Attached files
| file | filename |
|---|---|
| 8-K - FORM 8-K - Trius Therapeutics Inc | d276346d8k.htm |
 1
Tedizolid 112 Trial: Top Line Results
Update
January 3, 2012
Exhibit 99.1
Best-in-Class Anti-Infectives |
 2
Forward Looking Statements
Statements made in this presentation regarding matters that are not historical facts are
“forward-looking statements” within the meaning of the Private Securities
Litigation Reform Act of 1995. Because such statements are subject to risks and uncertainties,
actual results may differ materially from those expressed or implied by such forward-looking statements, and
the results of the 112 study are not necessarily indicative of the results of the 113 study.
Such statements include, but are not limited to, statements regarding Trius’ ability to
successfully complete its ongoing clinical trials and development programs and the expected
timing for reporting of top-line data for the TR701-113 study. Risks that contribute to the
uncertain nature of the forward-looking statements include: the success and timing of Trius’
preclinical studies and clinical trials; regulatory developments in the United States and
foreign countries; changes in Trius’ plans to develop and commercialize its product
candidates; the outcome of final analyses of data from recently-completed clinical trials of
tedizolid may vary from Trius’ initial analyses and the FDA may not agree with Trius’
interpretation of such results; additional ongoing or planned clinical trials of tedizolid may
produce negative or inconclusive results; Trius may decide, or the FDA may require Trius, to
conduct additional clinical trials or to modify Trius’ ongoing clinical trials; Trius may
experience delays in the commencement, enrollment, completion or analysis of clinical testing for its
product candidates, or significant issues regarding the adequacy of its clinical trial designs
or the execution of its clinical trials, which could result in increased costs and delays, or
limit Trius’ ability to obtain regulatory approval; the third parties with whom Trius has
partnered with for the development of tedizolid and upon whom Trius relies to conduct its clinical
trials and manufacture its product candidates may not perform as expected; tedizolid may not
receive regulatory approval or be successfully commercialized; unexpected adverse side effects
or inadequate therapeutic efficacy of tedizolid could delay or prevent regulatory approval or
commercialization; Trius’ ability to obtain and maintain intellectual property protection for its product
candidates; the loss of key scientific or management personnel, Trius’ ability to obtain
additional financing; and the accuracy of Trius’ estimates regarding expenses, future
revenues and capital requirements. These and other risks and uncertainties are described more
fully in Trius’ most recent Form 10-K, Forms 10-Q and other documents filed with the
United States Securities and Exchange Commission, including those factors discussed under the caption
“Risk Factors” in such filings. All forward-looking statements contained in this
press release speak only as of the date on which they were made. Trius undertakes no obligation
to update such statements to reflect events that occur or circumstances that exist after the
date on which they were made.
|
 3
Progression of ABSSSI Regulatory Environment for Phase 3
Studies 112 and 113
May 2010:
First in a series of FDA requested meetings of the FNIH (Foundation for the
National Institutes of Health) working group established to provide recommendations to
the
FDA
on
endpoints
for
clinical
trials
of
drugs
for
ABSSSI
and
other
indications
June
2010:
FDA
grants
Trius
a
“SPA
Letter
of
Agreement”
for
study
#112
August 2010:
–
FDA issues “Draft ABSSSI guidance”
that reflect changes from the 112 SPA including:
•
Carry-over of early failures at secondary outcome measurement and exclusion of
investigator- reported pain at EOT as a secondary outcome failure
criterion August 2011:
–
FDA grants Trius an SPA agreement for study #113 that reflects the recent draft
guidance –
FNIH submits to the FDA its recommendations on ABSSSI guidance (ABSSSI Docket ID:
FDA--2011--D- -0433). The recommendations include:
•
Removal of fever as a component of the primary endpoint leaving cessation of spread
& reduction in lesion size as the sole parameters for the primary efficacy
analysis at the 48-72hr visit
•
Recommendation to also use
20% reduction of lesion size at 48-72hrs as the primary
outcome
Trius has prospectively captured these suggested changes in the analyses of studies 112
and 113 Q2 2012: Expected issuance of final guidance on ABSSSI
|
 4
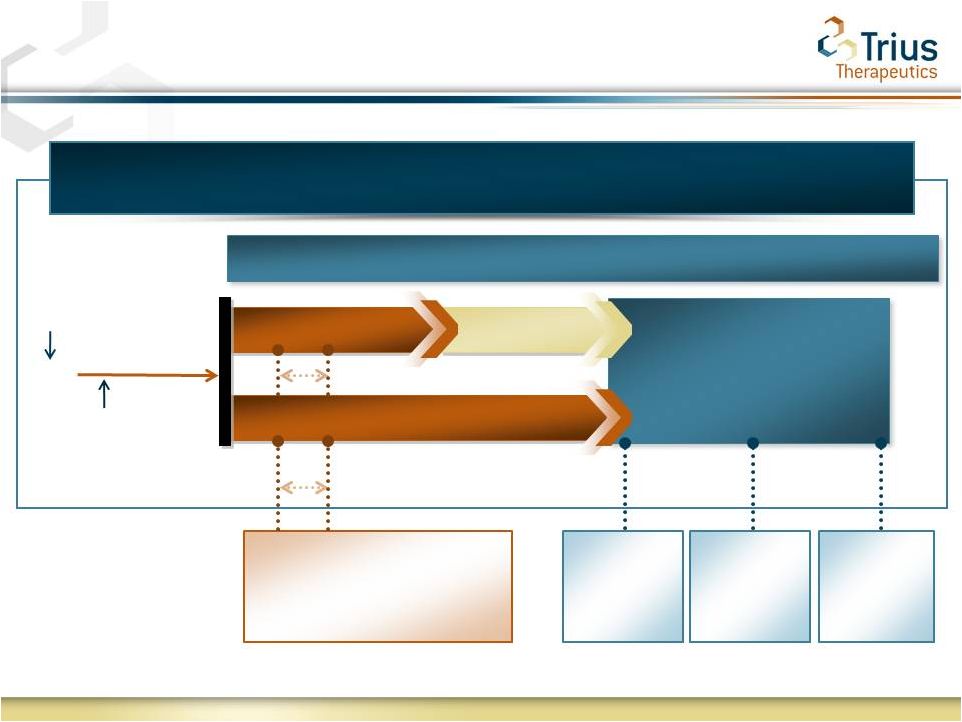
Tedizolid Phosphate Phase 3 Study Design:
Oral (112) and IV/Oral (113) Trials Under SPA
Post Treatment
Post Treatment
Evaluations
Evaluations
Patient
Screening
Randomization
Safety Analyses
Safety Analyses
Baseline
EMA
Primary
Endpoint at
PTE
FDA Primary Endpoint
48-72 hrs from Baseline
EMA
Secondary
Endpoint
LFU
FDA
Secondary
Endpoints
at EOT
667
6-Days Tedizolid QD
4-Days Placebo
10-Days Linezolid BID
FDA
FDA
and
and
EMA
EMA
Endpoints
Endpoints
for
for
Global
Global
Registration
Registration |
 5
Study 112 Primary & Secondary Endpoints: Capturing
FDA and EMA Endpoints
Primary Endpoint*:
Cessation of lesion spread & resolution of fever at 48-72 hour visit after
initiation of study drug (ITT analysis set) [FDA Primary Endpoint]
Secondary Endpoints:
Sustained clinical response at EOT in the ITT analysis set (days
11-13)
Sustained clinical response at EOT in the CE-EOT analysis set (days
11-13) Investigator’s
assessment of clinical success at PTE in the ITT analysis set
(days 17-24) [EMA Primary Endpoint]
Investigator’s
assessment of clinical success at PTE in the CE analysis set
(days 17-24)
*Study
112
was
90%
powered
for
a
10%
NI
margin
if
both
treatment
groups
had
~
81%
outcome
rate.
It
had 80% power for an outcome rate as low as 70%. |
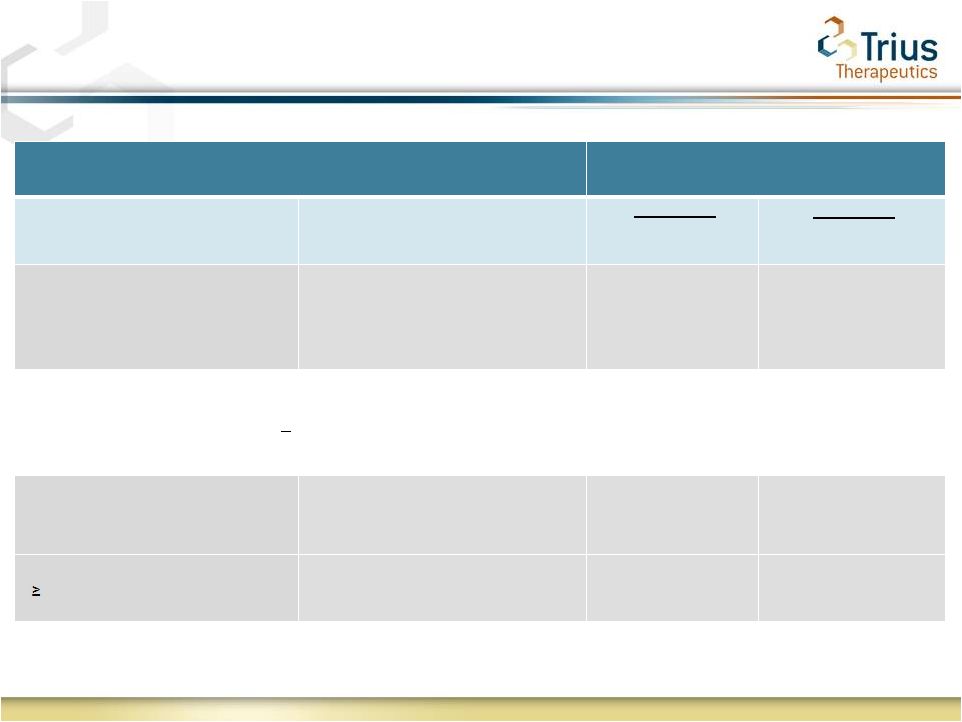 6
Primary Outcome at 48-72 hour visit
Treatment
Lesion Criteria
Fever Criteria
Tedizolid
(200 mg
QD 6 days)
Linezolid
(600 mg
BID 10 days)
No increase in lesion area from
baseline*
Temperature measurements
required within 24 hrs of
48-72 hr visit*
79.5%
79.4%
FNIH
recommended
to
FDA
to
exclude
temperature
as
a
component
of
the
primary
endpoint
and
to
assess
a
>20% reduction in lesion size at 48 to 72 hours.
Under these pre-specified analysis tedizold shows additional numerical separation
from linezolid No increase in lesion area from
baseline
Temperature excluded**
87.0%
85.4%
20% reduction of lesion area
from baseline**
Temperature excluded**
78.0%
76.1%
* Primary endpoint as agreed to under Study 112 and 113 SPA
** FNIH
recommendations
to
FDA:
ABSSSI
Docket
ID:
FDA--2010--D--0433
Primary Outcome: All Current and Contemplated Trial 112
Primary Endpoints Achieved in Pre-Specified Analyses |
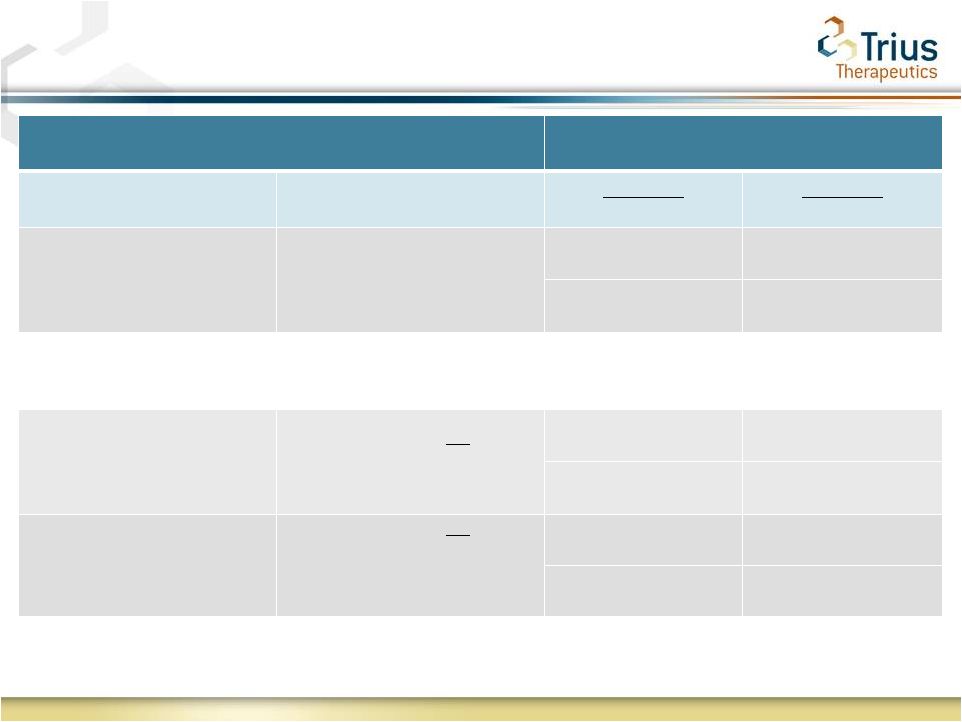 7
Secondary Outcomes: Tedizolid Demonstrates Comparable
Efficacy with Shorter Course of Therapy
Secondary Outcome at EOT or PTE
Treatment
Secondary Outcome
Criteria
Tedizolid
(200 mg QD 6 days)
Linezolid
(600 mg BID 10 days)
Clinical Response at EOT*
(Day 11)
Early clinical failures carried
forward to EOT*
69.3% (ITT)
71.9% (ITT)
80.2% (CE)
81.1% (CE)
In August 2010 draft guidance the FDA adopted changes to the secondary outcomes of
clinical response at the end of therapy (EOT). These were prospectively measured
in Study 112 sensitivity analyses and are captured in the Study 113 SPA.
Clinical Response at EOT*
(Day 11)
Early clinical failures not carried
forward to EOT**
80.7% (ITT)
80.9% (ITT)
87.5% (CE)
87.1% (CE)
Clinical Response at EOT*
(Day 11)
Early clinical failures not carried
forward to EOT** and
presence/absence of patient
reported pain at EOT excluded*/**
87.0% (ITT)
87.8% (ITT)
94.5% (CE)
95.1% (CE)
* Primary and secondary endpoints as agreed to under
Study 112 SPA ** Consistent with FDA draft ABSSSI Guidance
for Industry (August 2010) |
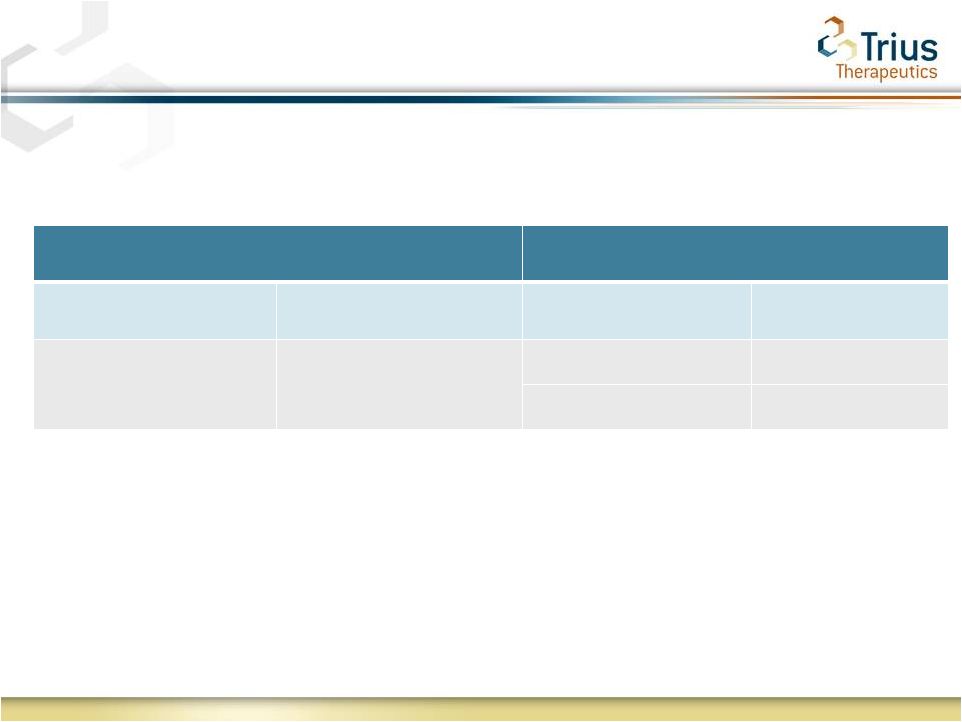 8
EMA Endpoint: A Once-Daily 200mg Dose of Tedizolid for 6 Days
Demonstrates Comparable Efficacy to Twice-Daily 600mg Dose of
Linezolid for 10 Days of Treatment
Secondary Outcome at PTE
Treatment
Secondary Outcome
Criteria
Tedizolid
(200 mg QD 6 days)
Linezolid
(600 mg BID 10 days)
Clinical Response at PTE* (Day
17-24)
Clinician
assessment
at
PTE
85.5% (ITT)
86.0% (ITT)
94.6% (CE)
95.0% (CE)
*
EMA
primary
endpoint
(EMA
Report
on
the
workshop
on
Antibacterials
issued
March
2011).
Captured
in
both
the
Study
112
and
113
SPA |
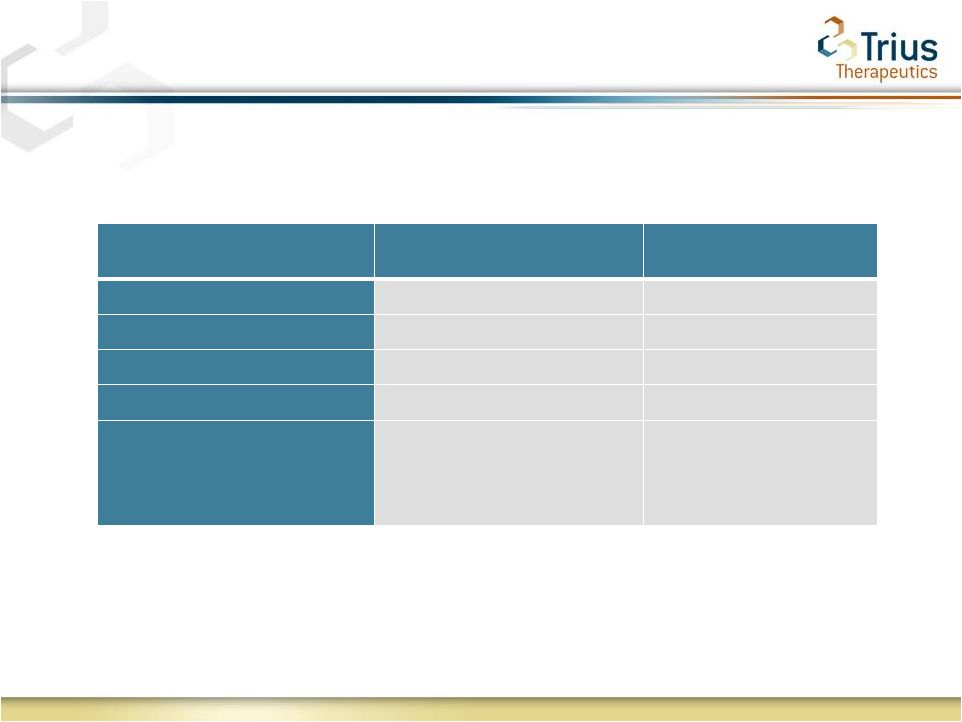 9
Shorter Course of Tedizolid Therapy Shows Comparable
Per Pathogen Clinical Response
Pathogen
Tedizolid
(200mg QD 6 days)
Linezolid
(600mg BID 10 days)
Staphylococcus
spp.
283/295 (96%)
300/302 (99%)
MRSA
73/78 (94%)
74/75 (99%)
MSSA
65/66 (99%)
75/75 (100%)
Other
7/7 (100%)
4/4 (100%)
Streptococcus
spp.
34/35 (97%)
27/29 (93%)
Clinical investigator’s assessment of clinical response at Post Treatment
Evaluation (days 17-24) in the Microbiological Evaluable analysis
set |
 10
Tedizolid was Well Tolerated with a Favorable AE Profile
Compared to Linezolid
Adverse Event
Tedizolid
(200mg QD 6 days)
Linezolid
(600mg BID 10 days)
Any Treatment
Emergent Adverse
Event (TEAE)
40.8%
43.3%
Any Drug Related TEAE
24.2%
31.0%
Gastrointestinal
Disorders*
16.3%**
25.4%
* Gastrointestinal AEs include:
Diarrhea, Nausea, Vomiting and Dyspepsia
** Statistically significant (p=0.004).
No Unexpected Safety Signals
•
Liver enzymes/function tests
•
QTc
Tedizolid had a numerically lower rate of drug-related treatment emergent
adverse events (TEAE) and
a
statistically
significant
lower
number
of
gastrointestinal
adverse
events |
 11
Hematology: Tedizolid had Significantly Lower Impact on
Platelets than Linezolid
Percent of Patients with Value below the
Lower Limit of Normal (LLN)
Hematology Parameter
Tedizolid
(200mg QD 6 days)
Linezolid
(600mg BID 10 days)
Platelets*
Below LLN
9.2%
14.9%
Platelets
-
Substantially
abnormal value
(<75% of LLN)
2.3%
4.9%
* Statistically significant (p=0.038) |
 12
Phase 3 trials conducted under new FDA ABSSSI guidance are
manageable
Study 112 design and outcomes will satisfy both FDA and EMA
regulatory requirements
All efficacy and safety objectives of Study 112 were successfully
achieved
–
Efficacy: All primary and secondary trial endpoints met with a once-daily short
course of therapy
–
Safety: statistically significant lower incidence in key tolerability and safety
parameters
Summary |
 13
Upcoming Milestones
Top line data from 112 Phase 3 ABSSSI trial
SPA for and initiation of Phase 3 pneumonia study
Potential partner for Europe
Initiation of clinical studies for Gyrase
Completion of enrollment for 113 Phase 3 ABSSSI trial |