Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Kips Bay Medical, Inc. | Financial_Report.xls |
| EX-10.52 - EX-10.52 - Kips Bay Medical, Inc. | a2208035zex-10_52.htm |
| EX-32.1 - EX-32.1 - Kips Bay Medical, Inc. | a2208035zex-32_1.htm |
| EX-31.1 - EX-31.1 - Kips Bay Medical, Inc. | a2208035zex-31_1.htm |
| EX-31.2 - EX-31.2 - Kips Bay Medical, Inc. | a2208035zex-31_2.htm |
| EX-32.2 - EX-32.2 - Kips Bay Medical, Inc. | a2208035zex-32_2.htm |
Use these links to rapidly review the document
Table of Contents
Item 8. Financial Statements and Supplementary Data
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2011 |
||
OR |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
||
KIPS BAY MEDICAL, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation) |
000-1460198 (Commission File Number) |
20-8947689 (IRS Employer Identification No.) |
| 3405 Annapolis Lane North, Suite 200 | ||
| Minneapolis, Minnesota | 55447 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant's telephone number, including area code: (763) 235-3540
Securities Registered Pursuant to Section 12(b) of the Act: Common Stock, $0.01 par value
Name of Each Exchange on Which Registered: The NASDAQ Stock Market LLC
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best or registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer o (Do not check if smaller reporting company) |
Smaller reporting company ý |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant's most recently completed second fiscal quarter: $10,800,000
As of March 9, 2012, there were 16,245,579 shares of the registrant's common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the proxy statement for the registrant's 2012 Annual Meeting of Stockholders are incorporated by reference into Items 10, 11, 12, 13 and 14 of Part III of this report.
1
Overview
We are a medical device company focused on developing, manufacturing and commercializing our proprietary external saphenous vein support technology, or eSVS MESH, for use in coronary artery bypass grafting ("CABG") surgery. Our eSVS MESH is a knitted, nitinol mesh sleeve that, when placed over a saphenous vein graft during CABG surgery, is designed to improve the structural characteristics and long-term performance of the vein graft. CABG is one of the most commonly performed surgeries in the United States, with the American Heart Association estimating that 416,000 CABG procedures were performed in the United States in 2009, the most recent year for which the American Heart Association has published such estimates. In a typical CABG procedure, surgeons harvest blood vessels, or conduits, including the left internal mammary artery from the chest and the saphenous vein from the leg, and attach the harvested vessels to bypass, or provide blood flow around, blocked coronary arteries. The effectiveness of the procedure, however, is often limited by the failure rate of saphenous vein grafts, which has been shown in various studies to range from 7% to 26% one year after surgery and 39% ten years after surgery. Failure of these grafts, typically evidenced by partial or complete blockage and reduced or stopped blood flow, can lead to the need for further coronary interventions up to and including performing additional, or re-do CABG procedures which are generally associated with mortality rates three to five times higher than the original CABG procedure. We believe the use of our eSVS MESH with saphenous vein grafts in CABG surgery can improve the long-term outcome of CABG procedures, including improved openness, or patency, and improved blood flow through the saphenous vein graft, resulting in a reduced need for costly and potentially complicated reoperations or revascularization procedures.
Physicians and patients may select among a variety of treatments to address coronary artery disease, including pharmaceutical therapy, balloon angioplasty, stenting with bare metal or drug-eluting stents, and CABG procedures, with the selection often depending upon the health of the individual patient, the stage of the disease and the presence of comorbidities such as valvular disease or heart failure, etc. The SYNTAX study, comparing CABG and implantation of drug-eluting stents, found that CABG is the more effective long-term treatment for coronary artery disease ("CAD") in patients with CAD in the left main coronary artery or in three or more coronary arteries, achieving the best long-term patient outcomes as measured by the composite rate of survival, myocardial infarction (heart attack), stroke or the need for re-intervention 12 to 36 months after surgery. Moreover, patients with severe and multi-vessel coronary artery disease often cannot be effectively treated with methods other than CABG. The prevalence of coronary artery disease and the success rates for CABG procedures versus other treatments for coronary artery disease has made CABG surgery one of the most commonly performed surgeries in the United States. It is generally accepted that the increased incidence and prevalence of diabetes, obesity, combined with aging populations and other environmental factors will mean that the addressable pool of patients eligible for CABG surgery will continue to endure into the future.
According to published reports, a typical CABG procedure involves an average of 3.3 bypass grafts. In CABG procedures the left internal mammary artery ("LIMA") is generally used for the first graft, generally to the left anterior descending artery ("LAD"), and saphenous veins are used for any remaining grafts. Saphenous vein grafts fail more frequently than LIMA grafts due to differences in structure and size of saphenous vein grafts as compared to LIMA grafts. Unlike the LIMA, which is a thick-walled artery intended to handle the high pressure blood flow from the heart, saphenous veins are thin-walled vessels that are intended for a low-pressure venous environment. Saphenous veins are also typically larger than the coronary arteries to which they are attached and this difference in size disrupts blood flow, adding stress to the vessel wall and increasing the risk of thrombosis, or blood clotting.
2
When the vein grafts used to bypass a blocked artery are exposed to the high pressure of arterial flow, there is significant stress on the thin wall of the veins. The vein responds to this injury by causing its inner walls to thicken, decreasing the inner diameter of the graft and often leading to failure of the bypass graft.
Our eSVS MESH is a nitinol mesh sleeve that is placed over the saphenous vein graft during CABG surgery and is designed to constrict the vein and prevent expansion of the vein graft and resulting injury due to increased pressure. The constriction of the vein graft also causes the diameter of the graft, or lumen, to more closely match the diameter of the target coronary artery to which it is attached, thereby reducing blood flow disruption. Our eSVS MESH is designed for quick deployment and is compatible with most current CABG surgery practices, including off-pump CABG, on-pump CABG and other less invasive methodologies. In addition, nitinol is commonly used in many other implantable medical devices.
In order to obtain authorization to apply the CE Mark to our product and begin sales in Europe, we conducted a 90 patient multi-center clinical trial outside the United States. The goals of this trial were to demonstrate that CABG surgery using our eSVS MESH was not inferior as to either safety or effectiveness as compared to traditional CABG surgery. We received our CE Mark in May 2010 based on data from angiographic studies nine to 12 months following surgery of the first 38 patients in the trial to complete such follow-up studies. Analysis of this data showed that the patency of vessels treated with our eSVS MESH was statistically equivalent and therefore non-inferior to the patency of untreated saphenous vein bypass vessels. The final results of the trial, which included angiographic data for 73 patients, differed from the results for the first 38 patients and were inconclusive as to whether the patency of eSVS MESH treated vessels was non-inferior to untreated vessels. Because our CE Mark submission was made pursuant to a protocol accepted by all participating clinical study sites and their respective Competent Authorities (government or government-appointed agencies in charge of approving medical device clinical studies prior to enrollment in such a study), and device and procedure safety have been demonstrated (no increase in adverse events as compared to published literature for CABG surgery), the final results do not impact the status or validity of our CE Mark. We began marketing and commenced shipments of our eSVS MESH in select European Union markets in June 2010, in the United Arab Emirates in October 2010 and in Turkey in January 2011. Our primary markets continue to be in select European countries and the United Arab Emirates.
In the trial, we evaluated the safety of our eSVS MESH by comparing the rate of major adverse cardiac and cerebral events, or MACCE, 30 days following surgery for patients treated with our eSVS MESH against the same rate reported in published literature for patients with traditional CABG surgery. We evaluated efficacy by comparing the patency of vessels treated with eSVS MESH against the patency of untreated saphenous vein bypass vessels as measured by angiographic studies nine to 12 months following implant. The safety data from this trial has indicated that our eSVS MESH and implant procedure do not result in an increase in patient complications during or after surgery. However, the effectiveness data from the trial is inconclusive primarily due to two complicating factors. First, one of the centers participating in the trial used implant methods incompatible with our eSVS MESH. Second, the amount of reduction in the diameter of the saphenous vein grafts, or downsizing, prescribed in our instructions for use and sizing tool was too aggressive, resulting in a higher than anticipated closure rate in saphenous vein grafts utilizing the eSVS MESH, particularly when our smallest device, 3.0 millimeters, was used. In response to this data, we have modified our instructions for use to provide clear direction on the surgical method to be used with our eSVS MESH, discontinued the use of our 3.0 millimeter eSVS MESH and reduced the amount of downsizing specified for other device sizes. We also exclude saphenous veins with walls thicker than 0.7 millimeters. We believe these steps have resolved the patency issues identified in the trial.
The U.S Food and Drug Administration ("FDA"), has reviewed and disapproved our most recent amendment to our application for an investigational device exemption ("IDE") in March 2011. At that
3
time, the FDA indicated that they intended to review our IDE information with outside experts before they provide further guidance to the Company. Due to internal delays, the FDA did not begin this review until August 2011. In September 2011 the FDA advised us that we had not provided sufficient data to support our request for an IDE for our eSVS MESH. Therefore, we intend to conduct a new feasibility trial in Europe based upon a protocol which incorporates additional guidance/requirements provided by the FDA. In November 2011 we began the process of recruiting study sites and expect to commence enrollment in the summer of 2012. Upon completion of this study, we expect to request an IDE for a pivotal study in the U.S. The results from this pivotal study are intended to provide the basis for filing of a pre-market approval application ("PMA"), which must be approved by the FDA prior to marketing the eSVS MESH in the United States. However, we could be delayed by adverse clinical results or regulatory complications, and we may never receive U.S. marketing approval.
In November 2011 we commenced enrollments in our first post-market study intended to support our international reimbursement and marketing activities with a clinical evaluation of the short-term (three to six months) and long-term (two years) post-implant patency of the eSVS MESH in the treatment of Saphenous Vein Grafts ("SVGs") used in Coronary Artery Bypass Grafting ("CABG"), as compared with prospective SVG CABG without the eSVS MESH. We intend to enroll up to 200 patients. Long term safety out to five years will also be assessed.
Our feasibility trial in Europe will be a multi-center, randomized study of external saphenous vein support using our eSVS MESH in CABG Surgery titled the "eMESH I" study. The objective of this study is to demonstrate the initial safety and performance of our eSVS MESH for use as an external SVG support device during coronary artery bypass procedures. The results of this study are intended to be used to obtain an approval from the FDA of an IDE for a pivotal study in the U.S. This feasibility study is also a prospective, randomized study and we will enroll a maximum of 120 patients.
See "Clinical Development of our eSVS MESH" below for additional information.
Our Strategy
Our objective is to achieve significant market adoption of our eSVS MESH technology in CABG and other vascular applications. Key elements of our strategy to achieve this objective include the following:
- •
- Work with respected medical centers and key thought leaders to demonstrate and communicate the potential benefits of our
eSVS MESH. We have commenced a post-market randomized study in Europe and are currently pursuing an additional feasibility
trial. The data from the feasibility study is intended to support our receipt of an approval for an IDE from the FDA for a pivotal study. The IDE approval will allow us to begin studying the eSVS MESH
clinically in the United States. We believe that it will be important to increase the awareness of our eSVS MESH by collaborating with key opinion leaders at leading academic and medical institutions
and supporting post-approval marketing studies and publication of peer-reviewed articles. We believe that we have formed strong relationships with surgeons at a growing number
of University-based and private cardiovascular surgery centers.
- •
- Commercialize our eSVS MESH in select European and other International markets. We received CE Mark approval in May 2010 and began marketing and commenced shipments of our eSVS MESH in select European markets in June 2010, in the United Arab Emirates in October 2010 and in Turkey in January 2011. We have engaged independent distributors for the United Arab Emirates and a number or European countries including: Switzerland, Italy, Spain, Belgium, the Netherlands, Luxembourg, the UK, Ireland, Greece, Denmark, Sweden, Norway, Turkey, Germany and, subsequent to year-end, France. We believe that we have engaged independent distributors experienced in their respective European markets to promote and sell our eSVS MESH. Concurrent with this effort, we have also engaged independent distributors in
4
- •
- Obtain regulatory approval and commercialize our eSVS MESH in the United States. The FDA has reviewed and disapproved our most recent amendment to our application for an IDE. As noted above, we intend to conduct a new feasibility trial in Europe based upon a protocol which incorporates additional guidance/requirements provided by the FDA. In November 2011 we began the process of identifying study sites and expect to commence enrollment in the summer of 2012. Upon completion of the feasibility study, we expect to request an IDE for a pivotal study in the U.S. In the U.S., a pivotal study is required to develop the clinical data to support an application for PMA approval from the FDA. If we receive the necessary regulatory approval, we plan to commercially introduce our eSVS MESH in the United States through independent distributors with access to key CABG centers and key physicians. However, we could be delayed by adverse clinical results or regulatory complications, and we may never receive U.S. marketing approval.
non-European countries and have commenced activities to seek regulatory approval to begin marketing in other international markets.
CABG Surgery
Coronary Artery Bypass Grafting involves the construction of an alternative path to bypass a narrowed or occluded coronary artery and restore blood flow from the aorta to an area past the blockage. This procedure is generally accomplished by harvesting and using saphenous veins from the patient's leg and the LIMA from the chest as bypass grafts. Most commonly, the LIMA is utilized for bypassing blockages in the left anterior descending artery ("LAD") of the heart, while saphenous veins are utilized for bypassing blockages in other coronary arteries.
For SVGs, one end of the harvested vessel is then generally attached to the aorta for blood inflow, and the opposite end is attached to the target coronary vessel. If a mammary artery is used as the bypass graft, it must be dissected from the chest wall, leaving one end in place on the aorta, while the opposite end is attached to the target vessel, providing uninterrupted blood flow from the arterial circulation. Once in place, these grafts provide blood flow to bypass the narrowed or occluded portion of the coronary artery. The following diagram illustrates the use of the internal mammary artery graft and SVGs in CABG surgery:
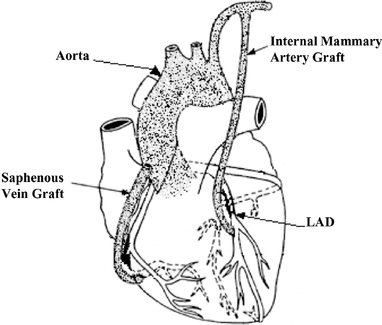
5
Current Disadvantages of Saphenous Vein Grafts
Since its first successful use in the 1960's, the SVG has been one of the most commonly used conduits in CABG surgery. Some of the main advantages of using the saphenous vein include its ease of accessibility, its ease of handling, and the number of grafts, typically three, that can be constructed from a single vein. Despite these advantages and the widespread use of saphenous veins in CABG surgery, several issues have been identified, such as:
- •
- Pressure normally exerted on veins is much lower than the pressure on arteries. Arterial pressure is normally
80-120 mm Hg while central venous pressure is normally about 3-7 mm Hg.
- •
- Veins do not have the strong muscular wall seen in arteries. Therefore, when placed under higher arterial pressures, the
veins typically dilate, or expand.
- •
- Veins have large lumens as compared to arteries, resulting in a mismatch of lumen diameters when an SVG is connected to a coronary artery. This size mismatch may result in slow, sluggish blood flow in the vein graft with more stress placed on the wall of the vein due to blood volume.
The higher pressure of arterial blood flow and the size mismatch that results when a saphenous vein is used as a graft in CABG surgery often cause the vein to expand, damaging the lining of the vein. The vein responds to this damage by causing its walls to thicken in a manner that often leads to failure of the bypass graft. Smooth muscle cells proliferate in the middle layer of the vein wall and migrate to the inner surface of the vein in a process known as neointimal hyperplasia. The resulting accumulation of activated smooth muscle cells secrete inflammatory and growth factors leading to a stenotic build-up, or constriction and narrowing of the graft, and graft failure over time. The failure rate of SVGs in CABG procedures is well documented in the scientific literature. A sampling of data from some of the larger benchmark studies is provided below:
| Saphenous Vein Graft Failure Rates | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
1 Year | 5 Year* | 10 Year* | |||||||||||||||||
Year
|
Author | Number of Patients |
Failure Rate |
Number of Patients |
Failure Rate |
Number of Patients |
Failure Rate |
||||||||||||||
1984 |
Barner, et al. |
248 | 7 | % | 112 | 26 | % | — | — | ||||||||||||
1996 |
Fitzgibbon, et al. |
3,993 | 19 | % | 1978 | 25 | % | — | — | ||||||||||||
2004 |
Goldman, et al. |
660 | 8 | % | 336 | 25 | % | 368 | 39 | % | |||||||||||
2005 |
Alexander |
2,000 | 26 | % | — | — | — | — | |||||||||||||
2009 |
Puskas, et al. |
183 | 18 | % | — | — | — | — | |||||||||||||
- *
- Five and ten year data is not available for those studies for which data is not presented in these columns.
Failure of these grafts typically evidenced by partial or complete blockage and reduced or stopped blood flow, can lead to chest pain or angina, congestive heart failure, irregular heartbeat, myocardial infarction, revascularization or death. A repeat of a CABG procedure to repair a failing or failed graft is a technically more difficult procedure with mortality rates three to five times higher than the original CABG procedure.
eSVS MESH—Our Solution
Our eSVS MESH is designed to improve the long-term outcome of CABG procedures by addressing limitations of unsupported saphenous veins. Our eSVS MESH is a highly flexible, semi-compliant, kink-resistant extra-vascular tubular prosthesis made of knitted nickel/titanium, or nitinol, wire mesh. Our eSVS MESH is designed to be fitted like a sleeve over vein grafts.
6
An artery has a thick muscular wall to handle higher pressures, and a relatively small lumen that produces higher blood velocities, offering less chance for blood to pool and clot. In contrast, a vein has a thinner, less muscular wall due to the lower pressures normally found in veins and a larger lumen designed to maintain these lower pressures. We believe that larger, thinner-walled veins will have greater potential benefit from our eSVS MESH.
Our eSVS MESH is designed to provide the vein graft with physiological attributes similar to those of an artery by reducing the lumen diameter and strengthening the vessel wall. We believe the key benefits of our eSVS MESH technology include:
- •
- Structural support designed to inhibit vessel expansion and resulting damage to the vessel, which can prevent a thickening
of the vessel wall over time, or hyperplasia, and resulting graft failure.
- •
- Radial constriction designed to cause the diameter of the graft, or lumen, to be consistent in size and more closely match
the diameter of the target coronary artery to which it is attached, thereby increasing blood flow velocities, reducing the potential for clot formation, and inhibiting hyperplasia.
- •
- Compatibility with current CABG procedures, including on-pump or off-pump procedures, and open or endoscopic saphenous vein harvest methods. On pump CABG procedures are performed on a non-beating heart with the patient on a heart-lung machine, and off-pump CABG procedures are performed on a beating heart. Open saphenous vein harvest involves a long incision in the leg to expose the entire length of vein being harvested, and endoscopic saphenous vein harvest involves only small slits at the beginning and end of the vein segment being harvested, with the use of an endoscopic device to harvest the vein segment. Except for the placement of our eSVS MESH on the SVG, the surgical steps to use a saphenous vein graft with our eSVS MESH are the same as would be performed for any coronary artery bypass procedure utilizing unsupported SVGs. We do not expect, nor have we seen, a significant increase in CABG procedure time due to eSVS MESH use.
Our eSVS MESH technology consists of the following:
- •
- eSVS MESH (25 cm length, and either 3.5, 4.0, or 4.5 mm in diameter);

- •
- INTRODUCER for use in placing our eSVS MESH on the saphenous vein (one for each diameter of our eSVS MESH);

- •
- SUTURE SNARE for use in loading our eSVS MESH onto the saphenous vein; and

7
- •
- SIZING TOOL for use in choosing the correct device size based on saphenous vein diameter.

Clinical Development of our eSVS MESH
European Post-Market Study
We are currently pursuing two post-market studies in Europe.
The first study is a prospective, multi-center, randomized, controlled study enrolling patients with multi-vessel coronary artery disease who require CABG of the right coronary artery ("RCA") and the left circumflex artery ("LCX") due to atherosclerotic coronary artery disease. Patients will serve as their own control, meaning that they will receive one SVG treated with our eSVS MESH and one untreated SVG. In addition, the use of the SVG treated with our eSVS MESH will be randomized between the RCA and the LCX.
The objective of this study is to support our international reimbursement and marketing activities with a clinical evaluation of the short (three to six months) and long-term (two years) post-implant patency of the eSVS MESH in the treatment of SVGs used in CABG surgery as compared with prospective SVG CABG without the eSVS MESH. Long term safety out to five years will also be assessed.
We intend to enroll up to 200 patients in this study at a maximum of six European hospitals. Clinical follow-up assessments consisting of a physical exam, laboratory testing, medication review, CT angiography for all enrolled patients will be performed at three or six months post procedure and coronary angiogram will be performed at 24 months. Additional yearly follow-ups through five years will be performed to assess safety.
Our second study will be performed at the University Hospital Basel, Clinic for Cardiac Surgery, in Basel, Switzerland. This study is also designed to evaluate the patency of SVGs treated with the eSVS MESH versus untreated SVGs 24 months after surgery. The study design is similar to the first study, except that patient follow-up will be at 30 days and six, 12 and 24 months after CABG and coronary angiograms will be performed at both the six and 24 month follow-up visits.
Feasibility Study for the FDA
We are currently recruiting study sites for a multi-center, randomized study of external saphenous vein support using our eSVS MESH in CABG Surgery. The title of this study is "The eMESH I Study" which will be performed at up to 10 sites in Europe.
The objective of this study is to demonstrate the initial safety and performance of our eSVS MESH for use as an external SVG support device during CABG surgery. The results of this study are intended to be used to obtain an approval from the FDA of an IDE for a pivotal study in the U.S. The study is a prospective, randomized study that will enroll a maximum of 120 patients.
8
The protocol for this study was reviewed by the FDA and is designed to collect the additional clinical data required by the FDA. As part of this study, each patient will be required to have two qualifying SVGs to be enrolled in the study. Additional SVGs are allowed but will not be included in the study evaluation. Each patient will serve as their own control, meaning that each study subject will be implanted with one SVG treated with our eSVS MESH and one untreated SVG. The two SVGs must be pre-specified during the procedure and the graft treated with our eSVS MESH will be randomized to either the right coronary artery ("RCA") or the left circumflex artery ("LCX") system.
The primary safety endpoint is the 30 day rate of major adverse coronary events (MACE) defined as the rate of the composite of total mortality, myocardial infarction (heart attack), and/or coronary target vessel revascularization (percutaneous coronary intervention or CABG) within 30 days of the procedure. The primary performance endpoint is the angiographic patency rate of the enrolled grafts, where patency is defined as < 75% stenosis, or blockage, of the SVG at six months.
Patients will be followed through hospital discharge, with follow-up visits at 30 days, three months, six months, one year and yearly thereafter through five years. Enrollment is expected to take nine to 12 months. Results through the six month follow-up visit will be submitted to the FDA with a request for an IDE approval of a pivotal study.
International Human Clinical Trial
The first human clinical trial of our eSVS MESH is a non-inferiority trial where each patient is randomized to receive an SVG with our eSVS MESH to bypass either the right coronary artery or the circumflex artery, two arteries commonly bypassed during CABG. The bypassed artery not chosen to receive our eSVS MESH serves as the control and receives a standard SVG. To ensure Good Clinical Practices compliance, outside resources are utilized for data collection and analysis, including a contract research organization for data entry and verification, a physician clinical events committee for the review and evaluation of adverse events, and an angiographic core lab for assessment of SVG patency.
Seven international centers enrolled 90 patients in this trial. Enrollment in this trial closed on July 21, 2009.
The international sites involved in this trial, and the number of patients enrolled at each site, is provided below:
Center Name
|
Number of Patients Enrolled |
|||
|---|---|---|---|---|
Schleswig-Holstein University Hospital, Kiel, Germany |
25 | |||
National University Hospital, Singapore |
21 | |||
University Of Cape Town, Cape Town, South Africa |
20 | |||
Hospital Regional De Sion, Sion, Switzerland |
9 | |||
Auckland City Hospital, Auckland, New Zealand |
8 | |||
Hospital Universitario 12 de Octubre, Madrid, Spain |
5 | |||
Prince Charles Hospital, Brisbane, Australia |
2 | |||
Total |
90 | |||
In this trial, our goal was to demonstrate that the use of our eSVS MESH results in no more major adverse cardiac and cerebral events, or MACCE, than standard CABG surgery. The primary safety endpoint of this trial was statistical non-inferiority based on the total rate of MACCE at 30-days post-implant as compared to published literature. MACCE is a composite of the following:
- •
- myocardial infarction, or heart attack;
- •
- stroke;
9
- •
- revascularization due to blocked vein grafts, including surgery or stenting; and
- •
- death.
In summary, there were four adverse events that met the protocol definition of MACCE, which compared favorably to the compilation of published literature that presented 30-day post-implant MACCE rates for CABG surgery patients, separating the MACCE category into the composite factors listed above.
A table summarizing these results is shown below:
| |
Trial Data | Objective Performance Criteria |
|||||
|---|---|---|---|---|---|---|---|
MI |
2(2.2 | )% | 2.8 | % | |||
Stroke |
2(2.2 | )% | 1.8 | % | |||
Revascularization |
0 | 0 | % | ||||
Death* |
0 | 4.8 | % | ||||
Total |
4(4.4 | )% | 9.4 | % | |||
- *
- One patient death eight months after surgery due to non-cardiac causes
The primary effectiveness endpoint of this trial is statistical non-inferiority of angiographic stenosis, or patency, of eSVS MESH vessels as compared to control vessels at nine to 12 months post-implant. A vessel is considered to be patent if there is less than 50% stenosis. This data has been inconclusive regarding the effectiveness of the eSVS MESH primarily due to the following two factors:
- •
- One center had implant methods incompatible with our eSVS MESH. Specifically, this center had issues with failure of the
proximal anastomotic site, resulting in graft closure. We have modified our instructions for use to provide clear direction to surgeons on how to make the proximal anastomotic site when using our eSVS
MESH.
- •
- The amount of reduction in the diameter of the SVGs, or downsizing, prescribed in our instructions for use and sizing tool was too aggressive, resulting in a higher than anticipated closure rate in SVGs utilizing the eSVS MESH, particularly when our smallest device, 3.0 millimeters, was used. This resulted in lumen diameters that were very small and did not remain patent. We have modified our instructions for use and sizing tool to decrease the amount of downsizing applied to SVGs by our eSVS MESH and discontinued the 3.0 millimeter size of our eSVS MESH.
Of the 90 patients participating in the study, 73 patients returned for angiographic studies nine to 12 months following their implant. In this group, 49% (36 of 73) of the eSVS MESH vessels were patent and 81% (59 of 73) of the untreated vessels were patent. If, however, we exclude eSVS MESH grafts implanted at the center with the incompatible treatment methods and grafts treated with our 3.0 millimeter eSVS MESH, at nine to 12 months following the implant, 73% (24 of 33) of the eSVS MESH vessels were patent and 81% (59 of 73) of the untreated vessels were patent, statistically equivalent results. The following table shows the patency of (i) vessels treated with eSVS MESH implanted at the center with incompatible treatment methods ("Site 3" in the table below), (ii) vessels treated with eSVS MESH implanted at all other centers and (iii) untreated saphenous veins used as
10
controls, for each of the 3.0, 3.5 and 4.0/4.5 millimeter eSVS MESH sizes. We combined the 4.0 and 4.5 sizes since only one patient received the 4.5 millimeter eSVS MESH.
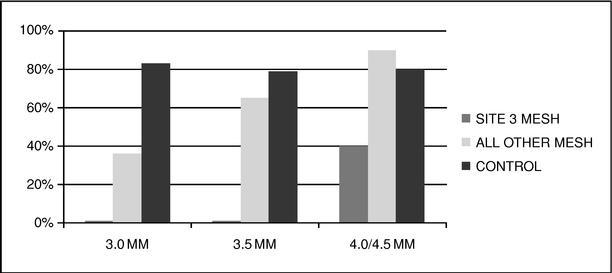
This trial formed the basis for our CE Mark application, which we submitted in February 2010 and received in May 2010. We began marketing and commenced shipments of our eSVS MESH in select European Union markets in June 2010, in the United Arab Emirates in October 2010 and in Turkey in January 2011.
European Trial to Support United States IDE Application
The FDA has reviewed and disapproved our most recent amendment to our application for an IDE in March 2011. At that time the FDA indicated that they intended to review our IDE information with outside experts before they provide further guidance to us. Due to internal delays, the FDA did not begin this review until August 2011. In September 2011, the FDA advised us that we had not provided sufficient data to support our request for an IDE for our eSVS MESH. Therefore, we intend to conduct a new feasibility trial in Europe based upon a protocol which incorporates additional guidance/requirements provided by the FDA. In November 2011, we began the process of recruiting study sites and expect to commence enrollment in the summer of 2012. Upon completion of this study, we expect to request an IDE for a pivotal study in the U.S. The results from this pivotal study are intended to provide the basis for filing of a PMA application, which must be approved by the FDA prior to marketing the eSVS MESH in the United States. However, we could be delayed by adverse clinical results or regulatory complications, and we may never receive U.S. marketing approval.
Pivotal Trial
The primary safety endpoint of a pivotal trial in the U.S. is expected to be statistical non-inferiority of major adverse cardiac events ("MACE") at 30-days post-implant in patients with SVGs treated with our eSVS MESH as compared with the reported total rate of MACE as published in prior CABG study reports. The primary effectiveness endpoint of this trial is expected to be statistical superiority of the patency of SVGs treated with our eSVS MESH vessels as compared to untreated SVG's at 12 months post-implant. This effectiveness endpoint is more rigorous than the effectiveness endpoint of our international trial, which was a non-inferiority comparison.
Until the requirements of the study are agreed to by the FDA, which includes the number of patients required, we cannot estimate the time required to complete enrollment on a pivotal study. However, prior to commercializing our eSVS MESH in the United States, we will be required to submit a PMA application to the FDA which includes the final results from a pivotal study. Approval
11
of a PMA by the FDA generally takes approximately one year after the application. We could be delayed by adverse clinical results or regulatory complications, and we may never receive marketing approval.
Preclinical Testing
Preclinical trials of our eSVS MESH technology have been presented in peer-reviewed journals, including The Journal of Thoracic and Cardiovascular Surgery in February 2008 and the Journal of Vascular Surgery in June 2009. Between 2002 and 2007, Medtronic, Inc. sponsored multiphase trials with the Cardiovascular Research Unit of the Christiaan Barnard Department of Cardiothoracic Surgery at the University of Cape Town in South Africa, or UCT, to evaluate the effects of various designs of external nitinol mesh sleeves on the vascular architecture of vein grafts used in CABG and peripheral bypass procedures. This multiphase research concluded that the use of our eSVS MESH showed a statistically significant decrease in intimal hyperplasia after six months of implantation. In addition to these trials, Medtronic, Inc. and UCT collaborated on stress, fatigue, durability, and finite element analysis of knitted eSVS MESH designs.
In October 2007, we acquired ownership of the core intellectual property relating to our eSVS MESH from Medtronic, Inc. and initiated additional work on the technology. This work included developing additional sizes of our eSVS MESH, completing required preclinical and biological testing of the product and accessories, developing packaging and labeling for our eSVS MESH, and creating product documentation intended to comply with relevant FDA and international standards.
In addition, we initiated and completed a series of animal trials utilizing sheep to confirm that our eSVS MESH, as manufactured by us, performed as expected, and produced the expected results. These animal trials showed a statistically significant inhibition of the formation of intimal hyperplasia when our eSVS MESH was used with an SVG in CABG procedures. However, sheep arterial pressures and vasculature differ from humans, and human clinical studies may not be consistent with animal trial results.
Additional eSVS MESH Applications
Additional development projects based on our eSVS MESH technology that we may explore and advance include:
Peripheral Grafts
In this clinical application, SVGs are used to bypass obstructed arterial vessels in the legs. We have begun initial preclinical trials for this application, utilizing SVGs with our eSVS MESH in place and have completed preliminary assessments of this procedure.
Coronary Allografts
In this clinical application, cadaver, or allograft, SVGs are used in CABG procedures for patients who do not have appropriate arterial or venous conduits. We have had discussions with suppliers of this allograft material to determine usage patterns.
Arteriovenous Fistula
In this clinical application, a fistula, or connection, is made between an artery and a vein, normally in the non-dominant arm, for circulatory system access in patients requiring chronic dialysis.
In light of and in response to developments with the FDA, our staff has been focused on advancing our clinical studies in Europe and developing the additional clinical information which the FDA has requested prior to allowing us to obtain an IDE approval for a US clinical trial. As a result,
12
we have not devoted substantial amounts of time to these development projects. We expect that our clinical trials will remain our primary focus for 2012 and we will devote time to the further development of our eSVS MESH and additional applications for the eSVS MESH as schedules and priorities permit.
Sales and Marketing
Europe and Other International Markets
On May 13, 2010, we obtained the CE Mark for our eSVS MESH. The CE Mark allows us to sell our eSVS MESH for use in CABG procedures in 32 countries within the European Union, the European Economic Area, and the European Free Trade Association. We began marketing and commenced shipments of our eSVS MESH in select European Union markets in June 2010. We have utilized independent distributors to commercialize our technology in Europe. We have entered into agreements with independent distributors for Switzerland, Italy, Spain, Belgium, the Netherlands, Luxembourg, the UK, Ireland, Greece, Denmark, Sweden, Norway, Turkey, Germany, and subsequent to year-end, France to conduct sales in these markets. These distribution agreements generally have terms of three years, restrict distributors from selling products competitive with our eSVS MESH and grant exclusivity within a territory, which is generally limited to a single country. In addition, we may terminate the distributor's exclusivity or the entire agreement if the distributor fails to achieve agreed upon sales targets. These distributors will be supported by our U.S.-based staff with regard to product and physician training and promotional materials.
We work with our distributors to drive clinical utilization in key centers within their respective territories. We have also identified other third parties that may be contracted to assist in obtaining country-specific product reimbursement where necessary. We were recently denied a government-sponsored supplemental reimbursement in Germany for our eSVS MESH for calendar year 2012, and will need to work with hospitals in Germany to obtain coverage for the eSVS MESH under a new or existing reimbursement code.
As the European cardiac surgery market is characterized by centralized, high-volume cardiac surgery centers, we believe this market can be effectively addressed through a small, highly-focused independent distributor network.
We are pursuing post-market clinical trials aimed at validating both the short and long-term outcomes of patients who receive our eSVS MESH. These post-market studies are designed so that a patient undergoing a three-vessel bypass surgical procedure will act as their own control, meaning they will receive the LIMA to the LAD and the eSVS MESH will be randomized to one of the two remaining vessels targeted for bypass. Follow-up imaging studies will then be used to compare patency rates of the untreated versus the eSVS MESH treated vein grafts. These studies are intended to show that bypass grafts treated with the eSVS MESH require less revascularization procedures than bypass grafts without the eSVS MESH, thereby also reducing the costs associated with revascularization procedures for the bypass grafts treated with the eSVS MESH. We envision that the results of these studies will be presented at scientific sessions and presented in peer-reviewed journals, thereby increasing the visibility and adoption of our eSVS MESH. These studies will also be used to support applications for public hospital reimbursement in those countries that require outcomes data for such reimbursement.
We believe that the CE Mark will allow us to begin regulatory submissions to obtain marketing approval in other select markets, including South Africa, Canada, New Zealand and Argentina. These markets require the CE Mark to begin the submission process, per their current medical device regulatory requirements. In addition, we have begun the regulatory submission process for Singapore, Hong Kong and Korea, and have entered into an agreement with an independent distributor for Singapore, Hong Kong, Thailand, Malaysia, Indonesia, the Philippines, Cambodia, Laos, Vietnam and
13
Brunei. We have also entered into an agreement and begun commercial sales with an independent distributor for the United Arab Emirates.
United States
We intend to utilize a combination of direct sales employees and independent distributors to commercialize our eSVS MESH in the U.S. We have identified and are in preliminary discussions with independent distributors that may be contracted to conduct sales, but we have not yet entered into any distribution agreements. We expect that these contracts will be on terms similar to those described above for our agreements with international distributors. These distributors will be supported by our staff with regard to training and promotional materials. We have contracted outside reimbursement experts to assist in obtaining Centers for Medicare & Medicaid Services, or CMS, product reimbursement.
We are required to obtain a PMA approval from the FDA in order to market our eSVS MESH in the United States. The timeline for obtaining such approval is subject to the requirements of the FDA. We may be delayed by adverse clinical results or regulatory complications, and we may never receive U.S. marketing approval.
Intellectual Property
As of March 1, 2012, we had six issued patents covering the eSVS MESH: two issued in the U.S. and one each issued in Japan, Europe, Canada and South Africa. The European patent has been validated and is enforceable in six European countries and we are presently engaged in the grant phase to validate this patent in two additional European countries. In addition, we have five patent applications pending in the U.S and eight patent applications pending in countries outside the U.S. covering various aspects of our eSVS MESH.
Our issued patents and pending patent applications include claims directed towards, among other things, the knitted, resilient structure of our eSVS MESH which is designed to provide structural support to inhibit vessel expansion and provide the vein graft with physiological attributes similar to those of an artery, and the surgical procedures relating to implanting our eSVS MESH.
During the examination process, the examiner assesses the patentability of the invention by comparing the pending claims to the relevant prior art. If the examiner determines that the claimed invention is unpatentable, the examiner will issue an "office action" providing the grounds for rejecting the claims. Such grounds for rejection can include, for example, that the claimed invention lacks novelty or is obvious in view of the relevant prior art. It is common for most U.S. patent applications to be rejected at least one time before issuing as a U.S. patent. To overcome the rejection, the applicant must generally reply by amending the claims and/or providing arguments to distinguish the claimed invention from the cited prior art. If the examiner is not persuaded by the amendments and/or arguments, the applicant can either continue to make amendments and/or arguments to the examiner, for example, by filing a Request for Continued Examination, or a RCE, or by appealing the examiner's decision to the Board of Patent Appeals and Interferences, or the BPAI.
Due to the indeterminate time frames in which patent examiners engage in prosecution and the uncertainty of how the examiners will respond to our submissions, it is difficult to accurately predict when the prosecution of our current patent applications will end. These pending patent applications may not issue as patents, or, if issued, may not issue in a form that is desirable or advantageous to us. Competitors can attempt to replicate some or all of the competitive advantages we derive from our eSVS MESH or design around our technology, and they might be able to market products and use manufacturing processes that are substantially similar to ours, each of which we believe would be highly likely if we are able to achieve significant market acceptance of our eSVS MESH.
14
In addition, third parties may assert that our eSVS MESH infringes the claims in their patents or seek to expand their patent claims to cover aspects of our eSVS MESH. An adverse determination in litigation or interference proceedings to which we may become a party could subject us to significant liabilities or require us to seek licenses. Although patent and intellectual property disputes in the medical device area have often been settled through licensing or similar arrangements, costs associated with these arrangements may be substantial and could include ongoing royalties. We may be unable to obtain necessary licenses on satisfactory terms, if at all, and we may be required to redesign our eSVS MESH to avoid infringement.
The core intellectual property relating to our eSVS MESH was sold to us by Medtronic, Inc. ("Medtronic") pursuant to an Assignment and License Agreement dated October 9, 2007. Pursuant to the Assignment and License Agreement, Medtronic also sold to us intellectual property relating to a brushed ePTFE vascular graft, or the Brushed Graft Product. As consideration for the sale of intellectual property relating to the eSVS MESH and the Brushed Graft Product and other rights granted by the Assignment and License Agreement, we have agreed to pay Medtronic an aggregate of $20.0 million upon the achievement of certain sales milestones relating to the eSVS MESH and the Brushed Graft Product and a royalty of 4% on sales of our eSVS MESH and the Brushed Graft Product. The royalty will terminate upon the earlier of the expiration of all of the patents and patent applications, or when the aggregate royalties paid reach $100.0 million.
Any or all licenses granted to us pursuant to our agreement with Medtronic may be terminated and potentially all of the core intellectual property and patent rights related to our eSVS MESH will revert to Medtronic, upon notice by Medtronic, if we become insolvent, make an assignment for the benefit of creditors, go into liquidation or receivership or otherwise lose legal control of our business.. Medtronic may also cause the core intellectual property and patent rights related to our eSVS MESH to revert to Medtronic if we determine to cease commercializing our eSVS MESH.
Competition
The development and commercialization of medical devices to treat cardiovascular disease is a highly competitive industry. Physicians and patients may select among a variety of treatments to address coronary artery disease, including pharmaceutical therapy, balloon angioplasty, stenting with bare metal or drug-eluting stents, and CABG procedures, with the selection often depending upon the health of the patient and the stage of the disease. If physicians or their patients choose alternative treatments to CABG surgery due to the disadvantages of CABG surgery, such as the failure rate of CABG surgery, or if additional alternative treatments for cardiovascular disease are developed, there may be a decrease in the number of CABG surgery procedures. The American College of Cardiology/ American Heart Association treatment guidelines state that CABG is the only recommended revascularization procedure for those patients with CAD in their left main coronary artery or CAD in three or more coronary arteries.
Our eSVS MESH is designed to improve the structural characteristics and long-term performance of vein grafts in CABG surgery. If our eSVS MESH is proven to do so successfully, we believe physicians may more frequently choose to perform CABG surgery over alternative treatments. We expect the primary competition for our eSVS MESH to be other products or techniques to improve the effectiveness of vein grafts in CABG surgery.
We are aware of three companies that have developed mesh devices to be used on the outside of blood vessels. Vascular Graft Solutions ("VGS"), an Israeli company is developing a product called the Fluent expandable external support system which is designed to reduce vein graft failures in CABG surgery. VGS is currently conducting its first in-human trial in the United Kingdom. According to information filed on ClinicalTrials.gov, VGS expects to enroll approximately 30 patients and complete data collection for their primary outcome measure in June 2012. Alpha Research, a Swiss company, has
15
developed a product known as the Biocompound Graft for use in coronary and peripheral bypass operations. The product is a stainless steel braided mesh, indicated for use in coronary or peripheral bypass with patients who have irregularly shaped veins. B. Braun, a German company, has developed a product known as ProVena for use in peripheral bypass operations. The product is a woven polymer mesh, indicated for use in peripheral artery bypass operations using vein grafts.
We believe that the VGS product will be a direct competitor to our eSVS MESH when and if the results of their on-going clinical trial in Great Britain allow VGS to secure a CE Mark approval for their product.
We believe that the Alpha Research and B. Braun products are not currently direct competitors to our eSVS MESH, and are not likely to become direct competitors in the near future, because the Biocompound Graft is intended for use only with irregularly shaped veins and the ProVena is intended for use with non-coronary procedures. However, it is possible that one or both of these companies, or other potential competitors, will seek approval to use these or similar devices for procedures with similar or identical indications for use as our eSVS MESH.
The key competitive factors affecting the success of our eSVS MESH are likely to be the effectiveness, safety profile and price of our eSVS MESH, as compared to existing methods for CABG surgery. We believe a potential disadvantage associated with our eSVS MESH is the possibility of allergic reaction to the implant materials. According to a July 2009 article in the Journal of Invasive Cardiology, nickel allergy after implantation of a nitinol-containing device is rare. This article described the rate of nickel allergy in cardiovascular implants to be between 0.002% and 0.02%. The article also stated that the patients with nickel allergy symptoms responded to medical management at the time of the reaction, did not require device explant, and no longer require medications for the reaction. In order to further safeguard against this rare occurrence, the eSVS MESH instructions for use state that it is contraindicated for patients with a known allergy to nitinol. We believe another potential disadvantage associated with our eSVS MESH is the possibility of damage to the saphenous vein during placement of our eSVS MESH. If the physician does not select the proper size eSVS MESH relative to the size of the vein, the saphenous vein may be damaged while placing our eSVS MESH over the vein. For example, if too small an eSVS MESH is chosen, there could be damage to the saphenous vein caused by stretching the vein while trying to place it inside our eSVS MESH. We have provided specific directions in the eSVS MESH instructions for use on how to properly size veins and place our eSVS MESH. In addition, we provide a sizing tool with our eSVS MESH to ensure proper vein sizing. The commercial success of our eSVS MESH will depend upon the results of clinical trials of the technology and experience with the technology in the commercial marketplace.
If the commercialization of our eSVS MESH technology is successful, we expect that other medical device companies, many of whom are larger and have greater financial resources than us, will seek to enter into this market by introducing competing technologies.
Manufacturing and Suppliers
We fabricate our eSVS MESH both at our facility and at a contract manufacturer. We conduct final assembly and packaging inside a controlled environment area within our facility that satisfies the requirements of a Class 10,000 level clean room. We have implemented systems to ensure that our manufacturing operations comply with relevant United States and International Good Manufacturing Practices requirements.
We have vendors for all of our key components and outsourced processes. We have identified alternate suppliers for each key component and outsourced process; however, in some cases, components are provided by single source suppliers at this time due to quality considerations, costs, or regulatory requirements. We have established redundancy for custom equipment used in the manufacture of our eSVS MESH. A third-party supplier performs sterilization services for our eSVS
16
MESH. We currently use four knitting machines that knit the mesh sleeve of our eSVS MESH, with three located at our facility and the fourth located off-site. We believe that these four machines will produce sufficient quantities of our eSVS MESH to meet our expected needs for the foreseeable future. In the event that one or all of our knitting machines were to become unavailable, we believe that we can obtain one or more replacement knitting machines, although the custom work required to enable the machines to produce our eSVS MESH may result in some delays in our production process.
Research and Development
During 2009, 2010 and 2011, we incurred $3.0 million, $2.5 million and $1.7 million, respectively, of research and development expenses. Research and development costs include the costs to design, develop, test, seek approval for, and enhance our eSVS MESH and production process. Expenses related to research and development consist primarily of personnel costs, including salaries, benefits and stock-based compensation, product development, pre-clinical and clinical trials, professional service fees, materials and supplies, and facilities-related costs. While our research and development expenses to date have been focused on product development and evaluating the feasibility of our eSVS MESH, we expect that a large percentage of our research and development expenses in the future will be incurred in support of our current and future clinical trials. As we develop further applications for our eSVS MESH, we intend to utilize internal resources, outside contract resources and facilities, and our Scientific Advisory Board.
Employees
As of March 1, 2012, we had 15 employees. We plan to continue to expand our research and development and commercialization activities. To support this growth, we will need to expand managerial, research and development, operations and other functions. None of our employees is represented by a labor union, and we consider our relationship with our employees to be good.
Government Regulation
United States Medical Device Regulation
The Federal Food, Drug, and Cosmetic Act, or FDCA, and the FDA's implementing regulations, govern medical device design and development, preclinical and clinical testing, pre-market clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, and post-market surveillance. Medical devices and their manufacturers are also subject to inspection by the FDA. The FDCA, supplemented by other federal and state laws, also provides civil and criminal penalties for violations of its provisions. We intend to manufacture and market a medical device that is regulated by the FDA, comparable state agencies and regulatory bodies in other countries.
Our eSVS MESH will require marketing authorization from the FDA prior to commercial distribution in the United States. The two primary types of FDA marketing authorization are pre-market notification (also called 510(k) clearance) and PMA approval. The type of marketing authorization applicable to a device—510(k) clearance or PMA approval—is generally linked to classification of the device. The PMA approval process is generally more stringent, time-consuming and expensive than the 510(k) clearance process.
The FDA classifies medical devices into one of three classes (Class I, II or III) based on the degree of risk FDA determines to be associated with a device and the extent of control deemed necessary to ensure the device's safety and effectiveness. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are deemed to pose the least risk and are subject only to general controls applicable to all devices, such as requirements for device labeling, pre-market notification, and adherence to the FDA's current Good Manufacturing Practice
17
requirements, as reflected in its Quality System Regulation, or QSR. Class II devices are intermediate risk devices that are subject to general controls and may also be subject to special controls such as performance standards, product-specific guidance documents, special labeling requirements, patient registries or post-market surveillance. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general or special controls, and include life-sustaining, life-supporting, or implantable devices, and devices not "substantially equivalent" to a device that is already legally marketed.
Most Class I devices and some Class II devices are exempted by regulation from the 510(k) clearance requirement and can be marketed without prior authorization from FDA. Class I and Class II devices that have not been so exempted are eligible for marketing through the 510(k) clearance pathway. By contrast, devices placed in Class III generally require PMA approval prior to commercial marketing. To obtain 510(k) clearance for a medical device, an applicant must submit a pre-market notification to the FDA demonstrating that the device is "substantially equivalent" to a predicate device legally marketed in the United States. A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and (1) the same technological characteristics, or (2) has different technological characteristics and the information submitted demonstrates that the device is as safe and effective as a legally marketed device and does not raise different questions of safety or effectiveness. A showing of substantial equivalence sometimes, but not always, requires clinical data. Generally, the 510(k) clearance process can exceed 90 days and may extend to a year or more.
After a device has received 510(k) clearance for a specific intended use, any modification that could significantly affect its safety or effectiveness, such as a significant change in the design, materials, method of manufacture or intended use, will require a new 510(k) clearance or (if the device as modified is not substantially equivalent to a legally marketed predicate device) PMA approval. While the determination as to whether new authorization is needed is initially left to the manufacturer, the FDA may review this determination and evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
Our coronary eSVS MESH has been designated a Class III product by the FDA and will be required to go through the PMA process. Other indications of our eSVS MESH, including peripheral and arteriovenous fistula applications, have not been classified at this time.
The FDA will require us to file a PMA application with respect to our eSVS MESH and there is no assurance that PMA approval will be granted. A PMA application requires the payment of significant User Fees, and must be supported by valid scientific evidence, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data, to demonstrate to the FDA's satisfaction the safety and effectiveness of the device. A PMA application also must include a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling. After a PMA application is submitted and found to be sufficiently complete, the FDA begins an in-depth review of the submitted information. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with the QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures.
18
FDA review of a PMA application is required by statute to take no longer than 180 days, although the process typically takes significantly longer, and may require several years to complete. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
- •
- the device may not be safe or effective to the FDA's satisfaction;
- •
- the data from our preclinical trials and clinical trials may be insufficient to support approval;
- •
- the manufacturing process or facilities we use may not meet applicable requirements; and
- •
- changes in FDA approval policies or adoption of new regulations may require additional data.
If the FDA evaluations of both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter, or an approvable letter, which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device for certain indications. If the FDA's evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the PMA. The PMA process can be expensive, uncertain and lengthy and a number of devices for which FDA approval has been sought by other companies have never been approved for marketing. Even if a PMA application is approved, the FDA may approve the device with an indication that is narrower or more limited than originally sought. The agency can also impose restrictions on the sale, distribution, or use of the device as a condition of approval, or impose post approval requirements such as continuing evaluation and periodic reporting on the safety, effectiveness and reliability of the device for its intended use.
New PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling, device specifications, materials or design of a device that is approved through the PMA process. PMA approval supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application and may not require as extensive clinical data or the convening of an advisory panel.
Clinical trials are almost always required to support a PMA application and are sometimes required for a 510(k) clearance. These trials generally require submission of an application for an investigational device exemption, or IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the trial protocol and informed consent are approved by appropriate institutional review boards at the clinical trial sites.
FDA approval of an IDE allows clinical testing to go forward, but does not bind the FDA to accept the results of the trial as sufficient to prove the product's safety and effectiveness, even if the trial meets its intended success criteria. With certain exceptions, changes made to an investigational plan after an IDE is approved must be submitted in an IDE supplement and approved by the FDA (and by governing institutional review boards when appropriate) prior to implementation. All clinical trials must be conducted in accordance with regulations and requirements collectively known as Good Clinical Practice, or GCP. GCPs include the FDA's IDE regulations, which describe the conduct of clinical trials with medical devices, including the recordkeeping, reporting and monitoring responsibilities of sponsors and investigators, and labeling of investigation devices. They also prohibit
19
promotion, test marketing, or commercialization of an investigational device, and any representation that such a device is safe or effective for the purposes being investigated. GCPs also include FDA's regulations for institutional review board approval and for protection of human subjects (informed consent), as well as disclosure of financial interests by clinical investigators.
Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended safety and effectiveness success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product. The commencement or completion of any of our clinical trials may be delayed or halted, or be inadequate to support approval of a PMA application or clearance of a pre-market notification for numerous reasons, including, but not limited to, the following:
- •
- the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial (or a change to a
previously approved protocol or trial that requires approval), or place a clinical trial on hold;
- •
- patients do not enroll in clinical trials or follow up at the rate expected;
- •
- institutional review boards and third-party clinical investigators may delay or reject our trial protocol or changes to
our trial protocol;
- •
- third-party clinical investigators decline to participate in a trial or do not perform a trial on our anticipated schedule
or consistent with the clinical trial protocol, investigator agreements, good clinical practices or other FDA requirements;
- •
- third-party organizations do not perform data collection and analysis in a timely or accurate manner;
- •
- regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to
undertake corrective action or suspend or terminate our clinical trials;
- •
- changes in governmental regulations or administrative actions;
- •
- the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness; and
- •
- the FDA concludes that our trial design is inadequate to demonstrate safety and effectiveness.
After a device is approved and placed in commercial distribution, numerous regulatory requirements apply. These include:
- •
- establishment registration and device listing;
- •
- the QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance
procedures;
- •
- labeling regulations, which prohibit the promotion of products for unapproved or "off-label" uses and impose
other restrictions on labeling;
- •
- medical device reporting regulations, which require that manufacturers report to the FDA if a device may have caused or
contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if malfunctions were to recur; and
- •
- corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA caused by the device that may present a risk to health.
20
Also, the FDA may require us to conduct post-market surveillance studies or order us to establish and maintain a system for tracking our eSVS MESH through the chain of distribution to the patient level. The FDA enforces regulatory requirements by conducting periodic, announced and unannounced inspections and market surveillance. Inspections may include the manufacturing facilities of our subcontractors.
Failure to comply with applicable regulatory requirements, including those applicable to the conduct of our clinical trials, can result in enforcement action by the FDA, which may lead to any of the following sanctions:
- •
- warning letters or untitled letters;
- •
- fines and civil penalties;
- •
- unanticipated expenditures;
- •
- delays in clearing or approving or refusal to clear or approve products;
- •
- withdrawal or suspension of FDA approval;
- •
- product recall or seizure;
- •
- orders for physician notification or device repair, replacement, or refund;
- •
- production interruptions;
- •
- operating restrictions;
- •
- injunctions; and
- •
- criminal prosecution.
We and our contract manufacturers, specification developers and suppliers are also required to manufacture our eSVS MESH in compliance with current Good Manufacturing Practice requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and includes extensive requirements with respect to quality management and organization, device design, buildings, equipment, purchase and handling of components, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing and record keeping. The FDA enforces the QSR through periodic announced and unannounced inspections that may include the manufacturing facilities of our subcontractors. If the FDA believes we or any of our contract manufacturers or regulated suppliers is not in compliance with these requirements, it can shut down our manufacturing operations, require recall of our eSVS MESH, refuse to clear or approve new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations, or assess civil and criminal penalties against us or our officers or other employees. Any such action by the FDA would have a material adverse effect on our business.
Fraud and Abuse
Our operations will be directly, or indirectly through our customers, subject to various state and federal fraud and abuse laws, including, without limitation, the FDCA, the federal Anti-Kickback Statute and the False Claims Act. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, these laws require us to screen individuals and other companies, suppliers and vendors in order to ensure that they are not "debarred" by the federal government and therefore prohibited from doing business in the healthcare industry. The association or conduct of business with a "debarred" entity could be detrimental to our operations and result in a negative impact on our business.
21
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as the Medicare and Medicaid programs. Several courts have interpreted the statute's intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The federal False Claims Act prohibits persons from knowingly filing or causing to be filed a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Various states have also enacted laws modeled after the federal False Claims Act.
In addition to the laws described above, the Health Insurance Portability and Accountability Act of 1996 created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
Voluntary industry codes, federal guidance documents and a variety of state laws address the tracking and reporting of marketing practices relative to gifts given and other expenditures made to doctors and other healthcare professionals. In addition to impacting our marketing and educational programs, internal business processes will be affected by the numerous legal requirements and regulatory guidance at the state, federal and industry levels.
If our operations are found to be in violation of any of the laws described above or other applicable state and federal fraud and abuse laws, we, as well as our employees, may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare programs, and the curtailment or restructuring of our operations. Individual employees may need to defend such suits on behalf of us or themselves, which could lead to significant disruption in our present and future operations. We cannot assure you that we will be able to comply with the above laws and regulations.
European Medical Device Regulation
The European Union has adopted directives and numerous standards that govern and harmonize the national laws and standards regulating the design, manufacture, clinical trials, labeling, adverse event reporting and post-market surveillance activities for medical devices that are marketed in member states.
Compliance with voluntary harmonized standards including ISO 13485 issued by the International Organization for Standards establishes the presumption of conformity with the essential requirements for a CE Mark. ISO certification is commonly a pre-requisite to use of the CE Mark and indicates that a quality system complies with standards applicable to activities ranging from initial product design and development through production and distribution.
Devices that comply with the requirements of a relevant directive will be entitled to bear the CE Mark and, accordingly, can be commercially distributed throughout the member states of the European Union, and other countries that comply with or have adopted these directives. The method of assessing conformity varies depending on the type and class of the product, but typically involves a combination
22
of self-assessment by the manufacturer and a third-party assessment by a "Notified Body," an independent and neutral institution appointed to conduct conformity assessment. This third-party assessment consists of an audit of the manufacturer's quality system and technical review of the manufacturer's product. For most classes of medical devices, an assessment by a Notified Body residing within the European Union is required in order for a manufacturer to commercially distribute the product throughout the European Union. The manufacturer's assessment will include a clinical evaluation of the conformity of the device with applicable regulatory requirements, which for our eSVS MESH will include clinical study results. The clinical data presented by us must provide evidence that the products meet the performance specifications claimed by us, provide sufficient evidence of adequate assessment of unwanted side effects and demonstrate that the benefits to the patient outweigh the risks associated with the device. We are subject to continued surveillance by the Notified Body and are required to report any serious adverse incidents to the appropriate authorities of the European Union member states.
Products intended for sale must bear the CE mark to show compliance with the Medical Devices Directive, or MDD. If a Notified Body is involved in the approval, the number of the Notified Body must also appear adjacent to the CE Mark.
The routes to compliance under the MDD depend on the classification of the product:
Class I devices are low risk, such as stethoscopes, hospital beds and wheelchairs. The manufacturer must produce a technical file, including product test results compared to relevant standards. In addition, manufacturers of sterile products and devices with a measuring function must apply to a Notified Body for certification of the aspects of manufacture relating to sterility or measurement.
Class IIa devices are low to medium risk, such as hearing aids, electrocardiographs and ultrasonic diagnostic equipment. As with Class I devices, the manufacturer produces a technical file, but a conformity assessment must be carried out by a Notified Body, according to one of the following routes, at the manufacturer's option:
- •
- examination and testing of each product or homogenous batch of products;
- •
- audit of the full quality assurance system;
- •
- audit of the production quality assurance system; or
- •
- audit of final inspection and testing.
Class IIb devices are medium-high risk devices, such as surgical lasers, infusion pumps, ventilators, intensive care monitoring equipment and many implantable devices. Routes to compliance are the same as for Class IIa devices, with the addition of required examination and testing of the product by the Notified Body; however, the full quality assurance route does not require type examination and testing.
Class III devices are high risk, such as balloon catheters and prosthetic heart valves. Our eSVS MESH is classified as a Class III device. Routes to compliance are:
- •
- audit of the full quality assurance system and examination of a design dossier by the Notified Body. A design dossier is a
submission similar to a PMA application with the FDA; or
- •
- examination and testing of the product, together with audit of the production quality assurance system.
We have obtained CE Mark approval to market our eSVS MESH in the European Union and other countries that accept the CE Mark.
23
Third Party Reimbursement
The availability of insurance coverage and reimbursement for newly approved medical devices is variable. The commercial success of our eSVS MESH in both domestic and international markets will be substantially dependent on whether third-party coverage and reimbursement is available for patients receiving bypass grafts with our eSVS MESH. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medical devices, and, as a result, they may not cover or provide additional payment for our eSVS MESH. In order to position our device for coverage by third-party payors, we may have to agree to a lower net sales price than we might otherwise charge. The continuing efforts of governmental and commercial third-party payors to contain or reduce the costs of healthcare may limit our revenue.
In many countries including the United States, third-party payors consist of both government funded insurance programs and private insurance programs who cover a significant portion of a patient's medical expenses. The trends toward managed healthcare in the U.S. and legislation intended to reduce the cost of government insurance programs will significantly influence the purchase of healthcare services and products, and could result in lower or no reimbursement for our eSVS MESH. Even before reimbursement may be obtained for our eSVS MESH in the United States, FDA approval will be required.
Providers have sought ways to manage costs, such as through the use of group purchasing organizations. It is our belief that the planned economic benefits provided by our eSVS MESH to physicians and hospitals through lower revascularization costs (PCI and/or CABG) will be viewed by providers and third-party payors as cost-effective. However, there remains uncertainty whether our eSVS MESH will be viewed positively in a cost-avoidance model so as to warrant adequate coverage and reimbursement levels.
Outside of the United States, there are many reimbursement programs through private payors as well as government programs. In some countries, government reimbursement is the predominant program available to patients and hospitals. While the majority of countries have existing reimbursement for CABG procedures and products, a number of countries may require us to gather additional clinical data before recognizing coverage and reimbursement for our eSVS MESH. It is our intent to complete the requisite clinical trials and obtain coverage and reimbursement approval in countries where it makes economic sense to do so.
European Union
Our reimbursement strategy for certain key European markets that we will initially pursue are briefly outlined below:
- •
- France: Initial sales of our eSVS MESH in France will be in the private sector. Concurrently, we intend to
conduct post-market studies to support the cost effectiveness of the device. These post-market studies will focus on the cost savings of decreased revascularization procedures
versus the cost of the device in order to justify device cost. We expect that our distributors, on our behalf, will then submit this analysis to the French reimbursement evaluation committee to list
our eSVS MESH on the reimbursement list applicable to France.
- •
- Germany: In advance of, or while awaiting a DRG, a company can apply for a "Neue Untersuchungs und Behandlungsmethode"("NUB"). This NUB allows payment for a new device or technology during the period in which the device or technology is used prior to placement into a DRG. The German reimbursement authority reviews NUB applications annually, publishing its decisions before March of each calendar year. Our most recent application was reviewed and denied with our eSVS MESH technology being categorized as "not meeting the
24
- •
- Italy: Initially, we expect that our distributors will sell our eSVS MESH in the private sector in Italy and
concurrently attempt to fit our eSVS MESH into an existing DRG code. If this is not effective in gaining public reimbursement, we expect that our distributors, on our behalf, may apply for a new DRG
for our eSVS MESH, utilizing the post-market study cost effectiveness data.
- •
- Spain: Initially, we expect that our distributors will sell our eSVS MESH in the private sector in Spain and
concurrently attempt to fit our eSVS MESH into an existing DRG code. If this is not effective in gaining public reimbursement, we expect that our distributors, on our behalf, may apply for a new DRG
for our eSVS MESH, utilizing post-market study cost effectiveness data.
- •
- United Kingdom: Initially, we expect that our distributors will sell our eSVS MESH in the private sector in
the UK, then attempt to fit our eSVS MESH into an existing DRG code. If this is not effective in gaining public reimbursement, we expect that our distributors, on our behalf, may apply for a new DRG
for our eSVS MESH, utilizing post-market study cost effectiveness data.
- •
- Switzerland: Initially, we expect that our distributors will sell our eSVS MESH in the private sector in Switzerland and concurrently attempt to fit our eSVS MESH into an existing DRG code. If this is not effective in gaining public reimbursement, we expect that our distributors, on our behalf, may apply for a new DRG for our eSVS MESH, utilizing post-market study cost effectiveness.
requirements for NUB reimbursement." While approval of the NUB would have allowed for specific German hospitals to apply for eSVS MESH reimbursement, we believe that customers interested in purchasing the eSVS MESH can still do so by securing internal funding from discretionary budgets within the hospital. We will need to work with hospitals in Germany to obtain coverage for our eSVS MESH under a new or existing reimbursement code.
United States
In the United States, governmental and private sector payors have instituted initiatives to limit the growth of healthcare costs, using, for example, price regulation or controls and competitive pricing programs. Some third party payors require pre-approval of coverage for new or innovative devices or therapies before they will reimburse healthcare providers who use such devices or therapies.
In the United States, CMS is the government entity responsible for oversight of the Medicare program. Medicare establishes coverage and reimbursement policies at a federal and local level for medical products and procedures, and such policies are periodically reviewed and updated. While private payors vary in their coverage and payment policies, the Medicare program is viewed as a benchmark.
There are established codes for CABG procedures and products that are payable for both Medicare and commercial payors. There are no assurances that our eSVS MESH technology would fall under existing policies or reimbursement codes. There are also no assurances that existing payment rates for such reimbursement codes will continue to hold at the current levels, such as if regulatory changes are implemented regarding the methodology for calculating hospital payments for current inpatient procedures. Medicare payment rates have decreased significantly for those procedures using drug-eluting stents since fiscal year 2007. In 2007, CMS also implemented a revised payment methodology that more accurately reflects the severity of the patient's condition.
Medicare reimburses hospital inpatient stays under the Medicare Severity Diagnosis-Related Group (MS-DRG) system. The MS-DRG system assigns individual cases to an MS-DRG according to the patient's diagnoses, the procedures performed, and the severity of a patient's condition as identified by
25
the presence or absence of complications and comorbidities, or CCs, or major CCs, or MCCs. MS-DRGs provide a single bundled payment which serves as reimbursement for all items and services provided to the Medicare beneficiary during a single hospitalization.
Additionally, a relative weight is calculated for each individual MS-DRG, which represents the average resources required to care for cases within a particular MS-DRG relative to the average resources required to treat cases in all MS-DRGs. Generally, MS-DRG relative weights are adjusted annually to reflect changes in medical practice in a budget neutral manner.
CMS has made no decisions with respect to MS-DRG assignment for patients who undergo CABG procedures in which our eSVS MESH would be used, and there can be no assurance that the MS-DRG to which such patients will be assigned will result in Medicare payment levels that are considered by hospitals to be adequate to further support purchase of our eSVS MESH. Under current CMS reimbursement policies, the agency offers a process to obtain add-on payment for a new medical technology when the existing MS-DRG prospective payment rate is inadequate. To obtain add-on payment, a technology must be considered "new," demonstrate substantial improvement above the current standard of care and exceed certain payment thresholds. Add-on payments are made for no less than two years and no more than three years. Following FDA approval in the United States, we intend to pursue an application for a hospital inpatient new technology add-on payment with CMS. We must demonstrate the safety and effectiveness of our eSVS MESH to the FDA in addition to the CMS requirements listed above before add-on payments will be approved. Should the clinical trial results or peer-reviewed publications prove that use of our eSVS MESH results in a lowering of revascularization rates, we believe there is a reasonable chance that CMS will grant our request. If CMS grants our request, we expect it will approve an add-on payment equal to 50% of the cost of labor and materials incurred. We do not expect that failure to receive approval for an add-on payment would have an adverse impact on our business because existing MS-DRGs already reimburse CABG procedures at a high level, and we believe that physicians and hospitals would be willing to use our eSVS MESH even if an add-on payment is not approved.
For reporting of physician services, the American Medical Association, or AMA, has developed a coding system known as Current Procedural Terminology, or CPT. CPT codes are established by the AMA and statutorily adopted by all government and commercial payors to describe and develop payment amounts for physician services. Physician services are reimbursed by Medicare based on a physician fee schedule whereby payment is based generally on the number of "relative value units" assigned by the AMA to each CPT code. No decision has been made concerning whether existing CPT codes would be appropriate for use in coding CABG procedures when our eSVS MESH is used or if separate, new CPT codes are required. We cannot assure you that codes used for submitting claims for CABG procedures using our eSVS MESH will result in incremental payment to physicians. Failure by physicians to receive what they consider to be adequate reimbursement for CABG procedures in which our eSVS MESH is used could harm our business, financial condition and results of operations.
26
You should carefully consider the following information about risks, together with the other information contained in this annual report on Form 10-K, before making an investment in our common stock. If any of the circumstances or events described below actually arises or occurs, our business, results of operations, cash flows and financial condition could be harmed.
Risks Related to Our Business and Strategy
We have a limited operating history, expect future losses, and may be unable to achieve or maintain profitability.
We were founded on May 1, 2007 and to date we have engaged primarily in development of and initial clinical trials of our external saphenous vein support system, or eSVS MESH. Accordingly, we have limited operating history on which to base an evaluation of our business and prospects. As of December 31, 2011, we had an accumulated deficit of $24.3 million. We have incurred net losses in each year since our inception, and we expect to continue to incur operating losses for the foreseeable future. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity and working capital. Because of the numerous risks and uncertainties associated with developing medical devices, we are unable to predict the extent of any future losses or when we will become profitable, if at all. To date, we have not generated sufficient product revenues to fund our operations and we have financed our operations and internal growth primarily through a public offering completed in February 2011 and private placements of equity securities and convertible promissory notes. Our prospects must be considered in light of the significant risks, expenses, and difficulties frequently encountered by medical device companies in their early stage of development. We may not be successful in addressing the risks we will encounter, and our failure to do so would likely harm our business and our ability to continue to operate.
The results of our initial human trial were inconclusive with respect to efficacy of our eSVS MESH and if we are unable to conclusively demonstrate the efficacy of our eSVS MESH through additional human trials, we may be unable to commercialize our eSVS MESH in the United States or other major markets or may experience significant delays in doing so, and our ability to generate revenue will be significantly delayed and our business will be harmed.
Our time and financial resources since our inception have largely been devoted to the development of our eSVS MESH. We have only completed one human clinical trial of 90 patients for our eSVS MESH, which was conducted outside of the United States. The safety data from this trial has indicated that our eSVS MESH and implant procedure do not result in an increase in patient complications during or in the 30 days after surgery. However, the effectiveness data from the trial is inconclusive primarily due to two complicating factors. First, one of the centers participating in the trial used implant methods incompatible with our eSVS MESH. Second, the amount of reduction in the diameter of the saphenous vein grafts, or downsizing, prescribed in our instructions for use and sizing tool was too aggressive, resulting in a higher than anticipated closure rate in saphenous vein grafts utilizing our eSVS MESH, particularly when our smallest device, 3.0 millimeters, was used. As a result, based on angiographic studies nine to 12 months following surgery, 49% (36 of 73) of the eSVS MESH vessels were patent or open and 81% (59 of 73) of the untreated vessels were patent. We are currently seeking FDA authorization to commence a trial in the U.S. involving a larger number of patients. In support of our request for such authorization, we have submitted to the FDA the results of our human clinical trial outside the U.S. as well as the results of our animal studies. The efficacy data from our trial outside the U.S. may cause the FDA to deny authorization for a larger human clinical trial in the U.S., in which case we would incur delays as we seek further efficacy data outside the U.S. If we are unable to demonstrate with human clinical data that our eSVS MESH is safe and improves the long term patency of saphenous vein grafts as compared to traditional CABG surgery, we will be unable to obtain
27
regulatory approval for, or successfully commercialize, our eSVS MESH. We have no other products ready for clinical testing or commercialization; therefore, our ability to remain in business would be doubtful if our eSVS MESH is not proven to be safe and effective.
If the data from our clinical trials is not adequate, we may not proceed with our planned filing of applications for regulatory approvals in the United States or other major markets, or we may be forced to delay these filings. Even if we file an application for approval with satisfactory clinical data, the FDA or foreign regulatory authorities may not accept our filing, or may request additional information, including data from additional clinical trials. Delays in collecting or analyzing our clinical trial data could result in delays in filing regulatory applications with the FDA or other regulatory authorities. The FDA or foreign regulatory authorities may also approve our eSVS MESH for very limited purposes with many restrictions on its use or in limited sizes, may delay approvals, or ultimately may not grant marketing approval for our eSVS MESH. Although we have obtained CE Mark approval in Europe and even if we do receive FDA or additional foreign regulatory approval, we may be unable to successfully commercialize our eSVS MESH in Europe, the United States, or other major markets, and our ability to generate revenue will be significantly impaired.
Our success depends on the coronary bypass graft market and the superior outcomes of coronary bypass surgery over competitive procedures, and such superior outcomes may not continue.
Physicians treat coronary artery disease with methods other than CABG procedures, including interventional techniques such as balloon angioplasty with or without the use of stents, pharmaceuticals, atherectomy catheters, and lasers. Several of these alternative treatments are widely accepted in the medical community and have a long history of use. In addition, technological advances may result in improvements in these alternative treatments or new therapies that produce superior treatment outcomes as compared to CABG surgery. The medical device industry is highly competitive and subject to rapid and profound technological change. Our success depends, in part, upon physicians continuing to perform a significant number of CABG procedures and our ability to achieve and maintain a competitive position in the development of technologies and products in the coronary artery bypass field. If physicians, patients, or hospitals opt to use our competitors' products, our commercial opportunity will be reduced and our potential revenues will suffer.
The market acceptance of new medical technologies is uncertain, and we may be unable to obtain market acceptance of our eSVS MESH.
Even if our clinical trials demonstrate that the use of our eSVS MESH provides equivalent or more effective results as compared to coronary bypass operations using only the unsupported saphenous vein grafts and if all regulatory approvals are obtained, the success of our eSVS MESH will depend upon the acceptance by cardiovascular and cardiothoracic surgeons of our eSVS MESH as equivalent or better than the current procedure using unsupported saphenous veins and other available treatments. We believe that physicians' recommendations will be essential for the development and successful marketing of our eSVS MESH, and physicians will not begin to use our eSVS MESH unless they determine that it is a safe and effective alternative to current treatment methods. The degree of physician and market acceptance of our eSVS MESH will depend on a number of factors, including:
- •
- the perceived effectiveness of our eSVS MESH relative to its cost;
- •
- the prevalence and severity of any side effects;
- •
- potential advantages over alternative treatments;
- •
- publication in peer-reviewed medical journals of data regarding the successful use and longer term clinical
benefits of our eSVS MESH;
- •
- development of new products and technologies by our competitors or new alternative treatments;
28
- •
- regulatory developments related to manufacturing, marketing and selling our eSVS MESH both within and outside the United
States;
- •
- perceived liability risks arising out of the use of new products;
- •
- the willingness of physicians to adopt new technologies and the ability of physicians to acquire the skills necessary to
use our eSVS MESH;
- •
- the effectiveness of our sales and marketing efforts; and
- •
- the adequacy of third-party coverage or reimbursement.
If our eSVS MESH does not achieve an adequate level of acceptance by physicians, healthcare payors, and patients, we may not generate meaningful revenue and we may not become profitable. In addition, we have not yet determined pricing for our eSVS MESH and our pricing policies could adversely impact market acceptance of our eSVS MESH as compared to competing products and treatments. Any of the foregoing factors, or other factors, could limit or detract from market acceptance of our eSVS MESH. If our eSVS MESH is not accepted by the market, our business would be harmed.
We will be subject to intense competition and the risk of obsolescence if our competitors develop products superior to our eSVS MESH.
We face competition from established medical technology, pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies, and private and public research institutions in the United States and abroad. The industry in which we operate has undergone, and is expected to continue to undergo, rapid and significant technological change, and we expect competition to intensify as technical advances are made. Our competitors may develop and commercialize medical device or pharmaceutical products that are safer or more effective, have fewer side effects or are less expensive than coronary artery bypass surgery. For example, we are aware of companies that are developing various other less invasive technologies for treating cardiovascular disease, which could make our technology obsolete. We also compete in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
Furthermore, companies with significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we have may be working on products similar to our eSVS MESH. Our eSVS MESH may not replace current surgical techniques and other products or techniques may render our eSVS MESH obsolete. In addition, our distributors will also face competition from established companies with significantly greater financial and marketing resources. Our competitors may produce more advanced products than ours or demonstrate superior safety of their products. Our ability to effectively compete depends on our ability to innovate successfully. There are few barriers that would prevent new or existing competitors from developing products that compete directly with ours. Demand for our eSVS MESH could be diminished by equivalent or superior products and technologies offered by competitors.
Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with, or mergers with or acquisitions by, large and established companies or through the development of novel products and technologies.
Our competitive position also depends on:
- •
- obtaining any necessary United States or foreign marketing approvals;
29
- •
- widespread awareness, acceptance and adoption by the cardiovascular and cardiothoracic markets of our eSVS MESH;
- •
- product coverage and reimbursement from third-party payors, insurance companies and others;
- •
- published studies supporting the effectiveness and safety and long-term clinical benefit of our eSVS MESH;
- •
- properly identifying customer needs and delivering new products or product enhancements to address those needs;
- •
- limiting the time required from proof of feasibility to routine production;
- •
- limiting the timing and cost of regulatory approvals;
- •
- our ability to attract and retain qualified personnel;
- •
- the extent of our patent protection or our ability to otherwise develop proprietary products and processes;
- •
- our ability to maintain adequate manufacturing capacity and to source the materials and equipment required to manufacture
our eSVS MESH; and
- •
- securing sufficient capital resources to expand our research and development, sales and marketing efforts, and manufacturing capacity.
If our eSVS MESH is not competitive based on these or other factors, our business would be harmed.
We have limited manufacturing resources and experience, and if our manufacturing facilities are unable to provide an adequate supply of our eSVS MESH, our growth could be limited and our business could be harmed.
We have limited experience in manufacturing our eSVS MESH and rely on outside vendors for several materials and processes. We currently manufacture our eSVS MESH for our clinical trials, research and development purposes and commercialization at our manufacturing facility in Minnesota. If our existing manufacturing facility experiences a disruption, we would have no other means of manufacturing our eSVS MESH until we are able to restore the manufacturing capability at our current facility or develop alternative manufacturing facilities.
If we are unable to produce sufficient quantities of our eSVS MESH for use in our current and planned clinical trials or for commercialization, or if our manufacturing process yields a substandard product, our regulatory, development and commercialization efforts would be delayed.
In order to produce our eSVS MESH in the quantities that will be required for commercialization, we will have to increase, or "scale up," the production process over the current level of production. Manufacturers often encounter difficulties in scaling up production, including problems involving yields, controlling and anticipating costs, quality control and assurance, supply and shortages of qualified personnel. If the scaled-up production process is not efficient or produces a product that does not meet quality and other standards, we may be unable to meet market demand and our revenues, business and financial prospects would be adversely affected. The contract vendors with which we are and will be developing relationships may not have the ability to produce the quantities of the materials needed for human clinical trials or commercial sales or may not do so at prices that allow our eSVS MESH to compete successfully in the market.
Additionally, any damage to or destruction of our facilities or our equipment, prolonged power outage or contamination at our facilities would significantly impair our ability to produce our eSVS MESH.
30
We depend upon third-party suppliers, making us vulnerable to supply problems and price fluctuations.
We rely on third-party suppliers to provide us certain components of our eSVS MESH. We depend on these suppliers to provide us with materials in a timely manner that meet our quality, quantity and cost requirements. These suppliers may encounter problems during manufacturing for a variety of reasons, including unanticipated demand from larger customers, failure to follow specific protocols and procedures, failure to comply with applicable regulations, equipment malfunction, quality or yield problems, and environmental factors, any of which could delay or impede their ability to meet our demand. Our reliance on these outside suppliers also subjects us to other risks that could harm our business, including:
- •
- interruption of supply resulting from modifications to, or discontinuation of, a supplier's operations;
- •
- delays in product shipments resulting from defects, reliability issues or changes in components from suppliers;
- •
- price fluctuations due to a lack of long-term supply arrangements for key components with our suppliers;
- •
- errors in manufacturing components, which could negatively impact the effectiveness or safety of our eSVS MESH or cause
delays in shipment of our eSVS MESH;
- •
- discontinued production of components, which could significantly delay our production and sales and impair operating
margins;
- •
- inability to obtain adequate supplies in a timely manner or on commercially acceptable terms;
- •
- difficulty locating and qualifying alternative suppliers for our sole-source supplies;
- •
- delays in production and sales caused by switching components, which may require product redesign and new regulatory
submissions;
- •
- delays due to evaluation and testing of products from alternative suppliers and corresponding regulatory qualifications;
- •
- non-timely delivery of components due to our suppliers manufacturing products for a range of customers; and
- •
- inability of suppliers to fulfill orders and meet requirements because of supplier financial hardships.
Other than existing, unfulfilled purchase orders, our suppliers have no contractual obligations to supply us with, and we are not contractually obligated to purchase from them, any of our supplies. Any supply interruption from our suppliers or failure to obtain additional suppliers for any of the components used in our eSVS MESH would limit our ability to manufacture our eSVS MESH and could have a material adverse effect on our business, financial condition and results of operations. We have no reason to believe that any of our current suppliers could not be replaced if they were unable to deliver components to us in a timely manner or at an acceptable price and level of quality. However, if we lost one of these suppliers and were unable to obtain an alternate source on a timely basis or on terms acceptable to us, our production schedules could be delayed, our margins could be negatively impacted, and we could fail to meet our customers' demand. Our customers will rely upon our ability to meet committed delivery dates and any disruption in the supply of key components would adversely affect our ability to meet these dates and could result in legal action by our customers, cause us to lose customers or harm our ability to attract new customers, any of which could decrease our revenue and negatively impact our growth. In addition, to the extent that our suppliers use technology or
31
manufacturing processes that are proprietary, we may be unable to obtain comparable materials or components from alternative sources.
Manufacturing operations are often faced with a supplier's decision to discontinue manufacturing a component, which may force us to make last time purchases, qualify a substitute part, or make a design change which may divert engineering time away from the development of new products.
Quality issues in our manufacturing processes could delay our clinical trials and our commercialization.
Even if we are able to contract with manufacturers for key materials or supplies, we may experience future manufacturing difficulties. Any difficulties in locating and hiring material manufacturers or in the ability of manufacturers to supply materials at the times and in the quantities we need, and at prices that allow us to compete, could have a material adverse effect on our business.
The production of our eSVS MESH must occur in a highly controlled, clean environment to minimize particles and other yield- and quality-limiting contaminants. In spite of stringent quality controls, weaknesses in process control or minute impurities in materials may cause a substantial percentage of defective products in a lot. In addition, we must meet certain lot release specifications before our eSVS MESH can be shipped to our clinical trial sites or to commercial markets. If a particular lot fails to meet lot release specifications, we will not be able to ship that lot to our clinical trial sites or to commercial markets. If we are not able to maintain stringent quality controls, if contamination problems arise or if we are not able to meet our lot release specifications, our clinical trials or sales efforts could be delayed, which would harm our business and our results of operations.
Our business is subject to risks relating to operating internationally.
As part of our product development and regulatory strategy, we intend to market our eSVS MESH internationally. There are a number of risks associated with conducting business internationally, including:
- •
- potential differences in treatment protocols and methods across the markets in which we expect to market our eSVS MESH;
- •
- potential differences in reimbursement levels and the requirements necessary to obtain such reimbursement;
- •
- general economic and political conditions in the markets in which we operate;
- •
- potential international conflicts, including terrorist acts;
- •
- potential increased costs associated with overlapping tax structures;
- •
- potential trade restrictions, exchange controls and legal restrictions on the repatriation of funds into the United
States;
- •
- difficulties and costs associated with staffing and managing foreign operations, including risks of violations of local
laws or the U.S. Foreign Corrupt Practices Act by employees overseas or the OECD Convention on Combating Bribery of Foreign Public Officials in International Business Transactions;
- •
- unexpected changes in regulatory requirements;
- •
- the difficulties of compliance with a wide variety of foreign laws and regulations;
- •
- unfavorable regulations in foreign jurisdictions regarding distributors;
- •
- the deferral of revenue recognition;
- •
- longer accounts receivable cycles in certain foreign countries; and
- •
- import and export licensing requirements.
32
Any of these risks could adversely affect our international operations or financial results, which would harm our business.
We could become subject to product liability claims, product recalls, other field actions and warranty claims that could be expensive, divert management's attention, and harm our business.
We face an inherent risk of exposure to product liability claims in the event that the use of our eSVS MESH results or is alleged to have resulted in adverse effects to a patient. In many jurisdictions, producers of medical products are strictly liable for personal injuries caused by medical devices. A product liability claim against us, even if we are ultimately successful in defending it, could have a material adverse effect on our business, results of operations and reputation.
We may be held liable if our eSVS MESH causes injury or death or is found otherwise unsuitable during usage. Because our eSVS MESH is designed to be used in complex surgical procedures, defects could result in a number of complications, including serious injury or death. It is also possible that defects in the design, manufacture or labeling of our eSVS MESH might necessitate a product recall or other field corrective action, which may result in warranty claims beyond our expectations and may harm our reputation. We believe potential disadvantages associated with our eSVS MESH include the possibility of allergic reaction to the implant materials and the possibility of damage to the saphenous vein during placement of our eSVS MESH. A product liability claim, regardless of its merit or eventual outcome, could result in significant legal defense costs. The coverage limits of our insurance policies may not be adequate to cover future claims. We may be unable to maintain product liability insurance in the future at satisfactory rates or with adequate amounts. A product liability claim, any product recalls or other field actions or excessive warranty claims, whether arising from defects in design or manufacture or otherwise, could divert management's attention from our core business, be expensive to defend and result in sizable damage awards against us, any of which could harm our reputation and business.
If third-party payors do not provide sufficient coverage or reimbursement to healthcare providers for the use of our eSVS MESH, our acceptance in the marketplace would be harmed.
The availability of insurance coverage and reimbursement for newly approved medical devices and procedures is uncertain. Our success depends upon the use of our eSVS MESH and whether third-party insurance coverage and reimbursement for the use of this product is available.
Our success in international markets depends upon the eligibility of reimbursement for our eSVS MESH through government-sponsored healthcare payment systems and third-party payors. Reimbursement and healthcare payment systems in international markets vary significantly by country and, within some countries, by region. In many international markets, payment systems may control reimbursement for procedures performed using new products as well as procurement of these products. As an example, we were recently denied government-sponsored reimbursement in Germany for our eSVS MESH for calendar year 2011. In addition, as economies of emerging markets develop, these countries may implement changes in their healthcare delivery and payment systems. Furthermore, healthcare cost containment efforts similar to those underway in the United States are prevalent in many of the other countries in which we intend to sell our eSVS MESH and these efforts are expected to continue. Market acceptance of our eSVS MESH in a particular country may depend on the availability and level of reimbursement in that country. In the event that our customers are unable to obtain adequate reimbursement for our eSVS MESH in international markets in which we are seeking to sell our eSVS MESH, market acceptance of our eSVS MESH would be adversely affected.
In the United States, our eSVS MESH would be purchased primarily by medical institutions, which would then bill various third-party payors, such as the Centers for Medicare & Medicaid Services, or CMS, which administer the Medicare program, and other government programs and private insurance
33
plans, for the healthcare services provided to their patients. The process involved in applying for coverage and incremental reimbursement from CMS is lengthy and expensive. Even if our eSVS MESH receives FDA and other regulatory approval, it may not be granted coverage and reimbursement in the foreseeable future, if at all. Moreover, many private payors look to CMS in setting their reimbursement policies and amounts. If CMS or other agencies limit coverage or decrease or limit reimbursement payments for doctors and hospitals, this may affect coverage and reimbursement determinations by many private payors.
CMS may not provide coverage and reimbursement for our eSVS MESH. If a medical device does not receive incremental reimbursement from CMS, then a medical institution would have to absorb the cost of our eSVS MESH as part of the cost of the procedure in which the products are used. Acute care hospitals are now generally reimbursed by CMS for inpatient operating costs under a Medicare hospital inpatient prospective payment system. Under the Medicare hospital inpatient prospective payment system, acute care hospitals receive a fixed payment amount for each covered hospitalized patient based upon the Diagnosis-Related Group, or DRG, to which the inpatient stay is assigned, regardless of the actual cost of the services provided. At this time, we do not know the extent to which medical institutions would consider insurers' payment levels adequate to cover the cost of our eSVS MESH. Failure by hospitals and physicians to receive an amount that they consider to be adequate reimbursement for procedures in which our eSVS MESH is used could deter them from purchasing our eSVS MESH and limit our revenue growth. In addition, pre-determined DRG payments may decline over time, which could deter medical institutions from purchasing our eSVS MESH. If medical institutions are unable to justify the costs of our eSVS MESH, they may refuse to purchase it, which would significantly harm our business.
We may not be able to attract and retain the technical, regulatory, and sales personnel necessary for our success, which may divert management's attention and negatively impact our operations.
We are highly dependent on our senior management, specifically Manny Villafaña, our Chairman and Chief Executive Officer. The loss of services of this individual would impair our ability to commercialize our eSVS MESH and develop new products and would harm our business. Our success will depend on our ability to retain our senior management and to attract and retain qualified personnel in the future. Competition for senior management personnel, as well as clinical and regulatory specialists, engineers and sales personnel is intense and we may not be able to retain our personnel. The loss of a member of our senior management or our professional staff would require the remaining senior executive officers to divert immediate and substantial attention to seeking a replacement. Each of our senior officers may terminate his employment at any time without notice and without cause or good reason. We do not carry key person life insurance on any of our employees. If we lose the services of any key personnel, our business, financial condition and results of operations may suffer.
We will need to increase the size of our organization and we may experience difficulties managing growth. If we are unable to manage the anticipated growth of our business, our future revenue and operating results may be adversely affected.
We expect to expand our sales support and marketing staff, and administrative and financial resources to meet anticipated growth in demand for our eSVS MESH. We may face difficulties in recruiting, training, managing and retaining an adequate number of qualified personnel to support this growth. Expansion in personnel may mean that less experienced people could be manufacturing and providing clinical and sales and marketing support for our eSVS MESH, and managing our administrative and financial functions, which could result in unanticipated costs and disruptions to our operations. If we cannot scale and manage our business appropriately, our anticipated growth may be impaired and our financial results will suffer.
34
Becoming a public company has caused us to incur increased costs and demands on our management and divert management's attention from our core business.
The obligations of being a public company, including substantial public reporting and auditing obligations, requires significant additional expenditures, places additional demands on our management and diverts management's time and attention away from our core business. These additional obligations may require us to hire additional personnel in order to ensure compliance with the regulatory requirements of the Securities and Exchange Commission and the NASDAQ Global Market. We are required to report on our internal controls, as required by Section 404 of the Sarbanes-Oxley Act. Our management may not be able to effectively and timely implement controls and procedures that adequately respond to the increased regulatory compliance and reporting requirements that are now applicable to us as a public company. If we fail to staff our accounting and finance function adequately or maintain internal controls adequate to meet the demands that will be placed upon us as a public company, including the requirements of the Sarbanes-Oxley Act, we may be unable to report our financial results accurately or in a timely manner and our business and stock price may suffer. The costs of being a public company, as well as diversion of management's time and attention, may harm our business, financial condition and results of operations.
Risks Related to Our Intellectual Property
If we are unable to protect our intellectual property rights, our ability to compete will be harmed.
As of March 1, 2012, we had six issued patents covering the eSVS MESH: two issued in the U.S. and one each issued in Japan, Europe, Canada and South Africa. The European patent has been validated and is enforceable in six European countries and we are presently engaged in the grant phase to validate this patent in two additional European countries. In addition, we have five patent applications pending in the U.S, including one for which were received a notice of allowance in February 2012, and eight patent applications pending in countries outside the U.S. covering various aspects of our eSVS MESH.
Any patents we have or obtain in the future might be invalidated or circumvented by third parties. If any challenges are successful, competitors might be able to market products and use manufacturing processes that are substantially similar to ours. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by consultants, vendors or former or current employees, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized use and disclosure of our intellectual property is difficult, and we do not know whether the steps we have taken to protect our intellectual property will be adequate. In addition, the laws of many foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. If our intellectual property is not adequately protected against competitors' products and methods, our competitive position could be adversely affected, as could our business.
Our commercial success, if any, will be significantly harmed if we infringe the patent rights of third parties or if we breach any agreements that we have entered into with regard to our technology or business. The medical device industry is characterized by frequent and extensive litigation regarding patents and other intellectual property rights. Many medical device companies with substantially greater resources than us have employed intellectual property litigation as a way to gain a competitive advantage. We may become involved in litigation, interference proceedings, oppositions, reexamination, protest or other potentially adverse intellectual property proceedings as a result of alleged infringement by us of the rights of others or as a result of priority of invention disputes with third parties. Third parties may also challenge the validity of any of our issued patents. Similarly, we may initiate proceedings to enforce our patent rights and prevent others from infringing our intellectual property rights. In any of these circumstances, we might have to spend significant amounts of money, time and
35
effort defending our position and we may not be successful. In addition, any claims relating to the infringement of third-party proprietary rights or proprietary determinations, even if not meritorious, could result in costly litigation, lengthy governmental proceedings, divert management's attention and resources, or require us to enter into royalty or license agreements that are not advantageous to us. We may not have sufficient resources to enforce our intellectual property rights or to defend our patents against a challenge.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We require our employees to execute appropriate confidentiality and assignment-of-inventions agreements with us. These agreements typically provide that all materials and confidential information developed or made known to the individual during the course of the individual's relationship with us be kept confidential and not disclosed to third parties except in specific circumstances and that all inventions arising out of the individual's relationship with us will be our exclusive property. Additionally, we seek to have our consultants and advisors execute similar confidentiality and assignment-of-inventions agreements with us, but in some instances these agreements have not included assignment-of-invention provisions. These agreements may be breached, and in some instances, we may not have an appropriate remedy available for breach of the agreements. Furthermore, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology.
Claims of infringement or misappropriation of the intellectual property rights of others could prohibit us from commercializing our eSVS MESH and harm our business.
The medical device industry has been characterized by extensive litigation regarding patents and other intellectual property rights, and companies in the industry have used intellectual property litigation to gain a competitive advantage. We may become a party to patent infringement claims and litigation or interference proceedings declared by the U.S. Patent and Trademark Office to determine the priority of inventions. The defense and prosecution of these matters are both costly and time consuming.
Additionally, we may need to commence proceedings against others to enforce our patents, to protect our trade secrets or know-how or to determine the enforceability, scope and validity of the proprietary rights of others. These proceedings would result in substantial expense to us and significant diversion of effort by our technical and management personnel.
We are aware of patents issued to third parties that contain subject matter related to our technology. These or other third parties may assert that our eSVS MESH infringes the claims in their patents or seek to expand their patent claims to cover aspects of our eSVS MESH. An adverse determination in litigation or interference proceedings to which we may become a party could subject us to significant liabilities or require us to seek licenses. In addition, if we are found to willfully infringe third-party patents, we could be required to pay treble damages in addition to other penalties. Although patent and intellectual property disputes in the medical device area have often been settled through licensing or similar arrangements, costs associated with these arrangements may be substantial and could include ongoing royalties. We may be unable to obtain necessary licenses on satisfactory terms, if at all. If we do not obtain necessary licenses, we may be required to redesign our eSVS MESH to avoid infringement, and it may not be possible to do so effectively. Adverse determinations in a judicial or administrative proceeding or failure to obtain necessary licenses could cause us to incur significant costs, place significant strain on our resources, divert management's attention from our business and harm our reputation and prevent us from commercializing our eSVS MESH or any other product we may develop, which would have a significant adverse impact on our business. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources.
36
If the core intellectual property relating to our eSVS MESH reverts to Medtronic, our business will be adversely affected.
The core intellectual property relating to our eSVS MESH, including five patent applications pending in the United States and nine patent applications pending in countries outside the United States, was sold to us by Medtronic pursuant to an Assignment and License Agreement dated October 9, 2007. Any or all licenses granted to us pursuant to our agreement with Medtronic may be terminated and potentially all of the core intellectual property and patent rights related to our eSVS MESH will revert to Medtronic, upon notice by Medtronic, if we become insolvent, make an assignment for the benefit of creditors, go into liquidation or receivership or otherwise lose legal control of our business. Medtronic may also cause the core intellectual property and patent rights related to our eSVS MESH to revert to Medtronic if we determine to cease commercializing our eSVS MESH. A dispute over the circumstances under which the core intellectual property rights revert to Medtronic could be expensive and time consuming, and any reversion of these rights to Medtronic would adversely affect our business.
Risks Related to Regulatory Approval and Other Government Regulations
We will be subject to government regulation, and we may not receive approval for our eSVS MESH in the United States on a timely basis, if at all.
Our eSVS MESH, product development activities and manufacturing processes are, and will continue to be, subject to extensive and rigorous scrutiny and regulation by the FDA and by comparable agencies in foreign countries. In the United States, the FDA regulates the introduction of medical devices, as well as manufacturing, labeling and record keeping procedures for such products. The process of obtaining marketing clearance or approval for new medical products from the FDA is costly and time consuming, and there can be no assurance that such approval will be granted for our eSVS MESH on a timely basis, if at all, or that the FDA review will not involve delays that would adversely affect our ability to commercialize our eSVS MESH. Even if regulatory clearance or approval to market a product is obtained from the FDA, this clearance or approval may entail limitations on the indicated uses of the product. Marketing clearance or approval can also be withdrawn by the FDA due to failure to comply with regulatory standards or the occurrence of unforeseen problems following initial clearance.
The FDA will require us to file a Pre-Market Approval, or PMA, application with regard to our eSVS MESH, and there is no assurance whatsoever that approval will be obtained. Even if approval is obtained, the process of obtaining a PMA is expensive, uncertain and lengthy, frequently requiring several years from the date of submission. Changing FDA policies and requirements for PMA products may add additional uncertainty. Significant delay or failure to obtain FDA approval to market our eSVS MESH would harm our business.
The FDA may not approve our investigational device exemption application for our eSVS MESH, which would prevent us from conducting our clinical trials in the United States, and even if the FDA does grant such approval, our clinical trials may be more costly and burdensome than we currently anticipate, which would limit or delay our ability to complete clinical trials and ultimately market our eSVS MESH in the United States.
The U.S Food and Drug Administration ("FDA"), has reviewed and disapproved our most recent amendment to our application for an investigational device exemption ("IDE") in March 2011. At that time the FDA indicated that they intended to review our IDE information with outside experts before they provide further guidance to the Company. Due to internal delays, the FDA did not begin this review until August 2011. In September 2011 the FDA advised us that we had not provided sufficient data to support our request for an IDE for our eSVS MESH. Therefore, we intend to conduct a new
37
feasibility trial in Europe. Upon completion of this study, we expect to file an application for an IDE approval of a pivotal study in the U.S.
If the FDA approves our IDE application, the clinical trials we conduct may have unanticipated complications and delays and may be more costly than we currently anticipate. The FDA may approve our IDE application with conditions relating to the scope or design of our clinical trials for which we have not planned. These conditions may require us to collect additional data, enroll more patients, spend more time and expend more resources than we currently anticipate, and these conditions may make a clinical trial in the United States more costly and time consuming than we currently plan. Any unanticipated costs and length of U.S. clinical trials would delay our ability to market our eSVS MESH in the United States, which would harm our business.
If the FDA does not approve our IDE application, we would be unable to conduct clinical trials of our eSVS MESH in the United States. If our IDE application is not approved and we are unable to conduct U.S. clinical trials, we would not be able to submit a PMA application and we would be unable to market our eSVS MESH in the United States, which would have an adverse effect on our business.
Even if our eSVS MESH is approved by regulatory authorities, if we fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our eSVS MESH, it could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continual review and periodic inspections by the FDA and other regulatory bodies. Even if regulatory approval of our eSVS MESH is granted in the United States, the approval may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for costly post-marketing testing and surveillance to monitor the safety or effectiveness of the product. Later discovery of previously unknown problems with our eSVS MESH, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturer or manufacturing processes, or failure to comply with regulatory requirements, may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recall, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
We will be highly dependent on third-party institutions to conduct our clinical testing, and the results of such testing may delay or prevent regulatory approval of our eSVS MESH.
We rely on clinical investigators and clinical trial sites to enroll patients in our clinical trials and other third parties to manage our trials and to perform related data collection and analysis. However, we are not able to control the amount and timing of resources that clinical trial sites devote to our clinical trials. If these clinical investigators and clinical trial sites fail to enroll a sufficient number of patients in our clinical trials or fail to ensure compliance by patients with clinical protocols, we will be unable to complete our planned trials, which could prevent us from obtaining regulatory approvals for our eSVS MESH. Our agreements with clinical investigators and clinical trial sites for clinical testing place substantial responsibilities on these parties and, if these parties fail to perform as expected, our planned trials could be delayed or terminated. If these clinical investigators, clinical trial sites, or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, the FDA's good clinical practice regulations or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our eSVS MESH.
38
In addition, the data obtained from human clinical testing is subject to varying interpretations that could delay, limit or prevent regulatory approval, and delays or rejection may be encountered based upon changes in FDA policy for device approval during the period of development.
Our facilities will be subject to inspection by the FDA and international authorities, and we could face penalties if we are found to be non-compliant with the regulations of the FDA or international authorities.
The FDA and various other authorities will inspect our facilities from time to time to determine whether we are in compliance with regulations relating to medical device manufacturing, including regulations concerning design, manufacturing, testing, quality control, product labeling, distribution, promotion, and record keeping practices. A determination that we are in material violation of such regulations could lead to the imposition of civil penalties, including fines, product recalls, product seizures or, in extreme cases, criminal sanctions. Even if regulatory approvals to market a product are obtained from the FDA, such approvals may contain limitations on the indicated uses of our eSVS MESH. The FDA could also limit or prevent the manufacture or distribution of our eSVS MESH and has the power to require the recall of products. FDA regulations depend heavily on administrative interpretation, and there can be no assurance that the future interpretations made by the FDA or other regulatory bodies with possible retroactive effect will not adversely affect us.
Our promotional and marketing activities will be subject to regulation by the FDA and international authorities, and we could face severe penalties if we are found to be promoting our eSVS MESH for an unapproved use.
If the FDA or international authorities determine that our promotional materials or activities constitute promotion of our eSVS MESH for an unapproved use, it could demand that we cease the use of or modify our promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider promotional or other materials to constitute promotion of our eSVS MESH for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
Our success will also be dependent on complying with foreign regulatory requirements, and our inability to do so could result in sales of our eSVS MESH being restricted internationally.
Our revenues will initially be dependent upon sales of our eSVS MESH outside the United States. Foreign regulatory bodies have established varying regulations governing product standards, packaging requirements, labeling requirements, import restrictions, tariff regulations, duties and tax requirements. We will rely heavily upon independent foreign distributors to comply with such foreign regulatory requirements. Our inability or failure or the inability or failure of such foreign distributors to comply with varying foreign regulation or the imposition of new regulations could restrict the sale of our eSVS MESH internationally and thereby harm our business.
Legislation may negatively affect coverage and reimbursement levels for our eSVS MESH.
Even if third-party payors provide adequate coverage and reimbursement for our eSVS MESH, adverse changes in third-party payors' general policies toward reimbursement could preclude market acceptance for our eSVS MESH and harm our potential sales and revenue growth, which in turn would harm our business. Recently, healthcare reform legislation was signed into law in the United States and we expect that there will continue to be legislative proposals for governmental controls over healthcare in the United States and other countries. Some third-party payors also require pre-approval of coverage or companies to demonstrate the superiority of their product before they will reimburse healthcare providers who use such devices or procedures.
39
The trend toward managed healthcare in the United States and other countries and legislation intended to reduce the cost of government insurance programs will significantly influence the purchase of healthcare services and products, and could result in lower or no reimbursement for our eSVS MESH. It is uncertain whether our eSVS MESH will be viewed as sufficiently cost-effective to warrant adequate coverage and reimbursement levels.
Our operations involve hazardous materials, and we must comply with environmental laws and regulations.
We are subject to a variety of state and local regulations relating to the use, handling, storage, disposal and human exposure to hazardous and toxic materials. We currently generate small quantities of waste alcohol and acids, classifying us as a Very Small Quantity Generator with the Minnesota Pollution Control Agency, which requires us to comply with county and state registration requirements. We expect that compliance costs will be less than $5,000 for 2012. However, environmental laws could become more stringent over time, and we may increase the use of hazardous materials in our operations in the future, which could impose greater compliance costs on us and increase the risks and penalties associated with violations, any of which could harm our business. Compliance with future environmental and safety laws and regulations could restrict our ability to expand our facilities, impair our research, development or production efforts, or require us to incur other significant expenses. We could incur costs, fines and civil and criminal sanctions, third-party property damage or personal injury claims, or could be required to incur substantial investigation or remediation costs, if we were to violate or become liable under environmental laws. There can be no assurance that violations of environmental laws or regulations will not occur in the future as a result of the inability to obtain permits, human error, accident, equipment failure or other causes.
Risks Related to Our Common Stock
Prior to our initial public offering, there had not been a public market for our common stock and our stock price may be volatile and decrease significantly in the future.
Prior to our initial public offering in February 2011, there had not been a public market for our common stock. We cannot predict the extent to which investors' interests will lead to an active trading market for our common stock or whether the market price of our common stock will be volatile in the future. If an active trading market does not develop or is not sustained, investors may have difficulty selling any of our common stock. In addition to the risk factors discussed elsewhere in this section, the following factors, most of which are outside of our control, could cause the market price of our common stock to decrease significantly in the future:
- •
- inconclusive or failed clinical trial outcomes of our eSVS MESH;
- •
- failure to achieve market acceptance for our eSVS MESH;
- •
- inability to manufacture our eSVS MESH in adequate quantities or to commercial standards;
- •
- departure of key personnel;
- •
- inability to hire, train and retain qualified personnel to support our growth;
- •
- variations in our quarterly operating results or those of companies that are perceived to be similar to us;
- •
- announcements by our competitors of significant technological developments;
- •
- changes in governmental regulations and standards;
- •
- litigation related to patent infringement and product liability claims;
- •
- changes to financial estimates by equity research analysts;
40
- •
- sales of common stock or other securities by us in the future;
- •
- decreases in market valuations of similar companies; and
- •
- fluctuations in stock market prices and volumes.
Each of these factors could cause the market price of our stock to decline significantly in the future.
In addition, the stock markets have been extremely volatile. Securities class action litigation is often initiated against a company following a period of volatility in the market price of the company's securities. If class action litigation is initiated against us, we would incur substantial costs and our management's attention would be diverted from our operations.
If equity research analysts do not publish research or reports about our business or if they issue unfavorable research or downgrade our common stock, the price of our common stock could decline.
The trading market for our common stock will rely in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts. Equity research analysts may elect not to provide research coverage of our common stock, which may adversely affect the market price of our common stock. If equity research analysts do provide research coverage of our common stock, the price of our common stock could decline if one or more of these analysts downgrade our common stock or if they issue other unfavorable commentary about us or our business. If one or more of these analysts ceases coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline.
Future sales of our common stock by our existing stockholders could cause our stock price to decline and cause you to lose part or all of your investment.
If our stockholders sell substantial amounts of our common stock in the public market, the market price of our common stock could decrease significantly. The perception in the public market that our stockholders might sell shares of our common stock could also depress the market price of our common stock. We may file registration statements with the SEC covering (a) any shares of our common stock acquired upon option exercises prior to the closing of our initial public offering, (b) all of the shares subject to options outstanding, but not exercised, as of the closing of our initial public offering and (c) all of the shares available for future issuance under our stock incentive plan upon the closing of our initial public offering. The market price of shares of our common stock may decrease significantly when the restrictions on resale by our existing stockholders lapse and our stockholders, warrant holders and option holders are able to sell shares of our common stock into the market. A decline in the price of shares of our common stock might impede our ability to raise capital through the issuance of additional shares of our common stock or other equity securities, and our ability to grow our business in the future. See "Management's Discussion and Analysis of Financial Condition and Results of Operation—Cash and Cash Equivalents" below for more information.
Our directors, executive officers and significant stockholders have substantial control over us and could limit stockholders' ability to influence the outcome of key transactions, including changes of control.
As of March 1, 2012, our directors and executive officers and their affiliated entities beneficially own 34.9% of our outstanding common stock. In addition, Kips Bay Investments, LLC beneficially owns 41.7% and Mr. Villafaña, our Chairman and Chief Executive Officer, beneficially owns 33.6% of our outstanding common stock, and together are able to control or influence significantly all matters requiring approval by our stockholders. Our directors, executive officers, significant stockholders and affiliated entities, if acting together, are able to control or influence significantly all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other significant corporate transactions. These stockholders may have interests that differ from other
41
stockholders, and they may vote in a way with which other stockholders disagree and that may be adverse to the interests of other stockholders. The concentration of ownership of our common stock may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company, and may affect the market price of our common stock. This concentration of ownership of our common stock may also have the effect of influencing the completion of a change in control that may not necessarily be in the best interests of all of our stockholders.
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable and our certificate of incorporation allows us to authorize and issue preferred stock with rights and preferences superior to our common stock without stockholder approval.
Provisions of our certificate of incorporation and amended and restated bylaws and applicable provisions of Delaware law may make it more difficult for or prevent a third party from acquiring control of us without the approval of our board of directors. These provisions:
- •
- set limitations on the removal of directors;
- •
- limit who may call a special meeting of stockholders;
- •
- establish advance notice requirements for nominations for election to our board of directors or for proposing matters that
can be acted upon at stockholder meetings;
- •
- do not permit cumulative voting in the election of our directors, which would otherwise permit less than a majority of
stockholders to elect directors;
- •
- prohibit stockholder action by written consent unless unanimous, thereby requiring all stockholder actions to be taken at
a meeting of our stockholders; and
- •
- provide our board of directors the ability to designate the terms of and issue a new series of preferred stock without stockholder approval.
In addition, Section 203 of the Delaware General Corporation Law generally limits our ability to engage in any business combination with certain persons who own 15% or more of our outstanding voting stock or any of our associates or affiliates who at any time in the past three years have owned 15% or more of our outstanding voting stock. These provisions may have the effect of entrenching our management team and may deprive stockholders of the opportunity to sell their shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
We do not intend to declare dividends on our common stock and stockholders should not expect to receive dividends on their common stock in the foreseeable future.
We currently intend to retain all future earnings for the operation and expansion of our business and, therefore, do not anticipate declaring or paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends on our common stock will be at the discretion of our board of directors and will depend upon our results of operations, earnings, capital requirements, financial condition, future prospects, contractual restrictions and other factors deemed relevant by our board of directors. Therefore, our stockholders should not expect to receive dividend income from shares of our common stock.
42
We anticipate future losses and may require additional financing, and our failure to obtain additional financing when needed could force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect to incur losses for the foreseeable future, and we may require future financing in order to satisfy our capital requirements. In particular, we may require additional capital in order to continue to conduct the research and development and obtain regulatory clearances and approvals necessary to bring any future products to market and to establish effective marketing and sales capabilities for existing and future products. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available on a timely basis, we may terminate or delay the development of our eSVS MESH, or delay establishment of sales and marketing capabilities or other activities necessary to commercialize our eSVS MESH.
Our future capital requirements will depend on many factors, including:
- •
- the costs of expanding our distribution network and our manufacturing operations;
- •
- the degree of success we experience in commercializing our eSVS MESH;
- •
- the number and types of future products we develop and commercialize;
- •
- the costs, timing and outcomes of regulatory reviews associated with our future product candidates;
- •
- the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual
property-related claims; and
- •
- the extent and scope of our general and administrative expenses.
Raising additional capital may cause dilution to our stockholders or restrict our operations.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, stockholders' ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect their rights as stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. Any of these events could adversely affect our ability to achieve our product development and commercialization goals and harm our business. We do not anticipate any adverse effects stemming from the lack of available credit facilities at this time.
Item 1B. Unresolved Staff Comments
None.
We lease approximately 5,000 square feet of office, laboratory, manufacturing and warehouse space at 3405 Annapolis Lane North, Suite 200, Minneapolis, Minnesota. In June 2010, we executed an amendment to our lease that extended the lease term through September 30, 2011. In May 2011, we executed a second amendment to our lease that extended the lease term through September 30, 2014. Under the terms of the second amendment, our base rent was reduced to the current market rate starting October 1, 2011 and will subsequently be increased by approximately 2.5% on October 1, 2012 and 2013. Our corporate offices, research and development facilities, prototype development, manufacturing, warehousing, and shipping facilities are located at this facility.
On June 2, 2011, we entered into an operating sub-lease agreement with New Horizon Enterprises, Ltd. for approximately 2,800 square feet of office space adjoining our current facility. The term of this lease runs from June 15, 2011 through September 30, 2014. The monthly base rent amount is fixed over the entire term of the sub-lease.
43
We are not a party to any pending or threatened litigation.
Item 4. Mine Safety Disclosures
None.
Executive Officers of the Registrant
The name, age and position of each of our executive officers as of the date of this annual report are as follows:
Name
|
Age | Position | |||
|---|---|---|---|---|---|
Manny Villafaña |
71 | Chairman of the Board and Chief Executive Officer | |||
Scott Kellen |
46 | Chief Financial Officer, Vice President of Finance, and Secretary | |||
Randall LaBounty |
49 | Vice President of Regulatory and Clinical Affairs | |||
Michael Reinhardt |
53 | Vice President of Sales and Marketing | |||
Manny Villafaña is our founder, and has been our Chairman of the Board and Chief Executive Officer since our inception in 2007. Prior to founding us and since 1999, Mr. Villafaña founded and served as Chairman of the Board and Chief Executive Officer of CABG Medical, Inc., formed to develop an artificial coronary graft for use in bypass surgery. From 1987 to 2004, Mr. Villafaña founded and served as Chairman of the Board and Chief Executive Officer of ATS Medical, Inc., which developed open-pivot mechanical heart valves. ATS Medical was subsequently acquired by Medtronic, Inc. From 1976 to 1982, Mr. Villafaña founded and served as President and Chairman of the Board of St. Jude Medical, Inc. From 1972 to 1976, Mr. Villafaña founded and served as President and Chairman of the Board of Cardiac Pacemakers, Inc., or CPI, a cardiac rhythm management company. CPI was ultimately acquired by Eli Lilly and Company, which spun out CPI as Guidant Corporation. Guidant was, in turn, purchased by Boston Scientific Corporation.
Mr. Villafaña has received numerous awards and honors, including the "Living Legend of Medicine" award from the International Society of Cardio Thoracic Surgeons, the Ellis Island Medal of Honor, the Grand Prize Recipient—Mediterranean Institute of Cardiology, the Ernst & Young LLP National Master Entrepreneur of the Year, the Boys and Girls Club of America Hall of Fame, induction into the Minnesota Business Hall of Fame and in 2010, was inducted into the Minnesota Science and Technology Hall of Fame.
Scott Kellen joined us as Chief Financial Officer, Vice President of Finance, and Secretary in February 2010. From 2007 to 2009, Mr. Kellen served as Director of Finance from 2007 to 2009 and Chief Financial Officer from February 1, 2009 to May 1, 2009 for Transoma Medical, Inc., including during the preparation of its proposed initial public offering, which was withdrawn in February 2008 due to deteriorated market conditions. From 2005 to 2007, Mr. Kellen served as the Corporate Controller for ev3 Inc. during the company's initial public offering and during additional follow-on offerings. From 2003 to 2005, Mr. Kellen served as Senior Audit Manager of Deloitte & Touche, LLP (now Deloitte LLP), providing auditing and consulting services to mid-size public companies after the passage of the Sarbanes-Oxley Act. Mr. Kellen has spent more than 15 years in the medical device industry, serving early stage and growth companies that produced Class II and III devices. Mr. Kellen began his career with Deloitte & Touche in 1987.
Randall LaBounty joined us in May 2011 as our Vice President of Regulatory and Clinical Affairs. Mr. LaBounty has over 20 years of experience in medical device research with 16 years of experience in cardiovascular medical devices. Prior to joining Kips Bay Medical, Mr. LaBounty served as Vice President of Clinical Research at Osprey Medical, Inc., a company focused on preventing contrast-
44
induced chronic kidney diseases in patients, from 2010 to 2011 and Vice President of Clinical and Regulatory Affairs at Lumen Biomedical, Inc. from 2005 to 2010. He has also held director level positions at Atritech, Inc., ev3, Inc. and IntraTherapeutics, Inc. Mr. LaBounty has extensive experience with Class II and Class III medical products. He has been involved in all phases of the medical device approval process including the development of clinical and regulatory strategies, clinical trial execution, obtaining FDA IDEs, PMAs and 510(k) submissions. Internationally, he has successfully brought medical devices from initial clinical trials through to CE Mark approval. Randy has a Master of Science degree in Biostatistics and Bachelor of Science degree in Genetics and Cell Biology from the University of Minnesota.
Michael Reinhart joined us in May 2011 as our Vice President of Sales & Marketing. He has nearly 25 years of experience in the medical device industry. From 2009 to 2010, Mr. Reinhart served as Vice President of Global Marketing for ATS Medical Inc. based in Minneapolis, MN which was focused on cardiac surgery and engaged in the development and commercialization of prosthetic heart valves, valvular repair devices and cryoablation products. ATS Medical was purchased in August, 2010 by Medtronic, Inc. From 2007 to 2009, Mr. Reinhardt served as an independent consultant providing strategic and operational planning and execution services to the medical device industry. Prior to 2007, Mr. Reinhardt had held several senior management level positions at companies including; St Jude Medical's Cardiac Surgery Division; C.R. Bard's Electrophysiology Division; Boston Scientific's Scimed Division; and Ethicon Endosurgery. He also has considerable International experience having worked extensively in the European and Asian markets. He has a broad spectrum of experiences including working in the fields of general surgery, laparoscopic surgery, interventional cardiology, electrophysiology and cardiothoracic surgery. Mr. Reinhardt holds a B.A. in Marketing from Ohio University and a M.B.A. from Baruch College in New York, New York.
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock began trading on the NASDAQ Global Market on February 11, 2011 under the symbol "KIPS" in connection with our initial public offering.
As of March 1, 2012, there were 68 registered holders of our common stock.
Sale Price Information
The following table reflects high and low sales price information for our common stock for each fiscal quarter in fiscal year 2011, as reported on the NASDAQ Global Market.
(Per share amounts)
|
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2011 High |
$ | 8.37 | $ | 6.37 | $ | 3.53 | $ | 1.94 | |||||
2011 Low |
5.71 | 2.85 | 1.06 | 1.00 | |||||||||
Dividends
We have never paid cash dividends on any of our securities. We currently intend to retain any earnings for use in operations and do not anticipate paying cash dividends in the foreseeable future.
45
Recent Sales of Unregistered Equity Securities
On October 24, 2011, we entered into a common stock purchase agreement (the "Purchase Agreement") with Aspire Capital Fund, LLC ("Aspire Capital"), which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $20.0 million of our shares of common stock over the term of the Purchase Agreement. In consideration for entering into the Purchase Agreement, concurrently with the execution of the Purchase Agreement, we issued to Aspire Capital a total of 378,788 shares of our common stock (the "Commitment Shares"). The issuance of the Commitment Shares and all other shares of common stock that may be issued from time to time to Aspire Capital under the Purchase Agreement is exempt from registration under the Securities Act, pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. The resale by Aspire Capital of the Commitment Shares and all other shares of common stock that may be issued from time to time to Aspire Capital under the Purchase Agreement was registered on a registration statement on Form S-1 under the Securities Act of 1933 (File No. 333-178019), which registration statement was declared effective by the SEC on January 26, 2012.
Purchase of Equity Securities by the Company
None.
Use of Proceeds
We completed our initial public offering of shares of common stock (the "Offering") during the first quarter of our 2011 fiscal year. The effective date of our registration statement relating to the Offering, filed on Form S-1 under the Securities Act of 1933 (File No. 333-165940), was February 10, 2011. A total of 2,062,500 shares of our common stock were registered and sold in the Offering. In addition, we granted Rodman & Renshaw, LLC ("Rodman"), the underwriter of the Offering, and its designees a 45 day over-allotment option to purchase 309,375 shares at $8.00 per share, less an underwriting discount of $0.56 per share. This option expired unexercised. We also granted to Rodman a warrant to purchase 103,125 shares of our common stock which becomes exercisable at a price of $10.00 per share on February 10, 2012 and expires on February 10, 2016.
The aggregate offering price of our securities sold was $16.5 million. The aggregate underwriting discount for shares sold in the offering was $1.2 million, none of which was or will be paid to our affiliates. We incurred approximately $1.7 million of offering costs in connection with the Offering, including a non-accountable expense allowance to Rodman of $165,000. We received net proceeds from this Offering of approximately $13.6 million.
Through December 31, 2011, we have used $5.0 million of the net proceeds to fund the first milestone payment payable for the acquisition of certain intellectual property rights to our eSVS MESH. We intend to use the remaining net proceeds from the Offering to fund the process of seeking regulatory approval to market our eSVS MESH in the United States which includes human clinical trials; expand regulatory approval abroad; fund the development and testing of additional applications of our eSVS MESH; and for working capital and general corporate purposes, including commercialization activities for our eSVS MESH in select European and other international markets. Pending the uses described above, we have invested the remaining net proceeds in a variety of short-term, interest-bearing, investment grade securities.
Item 6. Selected Financial Data
The following tables summarize our selected financial data for the periods and as of the dates indicated. The selected financial data should be read in conjunction with, and are qualified by reference
46
to, our financial statements and related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this annual report. The historical results are not necessarily indicative of results to be expected in any future period.
| |
|
|
|
|
Period from May 1, 2007 (Date of Inception) to December 31, 2007 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Year Ended December 31, | |||||||||||||||
| |
2011 | 2010 | 2009 | 2008 | ||||||||||||
Statements of Operations Data: |
||||||||||||||||
Net sales |
$ |
252 |
$ |
223 |
$ |
— |
$ |
— |
$ |
— |
||||||
Cost of sales |
(91 |
) |
(77 |
) |
— |
— |
— |
|||||||||
Gross profit |
161 | 146 | — | — | — | |||||||||||
Operating expenses: |
||||||||||||||||
Research and development |
1,675 | 2,521 | 3,004 | 2,635 | 196 | |||||||||||
Selling, general and administrative |
2,755 | 1,326 | 779 | 754 | 381 | |||||||||||
Milestone expense |
— | 5,000 | — | — | — | |||||||||||
Operating loss |
(4,269 | ) | (8,701 | ) | (3,783 | ) | (3,389 | ) | (577 | ) | ||||||
Interest income |
19 | 12 | 17 | 52 | 65 | |||||||||||
Interest expense |
— | (1 | ) | (181 | ) | (390 | ) | (164 | ) | |||||||
Realized gain on sale of short-term investments |
— | 46 | — | — | — | |||||||||||
Impairment of available for sale securities |
— | — | — | (85 | ) | — | ||||||||||
Change in fair value of investor stock purchase option |
— | (2,290 | ) | 610 | — | — | ||||||||||
Net loss before income taxes |
(4,250 | ) | (10,934 | ) | (3,337 | ) | (3,812 | ) | (676 | ) | ||||||
Provision for income tax benefit |
— | 42 | — | — | — | |||||||||||
Net loss attributable to common stockholders |
$ | (4,250 | ) | $ | (10,892 | ) | $ | (3,337 | ) | $ | (3,812 | ) | $ | (676 | ) | |
Basic and diluted net loss per share |
$ | (0.27 | ) | $ | (0.81 | ) | $ | (0.30 | ) | $ | (0.62 | ) | $ | (0.16 | ) | |
Weighted average common shares outstanding—basic and diluted |
15,557,969 | 13,431,661 | 11,069,342 | 6,100,767 | 4,106,557 | |||||||||||
(In thousands, except share and per share amounts)
| |
As of December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||
Balance Sheet Data: |
||||||||||||||||
Cash, cash equivalents and short-term investments |
$ | 9,168 | $ | 3,784 | $ | 3,417 | $ | 1,124 | $ | 2,370 | ||||||
Working capital |
9,940 | (18 | ) | 2,226 | 607 | 2,262 | ||||||||||
Total assets |
10,667 | 6,172 | 3,740 | 1,452 | 2,637 | |||||||||||
Long-term debt, net |
— | — | — | 2,862 | 2,770 | |||||||||||
Total stockholders' equity (deficit) |
$ | 10,407 | $ | 448 | $ | 2,512 | $ | (2,016 | ) | $ | (305 | ) | ||||
(in thousands)
47
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operation
You should read the following discussion and analysis of financial condition and results of operations together with our financial statements and the related notes included elsewhere in this annual report. This discussion and analysis contains forward-looking statements about our business and operations, based on current expectations and related to future events and our future financial performance, that involve risks and uncertainties. Our actual results may differ materially from those we currently anticipate as a result of many important factors, including the factors we describe under "Risk Factors" and elsewhere in this annual report.
Overview
Kips Bay Medical, Inc. ("we", "us", "our" or the "Company") was incorporated in the state of Delaware on May 1, 2007. We are a medical device company focused on manufacturing and commercializing our external saphenous vein support technology, or eSVS MESH, for use in coronary artery bypass grafting, or CABG, surgery. Our eSVS MESH is a nitinol mesh sleeve that, when placed over a saphenous vein graft during CABG surgery, is designed to improve the structural characteristics and long-term performance of the saphenous vein graft. CABG surgery is one of the most commonly performed surgeries in the United States. In CABG procedures, surgeons harvest blood vessels, including the internal mammary artery from the chest wall and the saphenous vein from the leg, and attach the harvested vessels to bypass, or provide blood flow around, blocked coronary arteries. We believe the use of our eSVS MESH with saphenous vein grafts in CABG surgery will improve the long-term outcome of CABG procedures, including improved openness, or patency, and improved blood flow characteristics through the saphenous vein graft, resulting in a reduced need for costly and potentially complicated reoperations or revascularization procedures.
During February 2011, we completed an initial public offering ("IPO") of 2,062,500 shares of our common stock at a purchase price of $8.00 per share. All shares sold in the IPO were newly issued by the Company. Gross proceeds from the IPO were $16.5 million. After deducting the underwriting commissions and other expenses, we realized net proceeds of approximately $13.6 million. As additional consideration for this transaction, we issued options to purchase 103,125 shares of our common stock to the underwriters and their designees. These options have a five year term, an exercise price of $10.00 per share or 125% of the purchase price of shares sold in the IPO, and become exercisable on February 10, 2012, one year after the effective date of the IPO.
In order to obtain authorization to apply the CE Mark to our product and begin sales in Europe, we conducted a 90 patient multi-center clinical trial outside the United States. The goals of this trial were to demonstrate that CABG surgery using our eSVS MESH was not inferior as to either safety or effectiveness as compared to traditional CABG surgery. Based on early results from this trial we received our CE Mark in May 2010. In the trial, we evaluated the safety of our eSVS MESH by comparing the rate of major adverse cardiac and cerebral events, or MACCE, 30 days following surgery for patients treated with our eSVS MESH against the same rate reported in published literature for patients with traditional CABG surgery. We evaluated efficacy by comparing the patency of vessels treated with eSVS MESH against the patency of untreated saphenous vein bypass vessels as measured by angiographic studies nine to 12 months following implant. The safety data from this trial has indicated that our eSVS MESH and implant procedure do not result in an increase in patient complications during or for 30 days after surgery. However, the effectiveness data from the trial was inconclusive, primarily due to two complicating factors. First, one of the centers participating in the trial used implant methods incompatible with our eSVS MESH. Second, the amount of reduction in the diameter of the saphenous vein grafts, or downsizing, prescribed in our instructions for use and sizing tool was too aggressive, resulting in a higher than anticipated closure rate in saphenous vein grafts utilizing the eSVS MESH, particularly when our smallest device, 3.0 millimeters, was used. In response to this data, we have modified our instructions for use to provide clear direction on the surgical
48
method to be used with our eSVS MESH, discontinued the use of our 3.0 millimeter eSVS MESH and reduced the amount of downsizing specified for other device sizes. We also exclude saphenous veins with walls thicker than 0.7 millimeters. We believe these steps will resolve the patency issues identified in the trial.
We began marketing and commenced shipments of our eSVS MESH in select international markets in June 2010. Our eSVS MESH is a novel product and we are not aware of the establishment of any specific or supplemental reimbursements for our eSVS MESH. Given the budgetary pressures in Europe, our sales to date have been limited. In November 2011 we commenced enrollments in our first post-market study of the eSVS MESH in Europe. The intent of this study is to develop clinical data to further support the marketing and reimbursement of the eSVS MESH.
The U.S Food and Drug Administration ("FDA") has reviewed and disapproved our most recent amendment to our application for an investigational device exemption, or IDE in March 2011. At that time the FDA indicated that they intended to review our IDE information with outside experts before they provide further guidance to the Company. Due to internal delays, the FDA did not begin this review until August 2011. In September 2011 the FDA advised us that we have not provided sufficient data to support our request for an IDE for our eSVS MESH. In response to this, we plan to conduct a new feasibility trial in Europe based upon a protocol which incorporates additional guidance/requirements provided by the FDA. We began the process of identifying study sites and expect to commence enrollment in the summer of 2012. Upon completion of this study, we expect to request an IDE for a pivotal study in the U.S. However, we could be delayed by adverse clinical results or regulatory complications, and we may never receive U.S. marketing approval.
Since our inception, we have generated losses. From inception to December 31, 2011, we had an accumulated deficit of $24.3 million. We expect to incur losses for the foreseeable future as we pursue the development and commercialization of our eSVS MESH. Our activities from inception through the receipt of CE Mark approval for our eSVS MESH in May 2010 consisted principally of acquiring product and technology rights, raising capital, performing research and development and conducting preclinical and clinical trials. Subsequently, our activities included commercialization efforts in select international countries which accepted the CE Mark.
Successful completion of our development programs and, ultimately, our ability to generate revenues and attain profitable operations are dependent on future trends or events, including:
- •
- The availability of adequate reimbursement levels in each jurisdiction. The trend toward managed healthcare in the United
States and other countries and legislation intended to reduce the cost of government insurance programs will significantly influence the purchase of healthcare services and products, and could result
in lower or no reimbursement for our eSVS MESH. If we are unable to obtain adequate reimbursement levels in a sufficient number of jurisdictions, our revenues and gross margins will be harmed.
- •
- The willingness of qualified distributors to agree to sell our eSVS MESH in each of the markets in which we are approved.
We have identified a number of independent distributors to conduct sales in Europe, and we have entered into agreements for distribution in Switzerland, Italy, Germany, Spain, Belgium, the
Netherlands, Luxembourg and the United Kingdom and, subsequent to the end of the fiscal year, France. We have also entered into an agreement with an independent distributor to conduct sales in the
United Arab Emirates. We may not be able to enter into additional distribution agreements on favorable terms or in a timely manner, which would harm our operating results.
- •
- Our ability to negotiate satisfactory pricing with qualified distributors. If we are unable to negotiate satisfactory pricing with qualified distributors in connection with their engagement, our revenues and gross margins will be harmed.
49
- •
- The pace at which we can train sales representatives of qualified distributors. We believe we have engaged and we intend
to further engage distributors that have experienced sales representatives who we expect to be able to train on the advantages and features of our eSVS MESH in a timely manner. In addition, our
clinical trial experience has shown that training of physicians can occur in a short period of time, normally less than two days, but may take longer.
- •
- Compliance with regulatory requirements for medical devices. These regulatory requirements are extensive, and we believe
they will continue to expand. We expect a substantial amount of our expenses will be used for compliance with these regulatory requirements, including in connection with conducting clinical trials,
regulatory submissions and ongoing compliance.
- •
- The level of acceptance of our eSVS MESH in the marketplace. If our eSVS MESH is unable to achieve market acceptance, our revenues will be limited.
Key Components of Our Results of Operations
Net Sales
We received CE Mark approval in May 2010 and began marketing and commenced shipments of our eSVS MESH in select European markets in June 2010. We sell our eSVS MESH to distributors who, in turn, sell to hospitals and clinics. The pricing in all distributor agreements is denominated in U.S. dollars and provides for the transfer of title when we ship our eSVS MESH to the distributors. We invoice shipping charges to our distributors and include them in net sales. We expense shipping costs at the time we report the related revenue and record them in cost of sales.
Cost of Sales
We fabricate our eSVS MESH both at our facility and at a contract manufacturer. We conduct final assembly and packaging at our facility. Our cost of sales consists primarily of purchased components, direct labor, allocated manufacturing overhead and royalties payable to Medtronic, Inc. ("Medtronic").
Research and Development Expenses
Since our inception, we have focused our activities on the development of our eSVS MESH. We expense both internal and external research and development costs as incurred. Research and development costs include the costs to design, develop, test, seek approval for, and enhance our eSVS MESH and production processes. Expenses related to research and development consist primarily of personnel costs, including salaries, benefits and stock-based compensation, product development, preclinical and clinical trials, professional service fees, materials and supplies and facilities-related costs. We expense amounts paid to obtain patents or acquire licenses, as the ultimate recoverability of the amounts paid is uncertain.
While our research and development expenses to date have been focused on product development and evaluating the feasibility of our eSVS MESH, we expect that a large percentage of our research and development expenses in the future will be incurred in support of our current and future clinical trials. These expenditures are subject to numerous uncertainties in timing and costs to complete. As we obtain results from clinical trials, we may elect to discontinue or delay clinical trials for certain product applications or programs in order to focus our resources on more promising product applications. Completion of clinical trials may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of a product. The cost of clinical trials may vary significantly over the life of the trial as a result of differences arising during the clinical trial, including:
- •
- the number of sites included in the clinical trials;
50
- •
- the length of time required to enroll suitable patient subjects;
- •
- the number of patients that participate in the clinical trials; and
- •
- the duration of patient follow-up.
Our expenses related to clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with multiple clinical trial sites and, in for certain trials, contract research organizations ("CROs") which administer clinical trials on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price. Payments under the contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. Expenses related to clinical trials generally are accrued based on contracted amounts and the achievement of milestones, such as number of patients enrolled. If timelines or contracts are modified based upon changes to the clinical trial design or scope of work to be performed, we modify our estimates of accrued expenses accordingly.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses consist primarily of salaries and benefits and other costs, including stock-based compensation, for our executive and administrative personnel; legal and other professional fees; travel, sales and marketing costs; insurance and other corporate costs. We have incurred a significant increase in selling general and administrative expenses as a result of becoming a public company in February 2011. These increases have include increased costs for insurance, costs related to quarterly, annual and other periodic filings with the SEC and payments to outside consultants, lawyers and accountants. We have also incurred significant costs to comply with the corporate governance, internal controls and similar requirements applicable to public companies.
Milestone Expense
As consideration for the purchase of the core intellectual property relating to our eSVS MESH, we have agreed to pay Medtronic an aggregate of up to $15.0 million upon the achievement of certain sales milestones. The milestones and related payments consist of $5.0 million due on the one-year anniversary of the first commercial sale of our eSVS MESH, $5.0 million due when our cumulative net sales reach $15.0 million and $5.0 million due when our cumulative net sales reach $40.0 million. We recorded our first commercial sale in June 2010 and recorded an expense for the first milestone obligation at that time.
Interest Income
Interest income consists of interest earned on investments in bank certificates of deposits, money market funds, commercial paper and corporate debt.
Interest Expense
Interest expense results primarily from interest associated with secured convertible notes in the aggregate principal amount of $3.0 million issued to Kips Bay Investments, LLC ("KBI"), or the Notes. Our reported interest expense includes interest payable in cash based upon the stated rate in the Notes and the amortization of discount recorded on the Notes created by the allocation of a portion of the Note proceeds to the fair value of the stock purchase options granted in conjunction with the issuance of the Notes and due to the beneficial conversion feature specified in the Notes. Beneficial conversion feature accounting rules require the recognition of the intrinsic value of the conversion feature at the time of the Notes' issuance, which is then amortized as additional interest expense over the life of the Notes.
51
Critical Accounting Policies and Estimates
Our management's discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
While our significant accounting policies are more fully described in Note 2 to our financial statements, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our reported financial results and affect the more significant judgments and estimates that we use in the preparation of our financial statements.
Revenue Recognition
We sell our eSVS MESH to international distributors, which subsequently resell it to hospitals and clinics. We recognize revenue in accordance with generally accepted accounting principles as outlined in SEC Staff Accounting Bulletin No. 104, Revenue Recognition, and Accounting Standards Codification ("ASC") 605-10-S99; specifically, when persuasive evidence of an arrangement exists, delivery of goods occurs through the transfer of title and risks and rewards of ownership, the selling price is fixed or determinable and collectability is reasonably assured.
We recognize revenue as products are shipped based on agreements with each of our distributors, which provide that title and risk of loss pass to the distributor upon shipment of the products to the distributor and do not provide the distributors a right of return.
Research and Development Expenses
We expense research and development costs, including clinical trial costs, when incurred, consistent with the guidance of ASC 730, Research and Development. All of our clinical trials are performed at clinical trial sites and certain trials are administered by contract research organizations ("CROs"). We accrue costs for clinical trials based on estimates of work performed under the contracts. Costs of setting up clinical trial sites are accrued immediately. Expenses related to clinical trials generally are accrued based on contracted amounts and the achievement of milestones, such as number of patients enrolled.
All material clinical trial and CRO contracts are terminable by us upon written notice and we are generally only liable for actual effort expended by the CROs and certain non-cancelable expenses incurred at any point of termination.
Stock-Based Compensation
Stock-based incentive awards are accounted for under the provisions of FASB ASC 718, Compensation—Stock Compensation, which requires companies to measure and recognize the cost of employee and non-employee services received in exchange for awards of equity instruments based on the grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the expected vesting period, which is considered to be the requisite service period. In addition, we are required to estimate the amount of expected forfeitures when calculating the compensation costs, instead of accounting for forfeitures as they occur. All of our
52
previously awarded options were classified as equity instruments and continue to maintain their equity classification.
The fair value of options is estimated at the date of grant using the Black-Scholes option pricing model with the assumptions described in the following sentences. Risk free interest rates are based upon U.S. Treasury rates appropriate for the expected term. Expected volatility and forfeiture rates are based on the volatility rates of a set of guideline companies, which consist of public and recently public medical technology companies. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term is determined using the simplified method allowed by SEC Staff Accounting Bulletin No. 110. Prior to the completion of our initial public offering in February 2011, the fair value of our common stock was determined by our Board of Directors at each award grant date based upon a variety of factors, primarily the most recent purchase prices of our common stock issued to third parties in arms-length transactions, but also the progress of our product development, the progress of our preclinical and clinical testing, and the risks associated with our business plan. If we had made different assumptions and estimates, the amount of our recognized and to be recognized stock-based compensation expense could have been materially different. We believe that we have used reasonable methodologies, approaches and assumptions in determining the fair market value of our common stock.
We grant options to employees and non-employees, including members of our Scientific Advisory Board. Option grants to employees have a maximum term of ten years and generally vest over four years at the rate of 25% of the total shares underlying the option each year. Options granted to non-employees have a maximum term of ten years and generally vest over three years with 25% of the total shares underlying the option vesting on the date of grant and 25% of the total shares vesting in each of the next three years.
Option grants to non-employees have been made in conjunction with and as sole consideration for their service as advisors to us. Certain of these advisors have also purchased shares of stock in our private placement offerings, but none beneficially own 5% or more of our outstanding common stock. The fair value of options granted to non-employees is measured at each reporting date until the option, or respective portion of the option, vests and the expense recorded by us is updated accordingly. See Note 8 to our financial statements included elsewhere in this annual report for additional information.
53
Results of Operations
Comparison of the Year Ended December 31, 2011 to the Year Ended December 31, 2010 (in thousands)
| |
Year Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percent Change |
|||||||||
| |
2011 | 2010 | ||||||||
Net sales |
$ | 252 | $ | 223 | 13.0 | % | ||||
Cost of sales |
(91 | ) | (77 | ) | 18.2 | |||||
Gross profit |
161 | 146 | 10.3 | |||||||
Operating expenses: |
||||||||||
Research and development |
1,675 | 2,521 | (33.6 | ) | ||||||
Selling, general and administrative |
2,755 | 1,326 | 107.8 | |||||||
Milestone expense |
— | 5,000 | — | |||||||
Total operating expenses |
4,430 | 8,847 | (49.9 | ) | ||||||
Other income (expense): |
||||||||||
Interest income |
19 | 12 | 58.3 | |||||||
Interest expense |
— | (1 | ) | — | ||||||
Realized gain on sale of short-term investment |
— | 46 | — | |||||||
Change in fair value of investor stock purchase option |
— | (2,290 | ) | — | ||||||
Net loss before income tax |
$ | (4,250 | ) | $ | (10,934 | ) | (61.1 | )% | ||
Cost of sales, research and development expenses and selling, general and administrative expenses include non-cash stock-based compensation expense as a result of our issuance of stock options and restricted stock grants. We expense the fair value of equity awards over their vesting periods. The terms and vesting schedules for share-based awards vary by type of grant and the employment status of the grantee. The equity awards granted through December 31, 2011 vest upon time-based conditions. We expect to record additional non-cash compensation expense in the future, which may be significant. The following table summarizes the stock-based compensation expense in our statements of operations for the years ended December 31, 2011 and 2010 (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | |||||
Cost of sales |
$ | 46 | $ | 12 | |||
Research and development |
153 | 520 | |||||
Selling, general and administrative |
293 | 93 | |||||
Total stock-based compensation |
$ | 492 | $ | 625 | |||
Net Sales and Gross Profit
Our net sales increased to $252,000 in 2011, an increase of 13.0% from $223,000 in 2010. Our gross profit increased to $161,000 in 2011, an increase of 10.3% from $146,000 in 2010. The increase in net sales and gross profit in 2011 results from having a full year of sales compared with 2010 in which we commenced commercial sales of our eSVS MESH in June 2010. We expect sales to continue at modest levels until additional clinical study data is available.
54
Research and Development
Our research and development ("R&D") expenses decreased 33.6% from $2.5 million in the year ended December 31, 2010 to $1.7 million for the year ended December 31, 2011. This decrease was driven by significant activities which were substantially completed in 2010, including product development for our eSVS MESH, the follow-up from our international feasibility trial, as well as the effects of the decline in the market value of our common stock during 2011 on the value of and recorded expense for unvested stock options awarded to non-employees. These changes caused decreases of approximately $465,000, $70,000 and $340,000, respectively. These decreases were partially offset by costs associated with the initiation of our European post-market studies, which began enrolling patients in November 2011. We expect that our research and development costs will increase to prior year levels now that our European post-market studies have begun to enroll patients and given that we expect to begin enrollments in a European feasibility study during the summer of 2012.
Selling, General and Administrative
Selling, general and administrative expenses increased 107.8% from $1.3 million in the year ended December 31, 2010 to $2.8 million in the year ended December 31, 2011. These increases were driven primarily by an additional $465,000 of costs incurred in conjunction with becoming a public company in February 2011. In addition, we expanded our management team to support our both the requirements associated with being a public company and to support our commercial sales activity. Increased costs related to the expansion of our management team and sales & marketing were approximately $530,000 and $284,000, respectively. We expect SG&A expenses to increase modestly as we continue our international sales and marketing efforts.
Milestone Expense
Our milestone expense for the year ended December 31, 2010 was $5.0 million. We recorded this milestone expense in conjunction with the first commercial sale of our eSVS MESH and it represents the first sales milestone under our Assignment and License Agreement with Medtronic. Under the provisions of this agreement, the payment of this milestone was due on the first anniversary of our first commercial sale of the eSVS MESH, or June 2011, and was paid at that time. No additional milestones under the Assignment and License Agreement were attained in 2011.
Interest Income
Interest income increased from $12,000 in the year ended December 31, 2010 to $19,000 in the year ended December 31, 2011. The increase resulted from the investment of the net proceeds from our February 2011 IPO partially offset by declines in short-term interest rates from 2010 to 2011.
Change in Fair Value of Investor Stock Purchase Option
The change in fair value of investor stock purchase option was a loss of $2.3 million in 2010. This loss resulted from a modification to the second Kips Bay Investments, LLC ("KBI") stock purchase option, which resulted in an increase in the estimated fair value of the option of $2.3 million. KBI exercised this option in February 2010.
55
Comparison of the Year Ended December 31, 2010 with the Year Ended December 31, 2009 (in thousands)
| |
Year Ended December 31, | |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percent Change |
|||||||||
| |
2010 | 2009 | ||||||||
Net sales |
$ | 223 | $ | — | — | |||||
Cost of sales |
(77 | ) | — | — | ||||||
Gross profit |
146 | — | — | |||||||
Operating expenses: |
||||||||||
Research and development |
2,521 | 3,004 | (16.1 | )% | ||||||
Selling, general and administrative |
1,326 | 779 | 70.2 | |||||||
Milestone expense |
5,000 | — | — | |||||||
Total operating expenses |
8,847 | 3,783 | 133.9 | |||||||
Other income (expense): |
||||||||||
Interest income |
12 | 17 | (29.4 | ) | ||||||
Interest expense |
(1 | ) | (181 | ) | — | |||||
Realized gain on sale of short term investment |
46 | — | ||||||||
Change in fair value of investor stock purchase option |
(2,290 | ) | 610 | — | ||||||
Net loss before income tax |
$ | (10,934 | ) | $ | (3,337 | ) | 227.7 | % | ||
Cost of sales, research and development expenses and selling, general and administrative expenses include non-cash stock-based compensation expense as a result of our issuance of stock options. We expense the fair value of stock options over their vesting periods. The terms and vesting schedules for share-based awards vary by type of grant and the employment status of the grantee. The awards granted through December 31, 2010 vest upon time-based conditions. We expect to record additional non-cash compensation expense in the future, which may be significant. The following table summarizes the stock-based compensation expense in our statements of operations for the years ended December 31, 2010 and 2009 (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | |||||
Cost of sales |
$ | 12 | $ | — | |||
Research and development |
520 | 390 | |||||
Selling, general and administrative |
93 | 47 | |||||
Total stock-based compensation |
$ | 625 | $ | 437 | |||
Research and Development
Our research and development ("R&D") expenses decreased 16.1% from $3.0 million in the year ended December 31, 2009 to $2.5 million for the year ended December 31, 2010. This decrease was driven by decreases in expenses related to our international feasibility trial, which completed enrollment in July 2009, and product development and testing related to our eSVS MESH of $420,000 and $243,000, respectively. We began commercial sales of our eSVS MESH in June 2010, and in conjunction with the start-up of commercial manufacturing, certain costs associated with personnel and facilities previously included in R&D were moved to manufacturing. This change resulted in a further decrease in R&D expenses of $248,000. These decreases were partially offset by increases in preclinical testing and stock-based compensation expenses of $310,000 and $130,000, respectively.
56
Selling, General and Administrative
Selling, general and administrative expenses increased 70.2% from $779,000 in the year ended December 31, 2009 to $1.3 million in the year ended December 31, 2010. This increase was primarily due to a $301,000 increase in salaries and benefits and a $45,000 increase in stock-based compensation costs, both related to the expansion of our management team, and a $89,000 increase in accounting and professional fees related to preparing the audited financial statements for our initial public offering. Prior to this offering, our financial statements were not audited. Sales and marketing travel and related costs were also higher by $144,000 as a result of commercialization activities we commenced after our receipt of the CE Mark for our eSVS MESH in May 2010. These increases were partially offset by a decrease of $67,000 related to the termination in July 2009 of a temporary consultant who performed marketing related work.
Milestone Expense
Our milestone expense for the year ended December 31, 2010 was $5.0 million. We recorded this milestone expense in conjunction with the first commercial sale of our eSVS MESH and it represents the first sales milestone under our Assignment and License Agreement with Medtronic. Under the provisions of this agreement, the payment of this milestone was due on the first anniversary of our first commercial sale of the eSVS MESH, or June 2011, and was paid at that time.
Interest Income
Interest income decreased from $17,000 in the year ended December 31, 2009 to $12,000 in the year ended December 31, 2010. Our interest income decreased due to the decline in rates paid on money market accounts, in which our cash and cash equivalents are primarily invested, offsetting the increase in our available cash and cash equivalents.
Interest Expense
Interest expense declined from $181,000 in the year ended December 31, 2009 to $1,000 in the year ended December 31, 2010. The Notes were converted into shares of common stock in February 2009, which eliminated all debt from our balance sheet. The year ended December 31, 2009 included two months of interest expense and the write-off of $138,000 of unamortized discount on the Notes at the time of their conversion.
Change in Fair Value of Investor Stock Purchase Option
The change in fair value of investor stock purchase option was a loss of $2.3 million in the year ended December 31, 2010, as compared to a gain of $610,000 in the year ended December 31, 2009. The loss in the year ended December 31, 2010 resulted from a modification to the second KBI stock purchase option, which resulted in an increase in the estimated fair value of the option of $2.3 million. KBI exercised this option in February 2010.
Income Taxes
Since inception, we have incurred operating losses and, accordingly, have not recorded a provision for income taxes for any of the periods prior to the current year. The state of Minnesota enacted a change to their tax code which made the Minnesota Research and Tax Credit a refundable credit for fiscal years starting on or after January 1, 2010. Based upon our research and development costs which qualify under the Minnesota tax code, we recorded an income tax benefit of $0 and $42,000 for the years ended December 31, 2011 and 2010, respectively.
57
As of December 31, 2011, we had net operating loss carryforwards for federal and state income tax purposes of approximately $13.5 million. We also had federal research and development tax credit carryforwards of approximately $230,000. If not utilized, the federal net operating loss and tax credit carryforwards will begin to expire in 2027. Utilization of net operating loss and credit carryforwards may be subject to a substantial annual limitation due to limitations provided by the Internal Revenue Code of 1986, as amended, that are applicable if we experience an "ownership change" that may occur, for example, as a result of our initial public offering of common stock in February 2011 aggregated with certain other sales of our stock before or after this offering. If not utilized, the state net operating loss carryforwards will begin to expire in 2022. The annual limitation may result in the expiration of our net operating loss and tax credit carryforwards before they can be used.
Liquidity and Capital Resources
The following table summarizes our liquidity and capital resources as of December 31, 2011 and 2010 and for each of fiscal years ended December 31, 2011, 2010 and 2009, and is intended to supplement the more detailed discussion that follows (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
Liquidity and Capital Resources
|
2011 | 2010 | |||||
Cash and cash equivalents |
$ | 6,211 | $ | 3,548 | |||
Short-term investments |
2,957 | 236 | |||||
Working capital |
9,940 | (18 | ) | ||||
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cash Flow Data
|
2011 | 2010 | 2009 | |||||||
Cash provided by (used in): |
||||||||||
Operating activities |
$ | (8,105 | ) | $ | (4,378 | ) | $ | (3,382 | ) | |
Investment activities |
(2,952 | ) | 471 | (840 | ) | |||||
Financing activities |
13,720 | 4,986 | 5,748 | |||||||
Net increase in cash and cash equivalents |
$ | 2,663 | $ | 1,079 | $ | 1,526 | ||||
Cash and Cash Equivalents
Our total cash resources, including short-term investments, as of December 31, 2011 were $9.2 million compared to $3.8 million as of December 31, 2010. As of December 31, 2011, we had $260,000 in current liabilities and $9.9 million in net working capital. As of December 31, 2010, we had $5.7 million in current liabilities and an $18,000 net working capital deficit. The increase in our total cash resources and reduction in current liabilities resulted from the net proceeds of our February 2011 IPO, partially offset by cash used in operations for 2011.
As we continue to pursue regulatory approvals, continue the process of commercialization in international markets, develop our manufacturing capabilities and develop additional applications for our eSVS MESH, we expect to continue to incur substantial and increasing losses, which will continue to generate negative net cash flows from operating activities.
Prior to our IPO, we had funded our operations primarily through private sales of common stock and convertible debt. As of December 31, 2010, we had received net proceeds of approximately $12.6 million from the sale of equity securities, and net proceeds of approximately $3.0 million from the issuance of the Notes, all of which have been converted into common stock.
In the IPO, we sold 2,062,500 shares of common stock receiving net proceeds of $13.6 million. We intend to use the net proceeds from our IPO, as further described in Item 2 of Part II of this annual
58
report, to seek regulatory approval to market our technology in the United States which will require human clinical trials; to expand regulatory approval abroad; to develop and test additional applications of our eSVS MESH; and for working capital and general corporate purposes.
On October 24, 2011, we entered into a common stock purchase agreement (the "Purchase Agreement") with Aspire Capital Fund, LLC, an Illinois limited liability company ("Aspire Capital"), which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $20.0 million of shares of our common stock (the "Purchase Shares") over a three year period at purchase prices determined in accordance with the Purchase Agreement. Concurrently with entering into the Purchase Agreement, we also entered into a registration rights agreement with Aspire Capital, dated as of October 24, 2011, in which we agreed to file one or more registration statements as permissible and necessary to register under the Securities Act of 1933, as amended, the sale by Aspire Capital of the shares of our common stock that have been and that may be issued to Aspire Capital under the Purchase Agreement. We agreed to file the initial registration statement with the SEC on or before November 21, 2011. This registration statement was declared effective by the SEC on January 26, 2012. In consideration for entering into the Purchase Agreement, concurrently with the execution of the Purchase Agreement, we issued to Aspire Capital 378,788 shares of our common stock as a commitment fee (the "Commitment Shares").
As of the date of this report, a total of 378,788 shares of common stock, representing the Commitment Shares, have been issued to Aspire Capital pursuant to the Purchase Agreement; we have not yet sold any shares of common stock to Aspire Capital pursuant to the Purchase Agreement.
In the future, we may seek, or have a need, to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. We cannot assure that we will be able to secure such additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs. We expect that the proceeds from the IPO will be sufficient to fund our planned operations for at least the next 12 months, and we have no current intention to enter into a credit facility or loan agreement. We do not anticipate any adverse effects stemming from the lack of available credit facilities at this time.
Net Cash Used in Operating Activities
Net cash used in operating activities was $8.1 million in 2011, $4.4 million in 2010, and $3.4 million in 2009. The net cash used in each of these periods primarily reflects the net loss for those periods, offset in part by depreciation, non-cash stock-based compensation, changes in the fair value of the KBI stock purchase option liability and the effects of changes in operating assets and liabilities.
Under the provisions of the Assignment and License Agreement with Medtronic, Inc., our first milestone payment obligation of $5.0 million was due and payable on the one year anniversary of the first commercial sale of our eSVS MESH. We accrued this milestone expense in June 2010 and remitted the payment in June 2011.
Net Cash Provided by (Used in) Investment Activities
Net cash (used in) provided by investment activities was ($3.0) million in 2011, $471,000 in 2010 and ($840,000) in 2009. Cash used in or provided by investment activities is related to purchases and sales of short-term investments and purchases of property and equipment.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $13.7 million in 2011, $5.0 million in 2010 and $5.7 million in 2009. Net cash provided by financing activities was primarily attributable to proceeds from our initial public offering, issuances of the Notes and private sales of our common stock.
59
Capital Requirements
We expect to incur substantial expenses and generate significant operating losses as we continue to execute our business strategy including:
- •
- seeking an IDE approval from the FDA to begin clinical trials in the United States;
- •
- commercializing our eSVS MESH in select European and other international markets;
- •
- obtaining regulatory approval and commercializing our eSVS MESH in the United States;
- •
- conducting pre-clinical and clinical trials to expand indications for our eSVS MESH and to pursue other product
development initiatives;
- •
- hiring additional personnel for managerial, research and development, operations and other functions;
- •
- expanding our facilities to increase our manufacturing and development capabilities; and
- •
- implementing new operational, financial and management systems to comply with SEC requirements.
Our future capital uses and requirements depend on numerous current and future factors. These factors include, but are not limited to, the following:
- •
- our ability to demonstrate safety and effectiveness of our eSVS MESH;
- •
- the selling price of our eSVS MESH to distributors and the price that distributors charge hospitals;
- •
- the rate of progress in establishing reimbursement arrangements with third-party payors;
- •
- the effect of competing technological and market developments;
- •
- the cost and delays in product development that may result from changes in regulatory oversight applicable to our eSVS
MESH;
- •
- the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims;
- •
- the cost of expanding our commercial operations, including our selling and marketing efforts;
- •
- our ability to establish and maintain effective relationships with independent distributors;
- •
- the rate at which physicians adopt our eSVS MESH for use in CABG surgery; and
- •
- the progress of preclinical and clinical trials required to support our applications for regulatory approvals, including our human clinical trials in the United States.
We expect that our existing resources as of the date of this annual report to be sufficient to fund our planned operations for at least the next 12 months. However, we may require significant additional funds earlier than we currently expect in order to conduct additional clinical trials to obtain regulatory approvals of our eSVS MESH. To the extent that we raise additional capital through the sale of equity or convertible debt securities, stockholders' interest will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of our stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. Any of these events could adversely affect our ability to achieve our product development and commercialization goals and harm our business.
60
If adequate funds are not available, we may be required to terminate, significantly modify or delay our development programs, reduce our planned commercialization efforts, or obtain funds through collaborators that may require us to relinquish rights to our technologies or product candidates that we might otherwise seek to develop or commercialize independently. We may elect to raise additional funds even before we need them if the conditions for raising capital are favorable.
Contractual Obligations, Commitments and Contingencies
To date, we have not entered into long-term minimum purchase commitments with suppliers. Our principal commitments consist of obligations under the lease for our facility in Minneapolis, Minnesota, and certain office equipment.
The following table summarizes our outstanding contractual obligations as of December 31, 2011 (in thousands):
| |
Total | Less than 1 Year |
1 - 3 Years | 4 - 5 Years | After 5 Years |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Operating lease obligations |
$ | 229 | $ | 84 | $ | 145 | $ | — | $ | — | ||||||
The operating lease obligation for our corporate facilities ends September 30, 2014.
Royalty Payments
The core intellectual property relating to our eSVS MESH was sold to us by Medtronic pursuant to an Assignment and License Agreement dated October 9, 2007. Pursuant to the Assignment and License Agreement, Medtronic also sold to us intellectual property relating to a brushed ePTFE vascular graft, or the Brushed Graft Product.
As consideration for the sale of intellectual property relating to the eSVS MESH and the Brushed Graft Product and other rights granted by the Assignment and License Agreement, we have agreed to pay Medtronic an aggregate of up to $20.0 million upon the achievement of certain sales milestones relating to our eSVS MESH and the Brushed Graft Product and a royalty of 4% on sales of our eSVS MESH and any future sales of the Brushed Graft Product. This royalty will terminate upon the earlier of the expiration of all of the patents and patent applications, or when the aggregate royalties paid reach $100.0 million. As of December 31, 2011, we have paid the first $5.0 million milestone payment and have recorded royalty expense of $10,000 and $9,000 for the years ended December 31, 2011 and 2010, respectively.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet activities as defined in Regulation S-K Item 303(a)(4).
Recent Accounting Pronouncements
In December 2011, FASB issued ASU 2011-12, Comprehensive Income, or ASU 2011-12, which amended ASU 2011-05 Presentation of Comprehensive Income. These accounting updates requires entities to present items of net income and other comprehensive income either in a single continuous statement, or in separate, but consecutive, statements of net income and other comprehensive income. The new requirements do not change which components of comprehensive income are recognized in net income or other comprehensive income, or when an item of other comprehensive income must be reclassified to net income. The updates are effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. Since this standard impacts disclosure requirements only, the Company does not expect its adoption will have a material impact on our consolidated results of operations or financial condition.
61
In May 2011, the FASB issued ASU 2011-04 which was issued to provide a consistent definition of fair value and ensure that the fair value measurement and disclosure requirements are similar between U.S. GAAP and International Financial Reporting Standards ("IFRS"). ASU 2011-04 changes certain fair value measurement principles and enhances the disclosure requirements particularly for Level 3 fair value measurements. This guidance is effective for us beginning on January 1, 2012. Its adoption is not expected to significantly impact our financial statements.
Special Note Regarding Forward-Looking Statements
This annual report contains forward-looking statements that involve risks and uncertainties. In some cases, you can identify forward-looking statements by the following words: "anticipate," "believe," "continue," "could," "estimate," "expect," "intend," "may," "ongoing," "plan," "potential," "predict," "project," "should," "will," "would," or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. The forward-looking statements in this annual report include, but are not limited to, statements regarding uses for and benefits of our technology, outlooks for our industry and our markets, the timing of and strategy for governmental approvals and product introductions and commercialization, expansion of and effects of domestic and foreign governmental regulations, use of research and development and SG&A expenditures, commencement, design and cost of preclinical and clinical trials, intellectual property protection, future competition, adequacy of our manufacturing and supply operations, reimbursement and distribution strategies and expectations, environmental compliance costs, dividend expectations, our expectations regarding future losses and future financing, milestone payments, increased costs as a public company, future tax benefits, use of proceeds from our initial public offering, the adequacy of our capital resources to fund future expenditures, and operating and capital requirement expectations. These statements involve known and unknown risks, uncertainties and other factors that may cause our or our industry's results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this annual report, we caution you that these statements are based on a combination of facts and factors currently known by us and our projections of the future, about which we cannot be certain. Many important factors affect our ability to achieve our objectives, including:
- •
- our ability to commercialize our eSVS MESH technology;
- •
- our ability to obtain and maintain foreign and domestic regulatory approval of our eSVS MESH technology;
- •
- our ability to obtain coverage and reimbursement from third-party payors for our eSVS MESH technology and the extent of
such coverage;
- •
- the successful development of our distribution and marketing capabilities;
- •
- our ability to attract and retain scientific, regulatory, and sales and marketing support personnel;
- •
- our ability to obtain and maintain intellectual property protection for our eSVS MESH technology;
- •
- any future litigation regarding our business, including product liability claims;
- •
- changes in governmental laws and regulations relating to healthcare;
- •
- the availability and cost of third-party products and the ability of our suppliers to timely meet our demands;
- •
- changes affecting the medical device industry;
- •
- general and economic business conditions; and
62
- •
- the other risks described under Item 1A. "Risk Factors" in this annual report.
You should read these risk factors and the other cautionary statements made in this annual report as being applicable to all related forward-looking statements wherever they appear in this annual report. We cannot assure you that the forward-looking statements in this annual report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. You should read this annual report completely. Other than as required by law, we undertake no obligation to update these forward-looking statements, even though our situation may change in the future.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Not required.
63
Item 8. Financial Statements and Supplementary Data
64
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures. We maintain controls and procedures designed to ensure that we are able to collect the information we are required to disclose in the reports we file with the SEC, and to process, summarize and disclose this information within the time periods specified in the rules of the SEC. Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Securities and Exchange Act of 1934, as amended). Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2011, our controls and procedures were effective.
Management's Annual Report on Internal Control Over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Securities and Exchange Act of 1934, as amended. Our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth in the report entitled Internal Control—Integrated Framework published by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO. Based on this assessment, management has concluded that, as of December 31, 2011, our internal control over financial reporting was effective.
Attestation Report of the Registered Public Accounting Firm. This annual report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to attestation by the Company's independent registered public accounting firm pursuant to Section 989G of the Dodd-Frank Wall Street Reform and Consumer Protection Act, which exempts smaller reporting companies from the auditor attestation requirement.
Changes in Internal Control over Financial Reporting. There were no changes in our internal control over financial reporting during the quarter ended December 31, 2011 that materially affected, or were reasonably likely to materially affect, our internal control over financial reporting.
None.
65
Item 10. Directors, Executive Officers and Corporate Governance
Other than the information included in Part I of this Form 10-K under the heading "Executive Officers of the Registrant," the information required by Item 10 is incorporated by reference to the sections labeled "Election of Directors," "Corporate Governance" and "Section 16(a) Beneficial Ownership Reporting Compliance," which will appear in our definitive proxy statement for our 2012 Annual Meeting.
Item 11. Executive Compensation
The information required by Item 11 is incorporated herein by reference to the sections entitled "Executive Compensation," "Director Compensation" and "Compensation Committee," which will appear in our definitive proxy statement for our 2012 Annual Meeting.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by Item 12 is incorporated herein by reference to the sections entitled "Principal Stockholders," which will appear in our definitive proxy statement for our 2012 Annual Meeting.
Equity Compensation Plan Information
The following table provides information concerning the Kips Bay Medical, Inc. 2007 Long-Term Incentive Plan, which was adopted by the Board of Directors and approved by stockholders in July 2007, as of December 31, 2011.
| |
Number of securities to be issued upon exercise of outstanding options |
Weighted average exercise price of outstanding options |
Number of securities remaining available for future issuance under equity compensation plans |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Equity compensation plans approved by security holders |
1,261,000 | $ | 3.73 | 515,500 | ||||||
Equity compensation plans not approved by security holders(1) |
103,125 | 10.00 | — | |||||||
Totals |
1,364,125 | $ | 4.20 | 515,500 | ||||||
- (1)
- Represents warrants to purchase our common stock issued to the underwriters and their designees in our initial public offering.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by Item 13 is incorporated herein by reference to the sections entitled "Corporate Governance—Independence" and "Certain Relationships and Related Party Transactions," which will appear in our definitive proxy statement for our 2012 Annual Meeting.
Item 14. Principal Accounting Fees and Services
The information required by Item 14 is incorporated herein by reference to the section entitled "Audit Fees" and "Audit Committee Pre-Approval Policies and Procedures," which will appear in our definitive proxy statement for our 2012 Annual Meeting.
66
Item 15. Exhibits, Financial Statements Schedules
- (a)
- Financial
Statements, Financial Statement Schedules, and Exhibits:
- (1)
- Financial Statements
- (2)
- Financial Statement Schedules
- (3)
- Exhibits
The financial statements filed as part of this filing are listed on the index to the Financial Statements and Supplementary Data, Item 8, on page 66.
Schedules not listed above have been omitted because they are not applicable or not required or the information required to be set forth therein is included in the Financial Statements and Supplementary Data, Item 8, or notes thereto.
Exhibits are listed in the exhibit index following the financial statement pages. The exhibits include management contracts, compensatory plans and arrangements required to be filed as exhibits to the Form 10-K by Item 601(10)(iii) of Regulation S-K.
67
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 15, 2012
| KIPS BAY MEDICAL, INC. | ||||
By: |
/s/ MANNY VILLAFAÑA Manny Villafaña Chairman of the Board and Chief Executive Officer |
|||
Each person whose signature appears below constitutes Scott Kellen as his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any or all amendments to this Annual Report on Form 10-K and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all said attorney-in-fact and agent, or his or her substitute or substitutes, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signatures
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ MANNY VILLAFAÑA Manny Villafaña |
Chairman and Chief Executive Officer (principal executive officer) | March 15, 2012 | ||
/s/ SCOTT KELLEN Scott Kellen |
Chief Financial Officer and Vice President of Finance (principal financial and accounting officer) |
March 15, 2012 |
||
/s/ ROBERT E. MUNZENRIDER Robert E. Munzenrider |
Director |
March 15, 2012 |
||
/s/ ARCH C. SMITH Arch C. Smith |
Director |
March 15, 2012 |
||
/s/ ROBERT J. SHEEHY Robert J. Sheehy |
Director |
March 15, 2012 |
68
Report of Independent Registered Public Accounting Firm
To
the Board of Directors and Stockholders
Kips Bay Medical, Inc.
We have audited the accompanying balance sheets of Kips Bay Medical, Inc. as of December 31, 2011 and 2010, and the related statements of operations, stockholders' equity (deficit), and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Kips Bay Medical, Inc. at December 31, 2011 and 2010, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young, LLP
Minneapolis,
Minnesota
March 15, 2012
F-1
Kips Bay Medical, Inc.
Balance Sheets
(In thousands, except share and per share amounts)
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | |||||
ASSETS |
|||||||
Current assets: |
|||||||
Cash and cash equivalents |
$ | 6,211 | $ | 3,548 | |||
Short-term investments, net |
2,957 | 236 | |||||
Accounts receivable, net of allowance for doubtful accounts of $14 and $0 as of December 31, 2011 and 2010, respectively |
40 | 56 | |||||
Inventories |
892 | 606 | |||||
Prepaid expenses and other current assets |
100 | 1,260 | |||||
Total current assets |
10,200 | 5,706 | |||||
Property and equipment, net |
467 | 466 | |||||
Total assets |
$ | 10,667 | $ | 6,172 | |||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||||
Current liabilities: |
|||||||
Accounts payable |
$ | 85 | $ | 180 | |||
Accrued liabilities |
171 | 539 | |||||
Accrued milestone and royalties |
4 | 5,005 | |||||
Total current liabilities |
260 | 5,724 | |||||
Commitments and contingencies (Note 6) |
|||||||
Stockholders' equity: |
|||||||
Undesignated stock, $0.01 par value, 10,000,000 shares authorized, no shares issued and outstanding as of December 31, 2011 and 2010, respectively |
— | — | |||||
Common stock, $0.01 par value, 40,000,000 shares authorized, 16,245,579 and 13,581,791 issued and outstanding as of December 31, 2011 and 2010, respectively |
162 | 136 | |||||
Additional paid-in capital |
34,591 | 20,405 | |||||
Accumulated other comprehensive loss |
(3 | ) | — | ||||
Accumulated deficit |
(24,343 | ) | (20,093 | ) | |||
Total stockholders' equity |
10,407 | 448 | |||||
Total liabilities and stockholders' equity |
$ | 10,667 | $ | 6,172 | |||
See accompanying notes to financial statements.
F-2
Kips Bay Medical, Inc.
Statements of Operations
(In thousands, except share and per share amounts)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Net sales |
$ | 252 | $ | 223 | $ | — | ||||
Cost of sales |
(91 | ) | (77 | ) | — | |||||
Gross profit |
161 | 146 | — | |||||||
Operating Expenses: |
||||||||||
Research and development |
1,675 | 2,521 | 3,004 | |||||||
Selling, general and administrative |
2,755 | 1,326 | 779 | |||||||
Milestone expense |
— | 5,000 | — | |||||||
Operating loss |
(4,269 | ) | (8,701 | ) | (3,783 | ) | ||||
Interest income |
19 | 12 | 17 | |||||||
Interest expense |
— | (1 | ) | (181 | ) | |||||
Realized gain on sale of short-term investments |
— | 46 | — | |||||||
Change in fair value of investor stock purchase option |
— | (2,290 | ) | 610 | ||||||
Net loss before income tax |
$ | (4,250 | ) | $ | (10,934 | ) | $ | (3,337 | ) | |
Income tax benefit |
— | 42 | — | |||||||
Net loss |
$ | (4,250 | ) | $ | (10,892 | ) | $ | (3,337 | ) | |
Basic and diluted net loss per share |
$ | (0.27 | ) | $ | (0.81 | ) | $ | (0.30 | ) | |
Weighted average shares outstanding—basic and diluted |
15,557,969 | 13,431,661 | 11,069,342 | |||||||
See accompanying notes to financial statements.
F-3
Kips Bay Medical, Inc.
Statements of Stockholders' Equity (Deficit)
(In thousands except share amounts)
| |
Common Stock | |
|
Accumulated Other Comprehensive Income |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-In Capital |
Accumulated Deficit |
Total Stockholders' Equity |
||||||||||||||||
| |
Shares | Amount | |||||||||||||||||
Balance at December 31, 2008 |
6,300,000 | $ | 63 | $ | 2,409 | $ | (4,488 | ) | $ | — | $ | (2,016 | ) | ||||||
Comprehensive loss |
|||||||||||||||||||
Net loss |
— | — | — | (3,337 | ) | — | (3,337 | ) | |||||||||||
Unrealized gain on investments, net |
— | — | — | — | 33 | 33 | |||||||||||||
Comprehensive loss |
(3,304 | ) | |||||||||||||||||
Cumulative effect adjustment for adoption of FASB ASC 815-40 |
(194 | ) | (1,376 | ) | (1,570 | ) | |||||||||||||
Common stock issued at $5.83 per share |
300,000 | 3 | 1,747 | — | — | 1,750 | |||||||||||||
Common stock issued upon conversion of note payable, conversion price of $0.625 |
4,800,000 | 48 | 2,952 | — | — | 3,000 | |||||||||||||
Common stock issued upon conversion of accumulated interest on note payable, conversion price of $0.625 |
347,389 | 4 | 213 | — | — | 217 | |||||||||||||
Common stock issued, $1.00 per share, employee exercise of incentive stock option |
1,000 | — | 1 | — | — | 1 | |||||||||||||
Common stock issued, $6.00 per share, under private placement offering (net of issuance costs of $29) |
516,241 | 5 | 3,063 | — | — | 3,068 | |||||||||||||
Common stock issued, $7.00 per share, under private placement offering (net of issuance costs of $11) |
134,289 | 1 | 928 | — | — | 929 | |||||||||||||
Stock-based compensation expense |
— | — | 437 | — | — | 437 | |||||||||||||
Balance at December 31, 2009 |
12,398,919 | 124 | 11,556 | (9,201 | ) | 33 | 2,512 | ||||||||||||
Comprehensive loss |
|||||||||||||||||||
Net loss |
— | — | — | (10,892 | ) | — | (10,892 | ) | |||||||||||
Unrealized gain on investments, net |
— | — | — | — | 13 | 13 | |||||||||||||
Reclassification of unrealized gain on short-term investments |
— | — | — | — | (46 | ) | (46 | ) | |||||||||||
Comprehensive loss |
(10,925 | ) | |||||||||||||||||
Common stock issued upon exercise of investor stock purchase option |
1,000,000 | 10 | 6,990 | — | — | 7,000 | |||||||||||||
Common stock issued, $7.00 per share, under private placement offering (net of issuance costs of $44) |
182,872 | 2 | 1,234 | — | — | 1,236 | |||||||||||||
Stock-based compensation expense |
— | — | 625 | — | — | 625 | |||||||||||||
Balance at December 31, 2010 |
13,581,791 | 136 | 20,405 | (20,093 | ) | — | 448 | ||||||||||||
Comprehensive loss |
|||||||||||||||||||
Net loss |
— | — | — | (4,250 | ) | — | (4,250 | ) | |||||||||||
Unrealized loss on investments, net |
— | — | — | — | (3 | ) | (3 | ) | |||||||||||
Comprehensive loss |
(4,253 | ) | |||||||||||||||||
Common stock issued in initial public offering, net of issuance costs |
2,062,500 | 20 | 13,612 | — | — | 13,632 | |||||||||||||
Issuance of common stock to Aspire Capital |
378,788 | 4 | (4 | ) | — | — | — | ||||||||||||
Restricted stock grants |
135,000 | 1 | (1 | ) | — | — | — | ||||||||||||
Stock-based compensation expense |
— | — | 492 | — | — | 492 | |||||||||||||
Exercise of stock options |
87,500 | 1 | 87 | — | — | 88 | |||||||||||||
Balance at December 31, 2011 |
16,245,579 | $ | 162 | $ | 34,591 | $ | (24,343 | ) | $ | (3 | ) | $ | 10,407 | ||||||
See accompanying notes to financial statements.
F-4
Kips Bay Medical, Inc.
Statements of Cash Flows
(In thousands)
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Cash flows from operating activities: |
||||||||||
Net loss |
$ | (4,250 | ) | $ | (10,892 | ) | $ | (3,337 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
Depreciation expense |
111 | 74 | 60 | |||||||
Stock-based compensation |
492 | 625 | 437 | |||||||
Amortization of premium on short-term investments |
132 | — | — | |||||||
Non-cash interest income |
(16 | ) | — | — | ||||||
Amortization of discount on secured convertible notes |
— | — | 138 | |||||||
Realized gain on sale of short-term investments |
— | (46 | ) | — | ||||||
Change in fair value of investor stock purchase option |
— | 2,290 | (610 | ) | ||||||
Changes in operating assets and liabilities: |
||||||||||
Accounts receivable |
16 | (56 | ) | — | ||||||
Inventories |
(286 | ) | (606 | ) | — | |||||
Prepaid expenses and other current assets |
1,160 | (1,223 | ) | 52 | ||||||
Accounts payable |
(95 | ) | 96 | 60 | ||||||
Accrued liabilities |
(368 | ) | 355 | 25 | ||||||
Accrued milestone and royalties |
(5,001 | ) | 5,005 | — | ||||||
Accrued interest payable |
— | — | (207 | ) | ||||||
Net cash used in operating activities |
(8,105 | ) | (4,378 | ) | (3,382 | ) | ||||
Cash flows from investing activities: |
||||||||||
Proceeds from sales and maturities of short-term investments |
4,029 | 966 | — | |||||||
Purchases of short-term investments |
(6,869 | ) | (241 | ) | (734 | ) | ||||
Purchase of property and equipment |
(112 | ) | (254 | ) | (106 | ) | ||||
Net cash (used in) provided by investing activities |
(2,952 | ) | 471 | (840 | ) | |||||
Cash flows from financing activities: |
||||||||||
Proceeds from sale of common stock in IPO, net of issuance costs of $2,868 |
13,632 | — | — | |||||||
Proceeds from exercise of investor option to purchase common stock |
— | 3,750 | 1,750 | |||||||
Proceeds from sale of common stock under private placement offerings, net of issuance costs |
— | 1,236 | 3,997 | |||||||
Proceeds from the exercise of employee stock options |
88 | — | 1 | |||||||
Net cash provided by financing activities |
13,720 | 4,986 | 5,748 | |||||||
Net increase in cash and cash equivalents |
2,663 | 1,079 | 1,526 | |||||||
Cash and cash equivalents at beginning of period |
3,548 | 2,469 | 943 | |||||||
Cash and cash equivalents at end of period |
$ | 6,211 | $ | 3,548 | $ | 2,469 | ||||
Supplemental disclosures: |
||||||||||
Interest paid in cash |
$ | — | $ | 1 | $ | 250 | ||||
Supplemental non-cash disclosures: |
||||||||||
Reclassification of investor stock purchase option liability to equity |
$ | — | $ | 3,250 | $ | — | ||||
Conversion of note payable into common stock |
$ | — | $ | — | $ | 3,000 | ||||
Accrued interest paid by conversion into common stock |
$ | — | $ | — | $ | 217 | ||||
See accompanying notes to financial statements.
F-5
Kips Bay Medical, Inc.
Notes To Financial Statements
1. Organization and Business
Kips Bay Medical, Inc. ("we", "us" or "our") was incorporated in the state of Delaware on May 1, 2007. We are a medical device company focused on manufacturing and commercializing our external saphenous vein support technology, or eSVS® MESH, for use in coronary artery bypass grafting ("CABG") surgery. Our eSVS MESH is a nitinol mesh sleeve that, when placed over a saphenous vein graft during CABG surgery, is designed to improve the structural characteristics and long-term performance of the saphenous vein graft. In CABG procedures, surgeons harvest blood vessels, including the internal mammary artery from the chest and the saphenous vein from the leg, and attach the harvested vessels to bypass, or provide blood flow around, blocked coronary arteries.
During 2011 we have commenced significant revenue generating activities and are focused on the manufacture and commercialization of our eSVS MESH. In previous years we were a development stage company.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts and disclosures in the combined consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and money market funds with original maturities of three months or less. The carrying value of these instruments approximates fair value. We have not experienced any losses on our cash and cash equivalents.
Short-term Investments
Short-term investments may consist of bank certificates of deposits, money market funds, commercial paper and corporate debt. Our investment policy seeks to manage these investments to achieve our goal of preserving principal, maintaining adequate liquidity at all times, and maximizing returns subject to our investment guidelines. Short-term investments have been classified and accounted for as available-for-sale securities and are reported on the balance sheet at fair value with unrealized gains or losses reported as a component of other comprehensive income. Interest earned on short-term investments is included in interest income. We periodically evaluate our investments in marketable securities for potential impairment due to declines in market value deemed by us to be other-than-temporary. If cost exceeds fair value, we consider, among other factors, the duration and extent to which cost exceeds fair value, the financial strength of the issuer, and our intent and ability to hold the investment to maturity. Once a decline in value is deemed to be other-than-temporary, an impairment charge is recorded and a new cost basis in the investment is established.
In 2011 our short-term investments include a $100,000 bank certificate of deposit which we pledged as collateral for a corporate purchasing card which we obtained during 2011 to facilitate certain corporate purchasing activity.
F-6
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Accounts Receivable
Accounts receivable are recorded at the invoiced amount and are due within 60 to 90 days of shipment. We grant credit to customers in the normal course of business and do not require collateral. Accounts receivable do not bear interest and are presented net of any allowances for doubtful accounts. We determine our allowance for doubtful accounts by considering a number of factors, including the length of time trade accounts receivable are past due, the customer's current ability to pay its obligation to us and the condition of the general economy and the industry as a whole.
Inventories
Inventories are stated at the lower of cost (first-in, first-out method) or market. Inventory cost includes purchased materials and overhead costs which are applied to work in process and finished goods based on annual estimates of production volume and overhead spending. Appropriate consideration is given to deterioration, obsolescence, excess inventory and other factors in evaluating net realizable value. Inventories consist of the following as of December 31 (in thousands):
| |
2011 | 2010 | |||||
|---|---|---|---|---|---|---|---|
Raw materials |
$ | 91 | $ | 102 | |||
Work in process |
392 | 231 | |||||
Finished goods |
409 | 273 | |||||
|
$ | 892 | $ | 606 | |||
Property and Equipment
Property and equipment is stated at cost less accumulated depreciation. Depreciation is computed based upon the estimated useful lives of the respective assets, or the lesser of the estimated useful life or the remaining life of the underlying facility lease for leasehold improvements, ranging from three to seven years, and is recorded using the straight-line method. Repairs and maintenance costs are expensed as incurred.
Impairment of Long-Lived Assets
In accordance with FASB ASC 360, Property, Plant and Equipment, long-lived assets, such as property and equipment, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, then an impairment charge is recognized as the amount by which the carrying amount of the asset exceeds the fair value of the asset. As of December 31, 2011 and 2010, no modification of the remaining useful lives or write-down of long-lived assets is required.
Revenue Recognition
We sell our eSVS MESH to international distributors in Europe and the United Arab Emirates, which subsequently resell it to hospitals and clinics. We recognize revenue when persuasive evidence of
F-7
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
an arrangement exists, delivery of goods occurs through the transfer of title and risks and rewards of ownership, the selling price is fixed or determinable and collectability is reasonably assured.
We recognize revenue as products are shipped based on agreements with each of our distributors, which provide that title and risk of loss pass to the distributor upon shipment of the products to the distributor and do not provide the distributors a right of return. We invoice shipping charges to our distributors and include them in net sales. We expense royalties and shipping costs at the time we report the related revenue and record them in cost of sales.
Research and Development Expenses
Research and development costs consist of costs incurred for internally sponsored research and development, direct expenses and research-related overhead. Research and development costs also include costs related to the execution of clinical trials and related efforts to obtain regulatory approval for our eSVS MESH.
We charge research and development costs, including clinical trial costs, to expense when incurred. Clinical trial costs are a significant component of research and development expenses. All of our clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from contract research organizations ("CRO"). Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials generally are accrued based on contracted amounts and the achievement of agreed upon milestones, such as number of patients enrolled. We monitor levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the CROs, and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual effort expended at each clinical trial site and by each CRO.
All material CRO contracts are terminable by us upon written notice and we are generally only liable for actual effort expended by the CROs and certain non-cancelable expenses incurred at any point of termination.
We expense costs incurred to obtain patents or acquire licenses as the ultimate recoverability of the amounts paid is uncertain.
Comprehensive Income/Loss
Comprehensive income/loss consists of net loss and the effect of unrealized gains and losses on available-for-sale securities.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances
F-8
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
are established when necessary to reduce deferred tax assets to the amount that is more likely than not to be realized.
Stock-Based Compensation
Stock-based incentive awards are accounted for under the provisions of FASB ASC 718, Compensation—Stock Compensation, which requires companies to measure and recognize the cost of employee and non-employee services received in exchange for awards of equity instruments based on the grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the expected vesting period, which is considered to be the requisite service period. We estimate pre-vesting award forfeitures when calculating the compensation costs and revise those estimates in subsequent periods if actual forfeitures differ from those estimates.
The fair value of stock based awards is estimated at the date of grant using the Black-Scholes option pricing model. Risk free interest rates are based upon U.S. Treasury rates appropriate for the expected term. Expected volatility and forfeiture rates are based primarily on the volatility rates of a set of guideline companies, which consist of public and recently public medical technology companies. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term is determined using the simplified method allowed by SEC Staff Accounting Bulletin No. 110.
Prior to the completion of our initial public offering in February 2011, the fair value of our common stock was determined by our Board of Directors at each award grant date based upon a variety of factors, primarily the most recent purchase prices of our common stock issued to third parties in arms-length transactions, but also the progress of our product development, the progress of our preclinical and clinical testing, and the risks associated with our business plan.
Stock-based compensation expense in our statements of operations is summarized as follows (in thousands):
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Cost of sales |
$ | 46 | $ | 12 | $ | — | ||||
Research and development |
153 | 520 | 390 | |||||||
Sales, general and administrative |
293 | 93 | 47 | |||||||
Total stock-based compensation |
$ | 492 | $ | 625 | $ | 437 | ||||
Net Loss per Share
We compute net loss per share by dividing our income or loss available to common stockholders (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. In addition, in computing the dilutive effect of convertible securities, the numerator is adjusted to add back the after-tax amount of interest recognized in the
F-9
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
period associated with any convertible debt. Diluted EPS is the same as basic EPS due to common equivalent shares being excluded from the calculation, as their effect is anti-dilutive.
The following table summarizes our calculation of net loss per common share for the periods (in thousands, except share and per share data):
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Net loss |
$ | (4,250 | ) | $ | (10,892 | ) | $ | (3,337 | ) | |
Weighted average shares outstanding—basic and diluted |
15,557,969 | 13,431,661 | 11,069,342 | |||||||
Net loss per share—basic and diluted |
(0.27 | ) | (0.81 | ) | (0.30 | ) | ||||
The following outstanding potential common shares are not included in diluted net loss per share calculations as their effects were not dilutive:
| |
|
December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Employee and non-employee stock options |
1,261,000 | 813,000 | 598,000 | |||||||
Common shares issuable to underwriters under option purchase agreements (See note 7) |
103,125 | — | — | |||||||
Common shares issuable under investor option purchase agreements (See note 7) |
— | — | 600,000 | |||||||
Fiscal Year
We operate on a manufacturing calendar with our fiscal year always ending on December 31. Each quarter is 13 weeks, consisting of two four-week and one five-week periods.
Reclassifications
Certain reclassifications have been made to prior years' financial statements to conform to the current year presentation. These reclassifications had no effect on previously reported results of operations or accumulated deficit.
New Accounting Pronouncements
In December 2011, FASB issued ASU 2011-12, Comprehensive Income, or ASU 2011-12, which amended ASU 2011-05 Presentation of Comprehensive Income. These accounting updates require entities to present items of net income and other comprehensive income either in a single continuous statement, or in separate, but consecutive, statements of net income and other comprehensive income. The new requirements do not change which components of comprehensive income are recognized in net income or other comprehensive income, or when an item of other comprehensive income must be reclassified to net income. The updates are effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. Since this standard impacts disclosure requirements only, the Company does not expect its adoption will have a material impact on our consolidated results of operations or financial condition.
F-10
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
In May 2011, the FASB issued ASU 2011-04 which was issued to provide a consistent definition of fair value and ensure that the fair value measurement and disclosure requirements are similar between U.S. GAAP and International Financial Reporting Standards ("IFRS"). ASU 2011-04 changes certain fair value measurement principles and enhances the disclosure requirements particularly for Level 3 fair value measurements. This guidance is effective for us beginning on January 1, 2012. Its adoption is not expected to significantly impact our financial statements.
3. Fair Value of Financial Instruments
We apply the provisions of FASB ASC 820, Fair Value Measurements and Disclosures, which defines fair value, establishes a framework for measuring fair value under US GAAP, and enhances disclosures about fair value measurements.
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The standard establishes a valuation hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizing the use of unobservable inputs by requiring that the most observable inputs be used when available.
The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value. Our assessment of the significance of a particular input to the fair value measurements requires judgment, and may affect the valuation of the assets and liabilities being measured and their placement within the fair value hierarchy. The fair value hierarchy of measurements is categorized into one of the following three levels based on the lowest level of significant input used:
Level 1— Quoted prices in active markets for identical assets or liabilities.
Level 2— Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3— Unobservable inputs that are supported by little or no market activity.
Our cash equivalents and short-term investments consist of bank deposits, money market funds, and corporate debt securities. Our money market funds are traded in active markets and are recorded at fair value based upon quoted market prices.
We determine the fair value of our bank certificate of deposit and corporate debt securities using other observable inputs which may include quoted prices for similar assets in active markets, quoted prices for identical or similar assets in markets which are not active, other quoted prices which are directly observable and/or market inputs that are not directly observable, but are derived from or corroborated by other observable market data. Accordingly, we have classified the valuation of these securities as Level 2.
Other financial instruments, including accounts receivable, accounts payable and accrued liabilities, are carried at cost, which we believe approximates fair value because of the short-term maturity of these instruments.
F-11
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
3. Fair Value of Financial Instruments (Continued)
A summary of financial assets (in thousands) measured at fair value on a recurring basis at December 31, 2011 and 2010 is as follows:
| |
December 31, 2011 | December 31, 2010 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Quoted Prices In Active Markets (Level 1) |
Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
Total | Quoted Prices In Active Markets (Level 1) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||||
Money market funds |
$ | 5,876 | $ | 5,876 | $ | — | $ | — | $ | 486 | $ | 486 | $ | — | ||||||||
Bank certificate of deposit |
100 | — | 100 | — | — | — | — | |||||||||||||||
Corporate debt securities |
2,621 | — | 2,621 | — | — | — | — | |||||||||||||||
Total |
$ | 8,597 | $ | 5,876 | $ | 2,721 | $ | — | $ | 486 | $ | 486 | $ | — | ||||||||
4. Short-Term Investments and Impairment
Short-term investments consist of bank certificates of deposits, money market funds and corporate debt securities. As of December 31, 2011, the remaining contractual maturities of all investments were less than 12 months. Due to the short-term nature of our investments, amortized cost approximates fair value for all investments.
During 2010 we sold our mutual fund investment and reclassified the unrealized gains of $46,000 to be reflected in the statements of operations.
5. Property and Equipment
At December 31, 2011 and 2010, property and equipment consist of the following (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | |||||
Furniture and fixtures |
$ | 56 | $ | 41 | |||
Machinery, equipment and tooling |
513 | 494 | |||||
Computers and software |
111 | 74 | |||||
Leasehold improvements |
90 | 49 | |||||
Accumulated depreciation |
(303 | ) | (192 | ) | |||
Property and equipment, net |
$ | 467 | $ | 466 | |||
Depreciation expense for the years ended December 31, 2011, 2010 and 2009 was $111,000, $74,000 and $60,000, respectively.
6. Commitments and Contingencies
Leases
On October 1, 2007, we entered into an operating lease agreement for our current facility which houses our corporate offices and manufacturing space. The term of this lease originally ran from October 1, 2007 through September 30, 2010. In June 2010, we executed an amendment to our lease that extended the lease term through September 30, 2011. In May 2011, we executed a second
F-12
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
6. Commitments and Contingencies (Continued)
amendment to our lease that extended the lease term through September 30, 2014. Under the terms of the second amendment, our base rent was reduced to the current market rate effective October 1, 2011 and will subsequently be increased by approximately 2.5% on October 1, 2012 and 2013.
On June 2, 2011, we entered into an operating sub-lease agreement for approximately 2,800 square feet of office space adjoining our current facility. The term of this lease runs from June 15, 2011 through September 30, 2014. The monthly base rent amount is fixed over the entire term of the sub-lease.
We also lease certain other office equipment under non-cancelable operating lease arrangements which are not recognized on our balance sheets.
Annual future minimum lease obligations under our operating lease agreements as of December 31, 2011 are as follows (in thousands):
2012 |
$ | 84 | ||
2013 |
82 | |||
2014 |
62 | |||
Total |
$ | 228 | ||
Rent expense was $88,000, $57,000 and $57,000 for each of the years ended December 31, 2011, 2010 and 2009, respectively.
Royalty Payments
The core intellectual property relating to our eSVS MESH was acquired from Medtronic, Inc. pursuant to an Assignment and License Agreement dated October 9, 2007. As consideration for the assignment of such intellectual property, we have agreed to pay Medtronic an aggregate of up to $15.0 million upon the achievement of certain sales milestones and a royalty of 4% on sales of our eSVS MESH. The milestones and related payments consist of $5.0 million due on the one-year anniversary of the first commercial sale of our eSVS MESH, $5.0 million due when our cumulative net sales reach $15.0 million and $5.0 million due when our cumulative net sales reach $40.0 million. We reached the first milestone in June 2010 and accrued an expense for the first milestone obligation at that time. This milestone obligation was paid in June 2011. Royalty obligations are payable 60 days after the end of each fiscal quarter, are recorded as a component of our cost of sales and will terminate upon the earlier of the expiration of all of the patents and patent applications acquired from Medtronic, Inc., or when the aggregate royalties paid reaches $100.0 million. We recognized royalty expense of $10,000 and $9,000 for the years ended December 31, 2011 and 2010, respectively.
Legal Proceedings
We are not currently engaged in any litigation.
Employment Agreements
We entered into an employment agreement with Manny Villafaña, our founder, Chief Executive Officer and, at that time, sole director, on July 19, 2007, which provides for a base salary and discretionary bonuses as determined by our Board of Directors. Mr. Villafaña is also entitled to
F-13
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
6. Commitments and Contingencies (Continued)
participate in any employee benefit plans we sponsor. If we terminate Mr. Villafaña's employment without cause, he is entitled to his base salary for the entire term of the employment agreement, which expires on July 1, 2012.
Mr. Villafaña's employment agreement contains usual and customary provisions relating to confidential information and assignment of inventions to us. In addition, the agreement contains certain provisions concerning Mr. Villafaña's post-employment activities. Pursuant to the agreement, he has agreed not to compete with us for a period of two years after the termination of his employment. Such two-year period will automatically be extended by one year increments, up to a total of five years, unless terminated by us. As consideration for this non-competition provision, we will make a monthly payment to Mr. Villafaña equal to one twelfth of his base salary at the time of termination, adjusted upward annually by the change in the Consumer Price Index, or CPI, beginning with the first month after termination of employment and continuing until the non-competition provision expires. Mr. Villafaña will also be entitled to continue his participation in our medical benefits plan for the term of the non-competition provision, provided he continues to pay the employee portion of the premium. Following the termination of his employment with us, Mr. Villafaña has also agreed to consult on non-confidential matters at the request of our Board of Directors.
We also entered into a change in control agreement with Mr. Villafaña on September 12, 2008. Under the terms of this agreement, if, within 24 months of a change in control, Mr. Villafaña's employment is terminated by us other than for cause, or if he resigns for good reason, Mr. Villafaña will be entitled to a prorated portion of any annual incentive bonus for the fiscal year in which the termination occurs and a severance benefit equal to three years of his base salary. The change in control agreement's expiration date was September 12, 2011, but it has been and will continue to be automatically extended by one-year increments unless either party provides written notice to the other of the intent not to extend the agreement.
We have entered into employment agreements with certain key employees providing for an annual salary and such benefits in the future as may be approved by the Board of Directors. We have also entered into change of control agreements with certain of our employees which provide that if the employee is terminated for a reason other than cause, or resigns for good reason, upon a merger, acquisition, sale of substantially all of our assets, or liquidation, the employee will receive severance payments equal to his or her monthly base salary for 12 to 36 months.
Indemnification Agreements
The Company, as permitted under Delaware law and in accordance with its Bylaws, indemnifies its officers and directors for certain events or occurrences, subject to certain limits, while the officer or director is or was serving at the Company's request in such capacity. The term of the indemnification period is for the officer's or director's lifetime. The Company may terminate the indemnification agreements with its officers and directors upon 90 days written notice, but termination will not affect claims for indemnification relating to events occurring prior to the effective date of termination. The maximum amount of potential future indemnification is unlimited; however, the Company has a director and officer insurance policy that limits its exposure and may enable it to recover a portion of any future amounts paid. The Company believes the fair value of these indemnification agreements is minimal. Accordingly, the Company has not recorded any liabilities for these agreements as of December 31, 2011 or 2010.
F-14
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
7. Convertible Promissory Notes and Equity Financing
Common Stock Offerings
On February 16, 2011, we issued 2,062,500 shares of our common stock in an initial public offering ("IPO") at $8.00 per share for gross proceeds of $16.5 million. We incurred $2.9 million in issuance costs for the IPO. In connection with our IPO, we entered into an underwriting agreement with Rodman & Renshaw, LLC ("Rodman") dated February 9, 2010, pursuant to which at closing on February 16, 2011, we paid Rodman fees of $1.3 million, which consisted of underwriting discounts of $1.16 million and non-accountable expense reimbursements of $165,000. In addition, we issued to the underwriters and their designees options to purchase an aggregate of 103,125 shares of our common stock at an exercise price of $10.00, or 125% of the purchase price of shares sold in the IPO. These options have a five year term and become exercisable on February 10, 2012, one year after the effective date of the IPO.
In October 2009, we commenced a private offering of 714,286 shares of our common stock to certain accredited investors at an offering price of $7.00 per share. We sold an aggregate of 317,161 shares of common stock in the private offering, which was completed in February 2010.
Common Stock Purchase Agreement
On October 24, 2011, we entered into a common stock purchase agreement (the "Purchase Agreement") with Aspire Capital Fund, LLC, an Illinois limited liability company ("Aspire Capital"), which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $20.0 million of shares of our common stock (the "Purchase Shares") over a three year period at purchase prices determined in accordance with the Purchase Agreement. Concurrently with entering into the Purchase Agreement, we also entered into a registration rights agreement with Aspire Capital, dated as of October 24, 2011, in which we agreed to file one or more registration statements as permissible and necessary to register under the Securities Act of 1933, as amended, the sale by Aspire Capital of the shares of our common stock that have been and that may be issued to Aspire Capital under the Purchase Agreement. We agreed to file the initial registration statement with the U.S. Securities and Exchange Commission (the "SEC") on or before November 21, 2011. This registration statement was declared effective by the SEC on January 26, 2012.
Pursuant to the Purchase Agreement, on any trading day on which the closing sale price of the Company's common stock exceeds $1.00 per share, we have the right, in our sole discretion, to present Aspire Capital with a purchase notice, directing Aspire Capital to purchase up to the lessor of 100,000 shares or $500,000 of the Company's common stock per trading day if the closing sale price of the Company's common stock is above $1.00 per share. The purchase price per Purchase Share will be equal to the lesser of (i) the lowest sale price of the Company's common stock on the purchase date or (ii) the arithmetic average of the three lowest closing sale prices for the Company's common stock during the twelve consecutive trading days ending on the trading day immediately preceding the purchase date. There are no trading volume requirements or restrictions under the Purchase Agreement. Aspire Capital has no right to require any sales by us, but is obligated to make purchases from us as we direct in accordance with the Purchase Agreement.
In addition, on any date on which we submit a Purchase Notice to Aspire Capital, we also have the right, in our sole discretion, to present Aspire Capital with a volume-weighted average price purchase notice (each, a "VWAP Purchase Notice") directing Aspire Capital to purchase an amount of
F-15
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
7. Convertible Promissory Notes and Equity Financing (Continued)
our common stock equal to up to 30% of the aggregate shares of common stock traded on the next business day (the "VWAP Purchase Date"), subject to a maximum number of shares determined by us (the "VWAP Purchase Share Volume Maximum"). The purchase price per Purchase Share pursuant to such VWAP Purchase Notice (the "VWAP Purchase Price") shall be the lower of (i) the closing sale price on the VWAP Purchase Date, or (ii) 95% of the volume weighted average price for all or a portion of our common stock traded on the VWAP Purchase Date. The 95% of the volume weighted average price shall be calculated based on the shares traded during (i) the VWAP Purchase Date if the aggregate shares to be purchased on such date does not exceed the VWAP Purchase Share Volume Maximum, or (ii) the portion of such business day until such time as the aggregate shares to be purchased will equal the VWAP Purchase Share Volume Maximum. Further, if the sale price of our common stock falls on the VWAP Purchase Date below the greater of (i) 90% of the closing price of our common stock on the business day immediately preceding the VWAP Purchase Date or (ii) a higher price set by us in the VWAP Purchase Notice (the "VWAP Minimum Price Threshold"), the VWAP Purchase Price will be determined using the percentage in the VWAP Purchase Notice of the total shares traded for such portion of the VWAP Purchase Date prior to the time that the sale price of our common stock fell below the VWAP Minimum Price Threshold and the volume weighted average price of our common stock sold during such portion of the VWAP Purchase Date prior to the time that the sale price of our common stock fell below the VWAP Minimum Price Threshold.
In consideration for entering into the Purchase Agreement, concurrently with the execution of the Purchase Agreement, we issued to Aspire Capital 378,788 shares of our common stock as a commitment fee (the "Commitment Shares"). The Purchase Agreement provides that we may not issue and sell more than 3,164,357 shares of our common stock, including the Commitment Shares (19.9% of the Company's outstanding shares as of October 24, 2011, the date of the Purchase Agreement), unless stockholder approval is obtained in compliance with the applicable listing maintenance rules of The NASDAQ Global Market. We currently intend to seek stockholder approval of the transactions contemplated by the Purchase Agreement.
As of December 31, 2011, we have not sold shares of common stock to Aspire Capital pursuant to the Purchase Agreement. As of December 31, 2011, a total of 378,788 shares of common stock, representing the Commitment Shares, have been issued to Aspire Capital pursuant to the Purchase Agreement.
The Purchase Agreement contains customary representations, warranties, covenants, closing conditions and indemnification and termination provisions by, among and for the benefit of the parties. The Purchase Agreement may be terminated by us at any time, at our discretion, without any cost or penalty to us. Aspire Capital has covenanted not to cause or engage in any manner whatsoever, any direct or indirect short selling or hedging of our shares. We did not pay Aspire Capital any expense reimbursement in connection with the transaction. There are no limitations on use of proceeds, financial or business covenants, restrictions on future fundings, rights of first refusal, participation rights, penalties or liquidated damages in the Purchase Agreement.
Investment Agreement with Kips Bay Investments, LLC
We are a party to an Investment Agreement dated July 19, 2007 with Manny Villafaña and Kips Bay Investments, LLC, or KBI, which had no relationship to us prior to entering into the Investment Agreement. Pursuant to the Investment Agreement, KBI sold us all of its right, title and interest to
F-16
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
7. Convertible Promissory Notes and Equity Financing (Continued)
certain intellectual property assets in exchange for a first secured promissory note dated July 19, 2007 with a principal amount of $100,000 and loaned to us $2.9 million in exchange for a second secured promissory note dated July 19, 2007. The $100,000 note and the $2.9 million note, collectively the Notes, accrued interest at a rate of 9% per annum. All principal and accrued interest under the Notes was convertible into shares of our common stock at a per share price of $0.625 per share.
In March 2009, KBI converted the entire principal amount of $3.0 million and partially converted $217,188 of $467,188 in accrued interest on the Notes into 5,147,389 shares of our common stock at a price of $0.625 per share, and we paid KBI the balance of $250,000 of accrued interest in cash.
The Investment Agreement also granted KBI two stock purchase options. The first stock purchase option granted KBI the right to purchase 600,000 shares of our common stock for $3.5 million following our determination that our eSVS MESH was suitable for human implantation. The second stock purchase option granted KBI the right to purchase an additional 600,000 shares of our common stock for $3.5 million following the first implantation of our eSVS MESH. The Investment Agreement also provides certain registration rights to KBI. The relative fair value of the stock purchase options and the beneficial conversion feature on the Notes were recorded as discounts on the Notes and were amortized over the term of the Notes using the effective interest method.
In April 2008, we determined that our eSVS MESH was suitable for human implantation, and KBI subsequently exercised its first stock purchase option under the Investment Agreement, purchasing an aggregate of 600,000 shares of our common stock for a purchase price of $3.5 million in nine installments from May 2008 to June 2009. In August 2008, the first implantation of our eSVS MESH took place thereby satisfying the condition to the second stock purchase option. KBI exercised the second option in February 2010, purchasing an additional 600,000 shares for a purchase price of $3.5 million.
Effective January 1, 2009, we adopted FASB ASC 815-40, (formerly EITF Issue No. 07-5, Determining Whether an Instrument (or Embedded Feature) is Indexed to an Entity's Own Stock), which clarified the determination of whether equity-linked instruments (or embedded features), such as the options to purchase our common stock granted to KBI, are considered indexed to our own stock, which would qualify as a scope exception under FASB ASC 815. As a result of adopting FASB ASC 815-40, the second option to purchase our common stock granted to KBI is considered a derivative instrument and is measured and reported at fair value. As noted above, the first option was exercised in 2008 and was not outstanding at the effective date for FASB ASC 815-40.
Prior to the exercise of the second option, the estimated fair value this stock purchase option liability was recorded as a current liability on our balance sheets. Changes in the estimated fair value of this liability were recorded in our statements of operations.
On January 1, 2009, the date of adoption, we estimated the fair value of the second option to be $1.6 million and this amount was recorded as a cumulative effect adjustment on January 1, 2009, which increased our accumulated deficit $1.4 million. We estimated the fair value of this option as of January 1, 2009 using a Black-Scholes valuation model using the following assumptions: fair value of our common stock: $6.00; dividend yield: 0%; volatility: 70%; risk free interest rate: 0.88%; and expected term: 2.5 years. The fair value of our common stock was determined based upon the sale price in our private placement offering that commenced in March 2009. The estimated dividend yield is zero as we have no intent to pay dividends in the foreseeable future. Volatility was estimated based
F-17
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
7. Convertible Promissory Notes and Equity Financing (Continued)
upon a portfolio of guideline companies in the same or similar lines of business. The risk free interest rate was determined based upon the yield of constant maturity U.S. Treasury bills with durations approximating the expected term. The expected term was based upon the term of the Notes.
The effect of adjusting this liability to its estimated fair value at December 31, 2009 resulted in a net decrease in the estimated fair value of this liability of $610,000, resulting in an estimated fair value of this liability of $960,000. We estimated the fair value of this option as of December 31, 2009 using a Black-Scholes valuation model using the following assumptions: fair value of our common stock: $7.00; dividend yield: 0%; volatility: 70%; risk free interest rate: 0.06%; and expected term: 0.25 years. These assumptions changed from January 1, 2009 due primarily to the commencement of our second private placement offering, under which we sold our common stock at $7.00 per share. In December 2009, we began discussions with an investment banker in order to prepare for a potential initial public offering of our common stock. As a result, we decreased the estimated expected term to coincide with the anticipated timing of an initial public offering.
In connection with KBI's exercise of the second stock purchase option in February 2010, we and KBI entered into an agreement whereby KBI repaid us the $250,000 in cash and we issued KBI 400,000 shares of our common stock at a price of $0.625 per share, the conversion price of the Notes. We accounted for this agreement and its effect on the second stock purchase option as an exchange of the original option for a new option. As the second stock purchase option was exercised concurrent with the repayment and conversion of interest, the fair value of the option was determined based upon the difference between the fair value of our common stock and the exercise price of the option. The fair value of our common stock at the date of modification was determined to be $7.00 per share, based upon the sale price of our common stock to unrelated third-party investors under a private offering which was completed in February 2010. We recorded a charge of $2.3 million as the change in fair value of investor stock purchase option, increasing the recorded investor stock purchase option liability to $3.25 million. This liability was then reclassified to additional paid in capital in conjunction with issuance of shares related to the exercise of the second stock purchase option.
8. Stock-Based Compensation
2007 Long-Term Incentive Plan
Our 2007 Long-Term Incentive Plan, or the Plan, was adopted by the Board of Directors and approved by our stockholders in July 2007. The Plan permits the granting of incentive and non-statutory stock options, restricted stock, stock appreciation rights, performance units, performance shares and other stock awards to eligible employee, directors and consultants. We grant options to purchase shares of common stock under the Plan at no less than the fair market value of the underlying common stock as of the date of grant. Options granted under the Plan have a maximum term of ten years and generally vest over four years for employees, at the rate of 25% of total shares underlying the option each year, and over three years for non-employees, with 25% vesting upon grant and 25% vesting each year thereafter. Under the Plan, a total of 2,000,000 shares of common stock were initially reserved for issuance. As of December 31, 2011, we have 515,500 shares of common stock available for issuance under the Plan.
F-18
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
8. Stock-Based Compensation (Continued)
A summary of option activity is as follows:
| |
Shares Under Option |
Weighted Average Exercise Price |
|||||
|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2009 |
598,000 | $ | 3.08 | ||||
Granted |
215,000 | 7.00 | |||||
Exercised |
— | — | |||||
Forfeited and cancelled |
— | — | |||||
Options outstanding at December 31, 2010 |
813,000 | $ | 4.12 | ||||
Granted |
688,000 | 3.41 | |||||
Exercised |
(87,500 | ) | 1.00 | ||||
Forfeited and cancelled |
(152,500 | ) | 5.93 | ||||
Options outstanding at December 31, 2011 |
1,261,000 | $ | 3.73 | ||||
A summary of the status of our unvested shares during the year ended and as of December 31, 2011 is as follows:
| |
Shares Under Option |
Weighted Average Grant-Date Fair Value |
|||||
|---|---|---|---|---|---|---|---|
Unvested at December 31, 2010 |
400,250 | $ | 2.67 | ||||
Granted |
688,000 | 1.57 | |||||
Vested |
(190,125 | ) | 1.82 | ||||
Forfeited and cancelled |
(137,125 | ) | 3.05 | ||||
Unvested at December 31, 2011 |
760,625 | $ | 1.86 | ||||
Information about stock options outstanding, vested and expected to vest as of December 31, 2011, is as follows:
| Outstanding, Vested and Expected to Vest | Options Vested | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Per Share Exercise Price |
Shares | Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
Options Exercisable |
Weighted Average Remaining Contractual Life (Years) |
||||||||||||
| $ | 1.00 | 234,000 | 5.86 | $ | 1.00 | 233,000 | 5.86 | ||||||||||
| 1.62 | 5,000 | 9.91 | 1.62 | — | — | ||||||||||||
| 2.00 | 418,000 | 9.72 | 2.00 | 35,625 | 9.04 | ||||||||||||
| 3.21 | 15,000 | 9.39 | 3.21 | — | — | ||||||||||||
| 5.25 | 130,000 | 9.33 | 5.25 | — | — | ||||||||||||
| 5.83 | 174,000 | 6.80 | 5.83 | 164,250 | 6.81 | ||||||||||||
| 6.00 | 30,000 | 7.68 | 6.00 | 15,000 | 7.68 | ||||||||||||
| 6.10 | 95,000 | 9.18 | 6.10 | — | — | ||||||||||||
| 7.00 | 160,000 | 8.07 | 7.00 | 52,500 | 8.07 | ||||||||||||
| 1,261,000 | 8.26 | 3.44 | 500,375 | 5.86 | |||||||||||||
F-19
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
8. Stock-Based Compensation (Continued)
The cumulative grant date fair value of employee options vested during the years ended December 31, 2011, 2010 and 2009 was $244,000, $118,000 and $67,000, respectively. The total intrinsic value of options exercised during the years ended December 31, 2011, 2010 and 2009 was $58,000, $0 and $5,000, respectively. Total proceeds received for options exercised during the year ended December 31, 2011, 2010 and 2009 were $87,500, $0 and $1,000, respectively. On an aggregated basis, as of December 31, 2011, there was no intrinsic value for our total outstanding options and for our options exercisable.
As of December 31, 2011, 2010 and 2009, total compensation expense related to unvested employee stock options not yet recognized was $944,000, $608,000 and $297,000, respectively, which is expected to be allocated to expenses over a weighted-average period of 2.72, 1.88 and 2.22 years, respectively.
The assumptions used in the Black-Scholes option-pricing model for the years ended December 31, 2011, 2010 and 2009 are as follows:
| |
December 31, | |||||
|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||
Risk free interest rate |
1.24% - 2.63% | 2.45 - 2.82% | 1.75 - 2.80% | |||
Dividend yield |
0% | 0% | 0% | |||
Expected volatility |
53% | 50% | 48% | |||
Expected term |
5.75 - 6.25 years | 5.75 - 6.25 years | 6.25 years | |||
Weighted average grant date fair value |
$1.76 | $3.56 | $2.91 | |||
Nonemployee Stock-Based Compensation
We account for stock options granted to nonemployees in accordance with FASB ASC 718. In connection with stock options granted to nonemployees we recorded $30,000, $388,000 and $342,000 for nonemployee stock-based compensation during the years ended December 31, 2011, 2010 and 2009, respectively. These amounts were based upon the fair value of the vested portion of the grants.
Amounts expensed during the remaining vesting period will be determined based on the fair value at the time of vesting.
Restricted stock awards
A summary of restricted stock award activity is as follows:
| |
Number of Shares |
Weighted Average Fair Value |
|||||
|---|---|---|---|---|---|---|---|
Awards outstanding at December 31, 2010 |
— | — | |||||
Granted |
135,000 | $ | 5.89 | ||||
Vested |
— | — | |||||
Cancelled |
— | — | |||||
Awards outstanding at December 31, 2011 |
135,000 | $ | 5.89 | ||||
F-20
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
8. Stock-Based Compensation (Continued)
The fair value of each restricted stock award is equal to the fair market value of our common stock at the date of grant. Restricted stock awards vest over a period of time that varies with the purpose of the individual award. As of December 31, 2011, outstanding awards vest over a range of one to four years. The estimated fair value of restricted stock awards, including the effect of estimated forfeitures, is recognized on a straight-line basis over the restricted stock's vesting period. We recorded stock-based compensation expense for restricted stock grants of $159,000 for the year ended December 31, 2011.
Common Stock Purchase Options issued to underwriters
In conjunction with the completion of our initial public offering in February 2011, we issued to the underwriters and their designees options to purchase an aggregate of 103,125 shares of our common stock at an exercise price of $10.00, or 125% of the purchase price of shares sold in the IPO. These options have a five year term and become exercisable on February 10, 2012, one year after the effective date of the IPO. These options were not issued under our 2007 Long-Term Incentive Plan.
9. Employee Benefit Plan
We maintain a simplified employee retirement plan, or SEP, which commenced on January 1, 2008. The SEP is a defined contribution plan; employee contributions are voluntary and are determined on an individual basis, limited by the maximum amounts allowable under federal tax regulations. We contribute up to 3% of each individual's base salary as required under the safe-harbor provisions of Internal Revenue Service rules governing SEP plans. Our contributions vest immediately and are expensed when paid. We have recorded contributions of $52,000, $46,000 and $38,000 for the years ended December 31, 2011, 2010 and 2009, respectively.
10. Income Taxes
We have incurred net operating losses since inception. We have not reflected the benefit of net operating loss carryforwards in the accompanying financial statements and have established a full valuation allowance against our deferred tax assets.
The state of Minnesota enacted a change to their tax code which made the Minnesota Research and Tax Credit a refundable credit for fiscal years starting on or after January 1, 2010. Based upon our research and development costs which qualify under the Minnesota tax code, we recorded an income tax benefit of $42,000 for the year ended December 31, 2010. We had no such qualifying expenses during 2011.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, and operating losses and tax credit carryforwards.
F-21
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
10. Income Taxes (Continued)
The significant components of our deferred tax assets and liabilities for are as follows (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | |||||
Deferred tax assets: |
|||||||
Net operating loss carryforwards |
$ | 5,468 | $ | 4,099 | |||
Intangible assets—patent prosecution costs |
138 | 119 | |||||
Stock-based compensation |
493 | 451 | |||||
Milestone obligation |
1,821 | 2,190 | |||||
Research credit carryforwards |
230 | 434 | |||||
Other |
42 | 79 | |||||
Total deferred tax assets |
8,192 | 7,372 | |||||
Deferred tax liabilities: |
(5 | ) | — | ||||
Total deferred taxes, net |
8,187 | 7,372 | |||||
Valuation allowance |
(8,187 | ) | (7,372 | ) | |||
Net deferred tax asset |
$ | — | $ | — | |||
A reconciliation of the statutory tax rates and the effective tax rates is as follows (in thousands):
| |
Year Ended December 31, |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Statutory rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||
Permanent differences |
(9.0 | ) | (8.8 | ) | (1.4 | ) | ||||
State and local income taxes |
6.0 | 7.5 | 9.4 | |||||||
Credits and other |
0.5 | 1.2 | 5.5 | |||||||
State tax rate true-up |
(12.3 | ) | — | — | ||||||
Valuation allowance |
(19.2 | ) | (33.5 | ) | (47.5 | ) | ||||
Effective rate |
0.0 | % | 0.4 | % | 0.0 | % | ||||
Realization of the future tax benefits is dependent on our ability to generate sufficient taxable income within the carry-forward period. Because of our history of operating losses, management believes that the deferred tax assets arising from the above-mentioned future tax benefits are currently not likely to be realized and, accordingly, we have provided a full valuation allowance. The net valuation allowance increased by $815,000 and $3.7 million for the years ended December 31, 2011 and 2010, respectively.
Net operating losses and tax credit carryforwards as of December 31, 2011, are as follows:
| |
Amount | Expiration Years | |||
|---|---|---|---|---|---|
| |
(In thousands) | |
|||
Net operating losses—federal |
$ | 13,513 | Beginning 2027 | ||
Tax credits—federal |
$ | 167 | Beginning 2027 | ||
F-22
Kips Bay Medical, Inc.
Notes To Financial Statements (Continued)
10. Income Taxes (Continued)
Utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, or the IRC, and similar state provisions. We have not performed a detailed analysis to determine whether an ownership change under Section 382 of the IRC has occurred. The effect of an ownership change would be the imposition of an annual limitation on the use of net operating loss carryforwards attributable to periods before the change.
We would classify interest and penalties related to uncertain tax positions in income tax expense, if applicable. There was no interest expense or penalties related to unrecognized tax benefits recorded through December 31, 2011. The tax years 2007 through 2011 remain open to examination by federal and state tax authorities.
11. Selected quarterly financial data (unaudited)
The following summarized unaudited quarterly financial data has been prepared using the unaudited quarterly financial statements of the Company (In thousands, except per share amounts):
| |
First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Fiscal 2011 |
|||||||||||||
Net sales |
$ | 110 | $ | 48 | $ | — | $ | 94 | |||||
Gross profit |
$ | 73 | $ | 27 | $ | — | $ | 61 | |||||
Operating loss |
$ | (913 | ) | $ | (1,049 | ) | $ | (1,194 | ) | $ | (1,113 | ) | |
Net loss |
$ | (909 | ) | $ | (1,044 | ) | $ | (1,188 | ) | $ | (1,109 | ) | |
Basic and diluted net loss per share |
$ | (0.06 | ) | $ | (0.07 | ) | $ | (0.08 | ) | $ | (0.07 | ) | |
Fiscal 2010 |
|||||||||||||
Net sales |
$ | — | $ | 48 | $ | 59 | $ | 116 | |||||
Gross profit |
$ | — | $ | 29 | $ | 37 | $ | 80 | |||||
Operating loss |
$ | (1,199 | ) | $ | (5,875 | ) | $ | (736 | ) | $ | (891 | ) | |
Net loss |
$ | (3,485 | ) | $ | (5,871 | ) | $ | (733 | ) | $ | (803 | ) | |
Basic and diluted net loss per share |
$ | (0.27 | ) | $ | (0.43 | ) | $ | (0.05 | ) | $ | (0.06 | ) | |
Fiscal 2009 |
|||||||||||||
Operating loss |
$ | (1,022 | ) | $ | (892 | ) | $ | (939 | ) | $ | (930 | ) | |
Net loss |
$ | (1,125 | ) | $ | (816 | ) | $ | (1,279 | ) | $ | (117 | ) | |
Basic and diluted net loss per share |
$ | (0.14 | ) | $ | (0.07 | ) | $ | (0.10 | ) | $ | (0.01 | ) | |
Quarterly calculations of basic and diluted loss per share are made independently during each fiscal quarter.
F-23
Kips Bay Medical, Inc.
Annual Report On Form 10-K
Exhibit Index
| Exhibit Number |
Description of Exhibit | ||
|---|---|---|---|
| 3.1 | Certificate of Incorporation of the Registrant—incorporated by reference to Exhibit 3.1 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. | ||
3.2 |
Amended and Restated Bylaws of the Registrant—incorporated by reference to Exhibit 3.2 to the Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2010 filed with the SEC on March 31, 2011. |
||
4.1 |
Form of Common Stock Certificate—incorporated by reference to Exhibit 4.1 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
||
4.2 |
Registration Rights Agreement by and between Registrant and Aspire Capital Fund, LLC, dated as of October 24, 2011—incorporated by reference to Exhibit 4.1 to the Registrant's Current Report on Form 8-K filed with the SEC on October 25, 2011. |
||
10.1 |
Lease Agreement by and between the Registrant and St. Paul Properties, Inc., as assigned to St. Paul Fire and Marine Insurance Company, dated as of July 26, 2007—incorporated by reference to Exhibit 10.1 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.2 |
Investment Agreement by and between the Registrant and Kips Bay Investments, LLC, dated as of July 19, 2007—incorporated by reference to Exhibit 10.2 to the Registrant's amendment no. 1 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on May 20, 2010. |
||
10.3 |
Loan and Security Agreement by and between the Registrant and Kips Bay Investments, LLC, dated as of June 19, 2007—incorporated by reference to Exhibit 10.3 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.4 |
First Secured Convertible Promissory Note executed by the Registrant in favor of Kips Bay Investments, LLC, dated as of July 19, 2007—incorporated by reference to Exhibit 10.4 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.5 |
Second Secured Convertible Promissory Note executed by the Registrant in favor of Kips Bay Investments, LLC, dated as of July 19, 2007—incorporated by reference to Exhibit 10.5 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.6 |
Agreement by and between the Registrant and Kips Bay Investments, LLC, dated as of February 12, 2010—incorporated by reference to Exhibit 10.6 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.7 |
Debt Conversion Agreement by and between the Registrant and Kips Bay Investments, LLC, dated as of February 12, 2010—incorporated by reference to Exhibit 10.7 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
| Exhibit Number |
Description of Exhibit | ||
|---|---|---|---|
| 10.8 | Assignment and License Agreement by and between the Registrant and Medtronic, Inc., dated as of October 9, 2007—incorporated by reference to Exhibit 10.8 to the Registrant's amendment no. 4 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on August 12, 2010. | ||
10.9 |
Assignment by Medtronic, Inc. to the Registrant, dated as of August 26, 2008—incorporated by reference to Exhibit 10.9 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.10 |
Trademark Transfer Agreement by Medtronic, Inc. to the Registrant, dated as of October 10, 2007—incorporated by reference to Exhibit 10.10 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
||
10.11 |
* |
Employment Agreement by and between the Registrant and Manuel A. Villafaña, dated as of July 19, 2007—incorporated by reference to Exhibit 10.11 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.12 |
* |
Employment Agreement by and between the Registrant and Scott Kellen, dated as of February 8, 2010—incorporated by reference to Exhibit 10.13 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.13 |
* |
Form of Indemnification Agreement between the Registrant and its Directors and Executive Officers—incorporated by reference to Exhibit 10.14 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
|
10.14 |
* |
Change in Control Agreement by and between the Registrant and Manuel A. Villafaña, dated as of September 12, 2008—incorporated by reference to Exhibit 10.15 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.15 |
* |
Change in Control Agreement by and between the Registrant and Scott Kellen, dated as of February 8, 2010—incorporated by reference to Exhibit 10.17 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.16 |
* |
2007 Long-Term Incentive Plan—incorporated by reference to Exhibit 10.18 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.17 |
* |
Form of Incentive Stock Option Agreement under the 2007 Long-Term Incentive Plan—incorporated by reference to Exhibit 10.19 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.18 |
* |
Form of Non-Qualified Stock Option Agreement under the 2007 Long-Term Incentive Plan—incorporated by reference to Exhibit 10.20 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.19 |
* |
Form of Restricted Stock Agreement under the 2007 Long-Term Incentive Plan—incorporated by reference to Exhibit 10.21 to the Registrant's registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on April 8, 2010. |
|
10.20 |
Letter by and between the Registrant and Kips Bay Investments, LLC, dated May 19, 2010—incorporated by reference to Exhibit 10.22 to the Registrant's amendment no. 1 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on May 20, 2010. |
||
10.21 |
Indemnification Agreement between the Registrant and Manny Villafaña—incorporated by reference to Exhibit 10.23 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
||
| Exhibit Number |
Description of Exhibit | ||
|---|---|---|---|
| 10.22 | Indemnification Agreement between the Registrant and Scott Kellen—incorporated by reference to Exhibit 10.25 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. | ||
10.23 |
Indemnification Agreement between the Registrant and Arch C. Smith—incorporated by reference to Exhibit 10.26 to the Registrant's amendment no. 9 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 18, 2011. |
||
10.24 |
Indemnification Agreement between the Registrant and Robert E. Munzenrider—incorporated by reference to Exhibit 10.27 to the Registrant's amendment no. 9 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 18, 2011. |
||
10.25 |
Indemnification Agreement between the Registrant and Robert J. Sheehy—incorporated by reference to Exhibit 10.28 to the Registrant's amendment no. 9 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 18, 2011. |
||
10.26 |
Assignment by Medtronic, Inc. to the Registrant, dated as of October 10, 2007—incorporated by reference to Exhibit 10.29 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
||
10.27 |
Consulting Agreement by and between the Registrant and Symbios Clinical, Inc., dated as of July 21, 2008—incorporated by reference to Exhibit 10.30 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
||
10.28 |
CRO Services Agreement by and between the Registrant and Symbios Clinical, Inc., dated as of March 25, 2010—incorporated by reference to Exhibit 10.31 to the Registrant's amendment no. 2 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on June 11, 2010. |
||
10.29 |
Amendment No. 1 to Lease by and between the Registrant and St. Paul Fire and Marine Insurance Company, dated as of June 14, 2010—incorporated by reference to Exhibit 10.32 to the Registrant's amendment no. 4 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on August 12, 2010. |
||
10.30 |
Distribution Agreement by and between the Registrant and LeviBiotech s.r.l., dated July 1, 2010—incorporated by reference to Exhibit 10.33 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.31 |
Distribution Agreement by and between the Registrant and Master Surgery Systems AS, dated November 22, 2010—incorporated by reference to Exhibit 10.34 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.32 |
Distribution Agreement by and between the Registrant and Pacific Medical Systems Ltd., dated October 12, 2010—incorporated by reference to Exhibit 10.35 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.33 |
Distribution Agreement by and between the Registrant and Systemed A.E., dated November 22, 2010—incorporated by reference to Exhibit 10.36 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
| Exhibit Number |
Description of Exhibit | ||
|---|---|---|---|
| 10.34 | Distribution Agreement by and between the Registrant and Transmedic PTE Ltd., dated August 2, 2010—incorporated by reference to Exhibit 10.37 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. | ||
10.35 |
Distribution Agreement by and between the Registrant and Advanced Biomedical Pty Ltd., dated July 22, 2010—incorporated by reference to Exhibit 10.38 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.36 |
Distribution Agreement by and between the Registrant and Biomed, S.A., dated July 12, 2010—incorporated by reference to Exhibit 10.39 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.37 |
Distribution Agreement by and between the Registrant and Calmedical Ltd., dated June 17, 2010—incorporated by reference to Exhibit 10.40 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.38 |
Distribution Agreement by and between the Registrant and Cardiac Services Ltd., dated November 22, 2010—incorporated by reference to Exhibit 10.41 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.39 |
Distribution Agreement by and between the Registrant and F.O.C.S. GmbH, dated June 17, 2010—incorporated by reference to Exhibit 10.42 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.40 |
Distribution Agreement by and between the Registrant and Fehling Instruments Middle East F.Z.C., dated September 1, 2010—incorporated by reference to Exhibit 10.43 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.41 |
Distribution Agreement by and between the Registrant and Krijnen Medical Innovations B.V., dated June 17, 2010—incorporated by reference to Exhibit 10.44 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.42 |
Distribution Agreement by and between the Registrant and ERA Foreign Trade Ltd., dated October 27, 2010—incorporated by reference to Exhibit 10.45 to the Registrant's amendment no. 8 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 7, 2011. |
||
10.43 |
Distribution Agreement by and between the Registrant and Sygan Medical GmbH, dated January 11, 2011—incorporated by reference to Exhibit 10.46 to the Registrant's amendment no. 9 to registration statement on Form S-1 (Reg. No. 333-165940) filed with the SEC on January 18, 2011. |
||
10.44 |
* |
Employment Agreement by and between the Registrant and Michael Reinhardt, dated as of May 2, 2011—incorporated by reference to Exhibit 10.47 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 filed with the SEC on May 12, 2011. |
|
| Exhibit Number |
Description of Exhibit | ||
|---|---|---|---|
| 10.45 | * | Employment Agreement by and between the Registrant and Randy LaBounty, dated as of May 2, 2011—incorporated by reference to Exhibit 10.48 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 filed with the SEC on May 12, 2011. | |
10.46 |
* |
Change in Control Agreement by and between the Registrant and Michael Reinhardt, dated as of May 2, 2011—incorporated by reference to Exhibit 10.49 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 filed with the SEC on May 12, 2011. |
|
10.47 |
* |
Change in Control Agreement by and between the Registrant and Randy LaBounty, dated as of May 2, 2011—incorporated by reference to Exhibit 10.50 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011 filed with the SEC on May 12, 2011. |
|
10.48 |
Amendment No. 2 to Lease by and between Registrant and St. Paul Fire and Marine Insurance Company, dated as of May 25, 2011—incorporated by reference to Exhibit 10.51 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended July 31, 2011 filed with the SEC on August 10, 2011. |
||
10.49 |
Sublease by and between the Registrant and New Horizon Enterprises, Ltd., dated as of June 2, 2011—incorporated by reference to Exhibit 10.52 to the Registrant's Quarterly Report on Form 10-Q for the quarter ended July 31, 2011 filed with the SEC on August 10, 2011. |
||
10.50 |
Common Stock Purchase Agreement by and between the Registrant and Aspire Capital Fund, LLC, dated as of October 24, 2011—incorporated by reference to Exhibit 10.1 to the Registrant's Current Report on Form 8-K filed with the SEC on October 25, 2011. |
||
10.51 |
Engagement and Fee Letter by and between the Registrant and Cohen and Company Capital Markets, LLC, dated as of October 13, 2011—incorporated by reference to Exhibit 10.2 to the Registrant's Current Report on Form 8-K filed with the SEC on October 25, 2011. |
||
10.52 |
+* |
2012 Director Stock Plan |
|
21 |
+ |
Subsidiaries of the Registrant—None. |
|
24.1 |
+ |
Powers of Attorney (see signature page). |
|
31.1 |
+ |
Chief Executive Officer Certification Pursuant to Rule 13a-14(a), of the Exchange Act, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
31.2 |
+ |
Chief Financial Officer Certification Pursuant to Rule 13a-14(a), of the Exchange Act, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
32.1 |
+ |
Chief Executive Officer Certification Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
32.2 |
+ |
Chief Financial Officer Certification Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
101 |
++ |
Financial statements from the annual report on Form 10-K of the Company for the year ended December 31, 2011, formatted in XBRL: (i) the Balance Sheets, (ii) the Statements of Operations, (iii) the Statements of Cash Flows, (iv) the Statements of Stockholders' Equity (Deficit), and (v) the Notes to Financial Statements tagged as blocks of text. |
|
- +
- Filed
herewith
- ++
- Furnished
herewith
- *
- Management Compensatory Plan or Arrangement