Attached files
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2011
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the transition period from to .
Commission File Number 000-23186
BIOCRYST PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| DELAWARE | 62-1413174 | |
| (State of other jurisdiction of incorporation or organization) |
(I.R.S. employer identification no.) |
4505 Emperor Blvd., Suite 200, Durham, North Carolina 27703
(Address of principal executive offices)
(919) 859-1302
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $.01 Par Value |
The NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
Title of each class
None
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ.
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ.
Indicate by a check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨.
Indicate by a check mark whether the registrant submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (Section 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (Section 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨ .
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ |
Accelerated filer þ | Non-accelerated filer ¨ | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by a check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yes ¨ No þ.
The Registrant estimates that the aggregate market value of the Common Stock on June 30, 2011 (based upon the closing price shown on the NASDAQ Global Select Market on June 30, 2011) held by non-affiliates was $109,536,705.
The number of shares of Common Stock, par value $.01, of the Registrant outstanding as of January 31, 2012 was 46,037,816 shares.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive Proxy Statement to be filed in connection with the solicitation of proxies for its 2012 Annual Meeting of Stockholders are incorporated by reference into Items 10, 11, 12, 13 and 14 under Part III hereof.
TABLE OF CONTENTS
PART I
| ITEM 1. | BUSINESS |
Forward-Looking Statements
This report includes forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created in Section 21E. In particular, statements about our expectations, beliefs, plans, objectives or assumptions of future events or performance are contained or incorporated by reference in this report. All statements other than statements of historical facts contained herein are forward-looking statements. We have based these forward-looking statements on our current expectations about future events. While we believe these expectations are reasonable, forward-looking statements are inherently subject to risks and uncertainties, many of which are beyond our control. Our actual results may differ materially from those suggested by these forward-looking statements for various reasons; including those discussed in this report under the heading “Risk Factors.” Given these risks and uncertainties, you are cautioned not to place undue reliance on our forward-looking statements. The forward-looking statements included in this report are made only as of the date hereof. We do not undertake, and specifically decline, any obligation to update any of these statements or to publicly announce the results of any revisions to any forward-looking statements to reflect future events or developments. When used in the report, unless otherwise indicated, “we,” “our,” “us,” the “Company” and “BioCryst” refers to BioCryst Pharmaceuticals, Inc.
Our Business
We are a biotechnology company that designs, optimizes and develops novel drugs that block key enzymes involved in the pathogenesis of diseases. We focus on therapeutic areas with unmet medical needs that are of interest to us and aligned with our capabilities and expertise. Our areas of interest and related development of drug candidates are determined by the scientific discoveries and the potential advantages that our experienced drug discovery group identifies, as well as by the associated potential commercial opportunity of those discoveries. We integrate the disciplines of biology, crystallography, medicinal chemistry and computer modeling to discover and develop small molecule pharmaceuticals through the process known as structure-guided drug design.
Structure-guided drug design is a drug discovery approach by which we design synthetic compounds from detailed structural knowledge of the active sites of enzyme targets associated with particular diseases. We use X-ray crystallography, computer modeling of molecular structures and advanced chemistry techniques to focus on the three-dimensional molecular structure and active site characteristics of the enzymes that control cellular biology. Enzymes are proteins that act as catalysts for many vital biological reactions. Our goal generally is to design a compound that will fit in the active site of an enzyme and thereby prevent its catalytic activity. Molecules in development by us and our partners are summarized in the table below:
| Drug/Drug Candidate |
Drug Class | Therapeutic Area(s) | Phase | Rights | ||||
| Peramivir | Intravenous Neuraminidase Inhibitor | Acute Influenza, hospital setting | Phase 3 | BioCryst (worldwide, except Japan, Taiwan, Korea and Israel) | ||||
| Seasonal Influenza | Approved (Japan) | Shionogi (Japan & Taiwan) | ||||||
| Approved (Korea) | Green Cross (Korea) | |||||||
| BCX4208 | Oral Purine Nucleoside Phosphorylase Inhibitor | Gout | Phase 2 |
BioCryst (worldwide) | ||||
| Forodesine | Oral Purine Nucleoside Phosphorylase Inhibitor | Oncology | Phase 2 | Mundipharma (worldwide) | ||||
| BCX5191 | Oral Nucleoside Analog, RNA Polymerase Inhibitor | Hepatitis C | Preclinical |
BioCryst (worldwide) | ||||
| BCX4161 | Oral Serine Protease Inhibitor Targeting Kallikrein | Hereditary angioedema | Preclinical |
BioCryst (worldwide) |
3
In addition to these drugs and drug candidates, we invest in drug discovery and retain rights to other compounds with various mechanisms of action for a number of therapeutic areas. We continue to evaluate, test and prioritize early compounds to identify assets that should be taken forward for further development.
Our Business Strategy
Our business strategy is to maximize sustainable value by moving our drug candidate portfolio from discovery through clinical development, registration and ultimately to the market. BioCryst was founded on the strength of its early stage discovery and development capabilities. Since 2006, we have expanded our late-stage development and regulatory capabilities. We may decide to market, distribute and sell our products in specific therapeutic areas. Alternatively, we may rely on partners, licensees and others to develop, market, distribute and/or sell our products in therapeutic areas where we have not developed the pre-requisite expertise or for which we do not intend to develop the commercial infrastructure to commercialize a product. The principal elements of our strategy are:
| Ÿ | Focusing on High Value-Added Structure-Guided Drug Design Technologies. We utilize structure-guided drug design in order to most efficiently develop new therapeutic candidates. Structure-guided drug design is a process by which we design a drug candidate through detailed analysis of the enzyme target, which the drug candidate must inhibit in order to stop the progression of the disease or disorder. We believe that structure-guided drug design is a powerful tool for the efficient development of small-molecule drug candidates that have the potential to be safe and effective. Our structure-guided drug design technologies typically allow us to design and synthesize multiple drug candidates that inhibit the same enzyme target, with the goal of establishing broad patent protection and formulating compounds with competitive advantages. |
| Ÿ | Selecting Inhibitors that are Promising Candidates for Commercialization. We test multiple compounds to identify those that are most promising for clinical development. We base our selection of drug candidates on desirable product characteristics, such as initial indications of safety and efficacy. We believe that this focused strategy allows us to eliminate less promising candidates from consideration sooner without incurring substantial clinical costs. In addition, our preference is to select drug candidates on the basis of their potential for relatively efficient Phase 1 and Phase 2 clinical trials. |
| Ÿ | Entering into Contractual Relationships. An important element of our business strategy is to control fixed costs and overhead through contracting and entering into license agreements with third parties. We maintain a streamlined corporate infrastructure that focuses our expertise. By contracting with other specialty organizations and the U.S. government, we believe that we can control costs, enable our drug candidates to reach the market more quickly and reduce our business risk. We generally plan to advance drug candidates through initial and early-stage drug development, and then may out-license drug candidates or continue later stage development, depending on the therapeutic area and our capabilities. We seek to retain U.S. rights to our drug candidates within specialty markets, while relying on collaborative arrangements with third parties for drug candidates within larger markets or outside our area of expertise. Potential third party alliances could include preclinical development, clinical development, regulatory approval, marketing, sales and distribution of our drug candidates. We believe partnerships are a good potential source of development payments, license fees, future event payments and royalties. Partnerships may reduce the costs and risks and increase the effectiveness of late-stage drug development, regulatory approval, manufacturing, and selling of our products. We are willing to license a drug candidate to a partner during any stage of the development process for which we determine it to be beneficial to us and to the ultimate development and commercialization of that drug candidate. |
We are a Delaware corporation originally founded in 1986. Our corporate headquarters is located at 4505 Emperor Blvd., Suite 200, Durham, North Carolina 27703 and the corporate telephone number is (919) 859-1302. Additionally, our Drug Discovery Center of Excellence is located in Birmingham, Alabama. For more information about us, please visit our website at www.biocryst.com. The information on our website is not incorporated into this Form 10-K.
4
Peramivir
Peramivir is a neuraminidase inhibitor for the treatment of patients with influenza. Influenza is a seasonal virus with highest infection rates generally observed in colder months. In Japan and Korea, where peramivir is currently approved for commercial sale, influenza occurs primarily throughout the September to April timeframe.
Intravenous (i.v.) peramivir, for the treatment of patients with influenza, has been approved in Japan and Korea. In addition, i.v. peramivir is currently in Phase 3 development to support filing of a New Drug Application (“NDA”) in the United States, and potentially other countries. We are currently developing i.v. peramivir through funding under a $234.8 million contract from the Biomedical Advanced Research and Development Authority within the United States Department of Health and Human Services (“BARDA/HHS”). See “Collaborations and In-License Relationships—BARDA/HHS” below for a further discussion of this development contract. We also have various regional collaborations for the development and commercialization of peramivir in Taiwan and Israel, as well as government stockpiling agreements in Europe, Russia, Canada and Singapore.
In January 2010, our partner Shionogi & Co., Ltd. (“Shionogi”) received the world’s first approval for i.v. peramivir and launched it under the commercial name RAPIACTA® in Japan. It was initially approved for the treatment of adults with uncomplicated seasonal influenza, as well as those at high-risk for complications associated with influenza. In October 2010, Shionogi received approval for an additional indication to treat children and infants with influenza in Japan. During 2011, Shionogi reported RAPIACTA sales of approximately 300 million Japanese Yen. In August 2010, Green Cross Corporation (“Green Cross”) received marketing and manufacturing approval from the Korean Food & Drug Administration (“KFDA”) for i.v. peramivir under the commercial name PERAMIFLU® to treat patients with influenza A & B viruses, including pandemic H1N1 and avian influenza.
Peramivir is an intravenously administered anti-viral agent that rapidly delivers high plasma concentrations to the sites of infection. Peramivir inhibits the interactions of influenza neuraminidase, an enzyme that is critical to the spread of influenza within the host. Peramivir is an inhibitor of influenza A and B viruses, including strains of influenza viruses that may be resistant to other available neuraminidase inhibitors. Because of the similarities of the neuraminidase active sites among the different strains of the influenza virus, peramivir is a potent broad-spectrum inhibitor and may be effective in the treatment and prevention of influenza irrespective of the strain of the virus. The availability of an i.v. neuraminidase inhibitor may be important in treating patients hospitalized with severe and potentially life-threatening influenza by ensuring that the appropriate dose is administered, which may be a concern with currently available oral or inhaled anti-influenza agents.
The influenza virus causes an acute viral disease of the respiratory tract. Unlike the common cold and some other respiratory infections, seasonal flu can cause severe illness, resulting in life-threatening complications. According to the Centers for Disease Control and Prevention (the “CDC”), an estimated 5% to 20% of the American population suffers from influenza annually, and there are approximately 3,000 to 49,000 flu-related deaths per year in the U.S. Most at risk are young children, the elderly and people with seriously compromised immune systems. With the concern of avian influenza and the possible threat of a pandemic, many governments throughout the world have been stockpiling antiviral drugs, such as oseltamivir (TAMIFLU®). We have several third-party commercial agreements to assist us should we receive any governmental stockpiling orders. There is interest by many of these governments, including the U.S. government, in finding additional vaccines and antivirals to address mutations to the influenza virus or a potential pandemic situation.
Clinical Trials
We are currently enrolling patients in our final i.v. peramivir Phase 3 trial, clinical trial 301. If successful, results from this clinical trial may be used to support an NDA with the U.S. Food & Drug Administration (“FDA”). The 301 clinical trial is an ongoing, multicenter, randomized, double-blind, controlled study to evaluate the efficacy and safety of 600 mg i.v. peramivir administered once-daily for five days in addition to standard of care (“SOC”), compared to SOC alone, in adults and adolescents who are hospitalized due to serious influenza.
5
The modification to our contract with BARDA/HHS and amended clinical trial 301 protocol announced in February 2011 provided for the following significant changes:
| Ÿ | Modifying the primary efficacy analysis population of the trial to focus on a subset of approximately 160 patients not treated with neuraminidase inhibitors as SOC, in order to provide the greatest opportunity to demonstrate a statistically significant peramivir treatment effect. |
| Ÿ | Increasing the total trial target enrollment to approximately 600 subjects from the prior target of 445 subjects. |
| Ÿ | Adding more clinical site locations in geographical regions where neuraminidase inhibitors are not widely used, including sites in India and possibly China. |
The time to reach completion of enrollment will depend on the prevalence and severity of influenza, as well as the ability of approximately 280 investigator sites to successfully enroll patients. Sites in Europe, North America and India are prepared to enroll patients during the 2012 Northern Hemisphere flu season. We intend to conduct a planned interim analysis, which will include an assessment of futility. This analysis is scheduled to be conducted at the earlier of the conclusion of the 2012 Southern Hemisphere flu season or reaching 70% of the current enrollment goal of 160 patients for the primary efficacy analysis population. If the analysis shows an efficacy trend in favor of peramivir, it is expected the clinical trial would continue toward either the current enrollment target or a higher target, depending on the trend. If, however, the new enrollment target to reach statistical significance is predicted to exceed 320 patients, we would terminate the clinical trial and evaluate the data in hand.
On January 13, 2011, we announced top-line results from our completed 303 clinical trial. This clinical trial was an open-label, randomized trial of the anti-viral activity, safety and tolerability of i.v. peramivir administered either as a once-daily infusion of 600 mg or a twice-daily infusion of 300 mg to adult and adolescent subjects hospitalized with confirmed or suspected influenza infection. Treatment was planned for 5 days with an extension to 10 days in patients who needed additional treatment. This completed Phase 3 safety and virology trial was one of the largest prospective clinical trials of an influenza anti-viral in the hospital setting completed to date. The clinical trial enrolled 234 patients aged 14 to 92 years during the 2009-2010 H1N1 pandemic.
Both dose regimens of i.v. peramivir evaluated in the 303 trial were generally safe and well-tolerated. The frequency and severity of adverse events were similar in the two groups, and were consistent with the profile of influenza patients hospitalized during the 2009-2010 H1N1 pandemic. Severe Adverse Events (“SAEs”) were reported in 20 percent of patients. Of the total SAEs reported, one case of elevated liver enzymes was attributed to the study drug and all other SAEs were attributed to other factors. The most common SAEs reported were respiratory failure, acute respiratory distress syndrome, septic shock and acute renal failure. Overall mortality within 28 days of initial peramivir treatment was 8.7 percent; no deaths were attributed to study drug. No safety signals were identified.
The primary endpoint of the 303 clinical trial was the change in influenza virus titer in nasopharyngeal samples, measured by TCID50. Forty-four patients had a positive baseline culture, 20 for the 300 mg twice-daily group and 24 for the 600 mg once-daily group. Similar reductions in log10 TCID50 viral titer were observed over the first 48 hours in the two treatment groups, -1.66 for 300 mg peramivir twice-daily and -1.47 for peramivir 600 mg once-daily.
The analysis of the combined Intent to Treat Infected (“ITTI”) population showed median time to resolution of fever was 25.3 hours; time to clinical resolution, 92.0 hours; time to alleviation of symptoms, 145 hours; and time to resumption of usual activities, 26.8 days.
Purine Nucleoside Phosphorylase (“PNP”) Inhibitors
PNP is a purine salvage pathway enzyme. Low doses of PNP inhibitors could be useful in reducing serum uric acid for the treatment of gout, while high doses of PNP inhibitors could be useful in the treatment of hematological malignancies. We have two PNP inhibitors that are in active development, BCX4208 for the treatment of gout and forodesine for the treatment of hematological malignancies.
6
BCX4208
BCX4208 is an oral PNP inhibitor with the potential for once-a-day dosing suitable for chronic administration. In September 2009, we announced the initiation of a clinical program to develop BCX4208 for the treatment of gout. Gout is a chronic inflammatory arthritis caused by monosodium urate crystal deposits in joints and the kidneys resulting from elevated serum uric acid (“sUA”) levels in the blood, a condition known as hyperuricemia. We believe that BCX4208 is a promising drug candidate to control gout because our Phase 2 clinical trials of BCX4208 confirmed a meaningful dose related reduction in sUA that was sustained for the duration of drug exposure. In addition, BCX4208 is generally safe and well tolerated through 24 weeks of treatment, when evaluated as an add-on therapy to allopurinol in gout patients who have not adequately responded to allopurinol alone.
Following the successful outcome of the Phase 2b 24-week BCX4208 clinical trial reported in January 2012, we are preparing for upcoming end of Phase 2 regulatory discussions, and we are actively evaluating potential partners to fund the Phase 3 development and commercialization of BCX4208.
Clinical Trials
On January 8, 2012, we reported positive long-term results from the extension phase of our randomized, placebo controlled Phase 2b trial 203 evaluating 5 mg, 10 mg, 20 mg and 40 mg of BCX4208 added to allopurinol in patients with gout who had failed to reach the serum uric acid sUA therapeutic goal of <6 mg/dL on allopurinol alone. The results of this 24-week, blinded safety extension confirmed that BCX4208 was generally safe and well-tolerated, and sustained sUA control over time. The longer-term safety profile of BCX4208 is consistent with the 12-week primary analysis results originally reported in October 2011. BCX4208 added to allopurinol was generally safe and well-tolerated at all doses studied, and responses to vaccines indicated healthy immune function. The types and rates of adverse events through 24 weeks, including infections, were similar between the groups treated with BCX4208 and placebo. No opportunistic or unusual infections were observed. As expected, a dose-dependent effect on lymphocyte counts was observed and this effect appeared to plateau within 12 weeks of treatment. Through 24 weeks of treatment, no patients from the placebo, 5 mg or 10 mg cohorts discontinued the study drug due to confirmed lymphocyte or CD4+ cell counts below certain pre-specified thresholds. Four patients were discontinued from the 20 mg group and 11 patients from the 40 mg group due to pre-specified stopping rules based on CD4+ cell counts. Following this analysis, the 40 milligram cohort was discontinued.
A healthy immune response was seen in all treatment arms in a vaccine challenge sub-study conducted in 84 patients. The vaccines were administrated at either 16 or 20 weeks of treatment, and responses were assessed by measuring changes in antibody titers 4 weeks later. The response rates to tetanus toxoid (50%-100%) and polyvalent pneumococcal vaccine (64%-67%) in patients treated with BCX4208 were similar to placebo-treated patients who received tetanus toxoid (50%) and pneumococcal vaccine (64%). The response rates for placebo-treated patients are consistent with responses in normal individuals reported in literature. Patients on BCX4208 doses of 5 mg, 10 mg, 20 mg and placebo were offered to continue treatment on blinded study drug through 52 weeks of treatment. The results of this analysis are expected in mid-2012.
On November 8, 2011, we presented during a late-breaker oral session at the American College of Rheumatology (ACR) positive top-line 12-week results from the Phase 2b BCX4208-203 trial. The clinical trial randomized 279 patients to five trial arms: BCX4208 at doses of 5 mg, 10 mg, 20 mg, 40 mg and placebo, administered once-daily for 12-weeks. Allopurinol 300 mg once-daily was administered in all trial arms. The primary endpoint of the trial was the proportion of patients with sUA <6 mg/dL at day 85. The primary endpoint of the trial was successfully achieved. When added to allopurinol 300 mg, BCX4208 was superior to allopurinol plus placebo (p=0.009 overall). BCX4208 doses evaluated in the trial showed response rates ranging from 33% to 49%, compared to 18% for placebo. Adding BCX4208 to allopurinol was generally safe and well-tolerated at all doses studied. Both the frequency and types of adverse events, including infections, were similar between the groups treated with BCX4208 and placebo. No opportunistic or unusual infections were reported in either the BCX4208 treated groups or placebo.
7
In May 2011, we presented results from our two completed, short-duration Phase 2 clinical trials of BCX4208 at the Annual European Congress of Rheumatology in London, U.K. We reported findings from the Company’s Phase 2 BCX4208-202 trial evaluating BCX4208 alone and in combination with allopurinol, a trial that utilized a factorial design to evaluate various doses of BCX4208 or placebo combined with various doses of allopurinol or placebo. The primary endpoint was change in sUA after 21 days of treatment compared to baseline concentration prior to treatment. A sUA dose-response was demonstrated for both BCX4208 and allopurinol, and the combination of BCX4208 and allopurinol was shown to be superior to either drug alone in sUA reduction. Combinations of lower doses of BCX4208 with allopurinol showed additive or synergistic effects in sUA reduction. The doses of BCX4208 alone and in combination with allopurinol were generally safe and well-tolerated. There were no pharmacokinetic drug-drug interactions between BCX4208 and either allopurinol or its active metabolite, oxypurinol.
In addition, we presented another poster with pooled safety results from the BCX4208-202 trial and the Phase 2 monotherapy trial BCX4208-201. We concluded that the adverse event profile was similar in recipients of BCX4208, allopurinol, placebo or both drugs combined, with the most common adverse events being diarrhea and headache. The rate of infections was similar between BCX4208 alone and in combination with allopurinol compared to placebo. The combination of BCX4208 and allopurinol did not alter the safety profile compared with either agent administered alone. In September 2009 and 2010, we reported positive results from the BCX4208-201 monotheapy trial. The trials’s primary endpoint was the change in sUA concentration after 21 days of treatment compared to baseline concentration prior to treatment. BCX4208 doses of 40 mg, 80 mg,120 mg, 160 and 240 mg per day showed a dose dependent response in sUA reduction. BCX4208 was generally safe and well-tolerated at the doses evaluated in both parts of this trial.
We are conducting a Phase 1 trial to evaluate the metabolic profile of BCX4208 and are also enrolling patients into the 12-week Phase 2 BCX4208-204 trial in patients with gout and moderately impaired renal function. We intend to complete these ongoing clinical trials and will continue our out-licensing conversations with potential partners in 2012. We expect to out-license BCX4208 prior to initiation of Phase 3 clinical trials, which are targeted to begin in the second half of 2012 after receiving guidance from regulatory agencies.
Forodesine
Forodesine is an orally-available transition-state analog PNP inhibitor that may be developed to treat variety of blood cancers, also known as hematological malignancies. Forodesine has been granted Orphan Drug status by the FDA for three indications: T-cell non-Hodgkin’s lymphoma, including cutaneous t-cell lymphoma, (CTCL); chronic lymphocytic leukemia (CLL) and related leukemias including T-cell prolymphocytic leukemia, adult T-cell leukemia, and hairy cell leukemia; and for treatment of acute B-lymphoblastic leukemia (B-ALL). The FDA has also granted “fast track” status to the development of forodesine for the treatment of relapsed or refractory T-cell leukemia, and Special Protocol Assessment (“SPA”) from the FDA for forodesine to conduct a pivotal clinical trial in CTCL with an oral formulation.
In February 2006, we entered into an exclusive, royalty bearing right and license agreement with Mundipharma International Corporation Limited, a subsidiary of Mundipharma International Holdings Limited (“Mundipharma”), for the co-development and commercialization of forodesine for use in the field of oncology (the “Original Agreement”). On November 11, 2011, we entered into an Amended and Restated License and Development Agreement (the “Amended and Restated Agreement”) with Mundipharma amending and restating the Original Agreement.
Under the terms of the Amended and Restated Agreement, Mundipharma was granted worldwide rights to forodesine in the field of oncology. Mundipharma controls all development and commercialization of forodesine and assumes all future development and commercialization costs. Mundipharma also purchased from us $0.9 million of forodesine drug substance. The Amended and Restated Agreement provides for the possibility of future event payments totaling $15.0 million for achieving specified regulatory events for certain indications. In addition, the Amended and Restated Agreement provides that we will receive tiered royalties ranging from mid to high single-digit percentages of net product sales in each country where forodesine is sold by Mundipharma.
8
See “Collaborations and In-License Relationships—Mundipharma” below for a further discussion of the terms and conditions of the Amended and Restated Agreement.
We licensed forodesine and other PNP inhibitors from Albert Einstein College of Medicine of Yeshiva University (“AECOM”) and Industrial Research, Ltd. (“IRL”) and will owe sublicense payments to AECOM/IRL based on the future milestone payments and royalties received by us from Mundipharma. On November 17, 2011, we further amended our agreements with AECOM/IRL whereby AECOM/IRL agreed to accept a reduction of one-half in the percentage of Net Proceeds (as defined in the license agreement) received by us under our Amended and Restated Agreement with Mundipharma. This reduction does not apply to royalty payments made as a result of sales of licensed products by our sublicensees.
Clinical Trials
In September 2010, we reported preliminary top-line results from our pivotal multinational, open-label, single-arm trial evaluating 200 mg once-daily oral forodesine in the treatment of relapsed or refractory CTCL. The clinical trial’s primary endpoint was objective response rate, defined as complete or partial cutaneous response that is sustained for at least 28 days, in patients with later stage (stage IIB, III and IVA) disease who had previously received at least three systemic therapies for their disease, one of which must have been oral bexarotene. Eleven of 101 (11%) later stage patients enrolled achieved a partial cutaneous response, while no patients achieved a complete response. Oral forodesine was generally safe and well-tolerated in this trial, and was administered daily for up to 839 days. The most common adverse events reported were peripheral edema, fatigue, insomnia, diarrhea, headache and nausea.
Also in September 2010, we reported interim results from our exploratory Phase 2 clinical trial to investigate the efficacy and safety of forodesine as monotherapy for CLL. In this open-label, single-arm, multi-center trial, forodesine was administered orally at 200 mg twice-daily for 28-day cycles in 25 previously treated CLL patients. The primary endpoint of the trial was overall response rate. An analysis later conducted after all patients were followed through 6 months showed that six of 23 response-evaluable patients demonstrated a partial response to forodesine, resulting in a response rate of 26%. Forodesine 200 mg orally-administered twice-daily was generally safe and well-tolerated in this trial. The pattern, frequencies and severity distribution of adverse events were generally consistent with CLL-associated poor bone marrow function and immunodeficiency, prior therapies and co-morbidities.
Pre-clinical Compounds
Our leading pre-clinical compounds include BCX4161, a potentially oral prophylactic drug for hereditary angioedema, and BCX5191, a novel adenine nucleoside analog targeting viral RNA polymerase for the potential treatment of hepatitis C. Both compounds are in pre-clinical toxicology studies and remain on track to be prepared to file Investigational New Drug applications (“IND”) during the second half of 2012.
In February 2012, we reported favorable preclinical study results for BCX5191. BioCryst successfully completed in vitro and in vivo studies in which BCX5191 exhibited potent and selective pan-genotypic antiviral activity against the isolated hepatitis C polymerase enzyme, while rapidly converting to the active triphosphate form in the liver. BCX5191 showed no inhibition of human RNA polymerase and no evidence of toxicity from standard in vitro screens. In preclinical models, BCX5191 demonstrates high oral bioavailability and its pharmacokinetic profile supports once-daily dosing in clinical studies. Additional BCX5191 non-clinical experiments are ongoing or planned, including Good Laboratory Practices (GLP) non-clinical safety studies and in vitro evaluation of BCX5191 in combination with ribavirin, to be prepared to file an IND during the fourth quarter of 2012.
Also in February 2012, the Company reported that it has confirmed the potency of BCX4161 in preclinical laboratory experiments using human plasma, and established a predicted therapeutic window for BCX4161 in the prevention of hereditary angioedema attacks. In addition, we have developed a formulation that we believe provides sufficient oral bioavailability to support clinical development. We are proceeding with additional IND-enabling evaluations and expect to be prepared to file an IND during the second half of 2012.
9
Collaborations and In-License Relationships
BARDA/HHS. In January 2007, BARDA/HHS awarded us a $102.6 million, four-year contract for the advanced development of peramivir for the treatment of influenza. Since the initial contract award, the contract has been amended to reflect modifications in the development plan of peramivir for influenza. During 2009, peramivir clinical development shifted to focus on intravenous delivery and the treatment of hospitalized patients. To support this change, the September 2009 contract modification was awarded to extend the i.v. peramivir program by 12 months and to increase funding by $77.2 million. The contract was further modified in February 2011 for an additional $55.0 million. The contract termination date is now December 31, 2013 and the total contract amount from BARDA/HHS is $234.8 million, which is expected to provide funding through completion of Phase 3 and support the filing of an NDA to seek regulatory approval for i.v. peramivir in the U.S. Through December 31, 2011, $174.7 million has been recognized as revenue under this contract.
In October 2010, the Company and BARDA/HHS began to explore certain changes to our currently ongoing Phase 3 i.v. peramivir clinical trial for the treatment of hospitalized patients with serious influenza, including increasing the size of the clinical trial. The necessity for a second pivotal study in acute, uncomplicated outpatient populations was discussed by BARDA/HHS and the FDA and was deemed unnecessary for a label indication for acute, complicated hospitalized patients. In January 2011, based on discussions between BARDA/HHS and the FDA, we submitted a revised contract proposal to BARDA/HHS seeking additional funding toward the completion of the Phase 3 development plan for i.v. peramivir. In the revised contract proposal, we identified changes to the design of our ongoing 301 clinical trial that could increase the likelihood of a positive clinical outcome.
On February 24, 2011, we reported that BARDA/HHS awarded us a $55.0 million contract modification, intended to fund completion of the Phase 3 development of i.v. peramivir for the treatment of patients hospitalized with influenza. This modification to our contract with BARDA/HHS (and amended protocol) provides for the following changes to the 301 clinical trial:
| Ÿ | Modifying the primary efficacy analysis population of the trial to focus on a subset of approximately 160 patients not treated with neuraminidase inhibitors as SOC, in order to provide the greatest opportunity to demonstrate a statistically significant peramivir treatment effect. |
| Ÿ | Increasing the total trial target enrollment to approximately 600 subjects from the prior target of 445 subjects. |
| Ÿ | Adding at least 45 more clinical site locations in geographical regions where neuraminidase inhibitors are not widely used, including sites in India and possibly China. |
In January 2006, the Company received FDA Fast Track designation for peramivir. In September 2009, we received a Request for Proposal (“RFP”) from BARDA/HHS for the supply of i.v. peramivir. In October 2009, the FDA granted an Emergency Use Authorization (“EUA”) for i.v. peramivir, which expired in June 2010, with the expiration of the declared emergency. On November 4, 2009, we received and shipped an order for 10,000 courses of i.v. peramivir (600 mg once-daily for five days) under the EUA for an aggregate purchase price of $22.5 million.
Shionogi. On February 28, 2007, we entered into a License, Development and Commercialization Agreement, as amended, supplemented or otherwise modified (the “Shionogi Agreement”), an exclusive license agreement with Shionogi to develop and commercialize peramivir in Japan for the treatment of seasonal and potentially life-threatening human influenza. Under the terms of the Shionogi Agreement, Shionogi obtained rights to injectable formulations of peramivir in Japan in exchange for a $14.0 million upfront payment. The license provided for development milestone payments (up to $21.0 million), which have all been paid, and for commercial milestone payments (up to $95.0 million) in addition to double digit (between 10% and 20%) royalty payments on product sales of peramivir. Generally, all payments under the Shionogi Agreement are nonrefundable and non-creditable, but they are subject to audit. Shionogi is responsible for all development, regulatory, and marketing costs in Japan. The term of the Shionogi Agreement is from February 28, 2007 until terminated.
10
Either party may terminate in the event of an uncured breach. Shionogi has the right of without cause termination. In the event of termination, all license and rights granted to Shionogi shall terminate and shall revert back to us. We developed peramivir under a license from UAB and have paid sublicense payments to UAB on the upfront payments and will owe sublicense payments on any future event payments and/or royalties received by us from Shionogi. In October 2008, we and Shionogi amended the Shionogi Agreement to expand the territory covered by the agreement to include Taiwan and to provide rights for Shionogi to perform a Phase 3 clinical trial in Hong Kong.
PhaRMA Notes and Currency Hedge Agreement
On March 9, 2011, we announced that JPR Royalty Sub LLC (“Royalty Sub”), a wholly-owned subsidiary of BioCryst, completed a private placement to institutional investors of $30.0 million in aggregate principal amount of its PhaRMA Senior Secured 14% Notes due 2020, (“PhaRMA Notes”). The PhaRMA Notes, which are obligations of Royalty Sub, are secured by (i) Royalty Sub’s rights to receive royalty payments from Shionogi in respect of commercial sales of RAPIACTA in Japan and, if approved for commercial sale, Taiwan, as well as future milestone payments payable by Shionogi under the Shionogi Agreement and all of Royalty Sub’s other assets, and (ii) a pledge by us of our equity interest in Royalty Sub. Royalty Sub’s obligations to pay principal and interest on the PhaRMA Notes are obligations solely of Royalty Sub and are without recourse to any other person, including us, except to the extent of our pledge of our equity interests in Royalty Sub in support of the PhaRMA Notes.
In connection with the issuance of the PhaRMA Notes by Royalty Sub, we entered into a purchase and sale agreement (the “Purchase and Sale Agreement”) dated as of March 9, 2011 between us and Royalty Sub. Under the terms of the Purchase and Sale Agreement, we transferred to Royalty Sub, among other things, (i) our rights to receive certain royalty and milestone payments from Shionogi arising under the Shionogi Agreement, and (ii) the right to receive payments under a Japanese yen/US dollar foreign currency hedge arrangement put into place by us in connection with the transaction. Of the $30.0 million in gross proceeds from the sale of the PhaRMA Notes by Royalty Sub, $3.0 million was used to fund an interest reserve account, and after fees and financing expenses in connection with the transactions, the net proceeds to us were approximately $22.7 million. See Note 3, Royalty Monetization, in the consolidated financial statements included in Item 8 in the Annual Report on Form 10-K for a further description of the terms and conditions of this financing transaction.
The Purchase and Sale Agreement includes customary representations, warranties and covenants by us and customary indemnification and other provisions typical for asset sale agreements in structured financing transactions for pharmaceutical royalty payments.
The PhaRMA Notes were issued by Royalty Sub under an Indenture, dated as of March 9, 2011 (the “Indenture”), by and between Royalty Sub and U.S. Bank National Association, as Trustee (the “Trustee”). Principal and interest on the PhaRMA Notes issued by Royalty Sub are payable from, and are secured by, the rights to royalty and milestone payments under the Shionogi Agreement transferred by us to Royalty Sub and payments, if any, made to Royalty Sub under the Currency Hedge Agreement (defined below). Payments may also be made from the interest reserve account and certain other accounts established in accordance with the Indenture. Principal on the PhaRMA Notes is required to be paid in full by the final legal maturity date of December 1, 2020, unless the PhaRMA Notes are repaid, redeemed or repurchased earlier. The PhaRMA Notes are redeemable by Royalty Sub beginning March 9, 2012 as described below. The PhaRMA Notes bear interest at the rate of 14% per annum, payable annually in arrears on September 1st of each year, beginning on September 1, 2011 (each, a “Payment Date”).
Various accounts have been established in accordance with the Indenture, including, among others, the interest reserve account as well as a collections account into which royalty and milestone payments under the Shionogi Agreement will be made. In addition, we may, but are not obligated to, make capital contributions to a capital account that may be used to redeem, or on up to one occasion pay any interest shortfall on, the PhaRMA Notes.
11
On each Payment Date in respect of the PhaRMA Notes, funds will be applied by the Trustee in the order of priority set forth below:
| Ÿ | first, to Royalty Sub for the payment of all taxes owed by Royalty Sub, if any; |
| Ÿ | second, to the payment of certain expenses of Royalty Sub not previously paid or reimbursed; |
| Ÿ | third, to the Trustee for distribution to the holders, all interest due and payable on the PhaRMA Notes, including any accrued and unpaid interest due on prior Payment Dates, and any accrued and unpaid interest on such unpaid interest, compounded annually, taking into account any amounts paid from the interest reserve account and capital account on such Payment Date; |
| Ÿ | fourth, as long as no event of default has occurred and is continuing, on the September 1, 2014 Payment Date, the September 1, 2015 Payment Date or the September 1, 2016 Payment Date, to the interest reserve account, the amount (if any) set forth in a written direction to the Trustee from Royalty Sub; provided, that such application of funds, together with any such prior application of funds, shall not exceed $2.1 million in the aggregate; |
| Ÿ | fifth, to the Trustee for distribution to the holders of the PhaRMA Notes, principal payments on the PhaRMA Notes (without premium or penalty), allocated pro rata among the holders of the PhaRMA Notes, until the outstanding principal balance of such PhaRMA Notes has been paid in full; |
| Ÿ | sixth, after the PhaRMA Notes have been paid in full, to the Trustee for the payment of principal of, and interest on, subordinated notes, if any, issued by Royalty Sub as permitted by the Indenture for the PhaRMA Notes in certain circumstances; |
| Ÿ | seventh, after the PhaRMA Notes have been paid in full, to the ratable payment of all other obligations under the Indenture for the PhaRMA Notes until all such amounts are paid in full; and |
| Ÿ | eighth, after the PhaRMA Notes and all amounts owing under the Indenture have been paid in full, to Royalty Sub, all remaining amounts. |
If the amounts available for payment on any Payment Date are insufficient to pay all of the interest due on a Payment Date, unless sufficient capital is contributed to Royalty Sub by us as permitted under the Indenture or the interest reserve account is available to make such payment, the shortfall in interest will accrue interest at the interest rate applicable to the PhaRMA Notes compounded annually. If such shortfall (and interest thereon) is not paid in full on or prior to the next succeeding Payment Date, an “Event of Default” under the Indenture will occur. Events of Default under the Indenture include, but are not limited to, the following:
| Ÿ | failure to pay interest on the PhaRMA Notes due on any Payment Date (other than the final legal maturity date or any redemption date) in full, on or prior to the next succeeding Payment Date, together with any additional accrued and unpaid interest on any interest not paid on the Payment Date on which it was originally due; |
| Ÿ | failure to pay principal and premium, if any, and accrued and unpaid interest on the PhaRMA Notes on the final legal maturity date, or failure to pay the redemption price when required on any redemption date; |
| Ÿ | failure to pay any other amount due and payable under the Indenture and the continuance of such default for a period of 30 or more days after written notice thereof is given to Royalty Sub by the Trustee; |
| Ÿ | failure by Royalty Sub to comply with certain covenants set forth in the Indenture or the PhaRMA Notes, provided, that, if the consequences of the failure can be cured, such failure continues for a period of 30 days or more after written notice of the failure has been given to Royalty Sub by the Trustee at the direction of holders of a majority of the outstanding principal balance of PhaRMA Notes, and, except in respect of a covenant, obligation, condition or provision already qualified in respect of Material Adverse Change (as defined in the Indenture), such failure is a Material Adverse Change; |
12
| Ÿ | Royalty Sub becomes subject to a Voluntary Bankruptcy or an Involuntary Bankruptcy (each as defined in the Indenture); |
| Ÿ | any judgment or order for the payment of money in excess of $1.0 million (not paid or covered by insurance) shall be rendered against Royalty Sub and either (i) enforcement proceedings have been commenced by any creditor upon such judgment or order or (ii) there is any period of 30 consecutive days during which a stay of enforcement of such judgment or order, by reason of a pending appeal or otherwise, shall not be in effect; |
| Ÿ | Royalty Sub is classified as a corporation or publicly traded partnership taxable as a corporation for U.S. federal income tax purposes; |
| Ÿ | Royalty Sub becomes an investment company required to be registered under the Investment Company Act of 1940, as amended; |
| Ÿ | we shall have failed to perform any of our covenants under the Purchase and Sale Agreement and such failure is a Material Adverse Change; or |
| Ÿ | the Trustee shall fail to have a first-priority perfected security interest in any of the collateral securing the PhaRMA Notes or in any of the equity in Royalty Sub pledged by us. |
The Indenture do not contain any financial covenants. Additionally, the Indenture includes customary representations and warranties of Royalty Sub, affirmative and negative covenants of Royalty Sub, the above-described Events of Default and related remedies, and provisions regarding the duties of the Trustee, indemnification of the Trustee, and other matters typical for indentures used in structured financings of this type.
Prior to March 9, 2012, the PhaRMA Notes will not be redeemable by Royalty Sub. Thereafter, the PhaRMA Notes will be redeemable at the option of Royalty Sub at any time at a redemption price equal to the percentage of the outstanding principal balance of the PhaRMA Notes being redeemed specified below for the period in which the redemption occurs, plus accrued and unpaid interest through the redemption date on the PhaRMA Notes being redeemed:
| Payment Dates (Between Indicated Dates) |
Redemption Percentage |
|||
| From and including March 9, 2012 to and including March 8, 2013 |
107.0 | % | ||
| From and including March 9, 2013 to and including March 8, 2014 |
103.5 | % | ||
| From and including March 9, 2014 and thereafter |
100.0 | % | ||
In association with the PhaRMA Notes, we entered into a Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar (the “Currency Hedge Agreement”). Under the Currency Hedge Agreement, we have the right to purchase dollars and to sell yen at a rate of 100 yen per dollar for which we may be required to pay a premium in each year from 2014 through 2020, provided the Currency Hedge Agreement remains in effect. A payment of $2.0 million will be required if, on May 18 of the relevant year, the U.S. dollar is worth 100 yen or less as determined in accordance with the Currency Hedge Agreement. In conjunction with establishing the Currency Hedge Agreement, we will be required to post collateral to the counterparty, which may cause us to experience additional quarterly volatility in our financial results. We will not be required at any time to post collateral exceeding the maximum premium payments remaining payable under the Currency Hedge Agreement. Subject to certain obligations we have in connection with the PhaRMA Notes, we have the right to terminate the Currency Hedge Agreement with respect to the 2016 through 2020 period by giving notice to the counterparty prior to May 18, 2014 and paying a $2.0 million termination fee.
Green Cross. In June 2006, we entered into an agreement with Green Cross to develop and commercialize peramivir in Korea. Under the terms of the agreement, Green Cross will be responsible for all development, regulatory, and commercialization costs in Korea. We received a one-time license fee of $0.25 million. The license provides that we will share in profits resulting from the sale of peramivir in Korea, including the sale of peramivir to the Korean government for stockpiling purposes. Furthermore, Green Cross will pay us a premium
13
over its cost to supply peramivir for development and any future marketing of peramivir products in Korea. Both parties have the right to terminate in the event of an uncured material breach. In the event of termination, all rights, data, materials, products and other information would be transferred to us.
In August 2010, we announced that Green Cross had received marketing and manufacturing approval from the Korean Food & Drug Administration for i.v. peramivir to treat patients with influenza A & B viruses, including pandemic H1N1 and avian influenza. Green Cross received the indication of single dose administration of 300 mg i.v. peramivir. Green Cross can launch peramivir under the commercial name PERAMIFLU ® in Korea at any time. As of December 31, 2011, PERAMIFLU has not been launched for sale in Korea.
Other Peramivir Collaborations. In addition to Shionogi and Green Cross, we have arrangements with several companies outside the U.S. to represent us and peramivir for government stockpiling purposes, including Merck KGaA for Europe, Russia, Canada, and Singapore, and Neopharm for Israel.
AECOM and IRL. In June 2000, we licensed a series of potent PNP inhibitors from AECOM/IRL. The license agreement was amended in July 2002, April 2005, December 2009, May 2010 and November 2011. The lead drug candidates from this collaboration are forodesine and BCX4208. We have obtained worldwide exclusive rights to develop these drug candidates for human PNP inhibition and ultimately to distribute these, or any other, drug candidates that might arise from research on these PNP inhibitors. We have the option to expand the agreement to include other inventions in the field made by the investigators or employees of AECOM/IRL. We have agreed to use commercially reasonable efforts to develop these drugs. This license agreement may be terminated by us at any time by giving 60 days advance notice or in the event of material uncured breach by AECOM/IRL.
In addition, we agreed to pay certain milestone payments for each licensed product, which range in the aggregate from $1.4 million to almost $4.0 million per indication, for future development of these inhibitors, single digit royalties on net sales of any resulting product made by us, and to share approximately one quarter of future payments received from third-party sublicensees of the licensed PNP inhibitors, if any. We also agreed to pay annual license fees ranging from $150,000 to $0.5 million, creditable against actual royalties and other payments due to AECOM/IRL.
In May 2010, we and AECOM/IRL agreed to further amend the license agreement. Under the terms of that amendment, AECOM/IRL agreed to accept a reduction of one-half in the percentage of future Net Proceeds (as defined in the license agreement). The reduction did not apply to any payment received by us under the license agreement dated February 1, 2006 with Mundipharma. Further, the reduction did not apply to royalty payments as a result of sales of licensed products by us or our sublicensees. In consideration for the May 2010 modification, we issued to AECOM/IRL shares of our common stock with an aggregate value of approximately $5.9 million and paid AECOM/IRL approximately $90,000 in cash. The value of this consideration began to be amortized to expense in May 2010 and will end in September 2027, which is the expiration date for the last-to-expire patent covered by the agreement. We also agreed to pay certain fees or commissions incurred by AECOM/IRL in connection with subsequent sales of the shares issued pursuant to the amendment.
On November 17, 2011, we further amended our agreements with AECOM/IRL whereby AECOM/IRL agreed to accept a reduction of one-half in the percentage of all Net Proceeds (as defined in the license agreement) received by us under our Amended and Restated Agreement with Mundipharma.
At our sole option and subject to certain agreed upon conditions, any future non-royalty payments due to be paid by us to AECOM/IRL under the license agreement may be made either in cash, in shares of our common stock, or in a combination of cash and shares.
Mundipharma. In February 2006, we entered into an exclusive, royalty bearing right and license agreement with Mundipharma for the development and commercialization of forodesine, a PNP inhibitor, for use in oncology (the “Original Agreement”). Under the terms of the Original Agreement, Mundipharma obtained rights to forodesine in markets across Europe, Asia, and Australasia in exchange for a $10.0 million up-front payment. In addition, Mundipharma contributed $10.0 million of the documented out-of-pocket development costs incurred by us in respect of the current and planned trials as of the effective date of the agreement, and Mundipharma would conduct additional clinical trials at their own cost up to a maximum of $15.0 million. The
14
Original Agreement provided for the possibility of future event payments totaling $155.0 million for achieving specified development, regulatory and commercial events (including certain sales level amounts following a product’s launch) for certain indications. In addition, the Original Agreement provided that we would receive royalties (ranging from single digits to mid teens) based on a percentage of net product sales, which varies depending upon when certain indications receive NDA approval in a major market country and can vary by country depending on the patent coverage or sales of generic compounds in a particular country. Generally, all payments under the Original Agreement were nonrefundable and non-creditable, but they are subject to audit. We licensed forodesine and other PNP inhibitors from AECOM/IRL and will owe sublicense payments to AECOM/IRL on all milestone payments and royalties received by us from Mundipharma.
On November 11, 2011, we entered into the Amended and Restated Agreement with Mundipharma. Under the terms of this Amended and Restated Agreement, Mundipharma obtained worldwide rights to forodesine in the field of oncology. Mundipharma will control the development and commercialization of forodesine and assume all future development and commercialization costs. Mundipharma also purchased from us certain drug substance for forodesine at a cost of approximately $0.9 million. The Amended and Restated Agreement provides for the possibility of future event payments totaling $15.0 million for achieving specified regulatory events for certain indications. In addition, the Amended and Restated Agreement provides that we will receive tiered royalties ranging from mid- to high-single-digit percentages of net product sales in each country where forodesine is sold by Mundipharma. These royalties are subject to downward adjustments based on the then-existing patent coverage and/or the availability of generic compounds in each country. Generally, all payments under the Amended and Restated Agreement are nonrefundable and non-creditable, but they are subject to audit.
Mundipharma will also have a right of exclusive negotiations with us for a limited period of time if they initiate the negotiations for a specified backup PNP inhibitor. Otherwise, they will be able to participate in the same negotiations process we enter into with another company for the backup PNP inhibitor. The Amended and Restated Agreement will continue for the commercial life of the licensed products, but may be terminated by either party following an uncured material breach by the other party or in the event the pre-existing third party license with AECOM/IRL expires. It may be terminated by Mundipharma upon 60 days written notice without cause or under certain other conditions as specified in the Amended and Restated Agreement. If Mundipharma terminates the Amended and Restated Agreement, Mundipharma would no longer have any rights in forodesine and the rights would revert back to us; provided, however, that in the event the we determine to subsequently use the data developed under the Amended and Restated Agreement for development and commercialization of forodesine in the field of oncology, then we would have to pay Mundipharma 150% of the cost of such data for such use. The Amended and Restated Agreement resolved all ongoing disputes between the parties and concluded ongoing negotiations.
Emory University (“Emory”). In June 2000, we licensed intellectual property from Emory related to the HCV polymerase target associated with hepatitis C viral infections. Under the original terms of the agreement, the research investigators from Emory provided us with materials and technical insight into the target. We have agreed to pay Emory single digit royalties on sales of any resulting product and to share in future payments received from other third party partners, if any. We can terminate this agreement at any time by giving 90 days advance notice. Upon termination, we would cease using the licensed technology.
The University of Alabama at Birmingham (“UAB”). We have had a close relationship with UAB since our formation. Our former Chairman, Dr. Charles E. Bugg, was the previous Director of the UAB Center for Macromolecular Crystallography, and our former Chief Operating Officer, Dr. J. Claude Bennett, was the former President of UAB, the former Chairman of the Department of Medicine at UAB and a former Chairman of the Department of Microbiology at UAB. Several of our early programs originated at UAB.
We currently have agreements with UAB for influenza neuraminidase and complement inhibitors. Under the terms of these agreements, UAB performed specific research for us in return for research payments and license fees. UAB has granted us certain rights to any discoveries in these areas resulting from research developed by UAB or jointly developed with us. We have agreed to pay single digit royalties on sales of any resulting product and to share in future payments received from other third-party partners. We have completed the research under both the complement and influenza agreements. These two agreements have initial 25-year terms, are
15
automatically renewable for five-year terms throughout the life of the last patent and are terminable by us upon three months notice and by UAB under certain circumstances. Upon termination each party shall cease using the other party’s proprietary and confidential information and materials, the parties shall jointly own joint inventions and UAB shall resume full ownership of all UAB licensed products. There is currently no activity between us and UAB on these agreements, but when we license this technology, such as in the case of the Shionogi and Green Cross agreements, or commercialize products related to these programs, we will owe sublicense fees or royalties on amounts we receive.
Government Contracts
On February 24, 2011, we announced that BARDA/HHS had awarded us a contract modification of $55.0 million, intended to fund completion of the Phase 3 development of i.v. peramivir for the treatment of patients hospitalized with influenza. This contract modification brings the total award from BARDA/HHS to $234.8 million and extends the contract term by 24 months through December 31, 2013, providing funding through completion of Phase 3 and to support the filing of an NDA to seek regulatory approval for i.v. peramivir in the U.S. Through December 31, 2011, approximately $174.7 million has been recognized as revenue under the contract.
Our contract with BARDA/HHS for the advanced development of peramivir is a milestone-driven, cost-plus-fixed-fee contract. BARDA/HHS will make periodic assessments of our progress, and the continuation of the contract is based on our performance, the timeliness and quality of deliverables, and other factors. The government has rights under certain contract clauses to terminate this contract. The contract is terminable by the government at any time for breach or for convenience. In addition, the government has the right to audit costs billed to them under the contract and routinely conducts audits on our contract. Any findings associated with these routine audits are generally reflected prospectively in our operating results upon the ultimate agreement and resolutions of the audit findings.
BARDA/HHS has indicated that antiviral drugs are an important element of their pandemic influenza preparedness efforts and that their strategy includes not only stockpiling of existing antiviral drugs, but also seeking out new antiviral medications to further broaden their capabilities to treat and prevent all forms of influenza. Peramivir is in the same class of neuraminidase inhibitors as oseltamivir (Tamiflu) and zanamivir (Relenza). We are committed to working with BARDA/HHS for the development of these parenteral formulations of peramivir which could be especially useful in hospital settings or pandemic situations due to the ability to achieve high levels of the drug rapidly throughout the body.
Under the defined scope of work in the contract with BARDA/HHS for the development of peramivir, a process was undertaken to validate a U.S.-based manufacturer and the related method for producing commercial batches of peramivir active pharmaceutical ingredient (“API”). As a required outcome of this validation process, large quantities of peramivir API were produced. In accordance with our accounting practices, we recorded all costs associated with this validation process as research and development expenses in our Consolidated Statement of Operations. Simultaneously, revenue from the BARDA/HHS contract was also recorded in our Consolidated Statement of Operations in 2009. BARDA/HHS subsequently reimbursed us for these costs and upon reimbursement from BARDA/HHS, the associated peramivir API became property of the U.S. government.
Under the terms of the contract, if we determine the amount of peramivir API produced under the contract is in excess of what is necessary to complete the contract, we can acquire any excess peramivir API at cost to use for our own purposes. We believe that as a result of the manufacturing campaign described above, more peramivir API has been produced than is required to support U.S. regulatory approval. If we use any excess API for our other contracts or activities, we will need to reconcile through an appropriate acquisition process with BARDA/HHS and to determine the appropriate acquisition process remuneration for this API.
Patents and Proprietary Information
Our success will depend in part on our ability to obtain and enforce patent protection for our products, methods, processes and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. We own or have rights
16
to certain proprietary information, proprietary technology, issued and allowed patents and patent applications which relate to compounds we are developing. We actively seek, when appropriate, protection for our products, proprietary technology and proprietary information by means of U.S. and foreign patents, trademarks and contractual arrangements. In addition, we rely upon trade secrets and contractual arrangements to protect certain of our proprietary information, proprietary technology and products.
The patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective patent claims and enforcing those claims once granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated, rendered unenforceable or circumvented, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our products. In addition, the rights granted under any issued patents may not provide us with competitive advantages against competitors with similar compounds or technology. Furthermore, our competitors may independently develop similar technologies or duplicate any technology developed by us in a manner that does not infringe our patents or other intellectual property. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our drug candidates or those developed by our partners can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
As of January 31, 2012, we have been issued 19 U.S. patents that expire between 2015 and 2025 and that relate to our PNP, serine protease and neuraminidase inhibitor compounds. We have licensed six different class of compounds representing new composition of matter patents from AECOM and IRL for our PNP inhibitors, plus additional manufacturing patents related to these PNP inhibitors and one patent from Emory related to hepatitis C. Additionally, we have approximately 28 PCT or U.S. patent applications pending related to PNP, neuraminidase, RNA or DNA polymerase, Janus Kinase and serine protease inhibitors. Our pending applications may not result in issued patents, and our patents may not provide us with sufficient protection against competitive products or otherwise be commercially viable.
Our success is also dependent upon the skills, knowledge and experience of our scientific and technical personnel, none of which is patentable. To help protect our rights, we require all employees, consultants, advisors and partners to enter into confidentiality agreements, which prohibit the disclosure of confidential information to anyone outside of our Company and, where possible, requires disclosure and assignment to us of their ideas, developments, discoveries and inventions. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information.
Competition
The pharmaceutical and biotechnology industries are intensely competitive. Many companies, including biotechnology, chemical and pharmaceutical companies, are actively engaged in activities similar to ours, including research and development of drugs for the treatment of cancer, infectious, autoimmune, and inflammatory disorders. Many of these companies have substantially greater financial and other resources, larger research and development staffs, and more extensive marketing and manufacturing organizations than we do. In addition, some of them have considerable experience in preclinical testing, clinical trials and other regulatory approval procedures. There are also academic institutions, governmental agencies and other research organizations that are conducting research in areas in which we are working. They may also market commercial products, either on their own or through collaborative efforts. We expect to encounter significant competition for any of the pharmaceutical products we plan to develop. Companies that complete clinical trials, obtain required regulatory approvals and commence commercial sales of their products before their competitors may achieve a significant competitive advantage.
The pharmaceutical market for products that prevent or treat influenza is very competitive. Key competitive factors for i.v. peramivir include, among others, efficacy, ease of use, safety, price and cost-effectiveness, storage
17
and handling requirements and reimbursement. A number of neuraminidase inhibitors are currently available in the U.S. and other counties, including Japan, for the prevention or treatment of influenza, including seasonal flu vaccines and Roche’s Tamiflu, GlaxoSmithKline’s (“GSK”) Relenza and Daiichi Sankyo’s Inavir. Roche’s neuraminidase inhibitor is also approved for prophylaxis of influenza, and both Roche and GSK have i.v. formulations in clinical trial development. In January 2011, GSK announced initiation of a multi-country Phase 3 study of intravenous zanamivir (the same active ingredient as in Relenza) in hospitalized patients with influenza. Various government entities throughout the world are offering incentives, grants and contracts to encourage additional investment into preventative and therapeutic agents against influenza, which may have the effect of further increasing the number of our competitors and/or providing advantages to certain competitors.
In addition to these companies with neuraminidase inhibitors, there are other companies working to develop additional antiviral drugs to be used against various strains of influenza. In addition, several pharmaceutical and biotechnology firms, including major pharmaceutical companies, have announced efforts in the field of structure-based drug design and in the therapeutic areas of cancer, infectious disease, autoimmune, and inflammatory disorders, as well as other therapeutic areas where we are focusing our drug discovery efforts.
Gout is a large, growing market with a trend of increasing prevalence that experts expect to carry into the foreseeable future. Over 17 million patients have been diagnosed with gout in the major industrial markets. Doctors seek to manage both acute gout attacks and the underlying cause of the disease chronically. BCX4208 is focused on the latter, with the objective of achieving and sustaining a reduced serum uric acid level at or below 6 mg/dL in patients who have failed to reach target on their current therapies.
There remains a high unmet medical need in the gout patient population and several companies are working to address it. More than half of the patients taking allopurinol, the most commonly prescribed urate lowering drug, fail to reach the treatment goal. Additionally, gout patients had suffered from the lack of improvements in treatment for nearly 40 years until the FDA approved Takeda Pharmaceuticals’ ULORIC® in 2009. During 2010, Savient Pharmaceuticals’ KRYSTEXXA® was approved for a severe, sub-population of gout patients. In 2012, there are several programs in late-stage clinical development, including BCX4208, to further improve the efficacy of urate lowering therapy in combination with allopurinol or Uloric.
In order to compete successfully in other therapeutic areas, we must develop proprietary positions in patented drugs for therapeutic markets that have not been satisfactorily addressed by conventional research strategies and, in the process, expand our expertise in structure-based drug design. Our products, even if successfully tested and developed, may not be adopted by physicians over other products and may not offer economically feasible alternatives to other therapies.
Research and Development
We initiated our research and development activities in 1986. We have assembled a scientific research staff with expertise in a broad base of advanced research technologies including protein biochemistry, X-ray crystallography, chemistry and pharmacology. Our research facilities include protein biochemistry and organic synthesis laboratories, testing facilities, X-ray crystallography, computer and graphics equipment and facilities to make drug candidates on a small scale for early stage clinical trials. Beginning in June 2006, we began building an internal clinical development and regulatory team based in North Carolina to manage the development strategy for our later stage products. During the years ended December 31, 2011, 2010, and 2009, our research and development expenses were $56.9 million, $83.9 million and $73.7 million, respectively.
Compliance
We conduct our business in an ethical, fair, honest and lawful manner. We act responsibly, respectfully and with integrity in our relationships with patients, health care professionals, collaborators, governments, regulatory entities, stockholders, suppliers and vendors.
In order to ensure compliance with applicable laws and regulations, our Chief Financial Officer, General Counsel and Vice President of Human Resources oversee compliance training, education, auditing and monitoring; enforce disciplinary guidelines for any infractions of our corporate polices; implement new policies
18
and procedures; respond to any detected issues; and undertake corrective action procedures. Our controls address compliance with laws and regulations that govern public pharmaceutical companies. including, but not limited to, the following: federal and state law, such as the Sarbanes-Oxley Act of 2002 and the U.S. Foreign Corrupt Practices Act of 1977; NASDAQ listing requirements; the Financial Industry Regulatory Authority; the Securities and Exchange Commission; the FDA; and the United States Department of Health and Human Services. Our standard operating procedures are designed to provide a framework for corporate governance in accordance with ethical standards and best legal practices.
Government Regulation
The FDA regulates the pharmaceutical and biotechnology industries in the U.S., and our drug candidates are subject to extensive and rigorous domestic government regulations prior to commercialization. The FDA regulates, among other things, the development, testing, manufacture, safety, efficacy, record-keeping, labeling, storage, approval, advertising, promotion, sale and distribution of pharmaceutical products. In foreign countries, our products are also subject to extensive regulation by foreign governments. These government regulations will be a significant factor in the production and marketing of any pharmaceutical products that we develop. Failure to comply with applicable FDA and other regulatory requirements at any stage during the regulatory process may subject us to sanctions, including:
| Ÿ | delays; |
| Ÿ | warning letters; |
| Ÿ | fines; |
| Ÿ | product recalls or seizures; |
| Ÿ | injunctions; |
| Ÿ | penalties; |
| Ÿ | refusal of the FDA to review pending market approval applications or supplements to approval applications; |
| Ÿ | total or partial suspension of production; |
| Ÿ | civil penalties; |
| Ÿ | withdrawals of previously approved marketing applications; and |
| Ÿ | criminal prosecutions. |
The regulatory review and approval process is lengthy, expensive and uncertain. Before obtaining regulatory approvals for the commercial sale of any products, we or our partners must demonstrate that our drug candidates are safe and effective for use in humans. The approval process takes many years, substantial expenses may be incurred and significant time may be devoted to clinical development.
Before testing potential candidates in humans, we carry out laboratory and animal studies to determine safety and biological activity. After completing preclinical trials, we must file an IND, including a proposal to begin clinical trials, with the FDA. We have filed 13 INDs to date and plan to file, or rely on future partners to file, additional INDs in the future as our potential drug candidates advance to that stage of development. Thirty days after filing an IND, a Phase 1 human clinical trial can start, unless the FDA places a hold on the trial.
Clinical trials to support NDAs are typically conducted in three sequential phases, but the phases may overlap.
Phase 1—During Phase 1, the initial introduction of the drug into healthy volunteers, the drug is tested to assess metabolism, pharmacokinetics and pharmacological actions and safety, including side effects associated with increasing doses.
19
Phase 2—Phase 2 usually involves trials in a limited patient population to: (1) assess the efficacy of the drug in specific, targeted indications; (2) assess dosage tolerance and optimal dosage; and (3) identify possible adverse effects and safety risks.
Phase 3—If a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 clinical trials, also called pivotal studies, major studies or advanced clinical trials, are undertaken to further demonstrate clinical efficacy and to further test for safety within an expanded patient population at geographically dispersed clinical trial sites. In general, the FDA requires that at least two adequate and well-controlled Phase 3 clinical trials be conducted.
Initiation and completion of the clinical trial phases are dependent on several factors including things that are beyond our control. For example, the clinical trials cannot begin at a particular site until that site receives approval from its Institutional Review Board (“IRB”), which reviews the protocol and related documents. This process can take from several weeks to several months. In addition, clinical trials are dependent on patient enrollment, but the rate at which patients enroll in the study depends on:
| Ÿ | willingness of investigators to participate in a study; |
| Ÿ | ability of clinical sites to obtain approval from their IRB; |
| Ÿ | the availability of the required number of eligible subjects to be enrolled in a given trial; |
| Ÿ | the availability of existing or other experimental drugs for the disease we intend to treat; |
| Ÿ | the willingness of patients to participate; and |
| Ÿ | the patients meeting the eligibility criteria. |
Delays in planned patient enrollment may result in increased expense and longer development timelines.
After successful completion of the required clinical testing, generally an NDA is submitted. The FDA may request additional information before accepting an NDA for filing, in which case the application must be resubmitted with the additional information. Once the submission has been accepted for filing, the FDA has 180 days to review the application and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the NDA to an appropriate advisory committee for review, evaluation and recommendation as to scientific issues relevant to whether the application should be approved, but the FDA is not bound by the recommendation of an advisory committee.
Based on its review of the NDA and associated support, such as the results from inspections of manufacturing and clinical sites, the FDA will either approve or refuse to approve the NDA, unless the FDA evaluation is inconclusive, in which case the FDA will issue a “complete response letter.” The complete response letter replaced the FDA’s “approvable” and “non-approvable” letters on August 11, 2008. A complete response letter will state that the FDA cannot approve the NDA in its present form and, usually, will describe all of the specific deficiencies that the FDA has identified in the application. The complete response letter, when possible, will include the FDA’s recommended actions to place the application in a condition for approval. Deficiencies can be minor (e.g., labeling changes) or major (e.g., requiring additional clinical trials). A complete response letter may also be issued before the FDA conducts the required facility inspection and/or reviews labeling, leaving the possibility that additional deficiencies in the original NDA could be subsequently cited. An applicant receiving a complete response letter is permitted to resubmit the NDA addressing the identified deficiencies (in which case a new two or six month review cycle will begin), or withdraw the NDA. The FDA may consider a failure to take action within one year of a complete response letter to be a request to withdraw, unless the applicant has requested an extension of time in which to resubmit. If the FDA approves an NDA, the marketing of the drug product will be limited to the particular disease states and conditions of use that are described in the product label.
We and all of our contract manufacturers are also required to comply with the applicable FDA current Good Manufacturing Practice, or cGMP, regulations during clinical development and to ensure that the product can be consistently manufactured to meet the specifications submitted in an NDA. The cGMP regulations include requirements relating to product quality as well as the corresponding maintenance of records and documentation.
20
Manufacturing facilities must be approved by the FDA before they can be used to manufacture our products. Based on an inspection, the FDA determines whether manufacturing facilities are in compliance with applicable regulations. Manufacturing facilities in non-U.S. countries that are utilized to manufacture drugs for distribution into the United States are subject to inspection by the FDA. Additionally, failure to comply with local regulatory requirements could affect production and availability of product in relevant markets.
Human Resources
As of January 31, 2012, we had 75 employees, of whom 52 were engaged in research and development and 23 were in general and administrative functions. Our research and development staff, 26 of whom hold Ph.D. or M.D. degrees, have diversified experience in biochemistry, pharmacology, X-ray crystallography, synthetic organic chemistry, computational chemistry, medicinal chemistry, clinical development and regulatory affairs.
Our employees are not represented by any collective bargaining agreements, and we have never experienced a work stoppage. Employees are required to execute confidentiality and assignment of intellectual property agreements. We consider our relations with our employees to be satisfactory.
Available Information
We have available a website on the Internet. Our address is www.biocryst.com. We make available, free of charge, at our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. We also make available at our website copies of our audit committee charter, compensation committee charter, corporate governance and nominating committee charter and our code of business conduct, which applies to all our employees as well as the members of our Board of Directors. Any amendment to, or waiver from, our code of business conduct will be posted on our website.
Financial Information
For information related to our revenues, profits, net loss and total assets, in addition to other financial information, please refer to the Financial Statements and Notes to Financial Statements contained in this Annual Report. Financial information about revenues derived from foreign countries is included in Note 1 to the Financial Statements contained in this Annual Report.
| ITEM 1A. | RISK FACTORS |
An investment in our stock involves risks. You should carefully read this entire report and consider the following uncertainties and risks, which may adversely affect our business, financial condition or results of operations, along with all of the other information included in our other filings with the Securities and Exchange Commission, before deciding to buy our common stock.
Risks Relating to Our Business
We have incurred substantial losses since our inception in 1986, expect to continue to incur such losses, and may never be profitable.
Since our inception in 1986, we have not achieved profitability. We expect to incur additional losses for the foreseeable future, and our losses could increase as our research and development efforts progress. To become profitable, we, or our collaborative partners, must successfully manufacture and develop drug product candidates, receive regulatory approval, and successfully commercialize and/or enter into profitable agreements with other parties. It could be several years, if ever, before we receive significant royalties from any current or future license agreements or revenues directly from product sales.
Because of the numerous risks and uncertainties associated with developing our drug candidates and their potential for commercialization, we are unable to predict the extent of any future losses. Even if we do achieve
21
profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. If we are unable to achieve and sustain profitability, the market value of our common stock will likely decline.
Our success depends upon our ability to advance our products through the various stages of development, especially through the clinical trial process.
To receive the regulatory approvals necessary for the sale of our drug candidates, we or our partners must demonstrate through preclinical studies and clinical trials that each drug candidate is safe and effective. The clinical trial process is complex and uncertain. Because of the cost and duration of clinical trials, we may decide to discontinue development of drug candidates that are unlikely to show good results in the trials, unlikely to help advance a product to the point of a meaningful collaboration, or unlikely to have a reasonable commercial potential. We may suffer significant setbacks in pivotal clinical trials, even after earlier clinical trials show promising results. Clinical trials may not be adequately designed or executed, which could affect the potential outcome and analysis of study results. Any of our drug candidates may produce undesirable side effects in humans. These side effects could cause us or regulatory authorities to interrupt, delay or halt clinical trials of a drug candidate. These side effects could also result in the FDA or foreign regulatory authorities refusing to approve the drug candidate for any targeted indications. We, our partners, the FDA or foreign regulatory authorities may suspend or terminate clinical trials at any time if we or they believe the trial participants face unacceptable health risks. Clinical trials may fail to demonstrate that our drug candidates are safe or effective and have acceptable commercial viability.
Our ability to successfully complete clinical trials is dependent upon many factors, including but not limited to:
| Ÿ | our ability to find suitable clinical sites and investigators to enroll patients; |
| Ÿ | the availability of and willingness of patients to participate in our clinical trials; |
| Ÿ | difficulty in maintaining contact with patients to provide complete data after treatment; |
| Ÿ | our drug candidates may not prove to be either safe or effective; |
| Ÿ | clinical protocols or study procedures may not be adequately designed or followed by the investigators; |
| Ÿ | manufacturing or quality control problems could affect the supply of drug product for our trials; and |
| Ÿ | delays or changes in requirements by governmental agencies. |
Clinical trials are lengthy and expensive. We or our partners incur substantial expense for, and devote significant time to, preclinical testing and clinical trials, yet cannot be certain that the tests and trials will ever result in the commercial sale of a product. For example, clinical trials require adequate supplies of drug and sufficient patient enrollment. Delays in patient enrollment can result in increased costs and longer development times. Even if we or our partners successfully complete clinical trials for our drug candidates, we or our partners might not file the required regulatory submissions in a timely manner and may not receive regulatory approval for the drug candidate.
Our clinical trials may not adequately show that our drugs are safe or effective.
Progression of our drug products through the clinical development process is dependent upon our trials indicating our drugs have adequate safety profiles and show positive therapeutic effects in the patients being treated by achieving pre-determined endpoints according to the trial protocols. Failure to achieve either of these could result in delays in our trials or even require the performance of additional unplanned trials. This could result in delays in the development of our drug candidates and could result in significant unexpected costs.
22
If we fail to reach milestones or to make annual minimum payments or otherwise breach our obligations under our license agreements, our licensors may terminate our agreements with them and seek additional remedies.
If we fail to meet payment obligations, performance milestones relating to the timing of regulatory filings, or development and commercial diligence obligations, fail to make milestone payments in accordance with applicable provisions, or fail to pay the minimum annual payments under our respective licenses, our licensors may terminate the applicable license. As a result, our development of the respective drug candidate or commercialization of the product would cease.
If we fail to obtain additional financing or acceptable partnership arrangements, we may be unable to complete the development and commercialization of our drug candidates or continue our research and development programs.
As our clinical programs continue to grow and patient enrollment increases, our costs will increase. Our current and planned clinical trials plus the related development, manufacturing, regulatory approval process requirements, and additional personnel resources and testing required for supporting the development of our drug candidates will consume significant capital resources. Our expenses, revenues and cash burn rate could vary significantly depending on many factors, including our ability to raise additional capital, the development progress of our collaborative agreements for our drug candidates, the amount of funding we receive from BARDA/HHS for peramivir, the amount of funding or assistance, if any, we receive from other governmental agencies or other new partnerships with third parties for the development of our drug candidates, including BCX4208, the amount or profitability of any orders for peramivir by any government agency or other party, the progress and results of our current and proposed clinical trials for our most advanced drug products, the progress made in the manufacturing of our lead products and the progression of our other programs.
We expect that we will be required to raise additional capital to complete the development and commercialization of our current drug candidates and we may seek to raise capital at any time we deem market conditions to be favorable. Additional funding, whether through additional sales of securities or collaborative or other arrangements with corporate partners or from other sources, including governmental agencies, in general and from any BARDA/HHS contract specifically, may not be available when needed or on terms acceptable to us. The issuance of preferred or common stock or convertible securities, with terms and prices significantly more favorable than those of the currently outstanding common stock, could have the effect of diluting or adversely affecting the holdings or rights of our existing stockholders. In addition, collaborative arrangements may require us to transfer certain material rights to such corporate partners. Insufficient funds or lack of an acceptable partnership may require us to delay, scale-back or eliminate certain of our research and development programs.
We expect that we will be required to raise additional capital or enter into one or more acceptable partnership arrangements in order to complete the development of BCX4208. The inability to raise such capital or enter into sufficient acceptable partnership arrangements may require us to delay or eliminate the development of BCX4208 for the treatment of gout.
If BARDA/HHS were to eliminate, reduce or delay funding from our contract, or dispute some of our incurred costs or other actions taken under the contract, this would have a significant negative impact on our revenues, cash flows and the development of peramivir.
Our projections of revenues and incoming cash flows are substantially dependent upon BARDA/HHS reimbursement for the costs related to our peramivir program. If BARDA/HHS were to eliminate, reduce or delay the funding for this program or disallow some of our incurred costs, we would have to obtain additional funding for development of this drug candidate or significantly reduce or stop the development effort. Further, BARDA/HHS may challenge actions that we have taken or may take under our contract, which could negatively impact our operating results and cash flows.
In contracting with BARDA/HHS, we are subject to various U.S. government contract requirements, including general clauses for a cost-reimbursement research and development contract, which may limit our reimbursement or if we are found to be in violation could result in contract termination. U.S. government
23
contracts typically contain extraordinary provisions which would not typically be found in commercial contracts. For instance, government contracts permit unilateral modification by the government, interpretation of relevant regulations (i.e., federal acquisition regulation clauses), and the ability to terminate without cause. In addition, U.S. government contracts are subject to an in process review, where the U.S. government will review the project and will consider its options under the contract. As such, we may be at a disadvantage as compared to other commercial contracts. U.S. government contracts are subject to audit and modification by the government at its sole discretion. If the government terminates its contract with us for its convenience or if we default by failing to perform in accordance with the contract schedule and terms, significant negative impact on our cash flows and operations could result.
Our contract with BARDA/HHS has special contracting requirements, which create additional risks of reduction or loss of funding.
We have entered into a contract with BARDA/HHS for the advanced development of our neuraminidase inhibitor, peramivir. In contracting with BARDA/HHS, we are subject to various U.S. government contract requirements, including general clauses for a cost-reimbursement research and development contract. U.S. government contracts typically contain unfavorable termination provisions and are subject to audit and modification by the government at its sole discretion, which subjects us to additional risks. These risks include the ability of the U.S. government to unilaterally:
| Ÿ | terminate or reduce the scope of our contract; and |
| Ÿ | audit and object to our contract-related costs and fees, including allocated indirect costs. |
The U.S. government may terminate its contracts with us either for its convenience or if we default by failing to perform in accordance with the contract schedule and terms. Termination for convenience provisions generally enables us to recover only our costs incurred or committed, and settlement expenses and profit on the work completed prior to termination. Termination for default provisions does not permit these recoveries.
As a U.S. government contractor, we are required to comply with applicable laws, regulations and standards relating to our accounting practices and are subject to periodic audits and reviews. As part of any such audit or review, the U.S. government may review the adequacy of, and our compliance with, our internal control systems and policies, including those relating to our purchasing, property, estimating, compensation and management information systems. Based on the results of its audits, the U.S. government may adjust our contract-related costs and fees, including allocated indirect costs. This adjustment could impact the amount of revenues reported on a historic basis and could impact our cash flows under the contract prospectively. In addition, in the event BARDA/HHS determines that certain costs and fees were unallowable or determines that the allocated indirect cost rate was higher than the actual indirect cost rate, BARDA/HHS would be entitled to recoup any overpayment as a result. In addition, if an audit or review uncovers any improper or illegal activity, we may be subject to civil and criminal penalties and administrative sanctions, including termination of our contracts, forfeiture of profits, suspension of payments, fines and suspension or prohibition from doing business with the U.S. government. We could also suffer serious harm to our reputation if allegations of impropriety were made against us. In addition, under U.S. government purchasing regulations, some of our costs may not be reimbursable or allowed under our contracts. Further, as a U.S. government contractor, we are subject to an increased risk of investigations, criminal prosecution, civil fraud, whistleblower lawsuits and other legal actions and liabilities as compared to private sector commercial companies.
If we fail to successfully commercialize or establish collaborative relationships to commercialize certain of our drug candidates or if any partner terminates or fails to perform its obligations under agreements with us, potential revenues from commercialization of our drug candidates could be reduced, delayed or eliminated.
Our business strategy is to increase the asset value of our drug candidate portfolio. We believe this is best achieved by retaining full product rights or through collaborative arrangements with third parties as appropriate. As needed, potential third-party alliances could include preclinical development, clinical development, regulatory approval, marketing, sales and distribution of our drug candidates.
24
Currently, we have established collaborative relationships with Mundipharma for the development and commercialization of forodesine and with each of Shionogi and Green Cross for the development and commercialization of peramivir. The process of establishing and implementing collaborative relationships is difficult, time-consuming and involves significant uncertainty, including:
| Ÿ | our partners may seek to renegotiate or terminate their relationships with us due to unsatisfactory clinical results, a change in business strategy, a change of control or other reasons; |
| Ÿ | our contracts for collaborative arrangements may expire; |
| Ÿ | our partners may choose to pursue alternative technologies, including those of our competitors; |
| Ÿ | we may have disputes with a partner that could lead to litigation or arbitration; |
| Ÿ | we do not have day to day control over the activities of our partners and have limited control over their decisions; |
| Ÿ | our ability to generate future event payments and royalties from our partners depends upon their abilities to establish the safety and efficacy of our drug candidates, obtain regulatory approvals and achieve market acceptance of products developed from our drug candidates; |
| Ÿ | we or our partners may fail to properly initiate, maintain or defend our intellectual property rights, where applicable, or a party may utilize our proprietary information in such a way as to invite litigation that could jeopardize or potentially invalidate our proprietary information or expose us to potential liability; |
| Ÿ | our partners may not devote sufficient capital or resources towards our drug candidates; and |
| Ÿ | our partners may not comply with applicable government regulatory requirements. |
If any partner fails to fulfill its responsibilities in a timely manner, or at all, our commercialization efforts related to that collaboration could be reduced, delayed or terminated, or it may be necessary for us to assume responsibility for activities that would otherwise have been the responsibility of our partner. If we are unable to establish and maintain collaborative relationships on acceptable terms, we may have to delay or discontinue further development of one or more of our drug candidates, undertake commercialization activities at our own expense or find alternative sources of funding. Any delay in the development or commercialization of our drug candidates would severely affect our business, because if our drug candidates do not progress through the development process or reach the market in a timely manner, or at all, we may not receive additional future event payments and may never receive milestone, product sales or royalty payments.
We have not commercialized any products or technologies and our future revenue generation is uncertain.
We have not commercialized any products or technologies, and we may never be able to do so. We currently have no marketing capability and no direct or third-party sales or distribution capabilities and may be unable to establish these capabilities for products we plan to commercialize. In addition, our revenue from collaborative agreements is dependent upon the status of our preclinical and clinical programs. If we fail to advance these programs to the point of being able to enter into successful collaborations, we will not receive any future event or other collaborative payments.
Our ability to receive revenue from products we commercialize presents several risks, including:
| Ÿ | we or our collaborators may fail to successfully complete clinical trials sufficient to obtain FDA marketing approval; |
| Ÿ | many competitors are more experienced and have significantly more resources and their products could be more cost effective or have a better efficacy or tolerability profile than our drug candidates; |
| Ÿ | we may fail to employ a comprehensive and effective intellectual property strategy which could result in decreased commercial value of our Company and our products; |
25
| Ÿ | we may fail to employ a comprehensive and effective regulatory strategy which could result in a delay or failure in commercialization of our products; |
| Ÿ | our ability to successfully commercialize our products are affected by the competitive landscape, which cannot be fully known at this time; |
| Ÿ | reimbursement is constantly changing which could greatly affect usage of our products; and |
| Ÿ | any future revenue directly from product sales would depend on our ability to successfully complete clinical studies, obtain regulatory approvals, manufacture, market and commercialize any approved drugs. |
If our development collaborations with third parties, such as our development partners and contract research organizations, fail, the development of our drug candidates will be delayed or stopped.
We rely heavily upon other parties for many important stages of our drug candidate development, including but not limited to:
| Ÿ | discovery of compounds that cause or enable biological reactions necessary for the progression of the disease or disorder, called enzyme targets; |
| Ÿ | licensing or design of enzyme inhibitors for development as drug candidates; |
| Ÿ | execution of some preclinical studies and late-stage development for our compounds and drug candidates; |
| Ÿ | management of our clinical trials, including medical monitoring and data management; |
| Ÿ | execution of additional toxicology studies that may be required to obtain approval for our drug candidates; and |
| Ÿ | manufacturing the starting materials and drug substance required to formulate our drug products and the drug candidates to be used in both our clinical trials and toxicology studies. |
Our failure to engage in successful collaborations at any one of these stages would greatly impact our business. If we do not license enzyme targets or inhibitors from academic institutions or from other biotechnology companies on acceptable terms, our drug development efforts would suffer. Similarly, if the contract research organizations that conduct our initial or late-stage clinical trials, conduct our toxicology studies, manufacture our starting materials, drug substance and drug products or manage our regulatory function breached their obligations to us or perform their services inconsistent with industry standards and not in accordance with the required regulations, this would delay or prevent the development of our drug candidates.
If we lose our relationship with any one or more of these parties, we could experience a significant delay in both identifying another comparable provider and then contracting for its services. We may be unable to retain an alternative provider on reasonable terms, if at all. Even if we locate an alternative provider, it is likely that this provider may need additional time to respond to our needs and may not provide the same type or level of service as the original provider. In addition, any provider that we retain will be subject to applicable FDA current Good Laboratory Practices (“cGLP”), current Good Manufacturing Practices (“cGMP”) and current Good Clinical Practices (“cGCP”), and comparable foreign standards. We do not have control over compliance with these regulations by these providers. Consequently, if these practices and standards are not adhered to by these providers, the development and commercialization of our drug candidates could be delayed, and our business, financial condition and results of operations could be materially adversely affected.
26
Our development of peramivir for influenza is subject to all disclosed drug development and potential commercialization risks and numerous additional risks. Any potential revenue benefits to us are highly speculative.
Further development and potential commercialization of peramivir is subject to all the risks and uncertainties disclosed in our other risk factors relating to drug development and commercialization. In addition, potential commercialization of peramivir is subject to further risks, including but not limited to the following:
| Ÿ | the peramivir i.v. currently in clinical development may not prove to be safe and sufficiently effective for market approval in the United States or other major markets; |
| Ÿ | necessary government or other third party funding and clinical testing for further development of peramivir may not be available timely, at all, or in sufficient amounts; |
| Ÿ | the flu prevention or pandemic treatment concerns may not materialize at all, or in the near future; |
| Ÿ | advances in flu vaccines or other antivirals, including competitive i.v. antivirals, could substantially replace potential demand for peramivir; |
| Ÿ | any substantial demand for pandemic or seasonal flu treatments may occur before peramivir can be adequately developed and tested in clinical trials; |
| Ÿ | peramivir may not prove to be accepted by patients and physicians as a treatment for seasonal influenza compared to the other currently marketed antiviral drugs, which would limit revenue from non-governmental entities; |
| Ÿ | numerous large and well-established pharmaceutical and biotech companies will be competing to meet the market demand for flu drugs and vaccines; |
| Ÿ | the only major markets in which patents relating to peramivir have issued or been allowed are the United States, Canada, Japan, Australia and many contracting and extension states of the European Union, while no patent applications or issued patents for peramivir exist in other potentially significant markets; |
| Ÿ | regulatory authorities may not make needed accommodations to accelerate the drug testing and approval process for peramivir; and |
| Ÿ | in the next few years, it is expected that a limited number of governmental entities will be the primary potential customers for peramivir and if we are not successful at marketing peramivir to these entities for any reason, we will not receive substantial revenues from stockpiling orders from these entities. |
If any or all of these and other risk factors occur, we will not attain significant revenues or gross margins from peramivir and our stock price will be adversely affected.
There are risks related to the potential emergency use or sale of peramivir.
To the extent that peramivir is used as a treatment for H1N1 flu (or other strains of flu), there can be no assurance that it will prove to be generally safe, well tolerated and effective. Emergency use of peramivir may create certain liabilities for us. There is no assurance that we or our manufacturers will be able to fully meet the demand for peramivir in the event of additional orders. Further, we may not achieve a favorable price for additional orders of peramivir in the U.S. or in any other country. Our competitors may develop products that could compete with or replace peramivir. We may face competition in markets where we have no existing intellectual property protection or are unable to successfully enforce our intellectual property rights.
There is no assurance that the non-U.S. partnerships that we have entered into for peramivir will result in any order for peramivir in those countries. There is no assurance that peramivir will be approved for emergency use or will achieve market approval in additional countries. In the event that any emergency use is granted, there is no assurance that any order by any non-U.S. partnership will be substantial or will be profitable to us. The sale of peramivir, emergency use or other use of peramivir in any country may create certain liabilities for us.
27
Because we have limited manufacturing experience, we depend on third-party manufacturers to manufacture our drug candidates and the materials for our drug candidates. If we cannot rely on third-party manufacturers, we will be required to incur significant costs and potential delays in finding new third-party manufacturers.
We have limited manufacturing experience and only a small scale manufacturing facility. We currently rely upon third-party manufacturers to manufacture the materials required for our drug candidates and most of the preclinical and clinical quantities of our drug candidates. We depend on these third-party manufacturers to perform their obligations in a timely manner and in accordance with applicable governmental regulations. Our third-party manufacturers may encounter difficulties with meeting our requirements, including but not limited to problems involving:
| Ÿ | inconsistent production yields; |
| Ÿ | product liability claims; |
| Ÿ | difficulties in scaling production to commercial and validation sizes; |
| Ÿ | interruption of the delivery of materials required for the manufacturing process; |
| Ÿ | scheduling of plant time with other vendors or unexpected equipment failure; |
| Ÿ | potential catastrophes, such as the recent earthquake in Japan, that could strike their facilities or have an effect on infrastructure; |
| Ÿ | potential impurities in our drug substance or drug products that could affect availability of product for our clinical trials or future commercialization; |
| Ÿ | poor quality control and assurance or inadequate process controls; and |
| Ÿ | lack of compliance with regulations and specifications set forth by the FDA or other foreign regulatory agencies. |
These contract manufacturers may not be able to manufacture the materials required or our drug candidates at a cost or in quantities necessary to make them commercially viable. We also have no control over whether third-party manufacturers breach their agreements with us or whether they may terminate or decline to renew agreements with us. To date, our third-party manufacturers have met our manufacturing requirements, but they may not continue to do so. Furthermore, changes in the manufacturing process or procedure, including a change in the location where the drug is manufactured or a change of a third-party manufacturer, may require prior review and approval in accordance with the FDA’s cGMPs and comparable foreign requirements. This review may be costly and time-consuming and could delay or prevent the launch of a product. The FDA or similar foreign regulatory agencies at any time may also implement new standards, or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. If we or our contract manufacturers are unable to comply, we or they may be subject to regulatory action, civil actions or penalties.
If we are unable to enter into agreements with additional manufacturers on commercially reasonable terms, or if there is poor manufacturing performance on the part of any of our third-party manufacturers, we may not be able to complete development of, or market, our drug candidates.
Our raw materials, drug substances, and drug products are manufactured by a limited group of suppliers and some at a single facility. If any of these suppliers were unable to produce these items, this could significantly impact our supply of drug candidate material for further preclinical testing and clinical trials.
Royalties and milestone payments from Shionogi under the Shionogi Agreement will be required to be used by Royalty Sub to service its obligations under its PhaRMA Notes, and generally will not be available to us for other purposes until Royalty Sub has repaid in full its obligations under the PhaRMA Notes.
In March 2011, our wholly-owned subsidiary Royalty Sub issued $30.0 million in aggregate principal amount of PhaRMA Notes. The PhaRMA Notes are secured principally by (i) certain royalty and milestone payments under the Shionogi Agreement, pursuant to which Shionogi licensed from us the rights to market
28
peramivir in Japan and, if approved for commercial sale, Taiwan, (ii) rights to certain payments under a Japanese yen/U.S. dollar foreign currency hedge arrangement put into place by us in connection with the issuance of the PhaRMA Notes and (iii) the pledge by us of our equity interest in Royalty Sub. Payments from Shionogi to us under the Shionogi Agreement will generally not be available to us for other purposes until Royalty Sub has repaid in full its obligations under the PhaRMA Notes. Accordingly, these funds will be required to be dedicated to Royalty Sub’s debt service and not available to us for product development or other purposes.
If royalties from Shionogi are insufficient for Royalty Sub to make payments under the PhaRMA Notes or if an event of default occurs under the PhaRMA Notes, investors may be able to foreclose on the collateral securing the PhaRMA Notes and our equity interest in Royalty Sub, in which case we may not realize the benefit of future royalty payments that might otherwise accrue to us following repayment of the PhaRMA Notes.
Royalty Sub’s ability to service its payment obligations in respect of the PhaRMA Notes, and our ability to benefit from our equity interest in Royalty Sub, is subject to numerous risks. Peramivir was first approved for marketing and manufacturing in Japan in October 2009 and has been offered for sale in Japan only since January 2010. As a result, there is very little sales history for peramivir in Japan, and there can be no assurance that peramivir will gain market acceptance in the Japanese market. In addition, Shionogi’s sales of peramivir are expected to be highly seasonal and vary significantly from year to year, and the market for products to treat or prevent influenza is highly competitive. Under our license agreement with Shionogi, Shionogi has control over the commercial process for peramivir in Japan and Taiwan. Royalty Sub’s ability to service the PhaRMA Notes may be adversely affected by, among other things, changes in or any termination of our relationship with Shionogi, reimbursement, regulatory, manufacturing and/or intellectual property issues, product returns, product recalls, product liability claims and allegations of safety issues, as well as other factors. In the event that for any reason Royalty Sub is unable to service its obligations under the PhaRMA Notes or an event of default were to occur under the PhaRMA Notes, the holders of the PhaRMA Notes may be able to foreclose on the collateral securing the PhaRMA Notes and our equity interest in Royalty Sub and exercise other remedies available to them under the indenture in respect of the PhaRMA Notes. In such event, we may not realize the benefit of future royalty payments that might otherwise accrue to us following repayment of the PhaRMA Notes and we might otherwise be adversely affected.
Shionogi’s failure to successfully market and commercialize peramivir in Japan would have a material adverse effect on Royalty Sub’s ability to service its obligations on the PhaRMA Notes.
The successful commercialization of peramivir in Japan depends on the efforts of Shionogi and is beyond the control of us or Royalty Sub. As discussed above, peramivir has only recently been introduced into the Japanese market, and there can be no assurance that peramivir will gain market acceptance in Japan. Future sales by Shionogi will depend on many factors, including the incidence and severity of seasonal influenza in Japan each year (both of which can vary very significantly from year to year), the perceived and actual efficacy and safety of peramivir, experience of physicians and patients with peramivir, continued market acceptance, continued availability of supply, competition, sales and marketing efforts, governmental regulation and pricing and reimbursement in Japan. Shionogi is responsible for the marketing and sale of peramivir in Japan, including with respect to the pricing of peramivir in that market. There are no minimum royalties, sales levels or other performance measures required of Shionogi under the Shionogi Agreement and Shionogi could in its sole discretion reduce or cease its sale efforts of peramivir in Japan, subject to its covenant in the Shionogi Agreement to use diligent efforts to commercialize peramivir in Japan. If Shionogi is unable to or fails to successfully market and commercialize peramivir, it would have a material adverse effect on Royalty Sub’s ability to service its obligations under the PhaRMA Notes and our ability to benefit from our equity interest in Royalty Sub.
29
We may be required to pay significant premiums under the foreign currency hedge arrangement entered into by us in connection with the issuance of the PhaRMA Notes. In addition, because our potential obligations under the foreign currency hedge are marked to market, we may experience additional quarterly volatility in our operating results and cash flows attributable to the foreign Currency Hedge Agreement.
In connection with the issuance by Royalty Sub of the PhaRMA Notes, we entered into a foreign Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. Under the Currency Hedge Agreement, we may be required to pay a premium in the amount of $2.0 million in each year beginning in May 2014 and, provided the Currency Hedge Agreement remains in effect, continuing through May 2020. Such payment will be required if, in May of the relevant year, the spot rate of exchange for Japanese yen-U.S. dollars (determined in accordance with the Currency Hedge Agreement) is such that the U.S. dollar is worth 100 yen or less. We will be required to mark-to-market our potential obligations under the currency hedge and post cash collateral, which may cause us to experience additional quarterly volatility in our operating results and cash flows as a result. Additionally, we may be required to pay significant premiums or a termination fee under the foreign Currency Hedge Agreement entered into by us in connection with the issuance of the PhaRMA Notes. As of December 31, 2011, we have realized a foreign currency hedge loss of approximately $4.0 million and posted cash collateral of approximately $3.5 million.
If we or our partners do not obtain and maintain governmental approvals for our products under development, we or our partners will not be able to sell these potential products, which would significantly harm our business because we will receive no revenue.
We or our partners must obtain regulatory approval before marketing or selling our future drug products. If we or our partners are unable to receive regulatory approval and do not market or sell our future drug products, we will never receive any revenue from such product sales. In the United States, we or our partners must obtain FDA approval for each drug that we intend to commercialize. The process of preparing for and obtaining FDA approval may be lengthy and expensive, and approval is never certain. Products distributed abroad are also subject to foreign government regulation and export laws of the U.S. Neither the FDA nor foreign regulatory agencies have approved any of our drug candidates. Because of the risks and uncertainties in biopharmaceutical development, our drug candidates could take a significantly longer time to gain regulatory approval than we expect or may never gain approval. If the FDA delays regulatory approval of our drug candidates, our management’s credibility, our company’s value and our operating results may suffer. Even if the FDA or foreign regulatory agencies approve a drug candidate, the approval may limit the indicated uses for a drug candidate and/or may require post-marketing studies.
The FDA regulates, among other things, the record keeping and storage of data pertaining to potential pharmaceutical products. We currently store most of our preclinical research data, our clinical data and our manufacturing data at our facility. While we do store duplicate copies of most of our clinical data offsite and a significant portion of our data is included in regular backups of our systems, we could lose important data if our facility incurs damage, or if our vendor data systems fail, suffer damage or are destroyed. If we receive approval to market our potential products, whether in the United States or internationally, we will continue to be subject to extensive regulatory requirements. These requirements are wide ranging and govern, among other things:
| Ÿ | adverse drug experience reporting regulations; |
| Ÿ | product promotion; |
| Ÿ | product manufacturing, including good manufacturing practice requirements; and |
| Ÿ | product changes or modifications. |
Our failure to comply with existing or future regulatory requirements, or our loss of, or changes to, previously obtained approvals, could have a material adverse effect on our business because we will not receive product or royalty revenues if we or our partners do not receive approval of our products for marketing.
30
We face intense competition, and if we are unable to compete effectively, the demand for our products, if any, may be reduced.
The biotechnology and pharmaceutical industries are highly competitive and subject to rapid and substantial technological change. We face, and will continue to face, competition in the licensing of desirable disease targets, licensing of desirable drug candidates, and development and marketing of our drug candidates from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies. Competition may also arise from, among other things:
| Ÿ | other drug development technologies; |
| Ÿ | methods of preventing or reducing the incidence of disease, including vaccines; and |
| Ÿ | new small molecule or other classes of therapeutic agents. |
Developments by others may render our drug candidates or technologies obsolete or noncompetitive.
We and our partners are performing research on or developing products for the treatment of several disorders including T-cell mediated disorders (T-cell cancers and other autoimmune indications), CTCL, CLL, influenza, gout, hereditary angiodema, and hepatitis C. We expect to encounter significant competition for any of the pharmaceutical products we plan to develop. Companies that complete clinical trials, obtain required regulatory approvals and commence commercial sales of their products before their competitors may achieve a significant competitive advantage. Such is the case with Eisai’s Targretin for CTCL and the current neuraminidase inhibitors marketed by GSK and F. Hoffman-La Roche, Ltd. and Hoffman-La Roche, Inc. (collectively “Roche”) for influenza. With respect to the neuraminidase inhibitors, these companies may develop i.v. formulations that could compete with peramivir. Further, several pharmaceutical and biotechnology firms, including major pharmaceutical companies and specialized structure-based drug design companies, have announced efforts in the field of structure-based drug design and in the fields of PNP, influenza, hepatitis C, and in other therapeutic areas where we have discovery efforts ongoing. If one or more of our competitors’ products or programs are successful, the market for our products may be reduced or eliminated.
Compared to us, many of our competitors and potential competitors have substantially greater:
| Ÿ | capital resources; |
| Ÿ | research and development resources, including personnel and technology; |
| Ÿ | regulatory experience; |
| Ÿ | preclinical study and clinical testing experience; |
| Ÿ | manufacturing and marketing experience; and |
| Ÿ | production facilities. |
Any of these competitive factors could reduce demand for our products.
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of those rights would diminish.
Our success will depend in part on our ability and the abilities of our partners to obtain, protect and enforce viable intellectual property rights including but not limited to trade name, trade mark and patent protection for our Company and its products, methods, processes and other technologies we may license or develop, to preserve our trade secrets, and to operate without infringing the proprietary rights of third parties both domestically and abroad. The patent position of biotechnology and pharmaceutical companies is generally highly uncertain, involves complex legal and factual questions and has recently been the subject of much litigation. Neither the United States Patent and Trademark Office (“USPTO”), the Patent Cooperation Treaty offices, nor the courts of the U.S. and other jurisdictions have consistent policies nor predictable rulings regarding the breadth of claims allowed or the degree of protection afforded under many biotechnology and pharmaceutical patents. Further, we do not have worldwide patent protection for our drug candidates and our intellectual property rights may not be
31
legally protected or enforceable in all countries throughout the world. The validity, scope, enforceability and commercial value of these rights, therefore, is highly uncertain.
Our success depends in part on avoiding the infringement of other parties’ patents and other intellectual property rights as well as avoiding the breach of any licenses relating to our technologies and products. In the U.S., patent applications filed in recent years are confidential for 18 months, while older applications are not published until the patent issues. As a result, avoiding patent infringement may be difficult and we may inadvertently infringe third-party patents or proprietary rights. These third parties could bring claims against us, our partners or our licensors that even if resolved in our favor, could cause us to incur substantial expenses and, if resolved against us, could additionally cause us to pay substantial damages. Further, if a patent infringement suit were brought against us, our partners or our licensors, we or they could be forced to stop or delay research, development, manufacturing or sales of any infringing product in the country or countries covered by the patent we infringe, unless we can obtain a license from the patent holder. Such a license may not be available on acceptable terms, or at all, particularly if the third party is developing or marketing a product competitive with the infringing product. Even if we, our partners or our licensors were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property.
If we or our partners are unable or fail to adequately initiate, protect, defend or enforce our intellectual property rights in any area of commercial interest or in any part of the world where we wish to seek regulatory approval for our products, methods, processes and other technologies, the value of the drug candidates to produce revenue would diminish. Additionally, if our products, methods, processes, and other technologies or our commercial use of such products, processes, and other technologies, including but not limited to any trade name, trademark or commercial strategy infringe the proprietary rights of other parties, we could incur substantial costs. The USPTO and the patent offices of other jurisdictions have issued to us a number of patents for our various inventions and we have in-licensed several patents from various institutions. We have filed additional patent applications and provisional patent applications with the USPTO. We have filed a number of corresponding foreign patent applications and intend to file additional foreign and U.S. patent applications, as appropriate. We have also filed certain trademark and trade name applications worldwide. We cannot assure you as to:
| Ÿ | the degree and range of protection any patents will afford against competitors with similar products; |
| Ÿ | if and when patents will issue; |
| Ÿ | if patents do issue we can not be sure that we will be able to adequately defend such patents and whether or not we will be able to adequately enforce such patents; or |
| Ÿ | whether or not others will obtain patents claiming aspects similar to those covered by our patent applications. |
If the USPTO or other foreign patent office upholds patents issued to others or if the USPTO grants patent applications filed by others, we may have to:
| Ÿ | obtain licenses or redesign our products or processes to avoid infringement; |
| Ÿ | stop using the subject matter claimed in those patents; or |
| Ÿ | pay damages. |
We may initiate, or others may bring against us, litigation or administrative proceedings related to intellectual property rights, including proceedings before the USPTO or other foreign patent office. Any judgment adverse to us in any litigation or other proceeding arising in connection with a patent or patent application could materially and adversely affect our business, financial condition and results of operations. In addition, the costs of any such proceeding may be substantial whether or not we are successful.
Our success is also dependent upon the skills, knowledge and experience, none of which is patentable, of our scientific and technical personnel. To help protect our rights, we require all employees, consultants, advisors and partners to enter into confidentiality agreements that prohibit the disclosure of confidential information to anyone outside of our company and require disclosure and assignment to us of their ideas, developments, discoveries and inventions. These agreements may not provide adequate protection for our trade secrets,
32
know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information, and if any of our proprietary information is disclosed, our business will suffer because our revenues depend upon our ability to license or commercialize our product candidates and any such events would significantly impair the value of such drug candidates.
There is a substantial risk of product liability claims in our business. If we are unable to obtain sufficient insurance, a product liability claim against us could adversely affect our business.
We face an inherent risk of product liability exposure related to the testing of our drug candidates in human clinical trials and will face even greater risks upon any commercialization by us of our product candidates. We have product liability insurance covering our clinical trials. Clinical trial and drug liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance or increase our existing coverage at a reasonable cost to protect us against losses that could have a material adverse effect on our business. An individual may bring a product liability claim against us if one of our products or drug candidates causes, or is claimed to have caused, an injury or is found to be unsuitable for consumer use. Any product liability claim brought against us, with or without merit, could result in:
| Ÿ | liabilities that substantially exceed our product liability insurance, which we would then be required to pay from other sources, if available; |
| Ÿ | an increase of our product liability insurance rates or the inability to maintain insurance coverage in the future on acceptable terms, or at all; |
| Ÿ | withdrawal of clinical trial volunteers or patients; |
| Ÿ | damage to our reputation and the reputation of our products, resulting in lower sales; |
| Ÿ | regulatory investigations that could require costly recalls or product modifications; |
| Ÿ | litigation costs; and |
| Ÿ | the diversion of management’s attention from managing our business. |
Insurance coverage is increasingly more costly and difficult to obtain or maintain.
While we currently have insurance for our business, property, directors and officers, and our products, insurance is increasingly more costly and narrower in scope, and we may be required to assume more risk in the future. If we are subject to claims or suffer a loss or damage in excess of our insurance coverage, we will be required to bear any loss in excess of our insurance limits. If we are subject to claims or suffer a loss or damage that is outside of our insurance coverage, we may incur significant uninsured costs associated with loss or damage that could have an adverse effect on our operations and financial position. Furthermore, any claims made on our insurance policies may impact our ability to obtain or maintain insurance coverage at reasonable costs or at all.
If our facility incurs damage or power is lost for a significant length of time, our business will suffer.
We currently store numerous clinical and stability samples at our facility that could be damaged if our facility incurred physical damage or in the event of an extended power failure. We have backup power systems in addition to backup generators to maintain power to all critical functions, but any loss of these samples could result in significant delays in our drug development process.
In addition, we currently store most of our preclinical and clinical data at our facilities. Duplicate copies of most critical data are secured off-site. Any significant degradation or failure of our computer systems could cause us to inaccurately calculate or lose our data. Loss of data could result in significant delays in our drug development process and any system failure could harm our business and operations.
33
If we fail to retain our existing key personnel or fail to attract and retain additional key personnel, the development of our drug candidates and the expansion of our business will be delayed or stopped.
We are highly dependent upon our senior management and scientific team, the unexpected loss of whose services might impede the achievement of our development and commercial objectives. Competition for key personnel with the experience that we require is intense and is expected to continue to increase. Our inability to attract and retain the required number of skilled and experienced management, operational and scientific personnel, will harm our business because we rely upon these personnel for many critical functions of our business.
Our stock price has been, and is likely to continue to be, highly volatile, which could result in the value of an investment to decline significantly.
The market prices for securities of biotechnology companies in general have been highly volatile and may continue to be highly volatile in the future. Moreover, our stock price has fluctuated frequently, and these fluctuations are often not related to our financial results. For the twelve months ended December 31, 2011, the 52-week range of the market price of our stock was from $2.29 to $5.34 per share. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
| Ÿ | announcements of technological innovations or new products by us or our competitors; |
| Ÿ | developments or disputes concerning patents or proprietary rights; |
| Ÿ | additional dilution through sales of our common stock or other derivative securities; |
| Ÿ | status of new or existing licensing or collaborative agreements and government contracts; |
| Ÿ | announcements relating to the status of our programs; |
| Ÿ | we or our partners achieving or failing to achieve development milestones; |
| Ÿ | publicity regarding actual or potential medical results relating to products under development by us or our competitors; |
| Ÿ | publicity regarding certain public health concerns for which we are or may be developing treatments; |
| Ÿ | regulatory developments in both the United States and foreign countries; |
| Ÿ | public concern as to the safety of pharmaceutical products; |
| Ÿ | actual or anticipated fluctuations in our operating results; |
| Ÿ | changes in financial estimate or recommendations by securities analysts; |
| Ÿ | changes in the structure of healthcare payment systems, including developments in price control legislation; |
| Ÿ | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| Ÿ | additions or departures of key personnel or members of our board of directors; |
| Ÿ | purchases or sales of substantial amounts of our stock by existing stockholders, including officers or directors; |
| Ÿ | economic and other external factors or other disasters or crises; and |
| Ÿ | period-to-period fluctuations in our financial results. |
34
Our existing principal stockholders hold a substantial amount of our common stock and may be able to influence significant corporate decisions, which may conflict with the interest of other stockholders.
As of January 31, 2012, our current 5% and greater stockholders and their affiliates beneficially owned approximately 30% of our outstanding common stock. These stockholders, if they act together, may be able to influence the outcome of matters requiring approval of the stockholders, including the election of our directors and other corporate actions such as:
| Ÿ | a merger or corporate combination with or into another company; |
| Ÿ | a sale of substantially all of our assets; and |
| Ÿ | amendments to our certificate of incorporation. |
Future sales and issuances of securities may dilute the ownership interests of our current stockholders and cause our stock price to decline.
Future sales of our common stock by current stockholders into the public market could cause the market price of our stock to fall. As of January 31, 2012, there were 46,037,816 shares of our common stock outstanding. We may from time to time issue securities in relation to a license arrangement, collaboration, merger or acquisition.
In addition, on June 28, 2011, we filed with the SEC a shelf registration statement on Form S-3. This shelf registration statement has been declared effective and allows us to sell up to $70 million of securities, including common stock, preferred stock, depository shares, stock purchase contracts and warrants, from time to time at prices and on terms to be determined at the time of sale.
As of January 31, 2012, there were 7,923,891 stock options and restricted stock units outstanding and 2,073,553 shares available for issuance under our Amended and Restated 2010 Equity Compensation Plan and equity compensation grants outside such plan. The shares underlying existing stock options and restricted stock units and possible future stock options, stock appreciation rights and stock awards have been registered pursuant to registration statements on Form S-8.
If some or all of such shares are sold or otherwise issued into the public market over a short period of time, our current stockholders’ ownership interests may be diluted and the value of all publicly traded shares is likely to decline, as the market may not be able to absorb those shares at then-current market prices. Additionally, such sales and issuances may make it more difficult for us to sell equity securities or equity-related securities in the future at a time and price that our management deems acceptable, or at all.
If, because of our use of hazardous materials, we violate any environmental controls or regulations that apply to such materials, we may incur substantial costs and expenses in our remediation efforts.
Our research and development involves the controlled use of hazardous materials, chemicals and various radioactive compounds. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and some waste products. Accidental contamination or injury from these materials could occur. In the event of an accident, we could be liable for any damages that result and any liabilities could exceed our resources. Compliance with environmental laws and regulations could require us to incur substantial unexpected costs, which would materially and adversely affect our results of operations.
Information Regarding Forward-Looking Statements
This filing contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created in Section 21E. All statements other than statements of historical facts contained in this filing, are forward-looking statements. These forward-looking statements can generally be identified by the use of words such as “may,” “will,” “intends,” “plans,” “believes,” “anticipates,” “expects,” “estimates,” “predicts,” “potential,” the negative of these words or similar expressions. Statements that describe our future plans, strategies, intentions, expectations, objectives, goals or prospects are also forward-looking statements. Discussions containing these forward-looking statements are
35
principally contained in “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, as well as any amendments we make to those sections in filings with the SEC. These forward-looking statements include, but are not limited to, statements about:
| Ÿ | the initiation, timing, progress and results of our preclinical testing, clinical trials, and other research and development efforts; |
| Ÿ | the potential funding from our contract with BARDA/HHS for the development of peramivir; |
| Ÿ | the potential for a stockpiling order or profit from any order for peramivir; |
| Ÿ | the potential use of peramivir as a treatment for H1N1 flu (or other strains of flu); |
| Ÿ | the further preclinical or clinical development and commercialization of our drug candidates, including peramivir, forodesine and other PNP inhibitor and hepatitis C development programs; |
| Ÿ | the implementation of our business model, strategic plans for our business, drug candidates and technology; |
| Ÿ | our ability to establish and maintain collaborations; |
| Ÿ | plans, programs, progress and potential success of our collaborations, including Mundipharma for forodesine and Shionogi and Green Cross for peramivir; |
| Ÿ | Royalty Sub’s ability to service its payment obligations in respect of the PhaRMA Notes, and our ability to benefit from our equity interest in Royalty Sub; |
| Ÿ | the foreign currency hedge agreement entered into by us in connection with the issuance by Royalty Sub of the PhaRMA Notes; |
| Ÿ | the scope of protection we are able to establish and maintain for intellectual property rights covering our drug candidates and technology; |
| Ÿ | our ability to operate our business without infringing the intellectual property rights of others; |
| Ÿ | estimates of our expenses, revenues, capital requirements and our needs for additional financing; |
| Ÿ | the timing or likelihood of regulatory filings and approvals; |
| Ÿ | our financial performance; and |
| Ÿ | competitive companies, technologies and our industry. |
These statements relate to future events or to our future financial performance and involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those listed under “Risk Factors.” Any forward-looking statement reflects our current views with respect to future events and is subject to these and other risks, uncertainties and assumptions relating to our operations, results of operations, industry and future growth. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
We lease offices in both Durham, North Carolina and Birmingham, Alabama. Our headquarters, including our clinical and regulatory operations, are based in Durham, while our principal research facilities are located in Birmingham. We lease approximately 17,256 square feet in Durham through December 31, 2014 and
36
approximately 50,125 square feet in Birmingham through June 30, 2015. We believe that our facilities are adequate for our current operations.
| ITEM 3. | LEGAL PROCEEDINGS |
None.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
37
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock trades on the NASDAQ Global Select Market under the symbol BCRX. The following table sets forth the low and high sales prices of our common stock as reported by the NASDAQ Global Select Market for each quarter in 2011 and 2010:
| 2011 | 2010 | |||||||||||||||
| Low | High | Low | High | |||||||||||||
| First quarter |
3.36 | 5.34 | 6.21 | 8.34 | ||||||||||||
| Second quarter |
3.21 | 4.02 | 5.79 | 8.37 | ||||||||||||
| Third quarter |
2.31 | 3.93 | 4.43 | 6.24 | ||||||||||||
| Fourth quarter |
2.29 | 3.28 | 4.65 | 5.86 | ||||||||||||
The last sale price of the common stock on January 31, 2012 as reported by the NASDAQ Global Select Market was $3.49 per share.
Holders
As of January 31, 2012, there were approximately 227 holders of record of our common stock.
Dividends
We have never paid cash dividends and do not anticipate paying cash dividends in the foreseeable future.
38
Stock Performance Graph
This performance graph is not “soliciting material,” is not deemed filed with the SEC and is not to be incorporated by reference in any filing by us under the Securities Act or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. The stock price performance shown on the graph is not necessarily indicative of future price performance.
PERFORMANCE GRAPH FOR BIOCRYST
Indexed Comparison Since 2006
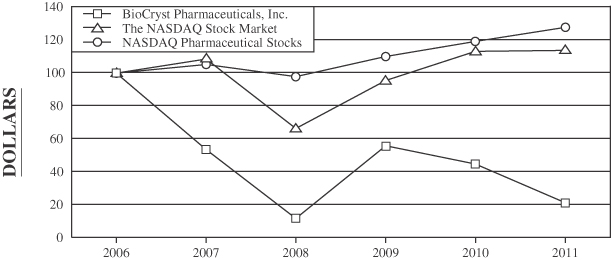
| |
Beginning 12/31/06 |
Investment at 12/31/07 |
Investment at 12/31/08 |
Investment at 12/31/09 |
Investment at 12/31/10 |
Investment at 12/31/11 |
||||||||||||||||||
| BioCryst Pharmaceuticals, Inc. |
$ | 100.00 | $ | 53.46 | $ | 11.85 | $ | 55.88 | $ | 44.72 | $ | 21.37 | ||||||||||||
| The NASDAQ Stock Market |
100.00 | 108.47 | 66.35 | 95.38 | 113.19 | 113.81 | ||||||||||||||||||
| NASDAQ Pharmaceutical Stocks |
100.00 | 105.17 | 97.85 | 109.95 | 119.19 | 127.71 | ||||||||||||||||||
The above graph measures the change in a $100 investment in our common stock based on its closing price of $11.56 on December 31, 2006 and its year-end closing price thereafter. Our relative performance is then compared with the CRSP Total Return Indexes for the NASDAQ Stock Market (U.S.) and NASDAQ Pharmaceutical Stocks.
Recent Sales of Unregistered Securities: None.
39
Issuer Purchases of Equity Securities
There were no repurchases of our common stock or shares surrendered to satisfy tax obligations during the fourth quarter of 2011.
| ITEM 6. | SELECTED FINANCIAL DATA |
The selected Statement of Operations Data and Balance Sheet data with respect to the years ended December 31, 2011, 2010, 2009, 2008, and 2007 set forth below are derived from our consolidated financial statements. The selected financial data set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operation contained in Item 7 below and our consolidated financial statements and the notes thereto appended to this annual report.
| Years Ended December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Total revenues |
$ | 19,643 | $ | 62,381 | $ | 74,590 | $ | 56,561 | $ | 71,238 | ||||||||||
| Cost of product sold |
— | 86 | 4,544 | — | — | |||||||||||||||
| Research and development expenses |
56,898 | 83,900 | 73,661 | 74,019 | 95,303 | |||||||||||||||
| General and administrative expenses |
12,332 | 12,752 | 10,122 | 9,707 | 8,215 | |||||||||||||||
| Loss from operations |
(49,587 | ) | (34,357 | ) | (13,737 | ) | (27,164 | ) | (32,280 | ) | ||||||||||
| Net loss |
(56,948 | ) | (33,853 | ) | (13,451 | ) | (24,732 | ) | (29,055 | ) | ||||||||||
| Amounts per common share: |
||||||||||||||||||||
| Basic and diluted net loss per share |
$ | (1.26 | ) | $ | (0.76 | ) | $ | (0.35 | ) | $ | (0.65 | ) | $ | (0.89 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average shares outstanding |
45,144 | 44,564 | 38,926 | 38,062 | 32,771 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and investments |
$ | 57,725 | $ | 66,341 | $ | 94,259 | $ | 63,314 | $ | 85,009 | ||||||||||
| Receivables |
5,831 | 30,227 | 33,722 | 11,982 | 39,128 | |||||||||||||||
| Inventory |
263 | 898 | 6,281 | — | — | |||||||||||||||
| Total assets |
82,208 | 109,447 | 142,190 | 84,692 | 142,717 | |||||||||||||||
| Long-term deferred revenue |
7,103 | 15,944 | 18,441 | 20,937 | 49,694 | |||||||||||||||
| Non-recourse notes payable |
30,000 | — | — | — | — | |||||||||||||||
| Accumulated deficit |
(353,520 | ) | (296,572 | ) | (262,719 | ) | (249,268 | ) | (224,536 | ) | ||||||||||
| Total stockholders’ equity |
14,806 | 65,503 | 86,266 | 46,426 | 64,905 | |||||||||||||||
40
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
This Annual Report on Form 10-K contains certain statements of a forward-looking nature relating to future events or the future financial performance of BioCryst. Such statements are only predictions and the actual events or results may differ materially from the results discussed in the forward-looking statements. Factors that could cause or contribute to such differences include those discussed below and elsewhere in this report, as well as those discussed in other filings made by BioCryst with the Securities and Exchange Commission.
The following Management’s Discussion and Analysis (“MD&A”) is intended to help the reader understand our results of operations and financial condition. MD&A is provided as a supplement to, and should be read in conjunction with, our audited Financial Statements and the accompanying notes to the financial statements and other disclosures included in this Annual Report on Form 10-K (including the disclosures under “Item 1A. Risk Factors”).
Cautionary Statement
The discussion herein contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created in Section 21E. Forward-looking statements regarding our financial condition and our results of operations that are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted within the United States, as well as projections for the future. The preparation of these financial statements requires our management to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. We evaluate our estimates on an ongoing basis. Our estimates are based on historical experience and on various other assumptions that are believed to be reasonable under the circumstances. The results of our estimates form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources.
We operate in a highly competitive environment that involves a number of risks, some of which are beyond our control. We are subject to risks common to biotechnology and biopharmaceutical companies, including risks inherent in our drug discovery, drug development and commercialization efforts, clinical trials, uncertainty of regulatory actions and marketing approvals, reliance on collaborative partners, enforcement of patent and proprietary rights, the need for future capital, competition associated with products, potential competition associated with our drug candidates and retention of key employees. In order for any of our drug candidates to be commercialized, it will be necessary for us, or our collaborative partners, to conduct clinical trials, demonstrate efficacy and safety of the drug candidate to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, and obtain market acceptance and adequate reimbursement from government and private insurers. We cannot provide assurance that we will generate significant revenues or achieve and sustain profitability in the future. In addition, we can provide no assurance that we will have sufficient funding to meet our future capital requirements. Statements contained in Management’s Discussion and Analysis of Financial Condition and Results of Operations and elsewhere in this report which are not historical facts are, or may constitute, forward-looking statements. Forward-looking statements involve known and unknown risks that could cause our actual results to differ materially from expected results. The most significant known risks are discussed in the section entitled “Risk Factors.” Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. We caution you not to place undue reliance on any forward-looking statements.
Our revenues are difficult to predict and depend on numerous factors, including the prevalence and severity of influenza in regions for which peramivir has received regulatory approval, as well as in those geographies that impact enrollment in our ongoing peramivir clinical trial. Furthermore, revenues related to our collaborative development activities are dependent upon the progress toward and the achievement of developmental milestones by us or our collaborative partners.
41
Our operating expenses are also difficult to predict and depend on several factors. Research and development expenses, drug manufacturing, and clinical research activities, depend on the ongoing requirements of our development programs, availability of capital and direction from regulatory agencies, which are difficult to predict. Management may be able to control the timing and level of research and development and general and administrative expenses, but many of these expenditures will occur irrespective of our actions due to contractually committed activities and/or payments.
As a result of these factors, we believe that period to period comparisons are not necessarily meaningful and you should not rely on them as an indication of future performance. Due to all of the foregoing factors, it is possible that our operating results will be below the expectations of market analysts and investors. In such event, the prevailing market price of our common stock could be materially adversely affected.
Overview
We are a biotechnology company that designs, optimizes and develops novel drugs that block key enzymes involved in the pathogenesis of diseases. We focus on therapeutic areas with unmet medical needs that are of interest to us and aligned with our capabilities and expertise. Our areas of interest and related development of drug candidates are determined by the scientific discoveries and the potential advantages that our experienced drug discovery group identifies, as well as by the associated potential commercial opportunity of those discoveries. We integrate the disciplines of biology, crystallography, medicinal chemistry and computer modeling to discover and develop small molecule pharmaceuticals through the process known as structure-guided drug design. Our strategy is to create a sustainable portfolio of commercial products and drug candidates whereby we out-license rights to drug candidates in geographies or therapeutic areas where we do not intend to and/or do not have the ability commercialize them.
Critical Accounting Policies and Estimates
The accompanying discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements and the related disclosures, which have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosure of contingent assets and liabilities. We evaluate our estimates, judgments and the policies underlying these estimates on a periodic basis, as situations change and regularly discuss financial events, policies, and issues with members of our audit committee and our independent registered public accounting firm. We routinely evaluate our estimates and policies regarding revenue recognition, administration, inventory and manufacturing, taxes, stock-based compensation, research and development, consulting and other expenses and any associated liabilities.
Recent Corporate Highlights
Peramivir
On February 24, 2011, BARDA/HHS awarded us a $55.0 million contract modification intended to fund completion of the Phase 3 development of i.v. peramivir for the treatment of patients hospitalized with influenza. This contract modification brings the total award from BARDA/HHS to $234.8 million and extends the contract term by 24 months through December 31, 2013. The contract, as it currently stands, provides for funding through completion of Phase 3 and to support the filing of an NDA to seek regulatory approval for i.v. peramivir in the U.S. Through December 31, 2011, approximately $174.7 million has been recognized as revenue under this contract.
This contract modification supports implementation of our proposed changes to our 301 clinical trial. Significant changes to the 301 clinical trial are as follows:
| (1) | Modifying the primary efficacy analysis population of the study to focus on a subset of approximately 160 patients not treated with neuraminidase inhibitors as SOC, in order to provide the greatest opportunity to demonstrate a statistically significant peramivir treatment effect; |
42
| (2) | Increasing the total study target enrollment to approximately 600 subjects from the prior target of 445 subjects; and |
| (3) | Adding more clinical site locations in geographical regions where neuraminidase inhibitors are not widely used, including sites in India and possibly China. |
These changes are expected to increase the amount of time required to complete enrollment in this ongoing study. The actual time to reach completion of enrollment will depend on the prevalence and severity of influenza, as well as the ability of the more than 265 investigator sites to successfully enroll patients.
In addition, we have agreed with the FDA and BARDA/HHS to conduct a planned interim analysis in our 301 clinical trial, which includes a futility assessment. This analysis is scheduled to be conducted at the earlier of the conclusion of the 2012 Southern Hemisphere flu season or reaching 70% of the current enrollment goal of 160 patients for the primary efficacy analysis population. If the interim analysis shows an efficacy trend in favor of peramivir, it is expected the clinical trial would continue toward either the current enrollment target or a higher target, depending on the trend. If, however, the new enrollment target to reach statistical significance is predicted to exceed 320 patients, we would expect to terminate the clinical trial and evaluate the data in hand.
On March 9, 2011, we completed a $30.0 million non-recourse financing transaction designed to monetize certain future royalty and milestone payments under our license agreement with Shionogi, pursuant to which Shionogi licensed from us the rights to market peramivir (RAPIACTA®) in Japan and, if approved for commercial sale, Taiwan. We formed Royalty Sub, a newly created wholly-owned subsidiary, which completed a private placement to institutional investors of $30.0 million in aggregate principal amount of PhaRMA Senior Secured 14.0% Notes. This private placement was exempt from registration under the Securities Act of 1933. The PhaRMA Notes, which are obligations of Royalty Sub, are secured by (i) Royalty Sub’s rights to receive royalty payments from Shionogi in respect of commercial sales of RAPIACTA in Japan and, if approved for commercial sale, Taiwan, as well as future milestone payments payable by Shionogi under the Shionogi Agreement and all of Royalty Sub’s other assets, and (ii) a pledge by us of our equity interest in Royalty Sub. Principal and interest on the PhaRMA Notes issued by Royalty Sub are payable from, and are secured by, the rights to royalty and milestone payments under the Shionogi Agreement. Principal on the PhaRMA Notes is required to be paid in full by the final legal maturity date of December 1, 2020, unless the PhaRMA Notes are repaid, redeemed or repurchased earlier. The PhaRMA Notes are redeemable by Royalty Sub beginning March 9, 2012 and bear interest at the rate of 14% per annum, payable annually in arrears on September 1st of each year, beginning on September 1, 2011. Royalty Sub’s obligations to pay principal and interest on the PhaRMA Notes are obligations solely of Royalty Sub and are without recourse to any other person, including us, except to the extent of our pledge of our equity interests in Royalty Sub in support of the PhaRMA Notes.
We received net proceeds of approximately $22.7 million after deducting transaction costs of $4.3 million and the establishment of a $3.0 million interest reserve account available to help cover future annual interest shortfalls. As of December 31, 2011, approximately $1.7 million remains in the interest reserve account.
In association with the PhaRMA Notes, we entered into a Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. Under this agreement, we have the right to purchase dollars and sell yen at a rate of 100 yen per dollar for which we may be required to pay a premium in each year from 2014 through 2020, provided the Currency Hedge Agreement remains in effect. A payment of $2.0 million will be required if, on May 18 of the relevant year, the U.S. dollar is worth 100 yen or less as determined in accordance with the Currency Hedge Agreement. In conjunction with establishing the Currency Hedge Agreement, we will be required to post collateral to the counterparty, which may cause us to experience additional quarterly volatility in our financial results. We will not be required at any time to post collateral exceeding the maximum premium payments remaining payable under the Currency Hedge Agreement. Subject to certain obligations we have in connection with the PhaRMA Notes, we have the right to terminate the Currency Hedge Agreement with respect to the 2016 through 2020 period by giving notice to the counterparty prior to May 18, 2014 and paying a $2.0 million termination fee. In advance of the May 18, 2014 termination date, we have a limitation on the maximum hedge collateral of approximately $5.9 million. The Currency Hedge Agreement does not qualify for hedge accounting treatment and therefore mark to market adjustments will be recognized in our Consolidated Statements of Operations. Cumulative mark to market adjustments for the year
43
ended December 31, 2011 resulted in a $4.0 million hedge loss and we posted $3.5 million in collateral based on defined thresholds in 2011. Our operating results will continue to be impacted by mark-to market adjustments while the Currency Hedge Agreement remains in effect.
BCX4208
On January 8, 2012, we reported positive long-term results from the extension phase of our randomized Phase 2b clinical trial of BCX4208 added to allopurinol in patients with gout who had failed to reach the serum uric acid (sUA) therapeutic goal of <6 mg/dL on allopurinol alone. The results of this 24-week, blinded safety extension confirmed that BCX4208 was generally safe and well-tolerated, and sustained sUA control over time. This longer-term safety profile of BCX4208 is consistent with the 12-week primary analysis results originally reported in October 2011. BCX4208 added to allopurinol was generally safe and well-tolerated at all doses studied, and responses to vaccines indicated healthy immune function. The types and rates of adverse events through 24 weeks, including infections, were similar between the groups treated with BCX4208 and placebo. No opportunistic or unusual infections were observed. The safety and efficacy data of BCX4208 at 12 weeks and through 24 weeks is sufficient to undertake end of Phase 2 interactions with regulatory authorities in the United States, European Union, and Japan to obtain guidance on the regulatory requirements to obtain approval to commercialize BCX4208 in those regions. Additionally, both sets of data allow us to continue in out-license discussions with potential partners for the continued Phase 3 development of the drug candidate and eventual commercialization on a worldwide basis. We expect these regulatory and out-license discussions to occur in 2012; however, we cannot predict the ultimate outcome of these discussions or the specific timing thereof.
Forodesine
On November 11, 2011, we entered into the Amended and Restated License and Development Agreement with Mundipharma, amending and restating the February 1, 2006 exclusive, royalty bearing Development and License Agreement for the development and commercialization of forodesine for use in the field of oncology. Under the terms of the Amended and Restated Agreement, Mundipharma obtained worldwide rights to forodesine, so they now control the worldwide development and commercialization of forodesine and assume all future development and commercialization costs. Mundipharma also purchased from us certain drug substance for forodesine at a cost of approximately $0.9 million in December 2011. Additionally, on November 17, 2011, we further amended our agreements with AECOM/IRL whereby AECOM/IRL agreed to accept a reduction of one-half in the percentage of Net Proceeds (as defined) received by us under our Amended and Restated Agreement with Mundipharma.
The Amended and Restated Agreement is a multiple element arrangement for accounting purposes in which we are required to deliver to Mundipharma both the worldwide rights to forodesine and the transfer of product data and know-how to permit Mundipharma to develop and commercialize forodesine (the “Knowledge Transfer”). The world-wide license rights were granted to Mundipharma upon execution of Amended and Restated Agreement and the Knowledge Transfer began in the fourth quarter of 2011. We expect to complete the Knowledge Transfer by June 30, 2012. We have accounted for these elements as a combined unit of accounting as neither one has stand-alone value to Mundipharma. Without completion of the Knowledge Transfer, Mundipharma will not be able to develop and commercialize forodesine in the U.S. Amortization of deferred revenue and expense items associated with the initial agreement with Mundipharma ceased in November 2011 when we were no longer responsible for the development of forodesine. The unamortized deferred revenue and deferred expense at December 31, 2011 was $7.8 million and $1.9 million, respectively, and will be recognized in our Statement of Operations upon completion of the Knowledge Transfer.
Results of Operations
Year Ended December 31, 2011 Compared to 2010
Total 2011 revenues decreased to $19.6 million compared to 2010 revenues of $62.4 million. Revenues in 2011 consisted primarily of reimbursement of collaboration expenses from BARDA/HHS with $17.1 million related to the continued development of i.v. peramivir and approximately $2.5 million associated with
44
collaborative revenue amortization from other corporate partnerships. Revenues in 2010 consisted primarily of reimbursement of collaboration expenses, including $42.5 million from BARDA/HHS for the continued development of i.v. peramivir and the sale of $8.3 million of peramivir active pharmaceutical ingredient (API) and other starting materials to Shionogi and Green Cross, as well as a $7.0 million milestone payment from Shionogi related to the marketing and manufacturing approval of RAPIACTA in Japan during the first quarter 2010.
Revenue associated with reimbursement from BARDA/HHS for the continued development of i.v. peramivir decreased $25.4 million in 2011 as compared to 2010. The decrease in revenue associated with our peramivir development program resulted from the completion of two clinical trials in 2010 and the realignment of ongoing clinical trials. In addition, the decrease was also partially related to an estimate revision of prior period expenses for a peramivir clinical trial associated with services performed by a contract research organization (“CRO”), and its subsequent revision of service costs in 2011 related to a final cost reconciliation. At the end of 2010, we estimated expenses related to this clinical trial and the associated revenue we expected to receive from BARDA/HHS from estimates provided to us by this CRO. Revisions to the estimated costs resulted in a $3.0 million reduction of peramivir expenses and a $3.6 million reduction to collaboration revenue during the first quarter of 2011, resulting in a net impact of $0.6 million to net loss.
Research and development (R&D) expenses decreased to $56.9 million in 2011 compared to $83.9 million for the prior year. The $27.0 million decrease was driven by lower development costs associated with our peramivir development program (as discussed above) and lower costs associated with our forodesine clinical programs. In connection with the Amended and Restated Agreement with Mundipharma, we ceased incurring all forodesine development costs in November 2011 and we received $0.9 million for previously expensed compound development costs. The decrease in aforementioned costs was partially offset by higher development costs associated with the BCX4208 program for the treatment of gout during 2011. Additionally, peramivir costs for 2010 included $8.2 million of manufacturing costs associated with peramivir API production for Shionogi and Green Cross.
The following table summarizes our R&D expenses for the years ended December 31, 2011, 2010 and 2009.
| 2011 | 2010 | 2009 | ||||||||||
| Research and development expenses by program: |
||||||||||||
| BCX4208 |
$ | 20,185 | $ | 13,174 | $ | 4,087 | ||||||
| Peramivir |
17,361 | 49,740 | 45,127 | |||||||||
| Forodesine |
759 | 7,277 | 14,758 | |||||||||
| Other research, preclinical and development costs |
18,593 | 13,709 | 9,689 | |||||||||
| Total research and development expenses |
$ | 56,898 | $ | 83,900 | $ | 73,661 | ||||||
Research and development expenses include all direct and indirect expenses and are allocated to specific programs at the point of development of a lead drug candidate. Direct expenses are charged directly to the program to which they relate and indirect expenses are allocated based upon internal direct labor hours dedicated to each respective program. Direct expenses consist of compensation for R&D personnel and costs of outside parties to conduct laboratory studies, develop manufacturing processes, manufacture the drug candidates, conduct and manage clinical trials, patent-related costs, as well as other costs related to our clinical and preclinical studies. Indirect R&D expenses consist of lab supplies and services, facility expenses, depreciation of development equipment and other overhead of our research and development efforts. R&D expenses vary according to the number of programs in clinical development and the stage of development of our clinical programs. Later stage clinical programs tend to cost more than earlier stage programs due to the longer length of time of the clinical trials and the higher number of patients enrolled in these clinical trials.
General and administrative (G&A) expenses decreased to $12.3 million for 2011 from $12.8 million in the prior year. The small change reflects timing of expenses between the years associated with the transition of our headquarters to Durham, North Carolina and cost containment procedures instituted in 2011.
45
Additionally, we incurred interest expense and losses on our foreign currency derivative during 2011, associated with our $30 million non-recourse debt financing transaction completed in March 2011 to monetize certain future royalty and milestone payments associated with a license agreement with Shionogi – see “Note 3 – Royalty Monetization” in our Notes to the Consolidated Financial Statements. We incurred $3.8 million in interest expense related to our PhaRMA Notes and recognized a $4.0 million mark to market loss related to our Currency Hedge Agreement. We entered into the foreign Currency Hedge Agreement to hedge changes in the value of the Japanese yen relative to the U.S. dollar. The currency hedge does not qualify for hedge accounting treatment and therefore mark to market adjustments will be recognized in our Consolidated Statements of Operations. Although we cannot predict the future yen/dollar exchange rate, we are aware that the applicable foreign currency rates have moved to increase the hedge loss in early 2012, and it is likely that additional cash collateral will be required in the first quarter of 2012. We are unable to predict future changes in the yen/dollar exchange rate or increases/decreases in our hedge loss associated with the Currency Hedge Agreement.
Year Ended December 31, 2010 Compared to 2009
Total revenues of $62.4 million consisted primarily of reimbursement of collaboration expenses, including $42.5 million from BARDA/HHS for the continued development of i.v. peramivir and the sale of $8.3 million of peramivir API and other starting materials to Shionogi and Green Cross, as well as a $7.0 million milestone payment from Shionogi related to the marketing and manufacturing approval of RAPIACTA in Japan during 2010. Full year 2009 total revenue of $74.6 million was significantly impacted by a $22.5 million product sale of i.v. peramivir for the treatment of critically ill influenza patients under an EUA to BARDA/HHS, and includes $37.9 million of peramivir development expense reimbursement from BARDA/HHS. In addition, we recognized less revenue from our collaboration with Mundipharma during 2010 compared to 2009.
Cost of products sold for the year ended December 31, 2010 was negligible due to the lower amount of product sale as compared to 2009. Cost of products sold for the year ended December 31, 2009 was approximately $4.5 million. Included in cost of products sold for the year ended December 31, 2009 is a $4.0 million provision for peramivir finished goods inventory. We expense costs related to the production of inventories as research and development expenses in the period incurred until such time it is believed that future economic benefit is expected to be recognized, which generally is reliant upon receipt of regulatory approval. Upon regulatory approval, we capitalize subsequent costs related to the production of inventories. We determined that the FDA’s granting of the EUA for peramivir in October 2009 was objective and persuasive evidence that supported capitalization of peramivir inventories manufactured after the issuance of the EUA. As a result, we recorded manufacturing costs of $4.0 million for peramivir finished goods inventory. However, we evaluated whether the costs capitalized as inventory would be recoverable in a future period. Given the lack of objective, reliable evidence to support future demand for peramivir, we concluded that there was no certainty that future sales would materialize and revenues would exceed the costs incurred. Therefore, the capitalized inventory was fully reserved.
Research and development expenses increased to $83.9 million for 2010 compared to $73.7 million for 2009. The $10.2 million increase was primarily due to higher development costs associated with the peramivir and the BCX4208 programs as well as our pre-clinical programs. These increases in R&D expenses were partially offset by a decrease in development costs associated with the forodesine program.
General and administrative expenses increased to $12.8 million for 2010 from $10.1 million for 2009. This increase was primarily due to higher consulting fees related to supply chain and other commercial activities, as well as legal fees, operating and personnel related costs.
Interest income for 2010 was $0.5 million as compared to $0.3 million for 2009, due to higher average cash and securities on hand during 2010 as compared to 2009. The increase in cash and securities primarily resulted from the sale of 5.0 million shares of common stock in November 2009 resulting in net proceeds of $47.5 million.
46
Liquidity and Capital Resources
Cash expenditures have exceeded revenues since our inception and we expect our 2012 operating expense to exceed our 2012 revenue. Our operations have principally been funded through public offerings and private placements of equity securities; cash from collaborative and other research and development agreements, including government contracts; and to a lesser extent, the PhaRMA Notes financing. On February 24, 2011, we announced that BARDA/HHS had awarded us a $55.0 million contract modification intended to fund completion of the Phase 3 development of i.v. peramivir, bringing the total award from BARDA/HHS to $234.8 million and extending the contract term by 24 months through December 2013. On March 9, 2011, we completed a $30.0 million PhaRMA Notes financing transaction designed to monetize certain future royalty and milestone payments under our license agreement with Shionogi. We received net proceeds from this transaction of approximately $22.7 million. Other sources of funding have included the following:
| Ÿ | other collaborative and other research and development agreements; |
| Ÿ | government grants; |
| Ÿ | equipment lease financing; |
| Ÿ | facility leases; |
| Ÿ | research grants; and |
| Ÿ | interest income. |
As of December 31, 2011, we had net working capital of approximately $26.6 million, a decrease of approximately $32.8 million from approximately $59.4 million at December 31, 2010. The decrease in working capital was principally due to the funding of our normal operating expenses associated with the development of our drug candidates. Our normal operating expenses were offset in 2011 by approximately $19.2 million of proceeds from the PhaRMA Notes (net proceeds less cash collateral posted against foreign currency losses) and approximately $1.0 million of net proceeds derived from the sale of approximately 437,000 shares of common stock through our At-the-Market financing facility under our Form S-3 shelf registration. Our principal sources of liquidity at December 31, 2011 were approximately $16.4 million in cash and cash equivalents; approximately $40.7 million in investments considered available-for-sale; and approximately $5.8 million in BARDA/HHS receivables.
We have attempted to contain costs and reduce cash flow requirements by closely managing our third party costs and headcount, renting scientific equipment and facilities, contracting with other parties to conduct certain research and development and using consultants. We expect to incur additional expenses, potentially resulting in significant losses, as we continue to pursue our research and development activities in general, and specifically related to our clinical trial activity. We also expect to incur substantial expenses related to the filing, prosecution, maintenance, defense and enforcement of patent and other intellectual property claims and additional regulatory costs as our clinical products advance through later stages of development. The objective of our investment policy is to ensure the safety and preservation of invested funds, as well as maintaining liquidity sufficient to meet cash flow requirements. We place our excess cash with high credit quality financial institutions, commercial companies, and government agencies in order to limit the amount of our credit exposure. We have not realized any significant losses on our investments.
During 2011, 2010, and 2009, we incurred capital costs of approximately $50,000, $0.3 million, and $0.6 million, respectively. At December 31, 2011, we had long-term operating lease obligations, which provide for aggregate minimum payments of approximately $0.9 million in 2012, $1.0 million in 2013, $1.0 million in 2014 and $0.3 million in 2015. These obligations include the future rental of our operating facilities.
We plan to finance our needs principally from the following:
| Ÿ | payments under our contract with BARDA/HHS; |
47
| Ÿ | our existing capital resources and interest earned on that capital; |
| Ÿ | payments under collaborative and licensing agreements with corporate partners; and |
| Ÿ | lease or loan financing and future public or private equity financing. |
As our clinical programs continue to progress and patient enrollment increases, our costs will increase. Our current and planned clinical trials plus the related development, manufacturing, regulatory approval process requirements and additional personnel resources and testing required for the continuing development of our drug candidates will consume significant capital resources and will increase our expenses. Our expenses, revenues and burn rate could vary significantly depending on many factors, including our ability to raise additional capital, the development progress of our collaborative agreements for our drug candidates, the amount and timing of funding we receive from BARDA/HHS for peramivir, the amount of funding or assistance, if any, we receive from other governmental agencies or other new partnerships with third parties for the development of our drug candidates, the progress and results of our current and proposed clinical trials for our most advanced drug candidates, the progress made in the manufacturing of our lead drug candidates and the progression of our other programs.
With the funds available at December 31, 2011 and future amounts that are expected to be received from BARDA/HHS, and our other financing sources, we believe these resources will be sufficient to fund our operations through 2012. However, this is a forward-looking statement, and there may be changes that would consume available resources significantly before such time.
Our long-term capital requirements and the adequacy of our available funds will depend upon many factors, including:
| Ÿ | our ability to perform under the contract with BARDA/HHS and receive reimbursement; |
| Ÿ | the progress and magnitude of our research, drug discovery and development programs; |
| Ÿ | changes in existing collaborative relationships or government contracts; |
| Ÿ | our ability to establish additional collaborative relationships with academic institutions, biotechnology or pharmaceutical companies and governmental agencies or other third parties; |
| Ÿ | the extent to which our partners, including governmental agencies will share in the costs associated with the development of our programs or run the development programs themselves; |
| Ÿ | our ability to negotiate favorable development and marketing strategic alliances for certain drug candidates; or a decision to build or expand internal development and commercial capabilities; |
| Ÿ | successful commercialization of marketed products by either us or a partner; |
| Ÿ | the scope and results of preclinical studies and clinical trials to identify and evaluate drug candidates; |
| Ÿ | our ability to engage sites and enroll subjects in our clinical trials; |
| Ÿ | the scope of manufacturing of our drug candidates to support our preclinical research and clinical trials; |
| Ÿ | increases in personnel and related costs to support the development of our drug candidates; |
| Ÿ | the scope of manufacturing of our drug substance and drug products required for future NDA filings; |
| Ÿ | competitive and technological advances; |
| Ÿ | the time and costs involved in obtaining regulatory approvals; and |
| Ÿ | the costs involved in all aspects of intellectual property strategy and protection including the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims. |
We expect that we will be required to raise additional capital to complete the development and commercialization of our current drug candidates and we may seek to raise capital at any time we deem market conditions to be favorable. Additional funding, whether through additional sales of equity or debt securities or collaborative or other arrangements with corporate partners or from other sources, including governmental
48
agencies in general and from the BARDA/HHS contract specifically, may not be available when needed or on terms acceptable to us. The issuance of preferred or common stock or convertible securities, with terms and prices significantly more favorable than those of the currently outstanding common stock, could have the effect of diluting or adversely affecting the holdings or rights of our existing stockholders. In addition, collaborative arrangements may require us to transfer certain material rights to such corporate partners. Insufficient funds may require us to delay, scale-back or eliminate certain of our research and development programs. Our future working capital requirements, including the need for additional working capital, will be largely determined by the advancement of our portfolio of drug candidates as well as rate of reimbursement by BARDA/HHS of our peramivir expenses. More specifically, our working capital requirements will be dependent on the number, magnitude, scope and timing of our development programs; regulatory approval of our drug candidates; obtaining and funding from collaborative partners; the cost, timing and outcome of regulatory reviews, regulatory investigations, and changes in regulatory requirements; the costs of obtaining patent protection for our drug candidates; the timing and terms of business development activities; the rate of technological advances relevant to our operations; the efficiency of manufacturing processes developed on our behalf by third parties; and the level of required administrative support for our daily operations.
Financial Outlook for 2012
Based upon planned strategic and development operations, we expect net operating cash usage to be in the range of $32 to $38 million, and expect our total operating expenses to be in the range of $57 to $69 million. Our operating cash forecast excludes any potential cash inflows from out-licensing or other sources. Our ability to remain within our operating expense and operating cash target ranges are subject to multiple factors, including unanticipated or additional general development and administrative costs and other factors described under the Risk Factors located elsewhere in this report. Furthermore, these ranges are highly dependent on peramivir-related operating expenses, which are reimbursed by BARDA/HHS. Our peramivir expenses are hard to predict and are largely a function of the rate of enrollment in the our ongoing 301 Phase 3 clinical trial, which in turn is dependent on the prevalence and severity of influenza in those geographies where we have enrolling clinical trial sites.
Off-Balance Sheet Arrangements
As of December 31, 2011, we are not involved in any unconsolidated entities or off–balance sheet arrangements.
49
Contractual Obligations
In the table below, we set forth our enforceable and legally binding obligations and future commitments and obligations related to all contracts that we are likely to continue regardless of the fact that they are cancelable as of December 31, 2011. Some of the amounts we include in this table are based on management’s estimates and assumptions about these obligations, including their duration, the possibility of renewal, anticipated actions by third parties, and other factors. Because these estimates and assumptions are necessarily subjective, the obligations we will actually pay in future periods may vary from those reflected in the table.
| Payments Due by Period (In thousands) |
||||||||||||||||||||
| Contractual Obligations |
Total | Less Than 1 Year |
1-3 Years | 3-5 Years | More Than 5 Years |
|||||||||||||||
| Operating lease obligations |
$ | 3,148 | $ | 933 | $ | 1,925 | $ | 290 | $ | — | ||||||||||
| Purchase obligations(1) |
33,795 | 33,795 | — | — | — | |||||||||||||||
| Contingent license obligations |
8,650 | 575 | 1,150 | 1,150 | 5,775 | |||||||||||||||
| Non-recourse notes payable(2) |
67,450 | 4,200 | 8,400 | 8,400 | 46,450 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 113,043 | $ | 39,503 | $ | 11,475 | $ | 9,840 | $ | 52,225 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | Purchase obligations include commitments related to clinical development, manufacturing and research operations and other purchase commitments. |
| (2) | Assumes the PhaRMA Notes will be repaid at maturity and the related interest costs will accrue and be paid annually through maturity. This assumption is based on the unpredictable nature of the royalty payments from Shionogi which are designated for both principal and interest payments on the PhaRMA Notes. |
Under the Currency Hedge Agreement, we have the right to purchase dollars and sell yen at a rate of 100 yen per dollar for which we may be required to pay a premium in each year from 2014 through 2020, provided the Currency Hedge Agreement is still in effect. A payment of $2.0 million will be required if, during the relevant year, the dollar is worth less than 100 yen. We have the right to terminate the Currency Hedge Agreement with respect to 2016 through 2020 by giving notice on May 18, 2014 and a payment of a $2.0 million termination fee. Prior to termination, the maximum amount of hedge collateral we may be required to post is $5.9 million. As of December 31, 2011, we have posted $3.5 million in hedge collateral. Because the posting of additional collateral and payment of annual premiums is contingent on the value of the yen relative to the dollar and other factors, such payments have been excluded from the foregoing table.
In addition to the above, we have committed to make potential future “sublicense” payments to third-parties as part of in-licensing and development programs. Payments under these agreements generally become due and payable only upon achievement of certain developmental, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, such contingencies have not been recorded on our balance sheet.
Critical Accounting Policies
We have established various accounting policies that govern the application of accounting principles generally accepted in the U.S., which were utilized in the preparation of our consolidated financial statements. Certain accounting policies involve significant judgments and assumptions by management that have a material impact on the carrying value of certain assets and liabilities. Management considers such accounting policies to be critical accounting policies. The judgments and assumptions used by management are based on historical experience and other factors, which are believed to be reasonable under the circumstances. Because of the nature of the judgments and assumptions made by management, actual results could differ from these judgments and estimates, which could have a material impact on the carrying values of assets and liabilities and the results of operations.
While our significant accounting policies are more fully described in Note 1 to our financial statements included in this Annual Report on Form 10-K for the year ended December 31, 2011, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our reported
50
financial results and affect the more significant judgments and estimates that we use in the preparation of our financial statements.
Inventory
Our inventories consist of peramivir finished goods and supplies for the manufacture of peramivir, which are valued at the lower of cost or market using the first-in, first-out (i.e., FIFO) method. Cost includes materials, labor, overhead, shipping and handling costs. Our inventories are subject to expiration dating. We regularly evaluate the carrying value of our inventories and provide valuation reserves for any estimated obsolete, short-dated or unmarketable inventories. In addition, we may experience spoilage of our raw materials and supplies. Our determination that a valuation reserve might be required, in addition to the quantification of such reserve, requires us to utilize significant judgment.
Accrued Expenses
We enter into contractual agreements with third-party vendors who provide research and development, manufacturing, and other services in the ordinary course of business. Some of these contracts are subject to milestone-based invoicing and services are completed over an extended period of time. We record liabilities under these contractual commitments when an obligation has been incurred. This accrual process involves reviewing open contracts and purchase orders, communicating with our applicable personnel to identify services that have been performed and estimating the level of service performed and the associated cost when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date based on the facts and circumstances known to us. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued expenses include:
| Ÿ | fees paid to CROs in connection with preclinical and toxicology studies and clinical trials; |
| Ÿ | fees paid to investigative sites in connection with clinical trials; |
| Ÿ | fees paid to contract manufacturers in connection with the production of our raw materials, drug substance and drug products; and |
| Ÿ | professional fees. |
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we will adjust the accrual accordingly. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of these costs, our actual expenses could differ from our estimates.
Revenue Recognition
The Company recognizes revenues from collaborative and other research and development arrangements and product sales. Revenue is realized or realizable and earned when all of the following criteria are met: (i) persuasive evidence of an arrangement exists; (ii) delivery has occurred or services have been rendered; (iii) the seller’s price to the buyer is fixed or determinable; and (iv) collectability is reasonably assured.
51
Collaborative and Other Research and Development Arrangements and Royalties
Revenue from license fees, royalty payments, event payments, and research and development fees are recognized as revenue when the earnings process is complete and we have no further continuing performance obligations or we have completed the performance obligations under the terms of the agreement. Fees received under licensing agreements that are related to future performance are deferred and recognized over an estimated period determined by management based on the terms of the agreement and the products licensed. In the event a license agreement contains multiple deliverables, we evaluate whether the deliverables are separate or combined units of accounting. Revisions to revenue or profit estimates as a result of changes in the estimated revenue period are recognized prospectively.
Under certain of our license agreements, we receive royalty payments based upon our licensees’ net sales of covered products. Generally, under these agreements, we receive royalty reports from our licensees approximately one quarter in arrears, that is, generally in the second month of the quarter after the licensee has sold the royalty-bearing product. We recognize royalty revenues when we can reliably estimate such amounts and collectability is reasonably assured.
Royalty revenue paid by Shionogi on their product sales is subject to returns. Peramivir is a newly introduced product and there is no historical experience that can be used to reasonably estimate product returns. Accordingly, we defer recognition of royalty revenue from Shionogi until a right of return no longer exists or until we have developed sufficient historical experience to estimate product returns.
Reimbursements received for direct out-of-pocket expenses related to research and development costs are recorded as revenue in the income statement rather than as a reduction in expenses. Event payments are recognized as revenue upon the achievement of specified events if (1) the event is substantive in nature and the achievement of the event was not reasonably assured at the inception of the agreement and (2) the fees are non-refundable and non-creditable. Any event payments received prior to satisfying these criteria are recorded as deferred revenue. Under the Company’s contract with BARDA/HHS, revenue is recognized as reimbursable direct and indirect costs are incurred.
Product Sales
Product sales are recognized net of estimated allowances, discounts, sales returns, chargebacks and rebates. Product sales recognized during 2010 and 2009 were not subject to a contractual right of return.
Research and Development Expenses
Our research and development costs are charged to expense when incurred. Advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are recognized as expense when the related goods are delivered or the related services are performed. Research and development expenses include, among other items, personnel costs, including salaries and benefits, manufacturing costs, clinical, regulatory, and toxicology services performed by CROs, materials and supplies, and overhead allocations consisting of various administrative and facilities related costs. Most of our manufacturing and clinical and preclinical studies are performed by third-party CROs. Costs for studies performed by CROs are accrued by us over the service periods specified in the contracts and estimates are adjusted, if required, based upon our on-going review of the level of services actually performed.
Additionally, we have license agreements with third parties, such as AECOM, IRL, and UAB, which require fees related to sublicense agreements or maintenance fees. We expense sublicense payments as incurred unless they are related to revenues that have been deferred, in which case the expenses are deferred and recognized over the related revenue recognition period. We expense maintenance payments as incurred.
At December 31, 2011, we had deferred collaboration expenses of approximately $7.7 million. These deferred expenses were sub-license payments, paid to our academic partners upon receipt of consideration from various commercial partners, and other consideration to our academic partners for modification to existing license agreements. These deferred expenses would not have been incurred without receipt of such payments or
52
modifications from our commercial partners and are being expensed in proportion to the related revenue being recognized. We believe that this accounting treatment appropriately matches expenses with the associated revenue.
We group our R&D expenses into two major categories: direct external expenses and indirect expenses. Direct expenses consist of compensation for R&D personnel and costs of outside parties to conduct laboratory studies, develop manufacturing processes and manufacture the drug candidate, conduct and manage clinical trials, patent related costs, and as well as other costs related to our clinical and preclinical studies. These costs are accumulated and tracked by program. Indirect expenses consist of lab supplies and services, facility expenses, depreciation of development equipment and other overhead of our research and development efforts. These costs apply to work on our clinical and preclinical candidates as well as our discovery research efforts.
Stock-Based Compensation
All share-based payments, including grants of stock option awards and restricted stock awards, are recognized in our Consolidated Statement of Operations based on their fair values. Stock-based compensation cost is estimated at the grant date based on the fair value of the award and is recognized as expense on a straight-line basis over the requisite service period of the award. Determining the appropriate fair value model and the related assumptions for the model requires judgment, including estimating the life of an award, the stock price volatility, and the expected term. We utilize the Black-Scholes option-pricing model to value our awards and recognize compensation expense on a straight-line basis over the vesting periods. The estimation of share-based payment awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from our current estimates, such amounts will be recorded as a cumulative adjustment in the period estimates are revised. Significant management judgment is also required in determining estimates of future stock price volatility and forfeitures to be used in the valuation of the options. Actual results, and future changes in estimates, may differ substantially from our current estimates.
Foreign Currency Hedge
In connection with our issuance of the PhaRMA Notes, we entered into a foreign Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. Under the Currency Hedge Agreement, we have the right to purchase dollars and sell yen at a rate of 100 yen per dollar for which we may be required to pay a premium in each year from 2014 through 2020, provided the Currency Hedge Agreement remains in effect. A payment of $2.0 million will be required if, on May 18 of the relevant year, the US dollar is worth 100 yen or less as determined in accordance with the Currency Hedge Agreement. In conjunction with establishing the Currency Hedge Agreement, we will be required to post collateral to the counterparty, which may cause us to experience additional quarterly volatility in our financial results. We will not be required at any time to post collateral exceeding the maximum premium payments remaining payable under the Currency Hedge Agreements. In establishing the hedge, we provided initial funds of approximately $2.0 million to support our potential hedge obligations. Subject to certain obligations we have in connection with the PhaRMA Notes, we have the right to terminate the Currency Hedge Agreement with respect to the 2016 through 2020 period by giving notice to the counterparty prior to May 18, 2014 and payment of a $2.0 million termination fee. Prior to this termination date, the maximum amount of hedge collateral we may be required to post is $5.9 million.
The Currency Hedge Agreement does not qualify for hedge accounting treatment and therefore mark to market adjustments will be recognized in our statement of operations. Cumulative mark to market adjustments for the year ended December 31, 2011 resulted in a $4.0 million loss. Mark to market adjustments are determined by quoted prices in markets that are not actively traded and for which significant inputs are observable directly or indirectly, representing the Level 2 in the fair value hierarchy as defined by generally accepted accounting principles. The Company is also required to post collateral in connection with the mark to market adjustments based on defined thresholds and as of December 31, 2011, $3.5 million was posted under the agreement.
53
Tax
We account for uncertain tax positions in accordance with accounting principles generally accepted in the United States. Significant management judgment is required in determining our provision for income taxes, deferred tax assets and liabilities and any valuation allowance recorded against net deferred tax assets. We have recorded a valuation allowance against all potential tax assets, due to uncertainties in our ability to utilize deferred tax assets, primarily consisting of certain net operating losses carried forward, before they expire. The valuation allowance is based on estimates of taxable income in each of the jurisdiction in which we operate and the period over which our deferred tax assets will be recoverable.
Impact of Inflation
We do not believe that our operating results have been materially impacted by inflation during the past three years. However, we cannot be assured that our operating results will not be adversely affected by inflation in the future. We will continually seek to mitigate the adverse effects of inflation on the services that we use through improved operating efficiencies and cost containment initiatives.
Recent Accounting Pronouncements
Note 11 to the Consolidated Financial Statements included in Item 8 of this Annual Report on Form 10-K discusses accounting pronouncements recently issued or proposed but not yet required to be adopted.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
Interest Rate Risk
We are subject to interest rate risk on our investment portfolio and borrowings under our PhaRMA Notes.
We invest in marketable securities in accordance with our investment policy. The primary objectives of our investment policy are to preserve capital, maintain proper liquidity to meet operating needs and maximize yields. Our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure to any single issue, issuer or type of investment. We place our excess cash with high credit quality financial institutions, commercial companies, and government agencies in order to limit the amount of credit exposure. Some of the securities we invest in may have market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate.
Our investment exposure to market risk for changes in interest rates relates to the increase or decrease in the amount of interest income we can earn on our portfolio, changes in the market value due to changes in interest rates and other market factors as well as the increase or decrease in any realized gains and losses. Our investment portfolio includes only marketable securities and instruments with active secondary or resale markets to help ensure portfolio liquidity. A hypothetical 100 basis point drop in interest rates along the entire interest rate yield curve would not significantly affect the fair value of our interest sensitive financial instruments. We generally have the ability to hold our fixed-income investments to maturity and therefore do not expect that our operating results, financial position or cash flows will be materially impacted due to a sudden change in interest rates. However, our future investment income may fall short of expectations due to changes in interest rates, or we may suffer losses in principal if forced to sell securities which have declined in market value due to changes in interest rates or other factors, such as changes in credit risk related to the securities’ issuers. To minimize this risk, we schedule our investments to have maturities that coincide with our expected cash flow needs, thus avoiding the need to redeem an investment prior to its maturity date. Accordingly, we do not believe that we have material exposure to interest rate risk arising from our investments. Generally, our investments are not collateralized. We have not realized any significant losses from our investments.
We do not use interest rate derivative instruments to manage exposure to interest rate changes. We ensure the safety and preservation of invested principal funds by limiting default risk, market risk and reinvestment risk. We reduce default risk by investing in investment grade securities.
54
Foreign Currency Risk
In connection with the issuance by Royalty Sub of the PhaRMA Notes, we entered into a Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. Under the Currency Hedge Agreement, we are required to post collateral based on our potential obligations under the Currency Hedge Agreement as determined by periodic mark to market adjustments. Provided the Currency Hedge Agreement remains in effect, we may be required to pay a premium in the amount of $2.0 million in each year beginning in May 2014 and continuing through May 2020. Such payment will be required if, in May of the relevant year, the spot rate of exchange for Japanese yen-U.S. dollars (determined in accordance with the Currency Hedge Agreement) is such that the U.S. dollar is worth 100 yen or less.
55
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
BIOCRYST PHARMACEUTICALS, INC.
(In thousands, except per share amounts)
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| ASSETS | ||||||||
| Cash and cash equivalents |
$ | 16,444 | $ | 13,622 | ||||
| Restricted cash |
625 | 625 | ||||||
| Investments |
25,274 | 40,323 | ||||||
| Receivables from collaborations |
5,831 | 30,227 | ||||||
| Interest reserve |
1,742 | — | ||||||
| Inventory |
263 | 898 | ||||||
| Prepaid expenses and other current assets |
378 | 1,005 | ||||||
| Deferred collaboration expense |
2,301 | 719 | ||||||
|
|
|
|
|
|||||
| Total current assets |
52,858 | 87,419 | ||||||
| Investments |
15,382 | 11,771 | ||||||
| Furniture and equipment, net |
1,098 | 1,929 | ||||||
| Deferred collaboration expense |
5,437 | 8,328 | ||||||
| Other assets |
7,433 | — | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 82,208 | $ | 109,447 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Accounts payable |
$ | 2,497 | $ | 8,201 | ||||
| Accrued expenses |
12,616 | 17,302 | ||||||
| Interest payable |
1,400 | — | ||||||
| Deferred collaboration revenue |
9,786 | 2,497 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
26,299 | 28,000 | ||||||
| Deferred collaboration revenue |
7,103 | 15,944 | ||||||
| Foreign currency derivative |
4,000 | — | ||||||
| Non-recourse notes payable |
30,000 | — | ||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; shares authorized — 5,000; no shares outstanding |
— | — | ||||||
| Common stock, $.01 par value; shares authorized — 95,000; shares issued and outstanding — 45,662 in 2011 and 44,959 in 2010 |
457 | 450 | ||||||
| Additional paid-in capital |
367,829 | 361,520 | ||||||
| Accumulated other comprehensive income |
40 | 105 | ||||||
| Accumulated deficit |
(353,520 | ) | (296,572 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
14,806 | 65,503 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 82,208 | $ | 109,447 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
56
BIOCRYST PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Revenues |
||||||||||||
| Product sales |
$ | — | $ | 325 | $ | 22,923 | ||||||
| Collaborative and other research and development |
19,643 | 62,056 | 51,667 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
19,643 | 62,381 | 74,590 | |||||||||
| Expenses |
||||||||||||
| Cost of products sold |
— | 86 | 4,544 | |||||||||
| Research and development |
56,898 | 83,900 | 73,661 | |||||||||
| General and administrative |
12,332 | 12,752 | 10,122 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
69,230 | 96,738 | 88,327 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(49,587 | ) | (34,357 | ) | (13,737 | ) | ||||||
| Interest and other income |
413 | 504 | 286 | |||||||||
| Interest expense |
(3,774 | ) | — | — | ||||||||
| Loss on foreign currency derivative |
(4,000 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (56,948 | ) | $ | (33,853 | ) | $ | (13,451 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per common share |
$ | (1.26 | ) | $ | (0.76 | ) | $ | (0.35 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average shares outstanding used in computing basic and diluted net loss per common share |
45,144 | 44,564 | 38,926 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
57
BIOCRYST PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (56,948 | ) | $ | (33,853 | ) | $ | (13,451 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation, amortization, and impairment |
886 | 2,267 | 1,613 | |||||||||
| Stock-based compensation expense |
4,772 | 6,302 | 5,525 | |||||||||
| Amortization of debt issuance costs |
356 | — | — | |||||||||
| Change in fair value of foreign currency derivative |
4,000 | — | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Receivables from collaborations |
24,396 | 3,495 | (21,740 | ) | ||||||||
| Inventory |
635 | 5,383 | (6,281 | ) | ||||||||
| Prepaid expenses and other assets |
626 | 51 | 81 | |||||||||
| Deferred collaboration expense |
1,309 | (220 | ) | 377 | ||||||||
| Accounts payable and accrued expenses |
(10,731 | ) | (9,483 | ) | 20,223 | |||||||
| Deferred collaboration revenue |
(1,552 | ) | (2,497 | ) | (2,565 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(32,251 | ) | (28,555 | ) | (16,218 | ) | ||||||
| Investing activities |
||||||||||||
| Acquisition of furniture and equipment |
(55 | ) | (325 | ) | (604 | ) | ||||||
| Change in restricted cash |
— | — | (625 | ) | ||||||||
| Purchases of investments |
(45,500 | ) | (55,909 | ) | (54,103 | ) | ||||||
| Sales and maturities of investments |
56,873 | 56,455 | 42,437 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
11,318 | 221 | (12,895 | ) | ||||||||
| Financing activities |
||||||||||||
| Sale of common stock, net |
1,027 | — | 45,740 | |||||||||
| Exercise of stock options |
278 | 553 | 2,117 | |||||||||
| Employee stock purchase plan sales |
300 | 283 | 194 | |||||||||
| Purchases of treasury stock |
(61 | ) | (5 | ) | (155 | ) | ||||||
| Issuance of non-recourse notes payable, net |
25,691 | — | — | |||||||||
| Payment of foreign currency derivative collateral |
(3,480 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
23,755 | 831 | 47,896 | |||||||||
|
|
|
|
|
|
|
|||||||
| Increase (decrease) in cash and cash equivalents |
2,822 | (27,503 | ) | 18,783 | ||||||||
| Cash and cash equivalents at beginning of year |
13,622 | 41,125 | 22,342 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of year |
$ | 16,444 | $ | 13,622 | $ | 41,125 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
58
BIOCRYST PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except per share amounts)
| Common Stock |
Additional Paid-In Capital |
Accumulated Other Comprehensive (Loss) Income |
Accumulated Deficit |
Total Stockholders’ Equity |
Comprehensive Loss |
|||||||||||||||||||
| Balance at December 31, 2008 |
$ | 383 | $ | 295,208 | $ | 104 | $ | (249,268 | ) | $ | 46,427 | |||||||||||||
| Net loss |
— | — | — | (13,451 | ) | (13,451 | ) | $ | (13,451 | ) | ||||||||||||||
| Unrealized loss on marketable securities available-for-sale |
— | — | (130 | ) | — | (130 | ) | (130 | ) | |||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
$ | (13,581 | ) | |||||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Exercise of stock options, 532 shares, net |
5 | 2,111 | — | — | 2,116 | |||||||||||||||||||
| Sale of common stock, 5,000 shares, net |
50 | 45,690 | — | — | 45,740 | |||||||||||||||||||
| Employee stock purchase plan sales, 123 shares |
1 | 193 | — | — | 194 | |||||||||||||||||||
| Purchases of treasury stock, 24 shares |
— | (155 | ) | — | — | (155 | ) | |||||||||||||||||
| Stock-based compensation expense |
— | 5,525 | — | — | 5,525 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2009 |
439 | 348,572 | (26 | ) | (262,719 | ) | 86,266 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net loss |
— | — | — | (33,853 | ) | (33,853 | ) | $ | (33,853 | ) | ||||||||||||||
| Unrealized gain on marketable securities available-for-sale |
— | — | 131 | — | 131 | 131 | ||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
$ | (33,722 | ) | |||||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Exercise of stock options, 240 shares, net |
2 | 550 | — | — | 552 | |||||||||||||||||||
| Employee stock purchase plan sales, 51 shares |
1 | 282 | — | — | 283 | |||||||||||||||||||
| Issuance of common stock, 761 shares, net |
8 | 5,819 | — | — | 5,827 | |||||||||||||||||||
| Purchases of treasury stock, 1 shares |
— | (5 | ) | — | — | (5 | ) | |||||||||||||||||
| Stock-based compensation expense |
— | 6,302 | — | — | 6,302 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2010 |
450 | 361,520 | 105 | (296,572 | ) | 65,503 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net loss |
— | — | — | (56,948 | ) | (56,948 | ) | $ | (56,948 | ) | ||||||||||||||
| Unrealized loss on marketable securities available-for-sale |
— | — | (65 | ) | — | (65 | ) | (65 | ) | |||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
$ | (57,013 | ) | |||||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Exercise of stock options, 184 shares, net |
2 | 276 | — | — | 278 | |||||||||||||||||||
| Employee stock purchase plan sales, 94 shares |
1 | 299 | — | — | 300 | |||||||||||||||||||
| Issuance of common stock, 437 shares, net |
4 | 1,023 | — | — | 1,027 | |||||||||||||||||||
| Purchases of treasury stock, 12 shares |
— | (61 | ) | — | — | (61 | ) | |||||||||||||||||
| Stock-based compensation expense |
— | 4,772 | — | — | 4,772 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2011 |
$ | 457 | $ | 367,829 | $ | 40 | $ | (353,520 | ) | $ | 14,806 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes to consolidated financial statements.
59
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(In thousands, except per share amounts)
Note 1 — Significant Accounting Policies
The Company
BioCryst Pharmaceuticals, Inc. (the “Company”) is a biotechnology company that designs, optimizes and develops novel drugs that block key enzymes involved in the pathogenesis of diseases related to therapeutic areas with unmet medical needs aligned with its capabilities and expertise. The Company was incorporated in Delaware in 1986 and its headquarters is located in Durham, North Carolina. Areas of interest for the Company are determined primarily by the scientific discoveries and the potential advantages that its experienced drug discovery group identifies, as well as by the associated potential commercial opportunity of those discoveries. The Company integrates the disciplines of biology, crystallography, medicinal chemistry and computer modeling to discover and develop small molecule pharmaceuticals through the process known as structure-guided drug design.
Basis of Presentation
Beginning in March 2011, the consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary, JPR Royalty Sub LLC (“Royalty Sub”). Royalty Sub was formed in connection with a $30,000 financing transaction the Company completed on March 9, 2011. See Note 3, Royalty Monetization, for a further description of this transaction. All intercompany transactions and balances have been eliminated.
The Company’s financial statements became consolidated beginning in March 2011 with the creation of Royalty Sub, and have been prepared in accordance with accounting principles generally accepted in the United States. Such financial statements reflect all adjustments that are, in management’s opinion, necessary to present fairly, in all material respects, the Company’s financial position, results of operations, and cash flows. There were no adjustments other than normal recurring adjustments.
Reclassifications
In the fourth quarter of 2011, the Company changed its classification of patent costs. This change resulted in $1,427 and $1,359 of patent expenses to be reclassified from general and administrative expense to research and development expense for the years ended December 31, 2010 and 2009, respectively. This reclassification had no effect on previously reported operating expenses or net loss amounts. Certain other balance sheet amounts as of December 31, 2010 have been reclassified to conform to the 2011 presentation.
Cash and Cash Equivalents
The Company generally considers cash equivalents to be all cash held in commercial checking accounts, money market accounts or investments in debt instruments with maturities of three months or less at the time of purchase. The carrying value of cash and cash equivalents approximates fair value due to the short-term nature of these items.
Restricted Cash
The Company is required to maintain $625 in an interest bearing money market account to serve as collateral for a corporate credit card program.
Investments
The Company invests in high-credit quality investments in accordance with its investment policy which minimizes the possibility of loss. The objective of the Company’s investment policy is to ensure the safety and
60
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
preservation of invested funds, as well as maintaining liquidity sufficient to meet cash flow requirements. The Company places its excess cash with high credit quality financial institutions, commercial companies, and government agencies in order to limit the amount of credit exposure. Per its policy, the Company is able to invest in marketable debt securities that may consist of U.S. government and government agency securities, money market and mutual fund investments, municipal and corporate notes and bonds, commercial paper and asset or mortgage-backed securities, among others. The Company’s investment policy requires it to purchase high-quality marketable securities with a maximum individual maturity of three years and requires an average portfolio maturity of no more than two years. Some of the securities the Company invests in may have market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. To minimize this risk, the Company schedules its investments with maturities that coincide with expected cash flow needs, thus avoiding the need to redeem an investment prior to its maturity date. Accordingly, the Company does not believe it has a material exposure to interest rate risk arising from its investments. Generally, the Company’s investments are not collateralized. The Company has not realized any significant losses from its investments.
The Company classifies all of its investments as available-for-sale. Unrealized gains and losses on investments are recognized in other comprehensive income/(loss), unless an unrealized loss is considered to be other than temporary, in which case the unrealized loss is charged to operations. The Company periodically reviews its investments for other than temporary declines in fair value below cost basis and whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company believes the individual unrealized losses represent temporary declines primarily resulting from interest rate changes. Realized gains and losses are reflected in other income/(expense), net in the Consolidated Statement of Operations and are determined using the specific identification method with transactions recorded on a settlement date basis. Investments with original maturities at date of purchase beyond three months and which mature at or less than 12 months from the balance sheet date are classified as current. Investments with a maturity beyond 12 months from the balance sheet date are classified as long-term. At December 31, 2011, the Company believes that the costs of its investments are recoverable in all material respects.
The following tables summarize the fair value of the Company’s investments by type. The estimated fair value of the Company fixed income investments are classified as Level 2 in the fair value hierarchy as defined in generally accepted accounting principles. These fair values are obtained from independent pricing services which utilize Level 2 inputs.
| December 31, 2011 | ||||||||||||||||||||
| Amortized Cost |
Accrued Interest |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
||||||||||||||||
| U.S. Treasury securities |
$ | 1,998 | $ | 2 | $ | 14 | $ | — | $ | 2,014 | ||||||||||
| Obligations of U.S. government and its agencies |
5,000 | 10 | — | — | 5,010 | |||||||||||||||
| Corporate debt securities |
10,924 | 80 | 15 | (9 | ) | 11,010 | ||||||||||||||
| Commercial paper |
10,939 | — | 2 | (1 | ) | 10,940 | ||||||||||||||
| Asset-backed securities |
611 | — | — | — | 611 | |||||||||||||||
| Certificate of deposit |
801 | 1 | — | — | 802 | |||||||||||||||
| Municipal obligations |
10,182 | 68 | 21 | (2 | ) | 10,269 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total investments |
$ | 40,455 | $ | 161 | $ | 52 | $ | (12 | ) | $ | 40,656 | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
61
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
| December 31, 2010 | ||||||||||||||||||||
| Amortized Cost |
Accrued Interest |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
||||||||||||||||
| U.S. Treasury securities |
$ | 7,505 | $ | 26 | $ | 24 | $ | — | $ | 7,555 | ||||||||||
| Obligations of U.S. government and its agencies |
12,065 | 92 | 13 | — | 12,170 | |||||||||||||||
| Corporate debt securities |
10,744 | 75 | 48 | — | 10,867 | |||||||||||||||
| Commercial paper |
14,572 | 2 | 7 | (1 | ) | 14,580 | ||||||||||||||
| Asset-backed securities |
1,079 | — | 1 | — | 1,080 | |||||||||||||||
| Certificate of deposit |
1,000 | 4 | 3 | — | 1,007 | |||||||||||||||
| Municipal obligations |
4,817 | 8 | 16 | (6 | ) | 4,835 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total investments |
$ | 51,782 | $ | 207 | $ | 112 | $ | (7 | ) | $ | 52,094 | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
The following table summarizes the scheduled maturity for the Company’s investments at December 31, 2011 and 2010.
| 2011 | 2010 | |||||||
| Maturing in one year or less |
$ | 25,274 | $ | 40,323 | ||||
| Maturing after one year through two years |
14,628 | 9,996 | ||||||
| Maturing after two years |
754 | 1,775 | ||||||
|
|
|
|
|
|||||
| Total investments |
$ | 40,656 | $ | 52,094 | ||||
|
|
|
|
|
|||||
Receivables from Collaborations
Receivables are recorded for amounts due to the Company primarily related to reimbursable research and development costs. These receivables are evaluated to determine if any reserve or allowance should be established at each reporting date. At December 31, 2011, the Company had the following receivables from collaborations.
| Billed | Unbilled | Total | ||||||||||
| U.S. Department of Health and Human Services |
$ | 1,146 | $ | 4,683 | $ | 5,829 | ||||||
| Shionogi & Co. Ltd. |
2 | — | 2 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total receivables from collaborations |
$ | 1,148 | $ | 4,683 | $ | 5,831 | ||||||
|
|
|
|
|
|
|
|||||||
During the third quarter of 2011, the Company received a payment of $2,884 from the U.S. Department of Health and Human Services (“BARDA/HHS”) related to indirect cost rate adjustments for calendar year 2010. This adjustment is calculated as the difference between the actual indirect costs incurred against the contract during the year and the indirect costs that are invoiced at a provisional billing rate during 2010. Because this adjustment amount represents actual costs incurred in performance of the contract and the costs are allowable, reasonable, and allocable to the contract, the Company recorded revenue accordingly in 2010. The Company’s calculations of its indirect cost rates are subject to an audit by the federal government.
Inventory
At December 31, 2011 and 2010, the Company’s inventory consisted of peramivir finished goods inventory and supplies for the manufacture of peramivir. Inventory is stated at the lower of cost, determined under the first-in, first-out (“FIFO”) method, or market. The Company expenses costs related to the production of
62
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
inventories as research and development expenses in the period incurred until such time it is believed that future economic benefit is expected to be recognized, which generally is reliant upon receipt of regulatory approval. Upon regulatory approval, the Company capitalizes subsequent costs related to the production of inventories.
During 2011, based on the annual variability of influenza, which impacts potential clinical and commercial demand and timing for peramivir administration, as well as the costs to store and maintain supplies, the Company decided for economic reasons to reduce its supplies inventory. This reduction resulted in a $635 charge in 2011.
The Company’s inventory consisted of the following:
| As of December 31 | ||||||||
| 2011 | 2010 | |||||||
| Supplies |
$ | 898 | $ | 898 | ||||
| Finished goods |
3,980 | 3,980 | ||||||
| Reserve for finished goods and supplies |
(4,615 | ) | (3,980 | ) | ||||
|
|
|
|
|
|||||
| Net inventories |
$ | 263 | $ | 898 | ||||
|
|
|
|
|
|||||
In October 2009, the Company determined that the U.S. Food and Drug Administration’s (“FDA”) granting of the Emergency Use Authorization (“EUA”) for peramivir was objective and persuasive evidence that supported capitalization of peramivir inventories manufactured after the issuance of the EUA. As a result, the Company recorded manufacturing costs of $3,980 for peramivir finished goods inventory. Prior to the issuance of the EUA, all costs associated with the manufacturing of peramivir were expensed as research and development expenses. During 2009, the Company evaluated whether the costs capitalized as inventory would be recoverable in a future period. Given the lack of objective, reliable evidence to support future demand for peramivir, management concluded that there was no certainty that future sales will materialize and revenues will exceed the costs incurred. Therefore in 2009, the capitalized inventory was fully reserved and such reserve was charged to cost of products sold.
Furniture and Equipment
Furniture and equipment are recorded at cost. Depreciation is computed using the straight-line method with estimated useful lives of five and seven years. Laboratory equipment, office equipment, and software are depreciated over a life of five years. Furniture and fixtures are depreciated over a life of seven years. Leasehold improvements are amortized over their estimated useful lives or the remaining lease term, whichever is less.
In accordance with generally accepted accounting principles, the Company periodically reviews its furniture and equipment for impairment when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values. Furniture and equipment to be disposed of are reported at the lower of carrying amount or fair value less cost to sell.
Patents and Licenses
The Company seeks patent protection on all internally developed processes and products. All patent related costs are expensed to research development expenses when incurred as recoverability of such expenditures is uncertain.
63
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Accrued Expenses
The Company enters into contractual agreements with third-party vendors who provide research and development, manufacturing, and other services in the ordinary course of business. Some of these contracts are subject to milestone-based invoicing and services are completed over an extended period of time. The Company records liabilities under these contractual commitments when it determines an obligation has been incurred, regardless of the timing of the invoice. This accrual process involves reviewing open contracts and purchase orders, communicating with its applicable personnel to identify services that have been performed and estimating the level of service performed and the associated cost when the Company has not yet been invoiced or otherwise notified of actual cost. The majority of service providers invoice the Company monthly in arrears for services performed. The Company makes estimates of accrued expenses as of each balance sheet date based on the facts and circumstances. The Company periodically confirms the accuracy of its estimates with the service providers and make adjustments if necessary. Examples of estimated accrued expenses include:
| Ÿ | fees paid to Clinical Research Organizations (“CROs”) in connection with preclinical and toxicology studies and clinical trials; |
| Ÿ | fees paid to investigative sites in connection with clinical trials; |
| Ÿ | fees paid to contract manufacturers in connection with the production of our raw materials, drug substance and drug products; and |
| Ÿ | professional fees. |
The Company bases its expenses related to clinical trials on its estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on the Company’s behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, the Company estimates the time period over which services will be performed and the level of effort expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust the accrual accordingly. If the Company does not identify costs that it has begun to incur or if it underestimates or overestimates the level of these costs, actual expenses could differ from estimates. Accrued expenses as of December 31, 2011 and 2010 included $8,622 and $13,827, respectively, of research and development costs.
Income Taxes
The liability method is used in the Company’s accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse.
Accumulated Other Comprehensive (Loss) Income
Accumulated other comprehensive (loss) income is comprised of unrealized gains and losses on investments available-for-sale and is disclosed as a separate component of stockholders’ equity.
64
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Revenue Recognition
The Company recognizes revenues from collaborative and other research and development arrangements and product sales.
Collaborative and Other Research and Development Arrangements and Royalties
Revenue from license fees, royalty payments, event payments, and research and development fees are recognized as revenue when the earnings process is complete and the Company has no further continuing performance obligations or the Company has completed the performance obligations under the terms of the agreement. Fees received under licensing agreements that are related to future performance are deferred and recognized over an estimated period determined by management based on the terms of the agreement and the products licensed. In the event a license agreement contains multiple deliverables, the Company evaluates whether the deliverables are separate or combined units of accounting. Revisions to revenue or profit estimates as a result of changes in the estimated revenue period are recognized prospectively.
Under certain of our license agreements, the Company receives royalty payments based upon our licensees’ net sales of covered products. Generally, under these agreements, the Company receives royalty reports from our licensees approximately one quarter in arrears, that is, generally in the second month of the quarter after the licensee has sold the royalty-bearing product. The Company recognizes royalty revenues when it can reliably estimate such amounts and collectability is reasonably assured.
Royalty revenue paid by Shionogi & Co. Ltd (“Shionogi”) on their product sales is subject to returns. RAPIACTA® is a newly introduced product and there is no historical experience that can be used to reasonably estimate product returns. Therefore, the Company defers recognition of RAPIACTA royalty revenue from Shionogi until the earlier of (1) a right of return no longer exists or (2) it has developed sufficient historical experience to estimate product returns. RAPIACTA royalty payments received from Shionogi in 2011 were $873.
Reimbursements received for direct out-of-pocket expenses related to research and development costs are recorded as revenue in the Consolidated Statement of Operations rather than as a reduction in expenses. Event payments are recognized as revenue upon the achievement of specified events if (1) the event is substantive in nature and the achievement of the event was not reasonably assured at the inception of the agreement and (2) the fees are non-refundable and non-creditable. Any event payments received prior to satisfying these criteria are recorded as deferred revenue. Under the Company’s contract with BARDA/HHS, revenue is recognized as reimbursable direct and indirect costs are incurred.
Product Sales
Sales are recognized when there is persuasive evidence that an arrangement exists, title has passed, the price was fixed and determinable, and collectability is reasonably assured. Product sales are recognized net of estimated allowances, discounts, sales returns, chargebacks and rebates. Product sales recognized during 2010 and 2009 were not subject to a contractual right of return.
65
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
The Company recorded the following revenues for the years ended December 31:
| 2011 | 2010 | 2009 | ||||||||||
| Product sales: |
||||||||||||
| U.S. Department of Health and Human Services |
$ | — | $ | — | $ | 22,500 | ||||||
| Neopharm Group (Israel) |
— | — | 398 | |||||||||
| NT Pharma Limited (Hong Kong) |
— | 250 | — | |||||||||
| Other |
— | 75 | 25 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total product sales |
— | 325 | 22,923 | |||||||||
| Collaborative and other research and development revenues: |
||||||||||||
| U.S. Department of Health and Human Services |
17,099 | 42,530 | 37,867 | |||||||||
| Shionogi (Japan) |
1,181 | 15,933 | 10,415 | |||||||||
| Mundipharma (United Kingdom) |
1,277 | 1,860 | 3,143 | |||||||||
| Grants (United States) |
86 | 978 | — | |||||||||
| Other |
— | 755 | 242 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total collaborative and other research and development revenues |
19,643 | 62,056 | 51,667 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
$ | 19,643 | $ | 62,381 | $ | 74,590 | ||||||
|
|
|
|
|
|
|
|||||||
Research and Development Expenses
The Company’s research and development costs are charged to expense when incurred. Research and development expenses include all direct and indirect development costs related to the development of the Company’s portfolio of drug candidates. Advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are recognized as expense when the related goods are delivered or the related services are performed. Research and development expenses include, among other items, personnel costs, including salaries and benefits, manufacturing costs, clinical, regulatory, and toxicology services performed by CROs, materials and supplies, and overhead allocations consisting of various administrative and facilities related costs. Most of the Company’s manufacturing and clinical and preclinical studies are performed by third-party CROs. Costs for studies performed by CROs are accrued by the Company over the service periods specified in the contracts and estimates are adjusted, if required, based upon the Company’s on-going review of the level of services actually performed.
Additionally, the Company has license agreements with third parties, such as Albert Einstein College of Medicine of Yeshiva University (“AECOM”), Industrial Research, Ltd. (“IRL”), and the University of Alabama at Birmingham (“UAB”), which require fees related to sublicense agreements or maintenance fees. The Company expenses sublicense payments as incurred unless they are related to revenues that have been deferred, in which case the expenses are deferred and recognized over the related revenue recognition period. The Company expenses maintenance payments as incurred.
Deferred collaboration expenses represent sub-license payments, paid to the Company’s academic partners upon receipt of consideration from various commercial partners, and other consideration paid to our academic partners for modification to existing license agreements. These deferred expenses would not have been incurred without receipt of such payments or modifications from the Company’s commercial partners and are being expensed in proportion to the related revenue being recognized. The Company believes that this accounting treatment appropriately matches expenses with the associated revenue.
66
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Stock-Based Compensation
All share-based payments, including grants of stock option awards and restricted stock awards, are recognized in the Company’s Consolidated Statement of Operations based on their fair values. The fair value of stock option awards is estimated using the Black-Scholes option pricing model. The fair value of restricted stock awards is based on the grant date closing price of the common stock. Stock-based compensation cost is recognized as expense on a straight-line basis over the requisite service period of the award.
Interest Expense and Deferred Financing Costs
Interest expense for the year ended December 31, 2011 was $3,774 and relates to the issuance of the PhaRMA Notes (defined in Note 3). Costs directly associated with the issuance of the PhaRMA Notes have been capitalized and are included in other non-current assets on the Consolidated Balance Sheet. These costs are being amortized to interest expense over the term of the PhaRMA Notes using the effective interest rate method. Amortization of deferred financing costs included in interest expense was $356 for the year ended December 31, 2011.
Currency Hedge Agreement
In connection with the issuance by Royalty Sub of the PhaRMA Notes, the Company entered into a Currency Hedge Agreement (defined in Note 3) to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. The Currency Hedge Agreement does not qualify for hedge accounting treatment; therefore mark to market adjustments are recognized in the Company’s Consolidated Statement of Operations. Cumulative mark to market adjustments for the year ended December 31, 2011 resulted in a $4,000 loss. Mark to market adjustments are determined by a third party pricing model which uses quoted prices in markets that are not actively traded and for which significant inputs are observable directly or indirectly, representing Level 2 in the fair value hierarchy as defined by generally accepted accounting principles. The Company is also required to post collateral in connection with the mark to market adjustments based on defined thresholds. As of December 31, 2011, $3,480 of hedge collateral was posted under the agreement.
Restructuring Activities
During the fourth quarter of 2010, the Company announced a restructuring plan to consolidate core facilities and outsource non-core activities. In connection with this plan, the Company recognized as general and administrative expense, approximately $302 in one-time termination benefits, of which approximately $144 was expensed in 2010 and the remaining balance was expensed in 2011. The Company also recognized approximately $890 in accelerated depreciation during the fourth quarter of 2010 for fixed assets no longer used by the Company.
Net Loss Per Share
Net loss per share is based upon the weighted average number of common shares outstanding during the period. Diluted loss per share is equivalent to basic net loss per share for all periods presented herein because common equivalent shares from unexercised stock options, outstanding warrants, and common shares expected to be issued under the Company’s employee stock purchase plan were anti-dilutive. The calculation of diluted earnings per share for the years ended December 31, 2011, 2010 and 2009 does not include 8,169, 6,937, and 5,965, respectively, of potential common shares, as their impact would be anti-dilutive.
67
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires the Company to make estimates and assumptions that affect the amounts reported in the financial statements. Actual results could differ from those estimates.
Concentration of Market Risk
The Company’s primary source of revenue is reimbursement of peramivir development expenses, which was earned under the cost-plus-fixed-fee contract with BARDA/HHS. The Company relies on the U.S. Government to reimburse predominantly all of the development costs for its peramivir program. Accordingly, reimbursement of these expenses represents a significant portion of the Company’s collaborative and other research and development revenues. The completion or termination of this program/collaboration could negatively impact the Company’s future Consolidated Statements of Operations and Cash Flows. The Company’s drug development activities are performed by a limited group of third party vendors. If any of these vendors were unable to perform their services, this could significantly impact the Company’s ability to complete its drug development activities.
Credit Risk
Cash equivalents and investments are financial instruments which potentially subject the Company to concentration of risk to the extent recorded on the Consolidated Balance Sheet. The Company deposits excess cash with major financial institutions in the United States. Balances may exceed the amount of insurance provided on such deposits. The Company believes it has established guidelines for investment of its excess cash relative to diversification and maturities that maintain safety and liquidity. To minimize the exposure due to adverse shifts in interest rates, the Company maintains a portfolio of investments with an average maturity of approximately 24 months or less.
Note 2 — Furniture and Equipment
Furniture and equipment consisted of the following at December 31:
| 2011 | 2010 | |||||||
| Furniture and fixtures |
$ | 596 | $ | 587 | ||||
| Office equipment |
1,500 | 1,470 | ||||||
| Software |
1,409 | 1,409 | ||||||
| Laboratory equipment |
6,033 | 6,033 | ||||||
| Leased equipment |
63 | 63 | ||||||
| Leasehold improvements |
5,267 | 5,251 | ||||||
|
|
|
|
|
|||||
| 14,868 | 14,813 | |||||||
| Less accumulated depreciation and amortization |
(13,770 | ) | (12,884 | ) | ||||
|
|
|
|
|
|||||
| Furniture and equipment, net |
$ | 1,098 | $ | 1,929 | ||||
|
|
|
|
|
|||||
Note 3 — Royalty Monetization
Overview
On March 9, 2011, the Company completed a $30,000 financing transaction to monetize certain future royalty and milestone payments under its license agreement with Shionogi (the “Shionogi Agreement”), pursuant
68
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
to which Shionogi licensed from the Company the rights to market peramivir in Japan and, if approved for commercial sale, Taiwan. The Company received net proceeds of $22,691 from the transaction after transaction costs of $4,309 and the establishment of a $3,000 interest reserve account by Royalty Sub, which will be available to help cover interest shortfalls in the future.
As part of the transaction, the Company entered into a purchase and sale agreement dated as of March 9, 2011 with Royalty Sub, whereby the Company transferred to Royalty Sub, among other things, (i) its rights to receive certain royalty and milestone payments from Shionogi arising under the Shionogi Agreement, and (ii) the right to receive payments under a Japanese yen/US dollar foreign currency hedge arrangement (as further described below, the “Currency Hedge Agreement”), put into place by the Company in connection with the transaction. Royalty payments will be paid by Shionogi in Japanese yen and milestone payments will paid in U.S. dollars. The Company’s collaboration with Shionogi was not impacted as a result of this transaction.
Non-Recourse Notes Payable
On March 9, 2011, Royalty Sub completed a private placement to institutional investors of $30,000 in aggregate principal amount of its PhaRMA Senior Secured 14.0% Notes due 2020 (the “PhaRMA Notes”). The PhaRMA Notes were issued by Royalty Sub under an Indenture, dated as of March 9, 2011 (the “Indenture”), by and between Royalty Sub and U.S. Bank National Association, as Trustee. Principal and interest on the PhaRMA Notes issued are payable from, and are secured by, the rights to royalty and milestone payments under the Shionogi Agreement transferred by the Company to Royalty Sub and payments, if any, made to Royalty Sub under the Currency Hedge Agreement. The PhaRMA Notes bear interest at 14% per annum, payable annually in arrears on September 1st of each year, beginning on September 1, 2011 (the “Payment Date”). The Company remains entitled to receive any royalties and milestone payments related to sales of peramivir by Shionogi following repayment of the PhaRMA Notes. Prorated interest for the period March 9, 2011 through the September 1, 2011 Payment Date totaled $2,018. Payment of such interest was made through $760 in royalty payments collected from Shionogi and a $1,258 draw-down from the interest reserve account. As of December 31, 2011, $1,742 remains in the interest reserve account for future interest payments.
Royalty Sub’s obligations to pay principal and interest on the PhaRMA Notes are obligations solely of Royalty Sub and are without recourse to any other person, including the Company, except to the extent of the Company’s pledge of its equity interests in Royalty Sub in support of the PhaRMA Notes. The Company may, but is not obligated to, make capital contributions to a capital account that may be used to redeem, or on up to one occasion pay any interest shortfall on, the PhaRMA Notes.
If the amounts available for payment on any Payment Date are insufficient to pay all of the interest due on a Payment Date, unless sufficient capital is contributed to Royalty Sub by the Company as permitted under the Indenture or the interest reserve account is available to make such payment, the shortfall in interest will accrue interest at the interest rate applicable to the PhaRMA Notes compounded annually. If such shortfall (and interest thereon) is not paid in full on or prior to the next succeeding Payment Date, an “Event of Default” as described in the Indenture will occur.
The Indenture does not contain any financial covenants. The Indenture includes customary representations and warranties of Royalty Sub, affirmative and negative covenants of Royalty Sub, Events of Default and related remedies, and provisions regarding the duties of the Trustee, indemnification of the Trustee, and other matters typical for indentures used in structured financings of this type.
As of December 31, 2011, the aggregate fair value of the PhaRMA Notes approximates its carrying value of $30,000 since the stated rate and terms are representative of current rates and terms available to the Company.
Prior to March 9, 2012, the PhaRMA Notes will not be redeemable by Royalty Sub. Thereafter, the PhaRMA Notes will be redeemable at the option of Royalty Sub at any time at a redemption price equal to the
69
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
percentage of the outstanding principal balance of the PhaRMA Notes being redeemed specified below for the period in which the redemption occurs, plus accrued and unpaid interest through the redemption date on the PhaRMA Notes being redeemed:
| Payment Dates (Between Indicated Dates) |
Redemption Percentage |
|||
| From and including March 9, 2012 to and including March 8, 2013 |
107.0 | % | ||
| From and including March 9, 2013 to and including March 8, 2014 |
103.5 | % | ||
| From and including March 9, 2014 and thereafter |
100.0 | % | ||
Foreign Currency Hedge
In connection with the issuance by Royalty Sub of the PhaRMA Notes, the Company entered into a Currency Hedge Agreement to hedge certain risks associated with changes in the value of the Japanese yen relative to the U.S. dollar. Under the Currency Hedge Agreement, the Company has the right to purchase dollars and sell yen at a rate of 100 yen per dollar for which the Company may be required to pay a premium in each year from 2014 through 2020, provided the Currency Hedge Agreement remains in effect. A payment of $1,950 will be required if, on May 18 of the relevant year, the U.S. dollar is worth 100 yen or less as determined in accordance with the Currency Hedge Agreement.
The Currency Hedge Agreement does not qualify for hedge accounting treatment; therefore mark to market adjustments are recognized in the Company’s Consolidated Statement of Operations. Cumulative mark to market adjustments in 2011 resulted in a $4,000 loss. The Company is also required to post collateral in connection with the mark to market adjustments based on defined thresholds. As of December 31, 2011, $3,480 was posted under the Currency Hedge Agreement. The Company will not be required at any time to post collateral exceeding the maximum premium payments remaining payable under the Currency Hedge Agreement. Subject to certain obligations the Company has in connection with the PhaRMA Notes, the Company has the right to terminate the Currency Hedge Agreement with respect to the 2016 through 2020 period by giving notice to the counterparty prior to May 18, 2014 and payment of a $1,950 termination fee.
Note 4 — Lease Obligations and Other Contingencies
The Company has the following minimum payments under operating lease obligations that existed at December 31, 2011:
| 2012 |
$ | 933 | ||
| 2013 |
954 | |||
| 2014 |
971 | |||
| 2015 |
290 | |||
| Thereafter |
— | |||
|
|
|
|||
| Total minimum payments |
$ | 3,148 | ||
|
|
|
The obligations in the preceding table are primarily related to the Company’s leases for buildings in Birmingham, Alabama and Durham, North Carolina. The lease for the building in Alabama expires June 30, 2015 and has an option to renew an additional five years at the current market rate on the date of termination. The lease for the building in Durham, North Carolina expires December 31, 2014. Rent expense for operating leases was $714, $771, and $763 in 2011, 2010, and 2009, respectively.
70
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Note 5 — Stockholders’ Equity
In June 2011, the Company entered into an At Market Issuance Sales Agreement (the “Agreement”) with McNicoll, Lewis & Valak (“MLV”) pursuant to which the Company may issue and sell $70,000 in shares of its common stock at current market prices under a Form S-3 registration statement with MLV acting as the sales agent. Subject to the terms and conditions of the Agreement, MLV will use commercially reasonable efforts to sell the Company’s common stock from time to time, based upon the Company’s instruction, including any price, time or size limits or other customary parameters or conditions the Company may impose. The Company will pay MLV an aggregate commission rate of 2 % or 3% of the gross proceeds of the sales price per share of any common stock sold under the Agreement depending on threshold of sales. On June 28, 2011, the Company filed a Registration Statement on Form S-3, which became effective on July 13, 2011, for the issuance and sale of up to $70,000 of equity or other securities. During 2011, The Company sold an aggregate of 437 shares of common stock at an average per share price of $2.65 pursuant to the Agreement for net proceeds of $1,027. Offering costs associated with the sale of these shares were $130.
In May 2010, the Company entered into an amendment to the License Agreement dated June 27, 2000, as subsequently amended (the “License Agreement”), by and among the Company and AECOM and IRL (the “Licensors”). The amendment further amended the License Agreement through which the Company obtained worldwide exclusive rights to develop and ultimately distribute any drug candidates that might arise from research on a series of PNP inhibitors, including forodesine and BCX4208. Under the terms of the amendment, the Licensors agreed to accept a reduction of one-half in the percentage of future payments received from third-party sublicensees of the licensed PNP inhibitors that must be paid to the Licensors. This reduction does not apply to (i) any milestone payments the Company may receive in the future under its license agreement dated February 1, 2006 with Mundipharma International Holdings Limited (“Mundipharma”) and (ii) royalties received from the Company’s sublicensees in connection with the sale of licensed products, for which the original payment rate will remain in effect. The rate of royalty payments to the Licensors based on net sales of any resulting product made by the Company remains unchanged.
In consideration for the modifications to the license agreement, the Company issued to the Licensors shares of its common stock with an aggregate value of $5,827 and paid the Licensors $90 in cash. The Company deferred the value of this consideration and is amortizing to research and development expense through September 2027, which is the date of expiration of the last-to-expire patent related to this agreement. Additionally, at the Company’s sole option and subject to certain agreed upon conditions, any future non-royalty payments due to be paid by the Company to the Licensors under the License Agreement may be made either in cash, in shares of its common stock, or in a combination of cash and shares.
In November 2009, the Company entered into an Underwriting Agreement with Morgan Stanley in connection with a registered offering of 5,000 shares of its common stock at a public offering price of $9.75 per share, resulting in proceeds net of offering costs of $45,740. The common stock was issued pursuant to a prospectus supplement filed with the Securities and Exchange Commission pursuant to Rule 424(b)(2) of the Securities Act of 1933.
In August 2007, the Company entered into a Stock and Warrant Purchase Agreement with a group of existing stockholders for the private placement of 8,316 shares of the Company’s common stock at a purchase price of $7.80 per share and warrants to purchase 3,160 shares of the Company’s common stock at a purchase price of $0.125 per warrant. The proceeds from the sale, net of offering costs, were $65,118. The exercise price of the warrants is $10.25 per share. All of the warrants remain outstanding as of December 31, 2011 and will expire in August 2012. The participants in the transaction included funds managed by Baker Brothers Investments, Kleiner Perkins Caufield & Byers, EHS Holdings, OrbiMed Advisors, Texas Pacific Group Ventures, and Stephens Investment Management, all of whom were shareholders of the Company at the time of the offering. Subsequent to the offering, the Company registered the shares and warrants under the Securities Act for resale.
71
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
In June 2002, the Company’s Board of Directors adopted a stockholder rights plan and, pursuant thereto, issued preferred stock purchase rights (“Rights”) to the holders of the Company’s common stock. The Rights have certain anti-takeover effects. If triggered, the Rights would cause substantial dilution to a person or group of persons who acquires more than 15% (19.9% for William W. Featheringill, a former Board of Director who owned more than 15% at the time the Rights were put in place) of the Company’s common stock on terms not approved by the Board of Directors. In August 2007, this plan was amended for a transaction involving funds managed by or affiliated with Baker Brother Investments such that they could purchase up to 25% without triggering the Rights. The Rights are not exercisable until the distribution date, as defined in the Rights Agreement by and between the Company and American Stock Transfer & Trust Company, as Rights Agent. The Rights will expire at the close of business on June 24, 2012, unless that final expiration date is extended or unless the rights are earlier redeemed or exchanged by the Company.
Each Right entitles the registered holder to purchase from the Company one one-thousandth of a share of Series B Junior Participating Preferred Stock (“Series B”), par value $0.001 per share, at a purchase price of $26.00, subject to adjustment. Shares of Series B purchasable upon exercise of the Rights will not be redeemable. Each share of Series B will be entitled to a dividend of 1 times the dividend declared per share of common stock. In the event of liquidation, each share of Series B will be entitled to a payment of 1times the payment made per share of common stock. Each share of Series B will have 1 vote, voting together with the common stock. Finally, in the event of any merger, consolidation, or other transaction in which shares of common stock are exchanged, each share of Series B will be entitled to receive 1 times the amount received per share of common stock. Effective in November 2008, the Company increased the authorized shares available under these rights to 95 to match the authorized common shares of 95,000 at that time. In addition, the Board of Directors has the authority to issue up to 4,905 shares of undesignated preferred stock and to determine the rights, preferences, privileges and restrictions of those shares without further vote or action by the Company’s stockholders.
Note 6 — Stock-Based Compensation
Stock Incentive Plan
As of December 31, 2011, the Company had two stock-based employee compensation plans, the Stock Incentive Plan (“Incentive Plan”) and the Employee Stock Purchase Plan (“ESPP”). The Incentive Plan was amended and restated in February 2011 and approved by the Company’s stockholders in May 2011, and the ESPP was amended and restated in March 2010 and approved by the Company’s stockholders in May 2010. During 2007, the Company made an inducement grant outside of the Incentive Plan and ESPP to recruit a new employee to a key position within the Company. Stock-based compensation expense of $4,772 ($4,589 of expense related to the Incentive Plan, $146 of expense related to the ESPP and $37 of expense related to the inducement grant) was recognized during 2011, while $6,302 ($5,960 of expense related to the Incentive Plan, $192 of expense related to the ESPP, and $149 of expense related to the inducement grant) was recognized during 2010 and $5,525 ($5,140 of expense related to the Incentive Plan, $235 of expense related to the ESPP, and $150 of expense related to the inducement grant) was recognized during 2009.
Under the Incentive Plan, the Company grants stock option awards and restricted stock awards to its employees, directors, and consultants. Stock option awards are granted with an exercise price equal to the market price of the Company’s stock at the date of grant. Prior to March 1, 2011, stock option awards granted to employees generally vest 25% after one year and monthly thereafter on a pro rata basis over the next three years until fully vested after four years. Commencing in March 2011, stock option awards granted to employees generally vest 25% each year until fully vested after four years. Stock option awards granted to non-employee directors of the Company generally vest over one year. All stock option awards have contractual terms of 10 years. The vesting exercise provisions of all awards granted under the Incentive Plan are subject to
72
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
acceleration in the event of certain stockholder-approved transactions, or upon the occurrence of a change in control as defined in the Incentive Plan. Under the Incentive Plan, the Company also grants shares of restricted common stock to employees that generally vest 25% each year until fully vested after four years.
Related activity under the Incentive Plan is as follows:
| Awards Available |
Options Outstanding |
Weighted Average Exercise Price |
||||||||||
| Balance December 31, 2008 |
1,114 | 5,478 | $ | 8.30 | ||||||||
| Plan amendment |
1,540 | — | — | |||||||||
| Stock option awards granted |
(1,559 | ) | 1,559 | 2.02 | ||||||||
| Stock option awards exercised |
— | (532 | ) | 3.98 | ||||||||
| Stock option awards canceled |
678 | (678 | ) | 12.04 | ||||||||
|
|
|
|
|
|||||||||
| Balance December 31, 2009 |
1,773 | 5,827 | 6.58 | |||||||||
| Plan amendment |
1,300 | — | — | |||||||||
| Stock option awards granted |
(1,550 | ) | 1,550 | 6.68 | ||||||||
| Stock option awards exercised |
— | (240 | ) | 2.30 | ||||||||
| Stock option awards canceled |
335 | (335 | ) | 8.42 | ||||||||
|
|
|
|
|
|||||||||
| Balance December 31, 2010 |
1,858 | 6,802 | 6.66 | |||||||||
| Plan amendment |
1,600 | — | — | |||||||||
| Restricted stock awards granted |
(211 | ) | — | — | ||||||||
| Restricted stock awards cancelled |
8 | — | — | |||||||||
| Stock option awards granted |
(1,830 | ) | 1,830 | 3.97 | ||||||||
| Stock option awards exercised |
— | (190 | ) | 1.57 | ||||||||
| Stock option awards canceled |
584 | (584 | ) | 5.99 | ||||||||
|
|
|
|
|
|||||||||
| Balance December 31, 2011 |
2,009 | 7,858 | $ | 6.21 | ||||||||
|
|
|
|
|
|||||||||
For stock option awards granted under the Incentive Plan during 2011, 2010 and 2009, the fair value was estimated on the date of grant using a Black-Scholes option pricing model and the assumptions noted in the table below. The weighted average grant date fair value of these awards granted during 2011, 2010 and 2009 was $2.64, $4.65 and $1.52 , respectively. The fair value of the stock option awards is amortized to expense over the vesting periods using a straight-line expense attribution method. The following explanations describe the assumptions used by the Company to value the stock option awards granted during 2011, 2010, and 2009. The expected life is based on the average of the assumption that all outstanding stock option awards will be exercised at full vesting and the assumption that all outstanding stock option awards will be exercised at the midpoint of the current date (if already vested) or at full vesting (if not yet vested) and the full contractual term. The expected volatility represents an average of the implied volatility on the Company’s publicly traded options, the volatility over the most recent period corresponding with the expected life, and the Company’s long-term reversion volatility. The Company has assumed no expected dividend yield, as dividends have never been paid to stock or option holders and will not be for the foreseeable future. The weighted average risk-free interest rate is the implied yield currently available on zero-coupon government issues with a remaining term equal to the expected term.
73
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Weighted Average Assumptions for Stock Option Awards Granted under the Incentive Plan
| 2011 | 2010 | 2009 | ||||||||||
| Expected Life |
5.5 | 5.5 | 5.6 | |||||||||
| Expected Volatility |
80.4 | % | 89.3 | % | 104.2 | % | ||||||
| Expected Dividend Yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Risk-Free Interest Rate |
2.2 | % | 2.4 | % | 2.1 | % | ||||||
The total intrinsic value of stock option awards exercised under the Incentive Plan was $374 during 2011, $1,169 during 2010, and $2,787 million during 2009. The intrinsic value represents the total proceeds (fair market value at the date of exercise, less the exercise price, times the number of stock option awards exercised) received by all individuals who exercised stock option awards during the period.
The following table summarizes, at December 31, 2011, by price range: (1) for stock option awards outstanding under the Incentive Plan, the number of stock option awards outstanding, their weighted average remaining life and their weighted average exercise price; and (2) for stock option awards exercisable under the Plan, the number of stock option awards exercisable and their weighted average exercise price:
| Outstanding | Exercisable | |||||||||||||||||||
| Range | Number | Weighted Average Remaining Life |
Weighted Average Exercise Price |
Number | Weighted Average Exercise Price |
|||||||||||||||
| $0 to 3 | 1,348 | 6.6 | $ | 1.48 | 977 | $ | 1.38 | |||||||||||||
| 3 to 6 | 2,570 | 7.9 | 3.95 | 880 | 3.73 | |||||||||||||||
| 6 to 9 | 2,276 | 6.5 | 7.34 | 1,634 | 7.58 | |||||||||||||||
| 9 to 12 | 841 | 3.8 | 11.37 | 839 | 11.37 | |||||||||||||||
| 12 to 15 | 817 | 4.7 | 12.54 | 788 | 12.54 | |||||||||||||||
| 15 to 18 | 4 | 4.0 | 15.45 | 4 | 15.45 | |||||||||||||||
| 18 to 21 | 2 | 4.1 | 18.99 | 2 | 18.99 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| $0 to 21 | 7,858 | 6.5 | $ | 6.21 | 5,124 | $ | 7.13 | |||||||||||||
|
|
|
|
|
|||||||||||||||||
The weighted average remaining contractual life of stock option awards exercisable under the Incentive Plan at December 31, 2011 was 5.3 years.
The aggregate intrinsic value of stock option awards outstanding and exercisable under the Incentive Plan at December 31, 2011 was $1,078. The aggregate intrinsic value represents the value (the period’s closing market price, less the exercise price, times the number of in-the-money stock option awards) that would have been received by all stock option award holders under the Incentive Plan had they exercised their stock option awards at the end of the year.
The total fair value of the stock option awards vested under the Incentive Plan was $4,775 during 2011, $4,441 during 2010, and $5,261 during 2009.
As of December 31, 2011, the number of stock option awards vested and expected to vest under the Incentive Plan is 7,209. The weighted average exercise price of these stock option awards is $6.42 and their weighted average remaining contractual life is 6.6 years.
74
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
The following table summarizes the changes in the number and weighted-average grant-date fair value of non-vested stock option awards during 2011:
| Non-Vested Stock Option Awards |
Weighted Average Grant-Date Fair Value |
|||||||
| Balance December 31, 2010 |
2,701 | $ | 3.39 | |||||
| Stock option awards granted |
1,830 | 2.64 | ||||||
| Stock option awards vested |
(1,523 | ) | 3.14 | |||||
| Stock option awards forfeited |
(275 | ) | 3.27 | |||||
|
|
|
|||||||
| Balance December 31, 2011 |
2,733 | $ | 3.04 | |||||
|
|
|
|||||||
During 2007, the Company granted 50 restricted stock awards under the Incentive Plan with a grant date fair value of $11.81. Vesting commenced during the first quarter of 2009 and continued ratably through the first quarter of 2011.
During the second quarter of 2008, the Company also granted 77 restricted stock awards under the Incentive Plan with a grant date fair value of $3.12. All of these restricted stock awards vested on December 31, 2009.
As of December 31, 2011, there was approximately $6,923 of total unrecognized compensation cost related to non-vested employee stock option awards and restricted stock awards granted by the Company. That cost is expected to be recognized as follows: $2,844 in 2012, $2,466 in 2013, $1,345 in 2014, and $267 in 2015.
Employee Stock Purchase Plan
The Company has reserved a total of 825 shares of common stock to be purchased under the ESPP, of which 137 shares remain available for purchase at December 31, 2011. Eligible employees may authorize up to 15% of their salary to purchase common stock at the lower of 85% of the beginning or 85% of the ending price during six-month purchase intervals. No more than 3 shares may be purchased by any one employee at the six-month purchase dates and no employee may purchase stock having a fair market value at the commencement date of $25 or more in any one calendar year.
There were 94, 51, and 123, shares of common stock purchased under the ESPP in 2011, 2010, and 2009, respectively, at a weighted average price per share of $3.21, $5.50, and $1.57, respectively. Expense of $146, $192, and $235 related to the ESPP was recognized during 2011, 2010, and 2009, respectively. Compensation expense for shares purchased under the ESPP related to the purchase discount and the “look-back” option were determined using a Black-Scholes option pricing model. The weighted average grant date fair values of shares granted under the ESPP during 2011, 2010, and 2009 were $1.33, $2.76, and $1.70, respectively.
75
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Note 7 — Income Taxes
The Company has incurred net losses since inception and, consequently, has not recorded any U.S. federal and state income tax expense or benefit. The differences between the Company’s effective tax rate and the statutory tax rate in 2011, 2010, and 2009 are as follows:
| 2011 | 2010 | 2009 | ||||||||||
| Income tax benefit at federal statutory rate (35%) |
$ | (19,932 | ) | $ | (11,457 | ) | $ | (4,708 | ) | |||
| State and local income taxes net of federal tax benefit |
(2,503 | ) | (1,092 | ) | (6,412 | ) | ||||||
| Permanent items |
890 | 1,753 | 834 | |||||||||
| Rate change |
(2,500 | ) | 5,178 | — | ||||||||
| Expiration of attribute carryforwards |
2,884 | 5,343 | 1,072 | |||||||||
| Research and development tax credits |
(2,108 | ) | (5,359 | ) | 292 | |||||||
| Other |
731 | 253 | (218 | ) | ||||||||
| Change in valuation allowance |
22,538 | 5,381 | 9,140 | |||||||||
|
|
|
|
|
|
|
|||||||
| Income tax expense (benefit) |
$ | — | $ | — | $ | — | ||||||
The Company recognizes the impact of a tax position in its financial statements if it is more likely than not that the position will be sustained on audit based on the technical merits of the position. The Company has concluded that it has one uncertain tax position pertaining to its research and development credit carryforwards. The Company has not yet conducted an in-depth study of its research and development credits. This study could result in an increase or decrease to the Company’s research and development credits. Until studies are conducted of the Company’s research and development credits, no amounts are being recorded as an unrecognized tax benefits, separate from the valuation allowance against deferred tax assets. Any future changes to the Company’s unrecognized tax benefits would be offset by an adjustment to the valuation allowance and there would be no impact on the Company’s financial statements.
Additionally, utilization of the Company’s net operating loss carryforwards could be subject to a substantial annual limitation due to ownership change limitations described in Section 382 of the Internal Revenue Code and similar state provisions. The Company has performed an analysis and has determined there have been no changes in control that would limit the use of the Company’s net operating losses through December 31, 2011.
Significant components of the Company’s deferred tax assets and liabilities are as follows:
| 2011 | 2010 | |||||||
| Deferred tax assets: |
||||||||
| Net federal and state operating losses |
$ | 97,054 | $ | 77,628 | ||||
| General business credits |
38,119 | 36,906 | ||||||
| Fixed assets |
1,265 | 1,202 | ||||||
| Reserve for inventories |
1,827 | 1,540 | ||||||
| Deferred revenue |
5,801 | 6,120 | ||||||
| Stock-based compensation |
5,915 | 5,018 | ||||||
| Foreign currency derivative |
1,584 | — | ||||||
| Other |
422 | 1,035 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
151,987 | 129,449 | ||||||
| Valuation allowance |
(151,987 | ) | (129,449 | ) | ||||
|
|
|
|
|
|||||
| Total deferred tax liabilities |
— | — | ||||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
76
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
The majority of the Company’s deferred tax assets relate to net operating loss and research and development carryforwards that can only be realized if the Company is profitable in future periods. It is uncertain whether the Company will realize any tax benefit related to these carryforwards. Accordingly, the Company has provided a full valuation allowance against the net deferred tax assets due to uncertainties as to their ultimate realization. The valuation allowance will remain at the full amount of the deferred tax assets until it is more likely than not that the related tax benefits will be realized. The Company’s valuation allowance increased by $22,538 in 2011, $5,381 in 2010 and $9,140 in 2009.
As of December 31, 2011, the Company had federal operating loss carryforwards of $244,667, state operating loss carryforwards of $289,350, and research and development credit carryforwards of $38,119, which will expire at various dates from 2012 through 2031.
The Company’s federal and state operating loss carryforwards include $4,776 of excess tax benefits related to a deduction from the exercise of stock options. The tax benefit of these deductions has not been recognized in deferred tax assets. If utilized, the benefits from these deductions will be recorded as adjustments to income tax expense and additional paid-in capital.
Tax years 2008-2010 remain open to examination by the major taxing jurisdictions to which the Company is subject. Additionally, years prior to 2008 are also open to examination to the extent of loss and credit carryforwards from those years. The Company recognizes interest and penalties accrued related to unrecognized tax benefits as components of its income tax provision. However, there were no provisions or accruals for interest and penalties in 2011, 2010, and 2009.
Note 8 — Employee 401(k) Plan
In January 1991, the Company adopted an employee retirement plan (“401(k) Plan”) under Section 401(k) of the Internal Revenue Code covering all employees. Employee contributions may be made to the 401(k) Plan up to limits established by the Internal Revenue Service. Company matching contributions may be made at the discretion of the Board of Directors. The Company made matching contributions of $391, $434, and $378, in 2011, 2010, and 2009, respectively.
Note 9 — Collaborative and Other Research and Development Contracts
U.S. Department of Health and Human Services (“BARDA/HHS”). In January 2007, the U.S. Department of Health and Human Services (“BARDA/HHS”) awarded the Company a $102,661, four-year contract for the advanced development of peramivir for the treatment of influenza. During 2009, peramivir clinical development shifted to focus on intravenous delivery and the treatment of hospitalized patients. To support this focus, a September 2009 contract modification was awarded to extend the intravenous (“i.v.”) peramivir program by 12 months and to increase funding by $77,191. On February 24, 2011, the Company announced that BARDA/HHS had awarded it a $55,000 contract modification, intended to fund completion of the Phase 3 development of i.v. peramivir for the treatment of patients hospitalized with influenza. This contract modification brings the total award from BARDA/HHS to $234,852 and extends the contract term by 24 months through December 31, 2013, providing funding through completion of Phase 3 and to support the filing of a new drug application (“NDA”) to seek regulatory approval for i.v. peramivir in the U.S.
The contract with BARDA/HHS is a cost-plus-fixed-fee contract. That is, the Company is entitled to receive reimbursement for all costs incurred in accordance with the contract provisions that are related to the development of peramivir plus a fixed fee, or profit. BARDA/HHS will make periodic assessments of progress and the continuation of the contract is based on the Company’s performance, the timeliness and quality of deliverables, and other factors. The government has rights under certain contract clauses to terminate this contract. The contract is terminable by the government at any time for breach or without cause.
77
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
Shionogi & Co., Ltd. (“Shionogi”). In March 2007, the Company entered into an exclusive license agreement with Shionogi to develop and commercialize peramivir in Japan for the treatment of seasonal and potentially life-threatening human influenza. Under the terms of the agreement, Shionogi obtained rights to injectable formulations of peramivir in Japan. The Company developed peramivir under a license from UAB and will owe sublicense payments to them on any future milestone payments and/or royalties received by the Company from Shionogi. In October 2008, the Company and Shionogi amended the license agreement to expand the territory covered by the agreement to include Taiwan and to provide rights for Shionogi to perform a Phase 3 clinical trial in Hong Kong. Shionogi has commercially launched peramivir under the commercial name RAPIACTA in Japan.
Green Cross Corporation (“Green Cross”). In June 2006, the Company entered into an agreement with Green Cross to develop and commercialize peramivir in Korea. Under the terms of the agreement, Green Cross will be responsible for all development, regulatory, and commercialization costs in Korea. The Company received a one-time license fee of $250. The license also provides that the Company will share in profits resulting from the sale of peramivir in Korea, including the sale of peramivir to the Korean government for stockpiling purposes. Furthermore, Green Cross will pay the Company a premium over its cost to supply peramivir for development and any future marketing of peramivir products in Korea.
Mundipharma International Holdings Limited (“Mundipharma”). In February 2006, the Company entered into an exclusive, royalty bearing right and license agreement with Mundipharma for the development and commercialization of forodesine, a PNP inhibitor, for use in oncology (the “Original Agreement”). Under the terms of the Original Agreement, Mundipharma obtained rights to forodesine in markets across Europe, Asia, and Australasia in exchange for a $10,000 up-front payment.
The Company deferred revenue recognition of the $10,000 up-front payment that was received from Mundipharma in February 2006 since the Company was involved in the continued development of forodesine. Amortization of this revenue commenced in February 2006 and was initially scheduled to end in October 2017, which is the date of expiration for the last-to-expire patent covered by the agreement. The Company also deferred revenue recognition of a $5,000 payment received from Mundipharma in connection with the initiation of a clinical trial in 2007. Amortization of this deferred revenue commenced in 2007 and was initially scheduled to end in October 2017. Under its agreement with AECOM/IRL, the Company paid sublicense payments related to these upfront cash payments received from Mundipharma. Expense recognition of these sublicense payment was deferred and recognized under the same term as the related deferred revenue.
On November 11, 2011, the Company entered into the Amended and Restated License and Development Agreement (the “Amended and Restated Agreement”) with Mundipharma, amending and restating the Original Agreement. Under the terms of the Amended and Restated Agreement, Mundipharma obtained worldwide rights to forodesine. Commencing on November 11, 2011, Mundipharma controls the development and commercialization of forodesine and assumes all future development and commercialization costs. Mundipharma also purchased from the Company certain drug substance for forodesine at a cost of approximately $901. The Amended and Restated Agreement provides for the possibility of future event payments totaling $15,000 for achieving specified regulatory events for certain indications and tiered royalties ranging from mid to high single-digit percentages of net product sales in each country where forodesine is sold by Mundipharma. These royalties are subject to downward adjustments based on the then-existing patent coverage and/or the availability of generic compounds in each country.
The Amended and Restated Agreement is a multiple element arrangement for accounting purposes in which the Company is required to deliver to Mundipharma both the worldwide rights to forodesine in the field of oncology and the transfer of product data and know-how to permit Mundipharma to develop and commercialize forodesine (the “Knowledge Transfer”). The worldwide license rights were granted to Mundipharma on November 11, 2011. The Knowledge Transfer commenced in 2011 with expected completion by June 30, 2012.
78
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
The Company accounted for these elements as a combined unit of accounting as they do not have stand-alone value to Mundipharma. Since the Company is no longer associated with the development of forodesine, amortization of the deferred revenue and expense associated with the Original Agreement ceased immediately. The unamortized deferred revenue and expense of $7,766 and $1,864, respectively, will be recognized in the Consolidated Statement of Operations upon completion of the Knowledge Transfer.
Albert Einstein College of Medicine of Yeshiva University and Industrial Research, Ltd. (“AECOM” and “IRL” respectively). In June 2000, the Company licensed a series of potent inhibitors of PNP from AECOM and IRL, (collectively, the “Licensors”). The lead drug candidates from this collaboration are forodesine and BCX4208. The Company has obtained worldwide exclusive rights to develop and ultimately distribute these, or any other, drug candidates that might arise from research on these inhibitors. The Company has the option to expand the Agreement to include other inventions in the field made by the investigators or employees of the Licensors. The Company agreed to use commercially reasonable efforts to develop these drugs. In addition, the Company has agreed to pay certain milestone payments for each licensed product (which range in the aggregate from $1,400 to almost $4,000 per indication) for future development of these inhibitors, single digit royalties on net sales of any resulting product made by the Company, and to share approximately one quarter of future payments received from other third-party partners, if any. In addition, the Company has agreed to pay annual license fees, which can range from $150 to $500, that are creditable against actual royalties and other payments due to the Licensors. This agreement may be terminated by the Company at any time by giving 60 days advance notice or in the event of material uncured breach by the Licensors.
In May 2010, the Company amended the licensee agreement through which the Company obtained worldwide exclusive rights to develop and ultimately distribute any drug candidates that might arise from research on a series of PNP inhibitors, including forodesine and BCX4208. Under the terms of the amendment, the Licensors agreed to accept a reduction of one-half in the percentage of future payments received from third-party sublicensees of the licensed PNP inhibitors that must be paid to the Licensors. This reduction does not apply to (i) any milestone payments the Company may receive in the future under its license agreement dated February 1, 2006 with Mundipharma and (ii) royalties received from its sublicensees in connection with the sale of licensed products, for which the original payment rate will remain in effect. The rate of royalty payments to the Licensors based on net sales of any resulting product made by the Company remains unchanged.
In consideration for these modifications in 2010, the Company issued to the Licensors shares of its common stock with an aggregate value of $5,911 and paid the Licensors $90 in cash. Additionally, at the Company’s sole option and subject to certain agreed upon conditions, any future non-royalty payments due to be paid by it to the Licensors under the license agreement may be made either in cash, in shares of its common stock, or in a combination of cash and shares.
On November 17, 2011, the Company further amended its agreements with the Licensors whereby the Licensors agreed to accept a reduction of one-half in the percentage of Net Proceeds (as defined) received by the Company under its Amended and Restated Agreement with Mundipharma that will be paid to AECOM/IRL.
The University of Alabama at Birmingham (“UAB”). The Company currently has agreements with UAB for influenza neuraminidase and complement inhibitors. Under the terms of these agreements, UAB performed specific research for the Company in return for research payments and license fees. UAB has granted the Company certain rights to any discoveries in these areas resulting from research developed by UAB or jointly developed with the Company. The Company has agreed to pay single digit royalties on sales of any resulting product and to share in future payments received from other third-party partners. The Company has completed the research under the UAB agreements. These two agreements have initial 25-year terms, are automatically renewable for five-year terms throughout the life of the last patent and are terminable by the Company upon three months notice and by UAB under certain circumstances. Upon termination both parties shall cease using the other parties’ proprietary and confidential information and materials, the parties shall jointly own joint inventions
79
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
and UAB shall resume full ownership of all UAB licensed products. There is currently no activity between the Company and UAB on these agreements, but when the Company licenses this technology, such as in the case of the Shionogi and Green Cross agreements, or commercialize products related to these programs, we will owe sublicense fees or royalties on amounts we receive.
Emory University (“Emory”). In June 2000, the Company licensed intellectual property from Emory related to the hepatitis C polymerase target associated with hepatitis C viral infections. Under the original terms of the agreement, the research investigators from Emory provided the Company with materials and technical insight into the target. The Company has agreed to pay Emory single digit royalties on sales of any resulting product and to share in future payments received from other third party partners, if any. The Company can terminate this agreement at any time by giving 90 days advance notice. Upon termination, the Company would cease using the licensed technology.
Note 10 — Quarterly Financial Information (Unaudited) (In thousands, except per share)
| First | Second | Third | Fourth | |||||||||||||
| 2011 Quarters |
||||||||||||||||
| Revenues |
$ | 5,435 | $ | 3,735 | $ | 5,249 | $ | 5,224 | ||||||||
| Net Loss |
(13,027 | ) | (16,271 | ) | (14,459 | ) | (13,191 | ) | ||||||||
| Diluted net loss per/share |
(.29 | ) | (.36 | ) | (.32 | ) | (.29 | ) | ||||||||
| 2010 Quarters |
||||||||||||||||
| Revenues |
$ | 26,071 | $ | 7,616 | $ | 12,000 | $ | 16,694 | ||||||||
| Net loss |
(2,595 | ) | (10,193 | ) | (10,864 | ) | (10,201 | ) | ||||||||
| Diluted net loss per share |
(.06 | ) | (.23 | ) | (.24 | ) | (.23 | ) | ||||||||
In the fourth quarter of 2010, $711 of royalty revenue related to Shionogi’s sales of RAPIACTA in Japan, which was originally recorded during the first quarter of 2010, was reversed. RAPIACTA received an accelerated Japanese approval in January 2010 so it could be made available as a treatment option during the H1N1 pandemic. At the time of approval, RAPIACTA stability testing was ongoing and as a result, the product sold during early 2010 had a short shelf life. During the fourth quarter of 2010, Shionogi chose to accept returns of the product shipped early in 2010. The adjustment had no impact on the second or third quarters of 2010 and had no impact on full year 2010 operating results.
Note 11 — Recent Accounting Pronouncements
In May 2011, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (“ASU”) 2011-04, “Fair Value Measurement (Topic 820): Amendments to Achieve Common Fair Value Measurements and Disclosure Requirement in U.S. GAAP and IFRS.” This ASU modifies the existing standards to include disclosure of all transfer between Level 1 and Level 2 asset and liability fair value categories. In addition, the ASU provides guidance on measuring the fair value of financial instruments managed within a portfolio and the application of premiums and discounts on fair value measurements. The ASU requires additional disclosure for Level 3 measurements regarding the sensitivity of fair value to changes in unobservable inputs and any interrelationships between those inputs. This guidance is effective for interim and annual periods beginning after December 15, 2011, with early adoption prohibited. The Company does not expect this ASU will have a material impact on its consolidated financial statements.
In June 2011, the FASB issued ASU 2011-05, “Comprehensive Income (Topic 220): Presentation of Comprehensive Income.” This ASU eliminates the current option to report other comprehensive income and its components in the statement of changes in equity. Under this new ASU, an entity can elect to present items of net
80
BIOCRYST PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
(In thousands, except per share amounts)
income, other comprehensive income and total comprehensive income in one continuous statement or in two separate, but consecutive statements. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. Early adoption is permitted, but retrospective application is required. The Company will adopt this ASU in the first quarter of 2012.
In December 2011, the FASB issued ASU 2011-12, “Comprehensive Income (Topic 220): Deferral of the Effective Date for Amendments to the Presentation of Reclassifications of Items Out of Accumulated Other Comprehensive Income in Accounting Standards Update No. 2011-05.” This ASU defers the requirement in ASU 2011-05 to present reclassification adjustments for each component of accumulated other comprehensive income in both net income and other comprehensive income on the face of the financial statements. This ASU does not affect the requirement to present items of net income, and other comprehensive income and total comprehensive income in one continuous statement or in two separate, but consecutive statements. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. Early adoption is permitted, but retrospective application is required. The Company will adopt this ASU in the first quarter of 2012.
81
Report of Independent Registered Public Accounting Firm on Consolidated Financial Statements
The Board of Directors and Stockholders
BioCryst Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of BioCryst Pharmaceuticals, Inc. as of December 31, 2011 and 2010, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of BioCryst Pharmaceuticals, Inc. at December 31, 2011 and 2010, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), BioCryst Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 6, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Raleigh, North Carolina
March 6, 2012
82
Report of Independent Registered Public Accounting Firm on Internal Control
The Board of Directors and Stockholders
BioCryst Pharmaceuticals, Inc.
We have audited BioCryst Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). BioCryst Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, BioCryst Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of BioCryst Pharmaceuticals, Inc. as of December 31, 2011 and 2010, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011 of BioCryst Pharmaceuticals, Inc. and our report dated March 6, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Raleigh, North Carolina
March 6, 2012
83
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We maintain a set of disclosure controls and procedures that are designed to ensure that information relating to BioCryst Pharmaceuticals, Inc. required to be disclosed in our periodic filings under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported in a timely manner under the Exchange Act of 1934. We carried out an evaluation as required by paragraph (b) of Rule 13a-15 or Rule 15d-15 under the Exchange Act, under the supervision and with the participation of management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) or Rule 15d-15 under the Exchange Act). Based upon that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2011, our disclosure controls and procedures are effective. We believe that our disclosure controls and procedures will ensure that information required to be disclosed in the reports filed or submitted by us under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission, and include controls and procedures designed to ensure that information required to be disclosed by us in such reports is accumulated and communicated to our management, including our Chairman and Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control Over Financial Reporting
Management of BioCryst Pharmaceuticals, Inc. is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. As defined by the Securities and Exchange Commission, internal control over financial reporting is a process designed by, or under the supervision of our principal executive and principal financial officers and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the financial statements in accordance with U.S. generally accepted accounting principles.
Our internal control over financial reporting is supported by written policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of the financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In connection with the preparation of our annual financial statements, management has undertaken an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO Framework). Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of those controls.
Based on this assessment, management has concluded that as of December 31, 2011, our internal control over financial reporting was effective. Management believes our internal control over financial reporting will
84
provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles.
Ernst & Young LLP, the independent registered public accounting firm that audited our financial statements included in this report, has issued an attestation report on the Company’s internal control over financial reporting, a copy of which appears on page 83 of this annual report.
Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2011 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this item is set forth under the captions “Items to be Voted on — 1. Election of Directors,” “Executive Officers,” “Section 16(a) Beneficial Ownership Reporting Compliance” and “Corporate Governance” in our definitive Proxy Statement for the 2012 Annual Meeting of Stockholders and incorporated herein by reference.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this item is set forth under the captions “Compensation Discussion and Analysis,” “Summary Compensation Table,” “Grants of Plan-Based Awards in 2011,” “Outstanding Equity Awards at December 31, 2011,” “2011 Option Exercises and Stock Vested,” “Potential Payments Upon Termination or Change in Control,” “Director Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report” in our definitive Proxy Statement for the 2012 Annual Meeting of Stockholders and incorporated herein by reference.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item is set forth under the captions “Equity Compensation Plan Information” and “ Security Ownership of Certain Beneficial Owners and Management” in our definitive Proxy Statement for the 2012 Annual Meeting of Stockholders and incorporated herein by reference.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this item is set forth under the captions “Certain Relationships and Related Transactions” and “ Corporate Governance “ in our definitive Proxy Statement for the 2012 Annual Meeting of Stockholders and incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required by this item is set forth under the caption “Items to be Voted on — Ratification of Appointment of Independent Registered Public Accountants” in our definitive Proxy Statement for the 2012 Annual Meeting of Stockholders and incorporated herein by reference.
85
PART IV
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
(a) Financial Statements
The following financial statements appear in Item 8 of this Form 10-K:
No financial statement schedules are included because the information is either provided in the consolidated financial statements or is not required under the related instructions or is inapplicable and such schedules therefore have been omitted.
(b) Exhibits. See Index of Exhibits.
86
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on March 6, 2012.
| BIOCRYST PHARMACEUTICALS, INC. | ||
| By: | /s/ Jon P. Stonehouse | |
| Jon P. Stonehouse Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated on March 6, 2012:
| Signature |
Title(s) | |
| /s/ Jon P. Stonehouse (Jon P. Stonehouse) |
President, Chief Executive Officer and Director (Principal Executive Officer) | |
| /s/ Thomas R. Staab II (Thomas R. Staab II) |
Senior Vice President, Chief Financial Officer and Treasurer | |
| /s/ Robert S. Lowrey (Robert S. Lowrey) |
Controller and Principal Accounting Officer | |
| /s/ George B. Abercrombie (George B. Abercrombie) |
Director | |
| /s/ Stanley C. Erck (Stanley C. Erck) |
Director | |
| /s/ John L. Higgins (John L. Higgins) |
Director | |
| /s/ Zola P. Horovitz (Zola P. Horovitz, Ph.D.) |
Director | |
| /s/ Peder K. Jensen (Peder K. Jensen, M.D.) |
Director | |
| /s/ Kenneth B. Lee, Jr. (Kenneth B. Lee, Jr.) |
Director | |
| /s/ Charles A. Sanders (Charles A. Sanders, M.D.) |
Director | |
| /s/ Nancy Huston (Nancy Huston, Ph.D.) |
Director | |
87
| Number |
Description | |
| 3.1 | Third Restated Certificate of Incorporation of Registrant. Incorporated by reference to Exhibit 3.1 to the Company’s Form 8-K filed December 22, 2006. | |
| 3.2 | Certificate of Amendment to the Third Restated Certificate of Incorporation of Registrant. Incorporated by reference to Exhibit 3.1 to the Company’s Form 8-K filed July 24, 2007. | |
| 3.3 | Certificate of Increase of Authorized Number of Shares of Series B Junior Participating Preferred Stock. Incorporated by reference to Exhibit 3.1 to the Company’s Form 8-K filed November 4, 2008. | |
| 3.4 | Amended and Restated Bylaws of Registrant effective October 29, 2008. Incorporated by reference to Exhibit 3.2 to the Company’s Form 8-K filed November 4, 2008. | |
| 4.1 | Rights Agreement, dated as of June 17, 2002, by and between the Company and American Stock Transfer & Trust Company, as Rights Agent, which includes the Certificate of Designation for the Series B Junior Participating Preferred Stock as Exhibit A and the form of Rights Certificate as Exhibit B. Incorporated by reference to Exhibit 4.1 to the Company’s Form 8-A filed June 17, 2002. | |
| 4.2 | Amendment to Rights Agreement, dated as of August 5, 2007. Incorporated by reference to Exhibit 4.2 of the Company’s Form 10-Q filed August 9, 2007. | |
| 4.3 | Indenture, dated as of March 9, 2011 by and between JPR Royalty Sub LLC and U.S. Bank National Association, as trustee. Incorporated by reference to Exhibit 4.3 of the Company’s Form 10-Q filed May 6, 2011. | |
| 10.1& | Amended and Restated Stock Incentive Plan dated February 17, 2011. Incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K, filed May 17, 2011. | |
| 10.2& | Employee Stock Purchase Plan, as amended and restated effective March 31, 2010. Incorporated by reference to Appendix B to the Company’s Definitive Proxy Statement, filed April 6, 2010. | |
| 10.3& | Form of Notice of Grant of Non-Employee Director Automatic Stock Option and Stock Option Agreement. Incorporated by reference to Exhibit 10.4 of the Company’s Form 10-K filed March 4, 2008. | |
| 10.4& | Form of Notice of Grant of Stock Option and Stock Option Agreement. Incorporated by reference to Exhibit 10.5 of the Company’s Form 10-K filed March 4, 2008. | |
| 10.5& | Annual Incentive Plan. Incorporated by reference to Exhibit 10.1 of the Company’s Form 10-K filed March 4, 2008. | |
| 10.6& | Executive Relocation Policy. Incorporated by reference to Exhibit 10.2 of the Company’s Form 10-K filed March 4, 2008. | |
| 10.7& | Amended and Restated Employment Letter Agreement dated February 14, 2007, by and between the Company and Jon P. Stonehouse. Incorporated by reference to Exhibit 10.12 to the Company’s Form 10-K for the year ended December 31, 2006, filed March 14, 2007. | |
| 10.8& | Employment Letter Agreement between BioCryst Pharmaceuticals, Inc. and Thomas R. Staab II, dated May 23, 2011. Incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K filed May 25, 2011. | |
| 10.9& | Employment Letter Agreement between BioCryst Pharmaceuticals, Inc. and William P. Sheridan dated June 12, 2008. Incorporated by reference to Exhibit 10.27 of the Company’s Form 10-Q filed August 8, 2008. | |
| 10.10& | Employment Letter Agreement between BioCryst Pharmaceuticals, Inc. and Peter L. McCullough dated December 11, 2009. Incorporated by reference to Exhibit 10.12 of the Company’s Form 10-K filed March 15, 2011. | |
| 10.11& | Employment Letter Agreement dated April 2, 2007, by and between the Company and David McCullough. Incorporated by reference to Exhibit 10.5 to the Company’s Form 10-Q filed May 10, 2007. |
88
| Number |
Description | |
| 10.12& | Retention Bonus Agreement between BioCryst Pharmaceuticals, Inc. and David McCullough dated May 21, 2008. Incorporated by reference to Exhibit 10.26 of the Company’s Form 10-Q filed August 8, 2008. | |
| 10.13& | Consulting Agreement between BioCryst Pharmaceuticals, Inc. and J. Claude Bennett, M.D. dated June 13, 2008. Incorporated by reference to Exhibit 10.28 of the Company’s Form 10-Q filed August 8, 2008. | |
| 10.14# | Agreement dated January 3, 2007, between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services, as amended by Amendment number 1 dated January 3, 2007 and Amendment number 2 dated May 11, 2007. (Portions omitted pursuant to request for confidential treatment.) Incorporated by reference to Exhibit 10.3 to the Company’s Form 10-Q filed August 9, 2007. | |
| 10.15 | Amendment #3 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services, dated October 2, 2007. Incorporated by reference to Exhibit 10.6 of the Company’s Form 10-K filed March 4, 2008. | |
| 10.16 | Amendment #4 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated April 3, 2008. Incorporated by reference to Exhibit 10.29 of the Company’s Form 10-Q filed August 8, 2008. | |
| 10.17 | Amendment #5 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated July 2, 2008. Incorporated by reference to Exhibit 10.30 of the Company’s Form 10-Q filed August 8, 2008. | |
| 10.18 | Amendment #6 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated August 18, 2008. Incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K filed November 7, 2008. | |
| 10.19 | Amendment #7 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated November 17, 2008. Incorporated by reference to Exhibit 10.12 of the Company’s Form 10-K filed March 6, 2009. | |
| 10.20 | Amendment #8 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated March 13, 2009. Incorporated by reference to Exhibit 10.13 of the Company’s Form 10-K filed March 9, 2010. | |
| 10.21 | Amendment #9 to the Agreement between BioCryst Pharmaceuticals, Inc. and the Department of Health and Human Services dated September 18, 2009. Incorporated by reference to Exhibit 10.1 of the Company’s Form 10-Q filed November 6, 2009. | |
| 10.22 | Amendment #10 to the Agreement between the Company and the U.S. Department of Health & Human Services, dated October 15, 2009. Incorporated by reference to Exhibit 10.2 of the Company’s Form 10-Q filed November 6, 2009. | |
| 10.23 | Amendment #11 to the Agreement between the Company and the U.S. Department of Health & Human Services, dated February 23, 2011. Incorporated by reference to Exhibit 10.25 of the Company’s Form 10-K filed March 15, 2011. | |
| 10.24 | Order for Supplies or Services from the U.S. Department of Health & Human Services, dated November 4, 2009. Incorporated by reference to Exhibit 10.16 of the Company’s Form 10-K filed March 9, 2010. | |
| 10.25# | License, Development and Commercialization Agreement dated as of February 28, 2007, by and between the Company and Shionogi & Co., Ltd. Incorporated by reference to Exhibit 10.4 to the Company’s Form 10-Q filed May 10, 2007. (Portions omitted pursuant to request for confidential treatment.) |
89
| Number |
Description | |
| 10.26# | First Amendment to License, Development and Commercialization Agreement, effective as of September 30, 2008, between the Company and Shionogi & Co., Ltd. Incorporated by reference to Exhibit 10.19 to the Company’s Form 10-K filed March 6, 2009. (Portions omitted pursuant to request for confidential treatment.) | |
| 10.27 | Warehouse Lease dated July 12, 2000 between RBP, LLC an Alabama Limited Liability Company and the Registrant for office/warehouse space. Incorporated by reference to Exhibit 10.8 to the Company’s Form 10-Q for the second quarter ending June 30, 2000 filed August 8, 2000. | |
| 10.28 | Third Amendment to Lease Agreement dated August 7, 2007, by and between Riverchase Capital LLC, a Florida limited liability company, Stow Riverchase, LLC, a Florida limited liability company, as successor landlord to RBP, LLC and the Company. Incorporated by reference to Exhibit 10.4 of the Company’s Form 10-Q filed August 9, 2007. | |
| 10.29 | Stock and Warrant Purchase Agreement dated as of August 6, 2007, by and among BioCryst Pharmaceuticals, Inc. and each of the Investors identified on the signature pages thereto. Incorporated by reference to Exhibit 4.1 of the Company’s Form 8-K filed August 7, 2007. | |
| 10.30 | Stock Purchase Agreement, dated as of February 17, 2005, by and among BioCryst Pharmaceuticals, Inc., Baker Bros. Investments, L.P., Baker Biotech Fund II, L.P., Baker Bros. Investments II, L.P., Baker Biotech Fund II (Z), L.P., Baker/Tisch Investments, L.P., Baker Biotech Fund III, L.P., Baker Biotech Fund I, L.P., Baker Biotech Fund III (Z), L.P. and 14159, L.P. Incorporated by reference to Exhibit 4.1 to the Company’s Form 8-K filed February 17, 2005. | |
| 10.31# | Development and License Agreement dated as of February 1, 2006, by and between BioCryst Pharmaceuticals, Inc. and Mundipharma International Holdings Limited. Incorporated by reference to Exhibit 10.2 to the Company’s Form 8-K/A filed May 2, 2006. (Portions omitted pursuant to request for confidential treatment.) | |
| (10.32*) | Amended and Restated Development and License Agreement, dated as of November 11, 2011, by and between BioCryst Pharmaceuticals, Inc. and Mundipharma International Corporation Limited. (Portions omitted pursuant to request for confidential treatment.) | |
| 10.33# | License Agreement dated as of June 27, 2000, by and among Albert Einstein College of Medicine, Industrial Research, Ltd. and BioCryst Pharmaceuticals, Inc., as amended by the First Amendment Agreement dated as of July 26, 2002 and the Second Amendment Agreement dated as of April 15, 2005. Incorporated by reference to Exhibit 10.1 to the Company’s Form 8-K filed November 30, 2005. (Portions omitted pursuant to request for confidential treatment.) | |
| 10.34# | Third Amendment Agreement by and among Albert Einstein College of Medicine, Industrial Research, Ltd. and BioCryst Pharmaceuticals, Inc., dated as of December 11, 2009. Incorporated by reference to Exhibit 10.33 to the Company’s Form 10-K filed March 9, 2010. (Portions omitted pursuant to request for confidential treatment.) | |
| 10.35# | Fourth Amendment Agreement by and among Albert Einstein College of Medicine, Industrial Research, Ltd. and BioCryst Pharmaceuticals, Inc., dated as of May 5, 2010. Incorporated by reference to Exhibit 10.1 to the Company’s Form 10-Q filed August 6, 2010. (Portions omitted pursuant to request for confidential treatment.) | |
| (10.36*) | Fifth Amendment Agreement by and among Albert Einstein College of Medicine, Industrial Research, Ltd. and BioCryst Pharmaceuticals, Inc., dated as of November 17, 2011. (Portions omitted pursuant to request for confidential treatment.) | |
| 10.37 | Stock Purchase Agreement, dated as of December 14, 2005, by and among BioCryst Pharmaceuticals, Inc., Kleiner Perkins Caufield & Byers, Texas Pacific Group Ventures and KPTV, LLC. Incorporated by reference to Exhibit 4.1 to the Company’s Form 8-K filed December 16, 2005. | |
| 10.38 | Nomination and Observer Agreement, dated as of December 16, 2005, by and between BioCryst Pharmaceuticals, Inc. and Kleiner Perkins Caufield & Byers. Incorporated by reference to Exhibit 4.2 to the Company’s Form 8-K filed December 16, 2005. |
90
| Number |
Description | |
| 10.39 | Purchase and Sale Agreement, dated as of March 9, 2011 between BioCryst Pharmaceuticals, Inc. and JPR Royalty Sub LLC. Incorporated by reference to Exhibit 10.1 of the Company’s Form 10-Q filed May 6, 2011. | |
| 10.40 | Pledge and Security Agreement, dated as of March 9, 2011 between BioCryst Pharmaceuticals, Inc. and U.S. Bank National Association, as trustee. Incorporated by reference to Exhibit 10.2 of the Company’s Form 10-Q filed May 6, 2011. | |
| 10.41 | Confirmation of terms and conditions of ISDA Master Agreement, dated as of March 7, 2011, between Morgan Stanley Capital Services Inc. and BioCryst Pharmaceuticals, Inc. dated as of March 9, 2011. Incorporated by reference to Exhibit 10.3 of the Company’s Form 10-Q filed May 6, 2011. | |
| 10.42 | At Market Issuance Sales Agreement, dated June 28, 2011, by and between BioCryst Pharmaceuticals, Inc. and McNicoll, Lewis & Vlak LLC. Incorporated by reference to Exhibit 1.2 of the Company’s Registration Statement on Form S-3 filed June 28, 2011 (File No. 333-175182). | |
| (21) | Subsidiaries of the Registrant. | |
| (23) | Consent of Ernst & Young, LLP, Independent Registered Public Accounting Firm. | |
| (31.1) | Certification of the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| (31.2) | Certification of the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| (32.1) | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (32.2) | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (101) | Financial statements from the Annual Report on Form 10-K of BioCryst Pharmaceuticals, Inc. for the year ended December 31, 2011, formatted in XBRL: (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Cash Flows, (iv) Consolidated Statements of Stockholders’ Equity and (v) Notes to Consolidated Financial Statements.† |
| * | Confidential treatment requested. |
| # | Confidential treatment granted. |
| & | Management contracts. |
| ( ) | Filed herewith. |
| † | In accordance with Rule 406T of Regulation S-T, the XBRL related information in Exhibit 101 to this Annual Report on Form 10-K shall not be deemed to be “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to the liability of that section, and shall not be part of any registration or other document filed under the Securities Act or the Exchange Act, except as shall be expressly set forth by specific reference in such filing. |
91