island Jewish / North Shore Hospital, we have completed the validation of MatBA®-SLL and are now offering MatBA®-SLL in our laboratory. We are also validating the MatBA® microarray in a variety of additional lymphoma subtypes, including mantle-cell lymphoma (“MCL”), follicular lymphoma (“FL”), and diffuse large B cell lymphoma (“DLBCL”). Collectively, these lymphomas represent over 70% of the mature B cell cancers (neoplasms) and over 66,000 newly diagnosed cancer cases each year in the United States. Our MatBA® array has been designed to measure genetic markers at 80 specific genomic sites where genetic alterations are associated with mature B cell neoplasms.
We are also developing microarray tests for the diagnosis, prognosis and theranosis of a range of urogenital cancers. These include the UroGenRA™ microarray for kidney, prostate and bladder cancers and the UGenRA™ microarray for endometrial (lining of the uterus), ovarian and cervical cancers. UroGenRA™ detects genomic changes in over 100 regions of the human genome with potential diagnostic and/or prognostic value in one or more of these types of cancer. We have initiated clinical validation for UroGenRA™ targeting kidney and prostate cancers in collaboration with Memorial Sloan-Kettering Cancer Center. In addition, we have initiated a clinical validation for UGenRA™ targeting kidney cancer in collaboration with the Cleveland Clinic. Our UGenRA™ microarray has been designed as a platform to detect genomic changes occurring in 83 regions of the human genome that have been linked to endometrial, ovarian and cervical cancers. In addition, we develop and manufacture a portfolio of fluorescence in situ hybridization (“FISH”) based DNA probes focused on blood-based and solid cancers that we currently sell outside the United States. We have filed five patent applications with the U.S. Patent and Trademark Office and two international (PCT) application covering our microarrays. We also have two issued U.S. patents, a U.S. patent application, a European application and a Canadian application (which has been allowed) covering our other proprietary probe products.
We are an early-stage company and recently launched our first proprietary microarray tests, MatBA®-CLL and MatBA®-SLL, for use in our CLIA-accredited clinical laboratory. To date, we have engaged in only limited sales and marketing activities and have generated most of our revenue through sales of our non-proprietary oncology testing services to a limited number of oncologists, pathologists and community hospitals located mostly in the eastern and midwestern United States. In 2011, we generated approximately 87% of our revenue from laboratory services, approximately 10% from government grants and approximately 3% from sales of our DNA probes, which are currently only sold outside the United States. In 2010, we generated approximately 95% of our revenue from our laboratory services, approximately 4% from government grants and approximately 1% from sales of our DNA probes. Our non-proprietary laboratory testing services include molecular testing, sequencing, mutational analysis, flow cytometry testing, histology testing and cytology testing and they are described in more detail in the section entitled “Description of the Business-Laboratory Services”. We also utilize our clinical laboratory to provide clinical trial services to biopharmaceutical companies and clinical research organizations to improve the efficiency and economic viability of their clinical trials. This service was branded “Select One™” in December 2011.
The non-proprietary testing services offered by us are entirely focused on specific oncology categories where we are developing our proprietary arrays and probe panels. We believe that there is significant synergy in developing and marketing a complete set of tests and services that are disease-focused and delivering those tests
-3-
Table of Contents
and services in a comprehensive manner to help with treatment decisions. The insights that we develop in delivering the non-proprietary services are often leveraged in the development of our proprietary programs and now increasingly in the validation of our proprietary programs (such as MatBA®) for clinical use.
In this prospectus, we use the terms “microarray test,” “oncology microarray” and “DNA microarray” interchangeably to refer to DNA-based tests that focus on multiple targets in the genomic sequence of a cancer cell. We use the terms “probe”, “DNA probe” or “FISH-based DNA probe” interchangeably to refer to DNA-based tests that focus on a single genomic abnormality. Finally, the terms “tests” and “tests and services” are used throughout this prospectus to refer to all of our laboratory tests, whether microarrays, probes, other genomic-based tests or other laboratory tests or services that we offer in our laboratory.
Our Strategy
Our objective is to be a leader in the development and commercialization of proprietary genomic tests and services. We aim to provide a full service solution for oncology professionals to improve the diagnosis, prognosis, theranosis and treatment of hematological, urogenital and HPV-associated cancers. To achieve this objective, we intend to:
| • | develop and commercialize additional proprietary genomic tests and services; |
| • | develop and expand our collaborations with leading universities and research centers; |
| • | continue to focus on rapidly applying genomic research to routine clinical cancer diagnostics (translational oncology) in order to expand and improve our proprietary genomic tests and services; |
| • | enhance our efforts in partnering with community hospitals in order to provide our tests to a broader patient base; |
| • | expand our scalable sales and marketing capabilities; |
| • | obtain protection for the intellectual property utilized in our proprietary tests and regulatory approvals and clearances required to sell our proprietary tests for use in other oncology testing centers; and |
| • | continue to reduce the costs associated with the development, manufacture and interpretation of our proprietary genomic tests and services by partnering with leading technology and service providers. |
We will continue offering our proprietary tests in the United States as LDTs and internationally as CE-marked in vitro diagnostic products. In addition, as part of our long term strategy, we plan to seek Food and Drug Administration (“FDA”) clearance or approval to expand the commercial use of our tests to other laboratories and testing sites. We believe it would likely take two years or more to conduct the studies and trials necessary to obtain approval from FDA to commercially launch MatBA®-CLL and MatBA®-SLL outside of our clinical laboratory. Our sales strategy is focused on direct sales to oncologists and pathologists at hospitals, cancer centers and physician offices in the United States, and expanding our relationships with leading distributors and medical facilities in emerging markets. We intend to emphasize partnering with community hospitals, where approximately 85% of all cancer patients in the United States are initially diagnosed, through our program called Expand Dx™, which was specifically designed to meet the needs of community hospitals. We believe our proprietary tests and services will enable community hospitals to optimize and expand their oncology services to better serve their cancer patients.
-4-
Table of Contents
Risks That We Face
An investment in our common stock involves a high degree of risk. You should carefully consider the risks summarized below. The risks are discussed more fully in the “Risk Factors” section of this prospectus immediately following this prospectus summary. These risks include, but are not limited to, the following:
| • | we are an early-stage company with a cumulative net loss through September 30, 2012 of approximately $44.9 million and we may never achieve sustained profitability; |
| • | our business depends upon our ability to increase sales of our laboratory tests and services; |
| • | we need to clinically validate our MatBA® pipeline of microarray tests currently in development; |
| • | our business depends on our ability to continually develop and commercialize novel and innovative diagnostic cancer tests and services; |
| • | our business depends on executing on our sales and marketing strategy for our proprietary tests and gaining acceptance of our tests in the market; |
| • | our business depends on satisfying United States (including FDA) and international regulatory requirements with respect to our tests and services and many of these requirements are new and still evolving; |
| • | our business depends on being able to obtain adequate reimbursement from governmental and other third-party payors for our tests and services (for the year ended December 31, 2011, approximately 24% of our revenues came from Medicare or Medicaid, approximately 12% of our revenue came from direct bill customers and 54% of our revenues came from private insurance carriers and other third party payors); |
| • | our business depends on our ability to effectively compete with other genomic-based diagnostic tests and services that now exist or may hereafter be developed; |
| • | we need to maintain our clinical collaborations and enter into new collaboration agreements with highly regarded organizations in the cancer field in order to, among other things, have access to both thought leaders in the field and samples to validate our proprietary tests; |
| • | we depend on our ability to attract and retain scientists, clinicians and sales personnel with extensive experience in oncology, who are in short supply; |
| • | we may need additional financing to meet our liquidity needs; |
| • | we need to obtain or maintain patents or other appropriate protection for the intellectual property utilized in our proprietary tests and services; and |
| • | approximately $5.0 million of the net proceeds of this offering will be used to repay outstanding indebtedness. |
Company Information
We maintain our principal executive offices at 201 Route 17 North, 2nd Floor, Rutherford, New Jersey 07070. Our telephone number is (201) 528-9200 and our website address is www.cancergenetics.com. The information contained in, and that can be accessed through, our website is not incorporated into and is not part of this prospectus.
-5-
Table of Contents
The Offering
| Common stock offered by us |
5,000,000 shares |
| Over-allotment option |
We have granted the underwriters a 45-day option to purchase up to 750,000 additional shares of our common stock from us at the initial public offering price less underwriting discounts and commissions. The option may be exercised only to cover any over-allotments. |
| Common stock outstanding after this offering |
11,414,665 shares |
| Use of proceeds |
We estimate that the net proceeds from our sale of shares of our common stock in this offering will be approximately $29.3 million, or approximately $34.2 million if the underwriters exercise their over-allotment option in full, based upon an assumed initial public offering price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Of these estimated offering expenses, we have already paid approximately $1.3 million and therefore will have approximately $1.3 million of additional proceeds from this offering available to us. We currently expect to use the net proceeds from this offering as follows: |
| • | approximately $5.0 million to repay certain outstanding indebtedness; |
| • | approximately $5.0 million to fund further research and development, potential regulatory submissions and potential commercial launch of our proprietary genomic-based diagnostic tests, and potential collaborations; |
| • | approximately $7.0 million to hire additional sales and marketing personnel and support increased sales and marketing activities; |
| • | $2.0 million to fund our initial contribution to our joint venture with Mayo; and |
| • | approximately $10.0 million to fund working capital for ongoing operations and expansion of the business. |
| Risk Factors |
See the section entitled “Risk Factors” beginning on page 10 of this prospectus for a discussion of factors you should carefully consider before deciding to invest in our common stock. |
| Proposed NASDAQ Capital Market symbol |
CGIX |
The number of shares of our common stock that will be outstanding immediately after this offering is based on 6,414,665 shares of common stock outstanding as of December 31, 2012, which number gives effect to the 1-for-2 reverse stock split described below (and is subject to adjustment for fractional shares), and assumes that convertible
-6-
Table of Contents
promissory notes in the amount of $6.0 million will convert at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, and all outstanding shares of our convertible preferred stock convert into shares of our common stock upon the closing of this initial public offering, and excludes:
| • | 1,383,350 shares of our common stock issuable upon the exercise of stock options as of December 31, 2012, with a weighted average exercise price of $5.10 per share, which includes 1,183,350 shares of our common stock issuable upon the exercise of stock options issued under our equity incentive plans and 200,000 shares of our common stock issuable upon the exercise of stock options issued outside of our equity incentive plans; |
| • | 3,942,198 additional shares of our common stock issuable upon the exercise of outstanding warrants as of December 31, 2012, at a weighted average exercise price of $6.15 per share; |
| • | 1,066,650 additional shares of our common stock reserved for future issuance under our equity incentive plans as of December 31, 2012, of which 133,750 shares have been reserved for option grants with an exercise price equal to the initial public offering price per share to be issued upon consummation of the initial public offering; and |
| • | 2,674 shares of common stock issuable to Cleveland Clinic pursuant to our license agreement with Cleveland Clinic. |
Except for historical financial information or as otherwise indicated herein, all information in this prospectus, including the number of shares that will be outstanding after this offering, assumes or gives effect to:
| • | the conversion of all outstanding shares of our convertible preferred stock into an aggregate of 2,182,680 shares of our common stock, assuming an initial public offering price of $7.00 per share, which is the midpoint of the range set forth in the cover page of this prospectus, and which will occur automatically upon the closing of this offering if we raise at least $15.0 million in this offering; |
| • | the conversion of convertible promissory notes in the amount of $6.0 million at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, into an aggregate of 857,143 shares of our common stock upon the closing of this offering; |
| • | the adoption of our amended and restated certificate of incorporation (“certificate of incorporation”) and our amended and restated by-laws, to be effective upon the closing of this offering; and |
| • | no exercise by the underwriters of their option to purchase up to 750,000 additional shares of our common stock from us in this offering. |
We anticipate effecting a 1-for-2 reverse stock split just prior to the date of this prospectus. Unless we indicate otherwise, all references to share numbers in this prospectus reflect the effects of this reverse stock split. In addition, if the initial public offering price were $6.00 per share, which is the bottom of the range set forth on the cover page of this prospectus, the number of shares outstanding as of December 31, 2012 would increase by 5.6% to 6,774,379 and the number of warrants outstanding as of December 31, 2012 would increase by 11.0% to 4,377,310 at a weighted average exercise price of $5.41 per share, as a result of certain anti-dilution provisions in certain preferred stock, convertible notes and warrants. If the initial public offering price were $8.00 per share, which is the top of the range set forth on the cover page of this prospectus, the number of shares outstanding as of December 31, 2012 would decrease by 4.2% to 6,144,879 and the number of warrants would decrease by 8.3% to 3,615,861 at a weighted average exercise price of $6.86 per share as a result of certain anti-dilution provisions in certain preferred stock, convertible notes and warrants.
-7-
Table of Contents
SUMMARY CONSOLIDATED FINANCIAL DATA
The following table sets forth our summary statement of operations data for the years ended December 31, 2011, 2010 and 2009 derived from our audited consolidated financial statements and related notes included elsewhere in this prospectus. Our financial statements are prepared and presented in accordance with generally accepted accounting principles in the United States. The unaudited selected consolidated statements of operations data for the nine months ended September 30, 2011 and 2012, and the unaudited consolidated balance sheet data as of September 30, 2012, are derived from our unaudited consolidated financial statements, which are included elsewhere in this prospectus. We have included all adjustments, consisting only of normal recurring adjustments, which we consider necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results to be expected for any future periods and our interim results are not necessarily indicative of the results to be expected for the full fiscal year. Pro forma net loss per common share and the pro forma balance sheet data have been calculated assuming the conversion of all outstanding shares of our preferred stock into 2,182,680 shares of common stock upon completion of this offering and the conversion of convertible promissory notes in the amount of $6.0 million, including $2.0 million issued after September 30, 2012, at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, into an aggregate of 857,143 shares of our common stock and the reclassification of certain warrants with a fair value of $4.9 million to equity due to the lapsing of anti-dilution provisions upon the offering date. Pro forma net loss per share does not include the recognition of noncash debt discounts and beneficial conversion amounts that would occur upon conversion of the promissory notes. The pro forma as adjusted balance sheet data reflects the balance sheet data at September 30, 2012 as adjusted to reflect our receipt of the net proceeds from the sale by us in this offering of 5,000,000 shares of common stock at an assumed initial public offering price of $7.00 per share, the midpoint of the estimated price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us and conversion of all of our outstanding shares of preferred stock into 2,182,680 shares of common stock upon completion of this offering, the conversion of $6.0 million of convertible promissory notes into 857,143 shares of common stock upon the completion of our initial public offering and the repayment of certain indebtedness. You should read this information together with the sections entitled “Capitalization,” “Selected Consolidated Financial Data,” “Management’s Discussion and Analysis of Financial Condition & Results of Operations” and our consolidated financial statements and related notes included elsewhere in this prospectus.
-8-
Table of Contents
| Nine Months Ended | Year Ended | |||||||||||||||||||
| September 30, | December 31, | |||||||||||||||||||
| 2012 | 2011 | 2011 | 2010 | 2009 | ||||||||||||||||
| (dollars in thousands, except share and per share data) | (unaudited) | |||||||||||||||||||
| STATEMENT OF OPERATIONS DATA: |
||||||||||||||||||||
| Revenue |
$ | 3,226 | $ | 2,144 | $ | 3,019 | $ | 2,522 | $ | 1,666 | ||||||||||
| Cost of revenues |
2,880 | 2,385 | 3,117 | 3,516 | 2,532 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Gross Profit |
346 | (241 | ) | (98 | ) | (995 | ) | (866 | ) | |||||||||||
| Operating Expenses |
||||||||||||||||||||
| Research and development |
1,552 | 1,442 | 2,074 | 1,167 | 1,336 | |||||||||||||||
| General and administrative |
3,475 | 3,160 | 4,439 | 3,446 | 1,845 | |||||||||||||||
| Sales and marketing |
1,050 | 1,200 | 1,574 | 716 | 239 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
6,077 | 5,802 | 8,087 | 5,329 | 3,420 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (Loss) income from operations |
(5,731 | ) | (6,044 | ) | (8,185 | ) | (6,323 | ) | (4,286 | ) | ||||||||||
| Total other income (expense) |
3,110 | (10,799 | ) | (11,702 | ) | (2,084 | ) | (3,042 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (Loss) income before income taxes |
(2,621 | ) | (16,843 | ) | (19,887 | ) | (8,407 | ) | (7,328 | ) | ||||||||||
| Reserve for income tax benefit |
— | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net (loss) |
$ | (2,621 | ) | $ | (16,843 | ) | $ | (19,887 | ) | $ | (8,407 | ) | $ | (7,328 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net (loss) per share: |
||||||||||||||||||||
| basic |
$ | (0.78 | ) | $ | (5.30 | ) | $ | (6.24 | ) | $ | (2.68 | ) | $ | (3.22 | ) | |||||
| diluted |
(2.66 | ) | (5.30 | ) | (6.24 | ) | (2.68 | ) | (3.22 | ) | ||||||||||
| Weighted average shares of common stock outstanding used in computing net (loss) per share: |
||||||||||||||||||||
| basic |
3,351,324 | 3,178,701 | 3,185,382 | 3,133,077 | 2,277,004 | |||||||||||||||
| diluted |
3,375,210 | 3,178,701 | 3,185,382 | 3,133,077 | 2,277,004 | |||||||||||||||
| Pro forma net (loss) per share of common stock: |
||||||||||||||||||||
| basic |
$ | (0.41 | ) | $ | (3.19 | ) | ||||||||||||||
| diluted |
(1.40 | ) | (3.19 | ) | ||||||||||||||||
| Weighted average shares of common stock outstanding used in computing pro forma net (loss) per share: |
||||||||||||||||||||
| basic |
6,391,147 | 6,225,205 | ||||||||||||||||||
| diluted |
6,415,033 | |
6,225,205 |
|
||||||||||||||||
| As of September 30, 2012 | ||||||||||||
| Actual | Pro Forma (unaudited) |
Pro Forma As Adjusted(1) (unaudited) |
||||||||||
| (dollars in thousands) | ||||||||||||
| BALANCE SHEET DATA: |
||||||||||||
| Cash and cash equivalents |
$ | 903 | $ | 2,903 | $ | 28,291 | ||||||
| Total Assets |
7,938 | 9,684 | 31,818 | |||||||||
| Total Liabilities |
28,095 | 22,357 | 16,251 | |||||||||
| Total Stockholders’ Equity (Deficit) |
$ | 20,157 | $ | (12,673 | ) | $ | 15,567 | |||||
(1) A $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share of our common stock, the midpoint of the estimated price range set forth on the cover of this prospectus, would increase (decrease) each of cash, total assets, and total stockholders’ equity (deficit) by $4.7 million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
-9-
Table of Contents
Risks Relating to Our Financial Condition and Capital Requirements
We are an early stage company with a history of net losses; we expect to incur net losses in the future, and we may never achieve sustained profitability.
We have historically incurred substantial net losses, including net losses of $2.6 million in the nine months ended September 30, 2012, $19.9 million in 2011, $8.4 million in 2010 and $7.3 million in 2009. From our inception in April 1999 through September 30, 2012, we had an accumulated deficit of $44.9 million. We expect our losses to continue as a result of ongoing research and development expenses and increased sales and marketing costs. These losses have had, and will continue to have, an adverse effect on our working capital, total assets and stockholders’ equity. Because of the numerous risks and uncertainties associated with our research, development and commercialization efforts, we are unable to predict when we will become profitable, and we may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations and cash flows.
We may need to raise additional capital.
Without the net proceeds from this offering, we believe our current cash resources, including the $2.0 million in debt financing we raised after September 30, 2012, are sufficient to satisfy our liquidity requirements at our current level of operations through February 28, 2013. If we do not consummate this offering prior to the end of February 2013, we expect that we will need to raise additional financing in the first quarter of 2013, which might not be available on favorable terms, if at all. We have had and will continue to have discussions with certain of our stockholders regarding potential additional financing, but we can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. Notwithstanding the receipt of proceeds from this offering, we may need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing, which is not in place at this time, may be from the sale of equity or convertible or other debt securities in a public or private offering, from an additional credit facility or strategic partnership coupled with an investment in us or a combination of both. We may be unable to raise sufficient additional financing on terms that are acceptable to us, if at all. Our failure to raise additional capital and in sufficient amounts may significantly impact our ability to expand our business. For further discussion of our liquidity requirements as they relate to our long-term plans, see the section entitled “Liquidity and Capital Resources—Capital Resources and Expenditure Requirements”.
Risks Relating to Our Business and Strategy
If we are unable to increase sales of our laboratory tests and services or to successfully develop and commercialize other proprietary tests, our revenues will be insufficient for us to achieve profitability.
We currently derive substantially all of our revenues from our laboratory testing services. We have recently begun offering our MatBA®-CLL microarray and our MatBA®-SLL microarray through our CLIA-accredited and state licensed laboratory. We are in varying stages of research and development for other diagnostic tests that we may offer. If we are unable to increase sales of our laboratory tests and services or to successfully develop and commercialize other diagnostic tests, we will not produce sufficient revenues to become profitable.
Our business depends on our ability to successfully develop and commercialize novel cancer diagnostic tests and services, which is time consuming and complex, and our development efforts may fail.
Our current business strategy focuses on discovering, developing and commercializing molecular diagnostic tests and services. We believe the success of our business depends on our ability to fully
-10-
Table of Contents
commercialize our existing diagnostic tests and services and to develop and commercialize new diagnostic tests. We have multiple tests in development, but research, development and commercialization of diagnostic tests is time-consuming, uncertain and complex. Our current diagnostic test pipeline includes: UroGenRA™ microarray, UGenRA™ microarray, LeukA™ microarray, FReCaD™ Renal Cancer Test, FHACT™ HPV-associated Cancer Test and expansion of the MatBA® microarray as a prognostic tool in FL and DLBCL. Tests such as these, or any additional technologies that we may develop, may not succeed in reliably diagnosing or predicting the recurrence of cancers with the sensitivity and specificity necessary to be clinically useful, and thus may not succeed commercially.
In addition, prior to commercializing our diagnostic tests, we must undertake time-consuming and costly development activities, sometimes including clinical studies, and obtain regulatory clearance or approval, which may be denied. This development process involves a high degree of risk, substantial expenditures and will occur over several years. Our development efforts may fail for many reasons, including:
| • | failure of the tests at the research or development stage; |
| • | difficulty in accessing archival tissue samples, especially tissue samples with known clinical results; or |
| • | lack of clinical validation data to support the effectiveness of the test. |
Tests that appear promising in early development may fail to be validated in subsequent studies, and even if we achieve positive results, we may ultimately fail to obtain the necessary regulatory clearances or approvals. There is substantial risk that our research and development projects will not result in commercial tests, and that success in early clinical trials will not be replicated in later studies. At any point, we may abandon development of a test or be required to expend considerable resources repeating clinical trials, which would adversely impact the timing for generating potential revenues from that test. In addition, as we develop tests, we will have to make significant investments in research, development and marketing resources. If a clinical validation study of a particular test then fails to demonstrate the outlined goals of the study, we might choose to abandon the development of that test. Further, our ability to develop and launch diagnostic tests will likely depend on our receipt of additional funding. If our discovery and development programs yield fewer commercial tests than we expect, we may be unable to execute our business plan, which may adversely affect our business, financial condition and results of operations.
If we are unable to obtain regulatory clearance or approvals in the United States, if we experience delays in receiving clearance or approvals, or if we do not gain acceptance from other laboratories of any cleared or approved diagnostic tests at their facilities, our growth strategy may not be successful.
We currently offer our proprietary tests in conjunction with our comprehensive panel of laboratory services in our CLIA-accredited laboratory. Because we currently offer these tests and services solely for use within our laboratory, we believe we may market the tests as LDTs. Under current FDA enforcement policies and guidance, LDTs generally do not require FDA premarket clearance or approval before commercialization, and we have marketed our LDTs on that basis. However, a key element of our long-term strategy is to place molecular diagnostic tests on-site with other laboratories to broaden access to our technology and increase demand for our tests and any future diagnostic tests that we may develop. FDA regulates diagnostic kits sold and distributed through interstate commerce as medical devices. Unless an exemption applies, generally, before a new medical device or a new use for a medical device may be sold or distributed in the United States, the medical device must receive either FDA clearance of a 510(k) pre-market notification or pre-market approval. As a result, before we can market or distribute our DNA probes or microarray tests in the United States for use by other clinical testing laboratories, we must first obtain pre-market clearance or pre-market approval from FDA. We have not yet applied for clearance or approval from FDA, and need to complete additional validations before we are ready to apply. We believe it would likely take two years or more to conduct the studies and trials necessary to obtain approval from FDA to commercially launch MatBA®-CLL and MatBA®-SLL outside of our
-11-
Table of Contents
clinical laboratory. Once we do apply, we may not receive FDA clearance or approval for the commercial use of our tests on a timely basis, or at all. If we are unable to achieve clearance or approval or if clinical diagnostic laboratories do not accept our tests, our ability to grow our business by deploying our tests could be compromised.
If we are unable to execute our marketing strategy for our cancer diagnostic tests and are unable to gain acceptance in the market, we may be unable to generate sufficient revenue to sustain our business.
We are an early-stage company and have engaged in only limited sales and marketing activities for the diagnostic tests and services offered in our clinical laboratory. To date, we have received very limited revenue from sales of our probes and microarrays. While we are in the process of launching several of our DNA probes outside of the United States, we have limited experience in marketing these probes and we need to develop relationships with third-party distributors in the emerging market countries where we are targeting our selling efforts.
Although we believe that our diagnostic tests represent promising commercial opportunities, our tests may never gain significant acceptance in the marketplace and therefore may never generate substantial revenue or profits for us. We will need to establish a market for our diagnostic tests and build that market through physician education and awareness programs. Gaining acceptance in medical communities requires publication in leading peer-reviewed journals of results from studies using our tests. The process of publication in leading medical journals is subject to a peer review process and peer reviewers may not consider the results of our studies sufficiently novel or worthy of publication. Failure to have our studies published in peer-reviewed journals would limit the adoption of our tests.
Our ability to successfully market the diagnostic tests that we may develop will depend on numerous factors, including:
| • | whether healthcare providers believe our diagnostic tests provide clinical utility; |
| • | whether the medical community accepts that our diagnostic tests are sufficiently sensitive and specific to be meaningful in patient care and treatment decisions; and |
| • | whether health insurers, government health programs and other third-party payors will cover and pay for our diagnostic tests and, if so, whether they will adequately reimburse us. |
Failure to achieve widespread market acceptance of our diagnostic tests would materially harm our business, financial condition and results of operations.
If we cannot develop tests to keep pace with rapid advances in technology, medicine and science, our operating results and competitive position could be harmed.
In recent years, there have been numerous advances in technologies relating to the diagnosis and treatment of cancer. There are several new cancer drugs under development that may increase patient survival time. There have also been advances in methods used to analyze very large amounts of genomic information. We must continuously develop new tests and enhance our existing tests to keep pace with evolving standards of care. Our tests could become obsolete unless we continually innovate and expand them to demonstrate benefit in patients treated with new therapies. New cancer therapies typically have only a few years of clinical data associated with them, which limits our ability to perform clinical studies and correlate sets of genes to a new treatment’s effectiveness. If we cannot adequately demonstrate the applicability of our tests to new treatments, sales of our tests and services could decline, which would have a material adverse effect on our business, financial condition and results of operations.
-12-
Table of Contents
If our tests do not perform as expected, our operating results, reputation and business will suffer.
Our success depends on the market’s confidence that we can provide reliable, high-quality diagnostic tests. We believe that our customers are likely to be particularly sensitive to test defects and errors. As a result, the failure of our tests or services to perform as expected would significantly impair our reputation and the public image of our tests and services, and we may be subject to legal claims arising from any defects or errors.
If our sole laboratory facility becomes damaged or inoperable, or we are required to vacate the facility, our ability to provide services and pursue our research and development efforts may be jeopardized.
We currently derive substantially all of our revenues from our laboratory testing services. We do not have any clinical reference laboratory facilities outside of our facility in Rutherford, New Jersey. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including fire, flooding and power outages, which may render it difficult or impossible for us to perform our tests or provide laboratory services for some period of time. The inability to perform our tests or the backlog of tests that could develop if our facility is inoperable for even a short period of time may result in the loss of customers or harm to our reputation or relationships with collaborators, and we may be unable to regain those customers or repair our reputation in the future. Furthermore, our facilities and the equipment we use to perform our research and development work could be costly and time-consuming to repair or replace.
Additionally, a key component of our research and development process involves using biological samples and the resulting data sets and medical histories, as the basis for our diagnostic test development. In some cases, these samples are difficult to obtain. If the parts of our laboratory facility where we store these biological samples are damaged or compromised, our ability to pursue our research and development projects, as well as our reputation, could be jeopardized. We carry insurance for damage to our property and the disruption of our business, but this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
Further, if our laboratory became inoperable we may not be able to license or transfer our proprietary technology to a third-party, with established state licensure and CLIA accreditation under the scope of which our diagnostic tests could be performed following validation and other required procedures, to perform the tests. Even if we find a third-party with such qualifications to perform our tests, such party may not be willing to perform the tests for us on commercially reasonable terms.
If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenues or achieve and sustain profitability.
Our principal competition comes from the existing mainstream diagnostic methods that pathologists and oncologists use and have used for many years. It may be difficult to change the methods or behavior of the referring pathologists and oncologists to incorporate our molecular diagnostic testing in their practices. We believe that we can introduce our diagnostic tests successfully due to their clinical utility and the desire of pathologists and oncologists to find solutions for more accurate diagnosis, prognosis and personalized treatment options for cancer patients.
We also face competition from companies that currently offer or are developing products to profile genes, gene expression or protein biomarkers in various cancers. Personalized genetic diagnostics is a new area of science, and we cannot predict what tests others will develop that may compete with or provide results superior to the results we are able to achieve with the tests we develop. Our competitors include public companies such as CombiMatrix Corporation, Quest Diagnostics, Abbott Laboratories, Inc., Johnson & Johnson, Roche Molecular Systems, Inc., bioTheranostics, Inc. (part of bioMérieux SA), Genomic Health, Inc., Myriad Genetics Inc., Qiagen N.V. and Response Genetics, Inc., and many private companies, including Agendia B.V., Pathwork Diagnostics, Inc. and Foundation Medicine, Inc. We expect that pharmaceutical and biopharmaceutical
-13-
Table of Contents
companies will increasingly focus attention and resources on the personalized diagnostic sector as the potential and prevalence increases for molecularly targeted oncology therapies approved by FDA along with companion diagnostics. For example, FDA has recently approved two such agents—Xalkori crizotinib from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc. and Zelboraf vemurafenib from Genentech USA Incorporated and Daiichi-Sankyo Inc. along with its companion B-RAF kinase V600 mutation test from Roche Molecular Systems, Inc. These two recent FDA approvals are only the second and third instances of simultaneous approvals of a drug and companion diagnostic, the first being the 1998 approval of Genentech, Inc.’s Herceptin trastuzumab for HER2 positive breast cancer along with the HercepTest from partner Dako A/S.
With respect to our clinical laboratory sciences business we face competition from companies such as Genoptix, Inc. (a Novartis AG Company), Clarient, Inc. (a division of GE Healthcare, a unit of General Electric Company), Bio-Reference Laboratories, Inc., and Genzyme Genetics (a LabCorp Specialty Testing Group).
Many of our present and potential competitors have widespread brand recognition and substantially greater financial and technical resources and development, production and marketing capabilities than we do. Others may develop lower-priced, less complex tests that payors, pathologists and oncologists could view as functionally equivalent to our tests, which could force us to lower the list price of our tests and impact our operating margins and our ability to achieve profitability. In addition, technological innovations that result in the creation of enhanced diagnostic tools may enable other clinical laboratories, hospitals, physicians or medical providers to provide specialized diagnostic services similar to ours in a more patient-friendly, efficient or cost-effective manner than is currently possible. If we cannot compete successfully against current or future competitors, we may be unable to increase market acceptance and sales of our tests, which could prevent us from increasing or sustaining our revenues or achieving or sustaining profitability.
If pathologists and oncologists decide not to order our diagnostic tests, we may be unable to generate sufficient revenue to sustain our business.
To generate demand for our molecular diagnostic tests and services, we will need to educate oncologists and pathologists on the clinical utility, benefits and value of each type of test we provide through published papers, presentations at scientific conferences and one-on-one education sessions by members of our sales force. In addition, we will need to assure oncologists and pathologists of our ability to obtain and maintain adequate reimbursement coverage from third-party payors. We may need to hire additional commercial, scientific, technical and other personnel to support this process. If we cannot convince medical practitioners to order our diagnostic tests or other future tests we develop, we will likely be unable to create demand for our tests in sufficient volume for us to achieve sustained profitability.
A small number of test ordering sites account for most of the sales of our tests and services. If any of these sites orders fewer tests from us for any reason, our revenues could decline.
Due to the early stage nature of our business and our limited sales and marketing activities to date, we have historically derived a significant portion of our revenue from a limited number of test ordering sites, although the test ordering sites that generate a significant portion of our revenue have changed from period to period. For example, there was one site which represented more than 10% of our revenue for the year ended December 31, 2010 that generated less than 10% of our revenue for the year ended December 31, 2011. Our test ordering sites are largely hospitals, cancer centers, reference laboratories and physician offices. Oncologists and pathologists at these sites order the tests on behalf of the needs of their oncology patients. For the nine months ended September 30, 2012, our top five test ordering sites accounted for 61% of our clinical testing volume with approximately 47% of the volume coming from community hospitals. For the year ended December 31, 2011, our top five test ordering sites represented approximately 63% of our clinical testing volume, with approximately 29% of the volume coming from community hospitals. Our top five test ordering sites for the year ended December 31, 2010 accounted for 60% of our clinical testing volumes, with approximately 15% of the volume
-14-
Table of Contents
coming from community hospitals. In particular, during the year ended December 31, 2010, there were three sites which each accounted for 10% or more of our revenue: one community hospital accounted for approximately 12% of our revenue; a regional reference laboratory accounted for approximately 11% of our revenue and a community oncology practice accounted for another approximately 11% of our revenue. For the year ended December 31, 2011, we generated revenue from two test ordering sites that represented 10% or more of our revenue: a community hospital accounted for approximately 18% of our revenue and a community oncology practice accounted for approximately 11% of our revenue. For the nine months ended September 30, 2012, three test ordering sites accounted for 10% or more of our revenue; a university teaching center accounted for approximately 15%; a clinical trial client accounted for approximately 12% and a community hospital network accounted for approximately 11%. We generally do not enter into formal written agreements with such test ordering sites and, as a result, we may lose these significant test ordering sites at any time.
We expect to continue to incur significant expenses to develop and market our diagnostic tests, which could make it difficult for us to achieve and sustain profitability.
In recent years, we have incurred significant costs in connection with the development of our diagnostic tests. For the year ended December 31, 2011, our research and development expenses were $2.1 million, which was 69% of our net revenues and our sales and marketing expenses were $1.6 million, which was 52% of revenue. For the year ended December 31, 2010, our research and development expenses were $1.2 million, which was 46% of revenue, and our sales and marketing expenses were $716,000, which was 28% of revenue. For the year ended December 31, 2009, our research and development expenses were $1.3 million, which was 80% of revenue, and our sales and marketing expenses were $239,000, which was 14% of revenue. We expect our expenses to continue to increase, in absolute dollars, for the foreseeable future as we seek to expand the clinical utility of our diagnostic tests, drive adoption of and reimbursement for our diagnostic tests and develop new tests. As a result, we will need to generate significant revenues in order to achieve sustained profitability.
We depend on certain collaborations with third parties for the supply of certain tissue samples and biological materials that we use in our research and development efforts. If the costs of such collaborations increase after we complete our initial public offering or our third party collaborators terminate their relationship with us, our business may be materially harmed.
Under standard clinical practice in the United States, tumor biopsies removed from patients are chemically preserved, embedded in paraffin wax and stored. Our clinical development relies on our ability to access these archived tumor biopsy samples, as well as information pertaining to their associated clinical outcomes. Other companies often compete with us for access. Additionally, the process of negotiating access to archived samples is lengthy, because it typically involves numerous parties and approvals to resolve complex issues such as usage rights, institutional review board approval, privacy rights, publication rights, intellectual property ownership and research parameters.
We have collaborative relationships with Memorial Sloan-Kettering Cancer Center, Mayo, North Shore - Long Island Jewish Health System, the National Cancer Institute and other institutions who provide us with tissue samples and other biological materials that we use in developing and validating our tests. We do not have any written arrangement with certain third party collaborators, and in many of the cases in which the arrangements are in writing, our collaborative relationships are terminable on 30 days notice or less. If one or more collaborators terminate their relationship with us, we will need to identify other third parties to provide us with tissue samples and biological materials, which could result in a delay in our research and development activities and negatively affect our business. In addition, as we grow, our collaborators that are research and academic institutions will begin to seek additional financial contributions from us, which may negatively affect our results of operations.
-15-
Table of Contents
We have a substantial amount of indebtedness, which could have a material adverse effect on our financial condition and our ability to fund operations, obtain additional financing and react to changes in our business.
We have substantial indebtedness for borrowed money. As of September 30, 2012, we had indebtedness for borrowed money in the aggregate principal amount of $17.2 million of which $9.0 million was outstanding under our existing lines of credit with Wells Fargo Bank, N.A. (“Wells Fargo”) and DAM Holdings, LLC (“DAM”), $6.0 million was outstanding under a secured term loan credit agreement dated December 2011 as amended in February 2012, with two of our directors or their affiliates and $2.0 million was outstanding under the restated credit agreement with one of our directors and an independent third party investor. In addition, subsequent to September 30, 2012, we closed an additional $1.0 million in debt financing under the restated credit agreement with Mr. Pappajohn, one of our directors, and an independent third party investor and an additional $1 million from Mr. Pappajohn on the same terms as the restated credit agreement. The $4.0 million outstanding principal amount automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share and our initial public offering price per share. Mr. Pappajohn agreed to convert $2.0 million of debt due pursuant to the terms of the December 2011 credit agreement to common stock at the initial offering price per share. We expect that approximately $8.0 million in indebtedness, which will consist of $6.0 million under our existing line of credit with Wells Fargo and $2.0 million under the December 2011 credit agreement with Mr. Pappajohn, which is held by his spouse, shall remain outstanding after this offering. Substantially all of our assets, including our intellectual property, are pledged as collateral under our existing lines of credit and the term loans. Our significant debt could limit our ability to satisfy our obligations, limit our ability to operate our business and impair our competitive position. For example, it could:
| • | require us to dedicate a substantial portion of our cash flow from operations to payments on our debt, reducing the availability of our cash flow from operations to fund working capital, capital expenditures or other general corporate purposes; |
| • | limit our flexibility in planning for, or reacting to, changes in our business and industry; |
| • | place us at a disadvantage compared to competitors that may have proportionately less debt; and |
| • | increase our cost of borrowing. |
We expect to use approximately $5.0 million of the proceeds from this offering to repay outstanding indebtedness which includes $2.0 million due to one of our directors and his affiliates under the December 2011 financing transaction and $3.0 million due to DAM. For more information, see the section entitled “Use of Proceeds”.
We may need to raise additional capital in the future to develop and commercialize new tests and technologies and expand our operations.
We expect capital outlays and operating expenditures to increase over the next several years as we expand our infrastructure, commercial operations and research and development activities. Specifically, we may need to raise capital to, among other things:
| • | increase our sales and marketing efforts to drive market adoption and address competitive developments; |
| • | fund development and marketing efforts of any future tests; |
| • | further expand our clinical laboratory operations; |
| • | expand our technologies into other types of cancer; |
-16-
Table of Contents
| • | acquire, license or invest in technologies; |
| • | acquire or invest in complementary businesses or assets; |
| • | if our joint venture with Mayo is formed and if formed it achieves certain operational milestones, fund our subsequent contributions of $4.0 million to our joint venture with Mayo; |
| • | repay the approximately $8.0 million in indebtedness that will remain outstanding after this offering and matures in April 2014; and |
| • | finance capital expenditures and general and administrative expenses. |
Our present and future funding requirements will depend on many factors, including:
| • | our ability to achieve revenue growth; |
| • | our rate of progress in establishing reimbursement arrangements with domestic and international third-party payors; |
| • | the cost of expanding our laboratory operations and offerings, including our sales and marketing efforts; |
| • | our rate of progress in, and cost of the sales and marketing activities associated with, establishing adoption of and reimbursement for our microarray tests and probes; |
| • | our rate of progress in, and cost of research and development activities associated with, products in research and early development; |
| • | the effect of competing technological and market developments; |
| • | costs related to international expansion; and |
| • | the potential cost of and delays in test development as a result of any regulatory oversight applicable to our tests. |
The various ways we could raise additional capital carry potential risks. If we raise funds by issuing equity securities, dilution to our stockholders could result. Any equity securities issued also could provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise funds by issuing debt securities, those debt securities would have rights, preferences and privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings pursuant to a credit agreement could impose significant restrictions on our operations. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technologies or tests, or grant licenses on terms that are not favorable to us.
The credit markets and the financial services industry have experienced a period of unprecedented turmoil and upheaval characterized by the bankruptcy, failure, collapse or sale of various financial institutions and an unprecedented level of intervention from the United States federal government. These events have generally made equity and debt financing more difficult to obtain. Accordingly, additional equity or debt financing might not be available on reasonable terms, if at all. If we cannot secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs or sales and marketing initiatives. In addition, we may have to work with a partner on one or more of our development programs, which could lower the economic value of those programs to us.
-17-
Table of Contents
The loss of our Chairman or key members of our executive management team could adversely affect our business.
Our success in implementing our business strategy depends largely on the skills, experience and performance of our Chairman of our board of directors, Dr. Raju Chaganti, key members of our executive management team and others in key management positions, including Panna L. Sharma, our Chief Executive Officer, Elizabeth A. Czerepak, our Chief Financial Officer, and Jane Houldsworth, Ph.D., our Vice President of Research and Development. The collective efforts of each of these persons working as a team will be critical to us as we continue to develop our technologies, tests and research and development and sales programs. As a result of the difficulty in locating qualified new management, the loss or incapacity of existing members of our executive management team could adversely affect our operations. If we were to lose one or more of these key employees, we could experience difficulties in finding qualified successors, competing effectively, developing our technologies and implementing our business strategy. Our Chief Executive Officer, Chief Financial Officer and Vice President of Research and Development have employment agreements, however, the existence of an employment agreement does not guarantee retention of members of our executive management team and we may not be able to retain those individuals for the duration of or beyond the end of their respective terms. We do not maintain “key person” insurance on any of our employees except our Chief Executive Officer.
In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
There is a scarcity of experienced professionals in our industry. If we are not able to retain and recruit personnel with the requisite technical skills, we may be unable to successfully execute our business strategy.
The specialized nature of our industry results in an inherent scarcity of experienced personnel in the field. Our future success depends upon our ability to attract and retain highly skilled personnel (including medical, scientific, technical, commercial, business, regulatory and administrative personnel) necessary to support our anticipated growth, develop our business and perform certain contractual obligations. Given the scarcity of professionals with the scientific knowledge that we require and the competition for qualified personnel among life science businesses, we may not succeed in attracting or retaining the personnel we require to continue and grow our operations. The loss of a key employee, the failure of a key employee to perform in his or her current position or our inability to attract and retain skilled employees could result in our inability to continue to grow our business or to implement our business strategy.
Our inability to attract, hire and retain a sufficient number of qualified sales professionals would hamper our ability to increase demand for our tests, to expand geographically and to successfully commercialize any other diagnostic tests or products we may develop.
Our success in selling our clinical laboratory services, diagnostic tests and any other tests or products that we are able to develop will require us to expand our sales force in the United States and internationally by recruiting additional sales representatives with extensive experience in oncology and close relationships with medical oncologists, surgeons, pathologists and other hospital personnel. To achieve our marketing and sales goals, we will need to substantially expand our sales and commercial infrastructure, with which to date we have had little experience. Sales professionals with the necessary technical and business qualifications are in high demand, and there is a risk that we may be unable to attract, hire and retain the number of sales professionals with the right qualifications, scientific backgrounds and relationships with decision-makers at potential customers needed to achieve our sales goals. We may face competition from other companies in our industry, some of whom are much larger than us and who can pay greater compensation and benefits than we can, in seeking to attract and retain qualified sales and marketing employees. If we are unable to hire and retain qualified sales and marketing personnel, our business will suffer.
-18-
Table of Contents
International expansion of our business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Our business strategy incorporates international expansion, including establishing and maintaining clinician marketing and education capabilities outside of the United States and expanding our relationships with distributors and manufacturers. Doing business internationally involves a number of risks, including:
| • | multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| • | failure by us or our distributors to obtain regulatory approvals for the sale or use of our tests in various countries, including failure to achieve “CE Marking,” a conformity mark which is required to market in vitro diagnostic medical devices in the European Economic Area and which is broadly accepted in other international markets; |
| • | difficulties in managing foreign operations; |
| • | complexities associated with managing multiple payor-reimbursement regimes or self-pay systems; |
| • | logistics and regulations associated with shipping tissue samples, including infrastructure conditions and transportation delays; |
| • | limits on our ability to penetrate international markets if our diagnostic tests cannot be processed by an appropriately qualified local laboratory; |
| • | financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations; |
| • | reduced protection for intellectual property rights; |
| • | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and |
| • | failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales and distributors’ activities. |
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, have a material adverse effect on our financial condition, results of operations and cash flows.
Our dependence on distributors for foreign sales of our FISH-based DNA probes could limit or prevent us from selling our probes in foreign markets and from realizing long-term international revenue growth.
We intend to grow our business internationally, and to do so we must enter into agreements with local distributors to sell our FISH-based DNA probes. These agreements generally contain exclusivity provisions and generally cannot be terminated without cause during the term of the agreement. We may need to attract additional distributors to expand the territories in which we sell our probes. These distributors may not commit the necessary resources to market and sell our probes to the level of our expectations, and we may be unable to locate suitable alternatives should we terminate our agreement with such distributors or if such distributors terminate their agreement with us. If current or future distributors do not perform adequately, or we are unable to locate distributors in particular geographic areas, we may not realize long-term international revenue growth.
-19-
Table of Contents
We currently rely on a single third-party to produce our microarrays and any problems experienced by this vendor could result in a delay or interruption in the supply of our microarrays to us until the problem is cured by such vendor or until we locate and qualify an alternative source of supply
The design of our microarrays is currently optimized on a family of instruments referred to as the Agilent Microarray Platform, which is currently produced solely by Agilent Technologies Inc. (“Agilent”). We currently purchase these components from Agilent under purchase orders and do not have a long-term contract with Agilent. If Agilent were to delay or stop producing our microarrays, or if the prices Agilent charges us were to increase significantly, we would need to identify another supplier and optimize our microarrays on a new technology platform. We could experience delays in manufacturing the microarrays while finding another acceptable supplier, which could impact our results of operations. The changes could also result in increased costs associated with migrating to the new technology platform and in increased manufacturing costs. Further, any prolonged disruption in Agilent’s operations could have a significant negative impact on the supply of our microarrays.
If we were sued for product liability or professional liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of our tests could lead to the filing of product liability claims were someone to allege that our tests failed to perform as designed. We may also be subject to liability for errors in the test results we provide to pathologists and oncologists or for a misunderstanding of, or inappropriate reliance upon, the information we provide. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend.
Although we believe that our existing product and professional liability insurance is adequate, our insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could damage our reputation, result in the recall of our tests, or cause current clinical partners to terminate existing agreements and potential clinical partners to seek other partners, any of which could impact our results of operations.
If we use biological and hazardous materials in a manner that causes injury, we could be liable for damages.
Our activities currently require the controlled use of potentially harmful biological materials and hazardous materials and chemicals. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject to, on an on going basis, federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become significant and could have a material adverse effect on our financial condition, results of operations and cash flows. In the event of an accident or if we otherwise fail to comply with applicable regulations, we could lose our permits or approvals or be held liable for damages or penalized with fines.
We may acquire other businesses or form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our offerings or distribution. For example, we agreed to enter into a joint venture in March 2013 with Mayo
-20-
Table of Contents
Foundation for Education and Research. We have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
If we cannot support demand for our tests, including successfully managing the evolution of our technology and manufacturing platforms, our business could suffer.
As our test volume grows, we will need to increase our testing capacity, implement increases in scale and related processing, customer service, billing, collection and systems process improvements and expand our internal quality assurance program and technology to support testing on a larger scale. We will also need additional certified laboratory scientists and other scientific and technical personnel to process these additional tests. Any increases in scale, related improvements and quality assurance may not be successfully implemented and appropriate personnel may not be available. As additional tests are commercialized, we will need to bring new equipment on line, implement new systems, technology, controls and procedures and hire personnel with different qualifications. Failure to implement necessary procedures or to hire the necessary personnel could result in a higher cost of processing or an inability to meet market demand. We cannot assure you that we will be able to perform tests on a timely basis at a level consistent with demand, that our efforts to scale our commercial operations will not negatively affect the quality of our test results or that we will respond successfully to the growing complexity of our testing operations. If we encounter difficulty meeting market demand or quality standards for our tests, our reputation could be harmed and our future prospects and business could suffer, which may have a material adverse effect on our financial condition, results of operations and cash flows.
We may encounter manufacturing problems or delays that could result in lost revenue.
We currently manufacture our proprietary DNA probes outside the United States at a third party fully compliant facility and intend to continue to manufacture our probes outside the United States. We currently have limited manufacturing capacity for our probes. If demand for our probes increases significantly, we will need to either expand our manufacturing capabilities or outsource to other manufacturers. If we or third party manufacturers engaged by us fail to manufacture and deliver our probes in a timely manner, our relationships with our customers could be seriously harmed. We cannot assure you that manufacturing or quality control problems will not arise as we attempt to increase the production of our probes or that we can increase our manufacturing capabilities and maintain quality control in a timely manner or at commercially reasonable costs. If we cannot manufacture our probes consistently on a timely basis because of these or other factors, it could have a significant negative impact on the supply of our DNA probes.
Declining general economic or business conditions may have a negative impact on our business.
Continuing concerns over United States health care reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the United States and other
-21-
Table of Contents
countries have contributed to increased volatility and diminished expectations for the global economy. These factors, combined with low business and consumer confidence and high unemployment, precipitated an economic slowdown and recession. If the economic climate does not improve or continues to deteriorate, our business, including our access to patient samples and the addressable market for diagnostic tests that we may successfully develop, as well as the financial condition of our suppliers and our third-party payors, could be adversely affected, resulting in a negative impact on our business, financial condition and results of operations.
We depend on our information technology and telecommunications systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant aspects of our operations. In addition, our third-party billing and collections provider depends upon telecommunications and data systems provided by outside vendors and information we provide on a regular basis. These information technology and telecommunications systems support a variety of functions, including test processing, sample tracking, quality control, customer service and support, billing and reimbursement, research and development activities and our general and administrative activities. Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems or those used by our third-party service providers could prevent us from processing tests, providing test results to pathologists, oncologists, billing payors, processing reimbursement appeals, handling patient or physician inquiries, conducting research and development activities and managing the administrative aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business.
Regulatory Risks Relating to Our Business
Healthcare policy changes, including recently enacted legislation reforming the U.S. health care system, may have a material adverse effect on our financial condition, results of operations and cash flows.
In March 2010, U.S. President Barack Obama signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “PPACA”), which makes a number of substantial changes in the way health care is financed by both governmental and private insurers. Among other things, the PPACA:
| • | Requires each medical device manufacturer to pay a sales tax equal to 2.3% of the price for which such manufacturer sells its medical devices, beginning in 2013. This tax may apply to some or all of our current products and products which are in development. |
| • | Mandates a reduction in payments for clinical laboratory services paid under the Medicare Clinical Laboratory Fee Schedule of 1.75% for the years 2011 through 2015. In addition, a productivity adjustment is made to the fee schedule payment amount. These changes in payments apply to some or all of the clinical laboratory test services we furnish to Medicare beneficiaries. |
| • | Establishes an Independent Payment Advisory Board to reduce the per capita rate of growth in Medicare spending. The Independent Payment Advisory Board has broad discretion to propose policies, which may have a negative impact on payment rates for services, including clinical laboratory services, beginning in 2016, and for hospital services beginning in 2020. |
-22-
Table of Contents
Although some of these provisions may negatively impact payment rates for clinical laboratory services, the PPACA also extends coverage to approximately 32 million previously uninsured people, which may result in an increase in the demand for our tests and services. The mandatory purchase of insurance has been strenuously opposed by a number of state governors, resulting in lawsuits challenging the constitutionality of certain provisions of the PPACA. On June 28, 2012, the Supreme Court upheld the constitutionality of the health care reform law, with the exception of certain provisions dealing with the expansion of Medicaid coverage under the law. Therefore, most of the law’s provisions will go into effect in 2013 and 2014. Congress has also proposed a number of legislative initiatives, including possible repeal of the PPACA. At this time, it remains unclear whether there will be any changes made to the PPACA, whether to certain provisions or its entirety.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. Recently, on August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, creates the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. The full impact on our business of the PPACA and the new law is uncertain. In addition, on February 22, 2012, the President signed the Middle Class Tax Relief and Job Creation Act of 2012 (“MCTRJCA”), which, among other things, mandated an additional change in Medicare reimbursement for clinical laboratory services. This legislation requires a rebasing of the Medicare clinical laboratory fee schedule to effect a 2% reduction in payment rates otherwise determined for 2013. This will serve as a base for 2014 and subsequent years. As a result of the changes mandated by PPACA and MCTRJCA, CMS projects laboratory services for 2013 will be reduced by approximately 3%.
Certain of our laboratory services are paid under the Medicare Physician Fee Schedule and, under the current statutory formula, the rates for these services are updated annually. For the past several years, the application of the statutory formula would have resulted in substantial payment reductions if Congress failed to intervene. In the past, Congress passed interim legislation to prevent the decreases. On November 1, 2012, the Centers for Medicare & Medicaid Services (CMS) issued its 2013 Physician Fee Schedule Final Rule (“the Final Rule”). In the Final Rule, CMS called for a reduction of approximately 26.5% in the 2013 conversion factor that is used to calculate physician reimbursement. However, the American Taxpayer Relief Act of 2012, which was signed into law on January 2, 2013, prevents this proposed cut and keeps the current conversion factor rate in effect until December 31, 2013. If Congress fails to act in future years to offset similar proposed reductions, the resulting decrease in payment could adversely impact our revenues and results of operations.
In addition, many of the Current Procedure Terminology (“CPT”) procedure codes that we use to bill our tests were recently revised by the AMA, effective January 1, 2013. In the Final Rule, CMS announced that it has decided to keep the new molecular codes on the Clinical Laboratory Fee Schedule (CLFS), rather than move them to the Physician Fee Schedule as some stakeholders had urged. CMS has also announced that for 2013 it will price the new codes using a “gapfilling” process by which it will refer the codes to the Medicare contractors to allow them to determine an appropriate price. In addition, it has also stated that it will not recognize certain of the new codes for Multi-analyte Assays for Algorithmic Assays (MAAAs) because it does not believe they qualify as clinical laboratory tests. Our reimbursement could be adversely affected by CMS’ action in this area. If it reduces reimbursement for the new test codes or does not pay for our new MAAA codes, then our revenues will be adversely affected. There can be no guarantees that Medicare and other payers will establish positive or adequate coverage policies or reimbursement rates.
We cannot predict whether future health care initiatives will be implemented at the federal or state level, or how any future legislation or regulation may affect us. The taxes imposed by the new federal legislation and the expansion of government’s role in the U.S. health care industry as well as changes to the reimbursement amounts paid by payors for our products or our medical procedure volumes may reduce our profits and have a materially adverse effect on our business, financial condition, results of operations and cash flows. Moreover,
-23-
Table of Contents
Congress has proposed on several occasions to impose a 20% coinsurance on patients for clinical laboratory tests reimbursed under the clinical laboratory fee schedule, which would require us to bill patients for these amounts. Because of the relatively low reimbursement for many clinical laboratory tests, in the event that Congress were to ever enact such legislation, the cost of billing and collecting for these services would often exceed the amount actually received from the patient and effectively increase our costs of billing and collecting.
Our commercial success could be compromised if third-party payors, including managed care organizations and Medicare, do not provide coverage and reimbursement, breach, rescind or modify their contracts or reimbursement policies or delay payments for our molecular diagnostic tests.
Pathologists and oncologists may not order our molecular diagnostic tests unless third-party payors, such as managed care organizations and government payors such as Medicare and Medicaid, pay a substantial portion of the test price. Coverage and reimbursement by a third-party payor may depend on a number of factors, including a payor’s determination that tests using our technologies are:
| • | not experimental or investigational; |
| • | medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; |
| • | supported by peer-reviewed publications; and |
| • | included in clinical practice guidelines. |
Uncertainty surrounds third-party payor reimbursement of any test incorporating new technology, including tests developed using our DNA probes and microarrays. Technology assessments of new medical tests and devices conducted by research centers and other entities may be disseminated to interested parties for informational purposes. Third-party payors and health care providers may use such technology assessments as grounds to deny coverage for a test or procedure. No technology assessments have been performed on our tests to date.
Because each payor generally determines for its own enrollees or insured patients whether to cover or otherwise establish a policy to reimburse our diagnostic tests, seeking payor approvals is a time-consuming and costly process. We cannot be certain that coverage for our tests will be provided in the future by additional third-party payors or that existing contracts, agreements or policy decisions or reimbursement levels will remain in place or be fulfilled under existing terms and provisions. If we cannot obtain coverage and reimbursement from private and governmental payors such as Medicare and Medicaid for our current tests, or new tests or test enhancements that we may develop in the future, our ability to generate revenues could be limited, which may have a material adverse effect on our financial condition, results of operations and cash flow. Further, we have experienced in the past, and will likely experience in the future, delays and temporary interruptions in the receipt of payments from third-party payors due to missing documentation and other issues, which could cause delay in collecting our revenue.
We depend on Medicare and a limited number of private payors for a significant portion of our revenues and if these or other payors stop providing reimbursement or decrease the amount of reimbursement for our tests, our revenues could decline.
For the year ended December 31, 2011, we derived approximately 54% of our total revenue from private insurance, including managed care organizations and other health care insurance providers, 24% from government payor programs, most of which was derived from Medicare and 12% from direct-bill customers, including hospitals and other laboratories. In addition, for the year ended December 31, 2011, we derived approximately 10% of revenue from grants. Medicare and other third-party payors may withdraw their coverage policies or cancel their contracts with us at any time, review and adjust the rate of reimbursement or stop paying for our tests altogether, which would reduce our total revenues.
-24-
Table of Contents
Payors have increased their efforts to control the cost, utilization and delivery of health care services. In the past, measures have been undertaken to reduce payment rates for and decrease utilization of the clinical laboratory industry generally. Because of the cost-trimming trends, third-party payors that currently cover and provide reimbursement for our tests may suspend, revoke or discontinue coverage at any time, or may reduce the reimbursement rates payable to us. Any such action could have a negative impact on our revenues, which may have a material adverse effect on our financial condition, results of operations and cash flows.
In addition, we are currently considered a “non-contracting provider” by a number of private third-party payors because we have not entered into a specific contract to provide our specialized diagnostic services to their insured patients at specified rates of reimbursement. If we were to become a contracting provider in the future, the amount of overall reimbursement we receive is likely to decrease because we will be reimbursed less money per test performed at a contracted rate than at a non-contracted rate, which could have a negative impact on our revenues. Further, we typically are unable to collect payments from patients beyond that which is paid by their insurance and will continue to experience lost revenue as a result.
Because of certain Medicare billing rules, we may not receive reimbursement for all tests provided to Medicare patients.
Under current Medicare billing rules, claims for our tests performed on Medicare beneficiaries who were hospital inpatients when the tumor tissue samples were obtained and whose tests were ordered less than 14 days from discharge must be incorporated in the payment that the hospital receives for the inpatient services provided. Accordingly, we must bill individual hospitals for tests performed on Medicare beneficiaries during these timeframes in order to receive payment for our tests. Because we generally do not have a written agreement in place with these hospitals that purchase these tests, we may not be paid for our tests or may have to pursue payment from the hospital on a case-by-case basis. In addition, currently we are permitted to bill globally for certain anatomic pathology services we furnish to grandfathered hospitals, i.e. we bill both the technical component and the professional component to Medicare. As part of the Middle Class Tax Relief and Job Creation Act of 2012, Congress extended the special provision for “grandfathered” hospitals through July 1, 2012. Therefore, as of that date we were required to bill the grandfathered hospitals for the technical component of all anatomic pathology services we furnish to their patients, which may be difficult and/or costly for us.
Further, the Medicare Administrative Contractors who process claims for Medicare also can impose their own rules related to coverage and payment for laboratory services provided in their jurisdiction. Recently, Palmetto GBA, the Medicare Administrative Contractor for California and surrounding areas, announced a comprehensive new billing policy and a coverage policy applicable to molecular diagnostic tests, such as ours. Under coverage policy, Palmetto will deny payment for molecular diagnostic tests, unless it has issued a positive coverage determination for the test. If any of our tests are subject to the Palmetto policy and/or the Palmetto policy is adopted by other contractors that process claims with hospitals or laboratories that purchase and bill for our tests, our business could be adversely impacted.
Complying with numerous regulations pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties.
We are subject to CLIA, a federal law regulating clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. Our clinical laboratory must be accredited under CLIA in order for us to perform testing on human specimens. In addition, our proprietary tests must also be recognized as part of our accredited programs under CLIA so that we can offer them in our laboratory. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. We have a current certificate of accreditation under CLIA to perform high complexity testing and our laboratory is accredited by the College of American Pathologists (“CAP”), one of six CLIA-
-25-
Table of Contents
approved accreditation organizations. To renew this certificate, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make periodic inspections of our clinical reference laboratory outside of the renewal process.
The law also requires us to maintain a state laboratory license to conduct testing in that state. Our laboratory is located in New Jersey and must have a New Jersey state license; as we expand our geographic focus, we may need to obtain laboratory licenses from additional states. New Jersey laws establish standards for day-to-day operation of our clinical reference laboratory, including the training and skills required of personnel and quality control. In addition, several other states require that we hold licenses to test specimens from patients in those states. Other states may have similar requirements or may adopt similar requirements in the future. Finally, we may be subject to regulation in foreign jurisdictions as we seek to expand international distribution of our tests.
If we were to lose our CLIA accreditation or New Jersey laboratory license, whether as a result of a revocation, suspension or limitation, we would no longer be able to offer our tests, which would limit our revenues and harm our business. If we were to lose our license in other states where we are required to hold licenses, we would not be able to test specimens from those states.
If FDA were to begin requiring approval or clearance of our tests, we could incur substantial costs and time delays associated with meeting requirements for pre-market clearance or approval or we could experience decreased demand for, or reimbursement of, our tests.
Although FDA maintains that it has authority to regulate the development and use of LDTs, such as ours, as medical devices, it has not exercised its authority with respect to most LDTs as a matter of enforcement discretion. FDA does not generally extend its enforcement discretion to reagents or software provided by third parties and used to perform LDTs, and therefore these products must typically comply with FDA medical device regulations, which are wide-ranging and govern, among other things: product design and development, product testing, product labeling, product storage, pre-market clearance or approval, advertising and promotion and product sales and distribution.
We believe that our DNA probe and microarray tests, as utilized in our laboratory testing, are LDTs. As a result, we believe that pursuant to FDA’s current policies and guidance that FDA does not require that we obtain regulatory clearances or approvals for our LDTs. The container we provide for collection and transport of tumor samples from a pathology laboratory to our clinical reference laboratory may be a medical device subject to FDA regulation but is currently exempt from pre-market review by FDA. While we believe that we are currently in material compliance with applicable laws and regulations, we cannot assure you that FDA or other regulatory agencies would agree with our determination, and a determination that we have violated these laws, or a public announcement that we are being investigated for possible violations of these laws, could adversely affect our business, prospects, results of operations or financial condition.
Moreover, FDA guidance and policy pertaining to diagnostic testing is continuing to evolve and is subject to ongoing review and revision. A significant change in any of the laws, regulations or policies may require us to change our business model in order to maintain regulatory compliance. At various times since 2006, FDA has issued guidance documents or announced draft guidance regarding initiatives that may require varying levels of FDA oversight of our tests. For example, in June 2010, FDA announced a public meeting to discuss the agency’s oversight of LDTs prompted by the increased complexity of LDTs and their increasingly important role in clinical decision-making and disease management, particularly in the context of personalized medicine. FDA indicated that it was considering a risk-based application of oversight to LDTs and that, following public input and discussion, it might issue separate draft guidance on the regulation of LDTs, which ultimately could require that we seek and obtain either pre-market clearance or approval of LDTs, depending upon the risk-based approach FDA adopts. The public meeting was held in July 2010 and further public comments were submitted to FDA through September 2010. FDA has stated it is continuing to develop draft guidance in this area.
-26-
Table of Contents
Section 1143 of the Food and Drug Administration Safety and Innovation Act, signed by the U.S. President on July 9, 2012, requires FDA to notify U.S. Congress at least 60 days prior to issuing a draft or final guidance regulating LDTs and provide details of the anticipated action.
We cannot provide any assurance that FDA regulation, including pre-market review, will not be required in the future for our tests, whether through additional guidance issued by FDA, new enforcement policies adopted by FDA or new legislation enacted by Congress. We believe it is possible that legislation will be enacted into law or guidance could be issued by FDA which may result in increased regulatory burdens for us to continue to offer our tests or to develop and introduce new tests. Given the attention Congress continues to give to these issues, legislation affecting this area may be enacted into law and may result in increased regulatory burdens on us as we continue to offer our tests and to develop and introduce new tests.
In addition, the Secretary of the Department of Health and Human Services requested that its Advisory Committee on Genetics, Health and Society make recommendations about the oversight of genetic testing. A final report was published in April 2008. If the report’s recommendations for increased oversight of genetic testing were to result in further regulatory burdens, they could negatively affect our business and delay the commercialization of tests in development.
The requirement of pre-market review could negatively affect our business until such review is completed and clearance to market or approval is obtained. FDA could require that we stop selling our tests pending pre-market clearance or approval. If FDA allows our tests to remain on the market but there is uncertainty about our tests, if they are labeled investigational by FDA or if labeling claims FDA allows us to make are very limited, orders or reimbursement may decline. The regulatory approval process may involve, among other things, successfully completing additional clinical trials and making a 510(k) submission, or filing a pre-market approval application with FDA. If FDA requires pre-market review, our tests may not be cleared or approved on a timely basis, if at all. We may also decide voluntarily to pursue FDA pre-market review of our tests if we determine that doing so would be appropriate.
Additionally, should future regulatory actions affect any of the reagents we obtain from vendors and use in conducting our tests, our business could be adversely affected in the form of increased costs of testing or delays, limits or prohibitions on the purchase of reagents necessary to perform our testing.
If we were required to conduct additional clinical trials prior to continuing to offer our proprietary genetic-based tests or any other tests that we may develop as LDTs, those trials could lead to delays or failure to obtain necessary regulatory approval, which could cause significant delays in commercializing any future products and harm our ability to achieve sustained profitability.
If FDA decides to require that we obtain clearance or approvals to commercialize our proprietary genetic-based tests, we may be required to conduct additional pre-market clinical testing prior to submitting a regulatory notification or application for commercial sales. In addition, as part of our long-term strategy we plan to seek FDA clearance or approval so we can sell our proprietary tests outside our laboratory; however, we need to conduct additional clinical validation activities on our proprietary tests before we can submit an application for FDA approval or clearance. Clinical trials must be conducted in compliance with FDA regulations or FDA may take enforcement action or reject the data. The data collected from these clinical trials may ultimately be used to support market clearance or approval for our tests. We believe it would likely take two years or more to conduct the studies and trials necessary to obtain approval from FDA to commercially launch MatBA®-CLL and MatBA®-SLL outside of our clinical laboratory. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our test claims or that FDA or foreign authorities will agree with our conclusions regarding our test results. Success in early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and studies. If we are required to conduct pre-market clinical trials, whether using prospectively acquired samples or archival samples, delays in the commencement or completion of clinical testing could significantly increase our test
-27-
Table of Contents
development costs and delay commercialization. Many of the factors that may cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to delay or denial of regulatory clearance or approval. The commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial. Moreover, the clinical trial process may fail to demonstrate that our tests are effective for the proposed indicated uses, which could cause us to abandon a test candidate and may delay development of other tests.
We may find it necessary to engage contract research organizations to perform data collection and analysis and other aspects of our clinical trials, which might increase the cost and complexity of our trials. We may also depend on clinical investigators, medical institutions and contract research organizations to perform the trials properly. If these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may have to be extended, delayed or terminated. Many of these factors would be beyond our control. We may not be able to enter into replacement arrangements without undue delays or considerable expenditures. If there are delays in testing or approvals as a result of the failure to perform by third parties, our research and development costs would increase, and we may not be able to obtain regulatory clearance or approval for our tests. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all. Each of these outcomes would harm our ability to market our tests or to achieve sustained profitability.
We are subject to federal and state healthcare fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
We are subject to health care fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These health care laws and regulations include, for example:
| • | the federal Anti-kickback Statute, which prohibits, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase order or recommendation of, any item or services for which payment may be made under a federal health care program such as the Medicare and Medicaid programs; |
| • | the federal physician self-referral prohibition, commonly known as the Stark Law, which prohibits physicians from referring Medicare or Medicaid patients to providers of “designated health services” with whom the physician or a member of the physician’s immediate family has an ownership interest or compensation arrangement, unless a statutory or regulatory exception applies; |
| • | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which established federal crimes for knowingly and willfully executing a scheme to defraud any health care benefit program or making false statements in connection with the delivery of or payment for health care benefits, items or services; |
| • | federal false claims laws, which, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, and which may apply to entities like us to the extent that our interactions with customers may affect their billing or coding practices; and |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
-28-
Table of Contents
We have adopted policies and procedures designed to comply with these laws, including policies and procedures relating to financial arrangements between us and physicians who refer patients to us. In the ordinary course of our business, we conduct internal reviews of our compliance with these laws. Our compliance is also subject to governmental review. The government alleged that we engaged in improper billing practices in the past and we may be the subject of such allegations in the future as the growth of our business and sales organization may increase the potential of violating these laws or our internal policies and procedures. See the section entitled “Legal Proceedings” for a detailed description of the government’s prior allegations. The risk of our being found in violation of these laws and regulations is further increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Further, the PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal health care fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Any action brought against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of these laws and regulations, we may be subject to any applicable penalty associated with the violation, including civil and criminal penalties, damages and fines, and/or exclusion from participation in Medicare, Medi-Cal or other state or federal health care programs, we could be required to refund payments received by us, and we could be required to curtail or cease our operations. Any of the foregoing consequences could seriously harm our business and our financial results.
We recently settled a government claim related to operations at our former Milford, Massachusetts laboratory from 2003 to 2004.
From 2000 to 2004, we operated a clinical laboratory in Milford, Massachusetts providing cancer screening services, principally chromosome karyotyping. The clinical laboratory participated in the Medicare program. The Office of the Inspector General of the U.S. Department of Health and Human Services and the United States Department of Justice (together, the “Government”) informed us in February 2009 that they were contemplating commencing a civil False Claims Act action against us with respect to certain alleged improper billing practices and overpayments relating to operations at the Milford, Massachusetts clinical laboratory. While we did not and do not admit any liability nor concede that the claims of the Government are well founded, we entered into a settlement agreement and paid the Government $1 million in exchange for a release only of all common law claims. No release is specifically given with respect to other liabilities, including liabilities under the False Claims Act, and administrative liabilities, including mandatory and permissive exclusion from federal health care programs. Based on our understandings with government officials with whom we have negotiated such settlement, we do not expect the government to pursue any further claims with respect to the matters described above, but no assurances can be given.
We are required to comply with laws governing the transmission, security and privacy of health information that require significant compliance costs, and any failure to comply with these laws could result in material criminal and civil penalties.
Under the administrative simplification provisions of HIPAA, the U.S. Department of Health and Human Services has issued regulations which establish uniform standards governing the conduct of certain electronic health care transactions and protecting the privacy and security of Protected Health Information used or disclosed by health care providers and other covered entities. Three principal regulations with which we are currently required to comply have been issued in final form under HIPAA: privacy regulations, security regulations and standards for electronic transactions.
The privacy regulations cover the use and disclosure of Protected Health Information by health care providers. It also sets forth certain rights that an individual has with respect to his or her Protected Health
-29-
Table of Contents
Information maintained by a health care provider, including the right to access or amend certain records containing Protected Health Information or to request restrictions on the use or disclosure of Protected Health Information. We have also implemented policies, procedures and standards to comply appropriately with the final HIPAA security regulations, which establish requirements for safeguarding the confidentiality, integrity and availability of Protected Health Information, which is electronically transmitted or electronically stored. The HIPAA privacy and security regulations establish a uniform federal “floor” and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing Protected Health Information. As a result, we are required to comply with both HIPAA privacy regulations and varying state privacy and security laws.
Moreover, the Health Information Technology for Economic and Clinical Health Act (“HITECH”), among other things, established certain health information security breach notification requirements. A covered entity must notify any individual whose protected health information is breached.
These laws contain significant fines and other penalties for wrongful use or disclosure of Protected Health Information. We have implemented practices and procedures to meet the requirements of the HIPAA privacy regulations and state privacy laws. In addition, we are in the process of taking necessary steps to comply with HIPAA’s standards for electronic transactions, which establish standards for common health care transactions. Given the complexity of the HIPAA, HITECH and state privacy restrictions, the possibility that the regulations may change, and the fact that the regulations are subject to changing and potentially conflicting interpretation, our ability to comply with the HIPAA, HITECH and state privacy requirements is uncertain and the costs of compliance are significant. To the extent that we submit electronic health care claims and payment transactions that do not comply with the electronic data transmission standards established under HIPAA and HITECH, payments to us may be delayed or denied. Additionally, the costs of complying with any changes to the HIPAA, HITECH and state privacy restrictions may have a negative impact on our operations. We could be subject to criminal penalties and civil sanctions for failing to comply with the HIPAA, HITECH and state privacy restrictions, which could result in the incurrence of significant monetary penalties.
Intellectual Property Risks Related to Our Business
Our rights to use technologies licensed from third parties are not within our control, and we may not be able to sell our products if we lose our existing rights or cannot obtain new rights on reasonable terms.
Our ability to market certain of our tests and services, domestically and/or internationally, is in part derived from licenses to intellectual property which is owned by third parties. As such, we may not be able to continue selling our tests and services if we lose our existing licensed rights or sell new tests and services if we cannot obtain such licensed rights on reasonable terms. In particular, we in-license a biomarker used in our FHACT™ probe from the National Cancer Institute.
We may also need to license other technologies to commercialize future products. As may be expected, our business may suffer if (i) these licenses terminate; (ii) if the licensors fail to abide by the terms of the license, properly maintain the licensed intellectual property or fail to prevent infringement of such intellectual property by third parties; (iii) if the licensed patents or other intellectual property rights are found to be invalid or (iv) if we are unable to enter into necessary licenses on reasonable terms or at all. In return for the use of a third-party’s technology, we may agree to pay the licensor royalties based on sales of our products as well as other fees. Such royalties and fees are a component of cost of product revenues and will impact the margins on our tests.
We cannot sell our probes or any other tests that we may develop using blocking DNA in the United States until patents held by third parties expire.
Vysis, a division of Abbott Laboratories, Inc., possesses an exclusive license from the University of California for a family of patents in the United States (“Abbott patents”) directed broadly to the usage of
-30-
Table of Contents
blocking DNA. The Abbott patents present a barrier to our penetrating the United States market with certain of our probe-related tests because our probes are configured to use blocking DNA. The Abbott patents are due to expire in or about 2017. Unless we obtain a license from Abbott Laboratories, Inc. for use of blocking DNA or otherwise cross-license other satisfactory third party technology, we will not be able to sell our probes in the United States until the Abbott patents expire. Our current business plan does not involve developing U.S.-based sales for our DNA probe products; rather, we are currently focused entirely on growing our DNA probe business in higher growth emerging markets and select European markets.
Our collaborators may assert ownership or commercial rights to inventions we develop from our use of the biological materials which they provide to us.
We rely on certain third parties to provide us with tissue samples and biological materials that we use to develop our tests. In some cases we have written agreements with collaborators that provide that we must negotiate ownership and commercial rights with the third party collaborator if our use of such collaborator’s materials results in an invention, or that limit our use of those materials to research/not for profit use. In other cases, we do not have written agreements, or the written agreements we have do not clearly deal with intellectual property rights. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s samples, we may be limited in our ability to capitalize on the market potential of these inventions.
The U.S. government may have “march-in rights” to certain of our probe related intellectual property.
Because federal grant monies were used in support of the research and development activities that resulted in our two issued U.S. patents, the federal government retains what are referred to as “march-in rights” to these patents.
In particular, the National Cancer Institute and the National Institutes of Health, each of which administered grant monies to us, technically retain the right to require us, under certain specific circumstances, to grant the U.S. government either a nonexclusive, partially exclusive or exclusive license to the patented invention in any field of use, upon terms that are reasonable for a particular situation. Circumstances that trigger march-in rights include, for example, failure to take, within a reasonable time, effective steps to achieve practical application of the invention in a field of use, failure to satisfy the health and safety needs of the public and failure to meet requirements of public use specified by federal regulations. National Cancer Institute and the National Institutes of Health can elect to exercise these march-in rights on their own initiative or at the request of a third-party.
If we are unable to maintain intellectual property protection, our competitive position could be harmed.
Our ability to protect our proprietary discoveries and technologies affects our ability to compete and to achieve sustained profitability. Currently, we rely on a combination of U.S. and foreign patents and patent applications, copyrights, trademarks and trademark applications, confidentiality or non-disclosure agreements, material transfer agreements, licenses, work-for-hire agreements and invention assignment agreements to protect our intellectual property rights. We also maintain certain company know-how, trade secrets and technological innovations designed to provide us with a competitive advantage in the market place as trade secrets. Currently, we have only two issued U.S. patents and twelve pending patent applications, which includes both U.S. and foreign patent applications, relating to various aspects of our technology. While we intend to pursue additional patent applications, it is possible that our pending patent applications and any future applications may not result in issued patents. Even if patents are issued, third parties may independently develop similar or competing technology that avoids our patents. Further, we cannot be certain that the steps we have taken will prevent the misappropriation of our trade secrets and other confidential information as well as the misuse of our patents and other intellectual property, particularly in foreign countries where we have not filed for patent protection.
-31-
Table of Contents
From time to time the U.S. Supreme Court, other federal courts, the U.S. Congress or the U.S. Patent and Trademark Office (“USPTO”) may change the standards of patentability and any such changes could have a negative impact on our business. For instance, on October 30, 2008, the Court of Appeals for the Federal Circuit issued a decision that methods or processes cannot be patented unless they are tied to a machine or involve a physical transformation. The U.S. Supreme Court later reversed that decision in Bilski v. Kappos, finding that the “machine-or-transformation” test is not the only test for determining patent eligibility. The Court, however, declined to specify how and when processes are patentable. Most recently, on March 20, 2012, in the case Mayo v. Prometheus, the U.S. Supreme Court reversed the Federal Circuit’s application of Bilski and invalidated a patent focused on a diagnostic process because the patent claim embodied a law of nature. It is unclear at this time whether the USPTO will amend its patent prosecution guidelines for determining patentability of diagnostic or other processes, and how lower courts will implement the decision. Some aspects of our technology involve processes that may be subject to this evolving standard and we can not guarantee that any of our pending process claims will be patentable as a result of such evolving standards.
More recently a suit brought in the U.S. District Court for the Southern District of New York by multiple plaintiffs, including the American Civil Liberties Union, against Myriad Genetics, Inc. and the USPTO may have an impact on the biotechnology industry. Specifically, the case involves certain of Myriad Genetics, Inc.’s U.S. patents related to the breast cancer susceptibility genes BRCA1 and BRCA2. Plaintiffs allege, among other things, that gene-related patents (as a whole) stifle diagnostic testing and research that could lead to cures in the future. In that regard, plaintiffs filed motions for summary judgment alleging, among other things, that breast cancer genes are not patentable subject matter. On March 29, 2010, the court granted summary judgment finding that BRCA1 and BRCA2 patents are invalid under the “machine-or-transformation” test discussed above. On July 29, 2011, the Federal Circuit upheld the lower court on the invalidity of all but one of the process claims as failing the “machine-or-transformation” test, but reversed the lower court’s decision as to isolated genes, holding them patentable. On March 26, 2012, the U.S. Supreme Court vacated the Federal Circuit decision, and ordered the appellate court to reconsider the case in light of the recent Supreme Court decision in Mayo v. Prometheus discussed above, and the validity of patents on isolated genes remains uncertain.
In addition, on February 5, 2010, the Secretary’s Advisory Committee on Genetics, Health and Society voted to approve a report entitled “Gene Patents and Licensing Practices and Their Impact on Patient Access to Genetic Tests.” That report defines “patent claims on genes” broadly to include claims to isolated nucleic acid molecules as well as methods of detecting particular sequences or mutations. The report also contains six recommendations, including the creation of an exemption from liability for infringement of patent claims on genes for anyone making, using, ordering, offering for sale or selling a test developed under the patent for patient care purposes, or for anyone using the patent-protected genes in the pursuit of research. The report also recommended that the Secretary should explore, identify and implement mechanisms that will encourage more voluntary adherence to current guidelines that promote nonexclusive in-licensing of diagnostic genetic and genomic technologies. It is unclear whether the U.S. Department of Health and Human Services will act upon these recommendations, or if the recommendations would result in a change in law or process that could negatively impact our patent portfolio or future research and development efforts.
We may face intellectual property infringement claims that could be time-consuming and costly to defend, and could result in our loss of significant rights and the assessment of treble damages.
From time to time we may face intellectual property infringement (or misappropriation) claims from third parties. Some of these claims may lead to litigation. The outcome of any such litigation can never be guaranteed, and an adverse outcome could affect us negatively. For example, were a third-party to succeed on an infringement claim against us, we may be required to pay substantial damages (including up to treble damages if such infringement were found to be willful). In addition, we could face an injunction, barring us from conducting the allegedly infringing activity. The outcome of the litigation could require us to enter into a license agreement which may not be pursuant to acceptable or commercially reasonable or practical terms or which may not be available at all.
-32-
Table of Contents
It is also possible that an adverse finding of infringement against us may require us to dedicate substantial resources and time in developing non-infringing alternatives, which may or may not be possible. In the case of diagnostic tests, we would also need to include non-infringing technologies which would require us to re-validate our tests. Any such re-validation, in addition to being costly and time consuming, may be unsuccessful.
Finally, we may initiate claims to assert or defend our own intellectual property against third parties. Any intellectual property litigation, irrespective of whether we are the plaintiff or the defendant, and regardless of the outcome, is expensive and time-consuming, and could divert our management’s attention from our business and negatively affect our operating results or financial condition.
Risks Relating to Our Common Stock and This Offering
The price of our common stock may be volatile, and the market price of our common stock after this offering may drop below the price you pay.
The initial public offering price per share may vary from the market price of our common stock after the offering. If an active market for our stock develops and continues, our stock price nevertheless may be volatile. Market prices for securities of early-stage life sciences companies have historically been particularly volatile. As a result of this volatility, you may not be able to sell your common stock at or above the initial public offering price per share. The factors that may cause the market price of our common stock to fluctuate include, but are not limited to:
| • | progress, or lack of progress, in developing and commercializing our proprietary tests; |
| • | favorable or unfavorable decisions about our tests or services from government regulators, insurance companies or other third-party payors; |
| • | our ability to recruit and retain qualified regulatory and research and development personnel; |
| • | changes in investors’ and securities analysts’ perception of the business risks and conditions of our business; |
| • | changes in our relationship with key collaborators; |
| • | changes in the market valuation or earnings of our competitors or companies viewed as similar to us; |
| • | changes in key personnel; |
| • | depth of the trading market in our common stock; |
| • | termination of the lock-up agreement or other restrictions on the ability of our existing stockholders to sell shares after this offering; |
| • | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| • | the granting or exercise of employee stock options or other equity awards; |
| • | realization of any of the risks described under this section entitled “Risk Factors”; and |
| • | general market and economic conditions. |
-33-
Table of Contents
In addition, the equity markets have experienced significant price and volume fluctuations that have affected the market prices for the securities of newly public companies for a number of reasons, including reasons that may be unrelated to our business or operating performance. These broad market fluctuations may result in a material decline in the market price of our common stock and you may not be able to sell your shares at prices you deem acceptable. In the past, following periods of volatility in the equity markets, securities class action lawsuits have been instituted against public companies. Such litigation, if instituted against us, could result in substantial cost and the diversion of management attention.
The shares you purchase in this offering will experience immediate and substantial dilution and may also be diluted by exercises of outstanding options and warrants.
The initial public offering price per share of our common stock will be substantially higher than the net tangible book value per share of our common stock immediately after the offering. At an initial public offering price of $7.00 per share, the midpoint of the range set forth on the cover of this prospectus, purchasers of our common stock will effectively incur dilution of $5.67 per share in the net tangible book value of their purchased shares. Conversely, the shares of our common stock that our existing stockholders currently own will receive a material increase in net tangible book value per share. As of December 31, 2012, we had outstanding options to purchase an aggregate of 1,383,350 shares of our common stock at a weighted average exercise price of $5.10 per share and warrants to purchase an aggregate of 3,942,198 shares of our common stock at a weighted average exercise price of $6.15 per share. In addition, if the initial public offering price were $6.00 per share, which is the bottom of the range set forth on the cover page of this prospectus, the number of shares outstanding as of December 31, 2012 would increase by 5.6% to 6,774,379 shares and the number of warrants outstanding as of December 31, 2012 would increase by 11.0% to 4,377,310 at a weighted average exercise price of $5.41 per share as a result of certain anti-dilution provisions in certain preferred stock, convertible notes and warrants. The exercise of such outstanding options and warrants will result in further dilution of your investment. In addition, you may experience additional dilution if we issue common stock in the future. As a result of this dilution, you may receive significantly less than the full purchase price you paid for the shares in the event of liquidation.
Future sales of our common stock, or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing well.
Sales of substantial amounts of our common stock in the public market after this offering, or the perception that these sales may occur, could materially and adversely affect the price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. The shares of common stock sold in this offering will be freely tradable, without restriction, in the public market, except for any shares sold to our affiliates.
In connection with this offering, we, along with our officers, directors and certain stockholders, have agreed prior to the commencement of this offering, subject to limited exceptions, not to sell or transfer any shares of common stock for 180 days after the date of this prospectus without the consent of Aegis Capital Corp. However, Aegis Capital Corp. may release these shares from any restrictions at any time. We cannot predict what effect, if any, market sales of shares held by any stockholder or the availability of shares for future sale will have on the market price of our common stock.
A total of 6,105,187 shares of common stock may be sold in the public market by existing stockholders on or about 181 days after the date of this prospectus, subject to volume and other limitations imposed under the federal securities laws. Sales of substantial amounts of our common stock in the public market after the completion of this offering, or the perception that such sales could occur, could adversely affect the market price of our common stock and could materially impair our ability to raise capital through offerings of our common stock. See the section entitled “Shares Eligible for Future Sale” for a more detailed description of the restrictions on selling shares of our common stock after this offering.
-34-
Table of Contents
In addition, as of December 31, 2012, we had outstanding options to purchase 1,383,350 shares of our common stock and outstanding warrants to purchase an aggregate of 2,755,442 shares of our common stock. (As a result of certain anti-dilution provisions in certain outstanding warrants, the number of shares of common stock underlying outstanding warrants would increase by 43.1% to 3,942,198 at an offering price of $7.00 per share.) We have also reserved an aggregate of 133,750 shares for option grants with an exercise price equal to the initial public offering price per share to be issued upon consummation of the initial public offering. We plan to register for offer and sale the shares of common stock that are reserved for issuance pursuant to outstanding options. Shares covered by such registration statements upon the exercise of stock options generally will be eligible for sale in the public market, except that affiliates will continue to be subject to volume limitations and other requirements of Rule 144 under the Securities Act of 1933, as amended, or the Securities Act. The issuance or sale of such shares could depress the market price of our common stock.
An active trading market may not develop for our common stock, and you may not be able to sell your stock at or above the initial public offering price per share.
There is no established trading market for our common stock, and the market for our common stock may be highly volatile or may decline regardless of our operating performance. Prior to this offering, you could not buy or sell our equity publicly. An active public market for our common stock may not develop or be sustained after this offering. We cannot predict the extent to which investor interest in our company will lead to the development of an active trading market in our common stock or how liquid that market might become. If a market does not develop or is not sustained, it may be difficult for you to sell your shares of common stock at the time you wish to sell them, at a price that is attractive to you, or at all.
The initial public offering price per share has been determined through negotiation between us and representatives of the underwriters, and may not be indicative of the market price for our common stock after this offering. You may not be able to sell your shares at or above the initial public offering price per share.
Reports published by securities or industry analysts, including projections in those reports that exceed our actual results, could adversely affect our common stock price and trading volume.
We currently expect that securities research analysts, including those affiliated with our underwriters, will establish and publish their own periodic projections for our business. These projections may vary widely from one another and may not accurately predict the results we actually achieve. Our stock price may decline if our actual results do not match securities research analysts’ projections. Similarly, if one or more of the analysts who writes reports on us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price could decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, our stock price or trading volume could decline. While we expect securities research analyst coverage, if no securities or industry analysts begin to cover us, the trading price for our stock and the trading volume could be adversely affected.
Our directors and executive officers will continue to have substantial influence over us after this offering and could delay or prevent a change in corporate control.
Our directors and executive officers, together with their affiliates, beneficially own approximately 67.4% of our outstanding common stock-based on the number of shares outstanding on December 31, 2012 and, upon the closing of this offering, assuming no exercise of the underwriters’ option to purchase additional shares, will beneficially own approximately 44.5% of our outstanding shares of common stock. These stockholders, acting together, have significant influence over the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our
-35-
Table of Contents
assets. In addition, these stockholders, acting together, have significant influence over our management and affairs. Accordingly, this concentration of ownership might harm the market price of our common stock by:
| • | delaying, deferring or preventing a change in control; |
| • | impeding a merger, consolidation, takeover or other business combination involving us; or |
| • | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
If we are unable to favorably assess the effectiveness of our internal control over financial reporting, or if our independent registered public accounting firm is not able to provide an unqualified attestation report on the effectiveness of our internal controls over financial reporting, investors may lose confidence in our financial reporting and our stock price could be materially adversely affected.
As a private company, we were not subject to the requirements of Section 404 of the Sarbanes-Oxley Act of 2002. After completion of this offering, we will be required to document and test our internal control over financial reporting. For the year ended December 31, 2011, our independent registered public accounting firm reported a material weakness in our internal control over financial reporting related to our monitoring of the performance of the third-party service providers we use in our revenue cycle. During 2011, we changed third-party service providers to improve our platform for future growth. After the conversion, we identified instances of delayed billings and collection efforts and procedural issues with the timely application of cash receipts. If we fail to remediate the material weaknesses identified or to remediate any significant deficiencies or material weaknesses that may be identified in the future, we may be unable to conclude that our internal control over financial reporting is effective and our independent registered public accounting firm may not be able to provide an attestation reporting on the effectiveness of our internal control over financial reporting to the extent such an attestation report would be required. On April 5, 2012, President Obama signed the JOBS Act. Under the JOBS Act, issuers that qualify as “emerging growth companies” under the JOBS Act will not be required to provide an auditor’s attestation report on internal controls for so long as the issuer qualifies as an emerging growth company. We currently qualify as an emerging growth company under the JOBS Act and we may choose not to provide an auditor’s attestation report on internal controls. However, if we cannot favorably assess the effectiveness of our internal control over financial reporting, or if we require an attestation report from our independent registered public accounting firm and that firm is unable to provide an unqualified attestation report on the effectiveness of our internal controls over financial reporting, investor confidence and, in turn, our stock price could be materially adversely affected. For a discussion on our remediation of our material weaknesses please see “Management’s Discussion and Analysis-Internal Control over Financial Reporting.”
We are an “emerging growth company,” and any decision on our part to comply only with certain reduced disclosure requirements applicable to “emerging growth companies” could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and, for as long as we continue to be an “emerging growth company,” we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies but not to “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as discussed above, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an “emerging growth company” for up to five years, or until the earliest of (i) the last day of the first fiscal year in which our annual gross revenues exceed $1 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our common stock that is held by non-affiliates exceeds
-36-
Table of Contents
$700 million as of the last business day of our most recently completed second fiscal quarter, or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three year period. Except for our decision to “opt out” of the extended transition periods available under the JOBS Act for complying with new or revised accounting standards, we have not made a decision whether to take advantage of any or all of these exemptions, and if we do take advantage of these exemptions, we cannot predict if investors will find our common stock less attractive as a result. If some investors find our common stock less attractive as a result of any choices to take advantage of these reduced disclosure obligations, there may be a less active trading market for our common stock and our stock price may be more volatile.
We will incur significantly increased costs and devote substantial management time as a result of operating as a public company particularly after we are no longer an “emerging growth company.”
As a public company and particularly after we cease to be an “emerging growth company”, we will incur significant legal, accounting and other expenses that we did not incur as a private company. For example, in addition to being required to comply with certain requirements of the Sarbanes-Oxley Act of 2002, we will be required to comply with certain requirements of the Dodd Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations subsequently implemented by the SEC, and the NASDAQ Capital Market, our stock exchange, including the establishment and maintenance of effective disclosure and financial controls and changes in corporate goverance practices. We expect that compliance with these requirements will increase our legal and financial compliance costs and will make some activities more time consuming and costly. In addition, we expect that our management and other personnel will need to divert attention from operational and other business matters to devote substantial time to these public company requirements.
However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may choose to take advantage of these reporting exemptions until we no longer qualify as an “emerging growth company.”
Under the JOBS Act, “emerging growth companies” can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to take advantage of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not “emerging growth companies.”
After we are no longer an “emerging growth company”, we expect to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 and the other rules and regulations of the SEC. We cannot predict or estimate the amount of additional costs we may incur as a result of becoming a public company or the timing of such costs. We also expect that operating as a public company will make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers.
Our management will have broad discretion over the use of the proceeds we receive in this offering, and may not apply the proceeds in ways that increase the value of your investment.
We estimate that net proceeds of the sale of the common stock that we are offering will be approximately $29.3 million, or $34.2 million, if the underwriters exercise their option to purchase additional shares in this offering in full, based on an assumed initial public offering price of $7.00 per share, the midpoint of
-37-
Table of Contents
the price range set forth on the cover page of this prospectus. We currently intend to use the net proceeds of the offering to fund further research and development, potential regulatory submissions and potential commercial launch of our proprietary genomic-based diagnostic tests and potential collaborations; to hire additional sales and marketing personnel and support increased sales and marketing activities; to fund ongoing operations and expansion of the business; to fund our initial contribution to our joint venture with Mayo; and to repay certain outstanding indebtedness. However, other than the repayment of $5.0 million in outstanding indebtedness, we will have broad discretion in the application of the net proceeds, and investors will be relying on our judgment regarding the application of the proceeds of this offering. The actual amounts and timing of our actual expenditures depend on numerous factors, including the success of our efforts to market MatBA®-CLL and MatBA® -SLL, to obtain regulatory approval to sell MatBA®-CLL and MatBA®-SLL outside our clinical laboratory, the timing and progress of our discovery, research and development activities for the tests in our pipeline, the success of our efforts to increase sales of our laboratory services, the success of our efforts to expand our international sales, our ability to continue to reduce manufacturing costs by leveraging operations in low cost countries, changes in regulatory requirements for LDTs, and other unforeseen regulatory or compliance costs. The costs and timing of test discovery and development activities, particularly conducting clinical validation studies and obtaining regulatory clearance or approval, are highly uncertain, subject to substantial risks and can often change. Depending on the outcome of these activities, our plans and priorities may change and we may apply the net proceeds of this offering differently than we currently anticipate. Moreover, you will not have the opportunity to influence our decision on how to use the proceeds from this offering. We may use the proceeds for corporate purposes that do not immediately enhance our prospects for the future or increase the value of your investment. See the Section entitled “Use of Proceeds.”
Anti-takeover provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our amended and restated certificate of incorporation and bylaws that will be in effect upon the completion of this offering could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors. These provisions also could limit the price that investors might be willing to pay in the future for our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions, among other things:
| • | allow the authorized number of directors to be changed only by resolution of our board of directors; |
| • | authorize our board of directors to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; |
| • | establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings; and |
| • | limit who may call a stockholder meeting. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
-38-
Table of Contents
Because we do not expect to pay cash dividends for the foreseeable future, you must rely on appreciation of our common stock price for any return on your investment. Even if we change that policy, we may be restricted from paying dividends on our common stock.
We do not intend to pay cash dividends on shares of our common stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend upon results of operations, financial performance, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Investors seeking cash dividends in the foreseeable future should not purchase our common stock.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Our ability to utilize our federal net operating loss, carryforwards and federal tax credit may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended. The limitations apply if an “ownership change,” as defined by Section 382, occurs. Generally, an ownership change occurs if the percentage of the value of the stock that is owned by one or more direct or indirect “five percent shareholders” increases by more than 50 percentage points over their lowest ownership percentage at any time during the applicable testing period (typically three years). If we have experienced an “ownership change” at any time since our formation, we may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other tax attributes to offset United States federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us.
-39-
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements, including statements regarding the progress and timing of our product development, the goals of our development activities, estimates of the potential markets for our product candidates, estimates of the capacity of manufacturing and other facilities to support our products, our expected further revenues, operations and expenditures and projected cash needs. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business.” These statements relate to future events of our financial performance and involve known and unknown risks, uncertainties and other factors that could cause our actual results, levels of activity, performance or achievement to differ materially from those expressed or implied by these forward-looking statements. Those risks and uncertainties include, among others:
| • | our ability to achieve profitability by increasing sales of our laboratory tests and services and to continually develop and commercialize novel and innovative genomic-based diagnostic tests and services for cancer patients; |
| • | our ability to clinically validate our pipeline of genomic microarray tests currently in development; |
| • | our ability to execute on our marketing and sales strategy for our genomic tests and gain acceptance of our tests in the market; |
| • | our ability to keep pace with a rapidly advancing market; |
| • | our ability to satisfy U.S. (including FDA) and international regulatory requirements with respect to our tests and services, many of which are new and still evolving; |
| • | our ability to obtain reimbursement from governmental and other third-party payors for our tests and services; |
| • | competition from clinical laboratory services companies, genomic-based diagnostic tests currently available or new tests that may emerge; |
| • | our ability to maintain our clinical collaborations and enter into new collaboration agreements with highly regarded organizations in the cancer field so that, among other things, have access to thought leaders in the field and to a robust number of samples to validate our genomic tests; |
| • | our ability to maintain our present customer base and retain new customers; |
| • | potential product liability or intellectual property infringement claims; |
| • | our dependency on third-party manufacturers to supply or manufacture our products; |
| • | our ability to attract and retain a sufficient number of scientists, clinicians, sales personnel and other key personnel with extensive experience in oncology, who are in short supply; |
| • | our ability to obtain or maintain patents or other appropriate protection for the intellectual property in our proprietary tests and services; |
| • | our dependency on the intellectual property licensed to us or possessed by third parties; |
| • | our ability to raise additional capital to meet our liquidity needs; |
-40-
Table of Contents
| • | our ability to expand internationally and launch our tests in emerging markets, such as India and Brazil; and |
| • | our ability to adequately support future growth. |
Forward-looking statements include all statements that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential,” or the negative of those terms, and similar expressions and comparable terminology intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. These forward-looking statements represent our estimates and assumptions only as of the date of this prospectus and, except as required by law, we undertake no obligation to update or review publicly any forward-looking statements, whether as a result of new information, future events or otherwise after the date of this prospectus. You should read this prospectus and the documents referenced in this prospectus and filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
-41-
Table of Contents
We estimate that we will receive net proceeds of approximately $29.3 million from the sale of shares of common stock offered in this offering, or approximately $34.2 million if the underwriters exercise their over-allotment option in full, based on an assumed initial public offering price of $7.00 per share (the midpoint of the price range set forth on the cover page of this prospectus) and after deducting the estimated underwriting discounts and commissions and estimated offering expenses. Of the estimated offering expenses, we already paid approximately $1.3 million and therefore will have approximately $1.3 million of additional net proceeds from this offering available to us.
Each $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share (the midpoint of the price range set forth on the cover page of this prospectus) would increase (decrease) the net proceeds to us from this offering, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, by approximately $4.7 million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same. We may also increase or decrease the number of shares we are offering. An increase (decrease) of 1,000,000 in the number of shares we are offering would increase (decrease) the net proceeds to us from this offering, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, by approximately $6.5 million, assuming the initial public offering price per share stays the same. An increase of 1,000,000 in the number of shares we are offering, together with a $1.00 increase in the assumed initial public offering price per share, would increase the net proceeds to us from this offering, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, by approximately $12.1 million. A decrease of 1,000,000 in the number of shares we are offering, together with a $1.00 decrease in the assumed initial public offering price per share, would decrease the net proceeds to us from this offering, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, by approximately $10.2 million. We do not expect that a change in the offering price per share or the number of shares by these amounts would have a material effect on our intended uses of the net proceeds from this offering, although it may impact the amount of time prior to which we may need to seek additional capital.
We currently intend to use the net proceeds of the offering as follows:
| • | approximately $5.0 million to repay certain outstanding indebtedness, as described below; |
| • | approximately $5.0 million to fund further research and development, potential regulatory submissions (including but not limited to the costs for seeking FDA approval to commercially launch our proprietary genomic-based diagnostic tests) and the potential commercial launch of our proprietary tests and potential collaborations; |
| • | approximately $7.0 million to hire additional sales and marketing personnel and support increased sales and marketing activities; |
| • | $2.0 million to fund our initial contribution to our joint venture with Mayo; and |
| • | approximately $10.0 million to fund ongoing operations and expansion of the business. |
The expected use of net proceeds of this offering represents our current intentions based upon our present plan and business conditions. As of the date of this prospectus, we cannot specify with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering. For example, if we identify opportunities that we believe are in the best interests of our stockholders, we may use a portion of the net proceeds from this offering to acquire, invest in or license complementary products, technologies or businesses although we have no current commitments, understandings or agreements to do so. Other than the obligation to repay outstanding indebtedness of approximately $5.0 million, we will have broad discretion in the application of the net proceeds, and investors will be relying on our judgment regarding the application of the proceeds of this
-42-
Table of Contents
offering. The actual amounts and timing of our actual expenditures depend on numerous factors, including the success of our efforts to market MatBA®-CLL and MatBA®-SLL, to obtain regulatory approval to sell MatBA®-CLL and MatBA®-SLL outside our clinical laboratory, the timing and progress of our discovery, research and development activities for the tests in our pipeline, the success of our efforts to increase sales of our laboratory services, the success of our efforts to expand our international sales, our ability to continue to reduce manufacturing costs by leveraging operations in low cost countries, changes in regulatory requirements for LDTs, and other unforeseen regulatory or compliance costs. The costs and timing of test discovery and development activities, particularly conducting clinical validation studies and obtaining regulatory clearance or approval, are highly uncertain, subject to substantial risks and can often change. Depending on the outcome of these activities, our plans and priorities may change and we may apply the net proceeds of this offering differently than we currently anticipate. For example, if our joint venture with Mayo does not close on or before March 31, 2013, we will not be required to make an initial $2.0 million capital contribution, or if once formed the joint venture fails to achieve certain operational milestones, we may not be required to make additional capital contributions.
As of September 30, 2012, an aggregate of $3.0 million in principal remained outstanding under the DAM credit facility. Effective January 1, 2012, the DAM debt bears interest at an annual rate of 10.0%, payable in equal monthly installments. The DAM debt is due on April 1, 2013 unless this offering or certain other maturity events occur prior to such date. We expect to repay the outstanding indebtedness to DAM with proceeds from this offering.
As of September 30, 2012, we had an aggregate of $6.0 million outstanding pursuant to a Credit Agreement dated December 21, 2011, as amended and restated as of February 13, 2012 with John Pappajohn, a member of our board of directors and a stockholder, Dr. Pecora (indirectly through an investment company), a member of our board of directors, and NNJCA Capital, LLC, a limited liability company of which Dr. Pecora is a member. The $6.0 million loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures in twelve months, with an option at our election, if there has been no event of default, to extend the loan term for an additional six months. The term loan is due on August 13, 2013 (except as with respect to amounts due to Mr. Pappajohn, as described below); provided, however, that the lenders may require repayment of the $2.0 million due to Dr. Pecora and NNJCA, plus accrued interest and prepayment premium within 30 days of consummation of this offering. We expect to repay the indebtedness due to Dr. Pecora and NNJCA with proceeds from this offering. Mr. Pappajohn will convert $2.0 million of the indebtedness due to him at the initial offering price per share upon consummation of this offering and we expect that the remaining $2.0 million due to Mr. Pappajohn will remain outstanding until April 1, 2014. We used a portion of the net proceeds from this financing to pay approximately $1.3 million in expenses incurred in connection with our initial public offering. See the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—December 2011 Financing Transaction.”
-43-
Table of Contents
We have never declared dividends on our equity securities, and currently do not plan to declare dividends on shares of our common stock in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our board of directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our board of directors.
-44-
Table of Contents
The following table sets forth our capitalization as of September 30, 2012:
| • | on an actual basis; |
| • | on a pro forma basis as of September 30, 2012, to reflect the automatic conversion prior to the closing of this offering of all outstanding shares of preferred stock into 2,182,680 shares of common stock and the conversion of convertible promissory notes in the amount of $6.0 million, including $2.0 million issued after September 30, 2012, at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, into an aggregate of 857,143 shares of our common stock upon the closing of this offering, the reclassification of certain warrants with a fair value of $4.9 million to equity due to the lapsing of anti-dilution provisions upon the offering date, and the resulting recognition of unamortized debt discount and fees of $3.4 million and a contingently recognizable beneficial conversion feature of $2.4 million; and |
| • | on a pro forma as-adjusted basis to give effect to the sale of 5,000,000 shares of the common stock we are offering at the initial public offering price of $7.00 per share, which is the midpoint of the estimated price range shown on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us and the repayment of outstanding indebtedness of approximately $5.0 million resulting in the recognition of unamortized debt discounts of $0.5 million and fees and prepayment penalties of $0.3 million. The pro forma as-adjusted column assumes no exercise by the underwriters of their over-allotment option. |
The information below is illustrative only and our capitalization following the completion of this offering will be adjusted based on the actual initial public price. You should read this table together with the sections entitled “Use of Proceeds” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” as well as our financial statements and the related notes, which appear elsewhere in this prospectus.
| As of September 30, 2012 | ||||||||||||
| (unaudited) | ||||||||||||
| Actual | Pro Forma |
Pro Forma As Adjusted(1) |
||||||||||
| ($ in thousands) | ||||||||||||
| Cash and cash equivalents |
$ | 903 | $ | 1,903 | $ | 27,291 | ||||||
|
|
|
|
|
|
|
|||||||
| Long term debt and lines of credit |
$ | 15,081 | $ | 12,426 | $ | 7,942 | ||||||
| Convertible preferred stock, Series A 588,000 shares authorized, 587,691 shares issued and outstanding, actual; 588,000 shares authorized, no shares issued and outstanding, pro forma; 588,000 shares authorized, no shares issued and outstanding, pro forma as adjusted(2) |
$ | — | $ | — | $ | — | ||||||
| Convertible preferred stock, Series B 2,000,000 shares authorized, 1,821,600 shares issued and outstanding, actual; 2,000,000 shares authorized, no shares issued and outstanding, pro forma; 2,000,000 shares authorized, no shares issued and outstanding, pro forma as adjusted(2) |
$ | — | $ | — | $ | — | ||||||
| Common stock, additional paid-in capital and treasury stock 24,000,000 shares authorized, 3,365,311 shares issued and outstanding, actual; 100,000,000 shares authorized, 6,405,134 shares issued and outstanding, pro forma; 100,000,000 shares authorized, 11,405,134 shares issued and outstanding, pro forma as adjusted |
$ | 24,733 | $ | 38,037 | $ | 67,082 | ||||||
| Accumulated deficit |
$ | (44,890 | ) | $ | (50,710 | ) | $ | 51,515 | ||||
|
|
|
|
|
|
|
|||||||
| Total stockholders’ equity (deficit) |
$ | (20,157 | ) | $ | (12,673 | ) | $ | 15,567 | ||||
|
|
|
|
|
|
|
|||||||
| Total capitalization |
$ | (5,076 | ) | $ | (247 | ) | $ | 23,509 | ||||
|
|
|
|
|
|
|
|||||||
-45-
Table of Contents
(1) A $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share of our common stock, the midpoint of the estimated price range set forth on the cover page of this prospectus, in this offering would increase (decrease) each of cash, total stockholders’ deficit and total capitalization by $4.7 million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
(2) The amounts presented in the “actual” and “pro forma” columns are the same due to rounding of dollar amounts. The pro forma column reflects the conversion of all outstanding shares of preferred stock into 2,182,680 shares of common stock.
The number of shares of common stock to be outstanding after the offering is based on the number of shares outstanding as of September 30, 2012, and excludes:
| • | 9,532 shares of common stock issued after September 30, 2012 and before the date of this prospectus upon the exercise of warrants to purchase such number of shares at an exercise price of $1.60 per share; |
| • | warrants to purchase 8,781 shares of common stock with an exercise price of $1.60 per share which expired unexercised after September 30, 2012; |
| • | warrants to purchase 331,723 shares of common stock with an exercise price of the lesser of $17.00 per share or our initial public offering price per share, which were issued after September 30, 2012 (net of warrants cancelled after September 30, 2012) and before the date of this prospectus; |
| • | stock options to purchase 133,750 shares of common stock to be issued upon the completion of this offering with an exercise price equal to our initial public offering price; |
| • | 1,383,350 shares of our common stock issuable upon the exercise of stock options as of September 30, 2012, with a weighted average exercise price of $5.10 per share, which includes 1,183,350 shares of our common stock issuable upon the exercise of stock options issued under our equity incentive plans and 200,000 shares of our common stock issuable upon the exercise of stock options issued outside of our equity incentive plans; |
| • | 2,442,032 additional shares of our common stock issuable upon the exercise of outstanding warrants as of September 30, 2012, at a weighted average exercise price of $9.12 per share; |
| • | 1,066,050 additional shares of our common stock to be reserved for future issuance under our equity incentive plans as of September 30, 2012; |
| • | 2,674 shares of common stock issuable to Cleveland Clinic pursuant to our license agreement with Cleveland Clinic; and |
| • | the number of shares of common stock issuable upon the exercise of outstanding warrants exclude the effects of anti-dilution provisions that will occur upon completion of this offering. At an offering price of $7.00 per share, an aggregate of 3,942,198 shares of common stock would be issuable upon exercise of all of the outstanding warrants described above. |
-46-
Table of Contents
If you invest in our common stock in this offering, your ownership interest will be diluted to the extent of the difference between the
initial public offering price per share of our common stock in this offering and our pro forma as adjusted net tangible book value per share immediately after this offering. We calculate net tangible book value per share by dividing our net
tangible book value, which is tangible assets less total liabilities less debt discounts related to debt to be paid or converted as a result of this offering, by the number of outstanding shares of our common stock. Prior to considering the effects
of the proceeds of this offering, but giving effect to the automatic conversion of our outstanding shares of Series A and Series B preferred stock into 2,182,680 shares of our common stock upon completion of this offering and the
conversion of convertible promissory notes in the amount of $6.0 million, including $2.0 million issued after September 30, 2012, at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of
this prospectus, into an aggregate of 857,143 shares of our common stock, our pro forma net tangible book value (deficit) as of September 30, 2012 was approximately $(16.8) million, or approximately $(2.63) per share. Upon completion
of this offering, our pro forma net tangible book value as of September 30, 2012 will be approximately $15.2 million or approximately
$1.33 per share. This represents an immediate increase in pro forma net tangible book value of $3.96
per share to our existing stockholders and an immediate dilution of $5.67 per share to new investors purchasing our common stock in this offering. The following table illustrates the per share dilution:
| Assumed initial public offering price per share |
$ | 7.00 | ||||||
| Pro forma net tangible book value per share as of September 30, 2012 |
$ | (2.63 | ) | |||||
| Increase in pro forma net tangible book value per share after this offering |
3.96 | |||||||
|
|
|
|||||||
| Pro forma as adjusted net tangible book value per share after this offering |
1.33 | |||||||
|
|
|
|||||||
| Dilution in pro forma net tangible book value per share to new investors |
$ | 5.67 | ||||||
|
|
|
Each $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share, the midpoint of the range set forth on the cover page of this prospectus, would increase (decrease) our pro forma net tangible book value by approximately $4.7 million, the pro forma net tangible book value per share by approximately $0.41 per share and the dilution to investors purchasing shares of our common stock in this offering by approximately $0.41 per share, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
The information above assumes that the underwriters do not exercise their over-allotment option. If the underwriters exercise their over-allotment option in full, the pro forma as adjusted net tangible book value will increase to $1.65 per share, representing an immediate increase to existing stockholders of $4.28 per share and an immediate dilution of $5.35 per share to new investors. If any shares are issued upon exercise of outstanding options or warrants, new investors will experience further dilution.
The following table summarizes, on a pro forma as adjusted basis as of September 30, 2012, the differences between the number of shares of common stock purchased from us, the total consideration and the average price per share paid by existing stockholders and by investors participating in this offering, after deducting estimated underwriting discounts and commissions and estimated offering expenses, at an assumed initial public offering price of $7.00 per share, the midpoint of the estimated price range shown on the cover page of this prospectus.
| Shares Purchased | Total Consideration | Average Price |
||||||||||||||||||
| Number | % | Amount | % | Per Share | ||||||||||||||||
| Existing stockholders |
6,405,134 | 56 | $ | 22,607,060 | 39 | $ | 3.53 | |||||||||||||
| New investors |
5,000,000 | 44 | 35,000,000 | 61 | 7.00 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
11,405,134 | 100 | $ | 57,607,060 | 100 | $ | 5.05 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
-47-
Table of Contents
The number of shares purchased from us by existing stockholders is based on 6,405,134 shares of our common stock outstanding as of September 30, 2012 after giving effect to the conversion of convertible promissory notes in the amount of $6.0 million held by Mr. Pappajohn and Mr. Oman, including $2.0 million issued after September 30, 2012, at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, and to the automatic conversion of all of our outstanding shares of Series A preferred stock and Series B preferred stock into common stock upon the completion of this offering. This number excludes:
| • | 9,532 shares of common stock issued after September 30, 2012 and before the date of this prospectus upon the exercise of warrants to purchase such number of shares at an exercise price of $1.60 per share; |
| • | warrants to purchase 8,781 shares of common stock with an exercise price of $1.60 per share which expired unexercised after September 30, 2012; |
| • | warrants to purchase 331,723 shares of common stock with an exercise price of the lesser of $17.00 per share or our initial public offering price which were issued after September 30, 2012 (net of warrants cancelled after September 30, 2012) and before the date of this prospectus; |
| • | stock options to purchase 133,750 shares of common stock to be issued upon the completion of this offering with an exercise price equal to our initial public offering price; |
| • | 1,383,350 shares issuable upon the exercise of outstanding stock options with a weighted average exercise price of $5.10 per share, which includes 1,183,350 shares of our common stock issuable upon exercise of stock options issued under our equity incentive plans and 200,000 shares of our common stock issuable upon exercise of stock options issued outside of our equity incentive plans; |
| • | 2,442,032 shares issuable upon the exercise of outstanding warrants as of September 30, 2012 with a weighted average exercise price of $9.12 per share; |
| • | 1,066,050 shares available for future issuance under our equity compensation plans; |
| • | 2,674 shares of common stock issuable to Cleveland Clinic pursuant to our license agreement with Cleveland Clinic; and |
| • | the number of shares of common stock issuable upon the exercise of outstanding warrants exclude the effects of anti-dilution provisions that will occur upon completion of the offering. At an offering price of $7.00 per share, an aggregate of 3,942,198 shares of common stock would be issuable upon exercise of all of the outstanding warrants described above. |
If the underwriters exercise their option to purchase additional shares from us in full, the number of shares held by new investors will increase to 5,750,000, or 47% of the total number of shares of common stock outstanding after this offering and the shares held by existing stockholders will remain at 6,405,134, but the percentage of shares held by existing stockholders will decrease to 53% of the total shares outstanding.
-48-
Table of Contents
SELECTED HISTORICAL FINANCIAL DATA
The following table summarizes our selected consolidated financial data for the periods and as of the dates indicated. Our selected statements of operations data for each of the years in the periods ended December 31, 2009, 2010 and 2011, and our selected consolidated balance sheet data as of December 31, 2010 and 2011, have been derived from our audited consolidated financial statements and their related notes, which are included elsewhere in this prospectus. Our selected consolidated statements of operations data for the year ended December 31, 2007 and 2008, and our selected consolidated balance sheet data as of December 31, 2008 and 2009 have been derived from audited consolidated financial statements that are not included in this prospectus. Our selected consolidated balance sheet data as of December 31, 2007 has been derived from unaudited consolidated financial statements that are not included in this prospectus. The unaudited selected consolidated statements of operations data for the nine months ended September 30, 2011 and 2012, and the unaudited consolidated balance sheet data as of September 30, 2012, are derived from our unaudited consolidated financial statements, which are included elsewhere in this prospectus. We have included all adjustments, consisting only of normal recurring adjustments, which we consider necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results to be expected for any future periods and our interim results are not necessarily indicative of the results to be expected for the full fiscal year. Our selected consolidated financial data should be read together with the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and with our consolidated financial statements and their related notes, which are included elsewhere in this prospectus.
| Nine Months Ended |
Year Ended | |||||||||||||||||||||||||||
| September 30, | December 31, | |||||||||||||||||||||||||||
| 2012 | 2011 | 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||||||||||
| (dollars in thousands, except for share and per share data) |
||||||||||||||||||||||||||||
| Statements of Operations Data: |
||||||||||||||||||||||||||||
| Net Revenues |
$ | 3,226 | $ | 2,144 | $ | 3,019 | $ | 2,522 | $ | 1,666 | $ | 1,680 | $ | 2,343 | ||||||||||||||
| Cost of revenues |
2,880 | 2,385 | 3,117 | 3,516 | 2,532 | 2,223 | 2,128 | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Gross Profit (loss) |
346 | (241 | ) | (98 | ) | (995 | ) | (866 | ) | (543 | ) | 215 | ||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||||||
| Research and development |
1,552 | 1,442 | 2,074 | 1,167 | 1,336 | 608 | 254 | |||||||||||||||||||||
| General and administrative |
3,475 | 3,160 | 4,439 | 3,446 | 1,845 | 1,390 | 1,642 | |||||||||||||||||||||
| Sales and marketing |
1,050 | 1,200 | 1,574 | 716 | 239 | 336 | 274 | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total operating expenses |
6,077 | 5,802 | 8,087 | 5,329 | 3,420 | 2,334 | 2,170 | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| (Loss) income from operations |
(5,731 | ) | (6,044 | ) | (8,185 | ) | (6,323 | ) | (4,286 | ) | (2,877 | ) | (1,955 | ) | ||||||||||||||
| Interest expense |
(3,260 | ) | (918 | ) | (1,314 | ) | (792 | ) | (2,092 | ) | (266 | ) | (77 | ) | ||||||||||||||
| Interest and other income (expense) |
— | — | |
— |
|
734 | 3 | 18 | 16 | |||||||||||||||||||
| Change in fair value of warrant liability |
6,370 | (9,881 | ) | (10,388 | ) | (2,026 | ) | (953 | ) | — | — | |||||||||||||||||
| Tax benefit (expense) |
— | — | — | — | — | — | (286 | ) | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net (loss) |
$ | (2,621 | ) | $ | (16,843 | ) | $ | (19,887 | ) | $ | (8,407 | ) | $ | (7,328 | ) | $ | (3,124 | ) | $ | (2,302 | ) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net (loss) per common share: |
||||||||||||||||||||||||||||
| Basic |
$ | (0.78 | ) | $ | (5.30 | ) | $ | (6.24 | ) | $ | (2.68 | ) | $ | (3.22 | ) | $ | (1.65 | ) | ||||||||||
| Diluted |
(2.66 | ) | (5.30 | ) | (6.24 | ) | (2.68 | ) | (3.22 | ) | (1.65 | ) | ||||||||||||||||
| Weighted-average common shares outstanding used in computing net loss per share: |
||||||||||||||||||||||||||||
| Basic |
3,351,324 | 3,178,701 | 3,185,382 | 3,133,077 | 2,277,004 | 1,891,298 | ||||||||||||||||||||||
| Diluted |
3,375,210 | 3,178,701 | 3,185,382 | 3,133,077 | 2,277,004 | 1,891,298 | ||||||||||||||||||||||
| Pro forma net loss per share of common stock: |
||||||||||||||||||||||||||||
| Basic |
$ | (.41 | ) | $ | (3.19 | ) | ||||||||||||||||||||||
| Diluted |
(1.40 | ) | (3.19 | ) | ||||||||||||||||||||||||
-49-
Table of Contents
(1) Gives effect to the conversion of our Series A and Series B preferred stock into 2,182,680 shares of common stock upon consummation of this offering and the conversion of convertible promissory notes in the amount of $6.0 million including the $2.0 million issued after September 30, 2012, at a conversion price of $7.00 per share, which is the mid point of the price range set forth on the cover page of this prospectus, into an aggregate of 857,143 shares of common stock. Pro forma net loss per shares does not include the recognition of noncash debt discounts and beneficial conversion amounts that would occur upon conversion of the promissory notes.
| As of | As of | |||||||||||||||||||||||
| September 30, | December 31, | |||||||||||||||||||||||
| 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||||||||||
| (unaudited) | ||||||||||||||||||||||||
| (dollars in thousands) | ||||||||||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||||||
| Cash and cash equivalents |
$ | 903 | $ | 2,417 | $ | 1,779 | $ | 30 | $ | 28 | $ | 1,068 | ||||||||||||
| Working capital |
(8,508 | ) | (1,078 | ) | 1,785 | (1,303 | ) | (341 | ) | 787 | ||||||||||||||
| Total assets |
|
7,938 |
|
7,031 | 5,302 | 2,590 | 2,567 | 2,768 | ||||||||||||||||
| Current and long-term notes payable |
6,330 | 2,012 | 100 | 245 | 140 | 140 | ||||||||||||||||||
| Lines of credit |
8,751 | 8,437 | 6,000 | 6,410 | 2,850 | — | ||||||||||||||||||
| Warrant liability |
8,421 | 11,113 | 4,270 | 1,436 | — | — | ||||||||||||||||||
| Accumulated deficit |
(44,890 | ) | (42,269 | ) | (22,382 | ) | (13,975 | ) | (6,436 | ) | (3,312 | ) | ||||||||||||
| Total stockholder’s equity (deficit) |
$ | (20,157 | ) | $ | (19,065 | ) | $ | (6,736 | ) | $ | (6,711 | ) | $ | (1,226 | ) | $ | 1,488 | |||||||
-50-
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
The following discussion of our financial condition and results of operations should be read together with our consolidated financial statements and related notes included elsewhere in the prospectus. This discussion contains forward-looking statements based upon our current plans, estimates, beliefs and expectations that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under the sections entitled “Risk Factors,” “Special Note Regarding Forward-Looking Statements” and elsewhere in this prospectus. The share numbers in the following discussion reflect a 1-for-2 reverse stock split that we anticipate effecting just prior to the date of this prospectus.
Overview
We are an early-stage diagnostics company focused on developing and commercializing proprietary genomic tests and services to improve and personalize the diagnosis, prognosis and response to treatment (theranosis) of cancer. Our proprietary tests target cancers that are complicated to prognose and for which it is difficult to predict treatment outcomes using currently available mainstream techniques. These cancers include hematological, urogenital and HPV-associated cancers. We provide our proprietary tests and services along with a comprehensive range of non-proprietary oncology-focused tests and laboratory services, to oncologists and pathologists at hospitals, cancer centers, reference laboratories and physician offices. To date, we have engaged in only limited sales and marketing activities and have generated most of our revenue through sales of our non-proprietary testing services to a limited number of oncologists, pathologists, and community hospitals located mostly in the eastern and midwestern United States. Our non-proprietary laboratory testing services include molecular testing, sequencing, mutational analysis, flow cytometry testing, histology testing and cytology testing. These tests are described in more detail in the section entitled “Description of the Business—Laboratory Services”. We are currently offering our tests and laboratory services in our 17,936 square foot state-of-the-art laboratory located in Rutherford, New Jersey, which has been accredited by the College of American Pathologists, which is one of six approved accreditation methods under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”), to perform high complexity testing.
Our proprietary tests are based principally on our expertise in specific cancer types, test development methodologies and proprietary algorithms correlating genetic events with disease specific information. We have commercially launched MatBA®-CLL, our first proprietary microarray test for chronic lymphocytic leukemia (“CLL”) for use in our CLIA-accredited clinical laboratory. In January 2012, we received CLIA approval for MatBA®-SLL, our proprietary microarray for risk stratification in small lymphocytic lymphoma (“SLL”), and we are currently offering MatBA®-SLL in our laboratory. In addition, we are developing a series of other proprietary genomic tests in our core oncology markets. Through December 31, 2011, revenues from MatBA®-CLL were immaterial to our results of operations. Revenues from our proprietary MatBA® test represented approximately 5% of our first nine months of 2012 revenues. However, due to the recent introduction of this test, the small numbers involved in our revenues, and the variability expected with the adoption of any new tests, no assurance or prediction can be given with respect to the level of revenues from our proprietary tests in the future.
We have established collaborative relationships with key thought leaders in oncology, which enable us to develop and validate the effectiveness and utility of our tests in a clinical setting and which provide us access to clinically-robust patient data. For example, we agreed to form a joint venture in March 2013 with Mayo Foundation for Medical Education and Research focused on developing oncology diagnostic services and tests utilizing next-generation sequencing. Additionally, we agreed to a research collaboration with the Cleveland Clinic to validate our renal-cancer microarray, VroGenRA™-Renal.
The non-proprietary testing services we offer are entirely focused on specific oncology categories where we are developing our proprietary arrays and probe panels. We believe that there is significant synergy in developing and marketing a complete set of tests and services that are disease-focused and delivering those tests
-51-
Table of Contents
and services in a comprehensive manner to help with treatment decisions. The insight that we develop in delivering the non-proprietary services are often leveraged in the development of our proprietary programs and now increasingly in the validation of our proprietary programs (such as MatBA®) for clinical use.
We believe that we can be successful by offering cancer professionals a fully-integrated menu of oncology-focused proprietary and non-proprietary tests and customized laboratory services. Based on our discussions with leading researchers in the oncology field and interactions with our collaborators, as well as information we learn through performing the non-proprietary genetic diagnostic testing services, which are focused on the specific oncology categories where we are developing our proprietary tests, we believe our proprietary tests provide superior diagnostic and prognostic values than currently available tests. In particular, our proprietary tests deliver a level of genomic information not provided by other currently available tests. For example, the majority of current cytogenetic analysis for CLL and SLL that is available in clinical laboratories today assesses gain and loss in genomic material at four specific sites. There are two other marketed arrays for CLL (Combimatrix and Quest) of which we are aware. Both of these arrays report out gains and losses at four to five genomic sites. MatBA®-CLL, on the other hand, reports out gains and losses at nine genomic sites and MatBA®-SLL reports out gains and losses at seven genomic sites. We believe our ability to rapidly translate research insights about the genetics and molecular mechanisms of cancer into the clinical setting will improve patient treatment and management and that this approach will become a key component in the standard of care for personalized cancer treatment.
We will offer our proprietary tests in the United States as laboratory developed tests (“LDTs”) and internationally as CE-marked in vitro diagnostic products. In addition, as part of our long-term strategy we plan to seek Food and Drug Administration (“FDA”) clearance or approval to expand the commercial use of our tests to other laboratories and testing sites. We believe it would likely take two years or more to conduct the studies and trials necessary to obtain approval from FDA to commercially launch MatBA®-CLL and MatBA®-SLL outside of our clinical laboratory. Our sales strategy is focused on direct sales to oncologists and pathologists at hospitals, cancer centers, and physician offices in the United States and expanding our relationships with leading distributors and medical facilities in emerging markets. We intend to emphasize partnering with community hospitals, where nearly 85% of all cancers are initially diagnosed, through our program called Expand Dx™, which was specifically designed to meet the needs of community hospitals. We believe our proprietary tests and services will enable community hospitals to optimize and expand their oncology services to better serve their cancer patients.
We expect to continue to incur significant losses for the near future. We incurred net losses of $2.6 million for the nine months ended September 30, 2012, $19.9 million for the year ended December 31, 2011, $8.4 million for the year ended December 31, 2010, and $7.3 million for the year ended December 31, 2009. Changes in fair value of some of our common stock warrants have significantly impacted our results in recent periods. In particular, changes in the fair value of some of our common stock warrants accounted for a large portion of our losses in 2011 and 2010, whereas in the nine months ended September 30, 2012 we recognized non-cash income as a result of the change in fair value of such warrants. Accounting rules require us to record certain of our warrants as a liability, measure the fair value of these warrants each quarter and record changes in that value in earnings. Due to the significant number of warrants that we have outstanding, we may be exposed to significant non-cash charges, or we may record significant amounts of non-cash income, as a result of this warrant exposure in future periods. For a more detailed description of our warrant liability, see the section entitled “Warrant Liability.” As of September 30, 2012, we had an accumulated deficit of $44.9 million.
Key Factors Affecting our Results of Operations and Financial Condition
Our overall long-term growth plan is predicated on our ability to develop and commercialize our proprietary tests outside of our clinical laboratory and to increase comprehensive oncology testing volumes in our laboratory. We launched MatBA®-CLL in the first quarter 2011 for use in our clinical laboratory, we received CLIA approval for MatBA®-SLL in January 2012 and we are developing additional proprietary tests. In order to market our tests to independent laboratories and testing facilities, we believe we will need to obtain
-52-
Table of Contents
approvals or clearances from the appropriate regulatory authorities, including FDA. For further discussion of the approvals and clearances that may be required, see the section entitled “Description of the Business—Governmental Regulations.” Without these approvals, the success of these commercialization efforts will be limited. To obtain these approvals and facilitate market adoption of our proprietary tests, we anticipate having to successfully complete additional studies with clinical samples and publish our results in peer-reviewed scientific journals. Our ability to complete such studies is dependent upon our ability to leverage our collaborative relationships with leading institutions to facilitate our research and obtain data for our quality assurance and test validation efforts.
We believe that the factors discussed in the following paragraphs have had and are expected to continue to have a material impact on our results of operations and financial condition.
Revenues
Our revenue in 2011 was generated principally through our clinical laboratory services, with approximately 10% of our revenue from government research grants such as the National Cancer Institute, and approximately 3% of our revenue from sales of our DNA probes, which are only sold outside the United States. The clinical laboratory industry is highly competitive, and our relationship with the decision-maker at hospitals, cancer centers or physician offices is a critical component of securing their business. Consequently, our ability to attract and maintain productive sales personnel that have and can grow these relationships will largely determine our ability to grow our clinical services revenue. In order to grow our clinical laboratory revenue, we must continue to pursue validation studies and work with oncology thought leaders to develop data that is helpful in supporting the need for our tests and services.
Due to the early stage nature of our business and our limited sales and marketing activities to date, we have historically derived a significant portion of our revenue from a limited number of test ordering sites, although the test ordering sites that generate a significant portion of our revenue have changed from period to period. For example, there was one site which represented more than 10% of our revenue for the year ended December 31, 2010 that generated less than 10% of our revenue for the year ended December 31, 2011. Our test ordering sites are largely hospitals, cancer centers, reference laboratories and physician offices. Oncologists and pathologists at these sites order the tests on behalf of the needs of their oncology patients. For the nine months ended September 30, 2012, our top five test ordering sites accounted for 61% of our clinical testing volume with approximately 47% of the volume coming from community hospitals. For the year ended December 31, 2011, our top five test ordering sites represented approximately 63% of our clinical testing volume, with approximately 29% of the volume coming from community hospitals. Our top five test ordering sites for the year ended December 31, 2010 accounted for 60% of our clinical testing volumes, with approximately 15% of the volume coming from community hospitals. In particular, during the year ended December 31, 2010, there were three sites which each accounted for 10% or more of our revenue: one community hospital accounted for approximately 12% of our revenue; a regional reference laboratory accounted for approximately 11% of our revenue and a community oncology practice accounted for another approximately 11% of our revenue. For the year ended December 31, 2011, we generated revenue from two test ordering sites that represented 10% or more of our revenue: a community hospital accounted for approximately 18% of our revenue and a community oncology practice accounted for approximately 11% of our revenue. For the nine months ended September 30, 2012, three test ordering sites accounted for 10% or more of our revenue; a university teaching center accounted for approximately 15%; a clinical trial client accounted for approximately 12% and a community hospital network accounted for approximately 11%. We generally do not enter into formal written agreements with such test ordering sites and, as a result, we may lose these significant test ordering sites at any time. The loss of any one of these test ordering sites would not materially adversely affect our results of operations.
We receive revenue for our clinical lab services from private insurance carriers and other non-Medicare payors (such as unions and self-insured plans), Medicare, “direct bill” customers, and grants. Direct bill customers are institutions that choose, generally at the beginning of our relationship, to pay for our laboratory
-53-
Table of Contents
services directly, as opposed to having patients (or their insurers) pay for those services and providing us with the patients’ insurance information. For instance, bio-pharmaceutical companies generally are direct bill customers. A hospital may elect to be a direct bill customer, and pay our bills directly, or may provide us with patient information so that their patients pay our bills, in which case we generally look to payment from their private insurance carrier or Medicare. In a few instances, we have arrangements where a hospital may have two accounts with us, so that certain tests are direct billed to the hospital, and certain tests are billed to and paid by a patient’s insurer. The billing arrangements generally are dictated by our customers and in accordance with state and federal law. In 2011, private insurance accounted for approximately 54% of our total revenue, Medicare accounted for approximately 24% of our total revenue, direct bill clients accounted for 12% of our total revenue and the balance of our revenue was attributable to grants. In 2010, private insurance accounted for approximately 55% of our total revenue, Medicare accounted for approximately 24% of our total revenue, direct-bill clients comprised approximately 16% of our total revenue and the balance of our revenue was attributable to grants. As we expand our portfolio of tests and services, our sales activities and our ExpandDX™ program, we expect the percentage of revenue from direct-bill customers may decrease over the long term. However, during the nine months ended September 30, 2012 we started working with a community hospital that preferred the direct bill model and a new direct bill clinical trial services customer, which resulted in a significant increase in direct bill customers as a percentage of revenue for the nine months ended September 30, 2012. It is too early in our development to predict whether our experience during the nine months ended September 30, 2012 indicates a reversal in the trend we had seen in prior years or simply a quarterly variation as we attempt to expand our business and introduce new community hospitals, regional laboratories or clinical trial services customers in a particular period. On average, we generate less revenue per test from direct-bill customers than from other third-party payors but we also have reduced sales cost associated with direct bill clients and significantly reduced collections risk from direct-bill customers and have not experienced any significant collection issues or expenses as a result. Typically, we negotiate discounts in the range of 5% to 20% with direct bill clients depending on the volume of business in a twelve month period.
Cost of Revenues
Our cost of revenues consists principally of internal personnel costs, including stock-based compensation, laboratory consumables, shipping costs, overhead and other direct expenses, such as specimen procurement and third party validation studies. We are pursuing various strategies to reduce and control our cost of revenues, including automating our processes through more efficient technology, attempting to negotiate improved terms with our suppliers and exploring relocating our manufacturing operations to a lower cost-base country.
Operating Expenses
We classify our operating expenses into three categories: research and development, sales and marketing, and general and administrative. Our operating expenses principally consist of personnel costs, including stock-based compensation, outside services, laboratory consumables and overhead, development costs, marketing program costs and legal and accounting fees.
Research and Development Expenses. We incur research and development expenses principally in connection with our efforts to develop our proprietary tests. Our primary research and development expenses consist of direct personnel costs, laboratory equipment and consumables and overhead expenses. We anticipate that research and development expenses will increase in the near-term, principally as a result of hiring additional personnel to develop and validate tests in our pipeline and to perform work associated with our research collaborations. In addition, we expect that our costs related to collaborations with research and academic institutions will increase. For example, we recently entered into an affiliation agreement to form a joint venture with the Mayo Foundation for Medical Education and Research. All research and development expenses are charged to operations in the periods they are incurred.
Sales and Marketing Expenses. Our sales and marketing expenses consist principally of personnel and related overhead costs for our sales team and their support personnel, travel and entertainment expenses, and other selling costs including sales collaterals and trade shows. We expect our sales and marketing expenses to
-54-
Table of Contents
increase significantly after we complete our initial public offering as we expand into new geographies and add new clinical tests and services.
General and Administrative Expenses. General and administrative expenses consist principally of personnel-related expenses, professional fees, such as legal, accounting and business consultants, occupancy costs, bad debt and other general expenses. We expect that our general and administrative expenses will increase as we expand our business operations. We further expect that general and administrative expenses will increase significantly due to increased information technology (“IT”), legal, insurance, accounting and financial reporting expenses associated with being a public company.
Seasonality
Our business experiences decreased demand during spring vacation season, summer months and the December holiday season when patients are less likely to visit their health care providers. We expect this trend in seasonality to continue for the foreseeable future.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates based on historical experience and make various assumptions, which management believes to be reasonable under the circumstances, which form the basis for judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Section 107 of the JOBS Act provides that an “emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
The notes to our audited consolidated financial statements, which are included elsewhere in this prospectus, contain a summary of our significant accounting policies. We consider the following accounting policies critical to the understanding of the results of our operations:
| • | Revenue recognition; |
| • | Accounts receivable and bad debts; |
| • | Stock-based compensation; and |
| • | Warrant liability. |
Revenue Recognition
Revenue is recognized in accordance with ASC 605, Revenue Recognition, and ASC 954-605 Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the client or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. In determining whether the price is fixed or determinable, we consider payment limits imposed by insurance carriers and Medicare and the amount of
-55-
Table of Contents
revenue recorded takes into account the historical percentage of revenue we have collected for each type of test for each payor category. Periodically, an adjustment is made to revenue to record differences between our anticipated cash receipts from insurance carriers and Medicare and actual receipts from such payors. For the periods presented, such adjustments were not significant. For direct bill customers, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, we consider whether we have sufficient payment history to reliably estimate a payor’s individual payment patterns. For new tests where there is no evidence of payment history at the time the tests are completed, we only recognize revenues once reimbursement experience can be established. We then recognize revenue equal to the amount of cash received. Sales of probes are recorded on the shipping date. We do not bill customers for shipping and handling fees and we do not collect any sales or other taxes.
Accounts Receivable and Bad Debts
Accounts receivable are carried at original invoice amount less an estimate for contractual adjustments and doubtful receivables based on a review of all outstanding amounts on a periodic basis. The estimate for doubtful receivables is determined from an analysis of the accounts receivable on a quarterly basis, and is recorded as bad debt expense. Accounts receivable are written off when deemed uncollectible. Recoveries of accounts receivable previously written off are recorded when received.
Stock-Based Compensation Expense
We account for stock-based compensation under the provisions of FASB ASC Topic 718, Compensation—Stock Compensation, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and directors based on estimated fair values on the grant date. We estimate the fair value of stock-based awards on the date of grant using the Black-Scholes option pricing model (“Black Scholes valuation model”). The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. We estimate forfeitures at the time of grant and revise our estimates in subsequent periods if actual forfeitures differ from those estimates. At September 30, 2012, we had unrecognized compensation cost related to nonvested employee stock options of approximately $933,095, which amount is expected to be recognized over the next 2.9 years.
We account for stock-based compensation awards to non-employees in accordance with FASB ASC Topic 505-50, Equity-Based Payments to Non-Employees (“ASC 505-50”). Under ASC 505-50, we determine the fair value of the warrants or stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. All issuances of equity instruments issued to non-employees as consideration for goods or services received by us are accounted for based on the fair value of the equity instruments issued. These awards are recorded in expense and additional paid-in capital in stockholders’ equity over the applicable service periods based on the fair value of the options at the end of each period. As of September 30, 2012, we had total unrecognized compensation cost related to nonvested stock options granted to non-employees of approximately $450,500, which amount is expected to be recognized over the next 0.8 years. The estimate of unrecognized non-employee compensation is based on the fair value of the nonvested options as of September 30, 2012.
Calculating the fair value of stock-based awards requires the input of highly subjective assumptions into the Black Scholes valuation model. Stock-based compensation expense is significant to our financial statements and is calculated using our best estimate, which involves inherent uncertainties, and the application of our management’s judgment. Significant estimates include the fair value of our common stock at the date of grant, the expected life of the stock option, stock price volatility, risk-free interest rate and forfeiture rates.
Common Stock Valuation
In the absence of a public trading market, our board of directors determined a reasonable estimate of the then-current fair value of our common stock for purposes of granting stock-based compensation based on input
-56-
Table of Contents
from management and valuation reports prepared by an independent third-party valuation specialist. We determined the fair value of our common stock utilizing methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants Practice Aid, “Valuation of Privately-Held-Company Equity Securities Issued as Compensation,” which we refer to as the AICPA Practice Aid. In addition, we exercised judgment in evaluating and assessing the foregoing based on several factors including:
| • | the nature and history of our business; |
| • | our historical operating and financial results; |
| • | the market value of companies that are engaged in a similar business to ours; |
| • | the lack of marketability of our common stock; |
| • | the price at which shares of our equity instruments have been sold; |
| • | our progress in developing our technology; |
| • | the overall inherent risks associated with our business at the time stock option grants or warrants were approved; and |
| • | the overall equity market conditions and general economic trends. |
We relied upon the option pricing model, or OPM, and the probability-weighted expected return method, or PWERM, to allocate our company value to each of our classes of stock. If the valuation date approximated the close of recent or expected financings by third parties, we placed greater reliance on the OPM. If no such recent or expected financings were available at the valuation date, we placed greater reliance on the PWERM.
Probability-Weighted Expected Return Method. PWERM values each class of equity based on an analysis of the range of potential future enterprise values of the Company and the manner in which those values would accrue to the owners of the different classes of equity. This method involves estimating the overall value of the subject company under various liquidity event scenarios and allocating the value to the various share classes based on their respective claim on the proceeds as of the date of each event. These different scenarios typically include an initial public offering, an acquisition, or a liquidation of the business, each resulting in a different value. For each scenario, the future value of each share class is calculated and discounted to a present value. The results of each scenario are then probability-weighted in order to arrive at an estimate of fair value for each share class as of a current date.
We used the PWERM to allocate our estimated enterprise value between our preferred stock and common stock. At certain periods, we also utilized the OPM as described below. Under the PWERM, we analyzed the value of our company using several scenarios, which included an initial public offering (“IPO Scenario”), reverse merger (“Reverse Merger Scenario”), acquisition (“Sale Scenario”), discounted cash flow method (“Private Company Scenario”) and a liquidation of assets (“Liquidation Scenario”).
The IPO Scenario and Reverse Merger Scenario were based on market multiples of comparable publicly traded companies. We selected a subset of public companies we considered to be most similar to our company. We determined that each of the selected public companies was comparable to our company at the respective valuation date because they are molecular diagnostic or genetic analysis companies, generally in the early stages of commercialization. As of each valuation date, we evaluated the multiples of projected revenue of these companies and applied these multiples to our projected revenue. We furthermore evaluated this indication of value in relation to valuation considerations provided by an investment banking firm.
-57-
Table of Contents
The Sale Scenario was based on multiples observed in mergers, acquisitions, and financings of comparable companies. In this analysis, we identified transactions involving certain comparable molecular diagnostic or genetic analysis companies and applied the median multiple of revenue from these transactions to our projected revenue.
The Private Company Scenario was based on an income approach. The income approach estimates the present value of future estimated cash flows, based upon forecasted revenues and costs. These future cash flows are discounted to their present values using a discount rate which is derived using the build-up approach and are consistent with the required rates of return described in the AICPA Practice Aid. Our discount rates for common stock decreased from 41.4% at December 31, 2010 to 29.0% at September 30, 2012 as our stage of development progressed.
The Liquidation Scenario was based on the asset accumulation method which contemplated the sale of assets and winding up of operations.
We determined the value of our preferred stock and common stock under each scenario by allocating the equity value to each class of stock and discounting the value back to the present using a risk-adjusted discount rate. In certain scenarios, a large portion of the equity value is allocated to the convertible preferred stock to incorporate higher aggregate liquidation preferences. We then weighted the present value of the common stock under each scenario based upon the probability of each scenario occurring in order to determine a final indication of value for the common stock.
Option Pricing Model. OPM uses option theory to value the various classes of a company’s securities in light of their respective claims to the enterprise value. Total shareholders’ equity value is allocated to the various share classes based upon their respective claims on a series of call options with strike prices at various value levels depending upon the rights and preferences of each class. A Black-Scholes closed form option pricing model is typically employed in this analysis, with an option term assumption that is consistent with our expected time to a liquidity event and a volatility assumption based on the estimated stock price volatility of a peer group of comparable public companies over a similar term.
We applied the OPM by using the price of preferred stock issued by us to sophisticated investors in arms-length transactions and the backsolve method to derive the value of common stock. We estimated the volatility of our shares at 90% based on the expected term to a liquidity event.
We granted stock options between January 1, 2010 and September 30, 2012 with exercise prices between $1.60 and $13.52 per share. Information regarding our stock option grants to our employees and certain consultants since January 1, 2010 is summarized as follows:
| Date of issuance |
Number of options granted |
Exercise price | Common stock value |
Option fair value(1) |
||||||||||||
| January 19, 2010 |
89,125 | $ | 1.92 | (2) | $ | 1.92 | $ | 1.32 - 1.42 | ||||||||
| January 19, 2010 |
19,375 | $ | 1.60 | (3) | $ | 1.92 | $ | 1.32 - 1.42 | ||||||||
| April 1, 2010 |
492,150 | $ | 5.00 | $ | 2.90 | $ | 1.82 | |||||||||
| June 10, 2010 |
5,875 | $ | 5.00 | $ | 2.90 | $ | 1.80 | |||||||||
| June 11, 2010 |
375 | $ | 5.00 | $ | 2.90 | $ | 1.80 | |||||||||
| August 15, 2010 |
50,000 | $ | 10.00 | $ | 4.70 | (4) | $ | 3.40 | ||||||||
| September 15, 2010 |
150,000 | $ | 10.00 | $ | 4.70 | (4) | $ | 3.40 | ||||||||
| October 12, 2010 |
25,000 | $ | 5.00 | $ | 4.70 | $ | 3.20 | |||||||||
| December 9, 2010 |
18,250 | $ | 5.00 | $ | 4.70 | $ | 3.24 | |||||||||
| December 9, 2010 |
1,500 | $ | 10.00 | $ | 4.70 | $ | 2.68 | |||||||||
| February 8, 2011 |
125,000 | $ | 10.00 | $ | 4.76 | $ | 2.72 | |||||||||
| April 1, 2011 |
5,750 | $ | 10.30 | (5) | $ | 10.30 | $ | 7.22 | ||||||||
-58-
Table of Contents
| Date of issuance |
Number of options granted |
Exercise price | Common stock value |
Option fair value(1) |
||||||||||||
| April 1, 2011 |
600 | $ | 10.00 | (6) | $ | 10.30 | $ | 7.22 | ||||||||
| October 5, 2011 |
17,687 | $ | 12.66 | $ | 12.66 | $ | 8.58 | |||||||||
| December 29, 2011 |
20,000 | $ | 13.52 | $ | 13.52 | $ | 9.02 | |||||||||
| February 9, 2012 |
6,000 | $ | 13.52 | $ | 13.52 | $ | 9.34 | |||||||||
| April 1, 2012 |
20,000 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| April 3, 2012 |
5,000 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| April 8, 2012 |
40,000 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| May 3, 2012 |
1,500 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| June 15, 2012 |
1,500 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| June 18, 2012 |
1,250 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| July 9, 2012 |
7,000 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| August 8, 2012 |
37,500 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| September 10, 2012 |
2,000 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| September 17, 2012 |
3,500 | $ | (7) | $ | (7) | $ | (7) | |||||||||
| September 24, 2012 |
3,500 | $ | (7) | $ | (7) | $ | (7) | |||||||||
(1) Option fair value determined using the Black-Scholes option pricing model using the input assumptions outlined above.
(2) These options were subsequently amended from an exercise price of $1.60 to $1.92. The option fair value was calculated using the exercise price prior to amendment. There was no measurement period adjustment because the exercise price increased.
(3) These options were not amended to change the exercise price as they were forfeited, exercised, or expired prior to such amendment.
(4) These options were granted to non-employees directors, with a ten year term. Non-employee directors options are subsequently valued as of each vesting date. This common stock value and option fair value reflect the grant date values.
(5) These options were subsequently amended from an exercise price of $10.00 to $10.30. The option fair value was calculated using the exercise price prior to amendment. There was no measurement period adjustment because the exercise price increased.
(6) These options were not amended to change the exercise price as they were forfeited or expired prior to such amendment.
(7) These grants have not yet been issued. These grants have been approved to be issued upon consummation of our initial public offering, subject to continued employment. The exercise price of these options will be equal to the initial public offering price per share of common stock.
The following table summarizes the significant assumptions we used in our valuations to determine the fair value of our common stock as of the date indicated.
| 12/31/09 | 3/31/10 | 6/30/10 | 9/30/10 | 12/31/10 | 3/31/11 | 6/30/11 | 9/30/11 | 12/31/11 | 3/31/12 | 6/30/12 | 9/30/12 | |||||||||||||||||||||||||||||||||||||
| Option Pricing Model |
20 | % | 30 | % | 70 | % | 80 | % | 60 | % | 10 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||
| Probability-Weighted |
||||||||||||||||||||||||||||||||||||||||||||||||
| Expected Return Method |
80 | % | 70 | % | 30 | % | 20 | % | 40 | % | 90 | % | 100 | % | 100 | % | 100 | % | 100 | % | 100 | % | 100 | % | ||||||||||||||||||||||||
| IPO |
10 | % | 10 | % | 15 | % | 15 | % | 15 | % | 35 | % | 55 | % | 65 | % | 80 | % | 95 | % | 95 | % | 95 | % | ||||||||||||||||||||||||
| Reverse Merger |
20 | % | 20 | % | 20 | % | 20 | % | 25 | % | 30 | % | 20 | % | 15 | % | 5 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||
| Acquisition |
5 | % | 5 | % | 5 | % | 5 | % | 10 | % | 10 | % | 5 | % | 10 | % | 10 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||
| Private Company |
55 | % | 55 | % | 50 | % | 50 | % | 45 | % | 20 | % | 15 | % | 10 | % | 5 | % | 5 | % | 5 | % | 5 | % | ||||||||||||||||||||||||
| Liquidation |
10 | % | 10 | % | 10 | % | 10 | % | 5 | % | 5 | % | 5 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||
| Lack of Marketability Discount |
30 | % | 30 | % | 30 | % | 30 | % | 30 | % | 25 | % | 15 | % | 10 | % | 10 | % | 2.5 | % | 4 | % | 4 | % | ||||||||||||||||||||||||
| Stock Value |
1.92 | 2.90 | 4.18 | 4.70 | 4.76 | 10.30 | 12.98 | 12.66 | 13.52 | 12.64 | 11.94 | 7.48 | ||||||||||||||||||||||||||||||||||||
We engaged an independent third-party valuation specialist to perform retrospective valuations as of December 31, 2009 and March 31, 2010 and contemporaneous valuations as of December 31, 2010, March 31, 2011, June 30, 2011, September 30, 2011, December 31, 2011, March 31, 2012, June 30, 2012 and September 30, 2012. For the value of common stock as of June 30, 2010 and September 30, 2010, we applied valuation approaches consistent, where appropriate, with those performed by the third-party valuation specialist. We used the common stock value of the valuation date closest to the option grant date, where appropriate, in our calculation of stock option expense.
-59-
Table of Contents
No single event caused the valuation of our common stock to increase or decrease from January 2010 to September 30, 2012, rather it has been a combination of the following factors that lead to the changes in the fair value of the underlying common stock.
December 31, 2009. By the fourth quarter of 2009, the U.S. capital markets had stabilized from the period of very high volatility in 2008 and 2009. We achieved a number of milestones in this quarter including a historical revenue high since moving to our new facility. We formed a subsidiary in Italy, CGI Italia, and began operations of our probe distributions. We began discussions on a preferred stock Series B offering and obtained further access to our Wells Fargo line of credit. We relied 20% on the OPM using anticipated pricing of the Series B preferred stock offering.
March 31, 2010. During this quarter, U.S. market indices trended positive. In addition, the subset of public companies we considered to be most similar to our company significantly outperformed the broader market. We made significant progress in this period by hiring Panna Sharma as our Chief Executive Officer on April 1, 2010. This is a critical milestone because we had not had a Chief Executive Officer since October 2009 when our previous Chief Executive Officer transitioned to general counsel. We prepared new projections based on our renewed strategy and timeline given the new Chief Executive Officer and management team’s experience. We entered into a Supply and Distribution Agreement on March 17, 2010 that enabled us to market our DNA probes outside the United States. The agreement also provided us with access to proprietary fluorescent dye labeling technology. The most significant technical milestone during this period was our receipt of the initial validation data and successful sample data set for our first proprietary test, MatBA®-CLL. As the preferred stock Series B offering was in progress in this period, we relied 30% on the OPM using the backsolve method. Under the PWERM approach, we assessed the likelihood of an initial public offering and reverse merger to have increased due to the milestones achieved and proceeds received from the Series B offering.
June 30, 2010. In the second quarter of 2010, the enterprise values of the subset of public companies we considered to be most similar to our company declined. However, we closed on an additional $5.6 million of Series B proceeds. Therefore, we relied 70% on the OPM using the backsolve method. We also hired a lab director in April 2010 which accelerated the improvement and competitive position of our lab operations. Therefore, we also increased the likelihood of an initial public offering. No options were granted using this common stock valuation; however, warrants were revalued at June 30, 2010 using this common stock valuation.
September 2010. The enterprise values of the subset of public companies we considered to be most similar to our company increased by double digits in the third quarter of 2010. We held additional Series B closings of approximately $1.0 million in July and August 2010 at $5.00 per share of Series B preferred stock. We elected to rely 80% on the OPM using the backsolve method.
December 2010. In the fourth quarter of 2010, we noted significant increases in the enterprise values of the subset of public companies we considered to be most similar to our company. In addition, we achieved certain milestones, including the approval of MatBA®-CLL by CLIA for use as an LDT on November 30, 2010. The final two Series B Preferred Stock closings occurred with gross proceeds of $2.5 million. We had several successful presentations and meetings to introduce the MatBA® microarray at the Annual Meeting and Exposition of the American Society of Hematology in early December 2010. December 2010 was the first month and the fourth quarter of 2010 was the first quarter of positive gross margin for us since entering this growth phase. We hired two key new microarray scientists, both of whom commenced employment in December 2010. In October 2010, we were awarded three grants in lieu of federal income tax credits under the Qualifying Therapeutic Discovery Project Program to help in further validation and commercialization of: (1) FHACT™, our proprietary FISH-based assay for detecting copy number changes that are often observed in HPV-associated cancers, (2) FReCaD™, our proprietary FISH-based assay, and (3) MatBA®, our microarray developed for the analysis of genomic copy number alterations in mature B-cell neoplasms. As the final Series B Preferred Stock offering had closed, we reduced our reliance on the OPM backsolve method to 60%. With the significant milestones achieved in this quarter, we increased our estimate of the likelihood of a reverse merger and acquisition.
-60-
Table of Contents
March 2011. The enterprise values of the subset of public companies we considered to be most similar to our company underperformed the market in this quarter. However, we made significant progress toward an initial public offering. We hired Elizabeth Czerepak as Chief Financial Officer, bringing 18 years of pharmaceutical industry experience and nine years of venture capital experience to our Company. We began discussions with various investment banking firms regarding services related to our anticipated initial public offering. Our first quarter revenue improved from the prior year and we prepared revised projections. The New York State Department of Health approved MatBA®-CLL and we commercially launched MatBA-CLL® for use in our clinical laboratory. Scientist Magazine listed our company as a Top 20 Place to Work. We initiated a community hospital outreach program—Expand Dx™. We completed a $3 million line of credit with DAM Holdings, LLC. We launched a marketing campaign and bulked up our sales force along the eastern coast of the United States. Based on these accomplishments, we assessed the likelihood of an initial public offering or reverse merger to have increased significantly. We reduced our reliance on the OPM backsolve method to 10%. In addition, we reduced the discount for lack of marketability to 25% due to the decreasing term to anticipated liquidity events.
June 2011. While the major U.S. indices were relatively flat this quarter, the subset of public companies we considered to be most similar to our company realized significant appreciation in their enterprise values. By the end of the quarter, we had engaged investment bankers to assist in an initial public offering. Our microarray for kidney, prostate and bladder cancers, UroGenRATM, entered clinical trials. We continued collaborations to evaluate FHACT™ with the Kamineni Hospital in India. We also signed a Material Transfer Agreement with University of Iowa Research Foundation. We engaged in negotiations on collaboration with Cleveland Clinic. In addition, we set up our first electronic medical records exchange with a physician’s office. Based on these milestones, we assessed the likelihood of an initial public offering continued to increase significantly while the likelihood of a reverse merger, acquisition, and remaining a private company declined. We further reduced our reliance on the OPM backsolve method due to the milestones achieved since the Preferred Stock Series B offering closed in the fourth quarter of 2010. We also reduced our discount for lack of marketability as the likelihood of a shorter term liquidity event increased.
September 2011. The enterprise values of the subset of public companies we considered to be most similar to our company continued to appreciate. However, we revised our projections primarily due to a change in projected timing of our initial public offering. We were drafting an S-1 in anticipation of an initial public offering. We had identified investors and were in negotiations for bridge financing. We received validation of 15 new probes, including FHACT™ for HPV-associated cancers. Another probe, FReCaD for renal cell carcinoma, was in patient trials. MatBA® products advanced in development. We signed a probe distribution agreement with Labomics S.A. (Belgium) for territories outside the United States. We were in negotiations with Mayo regarding an affiliation agreement. Robert Kaufman was added to our Board of Directors and appointed Chair of Audit Committee. We continued to increase our assessed likelihood of an initial public offering based on the progress made in the third quarter. We also decreased our discount for lack of marketability as the likelihood of a shorter term liquidity event increased.
December 2011. In the fourth quarter of 2011, the enterprise values of the subset of public companies we considered to be most similar to our company underperformed the market. However, we successfully filed an S-1 and closed on $3.0 million of bridge financing. We launched CLL Complete - a new, comprehensive set of tests for CLL. We were in advanced negotiations with Kamineni Hospital in India on probe manufacturing collaboration. We signed an affiliation agreement with Mayo. We completed conversion to XiFin Revenue Management System and related documentation of policies and procedures. Our assessment of the likelihood of an initial public offering continued to increase based on our milestones in the fourth quarter.
March 2012. In light of the performance of comparable public companies in the first quarter and capital market conditions in general we reviewed our assumptions regarding pricing for our upcoming initial public offering and lowered our expected initial public offering price per share. We also closed on an additional $3.0 million of bridge financing and received another CLIA approval for a proprietary microarray for SLL.
-61-
Table of Contents
June 2012. In the second quarter of 2012, the enterprise values of the subset of public companies we considered to be most similar to our company outperformed the market slightly. We successfully migrated probe manufacturing to India during the second quarter. We also expanded our relationship with Roche Servicios, S.A. We are now their service provider for biomarker based cancer treating services in 14 different locations covering Central America and the Caribbean. We did not revise our assessment of the likelihood of an initial public offering, but the delay in receipt of the net proceeds from our anticipated initial public offering did negatively impact our valuation. Furthermore, our valuation did not take into account changing conditions since June 30, 2012, including the negative market conditions we experienced subsequent to June 30, 2012 in trying to effect our initial public offering.
September 2012. In the third quarter of 2012, the enterprise values of the subset of public companies we considered to be most similar to our company were mixed with the majority underperforming the market. We increased our volume of business with a key clinical trials customer. However, we experienced negative market conditions for our initial public offering. We did not revise our assessment of the likelihood of an initial public offering, but the expected initial public offering price per share is significantly lower than in the second quarter of 2012 and the delay in receipt of the net proceeds from our anticipated initial public offering continued to negatively impact our valuation.
The expected life of a stock option represents the weighted average period over which we expect our stock options to remain outstanding. The expected life assumption is based on the Staff Accounting Bulletin 107 (“SAB 107”), simplified method. SAB 107 provides guidance related to share-based payment transactions with non-employees, and valuation methods, including assumptions such as expected volatility and expected term. As we have been operating as a private company since inception with no active market for our stock or stock options, it is not possible to use actual price volatility data. Therefore, we estimated the volatility of our common stock-based on the historical volatility of entities in our industry that have been public for a period of time and are comparable to us in terms of market capitalization and financial position. Using an expected volatility based on the average historical volatility of other entities may result in variability when compared to actual historical volatility once we have a public market for our common stock. We base the risk-free interest rate that we use in the option pricing model on the U.S. Treasury Yield Curve in effect at the time of grant. We have never paid and do not anticipate paying in the foreseeable future any cash dividends and therefore use an expected dividend yield of zero in the option pricing model. In order to properly attribute compensation expense, we estimate pre-vesting forfeitures at the time of grant and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. We use historical data to estimate pre-vesting forfeitures and record stock-based compensation expense only for those awards that are expected to vest. Due to the small number of employees and design of our option plan, we have used a forfeiture assumption of zero. If the actual forfeiture rate is materially different from our estimate, our stock-based compensation expense could be significantly different from what has been recorded. For stock options granted to employees, we allocate expense on a straight-line basis over the requisite service period.
Because other companies use different models, methods and assumptions, our comparisons to them may be of limited use. If factors change and we employ different assumptions than those described above in future periods, or if we decide to use a different valuation model, the stock-based compensation expense that we record in the future may differ significantly from what we have recorded and could materially affect our operating results. Once this offering is complete and our stock is listed on an exchange, we plan to use the closing price at the end of each reporting period in determining the amount of stock-based compensation expense to record.
Warrant Liability
We have issued certain warrants that include an exercise price adjustment feature in the event that we issue securities for consideration less than the warrants’ exercise price (referred to as “derivative warrants”). Effective January 1, 2009, the accounting guidance regarding derivative warrants changed and required that certain of our warrants be recorded as a liability and measured at fair value each quarter with changes in that
-62-
Table of Contents
value recorded in earnings. We record changes in the fair value of these warrants in our statement of operations in the line “change in fair value of warrant liability”. We measure the fair value of these warrants using the lattice-based binomial valuation model (the “Lattice valuation model”), using similar assumptions to those described above in the section entitled “Stock-Based Compensation Expense.” At September 30, 2012, there were exercisable warrants to purchase an aggregate of 2,442,032 shares of common stock outstanding, of which 1,707,988 contain an exercise price adjustment feature in the event that we issue securities for consideration less than the warrants’ exercise price. The average remaining life of all of our outstanding common stock warrants as of September 30, 2012 is approximately 2 years and 7 months.
We compute the fair value of the warrant liability at each reporting period and the change in the fair value is recorded as non-cash expense or non-cash income. The key component in the value of the warrant liability is our stock price, which is subject to significant fluctuation and is not under our control. Please see the section entitled “Stock-Based Compensation Expense - Common Stock Valuation” for a detailed description of how we determined the fair value of our common stock at each reporting date. The resulting effect on our net loss is therefore subject to significant fluctuation and will continue to be so until the warrants are exercised, amended or expire. Assuming all other fair value inputs remain constant, we will record non-cash expense when our stock price increases and non-cash income when our stock price decreases.
The assumptions used in determining fair value represent our management’s best estimates, but these estimates involve inherent uncertainties and the application of our management’s judgment. As a result, if factors change, including changes in the fair value of our common stock, our fair value estimates could be materially different in the future.
Internal Control over Financial Reporting
For the years ended December 31, 2010 and 2009, our independent registered public accounting firm identified the following material weaknesses in our internal control over financial reporting: (i) lack of sufficient segregation of duties within accounting functions; (ii) lack of sufficient, qualified accounting personnel to accurately and timely record and report our financial statements in accordance with generally accepted accounting principles and (iii) insufficient corporate record keeping related to equity transactions and contractual arrangements.
We have undertaken measures to remediate the material weaknesses identified above. Specifically, since January 1, 2011 we have hired a new chief financial officer, controller, part-time senior accountant and accounting clerk. In addition, we concluded that additional resources were needed for more complex matters and to assist us with certain financial reporting matters and we have retained such resources on a contract basis. We anticipate that contract resources will be replaced with in-house employees at or around the time we complete our initial public offering.
The addition of these resources has allowed us to properly segregate duties and to accurately and timely prepare financial statements in accordance with generally accepted accounting principles. We have remediated the weakness related to corporate records and equity transactions through a retrospective review of all such arrangements and have developed communication guidelines to ensure that all such arrangements and transactions are monitored and properly recorded and disclosed.
For the year ended December 31, 2011, our independent registered public accounting firm reported a material weakness in our internal control over financial reporting related to our monitoring of the performance of the third-party service providers we use in our revenue cycle. During 2011, we changed third-party service providers to improve our platform for future growth. After the conversion we identified instances of delayed billings and collection efforts and procedural issues with the timely application of cash receipts. To remediate this weakness, we have dedicated a full-time senior accountant to monitor the revenue recognition process and have improved our monitoring systems for data transmission to our third party billing service provider. We have
-63-
Table of Contents
also established a system with our bank to correct a deficiency in the bank’s system that prevented our timely review of electronic deposits.
Results of Operations
Nine Months Ended September 30, 2012 and 2011
The following table sets forth certain information concerning our results of operations for the periods shown:
| Nine Months Ended September 30, |
Change | |||||||||||||||
| 2012 | 2011 | $ | % | |||||||||||||
| (dollars in thousands) |
||||||||||||||||
| Revenue |
3,226 | 2,144 | 1,082 | 50 | % | |||||||||||
| Cost of revenues |
2,880 | 2,385 | 495 | 21 | % | |||||||||||
| Research and development expenses |
1,552 | 1,442 | 110 | 8 | % | |||||||||||
| Sales and marketing expenses |
1,050 | 1,200 | (150 | ) | -13 | % | ||||||||||
| General and administrative expenses |
3,475 | 3,160 | 315 | 10 | % | |||||||||||
| Total Operating Loss |
(5,731 | ) | (6,044 | ) | 313 | 5 | % | |||||||||
| Interest income (expense) |
(3,260 | ) | (918 | ) | (2,342 | ) | 255 | % | ||||||||
| Change in fair value of warrant liability |
6,370 | (9,881 | ) | 16,251 | 164 | % | ||||||||||
| Net Loss |
(2,621 | ) | (16,843 | ) | 14,222 | 84 | % | |||||||||
Revenue
Revenue increased 50%, or $1.1 million, to $3.2 million for the nine months ended September 30, 2012, from $2.1 million for the nine months ended September 30, 2011, principally due to an increase in test volume partially offset by decreased revenue per test due to a heavier concentration of direct bill clients in the payor mix. Our average revenue (excluding grant revenue and probe revenue) per test decreased by 20% to $545 per test for the nine months ended September 30, 2012, from $682 per test for the nine months ended September 30, 2011, principally due to an increase in test volume related to direct bill clients, which have a lower revenue per test on average. Our test volume increased by 80% to 4,937 for the nine months ended September 30, 2012 from 2,749 for the nine months ended September 30, 2011. Grant revenue increased 116%, or $255,000 to $475,000 for the nine months ended September 30, 2012, from $220,000 for the nine months ended September 30, 2011 principally due to achieving the scheduled milestones for grant drawdowns. Revenue from grants, as a percentage of revenue, increased to 15% for the nine months ended September 30, 2012, from 10% for the nine months ended September 30, 2011. MatBA® revenue for the nine months ended September 30, 2012 was $151,000 or 5% of revenue, compared to no MatBA® revenue for the nine months ended September 30, 2011. However, due to the recent introduction of this test, the small numbers involved in our revenues, and the variability expected with the adoption of any new tests, no assurance or prediction can be given with respect to the level of revenues from our proprietary tests in the future.
Revenue from private insurance carriers and other non-Medicare payors, decreased 11%, or $122,000, to $1.0 million for the nine months ended September 30, 2012, from $1.1 million for the nine months ended September 30, 2011. Revenue from private insurance carriers and other non-Medicare payors as a percentage of total revenue decreased to 31% of total revenue for the nine months ended September 30, 2012, from 53% of total revenue for the nine months ended September 30, 2011. Revenue from Medicare increased 12%, or $63,000, to $587,000 for the nine months ended September 30, 2012, from $524,000 for the nine months ended September 30, 2011. Revenue from Medicare as a percentage of total revenue decreased to 18% for the nine months ended September 30, 2012, from 24% for the nine months ended September 30, 2011. The changes in revenue from private insurance carriers, other non-Medicare payors and Medicare, were primarily due to a
-64-
Table of Contents
change in test mix. Revenue from direct bill customers increased 326%, or $886,000, to $1.2 million for the nine months ended September 30, 2012, from $272,000 for the nine months ended September 30, 2011, principally due to the introduction of our clinical trial services, which, to date, generally have been sold on a direct bill model and the addition of a large community hospital customer that preferred the direct bill model during this period. Revenue from direct bill customers as a percentage of total revenue increased to 36% for the nine months ended September 30, 2012, from 13% for the nine months ended September 30, 2011.
Cost of Revenues
Cost of revenues increased 21%, or $495,000 to $2.9 million for the nine months ended September 30, 2012, from $2.4 million for the nine months ended September 30, 2011, principally due to clinical supply costs related to higher test volumes.
Operating Expenses
Research and Development Expenses. Research and development expenses increased 8%, or $110,000 to $1.6 million for the nine months ended September 30, 2012, from $1.4 million for the nine months ended September 30, 2011, principally as a result of increased headcount and expenses related to our microarray and DNA probe pipeline.
Sales and Marketing Expenses. Sales and marketing expenses decreased 13%, or $150,000 to $1.1 million for the nine months ended September 30, 2012, from $1.2 million for the nine months ended September 30, 2011, principally due to employee turnover.
General and Administrative Expenses. General and administrative expenses increased 10%, or $315,000 to $3.5 million for the nine months ended September 30, 2012, from $3.2 million for the nine months ended September 30, 2011, principally as a result of costs relating to the settlement agreement we entered into with Mr. Maione.
Interest Income and Expense
Interest expense increased 255%, or $2.3 million to $3.3 million, of which approximately $761,000 million was cash interest payments for the nine months ended September 30, 2012, from $918,000, of which approximately $185,000 million was cash interest payments, for the nine months ended September 30, 2011, principally due to a $3.0 million new loan received at the end of March 2011, $3.0 million in new loans received in December 2011, $3.0 million in new loans received in February 2012, and $2.1 million in new loans received during the quarter ended September 30, 2012.
Change in Fair Value of Warrant Liability
The effect of the change in the fair value of our warrant liability caused a $16.3 million variance in a comparison of our results. We recorded income of $6.4 million for the nine months ended September 30, 2012 as compared to an expense of $9.9 million for the nine months ended September 30, 2011. The fair market value of certain of our outstanding common stock warrants that we are required to account for as liabilities decreased in the nine months ended September 30, 2012, which is principally the result of a decrease in our stock price from December 31, 2011 to September 30, 2012, resulting in non-cash income during this period. Because our stock price increased from December 31, 2010 to September 30, 2011, our warrant liability increased in that period, resulting in a significant non-cash expense. Taking into consideration the proposed price range for our initial public offering, we expect that the fair market value of these warrants may decrease further, and that as a result we will record non-cash income in the fourth quarter.
-65-
Table of Contents
Years Ended December 31, 2011 and 2010
The following table sets forth certain information concerning our results of operations for the periods shown:
| Year Ended December 31, | Change | |||||||||||||||
| 2011 | 2010 | $ | % | |||||||||||||
| (dollars in thousands) |
||||||||||||||||
| Revenue |
$ | 3,019 | $ | 2,522 | $ | 497 | 20 | % | ||||||||
| Cost of revenues |
3,117 | 3,516 | (399 | ) | (11 | )% | ||||||||||
| Research and development expenses |
2,074 | 1,167 | 907 | 78 | % | |||||||||||
| Sales and marketing expenses |
1,574 | 716 | 858 | 120 | % | |||||||||||
| General and administrative expenses |
4,439 | 3,446 | 993 | 29 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total Operating Loss |
(8,185 | ) | (6,323 | ) | (1,862 | ) | (29 | )% | ||||||||
| Interest income (expense) |
(1,314 | ) | (792 | ) | 522 | 66 | % | |||||||||
| Change in fair value of warrant liability |
(10,388 | ) | (2,026 | ) | 8,362 | 413 | % | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Qualifying Therapeutic Discovery Project Grants |
— | 733 | (733 | ) | (100 | )% | ||||||||||
| Net Loss |
$ | (19,887 | ) | $ | (8,407 | ) | $ | (11,480 | ) | (137 | )% | |||||
Revenue
Revenue increased 20%, or $ 497,000, to $3.0 million for the year ended December 31, 2011, from $2.5 million for the year ended December 31, 2010, principally due to an increase in our test volume as well as an additional $205,000 in grant revenue from National Institutes of Health and Small Business Innovation Research and an additional $75,000 of revenue from probes sales, partially offset by a decrease in revenue per test. Our average revenue (excluding grant revenue and probe revenue) per test decreased by 6% to $713 per test for the year ended December 31, 2011, from $758 per test for the year ended December 31, 2010, principally due to a reduction in the reimbursement level per test we receive for the UroVysion® testing services we provide, partially offset by a change in mix of tests ordered. Our test volume increased by 15% to 3,622 for the year ended December 31, 2011, from 3,146 for the year ended December 31, 2010. The increased sales volume was a result of new customer additions in the northeastern United States, new territory development in the southeastern United States and an increase in orders from certain of our existing test ordering sites. Our increased clinical laboratory capabilities also contributed to the increase in our test volumes. Revenues from our MatBA®-CLL tests in 2011 were immaterial.
Revenue from private insurance carriers and other non-Medicare payors increased 17%, or $239,000, to $1.6 million for the year ended December 31, 2011, from $1.4 million for the year ended December 31, 2010, principally due to an increase in test volume. Revenue from private insurance carriers and other non-Medicare payors was 54% of total revenue for the year ended December 31, 2011 and 55% of total revenue for the year ended December 31, 2010. Revenue from Medicare increased 18%, or $112,000, to $717,700 for the year ended December 31, 2011, from $606,000 for the year ended December 31, 2010, principally due to an increase in test volume. Revenue from Medicare as a percentage of total revenue remained relatively flat at 24% for the years ended December 31, 2011 and December 31, 2010. Revenue from direct bill customers decreased slightly to $350,000 for the year ended December 31, 2011, from $408,000 for the year ended December 31, 2010. Revenue from direct bill customers as a percentage of total revenue decreased to 12% for the year ended December 31, 2011, from 16% of total revenue for the year ended December 31, 2010, principally due to a shift in focus away from direct bill customers and toward third party payors.
-66-
Table of Contents
Cost of Revenues
Cost of revenues decreased by 11%, or $399,000, to $3.1 million for the year ended December 31, 2011, from $3.5 million for the year ended December 31, 2010. Even though revenue increased, the cost of revenues decreased as a result of operational efficiencies achieved during 2011.
Operating Expenses
Research and Development Expenses. Research and development expenses increased by 78%, or $907,000, to $2.1 million for the year ended December 31, 2011, from $1.2 million for the year ended December 31, 2010, principally as a result of increased headcount for additional research and development efforts related to our microarray and DNA probe pipeline.
Sales and Marketing Expenses. Sales and marketing expenses increased by 120%, or $858,000, to $1.6 million for the year ended December 31, 2011, from $716,000 for the year ended December 31, 2010. The increase in our sales and marketing expenses was principally due to the expansion of our sales and marketing activities, including hiring additional sales and marketing personnel and utilizing consultants in connection with the launch of our MatBA®-CLL, and the introduction of new DNA probes outside the United States.
General and Administrative Expenses. General and administrative expenses increased by 29%, or $993,000, to $4.4 million from the year ended December 31, 2011, from $3.5 million for the year ended December 31, 2010. This increase was principally due to the increase in our bad debt expense and the recruiting and hiring of additional personnel, including a Chief Financial Officer, Controller, and Director of IT, and a significant increase in professional fees as we prepared to become a public company. Bad debt expense was $373,000 for the year ended December 31, 2011 compared to $46,000 for the year ended December 31, 2010. This increase of $327,000 is principally due to a write down in receivables resulting from a changeover in our billing providers and resulting collection problems during third quarter 2011. We observed that our prior billing company slowed their invoicing activities and collection efforts during the months immediately preceding our transition, effective July 1, 2011, to the new billing company. This resulted in reduced receipts of cash relating to the invoices for that period, during the third quarter 2011. We do not expect any continuing collection problems in the future relating to our transition to a new billing company.
Interest Income and Expense
Interest expense increased by 66%, or $522,000, to $1.3 million for the year ended December 31, 2011, from $792,000 for the year ended December 31, 2010, due to amortization of the consideration paid to John Pappajohn for the guarantee of our loan from Wells Fargo Bank, N.A. (“Wells Fargo”) and interest related to the March 2011 loan from DAM Holdings, LLC (“DAM”).
Change in Fair Value of Warrant Liability
The expense we booked for the change in the fair value of our warrant liability increased by 413%, or $8.4 million to $10.4 million for the year ended December 31, 2011, from $2.0 million for the year ended December 31, 2010, due to an increase in the fair market value of certain of our outstanding common stock warrants that we are required to account for as liabilities, which is principally the result of an increase in our stock price.
-67-
Table of Contents
Years Ended December 31, 2010 and 2009
The following table sets forth certain information concerning our results of operations for the periods shown:
| Year Ended December | Change | |||||||||||||||
| 2010 | 2009 | $ | % | |||||||||||||
| (dollars in thousands) |
||||||||||||||||
| Revenue |
$ | 2,522 | $ | 1,666 | $ | 856 | 51 | % | ||||||||
| Cost of revenues |
3,516 | 2,532 | 984 | 39 | % | |||||||||||
| Research and development expenses |
1,167 | 1,336 | (169 | ) | (13 | )% | ||||||||||
| Sales and marketing expenses |
716 | 239 | 477 | 200 | % | |||||||||||
| General and administrative expenses |
3,446 | 1,845 | 1,601 | 87 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total Operating Loss |
(6,323 | ) | (4,286 | ) | (2,037 | ) | (48 | )% | ||||||||
| Interest expense |
(792 | ) | (2,089 | ) | 1,297 | 62 | % | |||||||||
| Change in fair value of warrant liability |
(2,026 | ) | (953 | ) | (1,073 | ) | (113 | )% | ||||||||
| Qualifying Therapeutic Discovery Project Grants |
733 | — | 733 | 100 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net Loss |
$ | (8,407 | ) | $ | (7,328 | ) | $ | (1,079 | ) | (15 | )% | |||||
Revenue
Revenue increased by 51%, or $856,000, to $2.5 million for the year ended December 31, 2010, from $1.7 million for the year ended December 31, 2009 as a result of increases in average revenue per test and test volumes. Our average revenue per test (excluding grant revenue and probe revenue) increased by 14% to $758 for the year ended December 31, 2010, from $667 for the year ended December 31, 2009. A shift in test ordering sites away from direct-bill customers and a shift to more comprehensive testing contributed to the increase in average revenue per test. Our test volume increased by 36% to 3,146 for the year ended December 31, 2010, from 2,321 for the year ended December 31, 2009. In 2010 we built a direct sales force focused on the northeastern and southeastern United States, with particular emphasis on oncology practices in New Jersey, New York, Pennsylvania, Georgia, and Florida, which resulted in new customers. The increase in test volumes was principally driven by such increased focus on sales and marketing activities as well as increased clinical laboratory capabilities, which allowed us to offer a broader range of testing services. As a result of our ability to offer a broader range of testing services, we were able to get additional business from existing test ordering sites and attract new physicians and clinical groups.
Revenue from private insurance carriers and other non-Medicare payors increased 86%, or $645,000 to $1.4 million for the year ended December 31, 2010, from $752,000 for the year ended December 31, 2009 principally due to an increase in test volume. Revenue from private insurance carriers and other non-Medicare payors as a percentage of total revenue increased to 55% for the year ended December 31, 2010, from 45% of total revenue for the year ended December 31, 2009. Revenue from Medicare increased 72%, or $253,000, to $606,000 for the year ended December 31, 2010, from $352,000 for the year ended December 31, 2009 principally due to an increase in test volume. Revenue from Medicare as a percentage of total revenue increased to 24% for the year ended December 31, 2010, from 21% of total revenue for the year ended December 31, 2009. Revenue from direct bill customers decreased 11%, or $53,000 to $408,000 for the year ended December 31, 2010, from $461,000 for the year ended December 31, 2009. Revenue from direct bill customers as a percentage of total revenue decreased to 16% for the year ended December 31, 2010, from 28% of total revenue for the year ended December 31, 2009 principally due to a shift in focus away from direct bill customers and toward third party payors.
-68-
Table of Contents
Cost of Revenues
Cost of revenues increased by 39%, or $984,000, to $3.5 million for the year ended December 31, 2010, from $2.5 million for the year ended December 31, 2009. The increase was largely a result of the increased headcount and testing supplies required to meet increased demand for our clinical laboratory services and an increase in test volume.
Operating Expenses
Research and Development Expenses. Research and development expenses decreased by 13%, or $169,000, to $1.2 million for the year ended December 31, 2010, from $1.3 million for the year ended December 31, 2009, principally due to adjustments to and timing of our research and development activities.
Sales and Marketing Expenses. Sales and marketing expenses increased by 200%, or $477,000, to $716,000 for the year ended December 31, 2010, from $239,000 for the year ended December 31, 2009. This increase was principally driven by our hiring of additional sales personnel.
General and Administrative Expenses. General and administrative expenses increased by 87%, or $1.6 million, to $3.6 million for the year ended December 31, 2010, from $1.9 million for the year ended December 31, 2009. General and administrative expenses for the year ended December 31, 2010 included one-time and non-recurring expenses of approximately $1.1 million, including an accrual related to a potential litigation liability with respect to our former operations in Massachusetts, severance expenses, and other expenses related to recruitment of our new Chief Executive Officer and of other staff, including sales and IT directors. Bad debt expense was $46,000 for the year ended December 31, 2010 compared to $23,000 for the year ended December 31, 2009.
Interest Expense
Interest expense decreased by 62%, or $1.3 million, to $792,000 for the year ended December 31, 2010, from $2.1 million for the year ended December 31, 2009, due to a decrease in the consideration payable by us to extend certain guarantees of our Wells Fargo loan.
Change in Fair Value of Warrant Liability
The expense we booked for the change in fair value of our warrant liability increased by 113%, or $1.1 million to $2.0 million for the year ended December 31, 2010, from $953,000 for the year ended December 31, 2009, due to an increase in the fair market value of certain of our outstanding common stock warrants that we are required to account for as liabilities, which is principally the result of an increase in our stock price.
Qualifying Therapeutic Discovery Project Grants
For the year ended December 31, 2010, we recognized other income of $733,000 for grants in lieu of federal income tax credits received in 2010 under the Qualifying Therapeutic Discovery Project Program. This is a one-time item.
Liquidity and Capital Resources
Sources of Liquidity
Our primary sources of liquidity have been funds generated from our debt financings and equity financings. In addition, we have generated funds from the following sources: (i) the grants received in lieu of federal income tax credits under the Qualifying Therapeutic Discovery Project Program; (ii) grants from the National Institutes of Health and (iii) cash payments generated from operations.
-69-
Table of Contents
Series-B Preferred Stock. In seven closings from April 2010 to November 2010, we raised aggregate gross proceeds of $9.1 million through an offering of our Series B preferred stock. We sold an aggregate of 1,821,600 shares of Series B preferred stock at a purchase price of $5.00 per share. We realized approximately $8.3 million in net proceeds from the offering after payment of stock issuance costs. In addition, we issued warrants to acquire an aggregate of 131,160 shares of our common stock to certain third parties in connection with the Series B preferred stock offering. The conversion price of our Series B preferred stock is $10.00 and, if our initial public offering price is less than $10.00 per share, will ratchet down to our initial public offering price per share. See the section entitled “Description of Capital Stock—Preferred Stock” for a more detailed description of our Series B Preferred Stock.
Wells Fargo Line of Credit. In April 2008, we entered into and thereafter fully utilized a line of credit with Wells Fargo in the amount of $1.5 million for the purposes of meeting operating expenses and working capital needs. In July 2008, we increased the line of credit with Wells Fargo to $3.5 million. In March 2009, we increased the facility to $4.5 million. In July 2009, we increased the facility to $5.5 million and in October 2009, we increased the facility to $6.0 million, which we have fully utilized. In July 2010, we extended the maturity date of the facility from July 31, 2010 to July 31, 2011. In June 2011, we extended the maturity date on the facility to July 31, 2012. In February 2012, we extended the maturity date of the facility to July 31, 2013. In October 2012, we extended the maturity date to April 1, 2014. The interest is computed at LIBOR + 1.75%, which was 1.96% as of September 30, 2012. Mr. Pappajohn, a member of our board of directors, has guaranteed the Wells Fargo Line of Credit.
DAM Line of Credit. In March 2011, we entered into, and since have fully utilized, a line of credit with DAM in the amount of $3.0 million for the purposes of meeting operating expenses, including expenses related to our initial public offering process. As consideration, we pay an annual interest rate of 3% on the borrowed funds on a monthly basis and issued DAM warrants to purchase an aggregate of 150,000 shares of common stock at an exercise price of $10.00 per share. Pursuant to our credit agreement entered into in connection with our line of credit with DAM, we are required to pay back the full amount outstanding under this line of credit upon certain enumerated maturity events, including the completion of an initial public offering that generates gross proceeds of $10.0 million or more. Our interest rate under this line of credit increased to 10% per annum, effective January 1, 2012, because such maturity events, including completion of an initial public offering, did not occur before January 1, 2012. In March 2012, we extended the maturity date of this facility to April 1, 2013, unless certain maturity events occur prior to April 1, 2013, in exchange for a grant of 37,500 warrants on the same terms as those issued in the December 2011 Financing Transaction described below. On October 16, 2012, DAM Holdings agreed to surrender 37,500 warrants in exchange for a cash interest payment of $52,500 at maturity. In addition, we amended the agreement to provide that the interest rate from January 1, 2012 until a maturity event occurs shall be 10%. After a maturity event occurs, interest begins to compound at a rate of 18% per annum until the balance is paid in full.
December 2011 Financing Transaction. We entered into a Credit Agreement dated as of December 21, 2011, as amended and restated as of February 13, 2012, with John Pappajohn and Andrew Pecora (indirectly through an investment company), both members of our board of directors, and NNJCA Capital, LLC (“NNJCA”), a limited liability company of which Dr. Pecora is a member, for a $6.0 million secured term loan. Mr. Pappajohn has provided $4.0 million of financing, NNJCA has provided $1.5 million of financing and Dr. Pecora has provided $500,000 of financing under the Credit Agreement. A prior lending party to the December 2011 Credit Agreement, European Trust Management (ETM), defaulted in its obligation to lend under the agreement. The Company is currently seeking to recover $50,000 paid to ETM in connection with this loan facility.
The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 13, 2013, with an option, at our election, if there has been no event of default, to extend the loan term for an additional six months. We exercised our option to extend the maturity date to August 13, 2013. The term loan due to Dr. Pecora and NNJCA has a prepayment penalty in the aggregate amount of
-70-
Table of Contents
$190,000, less interest previously paid, if we prepay the loan prior to February 12, 2013. The lenders may require that the loan be repaid within 30 days should we complete our initial public offering and receive gross proceeds of at least $15.0 million. In the event that any lender requires payment upon completion of our initial public offering and certain other maturity events, the annual interest rate on any unpaid balance shall increase to 12%. The loan is secured by all of our assets, including our intellectual property, subject to prior first and second liens in favor of Wells Fargo Bank and DAM. Pursuant to an intercreditor agreement, the lenders have agreed that all amounts due to DAM are to be paid prior to payment to the lenders under this Credit Agreement, but that as between such lenders, following an event of default, all of the security granted by us is to be applied first to repay obligations due to Dr. Pecora and NNJCA, and then to Mr. Pappajohn after they have been paid in full. As Mr. Pappajohn has guaranteed the Wells Fargo debt, in essence under the intercreditor agreement, NNJCA and Dr. Pecora will be junior only to DAM.
Upon funding the lenders received five-year warrants to purchase an aggregate of 211,764 shares of our common stock at the lower of (i) $17.00 per share and (ii) a 20% discount to the initial public offering price if the initial public offering price is less than $21.25 per share, with warrants being issued proportionately to Mr. Pappajohn, Pecora and NNJCA upon their funding $4.0 million, $0.5 million and $1.5 million, respectively. The lenders received an aggregate of 70,588 additional warrants on the same terms, with the warrants being issued proportionately to Mr. Pappajohn, Dr. Pecora and NNJCA, because we did not consummate our initial public offering within 181 days of funding. The warrant exercise price is subject to full ratchet anti-dilution protection in the event of issuances of more than $5.0 million of securities at prices below the exercise price prior to the completion of our initial public offering. The lenders may elect to net exercise their warrants. Mr. Pappajohn also had the option, exercisable up to two hours after he receives notice of the completion of our initial public offering, to convert the outstanding principal amount of his debt into shares of our common stock at a conversion price equal to the lesser of $17.00 per share and a 20% discount to our initial public offering price, whichever is lower. However, Mr. Pappajohn subsequently agreed to convert $2.0 million of the outstanding principal due to him to common stock at the initial public offering price per share upon consummation of this offering, to eliminate the conversion feature of the remaining $2.0 million in debt, which is held by his spouse, effective upon consummation of this offering and to extend the maturity date to August 15, 2013 for the $2.0 million principal amount which is not converted. Subsequently, the warrants issued to Mr. Pappajohn were amended to provide that the exercise price shall equal the lesser of $17.00 per share or our initial public offering price per share and to extend the $2.0 million dollars in non-convertible debt, which is held by his spouse, to April 1, 2014. In connection with this agreement, we issued to Mr. Pappajohn additional warrants to purchase an aggregate of 75,202 shares of our common stock on the Modified Bridge Form. The warrant exercise price on the Modified Bridge Form is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants on the Modified Bridge Form are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00, so that the initial grant would be for an aggregate of 182,633 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 213,072 shares and 159,804 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively. The conversion price of the notes and the exercise price of the warrants are subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. In addition, the warrants to purchase an aggregate of 94,117 shares of our common stock issued to Dr. Pecora and NNJCA in connection with this financing were cancelled and the promissory notes issued to Dr. Pecora and NNJCA were amended to increase the pre-payment penalties.
Shares that the lenders receive are subject to a lock-up agreement for 180 days after the consummation of our initial public offering on the same terms as other lock-up agreements in favor of the underwriters of this offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
-71-
Table of Contents
2012 Convertible Debt Financing Transaction
We entered into a restated Credit Agreement dated as of October 17, 2012 with John Pappajohn and Mark Oman for a $3.0 million convertible term loan. Mr. Pappajohn provided approximately $1.8 million of financing and Mr. Oman provided approximately $1.3 million of financing under the Credit Agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 26, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
The lenders received ten-year warrants to purchase an aggregate of 286,275 shares of our common (which were issued in proportion to their respective funding amounts), in each case at the lower of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering price per share is less than $17.00 per share. The lenders received additional ten-year warrants to purchase an aggregate of 17,648 shares of our common stock on the same terms because we did not consummate our initial public offering by November 27, 2012. The lenders will receive additional ten-year warrants to purchase an aggregate of 17,648 shares of our common stock (to be issued in proportion to their respective funding amounts) on the same terms in the event we do not consummate our initial public offering by February 22, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5.0 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00 per share, so that the initial grant would be for an aggregate of 695,237 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 811,110 shares and 608,333 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
On December 7, 2012, Mr. Pappajohn provided an additional $1.0 million of financing on the same terms as the restated credit agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on June 4, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
Mr. Pappajohn received ten-year warrants to purchase an aggregate of 58,824 shares of our common at the lesser of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering price per share is less than $17.00 per share. He will receive additional ten-year warrants to purchase an aggregate of 5,886 shares of our common stock on the same terms in the event we do not consummate our initial public offering by each the following dates: March 4, 2013 and June 4, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00 per share, so that the initial grant would be for an aggregate of 142,857 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 166,667 shares and 125,000 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
Shares that the lenders receive are subject to a lock-up agreement for 180 days after the consummation of our initial public offering on the same terms as other lock-up agreements in favor of the underwriters of this offering, but
-72-
Table of Contents
otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
Cash Flows
Our net cash flow from operating, investing and financing activities for the periods below were as follows:
| Nine Months Ended | Year Ended | |||||||||||||||||||
| September 30, | December 31, | |||||||||||||||||||
| 2012 | 2011 | 2011 | 2010 | 2009 | ||||||||||||||||
| (in thousands) | (unaudited) | |||||||||||||||||||
| Cash provided by (used in): |
||||||||||||||||||||
| Operating activities |
$ | (5,763 | ) | $ | (4,442 | ) | $ | (5,073 | ) | $ | (5,731 | ) | $ | (3,335 | ) | |||||
| Investing activities |
(268 | ) | 31 | (113 | ) | (168 | ) | (191 | ) | |||||||||||
| Financing activities |
4,517 | 2,825 | 5,824 | 7,648 | 3,528 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net increase (decrease) in cash and cash equivalents |
$ | (1,514 | ) | $ | (1,586 | ) | $ | 638 | $ | 1,749 | $ | 2 | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
We had cash and cash equivalents of $903,000 at September 30, 2012, $2.4 million at December 31, 2011, $1.8 million at December 31, 2010, and $30,000 at December 31, 2009. The $1.5 million decrease in cash and cash equivalents from December 31, 2011, to September 30, 2012, was principally the result of our $5.8 million net cash used in operations, including $761,000 in cash interest payments, payment of $1.2 million in equity issuance costs and $184,000 in patent costs offset by $5.7 million in net proceeds from borrowings under new notes payable and warrant exercises. The $638,000 increase in cash and cash equivalents from December 31, 2010, to December 31, 2011, principally was the result of our $19.9 million net loss during the period, offset by non-cash equity compensation of $1.1 million, the change in value of derivative warrants of $10.4 million and an increase in accounts payable and accrued expenses of $1.2 million, resulting in $5.1 million of cash used in operations offset by $6.0 million in net proceeds from borrowings under our line of credit and on a note payable. The $1.8 million increase in our cash and cash equivalents from December 31, 2009, to December 31, 2010, resulted principally from our $8.4 million net loss during the year, offset by approximately $3.5 million in noncash expenses, principally due to the change in value of derivative warrants, and $8.3 million in net proceeds from the issuance of the Series B preferred stock.
Cash Used in Operating Activities
Net cash used in operating activities was $5.8 million for the nine months ended September 30, 2012, consisting primarily of a $2.6 million net loss during the period, which includes $761,000 in cash interest payments, and non-cash income from a change in fair value of warrant liability of $6.4 million offset by non-cash debt costs of $2.5 million and $900,000 in equity and warrant-based non-cash compensatory transactions.
Net cash used in operating activities was $5.1 million for the year ended December 31, 2011 consisting primarily of a $19.9 million net loss, offset by the change in the fair value of the warrant liability of $10.4 million, depreciation of $357,000, bad debt expense of $373,000 due a decline in collections related to a change in our billing company, stock-based compensation of $1.1 million, amortization of a loan guarantee fee of $575,000, additional accretion of debt discount on newly outstanding debt of $481,000 and an increase in accounts payable and accrued expenses due to an increase in professional fees.
Net cash used in operating activities was $5.7 million for the year ended December 31, 2010 consisting of a $8.4 million net loss partially offset by $2.0 million change in the fair value of warrant liabilities, $477,000 in equity based compensation expense, $528,000 in amortization of loan guarantee fees, an increase in accounts payable of $447,000 due to cash conservation efforts, an increase in accounts receivable of $570,000 due to
-73-
Table of Contents
collection problems with our prior billing company, an increase in other current assets of $711,000 related to IRS research credit, and $317,000 in depreciation expense.
Net cash used in operating activities was $3.3 million for the year ended December 31, 2009 consisting of a $7.3 million net loss partially offset by $953,000 change in the fair value of warrant liabilities, $301,000 in equity based compensation expense, $1.9 million in amortization of loan guarantee fees, an increase in accounts payable of $430,000 due to cash conservation efforts, and $304,000 in depreciation expense.
Cash Used in Investing Activities
Net cash used in investing activities was $268,000 for the nine months ended September 30, 2012 due to an increase in our restricted cash related to a $50,000 increase in the Letter of Credit related to our lease as well as $184,000 in patent application costs. Pursuant to the terms of our lease for our Rutherford facility, we were required to maintain a letter of credit in the amount of $450,000 to use as a guarantee for the security deposit. In February 2011, we allowed the letter of credit to expire, which as discussed below, led to a decrease in our restricted cash for fiscal 2011. On April 6, 2012, we reached an agreement with the landlord which requires us to provide a letter of credit in the amount of $250,000 and in exchange, the landlord agreed to forebear taking action to enforce our obligation to maintain the $450,000 letter of credit. The landlord also agreed to reduce our security deposit requirement to a $250,000 letter of credit upon a capital raise of at least $20.0 million by July 31, 2012. We have continued to update the landlord on the expected timing of a capital raise. The landlord has verbally agreed to extend the time to complete a capital raise and has allowed us to continue to lease the property with a reduced security deposit of $250,000.
Net cash used in investing activities was $113,000 for the year ended December 31, 2011 due to purchases of fixed assets of $269,000 and patent costs of $83,000 offset by a decrease of $238,000 in restricted cash related to a letter of credit with our landlord.
Net cash used in investing activities was $168,000 for the year ended December 31, 2010 due to purchases of fixed assets and patent costs.
Net cash used in investing activities was $191,000 for the year ended December 31, 2009 due to purchases of fixed assets and patent costs.
Cash Provided by Financing Activities
Net cash provided by financing activities was $4.5 million for the nine months ended September 30, 2012, principally due to our receipt of $3.0 million in net proceeds from the December 2011 financing transaction which closed in February 2012, $620,000 in net proceeds from the exercise of certain warrants and $2.0 million in proceeds from a convertible debt transaction that closed in August 2012. We paid $1.2 million in equity issuance costs related to our initial public offering in the nine months ended September 30, 2012.
Net cash provided by financing activities was $5.8 million for the year ended December 31, 2011 due to the issuance of the $3.0 million DAM Holdings line of credit and the $3.0 million portion of the December 2011 financing transaction which closed in 2011.
Net cash provided by financing activities was $7.7 million for the year ended December 31, 2010 due to the issuance of the $9.1 million in preferred stock offset by $827,000 in issuance costs, and the net $410,000 payment of an existing line of credit.
Net cash provided by financing activities was $3.5 million for the year ended December 31, 2009 due to the $3.6 million borrowing on a line of credit.
Capital Resources and Expenditure Requirements
We expect to continue to incur substantial operating losses in the future. It may take several years, if ever, to achieve positive operational cash flow. We expect that we will use a portion of the net proceeds from this offering, our revenues from operations and our existing cash and cash equivalents to finance further research and development and commercialization of our technology and tests, to expand our clinical laboratory, to fund collaborations, to repay approximately $5.0 million of existing indebtedness and for general working capital and other corporate purposes, including the increased costs associated with being a public company. We may also use a portion of the net proceeds of this offering to acquire or invest in businesses, technologies, services or products, although we do not have any current plans to do so.
-74-
Table of Contents
Without the net proceeds from this offering, we believe our current cash resources, including the $2.0 million in debt financing we raised after September 30, 2012, are sufficient to satisfy our liquidity requirements at our current level of operations through February 28, 2013. If we do not consummate this offering prior to the end of February 2013, we expect that we will need to raise additional financing in the first quarter of 2013, which might not be available on favorable terms, if at all. We have had and will continue to have discussions with certain of our stockholders regarding potential additional financing, but we can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. Our forecast of the period of time through which our current financial resources will be adequate to support our operations and the costs to support our general and administrative, sales and marketing and research and development activities are forward-looking statements and involve risks and uncertainties. Actual results could vary materially and negatively as a result of a number of factors, including:
| • | the timing of and the costs involved in obtaining regulatory approvals and clearances for our tests; |
| • | the costs of operating and enhancing our laboratory facilities; |
| • | if our new diagnostic tests are approved, our commercialization activities; |
| • | the scope, progress and results of our research and development programs; |
| • | the scope, progress, results, costs, timing and outcomes of the clinical trials of our diagnostic tests; |
| • | our ability to manage the costs for manufacturing our microarrays and probes; |
| • | the costs of maintaining, expanding and protecting our intellectual property portfolio, including potential litigation costs and liabilities; |
| • | our ability to obtain adequate reimbursement from governmental and other third-party payors for our tests and services; |
| • | revenues received from sales of our tests, if approved by FDA and accepted by the market; |
| • | the costs of additional general and administrative personnel, including accounting and finance, legal and human resources, as a result of becoming a public company; |
| • | the costs of developing our anticipated internal sales, marketing and distribution capabilities; |
| • | our ability to secure financing and the amount thereof; |
| • | whether we determine to fund our initial contribution pursuant to our Affiliation Agreement with Mayo in March 2013; |
| • | ability to collect revenues; and |
| • | other risks discussed in the section entitled “Risk Factors”. |
Even with the proceeds from this offering, we may need to raise additional capital to expand our business to meet our long-term business objectives or to repay the approximately $8.0 million in indebtedness that matures in April 2014. We expect that our operating expenses and capital expenditures will increase in the future as we expand our business. We plan to increase our sales and marketing headcount to promote our new clinical tests and services and to expand into new geographies and to increase our research and development headcount to develop and validate the proprietary tests currently in our pipeline, to expand our pipeline and to
-75-
Table of Contents
perform work associated with our research collaborations. We also expect that our costs of collaborations with research and academic institutions will increase in the future as such institutions begin to view us as a commercial company. For example, in 2011 we entered into an affiliation agreement to form a joint venture with the Mayo Foundation for Medical Education and Research pursuant to which we expect to make capital contributions of $6 million over the next three years, subject to the joint venture entity’s achievement of certain operational milestones. Until we obtain regulatory approval to market our proprietary tests outside our laboratory, if ever, and can generate a sufficient amount of revenues to finance our cash requirements, which we may never do, we may seek to finance future cash needs through public or private equity offerings, debt financings, borrowings or strategic partnerships coupled with an investment in our company or a combination thereof. If we raise additional funds through the issuance of convertible debt securities, or other debt securities, these securities could be secured and could have rights senior to those of our common stock. In addition, any new debt incurred by the Company could impose covenants that restrict our operations. The issuance of any new equity securities will also dilute the interest of our current stockholders. Given the risks associated with our business, including our unprofitable operating history and our ability to develop additional proprietary tests, additional capital may not be available when needed on acceptable terms, or at all. If adequate funds are not available, we may need to curb our expansion plans or limit our research and development activities, which would have a material adverse impact on our business prospects and results of operations. For further discussion of the impact of our present indebtedness and access to future financing on our business, see the section entitled “Risk Factors—Risks Related to Our Business and Strategy—We have a substantial amount of indebtedness, which could have a material adverse effect on our financial condition and our ability to fund operations, obtain additional financing and react to changes in our business.”
Future Contractual Obligations
The following table reflects a summary of our estimates of future contractual obligations as of December 31, 2011. The information in the table reflects future unconditional payments and is based on the terms of the relevant agreements, appropriate classification of items under U.S. GAAP as currently in effect and certain assumptions, such as the interest rate on our variable debt that was in effect as of December 31, 2011. Future events could cause actual payments to differ from these amounts.
| Total | Less than 1 Year |
2-3 Years | 4-5 Years | More than 5 Years |
||||||||||||||||
| (dollars in thousands) |
||||||||||||||||||||
| Principal and interest under notes payable and lines of credit |
$ | 12,998 | $ | 816 | $ | 12,182 | $ | — | $ | — | ||||||||||
| Capital Lease obligations, including interest, for equipment |
70 | 43 | 27 | — | — | |||||||||||||||
| Operating lease obligations relating to corporate headquarters and clinical laboratory |
3,347 | 522 | 1,080 | 1,133 | 612 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 16,415 | $ | 1,381 | $ | 13,289 | $ | 1,133 | $ | 612 | ||||||||||
Income Taxes
Over the past several years we have generated operating losses in all jurisdictions in which we may be subject to income taxes. As a result, we have accumulated significant net operating losses and other deferred tax assets. Because of our history of losses and the uncertainty as to the realization of those deferred tax assets, a full valuation allowance has been recognized. We do not expect to report a provision for income taxes until we have a history of earnings, if ever, that would support the realization of our deferred tax assets.
-76-
Table of Contents
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off balance sheet activities as defined in Item 303(a)(4) of Regulation S-K.
Qualitative and Quantitative Disclosures about Market Risk
Our exposure to market risk is limited to our cash, cash equivalents and marketable securities, all of which have maturities of one year or less. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities that management believes to be of high credit quality. The securities in our investment portfolio are not leveraged, are classified as available for sale and are, due to their short-term nature, subject to minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have a material negative impact on the value of our investment portfolio.
We do not have any material foreign currency exposure.
-77-
Table of Contents
Company Overview
We are an early-stage diagnostics company focused on developing and commercializing proprietary genomic tests and services to improve and personalize the diagnosis, prognosis and response to treatment (theranosis) of cancer. Our proprietary tests target cancers that are difficult to prognose and predict treatment outcomes by using currently available mainstream techniques. These cancers include hematological, urogenital and HPV-associated cancers. We provide our proprietary tests and services, along with a comprehensive range of non-proprietary oncology-focused tests and laboratory services, to oncologists and pathologists at hospitals, cancer centers, and physician offices. To date, we have engaged in only limited sales and marketing activities and have generated most of our revenue through sales of our non-proprietary testing services to a limited number of oncologists, pathologists and community hospitals located mostly in the eastern and mid western United States. Our non-proprietary laboratory testing services include molecular testing, sequencing mutational analysis, flow cytometry testing, histology testing and cytology testing. These tests are described in more detail in the section entitled “Description of the Business—Laboratory Services”. We are currently offering our tests and laboratory services from our 17,936 square foot state-of-the-art laboratory located in Rutherford, New Jersey, which has been accredited by the College of American Pathologists, which is one of six approved accreditation methods under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”), to perform high complexity testing. CLIA certification and accreditation are required before any laboratory, including ours, may perform testing on human specimens for the purpose of obtaining information for the diagnosis, prevention, treatment of disease, or impairment of, or assessment of health.
Our proprietary tests are based principally on our expertise in specific cancer types, test development methodologies and proprietary algorithms correlating genetic events with disease specific information. During the first quarter of 2011, we commercially launched MatBA®-CLL, our first proprietary microarray test for chronic lymphocytic leukemia (“CLL”). In January 2012, we received CLIA approval for MatBA®-SLL, our proprietary microarray for risk stratification in small lymphocytic lymphoma (“SLL”), and we are currently offering MatBA®-SLL in our laboratory. In addition, we are developing a series of other proprietary genomic tests in our core oncology markets.
We have established collaborative relationships with key thought leaders in oncology, which enable us to develop and validate the effectiveness and utility of our tests in a clinical setting and which provide us access to clinically robust patient data. For example, we agreed to form a joint venture in March 2013 with Mayo Foundation for Medical Education and Research (“Mayo”) focused on developing oncology diagnostic services and tests utilizing next-generation sequencing. Additionally, we agreed to a research collaboration with Cleveland Clinic to validate our renal-cancer microarray, UroGenRA™ Renal.
The non-proprietary testing services we offer are entirely focused on specific oncology categories where we are developing our proprietary arrays and probe panels. We believe that there is significant synergy in developing and marketing a complete set of tests and services that are disease-focused and delivering those tests and services in a comprehensive manner to help with treatment decisions. The insight that we develop in delivering the non-proprietary services are often leveraged in the development of our proprietary programs and now increasingly in the validation of our proprietary programs (such as MatBA®) for clinical use.
We believe that we can be successful by offering cancer professionals a fully-integrated menu of oncology-focused proprietary tests and customized laboratory services. Based on our discussions with leading researchers in the oncology field and our interactions with our collaborators, as well as information we learn through performing the non-proprietary genetic diagnostic testing services, which are focused on the specific oncology categories where we are developing our proprietary tests we provide to our customers, we believe that our proprietary tests provide superior diagnostic and prognostic values than currently available tests and services. In particular, our proprietary tests deliver a level of genomic information not provided by other currently
-78-
Table of Contents
available tests. For example, the majority of current cytogenetic analysis for CLL and SLL that is available in clinical laboratories today assesses gain and loss in genomic material at four specific sites. There are two other marketed arrays for CLL (Combimatrix and Quest) of which we are aware. Both of these arrays report out gains and losses at four to five genomic sites. MatBA®-CLL, on the other hand, reports out gains and losses at nine genomic sites and MatBA®-SLL reports out gains and losses at seven genomic sites. We believe our ability to rapidly translate research insights about the genetics and molecular mechanisms of cancer into the clinical setting will improve patient treatment and management and that this approach will become a key component in the standard of care for personalized cancer treatment.
MatBA®-CLL is unique in its targeted, proprietary design and has been validated in two independent, clinically robust data sets of specimens in collaboration with Dr. Kanti R. Rai, a renowned CLL oncologist and Chief of the Division of Hematology and Oncology at Long Island Jewish / North Shore Hospital. MatBA®-CLL is, to our knowledge based on our informal communications with New York State Department of Health personnel, the first oncology microarray to be approved by the New York State Department of Health, one of the only state governmental agencies that reviews the performance characteristics and clinical utility of new laboratory developed tests (“LDTs”).
There are approximately 14,500 new cases of CLL diagnosed in the United States each year, and these cases require risk stratification and guidance on patient management and treatment issues at multiple points during the course of the disease. Prior to the introduction of MatBA®-CLL, clinicians had to rely on diagnostic tests that provided limited information on the genetic abnormalities associated with CLL. In contrast, MatBA®-CLL identifies a much broader range of genomic markers associated with CLL, providing improved diagnostic and prognostic value, as well as critical information about how to best structure a treatment regimen for a patient. We developed the MatBA® platform under the guidance of Dr. Raju Chaganti, our Chairman and one of our founders. Dr. Chaganti founded one of the earliest comprehensive clinical cytogenetic laboratories focused on cancer in the United States at Memorial Sloan-Kettering Cancer Center, where he is on the faculty of the Department of Medicine and is William E. Snee Chair.
In collaboration with Memorial Sloan-Kettering Cancer Center and Long Island Jewish / North Shore Hospital, we have completed the validation of MatBA®-SLL. MatBA®-SLL was CLIA approved in January 2012 and is now offered in our laboratory. We believe that MatBA®-SLL is the only microarray that will permit risk-stratification in this previously underserved cancer subtype. This adaptation of MatBA® for SLL has allowed us to develop a robust mechanism to analyze DNA that is derived from formalin-fixed paraffin-embedded (“FFPE”) biopsy material. This adaptation has been a critical development which will accelerate the development of our microarrays for other solid tumors or cancers that present themselves as a mass.
We are also in advanced stages of validating the MatBA® array for prognostic utilization in the two predominant types of mature B cell neoplasms, diffuse large B cell lymphoma (“DLBCL”) and follicular lymphoma (“FL”). Collectively, these lymphomas represent over 70% of the mature B cell cancers (neoplasms) and over 66,000 newly diagnosed cancer cases each year in the United States. Our MatBA® array has been designed to measure genetic markers at 80 specific genomic sites where genetic alterations are associated with mature B cell neoplasms. We have initiated a 200 specimen clinical validation study for DLBCL with Dr. Julie Teruya-Feldstein, Director of Memorial Sloan-Kettering’s Immunohistochemisty Laboratory and a member of that hospital’s Institutional Review Board. Clinical validations are also underway for mantle-cell lymphoma (“MCL”), which is an aggressive sub-type of lymphoma.
We are developing microarray tests for the diagnosis, prognosis and theranosis of a range of urogenital cancers. These include the UroGenRA™ microarray for kidney, prostate and bladder cancers and the UGenRA™ microarray for endometrial, ovarian and cervical cancers. UroGenRA™ detects genomic changes in over 100 regions of the human genome with potential diagnostic and/or prognostic value in one or more of these types of cancer. These microarrays have been designed to address specific needs associated with the management of each urogenital cancer. For example, UGenRA™ Ovarian is designed to address the critical issue of chemotherapeutic
-79-
Table of Contents
resistance while UGenRA™ Endometrial is designed to distinguish hyperplastic lesions that have a high risk of progression. We have initiated clinical validation for UroGenRA™ targeting kidney and prostate cancers in collaboration with Memorial Sloan-Kettering Cancer Center. In addition, we have initiated a clinical validation for UGenRA™ targeting kidney cancer in collaboration with Cleveland Clinic. Our UGenRA™ microarray has been designed as a platform to detect genomic changes occurring in 83 regions of the human genome that have been linked to endometrial, ovarian and cervical cancers.
Additionally, we develop and manufacture a portfolio of fluorescence in situ hybridization (“FISH”) based DNA probes focused on blood-based and solid cancers. We currently offer 32 CE marked probes and we are in the process of developing two proprietary probes.
We currently offer our proprietary tests in conjunction with our comprehensive panel of laboratory services in our CLIA-accredited laboratory. Our current laboratory services include:
| • | Proprietary Oncology Testing Services. These services are based on our proprietary microarray tests and are currently available only in our clinical laboratory. After completing the testing, we provide our customers with a comprehensive analysis of all tests performed for a specific patient, designed to help the physician make an informed and definitive diagnosis and guide the treatment of the patient. |
| • | Esoteric Oncology Testing Services. We offer a comprehensive suite of esoteric oncology testing services for hematological, urogenital and HPV-associated cancers, including conventional and molecular cytogenetic techniques such as G-banding and FISH, mutation and sequencing analysis, flow-cytometry and immunohistochemistry (“IHC”). |
| • | Clinical Trial Services. We also utilize our clinical laboratory to provide clinical trial services to biopharmaceutical companies and clinical research organizations to improve the efficiency and economic viability of clinical trials. Our clinical trials services leverage our knowledge of clinical oncology and molecular diagnostics and our laboratory’s fully integrated capabilities. By utilizing biomarkers, we intend to optimize the clinical trial patient selection. This may result in an improved success rate of the clinical trial and may eventually help biopharmaceutical companies to select patients that are most likely to benefit from a therapy based on their genetic profile. |
We intend to continue offering our proprietary tests in the United States as LDTs offered in our laboratory and internationally as CE-marked in vitro diagnostic medical devices. In addition, as part of our long-term strategy, we plan to seek Food and Drug Administration (“FDA”) clearance or approval to expand the commercial use of our tests to other laboratories and testing sites. We believe it would likely take two years or more to conduct the studies and trials necessary to obtain approval from FDA to commercially launch MatBA®-CLL and MatBA®-SLL outside of our clinical laboratory. Our sales strategy is focused on direct sales to oncologists and pathologists at hospitals, cancer centers, and physician offices in the United States, and expanding our relationships with leading distributors and medical facilities in emerging markets. We intend to continue to focus on partnering with community hospitals, where nearly 85% of all cancers are initially diagnosed, through our program called Expand Dx™, which was specifically designed to meet the needs of community hospitals. We believe our proprietary tests and services will enable community hospitals to optimize and expand their oncology services to better serve their cancer patients.
-80-
Table of Contents
Cancer Market Overview
Despite many advances in the treatment of cancer, it remains one of the greatest areas of unmet medical need. In 2008, the World Health Organization attributed 7.6 million deaths worldwide to cancer-related causes. The World Health Organization projects that by 2030 this number will rise to 11 million deaths per year. Within the United States, the North Carolina Central Cancer Registry projects cancer to surpass cardiovascular disease as the leading cause of death by 2015. The incidence and deaths caused by the major cancers are staggering. The following table published by The American Cancer Society in 2011 shows estimated new cases and deaths that occurred in 2010 in the United States for the major cancers:
| Cancer Type |
Estimated New Cases Per Year | Estimated Deaths Per Year | ||
| Bladder* |
69,250 | 14,990 | ||
| Breast |
232,620 | 39,970 | ||
| Cervical* |
11,270 | 4,290 | ||
| Colorectal |
141,210 | 49,380 | ||
| Endometrial* |
46,470 | 8,120 | ||
| Kidney* |
56,046 | 12,070 | ||
| Leukemia* |
44,600 | 21,780 | ||
| Lung |
221,130 | 156,940 | ||
| Melanoma |
70,230 | 8,790 | ||
| Non-Hodgkin’s Lymphomas* |
66,360 | 19,320 | ||
| Ovarian* |
21,550 | 15,460 | ||
| Pancreatic |
44,030 | 37,660 | ||
| Prostate* |
240,890 | 33,720 | ||
| Thyroid |
48,020 | 1,740 |
* Areas where we currently have active development programs.
In addition to the human toll, the financial cost of cancer is overwhelming. An independent study published in 2010 and conducted jointly by the American Cancer Society and LIVESTRONG ranked cancer as the most economically devastating cause of death in the world—estimated to be as high as $895 billion globally in 2008. According to the National Institutes of Health, the direct cost of cancer care in the United States was approximately $125 billion in 2010.
Cancer is a Genetically Driven Disease
Cancer constitutes a heterogeneous class of diseases characterized by uncontrollable cell growth, and results from a combination of both environmental and hereditary risk factors. It has only been in recent years that technology has progressed far enough to enable researchers to understand many cancers at a molecular level and attribute specific cancers to genetic bases.
Cancer cells contain modified genetic material compared to normal human cells. Common genetic abnormalities correlated to cancer include gains or losses of genetic material on specific chromosomal regions (loci) or changes in specific genes (mutations) that ultimately result in detrimental cellular changes followed by cancerous or pre-cancerous conditions. For example, multiple gains or losses on various chromosomes and movement of genetic material among chromosomes (chromosomal translocations), collectively, copy number variation, have been often observed in various lymphomas and leukemias. Such genetic alterations can be caused by multiple factors, including genetic predisposition, environmental or lifestyle factors or viral infections, such as with HPV-associated cancers. Understanding the differences in these genomic changes helps clinicians to identify and stratify different forms of cancer in order to optimize patient treatment and patient management.
-81-
Table of Contents
Therefore, understanding and analysis of cancer at the molecular level is not only useful for diagnostic purposes, but also plays an important role in prognosis and disease management. Technology that can apply this predictive information has the potential to dramatically improve treatment outcomes for patients suffering from cancer.
Limitations of Traditional Cancer Diagnostic Approaches
Cancer is difficult to diagnose and manage due to its heterogeneity at morphologic, genetic and clinical levels. Traditional methods of diagnosis, routinely used as the initial step in cancer detection, involve a pathologist examining a thin slice of potentially cancerous tissue under a microscope or smear of blood or bone marrow. A relatively new tissue sample must be used along with chemical staining techniques to view the biopsy. Through visual inspection, the pathologist determines whether the biopsy contains normal or cancerous cells; those that are deemed cancerous are graded on a level of aggressiveness. After the diagnosis, a clinical workup is performed according to established guidelines for the specific cancer type. From there, the physician determines the stage of progression of the cancer based on a series of clinical measures (i.e., size, grade, metastasis rates, symptoms and patient history) and decides on a treatment plan (i.e., surgery, watchful waiting, chemotherapy, stem cell transplant).
When deciding treatment and management options for the particular cancer, the physician uses a combination of clinical and pathological features (i.e., the tumor’s assigned grade and stage) which depend heavily upon human interpretation and can suffer from inter-institutional variability. Due to the relatively subjective nature of this diagnostic process, the qualitative results of the analysis may not correlate well to the molecular structure and individual nature of the patient’s cancer. This subjectivity creates a high risk situation of misclassification that can ultimately prove dangerous or deadly, resulting in over-treatment for some patients and under-treatment for others. For example, a patient with a mild form of cancer may be mistakenly assigned to highly aggressive treatment. Side effects associated with such misaligned treatment can result in detrimental side effects or risks more significant than those posed by the original tumor. In addition, it is now well established that patients respond differently to the same medication, and multiple studies have linked the differences in patients’ response to various cancer drugs to differences at the genetic level. As such, the level of personalized treatment required to optimize a patient’s treatment regimen is only possible through the use of biomarker analysis and molecular diagnostics.
With the trend in medical practice for less invasive procedures, overall less specimen material is routinely available for diagnostic purposes and often the specimen type for analysis is restricted to that used for morphologic analysis (formalin-fixed paraffin-embedded material). For leukemias, where the specimen type is usually blood or bone marrow, this does not present a problem, but enrichment for the cells of interest for analysis is a challenge. Several adaptations of current procedures are being undertaken to improve diagnostic procedures for these cancer types to allow maximum sensitivity and specificity. For solid tissue specimens, the formalin-fixed paraffin-embedded (“FFPE”) diagnostic material is often the only tissue available for study and recent technologies, including MatBA®-SLL, have had to accommodate such limitations previously not encountered. Another trend in medical practice is the increased use of fine needle aspiration or core biopsy for diagnostic purposes, often requiring image guidance. Morphologic analysis of such specimens is challenging especially where the architecture of the specimen has been damaged. Genome-based analysis of such specimens is one method by which diagnostic results can be obtained.
Use of Genomic-Based Analysis in Cancer Diagnosis and Treatment
Molecular diagnostic tests for cancer aim to remove subjectivity from the diagnostic phase, and add prognostic information, thus enabling personalized treatments based on cancer analysis at its most basic genetic level. To date, genomic-based testing has produced higher value and more accurate cancer diagnostic information than traditional analytical methods. These tests create a data set that can both define the cancer subtype and help determine the best course of treatment by detecting mutations, gene fusions and DNA copy number changes, all of which are possible causes of or precursors to malignant growth. As a result of the ability to produce such genomic data and increased adoption of molecular testing, we believe that genomic-based analysis is becoming the fastest growing segment within oncology testing.
-82-
Table of Contents
An important method of measuring changes in the genomic profile of cancer cells is copy number variation. This method measures the gain or loss of DNA within specific regions of chromosomes. Three primary techniques for quantifying copy number variations include the following:
| • | Oligonucleotide-based microarrays are a multiplex technology that allow the attachment of thousands of microscopic spots of DNA onto a surface. The DNA sequences on the microarray can read multiple genetic aberrations in more than one cancer type following hybridization with DNA from a specific cancer sample and can yield diagnostic and prognostic information of importance to the treatment of the patient. Microarrays provide a powerful approach to distinguishing cancer types and those more or less likely to recur, progress or respond to specific treatments based upon comprehensive sequence analysis and the ability of one microarray to interrogate multiple cancer types in parallel. Because of the large number of DNA sequences being tested by the microarray, analysis involves bioinformatics-based algorithms. Considering the current clinical and societal demand for minimally invasive procedures, the diagnostic and prognostic applications of microarrays are highly desirable. |
| • | FISH-based DNA probes are fluorescently labeled sequences of DNA complementary to a genomic region of interest, which when hybridized to chromosomes, give rise to signals revealing the presence or absence of a specific genomic abnormality with high sensitivity. One probe identifies one specific genomic region. To create higher levels of specificity, multiple probes may be required to identify multiple genomic aberrations in the same cancer cell. Depending on the color scheme and custom design of each FISH-based DNA probe, genomic gain/loss and rearrangements can be detected in cancer specimens of multiple tissue types. |
| • | Next-Generation Sequencing performs massively parallel sequencing of human cancers effectively permitting a highly sensitive analysis of not only the sequence of the genome in cancer cells to reveal mutations and other aberrations associated with a cancer, but can also reveal other genomic rearrangements previously unknown to occur in the cancer genome. Translation of these findings for clinical implementation can also be achieved with a high degree of sensitivity using deep-sequencing at specific nucleotide sequences and can be translated where applicable into FISH or microarray-based assays depending on the aberrations that need to be detected. |
To date molecular and genetic detection methods have been successfully utilized to provide diagnostic, prognostic and theranostic information for several cancers, including breast and colon. The discovery of breast cancer genes BRCA-1, BRCA-2 and TP53 and colon cancer genes AXIN2 and APC have highlighted cancer’s underlying genetic component. With the prognostic nature of next generation genomic tests, physicians and researchers have begun to optimize patient treatment, increase survival rates and reduce healthcare costs in these cancer categories. Meanwhile, there are no equivalent prognostic tests for many other forms of cancer, including lymphomas, leukemias and urogenital and HPV-associated cancers.
We seek to provide the cancer professional and cancer patient a fully integrated offering of high-value, proprietary tests and customized services in cancers where there are no equivalent prognostic tests, including lymphomas, leukemias, and urogenital and HPV-associated cancers. We believe that our integrated approach combined with our ability to rapidly translate research insights about the genetics and molecular mechanisms of cancer into the clinical setting will improve patient treatment and management and will become a key component in the standard of care for personalized cancer treatment.
Our approach is to develop and commercialize proprietary genomic tests and services to enable us to provide a full service solution to improve the diagnosis, prognosis and treatment of hematological, urogenital and HPV-associated cancers. To achieve this, we intend to:
Develop and commercialize additional proprietary genomic tests and services. We intend to continue the development of additional proprietary diagnostic and prognostic tests and services to provide information that
-83-
Table of Contents
is essential to personalized cancer treatment. We have launched our first proprietary genomic-based test, MatBA®- CLL, for use in our CLIA-accredited facility and we recently launched MatBA®-SLL. We are also developing a number of other microarray-based tests, including additional MatBA®-based tests for additional hematological malignancies, as well as UroGenRA™ and UGenRA™ microarray platforms for urogenital cancers. We plan to obtain the necessary regulatory approvals to allow us to commercialize these microarray tests for use outside of our clinical laboratory.
Develop and expand our collaborations with leading universities and research centers. We have established research collaborations and joint research initiatives with key thought leaders and clinical research facilities, including Mayo, the National Cancer Institute, Memorial Sloan-Kettering Cancer Center and the University of Iowa Cancer Center. Our collaborations enable us to validate the effectiveness and utility of our proprietary tests and service offerings in a clinical setting and provide us access to clinically well characterized and highly annotated patient data. These data accelerate our validation process and facilitate the testing and refinement of our microarray algorithms.
Continue our unique focus on translational oncology. Translational oncology refers to our focus on bringing novel research insights that characterize cancer at the genomic level directly and rapidly into the clinical setting with the overall goal of improving value to patients in the treatment and management of disease. We actively integrate the dual disciplines of clinical diagnosis and fundamental research to foster a unique, interdisciplinary approach. This interdisciplinary approach enables us to design our research programs with a clinical outcome in mind, allowing for the rapid deployment of our proprietary microarrays and DNA probes into a clinical setting. We believe that our unique multidisciplinary approach allows us to rapidly expand our test and service offerings, and differentiates us from other diagnostics and laboratory services providers in the marketplace.
Enhance our efforts in partnering with community hospitals. According to the American Hospital Association, there are approximately 5,000 community hospitals in the United States. Community hospitals represent a large target market for our genomic tests and services because approximately 85% of cancer patients in the United States are initially diagnosed in such hospitals as reported to the National Cancer Database. We intend to continue to focus on partnering with such hospitals by targeting our sales and marketing efforts on this important customer segment. Our branded Expand Dx™ program is a suite of diagnostic and consultative services offered on a collaborative basis. Expand Dx™ is intended to expand and optimize the oncology diagnostics services and personalization of cancer treatment provided by community hospitals so that such hospitals can optimize and expand their oncology services to better serve their cancer patients.
Expand our scalable sales and marketing capabilities. We currently have a specialized team of sales professionals with backgrounds in hematology, pathology, and laboratory services. We intend to expand our sales force in order to provide geographic coverage throughout the United States. Additionally, we intend to expand internationally, particularly in emerging markets, by seeking leading local partners such as Roche Servicios, S.A. in Central Americas and Caribbean, DASA, S. A. in Brazil and Kamineni Life Sciences in India, to market and sell our tests and services.
Continue to integrate the insights and experience obtained from our reference laboratory to expand and improve our proprietary genomic tests and services. Our laboratory provides us with unique access to patient cases and clinician issues as well as the challenges that arise from current laboratory operations. Such insights obtained from clinical operations inform and often guide strategic decisions for our microarray and DNA probe development.
Continue to reduce the costs associated with the development, manufacture, and interpretation of our proprietary genomic tests and services by partnering with leading technology and service providers. We intend to work closely with select key suppliers and partners to reduce the costs associated with key material components of our microarrays and DNA probes. We initiated a program in December 2010 to identify key
-84-
Table of Contents
material components and labor processes involved in the manufacturing of our DNA probes and to date have significantly reduced our overall costs while increasing manufacturing yields and flexibility.
Our Competitive Advantages
We believe that our competitive advantages are as follows:
Our proprietary and clinically relevant genetic tests are the first to address several complex cancers that are difficult to prognose and where it is difficult to predict treatment outcomes using currently available technologies. Our two marketed tests are the first to address several underserved, complex cancers. MatBA®-CLL is, to our knowledge based on our informal communications with New York State Department of Health personnel, the only microarray that has been approved by the New York State Department of Health for diagnostic treatment and management of CLL. FHACT, our HPV-associated cancer test, is the first multi-region DNA probe to identify and stage HPV-associated cancers, which includes cervical, anal and oropharyngeal cancers.
Collaborative relationships with Mayo and other leading research centers, medical centers and oncology groups. Our collaborations with leading cancer centers provide us with a number of benefits, including valuable access to patient samples. In particular, we entered into an agreement with Mayo whereby we agreed to form a joint venture with Mayo in March 2013 focused on developing oncology diagnostic services and tests utilizing next-generation sequencing. With respect to marketing, we can leverage the brand name recognition of our collaborators when selling to our customers. With regard to research, our collaborations provide us with the fundamental science and research that underpin the development of our diagnostic tests. Additionally, these collaborations provide us with insight to maximize the utility of our tests in the clinical setting.
Our tests provide more information than existing tests to enable a more personalized treatment plan. Our tests are designed to provide an earlier, more accurate and more complete diagnosis, which potentially leads to better treatment and lower healthcare costs. For example, MatBA®-CLL evaluates a set of five biomarkers not previously assessed in CLL and also allows a more accurate interpretation of the loss at chromosome 13q as a sole abnormality than previously possible.
Our tests are designed for a wide range of sample types and sample preparation methods. We can currently process specimen types that include blood, bone marrow and tissue, including fresh, frozen and FFPE tissue samples. The ability to interrogate a wide variety of sample types increases clinical adoption of our tests and allows the health care provider to quickly and efficiently integrate our tests into its established workflow. This integration with existing oncology and pathology workflow and tissue analysis methods is integral to ensuring near term adoption.
Ability to test on FFPE tissue samples accelerates the time required to validate, develop and patent new tests. For several reasons, we have designed our tests for FFPE tissue samples. For decades, Archival FFPE has routinely been used to preserve cancer samples and offers a wealth of information and collaboration potential in comparison with fresh or freshly prepared samples. Our use of FFPE has three important consequences. First, it significantly increases the datasets of samples that can be used to validate our products, leading to more robust and reliable diagnostic tools. Second, it permits utilization of FFPE in a clinical setting, where often it is the only specimen available for study. This is of particular importance to tumors diagnosed using minimally invasive technologies where often very small biopsy material is available for diagnostic and prognostic studies. Third, it affords enrichment of the sample to be analyzed, increasing the probability with which genomic aberrations will be detected for any given specimen.
Our genomic tests are not platform dependent. The biology and algorithms behind our tests are adaptable to multiple instrumentation platforms, allowing us to incorporate our tests into a variety of existing clinical laboratory infrastructures without additional capital investment. We have currently optimized our tests
-85-
Table of Contents
for the Agilent platform. However, we believe that we can migrate to other similar platforms, including next gen sequencing, without significant modification.
Consultative, oncology-centered laboratory and clinical trial services. Our specimens are tested and interpreted by highly qualified oncology-focused laboratory professionals, many of whom hold MDs and PhDs. Because our clinical staff is highly specialized in oncology, we are better positioned to consult with our oncologist customers to help them derive maximum value from the diagnostic and prognostic data generated by our tests.
Focus on servicing the comprehensive needs of community hospitals, where approximately 85% of all cancer patients in the United States are initially diagnosed. Through our Expand Dx™ program, we work with community hospitals to better service their oncology patients. Our proprietary tests, as well as our comprehensive cancer diagnostic testing services, are fully integrated into the Expand Dx™ program and help the community hospitals deliver a higher value of service to their cancer patients.
Our Proprietary Genomic Tests and Services
We currently develop and produce two types of DNA-based genomic tests: microarrays and probes. Both are directed at identifying specific genetic aberrations in cancer cells that serve as markers for diagnosis, prognosis and prediction of treatment outcomes (called theranosis). In addition, we agreed to form a joint venture with Mayo in March 2013 that will focus on developing oncology diagnostic services and tests utilizing next-generation sequencing.
We offer both microarrays and probes because each serves a unique diagnostic or prognostic function. FISH-based tests, or probes, offer great sensitivity while microarrays provide a more comprehensive analysis of the cancer genome. While we expect both platforms to be utilized in cancer diagnostics for the foreseeable future, we believe microarrays will become a significant factor in our growth as they offer a broader range of genomic information, are of a higher resolution and lend themselves to automation. Beyond microarrays, we believe that next generation sequencing will rapidly become a powerful tool for the personalized diagnosis and management of cancer.
FDA clearance or approval is not currently required to offer these tests in our laboratory once they have been clinically and analytically validated and approved by the appropriate regulatory bodies. We seek licenses and approvals for our laboratory facility and for our LDTs from the appropriate regulatory authorities, such as the CMS, which oversees CLIA, and various state regulatory bodies, including the New York State Department of Health. At the federal level, certain proprietary tests must be part of proficiency testing programs approved under CLIA in order for us to be able to bill government payor program beneficiaries, such as Medicare patients, for such tests. In addition, certain states, such as New York, require us to obtain approval of our proprietary tests in order for us to collect patient specimens from such state.
Through our subsidiary, Cancer Genetics Italia, S.r.l. (“CGI Italia”), based in Milan, Italy, we have obtained CE marking for 32 of our DNA probes, which entitles us to market these probes in the European Economic Area (which includes the 27 Member States of the EU plus Norway, Liechtenstein and Iceland). We anticipate that we will need to conduct additional developmental activities for each of these tests and to submit these tests for regulatory clearance or approval by FDA or other regulatory agencies prior to commercialization outside of our reference laboratory in each of the markets where we plan to introduce them.
-86-
Table of Contents
The following diagram portrays our proprietary programs:
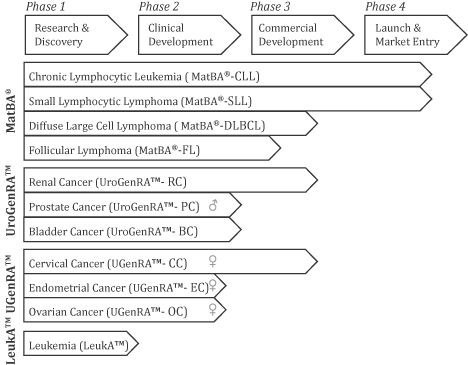
Hematological Cancer Arrays: MatBA®-CLL/SLL, Other MatBA® and LeukA™
MatBA® is the first targeted oligonucleotide-based microarray we developed for the analysis of genomic alterations in mature B-cell neoplasms to determine prognosis and theranosis. MatBA® incorporates a common architecture of specific genomic regions that can be applied across the seven major mature B-cell neoplasms. Mature B-cell neoplasms account for approximately 7% of all cancers diagnosed in the United States annually (approximately 110,280 expected in 2011) and for approximately 6% of all estimated cancer-related deaths (approximately 35,610 expected in 2011). They are the fifth most common malignancy in both males and females, and the incidence is rising.
As a group, hematologic cancers (cancers of the blood, bone marrow or lymph nodes) display significant clinical, pathologic and genetic complexity. Current diagnosis relies mostly on pathologic examination, flow cytometry and detection of only a few genetic markers. Importantly, the clinical course of the six main subtypes of these neoplasms ranges from indolent (follicular lymphoma) to aggressive (diffuse large B-cell lymphoma, mantle cell lymphoma and multiple myeloma), or mixed (chronic lymphocytic leukemia/small lymphocytic lymphoma, or CLL/SLL). Currently most risk-stratification for treatment decisions is based on clinical features of the disease. Few molecular prognostic biomarkers are utilized in a clinical setting. There is unmet medical need for robust biomarkers for the diagnosis, prognosis, theranosis and overall patient management in B-cell cancers. Given the higher frequency of these malignancies in the United States than in other countries, we expect significant clinical demand for MatBA®.
MatBA® is designed to detect genomic copy number changes in mature B-cell neoplasms either solely or in a unique combination, thus assisting the clinician in the management of a patient’s disease. The test relies on the comparative genomic hybridization of fluorescently differentially-labeled normal DNA and DNA extracted from the cancer specimen (array-CGH). Array-CGH utilizes minimal biopsy material and uses DNA as the analyte (the component whose properties are being measured), which is more stable, as compared to RNA
-87-
Table of Contents
used in other array detection methodologies. Both are important considerations for the ever increasing demand for less invasive procedures for diagnostic and prognostic purposes. Additionally, we have optimized the utility of the MatBA® array-CGH so that it can be routinely applied to the study of a range of specimen types including blood and bone marrow and FFPE biopsy specimens, which is often the only specimen available for analysis of FL, DLBCL and MCL. With the exception of CLL, biopsy/surgical procedures are rarely performed for B-cell neoplasms prior to the initiation of treatment, thus limiting the amount of tissue available for testing prior to deciding on the initial treatment regimen.
MatBA® was custom-designed to represent 80 regions of the human genome which have diagnostic and/or prognostic value in one or more of the mature B-cell neoplasm subtypes as identified through our research and analysis efforts. Unlike other technologies such as FISH, array-CGH using MatBA® simultaneously permits the detection of genomic gains and losses at multiple locations on a chromosome (loci) that characterize the mature B-cell neoplasm subtypes. For each subtype of B-cell neoplasm, cohorts of specimens with full clinical annotation are evaluated using MatBA® to identify novel associations between single and weighted combinations of genomic gains/losses and clinically relevant endpoints, including time to first treatment, treatment response, progression-free survival and overall survival, and to validate previously known associations. It is these associations, we believe, that provide valuable assistance to clinicians in risk stratification and guiding treatment plans for patients with these cancers.
MatBA®-CLL and SLL
We offer the first application of MatBA® for prognostication in one subtype of mature B-cell neoplasm, CLL, where about half of patients experience indolent disease, or slow progression, and the remaining half, a relatively aggressive progression. MatBA®-CLL provides important genetic-based information to guide clinical management of this disease. The test results are reported out in a unique format that allows ease of interpretation by the hematologist or oncologist. MatBA®-CLL is included in the tests we can provide under our New York laboratory and CLIA licenses, effective April 2011. New York is one of only a few states that separately and rigorously reviews LDTs for clinical and analytical validity. To date there are only a few companies that have commercially available oncology microarrays and, to our knowledge based on our informal communications with New York State Department of Health, MatBA®-CLL was the first oncology microarray approved for commercial use by the New York State Department of Health. We are currently working towards the development of a regulatory strategy to support the CE marking of MatBA®-CLL, which we expect to complete in the fourth quarter of 2012.
Approximately 14,500 new cases of CLL are expected to be diagnosed in the United States this year, and importantly, over time these cases undergo evolution, requiring risk stratification and guidance on patient management issues at multiple points during the course of the disease. Prior to the introduction of MatBA®, clinicians relied on the assessment of the gain or loss on only four chromosomal regions and potentially one gene mutation when testing for and stratifying a CLL patient. MatBA® improves on this by identifying information on five additional chromosomal regions, providing more valuable diagnostic data and critical information about the risk of progression and overall prognosis of the patient. In particular, because MatBA® has greater resolution than that available with prior tests, we can interrogate two different regions or loci on the 13q chromosome. We believe this type of genomic assessment of the patient’s cancer also saves the health care system thousands of dollars per year per patient as a result of improved patient management and more targeted therapeutic intervention. Loss of one specific locus or loss of both loci are in some circumstances believed to have differing prognostic value, hence the importance of being able to evaluate both loci. Also, loss of 13q as a sole abnormality is associated with a lower risk of progression and overall favorable outcome. With the increased capacity of MatBA® to assess abnormalities in multiple regions of the genome not usually assessed by other technologies, our studies have indicated that up to 23% of cases that would have shown 13q loss as a sole abnormality when assessed by FISH technologies do in fact have additional abnormalities. For these cases, the “favorable outcome” that would have been reported to the clinician was not accurate, leading to a change in the prognosis and consequently decision-making by the clinician regarding the management of these patients.
-88-
Table of Contents
We performed validation of these important new biomarkers in 317 CLL specimens in conjunction with Dr. Kanti Rai at Long Island Jewish / North Shore Hospital. We presented this data at the 2011 International Workshop on Chronic Lymphocytic Leukemiais and the American Society of Hematology’s 2011 Annual Meeting and Exposition. In 2011, we also presented a poster on the key methods involved in enabling the usage of DNA from FFPE material involved in certain sub-types of MatBA® at the Association for Molecular Pathology. In the poster, results from over 360 samples were reviewed and demonstrated highly accurate aberration detection as confirmed using Quantitative Polymerase Chain Reaction, an industry standard in molecular diagnostic measurement.
In addition we have identified novel biomarkers using MatBA® that are associated with a poor outcome in CLL. These include gains at 2p, 3q and 8q and a loss at 8p. Additional prognostic regions have been identified and are undergoing validation. These will be reported, further driving the value of more comprehensive genomic assessment of the patient’s cancer.
We validated MatBA®-SLL for risk stratification in SLL. In January 2012, MatBA®-SLL was approved under CLIA and accordingly may now be offered as an LDT by our laboratory. This adaptation of MatBA® for SLL has allowed us to develop a robust mechanism to analyze DNA that is derived from FFPE biopsy material and has been a critical development that we believe will accelerate the development of our microarrays for other solid tumors or cancers that present themselves as a mass.
Other MatBA® Microarrays in Development
We are now undergoing similar development of MatBA® as a prognostic tool in two of the other main subtypes of mature B-cell lymphomas, namely DLBCL and FL. FL is characterized by a slow progression that in up to approximately 60% of cases transforms to DLBCL, an aggressive lymphoma. Prognostic and theranostic biomarkers of therapeutic options are required for these diseases. We have identified several additional loci which we believe are relevant to the prognosis of DLBCL and FL and which cannot be assessed by currently available FISH tests alone. We are currently validating these extensions of MatBA®. MCL is another mature B-cell lymphoma for which MatBA® is in the clinical development stage. We believe MatBA® will provide increased management insight for patients with this type of lymphoma based on a more complete genomic assessment of the lymphoma.
LeukA™ Microarray
We are developing a proprietary leukemia microarray, LeukA™, for the diagnostic and prognostic evaluation of different types of leukemia. The microarray will detect genomic gains and losses in patient specimens using DNA extracted from blood and/or bone marrow. We are in the preliminary research and discovery stage with respect to this test with the potential inclusion of uniparental disomy assessment.
Urogenital cancer arrays: UroGenRA™, UGenRA™
There is a unmet clinical and patient need for improved diagnosis, prognosis and theranosis, including more detailed and staging information, in urogenital cancers, where biopsy materials are increasingly scarce. The cumulative number of annual new reported cases for kidney, prostate and bladder cancers is estimated to exceed 320,000 in 2011 according to the American Cancer Society. Gynecologic neoplasms contribute substantially to female mortality and morbidity in the United States and are an area where nearly 80,000 new cases are diagnosed each year. Although generally characterized by early stage detections, these cancers still represent a major health risk, a significant variability in patient outcome, which can be better managed through genomic assessment of the tumor(s), and a substantial medical cost burden to the public with the high rates of incidence and ongoing patient management needs.
-89-
Table of Contents
Developing sophisticated, state-of-the-art molecular tests that enable more accurate diagnosis and/or prognosis of these cancers will not only benefit the patients by offering more appropriate treatments, but also effectively reduce the unnecessary medical cost associated with surgery, long-term follow-up surveillance, or therapy after the treatment.
The UroGenRA™ microarray, which is being validated in collaboration with Memorial Sloan-Kettering Cancer Center, will provide diagnostic and prognostic analysis for kidney, bladder and prostate cancer. Our initial launch, UroGenRA™-Kidney, will target kidney cancer. We are also developing extensions of UroGenRA™ for bladder and prostate cancers. UGenRA™ will provide diagnostic, prognostic and theranostic information for the primary gynecological cancers, cervical, ovarian and endometrial.
UroGenRA™ for Kidney, Prostate and Bladder Cancers
UroGenRA™ is a proprietary CGH-based array which will serve as a platform for the diagnosis, prognosis and theranosis of kidney, prostate and bladder cancers. It was designed to detect gains and losses that frequently occur in genetic material in these three cancer types and has the potential to differentially diagnose and/or stratify patients to assist and guide clinical management. It represents 101 regions of the human genome potentially with diagnostic, prognostic and/or theranostic value in one or more of these types of cancers.
UroGenRA™-Kidney For kidney cancer, UroGenRA™ is specifically designed to classify renal tumors into the four main subtypes (clear cell, papillary, chromophobe and oncocytoma), which is critical to patient management and treatment protocols. This allows the clinician, especially in cases where there is limited biopsy material, to (i) diagnose renal cancer and accurately classify it into the correct subtype, (ii) provide rationale for selection among surgical and non-surgical intervention or ablation, (iii) stratify patients based on prognostic information for the advancement of renal cancer into local or regional cancer which then guides decisions on surgical intervention, and (iv) guide drug trial decisions in those with metastatic disease or “unclassified” renal cancers.
We are in the final stages of developing a study with two leading academic cancer centers for which we will obtain and use a group of 200 specimens comprising four kidney cancer subtypes to further develop and validate the algorithm of copy number variation known to be associated with these tumors that gives the best ability to differentiate among these four subtypes. These copy number changes are already known to minimally include loss in six regions of chromosomes among these four types and gain in three other regions, but we believe we will define additional and specific regional copy number variations. The derived proprietary renal cancer diagnostic algorithm or decision tree based on UroGenRA™ copy number alterations is to be validated for diagnostic potential in the IRB-approved study of over 50 image-guided needle biopsies and compared with the sensitivity and specificity obtained by our proprietary FISH-based assay, FReCaD™.
UroGenRA™-Kidney is in the commercial development stage. At the current time, validation of the clinical utility of UroGenRA™ is further advanced for kidney cancers than for prostate and bladder cancers, because we are able to leverage research and insights used in the clinical validation of FReCaD™ in our development activity for the UroGenRA™ indication for kidney cancer.
UroGenRA™-Prostate For prostate cancer, UroGenRA™ has the potential to use prostate core/needle biopsy to assess genomic variability of the cancer and help in the identification of biomarkers for assessment of the risk of recurrence, to assess treatment options for intermediate risk patients, and to explore the genomic aberrations of circulating tumor cells. In the case of recurrence, gain or loss in a limited number of regions represented on UroGenRA™ is considered informative. Application of the UroGenRA™ to circulating tumor cell genome scanning would require a modified version of the regions represented on UroGenRA™, but we believe it could be implemented considering the plasticity of the array platform. UroGenRA™-Prostate is in the commercial development stage.
-90-
Table of Contents
UroGenRA™-Bladder Newly diagnosed bladder cancers are defined by the fact or extent of invasion of the muscle. For non-muscle invasive bladder cancers, there is clinical need to identify the high proportion of patients in which the cancer will recur. The need in muscle-invasive tumors is to identify those patients most likely to benefit from treatment, considering that the survival benefit of peri-operative chemotherapy for such patients is only 5-10%. Genomic copy number alterations likely to be involved in the response of tumor cells to such therapy have been incorporated in UroGenRA™ for this specific application, and we are currently attempting to validate this microarray for this use. UroGenRA™-Bladder is in the clinical development stage.
UGenRA™ for Endometrial, Ovarian and Cervical Cancers
UGenRA™ was designed as a platform to detect gains and losses of genomic material in 83 regions of the chromosome associated with responses to particular therapies in patients with endometrial, ovarian and cervical cancers. We are committed to the development of UGenRA™ as a diagnostic tool that will assist in the screening, diagnosis and/or prognosis of these cancers. The use of UGenRA™ can be easily integrated into current clinical management protocols because it requires only small amounts of genetic material to test and can be performed on FFPE specimens.
UGenRA™-Endometrial Endometrial cancer is the fourth most common cancer in women in the United States representing approximately 6% of all newly diagnosed cancers in women in 2011. In this disease, endometrial hyperplasia is a precursor lesion of endometrioid endometrial carcinoma and since about 50% of women with atypical hyperplasia also have concurrent endometrioid endometrial carcinoma, it is important to identify those precursor lesions more likely to progress to cancer. UGenRA™ offers the opportunity to identify such specimens and potentially guide clinical management. Five regions of the chromosome interrogated by UGenRA™ have already been implicated to harbor gains and losses that, if detected in hyperplastic lesions, have a high likelihood of progression to cancer. We are in the process of clinically validating the use of UGenRA™ for these purposes, along with any novel regions that may be identified in the planned studies. Another potential application in endometrial cancer is to stratify those tumors likely to recur, permitting the identification of patients most likely to benefit from therapy. UGenRA™ Endometrial is in the clinical development stage.
UGenRA™-Ovarian There are approximately 21,550 cases of ovarian cancer diagnosed in the United States each year and approximately 15,460 people die from ovarian cancer each year in the United States. Risk-stratification of stage III/IV ovarian cancer patients after cytoreductive surgery (involving removal of only part of a malignant tumor) for a certain type of chemotherapy is a potential application for UGenRA™, and the design of UGenRA™ currently contains the sites of genomic gain/loss with such prognostic value. We believe we can validate these regions using the publicly available data copy number information from the Center for Applied Genomes for over 300 ovarian cancers with known response and overall outcome. This is a powerful resource for validation and would serve to confirm our test in a different cohort of patients than those used in the preliminary validations performed at our laboratory. UGenRA™ Ovarian is in the clinical development stage.
UGenRA™-Cervical There are approximately 11,270 cases of cervical cancer diagnosed and approximately 4,290 deaths from cervical cancer each year in the United States. With respect to cervical cancer, current clinical tests are unable to distinguish regressive cervical lesions from progressive lesions. Hence low-risk patients are treated the same way as high-risk patients, which increases health care costs. There is a great need for molecular-based diagnostic assays to address these questions, so that physicians can plan appropriate treatment strategies. We have designed UGenRA™-Cervical to distinguish among lesions which have a high likelihood of progression into cervical cancer versus those that do not have the genomic abnormalities related to progression to cervical cancer. UGenRA™ Cervical is in the clinical development stage.
Proprietary FISH-based DNA Probes
FHACT™ HPV-Associated Cancer Test
We have developed a proprietary, 4-color FISH-based DNA probe designed to identify the gain of the three most important chromosomal regions that have been implicated in cancers associated with HPV: cervical,
-91-
Table of Contents
anal and oropharyngeal. According to the National Cancer Institute, about 55 million PAP smear tests to detect HPV are performed in the United States each year. It is estimated that approximately 2 million patients have abnormal PAP smear test results and are referred for biopsy/colposcopy as a result of such tests. However, only 0.6%, or approximately 12,000, of these patients will develop cancer. It is believed that early detection of HPV-associated cancers could eliminate unnecessary biopsies/colposcopies and thereby reduce health care costs.
FHACT™ is designed to determine copy number changes of four particular genomic regions by FISH. These regions of DNA give specific information about the progression from HPV infection to cervical cancer, in particular the stage and subtype of disease. FHACT™ is designed to enable earlier detection of abnormal cells and can identify the additional biomarkers that allow for the prediction of cancer progression. FHACT™ is designed to leverage the same PAP smear sample taken from the patient during routine screening, thus reducing the burden on the patient while delivering greater genomic-based information to the clinician. We in-license a biomarker from the National Cancer Institute that is used in our FHACT™ probe.
In conjunction with the National Cancer Institute, we have begun a blinded study to evaluate the effectiveness of FHACT™ for both anal and cervical cancers associated with the HPV virus that will involve over 1,000 specimens. The FHACT™ study is ongoing. We have completed a blinded study of over 300 cervical specimens and the data has been provided to National Cancer Institute. This has been used for validation of the assay and development of automatic analysis for the FHACT™ probe. Upon review, National Cancer Institute will provide the remaining samples. We have yet to begin work with anal samples. We continue further clinical validations in collaborations that have been established with the University of Iowa and with Kamineni Hospital in Hyderabad, India to further strengthen the claims and data for use of FHACT™ as a staging and prognostic tool for cervical cancer in both the United States and in emerging markets. The sensitivity of FHACT™ was presented as a poster at the 27th International Pappillomavirus Conference in Berlin, Germany in September 2011. The publication demonstrated that by using FHACT™ over 90.9% sensitivity can be achieved as a screening tool for cervical intraepithelial neoplasia of 2nd degree or higher (known as CIN2+), which is a critical milestone in the development of cervical cancer.
In 2012, we expect to make FHACT™ available outside the United States as a diagnostic tool in certain emerging market countries, including India. This initial launch is applicable for detection and staging of cervical cancer, which is the third most common cancer among women worldwide, with one-fifth of the cases originating in India. The World Health Organization projects that cervical cancer deaths will rise to 320,000 in 2015 and 435,000 in 2030. In many emerging economies, cervical cancer is the most common cancer that affects women, and 80% of deaths from cervical cancer occur in these developing countries.
We continue to validate FHACT™ for anal and oropharyngeal cancers using specimens from the National Cancer Institute and are actively seeking collaborations to further validate the clinical utility of FHACT™ for anal and head and neck cancers.
Research for FHACT™ has been to date funded through a $763,958 grant awarded in 2009 from the National Cancer Institute. In October 2010, we were awarded a grant in lieu of a federal income tax credit under the Qualifying Therapeutic Discovery Project Program for approximately $244,500 to help in the further validation and commercialization of FHACT™.
FReCaD™ Renal Cancer Detection Test
We have developed a proprietary, novel and highly sensitive panel comprised of 20 FISH-based DNA probes for the detection of genomic abnormalities that differentially diagnose the four main subtypes of renal cell carcinoma—papillary, clear cell, chromophobe and oncocytoma. Our FReCaD™ panel provides precise classification of the subtypes of renal cell carcinoma using minimal biopsy material. The test detects chromosomal aberrations as molecular factors that are differentially observed in each of the four main subtypes of renal cancer. Differentiation between benign and malignant, and furthermore between the three malignant subtypes, is essential in treatment management for patients with suspect renal masses.
-92-
Table of Contents
FReCaD™ is based on the inherent differential genetic rearrangements of renal cell carcinoma rather than the form or structure of the cells (cell morphology). By detecting the inherent genomic rearrangements specific to renal cancer subtypes, we believe, the FReCaD™ panel allows a more accurate diagnosis. This results in better treatment decisions, higher remission rates and the prevention of disease progression among patients. A study of 145 ex-vivo core biopsies performed at our research laboratory clearly indicated the FReCaD™ panel combined with morphology improved the classification of renal needle biopsies by 16% above that of morphology alone and together the morphology and the FReCaD™ panel allowed the accurate detection of renal cell carcinoma in 89% of renal needle biopsies. FReCaD™ is in the commercial development stage.
FISH-based DNA Probes
We also develop FISH-based DNA probes for sale outside the United States. Our portfolio includes 32 CE-marked probes for hematopoietic neoplasms and solid tumors.
Our strategy is to sell conventional probes into emerging markets through Cancer Genetics Italia and local or regional partners. We have entered into an agreement with Labomics S.A., based near Brussels, Belgium, which will provide us with the manufacturing support, storage facilities, and fulfillment management of our FISH-based DNA probes to better serve European and global demand. We are in the process of moving these manufacturing operations to Kamineni Life Sciences in India. We expect initial manufacturing operations to commence there this year.
We plan for all of our probes to conform to the requirements of the European In Vitro Diagnostic Medical Devices Directive (98/79/EC IVDD). This entitles them to bear the CE marking, which enables us to market them in the European Economic Area and provides for clinical acceptance in other countries where the CE mark is valued.
Laboratory Services
We provide our complete suite of oncology-focused laboratory services to hospitals, cancer centers, oncologists and pathologists from our 17,936 square foot state-of-the-art, laboratory in Rutherford, New Jersey. At the federal level, clinical laboratories, such as ours, must be accredited under CLIA in order for us to perform testing on human specimens. Our laboratory is accredited by the College of American Pathology (“CAP”) which is one of six approved accreditation methods under CLIA. Our clinical laboratory is located in New Jersey and we hold the requisite licenses from the New Jersey State Department of Health to operate our laboratory. In addition certain states, such as New York, require out-of-state laboratories to obtain licenses in order to accept patient specimens from such states. In addition to New Jersey, we hold clinical laboratory licenses from the New York Department of Health, Florida Department of Health, Maryland Department of Health, and Pennsylvania Department of Health for all of our clinical departments. We are also qualified to accept specimens from all states in the United States, as well as from overseas locations.
Historically we have generated most of our revenue through our laboratory services. In 2011, we generated approximately 87% of our revenue from laboratory services, approximately 10% from government grants and approximately 3% from sales of our DNA probes, which are currently only sold outside the United States. In 2010, we generated approximately 95% of our revenue from our non-proprietary laboratory services, approximately 4% from government grants and approximately 1% from sales of our DNA probes.
Our comprehensive oncology-focused testing services for hematological, solid tumor urogenital cancers are utilized in the diagnosis, prognosis and theranosis of cancer patients and are growing rapidly as clinicians demand more precise and more comprehensive diagnostic evaluation of their patients. We utilize highly skilled scientists, pathologists and hematologists in our laboratory, including 15 individuals with doctorate degrees. These individuals assist our customers in integrating and technically assessing the testing results for their patients.
-93-
Table of Contents
The non-proprietary testing services that we offer are entirely focused on specific oncology categories where we are developing our proprietary arrays and probe panels. We believe that there is significant synergy in developing and marketing a complete set of tests and services that are disease focused and delivering those tests and services in a comprehensive manner to help with treatment decisions. The insight that we develop in delivering non-proprietary services are often leveraged in the development of our proprietary programs and now increasingly in the validation of our proprietary programs (such as MatBA®) for clinical use.
We currently offer a range of services in the following areas:
| • | Microarray based testing (MatBA®-CLL and MatBA®-SLL): our proprietary microarray test for the detection of chromosomal abnormalities observed in Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma; |
| • | Molecular testing: using quantitative methods, such as polymerase chain reaction, sequencing and mutahine analysis, to analyze DNA and RNA to follow progression of disease and response to therapy at the genetic level; |
| • | Cytogenetics testing: a series of methods that analyze human chromosomes in order to identify malignancy; |
| • | FISH testing: analysis of abnormalities at the chromosomal and gene levels using analyte specific reagents and FDA-cleared probes obtained from third parties performed on whole specimen or magnehell separated purified plasma cells; |
| • | Flow cytometry testing: Immuno pheno - type analysis of specific markers inside cells including specific cytosolic surface protiens, and on cell surfaces; |
| • | Histology testing: microscopic examination of stained tissue sections using various special staining techniques; |
| • | Cytology testing: non-gynecological fluid preparation for microscopic evaluations by a pathologist; and |
| • | IHC testing: analysis of the distribution of tumor antigens in specific cell and tissue types. |
We have developed the Summation™ Report which, we believe, provides an integrated view of a patient’s test results and diagnosis in a user-friendly, visually appealing format for clinicians. Our hematopathologists and laboratory directors prepare these Summation™ Reports based on the clinical information and diagnosis provided by our laboratory professionals. All our testing technologies are integrated into a Summation™ Report to allow oncologists to efficiently arrive at a definitive diagnosis and drive complete and effective decisions.
We expect to offer additional proprietary tests as LDTs in other areas of oncology and will seek the required CLIA and state approvals for these tests.
Clinical Trials Services
Industry research has shown many promising drugs have produced disappointing results in clinical trials. For example, a study by Princess Margaret Hospital in Toronto estimated that 85% of the phase III trials testing new therapies for solid tumors studied over a five-year period failed to meet their primary endpoint. Given such a high failure rate of oncology drugs under development, combined with constrained budgets for biopharmaceutical companies, there is a significant need for drug developers to utilize molecular diagnostics to
-94-
Table of Contents
decrease these failure rates. For specific molecular-targeted therapeutics, the identification of appropriate biomarkers potentially may help to optimize clinical trial patient selection and success rates by helping clinicians identify patients that are most likely to benefit from a therapy based on their individual genetic profile.
We recently launched our clinical trials services offering, which we have branded as Select One™, to help increase the efficiency and economic viability of clinical trials for biopharmaceutical companies and clinical research organizations. Our clinical trials services leverage our knowledge of clinical oncology and molecular diagnostics and our laboratory’s fully integrated capabilities. Our clinical trial services are aimed at developing customizable tests and techniques utilizing our proprietary microarrays and laboratory services to provide enhanced genetic signature and more comprehensive understanding of complex diseases at earlier stages. We leverage our knowledge of clinical oncology and molecular diagnostics and provide access to our genomic database and assay development capabilities for the development and validation of companion diagnostics. This enables companies to reduce the costs associated with development by determining earlier in the development process if they should proceed with additional clinical studies. We believe our clinical trial services may improve patient responder selection, thereby potentially increasing the likelihood our customer’s product is approved by FDA. Additionally, through our services we gain further insights into disease progression and the latest drug development that we can incorporate into our proprietary tests and services.
Test Development Process
Our proprietary microarrays and DNA probes have been, and continue to be, developed in conjunction with leading academic and clinical research centers to ensure that the needs of the clinical community are being met with the latest research on genomic alterations that cause, lead to, or are related to the development of cancer. We undergo a thorough research and validation process to ensure we are providing diagnostic and prognostic information that is clinically relevant and accurate. In our experience the time-frame for this process from design through development and market launch can be between 18 to 40 months based on complexity of the disease, the specific clinical claims being pursued and the availability of high quality samples with strong clinical correlations. We monitor and review the process in four stages as detailed below:
| • | Stage 1, Research and Discovery. We conduct extensive research of peer-reviewed publications and other disease-specific literature and public information databases. We gather the public information regarding genomic abnormalities as hallmarks and references for particular cancers and clinical correlations. Within a cancer type, the observed gains, losses or other aberrations and rearrangements of genetic material are recorded and noted when reported to have diagnostic or prognostic potential. |
| During this process, which is technology and platform agnostic, we extensively cross-analyze our findings in the literature with published data sets across a variety of technologies. Finally, we assess the merits of these findings internally with our research and development teams and with our scientific advisory board, when applicable, so that we can assure robust genomic coverage as we proceed into clinical development. |
| • | Stage 2, Clinical Development. We design a targeted array or probe panel based on the information gathered from the literature and database searches and review. A team of our scientists then seeks to refute the evidence compiled in the literature search process, serving as a system of checks and balances. Once that process is complete, we design an array based on its application within a particular cancer. For example, the kidney array is designed to subtype among the four main types of kidney cancers at various stages in the treatment of the patient. Within one array, we may be assessing three to four different subtypes of a cancer and for different applications, ranging from differential diagnosis to prognosis to prediction of therapeutic response. During this stage we select and refine the targeted regions and their potential suitability for analysis on the microarray. |
| • | Stage 3, Commercial Development. This process involves validating the performance characteristics of the microarray, as well as developing protocols for the use of the array or the DNA probe for the |
-95-
Table of Contents
| intended specimen. This quality assurance process notes reproducibility, accuracy, sensitivity, and specificity, and potential compliance to ranges of normalcy and reportability. We also compare data obtained for specimens and cell lines across different technology platforms to ensure accuracy of our processes. In this process, we confirm and validate the genomic biomarkers in independent clinically relevant datasets. During this process we also begin to develop the decision trees and algorithms, which are core to our intellectual property that guide the diagnostic and prognostic value of the microarray or other DNA probe. Once the initial decision tree and algorithm for the microarray and its use have begun development, we conduct trials which help to validate the design and usage of the tests. For this validation process, we partner with leading cancer institutions and regional cancer centers. |
| • | Stage 4, Market Entry and Launch. After commercial development is completed and prior to launch, we take several steps to prepare for marketing our tests as LDTs. We create standard operating procedures and quality assurance and quality control measures to ensure repeatability and high standards of quality. We train both our staff and the laboratory staff on the interpretation and use of the data. Licenses and approvals for our laboratory to use LDTs are obtained from the appropriate regulatory authorities, such as the Centers for Medicare and Medicaid Services (“CMS”), which oversees CLIA, and different state regulatory bodies. Before we CE mark our tests we also need to assess the conformity of our tests with the essential requirements of the European In Vitro Diagnostic Medical Devices Directive. As part of our long-term strategy, we plan to seek FDA clearance or approval to expand the commercial use of our tests to other laboratories and testing sites in the United States. We will also need to complete additional activities to submit each of these tests for regulatory clearance or approval prior to commercialization in each of the international markets where we plan to introduce them. |
Research and Development Expenses
We incurred research and development expenses of $2.1 million, which represents 69% of our net revenue, for the year ended December 31, 2011; $1.2 million, which represents 46% of our net revenue, for the year ended December 31, 2010; and $1.3 million, which represents 80% of our net revenue, for the year ended December 31, 2009. Research and development expenses represented 26% of our total operating expenses for the year ended December 31, 2011, 22% of our total operating expenses for the year ended December 31, 2010, and 39% for the year ended December 31, 2009. Major components of the research and development expenses included direct personnel costs, laboratory equipment and consumables and overhead expenses.
Sales and Marketing
Our sales and marketing efforts consist of (i) a direct sales force in the United States focused on developing direct channels to hospitals, cancer centers, pathologists and oncologists; and (ii) a channel approach outside the United States, specifically in the emerging markets, that is focused on partnering with leading distributors, medical facilities or medical service operators to develop and serve such regional oncology markets. We also sell our clinical trial services to biopharmaceutical companies and research organizations.
We currently have a dedicated and direct sales force consisting of five sales professionals focused on the eastern and midwestern United States with backgrounds in hematology, pathology, and laboratory services. Our sales professionals have an average of 20 years of experience in clinical oncology sales, esoteric laboratory sales from leading biopharmaceutical, pharmaceutical or specialty reference laboratory companies, including Laboratory Corporation of America Holdings, US LABS, Inc., Celgene Corporation and Genzyme, a Sanofi company, among others. We plan on growing this specialized, oncology-focused sales force and supporting it with clinical specialists who bring deep domain knowledge in the design and use of the microarrays that we plan on offering in the United States as LDTs.
-96-
Table of Contents
Our sales and marketing efforts are based on a three part go-to-market strategy:
| • | Collaborate with leading research universities and institutions that enable the validation of our new tests; |
| • | Work with community hospitals and community-based cancer centers that need a reliable and collaborative partner for genomic-based cancer testing; and |
| • | Build relationships with individual thought leaders in oncology, hematology and pathology to provide services that provide value to their patients. |
We also promote our tests and services through marketing channels commonly used by the biopharmaceutical and pharmaceutical industries, such as internet, medical meetings and broad-based publication of our scientific and economic data. In addition, we provide easy-to-access information to our customers through www.cgireports.com, www.cgimatba.com and www.cgiexpanddx.com. Our customers value easily accessible information in order to quickly review their patients’ information and begin developing a treatment protocol.
Expand Dx™ Program
According to American Hospital Association’s 2011 data, there are over 5,000 community hospitals registered in the United States, 998 of which are for profit. These hospitals are under pressure to create profitable cancer testing centers. However, community hospitals face numerous barriers, including rising costs of diagnostic technologies and treatments, complexity of new test validations and laboratory licensing requirements, difficulty in hiring, training and retaining qualified personnel and challenges in integrating new information technology systems. In particular, they generally do not have a dedicated pathologist or hematopathologist, and are not able to perform tests that provide an understanding of the genetic features of the tumor. Without this information, cancer specialists at these institutions are unable to plan an adequate course of treatment, which then limits the community hospital’s ability to adequately service their cancer patients. While nearly 85% of cancer patients in the United States are initially diagnosed in community hospitals, over half of these cancer patients are referred out to specialized cancer centers because community hospitals currently lack state-of-the-art oncology and pathology testing capabilities.
Our Expand Dx™ program for community hospitals is a suite of diagnostic and consultative services offered on a collaborative basis to expand and optimize the oncology diagnostics services and personalized cancer treatment provided by community hospitals so that such hospitals can retain their cancer patients. Our Expand Dx™ program focuses on enhancing the quality and increasing the efficiency of the community hospital’s oncology diagnostic process, including billing, turn around time for diagnostic tests, diagnostic procedures and training and assistance in the use of additional biomarkers for their routine cancer testing. We believe that through our Expand Dx™ solution, community hospitals and laboratories can get cost-effective access to leading-edge diagnostic tests and specialized testing capabilities of our clinical reference laboratory. Our Expand Dx™ solution provides the community hospitals with the necessary skills and services needed for comprehensive management of patients and their treatment while allowing laboratories to focus on efficient delivery of individual tests rather than comprehensive interpretation of specialized cases. Our focus on oncology allows the Expand Dx™ customers to intervene earlier and more comprehensively with their patients, thereby improving testing and treatment revenue.
Our sales force works with our laboratory directors as a team to market our Expand Dx™ solution to community hospitals, initially geographically focused on the eastern and mid-western United States.
Emerging Markets
We are initially targeting certain emerging markets, including India, Brazil, Turkey and Mexico, as an area of expansion of sales of our proprietary tests and probes. We sell in these countries through regional partners
-97-
Table of Contents
that have the ability to service both the cancer laboratories and doctors in that country. In February 2012 we entered an exclusive distribution agreement with Kamineni Life Sciences Pvt. Ltd for sale of our probes in India.
Roche Servicios, S.A., an affiliate of the Swiss drug maker Roche based in Costa Rica, recently selected us as their service provider for biomarker based cancer testing services. As part of our relationship with Roche Servicios, we will perform a variety of molecular, cytogenetic and immunohistochemistry based testing services to aid Roche Servicios, S.A. in delivering genetic testing results to hospitals and clinicians based in 14 different locations covering Central America and the Caribbean. Our testing will focus on assessing the biomarkers that are related to a variety of solid tumors and blood borne cancers which are the most prevalent cancers in those regions and which also correspond to therapeutics and treatments that are offered by Roche and Genentech.
In 2012, we plan to launch FHACT™ in India as a tool to provide specific information about the progression from HPV infection to cervical cancer. Cervical cancer is the third most common cancer among women worldwide, with more than 85% of cervical cancers and related deaths occurring in developing countries. Deaths from cervical cancer in India account for 27% of all deaths from cancer globally.
Key Research and Development Collaborations
We formally and informally collaborate with leading oncology centers and community-based hospitals to develop our proprietary diagnostic tests, and we work closely with leading cancer researchers at these institutions to develop proprietary tests tailored to their needs and specifications. Additionally, many of these centers have obtained Specialized Programs of Research Excellence status, as designated by the National Cancer Institute. Our collaborations with these centers give us access to large datasets of information that we use to develop our proprietary tests.
Below is a summary of our active key collaborations. In certain cases we have formal written agreements with collaborators and in other cases we have no written agreement with our collaborators or only informal written arrangements.
Joint Venture with Mayo Foundation for Medical Education and Research Focused on Next-Generation Sequencing and Oncology
On November 7, 2011, we entered into an affiliation agreement with the Mayo Foundation for Medical Education and Research, pursuant to which we agreed to form a joint venture with Mayo on August 1, 2012, or such earlier date as mutually agreed upon by Mayo and us (the “Closing Date”). We recently amended our agreement with Mayo to extend the Closing Date to March 31, 2013. The objectives of the joint venture entity will be to try to discover and validate biomarkers in specific hematologic and urogenital disorders utilizing next-generation sequencing with a possible expansion into other solid tumors, such as esophageal, head and neck, breast and lung cancers. Additionally, the joint venture entity would engage in biomarker discovery utilizing Mayo’s next-generation sequencing facility and the development of commercial products in the form of diagnostic products and services, as well as early stage therapeutic markers.
The joint venture entity will take the form of a limited liability company and will be governed by a board of governors consisting of six members, with three members to be appointed by us and three members to be appointed by Mayo. Initially, we will hold fifty percent of the issued and outstanding membership interests and Mayo will hold fifty percent of the issued and outstanding membership interests of the new entity. In exchange for our membership interests in the joint venture entity, we will make a capital contribution, to be paid in three equal installments of $2.0 million each with the first installment due on the Closing Date, and the second and third installments due on the first and second anniversaries of the Closing Date, respectively, subject to the joint venture entity’s achievement of certain operational milestones agreed upon by the board of governors of the joint venture entity (the “Milestones”). In addition, on November 14, 2011, we granted Mayo 50,000 shares of common stock, 33,000 of which are subject to certain forfeiture restrictions if the joint venture does not meet certain of the Milestones.
-98-
Table of Contents
On the Closing Date, we will enter into a three-year joint development intellectual property agreement with Mayo and the joint venture entity, pursuant to which we and Mayo will grant each other non-exclusive, non-transferable licenses to use certain intellectual property required for the performance of statements of work to be issued under such agreement. Also pursuant to the joint development intellectual property agreement, we, Mayo and the joint venture entity will agree that any intellectual property created by the joint venture entity shall be the property of the joint venture entity; however, the joint venture entity will grant us and Mayo licenses to commercialize such intellectual property in the form of diagnostic products and diagnostic lab services, respectively, at prices to be determined by the board of governors of the joint venture entity.
The board of governors will be advised by a six-member scientific review committee, which will also be composed of three members selected by us and three members selected by Mayo. The affiliation agreement may be terminated by mutual consent of the parties or by the non-breaching party upon a material breach of the affiliation agreement that remains uncured for a period of 90 days. Although we entered into the affiliation agreement with Mayo to form a joint venture in 2012, for a variety of reasons, including a mutual decision by the parties to terminate the affiliation agreement, the joint venture entity may never be formed or, if formed, may never achieve the anticipated research, development and commercial objectives currently contemplated by us and Mayo.
North Shore-Long Island Jewish Health System
In 2007, we started working with Drs. Kanti Rai and Nicholas Chiorazzi at the Feinstein Institute for Medical Research at the North Shore-Long Island Jewish Health System. Drs. Rai and Chiorazzi are leading clinicians and scientists in the study of chronic lymphocytic leukemia (“CLL”) and have provided over 300 clinical specimens and associated clinical and laboratory data for panels of CLL specimens that were used for clinical validation of MatBA®-CLL. We analyzed these samples at our clinical laboratory and published the resulting data jointly with Drs. Rai and Chiorazzi. We will use the same samples for additional collaborative studies involving the search for additional genomic-based biomarkers of CLL. This collaboration is not governed by a formal written agreement.
Memorial Sloan-Kettering Cancer Center
We have multiple research collaborations with Memorial Sloan-Kettering Cancer Center including
| • | In March 2008, we entered into a Biological Material Transfer Agreement with Memorial Sloan-Kettering Cancer Center, pursuant to which Dr. Victor Reuter at Memorial Sloan-Kettering Cancer Center provided us with slides of cells of over 140 renal tumor ex vivo core biopsies. These samples were used for validation of our FReCaD™ assay to evaluate the ability of the FISH-based assay to classify renal cortical neoplasms. In this study, we calculated the sensitivity and specificity of the core biopsy relative to that obtained by routine pathology of the core biopsy and the specimen proper. These studies are currently being written for joint publication. |
| • | In October 2009, we entered into a Biological Material Transfer Agreement with Dr. Julie Teruya-Feldstein at Memorial Sloan-Kettering Cancer Center, whereby Dr. Teruya-Feldstein provided us with 1,000 lymphoma specimens of varying histologies and with known clinical outcomes for clinical validations of MatBA® in all subtypes of mature B cell neoplasms. Dr. Teruya-Feldstein has provided cores of FFPE tumor material spear-heading and providing justification for the use of this tissue type in array-CGH. We evaluate the genomic gain or loss using MatBA® at our clinical reference laboratory and we are in the process of analyzing these specimens for clinical correlations. We will jointly publish the results of this collaboration with Dr. Teruya-Feldstein. |
| • | In June 2009 and March 2010, we entered into separate Biological Material Transfer Agreements with Dr. Raju S.K. Chaganti of Memorial Sloan-Kettering Cancer Center. Pursuant to the June 2009 |
-99-
Table of Contents
| agreement, Dr. Chaganti provided us with 50 follicular lymphoma and diffuse large B-cell lymphoma specimens. We used the specimens for purposes of validating a comparative genomic hybridization microarray-based assay in the diagnosis and prognosis of mature B-cell neoplasms. Pursuant to the March 2010, agreement, Dr. Chaganti provided us with 30 DNA samples. We used the samples for purposes of validating a comparative genomic hybridization microarray-based assay in the diagnosis and prognosis of genitourinary cancers. |
| • | In January 2011, we entered into a Biological Material Transfer Agreement with Dr. Jonathan Coleman at Memorial Sloan-Kettering Cancer Center to evaluate FISH-based and array-CGH tests in the diagnosis of renal mass aspirates/core biopsies. Dr. Coleman provided us with approximately 50 needle biopsy specimens. We will use the specimens to perform assays of prostate, bladder and kidney specimens using FReCaD™ and UroGenRA™ and compare the classification with that obtained by routine pathology. Any resulting data could be prepared for joint publication. |
National Cancer Institute
In July 2009 and December 2009, we entered into Simple Letter Agreements for the Transfer of Materials with the National Cancer Institute and began our collaboration with Dr. Nicolas Wentzensen at National Cancer Institute, an Institute of the National Institutes of Health, to interrogate the potential role of identification of host genomic abnormalities by FISH as a screening tool for the detection of HPV-associated pre-cancerous cells and cancerous cells. Dr. Wentzensen has provided us with liquid biopsy specimens for analysis by FISH using the FHACT™ DNA-FISH probe. In our first project together, National Cancer Institute provided cervical liquid biopsy specimens and in later collaborations, National Cancer Institute provided anal liquid biopsy specimens.
Kamineni Hospital
In November 2010, we began collaborating with Dr. Annie Hasan at the Kamineni Hospital in Hyderabad, India, to evaluate the FHACT™ DNA-FISH Probe as a screening tool for the identification of pre-cancerous and cancerous cervical cells. In this collaboration, we provide the FHACT™ DNA-FISH Probe to Dr. Hasan’s laboratory where the assay is performed on Pap smears obtained during routine health visits. We are analyzing the data from this collaboration jointly with Dr. Hasan and any resulting publications will be jointly produced. We anticipate providing Dr. Hasan with FReCaD™ for use as a screening tool in renal cancers to be performed on specimens obtained in Kamineni Hospital. This collaboration is not governed by a formal written agreement.
University of Iowa Hospitals and Clinics
In 2011, we entered into a Material Transfer Agreement with the University of Iowa Research Foundation, whereby Dr. Aaron Bossler will provide specimens useful to our studies evaluating the FHACT™ assay in cervical liquid biopsy specimens with known HPV type and clinical follow-up. In this study, specimens will be sent to us for FISH-based assays and the data analyzed jointly.
Aptiv Solutions
We recently developed a strategic relationship with Aptiv Solutions in order to deliver comprehensive solutions to biopharmaceutical and medical device firms conducting clinical trials for oncology therapeutics. Aptiv Solutions is a full-service clinical research organization that provides a portfolio of innovative services including adaptive trial design, early phase product strategy, regulatory services, pharmacovigilance, and the operational support of a global clinical research organization. We believe that together, we can provide biopharmaceutical firms with a better understanding of modes of action of a variety of therapeutics and the role biomarkers play in the oncology drug development process. We believe that this new relationship will provide
-100-
Table of Contents
biopharma companies with the insights and services necessary to bring oncology therapeutics to their patients faster and more efficiently. This relationship is not governed by a formal written agreement.
Cleveland Clinic
In May 2012, we entered into a collaborative research agreement and non-exclusive license arrangement with Cleveland Clinic to start development around a renal cell carcinoma diagnostic focused on validating genomic biomarkers from DNA. In connection with this collaboration, we have agreed to issue Cleveland Clinic shares of our common stock having an aggregate value of $20,000, which is 2,674 shares based upon the September 30, 2012 common stock valuation of $7.48 per share. In this relationship we will be working with Drs. Eric Klein and Magi Galluzzi at the Cleveland Clinic. Drs. Klein and Galluzzi are leading clinicians and scientists in the study of renal and urogenital cancers and will be providing over 200 clinical specimens and associated clinical and laboratory data for the validation of our UroGenRATM - Renal microarray. We will be analyzing these samples at our clinical laboratory and will have the opportunity to publish the resulting data jointly with Cleveland Clinic.
Stanford University
Stanford University recently granted us a worldwide, non-exclusive license under U.S. Patent No. 7,622,253 and U.S. Patent No. 7,332,280 directed to a method and an assay for the classification of diffuse large B-cell lymphoma (DLBCL) patients based on risk stratification and a predictive model for patient survival. The method and assay covered in these patents have been developed in the lab of Dr. Ronald Levy, a pioneer researcher in the area of lymphoma. DLBCL is an aggressive form of non-Hodgkin’s lymphoma with estimated 10,000 deaths annually in the United States. A current method of prognostication for DLBCL is done by using the International Prognostic Index score. However, a disadvantage to this method is that two individuals with an identical International Prognostic Index score can react differently to treatment thereby affecting their life expectancy and patient survival. The licensed technology analyzes the expression of six genes by a real-time PCR methodology in conjunction with an algorithm. This method and assay is not currently available in a clinical lab setting to our knowledge. We believe that developing and commercializing this licensed technology will allow us to provide a more accurate classification, stratification and prediction of the DLBCL patient population and also provide more personalized therapeutic options to this patient population. This assay will be added to our growing menu of tests (IHC, mutation analysis, etc.) to be offered under our Complete DLBCL™ program.
Scientific Advisory Board
Our Scientific Advisory Board is comprised of many preeminent scientists and physicians from the fields of cancer biology, cancer pathology, cancer medicine and molecular genetics. We have scientists and clinicians from leading cancer centers, including Memorial Sloan-Kettering Cancer Center, Mt. Sinai and the Institute for Cancer Genetics at Columbia University. These distinguished scientists and clinicians help oversee and review the scientific innovation, integrity and clinical relevancy of our program. The board of directors appoints members to the Scientific Advisory Board for terms of one year. Please see the section entitled “Management—Scientific Advisory Board” for the biographies of each member of our Scientific Advisory Board.
Competition
As a provider of genomic-based tests and services that provide personalized diagnostic and prognostic information for hematological, urogenital and HPV-associated cancers, we rely extensively on our ability to combine research insights with high-quality, state-of-the art clinical laboratory testing. We believe that we compete principally on the basis of:
| • | our ability to address complex cancers that are currently difficult to prognose and challenging to predict treatment outcomes using currently available technologies; |
| • | the ability of our proprietary tests and services to provide more information than existing tests with respect to the cancers we address; |
-101-
Table of Contents
| • | our ability to utilize a wide variety of sample types, accelerating the time-frame for clinical validation of our tests and allowing health care providers to readily integrate our tests into their established workflow; |
| • | our ability to perform clinical studies using FFPE samples to either validate or develop novel insights for our proprietary programs; |
| • | the quality of our services and our ability to collaborate with our customers on a consultative basis; |
| • | our research and clinical collaborations with key academic and clinical study groups; |
| • | the quality of our clinical reference laboratory, which enables consistent, comprehensive and reproducible results; |
| • | the level of disease specific knowledge and customer service we provide, both to academic centers and community based health care professionals; and |
| • | our workplace environment, recognized by being named #20 nationwide by The Scientist in “Best Places to Work Industry, 2011”, which increases our ability to attract both clinical and research talent. |
We believe that we compete favorably with respect to these factors, although we cannot assure you that we will be able to continue to do so in the future or that new products or tests that perform better than our proprietary tests and services will be introduced. We believe that our continued success depends on our ability to:
| • | expand and enhance our MatBA® tests to provide clinically meaningful information in additional indications; |
| • | continue to innovate and maintain scientifically advanced technology; |
| • | successfully market and sell our proprietary tests in the United States; |
| • | continue to obtain appropriate regulatory approvals in the United States and abroad; |
| • | continue to validate our pipeline of microarray tests and DNA probes; |
| • | continue to obtain positive reimbursement decisions from payors and from CMS; |
| • | continue to enter into partnerships with local distributors and/or manufacturers to expand into emerging markets, including India, Mexico and Brazil; |
| • | maintain existing and enter into new research and clinical collaborations with key academic and clinical study groups; |
| • | continue to attract and retain skilled scientific and clinical personnel; |
| • | obtain patents or other protection for our proprietary tests and services; and |
| • | obtain and maintain our clinical reference laboratory accreditations and licenses. |
Our principal competition comes from existing mainstream diagnostic methods that pathologists and oncologists use and have used for many years. It may be difficult to change the methods or behavior of the referring pathologists and oncologists to incorporate our molecular diagnostic testing in their practices. In addition, companies offering capital equipment and kits or reagents to local pathology laboratories represent another source of potential competition. These kits are used directly by the pathologist, which can facilitate adoption.
-102-
Table of Contents
We also face competition from companies that offer products or have conducted research to profile genes, gene expression or protein biomarkers in various cancers. In particular, Quest Diagnostics and CombiMatrix Corporation market arrays which are competitive to our MatBA®-CLL and MatBA®-SLL arrays. Personalized genetic diagnostics is a new area of science, and we cannot predict what tests others will develop that may compete with or provide results superior to the results we are able to achieve with the tests we develop. Our competitors include public companies such as CombiMatrix Corporation, Quest Diagnostics, Abbott Laboratories, Inc., Johnson & Johnson, Roche Molecular Systems, Inc., bioTheranostics, Inc. (part of the bioMérieux SA), Genomic Health, Inc., Myriad Genetics, Inc., Qiagen N.V. and Response Genetics, Inc. and many private companies, including Agendia B.V., Pathwork Diagnostics, Inc. and Foundation Medicine, Inc. We expect that pharmaceutical and biopharmaceutical companies will increasingly focus attention and resources on the personalized diagnostic sector as the potential and prevalence increases of molecularly targeted oncology therapies approved by FDA along with companion diagnostics. For example, FDA has recently approved two such agents—Xalkori crizotinib from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc. and Zelboraf vemurafenib from Genentech USA Incorporated and Daiichi-Sankyo Inc. along with its companion B-RAF kinase V600 mutation test from Roche Molecular Systems, Inc. These two recent FDA approvals are only the second and third instances ever of simultaneous approvals of a drug and companion diagnostic, the first being the 1998 approval of Genentech, Inc.’s Herceptin trastuzumab for HER2 positive breast cancer along with the HercepTest from partner Dako A/S. Our competitors may invent and commercialize technology platforms or tests that compete with ours.
With respect to our clinical laboratory services business we face competition from companies such as Genoptix, Inc. (a Novartis AG company), Clarient, Inc. (a division of GE Healthcare, a unit of General Electric Company), Bio-Reference Laboratories, Inc. and Genzyme Genetics (a LabCorp Specialty Testing Group).
Additionally, projects related to cancer genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at identifying targeted treatment options will be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our tests in countries where we did not apply for patents or where our patents have not issued and compete with us in those countries, including encouraging the use of their test by physicians or patients in other countries.
Third-Party Suppliers and Manufacturers
We maintain control, validation and quality assurance over our DNA microarrays and probes. Our microarrays are designed in our facility by our scientists and technicians using state of the art genomic mapping and analysis software. The specifications are sent to Agilent for final manufacturing. Agilent manufactures our microarrays under strict quality control and compliance with ISO 9001 and ISO 13485 at its Santa Clara, California facility. Agilent also has another manufacturing facility in Europe that can be made available for microarray printing. Upon manufacturing our custom, proprietary microarrays, Agilent ships them back to our Rutherford facility for testing and acceptance.
The DNA component of our DNA probes is produced in our Rutherford facility under strict adherence to Good Laboratory Practice. The DNA is shipped for final manufacturing to a third-party Good Manufacturing Practice compliant provider outside the United States. From 2009 to May 2011, the probe manufacturing was done in Milan, Italy under a contract with Technogenetics, the diagnostic subsidiary of Bouty S.p.A. In May 2011, we initiated a review of the manufacturing process to improve the manufacturing yields, cost structure and overall process flexibility. Currently we conduct final manufacturing in Belgium with Labomics S.A. while we pursue a dedicated manufacturing operation in India. In February 2012 we entered into an agreement with Kamineni Life Sciences to supply outsourced manufacturing services for the production of our DNA FISH probes. We expect the manufacturing operations to be fully operational by the end of 2012. We control the overall quality and process management and the final quality assurance in a manner that is CE compliant and adheres to our Quality Management System.
-103-
Table of Contents
Patents and Proprietary Technology
Our business is dependent upon our ability to develop and protect proprietary tests that enable oncologists and pathologists at hospitals, cancer centers and physician offices to properly diagnose and inform cancer treatment. We rely on a combination of patents, patent applications, trademarks, trademark applications, trade secrets, industry know-how as well as various contractual arrangements, in order to protect the proprietary aspects of our technology.
Our patent portfolio consists of two U.S. issued patents and several pending U.S. and foreign (PCT) applications. These patents and patent applications are related to various DNA-based probes and microarrays designed for detecting and correlating certain chromosomal markers associated with particular types of cancers. As of the date of this filing, we have two issued U.S. Utility Patents (U.S. Patent Nos. 7,585,964 and 7,964,345), which cover our probe technology. These patents cover probes and methodologies designed to detect and analyze particular chromosomal translocations (genetic lesions) associated with a wide range of cancers using a technique known as FISH and serve as the backbone for several of our other pending patent applications, which are more specifically geared towards other probes (and methodologies) directed to the detection, diagnosis and prognosis of specific types of cancers (e.g., kidney cancer and cancers associated with HPV).
We are also actively pursuing patents in the microarray space. We have prepared and filed U.S. and foreign (PCT) patent applications on a microarray directed to detecting (and distinguishing) particular types of mature B cell neoplasms present in typical non-Hodgkin’s lymphoma, Hodgkin’s lymphoma and chronic lymphocytic leukemia (U.S. Patent Application No. 12/980,480 and PCT Application No. US2010/062295). Developed under our trademark, MatBA®, the patent applications covering the MatBA® microarray are directed to both the microarray itself as well as associated methodologies designed to detect the particular type of mature B cell neoplasm present in a patient. These applications also cover the use of computer assisted means to facilitate and expedite that detection process. The MatBA® patent applications are the first of our family of applications in the microarray space. We recently filed a patent application on a detection method associated with HPV-associated cancers (US. Patent Application No. 13/227,027) and a patent application to cover our UGenRA™ microarray and methods of using the array in diagnosing and/or providing a prognosis for certain types of female gynecological cancers and pre-cancers (U.S. Patent Application No. 61/581,350). In addition, we are working on several additional patent filings directed to our other microarrays, namely UroGenRA™, and expect to file these applications with the U.S. Patent and Trademark Office at the end of 2012.
In addition to patents, we hold six U.S. registered trademarks, including a federal registration to “CGI” as well as four U.S. trademark applications and one foreign trademark registration for certain of our proprietary tests and services. Our strategic use of distinctive trademarks has garnered increased name recognition and brand awareness for our tests and services within the industry.
Through our clinical laboratory, we provide several clinical services that utilize our proprietary trade secrets. In particular, we maintain trade secrets with respect to specimen accessioning, sample preparation and certain aspects of cytogenetic analysis. All of our trade secrets are kept under strict confidence and we take all reasonable steps, including the use of non-disclosure agreements and confidentiality agreements, to ensure that our confidential information is not unlawfully disseminated. We also conduct training sessions on the importance of maintaining and protecting trade secrets with our scientific staff and laboratory directors and supervisors.
In addition to our proprietary intellectual property, we entered into a nonexclusive licenses with the National Cancer Institute for the use of its intellectual property relating to a 3q marker and with Stanford University for use and development of a diagnostic assay and predictive model that has been granted two patents for the stratification and risk prediction for DLBCL patients. Under the terms of the license, we are permitted to use the National Cancer Institute’s proprietary intellectual property for use in our patent pending FHACT™ DNA probe, which is directed to the diagnosis and prognosis of certain HPV-associated cancers.
-104-
Table of Contents
Operations and Production Facilities
Our research and development laboratories and our diagnostic laboratories are located in our Rutherford, New Jersey headquarters.
We work with electronic medical records providers to facilitate seamless communication between our laboratory and the oncologist or pathologist at the test ordering site. Currently, we have the ability to integrate with electronic medical record systems, as we have already done with MDL, an electronic medical record provider. We do this integration through utilizing HL7 interfaces, which are standard in health care information technology systems. We currently employ HL7 for its integration with a revenue cycle management company, Xifin, as well as with its electronic medical records partners such as MDL. The use of the HL7 interface allows systems written in different languages and running on different platforms to be able to talk to each other through the use of an abstracted data layer. This means that we do not have to spend significant extra time, often months, designing and developing common communications protocols when integrating with other electronic health records systems or billing systems providers.
When a customer takes a specimen from a patient for diagnostic testing, he or she will complete a requisition form (either by hand or electronically, or via electronic medical records technology), and package the specimen for shipment to us. Once we receive the specimen at our laboratory and we enter all pertinent information about the specimen into our clinical laboratory information system, one of our histotechnologists, cytotechnologists, flow technologists or molecular technologists prepares the specimen for diagnosis. The prepared specimen is sent to one of our pathologists or directors who is experienced in making the diagnosis requested by the referring oncologist or pathologist.
After diagnosis, our pathologist uses our laboratory information systems to prepare a comprehensive report, which includes any relevant images associated with the specimen. Our reporting portal, cgireports.com, allows a referring oncologist or pathologist to access his/her test results in real time in a secure HIPAA compliant manner. The reports are generated in industry standard PDF formats which allows for high definition color images to be reproduced clearly. This portal has been fully operational at our facilities for over the past eight quarters.
In most cases we provide both the technical analysis and professional diagnosis, although we also fulfill requests from oncologists and pathologists for only one service or the other. If an oncologist or pathologist at the hospital, cancer center, reference laboratory or physician office requires only the analysis, we prepare the data and then return it to the referring oncologist or pathologist for assessment and diagnosis.
Quality Assurance
Clinical Lab Services
We are committed to providing reliable and accurate diagnostic services to our customers. Accurate specimen identification, timely communication of diagnoses, and prompt correction of errors, is critical. We monitor and improve our performance through a variety of methods, including performance improvement indicators, proficiency testing (CAP and New York State), external audits and satisfaction surveys. All quality concerns and incidents are subject to root cause analysis and our procedures are put through annual evaluation to ensure that we are providing the best services possible to our patients and customers. Protection of patient results from misuse and improper access is imperative and thus electronic and paper results are guarded via password-protection and identification cards.
We have established a comprehensive Quality Assurance and Management Program for our laboratory designed to drive accurate and timely test results and to ensure the consistent high quality of our testing services. The Quality Assurance and Management Program documents the quality assurance/performance improvement
-105-
Table of Contents
plans and policies and the laboratory quality assurance and quality control procedures that are necessary to ensure that we offer the highest quality of diagnostic testing services. This program is designed to satisfy all the requirements necessary for local and state licensures by the New Jersey Health Department and the New York Department of Health Clinical Laboratory Evaluation Program and accreditation for clinical diagnostic laboratories by CAP. We follow the policies and procedures for patient and employee safety, hazardous waste disposal and fire codes stated in the general laboratory procedure manual. We believe that all pertinent regulations of CLIA, Occupational Safety and Health Administration (“OSHA”), Environmental Protection Agency and FDA are satisfied by following the established guidelines and procedures of our Quality Assurance and Management Program.
In addition to the compulsory proficiency programs and external inspections required by CMS and other regulatory agencies, we have developed a variety of internal systems and procedures to emphasize, monitor and continuously improve the quality of our operations. We maintain internal quality controls by routinely processing specimens with known diagnoses in parallel with patient specimens. We also have an extensive, internally administered program of specimen proficiency testing, in which our laboratory staff are blinded to the results.
We participate in numerous externally administered quality surveillance programs and our laboratory is accredited by CAP. The CAP accreditation program involves both unannounced on-site inspections of our laboratories and our participation in CAP’s ongoing proficiency testing program. CAP is an independent, non-governmental organization of board-certified pathologists that accredits laboratories nationwide on a voluntary basis and that has been recognized by CMS as an accreditation organization to inspect laboratories to determine adherence to the CLIA standards. Successful participation in CAP’s proficiency testing program satisfies the CLIA requirement for participation in proficiency testing programs administered by an external source.
Microarrays
We test each lot of microarrays that are manufactured for us by Agilent and maintain a log of all the hybridizations. We also have an extensive process of testing the hybridization results and comparing them to prior lots to ensure consistency and to review for potential changes. Any changes or results that are not consistent with expectations are logged and then immediately reviewed by our team, including the Vice President of Research & Development. In cases where a manufacturing problem is suspected, we immediately review the entire lot and prepare the results for review with Agilent.
FISH-based DNA Probes
We are committed to the highest level of quality in the development and manufacture of fluorescently-labeled DNA intended to compose our DNA-FISH probes. Our probes are manufactured to meet or exceed all established quality and performance specifications, and comply with relevant safety and regulatory requirements as defined in the European In Vitro Diagnostic Directive in order to qualify them for CE marking.
On behalf of our subsidiary, CGI Italia, we have created and implemented a Quality Management System applicable throughout the entire life cycle of our DNA-FISH probes. This Quality Management System maintains control over the quality of the goods manufactured by us or third parties employed by us and services provided to CGI Italia. This system addresses—within other procedures—the organizational structure, manufacturing process and related responsibilities, the systematic quality assurance and quality control of production, the means to monitor the performance of the quality system (internal/external audit) and the post-production vigilance.
-106-
Table of Contents
Third-party Payor Reimbursement
Revenues from our clinical laboratory tests are derived from several different sources. Depending on the billing arrangement and applicable law, parties that reimburse us for our services include:
| • | third-party payors that provide coverage to the patient, such as an insurance company, managed care organization or a governmental payor program; |
| • | physicians or other authorized parties (such as hospitals or independent laboratories) that order the testing service or otherwise refer the services to us; or |
| • | the patient. |
For the year ended December 31, 2011, we derived approximately 54% of our total revenue from private insurance, including managed care organizations and other health care insurance providers, 24% from government payor programs, 12% from direct-bill customers, including hospitals and other laboratories, and approximately 10% from other sources.
Where there is a coverage policy, contract or agreement in place, we bill the third-party payor, the hospital or referring laboratory as well as the patient (for deductibles and coinsurance or copayments, where applicable) in accordance with the policy or contractual terms. Where there is no coverage policy, contract or agreement in place, we pursue reimbursement on behalf of each patient on a case-by-case basis and rely on applicable billing standards to guide our claims.
We are reimbursed for three categories of tests: (1) genetic and molecular testing; (2) anatomic pathology and IHC and (3) general immunology and flow cytometry. Reimbursement under the Medicare program for the diagnostic services that we offer is based on either the Medicare Physician Fee Schedule or Medicare Clinical Laboratory Fee Schedule, each of which in turn is subject to geographic adjustments and is updated annually. Medical services provided to Medicare beneficiaries that require a degree of physician supervision or other involvement, such as pathology tests, are generally reimbursed under the Medicare Physician Fee Schedule, whereas clinical diagnostic laboratory tests are generally reimbursed under the Clinical Laboratory Fee Schedule. Most of the services that we provide are for genetic and molecular testing, which are reimbursed as clinical diagnostic laboratory tests.
Medicare fee schedule amounts are established for each billing code, or CPT code. In addition, for its laboratory fee schedule, Medicare also sets a cap on the amount that it will pay for any individual test. This cap, usually referred to as the National Limitation Amount, is set at a percentage of the median of all the contractor fee schedule amounts for each billing code. In the past, Congress has lowered the percentage of the median used to calculate the National Limitation Amount in order to achieve budget savings. Currently, the National Limitation Amount ceiling is set at 74% of the median for established tests and 100% of the median for certain new tests that were not previously reimbursed. In billing Medicare for clinical laboratory services, we are required to accept, as payment in full, the lowest of our actual charge, the fee schedule amount for the state or local geographical area or the National Limitation Amount.
Medicare also has policies that may limit when we can bill directly for our services and when we must instead bill another provider, such as a hospital. When the testing that we perform is done on a specimen that was collected while the patient was in the hospital, as either an inpatient or outpatient, we may be required to bill the hospital for our services, rather than the Medicare program, depending on whether or not the service was ordered more than 14 days after the patient’s discharge from the hospital. These requirements are complex and time-consuming and, depending on what they require, may affect our ability to collect for our services.
Our reimbursement rates can vary based on whether we are considered to be an “in-network” provider, a participating provider, a covered provider or an “out-of-network” provider. These definitions can vary from
-107-
Table of Contents
insurance company to insurance company, but we are generally considered an “out of network” or non-participating provider in the vast majority of our cases. It is not unusual for a company that offers highly specialized or unique testing to be an “out of network” provider. An “in-network” provider usually has a contracted arrangement with the insurance company or benefits provider. This contract governs, among other things, service-level agreements and reimbursement rates. In certain instances an insurance company may negotiate an “in-network” rate for our testing rather than pay the typical “out-of-network” rate. An “in-network” provider usually has rates that are lower per test than those that are “out-of-network”, and that rate can vary from a single digit percentage deduction discount to upwards of 25% to 30% percent lower than an “out-of-network” provider. The discount rate varies based on the insurance company, the testing type and the often times the specifics of the patient’s insurance plan.
In addition, as part of the Middle Class Tax Relief and Job Creation Act of 2012 (“MCTRJCA”), signed into law by the President on February 22, 2012, Congress extended the special billing rule that also allowed laboratories to bill Medicare for the technical component of certain pathology services furnished to patients of qualifying hospitals. Effective July 1, 2012, independent laboratories, like our laboratory, are required to bill for the technical component of these services in most instances.
Billing Codes for Third-party Payor Reimbursement
CPT codes are the main data code set used by physicians, hospitals, laboratories and other health care professionals to report separately-payable clinical laboratory tests for reimbursement purposes. The CPT coding system is maintained and updated on an annual basis by the American Medical Association. Although there is no specific code to report microarrays for oncology, such as our MatBA®-CLL, there are existing codes that describe all of the steps in our MatBA®-CLL testing process. We currently use a combination of different codes to describe the various steps in our testing process. Many of the CPT codes used to bill for molecular pathology tests such as ours have been significantly revised by the CPT Code Editorial Panel. These new codes replace the more general “stacking” codes that were previously used to bill for these services with more test-specific codes, which will be effective January 2013. In the Final Physician Fee Schedule Rule, which was issued in November 2012, CMS stated that it had determined it would pay for the new codes as clinical laboratory tests, which are payable on the Clinical Laboratory Fee Schedule (CLFS). CMS also stated that it plans to “gapfill” the new codes; that is, it will ask the contractors to determine a reasonable price for the new codes.
Among the new codes that were created by CPT were a specific subset of codes called Multi-analyte Assays with Algorithmic Analysis (MAAAs). These tests typically use an algorithm applied to certain specific components to arrive at a score that is used to predict a particular clinical outcome. CMS recently stated that it will not pay for some of these new codes, because it does not believe it is permitted to pay for the underlying algorithm. Instead, it will pay for only for the specific laboratory components that are performed as part of these tests. CMS also stated it has plans to seek additional information about these codes next year. It is not clear what position CMS will finally decide to take on the MAAA tests, but its decision could adversely affect future reimbursement for such tests, including those we may develop. Currently less than 10% of our revenue is derived from tests that may be considered MAAAs.
These changes in coding and reimbursement methods could have an adverse impact on our revenues going forward. However, we are currently working with our revenue consultants to determine what changes will be required by the new coding changes. The elimination of the “stacking” codes will require us to either use the new more specific codes where applicable beginning January 1, 2013, or to use other “Not Otherwise Classified” (NOC) codes when billing for some of our tests. If CMS decides not to reimburse for the algorithm included in the MAAA tests, then we would only be able to bill Medicare for the specific genetic examinations that we perform, without the algorithms. The introduction of the new codes, in combination with the other action being considered by CMS with regard to pricing, could result in a reduction in the payment that we receive for our tests and make it more difficult to obtain coverage from Medicare or other payers. There can be no guarantees that Medicare and other payers will establish positive or adequate coverage policies or reimbursement rates. We are moving forward with plans to obtain billing codes for our tests. A specific code for our tests, however, does not
-108-
Table of Contents
assure an adequate coverage policy or reimbursement rate. Please see the section entitled “Legislative and Regulatory Changes Impacting Clinical Laboratory Tests” for further discussion of certain legislative and regulatory changes to these billing codes and the impact on our business.
Coverage and Reimbursement for Our Microarray Tests
Although MatBA®-CLL is a relatively new test, some third-party payors have established coverage and reimbursement policies set for other microarray-based tests. We have been able to receive reimbursement for our tests from some payors based on their established policies, including major commercial third-party payors. In addition, microarray-based tests for congenital and prenatal diagnosis are routinely reimbursed by Medicare and commercial third-party payors. Similar processes can be used as a model for the reimbursement of microarray-based tests for oncology.
The current landscape with payors is generally as follows:
Commercial Third-party Payors and Patient Pay. Where there is a coverage policy in place, we bill the payor and the patient in accordance with the established policy. Where there is no coverage policy in place, we pursue reimbursement on behalf of each patient on a case-by-case basis. Our efforts in obtaining reimbursement based on individual claims, including pursuing appeals or reconsiderations of claims denials, take a substantial amount of time, and bills may not be paid for many months, if at all. Furthermore, if a third-party payor denies coverage after final appeal, payment may not be received at all. We are working to decrease risks of nonpayment by implementing a revenue cycle management system.
Medicare and Medicaid. We believe that as much as 30% to 40% of our future market for our tests may be derived from patients covered by Medicare and Medicaid.
We cannot predict whether, or under what circumstances, payors will reimburse our microarray tests. Payment amounts can also vary across individual policies. Denial of coverage by payors, or reimbursement at inadequate levels, would have a material adverse impact on market acceptance of our tests.
Legislative and Regulatory Changes Impacting Clinical Laboratory Tests
From time to time, Congress has revised the Medicare statute and the formulas it establishes for both the Medicare Clinical Laboratory Fee Schedule and the Physician Fee Schedule. The payment amounts under the Medicare fee schedules are important not only for our reimbursement under Medicare, but also because the schedule often is used as a basis for establishing the payment amounts set by other third party payors. For example, state Medicaid programs are prohibited from paying more than the Medicare fee schedule limit for clinical laboratory services furnished to Medicaid recipients.
Under the statutory formula for clinical laboratory fee schedule amounts, increases are made annually based on the Consumer Price Index for All Urban Consumers as of June 30 for the previous twelve-month period. From 2004 through 2008, Congress eliminated the Consumer Price Index for All Urban Consumers update in the Medicare Prescription Drug, Improvement and Modernization Act of 2003. In addition, for years 2009 through 2013, the Medicare Improvements for Patients and Providers Act of 2008 (“MIPPA”) mandated a 0.5% cut to the Consumer Price Index for All Urban Consumers. Accordingly, the update for 2009 was reduced to 4.5% and negative 1.9% for 2010. In March 2010, the President signed into law PPACA, which, among other things, imposed additional cuts to the Medicare reimbursement for clinical laboratories. The PPACA replaced the 0.5% cut enacted by MIPPA with a “productivity adjustment” that will reduce the Consumer Price Index update in payments for clinical laboratory tests. In 2011, the productivity adjustment was -1.2%. In addition, the PPACA includes a separate 1.75% reduction in the CPI update for clinical laboratories for the years 2011 through 2015. On February 22, 2012, President Obama signed the MCTRJCA, which mandated an additional change in reimbursement for clinical laboratory services payments. This legislation requires CMS to reduce the Medicare
-109-
Table of Contents
clinical laboratory fee schedule by 2% in 2013, which in turn will serve as a base for 2014 and subsequent years. CMS has projected that because of changes required by PPACA and MCTRJCA, payment for clinical laboratory services will go down by approximately 3% by 2013.
With respect to our diagnostic services for which we are reimbursed under the Medicare Physician Fee Schedule, because of the statutory formula, the rates would have decreased for the past several years if Congress failed to intervene. In the past, when the application of the statutory formula resulted in lower payment, Congress has passed interim legislation to prevent the reductions. On November 1, 2012, the Centers for Medicare & Medicaid Services (CMS) issued its 2013 Physician Fee Schedule Final Rule (“the Final Rule”). In the Final Rule, CMS called for a reduction of approximately 26.5% in the 2013 conversion factor that is used to calculate physician reimbursement. However, the American Taxpayer Relief Act of 2012, which was signed into law on January 2, 2013, prevents this proposed reduction and keeps the current conversion factor rate in effect until December 31, 2013. If Congress fails to act in future years to offset similar deductions, the resulting decrease in payment could adversely impact our revenues and results of operations. In addition, for 2012, CMS has requested that the American Medical Association’s Relative Value Scale Update Committee reexamine the relative values of certain pathology codes, including FISH codes. The Relative Value Scale Update Committee is an expert panel that provides relative value recommendations to CMS for use in annual updates to the Medicare Physician Fee Schedule. These relative values are used by CMS to determine payments and CMS seeks to assess whether such codes are misvalued and an adjustment is necessary. We cannot predict at this time whether the Relative Value Scale Update Committee will recommend any changes and/or whether CMS will accept those recommendations. For the year ended December 31, 2011 and the nine months ended September 30, 2012, approximately 24% and 18%, respectively, of our total revenues are derived from Medicare generally and any changes to the physician fee schedule that result in a decrease in payment could adversely impact our revenues and results of operations.
In addition, the Final Rule included a reduction of certain relative value units and geographic adjustment factors used to determine reimbursement for a number of commonly used pathology codes, including CPTs 88300, 88302, 88304, and 88305. In particular, the Final Rule implements a cut of approximately 33% in the global billing code for 88305 and a 52% cut in the Technical Component of that code. These codes describe services that we must perform in connection with our tests and we bill for these codes in connection with the services that we provide. The Company is currently studying the new rule and the impact on its revenues.
Further, with respect to the Medicare Program, Congress has proposed on several occasions to impose a 20% coinsurance on patients for clinical laboratory tests reimbursed under the clinical laboratory fee schedule, which would require us to bill patients for these amounts. Because of the relatively low reimbursement for many clinical laboratory tests, in the event that Congress were to ever enact such legislation, the cost of billing and collecting for these services would often exceed the amount actually received from the patient and effectively increase our costs of billing and collecting.
Some of our Medicare claims may be subject to policies issued by Palmetto GBA, the current Medicare Administrative Contractor for California, Nevada, Hawaii and certain U.S. territories. The Medicare contractor has recently issued a Local Coverage Decision that affects coverage, coding and billing of many molecular diagnostic tests. Under this Local Coverage Determination, Palmetto will not cover any molecular diagnostic tests, including our tests, unless the test is expressly included in a National Coverage Determination issued by CMS or a Local Coverage Determination or coverage article issued by Palmetto. Currently, laboratory providers may submit coverage determination requests to Palmetto for consideration and apply for a unique billing code for each test (which is a separate process from the coverage determination). In the event that a non-coverage determination is issued, the laboratory must wait six months following the determination to submit a new request. In addition, effective May 1, 2012, Palmetto implemented its new Molecular Diagnostic Services Program, under which, among other things, laboratories must use newly-assigned billing codes specific to the test. These new billing codes enable Palmetto to measure utilization and apply coverage determinations. Denial of coverage by Palmetto, or reimbursement at inadequate levels, would have a material adverse impact on market acceptance of our tests.
-110-
Table of Contents
Governmental Regulations
Clinical Laboratory Improvement Amendments of 1988 and State Regulation
As a diagnostic service provider, we are required to hold certain federal, state and local licenses, certifications and permits to conduct our business. As to federal certifications, in 1988, Congress passed the Clinical Laboratory Improvement Amendments (“CLIA”) establishing quality standards for all laboratories testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test was performed. The Company’s laboratory is CLIA accredited. As to state laws, we are required to meet certain laboratory licensing and other requirements. Our laboratory holds the required licenses and accreditations obtained from the applicable state agencies in which we operate. For more information on state licensing requirements, see the sections entitled “Description of the Business—Government Regulations—New Jersey and New York State Laboratory Licensing” and “Description of the Business—Government Regulations—Other States’ Laboratory Testing”.
Under CLIA, a laboratory is defined as any facility which performs laboratory testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease, or the impairment of, or assessment of health. CLIA also requires that we hold a certificate applicable to the type of work we perform and comply with certain standards. CLIA further regulates virtually all clinical laboratories by requiring they be accredited by the federal government and comply with various operational, personnel, facilities administration, quality and proficiency requirements intended to ensure that their clinical laboratory testing services are accurate, reliable and timely. CLIA compliance and accreditation is also a prerequisite to be eligible to bill for services provided to governmental payor program beneficiaries. CLIA is user-fee funded. Therefore, all costs of administering the program must be covered by the regulated facilities, including certification and survey costs.
We are subject to survey and inspection every two years to assess compliance with program standards, and may be subject to additional unannounced inspections. Laboratories performing high complexity testing are required to meet more stringent requirements than laboratories performing less complex tests. In addition, a laboratory like ours that is certified as “high complexity” under CLIA may obtain analyte specific reagents, which are used to develop diagnostic tests that are developed and validated for use in examinations the laboratory performs itself known as LDTs. Our laboratory is CLIA accredited and under our CLIA accreditation, we were allowed to first use MatBA®-CLL in November 2010 and MatBA®-SLL in the first quarter 2012.
In addition to CLIA requirements, we participate in the oversight program of CAP. Under CMS requirements, accreditation by CAP is sufficient to satisfy the requirements of CLIA. Therefore, because we are accredited by CAP, we are deemed to also comply with CLIA. CLIA also provides that a state may adopt laboratory regulations that are more stringent than those under federal law, and a number of states have implemented their own more stringent laboratory regulatory schemes. State laws may require that laboratory personnel meet certain qualifications, specify certain quality controls, or prescribe record maintenance requirements.
FDA
FDA regulates the sale or distribution, in interstate commerce, of medical devices under the FDCA, including in vitro diagnostic test kits, reagents and instruments used to perform diagnostic testing. Such devices must undergo pre-market review by FDA prior to commercialization unless the device is of a type exempted from such review by statute or pursuant to FDA’s exercise of enforcement discretion. FDA, to date, has decided not to exercise its authority to actively regulate the development and use of LDTs such as ours as medical devices and therefore we do not believe that our LDTs currently require pre-market clearance or approval. It is possible, perhaps likely, that FDA will decide to more actively regulate LDTs, which could lead to premarket and post-market obligations. Section 1143 of the Food and Drug Administration Safety and Innovation Act, signed by the President on July 9, 2012, requires FDA to notify Congress at least 60 days prior to issuing a draft or final guidance regulating LDTS and provide details of the anticipated action. We are monitoring developments and
-111-
Table of Contents
anticipate that our products (CGH-Microarrays and FISH Probes) will be able to comply with anticipated requirements. In the meantime, we maintain our CLIA accreditation, which permits the use of LDTs for diagnostics purposes. FDA regulations pertaining to medical devices govern, among other things, the research, design, development, pre-clinical and clinical testing, manufacture, safety, efficacy, storage, record-keeping, packaging, labeling, adverse event reporting, advertising, promotion, marketing, distribution and import and export of medical devices. Pursuant to the FDCA, medical devices are subject to varying degrees of regulatory control and are classified in one of three classes depending on the controls FDA determines necessary to reasonably ensure their safety and efficacy.
Class I devices are those for which reasonable assurance of the safety and effectiveness can be provided by adherence to FDA’s general regulatory controls for medical devices, which include compliance with the applicable portions of FDA’s Quality System Regulations, facility registration and product listing, reporting of adverse medical events and appropriate, truthful and non-misleading labeling, advertising and promotional materials, or general controls. Many Class I devices are exempt from pre-market regulation, however, some Class I devices require pre-market clearance by FDA through the 510(k) pre-market notification process described below.
Class II devices are subject to FDA’s general controls, and any other special controls as deemed necessary by FDA to provide reasonable assurance of the safety and effectiveness of the device. Pre-market review and clearance by FDA for Class II devices are generally accomplished through the 510(k) pre-market notification procedure. Pre-market notification submissions are subject to user fees, unless a specific exemption applies. To obtain 510(k) clearance for a medical device (or for certain modifications to devices that have received 510(k) clearance), a manufacturer must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or to a preamendment device that was in commercial distribution before May 28, 1976 (a “predicate device”) for which FDA has not yet called for the submission of a pre-market approval (“PMA”) application. In making a determination that the device is substantially equivalent to a predicate device, FDA compares the proposed device to the predicate device or predicate devices and assesses whether the subject device is comparable to the predicate device or predicate devices with respect to intended use, technology, design and other features which could affect the safety and effectiveness. If FDA determines that the subject device is substantially equivalent to the predicate device or predicate devices, the subject device may be cleared for marketing. FDA’s 510(k) clearance pathway generally takes from three to twelve months from the date the application is completed, but can take significantly longer. Moreover, in January 2011, FDA announced twenty-five specific action items it intended to take to improve transparency and predictability of the 510(k) program. We anticipate that the changes may also result in additional requirements with which manufacturers will need to comply in order to obtain or maintain 510(k) clearance for their devices. These additional requirements could increase the costs or time for manufacturers’ seeking marketing clearances through the 510(k) process. Moreover, the 510(k) process could result in a not-substantially equivalent determination, in which case the device would be regulated as a Class III device, discussed below.
Class III devices are those devices which are deemed by FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, have a new intended use, or use advanced technology that is not substantially equivalent to that of a legally marketed device. Reasonable assurance of the safety and effectiveness of Class III devices cannot be assured solely by the general controls and the other requirements described above. These devices are required to undergo the PMA process in which the manufacturer must demonstrate reasonable assurance of the safety and effectiveness of the device to FDA’s satisfaction. A PMA application must provide extensive preclinical and clinical trial data and also information about the device and its components regarding, among other things, device design, manufacturing and labeling. Premarket approval applications (and supplemental pre-market approval applications) are subject to significantly higher user fees than are 510(k) pre-market notifications. After approval of a PMA, a new PMA or PMA supplement is required in the event of a modification to the device, its labeling or its manufacturing process. The PMA process, including the gathering of clinical and nonclinical data and the submission to and review by FDA, can take several years.
-112-
Table of Contents
A clinical trial may be required in support of a 510(k) submission and generally is required for a PMA application. These trials generally require an effective Investigational Device Exemption from FDA for a specified number of patients, unless the product is exempt from Investigational Device Exemption requirements or deemed a non significant risk device eligible for more abbreviated Investigational Device Exemption requirements. The Investigational Device Exemption application must be supported by appropriate data, such as animal and laboratory testing results. Clinical trials may begin 30 days after the submission of the Investigational Device Exemption application unless FDA or the appropriate institutional review boards at the clinical trial sites place the trial on clinical hold.
After a device is placed on the market, regardless of the classification or pre-market pathway, it remains subject to significant regulatory requirements. Even if regulatory approval or clearance of a medical device is granted, FDA may impose limitations or restrictions on the uses and indications for which the device may be labeled and promoted. Medical devices may be marketed only for the uses and indications for which they are cleared or approved. Device manufacturers must also establish registration and device listings with FDA. A medical device manufacturer’s manufacturing processes and those of its suppliers are required to comply with the applicable portions of the Quality Systems Regulations, which cover the methods and documentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging and shipping of medical devices. Domestic facility records and manufacturing processes are subject to periodic unscheduled inspections by FDA. FDA also may inspect foreign facilities that export products to the United States.
Failure to comply with applicable regulatory requirements can result in enforcement action by FDA, which may include any of the following sanctions: warning letters, fines, injunctions, civil or criminal penalties, recall or seizure of current or future products, operating restrictions, partial suspension or total shutdown of production, denial of 510(k) clearance or PMA applications for new products, or challenges to existing 510(k) clearances or PMA applications.
We believe that our LDTs and, should we reach that point, our in vitro diagnostic test kits, would likely be regulated as either Class II or Class III devices. It is also possible that some may fall into one Class and some into the other. Accordingly, some level of premarket review—either a 510(k) or a PMA—would likely be required for each test. While the data requirements are typically greater for Class III devices, the data required for Class II devices has increased, and it is likely that some amount of clinical data (retrospective or prospective or both) would be required for either type of submission. Currently, FDA is undertaking a review of the adequacy of the 510(k) process. It is difficult to predict what changes may result, but it should be assumed that any changes will increase, not decrease, the regulatory requirements.
Health Insurance Portability and Accountability Act, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”)
Under the administrative simplification provisions of HIPAA, as amended by HITECH, the United States Department of Health and Human Services has issued regulations which establish uniform standards governing the conduct of certain electronic health care transactions and protecting the privacy and security of Protected Health Information used or disclosed by health care providers and other covered entities. For further discussion of HIPAA and the impact on our business, see the section entitled “Risk Factors—Risks Related to Our Business—We are required to comply with laws governing the transmission, security and privacy of health information that require significant compliance costs, and any failure to comply with these laws could result in material criminal and civil penalties.”
Federal, State and Foreign Fraud and Abuse Laws
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under a governmental payor
-113-
Table of Contents
program. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, waivers of co-payments, ownership interests and providing anything at less than its fair market value. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements within the health care industry, the U.S. Department of Health and Human Services has issued a series of regulatory “safe harbors.” These safe harbor regulations set forth certain provisions, which, if met, will assure health care providers and other parties that they will not be prosecuted under the federal Anti- Kickback Statute. Although full compliance with these provisions ensures against prosecution under the federal Anti-Kickback Statute, the failure of a transaction or arrangement to fit within a specific safe harbor does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Statute will be pursued. For further discussion of the impact of federal and state health care fraud and abuse laws and regulations on our business, see the section entitled “Risk Factors—Risks Related to Our Business—We are subject to federal and state health care fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.”
In addition to the administrative simplification regulations discussed above, HIPAA also created two new federal crimes: health care fraud and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from governmental payor programs such as the Medicare and Medicaid programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from governmental payor programs.
Finally, another development affecting the health care industry is the increased enforcement of the federal False Claims Act and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal governmental payor program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has defrauded the federal government by submitting a false claim to the federal government and permit such individuals to share in any amounts paid by the entity to the government in fines or settlement. In addition, various states have enacted false claim laws analogous to the federal False Claims Act, although many of these state laws apply where a claim is submitted to any third-party payor and not merely a governmental payor program. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $5,500 to $11,000 for each false claim.
Additionally, in Europe various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines, for individuals and/or companies committing a bribery offence. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United Kingdom, under the new Bribery Act 2010, which went into effect in July 2011, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act of 2010, faces imprisonment of up to 10 years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
Physician Referral Prohibitions
Under a federal law directed at “self-referral,” commonly known as the “Stark Law,” there are prohibitions, with certain exceptions, on Medicare and Medicaid payments for laboratory tests referred by
-114-
Table of Contents
physicians who personally, or through a family member, have an investment or ownership interest in, or a compensation arrangement with, the clinical laboratory performing the tests. A person who engages in a scheme to circumvent the Stark Law’s referral prohibition may be fined up to $100,000 for each such arrangement or scheme. In addition, any person who presents or causes to be presented a claim to the Medicare or Medicaid programs in violation of the Stark Law is subject to civil monetary penalties of up to $15,000 per bill submission, an assessment of up to three times the amount claimed and possible exclusion from participation in federal governmental payor programs. Bills submitted in violation of the Stark Law may not be paid by Medicare or Medicaid, and any person collecting any amounts with respect to any such prohibited bill is obligated to refund such amounts. Many states have comparable laws that are not limited to Medicare and Medicaid referrals.
Corporate Practice of Medicine
Numerous states have enacted laws prohibiting business corporations, such as us, from practicing medicine and employing or engaging physicians to practice medicine, generally referred to as the prohibition against the corporate practice of medicine. These laws are designed to prevent interference in the medical decision-making process by anyone who is not a licensed physician. Violation of these laws may result in civil or criminal fines, as well as sanctions imposed against us and/or the professional through licensure proceedings.
New Jersey and New York State Laboratory Licensing
Our laboratory is licensed and in good standing under both the New Jersey and the New York State Departments of Health standards. Our current licenses permit us to receive specimens obtained in those states.
New Jersey and New York state laws and regulations also establish standards for the day-to-day operations of clinical laboratories, including physical facility requirements and equipment and quality control. New York standards include proficiency testing requirements, even for a laboratory not located in New York. In addition, the New York Department of Health separately approves certain LDTs offered in New York State. The Company has obtained the requisite approvals for its LDTs. If we are found to be out of compliance with New Jersey or New York statutory or regulatory standards we may be subject to suspension, restriction or revocation of our laboratory license or assessed civil money penalties. A noncompliant laboratory may also be found guilty of a misdemeanor under New Jersey and New York laws. A finding of noncompliance, therefore, may result in harm to our business.
Other States’ Laboratory Testing
In addition to New York, several other states require the licensure of out-of-state laboratories that accept specimens from those states, even though we are physically located in New Jersey. We have obtained licenses in these states and believe we are in compliance with their applicable licensing laws.
From time to time, other states may require out of state laboratories to obtain licensure in order to accept specimens from the state. If we identify any other state with such requirements or if we are contacted by any other state advising us of such requirements, we intend to follow instructions from the state regulators as to how we should comply with such requirements.
Other Regulatory Requirements
Our laboratory is subject to federal, state and local regulations relating to the handling and disposal of regulated medical waste, hazardous waste and biohazardous waste, including chemical, biological agents and compounds, blood and bone marrow samples and other human tissue. Typically, we use outside vendors who are contractually obligated to comply with applicable laws and regulations to dispose of such waste. These vendors are licensed or otherwise qualified to handle and dispose of such waste.
-115-
Table of Contents
OSHA has established extensive requirements relating to workplace safety for health care employers, including requirements to develop and implement programs to protect workers from exposure to blood-borne pathogens by preventing or minimizing any exposure through needle stick or similar penetrating injuries.
Segment and Geographical Information
We operate in one reportable business segment and derive revenue from multiple countries, with 99% and 97% coming from the United States in fiscal year 2010 and 2011, respectively.
Employees
As of September 30, 2012, we had a total of 48 full-time and four part-time employees, 54% of whom hold graduate degrees including 15 doctorate degrees and 13 of whom are engaged in full-time research and development activities. We plan to expand production, sales and marketing and our research and development programs, and we plan to hire additional staff as these initiatives are implemented. None of our employees are represented by a labor union, and we consider our employee relations to be good. Our workplace environment was recognized by being named #20 nationwide by The Scientist in “Best Places to Work Industry, 2011”.
Properties
As of September 30, 2012, we had a lease for approximately 17,936 square feet of space in Rutherford, New Jersey for use as a clinical reference laboratory and corporate headquarters. This lease expires in February 2018. Based on our current operational plans, we believe that such facilities are adequate for our operations for the near future.
Legal Proceedings
Between 2000 and July 2004, we operated a clinical laboratory in Milford, Massachusetts. That clinical laboratory provided a variety of cytogenetic testing services, including analyzing cells for genetic indicators of certain cancers. In accordance with our then existing policies, as well as the guidelines of the American College of Genetics, certain of these analyses were performed by karyotyping the patient’s genetic material. The services we provided were paid for by a variety of third-party payors, including the Medicare program.
For services provided to patients who were covered by Medicare, we billed for the diagnostic tests performed on behalf of the Medicare beneficiaries. Between 2003 and 2005, we billed Medicare for approximately 1,400 diagnostic tests. These tests were billed using certain CPT codes that are intended to identify the services provided. Because no single CPT code comprehensively or accurately captures the services performed by us, we needed to use several different codes and modifiers to fully identify the services provided by us. On each occasion, the bill was for services actually performed by us. Claim forms with supporting documentation were submitted for processing and payment to NHIC Corporation, the CMS Medicare Part B contractor for New England. NHIC processed and approved, and CMS paid, all of such claims.
In February 2009, we were notified that the Office of the Inspector General of the U.S. Department of Health and Human Services and the United States Department of Justice (together, the “Government”) were considering commencing a civil False Claims Act action against us. The Government issued a subpoena to us in connection with this potential action. The Government contends that Medicare overpaid for the services provided by us, and that we filed erroneous claims in connection with the services rendered to Medicare beneficiaries. In particular, the Government contends that we billed Medicare for services that were medically unnecessary and that we improperly coded bills submitted from 2003 to 2004, thereby causing Medicare to overpay for the services rendered. The Government alleges that our conduct may have violated the federal False Claims Act. The Government has indicated that the alleged overpayment is equal to approximately $1.6 million in damages, plus interest. The False Claims Act contains a treble damages provision and provides for the payment of penalties in connection with each violation of the statute.
-116-
Table of Contents
The Government did not commence any litigation against us. In January 2012 we executed a Settlement Agreement with the United States Department of Justice. Pursuant to the Settlement Agreement, we neither admitted liability nor conceded that the claims of the Government are well founded. We paid the Government the sum of $1 million. In exchange for such payment, the Government agreed to release us from any civil monetary claim the Government has for the conduct described above under the common law theories of payment by mistake, unjust enrichment and fraud. No release is specifically given with respect to other liabilities, including liabilities under the False Claims Act, and administrative liabilities, including mandatory and permissive exclusion from federal health care programs; however, based on our understandings with government officials with whom we have negotiated such settlement, we do not expect the Government to pursue any further claims with respect to the matters described above. The Settlement Agreement was based in part on our representations to the Government about our financial condition as of September 30, 2011, and we have agreed that if we misrepresented our financial condition, the Government may pursue certain remedies against us.
On December 23, 2011, we also entered into a Settlement Agreement and Release with our insurer, RSUI Indemnity Company, whereby RSUI paid us $400,000 to release claims for coverage of the above matter. We used such settlement proceeds to fund part of the payment to the Government.
All claims that are associated with the Government’s contentions relate to our operations in Milford, Massachusetts prior to July 2004. We sold the Milford, Massachusetts laboratory in 2004 and subsequently relocated operations to New Jersey in 2005. None of the employees who worked in the Milford, Massachusetts laboratory are currently employed by us.
In the normal course of business, the Company may be involved in legal proceedings or threatened legal proceedings. We are not party to any legal proceedings or aware of any threatened legal proceedings which are expected to have a material adverse effect on our financial condition, results of operations or liquidity.
-117-
Table of Contents
Executive Officers and Directors
The following table sets forth the name, age and position of each of our directors and executive officers.
| Name |
Age | Position | Served as an Officer or Director Since | |||
| Raju S. K. Chaganti, Ph.D. |
79 | Chairman of the Board of Directors | 1999 | |||
| Panna L. Sharma |
41 | Chief Executive Officer and Director | 2010 | |||
| Elizabeth A. Czerepak |
57 | Chief Financial Officer | 2011 | |||
| Jane Houldsworth, Ph.D. |
53 | Vice President, Research and Development | 2007 | |||
| Edmund Cannon |
68 | Director | 2005 | |||
| Robert Kaufman |
63 | Director | 2011 | |||
| John Pappajohn |
84 | Director | 2008 | |||
| Andrew Pecora, M.D. |
55 | Director | 2004 | |||
| Franklyn G. Prendergast, M.D., Ph.D. |
67 | Director | 2012 | |||
| Tommy G. Thompson |
71 | Director | 2008 |
Our executive officers are elected by, and serve at the discretion of, our board of directors. There are no family relationships among any of our directors and executive officers. The business experience for the past five years (and, in some instances, for prior years) of each of our executive officers and directors is as follows:
Raju S.K. Chaganti, Ph.D.
Dr. Chaganti, 79, is our founder and has served as the chairman of our board of directors since the Company’s inception. Dr. Chaganti is an internationally recognized leader in cancer cytogenetics and molecular genetics. He is a co-discoverer of patents for the cloning of two genes rearranged in lymphoma translocations, BCL6 and BCL8, and an additional two patents for the detection of translocations for the FISH classification of kidney cancers. Dr. Chaganti currently is the incumbent of the William E. Snee Chair at the Memorial Sloan- Kettering Cancer Center, where he is on the faculty of the Department of Medicine and Cell Biology Program. He is a professor and the Gernster Sloan- Kettering Graduate School of Biomedical Sciences at Cornell University Medical College, New York, New York. He was the chief of Memorial Sloan-Kettering Cancer Center’s cytogenetics service, which he established in 1976 as one of the earliest genetically based cancer diagnostic services in the country.
Dr. Chaganti received a Ph.D. in biology (genetics) from Harvard University Graduate School of Arts and Sciences and completed his post-doctoral training at the Medical Research Council of Great Britain. Additionally, he completed a sabbatical in the Department of Tumor Biology at Karolinska Institute Stockholm, focusing on experimental murine and tumorgenesis as well as immunology. Dr. Chaganti is American Board of Medical Genetics certified in medical genetics, with a subspeciality in clinical cytogenetics.
We selected Dr. Chaganti to serve on our board of directors and as our chairman due to the perspective and extensive experience he brings as one of our founders, his over 35 years of experience in managing clinical cytogenetic laboratories and his renown as an international leader in the areas of cancer cytogenetics and molecular genetics.
Panna L. Sharma
Mr. Sharma, 41, became a member of our board of directors and our Chief Executive Officer in May of 2010. Mr. Sharma was at TSG Partners, a specialty life sciences consultancy and advisory company, from 2001
-118-
Table of Contents
to 2010, where he was the Managing Partner and founder. At TSG he led the development of strategic initiatives, corporate growth strategy and corporate turnarounds for both public and private companies. He also led over 70 buy and sell-side transactions for life sciences, healthcare and biopharma companies. At TSG, he established the Global Diagnostics Index, the Global Biotools Index and several other life science capital markets indices that are still used in the life science industry.
Prior to founding TSG, Mr. Sharma was the Chief Strategy Officer for iXL Enterprises, Inc. (“iXL”), a public e-business consultancy where he led strategy development and acquisitions activity and was part of the management team that aided in taking the company public in June 1999. At iXL, he also managed the specialty e-business strategy practices group that grew from under $4 million in revenue in 1998 to over $75 million in 2000. From 1996 to 1998, Mr. Sharma was a partner at Interactive Solutions, Inc., a marketing and strategy consultancy focused on health care and financial services in Cambridge, Massachusetts, that was sold to Omnicom, Inc., one of the largest global market analysis and marketing companies. Prior to that time, Mr. Sharma served as a consultant to Putnam Investment Management, LLC and Bank of America Corporation. Mr. Sharma has also served on the board of directors of EpicEdge, a health care and government focused IT services firm, from 2001 to 2003 and as chairman of the Advisory Board for EndoChoice, a global leader for the gastrointestinal treatment market from 2008 to 2010.
We selected Mr. Sharma to serve on our board of directors due to his extensive knowledge of our operations, competitive challenges and opportunities gained through his position as our President and Chief Executive Officer as well as his extensive experience as a strategic advisor to companies in the health care and life sciences industry.
Elizabeth A. Czerepak
Elizabeth A. Czerepak, 57, joined us as Chief Financial Officer in 2011. She has 18 years of pharmaceutical industry experience and nine years of venture capital experience in the biosciences industry. From 2009 to 2011, Ms. Czerepak founded and led BIOptima Advisors LLC, a consulting firm that provides business development, strategic planning and clinical advisory services to biotechnology and pharmaceutical companies. From 2000 to 2011, she was founding general partner of Bear Stearns Health Innoventures (“BSHI”), a $212 million venture capital fund that led investments in 13 biotechnology companies, seven of which she served as a board member. From 2000 - 2009, she also served as managing director of Bear, Stearns & Co., and later, JPMorgan, Inc. From 1982 to 2000, Ms. Czerepak held senior positions in licensing, business development and finance at BASF Pharma, Hoffmann-La Roche, Inc. and Merck & Co., Inc., where she led or supported over 30 licensing and M&A transactions. She holds an MBA from Rutgers University and a B.A. magna cum laude from Marshall University, and is a member of the Licensing Executives Society.
Jane Houldsworth, Ph.D.
Dr. Houldsworth, 53, joined us in 2007 as head of Research and Development and was promoted to Vice President of Research and Development in June 2011. From 2007 to June 2011, Dr. Houldsworth also served as a consultant to Memorial Sloan-Kettering Cancer Center. She has a long-standing interest in the biology and genetics of lymphoma and male germ cell tumors, with over 20 years’ experience in translational research. Dr. Houldsworth is a co-inventor on four patents. These patents relate to methods for detecting HPV-associated cancers, a panel for detecting and differentiating renal cortical neoplasms, tool for diagnosing and prognosing mature B-cell neoplasms and methods and tools for diagnosing urogenital cancers and precancers in females. Dr. Houldsworth has published 15 book chapters and more than 50 peer-reviewed papers and is a reviewer for several scientific journals. She actively consults on academic research projects and is an active member of the American Society of Hematology and the American Association for Cancer Research. Dr. Houldsworth has been awarded several grants from the National Institutes of Health, the Lance Armstrong Foundation and other private foundations. In 2005, Dr. Houldsworth attained a New York State Certificate of
-119-
Table of Contents
Qualification as a laboratory director for oncology, molecular and cellular tumor markers. Prior to joining us in 2007, she was an Associate Attending geneticist and an associate laboratory member at the Memorial Sloan-Kettering Cancer Center in Dr. Chaganti’s laboratory. Dr. Houldsworth has a Ph.D. in biochemistry from University of Queensland, Australia and received post-doctoral training in molecular biology at the California Institute of Technology, Pasadena.
Edmund Cannon
Edmund Cannon, 68, is a member of our board of directors and is founder and President of the Clinical Research Center of Cape Cod, which specializes in finding institutional review board approved, consented specimens for the diagnostics and pharmaceutical industries, and in setting up studies to support FDA submissions for pharmaceutical and biotechnology companies. Previously, Mr. Cannon was a marketing and operations consultant for Franey Medical Labs. Mr. Cannon also formerly had the most national sales for Pharmacia Diagnostics Inc., and was a vice president and co-founder of Alletess, Inc. Mr. Canon has a degree from Boston State College and attended a Master’s program at Providence College.
We selected Mr. Cannon to serve on our board of directors due to his extensive experience in working with hospitals and oncologists and his world class expertise in clinical trials. Mr. Cannon also serves on our audit committee.
Robert Kaufman
Robert Kaufman, 63, is a member of our board of directors and serves as President and Chief Operating Officer of Oakley Investments, Inc., a private income and consulting firm. From April 2007 to April 2012, he served as the Senior Vice President and Chief Operating Officer of Outcome Sciences, Inc., a wholly owned subsidiary of Quintiles Pharma, Inc. Since 2002, Mr. Kaufman has served as Director and Chairman of the audit committee of Berkshire Income Realty, Inc. From January 2003 through March 2007 Mr. Kaufman served as President and Chief Operating Officer of Oakley Investments, Inc. In 2000, Mr. Kaufman founded Medeview, Inc., a healthcare services company, and served as its Chief Executive Officer until 2002. From 1996 through 1999 Mr. Kaufman served as Chief Executive Officer of a senior housing company known as Carematrix Corp. Mr. Kaufman worked for Coopers & Lybrand, LLP (now known as PricewaterhouseCoopers, LLP), an international accounting and consulting firm, from 1972-1996. During his tenure at Coopers & Lybrand, Mr. Kaufman was a partner from 1981 to 1996, primarily serving the healthcare, retail and real estate industries, and served as a member of the national Board of Partners. In addition, while a partner at Coopers & Lybrand, Mr. Kaufman was a member of the Mergers and Acquisitions and Real Estate Groups, the Associate Chairman of the National Retail and Consumer Products Industry Group and was a National Technical Consulting Partner. Mr. Kaufman received his MBA from Cornell University and his B.A. from Colby College.
We selected Mr. Kaufman to serve on our board of directors and as chairman of our audit committee due to his extensive experience in accounting and auditing matters, his experience serving on the boards of public companies, and his experience in the healthcare industry. As a former partner of an international accounting firm, and in light of his service on the audit committee of another publicly traded company, Mr. Kaufman brings financial and accounting expertise to our board of directors and audit committee.
John Pappajohn
Mr. Pappajohn, 84, is a member of our board of directors and is a pioneer in the venture capital industry. In 1969, Mr. Pappajohn founded Equity Dynamics, Inc., a financial consulting entity, and Pappajohn Capital Resources, a venture capital firm, both in Des Moines, Iowa. Mr. Pappajohn has been involved in over 100 start-up companies and has served as a director of over 40 public companies, many in the bioscience and health-related industries. He currently serves on the boards of the following public companies: American CareSource Holdings, Inc., since 2004, and CNS Response, since 2009.
-120-
Table of Contents
Previously, Mr. Pappajohn served on the boards of ConMed Healthcare Management, Inc. from 2005 until August 2012, PharmAthene, Inc., from 2007 until July 2011, Careguide, Inc., from 1995 until 2010, and SpectraScience, Inc., from 2007 until 2009. Mr. Pappajohn has a BSC degree in business from the University of Iowa.
Mr. Pappajohn was selected to serve on our board of directors due to his extensive background and experience in the venture capital industry, providing guidance to a variety of private and public companies in the bioscience and health related industries.
Andrew Pecora, M.D.
Dr. Pecora, 55, is a member of our board of directors and currently serves at the John Theurer Cancer Center at Hackensack University Medical Center as Chief Innovations Officer, Professor and Vice President of Cancer Services. From 2001 to 2011, Dr. Pecora served as the Chairman and Director of the John Theurer Cancer Center at Hackensack University Medical Center. Since 1996, he has been Co-Managing Partner of the Northern New Jersey Cancer Center, which is a private physicians practice group affiliated with Hackensack University Medical Center. In January 2012, this Center was consolidated with other oncology-focused groups and is now part of a network called Regional Cancer Care Associates. Since August 17, 2011, Dr. Pecora has served as Chief Medical Officer of NeoStem, Inc., which acquired Progenitor Cell Therapy, LLC in 2011. Prior to such acquisition, Dr. Pecora had served from 1999 to 2011 as Chairman, Chief Executive Officer and Chief Medical Officer of Progenitor Cell Therapy and as a member of the board of managers of Progenitor Cell Therapy. In December 2011, Dr. Pecora was appointed to the board of directors of NeoStem.
Dr. Pecora has also been a Professor of Medicine at the University of Medicine and Dentistry of New Jersey since 2004. Additionally, Dr. Pecora is a scientific advisor for numerous state, national and international organizations. He is a Diplomate of the American Board of Internal Medicine, subspecialty of hematology and subspecialty of oncology, a member of the National Blue Cross and Blue Shield Quality Centers for Transplant Experts Panel, a fellow of the Academy of Medicine of New Jersey, a fellow of the American College of Physicians and a member of the American Society of Bone Marrow Transplantation, American Society of Clinical Oncology and American Society of Hematology. Dr. Pecora co-founded and served as Chairman and Chief Scientific Officer of Amorcyte, Inc., a biotechnology company developing cell therapies for cardiovascular disease that was acquired by NeoStem in 2011. He serves as chairman of the board of Tetralogics, Inc., a company developing small molecules to treat cancer. He has served on the board of directors of the American Society of Bone Marrow Transplant and Cytotherapy and was a member of Accreditation Committee of the Foundation for Accreditation of Hematopoietic Cell Therapy. He has been a member of several National Heart, Lung and Blood Institute/National Cancer Institute state of the science meetings in transplantation and stem cell therapies. Dr. Pecora is actively involved as principal investigator and coinvestigator in many national research studies. He has been invited to present his work at various scientific meetings and continues to contribute to the published literature. Dr. Pecora received his medical degree from the University of Medicine and Dentistry of New Jersey. He went on to complete his medical education in internal medicine at New York Hospital and in hematology and oncology at Memorial Sloan-Kettering Cancer Center. He is board certified in internal medicine, hematology and oncology.
We selected Dr. Pecora to serve on our board of directors due to his knowledge of the indications for diagnostic and prognostic genomic testing in medical oncology and hematology. He is also an expert in medical reimbursement policies of insurance companies developed through his role as chairman of the John Theurer Cancer Center, and is renowned as an expert in cancer care policy and has experience as a founding chief executive officer of a biopharma company focused on cell therapy services and development cell-based therapeutics.
Franklyn G. Prendergast, M.D., Ph.D.
Franklyn G. Prendergast, M.D., Ph.D., 67, is a member of our board of directors and is the Edmond and Marion Guggenheim Professor of Biochemistry and Molecular Biology and Professor of Molecular
-121-
Table of Contents
Pharmacology and Experimental Therapeutics at Mayo Medical School and the director of the Mayo Clinic Center for Individualized Medicine. From 1994 to 2006, he served as a director of Mayo Clinic Cancer Center. He has held several other teaching positions at the Mayo Medical School since 1975. Dr. Prendergast has served for the National Institute of Health on numerous study section review groups; as a charter member of the Board of Advisors for the Division of Research Grants, now the Center for Scientific Review; the National Advisory General Medical Sciences Council; and the Board of Scientific Advisors of the National Cancer Institute. He held a Presidential Commission for service on the National Cancer Advisory Board. Dr. Prendergast also has served in numerous other advisory roles for the National Institute of Health and the National Research Council of the National Academy of Sciences, and he is a member of the board of directors of the Translational Genomics Research Institute and the Infectious Disease Research Institute (IDRI). Dr. Prendergast has served on the board of directors of Eli Lilly & Co. since 1995 and is a member of the board’s science and technology and public policy and compliance committees. He also currently serves on the board of directors for DemeRx, Inc., a private, biotechnology drug development company, and Ativa Medical Corporation, a private, diagnostic technology company. Dr. Prendergast obtained his medical degree with honors from the University of West Indies and attended Oxford University as a Rhodes Scholar, earning an M.A. degree in physiology. He obtained his Ph.D. in Biochemistry at the University of Minnesota.
We selected Dr. Prendergast to serve on our board of directors due to his extensive experience and expertise as a medical clinician, researcher and academician, particularly, in the areas of oncology and personalized medicine, developed through his roles with Mayo Clinic, including serving as director of the Mayo Clinic Cancer Center and the Mayo Clinic Center for Individualized Medicine.
Tommy G. Thompson
Mr. Thompson, 71, is a member of our board of directors and is the former Health and Human Services Secretary of the United States. He served as the Governor of Wisconsin for four terms. Mr. Thompson is building on his experience as Health and Human Services Secretary to develop innovative solutions to the health care challenges facing American families, businesses, communities, states and the nation as a whole. From 2005 until 2009, he served as a senior advisor at the consulting firm Deloitte and Touche USA LLP and the founding independent chairman of the Deloitte Center for Health Solutions, which researches and develops solutions to some of our nation’s most pressing health care and public health related challenges. From 2005 to early 2012, Mr. Thompson served as a senior partner at the law firm of Akin, Gump, Strauss, Hauer, & Feld LLP. Mr. Thompson served as chairman of the board of Logistics Health, Inc. from January 2011 to June 2011, and served as president from February 2005 to January 2011. He also serves on the board of directors of the following public companies: CareView Communications, Inc., as chairman of the board since 2005, Centene Corporation, C.R. Bard, Inc., since 2005 and United Therapeutics Corporation, since 2010. Mr. Thompson was formerly a director of AGA Medical Corporation, CNS Response, Inc., PURE Bioscience, SpectraScience, Inc., VeriChip Corporation, Cytori Therapeutics, Inc. and TherapeuticsMD, Inc. Mr. Thompson received his B.S. and J.D. from the University of Wisconsin-Madison.
We selected Mr. Thompson to serve on our board of directors due to his extensive experience with and knowledge of the health care issues facing the United States and the operational activities of companies within the health care industry as well as his public company board experience. Mr. Thompson’s legal experience is also useful in the board’s oversight of our legal and regulatory compliance. Mr. Thompson also serves on our audit committee.
Director Independence
Upon the completion of this offering, our common stock will be listed on the Nasdaq Capital Market. Under the rules of The Nasdaq Stock Market, independent directors must comprise a majority of a listed company’s board of directors within twelve months of the completion of an initial public offering. In addition,
-122-
Table of Contents
the rules of The Nasdaq Stock Market require that, (i) on the date of the completion of the offering, at least one member of each of a listed company’s audit, compensation and nominating and corporate governance committees be independent, (ii) within 90 days of the date of the completion of the offering, a majority of the members of such committees be independent and (iii) within one year of the date of the completion of the offering, all the members of such committees be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under the rules of The Nasdaq Stock Market, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
In order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our board of directors undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our board of directors has determined that Mr. Cannon, Mr. Kaufman, Dr. Prendergast and Mr. Thompson, representing four of our eight directors, do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the rules of The Nasdaq Stock Market. We intend to seek additional independent directors to achieve a majority of independent directors within twelve months of the completion of this offering.
Our board of directors also determined that (i) Messrs. Kaufman, Cannon and Thompson, who compose our audit committee, (ii) two of the three directors who serve on our compensation committee, Messrs. Cannon and Kaufman, and (iii) one of the two directors who serve on our nominating and corporate governance committee, Mr. Kaufman, each satisfy the independence standards for those committees established by the applicable rules and regulations of the SEC and The Nasdaq Stock Market. In making this determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director. We intend to comply with the other independence requirements for committees within the time periods specified above.
Scientific Advisory Board
Our Scientific Advisory Board comprises many preeminent scientists and physicians from the fields of cancer biology, cancer pathology, cancer medicine and molecular genetics. We have scientists and clinicians from leading cancer centers, including Memorial Sloan-Kettering Cancer Center, Mt. Sinai Sloan Kettering Cancer Center and the Institute for Cancer Genetics at Columbia University. These distinguished scientists and clinicians help oversee and review the scientific innovation, integrity and clinical relevancy of our program. Our board of directors appoints members to the Scientific Advisory Board for a term of one year, renewable at their option. The current Scientific Advisory Board includes:
Andrea Califano, Ph.D.
Dr. Califano co-founded Therasis, Inc. in 2008, where he now serves as a director. Dr. Califano serves as associate director at the Herbert Irving Comprehensive Cancer Center at Columbia University. He serves as director of Center for the Multi-scale Analysis of Genetic and Cellular Networks. He has more than 20 years of experience in both industry and academia. Since 1998, he has been especially active in the development of integrative methodologies for the dissection of dysregulated pathways in human B-cell lymphomas. In 2000, he
-123-
Table of Contents
co-founded First Genetic Trust, Inc. and served as its Chief Technology Officer. Dr. Califano served as Head of Computational Biology and Structural/Functional Genomics at IBM. He served as a member of the Scientific Management Committee at TSC. Since 2003, he has been a professor of Biomedical Informatics at Columbia, where he also serves as the director and chair of the Columbia Initiative for Systems Biology, which includes the Sulzberger Columbia Genome Center and the Center for Computational Biology and Bioinformatics. Dr. Califano is an internationally recognized leader in computational biology and specifically, in cancer systems biology. He serves on numerous editorial and scientific advisory boards, including the Board of Scientific Advisors of the National Cancer Institute. Dr. Califano holds a Ph.D. in physics from the University of Florence.
Timothy A. Chan, M.D., Ph.D.
Dr. Chan is a member of Memorial Sloan-Kettering Cancer Center’s central nervous system disease-management team and of the Brain Tumor Center. He is a board certified radiation oncologist specializing in the use of stereotactic radiosurgery, intensity modulated radiation therapy and conformal radiation therapy. In addition to treating patients, Dr. Chan is a Principal Investigator at a cancer genetics laboratory in the Human Oncology and Pathogenesis Program at Memorial Sloan-Kettering Cancer Center, which is focused on the genetics of cancer and using its findings on the cancer genome to develop better diagnostic and treatment approaches that will improve the care of cancer patients.
Riccardo Dalla-Favera, M.D.
Dr. Dalla-Favera has been the director of the Institute for Cancer Genetics at Columbia University since 1999 and the Percy and Joanne Uris Professor of Pathology and Genetics and Development at Columbia University Medical School. He is a member of the scientific advisory boards of the Yale Cancer Center of the Albert Einstein College of Medicine Cancer Center and of the Lymphoma Research Foundation. From 2005 to October 2011, Dr. Dalla-Favera served as a member of our board of directors. From 2005 to April 2011, Dr. Dalla-Favera served as a director of Callisto Pharmaceuticals Inc., a public company. He also founded and served as a director on our board until October 2011 and as a director of Therasis, Inc., a privately held company, from 2008 through March 2011. Dr. Dalla-Favera earned an M.D. from the University of Milan.
Hans-Guido Wendel, M.D.
Dr. Wendel is a Principal Investigator at the Cancer Genetics Laboratory at Memorial Sloan-Kettering Cancer Center, which is focused on the genetics of cancer and using its findings on the cancer genome to develop better diagnostic and treatment approaches that will improve the care of cancer patients. His research is focused on modeling relevant genetic changes found in human cancer specimens in cell culture and in accurate mouse models, in particular, using the technique of adoptive transfer of retrovirally transduced hematopoietic stem cells.
Vundavalli V. Murty, Ph.D.
Dr. Murty is currently director of the Cancer Cytogenetic Laboratory and Molecular Pathology at Columbia University. He is an associate editor of the journal Molecular Cancer and a diplomate of the American Board of Medical Genetics in the subspecialty of Clinical Cytogenetics. Dr. Murty has authored over 100 original research papers, book chapters and reviews covering human and murine cancers. Dr. Murty’s laboratory investigates the genetic mechanisms involved in testicular germ cell tumor and cervical cancer.
Andrew D. Zelenetz, M.D., Ph.D.
Dr. Zelenetz is chief of lymphoma service and head of the Molecular Hemo-oncology Laboratory in the Department of Medicine at Memorial Sloan-Kettering Cancer Center. He has published nearly 100 papers in lymphoma research and is board certified in internal medicine and medical oncology. In addition to several national honors, Dr. Zelenetz serves on the committees of the Leukemia and Lymphoma Society of America,
-124-
Table of Contents
Lymphoma Research Foundation and the National Cancer Institute. He was listed by New York Magazine as one of the top cancer clinicians in the metropolitan New York area in 2008.
Key Consultants
Dr. Wang
Dr. Lan Wang is a key consultant and serves as the medical director for our reference laboratory. Upon joining us, Dr Wang monitored the set up of our laboratory. Our laboratory has grown significantly since Dr. Wang’s arrival, both in volume and testing. She assisted in maturing our focus to become a full-service laboratory that targets hematological oncologists and pathologists. As Medical Director, Dr. Wang is responsible for supervising all compliance and operational aspects of our reference laboratory, including order testing for summation cases based on clinical information, reflex testing based on results, interpreting and diagnosing all flow and surgical specimens, summation reporting, performing internal and external correlation studies, reviewing and approving all standard operating procedures and reviewing and approving all validations.
Dr. Wang began working with us in 2007. Her career focus is in diagnostic hematopathology, centered on lymphomas and leukemias. In 1999, Dr. Wang joined as a clinical fellow at Harvard Medical School. She received residency training in anatomical and clinical pathology from 1999 to 2003 at Massachusetts General Hospital. From 2003 to 2004, Dr. Wang finished her fellowship training in hematopathology with Dr. Nancy L. Harris at Massachusetts General Hospital. From 2004 to date, Dr. Wang has held the position of staff pathologist and hematophathologist and serves as a cancer liaison physician at Chilton Memorial Hospital in New Jersey.
Dr. Wang is an active member of the Society of Hematopathology, the United States and Canadian Academies of Pathology and the College of American Pathologists. Her work has been published in numerous peer-reviewed publications. Dr. Wang is certified by the American Board of Pathology in anatomical and clinical pathology, as well as hematopathology. In New Jersey, Dr. Wang holds a medical license and bioanalytical laboratory director license from the board of medical examiners. She also has a certificate of qualification from New York State as a laboratory director in histopathology, cytopathology, hematology, immunohematology, oncology-molecular and cellular tumor markers, and cellular immunology-malignant leukocyte immunophenotyping.
-125-
Table of Contents
Summary Compensation Table
The following table shows the compensation awarded to or earned by our principal executive officer and our two most highly compensated executive officers who were serving as executive officers as of December 31, 2012. The persons listed in the following table are referred to herein as the “named executive officers.”
| Name and Principal Position |
Year | Salary ($) |
Bonus $ |
Option Awards ($)(1) |
All Other Compensation ($) |
Total ($) |
||||||||||||||||||
| Panna L. Sharma Chief Executive Officer and President |
|
2012 2011 |
|
$ $ |
350,000 350,000 |
|
$ |
— 175,000 |
(3)
|
|
— — |
|
$ $ |
26,950 87,168 |
(4) (5) |
$ $ |
376,950 612,168 |
| ||||||
| Elizabeth Czerepak(2) Chief Financial Officer |
|
2012 2011 |
|
$ $ |
250,000 224,437 |
|
$ |
— 125,000 |
(3)
|
$ |
— 340,000 |
|
|
— — |
|
$ $ |
250,000 689,437 |
| ||||||
| Jane Houldsworth Vice President Research and Development |
|
2012 2011 |
|
$ $ |
200,000 192,339 |
|
$ |
— 20,000 |
(3)
|
|
— — |
|
|
— — |
|
$ $ |
200,000 212,339 |
| ||||||
(1) Represents the aggregate grant date fair value for grants made in 2012 and 2011 computed in accordance with FASB ASC Topic 718. Unlike the calculations contained in our financial statements, this calculation does not give effect to any estimate of forfeitures related to service-based vesting, but assumes that the executive will perform the requisite service for the award to vest in full. The assumptions we used in valuing options are described in note 12 to our financial statements included in this prospectus.
(2) Ms. Czerepak commenced employment on January 24, 2011.
(3) In accordance with the terms of their respective employment agreements, each of the named executive officers is eligible for an annual cash bonus up to a specified percentage of his or her base salary. See the section entitled “Executive Compensation—Employment Agreements and Consulting Agreements” for a discussion of the terms of the employment agreements. The Compensation Committee of the Board of Directors has not yet determined the amounts of the cash bonuses for the year ended December 31, 2012, which are to be paid to the named executive officers.
(4) Consists of relocation allowance of $26,100 and $850 for life insurance benefits.
(5) Consists of costs for leasing a company car in the amount of $3,220, relocation and temporary living allowances of $80,124 and $3,824 for life insurance benefits.
Outstanding Equity Awards
The following table sets forth certain information, on an award-by-award basis, concerning unexercised options to purchase common stock and common stock that has not yet vested for each named executive officer and outstanding as of December 31, 2012.
| Option Awards | ||||||||||||||||
| Name |
Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities Underlying Unexercised Options (#) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date |
||||||||||||
| Panna L Sharma(1) |
281,500 | 193,500 | 5.00 | 3/31/2020 | ||||||||||||
| Elizabeth Czerepak(2) |
46,000 | 79,000 | 10.00 | 2/7/2021 | ||||||||||||
| Jane Houldsworth(3) |
26,125 | 14,375 | 1.92 | 1/18/2020 | ||||||||||||
(1) 45,000 shares vested immediately on the grant date, April 1, 2010. The remaining shares vest in 60 equal monthly installments of 7,166 shares commencing on the grant date.
(2) The shares vest in annual installments as follows: 22,000 in 2011, 24,000 in each of 2012, 2013, 2014 and 2015 and 7,000 in 2016.
(3) 6,000 shares vested immediately on the grant date, January 19, 2010. 6,900 shares, which represents 20% of the remaining shares, vested on the one-year anniversary of the grant date. The remaining shares vest in 48 equal monthly installments of 575 shares commencing one month following the one year anniversary of the grant date.
-126-
Table of Contents
Employment Agreements and Consulting Arrangements
Panna L. Sharma
We entered into an employment agreement as of December 28, 2011, effective as of April 1, 2010 (“CEO Agreement”), with Panna L. Sharma in connection with his appointment as our Chief Executive Officer and President. The CEO Agreement provides for, among other things: (i) an annual base salary of $350,000, or such greater amount as may be determined by the board, (ii) eligibility for an annual cash bonus of up to 50% of base salary, (iii) a one-time cash bonus of $100,000 upon completion of an initial public offering with gross proceeds of at least $25.0 million no later than December 31, 2012, and (iv) the following post-termination benefits: (a) any performance bonus plan, then in effect, pro rata for his period of actual employment during the year, payable at the regular bonus payment time but only if other employees are then paid their bonus amounts, and continuation of medical/dental, disability and life benefits for a period of six months following termination of employment and (b) monthly payments equal to his base salary immediately prior to such termination for a period of twelve months in the event his employment is terminated without “cause” or Mr. Sharma resigns for “good reason” not in connection with a “change of control” or in the event his employment is terminated due to injury, illness or disability, or (c) a lump sum payment equal to eighteen months of his then base salary plus an amount equal to the prior year bonus in the event his employment is terminated for any reason within twelve months following a change of control. The CEO Agreement further provides that Mr. Sharma will not engage in competitive activity, as set forth in the CEO Agreement, for a period of twelve months following termination of employment for any reason. The CEO Agreement also provides for reimbursement of reasonable expenses for travel between his then-current place of residence in Georgia and our office in New Jersey and reimbursement of direct relocation expenses of up to $50,000. We have recently agreed that we will pay Mr. Sharma $2,900 per month for up to twelve months to reimburse him for additional expenses incurred in renting his former residence. The monthly payments began in April 2012 and will terminate early if Mr. Sharma sells his former residence prior to the expiration of twelve month period. The CEO Agreement had an initial term through April 30, 2012, and automatically renews for additional one-year terms.
On December 29, 2012, the Compensation Committee of the Board of Directors approved a one-time cash bonus of $100,000 in the event we consummate an initial public offering with gross proceeds of at least $25.0 million no later than March 31, 2013.
On June 19, 2009, we entered into agreements with TSG pursuant to which TSG agreed to provide us with strategic consulting services. Our current Chief Executive Officer, Panna Sharma, was the managing partner, founder and majority owner of TSG and was actively involved in the consulting services provided to us pursuant to the June 19, 2009, consulting agreement. We compensated TSG an aggregate of $130,750 (excluding expenses) in 2009 and $97,575 (excluding expenses) in 2010 pursuant to such agreement. We also issued TSG warrants to purchase an aggregate of 10,075 shares of common stock at an exercise price of $5.00 in connection with the 2009 consulting agreement. These warrants were granted on April 1, 2010.
On September 23, 2010, we entered into a three-month consulting agreement with TSG for a fixed fee of $60,000 ($45,000 payable in cash and $15,000 payable in warrants) plus up to $5,000 of expenses. While Mr. Sharma retains a majority ownership position with TSG, pursuant to certain agreements between Mr. Sharma and TSG, Mr. Sharma did not and will not profit from the services provided under such agreement, as the operating agreement for TSG was amended following Mr. Sharma’s appointment as our Chief Executive Officer to preclude his participation in profits for consulting engagements in the life sciences industry. The project was subsequently reduced in scope and a revised total payment of $22,500 in cash (excluding expenses) was agreed upon and paid; no additional warrants were issued.
Elizabeth Czerepak
In January 2011, Elizabeth Czerepak was named Chief Financial Officer. We entered into an employment agreement effective as of January 1, 2012 (“CFO Agreement”) with Ms. Czerepak. The CFO
-127-
Table of Contents
Agreement provides for, among other things: (i) an annual base salary of $250,000, or such greater amount as may be determined by the board, (ii) eligibility for an annual cash bonus of up to 50% of base salary, (iii) a one-time cash bonus of $50,000 upon completion of an initial public offering with gross proceeds of at least $25.0 million no later than December 31, 2012, and (iv) the following post-termination benefits: (a) any performance bonus plan, then in effect, pro rata for her period of actual employment during the year, payable at the regular bonus payment time but only if other employees are then paid their bonus amounts, and continuation of medical/dental, disability and life benefits for a period of six months following termination of employment and (b) monthly payments equal to her base salary immediately prior to such termination for a period of six months in the event her employment is terminated without “cause” or Ms. Czerepak resigns for “good reason” not in connection with a “change of control”, (c) monthly payments equal to her base salary immediately prior to such termination for a period of twelve months in the event her employment is terminated due to illness, injury or disability or (d) a lump sum payment equal to twelve months of her then base salary plus an amount equal to the prior year bonus in the event her employment is terminated for any reason within twelve months following a change of control. The CFO Agreement further provides that Ms. Czerepak will not engage in competitive activity, as set forth in the CFO Agreement, for a period of twelve months following termination of employment for “cause” or due to illness, injury or disability or for a period of six months following termination of employment without “cause” or resignation for “good reason” or without “good reason”. The CFO Agreement has an initial term of January 1, 2012 through December 31, 2012, and automatically renews for additional one-year terms.
On December 29, 2012, the Compensation Committee of the Board of Directors approved a one-time cash bonus in the amount of $50,000 in the event we consummate an initial public offering with gross proceeds of at least $25.0 million no later March 31, 2013.
Jane Houldsworth, Ph.D.
We entered into an employment agreement with Dr. Houldsworth effective as of January 1, 2012 (“VP Agreement”). The VP Agreement provides for, among other things: (i) an annual base salary of $200,000, or such greater amount as may be determined by the board, (ii) eligibility for an annual cash bonus of up to 25% of base salary, and (iii) the following post-termination benefits: (a) any performance bonus plan, then in effect, pro rata for her period of actual employment during the year, payable at the regular bonus payment time but only if other employees are then paid their bonus amounts, and continuation of medical/dental, disability and life benefits for a period of six months following termination of employment and (b) monthly payments equal to her base salary immediately prior to such termination for a period of six months in the event her employment is terminated without “cause” or Dr. Houldsworth resigns for “good reason” not in connection with a “change of control”, (c) monthly payments equal to her base salary immediately prior to such termination for a period of twelve months in the event her employment is terminated due to illness, injury or disability or (d) a lump sum payment equal to twelve months of her then base salary plus an amount equal to the prior year bonus in the event her employment is terminated for any reason within twelve months following a change of control. The VP Agreement further provides that Dr. Houldsworth will not engage in competitive activity, as set forth in the VP Agreement, for a period of twelve months following termination of employment for “cause” or due to illness, injury or disability or for a period of six months following termination of employment without “cause” or resignation for “good reason” or without “good reason”. The VP Agreement has an initial term of January 1, 2012 through December 31, 2012, and automatically renews for additional one-year terms.
On January 10, 2010, we issued a convertible promissory note in favor of Dr. Houldsworth, which obligated us to pay her the sum of $55,000, together with interest at the rate of 5.5% per annum, on or before January 10, 2011. The $55,000 represented fees owed to Dr. Houldsworth pursuant to a certain consulting agreement between Dr. Houldsworth and us. We have repaid the principal, plus interest in the amount of $969.82.
-128-
Table of Contents
Lan Wang, M.D., Consultant
We entered into that certain Medical Director Agreement dated as of October 9, 2009, with Lan Wang, M.D. (“Medical Director Agreement”). The Medical Director Agreement has an initial one-year term and automatically renews for additional one-year terms, unless terminated by either party, in writing, within 90 days of the end of the applicable term. The Medical Director Agreement provides for, among other things: (i) the engagement of Dr. Wang to provide consulting services as our Medical Director, including, without limitation, to supervise and monitor our commercial clinical cytogenetics and molecular laboratory to ensure compliance with applicable regulations and (ii) an annual fee of $225,000 payable in equal monthly increments of $18,750.
Director Compensation
We are in the process of evaluating possible director compensation plans that would be appropriate for us as a public company. The non-employee directors did not receive any cash or equity compensation during 2010 and, except for Mr. Kaufman, during 2011.
As compensation for his service as a board member and as chair of our audit committee we issued Mr. Kaufman options to purchase 20,000 shares of our common stock at a purchase price of $13.52 per share. Such option vests in three equal annual installments commencing in December 2012. The aggregate grant date fair value of such option computed in accordance with FASB ASC Topic 718 was $180,400.
Certain non-employee directors entered into consulting agreements with us, as described below, pursuant to which they received cash compensation and equity for consulting services provided during 2012.
Pecora
On August 15, 2010, we entered into a two-year consulting agreement with Dr. Pecora, a member of our board of directors, pursuant to which Dr. Pecora received $5,000 per month for providing consulting and advisory services in connection with the following activities: (i) strategic guidance on the development of our laboratory service business and laboratory service portfolio, (ii) strategic guidance and facilitation with payors, insurers and third-party agencies on reimbursement for common tests, laboratory-developed tests and our portfolio of proprietary tests and (iii) gaining access to clinical advisory boards, peer review groups and ad-hoc clinical expert panels for the purpose of obtaining feedback on the development of our proprietary tests and service offerings in the field of advanced oncology testing and personalized medicine. Pursuant to the same consulting agreement, Dr. Pecora also received an option under our 2008 Stock Option Plan to purchase a total of 50,000 shares of our common stock at an exercise price of $10.00 per share, which vested over the two-year period ended on August 16, 2012. In August 2011, Dr. Pecora agreed to forego the monthly cash payments and such cash payments terminated effective immediately. This agreement expired on its terms and was not renewed.
Chaganti
On September 15, 2010, we entered into a three-year consulting agreement with Dr. Raju Chaganti, our chairman, pursuant to which Dr. Chaganti provides us with consulting and technical support services in connection with our technical and business affairs, including oversight of our clinical diagnostic laboratory. In consideration for Dr. Chaganti’s services, we pay Dr. Chaganti $5,000 per month and he received an option to purchase 150,000 shares of our common stock at a purchase price of $10.00 per share in connection with his execution of the consulting agreement. Such option vests in 12 quarterly installments of 12,500 shares commencing on October 1, 2010. Pursuant to his consulting agreement, Dr. Chaganti also assigned to us all rights to any inventions which he may invent during the course of rendering consulting services to us. In exchange for this assignment, if the U.S. Patent and Trademark Office issues a patent for an invention on which Dr. Raju Chaganti is listed as an inventor, we agreed to pay Dr. Chaganti (i) a one-time payment of $50,000 and (ii) 1% of the net revenues we receive from any licensed sales of the invention.
-129-
Table of Contents
Employee Benefit and Stock Plans
We have two equity incentive plans: the 2008 Stock Option Plan and the 2011 Equity Incentive Plan. Each plan is described separately below, followed by a description of certain federal income tax consequences with respect to plans of these types.
2008 Stock Option Plan
The following is a summary of the material terms of our amended and restated 2008 Stock Option Plan. This description is not complete. For more information, we refer you to the full text of the amended and restated 2008 Stock Option Plan, which we filed as an exhibit to the registration statement of which this prospectus forms a part.
The purposes of the 2008 Stock Option Plan are: (i) to attract and retain highly competent employees, board members, consultants and other advisors to serve our company and its affiliates, (ii) to provide additional incentives to such persons by aligning their interests with those of our shareholders and (iii) to promote the success and business of our company.
The 2008 Stock Option Plan authorizes the grant of the following types of awards: nonqualified stock options (“NSOs”) and incentive stock options (“ISOs”). Awards may be granted to employees, officers, non-employee board members, consultants and other service providers of our company and its affiliates. However, ISOs may be granted only to employees.
We have authorized a total of 1,375,000 shares of common stock for issuance pursuant to all awards granted under the 2008 Stock Option Plan. The number of shares issued or reserved pursuant to the 2008 Stock Option Plan (or pursuant to outstanding awards) is subject to adjustment as a result of mergers, consolidations, reorganizations, stock splits, stock dividends and other changes in our common stock. Shares subject to awards that have been terminated, expired unexercised, forfeited or settled in cash do not count as shares issued under the 2008 Stock Option Plan.
Performance Criteria. Vesting of awards granted under the 2008 Stock Option Plan may be subject to the satisfaction of one or more performance goals established by the board of directors. The performance goals may vary from participant to participant, group to group, and period to period. Performance goals may be weighted for different factors and measures.
Transferability. Unless otherwise determined by the board of directors, awards granted under the 2008 Stock Option Plan will not be transferable other than by will or by the laws of descent and distribution.
Change of Control. In the event of a change of control (as defined in the 2008 Stock Option Plan), each outstanding option will be assumed by the successor company or an equivalent option will be substituted by the successor company. If the successor company does not agree to assume or substitute the options, the vesting will accelerate and the options will become exercisable in full. In that case, the option must be exercised prior to the consummation of the change of control or it will terminate upon the change of control. If the options are assumed or substituted and a participant is involuntarily terminated within 24 months following the change of control, the vesting of the participant’s options will accelerate and they will become exercisable in full immediately prior to the date of the involuntary termination.
Effectiveness of the 2008 Stock Option Plan; Amendment and Termination. The 2008 Stock Option Plan became effective on April 9, 2008. The 2008 Stock Option Plan will remain available for the grant of awards until the tenth anniversary of the effective date. The board may amend, alter or discontinue the 2008 Stock Option Plan in any respect at any time, but no amendment may materially and adversely affect the rights of a participant under any awards previously granted, without his or her consent, except that stockholder approval will
-130-
Table of Contents
be needed for any amendment that would increase the maximum number of shares available for awards, reduce the exercise price of outstanding options, change the class of eligible participants, or if otherwise required by applicable law or stock market requirements. The 2008 Stock Option Plan permits us to reprice any stock option granted under the plan without the approval of our stockholders.
2011 Equity Incentive Plan
The following is a summary of the material terms of our amended and restated 2011 Equity Incentive Plan. This description is not complete. For more information, we refer you to the full text of the amended and restated Equity Incentive Plan, which we filed as an exhibit to the registration statement of which this prospectus forms a part.
The purposes of the Equity Incentive Plan are: (i) to attract and retain highly competent employees, board members, consultants and other advisors to serve our company and its affiliates (ii) to provide additional incentives to such persons by aligning their interests with those of our shareholders and (iii) to promote the success and business of our company.
The Equity Incentive Plan authorizes the grant of the following types of awards: NSOs, ISOs, stock appreciation rights (“SARs”), restricted stock, restricted stock units (“RSUs”), performance shares, performance units, other cash-based awards and other stock-based awards. Awards may be granted to employees, officers, non-employee board members, consultants and other service providers of our Company and its affiliates. However, ISOs may be granted only to employees.
We have authorized a total of 875,000 shares of common stock for issuance pursuant to all awards granted under the Equity Incentive Plan, including the RSUs. The number of shares issued or reserved pursuant to the Equity Incentive Plan (or pursuant to outstanding awards) is subject to adjustment as a result of mergers, consolidations, reorganizations, stock splits, stock dividends and other changes in our common stock. Shares subject to awards that have been terminated, expired unexercised, forfeited or settled in cash do not count as shares issued under the Equity Incentive Plan. No person may receive awards of stock options or SARs during any calendar year for more than 125,000 shares of our common stock (subject to adjustment for recapitalization or other changes in our common stock).
Administration. The Equity Incentive Plan will be administered by the Compensation Committee. The Compensation Committee will have the discretion to determine the individuals to whom awards may be granted under the Equity Incentive Plan, the number of shares of our common stock subject to each award, the type of award, the manner in which such awards will vest and the other conditions applicable to awards. The Compensation Committee will be authorized to interpret the Equity Incentive Plan, to establish, amend and rescind any rules and regulations relating to the Equity Incentive Plan and to make any other determinations that it deems necessary or desirable for the administration of the Equity Incentive Plan. All decisions, determinations and interpretations by the Compensation Committee, and any rules and regulations under the Equity Incentive Plan and the terms and conditions of or operation of any award, are final and binding on all participants. The 2011 Equity Incentive Plan permits us to reprice any stock option granted under the plan without the approval of our stockholders.
Stock Options. The Compensation Committee will determine the exercise price and other terms for each option and whether the options will be NSOs or ISOs. The exercise price per share of each option will not be less than 100% of the fair market value of our common stock on the date of grant (or 110% of fair market value in the case of an ISO granted to a 10% stockholder), which, unless otherwise determined by the Committee, will be deemed to be the closing price of a share of our common stock on its principal exchange on the last trading day before the grant date. ISOs may be granted only to employees and are subject to certain other restrictions. To the extent an option intended to be an ISO does not qualify as an ISO, it will be treated as an NSO. A participant may exercise an option by written notice and payment of the exercise price in shares, cash or a combination of shares
-131-
Table of Contents
and cash, as determined by the Compensation Committee, including an irrevocable commitment by a broker to pay over the net proceeds from a sale of the shares issuable under an option, the delivery of previously owned shares and/or withholding of shares deliverable upon exercise. The maximum term of any option granted under the Equity Incentive Plan is 10 years from the grant date (or five years in the case of an ISO granted to a 10% stockholder). The Compensation Committee may, in its discretion, permit a holder of an NSO to exercise the option before it has otherwise become exercisable, in which case the shares of our common stock issued to the recipient will continue to be subject to the vesting requirements that applied to the NSO before exercise.
Stock Appreciation Rights. The Compensation Committee may grant SARs independent of or in connection with an option. The Compensation Committee will determine the other terms applicable to SARs. The exercise price per share of each SAR will not be less than 100% of the fair market value of our common stock on the grant date, which, unless otherwise determined by the Committee, will be deemed to be the closing price of a share of our common stock on its principal exchange on the last trading day before the grant date. The price will be subject to adjustment for recapitalization or other changes in our common stock. The maximum term of any SAR granted under the Equity Incentive Plan will be 10 years from the grant date. Generally, each SAR will entitle a participant upon exercise to an amount equal to:
| • | the excess of the fair market value on the exercise date of one share of our common stock over the exercise price, multiplied by |
| • | the number of shares of common stock covered by the SAR. |
Payment may be made in shares of our common stock, in cash or partly in common stock and partly in cash, all as determined by the Compensation Committee.
Restricted Stock and Restricted Stock Units. The Compensation Committee will have the authority to award restricted common stock and/or RSUs under the Equity Incentive Plan. Restricted stock awards consist of shares of stock that are transferred to a participant subject to restrictions that may result in forfeiture if specified conditions are not satisfied. RSUs confer the right to receive shares of our common stock, cash or a combination of shares and cash, at a future date upon or following the attainment of certain conditions specified by the Compensation Committee. The Compensation Committee will determine the restrictions and conditions applicable to each award of restricted stock or RSUs, which may include performance-based conditions. Although we do not expect to declare any dividends in the foreseeable future, dividends with respect to restricted stock may be paid to the holder of the shares as and when dividends are paid to shareholders or at the time that the restricted stock vests, as determined by the Compensation Committee. Unless the Compensation Committee determines otherwise at the time the restricted stock award is granted, holders of restricted stock will have the right to vote the shares. The Equity Incentive Plan authorizes us to withhold from participants shares of common stock having a fair market value equal to our withholding obligation with respect to restricted stock and/or RSUs.
Performance Shares and Performance Units. The Compensation Committee may award performance shares and/or performance units under the Equity Incentive Plan. Performance shares and performance units are awards, denominated in shares of our common stock, cash or a combination thereof, which are earned during a specified performance period subject to the attainment of performance criteria, as established by the Compensation Committee. The Compensation Committee will determine the restrictions and conditions applicable to each award of performance shares and performance units.
Other Stock-Based and Cash-Based Awards. The Compensation Committee may award other types of equity-based or cash-based awards under the Equity Incentive Plan, including the grant or offer for sale of shares of our common stock that do not have vesting requirements and the right to receive one or more cash payments subject to satisfaction of such conditions as the Compensation Committee may impose.
Performance Criteria. Vesting of awards granted under the Equity Incentive Plan may be subject to the satisfaction of one or more performance goals established by the Compensation Committee. The performance
-132-
Table of Contents
goals may vary from participant to participant, group to group, and period to period. Performance goals may be weighted for different factors and measures.
Transferability. Unless otherwise determined by the Compensation Committee, awards granted under the Equity Incentive Plan will not be transferable other than by will or by the laws of descent and distribution.
Change in Control. The Compensation Committee may, at the time of the grant of an award provide for the effect of a change in control (as defined in the Equity Incentive Plan) on any award, including: (i) accelerating or extending the time periods for exercising, vesting in, or realizing gain from any award, (ii) eliminating or modifying the performance or other conditions of an award or (iii) providing for the cash settlement of an award for an equivalent cash value, as determined by the Compensation Committee. The Compensation Committee may, in its discretion and without the need for the consent of any recipient of an award, also take one or more of the following actions contingent upon the occurrence of a change in control: (a) cause any or all outstanding options and SARs to become immediately exercisable, in whole or in part, (b) cause any other awards to become non-forfeitable, in whole or in part, (c) cancel any option or SAR in exchange for a substitute option, (d) cancel any award of restricted stock, RSUs, performance shares or performance units in exchange for a similar award of the capital stock of any successor corporation, (e) redeem any restricted stock, RSU, performance share or performance unit for cash and/or other substitute consideration with a value equal to the fair market value of an unrestricted share of our common stock on the date of the change in control, (f) cancel any option or SAR in exchange for cash and/or other substitute consideration based on the value of our common stock on the date of the change in control, and cancel any option or SAR without any payment if its exercise price exceeds the value of our common stock on the date of the change in control or (g) make such other modifications, adjustments or amendments to outstanding awards as the Compensation Committee deems necessary or appropriate.
Recoupment of Awards. Each award under the Equity Incentive Plan is subject, in the discretion of the Compensation Committee, to termination, rescission, recapture and/or reimbursement if the award was based on performance criteria, the participant benefited from a calculation that later proves to be materially inaccurate or engaged in fraud or misconduct causing the need for a financial restatement and a lower granting, vesting or payment with respect to the award would have occurred based on a correct calculation or restated financial results.
Effectiveness of the Equity Incentive Plan; Amendment and Termination. The Equity Incentive Plan was approved by our board of directors on June 30, 2011 and thereafter ratified by our stockholders. The Equity Incentive Plan will remain available for the grant of awards until the tenth anniversary of the effective date. The board may amend, alter or discontinue the Equity Incentive Plan in any respect at any time, but no amendment may materially and adversely affect the rights of a participant under any awards previously granted, without his or her consent, except that stockholder approval will be needed for any amendment that would increase the maximum number of shares available for awards, reduce the exercise price of outstanding options or SARs, change the class of eligible participants, or if otherwise required by applicable law or stock market requirements.
Federal Income Tax Consequences
Following is a summary of the federal income tax consequences of option and other awards under the 2008 Stock Option Plan and 2011 Equity Incentive Plan. Optionees and recipients of other rights and awards granted under the 2008 Stock Option Plan or the Equity Incentive Plan are advised to consult their personal tax advisors before exercising an option, stock appreciation right or award or disposing of any stock received pursuant to the exercise of an option, stock appreciation right or award. In addition, the following summary is based upon an analysis of the Code as currently in effect, existing laws, judicial decisions, administrative rulings, regulations and proposed regulations, all of which are subject to change and does not address state, local or other tax laws.
-133-
Table of Contents
Treatment of Options. The Code treats incentive stock options and nonstatutory stock options differently. However, as to both types of options, no income will be recognized to the optionee at the time of the grant of the options under the 2008 Stock Option Plan or the Equity Incentive Plan.
Generally, upon exercise of a nonstatutory (or non-qualified) stock option (including an option intended to be an incentive stock option but which has not continued to so qualify at the time of exercise), an optionee will recognize ordinary income tax on the excess of the fair market value of the stock on the exercise date over the option price. In general, if an optionee, in exercising a nonstatutory stock option, tenders shares of our common stock in partial or full payment of the option price, no gain or loss will be recognized on the tender. However, if the tendered shares were previously acquired upon the exercise of an incentive stock option and the tender is within two years after the date of grant or within one year after the date of exercise of the incentive stock option, the tender will be a disqualifying disposition of the shares acquired upon exercise of the incentive stock option.
For incentive stock options, there is no taxable income to an optionee at the time of exercise. However, the excess of the fair market value of the stock on the date of exercise over the exercise price will be taken into account in determining whether the “alternative minimum tax” will apply for the year of exercise. If the shares acquired upon exercise are held until at least two years from the date of grant and more than one year from the date of exercise, any gain or loss upon the sale of such shares, if held as capital assets, will be long-term capital gain or loss (measured by the difference between the sales price of the stock and the exercise price). Under current federal income tax law, a long-term capital gain will be taxed at a rate which is less than the maximum rate of tax on ordinary income. If the two-year and one-year holding period requirements are not met (a “disqualifying disposition”), an optionee will recognize ordinary income in the year of disposition in an amount equal to the lesser of (i) the fair market value of the stock on the date of exercise minus the exercise price or (ii) the amount realized on disposition minus the exercise price. The remainder of the gain will be treated as long-term capital gain, depending upon whether the stock has been held for more than a year. If an optionee makes a disqualifying disposition, he or she will be obligated to notify us.
In general, if an optionee, in exercising an incentive stock option, tenders shares of our common stock in partial or full payment of the option price, no gain or loss will be recognized on the tender. However, if the tendered shares were previously acquired upon the exercise of another incentive stock option and the tender is within two years after the date of grant or within one year after the date of exercise of the other option, the tender will be a disqualifying disposition of the shares acquired upon exercise of the other option.
As noted above, the exercise of an incentive stock option could subject an optionee to the alternative minimum tax. The application of the alternative minimum tax to any particular optionee depends upon the particular facts and circumstances which exist with respect to the optionee in the year of exercise. However, as a general rule, the amount by which the fair market value of the common stock on the date of exercise of an option exceeds the exercise price of the option will constitute an item of “adjustment” for purposes of determining the alternative minimum taxable income on which the alternative tax may be imposed. As such, this item will enter into the tax base on which the alternative minimum tax is computed and may therefore cause the alternative minimum tax to become applicable in any given year.
Treatment of Stock Appreciation Rights. Generally, the recipient of a stock appreciation right will not recognize any income upon grant of the stock appreciation right. Upon exercise of a stock appreciation right, the holder will recognize ordinary income equal to the fair market value of our common stock at that time.
Treatment of Stock Awards. Generally, absent an election to be taxed currently under Section 83(b) of the Code (a “Section 83(b) Election”), there will be no federal income tax consequences to the recipient upon the grant of a restricted stock award. At the expiration of the restriction period and the satisfaction of any other restrictions applicable to the restricted shares, the recipient will recognize ordinary income equal to the fair market value of our common stock at that time. If a Section 83(b) Election is made within 30 days after the date the restricted stock award is granted, the recipient will recognize an amount of ordinary income at the time of the
-134-
Table of Contents
receipt of the restricted shares equal to the fair market value (determined without regard to applicable restrictions) of the shares of our common stock at such time. If a Section 83(b) Election is made, no additional income will be recognized by the recipient upon the lapse of restrictions on the shares (and prior to the sale of such shares), but, if the shares are subsequently forfeited, the recipient may not deduct the income that was recognized pursuant to the Section 83(b) Election at the time of the receipt of the shares.
The recipient of an unrestricted stock award will recognize ordinary income equal to the fair market value of our common stock that is the subject of the award when the award is made.
The recipient of a restricted stock unit will recognize ordinary income as and when the units vest. The amount of the income will be equal to the fair market value of the shares of our common stock issued at that time. The recipient of a restricted stock unit will not be permitted to make a Section 83(b) Election with respect to such award.
Treatment of Performance Share Awards, Performance Unit Awards, Other Cash-Based Awards and Other Stock-Based Awards. The federal income tax consequences of performance share awards, performance unit awards, other cash-based awards and other stock-based awards will depend on the terms and conditions of those awards.
Tax Withholding. We have the right to deduct or withhold, or require a participant to remit to us, the amount required to satisfy minimum statutory withholding requirements of federal, state and local tax laws and regulations (domestic or foreign) with respect to any taxable event arising as a result of the 2008 Stock Option Plan or the Equity Incentive Plan.
Inapplicability of Code Sections and ERISA. Sections 401(a) and 401(k) of the Code and the provisions of the Employee Retirement Income Security Act of 1974 (commonly known as “ERISA”) are not applicable to the 2008 Stock Option Plan or the Equity Incentive Plan.
Compensation Committee Interlocks and Insider Participation
Messrs. Cannon, Kaufman and Pappajohn served on our compensation committee during 2012. See the section entitled “Certain Relationships and Related Party Transactions” for a discussion of the transactions between Mr. Pappajohn and us. Prior to that our full board of directors performed compensation committee functions. Mr. Sharma, our President and Chief Executive Officer, served on our board of directors and participated in discussions regarding compensation of executive officers (other than himself). No compensation committee interlocks between us and another entity existed during the year ended December 31, 2012.
-135-
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Other than compensation arrangements for named executive officers and directors, we describe below each transaction and series of similar transactions, during our last three fiscal years, to which we were a party or will be a party, in which:
| • | the amounts involved exceeded or will exceed $120,000; and |
| • | any of our directors, executive officers or holders of more than 5% of our common stock, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Compensation arrangements for our named executive officers and directors are described in the section entitled “Executive Compensation”.
2012 Convertible Debt Financing Transaction
We entered into a restated Credit Agreement dated as of October 17, 2012 with John Pappajohn and Mark Oman for a $3.0 million convertible term loan. Mr. Pappajohn provided approximately $1.8 million of financing and Mr. Oman provided approximately $1.3 million of financing under the Credit Agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 26, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
The lenders received ten-year warrants to purchase an aggregate of 286,275 shares of our common stock (which were issued in proportion to their respective funding amounts), in each case at the lower of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering price per share is less than $17.00 per share. The lenders received additional ten-year warrants to purchase an aggregate of 17,648 shares of our common stock on the same terms because we did not consummate our initial public offering by November 27, 2012. The lenders will receive additional ten-year warrants to purchase an aggregate of 17,648 shares of our common stock (to be issued in proportion to their respective funding amounts) on the same terms in the event we do not consummate our initial public offering by February 22, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00, so that the initial grant would be for an aggregate of 695,237 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 811,110 shares and 608,333 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
On December 7, 2012, Mr. Pappajohn provided an additional $1.0 million of financing on the same terms as the restated credit agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on June 4, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
Mr. Pappajohn received ten-year warrants to purchase an aggregate of 58,824 shares of our common at the lesser of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering
-136-
Table of Contents
price per share is less than $17.00 per share. He will receive additional ten-year warrants to purchase an aggregate of 5,886 shares of our common stock on the same terms in the event we do not consummate our initial public offering by each the following dates: March 4, 2013 and June 4, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00 per share, so that the initial grant would be for an aggregate of 142,857 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 166,667 shares and 125,000 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
Shares that the lenders receive are subject to a lock-up agreement for 180 days after the consummation of our initial public offering on the same terms as other lock-up agreements in favor of the underwriters of this offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
December 2011 Financing Transaction
We entered into a Credit Agreement dated as of December 21, 2011, as amended and restated as of February 13, 2012, with John Pappajohn and Andrew Pecora (indirectly through an investment company), both members of our board of directors, and NNJCA Capital, LLC (“NNJCA”), a limited liability company of which Dr. Pecora is a member, for a $6.0 million secured term loan. Mr. Pappajohn has provided $4.0 million of financing, NNJCA Capital has provided $1.5 million of financing and Dr. Pecora has provided $500,000 of financing under the Credit Agreement.
The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 13, 2013, with an option, at our election, if there has been no event of default, to extend the loan term for an additional six months. We exercised our option to extend the maturity date to August 13, 2013. The term loan due to Dr. Pecora and NNJCA has a prepayment penalty in the aggregate amount of $190,000, less interest previously paid, if we prepay the loan prior to February 12, 2013. The lenders may require that the loan be repaid within 30 days should we complete our initial public offering and receive gross proceeds of at least $15.0 million. In the event that any lender requires payment upon completion of our initial public offering and certain other maturity events, the annual interest rate on any unpaid balance shall increase to 12%. The loan is secured by all of our assets, including our intellectual property, subject to prior first and second liens in favor of Wells Fargo Bank and DAM Holdings, LLC (“DAM”). Pursuant to an intercreditor agreement, the lenders have agreed that all amounts due to DAM are to be paid prior to payment to the lenders under this Credit Agreement, but that as between such lenders, following an event of default, all of the security granted by us is to be applied first to repay obligations due to Dr. Pecora and NNJCA, and then to Mr. Pappajohn after they have been paid in full. As Mr. Pappajohn has guaranteed the Wells Fargo debt, in essence under the intercreditor agreement, NNJCA and Dr. Pecora will be junior only to DAM.
Upon funding, the lenders received five-year warrants to purchase an aggregate of 211,764 shares of our common stock at the lower of (i) $17.00 per share and (ii) a 20% discount to the initial public offering price per share if the initial public offering price is less than $21.25 per share, issued proportionately to Mr. Pappajohn, Dr. Pecora and NNJCA upon their funding $4.0 million, $0.5 million and $1.5 million, respectively. The lenders received an aggregate of 70,588 additional warrants on the same terms, with the warrants being issued proportionately to Mr. Pappajohn, Dr. Pecora and NNJCA, because we did not consummate our initial public offering within 181 days of funding. The warrant exercise price is subject to full ratchet anti-dilution protection in the event of issuances of more than $5.0 million of securities at prices below the exercise price prior to the completion of our initial public offering. The lenders may elect to net exercise their warrants. Mr. Pappajohn also had the option, exercisable up to two hours after he receives notice of the completion of our initial public offering, to convert the outstanding principal amount of his debt into shares of our common stock at a conversion price equal
-137-
Table of Contents
to $17.00 per share or at a 20% discount to our initial public offering price, per share whichever is lower. However, Mr. Pappajohn subsequently agreed to convert $2.0 million of the outstanding principal due to him to common stock at the initial public offering price per share upon consummation of this offering, to eliminate the conversion feature of the remaining $2.0 million in debt, which is due to his spouse, effective upon consummation of this offering and to extend the maturity date to August 15, 2013 for the principal amount which is not converted. Subsequently, the warrants issued to Mr. Pappajohn were amended to provide that the exercise price shall equal the lesser of $17.00 per share or our initial public offering price per share and to extend the $2.0 million dollars in non-convertible debt, which is held by his spouse, to April 1, 2014. In connection with this agreement, we issued to Mr. Pappajohn additional warrants to purchase an aggregate of 75,202 shares of our common stock on the Modified Bridge Form. The warrant exercise price on the Modified Bridge Form is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5.0 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants on the Modified Bridge Form are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00, so that the initial grant would be for an aggregate of 182,633 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 213,072 shares and 159,804 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively. The conversion price of the notes and the exercise price of the warrants are subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. Subsequently, the warrants to purchase an aggregate of 94,117 shares of our common stock issued to Dr. Pecora and NNJCA in connection with this financing were cancelled and the promissory notes issued to Dr. Pecora and NNJCA were amended to increase the pre-payment penalties.
Shares that the lenders receive are subject to a lock up agreement for 180 days after the consummation of our initial public offering on the same terms as other lock-up agreements in favor of the underwriters of this offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
Funding Letter
In assessing our liquidity needs for 2012, we projected cash flows under a variety of scenarios and estimated that we would likely need additional cash for 2012 operations if we do not consummate an initial public offering. We asked Mr. Pappajohn to provide us with a funding commitment letter in the amount of $1.25 million, which he agreed to provide. The letter provides that in the event that we should require additional funding to continue our operations through January 1, 2013, he will help raise, or provide us with the necessary additional funding, up to $1.25 million. As of August 27, 2012, Mr. Pappajohn satisfied the terms of the funding commitment letter.
Credit Facilities
Mr. Pappajohn personally guarantees our revolving line of credit with Wells Fargo Bank, N.A. (“Wells Fargo”). This facility matures on April 1, 2014 by its terms. The maximum amount of indebtedness that was owed by us under this facility at any time since January 1, 2010, is $6.0 million, which amount remains outstanding as of the date hereof. This facility requires monthly interest payments. The interest is computed at LIBOR plus 1.75%, which was 1.96% as of September 30, 2012. As consideration for his personal guarantee of this facility, as well as each of the first six extensions of this facility, Mr. Pappajohn received warrants to purchase an aggregate of 2,440,284 shares of our common stock. The warrants issued to Mr. Pappajohn in connection with such guarantees were on our long form warrant, which is described in the section entitled “Description of Capital Stock—Warrants”, and have exercise prices ranging from $1.60 per share to $12.98 per share. In connection with the seventh extension of this facility, Mr. Pappajohn received warrants to purchase an aggregate of 92,500 shares of our common stock on the same terms as the Bridge Financing Warrant, which is described in the section entitled “Description of Capital Stock—Warrants”. In connection with the eighth extension of this facility and Mr. Pappajohn’s agreement to amend the exercise price of the warrants received by him in connection with the seventh extension, Mr. Pappajohn received
-138-
Table of Contents
warrants to purchase an aggregate of 95,981 shares of common stock on the Modified Bridge Form. The warrant exercise price is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00 per share, so that the initial grant would be for an aggregate of 233,096 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 271,945 shares and 203,959 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively. In addition, we paid the legal fees incurred by Mr. Pappajohn in connection with the guarantees issued to Wells Fargo. Mr. Pappajohn also has rights of subrogation under all security agreements, financing statements, patent filings and other collateral documents given by us to Wells Fargo under the credit agreement governing this facility.
On March 23, 2011, we issued Mr. Thompson warrants to purchase an aggregate of 10,000 shares of common stock at an exercise price of $10.00 per share expiring March 23, 2016 for certain consulting services in connection with our credit facility with DAM. The warrant was issued as a long form warrant, which is described in the section entitled “Description of Capital Stock—Warrants.”
Pursuant to an intercreditor agreement we entered into on March 23, 2011 with Mr. Pappajohn and DAM (“Intercreditor Agreement”), we are required to use the proceeds from this offering to repay the full amount outstanding under our DAM credit facility. If the proceeds from this offering are insufficient to repay the full amount outstanding under the DAM credit facility, we are required, pursuant to the Intercreditor Agreement, to use the proceeds to repay the debt outstanding under the DAM credit facility before any proceeds can be used to repay any debt outstanding under the Wells Fargo Line of Credit. In the event Wells Fargo attempts to collect payment under the Wells Fargo Line of Credit before the debt outstanding under the DAM credit facility is repaid or brings a course of action to prevent payment under the DAM credit facility, Mr. Pappajohn is required, pursuant to the Intercreditor Agreement, to cause Wells Fargo to cease any such course of action, including by paying any amounts outstanding under the Wells Fargo Line of Credit pursuant to his guarantee thereof.
See the section entitled “Management’s Discussion and Analysis of our Results of Operations and Financial Condition—Liquidity and Capital Resources—Sources of Liquidity” for a description of our Wells Fargo Line of Credit and our credit facility with DAM.
Mr. Pappajohn also personally guaranteed our $1.0 million revolving line of credit with West Bank. We repaid in full the outstanding indebtedness under this facility on August 17, 2010. The maximum amount of indebtedness that we owed under this facility at any time since January 1, 2010 was $1.0 million. This facility required monthly interest payments at the rate of 6% per annum.
Promissory Notes
On May 19, 2006, we issued a convertible promissory note in favor of our Chairman and founder, Dr. Chaganti, which obligates us to pay him the sum of $100,000, together with interest at the rate of 8.5% per annum, on demand. Since January 1, 2009 through September 30, 2012, we have made interest payments on the note in the amount of $17,500. As of September 30, 2012, we owe Dr. Chaganti $34,000 in accrued interest. On September 7, 2011, the note was amended to extend its due date to December 31, 2012 and to eliminate any equity conversion feature. In October 2012, we extended the maturity date until April 1, 2014.
Series A Preferred Stock Transaction
In 2007, we closed an initial offering of our Series A preferred stock. We received aggregate gross proceeds of approximately $4.0 million in the offering and issued an aggregate 469,488 shares of Series A preferred stock at a purchase price of $8.46 per share and an initial conversion price of $8.46 (subsequently adjusted to $5.64). Subsequently, Mr. Pappajohn purchased 114,109 shares of Series A preferred stock and also received warrants to
-139-
Table of Contents
purchase an aggregate of 510,437 shares of common stock for an aggregate purchase price of $965,366. Certain of the investors in the initial offering elected to make an additional investment on the same terms and purchased an aggregate of 4,094 shares of Series A preferred stock and also received warrants to purchase an aggregate of 18,313 shares of common stock for an aggregate purchase price of $34,634. Mr. Pappajohn and these investors received long form warrants in connection with such investment, the terms of which are described in the section entitled “Description of Capital Stock—Warrants”, which had an initial exercise price of $8.46 per share (subsequently adjusted to $5.64). Mr. Pappajohn also purchased an additional 11,820 shares of Series A preferred stock from one of the investors in the initial offering. Mr. Pappajohn subsequently joined our board of directors in April 2008.
Consulting Agreements
TSG
On June 19, 2009, we entered into agreements with TSG pursuant to which TSG would provide us with strategic consulting services. Our current Chief Executive Officer, Panna Sharma, was the managing partner, founder and majority owner of TSG and was actively involved in the consulting services provided to us pursuant to such consulting agreement. We compensated TSG an aggregate of $130,750 (excluding expenses) in 2009 and $97,575 in 2010 (excluding expenses) pursuant to such consulting agreement. We also issued TSG warrants to purchase an aggregate of 10,075 shares of common stock at an exercise price of $5.00 in connection with the 2009 consulting agreement. These warrants were granted on April 1, 2010.
On September 23, 2010, we entered into a three-month consulting agreement with TSG for a fixed fee of $60,000 ($45,000 of which was payable in cash and $15,000 was payable in warrants) plus up to $5,000 of expenses. While Mr. Sharma retains a majority ownership position with TSG, he represented to us that he did not and will not profit from the services provided under such agreement, as the operating agreement for TSG was amended following Mr. Sharma’s appointment as our Chief Executive Officer to preclude his participation in profits for consulting engagements in the life sciences industry. The project was subsequently reduced in scope and a revised total payment of $22,500 in cash (excluding expenses) was agreed upon and paid; no warrants were issued.
Equity Dynamics
Beginning in August 2010, Equity Dynamics, Inc., a financial consulting entity that was founded by John Pappajohn, began providing financial consulting services to us. We pay Equity Dynamics a monthly fee of $10,000 plus expenses for such consulting services. From August 2010 through September 30, 2012 we incurred an aggregate of $260,000 in fees plus expenses under this agreement. We may terminate our consulting arrangement with Equity Dynamics upon 30 days written notice.
Equity Dynamics also serves as collateral agent pursuant to our credit agreement with John Pappajohn, NNJCA and Pecora.
Louis Maione
Louis Maione had served as our chief executive officer until 2009, and as a director, general counsel and director of European operations until June 2010. To resolve all outstanding or potential claims between Mr. Maione and us in connection with his employment or the termination thereof and a $40,000 promissory note held by Mr. Maione, we entered into a separation agreement and general release with Mr. Maione dated as of June 10, 2010, which provided, among other things, for a lump sum payment to Mr. Maione of $120,000. To ensure a smooth transition of roles and responsibilities to our other executives, we entered into a one year consulting agreement with Mr. Maione, which provided for four quarterly payments of $12,500 each for an aggregate of up to 125 hours of consulting services related to our probe operations and services in connection with certain pending claims, including the OIG investigation, and an additional $400 per hour for each hour of consulting services Mr. Maione provided above the 125 hour cap. The consulting arrangement terminated in January 2012.
-140-
Table of Contents
Mr. Maione was paid approximately $25,000, $62,760 and $9,650 for legal and consulting services in 2010, 2011 and 2012, respectively.
In April 2012, Mr. Maione filed two lawsuits against us, one lawsuit alleged that we owed Mr. Maione a bonus in connection with his consulting agreement and the other lawsuit alleged that Mr. Maione was defrauded as to the value of the Company in connection with the sale of his equity interests in the Company in June 2010. Both lawsuits were dismissed voluntarily by Mr. Maione. We subsequently entered into a settlement agreement with Mr. Maione pursuant to which we paid Mr. Maione a lump sum of $350,000 to settle all claims raised or which could be raised by Mr. Maione. Such amount was paid in July 2012.
Familial Relationships
Seeta Chaganti, Ph.D., wife of our Chairman, Dr. Raju Chaganti, serves as our Senior Scientist Probe Development. Dr. Seeta Chaganti received a Master’s degree in Statistics from Andhra University, a Master’s degree in Mathematics from Courant Institute of New York University and a Ph.D. in Biology (molecular genetics) from New York University. She worked as a biostatistician and molecular geneticist at Memorial Sloan-Kettering Cancer Center for more than 25 years. She has extensive research experience in advanced molecular cloning techniques. Dr. Seeta Chaganti joined us in January 2005 as our Director of Probe Development. She is responsible for laboratory operation and administration including writing standard operating procedures, DNA clones inventory and maintenance, DNA-FISH Probe design and development validation and DNA propagation and purification. She receives an annual base salary of $89,040.
Indemnification Agreements
We have entered into indemnification agreements with each of our current directors and executive officers. These agreements will require us to indemnify these individuals to the fullest extent permitted under Delaware law against liabilities that may arise by reason of their service to us, and to advance expenses incurred as a result of any proceeding against them as to which they could be indemnified. We also intend to enter into indemnification agreements with our future directors and executive officers.
Policies and Procedures for Related Party Transactions
In anticipation of becoming a public company upon completion this offering, we adopted a policy that our executive officers, directors, nominees for election as a director, beneficial owners of more than 5% of any class of our common stock, any members of the immediate family of any of the foregoing persons and any firms, corporations or other entities in which any of the foregoing persons is employed or is a partner or principal or in a similar position or in which such person has a 5% or greater beneficial ownership interest (collectively, “related parties”) are not permitted to enter into a transaction with us without the prior consent of our board of directors acting through the audit committee or, in certain circumstances, the chairman of the audit committee. Any request for us to enter into a transaction with a related party, in which the amount involved exceeds $120,000 and such related party would have a direct or indirect interest must first be presented to our audit committee, or in certain circumstances the chairman of our audit committee, for review, consideration and approval. In approving or rejecting any such proposal, our audit committee is to consider the material facts of the transaction, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances, the extent of the benefits to us, the availability of other sources of comparable products or services and the extent of the related person’s interest in the transaction.
-141-
Table of Contents
The following table sets forth the beneficial ownership of our common stock as of December 31, 2012 by:
| • | each person, or group of affiliated persons, who we know to beneficially own more than 5% of our common stock; |
| • | each of our named executive officers; |
| • | each of our executive officers; |
| • | each of our directors; and |
| • | all of our executive officers and directors as a group. |
The percentage ownership information shown in the table is based upon 6,414,665 shares of common stock outstanding as of December 31, 2012, after giving effect to the automatic conversion of all of our convertible Series A and Series B preferred stock into 2,182,680 shares of common stock and the conversion of $6.0 million principal amount of convertible promissory notes at a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus, into an aggregate of 857,143 shares of common stock, each of which will occur upon the closing of this offering, and 11,414,665 shares of common stock outstanding after completion of this offering, assuming no exercise of the underwriters’ overallotment option.
We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options or warrants that are either immediately exercisable or exercisable on or before March 1, 2013, which is 60 days after December 31, 2012. These shares are deemed to be outstanding and beneficially owned by the person holding those options or warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
-142-
Table of Contents
Except as otherwise noted below, the address for persons listed in the table is c/o Cancer Genetics, Inc., Route 17 North, 2nd Floor, Rutherford, New Jersey 07070.
| Number of Shares Beneficially Owned |
Percentage of Shares Beneficially Owned |
|||||||||||
| Name of Beneficial Owner |
Before Offering |
After Offering |
||||||||||
| 5% Stockholders |
||||||||||||
| Ann and Argyris Vassiliou |
559,286 | (1) | 8.0 | % | 4.7 | % | ||||||
| Named Executive Officers, Executive Officers and Directors: |
||||||||||||
| Panna Sharma |
313,075 | (2) | 4.7 | % | 2.7 | % | ||||||
| Jane Houldsworth, Ph.D. |
57,167 | (3) | * | * | ||||||||
| Elizabeth A. Czerepak |
46,000 | (4) | * | * | ||||||||
| Raju S.K. Chaganti, Ph.D. |
1,438,242 | (5) | 21.2 | % | 12.2 | % | ||||||
| Edmund Cannon |
52,236 | (6) | * | * | ||||||||
| John Pappajohn |
4,474,778 | (7) | 51.1 | % | 32.5 | % | ||||||
| Andrew Pecora, M.D. |
93,479 | (8) | 1.4 | % | * | |||||||
| Tommy G. Thompson |
51,786 | (9) | * | * | ||||||||
| Robert Kaufman |
6,667 | (10) | * | * | ||||||||
| Dr. Franklyn G. Prendergrast, M.D., Ph.D. |
— | (11) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| All current directors and executive officers as a group |
6,533,428 | 67.4 | % | 44.5 | % | |||||||
* denotes less than 1%.
(1) Includes 25,000 shares of our common stock held by AANA, Ltd., an investment partnership in which Ann and Argyris Vassiliou and their two minor children are the sole partners. Includes 267,143 shares of common stock underlying warrants exercisable on or before March 1, 2013 held by Ann and Argyris Vassiliou and 267,143 shares of common stock underlying warrants exercisable on or before March 1, 2013 held by NICALE Partners, an investment partnership for the two minor children of Ann and Argyris Vassiliou. Ann Vassiliou is the daughter of John Pappajohn. The address for Ann and Argyris Vassiliou is 45-10 Court Square - Floor 2, Long Island City, New York 11101.
(2) Includes 303,000 shares of common stock underlying options exercisable on or before March 1, 2013. Excludes 172,000 shares of common stock underlying options that are not exercisable on or before March 1, 2013. Includes 10,075 shares of common stock underlying warrants held by TSG.
(3) Includes 27,275 shares of common stock underlying options exercisable on or before March 1, 2013. Excludes 13,225 shares of common stock underlying options that are not exercisable on or before March 1, 2013.
(4) Includes 46,000 shares of common stock underlying options exercisable on or before March 1, 2013. Excludes 79,000 shares of common stock underlying options that are not exercisable on or before March 1, 2013.
(5) Includes 375,000 shares of common stock underlying options held by Dr. Raju Chaganti exercisable on or before March 1, 2013. Excludes 25,000 shares of common stock underlying options held by Dr. Raju Chaganti that are not exercisable on or before March 1, 2013. Also, includes 150,000 shares of common stock owned by Chaganti LLC, 244,565 shares of common stock owned by his wife, Dr. Seeta Chaganti, and 208,735 shares of common stock held by grantor retained annuity trusts of which Dr. Raju Chaganti and his wife are co-trustees and/or recipients.
(6) Includes 37,500 shares of common stock underlying options exercisable on or before March 1, 2013.
(7) Includes 500,000 shares of common stock owned by his wife. Includes 37,500 shares of common stock underlying options exercisable on or before March 1, 2013. Includes 2,300,947 shares of common stock underlying warrants exercisable on or before March 1, 2013. Includes 188,895 shares of common stock issuable upon conversion of the shares of Series A preferred stock. Includes 678,571 shares which will be issued upon conversion of a $4.75 million convertible note assuming a conversion price of $7.00 per share, which is the midpoint of the range set forth on the cover page of this prospectus.
(8) Includes 87,500 shares of common stock underlying options exercisable on or before March 1, 2013.
(9) Includes 51,786 shares of common stock underlying options and warrants exercisable on or before March 1, 2013.
(10) Includes 6,667 shares of common stock underlying options exercisable on or before March 1, 2013. Excludes 13,333 shares of common stock underlying options exercisable on or after March 1, 2013.
(11) Excludes 20,000 shares underlying options that we have agreed to issue Dr. Prendergast on consummation of our initial public offering.
-143-
Table of Contents
General
Our amended and restated certificate of incorporation that will be in effect upon the closing of this offering authorizes us to issue up to 100,000,000 shares of common stock, par value $0.0001 per share, and 9,764,000 shares of preferred stock, par value $0.0001 per share. In addition, we anticipate effecting a 1-for-2 reverse stock split just prior to the date of this prospectus. All share numbers in this prospectus reflect the 1-for-2 reverse stock split.
As of September 30, 2012, there were 3,365,311 shares of common stock outstanding, held of record by 48 stockholders. The number of shares of common stock outstanding as of September 30, 2012 does not include (i) 9,532 shares of common stock issued after September 30, 2012 and before the date of this prospectus upon the exercise of warrants to purchase such number of shares at an exercise price of $0.80 per share; (ii) 2,182,680 shares of common stock issuable upon the conversion of our outstanding shares of convertible preferred stock, (iii) 2,442,032 shares of common stock issuable upon the exercise of our outstanding warrants to purchase common stock as of September 30, 2012, (iv) 1,383,350 shares of common stock issuable upon the exercise of outstanding options to purchase common stock, (v) 331,723 shares of common stock underlying warrants which were issued after September 30, 2012 (net of warrants cancelled after September 30, 2012), (vi) 857,143 shares of common stock underlying $6.0 million of convertible promissory notes, assuming an initial public offering price of $7.00 per share, which is the midpoint of the price range on the cover of this prospectus, (vii) 133,750 shares of common stock underlying options to be issued upon completion of this initial public offering and (viii) the shares that will be issued in this offering. The number of shares of common stock issuable upon the exercise of outstanding warrants exclude the effects of anti-dilution provisions that could occur upon completion of our initial public offering. At an initial public offering price of $7.00 per share, an aggregate of 3,942,198 shares of common stock would be issuable upon exercise of the outstanding warrants described above. Certain shares of common stock were issued in exchange for shares previously issued to cure possible technical deficiencies with respect to the original issuances, but three holders of common stock have not responded to requests to exchange their shares, which represent approximately 1.2% of the common shares outstanding or 0.8% of our common stock assuming the conversion of our outstanding Series A, Series A-1 and Series B preferred stock.
As of September 30, 2012, there were 52,926 shares of Series A preferred stock outstanding, which will convert into 79,389 shares of common stock, 1,604,295 shares of Series A-1 which have identical rights and privileges as Series A and will convert into 802,147 shares of common stock, and 1,821,600 shares of Series B preferred stock, which will convert into 1,301,143 shares of common stock outstanding. All references to our Series A preferred stock in this registration statement refer collectively to the Series A preferred and Series A-1 preferred shares. We issued the Series A-1 in exchange for the Series A to cure possible technical deficiencies with respect to the original issuance of the Series A preferred stock, but five holders of Series A have not responded to requests to exchange their shares, which represent approximately 9.0% of the Series A and A-1 shares that are outstanding or 1.5% of our common stock assuming the conversion of our outstanding Series A, Series A-1 and Series B preferred stock. In accordance with the terms of our amended and restated certificate of incorporation, upon the closing of an underwritten offering pursuant to an effective registration statement in connection with initial public offering with gross proceeds of $15.0 million or more, our preferred stock will automatically convert into an aggregate of 2,182,680 shares of our common stock. If we receive proceeds of at least $15.0 million in this offering, each outstanding share of our Series A preferred stock will automatically convert into one and one-half shares of common stock and each outstanding share of our Series A-1 and our Series B preferred stock will automatically convert into one-half of one share of common stock.
The following descriptions of our capital stock and provisions of our amended and restated certificate of incorporation an amended and restated bylaws are summaries and are qualified by reference to the amended and restated certificate of incorporation and the amended and restated bylaws that will be in effect upon completion of this offering, and applicable law. Copies of these documents will be filed with the SEC as exhibits to our registration statement, of which this prospectus forms a part. The descriptions of the common stock and preferred stock reflect (i) changes to our capital structure that will occur upon the closing of this offering and (ii) Delaware law.
-144-
Table of Contents
Common Stock
The holders of our common stock are entitled to the following rights:
Voting Rights
Holders of our common stock are entitled to one vote per share in the election of directors and on all other matters on which stockholders are entitled or permitted to vote. Holders of our common stock are not entitled to cumulative voting rights.
Dividend Rights
Subject to the terms of any outstanding series of preferred stock, the holders of our common stock are entitled to dividends in the amounts and at times as may be declared by the board of directors out of funds legally available therefor.
Liquidation Rights
Upon liquidation or dissolution, holders of our common stock are entitled to share ratably in all net assets available for distribution to stockholders after we have paid, or provided for payment of, all of our debts and liabilities, and after payment of any liquidation preferences to holders of our preferred stock.
Other Matters
Holders of our common stock have no redemption, conversion or preemptive rights. There are no sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to the rights of the holders of shares of any series of preferred stock that we may issue in the future.
Preferred Stock
Our board of directors has the authority to issue preferred stock in one or more classes or series and to fix the designations, powers, preferences and rights, and the qualifications, limitations or restrictions thereof, including dividend rights, conversion right, voting rights, terms of redemption, liquidation preferences and the number of shares constituting any class or series, without further vote or action by the stockholders. Although we have no present plans to issue any other shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal. The preferred stock provides for an adjustment of the conversion price in the event of an issuance or deemed issuance at a price less than the applicable conversion price, subject to certain exceptions. The conversion price for our Series A preferred stock and Series B preferred stock is $5.64 per share and $10.00, respectively. As a result of certain anti-dilution adjustments, if the initial public offering price per share is less than the conversion prices, such conversion prices will adjust to the initial public offering price per share.
Options
As of September 30, 2012, we had outstanding options to purchase an aggregate of 1,383,350 shares of our common stock with exercise prices ranging from $1.60 to $13.52 per share, with an approximate weighted average exercise price of $5.10 per share. The shares of our common stock underlying all such options will be registered for sale with the SEC as promptly as practicable following the completion of this offering.
-145-
Table of Contents
Warrants
As of September 30, 2012, we had outstanding warrants to purchase an aggregate of 2,442,032 shares of our common stock with exercise prices ranging from $1.60 to $17.00 per share, consisting of:
| • | warrants to purchase an aggregate of 589,034 shares of our common stock at an exercise price of $1.60 per share; |
| • | warrants to purchase an aggregate of 10,075 shares of our common stock at an exercise price of $5.00 per share; |
| • | warrants to purchase an aggregate of 188,323 shares of our common stock at an exercise price of $5.64 per share; |
| • | warrants to purchase an aggregate of 1,071,660 shares of our common stock at an exercise price of $10.00 per share; |
| • | warrants to purchase an aggregate of 100,000 shares of our common stock at an exercise price of $12.98 per share; and |
| • | warrants to purchase an aggregate of 482,940 shares of our common stock at an exercise price of $17.00 per share. |
All of our outstanding warrants were or will be issued pursuant to one of the following seven forms of warrant:
Short Form Warrant
We no longer have any outstanding warrants to purchase shares of our common stock pursuant to our Short Form Warrant because all of such warrants have either been exercised or expired. The Short Form Warrant provided for an adjustment of the exercise price and shares underlying the warrant in the event of a forward or reverse stock split. Holders of our Short Form Warrant did not have registration rights for the shares underlying the warrant.
Short Form Cashless Exercise Warrant
As of September 30, 2012, outstanding warrants to purchase an aggregate of 163,323 shares of our common stock were issued pursuant to our Short Form Cashless Exercise Warrant. The Short Form Cashless Exercise Warrant provides for the same terms as the Short Form Warrant except that holders of Short Form Cashless Exercise Warrants can elect to exercise their warrants using the cashless exercise feature.
Medium Form Warrant
Outstanding warrants to purchase an aggregate of 281,660 shares of our common stock were issued pursuant to our Medium Form Warrant, including warrants to purchase an aggregate of 150,000 shares of our common stock issued in connection with the DAM Financing. The Medium Form Warrant provides for an adjustment of the exercise price in the event of forward and reverse stock splits, stock dividends, an issuance of rights or warrants to all holders of our common stock entitling them to subscribe for or purchase shares of our common stock at a price less than the exercise price, and other such similar transactions in which we issue shares of our common stock, or securities convertible into or exchangeable for shares of our common stock, at a price or a conversion price which is less than the exercise price (“adjustment issuances”). The Medium Form Warrant also provides for an adjustment of the shares underlying the warrant in the event of any adjustment resulting from forward and reverse stock splits, stock dividends or an issuance of rights or warrants to all holders of our common stock entitling them to subscribe for or purchase
-146-
Table of Contents
shares of our common stock at a price less than the exercise price. The Medium Form Warrant contains a cashless exercise option. In addition, in the event we distribute to our shareholders evidences of indebtedness, assets, subscription rights or warrants, holders of a Medium Form Warrant will be entitled to receive the amount of any property or other securities which would have been distributed to such holder had such holder been a holder of one share of our common stock on the record date of such distribution. In the event we undergo a reclassification, capital reorganization or other change in outstanding shares of our common stock or if we undergo a consolidation or merger or sell all of our assets, holders of our Medium Form Warrant have the right to purchase the kind and amount of shares of stock and other securities and property receivable upon such event that the holder might have purchased upon exercise of the warrant immediately prior to such event. Holders of our Medium Form Warrant also have registration rights with respect to the shares underlying their warrants.
Long Form Warrant
Outstanding warrants to purchase an aggregate of 1,514,108 shares of our common stock were issued pursuant to our Long Form Warrant. Subsequent to September 30, 2012, Long Form Warrants to purchase an aggregate of 9,532 shares of our common stock were exercised by the holders thereof and Long Form Warrants to purchase an aggregate of 8,781 shares of our common stock expired unexercised. Long Form Warrant holders have the same rights as Medium Form Warrant holders except that the Long Form Warrant also provides for an adjustment of the shares underlying the warrant in the event we issue shares of our common stock, except in certain limited circumstances, at a price less than the exercise price of the Long Form Warrant and securities convertible into or exchangeable for our common stock at prices or conversion prices less than the exercise price of the Long Form Warrant. As of September 30, 2012, holders of warrants to purchase an aggregate of 570,721 shares of our common stock pursuant to our Long Form Warrant have waived their right to anti-dilution adjustments in the event we issue shares of our common stock at a price less than the exercise price of the Long Form Warrant and securities convertible into or exchangeable for our common stock at prices or conversion prices less than the exercise price of the Long Form Warrant. As a result of these adjustment provisions, the number of shares of common stock underlying the warrants on the Long Form Warrant would increase to 1,938,108, if the offering price is $7.00 per share, which is the midpoint of the range on the cover page of this prospectus.
Bridge Financing Warrant
As of September 30, 2012, outstanding warrants to purchase an aggregate of 412,352 shares of our common stock were issued on our form of Bridge Financing Warrant. Subsequent to September 30, 3012, we cancelled warrants to purchase an aggregate of 131,617 shares of common stock on our form of Bridge Financing Warrant, leaving warrants to purchase an aggregate of 280,735 shares of common stock outstanding on our form of Bridge Financing Warrant. Bridge Financing Warrant holders have the same rights as Medium Form Warrant holders except that the Bridge Financing Warrant also provides for an adjustment of the exercise price in the event we issue shares of our common stock in our initial public offering at a price to the public of less than $21.25 per share.
Modified Bridge Financing Warrant
At September 30, 2012, outstanding warrants to purchase an aggregate of 70,589 shares of our common stock were issued on our form of Modified Bridge Financing Warrant. Subsequent to September 30, 2012, such warrants to purchase an aggregate of 70,589 shares of our common stock were cancelled and we issued warrants to purchase an aggregate of 171,185 shares of our common stock on our form of Modified Bridge Financing Warrant. Modified Bridge Financing Warrant holders have the same rights as Bridge Financing Warrant holders except that the Modified Bridge Financing Warrant also provides for an adjustment of the number of shares purchasable under the warrant in the event we issue shares of our common stock in our initial public offering at a price to the public of less than $17.00 per share or engage in certain other adjustment issuances prior to our initial public offering. As a result of these adjustment provisions, the number of shares of common stock underlying the warrants on the form of Modified Bridge Financing Warrant would increase to 415,729, if the offering price is $7.00 per share, which is the midpoint of the range on the cover page of this prospectus.
-147-
Table of Contents
October 2012 Warrant
At September 30, 2012, there were no warrants issued or outstanding on our form of October 2012 Warrant. Subsequent to September 30, 2012, we issued warrants to purchase an aggregate of 362,746 shares of our common stock on our form of October 2012 Warrant. October 2012 Warrant holders have the same rights as Modified Bridge Financing Warrant holders except that the October 2012 Warrant provides for a term of ten (10) years and includes a provision for automatic net issuance exercise immediately prior to stated expiration date. As a result of anti-dilution adjustments, the number of shares of common stock underlying the warrants on the October 2012 Warrant would increase to 880,954 if the initial public offering price per share is $7.00 which is the midpoint of the price range on the cover page of this prospectus.
Anti-Takeover Effects of Delaware law and Our Certificate of Incorporation and Bylaws
The provisions of Delaware law, our certificate of incorporation and our bylaws described below may have the effect of delaying, deferring or discouraging another party from acquiring control of us.
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| • | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
| • | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| • | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines business combination to include the following:
| • | any merger or consolidation involving the corporation and the interested stockholder; |
| • | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; |
| • | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
| • | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or |
| • | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
-148-
Table of Contents
In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
Certificate of Incorporation and Bylaws
Following the completion of this offering, our certificate of incorporation and bylaws will be amended to provide that:
| • | the authorized number of directors can be changed only by resolution of our board of directors; |
| • | our bylaws may be amended or repealed by our board of directors or our stockholders; |
| • | no action can be taken by stockholders except at an annual or special meeting of the stockholders called in accordance with our bylaws, and stockholders may not act by written consent, unless the stockholders amend the certificate of incorporation to provide otherwise; |
| • | stockholders may not call special meetings of the stockholders or fill vacancies on the board; |
| • | our board of directors will be authorized to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; |
| • | our stockholders do not have cumulative voting rights, and therefore our stockholders holding a majority of the shares of common stock outstanding will be able to elect all of our directors; and |
| • | our stockholders must comply with advance notice provisions to bring business before or nominate directors for election at a stockholder meeting. |
Potential Effects of Authorized but Unissued Stock
We have shares of common stock and preferred stock available for future issuance without stockholder approval. We may utilize these additional shares for a variety of corporate purposes, including future public offerings to raise additional capital, to facilitate corporate acquisitions or payment as a dividend on the capital stock.
The existence of unissued and unreserved common stock and preferred stock may enable our board of directors to issue shares to persons friendly to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise, thereby protecting the continuity of our management. In addition, the board of directors has the discretion to determine designations, rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences of each series of preferred stock, all to the fullest extent permissible under the Delaware General Corporation Law and subject to any limitations set forth in our certificate of incorporation. The purpose of authorizing the board of directors to issue preferred stock and to determine the rights and preferences applicable to such preferred stock is to eliminate delays associated with a stockholder vote on specific issuances. The issuance of preferred stock, while providing desirable flexibility in connection with possible financings, acquisitions and other corporate purposes, could have the effect of making it more difficult for a third-party to acquire, or could discourage a third-party from acquiring, a majority of our outstanding voting stock.
Limitations of Director Liability and Indemnification of Directors, Officers and Employees
Our amended and restated certificate of incorporation, which will become effective upon the closing of this offering, limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law
-149-
Table of Contents
provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
| • | breach of their duty of loyalty to us or our stockholders; |
| • | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or |
| • | transaction from which the directors derived an improper personal benefit. |
These limitations of liability do not apply to liabilities arising under the federal or state securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our amended and restated bylaws, which will become effective upon the closing of this offering, provide that we will indemnify our directors and officers to the fullest extent permitted by law, and may indemnify employees and other agents. Our amended and restated bylaws, which will become effective upon the closing of this offering, also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding.
We have obtained a policy of directors’ and officers’ liability insurance.
We plan to enter into separate indemnification agreements with our directors and officers. These agreements, among other things, require us to indemnify our directors and officers for any and all expenses (including reasonable attorneys’ fees, retainers, court costs, transcript costs, fees of experts, witness fees, travel expenses, duplicating costs, printing and binding costs, telephone charges, postage, delivery service fees) judgments, fines and amounts paid in settlement actually and reasonably incurred by such directors or officers or on his or her behalf in connection with any action or proceeding arising out of their services as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request provided that such person follows the procedures for determining entitlement to indemnification and advancement of expenses set forth in the indemnification agreement. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
At present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
Transfer Agent
The transfer agent and registrar for our common stock is Continental Stock Transfer & Trust Company. Its address is 17 Battery Place, 8th Floor, New York, NY 10004 and its telephone number is 212-509-4000.
-150-
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Before this offering, there was no public market in the United States for our securities and a significant public market for our securities may not develop or be sustained after this offering. As described below, approximately 6,118,759 shares currently outstanding will not be available for sale immediately after this offering due to certain contractual and securities law restrictions on resale. Sales of substantial amounts of our common stock in the public market after these restrictions lapse could cause the prevailing market price to decline and limit our ability to raise equity capital in the future.
Upon completion of this offering, we will have outstanding an aggregate of 11,414,665 shares of common stock (12,164,665 shares if the underwriters exercise their over-allotment option in full). In addition, we have reserved:
| • | 2,442,032 shares for issuance in connection with warrants outstanding as of September 30, 2012; |
| • | 1,383,500 shares for issuance in connection with options outstanding as of September 30, 2012; |
| • | 1,066,650 shares reserved for future issuance in under our equity incentive plans as of September 30, 2012 of which 133,750 shares have been reserved for option grants to be issued upon consummation of our initial public offering; |
| • | warrants to purchase a total of 331,723 additional shares of our common stock at an exercise price of $17.00 per share, which have been issued since September 30, 2012 (net of warrants cancelled after September 30, 2012); and |
| • | 2,674 shares of common stock issuable to Cleveland Clinic pursuant to our license agreement with Cleveland Clinic. |
Of these shares, the 5,000,000 shares sold in this offering (5,750,000 shares if the underwriters exercise their over-allotment option in full) will be freely transferable without restriction or further registration under the Securities Act, except for any shares that are acquired by affiliates as that term is defined in Rule 144 under the Securities Act (“Rule 144”). The remaining 6,414,665 shares of common stock held by existing stockholders are “restricted securities,” as that term is defined in Rule 144 under the Securities Act. Restricted securities may be sold in the public market only if registered or if their resale qualifies for exemption from registration described below under Rule 144 or 701 promulgated under the Securities Act.
As a result of contractual restrictions described below and the provisions of Rules 144 and 701, the shares sold in this offering and the restricted securities will be available for sale in the public market as follows:
| • | the 5,000,000 shares sold in this offering (5,750,000 shares if the underwriters exercise their over-allotment option in full) and 295,906 of the existing restricted shares will be eligible for immediate sale upon the completion of this offering; and |
| • | approximately 6,118,759 restricted shares will be eligible for sale in the public market upon expiration of lock-up agreements 180 days after the date of this prospectus, which date may be extended in specified circumstances, subject in certain circumstances to the volume, manner of sale and other limitations under Rule 144 and Rule 701. |
Rule 144
In general, under Rule 144 as currently in effect, once we have been subject to the reporting requirements under the Exchange Act for at least 90 days a person (or persons whose shares are aggregated) who is not deemed to have been an affiliate of ours at any time during the three months preceding a sale, and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months, would be entitled
-151-
Table of Contents
to sell those shares, subject only to the availability of current public information about us. A non-affiliated person who has beneficially owned restricted securities within the meaning of Rule 144 for at least one year would be entitled to sell those shares without regard to the provisions of Rule 144.
An affiliate of ours who has beneficially owned restricted shares of our common stock for at least twelve months (or six months, provided that such sale occurs after we have been subject to the reporting requirements under the Exchange Act for at least 90 days) would be entitled to sell, within any three-month period, a number of shares that does not exceed the greater of:
| • | 1% of shares of our common stock then outstanding; or |
| • | the average weekly trading volume of shares of our common stock on the NASDAQ during the four calendar weeks preceding the date on which notice of the sale is filed with the SEC. |
Sales under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to manner of sale provisions, notice requirements and the availability of current public information about us.
Rule 701
Under Rule 701, common stock acquired upon the exercise of certain currently outstanding options or pursuant to other rights granted under our stock plans may be resold, to the extent not subject to lock-up agreements, (a) by persons other than affiliates, beginning 90 days after the effective date of this offering, subject only to the manner-of-sale provisions of Rule 144 and (b) by affiliates, subject to the manner-of-sale, current public information and filing requirements of Rule 144, in each case, without compliance with the holding period requirement of Rule 144. The Rule 701 shares held by our executive officers, directors and substantially all of our stockholders are, however, subject to lock-up agreements and will only become eligible for sale upon the expiration of the contractual lock-up agreements. The underwriters may release all or any portion of the securities subject to lock-up agreements.
Lock-Up Agreements
In connection with this offering, our directors and officers and holders of approximately 95.39% of our outstanding common and preferred stock, on an as converted basis, and holders representing approximately 99.44% of our outstanding warrants and options agreed not to sell or otherwise dispose of any securities without the prior written consent of the underwriters for a period of 180 days after the date of this prospectus, subject to certain terms and conditions. See the section entitled “Underwriting” for more information regarding such restrictions.
Registration Rights
After the closing of this offering, certain holders of our securities will be entitled to rights with respect to the registration of their shares under the Securities Act. These registration rights are contained in our Amended and Restated Investors’ Rights Agreement, dated as of April 13, 2010, as amended on December 8, 2011 (“Investors’ Rights Agreement”), our registration rights agreement, dated as of March 23, 2011, with DAM, and our registration rights agreement, dated as of February 13, 2012, with John Pappajohn, NNJCA and Pecora and our registration rights agreement, dated October 17, 2012 with John Pappajohn and Mark Oman (together, the “Registration Rights Agreements”). Our Investors’ Rights Agreement provides registration rights to holders of our Series A preferred stock and our Series B preferred stock. The holders of warrants to purchase shares of our common stock issued pursuant to our Medium Form Warrant and Long Form Warrant have registration rights as set forth in the Investors’ Rights Agreement. The Registration Rights Agreements provide registration rights to holders of warrants to purchase shares of our common stock issued in connection with our DAM credit facility and the extension thereof (“DAM Warrants”) and in connection with our December 2011 Financing Transaction.
-152-
Table of Contents
The registration rights available under both our Investors’ Rights Agreement and our Registration Rights Agreements will terminate on the earlier of (i) the third anniversary following the closing of this offering, or (ii) with respect to any particular holder, when such holder is able to sell all of its shares pursuant to Rule 144 of the Securities Act or a similar exemption during any 90-day period without registration and without certain other restrictions set forth in Rule 144 regarding manner of sale and brokers’ transactions. Both agreements provide that we will pay the registration expenses (other than underwriting discounts, selling commissions and transfer taxes) of the holders of the shares registered pursuant to the registrations described below. In an underwritten offering, the managing underwriter, if any, has the right, subject to specified conditions, to limit the number of shares such holders may include in a registration statement.
Demand Registration Rights
Pursuant to the Investors’ Rights Agreement, at any time beginning 180 days after the closing of this offering, and, in the event of the Company’s merger with a public company, 90 days after the completion of such merger, the holders of a majority of the outstanding shares of Series B preferred stock are entitled to demand registration rights upon providing the Company with written notice.
Pursuant to the Registration Rights Agreements, at any time beginning 180 days after the closing of the initial public offering, and, in the event of the Company’s merger with a public company, 90 days after the completion of such merger, the holders of a majority of shares issued pursuant to the DAM Warrants or the warrants issued in connection with our December 2011 Financing Transaction, as applicable, are entitled to demand registration rights upon providing the Company with written notice.
Piggyback Registration Rights
Pursuant to the Investors’ Rights Agreement and the Registration Rights Agreements, after the closing of the initial public offering, if we propose to register the offer and sale of any of our securities under the Securities Act in connection with the public offering of such securities, the holders of Series A preferred stock, Series B preferred stock and shares of common stock issued pursuant to the DAM Warrants, the Wells Fargo Warrants and warrants issued in connection with the December 2011 Financing Transaction and 2012 convertible debt financing are entitled to certain “piggyback” registration rights. Such rights allow the holders to include their shares in such registration, subject to certain marketing and other limitations. As a result, whenever we propose to file a registration statement under the Securities Act, other than with respect to (i) a registration related to a company stock plan, (ii) a registration related to the exchange of securities in certain corporate reorganizations or certain other transactions and (iii) a registration of the issuance of common stock upon conversion of debt securities, the offer and sale of which are also being registered, the holders of these shares are entitled to notice of the registration and have the right, subject to limitations that the underwriters may impose on the number of shares included in the registration, to include their shares in the registration.
-153-
Table of Contents
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF OUR COMMON STOCK
The following discussion describes the material U.S. federal income tax consequences to non-U.S. holders (as defined below) of the acquisition, ownership and disposition of our common stock issued pursuant to the initial public offering. This discussion is not a complete analysis of all potential U.S. federal income tax consequences and does not address any tax consequences arising under any state, local or foreign tax laws, any income tax treaties, or any other U.S. federal tax laws, including U.S. federal estate and gift tax laws (except as specifically addressed herein with respect to U.S federal estate taxes). This discussion is based on the Internal Revenue Code of 1986, as amended (“Code”), U.S. Treasury Regulations promulgated thereunder, judicial decisions and published rulings and administrative pronouncements of the Internal Revenue Service (“IRS”), all as in effect on the date of the initial public offering. These authorities may change, possibly retroactively, resulting in tax consequences different from those discussed below. No rulings have been or will be sought from the IRS with respect to the matters discussed below, and there can be no assurance that the IRS will not take a different position regarding the tax consequences of a non-U.S. holder’s acquisition, ownership or disposition of our common stock or that any such position would not be sustained by a court.
This discussion is limited to non-U.S. holders who purchase our common stock pursuant to this offering and who hold our common stock as “capital assets” within the meaning of Code Section 1221 (generally, property held for investment). This discussion does not address all U.S. federal income tax consequences that may be relevant to a non-U.S. holder in light of the holder’s particular circumstances. It also does not consider any specific facts or circumstances that may be relevant to non-U.S. holders subject to special rules under the U.S. federal income tax laws, including, without limitation, U.S. expatriates, banks, financial institutions, insurance companies, regulated investment companies, real estate investment trusts, “controlled foreign corporations,” “passive foreign investment companies,” corporations that accumulate earnings to avoid U.S. federal income tax, brokers, dealers or traders in securities, commodities or currencies, partnerships or other pass-through entities (or investors in such entities), tax-exempt organizations, tax-qualified retirement plans, persons subject to the alternative minimum tax or the unearned income Medicare contribution tax, and persons holding our common stock as part of a straddle, hedge or other risk reduction strategy or as part of a conversion transaction or other integrated investment.
WE RECOMMEND THAT PROSPECTIVE INVESTORS CONSULT THEIR TAX ADVISORS REGARDING THE PARTICULAR U.S. FEDERAL INCOME TAX CONSEQUENCES TO THEM OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK, AS WELL AS ANY TAX CONSEQUENCES ARISING UNDER ANY STATE, LOCAL OR FOREIGN TAX LAWS, ANY APPLICABLE INCOME TAX TREATIES, OR ANY OTHER U.S. FEDERAL TAX LAWS (INCLUDING ESTATE AND GIFT TAX LAWS).
Definition of Non-U.S. Holder
As used in this discussion, a non-U.S. holder is any beneficial owner of our common stock who is not treated as a partnership for U.S. federal income tax purposes and is not:
| • | an individual citizen or resident of the United States; |
| • | a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) created or organized under the laws of the United States, any state thereof or the District of Columbia; |
| • | an estate, the income of which is subject to U.S. federal income tax regardless of its source; or |
| • | a trust if (i) a U.S. court is able to exercise primary supervision over its administration and one or more U.S. persons have authority to control all its substantial decisions or (ii) the trust was in existence on August 20, 1996, was treated as a U.S. person prior to that date and validly elected to continue to be so treated. |
-154-
Table of Contents
If any entity treated as a partnership for U.S. federal income tax purposes holds our common stock, the tax treatment of a partner generally will depend on the status of the partner and the activities of the partnership. Partnerships and their partners should consult their tax advisors as to the tax consequences to them of the acquisition, ownership and disposition of our common stock.
Distributions on Our Common Stock
As described in the section entitled, “Dividend policy,” we do not anticipate paying dividends on our common stock in the foreseeable future. If we make a distribution of cash or other property with respect to our common stock, the distribution generally will constitute a dividend for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income tax purposes will constitute a return of capital and will first be applied against and reduce a holder’s adjusted tax basis in its common stock, but not below zero. Any remaining excess will be treated as capital gain from the sale of property.
Dividends paid to a non-U.S. holder of our common stock that are not effectively connected to the holder’s conduct of a U.S. trade or business generally will be subject to U.S. federal withholding tax at a rate of 30% of the gross amount of the dividends, or a lower rate specified by an applicable tax treaty. To receive the benefit of a reduced treaty rate, a non-U.S. holder must furnish to us or our paying agent a valid IRS Form W-8BEN (or applicable successor form) certifying the holder’s qualification for the reduced rate. A non-U.S. holder may be required to obtain a U.S. taxpayer identification number to claim treaty benefits. This certification must be provided to us or our paying agent prior to the payment of dividends and may be required to be updated periodically. Non-U.S. holders that do not timely provide us or our paying agent with the required certification, but which qualify for a reduced treaty rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non-U.S. holders should consult their tax advisors regarding their entitlement to benefits under a relevant income tax treaty.
If a non-U.S. holder holds our common stock in connection with the conduct of a trade or business in the United States, and dividends paid on the common stock are effectively connected with the holder’s U.S. trade or business and, if an income tax treaty applies, the non-U.S. holder maintains a “permanent establishment” in the United States to which the dividends are attributable, the non-U.S. holder will be exempt from U.S. federal withholding tax, if the appropriate certification is provided. To claim the exemption for effectively connected income, the non-U.S. holder must furnish to us or our paying agent a properly executed IRS Form W-8ECI (or applicable successor form) prior to the payment of the dividends. Any dividends paid on our common stock that are effectively connected with a non-U.S. holder’s U.S. trade or business generally will be subject to U.S. federal income tax on a net income basis at the regular graduated U.S. federal income tax rates in the same manner as if the holder were a resident of the United States, unless the holder is entitled to the benefits of a tax treaty that provides otherwise. A non-U.S. holder that is a foreign corporation also may be subject to a branch profits tax equal to 30% (or a lower rate specified by an applicable tax treaty) of its effectively connected earnings and profits for the taxable year that are attributable to such dividends. Non-U.S. holders should consult any applicable tax treaties that may provide for different rules.
Gain on Disposition of Our Common Stock
Subject to the discussions below regarding backup withholding and foreign accounts, a non-U.S. holder generally will not be subject to U.S. federal income tax on any gain realized upon the sale or other disposition of our common stock unless:
| • | the gain is effectively connected with the non-U.S. holder’s conduct of a trade or business in the United States; |
-155-
Table of Contents
| • | the non-U.S. holder is a nonresident alien individual present in the United States for 183 days or more during the taxable year of the disposition and certain other requirements are met; or |
| • | our common stock constitutes a U.S. real property interest by reason of our status as a U.S. real property holding corporation at any time within the shorter of the five-year period preceding the disposition or the non-U.S. holder’s holding period for our common stock and certain other requirements are met. |
Unless an applicable tax treaty provides otherwise, gain described in the first bullet point above will be subject to U.S. federal income tax on a net income basis at the regular graduated U.S. federal income tax rates in the same manner as if the holder were a resident of the United States. Non-U.S. holders that are foreign corporations also may be subject to a branch profits tax equal to 30% (or a lower rate specified by an applicable tax treaty) of its effectively connected earnings and profits for the taxable year that are attributable to such gain. Non-U.S. holders should consult any applicable tax treaties that may provide for different rules.
Gain described in the second bullet point above will be subject to U.S. federal income tax at a flat 30% rate (or a lower rate specified by an applicable income tax treaty), but may be offset by U.S. source capital losses.
With respect to the third bullet point above, we believe we currently are not and will not become a U.S. real property holding corporation. However, because the determination of whether we are a U.S. real property holding corporation generally depends on whether the fair market value of our U.S. real property interests equals or exceeds 50% of the sum of the fair market value of our other trade or business assets and our worldwide real property interests, there can be no assurance that we will not become a U.S. real property holding corporation in the future. In the event we do become a U.S. real property holding corporation, as long as our common stock is regularly traded on an established securities market, our common stock will constitute a U.S. real property interest only with respect to a non-U.S. holder that actually or constructively holds more than five percent of our common stock at some time during the shorter of the five-year period preceding the disposition or the non-U.S. holder’s holding period for our common stock. Any taxable gain generally will be taxed in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business, except that the branch profits tax will not apply.
Information Reporting and Backup Withholding
We must report annually to the IRS and to each non-U.S. holder the amount of dividends on our common stock paid to the holder and the amount of any tax withheld with respect to those dividends. These information reporting requirements apply even if no withholding was required because the dividends were effectively connected with the holder’s conduct of a U.S. trade or business, or withholding was reduced or eliminated by an applicable tax treaty. This information also may be made available under a specific treaty or agreement with the tax authorities in the country in which the non-U.S. holder resides or is established.
Backup withholding, currently at a rate of 28%, generally will not apply to payments of dividends to a non-U.S. holder of our common stock provided the non-U.S. holder furnishes to us or our paying agent the required certification as to its non-U.S. status (typically, by providing a valid IRS Form W-8BEN or W-8ECI) or an exemption is otherwise established.
Payment of the proceeds from a non-U.S. holder’s disposition of our common stock made by or through a foreign office of a broker will not be subject to information reporting or backup withholding, except that information reporting (but generally not backup withholding) may apply to those payments if the broker does not have documentary evidence that the beneficial owner is a non-U.S. holder, an exemption is not otherwise established and the broker is:
| • | a U.S. person, as defined in the Code; |
| • | a controlled foreign corporation for U.S. federal income tax purposes; |
-156-
Table of Contents
| • | a foreign person 50% or more of whose gross income is effectively connected with a U.S. trade or business for a specified three-year period; or |
| • | a foreign partnership if at any time during its tax year (1) one or more of its partners are U.S. persons who hold in the aggregate more than 50% of the income or capital interest in the partnership or (2) it is engaged in the conduct of a U.S. trade or business. |
Payment of the proceeds from a non-U.S. holder’s disposition of our common stock made by or through the U.S. office of a broker generally will be subject to information reporting and backup withholding unless the non-U.S. holder certifies as to its non-U.S. status (such as by providing a valid IRS Form W-8BEN or W-8ECI) or otherwise establishes an exemption from information reporting and backup withholding.
Backup withholding is not an additional tax. Taxpayers may use amounts withheld as a credit against their U.S. federal income tax liability or may claim a refund if they timely provide certain information to the IRS.
U.S. Federal Estate Tax
Shares of common stock held (or deemed held) by an individual who is a non-U.S. holder at the time of his or her death will be included in such non-U.S. holder’s gross estate for U.S. federal estate tax purposes, unless an applicable estate tax treaty provides otherwise, and thus may be subject to U.S. federal estate tax.
Additional Withholding Tax Relating to Foreign Accounts
Legislation enacted in 2010 will generally impose a U.S. federal withholding tax of 30% on dividends and the gross proceeds of a disposition of our common stock paid after December 31, 2012 to a foreign financial institution (whether holding stock for its own account or on behalf of its account holders/investors) unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which includes certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). The legislation will also generally impose a U.S. federal withholding tax of 30% on dividends and the gross proceeds of a disposition of our common stock paid after December 31, 2012 to any other foreign entity unless such entity provides the withholding agent with a certification identifying the direct and indirect U.S. owners of the entity.
Recent administrative guidance provides, however, that such withholding would generally apply only to dividends paid on or after January 1, 2014, and to other “withholdable payments” (including payments of gross proceeds from a sale or other disposition of our common stock) made on or after January 1, 2017. Under certain circumstances, a holder might be eligible for refunds or credits of such taxes. Prospective investors are encouraged to consult with their own tax advisors regarding the possible impact of these rules on their investment in our common stock.
WE RECOMMEND THAT PROSPECTIVE INVESTORS CONSULT THEIR TAX ADVISORS REGARDING THE PARTICULAR U.S. FEDERAL INCOME TAX CONSEQUENCES TO THEM OF THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK, AS WELL AS ANY TAX CONSEQUENCES ARISING UNDER ANY STATE, LOCAL OR FOREIGN TAX LAWS, ANY APPLICABLE INCOME TAX TREATIES, OR ANY OTHER U.S. FEDERAL TAX LAWS (INCLUDING ESTATE AND GIFT TAX LAWS).
-157-
Table of Contents
Aegis Capital Corp. is acting as the sole manager of the offering and as representative of the underwriters, or the “Representative.” We have entered into an underwriting agreement, dated , 2013, with the Representative. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to each underwriter named below and each underwriter named below has severally and not jointly agreed to purchase from us, at the public offering price per share less the underwriting discounts set forth on the cover page of this prospectus, the number of shares of common stock listed next to its name in the following table:
| Name of Underwriter |
Number of Shares | |||
| Aegis Capital Corp. |
||||
| Feltl and Company, Inc. |
||||
|
|
|
|||
| Total |
5,000,000 | |||
|
|
|
|||
The underwriters are committed to purchase all the shares of common stock offered by us other than those covered by the option to purchase additional shares described below, if they purchase any shares. The obligations of the underwriters may be terminated upon the occurrence of certain events specified in the underwriting agreement. Furthermore, pursuant to the underwriting agreement, the underwriters’ obligations are subject to customary conditions, representations and warranties contained in the underwriting agreement, such as receipt by the underwriters of officers’ certificates and legal opinions.
We have agreed to indemnify the underwriters against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect thereof.
The underwriters are offering the shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel and other conditions specified in the underwriting agreement. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 45 days after the date of this prospectus, permits the underwriters to purchase a maximum of 750,000 additional shares (15% of the shares sold in this offering) from us to cover over-allotments, if any. If the underwriters exercise all or part of this option, they will purchase shares covered by the option at the public offering price per share that appears on the cover page of this prospectus, less the underwriting discount. If this option is exercised in full, the total price to the public will be $ and the total net proceeds, before expenses, to us will be $ .
Discount. The following table shows the public offering price, underwriting discount and proceeds, before expenses, to us. The information assumes either no exercise or full exercise by the underwriters of their over-allotment option.
| Per Share |
Total Without Over- Allotment Option |
Total With Over- Allotment Option |
||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discount (7%) |
$ | $ | $ | |||||||||
| Non-accountable expense allowance(1) |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to us |
$ | $ | $ | |||||||||
| (1) | The expense allowance of 1% is not payable with respect to the shares sold upon exercise of the underwriters’ over-allotment option. |
The underwriters propose to offer the shares offered by us to the public at the public offering price per share set forth on the cover of this prospectus. In addition, the underwriters may offer some of the shares to other
-158-
Table of Contents
securities dealers at such price less a concession of $ per share. If all of the shares offered by us are not sold at the public offering price per share, the underwriters may change the offering price per share and other selling terms by means of a supplement to this prospectus.
We have paid an expense deposit of $25,000 to the Representative, which will be applied against the accountable expenses that will be paid by us to the Representative in connection with this offering. The underwriting agreement provides that in the event the offering is terminated, the $25,000 expense deposit paid to the Representative will be returned to us to the extent that offering expenses are not actually incurred by the Representative.
We have also agreed to pay the Representative’s expenses relating to the offering, including (a) all fees, expenses and disbursements relating to background checks of our officers and directors in an amount not to exceed $2,500 per individual or $10,000 in the aggregate; (b) all filing fees incurred in clearing this offering with FINRA; (c) all fees, expenses and disbursements relating to the registration, qualification or exemption of securities offered under the securities laws of foreign jurisdictions designated by the underwriters; (d) the costs associated with bound or compact-disc volumes of the public offering materials as well as commemorative mementos and lucite tombstones, each of which we or our designee will provide within a reasonable time after the closing of the offering in such quantities as the Representative may reasonably request in an amount not to exceed $3,000 in the aggregate; (e) upon successfully completing this offering, $21,775 for the underwriters’ use of Ipreo’s book-building, prospectus tracking and compliance software for this offering; and (f) upon successfully completing this offering, up to $20,000 of the Representative’s actual accountable road show expenses for the offering.
We estimate that the total expenses of the offering payable by us, excluding underwriting discounts and commissions, will be approximately $3.3 million.
Discretionary Accounts. The underwriters do not intend to confirm sales of the securities offered hereby to any accounts over which they have discretionary authority.
Lock-Up Agreements. Pursuant to certain “lock-up” agreements, we, our executive officers and directors, and holders of substantially all of our outstanding shares of common stock on a fully diluted basis (including shares underlying options, warrants and convertible securities) have agreed, subject to certain exceptions, not to offer, sell, assign, transfer, pledge, contract to sell, or otherwise dispose of or announce the intention to otherwise dispose of, or enter into any swap, hedge or similar agreement or arrangement that transfers, in whole or in part, the economic risk of ownership of, directly or indirectly, engage in any short selling of any common stock or securities convertible into or exchangeable or exercisable for any common stock, whether currently owned or subsequently acquired, without the prior written consent of the underwriter, for a period of six (6) months from the date of effectiveness of the offering.
The lock-up period described in the preceding paragraph will be automatically extended if: (1) during the last 17 days of the restricted period, we issue an earnings release or announce material news or a material event; or (2) prior to the expiration of the lock-up period, we announce that we will release earnings results during the 16-day period beginning on the last day of the lock-up period, in which case the restrictions described in the preceding paragraph will continue to apply until the expiration of the 18-day period beginning on the date of the earnings release, unless the Representative waives this extension in writing; provided, however, that this lock-up period extension shall not apply to the extent that FINRA has amended or repealed NASD Rule 2711(f)(4), or has otherwise provided written interpretive guidance regarding such rule, in each case, so as to eliminate the prohibition of any broker, dealer, or member of a national securities association from publishing or distributing any research report, with respect to the securities of an emerging growth company (as defined in the JOBS Act) prior to or after the expiration of any agreement between the broker, dealer, or member of a national securities association and the emerging growth company or its shareholders that restricts or prohibits the sale of securities held by the emerging growth company or its shareholders after the initial public offering date.
-159-
Table of Contents
Right of First Refusal. Subject to certain limited exceptions, until twelve (12) months after the closing date of this offering, the Representative has a right of first refusal to purchase for its account or to sell for our account, or any subsidiary or successor, any securities of our company or any such subsidiary or successor which we or any subsidiary or successor may seek to sell in public or private equity and public debt offerings during such twelve (12)-month period.
Electronic Offer, Sale and Distribution of Shares. A prospectus in electronic format may be made available on the websites maintained by one or more of the underwriters or selling group members, if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses electronically. The Representative may agree to allocate a number of shares to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations. Other than the prospectus in electronic format, the information on these websites is not part of, nor incorporated by reference into, this prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or any underwriter in its capacity as underwriter, and should not be relied upon by investors.
Stabilization. In connection with this offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate-covering transactions, penalty bids and purchases to cover positions created by short sales.
| • | Stabilizing transactions permit bids to purchase shares so long as the stabilizing bids do not exceed a specified maximum, and are engaged in for the purpose of preventing or retarding a decline in the market price of the shares while the offering is in progress. |
| • | Over-allotment transactions involve sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing shares in the open market. |
| • | Syndicate covering transactions involve purchases of shares in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared with the price at which they may purchase shares through exercise of the over- allotment option. If the underwriters sell more shares than could be covered by exercise of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could be downward pressure on the price of the shares in the open market that could adversely affect investors who purchase in the offering. |
| • | Penalty bids permit the Representative to reclaim a selling concession from a syndicate member when the shares originally sold by that syndicate member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our shares or common stock or preventing or retarding a decline in the market price of our shares or common stock. As a result, the price of our common stock in the open market may be higher than it would otherwise be in the absence of these transactions. Neither we nor the underwriters make
-160-
Table of Contents
any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The NASDAQ Capital Market, in the over-the-counter market or otherwise and, if commenced, may be discontinued at any time.
Passive market making. In connection with this offering, underwriters and selling group members may engage in passive market making transactions in our common stock on The NASDAQ Capital Market in accordance with Rule 103 of Regulation M under the Exchange Act, during a period before the commencement of offers or sales of the shares and extending through the completion of the distribution. A passive market maker must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered below the passive market maker’s bid, then that bid must then be lowered when specified purchase limits are exceeded.
Other Relationships. Certain of the underwriters and their affiliates have provided, and may in the future provide, various investment banking, commercial banking and other financial services for us and our affiliates for which they have received, and may in the future receive, customary fees. However, except as disclosed in this prospectus, we have no present arrangements with any of the underwriters for any further services.
Offer restrictions outside the United States
Other than in the United States, no action has been taken by us or the underwriters that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to the offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter 6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer for the offeree under this prospectus.
China
The information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s Republic of China (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly to “qualified domestic institutional investors.”
-161-
Table of Contents
European Economic Area—Belgium, Germany, Luxembourg and Netherlands
The information in this document has been prepared on the basis that all offers of securities will be made pursuant to an exemption under the Directive 2003/71/EC (“Prospectus Directive”), as implemented in Member States of the European Economic Area (each, a “Relevant Member State”), from the requirement to produce a prospectus for offers of securities.
An offer to the public of securities has not been made, and may not be made, in a Relevant Member State except pursuant to one of the following exemptions under the Prospectus Directive as implemented in that Relevant Member State:
(a) to legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity that has two or more of (i) an average of at least 250 employees during its last fiscal year; (ii) a total balance sheet of more than €43,000,000 (as shown on its last annual unconsolidated or consolidated financial statements) and (iii) an annual net turnover of more than €50,000,000 (as shown on its last annual unconsolidated or consolidated financial statements);
(c) to fewer than 100 natural or legal persons (other than qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive) subject to obtaining the prior consent of the Company or any underwriter for any such offer; or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall result in a requirement for the publication by the Company of a prospectus pursuant to Article 3 of the Prospectus Directive.
France
This document is not being distributed in the context of a public offering of financial securities (offre au public de titres financiers) in France within the meaning of Article L.411-1 of the French Monetary and Financial Code (Code monétaire et financier) and Articles 211-1 et seq. of the General Regulation of the French Autorité des marchés financiers (“AMF”). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France.
This document and any other offering material relating to the securities have not been, and will not be, submitted to the AMF for approval in France and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in France.
Such offers, sales and distributions have been and shall only be made in France to (i) qualified investors (investisseurs qualifiés) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-1 to D.411-3, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation and/or (ii) a restricted number of non-qualified investors (cercle restreint d’investisseurs) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-4, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation.
Pursuant to Article 211-3 of the General Regulation of the AMF, investors in France are informed that the securities cannot be distributed (directly or indirectly) to the public by the investors otherwise than in accordance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 to L.621-8-3 of the French Monetary and Financial Code.
Ireland
The information in this document does not constitute a prospectus under any Irish laws or regulations and this document has not been filed with or approved by any Irish regulatory authority as the information has
-162-
Table of Contents
not been prepared in the context of a public offering of securities in Ireland within the meaning of the Irish Prospectus (Directive 2003/71/EC) Regulations 2005 (the “Prospectus Regulations”). The securities have not been offered or sold, and will not be offered, sold or delivered directly or indirectly in Ireland by way of a public offering, except to (i) qualified investors as defined in Regulation 2(l) of the Prospectus Regulations and (ii) fewer than 100 natural or legal persons who are not qualified investors.
Israel
The securities offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority, or the ISA, nor have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with the offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Italy
The offering of the securities in the Republic of Italy has not been authorized by the Italian Securities and Exchange Commission (Commissione Nazionale per le Societ—$$—Aga e la Borsa, “CONSOB” pursuant to the Italian securities legislation and, accordingly, no offering material relating to the securities may be distributed in Italy and such securities may not be offered or sold in Italy in a public offer within the meaning of Article 1.1(t) of Legislative Decree No. 58 of 24 February 1998 (“Decree No. 58”), other than:
| • | to Italian qualified investors, as defined in Article 100 of Decree no. 58 by reference to Article 34-ter of CONSOB Regulation no. 11971 of 14 May 1999 (“Regulation no. 1197l”) as amended (“Qualified Investors”); and |
| • | in other circumstances that are exempt from the rules on public offer pursuant to Article 100 of Decree No. 58 and Article 34-ter of Regulation No. 11971 as amended. |
Any offer, sale or delivery of the securities or distribution of any offer document relating to the securities in Italy (excluding placements where a Qualified Investor solicits an offer from the issuer) under the paragraphs above must be:
| • | made by investment firms, banks or financial intermediaries permitted to conduct such activities in Italy in accordance with Legislative Decree No. 385 of 1 September 1993 (as amended), Decree No. 58, CONSOB Regulation No. 16190 of 29 October 2007 and any other applicable laws; and |
| • | in compliance with all relevant Italian securities, tax and exchange controls and any other applicable laws. |
Any subsequent distribution of the securities in Italy must be made in compliance with the public offer and prospectus requirement rules provided under Decree No. 58 and the Regulation No. 11971 as amended, unless an exception from those rules applies. Failure to comply with such rules may result in the sale of such securities being declared null and void and in the liability of the entity transferring the securities for any damages suffered by the investors.
Japan
The securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an
-163-
Table of Contents
exemption from the registration requirements applicable to a private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly, in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition by any such person of securities is conditional upon the execution of an agreement to that effect.
Portugal
This document is not being distributed in the context of a public offer of financial securities (oferta pública de valores mobiliários) in Portugal, within the meaning of Article 109 of the Portuguese Securities Code (Código dos Valores Mobiliários). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in Portugal. This document and any other offering material relating to the securities have not been, and will not be, submitted to the Portuguese Securities Market Commission (Comissão do Mercado de Valores Mobiliários) for approval in Portugal and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in Portugal, other than under circumstances that are deemed not to qualify as a public offer under the Portuguese Securities Code. Such offers, sales and distributions of securities in Portugal are limited to persons who are “qualified investors” (as defined in the Portuguese Securities Code). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Sweden
This document has not been, and will not be, registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority). Accordingly, this document may not be made available, nor may the securities be offered for sale in Sweden, other than under circumstances that are deemed not to require a prospectus under the Swedish Financial Instruments Trading Act (1991:980) (Sw. lag (1991:980) om handel med finansiella instrument). Any offering of securities in Sweden is limited to persons who are “qualified investors” (as defined in the Financial Instruments Trading Act). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority.
This document is personal to the recipient only and not for general circulation in Switzerland.
United Arab Emirates
Neither this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates,
-164-
Table of Contents
nor has the Company received authorization or licensing from the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to market or sell the securities within the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. No services relating to the securities, including the receipt of applications and/or the allotment or redemption of such shares, may be rendered within the United Arab Emirates by the Company.
No offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
United Kingdom
Neither the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”)) has been published or is intended to be published in respect of the securities. This document is issued on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document, except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA.
This document should not be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in the United Kingdom.
Any invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply to Kips Bay.
In the United Kingdom, this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005 (“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”). The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
-165-
Table of Contents
The validity of the shares offered hereby will be passed upon for us by Lowenstein Sandler LLP, Roseland, New Jersey. Certain legal matters in connection with this offering will be passed upon for the underwriters by Reed Smith LLP, New York, New York.
McGladrey LLP, our independent registered public accounting firm, has audited our balance sheets as of December 31, 2010 and 2011, and the related statements of operations, changes in stockholders’ equity (deficit) and cash flows for the three years ended December 31, 2011, as set forth in their report. We have included our financial statements in this prospectus and in this registration statement in reliance on the report of McGladrey LLP given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1, including exhibits and schedules, under the Securities Act that registers the shares of our common stock to be sold in this offering. This prospectus does not contain all the information contained in the registration statement and the exhibits and schedules filed as part of the registration statement. For further information with respect to us and our common stock, we refer you to the registration statement and the exhibits and schedules filed as part of the registration statement. Statements contained in this prospectus as to the contents of any contract or other document are not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, we refer you to the copies of the contract or document that has been filed. Each statement in this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit.
Upon the consummation of this offering, we will file annual, quarterly and current reports, proxy statements and other information with the SEC under the Exchange Act. You can read our SEC filings, including the registration statement, at the SEC’s website at www.sec.gov.
You may read and copy this information at the SEC’s Public Reference Room at 100 F Street, N.E., Washington D.C. 20549, at prescribed rates. You may obtain information regarding the operation of the public reference room by calling the SEC at 1-800-SEC-0330. The SEC also maintains a website (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
Our website address is www.cancergenetics.com. The information contained in, and that can be accessed through, our website is not incorporated into and is not part of this prospectus.
-166-
Table of Contents
| Array-CGH | Array Comparative Genomic Hybridization; a technique that utilizes a DNA based microarray for the detection of genomic copy number changes; our proprietary microarrays are a type of array-CGH | |
| CE | Conformité Européenne; a conformity mark which is placed on all products including medical devices marketed in the European Economic Area | |
| CMS | Centers for Medicare & Medicaid Services; a U.S. federal agency that administers Medicare, Medicaid and the Children’s Health Insurance Program | |
| CLL | Chronic Lymphocytic Leukemia; the most common type of leukemia that affects B cell lymphocytes | |
| CLIA | Clinical Laboratory Improvement Amendments of 1988; federal regulatory standards that apply to all clinical laboratory testing performed on humans in the United States. | |
| CAP | College of American Pathologists; the leading organization of board-certified pathologists, serving patients, pathologists, and the public by fostering and advocating excellence in the practice of pathology and laboratory medicine worldwide | |
| CPT | Current Procedure Terminology | |
| DLBCL | Diffuse Large B Cell Lymphoma; an aggressive form of non-Hodgkin’s lymphoma | |
| FISH | Fluorescence in situ Hybridization; a molecular cytogenetic technique that is used to detect chromosomal aberrations that include deletions, amplifications and translocations; DNA FISH probes are fluorescently labeled segments of DNA that are complementary to specific sequences on a chromosome | |
| FL | Follicular Lymphoma; a sub type of non-hodgkin’s lymphoma that is indolent in nature | |
| FDCA | Food, Drug and Cosmetic Act | |
| FFPE | Formalin-Fixed Paraffin-Embedded; a type of specimen material typically available for solid tumors | |
| HITECH | Health Information Technology for Economic and Clinical Health Act | |
| HPV | Human papillomavirus; a group of viruses that contain more than one-hundred different strains responsible for causing sexually transmitted diseases in humans | |
| IHC | ImmunoHistoChemistry | |
| LDTs | Laboratory Developed Tests; assays developed in the laboratory for diagnostic or prognostic purposes | |
| MCL | Mantle-Cell Lymphoma; an aggressive sub type of B cell lymphoma with a poor long term prognosis | |
| MIPPA | Medicare Improvements for Patients and Providers Act of 2008 | |
-167-
Table of Contents
| Next gen sequencing |
A method of DNA sequencing using high-throughput sequencing technologies that parallelize the DNA sequencing process, producing thousands or millions of sequences at once. | |
| PPACA | Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act | |
| PMA | Pre-Market Approval; the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices | |
| SLL | Small Lymphocytic Lymphoma; a different manifestation of CLL that primarily manifests in the lymphatic system | |
-168-
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
| Consolidated Financial Report September 30, 2012 |
||||
| F-2 | ||||
| F-3 | ||||
| Consolidated Statements of Changes in Stockholders’ Equity (Deficit) (Unaudited) |
F-4 | |||
| F-5 | ||||
| F-6 - F-24 | ||||
| Consolidated Financial Report December 31, 2011 |
||||
| Report of Independent Registered Public Accounting Firm |
F-25 | |||
| F-26 | ||||
| F-27 | ||||
| Consolidated Statements of Changes in Stockholders’ Equity (Deficit) |
F-28 | |||
| F-29 | ||||
| F-31 - F-55 |
F-1
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Balance Sheets
| September 30, 2012 (Unaudited) |
December 31, 2011 |
|||||||
| ASSETS |
||||||||
| CURRENT ASSETS |
||||||||
| Cash and cash equivalents |
$ | 903,257 | $ | 2,417,256 | ||||
| Accounts receivable, net of allowance for doubtful accounts of 2012 $44,500; 2011 $24,050 |
839,378 | 688,980 | ||||||
| Other current assets |
452,072 | 269,269 | ||||||
|
|
|
|
|
|||||
| Total current assets |
2,194,707 | 3,375,505 | ||||||
| FIXED ASSETS, net of accumulated depreciation |
907,687 | 1,140,636 | ||||||
| OTHER ASSETS |
||||||||
| Security deposits |
1,564 | 1,564 | ||||||
| Restricted cash |
250,000 | 200,000 | ||||||
| Loan guarantee and financing fees, net of accumulated amortization of 2012 $454,746; 2011 $333,202 |
1,211,254 | 497,798 | ||||||
| Patents |
377,721 | 204,687 | ||||||
| Deferred IPO costs |
2,995,396 | 1,611,273 | ||||||
|
|
|
|
|
|||||
| 4,835,935 | 2,515,322 | |||||||
|
|
|
|
|
|||||
| Total Assets |
$ | 7,938,329 | $ | 7,031,463 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT |
||||||||
| CURRENT LIABILITIES |
||||||||
| Accounts payable and accrued expenses |
$ | 3,979,485 | $ | 4,317,121 | ||||
| Obligations under capital leases, current portion |
18,589 | 36,209 | ||||||
| Deferred revenue |
422,377 | — | ||||||
| Notes payable, current portion |
3,530,927 | 100,000 | ||||||
| Line of credit |
2,751,006 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
10,702,384 | 4,453,330 | ||||||
| Obligations under capital leases |
10,174 | 24,559 | ||||||
| Deferred rent payable |
162,675 | 156,311 | ||||||
| Notes payable, long-term |
2,798,696 | 1,912,365 | ||||||
| Line of credit |
6,000,000 | 8,437,255 | ||||||
| Warrant liability |
8,421,000 | 11,113,000 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
28,094,929 | 26,096,820 | ||||||
| STOCKHOLDERS’ DEFICIT |
||||||||
| Series A Preferred Stock, authorized 588,000 shares $0.0001 par value (purchase price liquidation preference of $4,971,866 or can elect to convert to common stock), 587,691 shares issued and outstanding |
59 | 59 | ||||||
| Series B Preferred Stock, authorized 2,000,000 shares $0.0001 par value (purchase price liquidation preference of $9,108,000 or can elect to convert to common stock), 1,821,600 shares issued and outstanding |
182 | 182 | ||||||
| Common stock, authorized 24,000,000 shares $0.0001 par value, 3,365,311 and 3,237,566 shares issued and outstanding as of September 30, 2012 and December 31, 2011 respectively |
337 | 324 | ||||||
| Additional paid-in capital |
24,750,612 | 23,220,478 | ||||||
| Treasury stock |
(17,442 | ) | (17,442 | ) | ||||
| Accumulated deficit |
(44,890,348 | ) | (42,268,958 | ) | ||||
|
|
|
|
|
|||||
| Total Stockholders’ Deficit |
(20,156,600 | ) | (19,065,357 | ) | ||||
|
|
|
|
|
|||||
| Total Liabilities and Stockholders’ Deficit |
$ | 7,938,329 | $ | 7,031,463 | ||||
|
|
|
|
|
|||||
See Notes to Unaudited Consolidated Financial Statements.
F-2
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Operations
(Unaudited)
| Nine Months Ended September 30, | ||||||||
| 2012 | 2011 | |||||||
| Revenue |
$ | 3,225,831 | $ | 2,143,827 | ||||
| Cost of revenues |
2,880,242 | 2,384,994 | ||||||
|
|
|
|
|
|||||
| Gross profit (loss) |
345,589 | (241,167 | ) | |||||
| Operating expenses: |
||||||||
| Research and development |
1,551,672 | 1,442,327 | ||||||
| General and administrative |
3,475,301 | 3,159,578 | ||||||
| Sales and marketing |
1,049,996 | 1,200,461 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
6,076,969 | 5,802,366 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(5,731,380 | ) | (6,043,533 | ) | ||||
| Other income (expense): |
||||||||
| Interest expense |
(3,260,010 | ) | (918,330 | ) | ||||
| Change in fair value of warrant liability |
6,370,000 | (9,881,000 | ) | |||||
|
|
|
|
|
|||||
| Total other income (expense) |
3,109,990 | (10,799,330 | ) | |||||
|
|
|
|
|
|||||
| Loss before income taxes |
(2,621,390 | ) | (16,842,863 | ) | ||||
| Income tax provision (benefit) |
— | — | ||||||
|
|
|
|
|
|||||
| Net loss |
$ | (2,621,390 | ) | $ | (16,842,863 | ) | ||
|
|
|
|
|
|||||
| Basic net loss per share |
$ | (0.78 | ) | $ | (5.30 | ) | ||
|
|
|
|
|
|||||
| Diluted net loss per share |
$ | (2.66 | ) | $ | (5.30 | ) | ||
|
|
|
|
|
|||||
| Basic Weighted Average Shares Outstanding |
3,351,324 | 3,178,701 | ||||||
|
|
|
|
|
|||||
| Diluted Weighted Average Shares Outstanding |
3,375,210 | 3,178,701 | ||||||
|
|
|
|
|
|||||
See Notes to Unaudited Consolidated Financial Statements.
F-3
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Changes in Stockholders’ Equity (Deficit)
For the year ended December 31, 2011, and the nine-months ended September 30, 2012 (Unaudited)
| Preferred Stock Series A |
Preferred Stock Series B |
Common Stock | Additional Paid-in Capital |
Treasury Stock |
Accumulated Deficit |
Total | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||
| Balance, January 1, 2011 |
587,691 | $ | 59 | 1,821,600 | $ | 182 | 3,178,701 | $ | 318 | $ | 15,662,368 | $ | (17,442 | ) | $ | (22,381,918 | ) | $ | (6,736,433 | ) | ||||||||||||||||||||
| Stock based compensation—employees |
— | — | — | — | — | — | 306,307 | — | — | 306,307 | ||||||||||||||||||||||||||||||
| Stock based compensation—non-employees |
— | — | — | — | — | — | 636,810 | — | — | 636,810 | ||||||||||||||||||||||||||||||
| Exercise of warrants |
— | — | — | — | 8,865 | 1 | 49,998 | — | — | 49,999 | ||||||||||||||||||||||||||||||
| Warrant liability reclassified to equity |
— | — | — | — | — | — | 6,415,000 | — | — | 6,415,000 | ||||||||||||||||||||||||||||||
| Issuance of common stock |
— | — | — | — | 50,000 | 5 | 149,995 | — | — | 150,000 | ||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (19,887,040 | ) | (19,887,040 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
| Balance, December 31, 2011 |
587,691 | 59 | 1,821,600 | 182 | 3,237,566 | 324 | 23,220,478 | (17,442 | ) | (42,268,958 | ) | (19,065,357 | ) | |||||||||||||||||||||||||||
| Stock based compensation—employees |
— | — | — | — | — | — | 256,302 | — | — | 256,302 | ||||||||||||||||||||||||||||||
| Stock based compensation—non-employees |
— | — | — | — | — | — | 509,865 | — | — | 509,865 | ||||||||||||||||||||||||||||||
| Warrant extension |
— | — | — | — | — | — | 144,000 | — | — | 144,000 | ||||||||||||||||||||||||||||||
| Exercise of warrants |
— | — | — | — | 127,745 | 13 | 619,967 | — | — | 619,980 | ||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (2,621,390 | ) | (2,621,390 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
| Balance, September 30, 2012 |
587,691 | $ | 59 | 1,821,600 | $ | 182 | 3,365,311 | $ | 337 | $ | 24,750,612 | $ | (17,442 | ) | $ | (44,890,348 | ) | $ | (20,156,600 | ) | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
See Notes to Unaudited Consolidated Financial Statements
F-4
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Cash Flows
(Unaudited)
| Nine Months Ended September 30, |
||||||||
| 2012 | 2011 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES |
||||||||
| Net loss |
$ | (2,621,390 | ) | $ | (16,842,863 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation |
266,489 | 270,355 | ||||||
| Amortization |
11,422 | 7,292 | ||||||
| Provision (recovery) for bad debts |
(528 | ) | 340,507 | |||||
| Equity-based consulting and compensation expenses |
766,167 | 680,037 | ||||||
| Extension of warrants |
144,000 | — | ||||||
| Derivative warrants issued for consulting services |
— | 69,000 | ||||||
| Change in fair value of warrant liability |
(6,370,000 | ) | 9,881,000 | |||||
| Amortization of loan guarantee and financing fees |
952,544 | 380,583 | ||||||
| Accretion of discount on debt |
1,559,009 | 352,731 | ||||||
| Deferred rent |
6,364 | 19,442 | ||||||
| Change in working capital components: |
||||||||
| Accounts receivable |
(149,870 | ) | (327,214 | ) | ||||
| Other current assets |
(182,803 | ) | 547,163 | |||||
| Accounts payable, accrued expenses and deferred revenue |
(144,773 | ) | 179,886 | |||||
|
|
|
|
|
|||||
| Net cash (used in) operating activities |
(5,763,369 | ) | (4,442,081 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES |
||||||||
| Purchase of fixed assets |
(33,540 | ) | (207,234 | ) | ||||
| Patent costs |
(184,456 | ) | — | |||||
| (Increase) decrease in restricted cash |
(50,000 | ) | 238,400 | |||||
|
|
|
|
|
|||||
| Net cash provided by (used in) investing activities |
(267,996 | ) | 31,166 | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES |
||||||||
| Principal payments on capital lease obligations |
(32,005 | ) | (32,463 | ) | ||||
| Payment of equity issuance costs |
(1,190,609 | ) | (142,933 | ) | ||||
| Proceeds from warrant exercises |
619,980 | — | ||||||
| Proceeds from borrowings on notes payable |
5,120,000 | — | ||||||
| Proceeds from borrowings on line of credit |
— | 3,000,000 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
4,517,366 | 2,824,604 | ||||||
|
|
|
|
|
|||||
| Net (decrease) in cash and cash equivalents |
(1,513,999 | ) | (1,586,311 | ) | ||||
| CASH AND CASH EQUIVALENTS |
||||||||
| Beginning |
2,417,256 | 1,779,251 | ||||||
|
|
|
|
|
|||||
| Ending |
$ | 903,257 | $ | 192,940 | ||||
|
|
|
|
|
|||||
| SUPPLEMENTAL CASH FLOW DISCLOSURE |
||||||||
| Cash paid for interest |
$ | 761,458 | $ | 184,917 | ||||
| SUPPLEMENTAL DISCLOSURE OF NONCASH |
||||||||
| INVESTING AND FINANCING ACTIVITIES |
||||||||
| Fixed assets acquired through capital lease arrangement |
$ | — | $ | 49,600 | ||||
| Warrants issued with notes payable and line of credit |
2,048,000 | 1,019,000 | ||||||
| Warrants issued for loan guarantee fee |
755,000 | 831,000 | ||||||
| Warrants issued for financing fees |
727,000 | — | ||||||
| Warrant liability reclassified to equity |
— | 6,415,000 | ||||||
| Accrued IPO costs |
1,384,123 | 618,198 | ||||||
| Accrued expenses recorded as financing fees |
184,000 | — | ||||||
| Accrued expenses reclassified as derivative warrant liability |
148,000 | — | ||||||
See Notes to Unaudited Consolidated Financial Statements.
F-5
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
Note 1. Organization, Description of Business, Reverse Stock Split and Charter Amendment
We were incorporated in the State of Delaware on April 8, 1999 and have offices and a laboratory located in Rutherford, New Jersey. Our wholly owned subsidiary, Cancer Genetics Italia SRL (“CGI Italia”), manufactures DNA probes. CGI Italia had approximately $304,000 in total assets at September 30, 2012 and approximately $60,000 and $49,000 in total revenue for the nine months ended September 30, 2012 and 2011, respectively.
We are a diagnostics company focused on developing and commercializing proprietary genomic tests and services to improve the diagnosis, prognosis and response to treatment of cancer (theranosis). Our proprietary tests target cancers where prognosis information is critical and where predicting treatment outcomes using currently available techniques is limited. These cancers include hematological, urogenital and HPV-associated cancers. We have commercially launched MatBA® -CLL and -SLL, our first proprietary microarray diagnostic tests, and seek to provide our tests and services to oncologists and pathologists at hospitals, cancer centers and physician offices.
On April 23, 2012, the Board of Directors approved a charter amendment giving the Board the authority to effect a split within the range of one to two shares for one share, which amendment was approved by the stockholders on May 3, 2012. The Board anticipates approving a 1-for-2 reverse stock split of the Company’s common stock to be effected immediately prior to the effectiveness of the Company’s registration statement. All shares and per share information referenced throughout the consolidated financial statements have been retroactively adjusted to reflect this 1-for-2 reverse stock split.
Our Board of Directors and stockholders have approved a charter amendment that reduces the threshold for automatic conversion of our Series A preferred stock, our Series A-1 preferred stock and our Series B preferred stock from $25.0 million to $15.0 million. If we receive gross proceeds of at least $15.0 million in this offering, each outstanding share of our Series A preferred stock will automatically convert into one and one-half shares of common stock and each outstanding share of our Series A-1 and our Series B preferred stock will automatically convert into one-half of one share of common stock.
Note 2. Significant Accounting Policies
Basis of presentation: The accompanying unaudited financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States of America for interim financial information and with the instructions for interim reporting as they are prescribed by the Securities and Exchange Commission. Accordingly, they do not include all of the information and footnotes required by accounting principles generally accepted in the United States of America for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring accruals) considered necessary to make the financial statements not misleading have been included. Operating results for the nine-month period ended September 30, 2012 are not necessarily indicative of the results that may be expected for the year ending December 31, 2012. These statements should be read in conjunction with the financial statements and related notes for the year ended December 31, 2011.
Liquidity: Our primary sources of liquidity have been funds generated from debt financing, the sale of shares of common and preferred stock, grants in lieu of federal income tax credits and National Institute of Health grants. We plan on raising capital in the near term in an initial public offering (“IPO”); however, without the proceeds from this offering, we believe our current cash resources, including the $1.0 million in debt
F-6
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
financing we raised after September 30, 2012, are sufficient to satisfy our liquidity requirements at our current level of operations through December 31, 2012. If we do not consummate this offering prior to the end of the year, we expect that we will need to raise additional financing in the first quarter of 2013, which might not be available on favorable terms, if at all. We have had and will continue to have discussions with certain of our stockholders regarding potential additional financing, but we can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. On a long-term basis and based on our current operating projections, we will need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing, which is not in place at this time, may be from the sale of equity or convertible or other debt securities in a public or private offering, from an additional credit facility or strategic partnership coupled with an investment in the Company or a combination of both. We may be unable to raise sufficient additional financing on terms that are acceptable, if at all. Our failure to raise additional capital and in sufficient amounts will severely impact our ability to expand our business.
Principles of consolidation: The accompanying consolidated financial statements include the accounts of Cancer Genetics, Inc. and our wholly owned subsidiary, Cancer Genetics Italia SRL. All significant intercompany account balances and transactions have been eliminated in consolidation.
Use of estimates and assumptions: The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates made by management include, among others, realization of amounts billed, realization of long-lived assets, realization of intangible assets, accruals for litigation and registration payments and assumptions used to value stock options and warrants. Actual results could differ from those estimates.
Risks and uncertainties: We operate in an industry that is subject to intense competition, government regulation and rapid technological change. Our operations are subject to significant risk and uncertainties including financial, operational, technological, regulatory and other risks, including the potential risk of business failure.
Cash and cash equivalents: Highly liquid investments with original maturities of three months or less when purchased are considered to be cash equivalents. Financial instruments which potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents. We maintain cash and cash equivalents with high-credit quality financial institutions. At times, such amounts may exceed insured limits. We have not experienced any losses in such accounts and believe we are not exposed to any significant credit risk on our cash and cash equivalents.
Revenue recognition: Revenue is recognized in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 605, Revenue Recognition, and ASC 954-605 Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence that an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the customer or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. In determining whether the price is fixed or determinable, we consider payment limits imposed by insurance carriers and Medicare and the amount of revenue recorded takes into account the historical percentage of revenue we have collected for each type of test for each payor category. Periodically, an adjustment is made to revenue to record differences between our anticipated cash receipts from insurance carriers and Medicare and actual receipts from such payors. For the
F-7
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
periods presented, such adjustments were not significant. For direct bill customers, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, we consider whether we have sufficient payment history to reliably estimate a payor’s individual payment patterns. For new tests where there is no evidence of payment history at the time the tests are completed, we only recognize revenues once reimbursement experience can be established. We then recognize revenue equal to the amount of cash received. Sales of probes are recorded on the shipping date. We do not bill customers for shipping and handling fees and do not collect any sales or other taxes.
Revenues from grants to support product development are recognized when costs and expenses under the terms of the grant have been incurred and payments under the grants become contractually due.
Deferred IPO costs: Deferred IPO costs represent legal, accounting and other direct costs related to our effort to raise capital through an IPO. Future costs related to our IPO activities will be deferred until the completion of the IPO, at which time they will be reclassified to additional paid-in capital as a reduction of the IPO proceeds. If we terminate our plan for an IPO, any deferred costs would be expensed at that time.
Warrant liability: We account for warrants that contain an exercise price adjustment feature as liabilities. These common stock purchase warrants do not trade in an active securities market, and as such, we estimate the fair value of these warrants using the binomial lattice valuation pricing model with the assumptions as follows: The risk-free interest rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve. The expected life of the warrants is based upon the contractual life of the warrants. Volatility is estimated based on an average of the historical volatilities of the common stock of four entities with characteristics similar to those of the Company. The measurement date fair value of the underlying common shares is based upon an external valuation of our shares. (See Notes 8 and 9).
We compute the fair value of the warrant liability at each reporting period and the change in the fair value is recorded as non-cash expense or non-cash income. The key component in the value of the warrant liability is our stock price, which is subject to significant fluctuation and is not under our control. The resulting effect on our net loss is therefore subject to significant fluctuation and will continue to be so until the warrants are exercised, amended or expire. Assuming all other fair value inputs remain constant, we will record non-cash expense when the stock price increases and non-cash income when the stock price decreases.
Registration payment arrangements: We account for our obligations under registration payment arrangements in accordance with ASC 825-20, Registration Payment Arrangements. ASC 825-20 requires us to record a liability if we determine a registration payment is probable and if it can reasonably be estimated. As of September 30, 2012 and December 31, 2011, we have an accrued liability of $263,000 and $150,000, respectively, related to the issuance of Series B preferred stock.
Stock-based compensation: Stock-based compensation is accounted for in accordance with the provisions of ASC 718, Compensation-Stock Compensation, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and directors based on estimated fair values on the grant date. We estimate the fair value of stock-based awards on the date of grant using the Black-Scholes option pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. See additional information in Note 6.
All issuances of stock options or other issuances of equity instruments to employees as the consideration for services received by us are accounted for based on the fair value of the equity instrument issued.
F-8
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
We account for stock-based compensation awards to non-employees in accordance with ASC 505-50, Equity Based Payments to Non-Employees. Under ASC 505-50, we determine the fair value of the warrants or stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. Stock-based compensation awards issued to non-employees are recorded in expense and additional paid-in capital in stockholders’ deficit over the applicable service periods based on the fair value of the awards or consideration received at the vesting date.
Subsequent events: We have evaluated potential subsequent events through November 15, 2012, which is the date the financial statements were issued.
Earnings (loss) per share: Basic earnings (loss) per share is computed by dividing net income (loss) available to common shareholders by the weighted average number of common shares assumed to be outstanding during the period of computation. Diluted earnings per share is computed similar to basic earnings per share except that the numerator is adjusted for the change in fair value of the warrant liability (only if dilutive) and the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common shares had been issued and if the additional common shares were dilutive using the treasury stock method, if dilutive.
Basic net loss and diluted net loss per share data were computed as follows:
| Nine Months Ended September 30, |
||||||||
| 2012 | 2011 | |||||||
| Numerator: |
||||||||
| Net (loss) for basic earnings per share |
$ | (2,621,390 | ) | $ | (16,842,863 | ) | ||
| Less change in fair value of warrant liability |
6,370,000 | — | ||||||
|
|
|
|
|
|||||
| Net (loss) for diluted earnings per share |
$ | (8,991,390 | ) | $ | (16,842,863 | ) | ||
|
|
|
|
|
|||||
| Denominator: |
||||||||
| Weighted-average basic common shares outstanding |
3,351,324 | 3,178,701 | ||||||
| Assumed conversion of dilutive securities: |
||||||||
| Common stock purchase warrants |
23,886 | — | ||||||
|
|
|
|
|
|||||
| Potentially dilutive common shares |
23,886 | — | ||||||
|
|
|
|
|
|||||
| Denominator for diluted earnings per share – adjusted weighted-average shares |
3,375,210 | 3,178,701 | ||||||
|
|
|
|
|
|||||
| Basic net loss per share |
$ | (0.78 | ) | $ | (5.30 | ) | ||
|
|
|
|
|
|||||
| Diluted net loss per share |
$ | (2.66 | ) | $ | (5.30 | ) | ||
|
|
|
|
|
|||||
The following table summarizes potentially dilutive adjustments to the weighted average number of common shares which were excluded from the calculation:
| Nine Months Ended September 30, |
||||||||
| 2012 | 2011 | |||||||
| Common stock purchase warrants |
2,388,644 | 2,124,831 | ||||||
| Stock options |
1,383,950 | 1,396,196 | ||||||
| Common shares issuable upon conversion of Series A Preferred Stock |
881,537 | 881,537 | ||||||
| Common shares issuable upon conversion of Series B Preferred Stock |
910,800 | 910,800 | ||||||
|
|
|
|
|
|||||
| 5,564,931 | 5,313,364 | |||||||
|
|
|
|
|
|||||
F-9
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
Note 3. Revenue and Accounts Receivable
Revenue by payor type for the nine months ended September 30 is comprised of the following:
| Nine Months Ended September 30, |
||||||||
| 2012 | 2011 | |||||||
| Medicare |
$ | 586,896 | $ | 523,813 | ||||
| Direct bill (including clinical trials) |
1,158,212 | 271,866 | ||||||
| Grants and royalty |
474,973 | 220,195 | ||||||
| Insurance carrier and all others |
1,005,750 | 1,127,953 | ||||||
|
|
|
|
|
|||||
| $ | 3,225,831 | $ | 2,143,827 | |||||
|
|
|
|
|
|||||
Accounts receivable by payor type at September 30, 2012 and December 31, 2011 consists of the following:
| September 30, 2012 |
December 31, 2011 |
|||||||
| Medicare |
$ | 171,175 | $ | 152,186 | ||||
| Direct bill |
446,968 | 64,183 | ||||||
| Insurance carrier and all others |
265,735 | 496,661 | ||||||
| Allowance for doubtful accounts |
(44,500 | ) | (24,050 | ) | ||||
|
|
|
|
|
|||||
| $ | 839,378 | $ | 688,980 | |||||
|
|
|
|
|
|||||
We have historically derived a significant portion of our revenue from a limited number of test ordering sites. The test ordering sites are largely hospitals, cancer centers, reference laboratories, physician offices and clinical trial clients. Oncologists and pathologists at these sites order the tests on behalf of the needs of their oncology patients. The top five test ordering sites during the nine months ended September 30, 2012 and 2011 accounted for 61% and 62%, respectively, of our clinical testing volumes, with 47% and 28%, respectively, of the volume coming from community hospitals. In particular, during the nine months ended September 30, 2012, there were three sites which each accounted for approximately 10% or more of our revenue. A university teaching center accounting for approximately 15%, a clinical trial client accounted for approximately 12%, and a community hospital accounted for approximately 11%. During the nine months ended September 30, 2011, there were three sites which accounted for more than 10% of our revenue: a community hospital accounted for approximately 12%, a community oncology practice accounted for approximately 11%, and a university teaching center accounted for approximately 10%. We generally do not enter into formal written agreements with such testing sites and, as a result, we may lose these significant test ordering sites at any time.
Accounts receivable are reduced by an allowance for doubtful accounts, the amount of which is determined by an analysis of individual accounts. Our policy for assessing the collectability of receivables is dependent upon the major payor source of the underlying revenue. For direct bill clients, an assessment of credit worthiness is performed prior to initial engagement and is reassessed periodically. If deemed necessary, an allowance is established on receivables from direct bill clients. For insurance carriers where there is not an established pattern of collection, revenue is not recorded until cash is received. For receivables where insurance carriers have made payments to patients instead of directing payments to us, an allowance is established for a portion of such receivables. After reasonable collection efforts are exhausted, amounts deemed to be
F-10
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
uncollectible are written off against the allowance for doubtful accounts. Since we only recognize revenue to the extent we expect to collect such amounts, bad debt expense related to receivables from patient service revenue is recorded in general and administrative expense in the consolidated statement of operations. The increase in the allowance from December 31, 2011 to September 30, 2012 was based upon the individual analysis of accounts receivable as described above.
Note 4. Notes Payable and Lines of Credit
Below is a summary of our short-term and long-term debt obligations as of September 30, 2012 and December 31, 2011:
| September 30, 2012 |
December 31, 2011 |
|||||||
| December 2011 Financing Transaction |
$ | 4,000,000 | $ | — | ||||
| Secured Note Payable, short-term |
76,378 | — | ||||||
| Other Note Payable |
— | 100,000 | ||||||
| Unamortized debt discount |
(545,451 | ) | — | |||||
|
|
|
|
|
|||||
| Notes Payable, Current Portion |
$ | 3,530,927 | $ | 100,000 | ||||
|
|
|
|
|
|||||
| Line of Credit, Principal Balance |
$ | 3,000,000 | $ | — | ||||
| Unamortized Debt Discount |
(248,994 | ) | — | |||||
|
|
|
|
|
|||||
| Line of Credit, Current Portion |
$ | 2,751,006 | $ | — | ||||
|
|
|
|
|
|||||
| December 2011 Financing Transaction |
$ | 2,000,000 | $ | 3,000,000 | ||||
| 2012 Convertible Debt Financing Transaction |
2,000,000 | — | ||||||
| Other Note Payable |
100,000 | |||||||
| Secured Note Payable |
43,622 | — | ||||||
| Unamortized debt discount |
(1,344,926 | ) | (1,087,635 | ) | ||||
|
|
|
|
|
|||||
| Notes Payable, Long-Term |
$ | 2,798,696 | $ | 1,912,365 | ||||
|
|
|
|
|
|||||
| Lines of Credit, Principal Balances |
$ | 6,000,000 | $ | 9,000,000 | ||||
| Unamortized Debt Discount |
— | (562,745 | ) | |||||
|
|
|
|
|
|||||
| Lines of Credit, Long-Term |
$ | 6,000,000 | $ | 8,437,255 | ||||
|
|
|
|
|
|||||
December 2011 Financing Transaction
As of September 30, 2012, we had $6 million outstanding under a Credit Agreement dated as of December 21, 2011, as amended and restated as of February 13, 2012. The Credit Agreement is with John Pappajohn and Andrew Pecora (indirectly through an investment company), both members of our board of directors, and NNJCA Capital, LLC (“NNJCA”), a limited liability company of which Dr. Pecora is a member, for a $6.0 million secured term loan. Mr. Pappajohn provided $4.0 million of financing, NNJCA provided $1.5 million of financing and Dr. Pecora provided $500,000 of financing under the Credit Agreement.
The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 13, 2013, with an option, at our election, if there has been no event of default, to extend the loan term for an additional six months. The term loan due to Pecora and NNJCA has a prepayment penalty in the aggregate amount of $190,000, less interest previously paid, if we prepay the loan prior to February 12, 2013. The lenders may require that the loan be repaid within 30 days should we complete our IPO and receive gross
F-11
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
proceeds of at least $15 million. In the event that any lender requires payment upon completion of our IPO and certain other maturity events, and we fail to make payment, the annual interest rate on any unpaid balance shall increase to 12%. The loan is secured by all of our assets, including our intellectual property, subject to prior first and second liens in favor of Wells Fargo Bank and DAM Holdings, LLC (“DAM”). Pursuant to an intercreditor agreement, the lenders have agreed that all amounts due to DAM are to be paid prior to payment to the lenders under this Credit Agreement, but that as between such lenders, following an event of default, all of the security granted by us is to be applied first to repay obligations due to Pecora and NNJCA, and then to Mr. Pappajohn after they have been paid in full. As Mr. Pappajohn has guaranteed the Wells Fargo debt, in essence under the intercreditor agreement, NNJCA and Pecora will be junior only to DAM.
The lenders received five-year warrants to purchase an aggregate of 211,764 shares of our common stock at an exercise price equal to the lesser of (i) $17.00 per share and (ii) a 20% discount to the IPO or merger price if the IPO or merger price is less than $21.25 per share, with 105,882 warrants issued to Mr. Pappajohn upon his fully funding $3.0 million on December 22, 2011 and an additional 105,882 warrants being issued proportionately to Mr. Pappajohn, Pecora and NNJCA upon their funding $1.0 million, $0.5 million and $1.5 million, respectively, on February 13, 2012. The lenders received an aggregate of 70,588 additional warrants on the same terms, with the warrants being issued proportionately to Mr. Pappajohn, Dr. Pecora and NNJCA because we did not consummate our IPO within 181 days of funding. The warrant exercise price is subject to anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO. The lenders may elect to net exercise their warrants. Mr. Pappajohn also had the option, exercisable up to two hours after he receives notice of the completion of our IPO, to convert the outstanding principal amount of his debt into shares of our common stock at a conversion price equal to the lesser of $17.00 per share or at a 20% discount to our IPO or merger price. However, Mr. Pappajohn subsequently agreed to convert $2.0 million of the outstanding principal due to him to common stock at the IPO price upon consummation of this offering and to eliminate the conversion feature of the remaining $2.0 million in debt effective upon consummation of this offering. The conversion price of the notes and the exercise price of the warrants are subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like.
We determined that the fair value of the warrants issued in connection with the notes to be approximately $2,060,000 which was recorded as a discount to the notes on the inception date and is being amortized to interest expense over the term of the notes using the effective interest method.
We determined that the fair value of the warrants issued as a consequence of not consummating our IPO within 180 days of funding to be approximately $421,000 which was recorded as a financing fee asset and is being amortized to interest expense over the term of the notes.
2012 Convertible Debt Financing Transaction
As of September 30, 2012, we had $2 million outstanding under a Credit Agreement dated August 27, 2012. The Credit Agreement is with Mr. Pappajohn and Mr. Oman, who each provided $1 million of financing. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 26, 2014. The Credit Agreement requires the lenders to convert the principal amount of the loan as of the closing of an IPO into shares of our common stock at a conversion price equal to the lesser of $17.00 or at a 20% discount to our IPO price.
The lenders received five-year warrants to purchase an aggregate of 70,588 shares of our common stock at an exercise price equal to the lesser of (i) $17.00 per share or (ii) a 20% discount to the IPO or merger price if
F-12
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
the IPO or merger price is less than $21.25 per share. The lenders will receive 11,765 additional warrants on the same terms in the event we do not consummate our IPO by each of the following dates: November 26, 2012 and February 24, 2013. The warrant exercise price is subject to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO. The lenders may elect to net exercise their warrants.
We determined that the fair value of the warrants issued in connection with the notes to be approximately $1,108,000 which was recorded as a discount to the notes on the inception date and is being amortized to interest expense over the term of the notes using the effective interest method.
Shares that the lenders receive are subject to a lock-up agreement for 180 days after the consummation of an IPO on the same terms as other lock-up agreements in favor of the underwriters of an offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Restated Credit Agreement.
Business Lines of Credit
At September 30, 2012 and December 31, 2011, we have fully utilized a line of credit with Wells Fargo Bank which provides for maximum borrowings of $6,000,000. Interest on the line of credit is due monthly equal to 1.75% above the Daily One Month LIBOR rate (1.96% at September 30, 2012). The line of credit requires the repayment of principal, and any unpaid interest, in a single payment due upon maturity. The line of credit is guaranteed by one of our stockholders and is collateralized by a first lien on all of our assets including the assignment of our approved and pending patent applications.
On February 15, 2012, we entered into a seventh addendum to the Credit Agreement with Wells Fargo which extended the maturity of the line of credit to July 31, 2013. The guarantee by John Pappajohn, one of our stockholders, on this line of credit continues through the new maturity date of July 31, 2013, and in consideration for this extension, in March 2012 the stockholder was granted warrants to purchase 92,500 shares of our common stock at the lesser of (i) $17.00 per share or (ii) a 20% discount to the IPO price.
On March 23, 2011, we entered into a line of credit agreement with DAM in which we received proceeds of $3 million. Pursuant to an intercreditor agreement between Mr. Pappajohn and DAM (the “Intercreditor Agreement”), we are required to use the proceeds from our IPO to repay the full amount outstanding under the DAM Loan Agreement before any proceeds can be used to repay any debt outstanding under the Wells Fargo Line of Credit. The DAM debt bears an annual interest rate of 10% payable in equal monthly installments and expires upon the earlier of the following: (i) April 1, 2013, (ii) the occurrence of an IPO of our equity securities in which we receive gross proceeds in the amount of $10,000,000 or more, or (iii) the consummation of a transaction in which we either merge with a reporting company under the Securities Exchange Act of 1934, as amended, or a public company acquires all or substantially all of our Company, and the survivor of such merger of the public company receives gross proceeds from the sale of the survivors securities or the public company’s securities in the amount of $10,000,000 or more. If certain maturity events do occur prior to April 1, 2013 and the line of credit is not repaid on the date of the maturity event, then the interest rate will increase to 18% per annum.
In connection with the acquisition of the line of credit, we issued 150,000 warrants to DAM with an exercise price of $5.00 per share expiring March 2016. We determined the fair value of the warrants issued in connection with the line of credit to be approximately $1,019,000 which was recorded as a debt discount on the
F-13
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
inception date of the line of credit and is being amortized to interest expense over the term of the facility using the effective interest method.
In March 2012 in consideration for the lender extending the maturity date of the loan to April 1, 2013, we granted DAM warrants to purchase 37,500 shares of our common stock at the lesser of (i) $17.00 per share or (ii) a 20% discount to the IPO price. See Note 7.
Subsequent Events
On October 15, 2012, Dr. Pecora and NNJCA agreed to surrender warrants to purchase an aggregate of 94,117 shares of our common stock issued in connection with the December 2011 Financing Transaction in exchange for an amendment to increase the pre-payment penalties by $130,000.
On October 16, 2012, DAM agreed to surrender 37,500 warrants issued in connection with their line of credit in exchange for a cash interest payment of $52,500 at the maturity date.
On October 17, 2012, the 92,500 warrants issued to Mr. Pappajohn in connection with the seventh extension of his guarantee of the Wells Fargo line of credit were amended to provide that the exercise price shall equal the lesser of (i) $17.00 per share and (ii) the IPO price per share. In exchange, we issued five-year warrants to purchase 25,393 shares of our common stock with an exercise price equal to the lesser of (i) $17.00 per share and (ii) the IPO price per share. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO (and in the event the IPO price is less than $17.00 or the consideration per share in a merger transaction is less than $17.00 per share).
On October 17, 2012, we entered into a Restated Credit Agreement with Mr. Pappajohn and Mr. Oman. Under the Restated Credit Agreement, we received an additional $750,000 from Mr. Pappajohn and $250,000 from Mr. Oman. Pursuant to the terms of the Restated Credit Agreement, the 70,588 warrants issued under the 2012 Convertible Debt Financing Transaction were cancelled and we issued ten-year warrants to purchase 286,275 shares of our common stock (which were issued in proportion to their respective funding amounts) in each case with an exercise price equal to the lesser of (i) $17.00 per share or (ii) the IPO or merger price per share. The lenders will receive additional ten-year warrants to purchase an aggregate of 17,648 shares of our common stock (to be issued in proportion to their respective funding amounts) on the same terms in the event we do not consummate our IPO by each the following dates: November 27, 2012 and February 22, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO. The Restated Credit Agreement requires the lenders to convert the principal amount of the loan as of the closing of an IPO into shares of our common stock at a conversion price equal to the lesser of $17.00 or at the IPO price.
On October 23, 2012, the 188,235 warrants issued to Mr. Pappajohn in connection with the December 2011 Financing Transaction were amended to provide that the exercise price shall equal the lesser of (i) $17.00 per share or (ii) our IPO or merger price per share. In exchange, we issued to Mr. Pappajohn additional warrants to purchase an aggregate of 51,673 shares of our common stock at an exercise price equal to the lesser of (i) $17.00 per share and (ii) the IPO or merger price per share. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the
F-14
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO (and in the event the IPO price is less than $17.00 or the consideration per share in a merger transaction is less than $17.00 per share).
On October 23, 2012, Mr. Pappajohn agreed to extend $2.0 million, which is held by his spouse, of the $4.0 million of notes payable to him in connection with the December 2011 Financing Transaction to April 1, 2014. Because of the extension provided by Mr. Pappajohn, we have classified $2.0 million of the December 2011 Financing Transaction as long-term. In exchange for this extension, we issued to Mr. Pappajohn additional warrants to purchase an aggregate of 23,530 shares of our common stock at an exercise price equal to the lesser of (i) $17.00 per share and (ii) the IPO or merger price per share. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO (and in the event the IPO price is less than $17.00 or the consideration per share in a merger transaction is less than $17.00 per share).
On October 18, 2012, we entered into an eighth amendment to the Credit Agreement with Wells Fargo, which extended the maturity date to April 1, 2014. Mr. Pappajohn also agreed to extend his guarantee of this credit facility to April 1, 2014. In exchange, we issued five-year warrants to purchase 70,589 shares of our common stock at an exercise price equal to the lesser of (i) $17.00 per share and (ii) the IPO or merger price per share. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our IPO (and in the event the IPO price is less than $17.00 or the consideration per share in a merger transaction is less than $17.00 per share).
Secured Note Payable
On September 25, 2012, we entered into a note payable secured by lab equipment due March 25, 2014. The note requires monthly payments of principal and interest at 18% per annum beginning October 25, 2012. At September 30, 2012, $120,000 was outstanding under the note.
Other Note Payable
At September 30, 2012 and December 31, 2011, notes payable included a note payable to Dr. Chaganti, our Chairman of the Board. The note was due on December 31, 2012 and bears interest at 8.5% per annum. At September 30, 2012 and December 31, 2011, $100,000 was outstanding under this note. Accrued interest at September 30, 2012 and December 31, 2011 was approximately $34,000 and $36,000, respectively. On October 11, 2012, this note was extended to April 1, 2014 and has been classified as non-current.
Note 5. Letter of Credit
In connection with the facility lease, the lessor required the establishment of a stand by letter of credit in the amount of $450,000 with Capital One Bank to use as a guarantee for a security deposit. In February 2011, we allowed the letter of credit to expire. On April 6, 2012, we reached an agreement with the landlord which requires us to provide a letter of credit in the amount of $250,000 and in exchange, the landlord agreed to forebear taking action to enforce our obligation to maintain the $450,000 letter of credit. The landlord also agreed to reduce our security deposit requirement to a $250,000 letter of credit upon a capital raise of at least $20
F-15
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
million by July 31, 2012. We are in violation of the terms of the agreement, but are in negotiations with our landlord to extend the forbearance agreement.
Note 6. Stock Option Plan
We have two equity incentive plans: the 2008 Stock Option Plan (the “2008 Plan”) and the 2011 Equity Incentive Plan (the “2011 Plan”, and together with the 2008 Plan, the “Stock Option Plans”). The 2011 Plan was approved by the Board of Directors on June 30, 2011 and was subsequently ratified by shareholders. The 2011 Plan authorizes the issuance of up to 375,000 shares of common stock under several types of equity awards including stock options, stock appreciation rights, restricted stock awards and other awards defined in the 2011 Plan. There have been no awards under the 2011 Plan.
The Board of Directors adopted the 2008 Plan on April 29, 2008 and reserved 628,689 shares of common stock for issuance under the plan. On April 1, 2010, the shareholders voted to increase the number of shares reserved by the plan to 1,375,000. The 2008 Plan is meant to provide additional incentive to officers, employees and consultants to remain in our employment. We are authorized to issue incentive stock options or nonstatutory stock options to eligible participants. Options granted are generally exercisable for up to 10 years.
At September 30, 2012, 191,050 shares remain available for future awards under the 2008 Plan and 375,000 shares remain available for future awards under the 2011 Plan. As of September 30, 2012, no stock appreciation rights had been awarded under the Stock Option Plans. We have also issued 200,000 options outside of the Stock Option Plans.
A summary of employee and nonemployee stock option activity for the year ended December 31, 2011 and the nine months ended September 30, 2012 is as follows:
| Options Outstanding | Weighted- Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value |
||||||||||||||
| Number of Shares |
Weighted- Average Exercise Price |
|||||||||||||||
| Outstanding January 1, 2011 |
1,279,150 | $ | 4.36 | 8.97 | $ | 1,693,760 | ||||||||||
| Granted |
169,038 | 10.69 | ||||||||||||||
| Cancelled or expired |
(48,213 | ) | 4.71 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding December 31, 2011 |
1,399,975 | $ | 5.14 | 8.10 | $ | 11,737,710 | ||||||||||
| Granted |
6,000 | 13.52 | ||||||||||||||
| Cancelled or expired |
(22,025 | ) | 9.49 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding September 30, 2012 |
1,383,950 | $ | 5.10 | 7.39 | $ | 4,294,594 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable, September 30, 2012 |
950,444 | $ | 4.27 | 7.18 | $ | 3,525,787 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Aggregate intrinsic value represents the difference between the estimated fair value of our common stock and the exercise price of outstanding, in-the-money options. The estimated fair value of our common stock was $7.48 and $13.52 as of September 30, 2012 and December 31, 2011, respectively. No options were exercised during the nine months ended September 30, 2012 and 2011.
As of September 30, 2012 and December 31 2011, total unrecognized compensation cost related to nonvested stock options granted to employees was $933,095 and $1,262,246, respectively, which we expect to recognize over the next 2.86 and 3.61 years, respectively.
F-16
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
As of September 30, 2012 and December 31, 2011, total unrecognized compensation cost related to nonvested stock options granted to non-employees was $450,500 and $1,061,270, respectively, which we expect to recognize over the next 0.75 and 1.40 years, respectively. The estimate of unrecognized nonemployee compensation is based on the fair value of the nonvested options as of September 30, 2012 and December 31, 2011.
The following table summarizes information about outstanding and vested stock options granted to employees and non-employees as of September 30, 2012 as follows:
| Options Outstanding | Options Vested and Exercisable | |||||||||||||||||||
| Exercise Price |
Number of Shares Outstanding |
Weighted- Average Remaining Contractual Life (in years) |
Weighted- Average Exercise Price |
Number of Shares |
Weighted- Average Exercise Price |
|||||||||||||||
| 1.60 |
437,500 | 6.58 | $ | 1.60 | 437,500 | $ | 1.60 | |||||||||||||
| 1.92 |
84,600 | 7.30 | 1.92 | 49,629 | 1.92 | |||||||||||||||
| 5.00 |
504,725 | 7.52 | 5.00 | 273,125 | 5.00 | |||||||||||||||
| 10.00 |
325,500 | 8.10 | 10.00 | 190,190 | 10.00 | |||||||||||||||
| 12.66 |
5,625 | 9.01 | 12.66 | — | — | |||||||||||||||
| 13.52 |
26,000 | 9.27 | 13.52 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
1,383,950 | 7.39 | $ | 5.10 | 950,444 | $ | 4.27 | |||||||||||||
|
|
|
|
|
|||||||||||||||||
The fair value of options granted to employees is estimated on the grant date using the Black-Scholes option valuation model. This valuation model for stock-based compensation expense requires us to make assumptions and judgments about the variables used in the calculation, including the fair value of our common stock (see Note 8), the expected term (the period of time that the options granted are expected to be outstanding), the volatility of our common stock, a risk-free interest rate, and expected dividends. We also estimate forfeitures of unvested stock options. To the extent actual forfeitures differ from the estimates, the difference will be recorded as a cumulative adjustment in the period estimates are revised. No compensation cost is recorded for options that do not vest. We use the simplified calculation of expected life described in the SEC’s Staff Accounting Bulletin No. 107, Share-Based Payment, and volatility is based on an average of the historical volatilities of the common stock of four entities with characteristics similar to those of the Company. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option. We use an expected dividend yield of zero, as we do not anticipate paying any dividends in the foreseeable future. Expected forfeitures are assumed to be zero due to the small number of plan participants and the plan design which has monthly vesting after an initial cliff vesting period.
The following table presents the weighted-average assumptions used to estimate the fair value of options granted to employees during the periods presented:
| Nine Months Ended September 30, | ||||||||
| 2012 | 2011 | |||||||
| Volatility |
77.39 | % | 76.02 | % | ||||
| Risk free interest rate |
1.43 | % | 3.11 | % | ||||
| Dividend yield |
0.00 | % | 0.00 | % | ||||
| Term (years) |
6.50 | 6.50 | ||||||
| Weighted-average fair value of options granted during the period |
$ | 9.34 | $ | 2.95 | ||||
F-17
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
The following table presents the weighted-average assumptions used to estimate the fair value of options reaching their measurement date for non-employees during the periods presented:
| Nine Months Ended September 30, | ||||||||
| 2012 | 2011 | |||||||
| Volatility |
75.01 | % | 75.75 | % | ||||
| Risk free interest rate |
1.31 | % | 3.17 | % | ||||
| Dividend yield |
0.00 | % | 0.00 | % | ||||
| Term (years) |
8.35 | 9.35 | ||||||
The following table presents the effects of stock-based compensation related to stock option awards to employees and nonemployees on our Statement of Operations during the periods presented:
| Nine Months Ended September 30, | ||||||||
| 2012 | 2011 | |||||||
| Cost of revenues |
$ | 9,557 | $ | 6,358 | ||||
| Research and development |
389,295 | 293,258 | ||||||
| General and administrative |
223,150 | 204,690 | ||||||
| Sales and marketing |
144,165 | 175,731 | ||||||
|
|
|
|
|
|||||
| Total stock-based compensation |
$ | 766,167 | $ | 680,037 | ||||
|
|
|
|
|
|||||
Subsequent Events
On October 12, 2012, the Board of Directors unanimously approved amendments to the Stock Option Plans to permit the re-pricing of stock options issued under the Stock Option Plans. The Board of Directors also voted to offer re-priced stock options to certain option holders on the following terms: those persons holding stock options with a strike price of $10.00 or more have the opportunity to exchange their options for 60% of the number of options currently held with an exercise price equal to the IPO price. Vesting of the re-priced options will be on the same terms and as of the same vesting start date as the exchanged options. This offer affects 357,125 option shares held by three board members, an executive officer, and seven other employees.
On October 19, 2012, the Board of Directors voted to amend the 2011 Plan. The amendment increases the number of shares available for grant under the 2011 Plan from 375,000 to 875,000 shares. This amendment has been ratified by our shareholders.
Note 7. Warrants
We have issued certain warrants which contain an exercise price adjustment feature in the event we issue additional equity instruments at a price lower than the exercise price of the warrant. The warrants are described herein as derivative warrants. These warrants are initially recorded as a warrant liability at fair value with a corresponding entry to the loan guarantee fee asset, additional paid-in capital or expense dependent upon the service provided in exchange for the warrant grant. Subsequently, any change in fair value is recognized in earnings until such time as the warrants are exercised, amended or expire.
In connection with the extension of a debt guaranty, we issued 92,500 warrants to Mr. Pappajohn, a member of our Board of Directors and shareholder, on March 9, 2012 (Note 4). These warrants were initially recorded at the issue date fair value of $755,000 as a loan guarantee fee and amortized over the period of the guarantee to interest expense.
F-18
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
On February 13, 2012, we granted 105,882 warrants proportionately to Mr. Pappajohn, Pecora and NNJCA in connection with a secured term loan of $3 million (see Note 4). The value of these warrants reduced the carrying value of the related debt and will be amortized over the period of the debt agreement to interest expense. These warrants were recorded at an initial estimated fair value of $940,000.
In connection with an extension of a line of credit, we granted 37,500 warrants to DAM on March 9, 2012. These warrants were initially recorded at fair value as a financing fee asset amortized over the period of the line of credit to interest expense. The issue date fair value of these warrants was $306,000.
In connection with the Credit Agreement (see Note 4), we granted 17,647 warrants on April 1, 2012 to Mr. Pappajohn. On the date we entered into the Credit Agreement these warrants were initially recorded in accrued expenses at fair value of $148,000 as a debt discount amortized over the period of the line of credit to interest expense using the effective interest method. Also in connection with the Credit Agreement, we granted 17,647 warrants proportionately to Mr. Pappajohn, Pecora and NNJCA on May 15, 2012, 17,647 warrants to Mr. Pappajohn on July 1, 2012, and 17,647 warrants proportionately to Mr. Pappajohn, Pecora and NNJCA on August 14, 2012. These warrants were initially recorded at fair value as a financing fee asset and will be amortized over the period of the note to interest expense. The issue date fair value of these warrants was $421,000.
In connection with a credit agreement (see Note 4), we granted five-year warrants to purchase an aggregate of 70,588 shares of our common stock to Mr. Pappajohn and Mr. Oman. The value of these warrants reduced the carrying value of the related debt and will be amortized over the period of the debt agreement to interest expense. These warrants were recorded at an initial estimated fair value of $1,108,000.
During the nine months ended September 30, 2012, warrants to purchase 127,745 shares were exercised and $619,980 was received and recorded as increases to common stock and additional paid-in capital. An additional 29,129 warrants expired unexercised.
On September 6, 2012, we extended the maturity date of warrants to purchase 176,496 shares of our common stock which were due to expire on September 10, 2012 to September 28, 2012. On September 27, 2012, we extended the maturity date of 163,322 of these warrants to September 10, 2013. In exchange, the warrants are now subject to a lock-up agreement for 180 days after the consummation of an IPO on the same terms as other lock-up agreements in favor of the underwriters of an offering. We determined the fair value of the warrant extensions to be approximately $144,000, which was recorded as an increase to general and administrative expense.
F-19
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
The following table summarizes the warrant activity for the nine months ended September 30, 2012:
| Issued With / For |
Exercise Price |
Warrants Outstanding January 1, 2012 |
Warrants Issued |
Warrants Exercised |
Warrants Expired |
Warrants Outstanding September 30, 2012 |
||||||||||||||||||||
| Debt Guarantee |
$ | 1.60 | 570,721 | — | — | — | 570,721 | |||||||||||||||||||
| Series A Pref. Stock |
4.30 | 75,000 | — | (75,000 | ) | — | — | |||||||||||||||||||
| Series A Pref. Stock |
5.64 | 223,035 | — | (46,539 | ) | (13,173 | ) | 163,323 | ||||||||||||||||||
| Financing |
5.64 | 22,162 | — | (6,206 | ) | (15,956 | ) | — | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| 2.50 | 890,918 | — | (127,745 | ) | (29,129 | ) | 734,044 | |||||||||||||||||||
| Series A Pref. Stock |
1.60 | A | 18,313 | — | — | — | 18,313 | |||||||||||||||||||
| Financing |
10.00 | A | 150,000 | — | — | — | 150,000 | |||||||||||||||||||
| Financing |
17.00 | AB | 105,882 | 284,558 | — | — | 390,440 | |||||||||||||||||||
| Debt Guarantee |
10.00 | A | 780,000 | — | — | — | 780,000 | |||||||||||||||||||
| Debt Guarantee |
12.98 | A | 100,000 | — | — | — | 100,000 | |||||||||||||||||||
| Debt Guarantee |
17.00 | AB | — | 92,500 | — | — | 92,500 | |||||||||||||||||||
| Series B Pref. Stock |
10.00 | A | 131,160 | — | — | — | 131,160 | |||||||||||||||||||
| Consulting |
5.00 | A | 10,075 | — | — | — | 10,075 | |||||||||||||||||||
| Consulting |
5.64 | A | 25,000 | — | — | — | 25,000 | |||||||||||||||||||
| Consulting |
10.00 | A | 10,500 | — | — | — | 10,500 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| 11.97 | 1,330,930 | 377,058 | — | — | 1,707,988 | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| $ | 9.12 | 2,221,848 | 377,058 | (127,745 | ) | (29,129 | ) | 2,442,032 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| A | These warrants are subject to fair value accounting and contain an exercise price adjustment feature. See Note 9. |
| B | As of September 30, 2012, the exercise price is the lower of $17.00 per share or a 20% discount to the IPO price if the IPO price is less than $21.25 per share, subsequently amended to remove the provision for the 20% discount. |
Note 8. Fair Value of Warrants
The following tables summarize the assumptions used in computing the fair value of derivative warrants subject to fair value accounting at the date of issue during the nine months ended September 30, 2012 and 2011 and at September 30, 2012 and December 31, 2011:
| As of September 30, 2012 |
As of December 31, 2011 |
|||||||
| Series A |
||||||||
| Exercise Price |
$ | 1.60 | $ | 1.60 | ||||
| Expected life (years) |
0.08 | 0.83 | ||||||
| Expected volatility |
42.92 | % | 64.13 | % | ||||
| Risk-free interest rate |
0.06 | % | 0.12 | % | ||||
| Expected dividend yield |
0.00 | % | 0.00 | % | ||||
F-20
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
| Issued During the Nine Months Ended September 30, |
As
of September 30, 2012 |
As
of December 31, 2011 |
||||||||||||||
| 2012 | 2011 | |||||||||||||||
| Debt Guarantee |
||||||||||||||||
| Exercise Price |
$ | 17.00 | $ | 12.98 | $ | 10.97 | $ | 10.34 | ||||||||
| Expected life (years) |
4.73 | 5.00 | 2.67 | 3.26 | ||||||||||||
| Expected volatility |
80.47 | % | 77.35 | % | 64.44 | % | 76.53 | % | ||||||||
| Risk-free interest rate |
0.90 | % | 1.76 | % | 0.29 | % | 0.51 | % | ||||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
| As of September 30, 2012 |
As of December 31, 2011 |
|||||||
| Series B |
||||||||
| Exercise Price |
$ | 10.00 | $ | 10.00 | ||||
| Expected life (years) |
3.17 | 3.92 | ||||||
| Expected volatility |
62.10 | % | 81.59 | % | ||||
| Risk-free interest rate |
0.31 | % | 0.36 | % | ||||
| Expected dividend yield |
0.00 | % | 0.00 | % | ||||
| Issued During the Nine Months Ended September 30, 2011 |
As of September 30, 2012 |
As of December 31, 2011 |
||||||||||
| Consulting |
||||||||||||
| Exercise Price |
$ | 10.00 | $ | 6.50 | $ | 6.50 | ||||||
| Expected life (years) |
5.00 | 2.73 | 3.48 | |||||||||
| Expected volatility |
78.45 | % | 62.24 | % | 81.87 | % | ||||||
| Risk-free interest rate |
2.07 | % | 0.25 | % | 0.47 | % | ||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | ||||||
| Issued During the Nine Months Ended September 30, |
As
of September 30, 2012 |
As
of December 31, 2011 |
||||||||||||||
| 2012 | 2011 | |||||||||||||||
| Financing |
||||||||||||||||
| Exercise Price |
$ | 17.00 | 10.00 | $ | 15.06 | $ | 12.90 | |||||||||
| Expected life (years) |
4.93 | 5.00 | 4.15 | 4.56 | ||||||||||||
| Expected volatility |
79.41 | % | 78.46 | % | 78.20 | % | 79.40 | % | ||||||||
| Risk-free interest rate |
0.79 | % | 2.07 | % | 0.53 | % | 0.83 | % | ||||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
The assumed Company stock price used in computing the warrant fair value for warrants issued during the nine months ended September 30, 2012 was $7.48 – $13.52 and $4.76 – $12.66 for the nine months ended September 30, 2011. In determining the fair value of warrants at each reporting date, the assumed company stock price was $7.48 at September 30, 2012 and $13.52 at December 31, 2011.
F-21
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
The following table summarizes the derivative warrant activity subject to fair value accounting for the nine months ended September 30, 2012:
| Issued with/for |
Fair value of warrants outstanding as of December 31, 2011 |
Fair value of warrants issued |
Change in fair value of warrants |
Fair value of warrants outstanding as of September 30, 2012 |
||||||||||||
| Series A Preferred Stock |
$ | 220,000 | $ | — | $ | (111,000 | ) | $ | 109,000 | |||||||
| Series B Preferred Stock |
1,160,000 | — | (725,000 | ) | 435,000 | |||||||||||
| Debt Guarantee |
6,993,000 | 755,000 | (3,318,000 | ) | 4,430,000 | |||||||||||
| Consulting |
447,000 | — | (257,000 | ) | 190,000 | |||||||||||
| Financing |
2,293,000 | 2,923,000 | (1,959,000 | ) | 3,257,000 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 11,113,000 | $ | 3,678,000 | $ | (6,370,000 | ) | $ | 8,421,000 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Note 9. Fair Value Measurements
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. The Fair Value Measurements and Disclosures Topic of the FASB Accounting Standards Codification requires the use of valuation techniques that are consistent with the market approach, the income approach and/or the cost approach. Inputs to valuation techniques refer to the assumptions that market participants would use in pricing the asset or liability. Inputs may be observable, meaning those that reflect the assumptions market participants would use in pricing the asset or liability developed based on market data obtained from independent sources, or unobservable, meaning those that reflect our own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. In that regard, the Topic establishes a fair value hierarchy for valuation inputs that give the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs.
The fair value hierarchy is as follows:
Level 1: Quoted prices (unadjusted) for identical assets or liabilities in active markets that we have the ability to access as of the measurement date.
Level 2: Significant other observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data.
Level 3: Significant unobservable inputs that reflect our own assumptions about the assumptions that market participants would use in pricing an asset or liability.
F-22
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
The following table summarizes the financial liabilities measured at fair value on a recurring basis segregated by the level of valuation inputs within the fair value hierarchy utilized to measure fair value:
| September 30, 2012 | ||||||||||||||||
| Total | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Warrant liability |
$ | 8,421,000 | — | — | $ | 8,421,000 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| December 31, 2011 | ||||||||||||||||
| Total | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Warrant liability |
$ | 11,113,000 | — | — | $ | 11,113,000 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The warrant liability consists of stock warrants we issued that contain an exercise price adjustment feature. In accordance with derivative accounting for warrants, we calculated the fair value of warrants and the assumptions used are described in Note 8, “Fair Value of Warrants”. Realized and unrealized gains and losses related to the change in fair value of the warrant liability are included in Other income (expense) on the Statement of Operations.
The following table reflects the activity for liabilities measured at fair value using Level 3 inputs for the nine months ended September 30:
| 2012 | 2011 | |||||||
| Balance as of January 1 |
$ | 11,113,000 | $ | 4,270,000 | ||||
| Derivative financial instruments reclassified to equity upon amendment |
— | $ | (6,415,000 | ) | ||||
| Issuances of derivative financial instruments |
3,678,000 | 1,919,000 | ||||||
| Unrealized gain (loss) related to change in fair value |
(6,370,000 | ) | 9,881,000 | |||||
|
|
|
|
|
|||||
| Balance as of September 30 |
$ | 8,421,000 | $ | 9,655,000 | ||||
|
|
|
|
|
|||||
Note 10. Joint Venture Agreement
On November 7, 2011, we entered into an agreement with the Mayo Foundation for Medical Education and Research (“Mayo”) pursuant to which we agreed to form a joint venture with Mayo in August 2012 (subsequently extended to December 2012). The joint venture will take the form of a limited liability company, with each party initially holding fifty percent of the issued and outstanding membership interests of the new entity (the “JV”). In exchange for the membership interests in the JV, we will make a capital contribution of $6 million, to be paid over a three year period. In exchange for its membership interests, Mayo’s capital contribution will take the form of cash, staff, services, hardware and software resources, laboratory space and instrumentation, the fair market value of which will be approximately equal to $6 million. Mayo’s continued contribution will also be conditioned upon the JV’s achievement of certain milestones. In addition, on November 14, 2011, without any additional consideration, we granted Mayo 50,000 shares of our common stock, 33,000 of which shall initially be subject to certain forfeiture restrictions if the joint venture does not meet certain milestones. We applied accounting for share-based payments to non-employees and determined that a performance commitment for the
F-23
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Unaudited Consolidated Financial Statements
33,000 shares subject to restriction was not present at the date of grant and therefore the remaining shares will be valued and expensed as the restrictions lapse. We recorded expense of $150,000 during the fourth quarter of 2011 related to the 17,000 shares that were fully vested at the date of grant.
Note 11. Related Party Transactions
John Pappajohn, a member of the Board of Directors and shareholder, personally guarantees our revolving line of credit with Wells Fargo Bank. As consideration for his guarantee, as well as each of the seven extensions of this facility through September 30, 2012, Mr. Pappajohn received warrants to purchase an aggregate of 2,532,784 shares of common stock.
In August 2010, we entered into a consulting agreement with Equity Dynamics, Inc., an entity controlled by John Pappajohn, pursuant to which Equity Dynamics, Inc. receives a monthly fee of $10,000 plus reimbursement of expenses. Total fees for the nine months ended September 30, 2012 under the consulting agreement was $90,000. As of September 30, 2012, we owed Equity Dynamics, Inc. $103,000.
In December 2011, we granted 105,882 warrants to John Pappajohn, a member of our Board of Directors and stockholder in connection with a secured term loan of $3 million. In February 2012, we granted 105,882 warrants proportionately to Mr. Pappajohn, Pecora and NNJCA upon their funding of $1.0 million, $0.5 million and $1.5 million, respectively, under a new Credit Agreement. (See Notes 4 and 8) On April 1, 2012 and on July 1, 2012, we issued 17,647 of these warrants to Mr. Pappajohn in connection with this loan. On May 15, 2012 and on August 14, 2012, we issued 17,647 of these warrants proportionately to Mr. Pappajohn, Pecora and NNJCA (Note 4).
In August 2012, we granted warrants to purchase 35,294 warrants to John Pappajohn, a member of our Board of Directors and stockholder in connection with a secured term loan of $1 million. We have also recorded $184,000 of accrued expenses at September 30, 2012 for contingently issuable warrants related to the 2012 Bridge Financing (See Note 4).
In October 2012, we consummated a series of transactions, some of which involved related parties, that resulted in warrant issuances, warrant cancellations, and debt issuances (See Note 4).
Note 12. Contingencies
In the normal course of business, the Company is involved in various claims and legal proceedings. In the opinion of management, the ultimate liability or disposition thereof is not expected to have a material adverse effect on our financial condition, results of operations or liquidity.
F-24
Table of Contents
The accompanying consolidated financial statements give effect to a 1-for-2 reverse stock split of the common stock of Cancer Genetics, Inc. which will take place immediately prior to the effectiveness of the registration statement. The following report is in the form which will be furnished by McGladrey LLP, an independent registered public accounting firm, upon completion of the 1-for-2 reverse split of the common stock of Cancer Genetics, Inc. described in the first the paragraph of Note 11 to the consolidated financial statements and assuming that from March 13, 2012 to the date of such completion no other material events have occurred that would affect the consolidated financial statements or the required disclosures therein.
/s/ McGladrey LLP
Des Moines, Iowa
May 7, 2012
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Cancer Genetics, Inc. and Subsidiary
We have audited the accompanying consolidated balance sheets of Cancer Genetics, Inc. and subsidiary as of December 31, 2011 and 2010, and the related consolidated statements of operations, changes in stockholders’ equity (deficit), and cash flows for each of the three years ended December 31, 2011, 2010 and 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Cancer Genetics, Inc. and subsidiary as of December 31, 2011 and 2010, and the results of their operations and their cash flows for each of the three years ended December 31, 2011, 2010 and 2009 in conformity with U.S. generally accepted accounting principles.
/s/ McGladrey LLP
Des Moines, Iowa
March 13, 2012, except for the
first paragraph of Note 11
as to which the date is
, 2012.
F-25
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
December 31, 2011 and 2010
| 2011 | 2010 | |||||||
| ASSETS |
||||||||
| CURRENT ASSETS |
||||||||
| Cash and cash equivalents |
$ | 2,417,256 | $ | 1,779,251 | ||||
| Accounts receivable, net of allowance for doubtful accounts of 2011 $24,050; 2010 $46,356 |
688,980 | 705,844 | ||||||
| Other current assets |
269,269 | 821,007 | ||||||
|
|
|
|
|
|||||
| Total current assets |
3,375,505 | 3,306,102 | ||||||
| FIXED ASSETS, net of accumulated depreciation |
1,140,636 | 1,179,007 | ||||||
|
|
|
|
|
|||||
| OTHER ASSETS |
||||||||
| Security deposits |
1,564 | 1,564 | ||||||
| Restricted cash |
200,000 | 438,400 | ||||||
| Loan guarantee fee, net of accumulated amortization of 2011 $333,202; 2010 $172,917 |
497,798 | 242,083 | ||||||
| Patents |
204,687 | 134,863 | ||||||
| Deferred IPO costs |
1,611,273 | — | ||||||
|
|
|
|
|
|||||
| 2,515,322 | 816,910 | |||||||
|
|
|
|
|
|||||
| Total Assets |
$ | 7,031,463 | $ | 5,302,019 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT |
||||||||
| CURRENT LIABILITIES |
||||||||
| Accounts payable and accrued expenses |
$ | 4,317,121 | $ | 1,484,187 | ||||
| Obligations under capital leases, current portion |
36,209 | 36,922 | ||||||
| Notes payable, current portion |
100,000 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
4,453,330 | 1,521,109 | ||||||
| OBLIGATIONS UNDER CAPITAL LEASES |
24,559 | 16,954 | ||||||
| DEFERRED RENT PAYABLE |
156,311 | 130,389 | ||||||
| NOTES PAYABLE, LONG-TERM |
1,912,365 | 100,000 | ||||||
| LINES OF CREDIT |
8,437,255 | 6,000,000 | ||||||
| WARRANT LIABILITY |
11,113,000 | 4,270,000 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
26,096,820 | 12,038,452 | ||||||
|
|
|
|
|
|||||
| COMMITMENTS AND CONTINGENCIES (Notes 5 and 16) |
||||||||
| STOCKHOLDERS’ DEFICIT |
||||||||
| Series A Preferred Stock, authorized 588,000 shares $0.0001 par value (purchase price liquidation preference of $4,971,866 or can elect to convert common stock), 587,691 shares issued and outstanding |
59 | 59 | ||||||
| Series B Preferred Stock, authorized 2,000,000 shares $0.0001 par value (purchase price liquidation preference of $9,108,000 or can elect to convert to common stock), 1,821,600 shares issued and outstanding |
182 | 182 | ||||||
| Common stock, authorized 24,000,000 shares $0.0001 par value, 3,237,566 and 3,178,701 shares issued and outstanding as of December 31, 2011 and 2010 respectively. |
324 | 318 | ||||||
| Additional paid-in capital |
23,220,478 | 15,662,368 | ||||||
| Treasury stock |
(17,442 | ) | (17,442 | ) | ||||
| Accumulated deficit |
(42,268,958 | ) | (22,381,918 | ) | ||||
|
|
|
|
|
|||||
| Total Stockholders’ Deficit |
(19,065,357 | ) | (6,736,433 | ) | ||||
|
|
|
|
|
|||||
| Total Liabilities and Stockholders’ Deficit |
$ | 7,031,463 | $ | 5,302,019 | ||||
|
|
|
|
|
|||||
See Notes to Consolidated Financial Statements.
F-26
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Operations
Years Ended December 31, 2011, 2010 and 2009
| 2011 | 2010 | 2009 | ||||||||||
| Revenue |
$ | 3,019,407 | $ | 2,521,579 | $ | 1,665,687 | ||||||
| Cost of revenues |
3,117,411 | 3,516,189 | 2,531,618 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit (loss) |
(98,004 | ) | (994,610 | ) | (865,931 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Operating expenses: |
||||||||||||
| Research and development |
2,073,661 | 1,166,553 | 1,336,037 | |||||||||
| General and administrative |
4,439,170 | 3,445,998 | 1,844,572 | |||||||||
| Sales and marketing |
1,574,088 | 715,966 | 239,459 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
8,086,919 | 5,328,517 | 3,420,068 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(8,184,923 | ) | (6,323,127 | ) | (4,285,999 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Other income (expense): |
||||||||||||
| Interest expense |
(1,314,234 | ) | (792,415 | ) | (2,091,807 | ) | ||||||
| Interest income |
117 | 763 | 2,514 | |||||||||
| Change in fair value of warrant liability |
(10,388,000 | ) | (2,026,000 | ) | (953,000 | ) | ||||||
| Qualifying Therapeutic Discovery Project Grant |
— | 733,438 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total other income (expense) |
(11,702,117 | ) | (2,084,214 | ) | (3,042,293 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(19,887,040 | ) | (8,407,341 | ) | (7,328,292 | ) | ||||||
| Income tax provision (benefit) |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (19,887,040 | ) | $ | (8,407,341 | ) | $ | (7,328,292 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (6.24 | ) | $ | (2.68 | ) | $ | (3.22 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted Average Shares Outstanding |
3,185,382 | 3,133,077 | 2,277,004 | |||||||||
|
|
|
|
|
|
|
|||||||
See Notes to Consolidated Financial Statements.
F-27
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Changes in Stockholders’ Equity (Deficit)
Years Ended December 31, 2011, 2010 and 2009
| Preferred Stock Series A |
Preferred Stock Series B |
Common Stock | Additional Paid-in Capital |
Treasury Stock |
Accumulated Deficit |
Total | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||
| Balance, December 31, 2008 |
587,691 | 59 | — | — | 1,818,201 | 182 | 5,227,346 | (17,442 | ) | (6,436,285 | ) | (1,226,140 | ) | |||||||||||||||||||||||||||
| Cumulative effect of change in accounting principle January 1, 2009, reclassification of embedded feature of equity—linked financial instrument to derivative liability, at fair value |
— | — | — | — | — | — | (711,000 | ) | — | (210,000 | ) | (921,000 | ) | |||||||||||||||||||||||||||
| Stock based compensation—employees |
— | — | — | — | — | — | 300,702 | — | — | 300,702 | ||||||||||||||||||||||||||||||
| Exercise of warrants |
— | — | — | — | 1,260,000 | 126 | 2,463,874 | — | — | 2,464,000 | ||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (7,328,292 | ) | (7,328,292 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
| Balance, December 31, 2009 |
587,691 | 59 | — | — | 3,078,201 | 308 | 7,280,922 | (17,442 | ) | (13,974,577 | ) | (6,710,730 | ) | |||||||||||||||||||||||||||
| Sale of series B Preferred Stock, net of stock issuance costs of $1,183,751 |
— | — | 1,821,600 | 182 | — | — | 7,902,981 | — | — | 7,903,163 | ||||||||||||||||||||||||||||||
| Stock based compensation—employees |
— | — | — | — | — | 344,275 | — | — | 344,275 | |||||||||||||||||||||||||||||||
| Stock based compensation—consultants |
— | — | — | — | 5,000 | — | 90,600 | — | — | 90,600 | ||||||||||||||||||||||||||||||
| Exercise of options |
— | — | — | — | 95,500 | 10 | 43,590 | — | — | 43,600 | ||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (8,407,341 | ) | (8,407,341 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
| Balance, December 31, 2010 |
587,691 | $ | 59 | 1,821,600 | $ | 182 | 3,178,701 | $ | 318 | $ | 15,662,368 | $ | (17,442 | ) | $ | (22,381,918 | ) | $ | (6,736,433 | ) | ||||||||||||||||||||
| Stock based compensation—employees |
— | — | — | — | — | — | 306,307 | — | — | 306,307 | ||||||||||||||||||||||||||||||
| Stock based compensation—non-employees |
— | — | — | — | — | — | 636,810 | — | — | 636,810 | ||||||||||||||||||||||||||||||
| Exercise of warrants |
— | — | — | — | 8,865 | 1 | 49,998 | — | — | 49,999 | ||||||||||||||||||||||||||||||
| Warrant liability reclassified to equity |
— | — | — | — | — | — | 6,415,000 | — | — | 6,415,000 | ||||||||||||||||||||||||||||||
| Issuance of common stock |
— | — | — | — | 50,000 | 5 | 149,995 | — | — | 150,000 | ||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | (19,887,040 | ) | (19,887,040 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
| Balance, December 31, 2011 |
587,691 | $ | 59 | 1,821,600 | $ | 182 | 3,237,566 | $ | 324 | $ | 23,220,478 | $ | (17,442 | ) | $ | (42,268,958 | ) | (19,065,357 | ) | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
See Notes to Consolidated Financial Statements.
F-28
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Cash Flows
Years Ended December 31, 2011, 2010 and 2009
| 2011 | 2010 | 2009 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES |
||||||||||||
| Net loss |
$ | (19,887,040 | ) | $ | (8,407,341 | ) | $ | (7,328,292 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation |
356,603 | 317,095 | 303,771 | |||||||||
| Amortization |
13,048 | 9,723 | 3,241 | |||||||||
| Provision for bad debts |
372,680 | 46,356 | 23,000 | |||||||||
| Equity-based consulting and compensation expenses |
1,093,117 | 476,875 | 300,702 | |||||||||
| Derivative warrants issued for consulting services |
69,000 | 65,000 | — | |||||||||
| Change in fair value of warrant liability |
10,388,000 | 2,026,000 | 953,000 | |||||||||
| Amortization of loan guarantee fee |
575,285 | 527,584 | 1,898,139 | |||||||||
| Accretion of discount on debt |
480,620 | — | — | |||||||||
| Deferred rent |
25,922 | 42,363 | 42,364 | |||||||||
| Change in working capital components: |
||||||||||||
| Accounts receivable |
(355,816 | ) | (570,162 | ) | 95,697 | |||||||
| Other current assets |
551,738 | (710,643 | ) | (57,072 | ) | |||||||
| Accounts payable and accrued expenses |
1,243,594 | 446,603 | 430,276 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) operating activities |
(5,073,249 | ) | (5,730,547 | ) | (3,335,174 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM INVESTING ACTIVITIES |
||||||||||||
| Purchase of fixed assets |
(268,632 | ) | (161,201 | ) | (160,583 | ) | ||||||
| Patent costs |
(82,872 | ) | (18,407 | ) | (30,226 | ) | ||||||
| Decrease in restricted cash |
238,400 | 11,600 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) investing activities |
(113,104 | ) | (168,008 | ) | (190,809 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM FINANCING ACTIVITIES |
||||||||||||
| Principal payments on capital lease obligations |
(42,708 | ) | (79,848 | ) | (136,926 | ) | ||||||
| Proceeds from issuance of preferred stock |
— | 9,108,000 | — | |||||||||
| Payment of equity issuance costs |
(182,933 | ) | (826,837 | ) | — | |||||||
| Proceeds from option exercises |
— | 1,600 | — | |||||||||
| Proceeds from warrant exercises |
49,999 | — | — | |||||||||
| Proceeds from borrowings on notes payable |
3,000,000 | — | — | |||||||||
| Proceeds from borrowings on line of credit |
3,200,000 | 590,000 | 3,560,000 | |||||||||
| Payments on line of credit |
(200,000 | ) | (1,000,000 | ) | — | |||||||
| Proceeds from notes payable |
— | — | 105,000 | |||||||||
| Principal payments on notes payable |
— | (145,000 | ) | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
5,824,358 | 7,647,915 | 3,528,074 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase in cash and cash equivalents |
638,005 | 1,749,360 | 2,091 | |||||||||
| CASH AND CASH EQUIVALENTS |
||||||||||||
| Beginning |
1,779,251 | 29,891 | 27,800 | |||||||||
|
|
|
|
|
|
|
|||||||
| Ending |
$ | 2,417,256 | $ | 1,779,251 | $ | 29,891 | ||||||
|
|
|
|
|
|
|
|||||||
(Continued)
F-29
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Consolidated Statements of Cash Flows (Continued)
Years Ended December 31, 2011, 2010 and 2009
| 2011 | 2010 | 2009 | ||||||||||
| SUPPLEMENTAL CASH FLOW DISCLOSURE |
||||||||||||
| Cash paid for interest |
$ | 253,996 | $ | 274,356 | $ | 164,407 | ||||||
| SUPPLEMENTAL DISCLOSURE OF NONCASH INVESTING AND FINANCING ACTIVITIES |
||||||||||||
| Fixed assets acquired through capital lease arrangement |
$ | 49,600 | $ | — | $ | 70,970 | ||||||
| Warrants issued for debt guarantee fee |
831,000 | 415,000 | 2,026,000 | |||||||||
| Warrants issued for equity issuance costs |
— | 328,000 | — | |||||||||
| Warrants issued with debt |
1,970,000 | — | — | |||||||||
| Cashless exercise of derivative warrants |
— | — | 2,464,000 | |||||||||
| Reclassification of embedded feature of equity-linked financial instruments to warrant liability upon adoption of Accounting Standards Codification Topic 815-40 |
— | — | 921,000 | |||||||||
| Warrant liability reclassified to equity |
6,415,000 | — | — | |||||||||
| Accrued IPO costs |
1,428,340 | — | — | |||||||||
| Accrued expenses recorded as a discount on debt |
161,000 | — | — | |||||||||
See Notes to Consolidated Financial Statements.
F-30
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Note 1. Organization and Description of Business
We were incorporated in the State of Delaware on April 8, 1999 and have offices and a laboratory located in Rutherford, New Jersey. Our wholly owned subsidiary, Cancer Genetics Italia SRL (“CGI Italia”), manufactures DNA probes. CGI Italia had approximately $236,000 in total assets at December 31, 2011 and approximately $103,000 in total revenue for the year ended December 31, 2011.
We are a diagnostics company focused on developing and commercializing proprietary genomic tests and services to improve the diagnosis, prognosis and response to treatment of cancer (theranosis). Our proprietary tests target cancers where prognosis information is critical and where predicting treatment outcomes using currently available techniques is limited. These cancers include hematological, urogenital and HPV-associated cancers. We have commercially launched MatBA®-CLL and -SLL, our first proprietary microarray diagnostic tests, and seek to provide our tests and services to oncologists and pathologists at hospitals, cancer centers and physician offices.
Note 2. Significant Accounting Policies
Basis of presentation: We prepare our financial statements on the accrual basis of accounting in accordance with accounting principles generally accepted in the United States of America. Revenues are recognized in the period in which they are earned. Expenses are recognized in the period in which the related liability is incurred.
Segment Reporting: Operating segments are defined as components of an enterprise about which separate discrete information is used by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. We view our operations and manage our business in one operating segment, which is the business of developing and selling diagnostic tests.
Liquidity: Our primary sources of liquidity have been funds generated from the sale of shares of common and preferred stock, debt financings, grants in lieu of federal income tax credits and National Institute of Health grants. We plan on raising capital in the near term in an IPO; however, without these proceeds, we have a potential shortfall in cash of approximately $1.0 million through the end of 2012. We have received a funding commitment letter from a significant stockholder and board member to help us raise or provide additional funding up to $1.25 million, should it become necessary. On a long-term basis and based on our current operating projections, we will need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing, which is not in place at this time, may be from the sale of equity or convertible or other debt securities in a public or private offering, from an additional credit facility or strategic partnership coupled with an investment in the Company or a combination of both. We may be unable to raise sufficient additional financing on terms that are acceptable, if at all. Our failure to raise additional capital and in sufficient amounts will severely impact our ability to expand our business.
Principles of consolidation: The accompanying consolidated financial statements include the accounts of Cancer Genetics, Inc. and our wholly owned subsidiary, Cancer Genetics Italia SRL. All significant intercompany account balances and transactions have been eliminated in consolidation.
Use of estimates and assumptions: The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the
F-31
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
reporting period. Significant estimates made by management include, among others, realization of amounts billed, realization of long-lived assets, realization of intangible assets, accruals for litigation and registration payments and assumptions used to value stock options and warrants. Actual results could differ from those estimates.
Risks and uncertainties: We operate in an industry that is subject to intense competition, government regulation and rapid technological change. Our operations are subject to significant risk and uncertainties including financial, operational, technological, regulatory and other risks, including the potential risk of business failure.
Cash and cash equivalents: Highly liquid investments with original maturities of three months or less when purchased are considered to be cash equivalents. Financial instruments which potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents. We maintain cash and cash equivalents with high-credit quality financial institutions. At times, such amounts may exceed insured limits. We have not experienced any losses in such accounts and believe we are not exposed to any significant credit risk on our cash and cash equivalents.
Revenue recognition: Revenue is recognized in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 605, Revenue Recognition, and ASC 954-605 Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence that an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the customer or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. In determining whether the price is fixed or determinable, we consider payment limits imposed by insurance carriers and Medicare and the amount of revenue recorded takes into account the historical percentage of revenue we have collected for each type of test for each payor category. Periodically, an adjustment is made to revenue to record differences between our anticipated cash receipts from insurance carriers and Medicare and actual receipts from such payors. For the periods presented, such adjustments were not significant. For direct bill customers, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, we consider whether we have sufficient payment history to reliably estimate a payor’s individual payment patterns. For new tests where there is no evidence of payment history at the time the tests are completed, we only recognize revenues once reimbursement experience can be established. We then recognize revenue equal to the amount of cash received. Sales of probes are recorded on the shipping date. We do not bill customers for shipping and handling fees and do not collect any sales or other taxes.
Revenues from grants to support product development is recognized when costs and expenses under the terms of the grant have been incurred and payments under the grants become contractually due. In 2010, we were awarded a federal grant in the amount of $733,438 under the Qualifying Therapeutic Discovery Project which funded research targeted at new therapies to treat areas of unmet medical need or prevent, detect or treat chronic or acute diseases and conditions, reduce the long-term growth of health care costs in the U.S. or significantly advance the goal of curing cancer within 30 years. The qualifying expenditures were incurred in 2009 and 2010 and we have presented this grant in other income.
Accounts receivable: Accounts receivable are carried at original invoice amount less an estimate for contractual adjustments and doubtful receivables based on a review of all outstanding amounts on a periodic basis. The estimate for doubtful receivables is determined from an analysis of the accounts receivable on a quarterly basis, and is recorded as bad debt expense. Accounts receivable are written off when deemed uncollectible. Recoveries of accounts receivable previously written off are recorded when received.
F-32
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Fixed assets: Fixed assets consist of diagnostic equipment, furniture and fixtures and leasehold improvements. Fixed assets are carried at cost and are depreciated over the estimated useful lives of the assets, which generally range from five to seven years. Leasehold improvements are amortized over the lesser of the lease term or the estimated useful lives of the improvements. The straight-line method is used for depreciation and amortization. Repairs and maintenance are charged to expense as incurred while improvements are capitalized. Upon sale, retirement or disposal of fixed assets, the accounts are relieved of the cost and the related accumulated depreciation or amortization with any gain or loss recorded to the consolidated statement of operations.
Fixed assets are reviewed for impairment whenever changes in circumstances indicate that the carrying amount of an asset may not be recoverable. These computations utilize judgments and assumptions inherent in our estimate of future cash flows to determine recoverability of these assets. If our assumptions about these assets were to change as a result of events or circumstances, we may be required to record an impairment loss.
Loan guarantee fee: Loan guarantee fees are amortized on a straight-line basis over the term of the guarantee.
Deferred IPO costs: Deferred initial public offering (“IPO”) costs represent legal, accounting and other direct costs related to our effort to raise capital through an IPO. Future costs related to our IPO activities will be deferred until the completion of the IPO, at which time they will be reclassified to additional paid-in capital as a reduction of the IPO proceeds. If we terminate our plan for an IPO, any deferred costs would be expensed at that time.
Warrant liability: We account for warrants that contain an exercise price adjustment feature as liabilities. Effective January 1, 2009, 839,034 of the issued and outstanding common stock purchase warrants previously treated as equity were no longer afforded equity treatment. As a result, we recorded a cumulative adjustment to reclassify $711,000 from additional paid-in capital and $210,000 from retained earnings and recorded a $921,000 long-term warrant liability to recognize the fair value of such warrants on January 1, 2009. These common stock purchase warrants do not trade in an active securities market, and as such, we estimate the fair value of these warrants using the binomial lattice valuation pricing model with the assumptions as follows: The risk-free interest rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve. The expected life of the warrants is based upon the contractual life of the warrants. Volatility is estimated based on an average of the historical volatilities of the common stock of four entities with characteristics similar to those of the Company. The measurement date fair value of the underlying common shares is based upon an external valuation of our shares. (See Notes 13 and 14).
We compute the fair value of the warrant liability at each reporting period and the change in the fair value is recorded as non-cash expense or non-cash income. The key component in the value of the warrant liability is our stock price, which is subject to significant fluctuation and is not under our control. The resulting effect on our net loss is therefore subject to significant fluctuation and will continue to be so until the warrants are exercised, amended or expire. Assuming all other fair value inputs remain constant, we will record non-cash expense when our stock price increases and non-cash income when our stock price decreases.
Income taxes: Income taxes are provided for the tax effects of transactions reported in the consolidated financial statements and consist of taxes currently due plus deferred income taxes. Deferred income taxes are recognized for temporary differences between the financial statement and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future. Deferred income taxes are also recognized for net operating loss carryforwards that are available to offset future taxable income and research and development credits.
F-33
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. We have established a full valuation allowance on our deferred tax assets as of December 31, 2011 and 2010, therefore we have not recognized any tax benefit or expense in the periods presented.
ASC 740, Income Taxes, clarifies the accounting for uncertainty in income taxes recognized in the financial statements. ASC 740 provides that a tax benefit from uncertain tax positions may be recognized when it is more-likely-than-not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits of the position. Income tax positions must meet a more-likely-than-not recognition threshold to be recognized. ASC 740 also provides guidance on measurement, derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. See Note 10 for a discussion of uncertain tax positions.
Our policy is to recognize interest and/or penalties related to income tax matters in income tax expense. There is no accrual for interest or penalties on our consolidated balance sheets at December 31, 2011 or 2010, and we have not recognized interest and/or penalties in the consolidated statements of operations for the years ended December 31, 2011, 2010 or 2009.
Patents: We account for intangible assets under ASC 350, Goodwill and Other Intangibles – 30 General Intangibles Other than Goodwill. Patents consist of legal fees incurred and are recorded at cost and amortized over the useful lives of the assets, using the straight-line method. Certain patents are in the legal application process and therefore are not currently being amortized. We review the carrying value of patents at the end of each reporting period. Based upon our review, there were no intangible asset impairments in 2011, 2010 or 2009. Accumulated amortization of patents as of December 31, 2011 and 2010 was approximately $24,000 and $13,000, respectively. Future amortization expense for legally approved patents is estimated at $13,100 per year through 2018 and approximately $9,800, $3,400, and $1,700 for 2019, 2020, and 2021, respectively.
Research and development: Research and development costs associated with service and product development include direct costs of payroll, employee benefits, stock-based compensation and supplies and an allocation of indirect costs including rent, utilities, depreciation and repairs and maintenance. All research and development costs are expensed as they are incurred.
Registration payment arrangements: We account for our obligations under registration payment arrangements in accordance with ASC 825-20, Registration Payment Arrangements. ASC 825-20 requires us to record a liability if we determine a registration payment is probable and if it can reasonably be estimated. As of December 31, 2011, we have accrued a liability of $150,000 with corresponding charge to earnings for a registration obligation related to the issuance of Series B preferred stock.
Stock-based compensation: Stock-based compensation is accounted for in accordance with the provisions of ASC 718, Compensation-Stock Compensation, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and directors based on estimated fair values on the grant date. We estimate the fair value of stock-based awards on the date of grant using the Black-Scholes option pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. See additional information in Note 12.
All issuances of stock options or other issuances of equity instruments to employees as the consideration for services received by us are accounted for based on the fair value of the equity instrument issued.
We account for stock-based compensation awards to non-employees in accordance with ASC 505-50, Equity Based Payments to Non-Employees. Under ASC 505-50, we determine the fair value of the warrants or
F-34
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. Stock-based compensation awards issued to non-employees are recorded in expense and additional paid-in capital in stockholders’ deficit over the applicable service periods based on the fair value of the awards or consideration received at the vesting date.
Fair value of financial instruments: The carrying amount of cash and cash equivalents, restricted cash, accounts receivable, accounts payable and accrued expenses, and capital leases approximate their estimated fair values due to the short-term maturities of those financial instruments. The fair value of notes payable and the line of credit is estimated based on anticipated interest rates which we believe would currently be available to us for similar issues of debt, taking into account our current credit risk and other market factors. We believe the carrying value of debt instruments approximates fair value. The fair value of warrants recorded as derivative liabilities is described in Note 14.
Earnings (loss) per share: Basic earnings (loss) per share is computed by dividing net income (loss) available to common shareholders by the weighted average number of common shares assumed to be outstanding during the period of computation. Diluted earnings per share is computed similar to basic earnings per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common shares had been issued and if the additional common shares were dilutive. For all periods presented, basic and diluted loss per share are the same, as any additional common stock equivalents would be antidilutive due to the net loss, and as such have been excluded from the calculation of the weighted average number of dilutive common shares.
The following table summarizes potentially dilutive adjustments to the weighted average number of common shares which were excluded from the calculation:
| 2011 | 2010 | 2009 | ||||||||||
| Common stock purchase warrants |
2,221,848 | 1,864,331 | 1,518,096 | |||||||||
| Stock options |
1,399,975 | 1,279,150 | 550,000 | |||||||||
| Common shares issuable upon conversion of Series A Preferred Stock |
881,537 | 881,537 | 881,537 | |||||||||
| Common shares issuable upon conversion of Series B Preferred Stock |
910,800 | 910,800 | — | |||||||||
| Common shares issuable upon conversion of note payable to shareholder |
— | 23,220 | 23,220 | |||||||||
|
|
|
|
|
|
|
|||||||
| 5,414,160 | 4,959,038 | 2,972,853 | ||||||||||
|
|
|
|
|
|
|
|||||||
Recent accounting pronouncements: In January 2010, the FASB issued Accounting Standards Update (“ASU”) 2010-06, Improving Disclosures about Fair Value Measurements. ASU 2010-06 provides amendments to ASC 820-10, Fair Value Measurements and Disclosures, that require entities to disclose separately the amounts of significant transfers in and out of Level 1 and Level 2 fair value measurements and describe the reasons for the transfers. In addition, ASU 2010-06 requires entities to present separately information about purchases, sales, issuances, and settlements in the reconciliation for fair value measurements using significant unobservable inputs (Level 3). The disclosures related to Level 1 and Level 2 fair value measurements became effective for us during 2010 and the disclosures related to Level 3 fair value measurements are effective for us in 2011. ASU 2010-06 required new disclosures only, and had no impact on our consolidated financial position, results of operations, or cash flows.
In May 2011, the FASB issued ASU 2011-04, “Fair Value Measurement (Topic 820) — Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. GAAP and IFRSs”. ASU 2011-04 generally represents clarification of Topic 820, but also includes instances where a particular principle
F-35
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
or requirement for measuring fair value or disclosing information about fair value measurements has changed. ASU 2011-04 results in common principles and requirements for measuring fair value and for disclosing information about fair value measurements in accordance with Generally Accepted Accounting Principles and International Financial Reporting Standards. ASU 2011-04 is effective for interim and annual periods beginning after December 15, 2011 and is to be applied prospectively. Early application is not permitted. We do not expect the adoption of ASU 2011-04 to have a material impact on our consolidated financial statements.
In July 2011, the FASB issued ASU 2011-07, “Health Care Entities (Topic 954): Presentation and Disclosure of Patient Service Revenue, Provision for Bad Debts and the Allowance for Doubtful Accounts for Certain Health Care Entities”. ASU 2011-07 requires certain health care entities to change the presentation in their statement of operations by reclassifying the provision for bad debts associated with patient service revenue from an operating expense to a deduction from patient service revenue (net of contractual allowances and discounts). Additionally, those health care entities are required to provide enhanced disclosure about their policies for recognizing revenue and assessing bad debts. The amendments also require disclosures of patient service revenue (net of contractual allowances and discounts) as well as qualitative and quantitative information about changes in the allowance for doubtful accounts. ASU 2011-07 is effective for fiscal years and interim periods within those fiscal years beginning after December 15, 2011, with early adoption permitted. We do not expect the adoption of ASU 2011-07 to have a material impact on our consolidated financial statements.
Subsequent events: We have evaluated potential subsequent events through March 13, 2012, which is the date the financial statements were issued.
Note 3. Revenue and Accounts Receivable
Revenue by payor type for the years ended December 31 is comprised of the following:
| 2011 | 2010 | 2009 | ||||||||||
| Medicare |
$ | 717,661 | $ | 605,776 | $ | 352,328 | ||||||
| Direct bill |
350,290 | 408,356 | 461,374 | |||||||||
| Grants and royalty |
315,195 | 110,000 | 99,565 | |||||||||
| Insurance carrier and all others |
1,636,261 | 1,397,447 | 752,420 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 3,019,407 | $ | 2,521,579 | $ | 1,665,687 | |||||||
|
|
|
|
|
|
|
|||||||
Accounts receivable by payor type at December 31 consists of the following:
| 2011 | 2010 | |||||||
| Medicare |
$ | 152,186 | $ | 173,996 | ||||
| Direct bill |
64,183 | 20,058 | ||||||
| Insurance carrier and all others |
496,661 | 558,146 | ||||||
| Allowance for doubtful accounts |
(24,050 | ) | (46,356 | ) | ||||
|
|
|
|
|
|||||
| $ | 688,980 | $ | 705,844 | |||||
|
|
|
|
|
|||||
We have historically derived a significant portion of our revenue from a limited number of test ordering sites. The test ordering sites are largely hospitals, cancer centers, reference laboratories and physician offices. Oncologists and pathologists at these sites order the tests on behalf of the needs of their oncology patients. The top five test ordering sites during 2011 and 2010 accounted for 63% and 60%, respectively, of our clinical testing volumes, with 29% and 15%, respectively, of the volume coming from community hospitals. For the year ended
F-36
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
December 31, 2011, we generated revenue from two test ordering sites that represented 10% or more of our revenue: a community hospital accounted for approximately 18% of our revenue and a community oncology practice accounted for approximately 11% of our revenue. During the year ended December 31, 2010, there were three sites which each accounted for 10% or more of our revenue: one community hospital accounted for approximately 12% of our revenue; a regional reference laboratory accounted for approximately 11% of our revenue and a community oncology practice accounted for another approximately 11% of our revenue. We generally do not enter into formal written agreements with such test ordering sites and, as a result, we may lose these significant test ordering sites at any time.
Note 4. Other Current Assets
At December 31, 2011 and 2010 other current assets consisted of the following:
| 2011 | 2010 | |||||||
| Grants receivable |
$ | — | $ | 733,438 | ||||
| Inventory probes and arrays |
203,507 | 41,546 | ||||||
| Prepaid expenses |
65,762 | 46,023 | ||||||
|
|
|
|
|
|||||
| Total other current assets |
$ | 269,269 | $ | 821,007 | ||||
|
|
|
|
|
|||||
The grants receivable consists of three Qualifying Therapeutic Discovery Project grants administered under Section 48D of the Internal Revenue Code which were awarded in October 2010. The qualifying expenses were incurred in 2010 and 2009.
Note 5. Lease Commitments
We lease our laboratory and research facilities and administrative office space in Rutherford, New Jersey under an escalating lease agreement that expires on October 6, 2017. The lease requires monthly rent for 10 years with periodic rent increases that vary from $1 to $2 per square foot of the rented premises per year. The difference between minimum rent and straight-line rent is recorded as deferred rent payable. The terms of the lease require that a $450,000 security deposit for the facility be held in a stand by letter of credit held in favor of the landlord (see Note 7).
We acquired office and scientific equipment under long term leases which have been capitalized at the present value of the minimum lease payments. The equipment under these capital leases had a cost of $142,196 and accumulated depreciation of $78,916, as of December 31, 2011.
F-37
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Minimum future lease payments under all capital and operating leases as of December 31, 2011 are as follows:
| Capital Leases |
Operating Leases |
Total | ||||||||||
| December 31, |
||||||||||||
| 2012 |
$ | 43,173 | $ | 521,983 | $ | 565,156 | ||||||
| 2013 |
19,391 | 523,478 | 542,869 | |||||||||
| 2014 |
7,739 | 556,361 | 564,100 | |||||||||
| 2015 |
— | 567,590 | 567,590 | |||||||||
| 2016 |
— | 564,984 | 564,984 | |||||||||
| Thereafter |
— | 612,066 | 612,066 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total of minimum lease payments |
70,303 | $ | 3,346,462 | $ | 3,416,765 | |||||||
|
|
|
|
|
|||||||||
| Less amount representing interest |
9,535 | |||||||||||
|
|
|
|||||||||||
| Present value of net minimum obligations |
60,768 | |||||||||||
| Less current obligation under capital lease |
36,209 | |||||||||||
|
|
|
|||||||||||
| Long-term obligation under capital lease |
$ | 24,559 | ||||||||||
|
|
|
|||||||||||
Rent expense for the years ended December 31, 2011, 2010, and 2009 was $517,667, $546,169, and $535,558, respectively.
Note 6. Notes Payable
As of December 31, 2011, we had $3 million outstanding under a Credit Agreement dated as of December 21, 2011, as amended and restated as of February 13, 2012. We borrowed an additional $3 million under this agreement in February 2012. The Credit Agreement is with John Pappajohn and Andrew Pecora (indirectly through an investment company), both members of our board of directors, and NNJCA Capital, LLC (“NNJCA”), a limited liability company of which Dr. Pecora is a member, for a $6.0 million secured term loan. Mr. Pappajohn has provided $4.0 million of financing, NNJCA has provided $1.5 million of financing and Pecora has provided $500,000 of financing under the Credit Agreement.
The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at December 31, 2011) and matures on February 13, 2013, with an option, at our election, if there has been no event of default, to extend the loan term for an additional six months. The term loan due to Pecora and NNJCA has a prepayment penalty in the aggregate amount of $190,000, less interest previously paid, if we prepay the loan prior to February 12, 2013. The lenders may require that the loan be repaid within 30 days should we complete our initial public offering and receive gross proceeds of at least $15 million. In the event that any lender requires payment upon completion of our initial public offering and certain other maturity events, the annual interest rate on any unpaid balance shall increase to 12%. The loan is secured by all of our assets, including our intellectual property, subject to prior first and second liens in favor of Wells Fargo Bank and DAM Holdings, LLC (“DAM”). Pursuant to an intercreditor agreement, the lenders have agreed that all amounts due to DAM are to be paid prior to payment to the lenders under this Credit Agreement, but that as between such lenders, following an event of default, all of the security granted by us is to be applied first to repay obligations due to Pecora and NNJCA, and then to Mr. Pappajohn after they have been paid in full. As Mr. Pappajohn has guaranteed the Wells Fargo debt, in essence under the intercreditor agreement, NNJCA and Pecora will be junior only to DAM.
F-38
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
The lenders received five-year warrants to purchase an aggregate of 211,764 shares of our common stock at the lower of (i) $17.00 per share and (ii) a 20% discount to the initial public offering price if the initial public offering price is less than $21.25 per share, with 105,882 warrants issued to Mr. Pappajohn upon his fully funding $3.0 million on December 22, 2011 and an additional 105,882 warrants being issued proportionately to Mr. Pappajohn, Pecora and NJCCA upon their funding $1.0 million, $0.5 million and $1.5 million, respectively, on February 13, 2012. The lenders will receive 17,647 additional warrants on the same terms in the event we do not consummate our initial public offering by each the following dates: April 1, 2012 (Mr. Pappajohn only); May 15, 2012 (each of the three lenders will receive a portion of the 17,647 warrants proportional to their contributions to the second $3.0 million tranche); July 1, 2012 (Pappajohn only) and August 14, 2012 (each of the three lenders will receive a portion of the 17,647 warrants proportional to the second tranche). The warrant exercise price is subject to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The lenders may elect to net exercise their warrants. Mr. Pappajohn also has the option, exercisable up to two hours after he receives notice of the completion of our initial public offering, to convert the outstanding principal amount of his debt into shares of our common stock at a conversion price equal to $17.00 per share or at a 20% discount to our initial public offering price, whichever is lower. There is a minimum contingent beneficial conversion feature of approximately $150,000 for each $1 million of debt which would result in a noncash charge in the event of an IPO. Such beneficial conversion amount would increase should the IPO price be below $21.25 per share. The conversion price of the notes and the exercise price of the warrants are subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like.
Shares that the lenders receive are subject to a lock up agreement for 180 days after the consummation of our IPO on the same terms as other lock up agreements in favor of the underwriters of this offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
At December 31, 2011 and 2010, notes payable included a note payable to a shareholder. The note is due on December 31, 2012 and bears interest at 8.5% per annum. At December 31, 2011 and 2010, $100,000 was outstanding under this note. Accrued interest at each of the years ended December 31, 2011 and 2010 was approximately $36,000.
Note 7. Letter of Credit
In connection with the facility lease described in Note 5, the lessor required the establishment of a stand by letter of credit in the amount of $450,000 with Capital One Bank to use as a guarantee for a security deposit. The amount of the letter of credit was reduced by $54,800 and $11,600 during the years ended December 31, 2011 and 2010, respectively, and provided there is no event of default under the lease agreement, it will be reduced by $54,800 as of each subsequent anniversary of the commencement date of the lease. In no event will it be reduced below $127,794. In February 2011, we allowed the letter of credit to expire and are in discussions with the landlord to avoid default on the lease agreement. We are negotiating a letter agreement that allows for a reduction of the amount required under the letter of credit in exchange for an option, contingent on completion of an initial public offering, to lease additional office space.
Note 8. Business Lines of Credit
At December 31, 2011, we have fully utilized a line of credit with Wells Fargo Bank which provides for maximum borrowings of $6,000,000. Interest on the line of credit is due monthly equal to 1.75% above the Daily One Month LIBOR rate (2.05% at December 31, 2011). The line of credit requires the repayment of principal,
F-39
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
and any unpaid interest, in a single payment due on July 31, 2012. The line of credit is guaranteed by one of our stockholders and is collateralized by a first lien on all of our assets including the assignment of our approved and pending patent applications (see Note 18). At December 31, 2011 and 2010, $6,000,000 was outstanding under the line of credit.
Subsequent to December 31, 2011, we entered into a seventh addendum to the Credit Agreement with Wells Fargo which extended the maturity of the line of credit to July 31, 2013. As a result of the extension of the maturity date, we have presented the line of credit as a long-term liability. The guarantee by one of our stockholders on this line of credit continues through the new maturity date of July 31, 2013, and in consideration for this extension in March 2012 the stockholder was granted warrants to purchase 92,500 shares of our common stock at the lower of (i) $17.00 per share or (ii) a 20% discount to the initial public offering price if the initial public offering price is less than $21.25 per share.
On March 23, 2011, we entered into a line of credit agreement with DAM Holdings, LLC (“DAM”) in which we received proceeds of $3 million. Pursuant to an intercreditor agreement between a member of our board of directors and DAM (the “Intercreditor Agreement”), we are required to use the proceeds from our IPO to repay the full amount outstanding under the DAM Loan Agreement before any proceeds can be used to repay any debt outstanding under the Wells Fargo Line of Credit. The DAM debt bears an initial annual interest rate of 3.0% payable in equal monthly installments and expires upon the earlier of the following: (i) September 23, 2012, (ii) the occurrence of an initial public offering of our equity securities in which we receive gross proceeds in the amount of $10,000,000 or more, or (iii) the consummation of a transaction in which we either merge with a reporting company under the Securities Exchange Act of 1934, as amended, or a public company acquires all or substantially all of our Company, and the survivor of such merger of the public company receives gross proceeds from the sale of the survivors securities or the public company’s securities in the amount of $10,000,000 or more. If certain maturity events do not occur prior to January 1, 2012, then the interest rate on the DAM debt will increase to 10% per annum and if certain maturity events do not occur prior to April 1, 2012 then the interest rate will increase to 15% per annum.
As of December 31, 2011, certain maturity events had not occurred and the interest rate on the DAM debt increased to 10% per annum on January 1, 2012. Subsequent to December 31, 2011, we entered into an agreement with DAM which extended the maturity of the line of credit to April 1, 2013 and provided that the interest rate from January 1, 2012 until a maturity event occurs will be 10% and after a maturity event occurs the interest rate adjusts to 18% until paid in full. In consideration for this extension, in March 2012, we granted DAM warrants to purchase 37,500 shares of our common stock at the lower of (i) $17.00 per share or (ii) a 20% discount to the initial public offering price if the initial public offering price is less than $21.25 per share.
Note 9. Fixed Assets
Fixed Assets are summarized by major classifications as follows:
| 2011 | 2010 | |||||||
| Equipment |
$ | 1,752,398 | $ | 1,434,166 | ||||
| Furniture |
461,119 | 461,119 | ||||||
| Leasehold improvements |
583,592 | 583,592 | ||||||
|
|
|
|
|
|||||
| 2,797,109 | 2,478,877 | |||||||
| Less accumulated depreciation |
(1,656,473 | ) | (1,299,870 | ) | ||||
|
|
|
|
|
|||||
| Net fixed assets |
$ | 1,140,636 | $ | 1,179,007 | ||||
|
|
|
|
|
|||||
F-40
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Note 10. Income Taxes
The provision for income taxes for the years ended December 31, 2011, 2010 and 2009 differs from the approximate amount of income tax benefit determined by applying the U.S. federal income tax rate to pre-tax loss, due to the following:
| For the Year Ended December 31, 2011 |
For the Year Ended December 31, 2010 |
For the Year Ended December 31, 2009 |
||||||||||||||||||||||
| Amount | % of Pretax Loss |
Amount | % of Pretax Loss |
Amount | % of Pretax Loss |
|||||||||||||||||||
| Income tax benefit at federal statutory rate |
$ | (6,960,000 | ) | 35.0 | % | $ | (2,943,000 | ) | 35.0 | % | $ | (2,565,000 | ) | 35.0 | % | |||||||||
| State tax provision, net of federal tax benefit |
(604,000 | ) | 3.0 | % | (437,000 | ) | 5.2 | % | (112,000 | ) | 1.5 | % | ||||||||||||
| Tax credits |
(57,000 | ) | 0.3 | % | (26,000 | ) | 0.3 | % | (6,000 | ) | 0.1 | % | ||||||||||||
| Stock option permanent differences |
43,000 | -0.2 | % | 33,000 | -0.4 | % | — | 0.0 | % | |||||||||||||||
| Derivative warrant permanent differences |
3,636,000 | -18.3 | % | 16,000 | -0.2 | % | 590,000 | -8.0 | % | |||||||||||||||
| Change in valuation allowance |
5,320,000 | -26.8 | % | 2,543,000 | -30.3 | % | 1,528,000 | -20.9 | % | |||||||||||||||
| Change in uncertain tax positions |
(1,395,000 | ) | 7.0 | 1,003,000 | -11.9 | % | 330,000 | -4.5 | % | |||||||||||||||
| Other |
17,000 | -0.1 | % | (189,000 | ) | 2.3 | % | 235,000 | -3.2 | % | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Provision for income taxes |
$ | — | 0.0 | % | $ | — | 0.0 | % | $ | — | 0.0 | % | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Approximate deferred taxes consist of the following components as of December 31, 2011 and 2010:
| 2011 | 2010 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 10,575,000 | $ | 5,867,000 | ||||
| Accruals and reserves |
568,000 | 392,000 | ||||||
| Non-qualified stock options |
575,000 | 246,000 | ||||||
| Research and development tax credits |
395,000 | 338,000 | ||||||
| Derivative warrant liability |
26,000 | 26,000 | ||||||
| Other |
9,000 | 10,000 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
12,148,000 | 6,879,000 | ||||||
| Less valuation allowance |
(12,089,000 | ) | (6,769,000 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
59,000 | 110,000 | ||||||
| Deferred tax liabilities: |
||||||||
| Fixed assets |
(59,000 | ) | (110,000 | ) | ||||
|
|
|
|
|
|||||
| Net deferred taxes |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
Due to a history of losses we have generated since inception, we believe it is more-likely-than-not that all of the deferred tax assets will not be realized as of December 31, 2011 and 2010. Therefore, we have recorded a full valuation allowance on our deferred tax assets.
F-41
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
We have net operating loss carryforwards for federal income tax purposes of approximately $26,000,000 as of December 31, 2011. The net operating loss carryforwards will begin to expire in 2026. Utilization of these carryforwards is subject to limitations due to ownership changes that may delay the utilization of a portion of the carryforwards.
Uncertain Tax Positions
The aggregate changes in the balance of unrecognized tax benefits were as follows:
| 2011 | 2010 | |||||||
| Balance, beginning of year |
$ | 1,395,000 | $ | 392,000 | ||||
| Increases for tax positions related to the current year |
— | 1,003,000 | ||||||
| Change in expected tax position |
(1,395,000 | ) | — | |||||
|
|
|
|
|
|||||
| Balance, end of year |
$ | — | $ | 1,395,000 | ||||
|
|
|
|
|
|||||
During 2011, we changed a tax position that was expected to be taken on future tax returns related to stock-based compensation to non-employees which reduced our unrecognized tax benefits to zero at December 31, 2011. Therefore, at December 31, 2011, we have no uncertain tax positions and do not expect any change in 2012.
Our federal and New Jersey tax returns remain open for examination for tax years 2006 to 2011 due to our net operating loss carryforward.
Note 11. Capital Stock
On December 29, 2011, the Board of Directors approved the filing of a registration statement with the Securities and Exchange Commission for an initial public offering of the Company’s common stock. On April 16, 2012, the Board of Directors approved a 1-for-2 reverse stock split of the Company’s common stock to be effected immediately prior to the effectiveness of the Company’s registration statement. All shares and per share information referenced throughout the consolidated financial statements have been retroactively adjusted to reflect this 1-for-2 reverse stock split.
We are authorized to issue 588,000 shares of Series A Convertible Preferred Stock (“Series A”) and 2,000,000 shares of Series B Convertible Preferred Stock (“Series B”). The holders of the Series A and Series B are entitled to participate in any dividend declared by the Board of Directors on the Common Stock on a pro-rata basis with holders of the Common Stock. The Series A holders have a liquidation preference equal to the original Series A purchase price of $8.46 per share (or $4,971,866) and the Series B holders have a liquidation preference equal to the Series B original purchase price of $5.00 per share (or $9,108,000) subject to adjustment for stock splits, stock dividends and combinations and similar adjustments to capitalization. Alternatively, the preferred stock shareholders can elect to convert the Series A and Series B into Common Stock and participate ratably with holders of Common Stock in the distribution of assets upon liquidation of the Company. The holders of Series A and Series B have the right to convert their shares into Common Stock at any time. The initial conversion rate is the original purchase price of the Series A or Series B shares divided by the conversion price in effect at the time of conversion. The initial conversion price equals the original purchase price paid by the holder for a share of Series A or series B, as applicable. The Series A shares will automatically convert into 881,537 shares of common stock and the Series B shares will automatically convert into 910,800 shares of common stock upon the closing of an underwritten public offering pursuant to an effective registration statement in connection with an initial public offering with gross proceeds of $25,000,000 or more and under certain other circumstances.
F-42
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
The conversion price is subject to adjustment in the event of stock dividends, stock splits, combinations and similar adjustments to capitalization. The prices of each series of preferred stock are also subject to adjustment in the event that we issue additional equity securities at a per share price less than the applicable conversion price for each such series of preferred stock. The current conversion price for the Series A is $5.64 and for the Series B is $10.00. All references to the Company’s Series A preferred stock in these financial statements refer collectively to the Series A and Series A-1 preferred shares. The Company issued the Series A-1 preferred stock in exchange for the Series A to cure possible technical deficiencies with respect to the original issuance of the Series A preferred stock, but five holders of Series A have not responded to requests to exchange their shares, which represent approximately 9.01% of the two Series A and A-1 that is outstanding.
The Series B purchase agreement provides for the payment of a semiannual cash penalty payable to each holder of Series B if we fail to complete a merger with a corporation whose shares were registered for issuance pursuant to the Securities Act of 1933, as amended (the “Securities Act”), within 9 months of the closing of the Series B offering (or August 19, 2011) (the “Merger Period”). The penalty shall be equal to 1% of the original purchase price for a share of Series B for the first 30 day period following the expiration of the Merger Period if a merger shall not have been consummated and an additional 2% of the original purchase price for a share of Series B for each successive 30 day period following the first penalty period, pro rated daily up to a maximum penalty of 11% of the original purchase price for a share of Series B per annum. The maximum potential consideration to be transferred under the terms of the purchase agreement is unlimited. In September 2011, we solicited holders of the Series B to execute an amendment and waiver to the Series B purchase agreement, which provided for (i) a waiver of the cash penalties and (ii) an amendment extending the Merger Period to March 31, 2012 and providing for (A) the tolling of the Merger Period if we file a registration statement with the Securities and Exchange Commission with respect to an lPO and (B) elimination of penalties if we consummate the lPO. As of December 31, 2011, holders of 1,522,600 shares of Series B Convertible Preferred Stock have agreed to the waiver.
Note 12. Stock Option Plan
We have two equity incentive plans: the 2008 Stock Option Plan (2008 Plan) and the 2011 Equity Incentive Plan (2011 Plan). The 2011 Plan was approved by the Board of Directors on June 30, 2011 and was subsequently ratified by shareholders. The 2011 Plan authorizes the issuance of up to 375,000 shares of common stock under several types of equity awards including stock options, stock appreciation rights, restricted stock awards and other awards defined in the 2011 Plan. There have been no awards under the 2011 Plan.
The Board of Directors adopted the 2008 Plan on April 29, 2008 and reserved 628,689 shares of common stock for issuance under the plan. On April 1, 2010, the stockholders voted to increase the number of shares reserved by the plan to 1,375,000. The 2008 Plan is meant to provide additional incentive to officers, employees and consultants to remain in our employment. We are authorized to issue incentive stock options or nonstatutory stock options to eligible participants. Options granted are generally exercisable for up to 10 years.
During 2011, 89,125 options were amended to increase the exercise price from $1.60 to $1.92 based upon a retrospective valuation of our common stock as of the date of grant. The increase in the exercise price of the options did not result in the recognition of incremental compensation cost.
F-43
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
A summary of employee and nonemployee stock option activity for the years ended December 31, 2011 and 2010 is as follows:
| Options Outstanding | Weighted- Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value |
||||||||||||||
| Number of Shares |
Weighted- Average Exercise Price |
|||||||||||||||
| Outstanding December 31, 2008 |
112,500 | $ | 1.60 | 9.83 | $ | — | ||||||||||
| Granted |
439,000 | 1.60 | ||||||||||||||
| Cancelled or expired |
(1,500 | ) | 1.60 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding December 31, 2009 |
550,000 | $ | 1.60 | 9.13 | $ | 176,400 | ||||||||||
| Granted |
851,650 | 5.74 | ||||||||||||||
| Exercised |
(113,500 | ) | 1.60 | |||||||||||||
| Cancelled or expired |
(9,000 | ) | 1.60 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding December 31, 2010 |
1,279,150 | $ | 4.36 | 8.97 | $ | 1,693,760 | ||||||||||
| Granted |
169,038 | 10.69 | ||||||||||||||
| Cancelled or expired |
(48,213 | ) | 4.71 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding December 31, 2011 |
1,399,975 | $ | 5.14 | 8.10 | $ | 11,737,710 | ||||||||||
| Exercisable, December 31, 2011 |
799,021 | $ | 3.71 | 7.74 | $ | 7,836,506 | ||||||||||
Aggregate intrinsic value represents the difference between the estimated fair value of our common stock and the exercise price of outstanding, in-the-money options. The estimated fair value of our common stock was $13.52, $4.76 and $1.92 as of December 31, 2011, 2010, and 2009, respectively. The total intrinsic value of options exercised was approximately $0, $293,350 and $0 for the years ended December 31, 2011, 2010 and 2009, respectively. During the year ended December 31, 2010, we received $1,600 from the exercise of options. During the year ended December 31, 2010, 18,000 shares were surrendered as part of a net-issue exercise.
As of December 31, 2011 and 2010, total unrecognized compensation cost related to nonvested stock options granted to employees was approximately $1,262,246 and $976,717, respectively, which we expect to recognize over the next 3.61 and 4.16 years, respectively.
As of December 31, 2011 and 2010, total unrecognized compensation cost related to nonvested stock options granted to non-employees was approximately $1,061,270 and $610,300, respectively, which we expect to recognize over the next 1.4 and 2.3 years, respectively. The estimate of unrecognized nonemployee compensation is based on the fair value of the nonvested options as of December 31, 2011 and 2010.
F-44
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
The following table summarizes information about outstanding and vested stock options granted to employees and non-employees as of December 31, 2011 as follows:
| Options Outstanding | Options Vested and Exercisable | |||||||||||||||||||
| Exercise Price |
Number of Shares Outstanding |
Weighted- Average Remaining Contractual Life (in years) |
Weighted- Average Exercise Price |
Number of Shares |
Weighted- Average Exercise Price |
|||||||||||||||
| 1.60 |
437,500 | 7.34 | $ | 1.60 | 437,500 | $ | 1.60 | |||||||||||||
| 1.92 |
89,125 | 8.05 | 1.92 | 40,401 | 1.92 | |||||||||||||||
| 5.00 |
505,725 | 8.27 | 5.00 | 204,503 | 5.00 | |||||||||||||||
| 10.00 |
331,250 | 8.86 | 10.00 | 116,617 | 10.00 | |||||||||||||||
| 12.66 |
16,375 | 9.76 | 12.66 | — | — | |||||||||||||||
| 13.52 |
20,000 | 9.99 | 13.52 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
1,399,975 | 8.10 | $ | 5.14 | 799,021 | $ | 3.71 | |||||||||||||
|
|
|
|
|
|||||||||||||||||
The fair value of options granted to employees is estimated on the grant date using the Black-Scholes option valuation model. This valuation model for stock-based compensation expense requires us to make assumptions and judgments about the variables used in the calculation, including the fair value of our common stock (see Note 14), the expected term (the period of time that the options granted are expected to be outstanding), the volatility of our common stock, a risk-free interest rate, and expected dividends. We also estimate forfeitures of unvested stock options. To the extent actual forfeitures differ from the estimates, the difference will be recorded as a cumulative adjustment in the period estimates are revised. No compensation cost is recorded for options that do not vest. We use the simplified calculation of expected life described in the SEC’s Staff Accounting Bulletin No. 107, Share-Based Payment, and volatility is based on an average of the historical volatilities of the common stock of four entities with characteristics similar to those of the Company. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option. We use an expected dividend yield of zero, as we do not anticipate paying any dividends in the foreseeable future. Expected forfeitures are assumed to be zero due to the small number of plan participants and the plan design which has monthly vesting after an initial cliff vesting period.
The following table presents the weighted-average assumptions used to estimate the fair value of options granted to employees during the periods presented:
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Volatility |
76.13 | % | 78.02 | % | 74.15 | % | ||||||
| Risk free interest rate |
2.67 | % | 3.20 | % | 1.75 | % | ||||||
| Dividend yield |
— | — | — | |||||||||
| Term (years) |
6.44 | 6.47 | 5.43 | |||||||||
| Weighted-average fair value of options granted during the period |
$ | 4.24 | $ | 1.84 | $ | 0.96 | ||||||
F-45
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
In 2010, we issued an aggregate of 200,000 options to non-employees with an exercise price of $10.00. The following table presents the weighted-average assumptions used to estimate the fair value of options reaching their measurement date for non-employees during the periods presented:
| Year Ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| Volatility |
75.45 | % | 77.89 | % | ||||
| Risk free interest rate |
2.85 | % | 2.76 | % | ||||
| Dividend yield |
— | — | ||||||
| Term (years) |
9.23 | 9.86 | ||||||
The following table presents the effects of stock-based compensation related to stock option awards to employees and nonemployees on our Statement of Operations during the periods presented:
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Cost of revenues |
$ | 9,603 | $ | 17,748 | $ | 1,380 | ||||||
| Research and development |
423,950 | 64,038 | 230,000 | |||||||||
| General and administrative |
263,693 | 323,380 | 69,322 | |||||||||
| Sales and marketing |
245,871 | 50,809 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation |
$ | 943,117 | $ | 455,975 | $ | 300,702 | ||||||
|
|
|
|
|
|
|
|||||||
Note 13. Warrants
In connection with financing obtained on January 22, 2007, we issued a warrant to purchase 22,163 shares of common stock (post split) at an exercise price of $5.64 per share expiring January 22, 2012. This warrant was recorded at estimated fair value using a Black-Scholes valuation model as a $12,854 discount on debt and was subsequently amortized to interest expense.
In connection with the issuance of Series A Preferred Stock on November 14, 2007, we issued to placement agents warrants to purchase 75,000 shares of common stock (post split) at an exercise price of $4.30 per share expiring December 31, 2011. We also issued to the same placement agents warrants to purchase 46,539 shares of common stock at an exercise price of $5.64 per share expiring December 31, 2011, subsequently extended to January 2012. These warrants were recorded at an estimated fair value using a Black-Scholes valuation model of $53,685 in additional paid-in capital.
In connection with financing obtained on January 31, 2008, we issued a warrant to purchase 8,865 shares of common stock (post split) at an exercise price of $5.64 per share expiring January 22, 2012. This warrant was recorded at estimated fair value using a Black-Scholes valuation model as a $3,191 discount on debt which was subsequently amortized to interest expense. The warrant was exercised during 2011 and we received $49,999 of cash proceeds.
In connection with the issuance of Series A Preferred Stock on February 4, 2008, we issued to placement agents warrants to purchase 142,074 shares of common stock (post split) at an exercise price of $5.64 per share expiring September 10, 2012. These warrants were recorded at an estimated fair value using a Black-Scholes valuation model of $59,671 in additional paid-in capital.
F-46
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
In connection with the issuance of Series A Preferred Stock on May 7, 2008, we issued to placement agents warrants to purchase 34,422 shares of common stock at an exercise price of $5.64 per share expiring September 10, 2012. These warrants were recorded at an estimated fair value using a Black-Scholes valuation model of $13,769 in additional paid-in capital.
Derivative Warrants
We have issued certain warrants which contain an exercise price adjustment feature in the event we issue additional equity instruments at a price lower than the exercise price of the warrant. The warrants are described herein as derivative warrants. These warrants are initially recorded as a warrant liability at fair value with a corresponding entry to the loan guarantee fee asset, additional paid-in capital or expense dependent upon the service provided in exchange for the warrant grant. Subsequently, any change in fair value is recognized in earnings until such time as the warrants are exercised, amended or expire.
In connection with debt guarantees and extensions, we issued 2,440,284 warrants to a member of our Board of Directors and shareholder at various dates (see Note 18). These warrants are initially recorded at fair value as a loan guarantee fee amortized over the period of the guarantee to interest expense. The aggregate issue date fair value of the debt guarantee warrants was $831,000 in 2011, $415,000 in 2010, and $2,026,000 in 2009.
In connection with the issuance of Series A Preferred Stock on November 16, 2007, we issued a warrant to purchase shares of common stock at an exercise price of $5.64 per share expiring November 16, 2012. The warrant contained an exercise price adjustment feature, which was triggered by the 2008 split and adjusted the number of shares underlying the warrant to 510,438 and the exercise price to $1.60 per share. The grant date fair value was $319,000. The warrant was recorded as an increase to the warrant liability and additional paid-in capital at the estimated fair value of $547,000 on the effective date of derivative accounting for warrants. This warrant was exercised as part of a net-issue exercise in August 2009.
In connection with the issuance of Series A Preferred Stock on January 8, 2008, we issued warrants to purchase shares of common stock at an exercise price of $5.64 per share expiring November 16, 2012. These warrants contain an exercise price adjustment feature, which was triggered by the 2008 split and adjusted the number of shares underlying the warrant to 18,313 and the exercise price to $1.60 per share. The grant date fair value was $11,000. These warrants were recorded as an increase to the warrant liability and additional paid-in capital at the estimated fair value of $20,000 on the effective date of derivative accounting for warrants.
In August 2009, 1,500,000 of the derivative warrants outstanding were net-issue exercised resulting in 1,260,000 shares of common stock issued to John Pappajohn.
In connection with consulting agreements dated April 1, 2010, we issued to consultants warrants to purchase 10,075 and 25,000 shares of common stock at an exercise price of $5.00 per share and $5.64 per share, respectively. These warrants expire on April 1, 2015. These warrants were recorded as an increase to the warrant liability and consulting expense at the estimated fair value on the grant date of $65,000.
In connection with the issuance of Series B Preferred Stock on November 18, 2010, we issued warrants to purchase 131,160 shares of common stock at an exercise price of $10.00 per share expiring November 18, 2015. These warrants were recorded at estimated fair value of $328,000 as a reduction of additional paid-in capital and an increase to the warrant liability.
F-47
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
In connection with debt guarantees and extensions, we issued 100,000 warrants to John Pappajohn, a member of our Board of Directors and stockholder, in June 2011. These warrants were recorded as an increase to the warrant liability and the loan guarantee fee at the estimated fair value of $831,000 on the grant date and expire in June 2016. The fair value of the loan guarantee fee is amortized over the period of the guarantee to interest expense.
We issued 500 warrants to a consultant for services provided in February and March 2011 and 10,000 warrants to a member of our Board of Directors in connection with the March 2011 line of credit agreement with DAM. These warrants were recorded as consulting expense at an initial estimated fair value of $69,000 and expire in March 2016.
In connection with the acquisition of a line of credit, we issued 150,000 warrants to DAM in March 2011. The value of these warrants reduced the carrying value of the related debt and will be amortized over the period of the debt agreement to interest expense. These warrants were recorded at an initial estimated fair value of $1,019,000 and expire in March 2016.
In December 2011, we granted 105,882 warrants to John Pappajohn, a member of our Board of Directors and stockholder in connection with a secured term loan of $3 million (see Note 6). The value of these warrants reduced the carrying value of the related debt and will be amortized over the period of the debt agreement to interest expense. These warrants were recorded at an initial estimated fair value of $951,000 and expire in December 2016.
On September 30, 2011, certain holders of derivative warrants to purchase 570,721 shares of common stock with an exercise price of $1.60 agreed to an amendment to their warrants to remove the exercise price adjustment feature. As of September 30, 2011, the fair value of these warrants of $6,415,000 was reclassified from the warrant liability to equity.
F-48
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
The following table summarizes the warrant activity for 2011, 2010 and 2009:
| Purpose |
Exercise Price |
Warrants Outstanding January 1, 2009 |
2009 Warrants Issued |
2009 Warrants Exercised |
Warrants Outstanding December 31, 2009 |
2010 Warrants Issued |
Warrants Outstanding December 31, 2010 |
2011 Warrants Issued |
2011 Warrants Exercised |
2011 Warrants Amended |
Warrants Outstanding December 31, 2011 |
|||||||||||||||||||||||||||||||||
| Debt Guarantee |
$ | 1.60 | — | — | — | — | — | — | — | — | 570,721 | 570,721 | ||||||||||||||||||||||||||||||||
| Series A Preferred Stock |
4.30 | 75,000 | — | — | 75,000 | — | 75,000 | — | — | — | 75,000 | |||||||||||||||||||||||||||||||||
| Series A Preferred Stock |
5.64 | 223,035 | — | — | 223,035 | — | 223,035 | — | — | — | 223,035 | |||||||||||||||||||||||||||||||||
| Financing |
5.64 | 31,027 | — | — | 31,027 | — | 31,027 | — | (8,865 | ) | — | 22,162 | ||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| 2.94 | 329,062 | — | — | 329,062 | — | 329,062 | — | (8,865 | ) | 570,721 | 890,918 | |||||||||||||||||||||||||||||||||
| Series A Preferred Stock |
1.60 | A | 528,750 | — | (510,437 | ) | 18,313 | — | 18,313 | — | — | — | 18,313 | |||||||||||||||||||||||||||||||
| Financing |
10.00 | A | — | — | — | — | — | — | 150,000 | — | — | 150,000 | ||||||||||||||||||||||||||||||||
| Financing |
17.00A | B | — | — | — | — | — | — | 105,882 | — | — | 105,882 | ||||||||||||||||||||||||||||||||
| Debt Guarantee |
1.60 | A | 310,284 | 1,250,000 | (989,563 | ) | 570,721 | — | 570,721 | — | — | (570,721 | ) | — | ||||||||||||||||||||||||||||||
| Debt Guarantee |
10.00 | A | — | 600,000 | — | 600,000 | 180,000 | 780,000 | — | — | — | 780,000 | ||||||||||||||||||||||||||||||||
| Debt Guarantee |
12.98 | A | — | — | — | — | — | — | 100,000 | — | — | 100,000 | ||||||||||||||||||||||||||||||||
| Series B Preferred Stock |
10.00 | A | — | — | — | — | 131,160 | 131,160 | — | — | — | 131,160 | ||||||||||||||||||||||||||||||||
| Consulting |
5.00 | A | — | — | — | — | 10,075 | 10,075 | — | — | — | 10,075 | ||||||||||||||||||||||||||||||||
| Consulting |
5.64 | A | — | — | — | — | 25,000 | 25,000 | — | — | — | 25,000 | ||||||||||||||||||||||||||||||||
| Consulting |
10.00 | A | — | — | — | — | — | — | 10,500 | — | — | 10,500 | ||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| 10.55 | 839,034 | 1,850,000 | (1,500,000 | ) | 1,189,034 | 346,235 | 1,535,269 | 366,382 | — | (570,721 | ) | 1,330,930 | ||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| $ | 7.50 | 1,168,096 | 1,850,000 | (1,500,000 | ) | 1,518,096 | 346,235 | 1,864,331 | 366,382 | (8,865 | ) | — | 2,221,848 | |||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| A | These warrants are subject to fair value accounting. See Note 14. |
| B | Exercise price at lower of $17.00 per share or a 20% discount to the IPO price if the IPO price is less than $21.25 per share. |
F-49
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Note 14. Fair Value of Warrants
The following tables summarize the assumptions used in computing the fair value of derivative warrants subject to fair value accounting at the date of issue during the years ended December 31, 2011, 2010, and 2009 and at December 31, 2011 and 2010:
| As of December 31, | ||||||||
| 2011 | 2010 | |||||||
| Series A |
||||||||
| Exercise Price |
$ | 1.60 | $ | 1.60 | ||||
| Expected life (years) |
0.83 | 1.83 | ||||||
| Expected volatility |
64.13 | % | 77.33 | % | ||||
| Risk-free interest rate |
0.12 | % | 0.61 | % | ||||
| Expected dividend yield |
0.00 | % | 0.00 | % | ||||
| Issued During the Year Ended December 31, | As of December 31, | |||||||||||||||||||
| 2011 | 2010 | 2009 | 2011 | 2010 | ||||||||||||||||
| Debt Guarantee |
||||||||||||||||||||
| Exercise Price |
$ | 12.98 | $ | 10.00 | $ | 4.32 | $ | 10.34 | $ | 6.46 | ||||||||||
| Expected life (years) |
5.00 | 5.42 | 5.00 | 3.26 | 3.74 | |||||||||||||||
| Expected volatility |
77.35 | % | 79.37 | % | 78.73 | % | 76.53 | % | 81.67 | % | ||||||||||
| Risk-free interest rate |
1.76 | % | 1.60 | % | 2.15 | % | 0.51 | % | 1.13 | % | ||||||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||||
| Issued During the Year Ended December 31, 2010 |
As of December 31, | |||||||||||
| 2011 | 2010 | |||||||||||
| Series B |
||||||||||||
| Exercise Price |
$ | 10.00 | $ | 10.00 | $ | 10.00 | ||||||
| Expected life (years) |
5.00 | 3.92 | 4.92 | |||||||||
| Expected volatility |
80.22 | % | 81.59 | % | 78.77 | % | ||||||
| Risk-free interest rate |
1.51 | % | 0.36 | % | 2.01 | % | ||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | ||||||
| Issued During the Year Ended December 31, |
As of December 31, | |||||||||||||||
| 2011 | 2010 | 2011 | 2010 | |||||||||||||
| Consulting |
||||||||||||||||
| Exercise Price |
$ | 10.00 | $ | 5.46 | $ | 6.50 | $ | 5.46 | ||||||||
| Expected life (years) |
5.00 | 5.00 | 3.48 | 4.25 | ||||||||||||
| Expected volatility |
78.45 | % | 81.12 | % | 81.87 | % | 80.31 | % | ||||||||
| Risk-free interest rate |
2.07 | % | 2.59 | % | 0.47 | % | 2.01 | % | ||||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
F-50
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
| Issued During the Year Ended December 31, 2011 |
As of December 31, 2011 |
|||||||
| Financing |
||||||||
| Exercise Price |
$ | 12.90 | $ | 12.90 | ||||
| Expected life (years) |
5.01 | 4.56 | ||||||
| Expected volatility |
78.45 | % | 79.40 | % | ||||
| Risk-free interest rate |
1.59 | % | 0.83 | % | ||||
| Expected dividend yield |
0.00 | % | 0.00 | % | ||||
The assumed Company stock price used in computing the warrant fair value for warrants issued during the year is as follows: 2011 - $ 4.76 - $13.52; 2010 - $1.92 - $4.76; 2009 - $1.54 - $1.92. In determining the fair value of warrants issued at each reporting date, the assumed company stock price was $13.52 at December 31, 2011 and $4.76 at December 31, 2010.
The following table summarizes the derivative warrant activity subject to fair value accounting for the twelve months ended December 31, 2011, 2010 and 2009:
| Issued with Series A Preferred Stock |
Issued With Series B Preferred Stock |
Issued For Debt Guarantee |
Issued For Consulting |
Issued For Financing |
Total | |||||||||||||||||||
| Beginning balance adjustment due to change in accounting treatment of derivative warrants, January 1, 2009 |
$ | 567,000 | $ | — | $ | 354,000 | $ | — | $ | — | $ | 921,000 | ||||||||||||
| Fair value of warrants issued |
— | — | 2,026,000 | — | — | 2,026,000 | ||||||||||||||||||
| Fair value of warrants exercised |
(788,000 | ) | — | (1,676,000 | ) | — | — | (2,464,000 | ) | |||||||||||||||
| Change in fair value of warrants |
248,000 | — | 705,000 | — | — | 953,000 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Fair value of warrants outstanding as of December 31, 2009 |
27,000 | — | 1,409,000 | — | — | 1,436,000 | ||||||||||||||||||
| Fair value of warrants issued |
— | 328,000 | 415,000 | 65,000 | — | 808,000 | ||||||||||||||||||
| Change in fair value of warrants |
36,000 | (14,000 | ) | 1,969,000 | 35,000 | — | 2,026,000 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Fair value of warrants outstanding as of December 31, 2010 |
63,000 | 314,000 | 3,793,000 | 100,000 | — | 4,270,000 | ||||||||||||||||||
| Fair value of warrants issued |
— | — | 831,000 | 69,000 | 1,970,000 | 2,870,000 | ||||||||||||||||||
| Warrants amended |
— | — | (6,415,000 | ) | — | — | (6,415,000 | ) | ||||||||||||||||
| Change in fair value of warrants |
157,000 | 846,000 | 8,784,000 | 278,000 | 323,000 | 10,388,000 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Fair value of warrants outstanding as of December 31, 2011 |
$ | 220,000 | $ | 1,160,000 | $ | 6,993,000 | $ | 447,000 | $ | 2,293,000 | $ | 11,113,000 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Note 15. Fair Value Measurements
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. The Fair Value Measurements and Disclosures Topic of the FASB Accounting Standards Codification requires the use of valuation techniques that are consistent with the market approach, the income approach and/or the cost approach. Inputs to valuation techniques refer to the assumptions that market participants would use in pricing the asset or liability. Inputs may be observable, meaning those that reflect the assumptions market participants would use in pricing the asset or liability
F-51
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
developed based on market data obtained from independent sources, or unobservable, meaning those that reflect our own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. In that regard, the Topic establishes a fair value hierarchy for valuation inputs that give the highest priority to quoted prices in active markets for identical assets or liabilities and the lowest priority to unobservable inputs.
The fair value hierarchy is as follows:
Level 1: Quoted prices (unadjusted) for identical assets or liabilities in active markets that we have the ability to access as of the measurement date.
Level 2: Significant other observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data.
Level 3: Significant unobservable inputs that reflect our own assumptions about the assumptions that market participants would use in pricing an asset or liability.
The following table summarizes the financial liabilities measured at fair value on a recurring basis segregated by the level of valuation inputs within the fair value hierarchy utilized to measure fair value:
| 2011 | ||||||||||||||||
| Total | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Warrant liability |
$ | 11,113,000 | — | — | $ | 11,113,000 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| 2010 | ||||||||||||||||
| Total | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant
Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Warrant liability |
$ | 4,270,000 | — | — | $ | 4,270,000 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The warrant liability consists of stock warrants we issued that contain an exercise price adjustment feature. In accordance with derivative accounting for warrants, we calculated the fair value of warrants and the assumptions used are described in Note 14, “Fair Value of Warrants”. Realized and unrealized gains and losses related to the change in fair value of the warrant liability are included in Other income (expense) on the Statement of Operations.
F-52
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
The following table reflects the activity for liabilities measured at fair value using Level 3 inputs for the twelve months ended December 31:
| 2011 | 2010 | |||||||
| Balance as of January 1 |
$ | 4,270,000 | $ | 1,436,000 | ||||
| Derivative financial instruments reclassified to equityupon amendment |
(6,415,000 | ) | — | |||||
| Issuances of derivative financial instruments |
2,870,000 | 808,000 | ||||||
| Unrealized loss related to change in fair value |
10,388,000 | 2,026,000 | ||||||
|
|
|
|
|
|||||
| Balance as of December 31 |
$ | 11,113,000 | $ | 4,270,000 | ||||
|
|
|
|
|
|||||
Note 16. Contingencies
In the normal course of business, the Company is involved in various claims and legal proceedings. In the opinion of management, the ultimate liability or disposition thereof is not expected to have a material adverse effect on our financial condition, results of operations or liquidity.
From 2000 to 2004, we operated a clinical laboratory in Milford, Massachusetts providing cancer screening services. The clinical laboratory participated in the Medicare program. The Office of the Inspector General of the U.S. Department of Health and Human Services and the United States Department of Justice informed us in February 2009 that they were contemplating commencing a civil False Claims Act action against us with respect to certain alleged improper billing practices and overpayments relating to operations at the Milford, Massachusetts clinical laboratory. In January 2012, we executed a settlement agreement with the United States Department of Justice. Pursuant to the settlement agreement, we neither admitted liability nor conceded that the claims of the United States are well founded. In January 2012, we paid to the United States the sum of $1,000,000. At December 31, 2011 and 2010, we accrued approximately $1,000,000 and $800,000 in connection with the investigation. We received $400,000 in December 2011 from our insurance carrier related to this matter. The net amount shown in our statement of operations is a net recovery of $200,000 in 2011 and an expense of $800,000 in 2010.
Note 17. Joint Venture Agreement
On November 7, 2011, we entered into an agreement with the Mayo Foundation for Medical Education and Research (“Mayo”) pursuant to which we agreed to form a joint venture with Mayo in August 2012. The joint venture will take the form of a limited liability company, with each party initially holding fifty percent of the issued and outstanding membership interests of the new entity (the “JV”). In exchange for the membership interests in the JV, we will make a capital contribution of $6 million, to be paid over a three year period. In exchange for its membership interests, Mayo’s capital contribution will take the form of cash, staff, services, hardware and software resources, laboratory space and instrumentation, the fair market value of which will be approximately equal to $6 million. Mayo’s continued contribution will also be conditioned upon the JV’s achievement of certain milestones. In addition, on November 14, 2011, without any additional consideration, we granted Mayo 50,000 shares of our common stock, 33,000 of which shall initially be subject to certain forfeiture restrictions if the joint venture does not meet certain milestones. We applied accounting for share-based payments to non-employees and determined that a performance commitment for the 33,000 shares subject to restriction was not present at the date of grant and therefore the remaining shares will be valued and expensed as the restrictions lapse. We recorded expense of $150,000 during 2011 related to the 17,000 shares that were fully vested at the date of grant.
F-53
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
Note 18. Related Party Transactions
John Pappajohn, a member of the Board of Directors and shareholder, personally guarantees our revolving line of credit with Wells Fargo Bank. As consideration for his guarantee, as well as each of the six extensions of this facility, Mr. Pappajohn received warrants to purchase an aggregate of 2,440,284 shares of common stock. Mr. Pappajohn extended his guarantee on this line of credit in March 2012 and received warrants to purchase 92,500 shares of common stock in consideration. See Note 8. Mr. Pappajohn also guaranteed our $1,000,000 revolving line of credit with West Bank which was repaid August 17, 2010.
On May 19, 2006, we issued a convertible promissory note in favor of our Chairman and founder, Dr. Chaganti, the holder, which obligates us to pay the holder the sum of $100,000, together with interest at the rate of 8.5% per annum, on demand of the holder. Interest expense totaled $8,400, $8,416 and $8,523 for the years ended December 31, 2011, 2010 and 2009, respectively. As of December 31, 2011 and 2010, we owe the holder $36,402 in accrued interest. On September 7, 2011, the promissory note was amended to extend its due date to December 31, 2012 and to eliminate any equity conversion feature.
On December 27, 2006, we issued a convertible promissory note in favor of our former Chief Executive Officer, the holder, which obligated us to pay the holder the sum of $40,000, together with interest at the rate of 8.5% per annum, on demand of the holder. Pursuant to the terms of the separation agreement between us, we issued a lump sum payment of $120,000 to the holder upon termination of his employment and the holder released us from any claims, including any claim for repayment of this note. Interest expense totaled $0, $1,407, and $3,400 for the years ended December 31, 2011, 2010 and 2009, respectively, and accrued interest totaled $0 as of December 31, 2011 and 2010, respectively.
On June 19, 2009, we entered into agreements with TSG Partners, LLC (“TSG”) pursuant to which TSG agreed to provide us with strategic consulting services. Our current Chief Executive Officer, Panna Sharma, was the managing partner, founder and majority owner of TSG and was actively involved in the consulting services provided pursuant to such consulting agreement. We compensated TSG an aggregate of $130,750 (excluding expenses) and warrants to purchase 10,075 shares of common stock under such consulting agreement in 2009, $97,575 (excluding expenses) in 2010, and $0 in 2011. On September 23, 2010, we entered into a three-month consulting agreement with TSG for a fixed fee of $60,000 ($45,000 of which was payable in cash and $15,000 was payable in warrants) plus up to $5,000 of expenses. As of December 31, 2011 and 2010, we had no amount due to TSG. The three-month consulting agreement between us and TSG was reduced in scope and a revised total payment of $22,500 (excluding expenses) was paid in cash and no warrants were issued. While Mr. Sharma retains a majority ownership position with TSG, pursuant to certain agreements between Mr. Sharma and TSG, Mr. Sharma did not and will not profit from the services provided under such agreement.
On January 10, 2010, we issued a convertible promissory note in favor of our Senior Scientific Officer, Dr. Houldsworth, the holder, which obligates us to pay the holder the sum of $55,000, together with interest at the rate of 5.5% per annum, on or before January 10, 2011. The $55,000 represents fees owed to Dr. Houldsworth pursuant to our consulting agreement with Dr. Houldsworth. We repaid the $55,000, plus interest in the amount of $970 in 2010.
On July 1, 2010, we entered into a one-year consulting agreement with Edmund Cannon, a member of our board of directors, pursuant to which Mr. Cannon receives $2,000 per calendar quarter for providing consulting services to us in connection with our clinical lab business. Pursuant to the same consulting agreement, Mr. Cannon also receives $100 per hour for each hour Mr. Cannon spends consulting on the development of new business opportunities for us. We can terminate this agreement for cause, as can Mr. Cannon, and expect the
F-54
Table of Contents
CANCER GENETICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
agreement to terminate upon consummation of an initial public offering. Total expense for 2011 and 2010 under the consulting agreement was $4,000 for each year.
On August 15, 2010, we entered into a two-year consulting agreement with Dr. Pecora, a member of our board of directors, pursuant to which Dr. Pecora receives $5,000 per month for providing consulting and advisory services. Dr. Pecora also received stock options under the consulting and advisory agreement to purchase a total of 50,000 shares of common stock at price of $10.00 per share which vests over a two year period. Total expense for 2010 under the consulting agreement was $20,000 paid in cash and $27,200 expensed under the stock option plan. Total expense for 2011 under the consulting agreement was $45,000 paid in cash and $235,560 expensed under the stock option plan. The cash component of this agreement was terminated by mutual consent in August 2011.
In August 2010, we entered into a consulting agreement with Equity Dynamics, Inc., an entity controlled by John Pappajohn, pursuant to which Equity Dynamics, Inc. receives a monthly fee of $10,000 plus reimbursement of expenses. Total expense for 2010 and 2011 under the consulting agreement was $50,000 and $120,000, respectively. As of December 31, 2010 and 2011, we owed Equity Dynamics, Inc. $30,000 and $0, respectively.
On September 15, 2010, we entered into a three-year consulting agreement with Dr. Chaganti, our Chairman, pursuant to which Dr. Chaganti receives $5,000 per month for providing consulting and technical support services. In addition, Dr. Chaganti received a non-qualified stock option to purchase 150,000 shares of common stock at a purchase price of $10.00 per share vesting quarterly beginning October 1, 2010. Also pursuant to the consulting agreement, Dr. Chaganti assigned to us all rights to any inventions which he may invent during the course of rendering consulting services to us. In exchange for this assignment, if the USPTO issues a patent for an invention on which Dr. Chaganti is listed as an inventor, we are required to pay Dr. Chaganti (i) a one-time payment of $50,000 and (ii) 1% of any net revenues we receive from any licensed sales of the invention.
We issued 10,000 warrants to Mr. Thompson, a member of our Board of Directors in connection with the March 2011 line of credit agreement with DAM. These warrants were recorded as consulting expense at an initial estimated fair value of $69,000 and expire in March 2016.
In December 2011, we granted 105,882 warrants to John Pappajohn, a member of our Board of Directors and stockholder in connection with a secured term loan of $3 million (see Note 6). These warrants were recorded at an initial estimated fair value of $951,000 and expire in December 2016. We have also recorded $161,000 of accrued expenses for contingently issuable warrants related to the secured term loan.
In February 2012, we entered into a new Credit Agreement with related warrants with Mr. Pappajohn, Dr. Pecora and entities affiliated with Dr. Pecora. See Note 6.
F-55
Table of Contents
Shares
Common Stock
PROSPECTUS
Aegis Capital Corp
Feltl and Company
, 2013
Until , 2013, all dealers that buy, sell or trade our common stock may be required to deliver a prospectus, regardless of whether they are participating in this offering. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the costs and expenses, other than underwriting discounts and commissions, payable in connection with the sale and distribution of the securities being registered. All amounts are estimated except the SEC registration fee, the FINRA filing fee and the NASDAQ Capital Market listing fee.
| Item |
Amount | |||
| SEC registration fee |
$ | 5,272 | ||
| FINRA filing fee |
6,710 | |||
| NASDAQ Capital Market listing fee |
75,000 | |||
| Legal fees and expenses |
1,950,000 | |||
| Accounting fees and expenses |
510,000 | |||
| Printing and engraving expenses |
240,000 | |||
| Transfer agent and registrar fees and expenses |
15,000 | |||
| Blue Sky fees and expenses |
5,000 | |||
| Non-accountable expense allowance |
350,000 | |||
| Miscellaneous fees and expenses |
118,018 | |||
|
|
|
|||
| Total* |
$ | 3,275,000 | ||
|
|
|
|||
* Approximately $1,250,000 of these expenses have already been paid by the Company.
Item 14. Indemnification of Directors and Officers
Section 145 of the Delaware General Corporation Law authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities, including reimbursement for expenses incurred, arising under the Securities Act of 1933, as amended, or the Securities Act.
Our amended and restated certificate of incorporation to be in effect upon the completion of this offering provides for indemnification of our directors and executive officers to the maximum extent permitted by the Delaware General Corporation Law, and our amended and restated bylaws to be in effect upon the completion of this offering provide for indemnification of our directors and executive officers to the maximum extent permitted by the Delaware General Corporation Law.
In addition, we have entered into indemnification agreements with each of our current directors and executive officers. These agreements will require us to indemnify these individuals to the fullest extent permitted under Delaware law against liabilities that may arise by reason of their service to us and to advance expenses incurred as a result of any proceeding against them as to which they could be indemnified. We also intend to enter into indemnification agreements with our future directors and executive officers.
In any underwriting agreement we enter into in connection with the sale of common stock being registered hereby, the underwriters will agree to indemnify, under certain conditions, us, our directors, our officers and persons who control us, within the meaning of the Securities Act, against certain liabilities.
II-1
Table of Contents
Item 15. Recent Sales of Unregistered Securities.
Since January 1, 2009, the Registrant made sales of the following unregistered securities:
Sales of Preferred Stock and Related Common Stock and Warrant Issuances
In seven closings from April 2010 to November 2010, the Registrant sold an aggregate of 1,821,600 shares of its Series B preferred stock to approximately 88 accredited investors at a purchase price of $5.00 per share, for aggregate gross proceeds of approximately $9.1 million. In addition, the Registrant issued 5,000 shares of its common stock and warrants to purchase an aggregate of 131,160 shares of its common stock at an exercise price of $10.00 per share to approximately 32 service providers and placement agents in connection with its Series B preferred stock offering. These warrants were issued on its medium form warrant, which is described in the section entitled “Description of Capital Stock–Warrants”.
Common Stock Issuances
On November 14, 2011, the Registrant issued 50,000 shares of common stock, subject to certain forfeiture restrictions, to the Mayo Foundation for Medical Education and Research in connection with the execution of the affiliation agreement between the Registrant and Mayo.
Convertible Promissory Notes and Related Warrant Issuances
Pursuant to the terms of the Credit Agreement dated as of December 21, 2011, the Registrant issued to Mr. Pappajohn, a member of the board of directors, a convertible promissory note in the amount of $4.0 million and to NNJCA Capital, a limited liability company of which one of our directors, Dr. Pecora is a member, a promissory note in the amount of $1.5 million and to Pecora and Company, a promissory note in the amount of $500,000. At Mr. Pappajohn’s option, his $4.0 million loan is convertible into shares of common stock at a conversion price equal to the lower of $17.00 per share or a 20% discount to the initial public offering price per share. In May 2012, Mr. Pappajohn agreed to convert $2.0 million of the outstanding principal due to him to common stock at the initial public offering price per share upon consummation of this offering, to eliminate the conversion feature of the remaining $2.0 million in debt effective upon consummation of this offering and to extend the maturity date to August 15, 2013 for the $2.0 million principal amount which was not converted. In connection with the issuance of the notes, the Registrant issued five year warrants to purchase shares of our common stock at the lower of $17.00 per share and a 20% discount to the initial public offering price per share if the initial public offering price is less than $21.25 per share as follows: 141,176 warrants to Mr. Pappajohn, 52,901 warrants to NNJCA Capital and 17,647 warrants to Pecora and Company. The lenders received an aggregate of 70,588 additional warrants on the same terms, with the warrants being issued proportionately to Mr. Pappajohn, Dr. Pecora and NNJCA, because we did not consummate our initial public offering within 181 days of funding. Subsequently, the warrants to purchase an aggregate of 94,117 shares of our common stock issued to Dr. Pecora and NNJCA in connection with this financing were cancelled and the promissory notes issued to Dr. Pecora and NNJCA were amended to increase the pre-payment penalties.
Subsequently, the warrants issued to Mr. Pappajohn were amended to provide that the exercise price shall equal the lesser of $17.00 per share or our initial public offering price per share and to extend the $2.0 million dollars in non-convertible debt, which is held by his spouse, to April 1, 2014. In connection with this agreement, we issued to Mr. Pappajohn additional warrants to purchase an aggregate of 75,202 shares of our common stock on the Modified Bridge Form. The warrant exercise price on the Modified Bridge Form is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share price is less than
II-2
Table of Contents
$17.00 per share, so that the initial grant would be for an aggregate of 182,633 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 213,072 shares and 159,804 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
2012 Convertible Debt Financing Transaction
We entered into a restated Credit Agreement dated as of October 19, 2012 with John Pappajohn and Mark Oman for a $3.0 million convertible term loan. Mr. Pappajohn provided approximately $1.8 million of financing and Mr. Oman provided approximately $1.3 million of financing under the Credit Agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on February 26, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
The lenders received ten-year warrants to purchase an aggregate of 286,275 shares of our common stock (which were issued in proportion to their respective funding amounts), in each case at the lower of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering price per share is less than $17.00 per share. On November 27, 2012, the lenders received additional warrants to purchase an aggregate of 17,648 shares of our common stock (issued in proportion to their funding amount) on the same terms because we did not consummate our initial public offering prior to November 27, 2012. The lenders will receive additional ten year warrants to purchase an aggregate of 17,648 shares of our common stock (to be issued in proportion to their respective funding amounts) on the same terms in the event we do not consummate our initial public offering by February 22, 2013. The warrant exercise price is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share price is less than $17.00 per share, so that the initial grant would will be for an aggregate of 695,237 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 811,110 shares and 608,333 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
On December 7, 2012, Mr. Pappajohn provided an additional $1.0 million of financing on the same terms as the restated credit agreement. The loan bears an annual interest rate equal to the prime rate plus 6.25% (9.50% at September 30, 2012) and matures on June 4, 2014. The outstanding principal amount of the loan automatically converts to common stock upon completion of our initial public offering. The conversion price is equal to the lesser of $17.00 per share or our initial public offering price per share. The conversion price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like. We may prepay the loan in whole or in part only with the prior consent of the lenders.
Mr. Pappajohn received ten-year warrants to purchase an aggregate of 58,824 shares of our common at the lesser of (i) $17.00 per share and (ii) the initial public offering price per share if the initial public offering price per share is less than $17.00 per share. He will receive additional ten-year warrants to purchase an aggregate of 5,886 shares of our common stock on the same terms in the event we do not consummate our initial public offering by each the following dates: March 4, 2013 and June 4, 2013. The warrant exercise price is subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the initial public offering price per share is less than $17.00 per share, so that the initial grant would be for an aggregate of 142,857 shares if the initial public offering price per share is $7.00, the midpoint of the range on the cover of this prospectus, and 166,667 shares and 125,000 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
II-3
Table of Contents
Shares that the lenders receive are subject to a lock-up agreement for 180 days after the consummation of our IPO on the same terms as other lock-up agreements in favor of the underwriters of this offering, but otherwise have registration rights pursuant to a registration rights agreement entered into simultaneously with the Credit Agreement.
The offers, sales and issuances of the securities described above under “Sales of Preferred Stock and Related Common Stock and Warrant Issuances”, “Common Stock Issuances” and “Convertible Promissory Note and Related Warrant Issuances” were exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act or Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The purchasers or recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof. The purchasers of securities or recipients in each of these transactions represented to the Registrant in connection with their purchase that they were accredited investors. The sales of these securities were made without any general solicitation or advertising. Appropriate legends were affixed to securities issued in these transactions.
Option and Common Stock Issuances
Since January 1, 2009, the Registrant granted to a total of approximately 63 employees, officers, directors, consultants and other service providers options to purchase an aggregate of 1,265,687 shares of common stock, at exercise prices ranging from $1.60 to $13.52 per share, under its 2008 Stock Option Plan. In addition the Registrant granted to two directors options to purchase 200,000 shares of common stock at an exercise price of $10.00 per share pursuant to certain consulting agreements.
Since January 1, 2009, the Registrant issued and sold to a former officer an aggregate of 113,500 shares of common stock upon the exercise of options under the 2008 Stock Option Plan at an exercise price of $1.60 per share, for an aggregate exercise price of $181,000, $1,600 paid in cash and the remainder paid in stock valued at $10.00 per share.
The offers, sales and issuances of the securities described above were exempt from registration under the Securities Act pursuant to Rule 701 as offers and sales of securities pursuant to certain compensatory benefit plans or contracts relating to compensation or Regulation D promulgated under Section 4(2) of the Securities Act. The recipients of such securities were employees, directors, consultants or other service providers of the Registrant. The securities were issued under our 2008 Stock Option Plan or pursuant to individual consulting agreements. Appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions represented their intention to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof. The sales of these securities were made without any general solicitation or advertising. Each of the recipients of securities in these transactions had adequate access, through their employment or business relationships, to information about the Registrant.
Warrant and Common Stock Issuances
Since January 1, 2009, as consideration for his personal guarantee of the Registrant’s credit facility with Wells Fargo and for each of the eight extensions of this facility since January 1, 2009, the Registrant issued to Mr. Pappajohn, a member of our board, warrants to purchase an aggregate of 2,532,783 shares of its common stock, of which 2,440,283 warrants were issued on its long form warrant, 92,500 were issued on its Bridge Financing Warrant Form, and warrants to purchase an aggregate of 95,981 shares of common stock on the Modified Bridge Form, each of which is described in the section entitled “Description of Capital Stock–Warrants”, and have exercise prices ranging from $1.60 to $17.00 per share. The warrant exercise price is subject to subject to standard anti-dilution protection in the event of stock splits, stock dividends, stock combinations, reclassifications and the like and to full ratchet anti-dilution protection in the event of issuances of more than $5 million of securities at prices below the exercise price prior to the completion of our initial public offering. The warrants are also subject to anti-dilution adjustment if the IPO price is less than $17.00 per share, so that the initial grant would be for an aggregate of 233,096 shares if the initial public offering price per share is $7.00, the
II-4
Table of Contents
midpoint of the range on the cover of this prospectus, and 271,945 shares and 203,959 shares in the event the initial public offering price per share is $6.00 or $8.00, respectively.
On March 23, 2011, the Registrant issued to DAM warrants to purchase 150,000 shares of its common stock at an exercise price of $10.00 per share in conjunction with the $3.0 million line of credit DAM provided to the Registrant. On March 9, 2012, in connection with the extension and amendment of this line of credit, the Registrant issued to DAM warrants to purchase 37,500 shares of its common stock, which warrants were subsequently cancelled in connection with an amendment to the note to increase interest payable thereunder. These warrants were issued on its medium form warrant, which is described in the section entitled “Description of Capital Stock–Warrants”.
Since January 1, 2009, the Registrant issued to one director and three new consultants, warrants to purchase an aggregate of 45,075 shares of common stock at exercise prices ranging from $5.00 to $10.00 per share on its long form warrant and 500 shares of common stock at an exercise price of $10.00 per share on its medium form warrant. The medium form warrant and long form warrant are described in the section entitled “Description of Capital Stock–Warrants.”
Since January 1, 2009, the Registrant issued and sold to Mr. Pappajohn, a member of our board, an aggregate of 1,500,000 shares of common stock upon the exercise of certain outstanding long form warrants at an exercise price of $1.60 per share, for an aggregate exercise price of $2,400,000 which was paid in stock at a value of $10.00 per share and 8,865 shares of common stock upon the exercise of certain outstanding short form warrants at an exercise price of $5.64 per share for an aggregate exercise price of $50,000.
Since January 1, 2009, the Registrant issued and sold to approximately five of the service providers and placement agents from its Series A preferred stock offering, an aggregate of 75,000 shares of common stock upon the exercise of certain outstanding short form warrants at an exercise price of $4.30 per share and an aggregate of 46,539 shares of common stock upon the exercise of certain outstanding short form warrants at an exercise price of $5.64 per share.
Since January 1, 2009, the Registrant issued and sold to an individual who financed a bridge loan to the Registrant in 2007 an aggregate of 6,205 shares of common stock upon the partial exercise of a Short Form Warrant at an exercise price of $5.64 per share, for an aggregate exercise price of approximately $35,000.
Since January 1, 2009, the Registrant issued and sold to five of its Series A preferred stock holders an aggregate of 9,532 shares of common stock upon the full exercise of Long Form Warrants at an exercise price of $1.60 per share, for an aggregate exercise price of approximately $15,251.
The offers, sales and issuances of the securities described above were exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act or Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof. Appropriate legends were affixed to securities issued in these transactions. Each of the recipients of securities in these transactions had adequate access, through their business relationships, to information about the Registrant.
II-5
Table of Contents
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits
EXHIBITS
| Exhibit |
Description of Exhibit | |
| 1.1# | Form of Underwriting Agreement between Cancer Genetics, Inc. and Aegis Capital Corp., as representative of the several underwriters. | |
| 3.1# | Amended and Restated Certificate of Incorporation of Cancer Genetics, Inc., as currently in effect. | |
| 3.2 | Form of Third Amended and Restated Certificate of Incorporation of Cancer Genetics, Inc., to be in effect upon the closing of this offering. | |
| 3.3# | Bylaws, as currently in effect. | |
| 3.4# | Form of Amended and Restated Bylaws of Cancer Genetics, Inc., to be in effect upon the closing of this offering. | |
| 3.5# | Certificate of Amendment of Amended and Restated Certificate of Incorporation of Cancer Genetics, Inc. | |
| 4.1# | Specimen Common Stock certificate of Cancer Genetics, Inc. | |
| 4.2# | Registration Rights Agreement, between Cancer Genetics, Inc. and DAM Holdings, LLC, dated March 23, 2011. | |
| 4.3# | Form of Amended and Restated Investors’ Rights Agreement, dated as of April 13, 2010, between Cancer Genetics, Inc. and certain investors named therein. | |
| 4.4# | Form of Amendment to Amended and Restated Investors’ Rights Agreement, dated as of December 8, 2011. | |
| 4.5# | Form of Amended and Restated Stockholders’ Agreement, dated April 13, 2010, between Cancer Genetics, Inc. and certain investors named therein. | |
| 4.6# | Form of Series B Convertible Preferred Stock Purchase Agreement, between Cancer Genetics, Inc. and certain purchasers named therein. | |
| 4.7# | Form of Amendment to Series B Convertible Preferred Stock Purchase Agreement, dated December 8, 2011. | |
| 4.8# | Form of Short Form Warrant. | |
| 4.9# | Form of Short Form Cashless Exercise Warrant. | |
| 4.10# | Form of Medium Form Warrant. | |
| 4.11# | Form of Long Form Warrant. | |
| 4.12# | Convertible Promissory Note, dated May 19, 2006, between Cancer Genetics, Inc. and R.S.K. Chaganti, as amended. | |
| 4.13# | Convertible Promissory Note, dated January 10, 2010, between Cancer Genetics, Inc. and Jane Houldsworth. | |
| 5.1# | Opinion of Lowenstein Sandler LLP. | |
II-6
Table of Contents
| Exhibit |
Description of Exhibit | |
| 10.1# | Amended and Restated 2008 Stock Option Plan. | |
| 10.2# | Form of Notice of Stock Option Grant under 2008 Stock Option Plan. | |
| 10.3# | Form of Stock Option Grant Agreement under 2008 Stock Option Plan. | |
| 10.4# | Form of Exercise Notice and Restricted Stock Purchase Agreement under 2008 Stock Option Plan. | |
| 10.5# | Amended and Restated 2011 Equity Compensation Plan. | |
| 10.6# | Form of Stock Option Grant Agreement under 2011 Stock Option Plan. | |
| 10.7# | Form of Indemnification Agreement. | |
| 10.8# | Consulting Agreement between Cancer Genetics, Inc. and TSG, LLC, dated June 19, 2009. | |
| 10.9# | Medical Director Agreement, between Cancer Genetics, Inc. and Lan Wang, M.D., dated October 9, 2009. | |
| 10.10# | Employment Agreement, between Cancer Genetics, Inc. and Louis Maione, dated October 21, 2009. | |
| 10.11# | Consulting Agreement, between Cancer Genetics, Inc. and Louis Maione, dated June 10, 2010. | |
| 10.12# | Termination Agreement, between Cancer Genetics, Inc. and Louis Maione, dated June 10, 2010. | |
| 10.13# | Consulting Agreement, between Cancer Genetics, Inc. and Edmund Cannon, dated July 1, 2010. | |
| 10.14# | Consulting Agreement, between Cancer Genetics, Inc. and Andrew Pecora, dated August 15, 2010. | |
| 10.15# | Consulting Agreement, between Cancer Genetics, Inc. and R.S.K. Chaganti, dated September 15, 2010. | |
| 10.16# | Consulting Agreement, between Cancer Genetics, Inc. and TSG, LLC, dated September 23, 2010. | |
| 10.17# | Employment Agreement, between Panna Sharma and Cancer Genetics, Inc., effective as of April 1, 2010. | |
| 10.18# | Employment Agreement, between Elizabeth Czerepak and Cancer Genetics, Inc., effective as of January 1, 2012. | |
| 10.19# | Employment Agreement, between Jane Houldsworth El Naggar, Ph.D. and Cancer Genetics, Inc., effective as of January 1, 2012. | |
| 10.20# | Office Lease Agreement, between Cancer Genetics, Inc. and Onyx Equities, LLC, dated October 9, 2007. | |
| 10.21# | Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated April 29, 2008. | |
| 10.22# | Security Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated April 29, 2008. | |
| 10.23# | First Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated July 7, 2008. | |
| 10.24# | Second Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated March 30, 2009. | |
| 10.25# | Third Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated July 2, 2009. | |
II-7
Table of Contents
| Exhibit |
Description of Exhibit | |
| 10.26# | Fourth Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated October 21, 2009. | |
| 10.27# | Fifth Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated July 29, 2010. | |
| 10.28# | Credit Agreement, between Cancer Genetics, Inc. and DAM Holdings, LLC, dated March 23, 2011. | |
| 10.29# | Inter-creditor Agreement, between Cancer Genetics, Inc., John Pappajohn and DAM Holdings, LLC, dated March 23, 2011. | |
| 10.30# | General Business Security Agreement, between Cancer Genetics, Inc. and DAM Holdings, LLC, dated March 23, 2011. | |
| 10.31# | Promissory Note, issued by Cancer Genetics, Inc. to DAM Holdings, LLC, dated March 23, 2011. | |
| 10.32# | Sixth Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated June 6, 2011. | |
| 10.33# | Amended and Restated Credit Agreement, by and among Cancer Genetics, Inc., John Pappajohn, Pecora and Company and NNJCA Capital, LLC dated February 13, 2012. | |
| 10.34# | Form of Promissory Note issued by Cancer Genetics, Inc. to John Pappajohn. | |
| 10.35# | Form of Promissory Note issued by Cancer Genetics, Inc. to NNJCA Capital, LLC and Pecora and Company. | |
| 10.36# | Form of Bridge Financing Warrant issued by Cancer Genetics, Inc. to John Pappajohn, NNJCA Capital, LLC, Pecora and Company and DAM Holdings, LLC. | |
| 10.37# | Inter-Creditor Agreement, between Cancer Genetics, Inc., John Pappajohn, DAM Holdings, LLC, Pecora and Company, NNJCA Capital, LLC and Equity Dynamics, Inc., dated February 13, 2012. | |
| 10.38# | Seventh Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated February 15, 2012. | |
| 10.39# | Amendment to Credit Agreement, between Cancer Genetics, Inc. and DAM Holdings, LLC, dated March 9, 2012. | |
| 10.40# | Affiliation Agreement, between Cancer Genetics, Inc. and Mayo Foundation for Medical Education and Research dated November 7, 2011. | |
| 10.41# | Consulting Agreement with Equity Dynamics, Inc. | |
| 10.42# | Funding Commitment Letter, from John Pappajohn, Equity Dynamics Inc. to the Company, dated April 14, 2012. | |
| 10.43# | Letter Agreement, between Panna Sharma and Cancer Genetics, Inc., dated March 29, 2012. | |
| 10.44# | Letter Agreement, between Meadows Office, L.L.C. and Cancer Genetics, Inc., dated January 10, 2008. | |
| 10.45# | Letter Agreement, between Meadows Office, L.L.C. and Cancer Genetics, Inc., dated April 6, 2012. | |
| 10.46# | Letter of Credit from JPMorgan Chase Bank, N.A., dated April 19, 2012. | |
| 10.47# | Letter Agreement between Cancer Genetics, Inc. and John Pappajohn | |
| 10.48# | Confidential Settlement Agreement and Release of All Claims, between and among Louis J. Maione, Esq., Cancer Genetics, Inc., John Pappajohn, Raju Chaganti, Andrew Pecora, Tommy Thompson, Edmund Cannon, Matthew Kinley, Panna Sharma, and GAP Partners, LLP, dated May 2012. | |
II-8
Table of Contents
| Exhibit |
Description of Exhibit | |
| 10.49# | Amendment No. 1 to Affiliation Agreement, between Cancer Genetics, Inc. and Mayo Foundation for Medical Education and Research, dated September 29, 2012. | |
| 10.50# | Form of Modified Bridge Warrant issued by Cancer Genetics, Inc. to John Pappajohn and Mark Oman. | |
| 10.51# | Restated Credit Agreement, between Mark Oman and John Pappajohn and Cancer Genetics, Inc., dated October 17, 2012. | |
| 10.52# | Form of Restated Promissory Note issued by Cancer Genetics, Inc. to John Pappajohn and Mark Oman. | |
| 10.53# | Form of October 2012 Warrant issued by Cancer Genetics, Inc. to John Pappajohn and Mark Oman. | |
| 10.54# | Restated Registration Rights Agreement, between Cancer Genetics, Inc., Mark Oman and John Pappajohn, dated October 17, 2012. | |
| 10.55# | Letter Agreement between Cancer Genetics, Inc. and Pecora. | |
| 10.56# | Letter Agreement between Cancer Genetics, Inc. and NNJCA Capital, LLC. | |
| 10.57# | Letter Agreement between Cancer Genetics, Inc. and DAM Holdings, Inc. | |
| 10.58# | Eighth Addendum to Credit Agreement, between Cancer Genetics, Inc. and Wells Fargo Bank, N.A., dated October 18, 2012. | |
| 10.59# | Credit Agreement between John Pappajohn and Cancer Genetics, Inc. dated December 4, 2012. | |
| 10.60# | Promissory Note issued by Cancer Genetics, Inc. to John Pappajohn dated December 4, 2012. | |
| 10.61 | Amendment No. 2 to Affiliation Agreement between Cancer Genetics, Inc. and Mayo Foundation for Medical Education and Research, dated January 4, 2013. | |
| 10.62 | Written Description of Amendment to Letter Agreement, between Meadows Office, L.L.C. and Cancer Genetics, Inc., dated April 6, 2012. | |
| 21.1# | Subsidiaries of Cancer Genetics, Inc. | |
| 23.1 | Consent of McGladrey LLP. | |
| 23.2# | Consent of Lowenstein Sandler LLP (included in Exhibit 5.1). | |
| 24.1# | Power of Attorney | |
| 24.2# | Power of Attorney | |
| * | To be filed by amendment |
| # | Previously Filed |
(b) Financial Statement Schedules
No financial statement schedules are provided because the information is not required or is shown either in the financial statements or the notes thereto.
Item 17. Undertakings
The undersigned Registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
II-9
Table of Contents
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by us of expenses incurred or paid by one of our directors, officers or controlling persons in the successful defense of any action, suit, or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes that:
(i) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; and
(ii) For purposes of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-10
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant has duly caused this Amendment No. 11 to Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the Rutherford, State of New Jersey, on the 8th day of January, 2013.
| CANCER GENETICS, INC. | ||
| By: | /s/ PANNA L. SHARMA | |
| Panna L. Sharma President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act, this Amendment No. 10 to Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Panna L. Sharma Panna L. Sharma |
President, Chief Executive Officer and Director (Principal Executive Officer) |
January 8, 2013 | ||
| /s/ Elizabeth A. Czerepak Elizabeth A. Czerepak |
Chief Financial Officer |
January 8, 2013 | ||
| * Raju S. K. Chaganti, Ph.D. |
Chairman of the Board of Directors | January 8, 2013 | ||
| * Edmund Cannon |
Director | January 8, 2013 | ||
| * John Pappajohn |
Director | January 8, 2013 | ||
| * Andrew Pecora, M.D. |
Director | January 8, 2013 | ||
| * Tommy G. Thompson |
Director | January 8, 2013 | ||
| * |
Director | January 8, 2013 | ||
| Robert Kaufman | ||||
| * Franklyn G. Prendergast, M.D., Ph.D. |
Director | January 8, 2013 | ||
| * /s/ Elizabeth A. Czerepak |
||||
| as attorney in fact | ||||
II-11