Attached files
| file | filename |
|---|---|
| EX-32 - SECTION 1350 CERTIFICATION - Vaxart, Inc. | d262743dex32.htm |
| EX-31.1 - RULE 13A-14(A)/15D-14(A) CERTIFICATION - Vaxart, Inc. | d262743dex311.htm |
| EXCEL - IDEA: XBRL DOCUMENT - Vaxart, Inc. | Financial_Report.xls |
| EX-23 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - Vaxart, Inc. | d262743dex23.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2011
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Nabi Biopharmaceuticals
(Exact name of registrant as specified in its charter)
| Delaware | 59-1212264 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
12270 Wilkins Avenue, Rockville, MD 20852
(Address of principal executive offices, including zip code)
(301) 770-3099
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $.10 per share
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ¨ Yes x No
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). x Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨ Smaller Reporting Company ¨
Indicate by check mark whether the Registrant is a shell company. ¨ Yes x No
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, as of the last business day of the Registrant’s most recently completed second fiscal quarter was: $216,835,372.
As of February 24, 2012, 42,892,605 shares of the Registrant’s common stock were outstanding.
Documents Incorporated by Reference
Portions of the Registrant’s definitive Proxy Statement for its Annual Meeting of Shareholders, which will be filed within 120 days after the close of the Registrant’s fiscal year ended December 31, 2011, are incorporated by reference into Part III.
Table of Contents
Nabi Biopharmaceuticals
| Page No. | ||||||
| Part I. |
||||||
| Item 1. |
1 | |||||
| Item 1A. |
12 | |||||
| Item 1B. |
15 | |||||
| Item 2. |
15 | |||||
| Item 3. |
15 | |||||
| Item 3a. |
15 | |||||
| Item 4. |
16 | |||||
| Part II. |
||||||
| Item 5. |
17 | |||||
| Item 6. |
19 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
20 | ||||
| Item 7A. |
26 | |||||
| Item 8. |
29 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
51 | ||||
| Item 9A. |
51 | |||||
| Item 9B. |
51 | |||||
| Part III. |
||||||
| Item 10. |
52 | |||||
| Item 11. |
52 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
52 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
52 | ||||
| Item 14. |
52 | |||||
| Part IV. |
||||||
| Item 15. |
Exhibits, Financial Statement Schedules | 53 | ||||
| 56 | ||||||
Table of Contents
Nabi Biopharmaceuticals
PART I
Overview
We are a biopharmaceutical company that has focused on the development of vaccines addressing unmet medical needs, including nicotine addiction. We have sought to leverage our experience and knowledge in powering the human immune system to target these serious unmet medical needs. We have been incorporated in Delaware since 1969 and our operations are located in Rockville, Maryland.
Our sole remaining product currently in development is NicVAX® [Nicotine Conjugate Vaccine], an innovative and proprietary investigational vaccine for the treatment of nicotine addiction and prevention of smoking relapse based on patented technology. We suffered a significant setback in 2011 when NicVAX did not achieve the primary endpoint in two Phase III efficacy trials conducted in the U.S. As a result, in November of 2011, we retained Piper Jaffray & Co. (Piper Jaffray) to assist with the exploration of strategic alternatives available to the company. This includes, but is not limited to, the sale, license or merger of all or part of the company or its assets, joint ventures, strategic alliances, recapitalization, or liquidation, and the process is ongoing. As of December 31, 2011, our remaining assets include the following: (i) $96.4 million of cash, cash equivalents and marketable securities, (ii) the potential residual value of NicVAX as well as any next-generation nicotine vaccine which was licensed to GlaxoSmithKline Biologicals S.A. (GSK) in 2010, (iii) the potential royalty from Phoslyra which was sold to Fresenius USA Manufacturing, Inc. (Fresenius) in 2006, and (iv) the potential value of our net operating losses (NOLs).
In the first quarter of 2010 we granted to GSK (i) an option to exclusively license NicVAX on a worldwide basis and (ii) a license to develop follow-on next-generation nicotine vaccines using our intellectual property combined with GSK proprietary technology including GSK proprietary adjuvants. GSK has not indicated that it is stopping the development of the next-generation nicotine vaccine. If GSK continues to develop the next-generation nicotine vaccine, Nabi may be eligible to receive milestones and royalties from that program. We continue to support an investigator initiated Phase IIb trial in the Netherlands of NicVAX in combination with Pfizer’s varenicline or Chantix®/Champix, the results of which are expected in the second half of 2012.
The smoking cessation market is estimated to exceed $4 billion annually and is currently considered to be a largely unmet medical need. Nicotine is a non-immunogenic small molecule that, upon inhalation into the lungs, quickly passes into the bloodstream and subsequently reaches the brain by crossing the blood-brain barrier. Once in the brain, the nicotine binds to specific nicotine receptors resulting in the release of substances, such as dopamine, a chemical which triggers the highly addictive pleasurable effects experienced by smokers and users of other nicotine products. NicVAX is designed to stimulate the immune system to produce highly specific antibodies that bind to nicotine in the bloodstream. A nicotine molecule attached to specific antibodies is too large to cross the blood-brain barrier and thus is unable to reach the receptors in the brain thereby reducing the pleasure experienced by the smoker making it easier to quit.
In March 2010, we closed an exclusive worldwide option and licensing agreement with GSK for NicVAX as well as for the development of follow-on nicotine addiction vaccines. Upon closing, we received a $40 million initial payment. Under the terms of the agreement, we granted to GSK (i) an option to obtain an exclusive worldwide license to develop, commercialize and manufacture NicVAX as it currently exists, as well as certain potential alternative forms of NicVAX together with an adjuvant other than a GSK proprietary adjuvant and/or with different presentation, dosage or administration (NicVAX Alternatives), and (ii) an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives). In addition to the $40 million received at closing, we are eligible to receive under the
1
Table of Contents
agreement up to $290 million in potential regulatory, development and sales milestones for any next generation nicotine vaccines. We are also eligible to receive royalties on global sales of any next generation of nicotine vaccines. Notwithstanding the failure to achieve the primary endpoint in the two Phase III trials, if GSK was to exercise its option under the agreement for NicVAX and any NicVAX alternatives, it will pay us $58 million upon exercise plus certain potential milestones and royalties over time.
During 2011 we completed our obligations to continue to develop PentaStaph™ [Pentavalent S.aureus Vaccine] under contract for GSK. PentaStaph is a new pentavalent vaccine designed to prevent S.aureus infections including those infections caused by the most dangerous antibiotic-resistant strains of S.aureus. In August 2009, we entered into an Asset Purchase Agreement (APA) with GSK for the sale of PentaStaph for a total consideration of $46 million including a $20 million up-front payment upon closing and $26 million payable upon achievement of certain milestones. The PentaStaph sale closed in November 2009, and we received payment from GSK of $21.5 million representing the up-front payment of $20 million, an additional $1 million for the sale of our Staphylococcus epidermidis vaccine program and an additional $0.5 million for transfer of certain specified materials. We also agreed to a Transition Services Agreement (TSA) to help GSK advance the program while in parallel transferring the technology to GSK. In 2011 we completed all of the efforts required of us from GSK under contract for PentaStaph. As a result, we have received the full $26 million in potential milestone payments under the APA, $5 million in 2011 and $21 million in 2010.
In 2006, we sold certain assets related to our PhosLo operations. Under the sale agreement, we received $65 million in cash at closing and are entitled to additional contingent milestone payments and royalties. The royalties relate to sales of a new product formulation over a base amount for 10 years after the November 14, 2006 closing date. In August of 2011, we announced that Fresenius had successfully launched the new product formulation, PhosLyra, and accordingly we received a $5 million milestone payment. We received $18 million of milestones through December 31 2011, and can also receive up to $67.5 million in additional milestone payments and royalties.
NICOTINE ADDICTION
Background
Smoking is a global healthcare problem. The World Health Organization estimates that there are over 1.3 billion smokers worldwide today and nearly five million tobacco-related deaths each year. If current smoking patterns continue, smoking will cause an estimated 10 million deaths each year by 2030. According to the U.S. Centers for Disease Control and Prevention (CDC), tobacco use is the single leading preventable cause of death in the U.S., responsible for approximately 443,000 deaths each year. In addition, it is estimated that smoking results in an annual health-related economic cost in the U.S. of approximately $193 billion. The CDC estimates that, among the 43.4 million adult smokers in the U.S., 70% want to quit, but less than five percent of those who try to quit remain smoke-free after 12 months.
Nicotine addiction is difficult to treat. Most current therapies involve the use of nicotine replacement products delivered via patches, lozenges or chewing gum. These therapies have shown only limited efficacy, particularly over the long term. Most smokers who stop smoking using current therapies resume their addiction after they stop therapy. Chantix, which is a prescription therapy introduced by Pfizer Inc. in 2006, acts by binding the nicotinic receptors in the brain and competing with inhaled nicotine for these receptors, while simultaneously partially activating these receptors, thereby breaking the addiction cycle. Data from the efficacy trials have shown that the short-term cessation rates were superior to other therapies for smokers receiving Chantix, although most individuals relapsed to smoking over the longer term. Significant neuropsychiatric adverse events have been reported including suicides and suicide ideation that led the FDA to require a warning (boxed warning) label on the drug in July 2009.
Nicotine is a small molecule that, upon inhalation or absorption into the body, quickly passes into the bloodstream and subsequently reaches the brain by crossing the blood-brain barrier. Once in the brain, the nicotine binds to specific nicotine receptors, resulting in the release of stimulants, such as dopamine, which
2
Table of Contents
provide the smoker with a positive sensation, leading to addiction. Because of its small size, nicotine on its own normally does not elicit the production of antibodies in humans. NicVAX is based on our proprietary conjugate technology whereby nicotine is attached to a carrier protein which renders the molecule immunogenic. Upon injection, NicVAX is capable of stimulating the immune system to produce nicotine-specific antibodies in the bloodstream that bind to nicotine from cigarette smoking or the use of other nicotine products and prevent it from crossing the blood-brain barrier and entering the brain. As a result, the brain does not release the positive-sensation stimulant dopamine. We believe NicVAX has safety advantages over existing treatment therapies, in part, because it does not act on the central nervous system.
NicVAX is our investigational vaccine designed as an aid to smoking cessation, as well as an aid to prevent relapse.
Clinical and Regulatory History
In March 2006, we announced that NicVAX received Fast Track Designation from the FDA. This designation is intended to facilitate the development of products that treat serious diseases where a significant unmet medical need exists. During 2006, we initiated and completed enrollment of a Phase IIb “proof-of-concept” study of 301 smokers who smoked an average of 24 cigarettes a day and thus, were highly addicted to smoking and who were randomly allocated to receive one of four administrations of NicVAX (two different doses according to two different schedules) or a placebo. This study was funded in part by the National Institute on Drug Abuse (NIDA).
The Phase IIb study was a double blind, placebo-controlled and dose-ranging study designed to establish proof-of-concept and the optimal dose for a Phase III program. This study, designed in collaboration with the FDA and other global regulatory agencies, incorporated the current clinical trial standards and protocol design for smoking cessation clinical research studies. The trial’s primary endpoint was the rate of carbon monoxide (CO)-confirmed continuous abstinence from smoking during weeks 19-26. In May 2007, we announced the trial’s six-month data, which showed that a statistically significant number of subjects in the high anti-nicotine antibody responder-group met the trial’s primary endpoint of eight weeks of continuous abstinence during weeks 19-26.
In November 2007, we announced final results from this trial. The trial demonstrated that higher levels of anti-nicotine antibodies correlated to higher smoking cessation rates and long-term continuous abstinence rates, demonstrating proof-of-concept that antibodies to nicotine generated through NicVAX immunization were useful as an aid to smoking cessation. The high-antibody responder group of vaccinated subjects showed continuous abstinence rates that were almost three times higher than the placebo group at 12 months. Moreover, those subjects in the NicVAX group with a high antibody response who continued to smoke showed a statistically significant reduction in cigarettes smoked over the full 12 months compared to placebo (p<0.022).
Importantly, for the first time, a statistically significant treatment effect was observed for a single intent-to-treat dose group (not stratified by antibody-response) of nicotine vaccine compared to placebo. The observed treatment effect was continuous long-term smoking abstinence to one year compared with placebo for the group receiving 5 injections of 400 mcg of NicVAX. This data demonstrated that nearly three times the number of subjects treated with the most effective dose and schedule tested were able to quit smoking and remained abstinent to 12 months as compared with placebo (p<0.038).
NicVAX was well tolerated with a low prevalence of side effects and an adverse event profile comparable to that seen with placebo and other similar vaccines. Additionally, no statistically significant evidence of compensatory smoking or increase in withdrawal symptoms has been observed in NicVAX treated subjects as compared to placebo at any stage of the trial.
3
Table of Contents
Based on the results of the Phase IIb study, we believed that NicVAX could help more smokers to stop smoking if they attempt to quit when higher levels of anti-nicotine antibodies are reached. Based on the profile of anti-nicotine antibodies achieved in the Phase IIb proof-of-concept trial, we reasoned that higher levels of antibodies could be achieved if an additional dose of NicVAX would be administered. Therefore, we initiated an immunogenicity study in January 2008, to further understand the potential of this improved dosing regimen. The results of this study confirmed our hypothesis that significantly higher antibodies could be achieved earlier, and in a higher percentage of volunteers, by including an additional dose of NicVAX. Those results were used to finalize the dosing schedule for the NicVAX Phase III program. The FDA agreed with our Phase III trial design and endpoints through a special protocol assessment (SPA), providing a clear, well-defined path for the approval of NicVAX. The SPA is an agreement with the FDA which is intended to reduce the regulatory risk of the program. In addition, we also sought and obtained Scientific Advice from the European Medicines Agency (EMA) which generally is well aligned with the SPA with the FDA regarding the trial design.
In November 2009 and March 2010, respectively, we announced the initiation of the first and second Phase III clinical trials of NicVAX that we are required to conduct in support of the NicVAX license, based on our SPA agreement with the FDA. Each of the two Phase III clinical trials recruited 1,000 subjects randomized equally between NicVAX and placebo. Results of both Phase III trials announced in 2011 indicated that NicVAX did not meet the primary endpoint. This was an unexpected and disappointing result. We do not know why NicVAX failed to achieve efficacy given that the Phase IIb results were encouraging. It may be that the anti-nicotine antibody response was not high enough and/or the target quit date was too late. It is possible that the anti-nicotine antibody response may be enhanced by including a better adjuvant. Such a second generation nicotine vaccine was licensed to and may be developed by our partner, GSK.
The remaining trial for NicVAX is an ongoing investigator-initiated combination clinical trial in the Netherlands with Pfizer’s varenicline (Chantix/Champix). The results of this trial are expected in the second half of 2012.
The NicVAX development program has been guided by a panel of outside experts providing input to the design and implementation of the Phase III clinical trials and the overall clinical development program.
4
Table of Contents
STRATEGIC TRANSACTIONS
In November 2009, we sold PentaStaph to GSK for a total consideration of up to $46 million, including a $20 million up-front payment. In addition, GSK paid us $1.5 million, $1 million of which was for the purchase of the results of an early research program for a vaccine against S.epidermidis and $0.5 million was for certain clinical materials. We received the remaining $26 million upon our achievement of certain milestones.
As part of this transaction, we entered into a TSA with GSK that required us to successfully transfer the PentaStaph technology and certain materials to GSK, as well as to manage, on their behalf, the relationship with the U.S. military and the conduct of a Phase I/II trial for two of the PentaStaph antigens. GSK has reimbursed us the full cost of such activities.
In November 2009, we signed an exclusive worldwide option and licensing agreement with GSK for NicVAX as well as for the development of a second-generation nicotine vaccine; this transaction closed in March 2010. Under the terms of the agreement:
| • | We granted to GSK (i) an option to obtain an exclusive worldwide license to develop, commercialize and manufacture NicVAX and NicVAX Alternatives, and (ii) an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives); and |
| • | GSK made a non-refundable $40 million up-front payment and GSK agreed to make certain additional option, milestone and royalty payments. Due to the failure of NicVAX to achieve the primary endpoint in the two Phase III efficacy trials, we believe GSK may not exercise the NicVAX option and we may not realize the associated option exercise payment, milestones and royalties. |
If GSK does not exercise the NicVAX option:
| • | We retain the right to commercialize and partner NicVAX and NicVAX Alternatives (but not certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives)); and |
| • | We are eligible to receive development milestones for future generation candidates of (i) up to an aggregate of $47 million based on Phase II and Phase III clinical trial-related milestones, and (ii) up to an aggregate of $34 million based on obtaining regulatory approval in certain major market countries, for future generation candidates. |
In addition, upon successful development of a future generation candidate, GSK may:
| • | Pay us certain tiered, sales-milestone payments up to an aggregate of $209 million based on aggregate annual sales of future generation candidates; and |
| • | Pay us royalties beginning at 5% and potentially increasing on incremental sales to as high as 7%, with the increase depending on whether aggregate annual net sales of future generation candidates meet or exceed specified annual sales targets in any calendar year ranging from $300 million to $600 million, if a future generation candidate is successfully commercialized. |
The economic terms of GSK’s license of NicVAX Alternatives and/or Complementary R&D (such as the combination trial in the Netherlands) are subject to mutual agreement between GSK and us. If the parties cannot mutually agree, then such economic terms will be determined through binding arbitration based on an agreed upon set of factors and principles relating to, among other things, the commercial potential of the NicVAX Alternatives and/or Complementary R&D subject to the option exercise and the relative contributions of us and GSK to the development of such NicVAX Alternatives.
During the fourth quarter of 2006, we sold certain assets related to our PhosLo operations. Under the sale agreement, we received $65 million in cash at closing and are entitled to additional contingent milestone payments and royalties. The royalties relate to sales of a new product formulation over a base amount through 2016. We have received $18 million of milestones through December 2011, and can also receive up to $67.5 million in additional milestone payments and royalties.
5
Table of Contents
CONTRACT MANUFACTURING AND PRODUCT DEVELOPMENT RELATIONSHIPS
Contract Manufacturing
In September 2010, we entered into a manufacturing agreement with Diosynth Biotechnology for the manufacture of NicVAX drug substance which is a step in the production of the vaccine. The drug substance ultimately would have been combined with an adjuvant and filled in syringes at another contract manufacturing organization to produce NicVAX. As a result of the recent NicVAX Phase III clinical trials, we are considering whether or not this agreement remains necessary. If we terminate the agreement because we determine it is unnecessary, we would be subject to a termination penalty.
Product Development
In preparation for the commercialization of NicVAX and prior to the failure of the NicVAX Phase III trials, we entered into relationships for certain products in development. Currently, these activities are inactive.
National Institute for Drug Abuse
We have received grants from NIDA that in the past have supported clinical development of NicVAX. In addition, in September 2009, we were awarded a $10 million grant from NIDA in support of the first of two Phase III efficacy trials of NicVAX. This agreement was completed in 2011.
National Institutes of Health
Under a license agreement with NIH, we have a non-exclusive, worldwide right to use certain rEPA carrier protein technology to develop, manufacture and commercialize vaccines against nicotine addiction. Under the terms of this rEPA agreement, NicVAX is subject to a 0.5% royalty.
Brookhaven National Labs
Under a license agreement with Brookhaven National Labs (Brookhaven), we have a non-exclusive right, with the right to sublicense, to patented T7 polymerase technology for research, development, and commercialization of vaccines for preventing and treating nicotine addiction, and for prevention and treatment of Enterococcal infections. Under the terms of this T7 agreement, NicVAX is subject to a 0.1% royalty upon commercialization, and any Enterococcal vaccine is subject to a 0.2% royalty upon its commercialization. The T7 license remains in effect until the expiration of the last-to-expire licensed patent, which is December 2, 2014, and no further payments or royalties will be due to Brookhaven for use of the subject technology after that date.
RESEARCH AND DEVELOPMENT PROGRAMS
The following table provides the estimated amounts spent during the last three fiscal years on our research and development programs:
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
|||||||||
| NicVAX |
$ | 17,765 | $ | 25,447 | $ | 14,583 | ||||||
| PentaStaph |
— | 505 | 1,843 | |||||||||
| Other programs |
— | 126 | 64 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total R&D programs |
$ | 17,765 | $ | 26,078 | $ | 16,490 | ||||||
|
|
|
|
|
|
|
|||||||
Research and development expenses related to the NicVAX program are reflected net of grant reimbursements of $0.3 million, $8.5 million and $1.5 million in 2011, 2010 and 2009, respectively. Research and development expenses related to the PentaStaph program are reflected net of grant reimbursements of $0.4 million and $0.1 million in 2010 and 2009, respectively (none in 2011).
6
Table of Contents
PATENTS AND PROPRIETARY RIGHTS
We depend on our ability to maintain our rights to our existing patent portfolio and our ability to obtain patent protection for product candidates. As of December 31, 2011, we retain rights to 99 patents and 46 patent applications pending worldwide.
Smoking Cessation
Our patent portfolio comprehends both compositions and therapeutic methodology for treating or preventing addiction. Our patent claims related to the NicVAX product are directed to compositions, or conjugates, that comprise a nicotine-like molecule linked to a carrier protein and to methods for the use of these conjugates to treat or prevent nicotine addiction. In particular, we hold four issued U.S. patents related to our conjugates, antibodies against the conjugates, and methods for using the conjugates and antibodies against nicotine addiction. These patents expire in December 2018. Another granted U.S. patent related to a method of making nicotine haptens expires in 2027. We hold granted patents in the U.S., Europe, Japan and various other countries and regions, relating to our conjugates and antibodies against our conjugates, for use in treating nicotine addiction. In addition, we also have pending foreign patent applications relating to our conjugate technology and to our method of making nicotine haptens in various countries and regions. We also have pending U.S., Patent Cooperation Treaty and foreign applications relating to antibody-based diagnostic kits and methods for smoking cessation. Another granted U.S. patent filed in cooperation with NIH is directed towards a method to decrease the toxic effects of nicotine on fetuses.
In July 2005, Cytos Biotechnology Ltd. filed an opposition against our European patent that covers NicVAX and its use in the treatment and prevention of nicotine addiction. The European Patent Office (EPO) originally issued this patent to Nabi in late 2004 with an expiration date of January 12, 2019. We filed our response to the opposition in December 2005, and in April 2008, the EPO upheld the patent, preserving our primary claim that protects our exclusive use of NicVAX for treating and preventing nicotine addiction but cancelled some ancillary claims in the patent and we continue our appeal of these ancillary claim cancellations.
In September 2008, Nabi, as well as four other entities, filed oppositions to invalidate all or a portion of the claims of two patents issued in May 2007 to Celtic Pharma (formerly Xenova), which covered hapten-carrier conjugates for use in drug abuse therapy including nicotine addiction. In a formal decision issued March 2010, the Opposition Division of the EPO revoked one of the patents in its entirety and Celtic Pharma subsequently filed an appeal that is under review by the EPO. The second patent was scheduled for oral proceedings in December 2010 but Celtic Pharma withdrew its request for oral proceedings in October 2010.
Gram-positive Program
Patents and patent applications within our Enterococcus patent portfolio relates to polysaccharide antigens E.faecalis and E.faecium,.
Also in the Gram-positive program portfolio are issued U.S., European, Canadian and Mexican patents that contain claims directed to a pharmaceutical composition containing a glucan and antibodies specific for a given pathogen like S.aureus. This combination produces an unexpected antimicrobial effect that is greater than that obtained when either the glucan or antibodies are used separately. Another related U.S. patent has been granted with claims to a pharmaceutical composition containing a glucan and intravenous hyperimmune globulin. In November 2009, we granted GSK an exclusive (even as to Nabi), royalty-free, fully paid-up, worldwide, perpetual, irrevocable right and license, with the right to grant sublicenses to the family of patents and patent applications directed to glycoconjugate vaccines or a component thereof, for diagnosis, prevention or treatment of Staphylococcus infections in humans.
7
Table of Contents
Our granted U.S. patent relating to a method of protecting a human being with a compromised immune system from Staphylococcal infection using Type 5 and Type 8 antigens is now owned by GSK under the APA with respect to PentaStaph. This patent expires in 2022. Corresponding patents have been granted in Australia, China, Eurasia, India, New Zealand and South Africa and applications are pending in other countries. GSK granted us an exclusive (even as to GSK), royalty-free, fully paid-up, worldwide, perpetual, irrevocable right and license, with the right to grant sublicenses for pending claims related to a method of protecting a human being with a compromised immune system from Enterococcus infection using the claimed CPS antigens of E.faecalis and E.faecium.
Trade Secrets and Trademarks
We rely on unpatented proprietary technologies in our development efforts. We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as those of our advisors, consultants and other contractors that cannot be patented. To help protect our proprietary know-how, we often use trade secret protection and confidentiality agreements to protect our interests. We require employees, consultants and advisors to enter into agreements that prohibit the disclosure of confidential information and where applicable require disclosure and assignment to us of the ideas, developments, discoveries and inventions that arise from their activities for us.
We own or license trademarks associated with each of our development products, including several international trademark registrations or common law rights.
GOVERNMENT AND INDUSTRY REGULATION
Our research, pre-clinical development and conduct of clinical trials are subject to regulation for safety and efficacy by numerous governmental authorities. In the U.S., the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and other federal and state statutes and regulations govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, transportation, approval, advertising and promotion of certain products. In addition, these statutes, regulations and policies may change and our products may be subject to new legislation or regulations.
Biopharmaceutical Products
In the United States, vaccines are classified as biological products under U.S. Food and Drug Administration (FDA) regulations and are subject to rigorous regulation by the FDA. All of our products will require regulatory approval by governmental agencies prior to commercialization. The process of obtaining these approvals and subsequent process of maintaining substantial compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. The steps required before a biological product may be marketed in the U.S. generally include pre-clinical laboratory tests, animal tests and formulation studies, and the submission of an Investigational New Drug (IND) application to the FDA, which must be accepted by the FDA before human clinical studies may commence, and adequate and well-controlled clinical trials to establish the potency, safety and efficacy of the biological product for each indication for which FDA approval is sought.
The clinical phase of development involves the activities necessary to demonstrate safety, tolerability, efficacy and dosage of the substance in humans, as well as the ability to produce the substance and finished biological product in accordance with the FDA’s current Good Manufacturing Practices (cGMP) requirements. Clinical trials to support the approval of a biological product are typically conducted in three sequential phases, Phases I, II and III, with Phase IV clinical trials sometimes conducted after marketing approval. The initial human clinical evaluation, called a Phase I clinical trial, generally involves administration of a product to a small number of normal, healthy volunteers to test for safety. Phase II clinical trials involve administration of a product
8
Table of Contents
to a limited number of patients with a particular disease to determine dosage, immunogenicity and safety. In some cases Phase II clinical trials may provide limited indications of efficacy. Multiple Phase II clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase III clinical trials. In some cases, a sponsor may decide to conduct what is referred to as a “Phase IIb” evaluation, which is a second, confirmatory Phase II clinical trial. Phase III clinical trials examine the efficacy and safety of a product in an expanded patient population. Phase IV clinical trials primarily monitor for adverse effects and are undertaken post-licensure, such as additional large-scale, long-term studies of morbidity and mortality. The FDA may require sponsors to conduct Phase IV clinical trials to study certain safety issues and/or efficacy. The FDA reviews the clinical plans and the results of trials and can stop the trials at any time if there are significant safety issues.
Success in early-stage clinical trials does not necessarily assure success in later-stage clinical trials. Data obtained from clinical activities is not always conclusive and may be subject to alternative interpretations that could delay, limit or even prevent regulatory approval. In addition, the FDA can request that additional clinical trials be conducted as a condition to product approval.
The results of all trials are included in a Biologics License Application (BLA). The BLA must be approved by the FDA prior to commencement of commercial sales. For BLA approval, the FDA requires that the sponsor demonstrate a favorable risk-benefit ratio. This often involves treatment of large numbers of patients, typically in double-blind, placebo-controlled or comparative randomized trials, followed for protracted periods of time. The actual size of the trials and the length of follow-up vary from indication to indication. If the FDA determines that a Risk Evaluation and Mitigation Strategy (REMS) is necessary to ensure that the benefits of the biological product outweigh the risks, a sponsor may be required to include as part of the application a proposed REMS, including a package insert directed to patients, a plan for communication with healthcare providers, restrictions on distribution, or a medication guide to provide better information to consumers about the risks and benefits of the biological product. In addition, the prospective manufacturer’s methods must conform to the agency’s cGMP regulations, which must be followed at all times. The prospective manufacturer must submit three conformance lots in support of the application. In complying with standards set forth in these regulations, manufacturers must continue to expend time, money and effort in the areas of production, compliance and quality control to ensure full regulatory compliance. The submission of the BLA is no guarantee that the FDA will find it complete and accept it for filing. The FDA reviews all applications submitted before it accepts them for filing. It may request additional information rather than accept the application for filing, in which case, the application must be resubmitted with the supplemental information. After the BLA is accepted for filing, the FDA reviews the application to determine, among other things, whether a product is safe and efficacious for its intended use. The approval process is affected by several factors, including the severity of the disease, the availability of alternative treatments, and the risks and benefits demonstrated in clinical trials. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its BLA. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically composed of a panel of physicians and other experts, for review, evaluation, and an approval recommendation. The FDA also may require post-marketing surveillance to monitor potential adverse effects of the product and/or efficacy. If required to conduct a post-approval study, periodic status reports must be submitted to the FDA. Failure to conduct such post-approval studies in a timely manner may result in substantial civil fines.
The overall regulatory process is similar within the EU insofar as the sponsor needs to demonstrate a favorable risk-benefit ratio of the biological product, as well as reproducible manufacturing methods. The European equivalent of the BLA is called the Marketing Authorization Application (MAA). There are two different procedures to file an MAA: the Centralized Registration Procedure and the Mutual Recognition Procedure. The Centralized Registration Procedure allows for simultaneous approval throughout the EU. The Mutual Recognition Procedure provides for initial approval in one country that can be used to seek approval in additional countries within the EU. There have been different requirements from country-to-country with regard to initiating clinical trials. However, that is also in the process of being standardized. A new standardized procedure, the Clinical Trials Application, was introduced in the EU during 2004.
9
Table of Contents
Fast Track Designation
NicVAX was granted Fast Track review designation for the indication aid to smoking cessation in 2006.
Fast Track designation refers to a process of interacting with the FDA during drug development and is intended for a combination of a product and a claim that addresses an unmet medical need. The Fast Track mechanism is described in the Food and Drug Administration Modernization Act of 1997. The benefits of the Fast Track designation include scheduled meetings to seek FDA input into development plans, the option of submitting a BLA in sections rather than all components simultaneously, and the option of requesting evaluation of studies using surrogate endpoints. Award of the designation does not ensure product approval by the agency, and the agency can withdraw the designation if the product, during development, no longer meets the standards for meeting an unmet medical need. The Fast Track mechanism is independent of Priority Review and Accelerated Approval, which are other regulatory programs to expedite product development and review.
Special Protocol Assessment (SPA)
The Company had an agreement with the FDA in an SPA for both pivotal Phase III trials of NicVAX. The SPA is a process that provides for an official FDA evaluation of Phase III clinical study protocols. The SPA provides trial sponsors with a binding written agreement that the design and analysis of the studies are adequate to support a license application submission if the study is performed according to the SPA parameters and the results are successful. The SPA agreement may only be changed by the sponsor company or the FDA by a written agreement, or if the FDA becomes aware of a substantial scientific issue essential to product efficacy or safety.
European Regulatory Scientific Advice (SA)
The Company announced in June 2009 that it had obtained SA from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) for NicVAX. Although not binding on the EMA, the SA is intended to optimize Research and Development, reduce uncertainty in regulatory outcomes and accelerate time to approval of a MAA. In efforts to further mitigate regulatory risk, Nabi elected to seek clarifying follow-on Scientific Advice from EMA and in December of 2010, Nabi received follow-on Scientific Advice from EMA confirming the adequacy of the existing Phase III trial designs and CMC plans for NicVAX. EMA recommended increasing the duration of the primary endpoint for the Phase III studies and agreed to the acceptability of a separate EU specific statistical analysis of a longer duration of assessment of abstinence. Nabi confirmed with EMA that no additional clinical studies are required prior to submission of an MAA for NicVAX.
Post-Approval Regulation
After approval, biological products are subject to ongoing review. The failure to comply with the applicable regulatory requirements may subject a company to a variety of administrative or judicially imposed sanctions. These sanctions could include FDA’s refusal to approve pending applications, withdrawal of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of operations, injunctions, fines, civil penalties or criminal prosecution.
Reimbursement
Future commercial sales of our products depend significantly on appropriate payments from federal and state government healthcare authorities, which regularly consider and implement coverage and payment reforms. An example of payment reform is the addition of an expanded prescription drug benefit for all Medicare beneficiaries known as Medicare Part D. This is a voluntary benefit that is being implemented through private plans under contractual arrangements with the federal government. Similar to pharmaceutical coverage through private health insurance, Medicare Part D plans establish formularies that govern the drugs, biologicals and vaccines that will be offered and the out-of-pocket obligations for such products. Medicare Part D plans often negotiate discounts from manufacturers for drugs that will be included on their drug formularies. Effective January 1, 2008, private Medicare Part D plans will pay physicians one payment that includes both the administration cost and the cost of the vaccine.
10
Table of Contents
COMPETITION
Existing prescription products in the smoking cessation marketplace consist of three general categories of therapeutic approach: direct nicotine replacement; anti-depressant therapy; and nicotine receptor partial agonists. Nicotine replacement therapies (NRTs) represent a first generation approach to assisting smokers to quit by substituting a less harmful form of nicotine than inhalation by smoking. NRTs are mildly effective and support smoking cessation in combination with behavioral modification counseling. NRTs come in a number of forms of administration: gums, patches, lozenges and inhalers. Many forms of NRTs are currently available over the counter. GSK’s Zyban (buproprion) is the only anti-depressant which is FDA approved specifically as an aid to smoking cessation acting mainly through a reduction in craving and withdrawal symptoms. Pfizer Inc.’s Chantix product, a nicotine receptor partial agonist, represents a new class of prescription therapeutic that blocks nicotine from interacting with the nicotine receptor in the brain and has defined a new standard of care. It has been reported that Chantix causes some untoward neuropsychiatric side effects including suicides, suicide ideations and other psychotic behaviors. This has led the FDA to require Pfizer to add a “boxed warning” on Chantix’s label.
Examples of other product candidates in development are additional selective glycine receptor antagonists (GlaxoSmithKline; Phase II) and additional nicotine-derived therapeutic vaccines. Nic-002 (Phase II), TA-Nic (Phase II), Niccine (Phase II) and SEL-068 (Phase I) are nicotine-derived therapeutic vaccines being developed by Cytos Biotechnology/Novartis Pharmaceuticals, Celtic Pharmaceuticals, Independent Pharmaceutica and Selecta Biosciences, respectively, which if successfully developed and registered, may directly compete with NicVAX. In addition, Pfizer Inc. stated that their pre-clinical nicotine vaccine was progressing to the clinic. Recently, Cytos announced that an interim analysis showed that Nic-002 did not achieve its primary endpoint of smoking cessation in a Phase II study. Results from a Phase II proof of concept of TA-Nic were expected in 2008 but so far no announcements have been made regarding the vaccine’s performance in the study. Finally, Independent Pharmaceutica’s Niccine is also engaged in a relapse prevention Phase II proof-of-concept study which completed enrollment in November of 2008 and results of which were anticipated at the end of 2009 or early 2010, but so far no announcements have been made regarding the vaccine’s performance in the study.
For a discussion of the risks associated with competition, see below under “Item 1A. Risk Factors.”
EMPLOYEES
We believe that relations between our management and our employees are generally good. None of our employees are covered by a collective bargaining agreement. We had a total of 16 employees at December 31, 2011.
FINANCIAL INFORMATION ABOUT SEGMENTS AND GEOGRAPHIC AREAS
We operate in one industry segment, and have no material operations in any country other than the U.S.
AVAILABLE INFORMATION
Our Internet address is http://www.nabi.com. We make available, free of charge, through our Internet website, our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission.
11
Table of Contents
Statements in this document that are not strictly historical are forward-looking statements and include statements about potential strategic transactions, products in development, results and analyses of clinical trials and studies, research and development expenses, cash expenditures, licensure applications and approvals, alliances and partnerships, among other matters. You can identify these forward-looking statements because they involve our expectations, intentions, beliefs, plans, projections, anticipations, or other characterizations of future events or circumstances. These forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties that may cause actual results to differ materially from those in the forward-looking statements as a result of any number of factors. These factors include, but are not limited to, risks relating to our ability to pursue and successfully complete a strategic transaction; realize any value for NicVAX in light of our two failed Phase III clinical trials; obtain a successful result in a remaining clinical trial for NicVAX; have GSK exercise its option for NicVAX and commercialize NicVAX; have GSK successfully develop and commercialize any future generation candidate nicotine vaccine; terminate existing NicVAX contract manufacturing and development agreements without significant penalties; collect any further milestones and royalty payments under the PhosLo agreement; maintain sufficient patent protection; avoid products liability claims; maintain sufficient insurance; and use our net operating loss carry forwards. These factors and others are more fully discussed below.
Each of the following risk factors could adversely affect our business, operating results and financial condition.
Our strategic alternatives process may not be successful.
With the assistance of Piper Jaffray we are exploring the full range of possible strategic alternatives available to us. This includes, but is not limited to, the sale, license or merger of all or part of the Company, liquidation and dissolution, joint ventures, strategic alliances or recapitalization. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions that will enhance shareholder value. Further, any strategic transaction that is completed ultimately may not deliver the anticipated benefits or enhance shareholder value.
Our remaining product candidate has had an unfavorable result from its two Phase III clinical trials. This has had and likely will continue to have a material adverse effect on us and our ability to realize any value for NicVAX.
Our remaining product candidate, NicVAX, did not meet the primary endpoint in its two confirmatory Phase III clinical trials the results of which we announced in 2011. This unfavorable clinical result has adversely affected our ability to commercialize and obtain FDA licensure of NicVAX, as well as GSK’s interest in exercising the NicVAX option and therefore our ability to realize any future value from NicVAX. In addition, the unfavorable clinical result could cause GSK to limit or cease development of a next-generation candidate under our exclusive license and option agreement with GSK. Any or all of these outcomes will adversely affect our future business, market valuation, financial condition and results of operations.
An unfavorable result in a remaining NicVAX clinical trial would have a material adverse effect on us.
NicVAX is being studied in an ongoing investigator-initiated combination clinical trial in the Netherlands with Pfizer’s varenicline (Chantix/Champix). An unfavorable result from this trial will adversely affect our ability to realize any remaining value for NicVAX.
GSK may not be successful in developing and commercializing the future generation candidate vaccine.
Under the option and license agreement for NicVAX, we granted GSK an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or
12
Table of Contents
treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives) and using their own proprietary technologies. We may be eligible for milestones and royalties from such development, if successful. If such development and commercialization by GSK does not progress or is not successful, we will not receive future milestones and royalties related to future generation candidate products which could have a material adverse effect on our future business, market valuation, financial condition and results of operations.
We depend upon third parties to manufacture and develop NicVAX under various agreements and may owe fees if we terminate these agreements or do not purchase required amounts of product.
We depend upon third parties to manufacture NicVAX. We entered into various development agreements with contract manufacturing organizations (CMOs) to both manufacture NicVAX and transfer the manufacturing technologies and know-how of NicVAX. If we terminate some or all of these agreements or do not purchase required amounts of product under them, we will be required to pay certain fees to the CMOs. Our ability to successfully partner our remaining rights to NicVAX may depend on our ability to successfully assign our rights under these agreements to any partner, and our inability to assign them on favorable terms or at all to any partner, would have a material adverse effect on our future business, financial condition and results of operations.
We may not collect any further milestone or royalty payments under the PhosLo Agreement.
We may not collect any further milestone or royalty payments under the PhosLo Agreement with Fresenius. We received $65 million in cash at closing and have received an additional $18 million of milestones as of December 31, 2011. We can also receive up to $67.5 million in additional milestone payments and royalties. The royalties relate to sales of a new product formulation over a base amount for 10 years after the November 14, 2006 closing date. There can be no assurance of the completion of any additional milestone or successful sales of the new product formulation. If any additional milestone is not completed or if sales of the new product formulation do not exceed the base amount, we will not collect the related future milestone or royalty payments under the PhosLo Agreement.
Our patents and proprietary rights may not provide sufficient protection, and patents of other companies could prevent us or our partners from developing and marketing NicVAX.
The patent positions of biopharmaceutical firms generally are highly uncertain and involve complex legal and factual questions. The ultimate degree of patent protection that will be afforded to biotechnology products and processes, including ours, in the U.S. and in other important markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts and lawmakers in these countries. There can be no assurance that existing patent applications will result in issued patents, that we will be able to obtain additional licenses to patents of others or that we will be able to develop additional patentable technology of our own. We cannot be certain that we were the first creator of inventions covered by our patents or pending patent applications or that we were the first to file patent applications for such inventions. There can be no assurance that any patents issued to us will provide us or our partners with competitive advantages or will not be challenged by others. Furthermore, there can be no assurance that others will not independently develop similar products, or, if patents are issued to us, others may design their patents around our patents.
A number of pharmaceutical companies, biotechnology companies, universities and research institutions have filed patents or patent applications or received patents relating to products or processes competitive with or similar to ours. Some of these applications or patents may compete with our applications or conflict in certain respects with claims made under our applications. Such a conflict could result in a significant reduction of the coverage of our patents, if issued. In addition, if patents that contain competitive or conflicting claims are issued to others and such claims are ultimately determined to be valid, we or our partners may be required to obtain licenses to these patents or to develop or obtain alternative technology. See “Business—Patents and Proprietary Rights—Smoking Cessation.”
13
Table of Contents
If any licenses are required, there can be no assurance that we will be able to obtain any such licenses on commercially favorable terms, if at all. Our failure to obtain a license to any technology that we may require in order to commercialize our products could have a material adverse effect on our future business, financial condition and results of operations.
Litigation may be necessary to enforce any patents issued to us or to determine the scope or validity of third-party proprietary rights or to defend against any claims that our business infringes on third-party proprietary rights. Patent litigation is expensive and could result in substantial cost to us or our partners. The costs of patent litigation and our ability to prevail in such litigation could have a material adverse effect on our future business, financial condition and results of operations.
We also rely on secrecy to protect our technology, especially where patent protection is not believed to be appropriate or obtainable. We maintain strict controls and procedures regarding access to and use of our proprietary technology and processes. However, there can be no assurance that these controls or procedures will not be violated, that we would have adequate remedies for any violation, or that our trade secrets will not otherwise become known or be independently discovered by competitors.
We or our partners may be subject to costly and damaging product liability and other claims in connection with the development and commercialization of NicVAX.
Pharmaceutical and biotechnology companies are subject to litigation, including class action lawsuits, and governmental and administrative investigations and proceedings, including with respect to product pricing and marketing practices. There can be no assurance that lawsuits will not be filed against us or our partners, or that we will be successful in the defense of these lawsuits. Defense of suits can be expensive and time consuming, regardless of the outcome, and an adverse result in one or more suits could have a material adverse effect on our future business, financial condition and results of operations.
We may not be able to maintain sufficient insurance, including products liability and directors and officers insurance, to cover claims against us.
Product liability and directors and officers insurance for the biopharmaceutical industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to maintain such insurance on acceptable terms or that existing or future claims against us will be covered by our insurance. Moreover, there can be no assurance that the existing coverage of our insurance policy and/or any rights of indemnification and contribution that we may have will offset any claims. A successful claim against us with respect to uninsured liabilities or in excess of insurance coverage and not subject to any indemnification or contribution could have a material adverse effect on our future business, financial condition and results of operations. Further, if we were unable to obtain directors and officers liability insurance, it could affect adversely our ability to attract and retain directors and officers.
Our ability to use our federal and state net operating loss carry forwards to reduce taxable income generated in the future could be substantially limited or eliminated.
Our ability to use our net operating losses is subject to an annual limitation due to ownership changes that may have occurred or that could occur in the future, as determined by Section 382 of the Internal Revenue Code of 1986, as amended, as well as similar state regulations. Depending on the actual amount of any limitation on our ability to use our net operating loss carry forwards, a significant portion of our future taxable income could be subject to federal and/or state income tax, creating federal and/or state income tax liabilities. Additionally, such limitation may result in our net operating losses expiring before we have the ability to use them. Moreover, the Internal Revenue Service may not agree with the amount or timing of prior losses, thereby further limiting our net operating loss carry forward. On August 25, 2011, we entered into a Rights Agreement with American Stock Transfer & Trust Company, LLC, as rights agent, in an effort to prevent an “ownership change” under Section 382 from occurring and thereby protect the value of our net operating losses. There can be no assurance, however, that the Rights Agreement will prevent an ownership change from occurring or protect the value of the net operating losses. In addition, any transaction that we may enter into as a result of our strategic alternatives process may significantly limit or eliminate our ability to realize any value from our net operating losses.
14
Table of Contents
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We lease office space in Rockville, Maryland on a month-to-month basis.
We are parties to legal proceedings that we believe to be ordinary, routine litigation, incidental to the business of present or former operations. It is management’s opinion, based on the advice of counsel, that the ultimate resolution of such litigation will not have a material adverse effect on our financial condition, results of operations, or cash flows.
ITEM 3(a). EXECUTIVE OFFICERS OF THE REGISTRANT
Our executive officers listed below:
| Name |
Age | Position | ||||
| Raafat E.F. Fahim, Ph.D. |
58 | Chief Executive Officer, President, Acting Chief Financial Officer and Director | ||||
| Paul Kessler, M.D. |
57 | Senior Vice President, Clinical, Medical and Regulatory Affairs and Chief Medical Officer | ||||
| Matthew W. Kalnik, Ph.D. |
49 | Senior Vice President, Strategic Planning and Business Operations | ||||
Dr. Fahim has served as Chief Executive Officer and President since January 22, 2008, and also as acting Chief Financial Officer since May 27, 2008. From July 2007 to January 2008, Dr. Fahim served as Senior Vice President, Research, Technical and Production Operations of the Company and Chief Operating Officer and General Manager of the Biologics SBU. Dr. Fahim is also non-executive Chairman of the Board of VM Farms, a Toronto, Canada-based private equity web hosting technology company. From March 2003 to July 2007, Dr. Fahim served as Senior Vice President, Research, Technical and Production Operations of the Company. From 2002 to 2003, Dr. Fahim was an independent consultant, working with Aventis Pasteur and other companies worldwide on projects that included product development, manufacturing, process improvement, quality operations and regulatory issues. From 2001 to 2002, he served as President and Chief Operating Officer of Lorus Therapeutics, Inc., a biopharmaceutical company. From 1987 to 2001, Dr. Fahim was employed by Aventis Pasteur where he was instrumental in developing several vaccines from early research to marketed products. During his employment with Aventis Pasteur, Dr. Fahim held the positions of Vice President, Industrial Operations; Vice President, Development, Quality Operations and Manufacturing; Director of Product Development and head of bacterial vaccines research/research scientist. He received his Ph.D. in Biochemistry from the University of Toronto.
Dr. Kessler has been the Senior Vice President, Clinical, Medical and Regulatory and Chief Medical Officer since March 2007. He joined Nabi Biopharmaceuticals in March 2005 as Senior Director, Clinical Research, and in April 2006, he was promoted to Vice President, Clinical Research. From 1998 to 2005, he served in several positions at GenVec, Inc., a gene therapy company, including Program Director, Director Clinical Research, Senior Director Clinical Research, and Executive Director Clinical Research. From 1989 to 1998, he was an Assistant Professor and later Associate Professor of Medicine at the Johns Hopkins University School of
15
Table of Contents
Medicine, where he conducted gene and cell therapy research and where he was an attending cardiologist on the Heart Failure and Transplant Service. He earned a B.S. from the University of Pittsburgh, a M.Sc. from the University of London, and an M.D. from Columbia University College of Physicians and Surgeons. He trained in Medicine and Cardiology at The Mount Sinai Hospital, New York, and Johns Hopkins.
Dr. Kalnik was appointed to the role of Senior Vice President, Strategic Planning and Business Operations in March 2009. He joined the Company as Vice President, Business Development and Project Management in July 2007. Prior to joining Nabi Biopharmaceuticals, Dr. Kalnik held senior management team positions at innovative biotechnology companies including Executive Vice President, Head of Business Development, at VistaGen Therapeutics and Senior Vice President, Business Development and Licensing, and corporate officer at Genaissance Pharmaceuticals. He has also served in an executive capacity in R&D and commercial development at global pharmaceutical companies Pfizer (Pharmacia) and Daiichi Medical Research including Executive Director, Commercial Development; Sr. Director, Development Technology, Medical Research; and Director, Technology Acquisitions, Discovery Research & Exploratory Development. Dr. Kalnik also founded Hedgerow Consulting and has authored more than a dozen primary research papers. He holds a Bachelor of Science in Chemistry from the University of North Carolina at Chapel Hill (1984) and an M.A, M.Sc. & Ph.D. in Molecular Biophysics from Columbia University (1989) and conducted his post-doctoral fellowship in Molecular Biology at The Scripps Research Institute in La Jolla, California.
ITEM 4. MINE SAFETY DISCLOSURES
None.
16
Table of Contents
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is quoted on the Nasdaq Global Select Market under the symbol “NABI”. The following table sets forth for each period the high and low sale prices for our common stock (based upon intra-day trading) as reported by the Nasdaq Global Select Market.
| High | Low | |||||||
| 2011: |
||||||||
| First Quarter ended March 26, 2011 |
$ | 5.98 | $ | 5.30 | ||||
| Second Quarter ended June 25, 2011 |
5.88 | 4.64 | ||||||
| Third Quarter ended September 24, 2011 |
5.82 | 1.55 | ||||||
| Fourth Quarter ended December 31, 2011 |
2.07 | 1.50 | ||||||
| 2010: |
||||||||
| First Quarter ended March 27, 2010 |
$ | 6.42 | $ | 4.70 | ||||
| Second Quarter ended June 26, 2010 |
5.98 | 4.40 | ||||||
| Third Quarter ended September 25, 2010 |
5.85 | 4.68 | ||||||
| Fourth Quarter ended December 25, 2010 |
5.75 | 4.75 | ||||||
The closing price of our common stock on February 24, 2012, was $1.85 per share. The number of record holders of our common stock on February 24, 2012, was 788.
No cash dividends have been paid on our common stock and none are anticipated in 2012.
Information regarding securities authorized for issuance under equity compensation plans is included in Item 12 of this Annual Report on Form 10-K.
We had no unregistered sales of equity securities in 2011.
17
Table of Contents
COMPARATIVE STOCK PERFORMANCE
The following graph and chart compare, during the five-year period commencing December 31, 2006 and ending December 31, 2011, the annual change in the cumulative total return of our common stock with the NASDAQ Stock Market (Composite) and the NASDAQ Biotechnology Index, assuming the investment of $100 on December 31, 2006 (at the market close), and the reinvestment of any dividends.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN
ASSUMES INITIAL INVESTMENT OF $100
DECEMBER 2011
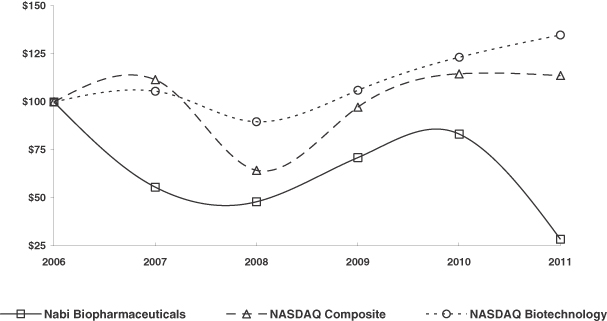
| Company/Market/Peer Group |
2006 | 2007 | 2008 | 2009 | 2010 | 2011 | ||||||||||||||||||
| Nabi Biopharmaceuticals |
$ | 100.00 | $ | 55.60 | $ | 48.08 | $ | 71.09 | $ | 83.26 | $ | 28.61 | ||||||||||||
| NASDAQ Composite Index |
$ | 100.00 | $ | 111.58 | $ | 64.44 | $ | 97.23 | $ | 114.59 | $ | 113.72 | ||||||||||||
| NASDAQ Biotechnology Index |
$ | 100.00 | $ | 105.61 | $ | 89.63 | $ | 105.97 | $ | 123.30 | $ | 134.75 | ||||||||||||
18
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA
The following table sets forth selected consolidated financial data for the five years ended December 31, 2011 that was derived from our audited Consolidated Financial Statements. The selected financial data should be read in conjunction with, and are qualified by reference to, our Consolidated Financial Statements and the Notes thereto and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| For the Years Ended | ||||||||||||||||||||
| (in thousands, except per share amounts) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
December 27, 2008 |
December 29, 2007 |
|||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenue: |
||||||||||||||||||||
| Revenue |
$ | 14,838 | $ | 35,005 | $ | 10,489 | $ | — | $ | — | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Cost of services |
1,463 | 3,951 | 1,988 | — | — | |||||||||||||||
| Research and development expense |
17,765 | 26,078 | 16,490 | 12,556 | 18,841 | |||||||||||||||
| General and administrative expense |
5,372 | 6,174 | 9,987 | 12,415 | 26,090 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
24,600 | 36,203 | 28,465 | 24,971 | 44,931 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Operating loss |
(9,762 | ) | (1,198 | ) | (17,976 | ) | (24,971 | ) | (44,931 | ) | ||||||||||
| Interest income |
194 | 230 | 368 | 4,579 | 6,026 | |||||||||||||||
| Interest expense |
— | (210 | ) | (1,071 | ) | (3,902 | ) | (9,007 | ) | |||||||||||
| Other income (expense), net |
37 | 291 | (48 | ) | (1,454 | ) | 446 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from continuing operations before income taxes |
(9,531 | ) | (877 | ) | (18,727 | ) | (25,748 | ) | (47,466 | ) | ||||||||||
| Benefit from income taxes |
2,018 | 1,765 | — | 2,765 | 14,265 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) from continuing operations |
(7,513 | ) | 878 | (18,727 | ) | (22,983 | ) | (33,201 | ) | |||||||||||
| Net income from discontinued operations |
2,982 | — | — | 4,245 | 71,587 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) |
$ | (4,531 | ) | $ | 878 | $ | (18,727 | ) | $ | (18,738 | ) | $ | 38,386 | |||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic income (loss) per share: |
||||||||||||||||||||
| Continuing operations |
$ | (0.18 | ) | $ | 0.02 | $ | (0.37 | ) | $ | (0.44 | ) | $ | (0.55 | ) | ||||||
| Discontinued operations |
$ | 0.07 | $ | — | $ | — | $ | 0.08 | $ | 1.19 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic income (loss) per share |
$ | (0.11 | ) | $ | 0.02 | $ | (0.37 | ) | $ | (0.36 | ) | $ | .64 | |||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Diluted income (loss) per share: |
||||||||||||||||||||
| Continuing operations |
$ | (0.18 | ) | $ | 0.02 | $ | (0.37 | ) | $ | (0.44 | ) | $ | (0.55 | ) | ||||||
| Discontinued operations |
$ | 0.07 | $ | — | $ | — | $ | 0.08 | $ | 1.19 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Diluted income (loss) per share |
$ | (0.11 | ) | $ | 0.02 | $ | (0.37 | ) | $ | (0.36 | ) | $ | .64 | |||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance Sheet Data (at year end): |
||||||||||||||||||||
| Cash, cash equivalents and marketable securities |
$ | 96,389 | $ | 110,667 | $ | 118,999 | $ | 130,338 | $ | 219,206 | ||||||||||
| Working capital |
91,629 | 92,093 | 95,783 | 134,540 | 205,893 | |||||||||||||||
| Total assets |
97,965 | 113,871 | 131,317 | 144,221 | 239,236 | |||||||||||||||
| Convertible senior notes, non current |
— | — | — | 15,202 | 64,450 | |||||||||||||||
| Total stockholders’ equity |
$ | 58,871 | $ | 60,570 | $ | 97,407 | $ | 121,382 | $ | 154,486 | ||||||||||
19
Table of Contents
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are a biopharmaceutical company that has focused on the development of vaccines addressing unmet medical needs, including nicotine addiction. We have sought to leverage our experience and knowledge in powering the human immune system to target these serious unmet medical needs. We have been incorporated in Delaware since 1969 and our operations are located in Rockville, Maryland.
Our sole remaining product currently in development is NicVAX® [Nicotine Conjugate Vaccine], an innovative and proprietary investigational vaccine for the treatment of nicotine addiction and prevention of smoking relapse based on patented technology. We suffered a significant setback in 2011 when NicVAX did not achieve the primary endpoint in two Phase III efficacy trials conducted in the U.S. As a result, in November of 2011, we retained Piper Jaffray & Co. (Piper Jaffray) to assist with the exploration of strategic alternatives available to the company. This includes, but is not limited to, the sale, license or merger of all or part of the company or its assets, joint ventures, strategic alliances, recapitalization, or liquidation, and the process is ongoing. As of December 31, 2011, our remaining assets include the following: (i) $96.4 million of cash, cash equivalents and marketable securities, (ii) the potential residual value of NicVAX as well as next-generation nicotine vaccine which was licensed to GlaxoSmithKline Biologicals S.A. (GSK) in 2010, (iii) the potential royalty from Phoslyra which was sold to Fresenius USA Manufacturing, Inc. (Fresenius) in 2006, and (iv) the potential value of our net operating losses (NOLs).
In the first quarter of 2010 we granted to GSK (i) an option to exclusively license NicVAX on a worldwide basis and (ii) a license to develop follow-on next-generation nicotine vaccines using our intellectual property combined with GSK proprietary technology including GSK proprietary adjuvants. GSK has not indicated that it is stopping the development of the next generation nicotine vaccine. If GSK continues to develop the next generation nicotine vaccine, Nabi may be eligible to receive milestones and royalties from that program. We continue to support an investigator initiated Phase IIb trial in the Netherlands of NicVAX in combination with Pfizer’s varenicline or Chantix®/Champix, the results of which are expected in the second half of 2012.
The smoking cessation market is estimated to exceed $4 billion annually and is currently considered to be a largely unmet medical need. Nicotine is a non-immunogenic small molecule that, upon inhalation into the lungs, quickly passes into the bloodstream and subsequently reaches the brain by crossing the blood-brain barrier. Once in the brain, the nicotine binds to specific nicotine receptors resulting in the release of substances, such as dopamine, a chemical which triggers the highly addictive pleasurable effects experienced by smokers and users of other nicotine products. NicVAX is designed to stimulate the immune system to produce highly specific antibodies that bind to nicotine in the bloodstream. A nicotine molecule attached to specific antibodies is too large to cross the blood-brain barrier and thus is unable to reach the receptors in the brain thereby reducing the pleasure experienced by the smoker making it easier to quit.
In March 2010, we closed an exclusive worldwide option and licensing agreement with GSK for NicVAX as well as for the development of follow-on nicotine addiction vaccines. Upon closing, we received a $40 million initial payment. Under the terms of the agreement, we granted to GSK (i) an option to obtain an exclusive worldwide license to develop, commercialize and manufacture NicVAX as it currently exists, as well as certain potential alternative forms of NicVAX together with an adjuvant other than a GSK proprietary adjuvant and/or with different presentation, dosage or administration (NicVAX Alternatives), and (ii) an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives). In addition to the $40 million received at closing, we are eligible to receive under the agreement up to $290 million in potential regulatory, development and sales milestones on any next generation nicotine vaccines. We are also eligible to receive royalties on global sales of any next generation of nicotine vaccines. Notwithstanding the failure to achieve the primary endpoint in the two Phase III trials, if GSK was to
20
Table of Contents
exercise its option under the agreement for NicVAX and any NicVAX alternatives, it will pay us $58 million upon exercise plus certain potential milestones and royalties over time.
During 2011 we completed our obligations to continue to develop PentaStaph™ [Pentavalent S.aureus Vaccine] under contract for GSK. PentaStaph is a new pentavalent vaccine designed to prevent S.aureus infections including those infections caused by the most dangerous antibiotic-resistant strains of S.aureus. In August 2009, we entered into an Asset Purchase Agreement (APA) with GSK for the sale of PentaStaph for a total consideration of $46 million including a $20 million up-front payment upon closing and $26 million payable upon achievement of certain milestones. The PentaStaph sale closed in November 2009, and we received payment from GSK of $21.5 million representing the up-front payment of $20 million, an additional $1 million for the sale of our Staphylococcus epidermidis vaccine program and an additional $0.5 million for transfer of certain specified materials. We also agreed to a Transition Services Agreement (TSA) to help GSK advance the program while in parallel transferring the technology to GSK. In 2011 we completed all of the efforts required of us from GSK under contract for PentaStaph. As a result, we have received the full $26 million in potential milestone payments, $5 million in 2011 and $21 million in 2010.
Results of Operations
The following discussion and analysis of our financial condition and results of operations for each of the three years ended December 31, 2011, December 25, 2010 and December 26, 2009, should be read in conjunction with our Consolidated Financial Statements and Notes thereto and with the information contained under “Risk Factors” in Item 1A. All amounts are expressed in thousands, except for per share and percentage data.
2011 as Compared to 2010
Revenue. Revenue reflects (i) the amortization of up-front fees received under our PentaStaph and NicVAX agreements, (ii) the completion of substantive milestones included in those agreements, and (iii) services provided to GSK. The amortization of up-front fees received under our PentaStaph and NicVAX agreements are being recognized as revenue ratably over the period of our participation on joint steering committees created under these agreements. The joint steering committee related to the NicVAX agreement is currently expected to operate for 190 months from the date of the agreement (or through December 2025). Our efforts under the joint steering committee related to the PentaStaph agreement were expected to continue for 20 months from the date of the agreement (or through June 2011); accordingly, we have fully recognized the up-front payment related to the PentaStaph agreement.
Total revenue in 2011 of $14.8 million included $7.8 million of deferred revenue amortization from the PentaStaph and NicVAX agreements, $5.0 million for a completed PentaStaph milestone, and $2.0 million for services under the PentaStaph and NicVAX agreements. Total revenue in 2010 of $35.0 million included $15.3 million of deferred revenue amortization from the PentaStaph and NicVAX agreements, $16.0 million for the completion of two PentaStaph milestones, and $3.7 million for services under the PentaStaph and NicVAX agreements. We expect our revenue to significantly decrease in 2012 as a result of the completion of our obligations under the PentaStaph agreement with GSK.
Cost of services. Cost of services was $1.4 million for 2011 compared to $4.0 million for 2010. These costs include internal labor, external contractors and allocated indirect costs. We do not expect to have any material cost of services in 2012 as a result of completing the PentaStaph agreement with GSK.
Research and development expenses. Research and development expenses were $17.8 million for 2011 compared to $26.1 million for 2010. The decrease of $8.3 million is due to a reduction of NicVAX manufacturing and related clinical trial activities in 2011. Approximately $0.3 million of the 2011 costs for NicVAX trials have been offset by grant funding compared to $8.8 million of costs for NicVAX and PentaStaph
21
Table of Contents
trials offset by grant funding in 2010. Research and development expenses in 2012 are expected to be materially less than 2011 levels since we completed the two phase III NicVAX trials and are winding down our one remaining NicVAX trial in combination with Chantix.
General and administrative expenses. General and administrative expenses, after an allocation of a portion of these expenses to cost of services and research and development expenses, were $5.4 million for 2011 compared to $6.2 million for 2010. The decrease of $0.8 million reflects our continued efforts to reduce overall expenses including reduction in work force as a result of the completion of the NicVAX Phase III trials. Excluding potential costs related to any strategic transaction, general and administrative expenses in 2012 are expected to decrease from 2011 levels, reflecting reduced headcount and lower level of activity.
Interest expense. We recorded no interest expense during 2011. Interest expense was $0.2 million for 2010 and consisted largely of interest expense associated with our Convertible Senior Notes. As of December 25, 2010, we had repurchased all of our Convertible Senior Notes.
Income tax expense (benefit). Because of the intra-period income tax allocation requirements, we recognized a benefit for income taxes from continuing operations of $2.0 million during 2011, offset in total by an identical income tax provision from discontinued operations. The intra-period income tax allocation considers discontinued operations for purposes of determining the amount of tax benefits that result from our loss from continuing operations. For the year ending 2010, as a result of recent tax law changes, we recognized an income tax benefit of $1.8 million relating to a refund of prior year alternative minimum tax payments.
Discontinued operations. During 2011, we recognized $3.0 million of income, net of tax of $2.0 million, from discontinued operations relating to the milestone of the first commercial sale of a new liquid formulation of PhosLo under our agreement with Fresenius (none in 2010).
2010 as Compared to 2009
Revenue. Revenue was $35.0 million for 2010 compared to $10.5 million for 2009. The increase of $24.5 million reflects amounts recognized under the PentaStaph and NicVAX agreements with GSK. Revenue includes amortization of up-front fees received under our PentaStaph and NicVAX agreements; those fees are being recognized as revenue ratably over the period of our participation on joint steering committees created under these agreements. The joint steering committee related to the NicVAX agreement is currently expected to operate for 190 months from the date of the agreement (or through December 2025). Our efforts under the joint steering committee related to the PentaStaph agreement was expected to operate for 20 months from the date of the agreement (or through June 2011); accordingly, we have fully recognized the up-front payment related to the PentaStaph agreement. The amount recognized in 2010 includes $13.2 million from the initial $21.5 million payment received from GSK for PentaStaph and $2.1 million from the initial $40.0 million payment received from GSK for NicVAX compared to $3.1 million recognized for PentaStaph in 2009. We also recognized $16.0 million of revenue related to the successful achievement of two PentaStaph performance milestones in 2010 compared to $5.0 million for the achievement of one performance milestone in 2009. We also recognized $3.2 million and $0.5 million related to services provided to GSK under the PentaStaph and NicVAX agreements, respectively, in 2010 compared to $2.4 million for services under the PentaStaph agreement in 2009.
Cost of services. Cost of services was $4.0 million for 2010 compared to $2.0 million for 2009. The increase of $2.0 million represents the cost incurred by us to perform under the PentaStaph and NicVAX agreements with GSK with respect to the transitional services, including performance of the PentaStaph Phase I/II clinical trial and associated activities. These costs include internal labor, external contractors and allocated indirect costs.
Research and development expenses. Research and development expenses were $26.1 million for 2010 compared to $16.5 million for 2009. The increase of $9.6 million is primarily due to our two ongoing Phase III trials for NicVAX and NicVAX manufacturing-related activities. The costs related to the PentaStaph Phase I/II
22
Table of Contents
clinical trial were reimbursed to us by GSK as they are conducted by us under contract for GSK. GSK’s payments for these costs are recognized as revenue. Approximately $8.8 million of the 2010 costs for NicVAX and PentaStaph trials have been offset by grant funding compared to $1.6 million of costs offset by grant funding in 2009.
General and administrative expenses. General and administrative expenses, net of an allocation of a portion of these expenses to cost of services, was $6.2 million for 2010 compared to $10.0 million for 2009. The decrease of $3.8 million reflects our continued efforts to reduce overall expenses and lower legal and facility costs.
Interest expense. Interest expense was $0.2 million and $1.1 million for 2010 and 2009, respectively, and consisted largely of interest expense associated with our Convertible Senior Notes. The decrease of $0.9 million reflects the impact of the repurchase of the remaining balance of our Convertible Senior Notes in the second quarter of 2010.
Income tax expense (benefit). During the third quarter 2010, as a result of recent tax law changes, we recognized an income tax benefit of $1.8 million relating to a refund of prior year alternative minimum tax payments (none in 2009).
Liquidity and Capital Resources
Our cash, cash equivalents and marketable securities at December 31, 2011 totaled $96.4 million as compared to $110.7 million at December 25, 2010. The decrease of $14.2 million is primarily the result of our net cash used in operating activities of $20.2 million offset in part by net cash provided by investing activities and the $5.0 million milestone payment received through discontinued operations related to the first commercial sale on the new liquid PhosLo formulation under our agreement with Fresenius.
Cash provided by (used in) operating activities from continuing operations was ($19.6) million, $40.5 million and ($3.6) million in 2011, 2010 and 2009, respectively. Cash used in operating activities from continuing operations in 2011 included cash expenditures for research and development expenses and general and administrative expenses, offset in part by cash received from GSK associated with the PentaStaph and NicVAX agreements. Cash provided by operating activities from continuing operations in 2010 was primarily related to the $66.3 million received from GSK associated with the PentaStaph and NicVAX agreements offset in part by cash used for research and development expenses and general and administrative expenses. In 2009, cash used in operating activities from continuing operations included cash expenditures for research and development expenses and general and administrative expenses, partially offset by $21.5 million received in connection with the closing of the PentaStaph sale to GSK. Cash provided by (used in) operating activities from discontinued operations was $4.5 million, ($0.6) million and $9.8 million for 2011, 2010 and 2009, respectively.
Cash provided by (used in) investing activities from continuing operations was $60.2 million, $2.3 million and ($35.6) million for 2011, 2010 and 2009, respectively, which consists largely of net proceeds from the maturities (purchases) of marketable securities.
Since December 2007, our Board of Directors has approved the repurchase of up to $115 million of our common stock in the open market or in privately negotiated transactions. Since the inception of the program in December 2007 through December 31, 2011, we have repurchased a total of 19.9 million shares for a total cost of $87.2 million, at an average price of $4.39 per share, leaving a balance of $27.8 million available for share repurchases under the current program. No shares were repurchased in 2011. Repurchased shares have been accounted for as treasury stock using the cost method.
23
Table of Contents
In 2005, we issued $112.4 million of our Convertible Senior Notes through a private offering to qualified institutional buyers. In 2010, we repurchased the remaining $6.1 million balance of our Convertible Senior Notes for a total of $6.1 million. In 2009, we repurchased $10.4 million of our Convertible Senior Notes for a total of $10.1 million.
We believe cash, cash equivalents and marketable securities on hand at December 31, 2011 will be sufficient to meet our anticipated cash requirements for operations for at least the next 12 months.
Aggregate Contractual Obligations
The following table provides information as of December 31, 2011 with respect to the amounts and timing of our known material contractual obligations as specified below (in thousands). As of December 31, 2011, there were no significant contractual obligations related to our discontinued operations.
| Total | Less than 1 Year |
1 to 3 Years |
3 to 5 Years |
More than 5 Years |
||||||||||||||||
| Open purchase orders |
$ | 1 | $ | 1 | $ | — | $ | — | $ | — | ||||||||||
| Manufacturing and clinical agreements |
1,710 | 1,710 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 1,711 | $ | 1,711 | $ | — | $ | — | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
The preceding table does not include information where the amounts of the obligations are currently not determinable, including contractual obligations in connection with clinical trials, which are payable on a per-patient basis.
Critical Accounting Policies and Estimates
We believe that the following policies and estimates are critical because they involve significant judgments, assumptions and estimates. We have discussed the development and selection of our critical accounting estimates with the Audit Committee of our Board of Directors and the Audit Committee has reviewed the disclosures presented below relating to those policies and estimates.
Accounting estimates: The preparation of financial statements in conformity with accounting principles generally accepted in the U.S. requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period, including such amounts related to discontinued operations. Actual results could differ from those estimates.
Revenue recognition: Our revenue generating arrangements may include multiple elements and deliverables, including licenses, options, research and development activities, participation on joint steering committees and contract manufacturing, among other elements.
For arrangements entered into or materially modified after December 25, 2010, when we determine that an element has stand-alone value to our customer, we allocate a portion of the total contract price to that element based on its relative selling price, determined pursuant to a selling price hierarchy, and recognize revenue for that element according to its characteristics.
Revenue consists of license fees, milestone payments, and payments for contractual services. License fees received are initially recorded as deferred revenue, and are subsequently recognized as revenue ratably over the period of our participation on joint steering committees. The joint steering committee related to the NicVAX agreement is currently expected to operate for 190 months from the date of the agreement (or through December 2025). Our efforts under the joint steering committee related to the PentaStaph agreement were completed in the second quarter of 2011; accordingly, we have fully recognized the up-front payment related to the PentaStaph agreement.
24
Table of Contents
For milestones that are deemed substantive, we recognize the contingent revenue when: (i) the milestones have been achieved; (ii) no further performance obligations with respect to the milestones exist; and (iii) collection is reasonably assured. A milestone is considered substantive if all of the following conditions are met: (i) the milestone is nonrefundable; (ii) achievement of the milestone was not reasonably assured at the inception of the arrangement; (iii) substantive effort is involved to achieve the milestone; and (iv) the amount of the milestone appears reasonable in relation to the effort expended with the other milestones in the arrangement and the related risk associated with achievement of the milestone. If a milestone is deemed not to be substantive, the Company would recognize the portion of the milestone payment as revenue that correlates to work already performed; the remaining portion of the milestone payment will be deferred and recognized as revenue as the Company completes its performance obligations.
Payments for contractual services are recognized as revenue when earned, typically when the services are rendered.
We analyze cost reimbursable grants and contracts to determine whether we should report such reimbursements as revenue or as an offset to research and development expenses incurred.
Collaborative arrangements: We are an active participant with exposure to significant risks and rewards of commercialization relating to the development of NicVAX and future generation nicotine vaccines based on NicVAX technology. For costs incurred and revenues generated from third parties where we are deemed to be the principal participant, we recognize revenues and costs using the gross basis of accounting; otherwise we use the net basis of accounting.
Research and development expenses: Research and development costs are expensed as incurred; advanced payments are deferred and subsequently expensed over the period of performance. Research and development expenses include direct labor costs as well as the costs of contractors and other direct and indirect expenses (including an allocation of the costs of facilities). We expense amounts payable to third parties under collaborative product development agreements at the earlier of the milestone achievement or as payments become contractually due. We recorded approximately $0.3 million, $8.8 million and $1.6 million for 2011, 2010 and 2009 respectively of cost reimbursements from government grants as an offset to research and development expenses.
Share-based compensation: We account for share-based compensation at fair value; accordingly we expense the estimated fair value of share-based awards made in exchange for employee services over the requisite employee service period. Share-based compensation cost for stock options is determined at the grant date using an option pricing model; share-based compensation cost for restricted stock is determined at the grant date based on the closing price of our common stock on that date. The value of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the employee’s requisite service period.
New accounting pronouncements: In October 2009, the Financial Accounting Standards Board, or the FASB, issued Accounting Standards Update 2009-13, “Revenue Recognition (Topic 605) Multiple-Deliverable Revenue Arrangements, a consensus of the FASB Emerging Issues Task Force,” or ASU 2009-13. ASU 2009-13 amends existing accounting guidance for separating consideration in multiple-deliverable arrangements. ASU 2009-13 establishes a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence if available, third-party evidence if vendor-specific evidence is not available, or the estimated selling price if neither vendor-specific evidence nor third-party evidence are available. ASU 2009-13 eliminates the residual method of allocation and requires that consideration be allocated at the inception of the arrangement to all deliverables using the “relative selling price method.” The relative selling price method allocates any discount in the arrangement proportionately to each deliverable on the basis of each deliverable’s selling price. ASU 2009-13 requires that a vendor determine its best estimate of selling price in a manner that is consistent with that used to determine the price to sell the deliverable on a stand-alone basis.
25
Table of Contents
We adopted the provisions of ASU 2009-13 effective December 26, 2010, for revenue arrangements entered into or materially modified in fiscal years beginning on or after that date. The adoption did not have any material effect on our consolidated balance sheets, consolidated statements of operations and consolidated statements of cash flows for any historical periods or as of, or for the year ended December 31, 2011 because we did not enter into or materially modify any revenue arrangements subsequent to December 26, 2010. We are not able to reasonably estimate the effect of adopting these standards on future periods because the impact will vary based on the nature and volume of new or materially modified revenue arrangements in any given period.
In April 2010, the FASB issued Accounting Standards Update 2010-17, “Revenue Recognition—Milestone Method (Topic 605) Milestone Method of Revenue Recognition, a consensus of the FASB Emerging Issues Task Force” or ASU 2010-17. ASU 2010-17 provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. For the milestone to be considered substantive, the consideration earned by achieving the milestone should meet all of the following criteria: (i) be commensurate with either the vendor’s performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from the vendor’s performance to achieve the milestone, (ii) relate solely to past performance, and (iii) be reasonable relative to all deliverables and payment terms in the arrangement. An individual milestone may not be bifurcated and an arrangement may include more than one milestone. Accordingly, an arrangement may contain both substantive and non-substantive milestones.
We adopted the provisions of ASU 2010-17 effective December 26, 2010, for milestones achieved on or after that date. Since our existing policies are consistent with those contained in ASU 2010-17, the adoption of ASU 2010-17 did not have any material effect on our consolidated balance sheets, consolidated statements of operations and consolidated statements of cash flows for any historical periods or as of, or for the year ended December 31, 2011. We believe that the effect of adopting these standards on future periods will not be material.
In June 2011, the FASB issued ASU 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income (ASU 2011-05). The accounting standard for other comprehensive income was amended to allow only two options for presenting the components of net income and other comprehensive income: (i) in a single continuous financial statement (statement of comprehensive income) or (ii) in two separate but consecutive financial statements, consisting of an income statement followed by a separate statement of other comprehensive income. In addition, items that are reclassified from other comprehensive income to net income must be presented on the face of the financial statements. This standard update requires retrospective application and is effective for our 2012 fiscal year. We do not expect the adoption of ASU 2011-05 to have a material effect on our consolidated financial statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We do not have “trading” or “other than trading” portfolios of market risk sensitive instruments, and we do not purchase hedging instruments that are likely to expose us to significant market risk, whether interest rate, foreign currency exchange, commodity price or equity price risk.
At December 31, 2011, we had cash, cash equivalents and marketable securities in the amount of $96.4 million. Our exposure to market interest rate risk relates solely to our cash, cash equivalents and marketable securities. Cash equivalents and marketable securities consist principally of money market funds placed with major financial institutions. Because of the nature of these funds and the short-term maturities of their investment securities, we do not believe that a change in market rates would have a material negative impact on the value of our investment portfolio. Interest income was $0.2 million for 2011.
26
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors
and Stockholders of Nabi Biopharmaceuticals
We have audited Nabi Biopharmaceuticals’ (the “Company”) internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Nabi Biopharmaceuticals’ management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Nabi Biopharmaceuticals maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2011 consolidated financial statements of Nabi Biopharmaceuticals and our report dated March 14, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
McLean, Virginia
March 14, 2012
27
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors
and Stockholders of Nabi Biopharmaceuticals
We have audited the accompanying consolidated balance sheets of Nabi Biopharmaceuticals as of December 31, 2011 and December 25, 2010, and the related consolidated statements of operations, changes in stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011. Our audits also included the financial statement schedule listed in the index at item 15(a). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Nabi Biopharmaceuticals at December 31, 2011 and December 25, 2010, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), internal control over financial reporting of Nabi Biopharmaceuticals as of December 31, 2011, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 14, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
McLean, Virginia
March 14, 2012
28
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Nabi Biopharmaceuticals
(In thousands, except share and per share data)
| December 31, 2011 |
December 25, 2010 |
|||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 94,310 | $ | 53,564 | ||||
| Marketable securities |
2,079 | 54,603 | ||||||
| Receivables |
995 | 1,030 | ||||||
| Prepaid expenses and other current assets |
497 | 829 | ||||||
|
|
|
|
|
|||||
| Total current assets |
97,881 | 110,026 | ||||||
| Marketable securities |
— | 2,500 | ||||||
| Property and equipment, net |
84 | 597 | ||||||
| Other assets |
— | 748 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 97,965 | $ | 113,871 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 146 | $ | 552 | ||||
| Accrued expenses and other current liabilities |
1,918 | 7,377 | ||||||
| Deferred revenue, current portion |
2,526 | 7,797 | ||||||
| Current liabilities of discontinued operations |
1,662 | 2,207 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
6,252 | 17,933 | ||||||
| Deferred revenue |
32,842 | 35,368 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
39,094 | 53,301 | ||||||
| Stockholders’ equity: |
||||||||
| Convertible preferred stock, par value $0.10 per share; 5,000,000 shares authorized; no shares outstanding |
— | — | ||||||
| Common stock, par value $0.10 per share; 125,000,000 shares authorized; 63,588,882 and 63,206,393 shares issued, respectively |
6,359 | 6,321 | ||||||
| Additional paid-in capital |
373,157 | 370,366 | ||||||
| Treasury stock, 20,696,277 shares at cost |
(92,567 | ) | (92,567 | ) | ||||
| Other comprehensive loss |
— | (3 | ) | |||||
| Accumulated deficit |
(228,078 | ) | (223,547 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
58,871 | 60,570 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 97,965 | $ | 113,871 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
29
Table of Contents
Nabi Biopharmaceuticals
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except share data)
| For the Years Ended | ||||||||||||
| December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
||||||||||
| Revenue: |
||||||||||||
| Revenue |
$ | 14,838 | $ | 35,005 | $ | 10,489 | ||||||
| Operating expenses: |
||||||||||||
| Cost of services |
1,463 | 3,951 | 1,988 | |||||||||
| Research and development expenses |
17,765 | 26,078 | 16,490 | |||||||||
| General and administrative expenses |
5,372 | 6,174 | 9,987 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
24,600 | 36,203 | 28,465 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating loss |
(9,762 | ) | (1,198 | ) | (17,976 | ) | ||||||
| Interest income |
194 | 230 | 368 | |||||||||
| Interest expense |
— | (210 | ) | (1,071 | ) | |||||||
| Other income (expense), net |
37 | 291 | (48 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Loss from continuing operations before income taxes |
(9,531 | ) | (887 | ) | (18,727 | ) | ||||||
| Benefit from income taxes |
2,018 | 1,765 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Income (loss) from continuing operations |
(7,513 | ) | 878 | (18,727 | ) | |||||||
| Income from discontinued operations, (net of tax provision of $2,018 in 2011) |
2,982 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) |
$ | (4,531 | ) | $ | 878 | $ | (18,727 | ) | ||||
|
|
|
|
|
|
|
|||||||
| Basic income (loss) per share: |
||||||||||||
| Continuing operations |
$ | (0.18 | ) | $ | 0.02 | $ | (0.37 | ) | ||||
| Discontinued operations |
$ | 0.07 | $ | — | $ | — | ||||||
| Diluted income (loss) per share: |
||||||||||||
| Continuing operations |
$ | (0.18 | ) | $ | 0.02 | $ | (0.37 | ) | ||||
| Discontinued operations |
$ | 0.07 | $ | — | $ | — | ||||||
| Basic weighted-average shares outstanding |
42,336 | 44,312 | 50,633 | |||||||||
|
|
|
|
|
|
|
|||||||
| Diluted weighted average-shares outstanding |
42,336 | 44,440 | 50,633 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
30
Table of Contents
Nabi Biopharmaceuticals
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(In thousands)
| Common Stock | Additional Paid-in Capital |
Treasury Stock | Accumulated Deficit |
Other Accumulated Comprehensive (Loss) Income |
Total Stockholders’ Equity |
|||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||
| Balance at December 27, 2008 |
62,397 | $ | 6,239 | $ | 363,003 | (10,882 | ) | $ | (42,187 | ) | $ | (205,733 | ) | $ | 60 | $ | 121,382 | |||||||||||||||
| Net loss |
— | — | — | — | — | (18,727 | ) | (18,727 | ) | |||||||||||||||||||||||
| Other comprehensive income |
(80 | ) | (80 | ) | ||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||
| Comprehensive loss |
(18,807 | ) | ||||||||||||||||||||||||||||||
| Stock options exercised |
140 | 14 | 417 | — | — | — | — | 431 | ||||||||||||||||||||||||
| Share-based compensation expense |
— | — | 2,463 | — | — | — | — | 2,463 | ||||||||||||||||||||||||
| Purchase of treasury stock |
— | — | — | (2,048 | ) | (8,080 | ) | — | — | (8,080 | ) | |||||||||||||||||||||
| Stock issued under Employee Stock Purchase Plan |
40 | 4 | 93 | — | — | — | — | 97 | ||||||||||||||||||||||||
| Restricted stock awards, net |
206 | 21 | (21 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Purchase of convertible senior notes |
— | — | (114 | ) | — | — | 35 | — | (79 | ) | ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 26, 2009 |
62,783 | $ | 6,278 | $ | 365,841 | (12,930 | ) | $ | (50,267 | ) | $ | (224,425 | ) | $ | (20 | ) | $ | 97,407 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net income |
— | — | — | — | — | 878 | 878 | |||||||||||||||||||||||||
| Other comprehensive income |
17 | 17 | ||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||
| Comprehensive income |
895 | |||||||||||||||||||||||||||||||
| Stock options exercised |
138 | 14 | 509 | — | — | — | — | 523 | ||||||||||||||||||||||||
| Share-based compensation expense |
— | — | 3,923 | — | — | — | — | 3,923 | ||||||||||||||||||||||||
| Purchase of treasury stock |
— | — | — | (7,766 | ) | (42,300 | ) | — | — | (42,300 | ) | |||||||||||||||||||||
| Stock issued under Employee Stock Purchase Plan |
22 | 2 | 90 | — | — | — | — | 92 | ||||||||||||||||||||||||
| Restricted stock awards, net |
257 | 26 | (26 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Directors fee paid in stock |
6 | 1 | 29 | — | — | — | — | 30 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 25, 2010 |
63,206 | $ | 6,321 | $ | 370,366 | (20,696 | ) | $ | (92,567 | ) | $ | (223,547 | ) | $ | (3 | ) | $ | 60,570 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net loss |
— | — | — | — | — | (4,531 | ) | (4,531 | ) | |||||||||||||||||||||||
| Other comprehensive income |
3 | 3 | ||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||
| Comprehensive income |
(4,528 | ) | ||||||||||||||||||||||||||||||
| Stock options exercised |
145 | 14 | 613 | — | — | — | — | 627 | ||||||||||||||||||||||||
| Share-based compensation expense |
— | — | 2,166 | — | — | — | — | 2,166 | ||||||||||||||||||||||||
| Stock issued under Employee Stock Purchase Plan |
15 | 2 | 34 | — | — | — | — | 36 | ||||||||||||||||||||||||
| Restricted stock awards, net |
223 | 22 | (22 | ) | — | — | — | — | — | |||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2011 |
63,589 | $ | 6,359 | $ | 373,157 | (20,696 | ) | $ | (92,567 | ) | $ | (228,078 | ) | $ | — | $ | 58,871 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
See accompanying notes to consolidated financial statements.
31
Table of Contents
Nabi Biopharmaceuticals
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| For the Years Ended | ||||||||||||
| December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
||||||||||
| Cash flow from operating activities: |
||||||||||||
| Income (loss) from continuing operations |
$ | (7,513 | ) | $ | 878 | $ | (18,727 | ) | ||||
| Adjustments to reconcile income (loss) from continuing operations to net cash provided by (used in) operating activities of continuing operations: |
||||||||||||
| Depreciation and amortization (including impairment |
237 | 376 | 502 | |||||||||
| Non-cash intra-period tax allocation |
(2,018 | ) | — | — | ||||||||
| Accretion of discount on convertible senior notes |
— | 99 | 483 | |||||||||
| Share-based compensation |
2,166 | 3,923 | 2,463 | |||||||||
| Loss (gain) on repurchase of convertible senior notes |
— | — | 302 | |||||||||
| Loss (gain) on sale of property and equipment |
47 | (4 | ) | 4 | ||||||||
| Changes in assets and liabilities: |
||||||||||||
| Receivables |
35 | 8,093 | (6,685 | ) | ||||||||
| Prepaid expenses and other assets |
1,080 | 753 | (2,713 | ) | ||||||||
| Accounts payable, accrued expenses and other |
(5,858 | ) | 1,705 | 2,298 | ||||||||
| Deferred revenue |
(7,797 | ) | 24,718 | 18,447 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) operating activities from continuing operations |
(19,621 | ) | 40,541 | (3,626 | ) | |||||||
| Net cash provided by (used in) operating activities from discontinued operations |
4,455 | (609 | ) | 9,843 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) operating activities |
(15,166 | ) | 39,932 | 6,217 | ||||||||
|
|
|
|
|
|
|
|||||||
| Cash flow from investing activities: |
||||||||||||
| Proceeds from maturities of marketable securities |
66,624 | 142,693 | 55,833 | |||||||||
| Purchases of marketable securities |
(11,598 | ) | (140,289 | ) | (91,471 | ) | ||||||
| Proceeds from sales of property and equipment |
224 | 50 | — | |||||||||
| Capital expenditures |
(1 | ) | (154 | ) | (4 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
55,249 | 2,300 | (35,642 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Cash flow from financing activities: |
||||||||||||
| Proceeds from issuance of common stock for employee benefit plans |
663 | 645 | 528 | |||||||||
| Purchase of common stock for treasury |
— | (42,773 | ) | (7,940 | ) | |||||||
| Repurchase of convertible senior notes |
— | (6,050 | ) | (10,091 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) financing activities |
663 | (48,178 | ) | (17,503 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
40,746 | (5,946 | ) | (46,928 | ) | |||||||
| Cash and cash equivalents at beginning of year |
53,564 | 59,510 | 106,438 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of year |
$ | 94,310 | $ | 53,564 | $ | 59,510 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
32
Table of Contents
Nabi Biopharmaceuticals
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 BUSINESS AND ORGANIZATION
We are a biopharmaceutical company that has focused on the development of vaccines addressing unmet medical needs, including nicotine addiction. We have sought to leverage our experience and knowledge in powering the human immune system to target these serious unmet medical needs. We have been incorporated in Delaware since 1969 and our operations are located in Rockville, Maryland.
Our sole remaining product currently in development is NicVAX® [Nicotine Conjugate Vaccine], an innovative and proprietary investigational vaccine for the treatment of nicotine addiction and prevention of smoking relapse based on patented technology. We suffered a significant setback in 2011 when NicVAX did not achieve the primary endpoint in two Phase III efficacy trials conducted in the U.S. As a result, in November of 2011, we retained Piper Jaffray & Co. (Piper Jaffray) to assist with the exploration of strategic alternatives available to the company. This includes, but is not limited to, the sale, license or merger of all or part of the company or its assets, joint ventures, strategic alliances, recapitalization, or liquidation, and the process is ongoing. As of December 31, 2011, our remaining assets include the following: (i) $96.4 million of cash, cash equivalents and marketable securities, (ii) the potential residual value of NicVAX as well next-generation nicotine vaccine which was licensed to GlaxoSmithKline Biologicals S.A. (GSK) in 2010, (iii) the potential royalty from Phoslyra which was sold to Fresenius USA Manufacturing, Inc. (Fresenius) in 2006, and (iv) the potential value of our net operating losses (NOLs).
In the first quarter of 2010 we granted to GSK (i) an option to exclusively license NicVAX on a worldwide basis and (ii) a license to develop follow-on next-generation nicotine vaccines using our intellectual property combined with GSK proprietary technology including GSK proprietary adjuvants. GSK has not indicated that it is stopping the development of the next generation nicotine vaccine. If GSK continues to develop the next generation nicotine vaccine, we may be eligible to receive milestones and royalties from that program. We continue to support an investigator initiated Phase IIb trial in the Netherlands of NicVAX in combination with Pfizer’s varenicline or Chantix®/Champix, the results of which are expected in the second half of 2012.
The smoking cessation market is estimated to exceed $4 billion annually and is currently considered to be a largely unmet medical need. Nicotine is a non-immunogenic small molecule that, upon inhalation into the lungs, quickly passes into the bloodstream and subsequently reaches the brain by crossing the blood-brain barrier. Once in the brain, the nicotine binds to specific nicotine receptors resulting in the release of substances, such as dopamine, a chemical which triggers the highly addictive pleasurable effects experienced by smokers and users of other nicotine products. NicVAX is designed to stimulate the immune system to produce highly specific antibodies that bind to nicotine in the bloodstream. A nicotine molecule attached to specific antibodies is too large to cross the blood-brain barrier and thus is unable to reach the receptors in the brain thereby reducing the pleasure experienced by the smoker making it easier to quit.
In March 2010, we closed an exclusive worldwide option and licensing agreement with GSK for NicVAX as well as for the development of follow-on nicotine addiction vaccines. Upon closing, we received a $40 million initial payment. Under the terms of the agreement, we granted to GSK (i) an option to obtain an exclusive worldwide license to develop, commercialize and manufacture NicVAX as it currently exists, as well as certain potential alternative forms of NicVAX together with an adjuvant other than a GSK proprietary adjuvant and/or with different presentation, dosage or administration (NicVAX Alternatives), and (ii) an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives). In addition to the $40 million received at closing, we are eligible to receive under the agreement up to $290 million in potential regulatory, development and sales milestones for any next generation nicotine vaccines. We are also eligible to receive royalties on global sales of any next generation of nicotine
33
Table of Contents
vaccines. Notwithstanding the failure to achieve the primary endpoint in the two Phase III trials, if GSK was to exercise its option under the agreement for NicVAX and any NicVAX alternatives, it will pay us $58 million upon exercise plus certain potential milestones and royalties over time.
During 2011 we completed our obligations to continue to develop PentaStaph™ [Pentavalent S.aureus Vaccine] under contract for GSK. PentaStaph is a new pentavalent vaccine designed to prevent S.aureus infections including those infections caused by the most dangerous antibiotic-resistant strains of S.aureus. In August 2009, we entered into an Asset Purchase Agreement (APA) with GSK for the sale of PentaStaph for a total consideration of $46 million including a $20 million up-front payment upon closing and $26 million payable upon achievement of certain milestones. The PentaStaph sale closed in November 2009, and we received payment from GSK of $21.5 million representing the up-front payment of $20 million, an additional $1 million for the sale of our Staphylococcus epidermidis vaccine program and an additional $0.5 million for transfer of certain specified materials. We also agreed to a Transition Services Agreement (TSA) to help GSK advance the program while in parallel transferring the technology to GSK. In 2011 we completed all of the efforts required of us from GSK under contract for PentaStaph. As a result, we have received the full $26 million in potential milestone payments, $5 million in 2011 and $21 million in 2010.
NOTE 2 SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of consolidation: The consolidated financial statements include the accounts of Nabi Biopharmaceuticals and our wholly-owned subsidiaries (referred to as “Nabi,” the “Company,” “us,” or “we” throughout this report). All significant inter-company accounts and transactions are eliminated in consolidation. All our wholly-owned subsidiaries are dormant or are otherwise non-operative.
Accounting estimates: The preparation of financial statements in conformity with accounting principles generally accepted in the U.S. requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of income and expenses during the reporting period, including such amounts related to discontinued operations. Actual results could differ from those estimates.
Fiscal year periods: Our fiscal year ends on the last Saturday of December. Consequently, we will periodically have a 53-week fiscal year, as was the case for 2011. The fiscal years ended for the periods presented in the accompanying consolidated financial statements are December 31, 2011, December 25, 2010, and December 26, 2009.
Financial instruments: The carrying amounts of financial instruments including cash equivalents, marketable securities, accounts receivable and accounts payable approximated fair value as of December 31, 2011 and December 25, 2010, because of the relatively short-term maturity of these instruments.
Cash, cash equivalents and marketable securities: Cash equivalents consist of investments in low risk, highly liquid securities with original maturities of 90 days or less. Marketable securities consist of low risk fixed income investment instruments such as government obligations, government agencies and FDIC backed notes with maturities typically less than eighteen months. Marketable securities are classified as available-for-sale and recorded at market value; unrealized gains and losses on those securities are reflected in other comprehensive income (loss). We assess the risk of impairment related to securities held in our investment portfolio on a regular basis and noted no “permanent” or “other than temporary” impairment during 2011, 2010 and 2009. Our investment policies and procedures are reviewed periodically including by management and our audit committee to minimize credit risk.
Concentration of Credit Risk: Financial instruments that potentially subject us to credit and liquidity risk consist primarily of cash, marketable securities and receivables. The Company maintains cash deposits at major financial institutions with high credit quality. The Company’s operating accounts exceed the Federal Deposit
34
Table of Contents
Insurance Corporation limits of $250,000. Cash equivalents primarily consist of short-term money market funds, which are deposited with reputable financial institutions. The Company’s short-term investments consist primarily of United States government agency securities as well as corporate debt securities and commercial paper. The Company’s investment policy limits investments to only investment-grade securities with the objective to preserve principal and maintain sufficient liquidity to meet operational objectives. The Company’s receivables are with one customer, who has a history of paying timely. The Company’s receivables are expected to be collected.
Property and equipment: Property and equipment are carried at cost. Depreciation is recognized on the straight-line method over the estimated useful lives of the assets as follows:
| Asset |
Estimated Useful Life | |
| Furniture and fixtures |
8 years | |
| Information systems |
3 - 7 years | |
| Machinery and equipment |
4 - 8 years | |
| Leasehold improvements |
Lesser of lease term or economic life |
Recoverability of Long-Lived Assets: Our policy is to evaluate our long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. When an evaluation indicates that an impairment has occurred, a loss is recognized and the asset is adjusted to its estimated fair value. In the fourth quarter of 2011, we recognized an impairment loss of approximately $75 thousand in loss before continuing operations before income taxes related to certain equipment that, as a result of our NicVAX Phase III clinical trials, is no longer used in operations.
Revenue Recognition: Our revenue generating arrangements may include multiple elements and deliverables, including licenses, options, research and development activities, participation on joint steering committees and contract manufacturing, among other elements.
For arrangements entered into prior to December 26, 2010, when we determine that an element has stand-alone value to our customer, we allocate a portion of the total contract price to that element based on its objectively determined and relative fair value, and recognize revenue for that element according to its characteristics. When we cannot reliably and objectively determine fair value of any delivered element, we combine that element with undelivered elements as a single unit of accounting.
For arrangements entered into or materially modified after December 25, 2010, when we determine that an element has stand-alone value to our customer, we allocate a portion of the total contract price to that element based on its relative selling price, determined pursuant to a selling price hierarchy, and recognize revenue for that element according to its characteristics. We did not enter into or materially modify any revenue arrangements subsequent to December 25, 2010.
Revenue consists of license fees, milestone payments, and payments for contractual services. License fees received are initially recorded as deferred revenue, and are subsequently recognized as revenue ratably over the period of our participation on joint steering committees. The joint steering committee related to the NicVAX agreement is currently expected to operate for 190 months from the date of the agreement (or through December 2025). Our efforts under the joint steering committee related to the PentaStaph agreement were completed in the second quarter of 2011; accordingly, we have fully recognized the up-front payment related to the PentaStaph agreement.
For milestones that are deemed substantive, we recognize the contingent revenue when: (i) the milestones have been achieved; (ii) no further performance obligations with respect to the milestones exist; and (iii) collection is reasonably assured. A milestone is considered substantive if all of the following conditions are met: (i) the milestone is nonrefundable; (ii) achievement of the milestone was not reasonably assured at the
35
Table of Contents
inception of the arrangement; (iii) substantive effort is involved to achieve the milestone; and (iv) the amount of the milestone appears reasonable in relation to the effort expended with the other milestones in the arrangement and the related risk associated with achievement of the milestone. If a milestone is deemed not to be substantive, the Company would recognize the portion of the milestone payment as revenue that correlates to work already performed; the remaining portion of the milestone payment will be deferred and recognized as revenue as the Company completes its performance obligations.
Payments for contractual services are recognized as revenue when earned, typically when the services are rendered.
We analyze cost reimbursable grants and contracts to determine whether we should report such reimbursements as revenue or as an offset to research and development expenses incurred.
Collaborative arrangements: We are an active participant with exposure to significant risks and rewards of commercialization relating to the development of NicVAX and future generation nicotine vaccines based on NicVAX technology. For costs incurred and revenues generated from third parties where we are deemed to be the principal participant, we recognize revenues and costs using the gross basis of accounting; otherwise we use the net basis of accounting.
Research and development expenses: Research and development costs are expensed as incurred; advanced payments are deferred and subsequently expensed over the period of performance. Research and development expenses include direct labor costs as well as the costs of contractors and other direct and indirect expenses (including an allocation of the costs of facilities). We expense amounts payable to third parties under collaborative product development agreements at the earlier of the milestone achievement or as payments become contractually due. We recorded approximately $0.3 million, $8.8 million and $1.6 million for 2011, 2010 and 2009 respectively of cost reimbursements from government grants as an offset to research and development expenses.
Income (loss) per share: Basic income (loss) per share is computed by dividing consolidated net income (loss) available to common shareholders by the weighted-average number of common shares outstanding during the period. For periods of net income when the effects are dilutive, diluted earnings per share is computed by dividing net income available to common shareholders (as adjusted for interest expense on our Convertible Senior Notes net of taxes when they were outstanding) by the weighted-average number of shares outstanding and the dilutive impact of all dilutive potential common shares. Dilutive potential common shares consist primarily of stock options and the common shares underlying our Convertible Senior Notes when they were outstanding. The dilutive impact of potential common shares resulting from stock options is determined by applying the treasury stock method. The dilutive impact of potential common shares resulting from our Convertible Senior Notes was determined by applying the “if converted” method. For all periods of net loss, diluted loss per share is calculated similarly to basic loss per share because the impact of all dilutive potential common shares is anti-dilutive due to the net losses; accordingly, diluted loss per share is the same as basic loss per share.
The Company’s unvested restricted shares contain non-forfeitable rights to dividends, and therefore are considered to be participating securities; the calculation of basic and diluted income per share excludes net income attributable to the unvested restricted shares from the numerator and excludes the impact of the shares from the denominator.
Because of our net income in 2010, the computation of diluted income per share differed from the computation of basic income per share as a result of a (i) numerator adjustment for net income allocated to participating securities and (ii) denominator adjustment related to stock options using the treasury stock method. A total of approximately 4.0 million potentially dilutive shares related to stock options were excluded from the calculation of diluted net income per share in 2010 as their inclusion would be anti-dilutive. For 2011 and 2009, a
36
Table of Contents
total of 4.0 million and 1.8 million potential diluted shares have been excluded in the calculation of diluted net loss per share, respectively.
The following table provides the computation of our basic and diluted earnings per share for 2010. Because of our net losses in 2011 and 2009, we did not present diluted earnings per share.
| (in thousands, except per share data) |
December 25, 2010 |
|||
| Numerator: |
||||
| Net income (loss) |
$ | 878 | ||
| Net income allocated to participating securities |
(6 | ) | ||
|
|
|
|||
| Numerator for basic income (loss) per share |
$ | 872 | ||
| Incremental allocation of net income to participating securities |
— | |||
|
|
|
|||
| Numerator for diluted income (loss) per share |
$ | 872 | ||
|
|
|
|||
| Denominator: |
||||
| Denominator for basic income (loss) per share—Weighted-average outstanding common shares |
44,312 | |||
| Dilutive effect of stock options |
128 | |||
|
|
|
|||
| Denominator for diluted income (loss) per share |
44,440 | |||
|
|
|
|||
| Income (loss) per share—basic |
$ | 0.02 | ||
| Income (loss) per share—diluted |
$ | 0.02 | ||
Share-based compensation: We expense the estimated fair value of share-based awards made in exchange for employee services over the requisite employee service period. Share-based compensation cost for stock options is determined at the grant date using an option pricing model; share-based compensation cost for restricted stock is determined at the grant date based on the closing price of our common stock on that date. The value of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the employee’s requisite service period.
Income taxes: We account for income taxes using the asset and liability approach, which requires, among other things, recognition of future tax benefits and liabilities measured at enacted rates attributable to temporary differences between financial statement and income tax bases of assets and liabilities and to tax net operating loss carryforwards to the extent that realization of these benefits is more likely than not. We periodically evaluate the realizability of our net deferred tax assets; a valuation allowance is established when the Company believes that it is more likely than not that its deferred tax assets will not be realized. Changes in valuation allowances from period to period are included in the Company’s tax provision in the period of change. We recognize a tax benefit from uncertain tax positions only if it is “more likely than not” that the position is sustainable based on its technical merits, and our policy is to recognize interest and penalties on uncertain tax positions as a component of income tax expense. We consider discontinued operations for purposes of determining the amount of tax benefits that result from a loss from continuing operations.
Segment information: We currently operate in a single business segment.
New accounting pronouncements: In October 2009, the Financial Accounting Standards Board, or the FASB, issued Accounting Standards Update 2009-13, “Revenue Recognition (Topic 605) Multiple-Deliverable Revenue Arrangements, a consensus of the FASB Emerging Issues Task Force,” or ASU 2009-13. ASU 2009-13 amends existing accounting guidance for separating consideration in multiple-deliverable arrangements. ASU 2009-13 establishes a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence if available, third-party evidence if vendor-specific evidence is not available, or the estimated selling price if neither vendor-specific evidence nor
37
Table of Contents
third-party evidence are available. ASU 2009-13 eliminates the residual method of allocation and requires that consideration be allocated at the inception of the arrangement to all deliverables using the “relative selling price method.” The relative selling price method allocates any discount in the arrangement proportionately to each deliverable on the basis of each deliverable’s selling price. ASU 2009-13 requires that a vendor determine its best estimate of selling price in a manner that is consistent with that used to determine the price to sell the deliverable on a stand-alone basis.
We adopted the provisions of ASU 2009-13 effective December 26, 2010, for revenue arrangements entered into or materially modified in fiscal years beginning on or after that date. The adoption did not have any material effect on our consolidated balance sheets, consolidated statements of operations and consolidated statements of cash flows for any historical periods as of, or for the year ended December 31, 2011 because we did not enter into or materially modify any revenue arrangements subsequent to December 26, 2010. We are not able to reasonably estimate the effect of adopting these standards on future periods because the impact will vary based on the nature and volume of new or materially modified revenue arrangements in any given period.
In April 2010, the FASB issued Accounting Standards Update 2010-17, “Revenue Recognition—Milestone Method (Topic 605) Milestone Method of Revenue Recognition, a consensus of the FASB Emerging Issues Task Force” or ASU 2010-17. ASU 2010-17 provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. For the milestone to be considered substantive, the consideration earned by achieving the milestone should meet all of the following criteria: (i) be commensurate with either the vendor’s performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from the vendor’s performance to achieve the milestone, (ii) relate solely to past performance, and (iii) be reasonable relative to all deliverables and payment terms in the arrangement. An individual milestone may not be bifurcated and an arrangement may include more than one milestone. Accordingly, an arrangement may contain both substantive and non-substantive milestones.
We adopted the provisions of ASU 2010-17 effective December 26, 2010, for milestones achieved on or after that date. Since our existing policies are consistent with those contained in ASU 2010-17, the adoption of ASU 2010-17 did not have any material effect on our consolidated balance sheets, consolidated statements of operations and consolidated statements of cash flows for any historical periods or as of, or for the year ended December 31, 2011. We believe that the effect of adopting these standards on future periods will not be material.
In June 2011, the FASB issued ASU 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income (ASU 2011-05). The accounting standard for other comprehensive income was amended to allow only two options for presenting the components of net income and other comprehensive income: (i) in a single continuous financial statement, statement of comprehensive income or (ii) in two separate but consecutive financial statements, consisting of an income statement followed by a separate statement of other comprehensive income. In addition, items that are reclassified from other comprehensive income to net income must be presented on the face of the financial statements. This standard update requires retrospective application and is effective for our 2012 fiscal year. We do not expect the adoption of ASU 2011-05 to have a material effect on our financial statements.
NOTE 3 DISCONTINUED OPERATIONS
In 2006, we sold our PhosLo (calcium acetate) product and the product’s related assets. In 2007, we sold certain assets related to our Biologics Strategic Business Unit (SBU) and certain corporate shared services assets. The assets and liabilities related to these businesses have identifiable cash flows that are largely independent of the cash flows of other groups of assets and liabilities, and we do not have a significant continuing involvement with the related products. Accordingly, the remaining assets and liabilities, and the results of operations, related to these businesses are presented as discontinued operations in all periods. In 2011, we received a $5 million
38
Table of Contents
milestone payment related to the sale of our PhosLo assets and recognized this payment as income from discontinued operations (net of $2.0 million of intra-period tax allocation).
NOTE 4 AVAILABLE FOR SALE INVESTMENTS
The amortized cost, gross unrealized gains, gross unrealized losses and estimated fair value of available-for-sale investments by security classification as of December 31, 2011 and December 25, 2010 are as follows:
| December 31, 2011 (in thousands) |
Amortized Costs | Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Values |
||||||||||||
| Government-sponsored securities |
$ | 993 | $ | — | $ | — | $ | 993 | ||||||||
| Corporate debt securities |
1,086 | — | — | 1,086 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total securities |
$ | 2,079 | $ | — | $ | — | $ | 2,079 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| December 25, 2010 (in thousands) |
Amortized Costs | Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Values |
||||||||||||
| Government-sponsored securities |
$ | 50,943 | $ | 15 | $ | (19 | ) | $ | 50,939 | |||||||
| Corporate debt securities |
6,163 | 1 | — | 6,164 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total securities |
$ | 57,106 | $ | 16 | $ | (19 | ) | $ | 57,103 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
During 2011, 2010 and 2009, we had no significant realized gains (losses) on sales of available-for-sale securities. Gains and losses on available-for-sale securities are based on the specific identification method.
The contractual maturities of available-for-sale investments by security classification as of December 31, 2011 and December 25, 2010 were as follows:
| December 31, 2011 (in thousands) |
Total | Less than 12 Months | 12 Months or More | |||||||||
| Government-sponsored securities |
$ | 993 | $ | 993 | $ | — | ||||||
| Corporate debt securities |
1,086 | 1,086 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total securities |
$ | 2,079 | $ | 2,079 | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| December 25, 2010 (in thousands) |
Total | Less than 12 Months | 12 Months or More | |||||||||
| Government-sponsored securities |
$ | 50,939 | $ | 48,439 | $ | 2,500 | ||||||
| Corporate debt securities |
6,164 | 6,164 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total securities |
$ | 57,103 | $ | 54,603 | $ | 2,500 | ||||||
|
|
|
|
|
|
|
|||||||
NOTE 5 PROPERTY AND EQUIPMENT
Property and equipment and related accumulated depreciation are summarized below:
| December 31, | December 25, | |||||||
| (in thousands) |
2011 | 2010 | ||||||
| Information systems |
$ | 1,745 | $ | 2,160 | ||||
| Leasehold improvements |
1,851 | 3,204 | ||||||
| Machinery and equipment |
488 | 4,420 | ||||||
| Furniture and fixtures |
236 | 239 | ||||||
|
|
|
|
|
|||||
| Property and equipment |
4,320 | 10,023 | ||||||
| Less accumulated depreciation |
(4,236 | ) | (9,426 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 84 | $ | 597 | ||||
|
|
|
|
|
|||||
39
Table of Contents
In 2011, we sold or disposed of property and equipment with a net book value of $271 thousand and recognized a loss on such sales and disposals of $47 thousand. We recorded depreciation expense in continuing operations related to property and equipment of $0.2 million, $0.4 million, and $0.5 million in 2011, 2010, and 2009, respectively.
NOTE 6 ACCRUED EXPENSES AND OTHER CURRENT LIABILITIES
Accrued expenses and other current liabilities consist of the following:
| December 31, | December 25, | |||||||
| (in thousands) |
2011 | 2010 | ||||||
| Employee compensation and benefits |
$ | 863 | $ | 2,504 | ||||
| Accrued clinical trial expenses |
595 | 4,411 | ||||||
| Other |
460 | 462 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 1,918 | $ | 7,377 | ||||
|
|
|
|
|
|||||
NOTE 7 SUPPLEMENTAL FAIR VALUE DISCLOSURES
We follow a three-tier fair value hierarchy which prioritizes the inputs used in measuring the fair value of our assets and liabilities. These tiers include (i) Level 1, defined as observable inputs such as quoted prices in active markets for identical assets; (ii) Level 2, defined as observable inputs other than Level 1 prices such as quoted prices for similar assets, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; and (iii) Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions.
All cash and cash equivalents, as well as available-for-sale marketable securities, are recorded at fair value at December 31, 2011 and December 25, 2010. The inputs used in measuring the fair value of these instruments are considered to be Level 1 and Level 2 in accordance with the three-tier fair value hierarchy. The inputs used in measuring the fair value of cash and cash equivalents are considered to be Level 1 in accordance with the three-tier fair value hierarchy and the fair values are based on period-end statements supplied by the various banks and brokers. The majority of our funds were deposited in institutional money market mutual funds with the remainder held in regular interest bearing and non-interest bearing depository accounts with commercial banks.
40
Table of Contents
The inputs used in measuring the fair value of our available-for-sale marketable securities are considered to be Level 2 in accordance with the three-tier fair value hierarchy. These securities are valued using a multi-dimensional pricing model that includes a variety of inputs, including quoted prices for similar assets and liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. To assess the fair value of these securities, we obtain fair values from an independent third-party valuation service provider. As we are responsible for the determination of fair value, we review the values provided by the independent third-party valuation service provider for reasonableness, which could include reviewing other publicly available information.
| Quoted Prices in Active Markets for Identical Assets |
Significant Other Observable Inputs |
Significant Unobservable Inupts |
||||||||||||||
| December 31, 2011 (in thousands) |
Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Cash and cash equivalents |
$ | 94,310 | $ | 94,310 | $ | — | $ | — | ||||||||
| Government-sponsored securities |
993 | — | 993 | — | ||||||||||||
| Corporate debt securities |
1,086 | 721 | 365 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 96,389 | $ | 95,031 | $ | 1,358 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Quoted Prices in Active Markets for Identical Assets |
Significant Other Observable Inputs |
Significant Unobservable Inupts |
||||||||||||||
| December 25, 2010 (in thousands) |
Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Cash and cash equivalents |
$ | 53,564 | $ | 50,564 | $ | 3,000 | $ | — | ||||||||
| Government-sponsored securities |
50,939 | 6,021 | 44,918 | — | ||||||||||||
| Corporate debt securities |
6,164 | 4,324 | 1,840 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 110,667 | $ | 60,909 | $ | 49,758 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
NOTE 8 CONVERTIBLE SENIOR NOTES
In 2005, we issued $112.4 million of our Convertible Senior Notes through a private offering to qualified institutional buyers. In 2010, we repurchased the remaining $6.1 million balance of our Convertible Senior Notes for a total of $6.1 million. In 2009, we repurchased $10.4 million of our Convertible Senior Notes for a total of $10.1 million resulting in a net loss of $0.3 million. Interest payments for 2010 and 2009 were $0.1 million and $0.2 million, which largely consisted of the interest payments for our Convertible Senior Notes. As of December 25, 2010, we had repurchased all of the Convertible Senior Notes.
NOTE 9 STOCKHOLDERS’ EQUITY
Preferred Stock
We have 5,000,000 shares of preferred stock authorized, 1,538,462 of which have been designated as “Series A Convertible Preferred Stock,” 750,000 of which have been designated “Series One Preferred Stock,” 125,000 of which have been designated “Series A Junior Participating Preferred Stock” and 2,586,538 of which remain available for future designation. Holders of preferred stock would normally be entitled to receive a preference payment in the event of any liquidation, dissolution or winding-up of us before any payment is made to the holders of common stock. Currently, there are no outstanding shares of preferred stock.
Treasury Stock
Since 2000, we have purchased our common stock in the open market or in privately negotiated transactions and have repurchased a total of 20.7 million shares for a total cost of $92.6 million, at an average price of $4.47 per share. In 2010, we purchased 7.8 million shares for $42.3 million at an average cost per share of $5.45. Under our current Board-approved repurchase plan, we have $27.8 million available for share repurchases. Repurchased shares have been accounted for as treasury stock using the cost method.
41
Table of Contents
NOTE 10 EMPLOYEE BENEFIT PLANS AND EQUITY-BASED COMPENSATION
We maintain several employee benefit plans for our employees. As of December 31, 2011, a total of 10.4 million shares of common stock were authorized for issuance under our stock option and employee benefit plans.
Retirement Savings Plan
We maintain a retirement savings plan which permits employees to contribute pre-tax annual compensation up to annual statutory limitations. The discretionary company match for employee contributions to the plan is 100% of up to the first 4% of the participant’s earnings contributed to the plan. Our matching contributions to the plan were approximately $0.2 million in 2011, 2010, and 2009. In 2000, the stockholders approved the issuance of up to 425,000 shares of our common stock to our employees participating in our retirement saving plan. In 2011, no shares were issued under this plan.
Employee Stock Purchase Plan
Under the Nabi Employee Stock Purchase Plan (ESPP), qualified employees may purchase our common stock at a price equal to 85% of the lower of the closing price at the beginning or end of each semi-annual stock purchase period. We issued 15,246 shares, 21,576 shares and 40,284 shares of common stock during 2011, 2010, and 2009, respectively, pursuant to this plan at an average price per common share of $2.37, $4.25 and $2.41, respectively. As a result of low employee participation, we discontinued the ESPP in 2011.
Incentive Stock Plan
In 2007, our shareholders approved the 2007 Omnibus Equity and Incentive Plan (2007 Stock Plan) which supersedes and replaces our previous incentive stock plans. All other incentive stock plans will remain in effect with respect to outstanding awards issued under those plans. Accordingly, we have one plan for both employees and directors related to both stock option and restricted stock awards. In connection with the approval of the 2007 Stock Plan, shareholders approved an additional 2.5 million shares of common stock and the transfer of all shares which were available for issuance under the prior incentive stock plans to be available for issuance under the new plan. As of December 31, 2011, we had 2.4 million shares of common stock available for issuance upon the exercise of outstanding options, future grants of options or restricted stock, or other awards under our incentive stock plans.
Under our incentive stock plans, we have granted options to employees and directors entitling them to purchase shares of common stock within seven to ten years of the date of grant. The options have generally been granted at exercise prices equal to the fair market value of the underlying common stock on the date of grant. Options granted to employees under our stock incentive plan typically become exercisable over four years in equal annual installments after the date of grant, and to non-employee directors become fully exercisable in equal quarterly installments over one year, subject to, in all cases, continuous service with the Company. Certain option awards are subject to accelerated vesting. Non-employee directors may elect to be paid their annual retainer as a director in whole or in part in shares of our common stock if approved in advance by our Board of Directors. The number of shares issued if this election is made is the director’s annual cash retainer divided by the closing price of our common stock on the date the annual retainer is awarded.
We also issue restricted stock awards; such awards generally vest over four years.
42
Table of Contents
Accounting for Share-Based Compensation
Share-based compensation expense for the three years ended December 31, 2011, was comprised of:
| For the Years Ended | ||||||||||||
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
|||||||||
| Stock option expense |
$ | 1,358 | $ | 2,854 | $ | 1,722 | ||||||
| Employee stock purchase plan expense |
7 | 37 | 49 | |||||||||
| Restricted stock expense |
801 | 1,032 | 692 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total share-based compensation |
$ | 2,166 | $ | 3,923 | $ | 2,463 | ||||||
|
|
|
|
|
|
|
|||||||
We estimate forfeitures of stock options and restricted stock awards and recognize compensation cost for only those awards expected to vest. Forfeiture rates are determined for three groups of non-employee directors, senior management and all other employees based on historical experience. Estimated forfeiture rates are adjusted from time to time based on actual forfeiture experience and expected future trends.
Our share-based compensation expense is reflected in our Consolidated Statements of Operations as follows:
| For the Years Ended | ||||||||||||
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
|||||||||
| Cost of services |
$ | 142 | $ | 827 | $ | 294 | ||||||
| General and administrative expense |
545 | 719 | 712 | |||||||||
| Research and development expense |
1,479 | 2,377 | 1,457 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total share-based compensation expense |
$ | 2,166 | $ | 3,923 | $ | 2,463 | ||||||
|
|
|
|
|
|
|
|||||||
Share-based compensation costs in 2011 and 2010 reflect $0.2 million and $1.3 million, respectively of share-based compensation expense relating to the modification of the terms of certain outstanding stock option and restricted stock awards. The modifications included accelerated vesting and extended exercise periods relating to certain employee terminations.
Stock Options
We determine the fair value of each stock option on the date of grant using the Black-Scholes option-pricing formula and recognize the resulting expense over the option’s vesting period using the straight-line attribution approach. Below are the calculated weighted-average fair values for 2011, 2010, and 2009 as well as the assumptions used in calculating those values:
| For the Years Ended | ||||||||||||
| December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
||||||||||
| Weighted-average fair value (per share) |
$ | 2.95 | $ | 3.51 | $ | 1.98 | ||||||
| Assumptions: |
||||||||||||
| Expected term (in years) |
4.5 - 6.3 | 4.5 - 6.3 | 4.5 - 6.3 | |||||||||
| Risk-free interest rate |
1.76% - 2.47% | 1.39% - 2.75% | 1.45% - 2.96% | |||||||||
| Expected volatility |
56.45% - 71.32% | 58.51% - 83.72% | 74.94% - 83.6% | |||||||||
| Expected dividend yield |
0% | 0% | 0% | |||||||||
| • | Expected Term: The expected term represents the period over which the share-based awards are expected to be outstanding based on the historical experience of our employees. |
43
Table of Contents
| • | Risk-Free Interest Rate: The risk-free interest rate is based on the implied yield currently available on U.S. Treasury zero-coupon issues with a remaining term equivalent to the stock option award’s expected term. |
| • | Expected Volatility: The expected volatility is based on the historical price of our stock over the most recent period commensurate with the expected term of the stock option award. |
| • | Expected Dividend Yield: We do not intend to pay dividends on common stock for the foreseeable future. Accordingly, we used a dividend yield of 0% in the assumptions. |
A summary of option activity under our stock plans as of December 31, 2011 and the changes during 2011 is presented below:
| Stock Options |
Number of Options |
Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (years) |
Aggregate Intrinsic Value ($000’s) |
||||||||||||
| Outstanding at December 25, 2010 |
4,125,394 | $ | 5.99 | 4.13 | $ | 3,704 | ||||||||||
| Granted |
853,350 | $ | 5.73 | |||||||||||||
| Exercised |
(144,788 | ) | $ | 4.33 | ||||||||||||
| Forfeited |
(377,568 | ) | $ | 5.28 | ||||||||||||
| Expired |
(402,736 | ) | $ | 6.31 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2011 |
4,053,652 | $ | 6.02 | 3.72 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Vested and expected to vest at December 31, 2011 |
3,464,295 | $ | 6.16 | 3.39 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable at December 31, 2011 |
2,874,938 | $ | 6.35 | 2.96 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
As of December 31, 2011 there was $2.8 million of unrecognized compensation cost related to the stock options granted under our stock plans which is expected to be recognized over a weighted-average period of four years. The total intrinsic value of stock options exercised was $0.2 million in 2011, 2010, and 2009.
Restricted Stock
A summary of the status of our restricted stock awards as of December 31, 2011 and changes during 2011 is presented below:
| Restricted Stock |
Number of Shares |
Weighted- Average Fair Value at Grant Date |
||||||
| Nonvested at December 25, 2010 |
437,051 | $ | 3.78 | |||||
| Granted |
226,268 | $ | 5.81 | |||||
| Vested |
(253,529 | ) | $ | 3.79 | ||||
| Forfeited |
(3,813 | ) | $ | 5.38 | ||||
|
|
|
|||||||
| Nonvested at December 31, 2011 |
405,977 | $ | 4.89 | |||||
|
|
|
|||||||
As of December 31, 2011, there was $1.7 million of total unrecognized compensation cost related to restricted stock awards granted under our stock plans. That cost is expected to be recognized over a weighted-average period of four years. The total fair value of shares vested during 2011, 2010 and 2009 was $1.0 million, $0.5 million, and $0.9 million, respectively.
44
Table of Contents
NOTE 11 INCOME TAXES
The provision (benefit) for income taxes from continuing operations consists of the following:
| For the Years Ended | ||||||||||||
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
|||||||||
| Current: |
||||||||||||
| Federal |
$ | — | $ | (1,765 | ) | $ | — | |||||
| State |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| — | (1,765 | ) | — | |||||||||
| Deferred: |
||||||||||||
| Federal |
453 | (1,692 | ) | (17,980 | ) | |||||||
| State |
123 | (8,587 | ) | (1,366 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| 576 | (10,279 | ) | (19,346 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total |
576 | (10,279 | ) | (19,346 | ) | |||||||
| Change in Valuation Allowance |
(576 | ) | 10,279 | 19,346 | ||||||||
|
|
|
|
|
|
|
|||||||
| Total, net before intra period allocation |
— | (1,765 | ) | — | ||||||||
| Intra-period tax allocation |
(2,018 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Total, net |
$ | (2,018 | ) | $ | (1,765 | ) | $ | — | ||||
|
|
|
|
|
|
|
|||||||
The following table includes deferred tax assets and liabilities from both continuing and discontinued operations as of December 31, 2011 and December 25, 2010:
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
||||||
| Deferred tax assets: |
||||||||
| Federal net operating loss carryforwards |
$ | 58,518 | $ | 62,325 | ||||
| State net operating loss carryforwards |
10,872 | 11,455 | ||||||
| Research and experimental tax credit |
12,302 | 12,708 | ||||||
| Income from sale of assets |
15,407 | 7,295 | ||||||
| Deferred research and experimental costs |
4,051 | 5,402 | ||||||
| Depreciation |
— | 1,237 | ||||||
| Alternative minimum tax credit |
854 | 854 | ||||||
| Equity-based and accrued compensation related costs |
3,477 | 6,399 | ||||||
| Other |
888 | 1,302 | ||||||
|
|
|
|
|
|||||
| Deferred tax assets |
106,369 | 108,977 | ||||||
| Deferred tax liabilities: |
||||||||
| Depreciation |
(110 | ) | — | |||||
| Other |
(204 | ) | (329 | ) | ||||
|
|
|
|
|
|||||
| Deferred tax liabilities |
(314 | ) | (329 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
106,055 | 108,649 | ||||||
| Valuation allowance |
(106,055 | ) | (108,649 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
As of December 31, 2011, we have gross federal net operating loss carryforwards of approximately $167.2 million that expire at various dates through 2031. We have federal research and experimental tax credit carryforwards of approximately $14.5 million ($12.3 million, net of unrecognized tax benefit) that expire in varying amounts through 2026. We have federal alternative minimum tax credit carryforwards of $0.9 million
45
Table of Contents
that are available to offset future regular tax liabilities and do not expire. Under Section 382 of the Internal Revenue Code of 1986, as amended (IRC), certain significant changes in ownership may restrict the future utilization of our tax loss carryforwards and tax credit carryforwards. The annual limitation is equal to the value of our stock immediately before the ownership change, multiplied by the long-term tax-exempt rate (i.e., the highest of the adjusted Federal long-term rates in effect for any month in the three-calendar-month period ending with the calendar month in which the change date occurs). In August 2011, in an effort to protect the use of its operating loss carryforwards, the Company adopted a “NOL” Rights Agreement that discourages significant changes in ownership of the Company’s stock that might limit the use of our operating loss carryforwards, as further discussed in Note 14. Based upon calculations performed for the period through December 31, 2011, the Company’s tax attributes are not currently limited under Section 382.
We have determined that a full valuation allowance is required against all our deferred tax assets that we do not expect to be offset by deferred tax liabilities. As a result, we recorded $106.1 million and $108.6 million valuation allowance as of December 31, 2011 and December 25, 2010, respectively.
The following table reconciles our losses from continuing operations before income taxes by jurisdiction:
| For the Years Ended | ||||||||||||
| (in thousands) |
December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
|||||||||
| Pre-tax (loss) income: |
|
|||||||||||
| U.S. |
$ | (9,491 | ) | $ | (821 | ) | $ | (18,720 | ) | |||
| Foreign |
(40 | ) | (66 | ) | (7 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | (9,531 | ) | $ | (887 | ) | $ | (18,727 | ) | |||
|
|
|
|
|
|
|
|||||||
The significant elements contributing to the difference between the federal statutory tax rate and the effective tax rate are as follows:
| For the Years Ended | ||||||||||||
| December 31, 2011 |
December 25, 2010 |
December 26, 2009 |
||||||||||
| Federal statutory rate |
(35.0 | )% | (35.0 | )% | (35.0 | )% | ||||||
| State income taxes, net of federal benefit |
(5.4 | ) | (5.0 | ) | (5.4 | ) | ||||||
| Foreign tax rate differential |
— | 2.6 | — | |||||||||
| Tax credits |
— | 194.7 | — | |||||||||
| Expiration of credits and other |
4.3 | — | — | |||||||||
| Valuation allowance |
(6.0 | ) | 1,114.7 | 105.0 | ||||||||
| Federal refund claim |
— | (199.0 | ) | — | ||||||||
| Return to provision |
42.1 | (1,305.0 | ) | (50.1 | ) | |||||||
| Other |
— | 3.0 | (14.5 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total before intra-period allocation |
— | % | (199.0 | )% | — | % | ||||||
|
|
|
|
|
|
|
|||||||
| Intra-period tax allocation |
(21.2 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Total |
(21.2 | )% | (199.0 | )% | — | % | ||||||
|
|
|
|
|
|
|
|||||||
During the third quarter of 2010, as a result of new legislation allowing for the carryback of Net Operating Losses generated in 2008 or 2009 for up to five years, we filed a refund claim with the IRS to recover taxes paid in 2007 and 2004 totaling approximately $1.8 million. We paid no income taxes in 2011 or 2009.
46
Table of Contents
Uncertain Income Tax Positions
We are subject to income taxes in the U.S., various states and numerous foreign jurisdictions. Significant judgment is required in evaluating our tax positions and determining our provision for income taxes. We establish reserves for tax-related uncertainties based on estimates of whether, and the extent to which, additional taxes will be due. These reserves are established when we believe that certain positions might be challenged despite our belief that our tax return positions are fully supportable. We adjust these reserves in light of changing facts and circumstances, such as the outcome of a tax audit. The provision for income taxes includes the impact of reserve provisions and changes to reserves that are considered appropriate.
We are subject to tax audits in all jurisdictions for which we file tax returns. Tax audits by their very nature are often complex and can require several years to complete. Under the tax statute of limitations applicable to the IRC, we are no longer subject to U.S. federal income tax examinations by the IRS for years before 2007. However, because we are carrying forward income tax attributes, such as net operating losses and tax credits from 2002 and earlier tax years, these attributes can still be audited when used on returns filed in the future. Tax attributes carried forward from 2002 and earlier tax years recently utilized in tax years for which the statue of limitations have not yet expired are also subject to audit. Under the statutes of limitation applicable to most state income tax laws, we are no longer subject to state income tax examinations by tax authorities for years before 2007 in states in which we have filed income tax returns. Certain states may take the position that we are subject to income tax in such states even though we have not filed income tax returns in such states and, depending on the varying state income tax statutes and administrative practices, the statute of limitations in such states may extend to years before 2007. We began foreign operations in 2004. We are subject to foreign tax examinations by tax authorities for all years of operation.
The following is a tabular reconciliation of the total amounts of unrecognized tax benefits:
| For the Years Ended | ||||||||||||
| December 31, | December 25, | December 26, | ||||||||||
| (in thousands) |
2011 | 2010 | 2009 | |||||||||
| Unrecognized tax benefit - opening balance |
$ | 3,917 | $ | 5,750 | $ | 8,150 | ||||||
| Gross increases |
— | — | 3,634 | |||||||||
| Gross decreases |
(443 | ) | (1,833 | ) | (6,034 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Unrecognized tax benefit - ending balance |
$ | 3,474 | $ | 3,917 | $ | 5,750 | ||||||
|
|
|
|
|
|
|
|||||||
As of December 31, 2011, any potential interest and penalties on unrecognized tax benefits were not significant. Unrecognized tax benefits are shown as a reduction in net deferred tax assets in the accompanying balance sheets.
NOTE 12 LEASES
All of our leases for facilities and equipment are considered operating leases and are renewed on a month-to-month basis. Rent expense for continuing operations was approximately $1.0 million for 2011, 2010, and 2009.
NOTE 13 LICENSES AND ROYALTY AGREEMENTS
We have entered into licenses and royalty agreements for our products in development.
PentaStaph: In November 2009, we sold our PentaStaph product candidate and related assets to GSK under an Asset Purchase Agreement for a total consideration of $46 million including a $20 million up-front payment and $26 million payable upon achievement of certain milestones, all of which we have received. At the same time, we received an additional $1 million for the sale of our Staphylococcus epidermidis vaccine program and an additional $0.5 million for transfer of certain specified materials. In 2011, 2010 and 2009, we recognized $5 million, $16 million and $5 million of development milestones, respectively. We determined the milestones to be substantive as their achievement required substantive efforts by us and was at risk until the milestones were ultimately achieved.
47
Table of Contents
Revenue under this arrangement consists of license fees, milestone payments, and payments for contractual services. License fees received are initially recorded as deferred revenue, and are subsequently recognized as revenue ratably over the period of our participation on joint steering committees. Our efforts under the joint steering committee related to the PentaStaph agreement were completed in the second quarter of 2011; accordingly, we have fully recognized the up-front payment related to the PentaStaph agreement. During the second quarter of 2011 we completed our work to help develop PentaStaph under contract with GSK.
NicVAX: In March 2010, we closed an exclusive worldwide option and licensing agreement with GSK for NicVAX as well as for the development of follow-on nicotine addiction vaccines. Upon closing, we received a $40 million initial payment. Under the terms of the agreement, we granted to GSK (i) an option to obtain an exclusive worldwide license to develop, commercialize and manufacture NicVAX as it currently exists, as well as certain potential alternative forms of NicVAX together with an adjuvant other than a GSK proprietary adjuvant and/or with different presentation, dosage or administration (NicVAX Alternatives), and (ii) an exclusive worldwide license to develop, commercialize and manufacture certain future generation candidate vaccines for the prevention or treatment of nicotine addiction based on our NicVAX intellectual property (other than NicVAX and NicVAX Alternatives). Notwithstanding the failure to achieve the primary endpoint in our two Phase III trials, if GSK were to exercise its option under the agreement for NicVAX and any NicVAX Alternatives, GSK would pay us $58 million upon exercise plus (i) up to $61 million in regulatory milestones based on obtaining regulatory approval for NicVAX in certain major market countries, and (ii) up to $42 million in development and regulatory milestones for future generation candidates based on (a) Phase II and Phase III clinical trial-related results (up to an aggregate of $21 million) and (b) obtaining regulatory approval in certain major market countries (up to an aggregate of $21 million). Alternatively, if GSK does not exercise the NicVAX option, for future generation candidates GSK will pay us (i) up to $47 million based on Phase II and Phase III clinical trial-related milestones, and (ii) up to an aggregate of $34 million based on obtaining regulatory approval in certain major market countries. Regardless of whether GSK exercises the NicVAX option, GSK will pay us certain tiered, sales-milestone payments up to an aggregate of $209 million based on aggregate annual sales of NicVAX, licensed NicVAX Alternatives and future generation candidates above certain net sales thresholds.
If GSK exercises the NicVAX option, it will make royalty payments to us on aggregate annual net sales of NicVAX, beginning at 10% and potentially increasing on incremental sales to as high as 15%, with the increase depending on whether aggregate annual net sales of NicVAX meet or exceed specified annual sales targets in any calendar year ranging from $300 million to $600 million. Whether or not GSK exercises the NicVAX option, it will pay us royalty payments on aggregate annual net sales of future generation candidates, beginning at 7% and potentially increasing on incremental sales to as high as 9%, with the increase depending on whether aggregate annual net sales of future generation candidates meet or exceed specified annual sales targets in any calendar year ranging from $300 million to $600 million. The royalties payable by GSK as described above (i) on future generation candidates are subject to certain reductions of up to 25% depending on improvements in the therapeutic effect and/or reduction in the dosing of future generation candidates relative to NicVAX, and (ii) on NicVAX and future generation candidates are subject to certain reductions if intellectual property license payments are owed to third parties. In either case, however, the minimum royalty rate on NicVAX will be 7.5% and the minimum royalty rate on future candidates will be 5%.
The economic terms of GSK’s license of NicVAX Alternatives (should GSK exercise the NicVAX option) are subject to mutual agreement between GSK and us. If the parties cannot mutually agree, then such economic terms will be determined through binding arbitration based on an agreed upon set of factors and principles relating to, among other things, the commercial potential of the NicVAX Alternatives subject to the option exercise and the relative contributions of us and GSK to the development of such NicVAX Alternatives.
We believe all of our future milestones under the NicVAX agreement to be substantive as they are at risk until ultimately achieved. The probability of us receiving future contingent milestones is uncertain as it is based on the achievement of various success-based development and regulatory approvals contingent upon the occurrence of various future events, most of which have a high degree of uncertainty of occurring.
48
Table of Contents
Revenue under the NicVAX agreement consists of license fees, milestone payments, and payments for contractual services. License fees received are initially recorded as deferred revenue, and are subsequently recognized as revenue ratably over the period of our participation on joint steering committees. The joint steering committee related to the NicVAX agreement is currently expected to operate for 190 months from the date of the agreement (or through December 2025).
Other: In November 2006, we sold certain assets related to our PhosLo operations. Under the sale agreement, we received $65 million in cash at closing and $18 million in milestones to date, of which $5 million was related to the first commercial sale of a new liquid formulation and was received in the third quarter of 2011. We also are entitled to additional contingent milestone payments of $2.5 million upon approval of a new indication for PhosLo and royalties of up to $65.0 million on annual sales of the new formulation over a base amount of $32 million for 10 years after the November 14, 2006 closing date. We believe future milestones to be substantive as they are at risk until ultimately achieved. The probability of us receiving the future contingent milestone is uncertain as it is based on the achievement of various success-based development and regulatory approvals contingent upon the occurrence of various future events, most of which have a high degree of uncertainty of occurring.
NOTE 14 NOL RIGHTS AGREEMENT
On August 25, 2011, our Board of Directors adopted a stockholder Rights Agreement with American Stock Transfer & Trust Company, LLC, as rights agent, in an effort to prevent an “ownership change” under Section 382 from occurring and thereby protect the value of our net operating losses. The Rights Agreement serves the interests of all stockholders by seeking to protect the use of our deferred tax assets to offset future tax liabilities. Under the Rights Agreement, the Board of Directors declared a non-taxable dividend of one preferred share purchase right for each outstanding share of common stock to be distributed to stockholders of record on August 25, 2011. These rights will become active if triggered under the Rights Agreement. If any person or group acquires 4.99 percent or more of the outstanding shares of common stock, the rights under the Rights Agreement would be triggered resulting in significant dilution in the ownership interest of such person or group in Nabi stock. Stockholders who currently beneficially own 4.99 percent or more of the outstanding shares of common stock would not trigger the preferred share purchase rights unless they acquire additional shares. The Rights Agreement has a three-year term and will be reviewed periodically by a committee of the Board to assess whether the agreement should be maintained.
NOTE 15 COMMITMENTS AND CONTINGENCIES
During 2006, we engaged an outside consultant to assess our pricing programs under Medicare/Medicaid and other governmental pricing programs during the period from 2002 through the second quarter of 2006. In connection with this review, we identified additional liabilities related to discontinued operations for possible overbilling under Medicare/Medicaid and other governmental pricing programs, of which our estimates of the remaining amounts due was approximately $1.0 million at December 31, 2011 and $1.5 million at December 25, 2010, which are included in the amounts recorded as current liabilities from discontinue operations. We intend to pay these obligations as they are rebilled to us.
In preparation for the commercialization of NicVAX and prior to the failure of the NicVAX Phase III trials, we had entered into various development agreements with CMOs to both manufacture NicVAX and transfer the manufacturing technologies know-how of NicVAX. If we terminate some or all of these agreements or do not purchase required amounts of product under them, we will be required to pay certain cancellation fees to the CMOs.
We have agreements with our employees that include certain cash payments and equity-based award modifications in the event of a termination of employment or a change in control of the Company.
49
Table of Contents
Litigation
We are parties to legal proceedings that we believe to be ordinary, routine litigation incidental to the business of present or former operations. It is management’s opinion, based on the advice of counsel, that the ultimate resolution of such litigation will not have a material adverse effect on our financial condition, results of operations or cash flows.
NOTE 16 SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
Due to rounding, the quarterly per share amounts may not add to the annual amount.
| For the Fiscal 2011 Quarters Ended | ||||||||||||||||
| March 26, | June 25, | September 24, | December 31, | |||||||||||||
| (in thousands, except per share data) |
2011 | 2011 | 2011 | 2011 | ||||||||||||
| Revenue |
$ | 9,173 | $ | 3,744 | $ | 1,086 | $ | 835 | ||||||||
| Operating income (loss) |
1,871 | (4,687 | ) | (4,937 | ) | (2,009 | ) | |||||||||
| Net income (loss) |
1,980 | (4,599 | ) | 78 | (1,990 | ) | ||||||||||
| Basic income (loss) per share |
$ | 0.05 | $ | (0.11 | ) | $ | — | $ | (0.05 | ) | ||||||
| Diluted income (loss) per share |
$ | 0.05 | $ | (0.11 | ) | $ | — | $ | (0.05 | ) | ||||||
| For the Fiscal 2010 Quarters Ended | ||||||||||||||||
| March 27, | June 26, | September 25, | December 25, | |||||||||||||
| (in thousands, except per share data) |
2010 | 2010 | 2010 | 2010 | ||||||||||||
| Revenue |
$ | 13,741 | $ | 4,849 | $ | 12,335 | $ | 4,080 | ||||||||
| Operating income (loss) |
5,392 | (3,487 | ) | 3,276 | (6,379 | ) | ||||||||||
| Net income (loss) |
5,480 | (3,406 | ) | 5,135 | (6,331 | ) | ||||||||||
| Basic income (loss) per share |
$ | 0.11 | $ | (0.08 | ) | $ | 0.12 | $ | (0.15 | ) | ||||||
| Diluted income (loss) per share |
$ | 0.11 | $ | (0.08 | ) | $ | 0.12 | $ | (0.15 | ) | ||||||
50
Table of Contents
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management has evaluated, with the participation of our Chief Executive Officer (who is also our acting Chief Financial Officer), the effectiveness of our disclosure controls and procedures as of December 31, 2011. Based upon this evaluation, our management has concluded that our disclosure controls and procedures were effective as of December 31, 2011.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended. Under the supervision and with the participation of our management, including our Chief Executive Officer (who is also our acting Chief Financial Officer), we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO criteria).
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2011, and this assessment identified no material weaknesses in our internal control over financial reporting as of that date. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2011. The effectiveness of our internal control over financial reporting as of December 31, 2011 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.
Changes in Internal Control Over Financial Reporting
There was no change in the Company’s internal control over financial reporting during the most recently completed quarter that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting.
None
51
Table of Contents
Nabi Biopharmaceuticals
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information called for by this Item and not already provided in Item 3(a) will be contained in our Proxy Statement, which we intend to file within 120 days following our fiscal year end, December 31, 2011, and such information is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information called for by this Item will be contained in our Proxy Statement, which we intend to file within 120 days following our fiscal year end, December 31, 2011, and such information is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information called for by this Item will be contained in our Proxy Statement, which we intend to file within 120 days following our fiscal year end, December 31, 2011, and such information is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information called for by this Item will be contained in our Proxy Statement, which we intend to file within 120 days following our fiscal year end, December 31, 2011, and such information is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information called for by this Item will be contained in our Proxy Statement, which we intend to file within 120 days following our fiscal year end, December 31, 2011, and such information is incorporated herein by reference.
52
Table of Contents
Nabi Biopharmaceuticals
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a) (1) FINANCIAL STATEMENTS
The following consolidated financial statements are filed as part of this report:
| Page No. | ||||
| 27 | ||||
| Consolidated Balance Sheets at December 31, 2011 and December 25, 2010 |
29 | |||
| 30 | ||||
| 31 | ||||
| 32 | ||||
| 33 | ||||
| (2) FINANCIAL STATEMENT SCHEDULES | ||||
| 57 | ||||
All other schedules omitted are not required, inapplicable or the information required is furnished in the financial statements or notes thereto.
(3) EXHIBITS
| 3.1 | Restated Certificate of Incorporation of Nabi Biopharmaceuticals, as amended (incorporated by reference to Exhibit 3.1 to our Quarterly Report on Form 10-Q for the quarter ended June 26, 2004) | |
| 3.2 | By-Laws of Nabi Biopharmaceuticals (incorporated by reference to Exhibit 3.1 to our Quarterly Report on Form 10-Q for the quarter ended June 28, 2003) | |
| 3.3 | Certificate of Designations of Series One Preferred Stock contained in the Restated Certificate of Incorporation of Nabi Biopharmaceuticals (incorporated by reference to Exhibit 3.1 to our Quarterly Report on Form 10-Q for the period ended June 26, 2004) | |
| 3.4 | Certificate of Designations of Series A Junior Participating Preferred Stock (incorporated by reference to Exhibit 3.1 to our Current Report on Form 8-K filed with the SEC on August 25, 2011) | |
| 4.1 | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.2 to our Annual Report on Form 10-K for the year ended December 30, 2006) | |
| 4.2 | Rights Agreement dated as of August 25, 2011 between Nabi Biopharmaceuticals and American Stock Transfer & Trust Company (incorporated by reference to Exhibit 3.2 to our Current Report on Form 8-K filed with the SEC on August 25, 2011) | |
| 10.1 | 2004 Stock Plan for Non-Employee Directors (incorporated by reference to Appendix C to our Definitive Proxy Statement dated April 9, 2004)+ | |
53
Table of Contents
| 10.2 | 1998 Non-Qualified Employee Stock Option Plan (incorporated by reference to Exhibit 10.22 to our Annual Report on Form 10-K for the year ended December 31, 1998)+ | |
| 10.3 | 2000 Equity Incentive Plan, as amended (incorporated by reference to Appendix B to our Definitive Proxy Statement dated April 9, 2004)+ | |
| 10.4 | 2000 Equity Incentive Plan Award Letter (incorporated by reference to Exhibit 10.8 to our Annual Report on Form 10-K for the year ended December 25, 2004)+ | |
| 10.5 | 2000 Equity Incentive Plan Special Award Letter (incorporated by reference to Exhibit 10.9 to our Annual Report on Form 10-K for the year ended December 25, 2004)+ | |
| 10.6 | 2007 Omnibus Equity and Incentive Plan (incorporated by reference to Appendix A of our Definitive Proxy Statement dated April 12, 2007)+ | |
| 10.7 | Nabi Biopharmaceuticals entered into an Indemnification Agreement in the form filed as Exhibit 10.24 to our Annual Report on Form 10-K for the year ended December 25, 2004, with the following named executive officers: Raafat E.F. Fahim, Ph.D., Matthew Kalnik, Ph.D. and Paul Kessler, M.D.+ | |
| 10.8 | Nabi Biopharmaceuticals has entered into an Indemnification Agreement with each of its directors in the form filed as Exhibit 10.24 to our Annual Report on Form 10-K for the year ended December 25, 2004)+ | |
| 10.9 | Asset Purchase Agreement between Nabi Biopharmaceuticals and Fresenius USA Manufacturing, Inc. dated October 11, 2006 (incorporated by reference to Exhibit 10.35 to our Annual Report on Form 10-K for the year ended December 30, 2006)++ | |
| 10.10 | Amendment No. 1 to Asset Purchase Agreement between Nabi Biopharmaceuticals and Fresenius USA Manufacturing, Inc. dated October 31, 2006 (incorporated by reference to Exhibit 10.36 to our Annual Report on Form 10-K for the year ended December 30, 2006) | |
| 10.11 | Amendment No. 2 to Asset Purchase Agreement between Nabi Biopharmaceuticals and Fresenius USA Manufacturing, Inc. dated November 14, 2006 (incorporated by reference to Exhibit 10.37 to our Annual Report on Form 10-K for the year ended December 30, 2006)++ | |
| 10.12 | Non-Competition and Non-solicitation Agreement between Nabi Biopharmaceuticals and Fresenius USA Manufacturing, Inc. dated November 14, 2006 (incorporated by reference to Exhibit 10.38 to our Annual Report on Form 10-K for the year ended December 30, 2006) | |
| 10.13 | Employment Agreement between Nabi Biopharmaceuticals and Paul Kessler, M.D. dated May 1, 2008 (incorporated by reference to Exhibit 10.1 to our Quarterly Report on Form 10-Q for the quarter ended June 28, 2008)+ | |
| 10.14 | Change of Control Severance Agreement between Nabi Biopharmaceuticals and Paul Kessler, M.D. dated August 21, 2007 (incorporated by reference to Exhibit 10.28 to our Annual Report on Form 10-K for the year ended December 27, 2008)+ | |
| 10.15 | Asset Purchase Agreement between Nabi Biopharmaceuticals and GlaxoSmithKline Biologicals S.A., dated August 5, 2009 (incorporated by reference to Exhibit 2.1 to our Quarterly Report on Form 10-Q for the quarter ended September 26, 2009)++ | |
| 10.16 | Employment Agreement between Nabi Biopharmaceuticals and Matthew W. Kalnik, Ph.D., dated March 17, 2009 (incorporated by reference to Exhibit 10.1 to our Quarterly Report on Form 10-Q for the quarter ended March 28, 2009)+ | |
| 10.17 | Change of Control Severance Agreement between Nabi Biopharmaceuticals and Matthew W. Kalnik, Ph.D., dated March 17, 2009 (incorporated by reference to Exhibit 10.1 to our Quarterly Report on Form 10-Q for the quarter ended March 28, 2009)+ | |
54
Table of Contents
| 10.18 | Exclusive Option and License Agreement between Nabi Biopharmaceuticals and GlaxoSmithKline Biologicals S.A., dated November 13, 2009 (incorporated by reference to Annex A to our Definitive Proxy Statement dated February 4, 2010)++ | |
| 10.19 | Amended and Restated Employment Agreement dated as of March 16, 2011, by and between Nabi Biopharmaceuticals and Raafat E.F. Fahim, Ph.D. 10.1 (incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on March 22, 2011)+ | |
| 10.20 | Amendment No. 1 to Change of Control Severance Agreement dated March 16, 2011 by and between Nabi Biopharmaceuticals and Matthew W. Kalnik, Ph.D. (incorporated by reference to Exhibit 10.2 to our Current Report on Form 8-K filed with the SEC on March 22, 2011)+ | |
| 10.21 | Amendment No. 1 to Change of Control Severance Agreement dated March 16, 2011 by and between Nabi Biopharmaceuticals and Paul Kessler, M.D. (incorporated by reference to Exhibit 10.3 to our Current Report on Form 8-K filed with the SEC on March 22, 2011)+ | |
| 23. | Consent of Independent Registered Public Accounting Firm* | |
| 31.1 | Rule 13a-14(a)/15d-14(a) Certification* | |
| 32. | Section 1350 Certification | |
| 101* | The following materials from the Nabi Biopharmaceuticals Annual Report on Form 10-K for the period ended December 31, 2011 formatted in Extensible Business Reporting Language (XBRL): (i) the Consolidated Balance Sheets at December 31, 2011 and December 25, 2010, (ii) the Consolidated Statements of Operations for the years ended December 31, 2011, December 25, 2010 and December 26, 2009, (iii) the Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2011, December 25, 2010 and December 26, 2009, (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2011, December 25, 2010 and December 26, 2009 and (v) related notes. | |
| * | Filed herewith |
| + | Management contract or compensatory plan or arrangement filed pursuant to Item 15(b) of Form 10-K. |
| ++ | The Company has requested confidential treatment of the redacted portions of this exhibit pursuant to Rule 24b-2, under the Securities Exchange Act of 1934, as amended, and has separately filed a complete copy of this exhibit with the Securities and Exchange Commission. |
55
Table of Contents
Nabi Biopharmaceuticals
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on this 14th day of March 2012.
| Nabi Biopharmaceuticals | ||
| By: | /S/ RAAFAT E.F. FAHIM, PH.D. | |
| Raafat E.F. Fahim, Ph.D. Chief Executive Officer, President, Acting Chief Financial Officer and Director | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signatures |
Title |
Date | ||
| /S/ RAAFAT E.F. FAHIM, PH.D. |
Chief Executive Officer, President, Acting |
March 14, 2012 | ||
| Raafat E.F. Fahim, Ph.D. | Chief Financial Officer and Director |
|||
| /S/ RONALD B. KOCAK |
Controller and Chief Accounting Officer |
March 14, 2012 | ||
| Ronald B. Kocak | ||||
| /S/ JASON ARYEH |
Director |
March 14, 2012 | ||
| Jason Aryeh | ||||
| /S/ DAVID L. CASTALDI |
Director |
March 14, 2012 | ||
| David L. Castaldi | ||||
| /S/ GEOFFREY F. COX, PH.D. |
Non-executive Chairman of the Board of |
March 14, 2012 | ||
| Geoffrey F. Cox, Ph.D. | Directors |
|||
| /S/ PETER B. DAVIS |
Director |
March 14, 2012 | ||
| Peter B. Davis | ||||
| /S/ RICHARD A. HARVEY, JR. |
Director |
March 14, 2012 | ||
| Richard A. Harvey, Jr. | ||||
56
Table of Contents
Nabi Biopharmaceuticals
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS AND RESERVES FROM TOTAL OPERATIONS
(In thousands)
| Additions | Deductions | |||||||||||||||||||||||
| Classification |
Balance at Beginning of Period |
Charged to Costs and Expenses |
Charged to Other Accounts |
Write-Offs Charged Against Reserve |
Other | Balance at End of Period |
||||||||||||||||||
| Year ended December 31, 2011: |
||||||||||||||||||||||||
| Net deferred tax asset valuation allowance |
$ | 108,649 | — | — | (2,594 | ) | — | $ | 106,055 | |||||||||||||||
| Year ended December 25, 2010: |
||||||||||||||||||||||||
| Net deferred tax asset valuation allowance |
$ | 98,494 | 10,155 | — | — | — | $ | 108,649 | ||||||||||||||||
| Year ended December 26, 2009: |
||||||||||||||||||||||||
| Net deferred tax asset valuation allowance |
$ | 86,634 | 11,860 | — | — | — | $ | 98,494 | ||||||||||||||||
Nabi Biopharmaceuticals
EXHIBIT INDEX
| Exhibit No. |
Description | |
| 23. | Consent of Independent Registered Public Accounting Firm | |
| 31.1 | Rule 13a-14(a)/15d-14(a) Certification | |
| 32. | Section 1350 Certification | |
| 101* | The following materials from the Nabi Biopharmaceuticals Annual Report on Form 10-K for the period ended December 31, 2011 formatted in Extensible Business Reporting Language (XBRL): (i) the Consolidated Balance Sheets at December 31, 2011 and December 25, 2010, (ii) the Consolidated Statements of Operations for the years ended December 31, 2011, December 25, 2010 and December 26, 2009, (iii) the Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2011, December 25, 2010 and December 26, 2009, (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2011, December 25, 2010 and December 26, 2009 and (v) related notes. | |
57