Attached files
| file | filename |
|---|---|
| EX-23.1 - InspireMD, Inc. | e609111_ex23-1.htm |
| EX-10.43 - InspireMD, Inc. | e609111_ex10-43.htm |
As filed with the Securities and Exchange Commission on December 1, 2011
SEC File No. 333-174948
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
______________________
AMENDMENT NO. 4
TO
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
______________________
|
InspireMD, Inc.
|
||
|
(Exact name of registrant as specified in its charter)
|
||
|
Delaware
|
3841
|
26-2123838
|
|
(State or other jurisdiction of
incorporation or organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer Identification No.)
|
|
3 Menorat Hamaor St.
Tel Aviv, Israel 67448
972-3-691-7691
|
||
|
(Address, including zip code, and telephone number,
including area code, of registrant’s principal executive offices)
|
||
|
Ofir Paz
Chief Executive Officer
InspireMD, Inc.
3 Menorat Hamaor St.
Tel Aviv, Israel 67448
972-3-691-7691
|
||
|
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
|
||
|
Copies of all communications, including communications sent to agent for service, should be sent to:
|
||
|
Rick A. Werner, Esq.
Haynes and Boone, LLP
30 Rockefeller Plaza, 26th Floor
New York, New York 10112
Tel. (212) 659-7300
Fax (212) 884-8234
|
||
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
(Check one):
|
Large accelerated filer o
|
Accelerated filer o
|
|
Non-accelerated filer o
|
Smaller reporting company x
|
|
(Do not check if a smaller reporting company)
|
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED DECEMBER 1, 2011
PRELIMINARY PROSPECTUS

InspireMD, Inc.
414,942 Shares of Common Stock Underlying Warrants
_________________
This prospectus relates to the resale of up to 414,942 shares of our common stock to be offered by the selling stockholders upon the exercise of outstanding common stock purchase warrants by the selling stockholders.
The selling stockholders may sell shares of common stock from time to time in the principal market on which our common stock is traded at the prevailing market price or in privately negotiated transactions. See “Plan of Distribution” which begins on page 60.
We will not receive any of the proceeds from the sale of common stock by the selling stockholders. However, we will generate proceeds in the event of a cash exercise of the warrants by the selling stockholders. We intend to use those proceeds, if any, for general corporate purposes. We will pay the expenses of registering these shares.
All expenses of registration incurred in connection with this offering are being borne by us, but all selling and other expenses incurred by the selling stockholders will be borne by the selling stockholders.
Our common stock is quoted on the regulated quotation service of the OTC Bulletin Board under the symbol “NSPR.OB”. On November 30, 2011, the last reported sale price of our common stock as reported on the OTC Bulletin Board was $2.25 per share.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
Investing in our common stock is highly speculative and involves a high degree of risk. You should carefully consider the risks and uncertainties in the section entitled “Risk Factors” beginning on page 4 of this prospectus before making a decision to purchase our stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2011
|
Page
|
|
|
1
|
|
|
5
|
|
|
19
|
|
|
19
|
|
|
20
|
|
|
20
|
|
|
20
|
|
|
27
|
|
|
44
|
|
|
47
|
|
|
50
|
|
| 51 | |
| 55 | |
| 55 | |
|
60
|
|
| 61 | |
|
61
|
|
|
61
|
|
|
F-1
|
You should rely only on the information contained in this prospectus. We have not authorized any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to sell these securities in any jurisdiction where offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
|
PROSPECTUS SUMMARY
The following summary highlights information contained elsewhere in this prospectus. It may not contain all the information that may be important to you. You should read this entire prospectus carefully, including the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and our historical financial statements and related notes included elsewhere in this prospectus or any accompanying prospectus supplement before making an investment decision. In this prospectus, unless the context requires otherwise, all references to “we,” “our” and “us” for periods prior to the closing of our share exchange transactions on March 31, 2011 refer to InspireMD Ltd., a private company incorporated under the laws of the State of Israel that is now our wholly-owned subsidiary, and its subsidiary, and references to “we,” “our” and “us” for periods subsequent to the closing of the share exchange transactions refer to InspireMD, Inc., a publicly traded Delaware corporation, and its direct and indirect subsidiaries, including InspireMD Ltd.
Overview
We are an innovative medical device company focusing on the development and commercialization of our proprietary stent platform technology, MGuard™. MGuard™ provides embolic protection in stenting procedures by placing a micron mesh sleeve over a stent (see photograph below of an MGuard™ Stent). Our initial products are marketed for use mainly in patients with acute coronary syndromes, notably acute myocardial infarction (heart attack) and saphenous vein graft coronary interventions (bypass surgery). According to the TYPHOON STEMI trial (New England Journal of Medicine, 2006) and the SOS SVG Trial (Journal of the American College of Cardiology, 2009), of patients with acute myocardial infarction and saphenous vein graft coronary interventions, 7.5% to 44% experience major adverse cardiac events, including cardiac death, heart attack, and restenting of the artery. When performing stenting procedures in patients with acute coronary symptoms, interventional cardiologists face a difficult dilemma in choosing between bare-metal stents, which have a high rate of restenosis (formation of new blockages), and drug-eluting (drug-coated) stents, which have a high rate of late thrombosis (formation of clots months or years after implantation), require administration of anti-platelet drugs for at least one year post procedure, are more costly than bare-metal stents and have additional side effects. We believe that MGuard™ is a simple, seamless and complete solution for these patients. For the year ended December 31, 2010, our total revenue was approximately $4.9 million and our net loss was approximately $3.4 million. For the nine months ended September 30, 2011, our total revenue was $4.7 million and our net loss was approximately $6.4 million.
MGuardTM Sleeve – Microscopic View
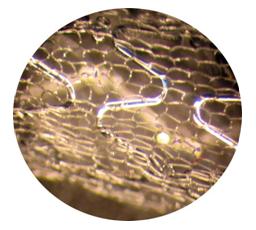 We intend to use our MGuard™ technology in a broad range of coronary related situations in which complex lesions are required and make it an industry standard for treatment of acute coronary syndromes. We believe that patients will benefit from a cost-effective alternative with a greater clinical efficacy and safety profile than other stent technologies. We believe that with our MGuard™ technology, we are well positioned to emerge as a key player in the global stent market.
We also intend to apply our technology to develop additional products used for other vascular procedures, specifically carotid (the arteries that supply blood to the brain) and peripheral (other arteries) procedures.
|
||||||
|
In October 2007, our first generation product, the MGuard™ Coronary, received CE Mark approval for treatment of coronary arterial disease in the European Union. CE Mark is a mandatory conformance mark on many products marketed in the European Economic Area and certifies that a product has met European Union consumer safety, health or environmental requirements. We began shipping our product to customers in Europe in January 2008 and have since expanded our global distribution network to Canada, Southeast Asia, India and Latin America.
Our initial MGuard™ products incorporated a stainless steel stent. We replaced this stainless steel platform with a more advanced cobalt-chromium based platform, which we refer to as MGuard Prime™. We believe the new platform will be superior because cobalt-chromium stents are generally known in the industry to provide better deliverability and possibly even a reduction in major adverse cardiac events. In particular, according to Jabara, et. al. (“A Third Generation Ultra-thin Strut Cobalt Chromium Stent: Histopathological Evaluation in Porcine Coronary Arteries,” EuroIntervention, November 2009), due to its greater density, cobalt-chromium enables the construction of stents that have both thinner struts and similar radial strength as stainless steel, with its thicker struts. In turn, Jabara, et. al. found that the reduced thickness of the struts provides more flexibility and lower crossing profiles, thereby reducing the inflammatory response and neointimal thickening, potentially lowering restenosis and target vessel revascularization rates.
MGuard Prime™ received CE Mark approval in the European Union in October 2010 for improving luminal diameter and providing embolic protection. We believe we can use and leverage the MGuard™ clinical trial results to market MGuard Prime™. However, we face a number of challenges to the further growth of MGuard™. For example, we face competition from numerous pharmaceutical and biotechnology companies in the therapeutics area, as well as competition from academic institutions, government agencies and research institutions. Most of our current and potential competitors have, and will continue to have, substantially greater financial, technological, research and development, regulatory and clinical, manufacturing, marketing and sales, distribution and personnel resources than we do. In addition, none of our products are currently approved by the U.S. Food and Drug Administration. Clinical trials necessary to support a pre-market approval application to the U.S. Food and Drug Administration for our MGuard™ stent will be expensive and will require the enrollment of a large number of patients, and suitable patients may be difficult to identify and recruit, which may cause a delay in the development and commercialization of our product candidates. Furthermore, our rights to our intellectual property with respect to our products could be challenged. Based on the prolific litigation that has occurred in the stent industry and the fact that we may pose a competitive threat to some large and well-capitalized companies that own or control patents relating to stents and their use, manufacture and delivery, we believe that it is possible that one or more third parties will assert a patent infringement claim against the manufacture, use or sale of our MGuard™ stent based on one or more of these patents. Additionally, there is a strong preference to use drug-eluting stents in some countries. Over the last decade, there has been an increasing tendency to use drug-eluting stents in percutaneous coronary intervention (PCI), commonly known as angioplasty (a therapeutic procedure to treat narrowed coronary arteries of the heart found in patients with heart disease), with a usage rate of drug-eluting stents in PCI approaching 70-80% in some countries, even though drug-eluting stents do not address thrombus management in acute myocardial infarction. Also, the use of other bare-metal stents is preferred over the use of MGuard™ products in certain circumstances, such as when placing the stent at the entrance to large side branches, known as jailing large side branches. Unless otherwise indicated, in this prospectus, references to MGuard™ are to both our initial product, MGuard™, and MGuard Prime™, as applicable.
Recent Events
On October 31, 2011, our stockholders authorized our board of directors to amend our amended and restated certificate of incorporation to effect a reverse stock split of our common stock at a ratio of one-for-two to one-for-four, at any time prior to our 2012 annual stockholders’ meeting, the exact ratio of the reverse stock split to be determined by the board. As of the date of this prospectus, we have not effected the reverse stock split and, as such, the information with respect to our common stock in this prospectus and the accompanying financial statements and related notes does not give effect to any reverse stock split.
On October 4, 2011, InspireMD Ltd., our wholly-owned subsidiary, entered into a clinical trial services agreement with Harvard Clinical Research Institute, Inc., pursuant to which Harvard Clinical Research Institute, Inc. will conduct a study entitled “MGuard Stent System Clinical Trial in Patients with Acute Myocardial Infarction” on our behalf. We will pay Harvard Clinical Research Institute, Inc. an estimated fee of approximately $10 million for conducting the study, subject to adjustment dependent upon changes in the scope and nature of the study, as well as other costs to be determined by the parties.
|
||
|
|
|||
|
On March 31, 2011, we completed a series of share exchange transactions pursuant to which we issued the shareholders of InspireMD Ltd. 50,666,663 shares of common stock in exchange for all of InspireMD Ltd.’s issued and outstanding ordinary shares, resulting in the former shareholders of InspireMD Ltd. holding a controlling interest in us and InspireMD Ltd. becoming our wholly-owned subsidiary.
Immediately following the share exchange transactions, we transferred all of our pre-share exchange operating assets and liabilities to our wholly-owned subsidiary, Saguaro Holdings, Inc., a Delaware corporation, and transferred all of Saguaro Holdings, Inc.’s outstanding capital stock to Lynn Briggs, our then-majority stockholder and our former president, chief executive officer, chief financial officer, secretary-treasurer and sole director, in exchange for the cancellation of 7,500,000 shares of our common stock held by Ms. Briggs.
After the share exchange transactions and the divestiture of our pre-share exchange operating assets and liabilities, we succeeded to the business of InspireMD Ltd. as our sole line of business, and all of our then-current officers and directors resigned and were replaced by some of the officers and directors of InspireMD Ltd.
Contemporaneously with the foregoing transactions, we completed a private placement pursuant to which we sold 6,454,002 shares of common stock and five-year warrants to purchase up to 3,226,999 shares of common stock at an exercise price of $1.80 per share for aggregate cash proceeds of $9,013,404 and the cancellation of $667,596 of indebtedness held by investors. In addition, on April 18, 2011 and April 21, 2011, we completed private placements pursuant to which we sold an aggregate of 983,334 shares of common stock and five-year warrants to purchase up to 491,667 shares of common stock at an exercise price of $1.80 per share for aggregate cash proceeds of $1,475,000.
Before the share exchange transactions, our corporate name was Saguaro Resources, Inc., and our trading symbol was SAGU.OB. On March 28, 2011, we changed our corporate name to InspireMD, Inc. and on April 11, 2011 our trading symbol was changed to NSPR.OB.
The Offering
|
|||
|
Common stock offered by the selling stockholders:
|
414,942 shares of our common stock to be offered by the selling stockholders upon the exercise of outstanding common stock purchase warrants.
|
||
|
Common stock outstanding prior to the offering:
|
68,178,947
|
||
|
Common stock outstanding after this offering:
|
68,593,889 (1)
|
||
|
Use of proceeds:
|
We will not receive any proceeds from the sale of the common stock offered by the selling stockholders. However, we will generate proceeds in the event of a cash exercise of the warrants by the selling stockholders. We intend to use those proceeds, if any, for general corporate purposes.
|
||
|
Offering Price:
|
All or part of the shares of common stock offered hereby may be sold from time to time in amounts and on terms to be determined by the selling stockholders at the time of sale.
|
||
|
OTC Bulletin Board symbol:
|
NSPR.OB
|
||
|
Risk factors:
|
You should carefully consider the information set forth in this prospectus and, in particular, the specific factors set forth in the “Risk Factors” section beginning on page 5 of this prospectus before deciding whether or not to invest in shares of our common stock.
|
||
|
________________
|
||||||
|
(1)
|
The number of shares of common stock outstanding after the offering is based upon 68,178,947 shares outstanding as of November 30, 2011 and assumes the exercise of all warrants with respect to those shares being registered for resale pursuant to the registration statement of which this prospectus forms a part.
|
|||||
|
The number of shares of common stock outstanding after this offering excludes:
|
||||||
|
·
|
7,723,583 shares of common stock issuable upon the exercise of currently outstanding warrants with exercise prices ranging from $1.23 to $1.80 per share and having a weighted average exercise price of $1.63 per share;
|
|||||
|
·
|
12,298,587 shares of common stock issuable upon the exercise of currently outstanding options with exercise prices ranging from $0.0 to $2.60 and having a weighted average exercise price of $1.09 per share; and
|
|||||
|
·
|
6,684,047 shares of common stock available for future issuance under our 2011 UMBRELLA Option Plan.
|
|||||
Investing in our common stock involves a high degree of risk. Before investing in our common stock, you should carefully consider the risks described below and the financial and other information included in this prospectus. If any of the following risks, or any other risks not described below, actually occur, it is likely that our business, financial condition, and/or operating results could be materially adversely affected. In such case, the trading price and market value of our common stock could decline and you may lose part or all of your investment in our common stock. The risks and uncertainties described below include forward-looking statements and our actual results may differ from those discussed in these forward-looking statements.
Risks Related to Our Business
We expect to derive our revenue from sales of our MGuardTM stent products and other products we may develop. If we fail to generate revenue from this source, our results of operations and the value of our business would be materially and adversely affected.
We expect our revenue to be generated from sales of our MGuard™ stent products and other products we may develop. Future sales of these products, if any, will be subject to the receipt of regulatory approvals and commercial and market uncertainties that may be outside our control. If we fail to generate such revenues, our results of operations and the value of our business and securities could be materially and adversely affected.
If we are unable to obtain and maintain intellectual property protection covering our products, others may be able to make, use or sell our products, which would adversely affect our revenue.
Our ability to protect our products from unauthorized or infringing use by third parties depends substantially on our ability to obtain and maintain valid and enforceable patents. Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering medical devices and pharmaceutical inventions and the scope of claims made under these patents, our ability to enforce patents is uncertain and involves complex legal and factual questions. Accordingly, rights under any of our pending patents may not provide us with commercially meaningful protection for our products or afford a commercial advantage against our competitors or their competitive products or processes. In addition, patents may not be issued from any pending or future patent applications owned by or licensed to us, and moreover, patents that may be issued to us in the future may not be valid or enforceable. Further, even if valid and enforceable, our patents may not be sufficiently broad to prevent others from marketing products like ours, despite our patent rights.
The validity of our patent claims depends, in part, on whether prior art references exist that describe or render obvious our inventions as of the filing date of our patent applications. We may not have identified all prior art, such as U.S. and foreign patents or published applications or published scientific literature, that could adversely affect the patentability of our pending patent applications. For example, patent applications in the U.S. are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the U.S. Patent and Trademark Office for the entire time prior to issuance as a U.S. patent. Patent applications filed in countries outside the U.S. are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in the scientific or patent literature often lags behind actual discoveries. Therefore, we cannot be certain that we were the first to invent, or the first to file patent applications relating to, our stent technologies. In the event that a third party has also filed a U.S. patent application covering our stents or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the U.S. Patent and Trademark Office to determine priority of invention in the U.S. It is possible that we may be unsuccessful in the interference, resulting in a loss of some portion or all of our position in the U.S. The laws of some foreign jurisdictions do not protect intellectual property rights to the same degree as in the U.S., and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties or are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
We may initiate litigation to enforce our patent rights on any patents issued on pending patent applications, which may prompt adversaries in such litigation to challenge the validity, scope or enforceability of our patents. If a court decides that such patents are not valid, not enforceable or of a limited scope, we may not have the right to stop others from using our inventions. Also, even if our patents are determined by a court to be valid and enforceable, they may not be sufficiently broad to prevent others from marketing products similar to ours or designing around our patents, despite our patent rights, nor provide us with freedom to operate unimpeded by the patent rights of others.
We also rely on trade secret protection to protect our interests in proprietary know-how and for processes for which patents are difficult to obtain or enforce. We may not be able to protect our trade secrets adequately. In addition, we rely on non-disclosure and confidentiality agreements with employees, consultants and other parties to protect, in part, trade secrets and other proprietary technology. These agreements may be breached and we may not have adequate remedies for any breach. Moreover, others may independently develop equivalent proprietary information, and third parties may otherwise gain access to our trade secrets and proprietary knowledge. Any disclosure of confidential data into the public domain or to third parties could allow competitors to learn our trade secrets and use the information in competition against us.
We have a history of net losses and may experience future losses
To date, we have experienced net losses. A substantial portion of the expenses associated with our manufacturing facilities are fixed in nature (i.e., depreciation) and will reduce our operating margin until such time, if ever, as we are able to increase utilization of our capacity through increased sales of our products. The clinical trials necessary to support our anticipated growth will be expensive and lengthy. In addition, our strategic plan will require a significant investment in clinical trials, product development and sales and marketing programs, which may not result in the accelerated revenue growth that we anticipate. As a result, there can be no assurance that we will ever generate substantial revenues or sustain profitability.
We have limited manufacturing capabilities and manufacturing personnel, and if our manufacturing facilities are unable to provide an adequate supply of products, our growth could be limited and our business could be harmed.
We currently manufacture our MGuard™ stent at our facilities in Tel Aviv, Israel, and we have contracted with QualiMed Innovative Medizinprodukte GmbH, a German manufacturer, to assist in production. If there were a disruption to our existing manufacturing facility, we would have no other means of manufacturing our MGuard™ stent until we were able to restore the manufacturing capability at our facility or develop alternative manufacturing facilities. If we were unable to produce sufficient quantities of our MGuard™ stent for use in our current and planned clinical trials, or if our manufacturing process yields substandard stents, our development and commercialization efforts would be delayed.
We currently have limited resources, facilities and experience to commercially manufacture our product candidates. In order to produce our MGuard™ stent in the quantities that we anticipate will be required to meet anticipated market demand, we will need to increase, or “scale up,” the production process by a significant factor over the current level of production. There are technical challenges to scaling-up manufacturing capacity, and developing commercial-scale manufacturing facilities will require the investment of substantial funds and hiring and retaining additional management and technical personnel who have the necessary manufacturing experience. We may not successfully complete any required scale-up in a timely manner or at all. If unable to do so, we may not be able to produce our MGuard™ stent in sufficient quantities to meet the requirements for the launch of the product or to meet future demand, if at all. If we develop and obtain regulatory approval for our MGuard™ stent and are unable to manufacture a sufficient supply of our MGuard™ stent, our revenues, business and financial prospects would be adversely affected. In addition, if the scaled-up production process is not efficient or produces stents that do not meet quality and other standards, our future gross margins may decline. Also, our current and planned personnel, systems, procedures and controls may not be adequate to support our anticipated growth. If we are unable to manage our growth effectively, our business could be harmed.
Additionally, any damage to or destruction of our Tel Aviv facilities or its equipment, prolonged power outage or contamination at our facility would significantly impair our ability to produce MGuard™ stents.
Finally, the production of our MGuard™ stent must occur in a highly controlled, clean environment to minimize particles and other yield and quality-limiting contaminants. In spite of stringent quality controls, weaknesses in process control or minute impurities in materials may cause a substantial percentage of defective products in a lot. If we are unable to maintain stringent quality controls, or if contamination problems arise, our clinical development and commercialization efforts could be delayed, which would harm our business and results of operations.
Clinical trials necessary to support a pre-market approval application will be lengthy and expensive and will require the enrollment of a large number of patients, and suitable patients may be difficult to identify and recruit. Any such delay or failure of clinical trials could prevent us from commercializing our stent products, which would materially and adversely affect our results of operations and the value of our business.
Clinical trials necessary to support a pre-market approval application to the U.S. Food and Drug Administration for our MGuard™ stent will be expensive and will require the enrollment of a large number of patients, and suitable patients may be difficult to identify and recruit, which may cause a delay in the development and commercialization of our product candidates. Clinical trials supporting a pre-market approval applications for the Cypher stent developed by Johnson & Johnson and the Taxus Express2 stent developed by Boston Scientific Corporation, which were approved by the U.S. Food and Drug Administration and are currently marketed, involved patient populations of approximately 1,000 and 1,300, respectively, and a 12-month follow up period. In some trials, a greater number of patients and a longer follow up period may be required. The U.S. Food and Drug Administration may require us to submit data on a greater number of patients or for a longer follow-up period than those for pre-market approval applications for the Cypher stent and the Taxus Express2 stent. Patient enrollment in clinical trials and the ability to successfully complete patient follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites, the eligibility criteria for the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and efficacy of our products, or they may be persuaded to participate in contemporaneous clinical trials of competitive products. In addition, patients participating in our clinical trials may die before completion of the trial or suffer adverse medical events unrelated to or related to our products. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may cause an increase in costs and delays or result in the failure of the clinical trial.
In addition, the length of time required to complete clinical trials for pharmaceutical and medical device products varies substantially according to the degree of regulation and the type, complexity, novelty and intended use of a product, and can continue for several years and cost millions of dollars. The commencement and completion of clinical trials for our products under development may be delayed by many factors, including governmental or regulatory delays and changes in regulatory requirements, policy and guidelines or our inability or the inability of any potential licensee to manufacture or obtain from third parties materials sufficient for use in preclinical studies and clinical trials.
Physicians may not widely adopt the MGuard™ stent unless they determine, based on experience, long-term clinical data and published peer reviewed journal articles, that the use of the MGuard™ stent provides a safe and effective alternative to other existing treatments for coronary artery disease.
We believe that physicians will not widely adopt the MGuard™ stent unless they determine, based on experience, long-term clinical data and published peer reviewed journal articles, that the use of our MGuard™ stent provides a safe and effective alternative to other existing treatments for coronary artery disease, including coronary artery bypass grafting balloon angioplasty, bare-metal stents and other drug-eluting stents, provided by Johnson & Johnson, Boston Scientific Corporation, Medtronic Inc., Abbott Laboratories and others.
We cannot provide any assurance that the data collected from our current and planned clinical trials will be sufficient to demonstrate that the MGuard™ stents are an attractive alternative to other procedures. If we fail to demonstrate safety and efficacy that is at least comparable to other drug-eluting stents or bare-metal stents that have received regulatory approval and that are available on the market, our ability to successfully market the MGuard™ stent will be significantly limited. Even if the data collected from clinical studies or clinical experience indicate positive results, each physician’s actual experience with our MGuard™ stent will vary. Clinical trials conducted with the MGuard™ stent have involved procedures performed by physicians who are technically proficient and are high-volume stent users. Consequently, both short-term and long-term results reported in these clinical trials may be significantly more favorable than typical results of practicing physicians, which could negatively affect rates of adoptions of our products. We also believe that published peer-reviewed journal articles and recommendations and support by influential physicians regarding our MGuard™ stent will be important for market acceptance and adoption, and we cannot assure you that we will receive these recommendations and support, or that supportive articles will be published.
In addition, currently, physicians consider drug-eluting stents to be the industry standard for treatment of coronary artery disease. While we believe that the MGuard™ stent is a safe and effective alternative, it is not a drug-eluting stent, which may further hinder its support and adoption by physicians.
Our products are based on a new technology, and we have only limited experience in regulatory affairs, which may affect our ability or the time required to navigate complex regulatory requirements and obtain necessary regulatory approvals, if such approvals are received at all. Regulatory delays or denials may increase our costs, cause us to lose revenue and materially and adversely affect our results of operations and the value of our business.
Because our products are new and long-term success measures have not been completely validated, regulatory agencies, including the U.S. Food and Drug Administration, may take a significant amount of time in evaluating product approval applications. For example, there are currently several methods of measuring restenosis and we do not know which of these metrics, or combination of these metrics, will be considered appropriate by the U.S. Food and Drug Administration for evaluating the clinical efficacy of stents. Treatments may exhibit a favorable measure using one of these metrics and an unfavorable measure using another metric. Any change in the accepted metrics may result in reconfiguration of, and delays in, our clinical trials. Additionally, we have only limited experience in filing and prosecuting the applications necessary to gain regulatory approvals, and our clinical, regulatory and quality assurance personnel are currently composed of only 5 employees. As a result, we may experience a long regulatory process in connection with obtaining regulatory approvals for our products.
In addition, the products we and any potential licensees license, develop, manufacture and market are subject to complex regulatory requirements, particularly in the U.S., Europe and Asia, which can be costly and time-consuming. There can be no assurance that such approvals will be granted on a timely basis, if at all. Furthermore, there can be no assurance of continued compliance with all regulatory requirements necessary for the manufacture, marketing and sale of the products we will offer in each market where such products are expected to be sold, or that products we have commercialized will continue to comply with applicable regulatory requirements. If a government regulatory agency were to conclude that we were not in compliance with applicable laws or regulations, the agency could institute proceedings to detain or seize our products, issue a recall, impose operating restrictions, enjoin future violations and assess civil and criminal penalties against us, our officers or employees and could recommend criminal prosecution. Furthermore, regulators may proceed to ban, or request the recall, repair, replacement or refund of the cost of, any device manufactured or sold by us. Furthermore, there can be no assurance that all necessary regulatory approvals will be obtained for the manufacture, marketing and sale in any market of any new product developed or that any potential licensee will develop using our licensed technology.
Even if our products are approved by regulatory authorities, if we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval in the U.S., along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continual review and periodic inspections by the U.S. Food and Drug Administration and other regulatory bodies. In particular, we and our suppliers will be required to comply with the U.S. Food and Drug Administration’s Quality System Regulation for the manufacture of our MGuard™ stent, which covers the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain marketing approval in the U.S. The U.S. Food and Drug Administration enforces the Quality System Regulation through unannounced inspections. We and our third-party manufacturers and suppliers have not yet been inspected by the U.S. Food and Drug Administration and will have to successfully complete such inspections before we receive U.S. regulatory approval for our products. Failure by us or one of our suppliers to comply with statutes and regulations administered by the U.S. Food and Drug Administration and other regulatory bodies, or failure to take adequate response to any observations, could result in, among other things, any of the following enforcement actions:
|
|
·
|
warning letters or untitled letters;
|
|
|
·
|
fines and civil penalties;
|
|
|
·
|
unanticipated expenditures;
|
|
|
·
|
delays in approving, or refusal to approve, our products;
|
|
|
·
|
withdrawal or suspension of approval by the U.S. Food and Drug Administration or other regulatory bodies;
|
|
|
·
|
product recall or seizure;
|
|
|
·
|
orders for physician notification or device repair, replacement or refund;
|
|
|
·
|
interruption of production;
|
|
|
·
|
operating restrictions;
|
|
|
·
|
injunctions; and
|
|
|
·
|
criminal prosecution.
|
If any of these actions were to occur, it could harm our reputation and could cause our product sales and profitability to suffer. Furthermore, key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements.
Even if regulatory approval of a product is granted in the U.S., the approval may be subject to limitations on the indicated uses for which the product may be marketed. If the U.S. Food and Drug Administration determines that our promotional materials, training or other activities constitutes promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
Moreover, any modification to a device that has received U.S. Food and Drug Administration approval that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, design or manufacture, requires a new approval from the U.S. Food and Drug Administration. If the U.S. Food and Drug Administration disagrees with any determination by us that new approval is not required, we may be required to cease marketing or to recall the modified product until approval is obtained. In addition, we could also be subject to significant regulatory fines or penalties.
Additionally, we may be required to conduct costly post-market testing and surveillance to monitor the safety or efficacy of our products, and we will be required to report adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements, such as Quality System Regulation, may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
Further, healthcare laws and regulations may change significantly in the future. Any new healthcare laws or regulations may adversely affect our business. A review of our business by courts or regulatory authorities may result in a determination that could adversely affect our operations. In addition, the healthcare regulatory environment may change in a way that restricts our operations.
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products in such jurisdictions.
We intend to market our products in international markets. In order to market our products in other foreign jurisdictions, we must obtain separate regulatory approvals from those obtained in the U.S. and Europe. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain CE Mark or U.S. Food and Drug Administration approval. Foreign regulatory approval processes may include all of the risks associated with obtaining CE Mark or U.S. Food and Drug Administration approval in addition to other risks. We may not obtain foreign regulatory approvals on a timely basis, if at all. CE Mark does not ensure approval by regulatory authorities in other countries. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in certain markets.
We operate in an intensely competitive and rapidly changing business environment, and there is a substantial risk our products could become obsolete or uncompetitive.
The medical device market is highly competitive. We compete with many medical service companies in the U.S. and internationally in connection with our current product and products under development. We face competition from numerous pharmaceutical and biotechnology companies in the therapeutics area, as well as competition from academic institutions, government agencies and research institutions. When we commercialize our products, we expect to face intense competition from Cordis Corporation, a subsidiary of Johnson & Johnson, Boston Scientific Corporation, Guidant, Medtronic, Inc., Abbott Vascular Devices, Terumo and others. Most of our current and potential competitors, including but not limited to those listed above, have, and will continue to have, substantially greater financial, technological, research and development, regulatory and clinical, manufacturing, marketing and sales, distribution and personnel resources than we do. There can be no assurance that we will have sufficient resources to successfully commercialize our products, if and when they are approved for sale. The worldwide market for stent products is characterized by intensive development efforts and rapidly advancing technology. Our future success will depend largely upon our ability to anticipate and keep pace with those developments and advances. Current or future competitors could develop alternative technologies, products or materials that are more effective, easier to use or more economical than what we or any potential licensee develop. If our technologies or products become obsolete or uncompetitive, our related product sales and licensing revenue would decrease. This would have a material adverse effect on our business, financial condition and results of operations.
We may become subject to claims by much larger and better capitalized competitors seeking to invalidate our right to our intellectual property.
Based on the prolific litigation that has occurred in the stent industry and the fact that we may pose a competitive threat to some large and well-capitalized companies that own or control patents relating to stents and their use, manufacture and delivery, we believe that it is possible that one or more third parties will assert a patent infringement claim against the manufacture, use or sale of our MGuard™ stent based on one or more of these patents. It is also possible that a lawsuit asserting patent infringement and related claims may have already been filed against us of which we are not aware. A number of these patents are owned by very large and well-capitalized companies that are active participants in the stent market. As the number of competitors in the stent market grows, the possibility of patent infringement by us, or a patent infringement claim against us, increases.
These companies have maintained their position in the market by, among other things, establishing intellectual property rights relating to their products and enforcing these rights aggressively against their competitors and new entrants into the market. All of the major companies in the stent and related markets, including Boston Scientific Corporation, Johnson & Johnson and Medtronic, Inc., have been repeatedly involved in patent litigation relating to stents since at least 1997. The stent and related markets have experienced rapid technological change and obsolescence in the past, and our competitors have strong incentives to stop or delay the introduction of new products and technologies. We may pose a competitive threat to many of the companies in the stent and related markets. Accordingly, many of these companies will have a strong incentive to take steps, through patent litigation or otherwise, to prevent us from commercializing our products.
If we fail to maintain or establish satisfactory agreements with suppliers, we may not be able to obtain materials that are necessary to develop our products.
We depend on outside suppliers for certain raw materials. These raw materials or components may not always be available at our standards or on acceptable terms, if at all, and we may be unable to locate alternative suppliers or produce necessary materials or components on our own.
Some of the components of our products are currently provided by only one vendor, or a single-source supplier. We depend on QualiMed Innovative Medizinprodukte GmbH, which manufactures the body of the stent, MeKo Laserstrahl-Materialbearbeitung for the laser cutting of the stent, Natec Medical Ltd. for the supply of catheters and Biogeneral Inc. for the fiber. We may have difficulty obtaining similar components from other suppliers that are acceptable to the U.S. Food and Drug Administration or foreign regulatory authorities if it becomes necessary.
If we have to switch to a replacement supplier, we will face additional regulatory delays and the interruption of the manufacture and delivery of our MGuard™ stent for an extended period of time, which would delay completion of our clinical trials or commercialization of our products. In addition, we will be required to obtain prior regulatory approval from the U.S. Food and Drug Administration or foreign regulatory authorities to use different suppliers or components that may not be as safe or as effective. As a result, regulatory approval of our products may not be received on a timely basis or at all.
We may be exposed to product liability claims and insurance may not be sufficient to cover these claims.
We may be exposed to product liability claims based on the use of any of our products, or products incorporating our licensed technology, in clinical trials. We may also be exposed to product liability claims based on the sale of any such products following the receipt of regulatory approval. Product liability claims could be asserted directly by consumers, health-care providers or others. We have obtained product liability insurance coverage; however such insurance may not provide full coverage for our future clinical trials, products to be sold, and other aspects of our business. We also have liability insurance for our ongoing clinical trial in Europe. Insurance coverage is becoming increasingly expensive and we may not be able to maintain current coverages, or expand our insurance coverage to include future clinical trials or the sale of products incorporating our licensed technology if marketing approval is obtained for such products, at a reasonable cost or in sufficient amounts to protect against losses due to product liability or at all. A successful product liability claim or series of claims brought against us could result in judgments, fines, damages and liabilities that could have a material adverse effect on our business, financial condition and results of operations. We may incur significant expense investigating and defending these claims, even if they do not result in liability. Moreover, even if no judgments, fines, damages or liabilities are imposed on us, our reputation could suffer, which could have a material adverse effect on our business, financial condition and results of operations.
We may implement a product recall or voluntary market withdrawal due to product defects or product enhancements and modifications, which would significantly increase our costs.
The manufacturing and marketing of our MGuard™ stent products involves an inherent risk that our products may prove to be defective. In that event, we may voluntarily implement a recall or market withdrawal or may be required to do so by a regulatory authority. A recall of one of our products, or a similar product manufactured by another manufacturer, could impair sales of the products we market as a result of confusion concerning the scope of the recall or as a result of the damage to our reputation for quality and safety.
The successful management of operations depends on our ability to attract and retain talented personnel.
We depend on the expertise of our senior management and research personnel, including our chief executive officer, Ofir Paz, and president, Asher Holzer, each of whom would be difficult to replace. The loss of the services of any of our senior management could compromise our ability to achieve our objectives. Furthermore, recruiting and retaining qualified personnel will be crucial to future success. There can be no assurance that we will be able to attract and retain necessary personnel on acceptable terms given the competition among medical device, biotechnology, pharmaceutical and healthcare companies, universities and non-profit research institutions for experienced management, scientists, researchers, and sales and marketing and manufacturing personnel. If we are unable to attract, retain and motivate our key personnel, our operations may be jeopardized and our results of operations may be materially and adversely affected.
We are an international business, and we are exposed to various global and local risks that could have a material adverse effect on our financial condition and results of operations.
We operate globally and develop and manufacture products in our research and manufacturing facilities in multiple countries. Consequently, we face complex legal and regulatory requirements in multiple jurisdictions, which may expose us to certain financial and other risks. International sales and operations are subject to a variety of risks, including:
|
|
·
|
foreign currency exchange rate fluctuations;
|
|
|
·
|
greater difficulty in staffing and managing foreign operations;
|
|
|
·
|
greater risk of uncollectible accounts;
|
|
|
·
|
longer collection cycles;
|
|
|
·
|
logistical and communications challenges;
|
|
|
·
|
potential adverse changes in laws and regulatory practices, including export license requirements, trade barriers, tariffs and tax laws;
|
|
|
·
|
changes in labor conditions;
|
|
|
·
|
burdens and costs of compliance with a variety of foreign laws;
|
|
|
·
|
political and economic instability;
|
|
|
·
|
increases in duties and taxation;
|
|
|
·
|
foreign tax laws and potential increased costs associated with overlapping tax structures;
|
|
|
·
|
greater difficulty in protecting intellectual property; and
|
|
|
·
|
general economic and political conditions in these foreign markets.
|
International markets are also affected by economic pressure to contain reimbursement levels and healthcare costs. Profitability from international operations may be limited by risks and uncertainties related to regional economic conditions, regulatory and reimbursement approvals, competing products, infrastructure development, intellectual property rights protection and our ability to implement our overall business strategy. We expect these risks will increase as we pursue our strategy to expand operations into new geographic markets. We may not succeed in developing and implementing effective policies and strategies in each location where we conduct business. Any failure to do so may harm our business, results of operations and financial condition.
If we fail to obtain an adequate level of reimbursement for our products by third party payors, there may be no commercially viable markets for our product candidates or the markets may be much smaller than expected.
The availability and levels of reimbursement by governmental and other third party payors affect the market for our product candidates. The efficacy, safety, performance and cost-effectiveness of our product candidates and of any competing products will determine the availability and level of reimbursement. Reimbursement and healthcare payment systems in international markets vary significantly by country, and include both government sponsored healthcare and private insurance. To obtain reimbursement or pricing approval in some countries, we may be required to produce clinical data, which may involve one or more clinical trials, that compares the cost-effectiveness of our products to other available therapies. We may not obtain international reimbursement or pricing approvals in a timely manner, if at all. Our failure to receive international reimbursement or pricing approvals would negatively impact market acceptance of our products in the international markets in which those approvals are sought.
We believe that future reimbursement may be subject to increased restrictions both in the U.S. and in international markets. There is increasing pressure by governments worldwide to contain health care costs by limiting both the coverage and the level of reimbursement for therapeutic products and by refusing, in some cases, to provide any coverage for products that have not been approved by the relevant regulatory agency. Future legislation, regulation or reimbursement policies of third party payors may adversely affect the demand for our products currently under development and limit our ability to sell our product candidates on a profitable basis. In addition, third party payors continually attempt to contain or reduce the costs of healthcare by challenging the prices charged for healthcare products and services. If reimbursement for our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, market acceptance of our products would be impaired and future revenues, if any, would be adversely affected.
In the U.S., our business could be significantly and adversely affected by recent healthcare reform legislation and other administration and legislative proposals.
The Patient Protection and Affordable Care Act and Health Care and Educational Reconciliation Act in the U.S. were enacted into law in March 2010. Certain provisions of these acts will not be effective for a number of years and there are many programs and requirements for which the details have not yet been fully established or consequences not fully understood, and it is unclear what the full impacts will be from the legislation. The legislation does levy a 2.3% excise tax on all U.S. medical device sales beginning in 2013. If we commence sales of our MGuard™ stent in the U.S., this new tax may materially and adversely affect our business and results of operations. The legislation also focuses on a number of Medicare provisions aimed at improving quality and decreasing costs. It is uncertain at this point what negative unintended consequences these provisions will have on patient access to new technologies. The Medicare provisions include value-based payment programs, increased funding of comparative effectiveness research, reduced hospital payments for avoidable readmissions and hospital acquired conditions, and pilot programs to evaluate alternative payment methodologies that promote care coordination (such as bundled physician and hospital payments). Additionally, the provisions include a reduction in the annual rate of inflation for hospitals starting in 2011 and the establishment of an independent payment advisory board to recommend ways of reducing the rate of growth in Medicare spending. We cannot predict what healthcare programs and regulations will be ultimately implemented at the federal or state level in the U.S., or the effect of any future legislation or regulation. However, any changes that lower reimbursements for our products or reduce medical procedure volumes could adversely affect our business and results of operations.
Our strategic business plan may not produce the intended growth in revenue and operating income.
Our strategies include making significant investments in sales and marketing programs to achieve revenue growth and margin improvement targets. If we do not achieve the expected benefits from these investments or otherwise fail to execute on our strategic initiatives, we may not achieve the growth improvement we are targeting and our results of operations may be adversely affected.
In addition, as part of our strategy for growth, we may make acquisitions and enter into strategic alliances such as joint ventures and joint development agreements. However, we may not be able to identify suitable acquisition candidates, complete acquisitions or integrate acquisitions successfully, and our strategic alliances may not prove to be successful. In this regard, acquisitions involve numerous risks, including difficulties in the integration of the operations, technologies, services and products of the acquired companies and the diversion of management’s attention from other business concerns. Although our management will endeavor to evaluate the risks inherent in any particular transaction, there can be no assurance that we will properly ascertain all such risks. In addition, acquisitions could result in the incurrence of substantial additional indebtedness and other expenses or in potentially dilutive issuances of equity securities. There can be no assurance that difficulties encountered with acquisitions will not have a material adverse effect on our business, financial condition and results of operations.
We may have violated Israeli securities law.
We may have violated section 15 of the Israeli Security Law of 1968. Section 15 to the Israeli Security Law of 1968 requires the filing of a prospectus with the Israel Security Authority and the delivery thereof to purchasers in connection with an offer or sale of securities to more than 35 parties during any 12 month period. We allegedly issued securities to more than 35 investors during certain 12-month periods, ending in October 2008. Our wholly-owned subsidiary, InspireMD Ltd, a private company incorporated under the laws of the State of Israel, applied for a no-action determination from the Israel Security Authority on February 14, 2011 in connection with the foregoing. To date, the Israel Security Authority has not responded to InspireMD Ltd.’s application for no-action determination and we are unable to predict when a response will be received. The maximum penalties for violating section 15 of the Israeli Security Law of 1968 are as follows: imprisonment of 5 years; a fine of up to approximately $317,000 to be paid by management of the violating company; and a fine of up to approximately $1,590,000 to be paid by the violating company, any of which penalties could result in a material adverse effect on our operations.
We will need to raise additional capital to meet our business requirements in the future and such capital raising may be costly or difficult to obtain and could dilute current stockholders’ ownership interests.
We will need to raise additional capital in the future, which may not be available on reasonable terms or at all. We recently raised approximately $10,500,000 and expect that such proceeds, together with our income, will be insufficient to fully realize all of our business objectives. For instance, we will need to raise additional funds to accomplish the following:
|
●
|
pursuing growth opportunities, including more rapid expansion;
|
|
●
|
acquiring complementary businesses;
|
|
●
|
making capital improvements to improve our infrastructure;
|
|
●
|
hiring qualified management and key employees;
|
|
●
|
developing new services, programming or products;
|
|
●
|
responding to competitive pressures;
|
|
●
|
complying with regulatory requirements such as licensing and registration; and
|
|
●
|
maintaining compliance with applicable laws.
|
Any additional capital raised through the sale of equity or equity backed securities may dilute current stockholders’ ownership percentages and could also result in a decrease in the market value of our equity securities.
The terms of any securities issued by us in future capital transactions may be more favorable to new investors, and may include preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect on the holders of any of our securities then outstanding.
Furthermore, any additional debt or equity financing that we may need may not be available on terms favorable to us, or at all. If we are unable to obtain such additional financing on a timely basis, we may have to curtail our development activities and growth plans and/or be forced to sell assets, perhaps on unfavorable terms, which would have a material adverse effect on our business, financial condition and results of operations, and ultimately could be forced to discontinue our operations and liquidate, in which event it is unlikely that stockholders would receive any distribution on their shares. Further, we may not be able to continue operating if we do not generate sufficient revenues from operations needed to stay in business.
In addition, we may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may adversely impact our financial condition.
It may be difficult for investors in the U.S. to enforce any judgments obtained against us or any of our directors or officers.
All of our assets are located outside the U.S. and we do not currently maintain a permanent place of business within the U.S. In addition, most of our directors and all of our officers are nationals and/or residents of countries other than the U.S., and all or a substantial portion of such persons’ assets are located outside the U.S. As a result, it may be difficult for investors to enforce within the U.S. any judgments obtained against us or any of our non-U.S. directors or officers, including judgments predicated upon the civil liability provisions of the securities laws of the U.S. or any state thereof. Consequently, you may be effectively prevented from pursuing remedies under U.S. federal and state securities laws against us or any of our non-U.S. directors or officers.
Risks Related to Our Organization and Our Common Stock
We are subject to financial reporting and other requirements for which our accounting, internal audit and other management systems and resources may not be adequately prepared.
On March 31, 2011, we became subject to reporting and other obligations under the Securities Exchange Act of 1934, as amended, including the requirements of Section 404 of the Sarbanes-Oxley Act. Section 404 will require us to conduct an annual management assessment of the effectiveness of our internal controls over financial reporting and to obtain a report by our independent auditors addressing these assessments. These reporting and other obligations will place significant demands on our management, administrative, operational, internal audit and accounting resources. We are presently upgrading our systems; implementing financial and management controls, reporting systems and procedures; implementing an internal audit function; and we have hired additional accounting, internal audit and finance staff. If we are unable to accomplish these objectives in a timely and effective fashion, our ability to comply with our financial reporting requirements and other rules that apply to reporting companies could be impaired. Any failure to maintain effective internal controls could have a material adverse effect on our business, operating results and stock price. Moreover, effective internal control is necessary for us to provide reliable financial reports and prevent fraud. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed.
Because we became public by means of a “reverse merger,” we may not be able to attract the attention of major brokerage firms.
There may be risks associated with us becoming public through a “reverse merger” with a shell company. Although the shell company did not have recent or past operations or assets and we performed a due diligence review of the shell company, there can be no assurance that we will not be exposed to undisclosed liabilities resulting from the prior operations of the shell company. Securities analysts of major brokerage firms and securities institutions may also not provide coverage of us because there were no broker-dealers who sold our stock in a public offering that would be incentivized to follow or recommend the purchase of our common stock. The absence of such research coverage could limit investor interest in our common stock, resulting in decreased liquidity. No assurance can be given that established brokerage firms will, in the future, want to cover our securities or conduct any secondary offerings or other financings on our behalf.
Our stock price may be volatile after this offering, which could result in substantial losses for investors.
The market price of our common stock is likely to be highly volatile and could fluctuate widely in response to various factors, many of which are beyond our control, including the following:
|
|
·
|
technological innovations or new products and services by us or our competitors;
|
|
|
·
|
additions or departures of key personnel;
|
|
|
·
|
sales of our common stock, particularly under any registration statement for the purposes of selling any other securities, including management shares;
|
|
|
·
|
limited availability of freely-tradable “unrestricted” shares of our common stock to satisfy purchase orders and demand;
|
|
|
·
|
our ability to execute our business plan;
|
|
|
·
|
operating results that fall below expectations;
|
|
|
·
|
loss of any strategic relationship;
|
|
|
·
|
industry developments;
|
|
|
·
|
economic and other external factors; and
|
|
|
·
|
period-to-period fluctuations in our financial results.
|
In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also significantly affect the market price of our common stock.
We are subject to penny stock rules which will make the shares of our common stock more difficult to sell.
We are subject to the Securities and Exchange Commission’s “penny stock” rules since our shares of common stock sell below $5.00 per share. Penny stocks generally are equity securities with a per share price of less than $5.00. The penny stock rules require broker-dealers to deliver a standardized risk disclosure document prepared by the Securities and Exchange Commission that provides information about penny stocks and the nature and level of risks in the penny stock market. The broker-dealer must also provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson, and monthly account statements showing the market value of each penny stock held in the customer’s account. The bid and offer quotations, and the broker-dealer and salesperson compensation information must be given to the customer orally or in writing prior to completing the transaction and must be given to the customer in writing before or with the customer’s confirmation.
In addition, the penny stock rules require that prior to a transaction the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. The penny stock rules are burdensome and may reduce purchases of any offerings and reduce the trading activity for shares of our common stock. As long as our shares of common stock are subject to the penny stock rules, the holders of such shares of common stock may find it more difficult to sell their securities.
There is, at present, only a limited market for our common stock and we cannot ensure investors that an active market for our common stock will ever develop or be sustained.
Our shares of common stock are thinly traded. Due to the illiquidity, the market price may not accurately reflect our relative value. There can be no assurance that there will be an active market for our shares of common stock either now or in the future. Because our common stock is so thinly traded, a large block of shares traded can lead to a dramatic fluctuation in the share price and investors may not be able to liquidate their investment in us at all or at a price that reflects the value of the business. In addition, our common stock currently trades on the OTC Bulletin Board, which generally lacks the liquidity, research coverage and institutional investor following of a national securities exchange like the NYSE Amex, the New York Stock Exchange or the Nasdaq Stock Market. While we intend to list our common stock on a national securities exchange once we satisfy the initial listing standards for such an exchange, we currently do not, and may not ever, satisfy such initial listing standards.
Our board of directors can authorize the issuance of preferred stock, which could diminish the rights of holders of our common stock, and make a change of control of us more difficult even if it might benefit our stockholders.
Our board of directors is authorized to issue shares of preferred stock in one or more series and to fix the voting powers, preferences and other rights and limitations of the preferred stock. Accordingly, we may issue shares of preferred stock with a preference over our common stock with respect to dividends or distributions on liquidation or dissolution, or that may otherwise adversely affect the voting or other rights of the holders of common stock. Issuances of preferred stock, depending upon the rights, preferences and designations of the preferred stock, may have the effect of delaying, deterring or preventing a change of control, even if that change of control might benefit our stockholders.
Offers or availability for sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
Sales of a significant number of shares of our common stock in the public market could harm the market price of our common stock and make it more difficult for us to raise funds through future offerings of common stock. Upon the effectiveness of the registration statement of which this prospectus forms a part, 414,942 shares of our common stock will become freely tradable. In addition, an additional approximately 59,278,947 shares of our common stock will become saleable under Rule 144 following April 6, 2012. As these shares and as additional shares of our common stock become available for resale in the public market, the supply of our common stock will increase, which could decrease the price of our common stock .
In addition, if our stockholders sell substantial amounts of our common stock in the public market, upon the expiration of any statutory holding period under Rule 144, upon the expiration of lock-up periods applicable to outstanding shares, or upon the exercise of outstanding options or warrants, it could create a circumstance commonly referred to as an “overhang” and in anticipation of which the market price of our common stock could fall. The existence of an overhang, whether or not sales have occurred or are occurring, could also make it more difficult for us to raise additional financing through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate.
We do not expect to pay dividends in the future. As a result, any return on investment may be limited to the value of our common stock.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. The payment of dividends on our common stock will depend on our earnings, financial condition and other business and economic factors as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on an investment in our common stock will only occur if our stock price appreciates.
Risks Related to Our Intended Reverse Stock Split
There can be no assurance that we will be able to meet all of the requirements for listing our common stock on the Nasdaq Capital Market or to meet the continued listing standards of the Nasdaq Capital Market after a reverse stock split.
The Nasdaq Capital Market has numerous initial listing requirements applicable to the listing of our common stock and its continued listing thereafter. While we believe we currently meet these standards, other than the minimum bid price requirement of more than $4.00 per share, we cannot assure you that our common stock will be accepted for listing on the Nasdaq Capital Market following the reverse stock split or that we will maintain compliance with all of the requirements for our common stock to remain listed. Moreover, there can be no assurance that the market price of our common stock after the reverse stock split will adjust to reflect the decrease in common stock outstanding or that the market price following a reverse stock split will either exceed or remain in excess of the current market price.
If the reverse stock split is implemented, the resulting per-share price may not attract institutional investors, investment funds or brokers and may not satisfy the investing guidelines of these investors or brokers, and consequently, the trading liquidity of common stock may not improve.
While we believe that a higher share price may help generate investor and broker interest in our common stock, the reverse stock split may not result in a share price that will attract institutional investors or investment funds or satisfy the investing guidelines of institutional investors, investment funds or brokers. A decline in the market price of our common stock after the reverse stock split may result in a greater percentage decline than would occur in the absence of the reverse stock split. If the reverse stock split is implemented and the market price of our common stock declines, the percentage decline may be greater than would occur in the absence of the reverse stock split. The market price of our common stock is also based on our performance and other factors, which are unrelated to the number of shares of common stock outstanding.
Special Note Regarding Forward-Looking Statements
This prospectus contains “forward-looking statements,” which include information relating to future events, future financial performance, strategies, expectations, competitive environment and regulation. Words such as “may,” “should,” “could,” “would,” “predicts,” “potential,” “continue,” “expects,” “anticipates,” “future,” “intends,” “plans,” “believes,” “estimates,” and similar expressions, as well as statements in future tense, identify forward-looking statements. Forward-looking statements should not be read as a guarantee of future performance or results and will probably not be accurate indications of when such performance or results will be achieved. Forward-looking statements are based on information we have when those statements are made or our management’s good faith belief as of that time with respect to future events, and are subject to risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to:
|
|
·
|
adverse economic conditions and/or intense competition;
|
|
|
·
|
loss of a key customer or supplier;
|
|
|
·
|
entry of new competitors and products;
|
|
|
·
|
adverse federal, state and local government regulation, in the U.S., Europe or Israel;
|
|
|
·
|
failure to adequately protect our intellectual property;
|
|
|
·
|
inadequate capital;
|
|
|
·
|
technological obsolescence of our products;
|
|
|
·
|
technical problems with our research and products;
|
|
|
·
|
price increases for supplies and components;
|
|
|
·
|
inability to carry out research, development and commercialization plans;
|
|
|
·
|
loss or retirement of key executives and research scientists and other specific risks; and
|
|
|
·
|
the uncertainty regarding the adequacy of our liquidity to pursue our complete business objectives.
|
You should review carefully the section entitled “Risk Factors” beginning on page 5 of this prospectus for a discussion of these and other risks that relate to our business and investing in shares of our common stock.
All shares of our common stock offered by this prospectus are being registered for the accounts of the selling stockholders and we will not receive any proceeds from the sale of these shares.
The shares of common stock offered by this prospectus are issuable upon the exercise of common stock purchase warrants. As such, if a selling stockholder exercises all or any portion of its warrants on a cash basis, we will receive the aggregate exercise price paid by such selling stockholder in connection with any such warrant exercise. The maximum amount of proceeds we would receive upon the exercise of all the warrants on a cash basis would be approximately $747,000.00. However, the selling stockholders may also exercise their warrants through a cashless exercise. In the event a selling stockholder exercises a warrant through a cashless exercise, we will not receive any proceeds from such exercise. We expect to use the proceeds received from the exercise of the warrants, if any, for general working capital purposes.
Market For Our Common Stock And Related Stockholder Matters
Our common stock has been quoted on the OTC Bulletin Board since April 11, 2011 under the symbol NSPR.OB. Prior to that date, there was no active market for our common stock. The following table sets forth the high and low bid prices for our common stock for the periods indicated, as reported by the OTC Bulletin Board. The quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions.
|
Fiscal Year 2011
|
High
|
Low
|
||
|
Second Quarter
|
$2.89
|
$1.75
|
||
|
Third Quarter
|
$2.74
|
$1.80
|
||
|
Fourth Quarter (through November 30, 2011)
|
$2.59
|
$1.62
|
The last reported sales price of our common stock on the OTC Bulletin Board on November 30, 2011, was $2.25 per share. As of November 30, 2011, there were approximately 197 holders of record of our common stock.
Dividend Policy
In the past, we have not declared or paid cash dividends on our common stock, and we do not intend to pay any cash dividends on our common stock. Rather, we intend to retain future earnings, if any, to fund the operation and expansion of our business and for general corporate purposes.
Overview
We are a medical device company focusing on the development and commercialization of our proprietary stent platform technology, MGuard™. MGuard™ provides embolic protection in stenting procedures by placing a micron mesh sleeve over a stent. Our initial products are marketed for use mainly in patients with acute coronary syndromes, notably acute myocardial infarction (heart attack) and saphenous vein graft coronary interventions (bypass surgery).
On March 31, 2011, we completed a series of share exchange transactions pursuant to which we acquired all of the capital stock of InspireMD Ltd., a company formed under the laws of the State of Israel, in exchange for an aggregate of 50,666,663 shares of our common stock. As a result of these share exchange transactions, InspireMD Ltd. became our wholly-owned subsidiary, we discontinued our former business and succeeded to the business of InspireMD Ltd. as our sole line of business.
The share exchange transactions are being accounted for as a recapitalization. InspireMD Ltd. is the acquirer for accounting purposes and we are the acquired company. Accordingly, the historical financial statements presented and the discussion of financial condition and results of operations herein are those of InspireMD Ltd., retroactively restated for, and giving effect to, the number of shares received in the share exchange transactions, and do not include the historical financial results of our former business. The accumulated earnings of InspireMD Ltd. were also carried forward after the share exchange transactions and earnings per share have been retroactively restated to give effect to the recapitalization for all periods presented. Operations reported for periods prior to the share exchange transactions are those of InspireMD Ltd.
Recent Events
On October 31, 2011, our stockholders authorized our board of directors to amend our amended and restated certificate of incorporation to effect a reverse stock split of our common stock at a ratio of one-for-two to one-for-four, at any time prior to our 2012 annual stockholders’ meeting, the exact ratio of the reverse stock split to be determined by the board. As of the date of this prospectus, we have not effected the reverse stock split and, as such, the information with respect to our common stock in this prospectus and the accompanying financial statements and related notes does not give effect to any reverse stock split.
On October 4, 2011, InspireMD Ltd., our wholly-owned subsidiary, entered into a clinical trial services agreement with Harvard Clinical Research Institute, Inc., pursuant to which Harvard Clinical Research Institute, Inc. will conduct a study entitled “MGuard Stent System Clinical Trial in Patients with Acute Myocardial Infarction” on our behalf. We will pay Harvard Clinical Research Institute, Inc. an estimated fee of approximately $10 million for conducting the study, subject to adjustment dependent upon changes in the scope and nature of the study, as well as other costs to be determined by the parties.
Critical Accounting Policies
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of sales and expenses during the reporting periods. Actual results could differ from those estimates.
As applicable to these consolidated financial statements, the most significant estimates and assumptions relate to revenue recognition including provision for returns, legal contingencies and estimation of the fair value of share-based compensation and convertible debt.
Functional currency
The currency of the primary economic environment in which our operations are conducted is the U.S. dollar (“$” or “dollar”). Accordingly, the functional currency of us and of our subsidiaries is the dollar.
The dollar figures are determined as follows: transactions and balances originally denominated in dollars are presented in their original amounts. Balances in foreign currencies are translated into dollars using historical and current exchange rates for non-monetary and monetary balances, respectively. The resulting translation gains or losses are recorded as financial income or expense, as appropriate. For transactions reflected in the statements of operations in foreign currencies, the exchange rates at transaction dates are used. Depreciation and changes in inventories and other changes deriving from non-monetary items are based on historical exchange rates.
Fair value measurement
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date.
In determining fair value, we use various valuation approaches, including market, income and/or cost approaches. Hierarchy for inputs is used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of us. Unobservable inputs are inputs that reflect our assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy is broken down into three levels based on the reliability of inputs.
Concentration of credit risk and allowance for doubtful accounts
Financial instruments that may potentially subject us to a concentration of credit risk consist of cash, cash equivalents and restricted cash which are deposited in major financial institutions in Germany and Israel, and trade accounts receivable. Our trade accounts receivable are derived from revenues earned from customers from various countries. We perform ongoing credit evaluations of our customers’ financial condition and, generally, require no collateral from our customers. We also have a credit insurance policy for some of our customers. We maintain an allowance for doubtful accounts receivable based upon the expected ability to collect the accounts receivable. We review our allowance for doubtful accounts quarterly by assessing individual accounts receivable and all other balances based on historical collection experience and an economic risk assessment. If we determine that a specific customer is unable to meet its financial obligations to us, we provide an allowance for credit losses to reduce the receivable to the amount our management reasonably believes will be collected. To mitigate risks, we deposit cash and cash equivalents with high credit quality financial institutions. Provisions for doubtful debts are netted against “Accounts receivable-trade.”
Inventory
Inventories include finished goods, work in process and raw materials. Inventories are stated at the lower of cost (cost is determined on a “first-in, first-out” basis) or market value. Our inventories generally have a limited shelf life and are subject to impairment as they approach their expiration dates. We regularly evaluate the carrying value of our inventories and when, in our opinion, factors indicate that impairment has occurred, we establish a reserve against the inventories’ carrying value. Our determination that a valuation reserve might be required, in addition to the quantification of such reserve, requires us to utilize significant judgment. Although we make every effort to ensure the accuracy of forecasts of future product demand, any significant unanticipated decreases in demand could have a material impact on the carrying value of our inventories and reported operating results. To date, inventory adjustments have not been material. In respect to inventory on consignment, see “Revenue recognition” below.
Revenue recognition
Revenue is recognized when delivery has occurred, evidence of an arrangement exists, title and risks and rewards for the products are transferred to the customer, collection is reasonably assured and when product returns can be reliably estimated. When product returns can be reliably estimated a provision is recorded, based on historical experience, and deducted from sales. The provision for sales returns and related costs are included in “Accounts payable and accruals - Other” under “current liabilities”, and “Inventory on consignment”, respectively.
When returns cannot be reliably estimated, both revenues and related direct costs are eliminated, as the products are deemed unsold. Accordingly, both related revenues and costs are deferred, and presented under “Deferred revenues” and “Inventory on consignment”, respectively.
We recognize revenue net of value added tax.
Research and development costs
Research and development costs are charged to the statement of operations as incurred.
Share-based compensation
Employee option awards are classified as equity awards and accounted for using the grant-date fair value method. The fair value of share-based awards is estimated using the Black-Scholes valuation model, which is expensed over the requisite service period, net of estimated forfeitures. We estimate forfeitures based on historical experience and anticipated future conditions.
We elected to recognize compensation expensed for awards with only service conditions that have graded vesting schedules using the accelerated multiple option approach.
We account for equity instruments issued to third party service providers (non-employees) by recording the fair value of the options granted using an option pricing model, at each reporting period, until rewards are vested in full. The expense is recognized over the vesting period using the accelerated multiple option approach. The expense relates to options granted to third party service providers with respect to successful investor introductions that are recorded at their fair value in equity, as issuance costs.
Uncertain tax and Value Added Tax positions
We follow a two-step approach to recognizing and measuring uncertain tax and value added tax positions. The first step is to evaluate the tax and value added tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit. The second step is to measure the tax and value added tax benefit as the largest amount that is more than 50% and 75%, respectively, likely of being realized upon ultimate settlement. Such liabilities are classified as long-term, unless the liability is expected to be resolved within twelve months from the balance sheet date. Our policy is to include interest and penalties related to unrecognized tax benefits within financial expenses.
Results of Operations
Three Months Ended September 30, 2011 Compared to Three Months Ended September 30, 2010
Revenues. For the three months ended September 30, 2011, total revenue increased approximately $0.8 million, or 62.4%, to approximately $2.0 million from approximately $1.2 million during the same period in 2010. The $0.8 million increase was due to an increase in volume of approximately $0.7 million, or approximately 55.9%, and by an increase of prices of approximately $0.1 million, or approximately 6.5%. The following is an explanation of the approximately $0.8 million increase in revenue broken down by its main two components, an increase in gross revenues of approximately $1.0 million offset by a net decrease in deferred revenues of approximately $0.2 million.
For the three months ended September 30, 2011, total gross revenue increased by approximately $0.9 million, or 87.8%, to approximately $2.0 million as compared to approximately $1.1 million during the same period in 2010. This increase in total gross revenue is predominantly volume based, accounting for approximately $0.8 million or approximately 80.3%, and an increase of prices of approximately $0.1 million, or approximately 7.5%. In general, we focused on opening new markets, such as Russia and the Ukraine, and also increasing sales in existing markets by presenting clinical data at conferences and individual presentations to doctors about the merits of MGuardTM. With respect to individual markets, this increase in gross revenue is mainly attributable to an increase of approximately $0.2 million of gross revenue from our distributor in Brazil, an increase of approximately $0.2 million of gross revenue from our distributor in Argentina, an increase of approximately $0.1 million of gross revenue from our new distributor in Russia, an increase of approximately $0.1 million of gross revenue from our new distributor in the Ukraine, an increase of approximately $0.1 million of gross revenue from our distributor in Mexico, an increase of approximately $0.1 million of gross revenue from our distributor in Italy, an increase of approximately $0.1 million of gross revenue from our distributor in Spain and an increase of approximately $0.1 million of gross revenue from our distributor in Israel. This increase was partially offset by a decrease of approximately $0.3 million in gross revenue from our distributor in Germany and a decrease of approximately $0.1 million from our distributor in Romania. We also shipped and recognized gross revenue for approximately $0.3 million more from our remaining distributors during the three months ended September 30, 2011, as compared to the same period in 2010.
For the three months ended September 30, 2011, net deferred revenue recognized during the period decreased by approximately $0.2 million, or 102.1%, to approximately $(4,000) and from approximately $0.2 million during the same period in 2010. The decrease was volume based. Revenue recognition out of deferred income had less of an impact in 2011 as compared to 2010 due to the fact that we deferred mainly shipments in 2008 and 2009 that were recognized in 2010. In 2010, no customers had revenues deferred until the three months ended September 30, 2011.
For the three months ended September 30, 2011, our net deferred revenue of $(4,000) consisted of only a provision for sales return included in "accounts payable and accrual - other." For the three months ended September 30, 2010, net deferred revenue of approximately $0.2 million was comprised mainly of shipments from 2008 and 2009 to our distributor in Israel of approximately $0.1 million and our distributor in Poland of approximately $50,000.
Gross Profit. For the three months ended September 30, 2011, gross profit (revenue less cost of revenues) increased approximately 79.0%, or approximately $0.5 million, to approximately $1.2 million from approximately $0.7 million during the same period in 2010. Gross margin increased from 54.1% in the three months ended September 30, 2010 to 59.7% in the three months ended September 30, 2011. We were able to improve our gross margin because of reduced production cost per stent driven by economies of scale, as well as an increase in average price per stent. For the three months ended September 30, 2011, our average selling price per stent recognized in revenue was $624, and we recognized the sale of 3,186 stents, compared to an average price of $577 per stent and 2,120 stents recognized in revenue for the same period in 2010. The higher average price per stent for the three months ended September 30, 2011 was driven by sales of MGuard Prime, which was launched in 2011 and is priced on average $171 more versus the average price of MGuard per stent. Our cost of goods sold per stent decreased from an average of $265 per stent recognized in revenue for the three months ended September 30, 2010 to an average of $251 per stent for the same period in 2011.
Research and Development Expense. For the three months ended September 30, 2011, research and development expense increased 179.1% to approximately $0.5 million from approximately $0.2 million during the same period in 2010. The increase in cost resulted primarily from higher clinical trial expenses of approximately $0.2 million, attributable mainly to the U.S. Food and Drug Administration clinical trial (approximately $0.1 million) and the MGuard for Acute ST Elevation Reperfusion Trial (MASTER Trial) (approximately $0.1 million), and an increase in R&D related salaries of approximately $0.2 million relating to the above mentioned clinical studies. Research and development expense as a percentage of revenue increased to 27.5% for the three months ended September 30, 2011 from 16.0% in the same period of 2010.
Selling and Marketing Expense. For the three months ended September 30, 2011, selling and marketing expense increased 8.2% to approximately $0.3 million, from approximately $0.28 million during the same period in 2010. The increase in cost resulted primarily from approximately $0.16 million of additional salaries and share based compensation of predominately newly hired sales personnel as we expand our sales activities worldwide. This increase was partially offset by a decrease of approximately $0.1 million in advertising, travel and other related expenses. Selling and marketing expense as a percentage of revenue decreased from 22.8% in 2010 to 15.2% in 2011.
General and Administrative Expense. For the three months ended September 30, 2011, general and administrative expense increased 175.0% to approximately $2.5 million from $0.9 million during the same period in 2010. The increase in cost resulted primarily from an increase in share based compensation of $1.3 million, which predominately pertains to directors’ compensation, approximately $0.2 million in legal fees related primarily to compliance with Securities and Exchange Commission standards, an increase in investor related activities of approximately $0.1 million due to us having been public during the three months ended September 30, 2011, but not during the same period in 2010, and an increase of $0.1 million in miscellaneous expenses. This increase was partially offset by a decrease of approximately $0.1 in audit and related expenses. General and administrative expense as a percentage of revenue increased to 125.2% in 2011 from 73.9% in 2010.
Financial Expenses. Financial expense remained relatively flat at $108,000 for the three months ended September 30, 2011, as compared to $121,000 during the same period in 2010. Our financial expenses reflect primarily changes in exchange rates, as well as interest related expenses. Financial expense as a percentage of revenue decreased to 5.4% in 2011, from 9.9% in 2010.
Tax Expenses. Tax expense remained relatively flat at $25,000 for the three months ended September 30, 2011, as compared to $9,000 during the same period in 2010. Our expenses for income taxes reflect primarily the tax liability due to potential tax exposure.
Net Loss. Our net loss increased by approximately $1.5 million, or 169.5%, to $2.3 million for the three months ended September 30, 2011 from $0.8 million during the same period in 2010. The increase in net loss resulted primarily from an increase in operating expenses of approximately $2.0 million (see above for explanations) and is partially offset by an increase of approximately $0.5 million in gross profit (see above for explanation).
Nine months Ended September 30, 2011 Compared to Nine months Ended September 30, 2010
Revenues. For the nine months ended September 30, 2011, total revenue increased approximately $0.5 million, or 11.4%, to approximately $4.7 million from approximately $4.2 million during the same period in 2010. The $0.5 million increase was due to an increase in volume of approximately $0.6 million or approximately 14.2%, offset by an approximately $0.1 million decrease, or approximately 2.7%, due to price decreases. The following is an explanation of the approximately $0.5 million increase in revenue broken down by its main two components, an increase in gross revenues of approximately $2.0 million offset by a net decrease in deferred revenues of approximately $1.5 million.
For the nine months ended September 30, 2011, total gross revenue increased by approximately $2.0 million, or 85.3%, to approximately $4.4 million from approximately $2.4 million during the same period in 2010. This increase in total gross revenue is predominantly volume based, accounting for approximately $2.0 million or approximately 87.3%, with price decreases accounting for the remaining approximately $45,000, or approximately 2.0%. In general, we focused on opening new markets, such as India, and also increasing sales in existing markets by presenting clinical data at conferences and individual presentations to doctors about the merits of MGuardTM. With respect to individual markets, this increase in gross revenue is mainly attributable to the first time shipment of approximately $1.2 million to our distributor in India during the first nine months of 2011, an increase of approximately $0.3 million of gross revenue from our distributor in Argentina, an increase of approximately $0.2 million of gross revenue from our distributor in Brazil, an increase of approximately $0.2 million of gross revenue from our distributor in Spain, an increase of approximately $0.2 million of gross revenue from our distributor in Israel, an increase of approximately $0.1 million of gross revenue from our new distributor in Russia, an increase of approximately $0.1 million of gross revenue from our new distributor in the Ukraine, an increase of approximately $0.1 million of gross revenue from our distributor in Mexico and approximately $0.1 million of gross revenue from our new distributor in the Netherlands. This increase was partially offset by a decrease of approximately $0.4 million in gross revenue from our distributor in Poland, a decrease of approximately $0.2 million in gross revenue from our distributor in Germany, a decrease of approximately $0.2 million from our distributor in Pakistan, and a decrease of approximately $0.1 million in gross revenue to our distributor in Kazakhstan. We also shipped and recognized gross revenue for approximately $0.4 million more from our remaining distributors during the nine months ended September 30, 2011, as compared to the same period in 2010.
For the nine months ended September 30, 2011, net deferred revenue recognized during the period decreased by approximately $1.5 million, or 81.0%, to approximately $0.4 million from approximately $1.9 million during the same period in 2010. The key driver of this decrease was volume based, accounting for approximately $1.4 million or approximately 77.4%, with the remaining approximately $0.1 million, or 3.6%, being driven by price decreases. Revenue recognition out of deferred income had less of an impact in 2011 as compared to 2010 due to the fact that we deferred mainly shipments in 2008 and 2009 that were recognized in 2010. In 2010, only a small set of customers had a large portion of their revenues deferred until 2011.
For the nine months ended September 30, 2011, our net deferred revenue consisted of approximately $0.2 million attributable to our distributor in Israel, approximately $0.1 million to our distributor in Brazil, approximately $0.1 million to our distributor in Poland, and approximately $0.05 million to our distributor in Italy, offset by approximately $0.1 million deferred for a shipment to our distributor in India. Our distributor in Israel had a contractual right to return all purchases to us within 18 months of the purchase date. Due to our inability to accurately estimate the amount of future returns, all sales to this distributor were deferred until this 18 month return period elapsed. On May 9, 2011, our distributor in Israel agreed to revoke its previous rights to return purchases, resulting in all future sales being final. The deferred revenue of approximately $0.2 million recognized during the nine months period ended September 30, 2011 accounted for all previous purchases by the distributor that the distributor no longer had a contractual right to return and were not yet recognized as revenues. Our distributor in Brazil has a contractual right to return all purchases for up to six months from the delivery date. Due to our inability to accurately estimate the amount of future returns by our distributor in Brazil, all sales made to it were also deferred until the six month return period elapsed. The deferred revenue of approximately $0.1 million recognized during the nine months period ended September 30, 2011 accounted for purchases made in December 2010 that were not returned by the Brazilian distributor and were not yet recognized as revenues.
For the first nine months of 2010, net deferred revenue of approximately $1.9 million was comprised mainly of shipments from 2008 and 2009 to our distributor in Poland of approximately $1.3 million, to our distributor in Brazil of approximately $0.4 million, to our distributor in Sri Lanka of approximately $0.1 million and approximately $0.1 million to miscellaneous distributors. For the nine months ended September 30, 2010, our distributor in Poland, subject to our sole discretion, had the right to return our products. Because we were unable to develop estimates for the level of returns, the $1.3 million worth of shipments made to the distributor in Poland that we recorded as deferred revenues was only recognized during the first nine months of 2010 as revenues. As noted above, our distributor in Brazil has a contractual right to return all purchases for up to six months from the delivery date. As also noted above, due to our inability to accurately estimate the rate of return by this distributor, all sales made to it were also deferred until the six month return period elapsed. The deferred revenue of approximately $0.4 million recognized during the nine months period ended September 30, 2010 accounted for purchases made in December 2009 that were not returned and were not yet recognized as revenues.
Gross Profit. For the nine months ended September 30, 2011, gross profit (revenue less cost of revenues) increased 28.1%, or approximately $0.5 million, to approximately $2.4 million from approximately $1.9 million during the same period in 2010. Gross margin increased from 43.8% in the nine months ended September 30, 2010 to 50.3% in the nine months ended September 30, 2011. In addition to an increase in sales, we were able to improve our gross margin because of reduced production cost per stent driven by economies of scale. For the nine months ended September 30, 2011, our average selling price per stent recognized in revenue was $570, and we recognized the sale of 8,261 stents, compared to an average price of $643 per stent and 6,566 stents recognized in revenue for the same period in 2010. Our cost of goods sold per stent decreased from an average of $362 per stent recognized in revenue for the nine months ended September 30, 2010 to an average of $283 per stent for the same period in 2011. The higher price per stent for the nine months ended September 30, 2010 was affected by the price of stents sold in 2008 and 2009 to one of our Europeans distributors in Euros when the Euro was much stronger than the U.S. dollar, at an average price of $997 when translated to U.S. dollars.
Research and Development Expense. For the nine months ended September 30, 2011, research and development expense increased 69.2% to approximately $1.6 million from approximately $1.0 million during the same period in 2010. The increase in cost resulted primarily from higher clinical trial expenses of approximately $0.8 million, attributable mainly to the U.S. Food and Drug Administration clinical trial (approximately $0.6 million) and the MGuard for Acute ST Elevation Reperfusion Trial (MASTER Trial) (approximately $0.2 million), offset by approximately $0.2 million reduction in miscellaneous expenses. Research and development expense as a percentage of revenue increased to 34.8% for the nine months ended September 30, 2011 from 22.9% in the same period of 2010.
Selling and Marketing Expense. For the nine months ended September 30, 2011, selling and marketing expense increased 47.1% to approximately $1.3 million, from approximately $0.9 million during the same period in 2010. The increase in selling and marketing expense resulted primarily from approximately $0.2 million of additional salaries and approximately $0.3 of share based compensation of predominately newly hired sales personnel as we expand our sales activities worldwide, and approximately $0.1 million of commissions pertaining mainly to our first time shipment of approximately $1.2 million to our distributor in India. This increase was partially offset by a decrease of approximately $0.1 million in advertising, and a decrease of approximately $0.1 million in miscellaneous expenses. Selling and marketing expense as a percentage of revenue increased to 28.6% in 2011 from 21.7% in 2010.
General and Administrative Expense. For the nine months ended September 30, 2011, general and administrative expense increased 141.9% to approximately $4.9 million from $2.0 million during the same period in 2010. The increase in cost resulted primarily from an increase in share based compensation of $1.1 million which predominately pertains to directors’ compensation, an increase of approximately $0.4 million in salary expenses (due to an increase in employee infrastructure to accommodate and comply with Securities and Exchange Commission standards and reporting), an increase in investor related activities of approximately $0.4 million (due to us having been a publicly reporting company during the nine months ended September 30, 2011, but not during the same period in 2010), an increase of approximately $0.5 million in litigation expenses (primarily due to a provision for our potential loss regarding a threatened lawsuit from a finder claiming a future success fee and commissions for assistance in finding our distributor in Brazil), and approximately $0.3 million in legal fees (also related primarily to compliance with Securities and Exchange Commission standards), and approximately $0.2 million increase in miscellaneous expenses. General and administrative expense as a percentage of revenue increased to 103.5% in 2011 from 47.7% in 2010.
Financial Expenses. For the nine months ended September 30, 2011, financial expense increased 496.7% to approximately $0.9 million from $0.2 million during the same period in 2010. The increase in expense resulted primarily from a one-time financial expense recording of approximately $0.6 million in the first quarter of 2011 pertaining to the revaluation of an outstanding convertible loan at fair value prior to redemption and approximately $0.2 million for the favorable impact of exchange rate differences for the nine months ended September 30, 2010 that did not occur during the nine months ended September 30, 2011. This increase was partially offset by a decrease of approximately $0.1 million in miscellaneous expenses. Financial expense as a percentage of revenue increased from 3.5% in 2010, to 19.0% in 2011.
Tax Expenses. Tax expense remained relatively flat at $45,000 for the nine months ended September 30, 2011, as compared to $39,000 during the same period in 2010. Our expenses for income taxes reflect primarily the tax liability due to potential tax exposure.
Net Loss. Our net loss increased by approximately $4.2 million, or 187.3%, to $6.4 million for the nine months ended September 30, 2011 from $2.2 million during the same period in 2010. The increase in net loss resulted primarily from an increase in operating expenses of approximately $4.0 million (see above for explanations) and an increase of approximately $0.7 million in financial expenses (see above for explanation). This increase was partially offset by an increase in gross profit of approximately $0.5 million.
Year Ended December 31, 2010 Compared to Year Ended December 31, 2009
Revenues. For the year ended December 31, 2010, total revenue increased 45.1% to $4.9 million from $3.4 million in 2009. The increase in revenue was primarily attributable to launching MGuard™ Coronary with bio-stable mesh in new markets around the world, particularly in Europe and Latin America.
Gross Margin. Our gross margin percentage for 2010 increased to 45.5% of revenues, compared to 32.8% during 2009. The increase in our gross margin resulted primarily from higher pricing, more efficient manufacturing and economies of scale due to the increase in sales volume.
Research and Development Expense. For the year ended December 31, 2010, research and development expense increased 0.6% to $1.338 million from $1.330 million in 2009. Research and development expense as a percentage of revenue decreased to 27.0% in 2010 from 39.0% in 2009.
Selling and Marketing Expense. For the year ended December 31, 2010, selling and marketing expense increased 18.8% to $1.2 million from $1.0 million in 2009. The increase in cost resulted primarily from additional promotional activities worldwide. Selling and marketing expense as a percentage of revenue decreased to 25.0% in 2010 from 30.5% in 2009.
General and Administrative Expense. For the year ended December 31, 2010, general and administrative expense increased 97.5% to approximately $2.9 million from $1.5 million in 2009. The increase in cost resulted primarily from a large increase in the amount of our share options being issued and the corresponding accounting charges and overall accounting and legal expenses. General and administrative expense as a percentage of revenue increased to 58.6% in 2010 from 43.0% in 2009.
Financial Expenses (Income). For the year ended December 31, 2010, financial expense increased to approximately $0.2 million from income of $0.04 million in 2009. The increase in expense resulted primarily from a one time financial income recording of $0.3 million in 2009 pertaining to the cancellation of the conversion feature of a convertible loan that was repaid in the same year. Financial expense as a percentage of revenue increased to 3.1% in 2010, compared to financial income as a percent of revenue of 1.2% in 2009.
Tax Expenses. Tax expense remained flat at $47,000 in 2010 and 2009. Our expenses for income taxes reflect primarily the tax liability due to potential tax exposure.
Net Loss. Our net loss increased 25.6% to $3.4 million in 2010 from $2.7 million in 2009.
Backlog. Our order backlog at December 31, 2010 was approximately $1.5 million, up 165% compared to approximately $0.6 million at December 31, 2009.
Liquidity and Capital Resources
Nine Months Ended September 30, 2011 Compared to Nine Months Ended September 30, 2010
General. At September 30, 2011, we had cash and cash equivalents of approximately $7.5 million, as compared to $0.6 million at December 31, 2010. The increase is attributable primarily to the private placement conducted in conjunction with the share exchange transactions on March 31, 2011 and other private equity issuances prior to and after the share exchange transactions. We have historically met our cash needs through a combination of issuance of new shares, borrowing activities and sales. Our cash requirements are generally for product development, clinical trials, marketing and sales activities, finance and administrative cost, capital expenditures and general working capital.
Cash used in our operating activities was approximately $3.9 million for the nine months ended September 30, 2011, and approximately $2.2 million for the same period in 2010. The principal reasons for the usage of cash in our operating activities for the nine months ended September 30, 2011 include a net loss of approximately $6.4 million, and a decrease in working capital of approximately $1.2 million, offset by approximately $2.8 million in non-cash share based compensation, and approximately $0.9 million in non-cash financial expenses related to the revaluation of a convertible loan.
Cash flow generated from investing activities was approximately $0.1 million during the nine months ended September 30, 2011, compared to approximately $0.1 million of cash used by investing activities during the same period in 2010. The principal reason for the increase in cash flow from investing activities was a decrease in restricted cash of approximately $0.2 million.
Cash flow generated from financing activities was approximately $10.8 million for the nine months ended September 30, 2011, and $2.7 million for the same period in 2010. The principal reason for the increase in cash flow from financing activities during 2011 was the private placement conducted in conjunction with the share exchange transactions on March 31, 2011 and other private equity issuances and exercise of options prior to and after the share exchange transactions in the aggregate amount of approximately $12.1 million, offset by the repayment of the non-converted portion of a convertible loan in the amount of approximately $1.0 million and the partial repayment of a long-term loan in the amount of approximately $0.3 million.
As of September 30, 2011, our current assets exceeded current liabilities by a multiple of 3.2. Current assets increased approximately $7.6 million during 2011, mainly due to cash raised from the private placements in 2011, while current liabilities decreased approximately $0.3 million during the same period. As a result, our working capital surplus increased by approximately $8.0 million to approximately $7.9 million during the nine months ended September 30, 2011.
Credit Facilities. As of September 30, 2011, we had a long term loan in the amount of approximately $0.2 million bearing interest at the three month U.S. Dollar LIBOR rate plus 4% per annum. The loan is payable in eight quarterly installments during a period of three years that began in April 2010 and ends in January 2012. According to the loan agreement, in case of an “exit transaction,” we will be required to pay to the bank an additional $0.25 million if the sum received in a “liquidity event” or the value of the company in an “IPO” is higher than $100 million.
Convertible Loans. Prior to September 30, 2011, we had a convertible loan with an aggregate principal amount outstanding of approximately $1.58 million that bore 8% interest. Following the share exchange transactions on March 31, 2011, $580,000 plus accrued interest converted into shares of our common stock. The remaining principle in the amount of $1.0 million was repaid on May 15, 2011.
Sales of Stock. For the nine months ended September 30, 2011, we issued an aggregate of 9,415,145 shares of common stock and warrants to purchase 6,709,073 shares of common stock for gross proceeds of approximately $12.0 million.
Year Ended December 31, 2010 Compared to Year Ended December 31, 2009
General. At December 31, 2010, we had cash and cash equivalents of approximately $636,000, as compared to $376,000 in 2009. We have historically met our cash needs through a combination of issuance of new shares, borrowing activities and sales. Our cash requirements are generally for product development, clinical trials, marketing and sales activities, finance and administrative cost, capital expenditures and overall working capital.
Cash used in our operating activities was approximately $2.7 million in 2010, and $1.5 million in 2009. The principal reasons for the decrease in cash flow from operations in 2010 included a $3.4 million net loss, a decrease of $1.6 million in deferred revenues offset by $1.6 million of non cash share based compensation expense and a $0.4 million increase in other working capital.
Cash used in investing activities was approximately $46,000 in 2010, and $0.3 million in 2009. The principal reasons for the decrease in cash flow from investing activities included $81,000 for plant and equipment purchases offset by a $52,000 decrease in restricted cash.
Cash flow generated from financing activities was approximately $3.0 million in 2010, and $0.7 million in 2009. The principal reasons for the increase in cash flow from financing activities during 2010 were the issuance of approximately $1.8 million in new shares and the issuance of a convertible loan of approximately $1.5 million, offset by the repayment of a long term loan in the amount of $0.3 million.
As of December 31, 2010, current assets were approximately equal with our current liabilities. Current assets decreased $0.2 million during 2010 while current liabilities decreased by $1.5 million during the same period. As a result, our working capital deficiency decreased by $1.2 million to approximately $53,000 during 2010.
Off Balance Sheet Arrangements
We have no off-balance sheet transactions, arrangements, obligations (including contingent obligations), or other relationships with unconsolidated entities or other persons that have, or may have, a material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Recent Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board issued amendments to the accounting and disclosure for revenue recognition. These amendments, effective for fiscal years beginning on or after June 15, 2010 (early adoption is permitted), modify the criteria for recognizing revenue in multiple element arrangements and require companies to develop a best estimate of the selling price to separate deliverables and allocate arrangement consideration using the relative selling price method. Additionally, the amendments eliminate the residual method for allocating arrangement considerations. We do not expect the standard to have material effect on its consolidated financial statements.
In January 2010, the Financial Accounting Standards Board updated the “Fair Value Measurements Disclosures”. More specifically, this update will require (a) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (b) information about purchases, sales, issuances and settlements to be presented separately (i.e. present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This update clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured at fair value, and requires disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. This update will become effective as of the first interim or annual reporting period beginning after December 15, 2009, except for the gross presentation of the Level 3 roll forward information, which is required for annual reporting periods beginning after December 15, 2010 and for interim reporting periods within those years. The adoption of the new guidance did not have a material impact on our consolidated financial statements.
In May 2011, the Financial Accounting Standards Board issued amended guidance and disclosure requirements for fair value measurements. These changes will be effective January 1, 2012 on a prospective basis. Early application is not permitted. These amendments are not expected to have a material impact to the consolidated financial results.
Factors That May Affect Future Operations
We believe that our future operating results will continue to be subject to quarterly variations based upon a wide variety of factors, including the cyclical nature of the ordering patterns of our distributors, timing of regulatory approvals, the implementation of various phases of our clinical trials and manufacturing efficiencies due to the learning curve of utilizing new materials and equipment. Our operating results could also be impacted by a weakening of the Euro and strengthening of the New Israeli Shekel, or NIS, both against the U.S. dollar. Lastly, other economic conditions we cannot foresee may affect customer demand, such as individual country reimbursement policies pertaining to our products.
Business
History
We were organized in the State of Delaware on February 29, 2008 as Saguaro Resources, Inc. to engage in the acquisition, exploration and development of natural resource properties. On March 28, 2011, we changed our name from “Saguaro Resources, Inc.” to “InspireMD, Inc.”
On March 31, 2011, we completed a series of share exchange transactions pursuant to which we issued the shareholders of InspireMD Ltd. 50,666,663 shares of common stock in exchange for all of InspireMD Ltd’s issued and outstanding ordinary shares, resulting in the former shareholders of InspireMD Ltd. holding a controlling interest in us and InspireMD Ltd. becoming our wholly-owned subsidiary.
Immediately following the share exchange transactions, we transferred all of our pre-share exchange operating assets and liabilities to our wholly-owned subsidiary, Saguaro Holdings, Inc., a Delaware corporation, and transferred all of Saguaro Holdings, Inc.’s outstanding capital stock to Lynn Briggs, our then-majority stockholder and our former president, chief executive officer, chief financial officer, secretary-treasurer and sole director, in exchange for the cancellation of 7,500,000 shares of our common stock held by Ms. Briggs.
After the share exchange transactions and the divestiture of our pre-share exchange operating assets and liabilities, we succeeded to the business of InspireMD Ltd. as our sole line of business, and all of our then-current officers and directors resigned and were replaced by some of the officers and directors of InspireMD Ltd.
Overview
We are an innovative medical device company focusing on the development and commercialization of our proprietary stent platform technology, MGuard™. MGuard™ provides embolic protection in stenting procedures by placing a micron mesh sleeve over a stent (see photograph below of an MGuard™ Stent). Our initial products are marketed for use mainly in patients with acute coronary syndromes, notably acute myocardial infarction (heart attack) and saphenous vein graft coronary interventions (bypass surgery). According to the TYPHOON STEMI trial (New England Journal of Medicine, 2006) and the SOS SVG Trial (Journal of the American College of Cardiology, 2009), of patients with acute myocardial infarction and saphenous vein graft coronary interventions, 7.5% to 44% experience major adverse cardiac events, including cardiac death, heart attack, and restenting of the artery. When performing stenting procedures in patients with acute coronary symptoms, interventional cardiologists face a difficult dilemma in choosing between bare-metal stents, which have a high rate of restenosis (formation of new blockages), and drug-eluting (drug-coated) stents, which have a high rate of late thrombosis (formation of clots months or years after implantation), require administration of anti-platelet drugs for at least one year post procedure, are more costly than bare-metal stents and have additional side effects. We believe that MGuard™ is a simple, seamless and complete solution for these patients.
MGuardTM Sleeve – Microscopic View
We intend to use our MGuard™ technology in a broad range of coronary related situations in which complex lesions are required and make it an industry standard for treatment of acute coronary syndromes. We believe that patients will benefit from a cost-effective alternative with a greater clinical efficacy and safety profile than other stent technologies. We believe that with our MGuard™ technology, we are well positioned to emerge as a key player in the global stent market.
We also intend to apply our technology to develop additional products used for other vascular procedures, specifically carotid (the arteries that supply blood to the brain) and peripheral (other arteries) procedures.
In October 2007, our first generation product, the MGuard™ Coronary, received CE Mark approval for treatment of coronary arterial disease in the European Union. CE Mark is a mandatory conformance mark on many products marketed in the European Economic Area and certifies that a product has met European Union consumer safety, health or environmental requirements. We began shipping our product to customers in Europe in January 2008 and have since expanded our global distribution network to Canada, Southeast Asia, India and Latin America.
Our initial MGuard™ products incorporated a stainless steel stent. We replaced this stainless steel platform with a more advanced cobalt-chromium based platform, which we refer to as MGuard Prime™. We believe the new platform will be superior because cobalt-chromium stents are generally known in the industry to provide better outcomes and possibly even a reduction in major adverse cardiac events. We believe we can use and leverage the MGuard™ clinical trial results to market MGuard Prime™. MGuard Prime™ received CE Mark approval in the European Union in October 2010 for improving luminal diameter and providing embolic protection. MGuard™ refers to both our initial products and MGuard Prime™, as applicable.
Our Industry
According to Fact Sheet No. 310/June 2011 of the World Health Organization, approximately 7.3 million people worldwide died of coronary heart disease in 2008. Physicians and patients may select from among a variety of treatments to address coronary artery disease, including pharmaceutical therapy, balloon angioplasty, stenting with bare metal or drug-eluting stents, and coronary artery bypass graft procedures, with the selection often depending upon the stage of the disease. A stent is an expandable “scaffold-like” device, usually constructed of a stainless steel material, that is inserted into an artery to expand the inside passage and improve blood flow.
According to the January 3, 2011 2011 MEDTECH OUTLOOK produced by the Bank of Montreal Investment Banking Group, known as BMO Capital Markets, after registering a compounded annual growth rate from 2002 to 2009 of approximately 13%, the revenues from global coronary stents market is predicted to remain relatively constant, although in volume of stents the market is predicted to continue to grow. The growth in volume is due to the appeal for less invasive percutaneous coronary intervention procedures and advances in technology coupled with the increase in the elderly population, obesity rates and advances in technology.
Coronary artery disease is one of the leading causes of death worldwide. The treatment of coronary artery disease includes alternative treatment methodologies, that is, coronary artery bypass grafting or angioplasty (percutaneous coronary intervention) with or without stenting. According to the January 3, 2011 2011 MEDTECH OUTLOOK produced by the BMO (Bank of Montreal) Investment Banking Group, the percutaneous coronary intervention procedures involving stents are increasingly being used to treat coronary artery diseases with an 88.3% penetration rate in 2009.
Our Products
The MGuardTM stent is an embolic protection device based on a protective sleeve, which is constructed out of an ultra-thin polymer mesh and wrapped around the stent. The protective sleeve is comprised of a micron level fiber-knitted mesh, engineered in an optimal geometric configuration and designed for utmost flexibility while retaining strength characteristics of the fiber material (see illustration below). The sleeve expands seamlessly when the stent is deployed, without affecting the structural integrity of the stent, and can be securely mounted on any type of stent.
MGuardTM Deployed in Artery
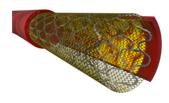
The protective sleeve is designed to provide several clinical benefits:
|
·
|
the mesh diffuses the pressure and the impact of deployment exerted by the stent on the arterial wall and reduces the injury to the vessel;
|
|
|
·
|
it reduces plaque dislodgement and blocks debris from entering the bloodstream during and post procedure (called embolic showers);
|
|
|
·
|
in future products, when drug coated, the mesh is expected to deliver better coverage and uniform drug distribution on the arterial wall and therefore potentially reduce the dosage of the active ingredient when compared to approved drug-eluting stents on the market; and
|
|
|
·
|
it maintains the standards of a conventional stent and therefore should require little to no additional training by physicians.
|
MGuardTM – Coronary Applications
Our MGuardTM Coronary with a bio-stable mesh and our MGuardTM Coronary with a drug-eluting mesh are aimed at the treatment of coronary arterial disease.
MGuardTM Coronary and MGuard Prime™ with a bio-stable mesh. Our first MGuardTM product, the MGuardTM Coronary with a bio-stable mesh, is comprised of our mesh sleeve wrapped around a bare-metal stent. It received CE Mark approval in October 2007 and, in January 2008, we started shipping this product to customers and distributors in Europe. MGuard Prime™ with a bio-stable mesh is comprised of our mesh sleeve wrapped around a cobalt-chromium stent. In comparison to a conventional bare-metal stent, we believe the MGuardTM Coronary and MGuard Prime™ with a bio-stable mesh provide protection from embolic showers. Results of clinical trials on the MGuardTM Coronary stent, including the MAGICAL, PISCIONE and MGuard international registry (iMOS) clinical trials described below (see “Business – Product Development and Critical Milestones - Comparison of Clinical Trial Results to Date with Results Achieved Using Bare Metal or Drug-Eluting Stents in the STEMI population” below), indicate positive outcomes and safety measures, as explained below (see “Business – Product Development and Critical Milestones - Comparison of Clinical Trial Results to Date with Results Achieved Using Bare Metal or Drug-Eluting Stents in the STEMI population” below). The results of these clinical trials for the MGuardTM Coronary stent suggest higher levels of myocardial blush grade 3 (occurrence in 73% of patients in the MAGICAL study and 90% of patients in the PISCIONE study, for the MGuardTM Coronary stent) and lower rates of 30 day and 1 year major adverse cardiac event rates, (2.4% and 5.9%, respectively, for the MGuardTM Coronary stent), as compared to the levels and rates of other bare-metal and drug-eluting stents, as reported by Svilaas, et. al. (“Thrombus Aspiration during Primary Percutaneous Coronary Intervention,” New England Journal of Medicine, Volume 358, 2008). As reported in the study by Svilaas, et. al., myocardial blush grade 3 occurred in 32.2% of patients with a bare-metal stent and 45.7% of patients with a bare-metal stent preceded by an aspiration procedure, and the 30 day and 1 year major adverse cardiac event rates were 9.4% and 20.3%, respectively, for patients with a bare-metal stent and 6.8% and 16.6%, respectively, for patients with a bare-metal stent preceded by an aspiration procedure. Furthermore, results from a recent HORIZONS-AMI trial demonstrated that 1 year major adverse cardiac event rates were 10.9% for patients with drug eluting stents. Myocardial blush grade refers to a 0-3 grade scale given to the adequacy of perfusion and blood flow through an area served by a coronary artery; the longer the blush persists, the poorer the blood flow and the lower the myocardial blush grade. Ndrepepa, et. al. (“5-Year Prognostic Value of No-Reflow Phenomenon After Percutaneous Coronary Intervention in Patients With Acute Myocardial Infarction,” Journal of the American College of Cardiology, Volume 55, Issue 21, 2010) reported that high myocardial blush grades correlate with higher survival rates among affected patients. Sustained performance by the MGuardTM Coronary stent with respect to contributing to higher levels of myocardial blush grade 3 and lower rates of 30 day and 1 year major adverse cardiac event rates would differentiate the MGuardTM Coronary stent from other bare-metal and drug-eluting stents that do not offer such benefits.
MGuardTM Coronary with a drug eluting bio-absorbable mesh. Based upon the clinical profile of MGuardTM Coronary, we anticipate that the MGuardTM Coronary with a drug-eluting bio-absorbable mesh will offer both the comparable myocardial blush grade 3 levels and 30-day and 1-year major adverse cardiac event rates as the MGuardTM Coronary with a bio-stable mesh, as described above, and a comparative restenosis rate, which is the rate at which patients experience formation of new blockages in their arteries, when compared to existing drug-eluting stents. The bio-absorbability of MGuardTM Coronary with a drug eluting bio-absorbable mesh is intended to improve upon the bio-absorbability of other drug-eluting stents, in light of the large surface area of the mesh and the small diameter of the fiber. We intend for the protective sleeve on the MGuardTM Coronary with a drug-eluting bio-absorbable mesh to improve uniform distribution of the applied drug to the vessel wall for improved drug therapy management compared to other drug-eluting stents, where the drug is distributed on the struts only. If this intended result is achieved with respect to the improved and uniform distribution of the applied drug to the vessel wall, the total dosage of the medication potentially could be reduced while increasing its efficacy. MGuardTM Coronary with a drug-eluting bio-absorbable mesh is expected to promote smooth and stable endothelial cell growth and subsequent attachment to the lumen of the vessel wall, which is essential for rapid healing and recovery. In addition, we believe bio-absorbable drug-eluting mesh may enable the use of more effective drug therapies that presently cannot be effectively coated on a metal-based stent due to their poor diffusion capabilities. Because the drug-eluting bio-absorbable mesh will be bio-absorbable, we anticipate that the mesh will completely dissolve after four months, which we expect will result in fewer of the chronic long term side effects that are associated with the presence of the drug.
MGuardTM – Carotid Applications
We intend to market our mesh sleeve coupled with a self-expandable stent (a stent that expands without balloon dilation pressure or need of an inflation balloon) for use in carotid-applications. We believe that our MGuardTM design will provide substantial advantages over existing therapies in treating carotid artery stenosis (blockage or narrowing of the carotid arteries), like conventional carotid stenting and endarterectomy (surgery to remove blockage), given the superior embolic protection characteristics witnessed in coronary arterial disease applications. We intend that the embolic protection will result from the mesh sleeve, as it traps emboli at their source. In addition, we believe that MGuardTM Carotid will provide post-procedure protection against embolic dislodgement, which can occur immediately after a carotid stenting procedure and is often a source of post-procedural strokes. Schofer, et. al. (“Late cerebral embolization after emboli-protected carotid artery stenting assessed by sequential diffusion-weighted magnetic resonance imaging,” Journal of American College of Cardiology Cardiovascular Interventions, Volume 1, 2008) have also shown that the majority of the incidents of embolic showers associated with carotid stenting occur immediately post-procedure.
MGuardTM – Peripheral Applications
We intend to market our mesh sleeve coupled with a self-expandable stent (a stent that expands without balloon dilation pressure or need of an inflation balloon) for use in peripheral applications. Peripheral Artery Disease, also known as peripheral vascular disease, is usually characterized by the accumulation of plaque in arteries in the legs, need for amputation of affected joints or even death, when untreated. Peripheral Artery Disease is treated either by trying to clear the artery of the blockage, or by implanting a stent in the affected area to push the blockage out of the way of normal blood flow.
The Peripheral Artery Disease market consists of three segments: Aortic Aneurysm, Renal, Iliac and Bilary and Femoral-Popliteal procedures. Aortic Aneurysm is a condition in which the aorta, the artery that leads away from the heart, develops a bulge and is likely to burst. This condition often occurs below the kidneys, in the abdomen. Renal, Iliac and Bilary procedures refer to stenting in the kidney, iliac arteries (which supply blood to the legs) and liver, respectively. Femoral-Popliteal procedures involve stenting in vessels in the legs.
As in carotid procedures, peripheral procedures are characterized by the necessity of controlling embolic showers both during and post-procedure. Controlling embolic showers is so important in these indications that physicians often use covered stents, at the risk of blocking branching vessels, to ensure that emboli does not fall into the bloodstream. We believe that our MGuardTM design will provide substantial advantages over existing therapies in treating peripheral artery stenosis (blockage or narrowing of the peripheral arteries).
Product Development and Critical Milestones
Below is a list of the products described above and our projected critical milestones with respect to each. As used below, “Q” stands for our fiscal quarter. While we currently anticipate seeking approval from the U.S. Food and Drug Administration for all of our products in the future, we have only outlined a timetable to seek U.S. Food and Drug Administration approval for our MGuardTM Coronary plus with bio-stable mesh product in our current business plan. We anticipate that our MGuardTM Coronary plus with bio-stable mesh product will be classified as a Class III medical device by the U.S. Food and Drug Administration. The use of the term “to be determined” in the table below with regard to certain U.S. Food and Drug Administration trial milestones indicates that the achievements of such milestones is unable to be accurately predicted as such milestones are too far in the future.
|
Product
|
Indication
|
Start
Development
|
CE Mark
|
European Union Sales
|
FDA Approval
|
U.S. Sales
|
|
MGuard™ Coronary Plus Bio-Stable Mesh
|
Bypass/ Coronary
|
2005
|
Oct. 2007
|
Q1-2008
|
Q4-2014
|
Q4-2014
|
|
MGuard™ Peripheral Plus Bio-Stable Mesh
|
Peripheral Arteries
|
Q1-2011
|
Q1-2012
|
Q2-2012
|
To be determined
|
To be determined
|
|
MGuard™ Carotid Plus Bio-Stable Mesh
|
Carotid
Arteries
|
Q1-2011
|
Q1-2012
|
Q2-2012
|
To be determined
|
To be determined
|
|
MGuard™ Coronary Plus Bio-Absorbable Drug-Eluting Mesh
|
Bypass/ Coronary
|
Q1-2013
|
Q3-2016
|
Q4-2016
|
To be determined
|
To be determined
|
We anticipate that our MGuardTM Coronary plus with bio-stable mesh product will be classified as a Class III medical device by the U.S. Food and Drug Administration.
Pre-Clinical Studies
We performed laboratory and animal testing prior to submitting an application for CE Mark approval for our MGuardTM Coronary with bio-stable mesh. We also performed all CE Mark required mechanical testing of the stent. We conducted pre-clinical animal trials at Harvard and MIT Biomedical Engineering Center BSET lab in July 2006 and August 2007. In these animal trials, on average, the performance of the MGuardTM Coronary with bio-stable mesh was comparable with the performance of control bare-metal stents. Analysis also indicated that in these animal trials the mesh produced levels of inflammation comparable with those levels produced by standard bare-metal stents. No human trials were conducted as part of these pre-clinical trials.
The table below describes our completed and planned pre-clinical trials. The use of the term “To be determined” in the table below with regard to milestone dates in our pre-clinical studies indicates that we have not yet decided when to schedule such milestones.
|
Product
|
Stent
Platform
|
Approval Requirement
|
Start of Study
|
End of Study
|
|
MGuardTM Coronary
|
Bare-Metal Stent Plus Bio-Stable
Mesh
|
CE Mark (European Union + Rest of World)
|
Q4-2006
|
Q3-2007
|
|
Drug-Eluting Mesh (Bare-Metal Stent Plus Drug-Eluting Mesh)
|
CE Mark (European Union + Rest of World)
|
Q3-2013
|
Q4-2014
|
|
|
FDA (U.S.)
|
To be determined
|
To be determined
|
||
|
Cobalt-Chromium Stent Plus Bio-Stable
Mesh
|
FDA
|
Q2-2011
|
Q4-2011
|
|
|
MGuardTM Peripheral/Carotid
|
Self Expending System Plus Mesh
|
CE Mark (European Union + Rest of World)
|
Q4-2011
|
Q1-2012
|
|
MGuardTM Carotid
|
Self Expending System Plus Mesh
|
FDA (U.S.)
|
Peripheral information on animals can be used
|
|
With respect to the preclinical studies for MGuardTM Coronary, the drug-eluting mesh trials have been either delayed or indefinitely suspended and the start of the cobalt-chromium stent plus bio-stable mesh trial was delayed from our previously announced target by one fiscal quarter due to a delay in our receipt of anticipated funding.
With respect to the preclinical studies for MGuard Peripheral/Carotid, the start of study of the Self Expending System Plus Mesh trial has been delayed from our previously announced target due to a delay in our receipt of anticipated funding.
Clinical Trials
The table below describes our completed and planned clinical trials. The use of the term “To be determined” in the table below with regard to milestone dates in our clinical trials indicates that we have not yet decided when to schedule such milestones. All milestone dates set forth in the table below are our best estimates based upon the current status of each clinical trial.
|
Product
|
Stent
Platform
|
Clinical
Trial Sites
|
Follow-up Requirement
|
Objective
|
Study Status
|
|||
|
No. of Patients
|
Start
|
End
Enrollment
|
End of Study
|
|||||
|
MGuardTM Coronary
|
Bare-Metal Stent Plus Bio-Stable
Mesh
|
Germany – two sites
|
12 months
|
Study to
evaluate safety and
performance of MGuardTM system
|
41
|
Q4-2006
|
Q4- 2007
|
Q2-2008
|
|
Brazil – one site
|
12 months
|
30
|
Q4-2007
|
Q1-2008
|
Q2-2009
|
|||
|
Poland – four sites
|
6 months
|
60
|
Q2-2008
|
Q3-2008
|
Q2-2009
|
|||
|
International MGuardTM Observational Study - worldwide - 50 sites
|
12 months
|
1,000
|
Q1-2008
|
Q4-2013
|
Q4-2013
|
|||
|
Israeli MGuardTM Observational Study - Israel - 8 sites
|
6 months
|
100
|
Q2-2008
|
Q3-2011
|
Q3-2012
|
|||
|
Master randomized control trial -
7 countries, 50 centers in South America, Europe and Israel
|
12 months
|
430
|
Q2-2011
|
Q1-2012
|
Q2-2013
|
|||
|
Brazil – 25 sites
|
12 months
|
500
|
Q3-2010
|
To be determined
|
To be determined
|
|||
|
FDA Study - 40 sites, U.S. and out of U.S.
|
12 months
|
Pilot study to
evaluate safety and
performance of
MGuardTM system for FDA approval
|
800
|
Q1-2012 -
Q2-2012 |
Q3-2013 -
Q1-2014 |
Q4-2014 -
Q2-2015 |
||
|
Drug-Eluting Stent (Bare-Metal Stent + Drug Eluting Mesh)
|
South America
and Europe – 10 sites
|
8-12 months
|
Pilot study to
evaluate safety and
performance of
MGuardTM system for FDA and CE Mark approval
|
500
|
To be determined
|
To be determined
|
To be determined
|
|
|
U.S. – 50 sites
|
12 months
|
2,000
|
To be determined
|
To be determined
|
To be determined
|
|||
|
Rest of World as a
registry study
|
8-12 months
|
Evaluation of safety
and efficacy for specific indications
|
400
|
To be determined
|
To be determined
|
To be determined
|
||
|
Study Status
|
||||||||
|
Product
|
Stent
Platform
|
Clinical
Trial Sites
|
Follow-up Requirement
|
Objective
|
No. of Patients
|
Start
|
End
Enrollment
|
End of Study
|
|
MGuardTM Peripheral
|
Self Expanding System + Mesh
|
South America and Europe – four sites
|
12 months
|
Pilot study to
evaluate safety and
performance of MGuardTM system for CE Mark approval
|
50
|
Q1-2012
|
Q3-2012
|
Q4-2014
|
|
South America and Europe – six sites
|
6 months
|
150
|
Q2-2010
|
Q4-2010
|
Q2-2011
|
|||
|
MGuardTM Carotid
|
Self Expanding System + Mesh
|
Rest of World as a registry study
|
6 months
|
Evaluation of safety and efficacy for specific indications post-marketing
|
200
|
Q2-2012
|
Q3-2013
|
Q3-2014
|
Each of the patient numbers and study dates set forth in the tables above are management’s best estimate of the timing and scope of each referenced trial. Actual dates and patient numbers may vary depending on a number of factors, including, without limitation, feedback from reviewing regulatory authorities, unanticipated delays by us, regulatory authorities or third party contractors, actual funding for the trials at the time of trial initiation and initial trial results.
With respect to the MGuardTM Coronary clinical trial for the Master randomized control trial, the start and end enrollment dates have been delayed from our previously announced target by a fiscal quarter and the end of study date has been delayed from our previously announced target by two fiscal quarters due to delays in the necessary approvals of the trial by local ethical committees in certain of the participant countries.
The MGuardTM Coronary clinical trials for the drug-eluting stent have been delayed from our previously announced target due to a delay in our receipt of anticipated funding.
With respect to the MGuardTM Peripheral clinical trial for the self expanding system + mesh, the start date has been delayed from our previously announced start date due to a delay in our receipt of anticipated funding.
Completed Clinical Trials for MGuardTM Coronary Bare-Metal Stent Plus Bio-Stable Mesh
As shown in the table above, we have completed five clinical trials with respect to our MGuardTM Coronary with bio-stable mesh. Our first study, conducted at two centers in Germany, included 41 patients with either saphenous vein graft coronary interventions or native coronary lesions treatable by a stenting procedure (blockages where no bypass procedure was performed). The MGuardTM Coronary rate of device success, meaning the stent was successfully deployed in the target lesion, was 100% and the rate of procedural success, meaning there were no major adverse cardiac events prior to hospital discharge, was 95.1%. At six months, only one patient (2.5% of participants) had major myocardial infarction (QWMI) and 19.5% of participants had target vessel revascularization (an invasive procedure required due to a stenosis in the same vessel treated in the study). This data supports MGuardTM’s safety in the treatment of vein grafts and native coronary legions.
Our 2007 study in Brazil included 30 patients who were candidates for a percutaneous coronary intervention (angioplasty) due to narrowing of a native coronary artery or a bypass graft. In all patients, the stent was successfully deployed with perfect blood flow parameters (the blood flow parameter is a measurement of how fast the blood flows in the arteries and the micro circulation system in the heart). There were no major cardiac events at the time of the follow-up 30 days after the deployment of the stents.
The study in Poland included 60 patients with acute ST-segment elevation myocardial infarction (the most severe form of a heart attack, referred to as “STEMI”). The purpose of the study was to confirm the clinical performance of MGuardTM Coronary with bio-stable mesh when used in STEMI patients where percutaneous coronary intervention is the primary line of therapy. Perfect blood flow in the artery was achieved in 90% of patients, perfect blood flow into the heart muscle was achieved in 73% of patients and complete restoration of electrocardiogram normality was achieved in 61% of patients. The total major adverse cardiac events rate during the six-month period following the deployment of the stents was 0%.
Ongoing Clinical Trials for MGuardTM Coronary Bare-Metal Stent Plus Bio-Stable Mesh.
Our ongoing observation study in Europe is an open registry launched in the first fiscal quarter of 2009. This registry is expected to enroll up to 1,000 patients and is aimed at establishing the performance of MGuardTM Coronary with bio-stable mesh in a “real world” population. To date, the primary countries to join are Austria, Czech Republic and Hungary. The primary endpoint that this registry will evaluate is the occurrence of major adverse cardiac events at six months following deployment of the stent, and the clinical follow-up will continue for a period of up to one year per patient. As of October 11, 2011, 467 patients of the prospective 1,000 have been enrolled in 28 sites.
Our ongoing observational study in Israel is an open registry launched in the fourth fiscal quarter of 2009. This registry is expected to enroll up to 100 patients. The purpose of this study is to support local Israeli regulatory approval. The primary endpoint that this registry will evaluate is the occurrence of major adverse cardiac events at 30 days following deployment of the stent, and the clinical follow-up will be conducted at six months following deployment of the stent. As of October 11, 2011, 74 patients of the prospective 100 have been enrolled.
In the third fiscal quarter of 2010, we launched a Brazilian registry to run in 25 Brazilian sites and enroll 500 patients. The primary endpoint that this registry will evaluate is the occurrence of major adverse cardiac events at six months following the deployment of the stent, and the clinical follow-up will continue for a period of up to one year per patient. As of October 11, 2011, 12 patients of the prospective 500 have been enrolled.
Comparison of Clinical Trial Results to Date with Results Achieved Using Bare Metal or Drug-Eluting Stents in the STEMI population
We conducted a meta-analysis of data from four clinical trials in which MGuardTM was used:
|
·
|
The MAGICAL study, a single arm study in which 60 acute ST-segment elevation myocardial infarction (the most severe form of a heart attack, referred to as STEMI) patients with less than 12 hours symptom onset were enrolled, as reported in “Mesh Covered Stent in ST-segment Elevation Myocardial Infarction” in EuroIntervention, 2010;
|
|
·
|
the PISCIONE study, a single arm study in which 100 STEMI patients were enrolled, as reported in “Multicentre Experience with MGuard Net Protective Stent in ST-elevation Myocardial Infarction: Safety, Feasibility, and Impact on Myocardial Reperfusion” in Catheter Cardiovasc Interv, 2009;
|
|
·
|
the iMOS study, a Registry on MGuard use in the “real-world” population, from a study whose data was not published; and
|
|
·
|
the Jain study, which looks at a small group of 51 STEMI patients, as reported in “Prevention of Thrombus Embolization during Primary Percutaneous Intervention Using a Novel Mesh Covered Stent” in Catheter Cardiovasc Interv, 2009.
|
Our meta-analysis included data from the following trials:
|
·
|
The CADILLAC (Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications) study, which found that primary stent implantation is a preferred strategy for the treatment of acute myocardial infarction, as reported in “A Prospective, Multicenter, International Randomized Trial Comparing Four Reperfusion Strategies in Acute Myocardial Infarction: Principal Report of the Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications (CADILLAC)” Trial in Journal of American College of Cardiology, 2001;
|
|
·
|
The EXPORT trial which was a randomized open-label study whose primary endpoint was to evaluate flow improvement in AMI patients using either conventional stenting or aspiration followed by stenting, as reported in “Systematic Primary Aspiration in Acute Myocardial Percutaneous Intervention: A Multicentre Randomised Controlled Trial of the Export Aspiration Catheter” in EuroIntervention, 2008;
|
|
·
|
The EXPIRA trial which was a single-center study aimed to explore pre-treatment with manual thrombectomy as compared to conventional stenting, as reported in “Thrombus Aspiration During Primary Percutaneous Coronary Intervention Improves Myocardial Reperfusion and Reduces Infarct Size: The EXPIRA (Thrombectomy with Export Catheter in Infarct-related Artery During Primary Percutaneous Coronary Intervention) Prospective, Randomized Trial” in Journal of American College of Cardiology, 2009;
|
|
·
|
The REMEDIA trial, whose objective was to assess the safety and efficacy of the EXPORT catheter for thrombus aspiration in STEMI patients, as reported in “Manual Thrombus-Aspiration Improves Myocardial Reperfusion: The Randomized Evaluation of the Effect of Mechanical Reduction of Distal Embolization by Thrombus-Aspiration in Primary and Rescue Angioplasty (REMEDIA) Trial” in Journal of American College of Cardiology, 2005;
|
|
·
|
The Horizons-AMI (Harmonizing Outcomes with RevascularIZatiON and Stents in Acute MI), which is the largest randomized trial which compared DES to BMS in MI patients, as reported in “Paclitaxel-Eluting Stents Versus Bare-Metal Stents in Acute Myocardial Infarction” in New England Journal of Medicine, 2009; and
|
|
·
|
The TAPAS Trial which showed that thrombus aspiration before stenting benefits MI patients, as reported in “Thrombus Aspiration During Primary Percutaneous Coronary Intervention” in New England Journal of Medicine, 2009.
|
The meta analysis of MGuardTM outcomes in STEMI population show comparable rates of thrombolysis in myocardial infarction (TIMI) 3 flow with no significant difference of the historical control as compared to MGuardTM (88.5% and 91.7%, respectively), while the rates of myocardial blush grade score 3 (37.3% for the historical control and 81.6% for MGuardTM) and ST segment resolution>70% (53.6% for the historical control and 79.1% for MGuardTM) are statistically significantly better with the MGuardTM. MGuardTM also appears consistently superior at the 30 days major adverse cardiac event (8.4% for the historical control and 2.4% for MGuardTM) and 1 year major adverse cardiac event (13.3% for the historical control and 5.9% for MGuardTM) endpoints. The data appears in the following tables.
|
NAME OF STUDY
|
|||||
|
MAGICAL
|
PISCIONE
|
iMOS
|
Jain
|
Average
|
|
|
Number of Patients
|
60
|
100
|
203
|
51
|
414 (Total)
|
|
Thrombolysis in myocardial infarction 0-1,%
|
0
|
0
|
1.2
|
0
|
0.6
|
|
Thrombolysis in myocardial infarction 3,%
|
90
|
85
|
93.5
|
100
|
91.7
|
|
Myocardial blush grade 0-1,%
|
3.3
|
0
|
--
|
--
|
1.2
|
|
Myocardial blush grade 3,%
|
73
|
90
|
80
|
--
|
81.6
|
|
ST segment resolution>70%,%
|
61
|
90
|
--
|
--
|
79.1
|
|
ST segment resolution>50%,%
|
88
|
--
|
85.4
|
96
|
87.6
|
|
30 day major adverse cardiac event,%
|
0
|
2.2
|
3.2
|
--
|
2.4
|
|
6 month major adverse cardiac events,%
|
0
|
4.5
|
6.0
|
--
|
4.6
|
|
1 year major adverse cardiac events,%
|
--
|
5.6
|
6.0
|
6.0
|
5.9
|
|
1 year target vessel revascularization
|
2.3
|
2.3
|
6.0
|
2.8
|
|
|
Acute Binary Resteonosis 6M,%
|
--
|
--
|
19.0*
|
--
|
19.0
|
|
Trial
|
CADILLAC
|
Horizons-AMI
|
Horizons-AMI
|
TAPAS
|
TAPAS
|
EXPORT
|
EXPORT
|
EXPIRA
|
EXPIRA
|
REMEDIA
|
REMEDIA
|
Historical comparison
|
MGuard
|
Level of Significance
|
|
Group
|
Stent + Abciximab
|
BMS
|
DES
|
Thrombus aspiration
|
control
|
control
|
TA
|
control
|
Thrombus aspiration
|
Thrombus aspiration
|
control
|
Average
|
Average
|
|
|
Number of Patients
|
524
|
749
|
2257
|
535
|
536
|
129
|
120
|
87
|
88
|
50
|
49
|
5124 (total)
|
414 (total)
|
|
|
Thrombolysis in myocardial infarction 0-1,%
|
--
|
--
|
--
|
--
|
--
|
3.9
|
2.4
|
1.1
|
0
|
--
|
--
|
2.1
|
0.6
|
|
|
Thrombolysis in myocardial infarction 3,%
|
96.9
|
87.6
|
89.8
|
86
|
82.5
|
76.9
|
82
|
--
|
--
|
--
|
--
|
88.5
|
91.7
|
|
|
Myocardial blush grade 0-1,%
|
48.7
|
--
|
--
|
17.1
|
26.3
|
31.6
|
27.6
|
40.2
|
11.4
|
32
|
55.1
|
35.2
|
1.2
|
*
|
|
Myocardial blush grade 3,%
|
17.4
|
--
|
--
|
45.7
|
32.2
|
25.4
|
35.8
|
--
|
--
|
--
|
--
|
37.3
|
81.6
|
**
|
|
ST segment resolution>70%,%
|
62
|
--
|
--
|
56.6
|
44.2
|
--
|
--
|
39.1
|
63.6
|
58
|
36.7
|
53.6
|
79.1
|
|
|
ST segment resolution>50%,%
|
--
|
--
|
--
|
--
|
--
|
71.9
|
85
|
--
|
--
|
--
|
--
|
78.2
|
87.6
|
|
|
30 day major adverse cardiac event,%
|
4.4
|
--
|
--
|
6.8
|
9.4
|
--
|
--
|
--
|
--
|
10
|
10.2
|
8.4
|
2.4
|
**
|
|
6 month major adverse cardiac events,%
|
10.2
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
10.2
|
4.6
|
|
|
1 year major adverse cardiac events,%
|
--
|
13.1
|
10.9
|
16.6
|
20.3
|
--
|
--
|
--
|
--
|
--
|
--
|
13.3
|
5.9
|
*
|
|
Acute Binary Resteonosis 6 month,%
|
20.8
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
20.8
|
19.0
|
|
|
1 year target vessel revascularization
|
7.4
|
4.6
|
12.9
|
11.2
|
||||||||||
|
Acute Binary Resteonosis 1 year,%
|
--
|
21
|
8.3
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
--
|
11.5
|
--
|
Future Clinical Trials for MGuardTM Coronary
We anticipate that additional studies will be conducted to meet registration requirements in key countries, particularly the U.S. We have currently budgeted $8.5 million for the U.S. Food and Drug Administration trial. We expect that post-marketing trials will be conducted to further establish the safety and efficacy of the MGuardTM Coronary with bio-stable mesh in specific indications. These trials will be designed to facilitate market acceptance and expand the use of the product. We anticipate that the MGuard for Acute ST Elevation Reperfusion Trial (MASTER Trial), for which we have budgeted $2.0 million, will serve to promote market acceptance of the product and expand its usage. The MASTER Trial is a multinational, randomized controlled trial of the MGuard™ mesh protective coronary stent that includes 432 patients in a two-arm, parallel design, with the intention of testing the MGuard™ stent against commercially approved bare-metal stents or drug-eluting stents with respect to myocardial reperfusion in primary angioplasty for the treatment of acute ST-elevation myocardial infarction. In other countries, we believe that we generally will be able to rely upon the CE Mark approval of the product, as well as the results of the U.S. Food and Drug Administration trial and MASTER Trial in order to obtain local approvals.
In the second fiscal quarter of 2011, we began a prospective, randomized study in Europe, South America and Israel to demonstrate the superiority of the MGuard™ stent over commercially-approved bare-metal and drug-eluting stents in achieving better myocardial reperfusion (the restoration of blood flow) in primary angioplasty for the treatment of acute STEMI. We anticipate that this trial will enroll 432 subjects, 50% of whom will be treated with an MGuard™ stent and 50% of whom will be treated with a commercially-approved bare-metal or drug-eluting stent. The primary endpoint of this study is the occurrence of the restoration of normal electrocardiogram reading. As of October 11, 2011, 28 patients of the prospective 432 have been enrolled.
We also plan to conduct a large clinical study for U.S. Food and Drug Administration approval in the U.S. We expect that this study will be a prospective, multicenter, randomized clinical trial. Its primary objective will be to compare the safety and the effectiveness of the MGuard™ stent in the treatment of de novo stenotic lesions in coronary arteries in patients undergoing primary revascularization (a surgical procedure for the provision of a new, additional, or augmented blood supply to the heart) due to acute myocardial infarction with the MultiLink Vision stent system from Abbott Vascular. We expect total enrollment of approximately 800 subjects, at up to 40 sites throughout the U.S. and Europe. The combined primary endpoint of this study will be the occurrence of Blush Score of 3, which would indicate that blood supply to the heart muscle is optimal, following the procedure, and the occurrence of target vessel failure (a composite endpoint of cardiac death, reoccurrence of a heart attack and the need for a future invasive procedure to correct narrowing of the coronary artery). This study is expected to start in 2012, and the enrollment phase is expected to last 18 months. We expect that subjects will be followed for 12 months with assessments at 30 days, six months and 12 months. This plan is tentative, and is subject to change to conform with U.S. Food and Drug Administration regulations and requirements.
Planned Trials for future MGuardTM Peripheral and Carotid Products
As shown in the table at the beginning of this section, we also plan to conduct clinical trials for our additional products in development in order to obtain approval for their use. We anticipate that local distributors in the countries in which such trials will take place will support many of these studies.
Growth Strategy
Our primary business objective is to utilize our proprietary technology to become the industry standard for treatment of acute coronary syndromes and to provide a superior solution to the common acute problems caused by current stenting procedures, such as restenosis, embolic showers and late thrombosis. We are pursuing the following business strategies in order to achieve this objective.
|
·
|
Successfully commercialize MGuardTM Coronary with bio-stable mesh. We have begun commercialization of MGuardTM Coronary with a bio-stable mesh in Europe, Asia and Latin America through our distributor network and we are aggressively pursuing additional registrations and contracts in other countries such as Russia, Canada, South Korea, Belgium, the Netherlands and certain smaller countries in Latin America. By the time we begin marketing this product in the U.S., we expect to have introduced the MGuardTM technology to clinics and interventional cardiologists around the world, and to have fostered brand name recognition and widespread adoption of MGuardTM Coronary. We plan to accomplish this by participating in national and international conferences, conducting and sponsoring clinical trials, publishing articles in scientific journals, holding local training sessions and conducting electronic media campaigns.
|
|
·
|
Successfully develop the next generation of MGuardTM stents. While we market our MGuardTM Coronary with bio-stable mesh, we intend to develop the MGuardTM Coronary with a drug-eluting mesh. We are also working on our MGuardTM stents for peripheral and carotid, for which we expect to have CE mark approval by the first quarter of 2012. In addition, we released our cobalt-chromium version of MGuardTM, MGuard Prime™, in 2010, which we anticipate will replace MGuardTM over the next couple of years.
|
|
·
|
Continue to leverage MGuardTM technology to develop additional applications for interventional cardiologists and vascular surgeons. In addition to the applications described above, we believe that we will eventually be able to utilize our proprietary technology to address imminent market needs for new product innovations to significantly improve patients’ care. We have secured intellectual property using our unique mesh technology in the areas of brain aneurism, treating bifurcated blood vessels and a new concept of distal protective devices. We believe these areas have a large growth potential given, in our view, that present solutions are far from satisfactory, and there is a significant demand for better patient care. We believe that our patents can be put into practice and that they will drive our growth at a later stage.
|
|
·
|
Work with world-renowned physicians to build awareness and brand recognition of MGuardTM portfolio of products. We intend to work closely with leading cardiologists to evaluate and ensure the efficacy and safety of our products. We intend that some of these prominent physicians will serve on our Scientific Advisory Board, which is our advisory committee that advises our board of directors, and run clinical trials with the MGuardTM Coronary stent. We believe these individuals, once convinced of the MGuardTM Coronary stent’s appeal, will be invaluable assets in facilitating the widespread adoption of the stent. In addition, we plan to look to these cardiologists to generate and publish scientific data on the use of our products, and to present their findings at various conferences they attend. Dr. Gregg W. Stone, director of Cardiovascular Research and Education at the Center for Interventional Vascular Therapy of New York Presbyterian Hospital/Columbia University Medical Center and the co-director of Medical Research and Education at The Cardiovascular Research Foundation is the study chairman for the MASTER Trial. Dr. Donald Cutlip, Executive Director of Clinical Investigation at the Harvard Clinical Research Institute, will provide scientific leadership of the U.S. Food and Drug Administration trials. On October 4, 2011, InspireMD Ltd., our wholly-owned subsidiary, entered into a clinical trial services agreement with Harvard Clinical Research Institute, Inc., pursuant to which Harvard Clinical Research Institute, Inc. will conduct a study entitled “MGuard Stent System Clinical Trial in Patients with Acute Myocardial Infarction” on our behalf. We will pay Harvard Clinical Research Institute, Inc. an estimated fee of approximately $10 million for conducting the study, subject to adjustment dependent upon changes in the scope and nature of the study, as well as other costs to be determined by the parties.
|
|
·
|
Continue to protect and expand our portfolio of patents. Our patents and their protection are critical to our success. We have filed ten separate patents for our MGuardTM technology in Canada, China, Europe, Israel, India, South Africa and the U.S. We believe these patents cover all of our existing products, and can be useful for future technology. We intend to continue patenting new technology as it is developed, and to actively pursue any infringement upon our patents. On October 25, 2011, one of our patent applications, U.S. patent application 11/582,354, was issued as U.S. Patent 8,043,323 .
|
|
·
|
Develop strategic partnerships. We intend to partner with medical device, biotechnology and pharmaceutical companies to assist in the development and commercialization of our proprietary technology. Although we have not yet done so, we plan to partner with a company in the U.S. to guide products through U.S. Food and Drug Administration approval and to support the sale of MGuardTM stents in the U.S.
|
As noted above, we previously filed patents for our MGuardTM technology in China, as part of our intended growth strategy. However, upon further consideration of the cost and resources required to achieve patent protection in China, we elected to prioritize our pursuit of growth opportunities in other countries and, as such, have ceased our growth efforts in China for the current time period. We intend to reevaluate our strategy towards commercialization of our MGuardTM technology in China in the future.
Competition
The stent industry is highly competitive. The bare-metal stent and the drug-eluting stent markets in the U.S. and Europe are dominated by Abbott Laboratories, Boston Scientific Corporation, Johnson & Johnson and Medtronic, Inc. Due to ongoing consolidation in the industry, there are high barriers to entry for small manufacturers in both the European and the U.S. markets. However, due to less stringent regulatory approval requirements in Europe, we believe that the European market is somewhat more fragmented, and small competitors appear able to gain market share with greater ease.
In the future, we believe that physicians will look to next-generation stent technology to compete with currently existing therapies. These new technologies will likely include bio-absorbable stents, stents that are customizable for different lesion lengths, stents that focus on treating bifurcated lesions, and stents with superior polymer and drug coatings. Some of the companies developing new stents are The Sorin Group, Xtent, Inc., Cinvention AG, OrbusNeich, Biotronik SE & Co. KG, Svelte Medical Systems, Inc., Reva Inc. and Stentys SA, among others. To address current issues with drug-eluting stents, The Sorin Group and Cinvention AG have developed stents that do not require a polymer coating for drug delivery, thereby expanding the types of drugs that can be used on their respective stents. OrbusNeich has addressed the problem differently, developing a stent coated with an antibody designed to eliminate the need for any drug at all. Xtent, Inc. has been concentrating on a stent that can be customized to fit different sized lesions, so as to eliminate the need for multiple stents in a single procedure. Biotronik SE & Co. KG is currently developing bio-absorbable stent technologies, and Abbott Laboratories is currently developing a bio-absorbable drug-eluting stent. These are just a few of the many companies working to improve stenting procedures in the future as the portfolio of available stent technologies rapidly increases. As the market moves towards next-generation stenting technologies, minimally invasive procedures should become more effective, driving the growth of the market in the future. We plan to continue our research and development efforts in order to be at the forefront of the acute myocardial infarction solutions.
According to the January 3, 2011 2011 MEDTECH OUTLOOK produced by the BMO (Bank of Montreal) Investment Banking Group, the worldwide stent market is dominated by four major players, with a combined total market share of approximately 96%. Within the bare metal stent market and drug-eluting stent market, the top four companies have approximately 92% and 98% of the market share, respectively. These four companies are Abbott Laboratories, Boston Scientific Corporation, Johnson & Johnson and Medtronic, Inc. To date our sales are not significant enough to register in market share. As such, one of the challenges we face to the further growth of MGuard™ is the competition from numerous pharmaceutical and biotechnology companies in the therapeutics area, as well as competition from academic institutions, government agencies and research institutions. Most of our current and potential competitors, including but not limited to those listed above, have, and will continue to have, substantially greater financial, technological, research and development, regulatory and clinical, manufacturing, marketing and sales, distribution and personnel resources than we do.
In addition to the challenges from our competitors, we face challenges related specifically to our products. None of our products are currently approved by the U.S. Food and Drug Administration. Clinical trials necessary to support a pre-market approval application to the U.S. Food and Drug Administration for our MGuard™ stent will be expensive and will require the enrollment of a large number of patients, and suitable patients may be difficult to identify and recruit, which may cause a delay in the development and commercialization of our product candidates. Furthermore, our rights to our intellectual property with respect to our products could be challenged. Based on the prolific litigation that has occurred in the stent industry and the fact that we may pose a competitive threat to some large and well-capitalized companies that own or control patents relating to stents and their use, manufacture and delivery, we believe that it is possible that one or more third parties will assert a patent infringement claim against the manufacture, use or sale of our MGuard™ stent based on one or more of these patents.
We note that an additional challenge facing our products comes from drug-eluting stents. Over the last decade, there has been an increasing tendency to use drug-eluting stents in percutaneous coronary intervention (PCI), with a usage rate of drug-eluting stents in PCI approaching 70-80% in some countries, even though drug-eluting stents do not address thrombus management in acute myocardial infarction. A recent HORIZONS-AMI trial that compared drug-eluting stents to bare-metal stents in STEMI patients failed to show any benefit of drug-eluting stents as compared to bare-metal stents with regard to safety (death, re-infarction, stroke, or stent thrombosis), but showed the 1 year target vessel revascularization (TLR) rate for drug-eluting stent patients was only 4.6%, as compared to 7.4% for patients with bare-metal stents. However, based on data from over 350 patients across three clinical trials, the TLR rate for MGuardTM was 2.8%. (This data is comprised of: (i) a TLR rate of 2.3% for a 100-patient study, as reported in “Multicentre Experience with MGuard Net Protective Stent in ST-elevation Myocardial Infarction: Safety, Feasibility, and Impact on Myocardial Reperfusion” in Catheter Cardiovasc Interv, 2009; (ii) a TLR rate of 2.3% for a sub-group of 203 STEMI patients from the International MGuardTM Observational Study; and (iii) a TLR rate of 6.0% for a group of 51 heart attack patients, as reported in “Prevention of Thrombus Embolization during Primary Percutaneous Intervention Using a Novel Mesh Covered Stent” in Catheter Cardiovasc Interv, 2009).
Another challenge facing the MGuardTM products is that placing the stent at the entrance to large side branches, known as jailing large side branches, is not recommended with the MGuardTM Coronary stent, because there is risk of thrombosis. Jailing requires the need to cross the stent with guidewire and to create an opening with the balloon to allow proper flow, which can be achieved with lower risk by using other bare-metal stents.
Research and Development Expenses
During each of 2010 and 2009, we spent approximately $1.3 million on research and development.
Sales and Marketing
Sales and Marketing
In October 2007, MGuardTM Coronary with a bio-stable mesh received CE Mark approval in the European Union, and shortly thereafter was commercially launched in Europe through local distributors. We are also in negotiations with additional distributors in Europe, Asia and Latin America and are currently selling our MGuardTM Coronary with a bio-stable mesh in more than 30 countries.
Until U.S. Food and Drug Administration approval of our MGuardTM Coronary with a bio-stable mesh, which we are targeting for 2014, we plan to focus our marketing efforts primarily on Europe, Asia and Latin America. Within Europe, we have focused on markets with established healthcare reimbursement from local governments such as Italy, Germany, Great Britain, France, Greece, Austria, Benelux, Denmark, Hungary, Poland, Slovenia, Czech Republic and Slovakia.
In addition to utilizing local and regional distributor networks, we are using international trade shows and industry conferences to gain market exposure and brand recognition. We plan to work with leading physicians to enhance our marketing efforts. As sales volume increases, we plan to open regional offices and manage sales activities more closely in each of our defined geographical regions, and to provide marketing support to local and regional distributors in each area.
Product Positioning
The MGuardTM Coronary has initially penetrated the market by entering market segments with indications that present high risks of embolic dislodgement, notably acute myocardial infarction and saphenous vein graft coronary interventions. The market penetration of the MGuardTM Coronary in 2010 was minimal, with total sales in the twelve months ended December 31, 2010 of approximately $5 million representing less than 1% of the total sales of the acute myocardial infarction solutions market.
When performing stenting procedures in patients with acute coronary symptoms, interventional cardiologists face a difficult dilemma in choosing between bare-metal stents, which have a high rate of restenosis, and drug-eluting stents, which have a high rate of late stent thrombosis, require administration of anti-platelet drugs for at least one year post procedure and are more costly than bare-metal stents. We are marketing our platform technology, MGuardTM, as a superior and cost effective solution to these currently unmet needs of interventional cardiologists. We believe our MGuardTM technology is clinically superior to bare-metal stents because it provides embolic protection during and post-procedure. We believe our MGuardTM technology is clinically superior to drug-eluting stents, due to its lower stent thrombosis rate and protection from embolic showers during and post-procedure.
In addition to the advantages of the MGuardTM technology that we believe to exist, the MGuardTM technology maintains the deliverability, crossing profile, and dilatation pressure of a conventional stent, and interventional cardiologists do not have to undergo extensive training before utilizing the product.
Insurance Reimbursement
In most countries, a significant portion of a patient’s medical expenses is covered by third-party payors. Third-party payors can include both government funded insurance programs and private insurance programs. While each payor develops and maintains its own coverage and reimbursement policies, the vast majority of payors have similarly established policies. All of the MGuardTM products sold to date have been designed and labeled in such a way as to facilitate the utilization of existing reimbursement codes, and we intend to continue to design and label our products in a manner consistent with this goal.
While most countries have established reimbursement codes for stenting procedures, certain countries may require additional clinical data before recognizing coverage and reimbursement for the MGuardTM products or in order to obtain a higher reimbursement price. In these situations, we intend to complete the required clinical studies to obtain reimbursement approval in countries where it makes economic sense to do so.
In the U.S., once the MGuardTM Coronary with bio-stable mesh is approved by the U.S. Food and Drug Administration, it will be eligible for reimbursement from the Centers for Medicare and Medicaid Services, which serve as a benchmark for all reimbursement codes. While there is no guarantee these codes will not change over time, we believe that the MGuardTM will be eligible for reimbursement through both governmental healthcare agencies and most private insurance agencies in the U.S.
Intellectual Property
Patents
We have filed ten separate patents for our MGuardTM technology in Canada, China, Europe, Israel, India, South Africa and the U.S. for an aggregate of 35 filed patents. These patents cover percutaneous therapy, knitted stent jackets, stent and filter assemblies, in vivo filter assembly, optimized stent jackets, stent apparatuses for treatment via body lumens and methods of use, stent apparatuses for treatment via body lumens and methods of manufacture and use, and stent apparatuses for treatment of body lumens, among others. In lay terms, these patents generally cover two parts of our products: the mesh sleeve, with and without a drug, and the delivery mechanism of the stent. On October 25, 2011, one of our patent applications, U.S. patent application 11/582,354, was issued as U.S. Patent 8,043,323. None of the other patents have been granted to date. We believe these patents, once issued, will cover all of our existing products and be useful for future technology. We also believe that the patents we have filed, in particular those covering the use of a knitted micron-level mesh sleeve over a stent for various indications, would create a significant barrier for another company seeking to use similar technology.
To date, we are not aware of other companies that have patent rights to a micron fiber, releasable knitted fiber sleeve over a stent. However, larger, better funded competitors own patents relating to the use of drugs to treat restenosis, stent architecture, catheters to deliver stents, and stent manufacturing and coating processes as well as general delivery mechanism patents like rapid exchange. Stent manufacturers have historically engaged in significant litigation, and we could be subject to claims of infringement of intellectual property from one or more competitors. Although we believe that any such claims would be un-founded, such litigation would divert attention and resources away from the development of MGuardTM stents. Other manufacturers may also challenge the intellectual property that we own, or may own in the future. We may be forced into litigation to uphold the validity of the claims in our patent portfolio, an uncertain and costly process.
Trademarks
We use the InspireMD and MGuard trademarks. We have registered these trademarks in Europe. The trademarks are renewable indefinitely, so long as we continue to use the mark in Europe and make the appropriate filings when required.
Government Regulation
The manufacture and sale of our products are subject to regulation by numerous governmental authorities, principally the European Union CE Mark, the U.S. Food and Drug Administration and other corresponding foreign agencies.
Sales of medical devices outside the U.S. are subject to foreign regulatory requirements that vary widely from country to country. These laws and regulations range from simple product registration requirements in some countries to complex clearance and production controls in others. As a result, the processes and time periods required to obtain foreign marketing approval may be longer or shorter than those necessary to obtain U.S. Food and Drug Administration market authorization. These differences may affect the efficiency and timeliness of international market introduction of our products. For countries in the European Union, medical devices must display a CE Mark before they may be imported or sold. In order to obtain and maintain the CE Mark, we must comply with the Medical Device Directive 93/42/EEC and pass an initial and annual facilities audit inspections to ISO 13485 standards by an European Union inspection agency. We have obtained ISO 13485 quality system certification and the products we currently distribute into the European Union display the required CE Mark. In order to maintain certification, we are required to pass annual facilities audit inspections conducted by European Union inspectors.
As noted below, we currently have distribution agreements for our products with distributors in the following countries: Italy, Germany, Austria, Czech Republic, Slovakia, France, Slovenia, Greece, Cyprus, Portugal, Spain, Poland, Hungary, Estonia, Lithuania, Ukraine, United Kingdom, Holland, Russia, Latvia, Brazil, Chile, Costa Rica, Mexico, Argentina, Colombia, India, Sri Lanka, South Africa, Pakistan and Israel. We are subject to governmental regulation in each of these countries and we are not permitted to sell all of our products in each of these countries. While each of the European Union member countries accepts the CE Mark as its sole requirement for marketing approval, some of these countries still require us to take additional steps in order to gain reimbursement rights for our products. Furthermore, while we believe that each of the above-listed countries that is not a member of the European Union accepts the CE Mark as its primary requirement for marketing approval, each such country requires additional regulatory requirements for final marketing approval for MGuard Prime™. Additionally, in Canada, we are required to pass annual facilities audit inspections performed by Canadian inspectors. Furthermore, we are currently targeting additional countries in Europe, Asia, and Latin America. We believe that each country that we are targeting also accepts the CE Mark as its primary requirement for marketing approval. We intend that the results of the MASTER Trial will satisfy any additional governmental regulatory requirements in each of the countries where we currently distribute our products and in any countries that we are currently targeting for expansion.
MGuard Prime™ received CE Mark approval in the European Union in October 2010 and marketing approval in Israel in September 2011. We are currently seeking marketing approval for MGuard Prime™ in Brazil, Malaysia, Mexico, Russia, Serbia, Singapore, Argentina, India, Sri Lanka and Pakistan. While each of these countries accepts the CE Mark as its primary requirement for marketing approval and does not require any additional tests, each country does require some additional regulatory requirements for marketing approval. More specifically, for the approval process in Malaysia, we need to submit an application for regulatory approval, which we anticipate will be granted in three months. For the approval process in Mexico, we need to submit an application for regulatory approval, which we anticipate will be granted in twelve months. For the approval process in Serbia, we need to submit an application for regulatory approval, which we anticipate will be granted in twelve months. For the approval process in Singapore, we need to submit an application for regulatory approval, which we anticipate will be granted in six months. For the approval process in Argentina, we need to submit an application for regulatory approval, which we anticipate will be granted in approximately twelve months. For the approval process in India, we need to submit an application for regulatory approval, which we anticipate will be granted in approximately twelve months. For the approval process in Sri Lanka, we need to submit an application for regulatory approval, which we anticipate will be granted in six to twelve months. For the approval process in Pakistan, we need to submit an application for regulatory approval, which we anticipate will be granted in six to twelve months. In Israel, where we received marketing approval in September 2011, we will be subject to annual renewal of our marketing approval. Regulators in Israel may request additional documentation or other materials and results of studies from medical device manufacturers such as us as part of the renewal process. Generally, however, the annual renewal of marketing approval is given automatically, barring a material change in circumstances or results.
For the approval process in Brazil, we must comply with Brazilian Good Manufacturing Practice, or GMP, quality system requirements. ANVISA, Brazil’s regulatory agency, must conduct an inspection of MGuard Prime™ to determine compliance with Brazil GMP regulations. Upon successful completion of an audit, ANVISA will then issue the GMP certificate necessary to register a medical device in Brazil. Once we receive the necessary GMP certificate, we can apply for regulatory approval. We anticipate that the approval process in Brazil will take between one and two years.
For the approval process in Russia, we must first provide test samples of MGuard Prime™ and then conduct government-authorized testing. We must then submit the test results together with our application for regulatory approval to the Russian regulatory authority. We anticipate that the approval process in Russia will take between five to twelve months.
Please refer to the table below setting forth the approvals for MGuard™ and MGuard Prime™ on a country-by-country basis.
|
APPROVALS OF MGUARD™ AND MGUARD PRIME™ ON A COUNTRY-BY-COUNTRY BASIS
|
|
Countries
|
MGuard™
|
MGuard Prime™
|
Countries
|
MGuard™
|
MGuard Prime™
|
|
|
Argentina
|
Y
|
N
|
Italy
|
Y
|
Y
|
|
|
Austria
|
Y
|
Y
|
Latvia
|
Y
|
N
|
|
|
Brazil
|
Y
|
N
|
Lithuania
|
Y
|
N
|
|
|
Chile
|
Y
|
N
|
Mexico
|
Y
|
N
|
|
|
Colombia
|
Y
|
N
|
Pakistan
|
Y
|
N
|
|
|
Costa Rica
|
Y
|
N
|
Poland
|
Y
|
Y
|
|
|
Cyprus
|
Y
|
N
|
Portugal
|
Y
|
Y
|
|
|
UK
|
Y
|
N
|
Russia
|
Y
|
N
|
|
|
Estonia
|
Y
|
Y
|
Slovakia
|
Y
|
N
|
|
|
France
|
Y
|
Y
|
Czech Rep
|
Y
|
N
|
|
|
Germany
|
Y
|
Y
|
Slovenia
|
Y
|
N
|
|
|
Greece
|
Y
|
Y
|
South Africa
|
Y
|
N
|
|
|
Holland (Netherlands)
|
N
|
Y
|
Spain
|
Y
|
Y
|
|
|
Hungary
|
Y
|
Y
|
Sri Lanka
|
Y
|
N
|
|
|
India
|
Y
|
N
|
Ukraine
|
Y
|
N
|
|
|
Israel
|
Y
|
Y
|
In the U.S., the medical devices that will be manufactured and sold by us will be subject to laws and regulations administered by the U.S. Food and Drug Administration, including regulations concerning the prerequisites to commercial marketing, the conduct of clinical investigations, compliance with the Quality System Regulation and labeling. We anticipate that our MGuardTM Coronary plus with bio-stable mesh product will be classified as a Class III medical device by the U.S. Food and Drug Administration.
A manufacturer may seek market authorization for a new medical device through the rigorous Premarket Approval application process, which requires the U.S. Food and Drug Administration to determine that the device is safe and effective for the purposes intended.
We will also be required to register with the U.S. Food and Drug Administration as a medical device manufacturer. As such, our manufacturing facilities will be subject to U.S. Food and Drug Administration inspections for compliance with Quality System Regulation. These regulations will require that we manufacture our products and maintain our documents in a prescribed manner with respect to design, manufacturing, testing and quality control activities. As a medical device manufacturer, we will further be required to comply with U.S. Food and Drug Administration requirements regarding the reporting of adverse events associated with the use of our medical devices, as well as product malfunctions that would likely cause or contribute to death or serious injury if the malfunction were to recur. U.S. Food and Drug Administration regulations also govern product labeling and prohibit a manufacturer from marketing a medical device for unapproved applications. If the U.S. Food and Drug Administration believes that a manufacturer is not in compliance with the law, it can institute enforcement proceedings to detain or seize products, issue a recall, enjoin future violations and assess civil and criminal penalties against the manufacturer, its officers and employees.
Customers
Our customer base is varied. We began shipping our product to customers in Europe in January 2008 and have since expanded our global distribution network to Canada, Southeast Asia, India and Latin America. Sixty six percent (66%) of our 2010 revenues were generated in Europe. Our major customer in 2010 was Hand-Prod Sp. Z o.o, a Polish distributor, that accounted for 29% of our revenues. We have an agreement with Hand-Prod Sp. Z o.o that grants Hand-Prod Sp. Z o.o the right to be the exclusive distributor of MGuardTM products in Poland until December 2012, subject to achievement of certain sales minimums.
Our major customers in the nine months ended September 30, 2011 were Kirloskar Technologies (P) Ltd., a distributor in India that accounted for 23% of our revenues, Izasa Distribuciones Tecnicas SA, a distributor in Spain that accounted for 11% of our revenues, and Tzamal Jacobsohn Ltd, a distributor in Israel that accounted for 10% of our revenues. Our agreement with Kirloskar Technologies (P) Ltd. grants Kirloskar Technologies (P) Ltd. the right to be the exclusive distributor of MGuardTM products in India until May 2013, subject to achievement of certain sales minimums. Under our agreement with Kirloskar Technologies (P) Ltd., Kirloskar Technologies (P) Ltd. must purchase from us 15,000 stents in 2011 and 20,000 stents in 2012, at a price per stent of $600, for total minimum order values of $9,000,000 in 2011 and $12,000,000 in 2012, respectively. Kirloskar Technologies (P) Ltd. will also be eligible to receive free stents representing 15% or 20% of the total value of stents purchased, depending upon the annual volume of the purchases of our stents. Our agreement with Tzamal Jacobsohn Ltd grants Tzamal Jacobsohn Ltd the right to be the exclusive distributor MGuardTM products in Israel until December 2012, subject to achievement of certain sales minimums. Under our agreement with Tzamal Jacobsohn Ltd, Tzamal Jacobsohn Ltd must achieve at least 85% of the following sales minimums: 1,400 stents in 2011 and 1,600 stents in 2012, at a price per stent of 450 Euros, for total minimum order values of 675,000 Euros in 2011 and 810,000 Euros in 2012, respectively. Tzamal Jacobsohn Ltd. will be granted options to purchase 8,116 shares of our common stock for each $100,000 in sales upon achievement of the sales minimums. Our agreement with Izasa Distribuciones Tecnicas SA grants Izasa Distribuciones Tecnicas SA the right to be the exclusive distributor of MGuardTM products in Spain until May 2012, subject to achievement of certain sales minimums. Under our agreement with Izasa Distribuciones Tecnicas SA, Izasa Distribuciones Tecnicas SA must purchase from us 4,000 stents in 2011, at a price per stent of 700 Euros, for total minimum order values of 2,800,000 Euros in 2011. Izasa Distribuciones Tecnicas SA will also be eligible to receive free stents representing 5% of the stents purchased free of charge. Pursuant to an amendment to our agreement with Izasa Distribuciones Tecnicas SA, Izasa Distribuciones Tecnicas SA, through its subsidiaries, was required to purchase 500 MGuard Prime™ stents at a price per stent of 700 Euros in February 2011, and received a bonus of 100 free stents. Izasa Distribuciones Tecnicas SA also agreed to partner with us in a study to be conducted in Spain entitled MGuard Prime Implementation in STEMI (acute myocardial infarction with ST elevation). In addition, other current significant customers are in Germany, Argentina, and Brazil.
Manufacturing and Suppliers
We manufacture our stainless steel MGuardTM stent through a combination of outsourcing and assembly at our own facility. Third parties in Germany manufacture the base stent and catheter materials, and we add our proprietary mesh sleeve to the stent. Our current exclusive product supplier is QualiMed Innovative Medizinprodukte GmbH. QualiMed Innovative Medizinprodukte GmbH is a specialized German stent manufacturer that electro polishes and crimps the stent onto a balloon catheter that creates the base for our MGuardTM stents. QualiMed Innovative Medizinprodukte GmbH has agreed to take responsibility for verifying and validating the entire stent system by performing the necessary bench test and biocompatibility testing. During the production process, QualiMed Innovative Medizinprodukte GmbH is responsible for integrating the mesh covered stent with the delivery system, sterilization, packaging and labeling. Our manufacturing agreement with QualiMed Innovative Medizinprodukte GmbH expires in September 2017, unless earlier terminated by either party in the event of breach of material terms of the agreement, liquidation of the other party, our failure to receive requested products for more than 60 days, a substantiated intellectual property claim is brought against the other party or the development agreement between the parties is terminated. The manufacturing agreement provides for a rebate program that rewards us for increases in sales of our products. Our proprietary mesh sleeve is supplied by Biogeneral, Inc., a San Diego, California-based specialty polymer manufacturer for medical and engineering applications. Natec Medical Ltd. supplies us with catheters that help create the base for our MGuardTM stents. Our agreement with Natec Medical Ltd., which may be terminated by either party upon six months notice, calls for non-binding minimum orders and discounted catheters upon reaching certain purchasing thresholds.
Our MGuard Prime™ cobalt-chromium stent was designed by Svelte Medical Systems Inc. We have an agreement with Svelte Medical Systems Inc. that grants us a non-exclusive, worldwide license for production and use of the MGuard Prime™ cobalt-chromium stent for the life of the stent’s patent, subject to the earlier termination of the agreement upon the bankruptcy of either party or the uncured default by either party under any material provision of the agreement. Our royalty payments to Svelte Medical Systems Inc. are determined by the sales volume of MGuard Prime™ stents. We will pay a royalty of 7% for all product sales outside of the U.S. and, for products sales within the U.S., a rate of 7% for the first $10 million of sales and a rate of 10% for all sales exceeding $10 million. We will also share with Svelte Medical Systems Inc. in the cost of obtaining the CE Mark approval, with our costs not to exceed $85,000, and the U.S. Food and Drug Administration approval, with our costs not to exceed $200,000. We have mutual indemnification obligations with Svelte Medical Systems Inc. for any damages suffered as a result of third party actions based upon breaches of representations and warranties or the failure to perform certain covenants in the license agreement, and Svelte Medical Systems Inc. will also indemnify us for any damages suffered as a result of third party actions based upon intellectual property or design claims against the MGuard Prime™ cobalt-chromium stent.
Our MGuard Prime™ cobalt-chromium stent is being manufactured and supplied by MeKo Laserstrahl-Materialbearbeitung. Our agreement with MeKo Laserstrahl-Materialbearbeitung for the production of electro polished L605 bare metal stents for MGuard Prime™ is priced on a per-stent basis, subject to the quantity of stents ordered. The complete assembly process for MGuard Prime™, including knitting and securing the sleeve to the stent and the crimping of the sleeve stent on to a balloon catheter, is done at our Israel manufacturing site. Once MGuard Prime™ has been assembled, it is sent for sterilization in Germany and then back to Israel for final packaging.
MGuardTM is manufactured from two main components , the stent and the mesh polymer. The stent is made out of stainless steel or cobalt chromium. Both of these materials are readily available and we acquire them in the open market. The mesh is made from polyethylene terephthalate (PET). This material is readily available in the market as well, because it is used for many medical applications. In the event that our supplier can no longer supply this material in fiber form, we would need to qualify another supplier, which could take several months. In addition, in order to retain the approval of the CE Mark, we are required to perform periodic audits of the quality control systems of our key suppliers in order to insure that their products meet our predetermined specifications.
Distributors
We currently have exclusive distribution agreements for our CE Mark-approved MGuard™ Coronary with bio stable mesh with medical product distributors based in Italy, Germany, Austria, Czech Republic, Slovakia, France, Slovenia, Greece, Cyprus, Portugal, Spain, Poland, Hungary, Estonia, Lithuania, Ukraine, United Kingdom, Holland, Russia, Latvia, Brazil, Chile, Costa Rica, Mexico, Argentina, Colombia, India, Sri Lanka, South Africa, Pakistan and Israel. We are currently in discussions with multiple distribution companies in Europe, Asia, and Latin America.
Current and future agreements with distributors stipulate that while we are responsible for training, providing marketing guidance, marketing materials, and technical guidance, distributors will be responsible for carrying out local registration, marketing activities and sales. In addition, in most cases, all sales costs, including sales representatives, incentive programs, and marketing trials, will be borne by the distributor. Under current agreements, distributors purchase stents from us at a fixed price. Our current agreements with distributors are for a term of approximately three years and automatically renew for an additional three years unless modified by either party.
Employees
As of November 30, 2011, we had 59 full-time employees. Our employees are not party to any collective bargaining agreements. We consider our relations with our employees to be good. We believe that our future success will depend, in part, on our continued ability to attract, hire and retain qualified personnel.
Properties
Our headquarters are located in Tel Aviv, Israel where we currently have an 825 square meter facility that employs 25 of our manufacturing personnel and currently has a capacity to manufacture and assemble 3,000 stents per month. We believe that our current facility is sufficient to meet anticipated future demand by adding additional shifts to our current production schedule.
Legal Proceedings
From time to time, we may be involved in litigation that arises through the normal course of business. As of the date of this filing, we are not a party to any material litigation nor are we aware of any such threatened or pending litigation, except for the matters described below.
On November 2, 2010, Eric Ben Mayor, a former senior employee of InspireMD Ltd., filed suit in Regional Labor Court in Tel Aviv, claiming illegal termination of employment and various amounts in connection with his termination, including allegations that he is owed salary, payments to pension fund, vacation pay, sick days, severance pay, commission for revenues and other types of funds. In total, Mr. Mayor is seeking $428,000, additional compensation for holding back wages, and options to purchase 2,029,025 shares of our common stock at an exercise price of $0.001 per share. We have filed a notice in Regional Labor Court indicating that the parties have rejected a court proposal for mediation and a second preliminary hearing was held on November 3, 2011. After requesting such from the court, the court granted us the opportunity to file motions regarding the disclosure procedure between the parties until December 15, 2011. No further hearing date has been set.
There are no proceedings in which any of our directors, officers or affiliates or any registered or beneficial shareholders is an adverse party or has a material interest adverse to our interest.
Executive Officers and Directors
The following table sets forth information regarding our executive officers and the members of our board of directors.
|
Name
|
Age
|
Position
|
|
Ofir Paz
|
46
|
Chief Executive Officer and Director
|
|
Asher Holzer, PhD
|
62
|
President
|
|
Craig Shore
|
50
|
Chief Financial Officer, Secretary and Treasurer
|
|
Eli Bar
|
47
|
Senior Vice President of Research and Development and Chief Technical Officer of InspireMD Ltd.
|
|
Sol J. Barer, PhD
|
63 |
Chairman of the Board of Directors
|
|
Paul Stuka
|
56 |
Director
|
|
Eyal Weinstein
|
56 |
Director
|
Our directors hold office until the earlier of their death, resignation or removal by stockholders or until their successors have been qualified. Our directors are divided into three classes. Sol Barer and Paul Stuka are our class 1 directors, with their terms of office to expire at our 2012 annual meeting of stockholders. Asher Holzer and Eyal Weinstein are our class 2 directors, with their terms of office to expire at our 2013 annual meeting of stockholders. Ofir Paz is our class 3 director, with his term of office to expire at our 2014 annual meeting of stockholders. At each annual meeting of stockholders, commencing with the 2012 annual meeting, directors elected to succeed those directors whose terms expire shall be elected for a term of office to expire at the third succeeding annual meeting of stockholders after their election, with each director to hold office until his or her successor shall have been duly elected and qualified.
Our officers are elected annually by, and serve at the pleasure of, our board of directors.
Executive Officers and Directors
Ofir Paz has served as our chief executive officer and a director since March 31, 2011. In addition, Mr. Paz has served as the chief executive officer and a director of InspireMD Ltd. since May 2005. From April 2000 through July 2002, Mr. Paz headed the Microsoft TV Platform Group in Israel. In this capacity, Mr. Paz managed the overall activities of Microsoft TV Access Channel Server, a server-based solution for delivering interactive services and Microsoft Windows-based content to digital cable set-top boxes. Mr. Paz joined Microsoft in April 2000 when it acquired Peach Networks, which he founded and served as its chief executive officer. Mr. Paz was responsible for designing Peach Networks’ original system architecture, taking it from product design to a viable product, and then managing and leading the company up to and after its acquisition, which was valued at approximately $100 million at the time of such acquisition. Mr. Paz currently serves on the board of directors of A. S. Paz Investment and Management Ltd., S.P. Market Windows Israel Ltd. and Peach Networks Ltd. Mr. Paz received a B.Sc. in Electrical Engineering, graduating cum laude, and a M.Sc. from Tel Aviv University. Mr. Paz’s qualifications to serve on the board include his prior experience in successfully establishing and leading technology companies in Israel. In addition, as chief executive officer, Mr. Paz’s position on the board ensures a unity of vision between the broader goals our company and our day-to-day operations.
Asher Holzer, PhD, has served as our president since March 31, 2011 and previously also served as our chairman from March 31, 2011 until November 16, 2011. In addition, Dr. Holzer has served as the president and chairman of the board of InspireMD Ltd. since April 2007. Previously, Dr. Holzer founded Adar Medical Ltd., an investment firm specializing in medical device startups, and served as its chief executive officer from 2002 through 2004. Dr. Holzer currently serves on the board of directors of Adar Medical Ltd., O.S.H.-IL The Israeli Society of Occupational Safety and Health Ltd., Ultra-Cure Ltd., GR-Ed Investment and Enterprise Ltd., Vasculogix Ltd., Theracoat Ltd., Cuber Stent Ltd., 2to3D Ltd., and S.P. Market Windows Cyprus. Dr. Holzer earned his PhD in Applied Physics from the Hebrew University. Dr. Holzer is also an inventor and holder of numerous patents. Dr. Holzer brings to the board his more than 25 years of experience in advanced medical devices, as well as expertise covering a wide range of activities, including product development, clinical studies, regulatory affairs, market introduction and the financial aspects of the stent business.
Craig Shore has served as our chief financial officer, secretary and treasurer since March 31, 2011. In addition, since November 10, 2010, Mr. Shore has served as InspireMD Ltd.’s vice president of business development. From February 2008 through June 2009, Mr. Shore served as chief financial officer of World Group Capital Ltd. and Nepco Star Ltd., both publicly traded companies on the Tel Aviv Stock Exchange, based in Tel Aviv, Israel. From March 2006 until February 2008, Mr. Shore served as the chief financial officer of Cellnets Solutions Ltd., a provider of advanced cellular public telephony solutions for low to middle income populations of developing countries based in Azur, Israel. Mr. Shore has over 25 years of experience in financial management in the U.S., Europe and Israel. His experience includes raising capital both in the private and public markets. Mr. Shore graduated with honors and received a B.Sc. in Finance from Pennsylvania State University and an M.B.A. from George Washington University.
Eli Bar has served as InspireMD Ltd.’s senior vice president of research and development and chief technical officer since February 2011. Prior to that, he served as InspireMD Ltd.’s vice president of research and development since October 2006 and engineering manager since June 2005. Mr. Bar has over 15 years experience in medical device product development. Mr. Bar has vast experience building a complete research and development structure, managing teams from the idea stage to an advanced marketable product. He has been involved with many medical device projects over the years and has developed a synthetic vascular graft for femoral and coronary artery replacement, a covered stent and a fully implantable Ventricular Assist Device. Mr. Bar has more than nine filed device and method patents and he has initiated two medical device projects. Mr. Bar is also a director of Blue Surgical Ltd., a medical device company based in Israel. Mr. Bar graduated from New Haven University in Connecticut with a B.Sc. in Mechanical Engineering.
Sol J. Barer, Ph.D., has served as a director since July 11, 2011 and has served as our chairman since November 16, 2011. Dr. Barer has over 30 years of experience with publicly traded biotechnology companies. In 1980, when Dr. Barer was with Celanese Research Company, he formed the biotechnology group that was subsequently spun out to form Celgene Corporation. Dr. Barer spent 18 years leading Celgene Corporation as president, chief operating officer and chief executive officer, culminating with his tenure as Celgene Corporation’s executive chairman and chairman beginning in May 2006 until his retirement in June 2011. Dr. Barer is also a director of Amicus Therapeutics, Inc. and Aegerion Pharmaceuticals, Inc. and serves as a senior advisor to a number of other biotechnology companies. Dr. Barer received a Ph.D. in organic chemistry from Rutgers University. Dr. Barer brings to the board significant scientific and executive leadership experience in the U.S. biotechnology industry and prior service on the board of directors of other publicly-held biopharmaceutical companies, as well as a unique perspective on the best methods of growth for a biotechnology company.
Paul Stuka has served as a director since August 8, 2011. Mr. Stuka has served as the managing member of Osiris Partners, LLC since 2000. Prior to forming Osiris Partners, LLC, Mr. Stuka, with 30 years experience in the investment industry, was a managing director of Longwood Partners, managing small cap institutional accounts. In 1995, Mr. Stuka joined State Street Research and Management as manager of its Market Neutral and Mid Cap Growth Funds. From 1986 to 1994, Mr. Stuka served as the general partner of Stuka Associates, where he managed a U.S.-based investment partnership. Mr. Stuka began his career in 1980 as an analyst at Fidelity Management and Research. As an analyst, Mr. Stuka followed a wide array of industries including healthcare, energy, transportation, and lodging and gaming. Early in his career he became the assistant portfolio manager for three Fidelity Funds, including the Select Healthcare Fund which was recognized as the top performing fund in the U.S. for the five-year period ending December 31, 1985. Mr. Stuka’s qualifications to serve on the board include his significant strategic and business insight from his years of experience investing in the healthcare industry.
Eyal Weinstein has served as a director since August 8, 2011. Mr. Weinstein is the chief executive officer of LEOREX Ltd., a company developing and marketing Dermo Cosmetic products. From 2001 to 2007, Mr. Weinstein worked as manager-partner of C.I.G., an economic and accounting consultancy, consulting for leading Israeli banks, including Leumi Bank, Hapoalim Bank, Discount Bank and Bank Hamizrachi. From 2000 to 2001, he was manager-partner of Exseed, a venture capital fund that invested in early-stage companies. Beginning in 1996, Mr. Weinstein was a partner and founder in the establishment of three high-tech companies that were ultimately sold, two to Microsoft Corporation. Mr. Weinstein brings to the board his considerable management and business experience as an executive of several companies and investment funds in Israel.
Agreements with Executive Officers
Ofir Paz
On April 1, 2005, InspireMD Ltd. entered into an employment agreement with Ofir Paz to serve as InspireMD Ltd.’s chief executive officer. Such employment agreement was subsequently amended on October 1, 2008 and March 28, 2011. Pursuant to this employment agreement, as amended, Mr. Paz was entitled to a monthly gross salary of $16,040. Mr. Paz was also entitled to certain social and fringe benefits as set forth in the employment agreement, which totaled 25% of his gross salary, as well as a company car. Mr. Paz was also entitled to a minimum bonus equivalent to three monthly gross salary payments based on achievement of objectives and board of directors approval. Mr. Paz was eligible to receive stock options pursuant to this agreement following its six month anniversary, subject to board approval. If Mr. Paz’s employment was terminated with or without cause, he was entitled to at least six months’ prior notice and would have been paid his salary and all social and fringe benefits in full during such notice period. If Mr. Paz’s employment was terminated without cause, Mr. Paz would also have been entitled to certain severance payments equal to the total amount that was contributed to and accumulated in his severance payment fund. 8.33% of Mr. Paz’s gross monthly salary was transferred to his severance payment fund each month.
On April 1, 2011, in order to obtain more favorable tax treatment in Israel, the employment agreement with Mr. Paz was terminated and InspireMD Ltd entered into a consulting agreement with A.S. Paz Management and Investment Ltd., an entity wholly-owned by Mr. Paz, through which Mr. Paz was retained to serve as InspireMD Ltd’s chief executive officer. Pursuant to this consulting agreement, Mr. Paz is entitled to a monthly consultancy fee of $21,563. Mr. Paz is also entitled to a minimum bonus equivalent to three monthly gross salary payments based on achievement of objectives and board of directors approval. If Mr. Paz’s employment is terminated without cause, he is entitled to at least six months’ prior notice and will be paid his consultancy fee during such notice period. If Mr. Paz’s employment is terminated without cause, he will also be entitled to certain severance payments equal to the total amount that has been contributed to and accumulated in his severance payment fund. The total amount accumulated in his severance payment fund as of September 20, 2011 was approximately $73,000, as adjusted for conversion from New Israeli Shekels to U.S. Dollars. No further contributions are provided for by the consulting agreement. Mr. Paz may be terminated with cause without any advance notice, and upon such termination would not be entitled to the amount that has been contributed to and accumulated in his severance payment fund.
Asher Holzer
On April 1, 2005, InspireMD Ltd. entered into an employment agreement with Dr. Asher Holzer to serve as InspireMD Ltd.’s president. Such employment agreement was subsequently amended on March 28, 2011. Pursuant to this employment agreement, as amended, Dr. Holzer was entitled to a monthly gross salary of $16,040. Dr. Holzer was also entitled to certain social and fringe benefits as set forth in the employment agreement, which totaled 25% of his gross salary, as well as a company car. Dr. Holzer was also entitled to a minimum bonus equivalent to three monthly gross salary payments based on achievement of objectives and board of directors approval. Dr. Holzer was eligible to receive stock options pursuant to this agreement following its six month anniversary, subject to board approval. If Dr. Holzer’s employment was terminated with or without cause, he was entitled to at least six months’ prior notice and would have been paid his salary and all social and fringe benefits in full during such notice period. If Dr. Holzer’s employment was terminated without cause, Dr. Holzer would also have been entitled to certain severance payments equal to the total amount that was contributed to and accumulated in his severance payment fund. 8.33% of Dr. Holzer’s gross monthly salary was transferred to his severance payment fund each month.
On April 29, 2011, effective April 1, 2011, in order to obtain more favorable tax treatment in Israel, the employment agreement with Dr. Holzer was terminated and InspireMD Ltd entered into a consulting agreement with The Israeli Society Ltd., an entity wholly-owned by Dr. Holzer, through which Dr. Holzer was retained to serve as InspireMD Ltd’s president. Pursuant to this consulting agreement, Dr. Holzer is entitled to a monthly consultancy fee of $21,563. Dr. Holzer is also entitled to a minimum bonus equivalent to three monthly gross salary payments based on achievement of objectives and board of directors approval. If Dr. Holzer’s employment is terminated without cause, he is entitled to at least six months’ prior notice and will be paid his consultancy fee during such notice period. If Dr. Holzer’s employment is terminated without cause, he will also be entitled to certain severance payments equal to the total amount that has been contributed to and accumulated in his severance payment fund. The total amount accumulated in his severance payment fund as of September 20, 2011 was approximately $79,000, as adjusted for conversion from New Israeli Shekels to U.S. Dollars. No further contributions are provided for by the consulting agreement. Dr. Holzer may be terminated with cause without any advance notice, and upon such termination would not be entitled to the amount that has been contributed to and accumulated in his severance payment fund.
Craig Shore
On November 28, 2010, InspireMD Ltd. entered into an employment agreement with Craig Shore to serve as InspireMD Ltd.’s vice president of business development. Pursuant to the employment agreement, Mr. Shore was entitled to a monthly gross salary of $8,750, which amount increased to $10,200 upon consummation of our share exchange transactions on March 31, 2011 and which further increased to $10,620 as of July 1, 2011. Mr. Shore is also entitled to certain social and fringe benefits as set forth in the employment agreement. Mr. Shore is also entitled to a grant of options to purchase 45,000 restricted ordinary shares of InspireMD Ltd. which were converted into options to purchase 365,223 options to purchase shares of our common stock following the consummation of our share exchange transactions on March 31, 2011; such options shall fully vest if Mr. Shore’s employment is terminated in connection with a change of control. If Mr. Shore’s employment is terminated without cause, Mr. Shore shall be entitled to at least 30 days’ prior notice and shall be paid his salary in full and all social and fringe benefits during such notice period. If a major change of control of InspireMD Ltd. occurs, Mr. Shore will be entitled to at least 180 days’ prior written notice and shall be paid his salary in full and all social and fringe benefits during such notice period. If Mr. Shore is terminated for cause, he is not entitled to any notice. In addition, if Mr. Shore’s employment is terminated without cause, Mr. Shore shall also be entitled to certain severance payments equal to the product obtained by multiplying the number of months Mr. Shore was employed by InspireMD Ltd. by 8.33% of his gross monthly salary.
Eli Bar
On June 26, 2005, InspireMD Ltd. entered into an employment agreement with Eli Bar to serve as InspireMD Ltd.’s engineering manager. Pursuant to this employment agreement, Mr. Bar is entitled to a monthly gross salary of $8,750, which amount increased to $10,620 as of July 1, 2011. Mr. Bar is also entitled to certain social and fringe benefits as set forth in the employment agreement including a company car. If Mr. Bar’s employment is terminated without cause, he is entitled to at least 60 days’ prior notice and shall be paid his salary in full and all social and fringe benefits during such notice period. If Mr. Bar’s employment is terminated without cause, Mr. Bar shall also be entitled to certain severance payments equal to the product obtained by multiplying the number of months Mr. Bar was employed by us by 8.33% of his current monthly salary.
Executive Compensation
Summary Compensation Table
The table below sets forth, for our last two fiscal years, the compensation earned by Ofir Paz, our chief executive officer, Asher Holzer, our president and former chairman of the board, Eli Bar, InspireMD Ltd.’s vice president of research and development, and Lynn Briggs, our former president, chief executive officer, chief financial officer, secretary and treasurer.
|
Name and Principal Position
|
Year
|
Salary
($)(1)
|
Bonus
($)(1)
|
Option
Awards(2)
|
All Other Compensation
($)(1)
|
Total
($)(1)
|
||
|
Ofir Paz(3)
Chief Executive Officer
|
2010
|
118,700
|
-
|
-
|
78,515
|
197,214
|
||
|
2009
|
104,301
|
-
|
-
|
57,755
|
162,057
|
|||
|
Asher Holzer(3)
President and Former Chairman
|
2010
|
122,412
|
-
|
-
|
74,813
|
197,225
|
||
|
2009
|
106,879
|
55,177
|
162,056
|
|||||
|
Eli Bar
Vice President, Research and Development of InspireMD Ltd.
|
2010
|
111,667
|
-
|
818,509
|
-
|
930,176
|
||
|
2009
|
106,001
|
-
|
-
|
-
|
106,001
|
|||
|
Lynn Briggs(4)
Former President, CEO, CFO, Secretary and Treasurer
|
2010
|
-
|
-
|
-
|
-
|
-
|
||
|
2009
|
-
|
-
|
-
|
-
|
-
|
|||
|
(1)
|
Compensation amounts received in non-U.S. currency have been converted into U.S. dollars using the average exchange rate for the applicable year. The average exchange rate for 2010 was 3.7319 NIS per dollar and the average exchange rate for 2009 was 3.9228 NIS per dollar.
|
|||||||
|
(2)
|
The amounts in this column reflect the dollar amounts recognized for financial statement reporting purposes with respect to the years ended December 31, 2009 and 2010, in accordance with SFAS 123(R).
|
|||||||
|
(3)
|
Both Mr. Paz and Dr. Holzer are directors but do not receive any additional compensation for their services as directors.
|
|||||||
|
(4)
|
Ms. Briggs resigned as our sole officer and director in connection with our share exchange transactions on March 31, 2011. She received no compensation for services, but was reimbursed for any out-of-pocket expenses that she incurred on our behalf.
|
|||||||
Outstanding Equity Awards at Fiscal Year-End
The following table shows information concerning unexercised options outstanding as of December 31, 2010 for each of our named executive officers.
|
Name
|
Number of securities underlying unexercised options (#) exercisable
|
Number of securities underlying unexercised options (#) unexercisable
|
Option exercise price ($)
|
Option expiration date
|
|
Ofir Paz
|
-
|
-
|
-
|
-
|
|
Asher Holzer
|
-
|
-
|
-
|
-
|
|
Eli Bar
|
243,481
|
-
|
0.001
|
10/28/2016
|
|
365,224
|
-
|
0.001
|
12/29/2016
|
|
|
152,177
|
456,530(1)
|
0.001
|
7/22/2020
|
|
|
20,290
|
60,871(1)
|
1.23
|
7/28/2020
|
|
(1)
|
These options were granted in July 2010 and vest one-twelfth quarterly commencing with the quarter in which they were granted.
|
2011 UMBRELLA Option Plan
On March 28, 2011, our board of directors and stockholders adopted and approved the InspireMD, Inc. 2011 UMBRELLA Option Plan, which was subsequently amended on October 31, 2011. Under the InspireMD, Inc. 2011 UMBRELLA Option Plan, we have reserved 15,000,000 shares of our common stock as awards to the employees, consultants, and service providers to InspireMD, Inc. and its subsidiaries and affiliates worldwide.
The InspireMD, Inc. 2011 UMBRELLA Option Plan currently consists of three components, the primary plan document that governs all awards granted under the InspireMD, Inc. 2011 UMBRELLA Option Plan, and two appendices: (i) Appendix A, designated for the purpose of grants of stock options and restricted stock to Israeli employees, consultants, officers and other service providers and other non-U.S. employees, consultants, and service providers, and (ii) Appendix B, which is the 2011 U.S. Equity Incentive Plan, designated for the purpose of grants of stock options and restricted stock awards to U.S. employees, consultants, and service providers who are subject to the U.S. income tax.
The purpose of the InspireMD, Inc. 2011 UMBRELLA Option Plan is to provide an incentive to attract and retain employees, officers, consultants, directors, and service providers whose services are considered valuable, to encourage a sense of proprietorship and to stimulate an active interest of such persons in our development and financial success. The InspireMD, Inc. 2011 UMBRELLA Option Plan is administered by our compensation committee. Unless terminated earlier by the board of directors, the InspireMD, Inc. 2011 UMBRELLA Option Plan will expire on March 27, 2021.
Since its adoption, we have granted options to purchase common stock under the InspireMD, Inc. 2011 UMBRELLA Option Plan that are currently outstanding to the following named executive officer:
|
Name
|
Shares Subject to Options
|
Exercise Price
|
Vesting Schedule
|
Expiration
|
||||
|
Eli Bar
|
200,000
|
1.93
|
One-third annually in 2012, 2013 and 2014 on the anniversary of the grant date
|
|
May 23, 2016
|
2010 Director Compensation
We did not provide any separate compensation to our sole director in 2010. The following table shows information concerning the directors of InspireMD Ltd., other than Ofir Paz and Asher Holder, during the fiscal year ended December 31, 2010.
|
Name
|
Fees Earned or
Paid in Cash
($)
|
Option Awards(1)(2)
($)
|
All Other Compensation
($)
|
Total
($)
|
|
David Ivry(3)
|
6,083
|
133,398
|
-
|
139,481
|
|
Robert Fischell(3)
|
3,783
|
133,398
|
-
|
137,181
|
|
Fellice Pelled (3)
|
5,885
|
133,398
|
-
|
139,283
|
|
(1)
|
Based on the fair market value of the stock awards on the date of grant in accordance with SFAS 123R.
|
|
(2)
|
As of December 31, 2010, the following directors owned the following number of outstanding options to purchase common stock: David Ivry (121,742), Fellice Pelled (121,742) and Robert Fischell (121,742).
|
|
(3)
|
Each of David Ivry, Robert Fischell and Fellice Pelled resigned as directors of InspireMD, Ltd. on March 31, 2011. Pursuant to the terms of the directors’ vested options, the vested options expired thirty days after the directors’ resignations. However, in connection with their resignation, we granted Mr. Ivry and Mr. Pelled replacement options with substantially similar terms to the expired options.
|
Other than Mr. Paz and Dr. Holzer, we previously paid each director $330 per meeting for each board meeting attended and $1,230 for each quarter served on the board of directors. We also granted annually to each director options to purchase 81,160 shares of our common stock at an exercise price per share equal to the fair market value of our common stock on the grant date. The options vest over four quarters from the grant date.
We do not currently provide cash compensation to our directors for acting as such, although we may do so in the future. We reimburse our directors for reasonable expenses incurred in connection with their service as directors. In addition, in 2011, we made the following option grants to the following directors. Each grant was made under the InspireMD, Inc. 2011 UMBRELLA Option Plan, unless otherwise noted.
|
Name
|
Shares Subject to Options
|
Exercise Price
|
Vesting Schedule
|
Expiration
|
||||
|
Sol J. Barer, Ph.D.
|
1,000,000(1)(2)
|
$1.50
|
Fully vested upon grant.
|
September 30, 2011(3)
|
||||
|
500,000(2)
|
$2.50
|
One-half annually in 2012 and 2013 on the anniversary of the date of grant, provided that if Dr. Barer is (i) not reelected as a director at our 2012 annual meeting of stockholders, or (ii) not nominated for reelection as a director at our 2012 annual meeting of stockholders, the option vests and becomes exercisable on the date of such failure to be reelected or nominated.
|
July 11, 2021
|
|||||
|
1,450,000(1)(4)
|
$1.95
|
In substantially equal monthly installments (with any fractional shares vesting on the last vesting date) on the last business day of each calendar month over a two year period from the date of grant, with the first installment vesting on November 30, 2011, provided that Dr. Barer is still providing services to us in some capacity as of each such vesting date.
|
November 16, 2021
|
|||||
|
725,000(1)
|
$1.95
|
Upon the date we become listed on a registered national securities exchange (such as the New York Stock Exchange, NASDAQ Stock Market, or the NYSE Amex), provided that such listing occurs on or before December 31, 2012, and provided further that Dr. Barer is still providing services to us in some capacity as of such vesting date.
|
November 16, 2021
|
|||||
|
725,000(1)(4)
|
$1.95
|
Upon the date that we receive research coverage from at least two investment banks that ranked in the top 20 investment banks in terms of underwritings as of their most recently completed fiscal year, and/or leading analysts, as ranked by either the Wall Street Journal, the Financial Times, Zacks Investment Research or Institutional Investor, provided that we receive such coverage on or before December 31, 2012, and, provided further that Dr. Barer is still providing services to us in some capacity as of such vesting date.
|
November 16, 2021
|
|||||
|
Paul Stuka
|
100,000(2)
|
$1.95
|
One-third annually in 2012, 2013 and 2014 on the anniversary of the date of grant, provided that if Mr. Stuka is (i) not reelected as a director at our 2012 annual meeting of stockholders, or (ii) not nominated for reelection as a director at our 2012 annual meeting of stockholders, the option vests and becomes exercisable on the date of such failure to be reelected or nominated.
|
August 8, 2021
|
||||
|
Eyal Weinstein
|
25,000(2)
|
$1.95
|
One-third annually in 2012, 2013 and 2014 on the anniversary of the date of grant, provided that if Mr. Weinstein is required to resign from the board due to medical reasons, the option vests and becomes exercisable on the date of Mr. Weinstein’s resignation for medical reasons.
|
August 8, 2021
|
____________________
(1) This option was issued outside the InspireMD, Inc. 2011 UMBRELLA Option Plan.
(2) This option was granted in connection with the appointment of this person to our board of directors.
(3) This option was exercised in full by Dr. Barer on September 28, 2011.
(4) This option was granted to Dr. Barer in connection with his appointment as chairman of our board of directors on November 16, 2011.
In addition to the foregoing, on November 16, 2011, in connection with his appointment as chairman of our board of directors, we issued Dr. Barer 2,900,000 shares our common stock, all of which were immediately vested.
Directors’ and Officers’ Liability Insurance
We currently have directors’ and officers’ liability insurance insuring our directors and officers against liability for acts or omissions in their capacities as directors or officers, subject to certain exclusions. Such insurance also insures us against losses which we may incur in indemnifying our officers and directors. In addition, we have entered into indemnification agreements with key officers and directors and such persons shall also have indemnification rights under applicable laws, and our certificate of incorporation and bylaws.
Code of Ethics
We intend to adopt a code of ethics that applies to our officers, directors and employees, including our principal executive officer and principal accounting officer, but have not done so to date due to our relatively small size. We intend to adopt a written code of ethics in the near future.
Board Committees
Our board of directors has established an audit committee and a compensation committee, each of which has the composition and responsibilities described below.
Audit Committee. Our audit committee is currently comprised of Messrs. Stuka and Weinstein and Dr. Barer, each of whom our board has determined to be financially literate and qualify as an independent director under Section 5605(a)(2) of the rules of the Nasdaq Stock Market. Mr. Stuka is the chairman of our audit committee and qualifies as a financial expert, as defined in Item 407(d)(5)(ii) of Regulation S-K. The audit committee’s duties are to recommend to our board of directors the engagement of independent auditors to audit our financial statements and to review our accounting and auditing principles. The audit committee will review the scope, timing and fees for the annual audit and the results of audit examinations performed by the internal auditors and independent public accountants, including their recommendations to improve the system of accounting and internal controls.
Compensation Committee. Our compensation committee is currently comprised of Messrs. Stuka and Weinstein and Dr. Barer. Mr. Weinstein is the chairman of our compensation committee. The compensation committee reviews and approves our salary and benefits policies, including compensation of executive officers. The compensation committee also administers our stock option plans and recommends and approves grants of stock options under such plans.
Compensation Committee Interlocks and Insider Participation
During the fiscal year ended December 31, 2010, we did not have a compensation committee and during such period, Ofir Paz, our chief executive officer, and Asher Holzer, our president and former chairman, participated in deliberations of the board of directors concerning executive officer compensation. None of our executive officers currently serves, or in the past year has served, as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving on our board of directors or compensation committee.
Security Ownership Of Certain Beneficial Owners And Management
The following table sets forth information with respect to the beneficial ownership of our common stock as of November 30, 2011 by:
|
|
·
|
each person known by us to beneficially own more than 5.0% of our common stock;
|
|
|
·
|
each of our directors;
|
|
|
·
|
each of the named executive officers; and
|
|
|
·
|
all of our directors and executive officers as a group.
|
The percentages of common stock beneficially owned are reported on the basis of regulations of the Securities and Exchange Commission governing the determination of beneficial ownership of securities. Under the rules of the Securities and Exchange Commission, a person is deemed to be a beneficial owner of a security if that person has or shares voting power, which includes the power to vote or to direct the voting of the security, or investment power, which includes the power to dispose of or to direct the disposition of the security. Except as indicated in the footnotes to this table, each beneficial owner named in the table below has sole voting and sole investment power with respect to all shares beneficially owned and each person’s address is c/o InspireMD, Inc., 3 Menorat Hamaor St., Tel Aviv, Israel 67448. As of November 30, 2011, we had 68,178,947 shares outstanding.
|
Name of Beneficial Owner
|
Number of Shares Beneficially Owned(1)
|
Percentage
Beneficially Owned(1)
|
||||||
|
5% Owners
|
||||||||
|
Yuli Ofer (2)
|
4,518,301 | 6.6 | % | |||||
|
Officers and Directors
|
||||||||
|
Ofir Paz
|
10,263,752 | (3) | 15.1 | % | ||||
|
Asher Holzer
|
10,300,437 | (4) | 15.1 | % | ||||
|
Eli Bar
|
953,638 | (5) | 1.4 | % | ||||
|
Sol J. Barer, Ph.D. (6)
|
4,020,834 | (7) | 5.9 | % | ||||
|
Paul Stuka (8)
|
2,000,000 | (9) | 2.9 | % | ||||
|
Eyal Weinstein (10)
|
0 | * | ||||||
|
All directors and executive officers as a group (7 persons)
|
27,660,402 | 39.5 | % | |||||
|
*
|
Represents ownership of less than one percent.
|
|
(1)
|
Shares of common stock beneficially owned and the respective percentages of beneficial ownership of common stock assumes the exercise of all options, warrants and other securities convertible into common stock beneficially owned by such person or entity currently exercisable or exercisable within 60 days of November 30, 2011. Shares issuable pursuant to the exercise of stock options and warrants exercisable within 60 days are deemed outstanding and held by the holder of such options or warrants for computing the percentage of outstanding common stock beneficially owned by such person, but are not deemed outstanding for computing the percentage of outstanding common stock beneficially owned by any other person.
|
|
(2)
|
Mr. Ofer’s address is 36 Hamesila Street, Herzeliya, Israel.
|
|
(3)
|
This amount does not include 372,528 shares of common stock that Mr. Paz presently holds as trustee for a family trust. Mr. Paz does not have either voting power or dispositive power over these shares and disclaims all beneficial ownership therein.
|
|
(4)
|
This amount does not include 58,923 shares of common stock that Dr. Holzer presently holds as trustee for a family trust. Dr. Holzer does not have either voting power or dispositive power over these shares and disclaims all beneficial ownership therein.
|
|
(5)
|
Represents options that are currently exercisable or exercisable within 60 days of November 30, 2011.
|
|
(6)
|
Dr. Barer’s address is 67 Park Place East, Suite 675, Morristown, NJ 07960.
|
|
(7)
|
Comprised of (i) 3,900,000 shares of common stock and (ii) options to purchase 120,834 shares of common stock exercisable within 60 days of November 30, 2011.
|
|
(8)
|
Mr. Stuka’s address is c/o Osiris Partners, LLC, 1 Liberty Square, 5th Floor, Boston, MA 02109.
|
|
(9)
|
Paul Stuka is the principal and managing member of Osiris Investment Partners, L.P., and, as such, has beneficial ownership of the (i) 1,333,333 shares of common stock and (ii) currently exercisable warrants to purchase 666,667 shares of common stock held by Osiris Investment Partners, L.P.
|
|
(10)
|
Mr. Weinstein’s address is c/o Leorlex Ltd., P.O. Box 15067 Matam, Haifa, Israel 31905.
|
Up to 414,942 shares of common stock issuable upon the exercise of warrants are being offered by this prospectus, all of which are being registered for sale for the accounts of the selling stockholders. These warrants were issued in connection with a series of private placements we conducted on March 31, 2011, April 18, 2011 and April 21, 2011, pursuant to which we issued 7,437,336 shares of common stock and five year warrants to purchase up to 3,718,666 shares of common stock at an exercise price of $1.80 per share for aggregate cash proceeds of $10,488,404 and the cancellation of $667,596 of indebtedness held by investors.
Each of the transactions by which the selling stockholders acquired their securities from us was exempt under the registration provisions of the Securities Act of 1933, as amended.
The shares of common stock referred to above are being registered to permit public sales of the shares, and the selling stockholders may offer the shares for resale from time to time pursuant to this prospectus. The selling stockholders may also sell, transfer or otherwise dispose of all or a portion of their shares in transactions exempt from the registration requirements of the Securities Act of 1933, as amended, or pursuant to another effective registration statement covering those shares. We may from time to time include additional selling stockholders in supplements or amendments to this prospectus.
The table below sets forth certain information regarding the selling stockholders and the shares of our common stock offered by them in this prospectus. The selling stockholders have not had a material relationship with us within the past three years other than as described in the footnotes to the table below or as a result of their acquisition of our shares or other securities. To our knowledge, subject to community property laws where applicable, each person named in the table has sole voting and investment power with respect to the shares of common stock set forth opposite such person’s name.
Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission. In computing the number of shares beneficially owned by a selling stockholder and the percentage of ownership of that selling stockholder, shares of common stock underlying warrants held by that selling stockholder that are convertible or exercisable, as the case may be, within 60 days of November 30, 2011 are included. Those shares, however, are not deemed outstanding for the purpose of computing the percentage ownership of any other selling stockholder. Each selling stockholder’s percentage of ownership of our outstanding shares in the table below is based upon 68,178,947 shares of common stock outstanding as of November 30, 2011. With respect to the warrants held by the selling stockholders, there exist contractual provisions limiting conversion and exercise to the extent such conversion or exercise would cause such selling stockholder, together with its affiliates or members of a “group,” to beneficially own a number of shares of common stock which would exceed 4.99% of our then outstanding shares of common stock following such conversion or exercise. The shares and percentage ownership of our outstanding shares indicated in the table below do not give effect to this limitation.
|
Ownership Before Offering
|
Ownership After Offering
|
||||||
|
Selling Stockholder
|
Number of
shares of
common stock
beneficially owned
|
Number of
shares
offered (1)
|
Number of
shares of
common stock
beneficially
owned
|
Percentage of
common stock
beneficially owned
|
|||
|
Platinum Partners Value Arbitrage Fund LP (2)
|
3,435,000 (3)
|
100,000
|
3,335,000 (4)
|
4.8%
|
|||
|
Osiris Investment Partners, L.P. (5)
|
2,000,000 (6)
|
66,667
|
1,933,333 (7)
|
2.8%
|
|||
|
Allan Pasternack
|
50,000 (8)
|
1,667
|
48,333 (9)
|
*
|
|||
|
Leon Frenkel
|
200,000 (10)
|
6,667
|
193,333 (11)
|
*
|
|||
|
CNH Diversified Opportunities Master Account, L.P. (12)
|
10,698 (13)
|
357
|
10,141 (14)
|
*
|
|||
|
Advanced Series Trust – AST Academic Strategies Asset Allocation Portfolio (15)
|
17,664 (16)
|
589
|
17,075 (17)
|
*
|
|||
|
AQR Opportunistic Premium Offshore Fund, L.P. (18)
|
17,904 (19)
|
597
|
17,307 (20)
|
*
|
|||
|
AQR Funds – AQR Diversified Arbitrage Fund (21)
|
203,734 (22)
|
6,791
|
196,943 (23)
|
*
|
|||
|
Joseph Kazarnovsky
|
360,000 (24)
|
12,000
|
348,000 (25)
|
*
|
|||
|
Fame Associates (26)
|
250,000 (27)
|
8,333
|
241,667 (28)
|
*
|
|||
|
American European Insurance Co. (29)
|
300,000 (30)
|
10,000
|
290,000 (31)
|
*
|
|||
|
Harborview Value Master Fund L.P. (32)
|
625,000 (33)
|
18,333
|
606,667 (34))
|
*
|
|||
|
The Corbran LLC (35)
|
1,535,862 (36)
|
8,333
|
1,527,529 (37))
|
2.2%
|
|||
|
David Stefansky (38)
|
1,937,863 (39)
|
21,666
|
1,916,197 (40)
|
2.8%
|
|||
|
Endicott Management Partners, LLC (41)
|
2,775,492 (42)
|
8,333
|
2,767,159 (43)
|
4.0%
|
|||
|
Ralph Rieder
|
80,000 (44)
|
2,667
|
77,333 (45)
|
*
|
|||
|
Harmony Finance Holdings Ltd. (46)
|
100,000 (47)
|
3,333
|
96,667 (48)
|
*
|
|||
|
Alan Kneller
|
15,000 (49)
|
500
|
14,500 (50)
|
*
|
|||
|
Alpha Capital Anstalt (51)
|
1,025,000 (52)
|
33,333
|
991,667 (53)
|
1.4%
|
|||
|
Fortis Business Holdings, LLC (54)
|
100,000 (55)
|
3,333
|
96,667 (56)
|
*
|
|||
|
Gedalya Shai
|
50,000 (57)
|
1,667
|
48,333 (58)
|
*
|
|||
|
Sandor Capital Master Fund, L.P. (59)
|
492,000 (60)
|
15,000
|
477,000 (61)
|
*
|
|||
|
Lev Michael
|
40,000 (62)
|
1,333
|
38,667 (63)
|
*
|
|||
|
Shmuel and Serena Fuchs Foundation (64)
|
115,000 (65)
|
3,333
|
111,667 (66)
|
*
|
|||
|
RPSMSS, LLC (67)
|
325,000 (68)
|
10,000
|
315,000 (69)
|
*
|
|||
|
Petr Gukovskiy
|
200,000 (70)
|
6,667
|
193,333 (71)
|
*
|
|||
|
LR Holdings Associates (72)
|
50,000 (73)
|
1,667
|
48,333 (74)
|
*
|
|||
|
Seth Padowitz
|
36,000 (75)
|
1,200
|
34,800 (76)
|
*
|
|||
|
Gary and Jane Klopfer
|
400,000 (77)
|
13,333
|
386,667 (78)
|
*
|
|||
|
Ronald A. Durando
|
25,000 (79)
|
833
|
24,167 (80)
|
*
|
|||
|
Palladium Capital Advisors, LLC (81)
|
99,268 (82)
|
9,927
|
89,341 (83)
|
*
|
|||
|
Reinder Hogeboom
|
50,000 (84)
|
1,667
|
48,333 (85)
|
*
|
|||
|
Moishe Hartstein (86)
|
294,205 (87)
|
29,421
|
264,784 (88)
|
*
|
|||
|
Abraham Biderman
|
8,500 (89)
|
850
|
7,650 (90)
|
*
|
|||
|
Jeffrey Frank
|
3,315 (91)
|
332
|
2,983 (92)
|
*
|
|||
|
The Benchmark Company, LLC (93)
|
8,840 (94)
|
884
|
7,956 (95)
|
*
|
|||
|
William Odenthal
|
9,945 (96)
|
995
|
8,950 (97)
|
*
|
|||
|
Cato Capital LLC (98)
|
6,667 (99)
|
667
|
6,000 (100)
|
*
|
|||
|
Eisenberg Family Foundation (101)
|
50,000 (102)
|
1,667
|
48,333 (103)
|
*
|
|||
________________________________________
*Less than 1%
(1) Number of shares offered represents number of shares of common stock issuable upon the exercise of a warrant
(2) Platinum Management (NY) LLC is the general partner of Platinum Partners Value Arbitrage Fund LP. Platinum Partners Value Arbitrage Fund LP has sole voting and dispositive power over the securities held for the account of this selling stockholder. Mark Nordlicht has the sole voting and investment power over the securities beneficially owned or that may be purchased by Platinum Partners Value Arbitrage Fund LP.
(3) Includes 1,000,000 shares of common stock issuable upon the exercise of warrants.
(4) Includes 900,000 shares of common stock issuable upon the exercise of warrants.
(5) Paul Stuka, Principal and Managing Member, has voting and dispositive power over the securities held for the account of this selling stockholder. Mr. Stuka disclaims beneficial ownership of these securities.
(6) Includes 666,667 shares of common stock issuable upon the exercise of warrants.
(7) Includes 600,000 shares of common stock issuable upon the exercise of warrants.
(8) Includes 16,667 shares of common stock issuable upon the exercise of warrants.
(9) Includes 15,000 shares of common stock issuable upon the exercise of warrants.
(10) Includes 66,667 shares of common stock issuable upon the exercise of warrants.
(11) Includes 60,000 shares of common stock issuable upon the exercise of warrants.
(12) CNH Partners, LLC, as the advisor of CNH Diversified Opportunities Master Account, L.P., has voting and dispositive power over the securities held for the account of this selling stockholder. CNH Partners, LLC is controlled indirectly by Todd Pulvino and Mark Mitchell, and accordingly, both Mr. Pulvino and Mr. Mitchell may each be deemed to share voting and dispositive power over the securities owned by CNH Diversified Opportunities Master Account, L.P.
(13) Includes 3,566 shares of common stock issuable upon the exercise of warrants.
(14) Includes 3,209 shares of common stock issuable upon the exercise of warrants.
(15) Advanced Series Trust — AST Academic Strategies Asset Allocation Portfolio is an affiliate of Prudential Investment Management Services LLC and Prudential Annuities Distributors, Inc., both of whom are broker-dealers registered under Section 15 of the Exchange Act. CNH Partners, LLC, as the sub-advisor of Advanced Series Trust — AST Academic Strategies Asset Allocation Portfolio, has discretionary voting and dispositive power over the securities held for the account of this selling stockholder. CNH Partners, LLC is controlled indirectly by Todd Pulvino and Mark Mitchell, and accordingly, both Mr. Pulvino and Mr. Mitchell may be deemed to share voting and dispositive power over the securities owned by Advanced Series Trust — AST Academic Strategies Asset Allocation Portfolio. These securities were purchased by Advanced Series Trust — AST Academic Strategies Asset Allocation Portfolio in the ordinary course of business, and at the time of the time of transfer, Advanced Series Trust — AST Academic Strategies Asset Allocation Portfolio had no agreements or understandings directly or indirectly with any person to distribute the shares of common stock underlying this warrant.
(16) Includes 5,888 shares of common stock issuable upon the exercise of warrants.
(17) Includes 5,299 shares of common stock issuable upon the exercise of warrants.
(18) CNH Partners, LLC, as the sub-advisor of AQR Opportunistic Premium Offshore, L.P., has discretionary voting and dispositive power over the securities held for the account of this selling stockholder. CNH Partners, LLC is controlled indirectly by Todd Pulvino and Mark Mitchell, and accordingly, both Mr. Pulvino and Mr. Mitchell may be deemed to share voting and dispositive power over the securities owned by AQR Opportunistic Premium Offshore Fund, L.P.
(19) Includes 5,968 shares of common stock issuable upon the exercise of warrants.
(20) Includes 5,371 shares of common stock issuable upon the exercise of warrants.
(21) CNH Partners, LLC, as the sub-advisor of AQR Funds — AQR Diversified Arbitrage Fund, has discretionary voting and dispositive power over the securities held for the account of this selling stockholder. CNH Partners, LLC is controlled indirectly by Todd Pulvino and Mark Mitchell, and accordingly, both Mr. Pulvino and Mr. Mitchell may be deemed to share voting and dispositive power over the securities owned by AQR Funds — AQR Diversified Arbitrage Fund.
(22) Includes 67,911 shares of common stock issuable upon the exercise of warrants.
(23) Includes 61,120 shares of common stock issuable upon the exercise of warrants.
(24) Includes 120,000 shares of common stock issuable upon the exercise of warrants.
(25) Includes 108,000 shares of common stock issuable upon the exercise of warrants.
(26) Abraham Fruchthandler, general partner of Fame Associates, has sole voting and dispositive power over the securities held for the account of this selling stockholder.
(27) Includes 83,333 shares of common stock issuable upon the exercise of warrants.
(28) Includes 75,000 shares of common stock issuable upon the exercise of warrants.
(29) Nachum Stein has sole voting and dispositive power over the securities held for the account of this selling stockholder.
(30) Includes 100,000 shares of common stock issuable upon the exercise of warrants.
(31) Includes 90,000 shares of common stock issuable upon the exercise of warrants.
(32) Harborview Advisors LLC is the general partner of Harborview Value Master Fund, L.P. Richard Rosenblum and David Stefansky are the managers of Harborview Advisors LLC and have shared voting and dispositive power over the securities held by Harborview Value Master Fund, LP. Mr. Rosenblum and Mr. Stefansky disclaim beneficial ownership of such securities.
(33) Includes 183,333 shares of common stock issuable upon the exercise of warrants.
(34) Includes 165,000 shares of common stock issuable upon the exercise of warrants.
(35) Richard Rosenblum exercises sole voting and dispositive power over the securities held for the account of this selling stockholder. The Corbran LLC provided us with advisory consulting services in connection with the structuring of our share exchange transactions. In consideration for such services, we issued The Corbran LLC a three-year warrant to purchase up to 625,000 shares of common stock at an exercise price of $1.50 per share.
(36) Includes 708,333 shares of common stock issuable upon the exercise of warrants.
(37) Includes 700,000 shares of common stock issuable upon the exercise of warrants.
(38) David Stefansky provided us with advisory consulting services in connection with the structuring of our share exchange transactions. In consideration for such services, we issued David Stefansky a three-year warrant to purchase up 625,000 shares of common stock at an exercise price of $1.50 per share.
(39) Includes 841,666 shares of common stock issuable upon the exercise of warrants.
(40) Includes 820,000 shares of common stock issuable upon the exercise of warrants.
(41) Ken Londoner exercises sole voting and dispositive power over the securities held for the account of this selling stockholder. Endicott Management Partners, LLC provided us with advisory consulting services in connection with the structuring of our share exchange transactions. In consideration for such services, we issued Endicott Management Partners, LLC a three-year warrants to purchase up to 1,250,000 shares of common stock at an exercise price of $1.50 per share.
(42) Includes 1,333,333 shares of common stock issuable upon the exercise of warrants and 93,000 shares of common stock held by Ken Londoner.
(43) Includes 1,325,000 shares of common stock issuable upon the exercise of warrants and 93,000 shares of common stock held by Ken Londoner.
(44) Includes 26,667 shares of common stock issuable upon the exercise of warrants.
(45) Includes 24,000 shares of common stock issuable upon the exercise of warrants.
(46) Independent Management Inc., as the sole director of Harmony Finance Holdings Ltd., has discretionary voting and dispositive power over the securities held for the account of this selling stockholder. Independent Management Inc. is controlled by Sean Breslin and Meral Baruh, who may be deemed to have voting and dispositive power over the securities held for the account of this selling stockholder.
(47) Includes 33,333 shares of common stock issuable upon the exercise of warrants.
(48) Includes 30,000 shares of common stock issuable upon the exercise of warrants.
(49) Includes 5,000 shares of common stock issuable upon the exercise of warrants.
(50) Includes 4,500 shares of common stock issuable upon the exercise of warrants.
(51) Konrad Ackemann exercises sole voting and dispositive power over the securities held for the account of this selling stockholder.
(52) Includes 333,333 shares of common stock issuable upon the exercise of warrants.
(53) Includes 300,000 shares of common stock issuable upon the exercise of warrants.
(54) Louis, Joel, and Sarah Kestenbaum have voting power of Fortis Business Holdings, LLC. Louis Kestenbaum, Margaret Kestenbaum, Joel Kestenbaum, and Sarah Rosenfeld also claim beneficial ownership of Fortis Business Holdings, LLC’s shares.
(55) Includes 33,333 shares of common stock issuable upon the exercise of warrants.
(56) Includes 30,000 shares of common stock issuable upon the exercise of warrants.
(57) Includes 16,667 shares of common stock issuable upon the exercise of warrants.
(58) Includes 15,000 shares of common stock issuable upon the exercise of warrants.
(59) John S. Lemak, as manager of this security holder, has voting and dispositive power over the securities held for the account of this selling stockholder and may be deemed to be the beneficial owner of these securities.
(60) Includes 150,000 shares of common stock issuable upon the exercise of warrants.
(61) Includes 135,000 shares of common stock issuable upon the exercise of warrants.
(62) Includes 13,333 shares of common stock issuable upon the exercise of warrants.
(63) Includes 12,000 shares of common stock issuable upon the exercise of warrants.
(64) The Shmuel & Serena Fuchs Foundation is a charitable trust and the trustees are Bernard and Hanna Fuchs.
(65) Includes 33,333 shares of common stock issuable upon the exercise of warrants.
(66) Includes 30,000 shares of common stock issuable upon the exercise of warrants.
(67) Richard P. Stadtmauer exercises sole voting and dispositive power over the securities held for the account of this selling stockholder.
(68) Includes 100,000 shares of common stock issuable upon the exercise of warrants.
(69) Includes 90,000 shares of common stock issuable upon the exercise of warrants.
(70) Includes 66,667 shares of common stock issuable upon the exercise of warrants.
(71) Includes 60,000 shares of common stock issuable upon the exercise of warrants.
(72) Leslie Rieder and Samuel J. Rieder have voting and dispositive power over the securities held for the account of this selling stockholder.
(73) Includes 16,667 shares of common stock issuable upon the exercise of warrants.
(74) Includes 15,000 shares of common stock issuable upon the exercise of warrants.
(75) Includes 12,000 shares of common stock issuable upon the exercise of warrants.
(76) Includes 10,800 shares of common stock issuable upon the exercise of warrants.
(77) Includes 133,333 shares of common stock issuable upon the exercise of warrants.
(78) Includes 120,000 shares of common stock issuable upon the exercise of warrants.
(79) Includes 8,333 shares of common stock issuable upon the exercise of warrants.
(80) Includes 7,500 shares of common stock issuable upon the exercise of warrants.
(81) Palladium Capital Advisors LLC is a registered broker-dealer. Joel Padowitz is the CEO of Palladium Capital Advisors LLC and, in such capacity, may be deemed to have voting and dispositive power over the securities held for the account of this selling stockholder. On July 18, 2010, we engaged Palladium Capital Advisors LLC to serve as our placement agent in connection with our March 31, 2011 and April 18, 2011 private placements. In connection with such private placements, we paid Palladium Capital Advisors LLC a fee of $757,170, expenses reimbursement of $15,000 and we issued it a five-year warrant to purchase 430,740 shares of our common stock, at an initial exercise price of $1.80 per share.
(82) All 99,268 shares of common stock issuable upon the exercise of warrants.
(83) All 89,341 shares of common stock issuable upon the exercise of warrants.
(84) Includes 16,667 shares of common stock issuable upon the exercise of warrants.
(85) Includes 15,000 shares of common stock issuable upon the exercise of warrants.
(86) Moishe Hartstein is an affiliate of Palladium Capital Advisors LLC, a registered broker-dealer. These securities were transferred to Mr. Hartstein by Palladium Capital Advisors LLC in the ordinary course of business, and at the time of the time of transfer, Mr. Hartstein had no agreements or understandings directly or indirectly with any person to distribute the shares of common stock underlying this warrant.
(87) All 294,205 shares of common stock issuable upon the exercise of warrants.
(88) All 264,784 shares of common stock issuable upon the exercise of warrants.
(89) All 8,500 shares of common stock issuable upon the exercise of warrants.
(90) All 7,650 shares of common stock issuable upon the exercise of warrants.
(91) All 3,315 shares of common stock issuable upon the exercise of warrants.
(92) All 2,983 shares of common stock issuable upon the exercise of warrants.
(93) The Benchmark Company, LLC is a registered broker-dealer. Mr. Adam Gordon and Mr. Richard Messina share voting and investment power over these securities. On March 31, 2011, we engaged The Benchmark Company, LLC to provide financial advisory services and other investment banking services to us for a period of six months. In connection with this engagement, we issued to The Benchmark Company, LLC 50,000 restricted shares of our common stock and a five-year warrant to purchase 50,000 shares of our common stock, at an initial exercise price of $1.50 per share and we are obligated to pay The Benchmark Company LLC a monthly fee of $8,000 and aggregate expenses over the period of the engagement not to exceed $10,000.
(94) All 8,840 shares of common stock issuable upon the exercise of warrants.
(95) All 7,956 shares of common stock issuable upon the exercise of warrants.
(96) All 9,945 shares of common stock issuable upon the exercise of warrants.
(97) All 8,950 shares of common stock issuable upon the exercise of warrants.
(98) Solomon Lax has voting and dispositive power over the securities held for the account of this selling stockholder.
(99) All 6,667 shares of common stock issuable upon the exercise of warrants.
(100) All 6,000 shares of common stocck issuable upon the exercise of warrants.
(101) Solomon Eisenberg has voting and dispositive power over the securities held for the account of this selling stockholder.
(102) Includes 16,667 shares of common stock issuable upon the exercise of warrants.
(103) Includes 15,000 shares of common stock issuable upon the exercise of warrants.
Certain Relationships and Related Party Transactions
On March 31, 2011, in connection with our share exchange transaction with the former shareholders of InspireMD Ltd. and succession to InspireMD Ltd.’s business as our sole line of business, we transferred all of our pre-share exchange operating assets and liabilities to Saguaro Holdings, Inc., a Delaware corporation and our wholly owned subsidiary. Immediately after this transfer, we transferred all of Saguaro Holdings, Inc.’s outstanding capital stock to Lynn Briggs, our then-majority stockholder and our former president, chief executive officer, chief financial officer, secretary-treasurer and sole director, in exchange for the cancellation of 7,500,000 shares of our common stock held by Ms. Briggs.
On May 6, 2008, InspireMD Ltd. entered into a consultancy agreement (the “2008 Consultancy Agreement”) for marketing services with Sara Paz, the wife of Ofir Paz, our chief executive officer. Pursuant to the 2008 Consultancy Agreement, Ms. Paz was paid by InspireMD Ltd. a fixed hourly fee of $45(154 New Israeli Shekels) in Israel and a fixed daily fee of $400 abroad with respect to her services. Under the 2008 Consultancy Agreement, either party was able to terminate the agreement, in whole or in part, without cause by submitting written notice of such termination to the other party at least 14 days prior to such termination. Under the 2008 Consultancy Agreement, InspireMD Ltd. paid Ms. Paz approximately $34,000, $72,600 and $103,000 in 2009, 2010 and the nine month period ended September 30, 2011, respectively. Under the 2008 Consultancy Agreement, as of December 31, 2010 and September 30, 2011, we recorded a provision of approximately $97,000 and $8,000, respectively. In addition, on September 1, 2011, effective April 1, 2011, the 2008 Consultancy Agreement was terminated and InspireMD Ltd. and Sara Paz Management and Marketing Ltd., an entity wholly-owned by Ms. Paz, entered into a new consultancy agreement (the “2011 Consultancy Agreement”) pursuant to which Ms. Paz was retained to serve as InspireMD Ltd.’s vice president of sales. Pursuant to the 2011 Consultancy Agreement, Ms. Paz was entitled to a monthly consultancy fee of $12,500 (42,684 New Israeli Shekels) from April 1, 2011 through June 30, 2011 and is entitled to a monthly consultancy fee of $15,500 (52,927 New Israeli Shekels) thereafter.
Description Of Securities
We have authorized 130,000,000 shares of capital stock, par value $0.0001 per share, of which 125,000,000 are shares of common stock and 5,000,000 are shares of “blank check” preferred stock. On November 30, 2011, there were 68,178,947 shares of common stock issued and outstanding and no shares of preferred stock issued and outstanding.
On October 31, 2011, our stockholders authorized our board of directors to amend our amended and restated certificate of incorporation to effect a reverse stock split of our common stock at a ratio of one-for-two to one-for-four, at any time prior to our 2012 annual stockholders’ meeting, the exact ratio of the reverse stock split to be determined by the board. As of the date of this prospectus, we have not effected the reverse stock split and, as such, the information with respect to our common stock in this prospectus and the accompanying financial statements and related notes does not give effect to any reverse stock split.
Common Stock
The holders of our common stock are entitled to one vote per share. Our certificate of incorporation does not provide for cumulative voting. The holders of our common stock are entitled to receive ratably such dividends, if any, as may be declared by our board of directors out of legally available funds; however, the current policy of our board of directors is to retain earnings, if any, for operations and growth. Upon liquidation, dissolution or winding-up, the holders of our common stock are entitled to share ratably in all assets that are legally available for distribution. The holders of our common stock have no preemptive, subscription, redemption or conversion rights. The rights, preferences and privileges of holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of any series of preferred stock, which may be designated solely by action of our board of directors and issued in the future.
Preferred Stock
The board of directors is authorized, subject to any limitations prescribed by law, without further vote or action by the stockholders, to issue from time to time shares of preferred stock in one or more series. Each such series of preferred stock shall have such number of shares, designations, preferences, voting powers, qualifications, and special or relative rights or privileges as shall be determined by the board of directors, which may include, among others, dividend rights, voting rights, liquidation preferences, conversion rights and preemptive rights.
Warrants
March $1.80 Warrants
On March 31, 2011 and on April 18, 2011, we issued certain investors five-year warrants to purchase up to an aggregate of 3,560,332 shares of common stock at an exercise price of $1.80 per share. We are prohibited from effecting the exercise of any such warrant to the extent that as a result of such exercise the holder of the exercised warrant beneficially owns more than 4.99% in the aggregate of the issued and outstanding shares of our common stock calculated immediately after giving effect to the issuance of shares of our common stock upon the exercise of the warrant. The warrants contain provisions that protect their holders against dilution by adjustment of the purchase price in certain events such as stock dividends, stock splits and other similar events. If at any time after the one year anniversary of the original issuance date of such warrants there is no effective registration statement registering, or no current prospectus available for, the resale of the shares of common stock underlying the warrant, then the holders of such warrants have the right to exercise the warrants by means of a cashless exercise. In addition, if (i) the volume-weighted average price of our common stock for 20 consecutive trading days is at least 250% of the exercise price of the warrants; (ii) the 20-day average daily trading volume of our common stock has been at least 175,000 shares; (iii) a registration statement providing for the resale of the common stock issuable upon exercise of the warrants is effective and (iv) the common stock is listed for trading on a national securities exchange, then we may require each holder to exercise all or a portion of its warrant pursuant to the terms described above within seven business days following the delivery of a notice of acceleration. Any warrant that is not exercised as aforesaid shall expire automatically at the end of such seven-day period.
April $1.80 Warrants
On April 18 and April 21, 2011, we issued certain investors five-year warrants to purchase up to an aggregate of 158,334 shares of common stock at an exercise price of $1.80 per share. We are prohibited from effecting the exercise of any such warrant to the extent that as a result of such exercise the holder of the exercised warrant beneficially owns more than 4.99% in the aggregate of the issued and outstanding shares of our common stock calculated immediately after giving effect to the issuance of shares of our common stock upon the exercise of the warrant. The warrants contain provisions that protect their holders against dilution by adjustment of the purchase price in certain events such as stock dividends, stock splits and other similar events. In addition, if (i) the volume-weighted average price of our common stock for 20 consecutive trading days is at least 250% of the exercise price of the warrants; (ii) the 20-day average daily trading volume of our common stock has been at least 175,000 shares; and (iii) a registration statement providing for the resale of the common stock issuable upon exercise of the warrants is effective, then we may require each holder to exercise all or a portion of its warrant pursuant to the terms described above within three business days following the delivery of a notice of acceleration. Any warrant that is not exercised as aforesaid shall expire automatically at the end of such three-day period.
Placement Agent Warrant
As consideration for serving as our placement agent in connection with certain private placements, we have issued Palladium Capital Advisors, LLC a five-year warrant to purchase up to 430,740 shares of common stock at an exercise price of $1.80 per share. The terms of this warrant are identical to the March $1.80 Warrants described above.
Employee Warrants
On March 31, 2011, for work performed in connection with the share exchange transactions and as bonus compensation, we issued Craig Shore, our chief financial officer, secretary and treasurer, a five-year warrant to purchase up to 3,000 shares of common stock at an exercise price of $1.80 per share. The terms of this warrant are identical to the April $1.80 Warrants described above.
Consultant Warrants
In connection with our March 31, 2011 private placement, we issued to Hermitage Capital Management, a consultant, a five-year warrant to purchase up to 6,667 shares of common stock at an exercise price of $1.80 per share, in consideration for consulting services. The terms of this warrant are identical to the April $1.80 Warrants described above.
In consideration for financial consulting services, we issued to The Benchmark Company, LLC, a consultant, a five-year warrant to purchase up to 50,000 shares of common stock at an exercise price of $1.50 per share. The terms of this warrant are identical to the April $1.80 Warrants described above, except that the exercise price for this warrant is $1.50 per share.
On March 31, 2011, we issued certain consultants five-year warrants to purchase up to an aggregate of 2,500,000 shares of common stock at an exercise price of $1.50 per share. The terms of these warrants are identical to the March $1.80 Warrants described above, except that the exercise price for these $1.50 warrants is $1.50 per share.
$1.23 Warrants
In connection with our share exchange transactions on March 31, 2011, we issued certain investors warrants to purchase up to an aggregate of 1,014,500 shares of our common stock at an exercise price of $1.23 per share. These warrants may be exercised any time on or before July 20, 2013 and were issued in exchange for warrants to purchase up to 125,000 ordinary shares of InspireMD Ltd. at an exercise price of $10 per share. We are prohibited from effecting the exercise of any such warrant to the extent that as a result of such exercise the holder of the exercised warrant beneficially owns more than 9.99% in the aggregate of the issued and outstanding shares of our common stock calculated immediately after giving effect to the issuance of shares of our common stock upon the exercise of the warrant. The warrants contain provisions that protect their holders against dilution by adjustment of the purchase price in certain events such as stock dividends, stock splits and other similar events. In addition, if at any time following the one year anniversary of the original issuance date of the warrants, (i) our common stock is listed for trading on a national securities exchange, (ii) the closing sales price of our common stock for 15 consecutive trading days is at least 165% of the exercise price of the warrants; (iii) the 15 day average daily trading volume of our common stock has been at least 150,000 shares and (iv) a registration statement providing for the resale of the common stock issuable upon exercise of the warrants is effective, then we may require each investor to exercise all or a portion of its warrant pursuant to the terms described above at any time upon at least 15 trading days prior written notice. Any warrant that is not exercised as aforesaid shall expire automatically at the end of the 15-day notice period.
Delaware Anti-Takeover Law and Provisions of our Certificate of Incorporation and Bylaws
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
|
|
·
|
prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
|
|
|
·
|
the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (i) shares owned by persons who are directors and also officers and (ii) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
|
|
|
·
|
on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder.
|
Section 203 defines a business combination to include:
|
|
·
|
any merger or consolidation involving the corporation and the interested stockholder;
|
|
|
·
|
any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation;
|
|
|
·
|
subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; or
|
|
|
·
|
the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
|
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with, or controlling, or controlled by, the entity or person. The term “owner” is broadly defined to include any person that, individually, with or through that person’s affiliates or associates, among other things, beneficially owns the stock, or has the right to acquire the stock, whether or not the right is immediately exercisable, under any agreement or understanding or upon the exercise of warrants or options or otherwise or has the right to vote the stock under any agreement or understanding, or has an agreement or understanding with the beneficial owner of the stock for the purpose of acquiring, holding, voting or disposing of the stock.
The restrictions in Section 203 do not apply to corporations that have elected, in the manner provided in Section 203, not to be subject to Section 203 of the Delaware General Corporation Law or, with certain exceptions, which do not have a class of voting stock that is listed on a national securities exchange or authorized for quotation on the Nasdaq Stock Market or held of record by more than 2,000 stockholders. Our certificate of incorporation and bylaws do not opt out of Section 203.
Section 203 could delay or prohibit mergers or other takeover or change in control attempts with respect to us and, accordingly, may discourage attempts to acquire us even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
Certificate of Incorporation and Bylaws
Provisions of our certificate of incorporation and bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our certificate of incorporation and bylaws:
|
|
·
|
permit our board of directors to issue up to 5,000,000 shares of preferred stock, without further action by the stockholders, with any rights, preferences and privileges as they may designate, including the right to approve an acquisition or other change in control;
|
|
|
·
|
provide that the authorized number of directors may be changed only by resolution of the board of directors;
|
|
|
·
|
provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum;
|
|
|
·
|
divide our board of directors into three classes, with each class serving staggered three-year terms;
|
|
|
·
|
do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose);
|
|
|
·
|
provide that special meetings of our stockholders may be called only by our board of directors; and
|
|
|
·
|
set forth an advance notice procedure with regard to the nomination, other than by or at the direction of our board of directors, of candidates for election as directors and with regard to business to be brought before a meeting of stockholders.
|
Indemnification of Directors and Officers
Section 145 of the General Corporation Law of the State of Delaware provides, in general, that a corporation incorporated under the laws of the State of Delaware, as we are, may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (other than a derivative action by or in the right of the corporation) by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe such person’s conduct was unlawful. In the case of a derivative action, a Delaware corporation may indemnify any such person against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification will be made in respect of any claim, issue or matter as to which such person will have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery of the State of Delaware or any other court in which such action was brought determines such person is fairly and reasonably entitled to indemnity for such expenses.
Our certificate of incorporation and bylaws provide that we will indemnify our directors, officers, employees and agents to the extent and in the manner permitted by the provisions of the General Corporation Law of the State of Delaware, as amended from time to time, subject to any permissible expansion or limitation of such indemnification, as may be set forth in any stockholders’ or directors’ resolution or by contract. Any repeal or modification of these provisions approved by our stockholders will be prospective only and will not adversely affect any limitation on the liability of any of our directors or officers existing as of the time of such repeal or modification.
We are also permitted to apply for insurance on behalf of any director, officer, employee or other agent for liability arising out of his actions, whether or not the General Corporation Law of the State of Delaware would permit indemnification.
Disclosure of Commission Position on Indemnification for Securities Act Liabilities
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, may be permitted to our directors, officers and persons controlling us, we have been advised that it is the Securities and Exchange Commission’s opinion that such indemnification is against public policy as expressed in the Securities Act of 1933, as amended, and is, therefore, unenforceable.
Plan Of Distribution
The selling stockholders may, from time to time, sell any or all of their shares of common stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These sales may be at fixed or negotiated prices. The selling stockholders may use any one or more of the following methods when selling shares:
|
|
·
|
ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers;
|
|
|
·
|
block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction;
|
|
|
·
|
purchases by a broker-dealer as principal and resale by the broker-dealer for its account;
|
|
|
·
|
an exchange distribution in accordance with the rules of the applicable exchange;
|
|
|
·
|
privately negotiated transactions;
|
|
|
·
|
short sales;
|
|
|
·
|
broker-dealers may agree with the selling stockholders to sell a specified number of such shares at a stipulated price per share;
|
|
|
·
|
a combination of any such methods of sale;
|
|
|
·
|
through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; or
|
|
|
·
|
any other method permitted pursuant to applicable law.
|
The selling stockholders may also sell shares under Rule 144 under the Securities Act of 1933, as amended, if available, rather than under this prospectus.
Broker-dealers engaged by the selling stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the selling stockholders (or, if any broker-dealer acts as agent for the purchaser of shares, from the purchaser) in amounts to be negotiated. The selling stockholders do not expect these commissions and discounts to exceed what is customary in the types of transactions involved. Any profits on the resale of shares of common stock by a broker-dealer acting as principal might be deemed to be underwriting discounts or commissions under the Securities Act of 1933, as amended. Discounts, concessions, commissions and similar selling expenses, if any, attributable to the sale of shares will be borne by a selling stockholder. The selling stockholders may agree to indemnify any agent, dealer or broker-dealer that participates in transactions involving sales of the shares if liabilities are imposed on that person under the Securities Act of 1933, as amended.
The selling stockholders may from time to time pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock from time to time under this prospectus after we have filed a supplement to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act of 1933, as amended, supplementing or amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus.
The selling stockholders also may transfer the shares of common stock in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus and may sell the shares of common stock from time to time under this prospectus after we have filed a supplement to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act of 1933, as amended, supplementing or amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus.
The selling stockholders and any broker-dealers or agents that are involved in selling the shares of common stock may be deemed to be “underwriters” within the meaning of the Securities Act of 1933, as amended, in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the shares of common stock purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act of 1933, as amended.
We have agreed to pay all fees and expenses incident to the registration of the shares of common stock. We have agreed to indemnify the selling stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act of 1933, as amended.
We do not believe that the selling stockholders have entered into any agreements, understandings or arrangements with any underwriters or broker-dealers regarding the sale of their shares of common stock, nor is there an underwriter or coordinating broker acting in connection with a proposed sale of shares of common stock by any selling stockholder. If we are notified by any selling stockholder that any material arrangement has been entered into with a broker-dealer for the sale of shares of common stock, if required, we will file a supplement to this prospectus. If the selling stockholders use this prospectus for any sale of the shares of common stock, they will be subject to the prospectus delivery requirements of the Securities Act of 1933, as amended.
The anti-manipulation rules of Regulation M under the Securities Exchange Act of 1934, as amended, may apply to sales of our common stock and activities of the selling stockholders.
Haynes and Boone, LLP, New York, New York, will pass upon the validity of the shares of our common stock offered by the selling stockholders under this prospectus.
Our financial statements as of December 31, 2009 and 2010 and for the years ended December 31, 2009 and 2010 included in this prospectus have been audited by Kesselman & Kesselman, Certified Public Accountants, a member of PricewaterhouseCoopers International Limited, an independent registered public accounting firm, as stated in its report appearing in the registration statement, and are included in reliance upon the report of such firm given upon its authority as experts in accounting and auditing.
We have filed with the Securities and Exchange Commission a registration statement on Form S-1, together with any amendments and related exhibits, under the Securities Act of 1933, as amended, with respect to our shares of common stock offered by this prospectus. The registration statement contains additional information about us and our shares of common stock that the selling stockholders are offering in this prospectus.
We file annual, quarterly and current reports and other information with the Securities and Exchange Commission under the Securities Exchange Act of 1934, as amended. Our Securities and Exchange Commission filings are available to the public over the Internet at the Securities and Exchange Commission’s website at http://www.sec.gov. You may also read and copy any document we file at the Securities and Exchange Commission’s public reference room located at 100 F Street, N.E., Washington, D.C. 20549. Please call the Securities and Exchange Commission at 1-800-SEC-0330 for further information on the public reference rooms and their copy charges. In addition, through our website, http://www.inspire-md.com, you can access electronic copies of documents we file with the Securities and Exchange Commission, including our Quarterly Report on Form 10-Q, and Current Reports on Form 8-K and any amendments to those reports. Information on our website is not incorporated by reference in this prospectus. Access to those electronic filings is available as soon as practicable after filing with the Securities and Exchange Commission. You may also request a copy of those filings, excluding exhibits, from us at no cost. Any such request should be addressed to us at: 3 Menorat Hamaor St., Tel Aviv, Israel 67448, Attention: Ofir Paz, Chief Executive Officer.
INSPIREMD LTD.
CONSOLIDATED FINANCIAL STATEMENTS
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated Balance Sheets at December 31, 2010 and 2009
|
F-3
|
|
Consolidated Statements of Operations for the Year Ended December 31, 2010 and 2009
|
F-5
|
|
Consolidated Statement of Changes in Equity (Capital Deficiency) for the Year Ended December 31, 2010 and 2009
|
F-6
|
|
Consolidated Statement of Cash Flows for the Year Ended December 31, 2010 and 2009
|
F-7
|
|
Notes to Consolidated Financial Statements (Two years ended December 31, 2010)
|
F-8
|
|
Condensed Consolidated Balance Sheets (Unaudited) at September 30, 2011 and December 31, 2010
|
F-36 |
|
Condensed Consolidated Statements of Operations (Unaudited) for Nine months ended September 30, 2011 and 2010
|
F-38 |
|
Condensed Consolidated Statements of Changes in Equity (Capital Deficiency) (Unaudited) for Nine months ended September 30, 2011 and 2010
|
F-39 |
|
Condensed Consolidated Statements of Cash Flows (Unaudited) for Nine months ended September 30, 2011 and 2010
|
F-40 |
|
Notes to Condensed Consolidated Financial Statements for the Nine months ended September 30, 2011
|
F-41 |
The amounts are stated in U.S. dollars in thousands
_______________
_________________________
_______________

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders of
InspireMD Ltd.
We have audited the accompanying consolidated balance sheets of InspireMD Ltd. (the “Company”) and its subsidiary as of December 31, 2010 and 2009 and the related consolidated statements of operations, changes in equity (capital deficiency) and cash flows for each of the two years in the period ended December 31, 2010. These consolidated financial statements are the responsibility of the Company’s Board of Directors and management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by the Company’s board of directors and management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company and its subsidiary as of December 31, 2010 and 2009 and the results of their operations, changes in equity (capital deficiency) and cash flows for each of the two years in the period ended December 31, 2010, in conformity with accounting principles generally accepted in the United States of America.
|
Tel-Aviv, Israel
|
/s/ Kesselman & Kesselman
|
|
March 31, 2011, except for notes 10 c(1) and 15 for which the date is June 13, 2011
|
Certified Public Accountants (Isr.)
A member firm of PricewaterhouseCoopers International Limited
|
|
|
INSPIREMD LTD.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands)
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
CURRENT ASSETS:
|
||||||||
|
Cash and cash equivalents
|
$ | 636 | $ | 376 | ||||
|
Restricted cash
|
250 | 302 | ||||||
|
Accounts receivable:
|
||||||||
|
Trade
|
852 | 1,189 | ||||||
|
Other
|
75 | 130 | ||||||
|
Prepaid expenses
|
3 | 39 | ||||||
|
Inventory:
|
||||||||
|
On consignment
|
371 | 1,093 | ||||||
|
Other
|
1,704 | 946 | ||||||
|
Total current assets
|
3,891 | 4,075 | ||||||
|
PROPERTY, PLANT AND EQUIPMENT, net of accumulated depreciation and amortization
|
282 | 292 | ||||||
|
NON-CURRENT ASSETS:
|
||||||||
|
Deferred debt issuance costs
|
15 | 29 | ||||||
|
Fund in respect of employee rights upon retirement
|
167 | 113 | ||||||
|
Total non-current assets
|
182 | 142 | ||||||
|
Total assets
|
$ | 4,355 | $ | 4,509 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD LTD.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands)
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
Liabilities net of capital deficiency
|
||||||||
|
CURRENT LIABILITIES:
|
||||||||
|
Current maturities of long-term loans
|
$ | 355 | $ | 281 | ||||
|
Accounts payable and accruals :
|
||||||||
|
Trade
|
1,103 | 907 | ||||||
|
Other
|
1,509 | 1,304 | ||||||
|
Advanced payment from customers
|
559 | 877 | ||||||
|
Loans from shareholders
|
20 | 20 | ||||||
|
Deferred revenues
|
398 | 1,975 | ||||||
|
Total current liabilities
|
3,944 | 5,364 | ||||||
|
LONG-TERM LIABILITIES:
|
||||||||
|
Long term loan
|
75 | 342 | ||||||
|
Liability for employees rights upon retirement
|
206 | 142 | ||||||
|
Convertible loan
|
1,044 | - | ||||||
|
Total long-term liabilities
|
1,325 | 484 | ||||||
|
COMMITMENTS AND CONTINGENT LIABILITIES (note 8)
|
||||||||
|
Total liabilities
|
5,269 | 5,848 | ||||||
|
CAPITAL DEFICIENCY:
|
||||||||
|
Common stock, par value $0.0001 per share; 125,000,000 shares authorized; 48,338,380 shares issued and outstanding at December 31, 2009 and 49,863,801 shares issued and outstanding at December 31, 2010
|
5 | 5 | ||||||
|
Additional paid-in capital
|
21,057 | 17,212 | ||||||
|
Accumulated deficit
|
(21,976 | ) | (18,556 | ) | ||||
|
Total capital deficiency
|
(914 | ) | (1,339 | ) | ||||
|
Total liabilities less capital deficiency
|
$ | 4,355 | $ | 4,509 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
Date of approval of financial statements: June 13, 2011
INSPIREMD LTD.
CONSOLIDATED STATEMENTS OF OPERATIONS
(U.S. dollars in thousands, except per share data)
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
REVENUES
|
$ | 4,949 | $ | 3,411 | ||||
|
COST OF REVENUES
|
2,696 | 2,291 | ||||||
|
GROSS PROFIT
|
2,253 | 1,120 | ||||||
|
OPERATING EXPENSES:
|
||||||||
|
Research and development
|
1,338 | 1,330 | ||||||
|
Selling and marketing
|
1,236 | 1,040 | ||||||
|
General and administrative
|
2,898 | 1,467 | ||||||
|
Total operating expenses
|
5,472 | 3,837 | ||||||
|
LOSS FROM OPERATIONS
|
(3,219 | ) | (2,717 | ) | ||||
|
FINANCIAL EXPENSES (INCOME), net
|
154 | (40 | ) | |||||
|
LOSS BEFORE TAX EXPENSES
|
(3,373 | ) | (2,677 | ) | ||||
|
TAX EXPENSES
|
47 | 47 | ||||||
|
NET LOSS
|
$ | (3,420 | ) | $ | (2,724 | ) | ||
|
NET LOSS PER SHARE - basic and diluted
|
$ | (0.07 | ) | $ | (0.06 | ) | ||
|
WEIGHTED AVERAGE NUMBER OF ORDINARY SHARES USED IN COMPUTING NET LOSS PER SHARE - basic and diluted
|
49,234,528 | 47,658,853 | ||||||
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD LTD.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY (CAPITAL DEFICIENCY)
(U.S. dollars in thousands)
|
Ordinary shares
|
||||||||||||||||||||
|
Number of shares
|
Par value
|
Additional paid-in capital
|
Accumulated deficit
|
Total equity (capital deficiency)
|
||||||||||||||||
|
BALANCE AT JANUARY 1, 2009
|
47,061,936 | $ | 5 | $ | 15,961 | $ | (15,832 | ) | $ | 134 | ||||||||||
|
CHANGES DURING 2009:
|
||||||||||||||||||||
|
Net loss
|
(2,724 | ) | (2,724 | ) | ||||||||||||||||
|
Exercise of options by employees
|
458,722 | * | * | * | ||||||||||||||||
|
Employee and non-employee share-based compensation expenses
|
594 | 594 | ||||||||||||||||||
|
Redemption of beneficial conversion Feature of convertible loan
|
(308 | ) | (308 | ) | ||||||||||||||||
|
Issuance of ordinary shares, net of $44 issuance costs
|
817,722 | * | 965 | 965 | ||||||||||||||||
|
BALANCE AT DECEMBER 31, 2009
|
48,338,380 | 5 | 17,212 | (18,556 | ) | (1,339 | ) | |||||||||||||
|
CHANGES DURING 2010:
|
||||||||||||||||||||
|
Net loss
|
(3,420 | ) | (3,420 | ) | ||||||||||||||||
|
Employee and non-employee share-based compensation expenses
|
1,640 | 1,640 | ||||||||||||||||||
|
Issuance of warrants, net of $23 issuance costs
|
424 | 424 | ||||||||||||||||||
|
Issuance of ordinary shares, net of $97 issuance costs
|
1,525,421 | * | 1,781 | 1,781 | ||||||||||||||||
|
BALANCE AT DECEMBER 31, 2010
|
49,863,801 | $ | 5 | $ | 21,057 | $ | (21,976 | ) | $ | (914 | ) | |||||||||
* Represents an amount less than $1
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD LTD.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||||||
|
Net loss
|
$ | (3,420 | ) | $ | (2,724 | ) | ||
|
Adjustments required to reconcile net loss to net cash used in
|
||||||||
|
operating activities:
|
||||||||
|
Depreciation and amortization of property, plant and equipment
|
91 | 89 | ||||||
|
Change in liability for employees right upon retirement
|
42 | 42 | ||||||
|
Financial expenses (income)
|
94 | (224 | ) | |||||
|
Share-based compensation expenses
|
1,620 | 562 | ||||||
|
Gains on amounts funded in respect of employee rights upon retirement, net
|
(11 | ) | (10 | ) | ||||
|
Changes in operating asset and liability items:
|
||||||||
|
Decrease (increase) in Prepaid expenses
|
36 | (32 | ) | |||||
|
Decrease (increase) in Trade receivables
|
337 | (969 | ) | |||||
|
Decrease (increase) in Other receivables
|
9 | (27 | ) | |||||
|
Decrease in Inventory on consignment
|
722 | 330 | ||||||
|
Increase in other inventories
|
(758 | ) | (241 | ) | ||||
|
Increase in Trade payables
|
196 | 612 | ||||||
|
Decrease in Deferred revenues
|
(1,577 | ) | (507 | ) | ||||
|
Increase (decrease) in Other payable
and advance payment from customers
|
(91 | ) | 1,554 | |||||
|
Net cash used in operating activities
|
(2,710 | ) | (1,545 | ) | ||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||
|
Decrease (increase) in restricted cash
|
52 | (272 | ) | |||||
|
Purchase of property, plant and equipment
|
(81 | ) | (34 | ) | ||||
|
Proceeds from sale of property, plant and equipment
|
4 | |||||||
|
Amounts funded in respect of employee rights upon retirement, net
|
(17 | ) | (44 | ) | ||||
|
Net cash used in investing activities
|
(46 | ) | (346 | ) | ||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||
|
Proceeds from issuance of shares, net of issuance costs
|
1,821 | 976 | ||||||
|
Proceeds from long-term loan, net of $41 issuance costs
|
419 | |||||||
|
Issuance of warrants, net of $23 issue costs
|
424 | |||||||
|
Proceeds from convertible loan at fair value through profit or loss,
|
||||||||
|
net of $60 issuance costs
|
1,073 | |||||||
|
Repayment of long term loan
|
(281 | ) | ||||||
|
Repayment of loans from shareholders
|
(20 | ) | ||||||
|
Repayment of Convertible loan
|
(720 | ) | ||||||
|
Net cash provided by financing activities
|
3,037 | 655 | ||||||
|
EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS
|
(21 | ) | 41 | |||||
|
INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS
|
260 | (1,195 | ) | |||||
|
BALANCE OF CASH AND CASH EQUIVALENTS AT BEGINNING OF YEAR
|
376 | 1,571 | ||||||
|
BALANCE OF CASH AND CASH EQUIVALENTS AT END OF YEAR
|
$ | 636 | $ | 376 | ||||
|
SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION:
|
||||||||
|
Taxes on income paid
|
$ | 56 | $ | - | ||||
|
Interest paid
|
$ | 30 | $ | 88 | ||||
|
SUPPLEMENTAL DISCLOSURE OF NON-CASH FINANCING ACTIVITIES -
|
||||||||
|
receivables on account of shares
|
$ | - | $ | 20 | ||||
* Represents an amount less than $1
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 - DESCRIPTION OF BUSINESS
InspireMD Ltd (the “Company"), an Israeli corporation, was incorporated and commenced operations in April 2005. InspireMD GmbH (the "Subsidiary") was incorporated on November 2007.
The Company and its Subsidiary, (collectively, the “Group”), develops, manufactures, markets and sells unique coronary stents.
The Group markets its products through distributers in international markets, mainly in Europe. The Company currently depends on a single manufacturer.
Management of the Company is in the opinion that as a result of the consummation of the reverse merger transaction described in note 15.f, the Company has sufficient cash to continue its operations into 2012. However, depending on the operating results in 2011, the Company may need to obtain additional cash in 2012 to continue to fund operations.
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES:
|
a.
|
Accounting principles
|
|
|
The consolidated financial statements are prepared in accordance with accounting principles generally accepted in the United States (“US GAAP”).
|
|
|
b.
|
Use of estimates
|
|
|
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of sales and expenses during the reporting periods. Actual results could differ from those estimates.
|
|
|
As applicable to these consolidated financial statements, the most significant estimates and assumptions relate to revenue recognition including provision for returns, legal contingencies, estimation of the fair value of share-based compensation and estimation of the fair value of a convertible loan.
|
|
c.
|
Functional currency
|
|
|
The currency of the primary economic environment in which the operations of the Company and its subsidiary are conducted is the U.S. dollar (“$” or “dollar”). Accordingly, the functional currency of the Company and of the subsidiary is the dollar.
|
The dollar figures are determined as follows: transactions and balances originally denominated in dollars are presented in their original amounts. Balances in foreign currencies are translated into dollars using historical and current exchange rates for non-monetary and monetary balances, respectively. The resulting translation gains or losses are recorded as financial income or expense, as appropriate. For transactions reflected in the statements of operations in foreign currencies, the exchange rates at transaction dates are used. Depreciation and changes in inventories and other changes deriving from non-monetary items are based on historical exchange rates.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES (continued):
|
d.
|
Principles of consolidation
|
|
|
The consolidated financial statements include the accounts of the Company and of its Subsidiary. Intercompany transactions and balances, have been eliminated upon consolidation.
|
|
e.
|
Cash and cash equivalents
|
|
|
The Group considers all highly liquid investments, which include short-term bank deposits (up to three months from date of deposit) that are not restricted as to withdrawal or use to be cash equivalents.
|
|
f.
|
Restricted cash
|
|
|
The Company maintains certain cash amounts restricted as to withdrawal or use, related mainly to long-term loan, see note 7. The restricted cash are denominated in U.S. dollars and NIS.
|
|
g.
|
Fair value measurement:
|
|
|
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (i.e., the “exit price”) in an orderly transaction between market participants at the measurement date.
|
In determining fair value, the Group uses various valuation approaches, including market, income and/or cost approaches. Hierarchy for inputs is used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Group’s assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy is broken down into three levels based on the reliability of inputs.
|
h.
|
Concentration of credit risk and allowance for doubtful accounts
|
|
|
Financial instruments that may potentially subject the Group to a concentration of credit risk consist of cash, cash equivalents and restricted cash which are deposited in major financial institutions in Germany and Israel, and trade accounts receivable. The Group’s trade accounts receivable are derived from revenues earned from customers from various counties. The Group performs ongoing credit evaluations of its customers’ financial condition and, generally, requires no collateral from its customers. The Group also has a credit insurance policy for part of its customers. The Group maintains an allowance for doubtful accounts receivable based upon the expected ability to collect the accounts receivable. The Group reviews its allowance for doubtful accounts quarterly by assessing individual accounts receivable and all other balances based on historical collection experience and an economic risk assessment. If the Group determines that a specific customer is unable to meet its financial obligations to the Group, the Group provides an allowance for credit losses to reduce the receivable to the amount management reasonably believes will be collected. To mitigate risks the Group deposits cash and cash equivalents with high credit quality financial institutions.
|
|
|
Provisions for doubtful debts are netted against “Accounts receivable-trade.”
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES (continued):
|
i.
|
Inventory
|
Inventories include finished goods, work in process and raw materials. Inventories are stated at the lower of cost (cost is determined on a “first-in, first-out” basis) or market value.
In respect to inventory on consignment, see note 2(l).
|
|
j.
|
Property, plant and equipment
|
|
|
Property, plant and equipment are stated at cost, net of accumulated depreciation and amortization. Depreciation is calculated using the straight-line method over the estimated useful lives of the related assets: over three years for computers and other electronic equipment, five years for vehicles and seven to fifteen years for office furniture and equipment, and machinery and equipment (mainly seven years). Leasehold improvements are amortized on a straight-line basis over the term of the lease, which is shorter than the estimated life of the improvements.
|
|
k.
|
Impairment of long-lived assets
|
The Group reviews all long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. If the sum of the expected future cash flows (undiscounted and without interest charges) of the long-lived assets is less than the carrying amount of such assets, an impairment loss would be recognized, and the assets would be written down to their estimated fair values.
To date, the Group has not recorded any impairment charges relating to its long-lived assets.
|
l.
|
Revenue recognition
|
|
|
Revenue is recognized when delivery has occurred, evidence of an arrangement exists, title and risks and rewards for the products are transferred to the customer, collection is reasonably assured and when product returns can be reliably estimated. When product returns can be reliably estimated a provision is recorded, based on historical experience, and deducted from sales. The provision for sales returns and related costs are included in “Accounts payable and accruals - Other” under “current liabilities", and "Inventory on consignment", respectively.
|
|
|
When returns cannot be reliably estimated, both revenues and related direct costs are eliminated, as the products are deemed unsold. Accordingly, both related revenues and costs are deferred, and presented under "Deferred revenues" and "Inventory on consignment", respectively.
|
|
|
The Company’s revenue arrangements may contain delivery of free products upon the achievement of sales targets. When free products are delivered in a different period than the related products that were fully paid by the distributor, the Company allocates revenue between the free products and the fully paid products based on the quantities of the free products and the fully paid products. Each period end, the Company estimates the amount of free products these certain distributors will be entitled to upon the expected achievement of sales targets and allocates revenue accordingly.
|
The Group recognizes revenue net of value added tax (VAT).
|
m.
|
Research and development costs
|
Research and development costs are charged to the statement of operations as incurred.
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES (continued):
|
n.
|
Share-based compensation
|
|
|
Employees option awards are classified as equity awards and accounted for using the grant-date fair value method. The fair value of share-based awards is estimated using the Black-Scholes valuation model, which is expense over the requisite service period, net of estimated forfeitures. The Company estimates forfeitures based on historical experience and anticipated future conditions.
|
|
|
The Company elected to recognize compensation expensed for awards with only service conditions that have graded vesting schedules using the accelerated multiple option approach.
|
The Company accounts for equity instruments issued to third party service providers (non-employees), by recording the fair value of the options granted using an option pricing model, at each reporting period, until rewards is vested in full. The expense is recognized over the vesting period using the accelerated multiple option approach. The expense relates to options granted to third parties service providers in respect of potential investor's introduction services to the Company in which the Company entered into an agreement with the investor (hereafter-Finder's services) is recorded at its fair value in Equity, as issuance costs.
|
|
o.
|
Uncertain tax positions
|
The Company follows a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon ultimate settlement. Such liabilities are classified as long-term, unless the liability is expected to be resolved within twelve months from the balance sheet date. The Company’s policy is to include interest and penalties related to unrecognized tax benefits within financial expenses.
|
p.
|
Deferred Income taxes
|
Deferred taxes are determined utilizing the “asset and liability” method based on the estimated future tax effects of differences between the financial accounting and tax bases of assets and liabilities under the applicable tax laws, and on tax rates anticipated to be in effect when the deferred taxes are expected to be paid or realized. Valuation allowance is provided if, based upon the weight of available evidence, it is “more likely than not” that a portion of the deferred tax assets will not be realized. The Company has established a valuation allowance against certain of its deferred tax assets because management believes that after considering all of the available evidence, historical and prospective, it is not more likely than not that such deferred tax assets will be realized within their recovery periods.
The Company may incur additional tax liability in the event of intercompany dividend distributions by its subsidiary. Such additional tax liability in respect of this non-Israeli subsidiary has not been provided for in these financial statements as it is the Company’s policy permanently to reinvest the subsidiary's earnings and to consider distributing dividends only when this can be facilitated in connection with a specific tax opportunity that may arise.
Taxes which would apply in the event of disposal of investments in non-Israeli subsidiary have not been taken into account in computing the deferred taxes, as it is the Company’s intention to hold, and not to realize, this investment.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES (continued):
|
q.
|
Advertising
|
|
|
Cost related to advertising and promotion of products is charged to sales and marketing expense as incurred. Advertising expenses for the end of the years 2009 and 2010 were $275 and $467 thousands, respectively.
|
|
r.
|
Net loss per share
|
Basic and diluted net loss per share is computed by dividing the net loss for the year by the weighted average number of ordinary shares outstanding during the year. The calculation of diluted net loss per share excludes potential ordinary shares as the effect is anti-dilutive. Potential ordinary shares are comprised of incremental ordinary shares issuable upon the exercise of share options, warrants or convertible loan.
For the years ended December 31, 2010 and 2009 all outstanding options, warrants and convertible loan have been excluded from the calculation of the diluted loss per share since their effect was anti-dilutive. The total number of ordinary shares related to outstanding options and convertible loan excluded from the calculations of diluted loss per share were 9,502,111 and 5,877,388 for the years ended December 31, 2010 and 2009, respectively.
|
s.
|
Segment reporting
|
|
|
The Company has one operating and reportable segment.
|
|
t.
|
Subsequent events
|
|
|
Subsequent events were evaluated through June 13, 2011.
|
|
u.
|
Newly issued accounting pronouncements
|
|
|
In October 2009, the FASB issued amendments to the accounting and disclosure for revenue recognition. These amendments, effective for fiscal years beginning on or after June 15, 2010 (early adoption is permitted), modify the criteria for recognizing revenue in multiple element arrangements and require companies to develop a best estimate of the selling price to separate deliverables and allocate arrangement consideration using the relative selling price method. Additionally, the amendments eliminate the residual method for allocating arrangement considerations. The Company does not expect the standard to have material effect on its consolidated financial statements.
|
|
|
In January 2010, the FASB updated the “Fair Value Measurements Disclosures”. More specifically, this update will require (a) an entity to disclose separately the amounts of significant transfers in and out of Levels 1 and 2 fair value measurements and to describe the reasons for the transfers; and (b) information about purchases, sales, issuances and settlements to be presented separately (i.e. present the activity on a gross basis rather than net) in the reconciliation for fair value measurements using significant unobservable inputs (Level 3 inputs). This update clarifies existing disclosure requirements for the level of disaggregation used for classes of assets and liabilities measured at fair value, and require disclosures about the valuation techniques and inputs used to measure fair value for both recurring and nonrecurring fair value measurements using Level 2 and Level 3 inputs. This will become effective as of the first interim or annual reporting period beginning after December 15, 2009, except for the gross presentation of the Level 3 roll forward information, which is required for annual reporting periods beginning after December 15, 2010 and for interim reporting periods within those years. The adoption of the new guidance will not have a material impact on the Company's consolidated financial statements.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES (continued):
|
v.
|
Factoring of receivables
|
|
|
During 2010, the Company factored some of its trade receivables. The factoring was executed through banking institution on a recourse basis, and through other non-banking institute on a non-recourse basis. As of December 31, 2010 the Company did not have financial assets relates to such transaction.
|
|
|
The resulting costs were charged to “financial expenses-net”.
|
NOTE 3 - FAIR VALUE MEASURMENT
|
a.
|
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date.
|
The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
Convertible loan was initially recorded at fair value of $1,133, then subsequently remeasured at fair value with the decrease in fair value of $89 included in the profit or loss as of December 31, 2010. This security is measured at fair value on a recurring basis and classified in the "Significant Unobservable inputs (Level 3)" category.
|
b.
|
The carrying amounts of cash and cash equivalents, accounts receivable, accounts payable and other accrued liabilities approximate their fair value either because these amounts are presented at fair value or due to the relatively short-term maturities of such instruments. The carrying amount of the Group’s other financial long-term assets and other financial long-term liabilities approximate their fair value.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 4 - PROPERTY, PLANT AND EQUIPMENT:
|
a.
|
Composition of assets, grouped by major classifications, is as follows:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Cost:
|
||||||||
|
Vehicles
|
$ | 44 | $ | 28 | ||||
|
Computer equipment
|
75 | 45 | ||||||
|
Office furniture and equipment
|
54 | 53 | ||||||
|
Machinery and equipment
|
416 | 384 | ||||||
|
Leasehold improvements
|
47 | 45 | ||||||
| 636 | 555 | |||||||
|
Less - accumulated depreciation and amortization
|
(354 | ) | (263 | ) | ||||
|
Net carrying amount
|
$ | 282 | $ | 292 | ||||
|
b.
|
Depreciation and amortization expenses totaled approximately $91 thousands and $89 thousands for the years ended December 31, 2010 and 2009, respectively.
|
NOTE 5 - LIABILITY FOR EMPLOYEES RIGHT UPON RETIREMENT
Israeli labor law generally requires payment of severance pay upon dismissal of an employee or upon termination of employment in certain other circumstances.
Pursuant to section 14 of the Israeli Severance Compensation Act, 1963, some of the Company's employees are entitled to monthly deposits, at a rate of 8.33% of their monthly salary, made in their name with insurance companies. Payments in accordance with section 14 relieve the Company from any future severance payments in respect of those employees.
The severance pay liability of the Company to the rest of its employees, which reflects the undiscounted amount of the liability, is based upon the number of years of service and the latest monthly salary, and is partly covered by insurance policies and by regular deposits with recognized severance pay funds. The Company may only make withdrawals from the amounts funded for the purpose of paying severance pay. The severance pay expenses (income) were $14 thousands and $(7) thousands in the years ended December 31, 2010 and 2009, respectively. Gain on amounts funded in respect of employee rights upon retirement totaled to $11 thousands and $10 thousands for the years ended December 31, 2010 and 2009, respectively.
The Company expects to contribute approximately $195 thousands in 2011 to the pension funds and insurance companies in respect of its severance and pension pay obligations.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 6 – CONVERTIBLE LOAN AND REVERSE MERGER AGREEMENTS
At the beginning of 2010, the Company started a process of undergoing a Share Exchange transaction into a US public shell company (the "Shell"). In July 2010 The Company entered into an agreement with an investment bank (the "Investment Bank") on a best effort basis to act as an agent in connection with (i) the issuance of convertible debentures ("Convertible Debenture Transaction") to certain investors in the aggregate amount of $1.58 million (the "Debentures") and 1,014,513 warrants which will be allocated to each investor pro rata to the principal amount of the debenture purchased by such investor as compared to the aggregate principal amount of all Debentures issued in the offering ("the Warrants") and (ii) the sale of at least $7.5 million and up to $10 million (after deducting $1.58 million and any accrued interest as of the transaction date to be repaid to investors in a Convertible debenture Transaction) of equity or equity linked securities of the Shell to a limited number of investors (the “Private Placement”).
The convertible debentures and the Warrants in total amount of $1.58 million were issued on July 22, 2010. The Debentures bear annual interest of 8% and are payable upon the later of (i) two months subsequent to the Borrower's receipt of a tax ruling or (ii) six months from issuance date of the Debentures (the "Original Maturity Date"). Provided an Event of Default (as stipulated in the agreement) has not occurred before the Original Maturity Date, then the borrower shall have the right, at its sole discretion, to extend the maturity date until nine months after the Original Maturity Date (the "Second Maturity Date"). An Event of Default includes, inter alia, breach of covenants (as stipulated in the agreement), breach of standard representations and warranties, obtaining an unfavorable tax ruling, Merger and bankruptcy (as stipulated in the agreement).
Provided that neither an Event of Default nor an execution of the Private Placement have occurred prior to the Second Maturity Date, the Debenture shall be converted into Company's equity (or in the event of a successful execution of the Private Placement the Convertible debenture shall be converted to the Shell's equity) at predefined conversion ratios.
As indicated above, the holders of the Debentures, shall, at their option, have the right to demand immediate payment of both principal and interest then remaining unpaid upon the occurrence of Event of Default or upon the execution of the Private Placement prior to the Second Maturity Date.
If the Debentures are repaid to by the Company upon execution of the Private Placement, the Investment Bank will be obligated to raise such amounts to be repaid in addition to the minimum net amount of $7.5 million as indicated above.
The warrants conditions are as follows:
|
-
|
Exercise price of $1.23 per warrant.
|
|
-
|
Expiration term of 3 years.
|
|
-
|
In the event the company has not completed a Share Exchange before the original maturity date, third of the warrants shall expire immediately.
|
The Company has elected to apply regarding the debentures the fair value option in accordance with Topic 825 (i.e. the Debenture will be measured at each balance sheet date at fair value and the changes in its fair value will be recorded in profit and loss).
The proceeds from the issuance were allocated to the debentures at their fair value with the residual proceeds ascribed to the warrants as follows:
Debenture at fair value - $1,133 thousands.
Warrants - $447 thousands, net of $23 thousands direct transaction costs.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 6 – CONVERTIBLE LOAN AND REVERSE MERGER AGREEMENTS (continued):
The issuance of warrants was recorded in the additional paid-in capital, net of $23 thousands direct transaction costs allocated to the warrants.
The Company adjusted the value of the Debenture to fair value at December 31, 2010 and recorded the decrease in the value of $89 thousand as a gain included in Financial Income in the year ended December 31, 2010.
On December 29, 2010 the Company entered into a Share Exchange agreement (the "agreement") with an American shell company named Saguaro Resource Inc (the "Shell").
The reverse merger will be executed by share exchange between the Company's shareholders, in way that the Company's shareholders who represents at least 80% of the Company's shares, shall transfer their shares free and clear of all liens, in exchange of the Shell's shares in an exchange ratio of at least 6.67 shares of the shell for every Company's share. The final exchange ratio agreed upon the closing of the transaction on March 31, 2011 was 8.1161 shares of the shell for every Company's share.
The closing of the transactions contemplated under the agreement (the "transactions") is subject to and conditioned upon investors irrevocably (i) committing to purchase such number of shares of Shell shares, on terms acceptable to the Company, that would result in an aggregate net proceeds to the Shell of at least $7,500,000 (the “Private Placement”) (excluding (i) all fees payable to brokers and any other third party, including the Company’s legal counsel in connection with the Private Placement and the Transactions; and (ii) the conversion of the Convertible Debentures (see note 5(a)) in the aggregate original principal amount of $1,580,000, together with any interest accrued thereon), and shall have placed such funds in escrow to be automatically released into the Shell’s bank account upon consummation of the Transactions. The closing is subject to a previous wide disclosure of all parties including the Company, the Company's shareholders and the Shell, and several additional conditions as stipulated in the agreement.
The closing of the Share Exchange and the private placement were completed on March 31, 2011, see also note 15f.
NOTE 7 - 2008 CONVERTIBLE LOAN
In April 2008 (hereafter - Closing date) the Company signed a convertible loan agreement with certain lenders. Under this agreement the lenders shall provide a convertible loan at an aggregated amount of $720 thousands, bearing annual interest of 10%. The loan does not bear a maturity date.
The principal of the loan together with the accrued interest should be paid on the lender’s demand in any event of default or breach of covenant as stipulated in the convertible loan agreement.
The loan will be automatically converted into ordinary shares of the Company in the event of investment in the Company in an aggregate amount of $1 million (hereafter - qualified financing), at the lower conversion price of:
a) $1.48; or b) at a discount of 30% on the price per share in such qualified financing.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 7 - 2008 CONVERTIBLE LOAN (continued):
The loan will be automatically converted into ordinary shares in the event of an Initial Public Offering (hereafter - IPO) or in the event of consolidation, merger or sale of all assets or shares the Company (hereafter - exit transaction), in the lowest conversion price of: a) $1.48; or b) at a discount of 20% on the price per share in such exit transaction.
The loan and the accumulated interest may be converted to ordinary shares of the Company at any time prior to the event of qualified financing, according to the conversion terms in the event of qualified financing.
In accordance with ASC 470-20 "Debt with Conversion and Other Options", the Company determined that a beneficial conversion feature existed at the Closing date, totaling $308 thousands. Because the Convertible loan do not have a stated redemption date (except on event of default or breach of covenant), and may be converted by the holder at any time, the beneficial conversion feature was recognized immediately at the closing date as a financial expense, in the consolidated statements of operations.
In March 2009 ("the Redemption Date") the convertible loan was fully repaid (principal and accrued interest) to the lenders due to breach of the covenants by the Company. The Company allocated the proceeds paid between the portion related to the redemption of the beneficial conversion feature and that related to the convertible loan, based on the guidance stipulated in ASC 470-20. The Company measured the portion allocated to the beneficial conversion feature based on the intrinsic value of the conversion feature at the extinguishment date, which amounting to $308 thousands (which equals the original beneficial conversion feature since the price of the Company's shares, from Closing date to Redemption date, were the same). Accordingly, the difference between the amount allocated to the beneficial conversion feature plus the loan's carrying amount, and the cash paid, was recognized as financial income in the consolidated statements of operations.
NOTE 8 - LONG-TERM LOAN
In January, 2009 the Company signed a loan agreement with Mizrahi Tefahot Bank (hereafter- the bank).
According to the agreement the Company will be entitled to receive the following:
|
a.
|
A loan (hereafter - the first loan) amounting to $750 thousands, bearing annual interest (quarterly paid) equal to Libor + 4% (as of December 31, 2009 – 0.2531%). The loan is payable in eight quarterly installments during a period of 3 years beginning April 2010.
|
|
b.
|
An additional loan (hereafter - the second loan) amounting to $750 thousands which will be received no later than August 3, 2009 and subject to certain terms. The Company did not meet the specific certain terms and therefore was not able to receive the second loan.
|
|
c.
|
A credit line amounting to $500 thousand for the purpose of financing export shipments. The credit line was not utilized by the Company.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 8 - LONG-TERM LOAN (continued):
In addition, According to the loan agreement, the Company has an obligation to pay additional $250 thousands in the following events:
|
a)
|
Liquidity Event of at least $100 million (as stipulated in the agreement) or
|
|
b)
|
IPO in which the Company's valuation is at least $100 million.
|
The Company granted to the bank a floating lien of all of its assets and a fixed lien of all its intellectual property and rights of future payments from the company’s clients. The Company also committed to maintain in its bank account a minimum of $250 thousands in order to support an estimated cash burn rate of 3 months of activity based on average monthly cash flow in the preceding 3 months. This amount was recorded in the consolidated balance sheet under "restricted cash". In November 2010 the Company was asked by the bank, pursuant to its loan agreement, to grant a fixed lien to the bank in the amount of $300 thousands that would replace the $250 thousands of restricted cash since the actual cash burn rate was higher than the cash amount maintained in the Company’s bank account. The bank effectuated the transaction in January 2011.
On February 2009 the Company received the first loan and according to the loan agreement issued 234,814 ordinary shares to the bank. Subsequently, the Company has estimated the fair value of the first loan, the second loan, the credit line and the 234,814 ordinary shares issued to the bank using the following assumptions:
|
1.
|
Capitalization rate of 25.13% per year calculated by using Altman-Z score model.
|
|
2.
|
Probability of realizing the second loan - 40%
|
|
3.
|
Probability of realizing the credit line - 80%
|
The relative fair value of each component based on the valuation report is as follows:
|
1.
|
The first loan - $540 thousands.
|
|
2.
|
The second loan option - $20 thousands.
|
|
3.
|
The credit line - $59 thousands.
|
|
4.
|
The 234,814 ordinary shares issued to the bank - $290 thousands
|
The first loan was subsequently measured at amortized cost on the basis of the effective interest method over the loan period.
The second loan option and the credit line have been recorded in the consolidated financial statements in "financial expenses" during 2009.
Direct transaction costs of $41 thousands are recorded as deferred debt issuance costs in the consolidated balance sheet and amortized over the first loan period.
The contractual maturities of the first loan are as follows:
|
December 31
|
||||
|
2010
|
||||
|
($ in thousands)
|
||||
|
2011
|
$ | 375 | ||
|
2012
|
94 | |||
| $ | 469 | |||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 - RELATED PARTIES TRANSACTIONS:
|
a.
|
In January 2009 the Company signed a sub-lease agreement with a company controlled by the Company's shareholders, for a period of 12.5 months, for a monthly rent payment of $1 thousands. In 2010 the rent period was extended for an additional year and the rent payments increased by 10%.
|
|
b.
|
In 2008 the Company entered into aconsultancy agreement for marketing services with one of the Company's controlling shareholders of which she entitled for a fixed hourly fee of 154 NIS in Israel and a fixed daily fee of $400 abroad in respect to her services.
|
|
c.
|
During 2007 the Company received a loan of $40 thousands from its controlling shareholders. Half of the loan was paid during 2009.
|
|
d.
|
During the second half of 2008 the Company has decreased the salaries for most of its employees due to the economic slowdown. The Company also decreased the salaries of its two senior employees, the president and the CEO, both are shareholders. Their salaries were decreased in 25% and additional 25% were accrued and recorded in "accounts payable-trade". The accrued amounts were fully paid as of the December 31, 2010.
|
According to the agreement with the president and the CEO, As of September 2009, the above salaries decrease of 25% was cancelled.
|
e.
|
In July 2010 the Company's board of directors approved new employment agreements for the Company's President and the company's CEO with the following terms:
|
|
-
|
monthly gross salary of NIS 55,000.
|
|
-
|
certain social and fringe benefits as set forth in the employment agreement, which total 15% of the gross salary.
|
|
-
|
company car.
|
|
-
|
minimum bonus equivalent to three monthly gross salaries based on achievement of objectives and board of directors approval.
|
|
-
|
stock options pursuant to this agreement following its six month anniversary, subject to board approval.
|
|
-
|
six months prior notice.
|
The agreements were approved by the Company's shareholders meeting in February 2011, and are effective only upon the occurrence of certain events, which as of the date of the financial statements were met.
|
f.
|
Balances with related parties:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Current liabilities:
|
||||||||
|
Trade payable
|
$ | 3 | $ | 156 | ||||
|
Other accounts payable
|
121 | 82 | ||||||
|
Loans from shareholders
|
20 | 20 | ||||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 - RELATED PARTIES TRANSACTIONS (continued):
|
g.
|
Transactions with related parties:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Expenses:
|
||||||||
|
Salaries and related expenses
|
$ | 241 | $ | 152 | ||||
|
Consulting Fee
|
226 | 194 | ||||||
|
Financial expenses
|
- | 1 | ||||||
|
Rent income
|
(15 | ) | (13 | ) | ||||
* Represents an amount less than $1 thousands.
NOTE 10 - COMMITMENTS AND CONTINGENT LIABILITIES:
|
a.
|
Lease commitments:
|
|
|
1)
|
The Company leases its premises for a period beginning February, 2007 and ending February, 2012.
|
Rent expenses included in the statement of operations totaled to approximately $131 thousands and $126 thousands for the years ended December 31, 2010 and 2009, respectively.
As of December 31, 2010, the aggregate future minimum lease obligations of office rent under non-cancelable operating leases agreements were as follows:
|
($ in thousands)
|
||||
|
Year Ended December 31:
|
||||
|
2011
|
$ | 120 | ||
|
2012
|
20 | |||
| $ | 140 | |||
|
|
2)
|
The Company leases the majority of its motor vehicles under non-cancelable operating lease agreements.
|
As of December 31, 2010, the aggregate future minimum lease obligations of car lease under non-cancelable operating leases agreements were as follows:
|
($ in thousands)
|
||||
|
2011
|
$ | 20 | ||
|
2012
|
20 | |||
|
2013
|
18 | |||
| $ | 58 | |||
|
b.
|
On March 2010 the Company entered into a new license agreement to use a unique stent design developed by an American company considered to be a related party ("MGuard Prime"). According to the agreement the licensor is entitled to receive 7% royalties for sales outside the USA and inside the USA as follows: 7% royalties for the first $10,000 of net sales and 10% royalties of net sales exceeding the first $10,000. The Company began manufacturing the MGuard Prime during the last quarter of 2010. As of December 31, 2010 the Company has not yet began selling the MGuard Prime.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 - COMMITMENTS AND CONTINGENT LIABILITIES (continued):
|
c.
|
Litigation:
|
|
1)
|
The Company is a party to various claims arising in the ordinary course of the Company’s operations in the aggregate amount of approximately $20,000. The Company has not recorded an expense related to damages in connection with these matters because management, after considering the views of its legal counsel as well as other factors, is of the opinion a loss to the Company is neither probable nor is an amount or range of loss that is estimable.
|
|
2)
|
In March, 2009, a service provider submitted in the magistrates court in Tel Aviv a claim against the Company in the amount of $150 thousands claiming a success fee for assistance in finding potential investors and lenders in respect for the loan agreement signed with a bank (see also note 8). As of December 31, 2010 the Company has not recorded an expense related to damages in connection with these matters because as of March 31, 2011, the release date of these financial statements, management, after considering the views of its legal counsel as well as other factors, is in the opinion that any potential loss is not currently probable. On April 11, 2011, the Company received a court ruling directing the Company to pay the service provider an amount of $105,000. The Company has recorded a provision of $105,000 in the financial statements in 2011. In June 2011 a settlement was reached between the parties in which the Company will pay $96 thousands and grant 18,785 shares of the Shell.
|
|
3)
|
In July 2009, a Finder submitted in the magistrates court in Tel Aviv a claim against the Company in the amount of $100 thousands claiming a success fee for assistance in finding potential investor. In March 2010 a settlement was reached between the parties in which he Company will pay $60 thousands and grant 30,435 options to purchase ordinary shares of the Company. A provision for the settlement payment has been included in the financial statements in 2008 and 2009.
|
|
4)
|
In November 2010, a former senior employee submitted a claim against the Company in the total amount of $430,000 and options to purchase 2,029,025 shares of the Company at an exercise price of $0.001 per share in the Magistrate’s Court in Tel Aviv, claiming unpaid back wages and commissions. The fair value of those options was estimated using the Black-Scholes valuation model at $2.5 million as of the period he claimed to be entitled to the options. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a provision of $20,000 in the financial statements in 2009 and is of the opinion an additional loss to the Company is neither probable nor is an amount or range of loss that is estimable.
|
|
5)
|
In November 2010, a former alleged founder and legal advisor of the Company submitted a claim against the Company for options to purchase 496,056 shares of the Company at an exercise price of $0.001 per share in the Magistrate’s Court in Tel Aviv. The fair value of those options was estimated using the Black-Scholes valuation model at $134,000 as of the grant date. It was during 2005 and 2006 that the Company first became aware of the events that gave rise to this litigation. Also during this time, the Company had discussions with the plaintiffs on an informal basis. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a share-based compensation expense of $134,000 recorded in the year ended December 31, 2006, in respect of services allegedly provided in 2005 and 2006.
|
|
6)
|
In November 2010, a former legal advisor of the Company submitted in the Magistrate’s Court in Tel Aviv a claim against the Company in the total amount of $53 thousands due to a breach of employment promise. It was during 2005 and 2006 that the Company first became aware of the events that gave rise to this litigation. Also during this time, the Company had discussions with the plaintiffs on an informal basis. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a provision amounting to $53 thousands recorded in the year ended December 31, 2006.
|
|
7)
|
In February 2011, representatives of a third party indicated that they intended to seek damages from the Company in connection with certain finders’ fees that they claimed were owed to them. The claimants’ demand was for approximately $1 million. The claimants’ most recent demand, conveyed in April 2011, was for a total of $250,000 in cash and 250,000 shares of the Company common stock. To date, no lawsuit has been filed and the Company has not accrued an expense in connection with this matter because the Company’s management, after considering the views of its legal counsel as well as other factors is of the opinion a loss to the Company is neither probable nor is an amount or range of loss that is estimable.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11 - SHARE-BASED COMPENSATION:
|
a.
|
In June 2006, the Company’s board of directors approved a stock options plan (the “2006 plan”) for employees and consultants. The Company had reserved 2,434,830 ordinary shares for issuance under the plan. The Company’s Board of Directors selected the capital gains tax track for options granted to the Company’s Israeli employees.
|
In accordance with the track chosen by the company and pursuant to the terms thereof, the company is not allowed to claim, as an expense for tax purposes, the amounts credited to employees as a benefit, including amounts recorded as salary benefits in the company’s accounts, in respect of options granted to employees under the Plan - with the exception of the work-income benefit component, if any, determined on the grant date.
|
b.
|
Each option of the 2006 plan can be exercised to purchase one ordinary share of USD 0.0001 par value of the Shell. Upon exercise of the option and issuance of ordinary shares, the ordinary shares issued will confer the holders the same rights as the other ordinary shares. The exercise price and the vesting period of the options granted under the plans were determined by the Board of Directors at the time of the grant. Any option not exercised within 10 years from the date of grant will expire, unless extended by the Board of Directors.
|
|
c.
|
In 2006, the Company’s board of directors approved an increase of 2,434,830 in the number of ordinary shares reserved for purpose of grants under the Company's share option plans.
|
|
d.
|
In 2007, the Company’s board of directors approved an additional increase of 4,869,660 in the number of ordinary shares reserved for purpose of grants under the Company's share option plans.
|
As of December 31, 2010 the Company's board of directors approved the grant of additional 610,347 options to employees and consultants of the company. The options agreements for those grants were not yet signed and therefore were not granted.
|
e.
|
As of December 31, 2010, the Company had reserved 9,739,320 ordinary shares for issuance under the plans. The following table summarizes information about share options:
|
|
2010
|
2009
|
|||||||||||||||
|
Number of options
|
Weighted
average
exercise price
|
Number of options
|
Weighted
average
exercise Price
|
|||||||||||||
|
Outstanding - beginning of year
|
5,797,338 | $ | 0.36 | 5,829,308 | $ | 0.28 | ||||||||||
|
Granted
|
2,864,983 | 0.84 | 585,017 | 0.96 | ||||||||||||
|
Forfeited
|
(462,618 | ) | 0.65 | (158,264 | ) | 0.85 | ||||||||||
|
Exercised during the period
|
- | - | (458,722 | ) | - | |||||||||||
|
Outstanding - end of year
|
8,199,703 | $ | 0.52 | 5,797,339 | $ | 0.36 | ||||||||||
|
Exercisable at the end of the year
|
6,840,119 | $ | 0.51 | 4,474,073 | $ | 0.16 | ||||||||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 11 - SHARE-BASED COMPENSATION (continued):
The following table provides additional information about all options outstanding and exercisable:
|
Outstanding as of December 31
|
||||||||||||||||||||||||||
|
2010
|
2009
|
|||||||||||||||||||||||||
|
Exercise price
|
Options outstanding
|
Weighted
average
remaining contractual life (years)
|
Options exercisable
|
Options outstanding
|
Weighted
average
remaining contractual life (years)
|
Options exercisable
|
||||||||||||||||||||
| 0-0.01 | 3,943,125 | 6.79 | 3,203,546 | 3,318,186 | 7.10 | 3,206,590 | ||||||||||||||||||||
| 0.1 | 52,755 | 7 | 52,755 | 52,755 | 8.00 | 52,755 | ||||||||||||||||||||
| 1.49 | 205,013 | 5.78 | 205,013 | 205,013 | 6.78 | 205,013 | ||||||||||||||||||||
| 1.53 | 467,000 | 5.4 | 467,000 | 467,000 | 6.40 | 467,000 | ||||||||||||||||||||
| 3.67 | 108,350 | 6 | 108,350 | 108,350 | 7.00 | 108,350 | ||||||||||||||||||||
| 8 | 584,359 | 7.25 | 584,359 | 584,359 | 8.25 | - | ||||||||||||||||||||
| 10 | 2,783,912 | 8.87 | 2,165,733 | 1,006,486 | 7.49 | 388,306 | ||||||||||||||||||||
| 12.5 | 40,581 | 6.83 | 40,581 | 40,581 | 7.83 | 40,581 | ||||||||||||||||||||
| 14 | 14,608 | 8 | 12,782 | 14,609 | 9.00 | 5,478 | ||||||||||||||||||||
| 8,199,703 | 7.42 | 6,840,119 | 5,797,339 | 7.23 | 4,474,073 | |||||||||||||||||||||
The weighted average of the remaining contractual life of total vested and exercisable options for the years ended December 31, 2010 and 2009 is 7.04 and 6.65 years, respectively.
Aggregate intrinsic value of the total outstanding options as of December 31, 2010 and 2009 is $5,854 thousands and $5,084 thousands respectively. The aggregate intrinsic value of the total exercisable options as of December 31, 2010 and 2009 is $4,942 thousands and $4,802 thousands, respectively.
The total intrinsic value of options exercised during the year ended December 31, 2009 was $565 thousand respectively. No options were exercised during the year ended December 31, 2010.
The total cash received from employees as a result of employee stock option exercises for the years ended December 31, 2009 was less than $1 thousands.
The weighted average fair value of options granted was approximately $0.82 and $0.96 for the years ended December 31, 2010 and 2009, respectively. The weighted average fair value of options granted was estimated by using the Black-Scholes option-pricing model.
NOTE 11 - SHARE-BASED COMPENSATION (continued):
|
f.
|
The following table sets forth the assumptions that were used in determining the fair value of options granted to employees for the years ended December 31, 2010 and 2009:
|
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
Expected life
|
5.25-6 years
|
5.54-6 years
|
||||||
|
Risk-free interest rates
|
1.93%-2.69 | % | 1.7%-2.49 | % | ||||
|
Volatility
|
79%-80 | % | 75%-79 | % | ||||
|
Dividend yield
|
0 | % | 0 | % | ||||
The following table sets forth the assumptions that were used in determining the fair value of options granted to non-employees for the years ended December 31, 2010 and 2009:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
Expected life
|
9.7-10 years
|
9-10 years
|
||||||
|
Risk-free interest rates
|
2.65%-3.01 | % | 3.4%-3.59 | % | ||||
|
Volatility
|
87 | % | 86%-91 | % | ||||
|
Dividend yield
|
0 | % | 0 | % | ||||
The expected term for most of the options granted was determined using the simplified method, which takes into consideration the option’s contractual life and the vesting periods (for non-employees the expected term is equal to the option’s contractual life). The Company continued to use the simplified method in 2010 as the Company does not have sufficient historical exercise data to provide a reasonable basis upon which to estimate expected term. The expected term for options granted that do not meet the conditions of the simplified method was determined according to management's best estimates. The Company estimates its forfeiture rate based on its employment termination history, and will continue to evaluate the adequacy of the forfeiture rate based on analysis of employee turnover behavior, and other factors (for non-employees the forfeiture rate is nil). The annual risk free rates are based on the yield rates of zero coupon non-index linked U.S. Federal Reserve treasury bonds as both the exercise price and the share price are in U.S. Dollar terms. The Company’s expected volatility is derived from historical volatilities of companies in comparable stages as well as companies in the industry. Each Company’s historical volatility is weighted based on certain factors and combined to produce a single volatility factor used by the Company.
NOTE 11 - SHARE-BASED COMPENSATION (continued):
|
g.
|
As of December 31, 2010, the total unrecognized compensation cost on employee and non employee stock options, related to unvested stock-based compensation amounted to approximately $659 thousands and $49 thousands, respectively. This cost is expected to be recognized over a weighted-average period of approximately 0.84 and 0.73 years, respectively. This expected cost does not include the impact of any future stock-based compensation awards.
|
The following table summarizes the allocation of total share-based compensation expense in the Consolidated Statements of Operations:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Cost of revenues
|
$ | 160 | $ | 49 | ||||
|
Research and development
|
536 | 356 | ||||||
|
Sales and marketing
|
55 | 92 | ||||||
|
General and administrative
|
869 | 65 | ||||||
| $ | 1,620 | $ | 562 | |||||
NOTE 12 - TAXES ON INCOME:
|
a.
|
Tax benefits under the Law for Encouragement of Capital Investments, 1959 (“Capital Investments Law”)
|
The production facilities of the Company have been granted “approved enterprise” status under Israeli law. The main tax benefits available during the two years period of benefits commencing in the first year in which the Company earns taxable income (which has not yet occurred) are:
|
|
1)
|
Reduced tax rates:
|
|
|
Income derived from the “approved enterprise” is tax exempt for a period of 2 years, not later than 12 years as of December 31, 2007, after which the income will be taxable at the rate of 25% for 5 years.
|
|
|
In the event of distribution of cash dividends from income which was tax exempt as above, the tax rate applicable to the amount distributed will be 25%.
|
|
|
2)
|
Accelerated depreciation:
|
|
|
The Company is entitled to claim accelerated depreciation for five tax years in respect of machinery and equipment used by the approved enterprise.
|
|
|
3)
|
Conditions for entitlement to the benefits:
|
|
|
The entitlement to the above benefits is conditional upon the Company’s fulfilling the conditions stipulated by the law, regulations published there under and the instruments of approval for the specific investments in approved enterprises. In the event of failure to comply with these conditions, the benefits may be cancelled and the Company may be required to refund the amount of the benefits, in whole or in part, with the addition of linkage differences and interest.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12 - TAXES ON INCOME (continued):
Amendment of the Law for the Encouragement of Capital Investments, 1959
The Law for Encouragement of Capital Investments, 1959 (hereafter - the law) was amended as part of the Economic Policy Law for the years 2011-2012, which was passed in the Knesset (the Israeli parliament) on December 29, 2010 (hereafter - the amendment). The amendment becomes effective as from January 1, 2011.
The amendment sets alternative benefit tracks to the ones currently in place under the provisions of the Law, as follows: investment grants track designed for enterprises located in national development zone A and two new tax benefits tracks (preferred enterprise and a special preferred enterprise), which provide for application of a unified tax rate to all preferred income of the company, as defined in the amendment.
The tax rates at company level, under the law:
|
Years
|
Development Zone A
|
Other Areas in Israel
|
|||||||
|
"Preferred enterprise"
|
|||||||||
| 2011-2012 | 10 | % | 15 | % | |||||
| 2013-2014 | 7 | % | 12.5 | % | |||||
|
2015 and thereafter
|
6 | % | 12 | % | |||||
|
"Special Preferred Enterprise"
|
|||||||||
|
commencing 2011
|
5 | % | 8 | % | |||||
The benefits granted to the preferred enterprises will be unlimited in time, unlike the benefits granted to special preferred enterprises, which will be limited for a period of 10 years. The benefits shall be granted to companies that will qualify under criteria set in the amendment; for the most part, those criteria are similar to the criteria that were set in the law prior to its amendment.
Under the transitional provisions of the amendment, a company will be allowed to continue and enjoy the tax benefits available under the law prior to its amendment until the end of the period of benefits, as defined in the law. The company will be allowed to set the "year of election" no later than tax year 2012, provided that the minimum qualifying investment commenced not later than the end of 2010. On each year during the period of benefits, the company will be able to opt for application of the amendment, thereby making available to itself the tax rates as above. Company's opting for application of the amendment is irrecoverable.
In accordance with income taxes (Topic 740) the measurement of current and deferred tax liabilities and assets is based on provisions of the enacted tax law at balance sheet date. Since, as at December 31, 2010, the Amendment had not yet been “enacted”, as defined in Topic 740, the measurement of the current and deferred taxes for the year ended December 31, 2010 is made without taking the aforementioned Amendment into consideration. The Company is currently evaluating the impact of the adoption of these amendments would have on its consolidated financial statements.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12 - TAXES ON INCOME (continued):
|
b.
|
Measurement of results for tax purposes under the Income Tax (Inflationary Adjustments Law), 1985 (“Inflationary Adjustments Law”)
|
Pursuant to the Israel Income Tax Law (Adjustments for Inflation), 1985 (hereinafter - the Adjustments Law), the results for tax purposes have been measured through 2007 on a real basis, based on changes in the Israel Consumer Price Index. The Company is taxed under this law.
Under the Israel Income Tax Law (Adjustments for Inflation) (Amendment No. 20), 2008 (hereinafter - the amendment), the provisions of the Adjustments Law will no longer apply to the Company in the 2008 tax year and thereafter, and therefore, the results of the Company will be measured for tax purposes in nominal terms. The amendment includes a number of transition provisions regarding the end of application of the Adjustments Law, which applied to the company through the end of the 2007 tax year.
|
c.
|
Tax rates
|
The regular corporate tax rate in Israel was 26% and 27%, in 2009 and 2008, respectively. The corporate tax rate is to be reduced to 25% in 2010. Income not eligible for “approved enterprise” benefits, mentioned above, is taxed at a regular rate.
On July 23, 2009, the Israel Economic Efficiency Law (Legislation Amendments for Applying the Economic Plan for the 2009 and 2010), 2009 (hereinafter – the 2009 amendment), became effective, stipulating, among other things, an additional gradual decrease in tax rate in 2011 and thereafter, as follows: 2011 – 24%, 2012 – 23%, 2013 – 22%, 2014 – 21%, 2015 – 20%, and 2016 and thereafter – 18%.
The subsidiary is taxed according to the tax laws in Germany. Accordingly, the applicable tax rates are corporate tax rate of 15.825% and trade tax rate of 15%.
|
d.
|
Carry forward tax losses
|
As of December 31, 2010, the Company had a net carry forward tax loss of approximately $14.2 million. Under Israeli tax laws, the carry forward tax losses of the Company can be utilized indefinitely. The subsidiary had a net carry forward tax loss of approximately $560 thousands. Under German tax laws, the carry forward tax losses of the subsidiary can be utilized indefinitely.
|
e.
|
Tax assessments
|
The Company and its subsidiary have not been assessed for tax purposes since incorporation.
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12 - TAXES ON INCOME (continued):
|
f.
|
The components of income (loss) before income taxes are as follows:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Loss before taxes on income:
|
||||||||
|
The Company in Israel
|
$ | (3,115 | ) | $ | (2,624 | ) | ||
|
Subsidiary in Germany
|
(258 | ) | (53 | ) | ||||
| $ | (3,373 | ) | $ | (2,677 | ) | |||
|
Current Taxes on income:
|
||||||||
|
In Israel
|
$ | 17 | $ | 17 | ||||
|
Outside Israel
|
30 | 30 | ||||||
| $ | 47 | $ | 47 | |||||
Following is a reconciliation of the theoretical tax expense, assuming all income is taxed at the Regular tax rates applicable to the company in Israel (see c. above), and the actual tax expense:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Loss before taxes on income, as reported in the statements of operations
|
$ | 3,373 | $ | 2,677 | ||||
|
Theoretical tax benefit
|
(843 | ) | (696 | ) | ||||
|
Increase in tax benefit resulting from permanent differences
|
431 | 92 | ||||||
|
Increase in taxes on income resulting from the computation of deferred taxes at a rate which is different from the theoretical rate
|
62 | 24 | ||||||
|
Increase in uncertain tax positions - net
|
30 | 30 | ||||||
|
Change in corporate tax rates, see c above
|
- | 481 | ||||||
|
Change in valuation allowance
|
367 | 116 | ||||||
| $ | 47 | $ | 47 | |||||
As of December 31, 2010 and 2009, the Company determines that it was more likely than not that the benefit of the operating losses would not be realized and consequently, management concluded that full valuation allowance should be established regarding the Company's deferred tax assets.
The changes in the valuation allowance for the year ended December 31, 2010:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Balance at the beginning of the year
|
$ | 2,829 | $ | 2,713 | ||||
|
Changes during the year
|
367 | 116 | ||||||
|
Balance at the end of the year
|
$ | 3,196 | $ | 2,829 | ||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12 - TAXES ON INCOME (continued):
|
g.
|
Accounting for Uncertain Tax position
|
Following is a reconciliation of the total amounts of the Company's unrecognized tax benefits during the year ended December 31, 2010:
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Balance at beginning of year
|
$ | 30 | $ | - | ||||
|
Increases in unrecognized tax benefits as a result
|
||||||||
|
of tax positions taken during the current year
|
30 | 30 | ||||||
|
Balance at end of year
|
$ | 60 | $ | 30 | ||||
All of the above amounts of unrecognized tax benefits would affect the effective tax rate if recognized.
A summary of open tax years by major jurisdiction is presented below:
|
Jurisdiction
|
Years
|
|
|
Israel
|
2006-2010
|
|
|
Germany
|
2008-2010
|
|
h.
|
Deferred income tax:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Short-term :
|
||||||||
|
Allowance for doubtful accounts
|
$ | 36 | $ | 2 | ||||
|
Provision for vacation and recreation pay
|
38 | 25 | ||||||
| 74 | 27 | |||||||
|
Long-term :
|
||||||||
|
R&D expenses
|
531 | 469 | ||||||
|
Carry forward tax losses
|
2,582 | 2,326 | ||||||
|
Accrued severance pay
|
9 | 7 | ||||||
| 3,122 | 2,802 | |||||||
|
Less-valuation allowance
|
(3,196 | ) | (2,829 | ) | ||||
| $ | - | $ | - | |||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 13 - SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION:
Balance sheets:
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
a. Accounts receivable:
|
||||||||
|
1) Trade:
|
||||||||
|
Open accounts
|
$ | 998 | $ | 1,195 | ||||
|
Allowance for doubtful accounts
|
(146 | ) | (6 | ) | ||||
| $ | 852 | $ | 1,189 | |||||
|
2) Other:
|
||||||||
|
Due to government institutions
|
$ | 56 | $ | 76 | ||||
|
Receivables on account of shares
|
*20 | |||||||
|
Fund in respect of employee right upon retirement
|
8 | 34 | ||||||
|
Other
|
11 | |||||||
| $ | 75 | $ | 130 | |||||
* The amount was subsequently paid in January 2010.
|
b.
|
Inventory on consignment
|
The changes in inventory on consignment during the years ended December 31, 2010 and 2009 are as follows:
As of December 31, 2010 and 2009 Inventory on consignment included an amount of $280 thousands and $1,002 thousands, respectively related to products sales for which product returns could not be reliably estimated with the remainder relating to products sales for which returns were reliably estimated.
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Balance at beginning of year
|
$ | 1,093 | $ | 1,423 | ||||
|
Costs of revenues deferred during the year
|
326 | 421 | ||||||
|
Costs of revenues recognized during the year
|
(1,048 | ) | (751 | ) | ||||
|
Balance at end of year
|
$ | 371 | $ | 1,093 | ||||
|
c.
|
Inventories:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Finished goods
|
$ | 957 | $ | 520 | ||||
|
Work in process
|
573 | 331 | ||||||
|
Raw materials and supplies
|
174 | 95 | ||||||
| $ | 1,704 | $ | 946 | |||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 13 - SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (continued):
|
d.
|
Accounts payable and accruals - others:
|
|
December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Employees and employee institutions
|
$ | 375 | $ | 395 | ||||
|
Accrued vacation and recreation pay
|
147 | 95 | ||||||
|
Accrued expenses
|
632 | 502 | ||||||
|
Due to government institutions
|
100 | 37 | ||||||
|
Liability for employees rights upon retirement
|
7 | 30 | ||||||
|
Provision for returns
|
150 | 144 | ||||||
|
Taxes payable
|
98 | 101 | ||||||
| $ | 1,509 | $ | 1,304 | |||||
|
e.
|
Deferred revenues
|
The changes in deferred revenues during the years ended December 31, 2010 and 2009 are as follows:
|
Year ended December 31
|
|||||||||
|
2010
|
2009
|
||||||||
|
($ in thousands)
|
|||||||||
|
Balance at beginning of year
|
$ | 1,975 | $ | 2,482 | |||||
|
Revenue deferred during the year
|
320 | 616 | |||||||
|
Revenue recognized during the year
|
(1,897 | ) | (1,123 | ) | |||||
|
Balance at end of year
|
$ | 398 | $ | 1,975 | |||||
Statements of Operation:
|
f.
|
Financial expenses (income), net:
|
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Bank commissions
|
$ | 83 | $ | 18 | ||||
|
Interest income
|
(1 | ) | (1 | ) | ||||
|
Exchange rate differences
|
(33 | ) | 30 | |||||
|
Interest expense
|
105 | 221 | ||||||
|
Gain on extinguishment of convertible loan
|
- | (308 | ) | |||||
| $ | 154 | $ | (40 | ) | ||||
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 14 - ENTITY WIDE DISCLOSURES
The Company operates in one operating segment.
Disaggregated financial data is provided below as follows:
(1) Revenues by geographic area and
(2) Revenues from principal customers.
Revenues are attributed to geographic areas based on the location of the customers. The following is a summary of revenues by geographic areas:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Israel
|
$ | 119 | $ | - | ||||
|
Pakistan
|
193 | 477 | ||||||
|
Poland
|
1,446 | |||||||
|
Italy
|
390 | 668 | ||||||
|
Other
|
2,801 | 2,266 | ||||||
| $ | 4,949 | $ | 3,411 | |||||
By principal customers:
|
Year ended December 31
|
||||||||
|
2010
|
2009
|
|||||||
|
($ in thousands)
|
||||||||
|
Customer A
|
8 | % | 19 | % | ||||
|
Customer B
|
4 | % | 14 | % | ||||
|
Customer C
|
- | 10 | % | |||||
|
Customer D
|
29 | % | - | |||||
|
|
All tangible long lived assets are located in Israel.
|
NOTE 15 - SUBSEQUENT EVENTS:
|
a.
|
During the first quarter of 2011 and prior to the Share Exchange, the Company raised approximately $990,000 and issued approximately 803 thousands ordinary shares through private placements.
|
|
b.
|
On April 18, 2011, the Company issued 666,667 shares of its common stock and five-year warrants to purchase 333,333 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $1,000,000 in a private placement.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 15 - SUBSEQUENT EVENTS (continued):
|
c.
|
On April 18, 2011, the Company issued 283,334 shares of its common stock and five-year term warrants to purchase 141,667 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $425,000 in a private placement.
|
|
d.
|
In connection with the above-referenced transactions, the Company paid placement agent fees of approximately $471,000 and five-year term warrants to purchase 57,000 shares of the Company common stock at an exercise price of $1.80 per share.
|
|
e.
|
On April 21, 2011, the Company issued 33,333 shares of its common stock, and five-year term warrants to purchase 16,667 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $50,000 in a private placement.
|
|
f.
|
Subsequent to December 31, 2010 Company’s board of directors approved the issuance of approximately 156 thousands common stocks and five-year term warrants to purchase approximately 60 thousands shares of the Shell's common stock at an exercise price of $1.80 per share.
|
|
g.
|
Subsequent to December 31, 2010 the Company granted approximately 2.8 million of stock options to employees and consultants at a cash exercise price from $1.23 to $2.75 per share. The options had terms of four to ten years.
|
|
h.
|
During January 2011, the Company entered into a convertible loan agreement with its distributer in Israel (hereafter - the lender), in the amount of $100 thousands with the following conditions:
|
|
a.
|
The convertible loan does not bear annual interest.
|
|
b.
|
In the event of transaction (as stipulated in the agreement), the lender shall have at its sole discretion the option to convert the loan according to the following terms:
|
|
i.
|
Shell's shares at $1.23 per share; or
|
|
ii.
|
Company's product at 400 euro per unit (which represents the market price for this distributer).
|
|
c.
|
In case the company does not close a transaction by June 1, 2011 than the lender shall have the right to extend the loan and its terms for up to additional 6 months.
|
|
d.
|
In no event the loan shall be repaid by the company.
|
Subsequent to the consummation of the Share Exchange on June 1, 2011, the Lender converted the loan in the amount of $100 thousands into 81,161 shares of the Shell’s common stock (included in the 156 thousands common stock mentioned in 15(f) above).
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 15 - SUBSEQUENT EVENTS (continued):
|
i.
|
In February, 2011 a Finder submitted in the magistrates in Tel Aviv a claim against the Company in the amount of $327 thousands claiming future success fee and a commission for assistance in finding the Company's distributer in Brazil. At December 31,2010 the company, based on advice from its legal counsel, due to the early stage, was not able to assess the lawsuit outcome. As of March 31, 2011 the Company still was not able to assess the outcome of this lawsuit. No provision for this matter has been included in the accounts, as of December 31, 2010. As of May 15, 2011 due to the recent developments at that claim the Company, based upon the opinion of its legal counsel, has recorded a provision of $327 thousands in the financial statements in 2011. The related expense has been recorded to "General and administrative" within the Condensed Consolidated Statements of Operatio
|
|
j.
|
During March 2011 the company granted a new fixed lien of $40 thousands to bank Mizrahi.
|
|
k.
|
On March 31, 2011, the Company completed the reverse merger transaction by and among the Company and the Shell. Subsequent to the date of execution of the transaction, shareholders of the Company, holding 100% of its issued and outstanding ordinary shares, executed a joinder to the Exchange Agreement and became parties thereto (the “InspireMD Shareholders”). Pursuant to the Exchange Agreement, on March 31, 2011, the InspireMD Shareholders transferred all of their ordinary shares in InspireMD to the Shell in exchange for 50,666,667 newly issued shares of common stock of the Shell, resulting in InspireMD becoming a wholly owned subsidiary of the Shell.
|
Pursuant to the terms and conditions of the Exchange Agreement:
|
|
1)
|
The InspireMD Shareholders transferred 6,242,754 ordinary shares of InspireMD (which represented 100% of InspireMD’s issued and outstanding capital stock immediately prior to the closing of the Share Exchange) to the Shell in exchange for 50,666,667 shares of the Shell’s common stock (the “Share Exchange”).
|
|
|
2)
|
The Shell assumed all of InspireMD’s obligations under InspireMD’s outstanding stock options. Immediately prior to the Share Exchange, InspireMD had outstanding stock options to purchase an aggregate of 937,256 shares of its ordinary shares, which outstanding options became options to purchase an aggregate of 7,606,770 shares of common stock of the Shell after giving effect to the Share Exchange. Neither the Shell nor InspireMD had any other options to purchase shares of capital stock outstanding immediately prior to the closing of the Share Exchange.
|
|
|
3)
|
Three-year warrants to purchase up to 125,000 ordinary shares of InspireMD at an exercise price of $10 per share were assumed by the Shell and converted into warrants to purchase 1,014,510 shares of the Shell’s common stock at an exercise price of $1.23 per share.
|
|
|
4)
|
The Shell assumed 8% convertible debentures in an aggregate principal amount of $1,580,000 from InspireMD as follows: $580 thousands plus accrued interest of $88 thousands were converted upon closing and the remainder in the amount of $1,000 will be paid in May 15, 2011.
|
INSPIREMD LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 15 - SUBSEQUENT EVENTS (continued):
In connection with the closing of the Share Exchange, the Shell sold 6,454,000 shares of its common stock at a purchase price of $1.50 per share and five-year warrants to purchase up to 3,227,000 shares of common stock at an exercise price of $1.80 per share in a private placement to accredited investors, resulting in aggregate gross proceeds of approximately $9,680 thousands (the “Private Placement”). As a result of the consummation of the Private Placement, $580 thousands of the principal of the Convertible loan plus $88 thousands accrued interest, converted into approximately 445,060 shares (included in the 6,454,000 shares mentioned above) of common stock at a conversion price of $1.50 per share and 222,530 warrants (included in the 3,227,000 warrants mentioned above).
The transaction is being accounted for as a reverse recapitalization, equivalent to the issuance of stock by the Company, for the net monetary assets of the Shell. Accordingly, while the exchange ratio was only affected on March 31, 2011, these consolidated financial statements have been retrospectively adjusted to give effect to the reverse recapitalization and giving effect to the 8.1161 share exchange ratio. The shares, per share, share options and warrants information included herein have been revised for this exchange ratio.
Palladium Capital Advisors, LLC served as the Company’s placement agent in the Private Placement and received a fee of aproximately $300 thousands and issued Palladium Capital Advisors a five-year warrant to purchase 387,240 shares of our common stock (equal to 6% of the common stock on which the cash fee is payable), at an exercise price of $1.80 per share, with terms identical to the warrants issued to investors in the Private Placement.
In connection with the Share Exchange, the shell issued to certain consultants in consideration for consulting services five-year warrants to purchase up to an aggregate of 2,500,000 shares of common stock at an exercise price of $1.50 per share. The terms of these warrants are identical to the $1.80 Warrants described above, except that the exercise price for the $1.50 Consultant Warrants is $1.50 per share.
On February 20, 2011 the Company have received a tax pre-ruling from the Israeli tax authorities according to section 103 of the israeli tax law, with regards to the share exchange of the Company's shares and options. According to the tax pre-ruling, the shares and options exchange will not resolve immediate tax event for the Company's shareholders, but a deferred tax event, subject to certain condition as stipulated in the tax pre-ruling. The main condition of the tax pre-ruling is restriction of the exchanged shares for two years from December 31, 2010.
_______________
_________________________
_______________
PART I - FINANCIAL INFORMATION
|
Item 1.
|
Financial Statements
|
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
CONSOLIDATED BALANCE SHEETS
(Unaudited)
(U.S. dollars in thousands)
|
September 30,
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
ASSETS
|
||||||||
|
CURRENT ASSETS:
|
||||||||
|
Cash and cash equivalents
|
$ | 7,485 | $ | 636 | ||||
|
Restricted cash
|
40 | 250 | ||||||
|
Accounts receivable:
|
||||||||
|
Trade
|
1,778 | 852 | ||||||
|
Other
|
117 | 75 | ||||||
|
Prepaid expenses
|
103 | 3 | ||||||
|
Inventory:
|
||||||||
|
On hand
|
1,905 | 1,704 | ||||||
|
On consignment
|
102 | 371 | ||||||
|
T o t a l current assets
|
11,530 | 3,891 | ||||||
|
PROPERTY, PLANT AND EQUIPMENT, net of accumulated depreciation and amortization
|
346 | 282 | ||||||
|
OTHER NON-CURRENT ASSETS:
|
||||||||
|
Deferred debt issuance costs
|
5 | 15 | ||||||
|
Funds in respect of employees rights upon retirement
|
189 | 167 | ||||||
|
T o t a l other non-current assets
|
194 | 182 | ||||||
|
T o t a l assets
|
$ | 12,070 | $ | 4,355 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
CONSOLIDATED BALANCE SHEETS
(Unaudited)
(U.S. dollars in thousands)
|
September 30,
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
LIABILITIES AND EQUITY (CAPITAL DEFICIENCY)
|
||||||||
|
CURRENT LIABILITIES:
|
||||||||
|
Current maturities of long-term loans
|
$ | 183 | $ | 355 | ||||
|
Accounts payable and accruals :
|
||||||||
|
Trade
|
562 | 1,103 | ||||||
|
Other
|
2,337 | 1,509 | ||||||
|
Advanced payment from customers
|
516 | 559 | ||||||
|
Loans from shareholders
|
20 | |||||||
|
Deferred revenues
|
398 | |||||||
|
T o t a l current liabilities
|
3,598 | 3,944 | ||||||
|
LONG-TERM LIABILITIES:
|
||||||||
|
Long term loan
|
75 | |||||||
|
Liability for employees rights upon retirement
|
257 | 206 | ||||||
|
Convertible loan
|
1,044 | |||||||
|
T o t a l long-term liabilities
|
257 | 1,325 | ||||||
|
COMMITMENTS AND CONTINGENT LIABILITIES (note 10)
|
||||||||
|
T o t a l liabilities
|
3,855 | 5,269 | ||||||
|
EQUITY (CAPITAL DEFICIENCY):
|
||||||||
|
Common stock, par value $0.0001 per share; 125,000,000 shares authorized; 65,278,946 shares issued and outstanding at September 30, 2011 and 49,863,801 shares issued and outstanding at December 31, 2010
|
6 | 5 | ||||||
|
Additional paid-in capital
|
36,617 | 21,057 | ||||||
|
Accumulated deficit
|
(28,408 | ) | (21,976 | ) | ||||
|
T o t a l equity (capital deficiency)
|
8,215 | (914 | ) | |||||
|
T o t a l liabilities and equity (capital deficiency)
|
$ | 12,070 | $ | 4,355 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
(U.S. dollars in thousands, except per share data)
|
3 months ended
|
9 months ended
|
Year ended
|
||||||||||||||||||
|
September 30
|
September 30
|
December 31
|
||||||||||||||||||
|
2011
|
2010
|
2011
|
2010
|
2010
|
||||||||||||||||
|
REVENUES
|
$ | 1,986 | $ | 1,223 | $ | 4,712 | $ | 4,228 | $ | 4,949 | ||||||||||
|
COST OF REVENUES
|
801 | 561 | 2,340 | 2,377 | 2,696 | |||||||||||||||
|
GROSS PROFIT
|
1,185 | 662 | 2,372 | 1,851 | 2,253 | |||||||||||||||
|
OPERATING EXPENSES:
|
||||||||||||||||||||
|
Research and development
|
547 | 196 | 1,640 | 969 | 1,338 | |||||||||||||||
|
Selling and marketing
|
302 | 279 | 1,347 | 916 | 1,236 | |||||||||||||||
|
General and administrative
|
2,486 | 904 | 4,877 | 2,016 | 2,898 | |||||||||||||||
|
Total operating expenses
|
3,335 | 1,379 | 7,864 | 3,901 | 5,472 | |||||||||||||||
|
LOSS FROM OPERATIONS
|
(2,150 | ) | (717 | ) | (5,492 | ) | (2,050 | ) | (3,219 | ) | ||||||||||
|
FINANCIAL EXPENSES, net
|
108 | 121 | 895 | 150 | 154 | |||||||||||||||
|
LOSS BEFORE TAX EXPENSES
|
(2,258 | ) | (838 | ) | (6,387 | ) | (2,200 | ) | (3,373 | ) | ||||||||||
|
TAX EXPENSES
|
25 | 9 | 45 | 39 | 47 | |||||||||||||||
|
NET LOSS
|
$ | (2,283 | ) | $ | (847 | ) | $ | (6,432 | ) | $ | (2,239 | ) | $ | (3,420 | ) | |||||
|
NET LOSS PER SHARE - basic and diluted
|
$ | (0.04 | ) | $ | (0.02 | ) | $ | (0.11 | ) | $ | (0.05 | ) | $ | (0.07 | ) | |||||
|
WEIGHTED AVERAGE NUMBER OF ORDINARY SHARES USED IN COMPUTING NET LOSS PER SHARE - basic and diluted
|
64,300,685 | 49,490,460 | 59,667,655 | 49,072,828 | 49,234,528 | |||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY (CAPITAL DEFICIENCY)
(Unaudited)
(U.S. dollars in thousands)
|
Ordinary shares
|
||||||||||||||||||||
|
Number of shares
|
Par value
|
Additional paid-in capital
|
Accumulated deficit
|
Total equity (capital deficiency)
|
||||||||||||||||
|
BALANCE AT JANUARY 1, 2011
|
$ | 49,863,801 | $ | 5 | $ | 21,057 | $ | (21,976 | ) | $ | (914 | ) | ||||||||
|
CHANGES DURING 9 MONTHS OF 2011:
|
||||||||||||||||||||
|
Net loss
|
(6,432 | ) | (6,432 | ) | ||||||||||||||||
|
Employee and non-employee share-based compensation
|
4,834 | 4,834 | ||||||||||||||||||
|
Issuance of ordinary shares, net of $185 issuance costs
|
896,651 | * | 805 | 805 | ||||||||||||||||
|
Issuance of ordinary shares and warrants, net of $2,835 issuance costs.
|
12,992,269 | 1 | 7,653 | 7,654 | ||||||||||||||||
|
Exercise of options
|
1,000,000 | * | 1,500 | 1,500 | ||||||||||||||||
|
Conversion of convertible loans
|
526,225 | * | 768 | 768 | ||||||||||||||||
|
BALANCE AT SEPTEMBER 30, 2011
|
$ | 65,278,946 | $ | 6 | $ | 36,617 | $ | (28,408 | ) | $ | 8,215 | |||||||||
|
BALANCE AT JANUARY 1, 2010
|
$ | 48,338,380 | $ | 5 | $ | 17,212 | $ | (18,556 | ) | $ | (1,339 | ) | ||||||||
|
CHANGES DURING 9 MONTHS OF 2010:
|
||||||||||||||||||||
|
Net loss
|
(2,239 | ) | (2,239 | ) | ||||||||||||||||
|
Employee and non-employee share-based compensation
|
1,796 | 1,796 | ||||||||||||||||||
|
Issuance of ordinary shares, net of $73 issuance costs
|
1,152,080 | * | 1,345 | 1,345 | ||||||||||||||||
|
BALANCE AT SEPTEMBER 30, 2010
|
$ | 49,490,460 | $ | 5 | $ | 20,353 | $ | (20,795 | ) | $ | (437 | ) | ||||||||
|
BALANCE AT JANUARY 1, 2010
|
$ | 48,338,380 | $ | 5 | $ | 17,212 | $ | (18,556 | ) | $ | (1,339 | ) | ||||||||
|
CHANGES DURING 2010:
|
||||||||||||||||||||
|
Net loss
|
(3,420 | ) | (3,420 | ) | ||||||||||||||||
|
Employee and non-employee share-based compensation
|
1,640 | 1,640 | ||||||||||||||||||
|
Issuance of warrants, net of $23 issuance costs
|
424 | 424 | ||||||||||||||||||
|
Issuance of ordinary shares, net of $97 issuance costs
|
1,525,421 | * | 1,781 | 1,781 | ||||||||||||||||
|
BALANCE AT DECEMBER 31, 2010
|
$ | 49,863,801 | $ | 5 | $ | 21,057 | $ | (21,976 | ) | $ | (914 | ) | ||||||||
* Represents an amount less than $1,000
The accompanying notes are an integral part of the consolidated financial statements.
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
(U.S. dollars in thousands)
|
9 months ended
|
Year ended
|
|||||||||||
|
September 30
|
December 31,
|
|||||||||||
|
2011
|
2010
|
2010
|
||||||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
||||||||||||
|
Net loss
|
$ | (6,432 | ) | $ | (2,239 | ) | $ | (3,420 | ) | |||
|
Adjustments required to reconcile net loss to net
|
||||||||||||
|
cash used in operating activities:
|
||||||||||||
|
Depreciation and amortization of property, plant and equipment
|
52 | 85 | 91 | |||||||||
|
Loss from sale of property, plant and equipment
|
15 | |||||||||||
|
Change in liability for employees right upon retirement
|
45 | 3 | 42 | |||||||||
|
Financial expenses
|
852 | 96 | 94 | |||||||||
|
Share-based compensation expenses
|
2,817 | 1,352 | 1,620 | |||||||||
|
Loss (Gains) on amounts funded in respect of
|
||||||||||||
|
employee rights upon retirement, net
|
7 | 38 | (11 | ) | ||||||||
|
Changes in operating asset and liability items:
|
||||||||||||
|
Decrease (increase) in prepaid expenses
|
(100 | ) | 28 | 36 | ||||||||
|
Decrease (increase) in trade receivables
|
(926 | ) | 508 | 337 | ||||||||
|
Decrease (increase) in other receivables
|
(50 | ) | (35 | ) | 9 | |||||||
|
Decrease in inventory on consignment
|
269 | 829 | 722 | |||||||||
|
Increase in inventory on hand
|
(201 | ) | (518 | ) | (758 | ) | ||||||
|
Increase (decrease) in trade payables
|
(541 | ) | (231 | ) | 196 | |||||||
|
Decrease in deferred revenues
|
(398 | ) | (1,783 | ) | (1,577 | ) | ||||||
|
Increase (decrease) in other payable
|
||||||||||||
|
and advance payment from customers
|
740 | (287 | ) | (91 | ) | |||||||
|
Net cash used in operating activities
|
(3,851 | ) | (2,154 | ) | (2,710 | ) | ||||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||||||
|
Decrease in restricted cash
|
210 | 52 | 52 | |||||||||
|
Purchase of property, plant and equipment
|
(98 | ) | (64 | ) | (81 | ) | ||||||
|
Proceeds from sale of property, plant and equipment
|
29 | |||||||||||
|
Amounts funded in respect of employee rights upon retirement
|
(21 | ) | (41 | ) | (17 | ) | ||||||
|
Net cash provided by (used in) investing activities
|
120 | (53 | ) | (46 | ) | |||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||||||
|
Proceeds from issuance of shares and warrants, net of issuance costs of $1,014 and $25 in the nine months ended September 30, 2011 and 2010, respectively and $78 in the year ended December 31, 2010
|
10,564 | 1,789 | 2,245 | |||||||||
|
Exercise of options
|
1,500 | |||||||||||
|
Repayment of convertible loan
|
(1,000 | ) | ||||||||||
|
Repayment of long term loan
|
(281 | ) | (188 | ) | (281 | ) | ||||||
|
Proceeds from convertible loan at fair value through profit or loss, net of $60 issuance costs
|
1,073 | 1,073 | ||||||||||
|
Repayment of loans from shareholders
|
(20 | ) | ||||||||||
|
Net cash provided by financing activities
|
10,763 | 2,674 | 3,037 | |||||||||
|
EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS
|
(183 | ) | 13 | (21 | ) | |||||||
|
INCREASE IN CASH AND CASH EQUIVALENTS
|
6,849 | 480 | 260 | |||||||||
|
BALANCE OF CASH AND CASH EQUIVALENTS
|
||||||||||||
|
AT BEGINNING OF THE PERIOD
|
636 | 376 | 376 | |||||||||
|
BALANCE OF CASH AND CASH EQUIVALENTS
|
||||||||||||
|
AT END OF THE PERIOD
|
$ | 7,485 | $ | 856 | $ | 636 | ||||||
|
(*)
|
During the 9 month period ended September 30, 2011:
|
|
a.
|
a convertible loan in the amount of $668,000 was converted into shares of the Company's common stock.
|
|
b.
|
93,785 shares were issued in relation to services provided.
|
The accompanying notes are an integral part of the consolidated financial statements.
F-40
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 1 - DESCRIPTION OF BUSINESS
InspireMD, Inc., formerly Saguaro Resources, Inc. (the “Company”), a public company, is a Delaware corporation formed on February 29, 2008. On March 28, 2011, the Company changed its name to InspireMD, Inc.
On December 29, 2010, the Company entered into a Share Exchange Agreement (the “Exchange Agreement”) by and among the Company and InspireMD Ltd., a limited company incorporated under the laws of the State of Israel in April 2005. Subsequent to the date of execution of the Exchange Agreement, shareholders of InspireMD Ltd., holding 91.7% of InspireMD Ltd.’s issued and outstanding ordinary shares, executed a joinder to the Exchange Agreement and became parties thereto (the “InspireMD Shareholders”). Pursuant to the Exchange Agreement, on March 31, 2011, the InspireMD Ltd. Shareholders transferred all of their ordinary shares in InspireMD Ltd. to the Company in exchange for 46,471,907 newly issued shares of common stock of the Company (the “Initial Share Exchange”). In addition, the remaining holders of InspireMD Ltd.’s ordinary shares separately transferred all of their ordinary shares of InspireMD Ltd. to the Company, in exchange for an aggregate of 4,194,756 newly issued shares of common stock of the Company (the “Follow Up Share Exchange” and, together with the Initial Share Exchange, the “Share Exchange”). As a result of the Share Exchange, InspireMD Ltd. became a wholly owned subsidiary of the Company.
The Share Exchange is being accounted for as a reverse recapitalization, equivalent to the issuance of stock by InspireMD Ltd., for the net monetary assets of the Company. Accordingly, the historical financial statements of the Company reflect the historical operations and financial statements of InspireMD Ltd.
The Company, together with its subsidiaries, is a medical device company focusing on the development and commercialization of its proprietary stent platform technology, MGuard™. MGuard™ provides embolic protection in stenting procedures by placing a micron mesh sleeve over a stent. The Company’s initial products are marketed for use in patients with acute coronary syndromes, notably acute myocardial infarction (heart attack) and saphenous vein graft coronary interventions (bypass surgery). The Company markets its products through distributers in international markets, mainly in Europe and Latin America.
In addition, the Company operates in Germany through its wholly-owned subsidiary InspireMD GmbH, a German limited liability company incorporated in November 2007, where the Company subcontracts the manufacturing of its stents.
The Company believes that it has sufficient cash to continue its operations into 2013. However, depending on the operating results in 2011 and 2012, the Company may need to obtain additional cash in 2013 to continue to fund operations.
NOTE 2 - BASIS OF PRESENTATION
The accompanying unaudited condensed consolidated financial statements have been prepared on the same basis as the annual consolidated financial statements. In the opinion of management, the financial statements reflect all adjustments, which include only normal recurring adjustments, necessary to present fairly the financial position and results of operations of the Company. These consolidated financial statements and notes thereto are unaudited and should be read in conjunction with the InspireMD Ltd.’s audited financial statements for the year ended December 31, 2010. The balance sheet for December 31, 2010 was derived from InspireMD Ltd.'s audited financial statements for the year ended December 31, 2010. The results of operations for the nine months ended September 30, 2011 are not necessarily indicative of results that could be expected for the entire fiscal year.
F-41
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 3 - RECENTLY ADOPTED AND ISSUED ACCOUNTING PRONOUNCMENTS
|
|
In October 2009, the FASB issued amendments to the accounting and disclosure for revenue recognition. These amendments, effective for fiscal years beginning on or after June 15, 2010 (early adoption is permitted), modify the criteria for recognizing revenue in multiple element arrangements and require companies to develop a best estimate of the selling price to separate deliverables and allocate arrangement consideration using the relative selling price method. Additionally, the amendments eliminate the residual method for allocating arrangement considerations. The adoption of the new guidance did not have a material impact on the Company's consolidated financial statements.
|
|
|
In May 2011, the FASB issued amended guidance and disclosure requirements for fair value measurements. These changes will be effective January 1, 2012 on a prospective basis. Early application is not permitted. These amendments are not expected to have a material impact to the consolidated financial results.
|
NOTE 4 – DEFERRED REVENUE
As of September 30, 2011, there is no deferred revenue in the balance sheet since, as of this date, the rate of returns can be reliably estimated and is recorded based on historical experience in “Accounts Payable and Accruals – Other.”
NOTE 5 - FACTORING OF RECEIVABLES
|
|
During the nine month period ended September 30, 2011, the Company entered into a factoring agreement amounting to $1.2 million with a certain banking institution on a non-recourse basis. The factoring of trade receivables under this agreement was accounted for as a sale. Under the terms of this factoring agreement, the Company transferred ownership of eligible trade receivables without recourse to the banking institution in exchange for cash. Proceeds on the transfer reflected the face value of the account less a discount. The discount, amounting to $12,000 during the nine month period ended September 30, 2011 was recorded to “financial expenses - net” within the Condensed Consolidated Statements of Operations.
|
|
|
The receivables sold pursuant to this factoring agreement are excluded from trade receivables on the Condensed Consolidated Balance Sheets and are reflected as cash provided by operating activities on the Condensed Consolidated Statements of Cash Flows. The banking institution had no recourse to the Company’s assets for failure of debtors to pay when due.
|
|
|
The related commissions on the sales of trade receivables sold under these factoring agreements amounting to $22,000 were recorded to “financial expenses - net” within the Condensed Consolidated Statements of Operations.
|
F-42
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 6 - CERTAIN TRANSACTIONS
|
|
During the first quarter of 2011 and prior to the Share Exchange, InspireMD Ltd. raised approximately $990,000 and issued approximately 803,000 ordinary shares through private placements.
|
|
|
During the first quarter of 2011 and prior to the Share Exchange, InspireMD Ltd. granted 600,294 stock options to employees and consultants at a cash exercise price of $1.23 per share. The options had terms of four to ten years.
|
|
|
On January 4, 2011, InspireMD Ltd. entered into a convertible loan agreement with its distributer in Israel (the “Lender”), in the amount of $100,000 subject to the following conditions:
|
|
|
·
|
the convertible loan did not bear annual interest;
|
|
|
·
|
in the event of a share exchange or similar transaction, the Lender would have, at its sole discretion, the option to convert the loan into either (i) shares of the Company’s common stock at a price of $1.23 per share ($10 as relates to InspireMD Ltd.), or (ii) the Company’s product at a price of 400 euro per unit (which represents the market price for the Lender);
|
|
|
·
|
in the event that the Company did not close a share exchange or similar transaction by June 1, 2011, the Lender had the right to extend the loan and its terms for up to an additional 6 months (as noted in Note 1, the Exchange Agreement was closed on March 31, 2011); and
|
|
|
·
|
in no event was cash required to be repaid by the Company.
|
|
|
On June 1, 2011, the Lender surrendered $100,000 of the convertible loan in exchange for 81,161 shares of common stock of the Company.
|
|
|
On February 20, 2011, the Company received a tax pre-ruling from the Israeli tax authorities according to section 103 of the Israeli tax law, with regards to the Share Exchange. According to the tax pre-ruling, the exchange of shares and options of InspireMD Ltd. For shares and options of the Company pursuant to the Share Exchange will not result in an immediate tax event for InspireMD Ltd.’s former shareholders, but a deferred tax event, subject to certain conditions as stipulated in the tax pre-ruling. The main condition of the tax pre-ruling is a restriction on the exchanged shares for two years from December 31, 2010 for shareholders holding over of 5%.
|
|
|
In March 2011, the Company granted a new fixed lien of $40,000 to Bank Mizrahi.
|
|
|
Pursuant to the Exchange Agreement, the Company assumed all of InspireMD Ltd.’s obligations under InspireMD Ltd.’s outstanding stock options. Immediately prior to the Share Exchange, InspireMD Ltd. had outstanding stock options to purchase an aggregate of 937,256 ordinary shares, which outstanding options became options to purchase an aggregate of 7,606,770 shares of common stock of the Company after giving effect to the Share Exchange. In addition, three-year warrants to purchase up to 125,000 ordinary shares of InspireMD Ltd. at an exercise price of $10 per share were assumed by the Company and converted into warrants to purchase 1,014,500 shares of the Company’s common stock at an exercise price of $1.23 per share.
|
|
|
In connection with the closing of the Share Exchange, the Company sold 6,454,002 shares of its common stock at a purchase price of $1.50 per share and five-year warrants to purchase up to 3,226,999 shares of common stock at an exercise price of $1.80 per share in a private placement to accredited investors (the “Private Placement”). As part of the Private Placement, certain holders of the 8% convertible debentures, in an aggregate principal amount of $1,580,000 (the “Bridge Notes”), surrendered $667,596 of outstanding principal and interest due under such Bridge Notes in exchange for 445,064 shares of common stock and warrants to purchase an aggregate of 225,532 shares of common stock (the “Debt Conversions”). The number of shares of common stock and warrants issued in connection with the Debt Conversions are included in the aggregate figures for the Private Placement.
|
F-43
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 6 - CERTAIN TRANSACTIONS (continued)
|
|
As a result, the Company received aggregate cash proceeds of $9,013,404 in the Private Placement. In addition, as a result of the Debt Conversions, there was $1,000,000 of unpaid principal outstanding under the Bridge Notes, which was repaid by the Company in May 2011.
|
In connection with the Private Placement, the Company paid placement agent fees of approximately $300,000 and issued five-year warrants to purchase 373,740 shares of the Company’s common stock at an exercise price of $1.80 per share to the placement agent for this Private Placement. The fair value of the warrants is $212,000.
|
|
In connection with the Share Exchange, the Company also entered into a stock escrow agreement with certain stockholders, pursuant to which these stockholders deposited 1,015,622 shares of common stock held by them into escrow. These shares will be released to the Company for cancellation or surrender to an entity designated by the Company should the Company have $10 million in consolidated revenue, as certified by the Company’s independent auditors, during the first 12 months following the closing of the Private Placement, yet fail, after a good faith effort, to have the Company’s common stock approved for listing on a national securities exchange. On the other hand, should the Company fail to record at least $10 million in consolidated revenue during the first 12 months following the closing of the Private Placement or have its common stock listed on a national securities exchange within 12 months following the closing on the Private Placement, these escrowed shares shall be released back to the stockholders.
|
|
|
The shares of the Company’s common stock issued to the InspireMD Ltd. shareholders in connection with the Exchange Agreement and the shares of common stock issued to the investors in the Private Placement were not registered under the Securities Act of 1933, as amended. These securities may not be offered or sold in the U.S. absent registration or an applicable exemption from the registration requirements. Certificates representing these shares contain a legend stating the restrictions applicable to such shares.
|
|
|
On March 31, 2011, the Company issued certain consultants five-year warrants to purchase up to an aggregate of 2,500,000 shares of common stock at an exercise price of $1.50 per share in consideration for consulting services related to the Share Exchange, which warrants have a fair value of $1,500,000. The expenses related to the issuance of the warrants are recorded as share-based compensation and treated as issuance costs.
|
On April 18, 2011, the Company issued 666,667 shares of its common stock and five-year warrants to purchase 333,333 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $1,000,000 in a private placement.
On April 18, 2011, the Company issued 283,334 shares of its common stock and five-year term warrants to purchase 141,667 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $425,000 in a private placement.
In connection with the above-referenced transactions from April 18, 2011, the Company paid placement agent fees of approximately $471,000 which were recorded as issuance costs and five-year term warrants to purchase 57,000 shares of the Company common stock at an exercise price of $1.80 per share to the placement agent in this private placement. The fair value of those warrants amounting to $67,000 is estimated using the Black-Scholes valuation model.
|
|
On April 21, 2011, the Company issued 33,333 shares of its common stock, and five-year term warrants to purchase 16,667 shares of the Company’s common stock at an exercise price of $1.80 per share, for an aggregate purchase price of $50,000 in a private placement.
|
F-44
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 6 - CERTAIN TRANSACTIONS (continued)
|
|
During the nine month period ended September 30, 2011, the Company entered into investor relations consulting agreements (the “Consulting Agreements”) with investor relations companies (the “Advisors”) to provide investor relations services. Pursuant to the Consulting Agreements, in addition to monthly fees in a range of $3,000 - $15,000, the Company issued to the Advisors:
|
|
|
·
|
a one-year warrant to purchase 81,161 shares of common stock of the Company at an exercise price of $1.23 per share, valued at $21,000
|
|
|
·
|
50,000 restricted shares of the Company’s common stock, valued at $62,000, and a five-year warrant to purchase 50,000 shares of common stock of the Company at an exercise price of $1.50 per share, valued at $30,000.
|
|
|
·
|
25,000 shares of the Company’s common stock, valued at $68,750.
|
|
|
The Company recorded share-based compensation expenses of $181,750 related to these issuances, during the nine month period ended September 30, 2011.
|
|
|
During the three month period ended June 30, 2011, the Company granted 1,087,225 stock options to employees and consultants at cash exercise prices of $1.23-$2.75 per share. The options had terms of five years. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 2.85-5 years in each year; expected volatility of 63%-71%; and risk-free interest rate of 0.19%-3.39%.
|
|
|
During the three month period ended September 30, 2011, the Company cancelled 200,000 stock options of an employee that had a cash exercise price of $2.75 per share, and in exchange, issued, to the same employee, 200,000 shares of stock options at cash exercise prices of $1.93 per share. The options had a term of five years, continuing from the original grant date. For accounting purposes, the above mentioned transaction was treated as a modification to the original grant. The Fair Value of the modification was $36,000 and used the following assumptions: dividend yield of 0% and expected term of 3-4 years in each year; expected volatility of 67%-70%; and risk-free interest rate of 0.33%-0.65%
|
|
|
In addition to the above mentioned stock option grant, the Company granted an additional 95,000 stock options to employees and consultants at cash exercise prices of $1.93-$2.00 per share during the three month period ended September 30, 2011. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 3-4 years in each year; expected volatility of 67%-70%; and risk-free interest rate of 0.33%-0.68%.
|
|
|
On March 28, 2011, the board of directors and stockholders of the Company adopted and approved the InspireMD, Inc. 2011 UMBRELLA Option Plan (the “Umbrella Plan”). Under the Umbrella Plan, the Company reserved 9,468,100 shares of the Company’s common stock as awards to the employees, consultants, and service providers to the Company and its subsidiaries and affiliates worldwide. At a special meeting of stockholders of the Company held on October 31, 2011, the stockholders approved an amendment to the Umbrella Plan to add an additional 5,531,900 shares of common stock.
|
|
|
The Umbrella Plan currently consists of three components, the primary plan document that governs all awards granted under the Umbrella Plan, and two appendices: (i) Appendix A, designated for the purpose of grants of stock options and restricted stock to Israeli employees, consultants, officers and other service providers and other non-U.S. employees, consultants, and service providers, and (ii) Appendix B, which is the 2011 U.S. Equity Incentive Plan, designated for the purpose of grants of stock options and restricted stock awards to U.S. employees, consultants, and service providers who are subject to the U.S. income tax.
|
F-45
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 6 - CERTAIN TRANSACTIONS (continued):
The Umbrella Plan is administered by the compensation committee of the board of directors. Unless terminated earlier by the board of directors, the Umbrella Plan will expire on March 27, 2021.
|
|
U.S. federal income tax consequences relating to the transactions described under the Umbrella Plan are set forth in Section 409A, which was added to the Internal Revenue Code of 1986, as amended (the “Code”) and treasury regulations in 2004 to regulate all types of deferred compensation. If the requirements of Section 409A of the Code are not satisfied, deferred compensation and earnings thereon will be subject to tax as it vests, plus an interest charge at the underpayment rate plus 1% and a 20% penalty tax. Certain stock options and certain types of restricted stock are subject to Section 409A of the Code.
|
|
|
Israel income tax consequences of awards of options under the Umbrella Plan is general and does not purport to be complete. Pursuant to the current Section 102 of the Ordinance, which came into effect on January 1, 2003, options may be granted through a trustee (i.e., Approved 102 Options) or not through a trustee (i.e., Unapproved 102 Options).
|
|
|
On July 11, 2011, the board of directors of the Company appointed a new director, (“Director A”), with a term expiring at the Company’s 2012 annual meeting of stockholders. In connection with his appointment, Director A was granted an option to purchase 1,000,000 shares of the Company’s common stock at an exercise price of $1.50 per share, (the “$1.50 Option”). The $1.50 Option was exercisable immediately and expired on September 30, 2011. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 0.11 years in each year; expected volatility of 53%; and risk-free interest rate of 0.17%.
|
|
|
In addition, in connection with his appointment, Director A was granted an option to purchase 500,000 shares of common stock at an exercise price of $2.50 per share, the closing price of the common stock on the date of grant (the “$2.50 Option”), subject to the terms and conditions of the 2011 U.S. Equity Incentive Plan, a sub-plan of the Company’s 2011 new Option Plan approved on March 28, 2011 (“2011 Umbrella Option Plan”). The $2.50 Option vests and becomes exercisable in three equal annual installments beginning on the one-year anniversary of the date of grant, provided that in the event that Director A is either (i) not reelected as a director at the Company’s 2012 annual meeting of stockholders, or (ii) not nominated for reelection as a director at the Company’s 2012 annual meeting of stockholders, the option vests and becomes exercisable on the date Director A fails to be reelected or nominated. The $2.50 Option has a term of 10 years from the date of grant. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 5.5-6 years in each year; expected volatility of 62%-63%; and risk-free interest rate of 1.67%-1.85%.
|
|
|
The fair value of the options granted to the above-mentioned new director, using the Black-Scholes option-pricing model was approximately $1,700,000.
|
|
|
On September 28, 2011, Director A, exercised the $1.50 Option to purchase 1,000,000 shares of common stock, resulting in gross proceeds to the Company of $1,500,000.
|
F-46
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 6 - CERTAIN TRANSACTIONS (continued):
|
|
On August 5, 2011 and effective August 8, 2011, the Board appointed another two new directors (“Director B” and “Director C”). Director B was appointed for with a term expiring at the Company’s 2012 annual meeting of stockholders and Director C was appointed for a term expiring at the Company’s 2013 annual meeting of stockholder. In connection with their appointment, the directors were each granted an option to purchase shares of Common Stock at an exercise price of $1.95 per share, the closing price of the Common Stock on the date of grant (the “$1.95 Options”). The grant to Director B was for 100,000 shares and is subject to the terms and conditions of the 2011 U.S. Equity Incentive Plan, a sub-plan of the Company’s 2011 Umbrella Option Plan. The grant to Director C was for 25,000 shares and is subject to the 2006 Employee Stock Option Plan, a sub-plan of the Company’s 2011 Umbrella Option Plan. The $1.95 Options vests and become exercisable in two equal annual installments beginning on the one-year anniversary of the date of grant. In the case of Director B’s option, in the event that the Director B is either (i) not reelected as a director at the Company’s 2012 annual meeting of stockholders, or (ii) not nominated for reelection as a director at the Company’s 2012 annual meeting of stockholders, the option vests and becomes exercisable on the date of Director B’s failure to be reelected or nominated. In the case of Director C’s option, in the event that Director C is required to resign from the Board due to medical reasons, the option vests and becomes exercisable on the date of Director C’s resignation for medical reasons. The $1.95 Options have terms of 10 years from the date of grant.
|
|
|
In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 3-4 years in each year; expected volatility of 67%-70%; and risk-free interest rate of 0.45%-0.78%.
|
|
|
The fair value of the options granted to the above-mentioned new directors, using the Black-Scholes option-pricing model is approximately $118,000.
|
|
|
In addition, on August 5, 2011, 324,644 stock options were granted to former directors at a cash exercise price of $1.23 per share replacing 324,644 stock options held by former directors that expired during the second quarter of 2011. The options had terms of five years. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 5 years in each year; expected volatility of 62%; and risk-free interest rate of 1.23%.
|
|
|
The fair value of the options granted to the above-mentioned former directors, using the Black-Scholes option-pricing model is approximately $445,000.
|
|
|
On July 20, 2011, Mizrahi Tefahot Bank approved the release of a fixed lien in the amount of $300,000. Following the approval, $300,000 of Restricted Cash was classified to cash and cash equivalents.
|
NOTE 7 - FAIR VALUE MEASUREMENT:
|
|
a.
|
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date.
|
The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
F-47
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 7 - FAIR VALUE MEASUREMENT (continued):
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
|
|
b.
|
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value. In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
|
|
|
The convertible loan was recorded at a fair value of $1,044 as of December 31, 2010, then subsequently remeasured at fair value with the increase in fair value of $624 included in the Consolidated Statements of Operations as of March 31, 2011. This security was measured at fair value on a recurring basis and classified in the "Significant Unobservable inputs (Level 3)" category.
|
|
|
c.
|
The carrying amounts of cash and cash equivalents, accounts receivable, accounts payable and other accrued liabilities approximate their fair value either because these amounts are presented at fair value or due to the relatively short-term maturities of such instruments. The carrying amount of the Group's other financial long-term assets and other financial long-term liabilities approximate their fair value.
|
NOTE 8 - INVENTORY ON HAND:
|
September 30
|
December 31,
|
|||||||
|
2011
|
2010
|
|||||||
|
($ in thousands)
|
||||||||
|
Finished goods
|
$ | 445 | $ | 957 | ||||
|
Work in process
|
1,222 | 573 | ||||||
|
Raw materials and supplies
|
238 | 174 | ||||||
| $ | 1,905 | $ | 1,704 | |||||
NOTE 9 - RELATED PARTIES TRANSACTIONS
In July 2010, the Company’s board of directors approved new agreements for the Company’s President and CEO. The agreements were approved at the Company’s shareholders meeting in March 2011, and are effective from April 1, 2011.
Pursuant to these agreements, the above mentioned executives are entitled to a set monthly fee, as well as a minimum bonus. If their employment is terminated without cause, they are entitled to at least six months’ prior notice and will be paid their monthly fee during such notice period. If their employment is terminated without cause, they will also be entitled to certain severance payments equal to the total amount that has been contributed to and accumulated in their severance payment funds. No further contributions are provided for by these agreements.
F-48
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 10 - COMMITMENT AND CONTINGENT LIABILITIES:
|
|
a.
|
Commitment
|
|
|
In March 2010, the Company entered into a license agreement to use a stent design (“MGuard Prime”). Pursuant to the agreement, the licensor is entitled to receive royalty payments of 7% of net sales outside the United States and, for sales within the United States, royalty payments as follows: 7% of net sales for the first $10,000,000 of net sales and 10% of net sales for net sales exceeding $10,000,000. The Company began manufacturing the MGuard Prime during the fourth quarter of 2010 and began selling the MGuard Prime in the first quarter of 2011.
|
|
|
b.
|
Litigation
|
|
|
The Company is a party to various claims arising in the ordinary course of its operations in the aggregate amount of $10,000. The Company has not recorded an expense related to damages in connection with these matters because management, after considering the views of its legal counsel as well as other factors, is of the opinion that a loss to the Company is neither probable nor is an amount or range of loss that is estimable.
|
In February 2011, representatives of a third party have indicated that they intend to seek damages from the Company in connection with certain finders’ fees that they claim are owed to them. The claimants’ demand was for approximately $1 million. The claimants’ most recent settlement demand, conveyed in April 2011, was for a total of $250,000 in cash and 250,000 shares of the company common stock. To date, no lawsuit has been filed and the Company has not accrued an expense in connection with this matter because the Company’s management, after considering the views of its legal counsel as well as other factors, is of the opinion a loss to the Company is neither probable nor is an amount or range of loss that is estimable.
In March 2009, a service provider submitted a claim against the Company in the amount of $150,000 in the Magistrate’s Court in Tel Aviv, claiming a success fee for assistance in locating potential investors and lenders with respect to a loan agreement entered into with a bank. On April 11, 2011, the Company received a court ruling directing the Company to pay the service provider an amount of $105,000. Since both parties had claims against the court ruling, they renegotiated and on June 5, 2011, signed a settlement agreement according to which the Company shall pay $96,000 and shall issue 18,785 shares of common stock valued at $51,000.
|
|
The Company has recorded an expense of $147,000 for the nine months ended September 30, 2011. The expenses have been recorded to “General and administrative” within the Condensed Consolidated Statements of Operations.
|
|
|
In November 2010, a former senior employee submitted a claim against the Company in the total amount of $430,000 and options to purchase 2,029,025 shares of the Company at an exercise price of $0.001 per share in the Magistrate’s Court in Tel Aviv, claiming unpaid back wages and commissions. The fair value of those options was valued using the Black-Scholes valuation model at $2.5 million as of the period he claimed to be entitled to the options. The Company’s management after considering the views of its legal counsel as well as other factors has recorded a provision of $20,000 in the financial statements in 2009 and is of the opinion an additional loss to the Company is neither probable nor is an amount or range of loss that is estimable.
|
F-49
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 10 - COMMITMENT AND CONTINGENT LIABILITIES (continued)
|
|
b.
|
Litigation
|
|
|
In November 2010, a former alleged founder and legal advisor of the Company submitted a claim against the Company for options to purchase 496,056 shares of the Company at an exercise price of $0.001 per share in the Magistrate’s Court in Tel Aviv. The fair value of those options was estimated using the Black-Scholes valuation model at $134,000 as of the grant date. It was during 2005 and 2006 that the Company first became aware of the events that gave rise to this litigation. Also, during this time, the Company had discussions with the plaintiffs on an informal basis. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a share-based compensation expense of $134,000 recorded in the year ended December 31, 2006, in respect of services allegedly provided in 2005 and 2006.
|
|
|
In November 2010, a former legal advisor of the Company submitted in the Magistrate’s Court in Tel Aviv a claim against the Company in the total amount of $53,000 due to a breach of employment promise. It was during 2005 and 2006 that the Company first became aware of the events that gave rise to this litigation. Also during this time, the Company had discussions with the plaintiff on an informal basis. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a provision in the amounting to $53,000 recorded in the year ended December 31, 2006. The Company, based upon the opinion of its legal counsel has recorded a provision of $53,000 allocated to the year ended December 31, 2006.
|
|
|
In February 2011, a finder submitted a claim against the Company in the amount of $327,000 in the Magistrate’s Court in Tel Aviv, claiming a future success fee and commission for assistance in finding the Company's distributer in Brazil. The Company’s management, after considering the views of its legal counsel as well as other factors, has recorded a provision of $327,000 in the financial statements in the first quarter of 2011. The related expense has been recorded to “General and administrative” within the Condensed Consolidated Statements of Operations. On October 5, 2011, the Company filed a counter claim against the plaintiff in the amount of $29,000.
|
|
|
In August 2011, a former senior employee submitted to the Regional Labor Court in Tel Aviv a claim against the Company for (i) a compensation of $118,000; and (ii) a declaratory ruling that he is entitled to exercise 486,966 options to purchase the Company’s shares of common stock at an exercise price of $0.001 per option. After consulting with its legal advisor the Company is unable to assess the probable outcome of this claim.
|
NOTE 11 - TAXES ON INCOME
Amendment of the Law for the Encouragement of Capital Investments, 1959
The Law for Encouragement of Capital Investments, 1959 (the “Law”) was amended as part of the Economic Policy Law for the years 2011-2012, which was passed in the Knesset (the Israeli parliament) on December 29, 2010 (the “amendment”). The amendment became effective January 1, 2011.
The amendment sets alternative benefit tracks to the ones currently in place under the provisions of the Law, as follows: investment grants track designed for enterprises located in national development zone A and two new tax benefits tracks (preferred enterprise and a special preferred enterprise), which provide for application of a unified tax rate to all preferred income of the company, as defined in the amendment.
F-50
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 11 - TAXES ON INCOME (continued):
The tax rates at company level, under the Law:
|
Years
|
Development Zone A
|
Other Areas in Israel
|
|||||||
|
"Preferred enterprise":
|
|||||||||
| 2011-2012 | 10 | % | 15 | % | |||||
| 2013-2014 | 7 | % | 12.5 | % | |||||
|
2015 and thereafter
|
6 | % | 12 | % | |||||
|
"Special Preferred Enterprise"
|
|||||||||
|
commencing 2011
|
5 | % | 8 | % | |||||
The benefits granted to the preferred enterprises will be unlimited in time, unlike the benefits granted to special preferred enterprises, which will be limited for a period of 10 years. The benefits shall be granted to companies that will qualify under criteria set in the amendment; for the most part, those criteria are similar to the criteria that were set in the law prior to its amendment.
Under the transitional provisions of the amendment, a company will be allowed to continue and enjoy the tax benefits available under the Law prior to its amendment until the end of the period of benefits, as defined in the Law. The Company will be allowed to set the "year of election" no later than tax year 2012, provided that the minimum qualifying investment commenced not later than the end of 2010. On each year during the period of benefits, the Company will be able to opt for application of the amendment, thereby making available to itself the tax rates as above. A company may not revoke it election for application of the Amendment.
In accordance with income taxes (Topic 740) the measurement of current and deferred tax liabilities and assets is based on provisions of the enacted tax law at balance sheet date. The amendment was "enacted" at the first quarter of 2011 and did not have an impact on the Company’s consolidated financial statements.
F-51
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 12 - ENTITY WIDE DISCLOSURE
The Company operates in one reportable segment.
Disaggregated financial data is provided below as follows:
(1) Revenues by geographic area and
(2) Revenues from principal customers.
Revenues are attributed to geographic areas based on the location of the customers. The following is a summary of revenues by geographic areas:
|
3 months ended
|
9 months ended
|
Year ended
|
||||||||||||||||||
|
September 30
|
September 30
|
December 31,
|
||||||||||||||||||
|
2011
|
2010
|
2011
|
2010
|
2010
|
||||||||||||||||
|
($ in thousands)
|
||||||||||||||||||||
|
Israel
|
$ | 124 | $ | 109 | $ | 479 | $ | 109 | $ | 119 | ||||||||||
|
Spain
|
233 | 122 | 523 | 308 | 343 | |||||||||||||||
|
Germany
|
119 | 428 | 257 | 467 | 497 | |||||||||||||||
|
India
|
- | - | 1,083 | - | - | |||||||||||||||
|
Brazil
|
204 | - | 312 | 360 | 360 | |||||||||||||||
|
Poland
|
- | - | 74 | 1,446 | 1,446 | |||||||||||||||
|
Argentina
|
234 | 60 | 353 | 115 | 150 | |||||||||||||||
|
Other
|
1,072 | 504 | 1,631 | 1,423 | 2,034 | |||||||||||||||
| $ | 1,986 | $ | 1,223 | $ | 4,712 | $ | 4,228 | $ | 4,949 | |||||||||||
By principal customers:
|
3 months ended
|
9 months ended
|
Year ended
|
||||||||||||||||||
|
September 30
|
September 30
|
December 31,
|
||||||||||||||||||
|
2011
|
2010
|
2011
|
2010
|
2010
|
||||||||||||||||
|
Customer A
|
6 | % | 9 | % | 10 | % | 3 | % | 2 | % | ||||||||||
|
Customer B
|
12 | % | 10 | % | 11 | % | 7 | % | 7 | % | ||||||||||
|
Customer C
|
6 | % | 35 | % | 5 | % | 11 | % | 10 | % | ||||||||||
|
Customer D
|
- | - | 23 | % | - | - | ||||||||||||||
|
Customer E
|
10 | % | - | 7 | % | 9 | % | 7 | % | |||||||||||
|
Customer F
|
- | - | 2 | % | 34 | % | 29 | % | ||||||||||||
|
Customer G
|
12 | % | 5 | % | 7 | % | 3 | % | 3 | % | ||||||||||
|
|
All tangible long lived assets are located in Israel.
|
NOTE 13 - SUBSEQUENT EVENTS
On October 31, 2011, the stockholders approved the authorization of the board of directors, in its discretion, to amend the Amended and Restated Certificate of Incorporation of the Company to effect a reverse stock split of the Company’s common stock at a ratio of one-for-two to one-for-four, such ratio to be determined by the board of directors (the “Reverse Stock Split”), which approval will allow the board of directors to effect the Reverse Stock Split any time prior to the Company’s annual meeting of stockholders in 2012.
F-52
INSPIREMD, INC.
(FORMERLY SAGUARO RESOURCES, INC.)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(UNAUDITED)
NOTE 13 - SUBSEQUENT EVENTS (continued)
On October 31, 2011, the stockholders approved the amendment of the Umbrella Plan to increase the number of shares of the Company’s common stock available for issuance pursuant to awards under the Umbrella Plan by 5,531,900 shares to a total of 15,000,000 shares, which would provide an incentive to attract and retain employees, consultants, and service providers.
On November 13th, 2011, a previous finder of InspireMD Ltd. (the "Subsidiary") submitted to the Magister Court in Tel Aviv a claim against the Company , the Subsidiary and the Company’s President and CEO for a declaratory ruling that it is entitled to convert 13,650 options to purchase the Subsidiary’s ordinary shares in an exercise price of USD3.67 per option into 110,785 of the Company's common stock at an exercise price of $0.45 per option, and to convert 4,816 options to purchase the Subsidiary’s ordinary shares in an exercise price of USD10 per option into 39,087 of common stock at an exercise price of $1.23 per option. The statement of claims in such proceedings was served to the Company today, the 14th of November, 2011. After consulting with its legal advisor and due to the lack of time to review all the materials related to this claim the Company is unable to assess the probable outcome of this claim.
On November 16, 2011 the Company’s board of directors approved the appointment of Mr. Sol J. Barer (Director A) as the chairman of the board of directors. In connection with his appointment as chairman of the board of directors, the Company issued Director A 2,900,000 shares of common stock and 2,900,000 stock options to purchase shares of Common Stock at an exercise price of $1.95 per share, the closing price of the Common Stock on the date of grant. The fair value of the above granted shares is approximately $5.7 million and will be recorded as an expense in the financial statements ended December 31, 2011. In calculating the fair value of options granted under share-based remuneration arrangements the Company used the following assumptions: dividend yield of 0% and expected term of 5.5 years in each year; expected volatility of 61.6%; and risk-free interest rate of 1.07%. The options have terms of 10 years from the date of grant, and the vesting terms are as follows: tranche A vests and become exercisable in twenty four equal monthly installments, tranches B and C - vests and become exercisable upon meeting certain performance conditions. The fair value of the options granted above, using the Black-Scholes option-pricing model was approximately $3.1 million.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
We are paying all of the selling stockholders’ expenses related to this offering, except that the selling stockholders will pay any applicable underwriting discounts and commissions. The fees and expenses payable by us in connection with this Registration Statement are estimated as follows:
|
SEC Registration Fee
|
$ | 126.22 | ||
|
Accounting Fees and Expenses
|
50,000.00 | |||
|
Legal Fees and Expenses
|
70,000.00
|
|||
|
Miscellaneous Fees and Expenses
|
9,873.78 | |||
|
Total
|
$ |
130,000.00
|
Item 14. Indemnification of Directors and Officers.
Section 145 of the General Corporation Law of the State of Delaware provides, in general, that a corporation incorporated under the laws of the State of Delaware, as we are, may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (other than a derivative action by or in the right of the corporation) by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe such person’s conduct was unlawful. In the case of a derivative action, a Delaware corporation may indemnify any such person against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification will be made in respect of any claim, issue or matter as to which such person will have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery of the State of Delaware or any other court in which such action was brought determines such person is fairly and reasonably entitled to indemnity for such expenses.
Our certificate of incorporation and bylaws provide that we will indemnify our directors, officers, employees and agents to the extent and in the manner permitted by the provisions of the General Corporation Law of the State of Delaware, as amended from time to time, subject to any permissible expansion or limitation of such indemnification, as may be set forth in any stockholders’ or directors’ resolution or by contract. Any repeal or modification of these provisions approved by our stockholders will be prospective only and will not adversely affect any limitation on the liability of any of our directors or officers existing as of the time of such repeal or modification.
We are also permitted to apply for insurance on behalf of any director, officer, employee or other agent for liability arising out of his actions, whether or not the General Corporation Law of the State of Delaware would permit indemnification.
Item 15. Recent Sales of Unregistered Securities.
On June 16, 2008, we completed an offering of 2,500,000 shares of our common stock at a price of $0.005 per share to Lynn Briggs, our former president, chief executive officer, chief financial officer, secretary and treasurer. The total amount received from that offering was $12,500. These shares were issued pursuant to Section 4(2) of the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering.
On March 31, 2011, pursuant to a share exchange agreement, we issued 46,471,907 shares of common stock to certain shareholders of InspireMD Ltd. in exchange for 91.7% of the issued and outstanding capital stock of InspireMD Ltd. Separately, we issued 4,194,756 shares of common stock to the remaining shareholders of InspireMD Ltd. in exchange for the remaining 8.3% of the issued and outstanding capital stock of InspireMD Ltd. In addition, in connection with the share exchange agreement, we (i) assumed three year warrants to purchase up to 125,000 ordinary shares of InspireMD Ltd. at an exercise price of $10 per share that were converted into newly issued warrants to purchase up to 1,014,500 shares of our common stock at an exercise price of $1.23 per share and (ii) options to purchase up to 937,256 ordinary shares of InspireMD Ltd. with a weighted average exercise price of $4.35 that were converted into options to purchase up to 7,606,770 shares of our common stock with a weighted average exercise price of $0.54 per share. The securities issued in the above described transactions were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold pursuant to the exemption from registration under the Securities Act of 1933, as amended, provided by either Regulation S under the Securities Act of 1933, as amended, or Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended. Each of the shareholders of InspireMD Ltd. who received shares of our common stock in the above described share exchange transactions were either accredited investors (as defined by Rule 501 under the Securities Act of 1933, as amended) or not a “U.S. person” (as that term is defined in Rule 902 of Regulation S) at the time of the share exchange transaction.
On March 31, 2011, we entered into a securities purchase agreement with 30 accredited investors (as defined by Rule 501 under the Securities Act of 1933, as amended), pursuant to which we issued 6,454,002 shares of common stock and five-year warrants to purchase up to 3,226,999 shares of common stock at an exercise price of $1.80 per share for aggregate cash proceeds of $9,013,404 and the cancellation of $667,596 of indebtedness held by investors. The securities sold in this offering were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration under the Securities Act of 1933, as amended, provided by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended.
On March 31, 2011, upon the consummation of the above described private placement, we issued a five-year warrant to purchase up to 373,740 shares of common stock at an exercise price of $1.80 per share, to Palladium Capital Advisors, LLC, our placement agent in the private placement. The warrant was not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and was offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering. Palladium Capital Advisors, LLC was an accredited investor (as defined by Rule 501 under the Securities Act of 1933, as amended) at the time of the private placement.
On March 31, 2011, for work performed in connection with the share exchange transactions and as bonus compensation, we issued Craig Shore, our chief financial officer, secretary and treasurer, a five-year warrant to purchase up to 3,000 shares of common stock at an exercise price of $1.80 per share. The warrant was not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and was offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering. Craig Shore was an accredited investor (as defined by Rule 501 under the Securities Act of 1933, as amended) at the time of the issuance of the warrant.
On March 31, 2011, upon the consummation of the private placement, we issued a five-year warrant to purchase up to 6,667 shares of common stock at an exercise price of $1.80 per share, to Hermitage Capital Management, a consultant. The warrant was not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and was offered and sold in reliance on the exemption from registration afforded by Section 4(2) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering.
In consideration for financial consulting services, we issued to The Benchmark Company, LLC, a consultant, a five-year warrant to purchase up to 50,000 shares of common stock at an exercise price of $1.50 per share. The warrant was not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and was offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering.
On March 31, 2011, we issued five-year warrants to purchase up to an aggregate of 2,500,000 shares of common stock at an exercise price of $1.50 per share, to Endicott Management Partners, LLC, The Corbran LLC and David Stefansky, in consideration for consulting services. The warrants were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering. Each of Endicott Management Partners, LLC, The Corbran LLC and David Stefansky was an accredited investor (as defined by Rule 501 under the Securities Act of 1933, as amended) at the time of the issuance of the warrant.
On April 18, 2011, we consummated a private placement with an investor pursuant to which we sold 666,667 shares of our common stock and a five-year warrant to purchase up to 333,333 shares of common stock at an exercise price of $1.80 per share for aggregate cash proceeds of $1,000,000. The securities sold in this offering were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration under the Securities Act of 1933, as amended, provided by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended. This investor was an accredited investor (as defined by Rule 501 under the Securities Act of 1933, as amended) at the time of the private placement.
On April 18, 2011, we consummated a private placement with 2 accredited investors (as defined by Rule 501 under the Securities Act of 1933, as amended), pursuant to which we sold 283,334 shares of our common stock and a five-year warrant to purchase 141,667 shares of our common stock at an exercise price of $1.80 per share, for aggregate cash proceeds of $425,000. The securities sold in this offering were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration under the Securities Act of 1933, as amended, provided by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended.
On April 18, 2011, upon the consummation of the above described April 18, 2011 private placements, we issued a five-year warrant to purchase up to 57,000 shares of common stock at an exercise price of $1.80 per share to Palladium Capital Advisors, LLC, our placement agent in the April 18, 2011 private placements. The warrant was not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and was offered and sold in reliance on the exemption from registration afforded by Section 4(2) and Regulation D (Rule 506) under the Securities Act of 1933, as amended, and corresponding provisions of state securities laws, which exempt transactions by an issuer not involving a public offering. Palladium Capital Advisors, LLC was an accredited investor (as defined by Rule 501 under the Securities Act of 1933, as amended) at the time of the private placement
On April 21, 2011, we consummated a private placement with Mr. Reinder Hogeboom pursuant to which we sold 33,333 shares of our common stock and a five-year warrant to purchase 16,667 shares of our common stock at an exercise price of $1.80 per share, for aggregate cash proceeds of $50,000. The securities sold in this offering were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration under the Securities Act of 1933, as amended, provided by Regulation S under the Securities Act of 1933, as amended. Reinder Hogeboom was not a “U.S. person” (as that term is defined in Rule 902 of Regulation S) at the time of the private placement.
On January 4, 2011, we entered into a convertible loan agreement with our distributer in Israel, in the amount of $100,000. On June 1, 2011, we issued 81,161 shares of common stock to the lender upon conversion of the note. These securities were not registered under the Securities Act of 1933, as amended, or the securities laws of any state, and were offered and sold in reliance on the exemption from registration under the Securities Act of 1933, as amended, provided by Regulation S under the Securities Act of 1933, as amended. The lender was not a “U.S. person” (as that term is defined in Rule 902 of Regulation S) at the time of the issuance.
Item 16. Exhibits and Financial Statement Schedules.
|
Exhibit No.
|
Description
|
|
2.1
|
Share Exchange Agreement, dated as of December 29, 2010, by and among InspireMD Ltd., Saguaro Resources, Inc., and the Shareholders of InspireMD Ltd. that are signatory thereto (incorporated by reference to Exhibit 10.1 to Saguaro Resources, Inc. Current Report on Form 8-K filed with the Securities and Exchange Commission on January 5, 2011)
|
|
2.2
|
Amendment to Share Exchange Agreement, dated February 24, 2011 (incorporated by reference to Exhibit 2.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
2.3
|
Second Amendment to Share Exchange Agreement, dated March 25, 2011 (incorporated by reference to Exhibit 2.3 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
3.1
|
Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 1, 2011)
|
|
3.2
|
Amended and Restated Bylaws (incorporated by reference to Exhibit 3.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 1, 2011)
|
|
5.1**
|
Opinion of Haynes and Boone, LLP.
|
|
10.1
|
Amended and Restated 2011 Umbrella Option Plan (incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 4, 2011)
|
|
10.2
|
Form of Stock Option Award Agreement (incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.3
|
Agreement of Conveyance, Transfer and Assignment of Assets and Assumption of Obligations, dated as of March 31, 2011 (incorporated by reference to Exhibit 10.3 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.4
|
Stock Purchase Agreement, by and between InspireMD, Inc. and Lynn Briggs, dated as of March 31, 2011 (incorporated by reference to Exhibit 10.4 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.5**
|
Securities Purchase Agreement, dated as of March 31, 2011, by and among InspireMD, Inc. and certain purchasers set forth therein
|
|
10.6
|
Form of $1.80 Warrant (incorporated by reference to Exhibit 10.6 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.7
|
Form of $1.23 Warrant (incorporated by reference to Exhibit 10.7 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.8
|
$1,250,000 Convertible Debenture, dated July 20, 2010, by and between InspireMD Ltd. and Genesis Asset Opportunity Fund, L.P. (incorporated by reference to Exhibit 10.8 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.9
|
Unprotected Leasing Agreement, dated February 22, 2007, by and between Block 7093 Parcel 162 Company Ltd. Private Company 510583156 and InspireMD Ltd. (incorporated by reference to Exhibit 10.9 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.10**
|
Securities Purchase Agreement, dated as of July 22, 2010, by and among InspireMD Ltd. and certain purchasers set forth therein
|
|
10.11**
|
Manufacturing Agreement, by and between InspireMD Ltd. and QualiMed Innovative Medizinprodukte GmbH, dated as of September 11, 2007
|
|
10.12**
|
Development Agreement, by and between InspireMD Ltd. and QualiMed Innovative Medizinprodukte GmbH, dated as of January 15, 2007
|
|
10.13**
|
License Agreement, by and between Svelte Medical Systems, Inc. and InspireMD Ltd., dated as of March 19, 2010
|
|
10.14
|
Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of April 1, 2005 (incorporated by reference to Exhibit 10.14 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.15
|
Amendment to the Employment Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of October 1, 2008 (incorporated by reference to Exhibit 10.15 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.16
|
Second Amendment to the Employment Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of March 28, 2011 (incorporated by reference to Exhibit 10.16 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.17
|
Personal Employment Agreement, by and between InspireMD Ltd. and Asher Holzer, dated as of April 1, 2005 (incorporated by reference to Exhibit 10.17 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.18
|
Amendment to the Employment Agreement, by and between InspireMD Ltd. and Asher Holzer, dated as of March 28, 2011 (incorporated by reference to Exhibit 10.18 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.19
|
Personal Employment Agreement, by and between InspireMD Ltd. and Eli Bar, dated as of June 26, 2005 (incorporated by reference to Exhibit 10.19 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.20
|
Employment Agreement, by and between InspireMD Ltd. and Bary Oren, dated as of August 25, 2009 (incorporated by reference to Exhibit 10.20 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.21
|
Employment Agreement, by and between InspireMD Ltd. and Craig Shore, dated as of November 28, 2010 (incorporated by reference to Exhibit 10.21 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.22**
|
Form of Indemnification Agreement between InspireMD, Inc. and each of the directors and executive officers thereof
|
|
10.23
|
Agreement with Bank Mizrahi Tefahot LTD. for a loan to InspireMD Ltd. in the original principal amount of $750,000 (incorporated by reference to Exhibit 10.23 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.24
|
Securities Purchase Agreement, dated as of April 18, 2011, by and among InspireMD, Inc. and certain purchasers set forth therein (incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 22, 2011)
|
|
10.25
|
Form of Warrant (incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 22, 2011)
|
|
10.26**
|
Agreement by and between InspireMD Ltd. and MeKo Laser Material Processing, dated as of April 15, 2010
|
|
10.27**
|
Agreement by and between InspireMD Ltd. and Natec Medical Ltd, dated as of September 23, 2009
|
|
10.28*
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Hand-Prod Sp. Z o.o, dated as of December 10, 2007
|
|
10.29**
|
Factoring Agreement by and between InspireMD Ltd. and Bank Mizrahi Tefahot Ltd., dated as of February 22, 2011
|
|
10.30
|
$1.50 Nonqualified Stock Option Agreement, dated as of July 11, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2011)
|
|
10.31
|
$2.50 Nonqualified Stock Option Agreement, dated as of July 11, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2011)
|
|
10.32
|
$1.95 Nonqualified Stock Option Agreement, dated as of August 5, 2011, by and between InspireMD, Inc. and Paul Stuka (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on August 11, 2011)
|
|
10.33
|
$1.95 Nonqualified Stock Option Agreement, dated as of August 5, 2011, by and between InspireMD, Inc. and Eyal Weinstein (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on August 11, 2011)
|
|
10.34**
|
Consultancy Agreement, dated as of April 1, 2011, by and between InspireMD Ltd. and Ofir Paz
|
|
10.35**
|
Consultancy Agreement, dated as of April 29, 2011, by and between InspireMD Ltd. and Asher Holzer
|
|
10.36**
|
Exclusive Distribution Agreement by and between InspireMD GmbH. and IZASA Distribuciones Tecnicas SA, dated as of May 20, 2009
|
|
10.37**
|
Amendment to the Distribution Agreement by and between InspireMD GmbH. and IZASA Distribuciones Tecnicas SA, dated as of February 2011
|
|
10.38**
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Tzamal-Jacobsohn Ltd., dated as of December 24, 2008
|
|
10.39**
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Kirloskar Technologies (P) Ltd., dated as of May 13, 2010
|
|
10.40**
|
Consultancy Agreement by and between InspireMD Ltd. and Sara Paz, dated as of May 6, 2008
|
|
10.41**
|
Consultancy Agreement by and between InspireMD Ltd. and Sara Paz Management and Marketing Ltd., dated as of September 1, 2011
|
|
10.42
|
Clinical Trial Services Agreement, dated as of October 4, 2011, by and between InspireMD Ltd. and Harvard Clinical Research Institute, Inc. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on October 11, 2011)
|
|
10.43*
|
Letter Agreement by and between InspireMD Ltd. and Tzamal-Jacobsohn Ltd., dated as of May 9, 2011.
|
|
10.44
|
Stock Award Agreement, dated as of November 16, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 18, 2011)
|
|
10.45
|
Nonqualified Stock Option Agreement, dated as of November 16, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 18, 2011)
|
|
21.1
|
List of Subsidiaries (incorporated by reference to Exhibit 21.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
23.1*
|
Consent of Kesselman & Kesselman, Certified Public Accountants
|
|
23.2**
|
Consent of Haynes and Boone, LLP (included in Exhibit 5.1)
|
_______________________
* Filed herewith.
** Previously filed.
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of the securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered that remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability of the undersigned registrant under the Securities Act to any purchaser in the initial distribution of the securities:
The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424 (§ 230.424 of this chapter);
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable.
In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
For the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (§ 230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Tel Aviv, State of Israel on December 1, 2011.
|
|
||
|
By:
|
/s/ Ofir Paz | |
|
Name: Ofir Paz
|
||
|
Title: Chief Executive Officer
|
||
In accordance with the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
| /s/ Ofir Paz |
Chief Executive Officer and Director
|
December 1, 2011
|
||
|
Ofir Paz
|
(principal executive officer)
|
|||
| * |
President and Chairman of the Board of Directors
|
December 1, 2011
|
||
|
Asher Holzer
|
||||
| * |
Chief Financial Officer, Secretary and Treasurer
|
December 1, 2011
|
||
|
Craig Shore
|
(principal financial and accounting officer)
|
|||
| * |
Director
|
December 1, 2011
|
||
|
Sol Barer
|
||||
| * |
Director
|
December 1, 2011
|
||
|
Paul Stuka
|
||||
| * |
Director
|
December 1, 2011
|
||
|
Eyal Weinstein
|
* Signed by Ofir Paz as agent.
EXHIBIT INDEX
|
Exhibit No.
|
Description
|
|
2.1
|
Share Exchange Agreement, dated as of December 29, 2010, by and among InspireMD Ltd., Saguaro Resources, Inc., and the Shareholders of InspireMD Ltd. that are signatory thereto (incorporated by reference to Exhibit 10.1 to Saguaro Resources, Inc. Current Report on Form 8-K filed with the Securities and Exchange Commission on January 5, 2011)
|
|
2.2
|
Amendment to Share Exchange Agreement, dated February 24, 2011 (incorporated by reference to Exhibit 2.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
2.3
|
Second Amendment to Share Exchange Agreement, dated March 25, 2011 (incorporated by reference to Exhibit 2.3 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
3.1
|
Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 1, 2011)
|
|
3.2
|
Amended and Restated Bylaws (incorporated by reference to Exhibit 3.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 1, 2011)
|
|
5.1**
|
Opinion of Haynes and Boone, LLP.
|
|
10.1
|
Amended and Restated 2011 Umbrella Option Plan (incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 4, 2011)
|
|
10.2
|
Form of Stock Option Award Agreement (incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.3
|
Agreement of Conveyance, Transfer and Assignment of Assets and Assumption of Obligations, dated as of March 31, 2011 (incorporated by reference to Exhibit 10.3 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.4
|
Stock Purchase Agreement, by and between InspireMD, Inc. and Lynn Briggs, dated as of March 31, 2011 (incorporated by reference to Exhibit 10.4 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.5**
|
Securities Purchase Agreement, dated as of March 31, 2011, by and among InspireMD, Inc. and certain purchasers set forth therein
|
|
10.6
|
Form of $1.80 Warrant (incorporated by reference to Exhibit 10.6 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.7
|
Form of $1.23 Warrant (incorporated by reference to Exhibit 10.7 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.8
|
$1,250,000 Convertible Debenture, dated July 20, 2010, by and between InspireMD Ltd. and Genesis Asset Opportunity Fund, L.P. (incorporated by reference to Exhibit 10.8 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.9
|
Unprotected Leasing Agreement, dated February 22, 2007, by and between Block 7093 Parcel 162 Company Ltd. Private Company 510583156 and InspireMD Ltd. (incorporated by reference to Exhibit 10.9 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.10**
|
Securities Purchase Agreement, dated as of July 22, 2010, by and among InspireMD Ltd. and certain purchasers set forth therein
|
|
10.11**
|
Manufacturing Agreement, by and between InspireMD Ltd. and QualiMed Innovative Medizinprodukte GmbH, dated as of September 11, 2007
|
|
10.12**
|
Development Agreement, by and between InspireMD Ltd. and QualiMed Innovative Medizinprodukte GmbH, dated as of January 15, 2007
|
|
10.13**
|
License Agreement, by and between Svelte Medical Systems, Inc. and InspireMD Ltd., dated as of March 19, 2010
|
|
10.14
|
Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of April 1, 2005 (incorporated by reference to Exhibit 10.14 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.15
|
Amendment to the Employment Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of October 1, 2008 (incorporated by reference to Exhibit 10.15 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.16
|
Second Amendment to the Employment Agreement, by and between InspireMD Ltd. and Ofir Paz, dated as of March 28, 2011 (incorporated by reference to Exhibit 10.16 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.17
|
Personal Employment Agreement, by and between InspireMD Ltd. and Asher Holzer, dated as of April 1, 2005 (incorporated by reference to Exhibit 10.17 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.18
|
Amendment to the Employment Agreement, by and between InspireMD Ltd. and Asher Holzer, dated as of March 28, 2011 (incorporated by reference to Exhibit 10.18 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.19
|
Personal Employment Agreement, by and between InspireMD Ltd. and Eli Bar, dated as of June 26, 2005 (incorporated by reference to Exhibit 10.19 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.20
|
Employment Agreement, by and between InspireMD Ltd. and Bary Oren, dated as of August 25, 2009 (incorporated by reference to Exhibit 10.20 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.21
|
Employment Agreement, by and between InspireMD Ltd. and Craig Shore, dated as of November 28, 2010 (incorporated by reference to Exhibit 10.21 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.22**
|
Form of Indemnification Agreement between InspireMD, Inc. and each of the directors and executive officers thereof
|
|
10.23
|
Agreement with Bank Mizrahi Tefahot LTD. for a loan to InspireMD Ltd. in the original principal amount of $750,000 (incorporated by reference to Exhibit 10.23 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
10.24
|
Securities Purchase Agreement, dated as of April 18, 2011, by and among InspireMD, Inc. and certain purchasers set forth therein (incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 22, 2011)
|
|
10.25
|
Form of Warrant (incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 22, 2011)
|
|
10.26**
|
Agreement by and between InspireMD Ltd. and MeKo Laser Material Processing, dated as of April 15, 2010
|
|
10.27**
|
Agreement by and between InspireMD Ltd. and Natec Medical Ltd, dated as of September 23, 2009
|
|
10.28*
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Hand-Prod Sp. Z o.o, dated as of December 10, 2007
|
|
10.29**
|
Factoring Agreement by and between InspireMD Ltd. and Bank Mizrahi Tefahot Ltd., dated as of February 22, 2011
|
|
10.30
|
$1.50 Nonqualified Stock Option Agreement, dated as of July 11, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2011)
|
|
10.31
|
$2.50 Nonqualified Stock Option Agreement, dated as of July 11, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on July 15, 2011)
|
|
10.32
|
$1.95 Nonqualified Stock Option Agreement, dated as of August 5, 2011, by and between InspireMD, Inc. and Paul Stuka (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on August 11, 2011)
|
|
10.33
|
$1.95 Nonqualified Stock Option Agreement, dated as of August 5, 2011, by and between InspireMD, Inc. and Eyal Weinstein (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on August 11, 2011)
|
|
10.34**
|
Consultancy Agreement, dated as of April 1, 2011, by and between InspireMD Ltd. and Ofir Paz
|
|
10.35**
|
Consultancy Agreement, dated as of April 29, 2011, by and between InspireMD Ltd. and Asher Holzer
|
|
10.36**
|
Exclusive Distribution Agreement by and between InspireMD GmbH. and IZASA Distribuciones Tecnicas SA, dated as of May 20, 2009
|
|
10.37**
|
Amendment to the Distribution Agreement by and between InspireMD GmbH. and IZASA Distribuciones Tecnicas SA, dated as of February 2011
|
|
10.38**
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Tzamal-Jacobsohn Ltd., dated as of December 24, 2008
|
|
10.39**
|
Exclusive Distribution Agreement by and between InspireMD Ltd. and Kirloskar Technologies (P) Ltd., dated as of May 13, 2010
|
|
10.40**
|
Consultancy Agreement by and between InspireMD Ltd. and Sara Paz, dated as of May 6, 2008
|
|
10.41**
|
Consultancy Agreement by and between InspireMD Ltd. and Sara Paz Management and Marketing Ltd., dated as of September 1, 2011
|
|
10.42
|
Clinical Trial Services Agreement, dated as of October 4, 2011, by and between InspireMD Ltd. and Harvard Clinical Research Institute, Inc. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on October 11, 2011)
|
|
10.43*
|
Letter Agreement by and between InspireMD Ltd. and Tzamal-Jacobsohn Ltd., dated as of May 9, 2011.
|
|
10.44
|
Stock Award Agreement, dated as of November 16, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 18, 2011)
|
|
10.45
|
Nonqualified Stock Option Agreement, dated as of November 16, 2011, by and between InspireMD, Inc. and Sol J. Barer, Ph.D. (Incorporated by reference to Exhibit 10.2 to Current Report on Form 8-K filed with the Securities and Exchange Commission on November 18, 2011)
|
|
21.1
|
List of Subsidiaries (incorporated by reference to Exhibit 21.1 to Current Report on Form 8-K filed with the Securities and Exchange Commission on April 6, 2011)
|
|
23.1*
|
Consent of Kesselman & Kesselman, Certified Public Accountants
|
|
23.2**
|
Consent of Haynes and Boone, LLP (included in Exhibit 5.1)
|
_______________________
* Filed herewith.
** Previously filed.