Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Pernix Sleep, Inc. | Financial_Report.xls |
| EX-4.8 - EX-4.8 - Pernix Sleep, Inc. | d286915dex48.htm |
| EX-32.2 - EX-32.2 - Pernix Sleep, Inc. | d286915dex322.htm |
| EX-31.2 - EX-31.2 - Pernix Sleep, Inc. | d286915dex312.htm |
| EX-23.1 - EX-23.1 - Pernix Sleep, Inc. | d286915dex231.htm |
| EX-31.1 - EX-31.1 - Pernix Sleep, Inc. | d286915dex311.htm |
| EX-32.1 - EX-32.1 - Pernix Sleep, Inc. | d286915dex321.htm |
| EX-10.35 - EX-10.35 - Pernix Sleep, Inc. | d286915dex1035.htm |
| EX-4.7 - EX-4.7 - Pernix Sleep, Inc. | d286915dex47.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2011
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 000-51665
Somaxon Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 20-0161599 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 10935 Vista Sorrento Parkway, Suite 250, San Diego, CA | 92130 | |
| (Address of principal executive offices) | (Zip Code) | |
(858) 876-6500
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.0001 per share | Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (check one):
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ¨ No x
As of June 30, 2011, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $78.6 million, based on the closing price of the registrant’s common stock on the Nasdaq Capital Market of $2.13 per share.
The number of outstanding shares of the registrant’s common stock, par value $0.0001 per share, as of February 15, 2012 was 48,108,251.
SOMAXON PHARMACEUTICALS, INC.
FORM 10-K — ANNUAL REPORT
For the Fiscal Year Ended December 31, 2011
i
Forward-Looking Statements
Any statements in this report and the information incorporated herein by reference about our expectations, beliefs, plans, objectives, assumptions or future events or performance that are not historical facts are forward-looking statements. You can identify these forward-looking statements by the use of words or phrases such as “believe,” “may,” “could,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “seek,” “plan,” “expect,” “should,” or “would.” Among the factors that could cause actual results to differ materially from those indicated in the forward-looking statements are risks and uncertainties inherent in our business including, without limitation, our ability, together with our strategic advisor Stifel Nicolaus Weisel, to successfully identify, evaluate and execute strategies to maximize stockholder value; our ability to successfully commercialize Silenor; the market potential for insomnia treatments, and our ability to compete within that market; the potential to enter into an agreement with The Procter & Gamble Distributing Company LLC, or P&G, relating to over-the-counter, or OTC, rights for Silenor; our ability, together with any partner, to receive approval from the U.S. Food and Drug Administration, or FDA, for an OTC version of Silenor; our ability to successfully hire, manage and utilize a sales force to market Silenor; our ability to successfully enforce our intellectual property rights and defend our patents, including any developments relating to the submission of abbreviated new drug applications for generic versions of Silenor 3 mg and 6 mg tablets and related patent litigation; the scope, validity and duration of patent protection and other intellectual property rights for Silenor; whether the approved label for Silenor is sufficiently consistent with such patent protection to provide exclusivity for Silenor; the possible introduction of generic competition of Silenor; our ability to ensure adequate and continued supply of Silenor to successfully meet anticipated market demand; our ability to raise sufficient capital to fund our operations, including patent infringement litigation, and the impact of any financing activity on the level of our stock price; the impact of any inability to raise sufficient capital to fund ongoing operations, including any patent infringement litigation; our ability to fully utilize the sales agreement with Citadel Securities LLC as a source of future financings, whether due to market conditions, our ability to satisfy various conditions required to sell shares under the agreement, Citadel Securities’s performance of its obligations under the agreement or otherwise; the impact on the level of our stock price, which may decline, in connection with the occurrence of any sales under the sales agreement; changes in healthcare regulation and reimbursement policies; our ability to operate our business without infringing the intellectual property rights of others; our reliance on our licensee, Paladin Labs, Inc., or Paladin, for critical aspects of the commercial sales process for Silenor outside of the United States; the performance of Paladin and its adherence to the terms of its contracts with us; inadequate therapeutic efficacy or unexpected adverse side effects relating to Silenor that could result in recalls or product liability claims; other difficulties or delays in development, testing, manufacturing and marketing of Silenor; the timing and results of post-approval regulatory requirements for Silenor, and the FDA’s agreement with our interpretation of such results; and other risks detailed below in Part I — Item 1A “Risk Factors.”
Although we believe that the expectations reflected in our forward-looking statements are reasonable, we cannot guarantee future results, events, levels of activity, performance or achievement. We undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, unless required by law.
Corporate Information
We were incorporated in Delaware in August 2003. Our principal executive offices are located at 10935 Vista Sorrento Parkway, Suite 250, San Diego, CA 92130, and our telephone number is (858) 876-6500. Our website address is www.somaxon.com. The information on, or accessible through, our website is not part of this report. Unless the context requires otherwise, references in this report and the information incorporated herein by reference to “Somaxon,” “we,” “us” and “our” refer to Somaxon Pharmaceuticals, Inc.
We have received a trademark registration from the U.S. Patent and Trademark Office, or USPTO, for our corporate name, SOMAXON PHARMACEUTICALS, for use in connection with pharmaceutical preparations for the treatment of neurological, psychiatric and rheumatologic disorders. We have obtained foreign trademark registrations for the trademark SOMAXON PHARMACEUTICALS in Europe, Canada, Japan and Australia. We have received trademark registrations for the trademark SILENOR in the U.S., Europe and Canada. All other trademarks, trade names and service marks appearing in this report are the property of their respective owners. Use or display by us of other parties’ trademarks, trade dress or products is not intended to and does not imply a relationship with, or endorsement or sponsorship of, us by the trademark or trade dress owners.
1
Overview
We are a specialty pharmaceutical company focused on the in-licensing, development and commercialization of proprietary branded products and late-stage product candidates to treat important medical conditions where there is an unmet medical need and/or high-level of patient dissatisfaction, currently in the central nervous system therapeutic area. In March 2010, the U.S. Food and Drug Administration, or FDA, approved our New Drug Application, or NDA, for Silenor® 3 mg and 6 mg tablets for the treatment of insomnia characterized by difficulty with sleep maintenance. Silenor was made commercially available by prescription in the United States in September 2010.
Our principal focus is on commercial activities relating to Silenor. We commercially launched Silenor in September 2010 with 110 sales representatives provided to us on an exclusive basis under our contract sales agreement with Publicis Touchpoint Solutions, Inc., or Publicis, managed by our sales management personnel, and an additional 105 sales representatives provided to us under our co-promotion agreement with P&G. In February 2011, we amended our agreement with Publicis to have Publicis deploy for us an additional 35 sales representatives. Because we did not believe that the growth of Silenor revenues throughout 2011 was sufficient to support sales and marketing expenses at then-current levels, we terminated our agreements with Publicis and P&G in December 2011 in an effort to conserve cash, and effective January 3, 2012, we hired a reduced sales force of 25 field-based sales representatives from Publicis as our employees to promote Silenor.
We have also established the manufacturing and distribution channel for Silenor through agreements with third-party suppliers and service providers, and we have established reimbursement coverage for Silenor with numerous private and government payors.
We believe that Silenor is highly differentiated from other available insomnia treatments, and could have significant advantages in the large insomnia market. Based on data from IMS Health, in 2011 the prescription market for the treatment of insomnia grew approximately 2.2% compared to 2010 to more than 66 million prescriptions.
In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. The exploration of strategic alternatives may not result in any agreement or transaction and, if completed, any agreement or transaction may not be successful or on attractive terms.
Silenor for Insomnia
It is estimated that approximately one-third, or 70 million, of adult Americans are affected by insomnia. One study has found that approximately 20% of those who suffer from insomnia are treated with prescription medications. Silenor was approved by the FDA for the treatment of insomnia characterized by difficulty with sleep maintenance in March 2010, and we commercially launched the product in the United States in September 2010. We believe that Silenor has the potential to offer significant benefits to patients with insomnia.
Silenor is an oral tablet formulation of doxepin at dosages of 3 mg and 6 mg. Doxepin has been marketed and used for over 35 years at dosages from 75 mg to 300 mg per day and is indicated for the treatment of depression and anxiety. However, the available dosages of doxepin for the treatment of depression and anxiety have historically been seldom used in the treatment of insomnia, as they leave many patients reporting next-day residual effects and other undesirable side effects. According to IMS Health data, doxepin accounted for less than 0.1% of the insomnia prescriptions written during 2011.
We believe that Silenor, which utilizes doxepin at low dosages of 3 mg and 6 mg, does not exhibit the same pharmacologic effects as high-dose doxepin. Our clinical development program for Silenor included four Phase 3 clinical trials, and the primary efficacy endpoint achieved statistical significance in each trial. Our clinical trials for Silenor also demonstrated a favorable safety and tolerability profile, including a low dropout rate, an adverse event profile comparable to placebo, no clinically meaningful next-day residual effects and no evidence of amnesia, complex sleep behaviors, hallucinations, tolerance or withdrawal effects.
Silenor binds to H1 receptors in the brain and blocks histamine, which is believed to play an important role in the regulation of sleep. The leading approved insomnia medications, Ambien, Lunesta and Sonata, work by binding and activating a different set of brain receptors known as gamma aminobutyric acid, or GABA, receptors. Currently approved GABA receptor-activating drugs are designated by the Drug Enforcement Administration, or DEA, as Schedule IV controlled substances, which require additional registration and administrative controls. Silenor is not designated as a controlled substance, and according to its FDA-approved labeling, Silenor does not appear to have any potential for dependency, addiction or abuse.
2
Our Strategy
Our goal is to create value for our shareholders through maximizing the potential of our marketed product Silenor, subject to our resource constraints. We also may potentially commercialize promising products that treat medical conditions where there is an unmet medical need or a high level of patient dissatisfaction.
Specifically, we intend to:
| • | Maximize the value of Silenor. Silenor was approved by the FDA for the treatment of insomnia characterized by difficulty with sleep maintenance in March 2010, and we commercially launched the product in the United States in September 2010. We believe that Silenor is highly differentiated from currently available insomnia treatments and could have significant advantages in a large market. We continue to strategically invest in sales and marketing activities to maximize revenue and market share, subject to our resource constraints, and we intend to engage in life cycle management activities relating to Silenor, including potential over-the-counter, or OTC, opportunities. In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. |
| • | Establish collaborations and outsourcing arrangements. We have entered into strategic collaborations and outsourcing arrangements to drive growth and profitability, and we will seek additional collaborations. We believe that leveraging the capabilities of third parties will allow us to add efficiency to our operations and expand our commercial reach, including potentially outside of the United States. |
| • | Selectively evaluate marketed products and product candidates that are differentiated. We selectively evaluate products and product candidates that are differentiated and meet unmet medical needs or address areas of patient dissatisfaction. |
| • | Fully leverage our commercial organization. We have capacity within our existing sales force that can be utilized to sell other products, with the goal of maximizing its productivity and profitability for the benefit of our promoted brand(s) and stockholder value. |
Silenor Market and Commercialization
Disease Background and Market Opportunity
Sleep is essential for human performance, general health and well-being. Insomnia, the most common sleep complaint across all stages of adulthood, is a condition characterized by difficulty falling asleep, waking frequently during the night or too early, or waking up feeling unrefreshed. It is estimated that approximately one-third, or 70 million, of adult Americans are affected by insomnia. One study has found that only approximately 20% of those who suffer from insomnia are currently treated with prescription medications. Chronic insomnia, insomnia lasting more than four weeks, is often associated with a wide range of adverse conditions, including mood disturbances, difficulties with concentration and memory, and certain cardiovascular, pulmonary and gastrointestinal disorders. Chronic sleep deprivation has also been associated with an increased risk of depression, diabetes and obesity, among other disorders. The National Institutes of Health 2005 State-of-the-Science Conference statement on the treatment of insomnia stated that estimates placed the direct and indirect annual costs of chronic insomnia at tens of billions of dollars, but cautioned that such estimates were based on many assumptions and varied extensively.
Based on data from IMS Health, in 2011 the prescription market for the treatment of insomnia grew approximately 2.2% compared to 2010 to more than 66 million prescriptions.
Limitations of Current Therapies
According to the National Sleep Foundation, 65% of respondents reported experiencing insomnia symptoms a few nights a week. In addition, 42% of respondents often experienced awakenings during the night or waking up too early without being able to go back to sleep, which is referred to as sleep maintenance, and 26% had difficulty falling asleep, which is referred to as sleep onset. Historically, insomnia therapies have addressed sleep onset rather than sleep maintenance and duration. Newer therapies have been approved with indications for sleep maintenance, although the ability of previously-available drugs to maintain sleep throughout the night without unwanted next-day residual effects remains limited.
3
The current market-leading prescription products for the treatment of insomnia include GABA-receptor agonists such as Ambien, zolpidem, the generic form of Ambien, in various formulations, Ambien CR, a controlled-release formulation of Ambien, zolpidem ER, the generic form of Ambien CR, Lunesta, Sonata and zaleplon, the generic form of Sonata, in various formulations, melatonin agonists such as Rozerem, several hypnotic benzodiazepines such as temazapam (Restoril) and flurazepam (Dalmane), and sedating antidepressants such as trazodone (Desyrel).
According to physicians that we surveyed in our market research, one of the primary reasons they prescribe sedating antidepressants for the treatment of insomnia is that they generally are not associated with the risk of dependency such as that associated with GABA-receptor agonists. As a result, sedating antidepressants are not Schedule IV controlled substances, and there are no restrictions on their duration of use. As an example, it is estimated that the majority of trazodone prescriptions are prescribed off-label for the treatment of insomnia.
With respect to patients, our pre-launch market research indicated that the market is still underserved due in large part to characteristics associated with many of the then-marketed products. For example, 41% of patients claimed that their medication did not provide them with a full night’s sleep, almost one-third of patients claimed they woke feeling groggy, and 33% claimed to have suffered from memory impairment at some time after taking medication, with almost 80% reporting that they found memory lapse somewhat or very scary. Additionally, 24% of patients on prescription insomnia medication claimed that they were dependent on their medication and could not sleep without it.
In addition, drugs prescribed for insomnia have been associated with many other unwanted side effects, such as dry mouth, unpleasant taste, blurred vision, residual next-day effects, amnesia, hallucinations, physical and psychological dependence, complex sleep behaviors such as sleep driving, hormonal changes and gastrointestinal effects. We believe that drugs with improved tolerability would be well received by both physicians and patients and have the potential to accelerate the growth in the market.
Silenor and its Benefits
We believe that Silenor offers a number of benefits:
| • | Non-scheduled. Because Silenor is not a Schedule IV controlled substance, it can be made available to physicians, facilitating initial physician and patient trial without the additional sampling regulation that applies to controlled substances. |
| • | Safety and tolerability. In our clinical trials for Silenor, there was a low dropout rate, an adverse event profile comparable to placebo and no clinically meaningful next-day residual effects, and we did not observe any amnesia, complex sleep behaviors, hallucinations, tolerance or withdrawal effects or any effect on QT interval prolongation. In addition, high-dose doxepin has been prescribed for over 40 years for depression at up to 50 times our proposed maximum dosage for the treatment of insomnia. |
| • | Efficacy. Silenor is indicated for the treatment of insomnia characterized by difficulty with sleep maintenance. Silenor is the first and only non-scheduled prescription sleep medication approved by the FDA for the treatment of the most commonly reported nighttime symptoms of insomnia: waking frequently during the night and/or waking too early and being unable to return to sleep. Silenor is approved for the treatment of both transient, or short term, and chronic, or long term, insomnia characterized by difficulty with sleep maintenance in both adults and elderly patients. |
Commercialization
Our commercial strategy is multi-faceted and geared toward maximizing the value of products we commercialize, subject to our resource constraints. We are committed to driving prescriptions and market share through experienced sales representatives providing in-person promotion to high-prescribing physicians. These efforts are complemented by on-line and other non-personal promotional initiatives that target both physicians and patients. We are also focused on ensuring broad patient access to our products by negotiating agreements with leading commercial managed care organizations and with government payors. The goals of these efforts are to achieve preferred formulary status relative to our branded competitors and to minimize patients’ out-of-pocket costs.
4
We believe that the commercial success of Silenor largely depends on gaining access to high prescribing physicians of insomnia treatments. We have built a sales and marketing infrastructure focused on timely and informative education of physicians and their office personnel in the effective and appropriate use of Silenor. We accomplish this through in-person promotional and educational programs, product sampling, and other non-personal promotional activities. Our field sales representatives each undergo a rigorous training program focused on our product attributes, disease background, competitive products and our sales techniques, as well as compliance with applicable laws.
We commercially launched Silenor in September 2010 with 110 sales representatives provided to us on an exclusive basis under our contract sales agreement with Publicis, managed by our sales management personnel, and an additional 105 sales representatives provided to us under our co-promotion agreement with P&G. In February 2011, we amended our agreement with Publicis to have Publicis deploy for us an additional 35 sales representatives. Because we did not believe that the growth of Silenor revenues throughout 2011 was sufficient to support sales and marketing expenses at then-current levels, we terminated our agreements with Publicis and P&G in December 2011 in an effort to conserve cash, and effective January 3, 2012, we hired a reduced sales force of 25 field-based sales representatives from Publicis as our employees to promote Silenor.
The efforts of our sales force are complemented by on-line and other non-personal promotional initiatives that target both physicians and patients. We are also focused on ensuring broad patient access to Silenor by negotiating agreements with leading commercial managed care organizations and with government payors. In addition, under our exclusive collaboration with Paladin, Paladin has the right to commercialize Silenor in Canada, South America and Africa, subject to the receipt of marketing approval in each such territory.
Technology In-Licenses
In a license agreement entered into in August 2003, which was amended and restated in September 2010, we acquired the exclusive, worldwide license from ProCom One, Inc., or ProCom, to certain patents to develop and commercialize low dosages of doxepin for the treatment of insomnia. Although patent protection for the current dosage form is limited to the United States, our license to these low-dose doxepin patents is a worldwide license. The term of the license extends until the last licensed patent expires, which is expected to occur no earlier than 2020. The license agreement is terminable at any time by us with 30 days’ notice if we believe that the use of the product poses an unacceptable safety risk or if it fails to achieve a satisfactory level of efficacy. Either party may terminate the agreement with 30 days’ notice if the other party commits a material breach of its obligations and fails to remedy the breach within 90 days, or upon the filing of bankruptcy, reorganization, liquidation, or receivership proceedings relating to the other party.
In October 2006, we entered into a supply agreement with JRS Pharma L.P., or JRS, under which we purchase from JRS all of our requirements for ProSolv®HD90, an ingredient used in our formulation for Silenor. In August 2008, we amended our supply agreement to provide us with the exclusive right to use this ingredient and any successor product to ProSolv®HD90 in combination with doxepin, as well as the right to list the U.S. patents owned by JRS and covering ProSolv®HD90 in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, relating to Silenor. JRS also agreed to enforce any such patents listed in the Orange Book on our behalf. The term of the agreement runs through January 1, 2013, but it will be automatically extended for additional one year periods unless we terminate the license upon written notice at least 90 days prior to the end of the term. Either party may terminate the agreement upon written notice to the other party if the other party commits a material breach of its obligations and fails to remedy the breach within 30 days, or upon the filing of bankruptcy, reorganization, liquidation, or receivership proceedings relating to the other party. We also have the right to terminate the agreement if JRS no longer has any valid patent claim in the U.S. covering ProSolv®HD90 or any successor product to ProSolv®HD90 used by us in combination with doxepin. Each of the current patents listed in the Orange Book by us relating to Silenor expires in 2015.
As part of the amendment, we made an upfront license payment of $0.2 million and are obligated to pay a royalty of less than 1% on net sales of Silenor beginning as of the expiration of the statutory exclusivity period for Silenor in each country in which a Silenor formulation containing ProSolv®HD90 or any successor product to ProSolv®HD90 is marketed, which with respect to U.S. sales of Silenor we expect to occur in March 2013. This royalty is only payable if one or more patents under the license agreement continues to be valid in each such country and a patent relating to our formulation for Silenor has not issued in such country.
5
Technology Out-License
In June 2011, we entered into a license agreement with Paladin under which Paladin has the right to commercialize Silenor in Canada, South America, the Caribbean and Africa, subject to the receipt of marketing approval in each such territory. We also granted to Paladin a right of first negotiation with respect to additional doxepin products we may develop in the licensed territories and, subject to the rights we have previously granted to P&G, a right of first negotiation relating to rights to develop and market Silenor as an over-the-counter medication in the licensed territories.
As consideration for the license, Paladin paid us an upfront payment of $0.5 million. Paladin also purchased 2,184,769 shares of our common stock for an aggregate purchase price of $5.0 million. Once Silenor is commercialized in the licensed territories, we will also be eligible to receive sales-based milestone payments of up to $128.5 million as well as a tiered double-digit percentage of net sales in the licensed territories. Paladin will be responsible for regulatory submissions for Silenor in the licensed territories and will have the exclusive right to commercialize Silenor in the licensed territories. Governance of the collaboration will occur through a joint committee. In February 2012, we announced that Paladin had filed a New Drug Submission that had been accepted for review by Health Canada for Silenor for the treatment of insomnia.
The term of the license agreement runs until the later of the last date on which Silenor is sold by Paladin in the licensed territories or 15 years from the first commercial sale of Silenor in the licensed territories. We may terminate the license agreement on a country-by-country basis in specified key countries upon 60 days’ prior written notice if the first commercial sale has not occurred in such country within 12 months of the date on which marketing approval was obtained in such country. We may also terminate the license agreement upon 60 days’ prior written notice if marketing approval in Canada has not been received by December 7, 2013 or if we are unable to license rights to a third party’s intellectual property and such failure would reasonably be expected to result in a claim from such third party alleging intellectual property infringement or misappropriation. Either party may terminate the license agreement upon an uncured material breach by the other party, upon the bankruptcy or insolvency of the other party, or a force majeure event that lasts for at least 120 days.
In connection with the license agreement, we also entered into a supply agreement with Paladin under which we will supply Paladin with all of its requirements for Silenor during the term of the license agreement or until Paladin procures its own supply of Silenor. Paladin may terminate the supply agreement upon 10 business days’ notice if we are materially unable to supply Silenor to Paladin’s requirements, and at any time if Paladin enters into a direct contractual relationship with our contract manufacturer for Silenor. We may terminate the supply agreement upon 180 days prior written notice if there is a regulatory change or safety consideration that would have a material adverse effect on the global supply chain and at any time on six months’ prior notice after April 30, 2013.
Intellectual Property
Silenor Patents and Patent Applications
We are the exclusive licensee of four U.S. patents from ProCom claiming the use of low dosages of doxepin and other antidepressants. U.S. Patent No. 6,211,229, “Treatment of Transient and Short Term Insomnia,” or the ’229 patent, covers dosages of doxepin from 0.5 mg to 20 mg for use in the treatment of transient insomnia and expires in February 2020.
U.S. Patent No. 5,502,047, “Treatment for Insomnia,” claims the treatment of chronic insomnia using doxepin and expires in March 2013. Due to some prior art that we identified, we initiated a reexamination of our “Treatment for Insomnia” patent. The reexamination proceedings terminated and the United States Patent and Trademark Office, or USPTO, issued a reexamination certificate narrowing certain claims, so that the broadest dosage ranges claimed by us are 0.5 mg to 20 mg for otherwise healthy patients with chronic insomnia and for patients with chronic insomnia resulting from depression, and 0.5 mg to 4 mg for all other chronic insomnia patients. We also requested reissue of this same patent to consider some additional prior art and to add intermediate dosage ranges below 10 mg. In two office actions relating to this reissue request, the USPTO raised no prior art objections to 32 of the 34 claims we were seeking and raised a prior art objection to the other two, as well as some technical objections.
6
Each of the claims objected to by the USPTO related to dosage ranges having an upper limit of approximately 10 mg or higher. After further review of the prior art submitted, the USPTO withdrew all of its prior art objections. We then determined that the proposed addition of the intermediate dosage ranges and the resolution of the technical objections no longer warranted continuation of the reissue proceeding. As a result, we elected not to continue that proceeding. Because Silenor’s approved indication is consistent with the subject matter of our patent claims, we believe that our licensed patents will restrict the ability of competitors to market doxepin with identical drug labeling.
Additionally, we have the exclusive license from ProCom to a third patent in the series, U.S. Patent No. 5,643,897, which is a divisional of the ’047 patent and claims the treatment of chronic insomnia using amitriptyline, trimipramine, trazodone and mixtures thereof in a daily dosage of 0.5 mg to 20 mg. This patent expires in March 2013. A fourth patent to which we have an exclusive license from ProCom, U.S. Patent No. 6,344,487, claims a method of treating insomnia with low dosage forms (0.5 mg to 10 mg) of nortriptyline. This patent expires in June 2020.
In addition, pursuant to our agreement with JRS, we have the exclusive right to use ProSolv®HD90 in combination with doxepin, and the exclusive license to the related patents and patent applications. We listed six of these issued patents in the FDA’s publication “Approved Drug Product with Therapeutic Equivalence Evaluations,” commonly known as the Orange Book.
In March 2011, we received U.S. Patent No. 7,915,307, “Methods of Improving the Pharmacokinetics of Doxepin,” or the ‘307 patent, which generally relates to dosing Silenor 3mg and 6mg tablets at least three hours after a meal to promote faster onset of action and reduce the potential for next-day residual sedation. The pharmacokinetic changes that result from dosing Silenor within three hours after a meal have important implications relating to both the efficacy and safety of the product that are described in the Silenor prescribing information. As a result, we have listed the patent in the Orange Book.
We have filed multiple other patent applications resulting from unexpected findings from our development program. A brief summary of the content of these patent applications includes:
| • | Formulations and manufacturing processes, |
| • | Methods of preventing early awakenings and improving sleep efficiency, |
| • | Methods of treating insomnia without rebound insomnia or weight gain, and |
| • | Methods of treating insomnia in the elderly. |
We have included these findings in our approved prescribing information and, if the patents issue, we expect to list them in the Orange Book. The combination of these patents, if issued, and our label could result in our patent protection being extended to 2028.
We have also filed multiple patent applications relating to potential future products containing doxepin for the treatment of insomnia. A brief summary of the content of these patent applications includes:
| • | Orally disintegrating formulations, |
| • | Combination drug formulations, and |
| • | Method of treating insomnia with ultra-low dose doxepin. |
Silenor Patent Litigation
We have received notices from each of Actavis Elizabeth LLC and Actavis Inc., or collectively, Actavis, Mylan Pharmaceuticals Inc. and Mylan, Inc., or collectively, Mylan, Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc., or collectively, Par, and Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Limited (d/b/a
7
Zydus Cadila), or collectively, Zydus, that each has filed with the FDA an Abbreviated New Drug Application, or ANDA, for a generic version of Silenor 3 mg and 6 mg tablets. The notices included “paragraph IV certifications” with respect to eight of the nine patents listed in the Orange Book for Silenor. A paragraph IV certification is a certification by a generic applicant that in the opinion of that applicant, the patent(s) listed in the Orange Book for a branded product are invalid, unenforceable, and/or will not be infringed by the manufacture, use or sale of the generic product.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent, by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent, by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action.
At this time, the other patents included in Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but we cannot predict the outcome of these matters. Any adverse outcome in this litigation could result in one or more generic versions of Silenor being launched before the expiration of the listed patents, which could adversely affect our ability to successfully execute our business strategy to increase sales of Silenor and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows.
Other Intellectual Property
Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties and confidential information and inventions agreements with employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect our rights. Enforcing a claim that a third party illegally obtained and is using our trade secrets or unpatented know-how is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secret information. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how, and we would not be able to prevent their use.
Third Party Intellectual Property
Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are operating. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our products or product candidates may infringe.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our products or product candidates infringe their intellectual property rights. If any of these intellectual property rights was found to cover our products or product candidates or their uses, we could be required to pay damages and could be restricted from commercializing our products or from using our proprietary technologies unless we obtained a license to the intellectual property rights. A license may not be available to us on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable right, which could prohibit us from making, using or selling our products.
8
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries generally. If a third party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including but not limited to:
| • | infringement and other intellectual property claims which, with or without merit, may be expensive and time-consuming to litigate and may divert our management’s attention from our core business; |
| • | substantial damages for infringement, including treble damages and attorneys’ fees, which we may be required to pay if a court decides that the product or product candidate at issue infringes on or violates the third party’s rights; |
| • | a court prohibiting us from selling or licensing the product or product candidate or using the proprietary technology unless the third party licenses its technology to us, which it is not required to do; |
| • | if a license is available from the third party, we may have to pay substantial royalties or fees or grant cross-licenses to our technology; and |
| • | redesigning our products or product candidates so they do not infringe, which may not be possible or may require substantial funds and time. |
No assurance can be given that patents issued to third-parties do not exist, have not been filed, or could not be filed or issued, which contain claims covering our products or product candidates or methods. Because of the number of patents issued and patent applications filed in our technical areas or fields, we believe there is a risk that third parties may allege that they have patent rights encompassing our products or product candidates or methods.
Research and Development
To date, our research and development expenses have consisted primarily of costs associated with our clinical trials managed by contract research organizations, or CROs, our non-clinical development program for Silenor, submitting and seeking approval of the NDA for Silenor, regulatory expenses, drug development costs, salaries and related employee benefits, and share-based compensation expense. During 2010 and 2011, our most significant research and development costs were salaries, benefits, and share-based compensation expense and costs associated with our development program for Silenor. During 2009, our most significant research and development costs were salaries, benefits, and share-based compensation expense, costs associated with the conduct of the continuing two-year carcinogenicity study for Silenor, costs associated with the resubmission of the Silenor NDA to the FDA and drug development costs pertaining to Silenor.
Silenor Competition
The FDA-approved products that are currently available for the treatment of insomnia consist of sedative hypnotics, including GABA-receptor agonists, hypnotic benzodiazepines and a melatonin agonist. In addition, products such as sedating antidepressants and other products which are not approved for the treatment of insomnia are sometimes prescribed for such use.
Ambien, a GABA-receptor agonist, and its generic equivalents have historically been the market share leaders in the insomnia segment. Generic versions of Ambien (zolpidem) entered the market in April 2007. According to data obtained from IMS Health, generic versions of Ambien accounted for approximately 65% of insomnia prescriptions in 2011. In September 2005, Sanofi-Synthélabo, Inc. launched Ambien CR, a controlled-release version of Ambien. Unlike Ambien, Ambien CR is indicated for the treatment of sleep maintenance insomnia and does not have a label restriction limiting the length of time of its use. Generic versions of Ambien CR, called zolpidem ER, entered the market in October 2010. Ambien CR accounted for approximately 1% of insomnia prescriptions in 2011, zolpidem ER contributed approximately 6% and branded Ambien accounted for approximately 1% of insomnia prescriptions in 2011, according to data obtained from IMS Health.
9
Lunesta, marketed by Sunovion Pharmaceuticals Inc., a wholly-owned subsidiary of Dainippon Sumitomo Pharma Co., Ltd., is a GABA-receptor agonist that was approved in December 2004 by the FDA and was launched in the second quarter of 2005. Lunesta accounted for approximately 7% of insomnia prescriptions in 2011 according to data obtained from IMS Health. Lunesta is indicated for the treatment of insomnia and has been shown to decrease sleep latency and increase sleep maintenance. It was the first of several products to have the short-term use restriction removed from its label.
Sonata, a GABA-receptor agonist sold by Pfizer Inc. for the treatment of insomnia, and its generic equivalents accounted for less than 0.1% of insomnia prescriptions in 2011 according to data obtained from IMS Health.
Rozerem was launched by Takeda Pharmaceuticals North America, Inc. in September 2005 and accounted for 0.6% of insomnia prescriptions in 2011 according to data obtained from IMS Health. Rozerem is indicated for the treatment of insomnia characterized by difficulty with sleep onset. It was the first drug approved for the treatment of insomnia that is not a Schedule IV controlled substance. With the exception of Rozerem, the approved medications for the treatment of insomnia all act on GABA receptors and are designated as Schedule IV controlled substances. Takeda Pharmaceuticals North America, Inc. conducted a clinical trial to evaluate the administration of a combination of Takeda’s product Rozerem and 3 mg of doxepin in patients with insomnia. We are unaware of the results of this trial.
A number of companies are marketing reformulated versions of previously approved GABA-receptor agonists. In March 2009, Meda AB and Orexo AB received approval from the FDA for Edluar, formerly known as Sublinox, a sublingual tablet formulation of zolpidem, for the short-term treatment of insomnia. Meda and Orexo launched this product in the U.S. in September of 2009.
In December 2008, NovaDel Pharma, Inc. received approval from the FDA for ZolpiMist, an oral mist formulation of zolpidem for the short-term treatment of insomnia characterized by difficulties with sleep initiation. In November 2009, NovaDel and ECR Pharmaceuticals Company, Inc., a wholly owned subsidiary of Hi-Tech Pharmacal Co., Inc., entered into an exclusive license and distribution agreement to commercialize and manufacture ZolpiMist in the United States and Canada. ECR Pharmaceuticals launched the product in the United States in February 2011.
In November 2011, Transcept Pharmaceuticals, Inc. received approval from the FDA for Intermezzo, a low-dose sublingual tablet formulation of zolpidem. Transcept and Purdue Pharmaceutical Products L.P. have entered into an exclusive license and collaboration agreement to commercialize Intermezzo in the United States. Purdue has publicly announced that it plans to launch Intermezzo in the second quarter of 2012.
The remaining market is comprised of older generic benzodiazepines and sedative antidepressants. In addition to the currently approved or off-label products for the treatment of insomnia, a number of new products may enter the insomnia market over the next several years. While the new entrants would bring additional competition to the insomnia market, they would also increase the awareness of insomnia and further expand the market. Additionally, we believe market growth will also be driven by the aging of the population and emerging awareness of the links between sleep, health and overall well-being.
Alexza Pharmaceuticals, Inc. has announced positive results from a Phase 1 clinical trial of an inhaled formulation of zaleplon, the active pharmaceutical ingredient in Sonata. In July 2010, Alexza announced that it was advancing this product candidate into Phase 2 clinical trials during the first half of 2011 for the treatment of insomnia in patients who have difficulty falling asleep, including those patients who awaken in the middle of the night and have difficulty falling back asleep. Somnus Therapeutics, Inc. has announced positive results from two Phase 1 clinical trials and one Phase 2 clinical trial of a delayed-release formulation of zaleplon.
Vanda Pharmaceuticals Inc. has completed two Phase 3 insomnia clinical trials of tasimelteon, a melatonin receptor agonist. Tasimelteon received orphan drug designation status for non-24 hour sleep/wake disorder in blind individuals with no light perception. Vanda has initiated a Phase 3 clinical trial for tasimelteon to treat this disorder. Vanda has announced that it plans to conduct additional clinical trials and plans to file an NDA with the FDA in the first half of 2013.
Merck & Co., Inc. has completed Phase 3 clinical trials for suvorexant, an orexin antagonist, for the treatment of insomnia and has MK-6096 and MK-3697 in Phase 2 clinical trials for the treatment of insomnia. Merck has announced that it plans to file regulatory applications for suvorexant in 2012.
10
Several other companies, including Sunovion Pharmaceuticals, are evaluating 5HT2 antagonists as potential hypnotics, and Eli Lilly and Company is evaluating a potential hypnotic that is a dual histamine/5HT2 antagonist. Additionally, several companies are evaluating new formulations of existing compounds and other compounds for the treatment of insomnia.
Upon the expiration of, or successful challenge to, our patents covering Silenor, generic competitors may introduce a generic version of Silenor at a lower price. Some generic manufacturers have also demonstrated a willingness to launch generic versions of branded products before the final resolution of related patent litigation, known as an “at-risk launch”.
We have received notices from Actavis, Mylan, Par, and Zydus that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in the Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but given the unpredictability of patent litigation, we cannot predict the outcome of these matters.
Manufacturing
We have entered into a non-exclusive manufacturing services agreement with Patheon for the manufacture of commercial quantities of our Silenor 3 mg and 6 mg tablets. Although we are not required to purchase any minimum quantity of Silenor under the agreement, we have agreed to purchase from Patheon not less than specified percentages of our total annual commercial requirements from all suppliers of Silenor, which vary depending upon annual volume. The agreement provides for an initial five-year term that began upon commencement of the manufacturing services, and thereafter automatically continues for successive twelve-month terms unless terminated by written notice at least eighteen months prior to the end of the then-current term. Either party may terminate the agreement upon written notice if the other party has failed to remedy a material breach of any of its representations, warranties or other obligations under the agreement within 60 days following receipt of written notice of such breach. In addition, either party may immediately terminate the agreement upon written notice if (1) the other party is declared insolvent or bankrupt by a court of competent jurisdiction, (2) a voluntary petition of bankruptcy is filed in any court of competent jurisdiction by such other party or (3) the agreement is assigned by such other party for the benefit of creditors. We have the right to terminate the agreement upon 30 days prior written notice in the event that any governmental agency takes any action, or raises any objection, that prevents us from importing, exporting, purchasing or selling the product candidate. In addition, we have the right to terminate the agreement upon twelve months’ prior written notice in connection with our partnering, collaboration, licensing, sublicensing, co-promotion, sale or divestiture of rights to the product candidate, provided that no such termination shall be effective before the third anniversary of the commencement date.
11
We have also entered into agreements with Plantex USA, Inc. to manufacture our supply of doxepin active pharmaceutical ingredient, with Anderson Packaging, Inc. to package Silenor finished products, and with JRS to supply us with ProSolv®HD90. In August 2008, we entered into an amendment to our supply agreement providing us with the exclusive right to use ProSolv®HD90 in combination with doxepin. Pursuant to the amended supply agreement, we made an upfront license payment of $0.2 million and are obligated to pay a royalty on worldwide net sales of Silenor beginning as of the expiration of the statutory exclusivity period for Silenor in each country in which Silenor is marketed. This royalty is only payable if one or more patents under the license agreement continue to be valid in each such country and a patent relating to our formulation for Silenor has not issued in such country.
We sell Silenor through pharmaceutical wholesalers, who in turn distribute the products to retail pharmacies, mail order pharmacies, hospitals and other institutional customers. We have retained third-party service providers to perform a variety of functions related to the distribution of our products, including logistics management, sample accountability, storage and transportation.
Government Regulation
Governmental authorities in the United States and other countries extensively regulate the testing, manufacturing, labeling, storage, record-keeping, advertising, promotion, export, marketing and distribution, among other things, of pharmaceutical products. In the United States, the FDA, under the Federal Food, Drug, and Cosmetic Act and other federal statutes and regulations, subjects pharmaceutical products to rigorous review. If we do not comply with applicable requirements, we may be fined, the government may refuse to approve our marketing applications or allow us to manufacture or market our products, and we may be criminally prosecuted.
We and our manufacturers and CROs may also be subject to regulations under other federal, state, and local laws, including the Occupational Safety and Health Act, the Environmental Protection Act, the Clean Air Act and import, export and customs regulations as well as the laws and regulations of other countries.
FDA Approval Process
To obtain approval of a new product from the FDA, we must, among other requirements, submit data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product and proposed labeling, including a proposed proprietary name for the product. The testing and collection of data and the preparation of necessary applications are expensive and time-consuming. The FDA may not act quickly or favorably in reviewing these applications, and we may encounter significant difficulties or costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing our products.
The process required by the FDA before a new drug may be marketed in the United States generally involves the following: completion of non-clinical laboratory and animal testing in compliance with FDA regulations, submission of an IND, which must become effective before human clinical trials may begin, performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use, and submission and approval by the FDA of an NDA. The sponsor typically conducts human clinical trials in three sequential phases, but the phases may overlap. In Phase 1 clinical trials, the product is tested in a small number of patients or healthy volunteers, primarily for safety at one or more dosages. In Phase 2 clinical trials, in addition to safety, the sponsor evaluates the efficacy of the product on targeted indications, and identifies possible adverse effects and safety risks in a patient population. Phase 3 clinical trials typically involve testing for safety and clinical efficacy in an expanded population at geographically-dispersed test sites.
Clinical trials must be conducted in accordance with the FDA’s good clinical practices requirements. The FDA may order the partial, temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The institutional review board, or IRB, generally must approve the clinical trial design and patient informed consent at each clinical site and may also require the clinical trial at that site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
12
The applicant must submit to the FDA the results of the non-clinical and clinical trials, together with, among other things, detailed information on the manufacture and composition of the product and proposed labeling, in the form of an NDA, including payment of a user fee, unless the applicant qualifies for a waiver of the user fee. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the policies agreed to by the FDA under the Prescription Drug User Fee Act of 1992, or PDUFA, the FDA has ten months in which to complete its review of a standard NDA and respond to the applicant. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the three months prior to the PDUFA goal date. If the FDA’s evaluations of the NDA and the clinical and manufacturing procedures and facilities are favorable, the FDA may issue an approval letter, authorizing commercial marketing of the drug for a specified indication. If the FDA is not sufficiently satisfied with the information in the NDA to issue an approval letter, the FDA may issue a complete response letter, which usually will describe all of the specific deficiencies that the FDA has identified in the NDA and when possible, recommend actions that the NDA sponsor may take to address the identified deficiencies.
On March 18, 2010, the FDA notified us that it approved our NDA for Silenor 3 mg and 6 mg tablets for the treatment of insomnia characterized by difficulty with sleep maintenance.
Section 505(b)(1) New Drug Applications
The approval process described above is premised on the applicant being the owner of, or having obtained a right of reference to, all of the data required to prove the safety and effectiveness of a drug product. This type of marketing application, sometimes referred to as a “full” or “stand-alone” NDA, is governed by Section 505(b)(1) of the Federal Food, Drug, and Cosmetic Act. A Section 505(b)(1) NDA contains full reports of investigations of safety and effectiveness, which includes the results of non-clinical studies and clinical trials, together with detailed information on the manufacture and composition of the product, in addition to other information.
Section 505(b)(2) New Drug Applications
As an alternate path to FDA approval for new indications, formulations or strengths of previously-approved products, a company may file a Section 505(b)(2) NDA, instead of a “stand-alone” or “full” NDA filing under Section 505(b)(1) as described above. Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The Hatch-Waxman Amendments permit the applicant to rely upon the FDA’s findings of safety and effectiveness for an approved product or on published information. We submitted our NDA for Silenor under Section 505(b)(2), and as such it relied, in part, on the FDA’s previous findings of safety and effectiveness for doxepin.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s findings for an already-approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (1) the required patent information relating to the listed patent has not been filed in the NDA for the approved product; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new product. A certification that the new product will not infringe the already approved product’s Orange Book-listed patents or that such patents are invalid is called a paragraph IV certification. If the applicant does not challenge the listed patents, the Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced product have expired. The Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired.
13
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If these companies successfully challenge the FDA’s interpretation of Section 505(b)(2), the FDA may be required to change its interpretation of Section 505(b)(2). This could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
Regulatory Status and Requirements
In connection with the approval of our NDA for Silenor, the FDA imposed certain requirements upon us. The FDA has required us to implement a risk evaluation and mitigation strategy, or REMS, consisting of a medication guide. We are also required to complete a standard clinical trial assessing the safety and efficacy of Silenor in children aged 6 to 16 pursuant to the Pediatric Research Equity Act of 2003, or PREA, and to submit the final results of this trial by March 2015.
The Hatch-Waxman Act
Under the Hatch-Waxman Act, newly-approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety. Hatch-Waxman prohibits the submission of an ANDA, or a Section 505(b)(2) NDA for another version of such drug during the five-year exclusive period; however, submission of an ANDA or Section 505(b)(2) NDA containing a paragraph IV certification is permitted after four years, which may trigger a 30-month stay of approval of the ANDA or Section 505(b)(2) NDA. Protection under Hatch-Waxman will not prevent the submission or approval of another full NDA; however, the subsequent applicant would be required to conduct its own non-clinical and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Act also provides three years of marketing exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) NDAs, for, among other things, new indications, formulations, or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application. We received three years of marketing exclusivity for Silenor.
We have received notices from Actavis, Mylan, Par, and Zydus that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in the Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but given the unpredictability of patent litigation, we cannot predict the outcome of these matters.
Pediatric Exclusivity
The Best Pharmaceuticals for Children Act, which was signed into law January 4, 2002, and which reauthorized Section 111 of the 1997 FDA Modernization Act, provides in some cases an additional six months of exclusivity for new or marketed drugs for specific pediatric studies conducted at the written request of the FDA. PREA authorizes
14
the FDA to require pediatric studies for drugs to ensure the drugs’ safety and efficacy in children. PREA requires that certain NDAs or supplements to NDAs contain data assessing the safety and effectiveness for the claimed indication in all relevant pediatric subpopulations. Dosing and administration must be supported for each pediatric subpopulation for which the drug is safe and effective. The FDA may also require this data for approved drugs that are used in pediatric patients for the labeled indication, or where there may be therapeutic benefits over existing products. The FDA may grant deferrals for submission of data, or full or partial waivers from PREA. We received a waiver from PREA requirements for children below age 6, and a deferral of submission of final pediatric study data until March 2015 for children aged 6 to 16.
Other Regulatory Requirements
Any approved product that we market may also be subject to a number of post-approval regulatory requirements. If we seek to make certain changes to an approved product, such as promoting or labeling a product for a new indication, making certain manufacturing changes or product enhancements or adding labeling claims, we will need FDA review and approval before the change can be implemented. While physicians may use products for indications that have not been approved by the FDA, we may not label or promote the product for an indication that has not been approved. Securing FDA approval for new indications or product enhancements and, in some cases, for manufacturing and labeling claims, is generally a time-consuming and expensive process that may require us to conduct clinical trials under the FDA’s IND regulations. Even if such studies are conducted, the FDA may not approve any change in a timely fashion, or at all. In addition, adverse experiences associated with use of the products must be reported to the FDA, and FDA rules govern how we can label, advertise or otherwise commercialize our products.
There are post-marketing safety surveillance requirements that we will need to meet to continue to market an approved product. The FDA also may, in its discretion, require additional post-marketing testing and surveillance to monitor the effects of approved products or place conditions on any approvals that could restrict the sale or use of these products. The FDA has required us to develop a REMS to ensure that the benefits of Silenor outweigh its risks. A REMS can include information to accompany the product, such as a patient package insert or a medication guide, a communication plan, elements to assure safe use, and an implementation system, and must include a timetable for assessment of the REMS. Our REMS for Silenor consists of a medication guide. In addition, the FDA may require modifications to the REMS at a later date if warranted by new safety information. Any future requirements imposed by the FDA may require substantial expenditures.
The FDA has also requested that all manufacturers of sedative-hypnotic drug products modify their product labeling to include stronger language concerning potential risks. These risks include severe allergic reactions and complex sleep-related behaviors, which may include sleep-driving. The FDA also recommended that the drug manufacturers conduct clinical studies to investigate the frequency with which sleep-driving and other complex behaviors occur in association with individual drug products. Our approved label for Silenor includes warnings relating to risks of complex sleep behaviors.
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal health care program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal health care programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
15
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn are used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
In addition, we and the manufacturers upon which we rely for the manufacture of our products are subject to requirements that drugs be manufactured, packaged and labeled in conformity with current good manufacturing practice, or cGMP. To comply with cGMP requirements, manufacturers must continue to spend time, money and effort to meet requirements relating to personnel, facilities, equipment, production and process, labeling and packaging, quality control, record-keeping and other requirements. The FDA periodically inspects drug manufacturing facilities to evaluate compliance with cGMP requirements.
Also, as part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. This practice is regulated by the FDA and other governmental authorities, including, in particular, requirements concerning record-keeping and control procedures.
In 2010, the U.S. Congress enacted legislation to reform the healthcare system. A major goal of the healthcare reform law was to provide greater access to healthcare coverage for more Americans. Accordingly, the healthcare reform law requires individual U.S. citizens and legal residents to maintain qualifying health coverage, imposes certain requirements on employers with respect to offering health coverage to employees, amends insurance regulations regarding when coverage can be provided and denied to individuals, and expands existing government healthcare coverage programs to more individuals in more situations. Among other things, the healthcare reform law specifically:
| • | established annual, non-deductible fees on any entity that manufactures or imports certain branded prescription drugs, beginning in 2011; |
| • | increased minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program, retroactive to January 1, 2010; |
| • | redefined a number of terms used to determine Medicaid drug rebate liability, including average manufacturer price and retail community pharmacy, effective October 2010; |
| • | extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations, effective March 2010; |
| • | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals beginning April 2010 and by adding new mandatory eligibility categories for certain individuals with income at or below 133 percent of the Federal Poverty Level beginning in 2014, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research; |
| • | required manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50 percent point-of-sale discounts off of negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D, beginning 2011; and |
| • | increased the number of entities eligible for discounts under the Public Health Service pharmaceutical pricing program, effective January 2010. |
16
Some states are also considering legislation that would control the prices of drugs, and state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and/or requiring prior authorization by the state program. It is likely that federal and state legislatures and health agencies will continue to focus on additional healthcare reform in the future.
There have been a number of other legislative and regulatory proposals aimed at changing the healthcare system and pharmaceutical industry, including reductions in the cost of prescription products, changes in the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products, proposals concerning reimportation of pharmaceutical products and proposals concerning safety matters. For example, in an attempt to protect against counterfeit drugs, the federal government and numerous states have enacted pedigree legislation. In particular, California has enacted legislation that requires development of an electronic pedigree to track and trace each prescription drug at the saleable unit level through the distribution system. California’s electronic pedigree requirement is scheduled to take effect beginning in January 2015.
Outside of the United States, our or our partners’ ability to market our products will also depend on receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country.
Third-Party Reimbursement and Pricing Controls
In the United States and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. It is and will continue to be time-consuming and expensive for us or our strategic collaborators to go through the process of seeking reimbursement from Medicare and private payors. Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
In many foreign markets, including the countries in the European Union, pricing of pharmaceutical products is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control.
While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Employees
As of February 15, 2012, we had 36 employees, consisting of manufacturing, regulatory affairs, sales, marketing and administration. We believe our relations with our employees are good.
Available Information
We make available free of charge on or through our internet website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission. Our internet address is www.somaxon.com. Information is also available through the Securities and Exchange Commission’s website at www.sec.gov or is available at the Securities and Exchange Commission’s Public Reference Room located at 100 F Street, NE, Washington DC, 20549. Information on the operation of the Public Reference Room is available by calling the Securities and Exchange Commission at 800-SEC-0330.
Investing in our common stock involves a high degree of risk. You should carefully consider the following risk factors, as well as the other information in this report, before deciding whether to invest in shares of our common stock. The occurrence of any of the following risks could harm our business, financial condition, results of operations or growth prospects. In that case, the trading price of our common stock could decline, and you may lose all or part of your investment.
17
Risks Related to Our Business
Our success is dependent on the success of Silenor (doxepin).
The majority of our resources are focused on the marketing and selling of Silenor. Our ability to generate revenue in the near term will depend solely on the success of this product. Accordingly, any disruption in our ability to generate revenues from the sale of Silenor or lack of success in generating Silenor sales will have a substantial adverse impact on our business.
We will require substantial additional funding and may be unable to raise capital when needed, which could force us to delay, reduce or eliminate planned activities or result in our inability to continue as a going concern.
We began generating revenues from the commercialization of Silenor late in the third quarter of 2010, and our operations to date have generated substantial needs for cash. We expect our negative cash flows from operations to continue until we are able to generate significant cash flows from sales of Silenor. Based on our recurring losses, negative cash flows from operations and working capital levels, we will need to raise substantial additional funds to finance our operations. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected. The report of our independent registered public accounting firm on our financial statements for the year ended December 31, 2011 contains an explanatory paragraph stating that our recurring losses raise substantial doubt about our ability to continue as a going concern.
We are responsible for the costs relating to the sales and marketing of Silenor in the United States. As a result, commercial activities relating to Silenor are likely to result in the need for substantial additional funds, including supporting the additional cost of the 25 field-based sales representatives we hired in January 2012. Our future capital requirements will depend on, and could increase significantly as a result of, many factors, including:
| • | our success in generating cash flows from sales of Silenor; |
| • | the costs of establishing or contracting for commercial programs and resources, and the scope of the commercial programs and resources we pursue; |
| • | the terms and timing of any future collaborative, licensing and other arrangements that we may establish; |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | the extent to which we acquire or in-license new products, technologies or businesses; |
| • | the rate of progress and cost of any future non-clinical studies, any future clinical trials and other development activities; |
| • | the scope, prioritization and number of development programs we pursue; and |
| • | the effect of competing technological and market developments. |
In August 2011, we entered into an at-the-market equity sales agreement with Citadel Securities LLC, or Citadel. However, there can be no assurance that Citadel will be successful in consummating sales under the agreement based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. Citadel or we are permitted to terminate the sales agreement at any time.
At December 31, 2011 we had cash and cash equivalents totaling $10.7 million. We will need to obtain additional funds to finance our operations. Actual financial results for the period of time through which our financial resources will be adequate to support our operations could vary based upon many factors, including but not limited to Silenor sales performance, the actual cost of commercial activities and any potential litigation expenses we may incur.
18
In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. The exploration of strategic alternatives may not result in any agreement or transaction and, if completed, any agreement or transaction may not be successful or on attractive terms. The inability to enter into a strategic transaction, or a strategic transaction that is not successful or on attractive terms, could accelerate our needs for cash and make securing funding on reasonable terms more difficult. In addition, if we raise additional funds through collaborations or other strategic transactions, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
In addition to the amounts available under our sales agreement with Citadel, we intend to obtain any additional funding we require through public or private equity or debt financings, strategic relationships, assigning receivables or royalty rights, or other arrangements and cannot assure that such funding will be available on reasonable terms, or at all. If we are unsuccessful in raising additional required funds, we may be required to delay, scale-back or eliminate plans or programs relating to our business, relinquish some or all rights to Silenor, or renegotiate less favorable terms with respect to such rights than we would otherwise choose or cease operating as a going concern. In addition, if we do not meet our payment obligations to third parties as they come due, we may be subject to litigation claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and distract management, and may result in unfavorable outcomes that could further adversely impact our financial condition.
If we raise additional funds by issuing equity securities, substantial dilution to existing stockholders would result. If we raise additional funds by incurring debt financing, the terms of the debt may involve significant cash payment obligations as well as covenants and specific financial ratios that may restrict our ability to operate our business. If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our financial statements, and it is likely that investors will lose all or a part of their investments.
We will need to retain qualified sales and marketing personnel and successfully manage our sales and marketing programs and resources in order to successfully generate sales of Silenor.
Prior to December 31, 2011, revenues we received from sales of Silenor largely depended upon the efforts of sales representatives employed by The Procter & Gamble Distributing Company LLC, or P&G, and Publicis Touchpoint Solutions, Inc., or Publicis. Because we did not believe that the growth of Silenor revenues throughout 2011 was sufficient to support sales and marketing expenses at then-current levels, we terminated our agreements with Publicis and P&G in December 2011 in an effort to conserve cash, and effective January 3, 2012, we hired a reduced sales force of 25 field-based sales representatives from Publicis as our employees to promote Silenor. We are now solely relying on our sales representatives to market and sell Silenor, and our sales in the short term may suffer as we make this transition and may continue to suffer in the long term if such transition is not successful. In addition, our strategy of focusing on an overall smaller, but more concentrated, geography may not be successful.
The efforts of our sales force are complemented by on-line and other non-personal promotional initiatives that target both physicians and patients. We are also focused on ensuring broad patient access to Silenor by negotiating agreements with leading commercial managed care organizations and with government payors. Although our goal is to achieve Silenor sales through the efficient execution of our sales and marketing plans and programs, we may not be able to effectively generate prescriptions and achieve broad market acceptance for Silenor on a timely basis, or at all.
If Silenor or any other product we commercialize does not achieve broad market acceptance, the revenues that we generate from its sale will be limited.
The commercial success of Silenor or any other product which we commercialize will depend upon the acceptance of the product by the medical community and reimbursement of the product by third-party payors, including government payors. The degree of market acceptance of any approved product will depend on a number of factors, including:
19
| • | the scope and effectiveness of our sales, marketing and distribution resources and strategies, or those of our collaborators, if any; |
| • | our ability to provide acceptable evidence of safety and efficacy and physician and patient understanding of differential indications for use, such as sleep maintenance; |
| • | pricing and cost effectiveness; |
| • | availability of alternative treatments, including, in the case of Silenor, a number of competitive products already approved for the treatment of insomnia or expected to be commercially launched in the future; |
| • | off-label substitution by chemically similar or equivalent products, including for Silenor the off-label use of higher-dose generic formulations of doxepin approved and available for the treatment of depression and anxiety; |
| • | prevalence and severity of any adverse side effects; |
| • | limitations or warnings contained in the U.S. Food and Drug Administration-, or FDA, approved labeling of Silenor or any other product that we commercialize; |
| • | relative convenience and ease of administration; and |
| • | our ability to obtain sufficient third-party coverage or reimbursement. |
If Silenor or any other product that we commercialize does not achieve an adequate level of acceptance by physicians, health care payors and patients, or adequate payment and reimbursement levels are not provided, or if those policies increasingly favor generic products, we may not generate sufficient revenue from the product, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of Silenor or any other product that we commercialize may require significant resources and may never be successful.
Restrictions on or challenges to our patent rights relating to our products and limitations on or challenges to our other intellectual property rights may limit our ability to prevent third parties from competing against us.
Our success will depend on our ability to obtain and maintain patent protection for Silenor and any other product candidate we develop or commercialize, preserve our trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others. The patent rights that we have in-licensed relating to Silenor are limited in ways that may affect our ability to exclude third parties from competing against us. In particular, we do not hold composition of matter patents covering the active pharmaceutical ingredient, or API, of Silenor. Composition of matter patents on APIs are a particularly effective form of intellectual property protection for pharmaceutical products, as they apply without regard to any method of use or other type of limitation. As a result, competitors who obtain the requisite regulatory approval can offer products with the same APIs as our products so long as the competitors do not infringe any method of use or formulation patents that we may hold.
The principal patent protection that covers, or that we expect will cover, Silenor consists of method of use patents. This type of patent protects the product only when used or sold for the specified method. However, this type of patent does not limit a competitor from making and marketing a product that is identical or similar to our product for an indication that is outside of the patented method. Moreover, physicians may prescribe such a competitive or similar identical product for off-label indications that are covered by the applicable patents. Some physicians are prescribing generic 10 mg doxepin capsules and generic oral solution doxepin for insomnia on such an off-label basis. In addition, some managed health care plans are requiring the substitution of these generic doxepin products for Silenor, and some pharmacies are suggesting such substitution. Although such off-label prescriptions may induce or contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
20
Because products with active ingredients identical to ours have been on the market for many years, there can be no assurance that these other products were never used off-label or studied in such a manner that such prior usage would not affect the validity of our method of use patents. Due to some prior art that we identified, we initiated a reexamination of one of the patents we have in-licensed covering Silenor, (specifically, U.S. Patent No. 5,502,047, “Treatment for Insomnia”) which claims the treatment of chronic insomnia using doxepin in a daily dosage of 0.5 mg to 20 mg and expires in March 2013. The reexamination proceedings terminated and the U.S. Patent and Trademark Office, or USPTO, issued a reexamination certificate narrowing certain claims, so that the broadest dosage ranges claimed by us are 0.5 mg to 20 mg for otherwise healthy patients with chronic insomnia and for patients with chronic insomnia resulting from depression, and 0.5 mg to 4 mg for all other chronic insomnia patients. We also requested reissue of this same patent to consider some additional prior art and to add intermediate dosage ranges below 10 mg. In two office actions relating to this reissue request, the USPTO raised no prior art objections to 32 of the 34 claims we were seeking and raised a prior art objection to the other two, as well as some technical objections. Each of the claims objected to by the USPTO related to dosages above 10 mg. After further review of the prior art submitted, the USPTO withdrew all of its prior art objections. We then determined that the proposed addition of the intermediate dosage ranges and the resolution of the technical objections no longer warranted continuation of the reissue proceeding. As a result, we elected not to continue that proceeding.
We also have multiple internally developed pending patent applications. No assurance can be given that the USPTO or other applicable regulatory authorities will allow pending applications to result in issued patents with the claims we are seeking, or at all.
Patent applications in the United States are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months. As a result, we cannot be certain that the inventors of issued patents to which we hold rights were the first to conceive of inventions covered by pending patent applications or that the inventors were the first to file patent applications for such inventions.
In addition, third parties may challenge issued patents to which we hold rights and any additional patents that we may obtain, which could result in the invalidation or unenforceability of some or all of the relevant patent claims, or could attempt to develop products utilizing the same APIs as our products that do not infringe the claims of our in-licensed patents or patents that we may obtain.
When a third party files an abbreviated new drug application, or ANDA, for a product containing doxepin for the treatment of insomnia at any time during which we have patents listed for Silenor in the FDA’s Orange Book publication, the applicant will be required to certify to the FDA concerning the listed patents. Specifically, the applicant must certify that: (1) the required patent information relating to the listed patent has not been filed in the NDA for the approved product; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new product. A certification that the new product will not infringe the Orange Book-listed patents for Silenor or that such patents are invalid is called a paragraph IV certification.
We have received notices from each of Actavis Elizabeth LLC and Actavis Inc., or collectively, Actavis, Mylan Pharmaceuticals Inc. and Mylan, Inc., or collectively, Mylan, Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc., or collectively, Par, and Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Limited (d/b/a Zydus Cadila), or collectively, Zydus, that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
21
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but we cannot predict the outcome of these matters. Any adverse outcome in this litigation could result in one or more generic versions of Silenor being launched before the expiration of the listed patents, which could adversely affect our ability to successfully execute our business strategy and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows. Such events could also significantly impact our ability to continue as a going concern.
Certain pharmaceutical companies’ patent settlement agreements with generic pharmaceutical companies have been challenged by the U.S. Federal Trade Commission alleging a violation of Section 5(a) of the Federal Trade Commission Act, and any patent settlement agreement that we may enter into with any generic pharmaceutical company may be subject to similar challenges, which will be both expensive and time consuming and may render such settlement agreements unenforceable. In addition, legislation has been proposed by Congress that, if passed, would subject patent settlement agreements to further restrictions.
We also rely upon unpatented trade secrets and improvements, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our collaborators, employees and consultants. We also have invention or patent assignment agreements with our employees and certain consultants. There can be no assurance that inventions relevant to us will not be developed by a person not bound by an invention assignment agreement with us. There can be no assurance that binding agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by our competitors.
Litigation or other proceedings to enforce or defend intellectual property rights is often very complex in nature, expensive and time-consuming, may divert our management’s attention from our core business and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with us.
The patent rights that we have in-licensed covering Silenor are limited to certain low-dosage strengths in the United States, and our market opportunity for this product may be limited by the lack of patent protection for higher dosage strengths for which generic formulations are available and the lack of patent protection in other territories.
Although we have an exclusive, worldwide license for Silenor for the treatment of insomnia through the life of the last issued patent to expire, we do not have patent protection for Silenor in any jurisdiction outside the United States. In addition, although our in-licensed patent for the treatment of transient insomnia is scheduled to expire in 2020 and our patent relating to the food effect of Silenor is scheduled to expire in 2027, our in-licensed patent for the treatment of chronic insomnia is scheduled to expire in March 2013. Accordingly, in the absence of additional patents or other alternatives to obtain additional exclusivity rights for Silenor, a competitor could attempt to market doxepin for a chronic insomnia indication as early as March 2013. Furthermore, the patent protection in the United States for Silenor for the treatment of insomnia is limited to dosages ranging from a lower limit of 0.5 mg to various upper limits up to 20 mg of the active ingredient, doxepin. Doxepin is prescribed at dosages ranging from 75 mg to 300 mg daily for the treatment of depression and anxiety and is available in generic form in strengths as low as 10 mg in capsule form, as well as in a concentrated liquid form dispensed by a marked dropper and calibrated for 5 mg. As a result, we may face competition from the off-label use of these or other dosage forms of generic doxepin. Off-label use occurs when a drug that is approved by the FDA for one indication is prescribed by physicians for a different, unapproved indication.
22
In addition, we do not have patent protection for Silenor in any jurisdiction outside the United States. Others may attempt to commercialize low-dose doxepin in the European Union, Canada, Mexico or other markets where we do not have patent protection for Silenor. Due to the lack of patent protection for doxepin in territories outside the United States and the potential for correspondingly lower prices for the drug in those markets, it is possible that patients will seek to acquire low-dose doxepin in those other territories. The off-label use of generic doxepin in the United States or the importation of doxepin from foreign markets could adversely affect the commercial potential for Silenor and adversely affect our overall business and financial results. We have submitted additional patent applications for Silenor in the United States and outside the United States, but we cannot assure that these will result in issued patents or additional protection in the United States or other jurisdictions.
We are subject to uncertainty relating to healthcare reform measures, reimbursement policies and regulatory proposals which, if not favorable to Silenor or any other product that we commercialize, could hinder or prevent our commercial success.
Our ability to successfully commercialize Silenor and any other product to which we obtain rights will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish appropriate coverage and reimbursement levels for the cost of our products and related treatments. Based on third party formulary data, we believe that 76% of patients in commercial health plans have coverage for Silenor.
The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may adversely affect:
| • | the ability to obtain a price we believe is fair for our products; |
| • | the ability to generate revenues and achieve or maintain profitability; |
| • | the future revenues and profitability of our potential customers, suppliers and collaborators; and |
| • | the availability of capital. |
The U.S. Congress has enacted legislation to reform the healthcare system. A major goal of this healthcare reform law was to provide greater access to healthcare coverage for more Americans. Accordingly, the healthcare reform law required individual U.S. citizens and legal residents to maintain qualifying health coverage, imposed certain requirements on employers with respect to offering health coverage to employees, amended insurance regulations regarding when coverage can be provided and denied to individuals, and expanded existing government healthcare coverage programs to more individuals in more situations. Among other things, the healthcare reform law specifically:
| • | established annual, non-deductible fees on any entity that manufactures or imports certain branded prescription drugs, beginning in 2011; |
| • | increased minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program, retroactive to January 1, 2010; |
| • | redefined a number of terms used to determine Medicaid drug rebate liability, including average manufacturer price and retail community pharmacy, effective October 2010; |
| • | extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations, effective March 2010; |
| • | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals beginning April 2010 and by adding new mandatory eligibility categories for certain individuals with income at or below 133 percent of the Federal Poverty Level beginning 2014, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research; |
23
| • | required manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50 percent point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D, beginning 2011; and |
| • | increased the number of entities eligible for discounts under the Public Health Service pharmaceutical pricing program, effective January 2010. |
While this legislation will, over time, increase the number of patients who have insurance coverage for our products, it also is likely to adversely affect our results of operations. Some states are also considering legislation that would control the prices of drugs, and state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and/or requiring prior authorization by the state program. It is likely that federal and state legislatures and health agencies will continue to focus on additional healthcare reform in the future.
As a result of these or other reform measures, we may determine to change our current manner of operation or change our contract arrangements, any of which could harm our ability to operate our business efficiently or on the scale we would like and our ability to raise capital. In addition, in certain foreign markets, the pricing of prescription drugs is subject to government control and reimbursement may in some cases be unavailable or insufficient.
Current healthcare reform measures and any future legislative proposals to reform healthcare or reduce government insurance programs may result in lower prices for Silenor and any other product that we commercialize or exclusion of Silenor or any such other product from coverage and reimbursement programs. Either of those could harm our ability to market our products and significantly reduce our revenues from the sale of our products.
Managed care organizations are increasingly challenging the prices charged for medical products and services and, in some cases, imposing restrictions on the coverage of particular drugs. Many managed care organizations negotiate the price of medical services and products and develop formularies which establish pricing and reimbursement levels. Exclusion of a product from a formulary can lead to its sharply reduced usage in the managed care organization’s patient population. The process for obtaining coverage can be lengthy and costly, and it can take several months before a particular payor initially reviews our product and makes a decision with respect to coverage. For example, third-party payors may require cost-benefit analysis data from us in order to demonstrate the cost-effectiveness of any product we might market and sell. For any individual third-party payor, we may not be able to provide data sufficient to gain reimbursement on a similar or preferred basis to competitive products, or at all.
In addition, many insurers and other healthcare payment organizations encourage the use of less expensive alternative generic brands and over the counter, or OTC, products through their prescription benefits coverage and reimbursement policies. The availability of generic prescription and OTC products for the treatment of insomnia has created, and will continue to create, a competitive reimbursement environment. Insurers and other healthcare payment organizations frequently make the generic or OTC alternatives more attractive to the patient by providing different amounts of reimbursement so that the net cost of the generic or OTC product to the patient is less than the net cost of a prescription branded product to the patient. Aggressive pricing policies by our generic or OTC product competitors and the prescription benefit policies of insurers could have a negative effect on our product revenues and profitability.
The competition among pharmaceutical companies to have their products approved for reimbursement also results in downward pricing pressure in the industry and in the markets where our products compete. In some cases, we may discount our products in order to obtain reimbursement coverage, and we may not be successful in any efforts we take to mitigate the effect of a decline in average selling prices for our products. Declines in our average selling prices would also reduce our gross margins.
In addition, once reimbursement at an agreed level is approved by a third-party payor, we may lose that reimbursement entirely. As reimbursement is often approved for a period of time, this risk is greater at the end of the time period, if any, for which the reimbursement was approved.
24
We may face additional challenges with regard to reimbursement which could affect our ability to successfully commercialize Silenor or any other product candidate that we commercialize, including:
| • | the variability of reimbursement rates likely to be caused by the use of miscellaneous drug codes and procedure codes may discourage physicians from providing Silenor or any other product candidate that we commercialize to certain or all patients depending on their insurance coverage; |
| • | the initial use of “miscellaneous drug codes” for billing Silenor or any other product candidate that we commercialize until such time as specific drug codes are approved could result in slow and/or inaccurate reimbursement and thereby discourage product use; |
| • | an increase in insurance plans that place more cost liability onto patients may limit patients’ willingness to pay for Silenor or any other product candidate that we commercialize and thereby discourage uptake; and |
| • | unforeseen changes in federal health care policy guidelines may negatively impact a physician practice’s willingness to provide novel treatments. |
If our products are not included within an adequate number of formularies or adequate reimbursement levels are not provided, or if those policies increasingly favor generic or OTC products, our overall business and financial condition would be adversely affected.
Further, there have been a number of legislative and regulatory proposals concerning reimportation of pharmaceutical products and safety matters. For example, in an attempt to protect against counterfeit drugs, the federal government and numerous states have enacted pedigree legislation. In particular, California has enacted legislation that requires development of an electronic pedigree to track and trace each prescription drug at the saleable unit level through the distribution system. California’s electronic pedigree requirement is scheduled to take effect beginning in January 2015. Compliance with California and future federal or state electronic pedigree requirements will likely require an increase in our operational expenses and will likely be administratively burdensome.
Generic pricing plans, such as those implemented by Wal-Mart, CVS, Rite Aid and Walgreens, may affect the market for our products.
In September 2006, Wal-Mart began offering certain generic drugs at $4 per prescription, and various other retailers including CVS, Rite Aid and Walgreens currently offer generic drugs at similar prices. Some retailers have also offered certain generic drugs for free on a limited basis. These and many other retailers have significant market presence, and any decision by them to make generic analogs of our products available at substantially lower prices may have a material adverse effect on the market for our products and our ability to generate revenues from their sale.
Even though Silenor received regulatory approval, it will still be subject to substantial ongoing regulation.
Even though U.S. regulatory approval has been obtained for Silenor, the FDA has imposed restrictions on its indicated uses or marketing and imposed ongoing requirements for post-approval studies or other activities. For example, the approved use of Silenor is limited to the treatment of insomnia characterized by sleep maintenance difficulty. In addition, the FDA has required us to implement a risk evaluation and mitigation strategy, or REMS, consisting of a medication guide. We are also required to complete a standard clinical trial assessing the safety and efficacy of Silenor in children aged 6 to 16 pursuant to the Pediatric Research Equity Act of 2003, or PREA, and to submit final results of this trial by March 2015. Any issues relating to these restrictions or requirements could have an adverse impact on our ability to achieve market acceptance of Silenor and generate revenues from its sale.
The FDA has also requested that all manufacturers of sedative-hypnotic drug products modify their product labeling to include stronger language concerning potential risks. These risks include severe allergic reactions and complex sleep-related behaviors, which may include sleep-driving. The FDA also recommended that the drug manufacturers conduct clinical studies to investigate the frequency with which sleep-driving and other complex behaviors occur in association with individual drug products. Our approved label for Silenor includes warnings relating to risks of complex sleep behaviors.
25
In addition, in August 2011 the FDA requested that we provide information about the pharmacokinetic and pharmacodynamic properties and adverse event profile of Silenor, including differences that might arise due to demographic factors, to enable the FDA to assess whether morning drug levels may remain high enough in some individuals or identifiable patient subgroups to impair driving to a degree that presents an unacceptable risk both to individuals and the public. The FDA’s request indicated that the same request was made to all sponsors of sedative hypnotic medications.
Silenor is also subject to ongoing FDA requirements for the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information. For example, the FDA may require modifications to our REMS for Silenor at a later date if warranted by new safety information. Any future requirements imposed by the FDA may require substantial expenditures.
In addition, all marketing activities associated with Silenor, as well as marketing activities related to any other products that we promote, are subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products. The FDA regulates post-approval promotional labeling and advertising to ensure that such activities conform to statutory and regulatory requirements. Such regulation and FDA review could require us to alter our marketing materials or strategy, incur additional costs or delay certain of our promotional activities.
In addition to FDA restrictions, the marketing of prescription drugs is subject to laws and regulations prohibiting fraud and abuse under government healthcare programs. For example, the federal health care program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn are used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, which apply regardless of the payor. If we fail to comply with applicable FDA regulations or other laws or regulations relating to the marketing of Silenor or any other product, we could be subject to criminal prosecution, civil penalties, seizure of products, injunction, or exclusion of such products from reimbursement under government programs, as well as other regulatory actions.
Approved products, manufacturers and manufacturers’ facilities are subject to continual review and periodic inspections. If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product or on us, including requiring withdrawal of the product from the market.
The distribution of pharmaceuticals is also regulated by state regulatory agencies, including the requirement to obtain and maintain distribution permits or licenses in many states. Compliance with these requirements may require the expenditure of substantial resources and could impact the manner and scope of our distribution operations. If we fail to comply with applicable state regulations relating to the distribution of Silenor or any other product we market, we could be subject to criminal prosecution, civil penalties, seizure of products, injunctions, or other regulatory actions.
We have implemented a comprehensive compliance program and related infrastructure, but we cannot provide absolute assurance that we are or will be in compliance with all potentially applicable laws and regulations.
26
If our operations relating to Silenor fail to comply with applicable regulatory requirements, a regulatory agency may:
| • | issue warning letters or untitled letters; |
| • | impose civil or criminal penalties, including fines; |
| • | require that we change the way the product is administered, change the labeling of the product or implement a new or amended REMS; |
| • | suspend regulatory approval; |
| • | impose requirements to conduct additional clinical, non-clinical or other studies; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve pending applications or supplements to approved applications filed by us; |
| • | impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products or require a product recall. |
We rely on third parties to perform many necessary services for our commercial products, including services related to the storage and distribution of our products.
We have retained third-party service providers to perform a variety of functions related to the sale and distribution of our products, key aspects of which are out of our direct control. For example, we rely on one third-party service provider to provide key services related to warehousing and inventory management, distribution, contract administration and chargeback processing, accounts receivable management and call center management, and, as a result, most of our inventory is stored at a single warehouse maintained by the service provider. We also utilize third parties to perform various other services for us relating to e-detailing, sample processing, and regulatory monitoring, including adverse event reporting, safety database management and other product maintenance services.
We place substantial reliance on the third-party providers that perform services for us. If these third-party service providers fail to effectively provide services to us, fail to comply with applicable laws and regulations, fail to meet expected deadlines, or otherwise do not carry out their contractual duties to us, or encounter physical or natural damage at their facilities, our ability to successfully commercialize Silenor would be significantly impaired, or we could be subject to regulatory sanctions. We do not currently have the internal capacity to perform these important commercial functions, and we may not be able to maintain commercial arrangements for these services on reasonable terms.
Our future reporting and payment obligations under governmental purchasing and rebate programs will be complex and may involve subjective decisions, and any failure to comply with those obligations could subject us to penalties and sanctions, which in turn could have a material adverse effect on our business and financial condition.
As a condition of reimbursement by various federal and state healthcare programs, we will need to calculate and report certain pricing information to federal and state healthcare agencies. The regulations regarding the reporting of such pricing information are complex. Our calculations and methodologies will be subject to review and challenge by the applicable governmental agencies, and it is possible that such reviews could result in material changes to our calculations. In addition, because our calculations and the judgments involved in making these calculations will involve subjective decisions and complex methodologies, these calculations are subject to the risk of errors. Any failure to comply with governmental reporting and payment obligations could result in civil and/or criminal sanctions.
27
We expect intense competition in the marketplace for Silenor and any other product to which we acquire rights, and new products may emerge that provide different and/or better therapeutic alternatives for the disorders that our products are intended to treat.
Silenor competes with well established drugs approved for the treatment of insomnia, including Lunesta, marketed by Sunovion Pharmaceuticals Inc., a wholly-owned subsidiary of Dainippon Sumitomo Pharma Co., Ltd., and the branded and generic versions of Sanofi-Synthélabo, Inc.’s Ambien and Ambien CR and Pfizer Inc.’s Sonata, all of which are GABA-receptor agonists, and Takeda Pharmaceuticals North America, Inc.’s Rozerem, a melatonin receptor antagonist.
A number of companies are marketing reformulated versions of previously approved GABA-receptor agonists. For example, in November 2011, Transcept Pharmaceuticals, Inc. received approval from the FDA for Intermezzo, a low-dose sublingual tablet formulation of zolpidem. Transcept and Purdue Pharmaceutical Products L.P. have entered into an exclusive license and collaboration agreement to commercialize Intermezzo in the United States. Purdue has publicly announced that it plans to launch Intermezzo in the second quarter of 2012. Meda AB and Orexo AB launched Edluar, formerly known as Sublinox, a sublingual tablet formulation of zolpidem in the third quarter of 2009. ECR Pharmaceuticals Company, Inc., a wholly owned subsidiary of Hi-Tech Pharmacal Co., Inc., launched NovaDel Pharma, Inc.’s ZolpiMist, an oral mist formulation of zolpidem, in the United States in February 2011.
In addition to the currently approved products for the treatment of insomnia, a number of new products may enter the insomnia market over the next several years. It has been reported that Neurim Pharmaceuticals Ltd. is seeking FDA approval of Circadin, a prescription form of melatonin that is already approved in the European Union and several other countries. Neurim also announced positive results from Phase 1 and 1b clinical trials for Neu-P11, a melatonin and serotonin agonist for the treatment of insomnia associated with pain.
Alexza Pharmaceuticals, Inc. has announced positive results from a Phase 1 clinical trial of an inhaled formulation of zaleplon, the API in Sonata. In July 2010, Alexza announced that it was advancing this product candidate into Phase 2 clinical trials during the first half of 2011 for the treatment of insomnia in patients who have difficulty falling asleep, including those patients who awake in the middle of the night and have difficulty falling back asleep, but has not yet done so. Somnus Therapeutics, Inc. has announced positive results from two Phase 1 clinical trials and one Phase 2 clinical trial of a delayed-release formulation of zaleplon.
Vanda Pharmaceuticals Inc. has completed two Phase 3 clinical trials of tasimelteon, a melatonin receptor agonist. Tasimelteon received orphan drug designation status for non-24 hour sleep/wake disorder in blind individuals with no light perception. Vanda has initiated Phase 3 clinical trials for tasimelteon to treat this disorder and plans to file an NDA with the FDA in the first half of 2013.
Merck & Co., Inc. has completed Phase 3 clinical trials for suvorexant, an orexin antagonist, for the treatment of insomnia and has MK-6096 and MK-3697 in Phase 2 clinical trials for the treatment of insomnia. Merck has announced that it plans to file regulatory applications for suvorexant in 2012.
Several other companies, including Sunovion Pharmaceuticals, are evaluating 5HT2 antagonists as potential hypnotics, and Eli Lilly and Company is evaluating a potential hypnotic that is a dual histamine/5HT2 antagonist. Additionally, several other companies are evaluating new formulations of existing compounds and other compounds for the treatment of insomnia.
Furthermore, generic versions of Ambien, Ambien CR and Sonata have been launched and are priced significantly lower than approved, branded insomnia products. Some managed health care plans require that patients try generic versions of these branded insomnia products before the patient can be reimbursed for Silenor. Sales of all of these drugs may reduce the available market for, and could put downward pressure on, the price we are able to charge for Silenor, which could ultimately limit our ability to generate significant revenues.
The active ingredient of Silenor is doxepin, which has been used at higher doses for over 40 years for the treatment of depression and anxiety. Doxepin is available generically in strengths as low as 10 mg in capsule form, as well as in a concentrated liquid form dispensed by a marked dropper and calibrated for 5 mg. Some physicians
28
are prescribing generic 10 mg doxepin capsules and generic oral solution doxepin for insomnia off-label for insomnia. In addition, some managed health care plans are requiring the substitution of these generic doxepin products for Silenor, and some pharmacies are suggesting such substitution. Such off label uses of generic doxepin may reduce the sales of Silenor and may put a downward pressure on the price we are able to charge for Silenor, which could ultimately limit our ability to generate significant revenues.
Upon the expiration of, or successful challenge to, our patents covering Silenor, generic competitors may introduce a generic version of Silenor at a lower price. Some generic manufacturers have also demonstrated a willingness to launch generic versions of branded products before the final resolution of related patent litigation, known as an “at-risk launch”. A launch of a generic version of Silenor could have a material adverse effect on our business and we could suffer a significant loss of sales and market share in a short period of time.
We have received notices from Actavis, Mylan, Par, and Zydus that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in the Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but we cannot predict the outcome of these matters. Any adverse outcome in this litigation could result in one or more generic versions of Silenor being launched before the expiration of the listed patents, which could adversely affect our ability to successfully generate sales of Silenor and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows, such events could also significantly impact our ability to continue as a going concern.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition in the development and marketing of Silenor or any other product candidate to which we acquire rights from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies. There can be no assurance that developments by others, including the development of other drug technologies and methods of preventing the incidence of disease, will not render Silenor or any other product candidate that we develop obsolete or noncompetitive.
Compared to us, many of our potential competitors have substantially greater:
| • | capital resources; |
| • | manufacturing, distribution and sales and marketing resources and experience; |
| • | research and development resources, including personnel and technology; |
| • | regulatory experience; |
| • | experience conducting non-clinical studies and clinical trials, and related resources; and |
| • | expertise in prosecution and enforcement of intellectual property rights. |
29
As a result of these factors, our competitors may develop drugs that are more effective and less costly than ours and may be more successful than we are in manufacturing, marketing and selling their products. Our competitors may also obtain patent protection or other intellectual property rights or seek to invalidate or otherwise challenge our intellectual property rights, limiting our ability to successfully market and sell products.
In addition, manufacturing efficiency and selling and marketing capabilities are likely to be significant competitive factors. We currently have no commercial manufacturing capability and more limited sales and marketing infrastructure than many of our competitors and potential competitors.
If the manufacturers upon whom we rely fail to produce our products in the volumes that we require on a timely basis, or to comply with stringent regulations applicable to pharmaceutical drug manufacturers, we may face delays in the development and commercialization of, or be unable to meet demand for, our products and may lose potential revenues.
We do not manufacture Silenor, and we do not plan to develop any capacity to do so. We have a contract with Patheon Pharmaceuticals Inc. to manufacture our future required clinical supplies, if any, of Silenor, and we have a contract with Patheon to manufacture our commercial supply of Silenor. We have also entered into agreements with Plantex USA, Inc. to manufacture our supply of doxepin API and with Anderson Packaging, Inc. to package Silenor finished products.
In addition, in October 2006, we entered into a supply agreement with JRS Pharma L.P., or JRS, under which we purchase from JRS all of our requirements for ProSolv®HD90, an ingredient used in our formulation for Silenor. In August 2008, we amended our supply agreement to provide us with the exclusive right to use this ingredient and any successor product to ProSolv®HD90 in combination with doxepin, as well as the right to list the U.S. patents owned by JRS and covering ProSolv®HD90 in the Orange Book relating to Silenor. JRS also agreed to enforce any such patents listed in the Orange Book on our behalf. The term of the agreement runs through January 1, 2013, but it will be automatically extended for additional one year periods unless we terminate the license upon written notice at least 90 days prior to the end of the term. Either party may terminate the agreement upon written notice to the other party if the other party commits a material breach of its obligations and fails to remedy the breach within 30 days, or upon the filing of bankruptcy, reorganization, liquidation, or receivership proceedings relating to the other party. We also have the right to terminate the agreement if JRS no longer has any valid patent claim in the U.S. covering ProSolv®HD90 or any successor product to ProSolv®HD90 used by us in combination with doxepin. Each of the current patents listed in the Orange Book by us relating to Silenor expires in 2015.
As part of the amendment, we made an upfront license payment of $0.2 million and are obligated to pay a royalty of less than 1% on net sales of Silenor beginning as of the expiration of the statutory exclusivity period for Silenor in each country in which a Silenor formulation containing ProSolv®HD90 or any successor product to ProSolv®HD90 is marketed, which with respect to U.S. sales of Silenor we expect to occur in March 2013. This royalty is only payable if one or more patents under the license agreement continues to be valid in each such country and a patent relating to our formulation for Silenor has not issued in such country.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up and validating initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Our manufacturers may not perform as agreed or may terminate their agreements with us. Additionally, our manufacturers may experience manufacturing difficulties due to resource constraints or as a result of labor disputes or unstable political environments. If our manufacturers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, our ability to sell Silenor or any other product candidate that we commercialize or provide any product candidates to patients in our clinical trials would be jeopardized. Any delay or interruption in the supply of clinical trial supplies could delay the completion of our clinical trials, increase the costs associated with maintaining our clinical trial program and, depending upon the period of delay, require us to commence new clinical trials at significant additional expense or terminate the clinical trials completely. In addition, any delay or interruption in our ability to meet commercial demand for Silenor will result in the loss of potential revenues.
30
In addition, all manufacturers of pharmaceutical products must comply with current good manufacturing practice, or cGMP, requirements enforced by the FDA through its facilities inspection program. The FDA is also likely to conduct inspections of our manufacturers’ facilities as part of their review of any marketing applications we submit. These cGMP requirements include quality control, quality assurance and the maintenance of records and documentation. Manufacturers of our products may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any quantities supplied is compromised due to our manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products.
Moreover, our manufacturers and suppliers may experience difficulties related to their overall businesses and financial stability, which could result in delays or interruptions of our supply of Silenor. We do not have alternate manufacturing plans in place at this time. If we need to change to other manufacturers, the FDA and comparable foreign regulators must approve these manufacturers’ facilities and processes prior to our use, which would require new testing and compliance inspections, and the new manufacturers would have to be educated in or independently develop the processes necessary for production.
Any of these factors could adversely affect the commercial activities for Silenor or suspend clinical trials, regulatory submissions, and required approvals for any other product candidate that we develop, or entail higher costs or result in our being unable to effectively commercialize our products. Furthermore, if our manufacturers failed to deliver the required commercial quantities of raw materials, including bulk drug substance, or finished product on a timely basis and at commercially reasonable prices, we would likely be unable to meet demand for our products and we would lose potential revenues.
We, Paladin or any other future licensee may never receive approval or commercialize our products outside of the United States, or our or their activities may not be effective or in compliance with applicable laws.
We have licensed to Paladin the rights to commercialize Silenor in Canada, South America, the Caribbean and Africa. Silenor has not been approved for marketing in any jurisdiction outside of the United States. Paladin will be responsible for regulatory submissions for Silenor in the licensed territories and will have the exclusive right to commercialize Silenor in the licensed territories. Paladin’s New Drug Submission filing in Canada was accepted for review by Health Canada in February 2012, but there is no assurance regulatory approval will be obtained in Canada or any of the other licensed territories. We may license rights to Silenor or other future products to others for territories outside the United States in the future.
Compared to a development and commercialization strategy for an ex-U.S. product that involves a third-party collaborator, the development and commercialization of such a product by us without a collaborator is likely to require substantially greater resources on our part and potentially adversely impact the timing and results of the development or commercialization of the product.
In order to market any products outside of the United States, we or our licensees must establish and comply with numerous and varying regulatory requirements regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. Any additional clinical studies that may be required to be conducted as part of the regulatory approval process may not corroborate the results of the clinical studies we have previously conducted or may have adverse results or effects on our ability to maintain regulatory approvals in the United States or obtain them in other countries. The time required to obtain approval might differ from that required to obtain FDA approval for Silenor.
The regulatory approval process in other countries may include all of the risks regarding FDA approval in the U.S. as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval
31
could limit the uses of the product candidate and have an adverse effect on potential royalties and product sales. Such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
In addition, any revenues we receive from sales of Silenor outside the United States will likely depend upon the efforts of Paladin or any other future licensees, as applicable, which in many instances will not be within our control. If we are unable to maintain our license agreements or to effectively establish alternative arrangements to market such products, or if Paladin or any future licensees do not perform adequately under such agreements or arrangements or comply with applicable laws, our business could be adversely affected and we could be subject to regulatory sanctions.
We or any collaborator may not be successful in developing, receiving approval for or commercializing an OTC product for Silenor.
In November 2011, we and P&G agreed that through the period ending on March 31, 2012, we would work together to evaluate the potential to develop and commercialize an OTC pharmaceutical product containing doxepin, including jointly developing a desired product profile for the OTC product and conducting market research on such product profile at P&G’s expense. During this period, we agreed not to negotiate rights in and to an OTC product with any third party. If P&G notifies us of its interest in negotiating for rights to the OTC product at any time prior to March 31, 2012, P&G will have the exclusive right to negotiate with us relating to such rights for 120 days from our receipt of the notice, or such longer period as may be mutually agreed by us and P&G. We cannot assure you that P&G will elect to negotiate with us with respect to OTC rights, that if P&G does not so elect that we will find another suitable third party interested in such rights, or that any such negotiations with P&G or another third party will result in a completed transaction or that such a transaction will be successful or on attractive terms. If we are unable to establish an OTC collaboration, we will require substantial additional funds, which may not be available, in order to develop and commercialize the product on our own.
Even if we are successful in establishing a collaboration with P&G or another third party for an OTC product, we or such third party must establish and comply with numerous and varying regulatory requirements regarding safety and efficacy of OTC products prior to selling the product, and we cannot give any assurance that any such OTC product will receive applicable regulatory approval or be successfully commercialized.
Our failure to successfully acquire, develop and market additional product candidates or approved products and integrate them into our operations may impair our ability to grow.
As part of our strategy, we may selectively evaluate products and product candidates that we believe may be a strategic fit with our business. Because we neither have, nor currently intend to establish, internal research capabilities, we would be dependent upon pharmaceutical and biotechnology companies, university scientists and other researchers to sell or license products to us. The success of this strategy will depend upon our ability to identify, select and acquire promising pharmaceutical product candidates and products. However, future acquisitions may entail numerous operational and financial risks, including:
| • | exposure to unknown liabilities; |
| • | disruption of our business and diversion of our management’s time and attention to develop acquired products or technologies; |
| • | incurrence of substantial debt or dilutive issuances of securities to pay for acquisitions; |
| • | higher than expected acquisition and integration costs; |
| • | increased amortization expenses; |
| • | difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel; |
32
| • | impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership; and |
| • | inability to retain key employees of any acquired businesses. |
We have limited resources to identify and execute the evaluation, acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. In particular, we may compete with larger pharmaceutical companies and other competitors in our efforts to establish new collaborations and in-licensing opportunities. These competitors likely will have access to greater financial resources than us and may have greater expertise in identifying and evaluating new opportunities. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts.
Further, any product candidate that we acquire may require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities. These efforts could be costly and divert resources from our Silenor commercial efforts and the remainder of our business. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot assure you that any products that we develop or approved products that we acquire will be manufactured or produced profitably, successfully commercialized or widely accepted in the marketplace.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in that litigation would have a material adverse effect on our business.
Our commercial success depends upon our ability, together with our collaborators, to manufacture, market and sell Silenor or any other product candidates that we develop and use our proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our collaborators are operating. As the biotechnology and pharmaceutical industry expands and more patents are issued, the risk increases that our operations may give rise to claims that our products infringe the patent rights of others. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our products or proprietary technologies may infringe.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our products and/or proprietary technologies infringe their intellectual property rights. If our products, proprietary technologies or their uses infringe any of these intellectual property rights, we or our collaborators could be required to pay damages and could be unable to commercialize our products or use our proprietary technologies unless we or they obtained a license. A license may not be available to us or our collaborators on acceptable terms, or at all. In addition, during litigation, the intellectual property rights holder could obtain a preliminary injunction or other equitable right which could prohibit us from making, using or selling our products, technologies or methods.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries generally. If a third party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including, but not limited to:
| • | infringement and other intellectual property claims which, with or without merit, may be expensive and time-consuming to litigate and may divert our management’s attention from our core business; |
| • | substantial damages for infringement, including treble damages and attorneys’ fees, which we may have to pay if a court decides that the product at issue infringes on or violates the third party’s rights; |
| • | a court prohibiting us from selling or licensing the product unless the third party licenses its product rights to us, which it is not required to do; |
33
| • | if a license is available from the third party, we may have to pay substantial royalties, fees and/or grant cross-licenses to our products; and |
| • | redesigning our products or processes so they do not infringe, which may not be possible or may require substantial funds and time. |
No assurance can be given that patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering our products, technology or methods. Because of the substantial number of patents issued and patent applications filed in our field, we believe there is a risk that third parties may allege they have patent rights encompassing our products, technology or methods.
We depend on a limited number of wholesaler customers for the retail distribution of Silenor, and if we lose significant wholesaler customers, our business could be harmed.
Our customers for our Silenor product include some of the nation’s leading wholesale pharmaceutical distributors, such as Cardinal Health, Inc., McKesson Corporation and AmerisourceBergen Corporation, and major drug chains. The loss of any of these companies as a wholesaler customer or a material reduction in their purchases or in the prices they are willing to pay for our products could harm our business, financial condition and results of operations.
Materials necessary to manufacture Silenor or any other product candidate that we develop or commercialize may not be available on commercially reasonable terms, or at all, which may delay development and commercialization.
Although we have contracted with suppliers of doxepin and other key raw materials for Silenor, we largely rely on our manufacturers to purchase from third-party suppliers the other materials necessary to produce our product candidates. Suppliers may not sell these materials to our manufacturers at the time we need them or on commercially reasonable terms. We do not have any control over the process or timing of the acquisition of these materials by our manufacturers. If our manufacturers or we are unable to purchase these materials for Silenor or any other product candidate that we commercialize, there would be a shortage in supply, which would materially affect our ability to generate sales revenues. If our manufacturers are unable to obtain these materials for our non-clinical studies or clinical trials of any other product candidate that we develop, product testing and potential regulatory approval could be delayed or suspended, significantly impacting our development programs.
Silenor or any other product candidate that we develop may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or commercialization.
Undesirable side effects caused by Silenor or any other product candidate that we develop could interrupt, delay or halt clinical trials, result in the denial or suspension of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, or cause us to evaluate the future of our development programs. Any of these occurrences could delay or prevent us from continuing to sell Silenor or from commercializing any product candidate that we develop. In addition, the FDA may require, or we may undertake, additional clinical trials to support the safety profile of Silenor or any proposed changes to the labeling for Silenor.
Silenor will be subject to continual review by the FDA, and we cannot assure you that newly discovered or developed safety issues will not arise. For example, in August 2011 the FDA requested that we provide information about the pharmacokinetic and pharmacodynamic properties and adverse event profile of Silenor, including differences that might arise due to demographic factors, to enable the FDA to assess whether morning drug levels may remain high enough in some individuals or identifiable patient subgroups to impair driving to a degree that presents an unacceptable risk both to individuals and the public. The FDA’s request indicated that the same request was made to all sponsors of sedative hypnotic medications.
With the use of any marketed drug by a wide patient population, serious adverse events may occur from time to time that initially do not appear to relate to the drug itself, and only if the specific event occurs with some regularity over a period of time does the drug become suspect as having a causal relationship to the adverse event. Any safety issues could cause us to suspend or cease marketing of our approved products, cause us to modify how we market our approved products, subject us to substantial liabilities, and adversely affect our revenues and financial condition.
34
In addition, if we or others identify undesirable side effects caused by Silenor or any other product candidate that we commercialize:
| • | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| • | regulatory authorities may withdraw their approval of the product or place restrictions on the way it is prescribed; |
| • | we may be required to change the way the product is administered, conduct additional clinical trials, change the labeling of the product or implement a new or amended REMS; |
| • | we may be subject to related liability; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the affected product, which in turn could delay or prevent us from generating significant revenues from its sale.
We have licensed Silenor from a third party. If we default on any of our obligations under that license, or if the licensor exercises a right to terminate the license, we could lose rights to Silenor.
We in-licensed rights to Silenor through an exclusive licensing arrangement, and we may enter into similar licenses in the future. Under our license agreement for Silenor, we are required to use commercially reasonable efforts to commercialize Silenor. In addition, our licensor for Silenor has the contractual right to terminate the license agreement upon the breach by or a specified insolvency event involving us. In the event that our licensor for Silenor terminates the license agreement, even though we would maintain ownership of our clinical data and the other intellectual property we have developed relating to Silenor, we would be unable to continue our commercialization activities relating to Silenor and our business and financial condition would be materially harmed.
We may experience difficulties in managing growth.
We have increased our headcount from 5 full-time employees at January 1, 2010 to 36 full-time employees as of February 15, 2012, including our field-based sales representatives. Our management and personnel, systems and facilities currently in place may not be adequate to support the growth in our number of employees. Our need to effectively manage our operations, growth and various projects requires that we:
| • | manage our commercial efforts effectively while carrying out our contractual obligations to collaborators and other third parties and complying with all applicable laws, rules and regulations; |
| • | continue to improve our operational, financial and management controls, reporting systems and procedures; and |
| • | attract, train and retain sufficient numbers of talented employees. |
We may be unable to successfully implement these tasks to achieve our commercial goals.
35
We face potential product liability exposure, and, if successful claims are brought against us, we may incur substantial liability for a product and may have to limit its commercialization.
The sale of approved products and the use of product candidates by us in clinical trials expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers, pharmaceutical companies or others selling our products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for our products; |
| • | impairment of our business reputation; |
| • | withdrawal of clinical trial participants; |
| • | costs of related litigation; |
| • | substantial monetary awards to patients or other claimants; |
| • | loss of revenues; and |
| • | the inability or lack of commercial rationale to continue development or commercial activities relating to Silenor or any other product candidate we develop. |
We have obtained product liability insurance coverage for commercial product sales of Silenor, including coverage for product liability claims, but our insurance coverage may not reimburse us at all or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel.
We may not be able to attract or retain qualified management, scientific, clinical and commercial personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses, particularly in the San Diego, California area. If we are not able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development or commercialization objectives, our ability to raise additional capital and our ability to implement our business strategy. In particular, if we lose any members of our senior management team, we may not be able to find suitable replacements, and our business may be harmed as a result.
We are highly dependent on the product acquisition, development, regulatory and commercialization expertise of our senior management. If we lose one or more of the members of our senior management team or other key employees, our ability to implement our business strategy successfully could be seriously harmed. Replacing key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel.
In addition, we have advisors who assist us in formulating our product development, clinical, regulatory and commercialization strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us, or may have arrangements with other companies to assist in the development or commercialization of products that may compete with ours.
36
Our clinical trials may fail to demonstrate the safety and efficacy of any future product candidates we develop, which could prevent or significantly delay their regulatory approval.
Even though Silenor has received regulatory approval, before obtaining regulatory approvals for the commercial sale of any other product candidate we develop, we must demonstrate through clinical trials that the product candidate is safe and effective for use in each target indication.
The results from clinical trials that we complete may not be predictive of results obtained in future clinical trials, and there can be no assurance that we will demonstrate sufficient safety and efficacy to obtain the requisite regulatory approvals or result in marketable products. A number of companies in the biotechnology and pharmaceutical industries have suffered significant setbacks in advanced clinical trials, even after promising results in earlier studies. If any product candidate that we develop is not shown to be safe and effective in clinical trials, or if the FDA does not deem the product candidate to be sufficiently safe and effective to warrant marketing approval, our business, financial condition and results of operations would be materially harmed.
We may be subject to claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is commonplace in our industry, we employ individuals who were previously employed at other biotechnology, specialty pharma or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Finances and Capital Requirements
Capital raising activities, such as issuing securities, incurring debt, assigning receivables or royalty rights or entering into collaborations or other strategic transactions, may cause dilution to existing stockholders or a reduction in our stock price, restrict our operations or require us to relinquish proprietary rights and may be limited by applicable laws and regulations.
Based on our recurring losses, negative cash flows from operations and working capital levels, we will need to raise substantial additional funds. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected. The report of our independent registered public accounting firm on our financial statements for the year ended December 31, 2011 contains an explanatory paragraph stating that our recurring losses raise substantial doubt about our ability to continue as a going concern.
Because we will need to raise additional capital to fund our business, among other things, we may conduct substantial equity offerings. For example, in August 2011 we entered into the sales agreement with Citadel pursuant to which we agreed to sell, at our option, up to an aggregate of $30.0 million in shares of our common stock through Citadel, as sales agent, of which we have sold $0.8 million to date. Sales of the common stock made pursuant to the sales agreement, if any, will be made on the NASDAQ Stock Market under our currently-effective Registration Statements on Form S-3 by means of ordinary brokers’ transactions at then-prevailing market prices. Additionally, under the terms of the sales agreement, we may also sell shares of our common stock through Citadel, on the NASDAQ Stock Market or otherwise, at negotiated prices or at prices related to the prevailing market price. However, there can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. We will not be able to make sales of our common stock pursuant to the sales agreement unless certain conditions are met, which include the accuracy of representations and warranties made to Citadel under the sales agreement; compliance with laws, and the continued listing of our stock on the Nasdaq Capital Market. On December 13, 2011, we received a letter from the Listing Qualifications Department of the Nasdaq Stock Market, or Nasdaq, informing us that because the closing bid price of our common stock listed on Nasdaq was below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2). If compliance is not demonstrated within the applicable timeframe, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market. Citadel or we are permitted to terminate the sales agreement at any time. Sales of shares pursuant to the sales agreement will have a dilutive effective on the holdings of our existing stockholders, and may result in downward pressure on the price of our common stock.
To the extent that we raise any required additional capital by issuing equity securities, our existing stockholders’ ownership will be diluted. Any such dilution of the holdings of our current stockholders may result in downward pressure on the price of our common stock.
Any debt, receivables or royalty financing we enter into may involve covenants that restrict our operations or conditions that require repayment.
Equity financing, debt financing, receivables assignments, royalty interest assignments and other types of financing are often coupled with an additional equity component, such as warrants to purchase stock. To the extent that any of our outstanding warrants or additional warrants that we may issue in the future, are exercised by their holders, dilution of our existing stockholders’ ownership interests will result.
37
In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. The exploration of strategic alternatives may not result in any agreement or transaction and, if completed, any agreement or transaction may not be successful or on attractive terms. The inability to enter into a strategic transaction, or a strategic transaction that is not successful or on attractive terms, could accelerate our needs for cash and make securing funding on reasonable terms more difficult. In addition, if we raise additional funds through collaborations or other strategic transactions, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
In addition, rules and regulations of the SEC or other regulatory agencies may restrict our ability to conduct certain types of financing activities, or may affect the timing of and the amounts we can raise by undertaking such activities. For example, under current SEC regulations, because the aggregate market value of our common stock held by non-affiliates, or our public float, is less than $75 million, the amount that we can raise through primary public offerings of securities in any twelve-month period using one or more registration statements on Form S-3 is limited to an aggregate of one-third of our public float.
We may not be able to sell shares of our common stock under our equity sales agreement with Citadel at times, prices or quantities that we desire and if such sales do occur, they may result in dilution to our existing stockholders.
In August 2011, we entered into the sales agreement with Citadel. Under the terms of the sales agreement, Citadel will use its commercially reasonable efforts to sell shares of our common stock designated by us. However, there can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. In addition, we will not be able to make sales of our common stock pursuant to the sales agreement unless certain conditions are met, which include the accuracy of representations and warranties made to Citadel under the sales agreement; compliance with laws; and the continued listing of our stock on the Nasdaq Capital Market. On December 13, 2011, we received a letter from the Listing Qualifications Department of the Nasdaq Stock Market, or Nasdaq, informing us that because the closing bid price of our common stock listed on Nasdaq was below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2). If compliance is not demonstrated within the applicable timeframe, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market. In addition, Citadel is permitted to terminate the sales agreement at any time. If we are unable to access funds through sales under the sales agreement, or it is terminated by Citadel, we may be unable to access capital on favorable terms or at all.
Should we sell shares pursuant to the sales agreement, it will have a dilutive effective on the holdings of our existing stockholders, and may result in downward pressure on the price of our common stock. If we sell shares under the sales agreement at a time when our share price is decreasing, we will need to issue more shares to raise the same amount than if our stock price was higher. Issuances in the face of a declining share price will have an even greater dilutive effect than if our share price were stable or increasing, and may further decrease our share price. During 2011, we sold an aggregate of 786,825 shares of our common stock and received gross proceeds of $0.8 million, and paid $0.3 million of legal and accounting fees associated with the execution of the sales agreement and commissions.
We have never been profitable and we may not be able to generate revenues sufficient to achieve profitability.
We only began generating revenues from the commercialization of Silenor late in the third quarter of 2010, we have not been profitable since inception, and it is possible that we will not achieve profitability. We incurred net losses of $59.3 million for 2011 fiscal year, and have accumulated losses totaling $276.2 million since inception. We expect to continue to incur significant operating losses and capital expenditures. As a result, we will need to generate significant revenues to achieve and maintain profitability. We cannot assure you that we will achieve significant revenues, or that we will ever achieve profitability. Even if we do achieve profitability, we cannot assure you that we will be able to sustain or increase profitability on a quarterly or annual basis in the future. If revenues
38
grow more slowly than we anticipate or if operating expenses exceed our expectations or cannot be adjusted accordingly, our business, results of operations and financial condition will be materially and adversely affected. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected and we may be unable to continue as a going concern. If we are unable to continue as a going concern, it is likely that investors will lose all or a part of their investment.
Our quarterly operating results may fluctuate significantly.
We expect our operating results to be subject to quarterly fluctuations. The revenues we generate, if any, and our operating results will be affected by numerous factors, including:
| • | the scope and effectiveness of commercial activities relating to Silenor or any other product that we may commercialize, alone or with a collaborator; |
| • | commercial activities of our competitors; |
| • | our entering into collaborations; |
| • | developments in our current intellectual property lawsuits with Actavis, Mylan, Par and Zydus; |
| • | any other intellectual property infringement lawsuit in which we may become involved; |
| • | our addition or termination of development programs or funding support; |
| • | variations in the level of expenses related to development of any product candidate that we develop; |
| • | non-cash charges which we incur, including relating to share-based compensation; and |
| • | regulatory developments. |
If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. We believe that quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
The use of our net operating loss and tax credit carryforwards may be limited.
Net operating loss carryforwards and research and development credits may expire and not be used. As of December 31, 2011, we had generated federal net operating loss carryforwards of approximately $234.1 million and state net operating loss carryforwards of approximately $226.1 million, the majority of which were generated in California. As of December 31, 2011, we had generated federal research and development tax credits of $4.3 million and California research and development tax credits of $2.0 million. Both federal net operating loss carryforwards and federal research and development tax credits have a 20-year carryforward period and begin to expire in 2023 and 2024, respectively. California net operating loss carryforwards have up to a 20-year carryforward period, depending on the date of origin, and begin to expire in 2013; however, California has currently suspended the use of net operating loss carryforwards to offset taxable income. California research and development tax credits have no expiration.
Pursuant to Sections 382 and 383 of the Internal Revenue Code, annual use of our net operating loss and credit carryforwards will be limited in the event a cumulative change in ownership of more than 50 percent occurs within a three-year period. We determined that such an ownership change occurred as of March 31, 2010 as a result of various stock issuances used to finance our development activities. This ownership change resulted in limitations on the utilization of our tax attributes, including our net operating loss carryforwards and tax credits. A portion of the remaining net operating losses limited by Section 382 becomes available for use each year.
39
If there are additional changes in ownership, then additional net operating loss carryforwards and research and development credit carryovers could be eliminated or restricted.
Our results of operations and liquidity needs could be materially negatively affected by market fluctuations and economic downturn.
Our results of operations and liquidity could be materially negatively affected by economic conditions generally, both in the United States and elsewhere around the world. Domestic and international equity and debt markets have experienced and may continue to experience heightened volatility and turmoil based on domestic and international economic conditions and concerns. In the event these economic conditions and concerns continue or worsen and the markets continue to remain volatile, our results of operations and liquidity could be adversely affected by those factors in many ways, including making it more difficult for us to raise funds if necessary, and our stock price may decline. In addition, our investment securities consist primarily of money market funds and corporate and United States government agency notes. While we do not believe that our investment securities have significant risk of default or illiquidity, we cannot provide absolute assurance that our investments are not subject to adverse changes in market value. If economic instability continues and the credit ratings of the security issuers deteriorate and any decline in market value is determined to be other-than-temporary, we would be required to adjust the carrying value of the investments through impairment charges. We also maintain significant amounts of cash and cash equivalents at one or more financial institutions that are not federally insured, and we cannot provide assurance that we will not experience losses on these investments.
Risks Relating to Securities Markets and Investment in Our Stock
There may not be a viable public market for our common stock, and market volatility may affect our stock price and the value of your investment.
Our common stock had not been publicly traded prior to our initial public offering, which was completed in December 2005, and an active trading market may not develop or be sustained. We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Therefore, investors will have to rely on appreciation in our stock price and a liquid trading market in order to achieve a gain on their investment. The market prices for securities of biotechnology and pharmaceutical companies have historically been highly volatile, and the market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. Since our initial public offering on December 15, 2005 through December 31, 2011, the trading prices for our common stock have ranged from a high of $21.24 to a low of $0.18.
The market price of our common stock may fluctuate significantly in response to a number of factors, most of which we cannot control, including:
| • | variations in our quarterly operating results; |
| • | events affecting our existing license agreements, and any future collaborations or other strategic transactions, commercial agreements and grants; |
| • | announcements of new products or technologies, commercial relationships or other events by us or our competitors; |
| • | developments in our current intellectual property lawsuits with Actavis, Mylan, Par and Zydus; |
| • | any other intellectual property infringement lawsuit in which we may become involved; |
| • | regulatory approval or other changes in the regulatory status of our products or product candidates; |
| • | decreased coverage and changes in securities analysts’ estimates of our financial performance; |
| • | our ability to maintain our listing on the Nasdaq Capital Market; |
40
| • | regulatory developments in the United States and foreign countries; |
| • | fluctuations in stock market prices and trading volumes of similar companies; |
| • | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders; |
| • | announcements concerning financing activities; |
| • | additions or departures of key personnel; and |
| • | discussion of us or our stock price by the financial and scientific press and in online investor communities. |
The realization of any of the risks described in these “Risk Factors” could have a dramatic and material adverse impact on the market price of our common stock. In addition, class action litigation has often been instituted against companies whose securities have experienced periods of volatility or declines in market price. Any such litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could hurt our business, operating results and financial condition.
If we are unable to comply with the minimum requirements for listing on the Nasdaq Capital Market, we may be delisted from the Nasdaq Capital Market, which would likely cause the liquidity and market price of our common stock to decline.
Our stock is listed on the Nasdaq Capital Market. In order to continue to be listed on the Nasdaq Capital Market, we must meet specific quantitative standards, including maintaining a minimum bid price of $1.00 for our common stock, a public float of $1.0 million, and either $2.5 million in stockholders equity or a market capitalization of $35 million. On December 13, 2011, we received a letter from the Listing Qualifications Department of Nasdaq informing us that because the closing bid price for our common stock had been below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market.
We have until June 11, 2012 to regain compliance with Nasdaq’s listing requirements by having the closing bid price of our common stock be at least $1.00 for at least 10 consecutive trading days. If we do not regain compliance within this time period but comply with all continued listing standards other than the closing bid price requirement, we may provide Nasdaq with written notice of our intention to cure the deficiency during a second compliance period of up to 180 days. If the Listing Qualifications Department of Nasdaq believes that we will be able to cure the deficiency during such additional period, it will grant us such period to do so.
If compliance is not demonstrated within the applicable compliance period, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market. However, we may appeal Nasdaq’s determination to delist our securities to a Nasdaq Hearings Panel. During any appeal process, shares of our common stock would continue to trade on the Nasdaq Capital Market.
If we were to be delisted from the Nasdaq Capital Market, trading, if any, in our shares may continue to be conducted on the Over-the-Counter Bulletin Board or in a non-Nasdaq over-the-counter market, such as the “pink sheets.” Delisting of our shares would result in limited release of the market price of those shares and limited analyst coverage and could restrict investors’ interest in our securities. Also, a delisting could have a material adverse effect on the trading market and prices for our shares and our ability to issue additional securities or to secure additional financing. In addition, if our shares were not listed and the trading price of our shares was less than $5.00 per share, our shares could be subject to Rule 15g-9 under the Exchange Act which, among other things, requires that broker/dealers satisfy special sales practice requirements, including making individualized written suitability determinations and receiving a purchaser’s written consent prior to any transaction. In such case, our securities could also be deemed to be a “penny stock” under the Securities Enforcement and Penny Stock Reform Act of 1990, which would require additional disclosure in connection with trades in those shares, including the delivery of a disclosure schedule explaining the nature and risks of the penny stock market. Such requirements could severely limit the liquidity of our securities and our ability to raise additional capital in an already challenging capital market.
41
If our executive officers, directors and largest stockholders choose to act together, they may be able to control our operations and act in a manner that advances their best interests and not necessarily those of other stockholders.
As of February 15, 2012, our executive officers, directors and holders of 5% or more of our outstanding common stock beneficially owned approximately 35% of our common stock. As a result, these stockholders, acting together, would likely be able to control all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of this group of stockholders may not always coincide with our interests or the interests of other stockholders, and they may act in a manner that advances their best interests and not necessarily those of other stockholders.
If we are unable to maintain an effective registration statement for the resale of shares under our July 2009 private placement, or if we are delisted from the Nasdaq Capital Market, Nasdaq Global Market, the New York Stock Exchange or the American Stock Exchange, we may be required to pay liquidated damages.
In July 2009, we issued 5.1 million shares of common stock and seven-year warrants to purchase up to 5.1 million additional shares of common stock, exercisable in cash or by net exercise at a price of $1.155 per share. In connection with the private placement, we agreed to register for resale both the shares of common stock purchased by the investors and the shares of common stock issuable upon exercise of the warrants. The resale registration statement was filed and declared effective by the SEC in August 2009. We also agreed to other customary obligations regarding registration, including matters relating to indemnification, maintenance of the registration statement and payment of expenses.
We may be liable for liquidated damages if we do not maintain the effectiveness of the registration statement or the listing of our common stock on the Nasdaq Capital Market, the Nasdaq Global Market, the New York Stock Exchange or the American Stock Exchange, in each case for a period of ten consecutive days or for more than thirty days in any 365-day period. The amount of the liquidated damages is one percent per applicable ten or thirty day period, subject to an aggregate maximum of eight percent per calendar year, of the aggregate purchase price of the common stock purchased in the private placement then held by each investor that are registrable securities.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws may delay or prevent an acquisition of us or a change in our management. These provisions include a classified board of directors, a prohibition on actions by written consent of our stockholders, and the ability of our board of directors to issue preferred stock without stockholder approval. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits stockholders owning in excess of 15 percent of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirors to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
We expend substantial costs and management resources as a result of laws and regulations relating to corporate governance matters.
As a public reporting company, we must comply with the Sarbanes-Oxley Act of 2002 and the related rules and regulations adopted by the SEC and by the Nasdaq Stock Market, including expanded disclosures, accelerated reporting requirements and more complex accounting rules. Compliance with Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, and other requirements has caused us to expend substantial costs and management resources and will continue to do so. Additionally, these laws and regulations could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or board committees or as executive officers. In June 2007, the Public Company Accounting Oversight Board approved Auditing Standard No. 5, and at the same time, the SEC issued guidance for management for complying with the requirements of Section 404. This auditing standard and the related management guidance provides a more risk-based approach to compliance and testing under Section 404. However, we still do and expect to continue to incur substantial costs and to devote significant resources to corporate governance matters.
42
We are also subject to changing rules and regulations of federal and state government as well as the stock exchange on which our common stock is listed. These entities, including the Public Company Accounting Oversight Board, the SEC and the Nasdaq Stock Market, have issued a significant number of new and increasingly complex requirements and regulations over the course of the last several years and continue to develop additional regulations and requirements in response to laws enacted by Congress. In July 2010, the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access, and the SEC has since issued final rules implementing “say on pay” measures. Our efforts to comply with corporate governance and related requirements have resulted in, and are likely to continue to result in, an increase in expenses and a diversion of management’s time from other business activities.
If we, or the third-party service providers on which we rely, fail to comply with Section 404 and the other corporate governance laws and regulations applicable to us, or if our independent registered public accounting firm cannot complete any required attestation of our evaluation of our internal controls in a timely manner, we could be subject to regulatory scrutiny and a loss of public confidence in our corporate governance or internal controls, which could have an adverse effect on our business and our stock price.
Item 1B. Unresolved Staff Comments
We do not have any unresolved staff comments relating to our periodic or current reports.
We lease approximately 12,000 square feet of space for our headquarters in San Diego, California subject to a lease arrangement that will expire in December 2016. We have no laboratory, research or manufacturing facilities.
We have received notices from Actavis, Mylan, Par, and Zydus that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed the ’229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in the Orange Book have not been asserted against these parties.
Item 4. Mine Safety Disclosures
Not applicable.
43
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock has been traded on the Nasdaq Stock Market since December 15, 2005 under the symbol SOMX. Prior to such time, there was no public market for our common stock. The following table sets forth the high and low sales prices per share of our common stock as reported on the Nasdaq Stock Market for the period indicated.
| Price Range | ||||||||
| High | Low | |||||||
| Year Ended December 31, 2010 |
||||||||
| First Quarter |
$ | 10.60 | $ | 1.10 | ||||
| Second Quarter |
$ | 9.10 | $ | 3.41 | ||||
| Third Quarter |
$ | 5.48 | $ | 2.65 | ||||
| Fourth Quarter |
$ | 3.98 | $ | 2.52 | ||||
| Year Ended December 31, 2011 |
||||||||
| First Quarter |
$ | 3.46 | $ | 2.65 | ||||
| Second Quarter |
$ | 2.94 | $ | 2.05 | ||||
| Third Quarter |
$ | 2.12 | $ | 0.87 | ||||
| Fourth Quarter |
$ | 1.09 | $ | 0.45 | ||||
As of February 22, 2012, there were approximately 8,364 holders of record of our common stock.
Performance Graph
The following graph illustrates a comparison of the total cumulative stockholder return on our common stock to two indices: the Nasdaq Composite Index and the Nasdaq Pharmaceuticals Index. The graph assumes an initial investment of $100 on January 1, 2007.
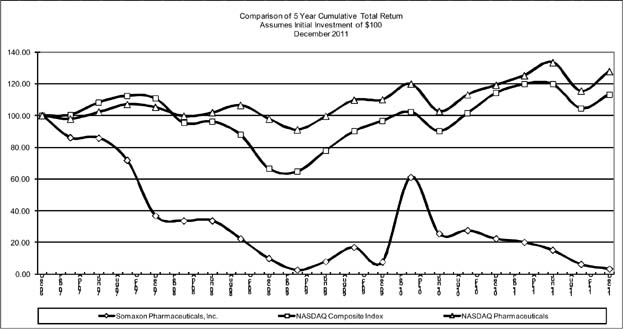
| December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| Somaxon Pharmaceuticals, Inc. |
$ | 3.17 | $ | 22.20 | $ | 7.61 | $ | 9.73 | $ | 36.72 | ||||||||||
| Nasdaq Composite Index |
$ | 113.16 | $ | 114.07 | $ | 96.54 | $ | 66.42 | $ | 110.65 | ||||||||||
| Nasdaq Pharmaceutical Index |
$ | 127.70 | $ | 119.18 | $ | 109.94 | $ | 97.85 | $ | 105.17 | ||||||||||
44
The foregoing graph and table are furnished solely with this report, and are not filed with this report, and shall not be deemed incorporated by reference into any other filing under the Securities Act of 1933, as amended, or the Securities Act, or the Securities Exchange Act of 1934, as amended, whether made by us before or after the date hereof, regardless of any general incorporation language in any such filing, except to the extent we specifically incorporate this material by reference into any such filing.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support operations and finance the growth and development of our business and do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors.
Securities Authorized for Issuance under Equity Compensation Plans
The following table summarizes securities available under our equity compensation plans as of December 31, 2011 (in thousands, except per share data).
| Weighted average per share exercise price of stock options |
Shares issuable upon exercise of outstanding stock options |
Shares issuable upon vesting of outstanding restricted stock units |
Total
shares issuable under current outstanding awards |
Number of securities available for future issuance |
||||||||||||||||
| Equity compensation plans approved by security holders: |
||||||||||||||||||||
| 2004 Equity Incentive Award Plan |
$ | 4.28 | 143 | — | 143 | — | ||||||||||||||
| 2005 Equity Incentive Award Plan |
3.01 | 5,120 | 233 | 5,353 | 2,054 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total Equity Incentive Award Plans |
$ | 3.05 | 5,263 | 233 | 5,496 | 2,054 | ||||||||||||||
| 2005 Employee Stock Purchase Plan |
— | 1,402 | ||||||||||||||||||
|
|
|
|
|
|||||||||||||||||
| Total Equity compensation plans approved by security holders |
$ | 3.05 | 5,263 | 233 | 5,496 | 3,456 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Equity compensation plans not approved by security holders: |
||||||||||||||||||||
| None. |
||||||||||||||||||||
We have share-based awards outstanding under the Somaxon Pharmaceuticals, Inc. 2004 Equity Incentive Award Plan and the 2005 Equity Incentive Award Plan for the benefit of our eligible employees, consultants, and directors. The 2004 Equity Incentive Award Plan was discontinued in November 2005 upon the adoption of the 2005 Equity Incentive Award Plan. No additional share-based awards will be granted under the 2004 Equity Incentive Award Plan and all share-based awards that are repurchased, forfeited, cancelled or expire will become available for grant under the 2005 Equity Incentive Award Plan. The 2005 Employee Stock Purchase Plan was adopted at the time of our initial public offering.
Stock options granted under the 2005 Equity Incentive Award Plan have an exercise price equal to the fair market value of the underlying common stock at the date of grant, have a ten year life and generally vest over a period of between one and four years for our employees and between one and three years for members of our board of directors, with some awards vesting upon the achievement of certain performance conditions.
Restricted shares of our common stock have also been granted under the 2005 Equity Incentive Award Plan. There were no restricted shares unvested as of December 31, 2010 and none were issued during 2011.
45
Awards of restricted stock units, or RSUs, which are convertible into an equivalent number of shares of common stock upon vesting, have also been granted under the 2005 Equity Incentive Award Plan and generally vest upon the achievement of certain performance or time-based conditions.
The 2005 Equity Incentive Award Plan and 2005 Employee Stock Purchase Plan contain “evergreen” provisions which allow for annual increases in the number of shares available for future issuance. The 2005 Equity Incentive Award Plan’s evergreen provision provides for annual increases in the number of shares available for grant equal to the lesser of: (i) 2,000,000 shares, (ii) 5% of the then-total outstanding shares of capital stock (48,063,000 shares were outstanding at December 31, 2011), or (iii) such lesser amount as determined by the board of directors. Pursuant to this evergreen provision, on January 1, 2012, the number of shares available for grant under the 2005 Equity Incentive Award Plan increased by 2,000,000 shares, resulting in a total of 4,054,000 shares available for grant at that time. The 2005 Employee Stock Purchase Plan’s evergreen provision provides for annual increases in the number of shares available for grant equal to the lesser of: (i) 300,000 shares, (ii) 1% of the then-total outstanding shares of capital stock (48,063,000 shares were outstanding at December 31, 2011), or (iii) such lesser amount as determined by the board of directors. Pursuant to this evergreen provision, on January 1, 2012, the number of shares available for grant under the 2005 Employee Stock Purchase Plan increased by 300,000 shares, resulting in a total of 1,702,000 shares available for grant at that time.
Recent Sales of Unregistered Securities
In December 2011, we relied on an exemption from registration contained in Section 4(2) of the Securities Act and Rule 506 of Regulation D, in connection with the issuance of warrants to Oxford Finance Corporation, or Oxford, and Silicon Valley Bank, or SVB, to purchase up to an aggregate of 150,000 shares of our common stock at an exercise price equal to $0.50 per share. The warrants issued to Oxford and SVB and the shares issuable upon the exercise of the warrants have not been registered under the Securities Act or state securities laws and may not be offered or sold in the United States without being registered with the Securities and Exchange Commission, or SEC, or through an applicable exemption from SEC registration requirements.
Issuer Repurchases of Equity Securities
None.
Item 6. Selected Financial Data
The selected statement of operations data for the years ended December 31, 2011, 2010 and 2009, and the balance sheet data as of December 31, 2011 and 2010 have been derived from our audited financial statements included elsewhere in this annual report. The selected statement of operations data for the years ended December 31, 2008 and 2007, and the balance sheet data as of December 31, 2009, 2008 and 2007 have been derived from audited financial statements which are not included in this Form 10-K. Historical results are not necessarily indicative of future results. The selected financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this annual report (amounts in thousands, except per share data).
| Year Ended December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Net product sales |
$ | 16,155 | $ | 1,382 | $ | — | $ | — | $ | — | ||||||||||
| Operating Costs and Expenses: |
||||||||||||||||||||
| Cost of sales |
2,493 | 244 | — | — | — | |||||||||||||||
| Selling, general and administrative |
69,758 | 36,579 | 10,874 | 18,809 | 15,614 | |||||||||||||||
| Research and development |
1,296 | 3,566 | 4,337 | 16,546 | 12,694 | |||||||||||||||
| License fees |
— | — | (999 | ) | 165 | 490 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating costs and expenses |
73,547 | 40,389 | 14,212 | 35,520 | 28,798 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(57,392 | ) | (39,007 | ) | (14,212 | ) | (35,520 | ) | (28,798 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Interest and other income |
52 | 262 | 30 | 903 | 2,387 | |||||||||||||||
| Interest and other expense |
(1,940 | ) | (68 | ) | (261 | ) | (2,610 | ) | — | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (59,280 | ) | $ | (38,813 | ) | $ | (14,443 | ) | $ | (37,227 | ) | $ | (26,411 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted net loss per share |
$ | (1.27 | ) | $ | (1.16 | ) | $ | (0.69 | ) | $ | (2.04 | ) | $ | (1.45 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Shares used to calculate net loss per share |
46,541 | 33,593 | 20,952 | 18,281 | 18,187 | |||||||||||||||
46
| As of December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 10,668 | $ | 54,817 | $ | 5,165 | $ | 14,290 | $ | 37,100 | ||||||||||
| Working capital |
5,107 | 52,407 | 3,404 | 4,258 | 34,385 | |||||||||||||||
| Total assets |
15,859 | 65,131 | 6,411 | 23,717 | 38,717 | |||||||||||||||
| Total debt |
— | — | — | 15,000 | — | |||||||||||||||
| Accumulated deficit |
(276,132 | ) | (216,852 | ) | (178,039 | ) | (163,596 | ) | (126,369 | ) | ||||||||||
| Total stockholders’ equity |
6,541 | 54,264 | 4,241 | 5,106 | 35,176 | |||||||||||||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with “Selected Financial Data” and our financial statements and related notes appearing elsewhere in this annual report. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties, and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including but not limited to those set forth under the caption “Risk Factors” in this annual report.
Overview
Background
We are a specialty pharmaceutical company focused on the in-licensing, development and commercialization of proprietary branded products and late-stage product candidates to treat important medical conditions where there is an unmet medical need and/or high-level of patient dissatisfaction, currently in the central nervous system therapeutic area. In March 2010, the U.S. Food and Drug Administration, or FDA, approved our New Drug Application, or NDA, for Silenor 3 mg and 6 mg tablets for the treatment of insomnia characterized by difficulty with sleep maintenance. Silenor was made commercially available by prescription in the United States in September 2010.
Our principal focus is on commercial activities relating to Silenor. We commercially launched Silenor in September 2010 with 110 sales representatives provided to us on an exclusive basis under our contract sales agreement with Publicis, managed by our sales management personnel, and an additional 105 sales representatives provided to us under our co-promotion agreement with P&G. In February 2011, we amended our agreement with Publicis to have Publicis deploy for us an additional 35 sales representatives. Because we did not believe that the growth of Silenor revenues throughout 2011 was sufficient to support sales and marketing expenses at then-current levels, we terminated our agreements with Publicis and P&G in December 2011 in an effort to conserve cash, and effective January 3, 2012, we hired a reduced sales force of 25 field-based sales representatives from Publicis as our employees to promote Silenor. The remainder of the Publicis sales force ceased promoting Silenor as of November 2, 2011. Our financial statements as of December 31, 2011 reflect all remaining contractual obligations owed to Publicis.
In June 2011, we entered into agreements with Paladin Labs Inc., or Paladin, under which Paladin has the right to commercialize Silenor in Canada, South America, the Caribbean and Africa, subject to the receipt of marketing approval in each such territory.
In order to reduce expenditures, we terminated the employment of 28 employees in the fourth quarter of 2011.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosures. Actual results could differ from those estimates. We believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
47
Revenue Recognition
Product Sales
We sell Silenor to wholesale pharmaceutical distributors. Our returned goods policy generally permits our customers to return products up to six months before and up to twelve months after the expiration date of the product. We authorize returns for expired products in accordance with our returned goods policy and issue credit to our customers for expired returned product. We do not exchange product from inventory for returned product. As of December 31, 2011, the dollar amount of returns received since we began commercial sales of Silenor has been negligible.
We recognize product revenue from product sales when it is realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) our price to the buyer is fixed or determinable; and (4) collectability is reasonably assured. Revenue from sales transactions where the buyer has the right to return the product is recognized at the time of sale only if (1) our price to the buyer is substantially fixed or determinable at the date of sale; (2) the buyer has paid us, or the buyer is obligated to pay us and the obligation is not contingent on resale of the product; (3) the buyer’s obligation to us would not be changed in the event of theft or physical destruction or damage of the product; (4) the buyer acquiring the product for resale has economic substance apart from that provided by us; (5) we do not have significant obligations for future performance to directly bring about resale of the product by the buyer; and (6) the amount of future returns can be reasonably estimated.
Prior to the second quarter of 2011, we were unable to reasonably estimate returns. We therefore deferred revenue recognition until the right of return no longer existed, which was the earlier of Silenor being dispensed through patient prescriptions or the expiration of the right of return. We estimated patient prescriptions dispensed using an analysis of third-party information. In order to develop a methodology to reliably estimate product returns and provide a basis for recognizing revenue on sales to customers at the time of product shipment, we analyzed many factors, including, without limitation, industry data regarding product return rates, and tracked the Silenor product return history, taking into account product expiration dating at the time of shipment and levels of inventory in the wholesale channel compared to prescription units dispensed and the sell-down of our launch inventory. During the second quarter of 2011, the sell-down of our launch inventory was completed, which we believe demonstrates sufficient market acceptance of our product for purposes of our revenue recognition analysis. In addition, since product launch, we have sold product to wholesale pharmaceutical distributors at standard commercial terms utilized in the industry. As a result, we believe we can analogize to industry data regarding product return rates. Based on the sell-down of our launch inventory and the industry and internal data gathered, we believe we have the information needed to reasonably estimate product returns. As a result, in the second quarter of 2011, we began recognizing revenue for Silenor sales at the time of delivery of the product to wholesale pharmaceutical distributors and our other customers.
License and Royalty Revenue
In June 2011, we entered into a license agreement with Paladin pursuant to which Paladin has the right to commercialize Silenor in Canada, South America, the Caribbean and Africa, subject to the receipt of marketing approval in each such territory. We received an upfront payment of $500,000 in connection with the execution of this agreement. We recorded the upfront payment as deferred revenue and are recognizing the upfront payment as license revenue over the period of our significant involvement under the agreement, which we are estimating to be 15 years. As of December 31, 2011, the deferred revenue balance associated with the license agreement is $481,000, of which $448,000 is recorded as a non-current liability and $33,000 is recorded as a current liability within accrued liabilities. We recognized $19,000 as revenue during the year ended December 31, 2011, which is recorded in interest and other income.
If Silenor is commercialized in the licensed territories, we would be eligible to receive sales-based milestone payments of up to $128.5 million as well as a tiered double-digit percentage of net sales in the licensed territories. Due to the uncertainty surrounding the achievement of these future sales-based milestones and royalties, these potential payments will not be recognized as revenue until they are realized and earned.
48
Product Sales Discounts and Allowances
We record product sales discounts and allowances at the time of sale and report revenue net of such amounts in the same period that product sales are recorded. In determining the amount of product sales discounts and allowances, we must make significant judgments and estimates. If actual results vary from our estimates, we may need to adjust these estimates, which could have an effect on product revenue in the period of adjustment. Our product sales discounts and allowances and the specific considerations we use in estimating these amounts include:
Prompt Pay Discounts. As an incentive for prompt payment, we offer a 2% cash discount to customers. We calculate the discount based on the gross amount of each invoice as we expect that all customers will comply with the contractual terms to earn the discount. We record the discount as an allowance against accounts receivable and a corresponding reduction of revenue. At December 31, 2011 and 2010, the allowance for prompt pay discounts was $39,000 and $114,000, respectively.
Patient Discount Programs. We offer discount programs to patients who are prescribed Silenor under which the patient receives a discount on his or her prescription. We reimburse pharmacies for these discounts through third-party vendors. We estimate the total amount that will be redeemed based on the dollar amount of the discounts, the timing and quantity of distribution and historical redemption rates. We accrue the discounts and recognize a corresponding reduction of revenue. At December 31, 2011 and 2010, the accrual for patient discount programs was $414,000 and $182,000, respectively.
Distribution Service Fees. We pay distribution services fees to each wholesaler for distribution and inventory management services. We accrue for these fees based on contractually defined terms with each wholesaler and recognize a corresponding reduction of revenue. At December 31, 2011 and 2010, the accrual for distribution service fees was $319,000 and $276,000, respectively.
Chargebacks. We provide discounts to federal government qualified entities with whom we have contracted. These federal entities purchase products from the wholesalers at a discounted price, and the wholesalers then charge back to us the difference between the current retail price and the contracted price the federal entity paid for the product. We accrue chargebacks based on contract prices and sell-through sales data obtained from third-party information. At December 31, 2011 and 2010, the accrual for chargebacks was $24,000 and $9,000, respectively.
Rebates. We participate in certain rebate programs, which provide discounted prescriptions to qualified insured patients. Under these rebate programs, we pay a rebate to the third-party administrator of the program. We accrue rebates based on contract prices, estimated percentages of product sold to qualified patients and estimated levels of inventory in the distribution channel. Our accrual consists of: (1) the amount expected to be incurred based on the current quarter’s product sold, (2) an accrual for unpaid rebates relating to prior quarters; and (3) an accrual for rebates relating to estimated inventory in the distribution channel. Our estimate of utilization is based on partial claims history data received, third-party data, and information about our expected patient population. At December 31, 2011 and 2010, the accrual for rebates was $1,896,000 and $6,000, respectively.
Product Returns. We estimate future product returns based upon actual returns history, product expiration dating analysis, estimated inventory levels in the distribution channel, and industry data regarding product return rates for similar products. There may be a significant time lag between the date we determine the estimated allowance and when we receive product returns and issue credits to customers. Due to this time lag, we may record adjustments to our estimated allowance over several periods, which would impact our operating results in those periods. At December 31, 2011, the allowance for product returns was $255,000.
Other Discounts and Allowances. From the initial launch of Silenor through September 30, 2010, we offered additional discounts to wholesale distributors for product purchased. At December 31, 2010, the allowance for product launch discounts was $277,000. At December 31, 2011, this balance has been settled in full.
49
The following table summarizes the activity for the years ended December 31, 2011 and 2010 associated with product sales discounts and allowances, with amounts shown in thousands:
| Prompt Pay Discounts |
Patient Discount Fees |
Distribution Service Fees |
Charge- backs and Rebates |
Product Returns & Other Discounts |
Total | |||||||||||||||||||
| Balance at January 1, 2010 |
$ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||
| 2010 period provision |
114 | 214 | 410 | 15 | 277 | 1,030 | ||||||||||||||||||
| Payments and other credits |
— | (32 | ) | (134 | ) | — | — | (166 | ) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2010 |
114 | 182 | 276 | 15 | 277 | 864 | ||||||||||||||||||
| Current period provision |
451 | 1,210 | 1,508 | 2,882 | 541 | 6,592 | ||||||||||||||||||
| Provision related to previously deferred sales |
(82 | ) | (158 | ) | (283 | ) | (14 | ) | (274 | ) | (811 | ) | ||||||||||||
| Payments and other credits |
(444 | ) | (820 | ) | (1,182 | ) | (963 | ) | (289 | ) | (3,698 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2011 |
$ | 39 | $ | 414 | $ | 319 | $ | 1,920 | $ | 255 | $ | 2,947 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Cost of Sales
Cost of sales includes the costs to manufacture, package, and ship Silenor, including personnel costs associated with manufacturing oversight, as well as royalties and amortization of capitalized license fees associated with our license agreement with ProCom One, Inc., or ProCom.
Inventory
Our inventories are valued at the lower of weighted average cost or net realizable value. We analyze our inventory levels quarterly and write down inventory that has become obsolete, or has a cost basis in excess of its expected net realizable value, as well as any inventory quantities in excess of expected requirements. Expired inventory is disposed of and the related costs are written off. We did not record a provision or write-down for excess or expired inventory as of December 31, 2010. We recorded a write-down of $0.6 million for excess inventory during the fourth quarter of fiscal 2011.
Capitalized License Fees
License fees related to our intellectual property are capitalized once technological feasibility has been established. Determining when technological feasibility has been achieved, and determining the related amortization period for capitalized intellectual property, requires the use of estimates and subjective judgment. Costs incurred to in-license our product candidates subsequent to FDA approval of our NDA for Silenor have been capitalized and recorded as an intangible asset. Capitalized amounts are amortized on a straight line basis over approximately ten years.
Share-based Compensation
Share-based compensation expense for employees and directors is recognized in the statement of operations based on estimated amounts, including the grant date fair value, the probability of achieving performance conditions and the expected service period for awards with conditional vesting provisions. For stock options, we estimate the grant date fair value using the Black-Scholes valuation model which requires the use of multiple subjective inputs including an estimate of future volatility and the expected terms of the awards. Our stock did not have a readily available market prior to our initial public offering in December 2005, creating a relatively short history from which to obtain data to estimate volatility for our stock price. Consequently, we estimate our expected future volatility based on a combination of both comparable companies and our own stock price volatility to the extent such history is available. Our future volatility may differ from our estimated volatility at the grant date. We estimate the expected term of our options using guidance provided by the Securities and Exchange Commission, or SEC, in Staff Accounting Bulletin, or SAB, No. 107 and SAB No. 110. This guidance provides a formula-driven approach for determining the expected term. Share-based compensation recorded in our statement of operations is based on awards expected to ultimately vest and has been reduced for estimated forfeitures. Our estimated forfeiture rates may differ from actual forfeiture rates which would affect the amount of expense recognized during the period. Estimated forfeitures are adjusted to actual amounts as they become known.
50
We recognize the value of the portion of the awards that are expected to vest on a straight-line basis over the awards’ requisite service periods. The requisite service period is generally the time over which our share-based awards vest. Some of our share-based awards have vested and may vest upon achieving certain performance conditions, generally pertaining to the commercial performance of Silenor or the achievement of other strategic objectives. Share-based compensation expense for awards with performance conditions is recognized over the period from the date the performance condition is determined to be probable of occurring through the time the applicable condition is met. If the performance condition is not considered probable of being achieved, then no expense is recognized until such time the performance condition is considered probable of being met. At that time, expense is recognized over the period during which the performance condition is likely to be achieved. Determining the likelihood and timing of achieving performance conditions is a subjective judgment made by management which may affect the amount and timing of expense related to these share-based awards. Share-based compensation is adjusted to reflect the value of options which ultimately vest as such amounts become known in future periods. As a result of these subjective and forward-looking estimates, the actual value of our stock options realized upon exercise could differ significantly from those amounts recorded in our financial statements.
Income Taxes
Our income tax expense consists of current and deferred income tax expense or benefit. Current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is recognized for the future tax consequences attributable to tax credits and loss carryforwards and to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. As of December 31, 2011, we have established a valuation allowance to fully reserve our net deferred tax assets. Tax rate changes are reflected in income during the period such changes are enacted. Changes in ownership may limit the amount of net operating loss carryforwards that can be utilized in the future to offset taxable income. In addition, the state of California has currently suspended the use of net operating loss carryforwards to offset taxable income.
Results of Operations
Comparisons of the Years Ended December 31, 2011, 2010 and 2009
Product Sales. Net product sales for the years ended December 31, 2011, 2010, and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, |
2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Gross product sales |
$ | 22,747 | $ | 1,704 | $ | — | $ | 21,043 | $ | 1,704 | 1,235 | % | N/A | |||||||||||||||
| Sales discounts and allowances |
||||||||||||||||||||||||||||
| Prompt pay discount |
(451 | ) | (32 | ) | — | (419 | ) | (32 | ) | 1,309 | % | N/A | ||||||||||||||||
| Patient discount programs |
(1,210 | ) | (56 | ) | — | (1,154 | ) | (56 | ) | 2,061 | % | N/A | ||||||||||||||||
| Distribution service fees |
(1,508 | ) | (118 | ) | — | (1,390 | ) | (118 | ) | 1,178 | % | N/A | ||||||||||||||||
| Chargebacks and rebates |
(2,882 | ) | (2 | ) | — | (2,880 | ) | (2 | ) | 144,000 | % | N/A | ||||||||||||||||
| Product returns and other discounts |
(541 | ) | (114 | ) | — | (427 | ) | (114 | ) | 375 | % | N/A | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total discounts and allowances |
(6,592 | ) | (322 | ) | — | (6,270 | ) | (322 | ) | 1,947 | % | N/A | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total net product sales |
$ | 16,155 | $ | 1,382 | $ | — | $ | 14,773 | $ | 1,382 | 1,069 | % | N/A | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Product sales represent sales of Silenor for which we have recognized revenue. We recognized net product sales of $16,155,000 and $1,382,000 for the years ended December 31, 2011 and 2010, respectively, and had no product sales for the year ended December 31, 2009 as sales of Silenor commenced in the third quarter of 2010. Sales discounts and allowances totaled $6,592,000, $322,000 and $0 for the years ended December 31, 2011, 2010, and 2009, respectively. As a percentage of gross sales, the reductions were 29.0% for the year ended December 31, 2011 and 18.9% for the year ended December 31, 2010. The net increases in gross product sales and sales discounts and allowances are due to growth of sales of Silenor since the commencement of Silenor sales in the third quarter of 2010. The increase in sales discounts and allowances as a percentage of gross product sales is primarily due to an expansion in our participation in payor rebate programs.
51
Cost of Sales. Cost of sales includes the costs to manufacture, package, and ship Silenor, including personnel costs associated with manufacturing oversight, as well as royalties associated with our license agreement. Cost of sales for the years ended December 31, 2011, 2010, and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Cost of sales |
$ | 2,493 | $ | 244 | $ | — | $ | 2,249 | $ | 244 | 922 | % | N/A | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
We recognized cost of sales of $2,493,000 and $244,000 for the years ended December 31, 2011 and 2010, respectively, relating to product with respect to which revenue was recognized. The net increase was due to growth of sales of Silenor since the commencement of Silenor sales in the third quarter of 2010. We had no cost of sales for the year ended December 31, 2009 as sales of Silenor commenced in the third quarter of fiscal 2010. Cost of sales for the year ended December 31, 2011 included a write-down of $0.6 million for potentially excess inventory. There was no similar write-downs during the year ended December 31, 2010. Gross profit was $13,662,000 and $1,138,000 for the years ended December 31, 2011 and 2010, respectively. Expressed as a percentage of net product sales, gross margin was 84.6% in 2011 and 82.3% in 2010.
Selling, General and Administrative Expense. Our selling, general and administrative expenses consist primarily of salaries, benefits, share-based compensation expense, advertising and market research costs, insurance and facility costs, and professional fees related to our marketing, administrative, finance, human resources, legal and internal systems support functions. Selling, general and administrative expenses for the years ended December 31, 2011, 2010 and 2009 are summarized in the following tables (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Sales and marketing |
$ | 51,327 | $ | 24,591 | $ | 1,461 | $ | 26,736 | $ | 23,130 | 109 | % | 1,583 | % | ||||||||||||||
| General and administrative |
18,431 | 11,988 | 9,413 | 6,443 | 2,575 | 54 | % | 27 | % | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total selling, general and administrative expenses |
$ | 69,758 | $ | 36,579 | $ | 10,874 | $ | 33,179 | $ | 25,705 | 91 | % | 236 | % | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Selling and marketing expenses increased $26.7 million for the year ended December 31, 2011 compared to the same period in 2010 primarily due to an increase in costs associated with the commercial activities of Silenor as Silenor was initially made commercially available during the third quarter of 2010. These costs included the costs of our sales representatives, royalties paid to our co-promotion partner, personnel costs and other promotional spending and consulting costs. The net increase was also partially due to additional share-based compensation expense being recorded during the second half of 2011 related to the accelerated vesting of certain share-based awards. General and administrative expenses increased $6.4 million for 2011 primarily due to an increase in salary and benefits expense resulting from an increase in overall headcount during 2011 compared to 2010 as well as an increase in legal expenses related to Silenor paragraph IV litigation. This increase was offset by a decrease in share-based compensation expense due to higher share-based compensation expense during 2010 as a result of vesting of performance-based equity awards upon FDA approval of the NDA for Silenor and the execution of the co-promotion agreement with P&G.
Selling and marketing expenses increased $23.1 million for the year ended December 31, 2010 compared to the same period in 2009 due to the costs associated with the commercial activities and launch of Silenor in 2010. Launch costs included the training and deployment of Somaxon’s sales representatives, sample distribution, and other promotional spending and consulting costs. General and administrative expenses increased $2.6 million for 2010 compared to 2009 primarily due to an increase in salary and benefits expense resulting from an increase in overall headcount in 2010 compared to 2009.
We expect our selling, general and administrative expenses to decrease in 2012 over 2011 levels due to the termination of our agreements with Publicis and P&G as well as a decrease in overall headcount for 2012 compared to 2011. We are unable to estimate with certainty our future selling, general and administrative expenses, in part because we cannot forecast with any degree of certainty the cost of commercial activities, the likelihood of commercial success, and to what degree such activities would affect our capital requirements.
52
Research and Development Expense. Our most significant research and development costs during 2011 were salaries, benefits, and share-based compensation expense related to our research and development personnel. Research and development expense for the years ended December 31, 2011, 2010 and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Personnel and other costs |
$ | 834 | $ | 2,272 | $ | 2,811 | $ | (1,438 | ) | $ | (539 | ) | (63 | )% | (19 | )% | ||||||||||||
| Share-based compensation |
462 | 1,294 | 1,526 | (832 | ) | (232 | ) | (64 | )% | (15 | )% | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total research and development expense |
$ | 1,296 | $ | 3,566 | $ | 4,337 | $ | (2,270 | ) | $ | (771 | ) | (64 | )% | (18 | )% | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Research and development expense decreased $2.3 million for the year ended December 31, 2011 compared to the same period in 2010 primarily due to lower personnel and other costs and share-based compensation expense. Personnel and other costs attributable to research and development personnel decreased primarily due to costs associated with the process validation for the packaging of Silenor which were incurred in 2010. Share-based compensation expense attributable to research and development personnel decreased due to recognition of compensation costs associated with the vesting of performance-based equity awards upon FDA approval of the NDA for Silenor in the first half of 2010 and the execution of the co-promotion agreement with P&G in 2010.
Research and development expense decreased $0.8 million for the year ended December 31, 2010 compared to the same period in 2009 primarily due to lower development expenses and share-based compensation expense. Silenor development expenses decreased because of the lower level of activity relating to both the NDA and non-clinical studies during 2010 as compared to 2009. Share-based compensation was lower in 2010 as 2009 included the additional cost of our one-time stock option exchange program that was completed in June 2009 and the impact of accelerated option vesting arrangements under severance-related agreements.
We are unable to estimate with certainty our future research and development expenses in part because we cannot forecast with any degree of certainty whether we will potentially pursue the development of other product candidates, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
License fees. License fees for the years ended December 31, 2011, 2010 and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| License fees |
$ | — | $ | — | $ | (999 | ) | $ | — | $ | 999 | 0 | % | (100 | )% | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
We had no license fees expense for the years ended December 31, 2011 and 2010. In March 2009, we entered into an agreement with a third party to mutually terminate our license for nalmefene. Pursuant to the termination agreement, the third party paid us a termination fee which we included as a reduction of license fees for the year ended December 31, 2009.
Interest and Other Income. Interest and other income for the years ended December 31, 2011, 2010 and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Interest and other income |
$ | 52 | $ | 262 | $ | 30 | $ | (210 | ) | $ | 232 | (80 | )% | 773 | % | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Interest and other income decreased $0.2 million for the year ended December 31, 2011 compared to the same period in 2010 primarily due to the one time grant of $0.2 million that we were awarded in 2010 under the Qualifying Therapeutic Discovery Project program included in the Patient Protection and Affordable Health Care Act of 2010.
53
Interest and other income increased $0.2 million for the year ended December 31, 2010 compared to the same period in 2009 primarily due to the receipt of this grant in 2010.
Interest and Other Expense. Interest and other expense for the years ended December 31, 2011, 2010 and 2009 are summarized in the following table (in thousands, except percentages).
| Dollar Change | Percent Change | |||||||||||||||||||||||||||
| Years Ended December 31, | 2011 vs. | 2010 vs. | 2011 vs. | 2010 vs. | ||||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2010 | 2009 | 2010 | 2009 | ||||||||||||||||||||||
| Interest and other expense |
$ | (1,940 | ) | $ | (68 | ) | $ | (261 | ) | $ | (1,872 | ) | $ | 193 | 2,753 | % | (74 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Interest and other expense increased $1.9 million for the year ended December 31, 2011 compared to the same period in 2010 primarily due to interest expense incurred in connection with our loan agreement with Oxford Finance Corporation, or Oxford, and Silicon Valley Bank, or SVB, which we entered into in August 2011 and repaid in full in December 2011.
Interest and other expense decreased $0.2 million for the year ended December 31, 2010 compared to the same period in 2009 due to the repayment in full of our debt obligations in March 2009.
Liquidity and Capital Resources
Since inception, our operations have been financed primarily through the sale of equity securities and the proceeds from the exercise of warrants and stock options. We have incurred losses from operations and negative cash flows since our inception, and we expect to continue to incur losses and have negative cash flows from operations in the foreseeable future as we continue our commercial activities for Silenor, commercialize any other products to which we obtain rights and potentially pursue the development of other product candidates. Based on our recurring losses, negative cash flows from operations and working capital levels, we will need to raise substantial additional funds to finance our operations. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected. The report of our independent registered public accounting firm on our financial statements for the year ended December 31, 2011 contains an explanatory paragraph stating that our recurring losses raise substantial doubt about our ability to continue as a going concern.
We are responsible for the costs relating to the sales and marketing of Silenor in the United States. We hired 25 sales representatives from the Publicis sales force, to promote Silenor as Somaxon employees effective January 3, 2012. The efforts of our sales force are complemented by on-line and other non-personal promotional initiatives that target both physicians and patients. We are also focused on ensuring broad patient access to Silenor by negotiating agreements with leading commercial managed care organizations and with government payors. Our commercial activities relating to Silenor are likely to result in the need for substantial additional funds. Our future capital uses and requirements depend on numerous forward-looking factors. These factors include but are not limited to the following:
| • | our success in generating cash flows from sales of Silenor; |
| • | the costs of establishing or contracting for commercial programs and resources, and the scope of the commercial programs and resources we pursue; |
| • | the terms and timing of any future collaborative, licensing and other arrangements that we may establish; |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | the extent to which we acquire or in-license new products, technologies or businesses; |
| • | the rate of progress and cost of any future non-clinical studies, any future clinical trials and other development activities; |
54
| • | the scope, prioritization and number of development programs we pursue; and |
| • | the effect of competing technological and market developments. |
In August 2011, we entered into an at-the-market equity sales agreement, or sales agreement, with Citadel Securities LLC, or Citadel. However, there can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. Citadel or the Company is permitted to terminate the sales agreement at any time. Sales of shares pursuant to the sales agreement will have a dilutive effective on the holdings of our existing stockholders, and may result in downward pressure on the price of our common stock.
As of December 31, 2011, we had $10.7 million in cash and cash equivalents which consist solely of cash. We will need to obtain additional funds to finance our operations. Actual financial results for the period of time through which our financial resources will be adequate to support our operations could vary based upon many factors, including but not limited to Silenor sales performance, the actual cost of commercial activities and any potential litigation expenses we may incur.
In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. The exploration of strategic alternatives may not result in any agreement or transaction and, if completed, any agreement or transaction may not be successful or on attractive terms. The inability to enter into a strategic transaction, or a strategic transaction that is not successful or on attractive terms, could accelerate our needs for cash and make securing funding on reasonable terms more difficult. In addition, if we raise additional funds through collaborations or other strategic transactions, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
In addition to the amounts available under our sales agreement with Citadel, we intend to obtain any additional funding we require through public or private equity or debt financings, strategic relationships, assigning receivables or royalty rights, or other arrangements and we cannot assure such funding will be available on reasonable terms, or at all. Additional equity financing will be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants.
If our efforts in raising additional funds when needed are unsuccessful, we may be required to delay, scale-back or eliminate plans or programs relating to our business, relinquish some or all rights to Silenor or renegotiate less favorable terms with respect to such rights than we would otherwise choose or cease operating as a going concern. In addition, if we do not meet our payment obligations to third parties as they come due, we may be subject to litigation claims. Even if we were successful in defending against these claims, litigation could result in substantial costs and be a distraction to management, and may result in unfavorable results that could further adversely impact our financial condition.
If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our financial statements, and it is likely that investors will lose all or a part of their investments. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We have two effective shelf registration statements on Form S-3 filed with the SEC under which we may offer from time to time any combination of debt securities, common and preferred stock and warrants. However, the rules and regulations of the SEC or other regulatory agencies may restrict our ability to conduct certain types of financing activities, or may affect the timing of and the amounts we can raise by undertaking such activities. For example, under current SEC regulations, because the aggregate market value of our common stock held by non-affiliates, or our public float, is less than $75 million, the amount that we can raise through primary public offerings of securities in any twelve-month period using one or more registration statements on Form S-3 is limited to an aggregate of one-third of our public float. Additional equity financing will be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants. There can be no assurance that we would be successful in selling securities under our shelf registration statements based on prevailing market conditions or in the quantities or at the prices that we deem appropriate.
55
Cash Flows
Net cash used in operations was $49.5 million for 2011 compared to $29.7 million in 2010. The increase in net cash used in operating activities was primarily due to an increase in our net loss in 2011 as compared to 2010.
Net cash provided by investing activities was $32.9 million for 2011 compared to net cash used in investing activities of $35.7 million in 2010. Results for 2011 reflect net sales and maturities of marketable securities of $33.7 million and payments for property and equipment of $0.4 million. Results for 2010 reflect a $1.0 million milestone payment to ProCom under our license agreement which became due as a result of the approval by the FDA of our NDA for Silenor and net purchases of marketable securities of $34.1 million.
Net cash provided by financing activities was $6.2 million for 2011 compared to $81.3 million in 2010. Results for 2011 reflect net proceeds of $14.1 million from the issuance of debt, net proceeds of $5.0 million received from Paladin from the sale by us of 2.2 million shares of our common stock, and net proceeds of $0.5 million from the sale of 786,825 shares of our common stock under our sales agreement with Citadel. This amount was offset by the repayment of debt of $14.1 million. Our 2010 results reflect cash proceeds of $77.6 million from our public offerings and proceeds of $3.8 million from the exercise of warrants and stock options.
Loan Agreement
In July 2011, we terminated our existing loan agreement with Comerica Bank and in August 2011, we entered into a new loan and security agreement, or the loan agreement, with Oxford and SVB under which we borrowed $15.0 million at an interest rate of 7.5% per annum. We were obligated to pay interest on the loan through December 31, 2011, and to thereafter pay the principal balance of the loan and interest in 24 equal monthly installments through December 31, 2013. As part of the financing, Oxford and SVB each received warrants to purchase up to an aggregate of 583,152 fully paid and non-assessable shares of our common stock at an exercise price equal to $1.5433 per share. The warrants will expire ten years from the date of the grant. At our option, we could prepay all or any part of the outstanding principal balance, subject to a pre-payment fee of $150,000. We paid to Oxford and SVB an initial fee of $150,000 upon the closing of the loan agreement. At the time of repayment, we were also obligated to make an additional final payment of $638,000.
In connection with the loan agreement, we granted Oxford and SVB a security interest in substantially all of our personal property then owned or thereafter acquired, excluding intellectual property. Under the loan agreement, we were subject to certain affirmative and negative covenants, including limitations on our ability to: undergo certain change of control events; convey, sell, lease, license, transfer or otherwise dispose of assets, other than in certain specified circumstances; create, incur, assume, guarantee or be liable with respect to certain indebtedness; grant liens; pay dividends and make certain other restricted payments; and make certain investments. In addition, under the loan agreement, subject to certain exceptions, we were required to maintain with SVB our primary cash and investment accounts, which accounts were also covered by control agreements for the benefit of Oxford and SVB. Prior to repayment of the loan, we met all of our obligations under the Loan Agreement.
We repaid the loan in full in December 2011. Upon such repayment of the loan in December 2011, we paid to Oxford and SVB $500,000 as partial payment of the prepayment and final payment fees required under the loan agreement. Oxford and SVB also accepted warrants to purchase up to an aggregate of 150,000 shares of our common stock at an exercise price equal to $0.50 per share in full satisfaction of the remaining prepayment and final payment fees. The warrants expire ten years from the date of grant. We no longer have any obligations under the loan agreement, and there are no further encumbrances on our assets under the loan agreement.
Equity Sales Agreement
In August 2011, we entered into the sales agreement with Citadel pursuant to which we agreed to sell, at our option, up to an aggregate of $30.0 million in shares of our common stock through Citadel, as sales agent. Sales of the common stock made pursuant to the sales agreement, if any, will be made on the NASDAQ Stock Market under our currently-effective Registration Statements on Form S-3 by means of ordinary brokers’ transactions at market prices. Additionally, under the terms of the sales agreement, we may also sell shares of our common stock through Citadel, on the NASDAQ Stock Market or otherwise, at negotiated prices or at prices related to the prevailing
56
market price. Under the terms of the sales agreement, we may also sell shares to Citadel as principal for Citadel’s own account at a price agreed upon at the time of sale pursuant to a separate terms agreement to be entered into with Citadel at such time. We pay Citadel a commission equal to 3% of the gross proceeds from the sale of shares of our common stock under the sales agreement. We or Citadel may terminate the sales agreement at any time. The offering of common stock pursuant to the sales agreement will terminate upon the earlier of (a) the sale of all of the common stock subject to the sales agreement or (b) the termination of the sales agreement by us or Citadel. There can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate.
We will not be able to make sales of our common stock pursuant to the sales agreement unless certain conditions are met, which include the accuracy of representations and warranties made to Citadel under the sales agreement; compliance with laws; and the continued listing of our stock on the Nasdaq Capital Market. On December 13, 2011, we received a letter from the Listing Qualifications Department of the Nasdaq Stock Market, or Nasdaq, informing us that because the closing bid price of our common stock listed on Nasdaq was below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2). If compliance is not demonstrated within the applicable timeframe, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market. During the year ended December 31, 2011, we sold an aggregate of 786,825 shares of our common stock under the sales agreement, received gross proceeds of $0.8 million, and paid $0.3 million of legal and accounting fees associated with the execution of the sales agreement and commissions.
Litigation
We have received notices from Actavis LLC and Actavis Inc., or collectively, Actavis, Mylan Pharmaceuticals Inc. and Mylan, Inc., or collectively Mylan, Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc., or collectively, Par, and Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Limited (d/b/a Zydus Cadila), or collectively, Zydus, that each has filed with the FDA an ANDA for a generic version of Silenor 3 mg and 6 mg tablets. The notices included paragraph IV certifications with respect to eight of the nine patents listed in the Orange Book for Silenor.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed U.S. Patent No. 6,211,229, or the ‘229 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed U.S. Patent No. 7,915,307, or the ’307 patent by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action. At this time, the other patents included in the Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but we cannot predict the outcome of these matters.
The prosecution of the lawsuits against Actavis, Mylan, Par and Zydus will increase our cash expenditures. Any adverse outcome in this litigation could result in one or more generic versions of Silenor being launched before the expiration of the listed patents, which could adversely affect our ability to successfully execute our business strategy to generate sales of Silenor and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows, such events could also significantly impact our ability to coutinue as a going concern.
Contractual Obligations
The following table describes our commitments to settle contractual obligations in cash as of December 31, 2011 (in thousands):
| Payments Due By Period | ||||||||||||||||||||
| 2012 | 2013 through 2014 |
2015 through 2016 |
After 2017 |
Total | ||||||||||||||||
| Non-cancellable purchase orders |
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Other contractual obligations |
150 | 100 | — | — | 250 | |||||||||||||||
| Operating lease obligations |
236 | 769 | 813 | — | 1,818 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 386 | $ | 869 | $ | 813 | $ | — | $ | 2,068 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| * | There can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. |
57
We entered into a license agreement with ProCom to acquire the rights to develop and commercialize Silenor. Pursuant to this agreement, we obtained exclusive, sub-licensable rights to the patents and know-how for certain indications. This license agreement required us to pay an upfront payment as well as additional payments upon the achievement of specific development and regulatory approval milestones. We are also obligated to pay royalties under the agreement until the expiration of the applicable patents. The royalty payments are recognized as an expense when the related shipment is recognized as revenue. Future royalty payments are not included in the table above because we cannot reasonably estimate them at this time.
In May 2011, we entered into a lease arrangement to rent approximately 12,100 square feet of office space, which we use as our corporate headquarters. The lease commenced on August 25, 2011. The lease will expire on December 24, 2016, and we will have the option to extend the term for an additional five years at the then-current fair market rental rate (as defined in the lease). We have paid the first month’s rent of approximately $30,000 and the monthly rent following lease commencement is approximately $30,000. However, the second through thirteenth month’s rent will be abated by one-half provided that we are not in default of the lease. After the first year, the monthly rent will increase by 3.5% per year, and we will provide reimbursements for our proportional share of any increases in operating expenses over the first year’s operating expenses (as defined in the lease). We also have a $200,000 letter of credit in favor of our landlord to secure our obligations under the lease.
In September 2011, we provided notice of termination to P&G of the co-promotion agreement, with such termination effective as of December 31, 2011. As a result of such termination, P&G is entitled to a low single digit royalty on net sales of Silenor for the 2012 fiscal year. Future royalty payments are not included in the table above because we cannot reasonably estimate them at this time.
We have contracted with various consultants and other vendors to assist in clinical trial work, pre-clinical studies, data analysis, and activities to support the marketing of Silenor. The contracts are generally terminable at any time, but obligate us to reimburse the providers for any time or costs incurred through the date of termination. We also have employment agreements with each of our current executive officers that provide for severance payments and accelerated vesting for certain share-based awards if their employment with us is terminated under specified circumstances.
Off-Balance Sheet Arrangements
We do not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Recent Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update , or ASU, No. 2009-13 “Revenue Recognition,” which provides guidance on recognizing revenue in arrangements with multiple deliverables. This standard impacts the determination of when the individual deliverables included in a multiple element arrangement may be treated as a separate unit of accounting. It also modifies the manner in which the consideration received from the transaction is allocated to the multiple deliverables and no longer permits the use of the residual method of allocating arrangement consideration. This accounting standard was effective for the first year beginning on or after June 15, 2010, with early adoption permitted. The adoption of ASU 2009-13, which occurred on January 1, 2011, did not have a material impact on our financial statements.
In December 2010, the FASB issued ASU No. 2010-27 “Other Expenses: Fees Paid to the Federal Government by Pharmaceutical Manufacturers,” which provides guidance concerning the recognition and classification of the annual fee payable by branded prescription drug manufacturers and importers on branded prescription drugs which was mandated under the health care reform legislation enacted in the United States in March 2010. Under this new authoritative guidance, the annual fee should be estimated and recognized in full as a liability upon the first qualifying commercial sale with a corresponding deferred cost that is amortized to operating expenses using a
58
straight-line method unless another method better allocates the fee over the calendar year in which it is payable. This new guidance was effective for calendar years beginning on or after December 31, 2010, when the fee initially becomes effective. The adoption of ASU 2010-27, which occurred on January 1, 2011, did not have a material impact on our financial statements.
In May 2011, the FASB issued ASU No. 2011-04 “Fair Value Measurement: Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. GAAP and IFRSs”. Some of the amendments clarify the FASB’s intent about the application of existing fair value measurement requirements. Other amendments change a particular principle or requirement for measuring fair value or for disclosing information about fair value measurements. This guidance is effective for interim and annual periods beginning after December 15, 2011. We are still evaluating the potential future effects of this guidance.
In June 2011, the FASB issued ASU No. 2011-05 “Comprehensive Income: Presentation of Comprehensive Income.” Under the new guidance, an entity has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. This guidance is effective for interim and annual periods beginning after December 15, 2011. We have evaluated the potential future effects of this guidance and do not expect the adoption of ASU 2011-05 to have a material impact on our financial statements.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
While our cash and cash equivalents at December 31, 2011 consisted primarily of cash, the primary objective of our investment activities, if any, is to preserve principal while maximizing the income we receive from our investments without significantly increasing risk. Historically, our primary exposure to market risk has been interest rate sensitivity. This means that a change in prevailing interest rates may cause the value of the investment to fluctuate. For example, if we purchase a security that was issued with a fixed interest rate and the prevailing interest rate later rises, the value of our investment will probably decline. Currently, our holdings are in cash, and therefore this interest rate risk is minimal. To minimize our interest rate risk going forward, we intend to continue to maintain our holdings in cash. If our cash balance increased significantly given our cash needs, we may also invest in cash equivalents and marketable securities such as money market funds and United States government debt securities, all with various maturities. In general, money market funds are not subject to market risk because the interest paid on such funds fluctuates with the prevailing interest rate. We also generally time the maturities of our investments to correspond with our expected cash needs, allowing us to avoid realizing any potential losses from having to sell securities prior to their maturities.
When our cash is invested, it is invested in accordance with a policy approved by our board of directors which specifies the categories, allocations, and ratings of securities we may consider for investment. We do not believe our cash and cash equivalents will have significant risk of default or illiquidity. While we intend that any future portfolio of cash, cash equivalents and short-term investments will be well diversified, we cannot provide absolute assurance that our investments will not be subject to future adverse changes in market value.
Item 8. Financial Statements and Supplementary Data
See the list of financial statements filed with this report under Part IV — Item 15 below.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Securities Exchange Act of 1934, as amended, or the Exchange Act, reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s, or SEC,
59
rules and forms and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(b), we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2011.
Management’s Report on Internal Control over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes those policies and procedures that: (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Management is responsible for establishing and maintaining adequate internal control over our financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting. Management has used the framework set forth in the report entitled “Internal Control—Integrated Framework” published by the Committee of Sponsoring Organizations of the Treadway Commission to evaluate the effectiveness of our internal control over financial reporting. Based on its evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2011, the end of our most recent fiscal year. The effectiveness of internal control over financial reporting as of December 31, 2011 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm.
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
60
Not applicable.
Item 10. Directors, Executive Officers and Corporate Governance
Board of Directors
At March 3, 2012, our board of directors consisted of the following members:
| Name |
Age | Position with the Company | ||
| David F. Hale |
63 | Chairman of the Board | ||
| Richard W. Pascoe |
48 | Director, President and Chief Executive Officer | ||
| Erle T. Mast |
49 | Director, Chairman of the Audit Committee | ||
| Kurt von Emster |
44 | Director, Chairman of the Nominating/Corporate Governance Committee | ||
| Terrell A. Cobb |
62 | Director | ||
| Michael L. Eagle |
64 | Director | ||
| Faheem Hasnain |
53 | Director | ||
| Thomas G. Wiggans |
60 | Director, Chairman of the Compensation Committee |
David F. Hale is one of our co-founders and has served as Chairman of the Board of Directors since our founding in August 2003. He also served as our interim Chief Executive Officer from January 2008 until August 2008. Mr. Hale has served as Executive Chairman of Biocept, Inc., a laboratory services company, since March 2011. He has served as Chairman and Chief Executive Officer of Hale BioPharma Ventures since May 2006. Mr. Hale served as President and Chief Executive Officer of CancerVax Corporation, a biotechnology company, from October 2000 until it merged with Micromet AG in 2006. He served as a director of CancerVax from December 2000 until the merger with Micromet Inc., and he served as Chairman of Micromet’s board of directors until Micromet’s acquisition by Amgen in March 2012. Prior to joining CancerVax, he was President and Chief Executive Officer of Women First HealthCare, Inc., a specialty pharmaceutical company, from January 1998 to May 2000. Mr. Hale served as President, Chief Executive Officer and Chairman of Gensia Inc., a pharmaceutical company which became Gensia Sicor, from May 1987 to November 1997. Prior to joining Gensia, Mr. Hale was President, Chief Operating Officer and Chief Executive Officer of Hybritech Inc. Mr. Hale serves as Chairman of the Board of Santarus, Inc. and the privately held companies SkinMedica, Inc., Neurelis, Inc. Ridge Diagnostics, Automedics, Inc., Advantar Laboratories, Crisi Medical Systems and Intrepid Therapeutics, Inc. He also was a co-founder and serves as a member of the Board of Directors of Conatus Pharmaceuticals, Inc., and served on the Board of Directors of Verus Pharmaceuticals, Inc. Mr. Hale was formerly a director of Metabasis Therapeutics, Inc. from 1998 through January 2010, including its Chairman of the Board from 2006 through January 2010.
Mr. Hale is a member of the boards of directors of industry organizations including BIOCOM/San Diego and the Biotechnology Industry Organization (BIO), is a member of the board of directors of Rady Children’s Hospital and Health Center and was Chairman of CONNECT from July 2004 to January 2011. Mr. Hale received a B.A. degree in Biology and Chemistry from Jacksonville State University. Mr. Hale’s depth and diversity of experience on boards of directors and in senior management of public and private specialty pharmaceutical companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Hale should serve as a director of the company and as its non-executive chairman of the board.
Richard W. Pascoe joined as our President and Chief Executive Officer in August 2008. Before joining us, Mr. Pascoe was the Chief Operating Officer at ARIAD Pharmaceuticals, an emerging oncology company, where he led commercial operations, manufacturing, information services, program and alliance management and business development. Mr. Pascoe held a series of senior management roles at King Pharmaceuticals, Inc., including senior vice president of neuroscience marketing and sales and vice president positions in both international sales and marketing and hospital sales. He also held positions in the commercial groups at Medco Research, Inc. (which was acquired by King), COR Therapeutics, Inc. (where he helped lead the successful launch of eptifibatide [Integrilin®]), B. Braun Interventional and the BOC Group. Mr. Pascoe served as a commissioned officer in the United States
61
Army following his graduation from the United States Military Academy at West Point where he received a B.S degree in Leadership. Mr. Pascoe’s appointment as our President and Chief Executive Officer and the board’s belief that the company’s chief executive officer should serve on the board, as well as Mr. Pascoe’s depth and diversity of experience in senior management of public specialty pharmaceutical companies and his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Pascoe should serve as a director of the company.
Erle T. Mast has served on our board of directors since June 2008. Mr. Mast currently serves as Executive Vice President and Chief Financial Officer and is a co-founder of Clovis Oncology, Inc., a biopharmaceutical company. Previously, Mr. Mast was Chief Financial Officer of Pharmion Corporation from 2002 until its acquisition by Celgene Corporation in March 2008. Mr. Mast was also an Executive Vice President of Pharmion from February 2007 until the acquisition. He was Vice President of Finance for Dura Pharmaceuticals, Inc. from 1997 until its acquisition by Elan Corporation, plc in 2000, and thereafter he was Chief Financial Officer of Elan’s Global Biopharmaceuticals business until 2002. Prior to that, Mr. Mast was a partner with Deloitte & Touche, LLP. Mr. Mast has been a director of Zogenix, Inc. since 2008 and was a director of Verus Pharmaceuticals, Inc. from 2007 to 2009. Mr. Mast graduated from California State University, Bakersfield with a degree in business administration. Mr. Mast’s depth and diversity of experience on boards of directors and in senior management of public and private specialty pharmaceutical companies, including his specific experience and skills that qualify Mr. Mast to be our “audit committee financial expert” as that term is defined in the rules and regulations established by the SEC, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Mast should serve as a director of the company.
Kurt von Emster, CFA has served as a member of our board of directors since August 2005. Mr. von Emster is currently Managing Director of venBio LLC. From November 2000 through February 2009, Mr. von Emster was a General Partner and Portfolio Manager for the MPM BioEquities Fund. Prior to joining MPM, Mr. von Emster spent 11 years with Franklin Templeton Group as a Vice President and Portfolio Manager where he managed over $2 billion in health and biotech funds. In his tenure at Franklin, Mr. von Emster was responsible for building the health care group and was responsible for conceiving and developing seven different life science investment products for Franklin. Mr. von Emster holds the Chartered Financial Analyst designation (CFA), is a member of the Association for Investment Management and Research and is a member of the Security Analysts of San Francisco. Mr. von Emster currently serves on the board of directors of Metabolex, a private drug development company. Mr. von Emster is a past director of Facet Biotech Corporation, a public biotech company sold to Abbott Labs in 2010 and past Board Observer at Acceleron Pharmaceuticals, a private biotechnology company. He has a degree from the University of California at Santa Barbara in Business and Economics and in 2010, completed the Director College Executive Education Program at the Rock Center for Corporate Governance at Stanford University. Mr. von Emster’s depth and diversity of experience on boards of directors of public and private biotechnology companies and in evaluating and investing in biotech companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. von Emster should serve as a director of the company.
Terrell A. Cobb has served as a member of our board of directors since August 2003. Mr. Cobb is the founder and currently serves as President of ProCom One, a drug development company, a position he has held since 1998. We license Silenor from ProCom One, Inc. From 1995 to the present, Mr. Cobb has served as a consultant focusing on business development activities in the pharmaceutical industry. Mr. Cobb previously spent 15 years in various positions at Johnson and Johnson. Mr. Cobb has founded four specialty pharmaceutical companies, has held senior management positions in several start-up organizations, including Pharmaco and Scandipharm, and has acted as an advisor and consultant to other drug development companies. He received a B.A. degree from Mercer University in Chemistry and Psychology. Mr. Cobb’s depth and diversity of experience on boards of directors and in senior management of public and private specialty pharmaceutical companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Cobb should serve as a director of the company. In addition, ProCom One has the right to designate one member of our board of directors, and ProCom One has designated Mr. Cobb to serve in such role.
Michael L. Eagle has served on our board of directors since May 2007. Mr. Eagle served as Vice President of Manufacturing for Eli Lilly and Company from 1994 through 2001 and held a number of executive management positions with Eli Lilly and its subsidiaries throughout his career there. Since retiring from Eli Lilly, he has served as a founding member of Barnard Life Sciences, LLC. Mr. Eagle earned his B. S. degree in mechanical engineering from Kettering University and an M.B.A. from the Krannert School of Management at Purdue University. He serves on the board of directors of Cadence Pharmaceuticals and Hansen Medical, Inc., and on the Board of Trustees of the La Jolla Playhouse and Futures for Children. Mr. Eagle’s depth and diversity of experience on boards of directors and in senior management of public and private pharmaceutical and medical device companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Eagle should serve as a director of the company.
62
Faheem Hasnain has served on our board of directors since September 2010. Mr. Hasnain is currently president, chief executive officer and a director of Receptos, Inc., a position he has held since December 2010. Previously, Mr. Hasnain was president, chief executive officer and a director of Facet Biotech Corporation from December 2008 until its acquisition by Abbott Laboratories in April 2010. Mr. Hasnain was president, chief executive officer and a director of PDL BioPharma, Inc. from October 2008 until Facet Biotech was spun off from PDL BioPharma in December 2008. From October 2004 to September 2008, Mr. Hasnain served at Biogen Idec Inc., most recently as executive vice president in charge of the oncology/rheumatology strategic business unit. Prior to Biogen Idec, Mr. Hasnain was president of Oncology Therapeutics Network, a subsidiary of Bristol-Myers Squibb Company, from March 2002 to September 2004. From 2000 to 2002, Mr. Hasnain served as vice president, global eBusiness, at GlaxoSmithKline and, from 1988 to 2000, he served in key commercial and entrepreneurial roles within GlaxoSmithKline and its predecessor organizations, spanning global eBusiness, international commercial operations, sales and marketing. Mr. Hasnain has been chairman of the board of Ambit Biosciences Corporation since November 2010. Mr. Hasnain received a B.H.K. and B.Ed. from the University of Windsor Ontario in Canada. Mr. Hasnain’s depth and diversity of experience on boards of directors and in senior management of public and private specialty and non-specialty pharmaceutical companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Hasnain should serve as a director of the company.
Thomas G. Wiggans has served on our board of directors since June 2008. Mr. Wiggans serves as Chief Executive Officer of Dermira, a company he co-founded in 2010. Mr. Wiggans served as Chief Executive Officer of Peplin, Inc. from August 2008 until Peplin’s acquisition by LEO Pharma A/S in November 2009, and as Chairman of Peplin’s Board of Directors from October 2007 until such acquisition. Mr. Wiggans served as Chief Executive Officer of Connetics Corporation from 1994 until December 2006 when Connetics was acquired by Stiefel Laboratories, Inc. Mr. Wiggans was also Chairman of the Board of Connetics from January 2006 until the acquisition. From 1992 to 1994, Mr. Wiggans served as President and Chief Operating Officer of CytoTherapeutics Inc. He served in various positions at Ares-Serono Group from 1980 to 1992, including President of its U.S. pharmaceutical operations and Managing Director of its U.K. pharmaceutical operations. Mr. Wiggans currently serves on the boards of directors of Sangamo Biosciences, Inc., Onyx Pharmaceuticals, Inc. and Lithera Inc. He is also on the Board of Trustees of the University of Kansas Endowment Association. In addition, Mr. Wiggans is Chairman of the Biotechnology Institute, a non-profit educational organization. Mr. Wiggans holds a B.S. in Pharmacy from the University of Kansas and an M.B.A. from Southern Methodist University. Mr. Wiggans’ depth and diversity of experience on boards of directors and in senior management of public and private specialty pharmaceutical companies, as well as his personal and professional integrity, ethics and values, led the board of directors to conclude that Mr. Wiggans should serve as a director of the company.
63
Director Nomination Process and Other Corporate Governance Matters
Director Qualifications
In evaluating director nominees, the nominating/corporate governance committee considers the following factors:
| • | personal and professional integrity, ethics and values; |
| • | experience in corporate management, such as serving as an officer or former officer of a publicly held company; |
| • | experience in our industry and with relevant social policy concerns; |
| • | experience as a board member of another publicly held company; |
| • | diversity of expertise and experience in substantive matters pertaining to our business relative to other board members; and |
| • | practical and mature business judgment. |
The nominating/corporate governance committee’s goal is to assemble a board of directors that brings to our company a variety of perspectives and skills derived from high quality business and professional experience.
Other than the foregoing, there are no stated minimum criteria for director nominees, although the nominating/corporate governance committee may also consider such other factors as it may deem to be in the best interests of our company and our stockholders. The nominating/corporate governance committee does, however, believe it appropriate for at least one, and preferably, several, members of our board of directors to meet the criteria for an “audit committee financial expert” as defined by SEC rules, and that a majority of the members of our board of directors meet the definition of “independent director” under the Nasdaq Stock Market qualification standards. The nominating/corporate governance committee also believes it appropriate for our President and Chief Executive Officer to serve as a member of our board of directors.
Identification and Evaluation of Nominees for Directors
The nominating/corporate governance committee identifies nominees for director by first evaluating the current members of our board of directors willing to continue in service. Current members with qualifications and skills that are consistent with the nominating/corporate governance committee’s criteria for board service and who are willing to continue in service are considered for re-nomination, balancing the value of continuity of service by existing members of our board of directors with that of obtaining a new perspective.
If any member of our board of directors does not wish to continue in service or if our board of directors decides not to re-nominate a member for re-election, the nominating/corporate governance committee identifies the desired skills and experience of a new nominee in light of the criteria above. The nominating/corporate governance committee generally polls our board of directors and members of management for their recommendations. The nominating/corporate governance committee may also review the composition and qualification of the boards of directors of our competitors, and may seek input from industry experts or analysts. The nominating/corporate governance committee reviews the qualifications, experience and background of the candidates. In making its determinations, the nominating/corporate governance committee evaluates each individual in the context of our board of directors as a whole, with the objective of assembling a group that can best perpetuate the success of our company and represent stockholder interests through the exercise of sound business judgment. After review and deliberation of all feedback and data, the nominating/corporate governance committee makes its recommendation to our board of directors. Historically, the nominating/corporate governance committee has not relied on third-party search firms to identify director candidates. The nominating/corporate governance committee may in the future choose to do so in those situations where particular qualifications are required or where existing contacts are not sufficient to identify an appropriate candidate.
64
We have not received director candidate recommendations from our stockholders, and we do not have a formal policy regarding consideration of such recommendations. However, any recommendations received from stockholders will be evaluated in the same manner that potential nominees suggested by board members, management or other parties are evaluated.
Board Leadership and Risk Oversight
We believe it is the chief executive officer’s responsibility to manage the company and the chairman’s responsibility to lead the board. As directors continue to have more oversight responsibilities than ever before, we believe it is beneficial to have an independent chairman whose sole job with respect to us is leading the board. By having another director serve as chairman of the board, Mr. Pascoe is able to focus his entire energy on managing the company.
We believe our chief executive officer and our chairman have an excellent working relationship. By clearly delineating the role of the chairman position in our corporate governance guidelines, we ensure there is no duplication of effort between the chief executive officer and the chairman. We believe this provides strong leadership for our board, while also positioning Mr. Pascoe as the leader of the company in the eyes of our collaborators, vendors, employees and other stakeholders.
Our board has five independent members and three non-independent members. A number of our independent board members are currently serving or have served as members of senior management of other public companies and have served as directors of other public companies. We have three board committees comprised solely of independent directors, each with a different independent director serving as chair of the committee. We believe that the number of independent, experienced directors that make up our board, along with the independent oversight of the board by the non-executive chairman, benefits our company and our stockholders.
Our audit committee is primarily responsible for overseeing the company’s risk management processes on behalf of the full board. The audit committee receives reports from management at least quarterly regarding the company’s assessment of risks. In addition, the audit committee reports regularly to the full board of directors, which also considers the company’s risk profile. The audit committee and the full board of directors focus on the most significant risks facing the company’s business and the company’s general risk management strategy, and also ensure that risks undertaken by the company are consistent with the board’s appetite for risk. While the board oversees the company’s risk management, company management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks facing our company and that our board leadership structure supports this approach.
Pursuant to our bylaws and our corporate governance guidelines, our board determines the best board leadership structure for our company from time to time. As part of our annual board self-evaluation process, we evaluate our leadership structure to ensure that the board continues to believe that it provides the optimal structure for our company and stockholders. We recognize that different board leadership structures may be appropriate for companies in different situations. We believe our current leadership structure, with Mr. Pascoe serving as chief executive officer and Mr. Hale serving as chairman of the board, is the optimal structure for our company at this time.
Audit Committee and Financial Expert
Our audit committee currently consists of Mr. Mast (chair), Mr. Eagle, and Mr. von Emster, each of whom our board of directors has determined is independent within the meaning of the independent directors standards of the SEC and the Nasdaq Stock Market, Inc. Our board of directors has determined that Mr. Mast qualifies as an “audit committee financial expert” as that term is defined in the rules and regulations established by the SEC.
65
Audit Committee Report
The audit committee oversees our financial reporting process on behalf of our board of directors. Management has the primary responsibility for the financial statements and the reporting process, including the systems of internal controls. In fulfilling its oversight responsibilities, the audit committee reviewed the audited financial statements in our annual report with management, including a discussion of any significant changes in the selection or application of accounting principles, the reasonableness of significant judgments, the clarity of disclosures in the financial statements and the effect of any new accounting initiatives.
The audit committee reviewed with PricewaterhouseCoopers LLP, who are responsible for expressing an opinion on the conformity of these audited financial statements with generally accepted accounting principles, their judgments as to the quality, not just the acceptability, of our accounting principles and such other matters as are required to be discussed with the audit committee under generally accepted auditing standards, including the Statement on Auditing Standards No. 114, The Auditor’s Communication With Those Charged With Governance. In addition, the audit committee has discussed with PricewaterhouseCoopers LLP their independence from management and our company, has received from PricewaterhouseCoopers LLP the written disclosures and the letter required by the Public Company Accounting Oversight Board, Rule 3526 Communication with Audit Committees Concerning Independence, and has considered the compatibility of non-audit services with the independence of our registered public accounting firm.
The audit committee met with PricewaterhouseCoopers LLP to discuss the overall scope of their audit. The meetings with PricewaterhouseCoopers LLP were held, with and without management present, to discuss the results of their examination, their comments on our internal controls and the overall quality of our financial reporting.
Based on the reviews and discussions referred to above, the audit committee has recommended to our board of directors that the audited financial statements be included in our annual report for the year ended December 31, 2011.
This Audit Committee Report is furnished solely with this report, and is not filed with this report, and shall not be deemed incorporated by reference into any other filing under the Securities Act of 1933, as amended, or the Securities Act, or the Securities Exchange Act of 1934, as amended, or the Exchange Act, whether made by us before or after the date hereof, regardless of any general incorporation language in any such filing, except to the extent we specifically incorporate this material by reference into any such filing.
The foregoing report has been furnished by the audit committee.
Erle T. Mast, Chairman
Michael L. Eagle
Kurt von Emster
Executive Officers
At March 3, 2012, our executive officers consisted of the following:
| Name |
Age | Position with the Company | ||
| Richard W. Pascoe |
48 | President and Chief Executive Officer | ||
| Tran B. Nguyen |
38 | Senior Vice President and Chief Financial Officer | ||
| Brian T. Dorsey |
43 | Senior Vice President, Technical Operations | ||
| Matthew W. Onaitis |
41 | Senior Vice President, General Counsel and Secretary |
See “Board of Directors” for the biography of Mr. Pascoe.
66
Tran B. Nguyen has served as our Vice President and Chief Financial Officer since April 2010 and as our Senior Vice President and Chief Financial Officer since February 2011. Mr. Nguyen brings to the Company more than 10 years of finance experience primarily focused in the life science industry. Previously, Mr. Nguyen was Vice President and Chief Financial Officer at Metabasis Therapeutics, Inc., a biopharmaceutical company, where he was responsible for managing all finance and accounting activities, and played a significant role in strategic and operating decisions from March 2009 until the company was sold to Ligand Pharmaceuticals Incorporated in January 2010. Prior to joining Metabasis, Mr. Nguyen was a Vice President in the Healthcare Investment Banking group at Citi Global Markets, Inc. from May 2007 until January 2009, where he was responsible for senior and junior relationship management of small-to-large-cap biotechnology and specialty pharma companies on the West Coast. Mr. Nguyen served in the Healthcare Investment Banking group at Lehman Brothers, Inc. as a Vice President from January 2006 until April 2007, and as an associate from July 2004 until December 2005 where he was responsible for executing various transactions including equity, equity-linked, debt, and mergers and acquisitions for small-to-large-cap biotechnology and specialty pharma companies. Mr. Nguyen received a B.A. in Economics and Psychology from Claremont McKenna College, and an M.B.A. from the Anderson School of Management at U.C.L.A.
Brian T. Dorsey joined us as Executive Director, Manufacturing and Program Management in March 2005. He was later promoted to Vice President, Manufacturing and Program Management in November 2006, named Vice President, Product Development in January 2007 and named Senior Vice President, Technical Operations in April 2010. From April 2002 to March 2005, Mr. Dorsey served as Head of Project Management, Medical Writing and Library Services at Maxim Pharmaceuticals Inc., a biopharmaceutical company. From May 2001 to April 2002, Mr. Dorsey served as Director, Head of Biopharmaceutical Project Management at Baxter Bioscience, a division of Baxter Healthcare Corporation. Previously, Mr. Dorsey served as a Global Project Leader / Project Director at Pfizer Global Research and Development (Agouron). Mr. Dorsey received his B.S. in chemistry and his Masters degree in executive leadership, both from the University of San Diego.
Matthew W. Onaitis joined us as Vice President, Legal Affairs and Secretary in May 2006. He was later promoted to Vice President, General Counsel and Secretary in January 2007 and named Senior Vice President, General Counsel and Secretary in April 2010. From January 2006 to May 2006, Mr. Onaitis served as Associate General Counsel at Biogen Idec Inc., a biopharmaceutical company. From June 2004 to December 2005, Mr. Onaitis was Director, Legal Affairs at Elan Corporation plc, a biopharmaceutical company. Mr. Onaitis practiced corporate and commercial law in private practice from 1998 to June 2004, which included a secondment to Elan from October 2003 to June 2004. Mr. Onaitis holds a J.D. from Stanford Law School and a B.S. in mechanical engineering from Carnegie Mellon University.
Section 16(a) Beneficial Ownership Reporting Compliance
Under Section 16(a) of the Exchange Act, directors, officers and beneficial owners of 10% or more of our common stock are required to file with the SEC on a timely basis initial reports of beneficial ownership and reports of changes regarding their beneficial ownership of our common stock. Officers, directors and 10% beneficial owners are required by SEC regulations to furnish us with copies of all Section 16(a) forms that they file.
Based solely on our review of the copies of such forms received and the written representations from certain reporting persons, we have determined that no officer, director or 10% beneficial owner known to us was delinquent with respect to their reporting obligations as set forth in Section 16(a) of the Exchange Act during the fiscal year ended December 31, 2011.
67
Code of Ethics
We have established a Code of Business Conduct and Ethics that applies to our officers, directors and employees which is available on our internet website at www.somaxon.com. The Code of Ethics contains general guidelines for conducting the business of our company consistent with the highest standards of business ethics, and is intended to qualify as a “code of ethics” within the meaning of Section 406 of the Sarbanes-Oxley Act of 2003 and Item 406 of Regulation S-K.
Item 11. Executive Compensation
Compensation Discussion and Analysis
Executive Summary
This Compensation Discussion and Analysis describes our overall compensation policies and practices and specifically analyzes the total compensation for the following executive officers, also referred to herein as our Named Executive Officers:
| • | Richard W. Pascoe, President, Chief Executive Officer and Director, |
| • | Tran B. Nguyen, Senior Vice President and Chief Financial Officer, |
| • | Brian T. Dorsey, Senior Vice President, Technical Operations, |
| • | Matthew W. Onaitis, Senior Vice President and General Counsel, and |
| • | Jeffrey W. Raser, our former Senior Vice President and Chief Commercial Officer. |
All of our compensation programs are designed to attract and retain key employees and to motivate them to achieve key strategic performance measures to create long-term shareholder value. With respect to the compensation of our Named Executive Officers, we believe that their compensation should largely reflect their success as a management team in meeting key corporate objectives, rather than as individual contributors. As a result, we believe that the performance of the executives in managing our company, considered in light of general economic and specific company, industry and competitive conditions, should be the main basis for determining their overall compensation.
Overview of Total Compensation and Process
Elements of total compensation for our Named Executive Officers include:
| • | base salary, which is initially negotiated with the executive when the executive is hired and is reviewed by the compensation committee annually, |
| • | annual cash bonus awards, which reward annual company performance based on pre-determined objectives, |
| • | performance-based equity awards, which reward company performance based on pre-determined criteria for key corporate objectives, |
| • | time based equity awards, which reward company performance as reflected in its stock price over time, and |
| • | other benefits, such as health insurance benefits. |
Each of these is described in more detail below.
68
The compensation committee has the primary authority to determine our company’s compensation philosophy and to establish compensation for our Named Executive Officers. Each year, generally in the first quarter, the compensation committee, which consists entirely of independent members of our board of directors, reviews the performance of each of our Named Executive Officers during the previous year. In connection with this review, the compensation committee typically reviews and resets base salaries for our Named Executive Officers, determines their annual cash bonuses relating to prior year performance, approves elements of the incentive bonus plan for the current year, including target bonuses and corporate objectives, and grants equity awards to our Named Executive Officers and certain other eligible employees. The compensation committee also has the discretion to make adjustments to executive compensation at other times during the year.
In making these compensation decisions, it has been the practice of our compensation committee to review the historical levels of each element of each Named Executive Officer’s total compensation (salary, bonus, equity awards and other benefits) and to compare each element with that of the executive officers in an appropriate group of comparable specialty pharmaceutical companies.
With respect to the compensation committee’s executive compensation review in early 2011, the committee authorized management to engage Barney & Barney Compensation Consulting, or Barney & Barney, to perform an assessment of our group of comparable specialty pharmaceutical companies. Our management reviewed with Barney & Barney the comparison group that we used in 2010, and this review resulted in a new comparison group for 2011 that possessed the following characteristics at the time of selection:
| • | market capitalizations between $100 and $500 million, |
| • | most advanced product in a pivotal clinical trial, NDA filed, FDA approved but awaiting launch, or marketed, |
| • | less than 250 employees, and |
| • | a reasonable expectation that we could compete with these companies to fill senior management positions. |
69
The compensation committee then approved the companies comprising the comparison group. The companies in the group were:
The changes made to the selection criteria for the 2011 comparison group reflected a desire on the part of the compensation committee to include additional companies having commercial operations as a result of the launch of Silenor in September 2010. The compensation committee intends to review compensation information relating to this sub-group both separately and as part of the entire comparison group, in each case in light of the company’s status with respect to commercialization efforts, in considering future compensation determinations.
Barney & Barney compiled competitive executive compensation information from each of the companies in this comparison group for the compensation committee to review and analyze in making executive compensation decisions during 2011.
With respect to Named Executive Officers for which the publicly available competitive information from the comparison group was not sufficient to provide meaningful analysis in 2011, Barney & Barney provided the compensation committee with competitive information relating to such officers’ positions from one or more executive compensation surveys. The surveys were national surveys of executive compensation in the life sciences industries that were selected to provide the most comparable, comprehensive comparative compensation information for our executives.
With respect to survey data not relating to our comparison groups that was reviewed by the compensation committee in 2011, the identities of the individual companies included in the surveys were not provided to the compensation committee, and the compensation committee did not refer to individual compensation information for such companies. Instead, the compensation committee only referred to statistical summaries of such surveys.
While we believe that comparisons to market data are a useful tool, we do not believe that it is appropriate to establish executive compensation levels based solely on a comparison to competitive data. While compensation paid by other companies is a factor that the compensation committee considers in assessing the reasonableness of compensation, the compensation committee does not rely entirely on that data to determine executive officer compensation. Instead, the compensation committee incorporates flexibility into our compensation programs and in the assessment process to respond to and adjust for the evolving business environment and specific circumstances relating to our company. As a result of this approach, there are no comparative guidelines, such as percentiles, used by our compensation committee in making compensation determinations relative to the compensation data from our comparison group. In addition, the compensation committee has discretion to make stock awards to executive officers that are outside of the ranges in previously-approved stock option grant guidelines. Our compensation committee relies upon the judgment of its members in making executive compensation decisions, after reviewing the following factors:
70
| • | our performance against corporate objectives for the previous year, |
| • | difficulty in achieving desired results in the previous year and the current year, |
| • | the value of the executive’s unique leadership and other skills and capabilities to support our long-term performance, |
| • | historical compensation versus performance, |
| • | status relative to similarly-situated executives from our comparison group or from compensation surveys, |
| • | the executive’s performance generally, including against individual objectives, if any, for the previous year, and |
| • | the impact that any compensation awards that are payable in cash would have on our cash position. |
With respect to the compensation committee’s executive compensation review in early 2012, because the committee did not intend to increase the base salaries of our executive officers, pay cash bonuses to our executive officers or make equity awards to our executive officers, in each case due to the company’s 2011 performance and/or our cash position, the committee instructed management not to update our comparison group at that time.
Historically, the data regarding the compensation history and the relevant comparison group for each Named Executive Officer are provided to our Chief Executive Officer, the Chairman of the Board and the compensation committee. Our Chief Executive Officer then makes compensation recommendations to the compensation committee with respect to the executive officers who report to him. Our Chairman of the Board makes compensation recommendations to the compensation committee with respect to the Chief Executive Officer. The compensation committee considers, but is not bound to accept, these recommendations with respect to executive officer compensation. No executive officer is present at the time that his or her compensation is being discussed or determined.
Our policy for allocating between long-term and currently paid compensation is to ensure adequate base compensation to attract and retain personnel, while providing incentives to maximize long-term value for our company and our stockholders. A significant percentage of total compensation is allocated to incentive compensation as a result of this philosophy. We have no pre-established policy or target for the allocation between either cash and non-cash or short-term and long-term incentive compensation. Rather, the compensation committee reviews historical and competitive information and applies its judgment in light of the company’s then-current circumstances regarding current and long-term goals to determine the appropriate level and mix of incentive compensation.
Elements of Executive Compensation
Summary Compensation Table
The following “Summary Compensation Table” summarizes the compensation received by the Named Executive Officers in the fiscal years ended December 31, 2011, 2010 and 2009. This table provides an all-inclusive presentation of the various cash and non-cash elements that comprise total compensation for each of the Named Executive Officers. Except as set forth below, no Named Executive Officer earned any pension or other nonqualified deferred compensation, or perquisites exceeding $10,000 during 2011, 2010 or 2009.
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) (1) |
Stock Awards ($) (2) |
Option Awards ($) (3) |
Non-Equity Incentive Plan Compensation ($) (4) |
All
Other Compensation ($) |
Total ($) | ||||||||||||||||||||||||
| Richard W. Pascoe, |
2011 | $ | 482,551 | $ | — | $ | 219,750 | $ | 508,276 | $ | — | $ | — | $ | 1,210,577 | |||||||||||||||||
| President, Chief Executive Officer and |
2010 | $ | 456,250 | $ | 112,050 | $ | 429,500 | $ | 1,283,536 | $ | 209,875 | $ | — | $ | 2,491,211 | |||||||||||||||||
| Director (5) |
2009 | $ | 415,000 | $ | — | $ | — | $ | 1,461,045 | $ | — | $ | — | $ | 1,876,045 | |||||||||||||||||
| Tran B. Nguyen, |
2011 | $ | 301,779 | $ | — | $ | 87,900 | $ | 203,310 | $ | — | $ | 22,998 | $ | 615,987 | |||||||||||||||||
| Senior Vice President and Chief Financial Officer (6) |
2010 | $ | 206,193 | $ | — | $ | 206,500 | $ | 1,234,226 | $ | 66,394 | $ | 42,163 | $ | 1,755,476 | |||||||||||||||||
| Brian T. Dorsey, |
2011 | $ | 302,878 | $ | — | $ | 87,900 | $ | 203,310 | $ | — | $ | — | $ | 594,088 | |||||||||||||||||
| Senior Vice President, |
2010 | $ | 284,500 | $ | 53,130 | $ | 257,700 | $ | 641,768 | $ | 91,609 | $ | — | $ | 1,328,707 | |||||||||||||||||
| Technical Operations |
2009 | $ | 253,000 | $ | — | $ | — | $ | 786,025 | $ | — | $ | — | $ | 1,039,025 | |||||||||||||||||
| Matthew W. Onaitis, |
2011 | $ | 302,878 | $ | — | $ | 87,900 | $ | 203,310 | $ | — | $ | — | $ | 594,088 | |||||||||||||||||
| Senior Vice President and |
2010 | $ | 280,313 | $ | 49,613 | $ | 257,700 | $ | 641,768 | $ | 90,261 | $ | — | $ | 1,319,655 | |||||||||||||||||
| General Counsel |
2009 | $ | 236,250 | $ | — | $ | — | $ | 957,609 | $ | — | $ | — | $ | 1,193,859 | |||||||||||||||||
| Jeffrey W. Raser, |
2011 | $ | 274,957 | $ | — | $ | 87,900 | $ | 203,311 | $ | — | $ | 341,287 | $ | 907,455 | |||||||||||||||||
| Former Senior Vice President and |
2010 | $ | 300,038 | $ | 56,732 | $ | 343,600 | $ | 962,652 | $ | 96,612 | $ | — | $ | 1,759,634 | |||||||||||||||||
| Chief Commercial Officer (7) |
2009 | $ | 270,150 | $ | — | $ | — | $ | 831,989 | $ | — | $ | — | $ | 1,102,139 | |||||||||||||||||
| (1) | Amounts listed under the “Bonus” column for 2010 reflect the discretionary bonuses paid to each of the Named Executive Officers in April 2010. |
71
| (2) | The “Stock Awards” column is the grant date fair value of stock awards issued during each respective year, adjusted where applicable for our assessment of the probability that performance conditions will be achieved. The grant date fair value was determined in accordance with the provisions of FASB ASC Topic 718. For information about the assumptions used in the calculation of grant date fair value, see the notes to our audited financial statements included in this report. The RSUs with performance conditions granted during 2010 were considered probable of achieving their vesting conditions at the date of grant and the full grant date fair value of such RSUs is included in the “Stock Awards” column above. The stock awards with performance conditions granted during 2009 and 2011 were not considered probable of achieving their vesting conditions at the date of grant. Therefore the grant date fair value of such performance awards for purposes of the Summary Compensation Table was zero. The full grant date fair value of all stock awards granted during 2009 and 2011 which vested upon achieving performance conditions is, however, provided in the table following the footnotes to this Summary Compensation Table. |
| (3) | The “Option Awards” column is the grant date fair value of stock options granted during each respective year, adjusted where applicable for our assessment of the probability that performance conditions will be achieved. The grant date fair value was determined in accordance with the provisions of FASB ASC Topic 718 using the Black-Scholes valuation model with assumptions described in more detail in the notes to our audited financial statements included in this report. None of the stock options with performance conditions that were granted in 2009 were considered probable of achieving their vesting conditions at the date of grant. Therefore the grant date fair value of such performance awards for purposes of the Summary Compensation Table was zero. No performance-based stock options were granted during 2010 or 2011. The full grant date fair value of stock options which vested upon achieving performance conditions is, however, provided in the table following the footnotes to the Summary Compensation Table. Amounts included in the “Option Awards” column for 2009 include the fair value of replacement awards granted in our stock option exchange program which was completed in June 2009. The fair value of the replacement awards granted in the stock option exchange program is the sum of the unrecognized expense from the original award at the time of the exchange, plus the incremental value from the replacement award. The incremental value is the difference between the fair value of the replacement award and the fair value of the original award at the time of exchange. |
| (4) | Amounts reflect the bonuses earned during 2010 under our 2010 Incentive Plan. Such bonuses were paid in February 2011. |
| (5) | Mr. Pascoe became our President and Chief Executive Officer in August 2009. Amounts included in “All Other Compensation” reflect the sum of: (a) a $25,000 signing bonus paid at the time Mr. Pascoe joined us, plus the gross-up for taxes of $18,668 on such signing bonus, and (b) amounts reimbursed to Mr. Pascoe of $102,276 incurred in connection with his relocation to San Diego, California from Massachusetts, plus $60,901 for the gross-up for taxes on these reimbursed expenses to the extent such amounts were taxable. Amounts reimbursed for his relocation include reimbursement for temporary living expenses in San Diego, reasonable expenses relating to the sale of Mr. Pascoe’s home in Massachusetts, closing costs associated with the purchase of a primary residence in San Diego and moving of household goods. |
| (6) | Mr. Nguyen became our Vice President and Chief Financial Officer in April, 2010, and Mr. Nguyen was promoted to Senior Vice President and Chief Financial Officer in February 2011. Amounts included in “All Other Compensation” for 2011 and 2010 reflect amounts reimbursed to Mr. Nguyen of $16,200 and $34,672, respectively, incurred in connection with his relocation to San Diego, California from San Mateo, California, plus $6,798 and $7,491, respectively, for the gross-up for taxes on these reimbursed expenses to the extent such amounts were taxable. Amounts reimbursed for his relocation include reimbursement for temporary living expenses in San Diego and moving of household goods. |
| (7) | Mr. Raser resigned from the company effective October 1, 2011. At the time of his resignation, Mr. Raser received a lump-sum payment of $341,287 for severance and the cost of 12 months of health care benefits and 12 months of the portion of the monthly premium for his life insurance and disability insurance coverage that was borne by us, which amount are included in “All Other Compensation.” |
72
The following table provides the full grant date fair values for all share-based awards that were excluded from the Summary Compensation Table (see footnotes 2 and 3 above) that were granted with performance-based vesting conditions, assuming that the highest level of performance would be achieved in each case.
| Name |
Year | Grant Date Fair Value of Stock Awards $ (1) |
Stock Award Vesting Conditions |
Grant Date Fair Value of Option Awards (2) |
Option Award Vesting Conditions |
|||||||||||||
| Richard W. Pascoe |
2011 | $ | 146,500 | (3) | $ | — | — | |||||||||||
| 2010 | $ | — | $ | — | — | |||||||||||||
| 2009 | $ | 100,001 | (3) | $ | 119,947 | (4) | ||||||||||||
| Tran B. Nguyen |
2011 | $ | 146,500 | (3) | $ | — | — | |||||||||||
| 2010 | $ | — | $ | — | — | |||||||||||||
| Brian T. Dorsey |
2011 | $ | 146,500 | (3) | $ | — | — | |||||||||||
| 2010 | $ | — | $ | — | — | |||||||||||||
| 2009 | $ | 43,864 | (3) | $ | 79,965 | (4) | ||||||||||||
| Matthew W. Onaitis |
2011 | $ | 146,500 | (3) | $ | — | — | |||||||||||
| 2010 | $ | — | $ | — | — | |||||||||||||
| 2009 | $ | 41,089 | (3) | $ | 79,965 | (4) | ||||||||||||
| Jeffrey W. Raser (5) |
2011 | $ | 146,500 | (3) | $ | — | — | |||||||||||
| 2010 | $ | — | $ | — | — | |||||||||||||
| 2009 | $ | 47,219 | (3) | $ | 79,965 | (4) | ||||||||||||
| (1) | The “Grant Date Fair Value of Stock Awards” is based on the closing stock price of our common stock on the date of grant and is determined in accordance with the provisions of FASB ASC Topic 718. For information about the assumptions used in the calculation of grant date fair value, see the notes to our audited financial statements included in this report. |
| (2) | The “Grant Date Fair Value of Option Awards” is the grant date fair value of stock options granted during each respective year. The grant date fair value was determined in accordance with the provisions of FASB ASC Topic 718 using the Black-Scholes valuation model with assumptions described in more detail in the notes to our audited financial statements included in this report. |
| (3) | Amounts reflect the value of RSUs (a) granted in 2011 which would have vested upon our attainment of certain Silenor sales objectives during 2011, and (b) granted in 2009 which would have vested upon completion of financing activities resulting in at least $25 million of non-restricted cash proceeds by December 31, 2009. These goals were not achieved, and all of the listed stock awards granted during 2011 and 2009 were subsequently forfeited. |
| (4) | Amounts reflect the value of stock options of which 50% vested upon FDA approval of the Silenor NDA and 50% vested upon the completion of a strategic relationship with regards to the commercialization of Silenor. |
| (5) | Mr. Raser resigned from the company effective October 1, 2011. |
Base Salary
Base salary is intended to provide a baseline of compensation that does not fluctuate, absent merit-based increases. As a general matter, the base salary for each Named Executive Officer is initially established through negotiation at the time the officer is hired, taking into account the officer’s qualifications, experience, prior salary and competitive salary information. Each of our Named Executive Officers has entered into an employment agreement with us that prohibits the compensation committee from decreasing his base salary as part of this annual review process.
Each year, the compensation committee reviews the base salary levels of each of the Named Executive Officers, which includes an analysis of the prior year’s individual and corporate performance, historical compensation awards, our budget for merit increases, anticipated contribution to the current year’s corporate goals and comparisons to data from the companies in our comparison group and compensation surveys, if applicable, with respect to each such executive officer. Salaries are also reviewed in the case of promotions or other significant changes in responsibilities.
73
In February 2011, the compensation committee determined to increase base salaries based on the criteria set forth above. The base salaries for each of 2010 and 2011 are as follows:
| Base Salary | ||||||||
| Named Executive Officer |
2010 | 2011 | ||||||
| Richard W. Pascoe |
$ | 470,000 | $ | 484,100 | ||||
| Tran B. Nguyen |
$ | 285,000 | $ | 303,850 | ||||
| Brian T. Dorsey |
$ | 295,000 | $ | 303,850 | ||||
| Matthew W. Onaitis |
$ | 295,000 | $ | 303,850 | ||||
| Jeffrey W. Raser (1) |
$ | 310,000 | $ | 319,300 | ||||
| (1) | Mr. Raser resigned from the company effective October 1, 2011. |
The percentage increase in base salaries for each of Messrs. Pascoe, Raser, Dorsey and Onaitis from 2010 to 2011 was 3%, reflecting the compensation committee’s determination that the base salaries for these Named Executive Officers were generally appropriate, subject to a merit increase that is consistent with industry averages provided by our compensation consultant. Mr. Nguyen’s base salary increased 6.6% from 2010 to 2011, reflecting an increase as a result of his promotion in February 2011 to Senior Vice President and Chief Financial Officer.
In March 2012, the compensation committee determined that, in order to reduce costs, the base salaries of our executive officers would be reduced, effective as of April 1, 2012. Mr. Pascoe’s base salary was reduced by 15%, and the base salaries of each or Messrs. Nguyen, Dorsey and Onaitis was reduced by 5%. The amount of such salary reduction for each executive officer will be paid to such executive officer on a quarterly basis in arrears in the form of RSUs. After each calendar quarter, each executive officer will receive a number of RSUs calculated by dividing the amount of such salary reduction relating to such quarter by the closing price of our common stock on the Nasdaq Capital Market on the last trading day of such quarter, and multiplying the result by 1.2. All of these RSUs will vest upon the earlier of March 15, 2013 or the first date included within an open trading window under our Insider Trading Policy following December 31, 2012, subject to the executive officer’s continued service to us on the vesting date. In the event of a change of control transaction involving us or our stock prior to the vesting of the RSUs, 100% of the unvested RSUs would vest upon the consummation of the change of control.
Bonus Awards
It is the compensation committee’s objective to have a significant percentage of each Named Executive Officer’s total compensation be contingent upon our performance and his contribution toward our performance. This allows our Named Executive Officers to receive bonus compensation in the event certain specified corporate performance objectives are achieved.
In February 2011, the compensation committee undertook its annual review of the compensation levels and performance of the company and each of the Named Executive Officers. In connection with this process, the committee assessed our overall performance and determined that we met our corporate objectives at the 92% level. This was based upon the determinations of the committee that each of our corporate goals relating to a strategic partnership relating to Silenor and financing activities were met at the 100% level, and our corporate goal relating to the commercialization of Silenor was met at the 80% level. Based upon this analysis, the compensation committee granted bonuses pursuant to the 2010 Incentive Plan as listed in the Summary Compensation Table above.
In February 2011, our compensation committee adopted the 2011 Incentive Plan. This plan is designed to reward our Named Executive Officers for the achievement of annual performance goals. Pursuant to the 2011 Incentive Plan, the Named Executive Officers are eligible to receive bonuses ranging from zero to 150% of their target bonuses based entirely on the achievement of corporate performance goals. For 2011, the target bonus percentage for our Chief Executive Officer was increased to 55% of his base salary, and the target bonus percentage for each other Named Executive Officer was increased to 40% of his base salary. Our 2011 corporate performance goals were established by our board of directors and include: (1) achieving revenues for Silenor that were sufficient to make the company cash flow positive for the fourth quarter of 2011, (2) securing an additional product or product candidate, (3) extending the Silenor franchise through partnering and intellectual property protection, and (4) effectively managing our operating expenses and sales force turnover.
In February 2012, the compensation committee undertook its annual review of the compensation levels and performance of the company and each of the Named Executive Officers. In connection with this process, the committee assessed our overall performance, including with respect to Silenor’s commercialization. In light of the importance to the company of Silenor commercial performance, and taking our cash position into account, our management recommended to the compensation committee that the payment of cash bonuses to our employees under the previously-approved 2011 Incentive Plan was not advisable. The compensation committee agreed with this recommendation, and determined that cash bonuses would not be paid under the 2011 Incentive Plan.
74
In February 2012, our compensation committee adopted the 2012 Incentive Plan. This plan is designed to reward our Named Executive Officers for the achievement of annual performance goals. Pursuant to the 2012 Incentive Plan, the Named Executive Officers are eligible to receive bonuses ranging from zero to 150% of their target bonuses based entirely on the achievement of corporate performance goals. For 2011, the target bonus percentage for our Chief Executive Officer is 55% of his base salary, and the target bonus percentage for each other Named Executive Officer is 40% of his base salary. Our 2012 corporate performance goals were established by our board of directors and include objectives relating to: (1) the commercial success of Silenor, (2) our intellectual property protection for Silenor, (3) extending the Silenor franchise, and (4) effectively managing corporate resources.
Equity-Based Awards
We generally provide equity-based incentive award compensation to our Named Executive Officers through grants of stock options and RSUs. We have also previously awarded restricted stock to our Named Executive Officers. Stock awards allow us to:
| • | enhance the link between the creation of stockholder value and long-term executive incentive compensation, |
| • | provide an opportunity for increased equity ownership by executives, and |
| • | maintain competitive levels of total compensation. |
Stock award grant levels are determined based on market data and vary among the Named Executive Officers based on their positions and performance. Newly hired or promoted Named Executive Officers also typically receive stock option grants in connection with those events. We have guidelines that provide ranges of options to be granted to our employees, including our Named Executive Officers, based on their positions and whether the grants are being made in connection with hiring or on a performance basis thereafter. These guidelines were adopted by our compensation committee after considering recommendations provided by our independent compensation consultant that were based upon option grant data from our comparison group and the statistical summaries of compensation data presented to them based on available surveys. Our compensation committee considers the ranges contained in our guidelines in making determinations regarding the size of option grants, but it is not bound to comply with these ranges.
In making determinations relating to the size of equity awards, the compensation committee takes into account a number of factors, such as the relative performance of the executive, individual scope of duties, the value of existing long-term incentive awards, prior contributions to company performance, the importance to the company of anticipated future performance, the size of prior grants and competitive market data. Based upon these factors, the compensation committee determines the size of equity awards at levels it considers appropriate to create a meaningful opportunity for reward predicated on the creation of long-term stockholder value.
In addition, the board of directors has implemented a policy regarding the grant of equity awards. With respect to equity awards that may be granted to our Named Executive Officers, this policy provides that:
| • | equity awards may be granted only at meetings of the compensation committee or the board of directors, |
| • | the grant date shall be the date that the meeting approving the equity award was held, or if later, the date of commencement of employment of a newly-hired executive officer, |
| • | the exercise price of a stock option may not be less than the fair market value of a share of our common stock on the grant date, |
| • | grants of equity awards to executive officers shall not be permitted if the compensation committee determines that at the time of grant its members are in possession of material, non-public information concerning the company, and |
| • | the material terms of each equity award are communicated to the executive officer in a timely manner. |
75
The policy also provides that equity awards can be granted outside the terms of the policy in the event of unique circumstances or when time is of the essence, but that we shall not have any program, plan or practice to coordinate the timing of equity awards with the release by us of material non-public information or any other investor relations activities.
The stock options that have been granted to our executive officers typically have a ten year term and vest over four years, with 25% vesting after one year and the remainder vesting in equal monthly installments over the subsequent three years. Prior to the exercise of an option, the holder has no rights as a stockholder with respect to the shares subject to such option, including voting rights and the right to receive dividends or dividend equivalents.
We do not have any security ownership requirements for our executive officers.
In February 2011, in connection with its annual review of the compensation levels and performance of the company and our Named Executive Officers, on February 18, 2011, the compensation committee approved awards of stock options to the Named Executive Officers, which stock option awards were granted effective March 7, 2011. The exercise price per share of such stock options was $2.93, which was the closing price per share of our common stock on March 7, 2011. The number of stock options received by each Named Executive Officer is as follows:
| Named Executive Officer |
Number of Stock Options Received |
|||
| Richard W. Pascoe |
250,000 | |||
| Tran B. Nguyen |
100,000 | |||
| Brian T. Dorsey |
100,000 | |||
| Matthew W. Onaitis |
100,000 | |||
| Jeffrey W. Raser (1) |
100,000 | |||
| (1) | Mr. Raser resigned from the company effective October 1, 2011. |
All of such stock options will vest based on our standard four-year vesting schedule described above.
Also in February 2011, the compensation committee approved two sets of RSU awards to each of our Named Executive Officers, which RSU awards were awarded effective March 7, 2011. One set of RSUs is subject to time-based vesting and vests as follows: one third of such RSUs vested on December 31, 2011, another one third of such RSUs will vest on December 31, 2012 and the remaining one-third of such RSUs will vest on December 31, 2013. The number of such time-based RSUs received by each Named Executive Officer is as follows:
| Named Executive Officer |
Number of
RSUs Received |
|||
| Richard W. Pascoe |
75,000 | |||
| Tran B. Nguyen |
30,000 | |||
| Brian T. Dorsey |
30,000 | |||
| Matthew W. Onaitis |
30,000 | |||
| Jeffrey W. Raser (1) |
30,000 | |||
| (1) | Mr. Raser resigned from the company effective October 1, 2011. |
The second set of RSUs was subject to performance-based vesting and was to vest during the first quarter of 2012 on the second business day following the public release of our audited financial statements for the fiscal year ended December 31, 2011, subject to the condition that the company achieved revenues for Silenor that were sufficient to make the company cash flow positive for the fourth quarter of 2011. This objective was not met, so each of these RSUs was forfeited. The number of such performance-based RSUs received and forfeited by each Named Executive Officer is as follows:
76
| Named Executive Officer |
Number of
RSUs Received |
|||
| Richard W. Pascoe |
50,000 | |||
| Tran B. Nguyen |
50,000 | |||
| Brian T. Dorsey |
50,000 | |||
| Matthew W. Onaitis |
50,000 | |||
| Jeffrey W. Raser (1) |
50,000 | |||
| (1) | Mr. Raser resigned from the company effective October 1, 2011. |
The awards of the options and RSUs to the Named Executive Officers in February 2011 reflected a desire on the part of the compensation committee to motivate the Named Executive Officers to drive the creation of stockholder value, and the vesting schedules of the awards are intended to provide incentive for the Named Executive Officers to maintain their employment with the company. In addition, the awards of the performance-based RSUs reflect a desire on the part of the compensation committee to provide incentive for the Named Executive Officers to cause us to achieve key corporate objectives.
In February 2012, the compensation committee undertook its annual review of the compensation levels and performance of the company and each of the Named Executive Officers. In light of the importance to the company of Silenor commercial performance, our management recommended to the compensation committee that the grant of equity awards to our employees was not advisable. The compensation committee agreed with this recommendation, and determined that equity awards would not be granted to the Named Executive Officers at this time.
For a description of the change of control provisions applicable to the stock awards granted to our Named Executive Officers, see “Severance Benefits and Change of Control Arrangements” below.
Outstanding Equity Awards at Fiscal Year End
The following table summarizes the outstanding stock options and RSUs as of December 31, 2011 held by our Named Executive Officers.
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||||||||||||
| Name |
Award Grant Date |
Number of Securities Underlying Unexercised Options (# Exercisable) (1) |
Number of Securities Underlying Unexercised Options (# Unexercisable) (1) |
Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) |
Option Exercise Price ($/Share) |
Option Expiration Date |
Number of Shares or Units of Stock that Have Not Vested (#)(2) |
Market Value of Stock that Has Not Vested ($)(3) |
Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights that Have not Vested (#) |
Equity Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights that Have not Vested ($) |
||||||||||||||||||||||||||||||
| Richard W. Pascoe |
6/9/2009 | (4) | 243,333 | — | $ | 1.23 | ||||||||||||||||||||||||||||||||||
| 7/16/2009 | (5) | 150,000 | — | $ | 1.17 | 7/15/2019 | ||||||||||||||||||||||||||||||||||
| 4/1/2010 | 83,332 | 116,668 | $ | 8.59 | 3/31/2020 | 33,334 | 15,000 | — | $ | — | ||||||||||||||||||||||||||||||
| 3/7/2011 | — | 250,000 | $ | 2.93 | 3/6/2021 | 50,000 | 22,500 | — | $ | — | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| 476,665 | 366,668 | — | 83,334 | $ | 37,500 | — | $ | — | ||||||||||||||||||||||||||||||||
| Tran B. Nguyen |
4/12/2010 | 83,332 | 116,668 | $ | 8.26 | 4/11/2020 | 16,667 | $ | 7,500 | — | $ | — | ||||||||||||||||||||||||||||
| 3/7/2011 | — | 100,000 | $ | 2.93 | 3/6/2021 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| 83,332 | 216,668 | — | 36,667 | $ | 16,500 | — | $ | — | ||||||||||||||||||||||||||||||||
| Brian T. Dorsey |
6/9/2009 | (4) | 74,708 | — | $ | 1.23 | ||||||||||||||||||||||||||||||||||
| 7/16/2009 | (5) | 100,000 | — | $ | 1.17 | 7/15/2019 | ||||||||||||||||||||||||||||||||||
| 4/1/2010 | 41,666 | 58,334 | $ | 8.59 | 3/31/2020 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
| 3/7/2011 | — | 100,000 | $ | 2.93 | 3/6/2021 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| 216,374 | 158,334 | — | 40,000 | $ | 18,000 | — | $ | — | ||||||||||||||||||||||||||||||||
| Matthew W. Onaitis |
6/9/2009 | (4) | 85,345 | — | $ | 1.23 | ||||||||||||||||||||||||||||||||||
| 7/16/2009 | (5) | 100,000 | — | $ | 1.17 | 7/15/2019 | ||||||||||||||||||||||||||||||||||
| 4/1/2010 | 41,665 | 58,335 | $ | 8.59 | 3/21/2020 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
| 3/7/2011 | — | 100,000 | $ | 2.93 | 3/6/2021 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| 227,010 | 158,335 | — | 40,000 | $ | 18,000 | — | $ | — | ||||||||||||||||||||||||||||||||
| Jeffrey W. Raser (6) |
3/2/2005 | 7,500 | — | $ | 2.40 | 3/1/2015 | ||||||||||||||||||||||||||||||||||
| 7/19/2005 | 83,333 | — | $ | 3.00 | 7/18/2015 | |||||||||||||||||||||||||||||||||||
| 6/9/2009 | (4) | 97,665 | — | $ | 1.23 | |||||||||||||||||||||||||||||||||||
| 7/16/2009 | (5) | 60,000 | — | $ | 1.17 | 7/15/2019 | ||||||||||||||||||||||||||||||||||
| 4/1/2010 | 150,000 | — | $ | 8.59 | 3/31/2020 | 13,334 | 6,000 | — | $ | — | ||||||||||||||||||||||||||||||
| 3/7/2011 | 81,249 | 18,751 | $ | 2.93 | 3/6/2021 | 20,000 | 9,000 | — | $ | — | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| 479,747 | 18,751 | — | 33,334 | $ | 15,000 | — | $ | — | ||||||||||||||||||||||||||||||||
| (1) | Except as described under footnotes 4 and 5 below, all of the stock options in this column vest such that 25% are vested one year after the vesting commencement date and 1/48th vest on the first day of each calendar month thereafter until all options are fully vested on the first day of the 48th month after grant. |
77
| (2) | Stock included under equity incentive plan awards include (a) RSUs granted in 2010 in which all of the amount included in the table above will vest on January 1, 2013, and (b) RSUs granted in 2011 in which 50% of the amount included in the table will vest on December 31, 2012, and the remainder will vest on December 31, 2013. |
| (3) | The value of stock awards is based on a closing stock price of $0.45 per share on December 31, 2011. |
| (4) | Denotes stock options that were granted under our stock option exchange program which was completed in June 2009. Such stock options vest such that one-third were vested at grant, with the remaining two-thirds vesting over the subsequent two years. The expiration dates for these stock options range between March 15, 2015 and February 16, 2019. |
| (5) | Denotes stock options that vested 50% upon FDA approval of our NDA for Silenor and 50% upon the completion of a strategic relationship regarding the commercialization of Silenor. |
| (6) | Mr. Raser resigned from the company effective October 1, 2011. |
Grants of Plan-Based Awards Table
The following table summarizes equity-based and incentive plan awards granted to our Named Executive Officers during the last fiscal year.
| Estimated Future Payouts
Under Non-Equity Incentive Plan Awards (1) |
Estimated Future Payouts
Under Equity Incentive Plan Awards |
All Other Stock Awards: Number of Shares of |
All Other Option Awards: Number of Securities |
Exercise or Base Option |
Grant Date Fair Value of Stock |
|||||||||||||||||||||||||||||||||
| Name |
Grant Date | Threshold ($) |
Target ($) |
Maximum ($) |
Threshold (#) |
Target (#) (2) |
Maximum (#) |
Stock
or Units (#) (3) |
Underlying Options (#) |
Awards ($/Share) |
and Option Awards (4) |
|||||||||||||||||||||||||||
| Richard W. Pascoe |
N/A | $ | 266,255 | $ | 399,383 | |||||||||||||||||||||||||||||||||
| 3/7/2011 | 50,000 | $ | 146,500 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 75,000 | $ | 219,750 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 250,000 | $ | 2.93 | $ | 508,276 | |||||||||||||||||||||||||||||||||
| Tran B. Nguyen |
N/A | $ | 121,540 | $ | 182,310 | |||||||||||||||||||||||||||||||||
| 3/7/2011 | 50,000 | $ | 146,500 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 30,000 | $ | 87,900 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 100,000 | $ | 2.93 | $ | 203,310 | |||||||||||||||||||||||||||||||||
| Brian T. Dorsey |
N/A | $ | 121,540 | $ | 182,310 | |||||||||||||||||||||||||||||||||
| 3/7/2011 | 50,000 | $ | 146,500 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 30,000 | $ | 87,900 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 100,000 | $ | 2.93 | $ | 203,310 | |||||||||||||||||||||||||||||||||
| Matthew W. Onaitis |
N/A | $ | 121,540 | $ | 182,310 | |||||||||||||||||||||||||||||||||
| 3/7/2011 | 50,000 | $ | 146,500 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 30,000 | $ | 87,900 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 100,000 | $ | 2.93 | $ | 203,310 | |||||||||||||||||||||||||||||||||
| Jeffrey W. Raser (5) |
N/A | $ | 127,720 | $ | 191,580 | |||||||||||||||||||||||||||||||||
| 3/7/2011 | 50,000 | $ | 146,500 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 30,000 | $ | 87,900 | |||||||||||||||||||||||||||||||||||
| 3/7/2011 | 100,000 | $ | 2.93 | $ | 203,310 | |||||||||||||||||||||||||||||||||
| (1) | Amount represents potential target and maximum bonus payments under our 2011 Incentive Plan. For more information about the 2011 Incentive Plan, please see the discussion under “—Bonus Awards” above. |
| (2) | Represents RSUs that were scheduled to vest on the second business day following the public release of our audited financial statements for the fiscal year ended December 31, 2011, subject to attainment of certain corporate performance objectives during 2011. The corporate performance objectives were not met, so each of these RSUs was forfeited. |
| (3) | Represents RSUs vesting as follows: one-third vested on December 31, 2011, one-third will vest on December 31, 2012 and one-third will vest on December 31, 2013. |
| (4) | For RSUs, the grant date fair value is calculated as the number of shares multiplied the stock price at the date of grant. For stock options, the grant date fair value is calculated in accordance with the provisions of FASB ASC Topic 718 using the Black-Scholes valuation model with the assumptions outlined in our audited financial statements included in this report. The RSUs with performance conditions granted during 2011 were not considered probable of achieving their vesting conditions at the date of grant. Therefore the grant date fair value of such performance awards for purposes of this was zero. The full grant date fair value of all stock awards granted during 2011 which vested upon achieving performance conditions is, however, provided in the table following the footnotes to this Summary Compensation Table. |
| (5) | Mr. Raser resigned from the company effective October 1, 2011. |
78
Stock Option Exercises and Stock Vested Table
The following table summarizes the exercises of stock options and the vesting of restricted stock and RSUs for our Named Executive Officers during our last fiscal year.
| Option Awards | Stock Awards | |||||||||||||||
| Name |
Number of Shares Acquired on Exercise (#) |
Value Realized on Exercise ($) (1) |
Number of Shares Acquired on Vesting (#) |
Value Realized on Vesting ($) (2) |
||||||||||||
| Richard W. Pascoe |
— | $ | — | 25,000 | $ | 11,250 | ||||||||||
| Tran B. Nguyen |
— | $ | — | 18,333 | $ | 28,249 | ||||||||||
| Brian T. Dorsey |
— | $ | — | 10,000 | $ | 4,500 | ||||||||||
| Matthew W. Onaitis |
— | $ | — | 10,000 | $ | 4,500 | ||||||||||
| Jeffrey W. Raser (3) |
— | $ | — | 23,333 | $ | 20,300 | ||||||||||
| (1) | The “Value Realized on Exercise” for an option award represents the stock price per share on the date of exercise less the exercise price per share of such option award multiplied by the number of shares for which such option award was exercised. |
| (2) | The “Value Realized on Vesting” for stock awards is based on a stock price on the date that the RSUs and restricted stock vested. |
| (3) | Mr. Raser resigned from the company effective October 1, 2011. |
Other Benefits
In order to attract, retain and pay market levels of compensation, we provide our Named Executive Officers and our other employees the following benefits and perquisites.
Medical Insurance
The company provides to each Named Executive Officer and their spouses and children such health, dental and vision insurance coverage as the company may from time to time make available to its other eligible employees.
Life, Disability and Long-term Care Insurance
We provide each Named Executive Officer such disability, life and/or long-term care insurance as we may from time to time make available to our other eligible employees.
Pension Benefits
We do not provide pension arrangements or post-retirement health coverage for our executives or employees. Our Named Executive Officers and other eligible employees are eligible to participate in our 401(k) defined contribution plan. We currently do not make matching contributions to the 401(k) plan.
Nonqualified Deferred Compensation
We do not provide any nonqualified defined contribution or other deferred compensation plans.
Perquisites
We generally limit the perquisites that we make available to our Named Executive Officers, particularly in light of recent developments with respect to corporate crime and abuse involving perquisites. Our executives are entitled to few benefits with de minimis value that are not otherwise available to all of our employees.
79
Post-Termination Benefits
Severance Benefits and Change of Control Arrangements
We believe that reasonable severance benefits for our Named Executive Officers are important because it may be difficult for our Named Executive Officers to find comparable employment within a short period of time. We also believe that it is important to protect our Named Executive Officers in the event of a change of control transaction involving us. In addition, it is our belief that the interests of stockholders will be best served if the interests of our senior management are aligned with them, and providing change of control benefits should eliminate, or at least reduce, the reluctance of senior management to pursue potential change of control transactions that may be in the best interests of stockholders. Accordingly, the employment agreements we have entered into with each of our Named Executive Officers provide for severance benefits in specified circumstances, as well as benefits in connection with a change of control.
Messrs. Pascoe, Nguyen, Dorsey, Onaitis and Raser entered into employment agreements with us upon joining the company.
The employment agreements, as amended to date, provide each executive with certain severance benefits in the event his employment is terminated as a result of his disability. Specifically, in the event of such a termination, each executive will receive any accrued but unpaid base salary and unused paid time-off as of the date of termination, a lump-sum severance payment equal to 12 months of base salary, and, in the discretion of our board of directors, a pro-rated bonus for the year in which the termination occurs.
The employment agreements also provide each executive with certain severance benefits in the event his employment is terminated by us other than for cause or if the executive resigns with good reason. Specifically, in the event of such a termination or resignation, each executive will receive any accrued but unpaid base salary and unused paid time-off as of the date of termination, a lump-sum severance payment equal to 12 months of base salary, 12 months of health care benefits continuation at our expense, and in the discretion of our board of directors a pro-rated bonus for the year in which the termination or resignation occurs. In addition, that portion of the executive’s stock awards, and any unvested shares issued upon the exercise of such stock awards, which would have vested if the executive had remained employed for an additional 12 months, will immediately vest on the date of termination or resignation and the executive will be entitled to exercise such stock awards for 180 days following the date of termination.
In the event of a change of control of the company, 50% of each executive’s unvested stock awards will immediately become vested and exercisable on the date of the change of control and any remaining unvested stock awards will become vested and exercisable on the one year anniversary of the date of the change of control. In addition, in the event an executive’s employment is terminated by us other than for cause or if the executive resigns with good reason, in each case within 12 months of a change of control, all of such executive’s unvested stock awards will immediately become vested and exercisable on the date of termination and the executive will be entitled to exercise such stock awards for 180 days following the date of termination.
In March 2012, in connection with the salary reductions of our executive officers, the employment agreements of our executive officers were amended to provide that the salary reductions would not amount to “good reason” under such agreements. The amendments also provide that in the event of either a later termination of the executive officer without cause or the executive’s later resignation for good reason, the base salary used to calculate the severance benefit would be the greater of the executive officer’s salary prior to the March 2012 salary reduction or the amount of base salary received by the executive officer in the 12 months preceding the termination or resignation.
Mr. Raser resigned from the company effective October 1, 2011. In connection with his resignation, Mr. Raser received the foregoing benefits under his employment agreement with us, including: (1) a lump sum severance payment of $315,954, (2) a lump sum payment equal to the cost of 12 months of health care benefits continuation at our expense, and (3) a lump sum payment equal to the cost of 12 months of the portion of the monthly premiums for Mr. Raser’s life insurance and disability insurance coverage that were borne by us. In addition, on October 1, 2011, the portion of Mr. Raser’s stock options and restricted stock units which would have vested if Mr. Raser had remained employed for an additional 12 months immediately vested.
Mr. Raser also entered into a consulting agreement us that will expire on June 30, 2013. This agreement was put into place primarily for the purpose of securing Mr. Raser’s assistance in our pending patent litigation relating to Silenor. We cannot estimate with any certainty the amounts that may be paid, if any, for consulting services under such agreement, but we do not currently expect the aggregate amount to be material. Mr. Raser’s remaining outstanding stock awards will continue to vest through the expiration of the consulting agreement, and he will be entitled to exercise such stock awards for 180 days following such expiration.
80
Definitions Applicable to All Executive Employment Agreements:
For purposes of all of the employment agreements with our Named Executive Officers, “cause” means, generally, the executive’s breach of the non-solicitation, nondisparagement or confidentiality provisions of the employment agreement, the executive’s conviction by, or entry of a plea of guilty or nolo contendere in, a court of competent and final jurisdiction for any crime involving moral turpitude or punishable by imprisonment in the jurisdiction involved, the executive’s commission of an act of fraud, the executive’s continuing, willful failure or refusal to perform his or her duties as required by the employment agreement, the executive’s gross negligence, insubordination or material violation of any duty of loyalty to us or any other material misconduct on the part of the executive, the executive’s commission of any act which is detrimental to our business or goodwill, or the executive’s breach of any other provision of the employment agreement after he or she has been afforded a specified period to correct the alleged breach.
For purposes of all of the employment agreements with our Named Executive Officers, “good reason” means, generally, a material diminution in the executive’s base compensation, a material diminution in the executive’s authority, duties or responsibilities, a material diminution in the authority, duties or responsibilities of the supervisor to whom the executive is required to report (or, in the case of Mr. Pascoe, a requirement that he reports to an employee rather than our board of directors), a material change in the geographic location at which the executive must perform his or her duties, or any other action or inaction that constitutes a material breach by us of our obligations to the executive under the employment agreement.
For purposes of all of the employment agreements with our Named Executive Officers, “change in control” has the same meaning as given to that term in our 2005 Equity Incentive Award Plan.
Potential Payments Upon Termination or Change in Control:
If the employment of each of our Named Executive Officers employed as of December 31, 2011 was terminated due to disability or was terminated without cause, or if each resigned for good reason, the estimated benefits that each would receive under their employment agreements as of December 31, 2011 would be as follows:
| Received if Terminated due to Disability | Received if Terminated Without Cause
or Resigned for Good Reason |
|||||||||||||||||||||||||||||||||||
| Name |
Salary ($) | Unused Paid Time-off ($) |
Total ($) | Salary ($) |
Unused Paid Time-off ($) |
Health Care Benefits ($) |
Intrinsic Value of Additional Vested Stock Options ($) (1) |
Intrinsic Value of Additional Vested Shares ($) (2) |
Total ($) | |||||||||||||||||||||||||||
| Richard W. Pascoe |
$ | 484,100 | $ | 48,424 | $ | 532,524 | $ | 484,100 | $ | 48,424 | $ | 20,219 | $ | — | $ | 18,750 | $ | 571,493 | ||||||||||||||||||
| Tran B. Nguyen |
$ | 303,850 | $ | 31,574 | $ | 335,424 | $ | 303,850 | $ | 31,574 | $ | 16,729 | $ | — | $ | 8,250 | $ | 360,403 | ||||||||||||||||||
| Brian T. Dorsey |
$ | 303,850 | $ | 35,059 | $ | 338,909 | $ | 303,850 | $ | 35,059 | $ | 19,952 | $ | — | $ | 9,000 | $ | 367,861 | ||||||||||||||||||
| Matthew W. Onaitis |
$ | 303,850 | $ | 35,059 | $ | 338,909 | $ | 303,850 | $ | 35,059 | $ | 19,945 | $ | — | $ | 9,000 | $ | 367,854 | ||||||||||||||||||
| (1) | The intrinsic value of additional vested stock options shown above is the difference between the closing stock price of $0.45 per share at December 31, 2011 and the exercise price. All of the options that would vest within 12 months of December 31, 2011 have an exercise price that exceeds the underlying stock price, so for those options, the intrinsic value at December 31, 2011 is zero and not reflected in the table above. |
| (2) | The intrinsic value of additional vested shares shown above is the number of shares that would vest within 12 months of December 31, 2011, multiplied by the closing stock price of $0.45 per share at December 31, 2011. All of our outstanding RSUs are time-based and vest upon continued service. |
81
If a change in control was consummated at December 31, 2011 and the employment of each of our Named Executive Officers employed as of such date was terminated without cause, or if an executive resigned for good reason, the estimated benefits that each would receive under their employment agreements would be as follows:
| Received if Terminated Without Cause or Resigned for Good Reason in Connection with a Change of Control |
||||||||||||||||||||||||
| Name |
Salary ($) | Unused Paid Time-off ($) |
Health Care Benefits ($) |
Intrinsic Value of Additional Vested Stock Options ($) (1) |
Intrinsic Value of Additional Vested Shares ($) (2) |
Total ($) | ||||||||||||||||||
| Richard W. Pascoe |
$ | 484,100 | $ | 48,424 | $ | 20,219 | $ | — | $ | 37,500 | $ | 590,243 | ||||||||||||
| Tran B. Nguyen |
$ | 303,850 | $ | 31,574 | $ | 16,729 | $ | — | $ | 16,500 | $ | 368,653 | ||||||||||||
| Brian T. Dorsey |
$ | 303,850 | $ | 35,059 | $ | 19,952 | $ | — | $ | 18,000 | $ | 376,861 | ||||||||||||
| Matthew W. Onaitis |
$ | 303,850 | $ | 35,059 | $ | 19,945 | $ | — | $ | 18,000 | $ | 376,854 | ||||||||||||
| (1) | The intrinsic value of additional vested stock options shown above is the difference between the closing stock price of $0.45 per share at December 31, 2011 and the exercise price. All of the options that would vest within 12 months of December 31, 2011 have an exercise price that exceeds the underlying stock price, so for those options, the intrinsic value at December 31, 2011 is zero and not reflected in the table above. |
| (2) | The intrinsic value of additional vested restricted shares, RSUs and stock options is based on a closing stock price of $0.45 per share at December 31, 2011. |
Policy Regarding Tax Deductibility of Compensation
Section 162(m) of the Internal Revenue Code of 1986, as amended, or the code, generally disallows a tax deduction to public companies for compensation in excess of $1 million paid to the corporation’s Chief Executive Officer and four other most highly paid executive officers. Qualifying performance-based compensation will not be subject to the deduction limitation if certain requirements are met.
The non-performance based compensation paid in cash to our executive officers in 2011 did not exceed the $1 million limit per officer, and the compensation committee does not anticipate that the non-performance based compensation to be paid in cash to our executive officers for 2012 will exceed that limit. In addition, our 2005 Equity Incentive Award Plan has been structured so that any compensation paid in connection with the exercise of option grants under that plan with an exercise price equal to the fair market value of the option shares on the grant date will qualify as performance-based compensation and it will not be subject to the $1 million deduction limitation.
We periodically review the potential consequences of Section 162(m) and may structure the performance-based portion of our executive compensation to comply with certain exemptions in Section 162(m). However, we reserve the right to use our judgment to authorize compensation payments that do not comply with the exemptions in Section 162(m) when we believe that such payments are appropriate and in the best interests of the stockholders, after taking into consideration changing business conditions or the officer’s performance.
Compensation Committee Report
We have reviewed and discussed with management the Compensation Discussion and Analysis provisions to be included in this report. Based on the reviews and discussions referred to above, we recommend to the board of directors that the Compensation Discussion and Analysis referred to above be included in this report.
Compensation Committee
Thomas G. Wiggans (Chair)
Michael L. Eagle
Faheem Hasnain
82
Compensation of our Board of Directors
We compensate non-employee directors for their service on our board of directors under our Director Compensation Policy.
Under the current Director Compensation Policy, each non-employee director other than our non-Executive Chairman of the Board is eligible to receive a quarterly retainer of $6,250, or $25,000 annually, for service on our board of directors. Our non-Executive Chairman of the Board is eligible to receive a quarterly retainer of $28,000, or $112,000 annually, for his service in such role.
Our non-employee directors also receive retainers for their service on board committees. The chairman of the audit committee of the board of directors receives a quarterly retainer of $2,500, or $10,000 per year. Each other member of our audit committee receives a quarterly retainer of $1,250, or $5,000 per year. Each member of the compensation committee of our board of directors receives a quarterly retainer of $625, or $2,500 per year, and each member of the nominating/corporate governance committee of our board of directors receives a quarterly retainer of $625, or $2,500 per year.
Each non-employee director is also eligible to receive an incremental stipend of $2,000 for each board meeting attended in person, or $1,000 for each board meeting attended by telephone, and $1,000 for each committee meeting attended in person, or $500 for each committee meeting attended by telephone. We reimburse our non-employee directors for their reasonable expenses incurred in attending meetings of our board of directors and committees of the board of directors.
In March 2012 our board of directors, upon the recommendation of the compensation committee, amended our Director Compensation Policy, retroactively to October 1, 2011, to provide that non-employee directors will receive their quarterly retainers for service on the board of directors or committees of the board and their fees for attending meetings of the board and committees of the board in RSUs. After each calendar quarter, each director will receive a number of RSUs calculated by dividing the total amount of such retainers and fees due to such director relating to such quarter, or the previous two quarters with respect to the grant of RSUs on March 31, 2012, by the closing price of our common stock on the Nasdaq Capital Market on the last trading day of such quarter, and multiplying the result by 1.2. All of these RSUs will vest upon the first date included within an open trading window under our Insider Trading Policy following December 31, 2012, subject to the director’s continued service to us on such date. In the event of a change of control transaction involving us or our stock prior to the vesting of the RSUs, 100% of the unvested RSUs would vest upon the consummation of the change of control.
Our directors may participate in our stock incentive plans and employee-directors may participate in our employee stock purchase plan. Any non-employee director who is elected to our board of directors is granted an option to purchase 35,000 shares of our common stock on the date of his or her initial election to our board of directors. In addition, on the date of each annual meeting of our stockholders, (1) each continuing non-employee director will be eligible to receive an option to purchase 15,000 shares of common stock, (2) the non-Executive Chairman of the Board will be eligible to receive an additional annual option to purchase 25,000 shares of common stock (for a total of 40,000 shares), (3) the chairman of our audit committee will be eligible to receive an additional annual option to purchase 5,000 shares of common stock (for a total of 20,000 shares) and (4) the chairmen of our nominating/corporate governance committee and our compensation committee will be eligible to receive an additional annual option to purchase 2,500 shares of common stock (for a total of 17,500 shares each). Such options will have an exercise price per share equal to the fair market value of our common stock on such date. The initial options granted to non-employee directors described above will vest over three years in 36 equal monthly installments on each monthly anniversary of the date of grant, subject to the director’s continuing service on our board of directors on those dates. The annual options granted to non-employee directors described above will vest in 12 equal monthly installments on each monthly anniversary of the date of grant, subject to the director’s continuing service on our board of directors (and, with respect to grants to a Chairman of the Board or board committee, service as Chairman of the Board or a committee) on those dates. The term of each option granted to a non-employee director shall be ten years.
83
Director Compensation Table
The following table summarizes our director compensation for each of our directors except Mr. Pascoe for the year ended December 31, 2011. Please see the tables relating to our Named Executive Officers for information relating to Mr. Pascoe.
| Name and Principal Position |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
Option Awards ($) (1) |
All Other Compensation ($) |
Total ($) | |||||||||||||||
| David F. Hale, Chairman of the Board |
$ | 129,000 | $ | — | $ | 57,909 | $ | — | $ | 186,909 | ||||||||||
| Erle T. Mast, Director, Chairman of the Audit Committee |
$ | 54,908 | $ | — | $ | 28,955 | $ | — | $ | 83,863 | ||||||||||
| Kurt von Emster, Director, Chairman of the Nominating / Corporate Governance Committee |
$ | 53,272 | $ | — | $ | 25,335 | $ | — | $ | 78,607 | ||||||||||
| Terrell A. Cobb, Director |
$ | 42,000 | $ | — | $ | 21,716 | $ | — | $ | 63,716 | ||||||||||
| Michael L. Eagle, Director |
$ | 52,544 | $ | — | $ | 21,716 | $ | — | $ | 74,260 | ||||||||||
| Faheem Hasnain, Director |
$ | 44,431 | $ | — | $ | 21,716 | $ | — | $ | 66,147 | ||||||||||
| Thomas G. Wiggans, Director, Chairman of the Compensation Committee |
$ | 47,658 | $ | — | $ | 25,335 | $ | — | $ | 72,993 | ||||||||||
| (1) | The “Option Awards” column is the grant date fair value of option awards issued to our non-employee directors during 2011, calculated in accordance with the provisions of FASB ASC Topic 718. See the assumptions used in the Black-Scholes model in the notes to our audited financial statements included in this report. During 2011, our non-employee directors were granted stock options to purchase the following numbers of shares of our common stock: Mr. Hale, 40,000 shares; Mr. Mast, 20,000 shares; Mr. von Emster, 17,500 shares; Mr. Cobb, 15,000 shares; Mr. Eagle, 15,000 shares; Mr. Wiggans, 17,500 shares; and Mr. Hasnain, 15,000 shares. At December 31, 2011, our non-employee directors held options to purchase the following number of shares of our common stock: Mr. Hale, 452,219 shares; Mr. Mast, 86,666 shares; Mr. von Emster, 110,831 shares; Mr. Cobb, 126,387 shares; Mr. Eagle, 78,333 shares; Mr. Wiggans, 73,333 shares; and Mr. Hasnain, 50,000 shares. |
Compensation Committee Interlocks and Insider Participation
Mr. Wiggans (chair) and Messrs. Eagle and Hasnain served on our compensation committee during the 2011 fiscal year. Mr. Von Emster also served on our compensation committee for part of 2011. No member of the compensation committee was at any time during the 2011 fiscal year or at any other time an officer or employee of the company. None of our executive officers serve, or in the past year has served as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving on our compensation committee. None of our executive officers serve, or in the past year has served, as a member of the compensation committee of any entity that has one or more executives serving on our board of directors.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth information regarding the beneficial ownership of our common stock as of February 15, 2012 for:
| • | each of our Named Executive Officers as defined in Part III — Item 11, “Executive Compensation” of this report; |
| • | each of our directors; |
| • | each person known by us to beneficially own more than 5% of our common stock; and |
| • | all of our Named Executive Officers and directors as a group. |
84
Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission and includes voting and investment power with respect to the securities. Except as indicated by footnote, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of common stock shown as beneficially owned by them. The number of shares of common stock used to calculate the percentage ownership of each listed person includes the shares of common stock underlying options held by such persons that are exercisable as of April 15, 2012, which is 60 days after February 15, 2012.
Percentage of beneficial ownership is based on 48,108,251 shares of common stock outstanding as of February 15, 2012. Unless otherwise indicated, the address for the following stockholders is c/o Somaxon Pharmaceuticals, Inc., 10935 Vista Sorrento Parkway, Suite 250, San Diego, CA 92130.
| Name and Address of Beneficial Owner |
Shares Beneficially Owned |
Percent Beneficially Owned |
||||||
| 5% Stockholders: |
||||||||
| Ingalls & Snyder LLC (1) 61 Broadway New York, NY 10006 |
5,957,000 | 12.4 | % | |||||
| Funds affiliated with MPM Capital, L.P. (2) 601 Gateway Boulevard, Suite 350 South San Francisco, CA 94080 |
5,909,155 | 12.0 | % | |||||
| Scale Venture Management I, LLC, (formerly BAVP, L.P.) (3) 950 Tower Lane, Suite 700 Foster City, CA 94404 |
3,002,858 | 6.2 | % | |||||
| Directors and Named Executive Officers: |
||||||||
| David F. Hale (4) |
745,505 | 1.5 | % | |||||
| Erle T. Mast (5) |
104,962 | * | ||||||
| Faheem Hasnain (6) |
30,972 | * | ||||||
| Kurt von Emster (7) |
177,546 | * | ||||||
| Terrell A. Cobb (8) |
155,909 | * | ||||||
| Michael L. Eagle (9) |
113,146 | * | ||||||
| Thomas G. Wiggans (10) |
96,942 | * | ||||||
| Richard W. Pascoe (11) |
633,433 | 1.3 | % | |||||
| Tran B. Nguyen (12) |
149,648 | * | ||||||
| Matthew W. Onaitis (13) |
317,463 | * | ||||||
| Brian T. Dorsey (14) |
286,242 | * | ||||||
| Jeffrey W. Raser (15) |
547,747 | 1.1 | % | |||||
| Named Executive Officers and directors as a group (12 persons) (16) |
3,359,515 | 6.6 | % | |||||
| * | Indicates beneficial ownership of less than 1% of the total outstanding common stock. |
| (1) | The voting and disposition of the shares held by Ingalls & Snyder was obtained from the Schedule 13-G/A filed by Ingalls & Snyder on February 7, 2012. |
| (2) | Funds affiliated with MPM Capital L.P. include the following holdings: |
| Shareholder Name |
Number of Shares |
Number of
Warrants Exercisable within 60 days of February 15, 2012 |
||||||
| MPM Asset Management Investors 2006 BVIII LLC |
68,399 | 18,848 | ||||||
| MPM BioVentures III GmbH & Co. Beteiligungs KG |
326,317 | 89,923 | ||||||
| MPM BioVentures III Parallel Fund, L.P. |
116,646 | 32,144 | ||||||
| MPM BioVentures III, L.P. |
259,654 | 71,553 | ||||||
| MPM BioVentures III-QP, L.P. |
3,861,546 | 1,064,125 | ||||||
|
|
|
|
|
|||||
| Total |
4,632,562 | 1,276,593 | ||||||
|
|
|
|
|
|||||
85
MPM BioVentures III GP, L.P. and MPM BioVentures III LLC are the direct and indirect general partners of MPM BioVentures III-QP, L.P., MPM BioVentures III GmbH & Co. Beteiligungs KG, MPM BioVentures III, L.P. and MPM BioVentures III Parallel Fund, L.P. The members of MPM BioVentures III LLC and MPM Asset Management Investors 2006 BVIII LLC are Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Dennis Henner, Nicholas Simon III, Michael Steinmetz and Kurt Wheeler, who disclaim beneficial ownership of these shares except to the extent of their pecuniary interest therein.
| (3) | Shares held by Scale Venture Management I, LLC include warrants to purchase 510,638 shares of our common stock within 60 days of February 15, 2011. The voting and disposition of the shares held by Scale Venture Management I, LLC was obtained from the Schedule 13-G/A filed by BAVP, L.P. on July 13, 2009. |
| (4) | Shares held by David F. Hale include 424,718 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (5) | Shares held by Erle T. Mast include 83,332 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (6) | Shares held by Faheem Hasnain include 30,972 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (7) | Shares held by Kurt von Emster include 107,914 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012 and warrants to purchase 42,553 shares of our common stock within 60 days of February 15, 2012. |
| (8) | Shares held by Terrell A. Cobb include 123,887 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (9) | Shares held by Michael L. Eagle include 75,833 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (10) | Shares held by Thomas G. Wiggans include 70,416 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (11) | Shares held by Richard W. Pascoe include 561,041 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (12) | Shares held by the Tran B. Nguyen include 127,082 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (13) | Shares held by Matthew W. Onaitis include 262,426 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (14) | Shares held by Brian T. Dorsey include 251,790 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (15) | Shares held by Jeffrey W. Raser include 524,414 shares of common stock subject to outstanding options which are exercisable within 60 days of February 15, 2012. |
| (16) | Includes 2,643,825 shares of common stock subject to outstanding options and 42,553 warrants which are exercisable within 60 days of February 15, 2012. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
We describe below transactions and series of similar transactions, since the beginning of fiscal year 2011, with respect to which we were a party, will be a party, or otherwise benefited, in which:
| • | the amounts involved exceeded or will exceed $120,000; and |
| • | a director, executive officer, holder of more than 5% of our common stock or any member of their immediate family had or will have a direct or indirect material interest. |
86
We also describe below certain other transactions with our directors, executive officers and stockholders. We believe that the terms obtained or consideration that we paid or received, as applicable, in connection with the transactions described below were comparable to terms available or the amounts that would be paid or received, as applicable, in arm’s-length transactions.
Pursuant to our Audit Committee Charter, the audit committee of our board of directors is responsible for reviewing and approving all transactions with related parties. We have not adopted written procedures for review of, or standards for approval of, these transactions, but instead the audit committee of our board of directors intends to review such transactions on a case by case basis. In addition, the compensation committee of our board of directors and/or our board of directors will review and approve all compensation-related policies involving our directors and executive officers.
Our board of directors has determined that the members of our board of directors, with the exception of Mr. Hale, Mr. Cobb and Mr. Pascoe, none of whom serve on our audit committee, compensation committee, or nominating and corporate governance committee, are independent within the meaning of the independent director standards of the SEC and the Nasdaq Stock Market, Inc. Additional information concerning the independence of the members of our board of directors and the committees of the board of directors is set forth under Part III — Item 10, “Directors, Executive Officers and Corporate Governance.”
Employment Agreements and Change of Control Arrangements
We have entered into employment agreements, which are described in Part III — Item 11, “Executive Compensation” of this report, with our executive officers.
Indemnification of Officers and Directors
Our restated certificate of incorporation and our bylaws provide that we will indemnify each of our directors and officers to the fullest extent permitted by the Delaware General Corporation Law. Further, we have entered into indemnification agreements with each of our directors and officers, and we have purchased a policy of directors’ and officers’ liability insurance that insures our directors and officers against the cost of defense, settlement or payment of a judgment under certain circumstances.
Other Transactions
The approval of the NDA for Silenor in March 2010 triggered a $1,000,000 milestone payment obligation to ProCom One. Mr. Cobb, a member of our board of directors, is a co-founder and President of ProCom One. Pursuant to the Amended and Restated License Agreement with ProCom One, ProCom also receives a royalty on the sales of Silenor. Mr. Cobb received 1,000 RSUs during 2010 as compensation for his services on our board of directors. These RSUs vested on December 1, 2010.
Mr. Raser resigned from the company effective October 1, 2011. In connection with his resignation, Mr. Raser received the foregoing benefits under his employment agreement with us, including: (1) a lump sum severance payment of $315,954, (2) a lump sum payment equal to the cost of 12 months of health care benefits continuation at our expense, and (3) a lump sum payment equal to the cost of 12 months of the portion of the monthly premiums for Mr. Raser’s life insurance and disability insurance coverage that were borne by us. In addition, on October 1, 2011, the portion of Mr. Raser’s stock options and restricted stock units which would have vested if Mr. Raser had remained employed for an additional 12 months immediately vested.
Mr. Raser also entered into a consulting agreement us that will expire on June 30, 2013. We cannot estimate with any certainty the amount that may be paid, if any, for consulting services under such agreement. Mr. Raser’s remaining outstanding stock awards will continue to vest through the expiration of the consulting agreement, and he will be entitled to exercise such stock awards for 180 days following such expiration.
Item 14. Principal Accountant Fees and Services
Audit and All Other Fees
The following table presents fees for services rendered by PricewaterhouseCoopers LLP, our independent registered public accounting firm, for 2011 and 2010 in the following categories:
| 2011 | 2010 | |||||||
| Audit fees (1) |
$ | 447,000 | $ | 315,000 | ||||
| Audit related fees (2) |
— | — | ||||||
| Tax fees (3) |
45,000 | 51,000 | ||||||
| All other fees (4) |
— | — | ||||||
|
|
|
|
|
|||||
| Total |
$ | 492,000 | $ | 366,000 | ||||
|
|
|
|
|
|||||
87
| (1) | Audit fees consist of fees for professional services performed by PricewaterhouseCoopers LLP for the audit of our annual financial statements, review of our quarterly financial statements, review of our registration statements on Forms S-3 and related services that are normally provided in connection with statutory and regulatory filings or engagements. |
| (2) | Audit related fees consist of fees billed for assurance and related services performed by PricewaterhouseCoopers LLP that are reasonably related to the performance of the audit or review of our financial statements. |
| (3) | Tax fees consist of fees for professional services performed by PricewaterhouseCoopers LLP with respect to tax compliance, tax advice and tax planning. |
| (4) | All other fees consist of fees for other permissible work performed by PricewaterhouseCoopers LLP that is not included within the above category descriptions. There were no such fees incurred during 2011 or 2010. |
The audit committee has considered whether the provision of non-audit services is compatible with maintaining the independence of PricewaterhouseCoopers LLP, and has concluded that the provision of such services is compatible with maintaining the independence of our registered public accounting firm.
Audit Committee Policy Regarding Pre-Approval of Audit and Permissible Non-Audit Services of Our Independent Registered Public Accounting Firm
The audit committee has established a policy that all audit and permissible non-audit services provided by our independent registered public accounting firm will be pre-approved by the audit committee, and all such services were pre-approved in accordance with this policy during the fiscal years ended December 31, 2011 and 2010. These services may include audit services, audit-related services, tax services and other services. The audit committee considers whether the provision of each non-audit service is compatible with maintaining the independence of our registered public accounting firm. Pre-approval is detailed as to the particular service or category of services and is generally subject to a specific budget. Our independent registered public accounting firm and management are required to periodically report to the audit committee regarding the extent of services provided by the independent registered public accounting firm in accordance with this pre-approval, and the fees for the services performed to date.
Item 15. Exhibits and Financial Statement Schedules
| (a) | Documents filed as part of this report. |
1. The following financial statements of Somaxon Pharmaceuticals, Inc. and Report of PricewaterhouseCoopers LLP, independent registered public accounting firm, are included in this report:
| • | Report of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
| • | Balance Sheets as of December 31, 2011 and 2010 |
| • | Statements of Operations for the years ended December 31, 2011, 2010 and 2009 |
| • | Statements of Cash Flows for the years ended December 31, 2011, 2010 and 2009 |
| • | Statements of Stockholders’ Equity and Comprehensive Loss for years ended December 31, 2011, 2010 and 2009 |
| • | Notes to Financial Statements |
2. List of financial statement schedules. All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
3. List of exhibits required by Item 601 of Regulation S-K. See part (b) below.
| (b) | Exhibits. |
88
| Exhibit Number |
Description | |
| 3.1(1) | Amended and Restated Certificate of Incorporation of the Registrant | |
| 3.2(2) | Amended and Restated Bylaws of the Registrant | |
| 4.1(3) | Form of the Registrant’s Common Stock Certificate | |
| 4.2(4) | Amended and Restated Investor Rights Agreement dated June 2, 2005 | |
| 4.3(5) | Warrant issued to Silicon Valley Bank dated May 21, 2008 | |
| 4.4(5) | Warrant issued to Oxford Finance Corporation dated May 21, 2008 | |
| 4.5(5) | Warrant issued to Kingsbridge Capital Limited dated May 21, 2008 | |
| 4.6(6) | Form of Warrant issued to certain Purchasers under the Securities Purchase Agreement dated July 2, 2009 | |
| 4.7 | Warrant issued to Silicon Valley Bank dated December 19, 2011 | |
| 4.8 | Warrant issued to Oxford Finance Corporation dated December 19, 2011 | |
| 4.9(7) | Warrant issued to Silicon Valley Bank dated August 2, 2011 | |
| 4.10(7) | Warrant issued to Oxford Finance Corporation dated August 2, 2011 | |
| 4.11(7) | Warrant issued to Oxford Finance Corporation dated August 2, 2011 | |
| 10.1(1) | Form of Director and Executive Officer Indemnification Agreement | |
| 10.2#(1) | Director Compensation Policy | |
| 10.3#(4) | 2004 Equity Incentive Award Plan and forms of option agreements thereunder | |
| 10.4#(8) | Amended and Restated 2005 Equity Incentive Award Plan | |
| 10.5#(9) | 2005 Employee Stock Purchase Plan and form of Offering Document thereunder | |
| 10.6#(10) | 2010 Incentive Plan | |
| 10.7#(11) | 2011 Incentive Plan | |
| 10.8#(12) | Form of Amended and Restated Employment Agreement | |
| 10.9#(13) | Employment Agreement between the Registrant and Richard W. Pascoe dated August 7, 2008 | |
| 10.10#(14) | Employment Agreement between the Registrant and Tran Nguyen dated April 12, 2010 | |
| 10.11#(15) | Amendment to Employment Agreement between the Registrant and Tran Nguyen dated September 15, 2010 | |
| 10.12#(16) | Form of Director Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement | |
| 10.13#(16) | Form of Employee Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement | |
| 10.14#(16) | Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement between the Registrant and David F. Hale dated November 28, 2008 | |
| 10.15#(12) | Form of Restricted Stock Purchase Agreement | |
| 10.16#(12) | Restricted Stock Purchase Agreement between the Registrant and David F. Hale dated October 8, 2007 | |
| 10.17(6) | Securities Purchase Agreement between the Registrant and the Purchasers identified therein dated July 2, 2009 | |
89
| Exhibit Number |
Description | |
| 10.18(15) | Amended and Restated License Agreement by and between the Registrant and ProCom One, Inc. dated September 15, 2010 | |
| 10.19(4) | Common Stock Purchase Agreement by and among the Registrant, ProCom One, Inc. and Terrell A. Cobb dated April 15, 2004 | |
| 10.20†(17) | Manufacturing Services Agreement between the Registrant and Patheon Pharmaceuticals Inc. dated February 1, 2006 | |
| 10.21†(18) | Professional Detailing Services Agreement between the Registrant and Publicis Touchpoint Solutions, Inc. dated July 14, 2010 | |
| 10.22†(19) | Supplement No. 1 to the Professional Detailing Services Agreement between the Registrant and Publicis Touchpoint Solutions, Inc. dated February 7, 2011 | |
| 10.23†(20) | Co-Promotion Agreement between the Registrant and The Procter & Gamble Distributing Company, LLC dated August 24, 2010 | |
| 10.24(21) | Loan and Security Agreement by and between the Registrant and Comerica Bank dated as of February 7, 2011 | |
| 10.25(21) | LIBOR Addendum to Loan and Security Agreement by and between the Registrant and Comerica Bank dated as of February 7, 2011 | |
| 10.26(22) | Lease Agreement between the Registrant and PRII Gateway Torrey Hills LLC dated as of April 22, 2010 | |
| 10.27(23) | Lease Agreement between the Registrant and TREA Pacific Plaza, LLC dated May 24, 2011 | |
| 10.28†(24) | License Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.29†(24) | Supply Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.30†(24) | Purchase Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.31(7) | At-the-Market Equity Offering Sales Agreement between the Registrant and Citadel Securities LLC dated August 1, 2011 | |
| 10.32(7) | Loan and Security Agreement between the Registrant, Oxford Finance LLC, as collateral agent, and Silicon Valley Bank and Oxford Finance LLC, as lenders, dated as of August 2, 2011 | |
| 10.33(25) | Letter Agreement dated September 30, 2011, between the Registrant and The Procter & Gamble Distributing Company LLC | |
| 10.34#(25) | Employment Agreement dated September 30, 2011, between the Registrant and Michael D. Allen | |
| 10.35# | Consulting Agreement dated October 1, 2011, between the Registrant and Jeffrey Raser | |
| 10.36(26) | Letter agreement dated November 23, 2011, between the Registrant and The Procter & Gamble Distributing Company LLC | |
| 10.37#(27) | Amendment dated November 1, 2011 to Employment Agreement between Registrant and Michael D. Allen | |
| 10.38†(27) | First Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated March 22, 2011 | |
| 10.39†(27) | Second Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated August 15, 2011 | |
| 10.40†(27) | Third Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated November 1, 2011 | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm | |
90
| Exhibit Number |
Description | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14 and Rule 15d-14 of the Securities Exchange Act of 1934, as amended | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14 and Rule 15d-14 of the Securities Exchange Act of 1934, as amended | |
| 32.1* | Certification of Chief Executive Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2003 | |
| 32.2* | Certification of Chief Financial Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2003 | |
| 101 | The following financial statements and footnotes formatted in XBRL: (i) Balance Sheets; (ii) Statements of Operations; (iii) Statements of Cash Flows; (iv) Statements of Stockholders’ Equity and Comprehensive Loss; and (v) Notes to Financial Statements, tagged as blocks of text. | |
| # | Indicates management contract or compensatory plan. |
| † | Confidential treatment has been granted or has been requested as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
| * | These certifications are being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and are not to be incorporated by reference into any filing of Somaxon Pharmaceuticals, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
| (1) | Filed with Amendment No. 3 to the Registrant’s Registration Statement on Form S-1 on November 30, 2005. |
| (2) | Filed with Registrant’s Current Report on Form 8-K on December 6, 2007. |
| (3) | Filed with Amendment No. 4 to the Registrant’s Registration Statement on Form S-1 on December 13, 2005. |
| (4) | Filed with the Registrant’s Registration Statement on Form S-1 on October 7, 2005. |
| (5) | Filed with Registrant’s Current Report on Form 8-K on May 22, 2008. |
| (6) | Filed with Registrant’s Current Report on Form 8-K on July 8, 2009. |
| (7) | Filed with Registrant’s Current Report on Form 8-K on August 2, 2011. |
| (8) | Filed with Registrant’s Definitive Proxy Statement on April 30, 2009. |
| (9) | Filed with the Registrant’s Registration Statement on Form S-8 on December 15, 2005. |
| (10) | Filed with Registrant’s Current Report on Form 8-K on April 7, 2010. |
| (11) | Filed with Registrant’s Current Report on Form 8-K on February 14, 2011. |
| (12) | Filed with Registrant’s Annual Report on Form 10-K on March 12, 2008. |
| (13) | Filed with Registrant’s Current Report on Form 8-K on August 7, 2008. |
| (14) | Filed with Registrant’s Current Report on Form 8-K on April 14, 2010. |
| (15) | Filed with the Registrant’s Quarterly Report on Form 10-Q on November 10, 2010. |
| (16) | Filed with Registrant’s Annual Report on Form 10-K on March 13, 2009. |
| (17) | Filed with the Registrant’s Quarterly Report on Form 10-Q on May 11, 2006 |
| (18) | Filed with Registrant’s Current Report on Form 8-K/A on July 21, 2010. |
91
| (19) | Filed with Registrant’s Current Report on Form 8-K/A on February 11, 2011. |
| (20) | Filed with Registrant’s Current Report on Form 8-K on August 25, 2010. |
| (21) | Filed with Registrant’s Current Report on Form 8-K on February 8, 2011. |
| (22) | Filed with Registrant’s Current Report on Form 8-K on April 28, 2010. |
| (23) | Filed with Registrant’s Current Report on Form 8-K on May 25, 2011. |
| (24) | Filed with Registrant’s Current Report on Form 8-K on June 13, 2011. |
| (25) | Filed with Registrant’s Current Report on Form 8-K on October 3, 2011. |
| (26) | Filed with Registrant’s Current Report on Form 8-K on November 25, 2011. |
| (27) | Filed with Registrant’s Quarterly Report on Form 10-Q on November 3, 2011. |
| (c) | Financial Statement Schedule. See Item 15(a)(2) above. |
92
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| SOMAXON PHARMACEUTICALS, INC. | ||||||
| Dated: March 9, 2012 | By: | /s/ Richard W. Pascoe | ||||
| Richard W. Pascoe | ||||||
| President and Chief Executive Officer | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Richard W. Pascoe |
President and Chief Executive Officer | March 9, 2012 | ||
| Richard W. Pascoe | (Principal Executive Officer) | |||
| /s/ Tran B. Nguyen Tran B. Nguyen |
Senior Vice President and Chief Financial Officer |
March 9, 2012 | ||
| (Principal Financial Officer and Principal Accounting Officer) |
||||
| /s/ David F. Hale |
Chairman of the Board of Directors | March 9, 2012 | ||
| David F. Hale | ||||
| /s/ Terrell A. Cobb |
Director | March 9, 2012 | ||
| Terrell A. Cobb | ||||
| /s/ Michael L. Eagle |
Director | March 9, 2012 | ||
| Michael L. Eagle | ||||
| /s/ Erle T. Mast |
Director | March 9, 2012 | ||
| Erle T. Mast | ||||
| /s/ Faheem Hasnain |
Director | March 9, 2012 | ||
| Faheem Hasnain | ||||
| /s/ Kurt von Emster |
Director | March 9, 2012 | ||
| Kurt von Emster | ||||
| /s/ Thomas G. Wiggans |
Director | March 9, 2012 | ||
| Thomas G. Wiggans | ||||
93
EXHIBIT INDEX
| Exhibit Number |
Description | |
| 3.1(1) | Amended and Restated Certificate of Incorporation of the Registrant | |
| 3.2(2) | Amended and Restated Bylaws of the Registrant | |
| 4.1(3) | Form of the Registrant’s Common Stock Certificate | |
| 4.2(4) | Amended and Restated Investor Rights Agreement dated June 2, 2005 | |
| 4.3(5) | Warrant issued to Silicon Valley Bank dated May 21, 2008 | |
| 4.4(5) | Warrant issued to Oxford Finance Corporation dated May 21, 2008 | |
| 4.5(5) | Warrant issued to Kingsbridge Capital Limited dated May 21, 2008 | |
| 4.6(6) | Form of Warrant issued to certain Purchasers under the Securities Purchase Agreement dated July 2, 2009 | |
| 4.7 | Warrant issued to Silicon Valley Bank dated December 19, 2011 | |
| 4.8 | Warrant issued to Oxford Finance Corporation dated December 19, 2011 | |
| 4.9(7) | Warrant issued to Silicon Valley Bank dated August 2, 2011 | |
| 4.10(7) | Warrant issued to Oxford Finance Corporation dated August 2, 2011 | |
| 4.11(7) | Warrant issued to Oxford Finance Corporation dated August 2, 2011 | |
| 10.1(1) | Form of Director and Executive Officer Indemnification Agreement | |
| 10.2#(1) | Director Compensation Policy | |
| 10.3#(4) | 2004 Equity Incentive Award Plan and forms of option agreements thereunder | |
| 10.4#(8) | Amended and Restated 2005 Equity Incentive Award Plan | |
| 10.5#(9) | 2005 Employee Stock Purchase Plan and form of Offering Document thereunder | |
| 10.6#(10) | 2010 Incentive Plan | |
| 10.7#(11) | 2011 Incentive Plan | |
| 10.8#(12) | Form of Amended and Restated Employment Agreement | |
| 10.9#(13) | Employment Agreement between the Registrant and Richard W. Pascoe dated August 7, 2008 | |
| 10.10#(14) | Employment Agreement between the Registrant and Tran Nguyen dated April 12, 2010 | |
| 10.11#(15) | Amendment to Employment Agreement between the Registrant and Tran Nguyen dated September 15, 2010 | |
| 10.12#(16) | Form of Director Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement | |
| 10.13#(16) | Form of Employee Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement | |
| 10.14#(16) | Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Award Agreement between the Registrant and David F. Hale dated November 28, 2008 | |
| 10.15#(12) | Form of Restricted Stock Purchase Agreement | |
| 10.16#(12) | Restricted Stock Purchase Agreement between the Registrant and David F. Hale dated October 8, 2007 | |
94
| Exhibit Number |
Description | |
| 10.17(6) | Securities Purchase Agreement between the Registrant and the Purchasers identified therein dated July 2, 2009 | |
| 10.18(15) | Amended and Restated License Agreement by and between the Registrant and ProCom One, Inc. dated September 15, 2010 | |
| 10.19(4) | Common Stock Purchase Agreement by and among the Registrant, ProCom One, Inc. and Terrell A. Cobb dated April 15, 2004 | |
| 10.20†(17) | Manufacturing Services Agreement between the Registrant and Patheon Pharmaceuticals Inc. dated February 1, 2006 | |
| 10.21†(18) | Professional Detailing Services Agreement between the Registrant and Publicis Touchpoint Solutions, Inc. dated July 14, 2010 | |
| 10.22†(19) | Supplement No. 1 to the Professional Detailing Services Agreement between the Registrant and Publicis Touchpoint Solutions, Inc. dated February 7, 2011 | |
| 10.23†(20) | Co-Promotion Agreement between the Registrant and The Procter & Gamble Distributing Company, LLC dated August 24, 2010 | |
| 10.24(21) | Loan and Security Agreement by and between the Registrant and Comerica Bank dated as of February 7, 2011 | |
| 10.25(21) | LIBOR Addendum to Loan and Security Agreement by and between the Registrant and Comerica Bank dated as of February 7, 2011 | |
| 10.26(22) | Lease Agreement between the Registrant and PRII Gateway Torrey Hills LLC dated as of April 22, 2010 | |
| 10.27(23) | Lease Agreement between the Registrant and TREA Pacific Plaza, LLC dated May 24, 2011 | |
| 10.28†(24) | License Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.29†(24) | Supply Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.30†(24) | Purchase Agreement between the Registrant and Paladin Labs Inc. dated June 7, 2011 | |
| 10.31(7) | At-the-Market Equity Offering Sales Agreement between the Registrant and Citadel Securities LLC dated August 1, 2011 | |
| 10.32(7) | Loan and Security Agreement between the Registrant, Oxford Finance LLC, as collateral agent, and Silicon Valley Bank and Oxford Finance LLC, as lenders, dated as of August 2, 2011 | |
| 10.33(25) | Letter Agreement dated September 30, 2011, between the Registrant and The Procter & Gamble Distributing Company LLC | |
| 10.34#(25) | Employment Agreement dated September 30, 2011, between the Registrant and Michael D. Allen | |
| 10.35# | Consulting Agreement dated October 1, 2011, between the Registrant and Jeffrey Raser | |
| 10.36(26) | Letter agreement dated November 23, 2011, between the Registrant and The Procter & Gamble Distributing Company LLC | |
| 10.37#(27) | Amendment dated November 1, 2011 to Employment Agreement between Registrant and Michael D. Allen | |
| 10.38†(27) | First Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated March 22, 2011 | |
| 10.39†(27) | Second Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated August 15, 2011 | |
| 10.40†(27) | Third Amendment to Supplement No. 1 to the Professional Detailing Services Agreement between Registrant and Publicis Touchpoint Solutions, Inc. dated November 1, 2011 | |
95
| Exhibit Number |
Description | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14 and Rule 15d-14 of the Securities Exchange Act of 1934, as amended | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14 and Rule 15d-14 of the Securities Exchange Act of 1934, as amended | |
| 32.1* | Certification of Chief Executive Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2003 | |
| 32.2* | Certification of Chief Financial Officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2003 | |
| 101 | The following financial statements and footnotes formatted in XBRL: (i) Balance Sheets; (ii) Statements of Operations; (iii) Statements of Cash Flows; (iv) Statements of Stockholders’ Equity and Comprehensive Loss; and (v) Notes to Financial Statements, tagged as blocks of text. | |
| # | Indicates management contract or compensatory plan. |
| † | Confidential treatment has been granted or has been requested as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
| * | These certifications are being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and are not to be incorporated by reference into any filing of Somaxon Pharmaceuticals, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
| (1) | Filed with Amendment No. 3 to the Registrant’s Registration Statement on Form S-1 on November 30, 2005. |
| (2) | Filed with Registrant’s Current Report on Form 8-K on December 6, 2007. |
| (3) | Filed with Amendment No. 4 to the Registrant’s Registration Statement on Form S-1 on December 13, 2005. |
| (4) | Filed with the Registrant’s Registration Statement on Form S-1 on October 7, 2005. |
| (5) | Filed with Registrant’s Current Report on Form 8-K on May 22, 2008. |
| (6) | Filed with Registrant’s Current Report on Form 8-K on July 8, 2009. |
| (7) | Filed with Registrant’s Current Report on Form 8-K on August 2, 2011. |
| (8) | Filed with Registrant’s Definitive Proxy Statement on April 30, 2009. |
| (9) | Filed with the Registrant’s Registration Statement on Form S-8 on December 15, 2005. |
| (10) | Filed with Registrant’s Current Report on Form 8-K on April 7, 2010. |
| (11) | Filed with Registrant’s Current Report on Form 8-K on February 14, 2011. |
| (12) | Filed with Registrant’s Annual Report on Form 10-K on March 12, 2008. |
| (13) | Filed with Registrant’s Current Report on Form 8-K on August 7, 2008. |
| (14) | Filed with Registrant’s Current Report on Form 8-K on April 14, 2010. |
| (15) | Filed with the Registrant’s Quarterly Report on Form 10-Q on November 10, 2010. |
| (16) | Filed with Registrant’s Annual Report on Form 10-K on March 13, 2009. |
| (17) | Filed with the Registrant’s Quarterly Report on Form 10-Q on May 11, 2006 |
| (18) | Filed with Registrant’s Current Report on Form 8-K/A on July 21, 2010. |
| (19) | Filed with Registrant’s Current Report on Form 8-K/A on February 11, 2011. |
| (20) | Filed with Registrant’s Current Report on Form 8-K on August 25, 2010. |
| (21) | Filed with Registrant’s Current Report on Form 8-K on February 8, 2011. |
| (22) | Filed with Registrant’s Current Report on Form 8-K on April 28, 2010. |
| (23) | Filed with Registrant’s Current Report on Form 8-K on May 25, 2011. |
| (24) | Filed with Registrant’s Current Report on Form 8-K on June 13, 2011. |
| (25) | Filed with Registrant’s Current Report on Form 8-K on October 3, 2011. |
| (26) | Filed with Registrant’s Current Report on Form 8-K on November 25, 2011. |
| (27) | Filed with Registrant’s Quarterly Report on Form 10-Q on November 3, 2011. |
96
SOMAXON PHARMACEUTICALS, INC.
INDEX TO FINANCIAL STATEMENTS
| F-1 | ||||
| Financial Statements |
||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
97
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Somaxon Pharmaceuticals, Inc.
In our opinion, the accompanying balance sheets and the related statements of operations, of stockholders’ equity and comprehensive loss and of cash flows present fairly, in all material respects, the financial position of Somaxon Pharmaceuticals, Inc. at December 31, 2011 and 2010, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2011 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our audits (which were integrated audits for 2011 and 2010). We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and negative cash flows that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regards to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
PricewaterhouseCoopers LLP
San Diego, California
March 9, 2012
F-1
SOMAXON PHARMACEUTICALS, INC.
(in thousands, except par value)
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| ASSETS |
||||||||
| Current assets |
||||||||
| Cash and cash equivalents |
$ | 10,668 | $ | 21,008 | ||||
| Short-term investments |
— | 33,809 | ||||||
| Current portion of restricted cash |
50 | — | ||||||
| Accounts receivable, net |
1,950 | 5,584 | ||||||
| Inventory |
264 | 991 | ||||||
| Other current assets |
1,003 | 1,882 | ||||||
|
|
|
|
|
|||||
| Total current assets |
13,935 | 63,274 | ||||||
| Long-term portion of restricted cash |
201 | — | ||||||
| Property and equipment, net |
634 | 755 | ||||||
| Intangibles, net |
1,089 | 1,102 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 15,859 | $ | 65,131 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities |
||||||||
| Accounts payable |
$ | 1,774 | $ | 1,709 | ||||
| Accrued liabilities |
7,054 | 5,699 | ||||||
| Deferred revenue |
— | 3,459 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
8,828 | 10,867 | ||||||
| Other long-term liabilities |
490 | — | ||||||
| Commitments and contingencies: (Notes 6 and 7) |
||||||||
|
|
|
|
|
|||||
| Total liabilities |
9,318 | 10,867 | ||||||
| Stockholders’ equity |
||||||||
| Preferred stock, $0.0001 par value; 10,000 shares authorized, none issued and outstanding |
— | — | ||||||
| Common stock, $0.0001 par value; 100,000 shares authorized; 48,063 and 45,004 shares outstanding at December 31, 2011 and 2010, respectively |
5 | 5 | ||||||
| Additional paid-in capital |
282,668 | 271,112 | ||||||
| Accumulated deficit |
(276,132 | ) | (216,852 | ) | ||||
| Accumulated other comprehensive loss |
— | (1 | ) | |||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
6,541 | 54,264 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 15,859 | $ | 65,131 | ||||
|
|
|
|
|
|||||
The Accompanying Notes are an Integral Part of these Financial Statements
F-2
SOMAXON PHARMACEUTICALS, INC.
(in thousands, except per share amounts)
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Revenue |
||||||||||||
| Net product sales |
$ | 16,155 | $ | 1,382 | $ | — | ||||||
| Operating costs and expenses |
||||||||||||
| Cost of sales |
2,493 | 244 | — | |||||||||
| Selling, general and administrative |
69,758 | 36,579 | 10,874 | |||||||||
| Research and development |
1,296 | 3,566 | 4,337 | |||||||||
| License fees |
— | — | (999 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total operating costs and expenses |
73,547 | 40,389 | 14,212 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(57,392 | ) | (39,007 | ) | (14,212 | ) | ||||||
| Interest and other income |
52 | 262 | 30 | |||||||||
| Interest and other expense |
(1,940 | ) | (68 | ) | (261 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (59,280 | ) | $ | (38,813 | ) | $ | (14,443 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (1.27 | ) | $ | (1.16 | ) | $ | (0.69 | ) | |||
|
|
|
|
|
|
|
|||||||
| Shares used to calculate net loss per share |
46,541 | 33,593 | 20,952 | |||||||||
|
|
|
|
|
|
|
|||||||
The Accompanying Notes are an Integral Part of these Financial Statements
F-3
SOMAXON PHARMACEUTICALS, INC.
(in thousands)
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Cash flows from operating activities |
||||||||||||
| Net loss |
$ | (59,280 | ) | $ | (38,813 | ) | $ | (14,443 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Share-based compensation expense |
5,248 | 6,706 | 6,163 | |||||||||
| Depreciation and amortization |
534 | 347 | 83 | |||||||||
| Amortization of intangible assets |
174 | 45 | — | |||||||||
| Amortization of investment discount or premium |
148 | 132 | (38 | ) | ||||||||
| Write-off of excess inventory |
570 | — | — | |||||||||
| Write-off of prepaid sales and marketing expenses |
1,339 | — | — | |||||||||
| Issuance of warrants related to repayment of loan |
59 | — | — | |||||||||
| Realized gain on sale of short-term investments |
1 | — | — | |||||||||
| Loss on disposal of equipment and technology development costs |
— | 119 | 2 | |||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Accounts receivable |
3,634 | (5,584 | ) | — | ||||||||
| Inventory |
38 | (991 | ) | — | ||||||||
| Other current and non-current assets |
(405 | ) | (1,213 | ) | 70 | |||||||
| Accounts payable |
65 | 1,354 | (1,470 | ) | ||||||||
| Accrued liabilities |
1,322 | 4,743 | 73 | |||||||||
| Deferred revenue and other liabilities |
(2,936 | ) | 3,459 | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(49,489 | ) | (29,696 | ) | (9,560 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities |
||||||||||||
| Purchases of property and equipment |
(413 | ) | (392 | ) | (74 | ) | ||||||
| Payments for intangible assets |
(161 | ) | (1,200 | ) | — | |||||||
| Purchases of marketable securities |
(3,508 | ) | (46,457 | ) | (2,505 | ) | ||||||
| Sales and maturities of marketable securities |
37,233 | 12,317 | 5,639 | |||||||||
| Restricted cash |
(251 | ) | — | 8,100 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
32,900 | (35,732 | ) | 11,160 | ||||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities |
||||||||||||
| Issue common stock, net of costs |
5,547 | 77,556 | 5,732 | |||||||||
| Net proceeds from issuance of debt |
14,050 | — | — | |||||||||
| Net proceeds from issuance of warrants |
701 | — | — | |||||||||
| Exercise of stock options |
1 | 2,285 | 173 | |||||||||
| Exercise of warrants |
— | 1,474 | 1,475 | |||||||||
| Purchase of treasury stock |
— | (44 | ) | — | ||||||||
| Repayment of debt |
(14,050 | ) | — | (15,000 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) financing activities |
6,249 | 81,271 | (7,620 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| (Decrease) increase in cash and cash equivalents |
(10,340 | ) | 15,843 | (6,020 | ) | |||||||
| Cash and cash equivalents at beginning of the period |
21,008 | 5,165 | 11,185 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of the period |
$ | 10,668 | $ | 21,008 | $ | 5,165 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing and financing activities |
||||||||||||
| Common stock issued to settle severance obligation |
$ | — | $ | 860 | $ | — | ||||||
| Issuance of warrants related to loan agreement |
— | — | 44 | |||||||||
| Supplemental cash flow information |
||||||||||||
| Cash paid for interest |
$ | 931 | $ | — | $ | 984 | ||||||
The Accompanying Notes are an Integral Part of these Financial Statements
F-4
SOMAXON PHARMACEUTICALS, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
(In thousands, except per share amounts)
| Common Stock | Additional Paid-in |
Accumulated | Other Accumulated Comprehensive |
|||||||||||||||||||||
| Shares | Amount | Capital | Deficit | Income (Loss) | Total | |||||||||||||||||||
| Balance at January 1, 2009 |
18,430 | $ | 2 | $ | 168,691 | $ | (163,596 | ) | $ | 9 | $ | 5,106 | ||||||||||||
| Net loss |
— | — | — | (14,443 | ) | — | (14,443 | ) | ||||||||||||||||
| Unrealized loss on available-for-sale securities |
— | — | — | — | (9 | ) | (9 | ) | ||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (14,452 | ) | |||||||||||||||||
| Warrants issued pursuant to loan payoff |
— | — | 44 | — | — | 44 | ||||||||||||||||||
| Issue common stock |
5,106 | 1 | 5,731 | — | — | 5,732 | ||||||||||||||||||
| Issue common stock pursuant to vesting of restricted stock units |
166 | — | — | — | — | — | ||||||||||||||||||
| Exercise of warrants for cash |
1,277 | — | 1,475 | — | — | 1,475 | ||||||||||||||||||
| Net share settlement of warrants |
183 | — | — | — | — | — | ||||||||||||||||||
| Exercise of stock options |
116 | — | 173 | — | — | 173 | ||||||||||||||||||
| Repurchase of restricted stock |
(30 | ) | — | — | — | — | — | |||||||||||||||||
| Share-based compensation |
— | — | 6,163 | — | — | 6,163 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2009 |
25,248 | 3 | 182,277 | (178,039 | ) | — | 4,241 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss |
— | — | — | (38,813 | ) | — | (38,813 | ) | ||||||||||||||||
| Unrealized loss on available-for-sale securities |
— | — | — | — | (1 | ) | (1 | ) | ||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (38,814 | ) | |||||||||||||||||
| Issue common stock |
15,700 | 2 | 77,554 | — | — | 77,556 | ||||||||||||||||||
| Issue common stock to settle severance obligations |
111 | — | 860 | — | — | 860 | ||||||||||||||||||
| Exercise of warrants for cash |
1,278 | — | 1,474 | — | — | 1,474 | ||||||||||||||||||
| Net share settlement of warrants |
330 | — | — | — | — | — | ||||||||||||||||||
| Exercise of stock options |
1,610 | — | 2,285 | — | — | 2,285 | ||||||||||||||||||
| Issue common stock pursuant to vesting of restricted stock units |
738 | — | — | — | — | — | ||||||||||||||||||
| Repurchase of restricted stock |
(11 | ) | — | (44 | ) | — | — | (44 | ) | |||||||||||||||
| Share-based compensation |
— | — | 6,706 | — | — | 6,706 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2010 |
45,004 | 5 | 271,112 | (216,852 | ) | (1 | ) | 54,264 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss |
— | — | — | (59,280 | ) | — | (59,280 | ) | ||||||||||||||||
| Unrealized gains in short-term investments |
— | — | — | — | 1 | 1 | ||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (59,279 | ) | |||||||||||||||||
| Issuance of common stock |
2,972 | — | 5,547 | — | — | 5,547 | ||||||||||||||||||
| Issue common stock pursuant to vesting of restricted stock units |
86 | — | — | — | — | — | ||||||||||||||||||
| Issuance of warrants related to loan agreement |
— | — | 760 | — | — | 760 | ||||||||||||||||||
| Exercise of stock options |
1 | — | 1 | — | — | 1 | ||||||||||||||||||
| Share-based compensation expense |
— | — | 5,248 | — | — | 5,248 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2011 |
48,063 | $ | 5 | $ | 282,668 | $ | (276,132 | ) | $ | — | $ | 6,541 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The Accompanying Notes are an Integral Part of these Financial Statements
F-5
SOMAXON PHARMACEUTICALS, INC.
Note 1. Organization
Business
Somaxon Pharmaceuticals, Inc. (“Somaxon”, “the Company”, “we”, “us” or “our”) is a specialty pharmaceutical company focused on the in-licensing, development and commercialization of proprietary branded products and late-stage product candidates to treat important medical conditions where there is an unmet medical need and/or high-level of patient dissatisfaction, currently in the central nervous system therapeutic area. In March 2010, the U.S. Food and Drug Administration (“FDA”) approved our New Drug Application (“NDA”) for Silenor® 3 mg and 6 mg tablets for the treatment of insomnia characterized by difficulty with sleep maintenance. Silenor was made commercially available by prescription in the United States in September 2010. We operate in one reportable segment, which is the development and commercialization of pharmaceutical products.
Capital Resources
Since inception, our operations have been financed primarily through the sale of equity securities and the proceeds from the exercise of warrants and stock options. We have incurred losses from operations and negative cash flows since our inception, and we expect to continue to incur substantial losses for the foreseeable future as we continue our commercial activities for Silenor, commercialize any other products to which we obtain rights and potentially pursue the development of other product candidates. Based on our recurring losses, negative cash flows from operations and working capital levels, we will need to raise substantial additional funds to finance our operations. If we are unable to maintain sufficient financial resources, including by raising additional funds when needed, our business, financial condition and results of operations will be materially and adversely affected.
In August 2011, we entered into an at-the-market equity sales agreement with Citadel Securities LLC (“Citadel”). However, there can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate. Citadel or the Company is permitted to terminate the sales agreement at any time. Sales of shares pursuant to the sales agreement will have a dilutive effect on the holdings of our existing stockholders, and may result in downward pressure on the price of our common stock.
We commercially launched Silenor in September 2010 with 110 sales representatives provided to us on an exclusive basis under our contract sales agreement with Publicis Touchpoint Solutions, Inc. (“Publicis”), managed by our sales management personnel, and an additional 105 sales representatives provided to us under our co-promotion agreement with The Procter & Gamble Distributing Company LLC (“P&G”). In February 2011, we amended our agreement with Publicis to have Publicis deploy for us an additional 35 sales representatives. Because we did not believe that the growth of Silenor revenues throughout 2011 was sufficient to support sales and marketing expenses at then-current levels, we terminated our agreements with Publicis and P&G in December 2011. At the conclusion of the contract term with Publicis, we were contractually obligated to assume financial responsibility for the remaining vehicle lease payments associated with our Publicis sales representatives. Accrued liabilities as of December 31, 2011, includes a balance due to Publicis of $0.6 million, which represents all amounts owed to Publicis and all other third parties in connection with the contract sales agreement. All of our obligations associated with our contract sales agreement with Publicis were settled in full in February 2012. Effective January 3, 2012, we hired a reduced sales force of 25 sales representatives from the Publicis sales force to promote Silenor as Somaxon employees. The remainder of the Publicis sales force ceased promoting Silenor as of November 2, 2011. In addition, we terminated the employment of 28 employees in the fourth quarter of 2011.
We will need to obtain additional funds to finance our operations. Until we can generate significant cash from our operations, we intend to obtain any additional funding we require through public or private equity or debt financings, strategic relationships, assigning receivables or royalty rights, or other arrangements and we cannot assure such funding will be available on reasonable terms, or at all. Additional equity financing will be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants.
F-6
In December 2011 we hired Stifel Nicolaus Weisel as a strategic advisor to assist us in identifying and evaluating strategies to maximize stockholder value by leveraging our rights in Silenor. The exploration of strategic alternatives may not result in any agreement or transaction and, if completed, any agreement or transaction may not be successful or on attractive terms. The inability to enter into a strategic transaction, or a strategic transaction that is not successful or on attractive terms, could accelerate our need for cash and make securing funding on reasonable terms more difficult. In addition, if we raise additional funds through collaborations or other strategic transactions, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
If our efforts in raising additional funds when needed are unsuccessful, we may be required to delay, scale-back or eliminate plans or programs relating to our business, relinquish some or all rights to Silenor or renegotiate less favorable terms with respect to such rights than we would otherwise choose or cease operating as a going concern. In addition, if we do not meet our payment obligations to third parties as they come due, we may be subject to litigation claims. Even if we were successful in defending against these claims, litigation could result in substantial costs and be a distraction to management, and may result in unfavorable results that could further adversely impact our financial condition.
If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our financial statements, and it is likely that investors will lose all or a part of their investments. These financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Note 2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates.
Cash and Cash Equivalents
All highly liquid investments with maturities of three months or less at the time of purchase are considered to be cash equivalents. Investments with maturities at the date of purchase greater than three months are classified as marketable securities. At December 31, 2010, our cash and cash equivalents consisted primarily of available-for-sale securities that have an original maturity date of three months or less. At December 31, 2011, our cash and cash equivalents consisted solely of cash.
Marketable Securities
Our investments in marketable securities are classified as available-for-sale securities. Available-for-sale securities are carried at fair market value, with unrealized gains and losses reported as a component of stockholders’ equity in accumulated other comprehensive income/loss. Interest and dividend income is recorded when earned and included in interest income. Premiums and discounts on marketable securities are amortized and accreted, respectively, to maturity and included in interest income. Marketable securities with a maturity date of less than one year as of the balance sheet date are classified as short-term investments. Marketable securities with a maturity of more than one year as of the balance sheet date are classified as long-term investments. We assess the risk of impairment related to securities held in our investment portfolio on a regular basis and noted no impairment during the year ended December 31, 2011.
Concentration of Credit Risk, Significant Customers and Sources of Supply
Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents, short-term investments, and accounts receivable. We maintain accounts in federally insured financial institutions in excess of federally insured limits. We also maintain investments in money market funds and similar
F-7
short-term investments that are not federally insured. However, management believes we are not exposed to significant credit risk due to the financial positions of the depository institutions in which these deposits are held and of the money market funds and other entities in which these investments are made. Additionally, we have established guidelines regarding the diversification of our investments and their maturities that are designed to maintain safety and liquidity.
We sell our product primarily to established wholesale distributors in the pharmaceutical industry. The following table sets forth customers who represented 10% or more of our revenues:
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Cardinal Health |
44 | % | — | — | ||||||||
| McKesson |
37 | % | — | — | ||||||||
| AmerisourceBergen |
12 | % | — | — | ||||||||
| Integrated Commercialization Solutions, Inc. |
— | 100 | % | — | ||||||||
The majority of our accounts receivable balance as of December 31, 2011 represents amounts due from these three wholesale distributors. Credit is extended based on an evaluation of the customer’s financial condition. Based upon the review of these factors, we did not record an allowance for doubtful accounts at December 31, 2011 or 2010.
We rely on third-party manufacturers for the production of Silenor and single source third-party suppliers to manufacture key components of Silenor. If our third-party manufacturers are unable to continue manufacturing Silenor, or if we lost our single source suppliers used in the manufacturing process, we may not be able to meet market demand for our product.
Inventory
Our inventories are valued at the lower of weighted average cost or net realizable value. We analyze our inventory levels quarterly and write down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value, as well as any inventory quantities in excess of expected requirements. We did not record a provision or write-down inventory as of December 31, 2010. We recorded a write-down of $0.6 million for excess inventory during the year ended December 31, 2011, which is included in cost of sales.
Intangible Assets
Our intangible assets consist of the costs incurred to in-license our product and technology development costs relating to our websites. Prior to the FDA approval of our NDA for Silenor, we had expensed all license fees and milestone payments for acquired development and commercialization rights to operations as incurred since the underlying technology associated with these expenditures related to our research and development efforts and had no alternative future use at the time. Costs related to our intellectual property are capitalized once technological feasibility has been established. Capitalized amounts are amortized on a straight line basis over the expected life of the intellectual property. License fees began being amortized upon the first sale of Silenor to our wholesaler in August 2010 and are being amortized over approximately ten years. Costs incurred in the planning stage of a website are expensed, while costs incurred in the development stage are capitalized and will be amortized over the expected life of the product associated with the website once the asset is placed in service. Costs incurred for other intangible assets to be used primarily on our website are capitalized and amortized over the expected useful life, which we estimate to be two years. The carrying values of our intangible assets are periodically reviewed to determine if the facts and circumstances suggest that a potential impairment may have occurred. We had no impairment of our intangible assets for the year ended December 31, 2011.
Property and Equipment
Property and equipment is stated at cost less accumulated depreciation. Depreciation is accounted for using the straight-line method over the estimated useful life of the asset or the shorter of the lease term or the estimated useful
F-8
life for leasehold improvements. Useful lives generally range from three years for computer equipment to five years for furniture, equipment and tooling. Leasehold improvements are amortized over the estimated useful life of the asset or the lease term, whichever is shorter.
Impairment of Long-Lived Assets
We assess the recoverability of our long-lived assets by determining whether the carrying value of such assets can be recovered through undiscounted future operating cash flows expected to result from the use of those assets. If impairment is indicated, we measure the amount of such impairment by comparing the fair value to the carrying value. We did not consider our long-lived assets to be impaired as of December 31, 2011.
Revenue Recognition
Product Sales
We sell Silenor to wholesale pharmaceutical distributors. Our returned goods policy generally permits our customers to return products up to six months before and up to twelve months after the expiration date of the product. We authorize returns for expired products in accordance with our returned goods policy and issue credit to our customers for expired returned product. We do not exchange product from inventory for returned product. As of December 31, 2011, the dollar amount of returns received in 2011 has been negligible.
We recognize product revenue from product sales when it is realized or realizable and earned. Revenue is realized or realizable and earned when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) our price to the buyer is fixed or determinable; and (4) collectability is reasonably assured. Revenue from sales transactions where the buyer has the right to return the product is recognized at the time of sale only if (1) our price to the buyer is substantially fixed or determinable at the date of sale, (2) the buyer has paid us, or the buyer is obligated to pay us and the obligation is not contingent on resale of the product, (3) the buyer’s obligation to us would not be changed in the event of theft or physical destruction or damage of the product, (4) the buyer acquiring the product for resale has economic substance apart from that provided by us, (5) we do not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (6) the amount of future returns can be reasonably estimated.
Prior to the second quarter of 2011, we were unable to reasonably estimate returns. We therefore deferred revenue recognition until the right of return no longer existed, which was the earlier of Silenor being dispensed through patient prescriptions or the expiration of the right of return. We estimated patient prescriptions dispensed using an analysis of third-party information. In order to develop a methodology to reliably estimate product returns and provide a basis for recognizing revenue on sales to customers at the time of product shipment, we analyzed many factors, including, without limitation, industry data regarding product return rates, and tracked the Silenor product return history, taking into account product expiration dating at the time of shipment and levels of inventory in the wholesale channel compared to prescription units dispensed and the sell-down of our launch inventory. During the second quarter of 2011, the sell-down of our launch inventory was completed, which we believe demonstrates sufficient market acceptance of our product for purposes of our revenue recognition analysis. In addition, since product launch, we have sold product to wholesale pharmaceutical distributors at standard commercial terms utilized in the industry. As a result, we believe we can analogize to industry data regarding product return rates. Based on the sell-down of our launch inventory and the industry and internal data gathered, we believe we have the information needed to reasonably estimate product returns. As a result, in the second quarter of 2011, we began recognizing revenue for Silenor sales at the time of delivery of the product to wholesale pharmaceutical distributors and our other customers.
License and Royalty Revenue
In June 2011, we entered into a license agreement with Paladin Labs Inc. (“Paladin”) pursuant to which Paladin has the right to commercialize Silenor in Canada, South America, the Caribbean and Africa, subject to the receipt of marketing approval in each such territory. We received an upfront payment of $500,000 in connection with the execution of this agreement. We recorded the upfront payment as deferred revenue and are recognizing the upfront payment as license revenue over the period of our significant involvement under the agreement, which we are estimating to be
F-9
15 years. As of December 31, 2011, the deferred revenue balance associated with the license agreement is $481,000, of which $448,000 is recorded as non-current and $33,000 is recorded as current within accrued liabilities. We recognized $19,000 as revenue during the year ended December 31, 2011, which is recorded in interest and other income.
If Silenor is commercialized in the licensed territories, we would be eligible to receive sales-based milestone payments of up to $128.5 million as well as a tiered double-digit percentage of net sales in the licensed territories. Due to the uncertainty surrounding the achievement of these future sales-based milestones and royalties, these potential payments will not be recognized as revenue until they are realized and earned.
Product Sales Discounts and Allowances
We record product sales discounts and allowances at the time of sale and report revenue net of such amounts in the same period that product sales are recorded. In determining the amount of product sales discounts and allowances, we must make significant judgments and estimates. If actual results vary from our estimates, we may need to adjust these estimates, which could have an effect on product revenue in the period of adjustment. Our product sales discounts and allowances and the specific considerations we use in estimating these amounts include:
Prompt Pay Discounts. As an incentive for prompt payment, we offer a 2% cash discount to customers. We calculate the discount based on the gross amount of each invoice as we expect that all customers will comply with the contractual terms to earn the discount. We record the discount as an allowance against accounts receivable and a corresponding reduction of revenue. At December 31, 2011 and 2010, the allowance for prompt pay discounts was $39,000 and $114,000, respectively.
Patient Discount Programs. We offer discount programs to patients of Silenor under which the patient receives a discount on his or her prescription. We reimburse pharmacies for these discounts through third-party vendors. We estimate the total amount that will be redeemed based on the dollar amount of the discounts, the timing and quantity of distribution and historical redemption rates. We accrue the discounts and recognize a corresponding reduction of revenue. At December 31, 2011 and 2010, the accrual for patient discount programs was $414,000 and $182,000, respectively.
Distribution Service Fees. We pay distribution services fees to each wholesaler for distribution and inventory management services. We accrue for these fees based on contractually defined terms with each wholesaler and recognize a corresponding reduction of revenue. At December 31, 2011 and 2010, the accrual for distribution service fees was $319,000 and $276,000, respectively.
Chargebacks. We provide discounts to federal government qualified entities with whom we have contracted. These federal entities purchase products from the wholesalers at a discounted price, and the wholesalers then charge back to us the difference between the current retail price and the contracted price the federal entity paid for the product. We accrue chargebacks based on contract prices and sell-through sales data obtained from third-party information. At December 31, 2011 and 2010, the accrual for chargebacks was $24,000 and $9,000, respectively.
Rebates. We participate in certain rebate programs, which provide discounted prescriptions to qualified insured patients. Under these rebate programs, we pay a rebate to the third-party administrator of the program. We accrue rebates based on contract prices, estimated percentages of product sold to qualified patients and estimated levels of inventory in the distribution channel. Our accrual consists of: (1) the amount expected to be incurred based on the current quarter’s product sold, (2) an accrual for unpaid rebates relating to prior quarters; and (3) an accrual for rebates relating to estimated inventory in the distribution channel. Our estimate of utilization is based on partial claims history data received, third-party data, and information about our expected patient population. At December 31, 2011 and 2010, the accrual for rebates was $1,896,000 and $6,000, respectively.
Product Returns. We estimate future product returns based upon actual returns history, product expiration dating analysis, estimated inventory levels in the distribution channel, and industry data regarding product return rates for similar products. There may be a significant time lag between the date we determine the estimated allowance and when we receive product returns and issue credits to customers. Due to this time lag, we may record adjustments to our estimated allowance over several periods, which would impact our operating results in those periods. At December 31, 2011, the allowance for product returns was $255,000.
F-10
Cost of Sales
Cost of sales includes the costs to manufacture, package, and ship Silenor, including personnel costs associated with manufacturing oversight, as well as royalties and amortization of capitalized license fees associated with our license agreement with ProCom One, Inc. (“ProCom”).
Share-Based Compensation Expense
Share-based compensation expense for employees and directors is recognized in the statement of operations based on estimated amounts, including the grant date fair value, the probability of achieving performance conditions and the expected service period for awards with conditional vesting provisions. We estimate the grant date fair value for our stock option awards using the Black-Scholes valuation model which requires the use of multiple subjective inputs including estimated future volatility and the expected terms of the stock option awards. Our stock did not have a readily available market prior to our initial public offering in December 2005, creating a relatively short history from which to obtain data to estimate the volatility of our stock price. Consequently, we estimate expected future volatility based on a combination of both comparable companies and our own stock price volatility to the extent such history is available. The expected term for stock options is estimated using guidance provided by the SEC in Staff Accounting Bulletin (“SAB”) No. 107 and SAB 110. This guidance provides a formula-driven approach for determining the expected term. Share-based compensation is based on awards expected to ultimately vest and has been reduced for estimated forfeitures. The estimated forfeiture rates may differ from actual forfeiture rates which would affect the amount of expense recognized during the period. Estimated forfeitures are adjusted to actual amounts as they become known.
We recognize the value of the portion of the awards that are ultimately expected to vest on a straight-line basis over the award’s requisite service period. The requisite service period is generally the time over which our share-based awards vest. Some of our share-based awards have vested and may vest upon achieving certain performance conditions, generally pertaining to the commercial performance of Silenor or the achievement of other strategic objectives. Share-based compensation expense for awards with performance conditions is recognized over the period from the date the performance condition is determined to be probable of occurring through the time the applicable condition is met. If the performance condition is not considered probable of being achieved, no expense is recognized until such time the meeting of the performance condition is considered probable.
Income Taxes
Our income tax expense consists of current and deferred income tax expense or benefit. Current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is recognized for the future tax consequences attributable to tax credits and loss carryforwards and to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. As of December 31, 2011, we have established a valuation allowance to fully reserve our net deferred tax assets. Tax rate changes are reflected in income during the period such changes are enacted. Changes in ownership may limit the amount of net operating loss carryforwards that can be utilized in the future to offset taxable income. In addition, the state of California has currently suspended the use of net operating loss carryforwards to offset taxable income.
Comprehensive Income (Loss)
Comprehensive income (loss) is net income (loss) plus certain other items that are recorded directly to stockholders’ equity, which for Somaxon consists of changes in unrealized gains and losses on marketable securities classified as available-for-sale. We report comprehensive income (loss) in the Statement of Stockholders’ Equity and Comprehensive Loss. In the event an available-for-sale security is sold prior to its maturity, the related unrealized gain or loss on the investment is recognized in the income statement on a specific identification basis. We had insignificant realized gains or losses on sales of available-for-sale securities for each of the years ended 2011 and 2009. We did not have any realized gains and losses on sales of available-for-sale securities for the year ended 2010.
F-11
Net Loss per Share
Basic earnings per share (“EPS”) excludes the effects of common stock equivalents. EPS is calculated by dividing net income or loss applicable to common stockholders by the weighted average number of common shares outstanding for the period, reduced by the weighted average number of unvested common shares outstanding subject to repurchase. Diluted EPS is computed in the same manner as basic EPS, but includes the effects of common stock equivalents to the extent they are dilutive, using the treasury-stock method. For us, basic and diluted net loss per share are equivalent because we have incurred a net loss in all periods presented, causing any potentially dilutive securities to be anti-dilutive.
Net loss per share was determined as follows (in thousands, except per share amounts):
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Numerator |
||||||||||||
| Net loss |
$ | (59,280 | ) | $ | (38,813 | ) | $ | (14,443 | ) | |||
|
|
|
|
|
|
|
|||||||
| Denominator |
||||||||||||
| Weighted average common shares outstanding |
46,541 | 33,615 | 21,077 | |||||||||
| Weighted average unvested common shares subject to repurchase |
— | (22 | ) | (125 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Denominator for basic and diluted net loss per share |
46,541 | 33,593 | 20,952 | |||||||||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (1.27 | ) | $ | (1.16 | ) | $ | (0.69 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average anti-dilutive securities not included in diluted net loss per share |
||||||||||||
| Weighted average stock options outstanding |
4,408 | 3,566 | 4,496 | |||||||||
| Weighted average warrants outstanding |
2,664 | 3,028 | 2,903 | |||||||||
| Weighted average restricted stock units outstanding |
479 | 715 | 1,198 | |||||||||
| Weighted average unvested common shares subject to repurchase |
— | 22 | 125 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total weighted average anti-dilutive securities not included in diluted net loss per share |
7,551 | 7,331 | 8,722 | |||||||||
|
|
|
|
|
|
|
|||||||
Recent Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2009-13 “Revenue Recognition,” which provides guidance on recognizing revenue in arrangements with multiple deliverables. This standard impacts the determination of when the individual deliverables included in a multiple element arrangement may be treated as a separate unit of accounting. It also modifies the manner in which the consideration received from the transaction is allocated to the multiple deliverables and no longer permits the use of the residual method of allocating arrangement consideration. This accounting standard was effective for the first year beginning on or after June 15, 2010, with early adoption permitted. The adoption of ASU 2009-13, which occurred on January 1, 2011, did not have a material impact on our financial statements.
In December 2010, the FASB issued ASU No. 2010-27 “Other Expenses: Fees Paid to the Federal Government by Pharmaceutical Manufacturers,” which provides guidance concerning the recognition and classification of the annual fee payable by branded prescription drug manufacturers and importers on branded prescription drugs which was mandated under the health care reform legislation enacted in the United States in March 2010. Under this new authoritative guidance, the annual fee should be estimated and recognized in full as a liability upon the first qualifying commercial sale with a corresponding deferred cost that is amortized to operating expenses using a straight-line method unless another method better allocates the fee over the calendar year in which it is payable. This guidance was effective for calendar years beginning on or after January 1, 2011, when the fee initially became effective. The adoption of ASU 2010-27 did not have a material impact on our financial statements.
F-12
In May 2011, the FASB issued ASU No. 2011-04 “Fair Value Measurement: Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. GAAP and IFRSs”. Some of the amendments clarify the FASB’s intent about the application of existing fair value measurement requirements. Other amendments change a particular principle or requirement for measuring fair value or for disclosing information about fair value measurements. This guidance is effective for interim and annual periods beginning after December 15, 2011. We are still evaluating the potential future effects of this guidance.
In June 2011, the FASB issued ASU No. 2011-05 “Comprehensive Income: Presentation of Comprehensive Income.” Under the new guidance, an entity has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. This guidance is effective for interim and annual periods beginning after December 15, 2011. We have evaluated the potential future effects of this guidance and do not expect the adoption of ASU 2011-05 to have a material impact on our financial statements.
Note 3. Fair Value of Financial Instruments
Cash and investment holdings, accounts receivable, accounts payable and accrued liabilities are presented in the financial statements at their carrying amounts, which are reasonable estimates of fair value due to their short maturities.
The fair value of financial assets and liabilities is measured under a framework that establishes “levels” which are defined as follows: Level 1 fair value is determined from observable, quoted prices in active markets for identical assets or liabilities. Level 2 fair value is determined from quoted prices for similar items in active markets or quoted prices for identical or similar items in markets that are not active. Level 3 fair value is determined using the entity’s own assumptions about the inputs that market participants would use in pricing an asset or liability. We did not have any investment holdings as of December 31, 2011. The fair value of our investment holdings as of December 31, 2010 is summarized in the following table (in thousands):
| December 31, 2010 | ||||||||||||||||
| Total Fair | Fair Value Determined Under: | |||||||||||||||
| Value | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Commercial paper |
$ | 18,415 | $ | — | $ | 18,415 | $ | — | ||||||||
| U.S. government agency securities |
30,628 | — | 30,628 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 49,043 | $ | — | $ | 49,043 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Commercial paper and U.S. government agency securities are classified as part of either cash and cash equivalents or short-term investments in the balance sheets. The carrying value of short-term investments consisted of the following as of December 31, 2010 (in thousands):
| Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Fair Market Value |
|||||||||||||
| Commercial paper |
$ | 18,415 | $ | — | $ | — | $ | 18,415 | ||||||||
| U.S. government agency securities |
30,629 | 2 | (3 | ) | 30,628 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 49,044 | $ | 2 | $ | (3 | ) | $ | 49,043 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
Available-for-sale marketable securities with maturities of three months or less from date of purchase have been classified as cash equivalents, and amounted to $15.2 million as of December 31, 2010.
F-13
Note 4. Composition of Certain Balance Sheet Items
Accounts Receivable
Accounts receivable, net consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Accounts receivable for product sales, gross |
$ | 1,989 | $ | 5,975 | ||||
| Allowances for discounts |
(39 | ) | (391 | ) | ||||
|
|
|
|
|
|||||
| Total accounts receivable |
$ | 1,950 | $ | 5,584 | ||||
|
|
|
|
|
|||||
Inventory
Inventory consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Work in process |
$ | 124 | $ | 207 | ||||
| Finished goods inventory held by the Company |
140 | 515 | ||||||
| Finished goods inventory held by others |
— | 269 | ||||||
|
|
|
|
|
|||||
| Total inventory |
$ | 264 | $ | 991 | ||||
|
|
|
|
|
|||||
Other Current Assets
Other current assets consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Deposits and other prepaid expenses |
$ | 903 | $ | 641 | ||||
| Prepaid sales and marketing expenses |
91 | 529 | ||||||
| Interest receivable |
— | 206 | ||||||
| Other current assets |
9 | 506 | ||||||
|
|
|
|
|
|||||
| Total other current assets |
$ | 1,003 | $ | 1,882 | ||||
|
|
|
|
|
|||||
During the fourth quarter of 2011, we expensed $1,339,000 of prepaid sales and marketing expenses which represented samples that we do not expect to be distributed prior to expiration.
Property and Equipment
Property and equipment consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Tooling |
$ | 867 | $ | 832 | ||||
| Computer equipment |
481 | 354 | ||||||
| Furniture and equipment |
241 | 66 | ||||||
| Leasehold improvements |
76 | — | ||||||
|
|
|
|
|
|||||
| Property and equipment, at cost |
1,665 | 1,252 | ||||||
| Less: accumulated depreciation and amortization |
(1,031 | ) | (497 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 634 | $ | 755 | ||||
|
|
|
|
|
|||||
As a result of the change in strategic direction described in footnote 1, we reduced our estimate of the useful lives of certain of our property and equipment during the fourth quarter of 2011 from 36 months to 7 to 15 months. Depreciation expense, which is included in sales, general and administrative expenses, was higher by approximately $0.2 million than it would have been had the useful lives of these assets not been shortened. The effect of this change on basic and diluted earnings per share for the year ended December 31, 2011 was to increase our net loss by $0.01 per share.
F-14
Depreciation and amortization expense was $534,000, $347,000 and $83,000 for the years ended 2011, 2010 and 2009, respectively.
Intangible Assets
Intangible assets consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| License fees |
$ | 1,000 | $ | 1,000 | ||||
| Technology development costs relating to websites |
147 | 147 | ||||||
| Other intangibles |
161 | — | ||||||
|
|
|
|
|
|||||
| Intangible assets, at cost |
1,308 | 1,147 | ||||||
| Less: accumulated amortization |
(219 | ) | (45 | ) | ||||
|
|
|
|
|
|||||
| Total intangible assets, net |
$ | 1,089 | $ | 1,102 | ||||
|
|
|
|
|
|||||
Amortization expense was $174,000, $45,000 and $0 for the years ended 2011, 2010 and 2009, respectively. We estimate the aggregate amortization expense to be $201,000 and $147,000 for 2012 and 2013, respectively. We estimate the aggregate amortization expense for 2014 through 2016 to be $120,000.
Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Accrued product discounts, allowances, and returns |
$ | 2,908 | $ | 473 | ||||
| Accrued fees and royalties |
1,904 | 1,566 | ||||||
| Accrued compensation and benefits |
427 | 1,329 | ||||||
| Accrued liability to third party sales organization |
614 | 842 | ||||||
| Accrued legal fees |
507 | — | ||||||
| Other accrued expenses |
694 | 1,489 | ||||||
|
|
|
|
|
|||||
| Total accrued liabilities |
$ | 7,054 | $ | 5,699 | ||||
|
|
|
|
|
|||||
Note 5. Loan and Security Agreement
In July 2011, we terminated our then existing loan agreement with Comerica Bank and in August 2011, and entered into a new loan and security agreement (the “Loan Agreement”) with Silicon Valley Bank (“SVB”) and Oxford Finance LLC (“Oxford”), collectively the “Lenders”, under which we borrowed $15.0 million. The term loan carried interest at 7.5% per annum. We were obligated to pay interest on the loan through December 31, 2011, and to thereafter pay the principal balance of the loan and interest in 24 equal monthly installments through December 31, 2013. At our option, we could prepay all or any part of the outstanding principal balance, subject to a pre-payment fee of $150,000. At the time of repayment, we were also obligated to make an additional final payment of $638,000. The loan was recorded at an initial carrying value less the debt discount of $900,000. In connection with this transaction, we also paid the lenders an initial fee of $150,000 and reimbursed them for certain transaction costs. As part of the financing, the Lenders received warrants to purchase up to an aggregate of 583,152 shares of our common stock at an exercise price equal to $1.5433 per share. The warrants will expire ten years from the date of grant. The value assigned to these warrants of $701,000 was determined using the Black-Scholes valuation method and was reflected in the debt discount and additional paid in capital in our balance sheet.
We repaid the loan in full in December 2011. Upon repayment of the loan, we paid to the Lenders $0.5 million as partial payment of the prepayment and final payment fees required under the Loan Agreement. The Lenders also accepted warrants to purchase up to an aggregate of 150,000 shares of our common stock at an exercise price equal to $0.5000 per share in full satisfaction of the remaining prepayment and final payment fees. The warrants will expire ten years from the date of grant. The value assigned to these warrants of $59,000 was determined using the Black-Scholes valuation method and is reflected in additional paid in capital. We no longer have any obligations under the Loan Agreement, and there are no further encumbrances on our assets under the Loan Agreement.
F-15
Note 6. License Agreements
Paladin. In June 2011, we entered into a license agreement, a supply agreement and a stock purchase agreement with Paladin. Under the license agreement, Paladin has the right to commercialize Silenor in Canada, South America, the Caribbean and Africa, subject to the receipt of marketing approval in each such territory. We received $500,000 in connection with the execution of the agreements, and Paladin purchased 2,184,769 shares of our common stock for an aggregate purchase price of $5.0 million. As of December 31, 2011, the deferred revenue balance associated with the license agreement is $481,000, of which $447,000 is recorded as non-current and $34,000 is recorded as current within accrued liabilities. We recognized $19,000 as revenue during 2011, which is recorded in interest and other income. Once Silenor is commercialized in the licensed territories, we will also be eligible to receive sales-based milestone payments of up to $128.5 million as well as a tiered double-digit percentage of net sales in the licensed territories. Paladin will be responsible for regulatory submissions for Silenor in the licensed territories and will have the exclusive right to commercialize Silenor in the licensed territories. Governance of the collaboration will occur through a joint committee. We have also granted to Paladin a right of first negotiation with respect to additional doxepin products we may develop in the licensed territories and, subject to the rights we have previously granted to P&G, a right of first negotiation relating to rights to develop and market Silenor as an over-the-counter medication in the licensed territories.
The term of the license agreement runs through the longer of the last date on which Silenor is sold by Paladin in the licensed territories or 15 years from the first commercial sale of Silenor in the licensed territories. We may terminate the license agreement on a country-by-country basis in specified key countries upon 60 days’ prior written notice if the first commercial sale has not occurred in such country within 12 months of the date on which marketing approval was obtained in such country. We may also terminate the license agreement upon 60 days’ prior written notice if marketing approval in Canada has not been received by December 7, 2013. Either party may terminate the license agreement upon an uncured material breach by the other party, upon the bankruptcy or insolvency of the other party, or a force majeure event that lasts for at least 120 days. We may also terminate the license agreement upon 60 days’ prior written notice and payment of a termination fee if we are unable to license rights to a third party’s intellectual property and such failure would reasonably be expected to result in a claim from such third party alleging intellectual property infringement or misappropriation.
In connection with the license agreement, we also entered into a supply agreement, under which we will supply Paladin all of its requirements for Silenor during the term of the license agreement or until Paladin procures its own supply of Silenor. Paladin may terminate the supply agreement upon 10 business days’ notice if we are materially unable to supply Silenor to Paladin’s requirements as defined in the supply agreement, and at any time if Paladin enters into a direct contractual relationship with our manufacturer of Silenor. We may terminate the supply agreement upon 180 days prior written notice if there is a regulatory change or safety consideration that would have a material adverse effect on the global supply chain and at any time on six months’ prior notice after April 30, 2013.
ProCom. In August 2003, we entered into an exclusive worldwide in-license agreement with ProCom to develop and commercialize Silenor for the treatment of insomnia. This agreement was amended and restated in September 2010. The term of the license extends until the last licensed patent expires, which is expected to occur no earlier than 2020. The license agreement is terminable by us at any time with 30 days’ notice if we believe that the use of the product poses an unacceptable safety risk or if it fails to achieve a satisfactory level of efficacy. Either party may terminate the agreement with 30 days’ notice if the other party commits a material breach of its obligations and fails to remedy the breach within 90 days, or upon the filing of bankruptcy, reorganization, liquidation, or receivership proceedings. Costs related to the licensed intellectual property incurred after approval of the Silenor NDA by the FDA in March 2010 have been capitalized and included in intangibles in our balance sheet as of December 31, 2011and 2010. Capitalized amounts are amortized on a straight line basis over approximately ten years. Royalty payments due under the terms of the agreement are recorded in accrued liabilities as of December 31, 2011 and 2010. The royalty payments are recognized as an expense in cost of sales when the related shipments of product are recognized as revenue.
Other Agreement. In October 2006, we entered into a supply agreement with JRS Pharma L.P. (“JRS”), under which we purchase from JRS all of our requirements for ProSolv®HD90, an ingredient used in the formulation for Silenor. In August 2008, this supply agreement was amended to provide us with the exclusive right to use this
F-16
ingredient in combination with doxepin. Pursuant to the amendment, we are obligated to pay a royalty on worldwide net sales of Silenor beginning as of the expiration of the statutory exclusivity period for Silenor in each country in which Silenor is marketed. Such royalty is only payable if one or more patents under the license agreement continue to be valid in each such country and a patent relating to our formulation for Silenor has not issued in such country.
Note 7. Commitments and contingencies
Commitments
Publicis Touchpoint Solutions, Inc. In July 2010, we entered into a professional detailing services agreement with Publicis under which Publicis agreed to provide sales support to promote Silenor in the U.S. through 110 full-time sales representatives, together with management coordination personnel, all of whom were employees of Publicis. In February 2011, we entered into an amended and restated agreement with Publicis under which Publicis deployed for us an additional 35 sales representatives that were exclusively promoting Silenor, together with management coordination personnel. We recognized the revenue from Silenor product sales generated by the promotional efforts of the sales organization. Under the terms of the agreement, we were required to pay a one-time startup fee, and we were required to pay a fixed monthly fee, which is subject to certain quarterly adjustments based on actual staffing and spending levels. During the term of the agreement, a portion of Publicis’ management fee was subject to payment by us only to the extent that specified performance targets were achieved. The performance targets related to initial scale-up activities, turnover and vacancy rates, call attainment and specified sales goals. In addition, we were obligated to reimburse the sales organization for approved pass-through costs, which primarily included bonuses, meeting and travel costs and certain administrative expenses.
On September 30, 2011, we provided notice of termination to Publicis of the services agreement, effective as of December 31, 2011. At the conclusion of the contract term with Publicis, we were contractually obligated to assume financial responsibility for the remaining vehicle lease payments associated with our Publicis sales representatives. Accrued liabilities as of December 31, 2011, includes a balance due to Publicis of $0.6 million, which represents all amounts owed to Publicis and all other third parties in connection with the contract sales agreement. All of our obligations associated with our contract sales agreement with Publicis were settled in full in February 2012. Effective January 3, 2012, we hired a reduced sales force of 25 sales representatives from the Publicis sales force to promote Silenor as Somaxon employees. The remainder of the Publicis sales force ceased promoting Silenor as of November 2, 2011.
Procter & Gamble. In August 2010, we entered into a co-promotion agreement with P&G under which P&G provided sales support to promote Silenor in the U.S. through its team of full-time sales representatives. We recognized the revenue from Silenor product sales generated by the promotional efforts of P&G. Under the terms of the agreement, we were required to pay P&G a fixed fee and a royalty fee as a percentage of U.S. net sales, in each case on a quarterly basis during the term of the agreement. The fees were recognized as a sales, general, and administrative expense. Each party was responsible for the costs of training, maintaining and operating its own sales force, and we were responsible for all other costs pertaining to the commercialization of Silenor.
On September 30, 2011, we provided notice of termination to P&G of the co-promotion agreement, with such termination effective as of December 31, 2011. As a result of such termination, P&G is entitled to a low single digit royalty on net sales of Silenor for the 2012 fiscal year. In addition, on September 30, 2011, we and P&G amended the surviving provision of such agreement relating to over-the-counter (“OTC”) rights for Silenor, and we and P&G further amended this provision on November 23, 2011. Pursuant to these amendments, through the period ending on March 31, 2012, we and P&G will work together to evaluate the potential to develop and commercialize an OTC pharmaceutical product containing doxepin, including jointly developing a desired product profile for the OTC product and conducting market research on such product profile at P&G’s expense. During this period, we will not negotiate rights in and to an OTC product with any third party. If P&G notifies us of its interest in negotiating for rights to the OTC product at any time prior to March 31, 2012, P&G will have the exclusive right to negotiate with us relating to such rights for 120 days from our receipt of the notice, or such longer period as may be mutually agreed by us and P&G.
F-17
Comerica Bank (“Comerica”). In February 2011, we entered into a $15.0 million loan agreement with Comerica. This agreement was terminated in July 2011 in connection with our loan and security agreement with the Lenders as discussed above in “Note 5. Loan and Security Agreement.”
Citadel Securities LLC. In August 2011, we entered into an at-the-market equity sales agreement with Citadel (the “Sales Agreement”) pursuant to which we may sell, at our option, up to an aggregate of $30.0 million in shares of our common stock through Citadel, as sales agent. Sales of the common stock made pursuant to the Sales Agreement, if any, will be made on the NASDAQ Stock Market under our currently-effective Registration Statements on Form S-3 by means of ordinary brokers’ transactions at then-prevailing market prices. Additionally, under the terms of the Sales Agreement, we may also sell shares of our common stock through Citadel, on the NASDAQ Stock Market or otherwise, at negotiated prices or at prices related to the prevailing market price. Under the terms of the Sales Agreement, we may also sell shares to Citadel as principal for Citadel’s own account at a price agreed upon at the time of sale pursuant to a separate terms agreement to be entered into with Citadel at such time. We will pay Citadel a commission equal to 3% of the gross proceeds from the sale of shares of our common stock under the Sales Agreement. The offering of common stock pursuant to the Sales Agreement will terminate upon the earlier of (a) the sale of all of the common stock subject to the Sales Agreement or (b) the termination of the Sales Agreement by us or Citadel. Either party may terminate the Sales Agreement in its sole discretion at any time upon written notice to the other party. There can be no assurance that Citadel will be successful in consummating such sales based on prevailing market conditions or in the quantities or at the prices that we deem appropriate.
We will not be able to make sales of our common stock pursuant to the sales agreement unless certain conditions are met, which include the accuracy of representations and warranties made to Citadel under the sales agreement; compliance with laws; and the continued listing of our stock on the Nasdaq Capital Market. On December 13, 2011, we received a letter from the Listing Qualifications Department of the Nasdaq Stock Market, or Nasdaq, informing us that because the closing bid price of our common stock listed on Nasdaq was below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2). If compliance is not demonstrated within the applicable timeframe, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market.
Lease. In May 2011, we entered into a new lease arrangement to rent approximately 12,100 square feet of office space, which we use as our corporate headquarters. The lease commenced on August 25, 2011. The lease will expire on December 24, 2016, and we will have the option to extend the term for an additional five years at the then-current fair market rental rate (as defined in the lease). We have paid the first month’s rent of approximately $30,000 and the monthly rent is approximately $30,000. However, the second through thirteenth month’s rent will be abated by one-half, provided that we are not in default of the lease. After the first year, the monthly rent will increase by 3.5% per year. We also have a letter of credit in the amount of $200,000 in favor of our landlord to secure our obligations under the lease which is recorded as restricted cash in our balance sheet as of December 31, 2011.
At December 31, 2011, the estimated future minimum lease payments for each of the years ended December 31, are as follows (in thousands):
| 2012 |
$ | 226 | ||
| 2013 |
373 | |||
| 2014 |
386 | |||
| 2015 |
399 | |||
| 2016 |
413 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 1,797 | ||
|
|
|
Rent expense for 2011, 2010 and 2009 was $286,000, $178,000 and $88,000, respectively. As discussed above, our facility operating lease contains rent abatement and escalation clauses. The Company recognizes rent expense on a straight line basis over the lease term. The difference between rent expense recorded and amounts paid under lease agreements is recorded as deferred rent and included in other long-term liabilities in the accompanying balance sheet.
Employee arrangements. In October 2011, we accepted the resignation of one of our officers, under which the individual received certain benefits including: (1) a lump sum severance payment; (2) a lump sum payment equal to the cost of 12 months of health care benefits continuation at our expense; and (3) a lump sum payment equal to the cost of 12 months of the portion of the monthly premiums for the individual’s life insurance and disability insurance coverage that are borne by us. In addition, on the resignation date, the portion of the stock options and restricted stock units which would have vested if the individual had remained employed for an additional 12 months immediately vested. The individual entered into a consulting agreement with us that will expire on June 30, 2013. We cannot estimate with any certainty the amount that may be paid, if any, for consulting services under such agreement. The remaining outstanding stock awards will continue to vest through the expiration of the consulting agreement, and the individual will be entitled to exercise such stock awards for 180 days following such expiration. We paid $0.3 million in 2011 for severance payments and recorded additional share-based compensation expense of $0.8 million as a result of accelerated vesting of the share-based awards.
F-18
In November 2011, we terminated the employment of 14 employees. Each of the terminated employees entered into a separation agreement with us. Under these agreements, two of the terminated employees received a lump sum severance payment equal to three months of base salary and the amount of the health insurance benefits paid by us for the previous three months, and the remaining terminated employees received a lump sum severance payment equal to two months of their base salary and the amount of the health insurance benefits paid by us for the previous two months. We paid $0.4 million for these severance payments in 2011 and do not owe any additional amounts as of December 31, 2011. We recorded additional share-based compensation expense of $0.1 million as a result of accelerated vesting for certain share-based awards for certain employees that entered into consulting agreements with us.
In December 2011, we committed to a plan of termination that resulted in a work force reduction of 16 employees. We commenced notification of employees affected by the workforce reduction on December 15, 2011, and the workforce reduction was completed by February 15, 2012. Two of the terminated employees entered into a separation agreement with us under which each such employee received a lump sum severance payment based upon the employment agreements such employees previously entered into with us. Both of these employees received a payment equivalent to six months of base salary, with one receiving an additional amount equal to the amount of benefits payable by us for six months, and the other receiving an additional amount equal to the amount of benefits payable by us for twelve months. Each of the other affected employees entered into a separation agreement with us under which each such employee received a lump sum severance payment equivalent to two months of their base salary and the amount of the healthcare benefits paid by us for the previous two months. We paid $0.6 million and $0.1 million for these severance payments in 2011 and 2012, respectively. We recorded additional share-based compensation expense of $0.3 million as a result of accelerated vesting for certain share-based awards for certain employees that entered into consulting agreements with us that will expire on December 15, 2012. We cannot estimate with any certainty the amounts that may be paid, if any, for consulting services under such agreements.
Other Commitments. We have contracted with various consultants, drug manufacturers, wholesalers, and other vendors to assist in clinical trial work, data analysis, and commercialization activities for Silenor. The contracts are terminable at any time, but obligate us to reimburse the providers for any time or costs incurred through the date of termination. We have employment agreements with certain of our current employees that provide for severance payments and accelerated vesting for certain share-based awards if their employment is terminated under specified circumstances.
Litigation
We have received notices from each of Actavis Elizabeth LLC and Actavis Inc. (collectively, “Actavis”), Mylan Pharmaceuticals Inc. and Mylan, Inc. (collectively, “Mylan”), Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc. (collectively, “Par”), and Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Limited (d/b/a Zydus Cadila) (collectively, “Zydus”) that each has filed with the FDA an Abbreviated New Drug Application (“ANDA”) for a generic version of Silenor 3 mg and 6 mg tablets. The notices included “paragraph IV certifications” with respect to eight of the nine patents listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, for Silenor. A paragraph IV certification is a certification by a generic applicant that in the opinion of that applicant, the patent(s) listed in the Orange Book for a branded product are invalid, unenforceable, and/or will not be infringed by the manufacture, use or sale of the generic product.
We, together with ProCom, have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus. The lawsuits allege that each of Actavis, Mylan, Par and Zydus have infringed U.S. Patent No. 6,211,229 (the “’229 patent”) by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
In addition, we have filed suit in the United States District Court for the District of Delaware against each of Actavis, Mylan, Par and Zydus alleging that such parties have infringed U.S. Patent No. 7,915,307 (the “’307 patent”) by seeking approval from the FDA to market generic versions of Silenor 3 mg and 6 mg tablets prior to the expiration of this patent.
F-19
Pursuant to the provisions of the Hatch-Waxman Act, FDA final approval of the Actavis and Mylan ANDAs can occur no earlier than May 3, 2013, FDA final approval of the Par ANDA can occur no earlier than June 23, 2013 and FDA final approval of the Zydus ANDA can occur no earlier than November 13, 2013, unless in each case there is an earlier court decision that the ’229 patent and the ’307 patent are not infringed and/or invalid or unless any party to the action is found to have failed to cooperate reasonably to expedite the infringement action.
At this time, the other patents included in Orange Book have not been asserted against these parties.
We intend to vigorously enforce our intellectual property rights relating to Silenor, but cannot predict the outcome of these matters.
Note 8. Stockholders’ Equity
Public Offerings of Common Stock
In March 2010, we issued 6,900,000 shares of common stock in a public offering of our stock. The net proceeds from the offering, after underwriting discounts and commissions and offering costs of $4,180,000, were approximately $52,745,000.
In November 2010, we issued 8,800,000 shares of common stock in a public offering of our stock. The net proceeds from the offering, after underwriting discounts and commissions and offering costs of $1,149,000, were approximately $24,811,000.
During the year ended December 31, 2011, we sold an aggregate of 786,825 shares of our common stock and received gross proceeds of $0.8 million under our Sales Agreement with Citadel. Our financial statements for the period ended December 31, 2011 reflect the gross proceeds from these transactions, which are reflected in stockholders’ equity net of $0.3 million of legal and accounting fees associated with the execution of the sales agreement and commissions.
Private Placements of Common Stock
In June 2011, we entered into a stock purchase agreement with Paladin pursuant to which Paladin purchased 2,184,769 shares of our common stock for an aggregate purchase price of $5.0 million (see Note 6, “License Agreements”) in a private placement pursuant to Rule 506 of the Securities Act of 1933, as amended.
In July 2009, we issued 5.1 million shares of common stock at $1.05 per share and seven-year warrants to purchase up to 5.1 million additional shares of common stock, exercisable in cash or by net exercise at a price of $1.155 per share, for aggregate gross proceeds of $6.0 million and net proceeds of $5.7 million after deducting offering costs of $0.3 million. In connection with the private placement, we agreed to register for resale both the shares of common stock purchased by the investors and the shares of common stock issuable upon exercise of the warrants. The resale registration statement was filed and declared effective by the SEC in August 2009. We also agreed to other customary obligations regarding registration, including matters relating to indemnification, maintenance of the registration statement and payment of expenses. We may be liable for liquidated damages if we do not maintain the effectiveness of the registration statement or the listing of our common stock on the Nasdaq Capital Market, the Nasdaq Global Market, the New York Stock Exchange or the American Stock Exchange, in each case for a period of ten consecutive days or for more than thirty days in any 365-day period. The amount of the liquidated damages is one percent per applicable ten or thirty day period, subject to an aggregate maximum of eight percent per calendar year, of the aggregate purchase price of the common stock purchased in the private placement then held by each investor that are registrable securities.
In order to continue to be listed on the Nasdaq Capital Market, we must meet specific quantitative standards, including maintaining a minimum bid price of $1.00 for our common stock, a public float of $1.0 million, and either $2.5 million in stockholders equity or a market capitalization of $35 million. On December 13, 2011, we received a letter from the Listing Qualifications Department of Nasdaq informing us that because the closing bid price for our common stock had been below $1.00 for 30 consecutive trading days, we did not comply with the minimum closing bid price requirement for continued listing on the Nasdaq Capital Market. We have until June 11, 2012 to regain compliance with Nasdaq’s listing requirements by having the closing bid price of our common stock be at least
F-20
$1.00 for at least 10 consecutive trading days. If we do not regain compliance within this time period but comply with all continued listing standards other than the closing bid price requirement, we may provide Nasdaq with written notice of our intention to cure the deficiency during a second compliance period of up to 180 days. If the Listing Qualifications Department of Nasdaq believes that we will be able to cure the deficiency during such additional period, it will grant us such period to do so. If compliance is not demonstrated within the applicable compliance period, Nasdaq will notify us that our securities will be delisted from the Nasdaq Capital Market. However, we may appeal Nasdaq’s determination to delist our securities to a Nasdaq Hearings Panel. During any appeal process, shares of our common stock would continue to trade on the Nasdaq Capital Market. We do not believe it is probable that we will be required to pay liquidated damages and have not recognized any amounts in the financial statements related to such potential liquidated damages.
Warrants
At December 31, 2011, we have warrants outstanding to purchase 3,150,784 shares of our common stock. These warrants are immediately exercisable, have remaining terms of two to ten years, exercise prices ranging from $0.5000 to $5.4175 per share, and a weighted average exercise price of $1.39 per share.
Preferred Stock
Concurrent with our initial public offering in December 2005, 10,000,000 shares of preferred stock were authorized, none of which have been issued or are outstanding as of December 31, 2011.
Note 9. Share-Based Compensation
We have issued and intend to continue to issue stock options, restricted stock units (“RSUs”) and restricted stock awards under our equity incentive award plans. We have equity awards outstanding under both our 2004 Equity Incentive Award Plan (the “2004 Plan”) and our 2005 Equity Incentive Award Plan (the “2005 Plan”). During 2011, we had the following types of equity awards outstanding:
| • | Stock Options: Stock options generally have ten-year terms and vest over a period of between one and four years and are service-based. The exercise price for our stock options is generally equal to the closing stock price at the date of grant. |
| • | Restricted Stock Units: RSUs, which are convertible into an equivalent number of shares of common stock upon vesting, have been granted to employees and members of our board of directors. The majority of our outstanding RSUs vested during 2011 upon achieving certain time-based criteria. |
We issue equity awards under our 2005 Plan. The 2005 Plan contains an “evergreen provision” that allows annual increases in the number of shares available for issuance on the first day of each year through January 1, 2015 in an amount equal to the lesser of: (i) 2,000,000 shares, (ii) 5% of the outstanding capital stock on each January 1, or (iii) an amount determined by our board of directors. As of December 31, 2011, an aggregate of 2,054,000 shares of common stock were authorized, available for issuance and not subject to previous awards under the 2005 Plan. Under the evergreen provision, on January 1, 2012, an additional 2,000,000 shares became available for issuance under the 2005 Plan.
We also have an employee stock purchase plan (“ESPP”) which allows employees to contribute up to 20% of their cash earnings, subject to certain maximums, to be used to purchase shares of our common stock on each semi-annual purchase date. The purchase price is equal to 95% of the market value per share on each purchase date. Our ESPP is non-compensatory pursuant to the provisions of generally accepted accounting principles for share-based compensation expense. The ESPP contains an “evergreen provision” with annual increases in the number of shares available for issuance on the first day of each year through January 1, 2015 equal to the lesser of: (i) 300,000 shares, (ii) 1% of the outstanding capital stock on each January 1, or (iii) an amount determined by our board of directors. As of December 31, 2011, an aggregate of 1,402,000 shares of common stock were authorized and available for issuance under the ESPP. Under the evergreen provision, on January 1, 2012, an additional 300,000 shares were authorized under our ESPP. No shares have been issued under the ESPP through December 31, 2011.
F-21
Share-Based Compensation Expense
The following table summarizes share-based compensation expense recognized in 2011, 2010, and 2009:
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Share-based compensation expense included in selling, general and administrative expense |
$ | 4,786 | $ | 5,412 | $ | 4,637 | ||||||
| Share-based compensation expense included in research and development expense |
462 | 1,294 | 1,526 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total share-based compensation expense |
$ | 5,248 | $ | 6,706 | $ | 6,163 | ||||||
|
|
|
|
|
|
|
|||||||
Included in the table for 2011 is the effect of the resignation or termination of employment for certain individuals which created an acceleration of share-based compensation expense. During 2011, 11 employees ceased employment and entered into a consulting agreement with us. Also, in 2011 upon separation from the Company, certain individuals received accelerated vesting of their share-based awards. As a result of such non-substantive consulting arrangements and accelerated vesting, we recognized $1.2 million of share-based compensation expense during 2011 on the dates of termination.
Included in the table for 2009 is the effect of the termination of employment for certain individuals which created an acceleration of share-based compensation expense. During 2009, 15 employees ceased employment and entered into a consulting agreement with us. Also, in 2009 upon separation from the Company, certain individuals received accelerated vesting of their share-based awards. As a result of such non-substantive consulting arrangements and accelerated vesting, we recognized $2.4 million of share-based compensation expense during 2009 on the dates of termination.
The table above also includes compensation costs of $0.7 million in 2009 from our one-time stock option exchange program that was completed in June 2009. Under the program, employees and directors as of March 1, 2009 were eligible to exchange their stock options having exercise prices above $1.00 for the grant of a lesser number of replacement awards having an exercise price of $1.23. In total, 4,320,000 stock options were tendered in exchange for 2,880,000 replacement awards. One-third of the replacement awards vested upon grant and the remaining two-thirds vested in equal monthly installments over the following two year period such that all the shares became fully vested in June 2011, subject to each participant’s continued service.
Stock Options
We use the Black-Scholes model to estimate the grant date fair value of stock options. To calculate these fair values, the following assumptions were used:
| Year Ended December 31, | ||||||
| 2011 | 2010 | 2009 | ||||
| Risk free interest rate |
1.0% to 2.6% | 2.5% to 3.0% | 1.9% to 2.9% | |||
| Expected term |
5.27 to 6.25 years | 5.25 to 6.25 years | 5.25 to 6.25 years | |||
| Expected volatility |
75% to 78% | 86% to 88% | 74% to 84% | |||
| Weighted average volatility |
77% | 87% | 78% | |||
| Expected dividend yield |
0% | 0% | 0% | |||
| Fair value of underlying stock |
$0.45 to $3.10 | $2.69 to $8.59 | $1.17 to $2.18 | |||
| Weighted average fair value of stock options granted |
$1.31 | $4.89 | $1.19 | |||
F-22
The following table summarizes our stock options as of December 31, 2011 and the activity for the year then ended (share and dollar amounts in thousands, except per-share exercise prices):
| Shares | Weighted Average Exercise Price |
Weighted Average Contractual Term (in years) |
Aggregate Intrinsic Value (in thousands) |
|||||||||||||
| Outstanding at December 31, 2010 |
3,382 | $ | 3.86 | |||||||||||||
| Granted |
2,348 | 1.99 | ||||||||||||||
| Exercised |
(1 | ) | 1.23 | $ | 2 | |||||||||||
| Forfeited |
(466 | ) | 3.67 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding at December 31, 2011 |
5,263 | $ | 3.05 | 8.1 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Vested and exercisable at December 31, 2011 |
2,734 | $ | 3.07 | 7.1 | $ | — | ||||||||||
The intrinsic value of an equity award is the difference between the fair value of the underlying stock and its exercise price. If the exercise price equals or exceeds the fair value of the underlying stock, then the award is considered to have a zero intrinsic value. The following table summarizes certain information for options (in thousands).
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Fair value of vested stock options |
$ | 4,713 | $ | 1,563 | $ | — | ||||||
| Intrinsic value of exercised stock options |
$ | 2 | $ | 5,883 | $ | 225 | ||||||
Unrecognized compensation expense related to non-vested stock options totaled $4.5 million as of December 31, 2011. Such compensation expense is expected to be recognized over a weighted-average period of 2.11 years.
We did not realize any tax benefits from option exercises during 2011, 2010 and 2009.
Restricted Stock Units and Awards
Restricted Stock Units — The following table summarizes our restricted stock units as of December 31, 2011 and the activity during the year then ended (share numbers in thousands):
| Employee and
Director Awards |
||||||||||||
| Weighted Average Grant Date Fair Value |
Total Awards |
|||||||||||
| # Shares | per Share | # Shares | ||||||||||
| December 31, 2010 |
125 | $ | 8.52 | 125 | ||||||||
| Granted |
445 | 2.93 | 445 | |||||||||
| Vested |
(87 | ) | 4.31 | (87 | ) | |||||||
| Forfeited |
(250 | ) | 2.93 | (250 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| December 31, 2011 |
233 | $ | 5.41 | 233 | ||||||||
|
|
|
|
|
|
|
|||||||
The intrinsic value of an RSU is equal to our stock price. The intrinsic value of vested RSUs was $72,000, $2,161,000, and $235,000 during 2011, 2010, and 2009, respectively. The weighted average grant date fair value per share for RSUs granted in 2010 and 2009 was $8.55 and $1.28, respectively. Unrecognized compensation expense related to non-vested RSUs totaled $0.4 million as of December 31, 2011. Such compensation expense is expected to be recognized over a weighted-average period of 1.60 years.
Restricted Stock Awards —At December 31, 2010, we did not have any non-vested restricted stock awards outstanding. We did not grant any restricted stock awards in 2011. The intrinsic value of restricted stock is equal to our stock price. The intrinsic value of vested restricted stock awards was $414,000 and zero in 2010 and 2009, respectively.
F-23
Note 10. Income taxes
We have incurred losses since inception, so no current income tax provision or benefit has been recorded. Significant components of our net deferred tax assets are shown in the table below (amounts are in thousands).
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Deferred Tax Assets: |
||||||||
| Net operating loss carryforwards |
$ | 90,864 | $ | 71,038 | ||||
| Research and development credits |
4,752 | 4,752 | ||||||
| Intangibles |
1,349 | 360 | ||||||
| Non-cash compensation expense |
4,340 | 3,270 | ||||||
| Other, net |
1,629 | 1,982 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
102,934 | 81,402 | ||||||
| Valuation allowance |
(102,934 | ) | (81,402 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
At December 31, 2011, we had generated federal net operating loss carryforwards of $234.1 million and state net operating loss carryforwards of $226.1 million on the respective tax return bases. We have generated windfall tax benefits from the settlement of certain share-based awards. These tax benefits have not been reflected in the table of deferred tax assets presented above since the tax deduction increases our net operating loss carryforward and does not result in a cash tax savings in the current year. The tax benefit will be recorded as a credit to additional paid-in capital in the year the deduction reduces income taxes payable. However, the net operating loss carryforwards related to these windfall tax benefits of approximately $1,199,000 are included in the federal and state net operating loss carryforward amounts of $234.2 million and $226.5 million, respectively. Unless previously utilized, the federal and state tax loss carryforwards will begin to expire in 2023 and 2013, respectively.
We have federal and state research and development tax credit carryforwards at December 31, 2011 of $4,282,000 and $1,954,000, respectively. The federal research and development credits will begin to expire in 2024 and the state research and development credits do not expire.
Pursuant to Sections 382 and 383 of the Internal Revenue Code (“IRC”), annual use of our net operating loss and credit carryforwards may be limited in the event a cumulative change in ownership of more than 50% occurs within a three-year period. We determined that such an ownership change occurred as of March 31, 2010 as defined in the provisions of Section 382 of the IRC as a result of various stock issuances used to finance our operations. Such ownership change resulted in annual limitations on the utilization of tax attributes, including net operating loss carryforwards and tax credits. We estimate that $111,000 and $376,000 of our state net operating loss carryforwards were effectively eliminated under Section 382 for federal and California, respectively. A portion of the remaining net operating losses limited by Section 382 become available each year. We have not performed a Section 382 analysis since December 31, 2010. There is a risk that additional changes in ownership could have occurred since that date. If a change in ownership were to have occurred, additional net operating loss carryforwards and research and development credit carryovers could be eliminated or restricted. If eliminated, the related asset would be removed from the deferred tax asset with a corresponding reduction in the valuation allowance.
The following table provides a reconciliation between income taxes computed at the federal statutory rate and our provision for income taxes (amounts are in thousands):
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Federal income taxes at 34% |
$ | (20,152 | ) | $ | (13,231 | ) | $ | (4,911 | ) | |||
| State income taxes, net of federal benefit |
(2,236 | ) | (1,779 | ) | (15 | ) | ||||||
| Research and development credits |
— | — | (116 | ) | ||||||||
| Share-based compensation expense |
793 | (1,741 | ) | 5,069 | ||||||||
| Tax effect of non-deductible expenses and credits |
64 | 44 | (3 | ) | ||||||||
| Increase in valuation allowance |
21,531 | 16,707 | (24 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Provision for income taxes |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
F-24
Income tax accounting and reporting may contain uncertain income tax positions. The accounting for uncertain income taxes recognized in an entity’s financial statements requires a recognition threshold and measurement of uncertain tax positions taken or expected to be taken on a tax return. The impact of an uncertain income tax position on the income tax return is recognized at the largest amount that is cumulatively more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
The following table summarizes our unrecognized tax benefit activity (amounts are in thousands):
| Year Ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| Unrecognized tax benefits at the beginning of the year |
$ | 910 | $ | 910 | ||||
| Gross decreases related to prior year tax positions |
0 | 0 | ||||||
| Gross increases related to current year tax positions |
0 | 0 | ||||||
| Settlements |
None | None | ||||||
| Lapse of statute of limitations |
None | None | ||||||
|
|
|
|
|
|||||
| Unrecognized tax benefits at year end |
$ | 910 | $ | 910 | ||||
|
|
|
|
|
|||||
The unrecognized tax benefits have been recorded as a reduction of the related deferred tax asset. Because our deferred tax assets are fully reserved, none of the amount included in the balance of unrecognized tax benefits would affect the effective tax rate if recognized. We are subject to taxation in each of the jurisdictions in which we operate. We are currently not under examination by the Internal Revenue Service or any other taxing authority. Our tax years from inception in 2003 and forward can be subject to examination by the tax authorities due to the carryforward of net operating losses and research and development credits. Our accounting policy is to record interest and penalties related to unrecognized tax benefits in income tax expense. No interest or penalties have been accrued as of December 31, 2011.
Note 11. Related Party Transactions
We have in-licensed certain intellectual property from ProCom (see Note 6, “License Agreements”). As part of the license agreement, ProCom has the right to designate one nominee for election to our board of directors. ProCom designated Terrell A. Cobb, a principal of ProCom, for nomination as a member of our board of directors. In 2010, we paid ProCom $1.0 million for license fees and $235,000 for royalty payments pursuant to the terms of this agreement. During 2011, we recognized in costs of sales $929,000 of ProCom royalty payments in connection with this arrangement. At December 31, 2011 and 2010, $239,000 and $16,000, respectively, is recorded in accrued liabilities for ProCom royalty payments.
The license agreement also provided a consulting arrangement for Dr. Neil Kavey, who is the other principal of ProCom. Under the consulting agreements, we paid an aggregate of $8,000, $59,000 and $135,000, for consulting services for the years ended 2011, 2010 and 2009, respectively. Payments to Dr. Kavey under that consulting arrangement ended in September 2011 and a subsequent consulting agreement was entered into which terminates in September 2012.
Mr. Cobb and Dr. Kavey have an aggregate of 149,000 stock options outstanding of which 141,000 were vested as of December 31, 2011. The weighted average exercise price of the outstanding options was $3.56 and the weighted average exercise price of the vested stock options was $3.63. None of the stock options had been exercised as of December 31, 2011. In addition, 49,000 RSUs granted to Mr. Cobb vested in 2010 in connection with the first commercial sale of Silenor in the United States.
In July 2009, we raised $6,000,000 through a private placement of 5,106,000 shares of our common stock and seven-year warrants to purchase up to 5,106,000 additional shares of our common stock. Among the investors in the private placement were: (1) a trust of which Kurt von Emster, a member of our board of directors, is a trustee and beneficiary; (2) investment funds affiliated with Jesse I. Treu, Ph.D., a member of our board of directors through June 2010, and (3) investment funds affiliated with Kurt C. Wheeler, a member of our board of directors through March 2010.
F-25
Note 12. Selected Quarterly Financial Information (Unaudited)
The following table presents our unaudited quarterly results of operations for 2011 and 2010 (in thousands, except per share data). The sum of the quarterly per share amounts may not equal the amounts presented for the full year due to differences in the weighted average number of shares outstanding as calculated on a quarterly compared to an annual basis.
| First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
Year | ||||||||||||||||
| 2011: |
||||||||||||||||||||
| Net product sales |
$ | 2,322 | $ | 6,242 | $ | 3,676 | $ | 3,915 | $ | 16,155 | ||||||||||
| Gross profit |
$ | 1,959 | $ | 5,581 | $ | 3,222 | $ | 2,900 | $ | 13,662 | ||||||||||
| Loss from operations |
$ | (17,053 | ) | $ | (14,949 | ) | $ | (15,121 | ) | $ | (10,269 | ) | $ | (57,392 | ) | |||||
| Net loss |
$ | (17,038 | ) | $ | (14,949 | ) | $ | (17,031 | ) | $ | (10,262 | ) | $ | (59,280 | ) | |||||
| Basic and diluted net loss per share |
$ | (0.38 | ) | $ | (0.33 | ) | $ | (0.36 | ) | $ | (0.21 | ) | $ | (1.27 | ) | |||||
| 2010: |
||||||||||||||||||||
| Net product sales |
$ | — | $ | — | $ | 38 | $ | 1,344 | $ | 1,382 | ||||||||||
| Gross profit |
$ | — | $ | — | $ | 35 | $ | 1,103 | $ | 1,138 | ||||||||||
| Loss from operations |
$ | (4,165 | ) | $ | (5,716 | ) | $ | (12,902 | ) | $ | (16,224 | ) | $ | (39,007 | ) | |||||
| Net loss |
$ | (4,165 | ) | $ | (5,721 | ) | $ | (12,900 | ) | $ | (16,027 | ) | $ | (38,813 | ) | |||||
| Basic and diluted net loss per share |
$ | (0.16 | ) | $ | (0.16 | ) | $ | (0.37 | ) | $ | (0.42 | ) | $ | (1.16 | ) | |||||
F-26